TopHat 2014.10.01 Mi-kyoung Seo

TopHat 2014.10.01 Mi-kyoung Seo. Today’s paper..TopHat Cole Trapnell at the University of Washington's Department of Genome Sciences Steven Salzberg Center.
Jan 01, 2016
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

TopHat
2014.10.01Mi-kyoung Seo

Today’s paper..TopHat
Cole Trapnellat the University of Washington's Department of Genome Sciences
Steven Salzberg Center for Computational Biology at Johns Hopkins University
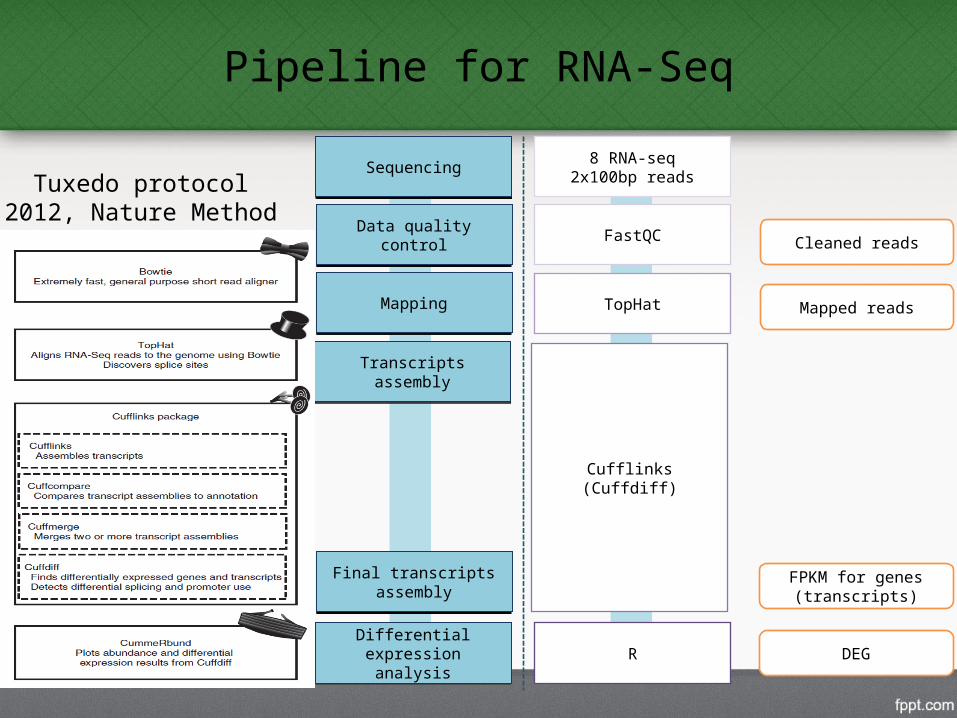
Pipeline for RNA-Seq
TopHat
FastQCData quality controlData quality control
MappingMapping
Differential expression analysis
Differential expression analysis
Cufflinks(Cuffdiff)
FPKM for genes (transcripts)
Cleaned reads
Mapped reads
R DEG
Transcripts assemblyTranscripts assembly
Final transcripts assembly
Final transcripts assembly
8 RNA-seq2x100bp readsSequencingSequencing
Tuxedo protocol2012, Nature Method

Software information
• Purpose– A spliced read mapper for RNA-Seq
• TopHat has a number of parameters and options, and their default values are tuned for processing mammalian RNA-Seq reads
• Software URL– http://ccb.jhu.edu/software/tophat/index.shtml
• Category– Aligner
• License– Open source and freely available under the Artistic license

Overview of RNA-Seq
5Garber, M., et al. (2011), Nature Methods, 8(6), 469–477.Microarray or cDNA/EST sequencing RNA-seq

QPALMA
• De Bona et al., 2008• Optimal spliced alignments of short sequence reads• Q-PALMA is based on a machine learning approach, in which
data from previously known splice junctions are used to train the software.
• Initial mapping phase uses Vmatch (Abouelhoda et al., 2004)• Vmatch is a flexible, fast aligner, but because it is not
designed to map short reads on machines with small main memories, it is substantially slower than other specialized short-read mappers.– Vmatch: 180,000 reads per CPU hour
– TopHat: 2.2 million reads per CPU hour

The Tophat pipeline
1. Find Exons- Mapping using BowtieMapped & IUM reads- Assemble exons using MAQPutative exons (islands)- Extend exons by 45bp- Gap
2. Find Splice Junctions

Mapping: Bowtie
TopHat uses Bowtie in order to initially map all reads to the reference genome while collecting all the unmapped reads for further analysis
Bowtie is reporting reads with no more than a few mismatches (default: 2) within s bp from the 5' end and the 3' end may have errors based on Phred-Quality-Weighted Hamming Distance (s default: 28)
TopHat allows reads from bowtie that map up to 10 locations (multireads)
read
s bp
5' 3'

Assembly: MAQ
Exon 2 Exon 3Exon 1
read
read
Exon 2Exon 1Transcript 1
Gene(DNA or reference)
read read
GU AG
read
read
readread
readread
read
readread
readread
island
consensus
read
read
Putative exons
IUM reads

Identification of read spanning junction
Exon 2 Exon 3Exon 1
read
GT AG AG AG AG
70 < potential Intron length < 20,000bp
IUM reads

Identification of read spanning junction
ERAGNE (annotation based pipeline)(Mortazavi et al., 2008)
Each island spanning coordinates i to jdm: depth of coverage at coordinate mn: the length of the reference genome
B3gat1 gene

Seed and extend strategy
To find reads that span junction
2K-mer seed

Running Operations
• Input– For Paired-end read, Read1.fastq, Read2.fastq– genome (Genome index by bowtie-build)– Option: genes.gtf (Gene annotation)
• Command– Usage: tophat [options]* <genome_index_base>
<reads1_1[,...,readsN_1]> [reads1_2,...readsN_2]– tophat –p 4 –o output/Normal –G genes.gtf BowtieIndex/genome
read1.fastq read2.fastq --library-type fr-unstranded• TopHat Output
– accepted_hits.bam– unmapped.bam– junctions.bed– insertion.bed– deletion.bed
Usage: tophat [options]* <genome_index_base> <reads1_1[,...,readsN_1]> [reads1_2,...readsN_2]$ tophat –p 4 –o output/Normal –r 100 –G genes.gtf BowtieIndex/genome read1.fastq read2.fastq

Input Parameters
• $ tophat [optioins] <bowtie_index_base> <read1_1> <read1_2>
• -o: output dir• -p: use this many threads to align reads [default: 1]• -r: this is the expected (mean) inner distance between mate
pairs [default: 50bp]– For, example, for paired end runs with fragments selected
at 300bp, where each end is 50bp, you should set -r to be 200. The default is 50bp
• -G: geneset
$ tophat –p 4 –o output/Normal –r 100 –G genes.gtf BowtieIndex/genome read1.fastq read2.fastq

Tophat output by IGV

Discussion
• Tophat2 default option 확인– r: 실험에 따라 다름– Bowtie2 for mapping
Related Documents











