Molecular and Cellular Endocrinology 250 (2006) 8–19 Transcriptome analyses of male germ cells with serial analysis of gene expression (SAGE) Wai-Yee Chan a,b,∗ , Tin-Lap Lee a , Shao-Ming Wu a , Lisa Ruszczyk a , Diana Alba a , Vanessa Baxendale a , Owen M. Rennert a a Laboratory of Clinical Genomics, National Institute of Child Health and Human Development, National Institutes of Health, 49 Convent Drive, MSC 4429, Bethesda, MD 20892-4429, United States b Department of Pediatrics, Georgetown University Medical Center, Washington, DC 20007, United States Abstract Serial analysis of gene expression (SAGE) provides an alternative with additional advantages to microarrays for studying gene expression during spermatogenesis. The digitized transcriptome provided by SAGE of purified mouse germ cells identified 27,504 species of transcripts expressed in type A spermatogonia, pachytene spermatocytes, and round spermatids. Over 2700 of these transcripts were novel. Computational analyses allowed the identification of clusters of co-regulated genes, cell-specific promoter modules, cell-specific biological processes, as well as “preferential” biological networks in different cell types. These analyses provided potential drug targets for interference of specific pathways at different stages of spermatogenesis. Analyses of the transcriptomes revealed the prominent role of cytochrome c oxidase in germ cells and suggest a novel role for this enzyme in cytochrome c-mediated apoptosis in spermatogonia. A number of genes were shown to undergo differential splicing during spermatogenesis giving rise to cell-specific splice variants. Published by Elsevier Ireland Ltd. Keywords: Germ cells; Transcriptome; Promoter; Network; Apoptosis; Splicing 1. Introduction Spermatogenesis is a complex and tightly regulated pro- cess during which spermatogonial stem cells undergo mitotic expansion, meiosis, and differentiation to yield mature sperma- tozoa. It provides an informative model system for investigat- ing the underlying molecular mechanisms contributing to the physiological changes in self-renewal and differentiation dur- ing germ cell development. Despite its biological importance, remarkably little is known about the underlying mechanisms of stage-specific gene regulation at different stages of spermato- genesis. There are important features of the intrinsic genetic program underlying the process of male germ cell development. Repression of translation is one of the characteristic features of mammalian spermatogenesis (Kleene, 2001). Meiotic and haploid spermatogenic cells exhibit atypical patterns of gene expression in which alternative splicing often gives rise to sper- matogenic cell-specific transcripts (Cataldo et al., 1999). There is also global inhibition of initiation of mRNA translation in ∗ Corresponding author. Tel.: +1 301 451 6621; fax: +1 301 480 4700. E-mail address: [email protected] (W.-Y. Chan). pachytene spermatocytes and round spermatids (Cataldo et al., 1999; Kleene, 2001, 2003). Information on the mechanisms of translational regulation in spermatogenic cells is lacking. Even the network of genes regulating spermatogenesis was largely unknown until recently. In the last 5 years, numerous efforts were spent to map the transcriptome of male germ cells. The pace of discovery of germ cell genes was accelerated with advances in functional genomic techniques. The repertoire of germ cell genes is rapidly expanding. The increasing knowledge of germ cell genes allows a better understanding of the biological path- ways that play critical roles during spermatogenesis. This has led to an enhanced recognition of transcriptional and translational regulation during this process. This explosion of information about the biology of spermatogenesis will allow better treatment of male reproduction in health and disease. 2. Mapping the transcriptome of male germ cells The NCBI Mouse UniGene database in 2004 contained less than 7000 different sequences identified to be from spermatogo- nium, spermatocyte, or spermatid libraries. However, most are ESTs and few have been confirmed to be expressed by other methods. An unknown fraction consists of non-overlapping 0303-7207/$ – see front matter. Published by Elsevier Ireland Ltd. doi:10.1016/j.mce.2005.12.018

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Molecular and Cellular Endocrinology 250 (2006) 8–19
Transcriptome analyses of male germ cells withserial analysis of gene expression (SAGE)
Wai-Yee Chan a,b,∗, Tin-Lap Lee a, Shao-Ming Wu a, Lisa Ruszczyk a,Diana Alba a, Vanessa Baxendale a, Owen M. Rennert a
a Laboratory of Clinical Genomics, National Institute of Child Health and Human Development, National Institutes of Health,49 Convent Drive, MSC 4429, Bethesda, MD 20892-4429, United States
b Department of Pediatrics, Georgetown University Medical Center, Washington, DC 20007, United States
Abstract
Serial analysis of gene expression (SAGE) provides an alternative with additional advantages to microarrays for studying gene expressionduring spermatogenesis. The digitized transcriptome provided by SAGE of purified mouse germ cells identified 27,504 species of transcriptsexpressed in type A spermatogonia, pachytene spermatocytes, and round spermatids. Over 2700 of these transcripts were novel. Computationalanalyses allowed the identification of clusters of co-regulated genes, cell-specific promoter modules, cell-specific biological processes, as well as“dadP
K
1
cetipirsgpRohemi
0d
preferential” biological networks in different cell types. These analyses provided potential drug targets for interference of specific pathways atifferent stages of spermatogenesis. Analyses of the transcriptomes revealed the prominent role of cytochrome c oxidase in germ cells and suggestnovel role for this enzyme in cytochrome c-mediated apoptosis in spermatogonia. A number of genes were shown to undergo differential splicinguring spermatogenesis giving rise to cell-specific splice variants.ublished by Elsevier Ireland Ltd.
eywords: Germ cells; Transcriptome; Promoter; Network; Apoptosis; Splicing
. Introduction
Spermatogenesis is a complex and tightly regulated pro-ess during which spermatogonial stem cells undergo mitoticxpansion, meiosis, and differentiation to yield mature sperma-ozoa. It provides an informative model system for investigat-ng the underlying molecular mechanisms contributing to thehysiological changes in self-renewal and differentiation dur-ng germ cell development. Despite its biological importance,emarkably little is known about the underlying mechanisms oftage-specific gene regulation at different stages of spermato-enesis. There are important features of the intrinsic geneticrogram underlying the process of male germ cell development.epression of translation is one of the characteristic featuresf mammalian spermatogenesis (Kleene, 2001). Meiotic andaploid spermatogenic cells exhibit atypical patterns of genexpression in which alternative splicing often gives rise to sper-atogenic cell-specific transcripts (Cataldo et al., 1999). There
s also global inhibition of initiation of mRNA translation in
pachytene spermatocytes and round spermatids (Cataldo et al.,1999; Kleene, 2001, 2003). Information on the mechanisms oftranslational regulation in spermatogenic cells is lacking. Eventhe network of genes regulating spermatogenesis was largelyunknown until recently. In the last 5 years, numerous effortswere spent to map the transcriptome of male germ cells. The paceof discovery of germ cell genes was accelerated with advancesin functional genomic techniques. The repertoire of germ cellgenes is rapidly expanding. The increasing knowledge of germcell genes allows a better understanding of the biological path-ways that play critical roles during spermatogenesis. This has ledto an enhanced recognition of transcriptional and translationalregulation during this process. This explosion of informationabout the biology of spermatogenesis will allow better treatmentof male reproduction in health and disease.
2. Mapping the transcriptome of male germ cells
The NCBI Mouse UniGene database in 2004 contained lessthan 7000 different sequences identified to be from spermatogo-nium, spermatocyte, or spermatid libraries. However, most are
∗ Corresponding author. Tel.: +1 301 451 6621; fax: +1 301 480 4700.E-mail address: [email protected] (W.-Y. Chan).
ESTs and few have been confirmed to be expressed by othermethods. An unknown fraction consists of non-overlapping
303-7207/$ – see front matter. Published by Elsevier Ireland Ltd.oi:10.1016/j.mce.2005.12.018

W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 9
ESTs from different regions of full-length cDNA sequences.A proportion of ubiquitously expressed genes are commonamong the different libraries. Zhang et al. (1997) estimatedthat up to 20,000 different transcripts are present in a givencell population. Thus, this number of genes is inadequate todefine the genetic regulatory pathways of spermatogenesis, norto abnormalities that occur in this process. Numerous investi-gators attempted to delineate the transcriptome of mouse malegerm cells. A series of mouse and rat testis cDNA librarieswere described in 1999 by McCarrey et al. (1999). Sequenc-ing of these cDNA libraries may generate information aboutgenes expressed in germ cells at different stages of develop-ment. Wang et al. (2001) using cDNA subtraction identified 25male germ cell-specific genes that are expressed in spermatogo-nia. A 950-gene DNA array was developed for examining geneexpression patterns in mouse testis (Rockett et al., 2001). Shaet al. (2002) using cDNA array hybridization identified 7500genes in mouse testis; 256 genes displayed differential expres-sion between the ages of 1 and 4 weeks. Among these, 101genes were identified as critically related to testicular devel-opment and possibly to spermatogenesis. Fifty-nine are knownfull-length cDNAs, while the full-length cDNAs of the other 42genes have not been published. Using cDNA microarray andrepresentational difference analysis Tanaka et al. (2002) identi-fied 20 known genes and 4 uncharacterized cDNAs expressed intesticular cells. More recently, using differential display, Anwayegii2efsc(amsacmgtopufsttt
afilo
since much effort will be spent in sequencing the most abundantcDNAs. With the application of microarrays, the expression of alarger number of genes can be interrogated simultaneously. Thismethod is fast and is normally reliable especially for genes withhigher abundance and greater changes in expression levels (Ishiiet al., 2000). However, microarray studies are confined to the ele-ments on the array which are limited by the knowledge of genesat the time of the fabrication of the microarrays. Consequently,discovery of novel germ cell genes is not possible. Furthermore,the interpretation of the expression data is affected by the controlused. It has been pointed out that results obtained using differentarray formats and data obtained in different laboratories are notalways concordant (Jordan, 2004). As an alternative to librarycDNA sequencing, microarray hybridisation, and other profilingtechnologies, serial analysis of gene expression (SAGE) pro-vides a global view of the transcriptome of cells and tissueswith specific advantages over the other methods.
Unlike microarray technology, SAGE provides a true statis-tical description of the mRNA population in the cells withoutprior selection of the genes to be analyzed. Since it providesdigital output of all genes examined, the data are free from biasintroduced by control RNA (Velculescu et al., 1995; Gorski etal., 2003). SAGE only requires sequencing of 10–14 bp of eachtranscript, the SAGE tag, thus the sequencing effort is simplifiedcompared to library cDNA sequencing. SAGE data are directlycomparable from one library to another by “virtual” subtraction.I3cadcpc
s(uI2wfiTgTlfssl
3
3
t
t al. (2003) identified five transcripts in developing testicularerm cells. A more recent report using a cDNA array contain-ng 1176 elements, identified several hundred genes expressedn germ cells at different stages of spermatogenesis (Yu et al.,003). Use of the mouse GeneFilter microarray containing morelements (5814), a report identified 79 genes which were dif-erentially expressed among type A spermatogonia, pachytenepermatocytes, and round spermatids in the mouse. This wasonfirmed by quantitative real-time polymerase chain reactionQPCR) (Pang et al., 2003). Another study used mouse testist different postnatal ages and the Affymetrix mouse genomeicroarray identified 351 germ cell-specific genes whose tran-
cript abundance increased with the onset of meiosis (Schultz etl., 2003). Almstrup et al. (2004) used a focused oligonucleotidehip containing 379 elements to interrogate gene expression inouse testis at 14–44 postnatal days. The expression of selected
enes in specific cell types was correlated by in situ hybridiza-ion experiments. They succeeded in identifying three clustersf genes with similar expression profiles from the start of theachytene phase to the release of spermatozoa. Another studysed a cDNA array with 1176 elements to interrogate 6 dif-erent germ cell types including type A spermatogonia, type Bpermatogonia, preleptotene spermatocytes, pachytene sperma-ocytes, round spermatids, and elongating spermatids. The inves-igators identified 23 genes that were differentially expressed inhese 6 stages of spermatogenesis (Guo et al., 2004).
Earlier attempts to identify germ cell genes only focused onfew spermatogonial genes, or the expression of the identi-
ed genes was not confirmed. Sequencing of germ cell cDNAibrary clones gives a detailed description of the transcriptomef germ cells. However, it is tedious and potentially wasteful
t is an extremely powerful tool to identify novel genes or novel′ end splice variants of predicted genes that are unique to a spe-ific cell type. The major shortcoming of SAGE is the occasionalmbiguity in gene assignment of tags. This is predominantlyue to the inherent problems in the construction of the UniGeneluster database. The other factor that contributes to the lesseropularity of SAGE as a profiling method is the lack of sophisti-ated computational methods available for SAGE data analyses.
SAGE was used in two recent reports to investigateenescence-dependent changes in testicular gene expressionYao et al., 2004) and expression of X-linked genes in testic-lar germ and somatic cells (Divina et al., 2005) of the mouse.n spite of the relatively small SAGE libraries (all libraries below5,000 tags) generated in both studies, interesting informationas obtained. No gross change in testicular gene expression pro-le was found between old and young mice (Yao et al., 2004).he other study showed a significant enrichment of X-linkedenes specific for testicular somatic cells (Divina et al., 2005).he size of the SAGE libraries and the use of the whole testis
imits the applicability of the data generated by these two studiesor examining global gene expression in germ cells at differenttages of spermatogenesis. This was achieved in a more exten-ive study using purified mouse germ cells and larger SAGEibraries (∼111,000 tags per library) (Wu et al., 2004).
. SAGE analyses of mouse germ cells
.1. Analysis of germ cell transcriptomes
In the SAGE transcriptome study (Wu et al., 2004), mouseype A spermatogonium (Spga), pachytene spermatocyte (Spcy),

10 W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19
and round spermatid (Sptd) SAGE libraries were sequencedto comparable depth (∼111,000 tags). The SAGE data weredeposited for public access at http://nichddirsage.nichd.nih.gov/publicsage/. The number of unique tags in the spermatogo-nium, spermatocyte and spermatid library is 31,514, 36,147, and34,000, respectively. A number of the SAGE tags in each libraryoccurred only once (singletons). These tags were considered tobe very rare transcripts or the products of sequencing errors, andwere not considered in the subsequent analyses. Consequently,there are 27,504 unique tags present as ≥2 copies among the 3germ cell libraries. These “non-singleton tags” were identifiedas transcripts expressed in the germ cells. Among these tags,2776 have no matching UniGene cluster and represent noveltranscripts. In this article, tags identify non-singleton tags unlessotherwise stated. Even though tags and transcripts are not alwaysequivalent, under the condition of the analysis described, theycan be used interchangeably.
The majority of tags occur at <0.01% (<12 copies) in thetotal library. Only spermatogonia and spermatocytes containtags present at >1% of the library (two tags in each). Thereare 22, 17, and 15 tags present at 0.25% or more in spermato-gonia, spermatocytes, and spermatids, respectively (Wu et al.,2004). An update of the SAGE data using a more recent NCBIUniGene Build (#145, released April 10, 2005) is shown inSupplementary Table S1 which can be accessed at the URL:http://lcg.nichd.nih.gov/MolCelEndo.html. The statistics of theSopopmwrttt
TS
S
R
T
T
SmnUiIhgar
matic decreases in the number of germ cell novel tags betweenthe publication of the data in 2004 (UniGene Cluster Build #134)and UniGene Cluster Build #145 indicating rapid progress in theidentification of mouse germ cell genes.
3.2. Major biological processes in each cell type
Supplementary Table S2 shows the 38 SAGE tags (tran-scripts) that are present in >200 copies (>0.18%) in mouse typeA spermatogonia. The three most abundant tags together consti-tute 6.55% and the remainder constitutes 11.20% of the library.Among these 38 tags, 3 match uncharacterized cDNAs, while 1represents a novel transcript. Two of the uncharacterized cDNAsare preferentially expressed in type A spermatogonia suggest-ing that our knowledge of spermatogonial genes is still limited.Almost half of the abundant tags (18) match ribosomal pro-tein UniGene clusters suggesting that protein biosynthesis is themajor biological process in these cells. Another abundantly rep-resented group of genes are the cytochrome c oxidase subunits.The putative biological process represented by these genes willbe discussed later.
For the pachytene spermatocyte library, only 23 SAGE tagsare present in >200 copies (>0.18% of library) (SupplementaryTable S3); together they constitute only 8.23% of the pachytenespermatocyte library. Among the abundant tags, only six are incommon with the type A spermatogonia library. They includecsDltpt(cpos
>(mtasatpboa(titat
AGE libraries is presented in Table 1. There are more speciesf SAGE tags in the spermatocyte and spermatid library com-ared to the type A spermatogonium library. The observationf the presence of more unique tags in spermatocytes com-ared to spermatogonia may not be accurate. Type A sper-atogonia used in the study was relatively pure while thereas cross-contamination between pachytene spermatocytes and
ound spermatids (Wu et al., 2004). Consequently, some of theags in the spermatocyte library may be contributed by the con-aminating spermatids. This cross-contamination also reduceshe number of cell-specific tags in both libraries. There are dra-
able 1tatistics of germ cell SAGE libraries
AGE tag Spga Spcy Sptd
eliable tags 110872 111384 111214
otal unique tags 31514 36147 34000Known genes 12508 13691 12368Uncharacterized cDNA 3020 4406 4234Novel 563 1058 1155
ranscripts identified 16091 19155 17757
pga: type A spermatogonia; Spcy: pachytene spermatocytes; Sptd: round sper-atids. Reliable tags are defined as SAGE tags that contain no ambiguous
ucleotide base. Tags matching known genes are defined as those that matchniGene clusters with at least one known mRNA; tags matching uncharacter-
zed cDNA include all tags that match UniGene clusters containing only RIKEN,MAGE, expressed clones, or any cDNAs without annotation. Novel tags do notave any matching UniGene cluster. The summation of these three categoriesives the number of transcripts identified. Number of total unique tags includeslso tags that are singletons. These data are based on the NCBI Build #145,elease April 10, 2005.
ytochrome b-245 beta polypeptide (Cbb), cytochrome c oxidaseubunit 5b (Cox5b), cytochrome c oxidase subunit 1 (Cox1),EAD (Asp–Glu–Ala–Asp) box polypeptide 5 (Ddx5), tubu-
in � 3/7 (Tuba3/7), and ribosomal protein S29 (Rps29). Sincehere is no cross-contamination in the type A spermatogonia andachytene spermatocyte preparations, these tags represent geneshat participate in cellular activities common between mitotictype A spermatogonia) and meiotic cells (pachytene spermato-ytes). There is no clearly discernable predominant process inachytene spermatocytes. The most significant tag may be thatf meiosis expressed gene 1 (Meig1) corroborating the meiotictate of the pachytene spermatocytes.
The spermatid library contains 28 tags that are present at200 copies and together they constitute 10.10% of the librarySupplementary Table S4). The two most abundant round sper-atid tags representing protamine 2 (Prm2) and transition pro-
ein 2 (Tnp2) are absent in type A spermatogonia. They arelso found at extremely low copy number in the pachytenepermatocyte library (1470 copies compared with 103 copiesnd 1286 copies compared with 155 copies); this may be con-ributed by the small number of spermatids contaminating theachytene spermatocyte preparation. Both genes are known toe involved in chromatin reorganization indicating that this isne of the major biological processes in the more differenti-ted round spermatids. Seventeen of the 28 most abundant tags>200 copies) in the spermatid library are in common withhose in the spermatocytes. Five of these tags are also presentn high abundance in the spermatogonia indicating their impor-ance in the survival of all mouse germ cells. Among the morebundant tags in spermatids are those matching sperm-specificranscripts consistent with the more differentiated state of round

W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 11
spermatids compared to type A spermatogonia and pachytenespermatocytes.
4. Computational analyses of SAGE data
4.1. Unsupervised clustering based on tag expression
The power of expression profiling is its capability for genediscovery using unguided approaches. Extremely importantresults have been obtained by applying cluster analyses inmicroarray experiments. One specific advantage of SAGE overmicroarray analysis is its ability to discover novel transcripts.Therefore, application of unsupervised clustering to SAGE datahas the potential of discovering novel genes that have biologicalactivities related to known genes or novel genes that are co-regulated by similar factors. However, cluster analyses had notbeen applied successfully to SAGE experiments until recently(Getz et al., 2000). Similar to this report, a two-way unsu-pervised clustering analysis was performed successfully withgerm cell SAGE data yielding interesting results (Lee, T.L.,Alba, D., Rennert, O.M., Chan, W.Y., submitted). Based on theglobe expression matrix obtained, the dendrogram clearly dis-tinguishes type A spermatogonia from pachytene spermatocytesand round spermatids, while the latter two are more alike (Fig. 1).The preferentially regulated genes in each stage are representedin one of the six clusters that show significantly up-regulated(tmStmatfacp((risddpi
4
tgnaeo
Fig. 1. Unsupervised SAGE clustering based on tag expression at differentstages of spermatogenesis. Hierarchical clustering dendrogram of 2384 tagshaving total tag count ≥20 in the three stages. From left to right, spermatogonia(Spga), spermatocytes (Spcy), and spermatids (Sptd). Cluster of co-regulatedgenes clearly distinguishable are numbered form 1 to 6. Spga clusters are high-lighted in red (clusters 1 and 2), Spcy clusters are labeled in blue (clusters 3and 4), and Sptd clusters are labeled in green (clusters 5 and 6). Relative tagexpression levels are indicated by color ranging from high (red) to low (green).(For interpretation of the references to color in this figure legend, the reader isreferred to the web version of this article.)
to affect spermatogenesis at a specific stage. Identification ofgene clusters in specific cell types provides the opportunity tostudy promoter modules specific for gene expression at thatstage of spermatogenesis. This can be achieved by aligning thepromoter sequences of the clustered genes with orthologous pro-moters from human and rat, discarding sequences that are notin common, and then analyze the promoter sequences usingFrameWorker (http://www.genomatrix.de) to look for two ormore transcription factor binding sites arranged in a defineddistance range as shown in Fig. 2. With this approach, a num-ber of distinct elements at each stage were identified (Lee, T.L.,Alba, D., Rennert, O.M., Chan, W.Y., submitted). Table 2 showsexamples of the cell-specific promoter modules identified withthis approach.
clusters 1, 3, and 5) or down-regulated (clusters 2, 4, and 6)ags with minimal changes in the other two stages. Unigenes
atching the representative tags for each cluster are shown inupplementary Table S5. A number of well-known genes in
hese clusters have been reported to play critical roles in sper-atogenesis. These include heat shock protein 70 (Christians et
l., 2003), histone 2A protein (Brown, 2003), and integrin pro-ein family member (Shinohara et al., 1999) in clusters 1 and 2rom type A spermatogonia, respectively; dynamin (Kamitani etl., 2002), ubiquitin-specific protease (Stouffs et al., 2005), andyclins (Ravnik and Wolgemuth, 1999) in clusters 3 and 4 fromachytene spermatocytes, respectively, and phospholipase A2Seigneurin-Berny et al., 2001) and high-mobility group box 2Ronfani et al., 2001) in clusters 5 and 6 from round spermatids,espectively. Comparison of the SAGE data with results obtainedn various microarray platforms including previously publishedtudies (Pang et al., 2003), public high-throughput expressionata available at SOURCE (Diehn et al., 2003) and multipleatabases at GermOnline (Wiederkehr et al., 2004) shows theercentage of concordant clustered genes to be 71, 67, and 63%n spermatogonia, spermatocytes, and spermatids, respectively.
.2. Discovery of stage-specific promoter modules
Regulation of gene expression begins at the level of transcrip-ion initiation controlled by transcription factors. The myriad ofene expression patterns is achieved through different combi-ations of transcription factors. Genes that are co-regulated inspecific type of germ cell are likely to be under the influ-
nce of the same group of transcription factors. The knowledgef such transcription factor groups will give one the ability
12 W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19
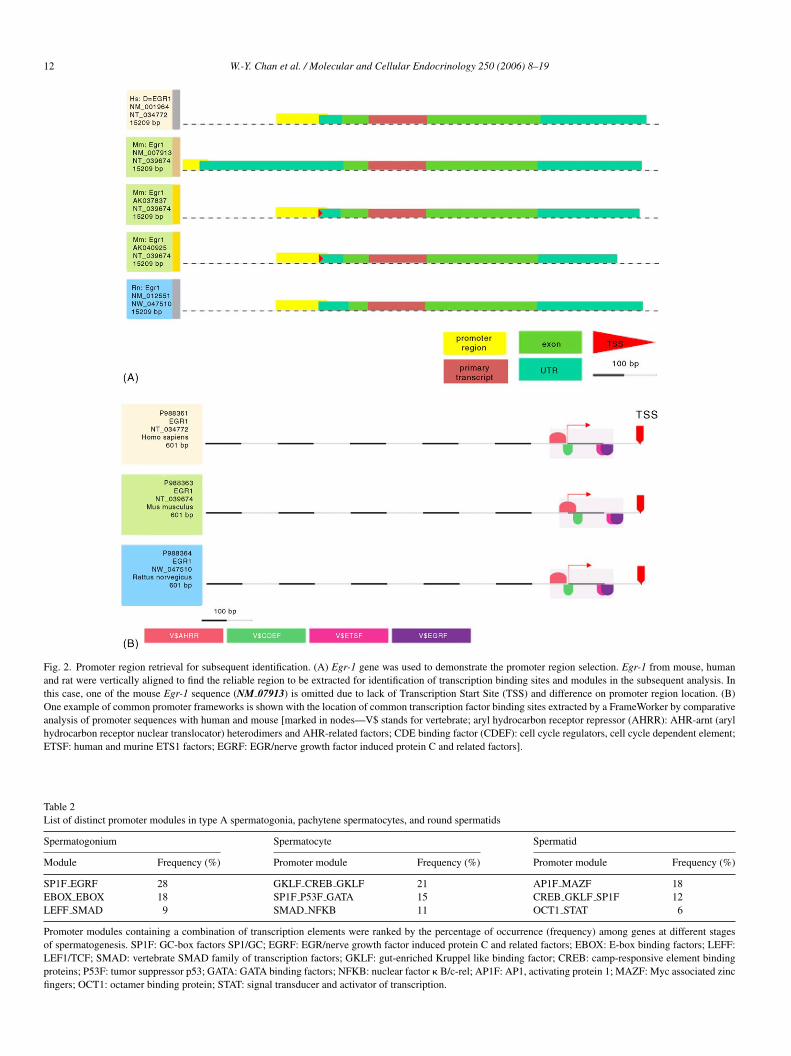
Fig. 2. Promoter region retrieval for subsequent identification. (A) Egr-1 gene was used to demonstrate the promoter region selection. Egr-1 from mouse, humanand rat were vertically aligned to find the reliable region to be extracted for identification of transcription binding sites and modules in the subsequent analysis. Inthis case, one of the mouse Egr-1 sequence (NM 07913) is omitted due to lack of Transcription Start Site (TSS) and difference on promoter region location. (B)One example of common promoter frameworks is shown with the location of common transcription factor binding sites extracted by a FrameWorker by comparativeanalysis of promoter sequences with human and mouse [marked in nodes—V$ stands for vertebrate; aryl hydrocarbon receptor repressor (AHRR): AHR-arnt (arylhydrocarbon receptor nuclear translocator) heterodimers and AHR-related factors; CDE binding factor (CDEF): cell cycle regulators, cell cycle dependent element;ETSF: human and murine ETS1 factors; EGRF: EGR/nerve growth factor induced protein C and related factors].
Table 2List of distinct promoter modules in type A spermatogonia, pachytene spermatocytes, and round spermatids
Spermatogonium Spermatocyte Spermatid
Module Frequency (%) Promoter module Frequency (%) Promoter module Frequency (%)
SP1F EGRF 28 GKLF CREB GKLF 21 AP1F MAZF 18EBOX EBOX 18 SP1F P53F GATA 15 CREB GKLF SP1F 12LEFF SMAD 9 SMAD NFKB 11 OCT1 STAT 6
Promoter modules containing a combination of transcription elements were ranked by the percentage of occurrence (frequency) among genes at different stagesof spermatogenesis. SP1F: GC-box factors SP1/GC; EGRF: EGR/nerve growth factor induced protein C and related factors; EBOX: E-box binding factors; LEFF:LEF1/TCF; SMAD: vertebrate SMAD family of transcription factors; GKLF: gut-enriched Kruppel like binding factor; CREB: camp-responsive element bindingproteins; P53F: tumor suppressor p53; GATA: GATA binding factors; NFKB: nuclear factor � B/c-rel; AP1F: AP1, activating protein 1; MAZF: Myc associated zincfingers; OCT1: octamer binding protein; STAT: signal transducer and activator of transcription.
W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 13
Sp1 is present among all three types of germ cells, suggest-ing its indispensable role during germ cell development. Indeed,Sp1 has been shown to play a critical role during the mainte-nance of germ cell development (Persengiev et al., 1996) and itis one of the most common transcription regulators involved inbiological processes in germ cells as revealed by network anal-ysis described in the later section. Sp1 binding GC-box domainsare commonly present in the promoters of developmental stage-specific genes in differentiating mouse germ cells. Recently, Sp1transcripts encoding the 60 and 90 kDa protein were reportedto mediate stage- and cell-type-specific gene expression duringmouse spermatogenesis (Thomas et al., 2005). Other than Sp1,regulatory elements functioning in multi-stages include Smad,Gklf, and Creb. Smad is present in spermatogonia and sperma-tocytes while Gklf and Creb are found in spermatogonia andspermatids. Functional deletion of Creb leads to complete arrestat the pachytene stage and causes male infertility (Bleckmann etal., 2002). On the other hand, involvement of Gklf in germ cellgene expression has not been reported. Cell-specific elementsincluded Egrf, Ebox, and Leff in spermatogonia, Gata and NfκBin spermatocyctes, Ap1F, Mazf, Oct1, and Stat in spermatids.Further examination on the interactions between these elementswill shed light on the finer details of cell-specific transcrip-
tional regulation. With the recent development of microarraystargeting interactions between transcription factors, it will sim-plify the simultaneous functional analysis of multiple eukaryotictranscription factors compared to conventional cumbersome gelmobility-shift assays.
4.3. Finding functional enrichment in different stages bygene ontology
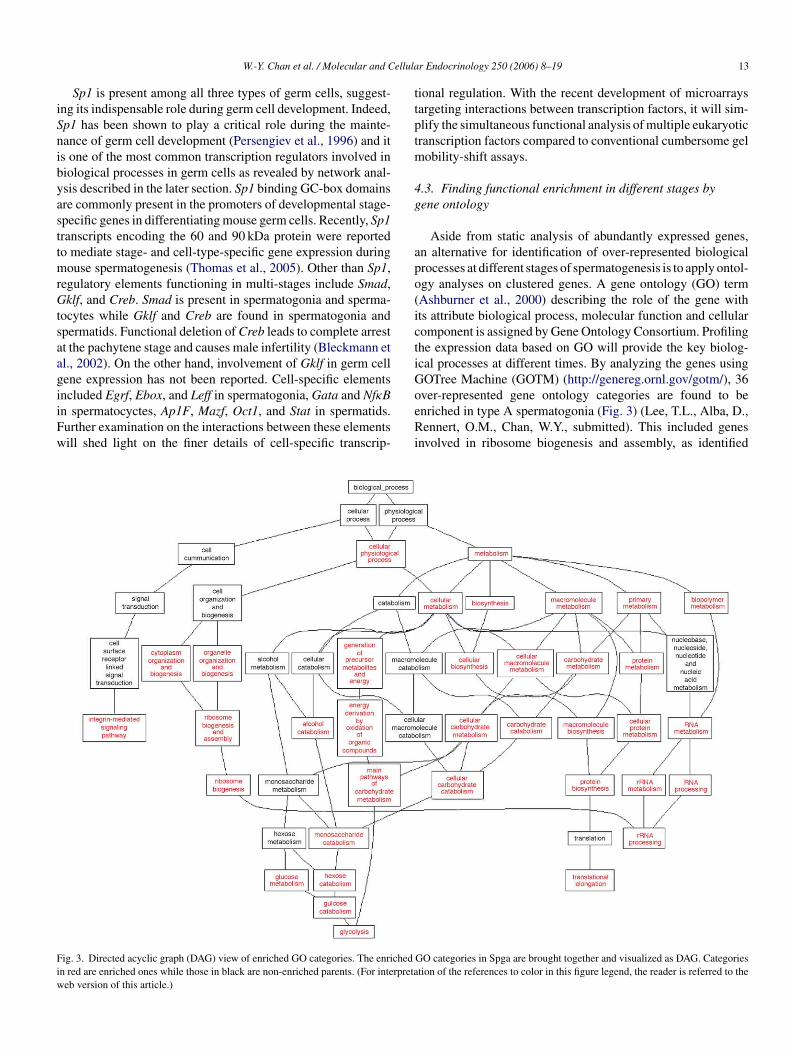
Aside from static analysis of abundantly expressed genes,an alternative for identification of over-represented biologicalprocesses at different stages of spermatogenesis is to apply ontol-ogy analyses on clustered genes. A gene ontology (GO) term(Ashburner et al., 2000) describing the role of the gene withits attribute biological process, molecular function and cellularcomponent is assigned by Gene Ontology Consortium. Profilingthe expression data based on GO will provide the key biolog-ical processes at different times. By analyzing the genes usingGOTree Machine (GOTM) (http://genereg.ornl.gov/gotm/), 36over-represented gene ontology categories are found to beenriched in type A spermatogonia (Fig. 3) (Lee, T.L., Alba, D.,Rennert, O.M., Chan, W.Y., submitted). This included genesinvolved in ribosome biogenesis and assembly, as identified
Fiw
ig. 3. Directed acyclic graph (DAG) view of enriched GO categories. The enrichedn red are enriched ones while those in black are non-enriched parents. (For interpretaeb version of this article.)
GO categories in Spga are brought together and visualized as DAG. Categoriestion of the references to color in this figure legend, the reader is referred to the

14 W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19
previously, and genes involved in the integrin signaling pathway,which consist of a disintegrin and metalloprotease domains 3 and5 (Adam3/5), integrin linked kinase (Ilk) and integrin, alpha E,epithelial associated (Itgae). Other interesting processes includecarbohydrate metabolism, protein biosynthesis and RNA pro-cessing. Similarly, 21 gene ontology categories are found tobe over-represented in pachytene spermatocytes. Some of theinteresting genes identified include protein phosphatase 4, regu-latory subunit 1 (Ppp4r1) involved in chromosome segregation,cyclin D3 (Ccnd3), cyclin L2 (Ccnl2), and cullin 4A (Cul4a)in cell cycle, tubulin � 3 (Tuba3) and ubiquitin-specific pro-tease 21 (Usp21) in cytoskeleton and ubiquitin regulation. The26 gene ontology categories enriched in round spermatids aremainly related to cellular physiological process and metabolicprocess related to the ubiquitin cycle, proteolysis and pepti-dolysis, cellular metabolism including arginine-tRNA-proteintransferase 1 (Ate1), proteasome (prosome, macropain) sub-unit, alpha type 3 (Psma3), ubiquitin-conjugating enzyme E2D2 (Ube2d2), ubiquitin-specific protease 5 (Usp5), etc. Thisinformation together with that obtained by analyzing the moreabundantly expressed genes provides a clearer picture of thebiochemical characteristics of cells at different stages of sper-matogenesis.
4.4. Linking genes to biological networks
stcweacaigdsa(tSfiDsmnwoimcitFa
highlighted with known antagonist (Fig. 4A and C). This infor-mation is valuable for the identification of potential targets forinterruption of spermatogenesis in male contraception and clin-ical intervention.
5. Cytochrome c oxidase in spermatogenesis
5.1. SAGE cloning of Cox subunits
Cytochrome c oxidase (Cox) subunits have a conspicuouspresence in germ cells. Initial analysis of the germ cell librariesshowed that the most abundant tag GTGGCTCACA matchestwo UniGene clusters while the third abundant tag, ATACT-GACAT matches a UniGene cluster of an uncharacterizedcDNA. Cloning of the transcripts corresponding to these twoSAGE tags using 5′-tag-specific primer and 3′-SAGE cloningprimer as described by Chen et al. (2000) and 3′ and 5′-Rapid Amplification of cDNA Ends (RACE) and nucleotidesequencing showed that the most abundant tag corresponds tocytochrome c oxidase subunit 5b (Cox5b), which is encodedby the nuclear genome. The third abundant tag correspondsto cytochrome c oxidase subunit 3 (Cox3), a mitochondrialgenome encoded subunit. Subsequently, the eighteenth mostabundant tag, GCTGCCCTCC, was shown to represent anothercytochrome c oxidase subunit, Cox1, by SAGE tag cloning(ouScAeedts
TT
S
CCCCCCCCCCCCC
SSpg
Another significant application of the knowledge of the tran-criptome is the identification of biological networks and poten-ial targets for interruption of spermatogenesis at a definedell stage. Biological network analysis has become a popularay for functional mining and illustration of high-throughput
xperiments with inherently noisy experimental datasets suchs microarray and SAGE. The expression data are analyzed andompared against curated databases containing protein–proteinnd protein–DNA interactions, transcriptional factors, signal-ng, metabolism, and bioactive molecules information. Inputenes are connected to each other directly or indirectly throughifferent intermediate objects such as neighbor genes, tran-cription factors or small molecules. Subjecting clusters of up-nd down-regulated genes of each cell stage to the MetaCorehttp://www.genego.com) program allows portraying the poten-ial biological pathways at different stages of spermatogenesis.elected biological networks of each germ cell stage identi-ed by such approach are shown in Fig. 4 (Lee, T.L., Alba,., Rennert, O.M., Chan, W.Y., submitted). The validity of
uch constructed pathways is demonstrated by the presence ofolecules known to be involved in spermatogenesis such as
itric oxide in spermatids (Fig. 4C) (O’Bryan et al., 2000). Path-ay analysis also allows cross-validation and complementationf other bioinformatic analyses. For instance, genes involvedn cell cycle and proliferation such as transcription factor E2F
embers, cyclins, proliferating cell nuclear antigen (Pcna), andell division protein kinase 2 (Cdk2) in spermatocytes are alsomplicated in the gene ontology results (Fig. 4B). More impor-antly, the construction of pathways allows drug target discovery.or example in a pathway identified for type A spermatogoniand round spermatids, the epidermal growth factor receptor is
Wu, S.M., Chan, W.Y., unpublished data). Further analysisf the germ cell data showed that the SAGE tags of all sub-nits of cytochrome c oxidase can be found in the germ cellAGE libraries. The copy number of the major tags of allytochrome c oxidase subunits in germ cells is shown in Table 3.ll cytochrome c oxidase subunits are quite abundant. With the
xception of three, all subunits show similar trend of differentialxpression with the highest level in type A spermatogonia andecrease towards round spermatids. This observation suggestshat cytochrome c oxidase is likely to play important roles inpermatogonia.
able 3he major SAGE tags of cytochrome c oxidase subunits in mouse germ cells
ubunit Tag sequence Spga Spcy Sptd Gene location
ox1 GCTGCCTCC 576 172 80 Mitochondriaox2 AGCAGTCCC 267 109 34 Mitochondriaox3 ATACTGACAT 1108 487 182 Mitochondriaox4i1 CTAATAAAAG 140 113 91 Nuclearox5a TAATAGTAAC 39 83 117 Nuclearox5b GTGGCTCACA 4735 773 631 Nuclearox6a1 TGAACCCACT 21 12 4 Nuclearox6b ATGCAACTAC 86 7 6 Nuclearox6c AATATGTGTG 103 114 65 Nuclearox7a2I TTTTCCACTT 7 5 2 Nuclearox7b TACTCATTAT 12 3 2 Nuclearox7c ACAAACTTAG 11 68 24 Nuclearox8a TATTGGCTCT 72 3 2 Nuclear
ubunit: subunit of cytochrome c oxidase; tag sequence: most abundant matchingAGE tag sequence; Spga/Spcy/Sptd: tag counts for type A spermatogonia,achytene spermatocytes, and round spermatids; gene location: location of theene on the nuclear or mitochondrial genome.

W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 15
Fig. 4. Biological network construction. Clustered gene sets from Spga (A), Spcy (B), and Sptd (C) were analyzed by GeneGO. Input gene sets are labeled in bluecircle. Objects with known drug target are labeled (EGFR in this example) with corresponding drug or compound targets. (For interpretation of the references tocolor in this figure legend, the reader is referred to the web version of this article.)

16 W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19
Fig. 4. (Continued )
5.2. Cytochrome c, Cox, and apoptosis
The well-known biological role of cytochrome c oxidase is asan active component of the electron transport chain. The chainis comprised of five complexes, namely NADH dehydrogenase,succinate dehydrogenase, ubiquinone-cytochrome c oxidore-ductase, cytochrome c oxidase, and ATP synthase, respectively.Each of these complexes contains multiple subunits (Lehningeret al., 1993). Theoretically for the chain to be active, all fivecomplexes, i.e. their constituent subunits, should be present andfollow a similar trend of change. However, with the exceptionof cytochrome c oxidase, most of the other complexes appearto be not active. For example, among the four subunits of succi-nate dehydrogenase, only three are present in the germ cells. Forubiquinone-cytochrome c oxidoreductase only 1 of 11 subunitsis present and for ATP synthase only 9 of 15 subunits are present.Among the subunits of NADH dehydrogenase only 12 of themfollow the same trend as cytochrome c oxidase. Even though thetranscripts encoding the subunits may just be present at levelstoo low to be detected in this study, it is safe to assume the activ-ities of the complexes of the electron transport chain to be low,with the exception of cytochrome c oxidase. Thus, cytochromec oxidase may have activities other than cellular respiration inmale germ cells.
One of the components of the electron transport chain iscytochrome c. There are two forms of cytochrome c: a testes-se1it
library, 4 copies in the spermatocyte library, and 2 copies in thespermatid library; a distribution pattern similar to the results ofimmunoelectron microscopic localization reported previously(Hess et al., 1993). This expression pattern is also similar tothat of the majority of the cytochrome c oxidase subunits. Thetestes-specific isoform is represented by the SAGE tag AAG-TAAAGTA which has a different pattern of distribution with 5copies in type A spermatogonia, 21 copies in spermatocytes and11 copies in spermatids.
Concurrent presence of high levels of cytochrome c oxidasesubunits and cytochrome c in type A spermatogonia suggestsimportant roles for these molecules in these cells. Besides its rolein electron transport, cytochrome c has been shown to play animportant role in the intrinsic pathway of apoptosis (Reed, 1997;Boehning et al., 2003; Mattson and Chan, 2003). In spermatoge-nesis, apoptosis is considered an important physiological mech-anism that regulates the number of sperm produced (Print andLoveland, 2000). During normal mitosis/differentiation in sper-matogonia there is substantial apoptosis (de Rooij and Russell,2000), particularly types A2 and A3 spermatogonia (Furuchiet al., 1996). Disruption in the mitochondria-mediated cascadeof apoptosis such as ectopic expression of Bcl-2 (Furuchi etal., 1996) or knockout of Bax in mice (Knudson et al., 1995)results in the accumulation of spermatogonia and the degenera-tion of germ cells. This cascade of apoptotic event is initiated bythe release of cytochrome c from the inter-membrane space ofmwbaP
pecific form only found in the testis and a somatic formxpressed in both somatic cells and testis (Goldberg et al.,977; Hecht, 1998). Both isoforms of cytochrome c are foundn the germ cells. The somatic form is represented by the SAGEag AATGGCTAGC which has 18 copies in the spermatogonia
itochondria (Strasser et al., 2000). Cytochrome c is associatedith cardiolipin in the inner mitochondrial membrane and solu-ilization of cytochrome c involves breaching of the electrostaticnd/or hydrophobic interactions between these two molecules.revious studies demonstrated that oxidation of cytochrome c

W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 17
is important for its release during apoptosis (Ghafourifar et al.,1999; Ott et al., 2002). It is tempting to speculate that the abun-dant cytochrome c oxidase in spermatogonia is responsible forthe oxidation of cytochrome c. This results in disrupting itsinteraction with cardiolipin and enhancing its release from mito-chondria leading to apoptosis. Similar involvement of caspaseand cytochrome c in sperm differentiation had also been demon-strated in Drosophila (Arama et al., 2003). Cytochrome c oxidasewill be less involved in apoptosis of meiotic and post-meioticcells since its expression levels in these cells is much lower thanin spermatogonia. Apoptosis in these stages of spermatogene-sis appears to occur mainly through the activation of the deathreceptor Fas and is cytochrome c independent (Lee et al., 1997,1999; Strasser et al., 2000).
6. Cell-specific alternative splicing variants
Alternative splicing of transcripts creates transcriptomediversification and is a major determinant for the functionaldiversity of proteins involved in human physiology, develop-ment, and behavior (Maniatis and Tasic, 2002; Black, 2003).Alternative transcripts are relatively common in germ cells andhave been assumed to be important for germ cell development(Walker et al., 1999; Venables, 2002). Until now, most of thegenome wide detection and classification of alternative splic-ing was based on the public EST data. Alternative splicing ofmai2osXdoutdteeptbbrr
sf2mviitg
give rise to 3′ end alternative splice variants (3′ AS) specific foreach type of germ cells. There are genes with cell-specific splic-ing variants in two cell types; 207 genes with 3′ AS expressed inboth type A spermatogonia and pachytene spermatocytes, 249 inboth spermatogonia and spermatids, and 158 in both spermato-cytes and spermatids. There are also 74, 58, and 62 genes with 3′AS in only type A spermatogonia, pachytene spermatocytes, andround spermatids, respectively (Lee, T.L., Alba, D., Chan, W.Y.,unpublished data). Novel splicing variants of genes involved indevelopmental and transcriptional controls were identified, suchas heat shock protein 4 (Hsp4), H3 histone, family 3B (H3f3b),and ubiquitin protein ligase E3A (Ube3a). Even though the bio-logical significance of cell-specific splicing of a gene is unclearat the present time, future functional studies on the cell-specificsplicing variants are expected to provide new insights into theregulation of germ cell development.
In spite of its usefulness in identifying splice variants, SAGEhas some significant limitations. The major limitations includethe inability to differentiate alternative transcripts with alter-native promoter or alternative internal exon usage, and tagswith multiple matching UniGene clusters. However, these short-comings can potentially be compensated by other tag-basedapproaches such as Cap Analysis Gene Expression (CAGE)(Shiraki et al., 2003) and 5′ SAGE (Hashimoto et al., 2004).Other than the unguided approach mentioned, the availabilityof alternative splicing microarray containing pre-defined sets ofscg
7
gtcfuumScdrgiaimmtatttoa
RNA is predicted by analyzing the exon linkage relationshipfter aligning the ESTs to the genome sequence and search-ng for splicing and polyadenylation evidence (Modrek et al.,001; Gupta et al., 2004). Although it allows the identificationf novel splicing variants and tissue-related splicing variants,uch as tumor-associated splicing isoforms (Wang et al., 2003;u and Lee, 2003; Hui et al., 2004), the results are totally depen-ent on the abundance of EST information. The limited numberf germ cell EST libraries makes the EST computational methodnsuitable and unreliable (Gupta et al., 2004). The identifica-ion of splice variants of Crem in the germ cell SAGE librariesemonstrates that SAGE offers an efficient approach to inves-igate the existence of alternatively processed transcripts (Wut al., 2004). SAGE provides a high-throughput means of gen-rating tissue-specific gene expression information that is notrone to potential orientation errors and sequence contamina-ions inherent in EST data. Putative splicing variants generatedy computational analysis can be experimentally confirmedy RT-PCR using reference primers amplifying the conservedegion of the gene and variant primers amplifying the variableegion.
Alternative splicing is often regulated in a temporal or tissue-pecific fashion giving rise to different protein isoforms in dif-erent tissues or at different developmental stages (Matter et al.,002). Even though alternative splice variants of genes are com-on in germ cells, little is known whether the different splice
ariants of a gene are cell- or stage-specific. Cell-specific splic-ng of a gene at different stages of germ cell development can bedentified by mapping and comparing the SAGE tags of a geneo the genomic sequence. With such approach, analysis of theerm cell SAGE libraries showed that there are 73 genes which
plicing variant of genes will also allow a genome-wide scan ofell-specific expression of the known splicing variants duringerm cell development.
. Conclusion
Knowledge of genes expressed at each stage of spermato-enesis is the first step towards understanding the mechanismshat allow a germ cell to progress from a spermatogonial stemell to become a healthy mature sperm. This information is ofundamental importance since it will help us understand the reg-latory network that causes a stem cell to replicate itself or tondergo differentiation. It will permit the design of better andore rational approaches to enhance and control male fertility.permatogenesis is a unique process since it is the only pro-ess in an adult organism that undergoes mitosis, meiosis, andifferentiation. It is likely that specific genes as well as theiregulation such as transcription and translation during spermato-enesis may be different from that found in somatic tissues. Its not surprising that a large number of uncharacterized cDNAsnd novel transcripts were found in the SAGE study. At present,n spite of the power of SAGE, microarray gene profiling has
ore sophisticated computational tools adapted for analyzingicroarray data. This SAGE study of germ cells is an attempt
o couple the advantage of SAGE with that of computationalnalyses. The result is encouraging as demonstrated by the iden-ification of a large number of stage-specific genes and processeshat will be useful for future research and for clinical applica-ions. The discovery of novel activity of known molecules andf novel biological processes corroborates the advantage of thispproach.

18 W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19
Acknowledgement
This research was supported by the Intramural ResearchProgram of the National Institute of Child Health and HumanDevelopment, National Institutes of Health.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version, at doi:10.1016/j.mce.2005.12.018.
References
Almstrup, K., Nielsen, J.E., Hansen, M.A., Tanaka, M., Skakkebaek, N.E.,Leffers, H., 2004. Analysis of cell-type-specific gene expression duringmouse spermatogenesis. Biol. Reprod. 70, 1751–1761.
Anway, M.D., Ravindranath, N., Dym, M., Griswold, M.D., 2003. Expressionof testicular germ cell genes identified by differential display. J. Androl.24, 173–184.
Arama, E., Agapite, J., Steller, H., 2003. Caspase activity and a specificcytochrome c are required for sperm differentiation in Drosophila. Dev.Cell 4, 687–697.
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M.,Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., Harris, M.A., Hill,D.P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J.C., Richardson,J.E., Ringwald, M., Rubin, G.M., Sherlock, G., 2000. Gene ontology:tool for the unification of biology. The Gene Ontology Consortium. Nat.
B
B
B
B
C
C
C
d
D
D
F
G
G
mitochondria. Importance of mitochondrial redox state. J. Biol. Chem.274, 6080–6084.
Goldberg, E., Sberna, D., Wheat, T.E., Urbanski, G.J., Margoliash, E., 1977.Cytochrome c: immunofluorescent localization of the testis-specific form.Science 196, 1010–1012.
Gorski, S.M., Chittaranjan, S., Pleasance, E.D., Freeman, J.D., Anderson,C.L., Varhol, R.J., Coughlin, S.M., Zuyderduyn, S.C., Jones, S.J.M.,Marra, M.A., 2003. A SAGE approach to discovery of genes involved inautophagic cell death. Curr. Biol. 13, 358–363.
Guo, R., Yu, Z., Guan, J., Ge, Y., Ma, J., Li, S., Wang, S., Xue, S., Han, D.,2004. Stage-specific and tissue-specific expression characteristics of dif-ferentially expressed genes during mouse spermatogenesis. Mol. Reprod.Dev. 67, 264–272.
Gupta, S., Zink, D., Korn, B., Vingron, M., Haas, S.A., 2004. Strengths andweaknesses of EST-based prediction of tissue-specific alternative splicing.BMC Genomics 28, 72.
Hashimoto, S., Suzuki, Y., Kasai, Y., Morohoshi, K., Yamada, T., Sese, J.,Morishita, S., Sugano, S., Matsushima, K., 2004. 5′-End SAGE for theanalysis of transcriptional start sites. Nat. Biotechnol. 9, 1146–1149.
Hecht, N., 1998. Molecular mechanisms of male germ cell differentiation.BioEssays 20, 555–561.
Hess, R.A., Miller, L.A., Kirby, J.D., Margoliash, E., Goldberg, E.,1993. Immunoelectron microscopic localization of testicular and somaticcytochromes c in the seminiferous epithelium of the rat. Biol. Reprod.48, 1299–1308.
Hui, L., Zhang, X., Wu, X., Lin, Z., Wang, Q., Li, Y., Hu, G., 2004. Iden-tification of alternatively spliced mRNA variants related to cancers bygenome-wide ESTs alignment. Oncogene 23, 3013–3023.
Ishii, M., Hashimoto, S., Tsutsumi, S., Wada, Y., Matsushima, K., Kodama,T., Aburatani, H., 2000. Direct comparison of GeneChip and SAGE onthe quantitative accuracy in transcript profiling analysis. Genomics 68,
J
K
K
K
K
L
L
L
M
M
M
M
M
Genet. 25, 25–29.lack, D.L., 2003. Mechanisms of alternative pre-messenger RNA splicing.
Annu. Rev. Biochem. 72, 291–336.leckmann, S.C., Blendy, J.A., Rudolph, D., Monaghan, A.P., Schmid, W.,
Schutz, G., 2002. Activating transcription factor 1 and CREB are impor-tant for cell survival during early mouse development. Mol. Cell Biol.22, 1919–1925.
oehning, D., Patterson, R.L., Sedaghat, L., Glebova, N.O., Kurosaki, T.,Snyder, S.H., 2003. Cytochrome c binds to inositol (1,4,5) triphosphatereceptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5,1051–1061.
rown, D.T., 2003. Histone H1 and the dynamic regulation of chromatinfunction. Biochem. Cell Biol. 81, 221–227.
ataldo, L., Mastrangelo, M.-A., Kleene, K.C., 1999. A quantitative sucrosegradient analysis of the distribution of 18 mRNA species in testes fromadult mice. Mol. Hum. Reprod. 5, 206–213.
hen, J.-J., Rowley, J.D., Wang, S.M., 2000. Generation of longer cDNAfragments from serial analysis of gene expression tags for gene identifi-cation. Proc. Natl. Acad. Sci. U.S.A. 97, 349–353.
hristians, E.S., Zhou, Q., Renard, J., Benjamin, I.J., 2003. Heat shock pro-teins in mammalian development. Semin. Cell Dev. Biol. 14, 283–290.
e Rooij, D.G., Russell, L.D., 2000. All you wanted to know about sper-matogonia but were afraid to ask. J. Androl. 21, 776–798.
iehn, M., Sherlock, G., Binkley, G., Jin, H., Matese, J.C., Hernandez-Boussard, T., Rees, C.A., Cherry, J.M., Botstein, D., Brown, P.O.,Alizadeh, A.A., 2003. SOURCE: a unified genomic resource of func-tional annotations, ontologies, and gene expression data. Nucleic AcidsRes. 31, 219–223.
ivina, P., Vlcek, C., Strnad, P., Paces, V., Forejt, J., 2005. Global tran-scriptome analysis of the C57BL/6J mouse testis by SAGE: evidence fornonrandom gene order. BMC Genomics 6, 29.
uruchi, T., Masuko, K., Nishimune, Y., Obinata, M., Matsui, Y., 1996.Inhibition of testicular germ cell apoptosis and differentiation in micemisexpressing Bcl-2 in spermatogonia. Development 122, 1703–1709.
etz, G., Levine, E., Domany, E., 2000. Coupled two-way clustering analysisof gene microarray data. Proc. Natl. Acad. Sci. U.S.A. 97, 12079–12084.
hafourifar, P., Klein, S.D., Schucht, O., Schenk, U., Pruschy, M., Rocha, S.,Richter, C., 1999. Ceramide induces cytochrome c release from isolated
136–143.ordan, B.R., 2004. How consistent are expression chip platforms? BioEssays
26, 1236–1242.amitani, A., Yamada, H., Kinuta, M., Watanabe, M., Li, S.A., Matsukawa,
T., McNiven, M., Kumon, H., Takei, K., 2002. Distribution of dynamicsin testis and their possible relation to spermatogenesis. Biochem. Biophys.Res. Commun. 294, 261–267.
leene, K.C., 2001. A possible meiotic function of the peculiar patternsof gene expression in mammalian spermatogenic cells. Mech. Dev. 106,3–23.
leene, K.C., 2003. Patterns, mechanisms, and functions of translation regu-lation in mammalian spermatogenic cells. Cytogenet. Genome Res. 103,217–224.
nudson, C.M., Tung, K.S., Tourtellotte, W.G., Brown, G.A., Korsmeyer,S.J., 1995. Bax-deficient mice with lymphoid hyperplasia and male germcell death. Science 270, 96–99.
ee, J., Richburg, J.H., Shipp, E.B., Meistrich, M.L., Boekelheide, K., 1999.The Fas system, a regulator of the germ cell apoptosis, is differen-tially up-regulated in Sertoli cell versus germ cell injury of the testis.Endocrinology 140, 852–858.
ee, J., Richburg, J.H., Younkin, S.C., Boekelheide, K., 1997. The Fas systemis a key regulator of germ cell apoptosis in the testis. Endocrinology 138,2081–2088.
ehninger, A.L., Nelson, D.L., Cox, M.M., 1993. Biochemistry, second ed.Worth Publishers, New York, pp. 551–555.
aniatis, T., Tasic, B., 2002. Alternative pre-mRNA splicing and proteomeexpansion in metazoans. Nature 418, 236–243.
atter, N., Herrlich, P., Konig, H., 2002. Signal-dependent regulation ofsplicing via phosphorylation of Sam68. Nature 420, 691–695.
attson, M.P., Chan, S.L., 2003. Calcium orchestrates apoptosis. Nat. CellBiol. 5, 1041–1043.
cCarrey, J.R., O’Brien, D.A., Skinner, M.K., 1999. Construction and pre-liminary characterization of a series of mouse and rat testis cDNAlibraries. J. Androl. 20, 635–639.
odrek, B., Resch, A., Grasso, C., Lee, C., 2001. Genome-wide detectionof alternative splicing in expressed sequences of human genes. NucleicAcids Res. 29, 2850–2859.

W.-Y. Chan et al. / Molecular and Cellular Endocrinology 250 (2006) 8–19 19
O’Bryan, M.K., Schlatt, S., Gerdprasert, O., Phillips, D.J., de Kretser, D.M.,Hedger, M.P., 2000. Inducible nitric oxide synthase in the rat testis:evidence for potential roles in both normal function and inflammation-mediated infertility. Biol. Reprod. 63, 1285–1293.
Ott, M., Robertson, J.D., Gogvadze, V., Zhivotovsky, B., Orrenius, S., 2002.Cytochrome c release from mitochondria proceeds by a two-step process.Proc. Natl. Acad. Sci. U.S.A. 99, 1259–1263.
Pang, A.L.Y., Taylor, H.C., Johnson, W., Alexander, S., Chen, Y., Su, Y.A.,Li, X., Ravindranath, N., Dym, M., Rennert, O.M., Chan, W.Y., 2003.Identification of differentially expressed genes in spermatogenesis in themouse. J. Androl. 24, 899–911.
Persengiev, S.P., Raval, P.J., Rabinovitch, S., Millette, C.F., Kilpatrick, D.L.,1996. Transcription factor Sp1 is expressed by three different develop-mentally regulated messenger ribonucleic acids in mouse spermatogeniccells. Endocrinology 137, 638–646.
Print, C.G., Loveland, K., 2000. Germ cell suicide: new insights into apop-tosis during spermatogenesis. BioEssays 22, 423–430.
Ravnik, S.E., Wolgemuth, D.J., 1999. Regulation of meiosis during mam-malian spermatogenesis: the A-type cyclins and their associated cyclin-dependent kinases are differentially expressed in the germ-cell lineage.Dev. Biol. 207, 408–418.
Reed, J.C., 1997. Cytochrome c: can’t live with it—can’t live without it. Cell9, 559–562.
Rockett, J.C., Luft, J.C., Garges, J.B., Krawetz, S.A., Hughes, M.R., Kim,K.H., Oudes, A.J., Dix, D.J., 2001. Development of a 950-gene DNAarray for examining gene expression patterns in mouse testis. GenomeBiol. 2, research0014.1–0014.9.
Ronfani, L., Ferraguti, M., Croci, L., Ovitt, C.E., Scholer, H.R., Con-salez, G.G., Bianchi, M.E., 2001. Reduced fertility and spermatogenesisdefects in mice lacking chromosomal protein Hmgb2. Development 128,1265–1273.
S
S
S
S
S
Stouffs, K., Lissens, W., Tournaye, H., Van Steirteghem, A., Liebaers, I.,2005. Possible role of USP26 in patients with severely impaired sper-matogenesis. Eur. J. Hum. Genet. 13, 336–340.
Strasser, A., O’Connor, L., Dixit, V.M., 2000. Apoptosis signaling. Annu.Rev. Biochem. 69, 217–245.
Tanaka, K., Tamura, H., Tanaka, H., Katoh, M., Futamata, Y., Seki, N.,Nishimune, Y., Hara, T., 2002. Spermatogonia-dependent expression oftesticular genes in mice. Dev. Biol. 246, 466–479.
Thomas, K., Sung, D.Y., Yang, J., Johnson, K., Thompson, W., Millette,C., McCarrey, J., Breitberg, A., Gibbs, R., Walker, W., 2005. Identifica-tion, characterization, and functional analysis of sp1 transcript variantsexpressed in germ cells during mouse spermatogenesis. Biol. Reprod. 72,898–907.
Velculescu, V.E., Zhang, L., Vogelstein, B., Kinsler, K.W., 1995. Serial anal-ysis of gene expression. Science 270, 484–487.
Venables, J.P., 2002. Alternative splicing in the testes. Curr. Opin. Genet.Dev. 12, 615–619.
Walker, W.H., Delfino, F.J., Habener, J.F., 1999. RNA processing and thecontrol of spermatogenesis. Front. Horm. Res. 25, 34–58.
Wang, P.J., McCarrey, J.R., Yang, F., Page, D.C., 2001. An abundance ofX-linked genes expressed in spermatogonia. Nat. Genet. 27, 422–426.
Wang, Z., Lo, H.S., Yang, H., Gere, S., Hu, Y., Buetow, K.H., Lee, M.P.,2003. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res. 63,655–657.
Wiederkehr, C., Basavaraj, R., Sarrauste de Menthiere, C., Hermida, L.,Koch, R., Schlecht, U., Amon, A., Brachat, S., Breitenbach, M., Briza, P.,Caburet, S., Cherry, M., Davis, R., Deutschbauer, A., Dickinson, H.G.,Dumitrescu, T., Fellous, M., Goldman, A., Grootegoed, J.A., Hawley,R., Ishii, R., Jegou, B., Kaufman, R.J., Klein, F., Lamb, N., Maro, B.,Nasmyth, K., Nicolas, A., Orr-Weaver, T., Philippsen, P., Pineau, C.,
W
X
Y
Y
Z
chultz, N., Hamra, F.K., Garbers, D., 2003. A multitude of genes expressedsolely in meiotic or postmeiotic spermatogenic cells offers a myriadof contraceptive targets. Proc. Natl. Acad. Sci. U.S.A. 100, 12201–12206.
eigneurin-Berny, D., Verdel, A., Curtet, S., Lemercier, C., Garin, J.,Rousseaux, S., Khochbin, S., 2001. Identification of components of themurine histone deacetylase 6 complex: link between acetylation and ubiq-uitination signaling pathways. Mol. Cell Biol. 21, 8035–8044.
ha, J., Zhou, Z., Li, J., Yin, L., Yang, H., Hu, G., Luo, M., Chan, H.C., Zhou,K., Spermatogenesis Study Group, 2002. Identification of testis develop-ment and spermatogenesis-related genes in human and mouse testes usingcDNA arrays. Mol. Hum. Reprod. 8, 511–517.
hinohara, T., Avarbock, M.R., Brinster, R.L., 1999. Beta1- and alpha6-integrin are surface markers on mouse spermatogonial stem cells. Proc.Natl. Acad. Sci. U.S.A. 96, 5504–5509.
hiraki, T., Kondo, S., Katayama, S., Waki, K., Kasukawa, T., Kawaji,H., Kodzius, R., Watahiki, A., Nakamura, M., Arakawa, T., Fukuda,S., Sasaki, D., Podhajska, A., Harbers, M., Kawai, J., Carninci, P.,Hayashizaki, Y., 2003. Cap analysis gene expression for high-throughputanalysis of transcriptional starting point and identification of promoterusage. Proc. Natl. Acad. Sci. U.S.A. 100, 15776–15781.
Rabitsch, K.P., Reinke, V., Roest, H., Saunders, W., Schroder, M., Schedl,T., Siep, M., Villeneuve, A., Wolgemuth, D.J., Yamamoto, M., Zickler,D., Esposito, R.E., Primig, M., 2004. GermOnline, a cross-species com-munity knowledgebase on germ cell differentiation. Nucleic Acids Res.32, D560–D567.
u, S.M., Baxendale, V., Chen, Y., Li, X., Pang, A.L.Y., Stitely, T., Munson,P.J., Leung, M.Y.K., Ravindranath, N., Dym, M., Rennert, O.M., Chan,W.Y., 2004. Analysis of mouse germ cell transcriptome at different stagesof spermatogenesis: biological significance. Genomics 84, 971–981.
u, Q., Lee, C., 2003. Discovery of novel splice forms and functional analy-sis of cancer-specific alternative splicing in human expressed sequences.Nucleic Acids Res. 31, 5635–5643.
ao, J., Chiba, T., Sakai, J., Hirose, K., Yamamoto, M., Hada, A., Kuramoto,K., Higuchi, K., Mori, M., 2004. Mouse testis transcriptome revealedusing serial analysis of gene expression. Mamm. Genome 15, 433–451.
u, Z., Guo, R., Ge, Y., Ma, J., Guan, J., Li, S., Sun, X., Xue, S., Han, D.,2003. Gene expression profiles in different stages of mouse spermatogeniccells during spermatogenesis. Biol. Reprod. 69, 37–47.
hang, L., Zhou, W., Velculescu, V.E., Kern, S.E., Hruban, R.H., Hamilton,S.R., Vogelstein, B., Kinzler, K.W., 1997. Gene expression profiles innormal and cancer cells. Science 276, 1268–1272.
Related Documents











