THIS IS UNFPA WHO/UNFPA Prequalification Programme Guidance for RH Devices (Condoms and IUDs)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

THIS IS UNFPA
WHO/UNFPA Prequalification Programme
Guidance for RH Devices (Condoms and IUDs)
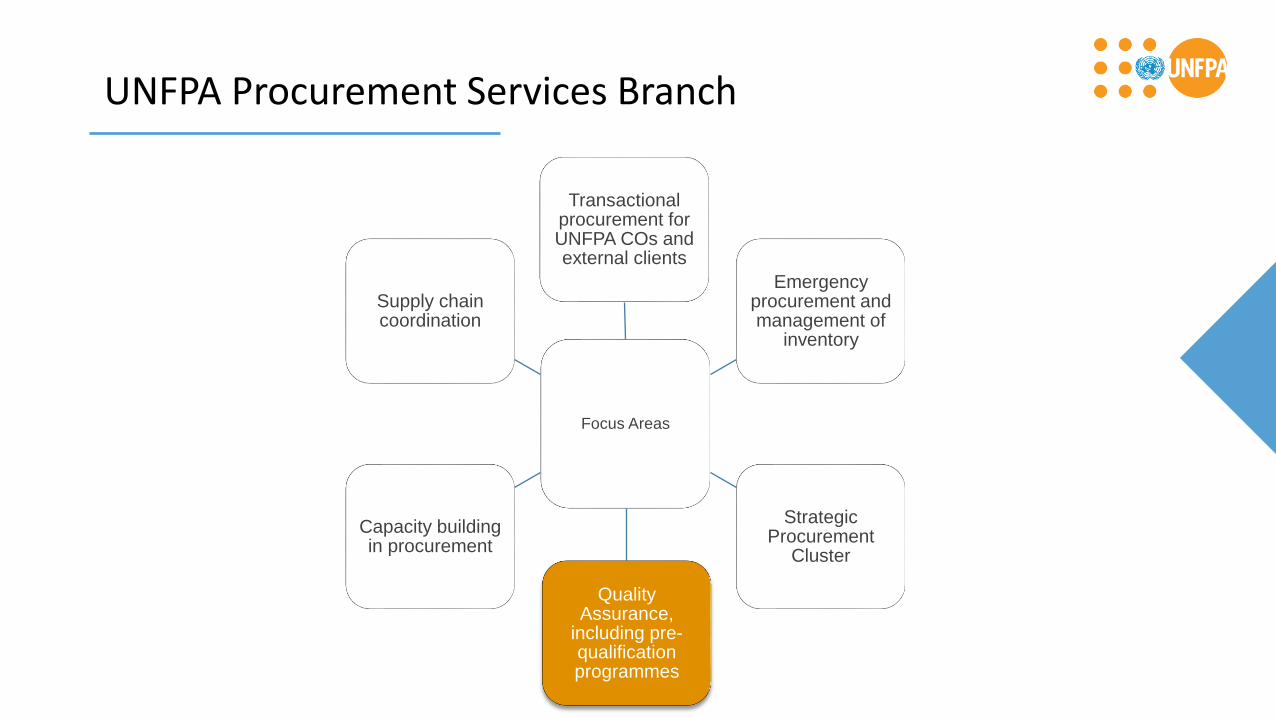
UNFPA Procurement Services Branch
Focus Areas
Transactional procurement for UNFPA COs and external clients
Emergency procurement and management of
inventory
Strategic Procurement
Cluster
Quality Assurance,
including pre-qualification programmes
Capacity building in procurement
Supply chain coordination

WHO/UNFPA Prequalification Programme
• Prequalification programmes started in 2001 (WHO’s normative and standard setting role)
• In 2008 it was agreed that UNFPA will manage the prequalification of condoms and intrauterine devices
• UNFPA aims to evaluate all suppliers that express interest

Aims of the Prequalification Programme
• Establish a system that promotes the procurement of good-
quality products that conform to the latest edition of the international
standard and the WHO/UNFPA Specification, and that retain their
effectiveness through out their stated shelf-life;
• Ensure that manufacturers have effective quality management
systems in place and meet quality standards
• Determine the physical capacity of manufacturers to deliver required
quantities of products
• Enhance confidence in ability of manufacturers to meet all
requirements and reduce the level of associated risk
• Save time and resources in identification of reliable manufacturers

1•Response to Expression of Interest and Documentation Submission
2• Screening of Submission
3•Document Review
4•On-site inspection
5•Product Testing
6 •Prequalifcation/Corrective Actions
7•Maintenance of Prequalification Status
Prequalification Programme StagesPrequalification Programme Stages

Changes to Prequalification Programme for RH Devices
• PQ Guidance document is separate from the technical specification and operational guidance for product
• Harmonize PQ document submission with Summary of Technical Documentation ( STED) • Separate Product Dossier and Site Master File no longer required • Only soft copies of document • To be implemented 2018 for male condoms ( based on draft document)
• Issuance of quality alerts and Notices of Concerns to notify National Regulatory Agencies and international procurers of quality and safety issues
• Public Inspection Reports (under development)

Summary of Technical Documentation
• New recommended format is based on the STED • Summary of Technical Documentation for Demonstrating Conformity to the
Essential Principles of Safety and Performance of Medical Devices) developed by the Global Harmonization Task Force (GHTF), a voluntary consortium of representatives from medical device Regulatory Authorities and Trade Associations from around the world
• Widely accepted by major regulatory agencies around the world
• The format has been amended to reflect the specific requirements of the WHO/UNFPA Prequalification process• New format essentially combines the Product Dossier and Site Master File• Reduces duplication of information• In line with conventional medical device technical files

Contents of STED (1)
• Characteristics of the product(s)
• Regulatory approvals
• Raw materials
• Product specifications
• Evidence of compliance with WHO/UNFPA general requirements
• Stability data
• Labelling and information
• Risk management analysis • Plan/Procedures• Report

Contents of STED (2)
• Manufacturing information • Site details• Certificates• Production processes• Premises and equipment• Personnel
• Quality Management System• Document management• Records• QA procedures• Distribution, complaints and product recall procedures• Internal audit procedures• CAPA procedures• Design and development procedures

Documentation Submission
Document Submission
• Manufacturers respond to Expression of Interest • Cover Letter
• Summary of Technical Documentation (STED)
• Copies of certificates/licenses
• Soft copies of documentation
• 10 Product samples may be required.

Screening
Purpose: Completeness of submissions
Resources and tools:
Screening template
WHO/UNFPA operational guidance on prequalification
Outcome communicated with manufacturer
If complete, proceed to detailed technical review
If incomplete, manufacturer will be asked to complete the documentation and resubmit within a specified period of time

Document Review and Inspection
Conducted by technical experts, two for each
submission
Assessors and Inspectors
• Experts with relevant years of experience in condom/IUD production, QA, Medical devices And
• Relevant qualifications in science e.g. chemistry, pharmacy
• Services procured through international bidding process
Confidentiality
Declaration of conflict of interest

Document Review
Purpose: To establish manufacturer has appropriate manufacturing
processes and quality management systems in place and conforms to
requirements of the respective WHO/UNFPA Specification and ISO
Standards
• ISO 4074 – male condoms
• ISO 25841- female condoms
• ISO 7439 – IUDs
- Biocompatibility testing according to ISO 10993
- Risk Management plan according to ISO 14971 and ISO 13485
- Confirm products have adequate shelf life data

On – Site Inspection
Verifies information from the document submission
Inspectors look at all parts of production process • Raw materials to final product warehouse
Suitability and state of the equipment are considered
Batch Documentation
Quality System
Validation
Maintenance
Laboratories
Shelf life • Detailed results reviewed on site and compared with submitted information
• In accordance with ISO standard? Data consistent? Protocols adequate?

ISO 13485 Audit and PQ Inspection Differences
Inspectors check certification but don’t fully
repeat ISO 13485 audit
PQ inspection is mainly technical inspection
Half of time is spent in factory or laboratory
Inspectors work through the manufacturing
process

Sampling and Testing
Product samples taken during the inspection from
randomly selected batches
Sample size in accordance with ISO 2859 standards
Range of tests conducted in accordance with relevant
WHO/UNFPA Specification and ISO standard
Testing conducted by independent, competent testing
laboratories (ISO 17025) appointed by UNFPA
Copy of test report provided to manufacturer

Inspection Report and Outcome
• Inspection report with test results provided to manufacturer
• Based on document review and inspection UNFPA recommends:•Prequalification without conditions
•Prequalification subject to specified corrective and preventative action ( CAPA)*
•Site ineligible for prequalification
• CAPA - Manufacturer Response
• Findings raised by inspectors will require a verified a CAPA before the factory can be prequalified
• CAPA report should be submitted within 90 days of final inspection report through UNFPA and cover all issues raised by inspectors
• CAPAs should contain evidence ( photographs, copies of amended documents etc) where appropriate to allow the CAPA to be verified

Outcomes of Prequalification
Outcome is posted on UNFPA’s website
• List of prequalified MLC manufacturers
• List of prequalified FC manufacturers
• List of prequalified IUD manufacturers

Maintenance of Prequalification
Notification of Changes:
Once manufacturing sites are prequalified, applicant/manufacturer is required to
advise UNFPA within 4 weeks of any matter that affects the information on which the
approval was based.
Reassessment:
Every 3 years prequalified factories must be re-assessed to maintain their
prequalification status. This re-assessment consists of:
1. Document review
2. On-site inspection
3. Product testing

Related Documents











