Cancer Therapy: Preclinical Tyrosine Phosphoproteomics Identifies Both Codrivers and Cotargeting Strategies for T790M-Related EGFR-TKI Resistance in Non–Small Cell Lung Cancer Takeshi Yoshida 1,5 , Guolin Zhang 1 , Matthew A. Smith 1 , Alex S. Lopez 2 , Yun Bai 1 , Jiannong Li 1 , Bin Fang 3 , John Koomen 3 , Bhupendra Rawal 4 , Kate J. Fisher 4 , Ann Y. Chen 4 , Michiko Kitano 5 , Yume Morita 5 , Haruka Yamaguchi 5 , Kiyoko Shibata 5 , Takafumi Okabe 5 , Isamu Okamoto 5 , Kazuhiko Nakagawa 5 , and Eric B. Haura 1 Abstract Purpose: Irreversible EGFR-tyrosine kinase inhibitors (TKI) are thought to be one strategy to overcome EGFR-TKI resistance induced by T790M gatekeeper mutations in non–small cell lung cancer (NSCLC), yet they display limited clinical efficacy. We hypothesized that additional resistance mechanisms that cooperate with T790M could be identified by profiling tyrosine phosphorylation in NSCLC cells with acquired resistance to reversible EGFR-TKI and harboring T790M. Experimental Design: We profiled PC9 cells with TKI-sensitive EGFR mutation and paired EGFR-TKI– resistant PC9GR (gefitinib-resistant) cells with T790M using immunoaffinity purification of tyrosine- phosphorylated peptides and mass spectrometry–based identification/quantification. Profiles of erlotinib perturbations were examined. Results: We observed a large fraction of the tyrosine phosphoproteome was more abundant in PC9- and PC9GR-erlotinib–treated cells, including phosphopeptides corresponding to MET, IGF, and AXL signaling. Activation of these receptor tyrosine kinases by growth factors could protect PC9GR cells against the irreversible EGFR-TKI afatinib. We identified a Src family kinase (SFK) network as EGFR-independent and confirmed that neither erlotinib nor afatinib affected Src phosphorylation at the activation site. The SFK inhibitor dasatinib plus afatinib abolished Src phosphorylation and completely suppressed downstream phosphorylated Akt and Erk. Dasatinib further enhanced antitumor activity of afatinib or T790M-selective EGFR-TKI (WZ4006) in proliferation and apoptosis assays in multiple NSCLC cell lines with T790M- mediated resistance. This translated into tumor regression in PC9GR xenograft studies with combined afatinib and dasatinib. Conclusions: Our results identified both codrivers of resistance along with T790M and support further studies of irreversible or T790M-selective EGFR inhibitors combined with dasatinib in patients with NSCLC with acquired T790M. Clin Cancer Res; 20(15); 4059–74. Ó2014 AACR. Introduction Despite the benefits shown with EGFR-tyrosine kinase inhibitor (EGFR-TKI) treatment in patients with non–small cell lung cancer (NSCLC) with TKI-sensitive EGFR muta- tions (1, 2), acquired resistance is a critical clinical problem. A secondary point mutation in exon 20 of EGFR that substitutes methionine for threonine at amino acid position 790 (T790M) was identified in patients with NSCLC who developed acquired resistance to gefitinib or erlotinib (3, 4). Nearly 50% of patients with NSCLC with acquired resis- tance to EGFR-TKIs have the T790M secondary mutation (5–7). Irreversible EGFR-TKIs, such as CL387,785 (8), PF00299804 (9), BIBW-2992 (afatinib; ref. 10), and HKI- 272 (11), are thought to be one strategy to overcome T790M-induced resistance. However, a number of studies have shown their limited activity in cells with T790M mutations given the increased affinity of ATP binding to T790M EGFR proteins or through mechanisms affecting other pathways such as MET activation (8, 9, 12–18). Clinical studies have also highlighted the limited efficacy of irreversible EGFR-TKIs. In the LUX-Lung 1 Trial, Authors' Affiliations: 1 Department of Thoracic Oncology, 2 Tissue Core, 3 Proteomics and Molecular Oncology Program, 4 Biostatistics Program, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida; and 5 Department of Medical Oncology, Kinki University Faculty of Medicine, Osaka-Sayama, Osaka, Japan Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). T. Yoshida and G. Zhang contributed equally to this article. Corresponding Author: Eric B. Haura, Department of Thoracic Oncology, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL 33612. Phone: 181-3745-6827; Fax: 181-3745-6817; E-mail: eric.haura@moffitt.org doi: 10.1158/1078-0432.CCR-13-1559 Ó2014 American Association for Cancer Research. Clinical Cancer Research www.aacrjournals.org 4059 on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559 on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559 on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript
-
Cancer Therapy: Preclinical
Tyrosine Phosphoproteomics Identifies Both Codrivers andCotargeting Strategies for T790M-Related EGFR-TKIResistance in Non–Small Cell Lung Cancer
Takeshi Yoshida1,5, Guolin Zhang1, Matthew A. Smith1, Alex S. Lopez2, Yun Bai1, Jiannong Li1, Bin Fang3,John Koomen3, Bhupendra Rawal4, Kate J. Fisher4, Ann Y. Chen4, Michiko Kitano5, Yume Morita5,Haruka Yamaguchi5, Kiyoko Shibata5, Takafumi Okabe5, Isamu Okamoto5, Kazuhiko Nakagawa5, andEric B. Haura1
AbstractPurpose: Irreversible EGFR-tyrosine kinase inhibitors (TKI) are thought to be one strategy to overcome
EGFR-TKI resistance induced by T790M gatekeeper mutations in non–small cell lung cancer (NSCLC), yet
they display limited clinical efficacy.Wehypothesized that additional resistancemechanisms that cooperate
with T790M could be identified by profiling tyrosine phosphorylation in NSCLC cells with acquired
resistance to reversible EGFR-TKI and harboring T790M.
Experimental Design: We profiled PC9 cells with TKI-sensitive EGFR mutation and paired EGFR-TKI–
resistant PC9GR (gefitinib-resistant) cells with T790M using immunoaffinity purification of tyrosine-
phosphorylated peptides and mass spectrometry–based identification/quantification. Profiles of erlotinib
perturbations were examined.
Results:We observed a large fraction of the tyrosine phosphoproteome was more abundant in PC9- and
PC9GR-erlotinib–treated cells, including phosphopeptides corresponding to MET, IGF, and AXL signaling.
Activation of these receptor tyrosine kinases by growth factors could protect PC9GR cells against the
irreversible EGFR-TKI afatinib. We identified a Src family kinase (SFK) network as EGFR-independent and
confirmed that neither erlotinib nor afatinib affected Src phosphorylation at the activation site. The SFK
inhibitor dasatinib plus afatinib abolished Src phosphorylation and completely suppressed downstream
phosphorylated Akt and Erk. Dasatinib further enhanced antitumor activity of afatinib or T790M-selective
EGFR-TKI (WZ4006) in proliferation and apoptosis assays in multiple NSCLC cell lines with T790M-
mediated resistance. This translated into tumor regression in PC9GR xenograft studies with combined
afatinib and dasatinib.
Conclusions: Our results identified both codrivers of resistance along with T790M and support further
studies of irreversible or T790M-selective EGFR inhibitors combinedwith dasatinib in patients withNSCLC
with acquired T790M. Clin Cancer Res; 20(15); 4059–74. �2014 AACR.
IntroductionDespite the benefits shown with EGFR-tyrosine kinase
inhibitor (EGFR-TKI) treatment in patients with non–small
cell lung cancer (NSCLC) with TKI-sensitive EGFR muta-tions (1, 2), acquired resistance is a critical clinical problem.A secondary point mutation in exon 20 of EGFR thatsubstitutesmethionine for threonine at aminoacid position790 (T790M) was identified in patients with NSCLC whodeveloped acquired resistance to gefitinib or erlotinib (3, 4).Nearly 50% of patients with NSCLC with acquired resis-tance to EGFR-TKIs have the T790M secondary mutation(5–7). Irreversible EGFR-TKIs, such as CL387,785 (8),PF00299804 (9), BIBW-2992 (afatinib; ref. 10), and HKI-272 (11), are thought to be one strategy to overcomeT790M-induced resistance. However, a number of studieshave shown their limited activity in cells with T790Mmutations given the increased affinity of ATP binding toT790M EGFR proteins or through mechanisms affectingother pathways such as MET activation (8, 9, 12–18).Clinical studies have also highlighted the limited efficacyof irreversible EGFR-TKIs. In the LUX-Lung 1 Trial,
Authors' Affiliations: 1Department of Thoracic Oncology, 2Tissue Core,3Proteomics and Molecular Oncology Program, 4Biostatistics Program, H.Lee Moffitt Cancer Center and Research Institute, Tampa, Florida; and5Department of Medical Oncology, Kinki University Faculty of Medicine,Osaka-Sayama, Osaka, Japan
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
T. Yoshida and G. Zhang contributed equally to this article.
Corresponding Author: Eric B. Haura, Department of Thoracic Oncology,H. LeeMoffitt Cancer Center andResearch Institute, 12902Magnolia Drive,Tampa, FL 33612. Phone: 181-3745-6827; Fax: 181-3745-6817; E-mail:[email protected]
doi: 10.1158/1078-0432.CCR-13-1559
�2014 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 4059
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/http://clincancerres.aacrjournals.org/http://clincancerres.aacrjournals.org/
-
conducted to compare afatinib treatment versus placebo inpatients with advanced NSCLC whose disease progressedafter receiving first-generation EGFR-TKIs (erlotinib, gefiti-nib), afatinib did not extend the primary endpoint ofoverall survival despite significant improvements in pro-gression-free survival (19). These preclinical and clinicalresults suggest that irreversible EGFR-TKIs as single agentsare insufficient to overcome resistance.
One strategy to improve on the limited efficacy of irre-versible EGFR-TKI is through combination with other path-way inhibitors. For example, studies that combined afatinibwith the anti-EGFR monoclonal antibody cetuximab (20)or the PI3K/mTOR inhibitor PI-103 (12) and HKI-272combined with mTOR inhibitor rapamycin (21) haveshown promise in overcoming T790M resistance. Anotherreason for the limited efficacy of agents targeting T790Mcould be mediated through other tyrosine kinases, such asreceptor tyrosine kinases (RTK), which provide additionalprotection against EGFR-TKIs (22). Recent studies haveshown that growth factor ligands can protect oncogene-addicted cells from molecularly targeted agents; thus,altered expression of these growth factor receptors couldfurther identify resistance pathways (23–25).
We explored the underlying ability of some growth factorligands to drive resistance to TKIs by examining the basaltyrosine phosphoproteome and the effects of EGFR-TKIs onother RTKs. In this study, we tested the hypothesis that aglobal evaluation of tyrosine phosphorylation (using massspectrometry) between the sensitive and resistant cells,along with EGFR perturbations, could identify additionalresistancemechanisms that could give insight into cotarget-ing strategies. Our results identified numerous coexpressedRTKs and non-RTKs that, under proper environmentalcircumstances, cooperate to drive resistance to EGFR-TKIs.We further showed that Src family kinase (SFK) signaling
was independent of EGFR signaling and that cotargetingSFKswith afatinib led to combined growth suppression in invitro and in vivo in cells with T790M. Globally, our resultssuggest that an unbiased mass spectrometry approach canidentify codrivers of resistance that can be cotargeted toenhance efficacy of targeted agents.
Materials and MethodsReagents
Gefitinib, erlotinib, afatinib, and WZ4002 were pur-chased from Chemie Tek (Indianapolis, IN). CL-387,785was purchased from AXXORA (San Diego, CA).
Cell cultureThe humanH1975, H460, A549, and H1299NSCLC cell
lines were obtained from American Type Culture Collec-tion. The human HCC4006 NSCLC cells were kindly pro-vided by Dr. Paul Bunn (University of Colorado, Aurora,CO). The human HCC827 NSCLC cells were provided byDr. Jon Kurie (MD Anderson Cancer Center, Houston, TX).The humanPC9NSCLC cell linewas kindly provided byDr.Hayata, Tokyo Medical University (Tokyo, Japan). PC9GRcells were generated by exposure of PC9 cells containing aTKI-sensitive EGFR mutation (exon 19; E746-A750) togradually increasing concentrations of gefitinib, beginningat 3 nM and up to 2 mM, for 3 months. HCC4006-T790Mand HCC827-T790M cells were generated as previouslydescribed (26). All cell lines have been maintained in acentral repository at Moffitt since 2008. All cell lines hadbeen authenticated by STR analysis (ACTG Inc, Wheeling,IL) as of September 2010, and all cells had been routinelytested and were negative for mycoplasma (PlasmoTest,InvivoGen, San Diego, CA). Cell viability was determined
using the CellTiter-Glo� Luminescent Cell Viability Assay(Promega, Madison, WI). Apoptosis assays were performedusing PE-conjugatedmonoclonal active caspase-3 antibodyapoptosis kit (BD Biosciences). Rescue experiments weredone as previously described (27).
GenotypingTotal genomic DNA from parental and resistant cells
was prepared using the DNeasy Blood & Tissue Kit (Qia-gen, Valencia, CA) in accordance with the product man-ual. Direct DNA sequencing was used to detect EGFRmutations as previously described (28). We also appliedthe PCR-invader assay to detect minor populations ofEGFR mutation, as previously described (29). MET genecopy number per cell was determined by fluorescence insitu hybridization with the use of the LSI D7S522 (7q31)Spectrum Orange and chromosome 7 centromere (CEP7)Spectrum Green probes (Vysis; Abbott), as previouslydescribed (30).
Tyrosine phosphoproteomicsTyrosine phosphopeptides were purified according to the
manufacturer’s recommendations for the Cell SignalingPhosphoScan kit (P-Tyr-100) (Cell Signaling Technology).Briefly, 2 � 108 cells were lysed in urea buffer; extracted
Translational RelevanceAcquired resistance to EGFR-tyrosine kinase inhibitor
(EGFR-TKI) is a critical clinical problem in patients withnon–small cell lung cancer (NSCLC) with TKI-sensitiveEGFR mutation. We applied mass spectrometry–basedtyrosine phosphoproteomics to paired TKI-sensitive andresistant cell lines to visualize molecular networks relat-ed to the acquired EGFR-TKI resistance. The resultssuggest that multiple receptors and signaling moleculessuch as MET, AXL, and IRS2 can collaborate to driveresistance to EGFR-TKI. We also identified Src familykinases (SFK) as a central signaling hub in TKI-resistantcells with T790M gatekeeper mutation. SFK phosphor-ylationwas also detected inhumanNSCLC sampleswithT790M. In vitro and in vivo experiments demonstratedthat irreversible EGFR-TKI (afatinib) or T790M-selectiveEGFR-TKI (WZ4006) combined with the SFK inhibitordasatinib overcameT790M-mediated resistance, therebynominating a new strategy for translation into the clinic.
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4060
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
proteins (40–80 mg) were then reduced by dithiothreitol,alkylated by iodoacetamide, and then digested by trypsin.Peptidemixture was isolated from lysate using Sep-Pak C18columns and then lyophilizated. Phosphorylated peptideswere immunoaffinity purified using phosphotyrosine anti-body after lyophilizated peptide mixture was dissolved.Volumesof phosphotyrosine peptideswere thendownsizedto 20 mL by vacuum drying for further experiments. Thefurther peptide mixture separation and phosphosite assign-ing have been previously described (31). To quantify eachtyrosine-contained peptide, we calculated peak area [alsocalled extraction ion chromatography (EIC)] using Label-free strategy and xCalibur as the tools. Identification andquantification of some obscure peptides were manuallyverified. After quantification, 774 phosphorylated tyrosinesites were identified. An in-house algorithmwas implemen-ted to identify unique phosphorylation tyrosine (pY) sites,remove redundant sites, and merge miss-cleaved peptidesby using protein ID, peptide sequence, and phosphoryla-tion start-site index,withquantificationof peak areas.Whenonly identifiable to the level of pairs of pYs (e.g., next to eachother or up to �11 amino acids apart), then the indepen-dent unit for analysis was the unique pY pair (instead ofsingle site). Mis-cleaved phosphopeptides or fragments ofthe samephosphopeptidesweremerged. Peptides sharedbymultiple proteins were annotated. Among which, two pairsof sequences were potential results of co-elution and there-fore not included in further analyses. A total of 524 uniquephosphotyrosine units (pYs) or pY pairs were identified.Quantification and stability of 5 MYG peptides acrosssamples were examined, with the average of 3 of them usedfor normalizing the peak ratio areas across 16 samples (8biological samples with technical duplicates) so that thenormalized quantities across samples were comparable.Reproducibility between technical replicates for each pYwas estimated using Pearson correlation. The correspond-ing P values were used to estimate false discovery rate. Hightechnical reproducibility of FDR �1% was used in ourstudy. In addition, if the pY was detected in at least halfof the samples in this study, i.e., at least 8 of 16 samples, itpassed theQC criteria. Among the 524 unique pY units, 403of them passed the QC criteria and were included in theanalyses. 377 of themwere unique pY sites while 26 of themwereuniquepYpairs. Averages of technical replicates from8biological samples were used in the analyses. We used asimple imputation (i.e., when one of technical replicates ismissing, the detected value from the other remaining tech-nical replicate was used). Data were analyzed in log2 scaleprior to parametric analyses and also for ease of interpre-tation. For example, the difference of 1 in log2 scale is a 2-fold change between two conditions. Two-way ANOVAwith the interaction term was performed to answer thefollowing three research questions: 1) Which tyrosine sitesare differentially phosphorylated between the cell lineswithand without drug resistance? 2) Which tyrosine sites aredifferentially phosphorylated between the control and erlo-tinib-treated groups? 3) Which pYs phosphorylationresponse to treatment is different between the resistant and
non-resistant cell line? To adjust for multiple hypothesistesting, the resulting P values for the main effects of cell lineand treatment as well as the cell line-by-treatment interac-tion termwere used to estimate false discovery rate.We usedFDR �20% to declare statistical significance. We furtherperformed network analysis based on these potential can-didates. Interactions among all identified tyrosine phos-phorylated proteins were retrieved from the MolecularInteraction database (MINT) (32); the IntAct database(33); the Database of Interacting Proteins (DIP) (34); theGeneral Repository for InteractionDatasets (BioGRID) (35)and the Biomolecular Interaction Network Database(BIND) (36) using InnateDB (37) and visualized in Cytos-cape 2.8.3 (38).
Protein expression analysisWestern blot analysis of whole cell lysates was performed
as described previously (27). Primary antibodies to EGFR,MET, pTyr 1234/1235 MET, IRS2, pTyr 1131 IGF1R, AXL,pTyr 702 AXL, Src, pTyr 416 Src, Akt, pSer 473 Akt, Erk,pThr202/Tyr204 Erk, and PARP were obtained from CellSignaling Technology. Primary antibodies to pY1068-EGFRwere obtained from Invitrogen (Carlsbad, CA). Primaryantibodies to b-actin were purchased from Sigma-Aldrich(St. Louis, MO).
Assessment of tumor growth inhibition in vivoAll animal procedures were approved by our Institutional
Animal Care and Use Committee. PC9GR cells (2�106)were injected subcutaneously into the flank of 7-week-oldfemale athymic nude mice. The mice were divided into 4treatment groups of 7 animals: those treated over 3weeks bydaily oral gavage of vehicle, afatinib (10 mg/kg), dasatinib(15 mg/kg), or both afatinib and dasatinib; 0.5% (wt/vol)aqueous solution of hydroxypropylmethylcellulose wasused as vehicle for afatinib, and 50% propylene glycol wasused as vehicle for dasatinib. Treatment was initiated whentumors in each group achieved an average volume of 100mm3, with tumor volume being determined twice weeklyfor 21 days after the onset of treatment from caliper mea-surement of tumor length (L) and width (W) according tothe formula LW2/2.
Src-Tyr416 immunohistochemistry stainingImmunohistochemistry staining was performed to mea-
sure the expression of phosphor-Src (Tyr416) in paraffintissues from 10 lung cancer patients with mutant-positiveEGFR T790M.
Slides were stained for phosphor-Src (Tyr416) (mousemonoclonal antibody; Millipore) using a Ventana DiscoveryXT automated system (Ventana Medical Systems, Tucson,AZ) following the manufacturer’s protocol with proprietaryreagents. Briefly, slidesweredeparaffinizedon the automatedsystem with EZ Prep solution (Ventana). Enzymatic retrievalmethod was used in protease 1 at 4 minute (Ventana), CC1Standard and CC2 standard conditions. The primary mono-clonal antibody (Millipore) reacts to secondary antibody atdifferent dilution-titrations. Both primary and secondary
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4061
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
antibodies were incubated following Ventana’s instructionand the antibody product recommendation. The intensity ofphosphor-Src expression was scored from 0–3 (0 ¼ noexpression, 1 ¼ weak expression, 2 ¼ moderate expression,and 3 ¼ high expression), while the cellularity was scoredfrom0–3 (0¼ 0%, 1¼ 1-33%, 2¼ 34-66%, and 3¼ >66%).The H scores formed by intensity of immunoactivity timingcellularity were stratified as low (0–2), intermediate (3–4),and high (6–9).
MET-pY100 proximity ligation and total METimmunofluorescence
Slides containing 5-mmsections were rehydrated throughxylene and graded alcohols. Heat-induced epitope retrievalwas carried out in Tris-EDTA (pH 9) in a pressure cooker for20 minutes and then allowed to cool for 20 minutes.Nonspecific binding was blocked by incubation with1.5% BSA, and primary antibodies were incubated over-night in 1.5% BSA-PBST.
For proximity ligation, antibodies were rabbit anti-MET(clone D1C2, Cell Signaling Technology) and mouse ant-pY100 (Cell Signaling Technology). PLA probes were anti-rabbit (-) andanti-mouse (þ) andwere incubated for 1hourin 0.15% BSA/PBST. Detection was carried out using theDuoLink in situ PLA Far Red kit (O-Link Biosciences,Uppsala, Sweden). AlexaFluor 488-conjugated anti-cyto-keratin was used to demarcate epithelial regions (cloneAE1/AE3, eBiosciences).
For immunofluorescence, antibodies were rabbit anti-MET (cloneD1C2, Cell Signaling Technology) and detectedviaAlexaFluor 647-labeled anti-rabbit secondary antibodies(Invitrogen). Murine pan-cytokeratin (clone AE1/AE3,Dako) was used to demarcate epithelial regions (tumormask) and detected via AlexaFluor 555-labeled anti-mousesecondary antibodies (Invitrogen). Images were acquiredon a PM2000.
Statistical methodsAnderson-Darling statistics and normal curves were
examined to assess whether tumor measurements werenormally distributed. A square-root transformation wasperformed on the tumor measurements to make themapproximately normal. ANOVA test was used to assesswhether there was a statistically significant difference ontumor sizes measured across treatment groups at each timepoint. Tukey-Kramer method was used to perform all pair-wise group comparisons. All statistical analyses were per-formed using SAS (version 9.2; SAS Institute; Cary, NC).
ResultsChronic gefitinib exposure of PC9 cells generatesstable cell-autonomous resistance to EGFR-TKIs withT790M
After generation of PC9GR cells, we identified single-cellclones of PC9GR cells that were highly resistant to erlotinib(Supplementary Fig. S1A). Although PC9GR cells are par-tially sensitive to the irreversible EGFR-TKI CL387,785 asexpected fromaprevious report (8), IC50 forCL387,785was
100-fold increased compared with parent PC9 cells (Sup-plementary Fig. S1A). This resistancewas stable as it was notreversed by culturing PC9GR cells for up to 6 months ingefitinib-free medium (data not shown). PC9GR cellsacquired T790M while retaining exon 19 E746-A750, asdeterminedbybothdirectDNAsequencing andPCR-invad-er assay (Supplementary Fig. S2A). In addition, we did notfind MET amplification by FISH analysis in PC9GR cells(Supplementary Fig. S2B), which is another mechanism ofacquired EGFR-TKI resistance in NSCLC (28). Erlotinib stillhas partial inhibitory effects on EGFR phosphorylation inPC9GR cells (Supplementary Fig. S1B), consistent withprior studies that T790M typically emerges as a minorpopulation and resistant cells retain drug-sensitive alleles(8, 33). However, erlotinib could not completely inhibitdownstream pAkt and pErk in PC9GR cells, consistent withresistance to EGFR-TKIs in the presence of T790M (Supple-mentary Fig. S1B).
System-level comparison of tyrosine phosphorylationidentifies common RTK pathways associated witherlotinib resistance
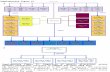
We hypothesized that erlotinib-resistant PC9GR cellscould collect additional mechanisms of resistance throughacquired alterations in tyrosine kinase signaling that couldcollaborate with T790M to codrive resistance to EGFR-TKI.We therefore profiled tyrosine kinase signaling by chartingtyrosine phosphorylated peptides in PC9 and PC9GR cells.As shown in our schema (Fig. 1), tryptic peptides werederived from cellular protein lysates and enriched withanti-phosphotyrosine (pTyr) antibodies followed by iden-tification and quantification using liquid chromatographycoupled with tandem mass spectrometry (LC/MS-MS;refs. 32, 34). Changes in peptides in PC9GR cells wereidentified and compared with PC9 cells, thus allowing usto determine additional changes beyond T790M that couldbe codrivers of TKI resistance. We perturbed EGFR-drivensignaling in erlotinib-sensitive PC9 and erlotinib-resistantPC9GR cells to identify EGFR-dependent pathways/net-works and potential pathways/networks independent ofEGFR signaling that could play a role in EGFR-TKI resis-tance. After 1-hour erlotinib treatment, cell pellets werecollected and pTyr peptides were identified in untreatedand treated PC9 and PC9GR cells. Changes in peptides wereidentified compared with control vehicle-treated cells ineach of the two cell lines. We hypothesized that thisapproachwould identify downstream signaling events driv-en by mutated EGFR but could also potentially identifyproteins or pathways activated by TKI or unaltered by TKIthat could, under the correct circumstances, potentiate drugresistance. In total, between the two cell lines, we identified403 pTyr peptides corresponding to 265 unique phospho-proteins. Examples of extracted ion chromatograms for pTyrpeptides corresponding to EGFR and MET are shown inSupplementary Figs. S3 and S4.
We next compared changes in pTyr abundance betweenPC9 and PC9GR cells (Fig. 2A; Supplementary Table S1).We found 110 unique pTyr peptides (76 proteins) that were
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4062
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
more abundant in the PC9GR cells, whereas 77 unique pTyrpeptides (55 proteins) were less abundant in PC9GR cellsthan in PC9 cells. Compared with PC9 cells, PC9GR cellsdemonstrated increased amounts of pTyr peptides corre-sponding to numerous RTKs, some of which could bepotential codrivers of resistance in PC9GR cells under thecorrect environmental circumstances. We observed a clearsubnetwork characterized by hyperactive MET signaling(Fig. 2A, right) despite the lack of MET gene amplification.We observed nearly 11-fold more MET pTyr peptidesin PC9GR than in PC9 cells. Similarly, we observed nearly>10-fold more pTyr peptides corresponding to ROR1 orneurotrophic tyrosine kinase receptor related-1 (pTyr-789,�13-fold; pTyr-828, �34-fold). ROR1 is a pseudokinasethat cooperates with MET to promote tumorigenesis (35).Tyrosine phosphorylation of the MET adaptor proteinsGab1 and Gab2 were also more abundant in PC9GR cells.In addition, pTyr peptides corresponding to the AXL
RTK were increased approximately 8-fold in PC9GR cells.AXL upregulation has recently been shown to be a mech-anism of acquired resistance of lung cancer cells to EGFR-TKI (36). Finally, increased abundance of multiple pep-tides corresponding to IRS2 (pTyr-675, 4.97-fold; pTyr-598, 5.47-fold; pTyr-823, 9.55-fold; pTyr-653, 19.93-fold;and pTyr-742, 21.29-fold), an adaptor protein linkinginsulin and insulin-like growth factor (IGF) signaling toPI3K signaling, was observed in PC9GR cells comparedwith parent PC9 cells. This suggested that either moreinsulin or IGF signaling exists in these cells or more IRS2protein is expressed. We confirmed higher levels of tyro-sine-phosphorylated MET and AXL in PC9GR than in PC9cells and also found more total IRS2 protein in PC9GRthan in PC9 cells (Fig. 2B). Despite the increased levels ofMET signaling, we found minimal effects of combinedMET-TKI (PHA665752) and EGFR-TKI (erlotinib or irre-versible CL387,785) in PC9GR cells (Fig. 2C). While MET
signaling is hyperactivated, in this context, it is notresponsible for affecting cell survival.
To examine whether changes of MET, IRS2, or AXL aredriven specifically by T790M, we examined phosphoryla-tion of these molecules in lung cancer cell lines (HCC4006and HCC827) engineered to express an exon 19 E746-A750þ T790M allele. (Fig. 2B). We observed less pMET, less totalMET, slightlymore abundant pAXL, and similar total AXL inHCC4006-T790M cells compared with parent HCC4006cells. We found equivalent pMET and total MET, less pAXLand equivalent total AXL inHCC827-T790Mcells comparedwith parent HCC827 cells. The levels of IRS2 protein wereunchanged across these HCC4006 and HCC827 cell linesunlike in PC9 and PC9GR cells. These results suggest thatchanges of MET, IRS2, or AXL are not dependent on EGFR-T790M but rather are likely to occur on a cell by cell basis.
Perturbations by EGFR-TKI identify downstreamproteins and proteins involved in adaptive andmicroenvironment-derived responses
Wenext compared alterations in pTyr peptide abundancein both cell lines following erlotinib exposure (Supplemen-tary Table S1). We identified pTyr peptides with >1.5-foldchange differences from control (P < 0.05). In PC9 cells, weobserved 31 less abundant and 45 more abundant uniquepTyr peptides following 1 hour of erlotinib treatment (Fig.3A). As expected, PC9GR cells displayed a more bluntedresponse to erlotinib than PC9 cells; nonetheless, we didobserve congruent changes in most pTyr peptides, thusincreasing our confidence that these pTyr peptides andpathways are downstream of mutant EGFR given theexpected biologic responses with cells harboring T790Mmutations. Among the reduced pTyr peptides, we observedMK01, SHC1, GAB1, EGFR, and ERBB3 consistent withtheir known roles in ERBB signaling. Interestingly, we alsoobserved reductions in peptides corresponding to Ras
Quantification (EIC)
Drug treatment Lysate Digestion
Tyrosine proteome
Anti-pTyr100 Ab
LC/MS-MS
PC9GRPC9 parental
DMSO DMSO DrugDrug
Drug
DMSO
–2.5
–2.0
–1.5
–1.0
–0.5
0.0
0.5
1.0
1.5
2.0
2.5
Drug response
Figure 1. Phosphoprotein networkassociated with mutant EGFR andT790M tyrosine kinase signaling.Workflow of quantitativephosphoproteomics analysis. EIC,extracted ion chromatography(used to quantify peptideabundance for each of theidentified tyrosine-containingpeptides).
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4063
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4064
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
signaling, including SYGP1 or SynGAP, which can affectERK and p38 MAPK functions in neurons (37, 38). Tyro-sine-phosphorylated Rab7A (also known as Ras-relatedprotein Rab-7a) was likewise reduced by erlotinib and hasbeen linked to EGFR trafficking in endosomes (39).Interestingly, we observed that nearly equal amounts of
pTyr peptides were increased by erlotinib compared withpeptides reduced by erlotinib (45 up and 31down in PC9; 26up and 30 down in PC9GR). We found more abundantpTyr peptides for IRS2,MET, YES, AXL, FAK, ERBB2, andBRK(PTK6) following erlotinib treatment, and this pattern wasconsistent across both PC9 and PC9GR cells (Fig. 3B). Wehypothesized that the increased levels of RTK identifiedthrough our approach could cooperate with exogenousligands and promote EGFR-TKI resistance. Recent studieshave highlighted the ability of growth factor ligands to pro-mote resistance to targeted agents (23–25). We thereforetested if the increased pTyr in key RTK could cooperate withgrowth factor ligands to drive resistance to EGFR-TKI. Weincubated PC9 and PC9GR cells with cognate ligands corre-sponding to the upregulated RTK in PC9GR, including IGF1,hepatocyte growth factor (HGF), and GAS6 (the ligand forAXL RTK), and determined the effects on erlotinib sensitivityin PC9 cells and afatinib sensitivity in PC9GR cells (Fig. 3C).Interestingly, HGF and IGF but not GAS6 had protectiveeffects on both PC9 cells exposed to erlotinib and PC9GRcells exposed to afatinib. In PC9 cells, the shift in IC50 wasrather modest; however, in PC9GR cells, the effect was moredramatic. This shift pattern was consistent between both celllines, with HGF having more of an effect than IGF1, whereasno effect was seen with activation of AXL by GAS6 in thesecells. UsingWestern blotting, we examined the effects of theseligands on EGFR signaling with or without EGFR-TKI in bothPC9 and PC9GR cells. HGF activated pMET in both PC9 andPC9GR cells (Supplementary Fig. S5A and S5B). This HGF-induced activation of pMET and downstream pAkt and pErkwere not inhibited by erlotinib in PC9 or by afatinib inPC9GR cells (Supplementary Fig. S5A and S5B). These resultsalso suggest thatMETactivation inPC9andPC9GRcells is notdependent on EGFR signaling.On the other hand, we did notobserve clear ligand-dependent activation of correspondingRTKs or sustained activation of pAkt and pErk in the presenceof EGFR-TKIs in IGF1-induced or Gas6-induced PC9 andPC9GR cells (Supplementary Fig. S5C–S5F). These results areconsistent with our data showing that IGF1 andGas6 had less
rescue effects compared with HGF in these cells (see Fig. 3C).These results suggest that altered RTK identified by phospho-proteomics can be codrivers of resistance under specific en-vironmental circumstances. Furthermore, the increased levelsof multiple RTKs in response to erlotinib suggest innatepriming of RTK, where RTKs are primed to cooperate withgrowth factor ligands through intracellular mechanisms.
Afatinib combined with dasatinib inhibits EGFRsignaling more efficiently than either agent alone inTKI-resistant NSCLC cells with T790M
We reexamined our data for pTyr peptides that were notperturbed by EGFR-TKI and were not different betweenthe PC9 and PC9GR cell lines. We hypothesized that thisanalysis may uncover parallel signaling pathways thatcooperate with EGFR to maintain cellular growth and/orsurvival. We identified 31 proteins that fulfilled thiscriterion, including multiple SFKs as well as CSK, PKCD,MAPK3, PIK3R2, SYK, TNK2, EPHB2, EPHA4, FAK, andPTK2B. We observed no changes in pTyr peptides corre-sponding to SFKs, including the pTyr peptide LIEDNEy-TAR corresponding to the common autocatalytic sitein c-SRC, YES, and FYN, following EGFR-TKI, suggestingthis as an EGFR-independent pathway. We linked SFKproteins to other proteins found in our entire datasetthrough interaction databases (Fig. 4A), identifying alarge group of proteins (N ¼ 28) with reported interac-tions with SFK proteins (gray circles) that were alsounchanged by erlotinib. In addition, we identified poten-tial interactions between SFK and proteins either alteredby erlotinib (gray parallelogram and diamond) or alteredin PC9GR compared with PC9 cells (gray V and dia-mond). For example, SRC can cooperate with EGFR, MET,ERBB3, SHC1, CBL, and STAT3 signaling nodes (grayparallelogram) that we previously identified as beingaltered by erlotinib and different between PC9 andPC9GR cells.
On the basis of this observation, we hypothesized thatcotargeting SFKs and EGFR T790M with dasatinib andafatinib, respectively, may produce additive or synergisticanti-tumor effects. Furthermore, our previous studies sug-gested that the antitumor effects of dasatinib are mediatedin part by direct EGFR inhibition that ismitigated by gain ofT790M in EGFR (32). However, these studies also suggestedthat irreversible EGFR-TKIs combined with dasatinib could
Figure 2. Phosphoproteins associated with T790M-mediated resistance. A, connectivity of MET protein was determined using protein–protein interactiondatabases tobetter aid in visualizingdifferentially expressedproteins thatmaybe associatedwithPC9GRcells. The left histograph showschangeof pTyr sitesin PC9GR cells compared with in PC9 cells. The fold change (P < 0.05, fold change > 1.5) of all tyrosine peptides were presented in log2 scale. Red barshows the tyrosine phosphosites of MET network proteins in PC9GR cells. Right, the MET network. Statistically decreased or increased pTyr peptides wereinput into Cytoscape 2.8.3, and protein–protein interactions were identified using InnateDB based on molecular interactions and functional relationsfrom public sources. Shapes reflect types of proteins shown in figure. Pink circle represents the pTyr peptides significantly different between PC9 andPC9GR cells and different between erlotinib-treated and control cells (P < 0.05; fold change >1.5). Color scale corresponds to fold change in Log2 scale. Theyellow lines represent the direct interaction with MET. B, Western blotting of selected proteins in PC9, PC9GR, HCC4006, HCC4006-T790M, HCC827,HCC827-T790M cells. Membranes were blotted with pTyr 1234/1235 MET, total MET, pTyr 702 AXL, total AXL, and total IRS2 antibodies in PC9, PC9GR,HCC4006, HCC4006-T790M, HCC827, HCC827-T790M cells with actin confirming equal protein loading. C, PC9GR cells were treated for 72 hours withincreasing concentrations of erlotinib alone, CL387,785 alone, PHA665752 alone, erlotinib þ PHA665752, or CL387,785 þ PHA665752. Data generated bycell viability assay (CellTiter-Glo) are expressed as a percentage of the value for untreated cells. Determinations were done in triplicate. Please view onlineversion for full details.
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4065
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
B C
Fo
ld c
han
ge
MET pY1234 AXL pY702 IRS2 pY675 IRS2 pY8230
5
10
15
20
25PC9PC9-ErlotinibPC9GRPC9GR-Erlotinib
Erlotinib-PC9GRErlotinib-PC9
–6.81 –0.58 1.63 4.84
GAB1 pY259
RAB7A pY183EGFR pY1197
CBL pY674SHC1 pY427
ERBB3 pY1276
EGFR pY1092ERBB3 pY1159EGFR pY1172EGFR pY998
CBLB pY889ERBB3 pY1307
GAB1 pY659
EGFR pY1138SHC1 pY349
MAPK1 pY187
Fold change (log2)–7 –6 –5 –4 –3 –2 –1 0 1 32 54 6 7
SHB pY114
ERBB2 pY1023
PTK6 pY114
PXN pY118
MET pY1234
TNS1 pY1254
TNS1 pY1404
NEDD9 pY166
GLUL pY185 TAGLN2 pY192
YES1 pY222
BCAR1 pY234
ANXA2 pY24
SHB pY268
NEDD9 pY317
NEDD9 pY345
HIPK3 pY359
STAT3 pY705
EPS8 pY774
PTK2 pY861
PXN pY88
INPPL1 pY886
NUP205 pY902
ACTB pY91
MAGED1 pY92
A
PC9GR
0.1 1 10 100 1,0000
25
50
75
100 CtrHGFIGF1GAS6
Erlotinib (nmol/L)
Rel
ativ
e ce
ll vi
alb
ility
0.1 1 10 100 1,0000
25
50
75
100 CtrHGFIGF1GAS6
Afatinib (nmol/L)
Rel
ativ
e ce
ll vi
alb
ility
Afatinib
PC9GR 193.00+ HGF 50 ng/mL >1000+ IGF1 50 ng/mL 422.30+ Gas6 800 ng/mL 174.00
ErlotinibIC50 nmol/L
IC50 nmol/L
PC9 6.94+ HGF 50 ng/mL 28.74+ IGF1 50 ng/mL 14.09+ Gas6 200 ng/mL 6.99
pY187
RAB7A
pY259
pY659
GAB1pY1138pY1172
SHC1
pY998CBL
pY1197
EGFR
pY674pY427
pY349
ERBB3
pY1276
pY1159
MAPK1
pY1307pY1172pY1197
CBLB
EGFR
pY1092
pY1138
pY1307
pY1159
pY1276
ERBB3
pY187MAPK1
pY427pY349
SHC1
pY259
pY659
GAB1
CBL
pY674
pY998
pY1092
pY1234METSTAT3
pY705
pY166pY317
pY774
NEDD9
pY345
pY114pY1404 pY268pY1254
pY91
ACTB
BCAR1
pY234
TNS1
EPS8
SHB
pY705
pY222YES1pY861
BCAR1
PTK2
pY1404
STAT3
TNS1pY234
pY24
pY91
TAGLN2
ACTB
PXN
pY114
ANXA2
ERBB2PTK6
pY1023
SHB
pY317
pY268
pY118
pY88
pY345
pY114
NEDD9
pY166
EPS8
pY185
MAGED1
pY192
pY902
pY774 HIPK3GLUL
pY92
NUP205
pY359
MET
pY1234pY886
INPPL1pY1254
Figure 3. Erlotinib perturbations inPC9andPCGRcell lines. A, effects of erlotinib on tyrosine containing phosphoproteomes inPC9 andPC9GRcell lines. Left,erlotinib-induced changes (P < 0.05; fold change >1.5) of pTyr sites in PC9 (bars) and PC9GR (�) cells. Four subnetworks were created within different catalogproteins (blue circle) based on increased or decreased pTyr sites in PC9 and PC9GR cells. Color scale represents the fold change of each pTyr site. B, pTyrpeptideabundancemeasuredbyEICacross erlotinib-treatedPC9andPC9GRcells forMET,AXL, and IRS2pTyr peptides.Y-axis indicates fold change abovePC9 untreated pTyr abundance. C, PC9 or PC9GR cells were seeded in 96-well plates for 24 hours and then exposed to HGF or IGF1 (50 ng/mL) or GAS6(200 ng/mL) and concomitantly exposed to increasing concentrations of relevant kinase inhibitor. After 72 hours, cell viability was assessed. IC50 wascalculated for each condition. Please view online version for full details.
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4066
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
inhibit EGFR T790M. Thus, we examined the effects oferlotinib, afatinib, dasatinib, or combined afatinib anddasatinib on EGFR signaling in PC9GR and H1975 cells,both resistant cell lines against EGFR-TKI because of T790M(Fig. 4B). In both cell lines, erlotinib inhibited neither EGFRnor SFKs, and no complete suppression of pAkt and pErk(both essential downstream molecules of EGFR) wasobserved. We found that afatinib could inhibit pEGFR;however, SFKs were again unaltered. These results witherlotinib and afatinib are consistent with our pTyr massspectrometry results that detected no changes in pTyr SFKsfollowing erlotinib exposure. We also found that combin-ing afatinib and dasatinib resulted in more efficient inhi-bition of pAkt and pErk in PC9GR and H1975 cells thaneach agent alone, suggesting that SFKs are cosignals relatedto EGFR T790M and that irreversible EGFR-TKIs combinedwith SFKs more efficiently block EGFR signaling in NSCLCcells harboring T790M.
Afatinib combined with dasatinib effectively inhibitscell growth and significantly increases apoptotic cellsin TKI-resistant NSCLC cells with T790MGiven that the combination of afatinib and dasatinib
efficiently inhibited EGFR signaling inNSCLC cells harboringT790M, we examined the effects of this combination on cellproliferation and apoptosis in PC9GR and H1975 cells. Wefound reduced IC50 levels in PC9GR and H1975 cells versuseither agent alone (Fig. 4C; Supplementary Tables S2 and S3).We examined the effects of combining afatinib plus dasatinibin other NSCLC cells with T790M (HCC4006-T790M andHCC827-T790M cells), cells with TKI-sensitive EGFR muta-tion only (PC9,HCC4006, andHCC827 cells), and cells withwild-type EGFR (H460, A549, andH1299 cells).Weobservedreduced IC50 for afatinib plus dasatinib versus either agentalone in HCC4006-T790M and HCC827-T790M cells (Sup-plementary Fig. S6), whereas curves for this combinationwere mostly overlapped with those for afatinib or dasatinibalone in PC9, HCC4006, and HCC827 cells with TKI-sensi-tive EGFR mutation only (Supplementary Fig. S7) or H460,A549, andH1299 cells with wild-type EGFR (SupplementaryFig. S8). We also found that dasatinib combined withCL387,785, another irreversible EGFR-TKI, reduced IC50versus either agent alone in PC9GR cells (SupplementaryFig. S9). Furthermore, we also detected more apparent PARPcleavage when agents were combined than when used aloneor when cells were erlotinib treated (see Fig. 4B). Similar toresults with afatinib, when we examined the effects of com-bined dasatinib with WZ4002, another T790M-specificEGFR-TKI (40), IC50 was reduced versus when agents wereused alone (Fig. 4D; Supplementary Tables S2 and S3).
Rescue experiments revealed that dasatinib enhancedantitumor effects of afatinib by inhibition of SRC andFYNAlthough our clustering approach and dasatinib results
strongly implicated SFKs as the key target, we examined thisin more detail given the extensive promiscuousness ofdasatinib. Using dasatinib-insensitive alleles expressed in
lentiviral vectors, we investigated whether dasatinib-resis-tant forms of key SFKs could rescue effects of dasatinib (32).To test which SFK is critical as a dasatinib target in NSCLCcells harboring T790M when combined with afatinib, weinfected cells with lentivirus expressing either wild-typekinases or kinase alleles with drug-resistant gatekeepermutations of SFKs (SRC, LYN, FYN, and FRK) and examinedcell viability in response to increasing concentrations ofdasatinib plus afatinib (Fig. 4E). Our results show that SRCand FYN were able to rescue PC9GR cells from dasatinibplus afatinib. However, no effects were observed with LYNand FRK, suggesting that SRC and FYN are essential SFKs asdasatinib targets in NSCLC cells with T790M.
Dasatinib enhances the antitumor activity of afatinibin vitro and in vivo
To evaluate more formally whether our combinationeffects were because of additional cell death, we measuredcaspase-3–positive cells following TKI treatment (Fig. 5A).In PC9GR cells, both erlotinib and dasatinib had noeffects on apoptosis, but the combination had modesteffects. Afatinib led to more apoptosis, which was furtherincreased when combined with dasatinib. Similar effectsof afatinib plus dasatinib on apoptosis were observed inHCC4006-T790M and HCC827-T790M cells (Supple-mentary Fig. S10). WZ4002 also induced apoptosis as asingle agent, which was potentiated when combined withdasatinib. In H1975 cells, similar effects were observed,with dasatinib increasing apoptosis when added to afa-tinib and WZ4002. These results indicate that Src inhi-bitors enhance antiproliferative and proapoptotic effectsof irreversible or T790M-selective EGFR-TKIs in NSCLCcells with T790M.
We hypothesized that the enhanced apoptosis withcombined afatinib and dasatinib would translate intoimproved in vivo effects on tumor growth. We examinedthe antitumor effects of this combination in mouse xeno-graft models with PC9GR cells. As single agents, afatinib(10 mg/kg) or dasatinib (15 mg/kg) had modest effectson inhibiting tumor growth in PC9GR xenografts;however, when combined, we observed significantly great-er inhibition of growth, including tumor regression con-sistent with our apoptosis results (Fig. 5B). These resultsdemonstrate that Src inhibitors effectively enhance anti-tumor effects of irreversible EGFR-TKI in gefitinib-resistantNSCLC xenografts with T790M, providing a rationale toevaluate this strategy in patients with NSCLC who haveacquired EGFR-TKI resistance related to T790M.
Tyrosine phosphoproteomes in lung adenocarcinomasamples with TKI-sensitive EGFR mutations
To validate that our cell line models and data are appli-cable to human lung cancer tissues, we conducted a massspectrometry tyrosine phosphoproteomics analysis on fourNSCLC tumor samples with TKI-sensitive EGFRmutations.In total, we identified 279 unique pTyr sites correspondingto 189 unique proteins across all four tumor samples. Foreach tumor, we identified 158, 153, 157, and 109 unique
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4067
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
pTyr sites corresponding to 117, 95, 111, and 87 proteins,respectively (Supplementary Table S4). Importantly, 83unique pTyr sites on 65 proteins identified from massspectrometry experiments in PC9 cells were also observedactive in human patient tissues (Supplementary Fig. S11and Supplementary Table S5). This included: EGFR(pTyr1172, 1197), MET (pTyr1234), SFKs including SRC(pTyr 419), FYN (pTyr 214, 420), LYN (pTyr 32, 193, 104,397), YES1 (pTyr223, 426), MK01 (pTyr 187), MK03 (pTyr204), STAT3 (pTyr 705), and AXL (pTyr 886). These resultsin human tumor tissues frompatients with EGFRmutationswell validate our findings gained from cell line model,
suggesting the potential of clinical application of our find-ings in this study.
Activation of Src or MET in human lung tumor sampleswith T790M gatekeeper EGFR mutations
To validate that Src phosphorylation is indeed observedas a target in human NSCLC samples with T790M gate-keeper mutation, we examined the expression of phos-phorylated Src (Tyr 416) in tumor samples with T790Musing immunohistochemical staining. We found pSrc inall EGFR T790M-positive tissue specimens, including 2paired samples of pre- and post-EGFR-TKI treatment, with
Proteins: no change in the PC9GR compared to PC9, also no response to erlotinib
B
CTTN
pY194
LYN
pY421
pY702
pY508
pY227
AXL
PTK2
pY293
pY341pY30
pY576
CTNNB1
GAB2
ITGB1PXN
pY88TYK2
pY292
pY397SDCBP
pY46
pY313
pY783PRKCD
pY780
TNS3pY296
pY359
SYKpY417
pY601pY163
PAG1TGM2
pY222pY904
pY334pY228
YES1
pY446
pY 334
pY193CTNND1
PTK2B
pY1138
pY869
pY1092
pY1172
pY1197pY998
EGFR
MCAM
TNK2
EPHA4BCAR1
MPZL1
FYN
CDCP1
CDKL5
PTPRA
PTPN11
PIK3R2
METpY1003
pY1234
pY427pY349
pY 204pY204
pY 427
MAPK3
SHC1
pY525
SHB
pY 774
pY774
pY 114
pY114
EPS8
pY214
pY345
pY317
pY261pY106
pY166
NEDD9
ELF2pY1307
pY91pY1159
pY1276
pY287
ERBB3 ACTB
PTPN6
EPHB2SRC
ERBB2
WASL
SHANK2
ASAP2
CSK
pY705
CBL
pY 705
pY674
pY 674
STAT3
pY 317pY802pY579 pY584pY369
pY241
pY491
pY629
pY268
pY177
pY 345
pTyr sites up-regulated by erlotinib or higher expression in PC9GR than PC9
A
pTyr sites down-regulated by erlotinib or lower expression in PC9GR than PC9
Proteins: altered by erlotinib only Proteins: altered in PC9GR compared to PC9
Proteins: altered by erlotinib and in PC9GR compared to PC9
PARP
PC9GR (Ex19del + T790M)
Actin
p-Erk
p-Akt
p-EGFR
p-Src
Akt
Erk
EGFR
Src
Erlo
tinib
100
nm
ol/L
Con
t
10 n
mol
/L
100
nmol
/L
Afatinib
10 n
mol
/L
100
nmol
/L
Dasatinib
10 n
mol
/L
100
nmol
/L
Combination
H1975 (L858R + T790M)
PARP
Actin
p-Erk
p-Akt
p-EGFR
p-Src
Akt
Erk
EGFR
Src
Con
t
10 n
mol
/L
100
nmol
/L
Afatinib
10 n
mol
/L
100
nmol
/L
Dasatinib
10 n
mol
/L
100
nmol
/L
Combination
Erlo
tinib
100
nm
ol/L
OCLNpY 287
Figure 4. Effects of dasatinib combined with afatinibon EGFR signaling and cell growth in gefitinib-resistant NSCLC cells with T790M. A, pTyr proteinscorresponding to pTyr peptides identified in PC9and PC9GR cells are linked to SFK using InnateDBto capture literature reports and displayed inCytoscape. Pink circle, pTyr sites upregulated byerlotinib or higher expression in PC9GR than in PC9cells. Blue circle, pTyr sites downregulated byerlotinib or less expression in PC9GR than in PC9cells. Yellow circles or V, SFK (SRC, YES1, FYN).Gray circle, pTyr proteins showing no differencebetween PC9 and PC9GR cells and no change witherlotinib treatment. Gray parallelogram, pTyrproteins altered by erlotinib across PC9andPC9GRdatasets. Gray V, pTyr proteins different betweenPC9 andPC9GR cells. Gray diamond, pTyr proteinsdifferent between PC9 and PC9GR cells andshowing changes with erlotinib treatment. B,PC9GR and H1975 cells were incubated for 6 hours(or 24 hours for PARP) in the absence or presence oferlotinib (100 nmol/L), afatinib (10 and 100 nmol/L),dasatinib (10 and 100 nmol/L), or afatinib anddasatinib in combination (10 and 100 nmol/L), asindicated. Cell lysates were subjected to proteinexpression analysis with antibodies to pEGFR,EGFR, pAkt, Akt, pErk, Erk, pSrc family, Src, orPARP along with antibodies to b-actin as a loadingcontrol. (Continued on the following page.)
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4068
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
E
C
D
Drug Sensitivity in PC9GR
1 10 100 1,000 10,0000
25
50
75
100
AfatinibErlotinib
Afatinib + DasatinibDasatinib
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
1 10 100 1,000 10,000
Drug concentration (nmol/L)
Cel
l via
bilit
y (%
)C
ell v
iabi
lity
(%)
Cel
l via
bilit
y (%
)
Cel
l via
bilit
y (%
)
Cel
l via
bilit
y (%
)
Cel
l via
bilit
y (%
)
Cel
l via
bilit
y (%
)
Drug Sensitivity in H1975
0
25
50
75
100
AfatinibErlotinib
Afatinib + DasatinibDasatinib
Cel
l via
bilit
y (%
)
Drug sensitivity in PC9GR
0
25
50
75
100
WZ4002Erlotinib
WZ4002 + DasatinibDasatinib
Drug Sensitivity in H1975
0
25
50
75
100
WZ4002Erlotinib
WZ4002 + DasatinibDasatinib
0
25
50
75
100
SRC-WT vs. Afatinib + Dasatinib
SRC-MT vs. Afatinib + Dasatinib
0
25
50
75
100
FYN-WT vs. Afatinib + Dasatinib
FYN-MT vs. Afatinib + Dasatinib
0
25
50
75
100
LYN-WT vs. Afatinib + Dasatinib
LYN-MT vs. Afatinib + Dasatinib
0
25
50
75
100
FRK-WT vs. Afatinib + Dasatinib
FRK-MT vs. Afatinib + Dasatinib
Figure 4. (Continued. ) C, PC9GR and H1975 cells were treated for 72 hours with increasing concentrations of erlotinib alone, afatinib alone, dasatinibalone, or afatinib þ dasatinib. D, PC9GR and H1975 cells were treated for 72 hours with increasing concentrations of erlotinib alone, WZ4002 alone,dasatinib alone, or WZ4002 þ dasatinib. Data generated by cell viability assay (CellTiter-Glo) are expressed as a percentage of the value for untreatedcells. Determinations were done in triplicate. E, PC9GR cells were infected with lentivirus expressing wild-type and mutant gatekeeper forms ofeach indicated Src family kinase for 48 hours. Subsequently, cells were exposed to increasing concentrations of afatinib plus dasatinib for 72 hours,after which cell viability was assessed by cell viability assay (CellTiter-Glo). Data are expressed as a percentage of the value for untreated cells.Determinations were done in triplicate. Please view online version for full details.
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4069
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
varied intensities and cellularity (Table 1; Fig. 5C). Theseresults confirmed results from cell lines that Src activitypersists in EGFR T790M-positive tumor tissues. We fur-ther examined changes in tyrosine phosphorylated METexpression in matched pre- and post-EGFR-TKI treatmentpatient tumor tissue specimens. We found evidence forincreased tyrosine phosphorylated MET in one patient(patient 10 in Table 1) that was not due to increased totalMET protein (Fig. 5D), whereas no evidence was foundin the second patient (patient 9 in Table 1) for which pre-and posttreatment biopsy tissues were available for study.These results provide further support using tumor tissuesthat MET tyrosine phosphorylation can occur in T790M-containing tissues and this can be independent of totalMET expression.
DiscussionWe applied tyrosine phosphorylation profiling using
LC/MS-MS to directly compare an EGFR-TKI–sensitivecell line versus its acquired resistance counterpart touncover additional resistance mechanisms and proposecotargeting strategies to enhance the effects of agentsspecifically targeting the T790M EGFR allele. To ourknowledge, this is the first such report to apply a massspectrometry–based phosphoproteomics approach tocompare the molecular networks between EGFR-TKI–sensitive and -resistant pairs. The driving force behindthis approach is the limited efficacy of irreversible EGFR-TKIs in targeting T790M, as shown in both preclinicaland clinical studies (8, 9, 12, 14–19), and the ability of
A
B
PC9GR (Ex19del +T790M) H1975 (L858R +T790M)
PC9GR
0
10
20
30
40
Treatment (48 h)
DMSO
E 10
0 nmo
l/L
A 10
0 nmo
l/L
D 10
0 nmo
l/L
E 10
0 nmo
l/L +
D 10
0 nmo
l/L
A 10
0 nmo
l/L +
D 10
0 nmo
l/L
W 10
0 nmo
l/L
W 10
0 nmo
l/L +
D 10
0 nmo
l/L
DMSO
E 10
0 nmo
l/L
A 10
0 nmo
l/L
D 10
0 nmo
l/L
E 10
0 nmo
l/L +
D 10
0 nmo
l/L
A 10
0 nmo
l/L +
D 10
0 nmo
l/L
W 10
0 nmo
l/L
W 10
0 nmo
l/L +
D 10
0 nmo
l/L
Treatment (48 h)
Cas
pas
e-3–
po
siti
ve c
ells
(%
)
0
10
20
30
40
Cas
pas
e-3–
po
siti
ve c
ells
(%
)
H1975
In vivo activity in PC9GR cells
0 2 4 6 8 10 12 14 16 18 20 220
50
100
150
200
250
300
350
400
450
500
550
600
650
700 Control
Afatinib
Dasatinib
Afatinib + Dasatinib
Days
Tu
mo
r vo
lum
e (m
m3 )
Figure 5. Effects of dasatinibcombined with afatinib onapoptosis and in vivo tumorregression in gefitinib-resistantNSCLC cells with T790M. A,apoptosis assay was carried outusing PE-conjugated caspase-3antibody, following incubation ofPC9GR or H1975 cells for 72 hourswith DMSO, erlotinib (E), dasatinib(D), erlotinib þ dasatinib, afatinib(A), afatinib þ dasatinib, WZ4002(W), and WZ4002 þ dasatinib.Values are expressed as apercentage of caspase-3-positivecells. Determinations were done intriplicate. Bars, SD. �, P < 0.001versus DMSO or each single agent(Student t test). B, Nude mice withtumor xenografts established bysubcutaneous implantation ofPC9GR cells were treated daily for21 days with vehicle (control),afatinib (10 mg/kg), dasatinib (15mg/kg), or afatinib þ dasatinib byoral gavage. Tumor volume wasdetermined at the indicated timesafter the onset of treatment. Points,mean of values from 5 mice/group;bars, SE. �, P < 0.05 for afatinibcombined with dasatinib versuscontrol or each agent alone byANOVA (Tukey–Kramercomparison). (Continued on thefollowing page.)
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4070
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
other tyrosine kinases, especially RTKs, to limit EGFR-TKIefficacy. Through a systematic interrogation of pTyr pep-tides and proteins using LC/MS-MS, we identified bothRTKs and non-RTKs able to be recruited to confer erlo-
tinib sensitivity. In PC9GR cells, we identified higherlevels of pTyr peptides corresponding to MET signaling,including more MET, ROR1, and Gab1/2 proteins. Inter-estingly, this coordinated activation of MET signalingwas not secondary to MET gene amplification, as ourFISH results revealed no amplification of the MET gene.The results with the basal phosphoproteome corre-sponded to HGF’s strong effects in protecting both PC9and PC9GR cells from erlotinib or afatinib, respectively.In addition, these results suggest a form of "lineage"addiction, whereby resistant cells with T790M can carryforward RTKs that can cooperate to drive resistance.Importantly, these results suggest that interrogating pro-tein activation status or network signaling may highlightproteins that play a role in protecting cells against EGFR-TKI, especially when in a microenvironment rich withcognate growth factor ligands. Despite observing moreAXL pTyr peptides in PC9GR cells, we demonstrated noability of AXL pathway activation by Gas6 ligand to driveresistance to either erlotinib or afatinib. The reasons forthis are not clear, but one limit of our approach was thelack of absolute measurements of pTyr peptides. It ispossible that, compared with MET or IRS2 pTyr peptides,pTyr peptides corresponding to AXL are far lower inabsolute amount and thus are inefficient to compete fordownstream signaling effectors. It will be interesting andimportant to determine how basal phosphoproteomemeasurements can predict the effects of growth factorprotection against targeted agents. As AXL signaling stillremains poorly understood, another explanation for ourresults could be the limited or absence of key adaptors orother effector proteins involved in AXL signaling.
Using pTyr peptide data obtained from both PC9 andPC9GR cells exposed to erlotinib, we identified proteinsdownstream of EGFR in these cells with mutant gain-of-function EGFR proteins. One of the more interestingfindings was that nearly half of the statistically significantpTyr peptides were increased in abundance followingerlotinib treatment. It is increasingly recognized thatsignaling pathways display large amounts of crosstalkand that adaptive resistance mechanisms have beenobserved in cells exposed to targeted agents (41). Ourresults match our investigations using purified Srchomology-2 domains to profile tyrosine kinase signalingin lung cancer cells, where we observed increased pTyrsignaling in multiple lung cancer cells exposed to TKIs(42). Similar events have also been observed in crizoti-nib-treated EML4-ALK cells and dasatinib-treated DDR2-mutant lung cancer cells, arguing that these paradoxicalchanges are consistent across multiple tumor types andkinase inhibitors (unpublished observations). Theunderlying mechanisms of these changes require addi-tional study, as they could be important in promotingadaptive resistance to targeted agents and could in somecases cooperate with microenvironmental factors, suchas growth factors, to limit TKI efficacy. Collectively, theseresults suggest that cell intrinsic (receptors, signalingproteins) and extrinsic (ligands) factors can collaborate
H score: 9
H score: 3
H score: 6
Pretreatment Posttreatment
ME
T-p
Y10
0 P
LAT
otal
ME
T IF
C
D
Figure 5. (Continued. ) C, evidence for activation of Src in the T790M-positive biopsy specimens. Immunohistochemical staining was used todetect expression of tyrosine phosphorylation Src (Tyr416) in 10 EGFRT790M-positive tissue specimens. H score (intensity � cellularity) wascalculated for each sample, with scores 0 and 9 representing the lowestand highest expression, respectively. Three representative�200 imagesshow the different levels of pSrc expression in EGFR T790M-positivehuman lung specimen. D, evidence for increased MET tyrosinephosphorylation in posttreatment T790M biopsy specimens. Proximityligation assays (PLA) for MET and pY100 were performed to assess METphosphorylation in serial biopsy specimens obtained from anadenocarcinomapatient (right lung, lower lobe, 33months apart). Originalbiopsy confirmed EGFR exon21 L858R mutation; rebiopsy confirmedL858R/T790M mutation. Top, evidence of MET-pY PLA signal inpretreatment biopsy, while clusters of highly phosphorylated MET areobserved in the posttreatment biopsy. MET-pY PLA was localized tocytokeratin (þ)-staining regions (data not shown). Bottom, increasedMET-pY PLA signal is not due to increased total MET protein.
Phosphoproteomics of EGFR-TKI Resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4071
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
to drive resistance to kinase inhibitors in a systems-levelmanner.
Our phosphoproteomics analyses in PC9 and PC9GRcells demonstrated that SFKs are also critical as an EGFR-independent cosignal in NSCLC cells with T790M. Theseresults were enabled by tyrosine phosphorylation profil-ing combined with analysis of proteins based on knownprotein–protein interactions. We validated the inferencesderived from the phosphoproteomics by showingthat afatinib combined with dasatinib resulted in antitu-mor activity regarding cell proliferation and apoptosis inPC9GR, H1975, HCC4006-T790M, and HCC827-T790Mcells (each harboring T790M). These results appear to begeneralized to additional T790M EGFR-TKIs, as dasatinibdemonstrated similar combination effects with theT790M-selective EGFR-TKI WZ4002 (40). We did notobserve combination effects with afatinib plus dasatinibcompared with either agent alone in cells with TKI-sen-sitive EGFRmutation only (PC9, HCC4006, and HCC827cells) or wild-type EGFR (H460, A549, and H1299 cells).The enhanced apoptosis with combined afatinib anddasatinib in the cells with T790M translated intoimproved in vivo effects on tumor growth in PC9GR cells.Collectively, our results suggest that dasatinib can begenerally used as a combination therapy with irreversibleor T790M-selective EGFR-TKIs for patients with NSCLCwho acquired EGFR-TKI resistance associated withT790M.
As our approach was limited to examining the tyrosinephosphoproteome, we were unable to detect serine/thre-onine signaling including mTOR/AKT or MEK/Erk path-ways both of which are also essential for carcinogenesis.Previous studies have indicated that mTOR inhibitorcombined with MEK inhibitor or irreversible EGFR-TKIis potential strategy to overcome T790M (21, 43). Furtherstudies examining the global phosphoproteome, such aswith immobilized metal affinity chromatography which
can detect serine/threonine phosphopeptides (44), couldidentify other proteins and pathways that may play rolesin EGFR TKI resistance.
Although single-agent dasatinib has no activity inpatients with NSCLC with TKI-sensitive EGFR mutationwho acquired resistance to EGFR-TKI (45), our resultssuggest a role for SFKs in maintaining downstream sig-naling despite irreversible EGFR-TKIs and support furtherstudies of irreversible EGFR-TKIs combined with dasati-nib in patients with NSCLC who acquire resistance toEGFR-TKI. Src is known to be both an upstream activatorand a downstream mediator of EGFR, and its phosphor-ylation is detected in about one-third of lung cancertumors (46, 47). Although MET activation might not bealways observed in the presence of T790M based on our invitro and tumor tissue analysis, pSrc seems to be generallydetected in our NSCLC tumor samples harboring T790M,consistent with our results of cell models. In addition, ourmass spectrometry data from tumor samples with TKI-sensitive EGFR mutation demonstrated a high degree ofoverlap with results from cell models, thereby validatingthe overall approach. These results from tumor samplessuggest that our results from lung cancer cell line modelsare applicable to translate in to the clinic. Our previouschemical and phosphoproteomic characterization iden-tified nearly 40 different kinase targets of dasatinib andshowed that SRC, FYN, and EGFR are relevant targets fordasatinib action in NSCLC (32). Our recent phase I/IIstudy showed that dasatinib combined with erlotinib istolerable, with 63% of patients with advanced NSCLCshowing disease control, including two having partialresponse and one having bone response (48). Anothergroup also showed that dasatinib combined with erloti-nib is safe and feasible in NSCLC (49).
On the basis of these clinical studies along with theexperiments reported here, dasatinib has potential clinicalactivity in NSCLC treatment, but this is limited to
Table 1. Src phosphorylation detected in human NSCLC samples with T790M gatekeeper mutation
Patient ID EGFR mutant H score Intensity Cellularity
1 T790M/L858R 3 1 32 T790M/19del(E746-A750) 4 2 23 T790M/19del(E746-A750) 6 2 34 T790M/L858R 6 2 35 T790M/19del(E746-A750) 6 2 36 T790M/19del(E746-A750) 9 3 37 T790M/19del(E746-A750) 9 3 38 T790M/L858R 9 3 39 (Pre-TKI) 19del(E746-A750) 9 3 39 (Post-TKI) T790M/19del(E746-A750) 9 3 310 (Pre-TKI) L858R 4 2 210 (Post-TKI) T790M/L858R 9 3 3
NOTE: Patient 9 was treated with "gefitinib and erlotinib." Patient 10 was treated with "gefitinib and erlotinib þ ARQ197 (MET-TKI)."
Yoshida et al.
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4072
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
combinationswith T790M-targeted agents and in genotype-specific patients. Thiswill be formally tested in a phase I trialof afatinib and dasatinib (NCT01999985). Our results alsohighlight the ability of phosphoproteomics to identify otherimportantmediators of drug sensitivity, and examinationofthese proteins may be important in clinical studies ofT790M-targeting agents.
Disclosure of Potential Conflicts of InterestE.B. Haura reports receiving a commercial research grant from Boehrin-
ger-Ingelheim. No potential conflicts of interest were disclosed by the otherauthors.
Authors' ContributionsConception and design: T. Yoshida, G. Zhang, E.B. HauraDevelopment of methodology: T. Yoshida, G. Zhang, M.A. Smith,A.S. Lopez, Y. Bai, J. Koomen, E.B. HauraAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): T. Yoshida, G. Zhang, M.A. Smith, B. Fang,J. Koomen, K. Nakagawa, E.B. HauraAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis):G. Zhang, M.A. Smith, A.S. Lopez, Y. Bai,B. Fang, B. Rawal, K.J. Fisher, A.Y. Chen, I. Okamoto, K. Nakagawa, E.B.HauraWriting, review, and/or revisionof themanuscript: T. Yoshida,G. Zhang,M.A. Smith, B. Fang, J. Koomen, B. Rawal, A.Y. Chen, K. Nakagawa,E.B. Haura
Administrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): T. Yoshida, G. Zhang, M.A. Smith,A.S. Lopez, Y. Bai, J. Li, M. Kitano, Y. Morita, H. Yamaguchi, K. Shibata,T. Okabe, I. Okamoto, K. Nakagawa, E.B. HauraStudy supervision: K. Nakagawa, E.B. Haura
AcknowledgmentsThe authors thank the Moffitt pY Group, the Kinki University Medical
Oncology Research Group, Tsutomu Iwasa, and Kunio Okamoto for helpfuldiscussions, Rasa Hamilton for editorial assistance, Fumi Kinose for assis-tance with cell culture, and Linda Ley and Carol Ulge for administrativeassistance. The authors also thank BML, Inc and SRL, Inc for technicalassistance.
Grant SupportThe work was partially funded by grants from the Moffitt Cancer Center
SPORE in Lung Cancer (P50-CA119997), the V Foundation for CancerResearch, and in part by the National Cancer Institute, part of the NIH,through grant number 2 P30-CA76292-14, which provide support to theProteomics Core, the Flow Cytometry Core, the Tissue Core, and the AnimalFacility at the H. Lee Moffitt Cancer Center and Research Institute, an NCI-designated Comprehensive Cancer Center.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received June 13, 2013; revised April 9, 2014; accepted April 23, 2014;published OnlineFirst June 11, 2014.
References1. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al.
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma.N Engl J Med 2009;361:947–57.
2. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J,et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growthfactor receptor (WJTOG3405): an open label, randomised phase 3 trial.Lancet Oncol 2010;11:121–8.
3. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, MeyersonM, et al. EGFRmutation and resistanceof non-small-cell lungcancer togefitinib. N Engl J Med 2005;352:786–92.
4. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al.Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib isassociated with a second mutation in the EGFR kinase domain. PLoSMed 2005;2:e73.
5. Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al.Analysis of epidermal growth factor receptor genemutation in patientswith non-small cell lung cancer and acquired resistance to gefitinib.Clin Cancer Res 2006;12:5764–9.
6. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB,Fidias P, et al. Genotypic and histological evolution of lung cancersacquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
7. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al.Analysis of tumor specimens at the time of acquired resistance toEGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.Clin Cancer Res 2013;19:2240–7.
8. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, GaleCM, et al. Allelic dilution obscures detection of a biologically significantresistance mutation in EGFR-amplified lung cancer. J Clin Invest2006;116:2695–706.
9. Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shi-mamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, iseffective in lung cancer models with EGFR and ERBB2 mutations thatare resistant to gefitinib. Cancer Res 2007;67:11924–32.
10. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR,et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effectivein preclinical lung cancer models. Oncogene 2008;27:4702–11.
11. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA,Brannigan BW, et al. Irreversible inhibitors of the EGF receptor maycircumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A2005;102:7665–70.
12. Sos ML, Rode HB, Heynck S, Peifer M, Fischer F, Kluter S, et al.Chemogenomic profiling provides insights into the limited activity ofirreversible EGFR inhibitors in tumor cells expressing the T790MEGFRresistance mutation. Cancer Res 2010;70:868–74.
13. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK,et al. The T790M mutation in EGFR kinase causes drug resistance byincreasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
14. Yu Z, Boggon TJ, Kobayashi S, Jin C, Ma PC, Dowlati A, et al.Resistance to an irreversible epidermal growth factor receptor (EGFR)inhibitor in EGFR-mutant lung cancer reveals novel treatment strate-gies. Cancer Res 2007;67:10417–27.
15. Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J,Maheswaran S, et al. The T790M "gatekeeper" mutation in EGFRmediates resistance to low concentrations of an irreversible EGFRinhibitor. Mol Cancer Ther 2008;7:874–9.
16. Shimamura T, Li D, Ji H, Haringsma HJ, Liniker E, Borgman CL,et al. Hsp90 inhibition suppresses mutant EGFR-T790M signalingand overcomes kinase inhibitor resistance. Cancer Res 2008;68:5827–38.
17. Yamada T, Matsumoto K, Wang W, Li Q, Nishioka Y, Sekido Y, et al.Hepatocyte growth factor reduces susceptibility to an irreversibleepidermal growth factor receptor inhibitor in EGFR-T790M mutantlung cancer. Clin Cancer Res 2010;16:174–83.
18. Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A,et al. Amplification of EGFRT790Mcauses resistance to an irreversibleEGFR inhibitor. Oncogene 2010;29:2346–56.
19. Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinibversus placebo for patients with advanced, metastatic non-small-celllung cancer after failure of erlotinib, gefitinib, or both, and one or twolines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial.Lancet Oncol 2012;13:528–38.
20. Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al.Dual targeting of EGFR can overcome a major drug resistance
www.aacrjournals.org Clin Cancer Res; 20(15) August 1, 2014 4073
Phosphoproteomics of EGFR-TKI Resistance
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
mutation in mouse models of EGFR mutant lung cancer. J Clin Invest2009;119:3000–10.
21. Li D, Shimamura T, Ji H, Chen L, Haringsma HJ, McNamara K, et al.Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combinationtherapy. Cancer Cell 2007;12:81–93.
22. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH,Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affectsthe response of tumor cells to targeted therapies. Science2007;318:287–90.
23. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al.Widespread potential for growth-factor-driven resistance to antican-cer kinase inhibitors. Nature 2012;487:505–9.
24. Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Sheppard KA, et al.Rescue screens with secreted proteins reveal compensatory potentialof receptor tyrosine kinases in driving cancer growth. Cancer Discov2012;2:948–59.
25. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al.Hepatocyte growth factor induces gefitinib resistance of lung adeno-carcinoma with epidermal growth factor receptor-activating muta-tions. Cancer Res 2008;68:9479–87.
26. Machida K, Eschrich S, Li J, Bai Y, Koomen J, Mayer BJ, et al.Characterizing tyrosine phosphorylation signaling in lung cancer usingSH2 profiling. PLoS ONE 2010;5:e13470.
27. Naoki K, Soejima K, Okamoto H, Hamamoto J, Hida N, Nakachi I, et al.The PCR-invadermethod (structure-specific 50 nuclease-basedmeth-od), a sensitive method for detecting EGFR gene mutations in lungcancer specimens; comparison with direct sequencing. Int J ClinOncol 2011;16:335–44.
28. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO,et al. MET amplification leads to gefitinib resistance in lung cancer byactivating ERBB3 signaling. Science 2007;316:1039–43.
29. Zhang G, Fang B, Liu RZ, Lin H, Kinose F, Bai Y, et al. Massspectrometrymapping of epidermal growth factor receptor phosphor-ylation related to oncogenic mutations and tyrosine kinase inhibitorsensitivity. J Proteome Res 2011;10:305–19.
30. Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, et al.InnateDB: facilitating systems-level analyses of the mammalian innateimmune response. Mol Syst Biol 2008;4:218.
31. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, et al.A travel guide to Cytoscape plugins. Nat Methods 2012;9:1069–76.
32. Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical andphosphoproteomic characterization of dasatinib action in lung cancer.Nat Chem Biol 2010;6:291–9.
33. Ogino A, Kitao H, Hirano S, Uchida A, Ishiai M, Kozuki T, et al.Emergence of epidermal growth factor receptor T790M mutationduring chronic exposure to gefitinib in a non small cell lung cancercell line. Cancer Res 2007;67:7807–14.
34. Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immu-noaffinity profiling of tyrosine phosphorylation in cancer cells. NatBiotechnol 2005;23:94–101.
35. Gentile A, Lazzari L, Benvenuti S, Trusolino L, Comoglio PM. Ror1 is apseudokinase that is crucial forMet-driven tumorigenesis. Cancer Res2011;71:3132–41.
36. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activationof the AXL kinase causes resistance to EGFR-targeted therapy in lungcancer. Nat Genet 2012;44:852–60.
37. Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P,Monti J, et al. SynGAP regulates ERK/MAPK signaling, synapticplasticity, and learning in the complex with postsynaptic density 95and NMDA receptor. J Neurosci 2002;22:9721–32.
38. Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulatessynaptic strength and mitogen-activated protein kinases in culturedneurons. Proc Natl Acad Sci U S A 2006;103:4344–51.
39. Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interactinglysosomal protein (RILP): the Rab7 effector required for transport tolysosomes. EMBO J 2001;20:683–93.
40. Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novelmutant-selective EGFR kinase inhibitors against EGFR T790M. Nature2009;462:1070–4.
41. Chandarlapaty S. Negative feedback and adaptive resistance to thetargeted therapy of cancer. Cancer Discov 2012;2:311–9.
42. MachidaK, ThompsonCM,DierckK, Jablonowski K, KarkkainenS, LiuB, et al. High-throughput phosphotyrosine profiling using SH2domains. Mol Cell 2007;26:899–915.
43. Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al.Differential induction of apoptosis in HER2 and EGFR addicted can-cers following PI3K inhibition. Proc Natl Acad Sci U S A 2009;106:19503–8.
44. Kim JY,Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, et al. Dissection ofTBK1 signaling via phosphoproteomics in lung cancer cells. Proc NatlAcad Sci U S A 2013;110:12414–9.
45. Johnson ML, Riely GJ, Rizvi NA, Azzoli CG, Kris MG, Sima CS,et al. Phase II trial of dasatinib for patients with acquired resistanceto treatment with the epidermal growth factor receptor tyrosinekinase inhibitors erlotinib or gefitinib. J Thorac Oncol 2011;6:1128–31.
46. Zhang J, KalyankrishnaS,WislezM, ThilaganathanN, Saigal B,WeiW,et al. SRC-family kinases are activated in non-small cell lung cancerand promote the survival of epidermal growth factor receptor-depen-dent cell lines. Am J Pathol 2007;170:366–76.
47. Leung EL, Tam IY, Tin VP, Chua DT, Sihoe AD, Cheng LC, et al.SRC promotes survival and invasion of lung cancers with epider-mal growth factor receptor abnormalities and is a potential can-didate for molecular-targeted therapy. Mol Cancer Res 2009;7:923–32.
48. Haura EB, Tanvetyanon T, Chiappori A, Williams C, Simon G, AntoniaS, et al. Phase I/II study of the Src inhibitor dasatinib in combinationwith erlotinib in advanced non-small-cell lung cancer. J Clin Oncol2010;28:1387–94.
49. Johnson FM, Tang X, Tran HT, McIntyre C, Price J, Lee JJ, et al. Aphase I/II study combining dasatinib (D) and erlotinib (E) in non-smallcell lung cancer. J Clin Oncol 31, 2013 (suppl; abstr 8102).
Clin Cancer Res; 20(15) August 1, 2014 Clinical Cancer Research4074
Yoshida et al.
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/
-
Correction
Correction: Tyrosine PhosphoproteomicsIdentifies Both Codrivers and CotargetingStrategies for T790M-Related EGFR-TKIResistance in Non–Small Cell Lung Cancer
In this article (ClinCancer Res 2014;20:4059–74),whichwaspublished in theAugust1, 2014, issue of Clinical Cancer Research (1), an author's name was misprinted.The corrected name should read as follows: "Y. Ann Chen." The authors regret thiserror.
Reference1. Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, et al. Tyrosine phosphoproteomics identifies
both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non–small celllung cancer. Clin Cancer Res 2014;20:4059–74.
Published online August 3, 2015.doi: 10.1158/1078-0432.CCR-15-1183�2015 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org 3571
-
2014;20:4059-4074. Published OnlineFirst June 11, 2014.Clin Cancer Res Takeshi Yoshida, Guolin Zhang, Matthew A. Smith, et al.
Small Cell Lung Cancer−Non Cotargeting Strategies for T790M-Related EGFR-TKI Resistance in
Tyrosine Phosphoproteomics Identifies Both Codrivers and
Updated version
10.1158/1078-0432.CCR-13-1559doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2014/06/16/1078-0432.CCR-13-1559.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/20/15/4059.full#ref-list-1
This article cites 48 articles, 25 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/20/15/4059.full#related-urls
This article has been cited by 11 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/20/15/4059To request permission to re-use all or part of this article, use this link
on June 19, 2021. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 11, 2014; DOI: 10.1158/1078-0432.CCR-13-1559
http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-13-1559http://clincancerres.aacrjournals.org/content/suppl/2014/06/16/1078-0432.CCR-13-1559.DC1http://clincancerres.aacrjournals.org/content/20/15/4059.full#ref-list-1http://clincancerres.aacrjournals.org/content/20/15/4059.full#related-urlshttp://clincancerres.aacrjournals.org/cgi/alertsmailto:[email protected]://clincancerres.aacrjournals.org/content/20/15/4059http://clincancerres.aacrjournals.org/
/ColorImageDict > /JPEG2000ColorACSImageDict > /JPEG2000ColorImageDict > /AntiAliasGrayImages false /CropGrayImages false /GrayImageMinResolution 200 /GrayImageMinResolutionPolicy /Warning /DownsampleGrayImages true /GrayImageDownsampleType /Bicubic /GrayImageResolution 300 /GrayImageDepth -1 /GrayImageMinDownsampleDepth 2 /GrayImageDownsampleThreshold 1.50000 /EncodeGrayImages true /GrayImageFilter /DCTEncode /AutoFilterGrayImages true /GrayImageAutoFilterStrategy /JPEG /GrayACSImageDict > /GrayImageDict > /JPEG2000GrayACSImageDict > /JPEG2000GrayImageDict > /AntiAliasMonoImages false /CropMonoImages false /MonoImageMinResolution 600 /MonoImageMinResolutionPolicy /Warning /DownsampleMonoImages true /MonoImageDownsampleType /Bicubic /MonoImageResolution 900 /MonoImageDepth -1 /MonoImageDownsampleThreshold 1.50000 /EncodeMonoImages true /MonoImageFilter /CCITTFaxEncode /MonoImageDict > /AllowPSXObjects false /CheckCompliance [ /None ] /PDFX1aCheck false /PDFX3Check false /PDFXCompliantPDFOnly false /PDFXNoTrimBoxError true /PDFXTrimBoxToMediaBoxOffset [ 0.00000 0.00000 0.00000 0.00000 ] /PDFXSetBleedBoxToMediaBox true /PDFXBleedBoxToTrimBoxOffset [ 0.00000 0.00000 0.00000 0.00000 ] /PDFXOutputIntentProfile (None) /PDFXOutputConditionIdentifier () /PDFXOutputCondition () /PDFXRegistryName () /PDFXTrapped /False
/CreateJDFFile false /Description > /Namespace [ (Adobe) (Common) (1.0) ] /OtherNamespaces [ > /FormElements false /GenerateStructure false /IncludeBookmarks false /IncludeHyperlinks false /IncludeInteractive false /IncludeLayers false /IncludeProfiles false /MarksOffset 18 /MarksWeight 0.250000 /MultimediaHandling /UseObjectSettings /Namespace [
Related Documents