Triggering of p73-dependent apoptosis in osteosarcoma is under the control of E2Fs–pRb2/p130 complexes Dario La Sala 1,2 , Marcella Macaluso 3 , Carmela Trimarchi 4 , Antonio Giordano 3 and Caterina Cinti* ,1,3 1 ITOI-CNR, Unit of Bologna, c/o IOR, 40136 Bologna, Italy; 2 Institute of Pathologic Anatomy and Histology, University of Siena, 53100 Siena, Italy; 3 Sbarro Institute for Cancer Research and Molecular Medicine, College of Science and Technology, Temple University, Philadelphia, PA 19107, USA; 4 Institute of Neuroscience, CNR, 56100 Pisa, Italy Mechanisms underlying multidrug resistance (MDR), one of the major causes of cancer treatment failure, are still poorly understood. We selected the osteosarcoma MDR HosDXR150 cell line by culturing Hos cells in the presence of increasing doxorubicin doses and showed that it is crossresistant to vinblastine. Similarly to the Hos parental cell line, HosDXR150 cells present mutated p53, functionally inactivated pRb/p105 and wild-type pRb2/ p130. Owing to p53 mutation, MDR-1 gene, codifying for P-glycoprotein, is upregulated. Evasion of apoptosis in HosDXR150 cells is only partially explained by drug extrusion because of P-glycoprotein overexpression. Analysis of gene expression level profiles showed that parental cell line undergoes apoptosis through an E2F1/ p73-dependent pathway while its resistant variant evades it. This result can be explained by the presence of distinct E2Fs–pRb2/p130 complexes on the p73 promoter. Namely, in Hos p73 transcription is activated by E2F1– Rb2/p130–p300 complexes, while in HosDXR150 it is kept repressed by E2F4–Rb2/p130–HDAC1 complexes. Oncogene (2003) 22, 3518–3529. doi:10.1038/sj.onc.1206487 Keywords: multidrug resistance; apoptosis; p53-inde- pendent pathways; pRb2/p130; p73; osteosarcoma Introduction Apoptosis is a fundamental mechanism of cell death that can be triggered by chemotherapeutic drugs. Understanding how the cell death program is engaged following an insult, and hence why it fails to be activated in certain settings, could offer a novel approach for overcoming the clinical problem of drug resistance. When a tumor does not respond to chemotherapeutic agents, this resistance is usually extended to more than one drug even structurally unrelated. This phenomenon is referred to as multidrug resistance (MDR) and it is the main cause of tumor treatment failure. Osteosarcoma is one of the most common primary tumors of bone, is highly malignant and typically affects children and adults. Osteosarcoma cells bear mutations of both p53 and pRb/p105 genes (Hansen, 1991). In total, 30–40% of these tumors show a resistance to drugs after a few courses of chemotherapeutic treatment, which deter- mines the failure of therapy. The mechanisms by which MDR is acquired in human osteosarcoma remain unclear. Overexpression of P-glycoprotein/p170, which works as an energy-dependent efflux pump for a diverse group of lipophilic compounds (Kane et al., 1990), has been described as a possible factor for MDR in osteosarcoma tumors (Serra et al., 1993). However, some other studies could not find any correlation between P-glycoprotein expression and clinical outcome in the patients with osteosarcoma (Gorlick et al., 1999). Moreover, lack of expression of this drug resistance- related protein does not appear to confer any advantage in terms of patient survival in osteosarcoma (Shnyder et al., 1998). Transcription of MDR1 gene, which encodes for P-glycoprotein (Chen et al., 1986; Gottes- man and Pastan, 1988), is negatively regulated by wild- type p53 (wt-p53) (Strauss et al., 1995) and the presence of mutated p53 correlates with high expression of MDR1 (Thottassery et al., 1997). p53 plays a pivotal role in modulating the cell response to various sources of damage and stress (Cinti et al., 2000; Vogelstein et al., 2000) by controlling the transcription of a large number of genes required for the apoptotic response (for a review, see Somasundaram and El-Deiry, 2000). Given that p53 plays a crucial and multivariate role in controlling cell growth and survival, it is not surprising that it has been found mutated in the majority of cancers (for a review, see Sigal and Rotter, 2000). Each cell is able to integrate both extra- and intracellular survival and death signals, thereby control- ling its own growth rate or, when harmful signals prevail, inducing its self-destruction. Mainly, this is achieved because of multiple interactions between the retinoblastoma family proteins (pRBs) pathway, whose main function is the control of G 1 to S progression, and the p53 pathway, which guards against genomic Received 23 December 2002; revised 6 March 2003; accepted 6 March 2003 *Correspondence: Caterina Cinti, ITOI-CNR, Unit of Bologna, c/o IOR, via di Barbiano 1/10, 40136 Bologna, Italy; E-mail: cinti@ area.bo.cnr.it Oncogene (2003) 22, 3518–3529 & 2003 Nature Publishing Group All rights reserved 0950-9232/03 $25.00 www.nature.com/onc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Triggering of p73-dependent apoptosis in osteosarcoma is under the control
of E2Fs–pRb2/p130 complexes
Dario La Sala1,2, Marcella Macaluso3, Carmela Trimarchi4, Antonio Giordano3 and CaterinaCinti*,1,3
1ITOI-CNR, Unit of Bologna, c/o IOR, 40136 Bologna, Italy; 2Institute of Pathologic Anatomy and Histology, University of Siena,53100 Siena, Italy; 3Sbarro Institute for Cancer Research and Molecular Medicine, College of Science and Technology, TempleUniversity, Philadelphia, PA 19107, USA; 4Institute of Neuroscience, CNR, 56100 Pisa, Italy
Mechanisms underlying multidrug resistance (MDR), oneof the major causes of cancer treatment failure, are stillpoorly understood. We selected the osteosarcoma MDRHosDXR150 cell line by culturing Hos cells in thepresence of increasing doxorubicin doses and showed thatit is crossresistant to vinblastine. Similarly to the Hosparental cell line, HosDXR150 cells present mutated p53,functionally inactivated pRb/p105 and wild-type pRb2/p130. Owing to p53 mutation, MDR-1 gene, codifying forP-glycoprotein, is upregulated. Evasion of apoptosis inHosDXR150 cells is only partially explained by drugextrusion because of P-glycoprotein overexpression.Analysis of gene expression level profiles showed thatparental cell line undergoes apoptosis through an E2F1/p73-dependent pathway while its resistant variant evadesit. This result can be explained by the presence of distinctE2Fs–pRb2/p130 complexes on the p73 promoter.Namely, in Hos p73 transcription is activated by E2F1–Rb2/p130–p300 complexes, while in HosDXR150 it iskept repressed by E2F4–Rb2/p130–HDAC1 complexes.Oncogene (2003) 22, 3518–3529. doi:10.1038/sj.onc.1206487
Keywords: multidrug resistance; apoptosis; p53-inde-pendent pathways; pRb2/p130; p73; osteosarcoma
Introduction
Apoptosis is a fundamental mechanism of cell deaththat can be triggered by chemotherapeutic drugs.Understanding how the cell death program is engagedfollowing an insult, and hence why it fails to be activatedin certain settings, could offer a novel approach forovercoming the clinical problem of drug resistance.When a tumor does not respond to chemotherapeuticagents, this resistance is usually extended to more than
one drug even structurally unrelated. This phenomenonis referred to as multidrug resistance (MDR) and it is themain cause of tumor treatment failure. Osteosarcoma isone of the most common primary tumors of bone, ishighly malignant and typically affects children andadults. Osteosarcoma cells bear mutations of both p53and pRb/p105 genes (Hansen, 1991). In total, 30–40%of these tumors show a resistance to drugs after a fewcourses of chemotherapeutic treatment, which deter-mines the failure of therapy. The mechanisms by whichMDR is acquired in human osteosarcoma remainunclear. Overexpression of P-glycoprotein/p170, whichworks as an energy-dependent efflux pump for a diversegroup of lipophilic compounds (Kane et al., 1990), hasbeen described as a possible factor for MDR inosteosarcoma tumors (Serra et al., 1993). However,some other studies could not find any correlationbetween P-glycoprotein expression and clinical outcomein the patients with osteosarcoma (Gorlick et al., 1999).Moreover, lack of expression of this drug resistance-related protein does not appear to confer any advantagein terms of patient survival in osteosarcoma (Shnyderet al., 1998). Transcription of MDR1 gene, whichencodes for P-glycoprotein (Chen et al., 1986; Gottes-man and Pastan, 1988), is negatively regulated by wild-type p53 (wt-p53) (Strauss et al., 1995) and the presenceof mutated p53 correlates with high expression ofMDR1 (Thottassery et al., 1997). p53 plays a pivotalrole in modulating the cell response to various sources ofdamage and stress (Cinti et al., 2000; Vogelstein et al.,2000) by controlling the transcription of a large numberof genes required for the apoptotic response (for areview, see Somasundaram and El-Deiry, 2000). Giventhat p53 plays a crucial and multivariate role incontrolling cell growth and survival, it is not surprisingthat it has been found mutated in the majority of cancers(for a review, see Sigal and Rotter, 2000).
Each cell is able to integrate both extra- andintracellular survival and death signals, thereby control-ling its own growth rate or, when harmful signalsprevail, inducing its self-destruction. Mainly, this isachieved because of multiple interactions between theretinoblastoma family proteins (pRBs) pathway, whosemain function is the control of G1 to S progression, andthe p53 pathway, which guards against genomic
Received 23 December 2002; revised 6 March 2003; accepted 6 March2003
*Correspondence: Caterina Cinti, ITOI-CNR, Unit of Bologna, c/oIOR, via di Barbiano 1/10, 40136 Bologna, Italy; E-mail: [email protected]
Oncogene (2003) 22, 3518–3529& 2003 Nature Publishing Group All rights reserved 0950-9232/03 $25.00
www.nature.com/onc

instability by inducing both arrest of the cell cycle andapoptosis. Moreover, it has recently been shown thatE2F1, the main target of pRb/p105 growth suppressivefunction, plays a dual role, on the one hand by inducingS-promoting genes transcription and, on the other one,by directly influencing apoptosis execution. Mutationsor functional inactivation because of post-translationalmodification of pRBs, p53 and/or of the other proteinsinvolved in their functional pathways can also influencecellular sensitivity to chemotherapeutic drugs allowingcancer cells to evade apoptosis after drug-induceddamage. Many links have been provided between bothp53-dependent and -independent pathways and thepRb/p105 pathway. The most direct evidence of pRb/p105 control on p53 proapoptotic activity is that it isable to prevent p53 degradation, mediated by MDM2,by forming a trimeric complex (Hsieh et al., 1999).Interestingly, pRb/p105 acetylation is needed for pRb/p105–MDM2 binding (Chan et al., 2001). pRb/p105deregulation determines unrestrained E2F1 expression,which may cause p53-dependent and -independentapoptosis (Hsieh et al., 1997; Johnson and Schneider-Broussard, 1998). Namely, E2F1 has been shown toinduce the accumulation of p53 (Hiebert et al., 1995;Kowalik et al., 1998), the transcription of p73 (Stieweand Putzer, 2000; Irwin et al., 2000) and the transcrip-tion of Apaf-1, a key element of apoptosome (Moroniet al., 2001). Moreover, it has been reported thatacetylation, dephosphorylation and consequent ubiqui-tin-mediated degradation of pRb/p105 retinoblastomaprotein could be early events in programmed cell death,possibly related to cell commitment to apoptosis (for areview, see Dou and An, 1998). As a consequence,failure to induce the interior cleavage of pRb/p105 canbe associated with drug resistance to proapoptoticanticancer treatments (An et al., 1996). Up to now nosimilar data have been reported for the retinoblastoma-related gene pRb2/p130. Tumors concomitantly lackingpRb/p105 and p53, such as osteosarcoma cells, are,however, still able to undergo apoptosis, thus suggestinga possible role in this process of another pRB familyprotein to regulate the p53-dependent and -independentapoptosis. Few data are available on the role of the tworetinoblastoma-related family members, pRb2/p130 andp107, in the apoptotic response even if a possible role ofpRb2/p130 in regulating the expression of the anti-apoptotic bcl-2 and of the proapoptotic p73 proteins hasrecently been indicated (Pucci et al., 2002). Moreover,an inverse correlation between pRb2/p130 expressionlevel and apoptotic index in retinoblastoma tumor hasbeen reported, suggesting a possible involvement ofpRb2/p130 in the apoptotic response (Bellan et al.,2002).
Many genes, involved in both DNA replication andcell cycle control, have been shown to contain E2F sitesin their promoter region (for a review, see Lavia andJansen-Durr, 1999; Ren et al., 2002), and it has beenshown that E2F-binding sites mediate both transcrip-tional activation and repression (Dyson, 1998). pRb2/p130 controls the transcription of E2F responsive genesby forming a repressive complex on the promoters.
pRb2/p130, in association with E2F4, is the mostabundant E2F complex found in resting or in G0quiescent cells, and this complex is thought to help inmaintaining a state of transcriptional silence (Zini et al.,2001). The presence of pRb2/p130–E2F4 DNA-bindingcomplexes correlates with E2F1 gene repression andoverexpression of pRb2/p130 inhibits transcription fromthe E2F1 promoter (Johnson, 1995). In this manner,pRb2/p130 also regulates the expression of pRb/p105and p107 genes because each contains E2F sites in theirpromoters. The pRB–E2F repressive complexes func-tion in association with histone deacetylase (HDAC),essentially repressing the transcription of E2F-depen-dent genes probably through deacetylation of histonetails that protrude from nucleosome (Ferreira et al.,1998; Cress and Seto, 2000). The shift from genesilencing to gene activation depends on the balancebetween nucleosome deacetylation and acetylation,possibly regulated by the replacement of HDAC withhistone acetyltransferase (HAT) in pRB–E2F complexesdirectly recruited to E2F-regulated promoters (Raymanet al., 2002; Macaluso et al., 2003). Interestingly, it hasbeen shown that p73 gene expression is positivelyregulated by E2F1 transcription factor, which bindsE2F sites on p73 promoter (Irwin et al., 2000; Seelanet al., 2002). Moreover, it has recently been reportedthat, in p53-deficient tumor cells, endogenous p73 a andb proteins are upregulated in response to exogenousE2F1 overexpression and repressed by ectopicallyexpressed pRb/p105 (Zaika et al., 2001; Seelan et al.,2002). Despite significant advances in our understandingof the mechanisms underlying p53-independent apopto-tic response and apoptosis evasion by MDR tumor cells,a complete characterization of the physiologicallyrelevant factors involved in these processes has not yetbeen achieved. The aim of the present study is, on theone hand, to characterize the p53-independent pathwayby which the osteosarcoma Hos cell line undergoesapoptosis and, on the other, to compare the results withthose obtained in its HosDXR150 MDR variant.
Results
Characterization of HosDXR150 as an MDR clone
We selected a cell line resistant to chemotherapeuticagents (HosDXR150) by culturing a human osteosarco-ma cell line (Hos) in the presence of increasing doses ofdoxorubicin hydrochloride (0.5–150 ng/ml). After appli-cation of 150 ng/ml doxorubicin, HosDXR150 growthrate slowed down initially (proliferation index 0.8 vs 1 inHos parental cell line) and then speeded up after 96 h oftreatment. In the same culturing conditions, the Hos cellline showed a marked decrease of the proliferation index(1 vs 0.2 after 96 h) (data not shown). To distinguishwhether doxorubicin induced cell death in the Hosparental cell line by necrosis or apoptosis, we processedour samples for in situ Nick Translation assay. Theapoptotic program is latent in virtually all cell types and,once triggered, unfolds following a precise sequence of
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3519
Oncogene
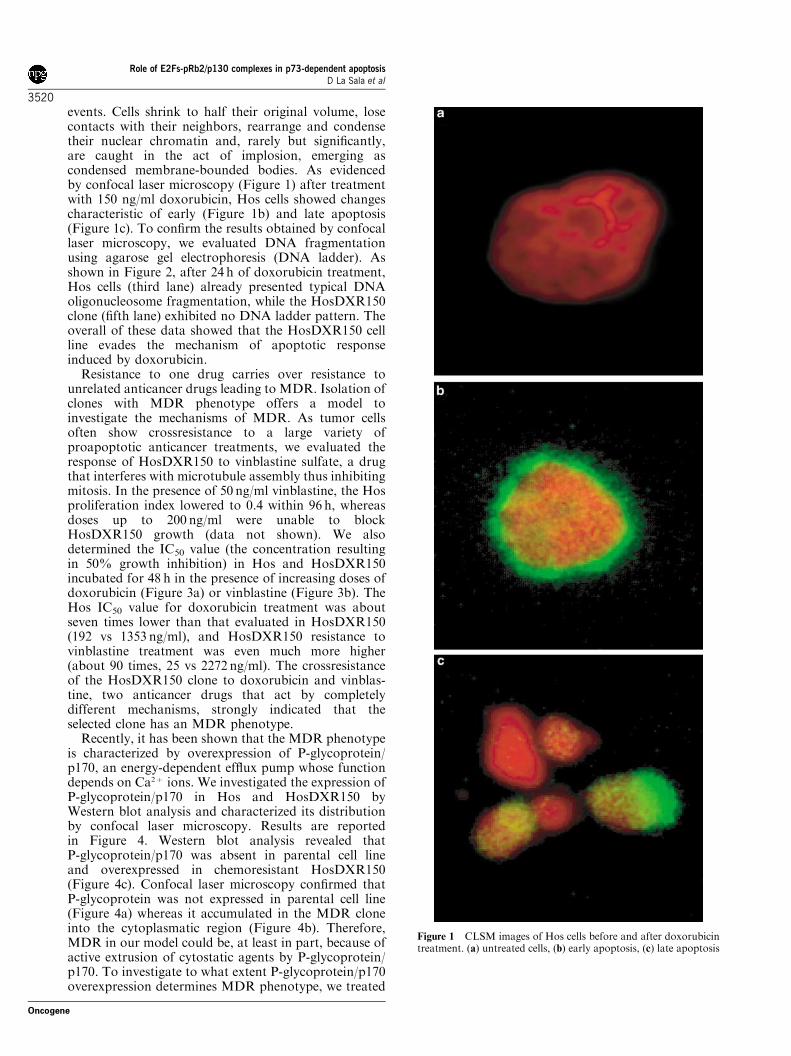
events. Cells shrink to half their original volume, losecontacts with their neighbors, rearrange and condensetheir nuclear chromatin and, rarely but significantly,are caught in the act of implosion, emerging ascondensed membrane-bounded bodies. As evidencedby confocal laser microscopy (Figure 1) after treatmentwith 150 ng/ml doxorubicin, Hos cells showed changescharacteristic of early (Figure 1b) and late apoptosis(Figure 1c). To confirm the results obtained by confocallaser microscopy, we evaluated DNA fragmentationusing agarose gel electrophoresis (DNA ladder). Asshown in Figure 2, after 24 h of doxorubicin treatment,Hos cells (third lane) already presented typical DNAoligonucleosome fragmentation, while the HosDXR150clone (fifth lane) exhibited no DNA ladder pattern. Theoverall of these data showed that the HosDXR150 cellline evades the mechanism of apoptotic responseinduced by doxorubicin.
Resistance to one drug carries over resistance tounrelated anticancer drugs leading to MDR. Isolation ofclones with MDR phenotype offers a model toinvestigate the mechanisms of MDR. As tumor cellsoften show crossresistance to a large variety ofproapoptotic anticancer treatments, we evaluated theresponse of HosDXR150 to vinblastine sulfate, a drugthat interferes with microtubule assembly thus inhibitingmitosis. In the presence of 50 ng/ml vinblastine, the Hosproliferation index lowered to 0.4 within 96 h, whereasdoses up to 200 ng/ml were unable to blockHosDXR150 growth (data not shown). We alsodetermined the IC50 value (the concentration resultingin 50% growth inhibition) in Hos and HosDXR150incubated for 48 h in the presence of increasing doses ofdoxorubicin (Figure 3a) or vinblastine (Figure 3b). TheHos IC50 value for doxorubicin treatment was aboutseven times lower than that evaluated in HosDXR150(192 vs 1353 ng/ml), and HosDXR150 resistance tovinblastine treatment was even much more higher(about 90 times, 25 vs 2272 ng/ml). The crossresistanceof the HosDXR150 clone to doxorubicin and vinblas-tine, two anticancer drugs that act by completelydifferent mechanisms, strongly indicated that theselected clone has an MDR phenotype.
Recently, it has been shown that the MDR phenotypeis characterized by overexpression of P-glycoprotein/p170, an energy-dependent efflux pump whose functiondepends on Ca2þ ions. We investigated the expression ofP-glycoprotein/p170 in Hos and HosDXR150 byWestern blot analysis and characterized its distributionby confocal laser microscopy. Results are reportedin Figure 4. Western blot analysis revealed thatP-glycoprotein/p170 was absent in parental cell lineand overexpressed in chemoresistant HosDXR150(Figure 4c). Confocal laser microscopy confirmed thatP-glycoprotein was not expressed in parental cell line(Figure 4a) whereas it accumulated in the MDR cloneinto the cytoplasmatic region (Figure 4b). Therefore,MDR in our model could be, at least in part, because ofactive extrusion of cytostatic agents by P-glycoprotein/p170. To investigate to what extent P-glycoprotein/p170overexpression determines MDR phenotype, we treated
Figure 1 CLSM images of Hos cells before and after doxorubicintreatment. (a) untreated cells, (b) early apoptosis, (c) late apoptosis
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3520
Oncogene

HosDXR150 with verapamil, a substance that antag-onizes P-glycoprotein activity through competitiveblock of calcium channels. Actually, contemporaryapplication of doxorubicin and 10 mm verapamil, a
concentration known not to have a cytotoxic effect, toHosDXR150 halved the IC50 value (734 vs 1353 ng/ml)but did not restore completely the apoptotic response(Figure 5).
Comparison between Hos and HosDXR150: mutationalstatus and gene expression profile
Overexpression of P-glycoprotein reflects MDR1 genetranscription that, in turn, is negatively regulated by wt-p53 and the presence of mutated p53 correlates withhigh expression of MDR1 gene (Thottassery et al.,1997). Therefore, we screened both Hos andHosDXR150 clones for mutations of p53 gene byPCR and sequencing of each p53 exon (Table 1). Weevidenced in both cell lines homozygous exon 5 pointmutation at codon 156, Arg vs Pro, due to thesubstitution of G with C (Figure 6). Loss of functionof both p53 and pRb/p105 (Lin et al., 1994; Weinberg,1995; Miller et al., 1996) in the Hos cell line determinesderegulated cell proliferation and altered apoptoticresponse. Since pRB family members (pRb/p105,pRb2/p130 and p107) could exhibit overlapping specifi-cities, we analysed the mutational status of Rb2/p130gene. Sequencing analysis of Rb2/p130 retinoblastoma-related gene did not reveal any mutations in both celllines (Table 2). This first series of experiments showedthat Hos and HosDXR150 cells do not show differencesin their mutational status.
An alternative explanation to the reported diffe-rences between the two clones in susceptibility to
Figure 2 DNA ladder of doxorubicin-treated Hos andHosDXR150 cells after 24 h (HosþDXR 24h) and 48 h(HosþDXR 48h)
Figure 3 IC50 value (concentration resulting in 50% growth inhibition) of Hos (dotted line) and HosDXR150 (continuous line)cultured for 48 h in the presence of increasing doses of doxorubicin (a) or vinblastine (b)
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3521
Oncogene

pharmacological treatments could derive fromchanges in the expression levels of genes codifyingfor proteins involved in both cell cycle arrest andapoptosis. Moreover, the presence of the same p53homozygous point mutation, and the consequentfunctional inactivation of p53 in both parental andresistant cell lines, claims a further investigationaimed to identify the p53-independent pathway(s)by which the Hos cell line undergoes apoptosiswhile the MDR clone does not. Therefore, we screenedthe Hos cell line, at baseline condition and at varioustimes after 150 ng/ml doxorubicin treatment, and theHosDXR150 clone for the expression of genesinvolved in apoptosis and/or cell cycle control (E2F1,p73, Apaf-1, bax, bcl-2, p21/Waf1, p27Kip1, pRb2/p130and E2F4). As shown in Figure 7, E2F1 expressionlevel increased upon doxorubicin treatment andkept high until 12 h. E2F1 amount in HosDXR150clone was comparable to what was observed inuntreated Hos cell line. On the other hand, Apaf-1,whose transcription is controlled by E2F1 (Moroniet al., 2001) during apoptosis execution, was undetect-able in both resistant and untreated sensitive osteosar-coma cell lines and its up-regulation was observedonly in Hos cells after 4–6 h of doxorubicin treatment.
Figure 4 P-glycoprotein/p170 expression in Hos andHosDXR150. CLSM of P-glycoprotein in parental cell line (a)and in the MDR clone (b). The p170 (green color) accumulatedin the cytoplasmatic region of MDR cells. (c) Westernblot analysis
Figure 5 IC50 value of HosDXR150 cultured for 48 h in thepresence of increasing doses of doxorubicin and 10 mm verapamil.The dotted line represents the HosDXR150 culture in the presenceof increasing doses of doxorubicin and 10 mm verapamil
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3522
Oncogene

Interestingly, E2F1 has recently been shown to directlyactivate also the transcription of p73, leading toactivation of p53-responsive genes and apoptosis.p73 level already increased in the Hos cell line after2 h of doxorubicin exposure and maintained a steadylevel for the next 6 h, decreasing below baseline levelafter 12 h. No difference in p73 expression level wasdetected between untreated parental and MDR celllines. Bax is one of the p73-target genes and exerts its
proapoptotic function by neutralizing bcl-2 anti-apoptotic activity. The increased level of p73 in Hoscells, after 4–6 h doxorubicin treatment, inducedbax overexpression and a corresponding bcl-2 down-regulation. On the other hand, no differences inbax and bcl-2 expression levels were detectable inHosDXR150 with respect to the untreated Hos cellline. These results indicate that the Hos cell lineundergoes apoptosis through an E2F1- and p73-dependent pathway while the HosDXR150 cells fail toactivate it.
Alterations of cell cycle regulators such as CDKinhibitors (i.e. p21/Waf1 and p27Kip1), pRB familyproteins (pRb/p105, pRb2/p130 and p107) and theirtranscription factors (E2F1-6) have been demonstratedto affect chemosensitivity by interfering with themechanisms controlling cell cycle arrest. Analysis ofthe expression of these cell cycle regulators showedthat p21/Waf1 did not change either in treated anduntreated Hos or in HosDXR150 cell lines, while p27Kip1
expression level increased after 4–6 h doxorubicintreatment only in Hos cells. Moreover, doxorubicinexposure induced an early evident decrease of pRb2/p130 expression (2–4 h after treatment) followed byan increase restricted to its inactive hypo- and hyper-phosphorylated forms (6 h) and exclusively to itsinactive hyperphosphorylated form after 12–24 h in
Table 1 Human p53 exons 2–11 primer sequences
Exon Primer Sequence Size of amplified fragment Primer location
2–3 PU2 50-TCCTCTTGCAGCAGCCAGACTGC-30 267 11678–11700PD3 50-AACCCTTGTCCTTACCAGAACGTTG-30 11942–11918
5 PU5 50-CTCTTCCTGCAGTACTCCCCTGC-30 211 13042–13065PD5 50-GCCCCAGCTGCTCACCATCGCTA-30 13253–13231
6 PU6 50-GATTGCTCTTAGGTCTGGCCCCTC-30 185 13308–13331PD6 50-GGCCACTGACAACCACCCTTAACC-30 13489–13466
7 PU7 50-GTGTTGTCTCCTAGGTTGGCTCGT-30 139 13986–14009PD7 50-CAAGTGGCTCCTGACCTGGAGTC-30 14124–14102
8 PU8 50-ACCTGATTTCCTTACTGCCTCTGGC-30 200 14404–14428PD8 50-GTCCTGCTTGCTTACCTCGCTTAGT-30 14603–14579
9 PU9 50-GCCTCTTTCCTAGCACTGCCCAAC-30 102 14668–14691PD9 50-CCCAAGACTTAGTACCTGAAGGGT-30 14769–14746
10 PU10 50-TGTTGCTGCAGATCCGTGGGCGT-30 131 17561–17583PD10 GAGGTCACTCACCTGGAGTGAGC-30 17690–17668
11 PU11 50-TGTGATGTCATCTCTCCTCCCTGC-30 153 18559–18583PD11 50-GGCTGTCAGTGGGGAACAAGAAGT-30 18712–18689
Figure 6 p53 gene sequence. Arrow indicates the homozygouspoint mutation on exon 5 at codon 156, Arg vs Pro, due to thesubstitution of G with C
Table 2 Human Rb2/p130 cDNA primer sequences
Fragment Primer Sequence Annealing Size of amplified fragment Primer location
1–6 S 50-TTCGCCGTTTGAATTGCTGCGG-30 481C 984 1–2212PC 50-AAGTTTCCTAATATAGGG-30 984–967
6–11 3 50-CCCCTTGTATCATTGAG-30 451C 751 884–9009R 50-AATACCAGATAAATCCATG-30 1635–1617
11–14 I10 50-GTTTTGCGGAGATGCTTTACTATAAAGTA-30 551C 543 1547–1575I13R 50-GGTGGGGAGCTGTACCTATCGTAT-30 2090–2067
15–18 I13 50-CTGATACTGGAGGACTTGGAAGGA-30 551C 610 2021–2044R34 50-GAGATCCCGAAGGCGGACAG-30 2631–2612
18–22 F20 50-GGAAGACCAGCTCTTTATCGC-30 551C 979 2561–258122R 50-AAGGCTGCTGCTAAACAGAT-30 3540–3521
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3523
Oncogene

the Hos cell line. On the contrary, both the activeand inactive forms of pRb2/p130 were highly expressedin HosDXR150. It is known that the pRb2/p130associates primarily with the E2F4 transcriptionfactor to modulate the transcription of specific cellcycle genes. We found that the E2F4 expression levelbegan to increase in the Hos cell line after 4 h reachinga peak after 6 h and subsequently decreasing to base-line level at 24 h. Even at its maximum, the E2F4expression level in the Hos cell line never reachedwhat was observed in HosDXR150. The differencesfound between the two clones in pRb2/p130 andE2F4 expression levels suggest that these twoproteins might play a role in response to doxorubicintreatment.
In vivo p73 promoter occupancy in Hos and HosDXR150cell lines
It has recently been shown that endogenous p73 a and bproteins are upregulated in osteosarcoma Saos cells,lacking both p53 and pRb/p105, in response to over-expression of E2F1 suggesting that E2F1 might regulatep73 expression (Stiewe and Putzer, 2000; Zaika et al.,2001). An important challenger in the study oftranscriptional control during cell cycle progressionand apoptosis in mammalian cells has been theidentification of proteins that bind and regulatepromoters under physiological conditions. Therefore,to study in vivo p73 promoter occupancy by E2Fs andpRb2/p130 in Hos and HosDXR150 cells, we performedchromatin immunoprecipitation (ChIP) assay in asyn-chronously growing Hos and HosDXR150 cells bothtreated with 150 ng/ml doxorubicin at various times.The immunoblottings were performed using antibodiesagainst E2F1, E2F4, p300, HDAC1 and pRb2/p130.The same blots were stripped and reprobed sequentially.As a negative control for chromatin immunoprecipita-tion specificity, we used an irrelevant antibody. In Hosparental cells, we detected a multimolecular complexformed by E2F1–pRb2/p130–p300 bound to p73promoter (Figure 8a), while in HosDXR150 cells amultimolecular complex formed by E2F4–pRb2/p130–HDAC1 was bound to the same p73 promoter region(Figure 8b). We performed PCR by using primersflanking the p73 promoter region. We were able toamplify the p73 promoter fragment containing theregion from �229 nt of the promoter to þ 81 nt of exon1, where þ 1 represents the first nt of exon 1, from thechromatin immunoprecipitated by ChIP (Figure 9).Direct sequencing of PCR products confirmed that theimmunoprecipitated DNA fragment corresponds to thep73 promoter (data not shown). This region confers thehighest promoter activity residues since it has beenidentified to contain five putative E2F elements and aTATA box (Seelan et al., 2002). The presence of E2F1–pRb2/p130–p300 complexes on the p73 promoter in theHos cells which showed an increased p73 expressionafter genotoxic damage suggests that it could act as anactivator of p73 transcription, thus triggering theapoptotic response. On the other hand, this proapopto-tic pathway seems to be impaired in HosDXR150 by thepresence of a repressive complex formed by E2F4–pRb2/p130–HDAC1 on the p73 promoter.
Discussion
In a first series of experiments, we characterized aputative MDR clone, HosDXR150, selected by cultur-ing an osteosarcoma cell line, Hos, in the presence ofincreasing doxorubicin doses. We showed that, while theHos parental cell line undergoes apoptosis upon drugtreatment, HosDXR150 does not. This acquired resis-tance extends, even in higher degree, also to otherpharmacological treatments, notwithstanding that theyact through completely different mechanisms. In parti-
Figure 7 Western blot analysis of genes involved in apoptosis and/or cell cycle control: E2F1, Apaf1, p73, bax, bcl2, p21/Waf1,p27Kip1, pRb2/p130 and E2F4. In total 40 mg of Hos whole celllysate at baseline condition and at various times after 150 ng/mldoxorubicin treatment, and of HosDXR150 cells were electro-phoretically fractionated. Western blots were normalized by usinga-actin antibody
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3524
Oncogene

cular, we showed that the HosDXR150 cells do notrespond to treatment by vinblastine sulfate, a drug,which exerts its anticancer effect by interfering withmitotic spindle assembly while doxorubicin carries outantitumoral activity, similarly to others anthracyclines,by intercalating DNA, thus preventing its synthesis.Therefore, HosDXR150 cells have been shown todisplay a typical MDR phenotype. The lack ofspecificity for any pharmacological treatment in MDRcan be explained by the observation that MDR tumorsoften overexpress P-glycoprotein/p170. P-glycoproteinacts as an energy-dependent efflux pump and extrudes alarge variety of lipophilic compounds, thus preventingtheir effect whatever it is. This allows tumor cells to
avoid the toxic effect of drugs and it is considered toconfer chemoresistance to cancer cells. We showed thatP-glycoprotein is overexpressed in HosDXR150 withrespect to the sensitive Hos parental cell line andaccumulates onto plasma membrane. However, activedrug extrusion can only partially explain MDR inHosDXR150 as application of verapamil, which acts asan antagonistic agent of P-glycoprotein blocking Ca2þ
channels, did not completely restore HosDXR150sensitivity to doxorubicin. P-glycoprotein is the productof a gene, MDR1, which is usually kept repressed by wt-p53, but can be activated by mutant p53 (Nguyen et al.,1994; Strauss and Haas, 1995; for a review, see Sigal andRotter, 2000). We screened Hos and HosDXR150 forp53 mutations and evidenced in both cell lines the samehomozygous point mutation at codon 156, inducing lossof p53 transactivation and DNA-binding abilities, thusconfirming similar results already reported in bothsensitive and MDR osteosarcoma cell lines (Park et al.,1994; Oda et al., 2000).
Anticancer treatments often switch on several path-ways in tumor cells. Besides activation of transmem-brane proteins extruding chemical substances from thecell, like P-glycoprotein, MDR may depend on dereg-ulation of genes and proteins involved in the control ofcell cycle progression and/or in apoptosis. This isparticularly evident in the case of osteosarcoma whoseprognosis and clinical outcome appear poorly correlatedto increased expression level of both MDR1/P-glyco-protein and mutant p53 (Gorlick et al., 1999). There-fore, since the sensitive osteosarcoma cell line showed anapoptotic response also in the presence of mutated p53,we investigated the possible p53-independent pathwaysunderlying programmed cell death and compared theresults with those obtained in its MDR variant in anattempt to identify alternative markers of MDR inosteosarcoma.
We compared the expression profile of genes involvedin both apoptosis and cell cycle control in the Hosparental cell line at baseline conditions and at varioustimes after doxorubicin treatment. Drug applicationinduced upregulation of E2F1, p73, Apaf-1 and bax anddownregulation of bcl-2. These results are in agreementwith recent data suggesting a model of E2F1-inducedapoptosis in which E2F1 overexpression directly reg-ulates the transcription of p73 and Apaf-1 (Irwin et al.,2000; Stiewe and Putzer, 2000; Moroni et al., 2001).High levels of p73 lead to the activation of p53-responsive target genes, like bax, while downregulationof bcl-2 moves the balance toward proapoptoticsignaling (Stiewe and Putzer 2000; Costanzo et al.,2002). E2F1 levels are higher than normal even incontrol Hos cells because of pRb/p105 functionalinactivation, which has been shown in osteosarcomaprimary tumors as well as in the Hos cell line and itsnickel-resistant variant (Lin et al., 1994; Weinberg, 1995;Miller et al., 1996). However, we report here a furtherincrease upon doxorubicin treatment. E2F1 transcrip-tion is under pRb2/p130 and E2F4 control, which havebeen previously reported to be negative regulators ofE2F1 expression (Johnson, 1995). Therefore, we
Figure 8 In vivo p73 promoter occupancy by pRb2/p130–E2F1–p300 and pRb2/p130–E2F4–HDAC1 in Hos (a) and HosDXR150(b), respectively. ChIP analysis was performed by using antibodyagainst E2F1 for Hos samples or E2F4 for HosDXR150 samples.The immunoblottings were performed using antibodies againstpRb2/p130, E2F1, E2F4, HDAC1 and p300. The same blot wasstripped and reprobed sequentially. The C lane corresponds to anegative control immunoprecipitation using an irrelevant antibody
Figure 9 PCR of the chromatin immunoprecipitated by ChIPfrom Hos and HosDXR150 cells. The DNA was amplified usingspecific primers to amplify the p73 promoter fragment. The totalchromatin (input), which represents 0.5% of the total amount ofchromatin, was used as a positive control in each PCR reaction.The mock lane corresponds to a negative control of PCR reactionperformed using DNA extracted by chromatin immunoprecipitatedwith an irrelevant antibody
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3525
Oncogene

screened untreated and treated Hos cell line also forpRb2/p130, which we showed to be wt in these cells, andE2F4. Initially, doxorubicin treatment induced a de-crease in pRb2/p130 expression followed by an increase,which, as time progress, is restricted to its inactivephosphorylated forms. Phosphorylation determines therelease of E2F4, whose expression level reaches amaximum after 6 h, and can also influence the proteinstability by determining the probability of ubiquitin-mediated degradation. Interestingly, it has been re-ported that, at an early stage of the apoptotic response,pRb/p105 first becomes dephosphorylated and immedi-ately cleaved (for a review, see Dou and An, 1998). Ourobservation of a decrease of pRb2/p130 expressionwithin the first 2 h after doxorubicin treatment wouldsuggest that this pRB family member also couldundergo a similar early degradation related to apoptosiscommitment. The major presence of pRb2/p130 inactiveforms at later stages, concomitant to E2F4 overexpres-sion, could result in removal of transcriptional repres-sion by pRb2/p130–E2F4 complexes and underlie E2F1overexpression. Analysis of the changes in the geneexpression profile following doxorubicin treatment inthe Hos-sensitive cell line strongly suggests that theapoptotic response is induced through an E2F1- andp73-dependent pathway. Moreover, we have shown thatthis same pathway is no more functional in the MDRHosDXR150. In fact, we did not observe any upregula-tion of E2F1 and p73 and, consequently, any change inbax/bcl-2 balance. pRb2/p130 is present with both itsactive and inactive forms and is therefore still able to actas a negative regulator of E2F1 transcription.
It has recently been reported that ectopic expressionof pRb2/p130 is associated with downregulation of theantiapoptotic factor bcl-2 and upregulation of p73(Pucci et al., 2002), suggesting that it might regulatep73 expression. We studied the in vivo occupancy of p73promoter by pRb2/p130 and our results suggest a directrole of this protein in regulating p73 expression. Wefound two distinct multimolecular complexes bound to aspecific region of the p73 promoter and formed byE2F1–pRb2/p130–p300 in Hos and by E2F4–pRb2/p130–HDAC1 in HosDXR150 cells. This result allowsone depict a model for both apoptosis induction in Hoscell line and apoptosis evasion in HosDXR150. Geno-toxic damage induced in Hos cells overexpression ofE2F1 and p73 upregulation, which would be determinedby an increase in its transcription due to binding ofE2F1–pRb2/p130–p300 to its promoter region. Onthe other hand, in HosDXR150, p73 transcriptionwould be maintained repressed by the presence ofE2F4–pRb2/p130–HDAC1 complexes, thus preventingtransactivation of p73 downstream proapoptotic genes.E2F-binding sites have been recently characterized inthe human p73 promoter (Seelan et al., 2002) and inmany other genes involved in both DNA replication andcell cycle control (for a review, see Lavia and Jansen-Durr, 1999). Moreover, it has been shown that E2F-binding sites mediate both transcriptional activation andrepression (Dyson, 1998). The pRB–E2F repressivecomplexes function in association with HDAC, essen-
tially repressing the transcription of E2F-dependentgenes probably through deacetylation of histone tailsthat protrude from nucleosome (Ferreira et al., 1998;Cress and Seto 2000). The shift from gene silencing togene activation depends on the balance betweennucleosome deacetylation and acetylation, possiblyregulated by replacement of HDAC with HAT, likep300, in pRB–E2F complexes directly recruited to E2F-regulated promoters (Rayman et al., 2002; Macalusoet al., 2003).
Our results provide an interesting link between E2Fs–pRb2/p130 complexes and specific chromatin-modifyingfactors in a physiological setting. In fact the recruitmentof p300 or HDAC1 by pRb2/p130 on p73 promotercould be associated with the transcriptional level of thisE2F-responsive gene. pRb2/p130 could modulate thestructure and accessibility of chromatin by maintainingthe balance between nucleosome acetylation and deace-tylation. In sensitive osteosarcoma cells, recruitment ofE2F1–Rb2/p130–p300 complexes on p73 promoterinduces its expression, thereby triggering cell death.The presence of E2F4–pRb2/p130–HDAC1 repressivecomplexes on p73 promoter leads to failure of theapoptotic response in MDR osteosarcoma cells andcould provide the mechanism by which this cell lineevades the proapoptotic effect of chemotherapeuticdrugs.
Materials and Methods
Chemotherapic drugs
Doxorubicin hydrochloride (DXR) was purchased fromPharmacia and Upjohn (Bentley, Australia), vinblastinesulfate (VLB) from Lilly SA (Fegersheim, France) andverapamil from Knoll Farmaceutici (Milan, Italy). TheDXR, VLB and verapamil stock solutions were 2, 1 and2.5mg/ml, respectively.
Cell culture
Human osteosarcoma cell line (Hos) was cultured in Dulbec-co’s modified eagle medium (DMEM) supplemented with 10%heat-inactivated FCS at 371C in humidified 5% CO2 atmo-sphere. Doxorubucin-resistant osteosarcoma cells(HosDXR150) were obtained by continuous exposure ofparental cells to increasing doses of DXR, 0.5–150 ng/ml.The drug concentration was raised anytime the growth rate ofresistant subclone matched with that of the parental cell line.Approximately, 4–5 weeks were required to establish adequategrowth at each new DXR concentration. DXR cells werecontinuously cultured in the presence of drug for at least 6weeks before starting the experiments. To rule out thehypothesis that the selection could induce the appearance ofhyperdiploid phenotype, the FACS analysis was performed.White blood cells were used as a diploid control. Cytofluori-metric analysis showed a normal diploid pattern in both celllines.
Cell viability
Quantitative cell viability was measured by colorimetric assayusing cell proliferation kit (MTT) (Roche Molecular Biochem-icals, Germany). In all, 5000 cells/well Hos and HosDXR150
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3526
Oncogene

were grown in microtiter plates (96-well) in a final volume of100ml culture medium. The incubation period of cells culturewas 24, 48, 72, 96 h in the presence or absence of the drug(150 ng/ml). After incubation period, 10 ml MTT labelingreagent was added to each well (final concentration 0.5 mg/ml). MTT is cleaved by growing cells to form purple formazancrystals and allows quantification of cell viability by spectro-photometric analysis (ELISA) at 550 nm. Cell viability wasexpressed as the percentage of the absorbance of drug-treatedcells relative to that of the control untreated cells.
IC50 index
To determine the DXR-IC50 value, the sensitive and resistantcells were incubated for 48 h in the presence of increasing dosesof doxorubicin (10–5.000 ng/ml). To assess the selection ofMDR phenotype, we determined the VLB-IC50 value of cellsin the presence of increasing doses of vinblastine (2–10 000 ng/ml). Quantitative cell viability was measured by the MTT kit.
Reversal DXR resistance
The effect of verapamil on the in vitro sensitivity todoxorubicin was analysed in noncytotoxic conditions (10 mm)only on the subline HosDXR150 showing the high expressionlevel of P-glycoprotein/p170. The decrease of DXR-IC50 valuein the presence of 10mm verapamil was detected after 48 h andreferred to as DXR-IC50 after 48 h of incubation with onlyincreasing doses of doxorubicin. To assess the cytotoxic effectof 10mm verapamil on Hos drug-resistant clone, proliferationtest was performed in the absence of doxorubicin.
Immunofluorescence and confocal laser scanning microscopy(CLSM)
Hos and HosDXR150 cells were grown on slides and fixedwith 4% paraformaldehyde in 1� PBS. To detect theexpression of P-glycoprotein/p170, the samples were incubatedwith primary monoclonal anti-P-glycoprotein (JSB1) antibody(Medac Diagnostika, Germany) and fluorescein isothiocyanate(FITC)-conjugated secondary antibody. DNA was counter-stained with propidium iodide (PI) and slides were observed bya Sarastro 1000 microscope equipped with an argon ion laser.For the image acquisition, FITC and PI were excitedsimultaneously with the blue line (488 nm) of the argon ionlaser. The emission signal was observed through a dichroicmirror (DM: 500 nm) followed by 595 nm DM and thecombination of a band-pass filter (530715 nm) and cutofffilter (600 nm) to detect FITC and PI signals, respectively. Forthe three-dimensional reconstruction, optical sections wereobtained at increments of 0.3 mm in the Z-axis, stored on thecomputer and the spatial projections were obtained by theVolume Analysis Software System (VANIS).
In situ nick translation
Cells were harvested by trypsin treatment, pelleted and fixed inmethanol/acetic acid (3 : 1). Cell suspensions were spread ontoslides and incubated with a solution containing: 2U DNApolymerase I, 10mm each of four deoxynucleotides, anddigoxigenin-11-dUTP in 50mm Tris-HCl pH 7.8, 5mm MgCl2,10mm 2-mercaptoethanol. The samples were treated with 5%trichloroacetic acid (TCA) at 41C to remove unincorporatednucleotides and washed in Buffer I (1m Tris base, 1m NaCl,20mm MgCl2.6H2O, pH 7.5). The reaction was blocked byincubation for 20min at 421C in Buffer I þ 2% BSA. Theslides, incubated with FITC-conjugated antidigoxigenin anti-
body, were counterstained with 1mg/ml PI and observed by aCLSM.
PCR analysis and sequencing
Genomic DNA was extracted from both Hos and HosDXR-150 cells. For p53 exons analysis, the PCR reaction mix (50 ml)contained genomic DNA at a final concentration of 4 ng/ml,0.2mm of each of the four deoxynucleotide triphosphates, 2Uof Taq I polymerase (Promega, USA) and p53 panel primers(Human p53 Amplimer Panels kit, Clontech, USA) at a finalconcentration of 0.4mm each. The sequence of primers isshown in Table 1. For p53 exon 4, new set of primers was used:P4ca:50-GGGAAGCGAAAATTCATGGGAC-30; P4cb:50-AGACTTCAATGCCTGGCCGTAT-30. In all, 35 cycles ofdenaturation (951C, 1min), annealing (661C, 1min) andextension (721C, 1min) linked to one cycle at 721C for 7minwere carried out. To amplify the entire Rb2/p130 cDNA, RT–PCR was performed using panel primers shown in Table 2.Sequences of the purified PCR products were carried out byautomated DNA sequencing using dideoxy-terminator reac-tion chemistry for sequence analysis on the Applied Biosystemmodel 373A DNA Sequencer.
Western blot assay
Whole-cell lysates were prepared by resuspending pelleted cellsin lysis buffer (50mm HEPES pH 7.5, 150mm NaCl, 10%glycerol, 1% Triton X-100, 1mm EGTA, 1.5mm MgCl2,100mm NaF, 10mm disodium pyrophosphate, 10 mg/mlaproptinin, 10mg/ml leupeptin). Proteins (40 mg) were dena-tured by boiling in 2� sample buffer (100mm Tris-HCl pH6.8, 200mm dithiothreitol, 2% SDS, 0.1% bromophenol blue,10% glycerol) and size fractioned by electrophoresis in SDS/polyacrylamide gel and transferred onto 0.2 mm nitrocellulosemembrane (BioRad, USA). After saturation with 3% fat-freemilk and 2% BSA solution, the membranes were incubatedwith the following monoclonal antibodies: anti-human-bax(2D2) (Kamiya Biomedical Company, USA) and anti-bcl-2a(100/D5) (Kamiya Biomedical Company, USA), anti-p53(Ab6) (Calbiochem, Germany), anti-P-glycoprotein (JSB1)(Medac Diagnostika, Germany), anti-pRb2/p130 (10) (Trans-duction Laboratories, USA), and anti-p21/Waf-1 (4D10)(Medac Diagnostika, Germany), anti-E2F1 (KH95) (SantaCruz Biotechnology, USA), anti-p27Kip1 (57) (TransductionLaboratories, USA). For E2F4, p73 and Apaf-1 expressionanalyses polyclonal antibodies were used: anti-E2F4 antibody(C20), anti-p73 (H79), anti-Apaf-1 (N19) (Santa Cruz Bio-technology, USA). To normalize Western blot analysis, theantiactin antibody (Sigma Chemical, MO, USA) was used.After three washings with PBS-Tween-20, the membranes wereincubated with secondary anti-mouse or anti-rabbit IgG,coupled with horseradish peroxidase (Amersham, Life Science,UK). Signal was detected using the ECL systemt (Amersham,Life Science, UK).
DNA ladder
Hos and HosDXR150 cells were harvested, pelleted andwashed with 1X PBS. The pellet was resuspended in a lysisbuffer (1% Nonidet P40, 10U/ml Proteinase K in 1X PBS)and incubated for 15min on ice. The samples were centri-fuged for 15min at 14 000 r.p.m., and the supernatant wereseparated and treated with 50mg/ml of RNasei A for 30min at371C. Cell extracts were analysed by electrophoresis in 2%agarose gel.
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3527
Oncogene

In vivo promoter occupancy by pRb2/p130, E2Fs, HDAC1 andp300 using chromatin immunoprecipitation (ChIP) in the Hosand HosDXR150 cell lines
We performed ChIP using previously published methods(Orlando et al., 1997; Takahashi et al., 2000; Macaluso et al.,2003). Exponentially growing Hos and HosDXR150 cells werefixed with formaldehyde and chromatin was solubilized bysonication. Immunoprecipitations were set up as indicatedpreviously, using 2–4mg of antibodies per immunoprecipita-tion. Antibodies against pRb2/p130, E2F1/4, HDAC1 andp300 were obtained from Santa Cruz Biotechnology andUpstate Biotechnology. An irrelevant antibody was used as anegative control in each immunoprecipitation. The immuno-complexes were recovered with protein A/G-Sepharose andimmunoblotting was performed using the specified antibodies.
DNAs were purified from these immunoprecipitatedchromatin/protein complexes, and PCR was performed usingspecific primers to amplify the p73 promoter (forward p73S: 50-CGA CTT GGA CGC GGC CAG CT-30 and reverse p73R:50-CGA CGC TGC CGA CGT CCA TC-30). The totalchromatin (input) was used as a positive control in eachPCR reaction.
Acknowledgements
We are grateful to Dr Fiorenzo Marinelli for the helpful andconstructive comments on the manuscript and Dr MassimoRiccio for helping in the acquisition of confocal laser scanningimages. This work was supported by CNR grant, MURST-LAG-CO3 grant, Sbarro health Research Organization andNIH grants.
References
An B, Jin JR, Lin P and Dou QP. (1996). FEBS Lett., 399,158–162.
Bellan C, Lazzi S, Ferrari F, Stumpo M, Bartolomei S, Toti P,Leoncini L, Tosi GM, Cevenini G, Cinti C, Trimarchi C,Giordano, Kraft R, Laissue J and Cottier H. (2002). Inv.Ophthal. Visual Sci., 43, 3602–3608.
Chan HM, Krstic-Demonacos M, Smith L, Demonacos C andLaThangue NB. (2001). Nat Cell Biol., 3, 667–674.
Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, GottesmannMM and Roninson IB. (1986). Cell, 47, 381–389.
Cinti C, Claudio PP, De Luca A, Cuccurese M, Howard CM,D’Esposito M, Paggi M, La Sala D, Azzoni L, HalazonetisT, Giordano A and Maraldi NM. (2000). Onocogene, 19,5098–5105.
Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V,Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, BlandinoG, Balsano C and Levrero M. (2002). Mol. Cell., 9,175–186.
Cress WW and Seto E. (2000). J. Cell Physiol., 184, 1–16.Dou QP and An B. (1998). Frontiers Biosci., 3, d419–d430.Dyson N. (1998). Gen. Dev., 12, 2245–2262.Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan Aand Trouche D. (1998). Proc. Natl. Acad. Sci. USA, 95,104–193.
Gorlick R, Huvos AG, Heller G, Aledo A, Beardsley GP,Healey JH and Meyers PA. (1999). J. Clin. Oncol., 17,2781–2788.
Gottesman MM and Pastan I. (1988). Trends Pharmacol. Sci.,9, 54–58.
Hansen MF. (1991). Clin. Orthop., 270, 237–246.Hiebert SW, Packham G, Strom DK, Haffner R, Oren M,Zambetti G and Cleveland JL. (1995). Mol. Cell. Biol., 15,6864–6874.
Hsieh JK, Chan FSG, O’Connor DJ, Mittnacht S, Zhong Sand Lu X. (1999). Mol. Cell., 3, 181–193.
Hsieh JK, Fredersdorf S, Kauzarides T, Martin K and Lu X.(1997). Genes Dev., 11, 1840–1852.
Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, LiuW, Flores ER, Tsai KY, Jacks T, Vousden KH and KaelinJr WG. (2000). Nature, 407, 645–648.
Johnson DG. (1995). Oncogene, 11, 1685–1692.Johnson DG and Schneider-Broussard R. (1998). Front.
Biosci., 3, d447–d448.Kane SE, Pastan I and Gottesman MM. (1990). J. Bioenerg.
Biomembr., 22, 593–618.
Kowalik TF, DeGregori J, Leone G, Jakoi L and Nevins JR.(1998). Cell. Growth Differ., 9, 113–118.
Lavia P and Jansen-Durr P. (1999). BioEssay, 21, 221–230.Lin X, Dowjat WK and Costa M. (1994). Cancer Res., 54,2751–2754.
Macaluso M, Cinti C, Russo G, Russo A and Giordano A.(2003). Oncogene, 22, 3511–3517.
Miller CW, Aslo A, Won A, Tan M, Lampkin B andKoeffler HP. (1996). J. Cancer Res. Clin. Oncol., 122,559–565.
Moroni MC, Hickman ES, Denchi LE, Caprara G, Colli E,Cecconi F, Muller H and Helin K. (2001). Nat. Cell. Biol., 3,552–558.
Nguyen KT, Liu B, Ueda K, Gottesman MM, Pastan I andChin KV. (1994). Oncol. Res., 6, 71–77.
Oda Y, Matsumoto Y, Harimaya K, Iwamoto Y andTsuneyoshi M. (2000). Oncol. Rep., 7, 859–866.
Orlando V, Strutt H and Paro R. (1997). Methods, 11,205–214.
Park DJ, Nakamura H, Chumakov AM, Said JW,Miller CW, Chen DL and Koeffler HP. (1994). Oncogene,9, 1899–1906.
Pucci B, Claudio PP, Masciullo V, Bellincampi L, Terrinoni A,Khalili K, Melino G and Giordano A. (2002). Oncogene, 21,5897–5905.
Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH,Catchpole S, Watson RJ, Riele H and Dynlacht BD. (2002).Gen. Dev., 16, 933–947.
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, YoungRA and Dynlacht BD. (2002). Gen. Dev., 16, 245–256.
Seelan RS, Irwin M, van der Stoop P, Qian C, Kaelin Jr WGand Liu W. (2002). Neoplasia., 4, 195–203.
Serra M, Scotlandi K, Manara MC, Maurici D, Lollini PL, DeGiovanni C, Toffoli G and Baldini N. (1993). AnticancerRes., 13, 323–329.
Shnyder SD, Hayes AJ and Pringle J. (1998). Br. J. Cancer, 78,757–759.
Sigal A and Rotter V. (2000). Cancer Res., 60, 6788–6793.Somasundaram K and El-Deiry WS. (2000). Front. Biosci., 5,d424–d437.
Stiewe T and Putzer BM. (2000). Nat. Genet., 26, 464–469.Strauss BE and Haas M. (1995). Biochem. Biophys. Res.
Commun., 217, 333–340.Strauss BE, Shivakumar C, Deb SP, Deb S and Haas M.(1995). Biochem. Biophys. Res. Commun., 217, 825–831.
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3528
Oncogene

Takahashi Y, Rayman JB and Dynlacht BD. (2000). Gen.Dev., 14, 804–816.
Thottassery JV, Zambetti GP, Arimori A, Shuetz EGand Shuetz JD. (1997). Proc. Natl. Acad. Sci. USA, 94,11037–11042.
Vogelstein B, Lane D and Levine AJ. (2000). Nature, 408,307–310.
Weinberg RA. (1995). Ann. NY Acad. Sci., 758, 331–338.Zaika A, Irwin M, Sansome C and Moll UM. (2001). J. Biol.
Chem., 276, 11310–11316.Zini N, Trimarchi C, Claudio PP, Stiegler P, Marinelli F,Maltarello MC, La Sala D, De Falco G, Russo G, AmmiratiG, Maraldi NM, Giordano A and Cinti C. (2001). J. CellPhysiol., 89, 34–44.
Role of E2Fs-pRb2/p130 complexes in p73-dependent apoptosisD La Sala et al
3529
Oncogene
Related Documents
![PROCEDURES MANUAL Manual July 2020.pdf · commentary below concerning P130 is for historical reference only.] P130 Ballots 90-267-1 and 90-267-2, passed in March 1, 1991, amended](https://static.cupdf.com/doc/110x72/5fa86abb3a8007612d7d51e8/procedures-manual-manual-july-2020pdf-commentary-below-concerning-p130-is-for.jpg)