1 Training Manual for Hemoglobinopathies and Hemophilia: Blood cell-MoHFW in collaboration with ICMR-National Institute of Immunohaematology Ministry of Health and Family Welfare, Government of India BLOOD CELL

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
Training Manual for
Hemoglobinopathies and
Hemophilia:
Blood cell-MoHFW in collaboration with
ICMR-National Institute of Immunohaematology
Ministry of Health and Family Welfare, Government of India
BBLLOOOODD CCEELLLL

2

3
4
List of Contributors
Aby Abraham Professor, Dept. of Haematology, Christian Medical College, Vellore
Anita Harinkhede Senior Resident, Government Medical College, Nagpur
Anita Nadkarni Scientist F, ICMR-National Institute of Immunohaematology, Mumbai
Arihant Jain
Assistant Professor, Dept. of Internal Medicine, Post Graduate Institute of Medical Education andResearch, Chandigarh
Ashutosh Panigrahi
Assistant Professor, Dept. of Medical Oncology/Hematology, All India Institute of Medical Sciences, Bhubaneshwar
Bipin Kulkarni Scientist D, ICMR-National Institute of Immunohaematology, Mumbai
Chandrakala S Professor& Head, Department of Haematology, KEM Hospital, Mumbai
Dipti Jain Professor, Dept of Pediatrics, Government Medical College,Nagpur
Jasmina Ahluwalia
Professor, Dept. of Hematology, Post Graduate Institute of Medical Education andResearch,Chandigarh
Jayashri Kale Professor, Department of Occupational Therapy, KEM Hospital, Mumbai
Kanjaksha Ghosh Former Director, ICMR-National Institute of Immunohaematology, Mumbai
Maitreyee Bhattacharyya Professor, Dept of Haematology, Calcutta Medical College, Kolkata
Malay Mukherjee
Former Scientist F& Head, Dept of Hematogenetics, ICMR-National Institute of Immunohaematology, Mumbai
Manisha Madkaikar Director, ICMR-National Institute of Immunohaematology, Mumbai
Meenu Bajpai Addl. Professor, Transfusion MedicineInstitiute of Liver and Biliary Science,New Delhi
5
Nikesh Kawankar Technical assistant, ICMR-National Institute of Immunohaematology, Mumbai
Pallavi Mehta Technician C, ICMR-National Institute of Immunohaematology, Mumbai
Pankaj Malhotra
Professor, Dept. of Internal Medicine, Post Graduate Institute of Medical Education and Research, Chandigarh
Prantar Chakravarty Former Professor, Dept. of Haematology, Nil Ratan Sircar Medical College, Kolkata
Prashant Sharma
Additional Professor, Dept of Hematology, Post Graduate Institute of Medical Education and Research,Chandigarh
Priya Hariharan Research Associate, ICMR-National Institute of Immunohaematology, Mumbai
Ravi Ranjan Scientist 1, AIIMS,New Delhi [email protected]
Reena Das
Professor, Dept. of Hematology, Post Graduate Institute of Medical Education andResearch,Chandigarh
Renu Saxena
Former Professor & Head Dept of Haematology, All India Institute of Medical Sciences, New Delhi
Richa Mohan
Clinical Psychologist, Empowering Minds Society for Research & Development, New Delhi
Roshan Colah Former Director In-Charge, ICMR-National Institute of Immunohaematology, Mumbai
Rucha Patil Scientist B, ICMR-National Institute of Immunohaematology, Mumbai
Sharda Shanbhag Technical assistant, ICMR-National Institute of Immunohaematology, Mumbai
Shrimati Shetty
Former Scientist F & Head Dept of Hemostasis, ICMR-National Institute of Immunohaematology, Mumbai
Shweta Sharma Dr. Shweta Sharma, Associate Professor, Dept of pediatrics, Gandhi Medical college, Bhopal
Soniya Nityanand
Professor& Head, Dept of Haematology, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow

6
Tulika Seth Professor, Dept of Haematology,All India Institute of Medical Sciences, New Delhi
V.P. Choudhry
Senior Consultant Hematology, Fortis Escorts Hospital, Batra Hospital & Medical Research Centre, Delhi,
Vikram Mathews Professor, Dept. of Haematology, Christian Medical College,Vellore
Vinita Srivastava
National Senior Consultant & Co-ordinator Incharge Blood cell - NHM, Nirman Bhavan, Ministry of Health & Family Welfare,GOI, New Delhi

7
Contents
Section A: Hemoglobinopathies
Chapters Page
no.
Introduction 6
Government policy on hemoglobinopathies 11
Abbreviations 15
Definitions of Various Levels of Training 17
1. Laboratory Diagnosis of Hemoglobinopathies and Counseling
Screening andDiagnosis of Hemoglobinopathies 20
Newborn screening for Sickle Cell Disease 51
MolecularAnalysis of Hemoglobinopathies 62
PrenatalDiagnosis of Hemoglobinopathies 88
Quality Assurance and Equipment Maintenance in Hemoglobinopathies 100
Genetic Counseling for Hemoglobinopathies 124
2.Management of Hemoglobinopathies:
i.Thalassemia Major
Basic Recognition of Symptoms and Signs of Thalassemia Major at Level 1
(PHC/CHC)
133
Initial Work Up (See diagnostic section too), Stabilization and Triage of Care of the
Sick Patient
136
Guidelines for Blood Transfusion in Hemoglobinopathies 148
Guidelines for Chelation Therapy in Thalassemia 155
Monitoring and Data Collection 168
Common Complications and their Management in Thalassemia major 174
Transfusion Reactions 188
ii.Sickle Cell Disease
Basic Recognition of Symptoms and Signs of Sickle Cell Disease at Level 1
(PHC/CHC)
194
Important Guidelines for Management of Sickle Cell Disease 196
Hydroxyurea Therapy-Principles and Guidelines 202
Common Complications and their Management 209
Exchange Transfusion and Simple Transfusion in Sickle Cell Disease 227
Management of Newborn and Pregnanacy 235
Alloimmunization 242
Hematopoietic Stem Cell Transplantation (HSCT) for Thalassemia Major 247
Hematopoietic Stem Cell Transplantation (HSCT) for Sickle Cell Disease 254

8
Alpha Thalassemias: Pathophysiology and Diagnosis 261
Role of Genetic Modifiers 271
Newer Modalities for Management of Hemoglobinopathies 277
Registry for Hemoglobinopathies 284

9
Introduction
The inherited disorders of hemoglobin constitute a major global health problem and broadly
include the and thalassemias, symptomatic structural hemoglobinopathies like sickle
cell disease and Hb E as well as co-inheritance of these genes. They have attained high
frequencies in regions where malaria has been endemic. They are also the commonest group
of single gene disorders in India with an autosomal recessive inheritance.
The thalassemia syndromes and sickle cell disease and HbE pose the major health burden
in India. The average prevalence of thalassemia carriers is 3.5- 4% which translates to
around 40 to 50million people. In some communities the prevalence is higher varying from 5-
17 %. It has been estimated that 10,000 to 15,000 babies with a major thalassemia
syndrome are born each year in Inda. More accurate numbers will become available once
there is a registry of all patients and carriers. Sickle cell disease is prevalent in central India
and in parts of western, southern and eastern India mainly among tribal and some non-tribal
populations. The prevalence of sickle cell carriers in some of these groups can be as high as
25- 35%. Hb E is seen mainly in the north eastern region where the prevalence of Hb E
carriers is as high as 40- 50 % in some populations and Hb E is common in West Bengal.
Accurate diagnosis of the inherited hemoglobinopathies is important to confirm a suspected
clinical diagnosis for appropriate management of patients, explain hematological
abnormalities like anemia or microcytosis, to identify carriers or heterozygotes of
thalassemia or other abnormal hemoglobin variants who are generally asymptomatic but
require counseling and to identify couples at- risk of having a child with a severe disorder to
give them the option of prenatal diagnosis to avoid the birth of an affected child. Accurate
diagnosis of sickle cell disease is also crucial in the newborn period to reduce morbidity and
mortality by providing early care, intervention and regular follow up of these babies.
A combination of different methods as described in this manual should be used for postnatal
and prenataldiagnosis of these disorders and strict internal quality assurance (IQA) and
external quality assurance (EQA) must be undertaken for each of these hematological,
biochemical and molecular methodswhichare crucial to avoid any misdiagnosis.
Introducing screening programmes at the National level on a large scale is a big challenge
and would require adequate resources for prevention programmes and long-term care of
patients. Efforts should be intensified to strengthen awareness in the population.

10
Training of Health Professionals and other Health Workers at all levels must be undertaken
for all the states. Uniform protocols need to be followed throughoutthe country as described
in this training manual. All patients of the thalassemia syndromes and sickle cell disease
should have access to clinical mannagement and multidisciplinary care with recommended
monitoring and treatment protocolsas described in thistraining manual. Those patients who
are eligible and have a matched donor should have the possibility of cure by access to bone
marrow/ stem cell transplant.
Information along with guidance and support are required for a successful control
programme. In a large and diverse country like India, screening cannot be offered to
everyone, even if it is desirable and targeting selected groups initially would be profitable and
cost effective. Such groups include newly married couples and couples during early
pregnancy and extended family members of affected or carrier individuals. Premarital
screening is by and large not acceptable due to the social stigmatization attached to being a
carrier of thalassemia or another hemoglobinopathy.However, those who come voluntarily
should be screened. Prenatal diagnosis facilities would be established and offered to all
couplesin medical colleges in each state.
Past experience in many programmes has shown that once only advice is insufficient and
genetic counseling needs to be a continuous process for which adequate well-
trainedcounselors are required.
Managing and controlling these disorders nationally is largely the responsibility of both the
Central and State Governments. This Training Manual describes in detail the training to be
imparted at different levels in the Public Health System to reach out and benefit both the
urban and rural population. National Institutes in the country with expertise in diagnosis and
management of hemoglobinopathies will serve as Training Centres for medical colleges to
impart the highest level of training. The success of such a huge initiative at the public health
level depends critically on dealing effectively with managerial challenges, availability of
adequate infrastructure, resources and commitment of trained staff. Public-Private
partnerships can also be developed and different NGOs working in this area could be
involved particularly for awareness generation in the population. An Expert Advisory Group
would periodically review and monitor the progress of this Comprehensive National
Programme for Prevention and Management of Hemoglobinopathies in the country

11
Government Guidelines on Hemoglobinopathies
Thalassemia and Sickle Cell Disease are two common genetic disorders that are chronic, life-
restricting and require long and specialized treatment.They cause distress and financial loss to
the family and are a great drain on the health resources of the country. With the fall in Infant
Mortality Rate due to control of communicable and nutritional disorders in the last decade in
India, these disorders have become important causes of morbidity and mortality.It is
estimated thatin India there are almost 42 million carriers of β-thalassemia, around 1-
1.5million patients of Thalassemia major and about 10,000 to 15,000 babies with β-
thalassemia major are born each year. The carrier frequency of Sickle cell gene varies from 1-
35% and hence there are huge numbers of people with Sickle cell disease.
There are various challenges related to prevention and management of Hemoglobinopathies
in India. The epidemiological data is incomplete, and the precise burden of these disorders is
unknown due to lack of awareness and problems related to diagnosis in rural & remote
areas.Treatment of Thalassemia requires repeated blood transfusions which should be safe
blood to avoid the transmission of transfusion transmitted infections such as HIV, hepatitis B
and C.The excess iron that gets into the body through the blood transfusions needs to be
removed by use of the expensive chelator medicines. Bone marrow transplantation is a
curative treatment that requires aHLA-matched donor, specific infrastructure and trained
doctors and nurses. The physicians need specialized training to treat, monitor and manage
the complications of Thalassemia & Sickle Cell Disease.
Recognizing the socio-economic burden these disorders place on the family, society and the
health services, the Government of India has formed this policy guidelines aimed at
informing and providing broad guidance on prevention and management of these disorders.
A Technical Committee was constituted comprising of all stakeholders including experts,
patient groups; civil society organizations etc., to formulate the policy guidelines on
Haemoglobinopathies.This policy guidelines is based on the recommendations made by

12
thecommittee and from evidence provided by Indian & International medical literature along
with the Thalassemia International FederationGuidelines & WHO Guidelines.
This policy guidelines encompasses the public health goal of reducing the birth of affected
children through carrier screening and prenatal diagnosis and providing evidence- based
treatment for those affected.
An appropriate mechanism is recommended under Blood cell NHM at the National
and the State level for monitoring and implementation. Periodic review and mid-
course corrections based on new data and information would be carried out in
consultation with ICMR and DGHS.
Public Health and Hospitals is a state subject and therefore the policy guidelines is
meant to provide vision and broad guidance for prevention and management of
hemoglobinopathies tostates and UTs.
The guiding principle recognizes that for prevention, the focus should be on creating
awareness of these disorders in the community for acceptance of carrier screening,
which is recommended for all pregnant mothers, school & college going students.
Pregnant women identified to be carriers, and their husbands should be screened and
in couples, where both the partners are carriers, prenatal diagnosis should be offered
to ensure that the child is not affected with a clinically significant hemoglobinopathy.
Premarital and preconception carrier screening should be instituted with appropriate
genetic counseling. All subjects should be informed their status, whether normal,
carrier or diseased through systems of coding. For Sickle Cell Disease, the policy
guidelines recommends initiation of newborn screening in areas of high prevalence.
Those detected to have sickle cell anemia (HbSS) or compound heterozygosity of
HbS and β-thalassemia should be provided appropriate prophylaxis and
immunizations, especially pneumococcal and Hib vaccine, and followed up carefully
In the rural areas, ASHAs/multipurpose workers are envisaged to be trained and
utilized to identify individuals with severe anemia who couldhaveThalassemia major
or Sickle Cell Disease. These patients will be referred to the appropriate health care
facilities (District hospitals & above) for further testing and confirmation. The
district Hospitals/ Medical colleges should be equipped with equipment to measure

13
the hemoglobin, red cell indices, to carry out carrier screening of β-thalassemia and
sickle celldisease.
The strategy focuses on providing comprehensive services for patients with
Hemoglobinopathies by strengthening public health systems and establishing ICHH
centers in Districts/ Medical colleges. Italso envisages creation of Centres of
Excellence in state medical colleges with advanced facilities required for
comprehensive care of patients with Thalassemia/Sickle Cell Disease as well as with
the facility for prenatal diagnosis and hematopoietic stem cell transplant. Such
Centers of Excellence to function as hubs for providing technical support for those
situated at district and below level health facilities.
The program recommends creation of an integrated unit for Hemoglobinopathies and
Hemophilia keeping patient load in mind, in government medical colleges and district
level hospitals inaphased manner. The policy guidelines envisages a system of
referral from district hospital to medical college/ Center of Excellence (COE)
including use of digital technology such as tele- consultations. The document links the
Haemoglobinopathies programme with other programmeseg. Hepatitis, HIV and
Malaria programme.
The policy guidelines also recommends creation of a web-based application at the
state and National level for providing information in simple language with translation
in the common Indian languages, about the disease, its complications, their
management, and the places where different facilities are available.The guidelines
advocates a multi-stakeholder approach with participation of patients, parent support
organizations academic institutions, not for profit organizations, and health care
facilities.
The plan recommends setting up of a patient registry for Thalassemia and Sickle Cell
Disease to obtain information on the number of persons affected and the number of
carriers to estimate patients who require various services. The data on carrier
screening performed in different regions should be collated to determine the burden of
hemoglobinopathies.
14
The strategy advocates promoting research to develop innovative treatments for
Thalassemia major and Sickle Cell Disease, and devise new diagnostic methods,
keeping in mind the continuously evolving technology in this field.
Thus, the policy guidelines helps as a guiding tool to the concerned states and will
help in implementation of the programme in an improved manner
Abbreviations
AH Area Hospital
ANC Antenatal Clinic
ANM Auxiliary Nurse Midwife
Apo B Apolipoprotein B
ARMS Amplification Refractory Mutation System
ASHA Accredited Social Health Activist
AWW AnganWadi Workers
BMT Bone Marrow Transplantation
Bp Base Pair
BT Blood Transfusion
CBC Complete Blood Count
CCl4 Carbon tetrachloride
CHC Community Health Centre
DEIC District Early Intervention Centre
DH District Hospital
DMER Directorateof Medical Education and Research
DMSO Dimethyl Sulphoxide
DNA Deoxyribo Nucleic Acid
dNTP Deoxyribo Nucleotide Triphosphate
D/W Distilled water
Hb Hemoglobin
HbA0 Adult Hemoglobin
HbF FetalHemoglobin
HPLC High Pressure Liquid Chromatography
EQAS External Quality Assurance Scheme
HSCT Hematopoietic Stem Cell Transplantation
IEC Institutional Ethical Committee
IVS Intervening sequences
Kb Kilobase
LT Laboratory Technologist
MCH Mean CorposcularHemoglobin
15
MCHC Mean Cell HemoglobinConcentration
MCV Mean Corpuscular Volume
MO Medical Officer
NBS Newborn Screening
NESTROFT Nacked Eye Single Tube Osmotic Fragility Test
NTDT Non Transfusion Dependent Thalassemia
PCR Polymerase Chain Reaction
PHC Primary Health Centre
PND Prenatal Diagnosis
POC Point of Care
RBSK Rashtriya Bal SwasthyaKaryakram
RFLP Restriction Fragment Length Polymorphism
CRDB Covalent Reverse Dot Blot Hybridization
SCD Sickle Cell Disease
TAE Tris Acetate EDTA buffer
TBE Tris Borate EDTA buffer
TD Transfusion Dependent
TOT Training of Trainers
TDT Transfusion Dependent Thalassemia
VNTR Variable Number of Tandam Repeats

16
Definition of Various Levels of Training
Level 1:
Primary Health Centre (PHC)
Community Health Centre (CHC)
Level 2: Integrated Day Care Centre for Hemoglobinopathies and Hemophilia
District Hospitals and District Early Intervention Centres (DEIC)
Government Medical Colleges
Level 3:Centres of Excellence
Government Medical College (1 to 2): States to decide
Level 4: National Training Institutes
ICMR-National Institute of Immunohaematology (NIIH), Mumbai
All India Institute of Medical Sciences(AIIMS), New Delhi
Sanjay Gandhi Postgraduate Istitute of Medical Science (SGPGIMS),Lucknow
Christian Medical College (CMC), Vellore
Post Graduate Institute of Medical Education and Research(PGIMER), Chandigarh
Kolkata Medical College, Kolkata
States to be covered by each National Training Institute for training
Name of the Institute States to be covered
NIIH, Mumbai Maharashtra, Gujarat, Goa, Daman and Diu, Dadra and Nagar
Haveli, Karnataka
AIIMS, New Delhi Delhi, Rajasthan, Madhya Pradesh, Chattisgarh
SGPGIMS, Lucknow Uttar Pradesh, Bihar, Jharkhand
CMC, Vellore Tamil Nadu, Kerala, Andhra Pradesh, Telangana, Pondicherry,
Andaman and Nicobar
PGIMER, Chandigarh Punjab, Haryana, Himachal Pradesh, Uttarakhand, Jammu and
Kashmir
Medical College, Kolkata West Bengal, Orissa, Assam, Arunachal Pradesh, Meghalaya,
Sikkim, Manipur, Nagaland, Tripura
Level 1: Primary Health Centre (PHC)
Personnel: Training will be imparted to ASHA, ANM and RBSK worker at Level 2 by TOT
Training to be imparted: CBC on automated cell counter, preparation of peripheral smear,
staining and evaluation, solubility test, point of care testing for SCD, counseling.

17
CBC and peripheral blood smear examination should be done for all ANC women and
suspected cases of hemoglobinopathies and blood samples should be sent to level 2 at nearest
Integrated Centre for Hemoglobinopathies and Hemophilia.
Newborn Screening by Point of Care Testing for sickle cell disease. Any baby who tests
positive will be confirmed by HPLC and family screening will be done at level 3 (Centre of
Excellence).After confirmation of diagnosis, these babies will be registered in the Integrated
Centre for HemoglobinopathiesandHemophilia.
Instruments: Three-part automated blood cell counters and microscope
Level 2: IntegratedCentre for Hemoglobinopathies and Hemophilia to be established at
District Hospitals and Govt. Medical colleges.
Personnel: Training will be imparted to medical officers (MO), laboratory technicians (LT),
pathologists, counselors and obstetricians and training of trainers (TOT).
Designated MOs and staff at the District Hospital (DH) and District Early Intervention
Centres (DEIC) will be imparted training.
Training to be imparted: CBC on automated cell counter, preparation of peripheral smear,
staining and evaluation, solubility test, point of care testing for SCD, HPLC analysis
counseling.
Instruments:
Three-part automated blood cell counters and microscope
D-10 HPLC/Electrophoresis
Newborn Screening by Point of Care Testingfor sickle cell disease. Any baby who tests
positive will be confirmed by HPLC and family screening will be done at level 3 (Centre of
Excellence).After confirmation of diagnosis, these babies will be registered in the Integrated
Centre for HemoglobinopathiesandHemophilia.
Level 3: Centre of Excellence- Government Medical College (One or two per state)
Personnel: Social counselor, TOT trainer, Pathologist, Obstetrician for CVSand
cordocentesis, Clinical Hematologist, Molecular Biologist.
Approximately 2500 square feet with lecture theaters and laboratory space
forimpartingtrainingfor Level 1 and 2 participants will be required.
Training to be imparted: CBC on automated cell counter, preparation of peripheral smear,
staining and evaluation, solubility test, point of care testing for SCD, HPLC analysis
counseling. Molecular technologies for prenatal diagnosis for pathologists and molecular
biologists. Fetal tissue sampling (CVS), amniocentesis and cordocentesis for obstetricians.
Clinical evaluation and management protocols for the patients for clinical hematologists.

18
Instruments:
1. Five-part automated blood cell counters
2. HPLCVariant 2
3. HPLC NBS
4. Hb electrophoresis system for cellulose acetate membranes
5. Molecular Diagnosis: PCR machine, Submergedelectrophoresis system, Gel
documentation system, Automated DNA sequencer, Inverted microscope for CVS
dissection
Quality Control: Both External Quality Assessment (EQAS) and Internal Quality Control
(IQC) to be done regularly.
Concept of Reagent Rental with maintenance of equipmentcould be used which will include
EQAS.
Level 4: National Training Institutes: NIIH, Mumbai; AIIMS, New Delhi; SGPGI,
Lucknow; CMC, Vellore; PGIMER, Chandigarh; Medical College Kolkata, Kolkata
Instruments:
1. Five-part automated blood cell counters
2. HPLC: Variant 2
3. Isoelectric focussing
Molecular Diagnosis: PCR machine, Submergedelectrophoresis system, Gel documentation
system, Automated DNA sequencer, Inverted microscope for CVS dissection.
Linkage Plan of action between all the Levels (1-4) will be assured by the National Training
Institutes.Level 4 will train the staff of level 3. Later Level 3 will train the staff of Levels 1
and 2. Patients from level 2 may be given preferential care and management at levels 3 and 4.
Voluntary thalassemia screening will be offered in all the centres
Additional staff to be provided as mentioned in the Guidelines at different levels by the
State/GOI

19
1. Laboratory Diagnosis of Hemoglobinopathies and Counseling
Screening and Diagnosis of Hemoglobinopathies
Introduction The hemoglobinopathies comprise of the inherited disorders of the structure or synthesis of
hemoglobin. They are one of the commonest groups of single gene disorders in the Indian
subcontinent and pose a major concern on our health resources.
The hemoglobinopathies can be broadly classified into three groups.
1. Hemoglobin variants, where there is a structural alteration in one of the globin chains
e.g.HbS, HbE, HbC, HbD etc.
2. The thalassemias, where there is impaired synthesis of normal globin chains
e.g.thalassemia, thalassemia, thalassemia, thalassemia and thalassemia.
3. A diverse group of conditions, in which there is a defect in developmental progression
from fetal to adult hemoglobin production e.g. hereditary persistence of fetalhemoglobin
(HPFH).
The abnormal hemoglobinsresultfrom mutations in the α, , or δ globin genes resulting
mainly in single amino acid substitutions. Both the thalassemias and abnormal hemoglobins
can be co-inherited to give compound heterozygous conditions.
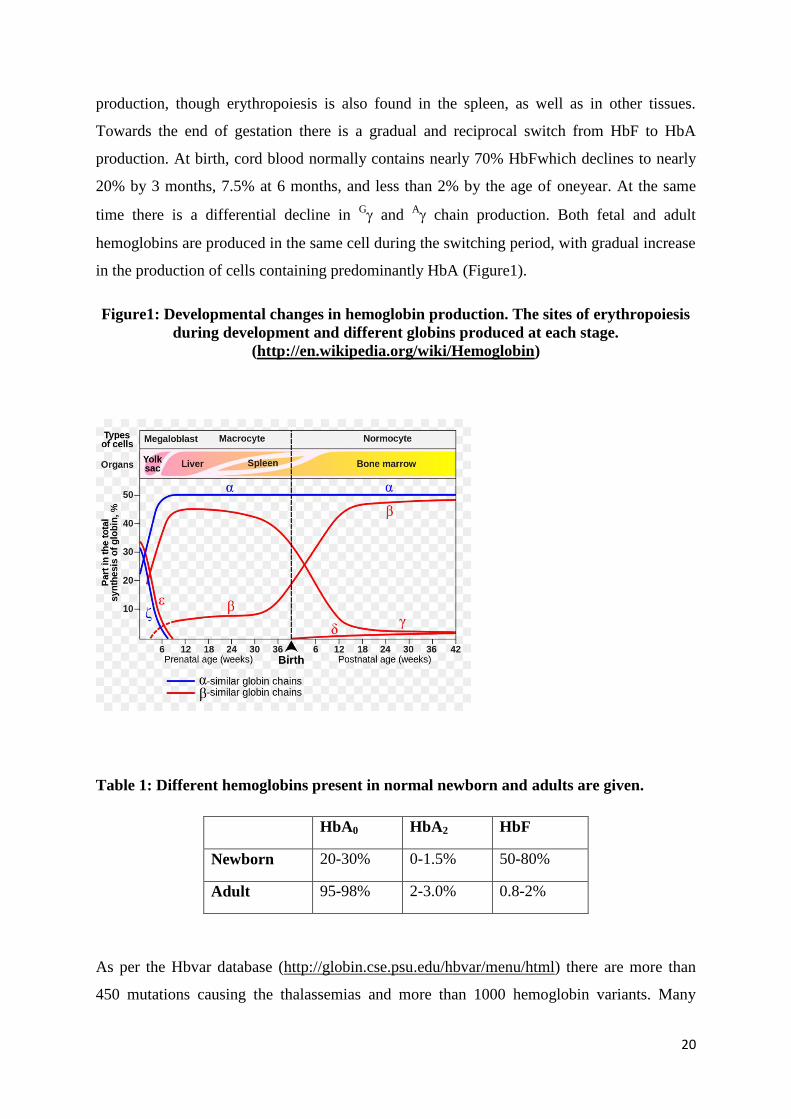
Human hemoglobin is heterogenous at all stages of development, beginning with the
youngest embryos that have been studied and continuing throughout adult life. These
hemoglobins are designated by the Greek letters alpha (α), beta (β), gamma (γ), delta (δ),
epsilon (ε) and zeta (ζ). In embryos, hemoglobin synthesis is confined to the yolk sac, where
hemoglobin Grower 1 (22), Grower 2 (α22) and Portland (22) are produced. Synthesis of
chains becomes detectable at about 6th
weeks of gestation, when it comprisesof nearly 1.5
% of the non alpha chains, increasing to 5% at the 7th
week and nearly 10% by the 10th
week.
At around 7-8 weeks of gestation the liver becomes the major site of erythropoiesis,
producing large enucleated red cells. Throughout most of fetal life HbF production
predominates, with a small amount (<10%) of adult hemoglobin (HbA). The different
chains are produced in a ratio of G to
A of 3:1, which remains constant until late in
gestation. At midterm the bone marrow begins to take over as the major site of red cell
20
production, though erythropoiesis is also found in the spleen, as well as in other tissues.
Towards the end of gestation there is a gradual and reciprocal switch from HbF to HbA
production. At birth, cord blood normally contains nearly 70% HbFwhich declines to nearly
20% by 3 months, 7.5% at 6 months, and less than 2% by the age of oneyear. At the same
time there is a differential decline in G and
A chain production. Both fetal and adult
hemoglobins are produced in the same cell during the switching period, with gradual increase
in the production of cells containing predominantly HbA (Figure1).
Figure1: Developmental changes in hemoglobin production. The sites of erythropoiesis
during development and different globins produced at each stage.
(http://en.wikipedia.org/wiki/Hemoglobin)
Table 1: Different hemoglobins present in normal newborn and adults are given.
HbA0 HbA2 HbF
Newborn 20-30% 0-1.5% 50-80%
Adult 95-98% 2-3.0% 0.8-2%
As per the Hbvar database (http://globin.cse.psu.edu/hbvar/menu/html) there are more than
450 mutations causing the thalassemias and more than 1000 hemoglobin variants. Many

21
variants of hemoglobin are harmless whereas some have significant clinical presentation.
Many of the Hb variants are picked up during population screening programmes; however,
some are identified while investigating cases of microcytosis, hemolyticanemia, cyanosis or
erythrocytosis. Few variants are unstable and may result in a thalassemia intermedia
phenotype.
Thalassemia minor individuals (carriers of thalassemia) are usually clinically asymptomatic
but occasionally they may have mild to moderate anemia. Children born with thalassemia
major usually develop the symptoms of severe anemia within the first year of life and are
unable to produce normal adult hemoglobin. They are chronically fatigued, fail to thrive, do
not grow normally and prolong anemia causes bone deformities.
All hemoglobinopathes are inherited in an autosomal recessive manner. If both parents are
thalassemia carriers, their children may be thalassemia carriers, or completely normal, or they
may have thalassemia major. In each pregnancy there is a one in four (25%) chance that their
child will be normal, a two in four (50%) chance that the child will have thalassemia minor or
a one in four (25%) chance that the child will have thalassemia major. Diagnosis and
management of these disorders both in the neonatal period or later using appropriate
approaches and uniform technology are extremely important.
The basic laboratory diagnoses for identification of thalassemia carriers are red cell indices
and morphology followed by hemoglobin electrophoresis. The cut-off value of HbA2 for
diagnosis of β-thalassemia carriers is generally taken as 3.5% along with the presence of
reduced red cell indices (MCV < 80 fl and MCH< 27 pg) and a relatively high RBC count
and normal RDW values. In α-thalassemia, HbA2can be lower than normal and it is
significant when iron deficiency is excluded.
The β-thalassemias and the common β chain variants are easily identified using an automated
HPLC machine. The atypical and or unusual ones require further investigations like
hemoglobin electrophoresis at alkaline and acidic pH, tests for hemoglobin stability like heat
stability and isopropanol stability test, oxygen dissociation curve and spectral analysis. Often
DNA sequencing of the respective globin genes is required for identification of the variant.
The common β-chain variants in India are Hb S, HbE and HbDPunjab
. Hemoglobin S is
predominantly seen in tribal and in a few nontribal populations in central, western and some
parts of eastern and southern India with the prevalence of heterozygous going as high as 35-

22
40% in some groups. Hemoglobin E is very common in the north-eastern region where
carrier frequencies go up to more than 50% in some groups. HemoglobinDPunjab
is mainly
seen in north-western India where the prevalence varies from 1-3%. Many clinically
significant α-chain variants are also seen amongst the Indian population which includes
HbQIndia
, HbSallanches, Hb Sun Prairie, Hb Evanston, Hb Jackson, Hb O Indonesia, Hb J
Paris (I), Hb J Meerut,
HbKoyaDora, Hb Hofu, and Hb Fontainbleau. Among these HbQIndia
is commonest and seen
mainly in the Sindhi community.
Individuals to be screened
All pregnant women (preferably in thefirst trimester but also those who come
later).
Husbands of all pregnant women who are carriers (heterozygous) of β-
thalassemia, Hb S, Hb E, δβ thalassemia.
Husbands of all pregnant women who are homozygous/compound
heterozygous for the following: HbShomozygous, HbS-β-thalassemia, HbE
homozygous, HbE-β-thalassemia, HbD-β-thalassemia and HbQ-β-thalassemia.
Extended family members of β-thalassemia major, Sickle cell disease patients
and siblings of carriers identified during screening.
All individuals who request for screening voluntarily.
High risks communities should be screened.
All couples where both husbands and wives are carriers should be referred for
molecular analysis and counseling with the option of prenatal diagnosis to be done at
the nearest Center of Excellence.

23
Investigations to be performed for diagnosis of the patients
CBC with peripheral blood smear examination
Reticulocyte count
HbH inclusion bodies and Heat/ Iso-propanol stability if required (If
suspecting HbH disease and unstable hemoglobin)
Solubility test
HPLC
Refer to center of excellence for molecular confirmation
For confirmation of alpha thalassemia and rare hemoglobin variants refer to
National Institutes.
Recommendations on Technologies to be used for screening
CBC to be done for all pregnant women and all the other groups of
individuals screened.
Along with CBC, solubility test for sickle hemoglobin to be done in regions
where HbS is prevalent.
All individuals with MCV< 80fl, MCH< 27pg and/or a positive test for HbS
require HPLC analysis which is recommended for diagnosis.
These positive samples should be sent to the Integrated Centre for
Hemoglobinopathiesand Hemophilia for HPLC for diagnosis
Hb electrophoresis on cellulose acetate at alkaline pH should be done to
differentiate hemoglobins which elute in the same window in HPLC (Hb
Lepore, HbE and HbDIran
)
Classical β-thalassemia carriers will have MCV<80, MCH<27, RDW -Normal, RBC
Count - Increased, HbA2> 3.5% and HbF 0.5-2.0%

24
Complete Blood Count
Principle: Complete blood count to determine red blood cell (RBC) indices is the most
common laboratory test and is usually carried out using blood collected in EDTAon an
automated electronic cell counterwithin a few hours of blood collection. Parameters such as
hemoglobin (Hb) concentration, mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH), RBC count and red cell distribution width (RDW) are strictly relevant
and useful for hemoglobinopathies screening (Table 2). Several automated cell counters are
available for doing a complete blood count. These counters work on the principle of electrical
impedance or laser light scattering. The counter needs to be calibrated daily with appropriate
material to obtain accurate results.
Interpretation:
MCV and MCH are variably reduced in thalassemia carriers. MCH is more reliable
than MCV, since the MCV does not remain stable due to a tendency for the red cells
to increase in size over timeduring storage.
The most widely used cut-off values of MCV and MCH for suspecting the presence
ofthalassemia carriersareMCV <80 fl and MCH <27 pgrespectively.
Silent β-thalassemia carriers may have normal MCV and MCH values.
RBC count is usually on the higher side in relation to the hemoglobin level in both
and thalassemia carriers.
RDW is normal in thalassemia carriers.
α-thalassemia carriers also have reduced MCV and MCH. However, α-thalassemia
carriers having asinglegene deletion may have near normal indices.
δβ-thalassemiacarriers have slightly reduced MCV and MCHvalues.

25
Table 2: Hematological parameters in thalassemia carriers and iron deficiency anemia.
Parameters with normal
range thalassemia
carriers
thalassemia
carriers
Iron deficiency
anemia
MCV (80-95 fl) Reduced Reduced Reduced
MCH (27-34 pg) Reduced Reduced Reduced
RBC count (4.5-5.6x 1012
/L) Increased Increased Reduced
RDW (11.5-14.5%) Normal Normal Increased
Red Blood Cell Morphology (Peripheral blood smear)
Principle: Morphological changes of red cells can be detected in most thalassemia carriers.
Theexamination of astained peripheral blood smear may be helpful in the evaluation of the
cases. Romanowsky stains are used for staining blood films. These are formulated by
blending methylene blue and eosin. Methylene blue is converted to it’s active form methyl
azures; when mixed with eosin, these stains are designated as polychromes because they
impart metachromatic qualities to the cell constituents.
Preparation of smear: EDTA anticoagulatedvenous blood or capillary blood is used to
prepare a blood film.
Reagents:
Leishman’s Stain:
0.2 g powdered Leishman’s dye is added to 100 mL methanol (Acetone free) and the
mixture is warmed to 500C in a shaking water bath for 15 minutes.
The solutionis then filtered and allowed to stand at room temperature for 24 hours.
This is stored at room temperature (250
C) in a dark bottle.
A pH of 6.8 is recommended for general use.
Method:
1. A small drop of blood is smearedon theslide using a spreader slide at an angle of 450.

26
2. The blood smear is driedat room temperature. Adequate drying is essential to preserve
the quality of the film.
3. By using alead pencil, the identification number is writtenon the slide.
4. The smear is covered with the staining solutionfor 2 minutes.
5. Distilled water is then added on the slide and the reaction mixture adequately
mixedby blowing on it using a pipette and allowed to standfor 20 minutes.
6. The slide is then rinsed in running tap water.
7. PBFis allowed to air dry and observed under theoil immersion lens of the microscope
(Figure 2).

27
Interpretation:
Figure 2: Red cell morphologyof a thalassemia carrier showing
anisocytosis,poikilocytosis, hypochromia and microcytosis
Microcytosis, hypochromia and anisopoikilocytosis are most typical changes in
thalassemia.
Other less common findings are basophilic stippling and presence of some target
cells.
Nucleated RBCs are indicative of bone marrow hyperactivity and can be found in
homozygous β-thalassemia.
Polychromasia is associated with the presence ofreticulocytosis.
Howell-Jolly bodies can be found after splenectomy or in the functional asplenic
condition in sickle cell syndromes, where sickle shaped cells are sometimes seen on
the stained films as well.
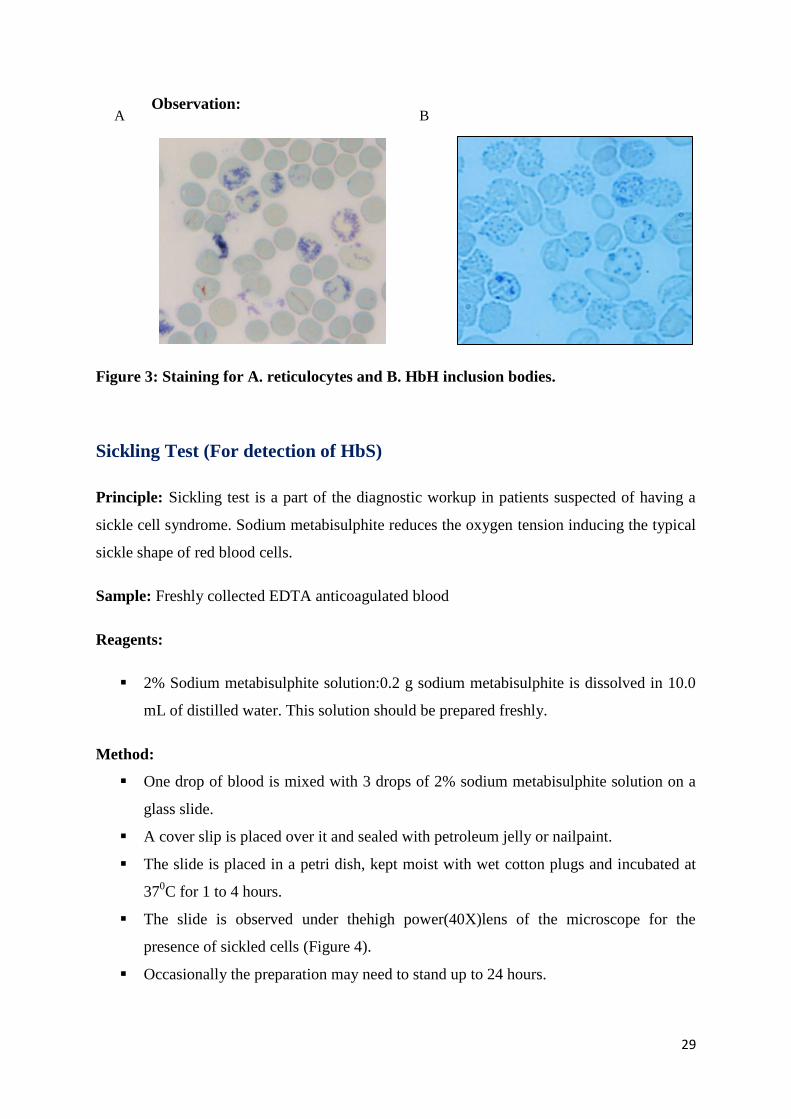
Reticulocyte Count and Hb H Inclusion Body Detection
Principle: For the detection of α-thalassemia, especially HbH disease, new methylene blue
stain will detect the characteristic HbH inclusion bodies. New methylene blue being a
supravital stain is able to stain residual mRNA in immature red blood cells (Reticulocytes).
Sample: EDTA anticoagulated blood

28
Reagents:
Iso-osmotic Phosphate buffer:
A. NaH2PO4.2H2O (150mM/L): 2.34g is dissolved in 100 mL of distilled
water (D/W)
B. Na2H PO4 (150mM/L): 2.13g is dissolved in 100 mL D/W
Note: Add Solution A- 18.0 mL and Solution B- 82 mL to prepare 100
mL of Iso-osmotic Phosphate buffer.
New Methylene Blue (NMB) stain: 1.0 gm (NMB) is dissolved in 100 mL of
Iso-osmotic phosphate buffer (pH- 7.4). The mixture is filtered and used.
Method:
1. Two drops of blood aremixed with one drop of stain in a tube and incubated at 370C
for 20 to 30 minutes.
2. A smear is then made on a grease free slide, air dried and observed under the oil
immersion lens of a microscope.
3. Reticulocytes are identified by their deep blue reticulated appearance.
4. A minimum of 1000 RBCs in successive fields are counted to determine the
percentage of reticulocytes.
5. Inclusion bodies can also be seen if the mixture of blood and dye is incubated for 1
hour at 370C and can be seen under the microscope (Figure 3)
Calculation:
Normal Values:
Adults and children: 0.2- 2.0 %
Newborn babies (cord blood/ full term baby): 2.0- 6.0 %
29
Observation:
Figure 3: Staining for A. reticulocytes and B. HbH inclusion bodies.
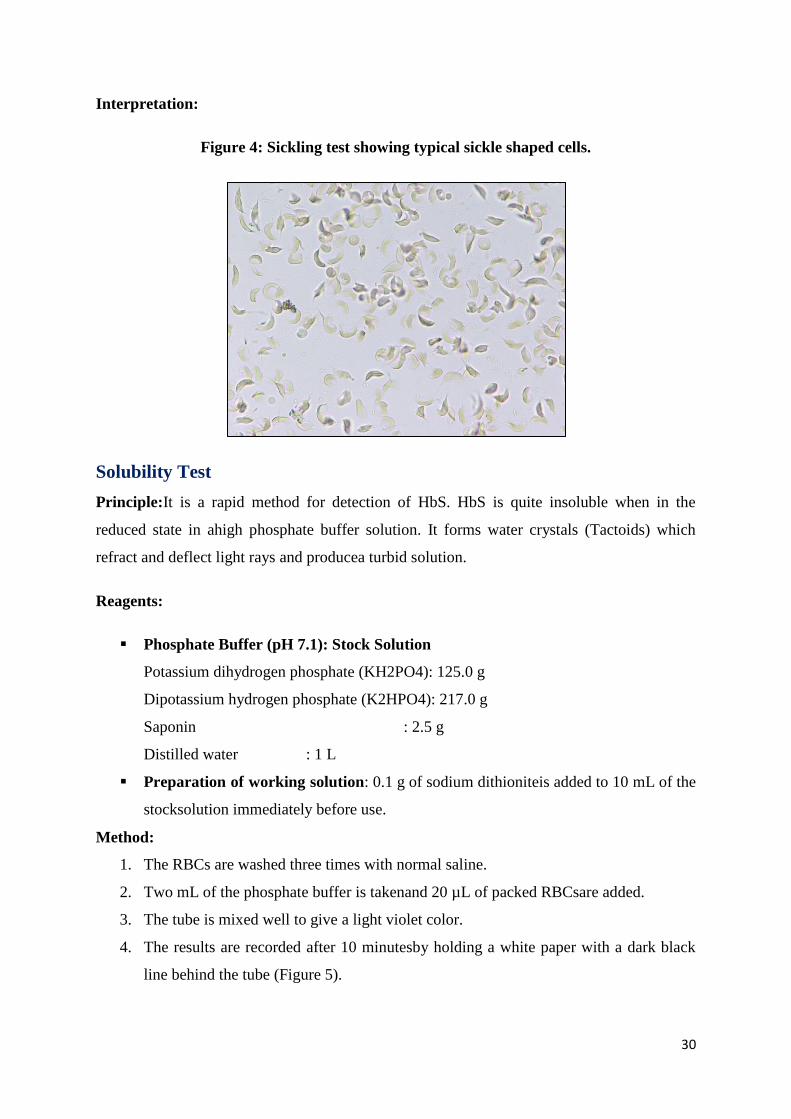
Sickling Test (For detection of HbS)
Principle: Sickling test is a part of the diagnostic workup in patients suspected of having a
sickle cell syndrome. Sodium metabisulphite reduces the oxygen tension inducing the typical
sickle shape of red blood cells.
Sample: Freshly collected EDTA anticoagulated blood
Reagents:
2% Sodium metabisulphite solution:0.2 g sodium metabisulphite is dissolved in 10.0
mL of distilled water. This solution should be prepared freshly.
Method:
One drop of blood is mixed with 3 drops of 2% sodium metabisulphite solution on a
glass slide.
A cover slip is placed over it and sealed with petroleum jelly or nailpaint.
The slide is placed in a petri dish, kept moist with wet cotton plugs and incubated at
370C for 1 to 4 hours.
The slide is observed under thehigh power(40X)lens of the microscope for the
presence of sickled cells (Figure 4).
Occasionally the preparation may need to stand up to 24 hours.
A B
30
Interpretation:
Figure 4: Sickling test showing typical sickle shaped cells.
Solubility Test
Principle:It is a rapid method for detection of HbS. HbS is quite insoluble when in the
reduced state in ahigh phosphate buffer solution. It forms water crystals (Tactoids) which
refract and deflect light rays and producea turbid solution.
Reagents:
Phosphate Buffer (pH 7.1): Stock Solution
Potassium dihydrogen phosphate (KH2PO4): 125.0 g
Dipotassium hydrogen phosphate (K2HPO4): 217.0 g
Saponin : 2.5 g
Distilled water : 1 L
Preparation of working solution: 0.1 g of sodium dithioniteis added to 10 mL of the
stocksolution immediately before use.
Method:
1. The RBCs are washed three times with normal saline.
2. Two mL of the phosphate buffer is takenand 20 µL of packed RBCsare added.
3. The tube is mixed well to give a light violet color.
4. The results are recorded after 10 minutesby holding a white paper with a dark black
line behind the tube (Figure 5).

31
Interpretation:
Positive result (+): the black line is not visible.
Negative result (-): the black line is clearly visible.
Doubtful result (+/-): the black line is partially visible.
Figure 5: Solubility test for Hb S detection
Preparation of Hemolysate for Hb Electrophoresis and Screening for
UnstableHemoglobins
Hemolysate preparation is required for two tests (Hb stability test and cellulose acetate
electrophoresis test) for detection of abnormal hemoglobin variants. For both the tests, the
initial step of red cell washing with saline is common. The difference lies in the addition of
distilled water to the packed RBC pellet. This is followed by the addition of Carbon
tetrachloride (CCl4) which is same for both the tests.
Requirements
Plastic tubes
Normal saline
Distilled water
CCl4
Centrifuge
Method
1. 1000 µL of blood is taken in a 1.5 mL tube and washed thrice with 0.9 % saline.

32
2. Lysate for heat stability test: 200 µL of packed RBCs are mixed with 400 µL of
distilled water in a fresh tube.
3. Lysate for Cellulose acetate electrophoresis test: 200 µl of packed RBCs are mixed
with 30 µL (2 drops) of distilled water in a fresh tube.
4. Tubes are vortexed thoroughly for 20 seconds to lyse the cells.
5. 200 µL of CCl4 is added to the above mixture to remove the cell membranes and other
cell debris.
6. The tubes are vortexed on a vortex mixer for 2 minutes and centrifuged at 10000 rpm
for 15 minutes.
7. The clear hemolysate from the top is pipetted out leaving behind the CCl4 (bottom
layer) and the cell membrane layer (middle layer).
Comments
Toluene can also be used instead of CCl4 in which case the hemolysate will be in the bottom
layer and pipetting out the lysate may be more difficult.
The hemolysate can be preserved at -200C after addition of 1-2 drops of 1 % potassium
cyanide (KCN) for cellulose acetate electrophoresis. Freshly prepared lysate without KCN is
used for the Hb stability test.
HemoglobinStability Test
Disruption of normal structure of the hemoglobin molecule can result in reduced stability,
which leads to precipitates in the erythrocyte causing it’s destruction. Amino acid
substitutions in the globin chains, particularly those involving non-polar amino acids that
constitute the heme pocket, may result in an unstable hemoglobin. However, if the substituted
amino acid is internal or the total charge of the molecule is unchanged, the hemoglobin
variant will not be detected by conventional electrophoresis. Therefore, if
anunstablehemoglobin is suspected clinically, a specific test for detection of the unstable
hemoglobin should be performed. Milder instability may also beassociated with Hb H
andHbvariants with altered oxygen affinity.
There are two stability testsIsopropanol stability test and Heat stability test.

33
Isopropanol stability test
Principle: The presence of isopropanol makes the buffer less polar, weakening the
hemoglobin hydrophobic binding that facilitate it’s denaturation and precipitation.
Reagents:
0.1 M Tris –HCL
12.1gmTris is dissolved in 600 mL distilled water, the pH is adjusted to 7.4 with 10 N
HCl and the volume made up to 1 liter.
17% Isopropanol- Tris buffer
17 mL of isopropanol is mixed with 83 mL of Tris-HCl buffer (pH 7.4). this buffer is
stable at room temperature.
Method:
1. 2 mL of isopropanol buffer is taken in a tube and equilibrated at 370C in a water bath
for 5 minutes.
2. 200 L of a freshly prepared hemolysate is then added and mixed properly. The tube
isagain re-incubated at 370C.
3. The tube is observed every 5 minutes to look for precipitation. For the unstable
hemoglobin the precipitate will form after 20 mins (Figure 6)
4. A normal control lysate is always put up simultaneously.
Interpretation:
Figure 6: Isopropanol stability test for unstable hemoglobin detection.
1. Normal Control: Clear solution after 1 hour of
incubation at 50oC.
2. Test sample: The solution turns turbid and a
flocculent precipitate forms within 20 minutes at
37oCwhich precipitates.
Precipitation

34
Heat stability test
Principle: Normal hemoglobin precipitates only slightly when incubated at 500C for 30
minutes, while an unstable hemoglobin under these conditions is completely denatured.
Sample: EDTA anticoagulated blood.
Reagents:
0.05 M Tris-HCL buffer
6.05gmTris is dissolved in about 600 mL distilled water. The pH is adjusted to 7.4
with 10 N HCl and the volume is made up to 1 liter.
Method:
1. 1.8 mL of 0.05 M Tris- HCL buffer is taken in a Kahn tube and equilibrated at 500C
in a water bath for 5 minutes.
2. 200 L of a freshly prepared hemolysate is then added and mixed properly. The tube
is re-incubated at 500C.
3. The tube is observed every 5 minutes to look for precipitation till 60 minutes.
4. A normal control lysate is always put up simultaneously.
Interpretation:Same as for isopropanol stability test
Hemoglobinpattern analysis
Different patterns of hemoglobin can be detected by using an electrophoretic method
(Cellulose acetate electrophoresis) and/or automated chromatographic methods (analysis on
D10 andHPLC Variant 2).
Cellulose Acetate Electrophoresis
Principle: Electrophoresis is a separation technique based on the mobility of ions in an
electric field. It is a classical method of identifying and quantifying the hemoglobin proteins.
At alkaline pH (8.4 to 8.6), hemoglobin is a negatively charged protein and migrates towards
Screening for unstable hemoglobin to be done at level 3 if required

35
the anode in an electrical field. During electrophoresis, various hemoglobins separate due to
charge differences caused by structural variations, thereby allowing their identification.
Cellulose acetate membranes demonstrate several features making them superior to filter
paper for hemoglobin separation. The absence of adsorption yields clean white backgrounds
between fractions and there is no adsorptive loss of protein during migration, hence theycan
be used to quantitate hemoglobinfractionson a spectrophotometer. The electro-endosmotic
flow is greater than in filter paper. This results in a continuous flow of buffer towards the
cathode and better separation of bands.
Reagents and materials:
Cellulose acetate membranes (stored in 30% methanol)
Tris EDTA Boric acid(TEB) buffer, pH 8.6
Tris : 14.4 g
EDTA (di sodium) : 1.5 g
Boric acid : 0.9 g
Distilled water to makethevolume to 1 L.
0.2% Ponceau S stain: 0.2 g of Ponceau S stain is dissolved in 100 mL of 3%
trichloroacetic acid
Destaining solution (2% Glacial acetic acid): 2 mL acetic acid made to 100 mL with
distilled water.
Paint brush
Horizontal electrophoresis tank
Filter paper wicks
Method:
1. Hemolysate is prepared from anticoagulated blood using theearlier mentioned
method.
2. The cellulose acetate membrane strips are soaked in TEB buffer for 30 minutes.
3. Excess buffer is removed by blotting the membranes between Whatman no. 1 filter
paper.
4. The membranes are placed across the bridge ofahorizontal electrophoresis chamber.
5. The membranes are secured using a double layer of Whatman no. 1 filter paper as
wicks. These wicks dip in the anode and cathode buffer compartments of the
chamber.
6. Hemolysate is applied at the cathode end of the strip using a fine tipped paint brush.

36
7. Electrophoresis is carried out at a constant voltage of 200-250 V (~ 1 mA/strip) for
1½ to 2 hours till adequate separation of bands is obtained.
8. The membranes are stained for 1 minute with Ponceau S stain in a petri dish and
destained in 2 % acetic acid till the background is clear.
9. The membrane can then be preserved in the destaining solution.
10. During every run it is advisable to electrophorese a known sample having a variant
hemoglobin as a control (Figure 7).
Interpretation:
Figure 7: Cellulose acetate electrophoresis at alkaline pH (8.9) (CA is carbonic
anhydrase)
The hemoglobins migrate on the cellulose acetate membrane from cathode to anode in the
following order: HbA2/ HbE, HbC, HbD/HbS, HbLepore, HbF, HbA0 and the fast
movinghemoglobin Bart’s and HbH.
HPLC for screening for hemoglobinopathies
Automated Cation exchange high performance liquid chromatography (CE-HPLC)
Principle: This method has emerged as the method of choice for quantification of HbA2,
HbF and for detection and quantitation of the Hb variants, particularly those which may
interact with β-thalassemia such as HbS, HbE, HbDPunjab
and Hb-Lepore. The Variant II
machineor the D10machine from BioRad laboratories has most commonly been used in
India, however, any other similar HPLC machine can be used. In this method phosphate
buffersof different concentration (mobile phase), pass under pressure through an ionic
+ -
N
HbA + HbS/D Punjab
HbS + HbF
HbE + HbF

37
exchange column (stationary phase). The stationary phase consists of a temperature
controlled analytical cartridge containing a resin of anionic or cationic particles (3-5 μm).
Two pumps and apre- programmed gradient control the elution buffer mixture (mobile phase)
passing through the analytical cartridge (stationary phase). As the ionic strength of the elution
buffer mixture increases, more strongly retained hemoglobins elute from the cartridge. A
photometer monitors the eluate and detects absorbance changes at 450 nm. Background
variations are compared by usingan additional filter at 690 nm. A chromatogram of
absorbance v/s time is displayed and printed. Windows are set for common hemoglobin
variants based on their retention times. Each sample takes 6.5 minutes for analysis and up to
100 samples can be loaded in the sampling chamber.
Sample:EDTA anticoagulated blood.
Analysis on D10 HPLC machine
Reagents:
Preparation of specimen
EDTA blood sample
If the blood sample is <2mL: 1.5 mL of wash/diluent solution and 5 µL of the
blood are mixed and used.
If the blood sample is≥ 2 mL: direct vacutainers can be placed in the machine.
Preparation of Primers
Reconstitute lyophilized primer with 1 mL of distilled water.
Allow to stand for 10-15 minutes; swirl gently to dissolve.
Stable for 1 day at 2-80C.
Preparation of the calibrator
Two calibrators (Level 1 and Level 2)
Reconstitute each vial with 7 mL of cold Calibrator Diluent.
Allow to stand for 5-10 minutes; swirl gently to dissolve.
Stable for 10 days at 2-80C.
Preparation of Controls
There are two sets of controls available (Level 1 and level 2)
Reconstitute each vial with 0.5 mL of distilled water
Allow to stand for 5-10 minutes; swirl gently to dissolve
Controls are stable for 21 days at 2-80C

38
Dilute 5µL of control in 1.5 mL of wash/Diluent solution.
Method:
1. Switch on the machine.
2. Place the system in sleep state
3. Go to LOT INFOscreen
4. Press the method box to display the select method screen.
5. Select A2/F METHOD
6. Press EXIST
7. Press YES to confirm the method change.
8. Press EXIT
9. The selected method is indicated in the status bar.
10. There is no need to perform a system flush unless a different lot of reagent is
installed.
11. A priming run is performed once per new cartridge and also following the
decontamination procedure.
12. Follow the instruction manual which comes along with the kit for priming.
13. Calibration is performed once when a new cartridgeis used.
14. Follow the instruction manual which comes along with the kit for calibration.
15. The calibration report is printed after the testing is complete. The slope and intercept
acceptable ranges are provided in calibrator diluents set insert.
16. Once the calibration is passed you can run the sample.
17. Once the cartridge has been calibrated there is no need to calibrate the cartridge again
when you switch on the machine next time.
18. Two different controls Level 1 and Level 2 are to be run every time before patient’s
samples.
19. Each sample will take 6.5 mins. to run.
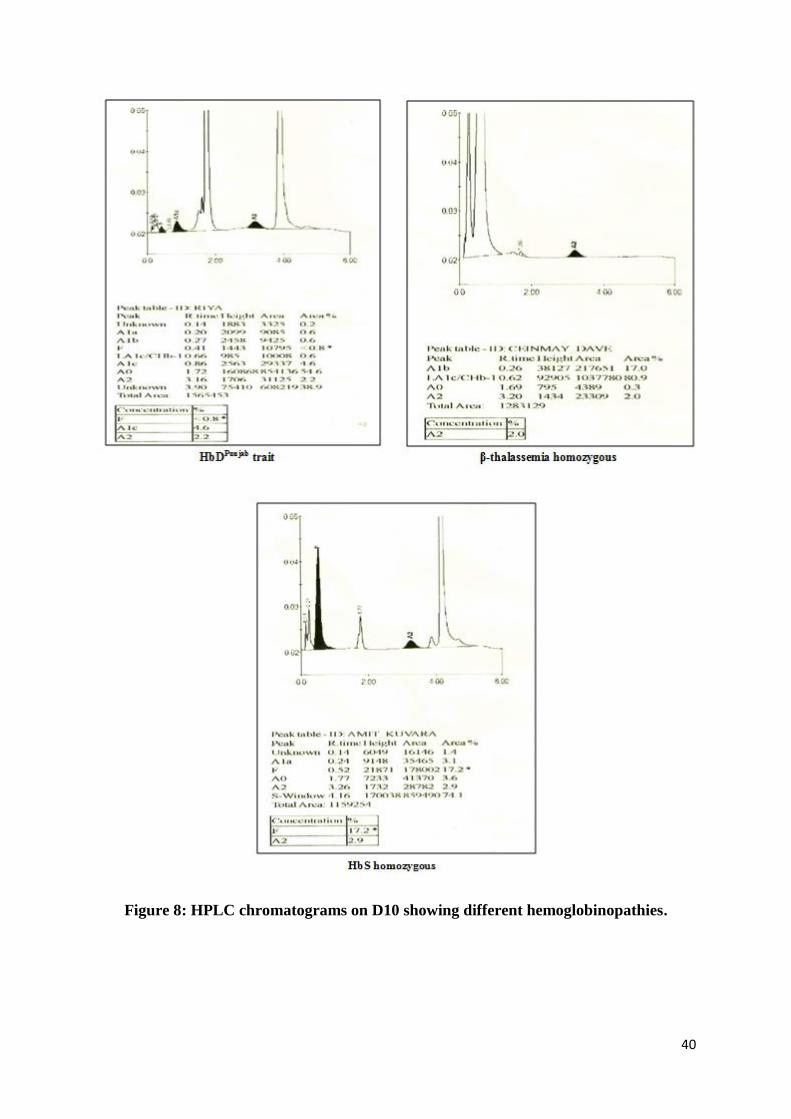
Interpretation: (Figure 8)
HbA2<3.5%: Normal
HbA2 3.6-3.9 %: Borderline HbA2[needs confirmation by DNA analysis]
HbA2> 4.0 %: β-thalassemia trait (BTT)
HbA2< 2.0 %: Can be α-thalassemia / δ-thalassemia heterozygote
Common Hb variants such as HbSare identified as a separate peak.

39
HbDPunjab
andHbQIndia
elutes as an unknown peak after HbA2witharetention time of 3.83 min
and4.37 min respectively
40
Figure 8: HPLC chromatograms on D10 showing different hemoglobinopathies.

41
Analysis on Variant II HPLC machine
Reagents:
Preparation of specimen
EDTA / heparinised blood sample
If the blood sample is < 2 mL: 1mL of hemolysing solution and 5 µL of the
blood are mixed and used.
If the blood sample is≥ 2 mL: direct vacutainers can be placed in the machine.
Preparation of the Primer
1 mL of distilled water is addedto the lyophilized primer and mixed properly.
It is allowedto stand at room temperature for 10 minutes at 15-300C.
Preparation of the calibrator
10 mL of calibrator diluent is added to the lyophilized calibrator whichcomes
with the kit. It ismixed properly and allowed to stand at room temperature for
10 minutes at 15-300C.
The reconstituted calibrator is stable for 10 days when stored in aliquots at 2-
80C.
Aliquotsare made of this prepared calibrator.
Preparation of the controls
A set of normal (HbF: 1-2%, HbA2: 1.8-3.2%) and abnormal (HbF: 5-10%,
HbA2: 4-6%) controls are to be prepared according to the instructions.
The controls should be run at the beginning of each group of test specimens.
Limitations:
HbLepore, HbE and HbD Iran are co-eluted with HbA2.
HbA2is falsely increased in thepresence of HbS.
HbA2 is falsely reduced in thepresence of HbD.
HbDPunjab
elutes in anunknown window
Elevated HbF in case of β-thalassemia major and intermedia will elute in two
windows A1b and LA1C/CHb. In such acase,you need to add the concentration of
Hb from both the windows to get thetotal HbFlevel.
Hb Bart’s show a sharp peak at the start of the chromatogram and HbH shows twin
peaks before 1 minute.

42
Method:
1. The machine is first switched onfollowed by the computer.
2. The software is startedby clicking on the CDM icon
3. The machine is allowed to attain it’ssteady state.
4. The buffer level, waste, column count and column temperatureare checked.
5. Theprimer is first run,followed by the blank and finally the calibrator.
6. After the completion of therun, the retention time of HbA2 of the calibrator is
checked.It should be between 3.63-3.67 minutes.
7. If not, then according to the instruction manual, the temperature of the columnis
changed.
8. Only proper calibration allows the samples to run.
9. Each sample takes 6 minutes to run.
Interpretation: (Figure 9)
Based on the level of HbA2, the interpretations are as follows:
HbA2< 3.5 %: Normal
HbA2 3.6-3.9 %: Borderline HbA2[needs confirmation by DNA analysis]
HbA2> 4.0 %: β-thalassemia trait
HbA2< 2.0 %: Can be α-thalassemia / δ-thalassemia heterozygote
Common Hb variants such as HbS, HbDPunjab
andHbQIndia
are identified as separate peaks in
different windows with specific retention times.
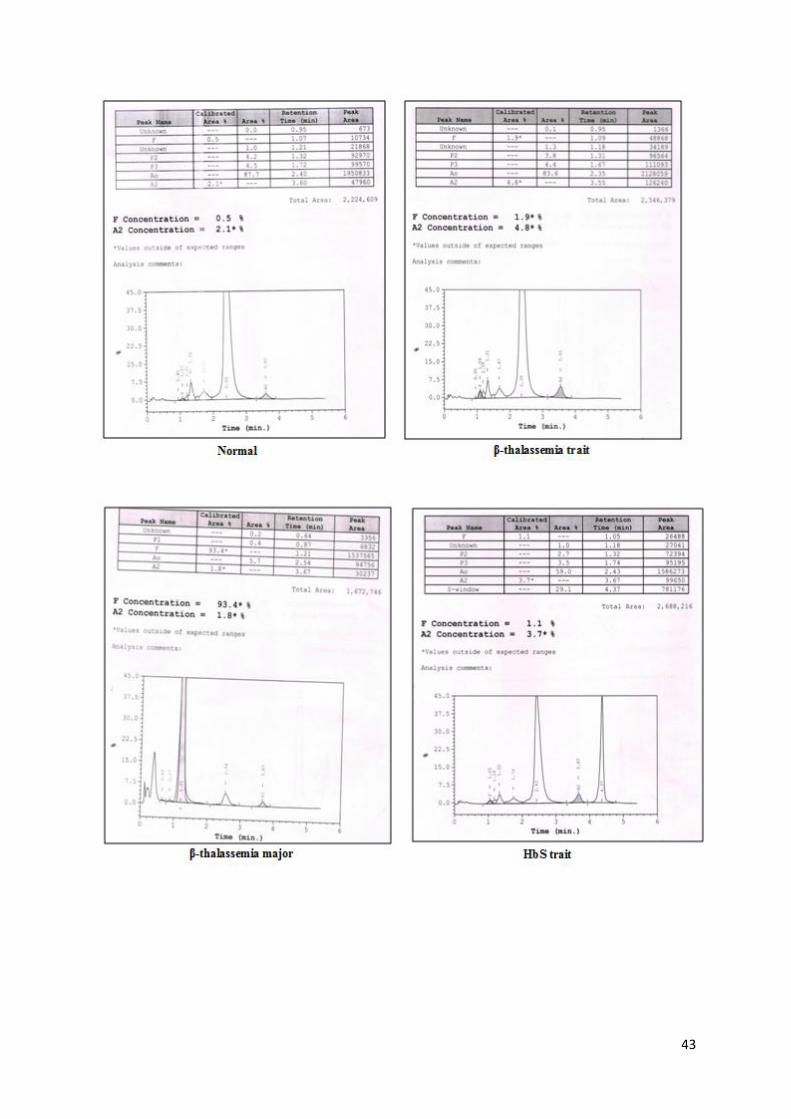
43
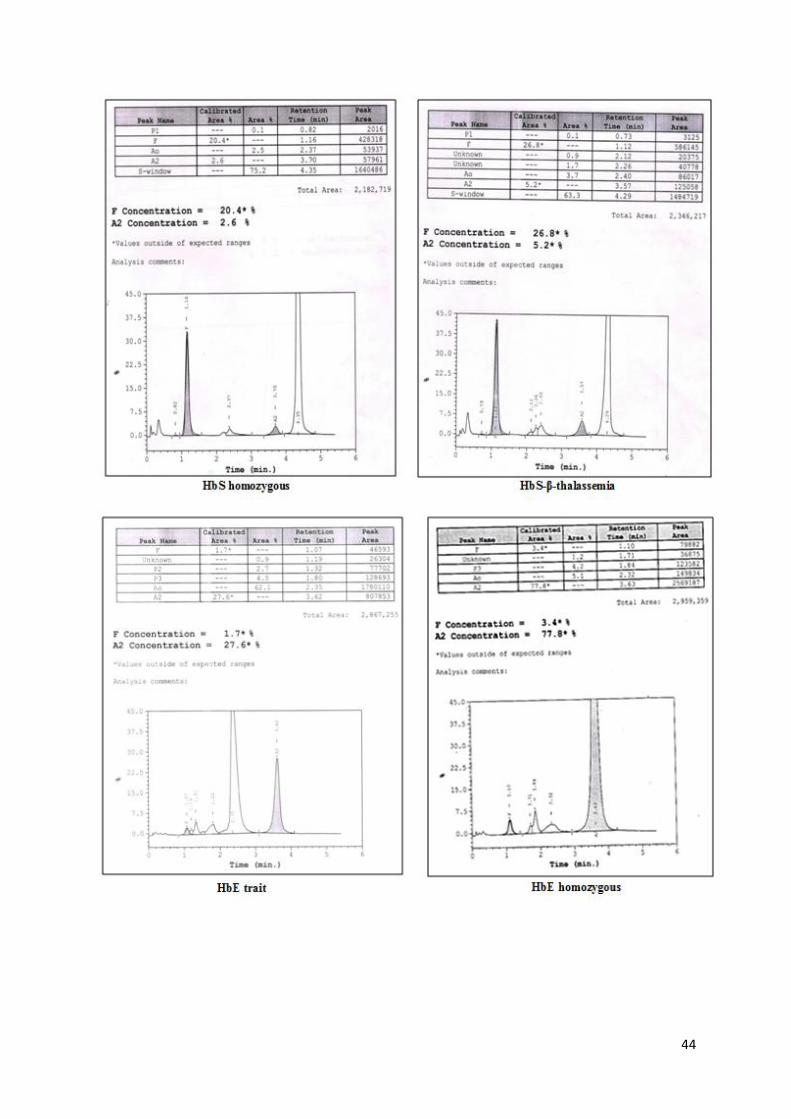
44
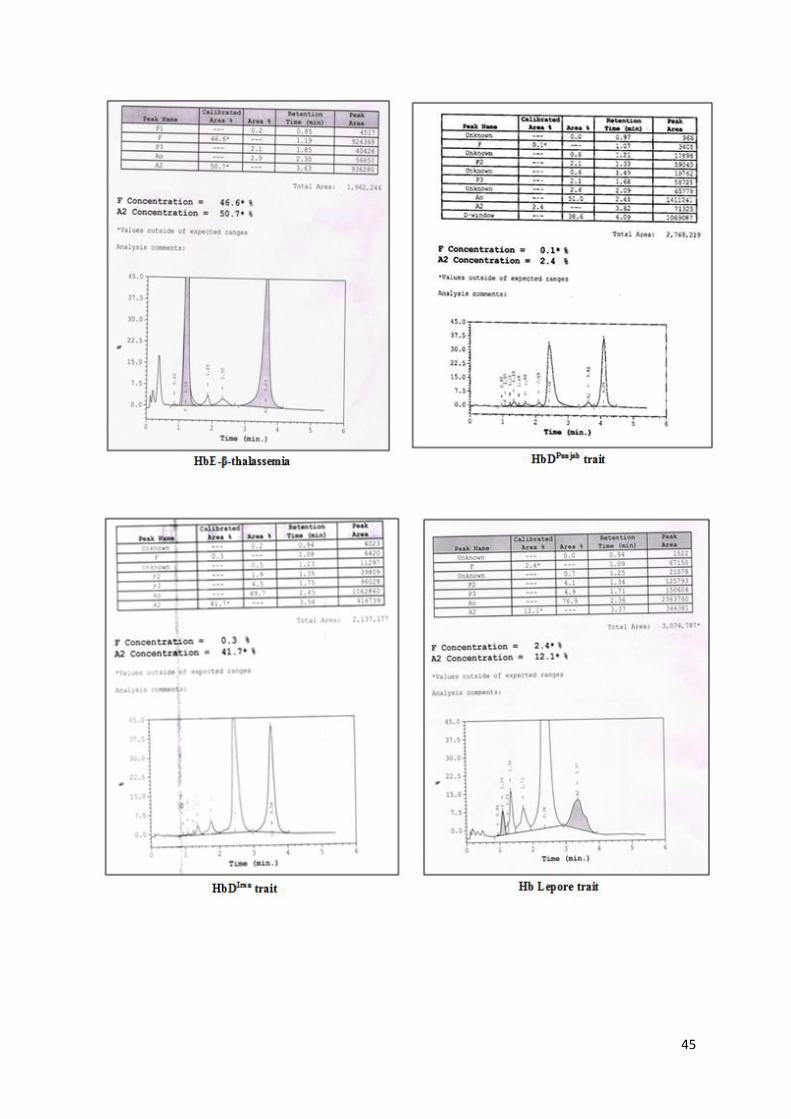
45
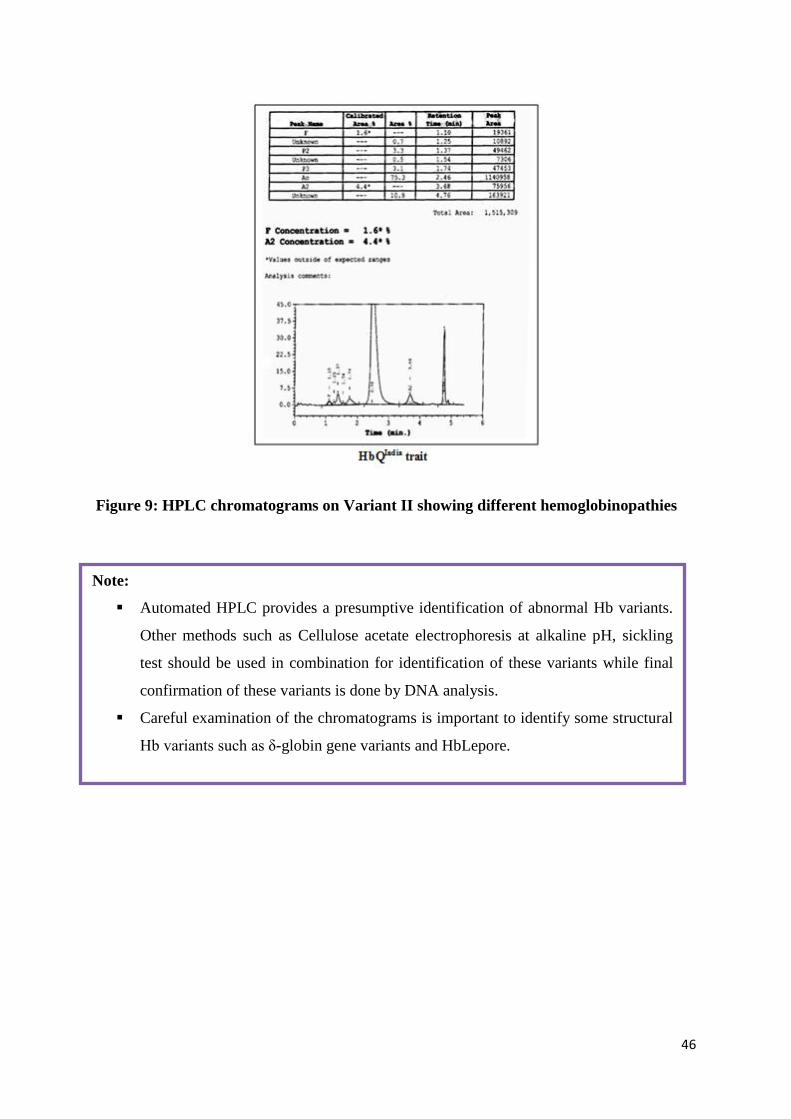
46
Figure 9: HPLC chromatograms on Variant II showing different hemoglobinopathies
Note:
Automated HPLC provides a presumptive identification of abnormal Hb variants.
Other methods such as Cellulose acetate electrophoresis at alkaline pH, sickling
test should be used in combination for identification of these variants while final
confirmation of these variants is done by DNA analysis.
Careful examination of the chromatograms is important to identify some structural
Hb variants such as δ-globin gene variants and HbLepore.

47
References:
1. Bain BJ, Lewis SM, Bates I. Basic haematological techiniques. In: Lewis SM, Bain
BJ, Bates I. Dacie and Lewis Practical Haematology, 10th
edition, Churchill
Livingstone p-25 (2006).
2. Itano HA and Pauling L. A rapid diagnostic test for sickle cell anemia. Blood
1949;4:66.
3. Huntsman R.G., Barclay G.P.T., Canning D.M. and Yawson G.I. A rapid whole blood
solubility test to diffrfentiate the sickle cell trait form sickle cell anemia. J. Clin.
Path.1970;23:781.
4. Gorakshakar AC, Colah R, Nadkarni A, Desai S: Evaluation of the single tube
osmotic fragility test in detection of beta thalassemia trait. Natl Med J India
1990;3:171.
5. Mohanty D, Colah R. Eds Laboratory Manual for Screening, Diagnosis and Molecular
analysis in Hemoglobinopathies and Red Cell Enzymopathies 1st Edition. Bhalani
Publishing House, Mumbai 2008.
6. Colah RB, Surve R, Sawant P, D’Souza E, Phanasgaonkar S, Nadkarni AH,
Gorakshakar AC. HPLC studies in hemoglobinopathies. Ind. J. Pediat. 2007;74:31.
Limitations:
HbLepore, HbE and HbD Iran co-elute with HbA2.
HbA2is falsely increased in thepresence of HbS.
HbA2 is falsely reduced in thepresence of HbD.
HbH and Hb Bart’s show a sharp peak at the start of the chromatogram but are not
identified accurately. They must be confirmed by other methods.

48
Patient information sheet
Thalassemia major
This is a life long blood disease that causes serious anemia, needing blood transfusion. It is an
inherited disease due to a minor blood mutation in both parents who do not show any disease.
This is why testing before pregnancy is very important. Prenatal testing can prevent this
disease.
If your child has thalassemia major then good medical care can prolong life and
decrease complications.
Regular blood transfusions are necessary, Keep hemoglobin more than 9 g/ dl before
the regular blood transfusion. If Hemoglobin goes to a very low level each month it
will result in more complications like big spleen, bone problems, poor growth etc.
It is important to check iron levels by serum ferritin. If this is high it is important to
take iron chelation (iron removal). There are 3 iron chelating medicines. Take the
medicine recommended by your doctor regularly. If iron overload is not treated it will
result in more problems e.g.hormone defects, affect the heart and liver and other
organs.
Special tests for better checking foriron overload like T2 * MRI are required. As the
child gets older different problems like bone problems may occur. If the special tests
are not available at your local centre, discuss with your doctor and you can be sent to
a higher centre for these tests and checkup.
Regular monitoring is very important for health, your doctor will give you full
information.
The only cure is Bone marrow transplant, but this may only be possible in some
patients. Also this procedure has risks including a chance of death, this may be higher
in older patients or those with complications. Ask your doctor if this is a suitable
option in your case.
With good blood transfusion and iron chelation medicines you can lead a productive
life.

49
Patient information sheet
Sickle cell disease (SCD)
Sickle cell disease (SCD) is a serious, inherited condition affecting the red blood cells. This causes”
sickling” (shaped like a crescent moon), of the usually round red blood cells, these sickle cells cause
pain and other symptoms and affect many organs of the body.
Sickle cell trait is not the same as sickle cell disease. Sickle cell trait means you carry a sickle cell
gene, but usually such people are not ill.
Diagnosis
We can make a diagnosis by the dried blood spot newborn test which can be confirmed by a
HPLC/Hb Capillary zone electrophoresis blood test. Early diagnosis means that the child will receive
early and appropriate care needed for management.
What problems can be seen?
Pain- mild to severe, pain crisis
Anemia
Jaundice,
Fever ( these children are at risk for serious infections)
Pneumonia etc
Acute chest syndrome
Stroke
Kidney problems etc
Treatment
Good treatment, started early in life, may help to prevent complications. See a specialist about the
disease at least annually.
Take pneumoccalvaccination and oral penicillin medicine to prevent some infections
Some things which can increase sickling, are cold weather, infection, lack of
fluid in the body (dehydration) or low oxygen.
The sickle cells containing mostly HbS are less flexible than normal red
blood cells and get stuck in small blood vessels and block them.
These sickle cells are destroyed more easily than normal red blood
cells. Patients will have a moderate anemia. Some other complications
include pain crisis due to sickling, trapping of blood in spleen,
increased infections, acute chest syndrome etc.

50
Take folic acid and Hydroxyurea as prescribed by the doctor. There is a national programme for
Sickle cell anemia through the government. Visit your state District hospital for details.

51
Newborn Screening for Sickle Cell Disease
Hemoglobinopathies are one of the commonest groups of single gene disorders in the Indian
subcontinent and pose a major drain on our health resources. Sickle cell disease (SCD) is an
important public health problem in Indiawith highest prevalence amongst the tribal and some
non-tribal ethnic groups. Sickle cell disease in India has a very varied clinical presentation
ranging from a severe to mild or asymptomatic condition. Early diagnosis and providing
comprehensive care are critical in SCD because of the possibility of lethal complications in
early infancy in pre-symptomatic children. Children with SCD have an increased
susceptibility to severe bacterial infection, particularly due to Streptococcus pneumoniae
which can occur as early as 4 months of age and carries a case fatality rate as high as 30%.
Acute splenic sequestration crisis also contributes to mortality in infancy. It has been
documented in different countries of the world that pneumococcal sepsis is a major cause of
early mortality in sickle cell disease and prophylactic penicillin therapy provided can
dramatically reduce the morbidity and mortality. Early introduction of penicillin can lead to a
dramatic decrease in early childhood mortality from pneumococcal sepsis. Newbornscreening(NBS) and comprehensive care have dramatically decreased morbidity and
improved survival of patients with SCD.Since the implementation of NBS in developed
countries, there has been a dramatic improvement in the survival of infants with sickle cell
disease.The aim of aNBS program for SCD is to prevent major
healthcomplicationsofSCDfrom early childhood onward. The approaches for a NBS program
could be “Targeted screening” which takes the ethnic ancestry of every newborn into account
or “universal screening” wherethe entire newborn population is screened irrespective of
family origins. Laboratory methods to be usedfor the detection of disease and carrier states should be very
sensitive and highly specific so that missing or falsely identifying patients will be minimized.
Technologies are based on the separation of hemoglobin (Hb) and the quantification of
respective hemoglobin fractions either from fresh cord blood samples or adried blood spot
(DBS) (Figure 10). Storage of cord blood shouldnot exceed seven days at 4°C and that of
dried blood spots should not exceed 1 month.Older. samples show lower elution of samples
from the DBS and, often, the degradation of hemoglobin,resulting in a high baseline,
increased noise, and unclear peaks with. difficulties in zone adjustmentand quantification,
especially in Hb species present at low percentage.

52
The commonly used technologies for NBS for SCD include high performance liquid
chromatography (HPLC), isoelectric focusing (IEF) and capillary electrophoresis (IEF).
Globally, isoelectric focusing (IEF) using eluates from dried blood spots was initially used
for screening of newborn babies for sickle cell disease but at many centres this had been
replaced by HPLC analysis.The Variant NBS machine. (BioRad laboratories) has been used
for hemoglobin analysis from dried blood spots or the Variant Hemoglobin Testing
System(BioRad laboratories) for cord blood samples using either the sickle cell short or the
thal short programmes. The thal short programme had the advantage of picking up other
hemoglobin abnormalities including some rare non deletional chain variants like Hb
Fontainebleau,Hb O Indonesia and Hb Koya Dora.
Figure 10: Newborn screening strategyfor sickle cell disorders
Specimens:
Anticoagulated (EDTA or Heparin) cord blood sample of newborn at birth
Heel prick sample on filter paper
Sample collection:
Cord bloodsample:

53
The blood sample is collected from the umbilical cord at the time of delivery making sure
there is no contamination with maternal blood.
Heel prick sample:
The newborn infant’s heel is cleaned with a disinfectant. The heel is allowed to air dry
and a puncture made using a sterile lancet (Figure 11).
The first drop of blood is gently wiped off with sterile cotton.
The newborn baby’s blood is collected from the second drop on filter paper by
touching the printed side of the filter paper to the blood drop.
The quantity of the blood should be quite enough to absorb and moist the paper from
both the sides (blood should not be applied on both the sides).
The filter paper is allowed to dry properly and kept away from direct sunlight and
heat.
One wet filter paper should never be superimposed on another before drying after
blood has been collected.
The dried filter paper cards can then be sent to the respective laboratories at the
earliest.
Samples collected on filter paper are run on another HPLC machine (NBS HPLC) and
can also be used forthe POC device.
Figure 11: Collection of blood sample from newborns by heel prick

54
Point of care (POC) Testing
Recently, several point-of-care devices have been developed for screening which are either
paper based screening protocols or antibody based rapid diagnostic devices based on lateral
flow immunoassay technologies. They are simple to use and relatively inexpensive as they do
not require any specific equipment or even electricity which is often not always available in
remote rural regions. These commercial devices have been validated for newborn screening
for SCD with a clinical accuracy of 97 to 100%. One such kit is based on competitive lateral-
flow immunoassay that uses monoclonal antibodies to detect hemoglobins A, S, and C in a
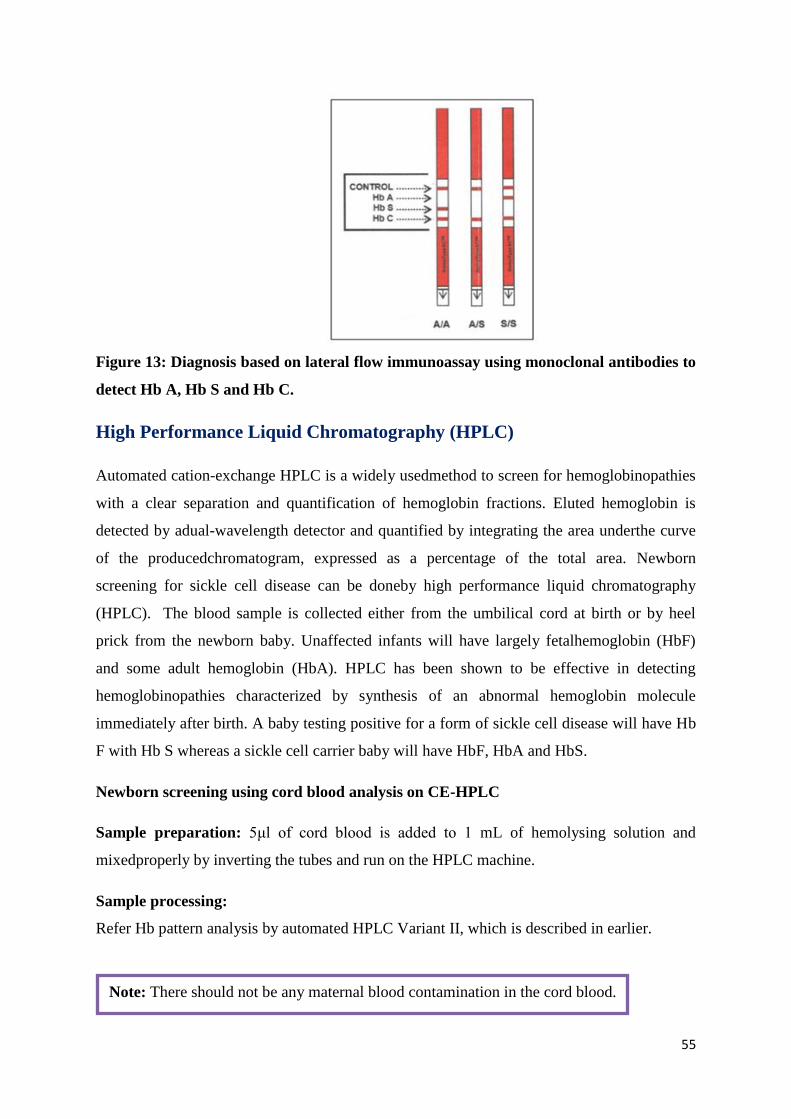
1.5-μL sample of whole blood(Figure 12 and 13).
Test procedure should be followed as per the manufacturer’s instruction with the kit
Figure 12: Test procedure for lateral flow immunoassay using monoclonal antibodies to
detect Hb A, Hb S and Hb C.
55
Figure 13: Diagnosis based on lateral flow immunoassay using monoclonal antibodies to
detect Hb A, Hb S and Hb C.
High Performance Liquid Chromatography (HPLC)
Automated cation-exchange HPLC is a widely usedmethod to screen for hemoglobinopathies
with a clear separation and quantification of hemoglobin fractions. Eluted hemoglobin is
detected by adual-wavelength detector and quantified by integrating the area underthe curve
of the producedchromatogram, expressed as a percentage of the total area. Newborn
screening for sickle cell disease can be doneby high performance liquid chromatography
(HPLC). The blood sample is collected either from the umbilical cord at birth or by heel
prick from the newborn baby. Unaffected infants will have largely fetalhemoglobin (HbF)
and some adult hemoglobin (HbA). HPLC has been shown to be effective in detecting
hemoglobinopathies characterized by synthesis of an abnormal hemoglobin molecule
immediately after birth. A baby testing positive for a form of sickle cell disease will have Hb
F with Hb S whereas a sickle cell carrier baby will have HbF, HbA and HbS.
Newborn screening using cord blood analysis on CE-HPLC
Sample preparation: 5μl of cord blood is added to 1 mL of hemolysing solution and
mixedproperly by inverting the tubes and run on the HPLC machine.
Sample processing:
Refer Hb pattern analysis by automated HPLC Variant II, which is described in earlier.
Note: There should not be any maternal blood contamination in the cord blood.
56
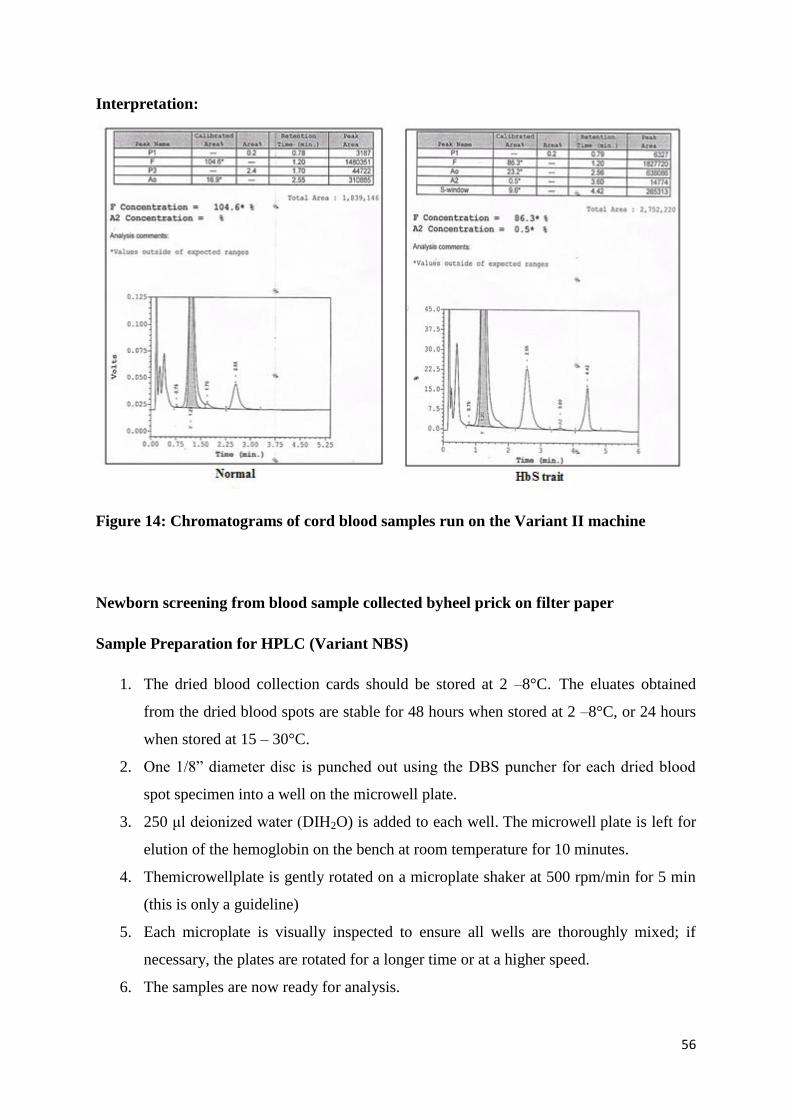
Interpretation:
Figure 14: Chromatograms of cord blood samples run on the Variant II machine
Newborn screening from blood sample collected byheel prick on filter paper
Sample Preparation for HPLC (Variant NBS)
1. The dried blood collection cards should be stored at 2 –8°C. The eluates obtained
from the dried blood spots are stable for 48 hours when stored at 2 –8°C, or 24 hours
when stored at 15 – 30°C.
2. One 1/8” diameter disc is punched out using the DBS puncher for each dried blood
spot specimen into a well on the microwell plate.
3. 250 μl deionized water (DIH2O) is added to each well. The microwell plate is left for
elution of the hemoglobin on the bench at room temperature for 10 minutes.
4. Themicrowellplate is gently rotated on a microplate shaker at 500 rpm/min for 5 min
(this is only a guideline)
5. Each microplate is visually inspected to ensure all wells are thoroughly mixed; if
necessary, the plates are rotated for a longer time or at a higher speed.
6. The samples are now ready for analysis.
57
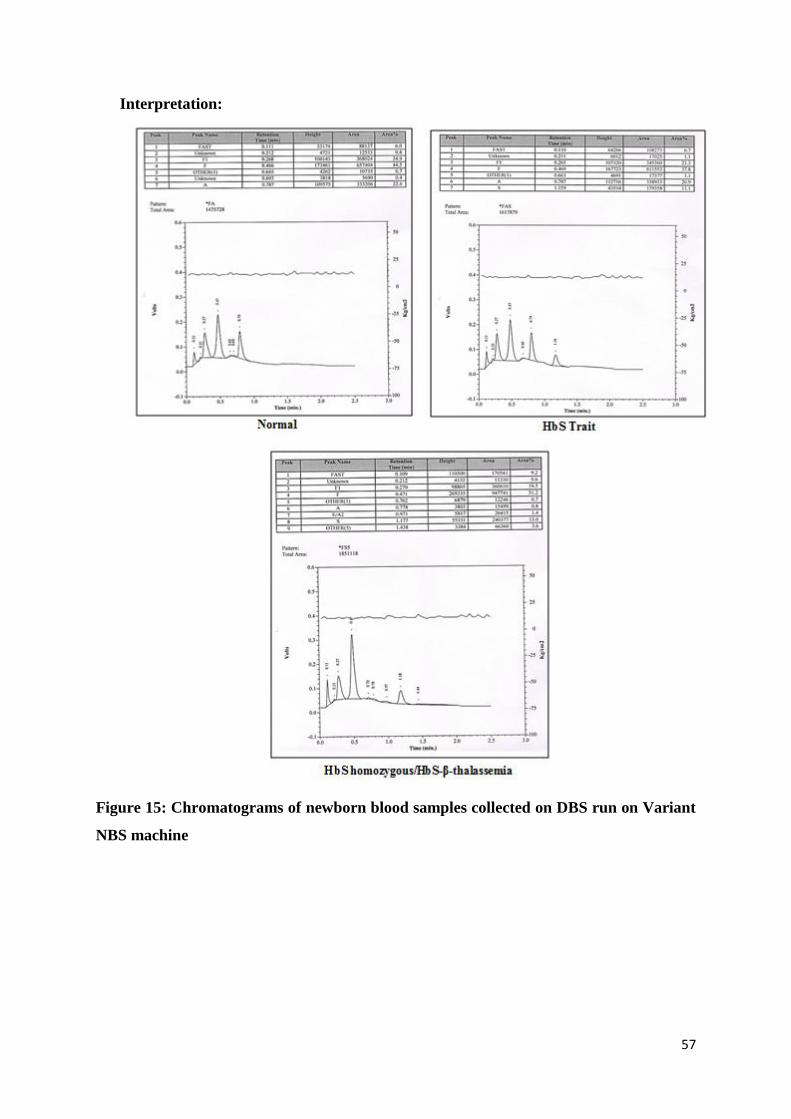
Interpretation:
Figure 15: Chromatograms of newborn blood samples collected on DBS run on Variant
NBS machine
58
Capillary electrophoresis (CE)
Capillary electrophoresis (CE) is a separation technique that separates molecules in an
electric field according to size and charge. CE is performed in a small glass tube called a
capillary that is filled with an electrolyte solution. CE combines two principles of separation
of hemoglobins, theelectrophoretic mobility in alkaline buffer and the electro-osmotic flow
resulting in excellentseparation. Detected hemoglobin fractions can be relativelyquantified
and produce an electropherogram. CE is able to detect and relatively quantify many Hb
variants including and chain abnormalities.
Hemoglobin analysis by CE
Capillary's Neonat Hb kit enables the efficient separation of hemoglobin fractions and
detection of a large number of hemoglobin variants and thalassemiaspatterns.When using
fresh cord blood, samples are prepared by mixing 5 μL of blood with 5 μL of hemolysis
solution. When using the dry blood spots, punched paper disks (3.8 mm) are obtained using
the DBS puncher from the dried blood spot. The disks are placed into the wells of a
microplate containing 50 μL of hemolysis solution and kept into a humid chamber for 2 hrs.
The samples are now ready for analysis.
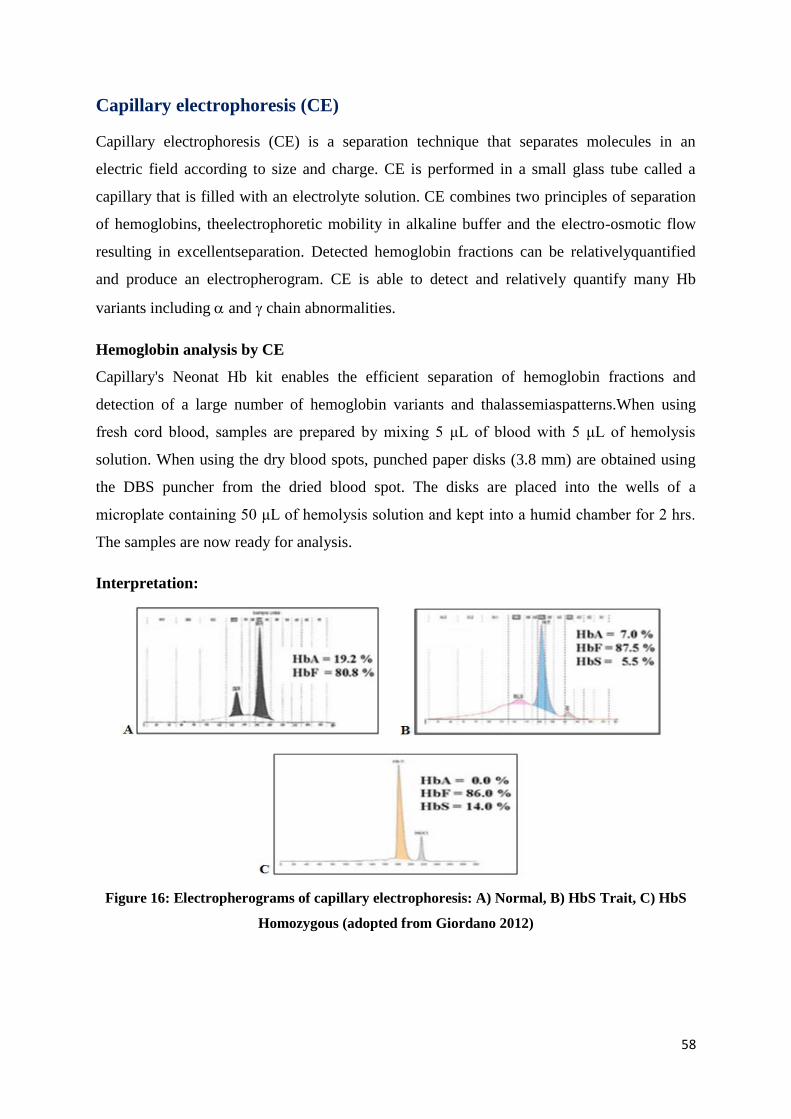
Interpretation:
Figure 16: Electropherograms of capillary electrophoresis: A) Normal, B) HbS Trait, C) HbS
Homozygous (adopted from Giordano 2012)

59
Isoelectric focusing (IEF)
IEF is a very sensitive method and is widely used at relatively low costs. IEF
separateshemoglobin species according to their isoelectric point on a gel medium with very
high resolution.Hemoglobin variants migrate in a pH gradient to the point where their net
charge becomes zero. Bands arenarrow compared to classical electrophoresis and give a
precise picture. HbF and HbA, aswell as relevant hemoglobins can be separated.
5 l of whole blood is placed in the wells of a microtitration plate containing 0.1 mL of
0.05% KCN. The samples are transferredwith a multiple syringe to a line of Whatman filter
paper placed on a strip of cellophane tape. The tape bearing the samples is then placed on the
cathodal side of the gel.The gel is run for around 2 hours using a cooling system at 40C with
1400 volts. The gel is then fixed in TCA for 15 minutes, stained for 10 minutes in
bromophenol blue and destained overnight till the background is clear. The gel can be either
dried on the plate itself or transferred to a Whatman no. 3 filter paper and dried for
preservation.
Interpretation:
AS SS AA
Figure 17: Isoelectric focusing gel picture (adapted from Claudia F, 2018)
References
1. Colah R, Mukherjee M, Ghosh K. Sickle cell disease in India. CurrOpinHematol.
2014;21:215-23.
2. Colombatti R, Montanaro M, Guasti F, Rampazzo P, Meneghetti G, Giordan M,
Basso G, Sainati L. Comprehensive care for sickle cell disease immigrant patients: a
reproducible model achieving high adherence to minimum standards of care.Pediatr
Blood Cancer. 2012; 59:1275-9.
3. Upadhye D, Das R, Ray J, Acharjee S, Ghosh K, Colah R, Mukherjee M. Newborn
screening for hemoglobinopathies and red cell enzymopathies in Tripura state: A
malaria endemic state in Northeast India. Hemoglobin 2018; ;42:43-46.

60
4. Upadhye DS, Jain D, Nair SB, Nadkarni AH, Ghosh K, Colah RB. First case of Hb
Fontainebleau with sickle haemoglobin and other non-deletional α gene variants
identified in neonates during newborn screening for sickle cell disorders.J.ClinPathol.
2012;65:654-9.
5. Eastman JW, Wong R, Liao CL, Morales DR. Automated HPLC screening of
newborns for sickle cell anemia and other hemoglobinopathies. Clin. Chem. 1996, 42,
704–710.
6. Upadhye DS, Jain DL, Trivedi YL, Nadkarni AH, Ghosh K, Colah RB. Newborn
screening for haemoglobinopathies by high performance liquid chromatography
(HPLC): Diagnostic utility of different approaches in resource-poor settings. Clin.
Chem. Lab. Med. 2014, 52, 1791–1796.
7. Giordano PC. Newborn screening for hemoglobinopathies using capillary
electrophoresis.Methods Mol Biol.2013;919:131-45.
8. Claudia F. Newborn Screening for Sickle Cell Disease and Other
Hemoglobinopathies: A Short Review on Classical Laboratory Methods—Isoelectric
Focusing, HPLC, and Capillary Electrophoresis. Int. J. Neonatal Screen.2018, 4, 39;
doi:10.3390/ijns4040039.
9. Steele C, SinskiA,AsibeyJ,et al. Point-of-care screening for sickle cell disease in low-
resource settings: A multi-center evaluation of HemoTypeSC, a novel rapid test. Am J
Hematol2019;94, 39-45.

61
Molecular Analysis of Hemoglobinopathies
β-thalassemiasarecaused by mutations that result in the reduced output or non-production of
β-globin chains. Depending on the output of the chains, they are designated as 0 or
+
thalassemia. The β-thalassemias are a very heterogeneous group of disorderswhere more than
200 mutations including deletional and non-deletional mutations causing -thalassemia (0 or
+) have been reported so far (http://globin.cse.psu.edu).Homozygosity and heterozygosity
for these mutations and their interaction with abnormal hemoglobins like HbS or HbE result
in variable clinical presentation or just a mild and asymptomatic hypochromic microcytic
anemia. The majority of the defects are single nucleotide substitutions, insertions or
deletions. Thus, molecular methods to identify mutations in β-globin genes often help to
understand the clinical heterogeneity of the thalassemia syndrome and may also help in
therapeutic interventions. Molecular characterization of the β-globin gene is important for
prenatal diagnosis to avoid the birth of affected babies.
At present there are various techniques available for detection of β-globin gene mutations.
Most of the procedures are based on amplification of the respective globin genes by PCR and
subsequent analysis of the PCR product. Here we have described few of these techniques.
1. Covalent Reverse Dot Blot Hybridization (CRDB): For detection of 6 common β-
globin gene mutations and two Hb variants (Hb S and HbE).
2. Gap PCR: For detection of deletional mutation (619 bp deletion).
3. Amplification Refrectory Mutation System (ARMS):Fordetection of common and
uncommon β-globin gene mutations.
4. Restriction Fragment Length Polymorphism –PCR (RFLP-PCR): For detection of Hb
Variants (Hb S and HbDPunjab
).
5. Direct DNA Sequencing (Sanger’s Sequencing): For detection of rare β-globin gene
mutations.
DNA extraction:
From blood using Phenol-chloroform method
Among the most convenient sources of DNA, leucocytes are considered best for the
extraction of human genomic DNA. It is estimated that 10 mL of whole blood yields about

62
250 μg of DNA. The phenol chloroform method was used for the extraction of DNA from the
peripheral venous blood.
Principle:
A phenol-chloroform extraction is a liquid-liquid extraction method, which separates
mixtures of molecules based on the differential solubilities of the individual molecules in two
different immiscible liquids. The RBCs to WBCs ratio is approximately 1000:1/ μl of blood.
So, the RBCs were lyzed by the freeze shock approach. The sample was placed at -200C and
then thawed at RT. The five basic steps for DNA extraction were as follows:
→ Chelation of divalent cations such as Mg2+
and Ca2+
to stop the function of DNase
enzymes and degradation of the DNA
→ Breaking open of the cells to get the intra cellular constituents in solution and remove
membrane lipids by adding a detergent.
→ Removal of cellular and histone proteins bound to the DNA by using a phenol- chloroform
extraction step.
→ Precipitation of the DNA in cold ethanol.
→ Solubilization of the DNA in a slightly alkaline buffer.
The lysis buffer added not only lyses the cells but also has EDTA and NaCl, which chelates
the divalent cations which inhibit the nuclease activity and shield the negative phosphate ends
of DNA that allow precipitation in ethanol respectively.
This was followed by addition is a biological detergent Sodium Dodecyl Sulphate (SDS), that
breaks down cell membranes by dissolving the lipids and proteins of the cell and disrupting
the bonds that hold the membrane together. The phenol: chloroform mixture was then added
in equal volumes to the aqueous DNA to form a biphasic mixture. The proteins precipitate
into the organic phase while the DNA remains in the aqueous phase. Ethanol precipitation is
a method used to concentrate DNA, as it is insoluble in the relatively non polar ethanol while
lipids and proteins are not. This property leads to easy separation of DNA.
Reagents:
Stock solutions:
5 M NaCl: Dissolve 292.5 gm of NaCl in 500 mL of D/W and makethe volumeup to
1000 mL with D/W.

63
0.5 M EDTA (pH 8.0): Dissolve Disodium EDTA -93.06 gm in 200 mL of D/W.
adjust pH to 8.0 with 10 N NaOH and make thevolume up to 500 mL with D/W.
1 M Tris: Dissolve Tris- 121.0 gm in 500 mL of D/W. Adjust pH to 8.0 with
concentrated HCl and make volume up to 1000 mL with D/W.
10 N NaOH:Dissolve NaOH 400 gm in 1000 mL of D/W
20 X SSC (pH 7.0):Dissolve NaCl- 175.3 gm and Sodium Citrate- 88.2 gm in 500 mL
of D/W. Adjust pH to 7.0 with 10 N NaOH and make volume up to 1000 mL with D/W.
10 % SDS:Dissolve 10 gm SDS in 100 mL D/W.
Working solution:
2X lysis buffer: 200 mM NaCl+ 50 mM EDTA
40 mL of 5 M NaCl+ 100 mL of 0.5 M EDTA. Make volume up to 1 litre with D/W.
1X lysis buffer: 100 mM NaCl + 25 mM EDTA
20 mL of 5 M NaCl + 50 mL of 0.5 M EDTA. Make volume up to 1 litre with D/W.
10 % SDS
Proteinase K: 20 mg/ml D/W (It was stored at -200C in aliquots).
Tris equilibrated phenol (pH 8.0)
Melt 100 mL of the distilled phenol at 680C in a water bath and add 8-hydroxy quinoline
to a final concentration of 0.1 %. First saturate phenol by adding an equal volume of
D/W and mix. After the phases separate on standing, aspirate the upper aqueous phase
and equilibrate the phenol with an equal volume of 1 M Tris (pH 8.0). Aspirate upper
aqueous layer and re-equiibrate phenol with 0.1 M Tris (pH 8.0). The pH of the phenolic
phase should be more than 7.8. Store this equilibrated phenol under 0.1 M Tris (pH 8.0)
in a dark bottle at 40C.
Chloroform: Octanol: Mix 24 mL of Chloroform with 1 mL of Octanol
Chilled ethanol
Tris- EDTA (TE) buffer (pH 7.4): 10 mM Tris + 1mM EDTA
1 mL of 1 M Tris (pH 7.5) + 0.2 mL of 0.5 M EDTA (pH 8.0).Adjustthe pH to 7.4 with
HCL and make the final volume up to 100 mL with D/W.
Procedure:
1. Keep 5-8 mL of EDTA blood sample at -200C overnight before the DNA extraction.
2. Allow the samples to thaw at RT for about 30 minutes to 1 hour.

64
3. Transfer blood samples to 50 mL polypropylene tubes and add 40 mL of 2X lysis buffer
and mix well the tubes.
4. Centrifuge tubes for 15 mins at 4000 rpm at 40C.
5. Drain off the supernatant by inverting the tubes and the WBC pellet retained.
6. Add 1.5 mL of 1X lysis buffer to the pellet and mix by vortexing.
7. Add 150 μl of 10 % SDS and 10 μl proteinase K and mix the tubes by vortexing.
8. Incubate samples overnight at 370C in a water bath.
9. Next day centrifuge the tubes at 4000 rpm for 2 minutes and transfer to 7 mL of plastic
tubes.
10. Add an equal volume of phenol to the sample; mix the tubes well by inversion and
centrifuge at 4000 rpm for 15 minutes.
11. Carefully transfer the upper aqueous layer into a new plastic tube. Repeat Step 10.
12. Following centrifugation again transfer the upper aqueous layer to a new plastic tube and
add anequal volume of Chloroform: Octanol.
13. Mix the tubes well by inversion and centrifuge at 4000 rpm for 5 minutes.
14. Take out the upper aqueous layer in another fresh tube.
15. To this add about 5 mL of chilled ethanol and mix by inversion.
16. Lift out the precipitated DNA fibre with a closed Pasteur pipette, air dry and dissolve in
200 to 500 μl of TE buffer (pH 7.4).
17. The quality of the DNA is checked as follows:
Checking for high molecular weight DNA:
Stock 50X TAE buffer: 242 g of Tris base + 100 mL of 0.5 M EDTA + 57.1 mL of
Glacial Acetic acid. Make thevolume up to 1000 mL with D/W.
1X TAE buffer (500 ml):Dilute 10 mL of 50X TAE buffer to 500 mL with D/W
Load 5 μl of DNA with 1 μl of loading dye on a 0.8% agarose gel with ethidium bromide
and run for 1 hour in 1X TAE buffer at 90 volts in a submergedgel electrophoresis tank.
Observe the gel under an ultraviolet transilluminator (UV). A single band near the well
fluoresces which is the DNA. When a smear is observed instead of the band it indicates
that the sample is degraded.

65
Quantification of the high molecular weight DNA:
The DNA concentration and purity is checked by diluting 5μl of DNA with 2ml of distilled
water and reading the O.D. at 260 and 280nm in a spectrophotometer.
1 OD AT 260 nm = 50 μg/ml of DNA
Concentration of DNA (50 μg/ml) = OD at 260 x 50 x dilution factor
OD ratio of 260 nm/280 nm should be between 1.6-1.8. If the ratio is less than 1.6, it
indicates protein contamination.
Polymerase Chain Reaction (PCR)
PCR is a technique used in molecular biology to amplify a region of DNA of interest to
generate thousands to millions of copies.
Principle:
It is based on the principle of thermal cycling which consists of repeated heating and cooling
of the reaction for DNA melting and enzymatic replication of DNA. Short DNA fragments
(primers) containing sequences complementary to the target region along with DNA
polymerase carry out the amplification process. The thermal cycling process physically
separates the two strands of the DNA double helix and then each strand is used as a template
to selectively amplify the region of interest. Thus, it is a chain reaction in which the DNA
template is exponentially amplified.
Components and reagents required:
DNA template: The region of interest to be amplified.
Primer set: Forward and Reserve sequences complementary to the 3’ ends of the sense
and antisense strands of the target DNA respectively.
Taq DNA polymerase: The enzymatic replication of DNA (the source of the enzyme is
the microorganism Thermusaquaticus).

66
Deoxyribonucleotide triphosphates (dNTPs): There are 4 dNTPs (dA,dT, dG, dC)
required for extension. These are building blocks from which the DNA polymerase
synthesizes the new strands.
Divalent cation generally Mg+2
: MgCl2
Buffer:Provides a suitable chemical environment for optimum activity and stability of
DNA polymerase.
PCR involves 3 main steps: (Figure 18)
Denaturation: This step is carried out at 940C- 98
0C for 20-30 sec. It causes the
denaturation of the DNA template by disrupting the hydrogen bonds between
complementary bases yielding separation of two strands of the DNA into two single
strands of DNA.
Annealing: In this step the temperature is lowered to 50-650C for 20-40 seconds. This
allows the annealing of the primers to the single stranded DNA template. The Taq DNA
polymerase binds to the primer-template hybrid and initiates DNA synthesis in the next
step.
Extension: The temperature is usually 720C for this step. The DNA polymerase
synthesizes a new DNA strand complementary to the DNA template by adding dNTPs in
the 5’ 3’ direction condensing the 5’ phosphate group of the dNTPs with the 3’
hydroxyl group at the end of the extending strand.
This cycle of 3 steps is repeated 30-35 times.
Final extension: This step is performed at a temperature of 720C (used in the extension step)
for 5-15 mins. This step ensures that any remaining single stranded DNA is fully extended.

67
Figure 18: Steps involved in PCR.
Mutation detection
I. Detection of the six common Indian β- thalassemia mutations and two
hemoglobinvariants, Hb S and Hb E by Covalent Reverse Dot Blot (CRDB)
Hybridization.
Summary and explanation of the test:
β- thalassemia is one of the commonest single gene disorders and is caused mainly by point
mutations in the β- globin gene. Six common Indian mutations namely IVS 1 nt 5 (G>C),
IVS 1 nt 1 (G>T), CD 8/9 (+G), CD 41/42 (-CTTT), CD 15 (G>A) and CD 30 (G>C) along
with two structural variants of haemoglobin, HbS and HbE are screened for by hybridization
with allele specific oligonucleotide probes followed by colour development. Using this
method we can distinguish between heterozygous, homozygous and compound heterozygous
states of β- thalassemia and these abnormal hemoglobins (Figure 19).
Reverse dot blot analysis was first described by Saiki et al in 1989 and then developed later
to screen many β-thalassemia mutations and for use in prenatal diagnosis. This method can
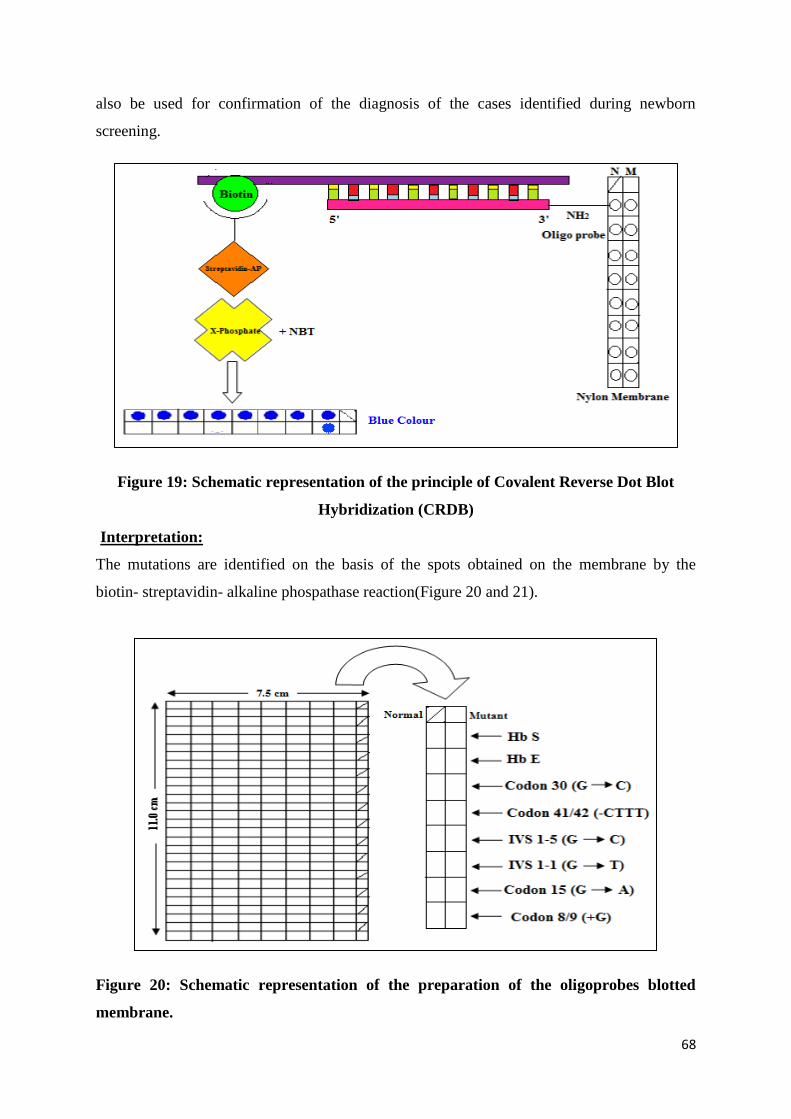
68
also be used for confirmation of the diagnosis of the cases identified during newborn
screening.
Figure 19: Schematic representation of the principle of Covalent Reverse Dot Blot
Hybridization (CRDB)
Interpretation:
The mutations are identified on the basis of the spots obtained on the membrane by the
biotin- streptavidin- alkaline phospathase reaction(Figure 20 and 21).
Figure 20: Schematic representation of the preparation of the oligoprobes blotted
membrane.

69
Figure 21: Covalent Reverse Dot Blot Hybridization showing different β-globin gene
mutations.
II. Detection of the 619 bp deletion causing β-thalassemia
The 619 bp deletion is another common β globin gene mutation in India. It is a β0 type of
mutation. It spans from exon 2 to IVS II of the β globin gene.
Principle:
A direct PCR and electrophoresis is used to detect this mutation. Two primers are used
named as 2a and 2b which amplify a 861 bp fragment from the 3’ end of the - globin gene.
Each primer lies on either side of the 619 bp deletion, therefore they directly detect this
mutation by amplifying a characteristic smaller fragment of 242 bp.
Reagents:
Primers: (Sigma Genosys, India)
Table 3: Primers used to amplify theregion of the -globin gene
Primers Sequence
2a 5’ CAA TGT ATC ATG CCT CTT TGC ACC 3’
2b 5’ GAG TCA AGG CTG AGA GAT GCA GGA 3’
Note: This method can be used as a second alternative when commercial kits become
available

70
dNTP mix (25mM)
PCR buffer
Taq Polymerase
Distilled water
Genomic DNA
PCR Reaction:
Table 4: Reagents used to detect the619 bp deletion
Reagents Concentration Volume
DNA 100-200 ng/ µl 2 µl
2a 10 picomoles/ µl 2 µl
2b 10 picomoles/ µl 2 µl
dNTP mix 25 mM 0.5μl
PCR buffer 10X containing 15mM MgCl2) 5.0μl
MgCl2 25mM 7.0μl
Taq Polymerase 5 units/ µl 0.25 μl
Distilled water -- 30.0μl
Total 50 μl
Procedure:
1. Set up PCR reactionin a single tube.
2. Add DNA
3. Vortex the tubes containing premix after addition of Taq polymerase and centrifuge. Place
the tubes in the PCR machine.
PCR Programme:
Table 5: PCR program to detect the619 bp deletion
Denaturation 940C for 5 mins
Annealing 550C for 3 mins
35 cycles of
Extension 720C for 2 min
Denaturation 940C for 1 min
Annealing 550C for 1 min
Final extension 720C for 7 mins
Hold 150C for
Check the result in 2% agarose gel.

71
Reagents
1X TAE Buffer
Gel loading dye
2% agarose gel
Procedure:
1. Add 2 µl of loading dye to the tube containing the PCR product.
2. Load the entire product on a 2% agarose gel.
3. Load the molecular weight marker VIII (Roche) side by side to check for specific
amplification.
4. Run the gel at 80-100 V for 1 hour and view under UV light on a gel documentation
system and record the results.
Interpretation:
Agarose gel electrophoresis
Figure 22: Schematic representation of detection of the 619 bp deletion mutation by
GAP- PCR.
Lane 1: DNA molecular weight marker
Lane 2: 619 bp del heterozygous
Lane 3: 619 bp del homozygous
Lane 4: Normal

72
Figure 23: 2% agarose gel electrophoresis showing the619 bp deletion.
III. Detection of β thalassemia mutations by Amplification Refractory Mutation System
(ARMS).
The amplification-refractory mutation system (ARMS) technique for detecting known point
mutations was first described by Newton et al in 1989. This method is also used to detect
known thalassemia mutations present among the Indian population.
Principle:
The technique is based on the principle of allele-specific priming of the PCR process. Allele
specific primers are used such that the nucleotide at the 3’ end of each primer is
complementary to the change of DNA sequence caused by the mutation that is being
screened. To enhance their specificity, a deliberate additional mismatch is introduced at
position - 4 from the 3’ end. Thus for every sample, two reactions are set up, one containing
the normal primer and the other containing the mutant primer. Along with these primers
another set of primers are also added which span a completely different part of the gene and
this serves as a control fragment thus confirming the amplification of the sample and that the
PCR has worked. Each of these primers is run along with the positive and negative control
DNA samples under uniform stringent PCR conditions to ensure that successful amplification
occurs only in the presence of the mutation (Figure 24).
Lane 1: DNA molecular weight marker
Lane 2: 619 bpdel heterozygous
Lane 3: 619 bpdel homozygous
Lane 4: Normal

73
Figure 24: Diagrammatic representation of principle of ARMS PCR.
Reagents:
dNTP stock Solutions (1.25mM)
60 μldATP (100 mM) + 60 µl dTTP (100 mM) + 60 µl dCTP (100 mM) + 60 µl dGTP
(100 mM) + 4760 µl of sterile D/W. Store at -200C.
ARMS stock Master Mix: Aliquots of this mix can be preserve at -20 0C
Table 6: Reagents required to prepare the mastermix for ARMS PCR
Reagents Volume
10X PCR Buffer containing 15 mM MgCl2 1000 µl
Sterile D/W 5400 µl
dNTP mix (1.25 mM) 1600 µl
Spermidin (1 M solution) Sigma 8 µl
Table 7: Primers used to detect different -globin gene mutations by ARMS
Fragment Primers Size (bp) Sequence (5’3’)
Set I
Control 2a 860 CAA TGT ATC ATG CCT CTT TGC ACC
2b GAG TCA AGG CTG AGA GAT GCA GGA
Common (Forward) CM 23 ACC TCA CCC TGT GGA GCC AC
Normal and Mutant
(Reverse)
IVS 1-1 (G>A) 281 N: TTA AAC CTG TCT TGT AAC CTT GAT ACC CAC
281 M: TTA AAC CTG TCT TGT AAC CTT GAT ACC GAT
Cd 16 (-C) 239 N: TCA CCA CCA ACT TCA TCC ACG TTC ACG TTG
238 M: TCA CCA CCA ACT TCA TCC ACG TTC ACG TTC
Cd 5 (-CT) 204 N: ACA GGG CAG TAA CGG CAG ACT TCT CCG CAG
202 M: ACA GGG CAG TAA CGG CAG ACT TCT CCG CGA
Cd 30 (G>A) 280 N: TAA ACC TGT CTT GTA ACC TTG ATA CCT ACC
280 M: TAA ACC TGT CTT GTA ACC TTG ATA CCT ACT
Cd 39 (C>T) 436 M: CAG ATC CCC AAA GGA CTC AAA GAA CCT GTA
IVS 1-5 (G>C) 285 N: CTC CTT AAA CCT GTC TTG TAA CCT TGT TAC
285 M: CTC CTT AAA CCT GTC TTG TAA CCT TGT TAG
IVS 1-1 (G>T) 281 M: TTA AAC CTG TCT TGT AAC CTT GAT ACG AAA
Cd 41/42
(-CTTT)
443 N: GAG TGG ACA GAT CCC CAA AGG ACT CAA AGA
439 M: GAG TGG ACA GAT CCC CAA AGG ACT CAA CCT
Cd 8/9 (+G) 214 N: CCT TGC CCC ACA GGG CAG TAA CGG CAC ACT
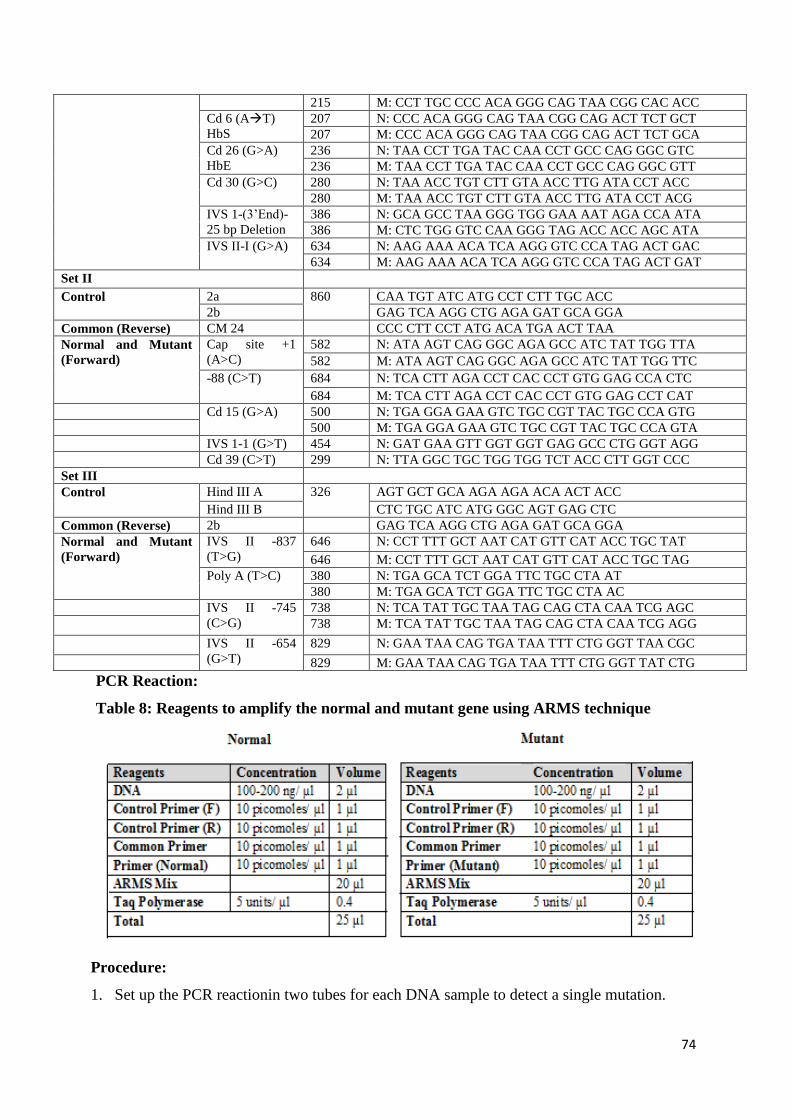
74
215 M: CCT TGC CCC ACA GGG CAG TAA CGG CAC ACC
Cd 6 (AT)
HbS
207 N: CCC ACA GGG CAG TAA CGG CAG ACT TCT GCT
207 M: CCC ACA GGG CAG TAA CGG CAG ACT TCT GCA
Cd 26 (G>A)
HbE
236 N: TAA CCT TGA TAC CAA CCT GCC CAG GGC GTC
236 M: TAA CCT TGA TAC CAA CCT GCC CAG GGC GTT
Cd 30 (G>C) 280 N: TAA ACC TGT CTT GTA ACC TTG ATA CCT ACC
280 M: TAA ACC TGT CTT GTA ACC TTG ATA CCT ACG
IVS 1-(3’End)-
25 bp Deletion
386 N: GCA GCC TAA GGG TGG GAA AAT AGA CCA ATA
386 M: CTC TGG GTC CAA GGG TAG ACC ACC AGC ATA
IVS II-I (G>A) 634 N: AAG AAA ACA TCA AGG GTC CCA TAG ACT GAC
634 M: AAG AAA ACA TCA AGG GTC CCA TAG ACT GAT
Set II
Control 2a 860 CAA TGT ATC ATG CCT CTT TGC ACC
2b GAG TCA AGG CTG AGA GAT GCA GGA
Common (Reverse) CM 24 CCC CTT CCT ATG ACA TGA ACT TAA
Normal and Mutant
(Forward)
Cap site +1
(A>C)
582 N: ATA AGT CAG GGC AGA GCC ATC TAT TGG TTA
582 M: ATA AGT CAG GGC AGA GCC ATC TAT TGG TTC
-88 (C>T) 684 N: TCA CTT AGA CCT CAC CCT GTG GAG CCA CTC
684 M: TCA CTT AGA CCT CAC CCT GTG GAG CCT CAT
Cd 15 (G>A) 500 N: TGA GGA GAA GTC TGC CGT TAC TGC CCA GTG
500 M: TGA GGA GAA GTC TGC CGT TAC TGC CCA GTA
IVS 1-1 (G>T) 454 N: GAT GAA GTT GGT GGT GAG GCC CTG GGT AGG
Cd 39 (C>T) 299 N: TTA GGC TGC TGG TGG TCT ACC CTT GGT CCC
Set III
Control Hind III A 326 AGT GCT GCA AGA AGA ACA ACT ACC
Hind III B CTC TGC ATC ATG GGC AGT GAG CTC
Common (Reverse) 2b GAG TCA AGG CTG AGA GAT GCA GGA
Normal and Mutant
(Forward)
IVS II -837
(T>G)
646 N: CCT TTT GCT AAT CAT GTT CAT ACC TGC TAT
646 M: CCT TTT GCT AAT CAT GTT CAT ACC TGC TAG
Poly A (T>C) 380 N: TGA GCA TCT GGA TTC TGC CTA AT
380 M: TGA GCA TCT GGA TTC TGC CTA AC
IVS II -745
(C>G)
738 N: TCA TAT TGC TAA TAG CAG CTA CAA TCG AGC
738 M: TCA TAT TGC TAA TAG CAG CTA CAA TCG AGG
IVS II -654
(G>T)
829 N: GAA TAA CAG TGA TAA TTT CTG GGT TAA CGC
829 M: GAA TAA CAG TGA TAA TTT CTG GGT TAT CTG
PCR Reaction:
Table 8: Reagents to amplify the normal and mutant gene using ARMS technique
Procedure:
1. Set up the PCR reactionin two tubes for each DNA sample to detect a single mutation.

75
2. Label them as normal (N) and mutant (M).
3. Add DNA.
4. Add a pair of control primers in each tube to see whether the PCR reaction is working.
5. Vortex the tubes containing premix after addition of Taq polymerase and centrifuge. Place
the tubes in the PCR machine.
PCR Programme:Table 9: PCR program for ARMS PCR
Denaturation 940C for 5 mins
25 cycles of
Denaturation 940C for 1 min
Annealing 650C for 1 min
Extension 720C for 1 min 30 sec
Final extension 720C for 3 mins
Hold 150C for
Check the result in 2% agarose gel.
Reagents
1X TAE Buffre
Gel loading dye
2% agarose gel
Procedure:
1. Add 2 µl of loading dye to the tube containing the PCR product.
2. Load the entire product on a 2% agarose gel.
3. Load the molecular weight marker VIII (Roche) side by side to check for specific
amplification.
4. Run the at 80-100 V for 1 hour and view under UV light on a gel documentation system
and record the results.
Interpretation:

76
Figure 25: Schematic representation of detection of mutations by ARMS
Figure 26: 2% agarose gel showing ARMS PCR for detection of Hb S
1: Molecular weight marker
2: Normal
3: Heterozygous
4: Homozygous
1: Molecular weight marker
2: Normal
3: HbSHeterozygous
4: HbSHomozygous
ARMS PCR will be used as the method of choice for detection of mutations causing
thalassemias and abnormal hemoglobins
77
III. Detection of Hemoglobin S and HemoglobinHbDPunjab
by Restriction Enzyme
Digestion: The main use of this PCR technique is for the diagnosis of the clinically important
Hb Variants Hb S and Hb DPunjab
.
Principle:
A small number of the β-thalassemia mutations create or abolish a restriction endonuclease
recognition site in the globin gene sequence. Hence direct detection of the mutation is
possible by amplification of a fragment of the gene containing the mutation followed by
restriction enzyme digestion and agarose gel electrophoresis (Old JM 2001).
Reagents:
Table 10: Primers used to amplify a specificregion of the-globin gene
Name of the Hb
Variant
Name of the
primer
Sequence (5’3’)
Hb S Hemat 1 F TGG TAT GGG GCC AAG AGA TA
Hemat 2 R AAC GAT CCT GAG ACT TCC ACA
Hb DPunjab
2a CAA TGT ATC ATG CCT CTT TGC ACC
2b GAG TCA AGG CTG AGA GAT GCA GGA
dNTP mix (25mM)
PCR buffer
Taq Polymerase
Distilled water
Genomic DNA
Restriction Enzymes
Dde 1: For Hb S
Eco RI: For HbDPunjab
PCR reaction:
Table 11: PCR reaction for detection of HbS and HbDPunjab
Reagents Concentration Volume
DNA 100 ng/ µl 2.5 µl
Forward primer: Hemat 1 (For HbS) 2a (For HbD) 10 pmoles/µl 2 µl
Reverse primer: Hemat 2 (For HbS) 2b (For HbD) 10 pmoles/µl 2 µl
dNTP Mix 25 mM 1 µl
PCR Buffer (Complete) 10 X 5 µl
MgCl2 25 mM 7 µl
Taq polymerase 5 u/ µl 0.5 µl
D/W 30 µl
Total 50 µl

78
Procedure:
1. Set up thePCR reaction in a single tube.
2. Add DNA
3. Vortex the tubes containing thepremix after addition of Taq polymerase and
centrifuge. Place the tubes in the PCR machine.
4. Always use known normal, homozygous and heterozygous samples as controls.
Table12: PCR programme for detection of HbS and HbDPunjab
Checking for amplification
Reagents
Stock solution: 50 X TAE buffer: 242 gm of Tris + 100 mL of 0.5 M NaCl + 57.1 mL of
Glacial acetic acid. Make thevolume up to 1 litre with D/W.
Working solution: 1 X
Gel loading dye: 125 mg of Xylene Cyanol+ 125 mg of Bromo phenol blue+ 20 gm of
Sucrose. Make thevolume up to 50 mL with D/W.
1% Agarose: Dissolve1gm of agarose in 100 mL of 1X TAE by boiling in a microwave.
Keep at RT to cool to a temperature of around 35-40 0C. Add 5 µl of Ethidium Bromide
to the molten agar and mix well. Pour the molten gel in a gel casting tray with the well
forming comb in place. Allow the gel to solidify at RT.
Procedure:
1. Mix 5 µl of the PCR product with 2 µl of gel loading dye in a loading tray.
2. Load 7 µl of total mixture (5 µl PCR product + 2 µl Dye) on 1% agarose gel.

79
3. Also load the molecular weight marker VIII (Roche) side by side to check for specific
amplification.
4. Run the gel at 80-100 V for half an hour and view under UV light on a Gel
Documentation System.
5. The remaining 45 µl of the PCR product will be used for restriction enzyme digestion.
Table 13: Restriction enzyme digestion
Reagents Concentration For HbS For HbDPunjab
Amplified Product -- 20.0 μl 10.0 μl
Buffer 10X 2.0 μl 2.0 μl
Enzyme 10U/μl 1.0 μl 1.0 μl
D/W -- 0.0 μl 7.0 μl
Total -- 23.0 μl 20.0 μl
1. Mix and centrifuge (pulse spin) the reaction mixture
2. Digest the product by keeping overnight at 370C
Checking the results in 3% agarose gel (for HbS) and 2% agarose gel (for HbDPunjab
)
Reagents
1X TAE buffer
Gel loading dye
3% and 2% agarose gel
Procedure:
1. Add 2 μl of loading dye to the digested products.
2. Load the products on the gel.
3. Load the molecular weight marker VIII (Roche) side by side to check specific band
size.
4. Always run one undigested product with the digested products to check the activity of
theenzyme.
5. Run the gel at 80-100 V for half an hour and view under UV light on a Gel
Documentation System.
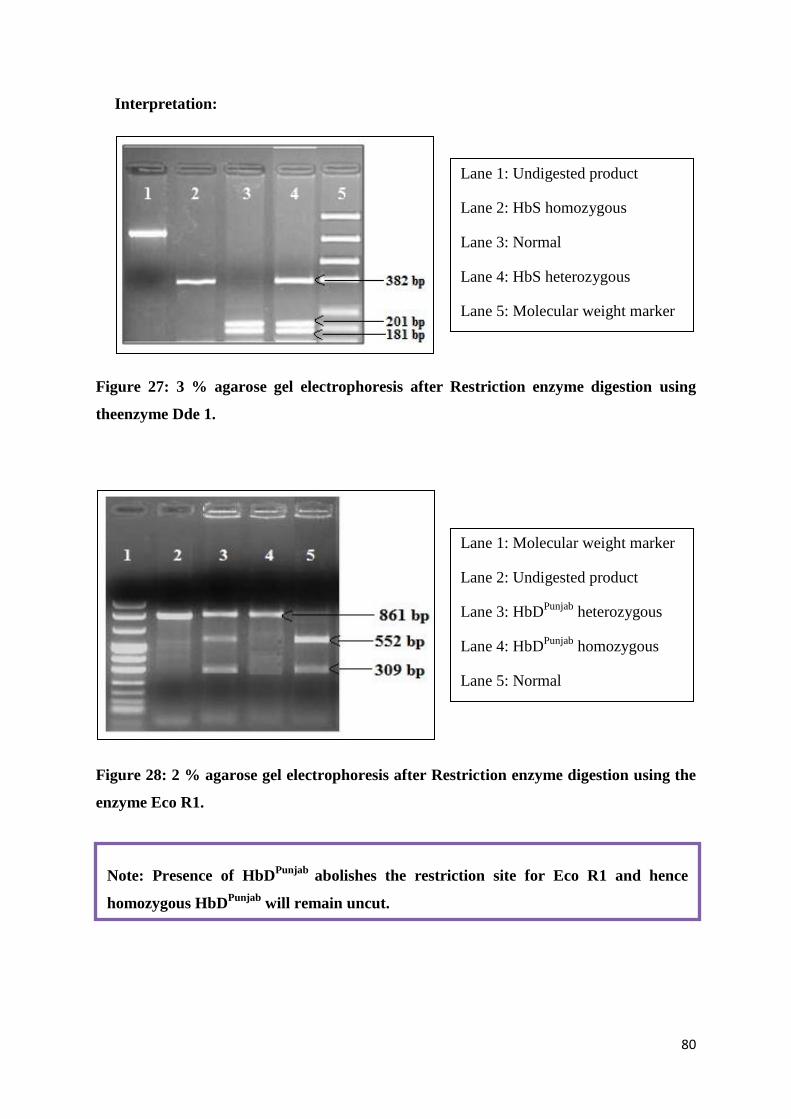
80
Interpretation:
Figure 27: 3 % agarose gel electrophoresis after Restriction enzyme digestion using
theenzyme Dde 1.
Figure 28: 2 % agarose gel electrophoresis after Restriction enzyme digestion using the
enzyme Eco R1.
Lane 1: Undigested product
Lane 2: HbS homozygous
Lane 3: Normal
Lane 4: HbS heterozygous
Lane 5: Molecular weight marker
Lane 1: Molecular weight marker
Lane 2: Undigested product
Lane 3: HbDPunjab
heterozygous
Lane 4: HbDPunjab
homozygous
Lane 5: Normal
Note: Presence of HbDPunjab
abolishes the restriction site for Eco R1 and hence
homozygous HbDPunjab
will remain uncut.
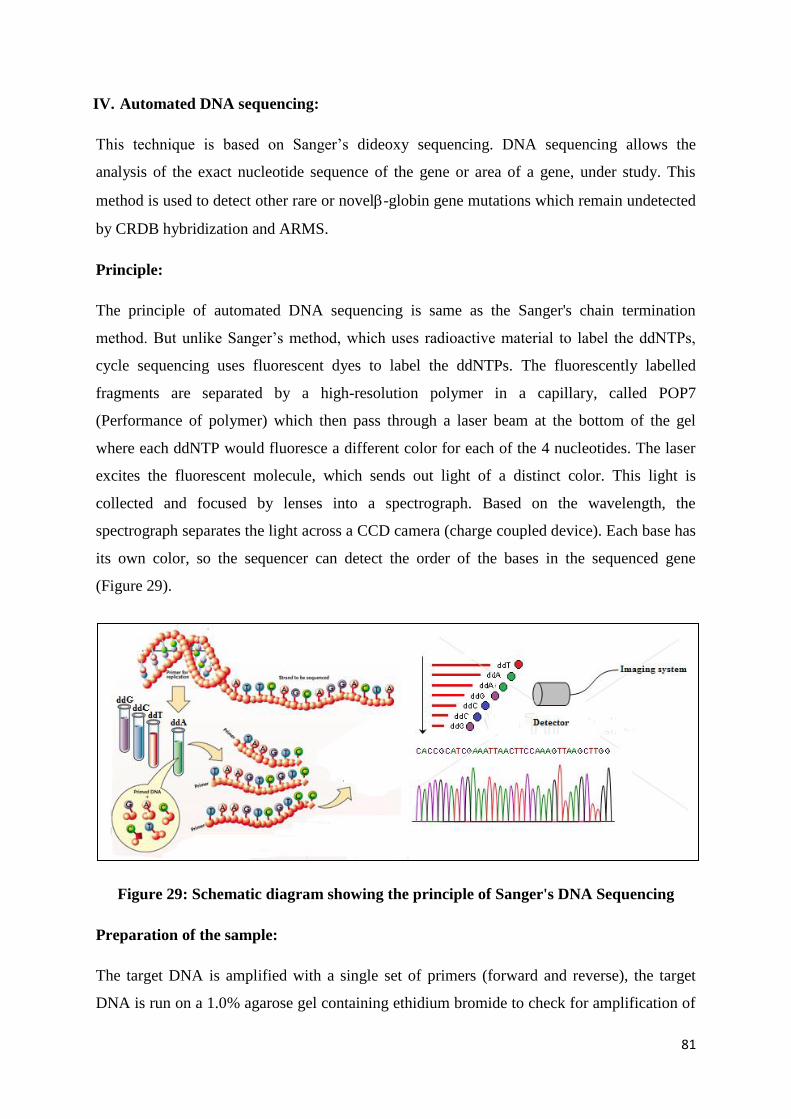
81
IV. Automated DNA sequencing:
This technique is based on Sanger’s dideoxy sequencing. DNA sequencing allows the
analysis of the exact nucleotide sequence of the gene or area of a gene, under study. This
method is used to detect other rare or novel-globin gene mutations which remain undetected
by CRDB hybridization and ARMS.
Principle:
The principle of automated DNA sequencing is same as the Sanger's chain termination
method. But unlike Sanger’s method, which uses radioactive material to label the ddNTPs,
cycle sequencing uses fluorescent dyes to label the ddNTPs. The fluorescently labelled
fragments are separated by a high-resolution polymer in a capillary, called POP7
(Performance of polymer) which then pass through a laser beam at the bottom of the gel
where each ddNTP would fluoresce a different color for each of the 4 nucleotides. The laser
excites the fluorescent molecule, which sends out light of a distinct color. This light is
collected and focused by lenses into a spectrograph. Based on the wavelength, the
spectrograph separates the light across a CCD camera (charge coupled device). Each base has
its own color, so the sequencer can detect the order of the bases in the sequenced gene
(Figure 29).
Figure 29: Schematic diagram showing the principle of Sanger's DNA Sequencing
Preparation of the sample:
The target DNA is amplified with a single set of primers (forward and reverse), the target
DNA is run on a 1.0% agarose gel containing ethidium bromide to check for amplification of

82
the PCR product. If there are no non-specific bands, the PCR product can be used directly for
sequencing, or it is purified using gel extraction (Bio Basics Canada INC. EZ-10 spin column
DNA, using the manufacturer’s instructions) or ethanol precipitation.
Amplification of target DNA:
Reagents
Primers:
Table 14: Primers used to amplify the region of interest
Primer Name Sequence (5'3') Size
(bp)
Region of the -
globin gene
amplified
β-fragment 1 F CGA TCT TCA ATA TGC TTA CCA AGC TGT GA 1400 Exon 1, IVS I,
Exon 2 and IVS II β-fragment 1 R TGG TGC AAA GAG GCA TGA TAC ATT GT
2a CAA TGT ATC ATG CCT CTT TGC ACC 860 IVS II and Exon 3
2b GAG TCA AGG CTG AGA GAT GCA GGA
dNTPs
PCR Buffer
MgCl2
TaqPolymarase
Distilled water
Genomic DNA
PCR reaction:
Table 15: Reagents required to amplify thefirst region of interest
Reagents Concentration Volume
DNA 100 ng/ µl 2.5 µl
Forward primer:β-fragment 1F/2a 10 pmoles/µl 2 µl
Reverse primer: β-fragment 1R/2b 10 pmoles/µl 2 µl
dNTP Mix 25 mM 1 µl
PCR Buffer (Complete) 10 X 5 µl
MgCl2 25 mM 7 µl
Taq polymerase 5 u/ µl 0.5 µl
D/W 30 µl
Total 50 µl
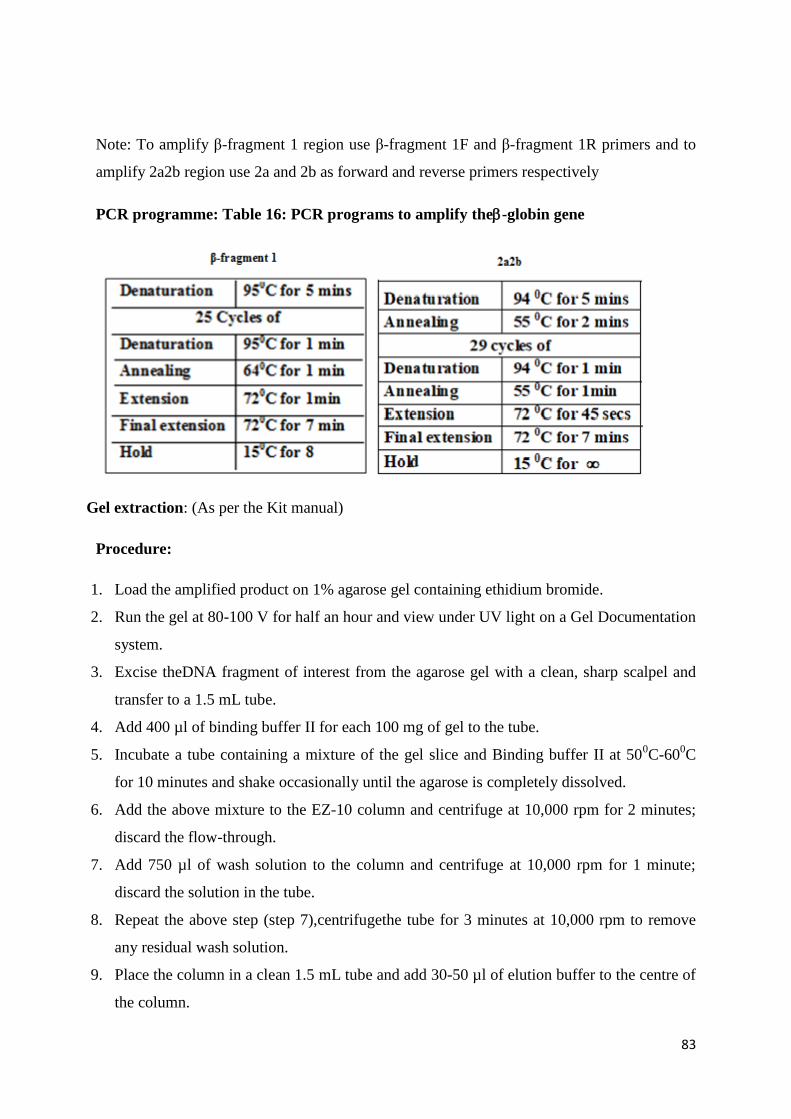
83
Note: To amplify β-fragment 1 region use β-fragment 1F and β-fragment 1R primers and to
amplify 2a2b region use 2a and 2b as forward and reverse primers respectively
PCR programme: Table 16: PCR programs to amplify the-globin gene
Gel extraction: (As per the Kit manual)
Procedure:
1. Load the amplified product on 1% agarose gel containing ethidium bromide.
2. Run the gel at 80-100 V for half an hour and view under UV light on a Gel Documentation
system.
3. Excise theDNA fragment of interest from the agarose gel with a clean, sharp scalpel and
transfer to a 1.5 mL tube.
4. Add 400 µl of binding buffer II for each 100 mg of gel to the tube.
5. Incubate a tube containing a mixture of the gel slice and Binding buffer II at 500C-60
0C
for 10 minutes and shake occasionally until the agarose is completely dissolved.
6. Add the above mixture to the EZ-10 column and centrifuge at 10,000 rpm for 2 minutes;
discard the flow-through.
7. Add 750 µl of wash solution to the column and centrifuge at 10,000 rpm for 1 minute;
discard the solution in the tube.
8. Repeat the above step (step 7),centrifugethe tube for 3 minutes at 10,000 rpm to remove
any residual wash solution.
9. Place the column in a clean 1.5 mL tube and add 30-50 µl of elution buffer to the centre of
the column.
84
10. Incubate the column for 15-20 minutes at room temperature and centrifuge at 10,000 rpm
for 3 minutes.
11. Store the purified DNA at -200C.
Cycle sequencing of the desired product:
Reagents:
Purified DNA
Big Dye Terminating sequencing Buffer (ABI 310)
Ready reaction Mix (ABI 310.)
Distilled water
PCR reaction:Table17: Reagents required forcycle sequencing
Reagents Concentration Amount
Purified DNA 100 ng 1-2 µl
Buffer 5X 2
Ready reaction mixture (RR) - 1
Primer (F/R) 3.5 picomole 1
D/W - 14
Total - 20
PCR programme:Table18: PCR program for Cycle sequencing
25 cycles
Initial Denaturation 96° C 10 secs
Annealing 60°C 4 mins
Final Hold 4°C -
Purification of the cycle sequencing product:
1. Add 80 μl of HPLC purified D/W to 20 μl of the product.
2. In a fresh eppendorf (1.5 ml), add 10.0 μl of 3M sodium acetate (pH-4.6) and 250 μl of
absolute ethanol. Add the above reaction mixture to the eppendorf and mix properly by
inverting the tubes.
3. Centrifuge the eppendorf at 14,000 rpm for 20 mins.
4. Discard the supernatant immediately after centrifugation.
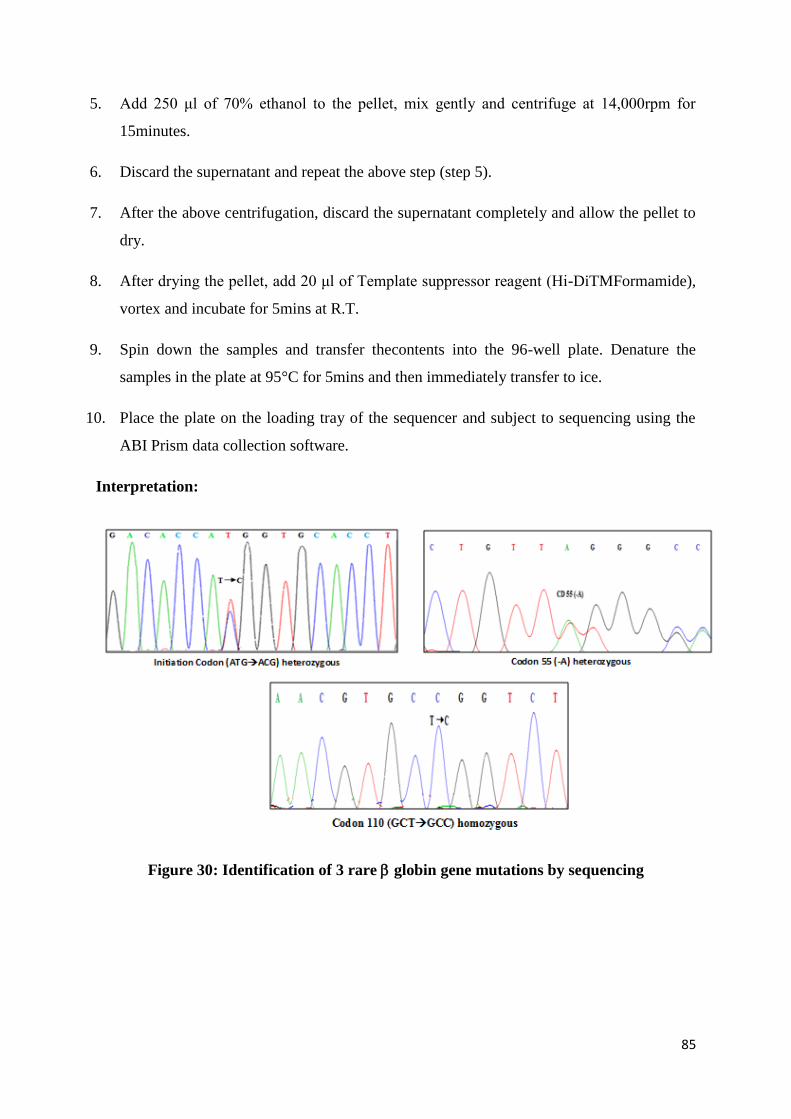
85
5. Add 250 μl of 70% ethanol to the pellet, mix gently and centrifuge at 14,000rpm for
15minutes.
6. Discard the supernatant and repeat the above step (step 5).
7. After the above centrifugation, discard the supernatant completely and allow the pellet to
dry.
8. After drying the pellet, add 20 μl of Template suppressor reagent (Hi-DiTMFormamide),
vortex and incubate for 5mins at R.T.
9. Spin down the samples and transfer thecontents into the 96-well plate. Denature the
samples in the plate at 95°C for 5mins and then immediately transfer to ice.
10. Place the plate on the loading tray of the sequencer and subject to sequencing using the
ABI Prism data collection software.
Interpretation:
Figure 30: Identification of 3 rare globin gene mutations by sequencing

86
Referances:
1. Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P. Inherited
Disorders of Hemoglobin. Weatherall D, Akinyanju O, Fucharoen S, Olivieri
N, Musgrove P.Editors In: Jamison DT, Breman JG, Measham AR, Alleyne
G, Claeson M, Evans DB, Jha P, Mills A, Musgrove P, editors. SourceDisease
Control Priorities in Developing Countries. 2nd edition. Washington (DC): World
Bank; 2006. Chapter 34.
2. http://globin.cse.psu.edu
3. Mohanty D, Colah R. Eds Laboratory Manual for Screening, Diagnosis and Molecular
analysis in Hemoglobinopathies and Red Cell Enzymopathies 1st Edition.Bhalani
Publishing House, Mumbai 2008.
4. Colah RB, Gorakshakar AC, Lu Cy, Nadkarni AH, Desai SN, Pawarf AR, Lulla CP,
Krishnamoorthy R and Mohanty D. Application of Covalent Reverse Dot Blot
Hybridization for Rapid prenatal diagnosis of the common Indian thalassemia
syndromes. Indian J Hematol Blood Transfus.1997;15:10.
5. Saiki RK, Walsh PS, Levenson CH, Erlich HA. Genetic analysis of amplified DNA
with immobilized sequence—specific oligonucleotide probes. Prot. Natl Acad Sci
USA. 1989;86: 6230-623.
6. Old JM. DNA based diagnosis of the hemoglobin disorders, In ‘Disorders of
hemoglobin: Genetics, pathophysiology and clinical management. Eds. Steinberg
MH, Forget BG, Higgs DR, Nagal RL, Cambridge University press, Cambridge pp
946-951. 2001.
7. Newton CR, Graham A and Heptinstall LE. Analysis of any point mutations in DNA.
The amplification refractory mutation system (ARMS). Nucl Acids Res 1989;17:
2503-2516
8. Sanger F, Nicfklen S, Carlson AR. DNA sequencing with chain terminating
inhibitors. Proc Natl Acad Sci 1977;74:5463-5467.
https://www.ncbi.nlm.nih.gov/pubmed?term=Akinyanju%20O%5BAuthor%5D&cauthor=true&cauthor_uid=21250308
https://www.ncbi.nlm.nih.gov/pubmed?term=Fucharoen%20S%5BAuthor%5D&cauthor=true&cauthor_uid=21250308
https://www.ncbi.nlm.nih.gov/pubmed?term=Akinyanju%20O%5BAuthor%5D&cauthor=true&cauthor_uid=21250308

87
Prenatal Diagnosis of Hemoglobinopathies
Prenatal diagnosis is a crucial component of a prevention programme for
hemoglobinopathies. It is an important option for high- risk couples where both partners are
carriers of hemoglobinopathy genes to have a normal child. The facilities for prenatal
diagnosis became available in India in mid 1980s. The techniques employed for prenatal
diagnosis include Chorionic Villus sampling (CVS) or Amniocentesis and DNA analysis or
Cordocentesis and fetal blood analysis.
Chorionic villus sampling (CVS)
This technique is used to obtain fetal tissue for prenatal diagnosis of hemoglobinopathies.
The sampling is usually done in the 1st trimester (10.5 -12 weeks of gestation). This
procedure can be done in two ways transcervical and transabdominal. The chances of
spontaneous miscarriage is more with the trancervical route hence transabdominal route is
highly preferable. CVS has an advantage that it is a first trimester procedure and hence
reduces both the emotional stress and the complications associated with 2nd
trimester
diagnosis. Also, CVS provides a good source of fetal DNA which allows quick results in few
days. The risk of fetal loss is minimal.
Amniocentesis:
Amniocentesis is done in the 2nd trimester between 14th
-16th
weeks of gestation. Around 30
to 35 mL of amniotic fluid is needed to get sufficient amount of fetal DNA for prenatal
diagnosis of hemoglobinopathies.
Amniocentesis is associated with 1% fetal loss. Sometimes there is insufficient DNA
obtained from the fluid which may require culturing of the cells and further delaying of the
diagnosis. Minor complications following amniocentesis include continuous leakage of
amniotic fluid, bleeding, and uterine irritability.
Cordocentesis:
The other name for this procedure is umbilical cord blood sampling or fetal blood sampling.
It is done between 18th
-20th
weeks of gestation for second trimester prenatal diagnosis of
hemoglobinopathies. This procedure is only done when couples at risk of having an affected
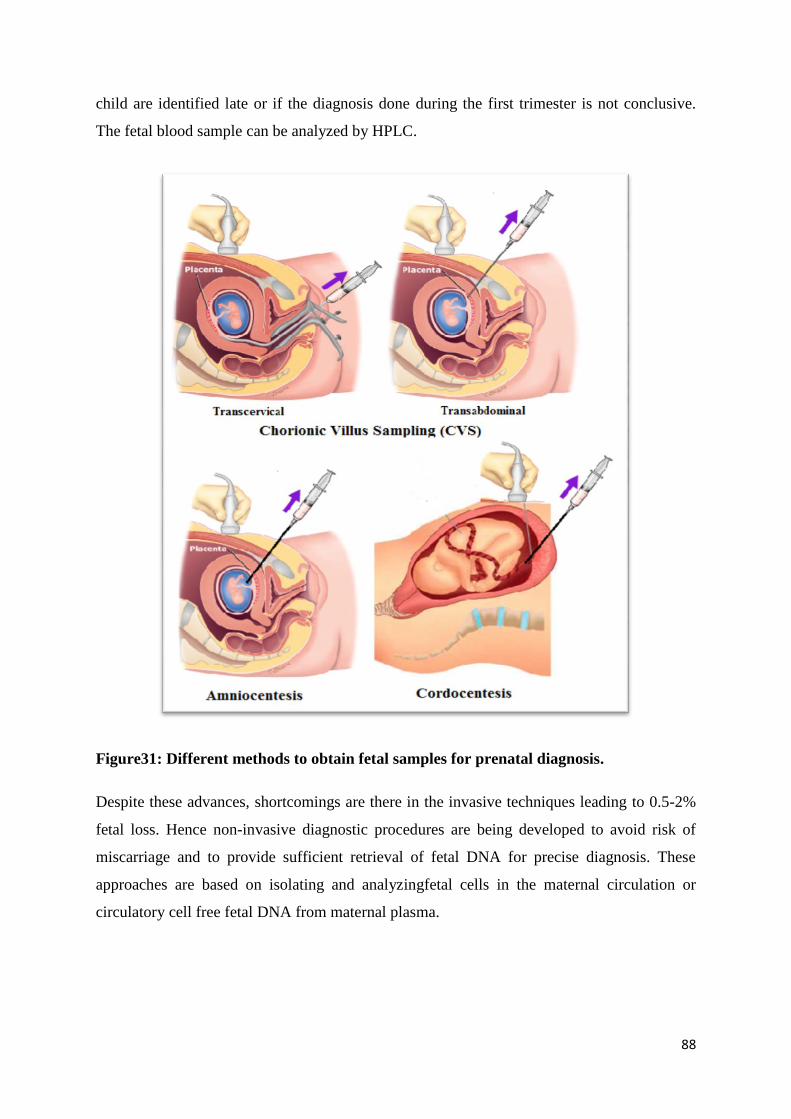
88
child are identified late or if the diagnosis done during the first trimester is not conclusive.
The fetal blood sample can be analyzed by HPLC.
Figure31: Different methods to obtain fetal samples for prenatal diagnosis.
Despite these advances, shortcomings are there in the invasive techniques leading to 0.5-2%
fetal loss. Hence non-invasive diagnostic procedures are being developed to avoid risk of
miscarriage and to provide sufficient retrieval of fetal DNA for precise diagnosis. These
approaches are based on isolating and analyzingfetal cells in the maternal circulation or
circulatory cell free fetal DNA from maternal plasma.

89
Sample Processing:
Chorionic villus sampling (CVS):
TheCVS sample isusually collected in sterile RPMI medium.
Once the sample is collected, it should be immediately sent to the respective
laboratories for further analysis.
As soon as the laboratory receives the sample, the first step is to clean the
CVS under aninverted dissecting microscope to remove maternal
contamination.
Cleaning of CVS should be done carefully since it can affect the diagnosis of
the fetus.
The cleaned CVStissueis resuspended in CVS Buffer ina 1.5 mL of
eppendorftube.
Preparation of CVS buffer
1.2 mL of 5 M NaCl + 2 mL of 0.5 M EDTA, and make thevolume up
to 40ml with sterile D/W.
Centrifuge the tube at 12000 rpm for 5 mins
Proceed further for DNA extraction
Amniotic fluid (AF):
Centrifuge the tube containing AF at 4000 rpm for 30 mins
Discard the supernatant and add normal saline ( 500μl to 1000μl) to the pelette
of amniocytes
Mix by tapping and transfer the saline with the cells in a1.5 mL eppendorftube
Centrifuge the tube at 12000 rpm for 3 mins
Discard the supernatant
Add 500-1000μl of normal saline to the cells.
Mix by tapping , Centrifuge the tube at 12000 rpm for 3 mins
Proceed further for DNA extraction
Cord blood sample/Fetal blood sample:
Cord blood sample of the fetusis collectedina heparin tube
Complete blood count should be done to know the value of MCV. It should be
>100 fl.

90
Fetal cell staining (Kleihauer –Betke method) is to be done to know the % of F
cells in the blood. There should not be any adult cell in the cord blood.
(Method is described below)
Presence of adult cells in the cord blood indicates that the cord blood is
contaminated with the maternal blood. Contaminated blood cannot be used for
further diagnosis. Repeat sampling has to be done.
The uncontaminated fetal blood is further processed for CE- HPLCto
determine the level of HbF,HbA0 and HbS (incase of Sickle cell) (Method and
interpretation are described below)
Proceed for DNA extraction
Fetal Cell Staining: Kleihauer –Betke Method
Principle: HbF is not uniformly distributed among red cells, except in the condition of
deletional HPFH. Cells with detectable amount of HbF are called F cells and they can be
detected on the blood smear by the Kleihauer-Betke acid elution test. This method is based on
elution of adult hemoglobin (HbA) from adult red cells, whereas the more acid-resistant
fetalhemoglobin (HbF) remains intact in fetal red cells. The F cells are stained pink in color
against the normal cells which appear as pale ghost cells and are colorless. It is a reliable
method for identification and quantification of maternal adult cells in the cord blood which is
aspirated in the second trimester of pregnancy for couples undergoing prenatal diagnosis by
fetal blood analysis. The Kleihauer-Betke test (KBT) is widely used for differentiating fetal
erythrocytes from the normal adult cells.
Reagents:
Fixative: 80 % ethanol: 80 mL of absolute ethanol is mixed with 20 mL of distilled
water.
Solution A: Haematoxylin:0.75 g Haematoxylin is dissolved in 100 mL of 96 %
ethanol.
Solution B: Ferric Chloride: 2.4 g Ferric Chloride (FeCl3 anhydrous) is dissolved in
distilled water, 2.0 mL of 25 % HCL is added and the volume is made up to 100 ml.

91
Elution solution :5.0mL of solution A is mixed with 1.0 mL of solution B and
filtered. This solution is prepared freshly and used for elution.
Counter Stain (2.5% Eosin): 2.5 g Eosin is dissolved in 100 mL of distilled water.
Method:
1. A smear is preparedusing a drop of fetal blood on a clean grease free slide and air
dried.
2. It is fixed in 80% ethanol in a coplin jar for 5 minutes.
3. It is then rinsed with tap water and air dried.
4. The slide is placed in the elution solution for 20 seconds; rinsed quickly with water
and counterstained with eosin in another coplin jar for exactly 2 minutes.
5. It is then rinsed with tap water and air dried.
6. The percentage of F- cells is calculated by counting 1000 cells in successive fields
under anoil immersion lens(100X)of the microscope.
Controls:
Mixed control: A drop of fetalblood is mixed with a drop of adult blood, mixed
properly and a smear is prepared.
Negative control: A drop of adult blood with very little HbF.
Positive control: A cord blood sample with a small amount of HbA.
Precaution:
Hematoxylin and FeCl3 can be prepared and stored at room temperature in dark
bottles.
The elution solution should be freshly prepared.
A thin smear is prepared to avoid cell clustering.
Interpretation: Adult cells appear as colourless ghost cells while fetal cells stain pink
A B
92
Figure 32: Fetal cell staining by Kleihauer-Betke method. A) Heterocellular B)
Pancellular
CE-HPLC forsecond trimester prenatal diagnosis of β-Thalassemia and
Sickle Cell Disease by fetal blood analysis
This method can also be used for prenatal diagnosis of β-thalassemia and Sickle cell disease
by fetal blood analysis. Based on the concentration of HbA0, HbF and HbS, one can give the
diagnosis.
Note: The fetal blood obtained by cordocentesis between 18-20 weeks of gestation
should contain 100% of fetal cells.
Preparation of the sample: (EDTA/ Heparinised blood sample)
1 mL of hemolysing solution is mixed with 5 µL of fetal blood and run on the machine.
Interpretation:
Type of hemoglobin Levels of hemoglobin Diagnosis
HbA0 0-<0.5% -thalassemia major
HbA0 >0.5- % -thalassemia trait or normal
HbS < HbA0 HbS trait
HbS >HbA0 Sickle cell disease
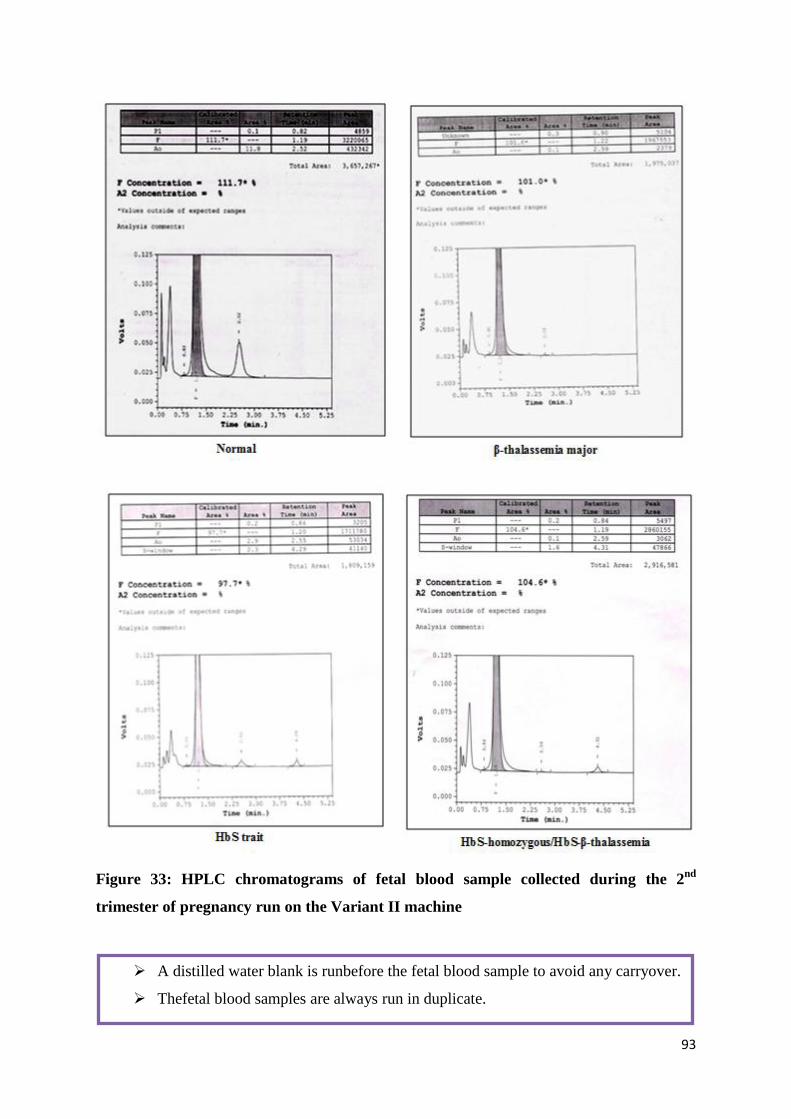
93
Figure 33: HPLC chromatograms of fetal blood sample collected during the 2nd
trimester of pregnancy run on the Variant II machine
A distilled water blank is runbefore the fetal blood sample to avoid any carryover.
Thefetal blood samples are always run in duplicate.

94
Extraction of DNA from CVS, AF and Cord blood
Variouskits for DNA extraction from tissues and cells are available commercially (eg.
Qiagen, Invitogen, Genetix etc)
Any of these kits can be used for DNA extraction for CVS, AF and cord blood.
The protocol of DNA extraction should be follow as per the manufacturer’s
instruction
The quality and quantity of the DNA can be checked by methods that are described
earlier in the chapter on Molecular diagnosis of hemoglobinopathies.
Molecular diagnosis of CVS, AF and cord blood
Depending on the mutations of the parents different molecular methods described
earlier such as ARMS, PCR-RFLP and direct DNA sequencing can be used for
detection of mutations in the fetus.
Analysis of Variable Number of Tandem Repeats (VNTRs) using PCR
A variable number tandem repeat (VNTR) is a location in a genome where a short nucleotide
sequence is organized as a tandem repeat. VNTRs are important genetic markers. Due to their
highly polymorphic nature, they are used in gene mapping, paternity analysis, and forensic
medicine and population diversity studies. Herewe describe the use of four different markers
Apo B, D1S80, ACTBP2 and Ig-JH to rule out maternal contamination in chorionic villus
samples for prenatal diagnosis.
Principle:
VNTR analysis is done by using specific pairs of primers complementary to the immediate
flanking region of these genes. It amplifies all the repeats along with the flanking region. The
product obtained by PCR is the function of a number of repeats and it can be resolved by
electrophoresis.
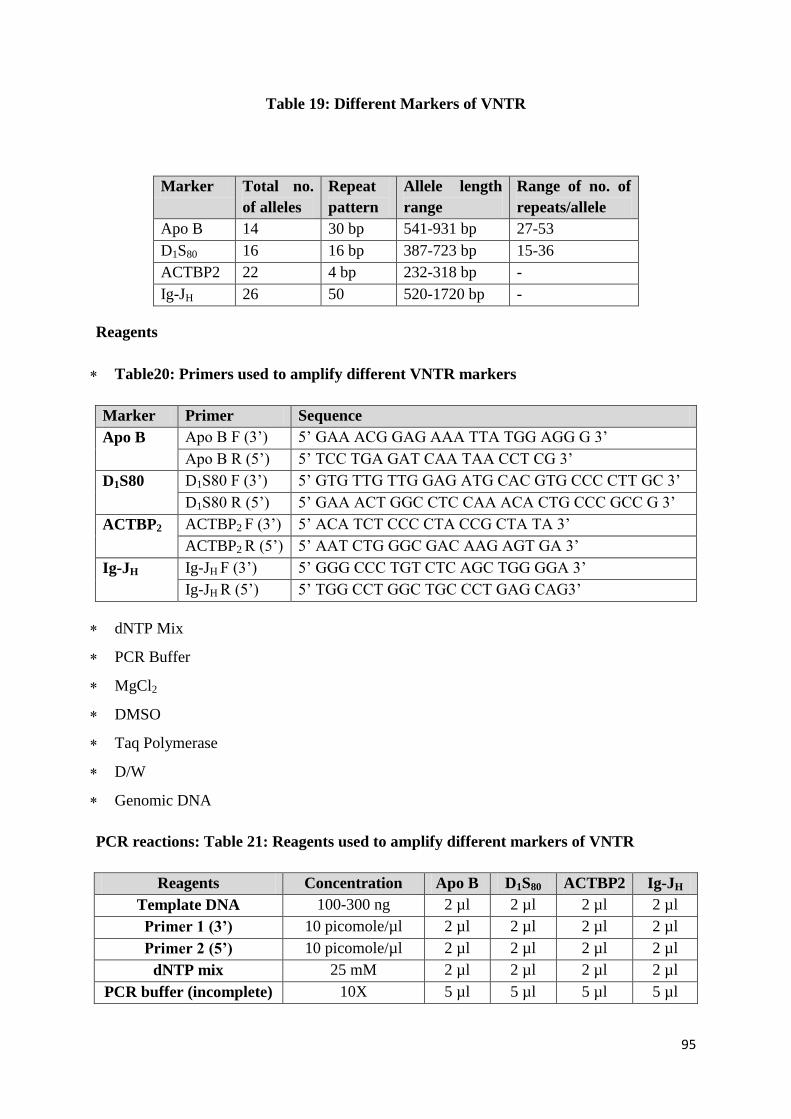
95
Table 19: Different Markers of VNTR
Marker Total no.
of alleles
Repeat
pattern
Allele length
range
Range of no. of
repeats/allele
Apo B 14 30 bp 541-931 bp 27-53
D1S80 16 16 bp 387-723 bp 15-36
ACTBP2 22 4 bp 232-318 bp -
Ig-JH 26 50 520-1720 bp -
Reagents
Table20: Primers used to amplify different VNTR markers
Marker Primer Sequence
Apo B Apo B F (3’) 5’ GAA ACG GAG AAA TTA TGG AGG G 3’
Apo B R (5’) 5’ TCC TGA GAT CAA TAA CCT CG 3’
D1S80 D1S80 F (3’) 5’ GTG TTG TTG GAG ATG CAC GTG CCC CTT GC 3’
D1S80 R (5’) 5’ GAA ACT GGC CTC CAA ACA CTG CCC GCC G 3’
ACTBP2 ACTBP2 F (3’) 5’ ACA TCT CCC CTA CCG CTA TA 3’
ACTBP2 R (5’) 5’ AAT CTG GGC GAC AAG AGT GA 3’
Ig-JH Ig-JH F (3’) 5’ GGG CCC TGT CTC AGC TGG GGA 3’
Ig-JH R (5’) 5’ TGG CCT GGC TGC CCT GAG CAG3’
dNTP Mix
PCR Buffer
MgCl2
DMSO
Taq Polymerase
D/W
Genomic DNA
PCR reactions: Table 21: Reagents used to amplify different markers of VNTR
Reagents Concentration Apo B D1S80 ACTBP2 Ig-JH
Template DNA 100-300 ng 2 µl 2 µl 2 µl 2 µl
Primer 1 (3’) 10 picomole/µl 2 µl 2 µl 2 µl 2 µl
Primer 2 (5’) 10 picomole/µl 2 µl 2 µl 2 µl 2 µl
dNTP mix 25 mM 2 µl 2 µl 2 µl 2 µl
PCR buffer (incomplete) 10X 5 µl 5 µl 5 µl 5 µl
96
MgCl2 25 mM 6 µl 7 µl 9 µl 9 µl
DMSO - - - 5 µl 5 µl
Taq Polymerase 5 Units/ µl 0.5 µl 0.5 µl 0.5 µl 0.5 µl
D/W - 30.5 µl 29.5 µl 22.5 µl 22.5 µl
Total - 50 µl 50 µl 50 µl 50 µl
PCR Programmes:
Table 22: PCR Program for Apolipoprotein B (Apo B)
Denaturation 940C for 5 Mins
25 cycles of
Annealing/Extension 580C for 3 Mins
Denaturation 94 0C for 1 Min
Annealing/ Extension 580C for 5 Mins
Final extension 600C for 10 Mins
Hold 15 0C for
Table 23: PCR program for D1S80(pMCT 118)
Denaturation 950C for 5 Mins
Annealing 650C for 3 Mins
25 Cycles of
Extension 700C for 8 Mins
Denaturation 950C for 1 Min
Annealing 65 0C for 1 Min
Final Extension 700C for 10 Mins
Hold 15 0C for
Table 24: PCR program for ACTBP2(Human - actin related pseudogene H beta
Ac-psi-2)
Denaturation 950C for 5 Mins
Annealing 550C for 2 Mins
30 Cycles of
Extension 720C for 1 Min
Denauration 940C for 20 Secs
Annealing 550C for 30 Secs
Final Extension 72 0C for 5 Mins
Hold 15 0C for
Table 25: PCR program for Ig-JH

97
Denaturation 940C for 5 Mins
Annealing 680C for 3 Mins
25 Cycles of
Denaturation 940C for 2 Mins
Annealing cum Extension 680C for 6 Mins
Hold 15 0C for
Procedure:
1. Add 2 μl of loading dye to the PCRproducts.
2. Load the entire products on a 3% agarose gel.
3. Load the molecular weight marker XIII (Roche) side by side to check specific band
size.
4. Run the gel at 80-100 V for 2-3 hour and view under UV light on a Gel
Documentation System.
Interpretation:
M: Mother; F: Father
Figure 34: 3% agarose gel electrophoresis showingtheD1S80 Marker Family 2: Non-
informative; Family 1,3,4 and 5: Informative (No maternal blood contamination)
References:
1. Kasai K, Nakamura Y, White R, Amplification of Variable number of Tandem Repeat
(VNTR) locus (pMCT118) by the polymerase chain reaction (PCR). And it’s
application to forensic science. J Forensic Sc 1990; 35:11
Centers in different states (Level 3) who will be undertaking prenatal diagnosis will
have to be registered as per the PCPNDT act in their respective states.

98
2. Klehaure E. Determination of fetalhemoglobin: elution technique. In: Schmidt RM,
Huisman THJ, Lehman H (eds). The detection of hemoglobinopathies, CRC Press,
Cleveland, OH. P 20 (1974)
3. Wadia M, Phanasgaonkar S, Nadkarni A, surve R, Gorakshakar A, colah R, Mohanty
D. Usefulness of automated chromatography for rapid fetal blood analysis for second
trimester prenatal diagnosis of beta thalassemia. Prenat. Diagn. 2002;22-153.
Quality Assurance and Equipment Maintenance in Hemoglobinopathies
Laboratory quality can be defined as accuracy, reliability and timeliness of reported test
results. The laboratory results must be as accurate as possible, all aspects of the laboratory
operations must be reliable, and reporting must be timely in order to be useful in a clinical or
public health setting.
Negative consequences of laboratory error
Laboratories produce test results that are widely used in clinical and public health settings,
and health outcomes depend on the accuracy of the testing and reporting. If inaccurate results
are provided, the consequences can be very significant, including:
unnecessary treatment
treatment complications
failure to provide the proper treatment
delay in correct diagnosis
additional and unnecessary diagnostic testing.
These consequences result in increased cost in time and personnel effort, and often in poor
patient outcomes.
Minimizing laboratory error
In order to achieve the highest level of accuracy and reliability, it is essential to perform all
processes and procedures in the laboratory in the best possible way. The laboratory is a
complex system, involving many steps of activity and many people. The complexity of the
system requires that many processes and procedures be performed properly. Therefore, the
quality management system model, which looks at the entire system, is very important for
achieving good laboratory performance.

99
Overview of the quality management system model
1. Organization
In order to have a functioning quality management system, the structure and management of
the laboratory must be organized so that quality policies can be established and implemented.
There must be a strong supporting organizational structure—management commitment is
crucial—and there must be a mechanism for implementation and monitoring.
As the laboratory moves from intent to action in the development of a quality management
system, the major organizational steps will be to assign responsibility for implementation,
allocate resources, develop and distribute a quality manual, begin implementation, and
monitor compliance with the quality policy and the quality management system requirements.
Successful implementation of a quality management system requires planning, management
commitment, understanding the benefits, engaging staff at all levels, setting realistic time
frames and looking for ways to continually improve.
2. Personnel
The most important laboratory resource is competent, motivated staff. The quality
management system addresses many elements of personnel management and oversight, and
reminds us of the importance of encouragement and motivation.
Management of personnel is critical to the success of a quality management programme.
Several elements are important in this management process. Job descriptions should reflect
all skills needed and accurately describe tasks, roles, and authorities. The competency of
personnel will need to be evaluated at the time of hiring and on a regular, recurring basis. A
very important part of the management process is to seek ways to attract qualified personnel,
and to provide motivation and appropriate benefits and working conditions so as to retain
staff.
3. Equipment
Many kinds of equipment are used in the laboratory, and each piece of equipment must be
functioning properly. Choosing the right equipment, installing it correctly, ensuring that new
equipment works properly, and having a system for maintenance are all part of the equipment
management programme in a quality management system.
4. Purchasing and inventory
The management of reagents and supplies in the laboratory is often a challenging task.
However, proper management of purchasing and inventory can produce cost savings in

100
addition to ensuring supplies and reagents are available when needed. The procedures that are
a part of management of purchasing and inventory are designed to ensure that all reagents
and supplies are of good quality, and that they are used and stored in a manner that preserves
integrity and reliability.
A well-managed laboratory will have a system for inventory maintenance and purchasing.
The system will require planning and monitoring to ensure that appropriate quantities of
supplies and reagents are always available, and to prevent wastage.
In implementing an inventory management system, the laboratory must assign responsibility
for the programme, analyze the needs of the laboratory and establish the minimum stock
needed for an appropriate time period. Appropriate logs and forms will be needed, as well as
a procedure for receiving, inspecting and storing supplies. The laboratory will need to
maintain an inventory system for all reagents and supplies used in the laboratory; this system
must include all areas where reagents and supplies are stored.
5. Process control
Process control is comprised of several factors that are important in ensuring the quality of
the laboratory testing processes. These factors include quality control for testing, appropriate
management of the sample, including collection and handling, and method verification
and validation. The elements of process control are very familiar to laboratorians; quality
control was one of the first quality practices to be used in the laboratory and continues to play
a vital role in ensuring accuracy of testing.
a) Sample management components
Written policies for sample management must be established and reflected in the laboratory
handbook. Components to be addressed include:
information needed on requisitions or forms;
handling urgent requests;
collection, labelling, preservation and transport;
safety practices (leaking or broken containers, contaminated forms, other
biohazards);
evaluating, processing and tracking samples;
storage, retention and disposal.
b) Sample processing

101
Verification of quality: Once a sample enters the laboratory, there are a number of steps
needed prior
to testing.
Verify the sample is properly labelled, adequate in quantity, in good condition and
appropriate for the test requested. The test request must be complete and include all
necessary information.
Record sample information into a register or log.
Enforce procedures for handling suboptimum samples, including sample rejection
when necessary.
Rejection of samples: The laboratory should establish rejection criteria and follow them
closely. It is sometimes difficult to reject a sample, but remember that a poor sample will not
allow for accurate results. It is the responsibility of the laboratory to enforce its policies on
sample rejection so that patient care is not compromised.
Management should regularly review the number of rejected samples and reasons for
rejections, conduct training on sample collection, and revise written procedures for sample
management as needed.
The following are examples of samples that should be rejected:
unlabelled sample;
broken or leaking tube/container;
insufficient patient information;
sample label and patient name on the test request form do not match;
haemolysed sample (depending on test requested);
non fasting samples, for tests that require fasting;
sample collected in wrong tube/container (e.g. using the wrong preservative or a
non-sterile container);
inadequate volume for the quantity of preservative;
insufficient quantity for the test requested;
prolonged transport time or other poor handling during transport.
Tracking system: The laboratory needs a system to allow for tracking a sample throughout
the laboratory from the time it is received until results are reported.
This can be done manually by careful keeping of records as follows.
Confirm receipt of samples and include date and time.

102
Label samples appropriately and keep with the test requisition until laboratory
identification is assigned.
Track aliquots—they should be traceable to the original sample.
If computers are available, maintain a database for tracking. The following information about
each sample should be entered into the database:
identification number
patient information
collection date and time
type of sample (e.g. urine, throat, cerebrospinal fluid for culture)
tests to be performed
name of ordering physician (or other health care provider)
location of patient (e.g. ward, clinic, outpatient)
diagnostic test results
time and date results are reported.
c) Sample storage, retention and disposal
Sample storage: Written policies should be developed that include:
description of what samples should be stored;
retention time;
location (consider ease of access);
conditions for storage, such as atmospheric and temperature requirements;
system for storage organization—one method is to store samples by day of receipt
or accession number.
Sample retention: Set a laboratory policy for retention of each type of sample. Some
samples can be quickly discarded and others may need to be retained for longer periods.
Monitor stored samples and do not keep for longer than necessary, as refrigerator and freezer
space may be limited. Sample freeze/thaw cycles must be monitored, as samples may
deteriorate under these conditions.
Sample Disposal: The laboratory is responsible for ensuring that disposal of all laboratory
waste is handled in a safe manner. To ensure proper disposal of patient samples:
Develop a policy for sample disposal; apply local as well as country regulations for
disposal of medical waste.
Establish and follow procedures to disinfect samples prior to disposal.

103
d) Sample transport
Need for transport: Frequently, samples are collected outside the laboratory and must be
transported for subsequent processing and testing. Transport may be for a short distance, but
sometimes a distant clinic or collection site requires the use of vehicles or aeroplanes. In
addition, it may be necessary for the laboratory to ship samples to referral laboratories. In all
cases, transport must be managed carefully in order to maintain integrity of the sample,
giving attention to temperature, preservation needs, special transport containers and time
limitations. It is also important to ensure the safety of those handling the material before,
during and after transport.
Accuracy and precision
If a measurement is repeated many times the result is a mean that is very close to the true
mean.
Accuracy is the closeness of a measurement to its true value.
Precision is the amount of variation in the measurements.
The less variation a set of measurements has, the more precise it is.
In more precise measurements, the width of the curve is smaller because the
measurements are all closer to the mean.
Bias is the difference between the expectation of a test result and an accepted reference
method.
The reliability of a method is judged in terms of accuracy and precision.
A simple way to portray precision and accuracy is to think of a target with a bull’s eye. The
bull’s eye represents the accepted reference value which is the true, unbiased value. If a set of
data is clustered around the bull’s eye, it is accurate.
The closer together the hits are, the more precise they are. If most of the hits are in the bull’s
eye, as in the figure on the left, they are both precise and accurate.
The values in the middle figure are precise but not accurate because they are clustered
together but not atthe bull’s eye. The figure on the right shows a set of hits that are
imprecise.

104
Measurements can be precise but not accurate if the values are close together but do not hit
the bull’s eye. These values are said to be biased. The middle figure demonstrates a set of
precise but biased measurements.
Figure 35: Shows the diagrammatical representation of Accuracy and Precidion.
A QC programme for quantitative tests is essential to ensuring accuracy and reliability of
laboratory testing. The laboratory must establish a QC programme that monitors all
quantitative tests. The programme will have written policies and procedures that are followed
by all laboratory staff.
The overall responsibility of managing the QC programme is usually assigned to the quality
manager, who monitors and reviews all QC data on a regular basis. The recording of the QC
data must be complete and easy to access.
For quantitative testing, statistical analysis can be used for the monitoring process, and the
use of Levey–Jennings charts provides a very useful visual tool for this monitoring.
When controls are out of range, corrective action and troubleshooting must be undertaken;
the problem must be fixed before reporting patient results. Therefore, good protocols for
troubleshooting and corrective action are an important part of the QC process.
Quality control of stains
Some stains can be purchased commercially, but others must be prepared by the laboratory,
following an established procedure. Once stains are made, their bottles should be labelled
with the following information:
name of the stain
concentration
date prepared
date placed in service
expiration date/shelf life

105
preparer’s initials.
Because of their importance, stains should be checked each day of use with positive and
negative QC materials, to make sure their reagents are active and they provide the intended
results. In most cases, positive and negative controls should be stained with each batch of
patients’ slides. All QC results must be recorded each time they are run.
Stains should also be examined to look for precipitation or crystal formation, and to check for
bacterial contamination. Careful maintenance and care of the stock and working solutions of
stains is an essential component in a system to provide good quality in microscopic
examinations.
6. Information management
The product of the laboratory is information, primarily in the form of test reporting.
Information (data) needs to be carefully managed to ensure accuracy and confidentiality, as
well as accessibility to the laboratory staff and to the health care providers. Information may
be managed and conveyed with either paper systems or with computers; both will be
discussed in the section on information management.
Information management is a system that incorporates all the processes needed for effectively
managing data—both incoming and outgoing patient information. The system can be entirely
paper-based, or it can be partly paper-based with some computer support, or it may be
entirely electronic. For either paper-based or computer systems, unique identifiers for patient
samples will be needed. Standardized test request forms, logs and worksheets are also
important to both systems. In helping to prevent transcription errors, a checking process is
beneficial.
When considering adding a computer-based system to a laboratory, cost is a big factor. In
implementation, careful planning and training will help to ensure good results.
A good information management system will:
ensure that all data—the fi nal product of the laboratory—is well managed;
consider all the ways laboratory data will be used when planning a system;
ensure the accessibility, accuracy, timeliness and security of data;
ensure confidentiality and privacy of patient information.
7. Documents and records

106
Many of the 12 quality system essentials overlap. A good example is the close relationship
between "Documents and records" and "Information management". Documents are needed in
the laboratory to inform how to do things, and laboratories always have many documents.
Records must be meticulously maintained so as to be accurate and accessible.
Documents include written policies, processes and procedures, and provide a framework for
the quality system. They need to be updated and maintained.
Records include information captured in the process of performing and reporting a laboratory
test. This information is permanent and does not require updating.
Having a good document control programme ensures that the most current version of a
document is used, and ensures availability and ease of access when a document is needed.
8. Occurrence management
An “occurrence” is an error or an event that should not have happened. A system is needed to
detect these problems or occurrences, to handle them properly, and to learn from mistakes
and take action so that they do not happen again.
Occurrence management is an integral component of laboratory quality management. It
establishes the methods for finding errors and preventing them from occurring again, and also
seeks to identify potential errors and prevent them from happening.
The laboratory should employ an active process for occurrence management and take a
positive approach. Make an effort to detect problems as early as possible, and then take
immediate remedial and corrective action. Be proactive and see opportunities to identify
potential error, thus preventing an occurrence. Finally, keep good records of all problems,
investigations and actions taken.
9. Assessment
The process of assessment is a tool for examining laboratory performance and comparing it to
standards, benchmarks or the performance of other laboratories. Assessment may be internal
(performed within the laboratory using its own staff) or it may be external (conducted by a
group or agency outside the laboratory).
Laboratory quality standards are an important part of the assessment process, serving as
benchmarks for the laboratory.

107
Auditing: The value of a well-designed audit is that it will reveal weaknesses in the pre-
examination, examination and post-examination phases. During audits, information is
gathered about:
processes and operating procedures
staff competence and training
equipment
environment
handling of samples
quality control and verification of results
recording and reporting practices.
The findings are compared with the laboratory’s internal policies and to a standard or external
benchmark. Any breakdown in the system or departure from procedures will be identified.
External quality assessment (EQA): The term EQA is used to describe a method that allows
for comparison of a laboratory’s testing to a source outside the laboratory
Several EQA methods or processes are commonly used. These include:
1. Proficiency testing—external provider sends unknown samples for testing to a set of
laboratories, and the results of all laboratories are analyzed, compared and reported to the
laboratories.
2. Rechecking or retesting—slides that have been read are rechecked by a reference
laboratory; samples that have been analyzed are retested, allowing for inter laboratory
comparison.
3. On-site evaluation—usually done when it is difficult to conduct traditional proficiency
testing or to use the rechecking/retesting method.
Participation in an EQA programme provides valuable data and information, which:
allows comparison of performance and results among different test sites;
provides early warning for systematic problems associated with kits or operations;
provides objective evidence of testing quality;
indicates areas that need improvement;
identifies training needs.
Proficiency testing:
gives a good, objective measure of the laboratory performance

108
can be organized to address most kinds of laboratory testing
is cost-effective and can therefore be used frequently.
Retesting/rechecking:
is useful when it is difficult or impossible to prepare samples to test all of the testing
process;
is expensive and uses considerable staff time.
On-site evaluation:
can give a true picture of a laboratory’s overall performance, and offer real-time guidance
for improvements that are needed;
is probably the most costly, requiring staff time, travel time and expenses of those
performing the evaluation.
10. Process improvement
The primary goal in a quality management system is continuous improvement of the
laboratory processes, and this must be done in a systematic manner. There are a number of
tools that are useful for process improvement.
The process for continual improvement includes:
identification of the problem;
analysis of the data and the processes;
determination of the root cause of the problem;
generation of ideas for solutions.
Continual improvement is the core of quality management, but it requires commitment,
planning, structure, leadership, participation and engagement.
11. Customer service
The concept of customer service has often been overlooked in laboratory practice. However,
it is important to note that the laboratory is a service organization; therefore, it is essential
that clients of the laboratory receive what they need. The laboratory should understand who
the customers are, and should assess their needs and use customer feedback for making
improvements.

109
Seeking customer satisfaction requires commitment from the laboratory management and
staff. It is important to remember that technical competency is not the only goal for the
laboratory. A programme for addressing customer satisfaction requires good planning, the
development of appropriate monitoring tools, and the knowledge to apply the tools to gain
usable information.
Customers or clients of the laboratory include physicians and other health care providers,
hospital and clinic staff, patients and their families, public health officials and the general
community.
Monitoring customer satisfaction requires some resources, primarily involving staff time.
Managers need to ensure that these resources are available.
12. Facilities and safety
Many factors must be a part of the quality management of facilities and safety.
These include:
Security—which is the process of preventing unwanted risks and hazards from entering the
laboratory space.
Containment—which seeks to minimize risks and prevent hazards from leaving the
laboratory space and causing harm to the community.
Safety—which includes policies and procedures to prevent harm to workers, visitors and
the community.
Ergonomics—which addresses facility and equipment adaptation to allow safe and healthy
working conditions at the laboratory site.
When designing a laboratory or organizing workflow, ensure that patients and patient
samples do not have common pathways. To identify where improvements in laboratory
design may be needed in order to prevent or reduce risks of cross contamination, follow the
path of the sample as it moves through the laboratory during the pre-examination,
examination and post-examination phases of testing. The design of laboratory work areas
should ensure proper ventilation and surfaces that can be cleaned and disinfected. In
establishing a safety management programme, it is important to appoint a responsible
supervisor. The laboratory should have a safety manual that establishes policy and describes
standard procedures for handling safety and emergency issues. Personnel need to be trained
in how to apply safety practices and techniques, and to be aware of potential hazards.

110
Quality Assurance for Hemoglobinopathy
1. Hematology Analyzer
For intra‐instrument quality control (internal quality control), the measurement of quality
control samples on a daily basis (or more often) is obligatory. Usually, these quality control
samples are obtained from the manufacturer and they consist of two or three levels (low,
medium, high). There are several pitfalls. First, these samples are usually manipulated to
lengthen the shelf life; therefore, they may behave differently than ordinary patient material.
Second, the manufacturer target's limits are often very broad, and subtle changes in
analyzerbehavior may thus be missed. Therefore, it is recommended to adjust the target range
after a run‐in period of several measurements, for example, to the mean ± 2 standard
deviations. A benefit in using these quality control samples is that they may be used to judge
the instrument precision over time (i.e., drift) using a Levey–Jennings graph; however, it
must be kept in mind that at the end of the shelf life the quality of the control samples may
deteriorate.
An attractive alternative approach is the use of the so‐called moving average. This statistical
method uses the fact that the analytical parameters of the CBC in a large population are stable
over time, changes in the average value thus possibly represent an analytical problem. This
method has proven to be very robust and cheap; however it cannot fully replace the use of
quality control samples.
If a laboratory has multiple HA and/or different analytical techniques that are used for the
same parameter, inter-instrument quality control should be performed. All methods should
lead to the same result in each sample. If different methods lead to unexpected discrepancies
between reported results, this may lead to misinterpretation and possibly unnecessary clinical
intervention. To assure harmonization of reported results between different analyzers and
methods, an inter-instrument quality control comparison is needed. Usually this can be
achieved by measuring patient samples (thereby avoiding so‐called matrix effect of stabilized
quality control samples) on these multiple systems and comparing the results. It is
recommended that multiple HA are compared at least on a weekly basis, using at least three
samples or more.
The participation in an External Quality Assessment (EQA) Scheme (also known as
proficiency testing scheme) is mandatory. The EQA samples should be handled in the same

111
way as routine patient samples to acquire a fair comparison. EQA usually uses manipulated
samples which are composed to resemble pathological samples (e.g., extremely high or low
cell counts). The samples are manipulated to lengthen the shelf life and reduce the sensitivity
of the sample to transport related problems (temperature fluctuations and vibration).
Therefore, direct comparison with patient results is not possible. Laboratories must use the
EQA results to compare their results with (inter)national consensus or reference results and
use these to improve their quality and to harmonize their HA with a consensus group. It must
be kept in mind that, in the absence of a reference method, the results should be compared
with a group of HA that use the same analytical technique, as consensus values may be
systematically biased by inaccuracy of most dominant group method. Moreover, EQA
material should not be used for calibration purposes (because a EQA consensus value and a
true value (calibrator) are not necessarily similar). In some countries, regulatory agencies
abuse the EQA results to evaluate the quality of the participating laboratory. This has a
negative influence on the self‐improvement of the laboratories, and this practice should be
discouraged.
2. High Pressure Liquid Chromatography
Quality assurance Controls should be used with each electrophoretic run, and should contain
the common haemoglobin variants as well as the normal haemoglobins. A control mixture of
HbF, HbA, HbS and HbC is widely used. UK NEQAS (UK National External Quality
Assessment Scheme) circulates samples for the identi¢cation of abnormal haemoglobins and
for the measurement of HbA2, HbF and HbS. Participation in these external quality
assessment programmes is a requirement for Clinical Pathology Accreditation of
haemoglobinopathy laboratories. Commercial controls are available from several
manufacturers for both HbA2 and HbF quantitation and as electrophoretic/chromatographic
markers for haemoglobins such as HbA, HbF, HbA2, HbS, HbC, HbD and HbE. Reference
materials certi¢ed by the World Health Organization are available from the National Institute
of Biological Standards and Controls to assist in the calibration of methods and instruments
used to quantitate HbA2 and HbF
Equipment Management

112
Equipment management is one of the essential elements of a quality management system.
Proper management of the equipment in the laboratory is necessary to ensure accurate,
reliable and timely testing.
The benefits of a good equipment management programme are many:
helps to maintain a high level of laboratory performance;
reduces variation in test results, and improves the technologist’s confidence in the
accuracy of testing results;
lowers repair costs, as fewer repairs will be needed for a well-maintained
instrument; lengthens instrument life;
reduces interruption of services due to breakdowns and failures;
increases safety for workers;
produces greater customer satisfaction.
A great deal of thought and planning should go into equipment management. As the
laboratory puts an equipment management programme in place, the following elements
should be considered.
Selection and purchasing—when obtaining new equipment, what criteria should be used to
select equipment? Should equipment be purchased or would it be better to lease?
Installation—For new equipment, what are the installation requirements and who will
install the new instrument?
Calibration and performance evaluation—What is needed to calibrate the equipment and
validate that it is operating correctly? How will these important procedures be conducted for
both old and new instruments?
Maintenance—What maintenance schedule is recommended by the manufacturer? Will
the laboratory need additional preventive maintenance procedures? Are current maintenance
procedures being conducted properly?
Troubleshooting—Is there a clear procedure for troubleshooting for each instrument?
Service and repair—What is the cost? Can the laboratory obtain the necessary service and
repair in its geographical area?
Retiring and disposing of equipment—What must be done to dispose of old equipment
when it needs to be replaced?
It is the responsibility of the laboratory director to:
oversee all the equipment management systems in the laboratory;

113
ensure that all persons who will be using the instruments have been appropriately trained
and understand how to both properly operate the instrument and perform all necessary routine
maintenance procedures.
Equipment management responsibility may be specifically assigned to a technologist in the
laboratory. In many laboratories, there is a person who has good skills with equipment
maintenance and troubleshooting. Giving this person the role of oversight of all equipment is
recommended.
Oversight of an equipment management programme includes:
assigning responsibilities for all activities
ensuring that all personnel are trained in operation and maintenance
monitoring the equipment management activities, including
- reviewing all equipment records routinely
- updating maintenance procedures as necessary
- ensuring that all procedures are followed.
Equipment Maintenance
1. Preventive maintenance
Preventive maintenance includes measures such as systematic and routine cleaning,
adjustment and replacement of equipment parts at scheduled intervals. Manufacturers
generally recommend a set of equipment maintenance tasks that should be performed at
regular intervals: daily, weekly, monthly or yearly.
Following these recommendations will ensure that the equipment performs at maximum
efficiency and will increase the lifespan of the equipment. This will also help to prevent:
inaccurate test results due to equipment failure
delays in reporting results
low productivity
large repair costs.
2. Equipment Calibration
Follow the manufacturer’s directions carefully when performing the initial calibration of the
instrument. It is a good idea to calibrate the instrument with each test run, when first putting
it into service. Determine how often the instrument will need to be recalibrated, based on its
stability and the manufacturer’s recommendation. It may be advantageous to use calibrators
provided by or purchased from the manufacturer.

114
3. Performance evaluation
Prior to testing patient specimens, it is important to evaluate the performance of new
equipment to ensure it is working correctly with respect to accuracy and precision. In
addition, test methods using kits or laboratory instruments need to be evaluated for the ability
to detect disease (sensitivity, specificity, positive and negative predictive value) and to
determine normal and reportable ranges.
Verification of manufacturers’ performance claims—Manufacturers provide performance
evaluations for testing methods using their kits or instruments, and include the information in
the package inserts or operator's manuals. However, laboratories need to verify the
manufacturer's performance claims, and demonstrate they can get the same results using the
kits or equipment in their laboratory, with their personnel.
Some of the steps that should be followed to verify performance include:
testing samples with known values and comparing the results to the expected or certified
value;
if equipment is temperature controlled, establishing the stability and uniformity of the
temperature.
Validation of new equipment and associated techniques—If the equipment and associated
techniques are new, validation processes will be important. Validation can be carried out by
running samples in parallel using both old and new equipment and methods for a period of
time to determine that the expected results can be obtained. These validation procedures
should be completely recorded.
4. Maintenance plan
A maintenance plan will include preventive maintenance procedures as well as provision for
inventory, troubleshooting and repair of equipment. When implementing an equipment
maintenance program, some of the initial steps will include:
assigning responsibility for providing oversight;
developing written policies and procedures for maintaining equipment, including routine
maintenance plans for each piece of equipment that specify the frequency with which all
maintenance tasks should be performed;
developing the format for records, creating logs and forms, and establishing the processes
to maintain records;

115
training staff on the use and maintenance of the equipment, and ensuring that all staff
understands their specific responsibilities.
It is recommended that a label is attached to the instrument indicating when the next
maintenance or service should be performed. The laboratory should keep an inventory log of
all equipment in the laboratory. The log should be updated with information on new
equipment and include documentation of when old equipment is retired. For each piece of
equipment, the equipment inventory log should have a record of:
instrument type, make and model number, and serial number so that any problems can be
discussed with the manufacturer;
date the equipment was purchased, and whether it was purchased new, used or
reconditioned;
manufacturer/vendor contact information;
presence or absence of documentation, spare parts and maintenance contract;
warranty’s expiration date;
specific inventory number indicating the year of acquisition (this is especially useful for
larger laboratories); for example, use the style “YY-number” (04-001, 04-002, etc.) where
“YY-number” equals the last two numbers of the year followed by a number attributed in the
year.
An inventory process must be conducted if the laboratory does not have an existing inventory
system for equipment. This could be conveniently organized following a model grid, room by
room; for example, conduct an inventory of equipment in the reception area, then the sample
collection area, the serology testing area, and the parasitology testing area. During the
inventory, the condition of the equipment should be documented as functional, partially
functional or nonfunctional. Equipment that is not functioning needs to be evaluated as
towhether or not it can be repaired. Nonrepairable equipment should be retired, and work
should be scheduled for equipment needing repair.
5. Inventory of spare parts
To ensure that the laboratory does not run out of spare parts, an inventory record of those
used most frequently should be kept for each piece of equipment.
The record should include:
part name and number;
average use of the part, and the minimum to keep on hand;

116
cost;
date when the part is placed into storage and when it is used (in and out stock log);
quantity of each part remaining in inventory.
6. Maintenance documentation
Equipment documents and records are an essential part of the quality system. The policies
and procedures for maintenance should be defined in appropriate documents, and keeping
good equipment records will allow for thorough evaluation of any problems that arise
Each major piece of equipment will have its own equipment maintenance document. Smaller,
commonly used equipment such as centrifuges and pipettes may be managed with an
equipment maintenance document or manual that deals with all such equipment in the
laboratory. An equipment maintenance document should include:
step-by-step instructions for routine maintenance, including frequency of performance and
how to keep records of maintenance;
instructions for carrying out function checks, frequency of performance, and how to record
the results;
directions for calibrating the instrument;
guide for troubleshooting;
any required manufacturer’s service and repair;
list of any specific items needed for use and maintenance, such as spare parts.
For major equipment, include identification of the specific instrument and perhaps
information on its performance.
Each piece of equipment should have a dedicated logbook documenting all characteristics
and maintenance elements, including:
preventive maintenance activities and schedule;
recording of function checks and calibration;
any maintenance performed by the manufacturer;
full information on any problem that the instrument develops, the subsequent
troubleshooting activity and follow-up information regarding resolution of the problem. In
recording problems, be sure to record
- date problem occurred and when equipment was removed from service;
- reason for breakdown or failure;
- corrective action taken, including a note about any service provided by the manufacturer;

117
- date returned to use;
- any changes to procedure for maintenance or function checks as a result of the problem.
Hematology Analyzer maintenance
Validation: According to ISO 15189, the laboratory should only implement HA that are
validated for the intended purpose.
First, certain parameters must be compared to a reference method. Most of the current
reference methods are outdated such as hematocrit, red blood cell count, white blood cell
count, and white blood cell differential. In such cases, improved performance of the new
method is compared or calibrated to a relatively poor standard, which leads to a poorer
performance than analytical and technically feasible. Unfortunately, in some countries, the
registration authorities demand a comparison with these reference methods and/or with a
current generation analyzer. This requirement could thus lead to poorer analytical
performance than necessary and also hampers innovation.
Second, it is challenging to compare the ‘flagging’ of a new instrument with an existing
method. Samples are for instance ‘flagged’ to identify pathological samples that need rerun in
a different mode, revision, or additional diagnostic work‐up (i.e., microscopic review or flow
cytometry). The underlying algorithms that generate these flags may be totally different,
making a comparison between two types of HA challenging. Usually manufacturers try to
make sure that all pathological samples are recognized, for example, the samples with
malignant WBCs (blasts). To avoid missing these pathological samples, the flagging
algorithm generates flags with a low positive predictive value (PPV) [and usually a high
negative predictive value (NPV)], that is, a lot of normal samples get a suspicious flagging
and need (microscopic) review, thereby unnecessary increasing the workload of the
laboratory.
Lastly, establishing reference intervals by manufacturers can be arduously, especially in the
pediatric population. Obtaining samples from healthy children and neonates, has shown to be
difficult. Reference intervals in the pediatric population are often based on publications from
decades ago, using technology that is no longer widely available, making it questionable
whether these reference ranges can be applied in the present time; or there are new
parameters or parameters using a new analytical technique for which there are no published

118
reference ranges available. Therefore, manufacturers should make considerable efforts to
generate reference intervals in these groups for their new HA.
Verification
The ISO 15189 states that the laboratory shall verify upon installation and before use that the
equipment is capable of achieving the necessary performance and that it complies with
requirements relevant to any examinations concerned. Both the ICSH and CSLI have
developed guidelines for the verification of HA. According to these guidelines, verification
studies should be performed on precision (reproducibility), accuracy (method comparison),
analytical sensitivity (limit of detection), analytical specificity including interfering
substances, reference ranges, patient correlation studies, linearity, limits of detection,
carryover, and the analytical measuring range. However, the ICSH and CLSI guidelines do
not specify the limits of acceptability of these studies throughout the different parameters.
Nor do these guidelines suggest a minimal range in which the reliability of the parameters
must be established. The extensiveness of the verification study depends upon the availability
of independent evaluation data (preferably published in the peer‐reviewed literature), the
extent of CBC parameters reported by the laboratory, and the range of samples available for
the evaluation of the equipment. As hematology parameters include cell differentiations and
cell indices, some (pre)analytical characteristics differ from more regular laboratory tests
expressed per concentration plasma. Therefore, some in‐depth knowledge about hematology
and hematology parameters is required to interpret the verification analyses.
Before starting a verification process, we should establish the medically allowable error. The
combination of different aspects of test performance forms the total analytical error. The total
analytical error reflects the analytic reliability of a test result. If a laboratory wants to verify
that it produces reliable results in a clinical perspective, there are several considerations.
Firstly, it is advised to establish which levels are clinically relevant. For instance, if platelet
transfusions are given below a platelet count of 10 × 109/L, it is sensible to verify the test
performance around this concentration. Secondly, the total analytical error around this cut off
point should be acceptable for clinical decision making. This medically allowable error may
depend on the expectations of the clinicians, the biological variation, and the current
state‐of‐the‐art performance.

119
Precision
Precision is the closeness of agreement between repeated measurements of a sample.
Within‐run precision (also known as reproducibility or repeatability) usually consists of a
single run of 20 measurements and is reported as a coefficient of variation [CV (%)].
Between‐batch precision [reported as CV (%)] is based on single measurements that are
repeated every day for a 20‐day period and is affected by random error and drift. Usually,
stabilized quality control samples are used to establish the between‐batch precision. The
manufacturers' claims about both within‐run and between‐batch precision must be verified. In
addition, we should always attempt to reach at least the current state‐of‐the‐art CV (%) for
reproducibility, so we can provide physicians with the most accurate information and thereby
provide opportunities to improve medical decision making and quality of care.
Accuracy and comparability
Accuracy (also known as trueness) is used to describe the closeness of a measurement to the
true value. The use of a ‘true value’ for the CBC is hard to apply in daily laboratory practice,
as reference methods only exists for a few selected parameters and these reference methods
tend to be rather impractical. For the leukocyte differential count, the microscopic evaluation
of a slide is currently regarded as the reference method (400‐cell manual differential).
However, this method suffers from several disadvantages such as statistical error, slide
distribution error, and morphological interpretation error. As a surrogate, usually in a
verification process, the HA under evaluation is compared with the instrument in routine. In
the comparability study (patient correlation studies), as many samples as possible (generally
400 samples or more for each parameter) should be compared using both HA. Furthermore, it
is of importance to include normal and abnormal samples in approximate equal proportions.
The difference between the HA under evaluation and the current analyzer or reference
method should be as low as possible. The parameter from the HA under evaluation can be
compared with the current HA using linear regression. Perfect concordance results in a
correlation coefficient (r value) of 1. Yet, if the new HA is much more accurate than the old
HA, this will negatively affect the correlation coefficient, and thus, the correlation coefficient
should be interpreted with caution. In addition, some parameters are known for their poor
correlation, such as the basophil count.
HPLC Maintenance

120
HPLC is the sensitive and high cost equipment. So, we need to care about regular
maintenance of HPLC. It leads to reducing the cost due to routine problems. The equipment
should be inspected weekly for signs of leaks. Prior to any analysis, a system suitability test,
which closely resembles the intended assay, should be performed to ensure that the system is
operating within establish criteria. Before using mobile phase solvents one should be
thoroughly familiar with hazards and safe handling practices. Observe the manufacturer
recommendations for use, storage and disposal. These recommendations are normally
provided in material safety data sheets supplied with the solvents. The storage condition is
one of the important parameter to maintain the HPLC. One should not operate the system in a
cold room or refrigerated area. The ambient temperature is 10-35◦C, ambient relative
humidity is 20-80%. It may vary depending on manufacturers. Some ofthe basic assumptions
to maintain HPLC are, the HPLC is plugged in and turned on, solvent is in the reservoir, The
detector hasa good lamp in it, the solvent bottle does not have a vacuum on it, should not use
acetone at 195nnm, should not use the water and hexane as gradient, should not use methanol
and water without degassing, solvent pH should not exceed 13 on a silica base column,
should not blow HCL vapours into HPLC, should not flush system with methanol after
running buffer, do not filter organic solvents through organic solvents, do not try to change
the column frits while it still has pressure in it, do not pump cyclohexane above 2000 psi and
don’ttighten the mobile phase container. The instrument should be frequently
calibratedusingtheappropriate procedure.
Calibrators and Controls
All HPLC runs are preceded by priming and then calibration of theinstrument. Separate
calibration factors are obtained for HbA2 and HbF as ratios of expected to obtained values.
Since the two values should ideally be equal (i.e., a ratio of 1) these are deemed to have
passed if they lie between 0.7 and 1.3. These calibration factors are then applied for all
subsequent patient samples. The retention time of HbA2 in the calibrator is also a useful
indicator of run reliability. Normally it lies between 2.60-2.70 min, and the instrument may
need temperature adjustments if wider deviations occur. This is especially common as the
column cartridge ages; usual cartridge lifetimes being around 250 injections. Bi-level
controls, one normal (HbF 1%-2%, HbA2 1.8%-3.2%) and one elevated (HbF 5%-10%,
HbA2 4%-6%), should be analyzed at the beginning as well as the end of each set of patient
specimens. The high control in case of Bio-Rad instruments also contains a variant peak that

121
must elute in the S-window. All peaks must be symmetrical, temperature variations being the
most common cause again of asymmetry.
References
1. ISO 9000:2005. Quality management systems–fundamentals and vocabulary. Geneva,
International Organization for Standardization, 2005.
2. CLSI. User protocol for evaluation of qualitative test performance, approved
guideline—2nd ed.
3. Cochran C. The five keys to a successful internal audit program. The Auditor 2:1.
Chico, CA, Paton Press, 2007 (http://www.dnvcert.com/DNV/Certifi
cation1/Resources1/Articles/NewsletterInfo/FiveKeystoaSuccessfulI/).
4. Kusum M, Silva P. Quality standards in health laboratories implementation in
Thailand: a novel approach. World Health Organization Regional Office for South-
East Asia, 2005 (http://www.searo.who.int/LinkFiles/Publications_SEA-HLM-
386__a4___2_.pdf
5. WHO.Accreditation of health laboratories in the countries of the SEA region: report
of a regional consultation, Bangkok, Thailand, 6–10 October, 2003. WHO Project:
ICP BCT 001, World Health Organization Regional Office for South-East Asia, 2004.
6. WHO Quality Assurance in Hematology 1998
7. Statistical methods for proficiency Testing ( NABL& Indian Statistical Institute)2003
8. Interlaboratory tests in :- Wenclawiak, Koch, Hadjicostas (eds). Quality Assurance in
Analytical Chemistry – Training and Teaching (2nd Edition) Springer verlag. Berlin
Heidelberg 2010
9. ISO 13528:2015, Statistical methods for use in proficiency testing by inter-laboratory
comparison: International standard.
10. Laboratory quality management system: handbook of WHO2011

122
Genetic Counseling for Hemoglobinopathies
Genetic counseling can be defined as:
A communication process by skilful counselors or medical social workers who have had
adequate training to provide often difficult to comprehend information and support to
individuals and families about genetic risks, testing and diagnosis of hemoglobinopathies,
consequences of the disorder and ways of preventing further birth of affected children with an
inherited hemoglobin disorder in the family.
Genetic counseling should help families understand and adapt to the medical,
psychological, and familial implications of the disease, or carrier status.
A trained genetic counselor can provide many important functions for the family;
however we have few such experts in the country. Armed with important and complete
information about the disorder, other trained healthcare professionals can also provide
this valuable support.
Genetic counseling is required:
Before undergoing screening for a hemoglobinopathy trait.
After diagnosis of a carrier state or major disease at different stages.
Prior to pregnancy and/or as early in pregnancy as possible.
After prenatal diagnosis to inform the couple about the diagnosis of the fetus.
Prior to and after newborn screening for a hemoglobinopathy eg sickle cell disease.
Pre and post hematopoietic stem cell transplant.
As an ongoing process rather than a onetime communication.
Awareness is the key to a successful programme:
Inspite of efforts since several years by many groups, awareness on the thalassemias and
sickle cell disorders is very limited in the population. Awareness generation is needed both
among medical professionals particularly pediatricians, obstetricians and family physicians,
among nurses, health care workers as well as different sections of society including school
and university students, young adults of marriageable age, newly married couples before
conception and during early pregnancy. With a better understanding of the inherited

123
hemoglobin disorders, their inheritance and consequences, screening and genetic counseling
will have greater meaning. An accurate diagnosis is a prerequisite for counseling.
Questions couples with an affected child have:
Why is our child suffering from the disease when we are both normal?
What treatment options are available and is there a cure for the disease?
What is the cost involved for treatment?
Can the next child also have a similar disease?
Is there any way to prevent it?
Components of genetic counseling:
Helping to proactively prepare the couple for the possibility of having a child with an
inherited hemoglobinopathy.
Interpretation of family and medical histories to assess the chance of disease
occurrence or recurrence.
Explaining the clinical presentation, severity and consequences of the disorder.
Explaining the autosomal recessive inheritance pattern.
Explaining to the family the risks and meaning of the carrier state.
Drawing a pedigree chart over three-generations to record family history, noting
consanguinity and ethnicity.
Promoting informed choices and adaptation to the risk or condition.
Explaining other options- providing information about natural, assisted, and non-
reproductive opportunities available for family planning.
Assessing risk for thalassemia, sickle cell disease or other compound heterozygous
conditions in family members.
Providing emotional and social support to reduce the family's anxiety.
Helping patients and parents to convey information about the genetic risk to other
family members.
Taking an informed consent.
Both pre and post-test counseling are important to enable the individual or family to
face the situation and take an appropriate decision.

124
During pre-test counseling, the clinical presentation of the condition the patient may be at
risk for is discussed, the pattern of inheritance of the condition, risk of recurrence, available
testing procedures with their advantages and limitations, reproductive options available and
the required follow up.
The inheritance patterns:
This should be explained by the counselor with the help of pedigree charts showing different
matings as shown below (Figures 36-40). The same inheritance pattern holds true for both the
thalassemias and sickle cell disease as well as other genetic combinations where one parent
is a carrier of thalassemia and the other is a carrier of a variant hemoglobin like HbS, HbE,
HbD or the uncommon HbLepore.
Figure 36 - One parent is normal and the other is a thalassemia carrier or a sickle cell
carrier. There is a 50% chance in every pregnancy that the baby will be normal and a
50% chance that the baby will be a thalassemia carrier or a sickle cell carrier.

125
Figure 37 -Both parents are thalassemia or sickle cell carriers. There is a 25% chance
in every pregnancy that the baby will be normal, a 50% chance that the baby will be a
carrier like the parents and a 25% chance that the baby will be affected with
homozygous thalassemia or sickle cell anemia.
Figure 38 - One parent is homozygous for β-thalassemia or has sickle cell anemia and
the other is normal. In every pregnancy the baby will be a β- thalassemia carrier or a
sickle cell carrier.

126
Figure 39 - One parent is homozygous for thalassemia or has sickle cell anemia and
the other is a thalassemia carrier or sickle cell carrier. There is a 50% chance in every
pregnancy that the baby will be a thalassemia carrier or a sickle cell carrier and a
50% chance that the baby will have homozygous thalassemia or sickle cell anemia.
Figure 40 -The very rare possibility that both parents have homozygous thalassemia
or sickle cell anemia. In such a situation in every pregnancy the baby will have
homozygous thalassemia or sickle cell anemia.
Due to the considerable phenotypic diversity of the thalassemia syndromes, it is imperative
that the genetic counselor has adequate knowledge of the molecular mechanisms involved in

127
this diversity of clinical presentation ranging from a very severe thalassemia major disorder
to a mild thalassemia intermedia presentation. The counselor should also be aware of other
ameliorating factors like the co-inheritance of thalassemia or gene triplication and
variations in the gene in modulating the disease apart from the inheritance of severe or mild
thalassemia mutations. These factors must also be discussed with the families seeking
advice.
Counseling couples at risk for sickle cell disease is also quite complex because of the variable
severity of sickle cell disorders, which can also vary from an early presentation with repeated
complications in some, to a relatively mild or even an almost asymptomatic presentation in
others. This must be clearly explained to the parents although it makes it considerably
difficult for them to take a decision on whether or not to opt for prenatal diagnosis.
Variable disease severity:
The table below (Table 26) shows the severity of homozygous or compound heterozygous
babies when different combinations of genes are inherited.
Table 26: The severity of homozygous or compound heterozygous babies
Homozygous thalassemia Usually severe thalassemia major; in 10 to 15% milder
thalassemia intermedia
Sickle cell anemia Variable clinical severity; more severe presentation in some
population groups than in others
Homozygous Hb E Mild clinical presentation
Homozygous HbD Very mild to asymptomatic presentation
HbS - thalassemia Usually a severe clinical presentation
HbE - thalassemia Often a severe clinical presentation
HbD - thalassemia Generally a mild presentation
Hb Q - thalassemia Very mild to asymptomatic presentation
thalassemia - thalassemia Moderate to severe clinical presentation
HbSD disease Severe clinical presentation
HbSE disease Usually a severe clinical presentation
HbDE disease Usually a milder clinical presentation
thalassemia – HPFH Mild clinical presentation

128
The general principles of counseling must be followed:
Ensure a quiet room where the parents of the child can sit and ask questions without
interruption.
Prepare the family by explaining that you have some important information regarding
the child. Include other family members if parents wish to have them present.
Explain in a language that the family can understand, avoid technical terms. If using
scientific terms or when explaining disease explain each term, repeat, ask if
understood before going on.
Pause and ask if any questions, frequently.
At the end of counseling, give written information sheet tothe family and ask them to
return it if any other questions.
Parents and families who are given a diagnosis of a genetic condition may have anger
and resentment. They often go through a period where they may be in denial.
Sometimes the anger may be targeted at the person giving them the information.
Hence information about diagnosis should be provided by the doctor /and an
additional staff.
Ethical and Religious Issues:
Apart from the information given by the genetic counselor, the acceptability of prenatal
diagnosis with selective termination of an affected fetus by couples is also guided by social,
cultural and religious factors as well as the influence of elders in the family. Genetic
counselling should be “non-directive”, where the genetic counselorshould present all the facts
and information and after understanding and considering all options, the couple or family
take a decision without being influenced by the counselor.
Ethical practice requires the following:
Protecting the right to autonomy of the individual or couple to make an informed
decision.
Their right to get complete information from the counselor.
Confidentiality should be maintained and their privacy protected to avoid
stigmatization.

129
Life-expectancy for individual’s withhalassaemia has improved significantly with
improved management protocols, and in most countries Quality of Life (QoL) is now
considered an important factor of effective health care. These issues on the available
treatment and QoL should also be discussed during genetic counseling.
Informed Consent:
Informed decision making is most often suboptimal particularly when the literacy level of the
individuals or family is low as is often the case in remote rural regions of the country. The
counselor has to bear this in mind while explaining the need for genetic testing and the
consequences of having a child with an inherited hemoglobin disorder.
Newborn screening for hemoglobinopathies allows presymptomatic diagnosis and timely
management of sickle cell disease. Counselors must explain the purpose of newborn
screening to the parents before taking an informed consent from them to test their newborn
baby. At the same time it provides an opportunity for genetic counseling of parents which
would help them to take reproductive decisions in subsequent pregnancies.
For couples opting for prenatal diagnosis, it is crucial to investigate and assess the couple
before pregnancy whenever possible. This will permit the laboratory to undertake molecular
analysis and know the mutations in the parents before the intervention is undertaken. It is also
important to explain and make the couple aware of the risks and complications of the fetal
sampling procedure and also about the occasional problems of the need of repeating the
procedure, if for technical reasons the laboratory is unable to give a report and also the very
occasional problem of a misdiagnosis.
The genetic counselor has to take an informed consent before any genetic screening or
testing is done or any intervention procedure is undertaken. Consent can be written, verbal or
non-verbal/implied as is followed in the UK and the written consent form serves as evidence
that consent has been given. Before this, the benefits, risks and other available options must
be explained to the patient. There are often privacy issues in a hospital environment and
counseling must be given to individual families in a confidential manner.
References
1. Colah R, Italia Y, Gorakshakar A. Burden of thalassemia in India: The road map for
control. PediatrHematol Oncol J. 2018; DOI: 10.1016/j.phoj.2017.10.002

130
2. Colah R, Italia K. Genetic Counseling for Hematological Disorders. In Fetal and
Neonatal Hematology, Oncology and Immunology Eds Lokeshwar MR, Suchdeva A,
Shah NK, Agarwal B, Manglani M. Jaypee Brothers Medical Publishers 2017; 28-42
3. Mohanty D, Das K. Genetic counselling in tribals in India. Indian J Med Res.
2011;134:561-71.
4. Puri RD, Verma IC. Screening, genetic counselling, prenatal diagnosis and pre-
implantation diagnosis for thalassemia. In: Sachdeva A (Ed). Thalassemia: National
guidelines for management of transfusion dependent and non-transfusion dependent
thalassemia, New Delhi. 2014. pp. 272-84.
5. Baker, DL, Schuette JL, Uhlmann WR. A Guide to Genetic Counseling. New York:
Wiley-Liss, 1998
6. Colah RB, Mukherjee MB, Ghosh K. Sickle cell disease in India. Current
OpinHematol. 2014;21:215-23.
7. Muthuswamy V. Ethical issues in genetic counselling with special reference to
haemoglobinopathies. Indian J Med Res. 2011;134:547-51.
8. Deka D, Malhotra N. Genetic counselling for obstetricians. Donald School J
Ultrasound Obstet Gynecol. 2010;4:447-53.
9. Bhat M. Social and cultural issues in genetic counselling. J Biosci. 2015;40:217-20.
10. Giordano PC. The Genetic Counselor. In: Carrier diagnostics and prevention of
hemoglobinopathies using high-performance liquid chromatography. Companion
handbood for the physician, the laboratory doctor and the genetic counselor. Hercules,
CA: Bio-Rad Laboratories Inc; 2006. pp. 81-93.

131
SECTION 2
MANAGEMENT OF HEMOGLOBINOPATHIES
132
2. Management of Hemoglobinopathies
i. Thalassemia Major
Basic recognition of symptoms and signs of thalassemias at level 1
(PHC/CHC)
Thalassemia is an inherited disease of the blood, passed from parents to children through the
genes. This hereditary disorder occurs as a result of mutations affecting the synthesis of the
hemoglobin molecule, found in the red blood cells (RBCs).
In ‘normal’ or adult hemoglobin (HbA), a protein is formed consisting of two alpha (α) and
two beta (β) chains. In thalassemia, there is a reduction in the production of either α- or β-
chains in the hemoglobin molecule.
In α-thalassemia, there is a decrease or lack of
α-chains.
The genes responsible for the production of α-
chains are the α-globin genes, located on
chromosome 16
In β-thalassemia, there is a decrease or lack of
β-chains.
The genes responsible for the production of β-
chains are the β-globin genes, found on
chromosome 11
Classification of Thalassemia disease states is based on severity: The diagnosis of
Thalassemia state has a wide range of clinical severity, however they are broadly divided into
two broad categories.

133
A. Thalassemia Major (Transfusion Dependant Thalassemia): Patients in this category
cannot survive for long after birth without regular blood transfusions and require
regular transfusions beginning at 6 to 24 months of age for sustaining their lives. A
later diagnosis ( >3 years)should make us suspect thalassemia intermedia. This is
important as some patients labeled as thalassmia major may respond to Hydroxyurea
if they are thalassemia intermedia
B. Thalassemia Intermedia[Non Transfusion Dependant Thalassemia (NTDT)]: Patients
in the second category, thalassemia intermedia, may survive with occasional or no
transfusions, or with chronic transfusions initiated at an older age. A patient initially
labeled as NTDT may later require transfusions and even regular transfusion therapy
if they have persistent growth retardation in consultation with a hemoglobinopathy
specialist.
Differences in the severity of thalassemia are usually due to genetic factors, such as
mutations that result in the production of more β-globin chains, or the co-inheritance of α-
thalassemia and/or other genetic factors, each of which contribute different mechanisms in
ameliorating the pathology and clinical outcome of the disease. For example, a given factor
may allow for the production of some fetalhemoglobin (HbF), leading to the partial
correction of the imbalance occurring between the α- and β-chains, which is the single most
important cause of the pathophysiology of thalassemia.
Table 1.
When to Suspect Thalassemia?
History :
Presentation :6 to 24 months age
Lethargy
Progressive Pallor
Failure to thrive
Feeding problems, abdominal distension
Irritability
Recurrent Fever
Family history of low Hemoglobin
sibling death or blood transfusionhistor
Examination :
History :
Presentation : 2 to 6 years of age or later
Mild Pallor
Mild Jaundice
Family history of low hemoglobin
Or sibling death
Examination :
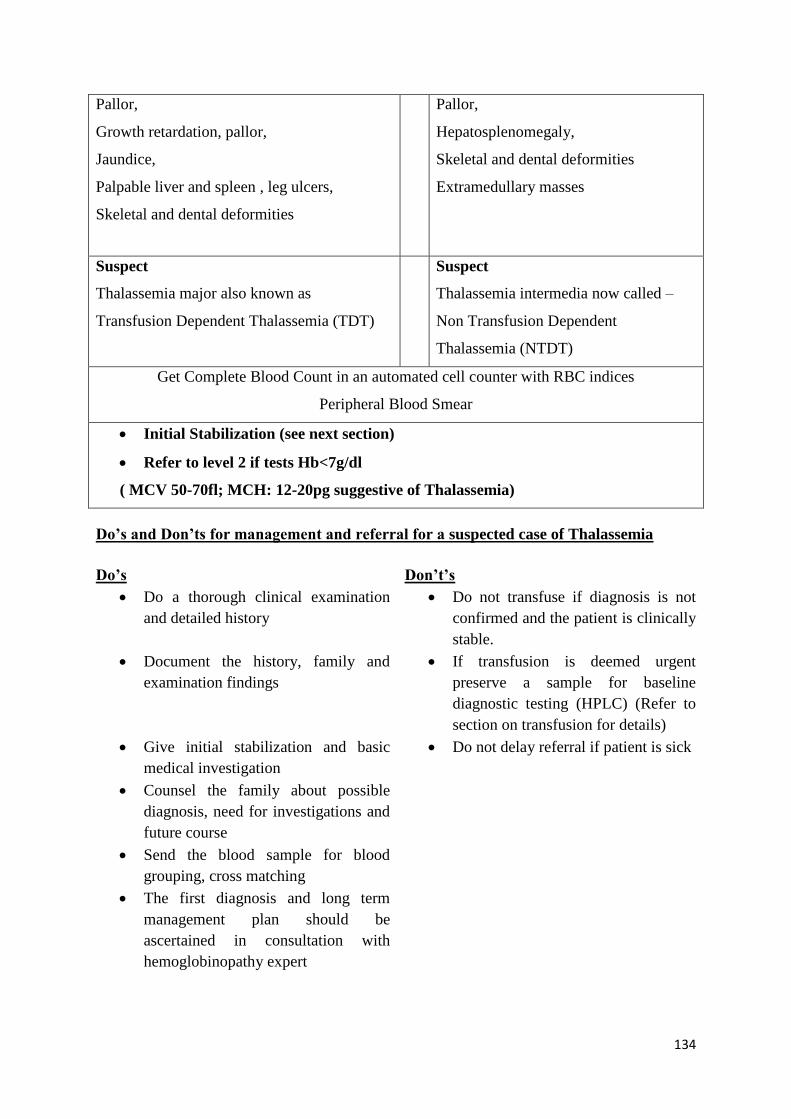
134
Pallor,
Growth retardation, pallor,
Jaundice,
Palpable liver and spleen , leg ulcers,
Skeletal and dental deformities
Pallor,
Hepatosplenomegaly,
Skeletal and dental deformities
Extramedullary masses
Suspect
Thalassemia major also known as
Transfusion Dependent Thalassemia (TDT)
Suspect
Thalassemia intermedia now called –
Non Transfusion Dependent
Thalassemia (NTDT)
Get Complete Blood Count in an automated cell counter with RBC indices
Peripheral Blood Smear
Initial Stabilization (see next section)
Refer to level 2 if tests Hb<7g/dl
( MCV 50-70fl; MCH: 12-20pg suggestive of Thalassemia)
Do’s and Don’ts for management and referral for a suspected case of Thalassemia
Do’s Don’t’s
Do a thorough clinical examination
and detailed history
Do not transfuse if diagnosis is not
confirmed and the patient is clinically
stable.
Document the history, family and
examination findings
If transfusion is deemed urgent
preserve a sample for baseline
diagnostic testing (HPLC) (Refer to
section on transfusion for details)
Give initial stabilization and basic
medical investigation
Do not delay referral if patient is sick
Counsel the family about possible
diagnosis, need for investigations and
future course
Send the blood sample for blood
grouping, cross matching
The first diagnosis and long term
management plan should be
ascertained in consultation with
hemoglobinopathy expert

135
Initial work up, stabilization and triage of care of the sick
patient(see diagnostic section toofor tests to be done)
Age of presentation:
Thalassemia major patients usually are diagnosed between 6 months to 2 years of age.Though
rarely symptoms may start as early as 3 months or even occur after two years of age.
Newborns are asymptomatic because of HbF, which is produced by gamma globin chains, at
the time of the normal switch in hemoglobins from fetal (HbF) to adult hemoglobin (HbA),
the defect manifests as anemia due to defect in beta globin synthesis required for production
of normal adult hemoglobin. If untreated the mortality of thalassemia major is very high, it is
estimated that 85 percent of untreated children will die before the age of five years.
Thalassemia intermedia patients usually present later in life due to milder phenotype than
major, the clinical features start to manifest over 2 to3 years of age, but may be diagnosed
even later.
Common presentation:
Thalassemia patients who are untreated or inadequately treated have symptoms
such as:
Pale or yellowish skin
Poor feeding, irritability
Abdominal swelling (spleen, liver enlargement)
Infections, fever
Slow growth
Weakness, delayed milestones
Fatigue
Facial bone deformities
The severity of clinical features correlates with the number of and the degree of loss of globin
chains. (Table 2, 3)
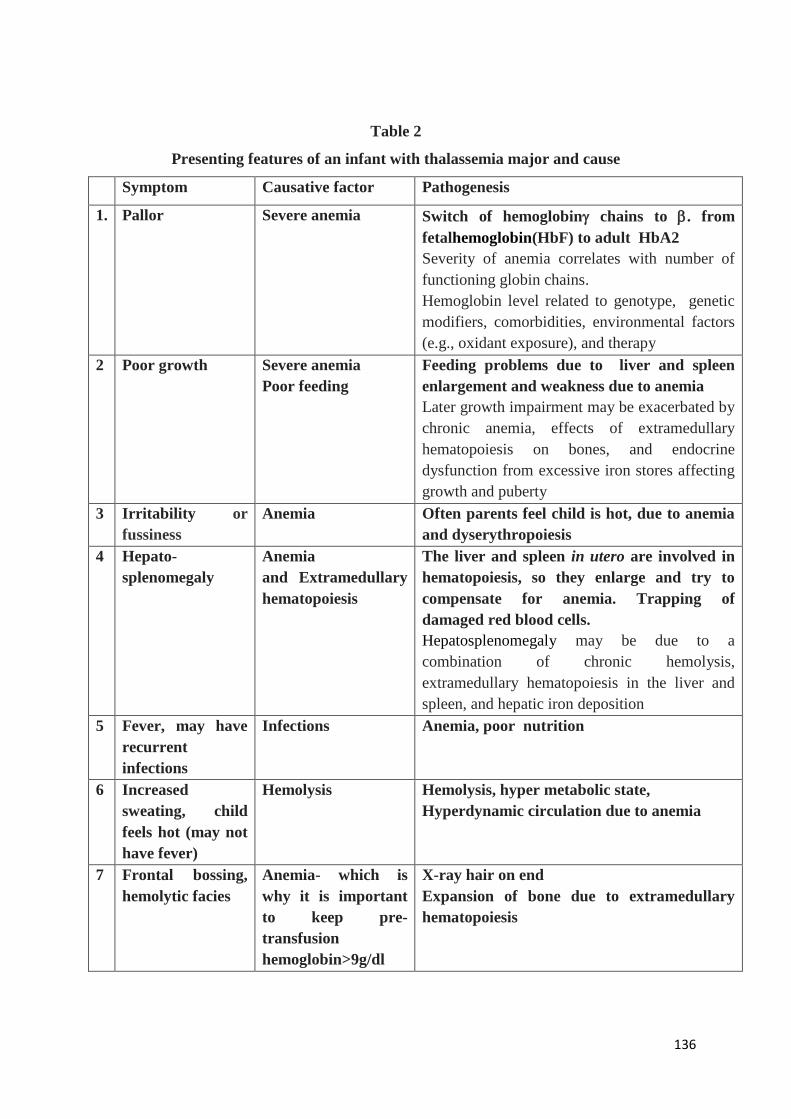
136
Table 2
Presenting features of an infant with thalassemia major and cause
Symptom Causative factor Pathogenesis
1. Pallor Severe anemia Switch of hemoglobin chains to . from
fetalhemoglobin(HbF) to adult HbA2
Severity of anemia correlates with number of
functioning globin chains.
Hemoglobin level related to genotype, genetic
modifiers, comorbidities, environmental factors
(e.g., oxidant exposure), and therapy
2 Poor growth Severe anemia
Poor feeding
Feeding problems due to liver and spleen
enlargement and weakness due to anemia
Later growth impairment may be exacerbated by
chronic anemia, effects of extramedullary
hematopoiesis on bones, and endocrine
dysfunction from excessive iron stores affecting
growth and puberty
3 Irritability or
fussiness
Anemia
Often parents feel child is hot, due to anemia
and dyserythropoiesis
4 Hepato-
splenomegaly
Anemia
and Extramedullary
hematopoiesis
The liver and spleen in utero are involved in
hematopoiesis, so they enlarge and try to
compensate for anemia. Trapping of
damaged red blood cells.
Hepatosplenomegaly may be due to a
combination of chronic hemolysis,
extramedullary hematopoiesis in the liver and
spleen, and hepatic iron deposition
5 Fever, may have
recurrent
infections
Infections Anemia, poor nutrition
6 Increased
sweating, child
feels hot (may not
have fever)
Hemolysis
Hemolysis, hyper metabolic state,
Hyperdynamic circulation due to anemia
7 Frontal bossing,
hemolytic facies
Anemia- which is
why it is important
to keep pre-
transfusion
hemoglobin>9g/dl
X-ray hair on end
Expansion of bone due to extramedullary
hematopoiesis
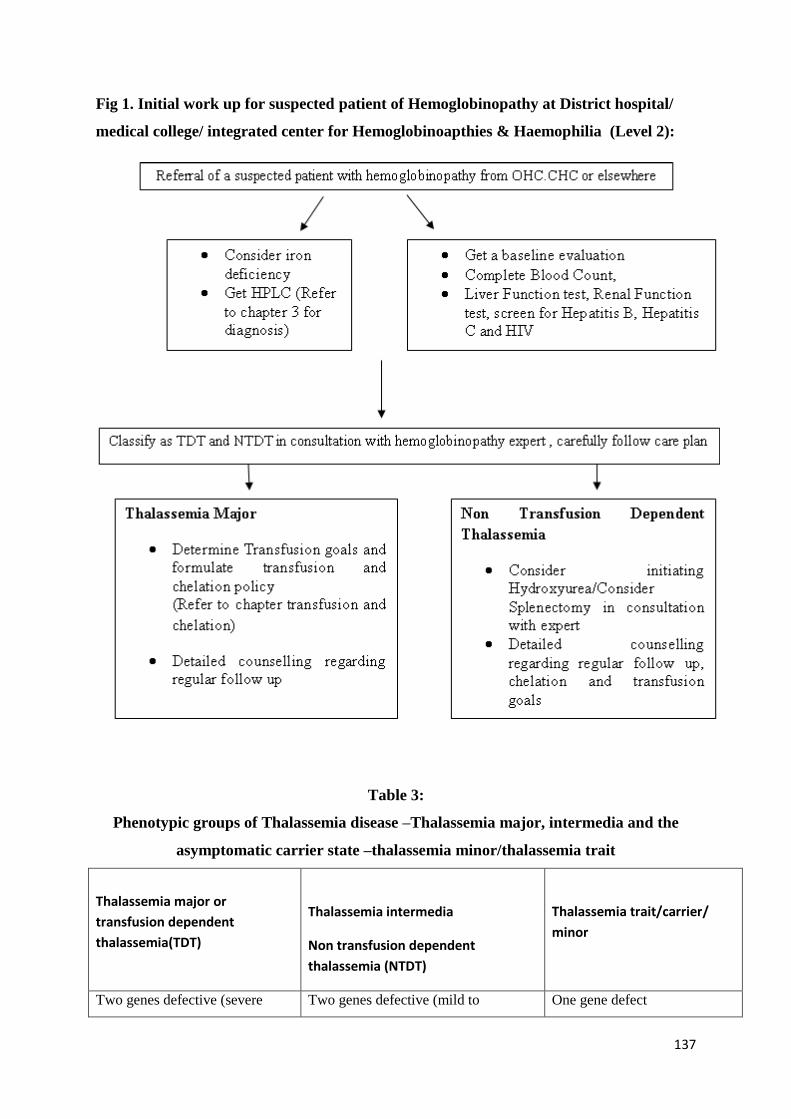
137
Fig 1. Initial work up for suspected patient of Hemoglobinopathy at District hospital/
medical college/ integrated center for Hemoglobinoapthies & Haemophilia (Level 2):
Table 3:
Phenotypic groups of Thalassemia disease –Thalassemia major, intermedia and the
asymptomatic carrier state –thalassemia minor/thalassemia trait
Thalassemia major or
transfusion dependent
thalassemia(TDT)
Thalassemia intermedia
Non transfusion dependent
thalassemia (NTDT)
Thalassemia trait/carrier/
minor
Two genes defective (severe Two genes defective (mild to One gene defect

138
decrease in
beta globin synthesis)
moderatedecrease in beta globin
synthesis)
Severe Pallor requires lifelong
blood transfusions,
irritability,
Hepato-splenomegaly, skeletal
abnormalities,
growth retardation,
Variable degrees of symptoms from
like thalassemia major to
asymptomatic like thalassemia minor,
most are in between.
Have pallor, jaundice,hepato-
splenomegaly, growth retardation,
some requirement of blood
transfusions.
Asymptomatic
Initial Stabilization for a case of suspected thalassemia major or a known case of
thalassemia patient who is ill:
1. The majority of medical management of thalassaemia takes place in the
outpatient clinic and day unit. However, patients occasionally present acutely
and when they do so they may be seriously ill. They need prompt assessment and
management, and will often need admission to hospital.
2. The health care personnel at the level 1 are expected to do the initial stabilization
prior to referral at higher center.
3. If transfusion is indicated in a sick infant and baseline HPLC has not been done,
attempt should be made to preserve the sample for diagnostic hemoglobinopathy
work up prior to transfusion.
Acute presentations may be of a general medical or surgical nature, coincident with the
diagnosis of thalassaemia, but there are also specific clinical presentations which occur
more frequently in thalassaemia and can result in rapid clinical deterioration with
associated morbidity and mortality if not recognised and treated promptly
Evaluation of a sick patient of thalassemia
HISTORY
Specific questions to be asked in history for admitted patient with TM/NTDT :-
i. Infection History
Is the patient feeling unwell, are there rigours, chills, fever? Ask leading questions
for severesystematic illness.
If there are symptoms of infection, consider:
Klebsiella sepsis, which is common in heavily iron-loaded patients. This can result in
septicaemia, urinary tract infection (UTI) or abscesses (cerebral, liver)

139
Yersinia enterocolitica, which may cause abdominal pain, diarrhoea and
lymphadenopathy (more likely with desferrioxamine therapy).
Has the patient been infected with HBV, HCV or HIV?
Thalassaemia patients are vulnerable to malaria infection. Is there malaria or dengue
outbreak in the community?
Does the patient have an indwelling IV device or catheter to provide intensive
chelation whichis a potential source of infection and/or thrombosis.
ii. Splenectomy History
Did the patient undergo splenectomy ?
If so, is he/she taking antibiotic prophylaxis?
(Pneumococcal, meningococcal and haemophilus infections are common in
splenectomised patients).
iii. Transfusion History
If this is the first transfusion, try to do an extended phenotype match. Give 10-
15 ml/kg of PRBC and either prestorageleucodepletedor give transfusion with
a bedside inline leucocyte filter.
Is the patient transfused regularly? If so at what age did this begin?
When was the patient last transfused? Answers may explain Hb levels;
consider the possibility of transfusion-related reactions (short-term or
delayed).
iv. Chelation History
Is the patient taking chelation therapy, and if so, which regime?
Consider the following:
Desferrioxamine- look for sites of sc or iv infusion
-watch for Yersinia and Klebsiellainfection
- ask about changes in eyesight and/or hearing
Deferiprone- check for consequences of reduced neutrophil counts and joint
pains
Deferasirox-investigate renal and hepatic function, -ask about gastrointestinal
disturbances.

140
v. Iron overload History
Adult thalassaemia patients usually know their iron assessment record, including
serum ferritin level and MRI results. Askthefollowing questions.
Has the patient not been adequately chelated recently?
What is the recent trend of serum ferritin? Is it increasing or decreasing?
Has the patient hadan MRI scan or used other tools to assess liver or heart iron
recently?
Does the patient have heart complications?
Is the patient demonstrating decreasing exercise tolerance or breathlessness?
Is there peripheral oedema?
Is the patient experiencing palpitations(awareness of heart beat)?
Are there any symptoms of liver distension with abdominal discomfort?
Does the patient have endocrine complications?
For diabetes, hypogonadism, hypothyroidism and hypoparathyroidism, check
blood and urine sugar, plasma calcium and thyroid function tests.
vi. Surgical history
Has the patient had a splenectomy?
Has the patient had gallstones or cholecystectomy?
vii. Fracture history:
Fractures are common in thalassaemia patients because of weakened bone structure.
Get radiologic evaluation as indicated. Was the patient investigated by DXA scan ?It
is important to know if the patient has a metal plate, which may exclude the
possibility of an MRI examination.
viii Medication history
Is the patient being treated for hepatitis? Many thalassaemia patients are receiving the
current standard for chronic HCV infection and therefore the relevant history needs to
be documented.
Is the patient on anticoagulants? Heart disease and thrombotic conditions (more
common in NTDT) may have necessitated anticoagulant treatment.
Is the patient on antiplatelet drugs?

141
Is the patient on hydroxyurea?
Is the patient on any medication for the heart?e.g. ACE inhibitors, amiodarone,
digoxin
Is the patient on medications for endocrine complications?
e.g. insulin, thyroxin, calcium, vitamin D, estrogens, testosterone,bisphosphonates
EXAMINATION
It is important to categorise the severity of the illness early. Look at the vital signs. Is the
patient in a critical condition?
General appearance
Skin may have a ‘tanned’ discolouration due to iron overload.
Pallor -From anemia.
Jaundice - from anaemia, liver complications or coexistence of Gilbert’s syndrome.
Short stature, short trunk and genu valgum (poor treatment in early life).
Skull and facial changes (signs of TI or poorly treated TM).
Skull enlargement with accentuation of the frontal and malar prominences and
depression of the bridge of the nose. Upper maxillary enlargement affecting teeth
spacing and leading to malocclusion.
Signs of previous surgery – splenectomy, cholecystectomy, fracture surgery.
Pubertal immaturity (as a sign of endocrine failure).
Vital signs
Blood pressure- many TM patients have low BP in steady state. However this may
also be a sign of cardiac decompensation or sepsis.
Pulse - an irregular pulse is an iron-related complication of the myocardium and
patients may be on anticoagulation for this.
Temperature - sepsis is common and life threatening, particularly in iron overloaded
and/or splenectomised patients. Respiratory rate, pulse oximetry, glucose check and
level of consciousness (Glasgow Coma Scale) should be checks of priority.
Hepato-splenomegaly
Most well-treated TM patients will not have had their spleen removed; assessment of
it'ssize may help guide diagnosis of the emergency problem.
Assessment of liver size is also essential. Enlarged liver may result from cirrhosis, right-sided
heart failure or extramedullary hematopoiesis.

142
Bone Marrow Expansion (BME)
BME is increasingly rare in well-transfused TM patients than in TI patients.
Consequences include
- Bone fractures, which are common in both TM and TI and often occur following
trivial injury.
- Micro-fractures –Tenderness in back, ribs, metatarsals due to March fractures of
feet.
- Masses/Weakness due to spinal cord compression
Sepsis -Potential sources/causes
Cholecystitis- Right upper Abdomen tenderness/Lump.
UTI- Renal Angle Tenderness.
Chest- Lung Crepts, Bronchial Breathing.
Teeth-gingivitis, dental abscess, dental cavity.
Endocarditis-Murmurs, Pericardial Rub.
Brain abscess- Papilledema, Focal deficit, Poor GCS.
Meningitis-neck stiffness, poor GCS.
Heart failure
Cardiovascular examination is essential: arrhythmias and biventricular heart failure
from myocardial iron may occur, usually after 10 years of age and can be diagnosed
by irregularities in pulse.
Clinical signs of congestive heart failure, including oedema, require emergency
intervention (iv Desferoxamine ± deferiprone orally).
Pulmonary hypertension is increasingly important, especially in TI with evidence of
heave and loud P2 .
Thrombosis:
Swelling of limb etc or DVT or low saturation hypoxia.
ECG changes of PE.
Endocrine complications
Hypothyroidism,
Hyperparathyroidism and
Diabetes mellitus and other endocrinopathies may be present which may complicate
management and require therapy.
Neurological complications
Extra medullary cord compression.
Brain Abscess and Stroke are common.

143
Look for weakness, focal deficits and evidence of raised intra cranial pressure.
MANAGEMENT
After Diagnosis and stabilization patient should be registered and referred to the day-
care centre for regular management.Routine blood transfusion of filtered PRBC, is the
mainstay of therapy as well as monitoring and appropriate management of iron
overload and evaluation for complications.
Criteria for Inpatient/Emergency management of Thalassemia
Table4:
Sepsis: Fever/suspected infection with Respiratory rate > 22/min or systolic
BP<100mm Hg or GlasGow Coma Scale<15 {qSOFA>2} or any other features of
hemodynamic instability/deranged vital parameters.
Anemia: Worsening anemia despite adequate transfusion or rapid fall from usual
steady state hemoglobin level /persistently increased transfusion requirement.
New onset shortness of breath (dyspnea)
New onset generalized swelling of body /feet (edema/anasarca)
Abdominal pain which is moderate to severe or persistent
Persistent or new onset severe headache
Syncope or altered level of consciousness
New onset focal neurologic deficit /weakness of any limb or face
Acute Back Pain
Trauma with suspected bone fracture or spleen rupture
Any other medical condition where admission is deemed necessary by the treating
physician
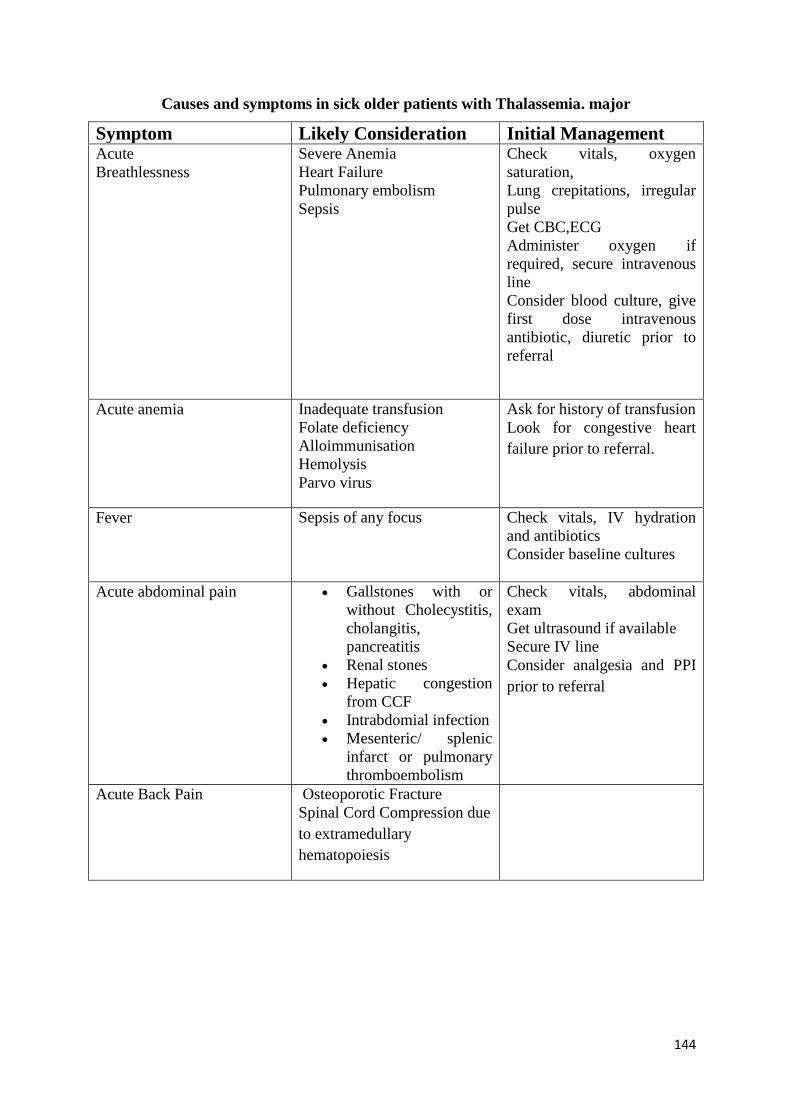
144
Causes and symptoms in sick older patients with Thalassemia. major
Symptom Likely Consideration Initial Management Acute
Breathlessness
Severe Anemia
Heart Failure
Pulmonary embolism
Sepsis
Check vitals, oxygen
saturation,
Lung crepitations, irregular
pulse
Get CBC,ECG
Administer oxygen if
required, secure intravenous
line
Consider blood culture, give
first dose intravenous
antibiotic, diuretic prior to
referral
Acute anemia Inadequate transfusion
Folate deficiency
Alloimmunisation
Hemolysis
Parvo virus
Ask for history of transfusion
Look for congestive heart
failure prior to referral.
Fever Sepsis of any focus Check vitals, IV hydration
and antibiotics
Consider baseline cultures
Acute abdominal pain Gallstones with or
without Cholecystitis,
cholangitis,
pancreatitis
Renal stones
Hepatic congestion
from CCF
Intrabdomial infection
Mesenteric/ splenic
infarct or pulmonary
thromboembolism
Check vitals, abdominal
exam
Get ultrasound if available
Secure IV line
Consider analgesia and PPI
prior to referral
Acute Back Pain Osteoporotic Fracture
Spinal Cord Compression due
to extramedullary
hematopoiesis
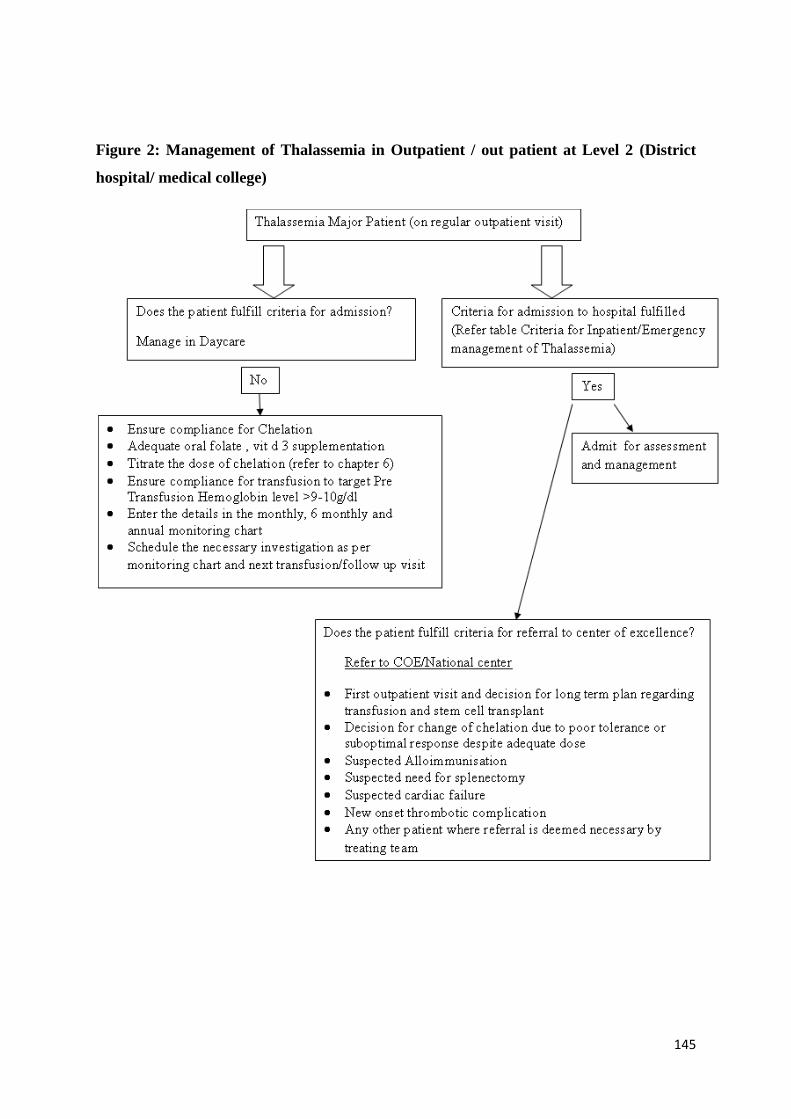
145
Figure 2: Management of Thalassemia in Outpatient / out patient at Level 2 (District
hospital/ medical college)

146
References
1. Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines for the Management
of Transfusion Dependent Thalassaemia (TDT). Guidelines for the Management of
Transfusion Dependent Thalassaemia (TDT). Thalassaemia International Federation; 2014.
2. Emergency management thalassemia - Books - NCBI [Internet]. [cited 2019 May 2].
Available from: https://www.ncbi.nlm.nih.gov/books/emergency management thallasemia
3. Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V, Weatherall SD.
Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT).
Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT).
Thalassaemia International Federation; 2013.

147
Guidelines for Blood Transfusion in Hemoglobinopathies
(Level II- District hospital/ medical colleges )
Thalassemia syndromes can be phenotypically classified into transfusion dependent (TDT)
and non-transfusion dependent thalassemias (NTDT). The former requires lifelong regular
transfusions, while NTDT syndromes affected patients require blood transfusion (BT) only
intermittently & do not depend on transfusions for survival.
‘One size fits all’ approach to transfusion of thalassemia patients, is not applicable. Indeed a
flexible approach crafted to the patient’s individual requirements and to the local availability
of safe blood products is needed for optimal outcomes. Guidelines aim to balance the benefits
of oxygenation and suppression of extra-medullary expansion with those of excessive iron
accumulation from over transfusion.
Goals of Blood Transfusion in Hemoglobinopathy
1. Achievement of appropriate haemoglobin level.
2. Achievement of normal growth & development in paediatric thalassemia patients.
3. Suppression of ineffective erythropoiesis.
4. Prevention of complications splenomegaly , skeletal abnormalities.
5. Minimising complications of transfusion.
Pre-requisite for blood transfusion ( BT )
1. Confirmation of diagnosis ( Mandatory )
2. Testing for HBsAg / HCV / HIV (Mandatory )
3. Vaccination for HBV( Mandatory )
4. Counselling of parents / patients for BT( Mandatory )

148
5. Extended red cell antigen typing that includes at least C, c, D, E, e, and Kell
(if facilities available )
Whom to transfuse?
In newly diagnosed children with thalassaemia, it is very essential to assess the patient
carefully over the first few months after the diagnosis is established and not to embark on
transfusion therapy, too hastily. Emphasize the importance of clinical assessment over
haemoglobin percentage alone. (In most of the cases the diagnosis is made during any
intercurrent illness for which haemoglobin drops from the baseline and the childisbrought for
consultation. ). While the child is under observation—
The Hb level should be checked at least monthly.
Wait till the steady state is reached.
During this period observe the child carefully for growth & activities.
Indication of Blood Transfusion (BT) in Thalassemia major or TDT
patients
Blood transfusion programme to be started if –
Hemoglobin percentage is below 7 g/dl on two occasions 1-2 weeks apart
OR
Any of thefollowing signs & symptoms are present ( Irrespective of haemoglobin
percentage )
Tachycardia
Sweating
Poor feeding
Poor growth
Inability to perform routine activities – attending school
Facial bony changes
Massive splenomegaly
Where possible, this decision should not be delayed until after the 3rd year of age, as the risk
of alloimmunization increases with subsequent difficulty in finding suitable RBC units.

149
Indication of Blood Transfusion (BT ) in NTDT patients
Hemoglobin level should not be a sole determinant of transfusion need except in
patients with considerably severe anemia (hemoglobin level <5 g/dl).
Blood transfusion, if initiated in patients with NTDT will require closer monitoring
and should be individually tailored to meet patient’s needs.
In a patient who never received BT before, if poor growth & development is noted,
start regular transfusions till maximum height is achieved, and bones are fused before
attempting to wean off and then stop regular transfusions.
In case of delayed pubertal growth, child should be on transfusion programme till
pubertal growth is complete.
It is important to maintain the child in good health and normal growth and
development with avoiding unnecessary transfusions in those with minor clinical
features.
Some children with NTDT, specifically with hemoglobin E- βthalassemia, have a
remarkable abilityfor adaptation to low haemoglobin levels and may not require
PRBC(endogenous erythropoiesis). InHbE- thalassemia hemoglobin may be
maintained at lower levels(7g/dl ) as it has beenshown that they do well with lower
Hb%.
Patients having sustained frequent transfusions for extended periods of time should be
managed as per the guidelines for transfusion-dependent β-thalassemia major patients
[28].
Patient should be on regular transfusion in the following settings:
o Falling hemoglobin level in parallel with profound enlargement of the spleen (at a
rate exceeding 3 cm/year in periods of maximal growth and development)
o Growth failure
o Poor school performance
o Diminished exercise tolerance
o Failure of pubertal development
o Signs of bony changes
o Frequent haemolytic crisis (haemoglobin H disease)
o Poor quality of life

150
Occasional blood transfusions should be considered in NTDT patients within any setting
with anticipated acute stress, hemoglobin drop, or blood loss; such as
o Pregnancy
o Surgery
o Infections
Transfusions may be considered for the primary prevention (in high-risk populations), ,
or secondary prevention of the following complications:
o Thrombotic or cerebrovascular disease
o Pulmonary hypertension
o Extramedullary haematopoiesis
o Leg ulcers
Once transfusion therapy is considered, two things need careful monitoring:
o Iron overload
o Risk of alloimmunization.
What is to be transfused?
o Packed Red Blood Cells (Only).
o Hemoglobin concentration of the bag shouldbe minimum 40%.
o Pretransfusion Leukocyte-depleted RBCs(Preferable ).
o If not available use leucocyte filter ( Preferable ).
o ABO and Rh(D) matched blood ( Mandatory ).
o Rh (C, c, E, e) and Kell matching (Preferable ).
o Before each transfusion, perform a full cross-match and screen for new antibodies (
Preferable ).
o Units stored less than 2 weeks (Preferable).
o Washed red cells for patients with repeated severe allergic transfusion reactions or
immunoglobulin A (IgA) deficiency.
In thalassemia major regular blood transfusion is essential and life saving
For any confusion regarding initiation of transfusion programme for thalassemia
intermedia patients, please send the patient to level 3 or 4 for opinion.

151
o Transfusion of blood from first degree relatives should be avoided; this is a bad blood
bank practise.
Transfusion Programme
o Base-line (pre transfusion) haemoglobin % should be maintained at 9-9.5 g% to
suppress hematopoiesis.
o The volume per transfusion is usually 10-15 ml/kg. (Desired – actual Hb) ×
weight × 3 / hematocrit of transfused unit = mL to be transfused.
o Does not waste blood,usual dose 10-15ml PRBC/ kg. Round up to nearest unit
o In uncomplicated patients the whole PRBC unit may be transfused over 1 hour.
o In patients with cardiac failure / very low Hb% e.g. haemoglobin 5 g/dl or below,
give PRBC at a very slow rate,only 1-2 ml/kg/hour.
o The post-transfusion target hemoglobin should be around 12 g/dland should not be
greater than 14 g/dl , though it is not routine practise to check post transfusion HCT.
o A careful record of transfused blood should be maintained for each patient, including
the volume of the administered units, pre-transfusion Hb%, any transfusion reaction.
o The transfusion record must note volume of blood given each time to the patient. If
volume of unit not given by your blood bank, then keep a weighing scale and weigh
bag, and note weight of bag in chart at each transfusion.
o Frequency of blood transfusions - 2-5 weeks depending on need of the child.
o Routine premedication not to be given.
o If patient is having recurrent febrile transfusion reaction – then may give paracetamol
before starting transfusion.
o If the child is in cardiac failure then give Inj Lasix 1 mg / kg before transfusion,
monitor closely during transfusion.
o No need to warm blood bag.
o Do not store blood bag in ward refrigerator.
If there is no increase in hemoglobin after BT on repeated occasion, suspectallo-
antibodies and send the patient to Level 3 or 4 centre for evaluation.
o If requirement of PRBC is> 250 mL / kg / year to maintain baseline haemoglobin 9-
9.5 g/dl, suspect hypersplenism, send to level 3 or 4 center.
152
o A higher target pre-transfusion haemoglobin level of 11-12 g/dl may be appropriate
for patients with heart disease, clinically significant extramedullary hematopoeisis or
other medical conditions
Transfusion Record
Date PreTransfus
ionHb (gm
%)
Volume
transfused
( may put
weight of
bag if
volume
not
known.)
Post
Transfusion
Hb(gm %)
Transfusion
reaction
Remarks
References
1. Sant-Rayn Pasricha, David M. Frazer, Donald K. Bowden and Gregory J. Anderson.
Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-
thalassemia major: a longitudinal study .Blood 2013 122:124-133;
2. Cazzola M, Borgna-Pignatti C, Locatelli F, et al. A moderate transfusion regimen may
reduce iron loading in beta- thalassemia major without producing excessive expansion
of erythropoiesis. Transfusion. 1997;37:135–140.
3. Cheng CK, Lee CK, Lin CK. Clinically significant red blood cell antibodies in
chronically transfused patients: a survey of Chinese thalassemia major patients and
literature review. Transfusion. 2010;52:2220–2224
4. Michail-Merianou V, Pamphili-Panousopoulou L, Piperi-Lowes L, et al.
Alloimmunization to red cell antigens in thalassemia: comparative study of usual
versus better-match transfusion programmes. Vox Sang. 1987;52:95–98.
5. Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines
fortheManagementofTransfusionDependentThalassaemia(TDT).Ed 3. Nicosia,
Cyprus: Thalassaemia International Federation; 2014.

153
6. VichinskyE, NeumayrL, TrimbleS, etal.Transfusioncomplicationsin thalassemia
patients: a report from the Centers for Disease Control and Prevention (CME).
Transfusion. 2014;54:972-981; quiz 1.
7. .Garbowski MV, et al.Residual erythropoiesis protects against myocardial
hemosiderosis in transfusion-dependent thalassemia by lowering labile plasma iron
via transient generation of apotransferrinHaematologica 102(10) 1640-1649 2017).
8. How I treat thalassemia Eliezer A, Patricia J. Blood. 2011;118:3479-88
9. Challenges of alloimmunization in patients with haemoglobinopathies Chou ST, Liem
RI, Thompson AA British journal of haematology. 2012;159(4):394-404.
10. Standards of care guidelines for thalassemia. Children’s Hospital & Research Center
Oakland; 2012 (http://thalassemia.com/treatmentguidelines-4.aspx). Accessed April
20, 2015.
11. Guidelines for the management of non transfusion dependent thalassaemia (NTDT):
Thalassaemia Taher A, Vichinsky E, Musallam K, Cappellini M-D, Viprakasit
V. International Federation, Nicosia, Cyprus; 2013.
12. Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent
thalassemia. Blood Rev 2012;26 Suppl1:S12-15.
13. Allen A, Fisher C, Premawardhena A, peto T, Allen S, Arambepola m, Thayalsutha
V, olivieri N, Weatherall D. Adaptation to anemia in hemoglobin E-ss thalassemia.
Blood 2010;116(24):5368-5370.

154
Guidelines for Chelation therapy in Thalassemia
At level 2 centers
(Comprehensive ICHH centers for hemoglobinopathies and hemophilia)
Iron overload is the main cause of morbidity and mortality is hemoglobinopathies.Iron
overload occurs when iron intake is increased over a sustained period of time, either as a
result of red blood cell transfusions in transfusion dependent thalassemia (TDT) or
thalassemia major or increased absorption of iron through the gastrointestinal (GI) tract in
non transfusion dependent thalassemia (NTDT) or thalassemia intermedia. Proper monitoring
of iron overload and chelation therapy is a very important component of treatment of
thalassemia patients. Chelation therapy aims to balance the rate of iron accumulation by
increasing iron excretion in urine and or faeces. The major challenge in chelation therapy is
to achieve regular adherence to treatment regimens. The goal is to equip the ICHH centers at
District hospitals or medical colleges to screen, and start iron chelation. Complicated cases or
non responders who need more evaluation should be referred to level 3 (COE) or 4 (National
referral center) for optimal investigation and care.
Monitoring of Iron Overload
Available methods-
Serum ferritin
Advantages
• Easy to assess
• Inexpensive
• Repeat serial measures are useful for monitoring chelation therapy
• Positive correlation with morbidity and mortality

155
Disadvantages
• Indirect measurement of iron burden
• Fluctuates in response to inflammation, abnormal liver function
• Most serum ferritin assays were developed mainly for detecting iron deficiency, and
the linear range of the assay at high SF values needs to be known. Need to dilute
samples with high values, to give readings within the linear range of the assay
• May not provide reliable indication of iron levels in cases of non transfusion
dependent thalassemia (NTDT) or thalassemia intermedia.
Liver iron concentration (LIC) measurement
Gold standard for iron assessment- liver biopsy
Methods – by liver biopsy
Advantages
• Direct measurement of LIC
• Quantitative, specific, and sensitive
• Provides information on liver histology/pathology
• Positive correlation with morbidity and mortality
Disadvantages
• Invasive, painful, potentially serious complications, eg, bleeding
• Practically not a feasible option.
Liver iron concentration (LIC) by T2* MRI
Advantages
• Assesses iron content throughout the liver
• Status of liver and heart can be assessed in parallel
• Validated relationship with LIC
• Allows longitudinal patient follow-up
Disadvantages
• Indirect measurement of LIC
• Requires MRI imager with dedicated imaging method,ensure the lab has proper
validation and standardization.
• Children younger than age 7 years may require a general anaesthetic

156
Cardiac iron by T2* MRI
Advantage
• Non-invasive
• Reproducible
• Have predictive value in identifying patients at high risk of developing
deterioration in LVEF
Disadvantages
• Indirect method
• Non-linear relationship with myocardial iron content ( MIC )
• Technically demanding ensure the lab has proper validation and
standardization.
Recommendations
Recommended method for monitoring iron overload is Ferritin ( at level 2)
Monitoring for Ferritin to be started after 2 years of age or after 10-20 BT in cases of
TDT
Monitoring to be started for all NTDT patients >10 years of age.
All patients may be screened by T2*MRI after age of >10 years annually or every two
years, they can be referred to the State center facility for this test.
T2* MRI for LIC to be done in NTDT patients with features of iron overload &
disproportionately low ferritin – patient to be referred to level 3 or 4
T2*MRI for MIC in patients with high Ferritin value or deteriorating cardiac function
– patient to be referred to level 3 or 4.
When to start chelation therapy?
Transfusion dependent thalassemia( TDT )
2 years of age or older,
1 to 2 years of chronic transfusions,
Serum ferritin level > 1000 ng/mL on 2 separate measurement obtained when the
child is well.
LIC of at least 7 mg/g dry weight( If available )
TDT patients < 2 years of age requiring chelation therapy to be referred to level 3 or
4 for opinion.

157
Non Transfusion Dependent Thalassaemia( NTDT )
More than 10 years of age orSerum ferritin level ≥750 ng / ml.
Liver iron concentration ≥5 mg Fe/g dry weight ( If available )
Goal of Chelation Therapy
To prevent the toxic effects of iron overload
To avoid chelator toxicities
To maximize adherence to therapy
To prevent organ dysfunction
o Liver iron concentration (LIC) <5mg/g dry weight,
o Cardiac T2* MRI >20ms
Recommendations
Chelation therapy to be started
1. TDT if Ferritin > 1000 ng / mL & the child is> 2 years old.
2. NTDT if Ferritin > 800 ng / mL & the child is 10 years of age
What Chelating agent to start?
Available iron chelation drugs
1. Deferroxamine (DFO)
2. Deferiprone (DFP)
3. Deferasirox (DFX)
Deferasirox – oral drug ( 1st line drug )
Thalassemia major Dose : 20 mg / kg/day, increase dose by 5-10 mg / kg every 3 months
based on Ferritin report ( see table 5, 6)
Dose in NTDT-10 mg/k/day
Escalation of dose in NTDT
At 1 month
No escalation if Ferritin < 1500 ng / mL ( Baseline )
To 15 mg /kg / day if Ferritin > 1500 < 3000 ng / ml, ( Baseline )

158
To 20 mg /kg/day if Ferritin > 3000 ng / mL ( Baseline )
At 6 months
Ferritin < 1500 ng / mL – no escalation
Ferritin > 1500 < 3000 ng / mL – escalate by 5 mg / kg/day ( maximum 20 mg / kg / day )
Ferritin > 3000 ng / mL – escalate by 10-15 mg / kg / day ( maximum 30mg / kg / day ) :
refer to level 3 or4
DFX is administration on empty stomach dissolved in a glass of water /fruitjuice, do
not use any metalic glass /spoon.
DFX can be given in two divided doses if the patient cannot tolerate once daily dose.
Twice daily dose is equally effective
Contraindicated: in patients with renal failure or significant renal dysfunction (see
below).
Caution is recommended for patients with advanced liver disease and hepatic
decompensation
Deferasirox to be discontinued if Ferritin < 300 ng / ml, but continue monitoring so
that it can be restarted as the ferritin will increase.
Table 5:
Monitoring for Deferasirox
Time Parameters
Before starting Deferasirox
Serum ferritin
Serum creatinine x2
Liver function tests
1st month of treatment Serum creatinine (weekly )
Monthly
Serum creatinine
Liver function tests ( every two weeks for first month )
3 monthly
Serum Ferritin
Annual Hearing and vision check up
Deferiprone : oral drug ( 2nd line drug )
Dose 75 mg/kg/day, given in three divided doses
Should not be started in patient < 6 years of age
Require monitoring of blood count

159
Each patient’s absolute neutrophil count should be measured before starting DFP
therapy and weekly during treatment. Should be stopped if ANC < 1500. Refer to
level 3 or 4
Deferiprone is not recommended in HIV positive or in immunocompromised patients,
due to its risk of agranulocytosis.
Arthropathy range from mild pain to severe arthritis, spontaneous recovery occurs
despite continuing therapy. If joint symptoms continue despite , reduce dose of
DFP, if not controlled after dose reduction , stop DFP. Refer to level 3 or 4
Increased ALT values usually asymptomatic and transient and ALT usually
returns to normal without discontinuation or dose decrease.
Deferiprone is renally excreted so there may be an increased risk of complications in
patients with impaired renal function
Desferrioxamine( DFO ) – 3rd line drug
Delivery: Subcutaneous or IV infusions
Dosage: 30-60mg/kg/day 12hours subcutaneous infusion 5-7 times per week
Average dose should not exceeds 40 mg / kg/ day until growth has ceased
IV dose only in severely iron over loaded patients with cardiac failure .
Needs special infusion pump
Disadvantages
Expensive, time-consuming, not comfortable
Local skin reactions
Infection with Yersinia enterocolitica
Severe hypersensitivity reaction
Hearing problems
Effects on the eye
Growth retardation
Skeletal changes
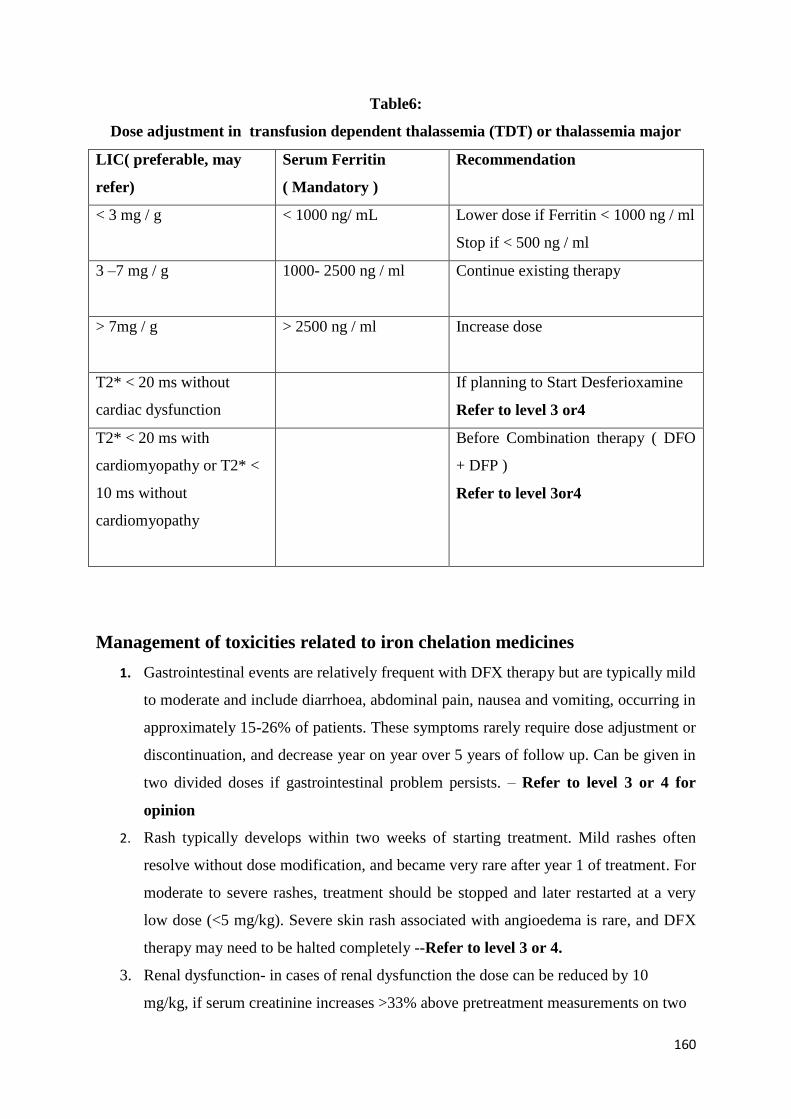
160
Table6:
Dose adjustment in transfusion dependent thalassemia (TDT) or thalassemia major
LIC( preferable, may
refer)
Serum Ferritin
( Mandatory )
Recommendation
< 3 mg / g < 1000 ng/ mL
Lower dose if Ferritin < 1000 ng / ml
Stop if < 500 ng / ml
3 –7 mg / g 1000- 2500 ng / ml
Continue existing therapy
> 7mg / g > 2500 ng / ml Increase dose
T2* < 20 ms without
cardiac dysfunction
If planning to Start Desferioxamine
Refer to level 3 or4
T2* < 20 ms with
cardiomyopathy or T2* <
10 ms without
cardiomyopathy
Before Combination therapy ( DFO
+ DFP )
Refer to level 3or4
Management of toxicities related to iron chelation medicines
1. Gastrointestinal events are relatively frequent with DFX therapy but are typically mild
to moderate and include diarrhoea, abdominal pain, nausea and vomiting, occurring in
approximately 15-26% of patients. These symptoms rarely require dose adjustment or
discontinuation, and decrease year on year over 5 years of follow up. Can be given in
two divided doses if gastrointestinal problem persists. – Refer to level 3 or 4 for
opinion
2. Rash typically develops within two weeks of starting treatment. Mild rashes often
resolve without dose modification, and became very rare after year 1 of treatment. For
moderate to severe rashes, treatment should be stopped and later restarted at a very
low dose (<5 mg/kg). Severe skin rash associated with angioedema is rare, and DFX
therapy may need to be halted completely --Refer to level 3 or 4.
3. Renal dysfunction- in cases of renal dysfunction the dose can be reduced by 10
mg/kg, if serum creatinine increases >33% above pretreatment measurements on two

161
consecutive visits and no other cause is found. Refer to level 3 or 4 for opinion as
thalassemia major patients can have renal dysfunction due to their disease..
4. Liver dysfunction- ALT if increased, chelation should be stopped & reintroduced at
low doses once ALT is normal. Refer to level 3 or 4 for opinion.
Recommendations
1. Recommended 1st line therapy is Deferasirox.
2. Chelation to be started after 2 years of age & ferritin > 1000 ng / mL in TDT &> 800
ng / ml& 10 years of age in NTDT.
3. Deferasirox can be given with food,not required to be on an empty stomach once or
twice a day regularly and daily.
4. Shift to twice daily dose of Deferasirox in cases of persistent gastrointestinal
symptoms.
5. In case of severe toxicity of DFX , DFP ( 2nd
line drug ) is recommended provided the
child is > 6 years old
6. In case of persistent or severe adverse effects patients to be referred to higher level
( COE ) or level 4 centerfor opinion
7. Before starting 2nd
or 3rd
line drug, patient to be referred to level 3 (COE) or
National level referral center (level 4 )for opinion.
8. Desferrioxamine IV injection require placement of a central venous catheter for
continuous infusion, in case of emergency it can be given through peripehral vein
diluted in at least 100ml of normal saline.
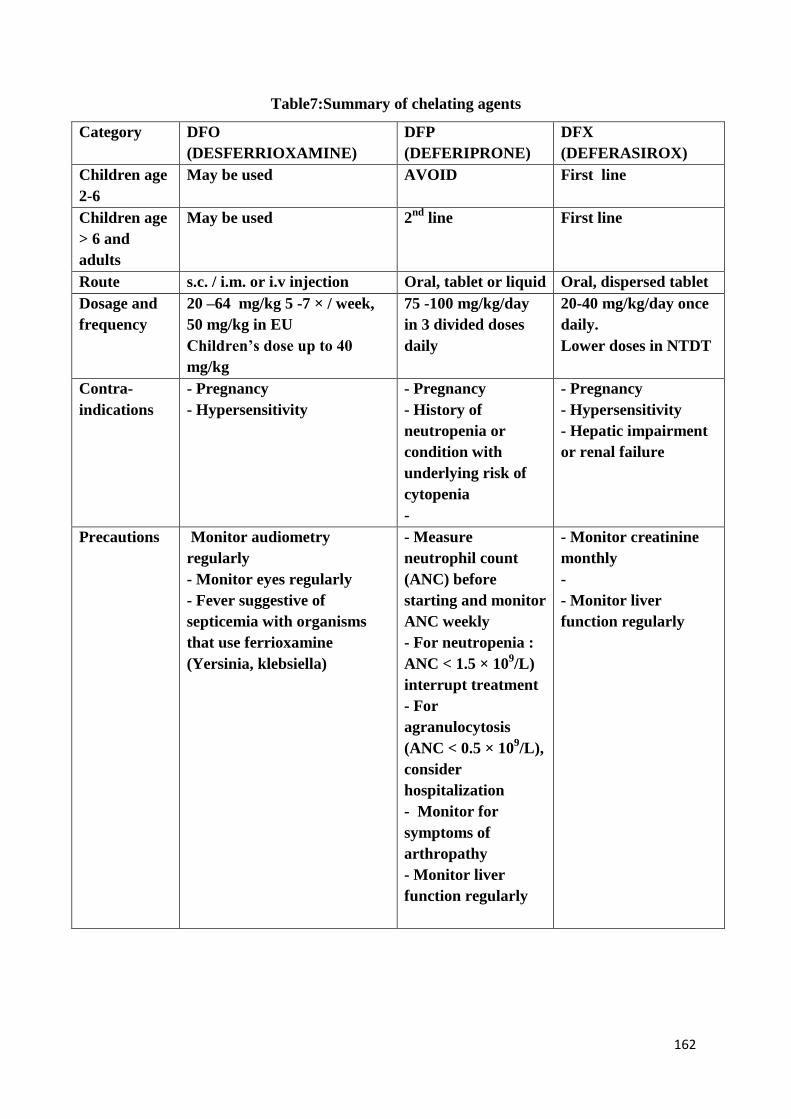
162
Table7:Summary of chelating agents
Category DFO
(DESFERRIOXAMINE)
DFP
(DEFERIPRONE)
DFX
(DEFERASIROX)
Children age
2-6
May be used AVOID First line
Children age
> 6 and
adults
May be used 2nd
line First line
Route s.c. / i.m. or i.v injection Oral, tablet or liquid Oral, dispersed tablet
Dosage and
frequency
20 –64 mg/kg 5 -7 × / week,
50 mg/kg in EU
Children’s dose up to 40
mg/kg
75 -100 mg/kg/day
in 3 divided doses
daily
20-40 mg/kg/day once
daily.
Lower doses in NTDT
Contra-
indications
- Pregnancy
- Hypersensitivity
- Pregnancy
- History of
neutropenia or
condition with
underlying risk of
cytopenia
-
- Pregnancy
- Hypersensitivity
- Hepatic impairment
or renal failure
Precautions Monitor audiometry
regularly
- Monitor eyes regularly
- Fever suggestive of
septicemia with organisms
that use ferrioxamine
(Yersinia, klebsiella)
- Measure
neutrophil count
(ANC) before
starting and monitor
ANC weekly
- For neutropenia :
ANC < 1.5 × 109/L)
interrupt treatment
- For
agranulocytosis
(ANC < 0.5 × 109/L),
consider
hospitalization
- Monitor for
symptoms of
arthropathy
- Monitor liver
function regularly
- Monitor creatinine
monthly
-
- Monitor liver
function regularly

163
NOTES
If any patient wants to conceive, chelation therapy should be stopped 3 months
before.
Adjunctive Vitamin C is not recommended.
I.V DFO along with blood transfusion not recommended.
IV DFO is recommended in continuous drip in patient with cardiac failure only(
Patient to be referred to level 3 or 4 for opinion before this )
Combination therapy
If a patient is failing on first line therapy, dose adjustment and attention to adherence
(practical as well as psychological support) are the next steps. If this fails then regime
adjustment can be considered, depending on the circumstances.
Failing this a combination therapy is recommended. Patient may bereferred to level 3
or 4 before startingthe combination therapy.
Following are the recommended combinations
Combined DFO and DFP : DFO given twice a week ( Week end ) combined with
DFP daily at standard doses (75 mg/kg/day).
DFX 20-40 mg/kg/d 7 days per week, plus DFO 40 mg/kg/d 5 days per week
Target of Iron chelation
Ferritin levels between< 500 ng/mL in TDT
Ferritin level < 300 ng / mL in NTDT
LIC - < 5 mg/g dry weight NTDT
LIC < 7 mg / g dry weight TDT
Cardiac T2* >20 ms.
Monitoring chelation therapy (seeTable7, 8)
Monthly biochemistry (creatinine and liver function tests) ( Mandatory )
3 monthly Serum ferritin (Ferritin levels may be inconsistent, especially in sickle ce
ll patients, so decisions must be based on trends over time) ( Mandatory )
Cardiac T2* MRI annually in selected patients (Refer to higher center)
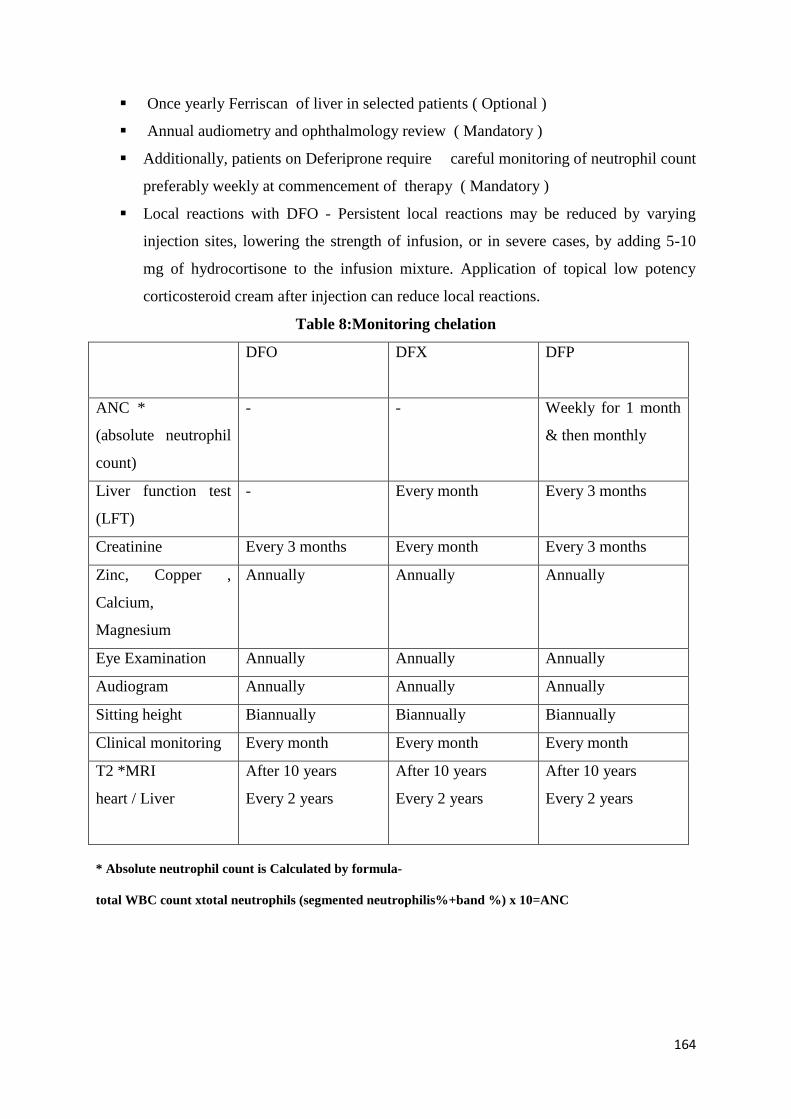
164
Once yearly Ferriscan of liver in selected patients ( Optional )
Annual audiometry and ophthalmology review ( Mandatory )
Additionally, patients on Deferiprone require careful monitoring of neutrophil count
preferably weekly at commencement of therapy ( Mandatory )
Local reactions with DFO - Persistent local reactions may be reduced by varying
injection sites, lowering the strength of infusion, or in severe cases, by adding 5-10
mg of hydrocortisone to the infusion mixture. Application of topical low potency
corticosteroid cream after injection can reduce local reactions.
Table 8:Monitoring chelation
DFO
DFX DFP
ANC *
(absolute neutrophil
count)
- - Weekly for 1 month
& then monthly
Liver function test
(LFT)
- Every month Every 3 months
Creatinine Every 3 months Every month Every 3 months
Zinc, Copper ,
Calcium,
Magnesium
Annually Annually Annually
Eye Examination Annually Annually Annually
Audiogram Annually Annually Annually
Sitting height Biannually Biannually Biannually
Clinical monitoring Every month Every month Every month
T2 *MRI
heart / Liver
After 10 years
Every 2 years
After 10 years
Every 2 years
After 10 years
Every 2 years
* Absolute neutrophil count is Calculated by formula-
total WBC count xtotal neutrophils (segmented neutrophilis%+band %) x 10=ANC

165
CONDITIONS WHEN PATIENT TO BE REFERRED TO LEVEL 3 OR 4
1. NTDT patients with features of iron overload & disproportionately low ferritin –
patient to be referred to level 3 or forT2* MRI for LIC.
2. Patients with high Ferritin value & deteriorating cardiac function – patient to be
referred to level 3 or 4 centersfor T2*MRI for MIC .
3. TDT patients < 2 years of age requiring chelation therapy to be referred to level 3
(COE) or 4 (National referral center) for opinion.
4. For any patient with chelation failure
5. For starting 2nd
or 3rd
line chelating agent
6. For starting combination of chelating agents
7. For persistent toxicity of chelating agent
References:
1. Adamkiewicz TV, Abboud MR, Paley C, et al. Serum ferritin level changes in children with
sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load
and liver injury. Blood. 2009;114:4632–4638.
2. Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the
early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–2179]
3. Kirk P, Roughton M, Porter JB, et al. Cardiac T2* magnetic resonance for prediction of cardiac
complications in thalassemia major. Circulation. 2009b;120:1961–1968.
4. Piga A, Galanello R, Forni GL, et al. Randomized phase II trial of deferasirox (Exjade, ICL670),
a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia
patients with transfusional iron overload. Haematologica. 2006;91:873–880.
5. Pongtanakul B, Viprakasit V. Twice daily deferasirox significantly improves clinical efficacy in
transfusion dependent thalassaemias who were inadequate responders to standard once daily dose.
Blood Cells Mol Dis. 2013;51:96–97.
6. Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron
stores in thalassemia major. N Engl J Med. 2000;343:327–331.
7. Aydinok Y, Kattamis A, Cappellini MD, et al. Deferasirox– Deferoxamine Combination Therapy
Reduces Cardiac Iron With Rapid Liver Iron Removal In Patients With Severe Transfusional Iron
Overload (HYPERION)(abstract). Blood. 2013:2257.
8. Olivieri NF, Buncic JR, Chew E, et al. Visual and auditory neurotoxicity in patients receiving
subcutaneous deferoxamine infusions. N Engl J Med. 1986;314:869–873.

166
9. Aydinok Y, Ulger Z, Nart D, et al. A randomized controlled 1-year study of daily deferiprone plus
twice weekly desferrioxamine compared with daily deferiprone monotherapy in patients with
thalassemia major. Haematologica. 2007;92:1599–1606.
10. Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-
transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the
THALASSA study. Annals of hematology. 2013;92:1485–1493. Cappellini MD, Bejaoui M,
Agaoglu L, et al. Iron chelation with deferasirox in adult and pediatric patients with thalassemia
major: efficacy and safety during 5 years’ follow-up. Blood. 2011;118:884–893.
11. Otto-Duessel M, Aguilar M, Nick H, et al. Comparison of twice-daily vs once-daily deferasirox
dosing in a gerbil model of iron cardiomyopathy. Exp Hematol. 2007;35:1069–1073.
12. Ceci A, Baiardi P, Felisi M, et al. The safety and effectiveness of deferiprone in a large-scale, 3-
year study in Italian patients. British journal of haematology. 2002;118:330–336.
13. Chiodo AA, Alberti PW, Sher GD, et al. Desferrioxamine ototoxicity in an adult transfusion-
dependent population. J Otolaryngol. 1997;26:116–122.
14. Cohen AR, Galanello R, Piga A, et al. Safety and effectiveness of long-term therapy with the oral
iron chelator deferiprone. Blood. 2003;102:1583–1587.]
15. Cohen AR, Mizanin J, Schwartz E. Rapid removal of excessive iron with daily, high-dose
intravenous chelation therapy. J Pediatr. 1989;115:151–155.
16. De Virgillis S, Congia M, Frau F, et al. Desferrioxamine-induced growth retardation in patients
with thalassaemia major. Journal of Pediatrics. 1988;113:661–669.

167
Monitoring and data collection
Routine Outpatient management of Thalassemia Major at level 2
All patients of Thalassemia need to follow up every month and monitored as per the
monitoring section 4, with the dedicated Thalassemia care doctor, nurses and counselor. This
must be supervised by a ICHH centre in charge who can be the pediatrician /medical officer
in charge of the ICHH, or Medical superintendent. This person is responsible for medical
care, record keeping and provision of transfusion, medicines, monitoring and timely referral.
The emphasis is on safe blood transfusion, monitoring of iron overload and documentation.
Register in hemoglobinopathy registry.
Children with thalassemia major or NTBT on regular blood transfusion and chelation
therapyrequire regular monitoring.
Medical staff at Hemoglobinopathy Center should maintain the records properly at regular
intervals as indicated on Table I – IV. Children need to be referred to higher center for tests
which can’t be undertaken at day care center. It is the responsibility of the staff of day care
center to get the appointment for tests or blood samples may to be sent to higher center in a
proper transport system for detailed investigation.
1. Adequacy of Blood transfusion therapy with packed red blood cells (pRBCs)
2. Monitoring for adequacy of Iron chelation for iron overload and adverse effects of
drugs

168
3. Monitoring of complications due to the disease and their treatment with special
emphasis on growth
4. Management of endocrine complications (endocrine, cardiac, skeletal etc.),
5. Discussion of Bone marrow transplantation (BMT)/ Hematopoietic stem cell
transplant (HSCT) if considered
6. Psychological support.
7. Growth assessment is important and for monitoring accurate maintenance of records
are necessary. Height & weight of all these children needs to be recorded on IAP
growth charts (Fig 3&4) to ensure normal growth velocity. Showing of growth
velocity is an early indication of growth retardation.
Integrated centres for hemoglobinopathies and hemophila.
Ensure all hemoglobinopathy patientsare enrolled in Registry as soon as
diagnosis is made.
Enroll at ICHH center for regular care and monitoring, get patient address,
contact number.
Make sure counselling of family for disease and complications is completed.
Make sure Genetic counselling is provided to family. Encourage cascade testing
of family members.
Thalassemia major patients are on regular blood transfusion and chelation
therapy so they need regular monitoring to see for efficacy of chelation, any
modification if needed and to evaluate for toxicities and complications.
Usually the monitoring and data collection is to be done at the integrated center
for haemophilia and hemoglobinopathies attached to Pediatrics/
Hematologydepartment in Medical college or District hospital.
Routine information to be captured for Hemoglobinopathies
Demographic-Complete name address, sex age, diagnosis
Contact number
Baseline tests- blood grouping extended, other important issues

169
Table 9:Monitoring at each visit/transfusion
Name, age, address, hospital number
Date Date Date
Weight
Pre transfusion hemoglobin
Note :- Routine post – transfusion Hb checking is not
recommended.
Liver size (by clinical examination)
Spleen size (by clinical examination)
Volume of PRBC given
Transfusion reaction
Immunizations up to date?
Current medications
1.Folic Acid
2.
3
Table 10:Monthly monitoring for patients
Date Date Date
If on deferasirox – SGPT, SGOT, BUN, S.creatinine, Urine
R/E
If on deferiprone, hydroxyurea
– Complete blood counts including DLC
Next date of transfusion
For SCD/T intermedia
Hydoxyurea
For sickle cell disease/ post splenectomy
Penicillin prophylaxis Other immunizations given
(Pneumococcal etc)
Table 11:Monitoring every 6 months for all patients
Date Date Date
S. ferritin (ng/ml)
SGPT
SGOT
BUN
S. Creatinine
Calcium
Phosphorus
Height
Weight
Growth velocity

170
Table 12: Monitoring every year
Date Date Date
Anti HBs antibody
HCV I gG antibody
HIV 1 & 2
The following tests to be performed annually after the age of 10 years
Blood sugar ( fasting) or GTT
TSH
ECG
Echocardiography
MRI T2* Heart/Liver (for all patients with iron overload)
DEXA Scan
Other endocrine work up as per Level 3 or 4 consultation
Additional tests for SICKLE CELL DISEASE PATIENTS ( SCD)
2-18 years :Trans cranial Doppler
>6 years : URINE Micro Albumin
>6 years : Pulmonary function test
While most of the above investigations for monitoring are available at district
hospitals or state medical colleges, patients may be referred to Institutes of National
importance for annual T2*MRI Heart/Liver at following centers:-
A few centers where these tests are standardized and available are given below. This
list will need updating periodically.
AIIMS, New Delhi
PGIMER, Chandiagarh
CMC,Vellore
SGPGI, Lucknow
Sri Chitra Tirunal for Medical Science and Technology, Thiruvananthapuram
Sri Jayadeva Institute of Cardiovascular Sciences and Research, Bangalore.
Children <10 years with Thalassemia do not routinely requireannual Dexa Scan and
T2*MRI Heart/Liver
171
Fig 3, IAP Boys growth chart
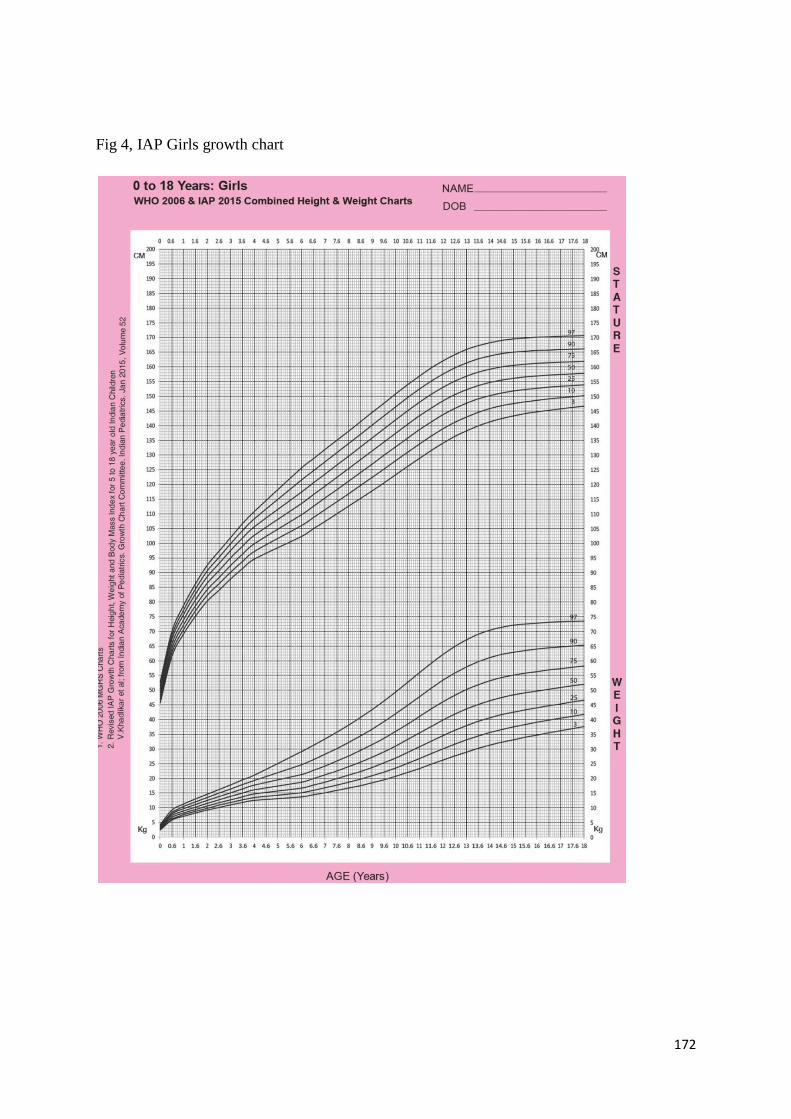
172
Fig 4, IAP Girls growth chart

173
Common Complications and their management in
Thalassemia major
Guidelines for management of complications and long term follow up at level 2/3
Contents
Alloimmunization and its management
Hypersplenism
Transfusion related infections
Cardiac complications
Hepatic complications
Endocrine complications
Indications for splenectomy
Long term follow up and monitoring
One must be aware, that despite adequate iron chelation therapy most of the patients will
develop complications. This is because of excessive iron which gets deposited in different
organs like heart, liver and various endocrine glands which results in their dysfunction /
toxicity of these organ systems. Iron deposition in various endocrine glands may cause
hypothyroidism, hypoparathyrodism, diabetes, growth and development retardation, bone
demineralization and skeletal deformities. Iron chelation if started appropriately and well
monitored for efficacy will reduce the severity of complications.
Patients are also at higher risk to develop transfusion transmitted infections such as
Hepatitis B, C or HIV.
These children need to be monitored for toxicity of iron chelation therapy. Therefore,
management of Thalassemia needsa multidisciplinary team approach consisting of
paediatrician, cardiologist, gastroenterologist, endocrinologist and many problems will
require a haematologist, if not already supervising.

174
There may be limitations of staff & facilities to investigate and diagnose various
complications. However medical staffat the integrated hemoglobinopathy centersshould
have adequate information & knowledge for earlysuspicionof these complications. Then
they need to get the tests performed or refer or make prompt arrangement for appropriate
evaluation at level 2 for their management.
A. Difficulty in maintaining Hb above 10 gm% despite adequate blood
transfusion
The common factors for inability to maintain pre transfusion average hemoglobin of
10g/dlshould be excluded (Table 13). Higher transfusion therapy usually decreases the spleen
enlargement & effect of hypersplenism. Thereis evidence that average transfusion
requirement is 20–30% higher in unsplenectomised than splenectomised thalassemia major
patients, but due to complications splenectomy is to be avoided, unless the hypersplenism is
causing serious compromise and higher transfusion is not manageable by chelation therapy.
1) Alloimmunization–
Children with thalassemia are at higher risk of developing allo-antibodies as a result of
mismatch of minor blood groups. The prevalence of allo-immunization in various counties
varies between 3 to 37 % in various studies. However its prevalence in our country varies
between 3.7 to 15.5%.
Alloimmunization occurs more frequently if blood transfusions are initiated between 18-24
months of age in thalassemia major (TM) or NTBT patients; after splenectomy and in those
who are not getting leucodepleted blood transfusion. Developmentofalloimmunization in
TDT can be prevented by (a) initiation of leucodepleted blood transfusion from the beginning
(b) extended blood group matching at the beginning and by providing proper cross matched
blood against major and minor blood group antigens from the beginning (c) by avoiding
splenectomy.
Development of alloantibodies should be suspected at the ICHH if -
(a) Child develops jaundice within 48 hours of blood transfusion
(b) Passing of high coloured urine
(c) Increased blood transfusion requirement which can be detected by maintaining
accurate amount of blood transfused every time
(d) Difficulty in maintaining the Hb levels
(e) Difficulty in cross matching
All these children need to be referred to level 4 or level 3 centers where the following
facilities are available.
(I) Antibody screening
(II) Identification of antibodies with adequate cell line panel

175
(III) Facilities for extended blood group match.
Children with allo-immunization need to be managed at level 3 or 4 centers. However blood
can be transfused if appropriate cross matched blood is made available from higher centerto
the ICHH centers. (Integrated centre for Hemoglobinopathies & Haemophilia )
Table 13:
Causes of Low Hemoglobin levels
2) Hypersplenism
This complication may be preventable if adequate transfusion is initiated from the beginning
and average pre-transfusion hemoglobin is maintained between 9.5 to 10.5g/dl. It can be
suspected if spleen size is gradually increasing, this can be easily detected in integrated
hemoglobinopathy center by recording the spleen size correctly during every visit. Blood
transfusion requirement exceeding 200ml/kg/year is another indication of hypersplenism.
In the day care center,amount of blood transfused at each visit should be recorded & average
requirement over the last 6 months needsto be calculated periodically at 6 months interval for
its early detection. Development of mono or bicytopenia further supports the possibility of
development of hypersplenism. Such children need to be referred to level 2 for further
evaluation & advice.
These children should be administered vaccines for pneumococcal, meningococcal & HIB
vaccine atleast 3 weeks prior to splenectomy. Children need to undergo splenectomy at level
2 or higher centers. Surgeon may decide laparoscopic or open splenectomy based upon on the
size of spleen and other related factors. All these children need to be given Penicillin
prophylaxis preferably life-long or at least till the age of 20 years.
General Poor Education of Parents & Patients Long Distance from House – DCC Doctors transfused blood only when Hb< 8 gm/dl Only one unit of blood transfused irrespective of Wt / Hb Poverty
Social Belief maintaining high Hb – Iron Overload Family functions (Marriages / House problems ) School exams Summer vacations Family Trips
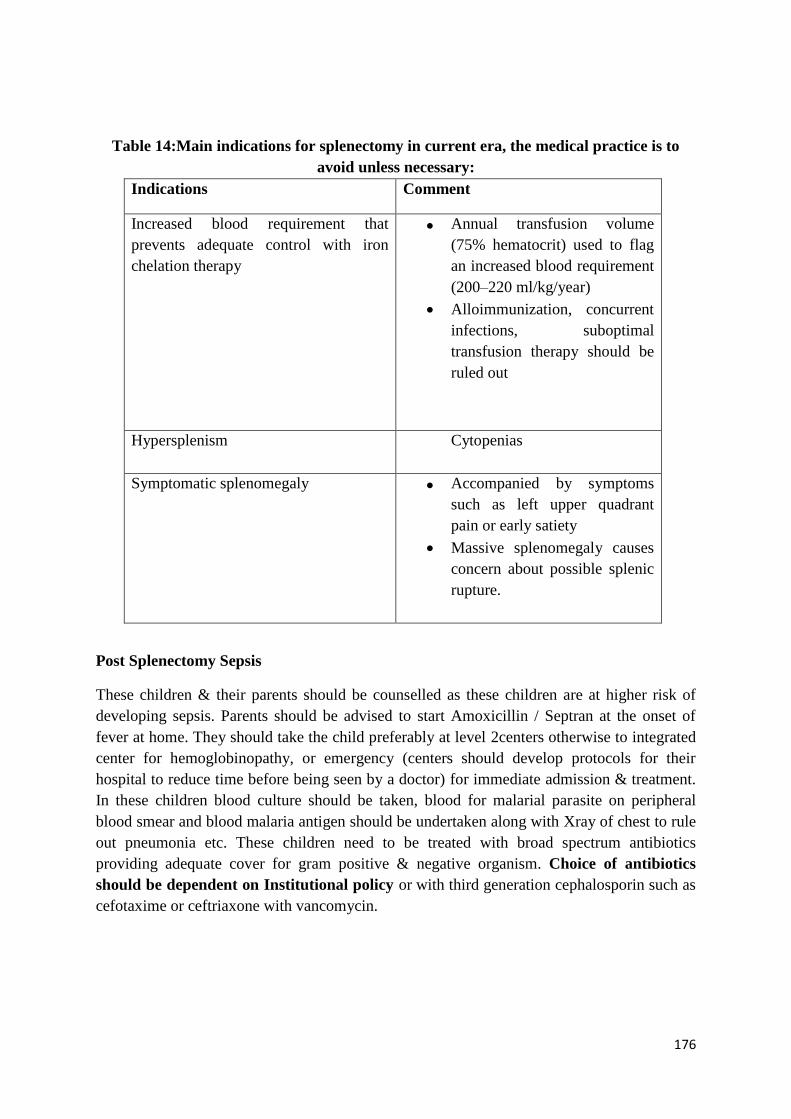
176
Table 14:Main indications for splenectomy in current era, the medical practice is to
avoid unless necessary:
Indications Comment
Increased blood requirement that
prevents adequate control with iron
chelation therapy
Annual transfusion volume
(75% hematocrit) used to flag
an increased blood requirement
(200–220 ml/kg/year)
Alloimmunization, concurrent
infections, suboptimal
transfusion therapy should be
ruled out
Hypersplenism
Cytopenias
Symptomatic splenomegaly Accompanied by symptoms
such as left upper quadrant
pain or early satiety
Massive splenomegaly causes
concern about possible splenic
rupture.
Post Splenectomy Sepsis
These children & their parents should be counselled as these children are at higher risk of
developing sepsis. Parents should be advised to start Amoxicillin / Septran at the onset of
fever at home. They should take the child preferably at level 2centers otherwise to integrated
center for hemoglobinopathy, or emergency (centers should develop protocols for their
hospital to reduce time before being seen by a doctor) for immediate admission & treatment.
In these children blood culture should be taken, blood for malarial parasite on peripheral
blood smear and blood malaria antigen should be undertaken along with Xray of chest to rule
out pneumonia etc. These children need to be treated with broad spectrum antibiotics
providing adequate cover for gram positive & negative organism. Choice of antibiotics
should be dependent on Institutional policy or with third generation cephalosporin such as
cefotaxime or ceftriaxone with vancomycin.

177
Early detection of TTI
Development of hepatitis B or C and HIV infection can be detected only by screening these
children for these infections at yearly interval. Children who are found to be positive need to
be referred to level 3 or 4 centers for detailed evaluation & appropriate management.
B) Transfusion related infections
Hepatitis C virus (HCV), hepatitis B virus (HBV), human immunodeficiency virus (HIV)
and syphilis are the infectious agents usually transmitted via packed red blood cell
(pRBC) transfusions.
Physicians should be aware of the potential life threatening infections in thalassemic
patients. All family needs to be counselled about these infections. Besides these common
TTI other infections which can also be transmitted following blood transfusions include
Cytomegalovirus (CMV), Parvo-B-19, West Nile Virus (WNV), Dengue etc.
Use of storage leucodepleted RBCs stored <14 days reduce risk of CMV, herpes viruses
(e.g Epstein Bar Virus) human T cell lymphotropic virus type I, Yersinia and protozoa
infections (Leishmania species &Trypanosomacruzi)
All patients with TDT should be protected by vaccination against HBV. As the protection
offered by vaccination is not absolute & it wears off over time. Therefore these children
should be given booster vaccine regularly at 5 years interval.
Almost these TTI, malaria may manifest with feverwhich may be associated with rigor
&chills. If any child develops fever within 7-10 days of blood transfusion. They need to
be investigated for TTI & should be treated at least at a level 2center.
C) Cardiaccomplications Cardiac complications secondary to iron overload remain the major cause of morbidity &
mortality. Heart failure is the major complication due to iron accumulation incardiac muscles.
However cardiac complications have been divided in two major groups (Fig. 5).
1) Iron Overload Complications
a) Reversible congestive heart failure
b) Arrhythmias, heart blocks
2) Non iron overload complication
a) Pulmonary hypertension
b) Arrhythmias such as atrial fibrillation (AF)
c) Thrombotic strokes related to AF
d) Cardiac functional changes due to restriction / diastolic dysfunction / fibrosis
e) Arterial changes due to loss of vascular compliance

178
Pathophysiology
Iron accumulation in the heart leads to cardiac dysfunction. Heart is exposed to high levels of
non-transferrin bound iron which results in cardiac damage. Labile iron is quickly bound to
ferritin and degraded to hemosiderin which gets deposited in the myocites. Accumulation of
labile iron in myocytes results in damage to the membrane, cardiac failure, arrhythmias and
fibrosis. Cardiac dysfunction occurs predominantly due to iron overload but development of
hypothyroidism, hypoparathyroidism, hypogonadism can exacerbate cardiac dysfunctions.
Similarly deficiency of thiamine, vitamin D & selenium can worsen cardiac dysfunctions.
MRI cardiac T2* can quantitate cardiac iron stores which allows the clinician to modify
theiron chelation before the development of any cardiac symptoms. Iron chelator should be
selected depending on site of excess iron overload. Consult with a level 3 or 4 center.
Clinical Symptoms
Early symptoms include breathiness on exertion, dyspnea, edema. Clinician needs to examine
the patient for signs of left heart failure such as rales, orthopnea etc. Children with right heart
failure may develop distended neck veins, peripheral oedema, hepatomegaly etc. Heart failure
due to iron overload recovers with intensive chelation therapy.
Symptoms of palpitation may lead to arrhythmia which may be due to myocardial iron
overload or myocardial dysfunction or myocardial fibrosis. These patients need to be
evaluated by ambulatory or exercise electro cardiogram (ECG). Echocardiography (ECHO) is
required to assess various dimensions of heart chambers along with their function, doppler
flow assessment and morphology. MRI T2* indicates the iron overload in the heart with
cardiac complication. These patients need to be evaluated at level 2 / 3centers where the
cardiologist & above facilities are available. Initial management need to be carried at these
centers. Once these patients are stabilised & subsequent management can be carried out at
DCC as per advice of higher centers.
Pulmonary Hypertension
It develops due to interaction of multiple mechanical & biochemical factors leading to
smooth muscle proliferation in pulmonary vasculature (Fig. 6)
Diagnosis & Management
Echocardiography, complete pulmonary functions, higher resolution CT & CT angiogram are
essential for complete evaluation. All these tests will be available at level 2 or level 3. Initial
evaluation & management need to be undertaken at these centers. Subsequent management
can be undertaken at the integrated center for hemoglobinopathies center in consultation with
higher centers.

179
Arrhythmias
Children may present with palpitation or may be asymptomatic. Arrhythmias are life
threatening in presence of heart failure. Presence of Ectopic may trigger more sustained
arrhythmias. The evaluation of these children and management should be undertaken at level
2 or level 3. The subsequent management can be carried at DCC after they have been
stabilised at higher centers.
Management of cardiac complications
Prevention of cardiac toxicity
• Combination therapy with deferiprone (75-100 mg/kg) and deferoxamine (40-50
mg/kg/day sc over 10 – 12 hours thrice a week) is an option to reduce cardiac iron
overload and stabilize cardiac function. Combination therapy needs to be monitored
closely for toxicity and needs to be continued till the cardiac functions improve. Should
be done in collaboration with level 3 or 4 center.
In cardiac failure
• Ensure monitoring for arrhythmias and start on continuous deferoxamine therapy (50
mg/kg/day )as long as the patient has adequate urine output.
• Deferiprone 75 mg/kg/day, should be added in divided doses (TID), as soon as the patient
stabilizes and starts taking orally.
• Management includes diuretics, pressors, and antiarrhythmic drugs in consultation with a
cardiologist.
Key recommendation
• Thalassemia major patients with heart failure should be managed at (or in close
consultation with) a tertiary center experienced in thalassemia.
• Screen and treat endocrine and metabolic co-morbidities in thalassamia major patients
with ventricular dysfunction.
• Combination of two chelation therapy is helpful.
• Routine cardiac T2* MRI assessment represents the best available tool to prevent cardiac
dysfunction
• Echocardiographic screening for pulmonary hypertension should be performed
annually.
• Lifestyle choices that promote vascular health (absence of smoking, regular physical
activity, weight control, vegetable and nitrate rich diet) should be vigorously promoted in
thalassemia patients.
(D) Hepatic dysfunction Liver represents a major target of iron overload in atransfusion dependent thalassemia patient
and majority of patients with non-transfusion dependant thalassemia (NTDT). It is essential

180
to remove excess of hepatic iron as early as possible to protect liver and other organs like
heart & endocrine glands. Hepatitis viruses, especially hepatitis C virus (HCV) and hepatitis
B virus (HBV), should not be underestimated, as these patients are more prone to catch these.
The aggravating role of hepatotoxic co-factors, such as dysmetabolism and alcohol, should
also be kept in mind. We should avoid development of chronic liver disease, as that is an
obvious risk for development of cirrhosis and hepatocellular carcinoma.
Recommendations for prevention of hepatic dysfunction
Hepatic iron excess should be evaluated using a non-invasive strategy based on combined
serum ferritin values and T2* MRI data.
Liver biopsy is often not necessary.
Deferasirox is effective in producing a negative iron balance and decreasing hepatic
damage.
Combination therapy may be used if mono therapy cannot meet its objectives.
Incidence of hepatitis B has gone down significantly by institution of hepatitis B
vaccination at diagnosis, while hepatitis C can be picked up by screening these children
regularly every year.
Diagnosis and treatment of HCV and/or HBV chronic hepatitis should be done in
consultation with hepatologist at level 2 or level 3.
E) Endocrine complications 1. Growth Retardation
Growth retardation occurs almost invariably in TM subjects.
Significant size retardation is observed in stature, sitting height, weight, and biacromial
(shoulder) and bicristal (iliac crest) breadths.
After the age of 4 years, the longitudinal growth patterns display rates of growth
consistently behind those of normal controls. The bone age is frequently delayed after the
age of 6–7 years.
Growth retardation becomes severe with the failure of the pubertal growth spurt.
Key contributing factors to stunted growth is chronic anaemia, transfusional iron overload
and chelation toxicity.
Other important contributing factors include nutritional deficiencies (protein-calorie
malnutrition, vitamin D and A, zinc and carnitine deficiencies), growth hormone
deficiency (GHD) /insufficiency (GHI), insulin like growth factor-I (IGF-I) deficiency,
chronic liver disease, hypogonadism, hypothyroidism and psychosocial stress.
Diagnosis and investigations
Diagnosis requires careful clinical evaluation to establish:
Short stature –
o height below the 3rd centile for sex and age (based on national growth
charts) , and/or
o Slow growth rates- growth velocity expressed in cm/year, below 1SD for
age and sex (based on growth velocity charts), and /or

181
o Signs of other pituitary hormone deficiencies (e.g., gonadotrophins, GHD,
TSH deficiency).
Signs of other possible causes of retarded growth (nutritional deficiencies, chronic hepatic
disease, chronic heart failure).
Routine investigations include: Biochemical analysis, thyroid function (TSH and FT4),
bone age (X-ray of wrist and hand) and bone mineral density (BMD)
Recommendation-
Regular (six-monthly intervals) and accurate measurement of standing and sitting
height, pubertal staging (Table 2) and bone age
Prevention and treatment of growth abnormalities in patients with TM should
include:
Proper blood transfusion to maintain pretransfusion haemoglobin level between 9.5 to 10
g/dl.
Proper chelation to attain serum ferritin < 1000 ng/ml and to maintain this level.
Use of iron-chelators (Deferasirox) with lower toxicity on the skeleton and with better
compliance. Combination therapy may be used to control the serum ferritin if mono therapy
is unsuccessful.
Correction of nutritional deficiencies (protein-calorie, folate, vitamin D, vitamin A, zinc,
carnitine) when suspected.
Oral zinc sulphate supplementation should be given to patients with proven zinc deficiency.
Correction of hypersplenism.
Refer to endocrinologist in level 2 or level 3centers for proper diagnosis and management
of growth delay and GH treatment in patients with GHD.
2. DELAYED PUBERTY AND HYPOGONADISM
Delayed puberty and hypogonadism are the most obvious clinical consequences of iron
overload.
Delayed puberty is defined as the complete lack of pubertal development in girls by the age
of 13 and in boys by the age of 14 year. (see table 15)
Hypogonadism is defined in boys as the absence of testicular enlargement (less than 4 ml)
and in girls as the absence of breast development by the age of 16 years.
All efforts should be made to detect early and refer the case to level 2 or level 3 for
diagnosis & management.
Routine investigations include: biochemical analysis, thyroid function (TSH and FT4),
bone age (X-ray of wrist and hand) and bone mineral density (BMD).

182
Proper and timely management of pubertal delay in boys and girls with TM and appropriate
induction of puberty to attain normal pubertal growth spurt and normal bone accretion.
Tanner staging should be done annually after the age of 10 years.
Treatment can be carried at DCC in consultation with an Endocrinologist who are available
at level 2 or level 3.
3.HYPOTHYROIDISM
Excessive iron gets deposited in the thyroid gland leading to thyroid dysfunctions
Presents with stunted growth, delayed puberty, cardiac failure and pericardial effusion.
Free T4 and TSH are the key investigations
Sub-clinical hypothyroidism is a combination of high TSH with normal FT4 levels.
Overt hypothyroidism is a combination of high TSH with low FT4.
Investigation should be performed annually, beginning at the age of 9 years (unless
symptomatic hypothyroidism is observed) at level 2. Blood samples for these tests may be
sent for early diagnoses.
Additional tests may include the following at level 3centers:
Thyroid autoantibodies: anti-thyroid peroxidase and antithyroglobulin autoantibodies.
Thyroid antibodies performed in selected cases and to exclude autoimmunity, but usually
negative.
Ultrasonography of thyroid gland - to evaluate structural thyroid abnormalities.
Bone age, in selected cases.
Biochemistry including lipid profile.
Serum ferritin.
ECG and Echocardiogram (especially in severe cases).
Hypothalamic-pituitary magnetic resonance imaging (MRI), in selected cases.
Treatment
Good compliance with chelation therapy may prevent or improve subclinical
hypothyroidism (basal TSH 5 to10 mUI/ml).
Patients with overt hypothyroidism should be treated with L-thyroxine which can be easily
given at DCC.
4. HYPOPARATHYROIDISM
Seen in second decade of life of transfusion dependent patients with thalassemia major
Majority of patients show a mild form of the disease accompanied by paraesthesia.
More severe cases may demonstrate tetany, seizures or cardiac failure.
These children may be sent to level 2 or 3 whichever is nearer to DCC.
Investigations
Investigations should begin from the age of 16 years
Serum Calcium, Serum Phosphate levels

183
In children with low serum calcium & higher phosphate level serum parathormone should
be measured
Management
Aim is to prevent acute and chronic complications of hypocalcemia.
Treatment includes:
Oral administration of Vitamin D or one of its analogues.(caution - High doses of Vitamin D
may lead to hypercalcemia) .
Calcitriol, 0.25-1.0 µg, twice daily, is usually sufficient.
In patients with persistently high serum phosphate levels, a phosphate binder (except
aluminium) may be considered.
Tetany and cardiac failure due to severe hypocalcaemia require intravenous administration of
calcium, under careful cardiac monitoring, followed by oral vitamin D.No special diet is
recommended, but few odification in diet can be tried e.g. Diet rich in calcium. - dairy
products, green leafy vegetables.
Diet low in phosphorus-rich items. - avoiding carbonated soft drinks, which contain
phosphorus in the form of phosphoric acid. Eggs and meats also tend to be high in
phosphorus.
Monitoring- At the start of the treatment, weekly blood tests are required. These are
followed by quarterly plasma and 24-hour urinary calcium and phosphate measurements
5.DIABETES MELLITUS (DM)
Impaired glucose tolerance (IGT) & DM accounts for nearly 30% of endocrine disorders.
Often it occurs in patients who have been poorly chelated. However this complication may
occur in children who are well chelated & transfused secondary to chronic anemia earlier,
zinc deficiency and increased collagen deposition. Development of IGT starts in early second
decade along with the development of puberty and DM follows. Development of DM can be
prevented if these children can be identified by oral glucose tolerance test (OGTT) and are
subjected to intensive chelation therapy and further avoidance of chronic hypoxia. Thus it is
essential to get the OGTT regularly from 10th
year of life onwards.
Diagnostic criteria for glucose tolerance test (OGTT):
Fasting glucose >126 mg/dl is diagnostic of diabetes mellitus.
OGTT serum glucose at 2 hours > 200 mg/dl is diagnostic of diabetes mellitus.
OGTT serum glucose at 2 hours > 140 < 200 mg/dl indicates glucose intolerance
Monitoring glycaemic control
Daily home capillary glucose monitoring.
Urine ketones if blood sugar is above 250 mg/dl.
Fructosamine estimation every month.

184
HbA1c is not a reliable indicator of glycaemic control
Assessment of renal function.
Urinary microalbumin and protein.
Evaluation for retinopathy of diabetes mellitus.
Management
Initially it should be at level 2center or higher. Subsequently it can be carried at integrated
center for hemoglobinopathies and haemophilia under the guidance of endocrinologist /
higher center. Measures which need to be followed include (a) strict diet control (b) regular
physical activity (c) intensive iron chelation with two drugs. Children with persistently
elevated blood sugar despite above measures need insulin therapy or oral anti diabetic drugs
under the guidance of an endocrinologist.
These children need to be counselled about the disease and dangers of hypoglycaemic attacks
as well as diabetic coma. These children should know the sign of hypoglycaemia as well as
hyperglycaemia. They can take appropriate measures at home & should reach an integrated
center for hemoglobinopathies and hemophiliaorlevel 2 or higher center immediately for
further management.
F). Osteoporosis
Definition
Osteoporosis is defined when bone mineral density (BMD) T-score is < -2.5 leading to higher
risk of fracture following trivial trauma or minor falls.
Osteopenia is defined when BMD score is between -1 and -2.5.
Normal BMD T score > -1.0
All bones are undergoing dynamic process of resorption and deposition which is controlled
by parathyroid hormone (PTH), vitamin D, growth hormone, steroids, calcitonin etc. In
thalassemia marrow expansion results in cortical thinning and fragility of bones which is
controlled by multiple genetic & acquired factors. Endocrine complications such as
hypothyroidism, hypoparathyroidism, DM hypogonadism result in osteopenia or
osteoporosis. Iron deposition impairs osteoid maturation leading to osteomalacia, osteopenia
& osteoporosis. Vitamin C deficiency in TM increases the risk of osteoporotic fractures.
Reduced physical activity in TM predisposes to bone loss & subsequent osteoporosis.
Symptoms
Bone pains, weakness are early symptoms. Development of fractures on trivial trauma or
minor falls suggests development of osteoporosis.
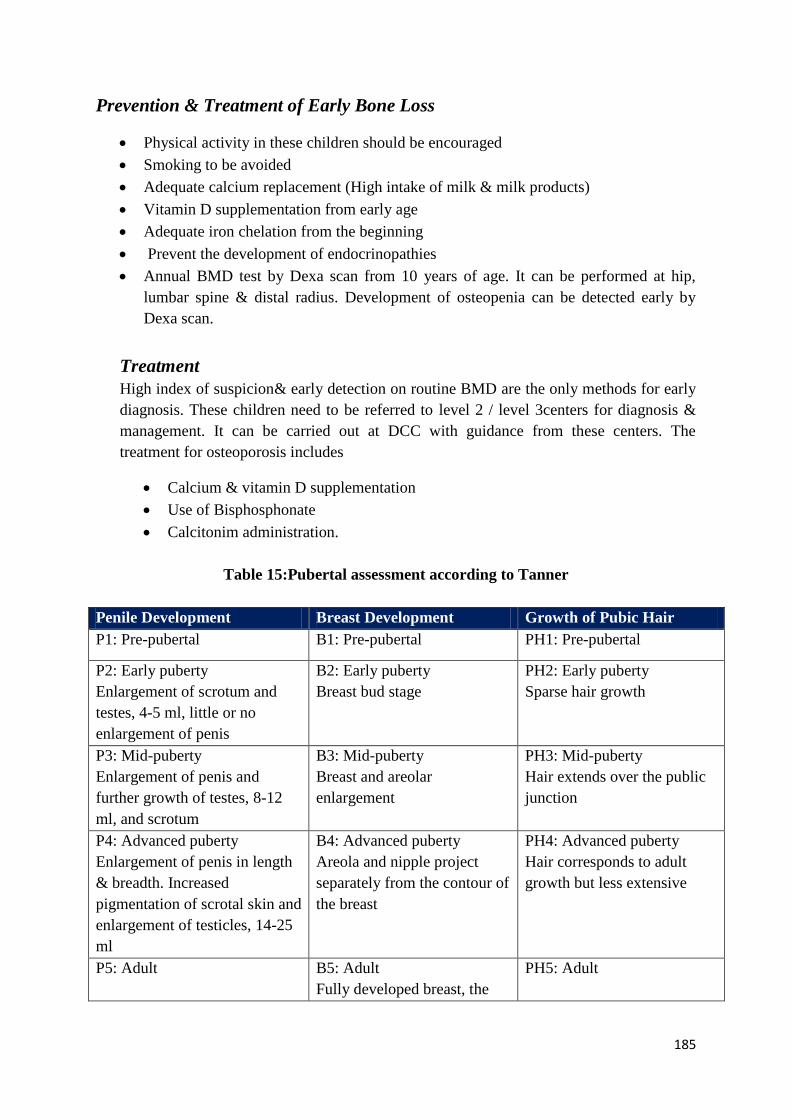
185
Prevention & Treatment of Early Bone Loss
Physical activity in these children should be encouraged
Smoking to be avoided
Adequate calcium replacement (High intake of milk & milk products)
Vitamin D supplementation from early age
Adequate iron chelation from the beginning
Prevent the development of endocrinopathies
Annual BMD test by Dexa scan from 10 years of age. It can be performed at hip,
lumbar spine & distal radius. Development of osteopenia can be detected early by
Dexa scan.
Treatment
High index of suspicion& early detection on routine BMD are the only methods for early
diagnosis. These children need to be referred to level 2 / level 3centers for diagnosis &
management. It can be carried out at DCC with guidance from these centers. The
treatment for osteoporosis includes
Calcium & vitamin D supplementation
Use of Bisphosphonate
Calcitonim administration.
Table 15:Pubertal assessment according to Tanner
Penile Development Breast Development Growth of Pubic Hair
P1: Pre-pubertal B1: Pre-pubertal PH1: Pre-pubertal
P2: Early puberty
Enlargement of scrotum and
testes, 4-5 ml, little or no
enlargement of penis
B2: Early puberty
Breast bud stage
PH2: Early puberty
Sparse hair growth
P3: Mid-puberty
Enlargement of penis and
further growth of testes, 8-12
ml, and scrotum
B3: Mid-puberty
Breast and areolar
enlargement
PH3: Mid-puberty
Hair extends over the public
junction
P4: Advanced puberty
Enlargement of penis in length
& breadth. Increased
pigmentation of scrotal skin and
enlargement of testicles, 14-25
ml
B4: Advanced puberty
Areola and nipple project
separately from the contour of
the breast
PH4: Advanced puberty
Hair corresponds to adult
growth but less extensive
P5: Adult B5: Adult
Fully developed breast, the
PH5: Adult
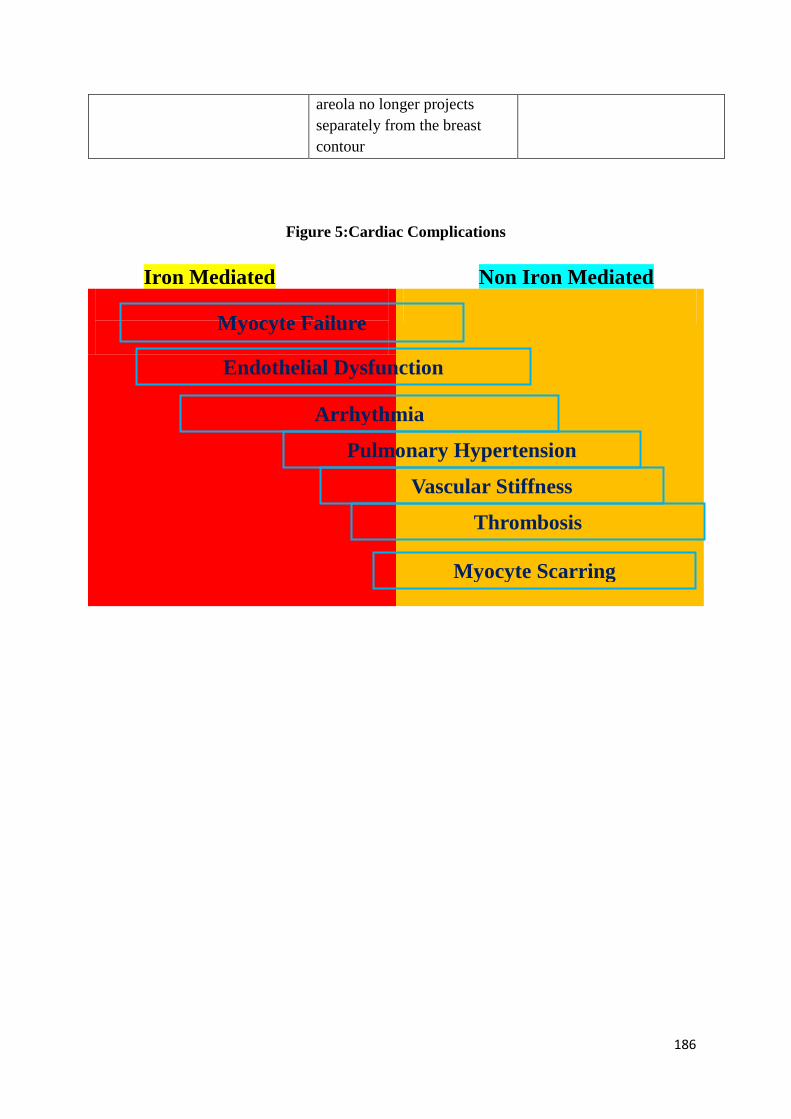
186
areola no longer projects
separately from the breast
contour
Figure 5:Cardiac Complications
Iron Mediated Non Iron Mediated
Myocyte Failure
Endothelial Dysfunction
Arrhythmia
Pulmonary Hypertension
Vascular Stiffness
Thrombosis
Myocyte Scarring
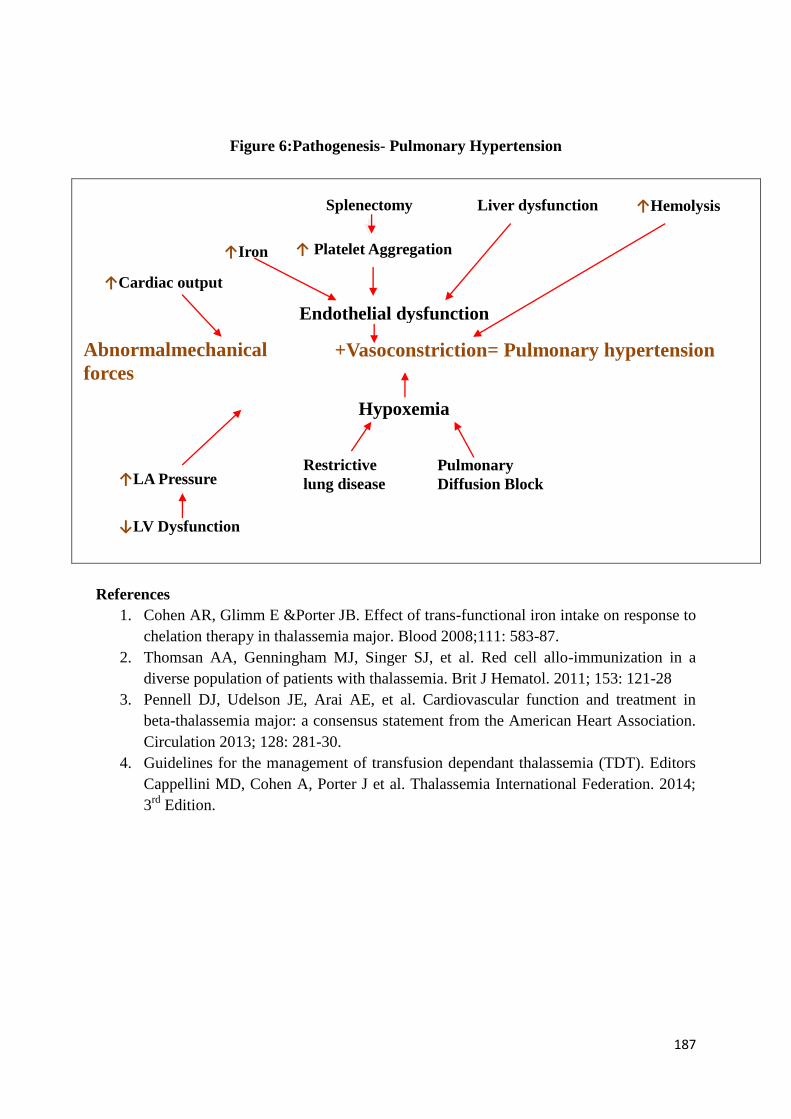
187
Figure 6:Pathogenesis- Pulmonary Hypertension
References
1. Cohen AR, Glimm E &Porter JB. Effect of trans-functional iron intake on response to
chelation therapy in thalassemia major. Blood 2008;111: 583-87.
2. Thomsan AA, Genningham MJ, Singer SJ, et al. Red cell allo-immunization in a
diverse population of patients with thalassemia. Brit J Hematol. 2011; 153: 121-28
3. Pennell DJ, Udelson JE, Arai AE, et al. Cardiovascular function and treatment in
beta-thalassemia major: a consensus statement from the American Heart Association.
Circulation 2013; 128: 281-30.
4. Guidelines for the management of transfusion dependant thalassemia (TDT). Editors
Cappellini MD, Cohen A, Porter J et al. Thalassemia International Federation. 2014;
3rd
Edition.
+Vasoconstriction= Pulmonary hypertension
↑Cardiac output
Abnormalmechanical
forces
↑Hemolysis Liver dysfunction Splenectomy
↑ Platelet Aggregation
Endothelial dysfunction
↑Iron
↑LA Pressure
↓LV Dysfunction
Hypoxemia
Restrictive
lung disease
Pulmonary
Diffusion Block

188
Transfusion Reactions
Blood is a biologic product and great safety must be taken during the blood transfusion
process. Documentation of transfusion records are vital, time of starting, interrupting or
finishing of transfusion, vitals of patient, verification of bag and matching with records , all
of the steps must be recorded with date time and initials or signature. Any symptoms during
transfusion must be noted. The health care providers must be aware of other complications
which may occur later, and report these complications as they may require attention.
Transfusion reaction (TR) can be defined as an unintended response in a patient to
transfusion of blood components which prolongs hospitalization, is disabling or
incapacitating and increases morbidity or causes mortality. TRs can be broadly classified as
(i) acute or delayed (depending upon the time of occurrence) and (ii) immune or non-immune
(depending upon the pathophysiology).
Multi-transfused patients such as thalassemics are at a high risk of having transfusion
reactions due to the multiple and repeated transfusions they receive.
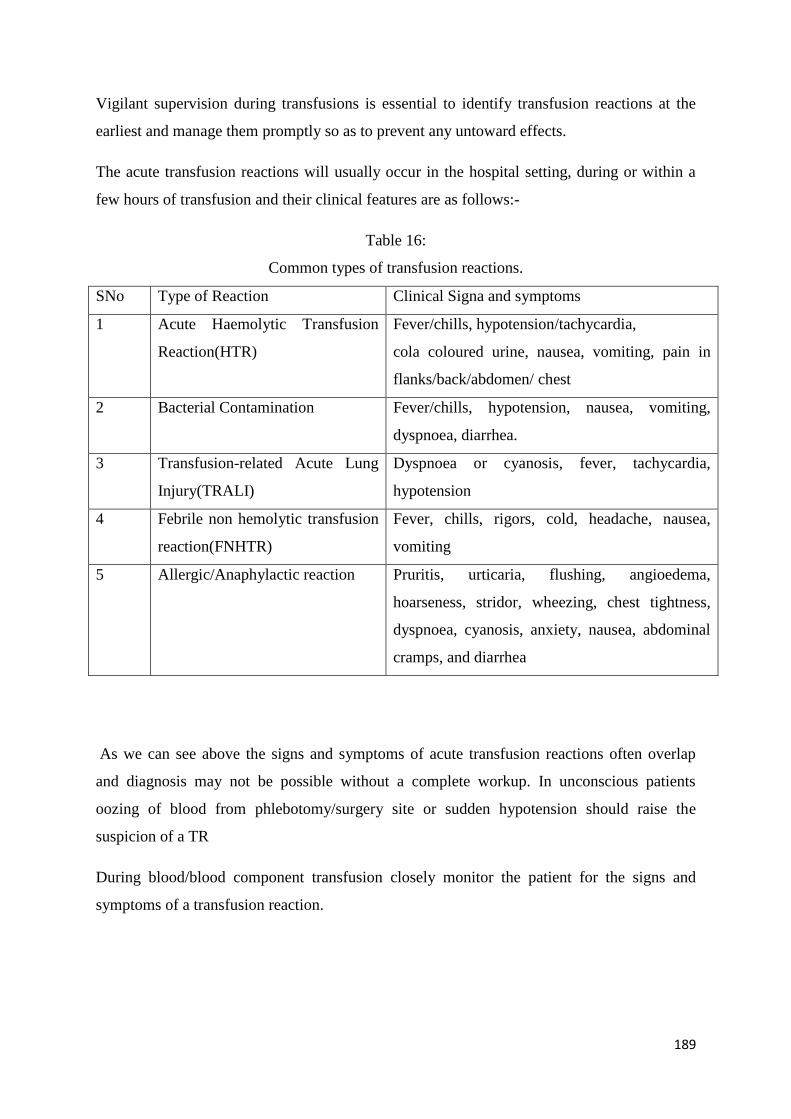
189
Vigilant supervision during transfusions is essential to identify transfusion reactions at the
earliest and manage them promptly so as to prevent any untoward effects.
The acute transfusion reactions will usually occur in the hospital setting, during or within a
few hours of transfusion and their clinical features are as follows:-
Table 16:
Common types of transfusion reactions.
SNo Type of Reaction Clinical Signa and symptoms
1 Acute Haemolytic Transfusion
Reaction(HTR)
Fever/chills, hypotension/tachycardia,
cola coloured urine, nausea, vomiting, pain in
flanks/back/abdomen/ chest
2 Bacterial Contamination Fever/chills, hypotension, nausea, vomiting,
dyspnoea, diarrhea.
3 Transfusion-related Acute Lung
Injury(TRALI)
Dyspnoea or cyanosis, fever, tachycardia,
hypotension
4 Febrile non hemolytic transfusion
reaction(FNHTR)
Fever, chills, rigors, cold, headache, nausea,
vomiting
5 Allergic/Anaphylactic reaction Pruritis, urticaria, flushing, angioedema,
hoarseness, stridor, wheezing, chest tightness,
dyspnoea, cyanosis, anxiety, nausea, abdominal
cramps, and diarrhea
As we can see above the signs and symptoms of acute transfusion reactions often overlap
and diagnosis may not be possible without a complete workup. In unconscious patients
oozing of blood from phlebotomy/surgery site or sudden hypotension should raise the
suspicion of a TR
During blood/blood component transfusion closely monitor the patient for the signs and
symptoms of a transfusion reaction.

190
IN CASE A REACTION IS SUSPECTED-
Stop the transfusion immediately and keep the IV line open with normal saline.
Institute immediate resuscitative care as per the nature of the transfusion reaction
Send the following to the Blood Bank
Blood bag and transfusion set
Post-transfusion blood sample-3-4ml inanEDTA vial
Reaction form with details of the nature of the reaction
If a hemolytic/ septic reaction is suspected: Send following investigations in addition-
Microbiology
1. Blood Culture-from patient within 1 hour of the onset of reaction
Hematology
1. Complete hemogram(CBC)
2. Plasma hemoglobin
3. Urine hemoglobin
4. Coagulation parameters
Biochemistry
1. Bilirubin(conjugated/unconjugated)
2. Serum electrolytes
3. Urea
4. S.Creatinine
5. LDH

191
Management of transfusion reactions-
1. FNHTR–Tab Paracetamol to be given and if this reaction occurs repeatedly, pre-
medication of antipyretic may be given for further transfusions. A cautionary note
being that an anti-pyretic may mask the signs/symptoms of other more serious TRs,
therefore vigilance becomes all the more important.
2. Allergic Reactions–An anti-allergic is to be given and transfusion re-attempted with a
fresh unit of the blood component.
3. Acute HTR–Give injection Lasix. Respiratory support as required and
Hydrocortisone if required.
4. Bacterial Contamination-Supportive care as per symptoms and antibiotics
5. TACO-Transfuse at a slower rate, give injection Lasix if required
6. TRALI- Respiratory support with supplemental oxygen, in severe cases intubation
and mechanical ventilation. Vasopressor agents in case of sustained hypotension.
There is no role of diuretics as the underlying pathophysiology is microvascular
injury.
At the Blood Bank, the following are done-
All documents are checked for any clerical error
In case of mix up, this prevents a second wrong unit from being transfused
Visual Inspection of blood bag
Direct Antiglobulin Test (DAT)
Repeat Grouping from pre and post Transfusion(Tx) sample
Repeat Cross-Match with pre and post-transfusion sample
Indirect Coomb’s Test (IAT)
Antibody identification if required
These tests will assess if the transfused was compatible or not and identification of antibody
if present. The blood bank will rule out any incompatible transfusion and identify any
incriminating antibody in case of incompatibility, which will prevent further reactions in case
of an immune hemolytic reactions. In the case of bacterial contamination, the organism
identified in patient and blood bag will be identical.
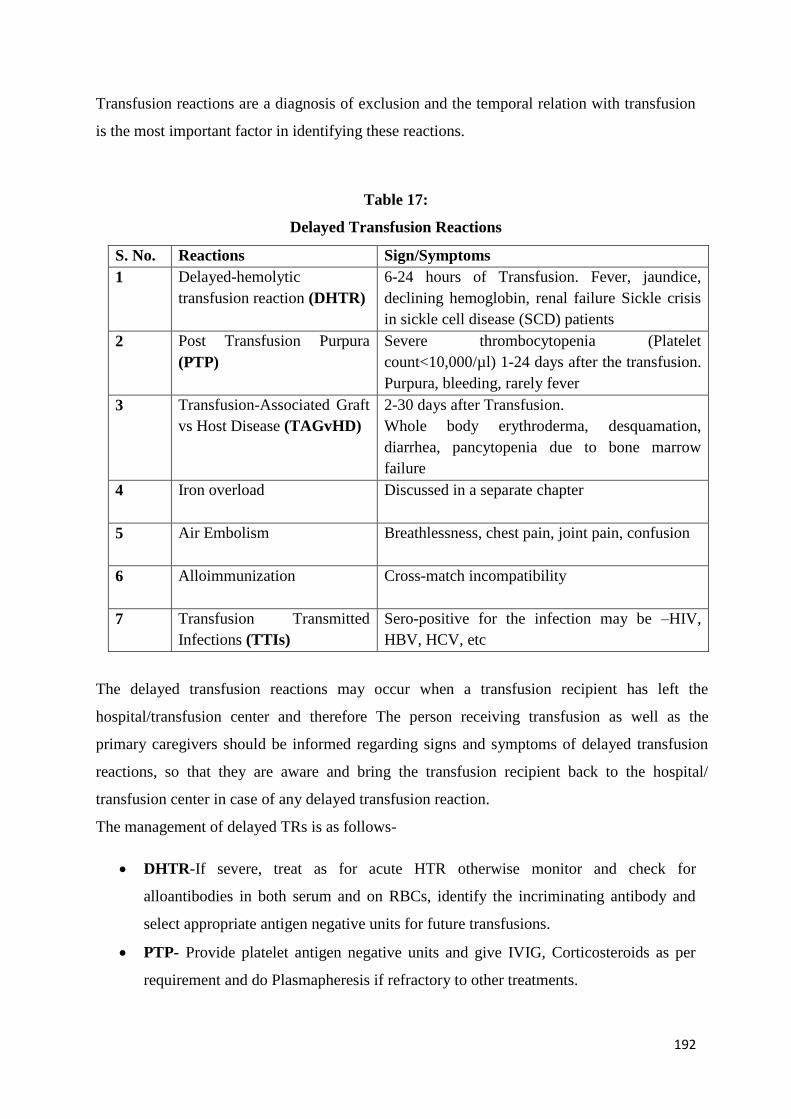
192
Transfusion reactions are a diagnosis of exclusion and the temporal relation with transfusion
is the most important factor in identifying these reactions.
Table 17:
Delayed Transfusion Reactions
S. No. Reactions Sign/Symptoms
1 Delayed-hemolytic
transfusion reaction (DHTR)
6-24 hours of Transfusion. Fever, jaundice,
declining hemoglobin, renal failure Sickle crisis
in sickle cell disease (SCD) patients
2 Post Transfusion Purpura
(PTP)
Severe thrombocytopenia (Platelet
count<10,000/µl) 1-24 days after the transfusion.
Purpura, bleeding, rarely fever
3 Transfusion-Associated Graft
vs Host Disease (TAGvHD)
2-30 days after Transfusion.
Whole body erythroderma, desquamation,
diarrhea, pancytopenia due to bone marrow
failure
4 Iron overload
Discussed in a separate chapter
5 Air Embolism
Breathlessness, chest pain, joint pain, confusion
6 Alloimmunization
Cross-match incompatibility
7 Transfusion Transmitted
Infections (TTIs)
Sero-positive for the infection may be –HIV,
HBV, HCV, etc
The delayed transfusion reactions may occur when a transfusion recipient has left the
hospital/transfusion center and therefore The person receiving transfusion as well as the
primary caregivers should be informed regarding signs and symptoms of delayed transfusion
reactions, so that they are aware and bring the transfusion recipient back to the hospital/
transfusion center in case of any delayed transfusion reaction.
The management of delayed TRs is as follows-
DHTR-If severe, treat as for acute HTR otherwise monitor and check for
alloantibodies in both serum and on RBCs, identify the incriminating antibody and
select appropriate antigen negative units for future transfusions.
PTP- Provide platelet antigen negative units and give IVIG, Corticosteroids as per
requirement and do Plasmapheresis if refractory to other treatments.

193
TAGvHD-Supportive therapy. The condition is difficult to treat and has a very high
mortality. Prevention by using irradiated blood components in at-risk patients should
be strictly adhered to.
Air Embolism- Secure airway, breathing, and circulation (ABC), including
cardiopulmonary resuscitation (CPR) when necessary. Shift to ICU setting for further
management
Alloimmunization- Identify the incriminating antibody and ensure the patient
receives antigen negative units for all future transfusions, whether the antibody is
detected or not in future testing. Give the Transfusion recipient an antibody card
stating the identity of the antigen against which the patient has developed an antibody.
TTIs- treat as per infection treatment protocols after due counseling
The early identification and management of TRs is extremely important to prevent morbidity
and mortality in the transfusion recipients. All transfusion reactions should be reported to the
blood transfusion services whether it impacts the management of the TR or not.
Prevention of transfusion reactions
Every hospital should have a hemo-vigilance programme.
1. Always recheck information and if possible recheck blood group prior to
administration. Positive patient identification is a step which has the highest
potential to prevent Acute TRs as most fatal Transfusions occur due to wrong blood
in tube (WBIT).
2. Do not leave blood after sent from blood bank or storage center, transfuse as soon as
possible. Maintenance of blood cold chain is essential to maintain the quality of
blood components. If a breach in the cold chain is suspected the component should
not be used for transfusion.
3. Always have a doctor on call to manage reactions.
4. Visual inspection of blood components prior to issue/transfusion for bloating,
discoloration, floaters, hemolysis, and leakage is essential.
5. Febrile non haemolytic transfusion reaction (FNHTR) – Due to presence of
leucocytes in the blood product. Use Leuco-depleted blood, if a patient is having
recurrent FNHTR give Paracetamol tablet before blood transfusion.

194
6. Allergic reactions – Urticaria, angioedema & severe cases anaphylaxis due to
plasma proteins, if recurrent , give washed RBC
7. Acute haemolytic transfusion reaction – Stop blood, start IV normal saline, give
injection Furesemide (lasix). Give oxygen, Hydrocortisone if required. Send sample
for complete hemogram, Direct Coombs test, PT, APTT, LFT, Urea, Creatinine.
Notify blood bank. Send blood bag & a fresh patients sample to the blood bank.
8. Volume overload & cardiac failure – slow transfusion, give injection Lasix if
required
9. TRALI (transfusion related acute lung injury) –This is acute lung injury by anti
neutrophilic antibodies present in donor blood. It causes capillary leak & ARDS
leading to tachycardia, dyspnoea, hypoxia within 4-6 hours of transfusion. Fatal
condition. Give oxygen, shift to Intensive care unit (ICU).
10. Transfusion transmitted infections (TTI) Check HBsAg, HCV & HIV every 6
months. NAT tested blood preferable.
11. Universal leucodepletion of blood components ( Platelet and RBC products).
12. Do not use plasma from multiparous women or persons with a history of blood
component transfusion (use only RBCs from this category of donors).
13. Patients with chronic anemia are at risk of volume overload and should be
transfused slowly to prevent TACO, also volume reduced blood components should
be used where ever possible. Never transfuse whole blood to raise hemoglobin.
Packed RBCs (PRBCs) should be used.
14. Irradiation in patients at risk of TAGvHD should be a part of the transfusion
protocol

195
ii) Sickle cell Disease
Basic recognition of symtoms and signs of sickle cell disease at level 1
(PHC/CHC)
Outline
Diagnosis
When to suspect?
Test to be done.
Referral to higher centre if required
Sickle cell disease is a group of inherited red blood cell disorders
When to suspect?
Any child presenting with one of the below mentioned symptoms should be screened for
sickle cell disease.

196
What to Investigate?
Screening test
Complete blood count( CBC) - Anemia with microcytic hypochromic red cell
indices
Peripheral smear- sickle shaped RBCs
Sickling test
Solubility test
Diagnostic test (if screeningtestpositive)
Hb electrophoresis/ HPLC test
What to do before referral
Maintain hydration
Treat Fever and Pain –with Acetaminophen or Non-steroidal anti-inflammatory
drugs (NSAIDs)
Infections (fever) - start antibiotic e.g. Amoxicillin
Anemia
Painful and swollen hands and feet
Recurrent unexplained body pain in chest, abdomen ,joints and bones
Frequent fever/infections
Jaundice
Bone pain
Sudden vision problems
Delayed growth
Stroke/paralysis
Large painful spleen with fever/increasing pallor
Non healing leg ulcer near ankle
Priapism
Non healing Leg ulcers
Family history

197
Important guidelines for management of Sickle Cell Disease
Sickle cell disease(SCD) is a group of inherited single-gene autosomal recessive disorders
caused by the ‘sickle’ gene, which affects haemoglobin structure and leads to complications
like pain, anemia, increased risk of infections, stroke etc.
If not recognized and managed, patients may even have life threatening situations.They
require lifelong treatment and monitoring to ensure the best health possible for the individual.
A suspected patient with sickle cell disease (SCD) will present with the following
complaints:
Persistent pallor , pain, fever, lethargy, jaundice, difficulty in breathing.
Complete physical examination of such a patient is done in the formof :
Vitals including B.P,SpO2 saturation
Hydration status.
Grading of pallor, presence of icterus.
Signs of infection including toxic look.
Systemic examination including Respiratory examination, splenic size, complete
neurological examination, examination of involved joint in pain crisis.
Initial investigationsto be done in a suspected case of sickle cell disease are:
CBC, Peripheral smear, Reticulocyte count, Solubility test.
HPLC analysis confirms the diagnosis and pattern of sickle cell disease.

198
Below ONE year of age only HPLC analysis should be done to diagnose Sickle
cell disease. Solubility. test and Hb Electrophoresis are not useful below 1 year of
age.
If transfusion given then no test for diagnosis of sickle cell disease should be
performed till 3 months after the transfusion.
Fig 7.
Evaluation of child with suspected sickle cell disease
Sodium metabisulfite slide test or solubility test.
(Cannot differentiate between sickle cell trait and disease)
(Not to be used in first year of life as a high level of ‘HbF’ is present)
Confirmation is done by Hb electrophoresis, isoelectric focusing,
highperformance liquid chromatography (HPLC), capillary electrophoresis or
DNA analysis.
(Diagnostic tests for SCD should not be done within 3 months of receiving blood
transfusions.)
Routine Outpatient Care, General Management and Monitoring
Enroll all newly diagnosed patients with sickle cell disease as well as newborns ofSCD
screening programme. Register in Hemoglobinopathy registry, enrol in integrated center
for hemoglobinopathies and haemophilia.
Ensure each family receives counselling for the disease and it'scomplications.
Ensure each family undergoes genetic counselling.
Obtain patient’s baseline clinical disease status via documenting crisis rate, usual
duration of pain crisis, usual home remedy and medication which parents give to subside
pain, and precipitating factors like past h/o acute chest syndrome and requirement of
ICU care and ventilation for it, transfusion received, baseline Hb level, episodes of
jaundice, documentation of co- morbidities, ongoing treatment.
Document HPLC pattern, HbF level.
The clinical evaluation and laboratory investigations should be done during the visit of
the patient to the clinic in the following fashion :-Keep monitoring as per assessment
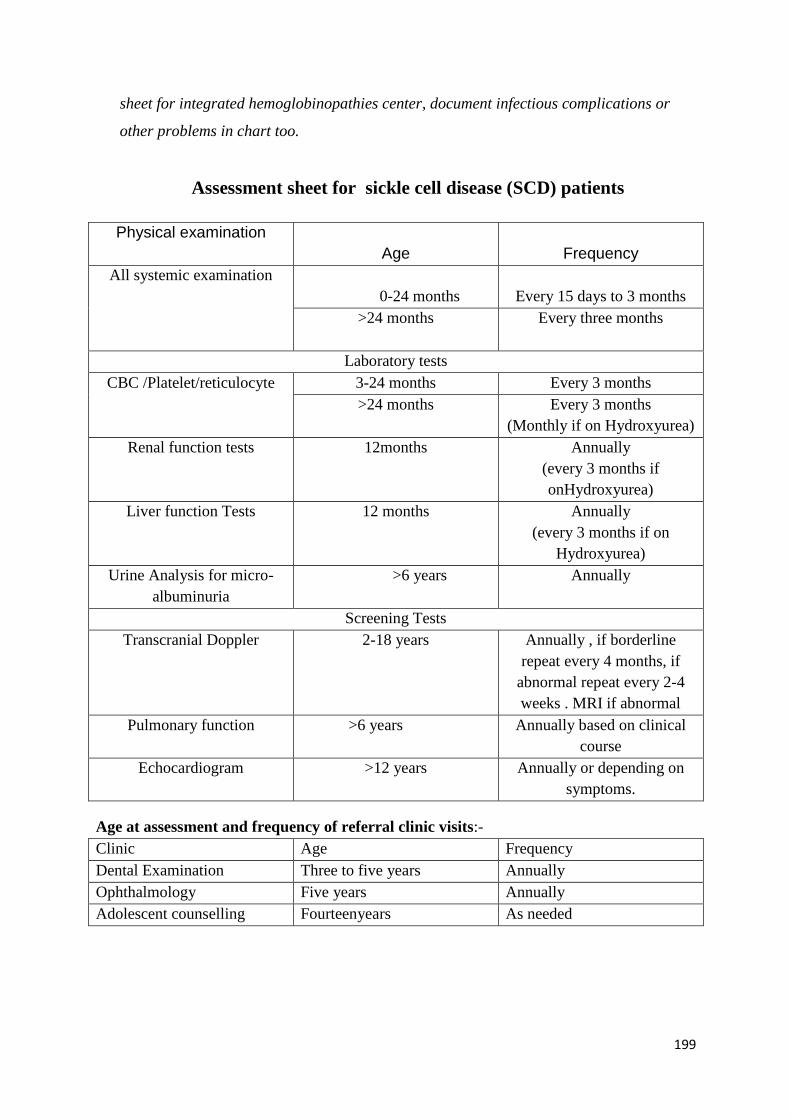
199
sheet for integrated hemoglobinopathies center, document infectious complications or
other problems in chart too.
Assessment sheet for sickle cell disease (SCD) patients
Physical examination
Age
Frequency
All systemic examination
0-24 months
Every 15 days to 3 months
>24 months Every three months
Laboratory tests
CBC /Platelet/reticulocyte 3-24 months Every 3 months
>24 months Every 3 months
(Monthly if on Hydroxyurea)
Renal function tests 12months Annually
(every 3 months if
onHydroxyurea)
Liver function Tests 12 months Annually
(every 3 months if on
Hydroxyurea)
Urine Analysis for micro-
albuminuria
>6 years Annually
Screening Tests
Transcranial Doppler 2-18 years Annually , if borderline
repeat every 4 months, if
abnormal repeat every 2-4
weeks . MRI if abnormal
Pulmonary function >6 years Annually based on clinical
course
Echocardiogram >12 years Annually or depending on
symptoms.
Age at assessment and frequency of referral clinic visits:-
Clinic Age Frequency
Dental Examination Three to five years Annually
Ophthalmology Five years Annually
Adolescent counselling Fourteenyears As needed

200
PENICILLIN PROPHYLAXIS :
Given only to children <5 years- 125 mg BD till 2 years , then 250mg BD after 2 years
till the age of 5 years.
Discontinue penicillin prophylaxis in children with HbSS at age 5 years unless they have
had splenectomy, then continue lifelong.
FOLIC ACID SUPPLEMENTATION:
o less than 1 year of age, 2.5 mg daily
o more than 1 year of age, 5 mg daily
CRITERIA FOR ADMISSION
Immunizations (In addition to routine vaccines)
Acute illness requiring immediate medical care, including emergencies that need to
be defined :-
a. Temperature >38 0C
b. Pain inadequately relieved by home measures
c. Significant respiratory symptoms( cough, shortness of breath, chest pain)
d. Abdominal pain, distention, acute enlargement of spleen.
e. Any neurological signs or symptoms if any
f. Significant increase in pallor, fatigue, lethargy
g. Significant vomiting and diarrhoea
Acute illness characterized by any one of the above signs or symptoms listed above
can prove rapidly life threatening.
Thus it is essential that SCD patients have unimpeded access to health care
providers at the community where appropriate measures can be taken.
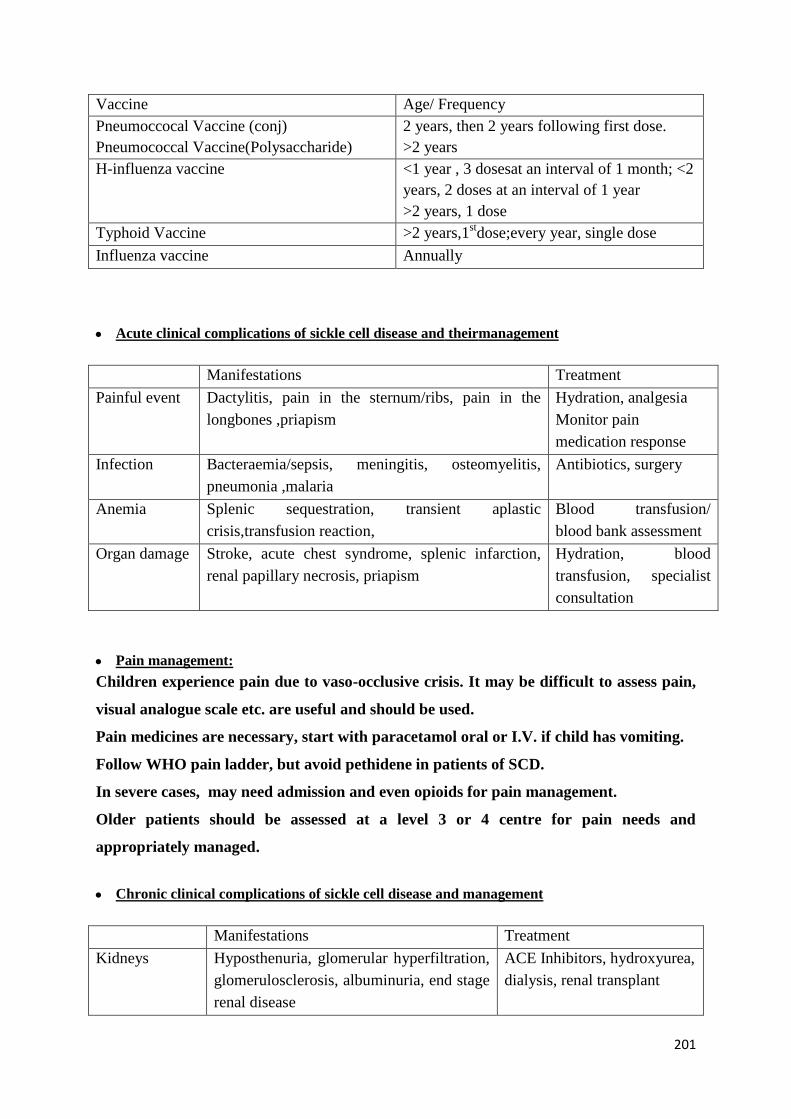
201
Vaccine Age/ Frequency
Pneumoccocal Vaccine (conj)
Pneumococcal Vaccine(Polysaccharide)
2 years, then 2 years following first dose.
>2 years
H-influenza vaccine <1 year , 3 dosesat an interval of 1 month; <2
years, 2 doses at an interval of 1 year
>2 years, 1 dose
Typhoid Vaccine >2 years,1stdose;every year, single dose
Influenza vaccine Annually
Acute clinical complications of sickle cell disease and theirmanagement
Manifestations Treatment
Painful event Dactylitis, pain in the sternum/ribs, pain in the
longbones ,priapism
Hydration, analgesia
Monitor pain
medication response
Infection Bacteraemia/sepsis, meningitis, osteomyelitis,
pneumonia ,malaria
Antibiotics, surgery
Anemia Splenic sequestration, transient aplastic
crisis,transfusion reaction,
Blood transfusion/
blood bank assessment
Organ damage Stroke, acute chest syndrome, splenic infarction,
renal papillary necrosis, priapism
Hydration, blood
transfusion, specialist
consultation
Pain management:
Children experience pain due to vaso-occlusive crisis. It may be difficult to assess pain,
visual analogue scale etc. are useful and should be used.
Pain medicines are necessary, start with paracetamol oral or I.V. if child has vomiting.
Follow WHO pain ladder, but avoid pethidene in patients of SCD.
In severe cases, may need admission and even opioids for pain management.
Older patients should be assessed at a level 3 or 4 centre for pain needs and
appropriately managed.
Chronic clinical complications of sickle cell disease and management
Manifestations Treatment
Kidneys Hyposthenuria, glomerular hyperfiltration,
glomerulosclerosis, albuminuria, end stage
renal disease
ACE Inhibitors, hydroxyurea,
dialysis, renal transplant

202
Heart/Lungs Restrictive lung disease, elevated tricuspid
jet velocity, pulmonary hypertension,
restrictive cardiomyopathy
Bronchodilators,
hydroxycarbamide,
transfusion
Brain Ischemic stroke, haemorrhagic stroke,
silent infarction, neurological decline
Transfusion,
hydroxycarbamide
Liver Jaundice, pigmented gall stones Cholecystectomy
Spleen Infarction, hypersplenism Splenectomy
Bones/skin Avascular necrosis, leg ulcers Physical therapy, cord
decompression, wound care.
Surgery
Eyes Retinopathy Laser therapy
Penis Impotence, infertility Surgery (if needed)
PARENTAL EDUCATION
a) Inheritance pattern, genetic risk
b) Splenic palpation
c) Sensitize to requirement of routine follow up and monitoring
d) Attention to fever and grade of fever
e) Vaccination
f) Recognition of pain
g) Home remedy for pain
h) Need of chronic medication like hydroxyurea
i) When to seek medical help
References
1. Zakari Y. Aliyu, Ashaunta R. Tumblin, and Gregory J. Kato. Current therapy of sickle
cell diseaseHaematologica. 2006 January ; 91(1): 7–10.
2. Expert panel report 2014. Evidence based management of Sickle cell disease
https://www.nhlbi.nih.gov/sites/default/files/media/docs/sickle-cell-disease-
report%20020816_0.pdf ( accessed May 2019)

203
Hydroxyurea therapy- principles and guidelines
Introduction Sickle cell disease (SCD) manifestations have great diversity, even though it isamonogenic
disease. In India SCD patients havethemilder Arab Indian haplotype with high HbFlevels as
compared to Africans. There are many factors responsible for diversity in the phenotype of
SCD, like co inheritance of alpha thalassemia and level of HbF. The overall mortality of
SCD patients in central India is 3.6 per 100 person years. The per patient mortality is 21.8 %
by 1.5 – 5 yrs of age in Gujarat whereas in central India it is higher, 82 % by 3 -4 yrs
Hydroxyurea isa ribonucleotide reductase inhibitor and it prevents the formation of
deoxyncleotides. It causes S phase arrest of all replicating cells. This leads to
myelosuppression,then stress erythropoiesis causesincrease in HbF production. HbF
ameliorates sickling improving both acute and chronic manifestations of SCD.
Even though many drugs lead to increase in HbF, HU has emerged as the most common drug
because of its advantages such as oral therapy, modest toxicity, predictable laboratory
evidence of increase in Hb F and proven clinical efficacy in acute sickle cell crisis,
prevention of organ dysfunction and survival benefits.
Hydroxyurea received US- FDA approval for the treatment of adults with severe SCD in
1998. On December 21, 2017, US -FDA granted regular approval to hydroxyurea for children
from 2 yrs of age with SCD. All the patients of SCD should be treated with HU as it not only
improves symptoms but also survival of these patients.

204
Studies of HU in both children and adults has shown reduced frequency of painful episodes,
Acute Chest Syndrome (ACS) events, less requirements for RBC transfusions and
hospitalizations compared to placebo. On long term follow up studies, it has also shown
survival advantage.
Recommendations:
1. Creating awareness and educatingall patients with SCA and their family members about
benefits of hydroxyurea therapy.
2. All children more than 9 months of age and all adults should be treated with
hydroxyurea. All the patients should be started on HU,however because cytopeniais one
of the preventable side effects, HU should be given wherever laboratory investigations
such as CBC is available and monitoring of SCD patients is possible .
3. To discontinue HU in pregnant or breast-feeding females.
4. To have established prescribing and monitoring protocol for Hydroxyurea therapy in
SCD patients.
5. Consider HU in patients with Hb S- β +thalassemia or Hb SC disease who have
recurrent sickle cell related pain that interferes with daily activities or quality of life.
6. As of now there is no evidence on the role of HU for primary stroke prevention in
adults (1D).
Note:Inthose. situations where the patient who is on hydroxyurea therapy is not likely
to follow up or cannot be monitored, the patient should be given fixed low dose
hydroxyurea therapy (10 mg/kg) without escalation.
Baseline investigation
Complete physical examination.
Hb electrophoresis and quantitative HbF % if available .
CBC with differential and reticulocyte count.
Liver function tests (AST, ALT Total protein).
Albumin, Total bilirubin.
Hydroxyurea Treatment Recommendations – modified as per NHLB 2014 and
BCSH guidelines

205
Renal function (BUN, Cr).
Pregnancy test for post- menarchal females.
Both adult male and female in reproductive age group should be counselled regarding
the need for contraception when on HU treatment
If clinically indicated may perform -
Test the Serum B-12 and folate (to ensure HU-related macrocytosis does not mask
deficiency).
Serum iron, TIBC, ferritin (to ensure HU related macrocytosis does not mask Fe-
deficiency).
Dosing
Optimal dosing of Hydroxyurea is still a debate.
Maximal therapeutic dose (MTD) is a dose which leads to greatest benefit without
toxicities or side effects where there is acceptable degree of marrow suppression.
As per our Indian study to start the patients on 10 – 15mg/kg/ day of hydroxyurea as
single morning dose which can be increased if there is no response or patient is still
symptomatic or patient has stroke and cannot be treated with regular transfusion
regimen.
Starting dosage of Hydroxyurea
Adults: 15 mg/kg/day (round up to the nearest 500 mg)
In patients with chronic kidney disease: 5–10 mg/kg/day
Infants and children: 10 -15 mg/kg/day
Dose escalation by 5 mg/kg every 4–6 weeks only in definite indication with all the
facility of regular follow up, laboratory monitoring and treating physician should be
able to manage the side effects. For dose escalation the flow chart has shown how to
monitor and change the dose of HU.
Essential Monitoring :
CBC and reticulocyte count at least every 4 weeks with the aim of maintaining ANC
>2 x10 9/L, platelet count ≥80,000/uL.
If neutropenia or thrombocytopenia, hold HU and monitor CBC weekly.
When the counts recover, restart the treatment at 5mg/kg/day lower than the previous
dose on which patient had cytopenias.

206
Dose escalation
Hydroxyurea dose escalation to the maximal tolerated dose (MTD) –this dose escalation is
only required if benefit not seen on the present dose.
Dose is escalated as tolerated to reach target absolute neutrophil count (ANC) of 2.0 X 10 9 /L
which is obtained during well or steady visits. Increase the dose by 5mg/kg/d every 8 weeks
up to maximum of 35mg/kg/day. During dose escalation CBC should be monitored at least
every 4 weeks
Once the HU dose is stabilized
CBC, with differential, reticulocyte count and platelet count every 2 – 3months should be
monitored
ALT, AST, BUN, S. Cr every 6 months.
Weight, BP and toxicity focused physical exam each visit.
Inquiry regarding side-effects and contraception.
Importance of adherence to the treatmentand the patient should be counselled frequently
How to prepare HU solution for paediatric patients (now liquid preparation also
commercially available)
Using sterile aseptic techniques- gloves etc empty the contents of the capsule and mix
with room temperature sterile water to get a concentration of 200 mg/ml. and the
solutions should be vigorously stirred.
Then the solution should be filtered to remove insoluble particles and flavoured syrup
can be added to produce a final concentration of 100 mg/ml.
Such preparations have chemical stability over 6 to 9 months in observation period in
a study. The Hospital pharmacy should be able to prepare this under strict aseptic
precaution.
Response to Hydroxyurea
Clinical response with HU treatment will take 3 to 6 months. Hence6 month trial
on the MTD is necessary before considering for drug discontinuation due to
failure of treatment.
Monitor RBC, increase in MCV and HbF levels (only if possible) for evidence of
consistent or progressive laboratory response. Increase in MCV can also be used
as evidence of compliance.

207
Majority of the non-response is mainly due to non- compliance to therapy or
failure to escalate to MTD.
HU should be continued during hospitalization unless patient has toxicity like
febrile neutropenia or bleeding due to thrombocytopenia
Fig 8 Toxicities of Hydroxyurea in SCD patients
Table 18. Short term toxicities of hydroxyurea
Common >10 % Occasional 1-10 % Rare <1% Very rare<<1%
Dose dependant
Neutropenia
Reticulocytopenia
Leukopenia
Thrombocytopenia Diarrhoea Allergic reaction
Anemia Gastritis Leg ulcer
Thinning of hairs Pancytopenia Increase in creatinine
Nausea Rash Increase in ALT
Non dose dependant
Nail / skin
hyperpigmentation
Anorexia Vomiting

208
Long term toxicities:
Leg ulcer is not shown to be common in patients on hydroxyurea but if the SCD
patienthas leg ulcer, he/sheshould be monitored carefully on HU treatment.
Hydroxyurea is not shown to increase the chance of myelodysplastic syndrome or
acute leukemia or any malignancy even after long term use of 17 years.
Fertility – On hydroxyurea many patients and partners had normal delivery.
Hydroxyurea should be stopped 3 months. prior to conception in females and in male
SCD patients whose partner is carrying, there is no need to discontinue. But if the
male partner has problems in conception it should be stopped for 3 months as some of
the studies have shown that HU affects the sperm motility.
Both male and female patients should be counselled regarding risks and benefits of
stopping Hydroxyurea prior to conception or in pregnancy.
To summarise, Hydroxyurea should be the standard of care for SCD patients and should be
included in the essentialdrugslist. Liquid formulation of hydroxyurea will be beneficial for
pediatric dosing. Early treatment will alter the natural course of the disease leading to
decrease in sickle associate acute events as well as chronic organ damage along with better
survival among these patients.
References
1. Bender MA. Sickle Cell Disease. 2003 Sep 15 [Updated 2017 Aug 17]. In: Adam MP,
Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA):
University of Washington, Seattle; 1993-2019.
2. McGann, Patrick T, and Russell E Ware. “Hydroxyurea therapy for sickle cell
anemia.” Expert opinion on drug safety vol. 14, 11 (2015): 1749-58.
3. Segal JB, Strouse JJ, Beach MC, et al. Hydroxyurea for the treatment of sickle cell
disease. Evid Rep Technol Assess. 2008; 165:1–95.
4. Amrana Qureshi,1 Banu Kaya,2 ShivanPancham, on behalf of the British Society for
Haematology. Guidelines for the use of hydroxycarbamide in children and adults
with sickle cell disease. A British Society for Haematology Guideline. British Journal
of Haematology, 2018, 181, 460–475.

209
5. Matthew M. Heeney, Matthew R. Whorton, Thad A. Howard, et al Chemical and
Functional Analysis of Hydroxyurea. Oral Solutions 2004. J PediatrHematol Oncol
2004;26:179–184.
6. Jain, D.L., Sarathi, V., Desai, S., Bhatnagar, M. &Lodha, A. (2012) Low fixed dose
hydroxyurea in severely affected children with sickle cell disease. Hemoglobin, 36,
323–332.
7. Evidence-Based Management of Sickle Cell Disease, Expert Panel Report, 2014:
Guide to Recommendations. U.S department of health and human services.National
Institute of health. National heart lung and blood institute.

210
Common complications in Sickle cell disease and their management
Clinical features of SCD are markedly variable among individuals, depending on the form of
the disease (HbSS, or HbSβ-thal).
Goal of Management of SCD:
To improve quality of life and life expectancy of the affected individuals.
Objectives of Clinical Management of SCD:
a) Prevent and reduce the number of crises and complications
b) Treat crises and complications promptly and effectively
c) Promote a healthy lifestyle
Clinical Presentations
The major acute complications of SCD include the following:
A. Infections
B. Severe anemia (eg, due to splenic sequestration, aplastic crisis, or hyperhemolysis)
C. Vaso-occlusive phenomena
1. Acute vaso-occlusive pain
2. Stroke
3. Acute chest syndrome
4. Renal infarction or medication toxicity
5. Dactylitis or bone infarction
6. Myocardial infarction
7. Complications related to pregnancy
8. Priapism

211
9. Venous thromboembolism
Major chronic manifestations are:
1. Chronic Pain
2. Anemia
3. Neurologic deficits or seizure disorder
4. Pulmonary conditions
5. Renal impairment and hypertension
6. Osteoporosis and complications of bone infarction
7. Cardiomyopathy with diastolic dysfunction
8. Hepatotoxicity and pigmented gallstones
9. Chronic leg ulcers
10. Proliferative retinopathy
Sickle Cell Crisis
This refers to a worsening, over a short period of time, of the symptoms and signs of SCD;
usually associated with pain and/or anemia.
Comprehensive management of the patient in crisis has two major aspects namely:
a) Treatment of acute clinical problems during the episode of crisis; and
b) Arrangements to ensure continued care of the person in (post-crisis) healthy state.
Predisposing Factors for Sickle Cell Crisis:
a) Exposure to cold / drenched by rain
b) Physical exertion
c) Dehydration
d) Injury (including surgical injury)
e) Psychological stress
f) Idiosyncratic (peculiar to the individual)
g) Idiopathic (unidentified)
h) Infections/infestations
Initial Evaluation of a SCD Patient in Crisis should include:
1. History of pain, self-assessment of pain, and prior treatment taken before arrival
atthehospital
2. History of usually effective analgesics
3. History of drug allergies

212
4. Assessment of vital signs: blood pressure, heart rate, respiratory rate, oxygen
saturation (administer oxygen if O2 saturation<90%) and temperature.
5. History of increasing jaundice and passage of coke-coloured urine
6. Assessment of areas of bone tenderness.
Table 19:Initial Treatment of Patient in Sickle Cell Crisis
Pain relief (preferably in a comfortable and quiet environment)
Optimal hydration
Identification and treatment of infections and/or other cor-morbid conditions
Blood transfusion if necessary
Address specific clinical problems e.g. stroke, acute chest syndrome, priapism.
Table 20:Investigationsthat Influence Immediate Management of Sickle Cell Crisis
Urgent Hb/PCV, WBC**, Platelet and Reticulocyte counts, ESR
Examination of Blood Film especially for malaria parasites.
Plasma Bilirubin Level (total & conjugated)
Serum Urea, Electrolytes, Creatinine & C-Reactive Protein (CRP)
Infection Screening (blood, urine, stool, sputum etc ) as necessary
Abdominopelvic ultrasound scan as necessary
Chest X-Ray
Pulse Oximetry (Arterial Blood Gases if SaO2 <92%)
Transcranial Doppler Ultrasonography
CT / MRI
ECG / ECHO
Although a rise in the plasma level of acute phase reactants (e.g. C-reactive protein) may
indicate the transition from steady-state to crisis, the diagnosis of crisis is made on clinical
grounds. The frequency of crisis varies from person to person and from time to time in the
same person.
Acute pain syndrome
This is caused by obstruction of blood vessels, which leads to tissue ischemia /infarction and
pain, which is of sudden onset, with or without history of a predisposing factor, hence also
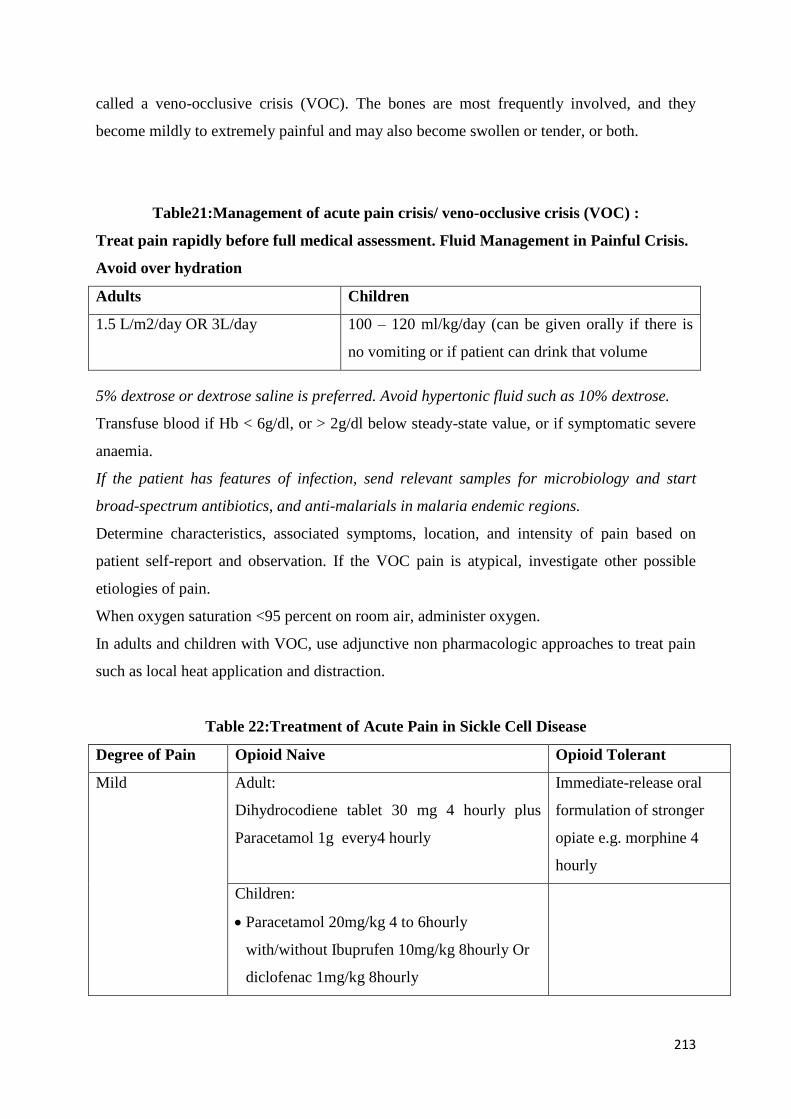
213
called a veno-occlusive crisis (VOC). The bones are most frequently involved, and they
become mildly to extremely painful and may also become swollen or tender, or both.
Table21:Management of acute pain crisis/ veno-occlusive crisis (VOC) :
Treat pain rapidly before full medical assessment. Fluid Management in Painful Crisis.
Avoid over hydration
Adults Children
1.5 L/m2/day OR 3L/day 100 – 120 ml/kg/day (can be given orally if there is
no vomiting or if patient can drink that volume
5% dextrose or dextrose saline is preferred. Avoid hypertonic fluid such as 10% dextrose.
Transfuse blood if Hb < 6g/dl, or > 2g/dl below steady-state value, or if symptomatic severe
anaemia.
If the patient has features of infection, send relevant samples for microbiology and start
broad-spectrum antibiotics, and anti-malarials in malaria endemic regions.
Determine characteristics, associated symptoms, location, and intensity of pain based on
patient self-report and observation. If the VOC pain is atypical, investigate other possible
etiologies of pain.
When oxygen saturation <95 percent on room air, administer oxygen.
In adults and children with VOC, use adjunctive non pharmacologic approaches to treat pain
such as local heat application and distraction.
Table 22:Treatment of Acute Pain in Sickle Cell Disease
Degree of Pain Opioid Naive Opioid Tolerant
Mild Adult:
Dihydrocodiene tablet 30 mg 4 hourly plus
Paracetamol 1g every4 hourly
Immediate-release oral
formulation of stronger
opiate e.g. morphine 4
hourly
Children:
Paracetamol 20mg/kg 4 to 6hourly
with/without Ibuprufen 10mg/kg 8hourly Or
diclofenac 1mg/kg 8hourly
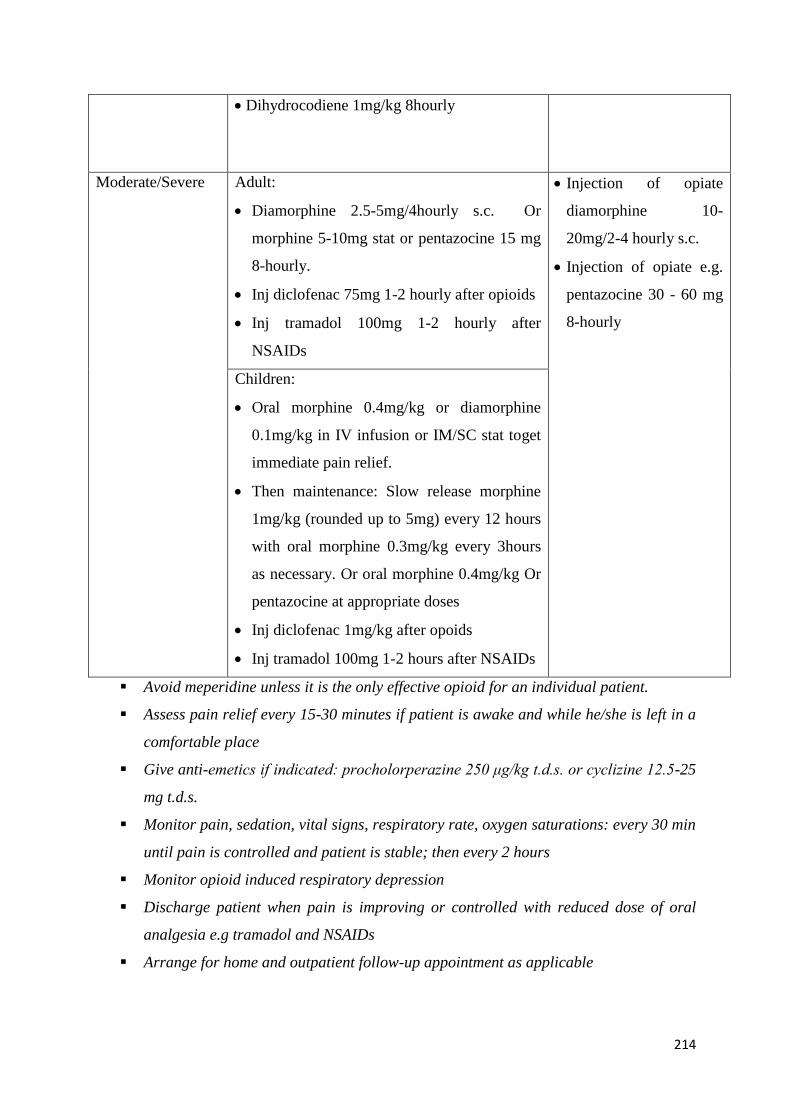
214
Dihydrocodiene 1mg/kg 8hourly
Moderate/Severe Adult:
Diamorphine 2.5-5mg/4hourly s.c. Or
morphine 5-10mg stat or pentazocine 15 mg
8-hourly.
Inj diclofenac 75mg 1-2 hourly after opioids
Inj tramadol 100mg 1-2 hourly after
NSAIDs
Injection of opiate
diamorphine 10-
20mg/2-4 hourly s.c.
Injection of opiate e.g.
pentazocine 30 - 60 mg
8-hourly
Children:
Oral morphine 0.4mg/kg or diamorphine
0.1mg/kg in IV infusion or IM/SC stat toget
immediate pain relief.
Then maintenance: Slow release morphine
1mg/kg (rounded up to 5mg) every 12 hours
with oral morphine 0.3mg/kg every 3hours
as necessary. Or oral morphine 0.4mg/kg Or
pentazocine at appropriate doses
Inj diclofenac 1mg/kg after opoids
Inj tramadol 100mg 1-2 hours after NSAIDs
Avoid meperidine unless it is the only effective opioid for an individual patient.
Assess pain relief every 15-30 minutes if patient is awake and while he/she is left in a
comfortable place
Give anti-emetics if indicated: procholorperazine 250 μg/kg t.d.s. or cyclizine 12.5-25
mg t.d.s.
Monitor pain, sedation, vital signs, respiratory rate, oxygen saturations: every 30 min
until pain is controlled and patient is stable; then every 2 hours
Monitor opioid induced respiratory depression
Discharge patient when pain is improving or controlled with reduced dose of oral
analgesia e.g tramadol and NSAIDs
Arrange for home and outpatient follow-up appointment as applicable

215
Sequestration Crisis
This is pooling (sequestration) of a large proportion of blood in the spleen or liver. Usually
occurs in children < 6 years and some HbSβ-thal adults; unusual in HbSS adults.
Features of sequestration crisis include:
a) Marked pallor
b) Precipitous fall in Hb Level
c) Sudden, progressive enlargement of the spleen / increase in abdominal girth
d) Hypovolaemic shock due to reduced circulating volume
e) Reticulocytosis in blood film (differentiate sequestration from aplastic crisis)
Treatment of Sequestration Crisis:
In consultation with a sickle cell expert, transfuse patientswho have acute splenic
sequestration and severe anemia to raise the hemoglobin to a stable level, while
avoiding over-transfusion.
In consultation with a sickle cell expert, address the performance and timing of
splenectomy in people with recurrent acute splenic sequestration.
Teaching parents, school teachers and care givers how to palpate for an enlarged spleen
or liver
Prevention of Sequestration Crisis:
This is advised after 2 episodes, using one of 2 approaches:
Splenectomy after two episodes of sequestration crises occurring in less than six months
apart
Hypertransfusion in malaria endemic regions.
Hyperhemolytic crisis
Hyperhemolytic crisis refers to the sudden exacerbation of hemolysis with worsening anemia
despite ongoing reticulocyte production. This is a rare complication. The proposed
mechanisms are:
1. Occult splenic sequestration or aplastic crisis detected during a period of resolving
reticulocytosis.
2. Delayed hemolytic transfusion reaction ("bystander hemolysis")

216
3. Accelerated hemolysis in association with acute vaso-occlusive events
4. Infections and/or drug exposure (e.g. co-existence of G6PD defeciency)
Hyperhemolytic crisis is potentially fatal if the cause of hemolysis is not addressed quickly.
Corticosteroids (methylprednisolone 500 mg/day for two days) and intravenous
immunoglobulin (IVIG; eg, 0.4 g/kg daily for five days) are the mainstays of treatment.
Additional compatible transfusions should be limited to situations of life-threatening anemia.
Stroke
Patient with SCD who presents with a new neurologic defect (focal or nonfocal) or altered
level of consciousness is considered to be having potential stroke.
The incidence of stroke is three hundred times higher in HbSS than HbAA children.
Risk Factors for Stroke in SCD:
a. Age (Children of 2-16 years of age are more commonly affected)
b. Blood velocity >200cm/s in distal internal carotid, middle or anterior cerebral
artery
c. Previous Transient Ischemic Attack
d. Steady state Hb < 7 g/dl or Neutrophils > 10 x 109/l
e. Nocturnal hypoxemia + Sleep apnoea
f. HbSS sibling with CVA (reflecting genetic factors)
Differential Diagnoses of Stroke in SCD:
a) Acute ischemic stroke
b) Acute hemorrhagic stroke
c) Transient ischemic attack (TIA)
d) Cerebral venous sinus thrombosis
e) Acute meningitis
f) Seizure, particularly when associated with prolonged postictal paralysis (Todd's)
Prevention of Primary Stroke:
a) All children aged 2-16 years should have trans-cranial ultrasonography to identify
those with a high risk of developing stroke. Repeat in 3 months in cases with a velocity
of 170-199cm/s. If blood velocity ≥ 200 cm/s in the anterior or middle cerebral artery

217
detected on two occasions 6 wks apart is an indication for a transfusion programme to
prevent Primary Stroke.
b) Another indication for Primary Stroke prevention is occurrence of transient ischemic
attacks (TIAs).
c) In those identified with the high risk, stroke should be prevented with hydroxyurea or
transfusion
d) If blood transfusion is used, to be effective, the circulating HbS should be reduced to
<30%
Features of Stroke in SCD:
e) Headache
f) Vomiting
g) Seizures, various sensory or motor neurological deficits: hemiplegia/paresis,
paraplegia/paresis, monoplegia /hemiparesis, loss of hearing, etc.
h) Rarely, sudden reduction or loss of consciousness, or sudden death
i) Magnetic Resonance Imaging (preferred over CT scan) may show acute infarct or
bleed in the brain.
Management of Acute Stroke in SCD:
a) Initial Assessment and Care:
Brief history and physical examination; including the nervous system to distinguish
between symptoms due to pain from those due to weakness (paresis or paraplegia)
Stabilization, support, and monitoring of vital signs as necessary; maintain normal
temperature
Good oxygenation
Careful hydration at two-third of the calculated maintenance volume in case the
Syndrome of Inappropriate ADH Secretion (SIADH) develops
Hydroxyurea can be used if hypertransfusion is not feasible.
b) Laboratory Evaluation (Investigations):
Full blood count with reticulocyte counts,grouping and extended cross-matching,
Blood electrolytes, creatinine, urea, tests for meningitis if suspected clinically.
c) Neuroimaging: MRI with MRV/MRA. DW MRI is highly sensitive in detecting early
ischemic changes.

218
d) Red Cell Transfusion:
Initially, simple (top-up) or exchange transfusion; to be followed by a regular
transfusion programme, preferably exchange, to keep the proportion of HbS below
30% in the patient’s blood; indefinitely, to prevent Secondary Stroke.
e) Iron Chelation: Start when the serum ferritin concentration is 1000 ug/L or greater.
f) Non-medical Interventions and Rehabilitation:
To improve quality of life of People Living With Sickle Cell Disease (PLWSCD), the
use of devices that facilitate mobility, education and self-help skills should be put in
place.
Priapism
This refers to a prolonged painful penile erection, not associated with sexual stimulation.
Types of Priapism:
a) Stuttering, lasts < 4 hr; resolves spontaneously
b) Major or fulminant, with duration of > 4 hrs ; less frequent.
Onset: Mostly in the early hours of the morning and at nights.
Complications:
a) Erectile dysfunction
b) Psychosocial problems.
During clinical assessment, ascertain if there is urinary retention and whether the glans penis
is soft or turgid. This influences treatment. If the patient is able to pass urine and the glans is
soft, then the corpus spongiosum is probably NOT affected, and a glans-cavernosa shunt may
confer clinical benefit.Otherwise, a surgical shunt between the dorsal vein of the penis and
the corpora cavernosa may be more beneficial.
Management of Stuttering (Minor) Priapism:
a) Give slow release tablets of etilefrine. The effect lasts for 8-9 hrs. Start with 25mg
daily taken by 10-11 pm. If clinical response is unsatisfactory after 2 weeks, increase
the dose to 50mg daily. If response still not satisfactory, increase the dose by 25 mg
every 2weeksup to a maximum dose of 100mg .

219
b) If the total daily dose of etilefrine is greater than 50mg, it is recommended that 50mg
be taken by 10-11 pm, and the rest by 4-5 pm.
c) Monitor BP and erectile function in people on etilefrine, because it is a vaso-
constrictor.
a) If etilefrine is not effective in preventing priapism, cyproterone(anti-androgen) is
added.
Management of Major Priapism in Sickle Cell Disease:
a) Give pain killers to comfort the patient
b) Give fluids (oral or intravenous)
c) Urosurgeons should aspirate and irrigate corpora cavernosa with 6-10 mg of
injectable etilefrine diluted in 20mls of saline. If there is no response (detumescence)
after 1 hour, repeat irrigation with etilefrine. Phenylephrine may be used in place of
etilefrine, but is less effective.
d) If repeat irrigation of cavernosa with etilefrine does not lead to detumescence after
1hour, the uro-surgeons should create shunts between the corpus spongiosum and
corpora cavernosa to drain deoxygenated and sickled blood sequestered in the
cavernosa into the spongiosum, and from there to the general circulation.
b) Exchange blood transfusion is usually not effective for established episodes of
fulminant priapis
Management of Chronic Leg Ulceration in Sickle Cell Disease
Chronic leg ulceration occurs in less than 10% of people with SCD, usually in patients who
are older than 12 years.
Clinical Features:
a) Typical ulcerations are situated around the medial or lateral malleolus of one or both
ankles
b) It arises spontaneously, although a history of preceding trauma may occasionally be
obtained
c) Healing is very slow and recurrence or breakdown of healed ulcers is very common.
Treatment:
a) Daily wound dressing
b) Rest and elevate the affected limb
c) Treat any associated infection

220
d) Autologous skin graft.
Factors that Promote Healing Leg Ulcers:
a) A clean wound
b) Daily dressings to keep the surface fresh and clean,
c) Rest.
Factors that reduce Healing of Leg Ulcers:
a) Walking and running long distances
b) Infection of ulcer surface
c) Scabs on the ulcer surface
d) Underlying osteomyelitis.
Infections
Globally, infection is the leading cause of death among SCD patients in developing countries.
It is critical that fever alone is taken seriously in these individuals and considered a potential
emergency situation. Fever associated with pain should not be considered a VOC until
infection is ruled out.
Predisposing factors:Functionalasplenia (hyposplenism), impaired complement activity,
zinc deficiency, iron overload, hypoventilation and the presence of necrotic tissue.
In addition, infection is a common precipitant of an acute crisis, and prompt treatment with
appropriate antibiotics is therefore essential.
Persons with SCD have normal T cell and B cell function, so the risk of acute infection is
generally limited to capsulated micro-organisms, salmonella, or other enteric pathogens.
Opportunistic infections are infrequent.
Viral infections may also be more virulent in individuals with SCD (eg, parvovirus, H1N1
influenza, Zika virus), possibly due to increased sickling and an enhanced inflammatory
response.
Management
1. In people with SCD and a temperature ≥101.3°F (38.5°C), immediately evaluate with
history and physical examination, complete blood count (CBC) with differential,
reticulocyte count, blood culture, and urine culture.

221
2. Promptly administer empiric parenteral antibiotics that provide coverage against
Streptococcus pneumoniae and gram-negative enteric organisms. Subsequent outpatient
management using an oral antibiotic is feasible in people who do not appear ill.
3. Hospitalize people with SCD and a temperature ≥103.1 °F (39.5 °C) and who appear ill
for close observation and intravenous antibiotic therapy.
4. In people with SCD whose febrile illness is accompanied by shortness of breath,
tachypnea, cough, and/or rales, manage according to the preceding recommendations
and obtain an immediate chest x ray to investigate for ACS.
5. In febrile people with SCD who have localized or multifocal bone tenderness, especially
when accompanied by erythema and swelling include bacterial osteomyelitis in the
differential diagnosis and manage accordingly.
6. Worldwide, malaria is a common cause of morbidity and mortality in children with
SCD.
Antibiotic prophylaxis
Penicillin VK 125 mg twice daily orally for children under 3 years of age and 250 mg bid for
children older than 3 years of age is recommended, and continued until adolescence.
Erythromycin is recommended for patients who are allergic to penicillin.
Immunisation
a) Pneumococcal vaccine. Adults and children >2 years of age should receive the
unconjugated vaccine (Pneumovax 23). Conjugated vaccine (Prevenar 13) is provided
to children under the age of 2 years as part of the national immunisation schedule.
Children who have missed the conjugated vaccine should have two doses of the
conjugated vaccine followed by a dose of the unconjugated vaccine, at 6 - 8-week
intervals.
b) A single dose conjugated meningococcal C vaccine should be given.
c) Haemophilus influenzae type B vaccine. If not given as part of the childhood
immunisation programme, a single dose should be given.
d) Annual influenza vaccination is recommended.
Hypersplenism
Hypersplenism classically refers to splenomegaly and any combination of anaemia,
leucopenia and/or thrombocytopenia, with compensatory bone marrow hyperplasia for a
sustained period, and improvement after splenectomy.

222
Because of pre-existing anemia; thrombocytopenia and ‘abnormally normal’ leucocyte count
are the main haematological signs. Transfusion efficiency is usually reduced in SCA patients
with hypersplenism.
Management of splenomegaly and hypersplenism:
Isolated mild splenomegaly warrants no specific management apart from parental education
on the risk of acute splenic sequestration.
When the spleen is markedly enlarged with biological signs of hypersplenism, along with
frequent life-threatening episodes of sequestration crisis, poor growth, bone marrow
hyperplasia and abdominal distension, a supportive transfusion programme can be initiated or
splenectomy performed if age allows. Violent sports should be restricted in order to avoid
traumatic splenic rupture.
Splenectomy in SCD
Splenectomy needs to be considered at an individual level, balancing relative risks and
benefits by taking into account the indication, the medical environment, parental wishes and
reliability, and the risks related to alternative treatment, such as chronic transfusions. In most
patients with SCD it is not required.
Laparoscopic splenectomy has become the procedure of choice for most children requiring
splenectomy. Partial splenectomy has also been proposed as an alternative treatment in very
young children. Following splenectomy, leucocytosis and thrombocytosis are common
findings and generally return to expected basal levels within a year.
Acute Chest Syndrome
ACS is the leading cause of death in SCD, and is characterised by:
Fever – temperature >38.5oC
Respiratory symptoms and signs, including cough, chest pain, dyspnoea or
wheezing
A new pulmonary infiltrate on chest X-ray (e.g. consolidation, segmental
changes, etc.)
ACS may begin as a pain crisis affecting the ribs, chest or shoulders.
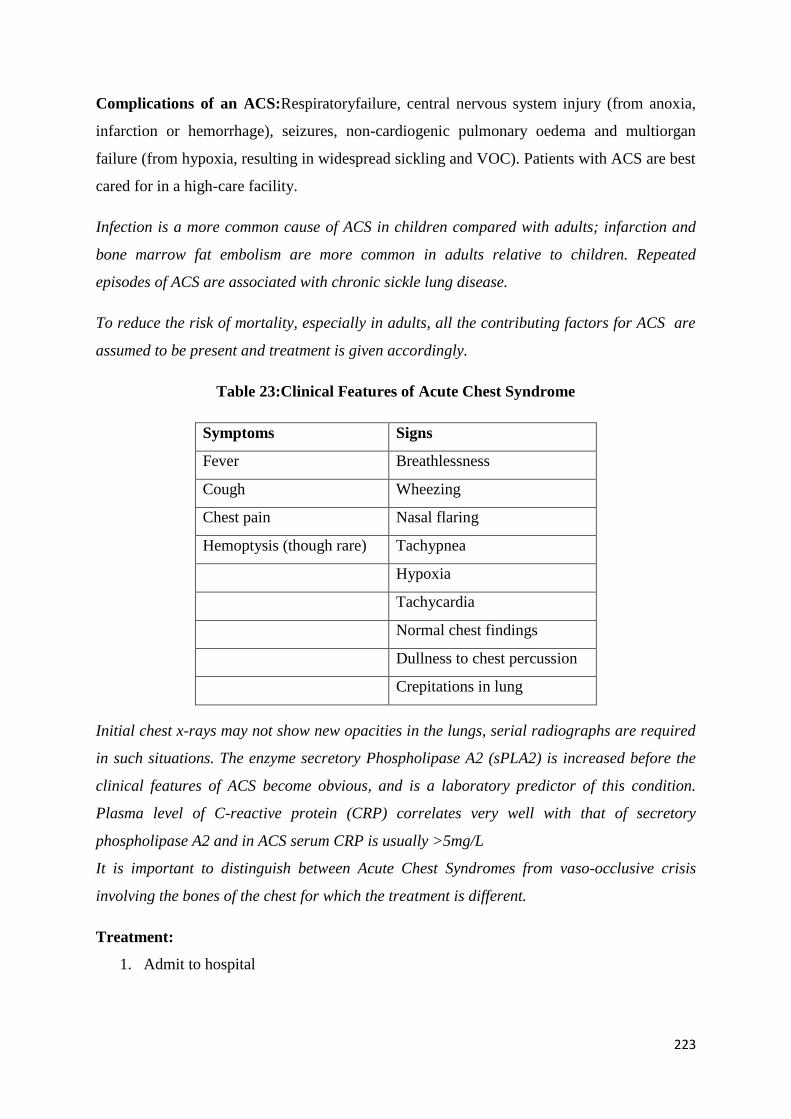
223
Complications of an ACS:Respiratoryfailure, central nervous system injury (from anoxia,
infarction or hemorrhage), seizures, non-cardiogenic pulmonary oedema and multiorgan
failure (from hypoxia, resulting in widespread sickling and VOC). Patients with ACS are best
cared for in a high-care facility.
Infection is a more common cause of ACS in children compared with adults; infarction and
bone marrow fat embolism are more common in adults relative to children. Repeated
episodes of ACS are associated with chronic sickle lung disease.
To reduce the risk of mortality, especially in adults, all the contributing factors for ACS are
assumed to be present and treatment is given accordingly.
Table 23:Clinical Features of Acute Chest Syndrome
Symptoms Signs
Fever Breathlessness
Cough Wheezing
Chest pain Nasal flaring
Hemoptysis (though rare) Tachypnea
Hypoxia
Tachycardia
Normal chest findings
Dullness to chest percussion
Crepitations in lung
Initial chest x-rays may not show new opacities in the lungs, serial radiographs are required
in such situations. The enzyme secretory Phospholipase A2 (sPLA2) is increased before the
clinical features of ACS become obvious, and is a laboratory predictor of this condition.
Plasma level of C-reactive protein (CRP) correlates very well with that of secretory
phospholipase A2 and in ACS serum CRP is usually >5mg/L
It is important to distinguish between Acute Chest Syndromes from vaso-occlusive crisis
involving the bones of the chest for which the treatment is different.
Treatment:
1. Admit to hospital

224
2. Oxygen is beneficial if there is hypoxemia; may not if blood oxygen saturation is
normal
3. Exchange Blood transfusion (EBT), but if not possible, do a top-up so as to facilitate
oxygen delivery to the tissues
4. Broad-spectrum antimicrobials, including clarithromycin for mycoplasma and
Chlamydia
5. Bronchodilators, such as nebulized salbutamol to improve oxygen delivery to the
Lungs
6. Anticoagulant therapy, especially if marrow fat or thromboembolism is suspected
7. Analgesics, to reduce chest pain which may inhibit breathing and impair oxygenation
8. Cautiously maintain optimal hydration (to prevent lung edema).
Prevention of ACS:
Hydroxyurea therapy or hypertransfusion should be commenced after recovery to prevent
recurrence.
Evidence-Based Recommendations for Use of Hydroxyurea Therapy
1. In adults with sickle cell who have ≥3 moderate to severe pain crises associated with
sickle cell disease (SCD) during a 12-month period, initiate treatment with
hydroxyurea.
2. In adults with SCD who have sickle cell–associated pain that interferes with daily
activities and quality of life, initiate treatment with hydroxyurea.
3. In adults with SCD who have a history of severe or recurrent acute chest syndrome
(ACS), initiate treatment with hydroxyurea.
4. In adults with SCD who have severe symptomatic chronic anemia that interferes with
daily activities or quality of life, initiate treatment with hydroxyurea.
5. In infants 9 monthsof age or older, in children, and in adolescents with SCD, offer
treatment with hydroxyurea regardless of clinical severity to reduce complications
(e.g, pain, dactylitis, ACS, anemia) related to SCD.
6. In adults and children with SCD who have chronic kidney disease and are taking
erythropoietin, add hydroxyurea therapy to improve anemia.
7. Discontinue hydroxyurea therapy in women who are pregnant or breastfeeding.
8. Use an established prescribing and monitoring protocol to ensure proper use of
hydroxyurea and maximize benefits and safety.

225
9. In persons with HbSβ+-thalassemia diseasewho have recurrent SCD-associated pain
that interferes with daily activities or quality of life, consult an SCD expert for
consideration of hydroxyurea therapy
10. In persons not demonstrating a clinical response to appropriate doses and duration of
hydroxyurea therapy, consult an SCD expert
Recommended Laboratory Tests before Starting Therapy
Complete blood count (CBC) with white blood cell (WBC) differential, reticulocyte
count, platelet countred blood cell count(RBC) and mean corpuscular volume (MCV)
Quantitative measurement of fetalhemoglobin if available (eg, hemoglobin
electrophoresis, high-performance liquid chromatography)
Renal and liver function tests
Pregnancy test for women
Initiating and Monitoring of Hydroxyurea Therapy
1. Baseline elevation of fetalhemoglobin should not affect the decision to initiate
hydroxyurea therapy.
2. Both males and females of reproductive age should be counseled regarding the need
for contraception while taking hydroxyurea.
3. Starting dosage for adults (500mg capsules): 15mg/kg/day (round up to the nearest
500mg); 5-10mg/kg/day if patient has chronic kidney disease.
4. Starting dosage for infants and children: 20mg/kg/day.
5. Monitor CBC count with WBC differential and reticulocyte count at least every 4
weeks when adjusting dosage.
6. Aim for a target absolute neutrophil count more or equal to 2000/μL; however,
younger persons with lower baseline counts may safely tolerate absolute neutrophil
counts down to 1250/μL.
7. Maintain platelet count more than or equal to 80 000/μL.
8. If neutropenia or thrombocytopenia occurs:
Temporarily stop hydroxyurea dosing.
Monitor CBC count with WBC differential weekly.
When blood counts have recovered, reinstitute hydroxyurea at a dose of 5
mg/kg/day lower than the dose given before onset of cytopenia

226
9. If dose escalation is warranted based on clinical and laboratory findings, proceed as
follows:
Increase by 5-mg/kg/day increments every 8weeks.
Give until mild myelosuppression (absolute neutrophil count of 2000-
4000/μL) is achieved, up to a maximum of 35mg/kg/day.
Once a stable dose is established, laboratory safety monitoring should include
CBC count with WBC differential, reticulocyte count, and platelet count every
2-3 months.
10. People should be reminded that the effectiveness of hydroxyurea depends on their
adherence to daily dosing; they should be counseled not to double up doses if a dose
is missed
11. A clinical response to treatment with hydroxyurea may take 3-6 months; therefore, a
6-month trial on the maximum tolerated dose is required prior to considering
discontinuation due to treatment failure (whether due to lack of adherence or failure to
respond to therapy).
Monitor RBC MCV and fetalhemoglobin levels for evidence of consistent or
progressive laboratory response.
12. A lack of increase in MCV, fetalhemoglobin, or both, is not an indication to
discontinue therapy.
13. For the patient who has a clinical response, long-term hydroxyurea therapy is
indicated.
14. Hydroxyurea therapy should be continued during hospitalizations or illness. In case
Patient doesn’t respond to Hydoxurea crizanlizuamb may be given to older patients
16 and above. Crizanlizumab is only recommended to the patients who are
experiencing severe adverse events when on Hydoxurea theraphy. Or on patients on
maximum dose of Hydoxurea but are stillexperiencing more than three VOCs per
annum
15. New data suggests lower doses of hydroxyurea may also be beneficial. Titrate to
response.
References:

227
1. Alli NA, Patel M Recommendations for the management of sickle cell disease in
South Africa. S Afr Med J. 2014 Nov;104(11):743-51.
2. Barbara P. Yawn, George R. Buchanan Management of Sickle Cell Disease Summary
of the 2014 Evidence-Based Report by Expert Panel Members. JAMA September 10,
2014 Volume 312, Number 10
3. http://www.nhlbi.nih.gov/health-pro/guidelines/current/management-sickle-cell-
disease.htm (Accessed on September 30, 2014).
4. John J Strouse. Is low dose hydroxyurea the solution to the global sickle cell crisis?
Pediatric Blood cancer 2015 June:62(6)929-30
Exchange transfusion and simple transfusion in Sickle Cell Disease
All patients with Sickle cell disease may not have anemia.
Avoid blood transfusion unlessanemia is symptomatic.
Transfusion therapy not indicated for management of uncomplicated pain crisis.
Patient should receive an annual check- up at level 3 or 4 center with an expert
or at anytime if there are complications.
Blood transfusion is a supportive therapy in Sickle cell disease
Blood transfusion therapy in SCD can be used regularly (prophylactically) to prevent
or manage complications like stroke or only in the case of an acute life threatening
complications.
Effect of blood transfusion in SCD patients due to-
o Dilution of HbS red blood cells by normal containing cells from normal
donors.

228
o Decrease in percentage of HbS, due to the longer circulating lifespan of HbA /
normal red blood cells.
o Increase in Hb oxygen saturation levels by approximately 1 to 6 %, this results
in increased oxygen delivery to tissues.
o Suppression of erythropoietin.
SCD patients are more likely to have complications of blood transfusion like-
alloimmunization (antibody to donor minor blood group antigens), febrile non-
hemolytic transfusion reactions etc.
BEFORE starting regular transfusion support consider performing or sending patient
for extended blood grouping.
Alloimmunization to red cell antigens is a common occurrence in transfused patients
with SCD. Alloimmunization occurs rapidly after the initiation of transfusions
without extended matching.
Alloimmunization to minor RBC antigens in patients with SCD can be associated
with vaso-occlusive episodes and life-threatening hemolysis. Equally important,
alloimmunization can result in a greater risk of future transfusion reactions and
greater difficulty identifying compatible units of blood
Strong recommendation for those patients with clinically significant antibody
inthepast to get matching for minor RBC antigens (C, E, and Kell) to reduce the
risk of alloimmunization,
DNA typing may be done at higher centers. genotyping can be performed after
transfusions, if needed, to determine sources of incompatibility.
Other precautions of any transfusion in SCD patients-
Use of prestorage leukodepletion or white blood cell filter to decrease the rate of
febrile non-hemolytic transfusion reactions.
Individuals with SCD should not receive blood transfusion therapy from
individuals with sickle cell trait as part of regular transfusion therapy program.
No evidence of PRBC Storage time linkage with outcomes.
Irradiation not required unless planning for BMT.
Immunize against hepatitis B.
Monitor for iron overload and start chelation therapy appropriately.
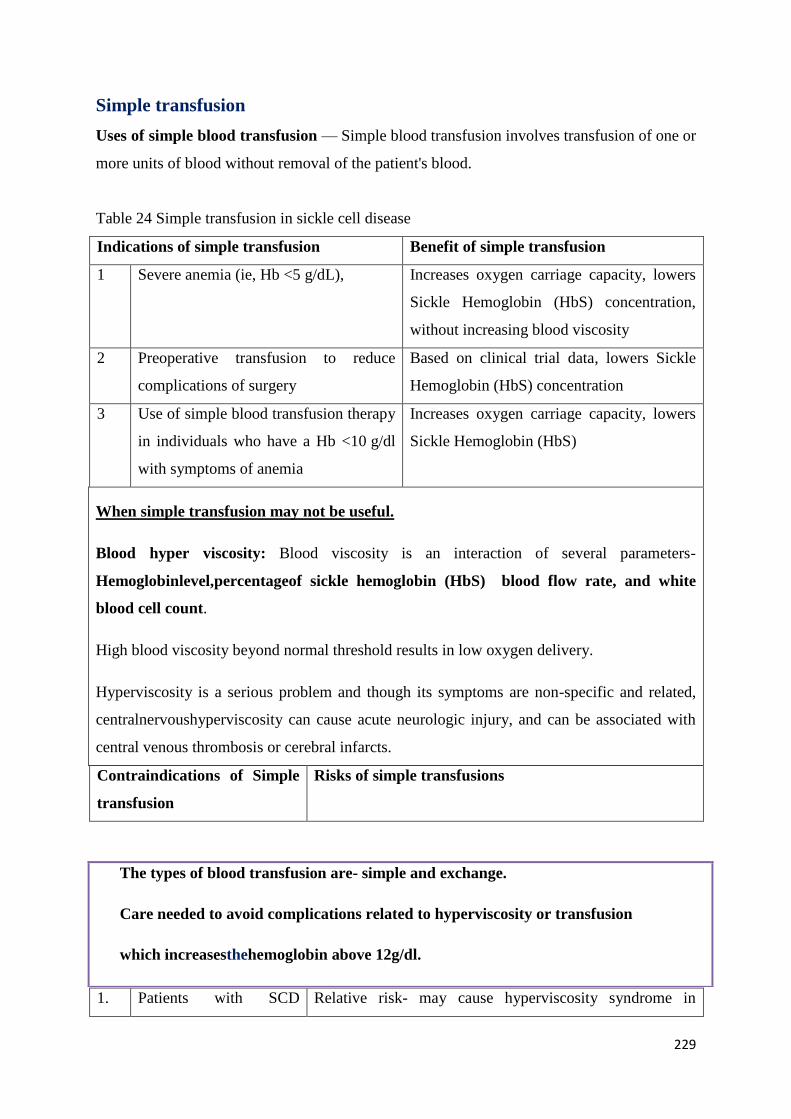
229
Simple transfusion
Uses of simple blood transfusion — Simple blood transfusion involves transfusion of one or
more units of blood without removal of the patient's blood.
Table 24 Simple transfusion in sickle cell disease
Indications of simple transfusion Benefit of simple transfusion
1 Severe anemia (ie, Hb <5 g/dL), Increases oxygen carriage capacity, lowers
Sickle Hemoglobin (HbS) concentration,
without increasing blood viscosity
2 Preoperative transfusion to reduce
complications of surgery
Based on clinical trial data, lowers Sickle
Hemoglobin (HbS) concentration
3 Use of simple blood transfusion therapy
in individuals who have a Hb <10 g/dl
with symptoms of anemia
Increases oxygen carriage capacity, lowers
Sickle Hemoglobin (HbS)
When simple transfusion may not be useful.
Blood hyper viscosity: Blood viscosity is an interaction of several parameters-
Hemoglobinlevel,percentageof sickle hemoglobin (HbS) blood flow rate, and white
blood cell count.
High blood viscosity beyond normal threshold results in low oxygen delivery.
Hyperviscosity is a serious problem and though its symptoms are non-specific and related,
centralnervoushyperviscosity can cause acute neurologic injury, and can be associated with
central venous thrombosis or cerebral infarcts.
Contraindications of Simple
transfusion
Risks of simple transfusions
1. Patients with SCD Relative risk- may cause hyperviscosity syndrome in
The types of blood transfusion are- simple and exchange.
Care needed to avoid complications related to hyperviscosity or transfusion
which increasesthehemoglobin above 12g/dl.
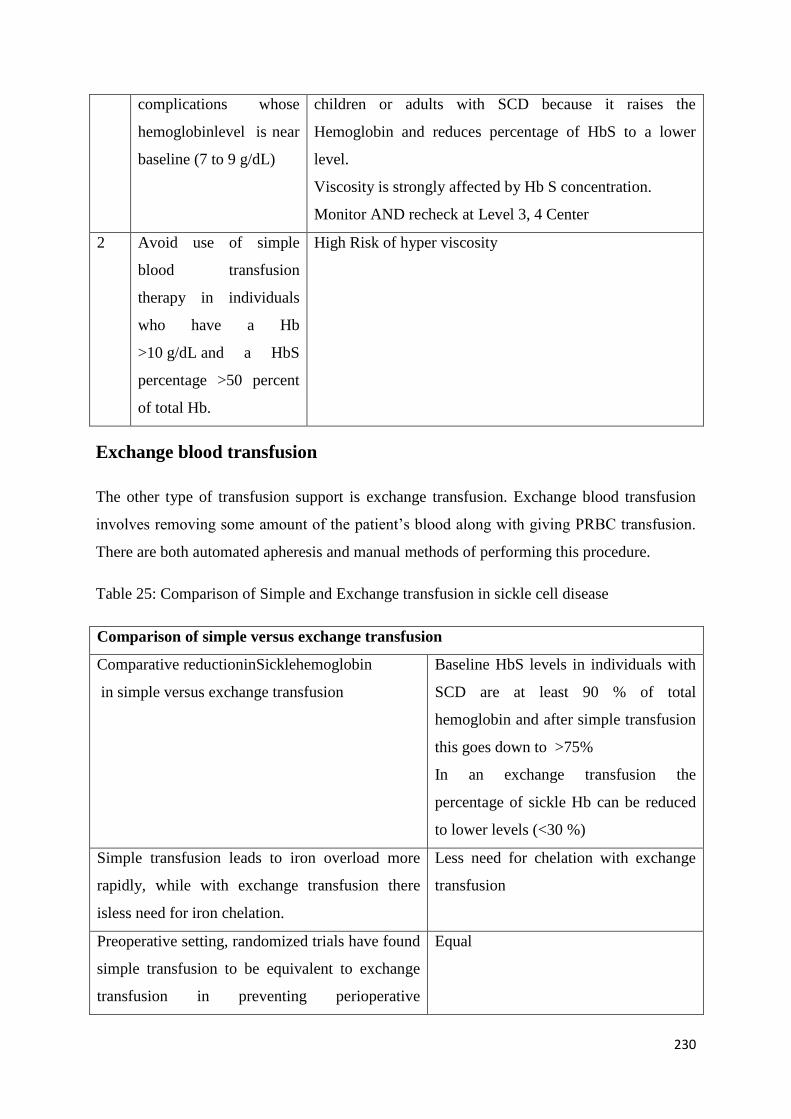
230
complications whose
hemoglobinlevel is near
baseline (7 to 9 g/dL)
children or adults with SCD because it raises the
Hemoglobin and reduces percentage of HbS to a lower
level.
Viscosity is strongly affected by Hb S concentration.
Monitor AND recheck at Level 3, 4 Center
2 Avoid use of simple
blood transfusion
therapy in individuals
who have a Hb
>10 g/dL and a HbS
percentage >50 percent
of total Hb.
High Risk of hyper viscosity
Exchange blood transfusion
The other type of transfusion support is exchange transfusion. Exchange blood transfusion
involves removing some amount of the patient’s blood along with giving PRBC transfusion.
There are both automated apheresis and manual methods of performing this procedure.
Table 25: Comparison of Simple and Exchange transfusion in sickle cell disease
Comparison of simple versus exchange transfusion
Comparative reductioninSicklehemoglobin
in simple versus exchange transfusion
Baseline HbS levels in individuals with
SCD are at least 90 % of total
hemoglobin and after simple transfusion
this goes down to >75%
In an exchange transfusion the
percentage of sickle Hb can be reduced
to lower levels (<30 %)
Simple transfusion leads to iron overload more
rapidly, while with exchange transfusion there
isless need for iron chelation.
Less need for chelation with exchange
transfusion
Preoperative setting, randomized trials have found
simple transfusion to be equivalent to exchange
transfusion in preventing perioperative
Equal
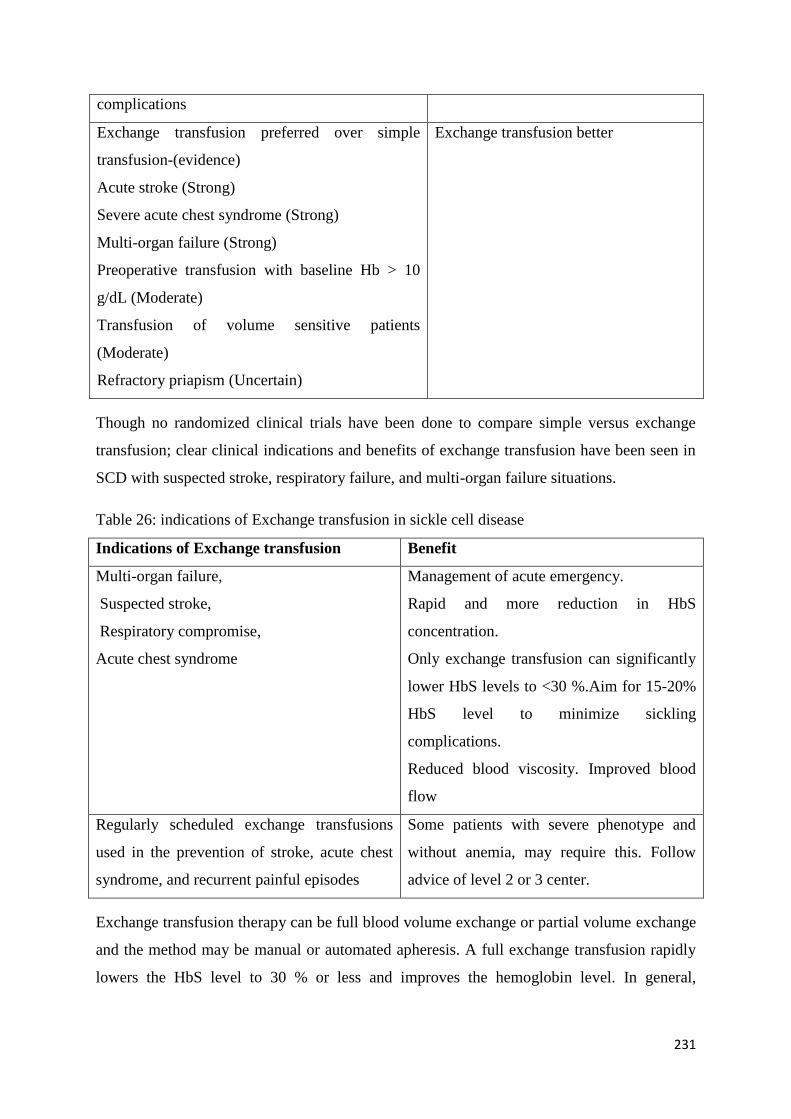
231
complications
Exchange transfusion preferred over simple
transfusion-(evidence)
Acute stroke (Strong)
Severe acute chest syndrome (Strong)
Multi-organ failure (Strong)
Preoperative transfusion with baseline Hb > 10
g/dL (Moderate)
Transfusion of volume sensitive patients
(Moderate)
Refractory priapism (Uncertain)
Exchange transfusion better
Though no randomized clinical trials have been done to compare simple versus exchange
transfusion; clear clinical indications and benefits of exchange transfusion have been seen in
SCD with suspected stroke, respiratory failure, and multi-organ failure situations.
Table 26: indications of Exchange transfusion in sickle cell disease
Indications of Exchange transfusion Benefit
Multi-organ failure,
Suspected stroke,
Respiratory compromise,
Acute chest syndrome
Management of acute emergency.
Rapid and more reduction in HbS
concentration.
Only exchange transfusion can significantly
lower HbS levels to <30 %.Aim for 15-20%
HbS level to minimize sickling
complications.
Reduced blood viscosity. Improved blood
flow
Regularly scheduled exchange transfusions
used in the prevention of stroke, acute chest
syndrome, and recurrent painful episodes
Some patients with severe phenotype and
without anemia, may require this. Follow
advice of level 2 or 3 center.
Exchange transfusion therapy can be full blood volume exchange or partial volume exchange
and the method may be manual or automated apheresis. A full exchange transfusion rapidly
lowers the HbS level to 30 % or less and improves the hemoglobin level. In general,
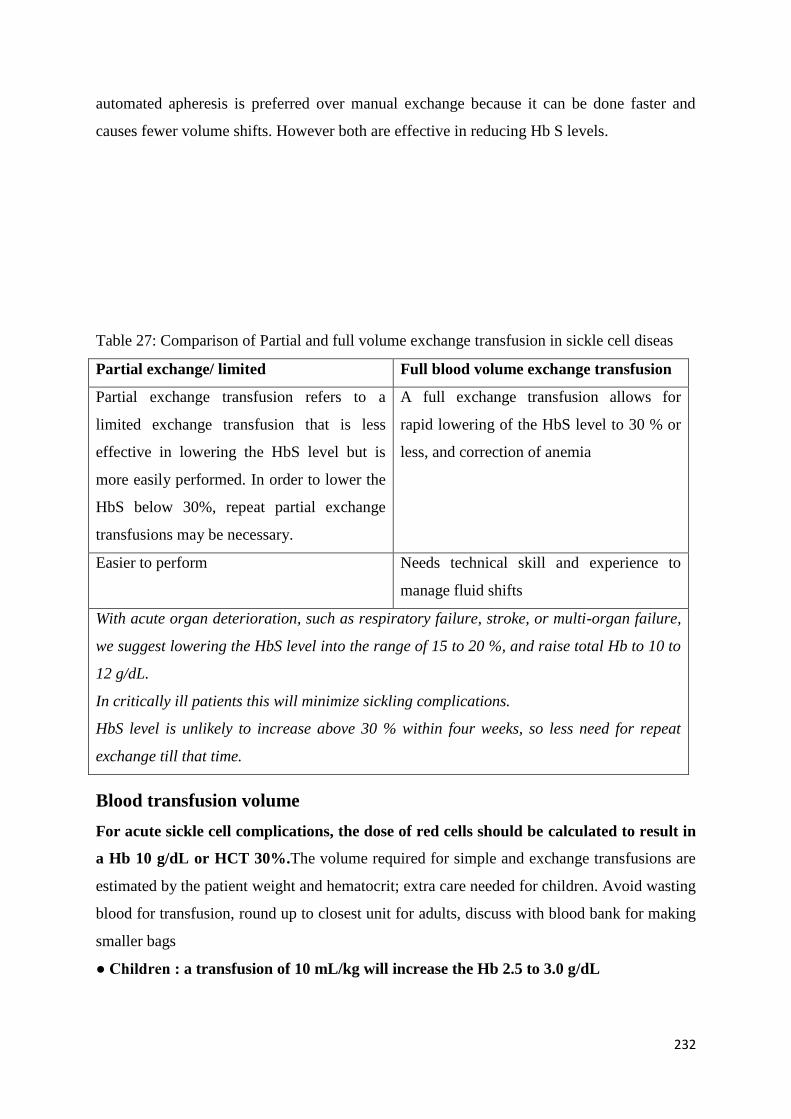
232
automated apheresis is preferred over manual exchange because it can be done faster and
causes fewer volume shifts. However both are effective in reducing Hb S levels.
Table 27: Comparison of Partial and full volume exchange transfusion in sickle cell diseas
Partial exchange/ limited Full blood volume exchange transfusion
Partial exchange transfusion refers to a
limited exchange transfusion that is less
effective in lowering the HbS level but is
more easily performed. In order to lower the
HbS below 30%, repeat partial exchange
transfusions may be necessary.
A full exchange transfusion allows for
rapid lowering of the HbS level to 30 % or
less, and correction of anemia
Easier to perform Needs technical skill and experience to
manage fluid shifts
With acute organ deterioration, such as respiratory failure, stroke, or multi-organ failure,
we suggest lowering the HbS level into the range of 15 to 20 %, and raise total Hb to 10 to
12 g/dL.
In critically ill patients this will minimize sickling complications.
HbS level is unlikely to increase above 30 % within four weeks, so less need for repeat
exchange till that time.
Blood transfusion volume
For acute sickle cell complications, the dose of red cells should be calculated to result in
a Hb 10 g/dL or HCT 30%.The volume required for simple and exchange transfusions are
estimated by the patient weight and hematocrit; extra care needed for children. Avoid wasting
blood for transfusion, round up to closest unit for adults, discuss with blood bank for making
smaller bags
● Children : a transfusion of 10 mL/kg will increase the Hb 2.5 to 3.0 g/dL

233
● Adults : each unit of PRBC will increase the Hb concentration by approximately
1 g/dL
1. Formula used for estimation of simple transfusion volumes:
Packed RBC volume for
SIMPLE transfusion (mL) = ([dHCT - iHCT] x TBV) ÷ RpHCT
[dHCT is the desired hematocritiHCT is the initial hematocrit (both given as percent;eg-
40percent) ];
[TBV is the estimated total blood volume in mL (e.g., 60 mL/kg in adult women,
70 mL/kg in adult men, 80 mL/kg in children, 100 mL/kg in infants)]
[RpHCT is the hematocrit of the replacement packed RBC (55 to 60%)].
2. Formula used for estimation of EXCHANGE transfusion volumes
Manual partial
exchange volume (mL) = ([dHCT - iHCT] x TBV) ÷ (RpHCT - [(iHCT + dHCT) ÷ 2])
[dHCT is the desired hematocritiHCT is the initial hematocrit (both given as percent;eg- 40
percent)]
[TBV is the estimated total blood volume in mL (e.g., 60 mL/kg in adult women,
70 mL/kg in adult men, 80 mL/kg in children, 100 mL/kg in infants)].
[RpHCT is the hematocrit of the replacement packed RBC (55 to 60%)].
Table 28: over transfusion management in SCD
In case hemoglobin level increases over 10g/dl, in patients after transfusion
in symptomatic SCD patients, necessary action
Use phlebotomy for all patients with a post-
transfusion Hb ≥13.0 and HbS>50%.
To reduce hyper viscosity
If a patient'shemoglobin has been raised to
above 12 g/dL
with simple transfusions and the HbS levels
are >50 % of total hemoglobin,
use phlebotomy to decrease the hemoglobin
to 10 g/dL
To reduce hyper viscosity
In adults the maximum amount of blood phlebotomized should not exceed two units

234
At any point of time consultation with level 3 or level 4 center is indicated for patient safety
and best management.
References
1. Ted Wun, Kathryn Hassell. Best practices for transfusion for patients with sickle cell.
Disease. Hematology Reviews 2009; volume 1:e22
2. Koehl B, Sommet J, Holvoet L, et al. Comparison of automated erythrocytapheresis
versus manual exchange transfusion to treat cerebral macrovasculopathy in sickle cell
anemia. Transfusion 2016; 56:1121.
3. Savage WJ, Reddoch S, Wolfe J, Casella JF. Partial manual exchange reduces iron
accumulation during chronic red cell transfusions for sickle cell disease. J
PediatrHematol Oncol 2013; 35:434.
4. Charache S. Treatment of sickle cell anemia. Annu Rev Med 1981; 32:195.
5. Carrara P, Balocco M, Pinto V, et al. Manual erythroexchange for chronic transfusion
therapy in patients with sickle cell syndromes unresponsive to hydroxyurea: a long-
term follow-up. Am J Hematol 2010; 85:974.
6. Nieburg PI, Stockman JA. Rapid correction of anemia with partial exchange
transfusion. Am J Dis Child 1977; 131:60.
7. Casella JF, King AA, Barton B, et al. Design of the silent cerebral infarct transfusion
(SIT) trial. PediatrHematol Oncol 2010; 27:69.

235
Management of newborn and pregnancy
Newborn screening
Rationale for newborn screening
The greatest chance of dying in sickle cell disease is the first year of life. Several of these
causes of early death may be prevented or more effectively treated. Prophylaxis and
education may only be put in place if the underlying diagnosis is known.
Technique of newborn screening:
• HPLC followed by DNA based tests
• Another option is isoelectric focussing.
Management
• Confirm diagnosis on capillary sample at age 1 month; once confirmed,children
should be registered in a sickle cell clinic for regular follow-up, education and
counselingofthefamily.
• Offer screening tests for parents to allow counseling on future risks of affected
children.
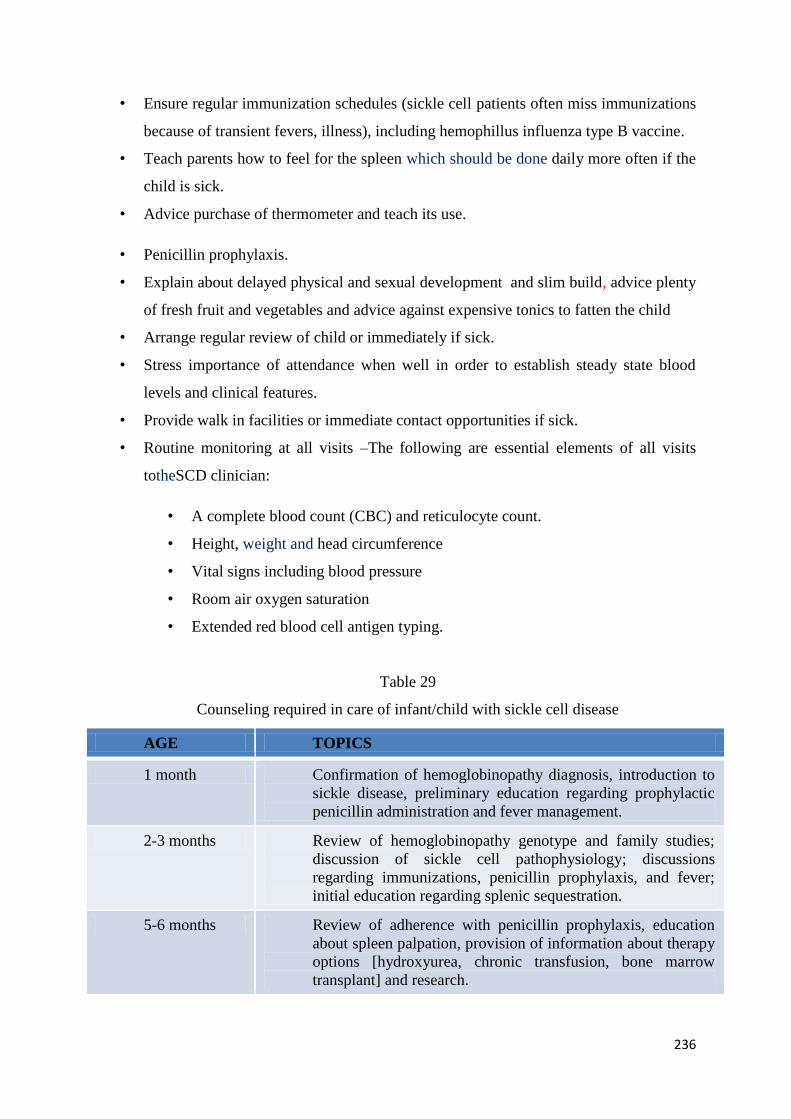
236
• Ensure regular immunization schedules (sickle cell patients often miss immunizations
because of transient fevers, illness), including hemophillus influenza type B vaccine.
• Teach parents how to feel for the spleen which should be done daily more often if the
child is sick.
• Advice purchase of thermometer and teach its use.
• Penicillin prophylaxis.
• Explain about delayed physical and sexual development and slim build, advice plenty
of fresh fruit and vegetables and advice against expensive tonics to fatten the child
• Arrange regular review of child or immediately if sick.
• Stress importance of attendance when well in order to establish steady state blood
levels and clinical features.
• Provide walk in facilities or immediate contact opportunities if sick.
• Routine monitoring at all visits –The following are essential elements of all visits
totheSCD clinician:
• A complete blood count (CBC) and reticulocyte count.
• Height, weight and head circumference
• Vital signs including blood pressure
• Room air oxygen saturation
• Extended red blood cell antigen typing.
Table 29
Counseling required in care of infant/child with sickle cell disease
AGE TOPICS
1 month Confirmation of hemoglobinopathy diagnosis, introduction to
sickle disease, preliminary education regarding prophylactic
penicillin administration and fever management.
2-3 months Review of hemoglobinopathy genotype and family studies;
discussion of sickle cell pathophysiology; discussions
regarding immunizations, penicillin prophylaxis, and fever;
initial education regarding splenic sequestration.
5-6 months Review of adherence with penicillin prophylaxis, education
about spleen palpation, provision of information about therapy
options [hydroxyurea, chronic transfusion, bone marrow
transplant] and research.

237
8-9 months Review of adherence with penicillin prophylaxis, review of
spleen palpation, discussion about dactylitis and pain events
12 months Discussion about acute chest syndrome, discussion about
hydroxyurea [and potential initiation of treatment ]
Pregnancy
SCD causes polymerisation of the abnormal hemoglobin in low-oxygensituations. This
causes it to become rigid and take on the fragile sickle-shaped red cells. These cells
causevaso-occlusion in the small blood vessels, leading to acute painful crises. stroke,
pulmonary hypertension, renal dysfunction, retinal disease, leg ulcers, cholelithiasis and
avascular necrosis. These cells are also likely to breakdown and result in hemolysis and
anemia. Women with sickle cell disease have increased complications for themselves and
their babies.
What are the additional risks to the woman and the baby?
Baby: Increased incidence of perinatal mortality, fetal growth restriction.
Mother:Premature labour and acute painful crises during pregnancy.
Some studies have shown increase in spontaneous miscarriage, maternal mortality,
infection, thromboembolic events and antepartum haemorrhage. Also,there are some
reports of increased risk of pre-eclampsia and pregnancy-induced hypertension .
What should be checked for women planning pregnancy?
When planning for pregnancy in women with SCD ensure they have received and are
compliant with basic SCD care and medications, immunizations , prophylactic
penicillin, folic acid etc.
They should have their routine assessment for complications of sickle cell disease at
their level 2 center and be referred to level 3 or 4 center if there are complications of
disease.
Screening for pulmonary hypertension with echocardiography, this may be associated
with increased mortality.
Blood pressure and urinalysis should be performed to identify women with
hypertension and/or proteinuria. Renal and liver function tests should be performed
annually to identify sickle nephropathy and/or deranged hepatic function.

238
Screening for iron overload, by serum ferritin, in multi-transfused patients,T2*
cardiac magnetic resonance imaging may be helpful to assess iron overload in heart
and liver. If high iron overload is aggressive,iron chelation before conception is
advisable.
Screening for red cell antibodies. Red cell antibodies may indicate an increased risk of
haemolytic disease of the newborn.
What test is required for the father?
If the mother has sickle cell disease or is a carrier for any hemoglobinopathy/ variant
hemoglobin trait, then the father should also be tested. If he is normal then likelihood
of baby being affected withaserioushemoglobin disorder is less (though silent
mutations cannot be ruled out).
If father is also a carrier of a hemoglobinopathy /variant hemoglobin trait, the genetic
counseling required and prenatal testing option to be given for serious disorders.
What medicines should be stopped?
Preconceptually Hydroxyurea should be stopped at least 3 months before conception.
Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers should
be stopped before conception.
Antenatal care
All routine antenatal tests and care are required.
Regular antenatal checkups required
Blood pressure and urinalysis should be performed at each consultation and
midstream urine for culture performed monthly.
Additionally
o influenza vaccine should be recommended.
o Women with SCD should aim to avoid precipitating factors of sickle cell
crises such as exposure to extreme temperatures, dehydration and

239
overexertion. Persistent vomiting can lead to dehydration and crisis.If this
occurs, should receive iv fluids.
o Iron supplementation should only be given if there is laboratory evidence of
iron deficiency.
o Low-dose aspirin 75 mg once daily from 12 weeks of gestation to reduce the
risk of developing pre-eclampsia.
o Women with SCD may be advised to receive prophylactic low-molecular-
weight heparin during antenatal hospital admissions.
o Non-steroidal anti-inflammatory drugs (NSAIDs) should be prescribed only
between 12 and 28 weeks of gestation owing to concerns regarding adverse
effects on fetal development.
Ultra-sound examination
o Offer a viability scan at 7–9 weeks of gestation.
o Offer routine first-trimester scan (11–14 weeks of gestation) and a detailed
anomaly scan at 20 weeks of gestation.
o May require serial fetal biometry scans (growth scans) every 4 weeks from 24
weeks of gestation.
Blood transfusion during pregnancy?
Though routine prophylactic transfusion is not recommended, if a woman with SCD
develops complications then an exchange transfusion is required. Such patients may
even require repeat transfusions every 4 weeks during pregnancy.
Blood should be matched for an extended phenotype including full rhesus typing (C,
D and E) as well as Kell typing. During pregnancy ensure leukodepletion of all blood
products.
Table 30:Indications for transfusion of blood during pregnancy in sickle cell disease.
Women who are on a chronic transfusion
regimen before pregnancy , continue to
prevent disease complications
Continue same transfusion regimen
Acute chest syndrome Exchange transfusion
Twin pregnancy Simple/ Exchange transfusion
Anemia- simple transfusion Simple transfusion

240
Women with previous serious medical,
obstetric or fetal complications
Exchange transfusion
How to manage acute pain crisis during pregnancy?
This is the commonest cause of admission with 27% and 50% of women having a painful
crisis during pregnancy. Advice to avoid excess heat/ cold environment, exercise,
dehydration is Tablerequired. If it occurs then-
Admit to the hospital or give daycare management under supervision.
A multidisciplinary team should assess for diagnosis of sickle pain crisis or other
medical complications, acute chest syndrome (ACS), sepsis or dehydration.
Urgent fluids
If pain crisis give analgesia within 30 minutes of reaching the hospital - The World
Health Organization analgesic ladder should be used, starting with paracetamol for
mild pain, for severe pain may need opioid. s (avoid pethidine in SCD patients -risk
of seizures).
o NSAIDS - if 12–28 weeks of gestation.
o Weak opioids such as Tramadol/codeine can be used for moderate
pain, and stronger opiates such as morphine can be used for severe
pain.
o If requiring opioids –admit
o Morphine can be given by the oral, subcutaneous, intramuscular or intravenous route
depending on the woman’s preference and local expertise.
o Parenteral opiates can be given by intermittent bolus or patient-
controlled administration systems.
o With opiods-Prescribe laxatives, antipruritic and antiemetic
o Monitor pain, sedation, vital signs, respiratory rate and oxygen
saturation every 20–30 minutes until pain is controlled and signs are
stable , then every 2 hours. Thromboprophylaxis should be given to
women admitted to hospital with acute painful crisis.
Type of delivery and timing?
Pregnant women with SCD with normal growing fetus- vaginal delivery after 38+0
weeks of gestation.

241
SCD is not considered a contraindication to attempting vaginal delivery. Obstetric
advice for c-section needed.
Blood should be cross-matched for delivery. Good hydration maintained at all times.
Ideally delivery should take place in center familiar with sickle cell disease patients.
Monitor for SCD complications in post partum time as well. Low-molecular-weight
heparin should be administered while in hospital and 7 days post-discharge following
vaginal delivery or for a period of 6 weeks following caesarean section.
What anesthesia to be offered to women with SCD?
Should have pre-anesthesia evaluation in third trimester of pregnancy.
Avoid the use of pethidine, though other opiates are safe to use.
Recommended to use Regional analgesia for caesarean section.
References
1. Afolabi BB, Iwuala NC, Iwuala IC, Ogedengbe OK. Morbidity and mortality in sickle
cell pregnancies in Lago, Nigeria: a case control study. J
ObstetGynaecol2009;29:104–6.
2. Rajab KE, Issa AA, Mohammed AM, Ajami AA. Sickle cell disease and pregnancy in
Bahrain. Int J GynaecolObstet2006;93:171–5.
3. National Institute for Health and Clinical Excellence. Hypertension in pregnancy.
The management of hypertensive disorders during pregnancy. NICE clinical guideline
107. London: NICE; 2010.
4. Howard RJ, Tuck SM, Pearson TC. Pregnancy in sickle cell disease in the UK: results
of a multicentre survey of the effect of prophylactic blood transfusion on maternal and
fetal outcome. Br J ObstetGynaecol 1995;102;947–51
5. Management of Sickle Cell Disease in Pregnancy. Green–top Guideline No. 61 July
2011.https://www.rcog.org.uk/globalassets/documents/guidelines/gtg_61.pdf
(accessed 6 May 2019)

242
Alloimmunization
All Multi-transfused patients are at risk of RBC allo-immunization i.e. they develop
antibodiestocertainRBC antigens. Though higher in patients of sickle cell disease and non
transfusion dependent thalassemia (NTDT), but may occur in any patient. Allo-immunization
puts these transfusion recipients at risk of hemolytic transfusion reactions if the antibody is
not detected at the time of pre-transfusion testing. Allo-immunization also shrinks the
precious donor pool for the allo-immunized patient as he can receive blood only from donors
who are negative for the antigen against which the recipient has developed antibodies.
Patients who develop one antibody are usually responders and have a propensity to develop
more allo-antibodies, this further shrinks the eligible donor pool sometimes making
compatible blood unavailable when required.

243
Allo-immunization should be suspected when pre-transfusion testing shows incompatibility
or the expected rise in hemoglobin is not seen and other causes for it are ruled out. A rise in
hemoglobin may be seen in some recipients but it is followed by a rapid fall in the levels.
Factors Impacting Alloimmunization
Rates of alloimmunization vary according to demography and heterogeneity of the
population group. Several contributing factors for alloimmunization are:
1. Responder Status
Every individual has a unique genetic makeup which makes them vulnerable to
several serological problems after chronic transfusion therapy. This genetic difference
helps us divide individuals into three broad categories:
Non-Responders – These are the individuals who never develop any allo antibodies
despite repeated exposures.
Responders – These are the individuals who develop at least one alloantibody with
one or more exposures.
Hyper-Responders – These are the individuals who develop multiple allo antibodies
with one or more exposures.
2. Antigenic [RBC and human leukocyte antigen (HLA)] disparity between the
blood donor and the recipient which will increase further if they both belong to
different ethnic groups.
3. Genetic Factors: Although the patient could produce antibodies against both red cell
antigens and/or Human leucocyte antigens separately after exposure to foreign
antigens, yet according to few researchers, formation of antibodies against blood
group antigens, for example, Rh, Kell, Kidd, Duffy, Diego could be accentuated
because of certain HLA phenotype of the individual. Thus some HLA phenotypes
make individuals more prone to alloimmunization making them responders or hyper-
responders.
4. Presence of leucocytes in Donor PRBCs/Platelets: Donor leucocytes are mediators
of direct antigen presentation. Leucodeplteted blood components (BCs) cause less
alloimmunization as compared to non-leucodeplted BCs.
5. Antigenic Dose: The higher the antigen dose, the greater is the risk of allo-
immunization.

244
6. Underlying Disorder: Persons with sickle cell anemia/HbD are at a higher risk of
alloimmunization as compared to patient cohorts.
7. Number of transfusions received: As the total number of BCs transfused increases,
so does the risk of alloimmunization.
8. Age at first transfusion: Starting PRBC transfusions at an early age confers some
immunotolerance and reduces the overall risk of alloimmunization.
9. Immunogenicity of antigen: Rh system antigens – D, C, E, c, and e and Kell antigen
K are the most immunogenic and most alloimmunized patients develop antibodies to
these RBC antigens. Transfusing PRBCs which are matched for extended Rh and K
phenotype has been proposed as a mechanism to reduce alloimmunization.
Implications of Alloimmunization:
1. Increased risk of delayed hemolytic transfusion reaction (DHTR) or delayed
serological hemolytic transfusion reaction (DSTR).
Allo-antibodies against cellular antigens or blood group antigens might disappear
from detection over time. If antigen re-exposure during subsequent transfusions
occurs, it can induce an anamnestic antibody response resulting in delayed
hemolytic transfusion reactions which could become life-threatening.
2. Decreased chances of finding a compatible blood product for future use thus
increasing turn-around time of blood. If alloantibody is detectable during
compatibility testing procedure, then antigen negative blood component unit has
to be found and this becomes more difficult if more than one alloantibodies are
present. Sometimes the alloantibody is masked by the presence of auto-antibodies
thus difficult to detect.
3. It will decrease post-transfusion survival of RBCs in the recipient. As the antibody
present in the recipient will cause lysis and thus the destruction of transfused
donor RBCs.
4. It might accelerate iron loading of tissues and organs as a higher number of RBCs
need to be transfused. Chronic transfusion and underlying inflammatory state of

245
the recipient cause elevations of the pro‐inflammatory cytokines TNF‐α and IL‐2
thus further accentuating iron overloading in these patients.
Approaches to Mitigate Risk of Alloimmunization:
1. Donors of similar ethnic descent should be considered to decrease the chances of
genetic disparity and hence alloimmunization.
2. Use of fresh blood (within one week of the collection): Due to metabolic and
rheological alteration of RBCs older blood units are associated with post-
transfusion hemolysis, decreased survival and alloimmunization.
3. Preventive phenotyping of the patient: Before starting transfusion therapy of
patients with hemoglobinopathies, ABO forward and reverse grouping along with
Rh and Kell typing should be done. It would be helpful in identifying antibodies
in case alloimmunization occurs.
4. Antibody screening could be performed using a sensitive method as a part of
routine pre-transfusion testing.
5. Leukoreduction of the blood components: Pre-storage or bedside leukoreduction
approaches could be helpful in preventing alloimmunization.
Management of Alloimmunization:
1. Antibody Screening using a three cell panel of reagent red cells with known
extended phenotypes ( commercially available) each time a patient's sample is
received.
2. In case the patient is antibody screen positive, the patient's plasma/serum
should be tested against an 11 cell panel of reagent RBCs of known extended
phenotypes( commercially available) to identify the antibody specificity.
3. Once the antibody specificity has been identified, donor RBCs should be typed
for the antigen of interest and antigen negative units should be selected for the
patient.
4. The antigen negative units should be cross-matched with the
recipient'splasma/serum and only cross-match compatible antigen negative
units should be issued for transfusion.
5. Antigen negative units should be provided for previously documented
antibodies even if the antibodies are undetectable at the time of antibody

246
screening/identification and cross-match in order to prevent immune HTRs
due to an anamnestic response.
6. Extended Rh and Kell antigen matched PRBCs should be provided when
possible especially in patients who have developed one or more alloantibodies.
Problems encountered:
1. Majority of blood banks have limited donor pools i.e. there is a lack of sufficient
phenotypically matched donors to provide blood to allo-immunized patients.
2. As patients of hemoglobinopathies are candidates of long term blood transfusion
therapy they might receive blood from different hospitals during their lifetime.
Different hospitals have different serological testing approaches for patients for
example, not all the institutes are providing phenotypically matched blood to their
patients and also some institutes are doing extended phenotype matching for all
the patients with hemoglobinopathies while others are only using for the patients
who have developed alloantibodies.
3. Undue delays could be faced by the patients in case we are not able to find
phenotypically matched blood for the patient and/or compatibility testing is
complicated by the presence of autoantibody.
4. Providing extended antigen-matched blood unit even in alloantibody negative
individuals might cause logistics issue at some centers.
References
1. Kruatrachue M., Sirisinha S., Pacharee P., Chandarayingyong D., Wasi P. An
association between thalassemia and autoimmune haemolytic anemia (AIHA)
Scandinavian Journal of Haematology. 1980;25(3):259–263.
2. Singer ST, Wu V, Mignacca R, Kuypers FA, Morel P, Vichinsky EP.
Alloimmunization and erythrocyte autoimmunization in transfusion-dependent
thalassemia patients of predominantly Asian descent. Blood 2000; 96: 3369–3373.
3. Spanos T, Karageorga M, Ladis V, et al. Red cell alloantibodies in patients with
thalassemia. Vox Sang 1990;58:50-5.
4. Datta SS, Mukherjee S, Talukder B, Bhattacharya P, Mukherjee K. Frequency of red
cell alloimmunization and autoimmunization in thalassemia patients: a report from
eastern India. Adv Hematol 2015; 2015:610931

247
5. Gehrie EA, Tormey CA. The Influence of Clinical and Biological Factors on
Transfusion-Associated Non-ABO Antigen Alloimmunization: Responders, Hyper-
Responders, and Non-Responders. Transfus Med Hemother. 2014;41(6):420–429.
6. Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in
sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood.
2012;120(3):528–537.
7. Gehrie EA, Ness PM, Bloch EM, Kacker S, Tobian AAR. Medical and economic
implications of strategies to prevent alloimmunization in sickle cell disease.
Transfusion. 2017;57(9):2267–2276. doi:10.1111/trf.14212
8. Bao W, Zhong H, Li X, et al. Immune regulation in chronically transfused allo-
antibody responder and nonresponder patients with sickle cell disease and β-
thalassemia major. Am J Hematol. 2011;86(12):1001–1006.
Hematopoietic Stem Cell Transplantation(HSCT) for Hemoglobinopathies
Hematopoietic Stem Cell Transplant for Thalassemia Major
Learning Objectives:
A. To discuss the need and anticipated outcomes of Hematopoietic Stem Cell Transplant
(HSCT) in Thalassemia major
B. Understand the risk categorization for patients undergoing HSCT and to recognize a
very high risk group.
C. Highlight the need of doing HSCT at a young age and preferably before the age of 7
years.
D. Address source of stem cells and alternative stem cell donor related issues in HSCT
for Thalassemia major
E. To understand the transplant procedure/protocol (that includes pre transplant work up,
preparative regimen and post-transplant follow up)
A]Need for HSCT in Thalassemia major:
Currently, an allogeneic hematopoietic stem cell transplant (HSCT) remains the only curative
option for patients with β thalassemia major. The use of HSCT is rapidly increasing in India
and other developing countries and is hence the most widely available and accessible curative

248
therapeutic strategy for this condition. The central concept revolves around the ability to
replace the hematopoietic stem cells (HSC) from a donor to a recipient resulting in a new
donor derived hematopoietic system in the recipient. Significant advances over the last two
decades have resulted in a steady improvement in clinical outcomes for patients with this
disorder undergoing such a procedure. Currently in patients with good risk features it is
reasonable to anticipate a greater than 90% chance of a successful transplant outcome. Even
among those with high risk features, success rates are approaching 80%. These improvements
have resulted from use of better risk stratified conditioning regimen and more effective
supportive care. With thalassemia being a significant public health problem in the country,
there is need for capacity building and increasing the centers capable of offering this therapy.
B] Pre-transplant risk assessment and stratification of patients with Thalassemia
major:
Risk stratification of patients with β thalassemia major undergoing a myelo-ablative
allogeneic (SCT) classifies them into three risk groups (Pesaro Class I, II and III) based on
liver size (>2cm), presence of liver fibrosis and inadequate iron chelation. Patients with none
of the above risk factors are classified as Class I, those with one or two of these risk factors
are Class II while those patients who have all three adverse risk factors are classified as Class
III (summarized in Figure 9). Patients in Class I and II our considered to be low risk and have
an excellent long term outcome following an allogeneic SCT. Class III patients on the other
hand are considered high risk and have inferior outcomes following a SCT. However, in a
population with poor medical treatment prior to SCT, the above risk stratification is limited
by its failure to recognize the significant heterogeneity among patients in Class III. The
Pesaro risk stratification does not recognize a very high risk subset of Class III in these
populations perhaps because such patients hardly exist in Western countries. With allo-SCT
being increasingly offered in many developing countries where this category of patients exist
in large numbers, recognizing this high risk group is extremely important to suitably modify
their HSCT protocols. Additionally, it must be stressed that HSCT is contraindicated in
patients who already have evidence of severe end organ damage from high iron overload
leading to organ dysfunction such as cardiac failure of cirrhosis, such patients even if
considered for HSCT should only be done in centers with substantial experience and
expertise with these procedures.
Figure 9: Risk factor and risk groups on pre-transplant evaluation of patients with
Thalassemia major undergoing an HSCT

249
C] Importance of doing HSCT at a young age and preferably before the age of 7 years:
Retrospective analysis of factors impacting clinical outcome on large data set from India
analyzed in 2007, helped identify age ≥7 years old and had a liver size ≥5 cm in a patient pre-
transplant to constitute what has been shown to be a very high risk subset of a conventional
Class III group (Class III high risk)[2]. The adverse impact of age (≥7 years) and liver size (>
2 cms) was further validated by an international collaborative analysis in 2010. The
significance of this differentiation has been emphasized by the fact that outcomes of HSCT
were clearly different in these two groups when treated with the same protocols. Class III and
more specifically Class III high risk subset have a high risk of graft rejection and regimen
related toxicity (RRT), especially sinusoidal obstruction syndrome (SOS) leading to multi-
organ failure and death. These complications are perhaps related to the high degree of allo-
immunization and iron over load related end organ damage in this cohort. When such a
population is transplanted even in a developed country with expertise in such transplants the
rejection rate is as high as 34%.
D] Source of stem cells and alternative stem cell donors for HSCT in Thalassemia
major:
Bone marrow (BM) has been the preferred choice of stem cells in non-malignant
hematological disorders to reduce the risk of GVHD, though the incidence of both acute and
chronic GVHD in this predominantly pediatric population is low. Peripheral blood stem cell
grafts (PBSC) when used have been reported to be associated with faster engraftment and
lower requirement of blood product support in the peri-transplant period and have also been
associated with a low incidence of graft rejection.

250
The best stem cell donor is a HLA identical sibling donor unfortunately, only about a third of
patients with thalassemia major have a HLA matched sibling donor which limits access to an
HSCT for these patients. There is a rare possibility of another related donor, such as a parent,
grandparent or cousin, being HLA identical though the prior probability of this is extremely
low.
Matched unrelated donors:Use of a matched unrelated donor (MUD) SCT has the potential
to overcome this. The initial results with this approach, prior to the advent of high resolution
HLA typing, were dismal with a 55% graft rejection. Since then, there has been significant
improvement in MUD SCT with high resolution molecular HLA typing becoming standard in
MUD selection. Outcome in malignant disorders with MUD is now comparable to that of
HLA matched sibling transplants. More recent data suggests that OS of 79% can be achieved
with this approach with thalassemia free survival of 66% and a 25% chance of treatment
related mortality [16]. This study also suggested that an extended haplotype match was
associated with a superior outcome. The overall data would suggest that MUD SCT should
only be considered in centers that have reasonable experience with this approach and is best
limited to low risk patients at present. High resolution HLA typing with a full (10 of 10)
match is the preferred donor and in addition they should ideally not have HLA-DP1
mismatches in the direction favoring graft rejection.
Cord blood transplants: Cord blood transplants over the last decade have been the most
rapidly growing source of stem cells for allogeneic SCT. However, for patients with
thalassemia major there is limited data. Using cord blood stem cells for allogeneic SCT in
thalassemia major must be considered under two categories, which are distinctly different in
terms of clinical outcome:
1. Related cord blood transplant: when a HLA matched or partly mismatched sibling is
the source of stem cells
2. Unrelated cord blood transplant: when an unrelated cord blood product is procured
from a cord blood bank as part of donor search for an allogeneic SCT. There could be
varying degrees of HLA mismatch.
Using related cord blood the Eurocord Transplant Group reported a 2 year probability of
thalassemia free survival of 79% in 33 patients with thalassemia.
Unrelated cord blood transplant genuinely increases the potential pool of donors for patients
with thalassemia major. Unfortunately the published data is limited. In a recent review over 6

251
studies a total of 19 patients were reported. After combining data from three different
registries, Ruggeri et al reported in 2011, that the cumulative graft failure rate was an
unacceptable 52%. At this time point one cannot recommend an unrelated cord blood
transplant outside the setting of a clinical trial.
Haplo-identical stem cell transplants:There is a lot of interest in haplo-identical stem cell
transplants in the world over the last few years. Novel conditioning and GVHD prophylaxis
regimens have resulted in dramatic improvements in clinical outcome even without T cell
depletion of the graft. There is however very limited data in thalassemia major. In one small
series (n=22) using T cell depletion grafts the graft rejection rate was 27% and the TFS about
67%. More recently the use of grafts with depletion of CD3αβ T cells looks promising with a
few successful reports. In a disease where several management options exist and newer ones
are on the horizon, whether a treatment option that gives less than ~80% thalassemia free
survival can or should be recommended needs further discussion. Haplo-identical SCT should
therefore, at this point of time, be done preferably in the setting of a clinical trial and only in
tertiary care centers.
E] Transplant procedure:
Prior to initiating the procedure a detailed pre-transplant evaluation of patient and donor is
done to confirm diagnosis and HLA match status and also to exclude any condition that is a
contraindication to proceeding with a transplant, in both the recipient and the donor. The
conventional conditioning regimen that is used is a combination of busulfan and
cyclophosphamide. More recently a number of novel conditioning regimens have been
introduced and popular alternative is a treosulfan based conditioning regimen that replaces
busulfan and is better tolerated with less adverse events. The first report on the use of
treosulfan being used as part of the conditioning regimen for thalassemia, in a small series of
20 patients of which 45% were Class III and 18 were matched unrelated stem cell transplants,
was by Bernado et al. Treosulfan based conditioning regimen is ideally suited for patients
with thalassemia major including very high risk patients. The low hepatic and other toxicity
profile over a wide range of AUCs obviates the need for drug dose monitoring and makes this
an attractive agent to use in the conditioning regimen.
Post-transplant patients need to be monitored carefully for engraftment. Patients continue to
remain under high risk for infections for the first year. An additional complication that can

252
occur in a proportion of cases and can occasionally be fatal includes graft rejection and graft
versus host disease, neither of which one can accurately predict.
The iron overload state continues to require attention post transplantation. Post a successful
transplant provided the patients is stable and his Hb is above 10mg/dl, then the preferred
method of iron removal is phlebotomy. It can be repeated once in 14 days and a volume of
6ml/kg can be removed in one sitting. If the hemoglobin level is not adequate or if
phlebotomy is not possible then the patient should be started on chelation therapy. It should
be continued (maybe required for years) till the ferritin level is <100ng/ml. The optimal
pharmacological agent(s) and chelation regimen post-transplant remains to be defined. In
addition to chelation these patients need close attention to immunization, endocrine and organ
dysfunction secondary to iron overload.
References:
1. Mathews, V., A. Srivastava, and M. Chandy, Allogeneic stem cell transplantation for
thalassemia major. Hematol Oncol Clin North Am, 2014. 28(6): p. 1187-200.
2. Mathews, V., et al., A new stratification strategy that identifies a subset of class III
patients with an adverse prognosis among children with beta thalassemia major
undergoing a matched related allogeneic stem cell transplantation. Biol Blood
Marrow Transplant, 2007. 13(8): p. 889-94.
3. Lucarelli, G., et al., Bone marrow transplantation in patients with thalassemia. N
Engl J Med, 1990. 322(7): p. 417-21.
4. Lucarelli, G., et al., Bone marrow transplantation in adult thalassemia. Blood, 1992.
80(6): p. 1603-7.
5. Hongeng, S., et al., Outcomes of transplantation with related- and unrelated-donor
stem cells in children with severe thalassemia. Biol Blood Marrow Transplant, 2006.
12(6): p. 683-7.
6. Fang, J.P. and L.H. Xu, Hematopoietic stem cell transplantation for children with
thalassemia major in China. Pediatr Blood Cancer, 2010. 55(6): p. 1062-5.

253
7. Sabloff, M., et al., HLA-matched sibling bone marrow transplantation for beta-
thalassemia major. Blood, 2010. 117(5): p. 1745-50.
8. Phrommintikul, A., et al., Splenectomy: a strong risk factor for pulmonary
hypertension in patients with thalassaemia. Heart, 2006. 92(10): p. 1467-72.
9. Chiesa, R., et al., Unpredictability of intravenous busulfan pharmacokinetics in
children undergoing hematopoietic stem cell transplantation for advanced beta
thalassemia: limited toxicity with a dose-adjustment policy. Biol Blood Marrow
Transplant, 2010. 16(5): p. 622-8.
10. Chandy, M., et al., Randomized trial of two different conditioning regimens for bone
marrow transplantation in thalassemia--the role of busulfan pharmacokinetics in
determining outcome. Bone Marrow Transplant, 2005. 36(10): p. 839-45.
11. Rajasekar, R., et al., Cellular immune reconstitution and its impact on clinical
outcome in children with beta thalassemia major undergoing a matched related
myeloablative allogeneic bone marrow transplant. Biol Blood Marrow Transplant,
2009. 15(5): p. 597-609.
12. Mathews, V., et al., Improved clinical outcomes of high risk beta thalassemia major
patients undergoing a HLA matched related allogeneic stem cell transplant with a
treosulfan based conditioning regimen and peripheral blood stem cell grafts. PLoS
One, 2013. 8(4): p. e61637.
13. Iravani, M., et al., Comparison of peripheral blood stem cell transplant with bone
marrow transplant in class 3 thalassemic patients. Exp Clin Transplant, 2010. 8(1): p.
66-73.
14. Ghavamzadeh, A., et al., Peripheral blood versus bone marrow as a source of
hematopoietic stem cells for allogeneic transplantation in children with class I and II
beta thalassemia major. Biol Blood Marrow Transplant, 2008. 14(3): p. 301-8.
15. Gaziev, D., et al., Bone marrow transplantation from alternative donors for
thalassemia: HLA-phenotypically identical relative and HLA-nonidentical sibling or
parent transplants. Bone Marrow Transplant, 2000. 25(8): p. 815-21.
16. La Nasa, G., et al., Unrelated donor bone marrow transplantation for thalassemia:
the effect of extended haplotypes. Blood, 2002. 99(12): p. 4350-6.
17. Fleischhauer, K., et al., Graft rejection after unrelated donor hematopoietic stem cell
transplantation for thalassemia is associated with nonpermissive HLA-DPB1
disparity in host-versus-graft direction. Blood, 2006. 107(7): p. 2984-92.

254
18. Pinto, F.O. and I. Roberts, Cord blood stem cell transplantation for
haemoglobinopathies. Br J Haematol, 2008. 141(3): p. 309-24.
19. Ruggeri, A., et al., Umbilical cord blood transplantation for children with
thalassemia and sickle cell disease. Biol Blood Marrow Transplant, 2011. 17(9): p.
1375-82.
20. Bertaina, A., et al., HLA-haploidentical stem cell transplantation after removal of
alphabeta+ T and B-cells in children with non-malignant disorders. Blood, 2014.
21. Bernardo, M.E., et al., Treosulfan-based conditioning regimen for allogeneic
haematopoietic stem cell transplantation in patients with thalassaemia major. Br J
Haematol, 2008. 143(4): p. 548-51.
Hematopoietic Stem Cell Transplant for Sickle Cell Disease
A.1.0 Need for HSCT in Sickle Cell Disease (SCD):
Sickle cell disease is the most common inherited hemoglobinopathy worldwide, and is a
devastating, life threatening disease with limited therapeutic options to reduce disease
Learning Objectives :
A. To discuss the need and outcome of Hematopoietic Stem Cell Transplant(HSCT) in sickle
cell disease (SCD)
B. To identify the candidates suitablefor transplant in SCD
C. To understand the transplant procedure/protocol (that includes pre transplant work up,
preparative regimen and post-transplant follow up)
D. To identify the warning signs in post-transplant patients.
E. To discuss special issues in HSCT for SCD

255
severity.Although more than 94% of children with SCD in well-resourced countries now
survive until the age of 18 years due to routine newborn screening, penicillin prophylaxis,
primary stroke prevention, and hydroxyurea (HU) therapy, chronic complications (that
include stroke, sickle lung disease, renal failure, RBC allo-immunization, etc.) severely
impact the quality of life (QoL), and mortality is still significant once patients reach
adulthood. Majority of patients develop end organ damage due to repeated sickling crisis
episodes, which ultimately severely cripple their young adult life. In India, the scenario is
even more dismal. Despite the fact that the sickle phenotype prevalent in our country is of
milder form as compared tothe African counterpart due to high HbF levels, it is estimated that
about 20 per cent of children with sickle disease died by the age of two as reported in one
ICMR survey, and 30 per cent children with SCD among the tribal community die before
they reach adulthood.1 Additionally as SCD is particularly prevalent in scheduled populations
in our country, which comprise the most socioeconomically disadvantaged communities with
lack of medical facilities to care for their disease, the estimates for chronic complications for
SCD are expected to be very high.2
Thus it is estimated that unlike inthewest, majority of
patients of SCD who reach adulthood in our country develop chronic complications, thereby
significantly compromising their QoL as well as adding liability to the already burdened
health care system of our country. In light of this, allogenic stem cell transplant is the only
treatment strategy that can actually cure this disease. All other treatment options (like HU
therapy, regular transfusion therapy) for the disease only ameliorate the disease severity
rather than actually curing it.
A.1.1 Transplant Outcome in Sickle Cell disease:
HSCT should be considered standard of care when a patient has an indication and an HLA-
identical sibling donor. The first successful cure of SCD after HSCT was reported in
a single pediatric patient in 1984 who had SCD and coexisting acute myeloid leukemia.3
Since then, over 1,000 patients have received an HLA-identical sibling HSCT worldwide
with a 5-year Event Free Survival (EFS) and Overall Survival (OS) of 91.4% and 92.9%,
respectively.4 EFS is lower with increasing age at transplantation and higher for
transplantations performed after 2006 given improvements in preparative regimens,
supportive care, and management of complications. The data on transplant outcome in SCD
from India is sparse, the reasons for which are numerous. Firstly, majority of patients
requiring Allo-SCT for their disease are either not referred to higher centre in a timely

256
manner or they do not have a matched sibling allogenic donor (found in only 10% of patients)
to undergo transplant. Additionally as previously mentioned, that the hot spot areas for the
severe variety of the disease requiring transplant is predominantly located in the tribal areas
of Central India and Orissa where the patients are very poor to afford this treatment. However
with the Government’s renewed interest in the prevention & treatment of this disease at the
national level, the financial roadblock for the transplant may be overcome in the near future.
B.1.0 Indications for Stem cell transplant in Sickle cell disease:
• Stroke or central nervous system event lasting longer than 24 h
• Impaired neuropsychological function with abnormal cerebral MRI scan
• 3 or more episodes per year of acute chest syndrome leading to recurrent
hospitalizations in patients on Hydroxyurea (HU) therapy for at least 9 months
• More than 3 episodes per year of vaso-occlusive crisis requiring hospitalizations in
patients on HU therapy for at least 9 months
• Stage I or II sickle lung disease (patients with pulmonary hypertension but
without/minimal limitation of physical activity)
• Sickle nephropathy (moderate or severe proteinuria defined as urinary protein to
creatinine ratio of >50 mg/mmol (442 mg/g) or a glomerular filtration rate 30 to 50%
of the predicted normal value)
• Bilateral proliferative retinopathy with major visual impairment in at least one eye
• Osteonecrosis of multiple joints
• Red-cell alloimmunization during long-term transfusion therapy
• TricuspidRegurgitantJetvelocity≥2.7 m/son2D-Echo
• RegularRBCtransfusiontherapy(≥8 transfusionsperyear. for ≥ 1 y)toprevent. vaso-
occlusivecomplications.
B.1.1 Contraindications for Stem cell transplant in Sickle cell disease:
• Karnofsky or Lansky functional performance score <50-70
• Acute hepatitis with evidence of intrahepatic cholestasis or cirrhosis on biopsy
• Severe renal impairment (GFR<30ml/min/1.73m2)
• Severe cardiac disease

257
• Stage III or IV sickle lung disease (Patients with pulmonary hypertension resulting in
marked limitation of physical activity).
• Demonstrated lack of compliance with medical care
B.1.2 Timing of Allogeneic SCT in Sickle cell Disease:
Young patients’preferably preschool age (2-5 yrs)with symptomatic SCD who have an HLA-
matched sibling donor should be transplanted as early as possible. This strategy is often
beneficial as it prevents permanent end organ damage, thereby also improving the transplant
outcome and quality of life.
B.1.3 Source of stem cell :
Unmanipulated Bone Marrow/Peripheral blood stem cell (PBSC)
C.1.0 Pre transplant Evaluation :
• History & Physical Examination
• Any pre-exisiting Co-morbidities
Routine :
• Complete Blood Count (CBC), Kidney Function Test (KFT), Liver Function
Test (LFT) , Urine Routine Examination
• Coagulation Screen (PT/APTT/Fibrinogen)
• Viral Serology: Hepatitis B, C ,HIV, CMV, EBV
• S. Ferritin (Quantitative)
• Surveillance Cultures (Blood, Urine, Stool. Throat swab)
• Chest X Ray
• HLA typing (High Resolution HLA-A,B,DQ, DR)
To Assess End Organ Damage:
• CNS: Cerebral magnetic resonance imaging (MRI) and magnetic resonance
angiography
• Pulmonary :Chest X Ray; PFT ((total lung capacity, forced vital capacity,
residual volume, and the ratio of forced expiratory volume to forced vital

258
capacity), HRCTindicated only in case of strong clinical suspicion of Sickle
lung disease
• Hepatic: Liver biopsy/R2 MRI if S.Ferritin>2500ng/ml to assess hepatic iron
overload
• Opthalmic : Fundus Examination (to r/o retinopathy)
• Renal: Urine protein/creatnine ratio, 24 hr. urine protein (to r/o proteinuria)
• 2-D Echo: to r/o pulmonary Hypertension
C.1.1. Preparative/Conditioning Regimen:
• Myeloablative. conditioning regimen comprising of myeloablative doses of Busulfan
(14-16 mg/kg in divided doses administered over 4 days); Cyclophosphamide (200
mg/kg in divided dosesadministered over 4 days) along with immunosuppressive
doses of Rabbit ATG (10-15mg/kg given over 3 days) with BM/PBSC as the HSC
source has resulted in excellent OS (91-100%) & EFS (82-100%) in several studies.5
The addition of ATG decreases the riskof graft rejection from 22.6% to 3% and
should be considered as standard of care in HSCT myeloablative preparative
regimens.6 Methotrexate (on day +1.+3,+6,+11) along with cyclosporine (maintaining
trough levels 200-300 ng/ml) is recommended as standard Graft versus Host Disease
(GVHD) prophylaxis. The overall rates of acute and chronic GVHD utilizing a
cyclosporine based immunosuppressive regimen range between 10-22%,though
GVHD was a main cause of treatment related mortality (TRM) in several
studies.7Prior HU therapy and young age at transplant are the strongest predictors of
successful treatment outcome.
C.1.2 Post Transplant Evaluation:
• Chimerism Studies – Quantitative restriction-fragment–length polymorphisms (RFLP)
or tandem repeats in DNA on Day 30, 60, 90 & 365.
• Cerebral MRI with angiography: at 180 and 365 dayspost transplantation.
• Hypertension monitoring at each OPD visit
• Iron Overload (S. Ferritin)
• Osteoporosis/ Avascular Necrosis

259
• Pulmonary function test (Annually)
• Renal: Urine Protein/ Creatinine Ratio, 24 Hr Urine Protein (Annually)
• Ophthalmic Examination (Annually)
To be monitored annually
• 2nd Malignancy
• Hypogonadism
• Dyslipidemia
• Thyroid function
Vaccinations : Inactivated or killed vaccines in all eligible patients between 6 and 12
months after transplantation
D.1.0 Warning Signs in Post transplant patients:
The presence of any of the following symptoms/signs in a post-transplant patient may suggest
GVHD/any serious infection and warrant urgent admission or referral to higher centre.
• Fever > 100.5 F lasting for >24 hrs.
• Loose motions
• Redness/rash over the skin
• Persistent vomiting
• Dryness of mouth/eyes
• Ulcerations in the mouth/ difficulty in swallowing
• Significant weight loss
• Recurrent fall in haemoglobin level
E.1.0 Special issues during HSCT in SCD:
• Seizures:It is one of the most dreaded complications during/after stem cell transplant
particularly in SCD patients. The etiology may be multifactorial that include
intracerebral haemorrhage, vasculopathy, hypoxic injury due to sickling, uncontrolled
hypertension or hypomagnesemia/electrolyte disturbances. Anticonvulsant
prophylaxis with phenytoin/levetiracitam should be routinely prescribed to all the
patients and continued for six months post transplantation. In addition other
supportive measures like intensive monitoring of blood pressure, magnesium levels
(maintained between 1.8-2.2 meq/dl), platelet count( maintained>20,000/cumm) and
haemoglobin levels (between 9-11gm/dl) should be done.

260
• Stable Mixed Chimerism (MC):Stable mixed chimerism with a reduction rather than
an elimination of hemoglobin S is sufficient to reverse the SCD phenotype as
erythropoiesis by a minority of engrafted donor cells can lead to a majority of
circulating normal erythrocytes with a survival advantage over short-lived sickle
RBCs. In these MAC regimens, donor chimerism values as low as 10% to 20% were
sufficient to improve Hb levels, HbS % and SCD-related complications.8
Younger age
(<10 years old), the use of ATG , were associated with more SCD patients achieving
mixed chimerism after HCT. Addition of ATG to the combination of busulfan and
cyclophosphamide increased the percentage of children with MC (50–95% of donor
cells) from 5% to 35% in children undergoing myeloablativeallo-HSCT, while
reducing the rejection rate.9
References
1. Serjeant GR, Ghosh K, Patel J. Sickle cell disease in India: A perspective. Indian J
Med Res. 2016;143:21-4.
2. Hockham C, Bhatt S, Colah R, Mukherjee MB, Penman BS, Gupta S, Piel FB.
Thespatial epidemiology of sickle-cell anaemia in India. Sci Rep. 2018; 8:17685.
3. Bernaudin F, Souillet G, Vannier JP, et al. Bone marrow transplantation (BMT) in 14
children with severesickle cell disease (SCD): the French experience. GEGMO.Bone
Marrow Transplant. 1993;12Suppl1:118-21.
4. Leonard A, Tisdale JF. Stem cell transplantation in sickle cell disease:therapeutic
potential and challenges faced. Expert Rev Hematol. 2018;11:547-565.
5. Glukman E, CapelliB, Bernaudin F, et al. Sickle cell disease: an international survey
of results of HLAidenticalsibling hematopoietic stem cell transplantation. Blood
2017:129:1548-56.
6. Walters MC, Patience M, Leisenring W, et al. Bone Marrow Transplantation for
Sickle Cell Disease. NEngl J Med. 1996 ;335:369–76.
7. Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell
transplantation inthalassemia major and sickle cell disease: indications and

261
management recommendations from aninternational expert panel. Haematologica.
2014;99:811-20.
8. Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism
after bone marrowtransplantation for sickle cell anemia. Biol Blood Marrow
Transplant. 2001;7:665–73.
9. Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell
transplantation for sicklecell disease. N Engl J Med. 2009 ;361:2309-17

262
Alpha Thalassemia: Pathophysiology and Diagnosis
Background:
Adult humans have the adult Hb (HbA) as their predominant hemoglobin (Hb), constituting
~97% of total Hb.The remainder is comprised of minor components of HbA2 (2-3.5%) and
fetalHb (HbF, <1%). The Hb molecule contains 4 polypeptide globin chains associated with 4
heme groups. Synthesis of the globin chains, defects in which forms the basis of the
thalassemias, requires two globin genes - α and β. The α-globin genes are duplicated and
located on the telomeric region of the short arm of chromosome 16 (16p13.3). These
duplicated α genes are present in a cluster which is shown in Figure 10 and contains an
embryonic α-like gene zeta (ζ), 3 pseudogenes (ψζ1, ψα1, ψα2), a theta (θ) gene of unknown
function along with two adult α-genes (α2 and α1). Expression of these α-globin genes is
regulated by sequences located in and around the structural genes and by aregion named HS-
40 that is located 40 kb upstream from the α-cluster. This region, contains an erythroid-
specific DNAase I hypersensitive site, multiple, small, highly-conserved binding sites for
trans-acting factors (NF-E2, GATA-1), several CACCC motifs and binding sites for YY1
transcription factors.
Figure 10: Spatial organisation of the human β- and α-globin gene clusters.
Disorders of globin chains can be classified into those associated with a quantitative
reduction in the polypeptide chain production, i.e. the thalassemias and those associated with
production of a varianthemoglobin like HbS, HbE etc., i.e. the hemoglobinopathies. A few

263
disorders show overlapping features and are designated thalassemichemoglobinopathies, for
e.g, HbLepore, HbE and Hb Constant Spring.
Thalassemias are further sub-classified based on the precise globin chain that is
underproduced, and thus include the α, β, δβ, δ, γ, γδβ, εγδβ and other rarer subtypes. This
manuscript deals exclusively with the α-thalassemias.
Nomenclature of α-Thalassemias
Alpha thalassemias can be α0 or α
+, deletional (-α) and non-deletional (α
T). Deletion or
inactivation of both the α-globin genes on one chromosome causes α0
thalassaemia. In the
heterozygous state, its genotype is represented as --/αα (heterozygous α0
thalassaemia) where
there is no output of α-globin from the affected chromosome. When only one of the two
linked α-genes (i.e. on the same chromosome 16) is inactivated, the condition is called α+
thalassaemia, and the genotype is written as -α/αα in cases in which one of the α-globin genes
is deleted, or αTα/αα if one of them is inactivated by a mutation.
Classification and Clinical Syndromes:Four phenotypic types of α-thalassemia can
occur, with loss of function of one to all 4 α-genes:
1) Single α-gene deletions, synonyms: silent carriers/ mild α-thalassemia/ heterozygous
α+ thalassemia (α-/αα): Individuals with a single defective α-globin gene, for e.g. the 4.2 kb
deletion (-α4.2
), are asymptomatic and can only be identified on DNA analysis. Neonates
show a mild increase (1-2%) in Hb Bart’s (γ4 tetramers), a feature that can be exploited for
screening.
2) Two α-gene deletions, i.e. either homozygous α+ thalassemia (α-/α-), or heterozygous
α0 thalassemia (--/αα): Here, the quantitative reduction of α-protein is greater. Individuals
with this condition have red cell microcytosis, hypochromia and erythrocytosis. Most remain
asymptomatic and are detected during family screening. There can however be mild anemia
with levels of Hb Bart’s in the range of 5-6%.
3) Hemoglobin H Disease (α-/--): In HbH diseasepatients, only one out of four α-globin
genes is functional. The magnitude of reduction of α-protein is however, highly
heterogeneous but may lead to moderate to severe anemia (TI phenotype), sometimes with
splenomegaly, bone deformities and chronic fatigue. HbH is formed due to excess β chains
(β4 tetramers).

264
4) Hb Bart’s HydropsFetalis or α-thalassemia major (--/--): Complete absence of α-globin
genes leads to a marked relative excess of γ-globin in the fetus. The abnormal Hb molecule
(γ4 tetramers) is called HbBart's. Patients with HbH disease die in utero or shortly after birth.
Both HbH and Bart’s are non-functional Hbs with very high oxygen affinity. Women
carrying a fetus with Hb Bart’s hydropsfetalis have an increased rate of pregnancy-related
hypertension and hydramnios and an increased rate of peripartum haemorrhage and retained
products of conception.
Interaction of the α-globin genotype with β-thalassemia: Several ethnic groups with a high
prevalence of β-thalassemia also have a high frequency of deletional α-thalassemia; hence
patients who have co-inherited both of these conditions are not uncommon. Homozygotes or
compound heterozygotes for β-thalassemia who co-inherit α-thalassemia have lesser excess
of free α-globin chains and may have less severe symptoms. While co-inheritance of a single
α-gene deletion does not have a remarkable effect on the β-thalassemia phenotype,
individuals with two α-gene deletions coinherited with homozygous β0/β+ thalassemia often
exhibit a mild form of β- thalassemia intermedia (TI). Patients whoco-inherit HbH disease
(i.e. have only one functioning α-gene) along with homozygous β-thalassemia also have a TI
phenotype.
Conversely, increased α-globin product in β-thalassemia trait (βTT) further exacerbates the
globin chain imbalance, converting a clinically asymptomatic βTT to βTI with worsening of
clinical severity. The effect of increased α-globin chain production by two extra α-globin
genes in homozygous triplicated α-globin genes (ααα/ααα) or heterozygous quadruplicated α-
globin genes (αααα/αα) individuals is clearly demonstrated in β-thalassemia heterozygotes,
by resulting in TI phenotype.
Genetic Mechanisms and Pathophysiology:
Deletional α-thalassemia: Molecular pathology of the common alpha globin gene deletions
is depicted in Figure 11. The α2 and α1 globin genes are embedded within two 4 kb
homologous units which have homologous sub-segments X, Y, Z. The Z segments are 3.7 kb
away and reciprocal recombination results in only one α-gene (rightward deletion -α3.7
) and
the other allele has three α-genes (ααα3.7
). Recombination between the X boxes which are 4.2
kb apart results in leftward deletion -α4.2
and an ααα4.2
allele.

265
Several variable length deletions completely abolish both α-genes leading to α0 alleles.
Deletions of upstream regulatory elements which leave the alpha globin genes intact can also
cause α0-thalassemia. This phenomenon in patients has shown that HS-40 (MCS-R2) is
essential as a regulatory element possibly due to recombinations between the rich Alu repeats
and subtelomeric rearrangements.
Figure 11: Duplicated X, Y, Z box arrangement containing the alpha genes and
crossovers leading to deletions and triplications.
Non-deletional types of α-thalassemia: Unlike β-thalassemias, point mutations are
uncommon in α-globin genes. The first non-deletional form was described in 1977 and since
then >70 mutations have been described. The overwhelming majority affect the α2 gene and a
few are described in cis forms with deletions (-αT). These mutations act at various levels of
gene regulation and expression and include RNA splicing variants, frameshifts, nonsense
mutations, poly(A) signal variations and chain-termination mutations which result in
extension of alpha chains by 31 amino acids. Many of the variants lead to variably unstable
Hbs. Hb Constant Spring (αCS, α
142Gln) is an extended variant which is found in 4% of Thais.

266
Epidemiology of Deletional and Non-Deletionalα-Thalassemia:
High frequencies of α+deletional forms are found in the tropical belt, especially amongst
endogamous communities. α0deletions are most common in south-east Asia and the Eastern
Mediterranean islands but are only sporadically seen in the Indian subcontinent and the
middle-East. Common α0
deletions are --MED
in Mediterranean region and --SEA
in south-east
Asia. In north Indians, α+ thalassemia was first documented in patients of βTI by Garewal et
al. in 1994. Their study showed that 12-13% of the population had α+ thalassemia with
triplicated α-genes in 3%. The vast majorities (98%) are -α3.7
and only 2% are -α4.2
allele.
A study on 17 HbH patients from India showed that seven cases had the genotype --SA
/-
α3.7
while three had --SA
/--α4.2
and --SA
was found to be not uncommon in Indians. Non-
deletional α-thalassemias are common in south-east Asia and India. The predominant group
are the termination codon mutations which results in incorporation of 31 codons leading to an
elongated unstable α-chain. HbConstant Spring α142, Term→Gln (TAA>CAA in α2) is found
in frequencies of 5-8% in South East Asia. HbKoya Dora α142, Term→Ser (TAA>TCA in
α2) has been described.
Laboratory Diagnosis of α-deletions, point mutations and triplications
Symptomatic α-thalassemia should be considered after excluding common conditions such as
β-thalassemia syndromes. Iron deficiency anemia should be excluded, since it is commonly
encountered in most populations where both β- and α-thalassemia are common. Automated
blood counts (Hb and red cell indices) and peripheral smear examination reveal hypochromic
microcytosis and red cell anisocytosis (Figure 12). Reticulocyte counts and screening for
HbH inclusions using supravital staining should be done for suspected HbH disease (Figure
13). Hb HPLC is very useful to identify cases of HbH disease since twin pre-integration
peaks (Figure 14) are present. In addition, HbA2 levels are lower than normal (values <2%).
Hemoglobin electrophoresis at alkaline pH shows a fast-moving band after the adult Hb.
Tests for unstable Hbs (isopropanol and heat instability tests) are positive with HbH disease.
Red cell indices in alpha-thalassaemia: It is difficult to classify any particular individual
based only on red cell indices. Mean values of MCV and MCH are significantly lower in both
α+-thalassemia heterozygotes and homogygotes when compared to normal genotype. MCV
and MCH values are however not reliable to be used as good screen for α thalassemia in
isolation. Individuals with microcytic hypochromic anaemia and red cell indices suggestive

267
of β- thalassaemia traits or iron deficiency anemia should be investigated for α- thalassemia
after excluding β- thalassemia trait and iron deficiency by special tests.
Screening tests:
Demonstration of HbH Inclusion Bodies
Reagent: One % Azure Blue or New methylene blue. New batches of stain must be tested
with a known positive control because the redox action of the dyes may vary from batch to
batch.
Method:
1. Mix equal volume of fresh blood (within 24 hours of collection) with staining
solution.
2. Incubate at 37°C for 2 hours or at room temperature for 4 hours.
3. Resuspend the cells and spread a thin blood film after 30 min, 60 min, 90 min and 120
min. If inclusions are not seen at 2 hours, incubate overnight and re-examine before
labelling the test negative
4. Examine the film as for a reticulocyte count. The inclusion bodies appear as multiple
greenish-blue dots, like the pitted pattern on a golf ball (Figure 13).
They can be readily distinguished from reticulocytes, which exhibit uneven reticular
material or infrequent fine dots.
Interpretation & Comments
o In α+ thalassemia trait, only a very occasional HbH body (1:1000 to 1:10,000) is usually
seen; they are more numerous in α0 thalassemia,
o The number of cells developing inclusions is not reliable in differentiating the various
gene deletion patterns seen in α thalassemia
o Absence of demonstrable inclusions does not preclude a diagnosis of α thalassemia trait.
o This test is most useful in Hb H disease, where inclusions are usually found in more than
30% of red cells.
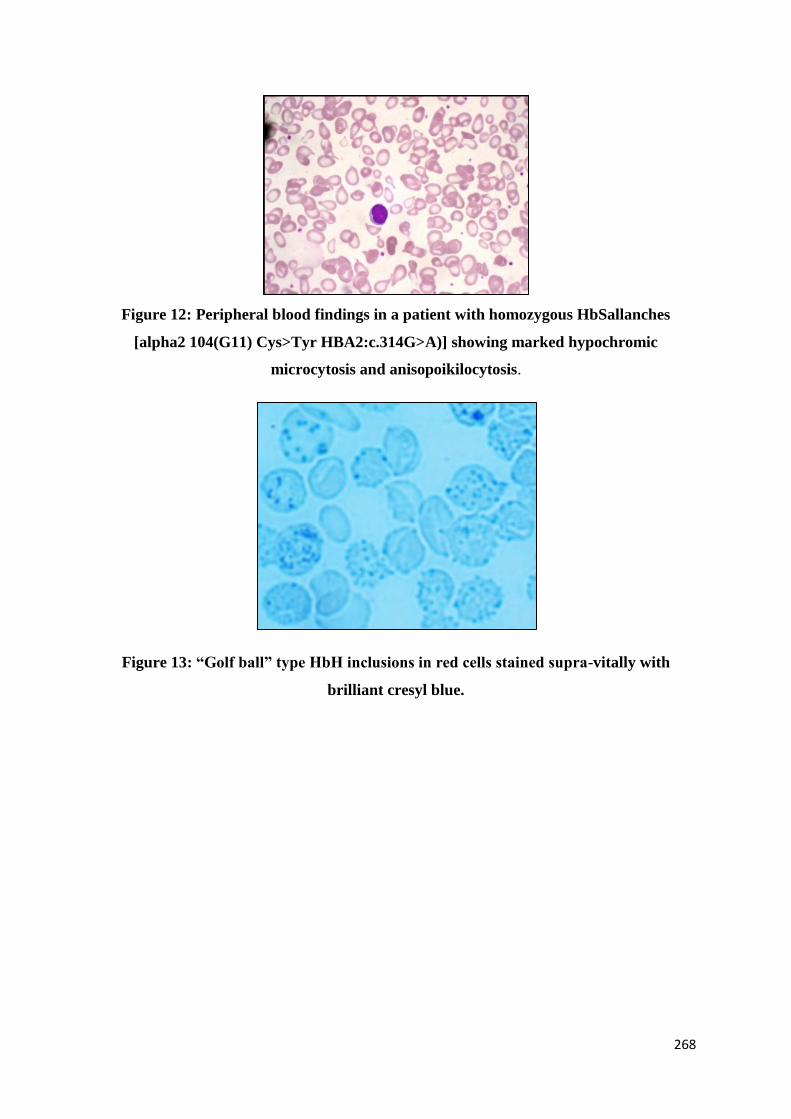
268
Figure 12: Peripheral blood findings in a patient with homozygous HbSallanches
[alpha2 104(G11) Cys>Tyr HBA2:c.314G>A)] showing marked hypochromic
microcytosis and anisopoikilocytosis.
Figure 13: “Golf ball” type HbH inclusions in red cells stained supra-vitally with
brilliant cresyl blue.
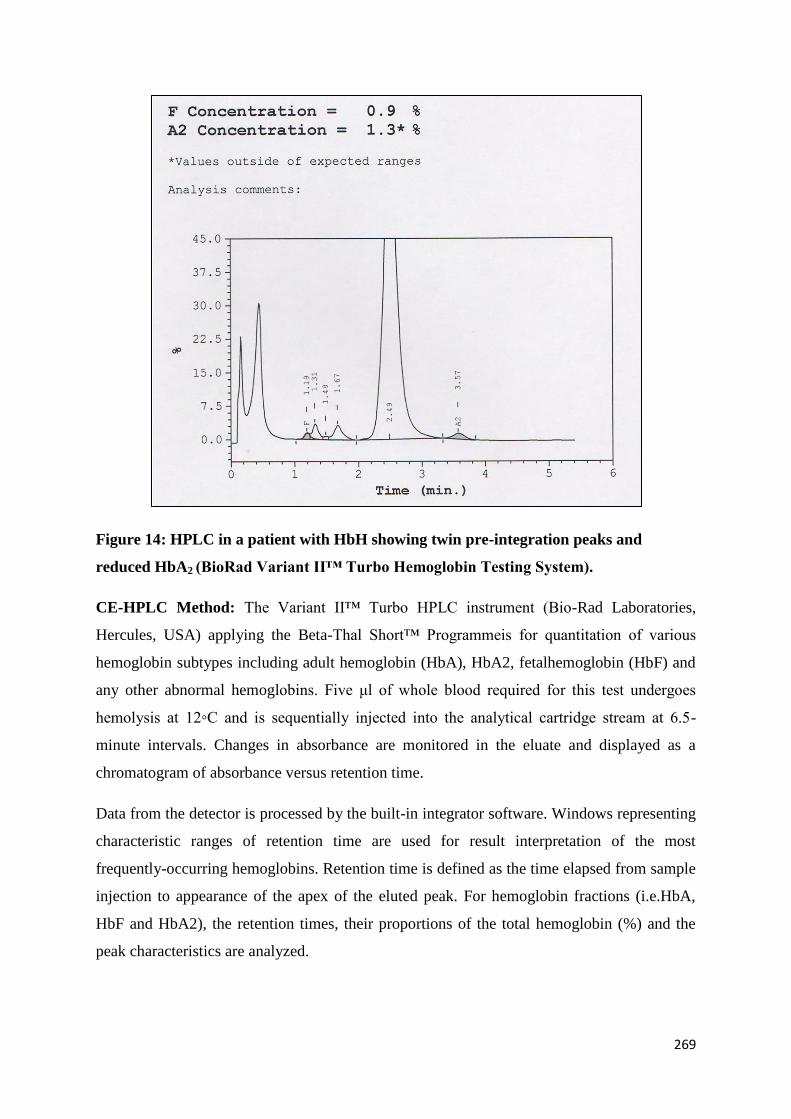
269
Figure 14: HPLC in a patient with HbH showing twin pre-integration peaks and
reduced HbA2 (BioRad Variant II™ Turbo Hemoglobin Testing System).
CE-HPLC Method: The Variant II™ Turbo HPLC instrument (Bio-Rad Laboratories,
Hercules, USA) applying the Beta-Thal Short™ Programmeis for quantitation of various
hemoglobin subtypes including adult hemoglobin (HbA), HbA2, fetalhemoglobin (HbF) and
any other abnormal hemoglobins. Five μl of whole blood required for this test undergoes
hemolysis at 12◦C and is sequentially injected into the analytical cartridge stream at 6.5-
minute intervals. Changes in absorbance are monitored in the eluate and displayed as a
chromatogram of absorbance versus retention time.
Data from the detector is processed by the built-in integrator software. Windows representing
characteristic ranges of retention time are used for result interpretation of the most
frequently-occurring hemoglobins. Retention time is defined as the time elapsed from sample
injection to appearance of the apex of the eluted peak. For hemoglobin fractions (i.e.HbA,
HbF and HbA2), the retention times, their proportions of the total hemoglobin (%) and the
peak characteristics are analyzed.

270
Cellulose Acetate Electrophoresis at Alkaline pH (pH 8.6):At alkaline pH,hemoglobin is
negatively charged. In an electric field it therefore migrates towards the anode (+). Structural
variants with surface charge difference will separate from HbA, however those without a
change in a charge will not. Cellulose acetate membranes are commercially available. They
contain about 80% air space in the form of pockets within the interlocking cellulose acetate
fibres. They come as dry, opaque, brittle films which crack easily if not handled properly.
When the film is placed in the buffer, the air spaces fill with liquid and then it can be easily
handled. These membranes can be made transparent for densitometry measurement of bands
using methanol and acetic acid mixture. The advantages of this technique are the speed of
separation (20-40 min) and the ability to store transparent membranes for longtime. Fully
automated instruments are now available for acid as well as alkaline pH electrophoresis
(Figure 15).
Figure 15. Automated Hb electrophoresis at alkaline pH (done on Genio S system,
Interlab, Rome) shows a fast moving relatively faint band of HbH in lane 2.
References:
1. Globin Gene Server: Laboratories of Computer Science, Engineering, Biochemistry
and Molecular Biology; Pennsylvania State University http://globin.cse.psu.edu
[accessed July 7, 2018]
2. Higgs DR, Weatherall DJ (2007) The alpha thalassaemias Cellular and Molecular Life
Sciences 1-9 DOI 101007/s00018-008-8529-9
3. Arnold SC, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex
polymerase chain reaction assay for α-thalassemia Blood 2001; 98,250-251.
4. Garewal G, Fearon CW, Warren TC, et al. The molecular basis of beta thalassaemia
in Punjabi and Maharashtrian Indians includes a multilocus aetiology involving
triplicated alpha-globin loci Br J Haematol1994;86:372-6.

271
5. Nadkarni A, Phanasgaonkar S, Colah R, Mohanty D, Ghosh K. Prevalence and
molecular characterization of alpha-thalassemia syndromes among Indians Genet Test
2008;12(2):177-80.
6. Nadkarni AH, Gorakshakar AC, Sawant PM, et al. The phenotypic and molecular
diversity of hemoglobinopathies in India: A review of 15 years at a referral centerInt J
Lab Hematol 2019;41(2):218-226.
7. Sen R, Chakrabarti S, Sengupta B, De M, Haldar A, Poddar S, et al. Alpha-
thalassemia among tribal populations of Eastern India Hemoglobin 2005;29(4):277-
80.
8. Shaji RV, Eunice SE, Baidya S, et al. Determination of the breakpoint and molecular
diagnosis of a common alpha-thalassaemia-1 deletion in the Indian population Br J
Haematol2003;123:942-7.
9. Sharma P, Das R, Trehan A, et al. Impact of iron deficiency on hemoglobin A2% in
obligate β-thalassemia heterozygotes Int J Lab Hematol2015;37(1),105-11.
10. Trehan U, Garewal G, Kaul D, Das R. Alpha thalassemia and alpha gene triplications
in Punjabis, with and without beta thalassemia trait. Haematology 2000; 6,153-160
11. Purohit P, Dehury S, Patel S, Patel DK. Prevalence of deletional alpha thalassemia
and sickle gene in a tribal dominated malarial endemic area of eastern India ISRN
Hematol 2014;745254

272
Role of Genetic Modifiers
Genetic modifiers can impact the phenotypic severity of -thalassemia at the primary level by
affecting the degree of globin chain imbalance and at the secondary level by modulating
complications of the disease. Hence, a major determinant at the primary level is the deficit in
β-globin production(milder or severe β-globin gene mutation), and this relates to the nature
and number of copies of the -thalassemia alleles. This is then followed by the 2 major
secondary modifiers which are coinheritance of α-thalassemia and the innate ability to
produce fetalhemoglobin (HbF, α2γ2), both of which directly reduce the globin chain
imbalance.
Primary Modifier of Hemoglobinopathies:
The pathophysiology of β-thalassemia involves imbalance between α and β globin chains of
hemoglobin. (Figure 16) Thus the reduced amount or absence of beta globin chains results in
an excess of unbound alpha globin chains which precipitate in erythroid precursors in the
bone marrow and interfere with the maturation of the red blood cells leading to their
destruction in the bone marrow and subsequent anemia. Thus the primary modifier of the
disease severity is the type of β-globin gene mutation (point changes to small deletions) that
the patient inherits, which further determines the extent of down regulation of the β-globin
gene.Accordingly the β-globin gene mutations are classified as: βo
or null mutations which
lead to complete absence of the β-globin on the affected allele, β+
mutations which cause
residual production of beta globin chains and β ++
(mildor silent mutations) which lead to very
mild reduction in beta globin production. Depending on the type of the β-globin gene
mutations inherited, the patients present variable clinical phenotypes.Similarly in other β-
thalassemia syndromes: HbS-β thalassemia and HbE-β thalassemia, the severity depends on
the type of the β –thalassemia mutation inherited along with the variant hemoglobin.The
milder β –thalassemia mutations leading to amelioration of the disease severity in the Indian
population are -88 ( C→T), Cap site +1 (A→C) and Poly A (T→C). Figure 16 shows the
basic pathophysiology of thalassemia and the role of secondary modifiers.

273
Figure 16: Basic pathophysiology of β-thalassemia. B: Correction of globin chain
imbalance in presence of secondary modifiers of hemoglobinopathies
Secondary Modifiers of Hemoglobinopathies:
The search for secondary modifiers of hemoglobinopathies was accelerated by the
observation that though all sickle cell anemia patients have the same mutation in the β-globin
gene, the patients display a varying degree of clinical severity. Similarly in β-thalassemics, it
is often observed that patients with the same β-globin gene mutations have diverse clinical
phenotypes. This led to the identification of potential secondary modifiers of
hemoglobinopathies, which are as follows:
a. Co-inheritance of α-thalassemia:
In many populations where -thalassaemia is prevalent, -thalassaemia also occurs at a high
frequency and hence it is not uncommon to co-inherit both conditions. Homozygotes or
compound heterozygotes for -thalassaemia who co-inherit -thalassaemia will have a lesser
imbalance of these globin chains and tend to have a less severe condition. The degree of
amelioration depends on the severity of the β-thalassaemia alleles and the number of
functional α-globin genes.
Alpha-thalassemia is caused most frequently by deletions involving one or both alpha
globin genes. The most common deletions remove a single alpha globin gene, resulting in
the mild alpha+-thalassemia phenotype. However the co-inheritance of α-thalassemia in
hemoglobinopathy patients is often found to be associated with a milder disease phenotype.
In β-thalassemia syndromes, associated α-thalassemia minimizes the excess precipitation of
the α-globin chains and reduces the globin chain imbalance. Similarly in sickle cell anemia,
the co-existing α-thalassemia reduces intracellular sickle hemoglobin concentration, thereby
reducing HbS polymerization, sickling and thus decreasing the hemolysis.

274
In -thalassemia heterozygotes, increased -globin production tilts the globin chain
imbalance, converting a typically clinically asymptomatic state to that of thalassemia
intermedia. This is related to the co-inheritance of triplicated () or quadruplicated -
globin genes () instead of the number of globin genes (seen in normal
individuals.
Apart from the number of α-globin genes and an inherent capacity for producing -globin,
the proteolytic capacity of the erythroid precursors in catabolising the excess chainshas
often been suggested, but this effect has been difficult to define. Alpha hemoglobin
stabilising protein(AHSP), a molecular chaperone of α-globin has been suggested as another
genetic modifier but so far studies have been inconclusive.
b. Genetic modifiers of fetalhemoglobin:
All -thalassemias, heterozygous or homozygous conditions, have a variable increase in HbF,
which relates to the expanded erythroid mass and selective survival of the red blood cell
precursors that contain HbF.
The clinical pathogenicity of hemoglobinopathies develops during the first year of life when
the -globin genes are gradually silenced with the activation of δ and β-globin genes. Thus
continued -globin gene expression could be considered as a new therapeutic strategy to treat
hemoglobinopathies. Since an elevated fetalhemoglobin (HbF) level ameliorates the clinical
severity of the disease, identification of the potential genetic markers that raise the HbF levels
is very important.Asin β-thalassemics, the main cause of the disease is altered globin chain
ratio, the continued -globin gene expression leads to the production of more globin chains
which combine with the excess unbound α-globin chains, thus further reducing the globin
chain imbalance. Similarly in sickle cell anemia patients, raised HbF (α22) levels reduces
intracellular HbS polymerization. Both HbF and its mixed hybrid tetramer (α β γ) cannot
enter the deoxy sickle hemoglobin polymer phase, thus circumventing the cellular damage
evoked by HbS polymers. Recent studies have shown that in humans, the HbF level is
regulated by 4 major quantitative trait loci (QTLs): HBB locus region (11p15, mainly the -
globin promoter region), BCL11A (2p16), HBS1L-MYB intergenic region (6q23), KLF1
(19p13) which brings about 20-50 % variance in HbF levels.

275
HBB loci[-globin promoter]:
The elevated HbF levels in an adult are generally due to large HBB cluster deletions
which lead to Hereditary Persistence of FetalHemoglobin (HPFH ) , δβ thalassemia or due to
single point mutations in the -globin gene promoter. The probable mechanism of non-
deletional forms of HPFH is increased competition for the Locus Control Region (LCR) to
the mutant -globin gene promoter consequently increasing the -globin gene transcription,
with respect to the β-globin gene. Alternatively the variations in the promoter region, ablate
the binding of repressors or increase the attachment of enhancers, thus affecting the gene
expression. The most extensively studied and the first QTL for HbFvariation is the sequence
variant (C→T) at -158 position in the G-globin gene promoter that alters the Xmn I
restriction enzyme recognition site, (HBG2 c.-211 C→T). This variation is reported to
influence the G-globin gene expression under stress erythropoietic conditions. The T allele
of the XmnI polymorphism is found to be linked to raised HbF levels and is frequently
observed in thalassemia intermedia patients. Similarly, SCA patients with the Senegal or
Arab-Indian βS
haplotype which is linked to the T allele of the XmnIpolymorphism,also show
increased HbF levels as compared to patients with the Bantu haplotype (with thewild type C
allele) which showed lowest HbF levels and a more severe clinical course, thus suggesting
the role of the mutant allele in HbF induction. Other variations in the -globin promoter
region, like -117 (G>A), -175 (T>C), -198 (T>C) and -202 (C>G) also generate HPFH
condition, as they are located in functionally active sites of the -promoter region.
B-cell CLL / lymphoma 11 A [BCL 11 A]:
B-cell CLL/ lymphoma11A, a zinc fingertranscription protein encoded by theBCL11A gene
located in chromosome 2p16, plays a crucial role in postnatal development and normal
lymphopoiesis. Recent studies have shown that BCL11A is a direct repressor of γ-globin
gene expression in adult erythroid progenitors.BCL11A along with other hematopoietic
transcription factors (GATA1, SOX6, ZFPM1/FOG) and thechromatin remodelling complex
plays a critical role in repressing -globin gene expression. Genome wide association studies
(GWAS)have identified BCL11A genetic polymorphisms located in intron 2 to be associated
with higher fetalhemoglobin levels in hemoglobinopathy patients. It has been observed that
the raised HbF-associated region in intron 2 of BCL11A is a distinct regulatory sequence for
binding of RNA polymerase II and GATA-1 transcription factor both of which regulate gene
expression. The intron 2 region of BCL11A also shows Histone 3 acetylation thus suggesting

276
the role in gene expression. Thus, several polymorphisms in this regioneg the C allele of
rs11886868 (C>T), T allele of rs1427407 (G>T), variant allele A of rs4671393 (A>G ) are
associated with increased HbF levels in different population groups. These results indicate
the importance of this region in elevating the HbF levels.
Tertiary Modifiers: Complications of –thalassemia
These modifiers do not affect globin imbalance directly but might moderate complications of
the disease in many different ways. They include genetic variants which affect bilirubin
metabolism, iron metabolism, bone disease and cardiac complications. For example, jaundice
and a propensity to cholelithiasis is a common complication of -thalassemia and is attributed
to the rapid turnover of the red blood cells. Increased bilirubin levels and predisposition to
gallstones in β-thalassemia is associated with a polymorphic variant in the UGT1A1
promoter. Individuals who are homozygous for 7 [TA] repeats, also referred to as Gilbert
syndrome, have higher levels of bilirubin and increased predisposition to gallstones, an
observation that has been validated at all levels of β-thalassaemia. Several genes involved in
iron homeostasis have now been characterised, including those encoding hemochromatosis
(HFE), transferrin receptor 2 (TFR2), ferroportin (SLC4OA1), hepcidin (HAMP) and
hemojuvelin (HFE2). The H63D variant, a common polymorphism in HFE, appears to have a
modulating effect on iron absorption. β-thalassemia carriers who are homozygous for the
HFE H63D polymorphism, have higher serum ferritin levels than carriers without the
polymorphism. Figure 17 shows the diagrammatic representation of genetic modifiers of
hemoglobinopathies.
Thus, improved understanding of the influence of modifier genes involved in modulating the
complex pathophysiology of hemoglobinopathies may allow prediction of the disease
phenotype and promises to improve patient management and treatment.

277
Figure 17: Schematic representation ofgenetic modifiers of hemoglobinopathies
References:
1. Bank A. Regulation of human foetal haemoglobin: new players, new
complexities. Blood. 2006;107:435-43.
2. Rund D, Fucharoen S. Genetic modifiers in hemoglobinopathies. CurrMol
Med.2008;8:600-8.
3. Thein S. Genetic basis and genetic modifiers of β-Thalassemia and sickle cell disease.
Gene and cell therapies for beta-globinopathies. New York, NY: Springer New York;
2017:27-57.
4. Uda M, Galanello R, Sanna S, Lettre G, Sankaran V, Chen W, et al. Genome-wide
association study shows BCL11A associated with persistent foetal haemoglobin and
amelioration of the phenotype of β-thalassaemia. ProcNatlAcad Sci USA.
2008;105:1620-25.

278
Newer Modalities of Management for Hemoglobinopathies
Symptomatic hemoglobinopathies present either as thalassemia syndromes where
unbalanced alpha and beta chain synthesis leads to ineffective erythopoiesis as a major
cause of anemiaand iron overload. Other clinical presentations in different
hemoglobinopathies are due to specific structural hemoglobin defects leading either to
(a)Sickling disorders (b) Anemia (c) Methaemoglobinaemia(d) Polycythemia (e)
Intravascular hemolysis, etc.
Majority of symptomatic hemoglobinopathies are autosomal recessive in nature hence
need to be inherited from both parents either as a homozygous or double heterozygous
state. Cases of dominantinheritanceandclinical presentation due to unstable hemoglobins
or rare subsets of dominant thalassemias due to unstable globin chains are known.
Certain hemoglobinopathies like HbS-Oman produce symptomatic sickling state even in
hetereozygous state.
Hence modern management of hemoglobinopathies can be described as :
a) Prevention of thebirth of babies with symptomatic hemoglobinopathies by premarital,
prenatal genetic counseling and intervention
b) Transfusion practice to treat different hemoglobinopathies and advances made in this
area
c) Management of iron overload arising out of chronic transfusion and increased iron
absorption.
d) Management of infectious complications of chronic transfusion.
e) Prevention and management of chronic organ damage and management of sickle cell
disease.
f) Management of other clinical presentations of symptomatic hemoglobinopathies
g) Recent advances in management of Sickle cell anaemia and thalassemia
a) Prevention of new births of babies withserious symptomatic
hemoglobinopathies

279
For the last 50 years premarital/ marital genetic counselling forpreventing marriage/
pregnancy between two heterozygote individualsled to significant reduction in the birth of
thalassemic children from Cyprus, Sicily and other Mediterranean Countries .
Development of various molecular techniques toidentify foetuses with severe
hemoglobinopathies in heterozygous couples at risk by antenatal diagnosis has reduced
thebirth of new patients with symptomatic hemoglobinopathies. Such facilities are
available in 5-6 government centres in India and many private laboratories, corporate
hospitals in this country. Antenatal diagnosis should be associated with pre and post
diagnosis genetic counselling so that the couple can decide about the fate of the affected
pregnancy.
Other alternatives of adoption, artificial insemination by donor sperm and in some
advanced centres detection of thepaternal mutation in the foetus using non invasive
diagnostic procedures should be discussed.Advances in assisted reproductive technology
may allow preconceptional genetic diagnosis (PGD) and implantation of non affected
zygotes leading to anormal pregnancywhich is the most advanced form of preventive
management and today it may be available in a handful of private medical centres in this
country.
b. Advances in Transfusion Practice to manage symptomatic hemoglobinopathies
including thalassemia syndromes
Red cell transfusion forms one of the major foundations to manage severe anemia of
thalassemia and other hemoglobinopathies. This mode of management is also used to
manage various complications of commonly prevalent sickle cell syndromes.
Red cell transfusion can be given in several ways. However red cell transfusion is a
double edged sword and when chronically used it could be associated with several serious
complications like
i) Alloimmunisations against multiple red cell antigens & HLA antigens
ii) Certain serious transfusion reactions
iii) Transfusion transmitted viral and bacterial infection (HIV, Hep B, Hep C)
Campylobacter infection, Yersiniosis to name a few. (this increases many fold in
splenctomised cases)
iv) Transfusion associated iron overload and its consequences.

280
All the above mentioned complications could be minimized by optimizing and proper
schedulingof red cell transfusions using safe blood under good medical supervision.
Advances in this area includes
i) Using packed red cells for transfusion rather than whole blood( 4 mlof packed
redcells/kgincreases post transfusion hemoglobin by 1 g/dL) and maintaining pre
transfusion haemoglobin level at around 9 g/dLand frequency of transfusion of
once in 2-4 weeks for transfusion dependent thalassaemia major. Sickle cell
disease cases can tolerate lower haemoglobin levels better and keep it around 7-9
g/dL.
ii) Use of leucocyte removing filter (Filtration during blood donation is better than
filtration during bed side transfusion- both are available now). Leucodepleted red
cells produce less frequent alloimmunisation and substantially reduce some of the
transfusion reactions.
iii) The patient’s red cells should also be phenotyped (genotyped) for extended blood
group antigen at least ABO, Rh CcDEe, Kell, Duffy, Kidd & MNSs needs to be
typed. Effort should be made at least to match for ABO, RhDCcEe and Kell
blood group antigen. If that is not possible (It is not possible in most centres in
India now) then patients extended phenotype may help in gettingthe right kind of
red cells for transfusion when the patient develop alloantibodies.
iv) Strict records should be kept on amount of packed red cells required per year to
maintainthe pre-transfusion hemoglobin at 9 g/dl (normally in multitransfused
thalassemia patients this should not exceed 200ml/kg/year). If the requirement
increasesto>300ml/kg/yr then the patient should be assessed for hypersplenism
and alloantibody formation.
v) Microbiologically safest blood should be used for these patients i.e the red cells
should be obtained from regular non remunerated voluntary blood donors who
have no risk factor for viral infection during donation. The blood also should be
tested using both ELISA (4th
generation) or chemiluminescence assay along with
Nucleic Acid Amplification test (NAT test) for the three common transfusion
viral infections (HIV 1+2, Hep B & Hep C infection). The patient should be
regularly tested for these viral infections and appropriate management measures
should be taken if found positive.

281
vi) For certain complications of hemoglobinopathies like sickling crises, sickle cell
associated with pregnancy, sickle cell patients with stroke or pre –operative
preparation for such patients, partial red cell exchange transfusion may be
prescribed to bring down circulating Hb S levels to below 35-40% (As is found in
asymptomatic Hb S heterozygote patients). Similar technique may be adopted in
patients presenting with severe methaemoglobinemia due to haemoglobinopathies.
vii) Management of iron overload for chronic transfusion dependent thalassemia and
other hemoglobinopathies
Iron Overload is an important complication of chronic RBC transfusion therapy.
This is universally seen in patients with transfusion dependent thalassemia
patients. It is also seen in non transfusion dependent thalassemia intermedia
patients where gastrointestinal iron absorption is the major cause of iron overload.
Traditionally this complication needed continuous (10-14 hours/day)
subcutaneous infusion of desferiroxamine (40 mg-50 mg/kg/day) usingan infusion
pump. This modality of management was inconvenient and extremely costly.
Introduction of oral iron chelators like L1 (Deferiprone)( 70 mg/kg) and
Deferasirox-(FCT), Deferosirox (AsunraR, Desorox
R, Defriset
R). They are now
universally used orally at a dose of 30-40 mg/kg/day. The iron level is controlled
by regular (3 monthly) measurement of the dose of iron chelators to maintain a
serum ferritin between 500-1000g/L , Liver function renal function , serum
ferritin levels and complete blood counts should be regularly monitored. Target
seum ferritin levels should be around 1000 g/L.
d. Management of Infectious Complications of hemoglobinopathies
Chronic transfusion therapy increases the risk of HIV 1+2, Hep B and Hepatitis C infection
and use ofsafe blood and immunization against Hepatitis B before beginning transfusion
therapy is desirable. However once these infections are detected in the patient during regular
follow up, the treatment of such patients should be by experts in this field as newer
antiretroviral drugs as combination therapy are regularly evolving so also the therapy of
established chronic hepatitis B and C infection. If the patient is splenectomised, the chances
of infection due to pneumococcus, and hemophilus infection increase tremendously. Hence
Pneumococcal and Hemophilus vaccination must precede splenectomy and thepatient should
be on regular penicillin prophylaxis.Anyfever should be aggressively investigated and
treated.

282
e. Prevention and management of organ damage
Proper iron chelation may prevent damage to liver, heart and various endocrine organs. Iron
overload of the liver can synergistically work to aggravate liver damage. Regular check up
and replacement therapy in chronically transfused thalassemia patients with the hormones
from failing endocrine glands (Pituitary ,Thyroid, Pancreas, testis, ovary, supraenal,) may
allow proper growth and development in these patients.
Amerorrhoea, failure of ovulation and testicular failure are not uncommon so also pancreatic
diabetics and growth failure in transfusion dependent thalassemia as well as iron overloaded
non transfusion dependent thalassemia intermedia patients.
f. Management of other clinical presentation of symptomatic hemoglobinopathies
(i) All patients in this group presenting with anemia should receive adequate vitamins and
minerals including regular folic acid replacement.
(ii) Patients presenting with methaemoglobinaemia should receive adequate doses of
ascorbic acid. In emergency situation with this complication exchange transfusion and
intravenous methylene blue is indicated.
(iii) Patients presenting with polycythemia may require regular venesection to maintain PCV
around 45% for preventing consequences of hyperviscosity
(iv) Many patients with hemolyticanemia and ineffective erythropoiesis as a part of
symptomatic hemoglobinopathies may also have hyperuricaemia and need to be treated with
Allopurinol or Febustat.
(v) Patients with sickle cell syndromes i.e sickle cell disease, sickle thalassemia, sickle
hemoglobin – D disease may present with diverse complications due to sickling process and
may need specialist treatment. However the general rules to treat sickle cell syndromesare as
follows:
Adequate hydration
Pain relief
Folic acid 5 mg/daily
Complete all immunizations including Pneumococcal pneumonia, hemophilus
influenza and hepatitis B vaccine.
Substantial advances in sickle cell disease therapy have taken place now

283
a. Hydroxyurea between 10-25 mg/kg/ day was found to improve hemoglobin, reduces
number of painful and other crises.
b. Crizanlizumab (SEG 101) an anti P-Selectin humanized antibody significantly
prevents painful crisis in sickle cell disease patients at a dose of 5mg/kg given
intravenously once every 3-4 weeks.
Indications and Usage :
Crizanlizumab is indicated to reduce the frequency of vaso occlusive crises (VOCs) in
adult and pediatric patients aged 16 years and older with sickle cell disease
Recommended Dosage
Administer Crizanlizumab 5 mg/kg by intravenous infusion over a period of 30 minutes at
Week 0, Week 2, and every 4 weeks thereafter.
Administer Crizanlizumab diluted solution by intravenous infusion over a period of 30
minutes through an intravenous line, which must contain a sterile, nonpyrogenic 0.2 micron
inline filter.
vi. For various thalassemia syndromes
a. Hydroxyurea has been assessed as therapy to reduce number of blood transfusions and
improve hemoglobin levels.
70% of thalassemia intermedia patients responds to hydroxyurea therapy and can
come out of intermittent red cell transfusion therapy. However the results are not so
satisfactory with thalassemia major or symptomatic Hb-E thalassemia patients. Our
study has shown 30% of such patients on hydroxyurea significantly reduce their
transfusion requirements. Molecular studies at two centres showed homozygosity of
the Xmn1 polymorphism(+/+) and co-inheritance of alpha thalassemia improves the
chances of positive results with hydroxyurea
b. Sotatercept and Luspatercept: These are new class of drugs used subcutaneously to
prevent apoptosis of late stage erythroblasts, a major cause of anemia in thalassemia
syndrome. These drugs act as TGF activating ligand traps. These drugs are
considered to be a significant advance to treat this disease
c. Allogenenic bone marrow transplantation with HLA matched sibling or unrelated
donor is already an accepted mode of therapy of severe thalassemia and sickle cell

284
disease. Significant side effects of this modality of therapy must be kept in mind
before advising to go for such therapy.
d. Gene Therapy. This mode of therapy has advanced significantly now and already
patients with thalassemia and sickle cell anemia have been treated with significantly
improved results in USA.
References:
1. Vrettou C, Kakourou G, Mamas T, Traeger-Synodinos J. Prenatal and
preimplantation diagnosis of hemoglobinopathies.Int J Lab Hematol. 2018 ;40
(S1):74-82.
2. Colah RB, Gorakshakar AC, Nadkarni AH.Invasive& non-invasive approaches
for prenatal diagnosis of haemoglobinopathies: experiences from India.Indian J Med
Res. 2011 ;134:552-60.
3. D.J.Weatherall and J.B.Clegg ,R.GibbonsD.R.HiggsJ.M.Old Nancy F.OlivieriSwee
Lay Thein W.G.Wood. The Thalassaemia Syndromes Fourth edition (2001)
Blackwell publication Oxford.Reprinted 2010.
4. Taher AT, CappelliniMD.How I manage medical complications of β-thalassemia in
adults.Blood. 2018 ;132:1781-1791.
5. Cappellini MD, Cohen A, Porter J, ( Eds).Guidelines for the Management of
Transfusion Dependent Thalassaemia (TDT) [Internet]. 3rd edition,
2014.Thalassaemia International Federation; Nicosia (CY).
6. Serjeant GR & Serjeant BE,(eds) Sickle cell Disease. 3rd
Ed 2001. Oxford University
Press.
7. GreerJP , Arber DA, Glader B, List AF, Means Jr.RT, Paraskevas F, Rodgers GM,
Foerster J . (Eds). Wintrobe’s ClinicalHematology. 13 th Ed 2013.Wolter Kluwer,
USA.
8. Torres L, Conran N.Emerging pharmacotherapeutic approaches for
the management of sickle cell disease.ExpertOpinPharmacother. 2019 ;20:173-186.
9. Taher AT, Weatherall DJ, CappelliniMD.Thalassaemia.Lancet. 2018 ;391:155-167.
10. Algiraigri AH, Wright NAM, Paolucci EO, Kassam A.Hydroxyurea for lifelong
transfusion-dependent β-thalassemia: A meta-analysis.PediatrHematol Oncol. 2017
;34:435-448.

285
Registry for Hemoglobinopathies
Hemoglobinopathies are the most prevalent genetic diseases in India. The national prevention
program is running successfully, however as the patients grow older, their management gets
more complicated. Paper based clinical notes are being used and there are some electronic
data banks mainly for financial matters.
The importance of ongoing systematic collection, analysis, and interpretation of data on
Hemoglobinopathies affecting the population, closely integrated with timely dissemination of
these data to those responsible for prevention and control will be quite useful. The registry is
meant to have a reliable data bank for epidemiologic and clinical data for the patients
suffering from blood disorders. This registry will be a source for health system decision
makers. The registry should consist of epidemiologic data, clinical history and important
physical examination points, laboratory results, a list of classic complications, list of
medications.
The registry needs to focus on:
• Assessing the burden of disease
• Monitoring trends in health
• Identifying emerging risks
Each record should be updated at least one time in each season. Electronic Registry should be
an online data bank for patients, physicians and policy makers for better research and
development in the field of Hemoglobinopathies.
The policy for Hemoglobinopathies envisages a robust system of data collection to be entered
systematically in a registry. Data should be collected both of patients to assess treatment
needs for blood and medicines etc. and carriers.
This data collection also helps in collecting secondary data:
1. Disease severity, co-morbidities, and chronic disease complications of persons with
Hemoglobinopathies.
2. Disease and treatment-related infections.
3. Reproductive and pregnancy outcomes of hemoglobinopathy patient populations.
4. Mortality rates, including case fatality rates for Hemoglobinopathies and complications.
5. Health care utilization, costs of care, and the geographic variation of specific service etc.
The policy recommends setting up of a patient registry for thalassemia and sickle cell
disease to obtain information on the number of persons affected and the number of
carriers to estimate patients who require various services.

286
The data on carrier screening performed in different regions should be collated to
determine the burden of hemoglobinopathies.
Creation of Central Hematopoietic Stem Cell donor registry, to facilitate bone marrow
transplants in those patients lacking sibling donors.
Generated across a variety of sources, data collection in hemoglobinopathies will also
encourage efficient communication between doctors and patients, and increase the
overall quality of patient care. As a response tothe digitization
of healthcare information and the rise of value-based care, the health care department
has taken advantage of this big data and analytics to make strategic decisions.

287
Section B: Hemophilia
Section Diagnosis of Management of Haemophilia Page
no.
1 Introduction 288
2 Abbreviations 290
3 Definition of various Levels of Training 293
4 Diagnosis of Hemophilia and other Bleeding Disorders 296
i Setting up a coagulation laboratory 297
ii Laboratory approach to bleeding disorders 301
iii Preanalytical variables and Quality Assurance in a coagulation laboratory 306
iv Laboratory Diagnosis of hemophilia and other bleeding disorders 315
v Genetic Counselling for haemophilia 348
vi Genetic Diagnosis of haemophilia 353
vii Women with Bleeding Disorders: Approach and laboratory Diagnosis 362
5 Management of Haemophilia and other Bleeding Disorders 369
i Clinical manifestations and management of Hemophilia 370
ii Management of hemophilia with inhibitors 385
iii Implementation of prophylaxis in hemophilia patients in India 391
iv Surgery in haemophilia 397
v Cautions to be exercised while using newer products/novel therapeutic
approaches for haemophilia
407
vi A comprehensive model for conservative management in haemophilia 414
vii Psychosocial Care and Support: Training Curriculum for
Psychologists/Social Workers/Nurses working in Bleeding Disorders
422

288
Diagnosis and
Management of
Hemophilia

289
Introduction
Hemophilia A and hemophilia B are inherited single gene disorders with an incidence of 1
per 10,000-30000 male births respectively. Besides hemophilia, there is a wide range of other
bleeding disorders which include von Willebrand disease (VWD), rare coagulation factor
defects, platelet function disorders and defects in the fibrinolytic pathway. These disorders
may manifest as spontaneous or post- trauma hemorrhagic episodes in patients. The
diagnostic and therapeutic aspects are important as specific therapy is now available for many
of these disorders.
The epidemiological data on the number of haemophilia patients in India is lacking which is
important for any public health intervention. Translating the incidence of haemophilia in the
Western World, it is anticipated that in India there may be approximately around 80000-
100000 haemophilia cases; however the number of registered patients in the disease registry
of Hemophilia Federation of India (HFI), a non-governmental Organization is only around
19000. As far as the global rank is concerned, India reports the second highest number of
patients with haemophilia A and 9 per cent of the total number of patients with haemophilia
A in the World. Though this data is generated by a very well organized network of more than
90 local Hemophilia Chapters across the country, there is still a gross unawareness and
underdiagnosis of haemophilia in this country.
The bleeding manifestations in hemophilia generally depend on the residual factor levels.
These patients are arbitrarily classified as severe (factor levels <1%), moderate (factor levels
1-5%) and mild (factor levels 5-40%). The appropriate treatment haemophilia patients is
replacement therapy i.e plasma derived or recombinant factor VIII or factor IX, which is
highly resource intensive. Due to the exorbitant cost and scanty availability of these products
in India, it used to be out of reach for many of our hemophilia patients. But thanks to the
Humanitarian Aid Program of World Federation of Hemophilia; in many of the Government
Hospitals and other places, there is a continuous supply of factor products which caters to not
only for “on demand” therapy but several children are on prophylactic treatment in several
Centers at no cost . This has also enabled to put few inhibitor positive patients on Immune
tolerance induction protocol. The HFI is mainly responsible for the data exchange,
requisition, import and distribution of treatment products, patient counselling, training ,
education, link patients to needed services and advocate for the rights of patients with
hemophilia and other bleeding disorders. Besides this, several State Governments are also
providing factors free of cost to haemophilia patients.

290
While more and more treatment products are becoming freely available, the training,
education and diagnostic facilities pose a major problem in this area. There are very few
comprehensive diagnostic facilities for bleeding disorders in the country, besides the fact that
even the medical personnel treating the haemophilia patients is not thoroughly educated about
the optimum treatment protocols within the existing resources. The only way to overcome all
these limitations is to implement a National program for diagnosis, care, support and
preventive strategies.

291
Abbreviations
AAP assembly activating protein
AAV Adeno-associated Virus
AD Autosomal dominant
ADP Adenosine diphosphate
ANM Auxiliary Nurse Midwife
aPCC Activated Prothrombin Complex Concentrates
APTT Activated Partial Thromboplastin time
AR Autosomal recessive
ASAP As soon as possible
ASHA Accredited Social Health Activist
AV Adenovirus
bp Base pair
BSC Biological safety cabinet
BSL Biosafety level
BSS Bernard Soulier Syndrome
BT Bleeding time
BU Bethesda Units
CBC Complete Blood count
CD Cluster of differentiation
CFC Clotting factor concentrates
CHC Community Health Centre
CHO-KLAT Canadian Hemophilia Outcomes: Kids’ Life Assessment Tool
CNS Central nervous system
CS Clot solubility
CVS Chorionic villus sampling
D/W Distilled water
DEIC District Early Intervention Centre
DH District Hospital
DIC Disseminated intravascular coagulation
DNA Deoxyribonucleic acid
dNTP Deoxyribonucleotide triphosphate
DRVVT Dilute Russell’s Viper venom time
EACA Epsilon aminocaproic acid
ED Exposure days
EDTA Ethylenediaminetetraacetic acid
ELISA Enzyme-linked immunosorbent assay
EQAS External Quality Assurance Scheme
F(Roman no) Factor
FDP Fibrinogen degradation product
FEIBA Factor eight inhibitor bypass activity

292
FGFR-1 Fibroblast growth factor receptor 1
FISH Functional Independence Score in Hemophilia
GI Gastrointestinal
GP Glycoprotein
GT Glanzmanns’s Thrombasthenia
HA Haemophilia A
HAL Haemophilia Activities List
HB Haemophilia B
HCL Hydrochloric acid
HCV Hepatitis C virus
HIV Human immunodeficiency virus
HPV Human Pappiloma virus
HR Health-related
HRP horseradish peroxidise
HSPG Heparan sulphate proteoglycans
HSV Herpes Simplex virus
HTC Haemophilia training center
IC Intracranial
IEC Institutional Ethical Committee
INR International normalized ratio
IQC Internal Quality Control
ITI Immune Tolerance Induction
ITR Inverted terminal repeats
IU International units
IVF In vitro fertilization
kb Kilo base
LA Lupus anticoagulants
LT Laboratory Technologist
Mab Monoclonal antibodies
mM Milli Molar
MMR Measles, mumps, and rubella
MO Medical Officer
MRI Magnetic resonance imaging
MSK Musculoskeletal
NPP Normal pool plasma
NSAIDS Nonsteroidal anti-inflammatory drugs
NV Non variceal
OBS Owren’s barbiturate saline
OD Optical density
OPD 1,2-o-Phenylenediamine dihydrochloride
ORF Open reading frames
PBS Phosphate buffer saline
PCR Polymerase Chain Reaction

293
PF3 Platelet factor 3
PFD Platelet function disorders
PGD Preimplantation genetic diagnosis
PHC Primary Health Centre
PK Prekallikrein
PNPP p-nitrophenyl phosphate
PPP Platelet poor plasma
PRP Platelet rich plasma
PSS Principles of Psychosocial Support
PT Prothrombin time
PWH Persons with haemophilia
QA Quality assurance
QC Quality control
QoL Quality of life
RBSK Rashtriya Bal SwasthyaKaryakarm
rFVIIa Recombinant activated factor VII
RG1 Risk Group classification 1
RIPA Ristocetin induced platelet aggregation
ROTEM Rotational thromboelastometry
RT Reptilase Time
RVVT Russell’s Viper venom time
SDS sodium dodecyl sulphate
SOP Standard operating procedure
TEG Thromboelastography
TMB 3,3',5,5'-Tetramethylbenzidine
TOT Training Of Trainers
TT Thrombin time
UK United Kingdom
VWF Von Willebrand factor
WFH World federation of haemophilia
μL Microliter

294
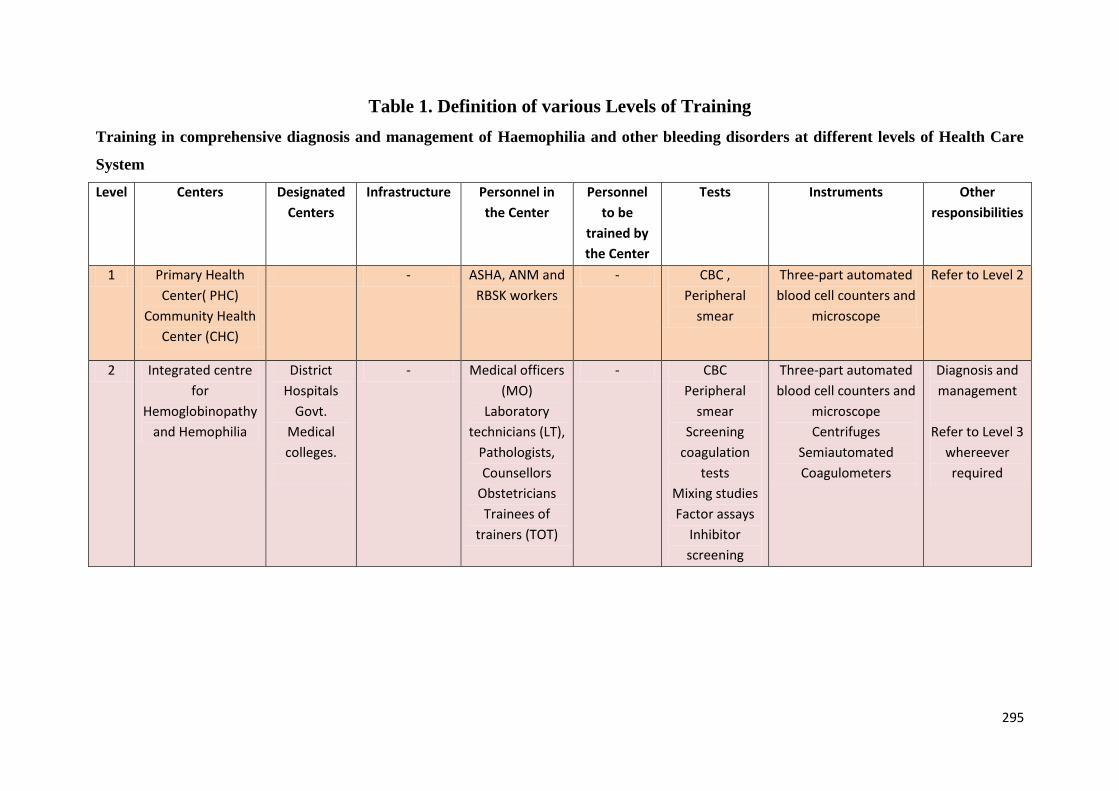
295
Table 1. Definition of various Levels of Training
Training in comprehensive diagnosis and management of Haemophilia and other bleeding disorders at different levels of Health Care
System
Level Centers Designated
Centers
Infrastructure Personnel in
the Center
Personnel
to be
trained by
the Center
Tests Instruments Other
responsibilities
1 Primary Health
Center( PHC)
Community Health
Center (CHC)
- ASHA, ANM and
RBSK workers
- CBC ,
Peripheral
smear
Three-part automated
blood cell counters and
microscope
Refer to Level 2
2 Integrated centre
for
Hemoglobinopathy
and Hemophilia
District
Hospitals
Govt.
Medical
colleges.
- Medical officers
(MO)
Laboratory
technicians (LT),
Pathologists,
Counsellors
Obstetricians
Trainees of
trainers (TOT)
- CBC
Peripheral
smear
Screening
coagulation
tests
Mixing studies
Factor assays
Inhibitor
screening
Three-part automated
blood cell counters and
microscope
Centrifuges
Semiautomated
Coagulometers
Diagnosis and
management
Refer to Level 3
whereever
required
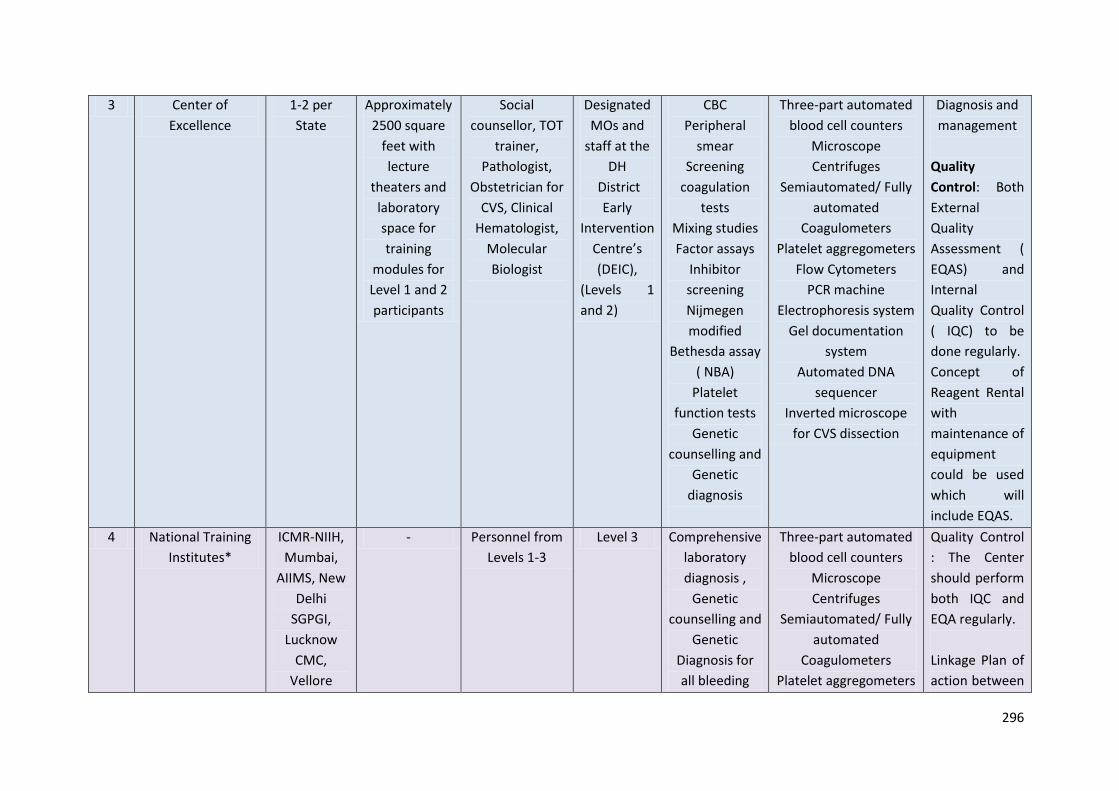
296
3 Center of
Excellence
1-2 per
State
Approximately
2500 square
feet with
lecture
theaters and
laboratory
space for
training
modules for
Level 1 and 2
participants
Social
counsellor, TOT
trainer,
Pathologist,
Obstetrician for
CVS, Clinical
Hematologist,
Molecular
Biologist
Designated
MOs and
staff at the
DH
District
Early
Intervention
Centre’s
(DEIC),
(Levels 1
and 2)
CBC
Peripheral
smear
Screening
coagulation
tests
Mixing studies
Factor assays
Inhibitor
screening
Nijmegen
modified
Bethesda assay
( NBA)
Platelet
function tests
Genetic
counselling and
Genetic
diagnosis
Three-part automated
blood cell counters
Microscope
Centrifuges
Semiautomated/ Fully
automated
Coagulometers
Platelet aggregometers
Flow Cytometers
PCR machine
Electrophoresis system
Gel documentation
system
Automated DNA
sequencer
Inverted microscope
for CVS dissection
Diagnosis and
management
Quality
Control: Both
External
Quality
Assessment (
EQAS) and
Internal
Quality Control
( IQC) to be
done regularly.
Concept of
Reagent Rental
with
maintenance of
equipment
could be used
which will
include EQAS.
4 National Training
Institutes*
ICMR-NIIH,
Mumbai,
AIIMS, New
Delhi
SGPGI,
Lucknow
CMC,
Vellore
- Personnel from
Levels 1-3
Level 3 Comprehensive
laboratory
diagnosis ,
Genetic
counselling and
Genetic
Diagnosis for
all bleeding
Three-part automated
blood cell counters
Microscope
Centrifuges
Semiautomated/ Fully
automated
Coagulometers
Platelet aggregometers
Quality Control
: The Center
should perform
both IQC and
EQA regularly.
Linkage Plan of
action between
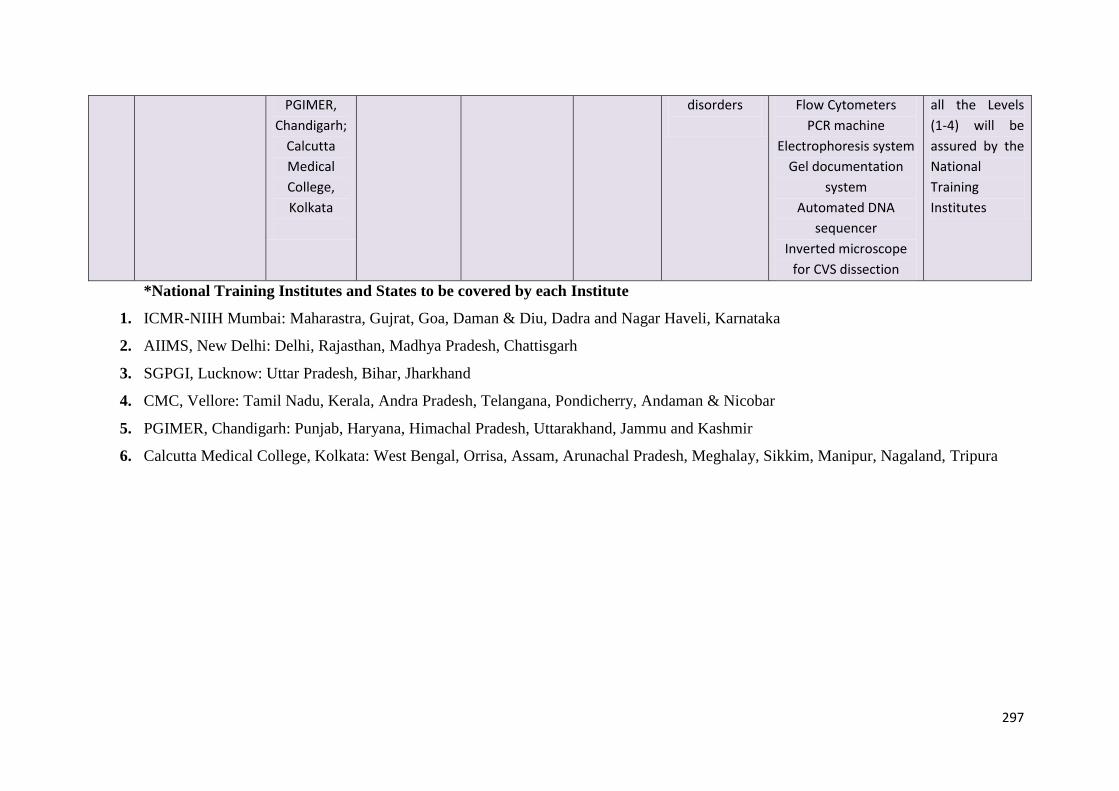
297
PGIMER,
Chandigarh;
Calcutta
Medical
College,
Kolkata
disorders
Flow Cytometers
PCR machine
Electrophoresis system
Gel documentation
system
Automated DNA
sequencer
Inverted microscope
for CVS dissection
all the Levels
(1-4) will be
assured by the
National
Training
Institutes
*National Training Institutes and States to be covered by each Institute
1. ICMR-NIIH Mumbai: Maharastra, Gujrat, Goa, Daman & Diu, Dadra and Nagar Haveli, Karnataka
2. AIIMS, New Delhi: Delhi, Rajasthan, Madhya Pradesh, Chattisgarh
3. SGPGI, Lucknow: Uttar Pradesh, Bihar, Jharkhand
4. CMC, Vellore: Tamil Nadu, Kerala, Andra Pradesh, Telangana, Pondicherry, Andaman & Nicobar
5. PGIMER, Chandigarh: Punjab, Haryana, Himachal Pradesh, Uttarakhand, Jammu and Kashmir
6. Calcutta Medical College, Kolkata: West Bengal, Orrisa, Assam, Arunachal Pradesh, Meghalay, Sikkim, Manipur, Nagaland, Tripura

298
Diagnosis of Hemophilia and other
Bleeding Disorders

299
Setting up a Coagulation Laboratory with available health care
infrastructure
Coagulation laboratory is an essential component of good health care delivery for every
hospital.These laboratories come with different levels of sophistication. Four Levels of such
laboratories can be conceived within the existing Infrastructure.
Level1 :Basic coagulation laboratory which can perform screening coagulation tests
including platelet count and complete haemogramalong with mixing studies.. This should be
available at CHC/ Rural hospitals / Taluk hospitals. Depending on how PHCs are improving
their service, these types of laboratories can be integrated to PHCs also.
Level2 : These are the laboratories with basic coagulation work up facilitiesalong with factor
assayusing one stage APTT based technique andscreening tests for inhibitors. This kind of
laboratory should be available at District / Civil hospitals.
Level3 :This laboratory should have all the facilities of a Tier 2 coagulation laboratoryand in
addition should be able to perform platelet function tests, von-Willebrand factor (VWF) assay
and inhibitor levels by Bethesda assay. The laboratory should also do all rare coagulation
factor assays. It is desirable that this facility in collaboration with Department of
Gynaecology and Obstetrics should be able to develop prenatal chorionic villus sampling /
amniotic fluid aspiration facility and at least do basic molecular biology procedures like DNA
extraction and transfer these samples to toLevel 4 laboratories for molecular biology based
detection of haemophilia and other rare bleeding disorders. Level 3 laboratories at present
should be located in Medical colleges/ Centers of Excellence ( 1-2 per state).Level 3
laboratories should also look after quality control and training of manpower for Level 1 and
Level 2 laboratories.
Level 4: These are the State Apex laboratories and should have all the facilities of previous
laboratories i.e.Level1 -3 laboratories.This laboratory will be a reference laboratory for that

300
State and is also responsible for the Quality control and assurance in Level 3 Laboratories. In
addition, these laboratories should be a comprehensive diagnostic Center for all hemostatic
and thrombotic investigations.Some of the advanced diagnostic facilities like Flow cytometry
for platelet receptor analysis, a full pledged molecular diagnosis facility for all types of
bleeding disorders , bioinformatics facility and so on.ThisCenter is also responsible for all
types of trouble shooting, holding interactive sessions with its peers and other National level
laboratories. The major responsibility of this Center is training manpower and develop human
resources. Appropriate training modules need to be developed by these Centers.Level 4
laboratories should be located in all AIIMS / or in the top medical colleges of the states.These
laboratories should also be engaged in active research, at least in various pilot projects and
operational research with practical applicability.
Basic requirements of laboratories from Levels 1-4
Space :Additional space of at least 300 sq.ft , 500 sqft , 600 sq ft, 1200 sq ft is required for
Level 1 , Level 2 , Level 3 and Level 4 laboratories respectively.
Location :Level 1 and Level 2 laboratories can work as part of Hematology/ Pathology
laboratories. For Level 3 and Level4 , independent modules of laboratory servicesshould be
available.
Manpower: All the laboratories should be under the supervision of a Hematopathologist/
Hematologist/ Pathologist / Laboratory medicine expert / Doctor with at least two years of
experience of working in a coagulation laboratory as the case may be. However for Level 3
and Level 4 laboratories, the working experience of more than two years in a coagulation
laboratory should be preferred. Exceptions should be made for exceptional candidates.
For Level 1, provision for one Medical Laboratory Technician with experience in specific
area should be employed. A trained technician if already available should be transferred to
this area and a new personmeanwhile can be trained. It is desirable that all technicians should
at least be able to do screening coagulation tests (PT and APTT) and complete blood count
(CBC) so that in emergency situations, they can do the job.For. Level 2 and Level 3
laboratories, at least two Medical Laboratory Techniciansshould be recruited for each Centre.
They should have a genetic counsellor too. For Level 4 laboratories,at least three Medical
Laboratory Technicians , one genetic counselor along witha Scientist with higher
qualificationswith expertise and experience inadvanced techniques like molecular biology,
flow cytometry and so on. This person should be a postgraduate in Micobiology/Applied
Biology/Biochemistry or similar subjectsPh.D degree holders should be desirable.

301
Instrumentation:FromLevels 1-4, all laboratories need to have the following basic
equipments.
Semiautomated 2- channel/ 4 - channel coagulometer
Blood Cell counter
Good quality microscope.
4 o C Refrigerator and -40/-70
oC Freezers
Refrigerated centrifuge
Water bath/ Thermometer/ Stop watch
Automated pipettes
Computers with wifi connection
Level 3 laboratories, in addition need the following equipments
Nano drop spectrophotometer.
Platelet aggregometer.
ELISA Reader
Dissecting microscope.
Whole blood coagulation analyser ( ROTEM,
Thromboelastography)
Level 4 laboratories need all the instruments mentioned above for Tiers 1-3 , but in
addition need the following equipments
Flowcytometer
A good Molecular Biology Laboratory with automated DNA
Sequencer
Reagents and consummables
The reagents and other consummables may vary from laboratory to laboratory; however all
laboratories need the following
Reagents to run automated blood cell counters.
Leishman stain.
PT/APTT/TT Reagents
Control normal lasma/ Deficient plasma
Calibrators with known factor levels
Disposable plasticwares/ Glasswares
Level 3 and 4 laboratories should also get fluorescence labelled antibodies and Molecular
Biology reagents

302
Tests to be available at different Levels of healthcare facility
Level 1
Screening coagulation tests ie PT, APTT and TT and CBC
Mixing studies
Level 2
All tests done at Tier 1
Factor VIII and IX assays
Screening for inhibitor including progressive inhibitor
D- dimer/ FDP screening
Level 3
All tests done at Tier 1-2
Quantitative inhibitor assays (Bethesda assay)
Thromboelastography
Platelet aggregometry
VWF assay
CVS and DNA extraction
Level 4
As this is the Reference Laboratory in the State, these laboratories should have a
comprehensive diagnostic facilities for diagnosis of all bleeding and thrombotic disorders.
These Centers also should look into developing novel simpler techniques for diagnosis. The
laboratory should also mmake efforts for diagnostic investigations to monitor altered newer
products in the marketwhich pose additional challenges of investigation for these apex
laboratories.Every laboratory should have a standard operating procedure ( SOP) manual.

303
Basics of Laboratory approach to bleeding disorders
Bleeding disorders constitute an important group of disorders in Hematology. The duties of
Laboratory Hematologists include diagnosis of the bleeding disorders by doing relevant tests
and interpret them correctly. It is important to have relevant clinical information before
interpreting the results. From the history, it is important to differentiate hereditary disorders
from acquired. Hereditary disorders generally start early in age, recurrent in nature and may
have a positive family history. On the other hand, acquired disorders occur at any age and
may have an underlying predisposing cause. History of medication including history of
administration of replacement therapy, warfarin or heparin also helps in interpretation of
tests. Depending on the type of bleed, it is important to differentiate between platelet and
coagulation factor deficiency disorders. In general, platelet disorder related bleeds present as
petichealhemorrhage, mucosal bleeds, whereas coagulation factor disorder presents as deep
abdominal bleeds, muscle bleeds, epistaxis and ecchymotic patches.
Laboratory Approach
Since a large number of tests are available for diagnosis of the spectrum of bleeding
disorders, it is important to go in a stepwise approach. A short screening coagulation should
be the starting point. Depending on the abnormalities observed in the screening tests,
specialized diagnostic tests can be performed.
Figure 1. The Coagulation Cascade

304
RVVT: Russel Viper venom time ; PF3: platelet factor 3; CS: clot solubility
Table 2. Screening tests in a patient with bleeding disorder
Screening test Normal Defective
Platelet count (PC) 150-450/cumm
Bleeding time (BT) 2-5 minutes PC
Platelet function
Vascular function
Prothrombin time (PT) 12-14 seconds Extrinsic pathway
(FVII deficiency)
Common Pathway (FI, II, V,
X deficiency)
Activated Partial
Thromboplastin time(APTT)
35-45 seconds Intrinsic pathway ( FVIII, IX,
XI, XII deficiency)
Common pathway (FI, II, V,
X deficiency)
Clot stability (CS) Stable upto 24 hours F XIII deficiency
Bleeding time
Bleeding time (BT) is prolonged in thrombocytopenia and platelet function disorders(PFD).
If it is prolonged and platelet count is normal, diagnostic tests for PFD such as platelet
aggregation with ADP, Adrenalin, Arachidonic acid, Collagen and Ristocetin can be
performed for specific diagnosis.
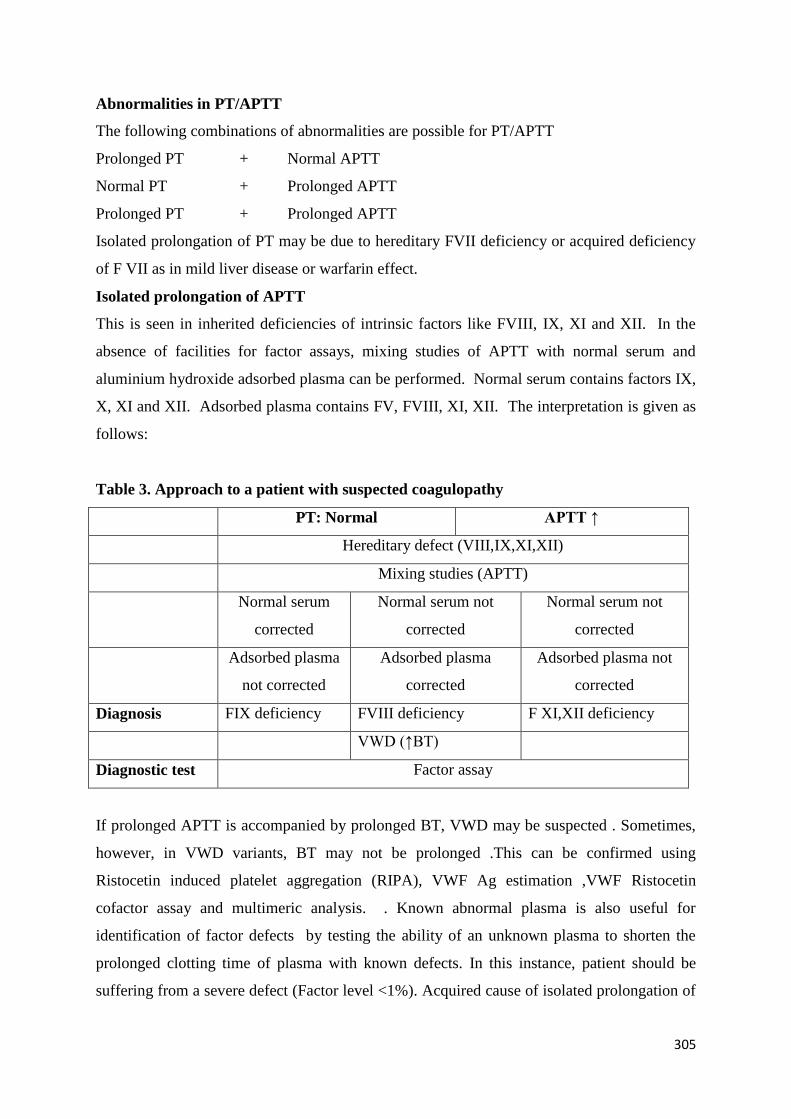
305
Abnormalities in PT/APTT
The following combinations of abnormalities are possible for PT/APTT
Prolonged PT + Normal APTT
Normal PT + Prolonged APTT
Prolonged PT + Prolonged APTT
Isolated prolongation of PT may be due to hereditary FVII deficiency or acquired deficiency
of F VII as in mild liver disease or warfarin effect.
Isolated prolongation of APTT
This is seen in inherited deficiencies of intrinsic factors like FVIII, IX, XI and XII. In the
absence of facilities for factor assays, mixing studies of APTT with normal serum and
aluminium hydroxide adsorbed plasma can be performed. Normal serum contains factors IX,
X, XI and XII. Adsorbed plasma contains FV, FVIII, XI, XII. The interpretation is given as
follows:
Table 3. Approach to a patient with suspected coagulopathy
PT: Normal APTT ↑
Hereditary defect (VIII,IX,XI,XII)
Mixing studies (APTT)
Normal serum
corrected
Normal serum not
corrected
Normal serum not
corrected
Adsorbed plasma
not corrected
Adsorbed plasma
corrected
Adsorbed plasma not
corrected
Diagnosis FIX deficiency FVIII deficiency F XI,XII deficiency
VWD (↑BT)
Diagnostic test Factor assay
If prolonged APTT is accompanied by prolonged BT, VWD may be suspected . Sometimes,
however, in VWD variants, BT may not be prolonged .This can be confirmed using
Ristocetin induced platelet aggregation (RIPA), VWF Ag estimation ,VWF Ristocetin
cofactor assay and multimeric analysis. . Known abnormal plasma is also useful for
identification of factor defects by testing the ability of an unknown plasma to shorten the
prolonged clotting time of plasma with known defects. In this instance, patient should be
suffering from a severe defect (Factor level <1%). Acquired cause of isolated prolongation of

306
APTT includes presence of lupus anticoagulants, intrinsic factor inhibitors or heparin
therapy. Lupus anticoagulant (LA) can be confirmed by Kaolin clotting assay and dilute
Russell viper venom test (DRVVT). Factor inhibitors can be confirmed by screening tests of
APTT followed by inhibitor assay (Bethesda assay).
Prolongation of both PT and APTT
This indicates deficiency in common pathway of coagulation (FI, II, V and X) or multiple
coagulation factor defects which may be inherited or acquired. When history suggests
hereditary defect, mixing studies with normal serum or adsorbed plasma are helpful in
differential diagnosis as given below:
Table 4. Interpretation of mixing studies
Causes of prolonged PT and APTT
PT↑ APTT ↑
Hereditary defect (V,X,II,I)
Mixing studies (APTT / PT)
Normal serum
corrected
Normal serum not
corrected
Normal serum not
corrected
Adsorbed plasma not
corrected
Adsorbed plasma
corrected
Adsorbed plasma not
corrected
FX deficiency FV deficiency TT
If normal: FII
deficiency
If ↑:
Hypofibrinogenemia
Dysfibrinogenemia
Perform specific Factor assay
Acquired conditions like Disseminated intravascular coagulation (DIC), fibrinolysis, liver
disease, vitamin K deficiency may also lead to prolongation of both PT and APTT. DIC is
confirmed by estimation of D-Dimer and/or FDP. Fibrinolysis shows normal D-Dimer but
abnormal FDP. Liver disease can be confirmed by liver function tests (SGOT/SGPT).
Vitamin K deficiency can be confirmed by repetition of PT, APTT 48 to 72 hrs after giving
5mg of vitamin K intravenously.
Despite such extensive testing, normal screening tests can be obtained in the following
conditions.
Disorders with normal screening tests
•Mild clotting factor deficiency
•Simple purpura

307
•Senile purpura
•Henoch Schonlein purpura
•Scurvy
•Hereditary hemorrhagictelengiectasia
Normal Control
These should be taken from healthy people not known to be suffering from any disease and
not from so called ‘normal’ patients (i.e. patients whose disease does not apparently seem to
affect coagulation system). Samples should not be collected from individuals under certain
conditions which might stimulate adrenoreceptors (e.g vigorous exercise such as running up
stairs ,donating blood transfusions etc.), which raise factor VIII in a few minutes or evoke
the ‘acute phase reaction’ raising fibrinogen, Factors V and VIII .Samples should not be
collected when a person has infection or from pregnant people.The wide range of normal
factor levels severely limits the usefulness of single random sample as control. Use of pooled
plasma from a number of healthy donors eliminates this variation to a considerable extent.
Abnormal Control
These are useful in the preparation of certain reagents. For instance the sensitivity of
phospholipid platelet substitute can be judged from the APTT performed on plasma from a
haemophilic with a mild defect(Factor VIII around 20%).
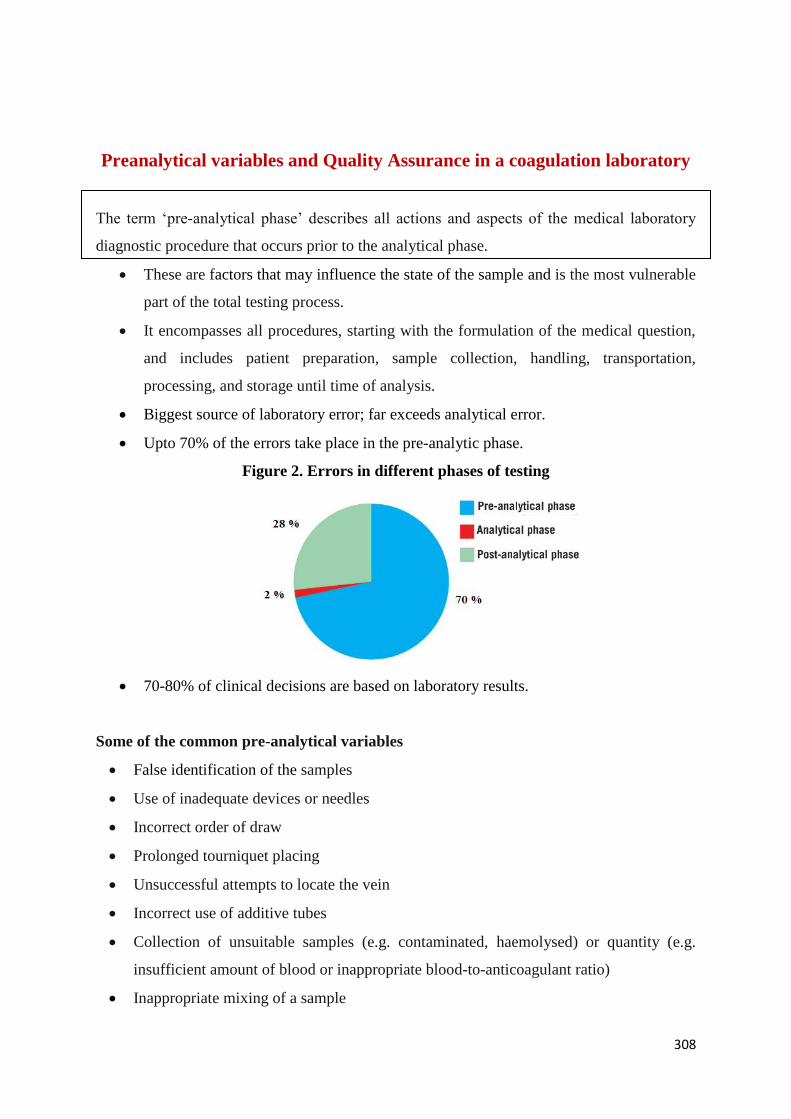
308
Preanalytical variables and Quality Assurance in a coagulation laboratory
The term ‘pre-analytical phase’ describes all actions and aspects of the medical laboratory
diagnostic procedure that occurs prior to the analytical phase.
These are factors that may influence the state of the sample and is the most vulnerable
part of the total testing process.
It encompasses all procedures, starting with the formulation of the medical question,
and includes patient preparation, sample collection, handling, transportation,
processing, and storage until time of analysis.
Biggest source of laboratory error; far exceeds analytical error.
Upto 70% of the errors take place in the pre-analytic phase.
Figure 2. Errors in different phases of testing
70-80% of clinical decisions are based on laboratory results.
Some of the common pre-analytical variables
False identification of the samples
Use of inadequate devices or needles
Incorrect order of draw
Prolonged tourniquet placing
Unsuccessful attempts to locate the vein
Incorrect use of additive tubes
Collection of unsuitable samples (e.g. contaminated, haemolysed) or quantity (e.g.
insufficient amount of blood or inappropriate blood-to-anticoagulant ratio)
Inappropriate mixing of a sample

309
Following are the areas where ‘Pre- analytical variations and errors’ can occur, which may be
addressed by following the suggested recommendations
Clinical history and physical examination
Bleeding history, bleeding score and family history
Drugs like aspirin or other antiplatelet drugs, NSAIDs, etc. Drug-induced effects on
platelet function should be considered when interpreting results.
Hormonal contraceptives
Bleeding history in relation to surgical, dental and obstetrical procedures.
Blood Group : Patients with O blood group are known to have lower levels of VWF.
Pregnancy status
Before sample collection
High fat meals interfere with light transmission aggregometry; 8- 12 hours fasting
samples are preferable
Smoking , consumption of alcohol, caffeine,physical activity , stress
During sample collection
Labeling must be done, before venipuncture, in the presence of the patient.
Blood sample should be collected directly from a peripheral vein (antecubital vein).
Tourniquet should not be used for over one minute.
The guage of the needle should preferably be between 19 and 22.
Blood samples for Haemostasis testing should not be obtained from an existing vascular
access device such as an intravenous (IV) line or a central line, which contain heparin.
Vacuum aspiration of blood into collection tubes may have adverse effect on platelet
function testing.
A syringe system permitting slow manual drawing of blood may be preferred.
Sample tubes and anticoagulants
Use siliconized glass or plastic (polypropylene) tubes.
Blood samples should be drawn into 105–109 mmol/L sodium citrate, buffered
anticoagulant.
The pH of the anticoagulated plasma should be between 7.3 and 7.45.
Collect venipuncture directly from a peripheral vein (antecubital vein).
The order of drawing blood during phlebotomy should be blood culture/sterile tubes,
then coagulation tubes, then plain tubes/gel tubes, then tubes containing additives.

310
Collect a discard tube when citrated plasma is obtained using butterfly needles.
The ratio of sodium citrate to whole blood must be 1:9.
In case of high haematocrit, the citrate volume should be adjusted maintain the citrate-
calcium ratio, and avoid plasma dilution with excess citrate.
Needed citrate volume (C, mL) may be calculated according to CLSI formula
C=(1.85x10-3) x(100-Hct)xV (where, V = original tube volume).
Figure 3. Citrate concentration in relation to hematocrit
Sample processing
Blood should be adequately, gently and promptly mixed by 4 to 6 complete end-over-end
inversions of the tubes in order to ensure even mixing of anticoagulant with blood.
Vigorous shaking, vortexing or agitation of blood samples should be avoided.
In case of high Haemocrit, Citrate volume adjustment must be done.
Examine blood sample tubes for presence of clots, precipitates or haemolysis, before
processing.
Blood sample transportation
Before transport, sample tubes should be examined for identification, safety conditions
and stability.
For local transportation, transport samples at ambient temperature (15–25°C), so as to
reach the testing laboratory within an hour of collection.
For long distance transportation, plasma should be separated, distributed in small
aliquots, and snap frozen in liquid nitrogen. The samples should be transported in frozen
condition.

311
Blood samples must remain capped for transport.
Sample rejection
Transported samples received in appropriate temperature conditions.
Inappropriate collection tubes and anti-coagulant.
Expired anti-coagulant tubes.
Patient identification ambiguity or lack of identification.
Inappropriate blood volume in relation to anti-coagulant.
Haemolysed samples.
Clot in the blood / Plasma sample.
Centrifugation
For obtaining platelet-rich plasma (PRP) for platelet function analysis, centrifugation
should be performed at 200–250 g for 10 min without application of a rotor brake.
Refrigerated centrifuge at 4°C should be used for processing samples for coagulation
assays.
Centrifuge the sample tube for coagulation testing at 1500g, 15 min.
The preparation of Platelet Poor Plasma (PPP) requires double centrifugation to obtain a
residual platelet count lower than 10x109/L.
Following initial centrifugation, carefully transfer the plasma to a non- activating plastic
centrifuge tube using an auto- pipette, and then centrifuged again for about 15 min.
There should be no vibrations or imbalance in the centrifuge.
Sample storage
Store samples at room temperature (15–25°C), to be processed and analyzed within 2- 4
hours of collection.
Perform whole blood assays < 4 hr after blood sampling and centrifugation < 1 hr.
Store PPP at −80 °C for long term storage (18- 24 months).
If whole blood sample is centrifuged within 1 hr of collection, the plasma can be left on
top of the cells at room temperature up to 4 hrs prior to assaying.
Time from sampling to analysis depends on analyte
Samples for PT/INR have longer stability (24 hrs) at room temperature (15–25°C).
Samples for APTT should be performed using fresh plasma < 4 hrs (<1 hr in patients
treated with unfractionated heparin).
For platelet function assays, samples should rest at room temperature for at least 15 min
before analysis. Testing should be completed within 3– 4 hrs of collection.

312
Factors V and VIII assays should be performed within 3 hrs of collection.
Protein S shows significant loss of activity at 8 hrs.
VWF appears to be stable at 15–25°C for 48 h.
Freezing and thawing
Centrifuge samples that cannot be tested within 4 hrs and freeze the plasma aliquot.
Use rapid freezing technique (liquid nitrogen).
Store samples at −70 °C (or below) rather than −20 °C.
Plasma samples frozen at minus 20 °C remain stable for 2 weeks.
Plasma frozen at minus 80 °C remains stable for 6 – 24 months.
Do not re-freeze samples (prepare sufficient number of aliquots).
Thaw samples rapidly at 37 °C (to prevent denaturing fibrinogen) at least 5 min in a
water bath at 37 °C and not at room temperature, or work bench or in a microwave oven.
Test immediately.
After thawing, mix the sample gently to re-suspend any cryoprecipitate. Do not vortex or
shake.
In a coagulation laboratory, it is essential to ensure that the right test is carried out with the
right blood sample; after which the correct results are issued to the specific patient without
any undue delay.
Quality control and assurance
Quality assurance (QA) is defined as the overall programme for achieving these objectives
whereas Quality control (QC) is defined as measures that needs to be included for each assay
performed to ensure that the test is working properly. The term ‘QC refers to the statistical
quality control that is commonly used in laboratories to monitor the routine performance of
testing processes, detect possible errors and correct problems before test results are reported.’
Quality Assurance Quality Control
An overall management plan to guarantee
the integrity of the data
A series of analytical measurements used to
assess the quality of the analytical data

313
Figure 4. The Quality Assurance Cycle
Components of Quality Control
The main aim of internal quality control procedures and external quality assessment programs
is to evaluate and improve quality in clinical laboratories.
Internal Quality control (IQC)
Monitoring all the laboratory tests done and includes measurements of specially
prepared materials and repeated measurements on routine specimens, along with daily
statistical analysis of the data.
It is primarily a demonstration of precision
It ensures continual evaluation of the reliability of the daily work of the laboratory
with validation of tests before reports are issued
Quality control for coagulation tests

314
Preparation and selection of reagents
Normal pooled plasma (NPP) preparation
For all coagulation factor assays, normal pooled plasma or unicalibrator with a known factor
level should be used as a control. Fresh control plasma from one healthy individual should
never be used as a control for factor assays.
Factor deficient plasma
Either “in house” deficient plasma or commercial deficient plasma may be used for factor
assays.
Reagent selection for PT and APTT
Calibration of centrifuge and pipettes
315
Schedule for quality control procedures
1. Calibration with reference standards
Instruments, pipettes After every 6 months or more frequently if
control chart or EQA indicates bias or
fluctuation in results
After any repair/ service
Others: thermometers, scales,
etc
Yearly
2. Control chart with control reagents
Every single day and with each batch of samples
For 2- 3 patient’s samples, duplicate tests needs to be done if there are
discrepancies
3. Patient’s test results analysis
Check previous tests and if changes in clinical state and analyse before issuing
report
4. EQAS performance
Monthly assessment
External Quality Control (EQA)
Evaluation by an outside agency of the laboratory and methods. This can be organized
nationally, regionally or internationally.
EQA ultimate and overarching goal is to improve the quality of laboratory services, along
with many other benefits as follows:
1. To enhance patient care and safety through improved laboratory practice
2. To characterize test bias and imprecision across multiple methods
3. To correlate specific method variables with bias and imprecision
4. To identify interfering substances and quantify their effects across multiple methods
5. To provide clinical laboratories with reliable information for replacing unsatisfactory
methodologies
6. To identify clinical laboratories that are at risk for poor performance

316
7. To satisfy accreditation and regulatory requirements
Source: Modified from Cunningham MT, Brandt JT, Chandler WL, et al. Quality assurance
in hemostasis: the perspective from the College of American Pathologists proficiency testing
program. Semin ThrombHemost2007;33:250–258.
Few of the Quality control exercises in Haemotology are as follows
CMC EQAS
UK NEQAS
EQAP
Suggested Reading
Westgard JO. Internal quality control: planning and implementation strategies. Ann Clin
Biochem 2003;40: 593–611.
Raiyani Ankit. Internal Quality control in coagulation lab. Sahayadri Speciality Hospital.
https://www.slideshare.net/adraiyani/internal-quality-control-iqc-in-coagulation-lab
Tietz. Fundamentals of Clinical Chemistry and Molecular Diagnostics - E-Book by Carl
A. Burtis, David E. Bruns. Chapter: Quality Management by George G klee and James
Westgard.
Cristiane F. Oliveira; Talma R. L. Fernandes. Analysis of the pre-analytical phase in a
private pathology laboratory of Maringá city-PR, Brazil. J Bras Patol Med Lab 2016; 52:.
78-83.
WHO Guidelines on Drawing Blood: Best Practices in Phlebotomy. Geneva; 2010.
http://www.euro.who.int/__data/assets/pdf_file/0005/268790/WHOguidelines-on
drawing-blood-best-practices-in-phlebotomy-Eng.pdf?ua=1.
Mackie I, Cooper P, Lawrie A, Kitchen S, Gray E, Laffan M, British Committee for
Standards in H. Guidelines on the laboratory aspects of assays used in haemostasis and
thrombosis. Int J Lab Hematol. 2013;35:1–13.
Harrison P, Mackie I, Mumford A, Briggs C, Liesner R, Winter M, Machin S. British
committee for standards in H. Guidelines for the laboratory investigation of heritable
disorders of platelet function. Br J Haematol. 2011;155:30–44.
Bonhomme F, Ajzenberg N, Schved JF, Molliex S, Samama CM, French A. Intensive
care committee on evaluation of routine preoperative T, French society of a, intensive C.
Pre-interventional haemostatic assessment: guidelines from the French society of
anaesthesia and intensive care. Eur J Anaesthesiol. 2013;30:142–62.
Magnette A, Chatelain M, Chatelain B, Ten Cate H, Mullier F. Pre-analytical issues in
the haemostasis laboratory: guidance for the clinical laboratories. Thromb J. 2016 Dec
12;14:49.
Favaloro EJ, Lippi G, Adcock DM. Preanalytical and PostAnalytical variables: The
leading cause of diagnostic error in hemostasis? Semin. Thromb. Hemost. 2008;34:612-
634.
CLSI. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-
Based Coagulation Assays and Molecular Hemostasis Assays; Approved Guideline-
Fifth Edition. CLSI document H21-A5. Wayne, PA: Clinical and Laboratory Standards
Institute; 2008.

317
DetailedLaboratory Diagnosis of Hemophilia and other Bleeding Disorders
Defects of the coagulation, platelet, endothelial or fibrinolytic systems may lead to bleeding
and thrombotic abnormalities. While acquired defects may involve multiple systems;
congenital defects are usually due to an abnormality of a single factor. For providing an
accurate and specific diagnosis application of specific laboratory tests along with an
appropriate history and clinical background.
A highly regulated cascade of reactions involving platelets and coagulation proteins result in
the formation of a blood clot following an injury. This is reversible and self-limited. Initially
platelets adhere to collagen through specific receptors in the presence of von Willebrand
factor (VWF) upon exposure of subendothelium resulting in subsequent activation and
aggregation of platelets which forms a “platelet plug” at the injury site. Subsequently the
coagulation cascade gets activated resulting in a stable fibrin clot. Specific inhibitors then
inhibits each of the coagulation factors preventing clot extension beyond the site of injury.
The final step involved the fibrinolytic step which involves the lysis of fibrin in the blood
clot; thus restoring the blood flow.
Three important parameters are necessary for the diagnosis of a bleeding patient: a) family
history b) clinical history and c) laboratory test results.
Family history & pedigree
This should mainly consist of details of the affected members in the family like gender of the
affected individuals, detailed history of bleeding during surgery or bleeding in any of the
family members, death due to unknown cause along with other details relevant to each
individual family, history of consanguinity among parents. An extended family pedigree
should also be drawn with details of both dead and live members in the family.
Clinical history
This includes type of bleeding (deep tissue or superficial bleeding, joint bleed), spontaneous
or traumatic, menorrhagia or excessive bleeding during dental extraction/ surgery. Other
details like number of bleeding episodes and factor infusion annually, sites of bleeding,
response to treatment should also be noted in the clinical proforma.

318
A detailed family history including family pedigree along with the clinical details will give a
hint of diagnosis in many of the cases.
Laboratory tests
Laboratory tests include both screening and confirmatory tests to detect the deficiency or
abnormality of each of these factors.Screening tests include peripheral smear, platelet count,
prothrombin time (PT), activated partial thromboplastin time (APTT) and thrombin time
(TT). The screening test results gives a clue as to which confirmatory or specific tests should
be chosen and done; making these tests highly cost effective. Confirmatory tests include
platelet aggregation tests, factor assays, platelet receptor studies, von Willebrand factor
(VWF) assay, VWF multimer studies, VWF-FVIII binding assay, VWF-ristocetin cofactor
assay, VWF-collagen binding assay, Nijmegen modified Bethesda assay for inhibitors. For
the diagnosis of rare platelet function defects, there are another battery of other tests which
are performed in specific cases.
Coagulation Screening Tests
A broad overview of hemostatic defects is provided by the coagulation screening tests which
further provide clues as to which specific tests need to be performed.
a. Prothrombin time (PT)
Principle
The extrinsic pathway of coagulation is measured by this test. Activation of extrinsic clotting
factors is done by adding tissue factor (extracted from brain tissue or synthetic),
phospholipids and calcium ions to plasma resulting in thrombin generation and fibrin clot
formation. The test is sensitive to vitamin K dependent factors i.e. factors II, VII, V, X as
well as fibrinogen. Primarily this test is used for monitoring of anticoagulant therapy.
Requirements
Control and patient citrated plasma
Thromboplastin reagent

319
Procedure
Test plasma (50 μL)
Incubate for 1 minute at 37°C
Thromboplastin reagent (100 μL)
Note the clotting time
Normal Range: 12 - 14 seconds
Interpretation
A prolonged PT may be observed in the following conditions:
a. Congenital factor VII deficiency in cases of isolated PT prolongation in the absence
of other acquired causes leads to the diagnosis of
Congenital deficiency of factors II, V, VII, X
Severe fibrinogen deficiency
Inherited or acquired defects due to consumption (e.g. DIC)
Liver disease (vitamin K deficiency or lack of synthesis)
Dilution (massive transfusion)
b. In the presence of lupus anticoagulant
c. In the presence of specific inhibitors to coagulation factors
d. In the presence of high levels of heparin
e. Very low levels of fibrinogen
f. Patients on warfarin
b.Prothrombin time – International normalized ratio (PT-INR) determination
The INR provides a standardized scale
For monitoring patients who are under stable oral anticoagulant therapy, the INR provides a
standardized scale. The International Sensitivity Index (ISI) is calculated according to the
following formula:
INR = RISI
Where R = Patient PT
Control PT
An ISI value is assigned by calibration against an International reference preparation
according to the WHO recommendation for every lot of thromboplastin reagent. For
mechanical and photo-optical coagulation instruments, Lot-specific ISI values are provided.

320
The recommended therapeutic range is between an INR of 2.0 and 4.5 for patients on oral
anticoagulant therapy.
c. Activated partial thromboplastin time (APTT)
Principle
In this test, the intrinsic coagulation pathway is triggered by an activator such as kaolin which
activates the contact factors. The contact factors cause a cascade of enzyme reactions in the
presence of calcium ions and phospholipids (partial thromboplastin) resulting in the
generation of thrombin and fibrin clot formation. An abnormal APTT usually indicates
deficiency of factors VIII or IX and occasionally of XI or XII; if prothrombin time is normal.
Requirements
Control and patient citrated plasma
APTT reagent
0.025M Calcium Chloride (CaCl2)
Procedure
Test plasma (50 μL) + APTT reagent (50 μL)
Mix and incubate at 37°C for 3 or 5 minutes
(as per the instructions)
Pre-warmed 0.025M CaCl2 (50 μL)
Note the clotting time
Normal range: 30 – 35 seconds
Interpretation
A prolonged APTT may be observed in the following conditions:
a. Isolated APTT prolongation in the absence of other acquired causes provides a diagnosis
of hemophilia A, hemophilia B or rarely of factor XI, factor XII or contact factor
deficiency.
b. Congenital deficiency of factors II, V, VIII, IX, X, XI, XII, prekallikrein (PK), high
molecular weight kininogen (HMWK) or fibrinogen
c. It may be acquired e.g. DIC, liver disease
d. Vitamin K deficiency or decreased synthesis, dilution (massive transfusion), or
anticoagulation (coumarin derivatives)
e. Patient on Heparin therapy
f. Presence of specific inhibitors

321
g. Presence of lupus anticoagulant
Differential Sensitivity of APTT reagents
APTT reagents are composed of synthetic phospholipids or rabbit brain phospholipid extracts
along with negatively charged activators like kaolin, ellagic acid, silica or celite. The in vitro
clot formation is triggered by the activation of factor XII when an APTT reagent is added to
plasma. The APTT reagent is responsive to deficiencies in the intrinsic and/or common
pathway. Generally factor deficiencies below 30IU/dL are detected by the APTT reagents.
APTT reagents are also used for monitoring the anticoagulant effect of unfractionated
heparin. For monitoring heparin therapy, laboratories generally establish a reagent-specific
APTT therapeutic range.For LA screening, one should use two APTT reagents i.e. one with
low phospholipid content and one with high phospholipid content. If the prolonged APTT
observed by using low phospholipid content is corrected by using high phospholipid reagent;
it means there is the presence of LA. This however needs to be confirmed with specific
assays. Different sensitivities have been seen in different APTT reagents not only for
different coagulation factors or in different clinical settings, but also to different levels of
factors
Note: Though PT and APTT values generally correspond to factor levels, this may not hold
true in all cases. High levels of one clotting factor can compensate for the lower levels of
other factor. Another important point to note is that in the presence of any history of bleeding,
it is justified to go ahead and do specific factor assays.
d. Mixing studies for prolonged PT and APTT
For plasma samples found to have prolonged screening tests (i.e. PT/ APTT), further
investigations are required to detect the specific factor deficiency by performing mixing tests.
However, it is important to rule out the presence of an inhibitor.
Principle
If prolonged PT and APTT does not get corrected or if there is <50% correction after mixing
patients plasma with NPP, the presence of an inhibitor is indicated. Factor deficiency will be
indicated by similar tests performed with specific factor deficient plasma samples will
indicate factor deficiency, if there is no correction with that specific deficient plasma.
Requirements
Citrated plasma of control and patient

322
Factor deficient plasma
APTT reagent
APTT mixing study results for detection of specific factor deficiency:
Reagent APTT (seconds)
NPP 29
Patient 82
Patient + NPP 30
Patient+FVIII deficient plasma 83
Patient +FIX deficient plasma 36
Diagnosis: FVIII deficiency
e. APTT screening test for contact factor deficiencies
Principle
When a sample with markedly prolonged APTT, incubated with APTT reagent for a longer
time i.e. at least 3 times the original incubation time, results in normalized APTT;
deficiencies of contact factors i.e. Prekallekrein (PK) and High molecular weight kininogen
(HMWK) is suggested.
Requirements
Citrated plasma of control and patient.
APTT reagent
0.025M Calcium Chloride (CaCl2)
Procedure
Test plasma (50 μL) + APTT reagent (50 μL)
Mix and incubate at 37°C for 15 minutes
(in place of 5 minutes)
Pre-warmed 0.025M CaCl2 (50 μL)
Note the clotting time
Interpretation
Contact factor deficiency is suggestive if the prolonged APTT gets normalized with
prolonged incubation. Deficiency of contact factors is not related to bleeding; however it is
still necessary to explain the prolonged APTT, especially during pre-surgical evaluation.

323
f. Thrombin time (TT)
Principle
The formation of a fibrin clot in plasma is measured by thrombin time test by the action of
thrombin on fibrinogen. Abnormal TT is observed in cases of fibrinogen deficiency,
abnormal fibrinogen or inhibition of conversion of fibrinogen to fibrin by heparin or fibrin
degeneration products. It is also seen in liver failure and intravascular clotting.
Requirements
Citrated plasma of control and patient
Thrombin reagent
Procedure
Test plasma (100 μL)
Incubate at 37°C for 1 minute
Thrombin reagent (100 μL) (3NIH U/mL)
Note the clotting time
Normal Range: 12-14 seconds
g. Thrombin time (TT) with protamine sulphate neutralization for the detection of
presence of heparin
In the presence of unfractionated heparin and sometimes even in the presence of low
molecular weight heparin, TT is prolonged. This can be checked by adding protamine
sulphate to the TT reagent. Protamine sulphate neutralizes heparin in the plasma sample. A
working solution of 40 mg% solution is prepared in normal saline. One part of this solution is
mixed with 9 parts of TT reagent and the TT test is repeated as mentioned above. If the
thrombin time gets normalized after this addition, it may be confirmed that the prolonged
thrombin time is due to the presence of heparin in the sample.
324
h. Reptilase Time (RT)
Principle
Reptilase is an enzyme which is extracted from the venom of the snake named Bothropsatrox.
This enzyme is not affected by the presence of heparin; thus used in a reagent and the test
performed whenever a patient is on heparin. Reptilase reagent is also more sensitive to
abnormal fibrinogen and is thus used as a test for dysfibrinogenemia in some of the
laboratories. The RT has been found much more prolonged than TT in case of
dysfibrinogenemia.
Requirements
Control and patient citrated plasma
Reptilase time reagent: 1.7 mg/mL stock solution of the Reptilase reagent is prepared in
Owren’s buffer and is preserved in -700C in small aliquots. Working solution is prepared
by diluting the stock 1:10 times in Owren’s buffer.
Note: Avoid any contact with the reagent; masks and gloves should be worn and care
should be taken not to inhale the reagent.
Procedure
Test plasma (150 μL)
Incubate at 37°C for 1 min
Reptilase time reagent (50 μL)
Note the clotting time
Normal Range: 15-18 seconds.
Table 5. Thrombin time and Reptilase time Interpretation
Reptilase time (RT) Thrombin time (TT) Interpretation
Prolonged Prolonged Afibrinogenemia,
Hypofibrinogenemia,
DIC
More prolonged than TT Prolonged Dysfibrinogenemia
Normal Prolonged Heparin
Note:
i. For all of the above tests, volume of all the reagents mentioned is for the semi-
automated coagulometers.
ii. The normal range may vary from laboratory to laboratory.
325
Table 6. Interpretation of Screening coagulation test results
Prolonged Factor deficiency Gene defect Acquired causes
PT FVII F7 Liver disease, Oral
anticoagulants, DIC, Sepsis
APTT FVIII, FIX, FXI, FXII &
Contact factors
F8, F9, F11 ,F12,
& contact factor
genes
Liver disease, Oral
anticoagulants, Sepsis, DIC,
Heparin
TT or RT Fibrinogen FGA, FGB, FGG Sepsis, Liver disease, DIC
PT and
APTT
FV, FX, Combined
deficiency of FV
&FVIII, MCFD
F5,F10,LMAN1,
MCFD2
Sepsis, Liver disease, DIC
PT,APTT
and TT
Fibrinogen, FII FGA, FGB, FGG,
F2
Sepsis, Liver disease, DIC
4. Fibrinogen estimation
Fibrinogen disorders are be of 3 types:
a) Afibrinogenemia: Grossly decreased fibrinogen levels is seen; with very low and
undetectable plasma fibrinogen. Prolonged PT, APTT and TT along with abnormal
platelet function is seen
b) Hypofibrinogenemia: Mild to moderate decreased fibrinogen levels
c) Dysfibrinogenaemia: Abnormal fibrinogen function.
The ratio of fibrinogen levels measured by clot based and immunological assays is
disproportionate. Some patients have both reduced levels of fibrinogen and abnormal
function: hypodysfibrinogenaemia.
a. Modified Clauss assay
Principle: The clotting time of diluted plasma has a direct bearing on plasma fibrinogen
levels in the presence of excess of thrombin. A graph prepared by plotting a series of
dilutions of a reference plasma sample of known fibrinogen concentrations is used to
compare the test result.
Requirements:
Control and patient citrated plasma
Glyoxaline buffer, Imidazole buffer or Owren’s buffer (pH 7.35)

326
Thrombin reagent (100 NIH Units/mL)
Procedure:
Prepare 1:5, 1:10, 1:20 dilutions of normal plasma and patient’s plasma in Owren’s buffer
Take 50 µL in the cuvette of the coagulometer
Incubate at 37°C for 60 seconds
Add 50 μL of pre-warmed thrombin reagent
Note the clotting time
Plot in a log-log graph paper with clotting time in Y-axis and fibrinogen concentration in Y-
axis.
Normal range: 150- 350 mg/dL.
b. ELISA - Enzyme linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) are also used for measuring Fibrinogen using
human fibrinogen ELISA kit. Fibrinogen concentration and not functional activity are
measured by immunological assays. They hold value in the investigation of congenital
dysfibrinogenemia where there is a discrepancy between antigen level and functional activity.
Diagnosis of afibrinogenemia is given when both fibrinogen antigen and activity level is less
than 50 mg/dL. In case of hypofibrinogenemia, fibrinogen level ranges between 50 to 150
mg/dL. Normal or abnormal screening coagulation tests with normal fibrinogen antigen
levels i.e. 150 to 350 mg/dL is seen in dysfibrinogenemia cases; however the functional
activity is abnormal.
Different ELISA kits with different working principles are available commercially for
estimating fibrinogen antigen.
5. Factor XIII (FXIII) estimation
The screening tests for FXIII i.e. clot solubility assays, FXIII activity tests and quantitative
FXIII antigen tests for total FXIII and for FXIII-A and FXIII-B subunits are the available
methods for FXIII deficiency. The most widely used test in routine hemostasis laboratories
for many years is the clot solubility test which is an old and inexpensive technique. But only
severe FXIII deficiency is detected in this test. For clinical use, the quantitative assays are
important and thus both antigenic and functional assays are performed in most of the
laboratories now.

327
a. FXIII screening techniques
i) Urea clot lysis technique (only severe F XIII deficiency detection)
Principle
Factor XIII (FXIII) is a transglutaminase enzyme serving as the fibrin-stabilizing factor. It
cross links fibrin strands within the clot. During the clotting of blood, FXIII is activated; for
which calcium ions and thrombin are necessary. The process of stabilization of fibrin clot by
activated FXIII is known as transamidation. This stable fibrin clot is insoluble in 5M Urea
solution, which is not the case in the clot formed from FXIII deficient plasma; these clots are
soluble within 1- 24 hours in 5M urea.
Requirements
Control and patient citrated plasma
0.025M CaCl2
5M Urea
Procedure
Add patient plasma and control plasma (200 μL) in 2 separate glass test tube
Add pre-warmed 0.025M CaCl2 solution (200 μL)
Mix & incubate at 37°C (water bath)
for 30 minutes
Add 3 mL of 5M Urea solution
Incubate at room temperature (RT) overnight
Check for clot lysis
Interpretation
Observation Interpretation
Fibrin clot stable FXIII 2-100%
Fibrin clot lysed Severe FXIII deficiency (FXIII<2% )
ii) Acetic acid clot lysis method (mild to moderate FXIII deficiency detection)
Principle

328
In the presence of thrombin, a stable fibrin clot in case of normal plasma is insoluble in 2%
acetic acid solution, whereas clots formed from FXIII deficient plasma (<1-18%) are soluble
within 24 hours
Requirements
Control and patient citrated plasma
Thrombin reagent, bovine thrombin (10 NIH Units/mL)
2% acetic acid solution
5M Urea
Procedure
Tube 1 Tube 2
Patient plasma/Control plasma (200 μL)
Pre-warmed thrombin solution (10 NIH
Units/mL) (200 μL)
Pre-warmed 0.025M CaCl2 solution
(200 μL)
Mix & incubate at 37°C (water bath) for 30 minutes
2% Acetic acid solution (3mL) 5M Urea solution (3mL)
Incubate at RT overnight
Check for clot lysis
Table 7. Two-tube technique interpretation for FXIII estimation
Thrombin+Urea Thrombin+Acetic acid Interpretation
+ + FXIII ≥ 18.8 IU/mL
+ - FXIII > 1 and < 18.8 IU/mL
- - FXIII < 18.8 IU/mL
Key: (-) clot lysed, (+) clot present
Mixing tests: These tests should be carried out with different proportions of patient and
normal plasma mixtures if clot solubility tests are positive to differentiate between a
congenital FXIII deficiency and acquired deficiency (presence of an inhibitor).
b. FXIII antigen estimation by ELISA
There are many ELISA kits with different working principle available in the market. In
routine laboratories generally the sandwich ELISA technique is employed wherein
monoclonal antibodies directed against each of the FXIII subunits i.e. FXIIIA and FXIIIB are

329
used as capturing and detecting antibodies. These tests detect only the FXIII tetramers; the
ones which are not complexed will be undetected. Plasma samples can be preserved in -700C
freezers and processed in batches. These ELISA assays are highly specific and sensitive.
One stage APTT-based factor assays (FVIII, FIX, FXI, and FXII)
For the measurement of factors VIII, IX, XI and XII, one stage APTT-based factor assay is
widely used. Factors X, V and II can also be assayed using this test, although for these
factors, a PT-based assay is more commonly used.
In hemophilia A patients, the severity of the bleeding disorder is depicted by the degree of
factor deficiency. Haemophilia patients are generally classified into three categories by their
factor activity: mild: 6-40% of normal; moderate: 1-5% of normal; and severe: < 1% of
normal.
Principle
The degree of correction obtained when the test plasma is added to specific factor deficient
plasma can determine the percent of factor present in the test plasma. APTT determines this
degree of correction. The results are compared to the degree of correction obtained when
normal plasma is added to same specific factor deficient plasma. Normal plasma is presumed
to give 100% correction.
Requirements
Citrated plasma of the patient
Standard plasma or normal pooled plasma (NPP)
Factor deficient plasma (substrate plasma): This is obtained either commercial or from a
hemophilic donor whose specific factor is <1 % of the standard plasma, absent for
inhibitors and transfusion transmitted diseases like HIV, HBsAg and HCV.
APTT reagent
0.025M CaCl2
Imidazole buffer pH 7.35 or Owren’s buffer/ Owren’s barbiturate saline (OBS), pH 7.35
i.e. barbiturate buffer pH 7.35 with 0.9% normal saline in 1:4 ratio
Procedure
Make serial dilutions (1:5, 1:10 and 1:20) of patient plasma and NPP in OBS or Imidazole
buffer
Diluted plasma (50 μL) + Factor deficient plasma (50 μL) + APTT reagent (50 μL)
Incubate for 3 minutes at 370C
0.025M CaCl2 (50 μL)

330
Normal range: 50- 150 % of NPP
The results are plotted on a semi-log graph paper with the time in seconds on the ordinate and
percent of dilution on the abscissa. By finding the point where the time obtained for the 1:5
dilution of the patient plasma intercepts the normal line, percent factor activity is read from
the abscissa of the graph. 100% factor is representation by the first dilution, i.e. 1:5 dilution
in this case, 1:10 represents 50% and 1:20 represents 25% factor of normal pooled plasma.
This assay is performed for factor VIII, IX, XI and XI assay using the respective deficient
plasma.
7. One stage PT- based factor assay (FVII)
For determining factor VII levels, a method based on PT is used. This same method can also
be employed for determining factor V, X and II levels.
Principle
The degree of correction obtained when the plasma is added to FVII deficient plasma helps
determine the percent of FVII present in test plasma. The PT determines the degree of
correction and this is compared to the degree of correction obtained when normal plasma is
added to the FVII deficient plasma. Normal plasma is presumed to give 100% correction.
Requirements
Citrated plasma of the patient
Standard plasma or NPP
FVII deficient plasma (substrate plasma): This is obtained either commercial or from a
FVII deficient donor whose FVII:C is <1 % of the standard plasma, absent for inhibitors
and transfusion transmitted diseases like HIV, HBsAg and HCV.
PT reagent.
0.025M CaCl2
Imidazole buffer pH 7.35 or OBS, pH 7.35
Procedure
Make serial dilutions (1:5, 1:10 and 1:20) of patient plasma and NPP in OBS or Imidazole
buffer
Diluted plasma (50 μL) + FVII deficient plasma (50 μL)
Incubate for 1 minute at 37°C
PT reagent (100 μL)
Normal range: 50- 150 % of NPP

331
100% factor VII is represented by the 1:5 dilution, 1:10 represents 50% and 1:20 represents
25% factor levels of normal pooled plasma. The graph is plotted in the same way as given
above in one stage APTT-based assay.
Either PT mode or APTT mode may be chosen using specific deficient plasma, for measuring
factors in the common pathway (factor II, V and X).
FVIII/FIX inhibitors
In patients having prolonged bleeding episodes or who do not respond well to the
conventional factor replacement therapy, inhibitors are generally suspected. Also before
surgical procedures, inhibitor screening tests are recommended so that the treatment products
can be kept handy. Patients once diagnosed with inhibitors need to be tested and monitored
periodically to ensure efficacy of treatment. Inhibitor screening assays are also advised when
patients are being treated with new factor products to check the immunogenicity of these
products. Immediate recognition of inhibitors is critical in most cases, since early therapy can
be life-saving.
When APTT is not corrected in mixing studies of a hemophilic patient plasma with NPP, it
indicates the presence of inhibitors. This could be against the coagulation factors or other
non-specific antibodies like LA. Patients could also have a heterogeneous mix of antibodies
which results in an accurate diagnosis.
Factor VIII / IX Inhibitor screening assay using NPP/FVIII concentrate
FVIII inhibitors are generally known to be time and temperature dependent i.e. detected after
incubation at 37°C over 2 hours. However, a heterogeneous mix of antibodies, including
lupus anticoagulants (LA) or even natural antibodies in NPP, could make diagnosis difficult
and give false-positive and non-specific results. Thus, using pure FVIII concentrates as the
‘control’, instead of NPP traditionally used to screen for FVIII inhibitors is more feasible. 1
IU/mL (100% FVIII: C) is prepared by diluting FVIII in Imidazole buffer pH 7.35. Instead of
NPP which is used to screen for FVIII inhibitors over 2 hours at 37°C, this FVIII is used. The
inhibitor screening protocol is as described below. The ‘incubated mix’ and ‘immediate mix’
APTT readings are noted and compared over 2 hours.
Reagents
Plasma-derived monoclonal purified FVIII or NPP
Test plasma
Reagents required for APTT test
Method
332
This screening assay involves incubating FVIII concentrate/ NPP, patient plasma and a 1:1
(or 3:1) NPP: patient plasma mix (incubated mix) in 3 tubes at 37°C for 2 hours. Most
importantly, to account for an increase in all the APTT readings after incubation at 37°C, the
APTT reading of the incubated mix is compared to that of a control immediate mix (1:1 mix
of the normal and patient plasma samples incubated separately). If the APTT reading of the
incubated mix is much higher than the immediate mix (generally >5 secs), the presence of an
inhibitor is indicated. This is subsequently confirmed by Bethesda assay.
Table 8: APTT- based Inhibitor Screen results of a positive sample
APTT 1 hour
APTT (seconds)
2 hour
APTT (seconds)
Incubated Mix 68 76
Separate Mix 58 60
Inhibitor screen test: Positive
Inhibitor Positive: Incubated Mix reading (seconds) > Immediate Mix reading (seconds).
However, this should be confirmed by Bethesda assay.
Inhibitor Negative: Incubated Mix reading (seconds) < Immediate Mix reading (seconds).
For screening for FIX inhibitors, same procedure is followed as for the Factor VIII
inhibitor assay by using NPP.
b. Classical Bethesda assay
This assay involves a two-hour incubation of a mixture of patient plasma with normal
pool plasma (NPP). The residual FVIII is compared with a similarly treated control
mixture containing buffer and NPP. This percent residual activity in the patient mix is
converted to Bethesda units (BU). One BU is defined as the amount of inhibitor which
will neutralize 50% of one unit of FVIII: C in normal plasma after 2 hours of incubation
at 37°C.
c. Nijmegen-modified Bethesda assay
The Nijmegen-modified Bethesda assay involves two modifications from the classical
Bethesda assay: i) To prevent loss of FVIII activity due to variation in pH, NPP is
buffered with 0.1 M imidazole pH 7.4 ii) factor deficient plasma is used in place of

333
imidazole buffer which helps to maintain constant protein concentration, thus
preventing any loss of FVIII activity.
Method
1. Doubling dilutions of patient plasma in FVIII deficient plasma is prepared. Depending
on the past or predicted inhibitor titer, the number of dilutions might vary.
2. Label standard tube and siliconized glass test tubes starting from 1:2,
1:4……….1:128….
3. Add 100 uL buffered NPP to all tubes. Add 100 uL FVIII deficient plasma to the buffer
blank tube and dilutions of patient plasma to the respective tubes.
4. Incubate for 2 hours at 370C
5. Place all tubes on ice at the end of 2 hours and measure residual FVIII activity in the
buffer blank standard and the first dilution. The first dilution residual FVIII assay will
provide a clue as which dilutions should be further analyzed.
6. That dilution of test plasma with residual FVIII closest to 50% of the buffer blank
standard is chosen for inhibitor titer calculation.
7. If the 1st dilution of test plasma and the second dilution of buffer blank standard do not
exactly match, the residual FVIII should be plotted in a log – log graph with FVIII in X-
axis vs inhibitor units in Y-axis. The actual FVIII inhibitor titer is calculated by
multiplying the residual FVIII with dilution factor.
Figure 5. The Bethesda assay graph (Dacie and Lewis 1999)

334
Buffering of NPP: Add solid Imidazole to NPP to make a final concentration of 0.1 M.
Adjust the pH to 7.4 slowly by adding 1N HCl with constant stirring at 40C.
ELISA assay for FVIII antibody detection
Below is the in-house method described by Shetty et al 2003 with minor modifications:
Reagents
Coating buffer consist of 15 mM phosphate, 150 mM NaCl, pH 7.4
Fixing solution consist of 0.05% Glutaraldehyde in coating buffer, immunoglobulin
standard.
Solid surface antigen consist of reconstituted lyophilized full-length recombinant FVIII in
coating buffer used at a concentration of 5U/well i.e. 5U/200µl.
Wash buffer contains phosphate 150 mM, 15 mM NaCl, 0.05% Tween 20, pH 7.4
Sample Dilution Buffer is 1% Gelatin in PBS.
Blocking solution is 3 % Gelatin in wash buffer.
Conjugate is human polyclonal IgG labeled with alkaline phosphatase.
Substrate solution consist of p-nitrophenyl phosphate (PNPP) 2 mg/ml in coating buffer.
Method
Pre- coat 96 wells flat bottomed ELISA plates with 0.05% glutaraldehyde overnight at 4°C
Wash with wash buffer 5 times
Coat the wells with 200 µL antigen i.e. recombinant FVIII
Cover tray, incubate overnight at 4°C
Wash with wash buffer 5 times
Remaining sites are blocked with 200 µL wash buffer with 3% gelatin for 1 hour, 20°C
Wash with wash buffer 3 times
Drain on absorbent paper
Add 200 µL of the test/ standard plasma in appropriate dilutions
Incubate for 2 hours (37°C)
Wash with wash buffer
Add 200 µL conjugated antibody which is diluted 1/10 times
Incubated for 2 hours
Wash with wash buffer
Incubate with 100 µL of substrate (PNPP)
Stop the reaction after 15 minutes using 3N NaOH

335
Take absorbance in an ELISA reader at 405 nm
Platelet aggregation tests
Principle
The adherence of one platelet to another is termed as Platelet aggregation. This phenomenon
can be induced by adding aggregating agents to PRP or whole blood where in the aggregation
depends on the presence of fibrinogen, Ca++, one or more plasmatic factors and an
aggregating agent. Each aggregating agent and their varying concentrations will give
different results in platelet aggregation test.

336
Procedure
Switch on aggregometer and recorder and wait for temperature display to indicate 37°C
Pipette 500 µL of test sample into the cuvette and add the stir bar
Pipette 500 µL of Reference (platelet poor plasma) into a second cuvette
Open the heater block cover and place the Sample into the well labeled PRP. Place the
Reference into well labeled PPP. Close the cover
After incubation for 2-3 minutes, add the agonist
Lower the recorder pens and switch “ON” the chart drive. Set the baselines by using the “Set
Baselines” button. Allow the chart to run for long enough to ensure the 0% (PRP) baseline
has stabilized
Open heater block and add appropriate agonist to the sample
Close heater block cover and allow the optical curve to run for the desired test time interval
At the end of the test time interval, raise the recorder pens and switch “OFF” the chart drive.
Platelet receptor studies
In this test platelets are labelled with fluorescent labeled monoclonal antibodies (Mab)
directed against surface membrane glycoproteins and then analyzed for their binding to
specific receptors by flow cytometry. It is a rapid and sensitive technique for diagnosis of
important platelet receptor defects such as Glanzmanns’s Thrombasthenia (GT) & Bernard
Soulier Syndrome (BSS). Whole blood or PRP can be used for testing. Additional tests for
confirmation may include gene sequencing to identify the causative mutation followed by
family studies.
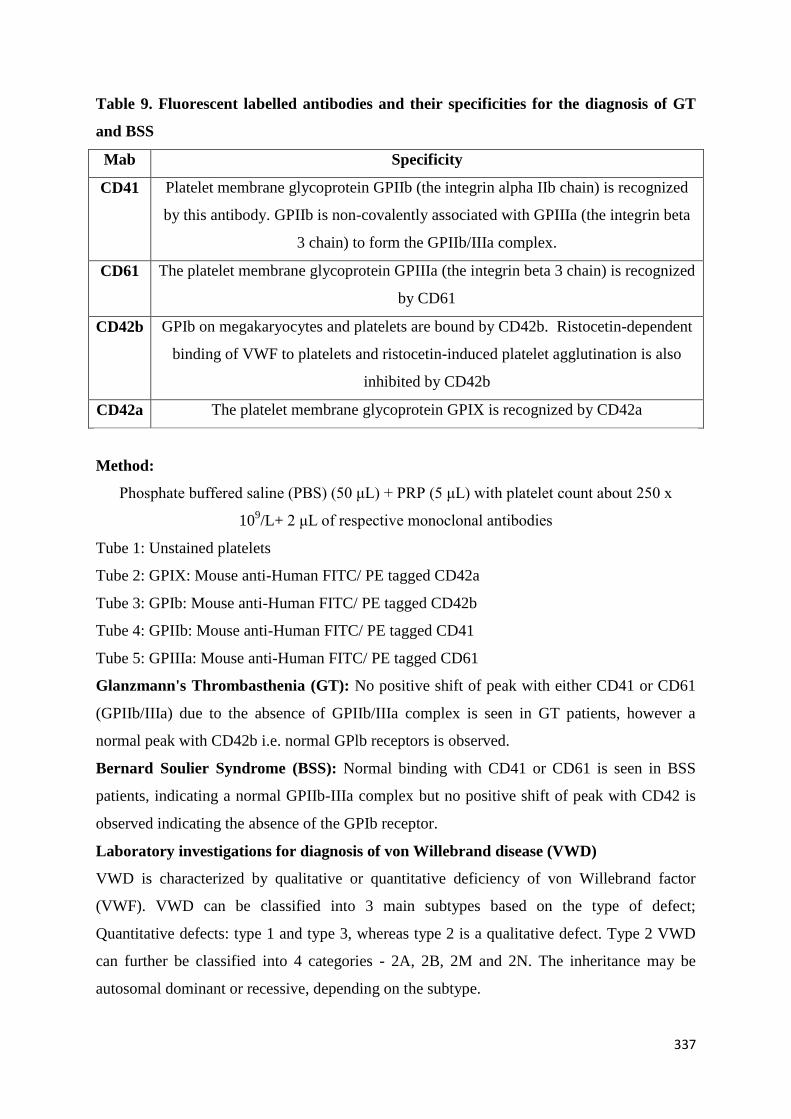
337
Table 9. Fluorescent labelled antibodies and their specificities for the diagnosis of GT
and BSS
Mab Specificity
CD41 Platelet membrane glycoprotein GPIIb (the integrin alpha IIb chain) is recognized
by this antibody. GPIIb is non-covalently associated with GPIIIa (the integrin beta
3 chain) to form the GPIIb/IIIa complex.
CD61 The platelet membrane glycoprotein GPIIIa (the integrin beta 3 chain) is recognized
by CD61
CD42b GPIb on megakaryocytes and platelets are bound by CD42b. Ristocetin-dependent
binding of VWF to platelets and ristocetin-induced platelet agglutination is also
inhibited by CD42b
CD42a The platelet membrane glycoprotein GPIX is recognized by CD42a
Method:
Phosphate buffered saline (PBS) (50 μL) + PRP (5 μL) with platelet count about 250 x
109/L+ 2 μL of respective monoclonal antibodies
Tube 1: Unstained platelets
Tube 2: GPIX: Mouse anti-Human FITC/ PE tagged CD42a
Tube 3: GPIb: Mouse anti-Human FITC/ PE tagged CD42b
Tube 4: GPIIb: Mouse anti-Human FITC/ PE tagged CD41
Tube 5: GPIIIa: Mouse anti-Human FITC/ PE tagged CD61
Glanzmann's Thrombasthenia (GT): No positive shift of peak with either CD41 or CD61
(GPIIb/IIIa) due to the absence of GPIIb/IIIa complex is seen in GT patients, however a
normal peak with CD42b i.e. normal GPlb receptors is observed.
Bernard Soulier Syndrome (BSS): Normal binding with CD41 or CD61 is seen in BSS
patients, indicating a normal GPIIb-IIIa complex but no positive shift of peak with CD42 is
observed indicating the absence of the GPIb receptor.
Laboratory investigations for diagnosis of von Willebrand disease (VWD)
VWD is characterized by qualitative or quantitative deficiency of von Willebrand factor
(VWF). VWD can be classified into 3 main subtypes based on the type of defect;
Quantitative defects: type 1 and type 3, whereas type 2 is a qualitative defect. Type 2 VWD
can further be classified into 4 categories - 2A, 2B, 2M and 2N. The inheritance may be
autosomal dominant or recessive, depending on the subtype.

338
a. RIPA: Ristocetin induced platelet aggregation
The interaction between VWF and platelet glycoprotein Ib (GPIb) is promoted by Ristocetin,
which is an antibiotic. The procedure is explained under Platelet aggregation section.
A concentration of 1.25 mg/mL Ristocetin is used for the test. However if type 2B VWD is
suspected, a low dose ristocetin i.e. 0.5 mg/mL should be used for the test. Presence of
aggregation with low dose ristocetin is suggestive of type 2B VWD.
b. von Willebrand Factor Antigen (VWF:Ag) ELISA
A modified method followed by Dacie and Lewis 1999 is used. A microtitre plate is coated
with a primary antibody to VWF:Ag. A suitable dilution of the test plasma is added to these
wells; VWF:Ag binds to the primary antibody. By washing the plate, excess antigen is
removed; the second antibody conjugated to an enzyme, usually peroxide, is added which
binds to the VWF:Ag already bound to the plate. A color change occurs on addition of a
specific substrate. The reaction is stopped with an acid and the optical density (OD) of the
wells is measured using an ELISA plate reader. The OD is directly proportional to the
amount of VWF:Ag present in the test plasma.
Reagents
Microtitre plates
0.05 M Carbonate buffer: composition: 1.59g Na2CO3, 2.93g NaHCO3, 0.2g NaN3 in 1L
of D/W (pH 9.6)
Phosphate buffered saline: composition: 0.39g NaH2PO4.2H2O, 2.68g Na2HPO4.12H2O,
8.47g NaCl in 1L of D/W (pH 7.2)
0.01M Citrate phosphate buffer: composition: 8.8g citric acid, 24.0 Na2HPO4.12H2O in
1L of D/W
3% gelatin and 1% gelatin solution
Anti VWF:Ag antiserum (Diluted in 0.05M carbonate buffer)
Anti VWF:Ag conjugate with peroxide (Diluted in PBS-Tween-1% gelatin)
100% Calibration plasma
Test and control platelet poor plasma
1,2-o- Phenylenediamine dihydrochloride (OPD, Urea Peroxide)
Tween 20
1M HCL
Microtitre plate reader
339
Procedure
Add diluted anti-human VWF:Ag (100μL) to each well
Incubate for 1 hour in a moist chamber
Discard antibody; wash 3 times with PBS
containing 0.5 mL/L tween for 2 minutes
Invert onto an absorbent paper
Add 3% gelatin (100μL) as blocking agent
Incubate for 1 hour at 37°C
Prepare dilutions of the 100% standard plasma (1:10, 1:20, 1:40, 1:80, 1:160) in PBS with
1mL/L tween and 1% gelatin
Dilute patient and control plasmas (1:10, 1:20 and 1:40) in the same way
Add 100 μL of each dilution in duplicate to the wells
Incubate for 2 hours at 37°C
Repeat the washings
Add diluted anti-human VWF:Ag peroxide conjugate (100μL) to each well
Incubate for 2 hours at 37°C
Repeat washings as before and was once with 0.1M citrate phosphate buffer
Dissolve the OPD tablet in 20 mL D/W (freshly prepared) and add 100 μL to each well
Stop the reaction by addition of 50 μL of 1M HCL when the color has reached intensity
where a mid-yellow ring is clearly visible at the bottom of the wells
Read the OD at 492 nm
Plot a standard curve on log-linear graph paper and obtain the VWF:Ag levels by reading
from the reference curve
Normal range: 50-200 IU/ dL
c. Determination of VWF:Ag by LaurellImmunoelectrophoresis
Principle

340
Into an agarose plate, a precipitating rabbit anti-serum to human VWF: Ag is incorporated;
the wells of which are filled with test/ standard plasma. After applying an electric current;
precipitation peaks are obtained; the height of which is proportional to the concentration of
VWF:Ag in the samples. The test is insensitive for mild to moderate deficiency of VWF.
Requirements:
Agarose: Low electro-endosmosis
Barbitone buffer: Composition: 0.05 mol/L, pH 8.4 (17 g of Barbital-Natrium powder and
3.722 g of EDTA and add HCL to adjust pH 8.4 ; add D/W to make up the volume to 2
L)
NaCl 30g/L
EDTA- dipotassium salt
Polyclonal Rabbit Anti-Human VWF
Hydrochloric acid about 35% pure
Coomassie Blue stain
Coomassie Brilliant Blue (R): Composition: 5gms is added to Methanol 96% (450 mL)
and 100 mL of Acetic Acid glacial (100%). This is incubated overnight at room
temperature and add distilled water (450 mL). This solution is filtered before use.
Destainer: 250 mL of 96% methanol, 100 mL of acetic acid, 450 mL of distilled water
Calibration plasma (VWF - 100%)
Platelet poor plasma (tests and controls)
Apparatus: Glass Plate – 9 x 11cm, Water bath (56•C), Well borer (to give 2mm well size),
Whatmann filter paper no. 1, Wicks, Horizontal electrophoresis tank, Power pack
Method:
Dissolve 200 mg of agarose in 18 mL of 1:1 diluted Barbitone Buffer pH 8.4 by boiling in a
conical flask. Transfer to a 56°C water bath
Clean the glass plates with spirit to remove dust and grease
Add 18 µL of antiserum to the boiled agarose (cooled till 55°C) and mixed gently
Pour the agarose carefully and spread quickly on the plate to form an even film. Allow to
cool and after it is set, keep in a moist chamber at 4°C until ready for use
Cut equidistant wells in the agarose plate

341
Add 5.5 µL of the sample (fresh sample or thawed at 37°C) to each of the wells
Place the plates in a tank containing barbitone buffer. Place the wells on the cathode side so
that the samples migrate towards the anode. The electrophoresis is carried out at 16 mA for
18 hours
Remove the glass plates and immerse in 3% NaCl for 24 hours to elute out any excess
protein. Immerse in 0.9% NaCl and finally in D/W
Cover the plates with filter paper and keep in a hot air oven for drying
Coomassie Blue is used for staining for 5-10 minutes. Remove the excess stain by using
destainer. Allow the plates to dry.
Measure the height in mm from the middle of the well to the tip of the peak and compare
with the peak of the control sample or the calibrator and calculate the VWF values of the test
samples against the calibrator values.
Normal range: 50-150%
d. VWF:RCo: VWF Ristocetin cofactor assay
Principle
Unless normal plasma is added as a source of VWF, washed platelets do not ‘agglutinate’ in
the presence of Ristocetin. Agglutination follows a dose response curve which is dependent
upon the amount of plasma added. In this assay, freshly washed platelets or formaldehyde
fixed platelets can be used. Though it takes longer to prepare fixed platelets, these are not
susceptible to ristocetin aggregation, and they can also be stored for emergency use. It is
quicker to prepare freshly washed platelets and retain a functional platelet membrane, but
they cannot be retained for later use. Commercial lyophilized washed platelet preparations
are also available. Depending on the work load and urgency of the test, the type of platelet
preparation which best suits an individual laboratory can be decided.
Reagents
Sodium citrate solution. 32g/L trisodium citrate (Na3C6H5O7.2H2O)
K2EDTA. 0.134 mol/L
0.2% paraformaldehyde in 9g/L NaCl
0.05% Sodium Azide
Ristocetinsulfate: 1mg/mL reconstituted in distilled water.

342
Normal pooled plasma (NPP)/ standard plasma
Patients platelet poor plasma
Procedure
Obtain citrated blood from a normal subject with a normal platelet count
Centrifuge the blood at 300g for 15 minutes at RT
Separate the PRP and add 9 volume of PRP to 1 volume of EDTA solution, incubate for 1
hour at 37ºC to reverse the effect of ADP released during the preparation
Add an equal volume of 0.2% paraformaldehyde and leave at 4ºC. Decant the supernatant
and recentrifuge at 250g for 20 minutes at 4ºC
Discard the supernatant and resuspend the platelet sediment in chilled saline (9 g/L)
Wash the platelets twice
After the final wash, resuspend the platelets in sodium azide solution. Adjust the platelet
count to 300-500 Χ 109/L. The suspension is stable for 1 month at 4ºC.
Method
Add 495 μL of washed platelets & 1 μL of ristocetin in platelet aggregometer cuvette. Zero
agglutination of the platelets should be seen. The absorbance taken represents zero (%)
agglutination (blank); which should not exceed 5 divisions on the chart paper (5-6%
agglutination). If it is greater, then the platelets should be washed again and the above
procedure repeated. Repeat the reading of this blank after one hour
Add 500 μL of citrate saline into another cuvette and place in the warming block. This
reading represents 100% agglutination
Make serial dilutions from 1:2 to 1:32 of NPP in citrate saline
Similarly test the patient plasma in two dilutions, depending on the expected concentration of
VWF in the plasma. Both dilutions should give agglutination within the range of that of the
standard curve

343
To each cuvette add the following for the assay:
0.4 mL platelet suspension + 0.1mL plasma dilution + 5μL Ristocetin
Reset 100% and zero agglutination for each patient
Plot a graph of agglutination on the linear scale and the concentration of VWF on the log
scale on semi-log paper
Read the patient’s VWF level directly from the standard curve, correct for the dilution factor
and average of the two results from the different dilutions are taken.
Note: Addition of paraformaldehyde should be very slow with constant and gentle mixing of
the solution to avoid platelet clumping.
Normal range: 50-200 U/Dl
e. VWF:CBA- VWF: Collagen binding assay
Principle
The assay involves the direct binding of the plasma VWF to a solid phase antigen i.e.,
collagen, which is subsequently detected using anti-human VWF- HRP conjugate.
Reagents:
Microtitre plates
Phosphate buffered saline: 0.39g NaH2PO4.2H2O, 2.68g NaH2PO4.12H2O, 8.47g NaCl in
1L D/W (pH 7.2)
Collagen; pepsin digested type III collagen from human placenta 100 μg/mL i.e.
10μg/well is added.
Gelatin
Anti-VWF:Ag conjugate with peroxidase
100% calibration plasma
Platelet poor plasma (tests and controls)
1,2-o-Phenylenediamine dihydrochloride (OPD, Urea peroxide)
Tween 20
1M HCL
Microtitre plate reader
Washing Buffer: 0.1% Tween –PBS pH 7.2
Dilution Buffer: 1% Gelatin in PBS pH 7.2

344
Procedure:
Add 100 μL of collagen to microtitre plate
Incubate overnight at 4°C.
Wash the plates five times by washing buffer
Add 100 mL of 3 % gelatin as a blocking agent.
Incubate at 37°C; repeat the washings
Dilute the standard plasma i.e., NPP in 1:5, 1:10, 1:20, 1:40, 1:80. 1:160 and 1:320 and the
patient plasma into 1:5, 1:10, 1:20 and 1:40 dilutions in 1% gelatin
Add 100 μL of diluted plasma to each well
Incubate at 37°C for 2 hours followed by 5
washings with washing buffer
Add 100 μL of anti-human VWF-HRP conjugate
Incubated for 1 hour at 37°C
Add 100 uL of the substrate i.e,. 1,2-o-Phenylenediamine dihydrochloride (OPD) to each well
Incubation for 30 minutes
Add 50 μL of 1N HCL to stop the reaction
Measure the color developed in a microtitre plate reader at 492 nm
Plot the standardized curve on a log-linear graph paper. Three different curves are obtained.
1:5 dilution of the standard plasma is considered as 100% activity and activity is obtained by
reading from the reference curve.
f. VWF multimer analysis
Principle: von Willebrand factor circulates in the plasma as dimmers and multimers. For
subtyping the VWD, determination of these dimers and multimers is essential. The precise
sub-type of VWD is determined by electrophoretic separation and visualization of the
multimeric components of VWF binding with a radio-labeled anti-VWF antibody which is
observed by exposure to X-ray film.
Agarose gel electrophoresis
Reagents: Glycine, TRIZMA Base, Urea, Sodium dodecyl sulphate (electrophoresis grade),
Agarose (Ultra pure DNA grade), Bromophenol Blue (1% Stock Solution), Sodium EDTA.

345
2H2O, Deionized Water, Electrophoresis tank, Cooling System, 60°C water Bath, Glass
Plates, Spacers, Well cutters, Well template, Whatmann 3mm filter paper, Scalpel.
Procedure
Stock solutions:
2M TRIZMA base: 121.1g in 500 mL water. Stable at 4°C for up to 3 months
3M HCl
0.01M sodium EDTA. 2H2O: 1.86 g in 500 mL water. Stable at 4°C for up to 3 months
Running Buffer (x4 Conc.): 37.5mL 2M TRIZMA base + 4.0 mL 3M HCl. Make up to
50 mL with water. Adjust pH to 8.8 with 3M HCl. Store at 4°C for up to 2 weeks
Stacking Buffer (x 4 conc.): 12.5 mL 2M TRIZMA base + 7.0 mL 3M HCl. Make up to
50 mL with water. Adjust pH to 6.8 with 3M HCl. Store at 4°C for up to 2 weeks.
Sample Buffer: 500 mL 2M TRIZMA base + 10 mL 0.01M sodium EDTA. Make up to
100 mL with water. Store at 4°C for up to 2 weeks. [N.B. This is stock solution. Urea and
SDS are added to make the working solution.]
10% SDS: 1g SDS in 10 mL water. Store at 4°C for up to 3 weeks. Warm at 37
°C to
dissolve SDS before use.
Electrophoresis:
On the day of electrophoresis, the following reagents were prepared:
1. Electrophoretic buffer: Glycine: 57.6 g, TRIZMA Base: 12.0 g, SDS: 2.0 g. Dissolve
in 1.5L of water then make up to 2L. Before use, cool the buffer to 4°C.
2. Working sample buffer: Urea: 9.61 g, SDS: 0.40 g. Dissolve in the sample buffer by
warming at 37°C. Make up to 20 mL and allow to cool to room temperature. Adjust the pH
to 8.0 with 1M HCl, gently adding one drop at a time.
3. Mould the gel assembled by mounting a 1mm spacer on a clean glass plate. Mount the
second plate and line them up so that all the edges are aligned. Clamp all together with
bulldog clips, leaving the top open for pouring the agarose.
4. Preparation of gel:
Running gel: 1.6% agarose, 10% SDS
Agarose 0.4 g
Running buffer 6.25 mL
GDW 18.75 mL
Dissolve the agarose by heating for 30-35 seconds at 70% power levels in the microwave
oven. Stir this and reheat for 5-10 seconds to dissolve the entire agarose and add 250 mL of

346
10% SDS. Pour this carefully into the gap between the glass plates, allow to cool at RT for
about 15-20 minutes and then transfer to 4°C.
Stacking gel, 0.8% agarose, 10% SDS
Agarose 0.16 g
Stacking Buffer 5.0 mL
GDW 14.8 mL
Dissolve the agarose in the stacking buffer by heating to 100°C and once dissolved, add 200
μL of 10% SDS. Pour this into the gap between the glass plates above the running gel.
Allow to cool at RT for about 15-20 minutes, before transferring to 4°C. The excess stacking
gel is kept in the 60°C water bath for later use. When the gel has fully set, remove the clamps
and slowly remove the top glass plate by sliding off the gel. Wells are made in the stacking
gel with a well cutter.
Sample Preparation
Dilute the plasma samples as follows:
25 uL sample buffer + 25 uL sample + 7.5 uL 1% bromophenol blue
Incubate these in a 60°C water bath for 30 minutes and keep at 4
°C prior to electrophoresis.
Electrophoresis: Load 20 uL of diluted samples into the wells. Trasnfer the gel into the
electrophoresis tank with the wells on the cathode (negative electrode). Add approximately
600 mL of the electrophoresis buffer into each side of the tank. Place filter paper wicks on
both sides of the gel so that it is in even, parallel contact with the gel surface, but not in
contact with the wells. The electrophoresis is done under refrigeration at a constant current
of 8 mA. After about 1 hour, when the dye has left the wells, re-fill the molten stacking gel
into the wells. The samples are run for approximately 18 hours. When the electrophoresis is
complete, fix the gel and stain using the radio labeled probe.
Iodination of Antibodies: For radiolabelling the antibodies, Chloramine T method of
Greenwood et al 1963, modified by McConhey and Dixon 1966, is used. Take 1 mg protein
in 100 mL 0.1M PBS in a small round bottomed polysterene vial, in which 1mCi (3.7 x
107Bq) Na125I is added. Place a vial in a beaker containing ice to keep the reaction mixture at
low temperature. Prepare 10 mL of 1% Chloramine T in 0.1 M PBS (Azide free); add and
incubate for 3 minutes with constant shaking. Add another 10 mL and stop reaction at a total
of 10 minutes. Prepare 50 mL of 2% Sodium meta bisulfate in PBS and add. Run the
mixture through a Sephadex G-25 column, which is previously saturated with 200 mL of
20% bovine serum albumin (BSA). Elute thirty fractions of 0.5 mL each using PBS. Check

347
each fraction for serological titre and also radioactive counts per minute (cpm) of 10 mL
suspension in a Gamma counter. Two peaks are always obtained, the first peak having high
radioactive counts, contains the radiolabeled antibody and the second peak contains free
iodine absorbed by albumin, present in the column.
g. VWF:FVIII binding ELISA for the diagnosis of type 2 VWD Normandy (Type 2N
VWD)
The characteristic of Type 2 Normandy (2N) which is a qualitative defect of VWF, are low
FVIII:C levels (caused by a reduced affinity of VWF for FVIII), but normal VWD related
investigations. Mutations responsible for type 2N VWD are found in the FVIII binding
domain of VWF. Phenotypically, patients with type 2N VWD resemble patients with mild
hemophilia with reduced FVIII:C, and often have normal VWF:RCo and VWF:Ag levels.
Type 2N VWD has an autosomal recessive pattern of inheritance. The differential diagnosis
between hemophilia A and VWD type 2N has very important implications for both treatment
and genetic diagnosis.
VWF- FVIII binding assay is an ELISA-based method to determine whether VWF binds
normally to FVIII. VWF is captured using a monoclonal antibody; to remove endogenous
FVIII from VWF calcium chloride is used and a known amount of recombinant FVIII is
added to the bound VWF. Chromogenic FVIII assay is used to measure the bound FVIII.
This assay is similar to the one described below in the initial analytical steps. Microtitre wells
coated with rabbit anti-VWF antibody is utilized, which binds VWF/FVIII from diluted
patient plasma. After removal of endogenous (patient) FVIII, recombinant FVIII is added.
This binds to the patient VWF depending on the nature of the patient’s VWF molecule. The
only difference in the method from the one described below is the method of detection of
bound FVIII.
The general procedure of ELISA is as follows:
The VWF present in the test plasma is captured by the rabbit antihuman VWF F(ab’)2 which
is coated on the internal walls of a plastic microplate well. The FVIII in the tested plasma
dissociated during the first step is removed. The recombinant FVIII is then added which
binds to VWF. Next step involved binding of mouse monoclonal anti-human FVIII antibody
coupled with peroxidase to the remaining free antigenic determinants of the bound FVIII. The
bound enzyme peroxidase is revealed by its action on the TMB substrate. After stopping the
reaction with a strong acid, the intensity of the color is directly proportional to the
concentration of VWF bound to FVIII.

348
The ELISA process and the procedure may vary with different commercial sources. For a
good interpretation of VWF:FVIIIB results, a VWF:Ag level greater than or equal to 15% is
necessary. An over-estimation or an under-estimation of VWF:Ag level consequently leads to
an error of VWF:FVIIIB level.
Principle: The assay involves the direct binding of plasma VWF to a solid phase antigen i.e.
recombinant FVIII. This is subsequently detected by using anti-human VWF-HRP conjugate.
Reagents
Microtitre plates
0.05% Glutaraldehyde
Phosphate buffered saline: 0.39g NaH2PO4.2H2O, 2.68g NaH2PO4.12H2O, 8.47g NaCl in
1L D/W (pH 7.2)
Recombinant FVIII
Gelatin
Anti-VWF:Ag conjugate with peroxidase
Test and control platelet poor plasma
1,2-o-Phenylenediamine dihydrochloride (OPD, Urea peroxide)
Tween 20
1M HCL
Microtitre plate reader
Dilution Buffer: 1% Gelatin in PBS, pH 7.2
Washing Buffer: 0.1% PBS-Tween, pH 7.2
Blocking Buffer: 3% Gelatin in PBS, pH 7.2

349
Procedure
Add 100 μL of 0.05% Glutaraldehyde to microtitre plate
Incubate overnight at 4°C
Wash the plates 5 times with washing buffer
Add 100 uL of recombinant FVIII (12.76 U)
Incubate overnight at 4°C
Wash the plates 5 times with washing buffer
Add 100 mL of 3% gelatin as a blocking agent
Incubate at 37°C
Wash the plates 5 times with washing buffer
Dilute the standard plasma i.e., NPP into 1:5, 1:10, 1:20, 1:40, 1:80. 1:160 and 1:320
dilutions and the patient plasma into 1:5, 1:10, 1:20 and 1:40 dilutions in 1% gelatin
Add 100 μL of diluted plasma to each well
Incubate at 37°C for 90 minutes
Wash the plates 5 times with PBS- Tween
Add 100 μL of anti-human VWF-HRP conjugate (1: 2000)
Incubated for 1 hour at 37°C
Add 100 uL of the substrate i.e.OPD to each well
Incubate for 30 minutes
Add 50 μL of 1N HCL to stop the reaction
Measure the color developed in a microtitre plate reader at 492 nm
Plot the standard curve on a log-linear graph paper. Three different curves are obtained. By
reading from the reference curve, activity is obtained. 1:5 dilution of the standard plasma is
considered as 100 % activity.
(Adapted from Practical Immunohaematology edited by Shrimati Shetty; published by Jaypee Brothers)
Suggested Reading
Dacie JV and Lewis SM . Practical Haematology. Elsevier Publications 1999

350
Karpati L, Penke B, Katona E, Balogh I, Vámosi G, Muszbek L.A modified, optimized
kinetic photometric assay for the determination of blood coagulation factor XIII activity
in plasma.Clin Chem 2000;46:1946-55.
Kasper CK, Aledort L, Aronson D, Counts R, Edson JR, van EJ, Fratantoni J, Green D,
Hampton J, Hilgartner M, Levine P, Lazerson J, McMillan C, Penner J, Shapiro S,
Shulman NR. Proceedings: A more uniform measurement of factor VIII inhibitors.
ThrombDiathHaemorrh 1975;34:869–72
Kershaw G. Performance of Activated Partial Thromboplastin Time (APTT):
Determining Reagent Sensitivity to Factor Deficiencies, Heparin, and Lupus
Anticoagulants.Methods Mol Biol 2017;1646:75-83.
Mazurier C, Gaucher C, Jorieux S, Parquet-Gernez A. Mutations in the FVIII gene in
seven families with mild haemophilia A. Br J Haematol1997; 96:426–7.
Sadler JE, Ginsburg D.A database of polymorphisms in the von Willebrand factor gene
and pseudogene. For the Consortium on von Willebrand Factor Mutations and
Polymorphisms and the Subcommittee on von Willebrand Factor of the Scientific and
Standardization Committee of the International Society on Thrombosis and
Haemostasis.ThrombHaemost 1993 ;69:185-91
Shanbhag S, Shetty S, Kulkarni B, Ghosh K. An improved, semi quantitative clot based
assay for factor XIII.Haemophilia2011 ;17:718-20.
Shetty S, Ghosh K, Mohanty D.AnELISAassay for the detection of factor VIII antibodies
- comparison with the conventional Bethesda assay in a large cohort of haemophilia
samples.ActaHaematol 2003;109:18-22.
Shetty S, Ghosh K, Mohanty D.Comparison of four commercially available activated
partial thromboplastin time reagents using a semi-automated coagulometer. Blood
Coagul Fibrinolysis. 2003 ;14:493-7.
Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-
BunschotenE.The Nijmegen modification of the Bethesda assay for factor VIII:C
inhibitors: improved specificity and reliability.ThrombHaemost. 1995 ;73:247-51.
Yatuv R, Dayan I, Baru M. A modified chromogenic assay for the measurement of very
low levels of factor VIII activity (FVIII:C). Haemophilia 2006;12:253-7.

351
Genetic Counselling for hemophilia
Genetic Counselling is an integral part of a Comprehensive Hemophilia Care Center which is
also a major resource for patients and their relatives to discuss about all available options
within the existing resources. The whole process involves explaining the families of the
clinical severity, existing facilities for diagnosis and the various inheritance patterns, taking
maximum caution to adapt to the culture, religion and other circumstantial factor to which the
family is exposed to. Though for each disease , most of the information delivered to the
families are universally applicable across different countries, there are cultural, religious and
often individual barriers affecting the decisions of the families. Genetic counseling has an
important role even in the absence of genetic testing.
Components of genetic counselling process
The genetic counselling process involves the following important components
Developing a rapport with the family: This is the first and the most important step of a
genetic counseling session. Patients or family members express a wide range of feelings
which include fear, anxiety and guilt. It is the responsibility of the Genetic Counselor to
convince and counsel in such a way that they feel free to express their feelings.
Education :Prior to the counseling session, it is important to understand whether the family
has an understanding about the disease, management , course of the disease, existing
treatment products and so on, since this may have an impact the decision of the family.
Patients and families should be provided with complete information which will help them
through the genetic counseling procedure and the decision-making process subsequent to a
new diagnosis of hemophilia or diagnosis of a carrier
Pretesting Education
Identify person to be tested
Do they want to be tested and know results?
What does being a carrier mean ?
How is haemophilia inherited ?
What does the test involve ?
Family planning
Possible options for pregnancies in case of a carrier

352
Figure 6. Inheritance patterns in haemophilia

353
Once the family decides to go through the genetic diagnostic process, the first step is to draw
an extensive family pedigree to know the relationship of the potential carriers in the family
with the index case. To get a clear picture, this should always be drawn in the form of a
pedigree chart rather than in descriptive terms.The obligate carriers will be identified by
drawing the family pedigree itself. All the remaining potential carriers may be assigned
carrier or noncarrier status by subjecting them to genetic diagnosis if the family decides to do
so.
Carriers of hemophilia face quite a few important and tough choices. They can accept the risk
of having a child with haemophilia, or perform prenatal diagnosis with the associated
problem of termination of pregnancy in case the fetus is found to be affected. A few carriers
do not go for prenatal diagnosis because they do not see haemophilia as a life threatening
disorder to justify termination. Religion, culture and the quality of life of haemophilia
patients in the family may be the key decisive factors . A diagnosis of hemophilia or carrier
may certainly confuse the patient or the relatives. At this stage the Counselor should provide
various alternate options available such as preimplantation diagnosis, noninvasive procedures
and the final decision should be taken by the family
Informed consent : Informed consent for genetic testing from the patient should be obtained
before any procedure if the family is interested in going ahead with carrier diagnosis and/or
prenatal diagnosis. It is always better to assign this responsibility to the person who has done
the genetic counseling
Post-procedure counseling
Results
Carrier or not
Children or not
Natural conception or IVF/PGD
When and options to test the pregnancy
Keep pregnancy or terminate
Frequently asked questions
1. Who are called “ Obligate Carriers”?
Ans : The obligate carriers are i. Daughters of hemophilic patients ii. A lady with two
children with hemophilia iii. A lady having onechild with hemophilia and another
patient in the maternal side. Such carriers do not need any genetic investigations and
the family pedigree itself will assign them the carrier status.

354
2. If a lady has an affected brother and two normal sons , is there a chance of her being a
carrier ?
Ans : Yes, Since a carrier has one normal X chromosome and one affected X
chromosome, even if she is a carrier, both her children might have inherited only
normal X chromosomes. However, this does not rule out the possibility of her being a
carrier.
Can there be a child with haemophilia in the family without any family history?
Ans : Yes, around 40% of the families do not have a family history. There are three
possibilities ; either the mutation might have arisen in the germ cells of the maternal
grand father , which is transmitted to the mother and then subsequently to the son or the
family history may still be positive, but due to female preponderance in the family or
due to chance factors , the hemophilic phenotype might not have been shown up in the
family. The last possibility is of a true “ de novo” mutation, where the mother may not
be a carrier and the mutation might have occurred in the affected child only. Under
these circumstances any indirect method of genetic diagnosis becomes highly
inaccurate.
3. During antenatal diagnosis , can the carrier status of the fetus be informed?
Ans : No. The carrier status of the fetus is not detected antenatally in a family with
hemophilia. The laboratory reports indicate the type of mutation in the index case and
just mentions whether the fetus is “ affected” or “ unaffected”. The “affected”fetus
means that the child has hemophilia. The “unaffected fetus” means normal male,
normal female or carrier female.The carrier status in case of a female child will only be
determined after birth.
4. Are we able to provide an accurate diagnosis in all the 100% of the families referred for
genetic diagnosis?
Ans : With direct identification of mutations, the reports are highly accurate; however
there are certain inherent problems in haemophilia genetics like somatic and germ line
mosaicism, wherein one can miss the mutation even by direct DNA sequencing.
However this is rare. In 1-2% of the families, the mutations are not found in the coding
regions, which are the regions generally scanned by the conventional DNA sequencing
techniques. The reasons may be the presence of the mutation in the deep intronic region
. Only the advanced next generation sequencing techniques can detect such variants
which cause the hemophilia phenotype.

355
5. Can the factor VIII or IX levels determine the carrier status of a female ?
Ans : No, there is a wide range of factor levels both for carriers and noncarriers and
there is a substantial overlap in the factor levels between these two groups. So factor
levels cannot be used to assign “ carrier” or “ noncarrier “ status to a lady belonging to
a hemophilia family
6. If the index case is not available, can a carrier diagnosis or prenatal diagnosis be done
in the family?
Yes, With the availability of direct mutation identification techniques, both carrier
diagnosis and prenatal diagnosis can be done even in the absence of affected member.
7. At what age should the carrier testing be done in a female belonging to a hemophilia
family?
Ans : There is no age limit as when a female should be tested. However, the families
are informed about the availability of genetic diagnosis techniques and counseled to get
the carrier status confirmed before getting married or before conception, in case the
family is opting for subsequent antenatal diagnosis .
Suggested Reading
Alabek M, Mohan R and Raia MH. Genetic Counselling for Hemophilia . WFH
special edition
Miller R . Genetic Counselling for Hemophilia . WFH special edition 2002.No 25

356
Genetic Diagnosis of Hemophilia
Hemophilia A: Factor VIII deficiency
Factor VIII gene (F8) mutations are highly heterogeneous and are not restricted to any
specific region. F8 is one of the largest genes in human genome with 26 exons and 25
introns, with the coding region being 9 kb in size. The exons range in size from 69 bp (exon
5) to 3106 bp ( exon 14), whereas the introns vary in size fr
om 200 bp (intron 17) to 32.4 kb ( intron 22).
Figure 7. General strategy for genetic diagnosis in severe hemophilia A
Linkage analysis or restriction fragment length polymorphism (RFLP) analysis can also be
applied for genetic diagnosis. However it is no longer recommended.
Inverse-PCR Technique for Intron 22 Inversion Analysis
The method is described by Rossetti et al 2005 in detail and involves:
Day 1: Stage 1: Digestion of genomic DNA with Bcl1

357
i. 0.5-2 µg of extracted genomic DNA is added to a labeled 1.5 mL microvial tube in a
total volume of 15 µL with sterile D/W.
ii. A Bcl1 restriction digestion master mix is prepared for the required number of
samples according to the calculation for 1X reaction:
iii.
Reagents 1x Volume
(uL)
D/W 5.0
Bcl1 10X buffer 2.5
Bcl1enzyme 2.5
Total 10
iv. 10 µL of the above Bcl1 master mix is added to each sample.
v. Vortex to mix well, spin briefly in a microfuge.
vi. Incubate at 55oC in a water bath for a minimum of 5 hours (maximum incubation
period overnight).
Stage 2: Ligation
i. Digestion reaction from water bath is removed and briefly spun in a microfuge.
ii. T4 DNA ligase master mix is prepared for the required number of samples according
to the calculation for a 1X reaction:
Reagents Volume 1x (uL)
D/W 199
10X T4 DNA ligase buffer 25
T4 DNA ligase(5U /mL) 1
Total 225
iii. Vortex to mix well, spin briefly in a microfuge.
iv. Incubate at 15oC in a cool block (thermal cycler) overnight.
Stage 3: Concentration or precipitation of the B-rings:
Following the ligation reaction the products have to be concentrated:
i. The reaction tubes are briefly spun in a microfuge.
ii. The entire volume is transferred to a labelled Ultracel YM-100 spin column sat in a
1.5mL tube (as supplied with the columns).
iii. Spin at 8,000rpm for 8 minutes.
iv. The spin column are inverted into a new, labelled tube (as supplied with the columns).

358
v. Spin at 12,000 rpm for 4 minutes to recover the product from the column filter.
or
i. 30 µL of 3M sodium acetate and 800 µL of chilled ethanol is added to the entire volume
of each ligated sample in a 1.5 mL microvial tube and gently mix by inverting the tubes.
ii. Incubate the tubes at -20°C for 1 hour.
iii. Centrifuge the tubes at 14000 rpm for 20 mins to recover the DNA rings.
iv. The pellet obtained was air dried and 15 µL of D/W was added and allowed to dissolve at
room temperature overnight.
Stage 4: PCR amplification:
This stage must be performed in the PCR laboratory. The complementary PCR is an optional
test, which is not required as a first line test for the investigation of males with severe
haemophilia A.
i. In a 1.5 mL tube a PCR master mix is prepared containing all components of the PCR
reaction except the ligated DNA. Volumes stated below are for 1 reaction. Sufficient
should be made for all reactions + 1 blank (negative control) +1 to allow for pipetting
variance.
ii. The PCR amplifications are performed in a reaction volume of 25 µL with 11 µL sterile
D/W, 2.5 µL of 10x complete buffer, 0.3 µL of 25mM dNTPs, 1.25 µL of each Primer
(10pM) and 0.2 µL of DNA Taq polymerase (5U/µL).
iii. For the Diagnostic PCR, Primers 1U, 2U, 3U and ID are used and for the Complementary
PCR, 1U, 2U, 3U and ED primers are used in the concentrations mentioned above.
iv. Vortex to mix and transfer 19 µL to each labeled 0.2 µL PCR tube
v. Add 6 µL of diluted ligation product, or 6 µL deionised water to the negative control
tube.
vi. Transfer to a PCR machine in the main laboratory and run the following conditions:
94◦C- 12 minutes X 1 cycle
94◦C- 30 seconds, 57◦C- 1 minute, 72◦C- 1 minute X 30 cycles
72◦C- 5 minutes X 1 cycle
4◦C- ∞
Approx. time taken for amplification: ~2.5 hours
Stage 5: Post PCR:
i. Post-PCR samples can be stored at 4oC prior to analysis if required. Samples are analyzed
by transferring 15 µL of PCR product into a clean 0.5 mL tube. 3 µL of loading buffer is
then added.
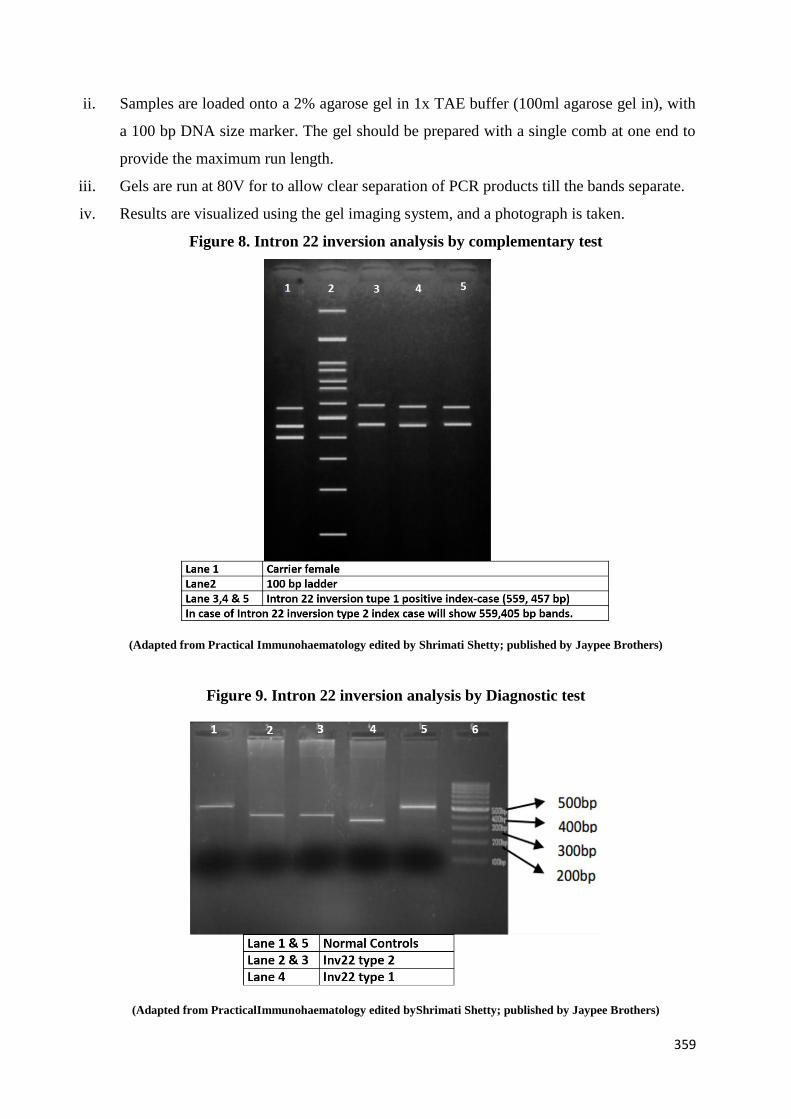
359
ii. Samples are loaded onto a 2% agarose gel in 1x TAE buffer (100ml agarose gel in), with
a 100 bp DNA size marker. The gel should be prepared with a single comb at one end to
provide the maximum run length.
iii. Gels are run at 80V for to allow clear separation of PCR products till the bands separate.
iv. Results are visualized using the gel imaging system, and a photograph is taken.
Figure 8. Intron 22 inversion analysis by complementary test
(Adapted from Practical Immunohaematology edited by Shrimati Shetty; published by Jaypee Brothers)
Figure 9. Intron 22 inversion analysis by Diagnostic test
(Adapted from PracticalImmunohaematology edited byShrimati Shetty; published by Jaypee Brothers)
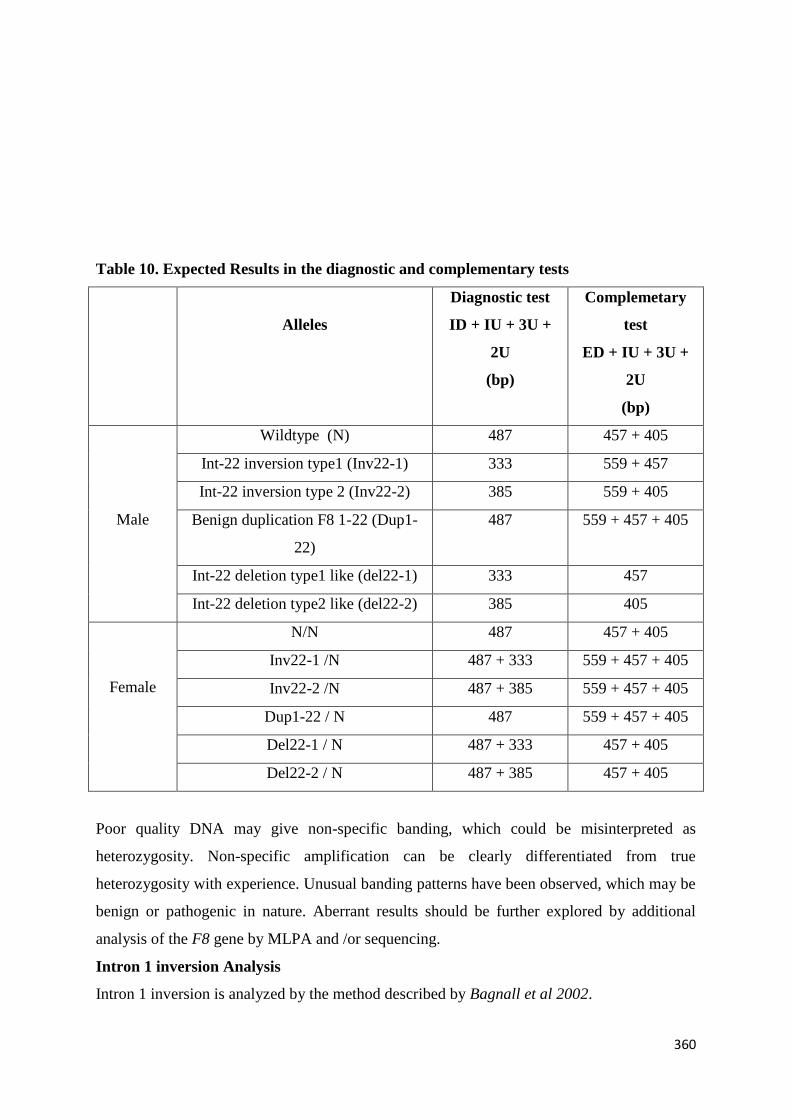
360
Table 10. Expected Results in the diagnostic and complementary tests
Alleles
Diagnostic test
ID + IU + 3U +
2U
(bp)
Complemetary
test
ED + IU + 3U +
2U
(bp)
Male
Wildtype (N) 487 457 + 405
Int-22 inversion type1 (Inv22-1) 333 559 + 457
Int-22 inversion type 2 (Inv22-2) 385 559 + 405
Benign duplication F8 1-22 (Dup1-
22)
487 559 + 457 + 405
Int-22 deletion type1 like (del22-1) 333 457
Int-22 deletion type2 like (del22-2) 385 405
Female
N/N 487 457 + 405
Inv22-1 /N 487 + 333 559 + 457 + 405
Inv22-2 /N 487 + 385 559 + 457 + 405
Dup1-22 / N 487 559 + 457 + 405
Del22-1 / N 487 + 333 457 + 405
Del22-2 / N 487 + 385 457 + 405
Poor quality DNA may give non-specific banding, which could be misinterpreted as
heterozygosity. Non-specific amplification can be clearly differentiated from true
heterozygosity with experience. Unusual banding patterns have been observed, which may be
benign or pathogenic in nature. Aberrant results should be further explored by additional
analysis of the F8 gene by MLPA and /or sequencing.
Intron 1 inversion Analysis
Intron 1 inversion is analyzed by the method described by Bagnall et al 2002.

361
PCR amplification of F8int1 regions and int1h repeats: 100 ng genomic DNA in a reaction
volume of 25µL with 2.5 μL 10 X buffer with MgCl2 (complete buffer), 1.5 mM MgCl2, 2
µM of each oligonucleotide primer, 25 mM of each dNTP, and 0.7 U DNA Taq polymerase.
Six primers are used in two sets for the multiplex PCR; group A: Primers 9F, 9cr and Int1h-
2F and group B: Primers Int1h-2F, Int1h-2R and 9F
Thermal cycling conditions
94°C- 5 minutes X 1
94°C- 30 seconds, 65°C- 40 seconds, 72°C- 2 minutes X 30 cycles
72°C- 5 minutes X 1
4°C- ∞
The PCR amplicons are analyzed on a 2% agarose gel.
Interpretation: The sample is normal in case, a 1.5 kb band is obtained in int1h-1 and a 1kb
band is obtained in int1h-2. The sample has intron 1 inversion mutation in case a 1 kb band is
obtained in int1h-1 and a 1.5 kb band is obtained in int1h-2. In a carrier both these bands are
observed in both the PCR tubes.
Figure 10.Intron 1 Inversion Analysis
(Adapted from Practical Immunohaematology edited by Shrimati Shetty; published by Jaypee Brothers)
Analysis of F8 mutations
The 26 exons of F8 may be scanned by 37 PCR reactions as given below:

362
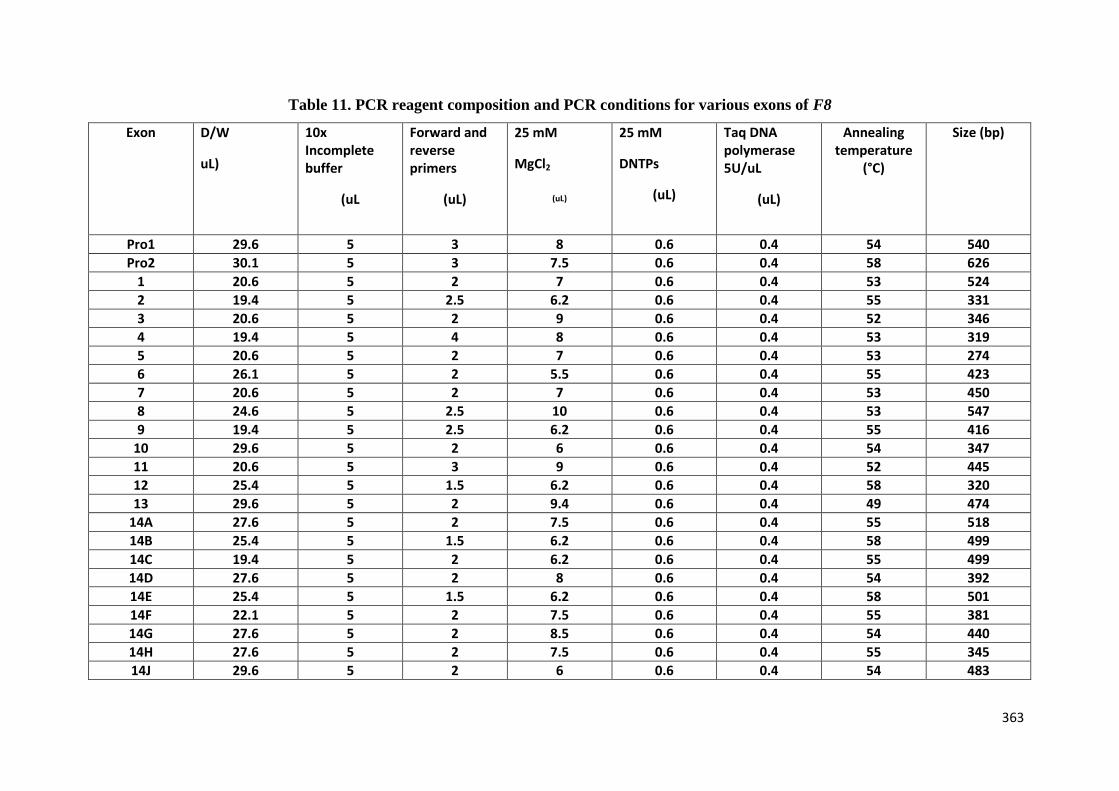
363
Table 11. PCR reagent composition and PCR conditions for various exons of F8
Exon D/W
uL)
10x Incomplete buffer
(uL
Forward and reverse primers
(uL)
25 mM
MgCl2
(uL)
25 mM
DNTPs
(uL)
Taq DNA polymerase 5U/uL
(uL)
Annealing temperature
(°C)
Size (bp)
Pro1 29.6 5 3 8 0.6 0.4 54 540
Pro2 30.1 5 3 7.5 0.6 0.4 58 626
1 20.6 5 2 7 0.6 0.4 53 524
2 19.4 5 2.5 6.2 0.6 0.4 55 331
3 20.6 5 2 9 0.6 0.4 52 346
4 19.4 5 4 8 0.6 0.4 53 319
5 20.6 5 2 7 0.6 0.4 53 274
6 26.1 5 2 5.5 0.6 0.4 55 423
7 20.6 5 2 7 0.6 0.4 53 450
8 24.6 5 2.5 10 0.6 0.4 53 547
9 19.4 5 2.5 6.2 0.6 0.4 55 416
10 29.6 5 2 6 0.6 0.4 54 347
11 20.6 5 3 9 0.6 0.4 52 445
12 25.4 5 1.5 6.2 0.6 0.4 58 320
13 29.6 5 2 9.4 0.6 0.4 49 474
14A 27.6 5 2 7.5 0.6 0.4 55 518
14B 25.4 5 1.5 6.2 0.6 0.4 58 499
14C 19.4 5 2 6.2 0.6 0.4 55 499
14D 27.6 5 2 8 0.6 0.4 54 392
14E 25.4 5 1.5 6.2 0.6 0.4 58 501
14F 22.1 5 2 7.5 0.6 0.4 55 381
14G 27.6 5 2 8.5 0.6 0.4 54 440
14H 27.6 5 2 7.5 0.6 0.4 55 345
14J 29.6 5 2 6 0.6 0.4 54 483
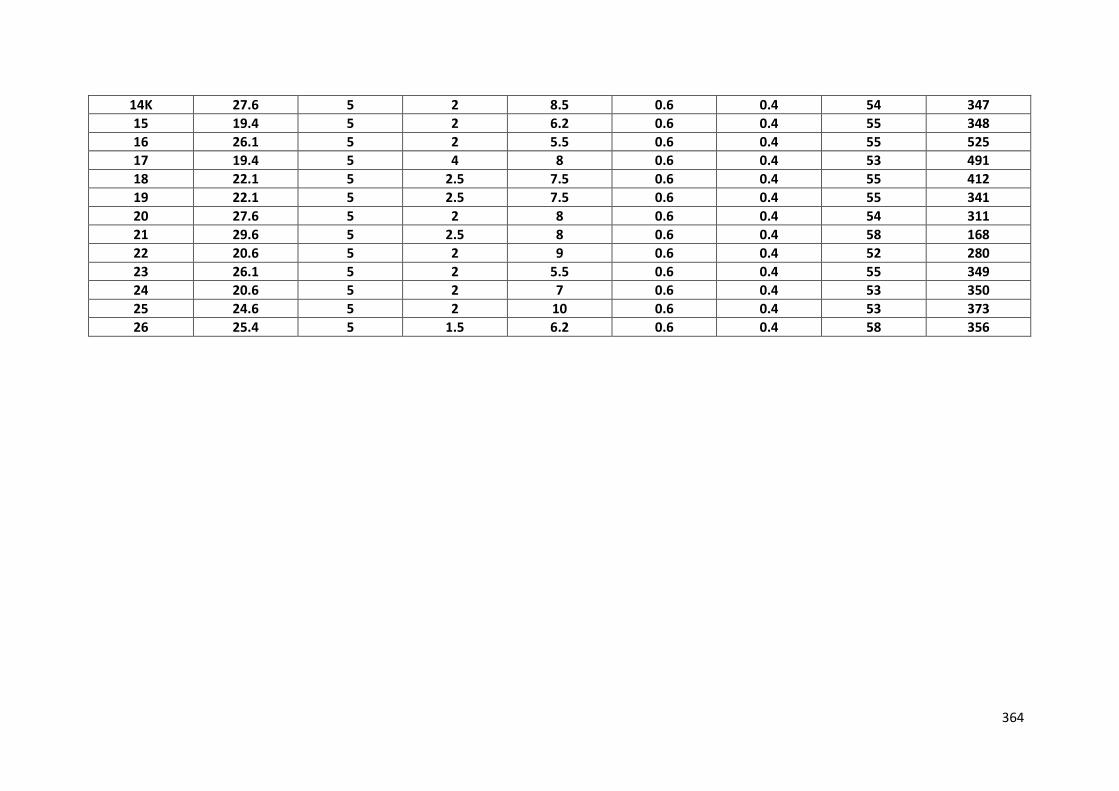
364
14K 27.6 5 2 8.5 0.6 0.4 54 347
15 19.4 5 2 6.2 0.6 0.4 55 348
16 26.1 5 2 5.5 0.6 0.4 55 525
17 19.4 5 4 8 0.6 0.4 53 491
18 22.1 5 2.5 7.5 0.6 0.4 55 412
19 22.1 5 2.5 7.5 0.6 0.4 55 341
20 27.6 5 2 8 0.6 0.4 54 311
21 29.6 5 2.5 8 0.6 0.4 58 168
22 20.6 5 2 9 0.6 0.4 52 280
23 26.1 5 2 5.5 0.6 0.4 55 349
24 20.6 5 2 7 0.6 0.4 53 350
25 24.6 5 2 10 0.6 0.4 53 373
26 25.4 5 1.5 6.2 0.6 0.4 58 356
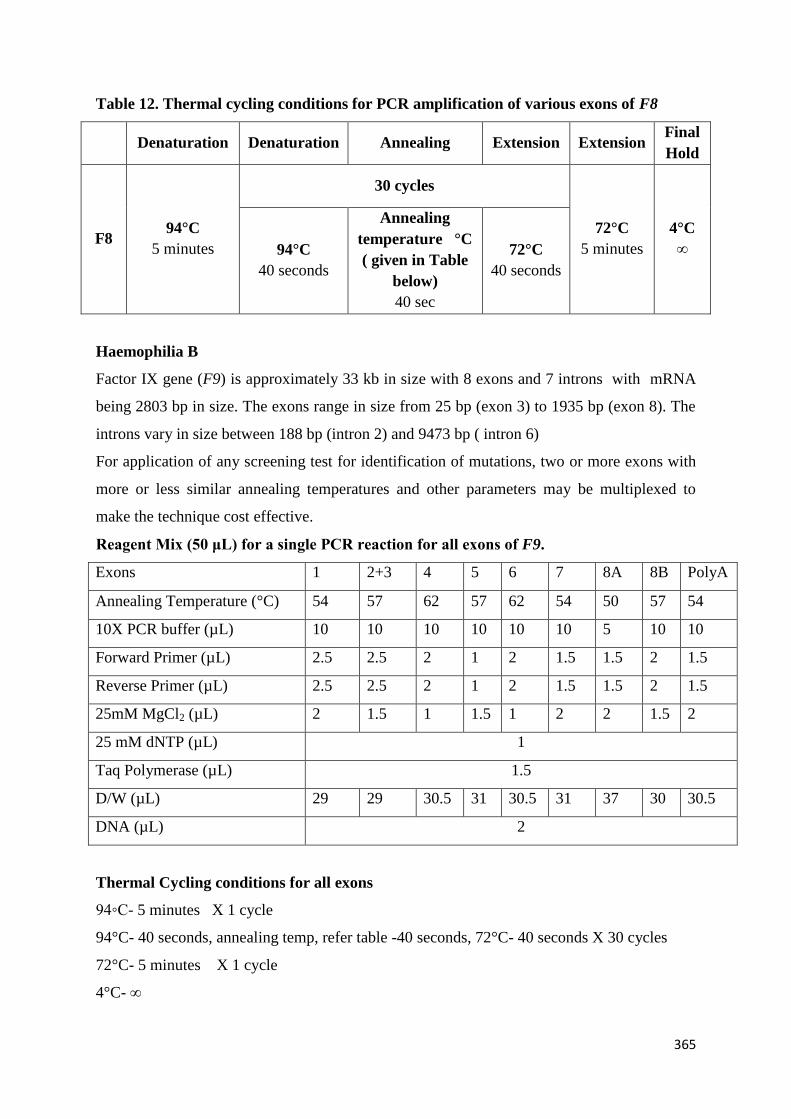
365
Table 12. Thermal cycling conditions for PCR amplification of various exons of F8
Denaturation Denaturation Annealing Extension Extension Final
Hold
F8 94°C
5 minutes
30 cycles
72°C
5 minutes
4°C
∞ 94°C
40 seconds
Annealing
temperature °C
( given in Table
below)
40 sec
72°C
40 seconds
Haemophilia B
Factor IX gene (F9) is approximately 33 kb in size with 8 exons and 7 introns with mRNA
being 2803 bp in size. The exons range in size from 25 bp (exon 3) to 1935 bp (exon 8). The
introns vary in size between 188 bp (intron 2) and 9473 bp ( intron 6)
For application of any screening test for identification of mutations, two or more exons with
more or less similar annealing temperatures and other parameters may be multiplexed to
make the technique cost effective.
Reagent Mix (50 μL) for a single PCR reaction for all exons of F9.
Exons 1 2+3 4 5 6 7 8A 8B PolyA
Annealing Temperature (°C) 54 57 62 57 62 54 50 57 54
10X PCR buffer (µL) 10 10 10 10 10 10 5 10 10
Forward Primer (µL) 2.5 2.5 2 1 2 1.5 1.5 2 1.5
Reverse Primer (µL) 2.5 2.5 2 1 2 1.5 1.5 2 1.5
25mM MgCl2 (µL) 2 1.5 1 1.5 1 2 2 1.5 2
25 mM dNTP (µL) 1
Taq Polymerase (µL) 1.5
D/W (µL) 29 29 30.5 31 30.5 31 37 30 30.5
DNA (µL) 2
Thermal Cycling conditions for all exons
94◦C- 5 minutes X 1 cycle
94°C- 40 seconds, annealing temp, refer table -40 seconds, 72°C- 40 seconds X 30 cycles
72°C- 5 minutes X 1 cycle
4°C- ∞

366
Women with bleeding disorders: Approach and laboratory diagnosis
It is important to recognise and diagnose bleeding disorders in women. The management of
bleeding would be most accurate when the etiology of the bleed is identified so that: Factor
replacement in factor deficiencies, platelet infusions for platelet factor defects and
antifibrinolytics in certain situations can be prescribed.
Important issues related to bleeding disorders in women-
a) Management of heavy menstrual blood flow especially in our country where adequate
provision of sanitary napkins is not a norm, leading to increased school dropout rates.
b) Worsening of already existing Iron deficiency anemia in women of poor socioeconomic
status and its attendant effects, which forms a bulk of the problem.
c) Life threatening bleeds in pregnancy and delivery with poor outcomes of unsupervised
pregnancies.
d) Increased load on a “stretched out” blood bank services.
e) Coexistent comorbidities may mask the bleeding to be a sign of a bleeding disorder and
delay its diagnosis.
Bleeding disorders that affect women
1) Von Willebrand Disease – Commonest coagulation disorder with varied phenotype –
presenting as mucosal wet/ dry bleeds
2) Haemophilia A and B (carriers generally do not bleed; however , few carriers have low
factor levels and they need to be investigated).
3) Platelet function disorders
4) Rare factor deficiency– Factor X, II, V, VII, X, XI, XIII
5) Collagen vascular diseases
Approach to a female with bleeding
A) Detailed history of bleeding
B) Family history
C) Laboratory tests
History of bleeding
Joint bleeds are infrequent and seen sometimes in severe VWD.
Bleeding in early life may indicate an inherited bleeding defect.
In infants – Intracranial bleeds, umbilical stump bleeding
In girls - epistaxis, easy bruising and dental bleeds

367
In adolescents and women – Menorrhagia, pregnancy related bleeds besides the other
bleeding complications
At any age- Post traumatic and post-surgical bleeds
Severity assessment can be made by using Bleeding Assessment Tools (BAT) or
questionnaires. Unlike the haemophilias in males, severe deficiency of other coagulation
factors (Factor II, V, VII, X XIII and fibrinogen) may not result in spontaneous bleeds,
thereby further delaying their diagnosis.
Comorbidities like liver disease, DIC, Vitamin K deficiency may confound the clinical
picture and results of tests, hence screening for these in the appropriate clinical setting is
advisable prior to undertaking extensive testing.
History of recent blood product therapy should be elicited since they can confound
laboratory results and mask an underlying severe defect.
Family history of bleeding
Most of the bleeding disorders in females are of autosomal recessive inheritance, wherein the
parents may be asymptomatic. Type 2 B vWD is inherited in an autosomal dominant fashion.
Carriers of X linked disorders such as Hemophilia A, may at times become symptomatic and
experience heavy bleeding and have low Factor VIII levels. This is the result of extreme
lyonisation of the normal X chromosome.
Family history may not be informative since current norms of bearing few children, results in
affected carriers not having any affected offspring by a turn of chance.
Laboratory Tests
Since most laboratory test for bleeding disorders are expensive a step wise screening and
confirmation is advisable.
1) Screening tests
2) Confirmatory tests
3) Specialised tests
4) Genetic tests

368
Screening tests
These tests will indicate if there is a coagulation or a platelet defect. These tests are sensitive
but not specific. The usual panel comprises
i) Prothrombin time (PT) – prolonged in Factor II, V, VII, X and fibrinogen deficiency
(in isolation, combination or as a part of liver disease, Vitamin K deficiency)
ii) Activated Partial Thromboplastin time (APTT)- prolonged in Factor II, V, X, VIII,
IX, XI, VWD deficiency
iii) Thrombin Time (TT) - Prolonged in fibrinogen deficiency or abnormality
iv) Fibrinogen assay- decreased in afibrinogenemia and hypofibrinogenemia
v) Platelet Count- decreased in Immune thrombocytopenias, DIC, Type 2BVWD,
Platelet Type vWD and some inherited macrothrombocytopenic syndromes
vi) Bleeding time – Prolonged in quantitative and qualitative platelet defects, VWD
vii) Urea clot solubility assay –prolonged in Factor XIII deficiency
viii) Platelet aggregation studies by light transmission aggregometry (LTA) – for
qualitative platelet defects – Glanzmann’s thrombasthenia , Bernard SoulierSyndome
viii) Screening for VWD- VWF antigen assay, VWF GPIbR assay, VWF Ristocetin
Cofactor assay, VWF Collagen Binding assay
Prolongation of the PT and APTT are followed by mixing studies with equal volumes of
normal plasma to establish a factor deficiency and exclude inhibitors.
The mixing tests can be modified to establishing the deficient factor by demonstrating failure
of normalisation of the clotting time on addition of individual deficient plasmas
Confirmatory tests
These tests are complex and expensive and are performed at tertiary centres with trained
manpower.
1) Once the deficient factor is identified then the degree of deficiency can be quantitated by a
specific factor assay e.g.- One stage clot-based Factor V assay for Factor V deficiency
2) Factor XIII assay by ammonia release methods
Specialised tests
These tests need expensive equipment and trained operators.
1) Platelet flowcytometry for CD41, CD6 and CD 42b, CD36 for confirming Glanzmann’s
thrombasthenia, Bernard Soulier syndrome, Collagen receptor defect
2) Electron microscopy – For confirming a decrease in the dense or alpha granule content in
storage pool disorders of platelets

369
3) vWF multimer analysis for confirming the subcategory of VWD
4) vWF / Factor VIII binding assay- For vWD Type 2N
5) Ristocetin induced platelet aggregation ( RIPA ) – For vWD Type 2B and Platelet Type 2B
6) VWF propeptide assay- for excluding acquired vWD in older females with bleeding
symptoms
Molecular tests
In women with bleeding disorders, molecular tests may be required to
1) Confirm the defect in cases where the phenotype of two disorders may be indistinguishable
on the currently available tests. The management of the two may be significantly different
e.g.- Type 2B VWD and Platelet Type VWD
2) For genetic counselling discuss the inheritance and likelihood of disease in offspring and
other related family members
3) For carrier screening and antenatal diagnosis in disorders associated with significant
bleeding
4) To understand the genetic basis of phenotypic variation in patients with similar diseases
Sequencing of the affected gene may be required for establishing the diagnosis. DNA
analysis of the family members is required to confirm the pattern of inheritance and provide
genetic counselling.
The acquired causes of bleeding especially liver disease, renal disease, DIC, hormonal
imbalance are far more common than any of the inherited bleeding disorders. While
investigating bleeding in women other than the tests specific for bleeding disorders it is
important to also correlate with results for the following tests to exclude the presence of a
comorbidity which may aggravate the bleeding.
1) Complete Blood count- For haematological causes for bleeding- aplastic anemia,
leukemia, DIC and for evidence of Iron deficiency
2) Examination of the peripheral blood film – Schistocytes in DIC, blasts in leukemias, giant
platelets in Bernard Soulier syndrome, washed out platelets in Grey Platelet Syndrome,
Nuclear inclusions in May Hegglin anomaly
3) Liver function tests
4) Renal Function test
5) Hormonal assays - Thyroid profile
6) Imaging studies- To exclude anatomical lesions e.g.- fibroids, polyps for menstrual
bleeding.
7) Tests for autoimmune disorders – Ds DNA

370
Hematochezia, hemoptysis and hematemesis are usually never associated with inherited
bleeding defects. Testing during pregnancy and puerperium may yield fallacious results since
levels of Factor VIII and VWF are raised in this time. Repeat testing is mandatory to avoid
misdiagnosis.Levels of Factor VII, V, IX, X are low in early infancy and may achieve
normal values only after 6 months of age. Hence the results of the girl child investigated in
infancy for these disorders must be compared with age related norms and reconfirmed again
after 6 months.Some polymorphisms in the VWF gene are associated with defective
Ristocetin binding but no bleeding symptoms. Some women may have low vWF antigen
levels with no bleeding.Hence the presence of an abnormal test should not be enough to label
a woman with a bleeding disorder in the absence of history of bleeding.
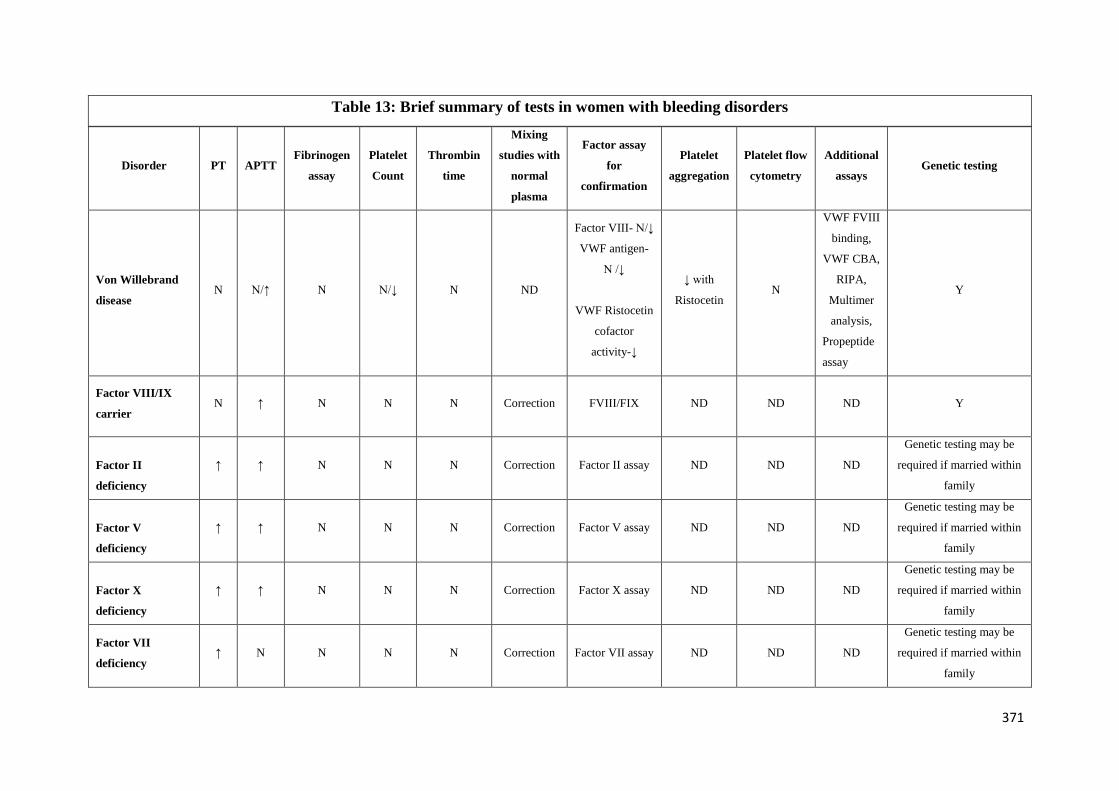
371
Table 13: Brief summary of tests in women with bleeding disorders
Disorder PT APTT Fibrinogen
assay
Platelet
Count
Thrombin
time
Mixing
studies with
normal
plasma
Factor assay
for
confirmation
Platelet
aggregation
Platelet flow
cytometry
Additional
assays Genetic testing
Von Willebrand
disease N N/↑ N N/↓ N ND
Factor VIII- N/↓
VWF antigen-
N /↓
VWF Ristocetin
cofactor
activity-↓
↓ with
Ristocetin N
VWF FVIII
binding,
VWF CBA,
RIPA,
Multimer
analysis,
Propeptide
assay
Y
Factor VIII/IX
carrier N ↑ N N N Correction FVIII/FIX ND ND ND Y
Factor II
deficiency
↑ ↑ N N N Correction Factor II assay ND ND ND
Genetic testing may be
required if married within
family
Factor V
deficiency
↑ ↑ N N N Correction Factor V assay ND ND ND
Genetic testing may be
required if married within
family
Factor X
deficiency
↑ ↑ N N N Correction Factor X assay ND ND ND
Genetic testing may be
required if married within
family
Factor VII
deficiency ↑ N N N N Correction Factor VII assay ND ND ND
Genetic testing may be
required if married within
family
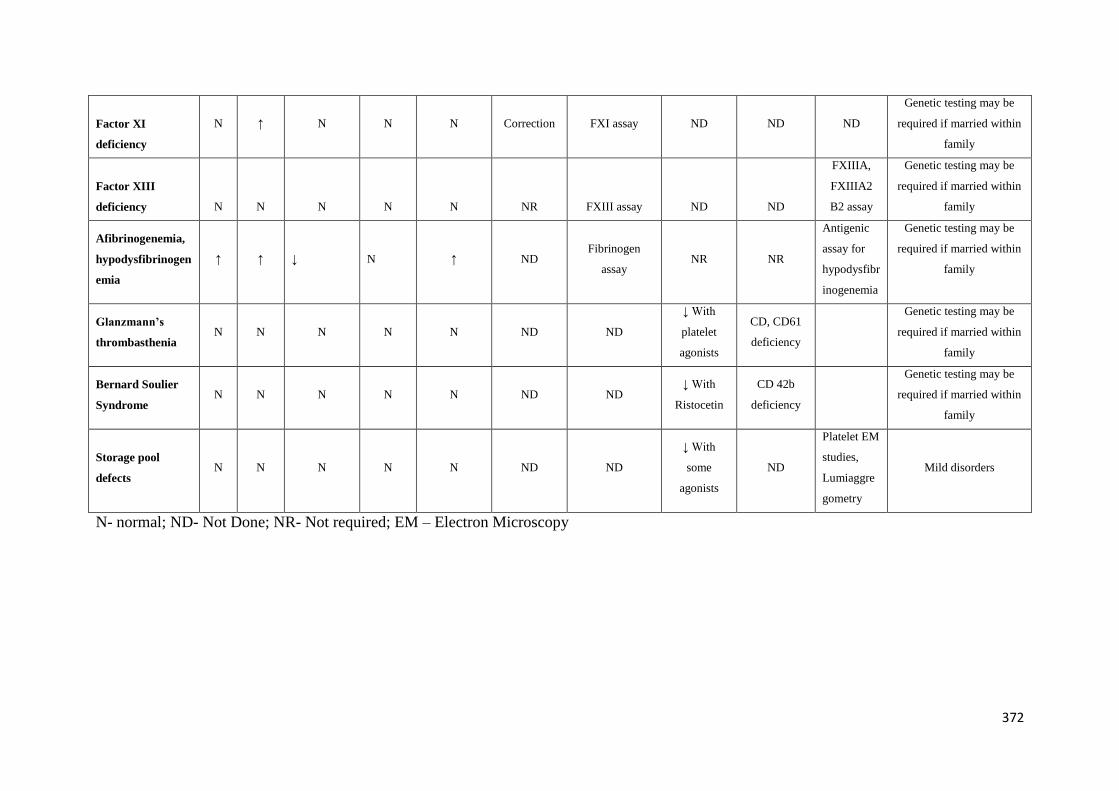
372
Factor XI
deficiency
N ↑ N N N Correction FXI assay ND ND ND
Genetic testing may be
required if married within
family
Factor XIII
deficiency N N N N N NR FXIII assay ND ND
FXIIIA,
FXIIIA2
B2 assay
Genetic testing may be
required if married within
family
Afibrinogenemia,
hypodysfibrinogen
emia
↑ ↑ ↓ N ↑ ND Fibrinogen
assay NR NR
Antigenic
assay for
hypodysfibr
inogenemia
Genetic testing may be
required if married within
family
Glanzmann’s
thrombasthenia N N N N N ND ND
↓ With
platelet
agonists
CD, CD61
deficiency
Genetic testing may be
required if married within
family
Bernard Soulier
Syndrome N N N N N ND ND
↓ With
Ristocetin
CD 42b
deficiency
Genetic testing may be
required if married within
family
Storage pool
defects N N N N N ND ND
↓ With
some
agonists
ND
Platelet EM
studies,
Lumiaggre
gometry
Mild disorders
N- normal; ND- Not Done; NR- Not required; EM – Electron Microscopy

373
Management of Hemophilia and
other Bleeding Disorders

374
Clinical manifestations and management of hemophilia
Hemophilia is a bleeding disorder caused by a deficiency of coagulation factor VIII or IX.
Deficiency of factor VIII is known as hemophilia A and factor FIX is known as in hemophilia B.
The deficiency is due to mutations of the respective clotting factor genes resulting in factor
deficiency. Hemophilia A is more common accounts for 80 – 85 % of total hemophilia
population and the approximate incidence of HA is 1 in 10,000 births and HB is one in 25000 to
30000 births.
Hemophilia mainly affects males, women are carriers. In one third of patients there will not be
any family history of hemophilia and it is due spontaneous mutation of F8 and F9 genes.
Affected males will pass on the hemophilia gene to their daughters. Most carriers are
asymptomatic. Carriers with clotting factor levels of 40–60% of normal may sometimes have an
increased bleeding tendency. Female carriers can have moderate or severe deficiency of factors
due to extreme lyonization Female haemophilia can be due Turners syndrome or they may be
offsprings of female carriers and haemophilic fathers. Hemophilia is classified as mild,moderate
and severe depending on the factor level.
Table 14. Classification of Hemophilia based on severity and its relationship to bleeding
manifestations
Severity Factor level Clinical manifestations and
bleeding episodes
Severe < 1 IU/dL (< 0.01 IU/mL) or <
1% of Normal
Spontaneous bleeding into
joints or muscles,
predominantly in the
absence of identifiable
hemostatic challenge
Moderate 1–5 IU/dL (0.01 –0.05
IU/mL) or 1–5% of normal
Occasional spontaneous
bleeding and prolonged
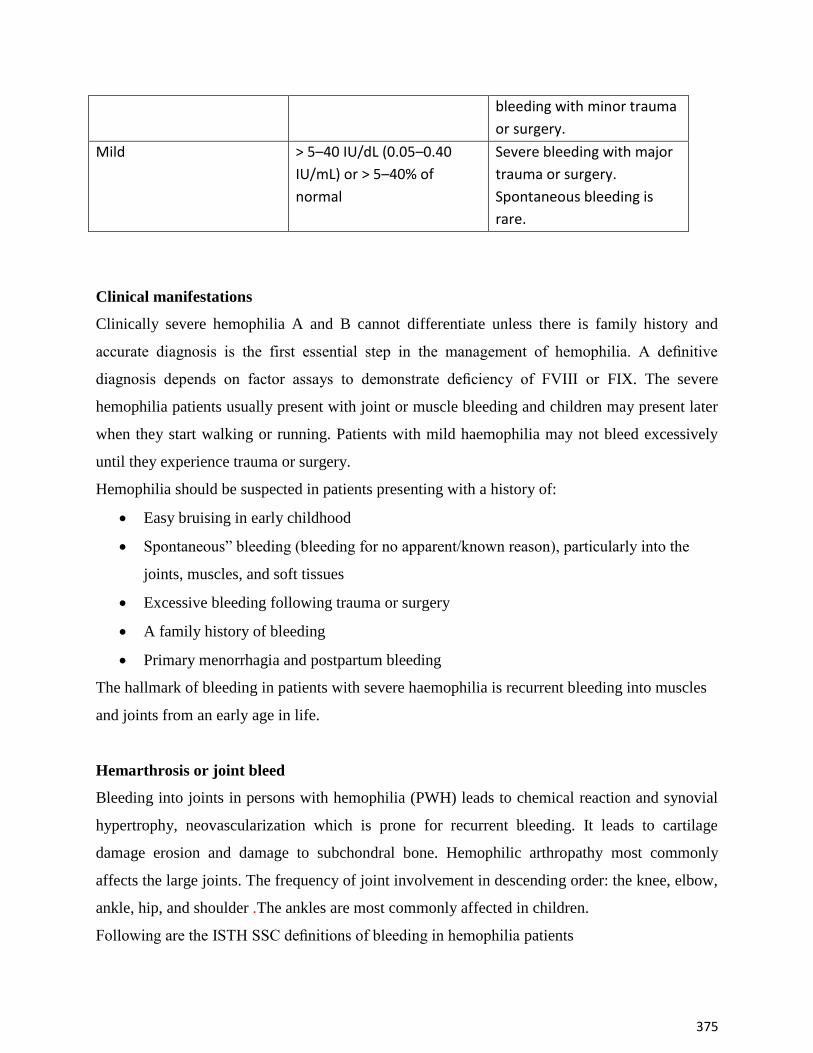
375
bleeding with minor trauma
or surgery.
Mild
> 5–40 IU/dL (0.05–0.40
IU/mL) or > 5–40% of
normal
Severe bleeding with major
trauma or surgery.
Spontaneous bleeding is
rare.
Clinical manifestations
Clinically severe hemophilia A and B cannot differentiate unless there is family history and
accurate diagnosis is the first essential step in the management of hemophilia. A definitive
diagnosis depends on factor assays to demonstrate deficiency of FVIII or FIX. The severe
hemophilia patients usually present with joint or muscle bleeding and children may present later
when they start walking or running. Patients with mild haemophilia may not bleed excessively
until they experience trauma or surgery.
Hemophilia should be suspected in patients presenting with a history of:
Easy bruising in early childhood
Spontaneous” bleeding (bleeding for no apparent/known reason), particularly into the
joints, muscles, and soft tissues
Excessive bleeding following trauma or surgery
A family history of bleeding
Primary menorrhagia and postpartum bleeding
The hallmark of bleeding in patients with severe haemophilia is recurrent bleeding into muscles
and joints from an early age in life.
Hemarthrosis or joint bleed
Bleeding into joints in persons with hemophilia (PWH) leads to chemical reaction and synovial
hypertrophy, neovascularization which is prone for recurrent bleeding. It leads to cartilage
damage erosion and damage to subchondral bone. Hemophilic arthropathy most commonly
affects the large joints. The frequency of joint involvement in descending order: the knee, elbow,
ankle, hip, and shoulder .The ankles are most commonly affected in children.
Following are the ISTH SSC definitions of bleeding in hemophilia patients

376
An acute joint bleed : an unusual sensation (“aura”) in the joint (such as a feeling of tingling or
pressure) in combination with any of the following:
Increasing swelling or warmth of the skin over the joint
Increasing pain
Progressive loss of range of motion or difficulty in using the limb as compared with
baseline.
The progression of these symptoms over a relatively short period of time is an important feature
of an acute joint bleed (hemarthrosis).
How to differentiate between acute joint bleed and chronic hemophilic arthropathy? – Figure 3
It is clinically difficult to differentiate between two but the following can be useful
Rapid resolution of pain following infusion of factor concentrates - typical of an acute
haemarthrosis.
Improvement of pain associated with activity soon after a period of rest - typical of
chronic arthritis.
Reluctance to use the limbs alone - indicative of an acute joint/muscle bleed in infants
and young children.
Target joint
Three or more spontaneous bleeds into a single joint within a consecutive 6-month period.
Significance of target joint
It is prone to spontaneous bleeding that may be difficult to control with medical
measures such as prophylaxis
Target joints may need to be managed with synovectomy, either with radioactive
agents (preferable) or chemicals or removed arthroscopically, if possible.
Muscle bleed
An episode of bleeding into a muscle, determined clinically and/or by imaging studies generally
associated with pain and/or swelling and loss of movement over baseline.
Significance of muscle hemorrhage

377
Muscle bleed can occur in any of the muscles and early identification and proper management is
very important to prevent as it may lead to neurovascular compromise leading to permanent
contracture or pseudotumour.
Iliopsoas muscle hematoma : risk of femoro-cutaneous, cruralandfemoral nervepalsy
Superior-posterior and deep posterior compartments of the lower leg : risk of posterior tibial and
deep peroneal nerve injury
Flexor group of forearm muscles - risk of Volkmann’s ischemic contracture
PWH need to be monitored continuously for signs and symptoms of neurovascular compromise
or radiological investigations. In case of muscle bleed, it is difficult to quantify and can cause
significant blood loss; hence hemoglobin should be monitored. Iliopsoas hematoma patients
usually present with pain in the lower abdomen, groin or lower back, and pain on extension (but
not on rotation) of the hip joint. Some of them also present with paraesthesia on the lateral part
of the thigh or other signs of femoral nerve compression such as loss of patellar reflex and
quadriceps weakness. The symptoms may mimic acute appendicitis.As soon as the pain subsides,
physiotherapy should be started and some of the patients with iliopsoas hematoma may require
skin traction to straighten the limb which may needs be covered with factor infusion.
Re-bleed into a muscle/ joint: Bleeding occurring for more than 72 hours after stopping treatment
for the original bleed for which treatment was initiated.
Central nervous system haemorrhage or head trauma: After head trauma patients can develop
intracranial hemorrhage or forehead hematoma. It is a medical emergency and life threatening.
Patients present with headache, with or without vomiting, altered sensorium or they may become
unconscious. These are the patients who are prone to have recurrence and prophylaxis for 3 to
6months can be given. Neurosurgery consultation should be taken as early possible.
Tables 15 and 16 shows sites and frequency of bleeding in different sites.
Table 15:
Sites of bleeding in hemophilia
Severity Sites of bleeding
Serious Joints (hemarthrosis)
Muscles, especially deep compartments
(iliopsoas, calf and forearm)
Mucous membranes in the mouth, gums,
nose and genitourinary tract

378
Life-threatening Intracranial
Neck or throat
Gastrointestinal
Table 16:
Frequency of bleeding in different sites
Sites of bleeding Approximate frequency (%)
Hemarthrosis
more common into hinged joints (i.e.
ankles, knees and elbows)
less common into multiaxial joints (i.e.
shoulders, wrists and hips)
70–80
Muscle 10–20
Other major bleeds 5–10
Central nervous system 5
Principles of care
Primary aim of care is to prevent and treat bleeding with the deficient clotting factor.
Should be treated with specific factor concentrate in specific factor deficiency
Acute bleeds should be treated as quickly as possible, preferably within 2 hours.
Patients usually recognize early symptoms of bleeding even before the manifestation of
physical signs. This is often described as a tingling sensation or “aura”.
In life-threatening bleeding especially in the head, neck, chest, and gastrointestinal tract,
treatment with factor should be initiated immediately, even before diagnostic tests
All persons with hemophilia (PWH)should carry an identification card mentioning the
diagnosis, severity and contact information of treating physician or hospital.
Veins are the lifelines for PWH and should be handled carefully. 23 or 25G butterfly are
preferred
Drugs that affect platelet function, particularly aspirin and NSAIDs should be avoided.

379
Good oral hygiene is essential to prevent periodontal disease and dental caries, which
predispose to gum bleeding
Regular exercise and other measures to stimulate normal psychomotor development
should be encouraged to promote strong muscles, develop balance and coordination, and
improve fitness
Dosage of factor concentrates
World Federation of Hemophilia (WFH) strongly recommends the use of viral inactivated
plasma derived or recombinant factor concentrates in preference to fresh frozen plasma or
cryoprecipitate for haemophilia. Selection of clotting factor concentrates should be made on very
well laid down WFH guidelines on selection and tendering of clotting factor concentrates (CFC).
When selecting plasma-derived concentrates, consideration needs to be given to both the plasma
quality and the manufacturing process. Among the manufacturing process factors, purity of the
product and viral inactivation or elimination should be considered as the most important.
Types of treatment
Clotting factor concentrates can be infused on demand/ episodic or prophylactic or regular
replacement therapy. Episodic (on demand) replacement therapy is factor replacement therapy
given at the time of clinically evident bleeding. Prophylaxis is infusion of clotting factor
concentrates to prevent bleeding. Principle of prophylaxis is from the observation that moderate
hemophilia patients with clotting factor level > 1 IU/ mL seldom experience spontaneous
bleeding. Prophylaxis prevents and decreases bleeding and joint destruction and preserve normal
musculoskeletal function
Dosage of factor for bleed management:
Calculation of FVIII concentrates
Each unit of factor VIII infusion will raise the plasma FVIII level by about 2 IU/dl or by 2 % in
the body.
The dose is calculated as follows
Patients weight (in Kg) X (Desired level – Patient level) X 0.5
FVIII should be infused by slow intravenous injection at a rate not greater than 3 mL/minute in
adults or as specified in the product information leaflet.
Half-life of FVIII : 8 – 12 hours

380
If the PWH requires second dose or symptoms subside or in case of severe bleeding the next
dose should be half of the first dose after 8 to 12 hours or depending on the FVIII level.
Calculation of FIX concentrates
Each unit of factor IX infusion will raise the plasma FIX level by about 1 IU/dl or by 1 % in the
body.
The dose is calculated as follows
Patients weight (in Kg) X (Desired level – Patient level)
FIX should be infused by slow intravenous injection at a rate not greater than 3 mL/minute in
adults or as specified in the product information leaflet.
Half-life of FVIII : 18 – 24 hours
FIX concentrates should be infused by slow intravenous injection at a rate not greater than 3
ml/minute in adults, or as recommended in the product information leaflet.
Management of Target joint
Good management of PWH can prevent target joints. Once a target joint occurs (3 or more
bleeds in a single joint within 6 months), recurrent bleeding into this joint will continue at a
higher rate.Conservative management: Short-term prophylaxis for 4– 8 weeks can be used to
interrupt the bleeding cycle with intensive physiotherapy
Surgical treatment:Synoviorthesis either Chemical synovectomy Oxytetracycline or radioactive
synovectomy, or arthroscopic synovectomy
Emergency management in hemophilia
PWH should be triaged urgently and delay in treatment can significantly affect the
morbidity and mortality.
There should not be any delay in administering factors if PWH have any signs or
symptoms of bleeding and it should be given first without waiting for radiological
confirmation. If transport to another hospital is planned always give a dose of factor to
cover bleeding episode before transport. Early factor administration is life saving.
After investigations maintenance treatment can be planned.
Indications for Factor replacement therapy

381
Suspected bleeding into a joint or muscle.
Any significant injury to the head, neck, mouth or eyes or evidence of bleeding in these
areas.
Any new or unusual headache, particularly one following trauma.
Severe pain or swelling at any site.
All open wounds requiring surgical closure, wound adhesive or steri-strips.
History of an accident or trauma that might result in internal bleeding.
Any invasive procedure or surgery
Heavy or persistent bleeding from any site
Gastrointestinal bleeding.
Acute fractures, dislocations and sprains.
Any other signs or symptoms suggestive of bleeding
WFH recommendations for maintenance plasma factor level and duration in different sites of
bleeding in resource constraints situations is shown in Table 17.
Table 17.Suggested plasma factor peak level and duration of administration (from WFH 2020 guidelines).
This gives doses in both practise patterns of lower dose ( useful in resource constrained settings and higher
practise patterns. The dose can be modified based on experience, supply and patient factors. If uncertain,
consult a hemtologist with experience in these issues.
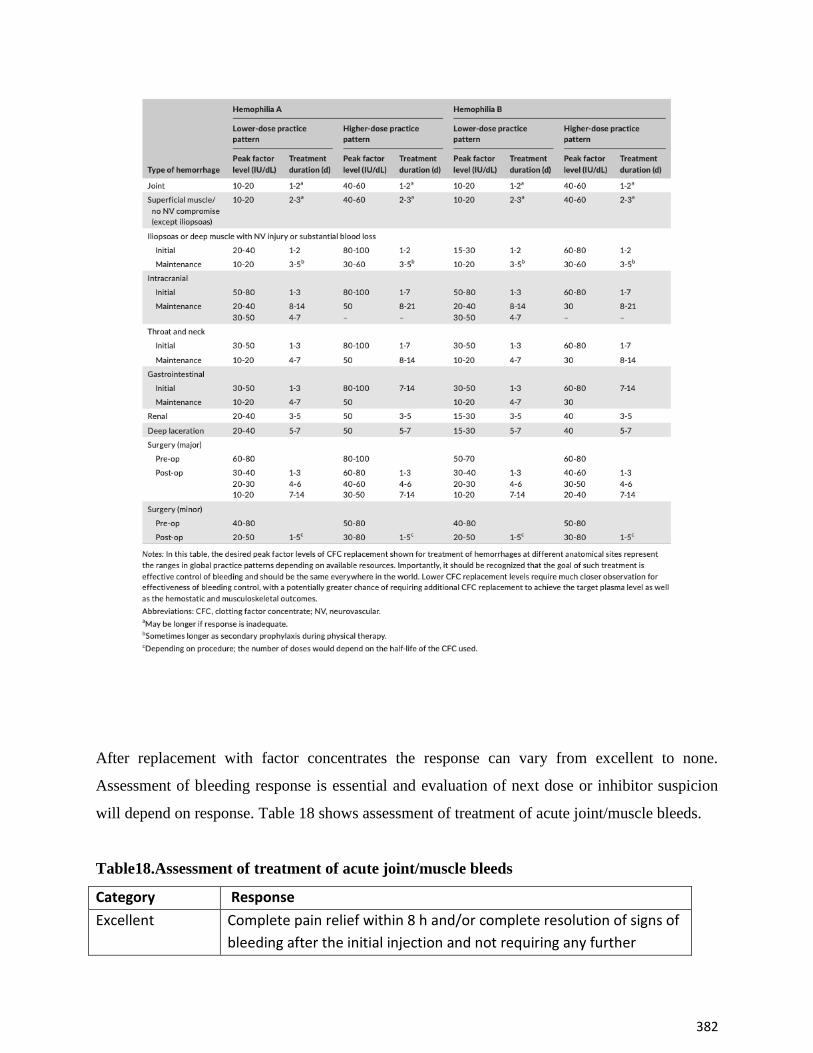
382
After replacement with factor concentrates the response can vary from excellent to none.
Assessment of bleeding response is essential and evaluation of next dose or inhibitor suspicion
will depend on response. Table 18 shows assessment of treatment of acute joint/muscle bleeds.
Table18.Assessment of treatment of acute joint/muscle bleeds
Category Response
Excellent Complete pain relief within 8 h and/or complete resolution of signs of
bleeding after the initial injection and not requiring any further
383
replacement therapy for relief of persistent symptoms and signs in
the same joint within 72 h.
Good Significant pain relief and/or improvement in signs of bleeding within
approximately 8 hours after a single injection, but requiring more
than one dose of replacement therapy within 72 h for complete
resolution.
Moderate Modest pain relief and/or improvement in signs of bleeding within
approximately 8 h after the initial injection and requiring more than
one injection within 72 h but without complete resolution.
None No or minimal improvement, or condition worsens within
approximately 8 hours after the initial injection
Physical activity
Physical activity should be promoted to improve physical fitness and normal
neuromuscular development and to concentrate on muscle strengthening, coordination,
general fitness, physical functioning, healthy body weight.
Contact sports such as cricket, tennis, football, basketball , boxing , wrestling etc should
be avoided
Non-contact sports such as swimming, walking table tennis and cycling under
observation
High contact and collision sports such as soccer, hockey, rugby, boxing, and wrestling,
motocross racing and skiing, should be avoided
Target joints can be protected with braces or splints during activity
Adjunctive treatment
For bleeding in muscles and joints
PRICE will relieve some symptoms and it is as follows
P – Protection with splints or compression bandages
R- Rest (new WFH 2020 guidelines have changed this to optimal loading (OL), meaning weight
bearing as soon as recovery occurs to prevent wasting from disuse of mucles. This changes the
acronym to POLICE)
I – Ice application

384
Ice or cold packs may be applied around the joint for 10–20 minutes every 2–4 hours for
pain relief.
Ice should never be applied in direct contact with skin.
Crushed ice wrapped in towel can be applied all over the joint
C-Compression
E-Elevation of the joint
Rehabilitation - Regular exercise is also the most important part in the management of PWH. As
soon as the pain and swelling begin to subside the patient should be encouraged to do isometric
exercises and then isotonic exercises with complete extension. Early active muscle control
mustbe encouraged, to minimise muscle atrophy and prevent chronic loss of joint motion
Antifibrinolytic drugs
Tranexamic acid is an antifibrinolytic agent that competitively inhibits the activation of
plasminogen to plasmin and it promoted clot stability. Oral Tranexamic acid tablets, 25 mg/Kg
or up to 1.5 g in adults, three to four times daily. Tranexamic acid is effective as adjunctive
treatment for mucosal bleeds and dental extractions. The use of tranexamic acid is
contraindicated for the treatment of haematuria because its use may prevent dissolution of clots
in the ureters, leading to serious obstructive uropathy and potential permanent loss of renal
function.
Epsilon amino caproic acid can also be used epsilon aminocaproic acid (EACA) 50 to
100 mg/kg every 6 hours in children and in adults loading dose of 4 to 5gm followed by lower
doses every 6 to 12 hourly(total suggested dose is 100 mg/kg/d).
Vaccinations
All bleeding disorders patients should be completely vaccinated including hepatitis A and
B
All the vaccines should be given subcutaneously
Intramuscular injections should be avoided.
Tetanus and Pertussis vaccine needs to be given intramuscularly
Infusion factor concentrates along with vaccine has not shown to increase inhibitor
chances however it is better to avoid CFC infusion prior to vaccination

385
Smallest gauge needle available (usually 25–27 G) should be used
An ice pack can be applied to the injection site for 5 mins and to apply pressure to the
injection site for at least 5 to 10 mins
Live virus vaccines (such as oral polio vaccine, MMR) may be contraindicated in those
with HIV infection
Pain management
Acute and chronic pain are common in patients with haemophilia. Adequate assessment of the
cause of pain is essential to guide proper management.
Pain caused by venous access : Local anaesthetic spray or cream can be used for paediatric
patients.
Pain caused by joint or muscle bleeding : Clotting factor concentrates should be infused as early
as possible to stop bleeding, other measures such as ice packs, splints crutches and pain
medication should be used
Postoperative pain: Intramuscular injection of analgesia should be avoided and pain strategy
should be followed.
Pain due to chronic hemophilic arthropathy: Functional training, adaptations and adequate
analgesia.
Table 19 .Strategies for pain management in patients with haemophilia.
No. Strategy
1 Paracetamol or acetaminophen If not effective, then use Strategy 2
2 COX-2 inhibitor (e.g. celecoxib, meloxicam and others) OR Paracetamol or
acetaminophen plus tramadol (3–4 times/day). If not effective, use Strategy 3
3 Opiate analgesia: use a slow-release product such as controlled-release
oxycodone, tramadol SR or controlled-release morphine
Dental care and management
Teeth should be brushed twice a day with a medium-texture brush to remove plaque
deposits
Good oral hygiene is essential to prevent periodontal disease and dental caries which may
predispose to gum bleeding

386
Dental examinations should be conducted regularly in all PWH
Dental floss or interdental brushes should be used wherever possible
Toothpaste containing fluoride should be used in places where fluoride content is not
present in water supply.
Toothpaste containing fluoride should be used in areas where natural fluoride is not
present in the water supply. Fluoride supplements may also be prescribed if appropriate.
Close liaison between the dental surgeon and the hemophilia team is important for good
dental care
Tranexamic acid or EACA is often used after dental procedures to reduce the need for
replacement therapy
Oral antibiotics should only be prescribed if clinically necessary.
Local hemostatic measures like fibrin glue or local tranexamic acid may also be used
Following a tooth extraction, the patient should be advised to avoid hot food and drinks
until normal feeling has returned.
Regular warm salt water mouthwashes (a teaspoon of salt in a glass of warm water)
should begin the day after treatment and continue for 5–7 days or until the mouth has
healed.
Prolonged bleeding and/or difficulty in speaking, swallowing, or breathing following
dental manipulation should be reported to treating hemophilia physician and dental
surgeon
Venous access
Veins must be treated with care
They are the lifelines for a person with haemophilia.
23- or 25-gauge butterfly needles are recommended.
Apply pressure for 3–5 min after venipuncture.
Venous access devices should be avoided whenever possible, but may be required in
some children.
Comprehensive care centre for Hemoglobinopathies & Haemophilia
Comprehensive care centre promotes physical and psychosocial health and quality of life while
decreasing morbidity and mortality.Hemophilia care is beyond just factor replacement and
management includes improvement in health and quality of life of patient and his family. The
wide-ranging needs of people with hemophilia and their families are best met through the

387
coordinated delivery of comprehensive care by a multidisciplinary team of healthcare
professionals.
The team consists of
Medical director (preferably a pediatric and/ or adult hematologist or a physician with
interest and expertise in hemostasis)
Nurse coordinator who coordinates the provision of care , educates patients and their
families ; acts as the first contact for patients with an acute problem or who require
follow-up and is able to assess patients and institute initial care where appropriate
Musculoskeletal expert (physiotherapist, occupational therapist, physiatrist, orthopaedics
surgeon, rheumatologist) who can address prevention as well as treatment.Experience
with PWH is preferred, or training can be undertaken.
A Hematologylaboratory specialist.
A psychosocial expert (preferably a social worker, or a psychologist) familiar with
available community resources
Members of the hemophilia care team: The comprehensive care team should also include or have
access to chronic pain specialist,dentist, geneticist, hepatologist, infectious disease specialist ,
immunologist, gynecologist/obstetrician and vocational counsellor etc.
Functions of a comprehensive care program
Provide or coordinate inpatient (i.e., during hospital stays) and outpatient (clinic and other
visits) care and services to patients and their family.
Patients should be seen by all core team members at least yearly (children every 6 months)
for a complete hematologic, musculoskeletal, and psychosocial assessment and to develop,
audit and refine an individual’s comprehensive management plan.
The plan of further management should be made and should be communicated to all
treaters and care facilities.
Communication among treaters
Advice to smaller centers and personal physicians and provide primary care and
management of some complications in frequent consultation with the comprehensive care
centre (particularly for patients who live a long distance from the nearest hemophilia
treatment centre).

388
Train and supervise home therapy where ever feasible
Educate patients, family members and other caregivers to ensure that the needs of the
patient are met
Collect data on sites of bleeds, types and doses of treatment given, assessment of long-term
outcomes (particularly with reference to musculoskeletal function), complications from
treatment, and surgical procedures. All this information should be recorded in registry.
Home therapy
Home therapy improves adherence to prophylaxis, allows immediate access to clotting factor
with early treatment resulting in decreased pain dysfunction and long-term disability improving
the quality of life, freedom to travel, less absenteeism and greater stability
What is the importance of Inhibitors?
Inhibitors are alloantibodies to factor VIII or IX and is an important complication of treatment.
Inhibitor testing should be done in a PWH who has inadequate response to optimum factor
correction and regularly in patients on prophylaxis which will be discussed in the next training
module. Neutralize the function of infused factor VIII or IX clotting factor concentrates, that is
the factor concentrates become ineffective to control bleeding.
Clinically relevant - Inhibitors should be documented on two separate occasions within a 1- to 4-
week period and should have a level of ≥ 0.6 Bethesda Units (BU/mL) using the Nijmegen
modification of the Bethesda assay. Inhibitors are discussed in the following section of training
module.
Documentation of treatment and data monitoring is the most important aspect in the management
of PWH. At least the annual bleed rate and school or parent absenteeism should be documented
to access the efficacy of any treatment. Following Joint assessment and functional activity
should be monitored at least on an annual basis
Clinical: WFH Physical Examination Score (aka Gilbert score), Hemophilia Joint Health
Score (HJHS)
Radiological: Pettersson score, MRI, and ultrasound scores
Activity: Hemophilia Activities List (HAL), Paediatric Hemophilia Activities List
(PedHAL), Functional Independence Score in Hemophilia (FISH)

389
Health-related quality of life: (HaemoQol, Canadian Hemophilia Outcomes: Kids’ Life
Assessment Tool [CHO-KLAT])
Figure 11.Images of different sites of hemarthroses and hematomas in patients with
Haemophilia
A. Patient with hemophilia with left elbow acute hemarthrosis
B. Child with hemophilia with forehead hematoma after trauma
C. Patient with hemophilia with left knee chronic hemarthrosis
Suggested Reading
Srivastava, A, Santagostino, E, Dougall, A, et al. WFH Guidelines for the Management of
Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl 6): 1‐ 158. https://doi.org/10.1111/hae.14046
Srivastava, A. K. Brewer, E. P. Mauser‐ Bunschoten et al. Guidelines for the management
of hemophilia. Haemophilia 2013: 19; e1-e. 47.
Victor S. Blanchette, Alok Srivastava. Definitions in Hemophilia: Resolved and Unresolved
Issues. Semin ThrombHemost 2015; 41:819–825.
Guidelines for emergency department management of individuals with hemophilia and other
bleeding disorders. MASAC Document #252 Replaces Doc #175
J. Hanley, a. Mckernan,† m. D. Creagh et al. Guidelines for the management of acute joint
bleeds and chronic synovitis in haemophilia A United Kingdom Haemophilia Centre
Doctors’ Organisation (UKHCDO) guideline. Haemophilia 2017; 1–10.
Guidelines for the management of haemophilia in Australia. Haemophilia Guidelines July
2016. National Blood Authority. Locked Bag 8430. Canberra ACT 2601

390
Management of hemophilia with inhibitors
Inhibitor development is a serious complication which occurs in some patients with hemophilia.
Approximately 33% patients with severe hemophilia A will develop inhibitors, some inhibitors
are transient but at other times they may persist.
When inhibitors occur, the factor replacement treatment becomes ineffective and risk
of life threatening hemorrhages again appear; these recurrent bleeds compromise joint
health and quality of life
At present in severe hemophilia A, only prophylaxis has been shown to possibly reduce the risk
of inhibitor development.
Some important factors which increase the risk of development of inhibitors are
Family history (siblings, cousins, uncles etc)
High risk mutations: null mutations, inversions,large deletions and mutations inducing
stop codons.
Intense exposure- High dose factor replacement during surgery
Infection
Early exposures, especially first 50 exposure days (ED) with a median time of inhibitor
presentation at about 10-15 EDs.
Hemophilia A: Age at first exposure does not appear to have an effect, though practically risk is
highest below the age of 5 years. There is an increased risk after the age of 60 years.
Haemophilia B: FIX inhibitors are associated with serious allergic reactions to FIX, especially in
those with gene deletions. Any reaction should prompt inhibitor testing before further FIX
exposure as even low-level FIX inhibitors may cause anaphylaxis

391
When to suspect inhibitors ?
Inhibitor testing should be performed if a patient has a poor clinical response to
concentrate or lower FVIII/IX levels than expected after concentrate infusion.
• Unsuspected increase in number or severity of bleeding episodes,
• Lesser response to previously used factor concentrates or less than expected recovery
(FVIII levels)
• Unusual bleeding episodes i.e. muco-cutaneous, GI, urogenital etc
When to measure inhibitors?
An inhibitor screen should be performed in patients with severe hemophilia frequently
till 30 exposures- recommended every fifth dose.
Thereafter, inhibitor testing should be undertaken every 3–6 months until 150 EDs.
Then annually or with increasing bleeding episodes.
How to measure inhibitors?
• The Specific assay - Bethesda assay (BA) or if available Nijmegen modified Bethesda assay
(NBA) should be done. The Inhibitor assay is positive when the titer is >0.6BU/mL.
If Bethesda assay/ Nijmegen modified Bethesda assay for inhibitors not available,
then an incubated mixing assay can be done.
If positive- then refer patient for inhibitor testing as soon as possible as this is a
serious problem.
Do not continue factor replacement till results are obtained.
More about inhibitors
• Titers of inhibitors: A low responding inhibitor is defined as an inhibitor level that is persistently
< 5 BU/mL, whereas a high responding inhibitor is defined by a level ≥ 5 BU/mL.
• High responding inhibitors tend to be persistent. If not treated for a long period, titer levels may
fall or even become undetectable, but there will be a recurrent anamnestic response in three to
five days when challenged again with anti - hemophilic factor products
• Some low titer inhibitors may be transient, disappearing within six months despite antigenic
challenge with factor concentrate.

392
· Very low titer inhibitors may not be detected by the Bethesda inhibitor assay, but by a poor
recovery and/or shortened half-life (T-1/2) following clotting factor infusions.
How to manage hemophilia patients with inhibitors?
The management / treatment of haemophilia patients with inhibitors has two components:
Treatment or prevention of acute bleeding
Eradication of inhibitors by Immune Tolerance Induction (ITI)
Treatment of acute bleed
If the inhibitor titre is low and low responding (inhibitortiter does not increase with factor
challenge, and historical inhibitor peak levels never exceeded 5 BU/mL), such cases can treated
with higher doses of factor VIII.
If this information not available or high titre inhibitor, then treat bleeds with bypassing
agents only.
Refer patient to Level 3 or Level 4 for complete laboratory evaluation and clinical
management.
High titer inhibitors: If high-titer, high-responding inhibitors (historical inhibitor peak levels that
exceeded 5 BU/mL at least once) are present, then FVIII/FIX concentrates are contra indicated.
Currently two treatment modalities are approved for this indication:
1. Bypassing Agents: aPCC (FEIBA®), rFVIIa (NovoSeven®)
2. Bispecific Monoclonal Antibody: Emicizumab.
Currently available and commonly used Bypassing agents:
Two by-passing agents are commercially available plasma-derived aPCC (FEIBA®), and rFVIIa
(NovoSeven®).
Both are clinically effective, however individual variations may occur, if the patient does not
respond to appropriate dose of one agent then the alternative agent is to be tried. Reasons for this
individual variation, is not known. Give BPA dose to stabilize and refer to higher center.

393
Table 20.
Characteristics of Bypassing agents
rFVIIa (NovoSeven®) aPCC (FEIBA®)
Mechanism of action
Half-life
Mode of Administration
Dosing and frequency
Acute Bleeding
Prophylaxis
Activates FX on the surface of
platelets
2-3 hours
IV
90-120µg/kg repeat doses
every 2-3 hourly
Maybe used three times a
week
Action of FXa and FII
6-12 hours
IV
50-100 U/kg 2-3 times daily [Max.
dose is 200 U/kg/day]
More evidence to support use.
Dose is usually
85 units /kg three times/week
Development of inhibitors leads to increased mortality and poor quality of life. Patient should be
seen in a center of excellence or tertiary care center (Level 3 or 4), The recommendations from
the center should be followed.
The standard Bypassing agents have been FEIBA and Recombinant factor 7a (rFVIIa)
• 2 doses of rFVIIais equivalent to one dose FEIBA in managing a joint bleed.
• Some patients respond better to one bypassing agent- switch if no response
rFVIIa:
• rFVIIa Dose 90mcg/kg iv. Every 2 -3 hours,
• Better response with 270mcg/kg single dose
FEIBA/APCC:
FEIBA dose 50-100 units/kg/day to manage bleeding,
• Max dose 200 units/kg/day, can be repeated every 6-12 hours.
• Risk of anamnestic increase in inhibitor (but more in FIX patients)

394
• To prevent bleeding in inhibitor patients FEIBA can be given on alternate days to prevent
bleeding, this is prophylactic use of bypassing agents. This is better than only treating
bleeds as study has shown better joint outcome.
Newer agents: Emicizumab is recombinant humanized bispecific monoclonal antibody that
bridges activated factor IX and factor X to replace the function of missing activated factor VIII,
thereby restoring hemostasis. This can be an effective agent for haemophilia patients with
inhibitors.Emicizumab is recombinant humanized bispecific monoclonal antibody that bridges
activated factor IX and factor X to replace the function of missing activated factor VIII, thereby
restoring hemostasis. The antibodies against FVIII do not bind or neutralize Emicizumab and it
bypasses the need for factor VIII hence, this is clinically effective in all individuals with
haemophilia A- those with inhibitors or those without inhibitors. Clinical trials suggest Ctrough
≥45 μg/Ml should result in a median ABR of 0 and Emicizumab is approved only for
Prophylactic treatment.
Table 21.Characteristics and dose of Emicizumab (Hemlibra)
EMICIZUMAB
Mechanism of action
Half-life
Mode of
Administration
Dosing and frequency
Prophylaxis
Replaces the action of FVIIIa by binding with both FIXa
and FX to trigger the activation of the rest of the
coagulation cascade
4 Weeks
SC (subcutaneous injection)
3mg/kg for 4 weeks loading Dose (to attain Ctrough ≥45
μg/Ml)
1.5 mg/ kg/ week or 3mg/kg once in 2 weeks
maintenance dose

395
Immune tolerance induction ( ITI)
Immune tolerance induction (ITI) is at present the only treatment which can eradicate
inhibitors. ITI consists of regular, frequent, and prolonged exposure of the patient to specific
factor concentrates to induce tolerance. Immune tolerance is reported to be highly effective and
about 70% of patients become inhibitor free. This therapy should only be performed at level 3/ 4
centers. Interruption of ITI, infections, and patients with high titers of inhibitors or long history
of persistent inhibitors have less chance of success. Success is considered when inhibitor titer
can no longer be measured, factor recovery is greater than 66% of normal and half-life of Factor
VIII is greater than 6 hours.
In ITI, regular replacement of factor is given at higher doses, with monitoring and management
of breakthrough bleeds with bypassing agents till the inhibitor disappears. This process may take
months to years. After eradication of inhibitors the patients need to be put on prophylaxis to
decrease recurrence of inhibitors in the patient.
There are several regimens for ITI, two protocols can be followed for ITI in consultation with
Level 3 or 4 center with experience with ITI treatment.
Bonn Protocol :
Hemophilia. A: FVIII 200-300 IU/kg daily + FEIBA 100 units/kg twice daily and Van
Crevald (Dutch or low dose): FVIII 25 -50 IU/kg on alternate days and escalated. Currently
many HTC use Factor VIII 50 units /kg three times a week and monitor. The higher dose ITI in
the International ITI study was shown to have less bleeding episodes.ITI therapy will take
6months or longer the remove the inhibitor. After ITI patients have to be put on prophylaxis, to
prevent recurrence. About 20 -30% patients may fail, then a different ITI regimen may be tried
or immunosuppressive/ immunomodulation therapy has been advocated, after failure of
conventional ITI by WFH.
Hemophilia B: Experience with ITI for hemophilia B inhibitor patients is limited. The principles
of treatment in these patients are similar, but the success rate is much lower, especially in
persons whose inhibitor is associated with an allergic diathesis. Hemophilia B inhibitor patients
with a history of severe allergic reactions to FIX may develop nephrotic syndrome during ITI,
which is not always reversible upon cessation of ITI therapy. Alternative treatment schedules,
including immunosuppressive therapies, are reported to be successful.

396
Suggested reading
Srivastava, A, Santagostino, E, Dougall, A, et al. WFH Guidelines for the Management of
Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl
6): 1‐ 158. https://doi.org/10.1111/hae.14046
Charles R. M. Hay and Donna M. DiMichele, on behalf of the International Immune Tolerance
Study. The principal results of the International Immune Tolerance Study: a randomized dose
comparison. Blood 2012; 119:1335-1344.
Minno GD, Santagostino E, Pratt K, Königs C.
New predictive approaches for ITI treatment.Haemophilia. 2014 Sep;20 Suppl 6:27-43.
Seth, Tulika. (2019). Experience of Immune Tolerance Induction Therapy for Hemophilia A
Patients with Inhibitors from a Single Center in India. Indian Journal of Hematology and Blood
Transfusion. 36. 10.1007/s12288-019-01218-2
Implementation of Prophylaxis for Persons with Hemophilia (PWH) in
India
1. What is prophylaxis?
Prophylaxis is the regular infusion of clotting factor concentrates in order to prevent bleeding.
The idea of prophylaxis came from the observation that people with moderate or mild
haemophilia (who have clotting factor levels of 1% or more) rarely experience spontaneous
bleeding. They also have less joint damage than people who have severe haemophilia.
2. What are the different types of prophylaxis?

397
Unlike episodic or “on demand” treatment, which is given at the time of a bleed to make it stop,
prophylaxis is given to prevent bleeding before it starts.
There are several types of prophylaxis. Continuous prophylaxis (primary, secondary, and
tertiary) is given regularly over a period of several months and often years. Intermittent or
periodic prophylaxis is given for shorter periods of time, usually a few weeks or months.
Table 22. Definitions of types of therapy for hemophilia
Type of Treatment Definition
Episodic (“on demand”) treatment Treatment given at the time of bleeding.
Continuous prophylaxis
Primary prophylaxis
Regular continuous treatment, started before the
second large joint bleed and age of 3 years.
Secondary prophylaxis
Regular continuous treatment started after 2 or
more large joint bleeds but before the onset of
joint disease.
Tertiary prophylaxis Regular continuous treatment started after the
onset of joint disease to prevent further damage.
Intermittent (“periodic”) prophylaxis
Treatment given to prevent bleeding for short
periods of time, such as during and after surgery
(not exceeding 45 weeks in a year)
Adapted from: Guidelines for the Management of Hemophilia, World Federation of Hemophilia, 2012.
Why prophylaxis is the way forward in the management of PWH in India?
The treating Physicians believed that if they could keep minimum factor levels around 1% with
regular infusions of clotting factor concentrates, they may reduce the risk of bleeding and
prevent joint damage. Since then, important studies have shown that children who receive
prophylaxis do have fewer bleeds and healthier joints. There is a false notion that prophylaxis is
more expensive than “on demand” therapy but studies have clearly shown that low dose
prophylaxis leads to less factor consumption and is more cost-effective than treating the bleeds

398
after they occur (ie. “on demand”). Prophylaxis is now the goal of treatment for people with
severe haemophilia, allowing them to remain active and participate more fully in daily life.
Caveat
Prophylaxis will not help repair joints that are already damaged. However, it will decrease the
frequency of bleeding, may slow progression of joint disease, and may improve quality of life.
Checklist before starting prophylaxis in your hospital
Are there motivated children and parents who would regularly visit your
hospital to take the injections? (unless carrying of factors to home and
storage for home therapy is permitted by local law)
Have you calculated the weekly requirement of total factors for the
group?
Have you ensured that the stock is adequate so that continuous
prophylaxis can be provided?
Have you sensitized the PWH that unless they perform regular exercise
the joint health will not improve?
Have you put in place a system for periodic screening for inhibitors as
per standard guidelines?
Are the monitoring charts for periodic assessment of joints and
functional activity ready?
Factors to consider when designing prophylaxis protocol
Age at which prophylaxis started (the earlier started, the better)
Current age (may be difficult to continue throughout life)
Venous access (butterfly needles with minimum damage to the veins)
Bleeding symptoms (frequent bleeders could be more benefitted)
Joint status (joints should not be damaged to such an extent that there is

399
Factors to consider when designing prophylaxis protocol
no reversible component)
Level and timing of physical activity (proper increment of factor levels
should be documented especially if the bleeding rate does not decrease)
Availability of clotting factor concentrates (uninterrupted supply is a
must)
Size(s) of vials of clotting factor concentrates available (larger dose vials
may be shared between multiple children to prevent wastage)
3. How to select the schedule and administer prophylaxis at your hospital ?
Studies are still underway to determine the best dosing schedule (also called a ‘protocol’). A
prophylaxis schedule should clearly outline:
The type of factor product to be used (Long acting factor concentrates are not available in
our country at this point of time)
The dose of factor administered with each injection
The frequency at which treatment is administered
The time (of day, or week) that treatment is administered
There are currently two protocols in use for which there is long-term data:
The Malmö protocol: Injections of 25-40 IU/kg, administered three times a week for those with
haemophilia A and twice a week for those with hemophilia B.
The Utrecht protocol: Injections of 15-30 IU/kg, administered three times a week for those with
haemophilia A and twice a week for those with hemophilia B.
However, these protocols are not practically feasible in most parts of the world including India
In countries with significant resource constraints like India, lower doses of prophylaxis given
more frequently (e.g., 10-15 IU/kg, 3 times per week) may be an effective option.[2-5]. The long
term benefits of these low dose prophylaxis regimens is not yet established and data should be
collected if using these newer regimens or it should be done under a clinical trial.Prophylaxis
should be administered to children to make it cost effective and ensure better outcomes.
In recent studies, it has even been proven that a low dose protocol as detailed below can
significantly reduce the annual bleeding rate and prevent joint damage. This kind of treatment

400
regimen is also associated with better school attendance and reduced hospitalization among
children with hemophilia.
Factor VIII concentrate may be given at a dose of 20-40 IU/kg in two divided doses per week for
hemophilia A (ie 10-20 IU/kg per dose).[6]
The dosage of factor IX concentrate for hemophilia B is 25- 40 IU/kg/wk.
The dosage is to be adjusted to the nearest available vial dose at the time of visit.
The frequency of administration can be increased to thrice a week for children with Haemophilia
A and twice a week for children with Hemophilia B if they have excessive bleeding.
Screening for inhibitors is an integral part of this management protocol. Inhibitor development is
particularly common in people with severe hemophilia within the first 75 treatments with
clotting factor concentrates. More than 50% of inhibitors develop within the first 15 exposure
days.
4. How do you assess the efficacy of the prophylaxis regimen?
More than three decades of research has shown that continuous prophylaxis is preferable to on-
demand therapy to reduce the frequency of bleeding and to prevent or delay joint damage.
People with haemophilia who are receiving prophylaxis should have an assessment on a regular
basis to ensure that the goals of therapy are being met and to make any necessary adjustments to
the treatment plan. These assessments should include an evaluation of:
Joint health/status
Bleed frequency
Limitations in activities
Psychosocial integration
5. What are the barriers and challenges to implementing prophylaxis in India?
Cost and access to treatment products
The biggest barrier to long-term prophylaxis is the cost of treatment. Prophylaxis is only possible
if significant resources are allocated to hemophilia care. However, it is cost-effective in the long-
term because it eliminates the high cost associated with subsequent management of damaged
joints and improves quality of life.

401
With NHM allocating significant resources for hemophilia care in India, prophylaxis is now a
reality for children with Hemophilia in India. It is very important to collect and provide scientific
evidence that justifies the high cost of treatment. Follow-up data of these patients will be very
important in confirming the effects on their joints. Cost-efficacy studies designed to identify
minimum dosage are necessary to allow access to prophylaxis across the country.
Venous access
Prophylaxis requires frequent injections and it can be difficult to find suitable veins in very
young children with hemophilia. In our country, all children can be administered early
prophylaxis without venous access devices provided good care of veins is taken.
Adherence
Adherence to (or compliance with) a treatment plan is generally defined as the extent to which
patients take medications as prescribed by their healthcare providers. According to the World
Health Organization, rates of non-adherence with any medication treatment may vary from 15%
to 93%, with an average estimated rate of 50%.
Adherence to a prophylaxis protocol is critical to its success. Prophylaxis is most effective if
factor levels are continuously maintained above the target level. Missing or skipping a dose can
cause clotting factor levels to fall below this target, which increases the risk of bleeding.
Bleeding that occurs while a patient is on prophylaxis is called ‘breakthrough bleeding’.
Patients and healthcare providers must work together to ensure that the protocol is manageable
for the person with haemophilia and their family. A patient’s adherence to the protocol should be
assessed regularly during clinic visits and strategies to improve adherence, including changes to
the protocol, should be explored wherever possible.
Suggested reading
Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S et al. Guidelines for
the management of hemophilia. Haemophilia, 2013; 19: p. e1-47.
Verma SP, Dutta TK, Mahadevan S, Nalini P, Basu D, Biswal N, et al. A randomized
study of very low-dose factor VIII prophylaxis in severe haemophilia - A success story from
a resource limited country. Haemophilia : the official journal of the World Federation of
Hemophilia. Haemophilia 2016; 22: 342-8.

402
Poon, .C and Lee A. Individualized prophylaxis for optimizing hemophilia care: can we
apply this to both developed and developing nations? Thrombosis Journal, 2016; 14: 32.
Dunkley S, Lam JCM, John MJ, Wong RSM, Tran H, Yang R, et al. Principles of
haemophilia care: The Asia-Pacific perspective. Haemophilia 2018; 24: 366-75.
Carcao M. and Srivastava A. Factor VIII/factor IX prophylaxis for severe hemophilia.
Seminars in Hematology 2016; 53: 3-9.
Sidharthan NR. Sudevan V. Narayana Pillai S. Mathew M. Raj D. Viswam C.et al. Low-
dose prophylaxis for children with haemophilia in a resource-limited setting in south India-A
clinical audit report. Haemophilia2017; 23: e382-e384.
Surgery in hemophilia
Surgery in hemophilia requires special care since the underlying problem of patients with
haemophilia is defective blood clotting. Only specialised centres should attempt surgical
procedures since a well equipped laboratory is a must.
What is the aim?
The goal is to achieve satisfactory hemostasis during the procedure as well as perioperative
period so that a stable blood clot is formed which will lead to wound healing.
Is it required to achieve normal level of factors for surgery?
Even though normal level of factors range from 50-150%, it is not required to achieve 100% for
effective hemostasis during surgery or during perioperative period.
Maximum levels are required during the first three days of surgery.
Once a stable blood clot forms by this time, lower levels are sufficient.

403
This in effect means higher doses for the first three days and lower doses subsequently.
What are the surgical procedures possible?
All surgeries done in a patient with normal haemostatic system can be done in a patient with
hemophilia. However the level of factor support required varies depending upon the type and
severity of hemophilia and the type of surgery planned.
How are surgeries classified as minor and major in patients with Hemophilia?
Although there are no strict definations, the level of surgical invasiveness is most
commonly used as a defining factor for major surgery, with criteria including: a
requirement for a general or spinal anesthetic, provision of respiratory assistance,
abdominal surgery, penetration of a major body cavity, orthopedic surgery (joints), and
extraction of more than 3 teeth .
World Federation of Hemophilia classifies “A major surgical procedure is defined as
one that requires hemostatic support for periods exceeding 5 consecutive days”.
Table 23. Classification of types of Surgery for Hemophilia
General Surgery Orthopedic Surgery
Major “-ectomy” procedures
“-otomy” procedures
Pseudotumor
Dental -Extraction >3 teeth
Osteotomy/arthrodesis Dentala
Joint replacement/ arthroplasty
Synovectomy/arthroscopic synovectomy
Bone fracture reduction
Osteosynthesis,Arthroscopy,Amputation
Minor Centralline insertion/removal
Liver biopsy
Prostate biopsy, colonoscopy
with biopsy and other
diagnostic biopsies
Abcess-drainage,
Wound suturing,circumcision
Chemical-/radio-/arthroscopic synovectomy
Mild (factor level 5-30%) and moderate (factor level 1-5%) hemophilia generally require
lower level of factor support compared to severe hemophilia (factor level <1%).
Hemophilia B requires lower level than hemophilia A

404
How long should factor support be given?
Generally factor support is required till wound healing or suture removal whichever is earlier.
This approximately corresponds to 3 days for minor procedures and 10 days for major
procedures. This can be longer depending on the particular type of surgery performed.
What are the pre requirements?
Additional requirements over and above in the usual setting
Laboratory should have the facility to do the following.
Factor assay
Bethesda assay
PT, APTT, fibrinogen if required post operatively
Ideally, full diagnostic testing of coagulation disorders
Clinical care should have the following human resources.
Hematologist
If not, a Physician well trained in managing coagulation disorders including support
during and after surgery
Ideally, Surgeons familiar with handling of patients with disorders of coagulation
How much factor support is required?
This will depend on the level and duration of factor support planned. Even though higher levels
are followed in countries without resource constraints, it has been shown that lower levels will
work equally well from studies done in developing countries, including India. Approximately
300 IU/kg of factor support will be required for a major procedure without complications.
How much blood and blood product support is required?
Like all surgical procedures, hemophilia patients will also require blood and blood product
support. If planned levels of factor are achieved, higher than anticipated blood loss is unlikely to
occur.
Is there any other support which will help?
Antifibrinolytic agents like tranexamic acid aid in forming a stable clot and should be routinely used.
However they are contraindicated in urological procedures.

405
What all things should be done prior to the planned surgery?
Correct diagnosis of the coagulation disorder, especially whether it is haemophilia A or
B.
If it is mild or moderate haemophilia, rule out von Willebrand disease.
Bethesda assay to look for inhibitor status, if high titre, factor will be ineffective and
patient will require bypassing agents like FEIBA or rVIIa.
If low titre, should look for recovery assay and for rise in titre 3-5 days following factor
infusion.
How do we know it is safe to do surgery after giving factors?
Factor levels should be checked after giving factor prior to the surgery, this is called a
recovery assay.
Subsequently it should be checked on days 1,3,5 and 7 of post operative period, these are
the trough levels.
Dose should be modified if satisfactory levels are not achieved or if higher than planned
levels are achieved.
Table 24.Recommended factor support for resource constrained settings
Day Target level Factor dose
Hemophilia A Hemophilia B Hemophilia A Hemophilia B
0 (Recovery) 80-100% 60-80% 40-50 IU/kg Q12H 60-80 IU/Kg OD
1-3 (Trough) 40-50% 30-40% 20-25 IU/kg Q12H 30-40 IU/kg OD
3-5(Trough) 30-40% 20-30% 15-20 IU/kg Q12H 20-30 IU/Kg OD
>5(Trough) 20-30% 10-20% 10-15 IU/kg Q12H 10-20 IU/kg OD
How are the peak levels and trough levels defined and how should they be monitored ?
Peak Level :- The peak factor level is the maximum factor level measured within 1 hour
of a bolus injection. Wherever feasible, peak level should be measured immediate pre op and

406
should be targeted to 60-80U/dl at the minimum and can be kept upto 100% in neurologic
surgeries.
Trough Level :- The trough factor level is the minimum factor level measured
immediately before the next bolus injection.. Wherever feasible the dosage in patients
undergoing major surgery should be titrated as per the trough which should be kept between 30-
50IU/dl for initial 2 to 3 days after major surgery and 20-50 after minor surgery.
Can patients with Hemophilia undergoing surgery be given continuous infusion regimes of
factor products? What are the necessary precautions to be taken while on continuous
infusion ?
Continuous infusions may provide improved coagulation factor cover, are associated with
improved bleeding outcomes and may use less coagulation factor than bolus regimes.
After an initial bolus injection of FVIII, an infusion is started to maintain coagulation according
to the following formula:
Infusion rate (IU/kg/h) = clearance (mL/kg/h) x steady state concentration (IU/mL)
Ideally, clearance can be calculated from pre-operative pharmacokinetic studies which can be
usedto guide initial infusion rates over several days.
Alternatively, an infusion rate of 3.0-5.0 IU/kg/h will produce a Factor level of
approximately 80 IU/dL. (See Table below for titrating the infusion regime)
Table 25.Guidelines for coagulation factor target levels in adults undergoing major surgery
using continuous infusion of recombinant factor VIII/IX
Day
Continuous Infusion
Target FVIII /IX steady state level
(%) Infusion rate dose (IU/kg/hour)
Pre-op(Bolus) 80-100 IU/kg load (round up to nearest vial size)
1-3 30-60 3.0-5.0 IU/kg/h
4-5 20-50 1.5.-4.0 IU/kg/h
5 and beyond Often change to bolus dosing

407
Continuous factor infusion must always be on an infusion pump. The following things should be
kept in mind:-
a. Always run a dedicated line
b. Factor should hang no longer than 12 hours, after 12 hours it should be
discarded and a new infusion set up.
c. Brand of the Factor product should always be checked for the use as continuous infusion. The
prerequisite is the factor should remain stable for the infusion hours after dissolution.
d. When one hour’s worth of factor left in the pump, a new infusion should be made ready
What are the preanaesthetic, intra-operative and post operative precautions to be kept in
mind for patients with bleeding disorders undergoing surgery?
The following factors need to be considered in Preoperative and Intraoperative and Postoperative
periods:-
Table 26. Preoperative anaesthetic assessment in patients with Hemophilia
History Examination Investigation
Nature of Bleeding disorder
Severity
Prior bleeding history
Prior treatment (factor therapy/Plasma
/transfusion etc/response to DDAVp)
Prior history of inhibitors Comorbidities
(especially HIV,HbsAg,Anti HCV) and
chronic liver disease or CD4 count
Availability of factor support and
haemophilia expert/haematologist
Dental hygiene and infections -treat
infection prior to orthopaedic surgery
Type of surgery:-
Elective-Always schedule on weekdays
Joint
deformities
Venous access
Airway
Blood Counts
Coagulation profile
including Fibrinogen
Specific Factor assay
if not already done
Inhibitor screen
within past 2 months
Ensure availability
of cross matched
blood products

408
with availability of full multidisciplinary
team)
Urgent-Ensure adequate factors
Table 27.Intraoperative Considerations during surgery in patients with Hemophilia
Peripheral lines are secured with utmost care.
Intramuscular injections and arterial punctures are avoided.
Smooth induction is given under deep plane of anesthesia.
Risk–benefits for neuraxial block and regional blocks need to be assessed individually and
in general are avoided.
To prevent muscle fasciculation (aggravate muscle and joint hematomas), use of
succinylcholine is avoided.
Utmost caution is done to do atraumatic tracheal intubation
Gentle suctioning of oropharynx should be done.
Extremities and pressure points should be padded.
Non-steroidal analgesics are not used as they cause gastrointestinal hemorrhage.
Hypothermia is avoided.
Hemodynamic conditions are maintained near normal,hypertension and tachycardia is
avoided
Small vessel hemostasis is achieved with minimal intraoperative blood loss
Tranexamic acid can be used at i.v dose of 10 mg kg 8th hourly). It should be avoided in
patients with haematuria due to obstructive uropathy.
Estimates of blood loss during surgery (Table 6) and data on pre- and post-operative
hemogloblin levels and the number of packed red blood cell units transfused should be
maintained to estimate surgical blood loss.
Surgical hemostasis should be assessed by an involved surgeon and/or anaesthetistand

409
Post-operative Considerations during surgery in patients with Hemophilia
Early mobilization, and physiotherapy should be instituted
Consider mechanical deep venous thrombosis prophylaxis using pneumatic
compression devices if available. Pharmacologic prophylaxis are best avoided
Adequate post operative analgesia should be given using tramadol, codeine and or
morphine. NSAIDs are avoided.
Drains, dressings and sutures should be removed at the time of peak factor
levels.(Within 1 hour of bolus dose of factor)
Patients with mild hemophilia A, as well as patients receiving intensive factor
replacement for the first time should be re-screened 4–12 weeks post-operatively
for development of inhibitors.
What are the precautions to be undertaken in female patients who are Hemophilia
Carriers while undergoing obstetric procedures / delivery?
Factor VIII and IX levels must be measured immediately before delivery and post-
partum with an aim to maintain above 0.5 IU ml(>50%) and accordingly supplemented
with factor VIII or Factor IX
Instrumental delivery is best avoided
The decision about caesarean section or vaginal delivery is as per obstetric indication
What are the precautions to be undertaken while undergoing dental procedures in patients
with Hemophilia ?
Close liaison between the dental surgeon and the hemophilia team is essential to provide
good comprehensive dental care.
Treatment can be safely carried out under local anesthesia using the full range of
techniques available to dental surgeons. Infiltration, intra- papillary, and intra-
records should be completed within 72 hours following surgery.

410
ligamentary injections, an inferior alveolar nerve block or lingual infiltrationare to be
done under factor cover of 20-40%.
Tranexamic acid or epsilon aminocaproic acid (EACA) is often used after dental
procedures to reduce the need for replacement therapy.
Following a tooth extraction, the patient should be advised to avoid hot food and drinks
until normal feeling has returned. Smoking should be avoided as this can cause problems
with healing.
Regular warm salt water mouthwashes (a teaspoon of salt in a glass of warm water)
should begin the day after treatment and continue for five to seven days or until the
mouth has healed.
What are the special precautions to be undertaken prior to undertaking orthopaedic
surgery in patients with Hemophilia?
Orthopedic surgeons should have had specific training in surgical management of persons
with hemophilia
If frequent bleeding is isolated to one target joint, minimally invasive procedures (e.g.
synovectomy) are preferred for young patients without significant joint damage.
Consider more aggressive and proactive strategies for patients in late teens to twenties as
they are more likely to have successful outcomes than older patients with significant joint
deterioration.
Consider patients with some preserved joint function and muscle mass as surgical
outcomes are more likely to be successful if joints are not already fixed with extensive
scar tissue and atrophied muscles.
Weigh benefits of multiple interventions and multiple site elective surgery using one
anaesthesia and favtor or bypassing agent coverage (cost saving) against the impact on
rehabilitation (easier vs. harder to rehabilitate and issues with assistive aids, such as
crutches) and stress to other arthritic joints.
What are the chances of inhibitor development during post operative period?
If the patient has had >50 exposure days to factor, the chances of developing inhibitor is
extremely low. Presence of inhibitor should be suspected if

411
There is unexplained bleeding despite adequate factor support
The trough levels of factor are below the expected range
A repeat Bethesda assay should be done if inhibitor development is suspected.
If inhibitor is confirmed to be present, switch to bypassing agents will be required.
Special situations
Patient with confirmed high titre inhibitor
In such a scenario, adequate FEIBA or rVIIa will be required . Since these are much
more expensive and in short supply compared to factor, adequate planning will be
required.
Factor assays will not be useful to monitor whether adequate haemstasis has been
achieved or not.
Tests of global haemostasis like TEG and Rotem might give some insight, however there
are no definite guidelines to modify dose based on the results.
Patients on newer agents
Newer agents like extended half life factors are useful in achieving surgical haemostasis
with the added advantage of less frequent doses.
This could vary from once a day for factor VIII products to once in 36-48 hours for factor
IX products, depending on the half life.
However standard factor assays might give falsely elevated or low level depending on the
reagents used. Therefore the assay should use reagents proven to give accurate factor
assay for the particular extended half life product.
Non factor agents like emicizumab and fitusiran are useful for prophylaxis of bleeds, not
for surgical hemostasis. However, if some one has already been on these agents and
requires emergency surgery, the dose of factor required will be much less.
If a patient with high titre inhibitor on emicizumab or fitusiran requires surgery, the dose
of bypassing agents used should be much less than standard recommendations.
In the above scenario, close monitoring should be done to look for thrombosis.
Suggested reading

412
1. Srivastava, A, Santagostino, E, Dougall, A, et al. WFH Guidelines for the Management of
Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl
6): 1‐ 158. https://doi.org/10.1111/hae.14046
2. Srivastava A et alLow-dose intermittent factor replacement for post-operative
haemostasis in haemophilia.Haemophilia1998 ;4:799-801.
3. Mahlangu J etal Long-acting recombinant factor VIII Fc fusion protein (rFVIIIFc) for
perioperative haemostatic management in severe haemophilia
ThrombHaemost2016;116:1-8
4. Rickard KA. Guidelines for therapy and optimal dosages of coagulation factors for
treatment of bleeding and surgery in haemophilia. Haemophilia. 1995 Jan;1 Suppl
1(S1):8–13.
5. Mathews V, Viswabandya A, Baidya S, George B, Nair S, Chandy M, et al. Surgery for
Hemophilia in Developing Countries. Semin Thromb Hemost. 2005 ;31:538–43.
6. Solimeno LP, Escobar MA, Krassova S, Seremetis S. Major and Minor Classifications
for Surgery in People With Hemophilia: A Literature Review. Clin Appl Thromb 2018
;24:549–59.
Cautions to be exercised while using newer products/novel therapeutic
approaches for haemophilia
Newer therapeutics in hemophilia include the extended half-life (EHL) products and the recently
approved substitution therapy (Emicizumab) while a range of haemostatic rebalancing therapies
and gene therapy are in various stages of clinical development.
Newer Extended Half Life Products
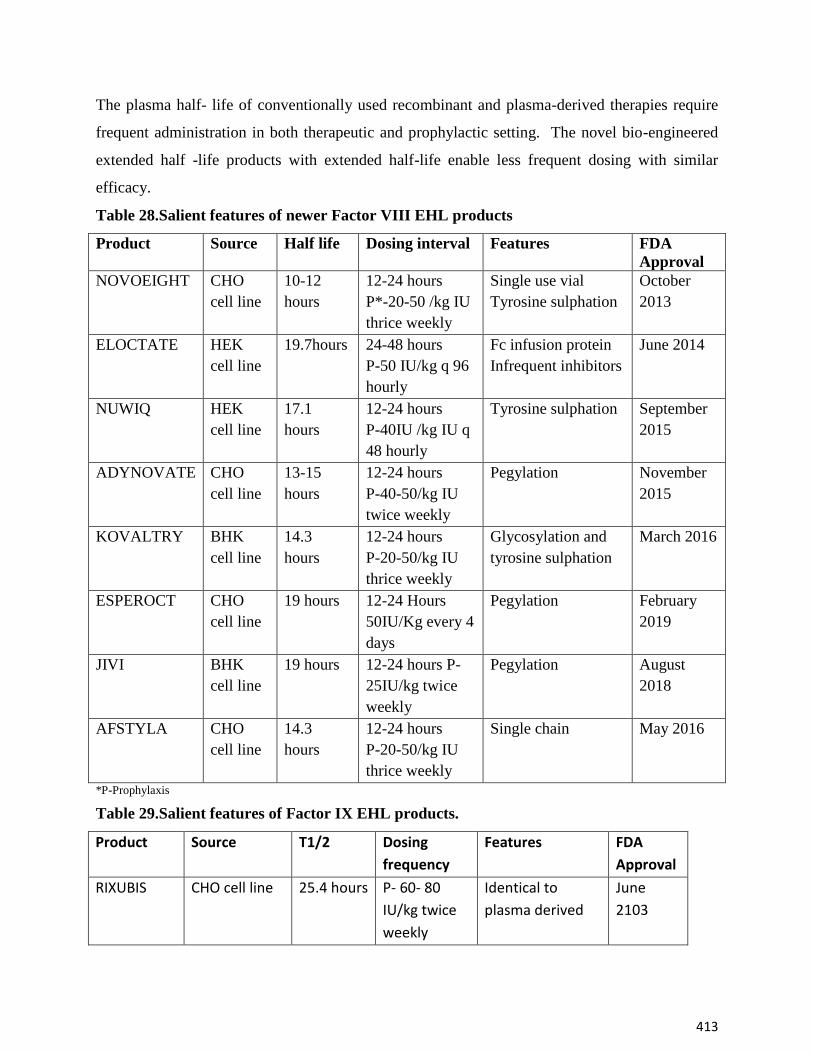
413
The plasma half- life of conventionally used recombinant and plasma-derived therapies require
frequent administration in both therapeutic and prophylactic setting. The novel bio-engineered
extended half -life products with extended half-life enable less frequent dosing with similar
efficacy.
Table 28.Salient features of newer Factor VIII EHL products
Product
Source Half life Dosing interval Features FDA
Approval
NOVOEIGHT
CHO
cell line
10-12
hours
12-24 hours
P*-20-50 /kg IU
thrice weekly
Single use vial
Tyrosine sulphation
October
2013
ELOCTATE
HEK
cell line
19.7hours 24-48 hours
P-50 IU/kg q 96
hourly
Fc infusion protein
Infrequent inhibitors
June 2014
NUWIQ
HEK
cell line
17.1
hours
12-24 hours
P-40IU /kg IU q
48 hourly
Tyrosine sulphation September
2015
ADYNOVATE
CHO
cell line
13-15
hours
12-24 hours
P-40-50/kg IU
twice weekly
Pegylation
November
2015
KOVALTRY
BHK
cell line
14.3
hours
12-24 hours
P-20-50/kg IU
thrice weekly
Glycosylation and
tyrosine sulphation
March 2016
ESPEROCT
CHO
cell line
19 hours 12-24 Hours
50IU/Kg every 4
days
Pegylation February
2019
JIVI BHK
cell line
19 hours 12-24 hours P-
25IU/kg twice
weekly
Pegylation August
2018
AFSTYLA CHO
cell line
14.3
hours
12-24 hours
P-20-50/kg IU
thrice weekly
Single chain May 2016
*P-Prophylaxis
Table 29.Salient features of Factor IX EHL products.
Product Source T1/2 Dosing
frequency
Features FDA
Approval
RIXUBIS CHO cell line 25.4 hours P- 60- 80
IU/kg twice
weekly
Identical to
plasma derived
June
2103
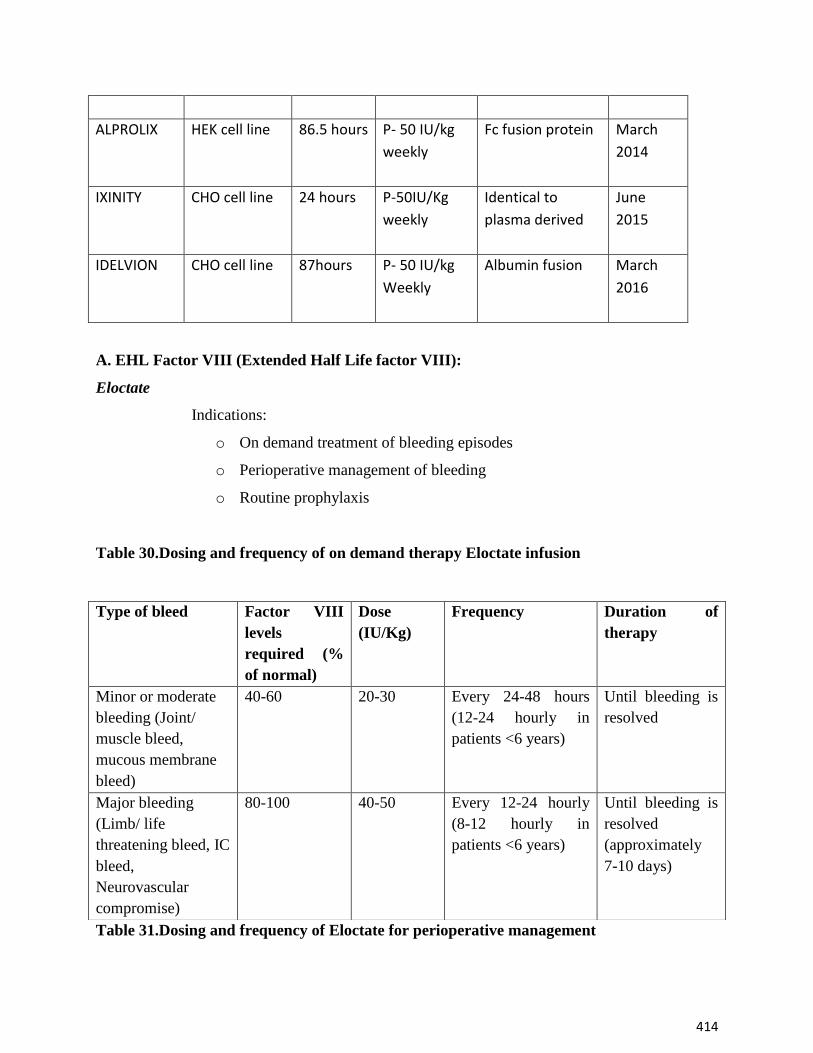
414
ALPROLIX HEK cell line 86.5 hours P- 50 IU/kg
weekly
Fc fusion protein March
2014
IXINITY CHO cell line 24 hours P-50IU/Kg
weekly
Identical to
plasma derived
June
2015
IDELVION CHO cell line 87hours P- 50 IU/kg
Weekly
Albumin fusion March
2016
A. EHL Factor VIII (Extended Half Life factor VIII):
Eloctate
Indications:
o On demand treatment of bleeding episodes
o Perioperative management of bleeding
o Routine prophylaxis
Table 30.Dosing and frequency of on demand therapy Eloctate infusion
Table 31.Dosing and frequency of Eloctate for perioperative management
Type of bleed Factor VIII
levels
required (%
of normal)
Dose
(IU/Kg)
Frequency Duration of
therapy
Minor or moderate
bleeding (Joint/
muscle bleed,
mucous membrane
bleed)
40-60 20-30 Every 24-48 hours
(12-24 hourly in
patients <6 years)
Until bleeding is
resolved
Major bleeding
(Limb/ life
threatening bleed, IC
bleed,
Neurovascular
compromise)
80-100 40-50 Every 12-24 hourly
(8-12 hourly in
patients <6 years)
Until bleeding is
resolved
(approximately
7-10 days)
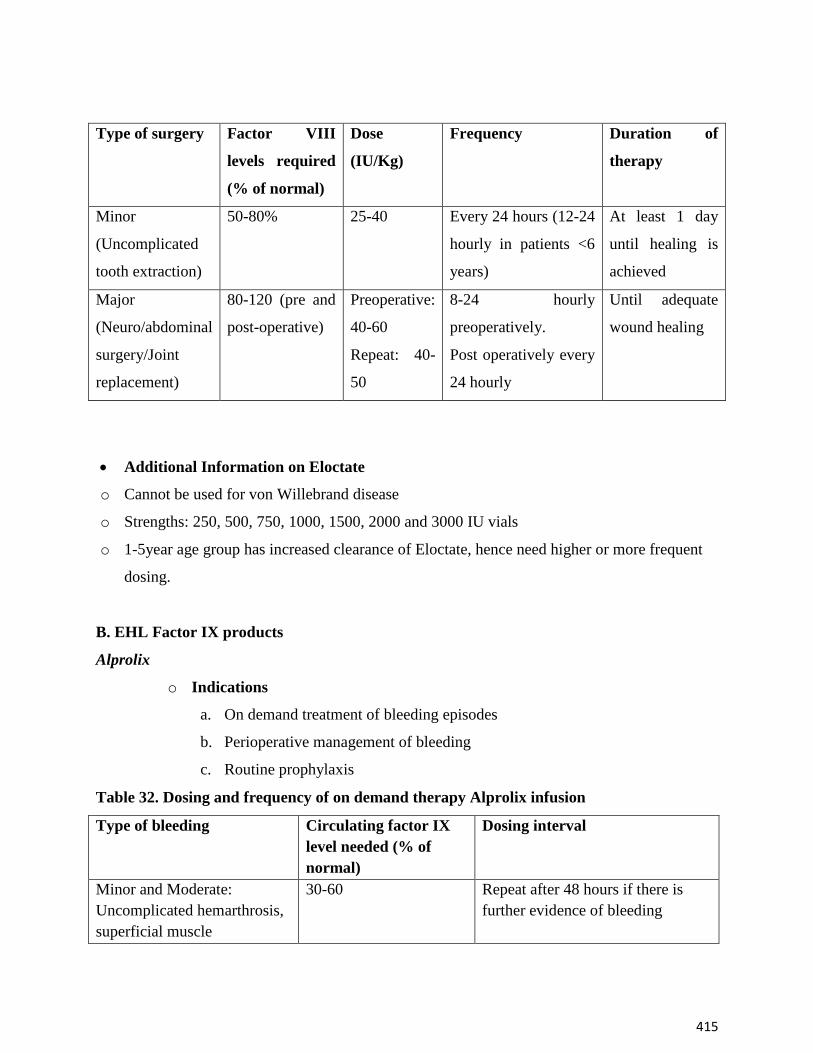
415
Type of surgery Factor VIII
levels required
(% of normal)
Dose
(IU/Kg)
Frequency Duration of
therapy
Minor
(Uncomplicated
tooth extraction)
50-80% 25-40 Every 24 hours (12-24
hourly in patients <6
years)
At least 1 day
until healing is
achieved
Major
(Neuro/abdominal
surgery/Joint
replacement)
80-120 (pre and
post-operative)
Preoperative:
40-60
Repeat: 40-
50
8-24 hourly
preoperatively.
Post operatively every
24 hourly
Until adequate
wound healing
Additional Information on Eloctate
o Cannot be used for von Willebrand disease
o Strengths: 250, 500, 750, 1000, 1500, 2000 and 3000 IU vials
o 1-5year age group has increased clearance of Eloctate, hence need higher or more frequent
dosing.
B. EHL Factor IX products
Alprolix
o Indications
a. On demand treatment of bleeding episodes
b. Perioperative management of bleeding
c. Routine prophylaxis
Table 32. Dosing and frequency of on demand therapy Alprolix infusion
Type of bleeding Circulating factor IX
level needed (% of
normal)
Dosing interval
Minor and Moderate:
Uncomplicated hemarthrosis,
superficial muscle
30-60 Repeat after 48 hours if there is
further evidence of bleeding

416
hematomas.
Major: Deep muscle
hematoma, neurovascular
compromise, CNS bleed,
Retroperitoneal bleed,
retropharyngeal bleed.
80-100 Consider a repeat dose after 6-10
hours and then every 24 hours for
the first 3 days. After 3 days
frequency may be adjusted every
48 hours or longer until bleeding
stops and healing is achieved
Additional Information on Alprolix
o Strength: 250, 500, 1000, 2000, 3000 and 4000 IU vials
o Not indicated for ITI
Cautions in laboratory monitoring of EHL products
The commonly used laboratory methods for measuring FVIII or FIX activity may not be
optimal for some modified rFVIII or
products.
The most consistent laboratory methods that can adequately measure modified rFVIII
products are based on chromogenic principles.
Any laboratory that measures factor activity in hemophilia patients should have knowledge
about (1) which replacement factor the patient is receiving and (2) whether their current
method(s) for determining factor activity provides an accurate (within 25% of target)
measurement of that particular replacement therapy.
For those laboratories that do not have the optimal measuring techniques, the treating
clinician must be informed about the potential biases (over- or underestimating factor
activity) and an alternative means of measuring accurate concentration provided.
C. Monoclonal antibody:Emicizumab (Monoclonal modified IgG4 with a bispecific antibody
structure) [ HEMILIBRA]
o Indications: Routine prophylaxis for bleeding episodes in patients with:
Hemophilia A with factor VIII inhibitors
Severe Hemophilia A without factor VIII inhibitors

417
Dosing: The total volume of Hemlibra to be injected subcutaneously is calculated as follows:
Total amount (mg) of emicizumab to be administered ÷ vial concentration (mg/mL) = total
volume of Hemlibra (mL) to be injected
Loading dose: 3mg/kg s/c once a week for first four weeks
Maintenance dose (from Week 5):
1.5mg/kg s/c once weekly OR,
3mg/kg s/c once in two weeks OR,
6mg/kg s/c once in four weeks.
Cautions:
Strengths available:
30mg/ml vial
60mg/0.4ml vial
105mg/0.7ml vial
150mg/ml vial
Treatment with bypassing agents (aPCC, rFVIIa){BPA} must be
discontinued 1 day before the start of therapy with hemlibra
Factor VIII PROPYLAXIS may be continued for the first 7 days of
therapy with hemlibra
The total volume of the dose must be injected subcutaneously only.(Not
intravenous or intramuscular)
Different hemlibra concentrations (30 mg/ml. 60 mg/ml) should not be
combined in the same syringe
A volume of >2 mL per injection should not be administered
Use of bypassing agents in patients who are on HEMILIBRA
Concomitant use of BPAs and hemlibra is a risk factor for thrombotic microangiopathy
(TMA) and thromboembolism because hemlibra increases the patient’s coagulation potential
BPAs must be discontinued 1day before the start of hemlibra therapy
If aPCC is indicated in patients on hemlibra, then the initial aPCC dose should not be
>50U/kg and laboratory monitoring is indicated (renal function, platelet count, evaluation

418
for thrombosis). The total dose of aPCC should not exceed >100 U/kg in the first 24 hours
of treatment
Recommendations on laboratory assays while on emicizumab
Although emicizumab mimics the function of activated factor VIII, it is biologically
different from activated factor VIII, having much lower affinity for factor IXa and X, not
requiring activation, and not deactivated by the protein C/S pathway.
These fundamental differences affect our interpretation of clotting assays
Laboratory monitoring of emicizumab is not required while on routine prophylactic dosing
APTT-based assays, including clot-based FVIII activity assays, should not be performed
while on emicizumab, as they will yield misleading results (ie. artifactually shortened aPTT
and elevated FVIII activity).
Chromogenic FVIII activity assays will only provide an assessment of emicizumab activity
if the assay includes all human reagents but these are not widely available. A chromogenic
FVIII assay that uses bovine reagents may be used to assay the FVIII activity of any
additional FVIII concentrate administered or endogenous FVIII.
The clot-based Bethesda assay cannot be utilized to assess FVIII inhibitor levels. However,
a bovine-reagent chromogenic-based inhibitor assay can report out FVIII inhibitor levels. If
samples are submitted to the CDC for central laboratory testing, they must be clearly
identified that the patient is on emicizumab.
Table 33.Coagulation tests affected and unaffected with Emicizumab
Results affected by Emicizumab
Results unaffected by Emicizumab
APTT - Bethesda assays (clotting-based) for
FVIII inhibitor titers
- One-stage, APTT-based, single-factor assays
- APTT-based activated protein C resistance
(APC-R)
- Activated clotting time (ACT)
-Bethesda assays (bovine chromogenic) for
FVIII inhibitor titers
- Thrombin time (TT)
- One-stage, prothrombin time (PT)-based,
single-factor assays
- Chromogenic-based single-factor assays other
than FVIII1
- Immuno-based assays (e.g. ELISA,
turbidimetric methods)
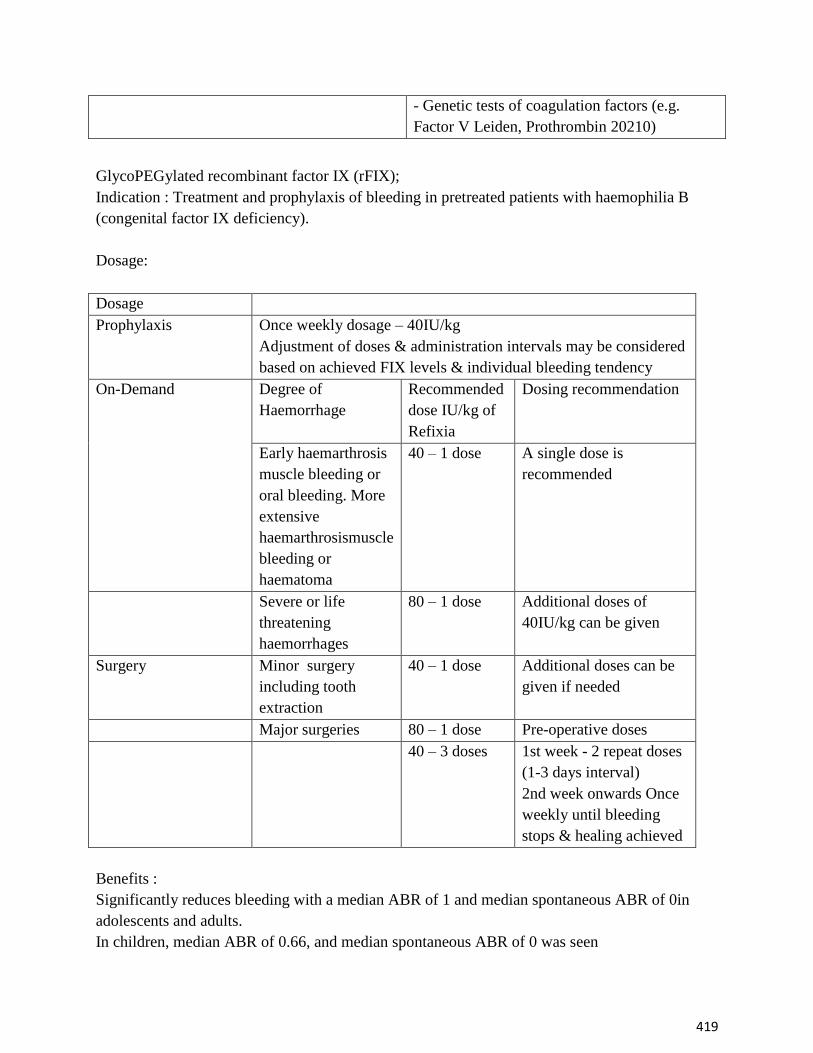
419
- Genetic tests of coagulation factors (e.g.
Factor V Leiden, Prothrombin 20210)
GlycoPEGylated recombinant factor IX (rFIX);
Indication : Treatment and prophylaxis of bleeding in pretreated patients with haemophilia B
(congenital factor IX deficiency).
Dosage:
Dosage
Prophylaxis Once weekly dosage – 40IU/kg
Adjustment of doses & administration intervals may be considered
based on achieved FIX levels & individual bleeding tendency
On-Demand Degree of
Haemorrhage
Recommended
dose IU/kg of
Refixia
Dosing recommendation
Early haemarthrosis
muscle bleeding or
oral bleeding. More
extensive
haemarthrosismuscle
bleeding or
haematoma
40 – 1 dose A single dose is
recommended
Severe or life
threatening
haemorrhages
80 – 1 dose Additional doses of
40IU/kg can be given
Surgery Minor surgery
including tooth
extraction
40 – 1 dose Additional doses can be
given if needed
Major surgeries 80 – 1 dose Pre-operative doses
40 – 3 doses 1st week - 2 repeat doses
(1-3 days interval)
2nd week onwards Once
weekly until bleeding
stops & healing achieved
Benefits :
Significantly reduces bleeding with a median ABR of 1 and median spontaneous ABR of 0in
adolescents and adults.
In children, median ABR of 0.66, and median spontaneous ABR of 0 was seen

420
Has the potential to prevent further bleeds in target joints by showing 100% resolution of target
joints at end of treatment
Less frequent injections:98.4% of bleeding episodes were controlled by 1 injection in
adolescents and adults and82% of bleedsin children.
Half-lifethat is five times longer than existing treatment meaning that Refixia® has the potential
to be incorporated into a once-weekly dosing schedule.
In patients undergoing surgery, a single preoperative dose provided effective haemostatic
coverage, and no patient required additional doses on the day of surgery.
Have a favourable safety profile with no inhibitorsor thromboembolic events or other adverse
effects
Significant improvement in the quality of life
Suggested reading
Weyand AC, Pipe SW. New therapies for hemophilia. Blood 2019 ;133:389 - 98.
Kitchen S, Tiefenbacher S, Gosselin R. Factor Activity Assays for Monitoring Extended
Half-Life FVIII and Factor IX Replacement Therapies. Semin Thromb Hemost. 2017
;43:331–7.

421
A Comprehensive model for conservative management in hemophilia
Hemophilia is known to cause bleeds in the joints and muscles, or bleeds in various systems of
the body-the brain, the renal system, the gastro-intestinal system. Bleeds may be spontaneous
bleed or due to injury. Every systemic bleed or bleed in the muscle or joint if not treated
appropriately and on time can lead to complications and compromise functioning.The
musculoskeletal [MSK] problems due to hemophilia are seen in the form of joint and muscle,
kinetic and kinematic dysfunction. The late effects on this system could be seen as-early joint
disintegration, joint deformities, loss in muscle mass, compromised muscle strength,
neurovascular complications, and pseudo tumors; ultimately a jeopardized function and quality
of life.
The PWH who do not receive adequate and consistent CFC have higher rate of mortality and
disability, most of the PWH suffer pain. In parts of the world where there is no or insufficient
factor supply the conservative management seems to be more affordable and feasible, and it is
this management that has to be have an evidence and scientific basis for management.Looking
at the international protocol of management of a bleed it is imperative to infuse factor in the
golden period after the bleed, that is ASAP, soon as the aura is experienced by the PWH, or
signs of bleed are noticed by the caregiver of the PWH in children who are very young.
Those on prophylaxis are seen to have lesser incidence of bleeds and an improved QoL versus
who are not on regular prophylaxis. Yet, there are incidences when the PWH on prophylaxis do
have bleeds due to one or the other reasons and in both [patients treated and untreated with
prophylaxis] the groups, issue remains managing an acute bleed. Especially for the group of
patients who are being managed on demand basis and have recurrent bleeds, immediate and
appropriate conservative management is essential.

422
I] Early infusion of factor concentrate: Thisis the most important aspect, however when not
possible to infuse, the managementmay start with step II.
Steps in management of an acute bleed when factor availability is not immediate or not
possible:
ii] Rest the joint and muscle
For joint bleeds, support the joint with a splint or rest the joint on a soft cushioned roll,
preferably in a position that relieves pain and stretch on the joint and then in the anti-deforming
positions, that is in the direction of extension range. This may be true depending on the site of
the bleed. For a flexed hip –knee position rest the knee on a roll under the knee so as to support
the hip in a pain free position.
In case of soft tissue bleeds ( muscle bleeds, compartment bleeds) which are seen on the body
surface, consider using a splint to the adjacent joint/s if deformed and splint the muscle
surface using a well-padded splint, covering a maximum area of bleed.
Infuse factor within the golden period
Before the bleed takes a large space
in the joint.
Before the joint temperature
Increases.
Soon as joint feels stiff or there is
difficulty in moving the joint.
Before the body part assumes a position
deviating from the normal alignment.
Benefits of early infusion
Reduces the chance of increase in
the bleed size and volume.
Enhances early absorption of the
bleed in the period following the
bleed.
Reduces pain.
Helps early mobility.
Helps early recovery and enhances
the healing process.
Figure 12: Resting in splint

423
Splints should be cost effective and
reusable.
Angle of the splint should be adjustable.
Depending on the site of bleed decision
to support a large surface area can be
made.
Consider splinting with a rigid material, but a soft and accommodative padding on the
surface that is in contact with the skin.
Splinting may be done using the three-point pressure principle, or may be simply
used as surface contact splints, with respect to the need.
Immobilization in splint helps to control unnecessary movements may prevent the clot
from dislodging.
Features of splinting
The joints have a tendency to recurrent bleeds, they can be used at the time of each
bleed.
With gradual resumption of the normal angle of joints, the splint angle should be
modifiable.
Rigid splints offer better rest; soft padding help to accommodate the change in joint
size in the splint, subsequent to the factor infusion.
The principle of three-point pressure can be more relevant to established anterior-
posterior deformities, where the intention is correction. Surface splints are options in
cases where there are muscle bleeds, or no joint static angulations observed.

424
III] Icing: Icing is the next step to be executed. Icing is a controversial approach, as evidence
proves thatsome patients are not able to tolerate icing due to aggravation of pain, some have
even reported about an increase in the size of the bleed. It is a myth that icing helps to clot
theblood, however it does not.
Procedure of icing
1.Do not use ice directly on the skin.
2.Crush and wrap ice in a thin cloth, cover the area to be cooled with a cloth, before
icing. Use ice cold water if icing cannot be tolerated.
3.Watch for skin color while icing.
4.Do not cool the part for more than 20 mins. Cold flex bandages could be preferred to,
if direct icing is not tolerated by the patient.
5. Use ice cold water alternated with normal room temperature water if cold water is not
tolerated. [Dose: 3 mins ice water/3 mins water at room temperature, repeat the procedure 5-
6 counts end with ice water].
6. Repeat the process two to three times a day if well tolerated.

425
Benefits of icing
1.It helps to cool the soft tissues and reduces the metabolic rate of tissues. Thus, reduces
the accumulation of the P ‘Factor at the site.
2.Icing can reduce pain in patients who tolerate it well, help to relax the muscle and
reduce the pain - spasm effect on the muscle
3.In advanced cases of arthropathy icing can aggravate pain, than reduce pain.
4.Watch for abrasions on the skin before soaking the wound with water due to condensation.

426
Figure 13. Use of ice cold water . If ice cubes are not tolerated cover skin with a thin cloth
or plastic while icing
IV] Compression: Compression can help better in immobilizing the part, application of
compression canalleviate pain, and reduce the swelling in the soft tissues. Do not continue with
tight, elastic, compression bandages for long after the bleed, especially over the muscle mass.
Constant and tight compression around the joint and soft tissues adds to aggravate muscle
wasting, habitual joint positioning and dependence.
How and when to use compression Benefits and ill effects
1.Continued compression under pressure
can cause muscle wasting and makes
muscle weak in the long run, due to
disuse atrophy.
2.Padded and protective cuffs should be a
better option to tight elastic wraps or tubi-
grips. As the padded cuffs allow optimal
contraction of the muscle fibers in
functional ranges, do not hold the joint and
muscle in static positions.
3.There is equal distribution of pressures
around the joint and on the muscle, in the
padded cuffs.
4. Padded cuffs provide protection and
proprioceptive input while the joint is
used.
1.Helps to support the joint during an
acute bleed.
2.Compression applied to the joint
helps to control the edema; in the soft
tissue helps reduce edema.
3.Compression helps to alleviate pain in
the acute stage.
4.It is best to apply compression with a
supporting splint as described above [ in
splinting], so as to avoid any deleterious
effects of external pressures, added to the
internal pressure caused by the bleed.
5.Do not continue to use compressionbandages
continuously after the bleed and pain subside.

427
IV] Elevation and Exercises: Elevating a joint to reduce the size of bleed may not be a good
option, especially if the bleed in the joint is in the intra-articular i.e. in the within the articular
capsule and not in the extra-articular space. For the muscle bleed elevation may be a good option
provided it is in the peripheral musculature, in deep muscle bleeds elevation may not be a
possible e.g.in case of the psoas bleed. In joint bleeds if there is effusion in the soft tissues
around the joint elevation can help reducing the swelling. Take care to keep the elevated part
above the level of heart when in supine lying.The `E’ also stands for exercises following a bleed;
Exercises should be started after appropriate and adequate amount of factor infusion, and
depending on the patients response to factor infusion. Exercises should be done in the expert
hands and under expert supervision of an MSK expert who may be a trained .
COMPRESSION
Figure 14: Compression.
Exercises have to be done after appropriate replacement of the CFC, after the pain subsides, and
after the joint movement can be initiated with support and assistance to the limb.
Initially focus on static isometric exercises in order to allow muscle fiber contractions with the
objective to prevent disuse atrophy.
Focus on ROM exercises so as to allow joint ranges to return to pre-episodic levels.
Exercises have to continue progressively to improve the muscle mass and strength.
Train the care givers to do simple exercises with hands on training so as to continue with home
programs along with home based prophylaxis.
Timely correction of compensated postures, exercise the agonist and antagonists to establish
symmetric balance of musculature.
Core muscle training, proprioceptive training to lower extremity joints for acquiring a near
normal gait pattern, fall prevention is an essential part of rehabilitation.
Weight bearing, static isometric contractions exercises to prevent bone demineralization are
equally important.
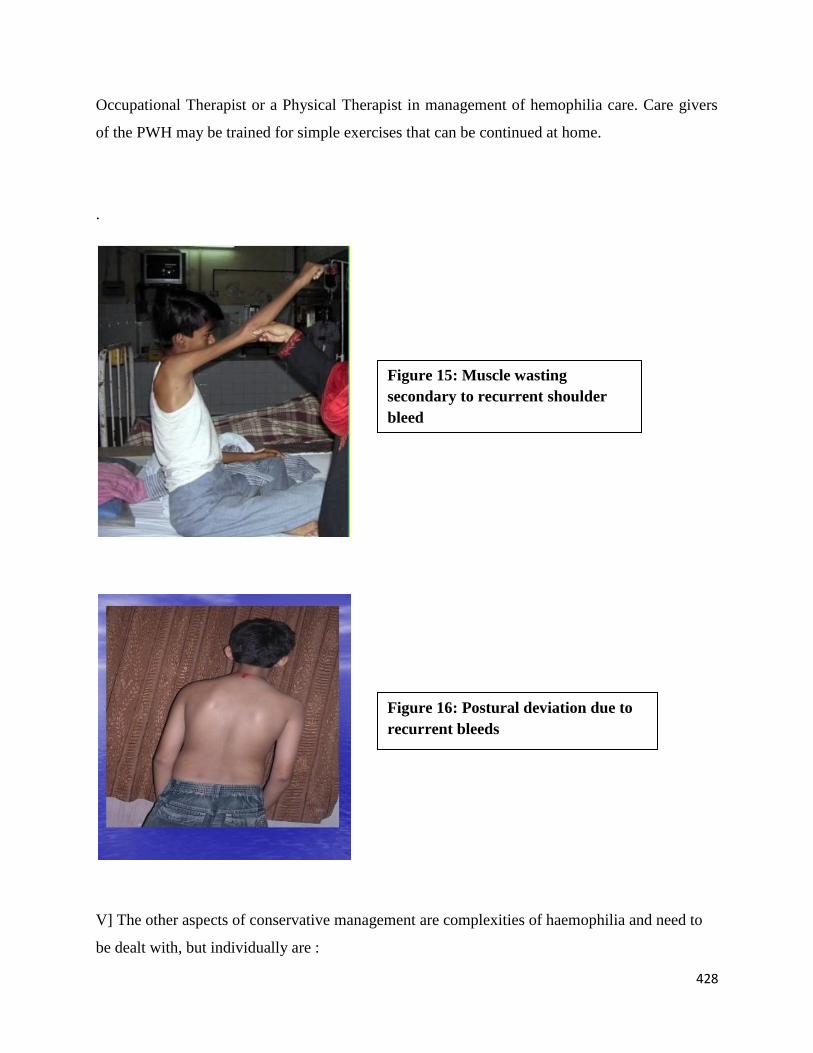
428
Occupational Therapist or a Physical Therapist in management of hemophilia care. Care givers
of the PWH may be trained for simple exercises that can be continued at home.
.
V] The other aspects of conservative management are complexities of haemophilia and need to
be dealt with, but individually are :
Figure 15: Muscle wasting
secondary to recurrent shoulder
bleed
Figure 16: Postural deviation due to
recurrent bleeds

429
Management of synovitis.
Management of patients with inhibitors.
Management of pseudo tumors and established
hematomas.
Management of psoas bleeds.
Management of soft tissue bleeds with residual
complications. Pain management.
Assessment tools for QoL.
Psycho-social aspects of the PWH related to the physical
limitations. Therapeutic intervention at different stages of the
arthropathy.
Conservative management of contractures and
deformities. Post joint replacement therapeutic
intervention.
Applicability of physical agent modalities in
hemophilia.
Compensatory devices for upper and lower extremity
adaptations. Clinical assessments and approaches to joint
mobilization.
Suggested Reading
Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A et al; Treatment
Guidelines Working Group on Behalf of The World Federation OfHemophilia. Guidelines for
the management of hemophilia.Haemophilia. 2013 ;19:e1-47

430
Psychosocial care and support: Training curriculum for Psychologists/Social
Workers/Nurses working with patients with bleeding disorders
Psychosocial impact in hemophilia is a lifelong condition affecting the PWH as well as the
immediate family members, friends and relatives. It affects them socially, physically and most
important emotionally/psychologically. Hemophilia is a not very common condition and as a
result, a large number of people are unaware about it. Specifically, Hemophilia population's need
of psychosocial support is underestimated due to myths, stigma and lack of knowledge.
However, psychosocial care and support in hemophilia are interventions and methods that
enhance the ability of children, families with Hemophilia, and communities to cope, in their own
context, and to achieve personal and social well-being; enabling PWH to experience love,
protection, and support that allows them to have a sense of self-worth and belonging with better
quality of life.Such interventions and methods are essential in order for children to learn, develop
life skills, participate actively, and have faith for the future.
Principles of Psychosocial Support (PSS) Program
There are different effective and sustainable ways to offer psychosocial support to PWH and his
family. However, the main principles of PSS program are not only important but also extremely
helpful for PWH in the following way:
* A Psychosocial Program as an integral part of the Comprehensive Care' program of the
hemophilia care centre plays an important role in taking in to account the developmental stages
of the child.
* The program acknowledges the critical role that potential care-givers play in the well-being of a
PWH and his family.
* The program offers continuity upon which the PWH can rely on. Follow up, support and
supervision are all of critical importance.

431
* Program focuses on empowering the PWH and his family, improving their self esteem,resilience
and enhancing their life skills.
* Under the program, assistance is also provided to enable the PWH and the family members to
ask for help when they need it and know whom to approach. This also includes putting in place a
referral system with qualified professionals.
* It also provides opportunities to promote the formation of support groups and encourage
interaction amongst them.
* The program educates and sensitizes the community in order to effectively seek participation for
help and support.
* The program identifies and trains a group of volunteers/lay counsellors from the affected
population and provide them back-up support in the form of a mental health expert.
Psychosocial tools and techniques
There can be various tools and techniques, such as psycho-education, counselling, groups for
children support, youth and women, relaxation techniques, behaviour management, stress
management, building coping skills and resilience life Skill Education, goal setting, self-esteem,
confidence building, Loss and grief etc. Each of these tools will be dealt in detail depending on
the requirement.
The Psychosocial Support Training Curriculum
This curriculum is designed to improve quality of care for people with bleeding disorders and to
increase their quality of life by making psychosocial care an integral part of a multi-disciplinary
approach to healthcare.
Aims
Raise awareness of psychosocial issues that affect people with bleeding disorders and
their families
Advocate nationally for comprehensive care
Ensure psychosocial participation in multidisciplinary hemophilia care teams
Provide support, information, and contacts for psychosocial workers
Objectives of Psychosocial Curriculum
The objectives of the Psychosocial Training Curriculum are:
To offer important psychosocial educational materials (sessions) for teaching and
training purposes

432
To design “user friendly” information curriculum that can be adapted culturally
To provide assistance to the access and understanding of the material
The Psychosocial Training Curriculum Modules
The modules are teaching tools for educating the psychosocial care professionals on the topics
provided in this Psychosocial Curriculum. The modules are identified and selected as important
topics for patients and families.
The Individual section of each module contains learning objectives, a short description, and
suggested supporting resources if any; to provide to participants.
Learning Objectives
Each module in the Psychosocial Curriculum has specific learning objectives that we hope to be
accomplished by the end of the module. It is important for the trainer to teach to the learning
objectives allowing for cultural modifications and any material adaptations to the targeted
audiences.
Interactive Element
It is helpful and valuable to the sessions by adding an interactive component that can be used in
the session presentation. The interactive elements are outlined in the sessions and are available
for the presenter to access. Interactive learning is very effective as a tool for teaching. One can
adapt the interactive piece as needed to gain the best learning experience for the audience.
Following are the important topics that forms the training curriculum under the section of
Psychosocial Training Curriculum:
Team Approach to Hemophilia Care
Coping with Chronic Pain
Assessing and enhancing coping strategies (resilience)
Adherence
Education and Employment
Hemophilia: Psychosocial impact on Carriers
Genetic Counselling -Basic level
Sexuality through the Life Stages
Coping with complications – HCV, HIV, Inhibitors, & Joint Disease
Disclosure

433
Women and Teenage Girls with Bleeding Disorders
Quality of Life Assessment Tools
Each module has been described as follows:
Module 1: Team Approach to Hemophilia Care
Estimated Duration
The presentation length is 30 minutes, allow additional time for questions
Description
The module discusses the importance of Comprehensive Care in the treatment of Hemophilia
and bleeding disorders with an emphasis on the composition of the care team. It describes the
role of Social Workers/psychologists/Nurses in Hemophilia Care and the various activities of
HTC and the foundation in general with an example to how to make a Psychosocial Action Plan
for their respective work places.
Learning Objectives
At the end of this session, participants will be able to:
Understand the comprehensive care model in the treatment of hemophilia
Discuss psychosocial care in hemophilia
Understand the role of social workers/psychologists/nurses in hemophilia care
Recall the goals of the social worker/psychologist/Nurses
Examine an example of a psychosocial action plan
Module 2: Coping with Chronic Pain
Estimated Duration
The presentation length is 30 minutes, allow additional time for questions.
Description
This module provides literature-based information which explains the physical and psychological
components of pain and offers practical suggestions on how to cope with chronic pain.
Topics covered include: the role of exercise, aids to relaxation, recognizing stress, depression,
physical and psychological dependency, complementary therapies, and tips for the family and
peer support.
Learning Objectives
At the end of this session, participants will be able to:
Describe the physical and psychological components
of pain

434
Understand how to cope physically and psychologically with chronic pain
Module 3: Quality of Life Assessment Tools
Estimated Duration
Presentation length for the introductory slide deck is 30 minutes, allow additional time for
questions.
Presentation length for the advanced slide deck is 45 minutes, allow additional time for
questions.
Description
There are two versions one is “introductory” (30 minute presentation) and the other is
“advanced” (45 minute presentation). Considerable flexibility is built into both.
Both the introductory and advanced sections introduces the audience to complications, from
bleeding and/or treatment, patients with hemophilia (PWH) experience. These complications
include: joint disease, inhibitors, and past exposure to viral infections prior the availability of
purified factor products. The potential impact of each complication in multiple physical and
mental health Quality of Life (QoL) domains is discussed. A higher level of detail is provided in
the advanced deck, especially related to social and psychosocial issues.
Learning Objectives
At the end of this session, participants will be able to:
Enhance participant understanding of the importance of quality of life (QoL) assessment
tools
Demonstrate the difference between types of outcome measures, with a specific emphasis
on patient-reported outcome measures
Enhance participant understanding of the dimensions and determinants of health-related
(HR) QoL assessment
Raise participant awareness of types of HRQoL questionnaires, especially hemophilia-
related QoL instruments
Demonstrate criteria for selecting an instrument
Highlight advantages of using multipronged assessment tools
Identify key QoL influences in the bleeding disorders community

435
Module 4: Assessing and Enhancing Coping Strategies (Resilience)
Estimated Duration
The presentation length is 30 minutes, allow additional time for questions
Description
This module discusses coping strategies both positive and maladaptive. It will provide some
examples and tips to for individuals, families and the community as a whole on coping skills.
The concept of resilience is discussed as a path to empowerment and a positive coping skill.
Learning Objectives
At the end of this session, participants will be able to:
Identify and define types of coping strategies
Describe stressors
Recognize maladaptive coping
Irrational belief systems
Describe positive coping skills
Adaptive coping skills
Provide tips for patients and families on coping within the larger community and
extended family
Recall psychosocial counselling options
Individual versus group counselling
Understand resilience as a path to empowerment
Recall assessment tools
Community psychosocial support
Module 5: Adherence
Estimated Duration
The presentation length is 45 minutes, allow additional time for questions
Description
This module provides literature-based information related to adherence in persons with
hemophilia (PWH). Potential barriers, primary motivators, and strategies to enhance adherence
are key components of this module. To enhance audience participation, assess baseline
knowledge, and to gain insight into adherence-related issues in the country, there are questions to
the participants.

436
Learning Objectives
At the end of this session, participants will be able to:
Understand issues related to adherence
Assess and address potential barriers to adherence for both providers and
patients/families
Understand the potential impact of cultural influences on adherence
Identify tools and psychosocial strategies to assess and enhance adherence
Module 6:Education and Employment
Estimated Duration
The presentation length is 60 minutes it is likely that a longer time will be necessary depending
on the Interactive elements, allow additional time for questions
Description
Topics covered in the Education section of the Module include: challenges, disclosure, educating
schools, Information Sheets for schools, the physical education class, after a bleed, the student
with arthritis, dealing with absences, overprotectiveness and bullying. Topics covered in the
Career Planning and Employment section of the Module include: career planning, job seeking,
developing a Curriculum Vitae, interview skills, networking, volunteering and work experience.
Within the education section of this Module, a Group Work interactive element is included in
order for participants to identify the schooling and education challenges, issues and problems for
two age groups – those 4 – 12 years and those aged 12 – 18 years within their local context.
Within the employment section of this Module, there is an option to interview a person with a
bleeding disorder from the local community to speak about their experience with employment.
Learning Objectives
At the end of this session, participants will be able to:
Understand the importance of education and employment for those with bleeding
disorders
Describe the challenges surrounding education for those with bleeding disorders
Identify strategies to promote participation in education for those with bleeding disorders
Describe the issues and challenges for those with bleeding disorders with regards to
employment

437
Identify strategies to assist those with bleeding disorders in career planning, job seeking
and employment
Module 7: Psychosocial Impact on Carriers
Estimated Duration
The presentation length is 60 to 90 minutes, allow additional time for questions
Description
The module on psychosocial impact on carriers is a very interactive module which defines the
types of Carriers and what it means. Then it discusses the Issues regarding testing and when to
test with example from developed and developing countries. It has a component of group work
which helps the individual to identify the centre specific issues and discussion thereof. The
module also discusses the stigma, emotional and concerns on being a carrier. It also touches
upon the Genetic Counselling and availability of reproductive choices with the factors affecting
reproductive choices & informed decision and cope mechanism to deal with the situation.
Learning Objectives
At the end of this session, participants will be able to understand:
Types of carriers and what this means
Issues regarding testing and when to test
Psychosocial issues and concerns about being a carrier
Genetic counselling and availability of reproductive choices
Factors affecting reproductive choices and informed decision
Coping with the situation
Module 8: Genetic Counselling
Estimated Duration
The presentation length is 45 minutes, allow additional time for questions
Description
The genetic counselling module is in two parts Basic and Advanced. The basic module defines
the genetic counselling and the role of genetic counsellor vis a vis psychosocial worker. It gives
the information about the inheritance of haemophilia and basic information relevant to

438
counselling a patient/family about hemophilia. It also makes the participants recognize the
cohesiveness between genetics and other health care providers within the HTC.
The module sheds light on how genetic counselling should take into account a patient’s
experience and perception, as well as the influence of social, cultural, and religious contexts,
whatever the level of facilities and services.
The advance module talks about how to draw the family tree and its interpretations as well as
availability of reproductive choices to the client.
Learning Objectives
At the end of this session, participants will be able to:
Understand the role of a genetic counsellor in the HTC
Recognize cohesiveness between genetic counselors and other health care providers
within the HTC
Identify key topics relevant to counselling a patient/family about hemophilia including
inheritance, factors that influence decision making, and family dynamics
Module 9A: Life Stages
Estimated Duration
The presentation length is 30 minutes, allow additional time for questions.
Description
This module provides literature-based information which explain the key issues, objectives and
strategies of each developmental stage from birth to 65 years. Topics covered include: -
Parents coping and normalizing, building trust, acceptance of hemophilia and emotional
reactions;
Child’s feelings, confidence and coping, understanding treatment and acceptance;
Teenager - learning to self-infuse, independence and responsibility, compliance issues, education
and career planning;
Young adult – transition, working, dating and relationships, genetic counselling;
Middle adulthood – career and family development, peer support, dealing with limitations;
Maturity – slowing down, age of wisdom.
There are notes for the trainers under some slides.

439
Learning Objectives
At the end of this session, participants will be able to understand the key issues and strategies for
each stage of psychosocial development
Module 9B: Sexuality
Estimated Duration
The presentation length is 20 minutes; allow additional time for questions.
Description
This module provides literature-based information which explain the key issues and strategies
regarding sexuality throughout the life stages. The trainer’s local knowledge of cultural
sensitivity, can decide which topics to include or exclude.
Topics covered include: early years, gratifying body experiences, attitudes to masturbation,
puberty and adolescence, body image, communication, stress and the libido, sexual intercourse
and bleeds, effects on sexual desire, sexual expression and relationships.
Learning Objectives
At the end of this session, participants will be able to understand key issues and strategies
regarding sexuality through the life stages
Module 10: Coping with Complications – HCV, HIV, Inhibitors and Joint Disease
Estimated Duration
The presentation length is 30 minutes (Introductory), allow additional time for questions. The
presentation length is 45 minutes (Advanced), allow additional time for questions.
Description
There are two versions of this module, one is “introductory” and the other is “advanced”.
Considerable flexibility is built into both. The presenter can opt to exclude any complication(s)
due to time constraints or to lack of relevance for the particular audience, without impacting the
flow of the presentation.
Both the introductory and advanced sections introduce the audience to complications, from
bleeding and/or treatment, patients with hemophilia (PWH) experience. These complications
include: joint disease, inhibitors, and past exposure to viral infections prior the availability of
purified factor products. The potential impact of each complication in multiple physical and
mental health Quality of Life (QoL) domains is discussed. A higher level of detail is provided in
the advanced deck, especially related to social and psychosocial issues.

440
Learning Objectives
At the end of this session, participants will be able to:
Identify/list hemophilia-related complications
Describe at least three ways these complications may impact the psychosocial
functioning of patients/families/caregivers
Appreciate how complex the psychosocial impact of these complications can be for
patients/families
Identify and describe psychosocial intervention strategies
Module 11: Disclosure
Estimated duration
The presentation length is 45 minutes, allow additional time for questions.
Description
This is a two-part session on decision making around disclosing a diagnosis of hemophilia,
including who, when, where, and why to tell.
Learning Objectives
At the end of this session, participants will be able to:
Appreciate the complexity of issues related to disclosure
Understand disclosure within the context of the participants’ culture
Assess and address potential risks and benefits to disclosure
Identify three psychosocial strategies to facilitate disclosure decision making
Module 12: Women and Teenage Girls with Bleeding Disorders
Estimated Duration
The presentation length is 20 minutes, allow additional time for questions.
Description
This module provides literature-based information which explain the key issues for women and
teenage girls with bleeding disorders. The trainer’s local knowledge of cultural sensitivity, can
decide which topics to include or exclude.
Topics covered include: quality of life, genetic counselling, carrier testing, emotional reactions
of carriers, coping with emotions, tips for teenage girls, living positively with bleeding disorders.

441
Learning Objectives
At the end of this session, participants will be able to:
Understand key issues for women and teenage girls with bleeding disorders
The above modules can be used, adapted depending upon the audience, their levels and the
training duration.
Evaluation process
Conducting an evaluation process for instance, “ Evaluation Form” is a key element to the
success and outcomes of the program. Without collecting feedback or an evaluation, it is very
difficult to have information that is useful to the facilitating/training team to see if they have
accomplished their goals and for future improvement.

442
Blood Collection Transportation Vans (BCTV)
IICCHHHH
((IInntteeggrraatteedd CCeenntteerrss ffoorr
HHeemmoogglloobbiinnooppaatthhiieess &&
HHeemmoopphhiilliiaa))
IT solution to connect, digitize and streamline the
blood banks
Training for Hemoglobinopathies
and Hemophilia
Related Documents











