Unraveling the Nature of the Oxide−Metal Interaction in Ceria-Based Noble Metal Inverse Catalysts Jesú s Graciani,* ,† Alba B. Vidal, ‡,§ Jose ́ A. Rodriguez, ‡ and Javier Fdez. Sanz † † Departamento de Química Física, Universidad de Sevilla, 41012 Sevilla, Spain ‡ Chemistry Department, Brookhaven National Laboratory, P.O. Box 5000, Upton, New York 11973-5000, United States ABSTRACT: Recently, it has been shown that model catalysts consisting of cerium oxide nanoparticles supported on noble metal surfaces are very convenient to clarify the reaction mechanism of important industrial reactions such as water−gas shift, CO oxidation, and methanol synthesis from CO 2 . Moreover, these special inverse catalysts are highly active, even more than the regular metal/oxide catalyst in some cases. However, there is a lack of fundamental knowledge about the electronic nature of the supported CeO x nanoparticles (NPs) and their interaction with the noble metal support. In this work we have carried out DFT calculations on CeO x /Cu(111), CeO x /Ag(111), and CeO x /Au(111) interfaces for both supported particles and periodic 2D film. The most stable and most reduced CeO x NPs are found when supported on Cu(111), followed by Ag(111) and Au(111). A similar behavior is observed for periodic 2D CeO 2 (111) films supported on the M(111) surfaces, although the reducibility is lower and the stability higher in extended films than in small particles. The charge transfer contribution from the support dominates the strength of the oxide(film)−metal interaction following the sequence Cu > Ag > Au. The high stability and reducibility of CeO x on Cu(111) and Ag(111) and the lower stability and reducibility on Au(111) are rationalized on the grounds of the metal−oxide/metal charge transfer, the electronic structure of the system, work function of the metal surfaces, structural mismatch, and the nature of the interface bonding. I. INTRODUCTION Inverse catalysts consisting of oxide nanoparticles (NPs) supported on metal surfaces have been used as model systems in which the reaction environment can be easily controlled and the arbitrary, complex, and undetermined phenomena that occur in the real powder catalyst can be avoided. In this controlled way it is possible to find some key intermediates of reactions and the role of each component (oxide, metal) in the process; i.e., an experiment-based reaction mechanism can be proposed at the molecular level. 1−8 Besides this important use in mechanistic studies, inverse catalysts have actual practical interest. Indeed, it has been shown that inverse catalysts achieve high catalytic activity, even at relatively low temperatures, for very important reactions such as water−gas shift (WGS, CO + H 2 O → H 2 + CO 2 ), CO oxidation (CO + 0.5O 2 → CO 2 ), and methanol synthesis from carbon dioxide (CO 2 + 3H 2 → CH 3 OH + H 2 O). 1,2,5−8 This is remarkable because, for example, neither Au(111) nor CeO 2 is able to catalyze the WGS reaction, but together they attain high activity. 2 Another significant example is the CeO x /Cu(111) system, which is able to catalyze the synthesis of methanol from carbon dioxide 200 times faster than reference Cu(111) and 14 times faster than the regular industrial catalyst Cu/ZnO. 1 Interestingly, it has been shown that sometimes inverse catalysts may be more active than the regular ones. This is the case of Cu and CeO 2 : CeO x /Cu(111) has higher catalytic activity than Cu/CeO 2 for the WGS reaction. 6 Coinage metals (Cu, Ag, and Au) have been widely used in catalysis for many key reactions, together with CeO 2 , mainly in the regular M/CeO 2 configuration. 9−13 The CeO x /Cu(111) and CeO x /Au(111) inverse catalysts have also been exper- imentally studied in great detail. 2,5,6 The oxide−metal interaction in CeO x /Cu(111) activates the system by generating numerous Ce 3+ species in the oxide particle through a charge transfer from the metal. 5,6 Both in CeO x /Au(111) and CeO x /Cu(111) the catalytic activity is related to the presence of these active Ce 3+ species. 2,5,6 In spite of that, to the best of our knowledge, the CeO x /Ag(111) model catalyst has not been yet studied. This is somewhat surprising since it has been found that both regular Ag/CeO 2 and inverse CeO 2 /Ag systems show remarkable catalytic activities for some interesting reac- tions. 14−19 From a theoretical point of view, there are only some preliminary and partial studies for CeO x /Cu(111) and CeO x /Au(111). 1,2,5,6,8 In this context, the comparative and systematic study reported in the present work on the nature of the interaction of cerium oxide species (particles and 2D films) supported on noble metal (111) surfaces results in additional interest. II. COMPUTATIONAL DETAILS The CeO x /M(111) catalytic system was represented using a model previously described 1,5,6,8 and designed to account for the small ceria particles seen in the STM experimental images that point to a O−Ce−O trilayer supported on M(111), 6 with Received: October 1, 2014 Revised: November 3, 2014 Published: November 3, 2014 Article pubs.acs.org/JPCC © 2014 American Chemical Society 26931 dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−26938

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Unraveling the Nature of the Oxide−Metal Interaction in Ceria-BasedNoble Metal Inverse CatalystsJesus Graciani,*,† Alba B. Vidal,‡,§ Jose A. Rodriguez,‡ and Javier Fdez. Sanz†
†Departamento de Química Física, Universidad de Sevilla, 41012 Sevilla, Spain‡Chemistry Department, Brookhaven National Laboratory, P.O. Box 5000, Upton, New York 11973-5000, United States
ABSTRACT: Recently, it has been shown that model catalysts consisting ofcerium oxide nanoparticles supported on noble metal surfaces are veryconvenient to clarify the reaction mechanism of important industrial reactionssuch as water−gas shift, CO oxidation, and methanol synthesis from CO2.Moreover, these special inverse catalysts are highly active, even more than theregular metal/oxide catalyst in some cases. However, there is a lack offundamental knowledge about the electronic nature of the supported CeOxnanoparticles (NPs) and their interaction with the noble metal support. In thiswork we have carried out DFT calculations on CeOx/Cu(111), CeOx/Ag(111), and CeOx/Au(111) interfaces for bothsupported particles and periodic 2D film. The most stable and most reduced CeOx NPs are found when supported on Cu(111),followed by Ag(111) and Au(111). A similar behavior is observed for periodic 2D CeO2(111) films supported on the M(111)surfaces, although the reducibility is lower and the stability higher in extended films than in small particles. The charge transfercontribution from the support dominates the strength of the oxide(film)−metal interaction following the sequence Cu > Ag >Au. The high stability and reducibility of CeOx on Cu(111) and Ag(111) and the lower stability and reducibility on Au(111) arerationalized on the grounds of the metal−oxide/metal charge transfer, the electronic structure of the system, work function of themetal surfaces, structural mismatch, and the nature of the interface bonding.
I. INTRODUCTION
Inverse catalysts consisting of oxide nanoparticles (NPs)supported on metal surfaces have been used as model systemsin which the reaction environment can be easily controlled andthe arbitrary, complex, and undetermined phenomena thatoccur in the real powder catalyst can be avoided. In thiscontrolled way it is possible to find some key intermediates ofreactions and the role of each component (oxide, metal) in theprocess; i.e., an experiment-based reaction mechanism can beproposed at the molecular level.1−8
Besides this important use in mechanistic studies, inversecatalysts have actual practical interest. Indeed, it has beenshown that inverse catalysts achieve high catalytic activity, evenat relatively low temperatures, for very important reactions suchas water−gas shift (WGS, CO + H2O → H2 + CO2), COoxidation (CO + 0.5O2 → CO2), and methanol synthesis fromcarbon dioxide (CO2 + 3H2 → CH3OH + H2O).
1,2,5−8 This isremarkable because, for example, neither Au(111) nor CeO2 isable to catalyze the WGS reaction, but together they attain highactivity.2 Another significant example is the CeOx/Cu(111)system, which is able to catalyze the synthesis of methanol fromcarbon dioxide 200 times faster than reference Cu(111) and 14times faster than the regular industrial catalyst Cu/ZnO.1
Interestingly, it has been shown that sometimes inversecatalysts may be more active than the regular ones. This isthe case of Cu and CeO2: CeOx/Cu(111) has higher catalyticactivity than Cu/CeO2 for the WGS reaction.6
Coinage metals (Cu, Ag, and Au) have been widely used incatalysis for many key reactions, together with CeO2, mainly in
the regular M/CeO2 configuration.9−13 The CeOx/Cu(111)and CeOx/Au(111) inverse catalysts have also been exper-imentally studied in great detail.2,5,6 The oxide−metalinteraction in CeOx/Cu(111) activates the system bygenerating numerous Ce3+ species in the oxide particle througha charge transfer from the metal.5,6 Both in CeOx/Au(111) andCeOx/Cu(111) the catalytic activity is related to the presenceof these active Ce3+ species.2,5,6 In spite of that, to the best ofour knowledge, the CeOx/Ag(111) model catalyst has not beenyet studied. This is somewhat surprising since it has been foundthat both regular Ag/CeO2 and inverse CeO2/Ag systems showremarkable catalytic activities for some interesting reac-tions.14−19 From a theoretical point of view, there are onlysome preliminary and partial studies for CeOx/Cu(111) andCeOx/Au(111).
1,2,5,6,8 In this context, the comparative andsystematic study reported in the present work on the nature ofthe interaction of cerium oxide species (particles and 2D films)supported on noble metal (111) surfaces results in additionalinterest.
II. COMPUTATIONAL DETAILS
The CeOx/M(111) catalytic system was represented using amodel previously described1,5,6,8 and designed to account forthe small ceria particles seen in the STM experimental imagesthat point to a O−Ce−O trilayer supported on M(111),6 with
Received: October 1, 2014Revised: November 3, 2014Published: November 3, 2014
Article
pubs.acs.org/JPCC
© 2014 American Chemical Society 26931 dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−26938

CeO2(111) structure and orientation.5 Basically it consists of aCe6O13 cluster supported on a (111) metal surface described bymeans of a 4√3 × 4√3 four-layer thick (192 Cu, Ag, or Auatoms) slab and a vacuum region of 15 Å between repeatedslabs (see Figure 1).The theoretical approach is based on density functional
theory (DFT) calculations and the generalized gradientapproximation. The Perdew−Wang 91 (PW91) functional20
was used for the exchange-correlation potential. The effect ofthe core electrons on the valence states was represented withthe projector-augmented wave (PAW) approach,21 as imple-mented in the Vienna ab initio simulation package (VASP5.3),22,23 with the valence states defined for each atom as Cu(3d,4s), Ag (4d, 5s), Au (5d, 6s), Ce (4f, 5s, 5p, 5d, 6s), and O(2s, 2p) electrons, while the remaining electrons were keptfrozen as core states. The valence electronic states are expandedin a basis of plane waves with a cutoff of 400 eV for the kineticenergy. In order to account for the strong localization of the Ce4f electrons, a Hubbard-like U term was used (GGA + U),according to the Dudarev et al. implementation, which makesuse of an effective parameter Ueff.
24 Several values of the Uparameter have been proposed for CeO2 and Ce2O3 bulk,ranging between 5 and 6 eV for LDA + U and between 3 and 5eV for GGA + U.9,25−32 Within this 3−5 eV range we adopted avalue for Ueff of 4.5 eV. This value was self-consistentlydetermined by Fabris et al.32 for the cerium atoms in ceriumoxides and used in our previous works dealing with supportedcerium oxide particles.1,5,6,8,33−35
The presence of Ce3+ was confirmed by three means: (i)appearance of the characteristic occupied 4f small peak in theband gap of the density of states (DOS) projected on aparticular Ce atom, (ii) the presence of a magnetization higherthan 0.9 in that Ce atom, and (iii) the Bader charge of the Ceatom compared to the charge of Ce in CeO2 and Ce2O3 purebulks: 2.27 and 2.02 |e−| for Ce4+ and Ce3+, respectively. In asimilar way, the presence of Ce4+ was checked by the absence ofthat characteristic occupied 4f peak in the band gap, theabsence of magnetization, and the Bader charge of the atom. Allthe calculations were performed at the Γ point of the Brillouinzone, and in the structural optimizations, the two bottom layersof the M(111) metal slab were kept at the bulk atomicpositions.The formation energies (Ef) of the CeOx particles on
M(111) were calculated as follows
=
− · − · −
E
E x E y E E
x
[Ce O /M(111)] [Ce] [O ] [M(111)]x y
f12 2
where E[CexOy/M(111)] is the energy of the whole system inwhich the particle is on the M(111) surface; E[Ce] is theenergy of an isolated Ce atom; E[O2] is the energy of anisolated O2 molecule; and E[M(111)] is the energy of the(111) surface metal slab. The formation energies of CeO2 andCe2O3 bulks were calculated analogously
=− · − ·
EE x E y E
x
[Ce O ] [Ce] [O ]x yf
12 2
In both cases the formation energies were normalizeddividing by the number of Ce atoms. The physical meaning ofthis definition could be expressed as the energy variation thattakes place when a unit formula of the system is formed in theactual physical−chemical conditions of the system on a Ceatom basis. This way to define the formation energy allows adirect comparison of Ef of cerium oxides with differentstoichiometry.Yet, it has been shown in previous works that the geometrical
expansion is essential to accommodate Ce3+ species in thestructure.25,26 If we start from the structure of the fully oxidizedCeO2, it is very difficult to converge to the solution in whichsome Ce atoms are reduced and some metal atoms from thesurface are oxidized. That is why it is necessary to force thegeometrical distortion before carrying out the full electronic-structural optimization of the system. VASP uses pseudopo-tentials (PAW method) to deal with the core electrons, whichremain “frozen” throughout the calculation. One can use apseudopotential that includes the 4f electron in the coreelectrons, so the Ce atom, to which that PAW potential isapplied, remains Ce3+ necessarily. With this potential weperformed geometrical optimizations forcing the accommoda-tion of the Ce3+ in the selected positions (structural expansion).Then, with that geometry frozen, we performed a pureelectronic calculation in which all the Ce atoms are definedwith the regular PAW potential, i.e., considering the 4felectrons as valence electrons, and now the convergence tothe electronic solution in which Ce3+ species appear at theintended positions is straightforward. Finally, we start a fullgeometrical-electronic optimization calculation, starting fromthe electronic configuration (reading the wave function and/orspace charge distribution) and geometry obtained in the
Figure 1. Optimized structures for CeOx/M(111) systems. CeOx/Cu(111) (left), CeOx/Ag(111) (middle), and CeOx/Au(111) (right). Top view(top), side view (bottom). Cerium (white), oxygen (red), copper (orange), silver (soft gray), and gold (golden).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826932
previous steps. In that way, the calculation converges to a localenergy minimum with the desired configuration.We have tested different ratios Ce3+/Ce4+ from full reduction
(all Ce are Ce3+) to full oxidation (all Ce are Ce4+). In theintermediate systems there are different electronic isomers,though we have seen that the differences in the total energiesare relatively small and that the observed trends are notchanged at all.
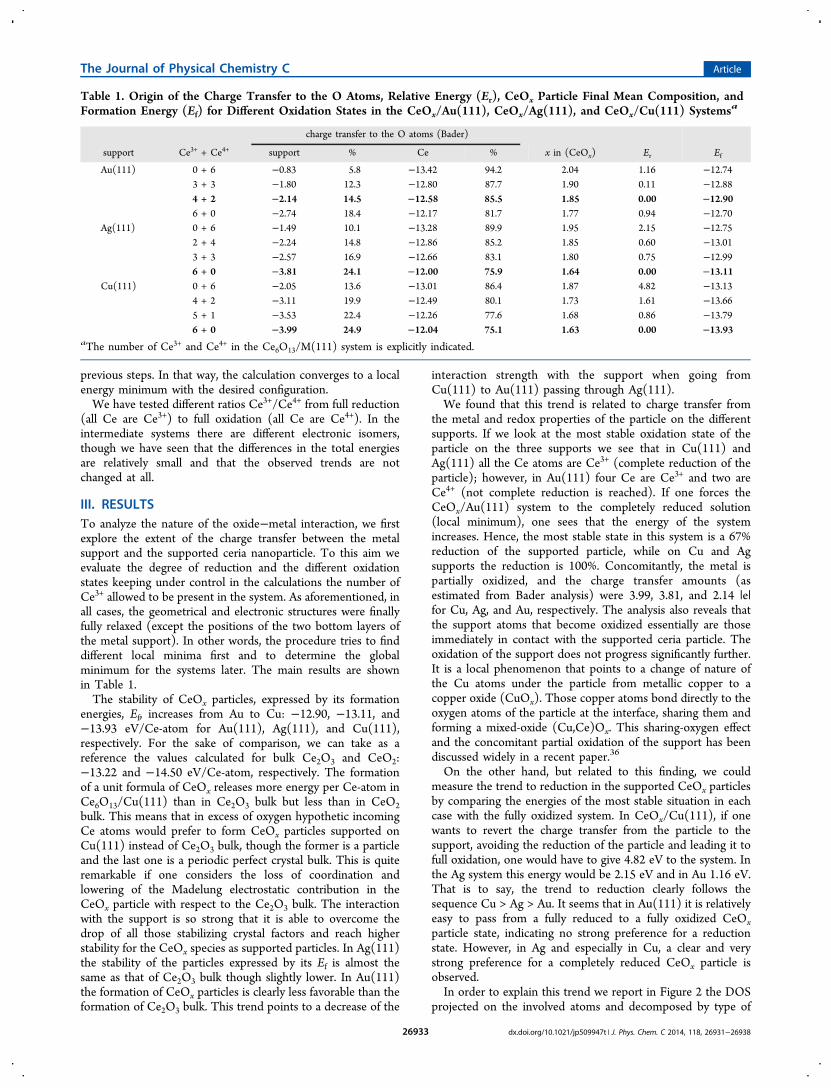
III. RESULTSTo analyze the nature of the oxide−metal interaction, we firstexplore the extent of the charge transfer between the metalsupport and the supported ceria nanoparticle. To this aim weevaluate the degree of reduction and the different oxidationstates keeping under control in the calculations the number ofCe3+ allowed to be present in the system. As aforementioned, inall cases, the geometrical and electronic structures were finallyfully relaxed (except the positions of the two bottom layers ofthe metal support). In other words, the procedure tries to finddifferent local minima first and to determine the globalminimum for the systems later. The main results are shownin Table 1.The stability of CeOx particles, expressed by its formation
energies, Ef, increases from Au to Cu: −12.90, −13.11, and−13.93 eV/Ce-atom for Au(111), Ag(111), and Cu(111),respectively. For the sake of comparison, we can take as areference the values calculated for bulk Ce2O3 and CeO2:−13.22 and −14.50 eV/Ce-atom, respectively. The formationof a unit formula of CeOx releases more energy per Ce-atom inCe6O13/Cu(111) than in Ce2O3 bulk but less than in CeO2bulk. This means that in excess of oxygen hypothetic incomingCe atoms would prefer to form CeOx particles supported onCu(111) instead of Ce2O3 bulk, though the former is a particleand the last one is a periodic perfect crystal bulk. This is quiteremarkable if one considers the loss of coordination andlowering of the Madelung electrostatic contribution in theCeOx particle with respect to the Ce2O3 bulk. The interactionwith the support is so strong that it is able to overcome thedrop of all those stabilizing crystal factors and reach higherstability for the CeOx species as supported particles. In Ag(111)the stability of the particles expressed by its Ef is almost thesame as that of Ce2O3 bulk though slightly lower. In Au(111)the formation of CeOx particles is clearly less favorable than theformation of Ce2O3 bulk. This trend points to a decrease of the
interaction strength with the support when going fromCu(111) to Au(111) passing through Ag(111).We found that this trend is related to charge transfer from
the metal and redox properties of the particle on the differentsupports. If we look at the most stable oxidation state of theparticle on the three supports we see that in Cu(111) andAg(111) all the Ce atoms are Ce3+ (complete reduction of theparticle); however, in Au(111) four Ce are Ce3+ and two areCe4+ (not complete reduction is reached). If one forces theCeOx/Au(111) system to the completely reduced solution(local minimum), one sees that the energy of the systemincreases. Hence, the most stable state in this system is a 67%reduction of the supported particle, while on Cu and Agsupports the reduction is 100%. Concomitantly, the metal ispartially oxidized, and the charge transfer amounts (asestimated from Bader analysis) were 3.99, 3.81, and 2.14 |e|for Cu, Ag, and Au, respectively. The analysis also reveals thatthe support atoms that become oxidized essentially are thoseimmediately in contact with the supported ceria particle. Theoxidation of the support does not progress significantly further.It is a local phenomenon that points to a change of nature ofthe Cu atoms under the particle from metallic copper to acopper oxide (CuOx). Those copper atoms bond directly to theoxygen atoms of the particle at the interface, sharing them andforming a mixed-oxide (Cu,Ce)Ox. This sharing-oxygen effectand the concomitant partial oxidation of the support has beendiscussed widely in a recent paper.36
On the other hand, but related to this finding, we couldmeasure the trend to reduction in the supported CeOx particlesby comparing the energies of the most stable situation in eachcase with the fully oxidized system. In CeOx/Cu(111), if onewants to revert the charge transfer from the particle to thesupport, avoiding the reduction of the particle and leading it tofull oxidation, one would have to give 4.82 eV to the system. Inthe Ag system this energy would be 2.15 eV and in Au 1.16 eV.That is to say, the trend to reduction clearly follows thesequence Cu > Ag > Au. It seems that in Au(111) it is relativelyeasy to pass from a fully reduced to a fully oxidized CeOxparticle state, indicating no strong preference for a reductionstate. However, in Ag and especially in Cu, a clear and verystrong preference for a completely reduced CeOx particle isobserved.In order to explain this trend we report in Figure 2 the DOS
projected on the involved atoms and decomposed by type of
Table 1. Origin of the Charge Transfer to the O Atoms, Relative Energy (Er), CeOx Particle Final Mean Composition, andFormation Energy (Ef) for Different Oxidation States in the CeOx/Au(111), CeOx/Ag(111), and CeOx/Cu(111) Systemsa
charge transfer to the O atoms (Bader)
support Ce3+ + Ce4+ support % Ce % x in (CeOx) Er Ef
Au(111) 0 + 6 −0.83 5.8 −13.42 94.2 2.04 1.16 −12.743 + 3 −1.80 12.3 −12.80 87.7 1.90 0.11 −12.884 + 2 −2.14 14.5 −12.58 85.5 1.85 0.00 −12.906 + 0 −2.74 18.4 −12.17 81.7 1.77 0.94 −12.70
Ag(111) 0 + 6 −1.49 10.1 −13.28 89.9 1.95 2.15 −12.752 + 4 −2.24 14.8 −12.86 85.2 1.85 0.60 −13.013 + 3 −2.57 16.9 −12.66 83.1 1.80 0.75 −12.996 + 0 −3.81 24.1 −12.00 75.9 1.64 0.00 −13.11
Cu(111) 0 + 6 −2.05 13.6 −13.01 86.4 1.87 4.82 −13.134 + 2 −3.11 19.9 −12.49 80.1 1.73 1.61 −13.665 + 1 −3.53 22.4 −12.26 77.6 1.68 0.86 −13.796 + 0 −3.99 24.9 −12.04 75.1 1.63 0.00 −13.93
aThe number of Ce3+ and Ce4+ in the Ce6O13/M(111) system is explicitly indicated.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826933

band (s, p, d, f) for the most stable state of the systems CeOx/Cu(111), CeOx/Ag(111), and CeOx/Au(111). We havedivided the atoms involved in six groups: metallic metalatoms (called M0 or M0Surf), oxidized metal atoms (calledMOx), O atoms above the Ce plane (O-Above), O atoms at theinterface between the Ce plane and the metal surface (O-Interface), Ce3+ ions and Ce4+ ions (when present). Manyaspects could be commented from this figure; however, wewould like to focus on the ones we consider more relevant:stability of the Ce3+ species (determined by the energy positionof the occupied 4f peaks under the Fermi level), formation of amixed-metal oxide (determined by the presence of a strongcovalency, established by the coincidence of peaks of differentatoms and different types of states at the same energy and withthe same shape), and the nature of the occupied electronicstates close to the Fermi level (most reactive electrons).The most stable Ce3+ species are found in the CeOx/
Cu(111) system, followed by CeOx/Au(111) and CeOx/Ag(111). This can be seen from the energy position of theoccupied 4f peak under the Fermi level; the most unstable isthe one belonging to CeOx/Ag(111) where this peak is close tothe Fermi level (between 0 and −0.5 eV). For CeOx/Cu(111)the peak appears at much lower energies (between −1 and−1.5 eV), and on Au(111) the peak lies in between (from −0.5to −1 eV).On the formation of (M,Ce)Ox mixed-oxides (where M may
be Cu, Ag, or Au), we look in the PDOS at the peaks in whichstates of O, Ce, and M match at the same energy and with thesame shape, and we marked them with vertical lines in Figure 2.
The more significant covalent feature can be seen in CeOx/Ag(111) where same-shape peaks of O atoms from above theCe plane, O atoms from the interface, Ce 5d and 4f states, andAg 4d states from both metallic and oxidized Ag coincide at thesame energy. This means that those electrons are shared by allof them, and a real (Ag,Ce)Ox mixed oxide is formed from anelectronic point of view. This mixed-oxide states fall above themain large 4d band of Ag. This covalent character seems tostabilize in such a way the CeOx particles on Ag(111) thatthough Ce3+ species on Ag(111) are the least stable of the threesystems the final formation energy of the oxide particle is largeron Ag(111) than on Au(111), and it is similar to that of thepure Ce2O3 bulk. In contrast, in CeOx/Cu(111) only somesmall covalent peaks are observed (from −4 to −5 eV), and thestates belonging to the O atoms above the Ce-plane (locatedmainly from −3.5 to −2.5 eV) do not contribute to thosecovalent states. Therefore, from an electronic point of view, the(Cu,Ce)Ox mixed-oxide consists only of Ce atoms, the Oatoms at the interface, and some Cu atoms underneath, and themixed electronic states lie in the bottom region of the mainlarge Cu 3d band from −4 to −6 eV. The extent of the covalentcharacter in CeOx/Cu is less than in CeOx/Ag; however, theinvolved states appear at lower energies (more stable) than inthe silver case, and the Ce3+ 4f peak lies also at lower energies.The coincidence of both beneficial factors (some covalentcontribution at very low energies and the high stability of theoccupied 4f Ce3+ states) may explain the highest stability ofCeOx on Cu(111) over the other systems. In Au(111) nosignificant covalent peaks are found in the PDOS, and the Ce3+
4f peak appears at higher energies, near the Fermi level. Thismay explain why the CeOx particles on Au(111) have thelowest stability of the three studied systems, since neither thecovalent contribution nor the occupied 4f Ce3+ state stability ispresent. It can be said, finally, that CeOx/Ag(111) is anintermediate case between CeOx/Cu(111) and CeOx/Au(111),in which the instability of the occupied 4f Ce3+ states iscompensated with a strong covalent contribution.If we look now at the nature of the most reactive electrons of
the three systems, i.e., the occupied states closest to the Fermilevel, we see that in the CeOx/Cu(111) system they mainlyarise from a mix of orbitals corresponding to copper−oxidebonding (O atoms at the interface and Cu atoms underneathand around). Only when we go deeper in energy, at −1 eV, the4f states of Ce3+ show up. Interestingly, in the CeOx/Ag(111)system the electrons closest to the Fermi level mainlycorrespond to Ce 4f states, indicating a very reactive Ce3+
species. If we go down in energy, from −0.5 to −1.5 eV, thestates start to form a mix mainly of AgOx silver oxide states (Oatoms at the interface and Ag states). If we go further we findthe states corresponding to the special (Ag,Ce)Ox mixed-oxidefrom −1.5 to −3 eV. Finally, in the CeOx/Au(111) system, thefirst states under the Fermi level correspond to metallic andoxidized Au, from 0 to −0.5 eV. From −0.5 to −1 eV the Ce3+
states appear, and last, from −1 to −1.75 eV, the statescorresponding to a mixing of O and Au states. Summarizing,the nature of the most reactive electrons in CeOx/Cu(111)corresponds to CuOx oxide states, in CeOx/Ag(111) to theCe3+ states, and in CeOx/Au(111) to metallic and oxidized Austates. This finding points to a different chemistry in the threesystems: (i) in CeOx/Cu(111) the most reactive sites would beat the interface (in the border of the supported particle, whereoxidized Cu and O at the interface appear together); (ii) inCeOx/Ag(111) they would be at the top of the supported oxide
Figure 2. Density of states projected on the atoms involved in theCeOx−support interaction for CeOx/Cu(111) (top), CeOx/Ag(111)(middle), and CeOx/Au(111) (bottom). O atoms located above theCe-plane are called O-Above; O atoms placed at the interface arecalled O-Interface; oxidized metal atoms are called MOx and metallicatoms M0 or M0Surf; oxidized Ce atoms are called Ce4+ and thereduced ones Ce3+ and 4f and 5d states are indicated. Since in Cu andAg all Ce atoms are Ce3+ they are simply called Ce. The Fermi level islocated at 0 eV in all cases. Vertical lines indicate the main covalentfeatures.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826934

particle (where the Ce3+ is present); and (iii) in CeOx/Au(111)they would be at the metal atoms around the supportedparticle.We have identified at least two more factors that influence
the stability of the CeOx particles in general and that of theoccupied 4f band in particular: (i) the work function of themetal surface gives an index of how likely the charge transfer isfrom the metal to the particle in order to get Ce3+ reducedspecies, i.e., the higher the work function, the higher theinstability of the Ce3+; and (ii) the matching of the equilibriumbond distances of metal support and supported CeOx particle atthe interface, i.e., the higher the structural mismatch, the higherthe instability of the Ce3+ species. The work functions ofCu(111) and Ag(111) are lower (4.94 and 4.74 eV,respectively)37 than that of Au(111) (5.31 eV),37 whichmeans that Ce3+ would be more stable on Cu and Ag thanon Au. On the other hand, Ag(111) and Au(111) have verysimilar structural parameters, significantly different from thoseof Cu(111). It is obvious that the interaction with the supportimposes some constraints to the structural relaxation of thesupported particle. This constraint would be different in Au andAg than in Cu since they have different lattice constants. As wesee in the PDOS (Figure 2), the 4f peak appears stabilized inCeOx/Cu(111) with respect to that of CeOx/Ag(111) andCeOx/Au(111). It seems that the structural parameters of theCu(111) are more favorable for the interaction with the CeOxthan those of Ag(111) and Au(111). Therefore, in Cu bothfactors (work function and geometry) are favorable; in Ag thework function is favorable but the structure is not; and in Auboth factors are unfavorable. That contributes to explain whythe most stable occupied 4f band appears in CeOx/Cu(111),and, at the same time, it implies that the structure is a moreimportant factor than the work function for the position of theoccupied 4f peak (stability of Ce3+ species) since Ag and Au,having very close structural parameters, have occupied 4f Ce3+
states at similar energies, although the work functions for bothmetals are different. The similar structure in Au and Ag seemsto determine the occupied 4f band stability over the differencesin the work function values.We mentioned above the formation of mixed oxides at the
interface particle/support. Using the charge given by thesupport and by the Ce atoms to the O atoms one coulddistribute how many O atoms belong to the surface and howmany to the Ce atoms. In that way we can approximate the realcomposition of the CeOx particle. For example, in CeOx/Cu(111) 75.1% of the charge received by the O atoms comesfrom Ce atoms, and 24.9% comes from the metal support. Onecould say that 75.1% of the O atoms belongs to the Ce atomsand 24.9% to the support. That would mean that from the 13 Oatoms of the supported particle only 9.76 O atoms belong toCe and the other 3.24 to the surface. As we have 6 Ce and 9.76O atoms in the CeOx particle, the resulting approximatecomposition would be CeO1.63, which is much closer to CeO1.5of Ce2O3 than to CeO2 and agrees well with the experimentalfinding in which Ce3+ is the main oxidation state of the CeOxparticles supported on Cu(111).6 If we follow the sameprocedure for the most stable configurations in Ag and Ausupports we get approximate compositions of CeO1.64 andCeO1.85, respectively, which agree well with a high reduction ofthe CeOx particle in Ag(111) and a partial reduction inAu(111) (see Table 1).In order to state whether or not this behavior is an effect of
the initial apparent nonstoichiometry of the isolated particle
(Ce6O13) or of its local nature and small size, we have testedthe behavior of a stoichiometric CeO2(111) 2D periodic film(one O−Ce−O atomic trilayer) on Cu(111), Ag(111), andAu(111). In order to avoid artifacts coming from a comprisedor expanded film model, we looked for a minimal mismatchbetween the CeO2(111) and M(111) surfaces. With a √7 ×√7 surface model for Ag(111) and Au(111) and a 2 × 2surface model for CeO2(111) (see Figure 3), the mismatch in a
per area basis was 0.27% and 1.03% for CeO2(111)/Ag(111)and CeO2(111)/Au(111), respectively. In the case ofCeO2(111)/Cu(111) a 4 × 4 surface model was chosen forthe Cu(111) and √7 × √7 for CeO2(111) (see Figure 3),being the resulting mismatch of 0.17%.Following the same methodology as in the case of supported
particles, we obtain the results shown in Table 2. Strongreduction of the oxide film still takes place by charge transferfrom the metal surface to the Ce atoms of the supportedstoichiometric CeO2 periodic film. However, the extent of thereduction differs from the case of supported particles. In theparticles, full reduction was achieved for CeOx/Ag(111) andCeOx/Cu(111), but in the stoichiometric periodic film it couldnot be achieved, the amount of formal Ce3+ with respect to thetotal number of Ce atoms being 75% and 86% for CeO2(111)/Ag(111) and CeO2(111)/Cu(111), respectively. In the case ofgold support, the partial reduction obtained for the supportedparticles (Ce3+: 67%) is lowered in the periodic film to 50%.The estimated final mean composition on an electronic basis ischanged from the particle to the periodic film, “x” being 1.87,1.74, and 1.67 for CeOx periodic film supported on Au(111),Ag(111), and Cu(111), respectively, compared to 1.85, 1.64,and 1.63 in the supported particles system. We can concludethat the reducibility of the supported one-trilayer-thick CeOxspecies is higher as small particles than as extended films,though it takes place in both.In all cases the stability of the supported CeOx species
increases when passing from particle to 2D periodic film, theformation energies being −13.54, −13.75, and −14.24 eV forthe film supported on Au(111), Ag(111), and Cu(111),respectively. So, now the formation of a unit formula ofCeOx liberates more energy per Ce atom in all the CeO2(111)-film/M(111) systems (M: noble metals) than in Ce2O3 bulkbut less than in CeO2 bulk. However, the formation energy ofthe film−oxide in the case of the CeOx/Cu(111) systemapproaches very close to the Ef for the CeO2 bulk (−14.50 eV).
Figure 3. Top view of the surface models used for CeO2(111) periodicfilms supported on the M(111) nobel metal surface. Unit cells areshown with dashed lines.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826935

This is quite remarkable if one considers the loss ofcoordination and Madelung field in the CeOx film with respectto the bulk. The interaction with the support is so strong that itis able to partially overcome the loss of all those stabilizingcrystal factors.In order to quantify this oxide−support interaction, we
calculate the adhesion energy of the film to the metal surface asfollows
=− −
EE E E[CeO (111)/M(111)] [CeO (111)] [M(111)]
surface area
adh
2 2
The resulting Eadh were −3.01, −4.23, and −7.95 eV/nm2 forAu(111), Ag(111), and Cu(111), respectively. There is acorrelation between the adhesion energy and the total chargetransfer from the metal to the film. So, it seems that theelectron transfer dominates the oxide(film)−metal interaction.Finally, we compared the electronic structure of CeOx NPs
and 2D films. We show the results in Figure 4. First, we can see
than NPs on Cu(111) and Ag(111) are fully reduced since noCe4+ (4f) peak appears, and only the two characteristic 4f bandsof Ce3+ appear (an occupied small one and an unoccupied bigone). However, for the oxide NPs supported on Au(111), inbetween the two bands corresponding to Ce3+ species, a thirdnew 4f band corresponding to the presence of Ce4+ speciesappears. This means that no full reduction is achieved for CeOx
NPs on Au(111). When we look at the PDOS of theCeO2(111)-film/M(111) systems, we see that there is nosystem able to achieve full reduction, and all of them have three4f bands: two of Ce3+ species and one of Ce4+ species. Thisagrees with the results shown in Table 2 commented before.Second, we can compare the stability of the occupied 4f bands(Ce3+) of the different systems. In the case of the CeOx/Ag(111) system the stability of the occupied 4f band (its energyposition in the PDOS) is equal for NP and extended 2D filmsupported on Ag(111) likely because the occupied 4f band isjust under the Fermi level. For Cu(111) a destabilization of theoccupied 4f band takes place when passing from CeOx NP tothe 2D extended film (∼0.5 eV to higher energies). ForAu(111) a similar behavior is observed, but the extent of thedestabilization is lower than in the case of copper, locating theoccupied 4f band just below the Fermi level, as in CeO2(111)/Ag(111). This destabilization of the occupied 4f band inperiodic films with respect to NPs can be explained consideringthat the periodic film is not able to relax the structure and bonddistances in the x−y plane at the same extent that an isolatedsmall NP does. Since the appearance of Ce3+ involves astructural expansion, this species would be more stable in asystem with a higher ability for structural relaxation.We can conclude that the trends observed for supported
particles are not induced by the apparent initial overoxidationof our model consisting of a Ce6O13 particle supported onM(111), and it is also not an effect of the borders of theparticle. We can state as a general behavior the charge transferfrom the noble metal (111) surface to one-layer-thickCeO2(111) species, small particles or extended 2D filmsbeing on the surface.
IV. CONCLUSIONS
In this work we have studied some oxidation−reduction trendsof one-trilayer-thick (O−Ce−O−) CeOx(111) species sup-ported on the noble metal (111) surface.Both Cu(111) and Ag(111) are able to fully reduce
supported small ceria particles, while Au(111) only can achievea partial reduction of them. However, the trend to thereduction of the supported oxide is much higher in CeOx/Cu(111) than in CeOx/Ag(111): −4.82 and −2.15 eV arereleased, respectively, when the system passes from completeoxidation to complete reduction of the supported ceriaparticles. As the charge transfer does, the stability of thesupported ceria particles on noble metals increases from Au toCu passing through the Ag support. Although in CeOx/Ag(111) the Ce3+ 4f states are the most unstable of the threestudied systems, the ceria particle as a whole is stabilized byforming a mixed-oxide with silver (Ag,Ce)Ox. In CeOx/Au(111), the Ce3+ 4f states are also very unstable, but nowno stabilizing mixed-oxide covalent contribution is observed,making this system the most unstable of the three examined. In
Table 2. Origin of the Charge Transfer to the O Atoms, Final Mean Composition (CeOx), and Formation Energy (Ef) for OneO−Ce−O Layer Thick CeO2(111) Periodic Film on Au(111), Ag(111), and Cu(111) Surfacesa
charge transfer to the O atoms composition
CeO2(111)-film/M(111) models Ce3+ (%) from metal support % from Ce atoms % x (CeOx) Ef
Ce4O8(111)/Au(111) (2 × 2)/(√7 × √7) 50 −0.63 6.6 −8.84 93.4 1.87 −13.54Ce4O8(111)/Ag(111) (2 × 2)/(√7 × √7) 75 −1.27 12.9 −8.56 87.1 1.74 −13.75Ce7O14(111)/Cu(111) (√7 × √7)/(4 × 4) 86 −2.93 16.7 −14.59 83.3 1.67 −14.24
aIn all models, the oxide is one-layer-thick CeO2(111) periodic film, and the metal slab is four layers thick.
Figure 4. Density of states projected on the atoms involved in theCeOx species supported on Cu(111) (left), Ag(111) (middle), andAu(111) (right) as Ce6O13 nanoparticles (NPs) (top figures) or asperiodic CeO2(111) film (bottom figures). O(2p) is represented inred, Ce(5d) in green, and Ce(4f) in blue. The Fermi level is located at0 eV in all cases. 4f peaks belonging to Ce3+ and Ce4+ species arelabeled in the figure (left-bottom).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826936

contrast, the Ce3+ 4f states in the CeOx/Cu(111) system arethe most stable, and some stabilizing covalent contribution isalso observed, leading this system to be the most stable of thethree here examined. Furthermore, two factors have beenpointed out to explain this behavior: (i) work function of thethree surfaces and (ii) structural match between the particleand the surface, the latter one being the most important tostabilize the 4f occupied states.For periodic 2D CeO2(111) films supported on the M(111)
surfaces we observed a similar behavior, although thereducibility is lower and the stability higher in extended filmsthan in small particles. The charge transfer dominates thestrength of the oxide(film)−metal interaction following thetrend Cu(111) > Ag(111 ) > Au(111). Whatever the type ofsystem is, small particles or periodic 2D films, the generalbehavior is that a charge transfer from the noble metal (111)surface toward the one-trilayer-thick (O−Ce−O−) CeO2(111)systems takes place.As a final remark, according to the high WGS reaction
catalytic activity observed for both CeOx/Cu(111) and CeOx/Au(111) systems, we may expect a high activity for the CeOx/Ag(111) catalyst since (1) this system is relatively stable, withan intermediate stability between CeOx/Cu(111) and CeOx/Au(111), and (2) it shows Ce3+ 4f states specially active, i.e.,very close to the Fermi level.
■ AUTHOR INFORMATION
Corresponding Author*Ph.: +34 954 55 71 75. E-mail: [email protected].
Present Address§Alba B. Vidal, Centro de Quımica, Instituto Venezolano deInvestigaciones Cientificas (IVIC), Apartado 21827, Caracas1020-A, Venezuela.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work was funded by the Ministerio de Economia yCompetitividad, Spain (grants MAT2012-31526 andCSD2008-0023), and EU FEDER. Computational resourceswere provided by the Barcelona Supercomputing Center/Centro Nacional de Supercomputacion (Spain). The researchcarried out at Brookhaven National Laboratory, was supportedby the Division of Chemical Sciences, Geosciences, andBiosciences, Office of Basic Energy Sciences of the U.S.Department of Energy under contract DE-AC02-98CH10886.
■ REFERENCES(1) Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A. E.; Evans, J.;Senanayake, S. D.; Stacchiola, D. J.; Liu, P.; Hrbek, J.; Sanz, J. F.; et al.Highly active copper-ceria and copper-ceria-titania catalysts formethanol synthesis from CO2. Science 2014, 345, 546−550.(2) Rodriguez, J. A.; Ma, S.; Liu, P.; Hrbek, J.; Evans, J.; M. Perez, M.Activity of CeOx and TiOx nanoparticles grown on Au(111) in thewater-gas shift reaction. Science 2007, 318, 1757−1760.(3) Senanayake, S. D.; Stacchiola, D.; Evans, J.; Estrella, M.; Barrio,L.; Perez, M.; Hrbek, J.; Rodriguez, J. A. Probing the reactionintermediates for the water−gas shift over inverse CeOx/Au(111)catalysts. J. Catal. 2010, 271, 392−400.(4) Rodriguez, J. A.; Hrbek, J. Inverse oxide/metal catalysts: Aversatile approach for activity tests and mechanistic studies. Surf. Sci.2010, 604, 241−244.
(5) Yang, F.; Graciani, J.; Evans, J.; Liu, P.; Hrbek, J.; Sanz, J. F.;Rodriguez, J. A. CO oxidation on inverse CeOx/Cu(111) catalysts:High catalytic activity and ceria-promoted dissociation of O2. J. Am.Chem. Soc. 2011, 133, 3444−3451.(6) Rodriguez, J. A.; Graciani, J.; Evans, J.; Park, J. B.; Yang, F.;Stacchiola, D.; Senanayake, S. D.; Ma, S.; Perez, M.; Liu, P.; et al.Water-gas shift reaction on a highly active inverse CeOx/Cu(111)catalyst: Unique role of ceria nanoparticles. Angew. Chem., Int. Ed.2009, 48, 8047−8050.(7) Suchorski, Y.; Wrobel, R.; Becker, S.; Strzelczyk, B.; Drachsel, W.;Weiss, H. Ceria nanoformations in CO oxidation on Pt(111):Promotional effects and reversible redox behavior. Surf. Sci. 2007,601, 4843−4848.(8) Mudiyanselage, K.; Senanayake, S. D.; Feria, L.; Kundu, S.; Baber,A. E.; Graciani, J.; Vidal, A. B.; Agnoli, A.; Evans, J.; Chang, R.; et al.Importance of the metal−oxide interface in catalysis: in situ studies ofthe water−gas shift reaction by ambient-pressure X-ray photoelectronspectroscopy. Angew. Chem., Int. Ed. 2013, 52, 5101−5105.(9) Zhang, C.; Michaelides, A.; Jenkins, S. J. Theory of gold on ceria.Phys. Chem. Chem. Phys. 2011, 13, 22−33.(10) Burch, R. Gold catalysts for pure hydrogen production in thewater−gas shift reaction: activity, structure and reaction mechanism.Phys. Chem. Chem. Phys. 2006, 8, 5483−5500.(11) Rodriguez, J. A.; Liu, P.; Hrbek, J.; Evans, J.; Perez, M. Water gasshift reaction on Cu and Au nanoparticles supported on CeO2(111)and ZnO(0001): Intrinsic activity and importance of supportinteractions. Angew. Chem., Int. Ed. 2009, 46, 1329−1332.(12) Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Activenonmetallic Au and Pt species on ceria-based water-gas shift catalysts.Science 2003, 301, 935−938.(13) Rodriguez, J. A.; Liu, P.; Wang, X.; Wen, W.; Hanson, J.; Hrbek,J.; Perez, M.; Evans, J. Water-gas shift activity of Cu surfaces and Cunanoparticles supported on metal oxides. Catal. Today 2009, 143, 45−50.(14) Machida, M.; Murata, Y.; Kishikawa, K.; Zhang, D.; Ikeue, K. Onthe reasons for high activity of CeO2 catalyst for soot oxidation. Chem.Mater. 2008, 20, 4489−4494.(15) Shimizu, K.; Kawachi, H.; Komai, S.; Yoshida, K.; Sasaki, Y.;Satsuma, A. Carbon oxidation with Ag/ceria prepared by self-dispersion of Ag powder into nano-particles. Catal. Today 2011,175, 93−99.(16) Farmer, J. A.; Campbell, C. T. Ceria maintains smaller metalcatalyst particles by strong metal-support bonding. Science 2010, 329,933−936.(17) Yamazaki, K.; Kayama, T.; Dong, F.; Shinjoh, H. A mechanisticstudy on soot oxidation over CeO2−Ag catalyst with ‘rice-ball’morphology. J. Catal. 2011, 282, 289−298.(18) Kayama, T.; Yamazaki, K.; Shinjoh, H. Nanostructured ceria−silver synthesized in a one-pot redox reaction catalyzes carbonoxidation. J. Am. Chem. Soc. 2010, 132, 13154−13155.(19) Zhang, J.; Li, L.; Huang, X.; Li, G. Fabrication of Ag−CeO2
core−shell nanospheres with enhanced catalytic performance due tostrengthening of the interfacial interactions. J. Mater. Chem. 2012, 22,10480−10487.(20) Perdew, J.; Wang, Y. Accurate and simple analyticrepresentation of the electron-gas correlation energy. Phys. Rev. B1992, 45, 13244−13249.(21) Kresse, G.; Joubert, J. From ultrasoft pseudopotentials to theprojector augmented-wave method. Phys. Rev. B 1999, 59, 1758−1775.(22) Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquidmetals. Phys. Rev. B 1993, 47, 558−561.(23) Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energycalculations for metals and semiconductors using a plane-wave basisset. Comput. Mater. Sci. 1996, 6, 15−50.(24) Dudarev, S. L.; Botton, G. A.; Savrasov, S. Y.; Humphreys, C. J.;Sutton, A. P. Electron-energy-loss spectra and the structural stability ofnickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505−1509.(25) Ganduglia-Pirovano, M. V.; Da Silva, J. L. F.; Sauer, J. Density-functional calculations of the structure of near-surface oxygen
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826937

vacancies and electron localization on CeO2(111). Phys. Rev. Lett.2009, 102, 026101.(26) Li, H.-Y.; Wang, H.-F.; Gong, X.-Q.; Guo, Y.-L.; Guo, Y.; Lu, G.;Hu, P. Multiple configurations of the two excess 4f electrons ondefective CeO2(111): Origin and implications. Phys. Rev. B 2009, 79,193401.(27) Nolan, M.; Parker, S. C.; Watson, G. W. The electronicstructure of oxygen vacancy defects at the low index surfaces of ceria.Surf. Sci. 2005, 595, 223−232.(28) Zhang, C.; Michaelides, A.; King, D. A.; Jenkins, S. J. Structureof gold atoms on stoichiometric and defective ceria surfaces. J. Chem.Phys. 2008, 129, 194708.(29) Herschend, B.; Baudin, M.; Hermansson, K. Oxygen vacancyformation for transient structures on the CeO2(110) surface at 300and 750 K. J. Chem. Phys. 2007, 126, 234706.(30) Nolan, M.; Grigoleit, S.; Sayle, D. C.; Parker, S. C.; Watson, G.W. Density functional theory studies of the structure and electronicstructure of pure and defective low index surfaces of ceria. Surf. Sci.2005, 576, 217−229.(31) Fabris, S.; Vicario, G.; Balducci, G.; de Gironcoli, S.; Baroni, S.Electronic and atomistic structures of clean and reduced ceria surfaces.J. Phys. Chem. B 2005, 109, 22860−22867.(32) Fabris, S.; de Gironcoli, S.; Baroni, S.; Vicario, G.; Balducci, G.Reply to “Comment on ‘Taming multiple valency with densityfunctionals: A case study of defective ceria’”. Phys. Rev. B 2005, 72,237102.(33) Park, J. B.; Graciani, J.; Evans, J.; Stacchiola, D.; Ma, S.; Liu, P.;Nambu, A.; Sanz, J. F.; Hrbek, J.; Rodriguez, J. A. High catalyticactivity of Au/CeOx/TiO2(110) controlled by the nature of the mixed-metal oxide at the nanometer level. Proc. Natl. Acad. Sci. U.S.A. 2009,106, 4975−4980.(34) Johnston-Peck, A. C.; Sanjaya, D. S.; Plata, J. J.; Kundu, S.; Xu,W.; Barrio, L.; Graciani, J.; Sanz, J. F.; Navarro, R. M.; Fierro, J. L.;et al. Nature of the mixed-oxide interface in ceria−titania catalysts:Clusters, chains, and nanoparticles. J. Phys. Chem. C 2013, 117,14463−14471.(35) Graciani, J.; Plata, J. J.; Sanz, J. F.; Liu, P.; Rodriguez, J. A. Atheoretical insight into the catalytic effect of a mixed-metal oxide at thenanometer level: The case of the highly active metal/CeOx/TiO2(110)catalysts. J. Chem. Phys. 2010, 132, 104703.(36) Graciani, J.; Sanz, J. F. Designing a new generation of catalysts:Water gas shift reaction example. Catal. Today 2014, DOI: 10.1016/j.cattod.2014.03.071.(37) Haynes, W. M., Ed., CRC Handbook of chemistry and physics,94th ed. (Internet version 2014); CRC Press/Taylor and Francis:Boca Raton, FL.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp509947t | J. Phys. Chem. C 2014, 118, 26931−2693826938
Related Documents











