Current Pharmaceutical Design, 2010, 16, 101-113 101 1381-6128/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd. Understanding Autophagy in Cell Death Control Francesca Platini 1 , Ricardo Pérez-Tomás 2, *, Santiago Ambrosio 3 and Luciana Tessitore 1,† 1 Department of Food Chemical, Pharmaceutical and Pharmacological Sciences (DISCAFF), University of East Piedmont “A. Avogadro”, Italy; 2 Pathology and Experimental Therapy Dept., University of Barcelona, Faculty of Medicine, Barcelona, Spain; 3 Department of Phisiological Sciences II, Biochemistry Unit, University of Barcelona, Barcelona, Spain Abstract: Autophagy is an evolutionarily conserved degradation pathway which primary functions as a cell survival adaptive mechanism during stress conditions. Autophagy is a tumor suppressor process and induction of the autophagic machinery can cause cell demise in apoptosis-resistant cancer. Thus, this metabolic pathway can act either to prevent or to promote carcinogenesis, as well as to modulate the response to anticancer therapies, included drug-induced apoptosis. Conventional therapies exert their cytotoxic activity mainly by inducing apoptosis. Massive activation of the apoptotic program in a tissue can result in cell loss providing a selective advantage for growth to displastic cells and tumor cell subpopulations with high levels of malignancy. This suggests that the activation of autophagy can counteract malignancy. On the contrary, therapeutic intervention-induced apoptosis can eliminate cells with pro-mutational biochemical alterations at risk for initiation, initiated cells and cells of focal and advanced preneoplastic and neoplastic lesions. Thus, pharmacological inhibition of autophagy may enhance apoptosis. Autophagy and apoptosis share common stimuli and signalling pathways, so that the final fate, life or death, depends on the cell response. Recently, accumulating data fuel novel potential therapeutic interventions to modulate autophagy to be beneficial in cancer therapy. This review highlights current knowledges aimed at unraveling the molecular interplay between autophagy and cell death as well as the possible therapeutic exploitation in cancer. Keywords: Autophagy, autophagic cell death, apoptosis, molecular swicth, oncogenesis, cancer therapy. 1. THE AUTOPHAGIC PATHWAY Autophagy literally means self-eating/digestion from greek in contrast to heterophagy that is eating from the outside. It is one part of everyday activities of the cell and a strategy evolved by eukaryotic cells to respond to stressful conditions. There are various types of autophagic processes: macroautophagy, microautophagy, chaperone-mediated autophagy to name a few Fig. (1) [1]. They differ in the substrates targeted, their regulation and selectivity as well as the conditions in which each of them is preferentially activated. However, the different types of autophagy share a common endpoint, the lysosome. Microautophagy participates in the continuous basal turnover of cellular components in normal cellular conditions and can remove the excess organelles when they are no longer needed, such as following peroxisome proliferators. To date, no identification of associations between microautophagy and particular human diseases are available due to the lack of efficient technologies to manipulate this process. Chaperone-mediated autophagy (CMA) is a pathway by which soluble cytosolic proteins are selectively targeted to lysosomes after interacting with a cytosolic chaperone, hsc70, the constitutive member of the 70kDa family of heat shock proteins. It is the only autophagic way to selectively degrade particular cytosolic proteins by lysosomes in mammals. Basal CMA activity is observed in most tissues, but it is maximally activated by different stressors such as oxidative, nutritional and toxic stresses. Perturbations in CMA have been associated with some lysosomal storage diseases, familial forms of Parkinson’s disease, chemical- induced nephropaties and in diabetes-associated kidney hyper- trophy [2]. Macroautophagy (which we refer to hereafter as autophagy) is an evolutionarily conserved, genetically controlled, homeostatic, multi-step process of “bulk” catabolism, by which cellular long-lived proteins and organelles are targeted to lysosomes *Address correspondence to this author at the Pathology and Experimental Therapy Dept., University of Barcelona, Faculty of Medicine, Barcelona, Spain; Pavelló Central, 5a planta, LR 5101 C/Feixa Llarga s/n, E 08907 L´Hospitalet, de Llobregat, Barcelona, Spain; Tel: +34 934024288; Fax: +34 934029082; E-mail: [email protected] † Prof. Lucina Tessitore is a principal author of this work who has dided tragically in summer 2008. [3-5]. In mammals, “housekeeping” levels of autophagy might occur to prevent the accumulation of superfluous/damaged/aged proteins and organelles and genome instability. Autophagy can be rapidly up-regulated when cells undergo architectural remodelling or any types of stressors. Autophagy is a unique form of membrane trafficking Fig. (2) [6]. A crescent-shaped isolation membrane forms the initial phagophore which expands and sequesters cytoplasmic macro- molecules and organelles. Then, the phagophore, known also as pre-autophagosomal structure (PAS), completes a closed double membrane structure giving life to the autophagosome, its defining feature by trasmission electron micrograph is a double-walled membrane. Autophagosomes then undergo a progressive stepwise process of maturation by fusion with endosomes or multi-lamellar bodies giving rise to a vesicle namely amphisome. In mammalian cells, the amphisome or directly the outer membrane of the auto- phagosome fuses with a lysosome to create an autophagolysosome, also called autolysosome, where the final breakdown takes place when lysosomal acidic hydrolases degrade the luminal content of the vacuole. Digestion of the sequestered material generates amino acids, free fatty acids and nucleotides that are recycled for ATP generation and macromolecular synthesis. The molecular machinery underlying autophagy has been extensively researched in the past decade and the genes partici- pating in this process, recently unified under the achronim Atg (- a ut ophag y-related) were found to be conserved from yeast to man [6]. Yet, the roles played by the individual gene products, namely Atg proteins, and their modes of action are still continuously in resolution. 2. AUTOPHAGY: LIFE OR DEATH? In human tumour cells, apoptosis, necrosis, autophagy and senescence are all found to contribute to overall cell death. As Bios and Thanatos, autophagy can promote both cell survival and cell death [7-9]. 2.1. Autophagic Cell Death Autophagy was first described in dying cells in the 1960s. Morphological evidence of accumulation of autophagosomes in

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Current Pharmaceutical Design, 2010, 16, 101-113 101
1381-6128/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
Understanding Autophagy in Cell Death Control
Francesca Platini1, Ricardo Pérez-Tomás
2,*, Santiago Ambrosio
3 and Luciana Tessitore
1,†
1Department of Food Chemical, Pharmaceutical and Pharmacological Sciences (DISCAFF), University of East Piedmont “A.
Avogadro”, Italy; 2Pathology and Experimental Therapy Dept., University of Barcelona, Faculty of Medicine, Barcelona, Spain;
3Department of Phisiological Sciences II, Biochemistry Unit, University of Barcelona, Barcelona, Spain
Abstract: Autophagy is an evolutionarily conserved degradation pathway which primary functions as a cell survival adaptive mechanism during stress conditions. Autophagy is a tumor suppressor process and induction of the autophagic machinery can cause cell demise in
apoptosis-resistant cancer. Thus, this metabolic pathway can act either to prevent or to promote carcinogenesis, as well as to modulate the response to anticancer therapies, included drug-induced apoptosis. Conventional therapies exert their cytotoxic activity mainly by
inducing apoptosis. Massive activation of the apoptotic program in a tissue can result in cell loss providing a selective advantage for growth to displastic cells and tumor cell subpopulations with high levels of malignancy. This suggests that the activation of autophagy
can counteract malignancy. On the contrary, therapeutic intervention-induced apoptosis can eliminate cells with pro-mutational biochemical alterations at risk for initiation, initiated cells and cells of focal and advanced preneoplastic and neoplastic lesions. Thus,
pharmacological inhibition of autophagy may enhance apoptosis. Autophagy and apoptosis share common stimuli and signalling pathways, so that the final fate, life or death, depends on the cell response. Recently, accumulating data fuel novel potential therapeutic
interventions to modulate autophagy to be beneficial in cancer therapy. This review highlights current knowledges aimed at unraveling the molecular interplay between autophagy and cell death as well as the possible therapeutic exploitation in cancer.
Keywords: Autophagy, autophagic cell death, apoptosis, molecular swicth, oncogenesis, cancer therapy.
1. THE AUTOPHAGIC PATHWAY
Autophagy literally means self-eating/digestion from greek in contrast to heterophagy that is eating from the outside. It is one part of everyday activities of the cell and a strategy evolved by eukaryotic cells to respond to stressful conditions. There are various types of autophagic processes: macroautophagy, microautophagy, chaperone-mediated autophagy to name a few Fig. (1) [1]. They differ in the substrates targeted, their regulation and selectivity as well as the conditions in which each of them is preferentially activated. However, the different types of autophagy share a common endpoint, the lysosome.
Microautophagy participates in the continuous basal turnover of cellular components in normal cellular conditions and can remove the excess organelles when they are no longer needed, such as following peroxisome proliferators. To date, no identification of associations between microautophagy and particular human diseases are available due to the lack of efficient technologies to manipulate this process. Chaperone-mediated autophagy (CMA) is a pathway by which soluble cytosolic proteins are selectively targeted to lysosomes after interacting with a cytosolic chaperone, hsc70, the constitutive member of the 70kDa family of heat shock proteins. It is the only autophagic way to selectively degrade particular cytosolic proteins by lysosomes in mammals. Basal CMA activity is observed in most tissues, but it is maximally activated by different stressors such as oxidative, nutritional and toxic stresses. Perturbations in CMA have been associated with some lysosomal storage diseases, familial forms of Parkinson’s disease, chemical-induced nephropaties and in diabetes-associated kidney hyper-trophy [2]. Macroautophagy (which we refer to hereafter as autophagy) is an evolutionarily conserved, genetically controlled, homeostatic, multi-step process of “bulk” catabolism, by which cellular long-lived proteins and organelles are targeted to lysosomes
*Address correspondence to this author at the Pathology and Experimental
Therapy Dept., University of Barcelona, Faculty of Medicine, Barcelona, Spain; Pavelló Central, 5a planta, LR 5101 C/Feixa Llarga s/n, E 08907
L´Hospitalet, de Llobregat, Barcelona, Spain; Tel: +34 934024288; Fax: +34 934029082; E-mail: [email protected] †Prof. Lucina Tessitore is a principal author of this work who has dided
tragically in summer 2008.
[3-5]. In mammals, “housekeeping” levels of autophagy might occur to prevent the accumulation of superfluous/damaged/aged proteins and organelles and genome instability. Autophagy can be rapidly up-regulated when cells undergo architectural remodelling or any types of stressors.
Autophagy is a unique form of membrane trafficking Fig. (2) [6]. A crescent-shaped isolation membrane forms the initial phagophore which expands and sequesters cytoplasmic macro-molecules and organelles. Then, the phagophore, known also as pre-autophagosomal structure (PAS), completes a closed double membrane structure giving life to the autophagosome, its defining feature by trasmission electron micrograph is a double-walled membrane. Autophagosomes then undergo a progressive stepwise process of maturation by fusion with endosomes or multi-lamellar bodies giving rise to a vesicle namely amphisome. In mammalian cells, the amphisome or directly the outer membrane of the auto-phagosome fuses with a lysosome to create an autophagolysosome, also called autolysosome, where the final breakdown takes place when lysosomal acidic hydrolases degrade the luminal content of the vacuole. Digestion of the sequestered material generates amino acids, free fatty acids and nucleotides that are recycled for ATP generation and macromolecular synthesis.
The molecular machinery underlying autophagy has been extensively researched in the past decade and the genes partici-pating in this process, recently unified under the achronim Atg (-autophagy-related) were found to be conserved from yeast to man [6]. Yet, the roles played by the individual gene products, namely Atg proteins, and their modes of action are still continuously in resolution.
2. AUTOPHAGY: LIFE OR DEATH?
In human tumour cells, apoptosis, necrosis, autophagy and senescence are all found to contribute to overall cell death. As Bios and Thanatos, autophagy can promote both cell survival and cell death [7-9].
2.1. Autophagic Cell Death
Autophagy was first described in dying cells in the 1960s. Morphological evidence of accumulation of autophagosomes in

102 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
dying cells has led to believe that autophagy may result in a nonapoptotic form of programmed cell death (PCD): autophagic cell death (ACD) or type II PCD [10]. Although the roles of the autophagic process in protein and organellar catabolism and in cellular protection during nutrient starvation are well accepted, the involvement of autophagy in PCD is more controversial. This is related, at least in part, to the application of the term ACD to two distinct observations, that is cell death associated with autophagy and cell death requiring autophagy. It is largely accepted that ACD occurs primarily when the developmental process or homeostatic
maintenance in adulthood need massive cell elimination as well as ACD is observed in pathologies such as cancer. Whether autophagy is responsible for death or a last attempt at survival or again a scavenger to reduce the phagocyte task after death is still a matter of question.
In some cases, autophagy appears as an effector mechanism of cell death because experiments of autophagy inhibition indicate prevention of ACD. Indeed, 3-methyladenine (3-MA), an inhibitor of autophagic sequestration, reduces the death of anti-estrogen exposed human mammary carcinoma cells [11]. The treatment with
Fig. (1). Autophagic and heterophagic pathways. Substrates are delivered to lysosomes from outside (heterophagy) or inside (autophagy) the cell.
Macroautophagy, microautophagy and CMA have been described as the main forms of autophagy.
Fig. (2). Autophagic machinery. Various genes are involved in the multistep autophagic process.

Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 103
both IFN- and the proteasome inhibitor MG132 induces ACD, which is inhibited by 3-MA. At any rate, to analyze the role of autophagy in cell death, the best approach appears to be the genetic approach, such as loss of function of autophagy genes. Evidence suggests that autophagic genes are indeed required for at least some of what have been referred to as ACD. z-VAD, a pan-caspase inhibitor, was found to induce, in L929 fibroblast cell line, a death that could be limited by down-regulating autophagy proteins [12]. Although less is known about the mediators of ACD, interestingly, this study also shows the involvement of receptor-interacting protein, RIP and Jun N-terminal kinase, JNK in the signalling pathway leading to death. Yet, they are not involved in the initiation of starvation-induced autophagy, suggesting that signalling pathways for autophagy and ACD may be in part different. Another study demonstrating that ACD is dependent on ATG genes shows that chemically-induced death in Bax and Bak deficient mouse embryo fibroblasts (MEF) is mediated by autophagy, since it does not occur in silenced Beclin 1 and Atg5 cells [13]. Taken together, these cell death studies show for the first time that silencing autophagy genes prevents both the accumulation of autophagic vacuoles and the cell death, indicating that autophagy is actually needed for the cell demise and is not a mere failed survival attempt.
2.2. Autophagy as a Survival Programm
Autophagy functions as a cellular guardian to avoid death waiting for better circumstances. The pro-survival function is an evolutionarily ancient self-defence response to exogenous stress, conserved from yeast to mammals. First of all, the nutrient limitation imposed by the sudden termination of the trans-placental nutrient supply physiologically activates massive autophagy soon after birth to maintain an adequate amino acids pool for energy metabolism until the nutrient supply from milk becomes steady [14]. Another classical example is the liver of mammals during complete food withdrawal. Rats exposed to fasting undergo a rapid activation of liver autophagy which can be delayed by intensive treatment with cycloheximide [15]. In vivo, the levels of autophagic vacuoles in starved liver are relatively low because of the high rates of clearance of autophagosomes rather than of the low rates of autophagosome formation. Mizushima et al. [16] show the first study on autophagy in mammals based on a transgenic approach to visualize the recruitment of LC3-labelled autophagic vacuoles in some organs, following the nutrient starvation-induced whole-body autophagic response. This suggests that other tissues besides liver, like muscle for its large mass, can contribute to produce substrates, which are required especially for the brain and erythrocytes to sustain life during fasting. However, in humans, prolongation of starvation for more than 3 days results in reduction of autophagy when ketone bodies substitute glucose as a source of fuel to avoid excessive breakdown of proteins essential for cell life. This also implies that ketone bodies can regulate autophagy.
To date, the concept of autophagy as a survival response is supported by evidence showing enhanced death in cells or organisms lacking gene products essential for autophagy. For example, mice lacking Beclin1 die in early embryogenesis [17] and mice lacking Atg5 die within one day after birth, unless newborns are immediately force-fed with milk, suggesting that autophagy is essential to overcome starvation. Nonetheless, mice lacking either Atg5 or Atg7 in neuronal cells develop age-related neuronal degeneration and shorten life span [18]. Autophagy genes are also necessary for maintaining cellular homeostasis and survival when cells are unable to introduce nutrients, such as during growth factor withdrawal [19]. Other mechanisms by which autophagy can promote cell survival include the removal of damaged organelles, the degradation of pathogens and large protein aggregates which cannot be removed by the ubiquitin-proteasome system. Thus, the relevance of autophagy in promoting survival during ageing, infections, neurodegenerative diseases and cancer.
What is the relationship between autophagy and cell cycle? According to Baserga, a cell undergoes the mass cycle, the DNA synthetic cycle and the division cycle in order to proliferate. Cell mass results mainly from the net balance between protein synthesis and degradation of long-lived proteins through the lysosomal autophagic pathway. We have previously reported that cell cycle progression largely depends on growth to such an extent that reaching the stationary phase for the ascites hepatoma AH-130 is entirely due to activation of the acidic vacuolar pathway for protein degradation with an increase in the volume fraction of early autophagic vacuoles, independently of the levels of lysosomal cathepsins B, D, H and L [20-22]. Consistently, the autophagic activity, directly measured as the sequestration of an endogenous cytosolic enzyme, is very low in actively proliferating AH-130 cells, and increases in quiescent stationary cells [23]. The enzyme sequestration is suppressed not only by ammonium chloride and leupeptin, but also by 3-MA, indicating that it represents authentic autophagy.
When does autophagy occurr during cell cycle progression? G1 is a period when many signals of diverse metabolic, stress and environmental cues are integrated and interpreted. To this end, during G1 the cell decides whether to self-renew, differentiate or die. In this contest, autophagy is one of signal transduction pathways that influence G1 progression. The capacity of cells to undertake the G1/S transition is restricted by starvation and oxidative stress, well known inducers of autophagy. The inhibition of mTOR by rapamycin, able to stimulate autophagy in normal culture conditions, stops cells at G1 phase [3]. Stress-induced activation of eIF2 kinases stimulates the autophagic proteolysis and triggers G1 arrest through the inhibition of cyclin 1 translation [24]. Intriguingly, inhibitors of autophagy, which inhibit PI3K like 3-MA, prevent the G1 arrest. On the other hand, drugs such as hydroxyurea, aphidicolin and mimosine, triggering cell cycle arrest in G1 and/or S phase, were also able to contribute to autophagy [25]. The autophagic process during G1 arrest can be useful for the cell to repair the damage and to avoid death when the damage stimulus is removed. DNA damage occurring in G1 is sensed by protein assemblies whose effector components are the protein kinases ATM (ataxia telangiectasia mutated) and ATR (ATM related) which in turn activate the transducer checkpoint kinases and then p53 and p21
cip. Moreover, another G1 checkpoint function
served by p53 and Rb is to supervise hyperactive Ras, Myc and E2F signalling. All these molecules, besides interfering with G1 checkpoint, are known to modulate autophagy.
2.3. Autophagy from Survival to Death
The relevance of the lysosomal pathway in cellular and tissue autolysis during necrosis as well as in the clean-up or self-clearance phase of apoptosis, resulting in engulfment and digestion of cells committed to die is well accepted. Yet, autophagy can have other roles.
2.3.1. Autophagy and Necrosis
Necrosis can result when cell metabolism and integrity are compromised by a nonphysiological insult. The programmed cascade of self-destruction includes activation of cathepsins and lysosomal rupture, thus, it is not surprising a link between autophagy and necrosis because they share the executioners of both processes [26]. Some studies underly a possible switch from autophagy to necrosis. A recent example indicates that hypoxia-induced autophagy reaches a point of no return in which acidic areas trigger necrotic cell death by means of a BNIP3-dependent action. In the protist Dictyostelium, the Atg1 inactivation suppresses vacuolization but not cell death, switching developmental ACD to a death very similar to if not identical to necrosis in mammalian cells [3]. Intriguingly, in the L929 mouse fibrosarcoma cell line, TNF through FADD, well known inducers of both apoptosis and autophagy, induces necrosis [5]. Knockout of RIP1 prevents the

104 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
TNF-mediated mitochondrial effects in L929 cells and Jurkat cells deficient in RIP1 are resistant to the Fas/TNF-R/TRAIL-R–induced necrosis. Cyclophilin D, a mitochondrial matrix protein, is another factor involved in necrosis and possibly in PCD, because it participates in mitochondrial permeability transition (MPT) and is the target of cyclosporin A. Cyclosporin A is able to inhibit necrosis, apoptosis and autophagy. Knockout of the gene encoding CypD induces resistance to ROS- or Ca
2+-mediated necrosis in
hepatocytes and in fibroblasts and to ischemic cardiac or cerebral injury in mice.
2.3.2. Autophagy and Apoptosis
Cell accumulation results from a positive net balance between the rate of cell proliferation and the rate of cell loss, that is mainly function of cell death. Recent evidence indicates that autophagy provides apoptotic cells with a signal to ensure their clearance by heterophagy [27]. Atg5 and Beclin 1 are necessary for the generation of the “eat-me” signal.
Autophagy and apoptosis are not mutually exclusive. They occur in the same cell, concurrently or sequentially in response to the same stimulus, both when autophagy is protective and when it participates in death. Differently from necrosis, the tissue inflammatory response is usually absent in ACD as well as in apoptosis, because of phagocytosis of dying cells. However, the traits of cells undergoing autophagy or apoptosis are markedly different. Since experimental models of autophagy detection are now available, not only morphological, but also biochemical features of ACD and apoptosis are distinct [3,8-11,26]. While in classical apoptosis (type I PCD) cytoskeletal elements early collapse and instead organelles are well preserved, conversely, ACD is characterized by early degradation of organelles with preservation of cytoskeletal constituents until late phase. In contrast with apoptosis, ACD is caspase-independent and DNA fragmen-tation is rarely required in the final stages. The consequences of their presence in the cell are different depending on the stimulus and kinetics of occurrence.
Already in 1996, Bursch and coworkers [11] described apoptosing breast cancer cells near cells undergoing autophagy and other cells with signs of both the processes following antiestrogen treatment by electron microscopy analysis. These observations could indicate that the same treatment causes distinct types of death in the same kind of cells, such as apoptosis, ACD and both together at the same time. Alternatively, some cells die by apoptosis: neiburgh vital cells engulf apoptotic bodies then undergo death. Other cells, after having activated autophagy then begin the switch to apoptosis but do not manage to end the process for lack of energy and show signs of both autophagy and apoptosis.
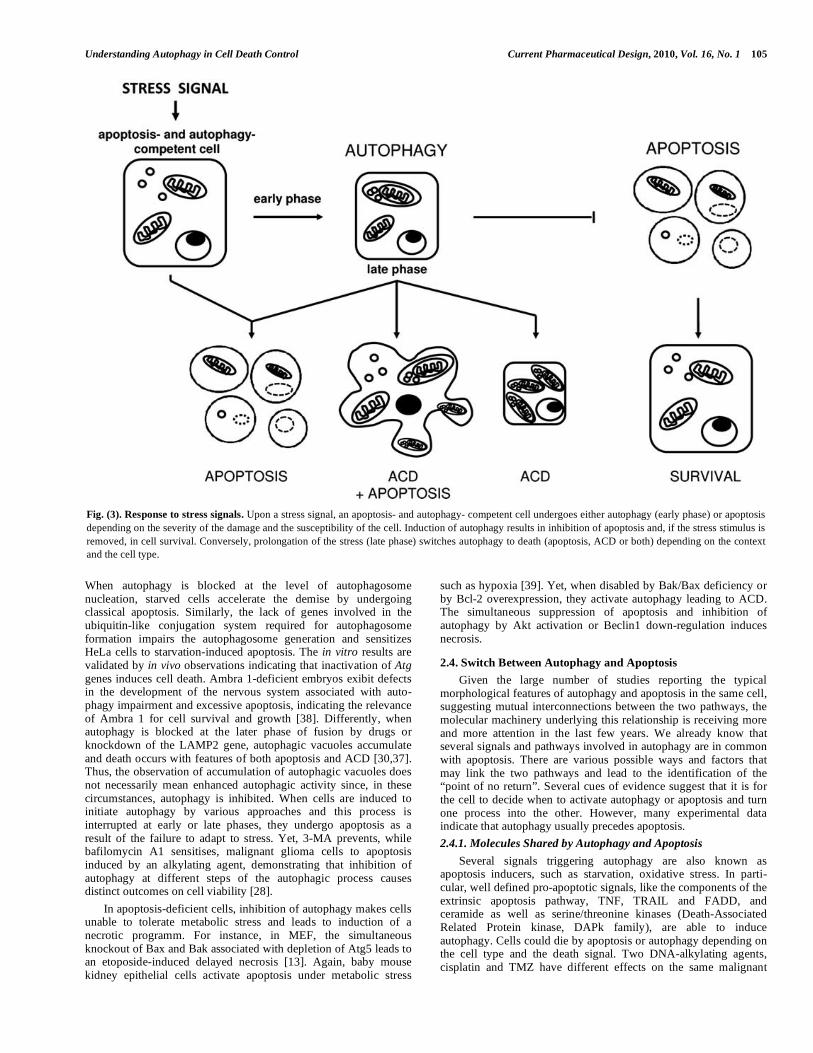
Studies in a variety of experimental systems indicate that the role of autophagy in cell death is likely to be context and cell type dependent Fig. (3). Autophagy can delay the onset of the apoptotic program, such as following starvation, DNA damage and hemody-mamic stress [28]. Exposure of rats to 1 day-fasting causes first liver autophagy but, when starvation is prolonged for a few days, hepatocytes die by apoptosis [29].
Haematopoietic cell lines withdrawn from the growth factor IL-3 activate first autophagy and eventually apoptosis [30]. This demonstrates the pro-survival function of Atg genes in cells with intact apoptotic machinery. Again, autophagy of p62/SQSTM1-labelled protein aggregates has a protective role on cell death, since a deletion mutant of p62 lacking the ubiquitin-binding ability domain impeded both the formation of p62-positive aggregates and enhanced cell death [5]. Other findings indicate that inhibition of autophagy prevents apoptosis and that autophagy can trigger apoptosis Fig. (3). When autophagy in breast, prostate and colon cancer as well as in malignant glioma cells is inhibited by 3-MA, these cells do not undergo apoptosis [28]. In line, resveratrol-induced autophagy switches to apoptosis which is prevented by
autophagy inhibition [31]. A recent study shows that autophagy acts upstream of apoptosis in death signals induced by HIV envelope glycoproteins in uninfected bystander CD4 T lymphocytes, since apoptosis is prevented by silencing Beclin1 and Atg7, suggesting that autophagy can trigger apoptosis under certain circustances. Atg1 overexpression in fly tissues induces autophagy, reduces TOR signalling, suppresses cell growth and these high levels of autophagy lead to apoptosis [32].
However, it remains unclear whether cells with intact apoptotic machinery can undergo autophagy as a death mechanism Fig. (3). Restoration of Beclin1 in breast cancer cells monoallelically deleted of Beclin1 enhanced autophagy [33]. In line with this, Atg5 has been found to contribute to ACD by interacting with Fas-associated protein with death domain (FADD) in IFN- -treated human cervical carcinoma HeLa cells [34]. Accordingly, cells might die by apoptosis unless exposed to harsh enough stimuli when any mechanism of death is likely. Consistently, autophagy appears to be primarily a pro-survival rather than a pro-death mechanism, at least in cells with intact apoptosis machinery and the presence of autophagy features in dying cells could be explained as a last attempt at survival. Among the many questions unanswered about ACD, the most intriguing is, if autophagy is a cellular protective program, what is the signal for the switch to initiate ACD?
Much evidence indicates that suppression of apoptosis induces autophagy and inhibition of autophagy causes apoptosis. On one hand, ACD is induced in MEFs or bone marrow immortalized cells or prostate cancer cells when the apoptotic pathway is impaired by defects in the pro-apoptotic proteins BAX and BAK [13,30]. Because autophagy is unveiled following a lethal stimulus when apoptosis is inhibited by either caspase inhibitors [12] or Bax and Bak double knockout [30], one possibility is that cells can decide to die by using apoptotic or autophagic effector mechanisms which are not inhibited in that specific circumstance. Indeed, growth factor withdrawal is also an apoptotic signal in normal mammalian cells. However, in the absence of the apoptotic proteins Bax and Bak, IL-3 growth dependent immortalized bone marrow cells can survive for weeks without IL-3, which is necessary for nutrient uptake. Surprisingly, these cells can fully recover and even begin to proliferate following addition of IL-3 to the culture medium after 6 weeks of depletion. Activation of autophagy is essential along this survival period resulting in a short-term escape from death, because inhibition of autophagy promotes cell death. How does autophagy overcome nutrient starvation? Autophagy inhibition-induced death is probably a result of bioenergetic catastrophe since cells are rescued by methylpyruvate, a possible oxidisable substrate for the few mitochondria that are still active. Besides metabolic stress other stimuli such as photodynamic therapy [35] and drugs [7] can maintain survival of apoptosis-blocked cells by inducing auto-phagy. However, if the death stimulus is not removed, cells undergo atrophy and eventually die of bioenergetic failure and self-consumption. The above findings support the concept of autophagy as a survival strategy towards various stress inducers when the apoptotic machinery is crippled. On the other hand, inhibition of the early phase of autophagy sensitised cells to stress-induced apoptosis. Inhibition of autophagy by 3-MA breaks the autophagic process induced by inhibition of p38 leading to ACD in colorectal cancer cells and triggers an apoptotic response [36]. These findings support autophagy as a survival response to damage stimuli which allows cell killing only after prolonged exposure, when suppression of autophagy substitutes the type of demise by turning ACD into apoptosis. In fact, removal of the death stimulus induces a reduction in number and size of autophagic vacuoles and re-entry into the cell cycle of colorectal cancer cells. Genetic studies also support a direct involvememt of autophagy proteins in the balance between life and death. Inhibition of nutrient deficiency-induced autophagy in HeLa cells by knockdowning autophagy genes triggers apoptosis which can be delayed by repression of the apoptotic machinery [37].
Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 105
When autophagy is blocked at the level of autophagosome nucleation, starved cells accelerate the demise by undergoing classical apoptosis. Similarly, the lack of genes involved in the ubiquitin-like conjugation system required for autophagosome formation impairs the autophagosome generation and sensitizes HeLa cells to starvation-induced apoptosis. The in vitro results are validated by in vivo observations indicating that inactivation of Atg genes induces cell death. Ambra 1-deficient embryos exibit defects in the development of the nervous system associated with auto-phagy impairment and excessive apoptosis, indicating the relevance of Ambra 1 for cell survival and growth [38]. Differently, when autophagy is blocked at the later phase of fusion by drugs or knockdown of the LAMP2 gene, autophagic vacuoles accumulate and death occurs with features of both apoptosis and ACD [30,37]. Thus, the observation of accumulation of autophagic vacuoles does not necessarily mean enhanced autophagic activity since, in these circumstances, autophagy is inhibited. When cells are induced to initiate autophagy by various approaches and this process is interrupted at early or late phases, they undergo apoptosis as a result of the failure to adapt to stress. Yet, 3-MA prevents, while bafilomycin A1 sensitises, malignant glioma cells to apoptosis induced by an alkylating agent, demonstrating that inhibition of autophagy at different steps of the autophagic process causes distinct outcomes on cell viability [28].
In apoptosis-deficient cells, inhibition of autophagy makes cells unable to tolerate metabolic stress and leads to induction of a necrotic programm. For instance, in MEF, the simultaneous knockout of Bax and Bak associated with depletion of Atg5 leads to an etoposide-induced delayed necrosis [13]. Again, baby mouse kidney epithelial cells activate apoptosis under metabolic stress
such as hypoxia [39]. Yet, when disabled by Bak/Bax deficiency or by Bcl-2 overexpression, they activate autophagy leading to ACD. The simultaneous suppression of apoptosis and inhibition of autophagy by Akt activation or Beclin1 down-regulation induces necrosis.
2.4. Switch Between Autophagy and Apoptosis
Given the large number of studies reporting the typical morphological features of autophagy and apoptosis in the same cell, suggesting mutual interconnections between the two pathways, the molecular machinery underlying this relationship is receiving more and more attention in the last few years. We already know that several signals and pathways involved in autophagy are in common with apoptosis. There are various possible ways and factors that may link the two pathways and lead to the identification of the “point of no return”. Several cues of evidence suggest that it is for the cell to decide when to activate autophagy or apoptosis and turn one process into the other. However, many experimental data indicate that autophagy usually precedes apoptosis.
2.4.1. Molecules Shared by Autophagy and Apoptosis
Several signals triggering autophagy are also known as apoptosis inducers, such as starvation, oxidative stress. In parti-cular, well defined pro-apoptotic signals, like the components of the extrinsic apoptosis pathway, TNF, TRAIL and FADD, and ceramide as well as serine/threonine kinases (Death-Associated Related Protein kinase, DAPk family), are able to induce autophagy. Cells could die by apoptosis or autophagy depending on the cell type and the death signal. Two DNA-alkylating agents, cisplatin and TMZ have different effects on the same malignant
Fig. (3). Response to stress signals. Upon a stress signal, an apoptosis- and autophagy- competent cell undergoes either autophagy (early phase) or apoptosis
depending on the severity of the damage and the susceptibility of the cell. Induction of autophagy results in inhibition of apoptosis and, if the stress stimulus is
removed, in cell survival. Conversely, prolongation of the stress (late phase) switches autophagy to death (apoptosis, ACD or both) depending on the context
and the cell type.

106 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
glioma cell line, cisplatin inducing apoptosis and TMZ ACD [28]. The same death stimulus, arsenic trioxide, induces apoptosis in human leukaemia cells, while conversely it triggers autophagy in malignant glioma cells. Autophagy and apoptosis share common components and inhibitory/activating signalling pathways in mammalian cells. A molecular link between autophagy and apop-tosis was first suggested by the similarity of the mammalian protein ASP (apoptosis-specific protein) and yeast Atg5 [35]. Ceramide can stimulate the expression of beclin-1, besides activating apoptosis. Accordingly, autophagy is suppressed by antiapoptotic signalling pathways, such as the class I PI3K/Akt/mTOR pathway. Well known regulators of apoptosis such as the members of the Bcl-2 family can directly affect autophagy execution proteins, such as Beclin1. Where does autophagy meet apoptosis?
p53, the tumour suppressor most commonly mutated in human cancer, induces cell cycle arrest, autophagy and apoptosis. The pro-autophagic function of p53 might be mediated by activating p21
cip1,
because p21cip1
overexpression induces autophagy [28], and/or by inhibiting mTOR via upregulation of the PTEN and TSC2 genes. In response to various stressors, most of the p53 accumulates in the nucleus to activate various target genes, including the damage-regulated autophagy modulator, DRAM, a protein required for p53-dependent autophagy [40]. Of interest, because DRAM is also required for p53-mediated PCD, it might be a candidate for turning autophagy into death. Intriguingly, p73, a p53 family member, also induces DRAM and autophagy in a p53 independent way [40]. Yet, p73-mediated activation of DRAM is not required for PCD and p73-induced autophagy does not contribute to PCD. Thus, DRAM appears to have different functions in p53 and p73 signalling pathways. The tumour suppressor protein human p14ARF/INK4a is known as an inducer of apoptosis and an activator of p53 by antagonizing the function of Mdm2. However, the short mitochondrial ARF, smARF, can function independently of Mdm2 and p53 [41]. It can dissipate mitochondrial membrane potential, independently of the action of the Bcl-2 family proteins and without mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c. The smARF-induced mitochondrial impairment leads to ACD, which is attenuated by silencing Atg5 or Beclin1. It has been suggested that full-length ARF stimulates autophagy by a p53-dependent mechanism and smARF by a p53-independent pathway. Yet, it is unclear how autophagy is triggered in smARF-expressing cells, what the role of ARF is in ACD and whether smARF stimulates autophagy to eliminate mitochondria through mitophagy. The best known growth inhibiting factors, TGF- and its family members are potent suppliers of extracellular signals that maintain tissue homeostasis, limit G1 progression and trigger apoptosis as well as autophagy. To this end, the TGF- cytostatic/cytotoxic program in epithelial cells involves the induction of cyclin-dependent kinase inhibitors, such as p21
Cip1 and
p15Ink4b
, and DAPk as well as repression of Myc, known to be involved in the autophagic control.
The main function of the DAPk family of proteins is to regulate cell death [42]. The DAPk family might have a role in autophagosome formation and ACD induction. DAPk is required for IF- -induced autophagy and cell death, but not for starvation- or rapamycin-stimulated autophagy. On the contrary, the lack of one of DAPk functions attenuates serum/amino acid or steroid withdrawal-induced autophagy. The DAPk-induced autophagy can involve p53 activation or CaMKK phosphorylation. Alternatively, DAPk behaves as a negative regulator of the pro-survival signals from MAPK/ERK pathway. The involvement of DAPk in auto-phagy is also suggested by its ability to modulate membrane trafficking, as shown by accumulation of early/late endosomes in its absence and interaction with components of the SNARE complex.
Another molecular puzzle sees as its “protagonist” p27kip1
which inhibits the kinase activity of cyclin-cdk holoenzymes resulting in cell cycle arrest, but it also can trigger not only
apoptosis, but also autophagy and ACD [28]. p27kip1
also activates PTEN, resulting in activation of autophagy. The activation of autophagy by metabolic stress requires stabilization of p27
kip1 via
its phosphorylation at Thr 198 by LKB1-AMPK pathway, which functions as an intracellular energy sensor [25]. Under adverse conditions of metabolic stress, the LKB1-AMPK-promoted p27
kip1
stability permits cells to survive through autophagy, whereas the loss of p27
kip1 below a level sufficient to maintain autophagy results
in apoptosis. The effect of p27kip1
on autophagy and cell survival has been related to its capacity to inhibit cyclin-cdks. Yet, under various autophagy inducers, p27
kip1 siRNA triggered apoptosis due
to the disruption of the intracellular energy balance, as methyl-pyruvate reduces apoptotic death. Thus, p27
kip1 accumulation acts
as a switch of whether quiescent cells enter the autophagic cell survival pathway or undergo quick apoptosis. Low levels of p27
kip1
might allow cell cycle progression, while lack of p27kip1
below the level necessary for maintaining autophagy might lead to apoptosis under adverse nutrient conditions. It cannot be ruled out that the subcellular localization of p27
kip1 is critical for its different
functions. Consistently, while nuclear p21cip1
can mediate cell cycle arrest, apoptosis and ACD [28], cytoplasmic p21
cip1 interacts with
apoptosis signal-regulating kinase 1 (ASK1) to inhibit apoptosis [43].
High levels of ROS are known to induce ACD [12]. Caspase 8 inhibition in L929 cells triggers selective autophagic degradation of catalase, a key enzymatic hydrogen peroxide scavenger, resulting in accumulation of ROS and ACD [12]. These data unravel a molecular pathway for autophagy as an inducer of ROS-mediated ACD by disrupting the balance between ROS production and degradation and shows how a mechanism originally promoting survival can turn into an efficient mediator of cell death. The same molecule, hydrogen peroxide, is a well known mediator of stress-induced, ROS-mediated apoptosis. Upon oxidative stress, the life span determinant p66Shc is phosphorylated by PKC b and possibly other kinases, recognized by the prolyl isomerase Pin1 and accumulate into mitochondria, where generates ROS and opens the MPT pore, leading to apoptosis. p66Shc or PKC might act as an integration point for many signalling pathways that affect mito-chondrial function and longevity. Thus, the levels of ROS produced inside a organelle can be critical for the cell to decide whether to activate senescence, autophagy, ACD, apoptosis or necrosis.
The negative control of autophagy by caspases has also been reported by at least other two studies. In NGF-deprived neuro-blastoma cells, autophagic sequestration of mitochondria is induced by blockade of caspase activities [44]. In cells lacking NF-kB activation, knockdown of Beclin1 and Atg7 expression reduces TNF -induced apoptosis. z-VAD inhibits apoptosis and enhances autophagy, both induced by TNF . Again, autophagy is upregulated in TNF -treated cells lacking NF-kB activation. Conversely, autophagy can selectively degrade NF-kB-inducing kinase and IkB kinase, resulting in limitation of both basal and inducible activation of NF-kB [45]. These observations suggest that the activation of caspases can regulate the initial autophagic activity and the repression of autophagy by NF-kB can represent a novel anti-apoptotic mechanism of this transcription factor.
Finally, autophagy proteins can activate apoptosis. Beclin1 overexpression in HeLa cells depresses proliferation and in vivo tumorigenesis in nude mice while it promotes autophagy-mediated cell death by enhancing caspase 9 expression [46]. Conversely, Beclin 1 siRNA promotes cell proliferation and reduces apoptosis and caspase 9 expression. The effect of Beclin1 can be related to its function to direct cathepsins to lysosomes from which they can be released into the cytosol to trigger apoptosis. Alternatively, Beclin1 might translocate into the nucleus and transactivate caspase 9. This indicates that caspases might be involved in the signalling or execution of the cell death downstream of or independent of autophagosome formation and this suggests the possibility of

Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 107
crosstalk from autophagy to the apoptotic pathway. Atg 5 has been linked to apoptosis through FADD, a multifunctional protein playing roles also in necrosis, ACD and growth. Thus, FADD may be a switching point that determines diverse signalling fates. Z-VAD-inhibitable proteases might be activated downstream FADD to execute Atg5-mediated IFN- -induced ACD, since z-VAD is not only a pan caspase inhibitor. IFN- -induced cell death in HeLa cells mediated by the interaction of Atg5 with FADD is suppressed by silencing FADD without inhibiting autophagosome formation. This indicates that Atg5, not autophagy, plays a crucial role in the signalling. Consistently, the cleavage of an autophagic specific protein, Atg5, can activate the apoptotic program. Several apoptotic stimuli trigger calpain-mediated cleavage of Atg5 [47]. The truncated form of Atg5 can bind the anti-apoptotic factor Bcl-xL in mitochondria, leading to apoptosis, indicating a novel non-autophagic function of Atg5 as a pro-apoptotic factor. These findings suggest that autophagy proteins can be directly involved in the apoptotic process and the function of other Atg proteins can also be modulated by post-translational modifications.
2.4.2. Meeting Points Between Autophagy and Apoptosis
Given the membrane and molecular trafficking interconnecting the various organelles, the subcellular compartment where auto-phagy and apoptosis could meet becomes relevant.
The role of the ER in the switch between autophagy and apoptosis has been recently described by a study on radiotherapy of cancer [48]. When the ER-associated degradation system is saturated by an excess of stress-induced substrates, in an attempt to survive cells can activate autophagy as an alternative proteolytic process. PERK (an ER transmembrane protein) responds to radiation by phosphorylating eIF2 to downregulate protein synthesis and activate autophagy. This is a cytoprotective survival mechanism. However, under prolonged or excessive stress, the activation of the PERK/eIF2 pathway leads to apoptosis via CHOP, JNK or BCL2 family members. Similarly to PERK, also IRE1 (an ER transmembrane protein) and ATF6 can trigger autophagy and promote apoptosis, via activation of the JNK pathway and BCL2 family proteins or potentiation of PERK signalling through CHOP.
One of the key functions of the ER is Ca2+
sequestration, thus it is not surprising that Ca
2+ might also be involved in the switch
between autophagy and apoptosis. How then does a cell decide whether to undergo apoptosis, autophagy or both, in response to Ca
2+ elevation? The irreversible oxidative damage to the SERCA2
pump is the “point of no return”, the decision to die in this model of oxidative stress is taken upstream of BAX/BAK-dependent MOMP. In the presence of BAX/BAK proteins autophagy is pre-empted by BAX/BAK-induced MOMP, leading to apoptosis. In the absence of BAX/BAK, ER damage triggers autophagy probably in order to remove the irreversibly damaged organelles and to preserve the cells from metabolic collapse, while excess and persistent levels of autophagy leads to cell killing. In wild type cells with intact apoptotic machinery, it is likely that lower doses of radiation and/or of hypericin can activate autophagy without the involvement of the apoptotic pathway. Only when the damage stimulus is prolonged or more intense, wild type cells undergo apoptosis.
Vitamin D and analogs or other agents mobilizing intracellular Ca
2+ trigger both apoptosis and autophagy in MCF7 breast cancer
cells. An increase in the cytosolic Ca2+
has been recently proposed as a potent inducer of autophagy and as a target for the anti-autophagic action of ER-located Bcl-2 [49]. The signalling pathway involves the Ca
2+/calmodulin dependent kinase kinase- (CaMKK-
), which directly activates AMPK of the AMPK-TSC2-RHEB pathway, a negative regulator of mTOR, leading to activation of autophagy. Bcl-2 located in ER might inhibit autophagy by redu-cing the levels of free ER Ca
2+ available for release and,
conversely, mitochondrial Bcl-2 can enhance autophagy by increasing the mitochondrial uptake of Ca
2+.
The Bcl-2 family which works in both ER and mitochondria to mediate survival and death might be also a good candidate to switch between autophagy and apoptosis. Down-regulation of Bcl-2 triggers both apoptotic and ACD, conversely, Bcl-2 over-expression retarded or blocked vacuole development and caspase-independent death. Recent studies underline that the balance between anti-apoptotic and pro-apoptotic Bcl-2 family members and between the anti-apoptotic Bcl-2 members and the pro-autophagic factor Beclin1 functions as a rheostat for both apoptosis and autophagy, indicating Bcl-2 and Bcl-xL as a commitment point [50]. The subcellular localization of Bcl-2 and Beclin1 might be critical to decide their function. Bcl-2 is predominantly found on the outer mitochondrial and ER membranes where over-expressed Beclin1 colocalized with Bcl-2. Conversely, Beclin1 has been reported to localize with its binding partner Class III PI3K predominantly to the TGN, where Bcl-2 should be absent. ER-targeted Bcl-2, but not mitochondrial-targeted Bcl-2, inhibited starvation-induced autophagy, indicating that the Bcl-2/Beclin1 interaction in the ER blocks a signal that is essential for autophagy induction and it is regulated by cellular nutrient status. It has been suggested that the phosphorylation status of Bcl-2 might be relevant for its anti-autophagic function because phosphorylated Bcl-2 localizes predominantly to the ER and Bcl-2 may be a target for autophagy-inhibitory signalling kinases involved in nutrient sensing. In the absence of Bcl-2 binding, Beclin1 mutants induce excessive autophagy and promote cell death. A novel BH3-like domain in Beclin1 could be critical for the interaction of Beclin1 with the anti-apoptotic members of the Bcl-2 family, possibly favouring apoptosis. Thus, Beclin1 is a new type of BH3 protein with a regulatory rather than an effector function. The pharmacological BH3 mimetic ABT737 was first descrived as an inducer of apoptosis by inhibiting the anti-apoptotic proteins of Bcl-2 family. It also inhibits the binding of Beclin1 and Bcl-xL in a competitive manner. Because the physiological function of Beclin1-Bcl-2 interaction is to control Beclin1-initiated autophagy, inhibition of the interaction would be expected to stimulate autophagy. ABT737, like nutrient deprivation, induces autophagy and, concomitantly, reduces the binding of Beclin1 and Bcl-xL in the ER, but not in mitochondria, indicating that only the ER-targeted pool of Bcl-2 is relevant to the suppression of autophagy. ABT737 also fails to induce major loss of MOMP, indicative of apoptosis or necrosis, unless Beclin1 is depleted. These findings support the concept that ABT737 can enhance autophagy without activation of cell death. This anti-autophagic function of Bcl-2 may help maintain autophagy at levels that are compatible with cell survival, rather than cell death.
On the whole, these data indicate that ER stress is a major inducer of the autophagic response, inhibition of autophagy enhancing apoptosis. However, in certain circumstances, autophagy inhibition can protect the cell from death.
The mitochondrion is a crucial site since it may serve as an integration point for signalling pathways that control longevity and cell death. Mitochondria can generate apoptotic signals, however, when they are damaged, they are removed by autophagy.
Autophagy can have a role in helping cells to escape from apoptosis. The sequestration of mitochondria can be a strategy to prevent the release of pro-apoptotic factors such as cytochrome c and AIF, resulting in a block of apoptosis. Lemasters et al. [51] first indicate autophagy as a primary response to apoptotic inducers and mitochondrial pro-apoptotic factors are released to activate the death program only when the autophagic capacity of the cell is overcome. Intriguingly, any stimulus of MOMP, including the strongest Ca
2+, followed by ROS, depletion of ATP and lipid stress
signals might be involved in the switch between autophagy and cell death. Of interest, a large portion of mTOR is associated with the

108 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
MOM, suggesting the existence of a mitochondrial stress-sensing pathway. At any rate, the mTOR-mediated inhibition of autophagy by amino acids does not involved mitochondria. The Bcl-2 protein is inserted in the MOM and protects mitochondria against MOMP. Bak pre-inserted in the MOM and Bax translocating from the cytosol to mitochondria are often required for apoptosis-specific MOMP. A yeast mitochondrial protein, Uth1 appears to be a potential link between autophagy and apoptosis. It is an ageing protein identified for stress resistance and longer life span of mutants. It is involved in mitochondrial biogenesis as well as in the autophagic degradation of mitochondria, its lack conferring resistance to rapamycin-induced autophagy. Mutants for the Uth1 gene, required for mitophagy, are resistant to Bax-induced cell death, indicating that Uth1 protein also contributes to Bax-mediated yeast cell death and suggesting that Bax also promotes mitophagy. Alternatively, Bcl-2 might modify mitochondrial autophagy by inhibiting BNIP3, the human Spin homologue localised, mainly, in mitochondrial membranes. The Bcl-2 homology 3 (BH3)-only subfamily of the Bcl-2 family proteins includes two members recently involved in the control of autophagy at the mitochondrial level BNIP3 and HSPIN1, a human homologue of the Drosophila melanogaster spin gene-product, also induces autophagy in HeLa cells [28]. In a hypoxic microenvironment the rapid induction of the pro-apoptotic factor BNIP3 by HIF contributes to cell survival because it is also a pro-autophagic factor. Several malignant glioma cell lines exposed to arsenic trioxide or ceramide up-regulate BNIP3 and undergo autophagy. BNIP3 over-expression in MCF7 and HeLa cells triggers autophagy and non-apoptotic death with features of necrosis, which is inhibited by Bcl-2 and Bcl-xL. Even if these atypical Bcl-2 family members activate autophagy and non apoptotic cell death, it is still unclear whether this caspase-independent cell death requires autophagy genes. Thus, the Bcl-2 family of proteins not only regulates apoptosis, but also controls autophagy, Bcl-2 protecting, while BNIP3 and HSPIN1 inducing the autophagic process.
Downstream of Bcl-2, MPT caused by pores in the mitochon-drial inner membrane may be a point of convergence of autophagic, apoptotic and necrotic pathways [51]. In rat hepatocytes, autophagy induced by serum deprivation or glucagon induces an increase in mitochondria sequestration rates, leading to mitochondrial depolari-zation in the formed autophagosome, followed by its digestion in the autolysosome. Laser-induced photo damage in hepatocytes triggers first mitochondrial depolarization followed by mitophagy [52]. Upon low intensity stresses by MPT inducers, limited MOMP onset might only enhance mitophagy to rid cells of damaged mitochondria as a repair mechanism. Medium stress overcomes mitophagy that releases pro-apoptotic factors from mitochondria undergoing MOMP, resulting in apoptosis. Overloaded autophagic machinery may release lysosomal enzymes/factors promoting the death pathway. Higher stress induces MOMP in all mitochondria, ATP drops and only necrosis can occur. This progression from autophagic repair through apoptosis to necrosis has been named necrapoptosis.
Lysosomes were first discovered by de Duve and co-workers in 1955 as intracellular organelles completely devoted to the degradation or recycling of intra- and extracellular components. Yet, lysosomes were initially shown to be involved in necrosis and, more recently, in apoptosis. The release of cathepsins into the cytosol following lysosomal membrane permeabilization (LMP) is believed to be an early event in the apoptotic cascade leading to activate the mitochondrial caspase-dependent cell death pathway. The signals of LMP, such as oxidative stress, lysosomotropic drugs and detergents induce not only necrosis, but also apoptosis and apoptosis-like cell death when the release of lysosomal enzymes into the cytoplasm is limited. In the presence of bafilomycin A1, U373-MG cells exposed to arsenic trioxide and malignant glioma cells treated with the alkylating agent temozolomide die more
rapidly. They shift from autophagy to apoptosis, probably because they accumulate lysosomes and are sensitised to bafilomycin A1-induced LMP and subsequent cathepsin-mediated apoptosis [53]. Again, intra-autophagosomal Ca
2+ might trigger the fusion between
autophagosome and lysosome as well as the release of cathepsins from the autolysosomes in the cytoplasm to activate cell death. Ca
2+
largely accumulates in lysosomes and phorbol myristate acetate, the ionophore A23187 and phentolamine modify lysosomal Ca
2+ levels
and modulate the total volume of autophagic vacuoles as well as NAADP mobilizes Ca
2+ from lysosome-related organelles [54].
Evidence has been provided about the signalling pathways by which apoptosis-inducers such as death receptor activation induces LMP [7]. Interestingly, caspase 8 is required for LMP induced by TNFR1, even if the way in which caspase 8 can trigger LMP is unknown. Indeed, caspase 8 might cleave BID to induce LMP, because TNF-triggered LMP is reduced in Bid depleted hepato-cytes. However, Bid deficiency does not rescue fibroblasts from TNF-triggered LMP, questioning a role for BID in LMP. On the contrary, BID might act downstream rather than upstream of LMP, since BID might be a target of lysosomal hydrolases and Bcl2 does not rescue from LMP and cell death where LMP is MOMP independent. Another link between TNF-induced cell death pathway and LMP might be the factor associated with neutral sphingomyelinase (NSM), which binds TNFR1 and activates NSM, since it has been shown that the production of ceramide mediated by this factor contributes to activation of caspase 8 upstream of LMP by using fibroblasts deficient for this factor. In this context, it has been suggested that the ceramide pathway can act directly on lysosomal membranes, because sphingosine, produced from ceramide through the lysosomal enzyme ceramidase, accumulates in lysosomes and can mediate LMP as a detergent. Alternatively, the molecular link between TNFR1 and LMP for the caspase-independent PCD might require the death domain-containing RIP1 and involve ROS, since intralysosomally-generated ROS might oxidise lysosomal membrane lipids, resulting in LMP.
The death signalling from TNFR1 to LMP depends on the cell type and the cellular context. Cells of the same type can respond to TNF differently depending on their activation state. Non transformed and immortalized cells, resistant to TNF effects, become susceptible to TNF-induced LMP and caspase-dependent apoptosis when exposed to inhibitors of transcription or translation, whereas cells undergo LMP and necrosis when TNF is combined with caspase inhibitors [5]. Indeed, TNF is also able to induce autophagy and ACD. The above data link the TNFR1 signalling pathway to LMP for both caspase-dependent and caspase-independent PCD. Interestingly, a physiological signal, TRAIL, induces the death of normal epithelial cells through the endogenous FADD protein which activates a novel cell death pathway involving both apoptosis and autophagy [55].
Another link between autophagy and apoptosis is represented by TMEM166, the novel lysosomal and ER transmembrane protein, which function as an adaptor recruiting or binding other specific proteins in the lysosome or ER [56]. TEM166 over-expression induces first autophagy, then, at a later stage, apoptosis, eventually leading to both ACD and apoptosis in HeLa cells. The authors suggest that TMEM166-induced autophagy occurs earlier than apoptosis and TMEM166 can mediate LMP resulting in release of cathepsins.
Taken together, these data suggest that for the prevention of cell death, it is likely to be relevant to identify the specific molecular linchpins of each available pathway of PCD, because blocking a single path will probably simply shift the cell to an alternative route to self killing. As a caveat, it should be noted that many molecules that were initially implicated specifically in apoptosis have turned out to be involved in normal cellular functions, such as metabolic control, differentiation and signal transduction [57]. Therefore, it

Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 109
seems unlikely that a particular set of proteins are exclusively implicated in autophagy control.
3. AUTOPHAGIC ALTERATIONS IN CANCER
Paradoxically, dual actions of autophagy in cancer have been proposed, because it functions either to promote or to prevent tumorigenesis [58]. Autophagy can have oncogenic effects by supporting cell survival and avoiding apoptosis as a protective mechanism against stressful conditions. Autophagy may serve to optimize oxygen and growth factor/nutrient utilization in pre-invasive and rapidly growing cancer cells when faced with hypoxic or metabolic stress similar to the starvation response [37]. As the tumour grows, cancer cells located in the central of the tumour mass are poorly vascularised and triggering autophagy allows cell survival. The tolerance of cancer cells to nutrient/O2/growth factor deficiency is extremely variable among cell types. High rates of autophagic proteolysis allow human colon cancer cells to survive long periods without nutrients and the response of human epider-moid lung carcinoma cells to nutrient deficiency can be similar to that of their non-transformed counterpart [28]. Again, in starved hepatoma cells the generation of amino acids through the autophagic process is needed to preserve the responsiveness of the mTOR dependent signalling to insulin, supporting a role for autophagy in the control of hormonal signalling pathway. Again, autophagy might aid in the degradation of organelles such as depolarized mitochondria that activate death pathways. The sequestration of mitochondria by autophagy as a protective mecha-nism against apoptosis might explain the tolerance of cancer cells to hypoxia. Autophagy might prevent cancer cells from accumulating free radical-induced damage to lethal levels by removing organelles that are sources or targets of such damage [56]. Autophagy may also represent the first defence mechanism for cancer cells towards therapeutic assault, i.e. chemo- and radio-therapies. Thus, these pro-survival effects of autophagy might foster tumour initiation and/or progression.
Alternatively, autophagy can have tumour suppressor functions, thus limiting tumour cell growth [5,57-59]. We have demonstrated that, in hepatoma cells, the rates of autophagic lysosomal proteolysis and LDH autophagic sequestration as well as the lysosomal enzyme activities, including cathepsins, are a degree of magnitude lower than in normal hepatocytes [21-23]. These observations suggest that the low proteolytic potential results from low autophagic capacity rather than from the low availability of lysosomal enzymes, possibly implying that changes in lysosomal capacity are a consequent adaptive mechanism to limited autophagic input. Again, differently from normal hepatocytes, in which autophagy is markedly suppressed by amino acids and hormones, the autophagic activity of the AH-130 cells is not inhibited by ascitic fluid, a relatively nutrient-rich body fluid. These findings indicate that the hepatoma cells display a decreased capacity to respond to the nutritional requirement of the whole organism. In line, neoplastic and transformed cells may be less prone to enhance their proteolytic rate in response to high cell density and serum/nutrient deprivation. Reduced protein catabolism is also largely responsible for cell growth during liver chemical carcinogenesis, since the rates of protein synthesis do not differ significantly between the various pre- and neoplastic lesions and their normal counterparts [60]. It is worth noting that proteolytic rates are similarly reduced in both nodules and hepatomas, indicating that restriction in protein degradation might be an early event in carcinogenesis. Consistently, we and others have shown that the decrease in proteolytic rates results from a reduction in the rates of autophagic sequestration already evident in the early steps of the carcinogenetic process [60,61]. A direct evidence of impairment of autophagy in carcinogenesis derives from a study that uses the carcinogen Lindane [62]. Lindane induces a MAPK/ ERK-mediated decrease in autophagic proteolysis, resulting in accumulation of unfunctional autolysosomes. Thus, the high levels
of ERK expression in some human cancers might provide a selective advantage by impairing the tumour suppression function of autophagy. A novel cell death pathway that combines apoptosis and autophagy is selectively inactivated at the early phase of immortalization of breast and prostate epithelial carcinogenesis. Thus, paradoxically the loss of the autophagic survival pathway favours tumour progression.
Why do preneoplastic or neoplastic cells reduce autophagy? It is largely accepted that limited autophagy is an adaptive mechanism to allow cancer cells to overcome limitation in nutrient supply [57]. In particular, the tolerance for nutrient deprivation has been proposed to be an important factor for tumour progression under hypovascular conditions and elimination of the tolerance might serve as a new strategy for cancer therapy. In line, tumour cells in culture grow with no or very low serum concentration. MCF7 human breast cancer cells lack Beclin1 expression and are unable to undergo nutrient deprivation-induced autophagy [63]. Consistently, no changes were observed in the expression levels of Beclin1 during amino acid deprivation of HepG2 liver cancer cells. What are the mechanisms by which tumour cells reduce the basal levels of autophagy? Up to now, evidence is available thank to ATG genes research, confirming the involvement of autophagy already at the earlier phase of tumour progression, the initiation. Recent data support that autophagy is a novel and unique tumour suppressor mechanism. Consistently, a single allele of BECN1 is often deleted in human mammary, ovarian and prostate cancer as well as in breast cancer cell lines and reduces their tumourigenic potential on xeno-transplantation into immunodeficient mice [15]. UVRAG is also a haploinsufficient tumour suppressor gene, being monoalleli-cally mutated at increased frequency in human colon cancers and it is required for Beclin1 function of suppressing tumourigenesis [64]. Also Bif-1, that interacts with Beclin 1 through UVRAG at the phagophore, negatively controls oncogenesis [65]. WIPI-1 , a member of a novel WD-repeat protein family involved in the assembly of multiprotein complexes, is an autophagy effector downstream class III PI3K hypoexpressed in human cancers [66]. Also LC3 is localized to a locus frequently deleted in liver, breast, prostate and ovarian cancer as well as human Atg7 lacks in lung cancer. Atg5 over-expression sensitizes tumour cells to various apoptotic stimuli [34]. In line, positive effectors of the autophagic process, such as Beclin1, function as tumour suppressors, while negative effectors of autophagy, such as Bcl-2, act as oncogenes and they might be responsible for the reduction of autophagic capacities frequently observed in cancer cells [57]. These data fit with previous observations from experimental carcinogenesis and indicate that reduction in autophagy could contribute to tumour development.
If early suppression of autophagy during oncogenesis actually contributes to tumour initiation, activation of a proto-oncogene or loss of a tumour suppressor gene in a normal cell should down-regulate the autophagic process. In line, Ras transformation depresses nutrient deficiency-induced autophagy by activating the class I PI3K pathway [67]. Intriguingly, over-expression of Myc stimulates autophagy in rat fibroblasts cultured in rich-nutrient conditions without any stress independently from its oncogenic or apoptogenic actions [68]. The double actions of Ras and the pro-autophagic function of Myc suggest that the interplay between the diverse signal transduction and the cellular contest with constitu-tively activated genes, controlling the main cellular functions, might account for these controversial findings, thus modulating the autophagic response in an unexpected way. Conversely, introduc-tion of a tumour suppressor gene might up-regulate the autophagic machinery. On the whole, proto-oncogenes such as class I PI3K, AKT, PDKI, Ras, mTOR, Bcl-2 negatively regulate autophagy while tumour suppressor genes such as PTEN, TSC , DAPK, p53, p27 positively control it [57].

110 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
How might autophagy limit carcinogenesis? An autophagy gene might contribute to restrict tumorigenesis presumably through its cellular housekeeping role in removing genotoxic substances that would otherwise induce mutations and altered cytoplasmic constituents, such as aggregated proteins and damaged organelles. ROS-producing, damaged mitochondria and other organelles might generate genotoxic stress and increase the probability of DNA damage and, as a consequence, the likelihood of oncogenic mutations. Thus, suppression of autophagy could contribute to development of cancer by allowing genotoxic free radicals to accumulate. It is likely that mitochondrial surveillance through autophagy plays an important role in tumour suppression, indicating that activation of autophagy may be a novel strategy for tumour prevention. Autophagy can suppress cell growth by degrading damaged mitochondria, but spares their normal counterparts [52]. Recently, White’s group [69] shows that autophagy functions to protect genoma during metabolic stress, providing a satisfactory explanation for how the loss of a survival programme can be one of the first events in carcinogenesis.
Again, autophagy favours cell differentiation, cell cycle arrest or promotes autophagic death of oncogenic cells. A massive autophagy kills the severely damaged cells as a safeguard mecha-nism against cancer. Conversely, defect in this pathway would confer a selective advantage to cells with cancer as consequence. Autophagy can limit oncogenesis through the involvement of the DAPk [42]. Also autophagy inhibits angiogenesis, limits necrosis and inflammation, and enhances tumour immunity, indicating that restriction of autophagy contributes to cell proliferation [39]. The tumour-suppressive function of Beclin1 has been related to its putative effector function in ACD. However, studies in Beclin1 deficient animals and cells suggest that the induction of death does not contribute to its function of tumour suppressor. Becn -/- mice die early during embryogenesis with massive cell death [17]. Mice heterozygous for Beclin1 display antigen-stimulated proliferation of B cells and enhanced proliferation in tissues which are prone to tumour formation, but not reduced cell death [62]. Similarly, embryonal stem cells knockout for Beclin1 are susceptible to death [17]. Beclin1 over-expression results in inhibition of cell proliferation, clonigenicity and tumorigenesis. Thus, restriction of autophagy might contribute to cell proliferation. This is supported by the fact that, differently from the most known tumour suppressor genes that usually display loss of heterozygosis in cancer, the wild-type BECN1 allele has never been found to be silenced or mutated in human cancers or in tumours developing in Becn1-heterozygous mice [63]. However, Beclin1 is not only the regulatory subunit of the Class III PI3K complex, but also the mammalian ortholog also involved in vacuolar protein sorting [57]. Thus, it remains unclear whether tumour cells need Beclin1 for autophagy or for autophagy-independent survival or growth. In line with the data on suppression of cell proliferation by Beclin1, high levels of Ambra 1, the novel Beclin1 interacting protein, besides elicits autophagy, decrease cell proliferation, an effect that needs the presence of Beclin1 [38]. Conversely, in Ambra 1-deficient embryos, neural tubes exhibit an increase in cell proliferation. Again, Atg7-deficient mice develop hepatomegaly [4], a preneoplastic lesion resulting from enhanced hepatocyte division and/or cell mass. Consistently, rapamycin induces autophagy and suppresses proliferation of malignant glioma [28]. It is worth noting that 3-MA increased the viability of hepatocytes isolated by normal rats to a level similar to that observed for the tumour cells [61]. This suggests that the decline in autophagic degradation contributes to the fast growth rates of cancer cells and confers them a selective advantage over the non-neoplastic hepatocytes. Taken together, these observations suggest that the function of autophagy proteins in tumour suppression implies inhibition of cell proliferation rather than induction of cell death. The data also indicate a mere explanation for the role of reduced autophagic rates in tumour development, the tumour requires higher levels of protein synthesis than protein catabolism
to grow, thus decrease in autophagy favours tumour growth. That means that autophagy can suppress cancer by controlling cell mass. In line, cell accumulation in liver cancer mainly occurs through reduced catabolic rates rather than enhanced protein synthesis [57] and highly proliferating hepatoma cells can display virtually no autophagy [20].
On one hand, growth factors and their signal transducers are known to suppress autophagy because they increase the uptake of nutrients and they activate the class I PI3K/Akt/mTOR signalling that both down-regulate the autophagic process, and they enhance cell anabolism and repress catabolism, thus leading to stimulation of cell growth and division [3]. Again, inhibition of autophagy by growth factors and transducers impairs the function of removing damages macromolecules and organelles, favouring accumulation of such damage and, thus, contribute to the onset and progression of cancer. On the other hand, growth inhibitory factors and their down stream transducers enhance the autophagic process. Intriguingly, it is worth noting that cancer cells may exploit any opportunity to lose autophagic and cytostatic responsiveness to the best known growth inhibitory factor TGF- and they use TGF- signalling to exacer-bate their own proliferative, invasive and metastatic behaviour. Thus, TGF- switches from a tumour suppressor into a tumour instigator. The several ways by which autophagy can suppress cancer onset and development support the concept of a cross-talk between autophagy, cell proliferation and death for carcinogenesis, which can be exploited for therapeutic approaches.
On the whole, the net balance between the pro-survival onco-genic functions and the pro-death tumour suppressor functions of autophagy might contribute to the knowledge of how to modify the autophagic process to counteract tumour progression. Further studies are needed to verify whether the decreased autophagic activity of cancer cells is instrumental in their active proliferation or, instead, a consequence better understanding of the tumour suppression functions of autophagy.
4. THERAPEUTIC IMPLICATIONS
The autophagic lysosomal pathway is a novel therapeutic target for cancer treatment. How might autophagy be manipulated to improve cancer therapy? Given the dual role of autophagy in cancer, it is not surprising that both inhibition and activation of autophagy can be benefical for cancer therapy [57,58]. Newly developed autophagy inhibitors and stimulators are potent anticancer drugs, yet their underlying molecular mechanism largely remains to be elucidated. The lack of autophagy can enhance susceptibility to death when cancer cells face stressful conditions, such as metabolic stress and therapeutic interventions. Kondo’s group showed for the first time the possibility of treating cancer cells by autophagy inhibition, since cancer cells respond to radiation, drug and hyperthermia by inducing autophagy [28]. Herman-Antosiewicz et al. [70] reported for the first time that the induction of autophagy by an isothiocyanate class of dietary chemopreventive agents and autophagy inhibition enhance apoptosis. More recently, autophagy inhibition has been shown to sensitize cells to p53-mediated or alkylating drug-induced apoptosis [71]. Consistently, autophagy down-regulation unmasks a caspase-dependent apoptotic response to DNA damage induced by camptothecin in MCF-7 cells [72]. The use of autophagy inhibitors for sensitizing tumour cells to BH3 mimetics or indirect activators of BH3-dependent apoptotic pathways might be useful [9]. These observations indicate that autophagy protects cells from drug-induced apoptosis, thus blocking autophagy might enhance the efficacy of pro-apoptotic chemotherapeutic strategies for the treatment of certain cancers. Because autophagy is commonly induced by hypoxia and it represents the ultimate nutritional source for tumour cells to survive low nutrient conditions [28], suppression of autophagy in combination with other treatments accelerate tumour death [19].

Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 111
On the other hand, stimulation of autophagy is a possible way to prevent/revert the neoplastic phenotype by limiting genome instability, since many tumours display lower autophagic activity and reduction of autophagic responses might represent one of the initial events during transformation. First, in tumour cells deficient in the autophagic pathway and resistant to ACD and autophagy-dependent apoptosis, replacement of autophagy-related proteins or positive regulators of autophagy might inhibit cell proliferation and revert the neoplastic phenotype or induce cell death. Current efforts are now focused on the identification of other methods that can activate autophagy. Because cancer cells frequently activate the PI3K/AKT/mTOR signalling which negatively controls autophagy, this pathway might be manipulated to activate autophagy in cancer cells. Rapamycin and its analogues are anticancer agents that specifically target mTOR already approved as a therapeutic drug in cancer clinical trials. They reduce tumour mass and stabilize disease in patients with metastasis. They are particularly useful for the cure of cancers which have inactivated the PTEN tumour suppressor and depend on the AKT pathway for survival. Rapa-mycin activates the autophagic process and induces ACD in cancer cells which have the autophagic machinery still functional even if down-regulated. Thus, it is likely that its anti-cancer effects are, at least in part, mediated by its action on the autophagic pathway. Indeed, rapamycin induces autophagy and suppresses lung adenocarcinoma growth and invasion [73]. It has been shown that small-molecule enhancers of the cytostatic effects of rapamycin (SMERs) enhance autophagy and might be helpful for cancer therapy. It is reasonable to think that autophagy-dependent death potential of cells resistant to apoptosis might be exploited for therapy by the administration of autophagy-inducing agents [3,28, 53,59]. Pharmacological inhibition of p38 has been recently proposed as a novel strategy to enhance autophagy as potential therapeutic tool for human cancers because of the possibility of local delivery of the drug [36].
Recently, several natural products have been found to trigger ACD. Apoptosis-resistant ovarian carcinoma cells respond to resveratrol by dying with autophagic vacuoles. Plumbagin, a quinonoid constituent isolated from the root of Plumbago zeylanica L., the “Chitrak” from Indian Medicine with anticarcinogenic properties inhibits AKT/mTOR pathway to induce autophagy and G2/M arrest [74]. Again, extracts of Solanum nigrum L., a plant indigenous to Asia, currently used in traditional oriental medicines for the cure of various kinds of tumours exert its cytotoxic effect on HepG2 cells by stimulating autophagic or apoptotic death, depending on the dose. In this context, autophagy is considered to be an alternative way to kill tumour cells when they are radio/ chemoresistant on the basis of ineffective apoptosis. Intriguingly, autophagy may interfere with cell death by modulating different pathways [75]. Curcumin, a natural product with low toxicity in normal cells, inhibits the Akt/mTOR/p70S6K pathway and activates ERK1/2 pathway, resulting in G2/M arrest and ACD in malignant glioma cells. However, activation of the Akt pathway suppresses drug-induced autophagy and death, while inhibition of ERK1/2 pathway inhibits drug-induced autophagy and triggers apoptosis, resulting in enhanced cytotoxicity.
New antitumour agents, able to overcome MDR, induce cell death in leukaemia cells via an autophagic process, with the absence of apoptosis, probably by interfering with mitochondrial function [76,77]. Induction of autophagy is also therapeutic because it can initiate an immune response by enhanced presentation of MHC class II antigens, thus promoting immunological recognition and elimination of cancer cells [78]. It is reasonable to think that autophagy might be initiated as an attempt to remove an irreversibly damaged molecule or organelle and to preserve the cells from a metabolic collapse and excess levels of autophagy while its persistence leads to death.
To induce a switch from necrosis to autophagy or ACD might be a therapeutic goal to avoid necrosis-mediated inflammation leading to enhancement of the original tissue damage, including cancer progression. Combined inhibition of executioners of apoptosis and essential mediators of the necrotic cascade associated to the activation of autophagy would truly prevent cell death [59].
On the whole, the above examples show the complexity of predicting the net effect of autophagy in cancer. Among the variables to be taken into account, the tumour progression, the sensitivity of the tumour cells to apoptosis and the specific genetic and epigenetic molecular alterations in the cells can be critical for the outcome.
5. CONCLUSION
The dual role of autophagy in cell survival and cell death has been known since de Duve discovered the catabolic machinery. In some cases the molecular machinery of autophagy is required during cell death as shown by genetic methods. Autophagy can either prevent or favour apoptosis. Thus, autophagy can function as a balance. The other paradox is the dual nature of autophagy in cancer. Evidence indicates that the failure of autophagy signalling is instrumental in malignant transformation and, in some circums-tances, activation of autophagy can allow cancer cells to survive, implying modulation of autophagy as a possible therapeutic target. For clinical use it becomes relevant to determine whether enhanced autophagy is a sign of drug responsiveness or resistance. To find out whether cancer cells use autophagy to survive therapy, autophagy inhibitors might improve the efficacy of anticancer therapy. Conversely, when autophagy contributes to oncogenesis, its activation might limit genome damage and revert the neoplastic phenotype. Novel therapeutic strategies combined drugs inhibiting/ enhancing autophagy with conventional chemotherapy, radio-therapy, hormonotherapy and anti-angiogenetic therapy might be the future approaches. One of the current challenges is to design new methods to modulate autophagy. Another is to determine when and how long this modulation can be effectively maintained. Tumour cells with signs of autophagic vacuoles should be analysed for actual detection of autophagic capacity to rule out an engulfment of cytoplasmic material without lysosomal breakdown. However, when autophagy is involved in drug resistance of cancer cells, inhibition of autophagy might be useful to sensitise cells to cell death induced by conventional therapies.
These challenges notwithstanding, the growing ability to develop new therapeutic strategies and explain why may work or fail indicates the progress achieved. The core machinery of autophagy has been delineated, at least in broadly. Many questions remain and many principles need to be established, but a problem that has long fascinated biologists is beginning to be unravelled. In the future, it will be important to understand the molecules involved in the autophagic pathway as autophagy proteins, positive and negative regulators and biochemical switches which direct a cell towards autophagy or apoptosis or necrosis. The modulation of intracellular switches by pharmaceuticals might optimize cancer treatment by increasing both conventional therapy sensitization and autophagy. Like all research at the cutting edge of a developing field, the recent findings, while providing novel insights about the role of autophagy in cell death and cancer therapy, also open new questions which stimulate further interest for future studies.
ACKNOWLEDGMENT
We thank RA Billington for correcting the English language, C Bottini and EL Failla for art graphic support. Work was supported by MIUR, CNR, AIRC, Lega Italiana per la Lotta contro i Tumori of Novara and University of East Piedmont “A. Avogadro”. This wprk was partially financed by: grant from the Ministerio de Sanidad y Consumo (FIS 06/1226) España.

112 Current Pharmaceutical Design, 2010, Vol. 16, No. 1 Tessitore et al.
REFERENCES
References 79-81 are related articles recently published.
[1] Cuervo AM. Autophagy: many paths to the same end. Mol Cell
Biochem 2004; 263: 55-72. [2] Martinez-Vicente M, Cuervo AM. Autophagy and neurodegene-
ration: when the cleaning crew goes on strike. Lancet Neurol 2007; 6: 352-61.
[3] Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115: 2679-88.
[4] Mizushima N. Autophagy: process and function. Genes Dev 2007; 21: 2861-73.
[5] Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132: 27-42.
[6] Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:
931-7. [7] Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death
control. Nat Rev Cancer 2005; 5: 886-97. [8] Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev
Mol Cell Biol 2005; 6: 505-10. [9] Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and
self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741-52.
[10] Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181: 195-213.
[11] Bursch W, Ellinger A, Kienzl H, Török L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and
ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996; 17: 1595-607.
[12] Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl
Acad Sci U S A 2006; 103: 4952-7. [13] Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-
Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy
genes. Nat Cell Biol 2004; 6: 1221-8. [14] Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H,
Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032-6.
[15] Baccino FM, Tessitore L, Cecchini G, Messina M, Zuretti MF, Bonelli G, et al. Control of cell protein catabolism in rat liver.
Effects of starvation and administration of cycloheximide. Biochem J 1982; 206: 395-405.
[16] Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation
using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101-11.
[17] Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a
haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100: 15077-82.
[18] Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature 2006; 441: 880-4. [19] Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans:
cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6: 439-48.
[20] Tessitore L, Bonelli G, Cecchini G, Amenta JS, Baccino FM. Regulation of protein turnover versus growth state: ascites
hepatoma as a model for studies both in the animal and in vitro. Arch Biochem Biophys 1987; 255: 372-84.
[21] Tessitore L, Bonelli G, Cecchini G, Autelli R, Amenta JS, Baccino FM. Regulation of protein turnover versus growth state. Studies on
the mechanism(s) of initiation of acidic vacuolar proteolysis in cells of stationary ascites hepatoma. Biochem J 1988; 251: 483-90.
[22] Pfeifer U, Tessitore L, Bonelli G, Baccino FM. Regulation of protein turnover versus growth state III. Growth cessation is
associated with activation of autophagy in Yoshida ascites hepatoma AH-340. Virchows Arch B Cell Pathol Incl Mol
Pathol1988; 55: 363-9. [23] Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE,
Baccino FM, et al. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells.
Carcinogenesis 1993; 14: 2501-5.
[24] Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P. Autophagy
signaling and the cogwheels of cancer. Autophagy 2006; 2: 67-73. [25] Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al.
The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or
apoptosis. Nat Cell Biol 2007; 9: 218-24. [26] Golstein P, Kroemer G. Cell death by necrosis: towards a molecular
definition. Trends Biochem Sci 2007; 32: 37-43. [27] Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al.
Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931-46.
[28] Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer
2005; 5: 726-34. [29] Tessitore L, Tomasi C, Greco M. Fasting-induced apoptosis in rat
liver is blocked by cycloheximide. Eur J Cell Biol 1999; 78: 573-9. [30] Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al.
Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120: 237-48.
[31] Trincheri NF, Follo C, Nicotra G, Peracchio C, Castino R, Isidoro C. Resveratrol-induced apoptosis depends on the lipid kinase
activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis 2008; 29: 381-9.
[32] Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr
Biol 2007; 17: 1-11. [33] Liang XH, Jackson S, Seaman M, Brown K, Kempkes B,
Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672-6.
[34] Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol 2006; 8: 1045-7.
[35] Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and
multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J
2006; 2: 756-8. [36] Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, et
al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death
Differ 2007; 14: 693-702. [37] Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P,
Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025-40.
[38] Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of
the nervous system. Nature 2007; 447: 1121-5. [39] Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in
cancer. Nat Rev Cancer 2007; 7: 961-67. [40] Crighton D, O'Prey J, Bell HS, Ryan KM. p73 regulates DRAM-
independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14: 1071-9.
[41] Codogno P. Autophagy and caspase-independent cell death: p19ARF enters the game. Dev Cell 2006; 10: 688-9.
[42] Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy 2006; 2: 74-9.
[43] Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1)
in monocytic differentiation. EMBO J1999; 18: 1223-34. [44] Guicciardi M, Leist M, Gores J. Lysosomes in cell death. Oncogene
2004; 23: 2881-90. [45] Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine
Growth Factor Rev 2007; 18: 233-43. [46] Wang ZH, Xu L, Duan ZL, Zeng LQ, Yan NH, Peng ZL. Beclin 1-
mediated macroautophagy involves regulation of caspase-9 expression in cervical cancer HeLa cells. Gynecol Oncol 2007; 107:
107-13. [47] Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T,
Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124-32.
[48] Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress?
Cell Cycle 2007; 6: 793-8. [49] Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M,
Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol
Cell 2007; 25: 193-205.

Understanding Autophagy in Cell Death Control Current Pharmaceutical Design, 2010, Vol. 16, No. 1 113
[50] Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et
al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927-39.
[51] Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in
cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998; 1366: 177-96.
[52] Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 2007; 462:
245-53. [53] Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy
2006; 2: 85-90. [54] Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S,
Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002; 111:
703-8. [55] Thorburn J, Moore F, Rao A, Barclay WW, Thomas LR, Grant
KW, et al. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in
immortal epithelial cells. Mol Biol Cell 2005; 16: 1189-99. [56] Wang L, Yu C, Lu Y, He P, Guo J, Zhang C, et al. TMEM166, a
novel transmembrane protein, regulates cell autophagy and apoptosis. Apoptosis 2007; 12: 1489-502.
[57] Levine B. Cell biology: autophagy and cancer. Nature 2007; 446: 745-7.
[58] Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23: 2891-906.
[59] Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007;
6: 304-12. [60] Canuto RA, Tessitore L, Muzio G, Autelli R, Baccino FM. Tissue
protein turnover during liver carcinogenesis. Carcinogenesis 1993; 14: 2581-7.
[61] Schwarze PE, Seglen PO. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes
isolated from carcinogen-treated rats. Exp Cell Res 1985; 157: 15-28.
[62] Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, et al. Disruption of autophagy at the maturation step by the
carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase
activity. Cancer Res 2006; 66: 6861-70. [63] Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al.
Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809-20.
[64] Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-
binding protein UVRAG. Nat Cell Biol 2006; 8: 688-99. [65] Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato
Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:
1142-51.
[66] Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A,
Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human
cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23: 9314-25.
[67] Furuta S, Hidaka E, Ogata A, Yokota S, Kamata T. Ras is involved in the negative control of autophagy through the class I PI3-kinase.
Oncogene 2004; 23: 3898-904. [68] Tsuneoka M, Umata T, Kimura H, Koda Y, Nakajima M, Kosai K,
et al. c-myc induces autophagy in rat 3Y1 fibroblast cells. Cell Struct Funct 2003; 28: 195-204.
[69] Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al.Autophagy suppresses tumor progression by limiting
chromosomal instability. Genes Dev 2007; 21: 1367-81. [70] Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane
causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res 2006; 66: 5828-35.
[71] Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326-36. [72] Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A.
Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14: 500-10.
[73] Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET Inhibitor, PHA665752, cooperates with
rapamycin. Clin Cancer Res 2005; 11: 2312-9. [74] Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not
only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res 2007; 31: 329-39.
[75] Kuo PL, Hsu YL, Cho CY. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin
pathway in breast cancer cells. Mol Cancer Ther 2006; 5: 3209-21. [76] Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y.
Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and
extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007; 72: 29-39.
[77] Kessel D, Reiners JJ Jr, Hazeldine ST, Polin L, Horwitz JP. The role of autophagy in the death of L1210 leukemia cells initiated by
the new antitumor agents, XK469 and SH80. Mol Cancer Ther 2007; 6: 370-09.
[78] Münz C. Autophagy and antigen presentation. Cell Microbiol 2006; 8: 891-8.
[79] Penaloza C, Orlanski S, Ye Y, Entezari-Zaher T, Javdan M, Zakeri Z. Cell death in mammalian development. Curr Pharm Des 2008;
14(2): 184-96. [80] Samara C, Tavernarakis N. Autophagy and cell death in
Caenorhabditis elegans. Curr Pharm Des 2008; 14(2): 97-115. [81] Tettamanti G, Salo E, Gonzalez-Estevez C, Felix DA, Grimaldi A,
de Eguileor M. Autophagy in invertebrates: insights into develop-ment, regeneration and body remodeling. Curr Pharm Des 2008;
14(2): 116-25.
Received: June 18, 2009 Accepted: June 29, 2009
Related Documents











