Tumor Microenvironment in Head and Neck Squamous Cell Carcinoma Joseph M. Curry, a John Sprandio, b David Cognetti, a Adam Luginbuhl, a Voichita Bar-ad, c Edmund Pribitkin, a and Madalina Tuluc d The tumor microenvironment (TME) of head and neck squamous cell carcinoma (HNSCC) is comprised of cancer-associated fibroblasts (CAFs), immune cells, and other supporting cells. Genetic changes in the carcinoma cells, such as alterations to TP53, NOTCH1, and specific gene expression profiles, contribute to derangements in cancer and microenvironment cells such as increased ROS, overproduction of cytokines, and epithelial to mesenchymal transition (EMT). CAFs are among the most critical elements of the TME contributing to proliferation, invasion, and metastasis. The adaptive immune response is suppressed in HNSCC through overexpression of cytokines, triggered apoptosis of T cells, and alterations in antigen processing machinery. Overexpression of critical cytokines, such as transforming growth factor-β (TGF-β), contributes to EMT, immune suppression, and evolution of CAFs. Inflammation and hypoxia are driving forces in angiogenesis and altered metabolism. HNSCC utilizes glycolytic and oxidative metabolism to fuel tumorigenesis via coupled mechanisms between cancer cell regions and cells of the TME. Increased understanding of the TME in HNSCC illustrates that the long-held notion of “condemned mucosa” reflects a process that extends beyond the epithelial cells to the entire tissue comprised of each of these elements. Semin Oncol 41:217-234 & 2014 Elsevier Inc. S quamous cell carcinoma comprises more than 90% of cancers of the head and neck and arises from the squamous lining of the mucosal surfa- ces of the upper aerodigestive tract, including the oral cavity, pharynx, larynx, and sinonasal tract. Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, and only 50%–60% of patients are alive at 5 years after diagnosis. 1,2 Treatment can be quite morbid and result in significant functional as well as aesthetic deficits, such as impair- ment of speech and swallowing and facial deformity. Treatment failure and locoregional recurrence are common and occur in up to 30% of patients and account for the majority of deaths. 3 The high rate of local recurrence produced the long-held notion of “condemned mucosa” or “field cancerization” initially described in the 1950s. 4 This concept underscores not only the difficulty in treating HNSCC but also denotes the complexity of the molecular conditions under which HNSCC develops and recurs. It is clear that the notion of the condemned mucosa reflects a “condemned tissue” composed of the cancerous cells, adjacent epithelial, stromal, and immune cells and their surrounding matrix. Together these elements comprise the tumor microenvironment (TME). In fact, this shift in thought from the concept that cancer is derived from a single cell type, to a disease occurring in a complex tissue, has led some investigators to suggest that the very definition of carcinoma be changed. 5 Tumorigenesis requires multiple elements outlined by Hanahan and Weinberg: (1) limitless replicative potential, (2) self-sufficiency in growth signals, (3) insen- sitivity to anti-growth signals, (4) ability to evade apoptosis, (5) increased angiogenesis, and (6) invasion and metastasis. 6 Knowledge of the mechanisms through which the cancer cells use the TME to execute these processes continues to evolve. 7,8 There is great interest in the downstream paracrine interactions with 0093-7754 & 2014 Elsevier Inc. http://dx.doi.org/10.1053/j.seminoncol.2014.03.003 Conflicts of interest: none. a Department of Otolaryngology Head and Neck Surgery, Thomas Jefferson University, Philadelphia, PA. b Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA. c Department of Radiation Oncology, Thomas Jefferson University, Philadelphia, PA. d Department of Pathology, Thomas Jefferson University, Philadelphia, PA. Address correspondence to Joseph M. Curry, MD, Department of Otolaryngology Head and Neck Surgery, Thomas Jefferson Uni- versity, Philadelphia, PA 19107. E-mail: joseph.curry@jefferson. edu Seminars in Oncology, Vol 41, No 2, April 2014, pp 217-234 217 Open access under CC BY-NC-ND license. Open access under CC BY-NC-ND license.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Tumor Microenvironment in Head and Neck SquamousCell Carcinoma
Joseph M. Curry,a John Sprandio,b David Cognetti,a Adam Luginbuhl,a Voichita Bar-ad,c
Edmund Pribitkin,a and Madalina Tulucd
The tumor microenvironment (TME) of head and neck squamous cell carcinoma (HNSCC) is
comprised of cancer-aGenetic changes in the
expression profiles, co
increased ROS, overprCAFs are among the m
metastasis. The adaptiv
cytokines, triggered aOverexpression of crit
to EMT, immune supp
forces in angiogenesmetabolism to fuel tu
cells of the TME. Incr
notion of “condemnedentire tissue comprise
Semin Oncol 41:217-2
0093-7754& 2014 Elshttp://dx.doi
Conflicts o
aDepartmenJefferson
bDepartmePhiladelp
cDepartmenPhiladelp
dDepartmePA.
Address coOtolarynversity,edu
Seminars
ssociated fibroblasts (CAFs), immune cells, and other supporting cells.carcinoma cells, such as alterations to TP53, NOTCH1, and specific gene
ntribute to derangements in cancer and microenvironment cells such as
oduction of cytokines, and epithelial to mesenchymal transition (EMT).ost critical elements of the TME contributing to proliferation, invasion, and
e immune response is suppressed in HNSCC through overexpression of
poptosis of T cells, and alterations in antigen processing machinery.ical cytokines, such as transforming growth factor-β (TGF-β), contributesression, and evolution of CAFs. Inflammation and hypoxia are driving
is and altered metabolism. HNSCC utilizes glycolytic and oxidativemorigenesis via coupled mechanisms between cancer cell regions and
eased understanding of the TME in HNSCC illustrates that the long-held
mucosa” reflects a process that extends beyond the epithelial cells to thed of each of these elements.
34 & 2014 Elsevier Inc. Open access under CC BY-NC-ND license.
Squamous cell carcinoma comprises more than90% of cancers of the head and neck and arises
from the squamous lining of the mucosal surfa-
ces of the upper aerodigestive tract, including the oralcavity, pharynx, larynx, and sinonasal tract. Head and
neck squamous cell carcinoma (HNSCC) is the sixth
most common cancer worldwide, and only 50%–60%of patients are alive at 5 years after diagnosis.1,2
Treatment can be quite morbid and result in significant
functional as well as aesthetic deficits, such as impair-ment of speech and swallowing and facial deformity.
evier Inc..org/10.1053/j.seminoncol.2014.03.003
f interest: none.
t of Otolaryngology Head and Neck Surgery, ThomasUniversity, Philadelphia, PA.
nt of Medical Oncology, Thomas Jefferson University,hia, PA.t of Radiation Oncology, Thomas Jefferson University,hia, PA.nt of Pathology, Thomas Jefferson University, Philadelphia,
rrespondence to Joseph M. Curry, MD, Department ofgology Head and Neck Surgery, Thomas Jefferson Uni-Philadelphia, PA 19107. E-mail: joseph.curry@jefferson.
in Oncology, Vol 41, No 2, April 2014, pp 217-234
Open access under CC BY-NC-ND license.
Treatment failure and locoregional recurrence arecommon and occur in up to 30% of patients and
account for the majority of deaths.3 The high rate of
local recurrence produced the long-held notion of“condemned mucosa” or “field cancerization” initially
described in the 1950s.4 This concept underscores not
only the difficulty in treating HNSCC but also denotesthe complexity of the molecular conditions under
which HNSCC develops and recurs. It is clear that
the notion of the condemned mucosa reflects a“condemned tissue” composed of the cancerous cells,
adjacent epithelial, stromal, and immune cells and their
surrounding matrix. Together these elements comprisethe tumor microenvironment (TME). In fact, this shift
in thought from the concept that cancer is derived
from a single cell type, to a disease occurring in acomplex tissue, has led some investigators to suggest
that the very definition of carcinoma be changed.5
Tumorigenesis requires multiple elements outlinedby Hanahan and Weinberg: (1) limitless replicative
potential, (2) self-sufficiency in growth signals, (3) insen-
sitivity to anti-growth signals, (4) ability to evadeapoptosis, (5) increased angiogenesis, and (6) invasion
and metastasis.6 Knowledge of the mechanisms
through which the cancer cells use the TME to executethese processes continues to evolve.7,8 There is great
interest in the downstream paracrine interactions with
217

J.M. Curry et al218
the stroma, immune interactions, and metabolic
changes and the role each plays in tumorigenesis.HNSCC is genetically heterogeneous, but a num-
ber of pathways have been found to be commonly
involved; the impact of several critical abnormalitieson the TME is highlighted below. The cellular
elements of the TME often coevolve with the tumor.
Stromal fibroblasts, T cells, macrophages, and othercell types develop abnormal phenotypes in a disor-
ganized response to the cancer (Figure 1). These non-
cancerous cells provide many of the paracrine signalsnecessary to turn on the pleotrophic abilities of
cancer cells.9 For example, fibroblasts become
cancer-associated fibroblasts and secrete factors suchas matrix metalloproteins (MMPs), contributing to
tumor invasiveness. Furthermore, as the chronic
inflammation of the TME remains unresolved, alter-ations in adaptive immune response such as apopto-
sis of cytotoxic T cells and activation of suppressor T
cells occurs.10 Additionally, tumors reprogram theirsurroundings creating a metabolically fertile environ-
ment to meet their high energy and anabolic require-
ments. This process was aptly described by Paget asthe “seed and soil” hypothesis.11 Fundamental tumor–non-tumor microenvironmental interactions such as
these represent potential points of intervention fortherapeutic strategies. Many critical targets, such as
nuclear factor-κB (NF-κB), hypoxia-inducible factor
(HIF)-1α, and vascular endothelial growth factor(VEGF), have been, and continue to be, explored as
therapeutic targets in the TME12–14 (Table 1).
IMPACT OF GENETIC AND EPIGENETICCHANGES OF THE EPITHELIUM ON THE TME
The initiating genetic alterations in the epithelial
cells of HNSCC are primarily the result of thecarcinogenic properties of tobacco and alcohol, and
in the oropharynx, oncogenic strains of the human
papilloma virus (HPV). Classically, HNSCC has beenthought of as a disease caused by tobacco and
alcohol, yet tobacco-related cancers are decreasing
Figure 1. Select elements and interaciotns of the TME. Theperivascular niche that commonly contain cancer stem cells ancompartment contains tumor cells that are glycolytic and less proDDR is shown between the leading edge tumor edge and normaadjacent to normal stroma and fibroblasts (yellow). CAFs expreprotein inducer (EMMPRIN) on cancer cells to activate MMP2activate TGF-β. CAFs and tumor cells produce elements like VEGinteractions between regulatory T cells (pink), cytotoxic T cells (r(blue/green) are shown. TGF-β and IL-10 produced by TAMs anMIF that recruits neutrophils. Regulatory T cells induce toleracytotoxic T cells in utlitzed by cancer cells to induce apoptosincrease angiogenesis and invasion by production of MMP-9, Vorange) are recruited to the TME by GM-CSF produced by cancTGF-β.
in incidence.15 Over the past several decades, onco-
genic strains of HPV have become apparent as anetiology for oropharyngeal squamous cell carcinoma
(OPSCC). HPV-related OPSCC accounts for up to 60%
of cases of oropharyngeal cancer in some regions;this has resulted in an increased incidence among
younger nonsmokers, and has been equated to an
epidemic by some investigators.16 Currently, this isthe second most common malignancy caused by
HPV.17 OPSCC is caused primarily by HPV16 (but
also HPV18, HPV31, and others), via the E6 and E7mechanisms established in cervical cancer.18
The most widely identified mutation in non–HPV-related HNSCC occurs in the tumor-suppressor geneTP53. This has been identified to occur in approx-
imately 50% of HNSCCs and is likely an early event, as
it is commonly found in premalignant lesions aswell.19–21 Mutations also have been shown to corre-
late with aggression and poor outcomes; for exam-
ple, p53 mutations have been found in 95% ofradioresistant tumors.22–24 Histologically negative
margins with p53 mutations have been shown to be
associated with a greater incidence of local recur-rence.25 Mutation of TP53 in tumor cells is associated
with increased migration of cancer-associated fibro-
blasts (CAFs) to the TME, while intact TP53 inhibitsmigration.26 Loss of functional p53 increases reactive
oxygen species (ROS) and reactive nitrogen species
(RNS) and may drive carcinogenesis via NF-κB andother inflammatory-mediated mechanisms. Altera-
tions in TP53 induce a DNA damage response
(DDR) in adjacent non-tumoral cells via productionof ROS. This effect was recently demonstrated in
esophageal SCC, and it increases with proximity to
and size of the primary tumor, with effects beingidentified several centimeters from the tumor.27–30
TP53 mutations also have been linked to abnormal
tumor metabolism, contributing to the Warburgeffect through increased activity of glucose trans-
porters and glycolytic enzymes furthering the pro-
duction of an acidic environment and high levels ofROS toxic to normal cells31 (Figure 1).
tumor is shown here with the leading tumor edge andd highly replicating tumor cells (blue). The more centralliferative (orange). Peritumoral epithelium demonstratingl epithelium. CAFs (purple) are shown in the tumor stromass MT-MMP that interacts with extrcellular metallomatrix. CSCs express CD144, which interacts with MMP9 toF, PGE2, and CXCL12 that trigger angiogenesis. Immuneed), M2 TAMs (green), and tumor-associated neurtrophilsd cancer cells suppress T-cell activity. TAMs also producence by cytotoxic T cells. The Fas receptor on activatedis. Tumor-associated neutrophils produce ROS, and alsoEGF, and HGF. CD34þ myeloid progenitor cells (yellow/er cells which in turn induce immunosuppression through
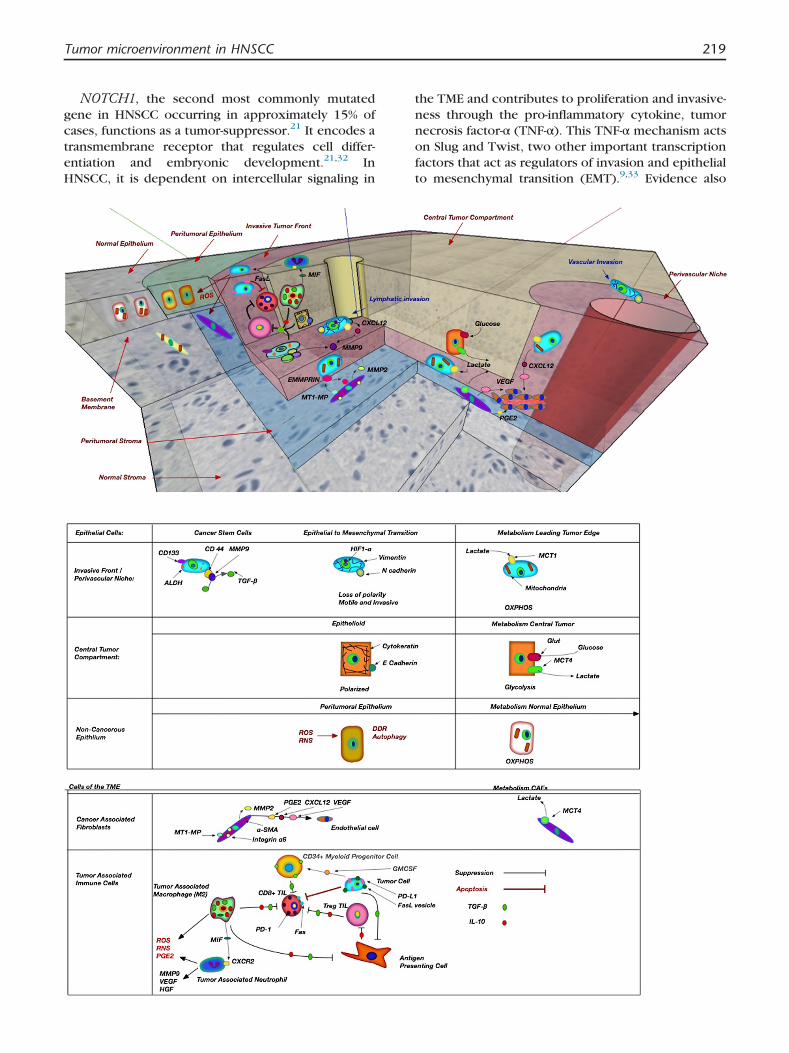
Tumor microenvironment in HNSCC 219
NOTCH1, the second most commonly mutated
gene in HNSCC occurring in approximately 15% ofcases, functions as a tumor-suppressor.21 It encodes a
transmembrane receptor that regulates cell differ-
entiation and embryonic development.21,32 InHNSCC, it is dependent on intercellular signaling in
the TME and contributes to proliferation and invasive-
ness through the pro-inflammatory cytokine, tumornecrosis factor-α (TNF-α). This TNF-α mechanism acts
on Slug and Twist, two other important transcription
factors that act as regulators of invasion and epithelialto mesenchymal transition (EMT).9,33 Evidence also

Table 1. Critical Cells of the TME in HNSCC
Cell Type Markers Secreted Factors Metabolism References
Squamous Cell CarcinomaKey Genetic Alterations: TP53,NOTCH1, EGFR, CDKN2a, STAT3,Cyclin D1, Rb
E-cadherin, cytokeratins,PD-L1, FasL
MMP 2, MMP 9, MMP 13,ROS,VEGF, CXCL1,CXCL8, PDGF, IL-8,FGF-2, TGF-β, TNF-α,IL-1, GMCSF
Tan et al,Koontongkaew S,Zhang Z et al,Smith A et al,Curry J et al, Feron O.
Central tumor compartment Glycolytic: (MCT4þ,MCT1�, TOMM20�,COX�)OXPHOS: (MCT1þ,MCT4-,TOMM20þ,COXþ)
Leading edge/invasive front,perivascular niche (proliferativecancer cells: high Ki-67)
Cancer stem cells CD33, CD144, ALDHEpithelial to mesenchymal
transitionN-cadherin, vimentin
Cancer-Associated Fibroblast α-SMA, integrin α6 HGF, CXCL12, TGF-β,MMP2, MMP9, PMF,PDGF, Type IV collagen,Col15-binding integrins,PGE2
Glycolytic: (MCT4þ,MCT1-LDH-Bþ)
Leef G, Curry J et al,Wheeler SE et al,Marsh D et al
Tumor-Infiltrating LymphocytesRegulatory T cells CD4þCD25þFoxP3þ IL-10, IL 12, TGF-β Young MR, Ferris RL
et al, Whiteside TLCytotoxic T cells CD8þ, TCR, Fas, PD-1 Perforin, granzymes,granulysin
Th2 suppressor cells CD4þ IL-4, IL-6, IL-10Myeloid progenitor cells CD34þ TGF-β
Tumor-Associated Macrophages(M2)
IL-10, TGF-β, MIF, EGF, CSF-1, MMP9, CXCL2,CXCL8,VEGF, ROS, RNS, PGEs
Lago Costa N et al,Dumitru C et al,Galdiero MR et al
Tumor-Associated Neutrophils MMP9, VEGF, HGF, elastase,ROS, PGEs
Galdiero MR et al,Dumtru C et al
Endothelial Cells Endothelins, CXCL1, CXCL8 Neiva KG et alAbbreviations: Rb, retinoblastoma gene; EGFR, epidermal growth factor receptor; CDKN2a, cyclin-dependent kinase inhibitor 2a; STAT 3, signal transducer and activator of transcription 3;
PD-L1, programmed death ligand-1; FasL, Fas ligand; MMP, matrix metalloprotein; ALDH, aldehyde dehydrogenase; ROS, reactive oxygen species; VEGF, vascular endothelial growthfactor; IL, inteleukin; FGF,fibroblast growth factor; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; PDGF, platelet-derived growth factor; GM-CSF, granulocye-macrophage colony-stimulating factor; EGF, epidermal growth factor; CSF-1, colony-stimulating factor-1; TCR, T-cell receptor; FoxP3, forked/winghead transcription factor; RNS, reactivenitrogen species; MCT, monocarboxylate transporter; PGE, protaglandin; PD-1, programmed death-1; TOMM20, translocase of outer mitochondrial membrane 20; COX, cytochrome Coxidase complex; LDH-B, lactate dehydrogenase B.
J.M.Curry
etal
220

Tumor microenvironment in HNSCC 221
has suggested that its activity is mediated through
MMPs and the inflammatory transcription factor, NF-κB, among other critical mechanisms34,35 (Figure 1).
EGFR is a membrane-bound tyrosine kinase recep-
tor that binds epidermal growth factor (EGF) and TGF-α. It is the target for the effective and widely used
monoclonal antibody, cetuximab. Mutation of the
EGFR gene is only present in about 10% of cases,but gene amplification is present in about 30% of cases
and overexpression has been identified in up to 90%
of cases. Increased expression and gene copy numbercorrelate with poor prognosis.36–39 After binding
one of its ligands, EGFR triggers multiple intracellular
signaling cascades that activate cell proliferation,survival, invasion, metastasis, and angiogenesis.21,40–43
It also allows for decreased response to radiotherapy
by enhancing proliferation, DNA-repair, and hypoxicresponses within the TME.44–46 Activation triggers
increased interleukin-8 (IL-8) and VEGF production,
promoting inflammation and angiogenesis.47,48
Cyclin-dependent kinase inhibitor 2a (CDKN2a) is
an important tumor-suppressor that is mutated in
9%–12% of HNSCC patients. The function of itsproduct, p16, is to block cell cycle progression from
the G1 to S phase, and is typically upregulated under
stress conditions in the cellular microenvironmentsuch as hypoxia.49 Mutations are common but alone
likely to be insufficient to result in tumorigenesis.21
Mutational loss of function correlates with a wors-ened prognosis, while overexpression of p16 is
common in HPV-related OPSCC and correlates with
an improved prognosis.45,48,50
Signal transducer and activation of transcription
(STAT) proteins are transcription factors that are
commonly overexpressed in cancer, and in HNSCC,STAT3 has been found to be commonly mutated and
overexpressed.5 STAT3 activation is linked to numer-
ous pathways, including TGF-β, IL-6, and EGFR, andit is involved in EMT, proliferation, apoptosis, and
inflammation.51 STAT3 is also central to mainte-
nance of self-renewal in cancer stem cells (CSCs).52
The STAT/JAK pathway is one of the critical targets
of cetuximab.53 Numerous other abnormalities have
been found to be prevalent in HNSCC; Tan et al hasaddressed many of these in an excellent review.21
Given the great variety of genetic abnormalities
that have been identified in HNSCC, gene expressionprofiles may offer greater accuracy for characteriza-
tion and diagnosis than analysis of single loci. Several
groups have established profiles that can differentiateHNSCC from surrounding normal tissues. For exam-
ple, in a early study Chung et al identified four
subtypes of SCC based on gene expression profiles,each with different survival and recurrence rates.54
They identified patterns that they classified as
(1) EGFR pathway subtype, (2) mesenchymal-enriched subtype, (3) normal-epithelium–like
subtype, and (4) high-antioxidant enzyme subtype.
The EGFR group had the worst outcome. The secondsubgroup had a high fibroblast component and
demonstrated evidence of EMT. The third group
demonstrated gene expression closest to normaltonsillar epithelium and had the best outcome. The
fourth group demonstrated patterns similar to that
induced by exposure to cigarette smoking with highlevels of antioxidant enzymes being expressed.54
Clatot et al recently published a series in which they
used high-throughput reverse transcriptase polymer-ase chain reaction to create a nine-gene model with
which they were able to classify patients with 90%
accuracy. Those in a cluster with higher expressionof the chemokine CXCL12 had significantly greater
disease-free survival compared to those in a low-
expression CXCL12 cluster. Among these nine genes,a high-fold change in survival was seen in the group
comprised of CXCL12, SCL16A4 (monocarboxylate
transporter 4, MCT4), and carbonic anhydrase IX(CA9). CXCL12 is an important cytokine in HNSCC
implicated in angiogenesis and other processes.55
SCL16A4/MCT4 is a lactate transporter that has beenshown to be overexpressed in response to hypoxia.
CA9 is also upregulated by hypoxia and functions to
regulate intracellular pH.56
A number of epigenetic changes have been found
to be common to HNSCC, including DNA methyla-
tion, histone modification, microRNA interference,and small interfering RNA. Epigenetic regulation such
as methylation of CDK2a and other genes has been
shown to occur.57 Methylation of death-associatedprotein kinase (DAPK) is associated with resistance
to anti-EGFR agents, like cetuximab.58 Jung et al
performed a combined analysis of the transcriptome,methylome, and miRNome of metastatic HNSCC and
non-metastatic HNSCC and identified a signature that
correlated with lower survival and metastatic pheno-type. The pathways involved in this group were
specifically related to cell–cell adhesion, EMT,
immune response, and apoptosis. For example, theyidentified decreased expression of desmoglein 3 (DSG
3), a component of desmosomes critical for cell–celladhesion. Desmosomes also have been shown to havetumor-suppressor function, and decreased expression
of DSG3 has been linked to a poor prognosis.59 They
also identified several elements significant in EMT,including upregulation of vimentin and downregula-
tion of cytokeratin intermediate fibers and activation
of TGF-β–related EMT pathways. Analysis of miRNAdemonstrated upregulation of pathways related to
DDR and immune response.60
CANCER-ASSOCIATED FIBROBLASTS
Normal squamous mucosal lining of the upperaerodigestive tract is organized into distinct

J.M. Curry et al222
compartments: the upper layer of differentiated
squamous or respiratory epithelial cells, a basalepithelial layer, the underlying basement membrane,
and stromal layer. Fibroblasts are abundant in the
stroma and are the primary element responsible forsecretion of the basement membrane proteins. They
secrete structural proteins such as type IV collagen
and laminin and also produce numerous cytokinesand paracrine signals. Accordingly, tumor- or cancer-
associated fibroblasts (CAFs) are among the most
critical cellular elements of the TME. CAFs arephenotypically altered fibroblasts, which are active
participants in the process of tumorigenesis, promot-
ing growth and metastasis.61
CAFs arise from the population of circulating
fibroblasts and co-evolve with the tumor developing
a distinct phenotype, and playing an active role incarcinogenesis.62–64 They produce a variety of con-
tractile proteins, giving them an “active” phenotype.Frequently, they demonstrate ultrastructural accu-mulation of α-smooth muscle actin (α-SMA), charac-
teristic of myofibroblastic (MF) differentiation.65–67
In HNSCC, CAFs frequently have this MF phenotypeand are associated with dense collagen deposition
and stromal desmoplasia.68,69 CAFs are also charac-
terized by expression of integrin α6, which is criticalto cell adhesion and surface signaling. It complexes
to bind laminins, components of the extrcellular
matrix, and interacts with CDKN1A, altering cellcycle progression. Lim et al demonstrated that
upregulation of α-SMA and integrin-α6 correlated
with worsened prognosis in oral cancer.70
CAFs express a variety of factors critical to
carcinogenesis, promoting cell motility by upregula-
tion of cytokines, such as paracrine motility factor,hepatocyte growth factor (HGF), CXCL12, and
TGF-β.71 HGF secreted by CAFs has been shown to
promote invasion and angiogenesis in HNSCC andesophageal SCC.65,72–74 CXCL12 binds to CXCR4;
this interaction plays a role in upregulation of MMP9,
EMT, and HIF-1α expression.75 TGF-β is a criticalelement in the TME that serves numerous functions,
including immunosuppression. Additionally, CAFs
directly contribute to extracellular matrix remodel-ing by secreting MMPs.76,77
Marsh et al demonstrated that the MF phenotype
seen in some oral carcinomas was strongly prognos-tic of a negative outcome.78 This study evaluated 282
oral HNSCC specimens and found that the presence
of MF stroma was the strongest prognostic variableassessed, as compared to surgical margins, extracap-
sular spread, and stage, among others. MF stroma
correlated with depth of invasion and with extrac-apsular spread in nodal metastasis. Interestingly,
tumor-containing lymph nodes with extranodal
spread were also surrounded with MF stroma. Inoral and lingual carcinoma cell lines, Lin et al were
able to demonstrate increased proliferation in asso-
ciation with CAFs.79,80 In a mouse model usingheterotopic injection of HNSCC cells with normal
fibroblasts or CAFs, Wheeler et al demonstrated that
HNSCC cells with CAFs resulted in increased growthof the primary tumor and nodal and distant meta-
stases compared to co-injection with normal
fibroblasts.61
CAFs are also critical to tumor metabolism. Recent
studies indicate that epithelial cancer cells may
derive nutrients from the CAFs via a coupled meta-bolic mechanism. Cancerous cells induce glycolysis
in adjacent stromal cells such as CAFs and then use
their high-energy byproducts, such as lactate andpyruvate.81 This is somewhat contrarian to the long
held belief of the Warburg effect, whereby tumors
are thought to rely on aerobic glycolysis to produceenergy for rapid growth. This has been labeled the
“reverse Warburg effect” and has been shown to be a
critical prognostic indicator in breast and otherhuman cancers. There is some evidence that sug-
gests this occurs in HNSCC as well.82
THE IMMUNE RESPONSE IN THE TME
The persistent unresolved inflammation associ-
ated with cancer results in a eventual decay and
malfunction of the normal immune processes, whichin turn contributes to tumorigenesis through
immune tolerance and suppression and also to
angiogenesis and production of ROS. Essentially,tumorigenesis is at least in part a byproduct of a
failure of the immune system.10,12,83,84 The adaptive
immune response contributes in a variety of ways totumorigenesis through the immune interactions in
the TME involving T lymphocytes, macrophages,
dendritic cells, and others.85
T Lymphocytes
T lymphocytes are the central component of theanti-tumor response. They serve to initiate and regulate
the adaptive immune response and to elicit the cyto-
toxic response to tumors.85 There is evidence thatdysfunction occurs at the local, regional, and systemic
levels in HNSCC. While a strong lymphocytic host
presence at the tumor interface is indicative of anadaptive immune response and correlates with an
improved survival,86–88 dysfunctional circulating T cells
and tumor-infiltrating T cells have been identified inHNSCC, suggesting that tumors can suppress a previ-
ously intact local and systemic immune response.84,89–93 Moreover on a regional level, metastatic lymph nodesof HNSCC show significantly decreased levels of CD8þ
lymphocytes.87,94 Common functional deficits of
tumor-infiltrating T cells include: (1) absent or lowexpression of a key molecule in the signaling receptor

Tumor microenvironment in HNSCC 223
receptor chain (CD3ζ), (2) decreased proliferation in
response to mitogens, (3) inability to kill tumor celltargets, (4) imbalance of their cytokine profile, and
(5) evidence of profound apoptotic features.84
Evasion of the adaptive response is executedthrough a variety of mechanisms such as decreased
expression of major histocompatibility complexes
(MHC I) or induction of apoptosis in T cells.Decreased expression of antigen-processing machi-
nery such as MHC glycoprotein allows escape of
subpopulations of tumor cells by avoiding activationof cell mediated immunity.95–97 This mechanism has
been demonstrated in HNSCC whereby tumor cells
produce gangliosides, which downregulate MHC I.98
Another means of evading detection is to induce
apoptosis in cytotoxic T cells. The FasL receptor
mechanism is expressed by activated cytotoxic T cells,which bind to FasL and typically result in triggering
the cytotoxic response. However, this also predis-
poses the T cell to apoptosis. Oral SCC cells have beenshown to contain membranous FasL-positive vesicles,
which trigger induction of T-cell apoptosis, circum-
venting the cytotoxic response.84,85,95
The cytotoxic response also can be dampened by
suppression. Intratumoral cytotoxic CD8þ T cells in
HNSCC show increased expression of programmeddeath-1 (PD-1), a marker of suppressed function.87,99
Its ligand, programmed death receptor ligand-1 (PD-
L1), is a surface protein that blocks function ofT lymphocytes and is expressed on malignant oral
SCC cells and also on CAFs.100 Cho et al demon-
strated that increased PD-L1 expression resulted inincreased apoptosis of intratumoral CD8þ TILs.101
Moreover, cytokines like, TGF-β, IL-10, and others
allow local naı̈ve T cells to be triggered to becomesuppressor T cells, while also exploiting the suppres-
sive functions of existing regulatory T cells.102PD-1 is
of particular interest in HPV-associated HNSCC, as alymphocytic infiltrate is one of the common features
of HPV-related OPSCC. Infiltration of the TME by PD-
1–positive T lymphocytes was correlated withimproved prognosis.103 While this is contrary to the
above findings, in the case of HPV-related OPSCC, the
PD-1–positive T lymphocytes, likely reflect an acti-vated chronic immune response due to long-standing
viral infection.103
ANTIGEN-PRESENTING CELLS AND TUMOR-ASSOCIATED MACROPHAGES
Dendritic cells are specialized antigen-presenting
cells (APCs) common in the TME of HNSCC.84,98,104
They have a high a capacity for antigen capture andalso stimulate T-cell maturation. In contrast, when
exposed to TGF-β and IL-10, they can promote
immune tolerance and differentiation of CD4þ Tcells into suppressive regulatory T cells.84,105–107
Langerhans cells are APCs located within the skin
and mucous membranes of the upper aerodigestivetract. They detect antigens in the mucosa and then
migrate to regional lymph nodes where they initiate
a primary immune response. Some evidence sug-gests that greater infiltration of HNSCC tumor sam-
ples with Langerhans cells correlates with improved
prognosis.84,108–110
Tumor-associated macrophages (TAMs) are
present with varying frequency in tumors, and are
common in HNSCC. TAMs are classified into twovarieties: proinflammatory (M1) and suppressive
(M2). Accordingly, studies in various cancers have
shown that TAMs can be associated with positive ornegative prognosis. M1 TAMs contribute to the anti-
tumor immune response via the production of
proinflammatory cytokines IL-12, IL-23, andinterferon-γ.84,111–113 While the M2 TAMs appear to
accumulate near blood vessels, promote angiogene-
sis,114,115 and produce a variety of suppressivecytokines such as IL-10 and TGF-β. They also serve
to promote tissue remodeling and inhibit anti-tumor
cytotoxic effects of M1 TAMs.84,111–113,116 Data inoral SCC suggest that TAMs are largely of the M2
type, as tumors with high levels of TAM infiltration
correlate with higher stage, lymph node metastasis,and extracapsular spread.114,117,118 Lago Costa et al
demonstrated that macrophages were increased in
the TME and the peripheral blood in HNSCC, andthat samples with increased TAMs showed increased
levels of TGF-β and its correlated immunosuppres-
sive effects.119 They produce ROS, RNS, and prosta-glandins (PGs), all of which can contribute to
inflammation and tumorigenesis. COX2 inhibitors
and nitric oxide synthase inhibitors (iNOS) havebeen used to antagonize these inflammatory agents
and their cytokines.120,121 TAMs in HNSCC also
produce significant levels of macrophage migrationinhibitory factor (MIF), which is an inflammatory
cytokine that stimulates neutrophils. MIF recruits
neutrophils to the tumor via a CXCR2 mechanismand then by feedback mechanisms increases inva-
siveness of the tumor cells.122 Neutrophils act on the
tumor in a variety of ways: inducing genetic insta-bility via ROS, increasing angiogenesis via MMP9 and
VEGF, and increasing invasion via HGF.123
THE BASEMENT MEMBRANE, INVASION, ANDMATRIX METALLOPROTIENASES
The basement membrane is barrier to tumor pro-
gression, and its degradation facilitates tumor invasion
and metastasis. For this to occur, cancer cells must(1) develop motility, (2) alter cell–cell adhesion, and(3) remodel the ECM.124 The basement membrane not
only serves as a structural framework for the overlyingepithelial cells but also provides paracrine signals that

J.M. Curry et al224
affect their behaviors such as differentiation and migra-
tion.125 Many of the key elements of the basementmembrane, including collagen type IV and fibronectin,
have been shown to be disregulated in HNSCC. MMPs
are most important group of proteolytic enzymes usedby cancer to degrade the ECM. MMPs in normal tissues
are expressed in balance with their inhibitors to main-
tain a well-organized system. MMPs are upregulated byNOTCH1 pathways, EGFR, TGF-β, HGF, and
granulocyte-macrophage colony-stimulating factor (GM-
CSF), which are commonly overexpressed inHNSCC.126–129 Among the most commonly identified
metalloproteinases in HNSCC are MMP-2, MMP-9 and
membrane-bound MMP (MT-MMP). MMP-2 and MMP-9are gelatinases and degrade collagen type IV, the most
critical step in degrading the BM.130 Increased levels of
MMP-2 and MMP-9 correlate with increased nodalmetastasis and poor prognosis.48 MMP-9 is the most
structurally complex and can degrade numerous ele-
ments of the TME, including elastin, fibrillin, laminin,gelatin, and types IV, V, XI, and XVI collagen.131,132
MMPs were initially thought to be produced solely by
the tumor cell, but further investigation has shownproduction also by the CAFs and surrounding inflam-
matory cells.48,133,134 CAFs are primarily responsible for
the increased production of MMP-2 in co-culture experi-ments.130 MT-MMP is critical in activating MMP-2.135,136
There are numerous other significant MMPs, such as
MMP-13, which participates in angiogenesis increasingthe level of VEGF at the invasive front.137
Importantly, the functions of MMPs extend
beyond protein degradation and invasion, as theytarget growth factors, growth factor receptors, and
cytokines.8 For example, MMP-9 also produces a
tolerogenic effect on dendritic APCs and also onregulatory T cells.43 Release of MMP-9 results in
endothelial cell invasion and vessel formation.12,55
MMPs impact differentiation and maturation of bonecells into osteoclasts, which is critical to the process
of bony invasion.138 HNSCC CSCs are characterized
by expression of CD44; CD44 is a surface proteinthat functions as a receptor for hyaluronic acid and
also is the docking receptor necessary for MMP-9
function.139 Given the broad significance of MMPsthey represent a possible target for therapy directed
at the TME. Interestingly, quercitin, a flavonoid
isolated from onions, inhibits MMP-2 and -9pathways.140
TGF-b AND EPITHELIAL–MESENCHYMALTRANSITION
A number of chemokines and cytokines providecritical paracrine signaling in the TME; here we focus
on TGF-β, as it broadly impacts many cellular behav-
iors in the TME. TGF-β has both growth-promotingand -suppressive effects on cells, and for some time
the role of TGF-β in malignancy had been controver-
sial. It typically inhibits epithelial cell proliferationand promotes secretion of matrix proteins and
proteases. Currently it is understood to act as a
tumor-suppressor early in tumorigenesis, then in laterphases it enhances the malignant phenotype.141 TGF-
β primarily acts through the SMAD family of tran-
scription factors and works in concert with mitogen-activated protein kinases (MAPKs), which regulate
diverse cellular activities such as mitosis, differentia-
tion, proliferation, cell survival, and apoptosis. Dys-regulation of TGF-β in malignancy occurs through
several mechanisms, including loss of response to its
ligand, defects in the transduction pathway, andothers.141 Oral SCC has been shown to be resistant
to the suppressive effects of TGF-β, secondary to
downregulation of TGF-β receptor II (TBRII).141,142
TGF-β is a primary factor triggering EMT in
HNSCC. EMT contributes to invasion allowing for
enhanced mobility via expression of a protein expres-sion patterns more characteristic of a mesenchymal
phenotype. Once established, nests of metastatic
tumor can transition back to a phenotype recapitulat-ing the original tumor in a distant site. EMT is
mediated through disruption of epithelial cell junc-
tions, remodeling of the actin cytoskeleton, andupregulation of mesenchymal markers like vimentin
and firbonectin.141 TGF-β pathways as well as those
triggered by the inflammatory cytokines TNF-α andIL-6 converge upon STAT3, upregulating it.52,143,144
STAT3 proteins are commonly overexpressed in
HNSCC. STAT3 interacts with Twist, Snail, and Slug(Snail2), transcription factors that contribute to EMT
in various cancers.52,144,145 Twist increases expres-
sion of N cadherin, a marker of a mesenchymalmotile phenotype, and decreases expression of E
cadherin, a marker of an epithelial phenotype. Slug
also decreases expression of E-cadherin. Loss ofE-cadherin and gained expression of N-cadherin is
critical to invasion and is referred to as cadherin
switching.146 Prime et al were able to demonstratemorphologic evidence of EMT and cadherin switch-
ing after several days exposure to TGF-β.138,147
Emerging evidence suggests that EMT is fundamentalto gaining “stemness” or the transition of cancer cells
to becoming CSCs. CSCs are thought to serve as a
fountainhead for tumors as they give rise to theremaining population of tumor cells, and contribute
to treatment resistance. CSCs accumulate at the
invasive front and perivascular spaces and are demar-cated by expression of markers such as CD133 and
CD44, and by aldehyde dehydrogenase activity.139
On the surface of CSCs, CD44 interacts with MMP-9and this allows for proteolytic activation of TGF-β.148
TGF-β extends beyond the epithelial cancer cells of
a tumor, and many of the effects have been describedabove. Lewis et al showed that TGF-β produced at the

Tumor microenvironment in HNSCC 225
invasive leading edge of the tumor induced a MF
phenotype in primary fibroblasts. They also showedthat this effect resulted in secretion of HGF by
myofibrblasts, which in turn promoted invasion
through the basement membrane. TGF-β serves toinhibit TH1 lymphocytes and cytotoxic T lympho-
cytes and the functions of natural killer cells.84
ANGIOGENESIS, INFLAMMATION, ANDHYPOXIA
Small tumor deposits of 1–3 mm can be supplied
by diffusion of nutrients from the surrounding tissue;
beyond this, the tumor is dependent on angiogenesisto supply its needs.149 A number of studies have
shown that angiogenesis is correlated with tumor
aggression.48,150–154 HNSCC often has large hypoxicareas of tumor necrosis where growth exceeds
angiogenesis.155–157 Hypoxic response and inflam-
mation are driving forces in angiogenesis.12,158 More-over, CSCs in HNSCC appear to be concentrated
along the invasive front of the tumor and in the
perivascular niche, an area within 100 μm of themicrovasculature. A variety of factors in the TME,
such as VEGF, NF-κB, and HIF-1α play central roles in
this process.VEGF enhances endothelial growth, migration of
endothelial precursors, and their differentiation. High
VEGF expression in oral SCC has been correlatedwith a poor prognosis, and a recent meta-analysis
suggested that VEGF overexpression could be a
useful prognostic marker.159 VEGF binds to its recep-tor, VEGFR1 in tumor cells, and induces expression
of Bcl-2, inducing chemokines like CXCL1 and
CXCL8. CXCL1 and CXCL8 promote endothelial cellproliferation and survival.160 Endothelial cells in turn
produce factors like EGF, which significantly increase
tumor cell survival and migration.161 VEGF and otherangiogenic factors such as IL-6 and IL-8 are increased
by a number of chemokines such as CXCL12, which
binds to chemokine receptors CXCR2 and CXCR4.High CXCR2 and CXCR4 levels have been shown to
be associated with increased microvessel density
within tumors.55,77,162
Chronic inflammation of the TME contributes to
tumor progression through a variety of mechanisms,
including production ROS and angiogenic factors.NF-κβ is an inflammatory signal transcription factor
playing a variety roles in invasion, proliferation, and
angiogenesis. Constitutive activation NF-κβ results inoverexpression of a variety of factors, including Il-6,
IL-8, and VEGF.43
There are many downstream inflammatorymarkers expressed as a result of NF-κβ and other
mechanisms, such as cyclooxygenases like COX-2.163
COX enzymes catalyze the production of PGs andlikely are the rate-limiting step in their synthesis.
COX-2 is usually overexpressed in inflammation and
preneoplastic lesions. PGs are increased in HNSCC,and PGE2 promotes invasion and angiogenesis and
inhibits apoptosis of cancer cells. COX-2 acts on
VEGF, fibroblast growth factor, and MMPs and is alsopro-angiogenic. COX-2 levels have been found to be
prognostic and selective COX-2 inhibitors have
been shown to increase the efficacy of radio-therapy in vitro.51,164 NF-κβ is the target of many
therapeutic interventions, such as curcumin, n-acetyl
cysteine (NAC), epigallocatechin gallate (EGCG), andothers.5
Intratumoral hypoxia is a key characteristic of
HNSCC, and is a negative prognostic factor, contri-buting to both chemotherapy and radiotherapy resist-
ance. Intratumoral hypoxia is generally accepted to
be a pO2 o10 mm Hg, and intratumoral pO2 levels≤2.5 mm Hg correlate with a worsened prognosis, as
does the overall volume of hypoxic tumor at the
primary site.165 HIF-1α is the most important factorinduced in adaptive response to hypoxia, and ele-
vated expression is also directly associated with a
poor prognosis.166 This transcription factor interactswith more than 100 genes to alter expression of
VEGF, CA9, lysyl oxidase, and many others.48,167 It
has been shown to alter cellular metabolism, and toincrease lymphatic vessel density and blood vessel
density in oral SCC.168,169 CA9 functions to regulate
pH homeostasis and alter the uptake of chemother-apeutic drugs, and also is purported to play a role in
proliferation and cell adhesion.165,170 Lysyl oxidase
catalyzes the crosslinking of collagens and elastins,and overexpression increases microvascular den-
sity.171,172 Agents such as reseveratrol, EGCG and
others may act by promoting degradation of HIF-1α.173–175 Resveratrol has been shown to decrease
expression of HIF-1α and VEGF in vitro.176
METABOLISM IN THE TME
Cancer cells have high bioenergetic requirementsneeded to maintain tumor growth. Tumor cells in
culture have long been demonstrated to rely heavily
on glycolysis with decreased, dysfunctional, or absentmitochondrial OXPHOS. Reliance on glycolysis in the
presence of oxygen is referred to as the Warburg
effect.177 This results in the generation of less ATPthan OXPHOS and yields high levels of pyruvate and
lactate. This is somewhat counterintuitive as there is
such a high bioenergetics requirement, yet OXPHOSis a more efficient means of energy generation than
glycolysis. Thus it is unclear why tumor cells would
thrive with a less efficient mechanism. It has beenhypothesized that glycolysis may confer a growth
advantage.178–180 Some normal, highly proliferative
cells, such as lymphocytes, favor aerobic glycolysisover oxidative metabolism, providing a rationale for

J.M. Curry et al226
the Warburg effect.181 Many cancer cells have defects
in critical components of the OXPHOS pathway, suchas the mitochondrial B-catalytic subunit of Hþ-ATPsynthase.182,183 Furthermore, when glycolytic flux is
high, the ATP yield can exceed that produced byOXPHOS.182,183 Additionally, the intermediates of
glycolytic metabolism can provide substrates for
amino acid, fatty acid, and nucleotide synthesis.167
The metabolic pressures induced by hypoxia in the
setting of rapid growth may then in turn select for
tumor cells which favor glycolytic metabolism evenin the presence of oxygen, as is suggested by the
frequent overexpression of HIF-1α in many cancers.
Hypoxic induction of HIF-1α favors this processspecifically inducing pyruvate dehydrogenase kinase
(PDK) and lactate dehydrogenase a (LDH-A). PDK
inactivates pyruvate dehydrogenase preventingimport of pyruvate to the mitochondria. LDH-A
restores NAD positivity and also uses pyruvate in
the cytosol, which together can reduce electron flowthough OXPHOS and also reduce oxidative stress.
Additionally, glycolytic metabolism results in the
acidotic efflux into the TME that assists in breakdownof the ECM and kills non-adapted normal cells.184
However, this is not likely the whole picture:
much recent evidence suggests that a metabolicsymbiosis exists within tumors cell between differ-
ent populations. Feron has likened this to the
coupling between fast and slow-twitch musclefibers. Fast-twitch glycolytic fibers release lactate
that is then taken up and utilized by slow twitch
fibers. MCT1 is a high-affinity transporter of lactate,which mediates influx into the cell; MCT4 is a low-
affinity transporter of lactate, which primarily medi-
ates efflux of lactate from cells. These transporterscouple cancer cells, so that hypoxic cells maintain
functioning glycolytic metabolism while aerobic
tumor cells recycle and utilize lactate and otherhigh-energy substrates produced by them. A similar
process in cancer would allow for an efficient intra-
tumoral metabolic coupling mechanism betweenoxygenated cells and hypoxic cells.179
Additional evidence favors multicompartmental
metabolism between the cancer cells and CAFs.Numerous co-culture experiments and in situ tumor
analyses have demonstrated this effect in breast and
other cancers.11,81,185 This work has brought to lighta “reverse Warburg effect”, where oxidative stresses
exerted by tumor cells induce aerobic glycolysis and
autophagy in CAFs. This, in turn, results in increasedlevels of intermediate catabolites such as lactate,
glutamine, and ketone bodies. These catabolites are
released into the TME and used for OXPHOS incarcinoma cells. This metabolically enriches the TME
and creates an environment that favors growth,
apoptosis resistance, invasion, and metastasis.81,185
This is Paget’s seed and soil hypothesis, a
phenomenon that may have been unnoticed in
previous homotypic culture experiments.11
Most studies on HNSCC cellular metabolism sug-
gest that the carcinoma cells are highly glycolytic
with high L-lactate generation, yet recent studiessuggest that metabolic heterogeneity and metabolic
coupling occur. Most HNSCC cells generate signifi-
cantly higher levels of lactate compared with normalhuman oral keratinocytes (NHOK), although several
cell lines generate significantly lower lactate levels
than NHOKs.186 It has been postulated that the cellswith decreased lactate production have increased
lactate uptake via MCTs, allowing them to utilize
OXPHOS.186 When some HNSCC cell lines that aretypically glycolytic are supplemented with excess
pyruvate, some of the effects were reversed, which
suggests that OXPHOS is important to supportHNSCC cell proliferation in the presence of a
catabolite-rich microenvironment.187 High tumor
lactate concentrations in HNSCC are associated withsubsequent nodal and distant metastatses.188,189
In our previously published work on oral SCC, we
demonstrated evidence of this multicompartmentmodel of metabolism. We have suggested that there
may be three metabolic compartments in HNSCC,
where the leading tumor edge relies on OXPHOSand the deeper layers of the tumor are more
glycolytic (aerobic or anaerobic) and tumor stroma
represents a third compartment undergoing aerobicglycolysis (Figure 2). This was demonstrated through
high expression of MCT4 in the stroma and deeper
tumor, while MCT1 was more highly expressed bythe leading tumor edge. We also confirmed OXPHOS
in the leading tumor edge with assays for TOMM20
and LDHb, both functional markers for mitochon-drial metabolism. This pattern of metabolic coupling
was demonstrated in a subset of our oral SCC
patients, and correlated with aggressive behaviorincluding a worsened disease-free survival and peri-
neural invasion. Interestingly, it also correlated with
increased specific uptake values (SUV) on positronemission tomography/computed tomography. We
further tested this metabolic coupling theory with
a squamous cell carcinoma line co-culture experi-ment. Using immortalized squamous cell lines we
were able to generate two divergent SCC popultions,
one RAS-dependent and another NF-κB–dependent.These cell lines were each able to induce metabolic
reprogramming of CAFs via oxidative stress. This
resulted in a lactate shuttling process that feeds thecancer cells fueling anabolic growth via and MCT1/
MCT4 metabolic couple between the tumor and the
stroma. Interestingly, this model also demonstratedthat the CAFs protected the cancer cells against
oxidative stress by reducing oxidative stresses within
the carcinoma cells. RAS-transformed cells were ableto reprogram adjacent epithelial cells, as well as
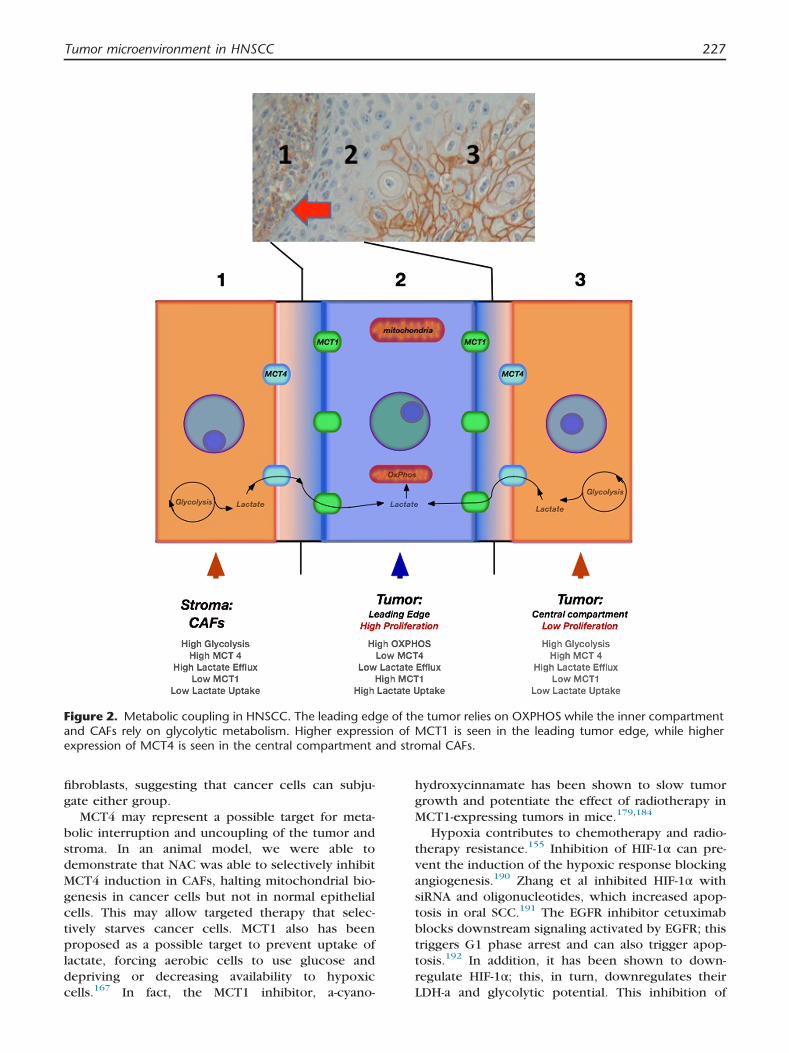
Figure 2. Metabolic coupling in HNSCC. The leading edge of the tumor relies on OXPHOS while the inner compartmentand CAFs rely on glycolytic metabolism. Higher expression of MCT1 is seen in the leading tumor edge, while higherexpression of MCT4 is seen in the central compartment and stromal CAFs.
Tumor microenvironment in HNSCC 227
fibroblasts, suggesting that cancer cells can subju-gate either group.
MCT4 may represent a possible target for meta-
bolic interruption and uncoupling of the tumor andstroma. In an animal model, we were able to
demonstrate that NAC was able to selectively inhibit
MCT4 induction in CAFs, halting mitochondrial bio-genesis in cancer cells but not in normal epithelial
cells. This may allow targeted therapy that selec-
tively starves cancer cells. MCT1 also has beenproposed as a possible target to prevent uptake of
lactate, forcing aerobic cells to use glucose and
depriving or decreasing availability to hypoxiccells.167 In fact, the MCT1 inhibitor, a-cyano-
hydroxycinnamate has been shown to slow tumorgrowth and potentiate the effect of radiotherapy in
MCT1-expressing tumors in mice.179,184
Hypoxia contributes to chemotherapy and radio-therapy resistance.155 Inhibition of HIF-1α can pre-
vent the induction of the hypoxic response blocking
angiogenesis.190 Zhang et al inhibited HIF-1α withsiRNA and oligonucleotides, which increased apop-
tosis in oral SCC.191 The EGFR inhibitor cetuximab
blocks downstream signaling activated by EGFR; thistriggers G1 phase arrest and can also trigger apop-
tosis.192 In addition, it has been shown to down-
regulate HIF-1α; this, in turn, downregulates theirLDH-a and glycolytic potential. This inhibition of

J.M. Curry et al228
glycolytic potential leads to inhibition of
proliferation.192
Metformin is a commonly used antihyperglycemic
drug in type 2 diabetics and has been proposed as a
potential anticancer therapy also that may impacttumor metabolism in the TME. Metformin has been
shown to inhibit cancer cell proliferation in several
human cancers, such as gastric, medullary thyroid,breast, and pancreatic cancers.193–196 Epidemiologic
studies also have shown significant effects from
metformin use in diabetics, lowering the risk ofcancer incidence and mortality.197 In oral SCC, Luo
et al demonstrated that metformin blocked cell cycle
progression at the G0/G1 phase and induced apop-tosis. Metformin triggered alterations in multiple
other pathways as well: increasing activation of the
adenosine monophosphate (AMP) kinase pathway,suppressing the mammalian target of rapamycin
(mTOR) pathway, decreasing cyclin D1 levels and
retinoblastoma (Rb) phosphorylation, and downregu-lating Bcl 2.198 They also were able to demonstrate
in vivo evidence of increased apoptosis in a xenograft
model. While this study demonstrated various effectson the cell cycle; the metabolic effects of metformin
on cancer have yet to be investigated.
CONCLUSION
Many elements of the TME beyond the cancerousepithelial cells impact progression of HNSCC.
Genetic alterations induced by tobacco and alcohol
or the HPV virus initiate the sequence of events thattrigger transformation of stromal cells, immune sup-
pression, and chronic inflammation. In turn,
unchecked growth, invasion, and metastasis prevail.The complexity of these processes reveals that the
long-held notion of “condemned mucosa” actually
reflects a “condemned tissue” comprised of many celltypes which have co-evolved during tumorigenesis.
REFERENCES1. Kamangar F, Dores GM, Anderson WF. Patterns of
cancer incidence, mortality, and prevalence across
five continents: defining priorities to reduce cancer
disparities in different geographic regions of the
world. J Clin Oncol. 2006;24:2137–50.
2. Marur S, Forastiere AA. Head and neck cancer:
changing epidemiology, diagnosis, and treatment.
Mayo Clin Proc. 2008;83:489–501.
3. Leemans CR, Tiwari R, Nauta JJ, van der Waal I, Snow
GB. Recurrence at the primary site in head and neck
cancer and the significance of neck lymph node
metastases as a prognostic factor. Cancer. 1994;73:
187–90.
4. Slaughter DP, Southwick HW, Smejkal W. Field
cancerization in oral stratified squamous epithelium;
clinical implications of multicentric origin. Cancer.
1953;6:963–8.
5. Albini A, Sporn MB. The tumour microenvironment
as a target for chemoprevention. Nat Rev Cancer.
2007;7:139–47.
6. Hanahan D, Weinberg RA. The hallmarks of cancer.
Cell. 2000;100:57–70.
7. Haddad RI, Shin DM. Recent advances in head and
neck cancer. N Engl J Med. 2008;359:1143–54.
8. Mbeunkui F, Johann DJ, Jr. Cancer and the tumor
microenvironment: a review of an essential relation-
ship. Cancer Chemother Pharmacol. 2009;63:571–82.
9. Weinberg RA. Twisted epithelial-mesenchymal tran-
sition blocks senescence. Nat Cell Biol. 2008;10:
1021–3.
10. Cavallo F, De Giovanni C, Nanni P, Forni G, Lollini
PL. The immune hallmarks of cancer. Cancer Immu-
nol Immunother. 2011;60:319–26.
11. Paget S. The distribution of secondary growths in
cancer of the breast. Cancer Metastasis Rev. 1989;8:
98–101.
12. Albini A, Tosetti F, Benelli R, Noonan DM. Tumor
inflammatory angiogenesis and its chemoprevention.
Cancer Res. 2005;65:10637–41.
13. Karin M. NF-kappaB and cancer: mechanisms and
targets. Mol Carcinog. 2006;45:355–61.
14. Bertl E, Bartsch H, Gerhauser C. Inhibition of angio-
genesis and endothelial cell functions are novel
sulforaphane-mediated mechanisms in chemopre-
vention. Mol Cancer Ther. 2006;5:575–85.
15. Leemans CR, Braakhuis BJ, Brakenhoff RH. The
molecular biology of head and neck cancer. Nat
Rev Cancer. 2011;11:9–22.
16. Ramqvist T, Dalianis T. An epidemic of oropharyng-
eal squamous cell carcinoma (OSCC) due to human
papillomavirus (HPV) infection and aspects of treat-
ment and prevention. Anticancer Res. 2011;31:
1515–9.
17. Syrjanen S, Lodi G, von Bultzingslowen I, Aliko A,
Arduino P, Campisi G, et al. Human papillomaviruses
in oral carcinoma and oral potentially malignant
disorders: a systematic review. Oral Dis. 2011;17
(Suppl 1):58–72.
18. Rautava J, Syrjanen S. Biology of human papilloma-
virus infections in head and neck carcinogenesis.
Head Neck Pathol. 2012;6(Suppl 1):S3–15.
19. Boyle JO, Hakim J, Koch W, van der Riet P, Hruban
RH, Roa RA, et al. The incidence of p53 mutations
increases with progression of head and neck cancer.
Cancer Res. 1993;53:4477–80.
20. el-Naggar AK, Lai S, Luna MA, Zhou XD, Weber RS,
Goepfert H, et al. Sequential p53 mutation analysis of
pre-invasive and invasive head and neck squamous
carcinoma. Int J Cancer. 1995;64:196–201.
21. Tan M, Myers JN, Agrawal N. Oral cavity and
oropharyngeal squamous cell carcinoma genomics.
Otolaryngol Clin North Am. 2013;46:545–66.
22. Koch WM, Brennan JA, Zahurak M, Goodman SN,
Westra WH, Schwab D, et al. p53 mutation and
locoregional treatment failure in head and neck
squamous cell carcinoma. J Natl Cancer Inst. 1996;
88:1580–6.
23. Alsner J, Sorensen SB, Overgaard J. TP53 mutation is
related to poor prognosis after radiotherapy, but not

Tumor microenvironment in HNSCC 229
surgery, in squamous cell carcinoma of the head and
neck. Radiother Oncol. 2001;59:179–85.
24. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy
JS, Beadle BM, et al. TP53 disruptive mutations lead
to head and neck cancer treatment failure through
inhibition of radiation-induced senescence. Clin Can-
cer Res. 2011;18:290–300.
25. Huang X, Pateromichelakis S, Hills A, Sherriff M,
Lyons A, Langdon J, et al. p53 mutations in deep
tissues are more strongly associated with recurrence
than mutation-positive mucosal margins. Clin Cancer
Res. 2007;13:6099–106.
26. Lin SY, Dolfi SC, Amiri S, Li J, Budak-Alpdogan T, Lee
KC, et al. P53 regulates the migration of mesenchy-
mal stromal cells in response to the tumor micro-
environment through both CXCL12-dependent and
-independent mechanisms. Int J Oncol. 2013;43:
1817–23.
27. He H, Tian D, Guo J, Liu M, Chen Z, Hamdy FC, et al.
DNA damage response in peritumoral regions of
oesophageal cancer microenvironment. Carcinogen-
esis. 2013;34:139–45.
28. Bhowmick NA, Neilson EG, Moses HL. Stromal
fibroblasts in cancer initiation and progression.
Nature. 2004;432:332–7.
29. Hu M, Polyak K. Microenvironmental regulation of
cancer development. Curr Opin Genet Dev. 2008;18:
27–34.
30. Szatrowski TP, Nathan CF. Production of large
amounts of hydrogen peroxide by human tumor
cells. Cancer Res. 1991;51:794–8.
31. Cairns RA, Harris IS, Mak TW. Regulation of cancer
cell metabolism. Nat Rev Cancer. 2011;11:85–95.
32. Bolos V, Grego-Bessa J, de la Pompa JL. Notch
signaling in development and cancer. Endocr Rev.
2007;28:339–63.
33. Yoshida R, Nagata M, Nakayama H, Niimori-Kita K,
Hassan W, Tanaka T, et al. The pathological signifi-
cance of Notch1 in oral squamous cell carcinoma.
Lab Invest. 2013;93:1068–81.
34. Liao S, Xia J, Chen Z, Zhang S, Ahmad A, Miele L,
et al. Inhibitory effect of curcumin on oral carcinoma
CAL-27 cells via suppression of Notch-1 and NF-
kappaB signaling pathways. J Cell Biochem. 2011;
112:1055–65.
35. Subramaniam D, Ponnurangam S, Ramamoorthy P,
Standing D, Battafarano RJ, Anant S, et al. Curcumin
induces cell death in esophageal cancer cells
through modulating Notch signaling. PLoS One.
2012;7:e30590.
36. Temam S, Kawaguchi H, El-Naggar AK, Jelinek J,
Tang H, Liu DD, et al. Epidermal growth factor
receptor copy number alterations correlate with
poor clinical outcome in patients with head and
neck squamous cancer. J Clin Oncol. 2007;25:
2164–70.
37. Chung CH, Ely K, McGavran L, Varella-Garcia M,
Parker J, Parker N, et al. Increased epidermal growth
factor receptor gene copy number is associated with
poor prognosis in head and neck squamous cell
carcinomas. J Clin Oncol. 2006;24:4170–6.
38. Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R,
Hammond EH, et al. Impact of epidermal growth
factor receptor expression on survival and pattern of
relapse in patients with advanced head and neck
carcinoma. Cancer Res. 2002;62:7350–6.
39. Rubin Grandis J, Melhem MF, Gooding WE, Day R,
Holst VA, Wagener MM, et al. Levels of TGF-alpha
and EGFR protein in head and neck squamous cell
carcinoma and patient survival. J Natl Cancer Inst.
1998;90:824–32.
40. Agrawal N, Frederick MJ, Pickering CR, Bettegowda
C, Chang K, Li RJ, et al. Exome sequencing of head
and neck squamous cell carcinoma reveals inactivat-
ing mutations in NOTCH1. Science. 2011;333:
1154–7.
41. Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC,
et al. Functional genomic analysis identified epider-
mal growth factor receptor activation as the most
common genetic event in oral squamous cell carci-
noma. Cancer Res. 2009;69:2568–76.
42. Kim S, Grandis JR, Rinaldo A, Takes RP, Ferlito A.
Emerging perspectives in epidermal growth factor
receptor targeting in head and neck cancer. Head
Neck. 2008;30:667–74.
43. Wang BQ, Zhang CM, Gao W, Wang XF, Zhang HL,
Yang PC. Cancer-derived matrix metalloproteinase-9
contributes to tumor tolerance. J Cancer Res Clin
Oncol. 2011;137:1525–33.
44. Nijkamp MM, Span PN, Bussink J, Kaanders JH.
Interaction of EGFR with the tumour microenviron-
ment: implications for radiation treatment. Radiother
Oncol. 2013;108:17–23.
45. Zimmermann M, Zouhair A, Azria D, Ozsahin M. The
epidermal growth factor receptor (EGFR) in head
and neck cancer: its role and treatment implications.
Radiat Oncol. 2006;1:11.
46. Rogers SJ, Harrington KJ, Rhys-Evans P, P OC, Eccles
SA. Biological significance of c-erbB family onco-
genes in head and neck cancer. Cancer Metastasis
Rev. 2005;24:47–69.
47. Kalyankrishna S, Grandis JR. Epidermal growth factor
receptor biology in head and neck cancer. J Clin
Oncol. 2006;24:2666–72.
48. Koontongkaew S. The tumor microenvironment
contribution to development, growth, invasion and
metastasis of head and neck squamous cell carcino-
mas. J Cancer. 2013;4:66–83.
49. Horiguchi M, Koyanagi S, Okamoto A, Suzuki SO,
Matsunaga N, Ohdo S. Stress-regulated transcription
factor ATF4 promotes neoplastic transformation by
suppressing expression of the INK4a/ARF cell sen-
escence factors. Cancer Res. 2012;72:395–401.
50. Bova RJ, Quinn DI, Nankervis JS, Cole IE, Sheridan
BF, Jensen MJ, et al. Cyclin D1 and p16INK4A
expression predict reduced survival in carcinoma
of the anterior tongue. Clin Cancer Res. 1999;5:
2810–9.
51. Wang F, Arun P, Friedman J, Chen Z, Van Waes C.
Current and potential inflammation targeted thera-
pies in head and neck cancer. Curr Opin Pharmacol.
2009;9:389–95.

J.M. Curry et al230
52. Chen YW, Chen KH, Huang PI, Chen YC, Chiou GY,
Lo WL, et al. Cucurbitacin I suppressed stem-like
property and enhanced radiation-induced apoptosis
in head and neck squamous carcinoma–derived
CD44(þ)ALDH1(þ) cells. Mol Cancer Ther. 2010;9:
2879–92.
53. Du Y, Peyser ND, Grandis JR. Integration of molec-
ular targeted therapy with radiation in head and neck
cancer. Pharmacol Ther. 2013.
54. Chung CH, Parker JS, Karaca G, Wu J, Funkhouser
WK, Moore D, et al. Molecular classification of head
and neck squamous cell carcinomas using patterns of
gene expression. Cancer Cell. 2004;5:489–500.
55. Benelli R, Morini M, Carrozzino F, Ferrari N, Min-
ghelli S, Santi L, et al. Neutrophils as a key cellular
target for angiostatin: implications for regulation of
angiogenesis and inflammation. Faseb J. 2002;16:
267–9.
56. Clatot F, Gouerant S, Mareschal S, Cornic M, Ber-
ghian A, Choussy O, et al. The gene expression
profile of inflammatory, hypoxic and metabolic
genes predicts the metastatic spread of human head
and neck squamous cell carcinoma. Oral Oncol.
2013;50:200–7.
57. Shaw R. The epigenetics of oral cancer. Int J Oral
Maxillofac Surg. 2006;35:101–8.
58. Ogawa T, Liggett TE, Melnikov AA, Monitto CL,
Kusuke D, Shiga K, et al. Methylation of death-
associated protein kinase is associated with cetux-
imab and erlotinib resistance. Cell Cycle. 2012;11:
1656–63.
59. Alaibac M. Targeting DSG3: from pemphigus to
squamous cell carcinoma. Expert Opin Ther Targets.
2013;17:477–9.
60. Jung AC, Job S, Ledrappier S, Macabre C, Abecassis J,
de Reynies A, et al. A poor prognosis subtype of
HNSCC is consistently observed across methylome,
transcriptome, and miRNome analysis. Clin Cancer
Res. 2013;19:4174–84.
61. Wheeler SE, Shi H, Lin F, Dasari S, Bednash J, Thorne
S, et al. Enhancement of head and neck squamous
cell carcinoma proliferation, invasion, and metastasis
by tumor-associated fibroblasts in preclinical models.
Head Neck. 2013.
62. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A.
Circulating fibrocytes define a new leukocyte sub-
population that mediates tissue repair. Mol Med.
1994;1:71–81.
63. Abe R, Donnelly SC, Peng T, Bucala R, Metz CN.
Peripheral blood fibrocytes: differentiation pathway
and migration to wound sites. J Immunol. 2001;166:
7556–62.
64. Franco OE, Shaw AK, Strand DW, Hayward SW.
Cancer associated fibroblasts in cancer pathogenesis.
Semin Cell Dev Biol. 2010;21:33–9.
65. Kawashiri S, Tanaka A, Noguchi N, Hase T, Nakaya
H, Ohara T, et al. Significance of stromal desmoplasia
and myofibroblast appearance at the invasive front in
squamous cell carcinoma of the oral cavity. Head
Neck. 2009;31:1346–53.
66. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C,
Brown RA. Myofibroblasts and mechano-regulation
of connective tissue remodelling. Nat Rev Mol Cell
Biol. 2002;3:349–63.
67. Tlsty TD, Hein PW. Know thy neighbor: stromal cells
can contribute oncogenic signals. Curr Opin Genet
Dev. 2001;11:54–9.
68. Chen Y, Satoh T, Sasatomi E, Miyazaki K, Tokunaga
O. Critical role of type IV collagens in the growth of
bile duct carcinoma. In vivo and in vitro studies.
Pathol Res Pract. 2001;197:585–96.
69. Kunz-Schughart LA, Knuechel R. Tumor-associated
fibroblasts (part I): Active stromal participants in
tumor development and progression? Histol Histo-
pathol. 2002;17:599–621.
70. Lim KP, Cirillo N, Hassona Y, Wei W, Thurlow JK,
Cheong SC, et al. Fibroblast gene expression profile
reflects the stage of tumour progression in oral
squamous cell carcinoma. J Pathol. 2011;223:
459–69.
71. Leef G, Thomas SM. Molecular communication
between tumor-associated fibroblasts and head and
neck squamous cell carcinoma. Oral Oncol. 2013;
49:381–6.
72. Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi
S, Klein-Szanto AJ, et al. Fibroblast-secreted hepato-
cyte growth factor plays a functional role in esoph-
ageal squamous cell carcinoma invasion. Proc Natl
Acad Sci U S A. 2010;107:11026–31.
73. Knowles LM, Stabile LP, Egloff AM, Rothstein ME,
Thomas SM, Gubish CT, et al. HGF and c-Met
participate in paracrine tumorigenic pathways in
head and neck squamous cell cancer. Clin Cancer
Res. 2009;15:3740–50.
74. Rousseau B, Larrieu-Lahargue F, Javerzat S, Guilhem-
Ducleon F, Beermann F, Bikfalvi A. The tyrp1-Tag/
tyrp1-FGFR1-DN bigenic mouse: a model for selec-
tive inhibition of tumor development, angiogenesis,
and invasion into the neural tissue by blockade of
fibroblast growth factor receptor activity. Cancer
Res. 2004;64:2490–5.
75. Ishikawa T, Nakashiro K, Klosek SK, Goda H, Hara S,
Uchida D, et al. Hypoxia enhances CXCR4 expres-
sion by activating HIF-1 in oral squamous cell
carcinoma. Oncol Rep. 2009;21:707–12.
76. De Wever O, Demetter P, Mareel M, Bracke M.
Stromal myofibroblasts are drivers of invasive cancer
growth. Int J Cancer. 2008;123:2229–38.
77. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F,
Delaunay T, Naeem R, et al. Stromal fibroblasts
present in invasive human breast carcinomas pro-
mote tumor growth and angiogenesis through ele-
vated SDF-1/CXCL12 secretion. Cell. 2005;121:
335–48.
78. Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper
C, Jerjes W, et al. Stromal features are predictive of
disease mortality in oral cancer patients. J Pathol.
2011;223:470–81.
79. Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT,
Chen YL. Targeting galectin-1 in carcinoma-
associated fibroblasts inhibits oral squamous cell
carcinoma metastasis by downregulating MCP-1/
CCL2 expression. Clin Cancer Res. 2011;17:
1306–16.

Tumor microenvironment in HNSCC 231
80. Lin J, Liu C, Ge L, Gao Q, He X, Liu Y, et al.
Carcinoma-associated fibroblasts promotes the pro-
liferation of a lingual carcinoma cell line by secreting
keratinocyte growth factor. Tumour Biol. 2011;32:
597–602.
81. Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power
surge: supporting cells “fuel” cancer cell mitochon-
dria. Cell Metab. 2012;15:4–5.
82. Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA,
Anantharaman A, Butera A, et al. Cancer metabolism,
stemness and tumor recurrence: MCT1 and MCT4
are functional biomarkers of metabolic symbiosis in
head and neck cancer. Cell Cycle. 2013;12:1371–84.
83. Dvorak HF. Angiogenesis: update 2005. J Thromb
Haemost. 2005;3:1835–42.
84. Duray A, Demoulin S, Hubert P, Delvenne P, Saussez
S. Immune suppression in head and neck cancers: a
review. Clin Dev Immunol. 2010;2010:701657.
85. Whiteside TL. Immunobiology of head and neck
cancer. Cancer Metastasis Rev. 2005;24:95–105.
86. Brandwein-Gensler M, Smith RV, Wang B, Penner C,
Theilken A, Broughel D, et al. Validation of the
histologic risk model in a new cohort of patients
with head and neck squamous cell carcinoma. Am J
Surg Pathol. 2010;34:676–88.
87. Maleki S, Schlecht NF, Keller C, Diaz J, Moss J,
Prystowsky MB, et al. Lymphocytic host response
to oral squamous cell carcinoma: an adaptive T-cell
response at the tumor interface. Head Neck Pathol.
2011;5:117–22.
88. Zancope E, Costa NL, Junqueira-Kipnis AP, Valadares
MC, Silva TA, Leles CR, et al. Differential infiltration
of CD8þ and NK cells in lip and oral cavity
squamous cell carcinoma. J Oral Pathol Med. 2010;
39:162–7.
89. Ferris RL, Hunt JL, Ferrone S. Human leukocyte
antigen (HLA) class I defects in head and neck
cancer: molecular mechanisms and clinical signifi-
cance. Immunol Res. 2005;33:113–33.
90. Albers A, Abe K, Hunt J, Wang J, Lopez-Albaitero A,
Schaefer C, et al. Antitumor activity of human
papillomavirus type 16 E7-specific T cells against
virally infected squamous cell carcinoma of the head
and neck. Cancer Res. 2005;65:11146–55.
91. Lopez-Albaitero A, Nayak JV, Ogino T, Machandia A,
Gooding W, DeLeo AB, et al. Role of antigen-
processing machinery in the in vitro resistance of
squamous cell carcinoma of the head and neck cells
to recognition by CTL. J Immunol. 2006;176:3402–9.
92. Hathaway B, Landsittel DP, Gooding W, Whiteside
TL, Grandis JR, Siegfried JM, et al. Multiplexed
analysis of serum cytokines as biomarkers in squa-
mous cell carcinoma of the head and neck patients.
Laryngoscope. 2005;115:522–7.
93. Hoffmann TK, Bier H, Whiteside TL. Targeting the
immune system: novel therapeutic approaches in
squamous cell carcinoma of the head and neck.
Cancer Immunol Immunother. 2004;53:1055–67.
94. Verastegui E, Morales R, Barrera JL, Mueller A, Guz-
man B, Meneses A, et al. Immunological approach in
the evaluation of regional lymph nodes of patients
with squamous cell carcinoma of the head and neck.
Clin Immunol. 2002;102:37–47.
95. Young MR. Protective mechanisms of head and neck
squamous cell carcinomas from immune assault.
Head Neck. 2006;28:462–70.
96. Ogino T, Shigyo H, Ishii H, Katayama A, Miyokawa N,
Harabuchi Y, et al. HLA class I antigen down-
regulation in primary laryngeal squamous cell carci-
noma lesions as a poor prognostic marker. Cancer
Res. 2006;66:9281–9.
97. Grandis JR, Falkner DM, Melhem MF, Gooding WE,
Drenning SD, Morel PA. Human leukocyte antigen
class I allelic and haplotype loss in squamous cell
carcinoma of the head and neck: clinical and immu-
nogenetic consequences. Clin Cancer Res. 2000;6:
2794–802.
98. Tourkova IL, Shurin GV, Chatta GS, Perez L, Finke J,
Whiteside TL, et al. Restoration by IL-15 of MHC
class I antigen-processing machinery in human den-
dritic cells inhibited by tumor-derived gangliosides. J
Immunol. 2005;175:3045–52.
99. Katou F, Ohtani H, Watanabe Y, Nakayama T, Yoshie
O, Hashimoto K. Differing phenotypes between
intraepithelial and stromal lymphocytes in early-
stage tongue cancer. Cancer Res. 2007;67:11195–201.
100. Strome SE, Dong H, Tamura H, Voss SG, Flies DB,
Tamada K, et al. B7-H1 blockade augments adoptive
T-cell immunotherapy for squamous cell carcinoma.
Cancer Res. 2003;63:6501–5.
101. Cho YA, Yoon HJ, Lee JI, Hong SP, Hong SD.
Relationship between the expressions of PD-L1 and
tumor-infiltrating lymphocytes in oral squamous cell
carcinoma. Oral Oncol. 2011;47:1148–53.
102. Ferris RL, Whiteside TL, Ferrone S. Immune escape
associated with functional defects in antigen-
processing machinery in head and neck cancer. Clin
Cancer Res. 2006;12:3890–5.
103. Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel
P, Benhamouda N, et al. PD-1-expressing tumor-
infiltrating T cells are a favorable prognostic bio-
marker in HPV-associated head and neck cancer.
Cancer Res. 2013;73:128–38.
104. Steinbrink K, Mahnke K, Grabbe S, Enk AH, Jonuleit
H. Myeloid dendritic cell: From sentinel of immunity
to key player of peripheral tolerance? Hum Immunol.
2009;70:289–93.
105. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K,
Rivera M, et al. Dendritic cells induce peripheral T
cell unresponsiveness under steady state conditions
in vivo. J Exp Med. 2001;194:769–79.
106. Yamazaki S, Inaba K, Tarbell KV, Steinman RM.
Dendritic cells expand antigen-specific Foxp3þCD25þ CD4þ regulatory T cells including suppres-
sors of alloreactivity. Immunol Rev. 2006;212:
314–29.
107. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH.
Induction of interleukin 10-producing, nonproliferat-
ing CD4(þ) T cells with regulatory properties by
repetitive stimulation with allogeneic immature
human dendritic cells. J Exp Med. 2000;192:
1213–22.

J.M. Curry et al232
108. Yilmaz T, Gedikoglu G, Celik A, Onerci M, Turan E.
Prognostic significance of Langerhans cell infiltration
in cancer of the larynx. Otolaryngol Head Neck Surg.
2005;132:309–16.
109. Ma CX, Jia TC, Li XR, Zhand ZF, Yiao CB. Langerhans
cells in nasopharyngeal carcinoma in relation to
prognosis. In Vivo. 1995;9:225–9.
110. Gallo O, Libonati GA, Gallina E, Fini-Storchi O,
Giannini A, Urso C, et al. Langerhans cells related to
prognosis in patients with laryngeal carcinoma. Arch
Otolaryngol Head Neck Surg. 1991;117:1007–10.
111. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L.
The origin and function of tumor-associated macro-
phages. Immunol Today. 1992;13:265–70.
112. Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-
associated macrophages are a distinct M2 polarised
population promoting tumour progression: potential
targets of anti-cancer therapy. Eur J Cancer. 2006;42:
717–27.
113. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-
1/M-2 macrophages and the Th1/Th2 paradigm. J
Immunol. 2000;164:6166–73.
114. Li C, Shintani S, Terakado N, Nakashiro K, Hamakawa
H. Infiltration of tumor-associated macrophages in
human oral squamous cell carcinoma. Oncol Rep.
2002;9:1219–23.
115. El-Rouby DH. Association of macrophages with
angiogenesis in oral verrucous and squamous cell
carcinomas. J Oral Pathol Med. 2010;39:559–64.
116. Zamarron BF, Chen W. Dual roles of immune cells
and their factors in cancer development and pro-
gression. Int J Biol Sci. 2011;7:651–8.
117. Marcus B, Arenberg D, Lee J, Kleer C, Chepeha DB,
Schmalbach CE, et al. Prognostic factors in oral cavity
and oropharyngeal squamous cell carcinoma. Cancer.
2004;101:2779–87.
118. Liu SY, Chang LC, Pan LF, Hung YJ, Lee CH, Shieh YS.
Clinicopathologic significance of tumor cell-lined
vessel and microenvironment in oral squamous cell
carcinoma. Oral Oncol. 2008;44:277–85.
119. Costa NL, Valadares MC, Souza PP, Mendonca EF,
Oliveira JC, Silva TA, et al. Tumor-associated macro-
phages and the profile of inflammatory cytokines in
oral squamous cell carcinoma. Oral Oncol. 2013;49:
216–23.
120. Karin M, Greten FR. NF-kappaB: linking inflammation
and immunity to cancer development and progres-
sion. Nat Rev Immunol. 2005;5:749–59.
121. Coussens LM, Werb Z. Inflammation and cancer.
Nature. 2002;420:860–7.
122. Dumitru CA, Gholaman H, Trellakis S, Bruderek K,
Dominas N, Gu X, et al. Tumor-derived macrophage
migration inhibitory factor modulates the biology of
head and neck cancer cells via neutrophil activation.
Int J Cancer. 2011;129:859–69.
123. Galdiero MR, Garlanda C, Jaillon S, Marone G,
Mantovani A. Tumor associated macrophages and
neutrophils in tumor progression. J Cell Physiol.
2013;228:1404–12.
124. Rosenthal EL, Matrisian LM. Matrix metalloproteases
in head and neck cancer. Head Neck. 2006;28:
639–48.
125. DeClerck YA, Mercurio AM, Stack MS, Chapman HA,
Zutter MM, Muschel RJ, et al. Proteases, extracellular
matrix, and cancer: a workshop of the path B study
section. Am J Pathol. 2004;164:1131–9.
126. Gutschalk CM, Yanamandra AK, Linde N, Meides A,
Depner S, Mueller MM. GM-CSF enhances tumor
invasion by elevated MMP-2, -9, and -26 expression.
Cancer Med. 2013;2:117–29.
127. Yu B, Wei J, Qian X, Lei D, Ma Q, Liu Y. Notch1
signaling pathway participates in cancer invasion by
regulating MMPs in lingual squamous cell carcinoma.
Oncol Rep. 2012;27:547–52.
128. Rosenthal EL, Hotary K, Bradford C, Weiss SJ. Role of
membrane type 1-matrix metalloproteinase and gelat-
inase A in head and neck squamous cell carcinoma
invasion in vitro. Otolaryngol Head Neck Surg.
1999;121:337–43.
129. Sinpitaksakul SN, Pimkhaokham A, Sanchavanakit N,
Pavasant P. TGF-beta1 induced MMP-9 expression in
HNSCC cell lines via Smad/MLCK pathway. Biochem
Biophys Res Commun. 2008;371:713–8.
130. Koontongkaew S, Amornphimoltham P, Monthanpi-
sut P, Saensuk T, Leelakriangsak M. Fibroblasts and
extracellular matrix differently modulate MMP acti-
vation by primary and metastatic head and neck
cancer cells. Med Oncol. 2012;29:690–703.
131. Murphy G, Docherty AJ. The matrix metalloprotei-
nases and their inhibitors. Am J Respir Cell Mol Biol.
1992;7:120–5.
132. Price SJ, Greaves DR, Watkins H. Identification of
novel, functional genetic variants in the human
matrix metalloproteinase-2 gene: role of Sp1 in
allele-specific transcriptional regulation. J Biol Chem.
2001;276:7549–58.
133. Rosenthal EL, McCrory A, Talbert M, Carroll W, Magnu-
son JS, Peters GE. Expression of proteolytic enzymes in
head and neck cancer-associated fibroblasts. Arch Oto-
laryngol Head Neck Surg. 2004;130:943–7.
134. Lynch CC, Matrisian LM. Matrix metalloproteinases in
tumor-host cell communication. Differentiation.
2002;70:561–73.
135. Kurahara S, Shinohara M, Ikebe T, Nakamura S,
Beppu M, Hiraki A, et al. Expression of MMPS, MT-
MMP, and TIMPs in squamous cell carcinoma of the
oral cavity: correlations with tumor invasion and
metastasis. Head Neck. 1999;21:627–38.
136. Birkedal-Hansen B, Pavelic ZP, Gluckman JL, Stam-
brook P, Li YQ, MMP Stetler-Stevenson WG. and
TIMP gene expression in head and neck squamous
cell carcinomas and adjacent tissues. Oral Dis. 2000;
6:376–82.
137. Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo
T, Subarnbhesaj A, et al. Matrix metalloproteinase-13
(MMP-13) directly and indirectly promotes tumor
angiogenesis. J Biol Chem. 2012;287:38716–28.
138. Quan J, Zhou C, Johnson NW, Francis G, Dahlstrom
JE, Gao J. Molecular pathways involved in crosstalk
between cancer cells, osteoblasts and osteoclasts in
the invasion of bone by oral squamous cell carci-
noma. Pathology. 2012;44:221–7.
139. Zhang Z, Filho MS, Nor JE. The biology of head and
neck cancer stem cells. Oral Oncol. 2012;48:1–9.

Tumor microenvironment in HNSCC 233
140. Lai WW, Hsu SC, Chueh FS, Chen YY, Yang JS, Lin JP,
et al. Quercetin inhibits migration and invasion of
SAS human oral cancer cells through inhibition of NF-
kappaB and matrix metalloproteinase-2/-9 signaling
pathways. Anticancer Res. 2013;33:1941–50.
141. Prime SS, Davies M, Pring M, Paterson IC. The role of
TGF-beta in epithelial malignancy and its relevance to
the pathogenesis of oral cancer (part II). Crit Rev
Oral Biol Med. 2004;15:337–47.
142. Paterson IC, Matthews JB, Huntley S, Robinson CM,
Fahey M, Parkinson EK, et al. Decreased expression
of TGF-beta cell surface receptors during progression
of human oral squamous cell carcinoma. J Pathol.
2001;193:458–67.
143. Cho KH, Jeong KJ, Shin SC, Kang J, Park CG, Lee HY.
STAT3 mediates TGF-beta1-induced TWIST1 expres-
sion and prostate cancer invasion. Cancer Lett.
2013;336:167–73.
144. Na YR, Lee JS, Lee SJ, Twist Seok SH Interleukin-6-
induced. and N-cadherin enhance melanoma cell
metastasis. Melanoma Res. 2013;23:434–43.
145. Whiteman EL, Liu CJ, Fearon ER, Margolis B. The
transcription factor snail represses Crumbs3 expres-
sion and disrupts apico-basal polarity complexes.
Oncogene. 2008;27:3875–9.
146. Nguyen PT, Kudo Y, Yoshida M, Iizuka S, Ogawa I,
Takata T. N-cadherin expression is correlated with
metastasis of spindle cell carcinoma of head and neck
region. J Oral Pathol Med. 2011;40:77–82.
147. Bonde AK, Tischler V, Kumar S, Soltermann A,
Schwendener RA. Intratumoral macrophages contrib-
ute to epithelial-mesenchymal transition in solid
tumors. BMC Cancer. 2012;12:35.
148. Yu Q, Stamenkovic I. Cell surface-localized matrix
metalloproteinase-9 proteolytically activates TGF-
beta and promotes tumor invasion and angiogenesis.
Genes Dev. 2000;14:163–76.
149. Folkman J. Role of angiogenesis in tumor growth and
metastasis. Semin Oncol. 2002;29:15–8.
150. El-Gazzar R, Macluskey M, Ogden GR. Evidence for
a field change effect based on angiogenesis in the
oral mucosa? A brief report Oral Oncol. 2005;41:
25–30.
151. El-Gazzar R, Macluskey M, Williams H, Ogden GR.
Vascularity and expression of vascular endothelial
growth factor in oral squamous cell carcinoma,
resection margins, and nodal metastases. Br J Oral
Maxillofac Surg. 2006;44:193–7.
152. Macluskey M, Chandrachud LM, Pazouki S, Green M,
Chisholm DM, Ogden GR, et al. Apoptosis, prolifer-
ation, and angiogenesis in oral tissues. Possible
relevance to tumour progression. J Pathol. 2000;
191:368–75.
153. Sedivy R, Beck-Mannagetta J, Haverkampf C, Battis-
tutti W, Honigschnabl S. Expression of vascular
endothelial growth factor-C correlates with the lym-
phatic microvessel density and the nodal status in
oral squamous cell cancer. J Oral Pathol Med. 2003;
32:455–60.
154. Williams MD. Integration of biomarkers including
molecular targeted therapies in head and neck can-
cer. Head Neck Pathol. 2010;4:62–9.
155. Perez-Sayans M, Somoza-Martin JM, Barros-Angueira
F, Diz PG, Rey JM, Garcia-Garcia A. Multidrug resist-
ance in oral squamous cell carcinoma: The role of
vacuolar ATPases. Cancer Lett. 2010;295:135–43.
156. Perez-Sayans M, Somoza-Martin JM, Barros-Angueira
F, Reboiras-Lopez MD, Gandara Rey JM, Garcia-Garcia
A. Genetic and molecular alterations associated with
oral squamous cell cancer (Review). Oncol Rep.
2009;22:1277–82.
157. Moulder JE, Rockwell S. Tumor hypoxia: its impact
on cancer therapy. Cancer Metastasis Rev. 1987;5:
313–41.
158. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi
LS, Sampaolesi M, et al. Tie2 identifies a hematopoi-
etic lineage of proangiogenic monocytes required for
tumor vessel formation and a mesenchymal popula-
tion of pericyte progenitors. Cancer Cell. 2005;8:
211–26.
159. Tse GM, Chan AW, Yu KH, King AD, Wong KT, Chen
GG, et al. Strong immunohistochemical expression of
vascular endothelial growth factor predicts overall
survival in head and neck squamous cell carcinoma.
Ann Surg Oncol. 2007;14:3558–65.
160. Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS,
Koch AE, et al. Unidirectional crosstalk between
Bcl-xL and Bcl-2 enhances the angiogenic phenotype
of endothelial cells. Cell Death Differ. 2007;14:
1657–66.
161. Neiva KG, Zhang Z, Miyazawa M, Warner KA, Karl E,
Nor JE. Cross talk initiated by endothelial cells
enhances migration and inhibits anoikis of squamous
cell carcinoma cells through STAT3/Akt/ERK signal-
ing. Neoplasia. 2009;11:583–93.
162. Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo
E, Tanghetti E, et al. CXCL1/macrophage inflamma-
tory protein-2-induced angiogenesis in vivo is medi-
ated by neutrophil-derived vascular endothelial
growth factor-A. J Immunol. 2004;172:5034–40.
163. Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB,
De Marzo AM. Cyclooxygenases in cancer: progress
and perspective. Cancer Lett. 2004;215:1–20.
164. Camacho M, Leon X, Fernandez-Figueras MT, Quer
M, Vila L. Prostaglandin E(2) pathway in head and
neck squamous cell carcinoma. Head Neck. 2008;30:
1175–81.
165. Li JZ, Gao W, Chan JY, Ho WK, Wong TS. Hypoxia in
head and neck squamous cell carcinoma. ISRN
Otolaryngol. 2012;2012:708974.
166. Roh JL, Cho KJ, Kwon GY, Ryu CH, Chang HW, Choi
SH, et al. The prognostic value of hypoxia markers in
T2-staged oral tongue cancer. Oral Oncol. 2009;45:
63–8.
167. Feron O. Pyruvate into lactate and back: from the
Warburg effect to symbiotic energy fuel exchange in
cancer cells. Radiother Oncol. 2009;92:329–33.
168. Liang X, Yang D, Hu J, Hao X, Gao J, Mao Z. Hypoxia
inducible factor-alpha expression correlates with
vascular endothelial growth factor-C expression and
lymphangiogenesis/angiogenesis in oral squamous
cell carcinoma. Anticancer Res. 2008;28:1659–66.
169. Mohamed KM, Le A, Duong H, Wu Y, Zhang Q,
Messadi DV. Correlation between VEGF and HIF-

J.M. Curry et al234
1alpha expression in human oral squamous cell
carcinoma. Exp Mol Pathol. 2004;76:143–52.
170. Teppo S, Sundquist E, Vered M, Holappa H, Parkki-
senniemi J, Rinaldi T, et al. The hypoxic tumor
microenvironment regulates invasion of aggressive
oral carcinoma cells. Exp Cell Res. 2013;319:376–89.
171. Shih YH, Chang KW, Chen MY, Yu CC, Lin DJ, Hsia
SM, et al. Lysyl oxidase and enhancement of cell
proliferation and angiogenesis in oral squamous cell
carcinoma. Head Neck. 2013;35:250–6.
172. Erler JT, Bennewith KL, Nicolau M, Dornhofer N,
Kong C, Le QT, et al. Lysyl oxidase is essential for
hypoxia-induced metastasis. Nature. 2006;440:1222–6.
173. Zhang Q, Tang X, Lu Q, Zhang Z, Rao J, Le AD. Green
tea extract and (-)-epigallocatechin-3-gallate inhibit
hypoxia- and serum-induced HIF-1alpha protein accu-
mulation and VEGF expression in human cervical
carcinoma and hepatoma cells. Mol Cancer Ther.
2006;5:1227–38.
174. Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y,
et al. Histone deacetylase inhibitors repress the
transactivation potential of hypoxia-inducible factors
independently of direct acetylation of HIF-alpha. J
Biol Chem. 2006;281:13612–9.
175. Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J.
Histone deacetylase inhibitors induce VHL and
ubiquitin-independent proteasomal degradation of
hypoxia-inducible factor 1alpha. Mol Cell Biol.
2006;26:2019–28.
176. Zhang Q, Tang X, Lu QY, Zhang ZF, Brown J, Le AD.
Resveratrol inhibits hypoxia-induced accumulation of
hypoxia-inducible factor-1alpha and VEGF expres-
sion in human tongue squamous cell carcinoma and
hepatoma cells. Mol Cancer Ther. 2005;4:1465–74.
177. Warburg O. On the origin of cancer cells. Science.
1956;123:309–14.
178. Lunt SY, Vander Heiden MG. Aerobic glycolysis:
meeting the metabolic requirements of cell prolifer-
ation. Annu Rev Cell Dev Biol. 2011;27:441–64.
179. Sonveaux P, Vegran F, Schroeder T, Wergin MC,
Verrax J, Rabbani ZN, et al. Targeting lactate-fueled
respiration selectively kills hypoxic tumor cells in
mice. J Clin Invest. 2008;118:3930–42.
180. Zu XL, Guppy M. Cancer metabolism: facts, fantasy,
and fiction. Biochem Biophys Res Commun. 2004;
313:459–65.
181. Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-
Hernandez A, Saavedra E. Energy metabolism in
tumor cells. Febs J. 2007;274:1393–418.
182. Guppy M, Greiner E, Brand K. The role of the
Crabtree effect and an endogenous fuel in the energy
metabolism of resting and proliferating thymocytes.
Eur J Biochem. 1993;212:95–9.
183. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and
competition in the evolution of ATP-producing path-
ways. Science. 2001;292:504–7.
184. Busk M, Walenta S, Mueller-Klieser W, Steiniche T,
Jakobsen S, Horsman MR, et al. Inhibition of tumor
lactate oxidation: consequences for the tumor micro-
environment. Radiother Oncol. 2011;99:404–11.
185. Pavlides S, Whitaker-Menezes D, Castello-Cros R,
Flomenberg N, Witkiewicz AK, Frank PG, et al. The
reverse Warburg effect: aerobic glycolysis in cancer
associated fibroblasts and the tumor stroma. Cell
Cycle. 2009;8:3984–4001.
186. Tripathi P, Kamarajan P, Somashekar BS, MacKinnon
N, Chinnaiyan AM, Kapila YL, et al. Delineating
metabolic signatures of head and neck squamous
cell carcinoma: phospholipase A2, a potential ther-
apeutic target. Int J Biochem Cell Biol. 2012;44:
1852–61.
187. Sandulache VC, Ow TJ, Pickering CR, Frederick MJ,
Zhou G, Fokt I, et al. Glucose, not glutamine, is the
dominant energy source required for proliferation
and survival of head and neck squamous carcinoma
cells. Cancer. 2011;117:2926–38.
188. Walenta S, Salameh A, Lyng H, Evensen JF, Mitze M,
Rofstad EK, et al. Correlation of high lactate levels in
head and neck tumors with incidence of metastasis.
Am J Pathol. 1997;150:409–15.
189. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough
RW, Dewhirst MW, et al. Elevated tumor lactate
concentrations predict for an increased risk of meta-
stases in head-and-neck cancer. Int J Radiat Oncol
Biol Phys. 2001;51:349–53.
190. Brennan PA, Mackenzie N, Quintero M. Hypoxia-
inducible factor 1alpha in oral cancer. J Oral Pathol
Med. 2005;34:385–9.
191. Zhang Q, Zhang ZF, Rao JY, Sato JD, Brown J,
Messadi DV, et al. Treatment with siRNA and anti-
sense oligonucleotides targeted to HIF-1alpha
induced apoptosis in human tongue squamous cell
carcinomas. Int J Cancer. 2004;111:849–57.
192. Li X, Fan Z. The epidermal growth factor receptor
antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-1alpha and Bcl-2 and
activating the beclin 1/hVps34 complex. Cancer Res.
2010;70:5942–52.
193. Kato K, Gong J, Iwama H, Kitanaka A, Tani J, Miyoshi
H, et al. The antidiabetic drug metformin inhibits
gastric cancer cell proliferation in vitro and in vivo.
Mol Cancer Ther. 2012;11:549–60.
194. Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH,
et al. Metformin inhibits cell proliferation, migration
and invasion by attenuating CSC function mediated
by deregulating miRNAs in pancreatic cancer cells.
Cancer Prev Res (Phila). 2012;5:355–64.
195. Klubo-Gwiezdzinska J, Jensen K, Costello J, Patel A,
Hoperia V, Bauer A, et al. Metformin inhibits growth
and decreases resistance to anoikis in medullary
thyroid cancer cells. Endocr Relat Cancer. 2012;19:
447–56.
196. Zhuang Y, Miskimins WK. Metformin induces both
caspase-dependent and poly(ADP-ribose) polymerase-
dependent cell death in breast cancer cells. Mol
Cancer Res. 2010;9:603–15.
197. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in
diabetic patients treated with metformin: a system-
atic review and meta-analysis. PLoS One. 2012;
7:e33411.
198. Luo Q, Hu D, Hu S, Yan M, Sun Z, Chen F. In vitro
and in vivo anti-tumor effect of metformin as a novel
therapeutic agent in human oral squamous cell
carcinoma. BMC Cancer. 2012;12:517.
Related Documents
![Heute Zahnersatz morgen krank.ppt [Kompatibilitätsmodus]€¦ · Periimplantitis Osteoporose Schleimhaut/Haut Kollagenase u.a. aMMP8 PGE2-Synthese Gewebe-/Kollagenabbau Parodontitis](https://static.cupdf.com/doc/110x72/5fa71d11a39507256565e16b/heute-zahnersatz-morgen-krankppt-kompatibilittsmodus-periimplantitis-osteoporose.jpg)










