Two-step reaction mechanism reveals new antioxidant capability of cysteine disulfides against hydroxyl radical attack Sarju Adhikari a , Ramon Crehuet b , Josep M. Anglada b,1 , Joseph S. Francisco c,1 , and Yu Xia a,d,1 a Department of Chemistry, Purdue University, West Lafayette, IN 47907; b Department of Biological Chemistry, Institute of Advanced Chemistry of Catalonia, IQAC-CSIC, E-08034 Barcelona, Spain; c Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104; and d Department of Chemistry, Tsinghua University, Beijing 100084, China Contributed by Joseph S. Francisco, May 30, 2020 (sent for review April 9, 2020; reviewed by Richard A. O’Hair and Henry F. Schaefer III) Cysteine disulfides, which constitute an important component in biological redox buffer systems, are highly reactive toward the hydroxyl radical ( • OH). The mechanistic details of this reaction, how- ever, remain unclear, largely due to the difficulty in characterizing unstable reaction products. Herein, we have developed a combined approach involving mass spectrometry (MS) and theoretical calcula- tions to investigate reactions of • OH with cysteine disulfides (Cys– S–S–R) in the gas phase. Four types of first-generation products were identified: protonated ions of the cysteine thiyl radical ( + Cys– S • ), cysteine ( + Cys–SH), cysteine sulfinyl radical ( + Cys–SO • ), and cys- teine sulfenic acid ( + Cys–SOH). The relative reaction rates and prod- uct branching ratios responded sensitively to the electronic property of the R group, providing key evidence to deriving a two-step reac- tion mechanism. The first step involved • OH conducting a back-side attack on one of the sulfur atoms, forming sulfenic acid (–SOH) and thiyl radical (–S • ) product pairs. A subsequent H transfer step within the product complex was favored for protonated systems, generat- ing sulfinyl radical (–SO • ) and thiol (–SH) products. Because sulfenic acid is a potent scavenger of peroxyl radicals, our results implied that cysteine disulfide can form two lines of defense against reac- tive oxygen species, one using the cysteine disulfide itself and the other using the sulfenic acid product of the conversion of cysteine disulfide. This aspect suggested that, in a nonpolar environment, cysteine disulfides might play a more active role in the antioxidant network than previously appreciated. disulfide bond | hydroxyl radical | reaction intermediate | mass spectrometry | antioxidant T he dynamic formation and cleavage of a disulfide linkage between two cysteine amino acid residues has profound im- portance in cell biology, in particular for redox sensing, allosteric regulation of protein functions, and enzyme catalysis (1, 2). Equally important is the disulfide (–SS–)/thiol (–SH)/thiyl radical (–S • ) redox system of low–molecular-weight sulfur compounds, such as the redox pairs of glutathione and cysteine, the molar ratios of which directly impact redox homeostasis in cells or plasma (3–5). Three types of reactions have been identified to be responsible for chemical transformations between the –SS–, –SH, and –S • functional groups: nucleophilic substitution-eliminations, specifically so-called thiol–disulfide interchange reactions (6), single-electron reactions of free thiols or disulfide bonds (7), and radical reactions (8). When the cell is under oxidative stress, low–molecular-weight sulfur compounds work synergistically with other antioxidants (e.g., ascorbic acid and tocopherol) in the “antioxidant network” to combat elevated concentrations of re- active oxygen species (ROS) (9, 10). The hydroxyl radical ( • OH) is among the most oxidative species produced in aerobic systems, typically generated from superoxide anion (O 2 •− ) and H 2 O 2 by Fenton and/or Haber–Weiss reactions (7). Besides, peroxynitrous acid (ONOOH) readily decomposes to • OH and NO 2 • at neutral pH in biological systems, thus serving as an in situ source of • OH (11, 12). Direct interaction between • OH and the thiol functional group leads to H-atom transfer (13); thus-formed thiyl radicals are cycled in the –SS–/–SH/–S • system via chemical reactions or en- zymatic regulation (3). Although disulfide is not considered to be an efficient antioxidant, due to its limited reactivity toward peroxyl radicals (e.g., • OOH and O 2 •− ), the rate of its reaction with • OH is under diffusion control (10); thus, its conversion to other sulfur compounds directly impacts the steady state of –SS–/–SH redox system. It is believed that, in aqueous solutions, single-electron oxidation of the disulfide bond followed by bond cleavage into –S • and –S + is the dominant pathway by which • OH reacts with disulfide bonds (14, 15). The disulfide cleavage products can further react with O 2 , • OH, OH − , or other ROS, forming a series of oxyacids of sulfur, including sulfenic acid (–SOH), sulfinic acid (–SO 2 H), and sulfonic acid (–SO 3 H) (3). For the reactions in nonpolar environments, such as at the interface of lipid membrane and plasma or in the hydrophobic regions of protein tertiary structures, the role of cysteine disulfides is less well understood. More relevant information here can be gleaned from reactions of • OH with organic disulfides in the gas phase. In general, • OH substitution at the disulfide bond occurs more readily than any possible H abstraction or C–S bond cleavage in gas-phase reac- tions (16, 17). Due to disagreement on the identities of the first- generation reaction products, different elementary steps have been proposed for the mechanism by which the disulfide bond is cleaved. For instance, the earliest description of the mechanism Significance In this work, we harnessed mass spectrometry for online reac- tion monitoring of hydroxyl radical ( • OH) attack to cysteine disulfide and acquired mechanistic details that had not been achieved previously. Our findings suggest that • OH substitution at the disulfide bond is a fast and predominant reaction channel in the gas phase, while subsequent hydrogen transfer within the product pairs can be favorable in protonated systems. Notably, by reacting with • OH cysteine disulfide converts itself to a more potent antioxidant, sulfenic acid (–SOH), thus forming two lines of defense against reactive oxygen species (ROS). These results provide insight into studying the antioxidant roles of cysteine disulfide in nonpolar biological environment, such as at the in- terface of lipid membrane and plasma. Author contributions: J.M.A., J.S.F., and Y.X. designed research; S.A., R.C., J.M.A., J.S.F., and Y.X. performed research; S.A., R.C., J.M.A., J.S.F., and Y.X. analyzed data; and S.A., R.C., J.M.A., J.S.F., and Y.X. wrote the paper. Reviewers: R.A.O., University of Melbourne; and H.F.S., University of Georgia. The authors declare no competing interest. Published under the PNAS license. 1 To whom correspondence may be addressed. Email: [email protected], frjoseph@sas. upenn.edu, or [email protected]. This article contains supporting information online at https://www.pnas.org/lookup/suppl/ doi:10.1073/pnas.2006639117/-/DCSupplemental. First published July 17, 2020. 18216–18223 | PNAS | August 4, 2020 | vol. 117 | no. 31 www.pnas.org/cgi/doi/10.1073/pnas.2006639117 Downloaded by guest on December 21, 2021

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Two-step reaction mechanism reveals new antioxidantcapability of cysteine disulfides against hydroxylradical attackSarju Adhikaria, Ramon Crehuetb, Josep M. Angladab,1, Joseph S. Franciscoc,1, and Yu Xiaa,d,1
aDepartment of Chemistry, Purdue University, West Lafayette, IN 47907; bDepartment of Biological Chemistry, Institute of Advanced Chemistry ofCatalonia, IQAC-CSIC, E-08034 Barcelona, Spain; cDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104; and dDepartment ofChemistry, Tsinghua University, Beijing 100084, China
Contributed by Joseph S. Francisco, May 30, 2020 (sent for review April 9, 2020; reviewed by Richard A. O’Hair and Henry F. Schaefer III)
Cysteine disulfides, which constitute an important component inbiological redox buffer systems, are highly reactive toward thehydroxyl radical (•OH). The mechanistic details of this reaction, how-ever, remain unclear, largely due to the difficulty in characterizingunstable reaction products. Herein, we have developed a combinedapproach involving mass spectrometry (MS) and theoretical calcula-tions to investigate reactions of •OH with cysteine disulfides (Cys–S–S–R) in the gas phase. Four types of first-generation productswere identified: protonated ions of the cysteine thiyl radical (+Cys–S•), cysteine (+Cys–SH), cysteine sulfinyl radical (+Cys–SO•), and cys-teine sulfenic acid (+Cys–SOH). The relative reaction rates and prod-uct branching ratios responded sensitively to the electronic propertyof the R group, providing key evidence to deriving a two-step reac-tion mechanism. The first step involved •OH conducting a back-sideattack on one of the sulfur atoms, forming sulfenic acid (–SOH) andthiyl radical (–S•) product pairs. A subsequent H transfer step withinthe product complex was favored for protonated systems, generat-ing sulfinyl radical (–SO•) and thiol (–SH) products. Because sulfenicacid is a potent scavenger of peroxyl radicals, our results impliedthat cysteine disulfide can form two lines of defense against reac-tive oxygen species, one using the cysteine disulfide itself and theother using the sulfenic acid product of the conversion of cysteinedisulfide. This aspect suggested that, in a nonpolar environment,cysteine disulfides might play a more active role in the antioxidantnetwork than previously appreciated.
disulfide bond | hydroxyl radical | reaction intermediate |mass spectrometry | antioxidant
The dynamic formation and cleavage of a disulfide linkagebetween two cysteine amino acid residues has profound im-
portance in cell biology, in particular for redox sensing, allostericregulation of protein functions, and enzyme catalysis (1, 2).Equally important is the disulfide (–SS–)/thiol (–SH)/thiyl radical(–S•) redox system of low–molecular-weight sulfur compounds,such as the redox pairs of glutathione and cysteine, the molarratios of which directly impact redox homeostasis in cells orplasma (3–5). Three types of reactions have been identified to beresponsible for chemical transformations between the –SS–, –SH,and –S• functional groups: nucleophilic substitution-eliminations,specifically so-called thiol–disulfide interchange reactions (6),single-electron reactions of free thiols or disulfide bonds (7), andradical reactions (8). When the cell is under oxidative stress,low–molecular-weight sulfur compounds work synergistically withother antioxidants (e.g., ascorbic acid and tocopherol) in the“antioxidant network” to combat elevated concentrations of re-active oxygen species (ROS) (9, 10). The hydroxyl radical (•OH) isamong the most oxidative species produced in aerobic systems,typically generated from superoxide anion (O2
•−) and H2O2 byFenton and/or Haber–Weiss reactions (7). Besides, peroxynitrousacid (ONOOH) readily decomposes to •OH and NO2
• at neutralpH in biological systems, thus serving as an in situ source of •OH(11, 12). Direct interaction between •OH and the thiol functional
group leads to H-atom transfer (13); thus-formed thiyl radicals arecycled in the –SS–/–SH/–S• system via chemical reactions or en-zymatic regulation (3). Although disulfide is not considered to bean efficient antioxidant, due to its limited reactivity toward peroxylradicals (e.g., •OOH and O2
•−), the rate of its reaction with •OHis under diffusion control (10); thus, its conversion to other sulfurcompounds directly impacts the steady state of –SS–/–SH redoxsystem. It is believed that, in aqueous solutions, single-electronoxidation of the disulfide bond followed by bond cleavage into–S• and –S+ is the dominant pathway by which •OH reacts withdisulfide bonds (14, 15). The disulfide cleavage products canfurther react with O2,
•OH, OH−, or other ROS, forming a seriesof oxyacids of sulfur, including sulfenic acid (–SOH), sulfinic acid(–SO2H), and sulfonic acid (–SO3H) (3). For the reactions innonpolar environments, such as at the interface of lipid membraneand plasma or in the hydrophobic regions of protein tertiarystructures, the role of cysteine disulfides is less well understood.More relevant information here can be gleaned from reactions of•OH with organic disulfides in the gas phase. In general, •OHsubstitution at the disulfide bond occurs more readily than anypossible H abstraction or C–S bond cleavage in gas-phase reac-tions (16, 17). Due to disagreement on the identities of the first-generation reaction products, different elementary steps havebeen proposed for the mechanism by which the disulfide bond iscleaved. For instance, the earliest description of the mechanism
Significance
In this work, we harnessed mass spectrometry for online reac-tion monitoring of hydroxyl radical (•OH) attack to cysteinedisulfide and acquired mechanistic details that had not beenachieved previously. Our findings suggest that •OH substitutionat the disulfide bond is a fast and predominant reaction channelin the gas phase, while subsequent hydrogen transfer within theproduct pairs can be favorable in protonated systems. Notably,by reacting with •OH cysteine disulfide converts itself to a morepotent antioxidant, sulfenic acid (–SOH), thus forming two linesof defense against reactive oxygen species (ROS). These resultsprovide insight into studying the antioxidant roles of cysteinedisulfide in nonpolar biological environment, such as at the in-terface of lipid membrane and plasma.
Author contributions: J.M.A., J.S.F., and Y.X. designed research; S.A., R.C., J.M.A., J.S.F.,and Y.X. performed research; S.A., R.C., J.M.A., J.S.F., and Y.X. analyzed data; and S.A.,R.C., J.M.A., J.S.F., and Y.X. wrote the paper.
Reviewers: R.A.O., University of Melbourne; and H.F.S., University of Georgia.
The authors declare no competing interest.
Published under the PNAS license.1To whom correspondence may be addressed. Email: [email protected], [email protected], or [email protected].
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2006639117/-/DCSupplemental.
First published July 17, 2020.
18216–18223 | PNAS | August 4, 2020 | vol. 117 | no. 31 www.pnas.org/cgi/doi/10.1073/pnas.2006639117
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1

described a relatively simple picture, involving addition of •OHonto the disulfide bond in R–S–S–R followed by rapid scission ofthe disulfide bond into RS• and RSOH (18). Butkovskaya andSetser (19) later detected two product pairs: CH3SH/CH3SO
• andCH3S
•/CH3SOH, with the former pair being found more favor-able from reactions of dimethyl disulfide with •OH. Bil et al.suggested H atom transfer in the product complex [CH3SOH–•SCH3] to be a key step to account for the detection of CH3SO
•/HSCH3 (20). However, the product pair CH3S
•/CH3SOH wasnot detected and thus ruled out in their mechanism. Obviously,the difficulties in identifying and quantifying the reactive inter-mediates have constituted a major hurdle for mechanistic studiesof radical reactions.While electron paramagnetic resonance and other spectro-
scopic methods are useful tools for detecting radical species (21),mass spectrometry (MS), which offers a distinct advantage inproviding detailed molecular information, is increasingly beingused to investigate the structure and reactivity of bio-radical ionsin the gas phase (22–25). The recently developed electrosprayionization (ESI)–MS and direct MS for online reaction moni-toring have provided additional powerful tools for elucidatingtransient radical species in solution reactions (26–29). Given theimportance of –SS–/–SH/–S• cycle in biology, we previously de-veloped an MS method to synthesize and characterize cysteinethiyl (Cys–S•), perthiyl (Cys–SS•), and sulfinyl (Cys–SO•) radicalsin the gas phase (30–32). The method is based on performingradical reactions in the plume region of a nanoelectrospray ioni-zation (nano-ESI) source right before the sampling interface of amass spectrometer. Such a setup provides a short sampling time(submicrosecond to microsecond) so that bio-radicals can be de-tected and subsequently characterized using MS. For instance, twopairs of reaction products, pep–S•/pep–SOH and pep–SH/pep–SO• resulting from disulfide bond cleavage in peptides (pep–S–S–pep), have been identified from reactions with •OH (33). Theseresults were consistent with the formation of two pairs of productsupon disulfide bond cleavage from gas-phase studies and alsosuggested the feasibility of using MS to monitor the presence ofsulfur radicals during reactions.Leveraging the detailed molecular information that MS can
provide for radical reactions, in the current work we aimed todelineate the mechanism of the reaction of •OH with the cysteinedisulfide bond in the gas phase. Gaining this type of knowledge isimportant for understanding the intrinsic reactivity of the disulfidebond, and it is relevant to reactions occurring in nonpolar mi-croenvironments of biological systems. A series of cysteine disul-fide derivatives was investigated (Cys–S–S–R, structure shown inScheme 1) using a combined experimental and theoretical ap-proach. The electronic character of the disulfide bond was mod-ulated by changing the inductive effect of the connecting R group.For MS experiments, we focused on optimizing the formation ofthe first-generation products and performing a cross-group com-parison of the reaction reactivities resulting from the R substitu-ents. We were interested in describing the effect of the electroniccharacter of the disulfide bond on its reactivity and the productbranching ratio. High-level theoretical calculations were carriedout to provide detailed descriptions of the thermodynamics andkinetics of the reactions. A two-step reaction mechanism wasderived, with this mechanism involving rapid •OH substitution oneither sulfur atom, leading to cleavage of the disulfide bond andformation of Cys–SOH/R–S• and Cys–S•/R–SOH. For protonatedreaction systems, subsequent transfer of H within the productcomplex was indicated to be a significant process, and to be assistedby a stabilizing effect of hydrogen bonding in the transition states.
Results and DiscussionReaction Phenomena of •OH and Cys–S–S–R. Experimental detailsfor conducting the reactions between •OH and Cys–S–S–R in theplume region of a nano-ESI source have been reported previously
and are supplied in SI Appendix. In brief, solutions containingequimolar amounts of the model compound and the internalstandard (O-ethylated cystine, I–S–S–I, 10 μM each in water) wereionized using nano-ESI in positive ion mode and subjected toonline reactions with •OH radicals produced by performing di-electric barrier discharge of helium in ambient air (SI Appendix,Fig. S1) (31). In this study, we chose to perform reactions inpositive ion mode because higher ionization efficiency for Cys–S–S–R was obtained than that from negative ion mode and theinternal standard could not be ionized in negative ion mode. Itshould be noted that similar reaction phenomena of •OH attack tocysteinyl disulfide bond were observed regardless of the ion po-larity (33). Although minor amounts of reactive species such as Hatoms, O atoms, singlet oxygen, and ozone were also expected tohave formed under the discharge conditions (34), their contribu-tions to the observed reaction phenomena were expected to beinsignificant. This expectation was due to a couple of factors: Hand O atoms having short (nanosecond-scale) lifetimes and beingquickly converted to •OH upon collisions with water molecules(35), and ozone (36) or singlet oxygen (37) reacting very sluggishlywith cystine disulfide. According to the ion evaporation model,developed to explain the ionization of small molecules by ESI(38), cysteine disulfides should most likely be in the form ofprotonated ions when encountering •OH. Note that because ofthe lack of basic functional groups for protonation in the tested Rgroups (except for R = cysteine), the disulfide cleavage productsinvolving the R side chain, viz. R–S•, R–SH, R–SO•, and R–SOH,could not be detected using MS. For this reason, the same types ofionic products were expected to be observed from the disulfidecleavages of all of the tested Cys–S–S–R compounds, albeit withperhaps different relative abundances of these ionic products dueto the impact of the R substituent. Fig. 1A summarizes theexpected main reaction products from •OH attack on differentsulfur atoms in the disulfide bond. The attack of •OH on the
Scheme 1. Online MS monitoring of reactions of OH• and cysteinyl disul-fides (Cys–S–S–R) for various R groups with different electronic properties.The Cys–S–S–R molecules became protonated at the amine group (+Cys–S–S–R) when ionized using nano-ESI in positive ion mode.
Adhikari et al. PNAS | August 4, 2020 | vol. 117 | no. 31 | 18217
CHEM
ISTR
Y
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1
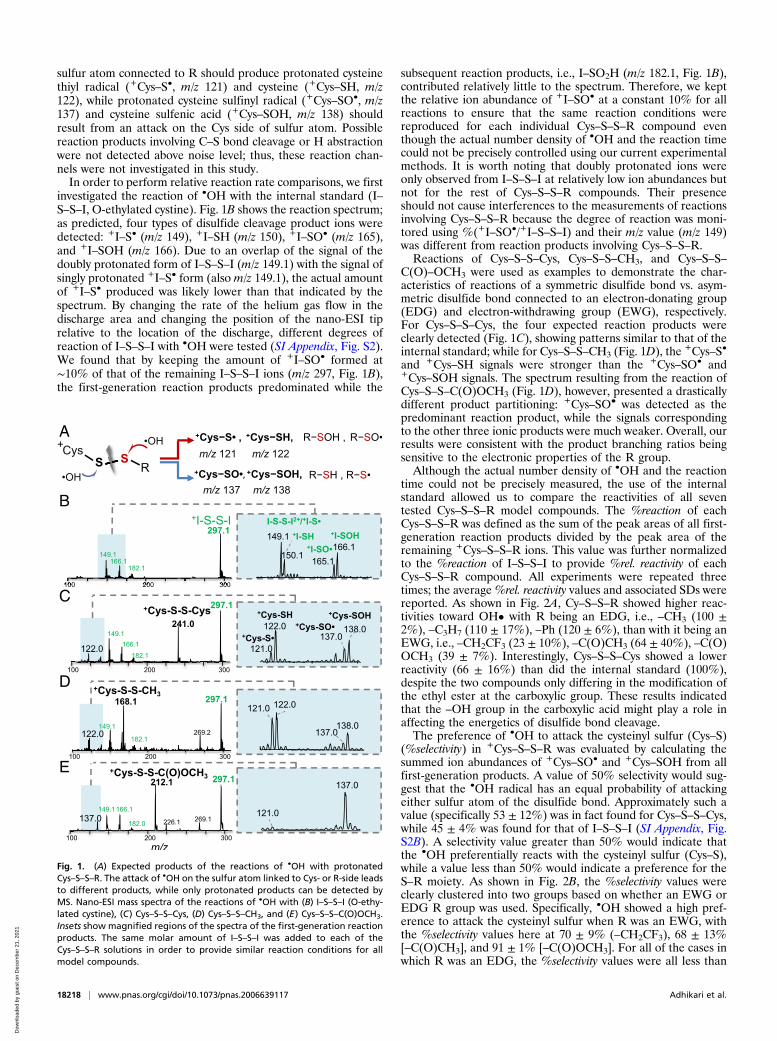
sulfur atom connected to R should produce protonated cysteinethiyl radical (+Cys–S•, m/z 121) and cysteine (+Cys–SH, m/z122), while protonated cysteine sulfinyl radical (+Cys–SO•, m/z137) and cysteine sulfenic acid (+Cys–SOH, m/z 138) shouldresult from an attack on the Cys side of sulfur atom. Possiblereaction products involving C–S bond cleavage or H abstractionwere not detected above noise level; thus, these reaction chan-nels were not investigated in this study.In order to perform relative reaction rate comparisons, we first
investigated the reaction of •OH with the internal standard (I–S–S–I, O-ethylated cystine). Fig. 1B shows the reaction spectrum;as predicted, four types of disulfide cleavage product ions weredetected: +I–S• (m/z 149), +I–SH (m/z 150), +I–SO• (m/z 165),and +I–SOH (m/z 166). Due to an overlap of the signal of thedoubly protonated form of I–S–S–I (m/z 149.1) with the signal ofsingly protonated +I–S• form (also m/z 149.1), the actual amountof +I–S• produced was likely lower than that indicated by thespectrum. By changing the rate of the helium gas flow in thedischarge area and changing the position of the nano-ESI tiprelative to the location of the discharge, different degrees ofreaction of I–S–S–I with •OH were tested (SI Appendix, Fig. S2).We found that by keeping the amount of +I–SO• formed at∼10% of that of the remaining I–S–S–I ions (m/z 297, Fig. 1B),the first-generation reaction products predominated while the
subsequent reaction products, i.e., I–SO2H (m/z 182.1, Fig. 1B),contributed relatively little to the spectrum. Therefore, we keptthe relative ion abundance of +I–SO• at a constant 10% for allreactions to ensure that the same reaction conditions werereproduced for each individual Cys–S–S–R compound eventhough the actual number density of •OH and the reaction timecould not be precisely controlled using our current experimentalmethods. It is worth noting that doubly protonated ions wereonly observed from I–S–S–I at relatively low ion abundances butnot for the rest of Cys–S–S–R compounds. Their presenceshould not cause interferences to the measurements of reactionsinvolving Cys–S–S–R because the degree of reaction was moni-tored using %(+I–SO•/+I–S–S–I) and their m/z value (m/z 149)was different from reaction products involving Cys–S–S–R.Reactions of Cys–S–S–Cys, Cys–S–S–CH3, and Cys–S–S–
C(O)–OCH3 were used as examples to demonstrate the char-acteristics of reactions of a symmetric disulfide bond vs. asym-metric disulfide bond connected to an electron-donating group(EDG) and electron-withdrawing group (EWG), respectively.For Cys–S–S–Cys, the four expected reaction products wereclearly detected (Fig. 1C), showing patterns similar to that of theinternal standard; while for Cys–S–S–CH3 (Fig. 1D), the +Cys–S•
and +Cys–SH signals were stronger than the +Cys–SO• and+Cys–SOH signals. The spectrum resulting from the reaction ofCys–S–S–C(O)OCH3 (Fig. 1D), however, presented a drasticallydifferent product partitioning: +Cys–SO• was detected as thepredominant reaction product, while the signals correspondingto the other three ionic products were much weaker. Overall, ourresults were consistent with the product branching ratios beingsensitive to the electronic properties of the R group.Although the actual number density of •OH and the reaction
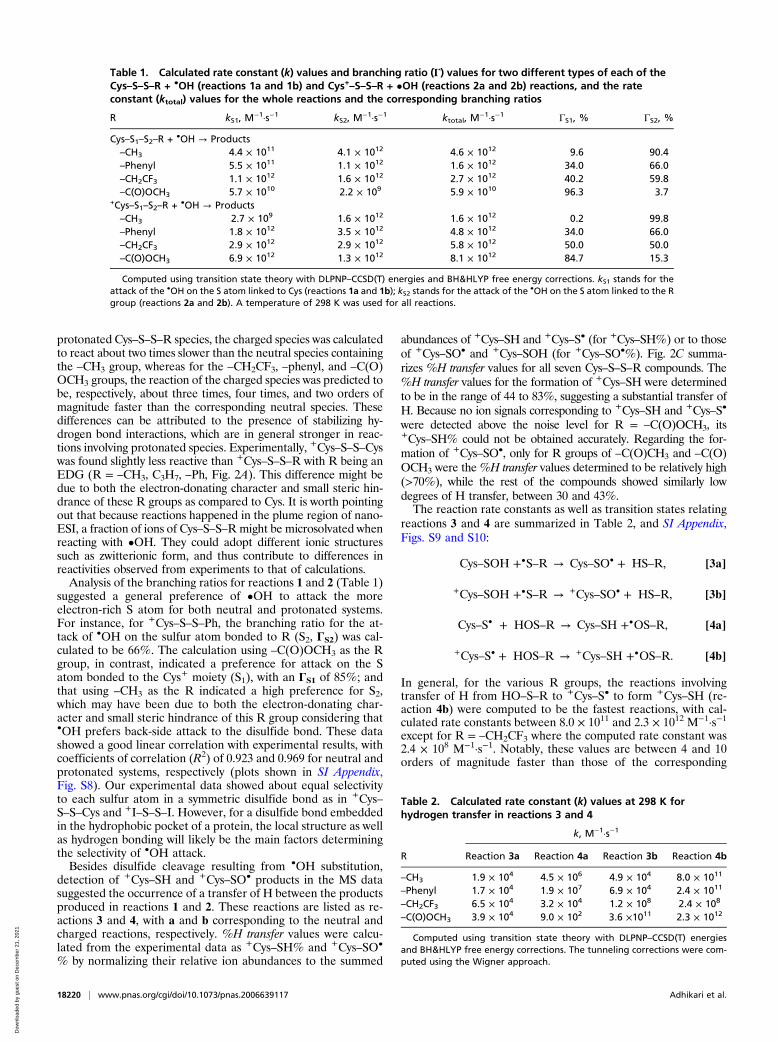
time could not be precisely measured, the use of the internalstandard allowed us to compare the reactivities of all seventested Cys–S–S–R model compounds. The %reaction of eachCys–S–S–R was defined as the sum of the peak areas of all first-generation reaction products divided by the peak area of theremaining +Cys–S–S–R ions. This value was further normalizedto the %reaction of I–S–S–I to provide %rel. reactivity of eachCys–S–S–R compound. All experiments were repeated threetimes; the average%rel. reactivity values and associated SDs werereported. As shown in Fig. 2A, Cy–S–S–R showed higher reac-tivities toward OH• with R being an EDG, i.e., –CH3 (100 ±2%), –C3H7 (110 ± 17%), –Ph (120 ± 6%), than with it being anEWG, i.e., –CH2CF3 (23 ± 10%), –C(O)CH3 (64 ± 40%), –C(O)OCH3 (39 ± 7%). Interestingly, Cys–S–S–Cys showed a lowerreactivity (66 ± 16%) than did the internal standard (100%),despite the two compounds only differing in the modification ofthe ethyl ester at the carboxylic group. These results indicatedthat the –OH group in the carboxylic acid might play a role inaffecting the energetics of disulfide bond cleavage.The preference of •OH to attack the cysteinyl sulfur (Cys–S)
(%selectivity) in +Cys–S–S–R was evaluated by calculating thesummed ion abundances of +Cys–SO• and +Cys–SOH from allfirst-generation products. A value of 50% selectivity would sug-gest that the •OH radical has an equal probability of attackingeither sulfur atom of the disulfide bond. Approximately such avalue (specifically 53 ± 12%) was in fact found for Cys–S–S–Cys,while 45 ± 4% was found for that of I–S–S–I (SI Appendix, Fig.S2B). A selectivity value greater than 50% would indicate thatthe •OH preferentially reacts with the cysteinyl sulfur (Cys–S),while a value less than 50% would indicate a preference for theS–R moiety. As shown in Fig. 2B, the %selectivity values wereclearly clustered into two groups based on whether an EWG orEDG R group was used. Specifically, •OH showed a high pref-erence to attack the cysteinyl sulfur when R was an EWG, withthe %selectivity values here at 70 ± 9% (–CH2CF3), 68 ± 13%[–C(O)CH3], and 91 ± 1% [–C(O)OCH3]. For all of the cases inwhich R was an EDG, the %selectivity values were all less than
E
D
C
B
A
Fig. 1. (A) Expected products of the reactions of •OH with protonatedCys–S–S–R. The attack of •OH on the sulfur atom linked to Cys- or R-side leadsto different products, while only protonated products can be detected byMS. Nano-ESI mass spectra of the reactions of •OH with (B) I–S–S–I (O-ethy-lated cystine), (C) Cys–S–S–Cys, (D) Cys–S–S–CH3, and (E) Cys–S–S–C(O)OCH3.Insets show magnified regions of the spectra of the first-generation reactionproducts. The same molar amount of I–S–S–I was added to each of theCys–S–S–R solutions in order to provide similar reaction conditions for allmodel compounds.
18218 | www.pnas.org/cgi/doi/10.1073/pnas.2006639117 Adhikari et al.
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1

50%, viz. 26 ± 5% for –CH3, 38 ± 5% for –C3H7, and 40 ± 3%for –Ph.
Theoretical Calculations. In order to rationalize these MS results,we carried out a series of theoretical calculations. These com-putational methods consisted of a combination of the molecularmechanics (MM) methods, ab initio density functional theorywith the BH&HLYP functional, and domain-based local pair-natural orbital coupled cluster including perturbative triplet ex-citation methods [DLPNO–CCSD(T)]. The kinetics calculationswere carried out in the framework of multiconformer transitionstate theory (MC-TST). The following elemental reactions wereenvisaged as the first steps of the disulfide bond cleavage:
Cys–S–S–R +•OH → Cys–SOH +•S–R, [1a]
Cys–S–S–R +•OH → Cys–S• + HOS–R. [2a]
We also considered protonated species (+Cys–S–S–R) in thereactions, with the proton bound to the primary amine of thecysteine moiety:
+Cys–S–S–R +•OH → +Cys–SOH +•S–R, [1b]
+Cys–S–S–R +•OH → +Cys–S• + HOS–R. [2b]
Regarding R substituents, –CH3 and –phenyl were chosen torepresent EDG groups, while –CH2CF3 and –C(O)OCH3 wereused as examples of weak and strong EWG groups, respectively.Previous reports showed that a carbon-centered radical or Hatom can attack either of the sulfur atoms in a disulfide bond,following either a back-side mechanism (with an attacking anglebetween 140° and 180°) or a front-side mechanism (∼90° attack-ing angle), but with the back-side attack being more favorable(39–42). The results of our calculations indicated the back-side
reaction mechanism to be the predominant pathway for bothneutral and protonated Cys–S–S–R compounds (SI Appendix,Figs. S3–S7). The distance between the approaching oxygenatom and the attacked sulfur atom was indicated from our cal-culations to, in general, range between 1.82 and 2.17 Å, and theOSS angle generally ranged between 132° and 155°, similar to thereaction of •OH with hydropersulfides (41). In addition, intra-molecular hydrogen bonds were indicated to play a role in sta-bilizing the transition states. Shorter hydrogen bonds, indicativeof stronger bonds, were calculated for the protonated species thanfor the corresponding neutral species. For example, the N–H. . .Obond in the low-lying transition state of reaction 2b of +Cys–S–S–CH3 showed a length of 1.794 Å (Fig. 3A), while O–H. . .O showeda length of 1.867 Å in that of 2a of Cys–S–S–CH3 (Fig. 3B).A new reaction channel, involving proton-coupled electron
transfer (PCET), was found to be competitive for the protonatedspecies, and to lead to the formation of [Cys–S–S–R] •+:
Cys+–S–S–R +•OH → [Cys–S–S–R]•+ + H2O (PCET).The calculations indicated a PCET branching ratio between 22%and 32%, but also indicated further dissociation at the S–S bondor S–R bond to not be energetically favorable. In addition, ionscorresponding to [Cys–S–S–R]•+ were not detected experien-tially. Therefore, this process was concluded to not contributesignificantly to disulfide bond cleavage.Table 1 summarizes kinetics data together with branching ra-
tios for reactions 1 and 2 from the calculations. When the neutralcysteine disulfides were considered, the summed rate constants fordisulfide cleavage (ktotal) ranged between 5.9 × 1010 and 4.2 × 1012
M−1·s−1, with R groups of –C(O)OCH3 and –CH3 yielding theslowest and fastest reactions, respectively. For reactions involving
A
B
C
Fig. 2. (A) %Rel. reactivity values of Cys–S–S–R toward •OH, i.e., normalizedto I–S–S–I. The Cys–S–S–R compound exhibited higher reactivities with –Rbeing an EDG. (B) %Selectivity values of OH attack on the Cys–S bond rel-ative to that on the S–R bond within Cys–S–S–R. (C) Values of %H transferleading to the formation of +Cys–SO• and +Cys–SH, respectively.
A B
C D
E F
Fig. 3. Relevant geometrical parameters for the low-lying transition statesof (A) reaction 2b for R = –CH3, (B) reaction 2a for R = –CH3, (C) reaction 3bfor R = –CH3, (D) reaction 3b for R = –C(O)OCH3, (E) reaction 4b for R = –CH3,and (F) reaction 4b for R = –C(O)OCH3. Distances are in angstroms and anglesin degrees.
Adhikari et al. PNAS | August 4, 2020 | vol. 117 | no. 31 | 18219
CHEM
ISTR
Y
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1
protonated Cys–S–S–R species, the charged species was calculatedto react about two times slower than the neutral species containingthe –CH3 group, whereas for the –CH2CF3, –phenyl, and –C(O)OCH3 groups, the reaction of the charged species was predicted tobe, respectively, about three times, four times, and two orders ofmagnitude faster than the corresponding neutral species. Thesedifferences can be attributed to the presence of stabilizing hy-drogen bond interactions, which are in general stronger in reac-tions involving protonated species. Experimentally, +Cys–S–S–Cyswas found slightly less reactive than +Cys–S–S–R with R being anEDG (R = –CH3, C3H7, –Ph, Fig. 2A). This difference might bedue to both the electron-donating character and small steric hin-drance of these R groups as compared to Cys. It is worth pointingout that because reactions happened in the plume region of nano-ESI, a fraction of ions of Cys–S–S–Rmight be microsolvated whenreacting with •OH. They could adopt different ionic structuressuch as zwitterionic form, and thus contribute to differences inreactivities observed from experiments to that of calculations.Analysis of the branching ratios for reactions 1 and 2 (Table 1)
suggested a general preference of •OH to attack the moreelectron-rich S atom for both neutral and protonated systems.For instance, for +Cys–S–S–Ph, the branching ratio for the at-tack of •OH on the sulfur atom bonded to R (S2, ΓS2) was cal-culated to be 66%. The calculation using –C(O)OCH3 as the Rgroup, in contrast, indicated a preference for attack on the Satom bonded to the Cys+ moiety (S1), with an ΓS1 of 85%; andthat using –CH3 as the R indicated a high preference for S2,which may have been due to both the electron-donating char-acter and small steric hindrance of this R group considering that•OH prefers back-side attack to the disulfide bond. These datashowed a good linear correlation with experimental results, withcoefficients of correlation (R2) of 0.923 and 0.969 for neutral andprotonated systems, respectively (plots shown in SI Appendix,Fig. S8). Our experimental data showed about equal selectivityto each sulfur atom in a symmetric disulfide bond as in +Cys–S–S–Cys and +I–S–S–I. However, for a disulfide bond embeddedin the hydrophobic pocket of a protein, the local structure as wellas hydrogen bonding will likely be the main factors determiningthe selectivity of •OH attack.Besides disulfide cleavage resulting from •OH substitution,
detection of +Cys–SH and +Cys–SO• products in the MS datasuggested the occurrence of a transfer of H between the productsproduced in reactions 1 and 2. These reactions are listed as re-actions 3 and 4, with a and b corresponding to the neutral andcharged reactions, respectively. %H transfer values were calcu-lated from the experimental data as +Cys–SH% and +Cys–SO•
% by normalizing their relative ion abundances to the summed
abundances of +Cys–SH and +Cys–S• (for +Cys–SH%) or to thoseof +Cys–SO• and +Cys–SOH (for +Cys–SO•%). Fig. 2C summa-rizes %H transfer values for all seven Cys–S–S–R compounds. The%H transfer values for the formation of +Cys–SH were determinedto be in the range of 44 to 83%, suggesting a substantial transfer ofH. Because no ion signals corresponding to +Cys–SH and +Cys–S•
were detected above the noise level for R = –C(O)OCH3, its+Cys–SH% could not be obtained accurately. Regarding the for-mation of +Cys–SO•, only for R groups of –C(O)CH3 and –C(O)OCH3 were the%H transfer values determined to be relatively high(>70%), while the rest of the compounds showed similarly lowdegrees of H transfer, between 30 and 43%.The reaction rate constants as well as transition states relating
reactions 3 and 4 are summarized in Table 2, and SI Appendix,Figs. S9 and S10:
Cys–SOH +•S–R → Cys–SO• + HS–R, [3a]
+Cys–SOH +•S–R → +Cys–SO• + HS–R, [3b]
Cys–S• + HOS–R → Cys–SH +•OS–R, [4a]
+Cys–S• + HOS–R → +Cys–SH +•OS–R. [4b]
In general, for the various R groups, the reactions involvingtransfer of H from HO–S–R to +Cys–S• to form +Cys–SH (re-action 4b) were computed to be the fastest reactions, with cal-culated rate constants between 8.0 × 1011 and 2.3 × 1012 M−1·s−1
except for R = –CH2CF3 where the computed rate constant was2.4 × 108 M−1·s−1. Notably, these values are between 4 and 10orders of magnitude faster than those of the corresponding
Table 1. Calculated rate constant (k) values and branching ratio (Γ) values for two different types of each of theCys–S–S–R + •OH (reactions 1a and 1b) and Cys+–S–S–R + •OH (reactions 2a and 2b) reactions, and the rateconstant (ktotal) values for the whole reactions and the corresponding branching ratios
R kS1, M−1·s−1 kS2, M
−1·s−1 ktotal, M−1·s−1 ΓS1, % ΓS2, %
Cys–S1–S2–R + •OH → Products–CH3 4.4 × 1011 4.1 × 1012 4.6 × 1012 9.6 90.4–Phenyl 5.5 × 1011 1.1 × 1012 1.6 × 1012 34.0 66.0–CH2CF3 1.1 × 1012 1.6 × 1012 2.7 × 1012 40.2 59.8–C(O)OCH3 5.7 × 1010 2.2 × 109 5.9 × 1010 96.3 3.7
+Cys–S1–S2–R + •OH → Products–CH3 2.7 × 109 1.6 × 1012 1.6 × 1012 0.2 99.8–Phenyl 1.8 × 1012 3.5 × 1012 4.8 × 1012 34.0 66.0–CH2CF3 2.9 × 1012 2.9 × 1012 5.8 × 1012 50.0 50.0–C(O)OCH3 6.9 × 1012 1.3 × 1012 8.1 × 1012 84.7 15.3
Computed using transition state theory with DLPNP–CCSD(T) energies and BH&HLYP free energy corrections. kS1 stands for theattack of the •OH on the S atom linked to Cys (reactions 1a and 1b); kS2 stands for the attack of the •OH on the S atom linked to the Rgroup (reactions 2a and 2b). A temperature of 298 K was used for all reactions.
Table 2. Calculated rate constant (k) values at 298 K forhydrogen transfer in reactions 3 and 4
k, M−1·s−1
R Reaction 3a Reaction 4a Reaction 3b Reaction 4b
–CH3 1.9 × 104 4.5 × 106 4.9 × 104 8.0 × 1011
–Phenyl 1.7 × 104 1.9 × 107 6.9 × 104 2.4 × 1011
–CH2CF3 6.5 × 104 3.2 × 104 1.2 × 108 2.4 × 108
–C(O)OCH3 3.9 × 104 9.0 × 102 3.6 ×1011 2.3 × 1012
Computed using transition state theory with DLPNP–CCSD(T) energiesand BH&HLYP free energy corrections. The tunneling corrections were com-puted using the Wigner approach.
18220 | www.pnas.org/cgi/doi/10.1073/pnas.2006639117 Adhikari et al.
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1

neutral compounds (reaction 4a, Table 2). Analysis of these datasuggested that protonated ions were likely responsible for thereaction products detected experimentally; moreover, the analy-ses indicated that formation of +Cys–SH should be detectable onthe same timescale as disulfide bond cleavage (reactions 1 and 2,Table 1). In addition, these high values of the rate constants maybe attributed to the combination of two factors, namely thestrength of the hydrogen bond interaction, between the proton-ated amine and the oxygen atom of the carbonyl group when R =–C(O)OCH3 (1.745 Å, Fig. 3F), and the early transition stateaccording the Hammond postulate, as in the case of reaction4b for R = –CH3, where the S–H distance of the H atom beingtransferred is quite large (1.736 Å, Fig. 3E). The differences inthe reactivity between compounds with different substitute inthis reaction are mainly due to different contribution of thesetwo effects. However, the reason for relatively low k4b associatedwith R = –CH2CF3 was not fully understood. The above-described predictions made were in good accordance with thesubstantial%H transfer observed for +Cys–SH formation accord-ing to the MS data (Fig. 2C). For reaction 3b, analysis of theresults of calculations predicted quite scattered values of the rateconstants, with a k value of 4.9 × 104 M−1·s−1 for R = –CH3 and3.6 × 1011 M−1·s−1 for R = –C(O)OCH3. These data were con-sistent with the experimental observations, in particular the high-est %H transfer observed for R = –C(O)OCH3 (88%, Fig. 2C).However, explaining such a large difference between the %Htransfer values of the different tested compounds by solely refer-ring to the different inductive effects of their R substituentswould be at odds with a previous study indicating a lack of anystrong effect of the identity of R of alkylthiols (RS–H) on theS–H bond dissociation energy (RS–H BDE: ∼360 kJ/mol) (43).Analysis of the low-lying transitions states of reaction 3b sug-gested an important role played by hydrogen bond interactionsin stabilizing the transition state, thus favoring the formation of+Cys–SO•. For instance, the hydrogen bond between the proton-ated amine and the carbonyl group was calculated to be 1.6 Å forthe compound containing an R of –C(O)OCH3 (an early transi-tion state, Fig. 3C), much shorter than the hydrogen bond inH2N–H ···S–CH3 (2.2 Å), with its R being –CH3 (Fig. 3D). Wenoticed that for Cys–S–S–Cys and I–S–S–I, which contained asymmetric disulfide bond, the detected %H transfer values werenot 50%. In fact, H transfer leading to the formation of +Cys–SH(70%) or +I–SH (71%, SI Appendix, Fig. S2B) was higher thanthat of +Cys–SO• (43%) or +I–S• (34%, SI Appendix, Fig. S2B).This is because the disulfide bond is no longer symmetrical inthese two compounds given that the proton can only reside onone side of the molecule. H transfer from +Cys–SOH to Cys–S•
leads to the formation of +Cys–SO• (reaction 3b), while H trans-fer from Cys–SOH to +Cys–S• leads to the formation of +Cys–SH (reaction 4b). Using the calculation results from R = –CH3 inCys–S–S–R (Table 2) as a simplified estimation for Cys–S–S–Cys, it is clear that reaction 4b is significantly enhanced relativeto reaction 3b when compared to their corresponding neutralreactions (reactions 4a and 3a). This difference might lead tothe higher %H transfer values for forming +Cys–SH or +I–SHthan +Cys–SO• or +I–SO• (SI Appendix, Scheme S2). Hydrogenbond interactions discovered in the cysteinyl disulfide systemalso presented a distinct difference from the calculated resultsof •OH reactions with small organic disulfides (20, 41).Combining the accumulated pieces of evidence from the ex-
perimental and computational studies, we derived a two-step re-action mechanism to account for •OH attack on cysteine disulfides(Scheme 2). According to this proposed mechanism, in the firststep, •OH performs a back-side attack on one of the sulfur atoms,with a preference for the electron-richer one—with this attackleading to •OH substitution and subsequent disulfide bond cleav-age and hence forming the sulfenic acid (–SOH) and thiyl radical(–S•) pair of products. Note that hydroxyl radical substitution was
indicated to be rapid for most neutral cysteine disulfides, with rateconstants on the order of 1012 M−1·s−1, but about two orders ofmagnitude slower for R being an EWG. For protonated com-pounds, however, the rate constant of •OH substitution on adisulfide bond connected to an EWG was indicated to be increasedto the same level as for R being an EDG, due to the stabilizingeffect of hydrogen bonding for the low-lying transition states. Hy-drogen bonding was also modeled to help stabilize the productcomplex and further facilitate rapid H transfer before productseparation in protonated systems (reaction 4b), explaining theobservation of +Cys–SO• and +Cys–SH in the MS experiments.Analysis of our calculation results suggested that H transfer wouldnot predominate for neutral species on the detection timescaleused in this study.
SummaryWe have developed a combined experimental and theoreticalapproach to study reactions of the •OH radical with a series ofcysteine disulfides (Cys–S–S–R) in the gas phase. The ability touse MS to perform online identification and relative quantitationof first-generation reaction products provided mechanistic de-tails that had not been achieved for this reaction previously. Atwo-step reaction mechanism has been proposed. In contrast tosolution reactions where single-electron transfer is prevalent,•OH substitution at the disulfide bond is a fast and predominantreaction channel in nonpolar environment, forming productpairs containing sulfenic acid (–SOH) and thiyl radical (–S•) atthe cleavage site (Scheme 2). For protonated cysteine disulfides,subsequent H transfer within each of the product pairs could becompetitive due to the stabilizing effect of hydrogen bonding inthe transition states. Both experimental and theoretical resultssupported the idea that reactions of •OH and cysteine disulfidecompounds produce four types of sulfur species, namely –SOH,–SO•, –SH, and –S•, which are important to the overall sulfurredox cycle. Of these species, –SOH is a potent scavenger forperoxyl radicals via H atom transfer (k = 3 × 107 M−1·s−1) (44,45), presenting a contrast to its disulfide bond precursor and thereduced thiol both of which show limited reactivity toward per-oxyl radicals (46). This implies that after the cysteine disulfidereacts directly with •OH, the in situ formed reaction product,–SOH, can join the antioxidant network of other peroxyl scav-engers (such as vitamin E), thus forming two lines of defense.Also of importance is the formation of a sulfinyl radical from Htransfer within [–SOH. . .•S–] product complex. Because thesulfinyl radical is much less reactive than the thiyl radical (32),this process basically detoxifies of thiyl radical. However, in anonpolar biological system, such as at the interface of lipidmembrane or in the hydrophobic regions of protein tertiarystructures, the local environment will largely modulate the re-activity of the cysteine disulfide toward •OH as well as the rate ofsubsequent H transfer. In summary, while cysteine disulfide hasbeen considered to be as an inefficient antioxidant, our study
Scheme 2. Two-step reaction mechanism for the •OH attack on the disul-fide bond in Cys–S–S–R. H transfer was found to be a competitive process for+Cys–S–S–R.
Adhikari et al. PNAS | August 4, 2020 | vol. 117 | no. 31 | 18221
CHEM
ISTR
Y
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1

suggested that by reacting it with •OH to form a more potentantioxidant in nonpolar environment, disulfide may be moreactively involved in the antioxidant network to combat elevatedlevels of ROS than previously appreciated.
MethodsMaterials. All reagents and solvents were purchased from commercial sourcesand were used without further purification. S-methyl methanethiosulfonate(MMTS), L-cysteine, DL-cysteine, thioacetic acid, methoxycarbonyl chloride,benzyl mercaptan, 1-propantethiol, 2,2,2-trifluoroethanethiol, acetyl chlo-ride, ethanol, anhydrous methanol, and trimethylamine were purchasedfrom Sigma-Aldrich. Syntheses of cysteine cysteinyl disulfides (Cys–S–S–R,Scheme 1) were achieved according to procedures described in the literature(details provided in SI Appendix) (47–49). The ethyl ester of cystine wassynthesized and used as an internal standard to compare the reaction ratesusing different cysteine disulfide derivatives. Working solutions for positivemode nano-ESI were prepared in deionized H2O (ultrapure purificationsystem at 0.03 μS·cm). The extents of reaction were online monitored byMS analysis.
MS and Online Radical Reactions. All MS data were collected on a 4000 QTRAPtriple quadrupole/linear ion trap (LIT) mass spectrometer equipped with anano-ESI source made in the laboratory. Analyst software 1.6.2 was used fordata acquisition, processing, and instrument control. Typical MS parametersused during the study were a spray voltage of ±1,500 to 1,800 V, curtain gaspressure of 10 psi, declustering potential of ±20 V, and scan rate of1,000 Da/s. MS1 mass analysis was performed in LIT mode in Q3. Beam-typecollision-induced dissociation (CID) was performed by performing precursor
ion selection in Q1 and ion acceleration into a Q2 collision cell followed byproduct analysis in a Q3 linear ion trap. Ion-trap CID consisted of precursorion selection in Q1 and ion transfer through Q2 to Q3 with minimum acti-vation energy followed by reisolation, accumulation, and application of di-polar excitation to effect CID in Q3. To induce radical reactions, the nano-ESIplume of disulfide was allowed to interact with •OH in the afterglow regionof an atmospheric-pressure helium low-temperature plasma enabled in aT-shaped glass tube placed in front of the entrance of the mass spectrometer(SI Appendix, Fig. S1) (16).
Computational Details. The computational methods employed in this workincludes a combination of MM methods (50), ab initio density functionaltheory with the BH&HLYP functional (51), domain-based local pair naturalorbital coupled cluster including perturbative triplet excitation [DLPNO–
CCSD(T)] methods (52), and kinetics calculations in the framework of MC-TST(53). A full detail of theoretical approaches used is discussed in SI Appendix,which also includes a detailed description of the electronic features of theprocesses investigated and the most relevant transition states of thedifferent reactions investigated.
Data Availability. All data are included in the manuscript and SI Appendix.
ACKNOWLEDGMENTS. Y.X. acknowledges financial support from NationalNatural Science Foundation of China (Grant 21722506). R.C. and J.M.A.acknowledge funding from the Ministry of Economic Affairs and DigitalTransformation (Grant CTQ2016-78636-P) and from the Generalitat deCatalunya (Grant 2017SGR348).
1. M. A. Wouters, S. W. Fan, N. L. Haworth, Disulfides as redox switches: From molecularmechanisms to functional significance. Antioxid. Redox Signal. 12, 53–91 (2010).
2. J. Chiu, P. J. Hogg, Allosteric disulfides: Sophisticated molecular structures enablingflexible protein regulation. J. Biol. Chem. 294, 2949–2960 (2019).
3. L. B. Poole, The basics of thiols and cysteines in redox biology and chemistry. FreeRadic. Biol. Med. 80, 148–157 (2015).
4. P. V. S. Oliveira, F. R. M. Laurindo, Implications of plasma thiol redox in disease. Clin.Sci. (Lond.) 132, 1257–1280 (2018).
5. M. Kemp, Y.-M. Go, D. P. Jones, Nonequilibrium thermodynamics of thiol/disulfideredox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 44,921–937 (2008).
6. R. D. Bach, O. Dmitrenko, C. Thorpe, Mechanism of thiolate-disulfide interchangereactions in biochemistry. J. Org. Chem. 73, 12–21 (2008).
7. S. B. Nimse, D. Pal, Free radicals, natural antioxidants, and their reaction mechanisms.RSC Advances 5, 27986–28006 (2015).
8. J. M. Anglada, M. Martins-Costa, J. S. Francisco, M. F. Ruiz-López, Interconnection ofreactive oxygen species chemistry across the interfaces of atmospheric, environmen-tal, and biological processes. Acc. Chem. Res. 48, 575–583 (2015).
9. R. Stocker, Antioxidant defenses in human blood plasma and extra-cellular fluids.Arch. Biochem. Biophys. 595, 136–139 (2016).
10. K. U. Ingold, D. A. Pratt, Advances in radical-trapping antioxidant chemistry in the21st century: A kinetics and mechanisms perspective. Chem. Rev. 114, 9022–9046(2014).
11. S. V. Lymar, R. F. Khairutdinov, J. K. Hurst, Hydroxyl radical formation by O–O bondhomolysis in peroxynitrous acid. Inorg. Chem. 42, 5259–5266 (2003).
12. G. Merényi, J. Lind, Thermodynamics of peroxynitrite and its CO2 adduct. Chem. Res.Toxicol. 10, 1216–1220 (1997).
13. M. Enescu, B. Cardey, Mechanism of cysteine oxidation by a hydroxyl radical: A the-oretical study. ChemPhysChem 7, 912–919 (2006).
14. R. Castañeda-Arriaga, J. R. Alvarez-Idaboy, Lipoic acid and dihydrolipoic acid. Acomprehensive theoretical study of their antioxidant activity supported by availableexperimental kinetic data. J. Chem. Inf. Model. 54, 1642–1652 (2014).
15. M. Bonifacic, K. Schaefer, H. Moeckel, K. D. Asmus, Primary steps in the reactions oforganic disulfides with hydroxyl radicals in aqueous solution. J. Phys. Chem. 79,1496–1502 (1975).
16. W. Wang, J. Xin, Y. Zhang, W. Wang, Y. Lu, Computational study on the mechanismfor the gas-phase reaction of dimethyl disulfide with OH. Int. J. Quantum Chem. 111,644–651 (2011).
17. P. H. Wine, N. M. Kreutter, C. A. Gump, A. R. Ravishankara, Kinetics of hydroxyl radicalreactions with the atmospheric sulfur compounds hydrogen sulfide, methanethiol,ethanethiol, and dimethyl disulfide. J. Phys. Chem. 85, 2660–2665 (1981).
18. S. Hatakeyama, H. Akimoto, Reactions of hydroxyl radicals with methanethiol, di-methyl sulfide, and dimethyl disulfide in air. J. Phys. Chem. 87, 2387–2395 (1983).
19. N. I. Butkovskaya, D. W. Setser, Mechanism for the reaction of hydroxyl radicals withdimethyl disulfide. Chem. Phys. Lett. 312, 37–44 (1999).
20. A. Bil, K. Grzechnik, K. Mierzwicki, Z. Mielke, OH-induced oxidative cleavage of di-methyl disulfide in the presence of NO. J. Phys. Chem. A 117, 8263–8273 (2013).
21. D. A. Stoyanovsky, A. Maeda, J. L. Atkins, V. E. Kagan, Assessments of thiyl radicals inbiosystems: Difficulties and new applications. Anal. Chem. 83, 6432–6438 (2011).
22. F. Turecek, R. R. Julian, Peptide radicals and cation radicals in the gas phase. Chem.Rev. 113, 6691–6733 (2013).
23. G. N. Khairallah et al., Radical formation in the gas-phase ozonolysis of deprotonatedcysteine. Angew. Chem. Int. Ed. Engl. 54, 12947–12951 (2015).
24. I. K. Chu et al., Are the radical centers in peptide radical cations mobile? The gen-eration, tautomerism, and dissociation of isomeric α-carbon-centered triglycine rad-ical cations in the gas phase. J. Am. Chem. Soc. 130, 7862–7872 (2008).
25. S. Osburn, B. Chan, V. Ryzhov, L. Radom, R. A. J. O’Hair, Role of hydrogen bonding onthe reactivity of thiyl radicals: A mass spectrometric and computational study usingthe distonic radical ion approach. J. Phys. Chem. A 120, 8184–8189 (2016).
26. A. Ray, T. Bristow, C. Whitmore, J. Mosely, On-line reaction monitoring by massspectrometry, modern approaches for the analysis of chemical reactions. Mass Spec-trom. Rev. 37, 565–579 (2018).
27. T. A. Brown, H. Chen, R. N. Zare, Identification of fleeting electrochemical reactionintermediates using desorption electrospray ionization mass spectrometry. J. Am.Chem. Soc. 137, 7274–7277 (2015).
28. C. Iacobucci, S. Reale, F. De Angelis, Elusive reaction intermediates in solution ex-plored by ESI-MS: Reverse periscope for mechanistic investigations. Angew. Chem. Int.Ed. Engl. 55, 2980–2993 (2016).
29. R. G. Cooks, X. Yan, Mass spectrometry for synthesis and analysis. Annu. Rev. Anal.Chem. (Palo Alto, Calif.) 11, 1–28 (2018).
30. Y. Xia, R. G. Cooks, Plasma induced oxidative cleavage of disulfide bonds in poly-peptides during nanoelectrospray ionization. Anal. Chem. 82, 2856–2864 (2010).
31. C. B. Love, L. Tan, J. S. Francisco, Y. Xia, Competition of charge- versus radical-directedfragmentation of gas-phase protonated cysteine sulfinyl radicals. J. Am. Chem. Soc.135, 6226–6233 (2013).
32. L. Tan, Y. Xia, Gas-phase reactivity of peptide thiyl (RS•), perthiyl (RSS•), and sulfinyl(RSO•) radical ions formed from atmospheric pressure ion/radical reactions. J. Am.Soc. Mass Spectrom. 24, 534–542 (2013).
33. L. Tan, Y. Xia, Gas-phase peptide sulfinyl radical ions: Formation and unimoleculardissociation. J. Am. Soc. Mass Spectrom. 23, 2011–2019 (2012).
34. G. C. Y. Chan et al., Spectroscopic plasma diagnostics on a low-temperature plasmaprobe for ambient mass spectrometry. J. Anal. At. Spectrom. 26, 1434–1444 (2011).
35. R. Ono, T. Oda, OH radical measurement in a pulsed arc discharge plasma observed bya LIF method. IEEE Trans. Ind. Appl. 37, 709–714 (2001).
36. T. Kotiaho, M. N. Eberlin, P. Vainiotalo, R. Kostiainen, Electrospray mass and tandemmass spectrometry identification of ozone oxidation products of amino acids andsmall peptides. J. Am. Soc. Mass Spectrom. 11, 526–535 (2000).
37. W. Lu, I. M. Tsai, Y. Sun, W. Zhou, J. Liu, Elucidating potential energy surfaces forsinglet O2 reactions with protonated, deprotonated, and di-deprotonated cystineusing a combination of approximately spin-projected density functional theory andguided-ion-beam mass spectrometry. J. Phys. Chem. B 121, 7844–7854 (2017).
38. L. Konermann, E. Ahadi, A. D. Rodriguez, S. Vahidi, Unraveling the mechanism ofelectrospray ionization. Anal. Chem. 85, 2–9 (2013).
39. C. H. Sohn et al., Mechanisms and energetics of free radical initiated disulfide bondcleavage in model peptides and insulin by mass spectrometry. Chem. Sci. (Camb.) 6,4550–4560 (2015).
40. F. Turecek, M. Polášek, A. Frank, M. Sadílek, Transient hydrogen atom adducts todisulfides. Formation and energetics. J. Am. Chem. Soc. 122, 2361–2370 (2000).
18222 | www.pnas.org/cgi/doi/10.1073/pnas.2006639117 Adhikari et al.
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1

41. J. M. Anglada, R. Crehuet, S. Adhikari, J. S. Francisco, Y. Xia, Reactivity of hydro-
persulfides toward the hydroxyl radical unraveled: Disulfide bond cleavage, hydrogen
atom transfer, and proton-coupled electron transfer. Phys. Chem. Chem. Phys. 20,
4793–4804 (2018).42. E. H. Krenske, W. A. Pryor, K. N. Houk, Mechanism of S(H)2 reactions of disulfides:
Frontside vs backside, stepwise vs concerted. J. Org. Chem. 74, 5356–5360 (2009).43. Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, (CRC
Press, Boca Raton, FL, 2003).44. V. Vaidya, K. U. Ingold, D. A. Pratt, Garlic: Source of the ultimate antioxidants—
sulfenic acids. Angew. Chem. Int. Ed. Engl. 48, 157–160 (2009).45. R. Amorati, P. T. Lynett, L. Valgimigli, D. A. Pratt, The reaction of sulfenic acids with
peroxyl radicals: Insights into the radical-trapping antioxidant activity of plant-
derived thiosulfinates. Chemistry 18, 6370–6379 (2012).46. J. H. B. Chenier, E. Furimsky, J. A. Howard, Arrhenius parameters for reaction of the
tert-butylperoxy and 2-ethyl-2-propylperoxy radicals with some nonhindered phe-
nols, aromatic amines, and thiophenols. Can. J. Chem. 52, 3682–3688 (1974).
47. D. J. Smith, E. T. Maggio, G. L. Kenyon, Simple alkanethiol groups for temporaryblocking of sulfhydryl groups of enzymes. Biochemistry 14, 766–771 (1975).
48. B. H. Rietman, R. F. R. Peters, G. I. Tesser, A facile method for the preparation of S-(Alkylsulfenyl)cysteines. Synth. Commun. 24, 1323–1332 (1994).
49. C. Starkenmann, Y. Niclass, M. Troccaz, Nonvolatile S-alk(en)ylthio-L-cysteine deriva-tives in fresh onion (Allium cepa L. cultivar). J. Agric. Food Chem. 59, 9457–9465(2011).
50. K. Roos et al., OPLS3e: Extending force field coverage for drug-like small molecules.J. Chem. Theory Comput. 15, 1863–1874 (2019).
51. A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories.J. Chem. Phys. 98, 1372–1377 (1993).
52. Y. Guo et al., Communication: An improved linear scaling perturbative triples cor-rection for the domain based local pair-natural orbital based singles and doublescoupled cluster method [DLPNO-CCSD(T)]. J. Chem. Phys. 148, 11101 (2018).
53. L. P. Vereecken, J. Peeters, The 1,5-H-shift in 1-butoxy: A case study in the rigorousimplementation of transition state theory for a multirotamer system. J. Chem. Phys.119, 5159–5170 (2003).
Adhikari et al. PNAS | August 4, 2020 | vol. 117 | no. 31 | 18223
CHEM
ISTR
Y
Dow
nloa
ded
by g
uest
on
Dec
embe
r 21
, 202
1
Related Documents











