c 2005 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 10.1002/14356007.a23 661 Silicates 1 Silicates Asbestos, Cement and Concrete, Clays, Glass, Glass Ceramics, Mica, Silica, Talc, and Zeolites are separate keywords. Gerhard Lagaly, Institute of Inorganic Chemistry, University of Kiel, Kiel, Federal Republic of Germany (Chap. 1) Werner Tufar, Department of Geosciences, Philipps University Marburg, Marburg, Federal Republic of Germany (Chaps. 1, 2) A. Minihan, Unilever Research Port Sunlight Laboratory, Bebington, Wirral, United Kingdom (Sections 3.1– 3.4 and 3.10) A. Lovell, Crosfield Chemicals, Warrington, Cheshire, United Kingdom (Sections 3.5 – 3.9) 1. Structural Chemistry of Silicates .. 2 1.1. Structural Classification ....... 3 1.2. Oligo- and Cyclosilicates ....... 4 1.3. Polysilicates ................ 4 1.4. Phyllosilicates .............. 6 1.4.1. Monophyllosilicates ........... 6 1.4.2. Diphyllosilicates ............. 8 1.4.3. Alkali Silicates .............. 8 1.4.4. Crystalline Silicic Acids ........ 8 1.4.5. Clay Minerals ............... 10 1.4.6. Intracrystalline Reactions ........ 13 1.5. Tectosilicates ............... 14 2. Natural Silicates ............. 15 2.1. Feldspar .................. 15 2.1.1. Structure and Composition ....... 15 2.1.2. Characterization of Individual Feldspars .................. 17 2.1.3. Production ................. 18 2.1.4. Properties .................. 19 2.1.5. Mineral Deposits and their Extraction 20 2.1.6. Processing and Quality Requirements 21 2.1.7. Uses and Economic Aspects ...... 21 2.2. Nepheline and Related Compounds 24 2.3. Leucite ................... 27 2.4. Olivine ................... 28 2.5. Andalusite ................. 31 2.6. Kyanite ................... 34 2.7. Sillimanite ................. 36 2.8. Mullite ................... 37 2.9. Vermiculite ................ 40 2.10. Perlite ................... 44 2.11. Pumice ................... 46 2.12. Basalt .................... 47 2.13. Wollastonite ................ 47 2.14. Toxicology ................. 52 3. Alkali Silicates .............. 52 3.1. Introduction ............... 52 3.2. Raw Materials .............. 53 3.3. Amorphous Anhydrous Alkali Sili- cates (Solid or Lump Glasses) .... 53 3.4. Silicate Solutions ............ 55 3.5. Hydrated Water-Soluble Silicates . 58 3.6. Crystalline Solids ............ 58 3.7. Uses and Applications ......... 59 3.8. Economic Aspects ............ 61 3.9. Storage, Safety, Labelling and Transportation ........... 61 3.10. Analysis .................. 62 4. References ................. 63 1. Structural Chemistry of Silicates More than 95 vol % of the earth’s crust is com- posed of quartz and a few rock-forming silicates (plagioclase 42 vol %, potash feldspar 22 vol %, quartz 18 vol %, amphibole 5 vol %, other sil- icates 12 vol %). Silicon compounds are also present in the hydrosphere, mainly as dissolved silica. A large number of silicates have been identified in extraterrestrial material [1]. The widespread abundance of silicate min- erals is accompanied by a diversity of practi- cal uses (Table 1). Silicates are components of technical materials such as cement (→ Cement and Concrete) and glass (→ Glass, Glass Ce- ramics). Granite and sandstone are used as building materials. Quartz (→ Silica) is the raw material for producing waterglass, silicon (→ Silicon), and silicon compounds (→ Silicon Compounds, Inorganic, → Silicon Compounds,

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

c© 2005 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim10.1002/14356007.a23 661
Silicates 1
Silicates
Asbestos, Cement and Concrete, Clays, Glass, Glass Ceramics, Mica, Silica, Talc, and Zeolites are separatekeywords.
Gerhard Lagaly, Institute of Inorganic Chemistry, University of Kiel, Kiel, Federal Republic of Germany(Chap. 1)
Werner Tufar, Department of Geosciences, Philipps University Marburg, Marburg, Federal Republic ofGermany (Chaps. 1, 2)
A. Minihan, Unilever Research Port Sunlight Laboratory, Bebington, Wirral, United Kingdom (Sections 3.1 –3.4 and 3.10)
A. Lovell, Crosfield Chemicals, Warrington, Cheshire, United Kingdom (Sections 3.5 – 3.9)
1. Structural Chemistry of Silicates . . 21.1. Structural Classification . . . . . . . 31.2. Oligo- and Cyclosilicates . . . . . . . 41.3. Polysilicates . . . . . . . . . . . . . . . . 41.4. Phyllosilicates . . . . . . . . . . . . . . 61.4.1. Monophyllosilicates . . . . . . . . . . . 61.4.2. Diphyllosilicates . . . . . . . . . . . . . 81.4.3. Alkali Silicates . . . . . . . . . . . . . . 81.4.4. Crystalline Silicic Acids . . . . . . . . 81.4.5. Clay Minerals . . . . . . . . . . . . . . . 101.4.6. Intracrystalline Reactions . . . . . . . . 131.5. Tectosilicates . . . . . . . . . . . . . . . 142. Natural Silicates . . . . . . . . . . . . . 152.1. Feldspar . . . . . . . . . . . . . . . . . . 152.1.1. Structure and Composition . . . . . . . 152.1.2. Characterization of Individual
Feldspars . . . . . . . . . . . . . . . . . . 172.1.3. Production . . . . . . . . . . . . . . . . . 182.1.4. Properties . . . . . . . . . . . . . . . . . . 192.1.5. Mineral Deposits and their Extraction 202.1.6. Processing and Quality Requirements 212.1.7. Uses and Economic Aspects . . . . . . 212.2. Nepheline and Related Compounds 242.3. Leucite . . . . . . . . . . . . . . . . . . . 272.4. Olivine . . . . . . . . . . . . . . . . . . . 28
2.5. Andalusite . . . . . . . . . . . . . . . . . 312.6. Kyanite . . . . . . . . . . . . . . . . . . . 342.7. Sillimanite . . . . . . . . . . . . . . . . . 362.8. Mullite . . . . . . . . . . . . . . . . . . . 372.9. Vermiculite . . . . . . . . . . . . . . . . 402.10. Perlite . . . . . . . . . . . . . . . . . . . 442.11. Pumice . . . . . . . . . . . . . . . . . . . 462.12. Basalt . . . . . . . . . . . . . . . . . . . . 472.13. Wollastonite . . . . . . . . . . . . . . . . 472.14. Toxicology . . . . . . . . . . . . . . . . . 523. Alkali Silicates . . . . . . . . . . . . . . 523.1. Introduction . . . . . . . . . . . . . . . 523.2. Raw Materials . . . . . . . . . . . . . . 533.3. Amorphous Anhydrous Alkali Sili-
cates (Solid or Lump Glasses) . . . . 533.4. Silicate Solutions . . . . . . . . . . . . 553.5. Hydrated Water-Soluble Silicates . 583.6. Crystalline Solids . . . . . . . . . . . . 583.7. Uses and Applications . . . . . . . . . 593.8. Economic Aspects . . . . . . . . . . . . 613.9. Storage, Safety, Labelling
and Transportation . . . . . . . . . . . 613.10. Analysis . . . . . . . . . . . . . . . . . . 624. References . . . . . . . . . . . . . . . . . 63
1. Structural Chemistry of Silicates
More than 95 vol% of the earth’s crust is com-posed of quartz and a few rock-forming silicates(plagioclase 42 vol%, potash feldspar 22 vol%,quartz 18 vol%, amphibole 5 vol%, other sil-icates 12 vol%). Silicon compounds are alsopresent in the hydrosphere, mainly as dissolvedsilica. A large number of silicates have beenidentified in extraterrestrial material [1].
The widespread abundance of silicate min-erals is accompanied by a diversity of practi-cal uses (Table 1). Silicates are components oftechnical materials such as cement (→Cementand Concrete) and glass (→Glass, Glass Ce-ramics). Granite and sandstone are used asbuilding materials. Quartz (→Silica) is theraw material for producing waterglass, silicon(→Silicon), and silicon compounds (→SiliconCompounds, Inorganic, →Silicon Compounds,

2 Silicates
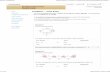
Figure 1. Oligosilicates and cyclosilicatesA) Monosilicates (forsterite, olivine, phenakite, garnets, zircon; formulae see text); B) Disilicates (thortveitite, Sc2[Si2O7];C) Monocyclosilicate: dreier single ring (benitoite, BaTi[Si3O9]); D) Monocyclosilicate: vierer single ring (taramel-lite, Ba4(Fe,Ti)4B2[Si4O12]2O5Clx ); E) Monocyclosilicate: sechser single ring (beryl, Be3Al2[Si6O18]; tourmalinesXY3Z6B3[Si6O18]O9(O, OH, F)4 (X =Na+, Ca2+; Z =Al3+, Mg2+; Y =Li+, Mg2+, Fe2+, Mn2+, Fe3+, Al3+); diop-tase, Cu6[Si6O18] · 6 H2O); F) Dicyclosilicate: sechser double ring (milarite, KCa2(Be2Al) [Si12O30] · 0.75 H2O)
Organic, →Silicones) and modern nonclay ce-ramics (SiC, Si3N4) (→Silicon Carbide). Mod-ern life is not only determined by the use of syn-thetic polymers but also by the widespread ap-plication of silicates and products derived fromthem. For example, silicon forms the basis ofmicroelectronics.
Table 1. Practical applications of natural silicates
Mineral(rock)
Uses Production,106 t/a
Kaolinite(kaolin)
paper industry, ceramics, chemical andpharmaceutical industries
25 (kaolin)
Smectite(bentonite)
foundry molding sands, iron orepelletizing, oil well drilling, buildingindustry, chemical and pharmaceuticalindustries
7(bentonite)
Talc,pyrophyllite
filler, electrical insulation, ceramics 6
Feldspars glass and ceramic industry, filler 3.5∗Micas filler, electrical industry 2.7Olivine,forsterite
refractory masses, fluxes, foundrymolding sands
2.3
Perlite lightweight material for thermal andsound insulation
2.5
Vermiculite thermal insulation, filler, lightweightmaterial
0.65
Sillimanite refractory masses 0.15Andalusite refractory massesKyanite refractory massesMullite refractory masses 0.005Zircon refractory material, ceramics, zirconium
compounds0.1
Spodumene,petalite
production of lithium
Beryl production of berylliumChrysotilasbestos
(high toxicity) 4 (1978)
∗ Including nepheline, leucite: 4.5×10 6 t/a.
1.1. Structural Classification
Silicates comprise the largest, most extensive,and manifold class of minerals. One reasonis that the bond energy of the Si –O bondis higher than that of the Si – Si and Si –Hbonds (bond energies, kJ/mol: Si –O, 452; Si –Si, 222; Si –H, 318). Comparison with the val-ues for carbon (C –O, 358; C –C, 346; C –H,413) reveals why the skeletons in silicates areSi –O – Si –O chains instead of the C –C –Cchains in carbon chemistry. The second reasonis the ease with which silicon – oxygen tetrahe-dra can be linked to form rings, chains, layers, orthree-dimensional frameworks. Diversity is in-creased by isomorphous substitution of siliconby small cations (mostly Al3+, but also othertri- and divalent cationsB3+, Fe3+, Ga3+, Be2+,Zn2+,Mg2+). In the 2 : 1 claymineral sauconiteZn3[Si4−xZnxO10(OH)2], Si4+ is substitutedby Zn2+. Synthetic mica phases in which Co3+
and Ni3+ replace Si4+ have been reported [1].The common coordination number of
silicon bonded to oxygen is four, but afew silicates together with the SiO2 mod-ification stishovite contain sixfold coordi-nated silicon, e.g., Mg[SiO3] (high-tem-perature modification), (NH4)2[SiP4O13],Ca3[Si(OH)6][SO4][CO3] · 12H2O (thauma-site), modifications of Si[P2O7].
Themost comprehensive classification of sil-icates is presented by Liebau [1–3] (Table 2).The term multiplicity (M ) denotes the numberof single polyhedra, rings, chains, or layers thatare linked to the complex anion. Dimensional-

Silicates 3
ity refers to the infinite extension of the anion:D = 0, groups;D = 1, chains;D = 2, layers;D = 3,framework. A silicate that contains double tetra-hedra, as in Sc2[Si2O7] (thortveitite, Fig. 1B), iscalled a disilicate (M = 2, D = 0). OrthoenstatiteMg2[Si2O6] (Fig. 2A) is amonopolysilicate thatcontains single chains of silicon – oxygen tetra-hedra.
Figure 2.MonopolysilicatesA) Zweier single chains (pyroxenes); B)Dreier single chains(wollastonite, Ca3[Si3O9]
Sometimes, particularly in the technical lit-erature, silicates of composition MO · 2 SiO2,MO · 3 SiO2 (M=metal) are called di- and trisil-icates, respectively. This practice is in con-tradiction to IUPAC rules and must be aban-doned. Also, the term “metasilicates” for sil-icates M2SiO3, MSiO3, M2(SiO3)3 should beabandoned because these silicates can be cyclo-or polysilicates.
Connectedness of [SiO4] tetrahedra is a fur-ther parameter that should be mentioned in clas-sifying silicate structures. A [SiO4] tetrahedroncan share common oxygen ions with 0, 1, 2,3, or 4 [SiO4] tetrahedra; this is indicated byQ0, Q1, Q2, Q3, or Q4, respectively. The con-nectedness of silicon – oxygen tetrahedra canbe obtained from magic angle spinning –NMR(MAS–NMR) measurements. This is particu-larly advantageous in the study of amorphous
materials or silicates with high levels of defectsor of unknown structure.
Mineralogists classify the silicates into neso-,soro-, cyclo-, ino-, phyllo-, and tectosilicates(Table 3).
In classifying silicates on the basis of theway in which the silicon – oxygen tetrahedra arelinked to each other, it should be rememberedthat the real structure is determined by the in-teraction between the [SiO4] tetrahedra and thenontetrahedral cations (Section 1.3).
1.2. Oligo- and Cyclosilicates
When not specified otherwise, references con-cerning structure, crystallographic data, or syn-thesis of the silicates mentioned are found in [1].
Single [SiO4] tetrahedra are found inmonosilicates (Fig. 1A) such as forsteriteMg2[SiO4], olivine (Mg, Fe)2[SiO4],phenakite Be2[SiO4], garnets M12
3 M62[SiO4]3
(M12 =Mg2+, Fe2+, Mn2+; M6 =Al3+, Fe3+,Cr3+), and zircon Zr[SiO4]. Because of thehigh charge of the [SiO4]4− anion, the stabilityof monosilicates increases in the order:
K4[SiO4]<Ca2[SiO4]<Ca3Al2[SiO4]3 <Zr[SiO4]
Examples of disilicates (Fig. 1 B) are thortveititeand barysilite MnPb8[Si2O7]3. SyntheticAg10[Si4O13] is a tetrasilicate. Oligosilicateswith unbranched chains M = 8, 9, 10 have beensynthesized.
A few monocyclosilicates (with single rings,M = 1) and a dicyclosilicate are shown in Fig-ure 1C – F.
Several mono- and cyclosilicates are ofpractical importance. Magnesium silicate,Mg2[SiO4], is used as a refractory material(forsterite products, Section 2.4). Olivine (ca.2.3×106 t/a) is mainly used as flux for slagsin blast furnaces but small amounts are neededas foundry molding sands (Section 2.4). Gar-nets are utilized as bearings, polishing powder,abrasive products, and precious stones. (Thegarnets used as ferrimagnetic materials are yt-trium and rare-earth iron garnets M3+
3 Fe5O12which consist of [FeO4] tetrahedra in place of[SiO4] tetrahedra.) Thortveitite (with scandiumions partially substituted by yttrium ions) is animportant scandium mineral.

4 Silicates
Table 2. Classification of silicates [1]
Dimensionality Multiplicity
1 2 3 4
0 Oligosilicates monosilicates disilicates trisilicates tetrasilicates0 Cyclosilicates monocyclosilicates dicyclosilicates tricyclosilicates tetracyclosilicates1 Polysilicates monopolysilicates dipolysilicates tripolysilicates tetrapolysilicates2 Phyllosilicates monophyllosilicates diphyllosilicates triphyllosilicates tetraphyllosilicates3 Tectosilicates tectosilicates
Table 3. Nomenclature of silicates used by mineralogists
Dimensionality Multiplicity
1 2 3 4
0 nesosubsilicates nesosilicates sorosilicates0 cyclosilicates1 inosilicates2 phyllosilicates3 tectosilicates
Several oligo- and cyclosilicates are valuedas semiprecious or precious stones: garnets,olivine (chrysolite), zircon (hyacinth, jargon),beryl (emerald, aquamarine, heliodor, dioptas),turmaline (rubellite, schorl, indigolit).
Zircon is an abundant accessory mineral, forexample in glass ceramics (→Glass Ceramics).The radioactivity of rocks is often caused by thethorium and uranium content in zircon.
1.3. Polysilicates
Monopolysilicates. Polysilicates are abun-dant minerals, several are of practical impor-tance. Single- chain polysilicates (dimensional-ity 1, multiplicity 1) are classified according tothe number of silicon – oxygen tetrahedra con-tained in the repeat unit of the chain (periodicity,P = 2, 3, etc.). Figure 2 shows polysilicate sin-gle chains with periodicities of two and three.Monopolysilicates are known with periodicitiesin the range 3 – 24 (P = 24 in synthetic Na24Y8[Si24O72] [1]).
The large group of pyroxenes (Fig. 2A)consists of monopolysilicates with zweiersingle chains. Examples include enstatite(Mg2[Si2O6]), diopside (CaMg[Si2O6]), andspodumene (LiAl[Si2O6]). Spodumene is asource for lithium (→Lithium and LithiumCompounds, Chap. 3.1.1.) and glass ceramicproduction (→Glass Ceramics). Enstatite anddiopside are sometimes found in ceramicmasses(→Glass Ceramics). Several calcium silicates
contain dreier single chains, for examplewollas-tonite (Ca3[Si3O9], modifications 1T, 2M, 7T,Fig. 2 B). Wollastonite is used as a ceramic ma-terial, as a filler in plastics and paints, and as asubstitute for asbestos.
Figure 3. DipolysilicatesA) Einer double chain (high-temperature sillimanite,Al6[AlSiO5] (hT)); B) Zweier double chain (amphiboles,e.g., tremolite, Mg5Ca2[Si4O11]2(OH)2); C) Dreier dou-ble chain (xonotlite, Ca6[Si6O17](OH)2; D) Dreier doublechain (epididymite, Na2Be2[Si6O15] ·H2O)
Dipolysilicates. The arrangement of thetetrahedra in sillimanite (high-temperaturemod-ification) is shown in Figure 3A. However,the tetrahedral Al3+ and Si4+ ions are al-most completely ordered so that sillimanite con-tains zweier double chains (Fig. 3 B) [1]. Zweierdouble chains are also the skeletons in am-

Silicates 5
phiboles (Fig. 3 B). Xonotlite and epididymiteare dipolysilicates with dreier double chains(Fig. 3 C, D).
Figure 4. Mutual adjustment of polysilicate anions andcolumns of [MO6] octahedraA) Enstatite, Mg2[Si2O6]; B) Wollastonite, Ca3[Si3O9](low-temperature modification); C) Synthetic Ba2[Si2O6](high-temperature modification) Na2Be2[Si6O15] ·H2O)
Oligopolysilicates. Synthetic oligopolysili-cates are Ba4[Si6O16] (zweier triple chain),Ba5[Si8O21] (zweier fourfold chain), andBa6[Si10O26] (zweier fivefold chain).
InfluenceofNontetrahedralCationson theStructure of Polysilicates. The nontetrahedralcations exert a significant influence on the struc-ture of polysilicates, in particular on their pe-riodicity [4]. The mutual adaption of the con-formation of the silicate chains to columns of[MOn] polyhedra is shown for enstatite, wol-lastonite, and synthetic Ba2[Si2O6] (high-tem-perature modification) in Figure 4. The dimen-sions of two [MgO6] octahedra (ionic radius ofMg2+: 0.072 nm) in enstatite (Fig. 4A) corre-spond to the repeat unit (identity period; twotetrahedra) of the stretched zweier single chain.In wollastonite (Fig. 4 B) the dreier single chainfits the columnof the considerably larger [CaO6]octahedra (two octahedra and three tetrahedra;ionic radius of Ca2+: 0.10 nm). A further in-crease of the ionic radius of the octahedral cation(Ba2+: 0.135 nm) allows the adjustment of two[SiO4] tetrahedra to one [BaO6] octahedron: the
polyanion assumes the form of a zweier singlechain with the identity period distinctly smallerthan in enstatite. Further examples of the adapta-tion of the silicate chain to edge-sharing [MO6]octahedra are shown for pyroxenes and pyrox-enoids in Figure 5 [4].
Figure 5. Adjustment of polysilicate anions to “slabs” of[MoO6] octahedraA) Ferrosilite-III, FeSiO3 (high-temperature, medium-pres-sure modification) neuner single chain and [FeO6] oc-tahedra; B) Pyroxmangite-type MnSiO3 (medium-pres-sure modification) (pyroxmangite = (Fe,Mn)7[Si7O21]),siebener single chain and [MnO6] octahedra; C) Rhodonite-typeMnSiO3 (low-pressuremodification) (rhodonite = (Mn,Ca5)[Si5O15]), single chain and [MnO6] octahedra
Other factors besides the size of the non-tetrahedral cations (e.g., electronegativity andvalency) also control the conformation of thepolysilicate anion [2,4].
1.4. Phyllosilicates
1.4.1. Monophyllosilicates
Layers of silicon – oxygen tetrahedra are formedwhen silicate single chains are linked with eachother. A simple possibility is to build up thezweier single layer (Fig. 6) by connecting zweiersingle chains. The term directedness indicatesthe way in which the apical oxygen ions of the[SiO4] tetrahedra are oriented. In Figure 6A allapical oxygen ions point in the same direction.

6 Silicates
Figure 6. Formation of a zweier single-layer phyllosilicate by linking zweier single chainsA) Unfolded layer with terminal oxygen ions pointing in the same direction; B) Folded layer with terminal oxygen ionspointing up and down: Li2[Si2O5]; C) Folded layer in petalite, Li[4]Al[4][Si2O5]2
In many other cases, however, they point in op-posite directions (Fig. 6 B, C; Fig. 7).
Figure 7. Folding of tetrahedral layers in anhydrousmonophyllosilicatesA) Li2[Si2O]5; B)α-Na2[Si2O5]; C)β-Na2[Si2O5];D) Sanbornite, Ba[Si2O5] (low-temperature modification)
The layers shown in Figure 6 are highly flex-ible, even when the terminal oxygen ions alter-nate between both sides. The layers can be sta-bilized in three ways:
1) The layers are folded2) The layers are linked to form diphyllosilicate
anions or structures with higher multiplici-ties
3) The layers are linked to octahedral layers ofnontetrahedral cations (Section 1.4.5)
Folded zweier single layers are found in sev-eral alkali and barium phyllosilicates (Fig. 7A –D). The degree of folding depends on the cation.The layers are highly folded in Li2[Si2O5](Fig. 7A). The occurrence of different modifi-cations of Na2[Si2O5] (Fig. 7 B, C) reveals thatthe convolution (degree of folding) is not onlydetermined by the nontetrahedral cation.
A divalent cation must balance the charge oftwo [SiO4] tetrahedra. This increases the fold-ing of the layer. The silicates Ca[Si2O5] andSr[Si2O5] do not exist because the folding ofthe layers, which in turn is a consequence ofthe electrical field of the cations, provides co-ordination numbers too small for calcium andstrontium ions. In the presence of barium ions(Fig. 7D), which have a larger ionic radius, thedegree of folding is not as large and providessites for barium ions with sufficiently high co-ordination numbers (9 in sanbornite Ba[Si2O5],low-temperature modification).
Anhydrous, single-layer silicates containingsolely trivalent cations are not known becauseeven strong convolution cannot produce a well-balanced charge distribution. However, Li+ andAl3+ form a very stable, anhydrous, layer sili-catewith highly folded layers (petalite, Li[4]Al[4]
[Si2O5]2, Fig. 6 C). The stability is related to thetetrahedral coordination of Li+ and Al3+ ionswhich is a consequence of the high degree oflayer folding induced by the trivalent aluminumion.

Silicates 7
Examples of monophyllosilicates withlayers composed of silicate chains withP> 2 are apophyllite (Fig. 8) and gillespiteBaFe[Si4O10] with vierer single layers. Syn-thetic CaCu[Si4O10], isostructural to gillespite,was used as a blue pigment in wall paintings(“Egyptian Blue”). The mineral name is cupror-ivaite.
Figure 8. Vierer single layer in apophyllite, KCa4[Si4O10]2(F, OH) · 8H2O
1.4.2. Diphyllosilicates
Stabilization of zweier single layers by linkageto diphyllosilicate anions is illustrated in hexac-elsian, Ba(AlSiO4)2 (high-temperature modifi-cation, Fig. 9). Further examples are vierer dou-ble layers in naujakasite and reyerite and evenneuner double layers in stilpnomelane.
Figure 9. Diphyllosilicates: zweier double layer in hexacel-sian, Ba[(Al, Si)O4]2 high-temperature modification
1.4.3. Alkali Silicates
See also Chapter 3. Among the anhydrous alkalisilicates only natrosilite, Na2[Si2O5], occurs asa mineral [5]; all others are synthetic.
Na4[Si2O6] is a monopolysilicate containingzweier single chains. Most of the alkali sili-cates aremonophyllosilicates with zweier singlelayers: Li2[Si2O5] (Fig. 7A); α-, β-Na2[Si2O5](Fig. 7 B, C); Na4[Si6O14]; KH[Si2O5] [6]; andK2[Si4O9]. Silicates with Si : O ratios < 2 : 5contain Q4 tetrahedra besides the Q3 tetrahe-dra. Potassium analogues of Na2[Si2O5] are un-known [1].
Anhydrous alkali silicates are formed whenquartz is melted with sodium or potassium car-bonate or hydroxide (alkali silicates, water-glass). The reaction product is dissolved in wa-ter to form waterglass solutions. δ-Na2[Si2O5]is considered as a builder and possible substitutefor zeolites in washing powders [7].
Several hydrated alkali silicates occur asmin-erals:
Kanemite NaH[Si2O5] · 3H2O [8]Makatite Na2[Si4O8](OH)2 · 4H2O [9]Magadiite Na2[Si14O29] · 11H2O∗ [10]Kenyaite Na2[Si20O41] · xH2O∗ [10]Revdite Na2[Si2O5] · 5H2O [11]Grumantite NaH[Si2O5] · 0.9H2O [12]Silinaite NaLi[Si2O5] · 2H2O [13]∗MAS–NMR measurements reveal that the formula should bewritten as Na2H2Si14O30 · xH2O, Na2H2Si20O42 · xH2O[14]. Eugster [10] reported the compositionNa2Si22O45 · 10H2O for kenyaite.
The structures of sodium monosilicatesNa2H2[SiO4] · xH2O (x = 1 – 8) [15], makatite[16], and silinaite have been determined [13].Makatite contains vierer single layers. Thezweier single layer in silinaite shows a directed-ness not observed previously: the slightly kinkedzweier single chains alternate such that in onechain the apices point up and in the adjoin-ing chain the apices point down. The differ-ent dimensions of the [LiO4] tetrahedra and[NaO2(OH2)4] octahedra which hold the lay-ers together, impose a slight wave in the silicatelayer.
The natural alkali silicates and several otheralkali silicates (Na2H2Si8O18 · xH2O [17];K2H2Si14O30 · xH2O; K2H2Si20O42 · xH2O)can be easily synthesized from dispersions ofSiO2 in aqueous NaOH or KOH solutions [17–20]. A characteristic property of these sili-

8 Silicates
Figure 10. Preparation of crystalline silicic acids from alkali phyllosilicates by exchanging protons for the interlayer cations
Figure 11. Clay mineral structureA) Zweier single layer (“tetrahedral sheets”); B), C) Layers of edge-sharing [MO6] octahedra (“octahedral sheets”)Clay minerals with octahedral sheets as in (B) are called “trioctahedral”, those with sheets (C) are called “dioctahedral”.
cates is their cation exchange capacity (see Sec-tion 1.4.4). Possible practical uses include ionexchangers, adsorbents, or builders in detergents[21].
1.4.4. Crystalline Silicic Acids
Exchanging protons for the cations sandwichedbetween the silicate layers transforms the alkaliphyllosilicates into crystalline acids (Fig. 10). Inspite of the fact that the first crystalline silicicacid was prepared from α-Na2Si2O5 in 1924
[22], most textbooks of inorganic chemistry re-fer to the orthosilicic acid Si(OH)4 as the onlyoxo acid of silicon. The crystal structures of twoH2Si2O5 acids prepared fromα-Na2Si2O5 weredetermined by Liebau [23].
Presently, more than 15 crystalline acidsare known and comprise at least six modifica-tions of H2Si2O5. The acids are derived fromthe natural and synthetic alkali silicates men-tioned above, from several synthetic and nat-ural cuprous silicates, and from a few othersilicates such as apophyllite, carletonite, andgillespite [24–26]. The acids derived from

Silicates 9
Na2H2Si14O30 · xH2O, K2H2Si14O30 · xH2O,and K2H2Si20O42 · xH2O have been consideredfor practical uses. These acids are interesting al-ternatives to various forms of silica. Practical ap-plications may be related to adsorption proper-ties [27] and the capability to intercalate neutralorganic molecules (Section 1.4.6). A few acidseven adsorb anionic surfactants (together withthe gegen ions) in the interlayer space [28].
Figure 12. Linking of zweier single layers to octahedralsheets (dotted areas) in clay mineralsA) Kaolinite, Al2[Si2O5(OH)4]; B) Antigorite, idealizedMg3[Si2O5(OH)4]
1.4.5. Clay Minerals
See also Chapter 2.In clay minerals (Table 4) zweier single lay-
ers (Fig. 11A) are linked to octahedral sheets(Fig. 11B, C). (In clay mineralogy the singletetrahedral and octahedral layers are called tetra-hedral and octahedral sheets; the assembly ofthese sheets is called a layer [29].) The structuresprovide a good example of different types of di-
rectedness. The terms “clays, kaolins and ben-tonites” are used somewhat differently by geolo-gists and in practical applications (see Chaps. 1,5, 7 in [31]). A clear distinction must be madebetween ”clay” (the rock) and “clay mineral”(the mineral) [32].
1: 1 Clay Minerals. In kaolinite(Al2[Si2O5(OH)4]) all terminal oxygen ions ofthe zweier single layer (tetrahedral sheet) pointto one side and simultaneously belong to the oc-tahedral sheet (Figs. 11A, 12A,13A). Thus, theoctahedra around the octahedral cations consistof these oxygen ions and additional OH− ions.
Figure 13. Linking of tetrahedral and octahedral sheets inclay mineralsA) 1 : 1 clay minerals (kaolinite, serpentines); B) 2 : 1 clayminerals (micas, vermiculites, smectites)
As the dimensions of the octahedral andthe tetrahedral sheets do not match exactly(boct = btetr, Fig. 11), mechanical stress arisesbetween both sheets (for details see [1,29]). Thiscauses curling of the crystals when their thick-ness decreases below a certain value [33]. Hal-loysite crystals are composed of kaolinite-typelayers separated by a water layer, and often as-sume a tubular morphology.
The misfit between both types of sheetsin the serpentine minerals (chrysotile, antig-orite, lizardite), which are the trioctahedralcounterparts of kaolinite (idealized formulaMg3[Si2O5(OH)4]), also leads to the curling of

10 Silicates
Table 4. Classification of 1 : 1 and 2 : 1 clay minerals [30]
ξ a Group Octahedral Predominant Speciescharacter b octahedral cation c
Layer type 1: 1kaolin – serpentine tri Mg2+ lizardite, chrysotile d, antigorite d
di Al3+ kaolinite, halloysite d
Layer type 2: 10 talc – pyrophyllite tri Mg2+ talc, willemseite
di Al3+ pyrophyllite0.2 – 0.6 smectite tri Mg2+ stevensite, saponite
Zn2+ sauconiteMg2+, Li+ hectorite
di Al3+ montmorillonite, beidelliteFe3+ nontroniteCr3+ volkonskite
0.6 – 0.9 vermiculite tri Mg2+ (trioctahedral) vermiculitedi Al3+ dioctahedral vermiculite
0.6 – 0.9 illites di Al3+
tri( ?)0.6 – 1 mica tri Mg2+ phlogopite
Mg2+, Fe2+, Fe3+ biotiteLi+, Al3+ lepidolite
di Al3+ muscovite, illiteFe3+, Al3+ glauconiteFe3+, Al3+, Mg2+,Fe2+
celadonite
ca. 1.8 – 2 brittle mica tri clintonitedi margarite
Variable chlorite tri Mg2+, Al3+ clinochloredi Al3+ donbasseitedi – tri e Al3+, Mg2+ sudoite
a Layer charge (equivalents/unit), see general formula in page 11;b Tri- or dioctahedral;c Exact compositions see [31, Chap. 2];d Nonplanar phyllosilicates;e Interlayer: dioctahedral, 2: 1 layer: trioctahedral.
the chrysotile crystals. This results in the forma-tion of tubes (tubular morphology of asbestos)or in the alternation of the terminal oxygen ionsin a bandlike manner as in antigorite (Fig. 12B).
The major mineral of this group is kaolin-ite, the most important mineral in kaolins. Totalworld production was ca. 25×106 t in 1992 (USBureau of Mines, 1992) (→Clays). It is used inthe ceramic industry (ca. 20%) and as a filler andcoating pigment in paper production (ca. 50%)[31,34]. Kaolin is also needed for the produc-tion of zeolites (→Zeolites) and ultramarines(→Pigments, Inorganic, Chap. 3.5.).
2: 1 Clay Minerals. 2 : 1 clay minerals con-sist of two zweier single layers (“tetrahedralsheets”)which enclose a central octahedral sheet(Fig. 13B). The diversity of this group of phyl-losilicates (Table 4) results from:
1) The occurrence of di- and trioctahedral lay-ers (Fig. 11B, C)
2) Isomorphous substitutions in the octahedralsheet
3) Isomorphous substitutions in the tetrahedralsheet, mainly Al3+ for Si4+
In addition, cationic sites can be vacant, andprotons may dissociate from OH− ions or as-sociate to them or to O2− ions. Depending onthe type and concentration of the substitutionsand defects, the layers can assume negativecharges. The number of charges per formula unit([(Si,Al)4O10] unit) is the most essential param-eter used to characterize the 2 : 1 clay minerals.
The chemical composition of the 2 : 1 clayminerals is represented by:

Silicates 11
Mν+ξ/γ (H2O)n
Interlayer space
(M3+, M2+, M+)(6−x)+2−3[Si4−yAlyO10(OH)2]ξ−
Layer
where ν is the valency of the interlayer cationand ξ is the layer charge.
The general formula indicates that the num-ber of octahedral cations (M3+, M2+, M+)varies between 2 and 3. Mostly, the occupancyis near 2 (dioctahedral minerals) or 3 (trioctahe-dral minerals). An extensive survey of chemicalcompositions is given in [31].
Figure 14. Structural relations between some silicatesA) Talc, Mg3[Si2O5]2(OH)2; B) Palygorskite, idealizedMg5[Si2O5]4(OH)2(OH2 )4· 4H2O, for exact formula see[29]; C) Sepiolite, idealized Mg4[Si2O5]3(OH)2 · 4H2O,for exact formula see [29]
In Table 4, the clay minerals are classi-fied according to their layer charge ξ andtheir di- or trioctahedral character. The sil-icates with uncharged layers are pyrophyl-lite (idealized Al2[Si4O10(OH)2]) and talc(Mg3[Si4O10(OH)2]). Talc (Fig. 14A) is a rawmaterial in the ceramic industry (→Talc). It
is used in the production of insulating materi-als (→ Insulation, Electric, Chap. 3.1.2.2.), as afiller (paper, paints, cosmetics, elastomers, ther-moplastics), and as a carrier material for pesti-cides. Pyrophyllite finds similar uses [31].
Montmorillonite is the most important smec-titic mineral (Table 4) of the bentonite rocks(→Clays). The various applications of ben-tonites (production ca. 8×106 t in 1992; US Bu-reau of Mines, 1992) are based on the specialproperties of montmorillonite, in particular itsfine particle size (< 2µm), intracrystalline reac-tivity (swelling inwater and organic liquids, ion-exchange properties, see Section 1.4.6), bind-ing of organic materials and colloidal behavior[31]. Only a few of the many hundreds of appli-cations (→Clays, Chap. 5.2.) [31,35] are men-tioned here. The special rheological propertiesof bentonites are exploited in the application as afoundry sandbinder, for oilwell drilling, iron orepelletizing, in the building industry (in partic-ular for subterranean curtains), in geotechnics,and as sealing materials; for pelletizing animalfeed, as fillers and adsorbents (e.g., in clarifyingand decolorizing mineral, animal and vegetableoils, beer, and fruit juices) and as a carrier mate-rial for pesticides. The profitable use as pet littershould not be overlooked.
Synthetic smectites are hectorite-type materi-als which are produced more easily than mont-morillonites under mild hydrothermal condi-tions. Hectorite is a very low- charged smectite(ξ ≈ 0.20 – 0.25 eq/unit) that contains Li+ ionsin the octahedral sheet. It has similar uses tobentonite [36]. The synthetic materials are usedin the pharmaceutical industry (e.g., as thicken-ers, thixotropic agents, gel-forming materials)and seem to provide suitable properties as com-ponents in muds for drilling very deep wells [31,35].
The largest amounts of vermiculite (produc-tion about 0.65×106 t/a) are used in the form oflight, expanded aggregates consisting of exfoli-ated crystals (see Section 2.9).
Illites are very fine, mica-type minerals, andessential components of soils.
Mica is used in the form of sheets, groundpowders, or micronized and is described in de-tail elsewhere (→Mica). Most synthetic prod-ucts are fluorinated micas. Formation of fluo-rophlogopite in glass ceramics gives this mate-

12 Silicates
rial a precision machinability with conventionalmetal-working tools (→Glass Ceramics) [37].
Chlorites. Chlorites are composed of silicatelayers (similar to the 2 : 1 clay minerals men-tioned above) that are separated by metal hy-droxide layers M(OH)2 or M(OH)3 [29]. Chlo-rites are not used on an industrial scale but areabundant in soils and are found in clays.
Mixed-Layer Minerals. Mixed-layer min-erals are very abundant in common clays andsoils. The crystals are composed of layers whicheither differ in the composition and layer chargeor are of different structural types. Inmost cases,the sequence of the layers is random. Typicalcombinations are [31] illite – smectite, mica –vermiculite, mica – chlorite, smectite – chlorite,chlorite – vermiculite, and kaolinite – smectite.
Some mixed-layer minerals also have a reg-ular sequence of layers [31]:
Rectorite = muscovite (or paragonite) –montmorillonite
Tosudite = dioctahedral chlorite – smectite
Corrensite = chlorite – vermiculite (or trioctahedral smectite)
Palygorskite and Sepiolite. Palygorskite(Fig. 14B) and sepiolite (Fig. 14C) providefurther examples of phyllosilicates with sin-gle layers of different directedness. The two-dimensional octahedral sheet is split into one-dimensional bands comprising eight or five octa-hedra. In the tetrahedral sheet some of the tetra-hedra are inverted. Bands of three zweier singlechains with different directedness alternate insepiolite whereas the bands in palygorskite arecomposed of two zweier single chains. Channelsare therefore formed in the structure which con-tain water molecules and exchangeable cations.The cations are bound as a consequence of iso-morphous substitutions in the tetrahedral and oc-tahedral sheets. Theminerals have zeolitic prop-erties and show a certain cation-exchange capac-ity. Sepiolite crystallizes as fibers with diame-ters of 5 – 30 nm and a length of 10 nm– 4µm.Fibers of palygorskite are 15 – 30 nm thick and0.5 – 4.5µm long.
Sepiolite and palygorskite are being increas-ingly used as thickeners, thixotropic agents, ad-sorbents, supports of pesticides and catalysts,and pet litter [31].
1.4.6. Intracrystalline Reactions
Kaolinite, 2 : 1 clay minerals, and alkali phyl-losilicates are intracrystalline-reactive, layeredmaterials [38]. The interlayer space is accessi-ble to guest molecules or ions (Fig. 15).
Figure 15. Intracrystalline reactions of phyllosilicatesA) Phyllosilicates with uncharged layers (kaolinite, crys-talline silicic acids); B) Phyllosilicates with charged layers(2 : 1 clay minerals, hydrated alkali silicates)
Kaolinite takes up various organic com-pounds (urea, hydrazine, dimethyl sulfoxide,short- chain acid amides, potassium and ammo-nium acetate) between the layers so that theyare separated by a mono- or bilayer of neutralguest molecules (Fig. 15A). Inmost cases, thesemolecules can be displaced by other molecules[31]. Intercalation of organic compounds pro-motes delamination of kaolinite particles andcan improve the mechanical stability of porce-lain. Kaolin modified by intercalation of ureawas used in China (mainly in the Sung period,960 – 1280) in the production of very thin porce-lain (egg-shell porcelain) [39].
The most striking difference between the be-havior of di- and trioctahedralminerals is that the

Silicates 13
trioctahedral 1 : 1 clay minerals such as antig-orite do not intercalate guest molecules. Pro-nounced intercalation capability is observed forthe crystalline silicic acids [28,38].
Intracrystalline-reactive phyllosilicates withcharged layers can undergo various reactions(Fig. 15B) [31]. Most important for technicalapplications is the intracrystalline swelling ofsmectites by interlamellar adsorption of water.The layer separation of calcium smectites doesnot normally exceed 1 nm. In the presence ofsodium ions, however, the layers separate com-pletely and form colloidal dispersions [31]. Thisprocess and the related changes in flow behav-ior are of great importance in many practical ap-plications (building industry, subterranean cur-tains, foundry sand binder, oil well drilling fluidsetc. [31]).
A further essential reaction is the exchange ofthe interlayer cations [31]. Soda activation is animportant technical process in which interlayercalcium ions are displaced by sodium ions. Fix-ation of potassium ions by more highly charged2 : 1 clay minerals plays an important role insoils. Clay minerals are considered as useful ad-sorbents in environmental technology becauseof their ability to bind heavymetal ions by cationexchange [31,40,41]. Radioactive nuclides maybe removed by clay adsorbents, for instance ce-sium ions by vermiculites and shales [42]. Evenradioactive iodide ions can be adsorbed bymod-ified clay minerals [43].
The third reaction of technical importance isthe binding of organic cations. Organo-modifiedbentonites are used inmany applications,mainlyas adsorbents, thickeners, or thixotropic agentsin organic systems (lubricants, greases, paints, orasphalt). In environmental technology, organicderivatives of clay minerals adsorb toxic sub-stances [31,40,41,44].
1.5. Tectosilicates
Almost all tectosilicates (framework silicates)contain quaternary tetrahedra such that the ra-tio of tetrahedral cations to oxygen is 1 : 2.The fundamental chain periodicities are P = 2,3, 4, 6: cristobalite, tridymite P = 2 (Fig. 16A);quartz, keatite P = 3; zeolites: laumontite, gis-mondine, harmotone P = 4; chabazite, silicalite-1 P = 6. Nephelines, (Na1−xKx)[AlSiO4], have
a tridymite structure (Section 2.2). The feldspars(Section 2.1) have loop-branched dreier frame-works (Fig. 16B) based on a branched dreier sin-gle chain. The black tetrahedra in Figure 16Bform a dreier single chain and additional tetrahe-dra (stippled) bridge two tetrahedra of this chain[3]. Cordierite, Mg2[(Al4Si5)O18], has an open-branched vierer framework.
Figure 16. Framework silicatesA) Zweier framework of tridymite [SiO2]; B) Loop-branched dreier framework of feldspars, e.g., orthoclaseK[AlSi3O8]
In Figure 16 the tetrahedral framework isthought to be composed of chains of a certainmultiplicity. Another more common view is to

14 Silicates
look at the polyhedrawhich form the framework,and the voids or polyhedral cavities generatedbetween the framework oxygen ions. The nota-tion [512], for example, indicates a polyhedroncomposed of 12 pentagonal faces (a pentagon-dodecahedron); [51262] is a polyhedron with 12pentagons and two hexagons (see Fig. 17). Ac-cording to the nature of the voids and polyhedralcavities tectosilicates can be divided into threegroups [45–47]:
1) Pyknosiles. Tectosilicates which have nopolyhedral cavities but small interstices bet-ween the framework oxygen ions; thesevoids can be occupied by cations and some-times water: quartz, cristobalite, tridymite,feldspars.
2) Clathrasils. Tectosilicates with polyhedralcavities. The windows of the cages aresmall and the encaged ions, molecules, ortheir decomposition products cannot diffusethrough them: melanophlogite, dodecasils,sodalite (→Silica, Chap. 8.2.2.).
3) Zeolites. The polyhedral cavities are inter-connected by large windows or tunnels, andthe enclosed ions or molecules can dif-fuse through the crystal: analcime, morden-ite, faujasite, zeolite A, silicalite (ZSM-5)(→Zeolites).
Under hydrothermal conditions silica crystal-lizes from aqueous solutions as quartz, cristo-balite, keatite, or coesite (→Silica) [47,48].In the presence of inorganic or organic guestcompounds, clathrasils are formed. The neutralmolecules become enclosed in the polyhedralcavities during synthesis. The guest compoundsact as templates for the formation of the polyhe-dra, thus determining the type of clathrasil [46,48]. Figure 17 shows polyhedra ofmelanophlog-ite and dodecasils occupied by guest molecules.The orientation of the molecules in the cageswas derived from single- crystal X-ray diffrac-tion [48].
Minerals of technical importance arethe feldspars orthoclase K[AlSi3O8], albiteNa[AlSi3O8], and anorthite Ca[Al2Si2O8] (Sec-tion 2.1). The largest amounts of feldspars (andnepheline) are used in the glass and ceramicindustries, mainly as fluxes. Cordierite glass ce-ramics have good thermal stability and shockresistance (→Glass Ceramics).
The most important synthetic tectosilicatesare the zeolites (→Zeolites) [49,50]. Their Si/Alratio varies considerably. The most startling de-velopment was the synthesis of the almost Al3+-free end members of silica-rich zeolites (ZSM-5 = silicalite-1, ZSM-11 = silicalite-2).
Figure 17. Guest molecules included in cages of the hostframework of clathrasilsA) Xenon in the [512] cage of melanophlogite; B) CO2 in[51262] of melanophlogite; C) Piperidine in [51264] of do-decasil 3C; D) Adamantylamine in [51268] of dodecasil 1H
2. Natural Silicates
2.1. Feldspar
Feldspars [68476-25-5] are anhydrousalkali/alkaline-earth aluminosilicates thatclosely resemble each other in structure andproperties. They are among the most commonand important mineral groups, constituting ca.60 – 65wt%of the Earth’s crust. Approximatelytwo-thirds of them occur as plagioclases.
2.1.1. Structure and Composition
The feldspars are tectosilicates (framework sil-icates, see Chap. 1). The [(Si, Al)O4] tetrahe-dra are linked at all four vertices yielding a

Silicates 15
framework [Si4−xAlxO8]x−. The voids withinthe tetrahedral framework contain alkali metalor alkaline-earth ions for charge compensation.The structure is composed of four-memberedrings, [(Si, Al)4O12], containing (Al Si3) or(Al2Si2) in each ring. The rings are linked bycommon oxygen atoms (two on each side) inthe a direction to two neighboring rings to formzigzag bands (or “double crank-shaft chains”)making use of three of the four common ver-tices. These bands are bonded in both the band c direction by the fourth common oxygenatom of each (Si, Al) atom (see Fig. 18: in theb direction in y = 0 or 1/2, in the c direction iny = 0.14 or 0.36) to form three-dimensional tetra-hedral frameworks. As a result, the (010) and(001) planes are rather weakly bonded and read-ily cleaved. This property is characteristic of allfeldspars. The cations K+, Na+, and Ca2+, andmore rarely Sr2+ and Ba2+, occupy the largespaces within the framework of tetrahedra, andare coordinated to oxygen in a fairly irregularmanner.
Figure 18. Structure of orthoclase, projection parallel to bon (010) showing two crank-shaft bands along a0
Three main types of feldspar (“molecules”)can be distinguished:
1) K[AlSi3O8] = potassiumfeldspar= orthoclase(Or)
2) Na[AlSi3O8] = sodiumfeldspar = albite (Ab)
3) Ca[Al2Si2O8] = calcium feldspar = anor-thite (An)
In nature, these three substances seldom oc-cur in a pure state. At high formation tempera-ture (e.g., in some volcanic rocks), a solid solu-tion series between Or and Ab exists: the al-kali feldspars. On slow cooling [i.e., in mostplutonic (igneous) rocks] an immiscibility gapbelow 650 C leads to partial unmixing (perthi-tization). Albite and anorthite form an almostinfinite solid solution series: the plagioclases.Between orthoclase and anorthite a wide immis-cibility gap exists (see Figs. 19, 20, 21).
Figure 19. Triangular phase diagram of orthoclase – albite –anorthite showing mixed crystal formation found in nature(at ca. 700 C)
Figure 20. Phase diagram of the alkali feldspars

16 Silicates
Pure potassium feldspar has the composition64.8wt%SiO2, 18.3wt%Al2O3 and 16.9wt%K2O. Potassium can be replaced by rubidium toa small extent, with the rubidium content in or-thoclases from pegmatites reaching ca. 3wt%.In the plagioclases, the Al2O3 content increaseson going from the acidic to the basic types: from>19wt% in albite to almost 37wt% in anorthitewith a concomitant decrease in the SiO2 contentfrom almost 69wt% in albite to 43wt% in anor-thite.
Figure 21. Phase diagram of plagioclases (dry mp can belowered by 500 C in the presence of water)
All feldspars have a high- and a low-tem-perature form. In the high form, the distribu-tion of [SiO4] and [AlO4] tetrahedra is at ran-dom, while in the low form there is a high de-gree of order. There are also intermediate typeswith varying degrees of Al/Si order. Orderingchanges the monoclinic orthoclase to triclinicmicrocline. The high forms of albite and anor-thite and of albite and orthoclase are completelymiscible, whereas the low forms are not.
On slow cooling of the (K,Na) feldspars, seg-regation (perthite formation) takes place due toselectivemigration of the small and large cations(Fig. 20). This leads to a regular intergrowth oforthoclase (or microcline), and albite, with char-acteristic formation of string- or worm-shapedbodies, which can be macroscopic (macrop-erthite), microscopic (microperthite) or submi-croscopic (cryptoperthite).Microcline – perthite
consists of microcline containing albite strings,and antiperthite consists of albitewith orthoclasestrings.
The feldspars are very widely distributed,mainly in igneous rocks (volcanic and plutonicrocks) and metamorphic rocks (contact and re-gional metamorphites). They also occur in peg-matitic and hydrothermal formations, sedimen-tary rocks and sediments. This means that thepetrographic classification of rocks is largelybased on the mass ratios and compositions ofthe individual feldspars and quartz. If there isa deficiency of SiO2, leucite K[AlSi2O6 ] (seeSection 2.3) or nepheline Na[AlSiO4 ] (see Sec-tion 2.2) is formed instead of the alkali feldspars.
2.1.2. Characterization of IndividualFeldspars
The alkali feldspars can havemonoclinic and tri-clinic crystal forms, but the plagioclases only tri-clinic forms. The most important feldspars fol-low.
PotassiumFeldspars. The term sanidine de-notes the metastable, glass- clear monoclinichigh form of K[AlSi3O8] with a random dis-tribution of (Al, Si) produced by rapid coolingin volcanic rocks (volcanites). During the veryslow cooling of plutonic rocks (plutonites) anAl/Si ordering takes place to a greater (triclinicmicrocline) or lesser (orthoclase) extent. Ortho-clase is not visibly triclinized despite prolongedcooling in plutonites. Sanidine melts incongru-ently with separation of leucite.
Sanidine (the high-temperature form ofK[AlSi3O8]) forms monoclinic – prismaticcrystals, with space group C3
2h –C 2/m; lat-tice constants: a0 = 0.856 nm, b0 = 1.303 nm,c0 = 0.718 nm (a0, b0, and c0 are somewhatsmaller when sodium is present); the axial ratiois a0 : b0 : c0 = 0.657 : 1 : 0.551; β = 11559′;Z = 4 (number of chemical formula units perunit cell).
Orthoclase (the intermediate form ofK[AlSi3O8]) forms monoclinic – prismaticcrystals, with space group C3
2h –C 2/m; lat-tice constants: a0 = 0.856 nm, b0= 1.300 nm,c0= 0.719 nm; the axial ratio is a0 : b0 : c0 =0.659 : 1 : 0.553; β = 11601′; Z = 4.

Silicates 17
Orthoclase often consists of thick plateletsparallel to [010], extended in the [001] directionor having columnar development in the [100]direction. Crystals may be several centimetersor even tens of centimeters in size. Twinning isvery characteristic.
Microcline (the low-temperature form ofK[AlSi3O8]) forms triclinic – pinacoidal crys-tals, with space groupC1
i –C 1; lattice constants:a0 = 0.857 nm, b0 = 1.298 nm, c0 = 0.722 nm;the axial ratio is a0 : b0 : c0 = 0.660 : 1 : 0.566;α = 9041′, β = 11559′, γ = 8730′; Z = 4.
Barium Feldspars.Hyalophane, (K, Ba) [Al(Al, Si)Si2O8],
forms monoclinic (or pseudomonoclin-ic?) crystals, with space group C3
2h –C 2/m; lattice constants: a0 = 0.854 nm,b0 = 1.298 nm, c0 = 0.715 nm; the axial ratiois a0 : b0 : c0 = 0.658 : 1 : 0.551; β = 11535′;Z = 4.
Celsian [001302-50-7], Ba[Al2Si2O8],forms monoclinic crystals, with spacegroup I 2/c; lattice constants: a0 = 0.865 nm,b0 = 1.313 nm, c0 = 2×0.730 nm; the axial ratiois a0 : b0 : c0 = 0.659 : 1 : 2×0.556; β = 11502′;Z = 8.
Plagioclases. Theplagioclases formca. 40%of the solid crust of the Earth and are widely dis-tributed. At high temperature (see Fig. 21) thereis complete miscibility between the end mem-bers anorthite, Ca[Al2Si2O8 ] (basic) and albite,Na[AlSi3O8 ] (acidic). At lower temperature,miscibility is incomplete. The plagioclases areusually twinned along [010], leading to lamellarreplication multiple twinning, and typical stria-tion.
The plagioclases have a structure similar tothat of sanidine, the replacement of K+ by thesmaller Na+ and Ca2+ ions leading to somedeformation with triclinic symmetry. The low-calcium plagioclases [albite (An0−10 ), oligo-clase (An10−30 ), and andesine (An30−50 )] havea cell size similar to that of sanidine, withC 1 symmetry. The high- calcium plagioclases(labradorite (An50−70 ), bytownite (An70−90 ),and anorthite (An90−100 )) show doubling of thec0 lattice constant, so that bytownite has I 1 sym-metry and anorthite P 1 symmetry. The plagio-clases are usually characterized by the content(in mol%) of anorthite (An), albite (Ab), and
orthoclase, whereby the content of orthoclase isusually negligibly small. An important charac-teristic of plagioclases is the degree of order, i.e.,not only of (Al, Si), but also of (Na, Ca) and of(Na, Ca, K) in high-temperature mixed crystalscontaining potassium. The high- and low-tem-perature plagioclases can be distinguished opti-cally. In the low-temperature plagioclases, thereis an ordered distribution of (Al, Si). For exam-ple, in pure anorthite, which has no high-tem-perature form, the [AlO4] and [SiO4] tetrahedraalternate, whereby each oxygen is neighboredby an aluminum and a silicon ion. Despite theAl/Si disorder in high-temperature plagioclases,Al –O –Al groupings do not occur. Also, be-cause the (Na, Ca) ions are smaller, the triclinicform is maintained at higher temperature, unlikesanidine. The triclinic high-temperature form ofalbite is also known as analbite. Monalbite isamonoclinic potassium-containing high-tempe-rature phase of albite.
Albite, Na[AlSi3O8], An0−10 orAb100An00 –Ab90An10; C 1 lattice constants:a0 = 0.814 nm, b0 = 1.2789 nm, c0 = 0.716 nm;the axial ratio is a0 : b0 : c0 = 0.636 : 1 : 0.559;α = 9419′, β = 11634′, γ = 8739′; Z = 4.
Oligoclase, An10−30 or Ab90An10 –Ab70An30; C 1 lattice constants: a0 = 0.817 nm,b0 = 1.284 nm, c0 = 0.713 nm; the axial ratio isa0 : b0 : c0 = 0.636 : 1 : 0.556, α = 9349′, β =11627′, γ = 8859′; Z = 4.
Andesine, An30−50 or Ab70An30 –Ab50An50; C 1 lattice constants: a0 = 0.818 nm,b0 = 1.288 nm, c0 = 0.711 nm; the axial ratio isa0 : b0 : c0 = 0.635 : 1 : 0.552; α = 9324′, β =11610′, γ = 9024′; Z = 4.
Labradorite, An50−70 or Ab50An50 –Ab30An70; C 1 lattice constants; a0 = 0.816 nm,b0 = 1.286 nm, c0 = 2×0.710 nm; the axial ratiois a0 : b0 : c0 = 0.635 : 1 : 2×0.552; α = 9334′,β = 11606′, γ = 8947′; Z = 8.
Bytownite, An70−90 or Ab30An70 –Ab10An90; I 1 lattice constants: a0 = 0.817 nm,b0 = 1.287 nm, c0 = 1.418 nm= 2×0.709 nm; theaxial ratio is a0 : b0 : c0 = 0.635 : 1 : 1.102;α = 9322′, β = 11558′, γ = 9031′; Z = 8.
Anorthite, Ca[Al2Si2O8], An90−100 orAb10An90 –Ab0An100; P 1 lattice constants:a0 = 0.818 nm, b0 = 1.288 nm, c0 = 1.417 nm=2×0.785 nm; the axial ratio is a0 : b0 : c0 =0.635 : 1 : 2×0.550; α = 9310′, β = 11551′,γ = 9113′; Z = 8.

18 Silicates
2.1.3. Production
Feldspar can be synthesized by solid-state or liq-uid-phase reactions:
1) Albite is formedbyheating amixture of 1molAl2O3, 6mol SiO2, an excess of Na2O, andsodium tungstate as a mineralizer at 900 –1000 C with a long reaction time (days).
2) Orthoclase is formed by heating a mixture of1mol Al2O3, 6mol SiO2, and a large excessof K2O, with tungstic acid for mineralizationat 900 – 1000 C, or by heating a mixture ofAl2O3, SiO2 and acidic potassium tungstateat 900 C. Crystals are obtained on treatmentwith boiling water.
3) Anorthite is obtained by fusing a mixtureof Al2O3, SiO2 and CaCO3. The tempera-ture is maintained slightly below the meltingpoint (≤ 1500 C), and the mixture is slowlycooled. Four modifications (α, β, γ, and δ)are known.
Preparative methods using hydrothermal re-actions in autoclaves or high-pressure reactorsare also known:
1) Albite is synthesized by heating precipitatedaluminum silicate and excess sodium silicateat ca. 500 C
2) Orthoclase is synthesized by heating a mix-ture of Al2O3, SiO2 andKOHwith water in aratio corresponding to K2O ·Al2O3 · 6 SiO2at 400 C, or from an aqueous slurry ofpotassium aluminate or Al(OH)3 with potas-sium silicate in the presence of CO2 at 360 –400 C
3) Anorthite is synthesized by heating amixtureof aqueous slurries of SiO2, Al2O3, and CaOin the presence of CaCl2 as a mineralizer at470 C
All synthetic feldspars are Al/Si disordered.Ordering requires geologic periods of time.Feldspars also occur in industrial products (ce-ramics). In synthetic feldspars, Si4+ can be com-pletely replaced by Ge4+, and Al3+ by Ga3+ orFe3+. This gives new end members that are onlyfound as synthetic compounds. For example, δ-anorthite, δ-Ca[Al2Si2O8], which correspondsto natural anorthite, occurs in fly ash and some-times in blast-furnace slags. It is formed whenCaO- containing slags attack the chamotte (firebricks) in blast furnaces and lime kilns.
The high-temperature form of albite, α-Na[AlSi3O8], occurs in chamottes that havebeen exposed to Na2O, e.g., in tank liningsand checker bricks used in furnaces for glass-making.
α-Celsian, α-Ba[Al2Si2O8], is the principalcomponent of special bricks used to line thearched roofs of the firing zones in electric tun-nel furnaces for glazing stoneware and sanitaryware. It is very resistant to vapors from lead andborate glazes. Other refractory materials reactwith these vapors to form liquid phases.
2.1.4. Properties
Physical Properties. Feldspars can be glassclear, transparent, translucent, cloudy, matt, col-orless, white, gray, or colored (e.g., green-ish, reddish, red, or brown) due to inclusions.Labradorite and orthoclase can be iridescent(labradorescent) and are used for decorative pur-poses. The feldspars can have a glassy or some-times nacreous luster. Fused feldspars, even iforiginally dark in color, usually solidify as atransparent, colorless glass.
Cleavage in the (001) plane is very good,moderate in the (010) plane, and just observablein the (110) plane [plagioclases also cleave inthe (110) plane]. Orthoclase is named from thecleavages in the mutually perpendicular (001)and (010) planes. The plagioclases form tricliniccrystals and owe their name to the fact that theangle between the (001) and (010) planes devi-ates by a few degrees from 90. The fracture isconchoidal, uneven, and splintered or brittle.
TheMohs hardness of albite is 6 – 6 1/2, andof anorthite 6 – 7. Densities (g/cm3) are: ortho-clase 2.53 – 2.56, albite ca. 2.62, and anorthiteca. 2.76.
In thin sections, the feldspars show low re-fraction of light and birefringence (double re-fraction).
The average specific heat (J g−1 K−1) of or-thoclase between 0 and 100 C is ca. 0.79, ofalbite 0.82, and of anorthite 0.80. The corre-sponding values for the range 0 – 1100 C are1.05, 1.1, and 1.12.
In the crystalline state the alkali feldspars be-come electrically conductive at moderate tem-peratures. Ionization is observed above 900 C.

Silicates 19
In the liquid phase, albite is one of the moststrongly dissociated silicates.
The rate of crystallization in alkali feldsparmelts is extremely small due to their high meltviscosity. The fusion range of natural orthoclaseis 1140 – 1300 C (incongruentmelting, see alsoSection 2.3). Fused anorthite has a compara-tively low viscosity, however, and crystallizesreadily and rapidly on cooling to form large,well-formed crystals. Themaximumcrystalliza-tion rate occurs 20 – 25 C below the tempera-ture at which melting starts. A glass is formedonly if the cooling rate is very rapid.
Chemical Properties. Chemical attack, in-cluding weathering, readily decomposes orchanges the feldspars. The prolonged action ofwater on pulverized feldspar leads to prefer-ential dissolution of the alkali ions, but alsodissolves CaO and Al2O3, eventually giving aresidue of polysilicic acids. Water containingdissolved CO2 is more aggressive; alkali met-als and Al2O3 are dissolved preferentially, andthe reaction proceedswith formation of kaolinite(Al2O3 · 2 SiO2 · 2H2O).
Strong mineral acids decompose alkalifeldspars slowly, producing a residue of polysili-cic acids. However, powdered anorthite dis-solves completely in hydrochloric acid at100 C. Under high pressure (i.e., in autoclavesat ca. 200 bar) potassium feldspars are convertedto kaolinite by 5%hydrofluoric acid at 225 Corby 0.5 – 1mol/L HCl at 320 – 330 C. This firstcauses complete hydrolysis of the starting mate-rial, followed by the formation of kaolinite andsilicic acid. At lower temperatures, the reactiononly produces a residue with a high silicic acidcontent. Anorthite is almost completely con-verted into kaolinite by treatmentwith 0.5mol/LHCl at 340 C.
Concentrated solutions of alkali hydrox-ide decompose the alkali feldspars completelyabove 110 C. Reaction with 1 – 2% solutionsof alkali hydroxide leads to the formation of sol-uble products.
2.1.5. Mineral Deposits and their Extraction
The most important raw materials are the potas-sium feldspars (orthoclase, microcline), fol-lowed by sodium feldspar (albite). The other
members of the plagioclase group are of verylittle value, although rocks with a high plagio-clase content has been tested as a raw materialfor aluminum production. Nepheline syenite isindustrially important (see Section 2.2), as areother rocks and sediments rich in feldspar (e.g.,granites, syenites, andporphyries). Feldspar alsooccurs in some apatite deposits (e.g., in the KolaPeninsula, Russia). Furthermore, feldspar is ex-tracted from sedimentary rocks (e.g., arkoses).
Economically useful deposits of high-gradefeldspars are of limited occurrence. They aremostly plutonic rocks, pegmatites, and volcanicrocks with feldspar inclusions. When exposed,feldspathic rocks are often partially or com-pletely kaolinized. Feldspar often arises as a by-product during the processing of kaolin or mica.World production of feldspar is ca. 6.7×106 t/a(all weights specified are in metric tons).
The demands of European industry can bemet from European deposits, the most importantbeing in Italy, Turkey, Germany, France, Spain,the Czech Republic, Portugal, Russia, Norway,Sweden, Poland, Hungary, Finland and Greece.
The most important producers are Germany:Amberger Kaolinwerke, Hirschau; Birken-felder Feldspatwerke Schweyer & Vollmer,Ellweiler; Gebruder Dorfner, Hirschau; Kali-Chemie, Hannover; Keramische RohstoffwerkeBauscher-Mandt, Weiden; Villeroy & Boch,Keramische Werke, Mettlach; Italy: GruppoMaffei SpA., Milano; Minerali Industriali SpA.;France: Denain Anzin Mineraux (Feldspathsdu Midi, Feldspaths du Morvan, FeldspathsBaux); Norway: Norfloat A/S; Spain: Unimin;Finland: Partek; Sweden: Svenska Forsham-mar AB, Gothenburg; Spain: Industrias delCuarzo and Lansa; Turkey: Esan; Elginkam; for-mer Soviet Union; United States: The FeldsparCorp., Spruce Pine/NC.; IMC Chemical GroupInc., Industrial Minerals Div., Boston/Mass.;Foote Minerals Co., Exton/Pa., Kings Moun-tain; Mica Co. Inc., Kings Mountain/NC.; PacerCorp., Custer/SD.; San Antonio Mica Mine,Ajo/Ariz.; Mexico: Materias Primas MonterreySA, Monterrey/N.L.; Argentina: Piedra GrandeSA, San Luis; Japan: Kinsei Kogyo Co Ltd., Os-aka; Morimura Bros. Inc., Tokyo; Nissho-IwaiCo. Ltd., Tokyo; Sumitomo Corp., Tokyo; In-dia: Golecha Palawat & Co., Rajastan; ShreeModi Levigated Kaolin Pvt. Ltd., Rajastan;South Africa: Ceramic Minerals (Pty.) Ltd.,

20 Silicates
Dunswart/Transvaal; General Overseas TradersS.A. (Pty.) Ltd., Johannesburg; Martin & Rob-son (Pty.) Ltd., Germiston; Otavi Mining Co.(Pty.) Ltd., Johannesburg; Rand London Corp.Ltd., Johannesburg; Rondebult Clayworks (Pty.)Ltd., Boksburg.
Opencast mining is used to extract sedimen-tary and pegmatitic deposits, although pegmatiteis also obtained by underground mining. Theusualmethods such as drilling, explosives or cut-ting are used for massive rock deposits. Scrapersand front loaders are used for sedimentary de-posits (loose rock), working in stepped faces.
2.1.6. Processing and Quality Requirements
Processing. Usually, processing of feldsparconsists of crushing and/or grinding with sub-sequent classification, but some feldspar is alsoprocessed by flotation (e.g., feldspar in graniticrocks). The methods used to process the mas-sive or loose rock depend on the end product,themineral composition and the available equip-ment. In the simplest case, the feldspathic rockis progressively broken down by jaw crushers,ball mills, roller mills or pin mills; classifiedby vibratory sieves; washed if necessary; anddried. It is then ground using impact mills orball mills to produce a coarse powder and classi-fied by air elutriation. The final product containsnot only feldspar, but also accompanying min-erals (e.g., quartz or mica). Marketable productsshould have a favorable mineral compositionwith a high feldspar content. A quartz contentof the product is not detrimental, since quartz isneeded in any case for many applications (e.g.,production of porcelain, glass and enamel). Anymica can be removed by air classification afterbreakdown of the rock. Accompanying miner-als with a high magnetic susceptibility can beremoved by magnetic separation. The contentof heavy metal ions in the end product can bereduced to a safe level by this method.
The flotation process was introduced for pro-cessing feldspar in 1960 by the Finnish companyLohja Oy. The rock is first crushed, sometimeswashed and dried and then ground.Mica, quartz,other accompanying minerals and feldspars areobtained in the froth or as a sediment in a seriesof flotation stages. Selectivity depends on thechoice and dosage of reagents (activators, col-
lectors, and foaming agents) and adjustment ofthe pH. Albite and potassium feldspar can alsobe separated from each other if necessary.
Feldspar also arises as a byproduct in the wetprocessing of kaolin. Quartz is first removedfrom an aqueous suspension of the crude ore.Kaolinite and feldspar are then separated inhydrocyclones, dewatered, and dried with rotaryor tray dryers (→Clays, Chap. 4.1.2.). The de-gree of separation (feldspar : quartz ratio) is ca.9%.
In another process, the kaolinite and ac-companying clay minerals are slurried, and thefeldspar – quartz concentrate (particle size 0.1 –0.5mm) is separated into feldspar and quartz byan electric field of 40 kV. The degree of separa-tion (feldspar : quartz ratio) is ca. 96%. High-intensitymagnetic separators are used to removeparamagnetic impurities. Grinding is performedin nonferrous equipment and is followed by airelutriation, giving products with a maximumgrain size of 40 – 150µm.
Quality requirements vary considerablyand depend on the end product consumer.High-quality products generally require high-purity rawmaterials of defined composition. Forwhite glasses, light- colored ceramics, glazesand enamels, the content of heavy metal ionsmust be extremely small (≤ 0.08wt%). Con-tents of Na2O and/or K2O and Al2O3 mustbe specified to ensure satisfactory physicaland chemical properties, especially strengths ofglasses. When the products are used as fluxes,the alkali oxide content must be increased tobring about the desired reduction of the melt-ing point. Important quality criteria for some ap-plications are the particle size, the particle sizedistribution, and the tolerance range for the frac-tions of over- and undersized particles.
Chemical and physical data of some feldspargrades are given in Table 5; some particle sizedistributions for feldspar sands and powders aregiven in Tables 6.
2.1.7. Uses and Economic Aspects
The glass and ceramic industries are the mainconsumers of sodium and potassium feldsparsand also of related minerals of lower quality

Silicates 21
Table 5. Chemical composition and physical data for selected feldspar grades
Theoretical Feldspar sand, AKW a Glass Norfloat c Forshammar d Maffei ed
FS 800 S FS 900L FS 960L FFF b K Na F 5 F 7 SE
Total feldsparcontent, wt%
100 100 81 94 92 90 96.5 86.5 67 80 90
K feldspar, wt% 100 76 87 43 45 65.5 16.5 41Na feldspar, wt% 100 5 7 43 42 28.5 61 25 80 90Ca feldspar, wt% 3 2.5 9 1Quartz, wt% 12 4 6 5 3 12 29 12 - 15 5Other silicates 7 2 2 2 0.5 1 4 5 - 8 5SiO2, wt% 64.8 68.7 67.5 65.8 67.7 67.5 65.4 69.2 75.5 70.9 70.10Al2O3, wt% 18.2 19.4 17.0 17.8 18.2 18.5 18.7 18.7 14.7 17.52 18.20Na2O 11.8 0.60 0.80 5.00 5.00 3.36 7.20 4.85 9.30 10.20K2O, wt% 16.9 12.70 14.65 7.20 8.12 11.10 2.80 4.25 0.65 0.30CaO, wt% 0.02 0.04 1.2 0.56 0.51 1.82 0.20 0.60 0.60MgO, wt% 0.04 0.02 0.03 0.10 0.10 0.10Fe2O3, wt% 0.12 0.042 0.08 0.10 0.06 0.11 0.13 0.30 0.10TiO2, wt% 0.50 0.044 0.03 0.03 0.05
Loss on ignition,wt%
1.0 0.25 0.20 0.29 0.19 0.45 0.75 0.50
Hardness (Mohs) 6 6 6 6 6 12 6 1
2Density, g/cm3 2.6 2.6 2.6 2.6 2.65 2.65Begin of sintering,C
1130 1175 1150 1140 1140 1135
End of sintering, C 1340 1365 1250 1270 1270Sintering interval, K 210 190 100 130 135Begin of fluxing, C 1430 1440 1330 1290 1300Softening interval, K 90 75 80 20 30Half-sphere point,C
1560 1570 1420 1460 1330 1365
End of fluxing, C 1630 1600 1580 1410 1500Fluxing interval, K 200 160 250 120 200
a AKW Amberger Kaolinwerke, Hirschau, Germany. b Glass-grade feldspar, floated, Lohja Oy, Virkkala, Finland.c H.Bjorum, K/S Norfloat, Oslo, Norway. d Svenska Forshammars AB, Gothenburg, Sweden. e GruppoMaffeiS.p.A., Monte Orno, Milan, Italy.
Table 6. Typical particle size distributions (in %) for selected feldspar sand grades
Screen analysis Grading
DIN 66 165, mm FS 800 S FS 900 SF FS 900S FS 900L 0305
> 1.0 0.10 0.200.5 – 1.0 0.05 0.05 0.20 30 650.1 – 0.5 18 21 85 70 350.063 – 0.1 25 29 11 0.100.040 – 0.063 22 23 3< 0.04 35 27 1
Dry screen residue Grind number
DIN 53 734, µm 3 6 8 10
> 125 6> 100 12> 90 18 5> 71 25 10 3> 63 34 18 8 3> 40 48 35 20 10

22 Silicates
with respect to heavy metal ions and accom-panying minerals (i.e., feldspathic pegmatites,aplites, phenolites and Cornish stone). See also→Glass, Chap. 5.1., →Ceramics, General Sur-vey, Chap. 2.4. The alkali feldspars have a flux-ing actiondue to their alkali-metal oxide content.They reduce the temperature at which meltingbegins (i.e., the sintering point of the raw mate-rial mixture) and thus shorten the time requiredfor melting or firing. This reduces energy con-sumption and increases production capacity andthe life of the refractory linings. On cooling themelt, the oxidic components of the feldspar forma glassy structure, which provides the glassyma-trix in ceramic products.
Al2O3 increases melt viscosity, facilitatingmechanical formingof the glass. It also increaseshardness and impact strength (toughness) andprevents crystallization (devitrification). In ce-ramic masses, the Al2O3 in the glassy matrix re-duces the tendency to fire cracking and increasesimpact strength.
Sodium feldspar is usually added at a level of0.2 – 17wt% as a raw material for glass. Potas-sium feldspar is generally employed for ceramicproducts, although sometimes calcium or cal-cium/sodium feldspar is also used.
Feldspar (admixtures of 4 – 20wt%) is usedin the coating for arc-welding electrodes to im-prove the flow of the molten slag. In blast fur-naces, the addition of feldspar also improves slagflow during tapping.
Powdered feldspar is used as a mild abra-sive due to its hardness, conchoidal fracture andplatelet-shaped particles. Light- colored alkalifeldspars, anorthite and plagioclase are added tocoatings in the form of fine sands or coarse pow-ders to give antiskid properties to floor coveringsand surfaces intended for vehicles and pedestri-ans.
Finely ground feldspar grades are good fillersfor exterior paints because of their chemical in-ertness, lack of photoreactivity and good drybrightness. As a filler feldpsar competes withother, often cheaper, raw materials such as cal-cite and dolomite. They are also used as fillersfor pigments, adhesives, rubber (especially foamrubber), thermoplasts and thermosets. They canbe used as fillers in large amounts approach-ing the critical pigment volume concentration(CPVC), giving transparent or nearly transpar-ent paints and polymer products, because the re-
fractive indices ofmany paint resins, plasticizersand polymers are almost the same as those of thefeldspars (n≈ 1.55).
Feldspathic rocks that can be dressed are alsoused as building stone, as hard core in roads,and as bright, reflective chippings in bitumen-bonded road surfaces.
In Western Europe, the approximated totalconsumption of feldspar is ca. 3.7×106 t/a andis divided as follows:
Ceramics 57%Glazes and enamels 4.5%Glass 34%Abrasives 1.5%Fillers 0.4%Others 2.6%
Alkali feldspars can be replaced completelyor partially by leucite (Section 2.3), nephelineor nepheline syenite (Section 2.2), thereby in-creasing the alkali oxide and Al2O3 contentand reducing the SiO2 content. If low levels ofaccompanying minerals and heavy metal ionsare unnecessary, other substitutes can be used:feldspathic pegmatites, aplites, phonolites andporphyries; and to a limited extent plagioclase,anorthosite (plutonic rocks containing ca. 90%plagioclases), Cornish stone (though this canliberate fluorine on thermal decomposition ofthe melts) and the blast furnace slags “Carolinastone” and “Calumite” (both consisting of kaoli-nite, feldspar, quartz and some fluorspar).
Worldwide extraction of feldspars is es-timated at > 6.7×106 t/a. Together withnepheline, nepheline syenite, and leucite, to-tal production exceeds 7.5×106 t. In 1990,247 000 t feldspar and 135 000 t pegmatite wereproduced in the Federal Republic of Germany.
The production figures of pegmatite in 1988 –1993 in the United States are ca. 670 000 t/a,in the CIS 310 000 t/a, in Italy ca. 295 000 t/a,in France ca. 195 000 t/a, in Mexico ca.127 000 t/a, in Brazil ca. 110 000 t/a, in Spainca. 90 000 t/a, in Turkey ca. 73 000 t/a, in Nor-way ca. 71 000 t/a, in Venezuela ca. 70 000 t/a, inFinland ca. 68 000 t/a, in Rumania ca. 60 000 t/a,in Sweden ca. 52 000 t/a, in India ca. 50 000 t/a,in Argentina and in the Republic of South Africaca. 47 000 t/a, respectively.
The total feldspar mining production in 1994amounted to 1 806 935 t in Italy, 765 202 t in theUnited States, 528 027 t in Thailand, 500 000 inTurkey, 385 000 t in France, 379 427 t in Ger-

Silicates 23
many, 319 658 t in South Korea, 274 789 t inVenezuela, 250 000 t in Spain, 170 000 t in theCzech Republic, 140 000 t in Brazil, 133 400 tin Mexico, 92 440 t in Portugal, 79 000 in Iran,76 188 t inColombia, 70 000 t inRussia, 70 000 tin India, 62 905 t in Norway, 55 000 t in Sweden,53 503 t in Japan, 50 000 in Argentina, 46 000 tin Poland, 43 805 t in the Philippines, 43 600 t inHungary, 41 389 t in Finland, 40 000 t in Cuba,39 745 t in Egypt, 37 156 t in the Republic ofSouth Africa, and 35 000 t in Greece. Further-more, the feldspar mining production amountedin 1994 e.g., to 23 500 t in Australia, to 8000 t inthe United Kingdom, and 4883 t in Austria.
2.2. Nepheline and Related Compounds
The alkali aluminosilicate nepheline,KNa3[AlSiO4]4, a feldspathoid, belongs to thenepheline group of tectosilicates without nonte-trahedral anions. The aluminum : silicon ratio is1 : 1.
Structure and Mineralogy. As in all tec-tosilicates, the oxygen ions at the vertices ofthe [AlO4] and [SiO4] tetrahedra in nephelineare linked to the four neighboring tetrahedra.This produces a three-dimensional open frame-work in which the relatively large Na+ and K+
cations are located in the spaces between thetetrahedra. In tectosilicates, these spaces can beoccupied by alkaline-earth ions, nontetrahedralanions (e.g., in the feldspathoids sodalite andscapolite), or water (in zeolites). In mineralsof the nepheline group the tetrahedra exhibita hexagonal or pseudohexagonal arrangement.In nepheline (Fig. 22), alternating [AlO4] and[SiO4] tetrahedra are linked together at com-mon vertices to form an easily distorted high-tridymite structure with six-membered rings.The apices of the [AlO4] tetrahedra point par-allel to the c axis, and those of the [SiO4] tetra-hedra point in the opposite direction. Unlike thehigh-tridymite structure, Si4+ ions in nephelineare replaced by Al3+ ions in half of the tetrahe-dral positions. In order to maintain charge neu-trality, 3 Na+ ions and 1 K+ ion are found performula unit in the centers of the channels par-allel to the c axis.
Figure 22. Idealized structure of nepheline,KNa3[AlSiO4]4, with [(Si, Al)O4] tetrahedra linkedto form six-membered ringsProjection parallel to c on (001)
In natural nepheline, the ratio of K+ to Na+
can vary considerably, and Ca2+ can also be in-corporated in the lattice. There is often an excessof SiO2, i.e., less than half the Si4+ is replacedby Al3+, so that less Na+ is required to maintaincharge neutrality.
Nepheline [012251-27-3], KNa3[AlSiO4]4,crystallizes in the hexagonal – pyramidal sys-tem with space group C6
6 – P 63 and has thefollowing lattice constants: a0 = 1.001 nm, c0 =0.841 nm; the axial ratio is c0/a0 = 0.840; Z = 2.= 2.65 g/cm3 (also potassium-free with a0=0.996 nm, c0= 0.836 nm).
The following minerals of the nephelinegroup have properties similar to those ofnepheline:
Kalsilite, K[AlSiO4] (Fig. 23), crystallizesin the hexagonal – pyramidal system withspace group D6
6 – P 63; lattice constants: a0 =0.518 nm, c0 = 0.869 nm; the axial ratio isc0/a0 = 1.678; Z = 2.
Trikalsilite, (Na, K)[AlSiO4], crystallizes inthe hexagonal system, probably with spacegroup C6
6 – P 63; lattice constants: a0 = 1.54 nm,c0 = 0.86 nm; the axial ratio is c0/a0 = 0.559;Z = 18.
Kaliophilite, K[AlSiO4], crystallizes inthe hexagonal – trapezohedral system withspace group D6
6 – P 6322; lattice constants:a0 = 2.706 nm, c0 = 0.861 nm; the axial ra-tio is c0/a0 = 0.318; Z = 54; pseudocell: a′
0 =1.562 nm= a0/3, c0 = 0.861 nm, c0/a0 = 0.551;Z = 18.

24 Silicates
Figure 23. Structure of kalsilite, K[AlSiO4]. The structureof nepheline, KNa3[AlSiO4]4, is obtained by replacementof K4 by KNa3
High eucryptite, Li[AlSiO4], only exists asa synthetic product, and is obtained by heatingnatural eucryptite (low eucryptite), LiAl[SiO4];it is stable above 962 C. It crystallizes in thehexagonal – trapezohedral system with a super-lattice structure, with space group D4
6 – P 6222or D5
6 – P 6422; lattice constants: a0 = 0.528 nm,c0 = 1.127 nm; the axial ratio is c0/a0 = 2.134;Z = 3.
Nepheline is a feldspathoid and occurs onlyin rocks with a low SiO2 content (< 50wt%SiO2); it has a lower SiO2 content than the al-kali feldspars. It is formed in alkaline rocks inplace of albite if the magma is sufficiently un-saturated with SiO2. It is often associated withother feldspathoids such as sodalite, cancrinite,or leucite, or less often with kalsilite. Nephelineis a typical main constituent of the Atlantic-type igneous rocks, plutonic rocks (e.g., foy-aites, theralites and essexites), hypabyssal andvolcanic rocks (e.g., phonolites, tephrites andnephelinites). In the Mediterranean suite, mi-nor amounts of nepheline are usually found withthe common leucite (see Section 2.3). Nephelinecrystallizes immediately after mafic minerals in
magmatic rocks, and at about the same time asalkali feldspars in plutonic rocks. In hypabyssaland effusive rocks, nepheline occurs as phe-nocryst and in the matrix.
The ratio potassium : (potassium+ sodium)in nepheline varies between 7% and 37% ineffusive rocks and also in plutonic rocks, inwhich nepheline was formed in the presence ofanorthoclase or sanidine. In plutonic rocks con-taining orthoclase and albite, nepheline contains14 – 23 atom% potassium. Igneous nepheline(slow cooling) has a cloudy or greasy luster andis known as elaeolite. On rapid cooling (effu-sive rock), the crystals remain clear. Albite canform isomorphous mixtures with nepheline insodium-rich solid solutions with a maximum al-bite content of ca. 20mol% at 700 C; the al-bite is often irregularly distributed, i.e., in zones.As with feldspar (see Section 2.1.2), the or-der/disorder behavior of Al/ Si in the nephelineis temperature dependent.
Occurrence in Industrial Products. In re-fractory technology nepheline occurs in fourmodifications that do not contain potassium,NaAlSiO4:
High carnegieite [001302-34-7], α-Na[AlSiO4], crystallizes in the cubic system,with space group T4 – P 213; lattice constants:a0 = ca. 0.74 nm, Z = 4. It has the cubic highcristobalite-type lattice with additional sodium,and can form mixed crystals with cristobalite toa limited extent. High carnegieite occurs onlyas a synthetic product; in mineralogy it is oftentermed β-carnegieite.
High nepheline [012251-27-3], β-Na[AlSiO4], is orthorhombic. Low nepheline[012251-27-3], γ-Na[AlSiO4], is hexagonal –pyramidal. It occurs, for example, in chamotteand SiO2- containing corundumbricks. It is con-verted into mullite in the walls of blast furnaces,glass-melting furnaces and checker bricks by theattack of Na2SO4 and Na2O. Low carnegieite,β-Na[AlSiO4], is triclinic.

Silicates 25
In refractory technology, five modificationsof the polymorphic substance KAlSiO4 areknown: hexagonal – trapezohedric kalsilite, or-thorhombic (high) kaliophilite, hexagonal kalio-philite (kalinepheline), natural hexagonal kalio-philite and anomalous kaliophilite. A character-istic feature of kalsilite is that it “polymerizes”:trikalsilite and tetrakalsilite are known.
Kalsilite occurs in incrustations on thechamotte walls of blast furnaces and in thechamotte checker bricks of Siemens –Martinfurnaces. Potassium – nepheline is formed at< 1540 C in chamotte bricks of industrial fur-naces that are exposed to K2O vapors (e.g., inthe brick linings of blast furnaces and tuyeres).High kaliophilite is stable above 1540 C. It alsooccurs in the chamotte linings of blast furnacesand tuyeres, as well as in the chamotte checkerbricks in Siemens –Martin furnaces and glassfurnaces used to melt optical K2O glasses.
Properties and Composition of IndustrialProducts. The following physical properties areimportant for industrial applications.
Density:Nepheline: 2.56 – 2.665 g/cm3
Kalsilite: 2.59 – 2.625 g/cm3
β-Carnegieite: 2.513 g/cm3
α-Carnegieite: 2.343 g/cm3
Mohs hardness:Nepheline: 5 1/2 – 6Kalsilite: 6
Specific heat at 20 – 100 C:Crystalline: 0.77 J g−1 K−1
Amorphous glass: 0.804 J g−1 K−1
Melting point:Sodium nepheline + albite, 100 – 70%NaAlSiO4: 1526 – 1410 CKalsilite (0 – 20% NaAlSiO4): 1300 – 1350 Cβ-Carnegieite (ca. 100% NaAlSiO4): 1526 CEutectic in the system nepheline – albite:1068 C
Transformation temperature:Nepheline→ α- carnegieite: 1248± 5 Cα-Carnegieite→ β- carnegieite: 687 – 692 CNepheline + 30% kalsilite→ α- carnegieite:1400 CNepheline + 35% anorthite→ α- carnegieite:900 C
Nepheline shows piezoelectric properties ina high-frequency field.
Compositions of commercially minednephelines are given in Table 7.
Nepheline has amarkedly alkaline reaction inwater. Dilute acids, especially HCl, decomposenepheline, producing gelatinous silicic acid.
Deposits. Only a small number of workablenepheline syenite deposits are known, the mostimportant of which are in Canada, Norway andRussia. The rock in a certain Canadian depositconsists of ca. 54% albite, 18% microcline,18% nepheline, < 4% biotite, 6% muscoviteand magnetite. In the Western world, only fourcompanies extract and process nepheline-con-taining rocks (only nepheline syenite), the com-bined output being ca. 1.2×106 t/a.
Extraction. The rock is usually extracted byopen- cast mining using explosives. Adheringimpurities are removed by sieve grates (griz-zlies) prior to size reduction. Ferromagnetic im-purities (e.g., biotite) are removed by means ofrotary magnets or high-intensity magnetic sepa-ration.
Quality Specifications. The quality requireddepends on the use, e.g., Grade A (Table 8) forthe glass industry, Grade B for glass wool manu-facture, ceramic grade for the ceramic industryand a lower grade for use as a filler. The gradesdiffer in iron content (Grade A≤ 0.08% Fe2O3,Grade B: ≤ 0.4% Fe2O3) and in color, whichvaries with heavy metal content and mean parti-cle size (in some applications).
Uses. The most important consumer is theglass industry, which accounts for ca. 70% ofthe nepheline marketed. Here, the silicate actsnot only as a source of SiO2 and Al2O3, butalso as a flux. Mixtures of raw materials for bot-tle manufacture can contain 5 – 15% nepheline.Nepheline can replace feldspars in most ce-ramic and glass-making applications (see Sec-tion 2.1.7) with only small adjustments to batchchemistry being necessary. The choice betweenfeldspar and nepheline (nepheline syenite) is of-ten a question of cost.
The ceramic industry consumes ca. 15% ofthe total nepheline output. The nepheline con-tent in sanitary and whiteware ceramics can be

26 Silicates
Table 7. Analyses of natural nephelines and kalsilites (in wt%)
A B C D E F G H
SiO2 44.65 41.88 40.20 38.47 38.48 63.36 60.20 60.8TiO2 0.00 0.03 0.05 0.00 0.05Al2O3 32.03 32.99 32.51 30.81 31.01 22.05 23.30 23.0Fe2O3 0.59 0.74 1.82 1.63 1.12 0.09 0.07 0.08MgO 0.00 0.00 0.10 0.63 0.00 0.03CaO 0.71 0.78 1.44 0.20 0.03 5.80 0.30 0.05Na2O 17.25 16.11 10.86 2.09 0.30 5.77 10.60 10.4K2O 3.66 6.82 12.22 25.65 28.33 2.43 5.10 4.6H2O (105 C) 0.21 0.03 0.00 0.00 0.7H2O (105 –1000 C)
0.96 0.71 0.20 0.20 0.67
Total 100.06 100.16 99.77 99.94 100.00 99.50 99.57 99.66nE 1.535 1.543 1.543 1.539 1.53n0 1.531 1.539 1.537 1.533 1.53Nepheline 81.9 76.1 54.7 9.9 1.4Kalsilite 12.9 23.6 45.3 88.9 96.8Others 5.2 0.3 1.2 1.8
A) Nepheline phonolite, New Zealand. B) Nepheline foyaite Transvaal. C) Nepheline potassium ankaratrite, Angola.D) Kalsilite, venanzite, Italy. E) Kalsilite, complex granular intergrowth of kalsilite and nepheline, Angola.F) Nepheline – aplite (whole rock), Feldspar Corp., USA. G) Nepheline – syenite (whole rock), Sobin Chem. Inc.,Canada. H) Nepheline Senite (Treminex 958), Quarzwerke GmbH, Frechen, Germany.
Table 8. Composition (wt%) of Canadian nepheline syenite products (Grade A)
Company SiO2 Al2O3 K2O Na2O CaO Fe2O3 LOI∗Indusmin 59.8 23.6 5.0 10.2 0.6 0.07 0.6IMC 60.2 23.3 5.1 10.6 0.3 0.08 0.4
∗Loss on ignition.
25 – 30%, in industrial chemical porcelain andtranslucent porcelain china 15 – 30%, and infloor and wall tiles 15 – 55%.
Finely ground nepheline and feldspar (seeSection 2.1.7) are becoming increasingly impor-tant as fillers for plastics [especially unsaturatedpolyester resins, poly(methyl methacrylate),and thermoplastic polyurethanes], rubber, adhe-sives, paints and pigments (Table 9). Since therefractive indices of nepheline and thematrix arealmost the same, e.g., in nepheline/poly(vinylchloride) compositions, transparency is main-tained, even at high pigment volume concentra-tions. The high surface and volume resistance ofnepheline mean that it can also be used as a fillerin insulating paints, casting resins for electricalapplications and other products.
In Russia, nepheline syenite is an importantrawmaterial for aluminum production. In amul-tistage process, the Al2O3 is extracted and thentreated by melt electrolysis. High activity silicagels can be produced from nepheline by decom-position with hot 22% sulfuric acid.
2.3. Leucite
The potassium aluminum silicate leucite, afeldspathoid, belongs to the analcime – leucitegroup of tectosilicates without nontetrahedralanions. The aluminum : silicon ratio is 1 : 2.
Structure and Mineralogy. As with all thetectosilicates, the oxygen ions at the verticesof the [AlO4] and [SiO4] tetrahedra are linkedto four neighboring tetrahedra. This produces athree-dimensional open framework inwhichK+
ions are located in the spaces within the frame-work. Whereas in the nepheline group the ar-rangement of tetrahedra is hexagonal or pseudo-hexagonal, theminerals of the analcime – leucitegroup have a cubic or pseudocubic arrangement.
Leucite [001302-34-7], K[AlSi2O6], is di-morphous. Below 605 C, it exists as low leucite(α-leucite) and above 605 C as high leucite (β-leucite). Low leucite crystallizes in the tetrago-nal – pseudocubic system, with the space groupC6
4h – I 41/a; lattice constants: a0 = 1.304 nm,

Silicates 27
Table 9. Physical data for nepheline fillers
Product∗ Particle diameter, µm Brightness Density,
D50 value Upper limit (555 nm) g/cm3
Minex 4 10.8 30 90 2.6Apex 400 9.1 32 90.5 2.6Minex 7 3.9 20 90 2.6Apex 700 5.9 25 92 2.6Minex 10 2.7 15 90 2.6
∗Trade names: Minex is produced by Indusmin and Apex by IMC.
c0 = 1.385 nm; the axial ratio is c0/a0 = 1.062;Z = 16. High leucite crystallizes in the cu-bic – hexakis-octrahedral system, with the spacegroup O10
h – Ia 3 d; a0 = 1.343 nm; Z = 16.Leucite is isotypical with anal-
cime, Na[AlSi2O6] ·H2O; pollucite,(Cs, Na)[AlSi2O6] ·H2O, and syntheticCs[FeSi2O6]; it is homeotypic with wairakite,Ca[AlSi2O6]2 · H2O. In leucite, K+ can bereplaced to a small extent by Na+, rarely byRb+, and very rarely by Ca2+; these minorsubstituents sometimes appear as enriched iso-morphous layers. The pseudocubic, pale greeniron(III) leucite, K[FeSi2O6], also known asa synthetic material, can occur in very smallquantities with the rock-forming leucite.
The apparently cubic crystals of leucite areanisotropic and consist of numerous twin lamel-lae, which are arranged parallel to the sur-faces of the apparent rhombic dodecahedron.In thin sections, leucite shows a complex twin-ning on 110 under crossed polarizers. Onraising the temperature to 605 C, leucite be-comes isotropic and is then genuinely cubic(high leucite). The transformation is reversible.
Leucite is the most characteristic componentof SiO2-unsaturated magmatites with a prepon-derance of potassium (Mediterranean rocks) andis typical of magmatic regions (e.g., the Romeregion in Central Italy). It is found in many foy-aitic and theralitic volcanic rocks.
Leucite occurs in refractory products, e.g., inchamotte bricks from blast furnaces and tuyeres.
High leucite melts congruently at 1686 C.When potassium feldspar is heated, it meltsincongruently above 1150 C forming a mix-ture of leucite crystals in a SiO2-rich melt. At1150 C, the melt contains 57.8wt% leucitecrystals, and at 1530 C all the leucite becomesmolten. On cooling, leucite crystallizes first, andthe melt therefore becomes richer in SiO2. The
leucite crystallization is completed at 1150 Cand potassium feldspar is then formed.
Properties. Leucite crystals are usuallywhite or gray, glassy, translucent, but very sel-dom clear. Cleavage is poor parallel to (110),the fracture being conchoidal and brittle. Thefollowing physical properties are important forthe use of leucite as a raw material for glass:
Density: leucite 2.47 – 2.50 g/cm3, moltenglass produced from leucite 2.41 g/cm3.
Mohs hardness: 5 1/2 – 6.Melting point: pure leucite 1686 C, impure
leucite 1298 – 1430 C.Heat of crystallization: 47.65 kJ/mol. The
tendency to crystallize is very low, and the melttherefore usually solidifies as a glass. Volumeincrease on solidification of the melt: 2.9%.
Specific heat (10 – 100 C): natural min-eral 0.8 J g−1 K−1, in the amorphous state0.733 J g−1 K−1.
Leucite can contain up to 1.5wt% Na2O and0.8wt%CaO. It is decomposed by concentratedhydrochloric acid with precipitation of SiO2.
Deposits and Processing. Leucite depositsoccur in the United States (Montana andWyoming) and in Italy (Mount Vesuvius). Pro-cessing and classification of the leucite gradesare very similar to that of feldspar and nepheline(see Sections 2.1.6 and 2.2).
Uses. Leucite, like feldspar, ismainly used asa raw material in glass manufacture. Other ap-plications include the production of potassiumfertilizers, alum and potassium nitrate.
2.4. Olivine
Olivine (peridot, chrysolite) [1317-71-1] is arock-forming nesosilicate (an orthosilicate, i.e.,

28 Silicates
a silicate with isolated [SiO4]4− tetrahedraheld together by metallic ions) belonging tothe olivine series of minerals (Mg, Fe)2[SiO4].There is an infinite series of solid solutions ofthe end members forsterite (Fo) [015118-03-3],Mg2[SiO4], and fayalite (Fa), Fe2[SiO4].The intermediate members are olivine(Mg90−70, Fe10−30)2[SiO4], hyalosiderite(Mg70−50, Fe30−50)2[SiO4], hortonolite(Mg50−30, Fe50−70)2[SiO4], and ferrohortono-lite (Mg30−10, Fe70−90)2[SiO4]. Other mem-bers of the olivine series include tephroiteMn2[SiO4], knebelite, (Mn, Fe)2[SiO4], andiron knebelite (Fe,Mn)2[SiO4].
Figure 24. Structure of olivine, projection parallel to b on(010)The [SiO4]4− tetrahedra point alternately upward anddownward
Structure andMineralogy. The minerals ofthe olivine series form orthorhombic crystals,space group D16
2h – Pmcn, Z = 4, with the follow-ing lattice constants (in nm):
Forsterite Olivine Hortonolite Fayalitea0 0.600 0.601 0.607 0.617b0 0.478 0.478 0.479 0.481c0 1.028 1.030 1.034 1.061
Olivine (Fig. 24) can include small amountsof other substituent elements (e.g., nickel).Weathering removes MgO and SiO2, formingthe important nickel serpentine ore mineral gar-nierite (nickel deposits of the New Caledoniatype).
Olivine occurs as intergrown, loose, pris-matic or thick tabular crystals and as granularaggregates. Cleavage in the (001) plane is good,and also takes place imperfectly in the (010)plane, the fracture being conchoidal.
Olivine crystallizes at an early stage from amagma. It is found typically as the main min-eral constituent in ultrabasic igneous rocks (peri-dotites and dunites), and as a residual material inthe serpentinites formed from them. In the ultra-basics, olivine is accompanied by pyroxene, etc.Olivine is often found in basic magmatites bothin plutonic and volcanic rocks (e.g., gabbros, di-abases, basalts and melaphyres). It also occursin meteorites. Olivine is a main constituent ofthe upper earth mantle.
Olivine of jewelry quality is knownas chryso-lite, and forms clear, pale green stones that canbe polished.
In the presence of water, olivine read-ily forms other silicates, e.g., serpentine (ser-pentinization – antigorite, chrysotile). Olivineoften then becomes pseudomorphosed toform serpentine, Mg6[(OH)8/Si4O10], and talc,Mg3[(OH)2/Si4O10]. The magnesium removedduring serpentinization occurs as brucite,Mg(OH)2, magnesite, MgCO3, and cryptocrys-talline magnesite of the Kraubath type (Styria,Austria). The iron present in olivine forms sec-ondary magnetite Fe3O4 [Fe3+(Fe2+Fe3+)O4]during serpentinization.
Synthesis. Synthetic olivine is produced bycalcination of chrysotile tailings from the as-bestos mines in the Thetford Mines area ofCanada. The tailings are calcined at 850 C ina rotary kiln to give a mixture of forsterite andsilica. A purer forsterite can be synthesized byadding magnesite during calcination. The pel-letization of magnesite and the tailings is fol-lowed by calcination in a rotary kiln at 800 Cand further heating to a temperature of 1350 Cin a static furnace, wherebymagnesite combineswith the silica to form forsterite.

Silicates 29
Forsterite in Industrial Products.Forsterite is a common component of refrac-tory magnesia products (especially chrome –magnesite bricks), and is the principal compo-nent of refractory forsterite bricks (→Glass,Chap. 5.7.). It is used in high-frequency insu-lators and also occurs in special metallurgicalslags and upon reaction of refractory magnesiaproducts with acidic slags.
Properties. Olivine has a glassy luster andis transparent or translucent, but seldom color-less (i.e., iron-free). The yellowish to green color(which gives it its name) varies from light to darkand may be opaque or even black with high ironcontent. The double refraction (birefringence)indices are usually high and depend on the mag-nesium : iron ratio.
The following physical properties are impor-tant for industrial applications:
Density: forsterite (Fo) 3.21 g/cm3,Fo90/Fa10 (90mol Fo/10mol Fa) 3.25 g/cm3,fayalite (Fa) 4.34 g/cm3.
Mohs hardness: forsterite 6 1/2 – 7, fayalite6 1/2.
Melting point: forsterite (no decomposition)1885 C (volume change 3%), fayalite 1205 C.
Temperature stability limit for magnesium-rich olivine in an oxidizing atmosphere 1750 C;softening temperature under a load of 2 kg/cm2
1600 – 1700 C; thermal shock resistance mod-erate.
Heat of fusion of forsterite 3.444 kJ/mol;specific heat (21 – 51 C) 0.791 kJ/g; thermalconductivity (kJm−1 h−1 K−1) at 100 C 19.3,at 500 C 11.3, and at 1000 C 8.8; meancoefficient of thermal expansion over therange 0 – 500 C 8.3×10−6K−1, 0 – 1000 C9.5×10−6K−1, 0 – 1500 C 11.0×10−6K−1.
Free energy of formation: forsterite− 1922 kJ/mol, fayalite − 1337.5 kJ/mol.
Electrical resistivity: at 800 C2×106 Ω · cm, at 1200 C104 Ω · cm, at 1400 C103 Ω · cm.
Ideal chemical compositions: forsterite(2MgO ·SiO2): 57.3wt% MgO, 42.7wt%SiO2; fayalite (2 FeO ·SiO2): 70.5wt% FeO,29.5wt% SiO2.
Olivine is insoluble in water, even at elevatedtemperature and pressure (620 C, 30.4MPa). Itis stable towards cold acid, but complete decom-position takes place on heating in dilute acid
(0.2mol/L HCl), forming silica gel. Olivine re-acts similarly with HNO3 and H2SO4, but dis-solves only slowly in 20% H2F2. Solutions ofthe alkali metal sulfates readily decompose it.Olivine is resistant to alkaline solutions.
The chemical resistance of olivine in refrac-tory materials is good towards reducing atmo-spheres, moderate toward carbon, poor towardacidic flux, good toward basic flux, and goodtoward molten metals.
The following data are characteristic of com-mercial olivines:
Density: 3.1 – 3.3 g/cm3
Bulk density (dry sand): 1.4 – 1.7 g/cm3
Tamped density (dry sand): 1.7 – 1.9 g/cm3
Bulk density (molding sand): 1.7 – 2.1 g/cm3
Moisture content (max.): 0.5%Softening point: > 1700 CSpecific heat: 0.84 – 1.38 J g−1 K−1
Coefficient of thermal expansion (20 –1200 C): 11.6×10−6 K−1
FeO content (max.): 8wt%pH: 9
The most useful properties of forsterite-richolivines are their high softening point, low andnearly linear thermal expansion, low thermalconductivity, high density, low abrasivity and(unlike silica) lack of toxicity.
Deposits and Commercial Products. Animportant economic factor in the extraction ofolivine is the fact that olivine-rich ultrabasicsor olivine rocks and the serpentinites derivedfrom them are associated with chromite, plat-inum, and nickel deposits. The principal olivinedeposits extracted today are in Norway, Japan,Austria, the United States, Italy, Spain and Pak-istan. Smaller producers are Mexico, Sweden,India and Brazil. Japan has a large production ofolivine and a production of serpentine which isseveral times higer than that of olivine. 6t. Thefirst deposit was extracted in 1948 in Aaheim(Norway).
World production of forsterite-rich olivineand dunite is ca. 6.8×106 t/a, of which ca.2.1×106 t/a are in Europe.
Commercial olivines usually contain at least85% forsterite. Their chemical composition isMgO ca. 45 – 50%, SiO2 ca. 40 – 43%, FeO ca.5 – 7%. Loss on ignition is ca. 1 – 2%.

30 Silicates
Rocks that contain higher proportions of de-rived minerals such as serpentine and talc areknown as dunite. The chemical composition isMgO ca. 36 – 42%, SiO2 ca. 36 – 39%, with acorrespondingly higher loss on ignition.
Extraction and Processing. Olivine is ex-tracted by both open- cast and undergroundmin-ing. The rock is usually broken up by explosives.
The mined rock is processed by multistagesize reduction (jaw crushers, cone crushers, andhammer mills), and classified by multiple-deckvibratory screens, with intermediate washing toremove any soft accompanying minerals (e.g.,pyroxene, serpentine, chlorite or talc) in theform of a slurry. After washing, the materialis mechanically dewatered by continuous cen-trifuges and dried by rotary or belt dryers (resid-ual moisture content < 0.5%).
Powdered olivine is produced by impact orhammer mills, giving products with narrow par-ticle size ranges and low dust contents.
Uses. Important uses for magnesium-richolivines are:
1) Ore additive in steel blast furnacesto condition the slag and improve itsflow. Here, olivine replaces dolomiteand silica and enables the ratio(CaO+MgO) : (SiO2 +Al2O3) to be ad-justed to ca. 0.8 – 1.2%. Also, less heat isrequired to melt the slag components.
2) Olivine is well suited for the slag condition-ing of furnace slag and for basicity controlin blast and electric arc furnaces. Olivine isadded in the form of lumps (10 – 40 mm),sinter (3 – 6 mm) and pellets.
3) Olivine sand is used in foundry sands forcasting manganese steel, high-alloy steels,brass, bronze and aluminum and in the pro-duction of gray iron and alloy steel, whichusually require a fine finish. Olivine sandshave several advantages over silica sands:the casting surfaces are smoother, fettlingand cleaning are easier. Olivine sands can bereused many times, and there is no dangerof silicosis. However, the basicity of olivineprevents its use in many applications, for ex-ample, with acid-catalyzed furan and phe-nolic no-bake binders.
4) Highly refractory forsterite bricks containingmagnesia binders and possibly chrome ore
(→Glass, Chap. 5.7.). The addedMgO com-bines with excess SiO2 to form forsterite.The addition of chrome ore improves ther-mal shock resistance and reduces the mod-ulus of distortion (V modulus). The mostimportant advantage compared with magne-sia bricks is that scale and slags with a highiron oxide content do not adhere to the wallsurfaces below 1400 C because the MgOis bonded. Also, forsterite bricks are less li-able to sulfate attack in the temperature range800 – 1100 C. However, olivine can be at-tacked by Al2O3, CaO, SiO2 and alkali dueto the formation of lower melting eutectics.For refractory-grade olivine anMg-rich com-position closer to that of forsterite is required.Refractory olivine is used in the steel indus-try and in the manufacture of precast refrac-tory linings for incinerators, where its heat-storage properties can result in more com-plete combustion, and its low expansion rateallows heat cycling without spalling of thelining.
5) Highly refractory cements (particle sizerange 0 – 0.2 or 0 – 1mm). These are formu-lated as for forsterite bricks, but with a highercontent of fluxing materials.
6) Highly refractory ramming mixtures withsimilar compositions to those of bricks.These are incorporated into furnace liningsby manual or compressed-air ramming.
7) Sand-blasting material which does not in-volve a silicosis hazard (unlike silica).Olivine is used to some extent as a blast-ing abrasive and as a grit in water-jet cutting.Together with other minerals, it is used incleaning buildings and occasionally for etch-ing structural steel.
8) Olivine bricks as heat reservoirs in electricalnight storage heaters.
2.5. Andalusite
Andalusite [12183-86-1] belongs to the neso-subsilicates (orthosilicates that contain isolated[SiO4] tetrahedra and additional nontetrahedralanions), and, like kyanite (Section 2.6) and silli-manite (Section 2.7), is amineral of the Al2SiO5group.

Silicates 31
Structure and Mineralogy. Al2SiO5 canform three different crystal structures, i.e.,Al2SiO5 exhibits polymorphism in the form ofandalusite, sillimanite and kyanite. In all threephases one aluminum atom (AlI, Fig. 25) alwayshas the coordination number six, i.e., it is sur-rounded octahedrally by six oxygen ions. The[AlIO6] octahedra are linked by common edgesto form chains parallel to the c axis. The c0 di-mension in all three minerals is therefore ap-proximately the same (ca. 0.55 nm). The otheraluminum atom (AlII, Fig. 25) in andalusite hasthe coordination number five, in sillimanite four,and in kyanite six. The chains of octahedrain andalusite are held together by [AlO5] and[SiO4] groups. In andalusite none of the bondsbreak preferentially, so that andalusite has ahigher Mohs hardness (7 1/2) than sillimaniteor kyanite. Libethenite Cu2[OH/PO4], adamineZn2[OH/AsO4] and eveite Mn2[OH/AsO4] areisotypical with andalusite.
The formulaof andalusite isAl[6]Al[5][O/SiO4].It forms orthorhombic – dipyramidal crystalswith space group D12
2h – Pnnm; lattice constants:a0 = 0.778 nm, b0 = 0.792 nm; c0 = 0.557 nm;the axial ratio is a0 : b0 : c0 = 0.982 : 1 : 0.703;Z = 4.
Some Mn3+ and Fe3+ and a small amountof Cr3+ and Ti4+ always replace the aluminumin andalusite (usually 2 – 3 atom% in total).Viridinite is a variety that contains several per-cent of Mn2O3 and Fe2O3 (a0 = 0.7808 nm,b0 = 0.5929 nm, c0 = 0.5567 nm). Manganeseandalusite is also known. Andalusite has agreater potential for ionic substitution (di-adochy) than kyanite and sillimanite.
Andalusite occurs rather widely as inter-grown, fairly large, thick columnar crystals. Itcan also occur in aggregates that may be granu-lar, in the form of bunches or tufts, as radially ar-ranged columns, and as coarsely tangled massesand lenses. Andalusite also occurs as chiasto-lite in argillaceous and spotted slates (Knoten-schiefer), forming yellowish-white crystals con-taining a gray to black carbonaceous impuritiesin a regular manner.
Andalusite usually has a matt, cloudy,translucent, or opaque appearance and can bered, green or reddish-gray with a vitreous luster.It shows a variable degree of cleavage parallel to110. Transparent crystals of gem quality arerare (California, Brazil).
Andalusite represents a typical example ofa mineral formed by contact metamorphism(hornfels, etc.). Typically, it often occurs inargillaceous rocks with a low lime content incontact aureoles around igneous intrusions. Inplaces, andalusite is found in granite when themagma resorbed aluminum-rich sedimentaryrocks. This mineral is also found in pegmatitesand in accidental blocks (volcanic ejecta). Fur-thermore, andalusite is observed in mesozonal,aluminum-richmica schists alongwith silliman-ite, muscovite, biotite, almandine, and in someplaces also with cordierite and staurolite, al-though never with microcline.
Properties. The following physical proper-ties are important for industrial applications:
Density: 3.09 – 3.16 g/cm3
Mohs hardness: 7 1/2Phase changes:
> 1350 C: decomposition to mullite andvitreous SiO2; on cooling, volume in-creases by 3 – 6%with respect to the start-ing material1840 C: incongruent melting point, con-version to corundum and vitreous SiO21920 C: complete fusion
Specific heat capacity (0 – 100 C):0.705 J g−1 K−1
Thermal conductivity: at0 C 2.26 kJm−1 h−1 K−1, at 189 C4.08 kJm−1 h−1 K−1
The ideal chemical composition is the sameas that of kyanite and sillimanite: 62.93wt%Al2O3, 37.07wt% SiO2.
Andalusite is not attacked by mineral acidsapart from hydrofluoric acid, which attacks itvery slowly in the cold, but readily when hot.Andalusite is stable towards dry HCl and Cl2.Fused KOH strongly attacks andalusite, as doesa solution of CO2 in water at 160 C and 0.6 –0.7MPa.
Deposits and Extraction. Andalusite de-posits are mined in France (Cotes du Nord), Por-tugal (Porto), the Republic of South Africa (inthree areas of the Transvaal: Groot Marico, Ly-denburg, Thabazimbi), the People’s Republic ofChina and the United States (White Mountains;Hawthorne, Nevada).
The overlying rocks above the andalusite-bearing schists and sands are not usually very

32 Silicates
Figure 25. Structure of andalusite, Al[6]Al[5][O/SiO4]A) Projection parallel to c on (001); B) [AlO6] octahedra along the c axis
thick. Therefore, extraction is mainly by open-cast mining using explosives, compressed airhammers and front loaders.
The French deposits contain schists in whichthe andalusite content, which is coarsely crys-talline, is ca. 15%. The andalusite crystals areup to several centimeters long in the direction ofthe c axis and 1 – 4mm wide in the direction ofthe a and b axes. Accompanyingminerals are bi-otite, iron-rich chlorite, and quartz. Thematerialis processed togive twogrades of andalusite con-centrate: 59%Al2O3 with 1% Fe2O3, and 53%Al2O3 with 1% Fe2O3. Processed andalusitesand has a particle size of 0.3 – 1.6mm and isslightly colored.
In the Republic of South Africa, the an-dalusite is interbeddedwith schists. The primarydeposits consist of andalusite-bearing schists inwhich the average content of extractable an-dalusite is 7 – 10%. The secondary deposits arealluvial andalusite sands (placer deposits), inwhich the content of extractable andalusite is10 – 50%, reaching 50 – 60% in the Burgersfortmine (Zimro).
In the White Mountains area of the UnitedStates 3 – 20m thick deposits containing 70 –80% andalusite occur. In Hawthorne, Nevada,the vein-like deposits are 0.5 – 1.5m thick and1000m long. They are extracted to a depth of30m.
Recently, a promising deposit was found nearTomduff in the Republic of Ireland. It consists ofa series of andalusite schists and quartz-biotiteschists, is 200m thick, and continues for 3000malong the strike.
OreTreatment andCommercial Products.The broken schists and sands are crushed to agrain size of 0.3 – 1.6mm (or to 0.6 – 3.35mmby Weedons Minerals). Biotite, chlorite andother ferromagnetic minerals are removed byhigh-intensity magnetic separators. Quartz andother low-densityminerals are removed by awetprocess using settling tanks. These processes aredescribed in detail elsewhere (→Magnetic Sep-aration; →Gravity Concentration). The driedandalusite concentrate is the commercial prod-uct. Commercial grades, with the amounts ofmajor impurities, are listed in Table 10.
The refractory properties of the mullite –vitreous SiO2 matrix (that is formed from theandalusite concentrate by reaction at high tem-perature) improve with increasing Al2O3 con-tent (ideally 62.93%). They also improve withdecreasing TiO2 content (max. 2%), decreasingFe2O3 content (< 1% if possible) and decreas-ing alkali-metal content (fluxing effect).
Uses. Andalusite, like sillimanite, kyaniteand synthetic mullite, is an important raw mate-rial for the production of highly refractory mate-rials and ismarketedwith sillimanite and kyaniteas “sillimanite minerals”. The increase in vol-ume when andalusite is fired above 1350 C issmall (3 – 6%), whereas the corresponding in-crease for kyanite is considerably greater.
Andalusite of the proper fineness is suitablefor use as a filler in coatings and plastics sub-jected to great mechanical or chemical stress,due to its crystal chemical properties and mor-

Silicates 33
Table 10. Grades of andalusite concentrates
Company∗ Grade Al2O3, wt% Fe2O3, wt%
Denain Anzin keraphalite 59 1.531Mineraux KB 53 1Cullinan Andalusite standard 52.5 2
premium 54.3 1.6Export minerals 56 < 1Marico Minerals normal 58.8 1.3
upgraded 61.6 0.6Zimro 59 1.2Lager Mining 57 1.2 – 2Hoogenoog Andalusite 59.9 – 60 < 1
∗ Denain Anzin Mineraux is located in Paris, all other companies are in the Republic of South Africa.
phology. The length : width ratio of the crystalsis 10 : 1 – 50 : 1. However, the energy requiredfor grinding is considerable owing to the hard-ness of the mineral.
Economic Aspects. The annual consump-tion of crude rock by the Federal Republic ofGermany, includingmaterial fired to> 1350 C,is ca. 51 000 t. In France the annual consump-tion of “sillimanite minerals” is ca. 30 000 t andin the Republic of South Africa ca. 135 000 t.The Western countries have a consumptionof ca. 170 000 t and the worldwide consump-tion of “sillimanite minerals” is ca. 300 000 –350 000 t/a in the last few years.
2.6. Kyanite
Kyanite [1302-76-7] (cyanite, disthene) belongsto the nesosubsilicates (orthosilicates that con-tain isolated [SiO4] tetrahedra and additionalnontetrahedral anions) and, like andalusite (Sec-tion 2.5) and sillimanite (Section 2.7), is a min-eral of the Al2SiO5 group.
Structure and Mineralogy. One aluminumatom (AlI, Fig. 26) always has the coordinationnumber six, the [AlIO6] octahedra being linkedby common edges to form chains parallel to thec axis. The other aluminum atom (AlII, Fig. 26)in kyanite also has the coordination number six.The chains of [AlIO6] octahedra are joined toother [AlIIO6] octahedra, which are attached al-ternately to the right and left and are also linkedvia isolated [SiO4] tetrahedra to form stable pla-nar structures parallel to [100]. Vacant octahedraform open channels parallel to (001). This ac-counts for the extension of the crystals in the c
direction [001], giving them a ruler-like appear-ance and, on the other hand, complete cleavagealong (100) and the anisotropic hardness, i.e.,4 – 4 1/2Mohs in the c [001] direction, and 6 –7Mohs perpendicular to this in the b [010] di-rection.
Figure 26. Structure of kyanite, Al[6]Al[6][O/SiO4], projec-tion parallel to c on (001)Due to the low degree of symmetry, there are four differenttypes of aluminum atoms, all with six- coordination. AlI andAlII in kyanite correspond to AlI in sillimanite (see Fig. 27)and andalusite (see Fig. 25) and form chains of octahedra.
The mineralogical formula of kyan-ite is Al[6]Al[6][O/SiO4]. It forms tri-clinic – pinacoidal crystals with space groupC1
i – P 1; lattice constants: a0 = 0.710 nm,b0 = 0.774 nm, c0 = 0.557 nm; the axial ratiois a0 : b0 : c0 = 0.917 : 1 : 0.720; α = 90051/2 ′,β = 10102 ′, γ = 105441/2 ′; Z = 4. The oxygenions in kyanite are approximately in cubic closepacking, yielding the highest density (3.53 –3.65) of the Al2SiO5 polymorphs.
Ionic substitution is possible to a somewhatlarger extent with kyanite than with sillimanite(Section 2.7), but is appreciably less than withandalusite (Section 2.5). Fe3+ can replace alu-

34 Silicates
minum in the lattice to a small extent, as canCr3+ in rare cases, which leads to an increasein the refractive indices. Low levels of alkalimetals and hydroxide have also been found incompletely transparent kyanite crystals.
Kyanite occurs mainly as radially arrangedlamellar aggregates thatmaybe tangled together.The crystals are intergrown, shaped like broadstalks or rulers that extend in the direction ofthe c (001) axis, and are frequently bent intoa slightly waved shape and transversely stri-ated due to translational movement. Kyanite istranslucent to transparent and is usually blue,but is oftenflecked and irregularly colored. It canalso bewhite or yellowish-white (rhatizite), pinkor pinkish, with a nacreous or vitreous luster.Cleavage parallel to 100 is fully developed,is well-marked parallel to 010 and is poor orfibrous parallel to 001. Cleavage parallel to001 has the character of a fibrous fracture,since 100 also represents a slip plane with[001] as the slip direction.
Kyanite is a typical mineral of (aluminum-rich) metamorphic rocks, especially of meso-zonal crystalline rocks, in which it is mainly as-sociated with rutile, staurolite, almandine, horn-blende, micas and corundum. It is occasionallyfound in gneisses, micaceous schists, granulitesand eclogites. In catazonal rocks, kyanite onlyoccurs in high-pressure formations because sil-limanite is stable at lower pressures. Kyanite isthe high-pressure form of Al2SiO5; andalusiteand sillimanite both crystallize at lower pres-sures, with sillimanite being the high-tempera-ture form.
On heating at atmospheric pressure, bothkyanite and andalusite are convertedmonotropi-cally to sillimanite. The conversion of kyanite tomullite and vitreous SiO2 is of industrial impor-tance, because these reactions take place aboveca. 1200 C.
Properties. The following physical proper-ties of kyanite are important for industrial appli-cations:
Density: 3.53 – 3.65 g/cm3
Mohs hardness: 4 12 – 7
Phase changes:< 1300 C: unstable with respect to silli-manite 1335 C: decomposition to mulliteand vitreous SiO2, on cooling, volume in-
creases by 14 – 16% with respect to theoriginal mineral1830 C: incongruent melting, formingcorundum and vitreous SiO21920 C: complete fusion
The ideal chemical composition is62.93wt% Al2O3, 37.07wt% SiO2.
Kyanite is stable towards strong mineralacids. Attack by chlorine and dry HCl is onlyslight at red heat. Hydrofluoric acid decomposesca. 84% of the material. Molten KOH stronglyattacks kyanite. However, there is little attack byaqueous solutions of alkali hydroxides.
Deposits and Extraction. Kyanite is a com-mon constituent of crystalline schists. Few de-posits can be extracted economically, and theseconsist mainly of schists that contain kyanitein crystal aggregates or as small coarse masses.Kyanite can also occur in lenses in pegmatitesand in similar formations or nest-like masses inquartz veins.
The world’s largest reserves are in the UnitedStates and stretch from northeast Georgia toSouth and North Carolina and Virginia in theAppalachians. Other large deposits are found inCanada and the Republic of South Africa. De-posits in the United States, Brazil, the Republicof South Africa, Zimbabwe, Spain and India areexploited commercially.
In the United States, kyanite- containingquartzite is extracted using explosives and exca-vation, followed by grinding and flotation. Theflotation concentrate contains 91% kyanite and2 – 5% iron oxides, with silica making up thebalance. The Fe2O3 content of the dried con-centrate is reduced to 0.16 – 0.94% by high-intensity magnetic separation. The commercialproduct contains 59.5 – 61.8% Al2O3.
A kyanite – sillimanite concentrate is pro-duced as a byproduct of the treatment of zir-conium sands in Australia.
In Spain, alluvial deposits of kyanite- con-taining gravel are dug by front-loading equip-ment.Afterwashing andwet tumbling, the kyan-ite is picked out by hand, calcined at 1480 C,crushed and classified into grain sizes of 0 – 1,1 – 3 and 3 – 5mm. Its composition is Al2O358 – 63%, Fe2O3 0.5 – 1.5%, TiO2 0.15 – 0.5%and alkali metals ca. 0.7%.

Silicates 35
Uses. Kyanite is an important raw materialfor refractory products (ceramics) and is mar-keted with sillimanite and andalusite as an in-dustrial mineral.
Unlike sillimanite, andalusite and mullite,kyanite undergoes a large volume increase (16 –18%) when heated above 1325 C. Powderedkyanite is therefore used in mixtures for ce-ramic and refractory products to compensate forshrinkage on firing. The refractory properties ofthe matrix of mullite and vitreous SiO2 formedfrom kyanite concentrates at high temperatureare a function of its chemical composition.
2.7. Sillimanite
Sillimanite (fibrolite) belongs to the nesosubsili-cates (orthosilicates that contain isolated [SiO4]tetrahedra and additional nontetrahedral anions)and, like andalusite (Section 2.5), kyanite (Sec-tion 2.6) andmullite, is a mineral of the Al2SiO5group.
Structure and Mineralogy. One aluminumatom (AlI, Fig. 27) always has the coordinationnumber six. The [AlIO6] octahedra are linkedby common edges to form chains parallel to thec axis. The other aluminum atom (AlII, Fig. 27)in sillimanite has the coordination number four.The chains of octahedra in sillimanite are lat-erally bonded by [AlIIO4] and isolated [SiO4]tetrahedra which form bands of [Al2Si2O10]tetrahedra from two linked chains of [AlSiO6](see Fig. 27). The four- coordinated aluminum insillimanite cannot be substituted by silicon. Par-allel to b (010), very strongly bonded layers areformed from the [AlO6] chains and [Al2Si2O10]bands. These can easily be cleaved from theneighboring layers by separation at the apices ofthe [AlO6] octahedra. This explains the perfectcleavage of sillimanite parallel to 010. Thechain structure also accounts for the generallyfibrous structure of sillimanite crystals (fibro-lite). As sillimanite has chains of tetrahedra, it isclassified as an inosilicate (chain silicates, i.e.,silicates with infinite chains of [SiO4]4− tetra-hedra).
The mineralogical formula of sillimaniteis Al[6]Al[4][O/SiO4]. It forms orthorhom-bic – dipyramidal crystals with space groupD16
2h – Pbnm; lattice constants: a0 = 0.744 nm,
b0 = 0.760 nm, c0 = 5.75 nm; the axial ratio isa0 : b0 : c0 = 0.979 : 1 : 0.757; Z = 4.
Figure 27.Structure of sillimanite,Al[6]Al[4][O/SiO4], pro-jection parallel to c on (001)
Sillimanite has low tolerance to ionic sub-stitution, the aluminum atoms in the lattice be-ing replaceable only by some Fe3+ and a smallamount of titanium. Although alkali metals andhydroxide are often reported, they either occupythe spaces in the lattice or are impurities in themineral.
Sillimanite usually forms columnar, fibrousormatted aggregates. Single crystals are rare, butlarge crystals (about the size of a fist) are pro-duced synthetically. Dense masses intergrownwith quartz are known as fibrous quartz. Sil-limanite is yellowish gray, gray, brownish orgrayish-green in color, is transparent to translu-cent and has a vitreous luster or a silky lusterwhen in fibrous form.
Sillimanite is usually a common minorconstituent of ortho- and paragneisses (e.g.,feldspar – quartz – cordierite gneisses), mica-ceous schists, granulites, eclogites, and of peg-matites and quartz veins that occur in theserocks. It also occurs in contact-metamorphicrocks, sometimes in the form of very finefibers. At high temperature (> 650 C theoret-ical, ca. 1480 C in practice), kyanite and an-dalusite are converted to sillimanite. At evenhigher temperature (1545 C), sillimanite is con-verted to mullite, which has a lower SiO2 con-tent, and vitreous SiO2. Accompanying miner-als include feldspars, quartz, cordierite, mus-covite, biotite, almandine, staurolite, corundum,andalusite, kyanite, rutile and ore minerals suchas magnetite and ilmenite.

36 Silicates
Properties. Sillimanite is themodification ofAl2[O/SiO4] that is stable at high temperatureand low pressure. Cleavage on (010) is fully de-veloped, but the fracture is uneven.
The following physical properties are impor-tant for industrial applications:
Density: 3.23 – 3.25 g/cm3 (2.95 g/cm3 afterheating at 1545 C)Mohs hardness: 6 – 7Phase changes:1545 C: decomposition to mullite and avitreous matrix containing ca. 95% SiO21810 C: liquid phase reaction to formcorundum and a vitreous phase1860 C: complete fusion> 1860 C: boiling of the melt, producinga vapor phasewith a high SiO2 content anda liquid phase with a high Al2O3 content
Temperature stability limit in an oxidizingatmosphere: 1600 CSoftening temperature under a load of2 kg/cm2: 1550 – 1650 CThermal coefficient of expansion: at20 – 100 C 2.8×10−6 K−1, at 300 C4.1×10−6 K−1, at 800 C 4.3×10−6 K−1,and at 1200 C 4.6×10−6 K−1.Specific heat capacity: at 300 C0.92 kJ kg−1 K−1, at 1200 C1.0 kJ kg−1 K−1
Thermal conductivity: at 300 C 5.4 –5.9 kJm−1 h−1 K−1, at 1200 C 4.6 –5.0 kJm−1 h−1 K−1
Sillimanite (Al2O3 · SiO2) has the idealchemical composition 62.93wt% Al2O3,37.07wt% SiO2. It is insoluble in water, isdecomposed by ammonium fluoride on heatingand is resistant to weathering.
Like the other Al2[O/SiO4] modifications,sillimanite is very difficult to melt and highlyresistant to acids. Its chemical resistance whenused as a refractory material is as follows:
Toward reducing atmospheres moderateToward carbon moderateToward acidic fluxes goodToward basic fluxes moderateToward molten metals good
Deposits and Extraction. Sillimanite de-posits occur in Australia, India, Namibia, theRepublic of South Africa, Brazil and Sri Lanka.The sillimanite is usually associated with quartzand kaolin (Australia) or with corundum (India,
Republic of South Africa). World production isca. 30 000 t/a.
The deposits are mainly extracted by open-cast mining. Due to the hardness of the rock,drilling and blasting are necessary. The rockis crushed and classified with screening equip-ment.
Quality Specifications (see Table 11). Silli-manite used in the productionof clay- containingrefractory products of group 1 (→RefractoryCeramics and ISO R 1109) must contain at least56% Al2O3 (ISO R 1109).
Uses. The most important use for sillimaniteis in refractory materials. It is normally precal-cined to ca. 1550 C to convert the sillimaniteto sinter-mullite (see Section 2.8), which has ahigh thermal shock resistance and a very low co-efficient of thermal expansion (5.6×10−6 K−1
at 20 – 1000 C). Sillimanite with a high corun-dum content is more resistant to slags with ahigh iron oxide content. Mechanically cut nat-ural rock and bonded bricks are only used forlining blast furnaces, glass melting furnaces androasting furnaces.
Coarsely ground material is used for highlyrefractory mortars and ramming compounds.Finely ground material is used in the productionof architecturally applied ceramics (e.g., sani-tary ware or wall and floor tiles).
Sillimanite is used as a filler in paints, coat-ings, adhesives and plastics, especially in sys-tems cross-linked by polyaddition due to the na-ture of its surface (functional SiOH groups forpolyaddition and polycondensation), high hard-ness, and ability to reinforce thematrix due to itsacicular (i.e., needle-like) or fibrous structure.
2.8. Mullite
Mullite [1302-93-8] belongs to the nesosubsili-cates (orthosilicates that contain isolated [SiO4]tetrahedra and additional nontetrahedral anions)and is very similar to sillimanite (Section 2.7) inits structure and properties.
Structure and Mineralogy. Mullite has asillimanite structure, but differs from sillimanitein having a deficiency of oxygen. Since some of

Silicates 37
Table 11. Sillimanite specifications (wt%)
Al2O3 SiO2 Fe2O3 TiO2 Na2O/K2O
Highly enriched 59 – 65 39 – 33 < 1 max. 2 max. 0.1With a high corundum content 65 – 78 33 – 20 < 1 max. 2 max. 0.1
the silicon atoms in the [SiO4] tetrahedra are re-placed by Al3+, some of the positions normallyoccupied by oxygen atoms are left unoccupied tomaintain charge balance. Therefore, mullite hasa higher aluminum content than sillimanite andhas a chemical composition between 3Al2O3· 2 SiO2 and 2Al2O3 · SiO2.
The mineralogical formula of mul-lite is Al[6]4 Al[4]4 [O3(O0.5, OH, F)/Si3AlO16]
or Al[6]4 Al[4]4 [O4−x/2/Si4−xAlxO16], wherex = 1.0 – 1.6. It forms orthorhombic –dipyramidal crystals, with space group D9
2h –Pbam; lattice constants: a0 = 0.7566 nm,b0 = 0.7682 nm, c0 = 0.2884 nm (i.e., halfof c0 for sillimanite); the axial ra-tio is a0 : b0 : c0 = 0.985 : 1 : 0.375;Z = 1. Synthetic mullite, 3Al2O3 · 2 SiO2,has lattice constants a0 = 0.7576 nm,b0 = 0.7688 nm, c0 = 0.2884 nm; the axial ra-tio is a0 : b0 : c0 = 0.981 : 1 : 0.375. Syn-thetic mullite, with the composition 2Al2O3· SiO2, has lattice constants a0 = 0.7576 nm,b0 = 0.7687 nm, c0 = 0.2883 nm; the axial ratiois a0 : b0 : c0 = 0.986 : 1 : 0.375.
Another modification of Al2SiO5 appearsto be a newly discovered phase in thesystem Al2O3 – SiO2 –H2O, crystallizing inthe orthorhombic – dipyramidal system, withspace group D9
2h – Pbam; lattice constants:a0 = 0.755 nm, b0 = 0.827 nm, c0 = 0.283 nm;the axial ratio is a0 : b0 : c0 = 0.913 : 1 : 0.342.
These cells of mullite are subcells (i.e., theordering effects of the oxygen defects and of thesilicon and aluminum ions are neglected). Thereare always six (silicon + aluminum) ions per sub-cell in mullite and twelve (silicon + aluminum)ions with doubled c lattice constants in silliman-ite. In a few cases, ordering is found, and addi-tional superlattice reflexes are observed corre-sponding to a larger unit cell, e.g., a0 = 3×a′
0,b0 = 8×b′
0, and c0 = 6×c′0, and a′
0 = 0.74 nm,b′0 = 0.76 nm and c′
0 = 0.29 nm. Sillimanite is or-dered, with c0 = 2×c′
0, and the same must alsobe true for “pragite”, 2Al2O3 · SiO2, which isonly known as a synthetic product.
In naturally-occurring mullite, Fe3+ or Ti4+
can replace Al3+ to a small extent (up to 1wt%Fe2O3 and up to 2wt% TiO2).
Naturalmullite forms small pseudotetragonalcolorless, white, yellowish or reddish crystalswith columnar or needle-like development in thec [001] direction. Cleavage is well developed on(010), but is difficult to observe due to the nor-mally small size of the crystals. Syntheticmulliteis colorless, but green when Al3+ is substitutedby Cr3+. In synthetic products (e.g., chamottesand porcelain bodies),mullite has extremely finecrystals (< 1µm). When these crystals comeinto contact with slags, larger prismatic crystalsoften form at the reaction interface.Melt-mulliteproducts contain even larger crystals.
Occurrence and Formation. Mullite is notvery often found in natural rocks. It greatly re-sembles sillimanite, with which it can easily beconfused. It occurs almost entirely in contact-metamorphosed formations in primary argilla-ceous sandstones and phyllites contained as in-clusions in basalts and also in a narrow exoge-nous contact zone (buchite) with anatexis at theedge of basalt pipes. The name mullite stemsfrom its occurrence in sintered lumps of sedi-mented clay in the basalt of the island of Mullin Scotland.
The formation of mullite in synthetic prod-ucts, such as chamotte products, porcelain andhigh-aluminum slags (e.g., from coal and coke)is of great industrial importance. It is formedwhen kaolinite is strongly heated or calcined to1200 – 1600 Cand is also producedwhen kyan-ite, andalusite, sillimanite or other aluminosil-icates are strongly heated, decomposing themintomullite and vitreous SiO2. At 1810 C,mul-lite dissociates into an equilibrium mixture ofcorundum and molten SiO2. However, some in-vestigations have shown that mullite can have acongruentmeltingpoint. In silicate slags,mulliteonly crystallizes if minor amounts of basic oxidecomponents are present. Very small amounts ofNa2O, etc., significantly reduce the dissociationtemperature of mullite.

38 Silicates
Properties. The following physical proper-ties are important for industrial applications:
Density: 3.10 – 3.16 g/cm3
Mohs hardness: 6 – 7Phase changes:1810 C: incongruent melting withdecomposition to corundum and a vit-reous phase1920 C: complete melting
Crystallization:Occurs readily from the melt, well-developed crystals (usually finely mattedtogether) are formed, even on very rapidcooling
Temperature stability limit in an oxidizingatmosphere: 1850 CThermal expansion coefficient:at 500 C 4.0×10−6 K−1,at 1000 C 4.5×10−6 K−1,at 1500 C 5.3×10−6 K−1
Specific heat capacity:at 25 C: 0.633 Jg−1 K−1
Thermal conductivity:at 100 C 22.2 kJm−1 h−1 K−1
at 500 C 15.5 kJm−1 h−1 K−1
at 1000 C 14.2 kJm−1 h−1 K−1
Electrical resistivity:at 500 C 3×10−5 Ω · cm,at 1000 C 7×10−3 Ω · cm,at 1500 C 5×10−2 Ω · cm
The ideal composition ofmullite is 71.8wt%Al2O3, 28.2wt% SiO2. Both natural and syn-thetic mullite often deviate from the composi-tion 3Al2O3 · 2 SiO2 (60mol% Al2O3), and anAl2O3 content of 63mol% is not uncommon.Mullite is obtained as a solid crystalline massby sintering a mixture of Al2O3 and SiO2 in themolar ratio 3 : 2 at 1775 C.
Water and strong mineral acids do not attackmullite even when heated. Mullite also resistsconcentrated hydrofluoric acid at room temper-ature for long periods, although rapid decompo-sition takes place on heating to 70 – 100 C. Re-sistance to molten metals and basic compoundsis moderate. To achieve complete breakdown,mullite must be fused with sodium peroxide ora mixture of sodium carbonate and borax glass.
The chemical resistance of mullite in refrac-tory materials is:
Toward reducing atmospheres moderateToward carbon moderateToward acidic fluxes goodToward basic fluxes moderateToward molten metals moderate
Deposits. Natural mullite is mined from onlyone significant deposit in northern Transvaalin the Republic of South Africa (Otavi MiningCompany). The production rate is ca. 5000 t/a.
Synthesis. Synthetic mullite is of consider-able economic importance. Outputs are as fol-lows: United States ca. 27 000 t/a , United King-dom: > 12 000 t/a. Producers in Germany Vere-inigte Aluminiumwerke and Hulst AG; in theUnited Kingdom Keith Ceramic Materials Ltd.;in Hungary Hungary “Motim” Alumina andElectrocorundum Factory; in the United StatesWashington Mills Electro Minerals Corp.; inBrazil Elfus Ltda.; in Japan Showa Denko KK.For the very great demands placed on refrac-tory materials, mullite is superior to the silli-manite minerals (sillimanite, andalusite, kyan-ite), which can be converted into crystallinemul-lite and a vitreous SiO2 matrix only after calci-nation.
Twomain grades of synthetic mullite are pro-duced: fused mullite (electromullite) and sintermullite. Fused zirconia mullite is also produced.In general, sintered mullite is manufactured ina rotary kiln, whereas fused mullite is producedeither in a Higgins furnace or using a tilt-pourelectric arc furnace.
Electromullite. A mixture of kaolinite andcalcined aluminum oxide in a precisely con-trolled ratio is melted in an electric arc furnace.The product is crushed and classified into vari-ous grain sizes. In the selectionof the rawmateri-als, careful attentionmust be paid to levels of im-purities, especially iron, other heavy metal ionsand alkali metals. Metallic contaminants gener-ated by abrasion of the crushing and classifyingequipment are removed by magnetic separators.
The reaction product contains up to 95%mullite and 76.4 – 75.2% Al2O3. The follow-ing composition is given for an English grade:mullite 95%, vitreous matrix and corundum5%, SiO2 23%, Al2O3 76.2%, Fe2O3 0.1%,CaO 0.2%, MgO 0.1%, K2O+Na2O 0.4%.The melting point (Seger cones) is ca. 1900 C,and thematerial withstands long-term loading at1750 C.

Silicates 39
Sinter mullite. The purity of this material issomewhat lower than that of electromullite. Bri-quettes of kaolinite, aluminum hydroxide andsome water for bonding are sintered in tunnelfurnaces at ≤ 1750 C and are then crushed andclassified into size fractions. The product con-tains ca. 86% mullite (i.e., 72.9% Al2O3), ca.0.7%Fe2O3 and small amounts of other impuri-ties. It withstands long-term loading at 1700 C.
The sinter mullite produced by the VereinigteAluminiumwerke (Germany) has the followingspecification: Al2O3 72%, SiO2 16%, density3.1 g/cm3, mean particle diameter 30µm, max-imum particle size 100µm, color white.
Uses. The main area of use for mullite is inhighly refractory materials for lining metallur-gical melting furnaces and glass melting tanks,along with other applications where stronglyacidic attack takes place at high temperatures.Mullite is also used as a raw material for porce-lain for electrical insulation, as an opacifier forhigh-quality enamels, as a polishing medium(due to its great hardness), as a filler in plasticsand paints, and as an abrasion-resistant reinforc-ing filler in coatings for surfaces and roadwaysthat are subject to severe mechanical stress.
Some of the synthetic mullite’s distinguish-ing attributes (e.g., excellent thermal shock re-sistance, good hot strength under load, volumestability at high temperatures, low shrinkage,high resistance to aggressive melts and slags,good abrasion resistance, and additionally highcreep resistance of fused mullite) also permitits wide use. Refractories for the steel and glassindustries are the most important market for sin-tered and fused mullite and fused zirconia mul-lite. Synthetic mullite is also used in the ceramicindustry.
2.9. Vermiculite
Structure and Mineralogy. Vermiculite be-longs to the phyllosilicates (sheet or layer sili-cates) and has a mica-like foliated structure. Itdisplays the typical properties of the montmoril-lonite – saponite group to a pronounced degree:the layers have a greater excess charge and agreater capacity for cation exchange than talcor pyrophyllite layers. The structure contains
layers similar to those found in talc, pyrophyl-lite (→Talc), biotite, and muscovite (→Mica,Chap. 2.). It comprises infinite, two-dimensionaldouble silicate layers 2
∞[Si4O10]4− (Fig. 28).Part of the Si4+ is substituted by Al3+, and theterminal oxygen ions of the [(Si, Al)O4] tetra-hedra are always on the same side (Fig. 28A).The hexagons formed by these oxygen ions havehydroxyl groups at their centers. In this dou-ble layer, the oxygen layers formed from O(bound to one Si) and OH groups face eachother directly, forming octahedral voids. Thesevoids are occupied mainly by Mg2+, but alsoby Fe3+ and Al3+ (Fig. 28B), which hold to-gether the two Si2O5/OH sheets. The octahe-dral voids are formed by four oxygen atomsand two hydroxyl groups. Due to the partial re-placement of Si4+ by Al3+, the double layerpocket Mg3[(SiAl)4O10/(OH)2] has a slight ex-cess negative charge, which is compensated byadditional cations, e.g., Mg. These cations, to-gether with water molecules, are interlayeredbetween the layer pockets, usually Mg2+ or,less frequently, Ca2+. This interlayer (interme-diate layer) has a positive excess charge and con-sists of a double layer of H2O–Mg2+ –H2O, inwhich the H2O locations may be only partiallyoccupied. EachH2O in this interlayer is linked toan oxygen atom in the neighboring silicate dou-ble layer by a hydrogen bond. Thus, vermiculitestructurally resembles a kind of talc expandedby H2O that has been formed from (hydrous)mica by potassium depletion.
The swelling capacity (ability to be hydrated)of vermiculite is due to the fact that the sili-cate layers have a slight excess negative chargeand thus repel each other; this facilitates thepenetration of water into the interlayer with itssmall positive excess charge. Unbonded watercan be removed from the vermiculite by heatingto < 110 C, but the water fixed by cations inthe interlayer is only released at > 300 C. Onheating, the water in the interlayer is released ei-ther gradually or, on sudden heating,with expan-sion and formation of worm-shaped structures.Exfoliation causes unidirectional expansion per-pendicular to the cleavage plane (001) and thevolume increases greatly (from 8- to 50-fold).If the trapped water is driven off slowly, evenat 250 C vermiculite shows no exfoliation. Ex-foliation of vermiculite can also be caused bytreatment with hydrogen peroxide, which leads

40 Silicates
Figure 28. Structure of vermiculliteA) The two-dimensional infinite silicate layer [(Si, Al)4O10]x−, projection parallel to c on (001); B) Intermediate H2O–Mg2+ –H2O layer between two silicate double layers, projection perpendicular to the a axis
to release of oxygen by reaction with interlayermagnesium ions.
The mineralogical formula of vermiculite is:
(Mg, Fe3+, Al)3[(OH)2/Al1.25Si2.75O10] ·Mg0.33(H2O)4
or
Mg2.36Fe3+0.48Al0.16[(OH)2/Al1.28Si2.72O10]0.64−
· Mg0.32(H2O)4
Vermiculite forms monoclinic crystals, withthe following lattice constants: a0 = 0.533 nm,b0 = 0.918 nm, c0 = 2×1.436 nm; β = 97; thedistance between the layers, d002 = 1/2c · sinβ,is ca. 1.425 nm.
Batavite and protovermiculite are related tovermiculite and have similar compositions andlattice constants. In nickel – vermiculite, a highproportion (10 – 25 atom%) of the Mg2+ ionsis substituted by Ni2+. The alkali metal contentof vermiculite is always very small, and highcontents are due to replacement of layers of ver-miculite by layers of biotite. An increase in thenumber of these biotite layers can eventuallylead to the formation of hydrobiotite. Another“mixed-layer” mineral is formed by interlayer-ing of the vermiculite with chlorite; when there
is a high iron content, this is known as jefferisite,andwhen the interlayering is regular, with a 1 : 1ratio, it is known as corrensite.
Crystals of vermiculite can reach a size ofseveral centimeters and are then easily confusedwith biotite. However, they are usually muchsmaller (2µm) and can be of the size of clayparticles when they occur in sediments. Ver-miculite forms monoclinic – pseudohexagonal,short, columnar platelets and mainly occurs inaggregates, which can be bent into worm-likeshapes. Fibrous formations can also occur, butare very rare. Vermiculite is brown, bronze-colored, yellow, green, or colorless. Cleavage on(001) is perfect and mica-like, but the plateletsare not elastic as in micas.
Deposits of vermiculite often occur in ultra-basic and basic rocks (e.g., pyroxenites, horn-blendites, peridotites and their metamorphicequivalents) that have undergone hydrothermalpotassium metasomatism and alteration to acti-nolite – biotite formations. These in turn can bealtered in the final stages potassium of this meta-somatism or by anhydrous processes to forma-tions of vermiculite and hydrobiotite with re-licts of phlogopite, biotite, tremolite and actino-lite. The alteration of biotite to vermiculite takes

Silicates 41
place by leaching of K+ from between the sili-cate layers by hydrothermal activity, percolatingground water or supergene weathering. Vermi-culite found as an alteration product of biotitein other metamorphic rocks, such as schists andgneisses, is of similar origin. Vermiculite is onlyrarely found as an alteration product of micain marine sediments. In places, vermiculite isfound in these sediments as an alteration productof, e.g., chlorite and amphibole. Vermiculite oc-curs occasionally in marble and in carbonatites.Of some economic importance are vermiculitedeposits in the contact zones between ultrabasicand basic rocks and acid intrusives.
Vermiculite is transformed into enstatite andtalc on heating to 650 C. Under increasinglysevere hydrothermal conditions (< 200 C,70MPa) it is converted into pseudochlorite,which is itself converted to chlorite and mont-morillonite at ca. 550 C. Vermiculite is easilyaltered into biotite by potassium metasomatism,although this process is only rarely observed innature.
Properties. The following physical proper-ties are of interest for industrial applications:
Density: 2.3 – 2.75 g/cmMohs hardness: 1 1/2 – 2Sintering temperature: ca. 1260 CFusion point: 2000 – 2400 Cmp: 1315 CSpecific heat capacity: 0.84 kJ kg−1 K−1
Thermal conductivity:0.062 – 0.065Wm−1 h−1 K−1
Deposits and Extraction. Deposits of eco-nomic importance are found in northernTransvaal (Republic of South Africa), at Libbyin Montana, at Louise in Virginia, and at En-coree in Laurens County in South Carolina(USA). Further deposits occur in Brazil, Zim-babwe, Kenya, Australia, India, China, Russiaand Japan. The vermiculite in these depositshas been formed from phlogopite and biotite byweathering (leaching of K) and hydrothermal al-teration. The vermiculite at Encoree in SouthCarolina occurs in metamorphic rocks in con-tact with acidic intrusions interspersedwith peg-matites. Accompanyingminerals include corun-dum, apatite, serpentine, chlorite, talc and mica.
At Palabora in the Transvaal (Republic ofSouth Africa), the Archean contains a Protero-
zoic intrusive alkali rock complex that consistsof, e.g., pyroxenite, syenite, ultrabasic pegma-toids and carbonatite. This intrusive complex isnot only well known for its large copper deposit,but also has an outstanding vermiculite deposit.The vermiculite is an alteration product of phl-ogopite and is free of asbestos.
The vermiculite bearing zones are selectivelyextracted by open-cast mining, The ore is loos-ened by blasting and removed by front loaders.The vermiculite ore is beneficiated by screening,jigging and classification.
Processing has three aims:
1) To separate the vermiculite from the accom-panying rock, including rudimentary biotiteand phlogopite
2) To delaminate the “books” formed from thevermiculite layers and to split the “leaves”(thickness > ca. 0.8mm) into “flakes”
3) To classify the flakes into commercial parti-cle size fractions
In the first stage, the rock is usually brokeninto < 20mm or < 16mm granules, and the ad-hering fine material is removed by washing andscreening. Further size reduction separates thevermiculite from the accompanying rock.
In a process operated by Palabora Mining(RSA), the vermiculite flakes are separated fromthe accompanying rock, includingmica, bymul-tistage screening in a horizontal stream of airusing various suspension techniques.
The products from both the United Statesand South Africa are separated into five or sixsize grades in the millimeter range. The chem-ical compositions are listed in Table 12. Com-pared with the North American material, SouthAfrican vermiculite has undergone a higher de-gree of physical transformation, gives a higheryield on thermal expansion, and therefore has ahigher market value. The difference can also beseen from the chemical composition.
Expansion. Vermiculite, similar to perlite(exhibiting three-dimensinal expansion), ex-pands when heated., but only in one direc-tion (one-dimensional) showing the unique phe-nomenon of exfoliation by sudden heating toca. 300 C or higher. Responsible for this ex-foliation is a double layer of water (i.e., an

42 Silicates
Table 12. Chemical composition of raw vermiculite (average values, wt%)
Component Libby, Montana Encoree, South Carolina Palabora (RSA)
SiO2 38.64 38.66 39.37MgO 22.68 20.04 23.37Al2O3 14.94 17.36 12.08Fe2O3 9.29 8.45 5.45K2O 7.84 5.24 2.46Na2O 0.80CaO 1.23 0.75 1.46Cr2O3 0.29 0.50MnO 0.10 0.11 0.30P2O5 Trace Trace 0.15S Trace Trace 0.18Cl 0.28 0.52 0.02H2O 5.29 8.71 14.24Total 100.58 100.34 99.88
H2O–Mg2+ – interlayer) trapped between thesilicate double layers and liberated upon heat-ing, whereby the rapidly generated vapor cannotbe set free without buckling and separating thestructural layers. The low thermal conductivityand low density of expanded vermiculite lead toits widespread use in thermal and acoustic insu-lation applications, aswell as in refrigeration en-gineering. Expansion occurs prerpendicular tothe cleavage plane (001).
The suitability of a particular vermiculite forconversion to expanded aggregates can be as-sessed by differential thermal analysis. How-ever, the temperature – time profile of the fur-nace used for expansion can only be establishedwith certainty by a laboratory-scale expansiontest.
For the expansion and exfoliation of vermi-culite various furnaces (e.g., rotary, cascade, vi-brating tray, injection tray) can be used. The ver-miculite concentrate is heated to between 430and 1100 C. The water in the vermiculite isliberated in three stages in 0.25 – 8 s, beginningwith a reversible reaction at ca. 150 C, followedby the onset of exfoliation above 500 C and va-porization of the residual water at 870 C. Thebulk density is reduced from about 640 – 1040to 64 – 176 kg/m3. In some industrial processes,the vermiculite flakes are expanded in 5 – 8 sat 1250 – 1500 C and then cooled immediatelyto 400 C. Since exfoliated vermiculite shows ahuge increase in bulk, only shipping of the ver-miculite ore or concentrate is economic. Plantsfor expansion of the vermiculite are thereforegenerally located close to the consumers.
Uses. In its most important applications, ver-miculite is used in its expanded state to exploitits low density, good thermal and acoustic in-sulating properties and high absorption capacityfor liquids.
Vermiculite is utilized in the construction in-dustry for thermal and acoustic insulation. Ther-mal insulation is the largest market.
Loose filling is widely used in the construc-tion industry and in industrial applications for in-sulating hollow spaces (e.g., cavity walls), ceil-ings, walls and roofs. Loose-fill vermiculite iscommonly a coarse grade with a particle size upto 16 mm and a bulk density of 56 – 72 kg/m3.At cryogenic temperatures perlite is a better in-sulator, but vermiculite is also used in limitedquantities for such purposes.
Vermiculite is also utilized in the manufac-ture of castable insulation products. For pro-ducing high-alumina vermiculite cement, a fast-hardening cement, alumina and lime are heatedin a furnace to a high temperature. The productis ground to a powder, and vermiculite is added.Vermiculite cement can also be manufacturedby combining it with Portland cement. Vermi-culite cement is used in the production of ultra-lightweight aggregates for the construction in-dustry (Table 13) and as lightweight insulation.
Vermiculite is utilized as a constituent in themanufacture of refractory blocks and fire bricksfor use in furnaces up to 1200 C.
In packaging technology, vermiculite is usedto cushion impacts, for thermal insulation andto absorb liquids in case of breakage of the con-tainer.

Silicates 43
Table 13. Physical properties of vermiculite lightweight concrete
Vermiculite/ cement Density, Compression strength, Thermal conductivity, Shrinkage on drying,ratio kg/m3 N/mm2 Wm−1 h−1 K−1 %
8 : 1 400 0.70 0.094 0.356 : 1 480 0.95 to to4 : 1 560 1.23 0.158 0.45
Inmetallurgy, it is used to covermoltenmetalfor thermal insulation during intermediate stor-age prior to casting.
Expanded vermiculite is also used with gar-den and house plants to store water and plant nu-trients (hydroculture), as a filter for swimmingpools and for absorbing spilled liquids.
Economic Aspects. Total world productionof crude vermiculite is ca. 512 500 t/a. Becauseof the short resources the production declined inthe United States to ca. 177 000 t/a. Vermiculiteis invariably expanded at a location close to theconsumer to facilitate transport. The largest con-sumer country is the United States. Europeanconsumption is modest, most being importedfrom the Republic of SouthAfrica. Although thetotal consumption of silicates used for thermaland acoustic insulation (including expanded per-lite, hydrated and expanded broken glass, glasswool and rock wool) continues to increase, thatof vermiculite is decreasing due to its compara-tively high price.
2.10. Perlite
Perlite of rhyolitic composition is a natural, vol-canic glass that is usually black or gray, butsometimes brownish red. It has curved shrinkagecracks and therefore breaks up into spheroidalgranules. It is usually produced by rapid cool-ing of rhyolitic melts (rhyolite being the vol-canic equivalent of granite). Being a volcanicglass, perlite contains few crystals and only asmall amount ofwater (2 – 6%combinedwater).Naturally-occurring glasses containing 3 – 8%water are known as pitchstone (hydrated glass).
Perlite was first mined and expanded in 1946in the United States. It has since been put toa wide range of uses in Europe. Thermally ex-panded perlite is increasing in importance as alightweight filler in constructional units for ther-mal and noise insulation.
Composition varies according to the type ofdeposit:
SiO2 72.0 – 76.0wt%Al2O3 11.0 – 17.0wt%K2O 4.0 – 5.0wt%Na2O 2.9 – 4.0wt%CaO 0.5 – 2.0wt%MgO 0.1 – 0.5wt%Fe2O3 0.5 – 1.5wt%TiO2 0.03 – 0.2wt%MnO2 0.03 – 0.1wt%SO3 0 – 0.2wt%H2O, chemically bound 2 – 7wt%Density 2.2 – 2.4 g/cm3
Softening temperature 810 – 1090 CMelting temperature 1260 – 1340 CSpecific heat 831 J g−1 K−1
Bulk density, broken, classified 1.45 kg/L
The most significant variations are in watercontent, which has an important influence on thephysical properties after expansion.
Deposits and Processing. The most impor-tant European deposits are found in Greece (Mi-los, Kos), Italy (Sardinia), Slovakia, Hungary,Bulgaria and Iceland. Further important de-posits are located in Turkey (Cumaovasi, Izmir),Canada, Australia, Japan and the United States(New Mexico). Perlite is commonly extractedby open-cast mining. It is then crushed (usu-ally by cone crushers) and classified using vibra-tory screens into several grain sizes with closelyspecified tolerances.
The industrial value of the classified ore de-pends on the temperature and time necessaryfor expansion, relative volume increase, ratio ofclosed to open bubbles (cells), nonexpandablefraction and compression strength of the prod-uct.
Expansion. Like vermiculite, perlite ex-pands when heated, but in three dimensions. Ex-panded perlite has excellent insulation proper-ties, comparable to those of vermiculite, whichit can substitute to some extent. Expansion isperformed in two types of furnace: (1) tilted

44 Silicates
horizontal rotary furnaces through which thefeed material passes in countercurrent flow tothe combustion gas and (2) vertical rotary orblast furnaces through which the expandinggrains and hot gases pass in cocurrent flow.Vertical expanders are more common. The ex-pansion results from shock heating the grainsto a temperature in the softening range (727 –1127 C), which results in bursting. Then thewater bound in the perlite vaporizes and the heat-softened grains expand into light, cellular par-ticles with up to 15 – 30 times the original vol-ume. The cellular structure consists of numeroustiny, closed, thin-walled, glass-sealed air bub-bles, which account for the excellent insulatingproperties of the expanded perlite. The optimumtemperature – time profile must be closely fol-lowed. Longer heating times lead to shrinkage ofthe heated aggregates, and higher temperaturescause shrinkage and/or melting. The expandedmaterial is classified in cyclones. In some cases,the expanded perlite is ground by pendulum orroller mills. If the product is to be used as a filleror additive, it is treated to make it hydrophobic;it may be silanized with dilute aqueous silanesuspensions, or coated with dilute, aqueous sil-icone oil emulsions.
Quality. Important quality criteria for the ex-panded material include bulk density, ratio ofclosed to open air bubbles (cells), unexpandedfraction, and, if used as a filler, compressionstrength of the bulk material and binder re-quirement or water absorption properties. Thebulk density of expanded perlite depends on theamount of chemically bonded water in the unex-panded material and on the temperature – timeprofile used for expansion and can be 0.03 –0.2 kg/L. The fraction of the material with opencells and the fraction of nonexpanded grainsshould not exceed 5%. These parameters areboth determinedby awater-immersion test usingmaterial that has not been rendered hydrophobic.
Properties. The most important propertiesfor the diverse applications of expanded perliteare its extremely low density, low thermal con-ductivity, low thermal expansion, good soundabsorption, adequate compression strength at abulk density of > 0.1 kg/L, and resistance toacids, weathering, and microbial attack. Theaverage properties of industrial perlite (from
Milos) after expanding and grinding to 98%< 40µm are given below:
Refractive index 1.49Brightness index Ry 82 – 85(according to DIN 53 163)
Bulk density 0.080 kg/LTamped density 0.240 kg/LOil absorption number 500 – 600 g/100 g(standard grade)
Water absorption number 400 – 500 g/100 g(standard grade)
Water absorption number 20 – 25 g/100 g(hydrophobic grade)
pH 8 – 9Thermal expansion coefficient 0.007K−1
(0 – 100 C)Thermal conductivity (25 C)Bulk density0.04 kg/L 0.04Wm−1 h−1 K−1
0.09 kg/L 0.05Wm−1 h−1 K−1
0.13 kg/L 0.06Wm−1 h−1 K−1
Thermal conductivity(bulk density 0.09 kg/L)20 C 0.05Wm−1 h−1 K−1
400 C 0.11Wm−1 h−1 K−1
800 C 0.22Wm−1 h−1 K−1
When perlite is used as a filler and as an ad-ditive to lightweight concrete, very high com-pression strength and resistance to abrasionare required during mixing. These grades havehigher densities (0.12 – 0.14 g/L) and are pro-duced from rhyolitic rock.
Uses. Expanded perlite is used as an ultra-lightweight aggregate in the construction in-dustry and as lightweight insulation. The mostimportant uses of expanded perlite (nor-mally treated to render it hydrophobic) includeloose-fill insulation, filled plaster board andwall elements, insulating silicate-bonded boardsfor thermal and flame protection, insulatingbitumen-bonded boards for back facing, insulat-ing boards and coating materials with polymerbinders, mortar (bonded with cement, plaster, orsilicate), polymer dispersion mortars and plas-ters for thermal insulation and prevention of con-densation on walls and ceilings (e.g., in swim-ming pools and high humidity rooms).
Coarse-grade expanded perlite (2 – 5 mm) isused in metallurgy to limit the loss of heat frommolten metal surfaces. A fine grade (0.15 – 1.8mm) is used as a thermal insulator in cryo-genic applications. In cryogenic applications,expanded perlite is normally uses unfer vaccumconditions.

Silicates 45
Expanded perlite, with a bulk density of0.04 – 0.06 kg/L, is used as an adsorptive filter-ing aid in precoated filters (e.g., for drinking wa-ter or medicinal products).
Economic Aspects. Next to the UnitedStates, Western Europe is now the largest con-sumer of perlite. World annual production ofraw perlite is estimated to be ca. 1.8×106 t.In Germany ca. 100 000 t/a perlite is expanded,of which ca. 70 000 t (corresponding to ca.580 000m3) is used as a raw material for thebuilding industry. The amount of perlite (fromItaly, Greece and Iceland) expanded in theUnited Kingdom is > 120 000 t/a. The total forFrance is ca. 90 000 t/a.
2.11. Pumice
Pumice [001332-09-8] is not a crystalline sili-cate mineral, but a light-colored, highly vesic-ulated, foamed volcanic glass of mainly acidcharacter (> 66% SiO2, rhyolite), with a highmelt viscosity, high pore volume (> 50%) andvariable water content. Rhyolite itself is an acidvolcanic rock consisting of feldspar, quartz, bi-otite, etc. and glassy groundmass. By rapid un-dercooling (quenching) of rhyoliticmagma, vol-canic rocks are produced which consist com-pletely or almost completely of volcanic glass,named obsidian (containing less than 1 wt%H2O and composed of more than 80 wt% ofvolcanic glass including mostly very negligi-ble amounts of minute inclusions of quartz,tridymite, feldspar, fayalite, etc.) or pumice, thelatter highly vesiculated. Volcanic lava of rhy-olitic composition releases enclosed gases (wa-ter vapor, carbon dioxide, nitrogen, sulfur diox-ide, chlorine, etc.) at ca. 1000 C explosively asa result of an abrupt pressure drop forming bub-bly pumice. Due to its high porosity, pumice isvery light and floats on water.
The specific heat capacity of pumice is1.00 J g−1 K−1 and its thermal conductivity at50 C is 0.274Wm−1 h−1K−1.
Crushed and classified pumice is employedas an additive in light insulating concrete usedto make constructional units. Due to its lim-ited compression strength, this buildingmaterialcan only be used for one- or two-storey build-ings or for cladding load-bearing frameworks.
The specific heat capacity of pumice concreteis 1.005 kJ kg−1 K−1 and its thermal conduc-tivity is 0.457Wm−1 K−1 for a density of ca.1.1 kg/L.
Building blocks (e.g., Bisotherm,Bisothermwerke Carl Riffer, Urmitz) consistof pure, natural pumice granules treated to re-move organic and inorganic impurities. Denserinorganic impurities such as fragments of obsid-ian and lavastone are removed by air separationand organic impurities by calcining at ca. 400 Cin rotary kiln (DIN 18 151, 18 152). They aremainly used to build houses.
Pumice in different industrial grades has awide range of applications: stonewashing ofdenim and cotton, in agriculture (soil substitute,additive), paint manufacture (non-skid coatings,acoustic insulating ceiling paints, fillers for tex-tured paint), chemical industry (filtration me-dia, chemical carriers, pesticide carriers), clean-ing and polishing of metals and plastics, com-pounders (powdered hand soaps, glass clean-ers), dental and cosmetic (polishing natural teethand dentures, smoothing rough skin), rubber(erasers, mold-release agents), glasses and mir-rors (TV tube processing, glass buffing and pol-ishing), furniture (hand-rubbed satin finishing,piano keys, picture frame gold leafing), leather(buffing), electronics (cleaning circuit boards)and pottery (filler).
Powdered pumice is used as an abrasive, asa polishing medium, as an antiskid material andas an additive to pumice soap. It is also used asan absorption medium due to its large surfacearea (e.g., in packages carrying breakable liquidcontainers).
As an insulator, pumice competes with ex-panded perlite (see Section 2.10) and expanded,exfoliated vermiculite (see Section 2.9).
The output of pumice in Germany is ca.2.4×106 t/a, worldwide ca. 6.5 – 7×106 t/a.
Important commercially exploited pumicedeposits occur in Germany (Koblenz-Neuwieddistrict), in Italy (isle of Lipari – northeast toSicily, and around Naples), on volcanic Greekislands (Dodecanese group: islands of Yali andNisyros, Cyclades group: Thera Island), in Spain(Province of Ciudad Real, Canadas area on isleof Tenerife), Portugal (on the volcanic island ofSao Miguel in the Azores), the United States (incentral New Mexico in the area of Santa Fe, atGate in Oklahoma, and in Idaho and California),

46 Silicates
onCaribbean islands (Dominicana,Martinique),on volcanic islands of Japan (southern parts ofHokkaido and Kyushu), and in New Zealand(North Island).
2.12. Basalt
Basalt is the most common volcanic rock, oc-cupying large areas of the Earth’s surface (e.g.,plateau basalt). It contains < 52% SiO2 andis the volcanic equivalent of the gabbro rockfamily. Basalt has a dense, compact, some-times granular or porphyric structure and isdark to black in color. The main constituents ofthe matrix are plagioclase (anorthite 50 – 75%)and pyroxene (mainly augite). Accessory min-erals include olivine, magnetite, ilmenite, al-kali feldspar, alkali pyroxene, alkali amphibole,feldspathoids and volcanic glass. Basalts areworldwide by far the most extensive exposedcrustal igneous rocks, and their formation canbe observed even now (e.g., numerous volca-noes in Asia, the United States, Europe and atthe mid-ocean ridges).
The most important types are the tholeiiticbasalts (often associated with some olivine and,when supersaturated with SiO2, even quartz)and the alkali basalts (feldspathoids, alkali py-roxene, alkali amphibole and olivine). Medium-to coarse-grained basalts are known as do-lerites, while olivine basalts (which containfeldspathoids as well as plagioclases) are knownas basanites, and anchimetamorphic basalts ofthe Permian/Carboniferous era are known asmelaphyres, diabases and greenstones.
The largest single basalt mass in Europe is inthe Vogelsberg region of Germany. Other largedeposits are in Hegau, the Eifel, the Westerwaldand the Rhon mountains. Another deposit ex-tends from the Oberpfalz (Bavaria), through theErzgebirge and as far as the Lausitz region.
Some basalts have high strengths and aretherefore used in the construction industry, e.g.,as hard core, paving stones and building mate-rials. In another important application, moltenbasalt is converted into refractory fibers (min-eral wool for insulation and fire protection)(→Fibers, 5. Synthetic Inorganic, Chap. 2.2.).
The physical properties required dependon the use intended for the basalt. Theseinclude high compression strength (2500 –
4000 kg/cm2, with a maximum of 5800 kg/cm2)and transverse strength (150 – 250 kg/cm2, DIN52 100). It shouldbe as homogeneous as possibleand should be resistant to frost, acids and wastegases (e.g., flue gas). The shapes of the naturally-occurring blocks of material also influence pos-sible uses, e.g., columnar masses with polygo-nal cross sections, having 4 – 7 sides (feldsparbasalts).
The density of basalt varies between 3.00 and3.15 g/cm3, the bulk density between 2.95 and3.00 g/cm3 and the bulk density of basalt lavabetween 2.20 and 2.35 g/cm3. The porosity ofbasalt varies between 0.2 and 0.9 vol%, and ofbasalt lava between 20 and 25 vol%. Thus, thewater absorption of basalt varies between 0.1and 0.3wt%, and of basalt lava between 4 and10wt%. The specific heat capacity (0 – 770 C)is ca. 1.09 J g−1 K−1, and the thermal conductiv-ity (0 – 100 C) is ca. 0.028Wcm−1 K−1. Thelinear expansion coefficient is ca. 9×10−6 K−1.Anhydrous basalt begins to melt at ca. 1280 C,but at only ca. 1100 C if it contains 1% water.Basalt wool is stable up to 900 C.
Sunburnt basalt is unsuitable for many appli-cations. It is affected by weathering very soonafter it is mined, especially by the heat of strongsunlight in the presence of moisture. Charac-teristic white spots are formed, and the rockcrumbles. This phenomenon is not limited tobasalts alone, but can also occur with othereffusive rocks (e.g., phonolite). The “sunburn-ing disease” of basalt is probably mainly dueto the conversion of nepheline into analcime(Na[AlSi2O6] · H2O), which is accompaniedby a volume increase of ca. 5.5%.
2.13. Wollastonite
Wollastonite [013983-17-0], Ca3[Si3O9 ], ispolymorphic. Three modifications occur natu-rally, the commonest being low wollastonitewhich has two polytypical structural modifica-tions: triclinic wollastonite (-1T) and monoclin-ic wollastonite (-2M, parawollastonite).
Structure and Mineralogy. In monoclinicwollastonite, the [SiO4] tetrahedra are linkedto form infinite one-dimensional dreier singlechains of [Si3O9]6− units (Fig. 29). These arebonded together by Ca2+ ions (which balance

Silicates 47
the electrical charge), and the chains lie parallelto b [010]. This explains why the crystals arealways extended in the direction of b [010]. Themonoclinic structure is derived from the triclinicby “inner twinning” on the (100) plane. Wollas-tonite (-1T) andwollastonite (-2M) are thereforeinosilicates (silicates containing [SiO4] tetrahe-dra in infinite chains).
Figure 29. Basic configuration of [SiO4 ] tetrahedra in theinfinite one-dimensional dreier single chains [Si3O9]
6−∞ ofwollastonite, Ca3[Si3O9 ]
Cyclowollastonite (high wollastonite orpseudowollastonite) is a cyclosilicate and is sta-ble above 1126 C. The [SiO4] tetrahedra arearranged in three-membered rings [Si3O9]6−,which are linked together and neutralized byCa2+ ions (Fig. 30). High-pressure Ca3[Si3O9]also contains rings of tetrahedra.
Figure 30. Basic configuration of [SiO4 ] tetrahedra inthe dreier single rings of [Si3O9 ]6− of cyclowollastonite,Ca3[Si3O9 ]
Wollastonite (-1T), Ca3[Si3O9], forms tri-clinic – pinacoidal crystals, with space groupC1
i – P 1, and has the following lattice constants:
a0 = 0.794 nm, b0 = 0.732 nm, c0 = 0.707 nm;the axial ratio is a0 : b0 : c0 = 1.084 : 1 : 0.966;α = 9002′, β = 9522′, γ = 10326′; Z = 2.
Wollastonite (-2M), (parawollastonite),Ca3[Si3O9], forms monoclinic – prismaticcrystals, with space group C5
2h –P 21/a; lattice constants: a0 = 1.536 nm,b0 = 0.729 nm, c0 = 0.708 nm; the axial ratiois a0 : b0 : c0 = 2.107 : 1 : 0.971; β = 95241/2 ′;Z = 4.
Cyclowollastonite (high wollastonite, pseu-dowollastonite), Ca[Si3O9], forms triclinic –pseudohexagonal crystals (b:a= 1.707 ≈√
3);lattice constants: a0 = 0.690 nm, b0 = 1.178 nm,c0 = 1.965 nm;the axial ratio is a0 : b0 : c0 = 0.586 : 1 : 1.668;α = γ = 90, β = 9048′; Z = 8.
High-pressure Ca3 [Si3O9] forms tricliniccrystals with lattice constants a0 = 0.6695 nm,b0 = 0.9257 nm, c0 = 0.6666 nm; the axial ratiois a0 : b0 : c0 = 0.723 : 1 : 0.720, α = 8638′, β =7608′, γ = 7023′.
In naturalwollastonite,Ca2+ canonly be sub-stituted to a very small extent (ca. 1 – 2 atom%)by Mg2+, Fe2+ and Mn2+. Alternatively, it canbe substituted by 2wt% Al2O3.
In the wollastonite (-1T) found in industrialproducts, (β-Ca3[Si3O9]), Ca2+ can be substi-tuted by Fe2+ up to 70wt% FeSiO3. In indus-trial cyclowollastonite, (α-Ca3[Si3O9]), Ca2+
cannot be substituted by Fe2+, but can be substi-tuted by Mn2+ up to 1 – 2 atom% and by Sr2+
over the entire concentration range.Wollastonite is usually intergrown and well-
formed crystals are very rare. It occurs mostlyin the form of thick plates parallel to 100and 001 and is always extended on the b axis[010]. It is often found in coarse masses, in flat,thin, divergently radiating masses, or in stalked,spiky, tabular, lamellar or finely fibrous aggre-gates and masses.
Magmatic formation of wollastonite is possi-ble where the magma resorbed calcium while incontact with limestones. This mineral is foundin some alkaline igneous rocks and in phono-lites, andesites, etc. Wollastonite is commonlya product of contact metamorphism, and some-times also of regional metamorphism. Under theconditions of contact metamorphism, wollas-tonite is formed at high temperatures fromquartzand calcite in siliceous limestones and by SiO2metasomatism in pure limestones. Cyclowollas-

48 Silicates
tonite is only rarely found in metamorphic rocks(e.g., south-west Iran, Kropfmuhl near Passau inBavaria).
Cyclowollastonite is formed when wollas-tonite is heated to > 1126 C. It melts con-gruently at 1544 C. In industrial products, cy-clowollastonite is the modification that is mostoften formed, e.g., in silicate bricks, in blast fur-nace slags and their reaction products with sili-cate bricks, in slightly basic open-hearth slags,or as a devitrification product in high-calciumbottle glasses.
Properties. The physical properties of inter-est for industrial applications are given in Ta-ble 14.Wollastonite is usually white, translucentand sometimes pale greenish, bluish or brown-ish. It has a vitreous luster, a nacreous luster onfractured surfaces and coarsemasses, and a silkyluster in fibrous aggregates. Cleavage is perfecton (100) and (001) and well developed but vari-able on (101) and (201). The Mohs hardness is4 1/2 – 5 and the density 2.8 – 2.9 g/cm3.
Cyclowollastonite usually forms irregulargrains and is colorless, with cleavage on (001).The Mohs hardness is 5 and the density2.91 g/cm3.
The lower limit of transparency to UV lightis at λ= 290 nm. Wollastonite shows a yellow-ish thermoluminescence at moderate light in-tensities. The phosphorescence is yellowish-white and is intensified by doping with 0.5wt%Nd2O3. Cathodic luminescence is influenced byvacuum, temperature and additives.
The electrical conductivity at 1300 C is0.18Ω−1 · cm−1 and the dielectric constant at20 C is 6.17.
The ideal chemical composition forCaO ·SiO2 is 48.29wt%CaO, 51.71wt%SiO2.Aqueous mineral acids decompose wollastoniteand pseudowollastonite, producing gelatinousSiO2. Alkaline solutions do not attack it. Itis resistant to weathering and industrial atmo-spheres.
Production. Pseudowollastonite (cy-clowollastonite) is produced industrially as asintered phase by a diffusion reaction:
The reaction is accelerated by the addition ofmineralizers (e.g., 1wt% NaCl, Na2WO4 orCaF2). The solid-state reaction between CaOand SiO2 is also accelerated in the presence ofwater vapor.
Finely ground CaO and SiO2 are melted to-gether in the molar ratio 1 : 1, the melt is thenrapidly cooled to form a glass, and the vitreousproduct is then heated to 800 – 1000 C.
Deposits and Processing. Although wollas-tonite (-1T) and wollastonite (-2M), which issomewhat rarer, are widely distributed, in thepast only a few deposits were extracted eco-nomically up to the 1980s, when the demandforwollastonite and its production increased anddoubled in less than 10 years. The most im-portant deposits are in the United States (Wills-boro and Harrisville in the State of New York,Riverside in California, Tonopah in Nevada),China (Meiyaoshan and Tiengongshan in Jilinprovince, Fulapu and Shijianfang, near Faku inLiaoning province, Xinyu in Jiangxi province,Daye in Hubei province, Changxing in Zhejiangprovince, Qinghai province), India (Belka inthe Sirohi District, Kheratarala in Udaipur Dis-trict), Finland (Lappeenranta), along with Mex-ico (La Blanca and Panfillo Natera in Zacate-cas State), Chile, Namibia (Usakos), Kenya (Ka-jiado), Turkey, Pakistan, North Korea, Russia,Canada, Spain, Greece, Morocco, New Zealandand Cuba.
In the Willsboro mines, the rock contains ca.60% wollastonite (-1T) and (-2M), ca. 40% al-mandine, and some diopside. It is crushed inthree stages to< 16 mesh (1.2mm). The alman-dine is then removed by magnetic separators.The wollastonite concentrate is finally groundand classified by air elutriation.
The rock from Lappeenranta contains an av-erage of 20% wollastonite (-1T) and (-2M) andaccompanying minerals: calcite, dolomite, andquartz. Treatment consists of wet grinding (rodmills), classification (hydrocyclones), flotation(the wollastonite becomes concentrated at thebottomof the flotation cell),magnetic separationof the wollastonite concentrate, filtration anddrying. The finished product consists of 90%wollastonite, 4% quartz, 4% calcite and 2%other silicates.
Table 15 gives the chemical compositionof commercial wollastonite products. Table 14

Silicates 49
Table 14. Physical properties of wollastonite
Property Wollastonite∗ Pseudowollastonite Vitreous CaO · SiO2
Density, g/cm3 1.8 – 2.9 2.91 2.9Mohs hardness 4 1/2 – 5 5Temperature of phase change, C 1190Enthalpy of phase change, kJ/mol 7.42mp, C 1512Heat capacity at 1400 C, kJ kg−1 1434Melt viscosity, kgm−1 s−1
at 1550 C 2.73×10−3
at 1600 C 2.40×10−3
at 1650 C 2.38×10−3
Specific heat capacity, J g−1 K−1
at 1100 C 1.09 1.08at 0 – 700 C 0.99
∗ (-1T) and (-2M).
Table 15. Chemical composition of commercial grades of wollastonite (-1T) and (-2M)
Component Finland United States India Theoretical
SiO2 51.80 50.90 52.00 51.79CaO 44.50 46.90 47.00 48.29Al2O3 0.44 0.25 0.14Fe2O3 0.22 – 0.3 0.55 0.31MnO max. 0.01 0.10TiO2 max. 0.05 0.05MgO 0.56 0.10Na2O 0.10K2O 0.01Loss on ignition 2.20 0.90 0.55
gives the physical properties of Finnish wollas-tonite (-1T) and (-2M).
Uses. Wollastonite is widely used in ceram-ics, plastics, asbestos substitution, metallurgy,paints and glass manufacturing. Wollastonite isclassifed into high aspect ratio wollastonite andmilled or powdered wollastonite. High aspectratio wollastonite has an aspect ratio of 1:10 to1:20 and is well suited as a reinforcing func-tional filler in plastics, rubber, paints, and coat-ings and as an asbestos substitute. In these ap-plications, wollastonite is characterized by en-hanced hardness, flexural strength, and impactresistance, and also improves the electrical prop-erties and the thermal and dimensional stabilityof plastics. Milled or powdered wollastonite isused as a filler in ceramics and in metallurgy.The milled wollastonite grades are cheaper thanthe high aspect ratio grades.
The highest volume use of high-grade wol-lastonite, i.e., modifications (-1T) and (-2M), isas a raw material for ceramic products (see Ta-bles 15 and 16). Teh considerable increase in
use o wollastonite can be attributed to the spreadof fast-firing techniques, significantly reducingthe consumption of energy and improving pro-ductivity. Its important advantages include itslack of chemically bonded water (which couldbreak down the structure and even cause ex-plosions during firing), the ease of water re-moval on drying via the capillaries associatedwith its acicular (i.e., needle-like) crystal struc-ture, the low shrinkage on drying and firing, thevery low coefficient of expansion, the fairly lowmelting point, the mechanical strengthening ofthe “green” molded articles due to the acicularstructure present until firing and the high break-ing strength and thermal shock resistance of thefinished product. The high lightness value (seeTable 16), which is maintained after firing, en-ables wollastonite (-1T) and (-2M) to be used inthe production of neutral, white ceramic prod-ucts (e.g., white tableware). The raw materialmixture should contain 20 – 50% wollastonite.Other uses include wall and floor tile bodiesand glazes, sanitary ware, earthenware, frits, frit

50 Silicates
glazes, and porcelain enamels and specializedapplications.
Table 16. Physical properties of Finnish wollastonite (-1T) and(-2M)
Property FW 200 FW 325
Density, g/cm3 2.9Mohs hardness 4.5 – 5Refractive index 1.63mp, C 1394 – 1410Coefficient of thermal expansion,K−1
1.5×10−6
pH of 10% aqueous suspension 9.0Particle size (<74µm), % 96.5 98.9Specific surface area, cm2/g 2980 3040Oil number, g oil/100 g substance 27 35Brightness value FMY/C∗, % 86 86Solubility in water(5min, 100 C), mg/100mL 0.0071 0.0076
Particle shape Columnarto needle-shaped
∗Tristimulus value Y measured with standard illuminantaccording to DIN 5033, color temperature 2856K, ca. 546.1 nmusing ELREPHO equipment.
The addition of wollastonite to frits for ce-ramic glazes and enamels reduces the firing tem-perature and the formation of hairline cracksand helps stop cracking, crazing, breaking andglaze defects. It also improves surface hardnessand luster. Blister formation due to outgassingof volatile constituents is also reduced. Wollas-tonite added in amounts of 1 – 3% to flint and/orflux reinforces semivitreous products and de-creases shrinkage and moisture expansion. Insome other ceramic applications, feldspar andquartz are substituted by 2 – 5% wollastonite invitrified bodies to lower vitrification tempera-tures and to decrease shrinkage.
The (-1T) and (-2M) modifications are pre-ferred for use as fillers in industrial chemicalproducts and plastics. The chain structure of thelattice leads to the formation of acicular crystalsand short fibers, which impart structural viscos-ity both to molten thermoplastics and to coatingmaterials (paints, plastisols, etc.) and improvethe mechanical properties of the solid matrix. Inpolymer emulsion paints, texture paints, plastersand putties, addition of wollastonite improvesabrasion resistance and weathering resistanceand reduces shrinkage cracking.
The plastics industry is a major consumer ofwollastonite. In plastics most grades of wollas-tonite can be utilized, but most significant arethose with high aspect ratio, fine and surface-
coated grades. Wollastonite is used in both ther-moplastics (polyamides, polyesters, liquid crys-tal polymers and engineering resins) and ther-mosets (phenolic molding compounds, epoxies,polyurethanes, polyurea and unsaturated poly-esters). Typical loadings of wollastonite in plas-tics are: nylon 50%, low-density polyethylene40%, polypropylene 23 – 28% and polystyrene30%. A further important market for wollas-tonite is automotive plastics, in which it actschiefly as a reinforcement, e.g., in reinforced re-action injection molded components.
In paints and coatings high aspect ratiowollastonite is used as a reinforcing filler. Inpaint films, wollastonite improves the mechani-cal strength, durability and resistance to weath-ering, cracking and other ageing effects. Wol-lastonite has a pH of 9.9 in water and hencefunctions as an effective alkaline buffer in la-tex paints, preventing destabilization due to pHdrift, particularly when acidic pigments and ex-tenders such as kaolin and aluminum silicate areutilized.
Acicularwollastonite can bemixedwithmin-eral filler materials exhibiting platelet-shaped,lamellar particle shapes (kaolinite, mica, talc,and pyrophyllite) to give filler mixtures with de-sirable rheological working properties (e.g., ad-justable thixotropy).
Wollastonite is an important substitute forshort-fiber asbestos and is used widely in re-inforcement applications, in fire-resistant wallboard and cement products and friction products(brake pistons, brake linings, clutches etc.).
In metallurgical applications the low temper-ature fluxing of wollastonite imparts better sur-face finish in continuously cast steel and inhibitssparking in welding.
Due to its luminescent properties, wollas-tonite, especially the synthetic variety, is the basematerial of many phosphors, whose characteris-tics can be varied by doping with heavy metaland rare earth ions.
Synthetic wollastonite is used in syntheticbone implants, to replace bone loss and as a ver-tebral prosthesis inmedical cases requiring load-bearing capacity. Synthetic wollastonite quicklyforms strong, biologically safe bonds with os-seous tissue.
The favorable electrical properties of wollas-tonite (high volume resistance and surface resis-tance, low dielectric loss angle) lead to its use

Silicates 51
as an additive to electrical insulating paints andcasting materials.
Acicular wollastonite particles can be mixedwith filler materials with platelet-shaped parti-cles (kaolinite, mica, talc and pyrophyllite) togive filler mixtures with desirable rheologicalworking properties (e.g., thixotropy that can beadjusted).
The most important producers and suppli-ers of natural and synthetic wollastonite arethe NYCO Minerals Corporation (Willsboro,New York, United States) and the Partek In-dustrial Minerals Oy (Lappeenranta, Finland).World production of natural wollastonite is esti-mated at 365 000 t/a. Of the 120 000 t/a of wol-lastonite that is expected to be produced in theUnited States, some 90 000 t is consumed bythe domestic market and the surplus is exportedmainly to Europe. The production of naturalwollastonite by Mexican producers is estimatedto be 30 000 t/a, by Indian ca. 60 000 t/a, byChina 120 000 t/a, and by Finnish producers ca.30 000 t/a. The European consumption is esti-mated to be well over 70 000 t/a, of which ca.65% is used in the ceramics industry.
2.14. Toxicology
Most of the silicates described in Chapter 2 arenot known to be toxic or carcinogenic. However,the inhalation of fine particles (< 5µm, fibro-genic dusts) of silicates and hydrated silicateswhich have an acicular or fibrous morphologyand are insoluble in the environment inside thelungs (pH < 7) should be filtered out by meansof effective industrial dust-removal equipmentor respiratory protection masks.
The MAK values for inert granular andplatelet-shaped silicate dusts is set at 6.0mg/m3
in view of possible effects on the respiratory or-gans. This figure applies to exposure to the dustfor one year, or, if each individual exposure todust is known and documented, for a total ex-posure time of one year spread over a period offive years.
In the case of exposure to fine wollastonitedust (respirable dust < 5µm), respiratory pro-tection is recommended. It is not yet knownwhether there is a health hazard from the inhala-tion of cyclowollastonite dust. However, res-
pirable wollastonite (-1T) and (-2M) dusts aresuspected to be carcinogenic.
3. Alkali Silicates
3.1. Introduction
Historical Aspects. The solubility in waterof a fused mixture of flint pebbles with potashwas first described by the Brussels physicianJean Baptiste van Helmont (1577 – 1644) inhis posthumous work “Ortus medicinae”, Ams-terdam 1648.
Independently, Johann Rudolph Glauber(1604 – 1670) studied the properties of a “thick”solution that he obtained from a fused mixtureof sand, flint pebbles, or crystalline quartz withpotash, and named “Liquor Silicum”. He rec-ommended its use as a curative agent, for theproduction of liquid fluxes in metal smelting,and for glazing earthenware vessels (Furni NoviPhilosophici, Amsterdam 1648 – 1650).
In 1768, during the course of his alchemicalstudies,Goethe became interested in potassiumsilicate [116].
Although there were many later reports, thewater-soluble silicates were seldom put to prac-tical use until about 1825 when Johann Nepo-muk von Fuchs in Munich investigated the in-dustrial production of water-soluble potassiumand sodium silicates, which he named “water-glasses” (i.e., water-soluble glasses), and pro-posed that their solutions might be used as ad-hesives, for sealing porous stone, and as bindersfor fresco painting [117]. He exchanged ideaswith Justus von Liebig and C. F. Kuhlmann.The latter first produced waterglass in France in1841 [118,119]. Soon afterwards, similar workwas undertaken in England byW.Gossage, andin various parts of the United States [120].
Waterglass was more widely used during thecourse of theAmericanCivilWar (1861 – 1865),when the northern states utilized it in soapmanu-facture as a replacement for rosin formerly ob-tained from the southern states.
Definitions. Commercial alkali silicates aregenerally specified according to:
1) Source of alkali (soda, potash, lithia).

52 Silicates
2) Ratio of silica to alkali-metal oxide. Theusual ratio given is the weight (wt%) ratioor modulus, RW. The equivalent molar ratio,RM, can be more useful, particularly whencomparing ratios of potassium and sodiumsilicates. For sodium silicates, the conversionfactor isRM = 1.032RW, while for potassiumsilicates, RM = 1.568 RW.
3) Water content of the silicate. For solid, crys-talline silicates, this corresponds to the wa-ter of crystallization. For example,sodiummetasilicate, Na2O ·SiO2, can be preparedin anhydrous form, or as the pentahydrateor nonahydrate [121]. Amorphous alkali sil-icate glasses are essentially anhydrous. Thewater content of silicate solutions is custom-arily defined indirectly by giving the densityof the solution, which together with the silicato alkali-metal oxide ratio, defines a uniquecomposition for the silicate solution.
Solid alkali-metal silicates used in industry aredivided into two groups:
1) Water-soluble silicates solidified as a glassfrom a melt, normally containing ca. 1.5 –4mol SiO2/mol M2O (Section 3.3). Thesesilicates and their aqueous solutions are com-monly referred to as waterglass (solid glassor liquid glass). The most common commer-cial products are concentrated waterglass so-lutions, which are usually produced by disso-lution of silicate glasses in water or by directdissolution of sand in sodium hydroxide (seeSection 3.4).
2) Solid, crystalline sodium silicates, oftenwithwater of crystallization. Commercial prod-ucts of this type are known forNa2O · nSiO2,where n = 0.5 (orthosilicate), 1.0 (metasili-cate), and 2.0 (disilicate) (see Section 3.6).
3.2. Raw Materials
The dominant silica component used in industryis quartz sand (grain size 0.1 – 0.5mm) whichhas been washed to remove clays and other im-purities. The most problematic impurity in suchsands is iron, normally present to the extent ofca. 300 ppm. To obtain extremely pure alkali sil-icates, mainly for scientific use, very finely di-vided, very pure silica (e.g., fume silica such as
Aerosil) is recommended. The alkali-metal ox-ides are provided in the form of their carbonatesor hydroxides. The purity of the starting materi-als is dictated by the purity requirements for theend products.
An appropriate sulfate may also be used asa cheap source of alkali in silicate production,but requires the presence of a reducing agent(normally coal dust). Very little sulfate-derivedglass is produced because the melting processis longer and this, together with the presenceof sulfur oxide gases, increases corrosion of thefurnace walls. In addition, residual alkali sulfateand ash from the coal result in a relatively impureproduct.
3.3. Amorphous Anhydrous AlkaliSilicates (Solid or Lump Glasses)
Properties. Sodium and potassium silicateglasses are prepared commercially as glasslumps or powders. They are normally coloredowing to the presence of impurities, the mostusual colors being blue to green or yellow-browncaused by the presence of di- or trivalent iron,respectively.
Alkali silicate glasses with composi-tions close to those of stoichiometric com-pounds (e.g., Na2O · 2 SiO2, K2O · 2 SiO2,K2O · 4 SiO2) can be made to crystallize byholding them just below their melting pointsfor long periods. In general, however, commer-cial alkali silicates are not stoichiometric com-pounds. Instead, their molar ratio, SiO2/M2O,usually fluctuates around average values atwhich stable solutions can be obtained (Ta-ble 17). The vast majority of commerciallyproduced alkali silicate glass is sodium silicate.Furthermore, sodium silicate liquorswith awiderange of RW values may be produced by blend-ing two different liquors with each other or withalkali hydroxide. Virtually all commercial glassproduction is of sodium silicate of RW = 2.0 (al-kaline glass) or RW = 3.3 (neutral glass) [122].A smaller amount of potassium silicate glassis manufactured and is normally restricted tothe ratios RW = 2.5 and RW = 2.0 – 2.1. Lithiumsilicate glasses are not readily watersoluble andhence are not produced on a commercial scale[123].

Silicates 53
Table 17. Typical, commercially available alkali silicate glasses
Product name Na2O, wt% K2O, wt% SiO2, wt% Weight ratio Mol ratio
PyramidAL 33.2 66.3 2.00 2.06PyramidNL 22.9 76.7 3.35 3.46HK40 32 68 2.1 3.3HK28/30 28 72 2.5 3.9
Table 18. Physical properties of commercial silicate solutions
Commercial name Density at20C, kg/m3
SiO2,wt%
Li2O,wt%
Na2O,wt%
K2O,wt%
Totalsolids,wt%
Weightratio
Mol ratio Viscosity at20C, cP
pH at20C
Crystal L29 1150 17.50 1.55 19.0 11.3 5.6 < 10 10.7Crystal 52 1260 22.2 5.8 28.0 3.85 3.97 20 10.5Crystal 79 1400 29.2 8.9 38.1 3.30 3.41 250 – 500 10.8Crystal 100S 1500 31.2 12.5 43.6 2.50 2.58 400 11.2Crystal 100A 1500 28.1 14.0 42.1 2.00 2.06 200 11.4PyramidK53 1260 21.3 8.6 29.9 2.48 3.89 180 10.9PyramidK66 1330 23.3 11.4 34.8 2.05 3.21 30 11.8PyramidK120 1600 30.8 21.6 52.4 1.43 2.24 200 12.6
Figure 31. SiO2 –Na2O system

54 Silicates
The structure of silicate glasses is based ontetrahedral SiO4 groups that are associated aspolysilicate anions by sharing corners of thetetrahedra. The environment of a single Si atomis referred to as Qn, where n is the number ofshared corners for the tetrahedron [124]. Thestructures of the glasses are discontinuous, withSiO2-rich domains separated by clusters of M+
ions.
Figure 32. SiO2 –K2O system
The melting points of the solid silicates maybe obtained from the liquidus diagrams of thesystems SiO2 –Na2O and SiO2 –K2O (Figs. 31and 32).
Sodium silicates with RW ≥ 3 are onlyslightly attacked by water at room temperature.Sodium silicates with RW = 2 are hygroscopicand cake on storage in moist air. At equiva-lent molar ratios, potassium silicate glasses aremore hygroscopic than their sodium counter-parts – potassium silicate ofRW = 2.0 (RM = 3.1)also cakes in contact with moist air. Additionof a large excess of hot water to silicate glasscauses hydrolysis and leaches out some of thealkali [125]. This alkali then attacks the residual
silica, causing dissolution. Solid silicates can bedissolved at temperatures in the region of theboiling point in concentrated circulating solu-tion, but silicate solutions are almost invariablyproduced by operating at pressure.
Production. Mixtures of pure silica sand(glass sand) and alkali carbonate in the requiredratio are continuously fed by a cooled screwconveyor into furnaces that are regenerativelyheated by oil or gas. The rawmaterial ratio mustallow for losses of alkali carbonate due to vapor-ization and attack on the material of the furnace.The temperature of the gas space is ca. 1600 C,the finalmelt temperature being 1300 – 1500 C.The carbon dioxide is driven off by reaction ofthe metal carbonate with the silica:
M2CO3 + nSiO2 −→ M2O · nSiO2 +CO2
The furnaces are all of similar construction andsize (capacity 150 t) to those used in the glassindustry (→Glass, Chap. 5.2.). They must belined with high-quality refractory bricks (e.g.,sillimanite) since the alkaline melts are very ag-gressive. Themelt flows through the furnace andleaves continuously via an overflow into smallmolds which are moved along in an endlesschain. The cast pieces (diameter ca. 8 cm, thick-ness 1 – 2 cm) cool as they are transported andoften shatter.
Silicates are also produced in a batch processin small furnaces or continuously in rotary fur-naces.
The cast blocks of silicate glass are dissolvedin water while still warm or after intermediatestorage (see Section 3.4). For some applications,they are ground and sieved.
3.4. Silicate Solutions
Properties. Solutions of sodium and potas-sium silicates with SiO2/M2O≥ 1.5 are color-less, water white, and viscous. The viscosityincreases with concentration and, for a givenconstant silica content, with the SiO2/M2O ra-tio. When a critical concentration is reached, itrises very rapidlywith further concentration (seeFig. 33). For a given ratio there is a limiting con-centration above which the solution becomes ei-ther too viscous or toounstable for industrial use.

Silicates 55
Figure 33. Change of viscosity with density at constant SiO2 : Na2O ratios
The structures of the silicate solutions havebeen probed by awide variety of techniques suchas IR/Raman spectroscopy, light scattering, andchromatography [126–130]. The most valuabletechniques are, however, trimethylsililation and29Si-NMR [129,131–133]. Alkali silicate solu-tions contain, in addition to Me+, OH−, andorthosilicate (HxSiO(4−x)−
4 ) ions, a wide va-riety of linear, cyclic, and highly cross-linkedpolysilicate ions. The degree of polymeriza-tion of the silicate ions increases with increas-ing concentration and increasing ratio of thesolution. Relatively small polysilicates (≤ 8 Siatoms) can be identified by comparing theirsilylated derivatives with derivatives of knownstructures from silicate minerals [134]. In ad-dition, 29Si-NMRalso allows identification ofoligomeric silicate ions in dilute solutions [131–133]. In more concentrated solutions, however,and particularly at high ratios, NMR signals ofindividual species cannot be observed, peaksin the NMR spectrum then correspond to theQn species (see Section 3.3) [135]. This is il-lustrated in Figure 34, which shows 29Si-NMRspectra for sodium silicates of RW = 2.0 and 3.3at typical commercial concentrations. The spec-trum of the material with RW = 2.0 is domi-nated by silicons of the Q2 type, indicating apreponderance of small cyclic or linear silicateions; in the material with RW = 3.3, on the otherhand, more than 50% of the silicon atoms arepresent as Q3 or Q4 species, indicating a higher
level of three-dimensional, branched polysili-cates. These large polysilicate anions are mainlyresponsible for the high viscosity of silicate so-lutions. The silicate anions are in dynamic equi-librium and addition of alkali hydroxide (tanta-mount to reducing the ratio) causes hydrolysisof the polysilicates, thus reducing the viscos-ity, although even highly alkaline silicate solu-tions still contain polysilicates. Extensive dilu-tionwith CO2-freewater also depolymerizes thepolysilicate anions. Awide variety of silicate so-lutions are available commercially. Sodium sil-icates are marketed with RW ranging from 1.5to 3.85, while potassium silicates are availablewith RW from 1.43 to 2.48. Lithium silicatesare also available commercially. Table 18 givesphysical data for a few typical commercial sili-cate solutions, while Table 19 lists typical com-position ranges for a commercial sodium silicate(RW = 3.3).
All silicate solutions are highly alkaline. Di-lution reduces the pH, but less than might beanticipated owing to the buffering action ofthe silicate. For example, dilution of Crystal 79(RW = 3.3, 25wt% silica) to a 1wt% solutionreduces the pH from 11.6 to 10.8.
Addition of acids and acid salts (includingcarbon dioxide and bicarbonates) to silicates lib-erates silica. This is the basis of the industrialpreparation of silica sols, silica gels, and precip-itated silicas, the type of silica being formed de-pending on the concentration, temperature and

56 Silicates
Figure 34. 29Si-NMR spectra for sodium silicatesA) Crystal 100A; B) Crystal 79
salt content of the solution (→Silica) [122,136].Salts of polyvalent cations yield mixtures ofsilica gel with metal hydroxide [123]. Well-defined silicates are usually formed only afterprolonged heating of the solution, sometimesunder pressure. Under these conditions, indus-trially valuable crystalline silicates such as zeo-lites (→Zeolites) or clays (e.g., kaolin) may beformed [136].
The addition of saturated sodium chlorideor sodium nitrate solutions to silicate solu-tions precipitates alkali- containing silica gelswhich redissolve on dilution [121]. Saturatedsodium sulfate and sodium carbonate solutionscan bemixedwith silicate solutions at room tem-perature without causing precipitation, as canstrongly alkaline salts such as trisodium phos-phate and sodium metaborate.
Most organic compounds are incompatiblewith soluble silicate solutions. Some polyhy-dric alcohols (e.g., glycerol) are compatiblewithsodium silicates and yield stable single-phasesolutions. Other organics, such as polyol esters,produce a homogeneous systemwith silicate so-lutions but slowly react to give a solid mass[137]. Such systems have found use as settingsystems (e.g., in the foundry industry and soilconsolidation). Heating of sodium and potas-sium silicates in the absence of carbon diox-ide initially produces very viscous solutions; as
water loss continues, solid foams are formedthat still contain significant amounts of water.Lithium silicates are stable at and around roomtemperature, but precipitate on heating to > ca.60 C. The effect is reversible upon cooling[138].
Table 19. Composition range of typical commercial sodiumsilicate solution (RW = 3.3)
Assay Content∗SiO2 29.2wt%Na2O 8.9wt%Li 0.2 – 0.5K 20 – 50Mg 5 – 20Ca 1 – 80Sr 1 – 5Ba < 1 – 5Al 50 – 200P < 1 – 10S 10 – 30Ti 30 – 80V 0.1 – 0.8Cr < 1Mn < 0.5 – 1Fe 25 – 100Co < 1Ni < 0.5Cu < 0.1 – 0.2Zn < 0.2 – 1.0La 0.2 – 1.0Ce < 0.3 – 2.0Zr 5 – 20W < 1 – 25
∗ ppm unless otherwise stated.

Silicates 57
Production. Cold or warm lump glass (thelatter to improve heat conservation) is usuallydissolved at ≥ 5 bar and ca. 150 C. Stationarysystems may be used, but rotating vessels orcirculation of the liquor accelerate dissolutionrates. Direct steam heating is usually employedin large (≥ 15m3) vessels. High glass : waterratios are used to give a satisfactory dissolutionrate. Since the alkali silicates do not have a def-inite solubility in water, prolonged heating canlead to setting of the glass – liquid mix into anintractable elasticmass [122]. An excess of solidglass is heated with water until the required con-centration is reached. The silicate solution, con-taining solid impurities (silicates of, for exam-ple, iron, titanium, alkaline earths) is removedfrom the residual excess glass. The solutions arethen clarifiedby settling at elevated temperaturesor by filtration. Some commercial silicate solu-tions are too viscous to be handled in this way,and are produced in amore dilute form, followedby evaporation of water under reduced pressure.
A variety of molar ratios and concentrationscan be produced by appropriate mixing of asmall number of silicate solutions produced di-rectly from glass, either with each other or withthe appropriate alkali hydroxide.
Sodium silicate solutions ofmolar ratio≤ 2.5can be prepared by direct dissolution of quartzsand in sodium hydroxide solution under pres-sure at ≤ ca. 150 C. Even higher ratios canbe obtained by this method if a more solubleform of silica is substituted for the quartz sand.Sodium silicates with RW up to 3.3 may be ob-tained by this method using silica sources suchas amorphous silica, diatomite, or cristobalite[123,139].
Processes of this kind are likely to increasein importance because they require less energythan glass formation followed by dissolution.
As lithium silicate glasses are insoluble,lithium silicate solutions are prepared commer-cially by dissolution of amorphous silica inlithium hydroxide. Owing to the temperature in-stability of lithiumsilicate solutions, this processis carried out at or near room temperature.
3.5. Hydrated Water-Soluble Silicates
Hydrated water-soluble silicates are normallymanufactured in the sodium form and are avail-
able as fine powders or as granules. In generalthey are prepared by evaporating silicate solu-tions to the point where solids are formed whichare sufficiently stable to allow commercial han-dling but which retain adequate solubility. Typi-cally this occurs at water contents of ca. 20%. Inprinciple a wide range of products with differentratios can be made, although in practice the ra-tio tends to be restricted to the range RW = 2.0 –3.3 where the products are amorphous. Unlikefinely divided silicate glasses, these materialsdissolve in water under normal conditions togive silicate solutions; the rate of dissolutionis a function of ratio, water content, and par-ticle size. Thus at low ratios (RW = 2.0), ma-terials in the size range 100 – 200µm dissolvecompletely at room temperature in a few min-utes, whereas a similar product with a higherratio (RW = 3.3) requires a higher temperature(≥ 50 C) to achieve the same dissolution rate.These are, however, both considerably fasterthan the anhydrous glass analogues, for whichdissolution takes many hours and may not gofully to completion.
In dissolving hydratedwater soluble silicates,too large an excess of water should be avoidedsince rapid leaching of the alkali hydroxide cangive a solution pH that is too low to attack theresidual silicic acid, thus leaving it as an insolu-ble residue.
Examples of commercial materials are listedin Table 20. The manufacturing processes em-ployed include spray or drum drying of silicatesolutions followed by sieving to the requiredsize. Granular materials are formed by mechan-ical compaction of the spray dried powders togive products which aremore easily handled andare less dusty.
3.6. Crystalline Solids
Numerous crystalline alkali silicates have beenreported both in anhydrous and hydrated forms[121]. The ratios vary typically from RW = 0.5to RW = 2.0 and they are often referred to by themore traditional names:
Common name RW
orthosilicate (SiO2 · 2Na2O) 0.5sesquisilicate (2SiO2 · 3Na2O) 0.67metasilicate (SiO2 · Na2O) 1.0disilicate (2SiO2 · Na2O) 2.0

58 Silicates
Table 20. Hydrated, water soluble sodium silicates
Commercial name Na2O, wt% SiO2, wt% Weight ratio Molar ratio Water, wt% Bulk density, g/L
Pyramid P 40 26.7 53.3 2.00 2.06 20.0 400 – 500Pyramid P 60 18.5 61.5 3.30 3.41 20.0 550 – 650Pyramid P 10 28.0 56.0 2.00 2.06 16.0 80 – 120Pyramid G 80 26.0 52.0 2.00 2.06 22.0 750 – 850Pyramid G 90 26.0 52.0 2.00 2.06 22.0 800 – 950
In addition, several higher ratio silicates havebeen discovered and synthesized with ratiossuch as 4 : 1, 8 : 1, 9.4 : 1, 14 : 1, and 22 : 1 [140,141]. Many of the latter are complex layeredstructures, often occurring naturally and hav-ing some cation exchange properties. The struc-tured principles of all of these crystalline silicatespecies have been reviewed [142].
The only crystalline silicates which arepresently of any commercial significance arethe metasilicate family represented by the gen-eral formula Na2O ·SiO2 · nH2O. Products areavailable with n = 0, 5, 8 and 9, and withinthis range the anhydrous silicate (n = 0) andthe pentahydrate (n = 5) are the most important.Metasilicate and its hydrates differ in structure.Whereas the anhydrous material contains infi-nite chains of general formula Na2SiO3, the hy-drates, more properly represented by the for-mula Na2[SiO2(OH)2](n− 1)H2O, contain the[SiO2(OH)2]2− ion as a discrete entity.
The metasilicates crystallize well and arereadily soluble in water. Anhydrous sodiummetasilicate melts congruently above 1018 C,and above 70 C it is the solid phase in equi-librium with aqueous solutions. Below this tem-perature, the hydrolyzed ion [SiO2(OH)2]2− isformed and the various hydrates can be crystal-lized. Although potassium metasilicates behavesimilarly, they are not commercial products.
Production. Anhydrous metasilicate is pro-duced by two general methods:
1) It can be prepared in a furnace by direct fu-sion of sand and sodium carbonate at tem-peratures above the melting point of metasil-icate (1088 C), followed by solidification,grinding, and sieving. In some cases a solid-state reaction has been used above the melt-ing point of sodium carbonate (850 C) butbelow that of themetasilicate in rotary or tun-nel kilns. The corrosive nature ofmetasilicate
melts on furnace refractoriesmakes this a lessattractive route.
2) Alternatively, it can be obtained by dryingsolutions at the metasilicate composition ina fluidized bed [143] or a drum granulator[144]. In the latter case the hot liquor issprayed on to the powder bed whose parti-cles grow and dry uniformly in a current ofhot air. At the end of the drum the beadedproduct is screened and the fines recycled toprovide powder for further growth.
Metasilicate hydrates can be obtained bydirect hydration of anhydrous metasilicate orby crystallization of a solution of metasili-cate with the appropriate composition. In thecase of metasilicate pentahydrate for examplethe heated solution of composition Na2O ·SiO2· 5H2O is kept above 72.2 C (its melting point)and is crystallized by spraying on to a bed ofparticles cooled to below this temperature in arotary drum granulator using a similar proce-dure to that described above for the anhydrousmetasilicate.
Typical products and properties of metasili-cates are shown in Table 21.
Of particular interest lately has been thepreparation of synthetic layered silicate struc-ture [145]. These materials of general compo-sition Na2O · 2 SiO2 have ion-exchange proper-ties.
3.7. Uses and Applications
The uses of alkali metal silicates fall into twobroad categories:
1) As raw materials from which further im-portant industrial products can be derived.This application utilizes the silicate anionsas building blocks for the formation of arange of silica- containing materials and re-presents the single largest use of soluble sil-icates. These derived products include silica

Silicates 59
Table 21. Typical properties of metasilicate products
Commercial name Chemical formula Water content, wt% Bulk density, g/L Average particle size, mm
Metso A 100 Na2O · SiO2 1200 0.8Metso 510 Na2O · SiO2· 5H2O 43 1000 0.8Metso 520 Na2O · SiO2· 5H2O 43 1000 1.0Metso 950 Na2O · SiO2· 9H2O 57 800 0.6
sols, silica gels, precipitated silicas, zeolites,aluminosilicates, magnesium silicates, syn-thetic clays, ceramics, and catalysts.
2) As functional additives in a wide range of ap-plications exploiting the varied properties ofthe silicates.
These properties include: a source of bufferedalkalinity; an inorganic polymeric system withadhesive, binder, and film-forming capabilities;nontoxic, nonflammable and environmentallybenign; wide range of available compositions;capable of forming hybrid systems with organ-ics (e.g., polymers) and other inorganics (phos-phates, borates); complexation or precipitationof ions in solution; intumescent properties onheating; low cost.
Detergents represent the largest functionaluse of silicates. In broad terms use is made ofthe alkalinity and buffering capacity to aid insoil removal, assist in the suspension of soil par-ticles, enhance the effectiveness of surfactantsby sequestering calcium and magnesium ions,and inhibit corrosion of metal surfaces. In fabricwashing powders [146] liquid silicates have tra-ditionally been incorporated into the spray driedpowders at levels of 5 – 15%, whereby the sili-cate also provides crisp and easily handled de-tergent granules. The levels of incorporation ofliquid silicates have been decreasing with thechange to zeolite-based powders and more useis now being made of powdered soluble silicatesadded after spray drying. In industrial cleans-ing agents and dishwasher detergents the silicateproduct of choice has been metasilicate, eitheranhydrous or pentahydrate. There are now someconcerns about the high alkalinity of such mate-rials in domestic products and amove to granularsoluble powders with higher ratio (RW ≥ 2.0) islikely to occur.
Foundries. Large quantities of liquid silicate(typically with RW = 2) are used in the foundry
industry to bind together sand molds and coresprior to pouring the molten metal. The silicate isset by reaction with CO2 [147] blown throughthe silicate – sand mixture, or by incorporationof glycerol esters which hydrolyze and cause thesetting reaction by release of organic acids [148].
Adhesives. Silicate liquors are used widelyas adhesives. The largest consumer is the paperand board industry for such duties as spiral tubewinding, corrugated boxboard, and fiber drums.Silicates are also used to bind insulating materi-als such as vermiculite in building panels, coaldust briquettes, roofing tiles, bricks and ceram-ics, refractory cements, and in the manufactureofwelding rods. In the latter case, lithiumsilicatehas proven to be particularly effective since it notonly provides the necessary strength and degreeof water resistance, it also reduces significantlythe amount of CrVI evolved during the welding,thus providing safety benefits [149,150].
Surface Coatings. Silicate liquors are usedin a variety of surface coating applications. Ex-amples include the sealing of porous surfacessuch as concrete; as a vehicle for the preparationof paints for masonry and glass surfaces and forgeneral fire proofing duties; and as a componentin spray coating systems based on concrete andglass fiber. Silicates are also used to coat TiO2pigment particles to improve stability.
PaperDeinkingandBleaching. Sodiumsil-icate liquors are used in the paper industry to pro-mote deinking of recycled newsprint and also toact as a promoter to achieve enhanced bleachingwith hydrogen peroxide. Both of these are en-vironmentally driven applications, and demandis expected to grow as pressure increases to re-cycle more paper and to switch from chlorine toperoxide- containing bleaches.
WaterTreatment. Silicates are added towa-ter, including potable water, in concentrations

60 Silicates
of a few ppm to prevent discoloration due todissolved iron. The precise mechanism is notfully understood, but it appears that the sili-cate forms a “soluble” iron – hydroxide – silicatecomplex which prevents the formation of visi-ble ironflocs.An additional benefit of suchwatertreatmentwith silicate is the corrosionprotectionafforded to metal pipes, presumably by similarcomplex formation at the metal surface. Otherwater treatment uses have included a coagula-tion aid in alum flocculation processes and forboiler water treatment.
Civil Engineering. Silicates are used to sta-bilize soils in many civil engineering projectsinvolving drilling, tunneling, and mining. Sil-icate solutions mixed with appropriate settingagents are pumped into the ground where theyharden the soil [151]. An interesting applicationof this technology is the Sanipor process for re-pairing sewers without the need to dig up cityroads and streets [152–154]. Other similar usesinclude sealing around landfill sites, waste fixa-tion, and coastline stabilization.
Enhanced Oil Recovery. Use of silicateswith considerable growth potential is in en-hanced or tertiary oil recovery, particularly inoffshore locations. Flooding the oil field withsilicate solutions promotes the formation in situof soaps and surfactants which improve the oilflow from the porous rock [155,156].
3.8. Economic Aspects
Estimated 1990 world silicate consumption (ex-pressed as dry glass) is ca. 4×106 t (includingcaptive use). The approximate geographic splitisUnited States, Japan andWesternEurope 20%each with the remaining 40% for the rest ofthe world. The latter figure is speculative sinceit does not include Eastern Europe and China.The sales value of the noncaptive volume isca. $1.5×109. Consumption is estimated at 65 –75% of installed capacity.
The volume of silicate supplied to the mer-chant market, expressed as 100% solids, is esti-mated to be:
United States 470 000 tWestern Europe 500 000 tJapan 500 000 t
Table 22 shows the breakdown of the totalvolume by application for Western Europe andthe United States.
Table 22. Silicate usage by industry application (in percent)
Application Western Europe United States
Detergents 28.1 32.6Paper 7.3 7.5TiO2 4.6 4.6Water treatment 2.9 3.5Civil engineering 7.7Miscellaneous 6.4 5.8Total 57 54Derivatives (silica, zeolitesetc)
43 46
Total 100 100
3.9. Storage, Safety, Labelling andTransportation
The CAS und EINECS numbers are shown inTable 23 for various soluble silicates.
A comprehensive review of the health andsafety aspects of silicates has been published[157,158]. The primary hazard of all soluble sil-icates is their alkalinity. Contact effects rangefrom irritation to corrosion. Ingested or inhaledsilicates are rapidly eliminated in the urine.Quoted LD50 (oral, rat) values are 1280mg/kgfor sodiummetasilicate, 1300mg/kg for sodiumsilicates with RW = 2.0, and 1600mg/kg forRW = 3.0 according to the Registry of ToxicEffects of Chemical Substances 1981 – 1982.There are no exposure limits quoted for sodiumor potassium silicates by any regulatory body.A limit of 2mg/m3 is given for caustic soda andcaustic potash by both ACGIH and the HSE (un-der the EH40/92 exposure regulations) and ex-posure to silicate should therefore be limited, ac-cording to its composition, to remain in linewiththe caustic soda and potash figures. For lithiumsilicates a similar situation prevails, but the ex-posure limit for lithium hydroxide is 1mg/m3.
Alkali silicates are not flammable or explo-sive. Due to their alkalinity, solutions attackaluminum, tin, and zinc, with evolution of hy-drogen, at concentrations above ca. 10%. Pow-dered silicates tend to be hygroscopic and pack-aging must therefore provide adequate protec-tion against water vapor ingress to avoid cak-ing. Avoidance of prolonged contact with CO2

Silicates 61
Table 23. CAS and EINECS numbers for various soluble silicates
Product Formula CAS registry no. EINECS no.
Sodium silicate solutions, Na2O(SiO2)x [1344-09-8] 2 156 874glasses, or solids x> 1.6
Sodium metasilicate and hydrate Na2O · SiO2(H2O)y [6834-92-0] 2 299 129y = 0, 5, 9
Sodium orthosilicate 2Na2O · SiO2 [13472-30-5] 2 367 413Sodium sesquisilicate 3Na2O · 2SiO2 [15593-82-5] 2 396 711Potassium silicates K2O(SiO2)x [1312-76-1] 2 151 991
x> 1.6Lithium silicate solutions Li2O(SiO2)x [12627-14-4] 2 357 300
6≤ x≤ 12
is also necessary since both solids and solutionswill readily absorb this from the air to formalkalimetal carbonates.
Under the rules of the EEC Dangerous Sub-stances Directive 67/548/EEC as amended by92/32/EEC all silicates with RW ≥ 1.6 are clas-sified as irritant. However, the labeling classi-fication for sodium silicates is under review inEurope and a new set of safety phrases will beagreed taking into account bothRW and concen-tration.
In the environment, commercial silicatesrapidly depolymerize on dilution to give molec-ular species indistinguishable from natural dis-solved silica in ground waters. The pH of mostsilicate solutions is however above the accept-able limit for direct discharge to rivers and sew-ers, and dilution or neutralization must be car-ried out first. For disposal to landfill, the clas-sification of silicates varies according to com-position and also by countries. In general themore alkaline the products the more likely it isto be classified as hazardous or special waste al-though concentration is also important. Thus inthe United Kingdom metasilicates above 0.1%and all silicate solutions above 10% qualify asspecial waste [159].
3.10. Analysis
The alkali-metal content of soluble silicates isdetermined by titration in dilute solution withhydrochloric or sulfuric acid using methyl or-ange as indicator. The silica is determined gravi-metrically. Alkali metals are removed as thechlorides by fuming the diluted silicate solu-tion several times with hydrochloric acid in aflat porcelain dish. The insoluble silica is then
filtered, washed, calcined, and then weighed.If very great accuracy is required, the silica isvolatilized by fuming with hydrofluoric and sul-furic acids. The sesquioxides remaining afterthis are accounted for in the calcination.
Potassium silicate glasses are dissolved andfumed with hydrochloric acid to give silica-freefiltrates whose sodium content is determinedby flame photometry. The potassium contentof sodium silicate glasses is determined anal-ogously.
The silica content of aqueous silicate so-lutions can be determined more convenientlyby titration. The solution is neutralized usingmethyl red as indicator, an excess of pure sodiumfluoride is then added, and the alkali that is lib-erated according to the equation
SiO2 + 6 F− + 2 H2O −→ SiF2−6 + 4 OH−
is backtitrated with hydrochloric acid.Chloride is titrated by the argentometric
method in a dilute aqueous solution of the sil-icate. Sulfate is determined by precipitation asbarium sulfate from the filtrate from which thesilica has been removed.
Other possible impurities (CO2, Al2O3,Fe2O3, TiO2, CaO, MgO) are determined bythe methods used for analyzing ordinary glass.Spectrophotometry is recommended, particu-larly for iron and titanium.
Alkali silicates that are normally insoluble inwater are dissolved by pressure dissolution andinvestigated as for soluble silicates. Remaininginsolublematter isweighed andmaybe analyzedas for water-insoluble silicates.
Since 1972, the following ISO specifica-tions for alkali silicate analysis were published,1686: general, 1687: density of silicate solu-tions, 1688: density of the dry substance, 1689:

62 Silicates
determination of ratio, 1690: gravimetric deter-mination of SiO2 content, 1691: gas volumetricdetermination of CO2 content, 1692: titration ofalkalinity, 2122: preparation of solutions of solidalkali silicates and determination of insolublecomponents, 2123: determination of dynamicviscosity of silicate solutions, 2124: titrimetricdetermination of SiO2 content, 3200: gravimet-ric determination of SO− 2
4 content of alkali sil-icates, 3201: photometric determination of ironcontent of alkali silicates.
4. References
1. F. Liebau: Structural chemistry of silicates(structure, bonding, classification), SpringerVerlag, Berlin 1985.
2. F. Liebau: “Die Systematik der Silicate,”Naturwissenschaften 49 (1962) 481 – 491;“Silicon” in K.H. Wedepohl (ed.): Handbookof Geochemistry, vol. II/3, chap. 14-A,Springer Verlag, Berlin 1972, pp. 1 – 32;“Classification of Silicates,” in P. H. Ribbe(ed.): Orthosilicates, reviews in mineralogy,vol. 5, Min. Soc. Am., 1980, pp. 1 – 24.
3. F. Liebau: “Silicates with Branched Anions: aCrystallochemically Distinct Class,” Am.Mineral. 63 (1978) 918 – 92.
4. F. Liebau: “The Influence of Cation Propertieson the Shape of Silicate Anions,” Z.Kristallogr. 155 (1981) 139 – 153.
5. M. Fleischer: “Natrosilite,” Am. Mineral. 61(1976) 339 – 340.
6. M. Th. Le Bihan, A. Kalt, R. Wey: “Etudestructurale de KHSi2O5 et H2Si2O5,” Bull.Soc. Fr. Mineral. Cristallogr. 94 (1971)15 – 23.
7. F.-J. Dany et al.: “Kristallines Schichtsilikat –ein neuer Builder,” Seifen ole Fette Wachse116 (1990) 805 – 808.
8. Z. Johan, G. F. Maglione: “La kanemite,nouveau silicate de sodium hydrate deneoformation,” Bull. Soc. Fr. Mineral.Cristallogr. 95 (1972) 371 – 382.
9. R. A. Sheppard, A. J. Gude, R. L. Hay:“Makatite, a new Hydrous Silicate Mineralfrom Lake Magadi, Kenya,” Am. Mineral. 55(1970) 358 – 366.
10. H. P. Eugster: “Hydrous Sodium Silicates fromLake Magadi, Kenya: Precursors of BeddedChert,” Science (Washington D.C.) 157(1967) 1177 – 1180.
11. M. Fleischer: “Revdite,” Am. Mineral. 67(1982) 1076.
12. J. Puziewicz: “Grumantite”, Am. Mineral. 73(1988) 440.
13. G.Y. Chao, J. D. Grice, R. A. Gault: “Silinaite,a new Sodium Lithium Silicate HydrateMineral from Mont Saint-Hilaire, Quebec,”Can. Mineral. 29 (1991) 359 – 362; J. D.Grice: “The Crystal Structure of Silinaite,NaLiSi2O5 · 2H2O: a Monophyllosilicate,”Can. Mineral. 29 (1991) 363 – 367; K.Benneke, P. Thiessen, G. Lagaly: “Synthesisand properties of sodium lithium silicateslinaite,” Inorg. Chem. 34 (1995) 900 – 907.
14. G. Scholzen, K. Beneke, G. Lagaly: “Diversityof Magadiite,” Z. Anorg. Allg. Chem. 597(1991) 183 – 196.
15. L. S. Dent Glasser, P. B. Jamieson: “SodiumSilicate Hydrates. The Structure of Na2O SiO2
· 8H2O,” Acta Crystallogr. Sect. B32 (1976)705 – 710.
16. H. Annehed, L. Falth, F. J. Lincoln: “CrystalStructure of Synthetic MakatiteNa2Si4O8(OH)2 · 4H2O,” Z. Kristallogr. 159(1982) 203 – 210.
17. G. Bergely et al.: “Chemical Characterization,Structural Features, and Thermal Behavior ofSodium Hydrogen Octosilicate,” Clays ClayMiner. 39 (1991) 490 – 497.
18. K. Beneke, G. Lagaly:“Kanemite – Intracrystalline Reactivity andRelations to Other Sodium Silicates,” Am.Mineral. 62 (1977) 763 – 771;“Kenyaite – Synthesis and Properties,” Am.Mineral. 68 (1983) 818 – 826; “A HydratedPotassium Layer Silicate and its CrystallineSilicic Acid,” Am. Mineral. 74 (1989)224 –229.
19. W. Schwieger, W. Heyer, F. Wolf, K.-H.Bergk: “Zur Synthese von kristallinenMetallsilicathydraten mit Schichtstruktur,” Z.Anorg. Allg. Chem. 548 (1987) 204 – 216.
20. K.H. Bergk, P. Grabner, W. Schwieger: “Diehydrothermale Kristallisation des Magadiits,”Z. Anorg. Allg. Chem. 600 (1991) 139 – 144.
21. K.-H. Bergk, W. Schwieger, M. Porsch:“Aluminiumfreie Schichtsilicathydrate –Synthese undEigenschafts-Anwendungs-Beziehungen,”Chem. Tech. (Leipzig) 39 (1987) 459 – 466,508 – 514; K.-H. Bergk, D. Kaufmann, M.Porsch, W. Schwieger: “Herstellung undVerwendung von Magadiit alsPhosphatsubstitut in Waschmitteln,” Seifenole Fette Wachse 113 (1987) 555 – 561.

Silicates 63
22. R. Schwarz, E. Menner: “Zur Kenntnis derKieselsauren,” Ber. Dtsch. Chem. Ges. 57(1924) 1477 –1481.
23. F. Liebau: “uber Kristallstrukturen zweierPhyllokieselsauren, H2Si2O5,” Z. Kristallogr.120 (1964) 427 – 449.
24. J.-L. Guth et al.: “Un nouveau type de silicehydratee cristallisee,” C.R. Hebd. SeancesAcad. Sci. Ser. D 286 (1978) 5 – 8.
25. C. Frondel: “Crystalline Silica Hydrates fromLeached Silicates,” Am. Mineral. 64 (1979)799 – 804.
26. G. Lagaly, R. Matouschek: “Die kristallinenKieselsauren aus Apophyllit, Carletonit undGillespit,” Neues Jahrbuch Miner. Abh. 138(1980) 81 – 93.
27. H.-H. Kruse, K. Beneke, G. Lagaly: “GasAdsorption by a Crystalline Silicic Acid,”Colloid Polymer Sci. 267 (1989) 844 – 852.
28. J. Doring, K. Beneke, G. Lagaly: “AdsorptionProperties of Crystalline Silicas: II. Adsorptionof Anionic Surfactants and Delamination,”Colloid Polym. Sci. 270 (1992) 609 – 616.
29. G.W. Brindley, G. Brown: Crystal Structuresof Clay Minerals and their X-rayIdentification, Mineralogical Society, London1980.
30. Report of the Clay Minerals Soc.Nomencalture Commitee, Clays Clay Miner.39 (1991) 333 – 335; 45 (1997) 298 – 300.
31. G. Lagaly, K. Jasmund (eds.): Tonmineraleund Tone. Struktur, Eigenschaften,Anwendungen und Einsatz in Industrie undUmwelt, Steinkopff Verlag, Darmstadt 1993.
32. “Definition of Clay and Clay Minerals,” ClaysClay Miner. 43 (1995) 255 – 256.
33. A. Weiss, J. Russow: “Uber das Einrollen vonKaolinkristallen,” in I. Th. Rosenqvist, P.Graff-Petersen (eds.): Proc. Int. Clay Conf.,Stockholm 1963, v. 1, 69 – 74.
34. W.B. Jepson: “Kaolins: their Properties andUses,” Philos. Trans. R. Soc. London Ser. A311 (1984) 411 –432.
35. I. E. Odom: “Smectite Clay Minerals:Properties and Uses,” Philos. Trans. R. Soc.London Ser. A 311 (1984) 391 – 409.
36. K.-H. Bergk, D. Wolf, W. Schwieger, M.Porsch: “Hectorit – ein kristallinesMagnesiumsilicat mit Zukunft,” Chem. Tech.(Leipzig) 41 (1989) 245 – 251.
37. W. Vogel, W.Holand: “Zur Entwicklung vonBioglaskeramiken fur die Medizin,” Angew.Chem. 99 (1987) 541 – 558.
38. G. Lagaly, K. Beneke: “Intercalation andExchange Reactions of Clay Minerals and
Nonclay Layer Compounds,” Colloid Polym.Sci. 269 (1991) 1198 –1211.
39. A. Weiss: “Ein Geheimnis des chinesischenPorzellans,” Angew. Chem. 75 (1963)755 – 762.
40. M. Stockmeyer: “Adsorption organischerSubstanzen an organophilen Bentoniten,” Z.Dtsch. Geol. Ges. 141 (1990) 445 – 451.
41. M. Stockmeyer, K. Kruse: “Adsorption of Znand Ni Ions and Phenol and Diethylketone byBentonites of Different Organophilicities,”Clay Miner. 26 (1991) 431 – 434.
42. S. Komarnini, D.M. Roy: “Shale as aRadioactive Waste Repository: the Importanceof Vermiculite,” J. Inorg. Nucl. Chem. 41(1979) 1793 – 1796.
43. J. Bors: “Sorption of Radioiodine inOrgano- clays and -Soils,” Radiochim. Acta51 (1990) 139 – 143.
44. L. J. Michor, T. J. Pinnavaia: “Adsorption ofChlorinated Phenols from Aqueous Solutionby Surfactant-modified Pillared Clays,” ClaysClay Miner. 39 (1991) 634 – 641.
45. F. Liebau: “Zeolites and Clathrasils – twoDistinct Classes of Framework Silicates,”Zeolites 3 (1983) 191 – 193.
46. F. Liebau: “Synthesis of Porous Tectosilicates:Parameters Controlling the Pore Geometry” inE. R. Corey, J. Y. Corey, P. P. Gasper (eds.):Silicon chemistry, chap. 29, Ellis Horwood,Chichester 1988, pp. 307 – 323.
47. F. Liebau: “Structural Similarities andDissimilarities between SiO2 and H2O,” in:R. A. B. Devine (ed.): The Physics andTechnology of Amorphous SiO2, Plenum Press,New York 1988, pp. 15 – 35.
48. H. Gies, B. Marler: “The Structure ControllingRole of Organic Templates for the Synthesis ofPorosils in the System SiO2/Template/H2O,”Zeolites 12 (1992) 42 – 49; H. Gies:“Clathrasils and Zeosiles: InclusionCompounds with Silica Host Frameworks” inJ. L. Atwood, J. E. D. Davies, D. D. MacNicol(eds): Inclusion Compounds, vol. 5, Univ.Press, Oxford 1991, pp. 1 – 36.
49. R.M. Barrer: Hydrothermal Chemistry ofZeolites, Academic Press, London 1982.
50. O. Szostak: Molecular Sieves. Principles ofSynthesis and Identification, van NostrandReinhold, New York 1989.
General References51. H. U. Bambauer, F. Taborszky, H. D. Trochim
in W. E. Troger (ed.): “Bestimmungstabellen,”

64 Silicates
Optische Bestimmung der gesteinsbildendenMinerale, Part 1, 4th ed., E.Schweizerbart’sche Verlagsbuchhandlung,Stuttgart 1971.
52. H.-R. Bosse et al.: “Untersuchungen uberAngebot und Nachfrage mineralischerRohstoffe,” XIX Industrieminerale,Bundesanstalt fur Geowissenschaften undRohstoffe, Hannover; Prognos AGEuropaisches Zentrum fur AngewandteWirtschaftsforschung Basel 1986.
53. W. E. Troger, H. U. Bambauer, F. Taborszky,H. D. Trochim in O. Braitsch (ed.): OptischeBestimmung der gesteinsbildenden Minerale,Part 2, E. Schweizerbart’scheVerlagsbuchhandlung, Stuttgart 1967.
54. B. Coope et al.: Industr. Minerals Directory,Metal Bulletin Books Ltd., Surrey 1977,p. 601.
55. D’Ans-Lax, Springer-Verlag, Heidelberg1967, pp. 1 - 217, 1 - 662, 1-681, 1-757, 3-493.
56. D’Ans-Lax, Springer-Verlag, Heidelberg1970, pp. 1 - 216, 1 - 341, 1 - 420, 1 - 754,1 - 1289, 3 - 492.
57. W.A. Deer, R. A. Howie, J. Zussman: “Ortho-and Ring Silicates,” Rock-Forming Minerals,vol. 1, Longmans, Green and Co., London1962.
58. W.A. Deer, R. A. Howie, J. Zussman: “SheetSilicates,” Rock-Forming Minerals, vol. 3,Longmans, Green and Co., London 1962.
59. W.A. Deer, R. A. Howie, J. Zussman:“Framework Silicates,” Rock-FormingMinerals, vol. 4, Longmans, Green and Co.,London 1963.
60. W.A. Deer, R. A. Howie, J. Zussman:“Disilicates,” Rock-Forming Minerals,vol. 1A, Longman Scientific and Technical,London 1997.
61. W.A. Deer, R. A. Howie, J. Zussman:“Orthosilicates,” Rock-Forming Minerals,vol. 1A, Longman Scientific and Technical,London 1997.
62. W.A. Deer, R. A. Howie, J. Zussman:“Disilicates,” Rock-Forming Minerals,vol. 1B, Longman Scientific and Technical,London 1997.
63. W.A. Deer, R. A. Howie, J. Zussman:“Single-chain Silicates,” Rock-FormingMinerals, vol. 2A, Longman Scientific andTechnical, London 1997.
64. W.A. Deer, R. A. Howie, J. Zussman:“Double-chain Silicates,” Rock-FormingMinerals, vol. 2B, Longman Scientific andTechnical, London 1997.
65. W.A. Deer, R. A. Howie, J. Zussman: AnIntroduction to the Rock-Forming Minerals,2nd ed., Longman Scientific and TechnicalGroup, London 1992.
66. H. Freund: Handbuch der Mikroskopie in derTechnik vol. 4, Umschau Verlag,Frankfurt/Main 1974.
67. Gmelin, System-Nr. 35, Al, [A], 35, 41, 47,57, 69.
68. Gmelin, System-Nr. 35, Al, [B], 312, 390,492.
69. Gmelin, System-Nr. 27, Mg, [A], Lief. 1, 92.70. Gmelin, System-Nr. 35, Al, [A], 41.71. Gmelin, System-Nr. 35, Al, [B], 313.72. Gmelin, System-Nr. 27, Mg, [B], 351.73. Gmelin, System-Nr. 1 5, Si, [8], 482.74. Ind. Miner. (London) 1970, 2, 11.75. Ind. Miner. (London) 1975, 7, 15.76. Ind. Miner. (London) 1975, 11, 9.77. Ind. Miner. (London) 1975, 12, 47.78. Ind. Miner. (London) 1976, 1, 22.79. Ind. Miner. (London) 1976, 9, 71.80. Ind. Miner. (London) 1977, 4, 17.81. Ind. Miner. (London) 1977, 5, 39.82. Ind. Miner. (London) 1977, 6, 41.83. Industrial Minerals Directory, 1st ed., Metal
Bulletin Books, Surrey 1977, pp. 374, 565,591, 620.
84. T. Hahn, M. J. Buerger: “The DetailedStructure of Nepheline, KNa3Al4Si4O16,” Z.Kristallogr. Kristallgeom. Kristallphys.Kristallchem. 106 (1955) 308.
85. H. Kittel in W.A. Colomb (ed.): Lehrbuch derLacke und Beschichtungen, vol. 2,Heenemann, Berlin 1974, p. 379.
86. O. Luckert: Pigment +Fullstoff Tabellen, M.u. O. Luckert, Laatzen 1980.
87. Landolt-Bornstein, 5th ed., p. 1259.88. F. Machatschki: Spezielle Mineralogie auf
geochemischer Grundlage, Springer Verlag,Wien 1953.
89. St. v. Naray-Szabo: “Die Struktur des PollucitsCsAlSi2O6 · xH2O,” Z. Kristallogr.Kristallgeom. Kristallphys. Kristallchem. 99(1938) no. 4, 277.
90. W. P. Pearson: Structure Reports for 1969,vol. 34A, Oosthoek, Scheltma & Holkema,Utrecht 1975, p. 366.
91. P. Ramdohr, H. Strunz: Klockmann’s Lehrbuchder Mineralogie, 16th ed., Enke Verlag,Stuttgart 1978.
92. Rheinisch Westfalisches ElektrizitatswerkAktiengesellschaft (RWE): Bau-HandbuchTechnischer Ausbau, 10th ed., Essen 1990.

Silicates 65
93. H. J. Rosler: Lehrbuch der Mineralogie, 3rded., VEB Deutscher Verlag furGrundstoffindustrie, Leipzig 1984.
94. H. Strunz: Mineralogische Tabellen, 7th ed.,Akademische Verlagsgesellschaft Geest &Portig, Leipzig 1977.
95. F. Trojer: Die oxydischen Kristallphasen deranorganischen Industrieprodukte, E.Schweizerbart’sche Verlagsbuchhandlung,Stuttgart 1963.
96. F. Trojer, K.-H. Obst, M. Munchberg:Mineralogie basischer Feuerfest-Produkte,vol. 12 in V. D. Ferchette et al. (eds.): AppliedMineralogy/Technische Mineralogie, SpringerVerlag, Wien –New York 1981.
97. L. Weber, G. Zsak: Welt-BergbauDaten –World Mining Data, Series A, vol. 13,Bundesministerium fur wirtschaftlicheAngelegenheiten, Vienna 1998.
98. H.G. F. Winkler: Petrogenesis of MetamorphicRocks, 4th ed., Springer Verlag, New York1976.
99. Winnacker-Kuchler, 2nd ed. 2, 403.100. J. K. Winter, S. Ghose: American Mineralogist,
vol. 64, Richmond, Virginia, 1979, p. 573.
Specific References101. “Feldspar and Nepheline Syenite – Rivals in a
Flux,” Ind. Miner. 1976 Jan., 15 – 26.102. “Feldspar Fluxes – the Rivalry Reviewed,”
Ind. Miner. 1981 April, 21 – 45.103. W.A. Deer, R. A. Howie, J. Zussman: An
Introduction to the Rock-Forming Minerals,Longmans Group, London 1952, p. 361. T. G.Sahama, Am. J. Sci. (1952) 460.
104. Ind. Miner. 1977 June, 55.105. “Vermiculite: A Market in Transition,” Ind.
Miner. 1970 Nov., 11.106. “Vermiculite, Increased Capacity Encourages
Optimism – but Construction Still the Key toDemand,” Ind. Miner. 1977 April, 19 – 23.
107. Ind. Miner. 1982 no. 175, 117.108. “Feldspar and Nepheline Syenite - At the
Mercy of Glass Markets,” Ind. Miner. 1990Aug., 21 – 33.
109. “Olivine – Long Live the Evolution,” Ind.Miner. 1995 Feb., 23 – 31.
110. “Sillimanite Minerals,” Ind. Miner. 1996 Nov.,63 – 67.
111. “Synthetic Mullite – Refractories ShockAbsorber,” Ind. Miner. 1991 Feb., 39 – 45.
112. “Minerals in Lightweight Insulation – Fillingthe Market,” Ind. Miner. 1991 Oct., 21 – 35.
113. “Pumice Markets – Volcanic Rise ofStonewashing,” Ind. Miner. 1990 May, 22 – 37.
114. “Synthetic Mineral Fibres – From Rocks toRiches,” Ind. Miner. 1986 Sept., 20 – 43.
115. “European Industrial Minerals – World Classbut Under Threat,” Ind. Miner. 1997 Aug.,55 – 59.
116. W. Goethe:Dichtung und Wahrheit, 1768, p. 8.117. J. von Fuchs, Dinglers Polytech. J. 17 (1825)
465.118. C. F. Kuhlmann, Ann. Chim. 41 (1842) 220.119. C. F. Kuhlmann, Ann. Chim. 64 (1848) 289.120. UK762, 1854, W. Gossage.121. J. G. Vail: Soluble Silicates, Reinhold, New
York 1952.122. D. Barby, T. Griffiths, A. R. Jacques, D.
Pawson: The Modern Inorganic ChemicalsIndustry, Chemical Society, London 1977,pp. 320 – 352.
123. Kirk-Othmer, 20, 855 – 880.124. G. Englehardt et al., Z. Anorg. Allg. Chem.
418 (1975) 17.125. T.M. El-Shamy, J. Lewins, R.W. Douglas,
Glass Technol. 13 (1972) 81.126. A. Marinangelli, M.A. Morelli, R. Simoni, A.
Bertoluzza, Can. J. Spec. 23 (1978) 173.127. A. J. Walker, N. Whitehead, J. Appl. Chem. 16
(1966) 230.128. P. K. Dutta, D. C. Shieh, Appl. Spectrosc. 39
(1985) 343.129. A. P. Brady, A.G. Brown, H. Huffer, J. Colloid
Sci. 8 (1953) 252.130. J. J. Fitzgerald, J. A. Hoffman, C. E. Nebo, J.
Chrom. Sci. 27 (1989) 186.131. G. Englehardt, H. Janck, D. Hoebbel, W.
Wieker, Z. Chem. 14 (1974) 109.132. R. K. Harris, C. T. G. Knight, J. Chem. Soc.
(Faraday II) 79 (1983) 1525.133. C. T. G. Knight, J. Chem. Soc. (Dalton) 1988,
1457.134. C.W. Lentz, Inorg. Chem. 3 (1964) 574.135. I. L. Svensson, S. Sjoberg, L. O. Ohlmann, J.
Chem. Soc. (Faraday I) 82 (1986) 3635.136. R. K. Iler, The Chemistry of Silica, John
Wiley, New York 1979.137. M. Roberts, Foundry Trade J. 133 (1972)
no. 2925, 783.138. J.M. Ordway, Am. J. Sci. 24 (1907) (ser. 4),
473.139. R. Novotny, A.Hoff, J. Schurtz, DE 3 902 754,
1990.140. J. H. Wills, J. Am. Chem. Soc. 72 (1952) 5369.141. L. McCulloch, J. Am. Chem. Soc. 74 (1950)
2453.142. D. Barby et al.: The Modern Inorganic
Chemicals Industry, Royal Soc. Chemistry,London 1977, p. 334.

66 Silicates
143. S. L. Bean, A.W. Mouton, US 3 748 103.144. C. L. Baker, P.W. Holloway, US 3 208 822.145. H. P. Rieck, US 4 585 642.146. R. S. Schreiber, ACS Symp. Ser. 194 (1982)
271.147. K. E. L. Nicholas: The CO2 Silicate Process in
Foundries, BCIRA, 1972.148. M. Roberts, Foundry Trade Journal 133
(1972) 783, 2925.149. T. Griffith, Welding Review 1989 .150. T. Griffith, EP 342 036.151. R. H. Karol: Chemical Grouting, Marcel
Dekker, New York 1990.152. T. Szekely et al., US 4 988 238.153. I. Clarke, NoDig International 1993, Feb., 8.
154. D. Bradshaw, Financial Times, 1990, Oct. 12.,p. 35.
155. L. Holm, S. D. Robertson, J. Pet. Tech. 30(1978) no. 1, 161.
156. W. L. Schleyer, J. G. Blumberg, ACS Symp.Ser. 194 (1982) 187 – 227.
157. W. L. Schleyer, J. G. Blumberg: “Health andEnvironmental Aspects of Soluble Silicates,”ACS Symp. Ser. 194 (1982) 49 – 69.
158. W. L. Schleyer, J. G. Blumberg: “CurrentRegulatory States of Soluble Silicates,” Am.Chem. Soc. 1982, 31 – 47.
159. Waste Management Paper no. 23, Departmentof Environment, London 1981.
Related Documents