T T H H È È S S E E En vue de l'obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Chimie - Biologie - Santé JURY Christophe Biot Remi Chauvin Jean-Jacques Girerd Armand Lattes Bernard Meunier Paul M. O'Neill Maître de Conférences à l'Université de Lille Professeur à l'Université de Toulouse Professeur à l'Université de Paris-Sud Orsay Professeur Emérite à l'Université de Toulouse PDG de la société Palumed, Castanet-Tolosan Professeur à l'Université de Liverpool Rapporteur Rapporteur Ecole doctorale : Science de la Matière Unité de recherche : Laboratoire de Chimie de Coordination - CNRS, Toulouse Directeur de Thèse : Dr Anne Robert Rapporteurs : Pr Jean-Jacques Girerd et Dr Christophe Biot Présentée et soutenue par Fatima Bousejra-El Garah Ingénieur ENSIACET Le 18 mars 2010 Titre : Role of metals in the mechanism of action of antimalarial peroxides

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

TTHHÈÈSSEE
En vue de l'obtention du
DDOOCCTTOORRAATT DDEE LL’’UUNNIIVVEERRSSIITTÉÉ DDEE TTOOUULLOOUUSSEE
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Chimie - Biologie - Santé
JURY
Christophe Biot Remi Chauvin Jean-Jacques Girerd Armand Lattes Bernard Meunier Paul M. O'Neill
Maître de Conférences à l'Université de Lille Professeur à l'Université de Toulouse Professeur à l'Université de Paris-Sud Orsay Professeur Emérite à l'Université de Toulouse PDG de la société Palumed, Castanet-Tolosan Professeur à l'Université de Liverpool
Rapporteur Rapporteur
Ecole doctorale : Science de la Matière
Unité de recherche : Laboratoire de Chimie de Coordination - CNRS, Toulouse Directeur de Thèse : Dr Anne Robert
Rapporteurs : Pr Jean-Jacques Girerd et Dr Christophe Biot
Présentée et soutenue par Fatima Bousejra-El Garah
Ingénieur ENSIACET
Le 18 mars 2010 Titre : Role of metals in the mechanism of action of antimalarial peroxides


A mes parents


Preface
Remerciements Mes premiers remerciements s’adressent à tous les membres du jury pour l’honneur qu’ils m’ont fait en acceptant de juger ce travail.
Je tiens à remercier Monsieur JeanJacques Girerd, Professeur et VicePrésident de l’Université ParisSud, et Monsieur Christophe Biot, Maître de Conférences à l’Université de Lille, pour avoir été rapporteurs de ce manuscrit. Merci à Monsieur Girerd d’avoir accepté de juger ce travail, et ce malgré ses nombreuses responsabilités. Je voudrais également lui exprimer ma reconnaissance pour avoir suscité chez moi, alors étudiante en licence, l’envie de me diriger vers la recherche.
Je remercie Monsieur Armand Lattes, Professeur Emérite de l’Université de Toulouse, de m’avoir fait l’honneur de présider le jury de thèse, ainsi que pour sa grande gentillesse.
Merci à Monsieur Remi Chauvin, Professeur de l’Université de Toulouse, pour l’intérêt qu’il a porté à ce travail depuis le premier jour, et pour l’enthousiasme qu’il a montré à chacune de nos discussions.
Je remercie également Monsieur Paul O’Neill, Professeur de l’Université de Liverpool, pour l’accueil chaleureux qu’il m’a offert dans son équipe, pour notre collaboration, ainsi que pour la confiance qu’il m’a accordée.
Enfin, mes remerciements chaleureux s’adressent à Monsieur Bernard Meunier, PDG de la société Palumed, pour avoir inspiré les différents projets de ce travail, pour le temps qu’il m’a consacré, et pour avoir stimulé ma curiosité visàvis de la modélisation moléculaire, entreautres. Travailler en sa collaboration fut un plaisir et un grand privilège.
Merci aux membres du jury pour l’intérêt qu’ils ont porté à ce travail et pour les commentaires chaleureux qu’ils en ont fait. Mes remerciements les plus profonds sont adressés à ma directrice de thèse, Madame Anne Robert, pour m’avoir permis de réaliser ce travail sur un sujet passionnant. Travailler à ces côtés fut extrêmement formateur et parmi ses nombreuses qualités, je soulignerai sa grande rigueur scientifique et son vif esprit critique. Anne, merci pour tout ce que vous m’avez appris. Merci de votre générosité, votre écoute, et vos encouragements continus qui m’ont été très bénéfiques, surtout les dernières semaines. Même dans mes moments de doutes, vous êtes toujours restée positive et confiante. Vous avez également toujours fait en sorte que je puisse travailler dans les meilleures conditions, y compris matérielles, et je vous en suis très reconnaissante.
Le travail effectué pendant ces années de thèse est le fruit de plusieurs collaborations, c’est pourquoi je tiens à remercier toutes les personnes qui ont pris part à ces projets :
Pour leur investissement essentiel dans le projet "adduits in vivo", je remercie Françoise BenoitVical, qui a réalisé les expériences in vivo, et Catherine Claparols pour son aide dans la partie analyse, ainsi que pour avoir accepté que j’injecte des "bouts de souris" en LCMS.

Preface
J’adresse ma profonde gratitude à JeanLuc Stigliani pour avoir pris le temps de m’initier à la modélisation moléculaire et pour son aide précieuse dans le projet de docking.
Merci à la société Palumed pour les mesures d’activité in vitro. Mes remerciements s’adressent plus particulièrement à Frédéric Coslédan pour notre collaboration, mais également pour sa gentillesse et ses encouragements.
Je tiens également à remercier Marguerite Pitié pour sa contribution importante dans le projet "cuivre", ainsi que pour les nombreuses discussions que nous avons eues.
Pour sa confiance et ses conseils avisés, je remercie Richard Amewu, de l’Université de Liverpool. Merci également au Pr Steve Ward et à Sant Muangnoicharoen, tous deux de la Liverpool School of Tropical Medicine, pour m’avoir permis d’utiliser leurs équipements analytiques.
Enfin, je remercie Yannick Coppel (RMN) et Laure Vendier (RX) pour leurs contributions respectives. Mes remerciements s’adressent également à l’ensemble des personnes que j’ai eu le plaisir de côtoyer quotidiennement, ou presque, au laboratoire.
Merci à tous les membres du groupe " Oxydations biomimétiques" (équipe K), successivement dirigé par Pr Jean Bernadou, que je remercie de son accueil, et Geneviève Pratviel merci Geneviève de m’avoir proposé de participer au Colloque Chimie&Terroir, ainsi qu’au MiniSymposium Marguerite Pitié, Vania BernardesGénisson, JeanLuc Stigliani, sans oublier Catherine Hemmert et Christophe Loup. Merci également à Peter Faller.
Je n’oublie pas les étudiants et postdocs avec lesquels j’ai passé de très bons moments au LCC et ce n’est pas fini : François B, Carmen Bonne chance pour la dernière ligne droite ! Jérôme, Vincent, Céline, Vanessa, Antonio, Tamara, François JBDD, Laurent, Irène, ... Mon séjour à Liverpool n’aurait pas été aussi agréable sans Edite, Chi, Zeyn, Richard, FrancescKiko, Olivier, James, Sunil & Rosanne, Vicki, Pete, David, Ally, Archana, Sant et tous les membres des groupes de Paul O’Neill et Steve Ward. Merci à Odile DechyCabaret de m’avoir orientée vers l’équipe K à l’époque où je cherchais un laboratoire pour mon stage de Master 2. Odile, merci pour ta gentillesse et ton amitié. Sois assurée de mon amitié réciproque.
Merci à Martine pour sa bonne humeur quotidienne – surtout ne change pas ! – ainsi qu’à toutes les personnes du LCC qui ont contribué, chacune à sa façon, à ce que ce travail soit réalisé dans les meilleures conditions. Je remercie le consortium européen Antimal pour avoir financé ces travaux d’une part, et pour les nombreuses formations/conférences auxquelles j’ai pu participer pendant ma thèse d’autre part. Merci également à Miriam Griesheimer pour son aide administrative et logistique.

Preface
Je remercie également ma famille toute entière, à laquelle j’associe ma bellefamille, qui m’a toujours accompagnée et encouragée. Merci également à mes amis, trop nombreux pour être tous cités. A mes sœurs et mon p’tit frère, qui m’avez soutenue dans mes études, même quand vous n’en voyiez plus la fin, je vous dis merci ! C’est de tout mon cœur que je remercie mon mari, Lhoussein, pour son soutien, ses encouragements continus et son affection. Merci d’avoir su trouver les mots pour me redonner confiance dans les moments difficiles. Cette thèse est aussi un peu la tienne… Enfin, je remercie les deux êtres les plus chers à mes yeux, mes parents. Vous avez toujours été là pour moi, et depuis le début vous avez toujours cru en moi, même lorsque je n’y croyais plus moimême ; c’est grâce à vous si je suis arrivée là aujourd’hui. Les mots ne seront jamais assez forts pour vous exprimer ma profonde gratitude, c’est pourquoi je vous dédie ce travail.
~~~~~~~~~~~~~~~~~~~~~


Preface
The work reported in this manuscript has been carried out under the supervision of Dr Anne Robert, in the Laboratoire de Chimie de Coordination at Toulouse, and Pr Paul M. O’Neill, in the Chemistry Department of the University of Liverpool, in collaboration with Dr Bernard Meunier (Palumed S.A.). The research leading to these results has received funding from AntiMal, an FP6funded integrated project under contract number LSHPCT20050188.


Role of metals in the mechanism of action of antimalarial peroxides
Names of drugs When existing, the trivial name of antimalarial drugs is used (e.g. chloroquine, …). In other cases, we use either the name given by the authors (e.g. OZ277, RKA182, PA1103, …) or a systematic numbering. List of acronyms and abbreviations ACN ACT ADME AQ CQ CQH+ CQH2
2+ Cys DDT DHA DFT DMPK DMPO DMSO DV EC50 EPR ESI Fe(II)PPIX FQ FV Glu Gly GSH GSK Hb HPLC HRMS HPRII HSA Hz IC50 IQ LCC
Acetonitrile Artemisinin-based Combination Therapy Absorption, Distribution, Metabolism and Excretion Amodiaquine Chloroquine mono-protonated Chloroquine di-protonated Chloroquine Cysteine Dichlorodiphenyltrichloroethane Dihydroartemisinin Density Functional Theory Drug Metabolism and Pharmacokinetics 5,5-Dimethyl-1-pyrroline N-Oxide Dimethylsulfoxide Digestive vacuole (= FV) 50% effective concentration (concentration of drug necessary to inhibit parasite growth by 50% in vivo) Electron Paramagnetic Resonance Electrospray ionization Ferroprotoporphyrin IX Ferroquine Food Vacuole (= DV) Glutamic acid Glycine Glutathione GlaxoSmithKline pharmaceuticals Hemoglobin High Performance Liquid Chromatography High resolution Mass Spectrometry Histidine Rich Protein II Human Serum Albumin Hemozoin 50% inhibitory concentration (concentration of drug necessary to inhibit parasite growth by 50% in vitro) Isoquine Laboratoire de Chimie de Coordination (Toulouse)

Role of metals in the mechanism of action of antimalarial peroxides
LC-MS LSTM MDR MMV MnTPC MQ MS MVI NMR NTBI P.f. PfATP6 PfCRT PPIX RBCs ROS SERCA TCTP TEMPO TG TPC TPP UV-Vis WHO
Liquid Chromatography-Mass Spectrometry Liverpool School of Tropical Medicine Multi-Drug Resistance Medicines for Malaria Venture Manganese tetraphenylchlorin Mefloquine Mass spectrometry Malaria Vaccine Initiative Nuclear Magnetic Resonance N-tert-butyl Isoquine Plasmodium falciparum Plasmodium falciparum ATPase 6 Plasmodium falciparum Chloroquine Resistance Transporter Protoporphyrin IX Red Blood Cells Reactive Oxygen Species Sarco/Endoplasmic Reticulum Calcium ATPase Translationally Controlled Tumor Protein 2,2,6,6-Tetramethylpiperidine-N-oxyl [radical] Thapsigargin Tetraphenylchlorin Tetraphenylporphyrin Ultra violet-Visible World Health Organization
Abbreviations for NMR data s singlet d doublet t triplet m multiplet bs broad singlet bd broad doublet dd doublet of doublet

Table of contents
1 1
1 1 3 4 44
6 6 7 7 9 9 10 11 13
13 15 17 18 18 19 20 21 21
21 22
22 27 8
228 32 33
34 36
36 36 36 38 38
Chapter 1. Alkylating properties of antimalarial peroxidecontaining drugs: A focused review I. Malaria and the Plasmodium parasite
day’s world situation
I.1. Plasmodium speciesI.2. Malaria: Brief history and to
e of Plasmodium I.3. Life cyclI.4. Clinical features of malaria I.5. Vaccine I.6. Host hemoglobin digestion and heme detoxification by Plasmodium
II. Quin century of use oline ugs: Over a
II.1. Co atment based drnventional trea. Quinine b. Chloroquine
II.2. Old pharmacophore for new drugs: N‐tert‐butyl Isoquine and Ferroquine c. Mefloquine
K369796) a. N‐tert‐butyl Isoquine (GSb. Ferroquine (SSR97193)
III. Fro gs: m the natural artemisinin to new synthetic peroxidecontaining dru
era in malaria chemotherapy g from the Chinese
A new III.1. Artemisinin (Qinghaosu): A natural peroxide dru
s and ACT traditional medicine
nthetic derivativeontaining drugs
III.2. Artemisinin semi‐syIII.3. Synthetic peroxide‐c
a. Arteflene b. Trioxanes c. Trioxolanes d. Tetraoxanes e. Trioxaquines IV. Fe( oxides and possible drug targets II)me
IV.1. Rediated reactivity of antimalarial per
s activity with inorganic salta. Oxidative stress and lipid peroxidation b. Reactivity with ferrous salt
IV.2. Rec. Reactivity with other transition metals ions activity with iron(II)‐heme and biological targets a. Heme as target for artemisinin and peroxide‐containing antimalarials
ity of artemisinin in vivo has been in malaria‐infected mice
b. The role of heme in the antimalarial activevidenced by identification heme‐artemisninc. Heme‐mediated reactivity of trioxolanes
xaquines IV.3. Pr emisinin
d. Heme‐mediated reactivity of triotl
oteins as possible targets for arive site modeitic proteins
a. GSH as enzyme actb. Alkylation of parasc. PfATP6 inhibition
V. Conclusion and scope of the thesis VI. Bibliography
T able of contents

Table of contents
59 59 1 4 66 71 71
5 7 89 89
3 9 115 115
18 1 125 125
32 32 1
1 147 147 151
53
151
53
11 161
161
Chapter 2. The antimalarial artemisone is an efficient heme g agent alkylatin
I. Résumé
l Garah, B. Meunier, and A. Robert, European Journal of 133‐2135
II. Publication: F. Bousejra‐EInorganic Chemistry 2008, 2III. Supplementary material Chapter 3. Alkylating ability of artemisinin after Cu(I)induced
n activatio I. Résumé II. Publication: F. Bousejra‐El Garah, M. Pitié, L. Vendier, B. Meunier, and A. Robert Journal of Biological Inorganic Chemistry 2009, 14(4), 601‐610 Chapter 4. Reactivity of antimalarial dispiro1,2,4,5tetraoxanes with
me and phospholipids: Implications for their mode of action Fe(II), he
ah, R.K. Amewu, I. Résumé II. Publication for submission: F. Bousejra‐El GarS. Muangnoicharoen, S.A. Ward, and P.M. O’Neill Chapter 5. The antimalarial trioxaquine DU1301 alkylates heme in
nfected mice malariai I. Résumé II. Publication: F. Bousejra‐El Garah, C. Claparols, F. Benoit‐Vical, B. Meunier, A. Robert, Antimicrobial Agents and Chemotherapy 2008, 52, 2966‐2969 Chapter 6. Docking study of structurally diverse antimalarial drugs targeting PfATP6: No correlation between in silico binding affinity
ro antimalarial activity and in vit I. Résumé II. Publication: F. Bousejra‐El Garah, J.‐L. Stigliani, F. Coslédan, B. Meunier, A. Robert. ChemMedChem 2009, 4(9), 1469‐1479 Conclusion Appendixes Appendix 1. Total synthesis of Quinine Appendix 2. Candidate Selection of a 1,2,4,5‐Tetraoxane Drug‐Development Candidate (RKA 182) with Superior Properties to the Semi‐Synthetic Artemisinin Based Antimalarials. Angew. Chem. Int. Ed. 2010 In press



Chapter 1
Alkylating properties of antimalarial peroxidecontaining drugs:
A focused review


Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
1
Chapter 1. Alkylating properties of antimalarial
peroxide-containing drugs: A focused review
I. Malaria and the Plasmodium parasite
I.1. Plasmodium species
Malaria is due to a blood infection by protozoan
parasites of the genus Plasmodium, which is transmitted
from a human to another by the bites of female
Anopheles mosquitoes. Five species of malaria parasite
can infect humans (Table 1). Plasmodium falciparum is
the most virulent species and is responsible for most of
the mortality associated with malaria, especially in
Africa.[1]
Plasmodium was often used in the early 1930s as a
pyretic agent for the treatment of neurosyphilis, caused
by a thermo-sensitive bacteria.[2] The first natural
infection of P. knowlesi in a human was reported in
1965,[3] but it was not considered an important public
health concern until 2004. Very recently, several fatal
cases of P. knowlesi have been reported in Southeast Asia.[4,5]
I.2. Malaria: Brief history and today’s world situation
Malaria has been present since ancient times and many cases of malaria are reported in writings
from old civilizations. Hippocrates described the various malaria fevers of man by 400 BC.[6]
It is believed that malaria has shaped the course of history of millennia.[7] Kings, popes, and
military leaders were struck down in their prime by malaria. Many great warriors succumbed to the
disease after returning from the warfront and in many conflicts, more troops were killed by malaria
than in combat.[7]
Until the twentieth century, malaria was widespread on all continents, including Europe and North
America.[8] After World War II, the Global Malaria Eradication Programme was launched by the
World Health Organization (WHO), with the objective to eliminate the disease totally.
Table 1: Species of Plasmodium that infect
humans
• Plasmodium falciparum: responsible of the most serious form of the disease, especially in Africa.
• Plasmodium vivax: most geographically widespread but produces less severe symptoms than P.falciparum.
• Plasmodium malariae: can persist in the blood for very long periods without producing symptoms.
• Plasmodium ovale: rare and generally infects in Africa.
• Plasmodium knowlesi: simian parasite, rare and limited to Southeast Asia but can be virulent and result in death.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
2
Widespread use of dichlorodiphenyltrichloroethane
(DDT) insecticide, coupled with the wide use of the
drug chloroquine, resulted in eradication of malaria in
Europe and North America. This initiative had
successes but, face to the emergence of resistances to
both DDT and chloroquine, the global eradication was
considered impossible and then abandoned in the
early 1970s. Since then, the burden of malaria has
increased substantially in many parts of the world.[1]
Today, malaria is curable and preventable but it is still the most important parasitic infection and
one of the major causes of mortality. According to recent estimates by the WHO, there are
approximately 300-500 million cases of malaria and about 1 million deaths each year, the vast
majority of which are African children under five.[9,10] Pregnant women are also especially
vulnerable. With more than a third of the world’s population living in endemic areas, malaria is a
serious global health and developmental challenge in Africa, South-East Asia and the Amazon
region of Latin America (figure 1).
Figure 1: Malaria endemic regions in 2004[11]
Global warming and other environmental factors, added to
migration of populations, may have contributed to the
spread of malaria into some previously malaria-free
regions. This situation could deteriorate even more as
available drugs are failing due to resistances.[9]
Table 2: Nobel Prizes in malaria
• Sir Ross, 1902: British bacteriologist, discovered the role of Anophele mosquitoes in the transmission of the disease
• Laveran, 1907: French army surgeon, discovered the Plasmodium parasite
• Wagner-Jauregg, 1927: Austrian professor in psychiatry, discovered the therapeutic value of malaria inoculation in the treatment of dementia paralytica
• Müller, 1948 : Swiss chemist, inventor of DDT
Table 3: Malaria world situation [11,12]
• 247 million malaria cases in 2006
• About 1 million deaths in 2006, mostly children under 5 years
• 3.3 billion people at risk
• 109 countries in endemic areas in 2008

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
3
I.3. Life cycle of Plasmodium
The malaria parasite has a complex life cycle[9] that requires two hosts: the intermediate human host
for the asexual stages, and the definite Anophele mosquito host, for the sexual stages (figure 2).
Human host: When the infected mosquito takes a blood meal, it injects saliva that contains
sporozoites, the infectious form of the parasite, into the bloodstream of its victim. The sporozoites
migrate to the liver where they multiply in the hepatocytes and develop into schizonts. After
approximately one week, the mature schizonts burst out and release merozoites that return to the
bloodstream and invade the erythrocytes. Once in the erythrocytes, the merozoites develop into
ring forms, then trophozoites (feeding stage) and form schizonts again. The schizonts mature,
rupture the cell, and release merozoites to the bloodstream, where they invade other red blood cells
(RBCs) to achieve a new asexual cycle. The synchronous rupture of RBCs (each 48 or 72 h
depending on the parasite species) is associated with the rythmic fever accesses and other symptoms
of malaria. Some of the newly formed merozoites do not develop into schizonts but change to the
sexual stage called gametocytes.
Figure 2: Life cycle of Plasmodium (adapted from[13])

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
4
Mosquito host: Gametocytes do not develop in humans but are extracted by a mosquito during
another blood meal. Inside the mosquito stomach, gametocytes produce male and female gametes.
The fertilization then produces oocytes filled with sporozoites. When the oocytes mature, they burst
out and sporozoites migrate to the salivary glands of mosquitoes, where the cycle can start over
again when the mosquito bites its next victim.
The initial liver stage of malaria infection is asymptomatic. As the rupture of infected RBCs and
reinvasion in responsible for the symptoms, many efforts have dealt with the erythrocytic stage of
the parasite. As a result, most of antimalarial drugs are active on this stage.
I.4. Clinical features of malaria
For most people, symptoms begin 10 days to 4 weeks after infection, although a person may feel ill
as early as 8 days or up to 1 year later. Clinical attacks of malaria normally begin with influenza-
like symptoms, fever often accompanied by a headache, muscle stiffness and shaking, sometimes
also vomiting and diarrhoea. Severe malaria includes anemia, organ failure, and cerebral malaria. If
untreated, severe malaria lead to coma and death.[14]
I.5. Vaccine
No vaccine is currently available to prevent malaria. However, many vaccine candidates are in
development, the most advanced to date being the RTS,S vaccine, developed by the pharmaceutical
company GlaxoSmithKline (GSK) in partnership with Malaria Vaccine Initiative (MVI).[15] RTS,S
has been designed to prevent from the pre-erythrocytic parasites i.e. invasion of the blood. Results
of RTS,S in endemic areas are very promising so far, as it reduces the risk of clinical malaria,
delays time to new infection, and reduces episodes of severe malaria for at least 18 months. RTS,S
has recently entered Phase III clinical trials.[16]
I.6. Host hemoglobin digestion and heme detoxification by Plasmodium
Once in the red blood cell, Plasmodium needs amino acids to synthesize its own proteins. A major
source of amino acids is provided by the digestion of the host hemoglobin.[17] Hemoglobin is one of
the major components of the erythrocyte with a concentration of 5 mM, corresponding to a
concentration of iron-heme of 20 mM (ferroprotoporphyrin IX, FePPIX). The malaria parasite
degrades up to 65% of the hemoglobin in the host cell, both for its development and to create space
with the digestive vacuole.[18,19] This digestion by protease enzymes releases peptides, degraded to
amino acids,[20] and a high concentration of free iron(II)-heme as a by-product. Like for every living

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
5
cell, iron(II)-heme is toxic to the parasite at micromolar concentration, i.e. several orders of
magnitude below the total heme concentration in the red blood cells, as it can reduce molecular
oxygen and generate reduced oxygen species (O2•- and H2O2) that produce HO•.[21] In order to
inhibit the toxic effect of heme, Plasmodium crystallizes it into hemozoin, an inert and insoluble
aggregate (also called malaria pigment, figure 3).[22,23] In hemozoin, iron is in oxidation state III,
i.e. no more able to reduce molecular oxygen.
Figure 3: Host hemoglobin degradation pathway and hemozoin formation
The exact mechanism of hemozoin formation, although still unclear, involves histidine-rich protein
II (HRPII),[24-26] a protein secreted by the parasite and mainly constituted of histidine residues
(34%).[26] Kannan et al. reported that HRPII can bind 17 molecules of heme in vitro at pH 5, the
pH of the parasite food vacuole, and up to fifty molecules at pH 7. This binding, probably by
coordination of histidine residues of HRPII as axial ligand of iron atom of heme, should result in
local concentration of heme favorable to initiate heme polymerization. The growing of chains is
then independent on the presence of HRPII, which is consequently not a true “polymerase”.
The structure of hemozoin has been a topic of debate for years. It has been finally reported that its
spectroscopic properties are identical to synthetic β-hematin, formed by polymerization of heme
under controlled conditions (pH, salt concentration, …). Structure determination using X-ray
powder diffraction by Pagola et al. has revealed that hemozoin is constituted by heme dimers,
interacting through hydrogen bonding.[27,28] Within the dimer, the heme units are linked to each

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
6
other by an iron-carboxylate bond: the propionate carboxylate side chain of one heme is coordinated
to the iron atom of the next heme (figure 4). Each heme then contains a pentacoordinated iron(III),
with no possibility to be reduced. Any perturbation of this heme detoxification process is expected
to have a drastic impact for the parasite survival.
Figure 4: Two unit cells of the crystal structure of ββββ-hematin dimers from Pagola et al.[27]
Because of their importance for the parasite survival, the hemoglobin degradation and hemozoin
formation processes are effective targets for therapeutic intervention.
Other processes of the asexual blood stage of the parasite have been identified (folate biosynthesis,
protein synthesis in the apicoplast, electron transport in the mitochondrion, etc). However in this
review we will only focus on drugs that target heme and interfere with its detoxification process.
II. Quinoline-based drugs: Over a century of use
II.1. Conventional treatment
Quinoline-containing drugs have been the mainstay of malaria treatment for the past 50 years.
Among them, quinine, chloroquine (CQ), and mefloquine (MQ ) are the most widely used
clinically.[29] This part summarizes the current knowledge on their common mechanism of action,
with a focus on the role of heme in the antimalarial activity of these three quinolines. [30,31]
Figure 5: Structures of quinoline drugs

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
7
It should be mentioned at this point that quinine, chloroquine, and mefloquine contain one or more
stereogenic centers. Only quinine, a natural product, is clinically used as a single optic isomer.
Chloroquine is used as a racemate and mefloquine, which contains 2 stereogenic centers, is also
used as a racemic mixture (11S,12R and 11R, 12S). Enantiomers of chloroquine and mefloquine
exhibit the same antimalarial activity, in the limits of experimental variability.[32]
a. Quinine
Quinine, an aryl-amino alcohol (figure 5), is the oldest agent used for the treatment of malaria. It is
extracted from the bark of the Cinchona tree, which was first imported into Europe from Peru for
antimalarial use in the seventeenth century.[33] Since then, quinine has been the treatment of choice
for intermittent fever throughout the world. In 1820, the French pharmacists Pelletier and Caventou
isolated quinine from cinchona bark, and its first total synthesis was reported in 1944 by Woodward
and Doering, but this claim has been a matter of debate until very recently (see Appendix 1).
Approximately 300–500 tons/year are produced commercially by extraction of the bark from
various cinchona species that are now widely cultivated. About 40% of the quinine goes into the
production of pharmaceuticals, while the remaining 60% is used by the food industry as the bitter
principle of soft drinks, such as bitter lemon and tonic water.[33]
Quinine has been widely used until the 1930s, when other synthetic drugs like chloroquine
appeared. However, quinine is still employed for the treatment of severe malaria,[34,35] although
there are reports on declining efficacy in South America and Africa.[36,37]
The mechanism of action of quinine is still not fully elucidated, but it has been established that this
quinoline interacts with heme in the food vacuole of the parasite.[38,39] This mechanism, common to
chloroquine, is further detailed in the next section.
b. Chloroquine
Chloroquine (CQ, figure 5) is a 7-chloroquinoline ring substituted by a N,N-diethyl-1,4-pentane
diamine in para position of the quinoline hydrogen. Thanks to its excellent properties, chloroquine
has been the drug of choice for more than forty years (1940s – 1980s): it is easy and cheap to
produce, well tolerated, safe for use by pregnant women and children, and used to be highly
effective against the blood stages of Plasmodium.[40]
Chloroquine, usually prepared as a diphosphate salt of the racemate, is a diprotic weak base (pKa1
= 8.1 cycle, pKa2 = 10.2 chain) that mainly exists in both mono- and di-protonated forms within the
biological pH. The neutral (CQ) and mono-protonated (CQH+) chloroquine species may enter in
the cytoplasm of the parasite (pH ∼ 7) and diffuse across the membranes to reach the acidic food

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
8
vacuole (pH ∼ 5).[41] In the food vacuole, the equilibrium is completely shifted toward the di-
protonated species CQH22+. The drug is then “trapped” in this compartment, where it rapidly
accumulates, potentially reaching millimolar concentrations (figure 6).[42]
Figure 6: Chloroquine accumulation in the food vacuole of the parasite
The total concentration of heme released in the food vacuole by hemoglobin digestion has been
estimated at 400 mM.[43] Chloroquine is thought to exert its antimalarial effect in the food
vacuole by binding heme and interfering with its detoxification process.[38,39] The ability of
chloroquine to inhibit hemozoin formation has been shown both in vitro and with P. falciparum
cultures.[44,45] Several studies, including modeling experiments, showed that the complex
chloroquine-heme is based on an energetically favorable ππππ-ππππ stacking interaction between the
quinoline heterocycle of CQ and the metalloporphyrin ring of hematin dimer.[31,38,46] Sullivan and
co-workers suggested that the chloroquine-heme complex incorporates into the growing hemozoin
crystal to terminate chain extension, blocking further sequestration of toxic heme.[43] The heme-
chloroquine complex itself has been shown to be toxic to the parasite.[47]
Unfortunately, massive use of chloroquine has resulted in the emergence of resistant P. falciparum
strains in the 1960s and their widespread in nearly every endemic region throughout Asia and then
Africa.[48] Today, more than 80% of field isolates of Plasmodium falciparum are resistant to CQ.[49]
The mechanism whereby P. falciparum becomes resistant to chloroquine is complex. Fitch and co-
workers showed that accumulation of CQ in resistant parasites is less important than in sensitive
strains.[29,50] This lower accumulation is due to a higher efflux of chloroquine: resistant parasites
have been found to release CQ up to 50 times more rapidly than the susceptible parasites.[51]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
9
The resistance is associated with mutations (K76T) in the Chloroquine Resistance Transporter
(PfCRT), a protein located on the membrane of the parasite digestive vacuole.[52-54]
c. Mefloquine
Mefloquine (MQ , figure 5) belongs to the 4-methanolquinolines subclass and is structurally related
to quinine. It was introduced in the 1980s to treat chloroquine-resistant malaria. Today, mefloquine
is used for prophylaxis (eg Lariam®) and to treat uncomplicated malaria, in combination with other
drugs. Mefloquine displays high activity against chloroquine-resistant strains.[55] However,
resistance to mefloquine is spreading.[56] In addition, mefloquine is quite expensive, then
unaffordable for many patients in Africa, and it is well established to be neurotoxic, for a review see [57]
and associated with psychiatric side effects, probably due to the presence of the (-)-erythro
diastereoisomer in the mixture. Clinical studies are ongoing with the pure (+)-erythro
diastereoisomer, to determine whether it has a better safety profile.[58]
As for chloroquine, MQ was shown to interact with heme, but it has also been proposed that
phospholipids are a second target for this quinoline.[59]
II.2. Old pharmacophore for new drugs: N-tert-butyl Isoquine and
Ferroquine
Because of the widespread of quinoline resistances, there is an urgent need of new antimalarial
drugs. One possibility is to re-design quinoline-based drugs and prepare chloroquine derivatives
that are still able to interact with heme by π-stacking, but can escape drug resistances.[60] In this
purpose, several chloroquine analogues with a shorter side chain have been prepared. They are
active against P. f. chloroquine-resistant strains (IC50s below 15 nM against CQR strains).[61]
However, the main drawback with these compounds is that they readily undergo metabolic N-
dealkylation in vivo, producing metabolites that are less potent than the parent drug.[62] For these
compounds, the in vitro activity is not translated into good in vivo activity. Other analogues with
metabolically more resilient side chain have been prepared. These second-generation compounds
display good activity both in vitro and in vivo.[63]
Another strategy is to develop drugs with modified quinoline structures. Several synthetic
quinoline-based drugs have been produced. The most important of these were amodiaquine (1951),
primaquine (1952), halofantrine (1966) and, in the past 30 years, piperaquine and lumefantrine. All
of these drugs interfere with the detoxification of heme, although there are important differences in
their activities. We report here the synthesis and pharmacological properties of two new promising
4-aminoquinoline derivatives: N-tert-butyl isoquine and ferroquine.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
10
a. N-tert-butyl Isoquine (GSK369796)
O’Neill, Ward and co-workers have designed and prepared a series of promising 4-aminoquinolines
antimalarials. Among them is the drug candidate N-tert-butyl isoquine (NTBI , figure 8). NTBI is
currently developed in a public/private partnership between GSK pharmaceuticals, Medicines for
Malaria Venture (MMV), and the University of Liverpool. This compound has completed
preclinical evaluation and entered phase I clinical trial in April 2008.[64]
NTBI is related to amodiaquine (AQ, figure 7), a drug active against chloroquine-resistant strains
of P. falciparum, but associated with potential cardiotoxicity and hepatotoxicity.[65] This toxicity
results from the P-450-catalyzed oxidation of the 1,4-aminophenol ring and the formation of
reactive quinine-imine (figure 7).[66]
Figure 7: Oxidation of amodiaquine to a reactive quinine-imine metabolite
In a first attempt to prevent this metabolism-dependent toxicity, the positions of the hydroxyl and
the diethylaminomethyl residue at the phenyl ring have been interchanged. The resulting 1,3-
aminophenol isoquine (IQ , figure 8) does not form quinine-imines and exhibits good oral in vivo
activity (ED50 = 3.7 mg/kg vs. P. berghei ANKA compared to amodiaquine ED50 = 7.65 mg/kg).[67]
Subsequent metabolism studies in the rat model demonstrated that isoquine does not undergo in
vivo bioactivation. However, a drawback of isoquine is its low oral bioavailability in animal
models, due to extensive metabolism of the N-diethylamino side chain. Further studies in this area
have revealed that replacement of the diethylamino moiety by a tert-butylamine avoids too rapid
biotransformation.
Figure 8: Structures of amodiaquine, isoquine and N-tert-butyl isoquine

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
11
The resulting N-tert-butyl isoquine displays potent in vitro and in vivo activity (ED50 = 2.8 mg/kg
and ED90 = 4.7 mg/kg against P. berghei ANKA). [68] Detailed study of drug metabolism and
pharmacokinetics (DMPK), in four animal species including primates, showed that NTBI has a
safer profile than chloroquine. In addition, no cross resistance with any clinically used antimalarial
drug has been observed so far.[69]
Synthesis of NTBI has been optimized and allows the preparation of the drug with fairly good yield
(57%) from the readily available 4,7-dichloroquinoline.[70] Molecular modeling of the interaction of
the NTBI with heme showed that, in the most
favorable complex, the aromatic quinoline ring is
parallel to the edge of the hematin, consistent with a π-
π stacking interaction. There is also a favorable
hydrogen-bonding network between the carboxylate
groups of the hematin and both the alcohol and
protonated amine of the Mannich side chain.
Model of the most favorable hematin-NTBI complex. For clarity, only hydrogens involved in hydrogen bonds are
shown.[68]
b. Ferroquine (SSR97193)
Inspired by work of Jaouen’s group on ferrocene-containing tamoxifen analogues,[71,72] Brocard and
co-workers have prepared several derivatives of antimalarial drugs, bearing a ferrocene
(dicyclopentadienyl iron (II), Fc) in their side chain.[73] Ferroquine (FQ, figure 9), a 4-
aminoquinoline linked to a ferrocenyl moiety, was selected from a first screening and is currently in
phase IIb clinical trials by Sanofi-Aventis.[74]
Figure 9: Structure of ferroquine
Ferroquine is active against various chloroquine-sensitive and chloroquine-resistant laboratory
strains (IC50 = 11 - 22 nM)[75] as well as various field isolates (IC50 = 0.4 - 47.0 nM).[76-81] In a
mouse model, ferroquine is curative at 10 mg/kg.[75]
Synthesis of ferroquine is simple from relatively inexpensive materials.[73,82] FQ exhibits planar
chirality inherent to the 1,2-substitution of the ferrocene cyclopentadienyl ring. Both the (1’R)-FQ

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
12
and (1’S)-FQ enantiomers have been prepared using bio-catalysis and tested against P. falciparum.
They exhibit the same activity in vitro and a slightly different activity in vivo, believed to be due to
different pharmacokinetic properties.[75] The overall close properties of the two enantiomers allow
ferroquine to be formulated as a racemic mixture for its development.
Investigations of the physicochemical properties showed that FQ is more lipophilic than CQ at
vacuolar pH, suggesting that its accumulation in the food vacuole is less significant than CQ.[83]
In vitro inhibition assays showed that the drug candidate FQ is a strong inhibitor of β-hematin
formation, in a higher extent than CQ.[83] These data suggest that the two drugs share a similar
mechanism of action e.g. inhibition of hemozoin formation by interaction with intra-vacuolar heme.
Furthermore, EPR and spin trapping experiments in the Fenton conditions put into evidence the
generation of hydroxyl radicals from the oxidation of the ferrocene into ferricinium.[84] In vivo,
radical concentration would be high enough to induce significant damage on the parasite
membranes.
It was also reported that FQ generates HO• in the presence of H2O2 and this redox activity of the
ferrocene moiety was suggested to be “a possible discriminating property from CQ in the
antimalarial activity” of the two drugs.[84] However, concentrations as high as 1 mM – i.e. 5 to 7
order of magnitude above the steady-state of H2O2 in living cells (10-7 to 10-9 M) - were required for
a hydroxyl radical production of 0.5 µM (calculated yield: 0.017%). In addition, at so low
concentration of hydroxyl radicals, a contamination by iron salts cannot be excluded.
Ruthenocene analogues of ferroquine have also been synthesized. First results showed similar in
vitro antimalarial activity, suggesting that the physical properties of ferroquine may be more
prevalent than the chemical reactivity of the metallocene moiety.[85]
As expected, the major metabolic pathway of ferroquine by P450s is demethylation of the side
chain amine leading to the mono- and the di-demethylated derivatives.[86] Interestingly, this
metabolites are still more active than CQ.[80]
Ferroquine exhibits good therapeutic index and did not show cross resistance with chloroquine.[75,87]
Because of the lipophilic ferrocenyl moiety, authors proposed that ferroquine does not fit into the
substrate binding site of the chloroquine resistance transporter (PfCRT). The mutational status of
the pfcrt gene did not influence the compound activity.
Using a strategy similar to the design of ferroquine, ferrocene conjugates of quinine and mefloquine
have been prepared, as well as artemisinin and atovaquone conjugates. However, the biological
activities of the new compounds were lower than that of the parent drugs.[88-91]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
13
III. From the natural artemisinin to new synthetic peroxide-
containing drugs: A new era in malaria chemotherapy
The discovery of artemisinin and its derivatives is another milestone in the history of antimalarial
drug research since the discovery of quinine. After a brief introduction on the chemistry and
pharmacological aspects of artemisinin and other peroxide-containing drugs, we will focus this
review on the radical chemistry and alkylation properties of artemisinin and synthetic peroxides. As
we will see, a good knowledge of the basis of mechanism of action of artemisinin and derivatives
allowed the rational design of new active compounds.
III.1. Artemisinin (Qinghaosu): a natural peroxide drug from the Chinese
traditional medicine
Artemisinin (figure 10) has a structure that differs from the classical quinoline-based drugs. In
1972, isolation and characterization of artemisinin revealed that the main structural feature of this
natural tetracyclic compound is the unusual 1,2,4-trioxane cycle.[92] Artemisinin, also called
qinghaosu, is extracted from the leaves of the Chinese wormwood Artemisia annua (Qing hao).[93]
This herb was specifically recommended for fevers in the Zhou Hou Bei Ji Fang, a Chinese
handbook of prescriptions published in 341 AD. Thereafter qing hao appears in several standard
Chinese texts as a treatment for febrile illnesses.[94] In a research effort apparently prompted by the
requests of Ho Chi Minh to the Chinese minister Zhou EnLai for antimalarial drugs to protect his
Vietnamese troops, the scientists identified the active antimalarial principle, characterized its
physicochemical properties, conducted in vitro and in vivo studies.[95]
It is known that the peroxide bond of artemisinin is essential for its antimalarial activity as
deoxyartemisinin (figure 10), for which the peroxide bond is replaced by an ether bridge, is totally
inactive.[92]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
14
Figure 10: Artemisinin, Artemisia annua, C10-derivatives and inactive derivatives of artemisinin. In vitro activity against the P.f. chloroquine-resistant strains K1 or W2 is reported
Artemisinin is the fastest acting antimalarial and kills all Plasmodium species that infect humans. In
particular, it is highly active against both chloroquine sensitive- and resistant-strains of P.
falciparum in the nanomolar range.[92,96] More important, artemisinin is active against all
erythrocytic stages of Plasmodium, including young rings and gametocytes, the sexual form of the
parasite.[97-99] Effect on young-ring stages prevent their development to the more pathological
parasites, while activity on gametocytes is important for the control of transmissibility. Indeed, the
introduction of artemisinin derivatives in routine treatment has been associated with a reduction in
the subsequent incidence of P. falciparum of 47% in Thailand.[98]
In addition of its strong antimalarial efficacy, artemisinin exhibits also some activity against
Schistosoma, although the required doses in infected mice are prohibitive for use in monotherapy
(200 to 400 mg/kg).[92,100] Artemisinin has been also shown to be potent against specific breast and
prostate cancer cell lines.[101]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
15
Total chemical synthesis of artemisinin is possible and several stereoselective methods have been
reported since 1983.[102-104] However, synthesis requires many intermediate steps (>10 steps) that
would make this approach too expensive for endemic countries.
In healthy volunteers, orally administrated artemisinin is mainly metabolized during the first hepatic
pass into inactive metabolites such as deoxyartemisinin and deoxydihydroartemisinin. This high
first pass clearance is responsible for the moderate oral bioavailability of the drug (32%) compared
to intramuscular oil.[105]
III.2. Artemisinin semi-synthetic derivatives and ACTs
In order to improve the solubility of artemisinin, C10-modified derivatives have been prepared.
Their semi-synthesis starts with the reduction of artemisinin lactone carbonyl at C10 with NaBH4 to
produce the lactol dihydroartemisinin (DHA , figure 10). DHA , itself highly active, is next used for
the preparation of the clinically used artemether and artesunate, obtained by methylation and
acylation with succinic acid, respectively (figure 10). Both compounds are more potent than
artemisinin but have short plasma half-lives (c.a. 40-60 min) and are rapidly metabolized to DHA ,
which can then be eliminated in urine as a glucuronide metabolite.[106] These short half-lives can be
associated to high recrudescence levels.
Artemisinin and derivatives have proved to be well tolerated and safe drugs. Based on animal
studies, a concern that has been raised was the possible neurotoxicity of artemisinin and
derivatives.[107,108] However, no significant toxicity has been reported in patients, although
artemisinin and derivatives have been extensively used in various formulations for years.[109-111]
There is evidence that artemisinin is safe for pregnant women; however the drug is not
recommended in the first trimester of pregnancy, except in severe malaria.[95]
Artemisinin and its derivatives are active against malaria parasites at nanomolar concentrations, but
micromolar concentrations are required for toxicity to mammalian cells. One reason for this
selectivity is the enhanced uptake of the trioxane drug by the parasite; P. falciparum infected
erythrocytes concentrate [3H]-dihydroartemisinin and [14C]-artemisinin to a >100-fold higher
concentration than do uninfected erythrocytes.[112,113]
Extensive work has been carried out to improve the pharmacokinetic properties of the first
generation analogues of artemisinin and several compounds, resulting from modifications on the C-
10 position, have emerged as potential next-generation artemisinin derivatives. [114-117] Among these
derivatives, artemisone (figure 10) has completed phase II clinical trials with Bayer and MMV.[118]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
16
The compound was not found to be dramatically superior to artemisinin.[119] However, artemisone
gave good results on patients who had showed longer parasite clearance times with artemisinin.[120]
Artemisinin Combination Therapy (ACT): To prevent the development of further resistances to
antimalarial drugs, the World Health Organization guidelines recommend the use of artemisinin in
combination with a more slowly eliminated antimalarial drug (ACT).[12]
The rationale for ACTs (3 days regimen) is that the short-acting but highly potent artemisinin
derivative delivers a rapid reduction in parasite biomass (reduction by a factor ∼ 108 in parasite
number), with the remaining parasites (up to 105) being removed by the less active but more slowly
eliminated partner drug, structurally unrelated to artemisinin.[121] Distinct modes of action of
artemisinin derivatives and partner drugs should, in theory, enable the combination to kill parasites
that manifest decreased sensitivity to one agent alone, and thus avoid or delay emergence of
resistances.[9] The first ACT to be evaluated was artesunate-mefloquine, but the fixed-dose
artemether-lumefantrine combination (Coartem, Novartis) represents 75% of the ACT market
today. Other used combinations are artesunate-amodiaquine (Coarsucam, Sanofi-Aventis),
artesunate-sulfadoxine-pyrimethamine, artesunate-mefloquine.[12,122]
Despite the high efficacy of ACT, a major obstacle to large-scale use of ACTs is their cost; they are
up to twenty times more expensive than monotherapy (artemether-lumefantrine costs around US$
0.9-1.4 for a child and US$ 2.4 per adult treatment dose, compared to US$ 0.34 for
chloroquine).[123,124] The use of artemisinin and derivatives has been limited: the only source of
artemisinin is the plant Artemisia, with short and unreliable supply. As already mentioned, the total
synthesis of the parent drug artemisinin is not an option as it would make them too costly for
developing countries. Studies are ongoing to improve and sustain artemisinin supply. They include
maximization of artemisinin production by genetic modification of the plant[125] and production of
the artemisinic acid precursor in engineered yeast.[126]
In addition to their cost, a decline of artesunate-mefloquine ACT efficacy against P.falciparum has
been observed in the Thai-Cambodian border region, the historical source of resistance to
antimalarial drugs (chloroquine, …). This reduced susceptibility might be due to high-level of
mefloquine resistance, as the drug was used for monotherapy long before the introduction of
ACT.[127] However, a more worrying decline of in vivo susceptibility to artesunate in the same
region, along with prolonged parasite time clearance, has been recently reported.[128-130] Although it
is not clinically relevant yet - no artemisinin-resistance isolate has been characterized - resistance of
P. falciparum to artemisinin derivatives appears to be a real threat.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
17
Toward synthetic peroxide drugs.
The disadvantage of all of the semisynthetic compounds described is that their production requires
artemisinin as starting material. To circumvent this problem, a number of groups have attempted to
produce totally synthetic peroxide analogues that are fast acting, highly potent against asexual blood
stage infections, non-toxic, and affordable to residents of endemic regions. Some of these
compounds demonstrate promising results. These include endoperoxide analogues such as arteflene,
simplified 1,2,4-trioxanes, the dispiro-trioxolanes and dispiro-tetraoxanes, and trioxaquines.
III.3. Synthetic peroxide-containing drugs
Mechanism-based rationale drug design has allowed the synthesis of numerous peroxides over the
last three decades. We will discuss here about the most advanced in terms of development and
knowledge of their reactivity. After a rapid presentation of their pharmacological activity, we will
focus on their common reactivity toward iron and heme.
Figure 11: Structures of peroxide-containing antimalarial drugs. In vitro activity against the P.f. chloroquine-sensitive (3D7) and -resistant strains FcB1, K1 or W2 is reported

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
18
a. Arteflene
Arteflene (figure 11) is a stable analogue of Yingzhaosu A, a natural endoperoxide extracted with
putative antimalarial properties isolated from the traditional Chinese herb, Yingzhao (Artabotrys
uncinatus L).[131] Arteflene contains the 2,3-dioxabicyclo[3.3.1]nonane core of Yingzhaosu
A.[132,133] It is active in vivo (ED50 = 2.6 mg/kg against P. berghei) and has lower rate of
recrudescence and a longer plasma half-life than artemether and artesunate.[133] Arteflene was
selected in the 1990s as the clinical candidate by Hoffman-LaRoche but has been discontinued after
phase III trials because of high production cost and lack of evident advantages over artemisinin
derivatives.[134]
b. Trioxanes
Several compounds were designed to see whether the tetracyclic framework in artemisinin is really
essential for its biological activity. Results of this structure-activity relationship suggested that (i)
small structural changes around the peroxide bond can have a significant effect on the activity,[135]
and (ii) neither the peroxide function, nor the 1,2,4-trioxane ring alone, are sufficient for maximum
efficacy.[136] However, (iii) ring A and lactone ring D are not essential for antimalarial activity of
artemisinin (see figure 10, p 14).[137]
Based on this observation, Jefford et al. have prepared a large series of cis-fused cyclopenteno-
1,2,4-trioxanes. They showed good activity on chloroquine-resistant Plasmodium strains. Among
them, the racemate Fenozan BO7 (figure 11) had the most promising activity profile and was
chosen for further development. The p-fluoro substitution of BO7 has been shown important for its
in vivo activity: BO7 is as equally active as artemether when administered by oral route (ED90 = 6.0
mg/kg).[138]
A series of tricyclic trioxanes was also synthesized by Posner et al. (figure 11).[139] Many of these
compounds were highly efficacious in vivo, against multidrug-resistant P. falciparum in Aotus
monkeys, confirming that the lactone ring of artemisinin is not essential for antimalarial activity. On
the basis of mechanistic observations (vide infra), new trioxanes for which the C3-methyl (trioxane
1 series) was replaced by a C3-phenyl were prepared by enantioselective synthesis (trioxane 2
series).[140,141] Among them, 3-fluorophenyltrioxane (figure 11) showed promising in vitro and in
vivo antimalarial activity.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
19
c. Trioxolanes
Identification of the trioxolane OZ277 (RBx-11160, also called arterolane, figure 11), by
Vennerstrom and co-workers was a significant breakthrough in antimalarial drug development
efforts during the past decade.[142,143] The key pharmacophore of OZ277 is the 1,2,4-trioxolane
cycle (secondary ozonide). Preliminary structure-activity relationship clearly showed that the
antimalarial activity of dispiro-1,2,4-trioxolanes decreases when the peroxide bond is too exposed
(3,5-bis-cyclohexane 3, figure 12), or sterically inaccessible (3,5-bis-adamantane 4). A middle
ground has been met with compound 5, for which one side of the trioxolane cycle is sterically
hindered by the adamantane moiety, shown to bring stability, while the other side leaves the
peroxide bond more accessible.[144]
Figure 12: Stability and activity of dispiro-trioxolanes
A large family of potent trioxolanes has been prepared using the Griesbaum coozonolysis reaction
with the symmetrical O-methyl 2-adamantanone oxime.[145] However, most of the first trioxolanes
had limited oral bioavailability due to poor aqueous solubility and high first pass metabolism.[146]
As the spiroadamantane trioxolane pharmacophore is inherently lipophilic, the polarity of
trioxolanes was adjusted by addition of an amine or amide side chain.[146] OZ277 has the best
combination of physic-chemical properties (lipophilicity, aqueous solubility), pharmacological
properties (metabolism) and antimalarial activity.[142,147]
OZ277 has in vitro and in vivo activity superior to artemether and artesunate, and completely cures
malaria-infected mice treated via oral route at 10 mg/day. Its half-life in plasma after intravenous
injection in the rat is 3 times longer than that of artesunate (1.4 h and 0.47, respectively). Equally
important is its low susceptibility to metabolic degradation by P450 monooxygenases that give a
biopharmaceutical profile better than that of artemisinin derivatives.[142]
Clinical development of OZ277 began in 2003 through collaboration between the University of
Nebraska, MMV, and Ranbaxy Laboratories Ltd., India.[143] Unfortunately, MMV stopped funding
the project in 2006, but Ranbaxy is still developing OZ277/arterolane and the trioxolane is
advancing through phase III in India, Africa and Thailand. MMV is currently supporting the
development of the next-generation trioxolane OZ439 (figure 11).[119,148]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
20
d. Tetraoxanes
While impressive activity profiles have been observed with the 1,2,4-trioxolane class, it has been
shown in parallel that the achiral 1,2,4,5-tetraoxane template is more stable compared to ozonides.
This prediction was confirmed by the observation that 3,5-dicyclohexyl trioxolane (3, figure 13) are
inactive and unstable, whereas the close tetraoxane 6 is relatively stable and expresses good
antimalarial activity.[142,149] Tetraoxanes are cyclic peroxides originally used for the production of
macrocyclic hydrocarbons and lactones.[150,151]
Figure 13: Rationale for dispiro-tetraoxanes
Work by the Vennerstrom’s group in the early 90’s showed that symmetrical dispiro-1,2,4,5-
tetraoxanes such as WR148999 possess high in vitro antimalarial activity.[152]
However, despite being more potent than artemisinin in vitro, these compounds
did not show a good activity when tested orally, probably because of an
extensive first-pass metabolism.[153]
Given the observation that introduction of adamantane group is capable of stabilizing the
endoperoxide 3,[154] O’Neill and co-workers explored 1,2,4,5-tetraoxanes that also incorporate this
stabilizing motif (7, figure 13). They have designed unsymmetrical dispiro-tetraoxanes, easily
prepared from inexpensive materials via acid-catalyzed cyclocondensation of bis(hydroperoxides)
with various ketones. Incorporation of water-soluble and polar functionalities via amide coupling
produces several simple and achiral analogues.[155] These analogues exhibit remarkable antimalarial
activities in vitro (IC50 1.5 – 29.4 nM against 3D7) and preliminary in vivo evaluation demonstrates
that they also have promising oral activities.[154,156]
From a library of over 150 tetraoxanes, RKA182 has been selected as candidate for full formal
preclinical development. RKA182 has outstanding in vitro and in vivo activity against P.
falciparum and shows improved pharmacokinetic characteristics compared to other peroxide drugs
(see Appendix 2).

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
21
e. Trioxaquines
Using a covalent bitherapy approach, Meunier’s
group has designed and synthesized a family of
chimeric molecules named trioxaquines (figure 11).
In this approach, two pharmacophores of known
modes of action are linked covalently to create a new
drug with the expectation of achieving a net increase
in therapeutic efficacy.[157] Trioxaquines combine a
1,2,4-trioxane, like in artemisinin, and a 4-
aminoquinoline, known from quinoline-based drugs
to facilitate penetration within infected red blood
cells and interaction with intra-vacuolar heme.[158]
The first synthesized trioxaquines were found highly
active in vitro on both chloroquine-sensitive and
resistant strains of P. falciparum with IC50 values ranging from 8 to 40 nM, depending on the
trioxane substituent. As an example, trioxaquine DU1102 was found to be very active in vitro on
field isolates (IC50 mean value = 43 nM).[159] Trioxaquines have also shown promising results on P.
berghei and P. vinckei in vivo.[158,160] Like artemisinin, trioxaquine are active against all erythrocytic
stages of Plasmodium, including gametocytes.[161]
Trioxaquines are currently developed by Palumed S.A. (www.palumed.fr). Among the 120
trioxaquines that have been prepared and tested so far, PA1103 (figure 11) was selected for further
development in January 2007 and is currently in pre-clinical trials in association with Sanofi-
Aventis.[162]
IV. Fe(II)-mediated reactivity of antimalarial peroxides and
possible drug targets
IV.1. Reactivity with inorganic salts
The development of previously described peroxide-containing drugs required knowledge of the
bases about the mechanism of action of artemisinin at a molecular level. In this part, we review the
results of biomimetic studies of artemisinin and synthetic derivatives, with ferrous iron. Similar
mechanistic issues are discussed with synthetic antimalarial peroxides.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
22
a. Oxidative stress and lipid peroxidation
Since peroxides may lead to the production of free radicals, it was first suggested that artemisinin
might kill the parasite by generation of activated oxygen species such as superoxide O2-• and
hydroxyl •OH radicals, known to cause cell damage.[163,164] Few studies have reported lipid
peroxidation and membrane thiol oxidation after membranes were treated with artemisinin or
artesunate.[163,165,166] However, the peroxidation products were observed only at very high
concentrations of the drugs (> 100 µM) while the effective concentration of artemisinin is in the
low nanomolar range, i.e. ∼10,000-fold lower. This observation implies that artemisinin and
derivatives must have a more selective toxic effect to kill the parasite, rather than random cell
damage caused by free radicals.[112] We will see below that this selectivity may be due to alkylation
of cell components and alter processes that are vital for the parasite.
b. Reactivity with ferrous salt
In the early 1990s, Meshnick and co-workers showed that iron, in the form of heme, catalyzes the
reductive decomposition of artemisinin and dihydroartemisinin in vitro.[167] It has been proposed
that free intraparasitic heme, released during hemoglobin digestion, might play an important role in
the selective toxicity of artemisinin toward the parasite.[168] Since then, Posner, Jefford and co-
workers have contributed in a major way to the comprehension of artemisinin chemistry and its
reductive activation by ferrous iron, used as models of biological iron sources.[169-174]
The reaction of artemisinin with iron(II) is a reduction reaction via single electron transfer
from the metal to the antibonding orbital σσσσ* of the peroxide bond. This reductive cleavage of a
peroxide by a low-valent transition metal ion/complex is a well known reaction of peroxides
chemistry, by which oxy radicals (O-centered) are generated.
Since artemisinin is an unsymmetrical peroxide, two different pathways are possible, according to
the oxygen atom of the peroxide bond (O1 or O2) coordinated on the metal (figure 14):
� Route 1: Coordination of Fe(II) on O1 provides the complexed oxy radical 8 (O2-centered)
that readily rearranges via C-C β-scission to the primary carbon-centered radical 9 (seco-
radical). In the absence of alkylable target (vide infra), intramolecular rearrangement of this radical
leads to the formation of the ring-contracted tetrahydrofuran 10, with expulsion of Fe(II) (figure
14). This rearrangement is energetically favored by the formation of the acetate group.[175]
Formation of a C-centered radical by cleavage of the adjacent C–C bond has also been evidenced by
Jefford, using BO7 as substrate (vide infra).[172]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
23
� Route 2: Alternatively, coordination on O2 provides the oxy radical 11 (O1-centered), that
produces the secondary carbon-centered radical 12 via a 1,5-H shift.[170] Route 2a leads to 4α-
hydroxy-deoxoartemisinin (14, figure 14) with formation of the intermediate 3,4-epoxide 13, first
postulated[176] and later isolated in low yield (1%).[177]
Figure 14: Fe-mediated activation of artemisinin

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
24
The 1,5-H shift proposition called into question because the distance between the H-C4 and O1
(2.478 – 2.803 Å)[173] was suspected to exceed the critical distance for 1,5-H abstraction (2.1
Å).[174,178] However, qualitative evidence for the formation of both the primary and secondary
carbon-centered radicals was provided by spin-trapping and EPR analysis.[177,179,180] Interestingly,
all the attempts to trap the intermediate oxy radicals fail, confirming that the rearrangement of these
species proceeds extremely rapidly (with a rate constant greater than that of typical reaction of
alkoxy radical with DMPO).
In some particular conditions, deoxyartemisinin (15, figure 14) was also identified as the major
product. Its formation was proposed to involve the electrophilic high-valent iron-oxo species
(Fe(IV)=O, route 2b).[115,171,181-183] However, the groups of Jefford[173,174] and Meunier have
contested this chemical mechanism on the basis that (i) the reported Raman resonance spectra
exhibited a signal/noise ratio for the Fe-O vibration below 2,[183] while a S/N up to 20 is expected
for well-characterized metal-oxo species;[184,185] (ii) no epoxidation of electron-rich olefin (such as
cyclohexene), characteristic of the presence of a metal-oxo species, have been observed;[172,186] and
(iii) formation of deoxyartemisinin 15 can be achieved without generation of ferryl-oxo species
(route 2c).
The ratio of Fe-mediated activation products has been found to be highly dependent of reaction
conditions (iron salt, solvent, …): route 1 is preferred when artemisinin is activated by FeCl2 in
acetonitrile while route 2 becomes significant when FeBr2 is used in THF.[176]
Despite the accumulation of evidence supporting the radical
pathways, Haynes and co-workers proposed that artemisinin acts
as a masked source of hydroperoxides, which would be generated
via iron-induced heterolytic cleavage of the O2-C3 bond at low
pH (SiO2/H2SO4 in CH2Cl2).[187-189] However, according to a
report by Robert et al., artemisinin peroxide bond is stable at pH
2.1.[190] In addition, the dissociation energy of the O-O bond
being very low, the homolytic cleavage of this bond is more
likely to occur, rather than a heterolytic C-O bond cleavage.
The formation of alkyl radicals is common to all active artemisinin derivatives that have been
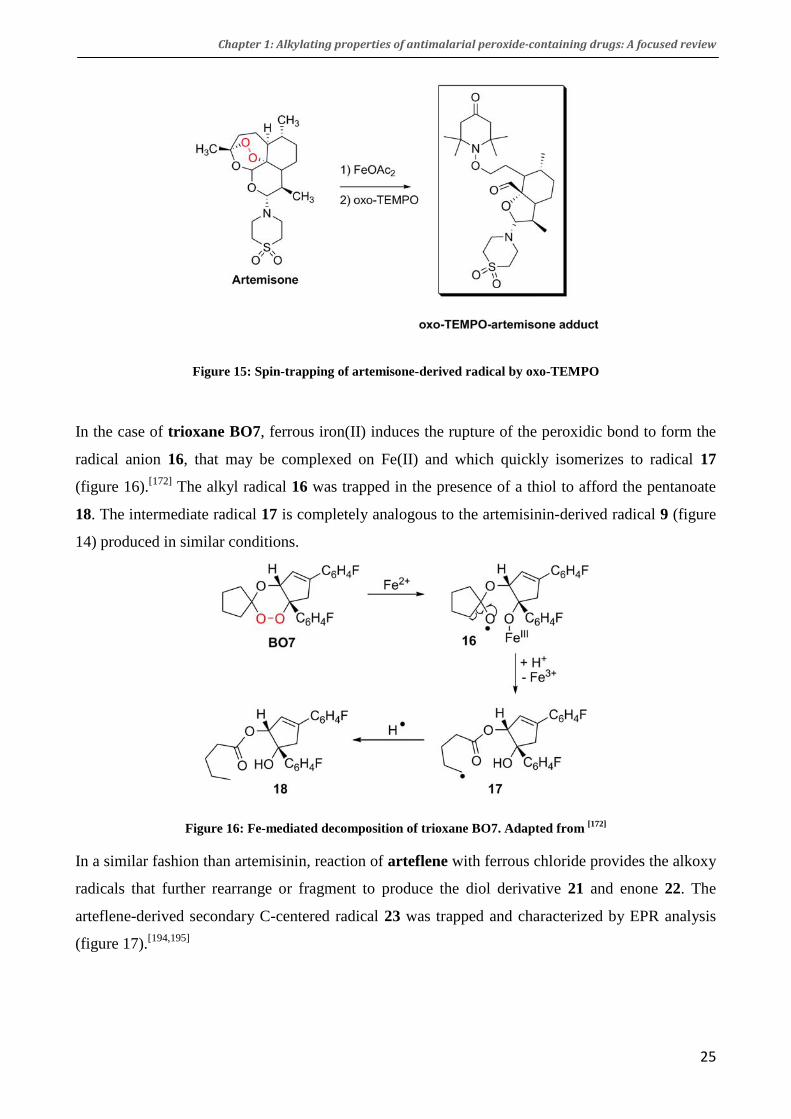
tested. Reaction of artemisone with ferrous acetate in presence of the alkyl radical spin trap 4-oxo-
2,2,6,6-tetramethyl-1-piperidine-1-oxyl (oxo-TEMPO)[191,192] provided a drug-TEMPO adduct by
trapping of the artemisone-derived seco-radical (figure 15).[193]
Table 4: Bond dissociation energy
(For information)
• RO-OR: 36 – 38 kcal/mol
• HO-OH: 51 kcal/mol
• HOO-H: 90 kcal/mol
• tBuO-H: 88 kcal/mol
• RC-O: 65 - 67 kcal/mol
Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
25
Figure 15: Spin-trapping of artemisone-derived radical by oxo-TEMPO
In the case of trioxane BO7, ferrous iron(II) induces the rupture of the peroxidic bond to form the
radical anion 16, that may be complexed on Fe(II) and which quickly isomerizes to radical 17
(figure 16).[172] The alkyl radical 16 was trapped in the presence of a thiol to afford the pentanoate
18. The intermediate radical 17 is completely analogous to the artemisinin-derived radical 9 (figure
14) produced in similar conditions.
Figure 16: Fe-mediated decomposition of trioxane BO7. Adapted from [172]
In a similar fashion than artemisinin, reaction of arteflene with ferrous chloride provides the alkoxy
radicals that further rearrange or fragment to produce the diol derivative 21 and enone 22. The
arteflene-derived secondary C-centered radical 23 was trapped and characterized by EPR analysis
(figure 17).[194,195]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
26
Figure 17: Fe-mediated activation of arteflene and radical spin-trapping.
The biomimetic Fe(II)-mediated reactivity of antimalarial dispiro-trioxolanes has been also
studied.[196,197] Reaction of trioxolane 3 with ferrous bromide produced lactone 30, bromoacid 31
and unsaturated acid 32 (figure 18) resulting from coordination of Fe(II) on O1 and subsequent C-C
β-scission of the adamantane to form the secondary radical 28. The reaction also produced 6-
bromohexanoic acid 29 resulting from coordination of Fe(II) on O2 and β-scission of
spirocyclohexanone. The reaction is strongly regioselective with a yield ratio of the products from
coordination of O1 vs. O2 of 73/3. The preferential coordination of Fe(II) is likely due to lower
steric hindrance around O1.[197]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
27
Figure 18: Fe-mediated activation of trioxolane 1.[197]
Formation of the secondary radical 28 was put into evidence by spin-trapping with nitroxide free
radical oxo-TEMPO to form the corresponding aminoxy acid 33 (figure 18). As expected by the
regioselectivity of the reaction, only adduct resulting from trapping of the secondary radical has
been isolated. Formation of TEMPO-adducts suggests that trioxolane-derived radicals survive long
enough to react with other molecules.[142,197]
The reaction of antimalarial tetraoxanes with iron has been less studied but the formation of C-
centered radicals has been reported in the literature.[198,199] In chapter 4, we further studied the Fe-
mediated reactivity of potent dispiro-1,2,4,5-tetraoxanes prepared by O’Neill’s group and
confirmed the formation of carbon-centered radicals. It should be also noted that Opsenica et al.
recently reported the spin trapping and EPR analysis of alkoxy radicals generated from steroidal
tetraoxanes.[200]
c. Reactivity with other transition metals ions
The reactivity of artemisinin with transition metal ions other than Fe is not as well documented as
with ferrous salts. Wu and co-workers studied the reaction of artemisinin with several low valent
transition metal salts, including copper salt - the only biologically relevant metal of the series. As
expected, these metals salts induce the reductive cleavage of artemisinin peroxide and all reactions
produced the same products as reported with Fe2+, despite in lower yields.[201,202]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
28
The Fe-mediated reactivity of artemisinin and synthetic peroxides depends on the same
mechanism, namely (i) reductive homolytic cleavage of the peroxide bond, (ii) formation of
oxy radicals and (iii) subsequent rearrangement to thermodynamically more stable carbon-
centered radicals.
All the reactions reported above have been carried out with iron/metals salts as models of possible
biological reactions and the results provided major insights in the mechanism of activation of
peroxide drugs. However, these models have a limited validity as “free” iron ions do not exist in
biological aerobic conditions. Due to the highly insoluble structure of [Fe(OH)3]n (Ks ∼ 10-39), iron
is indeed complexed in vivo, likely by proteins (ferritin, transferrin, heme proteins, …).
A more relevant iron complex to study the reactivity of proxide drugs is heme as (i) it is the most
abundant iron complex in the red blood cells (20 mM) and (ii) it is involved in a detoxification
process specific to Plasmodium.
IV.2. Reactivity with iron(II)-heme and biological targets:
There is no doubt that Fe-mediated reduction of artemisinin and synthetic antimalarial peroxides
generate carbon-centered radicals. How do these reactive species interact with biomolecules and
what are the implications for their antimalarial mechanism of action?
Extensive work has shown that heme could play a major role in the antimalarial activity of
artemisinin and other peroxide-containing drugs, both as activator and target. In this part, we review
results on heme alkylation by artemisinin and synthetic antimalarial peroxides. We also review the
alkylation/inhibition of parasitic proteins and enzymes that have been proposed as target for
artemisinin.
Figure 19: Bio-activation of artemisinin
a. Heme as target for artemisinin and peroxide-containing antimalarials
Alkylation of heme by artemisinin was first suggested by Meshnick after identification of heme-
drug adducts by mass spectrometry, both in solution and in parasite cultures treated with radio-
labeled artemisinin, but no structures were proposed for these adducts at that time.[203]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
29
After these first reports on the heme alkylation, Meunier and co-workers studied the reaction of
artemisinin and heme in vitro in order to identify the adducts proposed by Meshnick and investigate
the mechanism of this reaction.
In a preliminary study, the group used the synthetic manganese(II) tetraphenylporphyrin (MnIITPP)
to confirm the formation of adducts. MnTPP was first chosen as a simple heme model for its fourth-
order symmetry and eight equivalent β-pyrrolic positions that are possible alkylation sites. Another
practical reason was Mn porphyrins are easier to demetalate than their iron analogues.
Reaction of artemisinin with [MnIII(TPP)Cl], in the presence of borohydride to generate MnIITPP in
situ, gave the covalent porphyrin-drug adduct 37. This adduct is the result of alkylation by the
artemisinin-derived primary radical at the β-pyrrolic positions of the porphyrin cycle (figure
20).[204,205] Its formation can be explained by the mechanism proposed in figure 20.
Figure 20: Formation of the H2TCP-artemisinin adduct
The reductive activation of the artemisinin peroxide bond by MnIITPP produced the alkoxy radical
8 which quickly rearranged by homolytic cleavage of the C3–C4 bond to the non-sterically hindered
C-centered radical 9. The intramolecular addition of alkyl radical 9 on a β-pyrrolic carbon of the
porphyrin ring allowed the generation of the radical 35 on the adjacent β-pyrrolic position and then
the carbocation 36 by intramolecular electron transfer to Mn(III). The attack of borohydride at this

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
30
position led to the dihydropyrrole ring. Borohydride also mediated the reduction of the drug
fragment and the loss of the acetate function at C12 to provide the covalent manganese
tetraphenylchlorin (MnTPC) adduct. Further Mn removal under mild acidic conditions
(transmetalation from Mn(II) complex to its Cd(II) analogue, followed by in situ demetalation of
the cadmium complex) allowed full characterization of the resulting chlorin-type H2TPC adduct
37.[205]
It should be noted that the chlorin structure of this adduct is due to the use of borohydride and the
reduction by the latter of one of the pyrrole cycles. Borohydride here not only acts as a one-electron
reducing agent to reduce the Mn center, but also as a hydride donor toward the cation 36. Reaction
with other reducing agents leads to the porphyrin analogue of 37. The mechanistic issues of this
reaction have been discussed by the authors.[204]
Similar adducts have been prepared with artemether and the synthetic trioxane BO7.[186,206-209] To
investigate the importance of alkylation for the antimalarial activity of peroxide drugs, two other
synthetic peroxides prepared by Posner’s group were also used: trioxane 38 is highly active against
Plasmodium while its epimer 39 is totally inactive (figure 21). The difference between 38 and 39 is
the configuration at C4: in trioxane 39, the methyl substituent is on the same side than the peroxide
(with respect to the drug mean plane).
Figure 21: Activation of trioxanes 37 by an inner-sphere electron transfer: possible correlation between pharmacological activity and alkylating properties

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
31
No alkylation of MnTPP was observed with trioxane 39 while covalent adducts were characterized
with the epimer 38, suggesting that the methyl substituent at C4 prevents close interaction between
the metal and the peroxide, necessary for the reductive activation step. These data confirmed that (i)
drug activation occurs through an inner-sphere electron transfer from the low valent metal to the
peroxide bond, and (ii) more important, the alkylation ability correlates with the antimalarial
activity. Such a correlation has been shown for several other synthetic antimalarial peroxides.[190]
Artemisinin alkylates iron(II)-heme
Further investigations with iron(II)-heme showed the same reactivity of the peroxide as with
MnTPP. In few minutes at room temperature, reaction of artemisinin with iron(II)-heme led to the
formation, in excellent yield (>85%), of covalent heme-drug adducts Fe-40 (m/z 898.3 M+) and Fe-
41 (m/z 838.4 [M-CH3COOH]+). These adducts were formed by addition of the artemisinin-derived
C4-radical on the four α, β, δ, and γ meso-positions of the porphyrin, without regioselectivity
(figure 22).[210] The demetalation of the heme moiety allowed full NMR characterization of the
meso regioisomers.[210,211]
Figure 22: Alkylation of heme by artemisinin. For clarity, only the ββββ regioisomers are depicted.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
32
It should be also mentioned that a docking molecular modeling study that supports heme as a target
for artemisinin was reported. Results showed that in the most stable configuration, the endoperoxide
bridge of artemisinin is in close proximity to the iron center of heme.[212]
b. The role of heme in the antimalarial activity of artemisinin in vivo evidenced by the identification of heme-artemisnin adducts in malaria-infected mice.
Plasmodium vinckei-infected mice were treated with artemisinin by both oral (doses: 20, 50, or 100
mg/kg) and i.p. routes (doses: 100 or 200 mg/kg).[213] Mice were sacrificed 2 h after treatment,
organs were collected and extracts were analyzed by LC-MS, along with urine. The heme-drug
adducts Fe-40 and Fe-41, already known from in vitro reaction, were detected in the spleen
extracts. Spleen is the main organ involved in the elimination of damaged red blood cells.
Glucuroconjugated derivatives of these adducts were also detected in urine, indicating that these
compounds were metabolized in the liver (figure 23). Adducts were absent in non-infected mice
treated in the same conditions, indicating that alkylating ability of artemisinin in vivo is triggered by
the presence of the parasite. There is then a clear correlation between in vivo antimalarial
activity and heme alkylation.
Figure 23: Alkylation of heme by artemisinin within P. vinckei-infected mice[213]
Alkylation of heme within hemoglobin. Artemisinin is pharmacologically active on the early ring
stages of Plasmodium, when hemoglobin is not yet extensively degraded by the parasite. Meunier
and co-workers investigated the reactivity of artemisinin with intact human ferrous hemoglobin A0
(oxyHb) in order to see whether alkylation of heme within the protein can be related to its efficacy

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
33
against young ring parasites. The group reported that the heme co-factor of the protein was
efficiently alkylated by artemisinin and heme-drug adducts have been identified by comparison with
previously characterized adducts.[214] No alkylation was observed with ferric Met-hemoglobin
alone, while adducts were identified when a reducing agent was added, confirming the crucial role
of ferrous iron in artemisinin reactivity.
How heme-alkylation by artemisinin may induce the parasite death?
Artemisinin and derivatives form adducts with heme. Loup et al. reported that (i) heme-artemisinin
adducts inhibit the formation of β-hematin in vitro, in a better extent than chloroquine, and (ii) do
not dimerize themselves in the conditions of β-hematin formation.[215] A reasonable proposal for
this absence of dimerization is that the bulky artemisinin residues prevent the close stacking of the
porphyrin macrocycles. Heme-artemisinin adducts are then likely to accumulate in the food vacuole
of the parasite. Just like “free” heme, they contain a redox active iron and may generate oxidative
stress, and be responsible for the parasite death.
c. Heme-mediated reactivity of trioxolanes
Vennerstrom and co-workers studied the iron(II)-heme mediated reactivity of antimalarial
trioxolanes and reported that iron(II)-heme quickly reacts with the peroxide bond of these drugs.
The secondary alkyl radical 43 has been shown to form a covalent bond with the heme porphyrin on
the meso-positions and heme-drug adducts Fe-43 were characterized by LC-MS (figure 24).[216] A
correlation was found between the extent of heme alkylation and in vitro antimalarial activity,
suggesting that heme alkylation may be related to the mechanism of action for these
trioxolanes.[216,217]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
34
Figure 24: Formation of heme-trioxolane adduct Fe-43. For clarity, only regioisomer δδδδ is depicted
d. Heme-mediated reactivity of trioxaquines
As trioxaquines have been designed to combine the advantages of quinolines (accumulation in the
food vaccuole) and trioxane (alkylation), Meunier and co-workers evaluated the heme alkylating
ability of trioxaquines in vitro. Reaction of DU1301 with iron(II)-heme led to the formation of two
covalent heme-trioxaquine adducts Fe-47 and Fe-48, the second resulting from ester hydrolysis of
Fe-47 and loss of the terpene moiety (figure 25). These adducts were isolated, demetalated and fully
characterized.[218]
The evidence of heme alkylation by the trioxaquine DU1301 in malaria-infected mice is
reported in chapter 5.
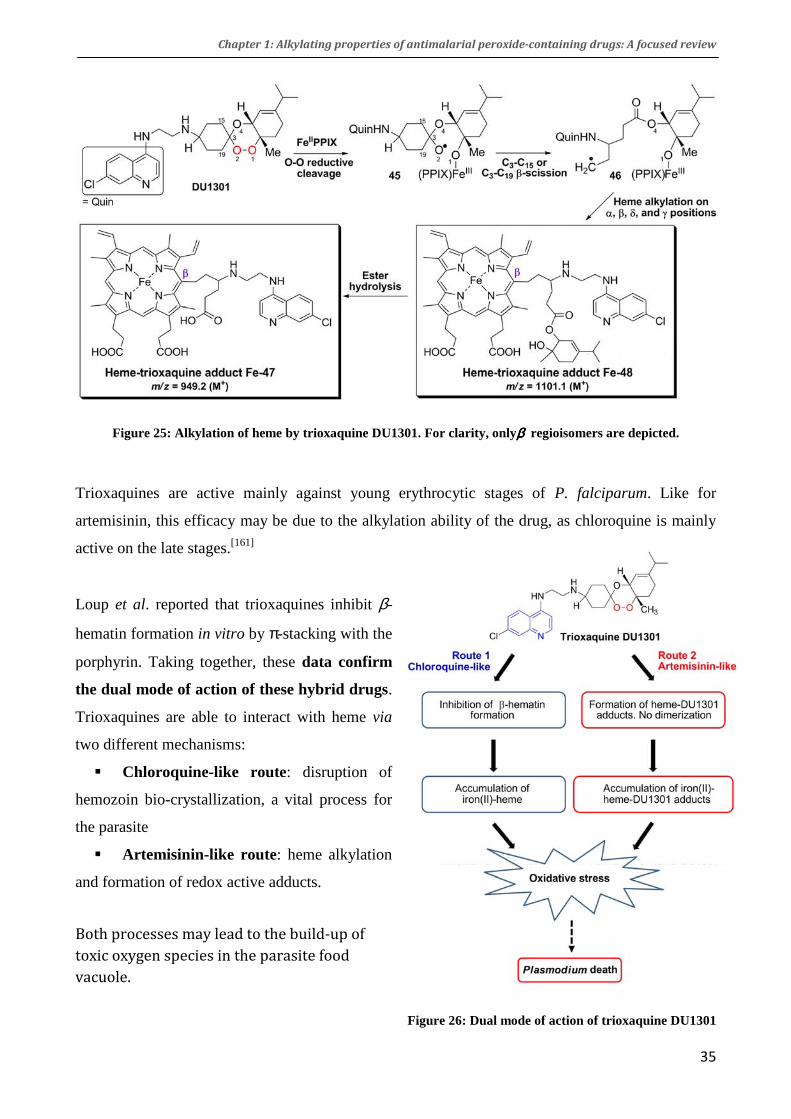
Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
35
Figure 25: Alkylation of heme by trioxaquine DU1301. For clarity, onlyββββ regioisomers are depicted.
Trioxaquines are active mainly against young erythrocytic stages of P. falciparum. Like for
artemisinin, this efficacy may be due to the alkylation ability of the drug, as chloroquine is mainly
active on the late stages.[161]
Loup et al. reported that trioxaquines inhibit β-
hematin formation in vitro by π-stacking with the
porphyrin. Taking together, these data confirm
the dual mode of action of these hybrid drugs.
Trioxaquines are able to interact with heme via
two different mechanisms:
� Chloroquine-like route: disruption of
hemozoin bio-crystallization, a vital process for
the parasite
� Artemisinin-like route : heme alkylation
and formation of redox active adducts.
Both processes may lead to the build-up of
toxic oxygen species in the parasite food
vacuole.
Figure 26: Dual mode of action of trioxaquine DU1301

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
36
IV.3. Proteins as possible targets for artemisinin
a. GSH as enzyme active site model
The heme alkylation is an intramolecular reaction, which would explain its high efficiency. To
study the possible alkylation of cysteine residues, Wu and co-workers presented a system where the
drug was activated with an iron-glutathione complex (γ-Glu_Cys-Gly). In these conditions, the
artemisinin-derived radicals reacted also in an intramolecular reaction with the thiyl radical of
amino acid chelates. Artemisinin-glutathione adducts were then identified.[199,202]
The alkylation of a protein via formation of a thioether bond is a plausible reaction for the
artemisinin.
b. Alkylation of parasite proteins
Protein alkylation was first reported by Meshnick in vitro with human serum albumin using
[14C]artemisinin and [3H]dihydroartemisinin. Mass spectrometry revealed that two molecules of
artemisinin where covalently attached to the protein.[219]
Later, the same group showed that when P. falciparum-infected erythrocytes are incubated with
[3H]dihydroartemisinin (DHA ), [3H]arteether and [14C]arteflene, besides heme, several malarial
proteins were covalently labeled.[220]
One of the major alkylated proteins was the translationally controlled tumor protein (TCTP), a 25
kDa parasitic protein that binds heme.[221] In P. falciparum, this protein – of which the function is
unknown - appears to be associated with food vacuolar membranes.[222] The covalent linkage
between DHA and the protein has been confirmed in vitro, with recombinant P. falciparum
TCTP.[221] It is dependent on the presence of heme and of the single cysteine residue in the TCTP
sequence.
c. PfATP6 inhibition
In addition to heme alkylation, an alternative mechanism of action for artemisinin has been
suggested. It involves PfATP6, a sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) orthologue
of P. falciparum and homologous to the mammalian SERCA1a. In 2003, Krishna and co-workers
reported that artemisinin inhibits PfATP6 overexpressed in Xenopus oocytes (Ki = 150 nM).[223]
Inhibition was also observed with artemisone (Ki = 1.7 nM)[224] and an antagonistic interaction was
also reported between artemisinin and thapsigargin (TG, figure 27), a specific mammalian SERCA
inhibitor,[225,226] although the antagonistic effect was not confirmed in a more recent publication.[227]

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
37
Figure 27: Structures of artemisinin and thapsigargine
Additional studies in Xenopus oocytes suggest that mutations in position 263 can modulate the
affinity of artemisinin for PfATP6.[224] Mutation S796N was also proposed to be related to
decreased sensitivity of the parasite to artemether, an artemisinin derivative.[228] However, no
mutations were found in atp6 genes of laboratory artemisinin-resistant P. falciparum and P.
chabaudi strains,[229] or in isolates of P. falciparum from Western Cambodia that showed a decrease
of susceptibility to artesuante in vivo.[128]
Krishna and coll. also reported that the inactive deoxyartemisinin, chloroquine and quinine do not
inhibit PfATP6 activity. Effect of artemisinin on PfATP6 was attenuated by the iron chelator
desferrioxamine, suggesting an iron-dependent activation mechanism, however no alkyation has
been proven with PfATP6. Trioxolane OZ277 showed poor inhibition of PfATP6 overexpressed in
Xenopus oocytes (Ki = 7700 nM).[230]
Although several reports do not support the direct implication of the protein in the mechanism of
action of artemisinin, the inhibition of PfATP6 has been relayed in a plethora of articles – the article
of Krishna and coll. in Nature 2003[223] is 7th most cited article of 2837 articles using “artemisinin”
as key-word on Web of science, accessed on 21/01/10 – and is often presented as the definite target
of artemisinin.
In order to provide information on the possible role of the PfATP6 calcium pump in the mechanism
of action of artemisinin and related compounds, we performed a docking study of a series of
structurally different antimalarial drugs into the thapsigargin-binding cleft of a PfATP6
model. In contrast to a recent report,[231] our study was not limited to artemisinin derivatives, but
included other peroxide- and quinoline-based antimalarial agents. Our objective was to determine
whether the binding affinity of the studied drugs to PfATP6 can be significantly correlated to their
in vitro antiplasmodial activity. The results of this study are reported in chapter 6.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
38
V. Conclusion and scope of the thesis
Alkylation of both heme and proteins has been reported to occur in vitro and/or in vivo. More work
is still needed to know which of these processes is the most relevant for artemisinin antiparasitic
effect. However, it is likely that both play a role in the impressive antimalarial activity of
artemisinin and derivatives.
In the following chapters, we confirm the alkylation properties of antimalarial peroxides toward
heme by studying the in vitro reactivity of artemisone and the new class of dispiro-tetraoxanes
(chapter 2 and 4, respectively).
We also confirmed the alkylation of heme by trioxaquine in infected mice (chapter 5).
In order to explore the possibility of reductive activation of artemisinin by other redox active metal
present in vivo, we have studied the alkylation ability of artemisinin with copper complexes
presenting possible alkylable sites (chapter 3).
Finally, we carried out a modeling study of PfATP6 with various antimalarial drugs and analogues,
to see whether the inhibition of protein observed in vitro is translated in terms of drug/protein
affinity in silico.
VI. Bibliography
[1] B. M. Greenwood, K. Bojang, C. J. M. Whitty, G. A. T. Targett. Malaria. Lancet 2005, 365, 1487-1498. [2] W. D. Nicol. Monkey Malaria in G.P.I. Br Med J 1935, 2, 760. [3] W. Chin, P. G. Contacos, G. R. Coatney, H. R. Kimball. A Naturally Acquired Quotidian-Type Malaria in Man Transferable to Monkeys. Science 1965, 149, 865-. [4] J. Cox-Singh, J. Hiu, S. Lucas, P. Divis, M. Zulkarnaen, P. Chandran, K. Wong, P. Adem, S. Zaki, B. Singh, S. Krishna. Severe Malaria - a Case of Fatal Plasmodium Knowlesi Infection with Post-Mortem Findings: A Case Report. Malar. J. 2010, 9, 10. [5] M. R. Galinski, J. W. Barnwell. Monkey Malaria Kills Four Humans . Trends Parasitol. 2009, 25, 200-204. [6] R. Sallares, A. Bouwman, C. Anderung. The Spread of Malaria to Southern Europe in Antiquity: New Approaches to Old Problems. Medical History 2004, 48, 311-328. [7] Malaria Site, History of Malaria During Wars . http://www.malariasite.com/malaria/history_wars.htm, accessed on 15/01/2010.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
39
[8] D. A. Joy, X. R. Feng, J. B. Mu, T. Furuya, K. Chotivanich, A. U. Krettli, M. Ho, A. Wang, N. J. White, E. Suh, P. Beerli, X. Z. Su. Early Origin and Recent Expansion of Plasmodium falciparum. Science 2003, 300, 318-321. [9] B. M. Greenwood, D. A. Fidock, D. E. Kyle, S. H. I. Kappe, P. L. Alonso, F. H. Collins, P. E. Duffy. Malaria: Progress, Perils, and Prospects for Eradication. J. Clin. Invest. 2008, 118, 1266-1276. [10] R. W. Snow, C. A. Guerra, A. M. Noor, H. Y. Myint, S. I. Hay. The Global Distribution of Clinical Episodes of Plasmodium falciparum Malaria. Nature 2005, 434, 214-217. [11] B. Greenwood. Between Hope and a Hard Place. Nature 2004, 430, 926-927. [12] WHO. Guidelines for the Treatment of Malaria. 2006. www.who.int/malaria/docs/TreatmentGuidelines2006.pdf [13] S. Parmet, C. Lynm, R. M. Glass. JAMA Patient Page. Malaria. JAMA 2007, 297, 2310. [14] S. C. Murphy, J. G. Breman. Gaps in the Childhood Malaria Burden in Africa: Cerebral Malaria, Neurological Sequelae, Anemia, Respiratory Distress, Hypoglycemia, and Complications of Pregnancy. Am. J. Trop. Med. Hyg. 2001, 64, 57-67. [15] A. Targett Geoffrey, M. Greenwood Brian. Malaria Vaccines and Their Potential Role in the Elimination of Malaria. Malar. J. 2008, 7 Suppl 1, S10. [16] W. R. Ballou. The Development of the RTS,S Malaria Vaccine Candidate: Challenges and Lessons. Parasite Immunol. 2009, 31, 492-500. [17] E. F. Roth, D. S. Brotman, J. P. Vanderberg, S. Schulman. Malarial Pigment-Dependent Error in the Estimation of Hemoglobin Content in Plasmodium falciparum-Infected Red-Cells: Implications for Metabolic and Biochemical Studies of Erythrocytic Phases of Malaria Am. J. Trop. Med. Hyg. 1986, 35, 906-911. [18] S. E. Francis, D. J. Sullivan, D. E. Goldberg. Hemoglobin Metabolism in the Malaria Parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997, 51, 97-123. [19] D. E. Goldberg, A. F. G. Slater, A. Cerami, G. B. Henderson. Hemoglobin Degradation in the Malaria Parasite Plasmodium falciparum: An Ordered Process in a Unique Organelle. Proc. Nat. Acad. Sci. U.S.A 1990, 87, 2931-2935. [20] M. Krugliak, J. M. Zhang, H. Ginsburg. Intraerythrocytic Plasmodium falciparum Utilizes Only a Fraction of the Amino Acids Derived from the Digestion of Host Cell Cytosol for the Biosynthesis of Its Proteins. Mol. Biol. Parasitol. 2002, 119, 249-256. [21] P. Browne, O. Shalev, R. P. Hebbel. The Molecular Pathobiology of Cell Membrane Iron: The Sickle Red Cell as a Model. Free Radical Biol. Med. 1998, 24, 1040-1048. [22] C. D. Fitch, P. Kanjananggulpan. The State of Ferriprotoporphyrin IX in Malaria Pigment. J. Biol. Chem. 1987, 262, 15552-15555.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
40
[23] T. J. Egan, J. M. Combrinck, J. Egan, G. R. Hearne, H. M. Marques, S. Ntenteni, B. T. Sewell, P. J. Smith, D. Taylor, D. A. van Schalkwyk, J. C. Walden. Fate of Haem Iron in the Malaria Parasite Plasmodium falciparum. Biochem. J. 2002, 365, 343-347. [24] D. J. Sullivan, Jr., I. Y. Gluzman, D. E. Goldberg. Plasmodium Hemozoin Formation Mediated by Histidine-Rich Proteins. Science 1996, 271, 219-222. [25] R. J. Howard, S. Uni, M. Aikawa, S. B. Aley, J. H. Leech, A. M. Lew, T. E. Wellems, J. Rener, D. W. Taylor. Secretion of A Malarial Histidine-Rich Protein (PFHRP-II) from Plasmodium Falciparum-Infected Erythrocytes. J. Cell Biol. 1986, 103, 1269-1277. [26] A. Lynn, S. Chandra, P. Malhotra, V. S. Chauhan. Heme Binding and Polymerization by Plasmodium falciparum Histidine Rich Protein II: Influence of Ph on Activity and Conformation. FEBS Lett. 1999, 459, 267-271. [27] S. Pagola, P. W. Stephens, D. S. Bohle, A. D. Kosar, S. K. Madsen. The Structure of Malaria Pigment Beta-Haematin. Nature 2000, 404, 307-310. [28] A. F. G. Slater, W. J. Swiggard, B. R. Orton, W. D. Flitter, D. E. Goldberg, A. Cerami, G. B. Henderson. An Iron-Carboxylate Bond Links the Heme Units of Malaria Pigment. Proc. Nat. Acad. Sci. U.S.A 1991, 88, 325-329. [29] C. D. Fitch. Ferriprotoporphyrin IX, Phospholipids, and the Antimalarial Actions of Quinoline Drugs. Life Sci. 2004, 74, 1957-1972. [30] M. Mungthin, P. G. Bray, R. G. Ridley, S. A. Ward. Central Role of Hemoglobin Degradation in Mechanisms of Action of 4-Aminoquinolines, Quinoline Methanols, and Phenanthrene Methanols. Antimicrob. Agents. Chemother. 1998, 42, 2973-2977. [31] D. J. Sullivan, H. Matile, R. G. Ridley, D. E. Goldberg. A Common Mechanism for Blockade of Heme Polymerization by Antimalarial Quinolines. J. Biol. Chem. 1998, 273, 31103-31107. [32] S. Fu, A. Bjorkman, B. Wahlin, D. Oforiadjei, O. Ericsson, F. Sjoqvist. Invitro Activity of Chloroquine, the 2 Enantiomers of Chloroquine, Desethylchloroquine and Pyronaridine against Plasmodium-Falciparum. Br. J. Clin. Pharmacol. 1986, 22, 93-96. [33] T. S. Kaufman, E. A. Ruveda. The Quest for Quinine: Those Who Won the Battles and Those Who Won the War. Angew. Chem. Int. Ed. 2005, 44, 854-885. [34] A. Yeka, J. Achan, U. D'Alessandro, A. O. Talisuna. Quinine Monotherapy for Treating Uncomplicated Malaria in the Era of Artemisinin-Based Combination Therapy: An Appropriate Public Health Policy? Lancet Infec. Dis. 2009, 9, 448-452. [35] S. R. Meshnick. Why Does Quinine Still Work after 350 Years of Use? Parasitol. Today 1997, 13, 89-90. [36] I. S. Adagu, D. C. Warhurst, W. N. Ogala, I. Abduaguye, L. I. Audu, F. O. Bamgbola, U. B. Ovwigho. Antimalarial Drug Response of Plasmodium falciparum from Zaria, Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1995, 89, 422-425.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
41
[37] M. G. Zalis, L. Pang, M. S. Silveira, W. K. Milhous, D. F. Wirth. Characterization of Plasmodium falciparum Isolated from the Amazon Region of Brazil: Evidence for Quinine Resistance. Am. J. Trop. Med. Hyg. 1998, 58, 630-637. [38] A. C. Chou, R. Chevli, C. D. Fitch. Ferriprotoporphyrin IX Fulfills the Criteria for Identification as the Chloroquine Receptor of Malaria Parasites. Biochemistry 1980, 19, 1543-1549. [39] A. F. G. Slater, A. Cerami. Inhibition by Chloroquine of a Novel Heme Polymerase Enzyme-Activity in Malaria Trophozoites. Nature 1992, 355, 167-169. [40] P. A. Winstanley, A. M. Breckenridge. Currently Important Antimalarial-Drugs . Ann. Trop. Med. Parasitol. 1987, 81, 619-627. [41] A. Yayon, Z. I. Cabantchik, H. Ginsburg. Identification of the Acidic Compartment of Plasmodium falciparum-Infected Human-Erythrocytes as the Target of the Antimalarial Drug Chloroquine. EMBO J. 1984, 3, 2695-2700. [42] C. D. Fitch, N. G. Yunis, R. Chevli, Y. Gonzalez. High-Affinity Accumulation of Chloroquine by Mouse Erythrocytes Infected with Plasmodium Berghei. J. Clin. Invest. 1974, 54, 24-33. [43] D. J. Sullivan, Jr., I. Y. Gluzman, D. G. Russell, D. E. Goldberg. On the Molecular Mechanism of Chloroquine's Antimalarial Action. Proc. Nat. Acad. Sci. U.S.A 1996, 93, 11865-11870. [44] T. J. Egan, D. C. Ross, P. A. Adams. Quinoline Antimalarial-Drugs Inhibit Spontaneous Formation of Beta-Hematin (Malaria Pigment). FEBS Lett. 1994, 352, 54-57. [45] A. Dorn, S. R. Vippagunta, H. Matile, C. Jaquet, J. L. Vennerstrom, R. G. Ridley. An Assessment of Drug-Haematin Binding as a Mechanism for Inhibition of Haematin Polymerisation by Quinoline Antimalarials. Biochem. Pharmacol. 1998, 55, 727-736. [46] S. Moreau, B. Perly, J. Biguet. Interactions between Chloroquine and Ferriprotoporphyrine IX. Nuclear Magnetic-Resonance Study. Biochimie 1982, 64, 1015-1025. [47] C. D. Fitch, R. Chevli, H. S. Banyal, G. Phillips, M. A. Pfaller, D. J. Krogstad. Lysis of Plasmodium falciparum by Ferriprotoporphyrin IX and a Chloroquine-Ferriprotoporphyrin IX Complex. Antimicrob. Agents Chemother. 1982, 21, 819-822. [48] S. J. Foote, A. F. Cowman. The Mode of Action and the Mechanism of Resistance to Antimalarial-Drugs . Acta Trop. 1994, 56, 157-171. [49] H. Ginsburg. Should Chloroquine Be Laid to Rest? Acta Trop. 2005, 96, 16-23. [50] C. D. Fitch. Chloroquine Resistance in Malaria: A Deficiency of Chloroquine Binding. Proc. Nat. Acad. Sci. U.S.A 1969, 64, 1181-1187. [51] D. J. Krogstad, I. Y. Gluzman, D. E. Kyle, A. M. J. Oduola, S. K. Martin, W. K. Milhous, P. H. Schlesinger. Efflux of Chloroquine from Plasmodium falciparum: Mechanism of Chloroquine Resistance. Science 1987, 238, 1283-1285.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
42
[52] V. Lakshmanan, P. G. Bray, D. Verdier-Pinard, D. J. Johnson, P. Horrocks, R. A. Muhle, G. E. Alakpa, R. H. Hughes, S. A. Ward, D. J. Krogstad, A. B. S. Sidhu, D. A. Fidock. A Critical Role for PfCRT K76T in Plasmodium falciparum Verapamil-Reversible Chloroquine Resistance. EMBO J. 2005, 24, 2294-2305. [53] P. G. Bray, R. E. Martin, L. Tilley, S. A. Ward, K. Kirk, D. A. Fidock. Defining the Role of PfCRT in Plasmodium falciparum Chloroquine Resistance. Mol. Microbiol. 2005, 56, 323-333. [54] R. E. Martin, R. V. Marchetti, A. I. Cowan, S. M. Howitt, S. Broer, K. Kirk. Chloroquine Transport via the Malaria Parasite's Chloroquine Resistance Transporter. Science 2009, 325, 1680-1682. [55] P. Ringwald, E. C. M. Eboumbou, J. Bickii, L. K. Basco. In Vitro Activities of Pyronaridine, Alone and in Combination with Other Antimalarial Drugs, against Plasmodium falciparum. Antimicrob. Agents Chemother. 1999, 43, 1525-1527. [56] A. L. Nelson, A. Purfield, P. McDaniel, N. Uthaimongkol, N. Buathong, S. Sriwichai, R. S. Miller, C. Wongsrichanalai, S. R. Meshnick. pfmdr1 Genotyping and in Vivo Mefloquine Resistance on the Thai-Myanmar Border. Am. J. Trop. Med. Hyg. 2005, 72, 586-592. [57] S. Toovey. Mefloquine Neurotoxicity: A Literature Review . Travel Med Infect Dis 2009, 7, 2-6. [58] T. N. C. Wells, P. L. Alonso, W. E. Gutteridge. New Medicines to Improve Control and Contribute to the Eradication of Malaria. Nature Rev. Drug. Discov. 2009, 8, 879-891. [59] R. Chevli, C. D. Fitch. The Antimalarial Drug Mefloquine Binds to Membrane Phospholipids. Antimicrob. Agents Chemother. 1982, 21, 581-586. [60] M. Foley, L. Tilley. Quinoline Antimalarials: Mechanisms of Action and Resistance. Int. J. Parasitol. 1997, 27, 231-240. [61] D. De, F. M. Krogstad, F. B. Cogswell, D. J. Krogstad. Aminoquinolines That Circumvent Resistance in Plasmodium falciparum in Vitro. Am. J. Trop. Med. Hyg. 1996, 55, 579-583. [62] R. Ridley, W. Hofheinz, H. Matile, C. Jaquet, A. Dorn, R. Masciadri, S. Jolidon, W. Richter, A. Guenzi, M. Girometta, H. Urwyler, W. Huber, S. Thaithong, W. Peters. 4-Aminoquinoline Analogs of Chloroquine with Shortened Side Chains Retain Activity against Chloroquine-Resistant Plasmodium falciparum. Antimicrob. Agents Chemother. 1996, 40, 1846-1854. [63] P. A. Stocks, K. J. Raynes, P. G. Bray, B. K. Park, P. M. O'Neill, S. A. Ward. Novel Short Chain Chloroquine Analogues Retain Activity against Chloroquine Resistant K1 Plasmodium falciparum. J. Med. Chem. 2002, 45, 4975-4983. [64] P. M. O'Neill, A. E. Shone, D. Stanford, G. Nixon, E. Asadollahy, B. K. Park, J. L. Maggs, P. Roberts, P. A. Stocks, G. Biagini, P. G. Bray, J. Davies, N. Berry, C. Hall, K. Rimmer, P. A. Winstanley, S. Hindley, R. B. Bambal, C. B. Davis, M. Bates, S. L. Gresham, R. A. Brigandi, F. M. Gomez-de-las-Heras, D. V. Gargallo, S. Parapini, L. Vivas, H. Lander, D. Taramelli, S. A. Ward. Synthesis, Antimalarial Activity, and Preclinical Pharmacology of a Novel Series of 4'-Fluoro and 4'-Chloro Analogues of Amodiaquine. Identification of a Suitable "Back-Up" Compound for N-tert-Butyl Isoquine. J. Med. Chem. 2009, 52, 1828-1844.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
43
[65] S. R. Hawley, P. G. Bray, B. K. Park, S. A. Ward. Amodiaquine Accumulation in Plasmodium falciparum as a Possible Explanation for Its Superior Antimalarial Activity over Chloroquine. Mol. Biol. Parasitol. 1996, 80, 15-25. [66] J. L. Maggs, M. D. Tingle, N. R. Kitteringham, B. K. Park. Drug-Protein Conjugates .14. Mechanisms of Formation of Protein-Arylating Intermediates from Amodiaquine, A Myelotoxin and Hepatotoxin in Man. Biochem. Pharmacol. 1988, 37, 303-311. [67] P. M. O'Neill, A. Mukhtar, P. A. Stocks, L. E. Randle, S. Hindley, S. A. Ward, R. C. Storr, J. F. Bickley, I. A. O'Neil, J. L. Maggs, R. H. Hughes, P. A. Winstanley, P. G. Bray, B. K. Park. Isoquine and Related Amodiaquine Analogues: A New Generation of Improved 4-Aminoquinoline Antimalarials . J. Med. Chem. 2003, 46, 4933-4945. [68] P. M. O'Neill, B. K. Park, A. E. Shone, J. L. Maggs, P. Roberts, P. A. Stocks, G. A. Biagini, P. G. Bray, P. Gibbons, N. Berry, P. A. Winstanley, A. Mukhtar, R. Bonar-Law, S. Hindley, R. B. Bambal, C. B. Davis, M. Bates, T. K. Hart, S. L. Gresham, R. M. Lawrence, R. A. Brigandi, F. M. Gomez-Delas-Heras, D. V. Gargallo, S. A. Ward. Candidate Selection and Preclinical Evaluation of N-tert-Butyl Isoquine (GSK369796), an Affordable and Effective 4-Aminoquinoline Antimalarial for the 21st Century. J. Med. Chem. 2009, 52, 1408-1415. [69] C. B. Davis, R. Bambal, G. S. Moorthy, E. Hugger, H. Xiang, R. K. Park, A. E. Shone, P. M. O'Neill, S. A. Ward. Comparative Preclinical Drug Metabolism and Pharmacokinetic Evaluation of Novel 4-Aminoquinoline Anti-Malarials. J. Pharm. Sci. 2009, 98, 362-377. [70] R. M. Lawrence, K. C. Dennis, P. M. O'Neill, D. U. Hahn, M. Roeder, C. Struppe. Development of a Scalable Synthetic Route to GSK369796 (N-tert-Butyl Isoquine), a Novel 4-Aminoquinoline Antimalarial Drug. Org. Process Res. Dev. 2008, 12, 294-297. [71] S. Top, J. Tang, A. Vessieres, D. Carrez, C. Provot, G. Jaouen. Ferrocenyl Hydroxytamoxifen: A Prototype for a New Range of Oestradiol Receptor Site-Directed Cytotoxics. J. Chem. Soc., Chem.Commun. 1996, 955-956. [72] S. Top, B. Dauer, J. Vaissermann, G. Jaouen. Facile Route to Ferrocifen, 1-[4-(2-Dimethylaminoethoxy)]-1-(Phenyl-2-Ferrocenyl-but-1-Ene), First Organometallic Analogue of Tamoxifen, by the McMurry Reaction. J. Organomet. Chem. 1997, 541, 355-361. [73] J. Brocard, J. Lebibi, L. Maciejewski. Preparation of Antimalarial Organometallic Iron Complexes. International Patent PCT/FR 96/00721. 1996. [74] D. Dive, C. Biot. Ferrocene Conjugates of Chloroquine and Other Antimalarials: The Development of Ferroquine, a New Antimalarial. ChemMedChem 2008, 3, 383-391. [75] L. Delhaes, C. Biot, L. Berry, P. Delcourt, L. A. Maciejewski, D. Camus, J. S. Brocard, D. Dive. Synthesis of Ferroquine Enantiomers: First Investigation of Effects of Metallocenic Chirality Upon Antimalarial Activity and Cytotoxicity. ChemBioChem 2002, 3, 418-423. [76] B. Pradines, T. Fusai, W. Daries, V. Laloge, C. Rogier, P. Millet, E. Panconi, M. Kombila, D. Parzy. Ferrocene-Chloroquine Analogues as Antimalarial Agents: In Vitro Activity of Ferrochloroquine against 103 Gabonese Isolates of Plasmodium falciparum. J. Antimicrob. Chemother. 2001, 48, 179-184.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
44
[77] B. Pradines, A. Tall, C. Rogier, A. Spiegel, J. Mosnier, L. Marrama, T. Fusai, P. Millet, E. Panconi, J. F. Trape, D. Parzy. In Vitro Activities of Ferrochloroquine against 55 Senegalese Isolates of Plasmodium falciparum in Comparison with Those of Standard Antimalarial Drugs. Trop. Med. Int. Health 2002, 7, 265-270. [78] C. Atteke, J. M. M. Ndong, A. Aubouy, L. Maciejewski, J. Brocard, J. Lebibi, P. Deloron. In Vitro Susceptibility to a New Antimalarial Organometallic Analogue, Ferroquine, of Plasmodium falciparum Isolates from the Haut-Ogooue Region of Gabon. J. Antimicrob. Chemother. 2003, 51, 1021-1024. [79] P. Chim, P. Lim, R. Sem, S. Nhem, L. Maciejewski, T. Fandeur. The in Vitro Antimalarial Activity of Ferrochloroquine, Measured against Cambodian Isolates of Plasmodium falciparum. Ann. Trop. Med. Parasitol. 2004, 98, 419-424. [80] M. Barends, A. Jaidee, N. Khaohirun, P. Singhasivanon, F. Nosten. In Vitro Activity of Ferroquine (SSR 97193) against Plasmodium falciparum Isolates from the Thai-Burmese Border. Malar. J. 2007, 6. [81] W. Daher, C. Biot, T. Fandeur, H. Jouin, L. Pelinski, E. Viscogliosi, L. Fraisse, B. Pradines, J. Brocard, J. Khalife, D. Dive. Assessment of Plasmodium falciparum Resistance to Ferroquine (SSR97193) in Field Isolates and in W2 Strain under Pressure. Malar. J. 2006, 5. [82] C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo, D. Dive, J. Lebibi. Synthesis and Antimalarial Activity in Vitro and in Vivo of a New Ferrocene-Chloroquine Analogue. J. Med. Chem. 1997, 40, 3715-3718. [83] C. Biot, D. Taramelli, I. Forfar-Bares, L. A. Maciejewski, M. Boyce, G. Nowogrocki, J. S. Brocard, N. Basilico, P. Olliaro, T. J. Egan. Insights into the Mechanism of Action of Ferroquine. Relationship between Physicochemical Properties and Antiplasmodial Activity. Mol. Pharm. 2005, 2, 185-193. [84] N. Chavain, H. Vezin, D. Dive, N. Touati, J. F. Paul, E. Buisine, C. Biot. Investigation of the Redox Behavior of Ferroquine, a New Antimalarial. Mol. Pharm. 2008, 5, 710-716. [85] P. Beagley, M. A. L. Blackie, K. Chibale, C. Clarkson, J. R. Moss, P. J. Smith. Synthesis and Antimalarial Activity in Vitro of New Ruthenocene-Chloroquine Analogues. J. Chem. Soc., Dalton Trans. 2002, 4426-4433. [86] W. Daher, L. Pelinski, S. Klieber, F. Sadoun, V. Meunier, M. Bourrie, C. Biot, O. Guillou, E. Fabre, J. Brocard, L. Fraisse, J. P. Maffrand, J. Khalife, D. Dive. In Vitro Metabolism of Ferroquine (SSR97193) in Animal and Human Hepatic Models and Antimalarial Activity of Major Metabolites on Plasmodium falciparum. Drug Metab. Dispos. 2006, 34, 667-682. [87] A. Kreidenweiss, P. G. Kremsner, K. Dietz, B. Mordmuller. In Vitro Activity of Ferroquine (SAR97193) Is Independent of Chloroquine Resistance in Plasmodium falciparum. Am. J. Trop. Med. Hyg. 2006, 75, 1178-1181. [88] C. Biot, L. Delhaes, L. A. Maciejewski, M. Mortuaire, D. Camus, D. Dive, J. S. Brocard. Synthetic Ferrocenic Mefloquine and Quinine Analogues as Potential Antimalarial Agents. Eur. J. Med. Chem. 2000, 35, 707-714.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
45
[89] S. Paitayatat, B. Tarnchompoo, Y. Thebtaranonth, Y. Yuthavong. Correlation of Antimalarial Activity of Artemisinin Derivatives with Binding Affinity with Ferroprotoporphyrin IX . J. Med. Chem. 1997, 40, 633-638. [90] L. Delhaes, C. Biot, L. Berry, L. A. Maciejewski, D. Camus, J. S. Brocard, D. Dive. Novel Ferrocenic Artemisinin Derivatives: Synthesis, in Vitro Antimalarial Activity and Affinity of Binding with Ferroprotoporphyrin IX . Bioorg. Med. Chem. 2000, 8, 2739-2745. [91] A. Baramee, A. Coppin, M. Mortuaire, L. Pelinski, S. Tomavo, J. Brocard. Synthesis and in Vitro Activities of Ferrocenic Aminohydroxynaphthoquinones against Toxoplasma Gondii and Plasmodium falciparum. Bioorg. Med. Chem. 2006, 14, 1294-1302. [92] D. L. Klayman. Qinghaosu (Artemisinin): An Antimalarial Drug from China. Science 1985, 228, 1049-1055. [93] H. J. Woerdenbag, C. B. Lugt, N. Pras. Artemisia Annua L: A Source of Novel Antimalarial-Drugs . Pharm. Weekbl. Sci. Ed. 1990, 12, 169-181. [94] T. T. Hien, N. J. White. Qinghaosu. Lancet 1993, 341, 603-608. [95] N. J. White. Qinghaosu (Artemisinin): The Price of Success. Science 2008, 320, 330-334. [96] N. White. Assessment of the Pharmacodynamic Properties of Antimalarial Drugs in Vivo. Antimicrob. Agents Chemother. 1997, 41, 1413-1422. [97] T. S. Skinner, L. S. Manning, W. A. Johnston, T. M. E. Davis. In Vitro Stage-Specific Sensitivity of Plasmodium falciparum to Quinine and Artemisinin Drugs. Int. J. Parasitol. 1996, 26, 519-525. [98] R. N. Price, F. Nosten, C. Luxemburger, F. O. ter Kuile, L. Paiphun, T. Chongsuphajaisiddhi, N. J. White. Effects of Artemisinin Derivatives on Malaria Transmissibility. Lancet 1996, 347, 1654-1658. [99] P. Q. Chen, G. Q. Li, X. B. Guo, K. R. He, Y. X. Fu, L. C. Fu, Y. Z. Song. The Infectivity of Gametocytes of Plasmodium Falciparum from Patients Treated with Artemisinin. Chin. Med. J. 1994, 107, 709-711. [100] X. Shu-Hua, J. Utzinger, J. Chollet, M. Tanner. Effect of Artemether Administered Alone or in Combination with Praziquantel to Mice Infected with Plasmodium Berghei or Schistosoma Mansoni or Both. Int. J. Parasitol. 2006, 36, 957-964. [101] J. A. Willoughby, S. N. Sundar, M. Cheung, A. S. Tin, J. Modiano, G. L. Firestone. Artemisinin Blocks Prostate Cancer Growth and Cell Cycle Progression by Disrupting Sp1 Interactions with the Cyclin-Dependent Kinase-4 (Cdk4) Promoter and Inhibiting Cdk4 Gene Expression. J. Biol. Chem. 2009, 284, 2203-2213. [102] G. Schmid, W. Hofheinz. Total Synthesis of Qinghaosu. J. Am. Chem. Soc. 1983, 105, 624-625. [103] M. A. Avery, C. Jenningswhite, W. K. M. Chong. The Total Synthesis of (+)-Artemisinin and (+)-9-Desmethylartemisinin. Tetrahedron Lett. 1987, 28, 4629-4632.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
46
[104] J. S. Yadav, R. Satheesh Babu, G. Sabitha. Total Synthesis of (+) Artemisinin. ARKIVOC 2003, III, 125-139. [105] H. A. C. Titulaer, J. Zuidema, P. A. Kager, J. Wetsteyn, C. B. Lugt, F. Merkus. The Pharmacokinetics of Artemisinin after Oral, Intramuscular and Rectal Administration to Volunteers. J. Pharm. Pharmacol. 1990, 42, 810-813. [106] J. L. Maggs, S. Madden, L. P. Bishop, P. M. Oneill, B. K. Park. The Rat Biliary Metabolites of Dihydroartemisinin, an Antimalarial Endoperoxide. Drug Metab. Dispos. 1997, 25, 1200-1204. [107] D. L. Wesche, M. A. DeCoster, F. C. Tortella, T. G. Brewer. Neurotoxicity of Artemisinin Analogs in Vitro. Antimicrob. Agents Chemother. 1994, 38, 1813-1819. [108] B. K. Park, P. M. O'Neill, J. L. Maggs, M. Pirmohamed. Safety Assessment of Peroxide Antimalarials: Clinical and Chemical Perspectives. Br. J. Clin. Pharmacol. 1998, 46, 521-529. [109] E. Kissinger, T. Hien, N. Hung, N. Nam, N. Tuyen, B. Dinh, C. Mann, N. Phu, P. Loc, J. Simpson, N. White, J. Farrar. Clinical and Neurophysiological Study of the Effects of Multiple Doses of Artemisinin on Brain-Stem Function in Vietnamese Patients. Am. J. Trop. Med. Hyg. 2000, 63, 48-55. [110] T. T. Hien, G. D. H. Turner, N. T. H. Mai, N. H. Phu, D. Bethell, W. F. Blakemore, J. B. Cavanagh, A. Dayan, I. Medana, R. O. Weller, N. P. J. Day, N. J. White. Neuropathological Assessment of Artemether-Treated Severe Malaria. Lancet 2003, 362, 295-296. [111] M. van Vugt, B. Angus, R. Price, C. Mann, J. Simpson, C. Poletto, S. Htoo, S. Looareesuwan, N. White, F. Nosten. A Case-Control Auditory Evaluation of Patients Treated with Artemisinin Derivatives for Multidrug-Resistant Plasmodium falciparum Malaria. Am. J. Trop. Med. Hyg. 2000, 62, 65-69. [112] S. R. Meshnick, T. E. Taylor, S. Kamchonwongpaisan. Artemisinin and the Antimalarial Endoperoxides: From Herbal Remedy to Targeted Chemotherapy. Microbiol. Rev. 1996, 60, 301-315. [113] H. M. Gu, D. C. Warhurst, W. Peters. Uptake of [3H]-Dihydroartemisinine by Erythrocytes Infected with Plasmodium Falciparum in Vitro. Trans. R. Soc. Trop. Med. Hyg. 1984, 78, 265-270. [114] K. Borstnik, I.-h. Paik, T. A. Shapiro, G. H. Posner. Antimalarial Chemotherapeutic Peroxides: Artemisinin, Yingzhaosu A and Related Compounds. Int. J. Parasitol. 2002, 32, 1661-1667. [115] P. M. O'Neill, G. H. Posner. A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides. J. Med. Chem. 2004, 47, 2945-2964. [116] P. M. O'Neill. The Therapeutic Potential of Semi-Synthetic Artemisinin and Synthetic Endoperoxide Antimalarial Agents. Expert Opinion on Investigational Drugs 2005, 14, 1117-1128. [117] B. Pacorel, S. C. Leung, A. V. Stachulski, J. Davies, L. Vivas, H. Lander, S. A. Ward, M. Kaiser, R. Brun, P. M. O'Neill. Modular Synthesis and in Vitro and in Vivo Antimalarial

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
47
Assessment of C-10 Pyrrole Mannich Base Derivatives of Artemisinin. J. Med. Chem. 2009, 53, 633-640. [118] R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H.-D. Heilmann, H.-W. Chan, M.-K. Cheung, W.-L. Lam, H.-N. Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters, B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle, B. Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Wollborn, K. Schmeer, A. Römer. Artemisone - A Highly Active Antimalarial Drug of the Artemisinin Class . Angew. Chem. Inter. Ed. 2006, 45, 2082-2088. [119] P. Olliaro, T. N. C. Wells. The Global Portfolio of New Antimalarial Medicines under Development. Clin. Phamacol. Ther. 2009, 85, 584-595. [120] Information from Medecines for Malaria Venture website: www.mmv.org Accessed 22 January 2010. [121] F. Nosten, N. J. White. Artemisinin-Based Combination Treatment of Falciparum Malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181-192. [122] R. N. Price, N. M. Douglas. Artemisinin Combination Therapy for Malaria: Beyond Good Efficacy. Clin. Infect. Dis. 2009, 49, 1638-1640. [123] C. J. M. Whitty, R. Allan, V. Wiseman, S. Ochola, M. V. Nakyanzi-Mugisha, B. Vonhm, M. Mwita, C. Miaka, A. Oloo, Z. Premji, C. Burgess, T. K. Mutabingwa. Averting a Malaria Disaster in Africa - Where Does the Buck Stop? Bull. World Health Organ. 2004, 82, 381-384. [124] T. K. Mutabingwa. Artemisinin-Based Combination Therapies (ACTs): Best Hope for Malaria Treatment but Inaccessible to the Needy! Acta Trop. 2005, 95, 305-315. [125] I. A. Graham, K. Besser, S. Blumer, C. A. Branigan, T. Czechowski, L. Elias, I. Guterman, D. Harvey, P. G. Isaac, A. M. Khan, T. R. Larson, Y. Li, T. Pawson, T. Penfield, A. M. Rae, D. A. Rathbone, S. Reid, J. Ross, M. F. Smallwood, V. Segura, T. Townsend, D. Vyas, T. Winzer, D. Bowles. The Genetic Map of Artemisia Annua L. Identifies Loci Affecting Yield of the Antimalarial Drug Artemisinin . Science 2010, 327, 328-331. [126] D.-K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong, J. D. Keasling. Production of the Antimalarial Drug Precursor Artemisinic Acid in Engineered Yeast. Nature 2006, 440, 940-943. [127] C. Wongsrichanalai, S. R. Meshnick. Declining Artesunate-Mefloquine Efficacy against Falciparum Malaria on the Cambodia-Thailand Border. Emerg. Infec. Dis. 2008, 14, 716-719. [128] A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. J. Day, N. Lindegardh, D. Socheat, N. J. White. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455-467. [129] S. M. Taylor, J. J. Juliano, S. R. Meshnick. Artemisinin Resistance in Plasmodium falciparum Malaria. N Engl J Med 2009, 361, 1807; Author reply 1808.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
48
[130] A. M. Dondorp, F. Nosten, N. J. White. Artemisinin Resistance in Plasmodium falciparum Malaria - Reply. N. Engl. J. Med. 2009, 361, 1808. [131] X.-X. Xu, J. Zhu, D.-Z. Huang, W.-S. Zhou. Total Synthesis of (+)-Yingzhaosu A. Tetrahedron Lett. 1991, 32, 5785-5788. [132] W. Hofheinz, H. Burgin, E. Gocke, C. Jaquet, R. Masciadri, G. Schmid, H. Stohler, H. Urwyler. Ro 42-1611 (Arteflene), A New Effective Antimalarial - Chemical-Structure and Biological-Activity. Trop. Med. Parasitol. 1994, 45, 261-265. [133] C. Jaquet, H. R. Stohler, J. Chollet, W. Peters. Antimalarial Activity of the Bicyclic Peroxide Ro 42-1611 (Arteflene). Trop. Med. Parasitol. 1994, 45, 266-271. [134] Y. Tang, Y. Dong, J. L. Vennerstrom. Synthetic Peroxides as Antimalarials. Med. Res. Rev. 2004, 24, 425-448. [135] M. A. Avery, F. Gao, W. K. M. Chong, T. F. Hendrickson, W. D. Inman, P. Crews. Synthesis, Conformational Analysis, and Antimalarial Activity of Tricyclic Analogs of Artemisinin . Tetrahedron 1994, 50, 957-972. [136] C. W. Jefford, E. C. McGoran, J. Boukouvalas, G. Richardson, B. L. Robinson, W. Peters. Synthesis of New 1,2,4-Trioxanes and Their Antimalarial Activity. Helv. Chim. Acta 1988, 71, 1805-1812. [137] C. W. Jefford, J. A. Velarde, G. Bernardinelli, D. H. Bray, D. C. Warhurst, W. K. Milhous. Synthesis, Structure, and Antimalarial Activity of Tricyclic 1,2,4-Trioxanes Related to Artemisinin . Helv. Chim. Acta 1993, 76, 2775-2788. [138] W. Peters, B. L. Robinson, G. Tovey, J. C. Rossier, C. W. Jefford. The Chemotherapy of Rodent Malaria. L. The Activities of Some Synthetic 1,2,4-Trioxanes against Chloroquine-Sensitive and Chloroquine-Resistant Parasites. Part 3: Observations on 'Fenozan-50f', a Difluorinated 3,3'-Spirocyclopentane 1,2,4-Trioxane. Ann. Trop. Med. Parasitol. 1993, 87, 111-123. [139] G. H. Posner, C. H. Oh, L. Gerena, W. K. Milhous. Extraordinarily Potent Antimalarial Compounds - New, Structurally Simple, Easily Synthesized, Tricyclic 1,2,4-Trioxanes. J. Med. Chem. 1992, 35, 2459-2467. [140] G. H. Posner, J. N. Cumming, S. H. Woo, P. Ploypradith, S. J. Xie, T. A. Shapiro. Orally Active Antimalarial 3-Substituted Trioxanes: New Synthetic Methodology and Biological Evaluation. J. Med. Chem. 1998, 41, 940-951. [141] P. M. O'Neill, A. Miller, J. F. Bickley, F. Scheinmann, H. O. Chang, G. H. Posner. Asymmetric Syntheses of Enantiomeric 3-P-Fluorophenyl 1,2,4-Trioxane Analogues of the Antimalarial Artemisinin . Tetrahedron Lett. 1999, 40, 9133-9136. [142] J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A. Charman, F. C. K. Chiu, J. Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padmanilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler, S. Wittlin, W. N. Charman. Identification of an Antimalarial Synthetic Trioxolane Drug Development Candidate. Nature 2004, 430, 900-904.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
49
[143] K. M. Muraleedharan, M. A. Avery. Progress in the Development of Peroxide-Based Anti-Parasitic Agents. Drug Discov. Today 2009, 14, 793-803. [144] Y. Dong, J. Chollet, H. Matile, S. A. Charman, F. C. Chiu, W. N. Charman, B. Scorneaux, H. Urwyler, J. Santo Tomas, C. Scheurer, C. Snyder, A. Dorn, X. Wang, J. M. Karle, Y. Tang, S. Wittlin, R. Brun, J. L. Vennerstrom. Spiro and Dispiro-1,2,4-Trioxolanes as Antimalarial Peroxides: Charting a Workable Structure-Activity Relationship Using Simple Prototypes. J. Med. Chem. 2005, 48, 4953-4961. [145] K. Griesbaum, X. J. Liu, A. Kassiaris, M. Scherer. Ozonolyses of O-Alkylated Ketoximes in the Presence of Carbonyl Groups: A Facile Access to Ozonides. Liebigs Ann. Recl. 1997, 1381-1390. [146] Y. Dong, Y. Tang, J. Chollet, H. Matile, S. Wittlin, S. A. Charman, W. N. Charman, J. S. Tomas, C. Scheurer, C. Snyder, B. Scorneaux, S. Bajpai, S. A. Alexander, X. Wang, M. Padmanilayam, S. R. Cheruku, R. Brun, J. L. Vennerstrom. Effect of Functional Group Polarity on the Antimalarial Activity of Spiro and Dispiro-1,2,4-Trioxolanes. Bioorg. Med. Chem. 2006, 14, 6368-6382. [147] C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 1997, 23, 3-25. [148] J. L. Vennerstrom, Y. Dong, S. A. Charman, S. Wittlin, J. Chollet, D. J. Creek, X. Wang, K. Sriraghavanm, L. Zhou, H. Matile, W. N. Charman. Dispiro 1,2,4-Trioxolane Antimalarials. 2008. US 2008/0125411 A1 [149] K. J. McCullough, J. K. Wood, A. K. Bhattacharjee, Y. Dong, D. E. Kyle, W. K. Milhous, J. L. Vennerstrom. Methyl-Substituted Dispiro-1,2,4,5-Tetraoxanes: Correlations of Structural Studies with Antimalarial Activity. J. Med. Chem. 2000, 43, 1246-1249. [150] P. R. Story, D. D. Denson, C. E. Bishop, B. C. Clark, Jr., J. C. Farine. A New General Synthesis of Macrocyclic Compounds. J. Am. Chem. Soc. 1968, 90, 817-818. [151] M. J. C. Harding, D. M. Whalen. Synthesis of Hexadecanolide. Ind. Eng. Chem. Prod. Res. Dev. 1975, 14, 232-239. [152] J. L. Vennerstrom, H. N. Fu, W. Y. Ellis, A. L. Ager, Jr., J. K. Wood, S. L. Andersen, L. Gerena, W. K. Milhous. Dispiro-1,2,4,5-Tetraoxanes: A New Class of Antimalarial Peroxides. J. Med. Chem. 1992, 35, 3023-3027. [153] J. L. Vennerstrom, A. L. Ager, S. L. Andersen, J. M. Grace, V. Wongpanich, C. K. Angerhofer, J. K. Hu, D. L. Wesche. Assessment of the Antimalarial Potential of Tetraoxane WR 148999. Am. J. Trop. Med. Hyg. 2000, 62, 573-578. [154] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. Shone, D. Stanford, P. Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer, S. Lozanom, M. Jesús, D. Gargallo, S. A. Ward, P. M. O'Neill. Two-Step Synthesis of Achiral Dispiro-1,2,4,5-Tetraoxanes with Outstanding Antimalarial Activity, Low Toxicity, and High-Stability Profiles . J. Med. Chem. 2008, 51, 2170-2177.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
50
[155] R. Amewu, P. M. O'Neill, A. Stachulski, G. Ellis, S. A. Ward. Preparation of Dispiro-Tetraoxane Compounds for the Treatment of Malaria and/or Cancer. 2008. WO/2008/038030 [156] R. Amewu, A. V. Stachulski, S. A. Ward, N. G. Berry, P. G. Bray, J. Davies, G. Labat, L. Vivas, P. M. O'Neill. Design and Synthesis of Orally Active Dispiro 1,2,4,5-Tetraoxanes; Synthetic Antimalarials with Superior Activity to Artemisinin . Org. Biomol. Chem. 2006, 4, 4431-4436. [157] B. Meunier. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2007, 41, 69-77. [158] O. Dechy-Cabaret, F. Benoit-Vical, A. Robert, B. Meunier. Preparation and Antimalarial Activities Of "Trioxaquines", New Modular Molecules with a Trioxane Skeleton Linked to a 4-Aminoquinoline. ChemBioChem 2000, 1, 281-283. [159] L. K. Basco, O. Dechy-Cabaret, M. Ndounga, F. S. Meche, A. Robert, B. Meunier. In Vitro Activities of DU-1102, a New Trioxaquine Derivative, against Plasmodium falciparum Isolates. Antimicrob. Agents Chemother. 2001, 45, 1886-1888. [160] O. Dechy-Cabaret, F. Benoit-Vical, C. Loup, A. Robert, H. Gornitzka, A. Bonhoure, H. Vial, J.-F. Magnaval, J.-P. Séguéla, B. Meunier. Synthesis and Antimalarial Activity of Trioxaquine Derivatives. Chem. Eur. J. 2004, 10, 1625-1636. [161] F. Benoit-Vical, J. Lelièvre, A. Berry, C. Deymier, O. Dechy-Cabaret, J. Cazelles, C. Loup, A. Robert, J.-F. Magnaval, B. Meunier. Trioxaquines Are New Antimalarial Agents Active on All Erythrocytic Forms, Including Gametocytes. Antimicrob. Agents. Chemother. 2007, 51, 1463-1472. [162] F. Coslédan, L. Fraisse, A. Pellet, F. Guillou, B. Mordmüller, P. G. Kremsner, A. Moreno, D. Mazier, J.-P. Maffrand, B. Meunier. Selection of a Trioxaquine as an Antimalarial Drug Candidate. Proc. Nat. Acad. Sci. U.S.A 2008, 105, 17579-17584. [163] S. R. Meshnick, T. W. Tsang, F. B. Lin, H. Z. Pan, C. N. Chang, F. Kuypers, D. Chiu, B. Lubin. Activated Oxygen Mediates the Antimalarial Activity of Qinghaosu. Prog. Clin. Biol. Res. 1989, 313, 95-104. [164] S. R. Krungkrai, Y. Yuthavong. The Antimalarial Action on Plasmodium Falciparum of Qinghaosu and Artesunate in Combination with Agents Which Modulate Oxidant Stress. Trans. R. Soc. Trop. Med. Hyg. 1987, 81, 710-714. [165] P. A. Berman, P. A. Adams. Artemisinin Enhances Heme-Catalysed Oxidation of Lipid Membranes. Free Radical Biol. Med. 1997, 22, 1283-1288. [166] M. D. Scott, S. R. Meshnick, R. A. Williams, D. T.-Y. Chiu, H. C. Pan, B. H. Lubin, F. A. Kuypers. Qinghaosu-Mediated Oxidation in Normal and Abnormal Erythrocytes. J. Lab. Clin. Med. 1989, 114, 401-406. [167] F. Zhang, D. K. Gosser, S. R. Meshnick. Hemin-Catalyzed Decomposition of Artemisinin (Qinghaosu). Biochem. Pharmacol. 1992, 43, 1805-1809.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
51
[168] S. R. Meshnick, A. Thomas, A. Ranz, C. M. Xu, H. Z. Pan. Artemisinin (Qinghaosu): The Role of Intracellular Hemin in Its Mechanism of Antimalarial Action . Mol. Biol. Parasitol. 1991, 49, 181-189. [169] G. H. Posner, C. H. Oh. A Regiospecifically O-18 Labeled 1,2,4-Trioxane - A Simple Chemical-Model System to Probe the Mechanism(s) for the Antimalarial Activity of Artemisinin (Qinghaosu). J. Am. Chem. Soc. 1992, 114, 8328-8329. [170] G. H. Posner, C. H. Oh, D. S. Wang, L. Gerena, W. K. Milhous, S. R. Meshnick, W. Asawamahasadka. Mechanism-Based Design, Synthesis, and in Vitro Antimalarial Testing of New 4-Methylated Trioxanes Structurally Related to Artemisinin - the Importance of A Carbon-Centered Radical for Antimalarial Activity. J. Med. Chem. 1994, 37, 1256-1258. [171] G. H. Posner, D. S. Wang, J. N. Cumming, C. H. Oh, A. N. French, A. L. Bodley, T. A. Shapiro. Further Evidence Supporting the Importance of and the Restrictions on A Carbon-Centered Radical for High Antimalarial Activity of 1,2,4-Trioxanes Like Artemisinin . J. Med. Chem. 1995, 38, 2273-2275. [172] C. W. Jefford, F. Favarger, M. Vicente, Y. Jacquier. The Decomposition of Cis-Fused Cyclopenteno-1,2,4-Trioxanes Induced by Ferrous Salts and Some Oxophilic Reagents. Helv. Chim. Acta 1995, 78, 452-458. [173] C. W. Jefford, M. G. H. Vicente, Y. Jacquier, F. Favarger, J. Mareda, P. Millasson-Schmidt, G. Brunner, U. Burger. The Deoxygenation and Isomerization of Artemisinin and Artemether and Their Relevance to Antimalarial Action. Helv. Chim. Acta 1996, 79, 1475-1487. [174] C. W. Jefford. Why Artemisinin and Certain Synthetic Peroxides Are Potent Antimalarials. Implications for the Mode of Action . Curr. Med. Chem. 2001, 8, 1803-1826. [175] C. W. Jefford, S. Kohmoto, D. Jaggi, G. Timari, J. C. Rossier, M. Rudaz, O. Barbuzzi, D. Gerard, U. Burger, P. Kamalaprija, J. Mareda, G. Bernardinelli, I. Manzanares, C. J. Canfield, S. L. Fleck, B. L. Robinson, W. Peters. Synthesis, Structure, and Antimalarial Activity of Some Enantiomerically Pure, Cis-Fused Cyclopenteno-1,2,4-Trioxanes. Helv. Chim. Acta 1995, 78, 647-662. [176] G. H. Posner, J. N. Cumming, P. Ploypradith, H. O. Chang. Evidence for Fe(IV)=O in the Molecular Mechanism of Action of the Trioxane Antimalarial Artemisinin . J. Am. Chem. Soc. 1995, 117, 5885-5886. [177] W. M. Wu, Y. K. Wu, Y. L. Wu, Z. J. Yao, C. M. Zhou, Y. Li, F. Shan. Unified Mechanistic Framework for the Fe(II)-Induced Cleavage of Qinghaosu and Derivatives/Analogues. The First Spin-Trapping Evidence for the Previously Postulated Secondary C-4 Radical. J. Am. Chem. Soc. 1998, 120, 3316-3325. [178] J. D. Gu, K. X. Chen, H. L. Jiang, J. Leszczynski. The Radical Transformation in Artemisinin: A DFT Study. J. Phys. Chem. A 1999, 103, 9364-9369. [179] A. R. Butler, B. C. Gilbert, P. Hulme, L. R. Irvine, L. Renton, A. C. Whitwood. EPR Evidence for the Involvement of Free Radicals in the Iron-Catalysed Decomposition of Qinghaosu (Artemisinin) and Some Derivatives; Antimalarial Action of Some Polycyclic Endoperoxides. Free Radical Res. 1998, 28, 471-476.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
52
[180] P. M. O'Neill, A. Miller, L. P. D. Bishop, S. Hindley, J. L. Maggs, S. A. Ward, S. M. Roberts, F. Scheinmann, A. V. Stachulski, G. H. Posner, B. K. Park. Synthesis, Antimalarial Activity, Biomimetic Iron(II) Chemistry, and in Vivo Metabolism of Novel, Potent C-10-Phenoxy Derivatives of Dihydroartemisinin. J. Med. Chem. 2001, 44, 58-68. [181] J. N. Cumming, P. Ploypradith, G. H. Posner. Antimalarial Activity of Artemisinin (Qinghaosu) and Related Trioxanes: Mechanism(s) of Action. Adv. Pharmacol. 1997, 37, 253-297. [182] S. Kapetanaki, C. Varotsis. Fourier Transform Infrared Investigation of Non-Heme Fe(III) and Fe(II) Decomposition of Artemisinin and of a Simplified Trioxane Alcohol. J. Med. Chem. 2001, 44, 3150-3156. [183] S. Kapetanaki, C. Varotsis. Ferryl-Oxo Heme Intermediate in the Antimalarial Mode of Action of Artemisinin . FEBS Lett. 2000, 474, 238-241. [184] R. S. Czernuszewicz, Y. O. Su, M. K. Stern, K. A. Macor, D. Kim, J. T. Groves, T. G. Spiro. Oxomanganese(IV) Porphyrins Identified by Resonance Raman and Infrared Spectroscopy. Weak Bonds and the Stability of the Half-Filled t2g Subshell. J. Am. Chem. Soc. 1988, 110, 4158-4165. [185] A. Robert, B. Meunier. Is Alkylation the Main Mechanism of Action of the Antimalarial Drug Artemisinin? Chem. Soc. Rev. 1998, 27, 273-274. [186] J. Cazelles, A. Robert, B. Meunier. Alkylating Capacity and Reaction Products of Antimalarial Trioxanes after Activation by a Heme Model. J. Org. Chem. 2002, 67, 609-619. [187] R. K. Haynes, H. H. O. Pai, A. Voerste. Ring Opening of Artemisinin (Qinghaosu) and Dihydroartemisinin and Interception of the Open Hydroperoxides with Formation of N-Oxides - A Chemical Model for Antimalarial Mode of Action. Tetrahedron Lett. 1999, 40, 4715-4718. [188] R. K. Haynes, S. C. Vonwiller. The Behaviour of Qinghaosu (Artemisinin) in the Presence of Heme Iron(II) and (III). Tetrahedron Lett. 1996, 37, 253-256. [189] R. K. Haynes, S. C. Vonwiller. The Behaviour of Qinghaosu (Artemisinin) in the Presence of Non-Heme Iron(II) and (III). Tetrahedron Lett. 1996, 37, 257-260. [190] A. Robert, O. Dechy-Cabaret, J. Cazelles, B. Meunier. From Mechanistic Studies on Artemisinin Derivatives to New Modular Antimalarial Drugs . Acc. Chem. Res. 2002, 35, 167-174. [191] J. Chateauneuf, J. Lusztyk, K. U. Ingold. Absolute Rate Constants for the Reactions of Some Carbon-Centered Radicals with 2,2,6,6-Tetramethyl-1-Piperidinoxyl. J. Org. Chem. 1988, 53, 1629-1632. [192] K. S. Root, C. L. Hill, L. M. Lawrence, G. M. Whitesides. The Mechanism of Formation of Grignard Reagents: Trapping of Free Alkyl Radical Intermediates by Reaction with Tetramethylpiperidine-N-Oxyl . J. Am. Chem. Soc. 1989, 111, 5405-5412. [193] R. K. Haynes, W. C. Chan, C.-M. Lung, A.-C. Uhlemann, U. Eckstein, D. Taramelli, S. Parapini, D. Monti, S. Krishna. The Fe2+-Mediated Decomposition, PfATP6 Binding, and

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
53
Antimalarial Activities of Artemisone and Other Artemisinins: The Unlikelihood of C-Centered Radicals as Bioactive Intermediates. ChemMedChem 2007, 2, 1480-1497. [194] P. M. O'Neill, L. P. D. Bishop, N. L. Searle, J. L. Maggs, R. C. Storr, S. A. Ward, B. K. Park, F. Mabbs. Biomimetic Fe(II)-Mediated Degradation of Arteflene (Ro-42-1611). The First EPR Spin-Trapping Evidence for the Previously Postulated Secondary Carbon-Centered Cyclohexyl Radical. J. Org. Chem. 2000, 65, 1578-1582. [195] P. M. O'Neill, L. P. Bishop, N. L. Searle, J. L. Maggs, S. A. Ward, P. G. Bray, R. C. Storr, B. Kevin Park. The Biomimetic Iron-Mediated Degradation of Arteflene (Ro-42-1611), an Endoperoxide Antimalarial: Implications for the Mechanism of Antimalarial Activity. Tetrahedron Lett. 1997, 38, 4263-4266. [196] D. J. Creek, W. N. Charman, F. C. Chiu, R. J. Prankerd, K. J. McCullough, Y. Dong, J. L. Vennerstrom, S. A. Charman. Iron-Mediated Degradation Kinetics of Substituted Dispiro-1,2,4-Trioxolane Antimalarials . J. Pharm. Sci. 2007, 96, 2945-2956. [197] Y. Tang, Y. Dong, X. Wang, K. Sriraghavan, J. K. Wood, J. L. Vennerstrom. Dispiro-1,2,4-Trioxane Analogues of a Prototype Dispiro-1,2,4-Trioxolane: Mechanistic Comparators for Artemisinin in the Context of Reaction Pathways with Iron(II). J. Org. Chem. 2005, 70, 5103-5110. [198] Y. Dong, J. L. Vennerstrom. Formation of Primary and Secondary Carbon-Centered Radicals in the Iron (II)-Mediated Decomposition of Antimalarial 1,2,4,5-Tetraoxanes. Abstracts of Papers 2006, 232nd ACS National Meeting, San Francisco, CA, United States, Sept. 10-14 2006. [199] H. H. Liu, Y. K. Wu, X. Shen. Alkylation of Sulfur Ligand in Cysteinate-Iron Chelates by a 1,2,4,5-Tetraoxane. Chin. J. Chem . 2003, 21, 875-877. [200] I. Opsenica, N. Terzic, D. Opsenica, G. Angelovski, M. Lehnig, P. Eilbracht, B. Tinant, Z. Juranic, K. S. Smith, Y. S. Yang, D. S. Diaz, P. L. Smith, W. K. Milhous, D. Dokovic, B. A. Solaja. Tetraoxane Antimalarials and Their Reaction with Fe(II). J. Med. Chem. 2006, 49, 3790-3799. [201] W. M. Wu, Y. L. Wu. Chemical and Electro-Chemical Reduction of Qinghaosu (Artemisinin) . J. Chem. Soc., Perkin Trans. 1 2000, 4279-4283. [202] Y. Wu, H.-H. Liu. Cleavage of Qinghaosu (Artemisinin) Induced by Non-Iron Transition-Metal Ions in the Presence of Excess Cysteine. Helv. Chim. Acta 2003, 86, 3074-3080. [203] Y. L. Hong, Y. Z. Yang, S. R. Meshnick. The Interaction of Artemisinin with Malarial Hemozoin. Mol. Biol. Parasitol. 1994, 63, 121-128. [204] A. Robert, B. Meunier. Alkylating Properties of Antimalarial Artemisinin Derivatives and Synthetic Trioxanes When Activated by a Reduced Heme Model. Chem. Eur. J. 1998, 4, 1287-1296. [205] A. Robert, B. Meunier. Characterization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968-5969.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
54
[206] J.-F. Berrien, O. Provot, J. Mayrargue, M. Coquillay, L. Cicéron, F. Gay, M. Danis, A. Robert, B. Meunier. Alkylation of Manganese (II) Tetraphenylporphyrin by a Synthetic Antimalarial Trioxane . Org. Biomol. Chem. 2003, 1, 2859-2864. [207] J. Cazelles, B. Camuzat-Dedenis, O. Provot, A. Robert, J. Mayrargue, B. Meunier. Alkylating Properties of Synthetic Trioxanes Related to Artemisinin. J. Chem. Soc., Perkin Trans. 1 2000, 1265-1270. [208] O. Provot, B. Camuzat-Dedenis, M. Hamzaoui, H. Moskowitz, J. Mayrargue, A. Robert, J. Cazelles, B. Meunier, F. Zouhiri, D. Desmaële, J. d'Angelo, J. Mahuteau, F. Gay, L. Cicéron. Structure-Activity Relationships of Synthetic Tricyclic Trioxanes Related to Artemisinin: The Unexpected Alkylative Property of a 3-(Methoxymethyl) Analog. Eur. J. Org. Chem. 1999, 1999, 1935-1938. [209] M. Rodriguez, D. Bonnet-Delpon, J.-P. Bégué, A. Robert, B. Meunier. Alkylation of Manganese (II) Tetraphenylporphyrin by Antimalarial Fluorinated Artemisinin Derivatives . Bioorg. Med. Chem. Lett. 2003, 13, 1059-1062. [210] A. Robert, Y. Coppel, B. Meunier. NMR Characterization of Covalent Adducts Obtained by Alkylation of Heme with the Antimalarial Drug Artemisinin . Inorg. Chim. Acta 2002, 339, 488-496. [211] A. Robert, J. Cazelles, B. Meunier. Characterization of the Alkylation Product of Heme by the Antimalarial Drug Artemisinin . Angew. Chem. Inter. Ed. 2001, 40, 1954-1957. [212] K. L. Shukla, T. M. Gund, S. R. Meshnick. Molecular Modeling Studies of the Artemisinin (Qinghaosu)-Hemin Interaction: Docking between the Antimalarial Agent and Its Putative Receptor. J. Mol. Graphics 1995, 13, 215-222. [213] A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier. The Antimalarial Drug Artemisinin Alkylates Heme in Infected Mice. Proc. Nat. Acad. Sci. U.S.A 2005, 102, 13676-13680. Erratum, 2006, 103, 3943. [214] K. Selmeczi, A. Robert, C. Claparols, B. Meunier. Alkylation of Human Hemoglobin A0 by the Antimalarial Drug Artemisinin . FEBS Lett. 2004, 556, 245-248. [215] C. Loup, J. Lelievre, F. Benoit-Vical, B. Meunier. Trioxaquines and Heme-Artemisinin Adducts Inhibit the in Vitro Formation of Hemozoin Better Than Chloroquine. Antimicrob. Agents Chemother. 2007, 51, 3768-3770. [216] D. J. Creek, W. N. Charman, F. C. K. Chiu, R. J. Prankerd, Y. Dong, J. L. Vennerstrom, S. A. Charman. Relationship between Antimalarial Activity and Heme Alkylation for Spiro- and Dispiro-1,2,4-Trioxolane Antimalarials. Antimicrob. Agents Chemother. 2008, 52, 1291-1296. [217] X. Wang, D. J. Creek, C. E. Schiaffo, Y. Dong, J. Chollet, C. Scheurer, S. Wittlin, S. A. Charman, P. H. Dussault, J. K. Wood, J. L. Vennerstrom. Spiroadamantyl 1,2,4-Trioxolane, 1,2,4-Trioxane, and 1,2,4-Trioxepane Pairs: Relationship between Peroxide Bond Iron(II) Reactivity, Heme Alkylation Efficiency, and Antimalarial Activity. Bioorg. Med. Chem. Lett. 2009, 19, 4542-4545.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
55
[218] S. A. L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meunier. Heme Alkylation by Artesunic Acid and Trioxaquine DU1301, Two Antimalarial Trioxanes. ChemBioChem 2005, 6, 653-658. [219] Y. Z. Yang, W. Asawamahasakda, S. R. Meshnick. Alkylation of Human Albumin by the Antimalarial Artemisinin . Biochem. Pharmacol. 1993, 46, 336-339. [220] W. Asawamahasakda, I. Ittarat, Y. M. Pu, H. Ziffer, S. R. Meshnick. Reaction of Antimalarial Endoperoxides with Specific Parasite Proteins. Antimicrob. Agents Chemother. 1994, 38, 1854-1858. [221] J. Bhisutthibhan, X. Q. Pan, P. A. Hossler, D. J. Walker, C. A. Yowell, J. Carlton, J. B. Dame, S. R. Meshnick. The Plasmodium falciparum Translationally Controlled Tumor Protein Homolog and Its Reaction with the Antimalarial Drug Artemisinin. J. Biol. Chem. 1998, 273, 16192-16198. [222] J. Bhisutthibhan, M. A. Philbert, H. Fujioka, M. Aikawa, S. R. Meshnick. The Plasmodium falciparum Translationally Controlled Tumor Protein: Subcellular Localisation and Calcium Binding. Eur. J. Cell Biol. 1999, 78, 665-670. [223] U. Eckstein-Ludwig, R. J. Webb, I. D. A. Van Goethem, J. M. East, A. G. Lee, M. Kimura, P. M. O'Neill, P. G. Bray, S. A. Ward, S. Krishna. Artemisinins Target the SERCA of Plasmodium falciparum. Nature 2003, 424, 957-961. [224] A.-C. Uhlemann, A. Cameron, U. Eckstein-Ludwig, J. Fischbarg, P. Iserovich, F. A. Zuniga, M. East, A. Lee, L. Brady, R. K. Haynes, S. Krishna. A Single Amino Acid Residue Can Determine the Sensitivity of SERCAs to Artemisinins. Nature Struct. Mol. Biol. 2005, 12, 628-629. [225] J. Lytton, M. Westlin, M. R. Hanley. Thapsigargin Inhibits the Sarcoplasmic or Endoplasmic Reticulum Ca-ATPase Family of Calcium Pumps. J. Biol. Chem. 1991, 266, 17067-17071. [226] O. Thastrup, P. J. Cullen, B. K. Drøbak, M. R. Hanley, A. P. Dawson. Thapsigargin, a Tumor Promoter, Discharges Intracellular Ca2+ Stores by Specific Inhibition of the Endoplasmic Reticulum Ca2+-ATPase. Proc. Nat. Acad. Sci. U.S.A 1990, 87, 2466-2470. [227] M. d. P. Crespo, T. D. Avery, E. Hanssen, E. Fox, T. V. Robinson, P. Valente, D. K. Taylor, L. Tilley. Artemisinin and a Series of Novel Endoperoxide Antimalarials Exert Early Effects on Digestive Vacuole Morphology. Antimicrob. Agents. Chemother. 2008, 52, 98–109. [228] R. Jambou, E. Legrand, M. Niang, N. Khim, P. Lim, B. Volney, M. T. Ekala, C. Bouchier, P. Esterre, T. Fandeur, O. Mercereau-Puijalon. Resistance of Plasmodium falciparum Field Isolates to in Vitro Artemether and Point Mutations of the SERCA-Type PfATPase6. Lancet 2005, 366, 1960-1963. [229] A. Afonso, P. Hunt, S. Cheesman, A. C. Alves, C. V. Cunha, V. do Rosário, P. Cravo. Malaria Parasites Can Develop Stable Resistance to Artemisinin but Lack Mutations in Candidate Genes atp6 (Encoding the Sarcoplasmic and Endoplasmic Reticulum Ca2+ ATPase), tctp, mdr1, and cg10. Antimicrob. Agents Chemother. 2006, 50, 480-489.

Chapter 1: Alkylating properties of antimalarial peroxide-containing drugs: A focused review
56
[230] A.-C. Uhlemann, S. Wittlin, H. Matile, L. Y. Bustamante, S. Krishna. Mechanism of Antimalarial Action of the Synthetic Trioxolane Rbx11160 (OZ277). Antimicrob. Agents. Chemother. 2007, 51, 667-672. [231] M. Jung, H. Kim, K. Y. Nam, K. T. No. Three-Dimensional Structure of Plasmodium falciparum Ca2+-ATPase (PfATP6) and Docking of Artemisinin Derivatives to PfATP6. Bioorg. Med. Chem. Lett. 2005, 15, 2994-2997.



Chapter 2
The antimalarial artemisone is an efficient heme alkylating agent


Chapter 2: The antimalarial artemisone is an efficient heme alkylating agent
Résumé en français de la publication :
The antimalarial artemisone is an efficient heme alkylating agent
F. Bousejra-El Garah, B. Meunier, and A. Robert, European Journal of Inorganic Chemistry 2008,
2133-2135.
L’artémisone, antipaludique dérivé de l’artémisinine, est un agent alkylant efficace de l’hème.
L’artémisone est un dérivé hémi-synthétique de l’artémisinine, plus précisément de la dihydro-
artémisinine qui est substituée en position C-10α par un groupement thiomorpholine-1,1-dioxyde
(figure ci-dessous). Comme pour l’artémisinine, la liaison peroxyde de l’artémisone peut être
réduite par le fer(II) et former des radicaux alkyles qui ont été piégés par 4-oxo-TEMPO.[1] Compte-
tenu du rôle de l’hème dans le paludisme, nous nous sommes intéressés à la réactivité de
l’artémisone vis-à-vis de l’hème, d’autant que, selon des travaux publiés, l’artémisone et tous les
dérivés en C-10α de l’artémisinine, seraient incapables d’alkyler le cycle porphyrinique de
l’hème.[2]
Nous avons donc fait réagir l’artémisone avec l’hème-fer(II) généré in situ par réduction de l’hème-
fer(III) en présence de dithionite. Lors de cette réaction, 93% de l’hème de départ est converti en 1
h. L’analyse par LC-MS a permis de caractériser les produits de couplage covalent hème-drogue 6
et 7, l’un et l’autre issus de réarrangements de l’adduit hème-artémisone 5 (figure ci-après) et
présents sous la forme de 4 régio-isomères, dus à la substitution du fragment artémisone en position
α, β, γ et δ de la porphyrine de l’hème. L’adduit 6 (m/z 957.5 M+) est issu du couplage covalent
entre l’hème et un radical alkyle dérivé de l’artémisone après réarrangement. Ce même radical avait
précédemment été piégé par 4-oxo-TEMPO.[1]
Ce résultat montre que la substitution en C-10α de la dihydro-artémisinine par le groupement
thiomorpholine-1,1-dioxyde n’affecte pas les propriétés alkylantes de ce peroxyde très actif.
L’artémisone, comme tous les dérivés actifs de l’artémisinine, est un alkylant efficace de l’hème et
cette réactivité pourrait être liée à son mécanisme d’action antipaludique. Le caractère alkylant de
l’artésunate, de la 10-déoxoartémisinine et de la 10α-(4-benzylpiperazinyl)artémisinine qui est un
autre dérivé de l’artémisinine substitué en C10α, avait précédemment été décrit.[3]
59
Chapter 2: The antimalarial artemisone is an efficient heme alkylating agent
60
Alkylation de l’hème par l’artémisone 3
Bibliographie
[1] R. K. Haynes, W. C. Chan, C.-M. Lung, A.-C. Uhlemann, U. Eckstein, D. Taramelli, S. Parapini, D. Monti, S. Krishna. The Fe2+-Mediated Decomposition, PfATP6 Binding, and Antimalarial Activities of Artemisone and Other Artemisinins: The Unlikelihood of C-Centered Radicals as Bioactive Intermediates. ChemMedChem 2007, 2, 1480-1497. [2] R. K. Haynes, W. Y. Ho, H. W. Chan, B. Fugmann, J. Stetter, S. L. Croft, L. Vivas, W. Peters, B. L. Robinson. Highly Antimalaria-Active Artemisinin Derivatives: Biological Activity Does Not Correlate with Chemical Reactivity. Angew. Chem. Int. Ed. 2004, 43, 1381-1385. [3] S. A. L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meunier. Heme Alkylation by Artesunic Acid and Trioxaquine DU1301, Two Antimalarial Trioxanes. ChemBioChem 2005, 6, 653-658.

SHORT COMMUNICATION
DOI: 10.1002/ejic.200800129
The Antimalarial Artemisone Is an Efficient Heme Alkylating Agent
Fatima Bousejra-El Garah,[a] Bernard Meunier,[b] and Anne Robert*[a]
Keywords: Alkylation / Artemisone / Heme / Malaria / Trioxane
The reductive activation by iron(II)-heme of artemisone, a C-10 substituted derivative of the antimalarial artemisinin, gen-erates covalent heme-drug adducts in high yields. This resultconfirms that the substitution at C-10 of artemisinin does not
Togetherwith AIDS and tuberculosis, malaria is one ofthe three main causes of mortality by infectious diseasesin the world. The naturally occurring artemisinin, and itssemisynthetic derivatives artemether and artesunate arenowadays the most efficient and rapidly acting antimalarialdrugs. Despite a wide use for more than 30 years, no clini-cally relevant parasite resistance to these drugs has beenreported up to now.[1–3]
The peroxide function of artemisinin (1; Figure 1) andsynthetic trioxanes is known to play a key role in their anti-malarial activity. The possible formation of alkylating spe-cies after reductive activation of the O–O bond by heme-or non-heme-iron species was early suspected.[4,5] The fullcharacterization of a covalent porphyrin-artemisinin adductafter reductive activation with a heme model confirmed thestrong alkylating ability of artemisinin.[6,7] Covalent heme-artemisinin adducts were also isolated and characterized.[8]
Furthermore, these heme-drug adducts have been detectedin the spleen and the urine of malaria-infected mice,whereas they are absent from healthy mouse organs treatedunder the same conditions. This experimental fact indicatesthat alkylation of heme by artemisinin is triggered by thepresence of the parasite within erythrocytes.[9] In fact, thereductive activation of peroxide-containing antimalarials toproduce heme-drug adducts has been found to correlatewell with their biological efficacy.[10,11] The active semisyn-thetic derivatives of artemisinin substituted at C-10 are alsoefficient heme-alkylating agents,[12,13] despite a claim byR. K. Haynes et al. to the contrary.[14]
Artemisone (3; Figure 1) is a promising artemisinin de-rivative substituted at the C-10α position by a thiomorpho-line 1,1-dioxide residue.[15] This compound was recently re-
[a] Laboratoire de Chimie de Coordination du CNRS,205 route de Narbonne, 31077 Toulouse cedex 4, France
[b] Palumed, Rue Pierre et Marie Curie,B. P. 28262, 31682 Labège cedex, FranceFax: +33-5-61553003E-mail: [email protected] information for this article is available on theWWW under http://www.eurjic.org or from the author.
Eur. J. Inorg. Chem. 2008, 2133–2135 © 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 2133
alter the alkylating ability of artemisinin derivatives whenactivated by a redox-active metal center.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim,Germany, 2008)
Figure 1. Synthesis of artemisone 3; (a) TMSBr, 25 min, 0 °C; (b)thiomorpholine 1,1-dioxide, 1,1,1,3,3,3-hexafluoropropan-2-ol(HFIP), 75 min, 0 °C. TMS = trimethysilyl.
ported to extensively react with iron(II) salts. This reactionproduced a C-4-centered radical which has been trapped by4-oxo-TEMPO in a rather low yield [10% when activationoccurred in the presence of Fe(OAc)2].[16] For this reason,the authors considered that “the structural flexibility of theC radicals from artemisinins allows facile extrusion of Fe2+
and collapse to benign isomerization products”. Then, theyrule out the possible importance of these radicals for theantimalarial activity, and state that “as an explanation forthe antimalarial activity of artemisinins (...) the C-radicalhypothesis is not feasible”. In fact, under the conditionsused by Haynes et al. (activation by an iron salt), the experi-mental mixture does not contain any component that couldbe considered as a putative biological target of the C-cen-tered radical.
Therefore, we decided to evaluate the alkylating capacityof artemisone in the presence of a potential target such asheme. We found that the alkylation of iron(II)-heme by art-emisone is indeed achieved in high yield as reported below.
61

F. Bousejra-El Garah, B. Meunier, A. RobertSHORT COMMUNICATIONA patent reports the large-scale synthesis of artemisone
(3) by using gazeous HCl.[17] In ref.[15b] is also reported thelaboratory-scale synthesis and the characterization of thiscompound. However, we prepared artemisone from dihy-droartemisinin using the route that we chose for the prepa-ration of the 10α-benzylpiperazinyl derivative of artemisi-nin.[12] The 10β-trimethylsilyl ether derivative 2 was firstprepared by reaction of chlorotrimethylsilane with dihy-droartemisinin in pyridine as solvent. Reaction of bromotri-methylsilane (1.2 mol-equiv.) with compound 2 in dichloro-methane for 25 min, followed by addition of thiomorpho-line 1,1-dioxide (10 mol-equiv.) in the presence of1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) at 0 °C providedartemisone 3 (Figure 1). After purification by chromatog-raphy and recrystallization (from ethyl acetate and hexane),the yield of 3 was 40% with respect to dihydroartemisinin,close to the reported value (44%).[15b] Elimination product4 was also isolated in 19% yield. The (R) configuration atC-10 of artemisone was assessed by the 1H NMR couplingconstant 9-H,10-H (J = 10.3 Hz).[18]
Iron(II)-heme, generated in situ by reduction of itsiron(III) analogue with sodium dithionite, reacted quicklywith artemisone (3) to provide the covalent heme-drug ad-duct 5 (heme/3/dithionite = 1:1:5 molar ratio, room temp.,DMSO). As previously described for artemisinin, reductiveactivation of the peroxide bond produces an alkoxy radicalcentered at O-2. Rapid and subsequent C-3–C-4 β-scissionleads to the alkyl radical centered at C-4. This radicalquickly alkylates the meso positions of the porphyrinmacrocycle by an intramolecular reaction. The reaction wasmonitored by high-pressure liquid chromatography (C18RP column). After 1 h, the conversion of heme monitoredby HPLC was 93% (Rt = 18.2 min, λmax = 398 nm). Seven
Figure 2. Alkylation of iron(II)-heme by artemisone. The oval stands for the protoporphyrin-IX macrocycle. Inset: adduct between artemi-sone and 4-oxo-TEMPO, reported in ref.[16]
www.eurjic.org © 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Inorg. Chem. 2008, 2133–21352134
resolved peaks were detected with retention times between23 and 26 min, and λmax = 405–407 nm. These peaks canbe assigned to the four regioisomers of the two differentheme-drug adducts, resulting from the alkylation of the fourmeso positions of heme without regioselectivity (eight peaksexpected, but two peaks overlapped; Figure S2, SupportingInformation). The crude reaction product was analyzed byESI+-MS after dilution with acetonitrile. The complete ad-duct 5 (Figure 2) was not detected; however, its formationwas assessed by the detection of adducts 6 and 7. Com-pound 6 (Figure 2; at m/z = 957.5) is generated by re-arrangement of the artemisone-derived moiety of 5. Thedrug-derived fragment of adduct 6 has been previouslytrapped using 4-oxo-TEMPO (Figure 2, inset), and a path-way has been proposed for its formation.[16] Adduct 7, de-tected at m/z = 840.3, arose from the hydrolysis of the C-12–O bond of 5, followed by the intramolecular attack ofthe hydroxy function at C-12 onto the C-10 and the releaseof the thiomorpholine dioxide residue (Figure 3). As an ad-ditional identification of compound 7, dilution with meth-anol caused hydration of the aldehyde at C-12 and methyl-ation of the alcohol function at C-10 (m/z = 872.5, com-pound 8; Figure 2). These analytical results are fully consis-tent with those reported for the adducts between heme andother artemisinin derivatives (higher retention times thanheme itself, and a bathochromic shift of absorbance dueto the substitution at the meso position of the porphyrinmacrocycle).[8,12] In particular, the alkylation of heme withanother C-10-substituted artemisinin derivative also led toadduct 7 after release of the C-10 substituent.[12] To confirmthese results, a similar reaction was carried out by usingheme dimethyl ester instead of heme. Under these condi-tions, covalent heme-drug adducts were detected at m/z =
62

The Antimalarial Artemisone Is an Efficient Heme Alkylating Agent
985.5, 868.4, and 900.5, corresponding to the dimethyl esteranalogues of adducts 6, 7, and 8, respectively. From HPLCand mass spectra, the yield of heme-drug adducts was rang-ing from 80 to 90% with respect to starting heme.
Figure 3. Mechanism of rearrangement of the covalent heme-arte-misone adduct 5.
There is no doubt that the substitution of dihydroartemi-sinin by the thiomorpholine dioxide moiety at C-10α doesnot prevent the reductive activation of the peroxide bondof artemisone by iron(II)-heme, leading to the alkylation ofthe four meso positions of heme. Artemisone, as all theother active artemisinin derivatives (artemether, artesunate,...) is a very efficient alkylating agent toward heme. Thisproperty of artemisone is an experimental fact that shouldbe taken into consideration in discussions about possiblemechanisms of action of this antimalarial drug.
Supporting Information (see footnote on the first page of this arti-cle): Experimental conditions of the alkylation reaction andcharacterization of the heme-drug adducts 6, 7, and 8 by mass spec-trometry; 13C NMR spectrum of 3 (Figure S1), and HPLC moni-toring of the alkylation reaction (Figure S2).
Acknowledgments
F. B.-E. G. is indebted to the EU-AntiMal program for a PhD fel-lowship (grant no: LSHP-CT-2005-018834). The Centre Nationalde la Recherche Scientifique (CNRS) and the Agence Nationale dela Recherche (ANR) are acknowledged for financial support (grantno: ANR-06-RIB-020-02).
[1] C. W. Jefford, Drug Discov. Today 2007, 12, 487–495.[2] C. Wongsrichanalai, A. L. Pickard, W. H. Wernsdorfer, S. R.
Meshnick, Lancet Infect. Dis. 2002, 2, 209–218.[3] P. M. O’Neill, G. H. Posner, J. Med. Chem. 2004, 47, 2945–
2964.[4] Y.-L. Hong, Y.-Z. Yang, S. R. Meshnick, Mol. Biochem. Parasi-
tol. 1994, 63, 121–128.
Eur. J. Inorg. Chem. 2008, 2133–2135 © 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjic.org 2135
[5] C. W. Jefford, F. Favarger, V. H. Maria da Graca, Y. Jacquier,Helv. Chim. Acta 1995, 78, 452–458.
[6] A. Robert, B. Meunier, J. Am. Chem. Soc. 1997, 119, 5968–5969.
[7] A. Robert, B. Meunier, Chem. Soc. Rev. 1998, 27, 273–279.[8] A. Robert, Y. Coppel, B. Meunier, Chem. Commun. 2002, 414–
415.[9] A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier, Proc.
Natl. Acad. Sci. USA 2005, 102, 13676–13680.[10] J. Cazelles, A. Robert, B. Meunier, J. Org. Chem. 2002, 67, 609–
619.[11] A. Robert, O. Dechy-Cabaret, J. Cazelles, B. Meunier, Acc.
Chem. Res. 2002, 35, 167–174.[12] S. A.-L. Laurent, A. Robert, B. Meunier, Angew. Chem. Int.
Ed. 2005, 44, 2060–2063.[13] S. A.-L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meun-
ier, ChemBioChem 2005, 6, 653–658.[14] R. K. Haynes, W.-Y. Ho, H.-W. Chan, B. Fugmann, J. Stetter,
S. L. Croft, L. Vivas, W. Peters, B. L. Robinson, Angew. Chem.Int. Ed. 2004, 43, 1381–1385.
[15] a) R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H.-D. Heilmann, H.-W. Chan, M.-K. Cheung, W.-L. Lam, H.-N.Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters,B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle, B.Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke,U. Wollborn, K. Schmeer, A. Römer, Angew. Chem. Int. Ed.2006, 45, 2082–2088; b) Supporting Information of ref.[15a]
[16] R. K. Haynes, W.-C. Chan, C.-M. Lung, A.-C. Uhlemann, U.Eckstein, D. Taramelli, S. Parapini, D. Monti, S. Krishna,ChemMedChem 2007, 2, 1480–1497.
[17] B. Hölzer, European Patent EP 1,604,992 A1, 14 December2005.
[18] Analytical data for 3: MS (DCI/NH3+): m/z = 402.5 [M + H+],
419.5 [M + NH4+]. C19H31NO6S (401.5): calcd. C 56.84, H
7.78, N 3.49; found C 56.67, H 7.61, N 3.41. 1H NMR(500 MHz, CDCl3): δ = 5.31 (s, 1 H, 12-H), 4.24 (d, 3J10-H,9-H
= 10.3 Hz, 1 H, 10-H), 3.54–3.49 (m, 2 H, CH2), 3.43–3.38 (m,2 H, CH2), 3.23 (m, 4 H, 2 CH2), 2.61 (m, 1 H, 9-H), 2.38 (m,1H, 4-H), 2.03 (m,1 H, 4-H), 1.90 (m, 1 H, 5-H), 1.78–1.73 (m,2 H, 7-H, 8-H), 1.60 (m,1 H, 8a-H), 1.56–1.44 (m, 1 H, 5-H),1.40 (s, 3 H, 3-CH3), 0.98 (d, 3J = 6.3 Hz, 3 H, 6-CH3), 0.83(d, 3J = 7.2 Hz, 3 H, 9-CH3) ppm. 13C NMR (125.7 MHz,CDCl3): δ = 104.30 (C-3), 92.14 (C-10), 91.03 (C-12), 80.18 (C-12a), 51.91 (N-CH2), 51.46 (C-6a), 47.02 (S-CH2), 45.63 (C-8a), 37.49 (C-6), 36.19 (C-4), 34.22 (C-7), 29.07 (C-9), 25.91 (3-CH3), 24.83 (C-5), 21.63 (C-8), 20.22 (6-CH3), 13.45 (9-CH3)ppm. The 13C NMR spectrum is available as Supporting Infor-mation. There is a discrepancy between the 13C data and thosereported in ref.[15b] The 13C NMR spectrum reported byHaynes et al. exhibits 18 signals for only 17 different carbonatoms in compound 3 (no assignment was provided); the signalat δ = 174.2 ppm (absent from our spectrum), cannot be likelyassigned to a carbon atom of artemisone. Such a chemical shiftis usual for a carbonyl function (acid, ester, amide) which doesnot exist in artemisone. In addition, we detected only two sig-nals at δ = 90–92 ppm, and not three as reported by Haynes etal. These signals have been assigned by 2D correlations to C-10 and C-12 (δ = 92.14 and 91.03 ppm, respectively). The signalof C-12a, at the junction of four cycles, was detected at δ =80.18 ppm. The article by Haynes et al. does not report anysignal at this chemical shift.
Received: February 1, 2008Published Online: March 31, 2008
(Since its publication in Early View,a few minor changes have been made.)
63

SUPPORTING INFORMATION
Alkylation of heme by artemisone
To a solution of Fe(PPIX)Cl (16.16 mg, 24.8 μmol) and artemisone (10.26 mg, 25.6 μmol, 1 mol.
equiv.) in DMSO (0.5 mL), was added Na2S2O4 (21.7 mg in 40 μL of water, 124.6 μmol, 4.3 mol.
equiv.).
At t = 0 (just before addition of Na2S2O4) and t = 1 h, an aliquot of the reaction mixture (20 μL) was
withdrawn and diluted with 980 μL of methanol/water/trifluoroacetic acid, 70/30/0.05, v/v. This
solution was then diluted 100 x in the same solvent, and 100 μL of this latter diluted solution were
injected for HPLC analysis on a 5 μm C18 Nucleosil column. The eluent gradient was from
methanol/water/trifluoroacetic acid, 70/30/0.05, v/v to methanol/water/trifluoroacetic acid,
100/0/0.05, v/v. The flow was 0.5 mL/min, and detection at 400 and 410 nm with a diode array
detector.
After 1 h, the peak of heme (Rt = 18.2, λmax = 398 nm) was decreased by 93% with respect to its
intensity at t = 0. The major products were detected as seven peaks having Rt = 23.1, 23.8, 24.1,
24.6, 25.0, 25.5, and 25.9 min. All these peaks have λmax = 405-407 nm, and the sum of their
intensities corresponds to ca. 80-90% of those of the heme derived material (Figure S2).
ESI+-MS after dilution of the crude reaction mixture in acetonitrile: m/z (relative intensity): 957.5
(6, M+, 13), 862.5 (7, MNa+, 14), 858.3 (hydrate derivative of 7, M+, trace), 840.3 (7, M+, 100),
616.2 (unreacted heme, M+, 16).
ESI+-MS after dilution in methanol: m/z (relative intensity): 950.5 (8•DMSO, M+, 100), 918.4
(7•DMSO, M+, 24), 872.5 (8, M+, 100), 840.5 (7, M+, 14), 694.5 (unreacted heme•DMSO, M+, 73).
64

Figures
S1 13C (125.7 MHz, CDCl3) NMR spectrum of artemisone 3
65

S2. HPLC chromatogram of heme alkylation by 3.
66



Chapter 3
Alkylating ability of artemisinin after Cu(I)induced activation


Chapter 3: Alkylating ability of artemisinin after Cu(I)induced activation
Résumé en français de la publication :
Alkylating ability of artemisinin after Cu(I)induced activation
F. Bousejra-El Garah, M. Pitié, L. Vendier, B. Meunier, and A. Robert Journal of Biological
Inorganic Chemistry 2009, 14(4), 601-610.
Capacité alkylante de l’artémisinine après réduction par le cuivre(I)
Les érythrocytes contiennent environ 20 µM de cuivre, principalement sous la forme de SOD
Cu/Zn.[1] La digestion de cette SOD par Plasmodium libère du cuivre qui pourrait être utilisé par le
parasite.[2] Comme le fer, le cuivre est un métal oxydo-réducteur biologique, capable de catalyser la
formation de radicaux par la réaction de Fenton. C’est pourquoi nous avons étudié l’activation de
l’artémisinine par ce métal, bien que sa concentration dans les globules rouges soit environ mille
fois moins importante que celle du fer contenu dans l’hème.
Pour étudier la réactivité de l’artémisinine vis-à-vis du cuivre, nous avons utilisé des complexes de
cuivre(I) dont les ligands présentent des sites susceptibles d’être alkylés par les radicaux de
l’artémisinine. Ainsi, des ligands peptidiques, Clip-Phen,[3] et dipyrrine ont été utilisés.
1. Activation de l’artémisinine par un sel de cuivre(I)
Il a été montré que des sels de métaux de transition autres que le fer peuvent réduire le peroxyde de
l’artémisinine pour donner les mêmes produits de dégradation que ceux générés en présence de sels
de fer(II).[4,5] Dans un travail préliminaire, nous nous sommes donc intéressés à l’activation de
l’artémisinine en présence de chlorure de cuivre(I). L’analyse des produits de réaction a montré la
formation de la déoxyartémisinine 4 et du dérivé furane-acétate 5, également générés en présence
de fer(II). Le composé 6 a également été caractérisé, sous la forme de deux épimères. A notre
connaissance, la formation de ce composé n’a pas été décrite dans la littérature. Un mécanisme de
sa formation est proposé dans la publication ci-après.
71

Chapter 3: Alkylating ability of artemisinin after Cu(I)induced activation
Activation de l’artémisinine en présence de cuivre(I) et produits de réarrangement
2. Activation de l’artémisinine par le cuivre(I) en présence de glutathion (γGluCysGly, GSH)
L’activation réductrice de l’artémisinine a pu être réalisée avec le complexe cuivre(I)-glutathion
généré in situ. L’analyse par spectrométrie de masse du mélange réactionnel a permis de
caractériser les adduits artémisinine-glutathion 7, 8 et 9 provenant de la liaison covalente entre le
radical thiyl du glutathion et le radical alkyle issu de l’artémisinine. Ce résultat est en accord avec
ceux décrits pour la réaction avec le fer(II) en présence de glutathion.[6]
Adduits covalents GS-artémisinine
3. Activation de l’artémisinine par le complexe CuI(ClipPhen)
Le complexe CuII(2-Clip-Phen)Cl2, synthétisé dans notre équipe,[3] contient deux groupements
phénanthroline, reliés par un pont 2-amino-1,3-propanediol. Après réduction in situ par le
dithionite, nous avons utilisé le complexe CuI(Clip-Phen) à la fois comme activateur et cible
potentielle de l’artémisinine. Un produit de masse m/z 791.3, correspondant à un produit de
couplage covalent entre le complexe Cu(Clip-Phen) et l’artémisinine, a été caractérisé par
spectrométrie de masse. La masse et la figure isotopique de cet ion sont compatibles avec les
structures proposées ci-dessous. En effet, la masse de cet ion correspond à la somme des masses du
complexe Cu(Clip-Phen) et de l’artémisinine. Par ailleurs, la répartition isotopique de cet ion est
caractéristique de la présence d’un atome de cuivre dans sa structure.
72

Chapter 3: Alkylating ability of artemisinin after Cu(I)induced activation
Propositions de structure pour l’adduit Cu(Clip-Phen)-artémisinine
4. Activation de l’artémisinine par le complexe CuI(dipyrrin)2
L’artémisinine étant capable de former des adduits avec la tétraphénylporphyrine (TPP) de
manganèse,[7] utilisée comme modèle de l’hème, nous avons dans un premier temps utilisé la
tétraphénylporphyrine de cuivre pour activer l’artémisinine. Cependant, compte-tenu de
l’impossibilité de réduire le cuivre dans le ligand porphyrinique plan-carré rigide, nous avons
ensuite utilisé le ligand meso-phényldipyrromethène (dipyrrine), plus souple, pour chélater le
cuivre. Le choix de ce ligand, qui peut être vu comme une demi-TPP, permet en effet la
coordination tétrahédrique requise pour le cuivre(I).
La réactivité de l’artémisinine vis-à-vis du complexe CuI(dipyrrine)2 a été
étudiée, et comparée à celle vis-à-vis du complexe FeII(dipyrrine)3. Dans les deux
cas, l’adduit covalent dipyrrine-artémisinine 14 (m/z 503.2, MH+) – démétallé
dans les conditions analytiques - a été identifié par spectrométrie de masse.
D’après les résultats obtenus avec MnTPP, on s’attend à ce que l’alkylation ait
lieu sur les positions pyrroliques.
Bien que la formation de l’adduit 14 ait été mise en évidence avec les deux
complexes Cu(dipyrrine)2 et Fe(dipyrrine)3, les rendements obtenus indiquent que
la réactivité de l’artémisinine est meilleure avec le fer(II) qu’avec le cuivre(I).
5. Conclusion
Ce travail nous a permis d’explorer une voie nouvelle dans le mécanisme d’action de l’artémisinine,
mettant en jeu le cuivre contenu dans les érythrocytes.
En présence de complexes de cuivre(I), l’activation réductrice de l’artémisinine a lieu. La formation
de produits d’alkylation a été mise en évidence avec tous les complexes étudiés. Toutefois, nous
avons également observé que, en présence de complexes de cuivre, le réarrangement intra-
moléculaire des radicaux de l’artémisinine, qui est observé avec un sel de cuivre, est
73

Chapter 3: Alkylating ability of artemisinin after Cu(I)induced activation
74
systématiquement favorisé par rapport à la formation d’adduits, et que l’alkylation du ligand reste
un phénomène minoritaire.
De plus, la formation d’adduits en présence de dipyrrine de fer indique qu’un complexe de fer non
héminique peut également être activateur et cible de l’artémisinine.
Bibliographie
[1] D. Rasoloson, L. Shi, C. R. Chong, B. F. Kafsack, D. J. Sullivan. Copper Pathways in Plasmodium falciparum Infected Erythrocytes Indicate an Efflux Role for the Copper P-ATPase. Biochem. J. 2004, 381, 803-811. [2] P. L. Olliaro, D. E. Goldberg. The Plasmodium Digestive Vacuole: Metabolic Headquarters and Choice Drug Target. Parasitol. Today 1995, 11, 294-297. [3] M. Pitié, B. Donnadieu, B. Meunier. Preparation of the New Bis(Phenanthroline) Ligand "Clip-Phen" And Evaluation of the Nuclease Activity of the Corresponding Copper Complex. Inorg. Chem. 1998, 37, 3486-3489. [4] W. M. Wu, Y. L. Wu. Chemical and Electro-Chemical Reduction of Qinghaosu (Artemisinin). J. Chem. Soc., Perkin Trans. 1 2000, 4279-4283. [5] Y. Wu, H.-H. Liu. Cleavage of Qinghaosu (Artemisinin) Induced by Non-Iron Transition-Metal Ions in the Presence of Excess Cysteine. Helv. Chim. Acta 2003, 86, 3074-3080. [6] W.-M. Wu, Z.-J. Yao, Y.-L. Wu, K. Jiang, Y.-F. Wang, H.-B. Cehn, F. Shan, Y. Li. Ferrous Ion Induced Cleavage of the Peroxy Bond in Qinghaosu and Its Derivatives and the DNA Damage Associated with This Process. Chem. Commun. 1996, 2213-2214. [7] A. Robert, B. Meunier. Characterization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968-5969.

ORIGINAL PAPER
Alkylating ability of artemisinin after Cu(I)-induced activation
Fatima Bousejra-El Garah Æ Marguerite Pitie ÆLaure Vendier Æ Bernard Meunier ÆAnne Robert
Received: 31 October 2008 /Accepted: 23 January 2009 / Published online: 7 February 2009! SBIC 2009
Abstract The reductive activation of artemisinin bycopper(I)-dipyrrin or copper(I)-(2-Clip-Phen) complexes
generates an artemisinin derived alkylating species leading
to covalent artemisinin–copper complex adducts. Thereactivity of the peroxide function of artemisinin toward
Cu(I) complexes is similar to that of Fe(II) analogues, even
though the reaction is more sluggish and product distri-bution slightly different.
Keywords Alkylation ! Artemisinin ! Copper !Malaria ! Non-heme complexes
Introduction
Faced with the burden of malaria, efforts to control thedisease are currently based on anti-vector measures asso-
ciated with drug combinations containing an artemisinin
derivative [1]. The recent success of these measures in afew specific areas of Africa, such as KwaZulu-Natal in
South Africa, Eritrea, and the Tanzanian island of Zanzi-bar, prompted renewed calls for global eradication [2].
Artemether and artesunate, now recommended by the
WHO, are semi-synthetic derivatives of artemisinin 1(Fig. 1), a 1,2,4-trioxane extracted from Artemisia annua.
Although the exact mechanism of action of artemisininand its derivatives is still a matter of debates, it is known
that the key pharmacophore of this highly efficient anti-
malarial is its 1,2,4-trioxane unit, and in particular itsperoxide bond [3]. Peroxides are known to react with
transition metals of low valency such as iron(II) [4]. In
particular, the crucial role of iron(II)-heme in the diges-tion/polymerization process in infected erythrocytes has
led to investigations of its possible interaction with
artemisinin.Since the initial work of Meshnick and coworkers [5] on
the heme-catalyzed reductive activation of artemisinin and
on the alkylation of heme and specific parasite proteins byartemisinin [6, 7], the radical chemistry of this antimalarial
drug has been investigated. In the same period, Posner,
Jefford and coworkers have also reported the chemicaldecomposition of artemisinin by simple iron(II) salts, and
have pointed out the importance of an artemisinin derived
alkyl radical centered at C4 [8, 9]. In the presence of iro-n(II), the peroxide bond of artemisinin is cleaved, leading
to the formation of oxy-radicals that rapidly rearrange viaeither hydrogen abstraction or b-scission to form carbon-
centred radicals [10, 11]. These latter radicals are able to
alkylate biomolecules such as heme or specific parasiticproteins. Covalent adducts between artemisinin and heme
have been characterized both in vitro and in malaria-
infected mice [12, 13]. On the basis of artemisinin reac-tivity, many synthetic peroxides have been synthesized as
potential antimalarial drugs (one of them, trioxaquine
PA1103, is currently under pre-clinical development[14, 15]) and the possible interaction of heme with the
peroxide function of the drugs has been investigated
[16–20]. Like artemisinin, the alkylating ability of thetrioxaquine DU1301 on heme has been observed in vivo
[21]. In addition, the in vitro alkylating ability of several
F. Bousejra-El Garah ! M. Pitie ! L. Vendier ! A. Robert (&)Laboratoire de Chimie de Coordination du CNRS,205 route de Narbonne,31077 Toulouse cedex 4, Francee-mail: [email protected]
B. MeunierPalumed, 3 rue de l’Industrie,Z.I. de Vic, 31320 Castanet-Tolosan, France
123
J Biol Inorg Chem (2009) 14:601–610
DOI 10.1007/s00775-009-0474-z
75

1,2,4-trioxolanes (family of OZ277) has been correlatedwith their antimalarial activity [22].
Moreover, the artemisinin-induced inhibition of two
parasitic proteins, a TCTP (translationally controlled tumorprotein) homolog [23] and the calcium-dependent ATP-ase
PfATP6 (Ca-ATP-ase of Plasmodium falciparum) [24],
was clearly reported as being iron-dependent. Alkylation ispossible, although adducts between artemisinin and these
proteins have never been identified in the biological
models.Like iron, copper is a biologically relevant metal which
can catalyze peroxide reduction, and thus participate inthe free-radical-producing Fenton reaction [25]. Many
copper–sulfur proteins participate to in vivo electron
transfer reactions. For example, one of the four copperatoms of ascorbate oxidase has an environment quite
similar to that of plastocyanins, chelated by two nitrogenand two sulfur atoms (contained in methionine and cys-
teine). Human erythrocytes contain about 20 lM copper,
of which 70% is found in the Cu/Zn superoxide dismutase(SOD). During the hemozoin formation process in the
food vacuole of Plasmodium, toxic ferrous heme is oxi-
dized to the ferric state. This oxidation reaction results inthe generation of reduced oxygen species, a major source
of oxidative stress that can be fatal for the parasite. To
counter this, Plasmodium has an anti-oxidant defense thatis not completely understood. Nevertheless, it seems that
Plasmodium concentrates the host Cu/Zn SOD in itsdigestive vacuole, and may use it to convert superoxide
radical to hydrogen peroxide [26]. The H2O2 is then
cleaved by a catalase activity whose origin and locationare unknown [27]. Like hemoglobin, this Cu/Zn SOD is
Fig. 1 Reductive activation ofartemisinin 1 by CuCl2/Na2S2O4, giving rise torearrangement products 4, 5,and 6 (isolated yields areindicated)
602 J Biol Inorg Chem (2009) 14:601–610
123
76

also digested inside the parasite food vacuole, a process
that may be one of the possible sources of copper for thePlasmodium [28].
To date, few studies have mentioned the reaction of
artemisinin with copper(I) salts [29]. The aim of thepresent study was to induce the copper(I) reductive acti-
vation of the artemisinin peroxidic bond and examine the
possible alkylation of copper complexes. Beyond thecharacterization of artemisinin rearrangement products, we
expected to detect possible covalent adducts by formationof C–C or C–heteroatom bonds between artemisinin and
copper complexes.
Results and discussion
After checking the reductive activation of artemisinin by
copper(I) salts, we then studied the reaction of artemisinin
with copper(I) complexes of (1) amino acids or peptides,(2) 2-Clip-Phen ligand, and (3) meso-diphenyldipyrro-methene (dipyrrin for short). In these three cases, we
detected covalent adducts between artemisinin derived C4radical and the ligand of copper, either through a C–S or a
C–C bond.
Activation of artemisinin by copper(I) salt
Wu and coworkers reported that, in acetonitrile/water,copper sulfate and other non-iron transition metal salts
induce the cleavage of artemisinin to produce the rear-
rangement products that have already been reported withFe2? ions [29, 30]. In our hands, reaction of artemisinin
with copper(II) chloride reduced in situ by sodium
dithionite in dimethylsulfoxide mainly produced deox-yartemisinin 4 in 57% yield with respect to the starting
amount of artemisinin (Fig. 1). Compound 4 resulted
from initial coordination of Cu(I) on O2 and homolysis ofthe peroxide to generate an alkoxy radical on O1 2a. Ithas been proposed that 2a rearranged via a stereospecific
1,5-H shift from H4a, leading to the secondary alkylradical 3a [8]. From 3a, deoxyartemisinin 4 might then be
formed by capture of H! from the DMSO used as solvent,
and release of CuII–OH. However, the formation of 4could also occur from 2a via capture of H! to generate an
hydroxyl function at C12a, followed by release of CuII–
OH, without involving the secondary alkyl radical 3a. Infact, the initial alkoxy radical 2a is better at abstracting a
hydrogen atom from an external source than the alkyl
radical 3a (this has been evidenced in autoxidation reac-tion). In addition, the distance for the H-atom transfer
from C4 to O1 probably exceeds the critical limit distance
of 2.1 A for such a migration [31]. Direct formation of4 from 2a is then more probable.
Compound 5 was also obtained in 9% yield. This
compound was produced by homolysis of the peroxidebond after initial coordination of Cu(I) on O1, followed by
C3–C4 bond cleavage and subsequent ring closure. Char-
acterisation of 4 and 5 was consistent with data reported inreferences [32] and [33], respectively. These two reaction
pathways, resulting of the coordination of a reduced metal
either on O1 or on O2 of the peroxide bond, have alreadybeen reported after activation of artemisinin by iron(II) salt
[18, 34, 35]. In Wu’s conditions (water/acetonitrile, 1/1),compound 4 was only obtained in 1 or 2% yield with
stoichiometric FeII or CuI, respectively, whereas the tetra-
hydrofurane derivative 5 was the major product (41%)[30]. Other reports suggest a small preference for attack of
Fe(II) on the nonketal peroxide oxygen atom of artemisinin
(O1) [36]. Anyway, as already pointed out with iron, dataobtained with copper suggest that the formation of the
alkoxy radical either on O1 or on O2 (Fig. 1, pathways (a)and (b), respectively), and subsequent product distribution,may depend on the reaction conditions [37].
In the present study, compound 6 (Fig. 1) was also
isolated in 6% yield. To our knowledge, this compound hadnever been isolated from artemisinin degradation up to
now. It was characterized by 2D NMR as a mixture of two
epimers at C12a (A:B = 60:40). A significant difference inthe chemical shifts (up to 0.6 ppm in 1H-NMR and
7.7 ppm in 13C-NMR) was observed for the two stereo-
isomers, indicating two markedly different conformations,keto-butyl at C5a and hydroxyl at C12a being either cis- ortrans-substituents with respect to the cyclohexyl ring. A
possible intramolecular H-bonding between O=C3 andHO–C12a giving rise to an eight-membered ring (for the
trans isomer), or the sterically hindered free rotation of the
keto-butyl chain (for the cis isomer) result in diastereotopicbehavior of the C4 and C5-methylene groups. A possible
pathway for the formation of 6, by homolysis of the C3–
O13 bond of the alkoxy radical 2b, is proposed in Fig. 1. Inprinciple, compound 6 can exist as a cyclic form 60.However, we failed to isolate 60, and the ‘‘open’’ structure
of 6 was confirmed by the 13C chemical shift of carbonylC3, detected at 208.8 and 212.6 ppm for epimers A and B,
respectively.
The studies described above, focused on copper ions,allowed only the characterisation of artemisinin rear-
rangement products. To explore the possible alkylation
reaction between artemisinin and a putative target, it ismore favorable to have an alkylable target in the close
vicinity of the metal center. In fact, an intramolecular
alkylation reaction (as in the case of alkylation of heme byartemisinin) should occur rather than an intermolecular
reaction. For this purpose, we used redox-active copper
chelates, with ligands having an alkylable position in theclose vicinity of the metal center.
J Biol Inorg Chem (2009) 14:601–610 603
123
77

Activation of artemisinin by copper in the presence
of glutathione
We activated artemisinin with copper complexes of cys-
teine-containing peptides. In particular, glutathione (c-Glu-Cys-Gly, GSH) is a physiological reducing agent occurringin high concentrations in red blood cells (5 mM). The
coordination of sulfur onto the metal center would place (1)
the reduced metal, (2) artemisinin, and (3) the peptidetarget in close proximity, thus increasing the probability of
peptide alkylation. So, the peptide may play the role of
both the reducing agent of the metal center, and the alky-lable target.
When artemisinin reacted with copper (I), generated in
situ from copper(II) chloride and glutathione, we detectedby mass spectrometry minor amounts of a ‘‘complete’’
covalent adduct between glutathione and artemisinin, that
retains the full fragments of glutathione and artemisinin.In fact, the exact mass of this compound (589) corresponds
to the addition of the masses of glutathione (307) and
artemisinin (282). This adduct may be formed by recom-bination of the S-centered radical of glutathione with the
C4-centered primary radical of artemisinin 3b, giving rise
to a thioether bond (GS-artemisinin adduct 7, Fig. 2,m/z = 590.3 and 612.3 for MH? and MNa?, respectively).
In fact, the production of the thiyl radical is also supported
by the detection of oxidized glutathione GS–SG (m/z =613.4, MH?). In addition, the formation of compound 10(m/z = 285.4, MH?), resulting from the capture of H! byradical 3b confirms the possibility of homolytic cleavage ofthe thiol function.
Compound 8, resulting from the hydrolysis of the lac-
tone in 7 and loss of an acetic acid unit, was also detected(m/z = 548.2, MH?) along with the 5-membered lactone
(9, m/z = 530.2, MH?). A similar reactivity of the lactone
ring of the drug-derived fragment has been reported forheme-artemisinin adducts [12, 13]. Compounds 4, 5 and 6were also detected.
A similar alkylation reaction occurred when h-cysteinewas used instead of glutathione, giving rise in low yield,
to the adduct between h-cysteine and artemisinin
(m/z = 418.4, MH?, see ‘‘Materials and methods’’).
Compound 8 was isolated by Wu and coworkers in the
presence glutathione and Fe2? [38]. The same researchgroup also reported the formation of a cysteine–artemisinin
adduct in the presence of FeSO4 [39]. These authors carried
out a similar reaction with copper salt. However, the yieldreported for the cysteine–artemisinin adduct in the latter
case (as high as 37%) was not reliable, being only ‘‘esti-
mated from missing amounts’’ of products in 1H-NMRspectrum of the crude product mixture [29]. On the con-
trary, our experience indicates that the yield of this C–Scoupling reaction is probably very low (\1%).
Activation of artemisinin by CuI(2-Clip-Phen)
Bis-phenanthroline ‘‘2-Clip-Phen’’ complexes, containing
two phenanthroline units linked by a serinol bridge in theortho position of a phenanthroline nitrogen, have been
designed as redox copper chelates with nuclease activity
[40–42]. In the present study, the copper complex of2-Clip-Phen was used both as an activator of artemisinin
and its putative alkylation target. The metal center of
the CuII(2-Clip-Phen)Cl2 complex (11-CuII, Fig. 3) wasreduced in situ by sodium dithionite under an argon
atmosphere and the formation of the diamagnetic copper(I)
complex was followed by 1H-NMR. Artemisinin was thenadded. After 1 h, TLC monitoring showed the disappear-
ance of artemisinin. 1H-NMR of the reaction mixture
suggested the presence of compounds 4 and 5, their H12resonances being detected as sharp singlets at 5.86 and
6.51 ppm, respectively, in DMSO-d6. The presence of 5was confirmed by the resonance of the methylene group atC4 (4.73 and 3.83 ppm). A modication of the –O–CH2–
signal of the 2-Clip-Phen ligand was also observed
(4.66 ppm compared with 4.95 ppm for the starting 11-CuI
complex). However, all NMR signals of the phenanthroline
residue were broadened, as already described for cuprous
Clip-Phen complexes, which consist of mixtures of speciesin rapid exchange at the NMR time scale [41]. In addition,
the region of aliphatic protons was very complicated due to
the overlapping of several artemisinin-derived fragments.Therefore, the characterisation of a 2-Clip-Phen–artemisi-
nin adduct could not be achieved by NMR. However, the
Fig. 2 Alkylation ofglutathione by artemisinin 1 inthe presence of copper(I), givingrise to GS-artemisinin covalentadducts 7, 8, and 9
604 J Biol Inorg Chem (2009) 14:601–610
123
78
mass spectrum of the crude reaction mixture indicated the
presence of a new compound with m/z = 791.3, along withcompounds 4, 5 and 6 as major products. This peak at
791.3 is the exact mass of Cu(2-Clip-Phen) (510.1) plus themass of artemisinin (282.2) minus one hydrogen atom (1).
Thus, a compound with this mass strongly suggests cova-
lent coupling between Cu(2-Clip-Phen) and artemisininwithout loss of a moiety. The MS isotopic pattern showed
the presence of one copper atom (M ? 2 due to 65Cu,
Fig. 3). When a similar reaction was made using Cu(phe-nanthroline)2Cl2 (lacking the serinol bridge) instead of 11-Cu, only rearrangement products 4, 5, and 6 were detected.
This result suggests that alkylation of 11-Cu by artemisinin
may occur on the serinol linker, rather than on the phe-nanthroline part.
The structure proposed in Fig. 3 (12-Cu) is consistentwith these results. As an alternative, a free hydroxyl
function at C12 and coordination of the deprotonated ali-
phatic amine on copper cannot be ruled out (120-Cu). Wetried to clarify the alkylation position of the 2-Clip-Phen
ligand. As mentioned above, 1H-NMR analysis of the crude
reaction mixture allowed the detection of methylene groupsH2C1
0, with a correct integration value with respect to
the phenanthroline protons. In contrast, HC20 was not
Fig. 3 Alkylation of Cu(2-Clip-Phen) by artemisinin 1.ESI?-MS characterisation andproposed structure of theCu(2-Clip-Phen)–artemisinincovalent adduct 12-Cu
J Biol Inorg Chem (2009) 14:601–610 605
123
79

unambiguously detected. In addition, the possible radical
alkylation at the a-position of aliphatic amines has beenreported [43], whereas radical N-alkylation has not. It can
therefore be proposed that the coupling between the 2-Clip-
Phen and the artemisinin fragments occurred throughC20–C4 bonding.
On the basis of the mass spectrometry of the crude
reaction mixture (triplicate), the relative intensity of theadduct 12-Cu with respect to the total amount of products
4 ? 5 ? 6 ? 12-Cu was c.a. 2%.
Activation of artemisinin by CuI(dipyrrin)
As artemisinin efficiently reacts with iron(II)-heme and
manganese(II) tetraphenylporphyrin, at meso- or b-pyrrolepositions, respectively, we first tried to activate artemisininwith copper tetraphenylporphyrin (CuTPP) or with the
copper analogue of heme (CuPPIX). These complexes are
stable Cu(II) species, and their cuprous analogues must begenerated in situ to induce artemisinin activation. How-
ever, we failed to generate CuITPP or CuIPPIX. In fact, the
most preferable coordination environments of Cu(II) andCu(I) are different. The d9 Cu(II) complexes generally
adopt Jahn-Teller distorted six-coordinate or five-coordi-
nate geometries which fit well with the porphyrin ligandswhen a solvent molecule or a counter-ion ligation is
included. The d10 Cu(I) complexes generally prefer lower
coordination numbers such as four-coordinate tetrahedralenvironments, which is an unfavorable situation for the
rigid square planar structure of porphyrins. Indeed, it has
been reported that the first one-electron reduction of Cu(II)porphyrins usually occurs at the ligand, resulting in the
formation of the p-radical anion of Cu(II) porphyrins
[44–46]. The relatively good fit of Cu(II) into the porphyrinhole, compared to the large radius of Cu(I), may also
explain the lack of any Cu(I) porphyrin complexes [47].
Copper porphyrins were therefore not suitable to induceartemisinin activation.
We then used meso-phenyldipyrromethene (dipyrrin, for
short) as ligand. This kind of ‘‘half-porphyrin’’ is expected
to have a similar reactivity to that of TPP at b-pyrrolepositions; but a non-square planar geometry should beallowed for a coordinated metal ion and dipyrrin should
then be able to coordinate Cu(I). Then, we prepared di-
pyrrin ligand [48] and its corresponding copper complexCu(dipyrrin)2, 13-Cu (Fig. 4) [modification from reference
49]. The reactivity of artemisinin with 13-Cu was then
investigated in the presence of 2,3-dimethylhydroquinoneas reducing agent. In comparison, a similar reaction was
made with the iron analogue Fe(dipyrrin)3 13-Fe [50]. Inboth cases, a compound with a molecular peak at m/z = 503.2 was detected by ESI?-MS, along with the
molecular peak of the dipyrrin ligand at m/z = 221.1. Theexact mass of this new compound (502) corresponds to the
mass of dipyrrin (220) ? the mass of artemisinin (282). It
is therefore consistent with the presence of a dipyrrin–ar-temisinin covalent adduct obtained after substitution of one
proton of dipyrrin by the drug residue (14, Fig. 4). Thealkylation site was not determined, however the reaction isexpected to occur in the same fashion as with Mn-TPP, e.g.at b-pyrrolic positions [11]. In this regard, the structure
proposed for adduct 14 is the most probable. It should bementioned that mass spectrometry of the crude reaction
mixtures did not allow the detection of the iron or copper
complexes of the dipyrrin–artemisinin adduct. In a similarway, when Fe(dipyrrin)3 13-Fe and Cu(dipyrrin)2 13-Cuwere analyzed by ESI?-MS, demetallation occurred in the
spectrometer and only the dipyrrin ligand was detected.Beside adduct 14, compounds 4, 5, and 6 were also formed
as by-products of the reaction.
On the basis of mass spectrometry, the relative ratio ofthe covalent dipyrrin–artemisinin adduct (m/z = 503.2)
with respect to the total amount of dipyrrin (e.g.221.1 ? 503.2) was 47 and 18% when starting from theiron and copper dipyrrin complexes, respectively. Mass
spectrometry is not, strictly speaking, a quantitative tech-
nique. However, this result indicates that the alkylationyield of Cu(dipyrrin)2 by artemisinin, although moderate, is
probably the highest yield reported up to now with a copper
complex. In the presence of Fe(dipyrrin)3, a yield of 47%
Fig. 4 Alkylation ofCu(dipyrrin)2 and Fe(dipyrrin)3by artemisinin 1, giving rise tothe dipyrrin–artemisinincovalent adduct 14. ORTEPview of Cu(dipyrrin)2
606 J Biol Inorg Chem (2009) 14:601–610
123
80

of dipyrrin–artemisinin adduct 14 was obtained after 4.5 h
at 38 "C, which is comparable to the yields reported for thealkylation of Mn(TPP) by artemisinin (25% after 80 min)
or several other antimalarial peroxides [51]. This result
indicates that the extended conjugation of the porphyrinmacrocycle is not required for an efficient alkylation
reaction by the artemisinin-derived C4-radical entity.
Conclusion
In the presence of copper(I) ions and chelates, the
reductive activation of the peroxidic bond of artemisininis observed. In all cases, the intra-molecular collapse of
the radicals generated by artemisinin reduction was
favored with respect to the alkylation of metal ligands.Nevertheless, this study documents the alkylation of a
target by an artemisinin-derived radical in the vicinity of
the reduced copper center, with formation of C–C or C–Sbonds. In addition, the intra-erythrocytic concentration of
copper being 1,000 times lower than that of iron, in vivo
activation of artemisinin by copper should be consideredas a minor reaction pathway compared to the activation
by heme.
The Fe(dipyrrin)3 complex was also alkylated by arte-misinin, in significantly higher yield than Cu(dipyrrin)2.
The formation of a covalent adduct between the Fe(di-
pyrrin)3 complex and artemisinin indicates that non-hemeiron complexes are able to activate artemisinin, leading to
covalent adducts between the drug and the iron ligand.
Such a reaction may occur with biomolecules containingnon-heme iron sites. Note that parasitic proteins such as
TCTP or PfATP6, both reported as putative artemisinin
targets, are inhibited by artemisinin in an iron-dependentmanner [23, 24].
Materials and methods
Chemicals
Artemisinin was supplied from Sigma-Aldrich. CuCl2!2H2O and Cu(CH3COO)2!H2O were purchased from Fluka;DMSO 99.9% and Na2S2O4 were from Acros. Chelex#
100, 100–200 mesh, was purchased from Bio-Rad.
Dichloromethane and hexane were supplied by Fluka andhad a low content of evaporation residue (B0.0005%).
Unless noted, all solvents and reagents were purchased
from commercial suppliers and used without furtherpurification. Solvents were freshly vacuum degassed and
purged with argon to remove oxygen prior to use. All
alkylation reactions were carried out under an argonatmosphere.
Instrumentation
1H- and 13C-NMR spectra were recorded on BrukerAvance 500 or Avance 300 instruments. Significant1H-NMR data are written following the order: multiplicity
(b, broad; s, singlet; d, doublet; t, triplet; q, quartet; m,multiplet), number of protons, coupling constants in Hertz,
assignment. Chemical shifts are expressed in ppm (d)relative to tetramethysilane as external reference. Massspectrometry was performed on a QTrap Applied Biosys-
tems or on an LCT Waters mass spectrometer, by direct
injection and detection in ESI or DCI/NH3 mode. Analyt-ical TLC was performed on aluminium sheets precoated
with silica gel (Merck). Visualization was accomplished
using phosphomolybdic acid. Elution conditions werehexane/AcOEt, 70/30, v/v, unless otherwise indicated.
X-ray crystallographic analysis
Data were collected at low temperature (180 K) on a
Bruker Kappa Apex II diffractometer using graphite-monochromated Mo-Ka radiation (k = 0.71073 A) and
equipped with an Oxford Cryosystems Cryostream Cooler
Device. The final unit cell parameters were obtained bymeans of a least-squares refinement performed on a set of
2,557 well measured reflections. The structures were
solved by direct methods using SIR92 [52], and refined bymeans of least-squares procedures on an F2 with the aid of
the program SHELXL97 [53] included in the software
package WinGX version 1.63 [54]. The atomic scatteringfactors were taken from International Tables for X-Ray
Crystallography [55]. All hydrogen atoms were located
geometrically, and refined by using a riding model. Allnon-hydrogen atoms were anisotropically refined, and in
the last cycles of refinement a weighting scheme was used,
where weights were calculated from the following formula:w = 1/[r2(Fo
2) ? (aP)2 ? bP] with P = (Fo2 ? 2Fc
2)/3.
Molecules were drawn with the program ORTEP32 [56]
with 30% probability displacement ellipsoids for non-hydrogen atoms.
Crystallographic data (without structure factors) for the
structure of Cu(dipyrrin)2 13-Cu were deposited with theCambridge Crystallographic Data Centre (CCDC) as sup-
plementary publication no. CCDC-706790. Copies of the
data can be obtained free of charge from the CCDC (12Union Road, Cambridge CB2 1EZ, UK; e-mail: depos-
[email protected]; Web site: http://www.ccdc.cam.ac.uk.
Reaction of artemisinin with copper(I) salt
A solution of artemisinin (51 mg, 181 lmol, 1.0 equiv),
CuCl2!2H2O (31 mg, 182 lmol, 1.0 equiv) and Na2S2O4
(44 mg, 2.26 lmol, 1.4 equiv) in 1.0 mL of degassed
J Biol Inorg Chem (2009) 14:601–610 607
123
81

DMSO was left to stir at 37 "C under argon atmosphere.
Reaction of artemisinin was complete after 1 h (TLCmonitoring). Water (15 mL) was then added and extraction
was achieved with CH2Cl2 (3 9 15 mL). The organic layer
was washed with 15 mL of water to remove traces ofDMSO, dried on Na2SO4, filtered and concentrated. The
crude mixture of products was separated by flash chro-
matography on silica (eluent: hexane/ethyl acetate, 80/20to 0/100, v/v). Yields: 57% of 4, 9% of 5 and 6% of 6.
Analyses for 4
1H-NMR (CDCl3, 500 MHz) d (ppm) 5.72 (s, 1H, H12),3.20 (dq, 1H, J = 7.3 and 4.7 Hz, H9), 2.03 (dt, 1H,
J = 13.1 and 4.5 Hz, H8a), 1.94 (m, 2H, H5eq and H8eq),
1.81 (m, 2H, H4eq and H7eq), 1.65 (m, 1H, H4ax), 1.55 (s,3H, 3-CH3), 1.30 (m, 3H, H5ax, H5a and H6), 1.22 (d, 3H,
J = 7.2Hz, 9-CH3), 1.11 (m, 1H, H7ax), 1.02 (m, 1H,
H8ax), 0.93 (d, 3H, J = 6.0 Hz, 6-CH3).13C-NMR (CDCl3,
125 MHz) d (ppm): 171.8 (C10), 109.2 (C3), 99.7 (C12),
82.4 (C12a), 44.7 (C5a), 42.5 (C8a), 35.4 (C6), 34.0 (C4),
33.5 (C7), 32.8 (C9), 24.0 (3-CH3), 23.5 (C8), 22.0 (C5),18.6 (6-CH3), 12.6 (9-CH3). MS (ESI?) m/z 267.4 (MH?),
289.3 (MNa?). MS (DCI/NH3) m/z 267.4 (MH?), 284.3
(MNH4?).
Analyses for 5
1H-NMR (CDCl3, 500 MHz) d (ppm): 6.67 (s, 1H, H12),
4.24 (bt, 1H, H4eq), 3.96 (bq, 1H, H4ax), 3.19 (qd, 1H,
J = 7.6 and 4.6 Hz, H9), 2.18 (s, 3H, CH3–CO), 2.06 (m,1H, H5eq), 2.02 (m, 1H, H7eq), 1.96 (m, 1H, H8eq), 1.90 (dt,
1H, J = 4.6 and 12.9 Hz, H8a), 1.75 (m, 1H, H6), 1.61 (m,
1H, H5ax), 1.49 (ddd, J = 8.2, 9.5 and 17.1 Hz, H5a), 1.24(d, 3H, J = 7.2Hz, 9-CH3), 1.09 (qd, 1H, J = 2.5 and
6.1 Hz, H7ax), 1.01 (d, 1H, J = 6.0 Hz, 6-CH3).13C-NMR
(CDCl3, 125 MHz) d (ppm) 171.7 (C10), 168.5 (C3), 93.0(C12), 79.4 (C12a), 69.3 (C4), 54.8 (C5a), 426.7 (C8a),
35.0 (C9), 34.6 (C7), 30.9 (C6), 27.7, (C5), 24.3 (C8), 21.2
(CH3–COO), 20.4 (6-CH3), 12.5 (9-CH3). MS (ESI?) m/z283.4 (MH?), 305.3 (MNa?). MS (DCI/NH3) m/z 283.4
(MH?), 300.3 (MNH4?).
Analyses for 6
The two epimers A and B were not separated. 1H-NMR(CDCl3, 500 MHz) d (ppm) 6.86 (s, 1H, OH B), 3.40 (qt,
1H, H9 B), 2.91 (m, 1H, H9 A), 2.82 (m, 1H, H4eq B),
2.68–2.62 (m, 2H, H4ax B and H8a A), 2.57 (m, 1H, H4eqA), 243–2.39 (m, 1H, H4ax A), 2.21–2.27 (m, 2H, H5eq B
and H8a B), 2.23 (s, 3H, CH3CO B), 2.20–2.12 (m, 2H,
H5a A and H8eq A), 2.15 (s, 3H, CH3CO A), 1.94–1.91
(m, 1H, H7eq A), 1.90–1.84 (m, 1H, H5eq A), 1.82–1.78 (m,
2H, H5ax A and H7eq B), 1.74 (m, 1H, H8eq B), 1.64 (m,1H, H6 A), 1.59–1.51 (m, 4H, H7ax A, H5a B, H8ax A and
H5ax B), 1.38–1.34 (m, 1H, H6 B), 1.24 (bd, 3H,
J = 6.7 Hz, 9-CH3 A), 1.14 (bd, 3H, J = 9.0 Hz, 9-CH3
B), 1.12 (bd, 3H, J = 6.0 Hz, 6-CH3 A), 1.09–0.95 (m, 2H,
H7eq B and H8ax B), 0.98 (bd, 3H, J = 6.6 Hz, 6-CH3 B).13C-NMR (CDCl3, 125 MHz) d (ppm) 212.6 (C3 B), 208.8(C3 A), 179.9 (C10 A), 179.8 (C10 B), 56.64 (C5a A),
53.67 (C8a A), 50.87 (C5 B), 46.13 (C8a B), 43.12 (C4 B),41.09 (C4 A), 40.55 (C6 A), 39.31 (C9 B), 38.53 (C9 A),
35.68 (C6 B), 34.34 (C7 A), 32.50 (C7 B), 30.05 (C8 A),
29.99 (CH3CO A), 29.91 (CH3CO B), 24.97 (C8 B), 21.12(C5 B), 20.51 (6-CH3 A), 20.05 (C5 A), 19.82 (6-CH3 B),
14.79 (9-CH3 A), 8.85 (9-CH3 B). MS (ESI?) m/z 255.4
(MH?), 277.3 (MNa?). MS (DCI/NH3) m/z 255.4 (MH?),272.4 (MNH4
?).
Reaction of CuCl2 with artemisinin in the presenceof glutathione
To a solution of CuCl2!2H2O (4.4 mg, 26 lmol, 1 equiv)and artemisinin (20.7 mg, 72 lmol, 3 equiv) in degassed
DMSO (1 mL), was added glutathione (26.7 mg, 87 lmol,
3.4 equiv) as a solid. The solution was stirred at 38 "Cunder argon for 32 h. Water (3 mL) was then added, and
the reaction mixture was extracted with CH2Cl2 (2 mL).
The aqueous phase was diluted with CH3OH (1/1) andtreated overnight in the presence of Chelex# 100, 100–
200 mesh.
The organic phase was diluted with methanol and ana-lyzed by ESI?-MS: m/z (relative intensity) 267.4 (4, MH?,
21), 277.3 (6, MNa?, 12), 289.3 (4, MNa?, 100), 305.4 (5,MNa?, 35), 307.3 (10, MNa?, 43).
The aqueous phase was diluted with methanol contain-
ing 10 vol% formic acid, and analyzed by mass
spectrometry. ESI?-MS m/z 530.2 (9, MH?), 548.2 (8,MH?), 590.3 (7, MH?), 612.3 (7, MNa?), 613.4 (GS–SG).
ESI--MS m/z 588.2 [7, (M-H)-].
Reaction of CuCl2 with artemisinin in the presence
of h-cysteine
To a solution of CuCl2!2H2O (3.1 mg, 18 lmol, 1 equiv)
and artemisinin (20.2 mg, 71 lmol, 4 equiv) in degassed
CH3CN (1 mL) and H2O (0.5 mL), was added h-cysteine(10.0 mg, 35 lmol, 2 equiv) as a solid. The solution was
stirred overnight at 38 "C under argon. Only a small por-
tion of the starting artemisinin was converted (\10%). Thecrude reaction mixture was diluted in methanol for ESI?-
MS analysis: m/z 418.4 (MH? for the h-cysteine–artemis-
inin adduct, trace amount).
608 J Biol Inorg Chem (2009) 14:601–610
123
82

Reaction of artemisinin with CuI(2-Clip-Phen)
2-Clip-Phen ligand was prepared according to reference 41.
Preparation of CuI(2-Clip-Phen)
A solution of CuCl2!2H2O (2.08 mg, 12 lmol, 1 equiv),
2-Clip-Phen (5.42 mg, 12 lmol, 1 equiv) in 0.6 mL
DMSO-d6 was left to stir at 38 "C under argon. Na2S2O4
(10.82 mg, 62 lmol, 5 equiv) was then added, and the
color of the reaction mixture shifted instantaneously from
green (CuII complex) to red (CuI complex). After 45 min ofreaction, the mixture was transferred to an NMR tube under
argon for 1H-NMR analysis.1H-NMR (300 MHz, DMSO-d6) of Cu
I(2-Clip-Phen) d(ppm): 9.04 (dd, 2H, J = 1.7 and 4.4 Hz, H900), 8.49 (dd,
2H, J = 1.7 and 8.2 Hz, H700), 8.45 (d, 2H, J = 8.7 Hz,
H400), 7.97 (d, 2H, J = 8.8 Hz, H600),7.88 (d, 2H,J = 8.8 Hz, H500), 7.75 (dd, 2H, J = 4.4 and 8.2 Hz, H800),
7.31 (d, 2H, J = 8.7 Hz, H300), 4.95 (4H, d, J = 3.8 Hz,
H2C10), 4.22 (m, 1H, HC20).
Reaction of CuI(2-Clip-Phen) with artemisinin
A solution of CuCl2!2H2O (2.12 mg, 12 lmol, 1 equiv)
and 2-Clip-Phen (5.48 mg, 12 lmol, 1.0 equiv) in
0.5 mL DMSO-d6 was left to stir at 38 "C for 10 minunder argon to generate the CuII complex of 2-Clip-Phen
Artemisinin (10.43 mg, 37 lmol, 3 equiv) was the added,
followed by Na2S2O4 (10.67 mg, 61 lmol, 5 equiv). Thecolor of the solution changed from green to red. The
reaction mixture was stirred at 38 "C for 45 min and
then transferred in an NMR tube under argon. 1H-NMRanalysis shows that the 2-Clip-PhenCuI complex was
modified after reaction with artemisinin. Deoxyartemisi-
nin 4 and the tetrahydrofurane derivative 5 were alsoidentified by their H12 protons.
1H-NMR (300 MHz, DMSO-d6, selected data): d (ppm)
9.91 (s, 0.5H, exchangeable H), 9.60 (bs, 0.5H,exchangeable H) 9.05 (2H, H900), 8.47 (d, 2H, J = 8.0 Hz,
H700), 8.41 (d, 2H, J = 8.5 Hz, H400), 7.91 (2H, H600), 7.82
(2H, H500), 7.72 (dd, 2H, J = 4.3 and 8.4 Hz, H800), 7.25(d, 2H, J = 8.4 Hz, H300), 6.50 (s, H12 of 5), 5.87 (s, H12
of 4), 4.66 (m, 4H, H2C10), 4.12 and 3.83 (H2C4 of 5).
Ratio 2-Clip-Phen/4/5 = 55/38/7. ESI?-MS m/z 791.3(11-Cu, M?).
Reaction of artemisinin with copper- and ironcomplexes of dipyrrin
Dipyrrin was prepared as already published [48] and
analyses were compliant.
Preparation of Cu(dipyrrin)2
The Cu(dipyrrin)2 complex 13-Cu was prepared by modi-fication of reference [49] as follows: To a solution of
dipyrrin (91 mg, 41 lmol, 2.2 equiv) and Cu(OAc)2!H2O
(41 mg, 21 lmol, 1.1 equiv) in methanol (5 mL), wasadded 0.5 mL of NH4OH. The complex precipitated
instantaneously as a dark solid. The crude product was
purified on a short chromatography column (SiO2, dichlo-romethane/hexane, 1/1, v/v). The first bright orange band
was collected and evaporated to dryness to give 91 mg of
complex as a dark green crystalline solid (yield: 88%).Red-green dichroic single crystals of Cu(dipyrrin)2 were
grown by slow evaporation of a solution of the complex in
dichloromethane/methanol, and analyzed by X-ray dif-fraction. Anal. calcd for C30H22CuN4: C, 71.77; H, 4.42; N,
11.16. Found: C, 71.70; H, 4.33; N, 11.07. The solid state
structure of this complex shows the metal center to possessa highly distorted square planar geometry which is typical
of bis(dipyrrin) metal complexes. This distortion has been
attributed to a steric clash between the a-pyrrole protons onopposing ligands [57].
Reaction of artemisinin with Cu(dipyrrin)2
A solution of 13-Cu (9.0 mg, 18 lmol, 1 equiv), artemisinin
(10.1 mg, 35 lmol, 2 equiv) and 2,3-dimethylhydroquinone(43.0, 311 lmol, 17 equiv) in degassed dichloromethane
(4 mL) was left to stir under an argon atmosphere at 38 "Cfor 24 h. The reaction mixture was diluted in CH2Cl2 andanalyzed by mass spectrometry. ESI?-MS m/z 503.3 (14,MH?), 221.1 (dipyrrin ligand, MH?). Intensity: 503.3/
(503.3 ? 221.1) = 0.18.
Preparation of Fe(dipyrrin)3
The Fe(dipyrrin)3 complex 13-Fe was prepared according
to reference 49. Anal. calcd for C45H33FeN6: C, 75.74; H,
4.66; N, 11.78. Found: C, 75.67; H, 4.86; N, 11.80.
Reaction of artemisinin with Fe(dipyrrin)3
A solution of 13-Fe (24.3 mg, 34 lmol, 1 equiv),
artemisinin (30.0 mg, 106 lmol, 3 equiv) and 2,3-dim-
ethylhydroquinone (16.6, 106 lmol, 3 equiv) in degasseddichloromethane (1 mL) was left to stir under an argon
atmosphere at 38 "C for 4.5 h. The reaction mixture was
diluted in CH2Cl2 and analyzed by mass spectrometry.ESI?-MS m/z 503.3 (14, MH?), 221.1 (dipyrrin ligand,
MH?). Intensity: 503.3/(503.3 ? 221.1) = 0.47.
Acknowledgments This work was supported by Palumed Co. andANR (ANR-06-RIB-020-02). F.B.-E.G. is indebted to the Antimal
J Biol Inorg Chem (2009) 14:601–610 609
123
83

program (EU-PF6) for Doctoral fellowship. The authors also thank F.Cosledan (Palumed Co.) for fruitful discussion, and Y. Coppel and C.Claparols (LCC-CNRS) for NMR and MS analyses, respectively.
References
1. Schlitzer M (2007) Chem Med Chem 2:944–9862. Greenwood BM, Fidock DA, Kyle DE, Kappe SHI, Alonso PL,
Collins FH, Duffy PE (2008) J Clin Invest 118:1266–12763. Klayman DL (1985) Science 228:1049–10554. Kochi JK (1967) Science 155:415–4245. Zhang F, Gosser DK Jr, Meshnick SR (1992) Biochem Phar-
macol 43:1805–18096. Asawamahasakda W, Ittarat I, Pu YM, Ziffer H, Meshnick SR
(1994) Antimicrob Agents Chemother 38:1854–18587. Hong YL, Yang YZ, Meshnick SR (1994) Mol Biochem Parasitol
63:121–1288. Posner GH, Ho CH, Wang D, Gerena L, Milhous WK, Meshnick
SR, Asawamahasakda W (1994) J Med Chem 37:1256–12589. Jefford CW, Favarger F, Maria da Graca VH, Jacquier Y (1995)
Helv Chim Acta 78:452–45810. Robert A, Dechy-Cabaret O, Cazelles J, Meunier B (2002) Acc
Chem Res 35:167–174 (and references therein)11. Robert A, Meunier B (1997) J Am Chem Soc 119:5968–596912. Robert A, Coppel Y, Meunier B (2002) Chem Commun (5):414–
41613. Robert A, Benoit-Vical F, Claparols C, Meunier B (2005) PNAS
102:13676–13680 (erratum: PNAS 2006, 103:3943)14. Dechy-Cabaret O, Benoit-Vical F, Loup C, Robert A, Gornitzka
H, Bonhoure A, Vial H, Magnaval JF, Seguela JP, Meunier B(2004) Chem Eur J 10:1625–1636
15. Cosledan F, Fraisse L, Pellet A, Guillou F, Mordmuller B,Kremsner PG, Moreno A, Mazier D, Maffrand J-P, Meunier B(2008) PNAS 105:17579–17584
16. Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, ChiuFCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H,McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C,Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN(2004) Nature 430:900–904
17. Tang Y, Dong Y, Vennerstrom JL (2004) Med Res Rev 24:425–448
18. O’Neill PM, Posner GH (2004) J Med Chem 47:2945–296419. Jefford CW (2007) Drug Discov Today 12:487–49520. Benoit-Vical F, Lelievre J, Berry A, Deymier C, Dechy-Cabaret
O, Cazelles J, Loup C, Robert A, Magnaval JF, Meunier B (2007)Antimicrob Agents Chemother 51:1463–1472
21. Bousejra-El Garah F, Claparols C, Benoit-Vical F, Meunier B,Robert A (2008) Antimicrob Agents Chemother 52:2966–2969
22. Creek DJ, Charman WN, Chiu FCK, Prankerd RJ, Dong Y,Vennerstrom JL, Charman SA (2008) Antimicrob AgentsChemother 52:1291–1296
23. Bhisutthibhan J, Pan XQ, Hossler PA, Walker DJ, Yowell CA,Carlton J, Dame JB, Meshnick SR (1998) J Biol Chem273:16192–16198
24. Eckstein-Ludwig U, Webb RJ, van Goethem IDA, East JM, LeeAG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S(2003) Nature 424:957–961
25. Kochi JK, Mains HE (1965) J Org Chem 30:1862–1872
26. Fairfield AS, Meshnick SR, Eaton JW (1983) Science 221:764–766
27. Olliaro PL, Goldberg DE (1995) Parasitol Today 11:294–29728. Rasoloson D, Shi L, Chong CR, Kafsack BF, Sullivan DJ Jr
(2004) Biochem J 381:803–81129. Wu Y, Liu HH (2003) Helv Chim Acta 86:3074–308030. Wu WM, Wu YL (2000) J Chem Soc Perkin Trans 1, pp 4279–
428331. Jefford CW, Vicente MGH, Jacquier Y, Favarger F, Mareda J,
Millasson-Schmidt P, Brunner G, Burger U (1996) Helv ChimActa 79:1475–1487
32. Jung M, ElSohly HN, Croom EM, McPhail AT, McPhail DR(1986) J Org Chem 51:5417–5419
33. Lin AJ, Klayman DL, Hoch JM (1985) J Org Chem 50:4504–4508
34. Cumming JN, Ploypradith P, Posner GH (1997) Adv Pharmacol37:253–297
35. Robert A, Meunier B (1998) Chem Soc Rev 27:273–27936. Tang Y, Dong Y, Wang X, Sriraghavan K, Wood JK, Venner-
strom JL (2005) J Org Chem 70:5103–511037. Creek DJ, Chiu FCK, Prankerd RJ, Charman SA, Charman WN
(2005) J Pharm Sci 94:1820–182938. Wang DY, Wu YL (2000) Chem Commun 2193–219439. Wu Y, Yue ZY, Wu YL (1999) Angew Chem Int Ed 38:2580–
258240. Pitie M, Donnadieu B, Meunier B (1998) Inorg Chem 37:3486–
348941. Pitie M, Boldron C, Gornitzka H, Hemmert C, Donnadieu C,
Meunier B (2003) Eur J Inorg Chem 528–54042. Pitie M, Boldron C, Pratviel G (2006) In: van Eldik R, Reedijk
J (eds) Advances in inorganic chemistry, vol 58. Elsevier,Academic Press, Amsterdam, pp 77–130
43. Urry WH, Juveland OO (1958) J Am Chem Soc 80:3322–332844. Felton RH, Linschitz H (1966) J Am Chem Soc 88:1113–111645. Giraudeau A, Louati A, Gross M, Callot HJ, Hanson LK, Rhodes
RK, Kadish KM (1982) Inorg Chem 21:1581–158646. Kumar Manmohan, Neta P, Sutter TPG, Hambright P (1992)
J Phys Chem 96:9571–957547. Sanders JKM, Bampos N, Clyde-Watson Z, Darling SL, Hawley
JC, Kim H-J, Mak CC, Webb SJ (2000) In: Kadish KM, SmithKM, Guilard R (eds) The porphyrin handbook, vol 3, chap 15.Academic Press, Dublin, pp 1–48
48. Reese CB, Yan H (2001) Tetrahedron Lett 42:5545–554749. Bruckner C, Karunaratne V, Retting SJ, Dolphin D (1996) Can
J Chem 74:2182–219350. Bruckner C, Zhang Y, Rettig SJ, Dolphin D (1997) Inorg Chim
Acta 263:279–28651. Robert A, Meunier B (1998) Chem Eur J 4:1287–129652. Altomare A, Cascarano G, Giacovazzo C, Guagliardi A (1993)
J Appl Crystallogr 26:343–35053. SHELX97 [Includes SHELXS97, SHELXL97, CIFTAB]—Pro-
grams for crystal structure analysis (Release 97–2). SheldrickGM, Institut fur Anorganische Chemie der Universitat, Tam-manstrasse 4, D-3400 Gottingen, Germany, 1998
54. Farrugia L (1999) J Appl Crystallogr 32:837–83855. International Tables for X-Ray Crystallography (1974), vol IV.
Kynoch Press, Birmingham56. Farrugia LJ (1997) J Appl Crystallogr 30:56557. Halper SR, Malachowski MR, Delaney HM, Cohen SM (2004)
Inorg Chem 43:1242–1249
610 J Biol Inorg Chem (2009) 14:601–610
123
84



Chapter 4
Reactivity of antimalarial dispiro1,2,4,5tetraoxanes with Fe(II),
heme and phospholipids: Implications for their mode of action


Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: implications in mode of action
Résumé en français de la publication en cours de rédaction :
Reactivity of antimalarial dispiro1,2,4,5tetraoxanes with Fe(II), heme and phospholipids: Implications for their mode of action
F. Bousejra-El Garah, R.K. Amewu, S. Muangnoicharoen, S.A. Ward, and P.M. O’Neill
Réactivité de tétraoxanes antipaludiques avec un sel de fer(II), l’hème ou les phospholipides : Relation avec leur mode d’action
Dans la recherche de nouvelles molécules synthétiques pour le traitement du paludisme, les dispiro-
1,2,4,5-tétraoxanes (ensuite dénommés tétraoxanes) se sont révélés très prometteurs. Le groupe du
Pr. O’Neill a mis au point une série de tétraoxanes stables, achiraux, peu coûteux à synthétiser et
très actifs.[1,2]
Nous nous sommes intéressés à la réactivité de ces peroxydes avec le fer (II) en présence du radical
2,2,6,6-tetramethyl-1-piperidine 1-oxyl (TEMPO) d’une part, et de phospholipides d’autre part.
Nous avons comparé cette réactivité à celle de trioxolanes analogues, qui sont également des
antipaludiques actifs. Nous nous sommes ensuite intéressés à la réaction des tétraoxanes avec
l’hème, principale source de fer dans les érythrocytes infectés.
Ce travail a été effectué au sein de l’équipe du Pr O’Neill, à l’Université de Liverpool, lors d’un
stage doctoral de 12 mois (de octobre 2008 à septembre 2009).
Reactivité des tétraoxanes avec le sulfate de fer en présence de TEMPO et de la phosphatidylcholine
Réactivité avec un sel de Fe2+ : Piégeage des radicaux alkyles par TEMPO
Afin de mieux comprendre le mécanisme de la réaction des tétraoxanes avec le fer (II) et de
confirmer la formation de radicaux, nous avons étudié cette réaction en présence du piège de
radicaux alkyles TEMPO. Comme attendu et comme dans le cas de l’artémisinine, la coordination
du fer (II) sur l’oxygène O2 induit la rupture homolytique de la liaison peroxyde, et conduit à la
formation du radical alkoxy 10, qui se réarrange par β-scission pour donner le radical alkyle
secondaire 12. Ce radical a été piégé par TEMPO pour former l’adduit 8 (figure ci-dessous). De la
même façon, la coordination du fer sur O1 génère le radical alkoxy 11, qui se réarrange en radical
alkyle primaire 13, également piégé par TEMPO pour former l’adduit 9. Les adduits 8 et 9 ont été
89

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: implications in mode of action
caractérisés par spectrométrie de masse. Une réaction analogue avec des trioxolanes également
actifs conduits à des résultats similaires.[3]
La formation de radicaux alkyles indique que cette réactivité est un caractère général des peroxydes
antipaludiques testés jusqu’à présent.
Activation réductrice du tétraoxane 2 et formation des adduits TEMPO-drogue 8 et 9
Réactivité avec un sel de Fe2+ en présence de phosphatidylcholine (PC)
Nous nous sommes intéressés à la réaction des tétraoxanes dans les conditions de Fenton en
présence de phosphatidylcholine, utilisée comme modèle de phospholipides. Nous avons également
comparé cette réactivité avec celle de trioxolanes analogues. Les résultats nous ont montré que ces
deux familles de peroxydes étaient capables d’induire l’oxydation de PC et plusieurs produits
d’oxydation ont été identifiés par spectrométrie de masse. Ce résultat confirme celui obtenu
récemment avec des tétraoxanes hydrosolubles.[4] Cette réactivité des tétraoxanes et trioxolanes
diffère de celle de l’artémisinine, pour laquelle aucun produit de dégradation de PC n’a été détecté.
Réactivité des tétraoxanes avec l’hèmefer(II) : mise en évidence de la formation d’adduits hèmetétraoxanes
La réactivité des trioxanes ou trioxolanes antipaludiques vis-à-vis de l’hème a été bien étudiée et,
dans plusieurs cas, des adduits hème-drogue ont été caractérisés.[5-8] Nous nous sommes donc
intéressés à l’étude de la réactivité des tétraoxanes avec l’hème.
90

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: implications in mode of action
La réaction de différents tétraoxanes antipaludiques, tous très actifs in vitro, avec l’hème-fer(II),
généré in situ par réduction de l’hème-fer(III), a été réalisée et suivie par analyse LC-MS. Comme il
vient d’être décrit pour un sel de fer (II), nous montrons que l’hème-fer(II) induit la coupure
homolytique de la liaison peroxyde des tétraoxanes pour former une espèce radicalaire qui alkyle le
ligand porphyrinique en positions meso. Ainsi, l’adduit (15, m/z 783.2) issu du couplage covalent
entre la porphyrine et le radical alkyle secondaire dérivé des tétraoxanes (figure ci-dessous) a été
caractérisé par LC-MS.
Mécanisme de formation des adduits hème-tétraoxanes. Seul le régio-isomère issu de l’alkylation en position δ est représenté. Les adduits en position α, β et γ sont également présents.
Des produits de couplages avec les porphyrines FeTPP et MnTPP ont également été identifiés, ce
qui confirme les propriétés alkylantes des tétraoxanes antipaludiques.
Une étude comparative de deux tétraoxanes et de leurs analogues trioxolanes a montré que ces deux
familles de molécules antipaludiques ont des réactivités similaires vis-à-vis de l’hème.
Conclusion
La formation de radicaux alkyles après activation des tétraoxanes par le fer (II) a été mise en
évidence par la caractérisation d’adduits avec TEMPO, avec l’hème et avec des métalloporphyrines
synthétiques.
Les tétraoxanes présentent une réactivité avec le fer qui est semblable à celle des trioxolanes ; ces
deux familles de molécules peuvent induire l’oxydation de phospholipides modèles.
91

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: implications in mode of action
92
Les tétraoxanes antipaludiques sont capables de se lier à l’hème de façon covalente. Cette
caractéristique est commune à tous les peroxydes actifs étudiés.
Bibliographie
[1] R. Amewu, A. V. Stachulski, S. A. Ward, N. G. Berry, P. G. Bray, J. Davies, G. Labat, L. Vivas, P. M. O'Neill. Design and Synthesis of Orally Active Dispiro 1,2,4,5-Tetraoxanes; Synthetic Antimalarials with Superior Activity to Artemisinin. Org. Biomol. Chem. 2006, 4, 4431-4436. [2] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. Shone, D. Stanford, P. Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer, S. Lozanom, M. Jesús, D. Gargallo, S. A. Ward, P. M. O'Neill. Two-Step Synthesis of Achiral Dispiro-1,2,4,5-Tetraoxanes with Outstanding Antimalarial Activity, Low Toxicity, and High-Stability Profiles. J. Med. Chem. 2008, 51, 2170-2177. [3] Y. Tang, Y. Dong, X. Wang, K. Sriraghavan, J. K. Wood, J. L. Vennerstrom. Dispiro-1,2,4-Trioxane Analogues of a Prototype Dispiro-1,2,4-Trioxolane: Mechanistic Comparators for Artemisinin in the Context of Reaction Pathways with Iron(II). J. Org. Chem. 2005, 70, 5103-5110. [4] N. Kumura, H. Furukawa, A. N. Onyango, M. Izumi, S. Nakajima, H. Ito, T. Hatano, H. S. Kim, Y. Wataya, N. Baba. Different Behavior of Artemisinin and Tetraoxane in the Oxidative Degradation of Phospholipid. Chem. Phys. Lipids 2009, 160, 114-120. [5] S. A. L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meunier. Heme Alkylation by Artesunic Acid and Trioxaquine Du1301, Two Antimalarial Trioxanes. ChemBioChem 2005, 6, 653-658. [6] A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier. The Antimalarial Drug Artemisinin Alkylates Heme in Infected Mice. Proc. Nat. Acad. Sci. U.S.A 2005, 102, 13676-13680. Erratum, 2006, 103, 3943. [7] A. Robert, Y. Coppel, B. Meunier. Alkylation of Heme by the Antimalarial Drug Artemisinin. Chem. Commun. 2002, 414-415. [8] D. J. Creek, W. N. Charman, F. C. K. Chiu, R. J. Prankerd, Y. Dong, J. L. Vennerstrom, S. A. Charman. Relationship between Antimalarial Activity and Heme Alkylation for Spiro- and Dispiro-1,2,4-Trioxolane Antimalarials. Antimicrob. Agents Chemother. 2008, 52, 1291-1296.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
Reactivity of antimalarial dispiro1,2,4,5tetraoxanes with Fe(II), heme and phospholipids: Implications for their mode of action
Fatima Bousejra-El Garah,*[a] Richard K. Amewu,[a] Sant Muangnoicharoen,[b] Stephen A. Ward,[b] and Paul M. O’Neill*[a]
[a] F. Bousejra-El Garah, Dr. R.K. Amewu, Prof. P.M. O’Neill, University of Liverpool, Department of Chemistry, Liverpool L697ZD (UK) Fax: (+)44 151-794-3553, E-mail1: [email protected]; Email2: [email protected]
[b] Dr. S. Muangnoicharoen, Prof. S.A. Ward, Liverpool School of Tropical Medicine, Pembroke Place, Liverpool L35QA (UK) Fax: (+44) 151-794-3588, E-mail: [email protected]
Abstract
Dispiro-1,2,4,5-tetraoxanes are an attractive class of synthetic antimalarial peroxides due to their
structural simplicity, good stability, and impressive activity. We investigated the reactivity of a
series of potent tetraoxanes with FeSO4/TEMPO, FeSO4/phosphatidylcholine and heme to gain
knowledge on their possible mechanism of action. Spin trapping experiments showed that Fe(II)-
mediated peroxide reduction gives a pair of primary and secondary alkyl radicals. Reaction with
unsaturated phosphatidylcholine under Fenton reaction conditions showed that tetraoxanes and
trioxolanes share a common reactivity in phospholipid oxidation that differs with that of
artemisinin. The implication in terms of mechanism of action is discussed. The tetraoxane-derived
secondary radical was also showed to quickly react with porphyrins and coupling products were
identified by LC-MS analysis as regio-isomers of the alkylated heme adduct. This alkylating ability
was confirmed with FeTPP and MnTPP. Results for tetraoxanes are consistent with previous reports
on trioxanes or trioxolanes Fe(II)/heme-mediated reactivity and the heme alkylation is a process
that may be involved in the mechanism of action of this promising class of antimalarials.
Introduction
According to the World Health Organization (WHO), more than one million people die of malaria
every year, most of them in African countries where children and pregnant women are especially at
risk.[1] The spread of chloroquine and sulfadoxine-pyrimethamine resistances have been implicated
in the re-emergence of malaria in areas where the disease had been eradicated. In order to control
93

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
the burden of malaria, artemisinin-based combination therapies are now recommended as first-line
treatment in many endemic regions.[2]
Artemisinin (figure 1), a sesquiterpene lactone extracted from the Chinese herb Artemisia Annua, is
highly active against both chloroquine sensitive- and resistant-strains of Plasmodium falciparum,
the most dangerous form of the parasitic agent responsible for malaria.[3]
It is known that the key pharmacophore of this highly efficient antimalarial is its 1,2,4-trioxane unit,
and in particular its peroxide bond.[3] It is also well established that iron(II) salts reduces the
peroxide bond of artemisinin leading to the formation of a pair of oxy-radicals that rapidly
rearrange via either hydrogen abstraction or β-scission to form more stable carbon-centered
radicals.[4-6] These alkyl radicals may be formed in vivo by reaction of artemisinin with iron(II)-
heme, the most abundant source of iron in Plasmodium.[7,8] They are able to interact with vital
cellular components such as specific parasitic proteins,[9,10] heme, and other targets[11] that may
result in the parasite death (figure 1).
Figure 1: Fe-mediated activation of artemisinin
Heme, in particular, seems to play a role in the mechanism of action of the drug since heme-
artemisnin adducts were chemically prepared and isolated,[12-14] but were also detected in malaria-
infected mice, treated with artemisinin.[15] Although artemisinin and clinically used derivatives
artemeter and artesunate are very potent antimalarials, they also exhibit high extraction costs and
poor pharmacokinetic properties. These drawbacks have driven the development of synthetic
antimalarials that incorporate the key peroxide pharmacophore.[16,17] Among them are found
dispiro-1,2,4,5-tetraoxanes, a promising class of antimalarials.
Tetraoxanes are cyclic peroxides originally used for the production of macrocyclic hydrocarbons
and lactones.[18] Work by the Vennerstrom group in the early 90’s showed that dispiro-tetraoxanes
possess high in vitro antimalarial activity.[19] More recently, we have designed and prepared simple,
94

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
achiral and highly potent dispiro-tetraoxanes that incorporate the adamantyl group, known to bring
stability to the endoperoxide motif.[20,21]
The first aim of this study was to investigate the radical pathway after reductive activation of highly
potent tetraoxanes with inorganic iron(II). In fact, formation of C-centered radicals with tetraoxanes
has been reported,[22,23] but Opsenica et al. also reported that steroidal tetraoxanes generate RO• that
do not undergo any further rearrangements.[24] These two contradicting observations call for further
investigation into intermediates that might be responsible for the antimalarial activity of these
endoperoxides. We were also interested to know whether the oxidative degradation of
phospholipids, recently reported for water soluble tetraoxanes,[25] was observed with adamantane
functionalized tetraoxanes 1-5 (figure 2) and trioxolane analogues. Finally, we investigated the
heme-mediated reactivity of tetraoxanes to determine whether the reaction with heme may be
related to their impressive antimalarial activity.
Figure 2: Structures of antimalarial tetraoxanes 1-5 with their in vitro activities vs. 3D7
Results and Discussion
Antimalarial activity
The in vitro antimalarial activity of tetraoxanes 2 and 3 was measured versus the chloroquine-
sensitive 3D7 strain of Plasmodium falciparum. Both tetraoxanes are very potent in vitro with IC50
of 1.35 nM and 0.44 nM, respectively. 6, the trioxolane equivalent of 2, was also tested against the
same strain and was found to be slightly less active than 2, with an IC50 of 13.24 nM.
Reactivity with Fe(II) and TEMPO spin trapping
Since tetraoxanes contain a peroxide bond like artemisinin, we were interested in the reaction
pathway of these peroxides with iron (II) and compare this reactivity with trioxolane analogues.
95
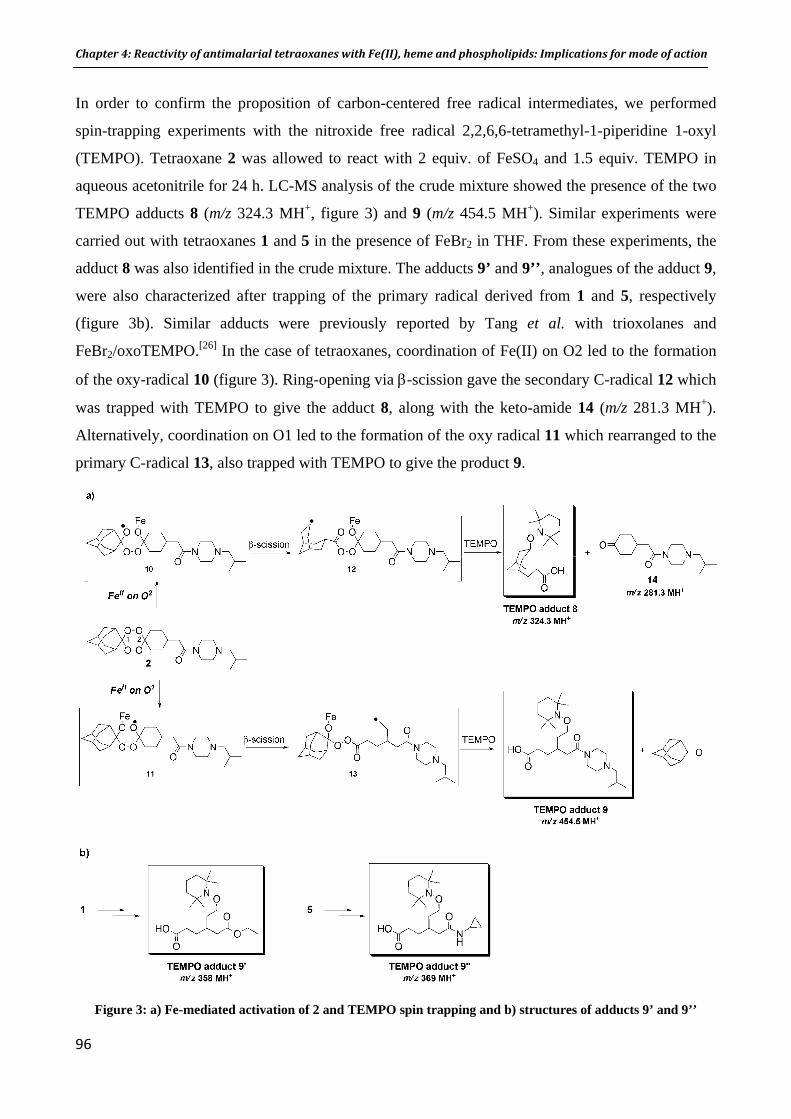
Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
In order to confirm the proposition of carbon-centered free radical intermediates, we performed
spin-trapping experiments with the nitroxide free radical 2,2,6,6-tetramethyl-1-piperidine 1-oxyl
(TEMPO). Tetraoxane 2 was allowed to react with 2 equiv. of FeSO4 and 1.5 equiv. TEMPO in
aqueous acetonitrile for 24 h. LC-MS analysis of the crude mixture showed the presence of the two
TEMPO adducts 8 (m/z 324.3 MH+, figure 3) and 9 (m/z 454.5 MH+). Similar experiments were
carried out with tetraoxanes 1 and 5 in the presence of FeBr2 in THF. From these experiments, the
adduct 8 was also identified in the crude mixture. The adducts 9’ and 9’’, analogues of the adduct 9,
were also characterized after trapping of the primary radical derived from 1 and 5, respectively
(figure 3b). Similar adducts were previously reported by Tang et al. with trioxolanes and
FeBr2/oxoTEMPO.[26] In the case of tetraoxanes, coordination of Fe(II) on O2 led to the formation
of the oxy-radical 10 (figure 3). Ring-opening via β-scission gave the secondary C-radical 12 which
was trapped with TEMPO to give the adduct 8, along with the keto-amide 14 (m/z 281.3 MH+).
Alternatively, coordination on O1 led to the formation of the oxy radical 11 which rearranged to the
primary C-radical 13, also trapped with TEMPO to give the product 9.
Figure 3: a) Fe-mediated activation of 2 and TEMPO spin trapping and b) structures of adducts 9’ and 9’’
96

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
The identification of the two TEMPO adducts 8 and 9 provided evidence of the formation of two
carbon-centered radicals after iron(II) activation of tetraoxanes. This result is not in accordance with
a previous report where the RO• generated by steroidal tetraoxanes did not undergo any further
rearrangements.[24] It should be noted that only the secondary radical has been trapped with
trioxolanes like OZ277. The formation of alkyl radicals from tetraoxanes is a common feature with
artemisinin, although their reactivity is quite different in the sense that the high energy primary
carbon centered radical of artemisinin has been trapped whereas the secondary C-radical has not
(vide infra).
Fe(II)-mediated reactivity: tetraoxane vs. trioxolanes
We prepared the trioxolane analogues of 2 and 3 to compare the stability and reactivity of the
1,2,4,5-tetraoxane pharmacophore to the ozonide core in the presence of iron (II) using standard
conditions (figure 4).
Figure 4: Tetraoxanes 2 and 3, and their trioxolanes analogues 6 and 7, respectively with their IC50 vs. 3D7
Tetraoxanes and trioxolanes were subjected to 20 equiv. of ferrous sulfate in aqueous acetonitrile
under nitrogen, for a 7 h period of time. Iron-mediated decomposition was observed for all the
tested compounds. The time-dependent degradation studies, conducted by monitoring the loss of
parent endoperoxide by LC-MS, showed a linear decrease of tetraoxanes concentration (figure 5).
Similar reaction profiles were observed for tetraoxanes and trioxolanes, indicating that the stability
of the two classes of drugs with inorganic Fe(II) is comparable.
97

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
Figure 5: Time-dependence profile for the decomposition of peroxides by 20 equiv. of FeSO4 in aqueous acetonitrile
Reactivity of tetraoxanes with Fe(II) and phosphatidylcholine (PC)
The radicals produced after tetraoxanes activation may react in vivo with cell components such as
lipids of the membrane bilayer, a well known target for ROS, leading to cell damage and death.
Kumara and coll. have recently reported that water soluble tetraoxanes can induce olefins oxidative
degradation in a phospholipid model in the presence of a Fe(II) salt.[25] Several phosphatidylcholine
degradation products were identified, while they were absent when artemisinin was used.
We studied the reactivity of tetraoxanes 2 and 3 with phosphatidylcholine, in the Fenton reaction
conditions, and we compared this reactivity with that of trioxolane analogues, 6 and 7, respectively.
2-linoleoyl-1-palmitoyl-sn-phosphatidylcholine (PC) is composed of a phosphocholine polar head
linked to the glycerol moiety with palmityl acid, a saturated fatty acid chain at the sn-1 position and
linoleic acid, an unsaturated fatty acid chain at the sn-2 position (figure 6).
Figure 6: Structure of 2-linoleoyl-1-palmitoyl-sn-phosphatidylcholine (PC)
Reactions of PC with 8 equiv. of tetraoxanes and FeSO4 were carried out in H2O/ethanol (2:1) at
38°C, for 24 h under N2 atmosphere. The crude mixture and the organic extract were analyzed by
ESI-MS and LC-MS. Reaction with 2 and 3 gave several peaks other than the native PC, observed
at M+ (m/z 758) and [MNa]+ (m/z 780). The predominant degradation products were found at m/z
622.4, 666.4, 788.6 (810.5), and 808.5. All these ions, expect m/z 808.5, were detected by
Kumura.[25] The structures proposed by the authors for the PC oxidation products are depicted in
98

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
figure 7. The bis-allylic hydrogen at C11 of the linoleic acid is more likely to be abstracted by the
tetraoxane-derived radicals.[27] The degradation products are the result of subsequent β-cleavage
and lipid peroxidation.
Figure 7: Proposed mechanism for tetraoxane-mediated lipid peroxidation with structures suggested in ref 25
The reaction with PC was carried out with trioxolanes 6 and 7 in the same conditions. Product
analysis indicated that the reaction pathway observed for trioxolanes was qualitatively the same
than for tetraoxanes. The same PC degradation products were observed, in a slightly higher extent,
indicating that in these conditions, the two classes of antimalarials exhibit the same reactivity
towards phospholipids.
After reaction with artemisinin in the same conditions, the native PC was found intact, confirming
that this natural peroxide is not responsible for phospholipid oxidation in these conditions. In fact,
artemisinin was showed to enhance the heme-mediated lipid membrane damage[28] but does not
affect PC in our heme-free conditions. This difference of reactivity between artemisinin and
tetraoxanes/trioxolanes here is likely due to the difference of stability of the generated radicals. The
99

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
artemisinin-derived radicals are very reactive and the intramolecular rearrangement of these radicals
is too rapid. The adamantane-derived secondary radical derived from trioxolanes/tetraoxanes, stable
enough to be trapped with TEMPO, would abstract the bis-allylic hydrogen and initiate the lipid
peroxidation process of PC.
Reactivity of tetraoxanes with heme
We studied the reactivity of tetraoxanes with heme, a more biologically relevant source of iron.
Several studies have showed that iron(II)-heme quickly reacts with the peroxide bond of various
antimalarial trioxanes and trioxolanes. The pathway is the same as described with ferrous ions and
the generated C-centered radicals were shown to bind to the porphyrin macrocycle and form heme-
drug adducts. Such adducts were isolated and/or characterized with artemisinin[12,13,15] and
derivatives,[29,30] hybrid drugs trioxaquines[31,32] and more recently with trioxolanes derivatives.[33]
We investigated the heme-mediated reactivity of tetraoxanes 2 and 3 to provide further insight into
the mechanism of action for this promising class of antimalarials. We next compared this reactivity
with that of their trioxolane analogues.
Tetraoxanes were allowed to react with iron(III)-hemin, in presence of an excess of dithionite to
generate iron(II)-heme in situ, in ACN/NaOH 0.1 M (50/50). In these conditions, iron(II)-heme
quickly reacts with the tetraoxanes. In the case of 2, HPLC analysis of the crude mixture showed
that most of the starting heme has reacted within 30 min, giving at least 3 products with higher
retention times than heme itself (figure 8a), and a maximum absorption of the Soret band at 430 nm,
instead of 398 nm for heme. A blue-shift of the Soret band has been also observed with artemisinin,
although to a lower extent.[13]
100

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
101
Figure 8: a) UV-Vis trace and b) extracted ion chromatogram at m/z 783.2 of reaction of heme with tetraoxane 2
LC-MS analysis showed that these three compounds exhibit a m/z 783.2 (M+), which is consistent
with a coupling product between heme (mass 616) and the tetraoxane-derived fragment previously
trapped with TEMPO (mass 167).
Both the maximum absorbance and the m/z ratio strongly suggest that these compounds result from
the covalent bonding of the tetraoxane-derived secondary C-centered radical 12 and the heme
porphyrin (15, figure 9). The same alkylated heme adducts have been reported with trioxolanes in
similar conditions by Creek et al.[33] The extracted ion chromatogram at m/z 783.2 showed four
peaks (figure 8b), expected to be the four possible regio-isomers of the alkylated heme adduct 15, as
reported by Robert et al. for heme-artemisinin adducts.[34]

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
102
Figure 9: Proposed mechanism for the alkylation of heme (Fe(II)-PPIX) by tetraoxane 2, adapted from [33]
Similar results were obtained with all the active antimalarial tetraoxanes tested, including 3 and 4
(results not shown). The ketoamide, a co-product of the reaction, was also detected in all cases.
Using a LC-MS calibration, we monitored the conversion of heme during the reaction (figure 10).
As a control, the reaction was carried out without drug, to estimate the loss of heme due to
degradation by residual oxygen. Reaction with artemisinin showed that c.a. 70% of the starting
heme was converted within 15 min. Reaction with tetraoxanes 2 and 3 was also rapid, and resulted
in conversion of the starting heme of 53% and 38%, respectively after 5 min only. Concentration-
time profiles of heme showed that the reaction is complete within 15 min, as the absorbance of the
Soret band remains constant beyond.
Figure 10: Time-dependence profile for the conversion of heme with peroxides

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
103
Reaction of trioxolanes with heme in the same conditions showed a very similar profile and the
same coupling products were identified (figure 11). However, the adduct formation appeared to be
more rapid with trioxolane 6 and the peak ratio is different.
Figure 11: LC trace of reaction mixture of tetraoxane 2 (top) and and trioxolane 6 (bottom) with heme
Due to their low stability, quantification of the adducts was not possible. However, estimated yield
of adducts after 15 min for 2 is 34%, which is about half of the yield observed with 6 in the same
conditions (71%).[35] No significant differences were observed between 3 and 7 (data not shown).
As reported with artemisinin, we also studied the alkylating activity of tetraoxanes with iron (II)
tetraphenylporphyrin and manganese (II) tetraphenylporphyrin, two symmetrical synthetic
porphyrin, to confirm results observed with heme.[36] Tetraoxane 2 was allowed to react with
MnIITPP in CH2Cl2 for 3h, in presence of tetrabutylammonium borohydride as a reducing agent.
Analysis by LC-MS of the crude mixture showed the presence of a porphyrin-drug adduct with m/z
835.10 (M+), consistent with a chlorin-type adduct (16), which result from alkylation on one of the
eight β-pyrrolic positions of the macrocycle[36] by the tetraoxane-derived secondary radical. The
same chlorin-type adduct was identified after reaction with FeIITPP. After work-up and Mn
removal,[36] the demetalated adduct (17, figure 12) was also identified (m/z 783.19, MH+).
These results with the tetraphenylporphyrins confirm the alkylating capacity of antimalarial
tetraoxanes observed with heme.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
104
Figure 12: Expected structures of MnTCP- and H2TCP-tetraoxane adducts 16 and 17
One can note that, although the primary and the secondary radical can possibly be generated after
reduction by Fe2+ salt, only the tetraoxane-derived secondary radical (12) was generated in the
presence of heme or tetraphenylporphyrins. In the case of artemisinin and trioxaquines, only
adducts resulting from the primary radical were reported.[12,13,32] This could be explained by the
relative positions of the peroxide bond and the porphyrin ligand. Contrary to “free” iron which can
coordinate both oxygen atoms, iron in the heme porphyrin would less easily coordinate O1, which
is close to the bulky adamantane group. However, we cannot rule out the possibility that the
primary radical is also formed, but does not react with the porphyrin. To confirm this hypothesis, a
modeling study of the interaction between heme and the peroxide bond of tetraoxanes would be
useful to predict which oxygen (O1 or O2) is the most favorable for association with iron in heme.
The reaction with heme occurs much faster than reaction with ferrous sulfate. Indeed, all
tetraoxanes were degraded in few minutes, while reaction with inorganic iron needs several hours.
This increased reaction rate, also observed with trioxanes[32] and trioxolanes, was discussed by
Creek et al.[33]
Conclusion
Iron(II)-mediated activation of tetraoxanes results in the formation of a primary and a secondary C-
centered radical. Stability of tetraoxanes with ferrous sulfate was shown to be comparable with that
of their trioxolane analogues. Both classes of drugs react with phosphatidylcholine to give
peroxidation products after reductive activation by Fe2+.
All the tetraoxanes used in this study were shown to rapidly react with Fe(II)-heme. The major
reaction products are alkylated heme adducts, resulting from addition of the adamantane-derived
secondary radical intermediate.
The high reactivity with heme and the formation of covalent heme-drug adducts is a feature shared
by all tested active trioxanes, trioxolanes and tetraoxanes derivatives.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
105
Experimental section
LC-MS analysis
Mass spectrometry analysis was performed on a Thermo TSQ Quantum Access triple quadrupole
mass spectrometer connected to LC device Thermo Accela high pressure pump, auto sampler and
PDA detector. Analytical separation were performed on a 150 x 3 mm Thermo Hypersyl
HyPURITY® C18 column (5 µm). Data were captured in full MS scan mode and proceeded using
Xcalibur software (version 2.0.7) and Thermo Quan browser (version 2.0.7).
Chemistry
All reagents were purchased from Sigma Aldrich UK and were used without purification. NMR
spectra were recorded on a Brucker AMX 400 (1H, 400 MHz; 13C, 100 MHz) spectrometer.
Chemical shifts are described in ppm (δ) downfield from an internal standard of trimethylsilane.
Tetraoxanes 1, 4 and 5 were prepared according the procedure reported by Amewu et al.
Synthesis of tetraoxanes 2 and 3
Preparation of adamantane-2-spiro-3'-9'-[1-(2-methylpropyl)piperazino]-1',2',4',5’-
tetraoxaspiro[5.5]undecane (2): To a solution of adamantane-2-spiro-3'-9'-(carboxymethyl)-
1',2',4',5’-tetraoxaspiro[5.5]undecane (0.50 g, 1.48 mmol) in dry dichloromethane (50 mL), were
added anhydrous triethylamine (0.4 mL, 3.0 mmol, 2 equiv.) and methyl chloroformate (0.11 mL,
1.48 mmol, 1 equiv.). The mixture was stirred under N2 atmosphere at 0°C for 1 h. 1-(2-
methylpropyl)piperazine (0.21 mL, 1.48 mmol, 1 equiv.) was added, and the mixture was allowed
to stir at 0°C for 30 min and at room temperature for further 90 min. The reaction mixture was
diluted with water (60 mL) and extracted with dichloromethane. (3 x 60 mL). The organic layers
were combined, washed with brine, dried over anhydrous Na2SO4 and evaporated under reduced
pressure. The crude product was purified by flash chromatography to give 145 mg of 2 as a white
solid (21%).
1H NMR (400 MHz, CDCl3) δ 3.62 (t, 2H, J = 4.7 Hz, NCH2), 3.45 (t, 2H, J = 4.7 Hz, NCH2), 2.35
(t, 4H, CH2N), 2.27-2.20 (m, 2H, NCH2), 2.08 (d, 2H, J = 7.4 Hz, CH2CO), 2.04-1.90 (m, 6H, CH),
1.86 bs, 2H, CH2), 1.84-1.50 (m, 14H, adamantylidene/CH2), 1.36-1.17 (m, 2H, adamantylidene),
0.89 (m, 6H, J = 6.5 Hz, CH3); 13C NMR (100 MHz, CDCl3), δ 170.7, 110.8, 108.2, 67.2, 54.2,
53.7, 46.3, 42.1, 39.4, 37.3, 34.4, 33.5, 27.4, 25.8, 21.2; HRMS calculated for C26H43O5N2:
463.3172, found 463.3187. Elemental analysis C: 67.62, H: 9.18, N: 6.09 (required values C: 67.50,
H: 9.15, N: 6.06).

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
106
Preparation of adamantane-2-spiro-3'-9'-[1-methyl-4-(4-piperidino)piperazino]-1',2',4',5’-
tetraoxaspiro[5.5]undecane (3): To a solution of adamantane-2-spiro-3'-9'-(carboxymethyl)-
1',2',4',5’-tetraoxaspiro[5.5]undecane (0.3 g, 0.89 mmol) in dry dichloromethane (20 mL), were
added anhydrous triethylamine (0.18 mL, 1.34 mmol, 2 equiv.) and ethyl chloroformate (0.08 mL,
0.89 mmol, 1equiv.). The mixture was stirred for 1 h at 0°C. 1-methyl-4-(4-piperidino)piperazine
(163 mg, 0.89 mmol, 1 equiv.) was added, and the mixture was allowed to stir at 0°C for 30 min
and at room temperature for further 90 min. The reaction mixture was diluted with water (30 mL)
and extracted with dichloromethane. The organic layers were combined, washed with brine, dried
over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by
flash chromatography to give 397 mg of 3 as a white solid (86%).
1H NMR (400 MHz, CDCl3) δ 4.65 (d, 2H, J = 13.4 Hz, CH2), 3.90 (d, 2H, J = 11.0 Hz, CH2), 3.00
(t, 2H, J = 11.1 Hz, CH2), 2.36-2.68 (m, 7H, CH/CH2), 2.28 (s, 3H, NCH3), 2.23 (bs, 2H, CH2),
1.20-2.07 (m, 27H, CH/CH2). 13C NMR (100 MHz, CDCl3), δ 170.5, 110.8, 108.2, 62.1, 55.7, 49.5,
46.4, 45.5, 41.5, 39.4, 37.3, 34.4, 33.5, 29.6, 28.6, 27.5. MS (ESI+), [M+H ] + (100) 504.4. HRMS
calculated for C28H46O5N3: 504.3437; found 504.3426. Elemental analysis C: 66.44, H: 9.06, N:
8.34 (required values C: 66.77, H: 9.04, N: 8.30).
Synthesis of trioxolanes 6 and 7
cis-adamantane-2-spiro-3’-8’-(carboxymethyl)-1’,2’,4’-trioxaspiro[4.5]decane was prepared
according to a reported method.[37]
Preparation of cis-adamantane-2-spiro-3'-8'-[1-(2-methylpropyl)piperazino]-1',2',4'-
trioxaspiro[4.5]decane 6: To a solution of cis-adamantane-2-spiro-3’-8’-(carboxymethyl)-1’,2’,4’-
trioxaspiro[4.5]decane (0.5 g, 1.55 mmol) in dry CH2Cl2 (50 mL) at 0°C, were added Et3N (0.44
mL, 3.10 mmol, 2 equiv.) and methyl chloroformate (0.12 mL, 1.55 mmol, 1 equiv.) under N2. The
mixture was stirred at 0°C for 1 h and 1-(2-methylpropyl)piperazine was added (0.22 mL, 1.55
mmol, 1.0 equiv.). The reaction mixture was stirred at 0°C for 45 min and at rt for other 2 h. The
reaction mixture was diluted with 50 mL of water and extracted with CH2Cl2 (2 x 60 mL). The
organic layers were combined and washed with water (40 mL) and brine (40 mL), dried on Na2SO4
and evaporated under vaccuo. The crude product was purified by flash chromatography (silica gel,
eluent: ethyl acetate/CH2Cl2 50/50) to afford trioxolane 6 (0.48 g, 70%) as a white solid.
1H NMR (500 MHz, 313 K, CDCl3) δ: 3.64 (bs, 2H), 3.48 (bs, 2H), 2.39 (bs, 4H), 2.23 (d, J = 6.9
Hz, 2H), 2.10-1.60 (m, 24H), 1.26 (dq, J = 13.2 Hz and 5.4 Hz, 2H), 0.93 (d, J = 6.6 Hz, 6H); 13C
NMR (125 MHz, 313 K, CDCl3) δ: 170.38, 111.09, 108.64, 66.69, 53.85, 53.28, 45.82, 41.64,

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
107
39.14, 36.85, 36.46, 34.82, 34.09, 33.40, 30.30, 26.93, 26.55, 25.41, 20.78; HRMS calculated for
C26H43N2O4 [M+H]+: 447.3223; found: 447.3221.
Preparation of cis-adamantane-2-spiro-3'-8'-[1-methyl-4-(4-piperidino)piperazino]-1',2',4'-
trioxaspiro[4.5]decane (7): To a solution of cis-adamantane-2-spiro-3’-8’-(carboxymethyl)-1’,2’,4’-
trioxaspiro[4.5]decane (1.5 g, 4.65 mmol) in dry CH2Cl2 (150 mL) at 0°C, were added Et3N (1.32
mL, 9.30 mmol, 2 equiv.) and methyl chloroformate (0.36 mL, 4.65 mmol, 1 equiv.) under N2. The
mixture was stirred at 0°C for 1 h and 1-methyl-4-(4-piperidino)piperazine was added (860 mg,
4.69 mmol, 1 equiv.). The reaction mixture was stirred at 0°C for 30 min and at rt for other 2 h. The
reaction mixture was diluted with 150 mL of water and extracted with CH2Cl2 (3 x 100 mL). The
organic layers were combined and washed with water (100 mL) and brine (100 mL), dried on
Na2SO4 and evaporated under vaccuo. The crude product was purified by flash chromatography
(silica gel, eluent: CHCl3/methanol 100/0 to 90/10) to afford trioxolane 7 (418 mg, 20%) as a white
solid.
1H NMR (500 MHz, 313 K, CDCl3) δ: 4.65 (bd, J = 12.8 Hz, 2H, CH2), 3.92 (bd, J = 13.3 Hz, 2H,
CH2), 3.01 (t, J = 12.8 Hz, 2H), 2.70-2.39 (m, 7H, CH/CH2), 2.31 (s, 3H, NCH3), 2.23 (d, J = 6.9
Hz, 2H), 2.03-1.25 (m, 27H, CH/CH2); 13C NMR (125 MHz, 313 K, CDCl3) δ: 170.21, 111.28,
108.65, 61.66, 55.40, 49.03, 45.88, 45.09, 41.06, 39.20, 36.85, 36.45, 34.81, 34.09, 33.41, 30.30,
29.18, 28.25, 26.93, 56.55; HRMS calculated for C28H46N3O4 [M+H]+: 488.3488; found: 488.3487.
General procedure for the reaction of tetraoxanes with Fe2+ and TEMPO
A solution of tetraoxane (0.5 mmol), FeSO4 or FeBr2 (2 equiv.), TEMPO (2 equiv.) in THF or
CH2Cl2/ CH3CN 50:50 (10mL) was stirred at ambient temperature under nitrogen atmosphere for
24 h and concentrated. The crude product was dissolved in ethyl acetate, washed with water and
brine, dried over MgSO4, filtered and concentrated. The crude product was analyzed by LC-MS and
TEMPO adducts were identified in all cases.
General procedure for the reaction of antimalarial peroxides with ferrous sulfate:
Stock solution of peroxide (10 mM in absolute ethanol) and ferrous sulfate (100 mM in water) were
freshly prepared and degassed prior to use.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
108
A solution of ferrous sulfate (1.0 mL, 18.2 mM) was added to a solution of peroxide (1.0 mL, 1
mM) in ACN/H2O 1:1 (final volume 5.5 mL). The reactions were allowed to stir at room
temperature under nitrogen with LC-MS monitoring.
LC-MS analysis: Compounds were eluted using a ternary solvent system consisting of 50% MeOH,
35% acetonitrile and 15% 0.1 M ammonium acetate, in isocratic mode (flow rate: 0.5 mL/min).
Reaction of antimalarial peroxides with FeSO4 and PC:
Stock solutions of PC, FeSO4 and peroxides were degassed prior to use. To a solution of PC in
water (500 µL, 0.83 µmol) were added an aqueous solution of ferrous sulfate (500 µL, 7.2 µmol,
8.7 equiv.) and a solution of peroxide in absolute ethanol (500 µL, 6.81 µmol, 8.2 equiv.). For
control reaction, the peroxide solution was replaced by absolute ethanol (500 µL). The cloudy
mixtures were stirred at 38°C for 24 h under a nitrogen atmosphere. An aliquot of each crude
mixture was taken for ESI/MS analysis before reactions were quenched with 50% phytic acid
solution (10 µL), and a trace of BHT, according to the procedure reported by Kumura et al. The
reaction mixtures were extracted with CHCl3/CH3OH (1:2, 1 mL) and the chloroform layers were
analyzed by ESI/MS.
General procedure for the reaction of antimalarial peroxides with heme:
Hemin stock solution (10 mM in 0.1 M NaOH) was freshly prepared and degassed with acetonitrile
prior to use. Hemin solution (2.0 mL, 0.02 mmol) was added to a solution of peroxide (0.02 mmol,
1 equiv.) and dithionite (41 mg, 0.20 mmol, 10 equiv.) in acetonitrile (2.0 mL) under argon. The
mixture was stirred at room temperature and monitored by LC-MS.
LC-MS analysis: Compounds were eluted using a binary gradient solvent system consisting of
MeOH, 1% TFA (solvent A) and H2O, 1% TFA (solvent B). The gradient used was 70% to 80% of
solvent A over 10 min (flow rate: 0.5 mL/min). Retention times were 3.24 min for heme and 5.39 to
8.15 min for the heme-tetraoxane adducts.
Acknowledgements
We are thankful to Dr Jill Davies (LSTM) for IC50 values and Dr Allan Mills (University of
Liverpool) for MS analysis. This work was supported by a grant from the EU (Antimal PhD
fellowship and FP6 Malaria Drugs Initiative)

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
109
Keywords
Tetraoxanes; heme alkylation; phospholipids oxidation; reactivity; malaria
Bibliography
[1] World Health Organization. World Malaria Report 2008 (Geneva, Switzerland). The WHO Website: Http://who.int/malaria/wmr2008/malaria2008.pdf, 2008. [2] B. M. Greenwood, D. A. Fidock, D. E. Kyle, S. H. I. Kappe, P. L. Alonso, F. H. Collins, P. E. Duffy. Malaria: Progress, Perils, and Prospects for Eradication. J. Clin. Invest. 2008, 118, 1266-1276. [3] D. L. Klayman. Qinghaosu (Artemisinin): An Antimalarial Drug from China. Science 1985, 228, 1049-1055. [4] G. H. Posner, C. H. Oh, D. Wang, L. Gerena, W. K. Milhous, S. R. Meshnick, W. Asawamahasadka. Mechanism-Based Design, Synthesis, and in Vitro Antimalarial Testing of New 4-Methylated Trioxanes Structurally Related to Artemisinin: The Importance of a Carbon-Centered Radical for Antimalarial Activity. J. Med. Chem. 1994, 37, 1256-1258. [5] G. H. Posner, D. Wang, J. N. Cumming, C. H. Oh, A. N. French, A. L. Bodley, T. A. Shapiro. Further Evidence Supporting the Importance of and the Restrictions on a Carbon-Centered Radical for High Antimalarial Activity of 1,2,4-Trioxanes Like Artemisinin. J. Med. Chem. 1995, 38, 2273-2275. [6] G. H. Posner, C. H. Oh. A Regiospecifically-18 Labeled 1,2,4-Trioxane - A Simple Chemical-Model System to Probe the Mechanism(s) for the Antimalarial Activity of Artemisinin(Qinqhaosu). J. Am. Chem. Soc. 1992, 114, 8328-8329. [7] Y. L. Hong, Y. Z. Yang, S. R. Meshnick. The Interaction of Artemisinin with Malarial Hemozoin. Mol. Biol. Parasitol. 1994, 63, 121-128. [8] S. R. Meshnick, A. Thomas, A. Ranz, C. M. Xu, H. Z. Pan. Artemisinin (Qinghaosu): The Role of Intracellular Hemin in Its Mechanism of Antimalarial Action. Mol. Biol. Parasitol. 1991, 49, 181-189. [9] J. Bhisutthibhan, X. Q. Pan, P. A. Hossler, D. J. Walker, C. A. Yowell, J. Carlton, J. B. Dame, S. R. Meshnick. The Plasmodium falciparum Translationally Controlled Tumor Protein Homolog and Its Reaction with the Antimalarial Drug Artemisinin. J. Biol. Chem. 1998, 273, 16192-16198. [10] W. Asawamahasakda, I. Ittarat, Y. M. Pu, H. Ziffer, S. R. Meshnick. Reaction of Antimalarial Endoperoxides with Specific Parasite Proteins. Antimicrob. Agents Chemother. 1994, 38, 1854-1858. [11] U. Eckstein-Ludwig, R. J. Webb, I. D. A. Van Goethem, J. M. East, A. G. Lee, M. Kimura, P. M. O'Neill, P. G. Bray, S. A. Ward, S. Krishna. Artemisinins Target the SERCA of Plasmodium falciparum. Nature 2003, 424, 957-961.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
110
[12] A. Robert, J. Cazelles, B. Meunier. Characterization of the Alkylation Product of Heme by the Antimalarial Drug Artemisinin. Angew. Chem. Inter. Ed. 2001, 40, 1954-1957. [13] A. Robert, Y. Coppel, B. Meunier. Alkylation of Heme by the Antimalarial Drug Artemisinin. Chem. Commun. 2002, 414-415. [14] A. Robert, B. Meunier. Alkylating Properties of Antimalarial Artemisinin Derivatives and Synthetic Trioxanes When Activated by a Reduced Heme Model. Chem. Eur. J. 1998, 4, 1287-1296. [15] A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier. The Antimalarial Drug Artemisinin Alkylates Heme in Infected Mice. PNAS 2005, 102, 13676-13680. Erratum, 2006, 103, 3943. [16] J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A. Charman, F. C. K. Chiu, J. Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padmanilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler, S. Wittlin, W. N. Charman. Identification of an Antimalarial Synthetic Trioxolane Drug Development Candidate. Nature 2004, 430, 900-904. [17] F. Coslédan, L. Fraisse, A. Pellet, F. Guillou, B. Mordmüller, P. G. Kremsner, A. Moreno, D. Mazier, J.-P. Maffrand, B. Meunier. Selection of a Trioxaquine as an Antimalarial Drug Candidate. P.N.A.S. 2008, 105, 17579-17584. [18] P. R. Story, D. D. Denson, C. E. Bishop, B. C. Clark, Jr., J. C. Farine. A New General Synthesis of Macrocyclic Compounds. J. Am. Chem. Soc. 1968, 90, 817-818. [19] J. L. Vennerstrom, H. N. Fu, W. Y. Ellis, A. L. Ager, Jr., J. K. Wood, S. L. Andersen, L. Gerena, W. K. Milhous. Dispiro-1,2,4,5-Tetraoxanes: A New Class of Antimalarial Peroxides. J. Med. Chem. 1992, 35, 3023-3027. [20] R. Amewu, A. V. Stachulski, S. A. Ward, N. G. Berry, P. G. Bray, J. Davies, G. Labat, L. Vivas, P. M. O'Neill. Design and Synthesis of Orally Active Dispiro 1,2,4,5-Tetraoxanes; Synthetic Antimalarials with Superior Activity to Artemisinin. Org. Biomol. Chem. 2006, 4, 4431-4436. [21] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. Shone, D. Stanford, P. Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer, S. Lozanom, M. Jesús, D. Gargallo, S. A. Ward, P. M. O'Neill. Two-Step Synthesis of Achiral Dispiro-1,2,4,5-Tetraoxanes with Outstanding Antimalarial Activity, Low Toxicity, and High-Stability Profiles. J. Med. Chem. 2008, 51, 2170-2177. [22] Y. Dong, J. L. Vennerstrom. Formation of Primary and Secondary Carbon-Centered Radicals in the Iron (II)-Mediated Decomposition of Antimalarial 1,2,4,5-Tetraoxanes. Abstracts of Papers 2006, 232nd ACS National Meeting, San Francisco, CA, United States, Sept. 10-14 2006. [23] H. H. Liu, Y. K. Wu, X. Shen. Alkylation of Sulfur Ligand in Cysteinate-Iron Chelates by a 1,2,4,5-Tetraoxane. Chin. J. Chem . 2003, 21, 875-877.

Chapter 4: Reactivity of antimalarial tetraoxanes with Fe(II), heme and phospholipids: Implications for mode of action
111
[24] I. Opsenica, N. Terzic, D. Opsenica, G. Angelovski, M. Lehnig, P. Eilbracht, B. Tinant, Z. Juranic, K. S. Smith, Y. S. Yang, D. S. Diaz, P. L. Smith, W. K. Milhous, D. Dokovic, B. A. Solaja. Tetraoxane Antimalarials and Their Reaction with Fe(II). J. Med. Chem. 2006, 49, 3790-3799. [25] N. Kumura, H. Furukawa, A. N. Onyango, M. Izumi, S. Nakajima, H. Ito, T. Hatano, H. S. Kim, Y. Wataya, N. Baba. Different Behavior of Artemisinin and Tetraoxane in the Oxidative Degradation of Phospholipid. Chem. Phys. Lipids 2009, 160, 114-120. [26] Y. Tang, Y. Dong, X. Wang, K. Sriraghavan, J. K. Wood, J. L. Vennerstrom. Dispiro-1,2,4-Trioxane Analogues of a Prototype Dispiro-1,2,4-Trioxolane: Mechanistic Comparators for Artemisinin in the Context of Reaction Pathways with Iron(II). J. Org. Chem. 2005, 70, 5103-5110. [27] M. R. M. Domingues, A. Reis, P. Domingues. Mass Spectrometry Analysis of Oxidized Phospholipids. Chem. Phys. Lipids 2008, 156, 1-12. [28] P. A. Berman, P. A. Adams. Artemisinin Enhances Heme-Catalysed Oxidation of Lipid Membranes. Free Radical Biol. Med. 1997, 22, 1283-1288. [29] F. Bousejra-El Garah, B. Meunier, A. Robert. The Antimalarial Artemisone Is an Efficient Heme Alkylating Agent. Eur. J. Inorg. Chem. 2008, 2008, 2133-2135. [30] S. A. L. Laurent, A. Robert, B. Meunier. C10-Modified Artemisinin Derivatives: Efficient Heme-Alkylating Agents. Angew. Chem. Inter. Ed. 2005, 44, 2060-2063. [31] F. Bousejra-El Garah, C. Claparols, F. Benoit-Vical, B. Meunier, A. Robert. The Antimalarial Trioxaquine DU1301 Alkylates Heme in Malaria-Infected Mice. Antimicrob. Agents Chemother. 2008, 52, 2966-2969. [32] S. A. L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meunier. Heme Alkylation by Artesunic Acid and Trioxaquine Du1301, Two Antimalarial Trioxanes. ChemBioChem 2005, 6, 653-658. [33] D. J. Creek, W. N. Charman, F. C. K. Chiu, R. J. Prankerd, Y. Dong, J. L. Vennerstrom, S. A. Charman. Relationship between Antimalarial Activity and Heme Alkylation for Spiro- and Dispiro-1,2,4-Trioxolane Antimalarials. Antimicrob. Agents Chemother. 2008, 52, 1291-1296. [34] A. Robert, Y. Coppel, B. Meunier. NMR Characterization of Covalent Adducts Obtained by Alkylation of Heme with the Antimalarial Drug Artemisinin. Inorg. Chim. Acta 2002, 339, 488-496. [35] Yields of Adducts Were Estimated by Measuring Their Concentration Using the Heme Calibration Plot, Considering That the Response Factor Would Be Similar. [36] A. Robert, B. Meunier. Characterization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968-5969. [37] J. L. Vennerstrom, Y. Dong, J. Chollet, H. Matile. Preparation of Spiro/Dispiro-1,2,4-Trioxolanes as Antimalarial Agents, US Patent 6,486,199 2002.


Chapter 5
The antimalarial trioxaquine DU1301 alkylates heme in malaria
infected mice


Chapter 5: The antimalarial trioxaquine DU1301 alkylates heme in malariainfected mice
Résumé de la publication :
The antimalarial trioxaquine DU1301 alkylates heme in malariainfected mice
F. Bousejra-El Garah, C. Claparols, F. Benoit-Vical, B. Meunier, A. Robert, Antimicrobial Agents
and Chemotherapy 2008, 52, 2966-2969.
La trioxaquine DU1301 alkyle l’hème chez la souris impaludée
Les trioxaquines ont été conçues de façon à être des alkylants potentiels de l’hème grâce à la
réactivité du cycle 1,2,4-trioxane. La capacité alkylante de la trioxaquine prototype DU1301 avait
été mise en évidence chimiquement avec la caractérisation des adduits hème-DU1301.[1] Nous nous
sommes intéressés ici à la réactivité de cette trioxaquine vis-à-vis de l’hème in vivo, et au lien
possible entre son caractère alkylant et son activité antipaludique. Pour cela, nous avons évalué les
propriétés alkylantes de la DU1301 chez la souris infectée par Plasmodium, en recherchant les
adduits covalents heme-trioxaquine dans les organes et fluides biologiques.
Des souris infectées par la souche P.vinckei petteri ont été traitées par voie orale, avec une dose
efficace de trioxaquine DU1301 (lot 1, dose : 100 mg/kg/100 µL DMSO).
A titre de contrôle,
des souris non infectées ont été traitées avec DU1301 dans les mêmes conditions que les
souris impaludées (lot 2).
des souris infectées ont été « traitées » au DMSO pur (lot 3).
Les souris ont été sacrifiées 2 h après le traitement ; les organes ont été broyés et extraits à la
pyridine. L’analyse par LC-MS des extraits de rate de souris impaludées et traitées à la DU1301, a
permis de mettre en évidence la présence des adduits hème-trioxaquine 1 (m/z 1101.1) et 2 (m/z
949.2). Ces adduits, présents dans les rates de toutes les souris du lot 1, ont été détectés avec des
temps de rétention identiques à ceux des adduits préparés chimiquement. De plus, les figures
isotopiques des deux ions m/z 1101.1 et 949.2 (figure ci-dessous) sont caractéristiques de la
présence d’un atome de fer et de chlore.
Les adduits hème-trioxaquine n’ont pas été détectés dans les rates de souris saines traitées dans les
mêmes conditions (lot 2), ni dans les rates de souris impaludées non traitées (lot 3), ce qui montre
que la réactivité alkylante de la trioxaquine in vivo est induite par la présence du parasite.
115

Chapter 5: The antimalarial trioxaquine DU1301 alkylates heme in malariainfected mice
Adduit hème-trioxaquine 2 m/z 949.2 M+
Adduit hème-trioxaquine 1 m/z 1101.1 M+
Figures isotopiques des ions m/z 1101.1 et 949.2, correspondant aux adduits hème-trioxaquine 1 et 2, respectivement
C’est également dans la rate que les adduits entre l’hème et l’artémisinine avaient été détectés.[2] En
effet, la rate étant le principal organe d’élimination des érythrocytes infectés, il est normal que l’on
y retrouve les adduits hème-drogue, eux-mêmes formés dans ces érythrocytes malades.
Lors de la recherche d’adduits hème-artémisinine chez la souris impaludée, des adduits glucuro-
conjugués avaient été détectés dans l’urine. Dans le cas présent, les adduits n’ont pas pu être
détectés dans les reins, ni l’urine. Ceci peut être dû à deux raisons : 1) une concentration des adduits
inférieure à la limite de détection ou 2) la métabolisation des adduits étant un processus dynamique,
ils n’ont peut-être pas encore atteint les reins pour être éliminés dans l’urine.
Conclusion
La trioxaquine DU1301 est un alkylant efficace de l’hème chez la souris impaludée. La mise en
évidence des adduits hème-drogue montre que la réactivité des trioxaquines vis-à-vis de l’hème in
vivo est similaire à celle de l’artémisinine. Cette propriété d’alkylation pourrait être liée à leur haute
efficacité antipaludique, d’autant plus que ces deux molécules ont montré des moments d’action sur
le cycle du parasite identiques.[3]
116

Chapter 5: The antimalarial trioxaquine DU1301 alkylates heme in malariainfected mice
117
Bibliographie
[1] S. A. L. Laurent, C. Loup, S. Mourgues, A. Robert, B. Meunier. Heme Alkylation by Artesunic Acid and Trioxaquine DU1301, Two Antimalarial Trioxanes. ChemBioChem 2005, 6, 653-658. [2] A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier. The Antimalarial Drug Artemisinin Alkylates Heme in Infected Mice. Proc. Nat. Acad. Sci. U.S.A 2005, 102, 13676-13680. Erratum, 2006, 103, 3943. [3] F. Benoit-Vical, J. Lelièvre, A. Berry, C. Deymier, O. Dechy-Cabaret, J. Cazelles, C. Loup, A. Robert, J.-F. Magnaval, B. Meunier. Trioxaquines Are New Antimalarial Agents Active on All Erythrocytic Forms, Including Gametocytes. Antimicrob. Agents. Chemother. 2007, 51, 1463-1472.

ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, Aug. 2008, p. 2966–2969 Vol. 52, No. 80066-4804/08/$08.00�0 doi:10.1128/AAC.00165-08Copyright © 2008, American Society for Microbiology. All Rights Reserved.
The Antimalarial Trioxaquine DU1301 Alkylates Heme inMalaria-Infected Mice�
Fatima Bousejra-El Garah,1 Catherine Claparols,1 Francoise Benoit-Vical,1,2
Bernard Meunier,3 and Anne Robert1*Laboratoire de Chimie de Coordination du CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 4, France1; Service de
Parasitologie et Mycologie, CHU Rangueil, Universite de Toulouse, TSA 50032, 31059 Toulouse Cedex 9, France2;and Palumed, rue Pierre et Marie Curie, BP 28262, 31682 Labege Cedex, France3
Received 5 February 2008/Returned for modification 6 March 2008/Accepted 3 June 2008
The in vivo alkylation of heme by the antimalarial trioxaquine DU1301 afforded covalent heme-drug adductsthat were detected in the spleens of Plasmodium sp.-infected mice. This result indicates that the alkylationcapacities of trioxaquines in mammals infected with Plasmodium strains are similar to that of artemisinin, anatural antimalarial trioxane-containing drug.
Artemisinin and its derivatives have emerged as antimalarialdrugs over the past three decades. With a pharmacophorebased on a 1,2,4-trioxane, these drugs are highly active againstmultidrug-resistant Plasmodium strains and are nontoxic, evenfor children and pregnant women. In addition, no clinicallyrelevant parasite resistance has been reported so far (7, 24, 25).Many synthetic peroxides have been synthesized as artemisininmodels in the last 15 to 20 years (for recent reviews, seereference 8, references therein, and references 15 and 22).Among them, trioxaquines have a unique structure, containingboth an aminoquinoline moiety (as in chloroquine) and a syn-thetic 1,2,4-trioxane entity (as an artemisinin mimic) (2, 4). Aspreviously described (4), trioxaquine DU1301 has been ob-tained by convergent synthesis from 7-chloro-4-(2-aminoethyl-amino)quinoline and a 1,2,4-trioxane derived from ascaridole(20, 10). Trioxaquines are able to cure Plasmodium-infectedmice treated orally at 15 to 20 mg/kg of body weight/day for 4days (as demonstrated by the “4-day suppressive test”) (seereference 16). Trioxaquines are being developed by Palumed,and one of them (PA1103-SAR116242) is currently under reg-ulatory preclinical development with Sanofi-Aventis (reference13 and references therein).
The mechanism of action of antimalarial 1,2,4-trioxanes hasbeen intensively studied during the last two decades. The cru-cial role of heme digestion-aggregation processes in infectederythrocytes has led to investigations of the possible interac-tion of heme with artemisinin. In the initial work, it was re-ported that heme-catalyzed peroxide cleavage is responsiblefor the alkylation of heme and specific parasite proteins (1, 6).Such iron(II)-mediated cleavage of the endoperoxide functiongenerates, in particular, an alkyl radical centered at positionC-4 of artemisinin or synthetic trioxanes (9, 17). The concen-tration of free iron ions in living cells is close to zero, whereasthe hemoglobin concentration in red blood cells is 5 mM (cor-responding to a heme concentration of 20 mM). In addition,
the extensive ingestion of host hemoglobin into the food vacu-oles of Plasmodium parasites leads to vacuolar heme concen-trations of about 400 mM (21). Bearing in mind the importantrole of free heme in infected erythrocytes, we reported previ-ously that artemisinin is efficiently activated by heme in vitroand also in vivo, leading to the alkylation of the heme macro-cycle (18, 19). Covalent heme-drug adducts resulting from al-kylation by the drug were detected in spleens and urine sam-ples from Plasmodium-infected mice (18). Yet, the exact roleof heme-artemisinin adducts in the death of the parasite willprobably remain a matter of debate. Nevertheless, it is nowwell established that artemisinin alkylates heme with its C-4-centered radical, and as underlined in a recent publication,free heme or hemoglobin heme alkylations are “possibly theonly malaria-parasite-relevant fully characterized alkylation re-actions reported so far for [artemisinin]” (23).
Trioxaquine DU1301 was designed as a potential heme-alkylating agent, and such an alkylating ability has been evi-denced previously in vitro (11). To confirm that the formationof heme-trioxaquine adducts is a biologically relevant process,we examined the in vivo alkylating ability of DU1301, a rep-resentative member of this new class of antimalarial drugs, inparasite-infected mice.
To address the question of the in vivo alkylating propertiesof trioxaquines, Plasmodium vinckei petteri-infected mice weretreated orally with DU1301, a trioxaquine prototype (Fig. 1). Aset of three Swiss female albino mice (25 to 30 g) were intra-peritoneally inoculated with erythrocytes parasitized by P.vinckei petteri. When the level of parasitemia (evaluated mi-croscopically on Giemsa-stained thin blood smears) was higherthan 40% (after 3 days), mice were treated orally with a singledose of trioxaquine DU1301 (2.5 mg diluted in 100 �l ofdimethyl sulfoxide [DMSO], corresponding to a dose of 100mg/kg). As control experiments, a set of three healthy micereceived the same dose of DU1301 and a set of infected micereceived pure DMSO (no treatment). Mice were sacrificed 2 hafter treatment, and organs (spleens, livers, and kidneys) werecollected.
The detection of heme-DU1301 adducts in mouse organextracts first required the efficient extraction of these hydro-
* Corresponding author. Mailing address: Laboratoire de Chimie deCoordination du CNRS, 205 route de Narbonne, 31077 ToulouseCedex 4, France. Phone: 33 5 61 33 31 26. Fax: 33 5 61 55 30 03. E-mail:[email protected].
� Published ahead of print on 16 June 2008.
2966
by Jean-Francois M
agnaval on July 28, 2008 aac.asm
.orgD
ownloaded from
118

phobic compounds from biological tissues. Each organ wascrushed with sand in a mortar. Heme-DU1301 adducts arepoorly soluble in common aqueous and organic solvents, andthe stacking of heme with quinoline moieties may account forthis low solubility (11). Strong �-� interactions between hemeand the quinoline ring of antimalarials has been evidencedpreviously by nuclear magnetic resonance analyses (14). Be-cause of the low solubilities of these heme-DU1301 adducts,pyridine (1.0 to 1.5 ml) was found to be the best solvent fortheir extraction (by subjecting the tissues to a vortex for 20min). The organ extracts were analyzed by liquid chromatog-raphy-mass spectrometry (LC-MS) without any additionaltreatment.
The characterization of small amounts of poorly solublehydrophobic adducts in biological extracts was much morecomplicated than the usual in vitro characterization of adductsprepared on the bench on a 10- to 100-mg scale (11). Becauseof the difficulties with the organ extracts, it was essential tooptimize the analytical conditions by using chemically pre-pared adducts (11) in order to have reliable, accurate, andsensitive detection of the in vivo adducts. Analytical separa-tions were performed on a 5-�m C18 Modulo-Cart QS Upti-sphere column (250 by 4.6 mm) equipped with a precolumncontaining the same packing material. Compounds were elutedunder the following conditions: solvent A consisted of metha-nol-H2O-trifluoroacetic acid, 70/30/0.05; solvent B consisted ofmethanol-trifluoroacetic acid, 100/0.05; the gradient from anA/B ratio of 100/0 to an A/B ratio of 0/100 over 25 min wasfollowed by 10 min at an A/B ratio of 0/100; the elution ratewas 0.5 ml/min; and UV-visible light detection was performedat 340 and 400 nm by using a diode array detector. The volumeof pyridine extract injected was 100 �l. Positive-ion electros-pray MS was performed on a Q-Trap AB Sciex quadrupoleinstrument (Fig. 2).
The LC-MS analyses of spleen extracts from a Plasmodium-infected mouse treated with trioxaquine DU1301 (100 mg/kg in100 �l of DMSO administered per os) are summarized in Fig.
3a to c. Currents of ions with m/z 1,101.0 and 949.2, whichcorrespond to adducts 1 and 2, respectively, were detected withretention times of 18.5 to 21.7 min, along with heme (retentiontime, 16.5 min). Retention times and mass spectra were con-sistent with those observed with chemically prepared adducts(Fig. 2) (11). Furthermore, the isotopic patterns of signals atm/z 1,101.1 and 949.2 clearly showed the presence of both oneiron atom (M � 2 due to 54Fe) and one chlorine atom (M �2 due to 37Cl) (Fig. 3). This result is fully consistent with thereductive activation of the peroxide bond of trioxaquineDU1301 by iron(II)-heme within infected erythrocytes. Thisreaction leads to the formation of an oxygen-centered radicalthat rapidly rearranges to give an alkyl radical (Fig. 1) able toalkylate heme at meso positions, giving rise to the “complete”covalent adduct heme-DU1301, adduct 1. Heme-drug adduct2, generated by hydrolysis and the loss of the terpene moiety,probably in the spectrometer, was also detected.
Adducts 1 and 2 were both detected in all infected mice
FIG. 1. Reductive activation of trioxaquine DU1301 by iron(II)-heme, leading to the covalent heme-drug adducts 1 and 2. Quin stands for theaminoquinoline fragment, and FeIIIPPIX stands for ferriprotoporphyrin IX.
FIG. 2. LC-MS analyses of chemically prepared heme-trioxaquineadducts. (a) UV-visible (vis.) trace at 400 nm; (b and c) extracted ioniccurrent (XIC) traces for the indicated m/z.
VOL. 52, 2008 NOTES 2967
by Jean-Francois M
agnaval on July 28, 2008 aac.asm
.orgD
ownloaded from
119

treated with DU1301. In the control experiments, these heme-trioxaquine adducts were undetectable in all spleen extractsfrom healthy mice treated under the same conditions as in-fected mice (Fig. 3d and e) and in all spleen extracts fromuntreated Plasmodium-infected mice (which received only ex-cipient) (data not shown).
Heme-DU1301 adducts remained below detectable levels inthe kidneys and urine samples from Plasmodium-infected micethat had been treated with trioxaquine. However, it should beconsidered that the spleen, an organ devoted to the elimina-tion of damaged red blood cells, is the first candidate for theaccumulation of heme-drug adducts. Further metabolism ofthese adducts is a dynamic process, whereas their distribution
may be responsible for the fact that they remained below thedetection limit in kidneys and urine samples.
The alkylation of heme by DU1301 occurred only in infectedmice treated at pharmacologically relevant doses. The pres-ence of these heme-drug adducts should therefore be consid-ered as evidence of the alkylating capacities of these trioxane-containing drugs triggered by the presence of the parasite inmice. The detection of heme-DU1301 adducts within a Plas-modium-infected mammal indicates that heme alkylation byDU1301 is an efficient reaction that occurs in vivo and may beconsidered to be a key element concerning the mechanism ofaction of this antimalarial agent. This result underlines theimportance of the alkylating abilities of trioxane-based anti-malarial drugs and also confirms that artemisinin and trioxa-quines share the same heme-alkylating properties. In addition,since artemisinin and the citrate salt of DU1301 are activeagainst the same stages of synchronized P. falciparum parasites(ring, trophozoite, and gametocyte stages) (2), these pharma-cological features strongly suggest that this common alkylatingreactivity may be one of the key factors of their antimalarialactivities.
In addition, aminoquinolines (such as chloroquine) are con-sidered to inhibit the formation of hemozoin by �-� stacking(21, 3, 5). It has been reported recently that trioxaquineDU1301 efficiently prevents the in vitro formation of �-hema-tin at a lower concentration than chloroquine itself, whereasthe synthetic trioxane precursor of DU1301 does not inhibitthe dimerization of hemin (12). On the other hand, heme-artemisinin adducts also inhibit �-hematin dimerization andare themselves unable to dimerize (12). These results suggestthat, besides the alkylating ability of the peroxide moiety, tri-oxaquines may have the ability to prevent heme aggregationwithin the parasite either (i) by the stacking of their quinolinefragment with heme or (ii) by the formation of unpolymeriz-able heme-drug adducts. Thus, trioxaquines should be consid-ered as hybrid antimalarial molecules with a dual mode ofaction: heme alkylation and the inhibition of heme polymer-ization.
F.B.-E.G. is indebted to the EU-AntiMal program for a Ph.D. fel-lowship. CNRS, INSERM, and ANR are acknowledged for financialsupport.
This study was approved by the French Institutional Animal Exper-imentation Ethics Committee (approval no. MP/R/06/33/11/07).
Guy Lavigne (LCC-CNRS, Toulouse) is acknowledged for Englishediting.
REFERENCES
1. Asawamahasakda, W., I. Ittarat, Y.-M. Pu, H. Ziffer, and S. R. Meshnick.1994. Reaction of antimalarial endoperoxide with specific parasite proteins.Antimicrob. Agents Chemother. 38:1854–1858.
2. Benoit-Vical, F., J. Lelievre, A. Berry, C. Deymier, O. Dechy-Cabaret, J.Cazelles, C. Loup, A. Robert, J.-F. Magnaval, and B. Meunier. 2007. Tri-oxaquines are new antimalarial agents active on all erythrocytic forms, in-cluding gametocytes. Antimicrob. Agents Chemother. 51:1463–1472.
3. Bray, P. G., O. Janneh, K. J. Raynes, M. Mungthin, H. Ginsburg, and S. A.Ward. 1999. Cellular uptake of chloroquine is dependent on binding toferriprotoporphyrin IX and is independent of NHE activity in Plasmodiumfalciparum. J. Cell Biol. 145:363–376.
4. Dechy-Cabaret, O., F. Benoit-Vical, C. Loup, A. Robert, H. Gornitzka, A.Bonhoure, H. Vial, J.-F. Magnaval, J.-P. Seguela, and B. Meunier. 2004.Synthesis and antimalarial activity of trioxaquine derivatives. Chem. Eur. J.10:1625–1636.
5. Egan, T. J., and H. M. Marques. 1999. The role of haem in the activity ofchloroquine and related antimalarial drugs. Coord. Chem. Rev. 190-192:493–517.
FIG. 3. (a to c) LC-MS analyses of the spleen extracts from amouse infected with P. vinckei petteri and treated orally with DU1301(single dose of 100 mg/kg). (a) UV-visible (vis.) trace at 400 nm; (b andc) extracted ionic current (XIC) traces for the indicated m/z; (d and e)LC-MS analyses of the spleen extracts from a healthy mouse treatedorally with DU1301 (single dose of 100 mg/kg).
2968 NOTES ANTIMICROB. AGENTS CHEMOTHER.
by Jean-Francois M
agnaval on July 28, 2008 aac.asm
.orgD
ownloaded from
120

6. Hong, Y.-L., Y.-Z. Yang, and S. R. Meshnick. 1994. The interaction ofartemisinin with malarial hemozoin. Mol. Biochem. Parasitol. 63:121–128.
7. Ittarat, W., A. L. Pickard, P. Rattanasinganchan, P. Wilairatana, S.Looareesuwan, K. Emery, J. Low, R. Udomsangpetch, and S. R. Meshnick.2003. Recrudescence in artesunate-treated patients with falciparum malariais dependent on parasite burden not on parasite factors. Am. J. Trop. Med.Hyg. 68:147–152.
8. Jefford, C. W. 2007. New developments in synthetic peroxidic drugs as arte-misinin mimics. Drug Discov. Today 12:487–495.
9. Jefford, C. W., F. Favarger, V. H. Maria da Graca, and Y. Jacquier. 1995.The decomposition of cis-fused cyclopenteno-1,2,4-trioxanes by ferrous saltsand some oxophilic reagents. Helv. Chim. Acta 78:452–458.
10. Jefford, C. W., A. Jaber, and J. Boukouvalas. 1989. The regio- and stereo-controlled synthesis of cis-p-menth-3-ene-1,2-diol by means of a 1,2,4-triox-ane intermediate. J. Chem. Soc. Chem. Commun. 1989:1916–1918.
11. Laurent, S. A.-L., C. Loup, S. Mourgues, A. Robert, and B. Meunier. 2005.Heme alkylation by artesunic acid and trioxaquine DU1301, two antimalarialtrioxanes. Chembiochem 6:653–658.
12. Loup, C., J. Lelievre, F. Benoit-Vical, and B. Meunier. 2007. Trioxaquinesand heme-artemisinin adducts inhibit the in vitro formation of hemozoinbetter than chloroquine. Antimicrob. Agents Chemother. 51:3768–3770.
13. Meunier, B. 2008. Hybrid molecules with a dual mode of action: dream orreality? Acc. Chem. Res. 41:69–77.
14. Moreau, S., B. Perly, C. Chachaty, and C. Deleuze. 1985. A nuclear magneticresonance study of the interactions of antimalarial drugs with porphyrins.Biochim. Biophys. Acta 840:107–116.
15. O’Neill, P. M., and G. H. Posner. 2004. A medicinal perspective on arte-misinin and related endoperoxides. J. Med. Chem. 47:2945–2964.
16. Peters, W., J. H. Portus, and B. L. Robinson. 1975. Chemotherapy of rodent
malaria. XXII. Value of drug-resistant strains of Plasmodium berghei inscreening for blood schizontocidal activity. Ann. Trop. Med. Parasitol. 69:155–171.
17. Posner, G. H., C. H. Ho, D. Wang, L. Gerena, W. K. Milhous, S. R. Meshnick,and W. Asawamahasakda. 1994. Mechanism-based design, synthesis, and invitro antimalarial testing of new 4-methylated trioxanes structurally relatedto artemisinin: the importance of a carbon-centered radical for antimalarialactivity. J. Med. Chem. 37:1256–1258.
18. Robert, A., F. Benoit-Vical, C. Claparols, and B. Meunier. 2005. The anti-malarial drug artemisinin alkylates heme in infected mice. Proc. Natl. Acad.Sci. USA 102:13676–13680. (Erratum, 103:3943, 2006.)
19. Robert, A., and B. Meunier. 1997. Characterization of the first covalentadduct between artemisinin and a heme model. J. Am. Chem. Soc. 119:5968–5969.
20. Schenck, G. O., and K. Ziegler. 1944. Die Synthese des Ascaridols. Natur-wissenschaften 32:157.
21. Sullivan, D. J., Jr., I. Y. Gluzman, D. G. Russel, and D. E. Goldberg. 1996.On the molecular mechanism of chloroquine’s antimalarial action. Proc.Natl. Acad. Sci. USA 93:11865–11870.
22. Tang, Y., Y. Dong, and J. L. Vennerstrom. 2004. Synthetic peroxides asantimalarials. Med. Res. Rev. 24:425–448.
23. Tang, Y., Y. Dong, X. Wang, K. Sriraghavan, J. K. Wood, and J. L. Venner-strom. 2005. Dispiro-1,2,4-trioxane analogues of a prototype dispiro-1,2,4-trioxolane: mechanistic comparators for artemisinin in the context of reac-tion pathways with iron(II). J. Org. Chem. 70:5103–5110.
24. White, N. J. 2008. Qinghaosu (artemisinin): the price of success. Science320:330–334.
25. Wongsrichanalai, C., A. L. Pickard, W. H. Wernsdorfer, and S. R. Meshnick.2002. Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2:209–218.
VOL. 52, 2008 NOTES 2969
by Jean-Francois M
agnaval on July 28, 2008 aac.asm
.orgD
ownloaded from
121


Chapter 6
Docking study of structurally diverse antimalarial drugs targeting PfATP6: No correlation between in silico binding affinity and in vitro
antimalarial activity


Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
Résumé de la publication :
Docking study of structurally diverse antimalarial drugs targeting PfATP6: No correlation between in silico binding affinity and in vitro antimalarial activity
F. Bousejra-El Garah, J.-L. Stigliani, F. Coslédan, B. Meunier, A. Robert. ChemMedChem 2009,
4(9), 1469-1479.
Calcul d’interaction entre divers antipaludiques et la protéine parasitaire PfATP6 : Absence de corrélation entre l’affinité calculée et l’activité antipaludique
L’un des mécanismes envisagés pour l’artémisinine 1 repose sur l’inhibition de PfATP6, une
protéine de type SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) de Plasmodium
falciparum. En effet, Krishna et ses collaborateurs ont montré que l’artémisinine pouvait inhiber
l’activité de PfATP6 surexprimée dans des ovocytes de xenopes.[1] Cette interaction de
l’artémisinine avec PfATP6 serait antagoniste de celle de la thapsigargine (TG), inhibiteur
naturel des SERCA de mammifères. Cependant, on peut noter que les deux molécules présentent
des structures très différentes, une des principales différences étant que TG est dépourvue de la
fonction peroxyde nécessaire à l’activité antipaludique de l’artémisinine. TG n’est d’ailleurs pas
active sur Plasmodium.
Structures de l’artémisinine et de la thapsigargine
Depuis la publication de cette étude, PfATP6 est considérée par certains auteurs comme la cible
responsable de l’activité de l’artémisinine et de ses dérivés hémi-synthétiques. Dans le but de
mieux comprendre le possible rôle de cette protéine dans le mécanisme d’action de
125

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
l’artémisinine, nous avons réalisé une étude de docking entre un modèle de PfATP6 et
différentes molécules antipaludiques de structures variées. L’objectif de ce travail est de savoir
s’il existe une corrélation significative entre l’affinité de ces molécules pour PfATP6, prédite
par le logiciel AutoDock, et leur activité antipaludique mesurée in vitro.
1. Modélisation par homologie. Validation du modèle et du protocole de docking
La séquence d’acides aminés de PfATP6 étant connue, nous avons reconstitué sa structure
tertiaire par homologie avec une SERCA de mammifère, protéine dont la séquence est la plus
proche de celle de PfATP6 (SERCA1a, référence pdb de la structure cristalline utilisée : 2DQS).
Dans la structure 2DQS, la protéine a été co-cristallisée avec la thapsigargine. La construction
du modèle de PfATP6 a donc été faite en plusieurs étapes, en utilisant plusieurs outils de
modélisation moléculaire : 1) alignement des séquences avec le logiciel ClustalW, 2)
construction en 3D de la protéine avec le logiciel Modeller et 3) vérification de sa qualité
stéréochimique avec Procheck. Enfin, la superposition de la structure cristalline de la SERCA1a
et de notre modèle PfATP6 montre un arrangement très proche des hélices M1 à M10, où est
localisé le site d’inhibition de TG.
Superposition de PfATP6 (bleu) et 2DQS (vert) avec TG (rose) dans son site
Nous avons ensuite utilisé le logiciel AutoDock4 pour calculer l’affinité de diverses molécules
antipaludiques pour la protéine PfATP6.
La validation préalable de la construction du modèle de PfATP6 et du protocole de docking par
l’utilisation des inhibiteurs connus de SERCA1a est également décrite ci-après.
126

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
2. Etude de docking de molécules antipaludiques et leurs dérivés avec la protéine PfATP6
Les molécules antipaludiques étudiées ici peuvent être divisées en deux groupes principaux :
celles comportant une liaison peroxyde (artémisinine et ses dérivés, trioxolanes, tétraoxanes) et
celles comportant un noyau quinoléine (chloroquine, quinine, méfloquine). Les trioxaquines,
contenant à la fois un trioxane et une 4-aminoquinoléine, appartiennent à ces deux groupes. A
titre de contrôle, nous avons également considéré des analogues inactifs de certaines molécules
antipaludiques (figure ci-dessous).
Pour l’analyse des résultats, nous nous sommes intéressés à l’énergie d’association (Ebind,
kcal/mol) et à la constante d’affinité (Ki, µM) de chaque drogue pour PfATP6. Une différence
d’énergie d’association a été considérée comme significative à partir de 2.0 à 2.5 kcal/mol.[2]
L’affinité des molécules testées a été corrélée avec leur activité in vitro sur Plasmodium
falciparum.
Quelques structures utilisées pour l’étude de docking avec PfATP6
127

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
Artémisinine et dérivés
L’étude de docking de l’artémisinine 1, ainsi que de ses dérivés 2 à 5, avec la protéine PfATP6
indique que ces molécules se positionnent préférentiellement dans le site de TG. Les dérivés de
l’artémisinine, dont la deoxyartémisinine (5) qui ne possède pas de peroxyde, ont tous la même
localisation et adoptent des orientations très similaires. Par ailleurs, l’af
la protéine est essentiellement basée sur une interaction de type
hydrophobe, quelle que soit la structure du composé. Aucune
corrélation n’a pu être établie entre l’affinité prédite pour
PfATP6 et l’activité in vitro des dérivés étudiés. En particulier,
la deoxyartémisinine (5, Ebind -6.9 kcal/mol), molécule
dépourvue d’activité antipaludique, présente la même affinité
que celle prédite pour l’artémisone (3, Ebind -6.7 kcal/mol), l’un
des dérivés les plus actifs de l’artémisinine.
Il est également intéressa
finité de ces dérivés pour
nt de noter que l’affinité de l’artémisinine et de ses dérivés pour
oquine (15), dans leurs différents états de
) et le tétraoxane RKA216 (9), deux peroxydes synthétiques très
comparable à celle de
Superposition de l’artémisinine et de la déoxyartémisinine (en
clair) dans le site de TG
PfATP6 est significativement moins importante que celle de TG (Ebind -10.0 kcal/mol).
Antipaludiques comportant un noyau quinoléine
La chloroquine (13), la quinine (14) et la méfl
protonation, présentent une affinité pour la protéine qui est du même ordre que celle de
l’artémisinine (Ebind -7.0 à -8.3 kcal/mol). Ce résultat est assez inattendu, dans la mesure où
PfATP6 n’est pas impliquée dans leur mécanisme d’action antipaludique. L’interaction de ces
molécules dans le site de TG est principalement de type hydrophobe.
Peroxydes synthétiques
Le trioxolane OZ277 (7
efficaces, ont été conçus dans le but de mimer l’activité de l’artémisinine.[3,4] C’est la raison
pour laquelle nous les avons inclus dans notre étude de docking.
Le trioxolane OZ277, en particulier, présente une activité in vitro
l’artémisinine (IC50 2.7 et 11 nM, respectivement). L’étude de docking montre que OZ277
présente une énergie de liaison avec PfATP6 très proche de celle de l’artémisinine (-7.9
kcal/mol). Par contre, OZ277 n’inhibe pas PfATP6 lorsque celle-ci est surexprimée dans des
ovocytes.[5] Il n’y a donc pas de corrélation entre l’affinité prédite de OZ277 pour la protéine et
l’inhibition mesurée dans les ovocytes.
128

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
Trioxaquines
Selon la position relative du peroxyde et de l’amine par rapport au plan moyen du cyclohexane,
la trioxaquine PA1103 existe sous la forme de deux diastéréo-isomères séparables (cis ou trans).
La même diastéréo-isomérie existe pour la trioxaquine DU1301 : dans l’isomère cis, la liaison
peroxyde se trouve en position axiale et l’amine se trouve en position équatoriale. Dans
l’isomère trans, ils sont tous les deux en positions équatoriales. Chaque diastéréo-isomère de la
trioxaquine DU1301 se présente sous la forme d’un couple d’énantiomères, en fonction de la
configuration des atomes de carbone de jonction des cycles trioxanes et cyclohexene ((RS ou
SR)). Les diastéréo-isomères de PA1103 et DU1301 ont été séparés et ont montré la même
activité in vitro.[6,7]
L’énergie de liaison calculée pour les diastéréo-isomères cis et trans de la trioxaquine DU1301
(10a et 10b) est de -9.9 et -9.3 kcal/mol, respectivement. Ces énergies sont très proches de celle
de TG (-10.0 kcal/mol) et significativement plus importantes que celle de l’artémisinine (-7.6
kcal/mol).
Les deux diastéréo-isomères de la trioxaquine PA1103, qui ont la même activité in vitro,
adoptent des conformations très différentes dans le site de TG (figure ci-dessous). Dans le cas
de la cis-PA1103, le noyau quinoléine se trouve à l’emplacement de l’artémisinine et le trioxane
à l’extérieur du site ; pour la trans-PA1103, c’est le trioxane qui occupe la cavité intérieure et le
noyau quinoléine est totalement sorti de la protéine.
Conformations prédites par AutoDock4 pour la trioxaquine cis-PA1103 (gauche) et la trans-PA1103 (droite)
Les résultats de docking obtenus pour les trioxaquines ne sont pas corrélés pas à leur activité
antipaludique mesurée in vitro. Cette étude laisse penser qu’une protéine avec un site stéréo-
spécifique telle que PfATP6 n’est pas une cible pour les trioxaquines.
129

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
3. Conclusion
Notre étude ne confirme pas l’existence d’une corrélation entre l’affinité calculée pour la
protéine PfATP6 et l’activité in vitro des molécules testées. De plus, les constantes d’inhibition
mesurées dans les ovocytes, en particulier en ce qui concerne l’artémisinine et ses dérivés, ne
peuvent être reliées à l’affinité pour PfATP6 calculée par AutoDock4.
L’inhibition de PfATP6 par l’artémisinine a été présentée dans la littérature comme dépendante
du fer.[1] D’autres travaux décrivant des coupures oxydantes de SERCA1a en présence de fer et
de H2O2 ont été publiés.[8] Sur cette base, une étude de docking prenant en compte le rôle du fer
dans l’inhibition de PfATP6 pourrait être envisagée.
Toutefois, afin de valider PfATP6 comme cible de l’artémisinine et de ses dérivés, les résultats
obtenus dans les membranes d’ovocytes de xenopes, qui constituent un milieu mal défini et
difficilement reproductible, devraient être confirmés par des mesures d’inhibition sur la protéine
isolée et purifiée.
Bibliographie
[1] U. Eckstein-Ludwig, R. J. Webb, I. D. A. Van Goethem, J. M. East, A. G. Lee, M. Kimura, P. M. O'Neill, P. G. Bray, S. A. Ward, S. Krishna. Artemisinins Target the SERCA of Plasmodium falciparum. Nature 2003, 424, 957-961. [2] R. Huey, G. M. Morris, A. J. Olson, D. S. Goodsell. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J. Comput. Chem. 2007, 28, 1145-1152. [3] J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A. Charman, F. C. K. Chiu, J. Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padmanilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler, S. Wittlin, W. N. Charman. Identification of an Antimalarial Synthetic Trioxolane Drug Development Candidate. Nature 2004, 430, 900-904. [4] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. Shone, D. Stanford, P. Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer, S. Lozanom, M. Jesús, D. Gargallo, S. A. Ward, P. M. O'Neill. Two-Step Synthesis of Achiral Dispiro-1,2,4,5-Tetraoxanes with Outstanding Antimalarial Activity, Low Toxicity, and High-Stability Profiles. J. Med. Chem. 2008, 51, 2170-2177. [5] A.-C. Uhlemann, S. Wittlin, H. Matile, L. Y. Bustamante, S. Krishna. Mechanism of Antimalarial Action of the Synthetic Trioxolane Rbx11160 (OZ277). Antimicrob. Agents. Chemother. 2007, 51, 667-672. [6] F. Benoit-Vical, J. Lelièvre, A. Berry, C. Deymier, O. Dechy-Cabaret, J. Cazelles, C. Loup, A. Robert, J.-F. Magnaval, B. Meunier. Trioxaquines Are New Antimalarial Agents
130

Chapter 6: Docking study of structurally diverse antimalarial drugs targeting PfATP6
131
Active on All Erythrocytic Forms, Including Gametocytes. Antimicrob. Agents. Chemother. 2007, 51, 1463-1472. [7] F. Coslédan, L. Fraisse, A. Pellet, F. Guillou, B. Mordmüller, P. G. Kremsner, A. Moreno, D. Mazier, J.-P. Maffrand, B. Meunier. Selection of a Trioxaquine as an Antimalarial Drug Candidate. Proc. Nat. Acad. Sci. U.S.A 2008, 105, 17579-17584. [8] C. Montigny, C. Jaxel, A. Shainskaya, J. Vinh, V. Labas, J. V. Møller, S. J. D. Karlish, M. le Maire. Fe2+-Catalyzed Oxidative Cleavages of Ca2+-ATPase Reveal Novel Features of Its Pumping Mechanism. J. Biol. Chem. 2004, 279, 43971-43981.

132

DOI: 10.1002/cmdc.200900200
Docking Studies of Structurally Diverse Antimalarial DrugsTargeting PfATP6: No Correlation between in silico BindingAffinity and in vitro Antimalarial Activity.Fatima Bousejra-El Garah,[a] Jean-Luc Stigliani,[a] Fr!d!ric Cosl!dan,[b] Bernard Meunier,[b] andAnne Robert*[a]
Introduction
Artemisinin (1), a 1,2,4-trioxane-derived antimalarial drug ex-tracted from the Chinese herb Artemisia annua, is highly activeagainst both chloroquine-sensitive and chloroquine-resistantstrains of Plasmodium falciparum (Pf), the most virulent speciesof Plasmodium that causes malaria in humans.[1,2] In order todelay the emergence of drug resistances, and to interrupt thetransmission of P. falciparum, a drug regime using artemisininderivatives, artesunate or artemether, in combination withmore slowly eliminated antimalarial drugs is now recommend-ed (artemisinin combination therapy, ACT).[3, 4] In spite of theseefforts, recent clinical and molecular studies suggest the emer-gence of ACT-resistant P. falciparum infections.[5]
Although the antimalarial mechanism of action of artemisi-nin (1) and related synthetic peroxides is still a matter ofdebate, it was established that the peroxide bond of artemisi-nin is essential for its activity.[1, 2] In the 1990s, it was shownthat iron(II) ions catalyze the reductive cleavage of the perox-ide bond, leading to the formation of oxy radicals that rapidlyrearrange to more stable carbon-centered radicals.[6,7] Thesemore stable radicals were thought to covalently bind heme, aproduct of host cell hemoglobin digestion.[8]
Supporting this hypothesis, covalent heme–artemisinin ad-ducts have been isolated and characterized.[9,10] The involve-ment of iron(II) heme in the mode of action of artemisinin wasfurther supported by in vivo studies. Heme–artemisinin ad-ducts were identified in the spleen and urine of P. vinckei in-fected mice orally treated with artemisinin, however, these ad-ducts were absent in the spleen and urine of healthy micetreated with artemisinin under the same condition.[11] Addition-ally, heme–artemisinin adducts are able to inhibit in vitro hemepolymerization and are themselves unable to polymerize,[12] aprocess that also contributes to parasite death. These resultsconfirm the importance of the alkylating ability of peroxide-based antimalarial drugs. Artemisinin (1) also interacts with
specific parasite proteins,[13] however, protein–drug couplingproducts have yet to be characterized.
In addition to heme alkylation, an alternative mechanism ofaction for artemisinin has been suggested. It involves PfATP6, aPf-sarco/endoplasmic reticulum calcium ATPase, homologousto mammalian sarco/endoplasmic reticulum calcium ATPase(SERCA1a). Krishna and co-workers reported that artemisinininhibits PfATP6 overexpressed in Xenopus oocytes.[14] An antag-onistic interaction was also reported between artemisinin andthapsigargin (TG), a specific SERCA inhibitor,[14] however, theseresults were not confirmed by a more recent publication.[15]
Additional studies in Xenopus oocytes suggest that mutationsin position 263 can modulate the affinity of artemisinin forPfATP6.[16] Mutation S796N was also proposed to be related todecreased sensitivity of the parasite to artemether, an artemisi-nin derivative.[17] However, a genetic analysis indicated the ab-sence of mutation in atp6 genes of artemisinin-resistant P. falci-parum and P. chabaudi, thus contradicting this assumption.[18]
In order to provide information on the possible role of thePfATP6 calcium pump in the mechanism of action of artemisi-nin and related compounds, we performed a docking study ofa series of structurally different antimalarial drugs into thethapsigargin-binding cleft of a PfATP6 model. In contrast to arecent report,[19] our study was not limited to artemisinin deriv-atives, but included other peroxide- and quinoline-based anti-malarial agents. The mechanism of action of the latter class ofantimalarial drugs does not involve PfATP6 inhibition, because
PfATP6, a calcium-dependent ATPase of Plasmodium falcipa-rum, is considered the putative target of the antimalarial drugartemisinin and its derivatives. Herein, the 3D structure ofPfATP6 was modeled on the basis of the crystal structure ofSERCA1a, the mammalian homologue. Model validation wasachieved using protein structure checking tools. AutoDock4
was used to predict the binding affinities of artemisinin (andanalogues) and various other antimalarial agents for PfATP6,for which in vitro activity is also reported. No correlation wasfound between the affinity of the compounds for PfATP6 pre-dicted by AutoDock4 and their antimalarial activity.
[a] F. B.-E. Garah, Dr. J.-L. Stigliani, Dr. A. RobertCNRS–LCC (Laboratoire de Chimie de Coordination)205, route de Narbonne, 31077 Toulouse (France)Fax: (+33)561-553-003E-mail : [email protected]
[b] Dr. F. Cosl!dan, Dr. B. MeunierPalumed S.A. , Zone Industrielle de Vic de Graves3 rue de l’Industrie, 31320 Castanet–Tolosan (France)
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim 1469
133

quinoline derivatives interact with heme, thus preventing he-mozoin formation.[12,20,21] Our objective was to determinewhether the binding affinity of the studied drugs to PfATP6can be significantly correlated to their in vitro antiplasmodialactivity.
Herein, we report the construction of a PfATP6 model from amammalian SERCA1a X-ray crystal structure, and the results ofthe docking study of structurally different antimalarial agentswith this PfATP6 model.
Results and Discussion
Homology modeling
The peptide sequence of PfATP6 is known (1228 amino acids)and available from the PlasmoDB website,[22] but no crystalstructure has been obtained so far. PfATP6 presents the high-est sequence homology with mammalian SERCAs (43.5%). Thetertiary structure of PfATP6 was therefore modeled using thecrystal structure of the SERCA1a homologue, extracted fromrabbit skeletal muscle (PDB:2DQS, resolution 2.5#). [23] In the2DQS structure, the protein was co-crystallized with thapsigar-gin (TG), a sesquiterpene lactone extracted from the plant spe-cies Thapsia garganica, which is a highly specific inhibitor ofCa2+-dependent pumps.[24,25] SERCA proteins actively transportCa2+ ions from the cytosol to the sarco/endoplasmic reticulumagainst a large concentration gradient.[26] SERCA1a contains994 amino acids, organized in ten transmembrane helices (M1to M10) and three cytosolic domains.
In SERCA1a, a thapsigargin-binding site was identified as acavity located in the transmembrane region and formed bythe M3, M5 and M7 helices. During its catalytic cycle, SERCAundergoes significant conformational changes that involveACHTUNGTRENNUNGinterconversion between the two conformations E1 and E2.[26]
In the E1 conformation, the ATPase presents two high-affinitybinding sites for two Ca2+ ions. These two sites are associatedwith protons in the E2 conformation. Thapsigargin inhibits theCa2+ ATPase by locking the E2 conformation,[27] predominantlythrough hydrophobic interactions.[28]
The sequences of PfATP6 and the rabbit muscle SERCA1awere aligned using ClustalW 2.0.1.[29] The 3D reconstruction ofPfATP6 was performed using Modeller 9 (version 2).[30,31] Thequality of the refined PfATP6 model was assessed with PRO-CHECK.[32] The distribution of the Psi/Phi torsion angles of thebest model is represented by a Ramachandran plot, whichshows 87.0% of residues in most favored regions, 10.0% in ad-ditional allowed regions, 1.6% (18) in tolerated regions and1.4% (15) in disallowed regions (Figure 1). The calculated Ram-
achandran Z-score is !0.168, meaning that there is a goodagreement between the PfATP6 model and the SERCA1a tem-plate. Superposition of the PfATP6 model with the SERCA1acrystal structure (PDB:2DQS) gave an root mean square devia-tion (RMSD) of 0.67 # for the 3880 Ca atoms, indicating highlysimilar arrangements of the M1 to M10 helices, where the TGsite is located (Figure 2).
Model and docking protocol validation
Both the validity of the model construction and the accuracyof the docking protocol was determined by assessing howclosely the lowest predicted energy conformation resemblesthe crystal-binding mode. In a first assay, AutoDock4[33] suc-cessfully found the TG-binding site in PfATP6 and was able togenerate positions and conformations closely related to theTG-binding pose observed in the 2DQS crystal structure. How-ever, because of deviations in the orientation of the octyl sidechain of TG, the lowest energy conformation of TG docked inPfATP6 did not exactly match the TG crystallographic positionin the TG–SERCA1a complex. However, this result can be
Figure 1. Ramachandran plot of PfATP6 model (PROCHECK). Red, mostACHTUNGTRENNUNGfavored regions; yellow, allowed regions, beige, tolerated regions; white,ACHTUNGTRENNUNGdisallowed regions.
1470 www.chemmedchem.org " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemMedChem 2009, 4, 1469 – 1479
MED A. Robert et al.
134

ACHTUNGTRENNUNGexplained; AutoDock4 is known to fail when ligands containmore than 15–20 degrees of freedom, since TG contains 19ACHTUNGTRENNUNGrotatable bonds the lack of correlation is not surprising.[33]
To circumvent this difficulty, TG was docked using a two-step procedure. In a first calculation, a TG derivative lackingthe octyl chain was docked (nine degrees of freedom). In asecond step, the octyl chain was added with full flexibility tothe best ranked pose previously found (11 rotatable bonds, in-cluding 8 in the side chain). This method led to a TG confor-mation very close to the X-ray structure of TG in SERCA1a(RMSD=1.68# ). From the result, AutoDock4 local minimiza-tion option was then applied for overall relaxation of the TGligand in the cavity of PfATP6 (Figure 3a). The polycyclic skele-ton of TG is located in a cavity in PfATP6, while the propylacyl-
oxy and 2-buten-2-acyloxy substituents are deeply buried inchannels. No H bond was detected between TG and the pro-tein, but interactions closer than 3 # are possible between thepolycyclic skeleton of the drug and many hydrophobic aminoacids: Ile261, Leu263, Phe264, and Leu268 on one side, andIle973, Ile977, Ile981, Leu1040, Ile1041, Leu1046, Ile1050 onthe other side.
AutoDock4 local minimization of TG in the binding cleft ofSERCA1a was also performed. The predicted inhibition con-stants (Ki) of TG against PfATP6 and SERCA1a were in the samerange: 50 and 24 nm, respectively. The same procedure of cal-culating the docking conformation followed by local minimiza-tion was used to allow comparison of results with all com-pounds tested.
In order to validate our model and docking protocol, wealso docked cyclopiazonic acid (CPA) and ouabain (OUA) intothe TG site of PfATP6. CPA, a secondary metabolite found incertain fungi, is also a Ca2+ ATPase inhibitor, but the CPA-bind-ing site is different to that of TG.[34] OUA is a highly polar ste-roid derivative and a specific Na+/K+ ATPase inhibitor.[35] CPAand OUA interaction in the TG site can therefore be considerednegative controls. As expected, docking results showed thatCPA and OUA exhibit low binding affinities for PfATP6 andSERCA1a. Their predicted Ki values were more than threeorders of magnitude higher than that of TG against PfATP6(CPA, Ki=99 mm; OUA, 512 mm) and SERCA1a (CPA, 124 mm;OUA, 2.4 mm). These preliminary results with TG, CPA and OUAsupport the validity of both the constructed PfATP6 model andthe docking protocol.
Docking studies with antimalarial drugs
Despite very high antimalarial activities of artemisinin and itssemisynthetic derivatives, their half-lives in plasma are short,and combination therapy with an antimalarial drug that iseliminated slowly is required to maximize cure rates.[2b] To cir-cumvent the drawbacks of artemisinin, many research groupshave designed and prepared fully synthetic peroxide-contain-ing analogues. Some of these compounds are highly potentantimalarial agents; among them are the trioxaquines,[36–38]
1,2,4-trioxolanes, such as OZ277,[39,40] and 1,2,4,5-tetraox-anes.[41,42] As these compounds were designed to mimic arte-misinin reactivity, we studied their interaction with PfATP6.
The compounds used in this study can be divided into twomajor groups based on their chemical structures and theirACHTUNGTRENNUNGreactivities toward iron: peroxide-containing drugs (1,2,4-tri-ACHTUNGTRENNUNGoxane, 1,2,4-trioxolane and 1,2,4,5-tetraoxane derivatives) con-sidered as artemisinin mimics, and quinoline-based drugs. Tri-oxaquines DU1301 (10), DU1302 (11) and PA1103 (12) arehybrid molecules with a dual antimalarial mode of action, con-taining both a 1,2,4-trioxane and a 4-aminoquinoline moiety.[43]
Inactive analogues of antimalarial drugs, such as deoxyartemi-sinin (5) or carbaOZ277[44] (8), were also studied for compari-son. For amine-containing drugs, we considered both the freebase and protonated forms, which are the major species atphysiological pH.
Figure 2. Superimposition of PfATP6 model (blue), SERCA1a (pink) with TGin its binding site.
Figure 3. a) Best ranked docked conformation of TG in its binding site inPfATP6; b) Best-ranked docked conformation of artemisinin in TG site ofPfATP6; c) The lipophylic aminoacids surrounding artemisinin in PfATP6.H bond is depicted as a green dashed line.
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemmedchem.org 1471
Docking Studies of Antimalarial Drugs
135

The predicted binding affinities of these compounds withPfATP6 and SERCA1a were evaluated using their binding freeenergies (Ebind, kcalmol!1) and inhibition constants (Ki, mm). Weconsidered the binding free energy of the best ranked confor-mations as the main parameter for analysis of AutoDock4ACHTUNGTRENNUNGresults. A difference of 2.0–2.5 kcalmol!1 was considered signif-icant.[33] Data are reported in Table 1. The affinity values werecorrelated with the in vitro antimalarial activity of the drugs,evaluated on FcM29, a chloroquine-resistant strain of P. falci-parum.
Artemisinin and derivatives
Docking of artemisinin (1) to the PfATP6 model indicated thatthe drug binds the protein preferentially in the TG-binding site(Figure 3b). However, as artemisinin is a smaller molecule thanTG, it occupies only a small portion of the TG site. Four semi-synthetic artemisinin derivatives (2–5) were also docked in thePfATP6 model. Three of these derivatives are active against Pf:
dihydroartemisinin (2),[45] which is the biological metabolite ofartemether and artesunate, artemisone (3), a highly activesecond-generation derivative currently under clinical develop-ment,[46, 47] and MP118 (4), a C10-substituted aryl artemisininderivative with a good antimalarial activity.[48] Deoxyartemisinin(5), which lacks the crucial peroxide bond, is inactive.[1]
The docking studies with artemisinin and its derivatives 2–5(a single cluster) gave protein binding in exactly the same loca-tion and with consensus orientations. An extensive network ofhydrophobic interactions was observed between artemisininand the protein TG site (Figure 3c). The C4 of artemisinin wasclose to NH of Ile1041 (2.8# ), while the C8 and C9 methylgroup were close to the Cg of Ile977 (3.1 and 3.3# , respective-ly). C8 was also close to Phe264 (3.1# ). The distance betweenthe C9 methyl group and Ca of Ile977 was 2.8# . The distancesbetween C6 methyl group of artemisinin and the methylgroups of Ile261 and Leu1040 were 3.2 and 3.5# , respectively.The C5 of artemisinin was in the vicinity of the hydrophobicside chains (methylene groups) of Asn1039 and Lys260 (3.5
1472 www.chemmedchem.org " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemMedChem 2009, 4, 1469 – 1479
MED A. Robert et al.
136

and 3.1# , respectively, from Cb). The distance between the O1and O2 and one of the Leu1046 methyl groups were 3.2 #and3.7# , respectively, while O3 was 3.0 # from the Leu263 methylgroup. In addition, an H bond was proposed between the O1of the peroxide function of artemisinin and the NH of Ile1041.All the tested artemisinin derivatives, including the inactive de-oxyartemisinin (5), were predicted by AutoDock4 to interactwith PfATP6 in a similar manner to artemisinin. The peroxidebond in compounds 1–4 is orientated toward the inner part ofthe cavity. Superimposition of docked conformations showsthat the ether oxygen atom of deoxyartemisinin (5) is locatedbetween the two oxygen atoms of the artemisinin peroxidebond (Figure 4).
As predicted by AutoDock4, artemisinin (1) interacts withPfATP6 with a binding energy of !7.6 kcalmol!1 and an inhibi-tion constant of 2.66 mm (Table 1, line 2). The affinities of arte-misinin (1) for PfATP6 and SERCA1a were compared (for clarity,affinity parameters for SERCA1a are not shown in Table 1). No-tably, the binding energy of artemisinin for SERCA1a wasfound to be !7.2 kcalmol!1, which is not significantly differentthan the energy calculated for PfATP6 (!7.6 kcalmol!1), al-though artemisinin reportedly does not inhibit SERCA1a.[14]
Artemisinin (1) was found to exhibit a binding affinity forPfATP6 higher than that of artemisone (3) (1, Ki=2.66 mm; 3,Ki=11.78 mm, Table 1, line 4), although the latter is more activeagainst the FcM29 strain of P. falciparum (1, IC50=11 nm; 3,IC50=0.7 nm). It has also been reported that artemisone (3) ismore efficient than artemisinin (1) at inhibiting PfATP6 over ex-pressed in Xenopus oocytes (1, Ki=170 nm; 3, Ki=1.7 nm).[49]
Interestingly, AutoDock4 data suggest similar binding energiesof artemisone (3) and deoxyartemisinin (5) (3, Ebind=!6.7 kcalmol!1; 5, Ebind=!6.9 kcalmol!1), without significantdifference from artemisinin (1) (Ebind=!7.6 kcalmol!1), despitethe fact that derivative 3 is highly active against Plasmodium,and derivative 5 is totally inactive.An artemisinin linked to an acridine moiety at the C10 posi-
tion (compound 6) was used to image the distribution of arte-misinin in infected erythrocytes.[14] In attempt to evaluate therelevance of such an acridine-labeled fluorescent derivative,we included the monoprotonated form of this artemisinin–acri-dine conjugate 6H in the present study (the alkylamine func-tion should indeed be protonated at physiological pH). The
top ranked conformations of artemisinin–acridine conjugate6H exhibited a binding affinity for the protein very close tothat of thapsigargin itself (Ki=0.06 cf. 0.05 mm, Ebind=!9.8 cf.!10.0 kcalmol!1, for 6H and TG, respectively, Table 1, lines 1and 7), and significantly higher than that of artemisinin (1, Ki=2.66 mm, and Ebind=!7.6), despite the fact that artemisinin andartemisinin–acridine conjugate exhibit similar in vitro antima-larial activities.[50] Note that the docking results obtained for6H were rather scattered (high number of clusters) due to thehigh degree of freedom in the molecule. However, in all cases,the artemisinin fragment of 6H was located in the TG site in ahighly conserved position, and in an environment similar tothat of the parent artemisinin, i.e. close to Leu263, Phe264,Leu1040 and Ile1041. The top-ranked conformations of 6H ex-hibited hydrophobic interactions between the acridine moietywith Ile977 and Ile981, Gly1043, Leu1046, and Leu1047 closerthan 4# . In clusters with a lower affinity (Ebind~!9.5 kcalmol!1), the acridine moiety of 6H was shifted and thesurrounding environment consisted of Ile275, Ile981, Gly982,
Figure 4. Superimposition of docked artemisinin (dark grey) and deoxyarte-misinin (light grey).
Table 1. IC50 values against the FcM29 P. falciparum strain and AutoDock4predicted binding affinities and inhibition constants for the PfATP6 mod-els.[a]
in vitro antimalarial activity AutoDock4 resultsDrug IC50 [nm] PfATP6
ligand[b]
Ebind[kcalmol!1]
PredictedKi [mm]
1 Thapsigargin 4300 TG !10.0 0.052 Artemisinin 11 1 !7.6 2.663 Dihydroartemisinin 2.4[c] 2 !7.1 6.44 Artemisone 0.7 3 !6.7 11.785 MP118 25[d] 4 !7.4 3.726 Deoxyartemisinin 5000–
100005 !6.9 8.71
7 Artemisinin–acridine 8–20[e] 6H !9.8 0.068 OZ277 2.7[f] 7 !7.9 1.569 OZ277·mesylate 2 7H !8.2 1.04
10 carbaOZ277 6400[g] 8 !7.7 2.2111 RKA216 5.2[h] 9 !8.3 0.8912 cis-DU1301 8 10a !9.9 0.0613 trans-DU1301 8 10b !9.3 0.1614 cis-DU1302·citrate 8 11a !9.6 0.0915 trans-DU1302·citrate 8 11b !9.8 0.0716 cis-PA1103 12 12a !7.9 1.717 trans-PA1103 8 12b !7.6 2.718 Chloroquine 420[i] 13 !8.3 0.8319 Chloroquine·diphosphate 518 13H !7.0 7.020 13H2 !6.1 33.921 Quinine 14 !7.7 2.4022 Quinine·chlorhydrate 217 14H !7.5 3.2623 Mefloquine 15 !7.0 7.224 Mefloquine·chlorhydrate 6[j] 15H !7.6 2.73
[a] Unless noted, IC50 values were evaluated in the present study on thechloroquine-resistant FcM29-Cameroon P. falciparum strain. [b] For agiven ligand, for example 13, 13H, and 13H2 stand for the free base, mo-noprotonated, and diprotonated species, respectively. [c] Measured onW2-Indochina cloroquine resistant strain.[44b] [d] Pf strain not given.[14]
[e] Reference [49] .[f] Measured on K1-Thailand chloroquine resistantstrain.[39] [g] Measured on K1-Thailand chloroquine resistant strain.[44]
[h] Measured on 3D7 chloroquine sensitive strain.[41] [i] Measured for theracemic drug.
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemmedchem.org 1473
Docking Studies of Antimalarial Drugs
137

Val984 and Ala985, which are amino acids located at the sur-face of the protein.
It should be noted that in one predicted conformation of6H that had binding energy and inhibition constant valuesclose to artemisinin (Ebind=!7.6 kcalmol!1, Ki=2.76 mm), theacridine moiety was located completely outside the protein,and had no significant interactions with amino acids. Thus, thehydrophobic interaction of the acridine residue with thePfATP6 amino acids seems to be the main reason for the high-est binding energy of !9.8 kcalmol!1 for the protonated arte-misinin–acridine conjugate 6H.[51] This result indicates thatusing an acridine probe to localize a small molecule such as ar-temisinin may not be appropriate, and may provide biaseddata concerning the interaction of an antimalarial drug with abiological target.
The affinities of artemisinin derivatives 1–6H for PfATP6, ascalculated using AutoDock4, clearly does not correlate withthe antimalarial activities of these drugs.
Quinoline-based antimalarial drugs
Chloroquine is clinically used as a racemate (R & S). Mefloquine,which contains two stereogenic centers, is also used as a race-mic mixture (11S,12R & 11R,12S). This is possible as both enan-tiomers of quinoline antimalarial drugs have the same antima-larial activity within the limits of experimental variability.[52] TheS enantiomer of chloroquine, and the (11R,12S)-mefloquineACHTUNGTRENNUNGenantiomer in which the C-OH is in the R configuration as inACHTUNGTRENNUNGquinine (14), were selected for docking studies.
The dibasic drug, chloroquine, was tested as the free base(13), monoprotonated on the tertiary alkyl amine (13H, pKa=10.2) and diprotonated (13H2, 4-aminoquinoline group pKa=8.2). Chloroquine (13) exhibits a predicted binding energy toPfATP6 of !8.3 kcalmol!1, comparable to that of artemisinin(Table 1, line 18). There is good shape complementarity be-tween the TG site and the top-ranked conformation; the quin-oline moiety, stacked with Phe264, occupies the cavity whereartemisinin (1) was previously found. The C7 chloro group is inthe place of C6 methyl group of artemisinin close to Ile261.The diethylamino chain of chloroquine interacts with the hy-drophobic channel where the octyl chain of TG is located.Minimized binding energies obtained with 13H and 13H2 are!7.0 and !6.1 kcalmol!1, respectively, indicating a slight butprobably significant decrease in affinity of chloroquine for theprotein induced by protonation. Ki values of 0.83, 7.0, and33.0 mm for 13, 13H and 13H2, respectively, support this analy-sis.
Docking conformations for the quinine free base (14) and itsquinuclidine-protonated derivative 14H were found to be su-perimposable. As in the case of chloroquine (13), the quinolinemoiety of quinine resides between the two channels of theTG-binding site, while the quinuclidine residue is located deepin the cavity. No H bond was identified. Calculated binding en-ergies were !7.7 and !7.5 kcalmol!1, respectively, and Kivalues were 2.40 and 3.26 mm for 14 and 14H, respectively.Calculated binding conformations of neutral mefloquine (15)
and its protonated piperidine form (15H) were totally superim-
posable with binding values of !7.0 and !7.6 kcalmol!1, re-spectively. Unlike quinine, the hydroxy group of derivative 15and 15H forms an H bond with the carbonyl of Ile1041. Calcu-lated binding energies were !7.0 and !7.6 kcalmol!1, and Kivalues were 7.20 and 2.73 mm for 15 and 15H, respectively.In summary, all quinoline drugs, either in the base or mo-
noprotonated form, exhibit similar binding affinity values forthe protein (Ebind=!7.0 to !8.3 kcalmol!1), with predominanthydrophobic interactions. Surprisingly, these affinity valueswere not significantly different to that of artemisinin (1, Ebind=!7.6 kcalmol!1) despite the fact that antimalarial quinolines donot target PfATP6. Indeed, it is accepted that this class of com-pounds exert their activity by interacting with heme, prevent-ing hemozoin formation.[12,53]
Synthetic peroxide-containing drugs
Among synthetic peroxides, OZ277 (7) is a highly potent anti-malarial agent. The pharmacophore of this drug is a 1,2,4-triox-olane, rather than the 1,2,4-trioxane ring of artemisinin (1).[39]
In our study, no significant difference was found betweenthe conformation of OZ277 (7), and its protonated derivative7H. Both drugs were predicted to interact with PfATP6 in theTG-binding pocket, predominantly through hydrophobic inter-actions (no H bond were identified), with the adamantanemoiety located in the artemisinin site. In contrast to artemisi-nin derivatives, the peroxide bond of 7 is directed towards thebinding site opening. As reported in Table 1 (lines 2, 8 and 9),docking with PfATP6 gave close binding energies for both neu-tral (7) and protonated (7H) forms of the trioxolane (7, Ebind=!7.9 kcalmol!1; 7H, Ebind=!8.2 kcalmol!1). Again, the bindingvalue is similar to that of artemisinin. However, OZ277 showedpoor inhibition of PfATP6 over expressed in Xenopus oocytes(7, Ki=7700 nm; 1, Ki=80–170 nm).[54]
As the peroxide bond of OZ277 was shown to be crucial forits antiplasmodial activity, we also docked its inactive ana-logue, 1,3-dioxolane carbaOZ277 (8), in which one of the twooxygen atoms of the peroxide bond was replaced by a carbonatom.[44] The adamantane and 5-membered ring are closely su-perimposed, whereas the p-substituted cyclohexane residueadopts a slightly different orientation compared with those in7 and 7H. The binding energies of OZ277 (7) and carbaOZ277(8) are similar (7, Ebind=!7.9 kcalmol!1; 8, Ebind=!7.7 kcalmol!1). Again, the binding affinity for the PfATP6 pro-tein does not correlate with the antimalarial activity of OZ277.
The 1,2,4,5-tetraoxane moiety has also received attention re-cently as an interesting pharmacophore for antimalarial agents.One such derivative, RKA216 (9), has in vitro and in vivo activi-ty similar to that of artemisinin (1).[41] RKA216 (9) was dockedwith PfATP6; the results gave a binding affinity close to that ofartemisinin (1, Ebind=!8.3 kcalmol!1; 9, Ebind=!7.6 kcalmol!1,Table 1, line 11). On the other hand, the affinity of RKA216 isalso in the same range as that of OZ277 (7, Ebind=!7.9 kcalmol!1). Interestingly, RKA216 has a spiroadamantaneresidue adjacent to the peroxide bond, as is the case in OZ277.In both cases, the hydrophobic adamantane occupies thesame location in the binding site (Figure 5).
1474 www.chemmedchem.org " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemMedChem 2009, 4, 1469 – 1479
MED A. Robert et al.
138

Trioxaquines
The same docking protocol was used to predict the binding af-finities of trioxaquines DU1301 (10) and PA1103 (12) forPfATP6, which are highly active against all erythrocytic stagesof both sensitive and resistant strains of P. falciparum, and areable to cure orally treated infected mice.[36–38,55] DU1302 (11),which is the monoprotonated derivative of DU1301, was alsodocked with PfATP6 for comparison. Trioxaquines are synthetichybrid molecules built with both a 1,2,4-trioxane ring and an
aminoquinoline residue.[43] It should be noted that compounds10–12 exist as cis (10a–12a) and trans (10b–12b) diastereo-isomers, the amine and the peroxide substituents being eitheron the same side or on the opposite side of the cyclohexanemean plane. For all trioxaquines, the two diastereoisomers aand b have the same in vitro activity against Plasmodiumstrains.[37,38,55] Furthermore, the four stereoisomers of DU1301(10), two enantiomers of 10a and two enantiomers of 10b,were also tested separately and they were shown to exhibitthe same activity in vitro.[55]
Note that each diastereoisomer of DU1301 (10) and DU1302(11), may in principle exist as two conformers, depending onwhether the peroxide and the amine are axial or equatorialwith respect to the cyclohexane ring. Docking studies of cis-DU1301 and cis-DU1302 were carried on the conformers withthe peroxide in the axial position and the amine equatorial,while docking of trans-DU1301 and trans-DU1302 were carriedon the conformers with both amine and peroxide in equatorialpositions.[37] Similarly, the conformer with the axial peroxideand equatorial amine was considered for cis-PA1103 (12), andthe conformer with both amine and peroxide in equatorialACHTUNGTRENNUNGpositions was considered for trans-PA1103. Notably, the 1,4-di-ACHTUNGTRENNUNGaminocyclohexane moiety of PA1103 has a trans configurationwith the two amines in equatorial positions.[38]
Calculated binding energies of the best-ranked conforma-tions of the free bases cis- and trans-DU1301 (10) and the mo-noprotonated forms cis- and trans-DU1302 (11) were in therange of Ebind=!9.3 to !9.9 kcalmol!1 (Table 1, lines 12–15),close to that of TG (Ebind=!10.0 kcalmol!1) and significantlyhigher than artemisinin (Ebind=!7.6 kcalmol!1). Moreover, theKi values were 0.06 and 0.16 mm for the cis- and trans- isomersof DU1301, respectively, compared to 2.66 mm for artemisinin(1), confirming that the affinity of these drugs for PfATP6 ishigher than the affinity of artemisinin.
In the best-ranked conformation, the fused trioxane and cy-clohexene moieties of trans-DU1301 (10b) are located in thepart of the TG site that is occupied by artemisinin (1), whilethe quinoline moiety is outside of the TG cavity. While noH bonds were seen, close hydrophobic interactions betweenthe quinoline ring and methylene residues of the side chainsof Gln267 and Leu268, two amino acids located at the periph-ery of the TG site, were observed. However, several clusters arefound with very close Ebind values, corresponding to conservedconformations of the trioxane moiety, but different positionsof the quinoline residue, due to a possible rotation around the!NH!ACHTUNGTRENNUNG(CH2)2!NH! tether. The conformation of the protonatedderivative 11b is similar to that of 10b.The overall conformation of lowest binding energy of cis-
DU1301 (10a) is similar to that of the trans stereoisomer 10b.In both cases, the positions of the trioxane ring are close al-though the peroxide bond of the cis isomer 10a was turnedtowards the outside the cavity, while that of the trans isomer10b was located deeper inside the pocket. The C7 chlorogroup of derivative 10a is 3.6 # from the Ca of Ala313, andthe quinoline is retained in the hydrophobic cage of Leu268,Ile271 and Ile981 (where the acridine of compound 6H wasACHTUNGTRENNUNGlocated), whereas the quinoline of compound 10b comes out
Figure 5. Best ranked docked conformation of a) OZ277 (7) and b) RKA216(9) in the TG-binding site.
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemmedchem.org 1475
Docking Studies of Antimalarial Drugs
139

of the binding pocket (Figure 6). The conformation of the pro-tonated cis-DU1302 (11a) is very similar to 10a.
Both diastereoisomers of PA1103 (12) were tested. Bindingparameters to PfATP6 were Ebind=!7.9 and !7.6 kcalmol!1
and Ki=1.7 and 2.7 mm for 12a and 12b, respectively. Theseresults are very close to those of artemisinin (1). In contrast toderivative 10a, the best-ranked conformation of 12a indicatesquinoline, and not trioxane, binding in the same location as ar-temisinin (1) in the TG site. There is a H bond between the car-bonyl group of Leu1046 and the cyclohexyl-NH-cyclohexyl of12a, while the trioxane residue is directed out of the bindingsite (Figure 7a).
In the lowest energy cluster for trioxaquine 12b, the quino-line ring is found in the same region of artemisinin (1). Themethyl substituents of the trioxane are at 3.1 and 3.7 #fromLys270 Cb and Ile271 Ca, respectively, and O2 is close to theGln267 carbonyl group (3.0# ). However, for both 12a and12b, other clusters corresponding to very different conforma-tions, that have the trioxane in the artemisinin-binding site, arealso found. These clusters, which correspond to a moleculethat is turned head to tail, have a binding energy value closeto that of the best-ranked conformation (12b, DEbind=0.2–0.3 kcalmol!1; 12a, DEbind=0.6–0.8 kcalmol!1). Two differentposes of 12b are depicted in Figure 8. Compound 12 adoptscompletely different poses in PfATP6 with close bindingenergy values; this same ability was not found for trioxaquine10a. It occurred to a much lower extent for derivative 10b(DEbind "2 kcalmol!1). This feature may be due to the in-creased rigidity and hindrance of the 1,4-diaminocyclohexanelinker of 12 compared to the 1,2-diaminoethane of 10. Trioxa-quine 10 may thus adopt more suitable poses, with lowerbinding energies.
Figure 6. Best-ranked docked conformation of a) cis-DU1301 (10a) andb) trans-DU1301 (10b) in the TG-binding site.
Figure 7. Best-ranked docked conformation of a) cis-PA1103 (12a) andb) trans-PA1103 (12b) in the TG-binding site.
Figure 8. Two different poses of 12b into the TG site of PfATP6.
1476 www.chemmedchem.org " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemMedChem 2009, 4, 1469 – 1479
MED A. Robert et al.
140

Notably, the binding affinities of compounds 10–12 forPfATP6 could not be correlated with their stereochemistry. Thissupports the proposal of binding driven mainly by hydropho-bic interactions and not correlated with stereospecific interac-tions with a chiral protein pocket. Indeed, all the stereoisomersof each trioxaquine have the same in vitro and in vivo anti-ACHTUNGTRENNUNGmalarial activity.[37,55]
Trioxaquines, containing a 1,2,4-trioxane (like artemisininand artemisone) and a 4 aminoquinoline (like chloroquine), aredesigned to kill the Plasmodium parasite via a dual mode ofaction. The endoperoxide bond of the trioxane cycle is cleavedby an iron(II)-catalyzed reduction, generating oxy-radicals thatfurther rearrange into alkyl radicals able to efficiently alkylateheme.[56] Heme–trioxaquine adducts were detected in thespleen of malaria infected mice, orally treated with DU1301 orPA1103.[38,57] This in vivo reactivity is a feature shared with arte-misinin (1)[11] that underlines the relevance of heme alkylationin the mechanism of action of these 1,2,4-trioxane-containingantimalarial drugs. In addition, it has also been reported that,like chloroquine, DU1301 and PA1103 prevent b-hematin for-mation, thus being able to interfere with the heme detoxifica-tion process.[12,38]
Conclusions
In the present work, we generated a model of PfATP6, a sarco/endoplasmic reticulum ATPase of P. falciparum, by homologymodeling. This model was used in docking calculations with aset of structurally diverse antimalarial agents. Most notably, thepredicted binding affinities of these compounds for PfATP6 donot correlate with either their antimalarial activity or the re-ported inhibition of the protein overexpressed in Xenopuslaevis oocytes.
Artemisinin (1) and artemisone (3) are known to react withheme to form covalent heme–drug adducts, which is anotherpossible mode of action for these agents.[58] Alkylation ofheme by OZ277 (7) and other 1,2,4-trioxolanes has also beencorrelated with antimalarial activity, suggesting that heme alky-lation could play a significant role in the mechanism of actionof these peroxide antimalarial agent.[59] Since their capacity toalkylate heme in infected mammals has been demonstrated,the importance of this process for the biological activity ofdrugs remains a credible hypothesis.
It should be noted that the PfATP6 inhibition by artemisininin Xenopus oocytes was reported by Krishna et al. to be irondependent, although the role of iron was not investigated inthe report.[14] Results presented here deal with the affinity ofthese drugs for PfATP6, however, these results do not take intoaccount the possible iron(II)-induced reaction of peroxide-containing drugs with PfATP6.
When discussing the role of iron in Ca2+ pump inhibition,Karlish and co-workers carried out the iron(II)-catalyzed oxida-tive cleavage of a mammalian SERCA by the Fenton reac-tion.[60,61] Analysis of the degradation patterns provided evi-dence for two iron ion sites, one being located in the trans-membrane region, near the TG-binding site. Taking into ac-count these results, a future docking study should include a
possible iron site in the protein structure. Work is in progressto investigate more precisely the role of iron in the putativeACHTUNGTRENNUNGinhibition of PfATP6 by artemisinin and artemisone.
Experimental Section
Artemisinin, thapsigargin, chloroquine diphosphate, quinine chlor-ACHTUNGTRENNUNGhydrate, mefloquine chlorhydrate, cyclopiazonic acid and ouabainwere purchased from Sigma–Aldrich. Trioxaquines DU1301 andPA1103, deoxyartemisinin and artemisone were prepared accordingto published methods.[37,38, 58,62] The synthesis of trioxolane OZ277was reported in Reference [63].
Antimalarial cell-based assay
Chloroquine-resistant P. falciparum strain FcM29-Cameroon waschosen for this study. The parasites were cultured using the modi-fied Trager’s method,[64] in 5% CO2 humid atmosphere at 37 8C,and maintained in vitro in human red blood cells (O#) that werediluted to 2% hematocrit in RPMI 1640 complemented with 5%human AB mixed serum.
The antiplasmodial activities of the test compounds were evaluat-ed by the radioactive microdilution method described by Desjar-dins et al.[65] Drug dilutions were tested in triplicate in 96-wellplates containing cultures of various P. falciparum strains at variousstages of 1% parasitemia and 2% hematocrit. For each test, platesof parasite culture were incubated with drugs at a fixed concentra-tion for 48 h in 5% CO2 humid atmosphere at 37 8C, and radioac-tive hypoxanthine was added to the medium 24 h after incubationcommenced. Artemisinin or artesunate were used as referencedrug. Stock solutions of artemisinin and tested compounds wereprepared in DMSO. All the dilutions were done in DMSO exceptthe last one (1/50), which used RPMI 1640 complemented with 5%human serum at 37 8C. Diluted solutions were added to 96-wellplates containing P. falciparum cultures. Parasite growth was esti-mated by [3H]hypoxanthine incorporation compared to the para-site growth without drug (=100%). Each drug dilution was testedin three separate wells on the same 96-well plate.
Based on data obtained using the scintillation spectrometer, thefollowing formula was used to calculate the inhibitory effect (%):
Inhibition (%)=100!( X!BnCo!Bn )$100
where X=cpm (counts per minute) for one well with the drug,Bn=background noise, Co=control (cpm for one well withoutdrug). The concentration that inhibited 50% parasite growth (IC50)was determined by plotting the drug concentration versus the per-centage of parasite growth inhibition at 48 h of incubation usingGraphPad Prism 4 software.[66]
Computational method
Resources : Docking calculations were performed on a SGIAltix 3700 cluster at the Toulouse University Computer Center(France)
Protein modeling and refinement : The PfATP6 sequence was ob-tained from PlasmoDB.[22] The BLAST server[67] was used to searchthe closest sequence of PfATP6 in the RCSB Protein Data Bank. Thehigh degree of primary sequence identity between the targetPfATP6 and mammalian SERCA1a (43.5% homology) indicates thatcrystal structures of the latter are good models, usable as tem-
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemmedchem.org 1477
Docking Studies of Antimalarial Drugs
141

plates (SERCA1a, EC 3.6.3.8). The SERCA1a 3D structure (in the E2conformation) was obtained from the RCSB Protein Data Bank(PDB: 2DQS).[23] The sequence alignment of PfATP6 with SERCA1aas a reference was performed using ClustalW 2.0.1, applying thedefault parameters.[29] Protein reconstruction was achieved withModeller 9 (version 2) on the entire PfATP6 sequence.[30,31] The re-finement was made with the loopmodel class. To maintain goodcomplementarity between the protein and the ligands, both thap-sigargin and the ACP cofactor, included in the 2DQS PDB file, wereretained during the refinement using the “env.io.hetatm=True”keyword. The quality of each Modeller refined model was evaluat-ed with PROCHECK (version 3.5.4).[32, 68] The final model was chosenfrom both the PROCHECK results and the Modeller energy score.Hydrogen atoms were added and the PfATP6 structure was mini-mized using the Amber (PARM99) force field.
Ligand preparation : When available, the coordinates of antimalarialcompounds were obtained from the Cambridge Structural Data-base (CSD), otherwise they were built and energy minimized usingthe AMMP force field[69] implemented on the VEGA ZZ molecularmodeling package.[70] Prior to docking, all the drugs were energyminimized using the semiempirical PM6 hamiltonian[71] and calcu-lated partial charges. Structures were placed inside the grid boxfor the docking studies.
Docking protocol : Thapsigargin, SERCA inhibitors and antimalarialdrugs were docked into the TG-binding site of the homology-mod-eled PfATP6 using AutoDock4.0.[33] The graphical interface Auto-Dock Tools[72] was used to keep polar hydrogens and add partialcharges for protein using the Gasteiger United charges. The gridmaps for each atom type found in the ligand structures were cal-culated using the auxiliary program AutoGrid4.0. The grid size wasset to 80$80$80 points with 0.375 # spacing, and the grid boxwas centered on the TG-binding site. The Lamarckian genetic algo-rithm (LGA) was selected for ligand conformational searching. De-fault parameters were used, except for the number of energy eval-uations and docking runs, which were set to 25,000,000 and 100,respectively. The resulting docked conformations were clusteredinto families of similar conformations, with a RMSD clustering toler-ance of 2# . Flexible torsion angles in the ligands were assignedwith Autotors, an auxiliary module of AutoDock Tools. The topranked conformation was extracted and redocked using the Auto-Dock4 local minimization option.
Acknowledgements
This work was supported by CNRS and Palumed. F. B.-E.G. is in-debted to the EU-Antimal program for a PhD fellowship. C!lineBerrone and Katia Jonot (Palumed) are acknowledged for techni-cal assistance in evaluation of antimalarial activities. ChristineSalle (Palumed) is acknowledged for the preparation of differentantimalarial drugs. We thanks CALMIP (Calcul Intensif en Midi-Pyr!n!es, Toulouse) for computing facilities.
Keywords: antimalarial agents · artemisinin · enzymes ·inhibitors · molecular modeling
[1] D. L. Klayman, Science 1985, 228, 1049–1055.[2] a) S. R. Meshnick, T. E. Taylor, S. Kamchonwongpaisan, Microbiol. Rev.
1996, 60, 301–315; b) N. J. White, Science 2008, 320, 330–334.[3] N. J. White, F. Nosten, S. Looareesuwan, W. M. Watkins, K. Marsh, R. W.
Snow, G. Kokwaro, J. Ouma, T. T. Hien, M. E. Molyneux, T. E. Taylor, C. I.
Newbold, T. K. Ruebush, M. Danis, B. M. Greenwood, R. M. Anderson, P.Olliaro, Lancet 1999, 353, 1965–1967.
[4] F. Nosten, P. Brasseur, Drugs 2002, 62, 1315–1329.[5] C. Wongsrichanalai, S. R. Meshnick, Emerging Infect. Dis. 2008, 14, 716–
719.[6] G. H. Posner, C. H. Oh, D. Wang, L. Gerena, W. K. Milhous, S. R. Meshnick,
W. Asawamahasadka, J. Med. Chem. 1994, 37, 1256–1258.[7] C. W. Jefford, F. Favarger, M. G. H. Vicente, Y. Jacquier, Helv. Chim. Acta
1995, 78, 452–458.[8] S. R. Meshnick, A. Thomas, A. Ranz, C. M. Xu, H. Z. Pan, Mol. Biochem.
Parasitol. 1991, 49, 181–189.[9] A. Robert, B. Meunier, J. Am. Chem. Soc. 1997, 119, 5968–5969.
[10] A. Robert, J. Cazelles, B. Meunier, Angew. Chem. 2001, 113, 2008–2011;Angew. Chem. Int. Ed. 2001, 40, 1954–1957.
[11] a) A. Robert, F. Benoit-Vical, C. Claparols, B. Meunier, Proc. Natl. Acad. Sci.USA 2005, 102, 13676–13680; erratum: b) A. Robert, F. Benoit-Vical, C.Claparols, B. Meunier, Proc. Natl. Acad. Sci. USA 2006, 103, 3943.
[12] C. Loup, J. Leli%vre, F. Benoit-Vical, B. Meunier, Antimicrob. Agents Chem-other. 2007, 51, 3768–3770.
[13] W. Asawamahasakda, I. Ittarat, Y. M. Pu, H. Ziffer, S. R. Meshnick, Anti-microb. Agents Chemother. 1994, 38, 1854–1858.
[14] U. Eckstein-Ludwig, R. J. Webb, I. D. A. Van Goethem, J. M. East, A. G.Lee, M. Kimura, P. M. O’Neill, P. G. Bray, S. A. Ward, S. Krishna, Nature2003, 424, 957–961.
[15] M. D. P. Crespo, T. D. Avery, E. Hanssen, E. Fox, T. V. Robinson, P. Valente,D. K. Taylor, L. Tilley, Antimicrob. Agents Chemother. 2008, 52, 98–109.
[16] A. C. Uhlemann, A. Cameron, U. Eckstein-Ludwig, J. Fischbarg, P. Isero-vich, F. A. Zuniga, M. East, A. Lee, L. Brady, R. K. Haynes, S. Krishna, Nat.Struct. Mol. Biol. 2005, 12, 628–629.
[17] R. Jambou, E. Legrand, M. Niang, N. Khim, P. Lim, B. Volney, M. T. Ekala,C. Bouchier, P. Esterre, T. Fandeur, O. Mercereau-Puijalon, Lancet 2005,366, 1960–1963.
[18] A. Afonso, P. Hunt, S. Cheesman, A. C. Alves, C. V. Cunha, V. do Rosario,P. Cravo, Antimicrob. Agents Chemother. 2006, 50, 480–489.
[19] M. Jung, H. Kim, K. Y. Nam, K. T. No, Bioorg. Med. Chem. Lett. 2005, 15,2994–2997.
[20] D. J. Sullivan Jr. , I. Y. Gluzman, D. G. Russell, D. E. Goldberg, Proc. Natl.Acad. Sci. USA 1996, 93, 11865–11870.
[21] D. J. Sullivan Jr. , H. Matile, R. G. Ridley, D. E. Goldberg, J. Biol. Chem.1998, 273, 31103–31107.
[22] http://plasmodb.org/plasmo/ (Last accessed, June 19, 2009).[23] a) http://www.rcsb.org/pdb/explore/explore.do?structureId=2DQS;
b) A previous crystal structure was reported by the same authors withpdb code 1VFP, resolution 2.9# : C. Toyoshima, T. Mizutani, Nature 2004,430, 529–535.
[24] O. Thastrup, P. J. Cullen, B. K. Drøbak, M. R. Hanley, A. P. Dawson, Proc.Natl. Acad. Sci. USA 1990, 87, 2466–2470.
[25] J. Lytton, M. Westlin, M. R. Hanley, J. Biol. Chem. 1991, 266, 17067–17071.
[26] C. Toyoshima, H. Nomura, Nature 2002, 418, 605–611.[27] Y. Sagara, G. Inesi, J. Biol. Chem. 1991, 266, 13503–13506.[28] S. Paula, W. J. Ball, Proteins Struct. Funct. Bioinf. Proteins 2004, 56, 595–
606.[29] M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H.
McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson,T. J. Gibson, D. G. Higgins, Bioinformatics 2007, 23, 2947–2948.
[30] A. Sali, T. L. Blundell, J. Mol. Biol. 1993, 234, 779–815.[31] A. Fiser, R. K. Do, A. Sali, Protein Sci. 2000, 9, 1753–1773.[32] R. A. Laskowski, M. W. MacArthur, D. S. Moss, J. M. Thornton, J. Appl.
Crystallogr. 1993, 26, 283–291.[33] R. Huey, G. M. Morris, A. J. Olson, D. S. Goodsell, J. Comput. Chem. 2007,
28, 1145–1152.[34] K. Moncoq, C. A. Trieber, H. S. Young, J. Biol. Chem. 2007, 282, 9748–
9757.[35] A. Kawamura, L. Abrell, F. Maggiali, N. Berova, K. Nakanishi, J. Labutti, S.
Magil, G. Haupert, J. Hamlyn, Biochemistry 2001, 40, 5835–5844.[36] O. Dechy-Cabaret, F. Benoit-Vical, A. Robert, B. Meunier, ChemBioChem
2000, 1, 281–283.[37] O. Dechy-Cabaret, F. Benoit-Vical, C. Loup, A. Robert, H. Gornitzka, A.
Bonhoure, H. Vial, J. Magnaval, J. S!gu!la, B. Meunier, Chem. Eur. J.2004, 10, 1625–1636.
1478 www.chemmedchem.org " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim ChemMedChem 2009, 4, 1469 – 1479
MED A. Robert et al.
142

[38] F. Cosl!dan, L. Fraisse, A. Pellet, F. Guillou, B. Mordm&ller, P. G. Kremsner,A. Moreno, D. Mazier, J.-P. Maffrand, B. Meunier, Proc. Natl. Acad. Sci.USA 2008, 105, 17579–17584.
[39] J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A. Charman, F. C. K. Chiu, J.Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padma-nilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler,S. Wittlin, W. N. Charman, Nature 2004, 430, 900–904.
[40] X. Wang, Y. Dong, S. Wittlin, D. creek, J. Chollet, S. A. Charman, J. SantoTomas, C. Scheurer, C. Snyder, J. L. Vennerstrom, J. Med. Chem. 2007, 50,5840–5847.
[41] R. Amewu, A. V. Stachulski, S. A. Ward, N. G. Berry, P. G. Bray, J. Davies, G.Labat, L. Vivas, P. M. O’Neill, Org. Biomol. Chem. 2006, 4, 4431–4436.
[42] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. Shone, D. Stanford, P.Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer,S. Lozanom, M. Jes's, D. Gargallo, S. A. Ward, P. M. O’Neill, J. Med. Chem.2008, 51, 2170–2177.
[43] B. Meunier, Acc. Chem. Res. 2008, 41, 69–77.[44] M. Kaiser, S. Wittlin, A. Nehrbass-Stuedli, Y. Dong, X. Wang, A. Hemphill,
H. Matile, R. Brun, J. L. Vennerstrom, Antimicrob. Agents Chemother.2007, 51, 2991–2993.
[45] A. J. Lin, M. Lee, D. L. Klayman, J. Med. Chem. 1989, 32, 1249–1252.[46] R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H. Heilmann, H.
Chan, M. Cheung, W. Lam, H. Wong, S. L. Croft, L. Vivas, L. Rattray, L.Stewart, W. Peters, B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle,B. Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Woll-born, K. Schmeer, A. Rçmer, Angew. Chem. 2006, 118, 2041–2041;Angew. Chem. Int. Ed. Engl. 2006, 45, 1989–1989.
[47] J. Nagelschmitz, B. Voith, G. Wensing, A. Roemer, B. Fugmann, R. K.Haynes, B. M. Kotecka, K. H. Rieckmann, M. D. Edstein, Antimicrob.Agents Chemother. 2008, 52, 3085–3091.
[48] P. M. O’Neill, A. P. Higson, S. Taylor, E. Irving, (UFC Ltd. , Manchester, UK),WO/03/048167A1, 2003.
[49] R. K. Haynes, W. C. Chang, C.-M. Lung, A.-C. Uhlemann, U. Eckstein, D.Taramelli, S. Parapini, D. Monti, S. Krishna, ChemMedChem 2007, 2,1480–1497.
[50] The antimalarial activity of compound 6 is not reported in Reference[14] . However, the antimalarial activity of artemisinin–acridine conju-gates with similar structures has recently been reported in: N. C. P.Ara'jo, V. Barton, M. Jones, P. A. Stocks, S. A. Ward, J. Davies, P. G. Bray,A. E. Shone, M. L. S. Cristiano, P. M. O’Neill, Bioorg. Med. Chem. Lett.2009, 19, 2038–2043. (IC50=8–20 nm against chloroquine resistant K1P. falciparum).
[51] LogP calculated by Crippen’s fragmentation (V. N. Viswanadhan, A. K.Ghose, G. R. Revankar, R. K. Robins, J. Chem. Inf. Comput. Sci. 1989, 29,163–172) using ChemDraw Pro software: TG, 3.67; artemisinin, 3.17;artemisone, 2.08; acridine–artemisinin conjugate, 8.13.
[52] S. Fu, A. Bjçrkman, B. W(hlin, D. Ofori-Adjei, O. Ericsson, F. Sjçqvist, Br. J.Clin. Pharmacol. 1986, 22, 93–96.
[53] D. J. Sullivan, I. Y. Gluzman, D. G. Russell, D. E. Goldberg, Proc. Natl.Acad. Sci. USA 1996, 93, 11865–11870.
[54] A. Uhlemann, S. Wittlin, H. Matile, L. Y. Bustamante, S. Krishna, Anti-microb. Agents Chemother. 2007, 51, 667–672.
[55] F. Benoit-Vical, J. Leli%vre, A. Berry, C. Deymier, O. Dechy-Cabaret, J. Ca-zelles, C. Loup, A. Robert, J. Magnaval, B. Meunier, Antimicrob. AgentsChemother. 2007, 51, 1463–1472.
[56] A. Robert, O. Dechy-Cabaret, J. Cazelles, B. Meunier, Acc. Chem. Res.2002, 35, 167–174.
[57] F. Bousejra-El Garah, C. Claparols, F. Benoit-Vical, B. Meunier, A. Robert,Antimicrob. Agents Chemother. 2008, 52, 2966–2969.
[58] F. Bousejra-El Garah, B. Meunier, A. Robert, Eur. J. Inorg. Chem. 2008,2133–2135.
[59] D. J. Creek, W. N. Charman, F. C. K. Chiu, R. J. Prankerd, Y. Dong, J. L. Ven-nerstrom, S. A. Charman, Antimicrob. Agents Chemother. 2008, 52, 1291–1296.
[60] J. M. Shin, R. Goldshleger, K. B. Munson, G. Sachs, S. J. Karlish, J. Biol.Chem. 2001, 276, 48440–48450.
[61] C. Montigny, C. Jaxel, A. Shainskaya, J. Vinh, V. Labas, J. V. Møller, S. J. D.Karlish, M. le Maire, J. Biol. Chem. 2004, 279, 43971–43981.
[62] C. W. Jefford, M. G. H. Vicente, Y. Jacquier, F. Favarger, J. Mareda, P. Mill-asson-Schmidt, G. Brunner, U. Burger, Helv. Chim. Acta 1996, 79, 1475–1487.
[63] J. L. Vennerstrom, Y. Dong, J. Chollet, H. Matile, (Medicines for MalariaVenture, International Centre Cointrin, Geneva, Switzerland), US/02/6486199B1, 2002.
[64] W. Trager, J. B. Jensen, Science 1976, 193, 673–675.[65] R. E. Desjardins, C. J. Canfield, J. D. Haynes, J. D. Chulay, Antimicrob.
Agents Chemother. 1979, 16, 710–718.[66] GraphPad Prism 4.0.0, GraphPad software, Inc. , San Diego, CA (USA);
www.graphpad.com.[67] S. F. Altschul, T. L. Madden, A. A. Sch)ffer, J. Zhang, Z. Zhang, W. Miller,
D. J. Lipman, Nucleic Acids Res. 1997, 25, 3389–3402.[68] A. L. Morris, M. W. MacArthur, E. G. Hutchinson, J. M. Thornton, Proteins
1992, 12, 345–364.[69] R. W. Harrison, J. Comput. Chem. 1993, 14, 1112–1122.[70] A. Pedretti, L. Villa, G. Vistoli, J. Mol. Graphics Modell. 2002, 21, 47–49.[71] J. J. P. Stewart, J. Mol. Model. 2007, 13, 1173–1213.[72] M. F. Sanner, B. S. Duncan, C. J. Carrillo, A. J. Olson, Pac. Symp. Biocom-
put. 1999, 401–412.
Received: May 15, 2009Revised: June 16, 2009Published online on July 30, 2009
ChemMedChem 2009, 4, 1469 – 1479 " 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.chemmedchem.org 1479
Docking Studies of Antimalarial Drugs
143


Conclusion


Conclusion
Conclusion
This thesis project was part of the Antimal European Programme and involved close
collaboration with Palumed S.A.
In a first review, we introduced the importance of heme for Plasmodium and presented
quinoline drugs, which have been the mainstay of malaria treatment for the last 6 decades. We
underlined the problem of chemo-resistance, particularly important with chloroquine, and
introduced two very promising quinoline-based drug candidates: N-tert-butyl Isoquine and
Ferroquine.
Artemisinin, a natural peroxide from the traditional Chinese medicine, and its derivatives have
become essential for malaria treatment. They are highly active on both early erythrocytic stages
and gametocytes of Plasmodium. Moreover, they are fast-acting and have proven to be safe.
However, one of the major issues of artemisinin derivatives is their short and variable supply.
The design of fully synthetic peroxides requires a good knowledge of artemisinin mode of
activation. Extensive work in the early 90’s on iron-mediated reactivity of artemisinin has
clearly demonstrated the formation of C-centered radicals. Significant progress has also been
made in the identification of potential biological targets and artemisinin-derived radicals
revealed to be powerful alkylating agents of both heme and parasite proteins. In particular,
alkylation of heme by the artemisinin-derived radicals may be directly involved in the parasite
death through the accumulation of non-dimerizable redox-active heme adducts.
This knowledge of the molecular mechanism of artemisinin activation has contributed to the
rational design of new synthetic antimalarial peroxide-containing drugs, including trioxolanes,
trioxaquines and tetraoxanes, which exhibit outstanding antimalarial activity and improved
DMPK and toxicological profiles.
In the present work, we studied the two main mechanisms proposed in the literature for
artemisinin, namely heme alkylation and PfATP6 inhibition. In addition, we also explored a
new hypothesis by studying the possible bio-activation of artemisinin by copper enzymes.
In the second chapter, we reported the heme-mediated reactivity of artemisone, an artemisinin
derivative, substituted at the C-10α position by a thiomorpholine-1,1-dioxide residue. The
characterization of heme-artemisone adducts confirmed that substitution at the C-10α position
does not prevent the reductive activation of the peroxide bond nor the alkylation of heme.
147

Conclusion
Artemisone, as all the other active artemisinin derivatives (artemether, artesunate, ...) is a very
efficient alkylating agent.
After iron, copper is the second biological metal in Plasmodium. In the third chapter, we
studied the reactivity of artemisinin with copper-complexes as models for protein active sites.
As expected, artemisinin can be activated by copper(I) complexes, in the same way than with
iron(II). We could characterize covalent coupling products and confirm the alkylating
properties of artemisinin with non-iron complexes. However, the reactivity with copper being
much more sluggish than with iron, this alternative mechanism is probably not significant for
artemisinin activation in vivo.
We next turned our attention to the heme-mediated reactivity of synthetic peroxides.
Hybrid trioxaquines have been shown to alkylate heme in vitro. In collaboration with Dr F.
Benoit-Vical (Inserm, LCC, Toulouse) and Dr C. Claparols (LCC, Toulouse), we confirmed
that the heme alkylation reaction by the trioxaquine DU1301 occurs in vivo. We could
characterize heme-drug adducts in the spleen of malaria-infected mice, while they were not
detected in non-infected mice, treated in the same conditions. This result confirmed that heme-
alkylation does not only occur in vitro, but is also relevant in vivo. Trioxaquine and artemisinin
share a common reactivity toward heme in vivo, which may be related to the high activity of
the two drugs.
The work reported in Chapter 4 was done in the group of Pr P. O’Neill, in the University of
Liverpool, in collaboration with Dr R. Amewu. LC-MS analysis was done in collaboration
with Dr S. Muangnoicharoen, in the Liverpool School of Tropical Medicine (LSTM). During
this stay, we confirmed the formation of carbon-centered radicals by Fe-mediated activation of
potent antimalarial tetraoxanes. We went further and characterized covalent coupling products,
both with heme, and tetraphenylporphyrins of iron and manganese. Modeling studies would be
useful to see whether the regioselectivity of the reaction toward the formation of the secondary
radical can be predicted. We also studied the reactivity of two potent tetraoxanes in presence of
a phospholipid model and confirmed the formation of phospholipid oxidation products. This
reactivity is common with trioxolanes analogues, but not with artemisinin. This work might
need to be confirmed with other tetraoxanes.
Overall, our results with heme confirmed that the alkylating properties of artemisinin, in
particular in malaria-infected mice, are not limited to this natural compound, but are shared
148

Conclusion
149
with other potent endoperoxide-containing drugs. Indeed, all the potent antimalarial peroxides
used in this study are also efficient heme-alkylating agents. It is likely that heme alkylation
plays a very important role in their antiplasmodial mechanism of action.
In this work, we also considered an alternative mechanism of action for artemisinin, based on
the inhibition of PfATP6. We generated a model of PfATP6 by homology modeling from its
SERCA1a homologue crystal structure and used this model in docking calculations with
structurally diverse antimalarials. The main result is that the predicted binding affinity of the
tested compounds does not correlate with their in vitro antiplasmodial activity. However,
PfATP6 inhibition by artemisinin in Xenopus oocytes has been shown to be iron-dependent.
Keeping this in mind, further docking studies that include an iron site in the protein might be
considered.
Working on the PfATP6 project was the good opportunity to gain useful experience in
homology modeling and docking calculation tools.
The precise mechanism of parasite killing process by artemisinin and antimalarial peroxides
will probably remain for a long time a matter of debates. Nevertheless, a good understanding of
the key features of its mode of action could help to develop efficient synthetic antimalarial
drugs.
As the alkylating ability is probably required for the antimalarial activity of this class of drugs,
the extent of the alkylation reaction could be a way of screening the potential antimalarial
activity of new compounds, before in vivo testing.


Appendix 1
Total synthesis of Quinine
151


Annex 1: Total synthesis of Quinine
Appendix 1: Total synthesis of Quinine
Quinine is a challenging synthetic target: it is an alkaloid with a
complicated, multi-ring structure that has four stereogenic centers (and
thus 15 other stereoisomers). In this appendix, we report the key events
of quinine total synthesis, reported in the very complete review of
Kaufman and Rùveda.[1]
1817: Pelletier and Caventou isolated quinine from cinchona tree.
1853: Pasteur obtained erization of quinine.
Figure 1: Formation of d-quinotoxine by acid-catalyzed degradation of quinin . Adapted from[2]
1908: Rabe, a German chemist, suggested the first correct connectivity of quinine
ce for the
d-quinotoxine (2) by acid-catalized isom
e
1918: In a very brief publication, Rabe and Kindler reported a synthetic sequen
reconstruction of quinine (and quinidine) from d-quinotoxine (figure 2). This three-step sequence
involved the construction of the C8-N bond (C8-N approach) and started with addition of sodium
hypobromite to d-quinotoxine to give the N-bromoquinotoxine. This intermediate was oxidized with
sodium ethoxide in ethanol to give quininone, which interconverts with quinidinone via the
formation of an enol intermediate in basic conditions. In the third step the ketone group is reduced
with aluminum powder and sodium ethoxide in ethanol. Quinine (12%) and quinidinine (6%) were
identified as major products.[3]
153

Annex 1: Total synthesis of Quinine
Figure 2: Rabe and Kindler three-step convertion of d-quinotoxine to (R-C8, S-C9) quinine and (S-C8, R-C9)
quinidine. Adapted from[1]
This transformation was the first major step towards the synthesis of quinine. However, perhaps
because of wartime pressures, Rabe’s procedure was not cautiously reviewed and the key procedure
for the reduction of quininone to quinine was detailed 14 years later, by the reduction of
dihydrocinchoninone to dihydrocincho-nine.[1]
1943: Prostenik and Prelog made a notable step forward when they degraded the natural
product cinchotoxine to optically active homomeroquinene (4), and used 4 to prepare d-quinotoxine
(2). The sequence included a key condensation with ethyl quininate (6).
Figure 3: Degradation and reconstitution of quinotoxine by Prostenik and Prelog. Adapted from[1]
1944/1945: Woodward and Doering (Harvard University) published the “formal” Total
Synthesis of Quinine.[4,5] In the 1940s, the synthesis of quinine became urgent when the war in
southeast Asia cut off supplies of the drug, thus causing thousands of soldiers to die after becoming
154

Annex 1: Total synthesis of Quinine
infected with malaria during the campaigns in Africa and the Pacific. Moreover, the cinchona
plantations established in Java by the Dutch were the major sources of the European reserves of
quinine, which were stored in Amsterdam. However, the German capture of Holland in 1940 and
the Japanese military invasion of Java in 1942 abruptly cut these vital supplies.[1]
Woodward ingeniously visualized that the basic homomeroquinene skeleton could be accessed
from an isoquinoline. His synthesis included transformation of 3-hydroxybenzaldehyde (8) into
isoquinolin-7-ol (10) via Schiff base 9 (figure 4). This starting isoquinoline was converted into its
8-methyl derivative 12 through the intermediacy of piperidine 11. In turn, 12 was partially
catalytically hydrogenated to the tetrahydroisoquinoline 13, which was isolated as its N-acetyl
derivative 14, while a second catalytic hydrogenation furnished 15, as a complex diastereomeric
mixture. This mixture was simplified by oxidation to the related ketones, with concomitant
epimerization of the tertiary carbon center next to the carbonyl group. Separation of the
diastereomers was aided by the formation of the hydrate of compound 16 with a cis ring junction.[1]
Figure 4: Preparation of the homomeroquinene derivative by Woodward and Doering.[1] Reagents and conditions: a) H2NCH(OEt)2 (94%); b) 1. 80% H2SO4 ; 2. NaOH, crystallization then H+ (64%); c) piperidine, HCHO, EtOH (61%); d) NaOMe, MeOH, 220°C, 16 h (65%); e) H2, Pt, AcOH; f) Ac2O (95%); g) H2, Raney nickel, EtOH, 150°C, 205 bar, 16 h [1:1 cis(crystalline)/trans(oil)]; h) H2Cr2O7, AcOH; Et2O/H2O, diastereomer separation (28 %).
Ring opening of 16 through preferential nitrosation of the tertiary carbon atom next to the carbonyl
group furnished the oxime 17 (figure 5). Conservation of the crucial cis geometry of the substituents
on the piperidine ring in 17 marked the success of the strategy for building both adjacent side chains.
155
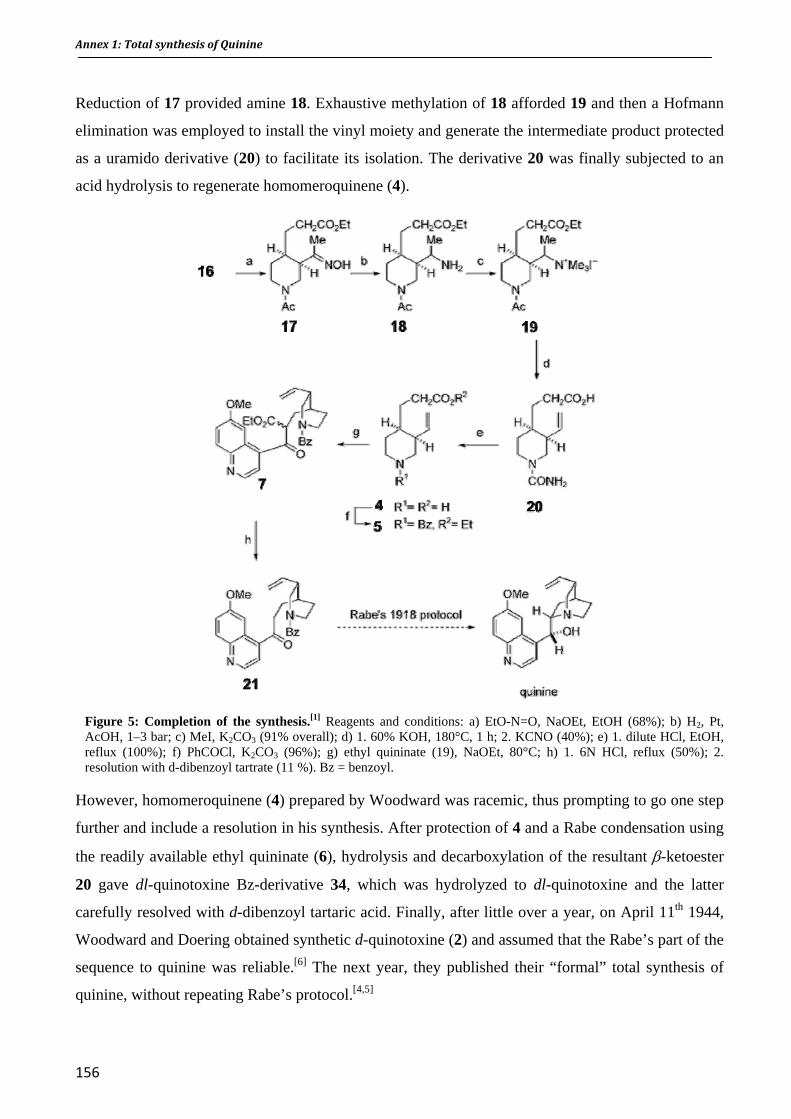
Annex 1: Total synthesis of Quinine
Reduction of 17 provided amine 18. Exhaustive methylation of 18 afforded 19 and then a Hofmann
elimination was employed to install the vinyl moiety and generate the intermediate product protected
as a uramido derivative (20) to facilitate its isolation. The derivative 20 was finally subjected to an
acid hydrolysis to regenerate homomeroquinene (4).
Figure 5: Completion of the synthesis.[1] Reagents and conditions: a) EtO-N=O, NaOEt, EtOH (68%); b) H2, Pt, AcOH, 1–3 bar; c) MeI, K2CO3 (91% overall); d) 1. 60% KOH, 180°C, 1 h; 2. KCNO (40%); e) 1. dilute HCl, EtOH, reflux (100%); f) PhCOCl, K2CO3 (96%); g) ethyl quininate (19), NaOEt, 80°C; h) 1. 6N HCl, reflux (50%); 2. resolution with d-dibenzoyl tartrate (11 %). Bz = benzoyl.
However, homomeroquinene (4) prepared by Woodward was racemic, thus prompting to go one step
further and include a resolution in his synthesis. After protection of 4 and a Rabe condensation using
the readily available ethyl quininate (6), hydrolysis and decarboxylation of the resultant β-ketoester
20 gave dl-quinotoxine Bz-derivative 34, which was hydrolyzed to dl-quinotoxine and the latter
carefully resolved with d-dibenzoyl tartaric acid. Finally, after little over a year, on April 11th 1944,
Woodward and Doering obtained synthetic d-quinotoxine (2) and assumed that the Rabe’s part of the
sequence to quinine was reliable.[6] The next year, they published their “formal” total synthesis of
quinine, without repeating Rabe’s protocol.[4,5]
156

Annex 1: Total synthesis of Quinine
The synthesis was plagued by low yields, did not allow control of the stereochemistry, and could not
have afforded commercial quantities. Indeed, in spite of wishful thinking surrounding the synthesis,
commercial production of quinine by the newly devised strategy
would have cost approximately 200 times more than its natural
equivalent.[1]
Nevertheless, the synthesis was viewed as a scientific milestone
and was the beginning of a successful carrier for Woodward,
who won the Nobel Prize in Chemistry in1965 for "his
outstanding achievements in the art of organic synthesis”.
Figure 6: Robert Woodward (left) and William
Doering (right)
2001: Several synthesis of quinine were proposed since Woodward and Doering publication
(reviewed in ref[1]). In 2001, Stork et al. (Columbia University) published the first stereoselective
synthesis of quinine – using the C6-N approach - and questioned the validity of the
Woodward/Doering strategy,[7] which is called “a widely believed myth”.[8]
2008: Smith and Williams reproduced the Rabe/Kindler protocol, and showed that the contentious
‘missing step’ in Woodward and Doering’s synthesis, but previously reported by Rabe and Kindler
in 1918, is indeed possible under the conditions sketchily described in that early work.[9] By
validating the Rabe and Kindler route, Smith and Williams wrote the last chapter of the quinine
saga, at least for now …
Thus, taken together, the total synthesis of d-quinotoxine by Woodward and Doering in 1944 with
the previously reported synthesis of homomeroquinene by Prostenik and Prelog, and the conversion
of d-quinotoxine into quinine by Rabe and Kindler in 1918 constitute the Woodward-
Doering/Prostenik-Prelog/Rabe-Kindler total synthesis of quinine (or Rabe-Kindler/Prostenik-
Prelog/Woodward-Doering depending if we consider the chemical order or the chronological order).
157

Annex 1: Total synthesis of Quinine
158
Figure 7: The Woodward-Doering / Prostenik-Prelog / Rabe-Kindler total synthesis of quinine. Adapted from[2]
Bibliography
[1] T. S. Kaufman, E. A. Ruveda. The Quest for Quinine: Those Who Won the Battles and Those Who Won the War. Angew. Chem. Int. Ed. 2005, 44, 854-885.
[2] J. I. Seeman. The Woodward-Doering/Rabe-Kindler Total Synthesis of Quinine: Setting the Record Straight. Angew. Chem. Int. Ed. 2007, 46, 1378-1413.
[3] P. Rabe, K. Kindler. Ber. Ctsch. Chem. Ges. 1918, 466. Cited in Kaufman and Rùvela Angew. Chem. Int. Ed. 2005.
[4] R. B. Woodward, W. E. Doering. The Total Synthesis of Quinine. J. Am. Chem. Soc. 1944, 66, 849.
[5] R. B. Woodward, W. E. Doering. The Total Synthesis of Quinine. J. Am. Chem. Soc. 1945, 67, 860-874.
[6] S. M. Weinreb. Chemistry: Synthetic Lessons from Quinine. Nature 2001, 411, 429-431.
[7] G. Stork, D. Niu, A. Fujimoto, E. R. Koft, J. M. Balkovec, J. R. Tata, G. R. Dake. The First Stereoselective Total Synthesis of Quinine. J. Am. Chem. Soc. 2001, 123, 3239-3242.
[8] G. Stork. Quinine Quandary. Chem. Eng. News 2000, 78, 8.
[9] A. C. Smith, R. M. Williams. Rabe Rest in Peace: Confirmation of the Rabe-Kindler Conversion of d-Quinotoxine into Quinine: Experimental Affirmation of the Woodward-Doering Formal Total Synthesis of Quinine. Angew. Chem. Int. Ed. 2008, 47, 1736-1740.

Appendix 2
Candidate Selection of a 1,2,4,5Tetraoxane DrugDevelopment Candidate (RKA 182) with Superior Properties to the Semi
Synthetic Artemisinin Based Antimalarials


1
Antimalarial Agents DOI: 10.1002/anie.200((will be filled in by the editorial staff))
Identification of the First 1,2,4,5-Tetraoxane Antimalarial Drug-Development Candidate (RKA 182) with Superior Properties to the Semi-Synthetic Artemisinins
Paul M. O’ Neill*, 1, 2 Richard K. Amewu, 1 Gemma L. Nixon, 4 Fatima Bousejra El-Garah, 1 Mathirut Mungthin, 3 James Chadwick, 1,2 Alison E. Shone, 4 Livia Vivas, 6 Hollie Lander, 6 Victoria Barton,1 Sant Muangnoicharoen , 4 Patrick G. Bray, 4 Jill Davies,4 B. Kevin Park, 2 Sergio Wittlin,5 Reto Brun,5 Michael Preschel, 7 Kesheng Zhang7 and Stephen A. Ward 4
((Dedication----optional))
Artemisinin 1 is an extract of the Chinese wormwood Artemisia annua and has been used since ancient times to treat malaria.[1] Today, semi-synthetic derivatives artesunate 2 and artemether 3 are used clinically in drug combinations (ACT).[2]. However, first generation analogues (eg. 2 and 3) have a limited availability,[3] high cost[4] and poor oral bioavailability (Figure 1a).[5] In addition to these drawbacks there have been recent reports of high failure rates associated with ACTs suggesting the possibility of clinical artemisinin resistance along the Thai–Cambodian border.[6] In the
light of these observations there is an urgent need to develop alternative endoperoxide based therapies.[7]
Figure 1. a. Artemisinin and its semi-synthetic analogues, b. Comparison of tetraoxanes with trioxolane based antimalarials.
The crucial structural functionality within artemisinin and synthetic 1,2,4-trioxanes [8] is the endoperoxide-bridge. Recently a series of molecules based on an ozonide structure were developed from which a candidate OZ277[9] was shown to have impressive antimalarial activity profiles in vitro and in rodent models of malaria. However, the recent development of OZ 277 has been hampered as this molecule was found to be unstable in the plasma of malaria patients during a Phase II dose ranging study. This instability stems from the inherent reactivity of the 1,2,4-trioxolane core structure in the presence of iron released during malaria infection.[10a, 10b] In our hands, studies of endoperoxide stability have shown that 1,2,4,5-tetraoxanes (eg. 6) have significantly higher stability than their 1,2,4-trioxolane (4) or 1,2,4-trioxane counterparts.[11] To exemplify further the chemical and biological differences between these two heterocycles it has been noted that the simple dispiro 1,2,4-trioxolane 4 is antimalarially inactive and unstable whereas the close chemically stable tetraoxane analogue 6 expresses antimalarial activity in the nanomolar range (IC50 = 25nM) (Figure 1b). For good levels of antimalarial activity in the ozonide series fusion of the 1,2,4-trioxolane-ring system to an adamantane core (see for example
[*] 1 Prof P. M. O’Neill, Dr R. Amewu, Dr J. Chadwick, F. Bousejra El-Garah, Dr V. Barton. Department of Chemistry University of Liverpool Liverpool, L69 7ZD, UK E-mail: [email protected] 2 Prof B. K. Park, Prof P. M. O’Neill MRC Centre for Drug Safety Science Department of Pharmacology University of Liverpool Liverpool, L69 3GE, UK 3 M. Mungthin Department of Parasitiology Phramongkutklao College of Medicine Bangkok
4 Dr G. L. Nixon, Dr A. E. Shone, Dr. S. Muangnoicharoen, Dr P. G. Bray, J. Davies, Prof. S. A. Ward. Liverpool School of Tropical Medicine Pembroke Place Liverpool, L3 5QA, UK 5 Dr S. Wittlin, Prof R. Brun Swiss Tropical and Public Health Institute Parasite Chemotherapy Socinstr. 57, P.O. Box CH-4002 Basel, Switzerland
6 Dr L Vivas, H. Lander Department of Infectious and Tropical Disease London School of Hygiene and Tropical Medicine Keppel Street London, WC1E 7HT, UK
7 Dr M. Preschel, Dr K. Zhang Carbogen AMCIS Neulandweg 5 CH-5502, Hunzenschwil, Switzerland
[∗∗] ((Acknowledgements))
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.((Please delete if not appropriate))
O
O
H
H
OO
R
H
Artemisinin 1, R = =OArtesunate 2, R = α-OC(O)CH2CH2CO2NaArtemether 3, R = β-OMe
O
OO
O O
OO
4 5Inactive, Unstable Reactive
Highly potent IC50 = 25nM Achiral, Stable
a
bO O
OR
Replacement ofcyclohexyl withadamantane
OZ Series (eg OZ 277) Stability issues in Phase 2Clinical Trials
Further enhancedstability
O O
OOR
Replacement ofcyclohexyl withadamantane
6 7
161

2
5, Figure 1)) was found to be essential.[9] Given the foregoing observations we reasoned that similar substitution of the cylohexyl group in antimalarially active analogue 6 would generate a new series of molecules with improved stability profiles and improved oral and PK profiles through optimization of the side chain in generic structure 7 (R = polar water-solubilising group). Identification of the optimal, metabolically stable polar side chain to counterbalance the lipophilicity of the adamantane functional group was the primary focus of the medicinal chemistry optimization.
Figure 2. Tetraoxane candidate development and SAR. [13] In order to candidate select a 1,2,4,5 tetraoxane we set a rigorous target product profile from the onset of our medicinal chemistry optimisation and for the candidate selection of RKA 182 (15) over 150 novel 1,2,4,5-tetraoxanes were synthesised and screened from two independent hit series (Figure 2). After extensive in vitro/in vivo and DMPK studies on Hit Series 1 this template was selected over series 2 for lead optimisation which ultimately led to the synthesis, profiling and selection of RKA 182 15 (vide infra) as the development candidate.
Synthesis of lead tetraoxanes (Hit series 1) is depicted in Scheme 1. The synthetic route is high yielding, only 5 steps and
divergent in the last step making expedient parallel synthesis possible. (supporting information)[12a]. In vitro and in vivo data of the most potent compounds synthesised in Hit Series 1 can be seen in Table 1. These molecules exhibit an IC50 of less than 6 nM against both chloroquine sensitive 3D7 and chloroquine resistant K1 strains of P.falciparum with the lowest IC50 being 0.8 nM (Table S1, supporting information).[14] Tetraoxane analogues also have ED50/ED90 values of less than 3.5/9.5 mg/Kg, with the lowest values being ED50/ED90 = 0.99/1.41 mg/Kg (tetraoxane 17) when tested in mice infected with the P.berghei ANKA parasite. The activity of 17, in terms of ED50 and ED90 in the 4-day test,[15] surpasses that of any synthetic endoperoxide reported in the literature to date after oral administration. Piperidinyl piperazine functionalised tetraoxane 15, when formulated as a tosylate salt, has outstanding in vitro activity (< 1nM) and in vivo activity with an ED50/ED90 of 1.33/4.18 mg/Kg which is superior to artemether, artesunate and comparable to Artemisone, the leading next generation semi-synthetic artemisinin which is currently undergoing Phase 2 clinical trials.[16] In studies conducted on mouse survival, mice treated at 3 x 10 mg/kg per day with 15 survived 22 days in comparison with only 9 days for artesunate. (Supporting information)
EtO
O
O O
OO OEt
O
30% H2O2HCOOH
O O
OO OH
O
O O
OO R1
O
OHO
O OHEtO
O
O
2-adamantanone,HBF4, EtOAc or2-adamantanone,Re2O7, DCM
KOH
1. Et3N, ClCO2C2H5. DCM, OoC, 1 hr.
2. Amine, 90 min, 0oC-rt
N N
N O
N N N
N N N
R1= R1=
R1=
R1=
12, 87%
13, 81%
15 (RKA 182), 86%
16, 88%
80%
10, 45-75%80%
N N OR1= 14, 88%
8 9
11
12-18
R1=
R1=
HN N 17, 76%
N N 18, 82%
Scheme 1. Synthesis of polar tetraoxane derivatives 12-18.
Table 1. In vitro and in vivo15 data for tetraoxanes 12-18 and Pharmocokinetic parameters after a single intravenous (1mg/kg) and single oral (10 mg/kg) administration.
IC50 (nM) Compound 3D7 K1
% Inhibition 30 mg/Kga
ED50 (mg/Kg)
ED90 (mg/Kg)
1 mg/kg (iv)
T1/2 (h)
10mg/kg (po)
T1/2 (h)
Fb (%)
12 1.4 0.9 100 3.47 5.40 0.39 1.64 9 13 5.2 0.8 100 3.18 3.88 1.16 5.89 11 14 2.5 2.8 99.7 3.02 9.25 0.54 1.72 9 15 4.9 1.9 100 1.82 8.38 0.61 3.53 24
15 (tosylate salt) 0.87 1.1 100 1.33 4.18 0.80 2.38 38 16 6.0 1.5 100 2.23 5.12 0.64 NC 36 17 1.2 0.9 100 0.99 1.41 0.57 1.08 9 18 1.2 0.9 100 1.60 2.91 0.40 1.23 23
Artesunate 1.8 1.6 100 3.96 11.72 0.26 NC NC Artemether 7.8 3.2 100 3.80 12.24 ND 1.4d
Chloroquineb 12.5 250.0 100 2 4.5 - - - [a] Parasitaemia was determined by microscopic examination of Giemsa stained blood films taken on day 4. Microscopic counts of blood films from each mouse were processed using spreadsheet (Microsoft Corp.) and expressed as percentage of inhibition from the arithmetic mean parasitaemias of each group in relation to the untreated group. [b] indicates F(oral bioavailability) calculated using AUC0-t and actual doses (see supporting online information). [d] Data taken from Vennerstrom et al.9 NC = Not calculable. ND = Not determined
162

3
Pharmacokinetic parameters and oral bioavailability were determined after intravenous (1mg/kg) and oral administration (10 mg/kg) in Sprague-Dawley rats (Table 1) (For in vitro metabolism data see supporting information). The most significant observation from this study is that tetraoxanes 15, 16 and 18 have oral bioavailabilities of greater than 20%, superior to OZ277 which had an oral bioavailability of less than 10% in the comparator arm (Table S2 and S3, supporting information). Taking this information together with the antimalarial activity data it was clear that although the activity of 12 and 17 in the 4-day test was very good, the poor bioavailability of 17 and 12’s complicated in vivo metabolic profile meant that these molecules were not considered further. Compounds 13 and 18 were also ruled out at this point due to relatively poor bioavailability and a very short half-life predicted for the latter tetraoxane. Based on the outstanding in vitro and in vivo antimalarial activity and pharmacokinetic profile 15 was selected as the lead candidate. Formulation work on 15 delivered the compound as a di-tosylate salt which had improved oral bioavailability of 38% in the rat (Table S4, supporting information).
Having selected 15 as the drug candidate a scalable, industrial synthesis was sought. The industrial synthesis (Carbogen AMCIS) of 15 is only four steps (due to the synthesis of 8 in one step from direct hydrogenation of phenol ester 19) involving a single chromatography step and has a projected low cost of goods. The key step in the scale up of this potentially hazardous chemistry involved the in situ generation of the keto ester 10 without isolation of the gem dihydroperoxide precursor followed by hydrolysis (of the ester function) to provide the acid 11 in acceptable yields on a 1.2 kg scale. The application of Dussault procedure (employing Re2O7 as a mild Lewis acid)12b for the key tetraoxane ring-forming step was also crucial to the scale-up of this chemistry.
As a part of our preclinical development 15 was also tested against eleven different South East Asian isolates from patients who had failed ACT combination chemotherapy (Figure 3) and clearly demonstrated superiority over mefloquine, artesunate and artemisinin with all measured IC50’s below 5nM.
To assess the speed of action a single oral dose of 30 mg/kg 15 was administered to mice infected with 4 x 106 P. berghei ANKA infected red cells two days post-infection (Figure 4). Parasitaemias rapidly decreased to undetectable levels 24h after treatment with RKA-182 (15) whereas treatment with the same dose of artesunate (ASN) reduced parasitaemias up to 5% 8h post-treatment increasing rapidly thereafter.
Figure 3. IC50 data for 15 (RKA182) compared with other antimalarials
O
OO
OO
O
O
OO
OOH
O
O
OO
ON
O
NN
CH3
HN N N CH3
1. 30% H2O2, HCOOH, ACN, DCM2. 2-adamantanone, Re2O7, DCM
(telescope)
3. 2 eq LiOH, THF, 40°C4. HCl
5. crystallization from ethanol/water
32% over 2 steps
1. methyl chlorformate, Et3N, toluene, 0°C2.
3. crystallization from toluene/heptane
70%
OO
OO
O
HO H2, 10% Pd/Cdioxane, 90°C
50-70%19 8 10
11
15 (RKA182)
tPSA: 63.71CLogP: 3.99
Scheme 2. Industrial synthesis of tetraoxane 15 (RKA182).
Figure 4. Speed of action of 15 (RKA 182) following a single oral dose of 30mg/kg.
To demonstrate the remarkable stability of tetraoxane 15 in both non-infected and infected red blood cells, in vitro studies were carried out to assess the percentage recovery of drug at set time points in non-infected and infected blood (Figure 5). It can be seen from this data that OZ277 rapidly degrades in infected blood cells giving no recovery of drug after only 35 minutes. In sharp contrast, RKA182 (15) shows 79% recovery after 4 hours in infected blood. In vivo pharmacokinetic analysis was also performed to demonstrate the impact of infection on the exposure profile of 15 in non-infected mice and mice infected with P.berghei (Table S6 in supporting information) and the data clearly demonstrates equivalent exposure (AUC) in both infected and non-infected mice. Taken together, these data suggests 15 is significantly more stable than OZ277 in which malaria infection was associated with a significant reduction in drug plasma concentrations in Phase II trials. [17]
In order to characterise the potential mediators of the antimalarial activity of RKA182 we performed mechanistic studies with ferrous (II) bromide in THF in the presence of the spin-
163

4
trapping agent 2,2,6,6-tetramethyl-1-piperidine-1-oxyl (TEMPO). From these studies, we were able to intercept both the primary and secondary carbon centred radicals to produce two TEMPO adducts A and B (Scheme 3).
Figure 5: Stability of 15 in non-infected and infected red blood cells in comparison with OZ277.
O O
OO
R
O
OHO
O N
RO
O
O O
OFe
RO
O
R
OHO
ON
O
O
FeBr2 (O2)THFTHF
FeBr2 (O1)
O O
OO
R
OFe
O O
O
R
OFe
O
O O
O
R
OFe
O
β-scission
3+
3+
3+
3+
ADDUCT A
1 2
12
m/z = 454.5 MH+ m/z = 324.3 MH+ADDUCT B
TEMPO TEMPO
β-scission
Scheme 3. TEMPO spin-trapping of C-radical intermediates generated following from 12 Fe(II) Activation . (R = N(CH2CH2)2NCH2iPr)
The behavior of the tetraoxanes reported here is distinct from 1,2,4,-trioxolanes since only the secondary carbon centered radical species has been characterized from OZ277 and other 1,2,4-trioxolanes. [9] Since heme alkylation is believed to play a vital role in the mechanism of action of endoperoxide antimalarials [17] (Scheme 4) we examined the reactivity of 12 with ferrous heme LC-MS analysis confirmed an m/z 782.3 Da for three adducts (maximum absorption of the Soret band at 430 nm) that result from the covalent bonding of the tetraoxane-derived secondary C-
centered radical and the heme porphyrin ; this process may play an important role in the molecular mechanism of action of these derivatives.
O O
OO
ON N
N N
N N
COOHHOOC
Fe β-scission
O O
OO
ON N
N N
N N
COOHHOOC
Fe
HOO
N N
N N
COOHHOOC
FeO
N NO
+
Ketoamide
12
O-O bondhomolysis
Fe(II)-PPIX
FePPIX-12 adduct
+
δ
Scheme 4. Proposed mechanism of the heme alkylation reaction with tetraoxane 12 (RKA099). For clarity, only the δ–regioisomer of the adduct is shown. 12 was allowed to react with iron(III)-hemin, in presence of an excess of dithionite to generate iron(II)-heme in situ, in ACN/NaOH 0.1 M (50/50)
In conclusion, we have identified the first water soluble 1,2,4,5-tetraoxane drug candidate that has outstanding antimalarial activity, stability, low toxicity (see supporting information) and ADME properties that overcome most of the problems encountered previously with the synthetic and semi-synthetic antimalarial endoperoxide drugs that have progressed into pre-clinical development. This work firmly establishes the potential of this class of tetraoxane to provide the next generation of synthetic drugs for deployment in the control and eradication of malaria as a component of combination chemotherapy.
Keywords: antimalarial agents · endoperoxide · tetraoxane · stability · candidate selection
[1] P. M. O'Neill, G. H. Posner, J Med Chem. 2004, 47, 2945 [2] Report of a WHO Informal Consultation. Geneva, World
Health Organization, 2001 (WHO/CDS/RBM/2001.33) ; [3] T. K. Mutabingwa, Acta Tropica 2005, 95, 305. [4] B. M. Greenwood, K. Bojang, C. J. M. Whitty, G. A. T.
Targett, Lancet 2005, 365, 1487. [5] R. K. Haynes, Curr Opin Infect Dis 2001, 14, 719. [6] A. M. Dondorp, F. Nosten, N. J. White, New Engl J Med.
2009, 361, 1808. [7] a) N. J. White, J Clin Invest 2004, 113, 1084 b) J. Wiesner,
R. Ortmann, H. Jomaa, M. Schlitzer, Angew Chem Int Ed Engl 2006, 43, 5274
[8] C. W. Jefford, Drug Discov Today. 2007, 12, 487 [9] J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A.
Charman, F. C. Chiu, J. Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padmanilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler, S. Wittlin, W. N. Charman, Nature 2004, 430, 900.
[10] a) P. Olliaro, T. N. C. Wells, Clin Pharmacol Ther. 2009, 85, 584K. b) A second generation 1,2,4-trioxolane OZ 439
164

5
is under development with the MMV see; http://www.mmv.org/article.php3?id_article=528
[11] G. L. Ellis, R. Amewu, S. Sabbani, P. A. Stocks, A. E. Shone, D. Stanford, P. Gibbons, J. Davies, L. Vivas, S. Charnaud, E. Bongard, C. Hall, K. Rimmer, S. L. María Jesús, D. Gargallo, S. A. Ward and P. M. O’Neill, J. Med. Chem., 2008, 51, 2170
[12] a) R. Amewu, A. V. Stachulski, S. A. Ward, N. G. Berry, P. G. Bray, J. Davies, G. Labat, L. Vivas, P. M. O'Neill, Org Biomol Chem. 2006, 4, 4431b) P. Ghora, P.H. Dussault, Org. Lett., 2009, 11, 213
[13] G. M. Sheldrick, Acta Cryst. 2008, A64, 112. [14] M. Smilkstein, N. Sriwilaijaroen, J. X. Kelly, P. Wilairat,
M. Riscoe, Antimicrob Agents Ch. 2004, 48, 1803.
[15] W. Peters, S. L. Fleck, B. L. Robinson, L. B. Stewart, C. W. Jefford, Ann. Trop. Med. Parasitol. 2002, 96, 559.
[16] R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H. D. Heilmann, H. W. Chan, M. K. Cheung, W. L. Lam, H. N. Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters, B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle, B. Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Wollborn, K. Schmeer, A. Romer, Angew Chem Int Ed Engl., 2006, 45, 2082.
[17] A. Robert, J. Cazelles, B. Meunier, Angew Chem Int Ed Engl., 2001, 40, 1954.
165

6
Identification of the First 1,2,4,5-Tetraoxane Antimalarial Drug-Development Candidate (RKA 182) with Superior Properties to the Semi-
Synthetic Artemisinins
Paul M. O’ Neill*, 1, 2 Richard K. Amewu, 2 Gemma L. Nixon, 4Fatima Bousejra El-Garah 1, Mathirut Mungthin, 3 James Chadwick, 1,2 Alison E. Shone, 4 Livia Vivas, 5 Hollie Lander, 5 Victoria Barton,1 Sant Muangnoicharoen , 4 Patrick G. Bray, 4 Jill Davies,4 B. Kevin Park, 2 Sergio Wittlin,6 Reto Brun,6 Michael Preschel7, Kesheng Zhang7and Stephen A. Ward 3
1 Dept. of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK.2 MRC Centre for Drug Safety Science, Dept. of Pharmacology, School of Biomedical Sciences, University of Liverpool, Liverpool L69 3GE, U.K, 3 Dept. of Parasitology, Phramongkutklao College of Medicine, Bangkok, 4 Liverpool School of Tropical Medicine, Pembroke Place, Liverpool, L3 5QA, UK 5 Swiss Tropical Institute, Parasite Chemotherapy, Socinstr. 57 P.O. Box CH-4002, Basel, Switzerland 6 Department of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, U.K., 7 Carbogen AMCIS, Neulandweg 5, CH-5502 Hunzenschwil, Switzerland
Supporting Information
166
7
Supplementary Tables Table S1: In Vitro Antimalarial Activities versus Chloroquine Sensitive and Resistant Strain of P. falciparum and Multidrug Resistant Cancer Cell-line for all analogues.
Compound 3D7 (nM) K1(nM) KB(nM) ClogP 12 1.4 - - 4.21 (± 0.85) 13 5.2 nd nd 3.47 (± 0.77) 14 2.5 2.8 72018.7 3.98 (± 0.82) 15 4.9 1.9 30458.8 3.99 (± 0.98) 16 6.0 1.5 21284.8 3.99 (± 0.88) 17 7.7 nd nd 4.09 (± 0.76) 18 1.2 0.9 111446.4 4.81 (± 0.94)
Chloroquine 12.5 250.0 nd - Artemether 2.6 nd nd -
POD nd nd 15.9 - nd = Not determined Table S2: Pharmocokinetic Parameters after a single Intravenous (1mg/kg) Administration.
Analyte Actual Dose
(mg/kg)
C0 (ng/mL)
AUC0-t (ng.h/mL)
AUC0-∞ (ng.h/mL)
T½ (h)
CL (mL/h/kg)
Rsq
12 1.124 280 144.1 147.9 0.39 7601 1.00 13 0.984 418 201.3 204.3 1.16 4816 0.96 14 1.093 262 140.6 149.8 0.54 7298 1.00 15 1.094 138 83.87 92.53 0.61 11820 0.98 16 1.047 60.7 31.85 35.17 0.64 29770 0.97 17 1.087 37 552.0 558.1 0.57 14793 1.00 18 1.032 66.1 39.09 40.26 0.40 25640 1.00
OZ277 0.721 52.1 32.16 32.59 0.32 22130 0.99
Table S3: Pharmocokinetic Parameters after a single Oral (10mg/kg) Administration.
* indicates F calculated using AUC0-t and actual doses, NC = not calculable. Table S4: In vivo Pharmacokinetic parameters of RKA 182 (tosylate salt) after p.o and i.v administrations (n=3).
i.v (1 mg/kg)
p.o. (10 mg/kg)
Compound Parameter
Mean SE Mean SE
F (%)
15 (tosylate salt)
AUC (min*µg/mL) 7.37 0.29 27.9 1.83 37.9
AUC 0-6h (min*µg/mL) 7.34 0.3 20.5 3.4 cmax (µg/mL) 0.11 0.01 0.09 0.02 tmax (min) 15 0 200 80 t½ (min) 48.2 2.11 143 54.5 CL/F (mL/min/kg) 136 5.3 360 23.6 CL (mL/min/kg) 136 5.3 136 8.93 Vd/F (L/kg) 9.48 0.69 76 33.2
Analyte Actual Dose
(mg/kg)
Cmax(obs) (ng/mL)
Tmax(obs) (h)
AUC0-t (ng.h/mL)
AUC0-∞ (ng.h/mL)
T½ (h)
CL/F (mL/h/kg) F* Rsq
12 8.912 34.2 1.00 103.4 113.5 1.64 78550 0.09 0.99 13 8.826 74.9 0.50 200.9 370.2 5.89 23840 0.11 0.80 14 9.797 29.5 2.00 116.7 131.3 1.72 74630 0.09 0.97 15 8.792 38.0 2.00 159.6 247.9 3.53 35470 0.24 0.94 16 7.360 20.5 4.00 79.69 NC NC NC 0.36 NC 17 8.981 82.0 0.25 636.1 645.1 2.77 3900 0.09 0.99 18 9.257 21.2 2.00 80.30 84.23 1.23 110100 0.23 0.90
OZ277 6.113 2.75 2.00 12.68 15.49 2.33 394600 0.05 0.78
167

8
Vd (L/kg) 9.48 0.69 28.8 12.6 MRT (min) 46.6 3.97 248 79.2
SE = standard error Table S5: Average Survival and Percentage Inhibition of Mice Infected With P. berghei ANKA in a Single Oral Dose of 1 x 30 mg/kg and an Oral Dose of 3 x 10mg/kg.
Comp. Avg. Survival (days) post-infection (3 x10 mg/kg)
% of inhibition at day 4
post-infection
Avg. Survival (days) post-infection (1 x 30 mg/kg)
13 nd nd 7.6 (7, 7, 7, 8, 9) 14 15.4 (14, 14, 21, 14, 14) 99.99 9.4 (8, 8, 15, 8, 8) 15 21.8 (20, 21, 23, 22, 23) 99.99 12.4 (21, 7, 6, 22, 6) 16 27.6 (30, 30, 20, 30, 28) 99.98 11.4 (15, 14, 7, 7, 14) 17 nd nd 7.6 (7, 7, 8, 8, 8)
Artesunate 9.0 (7, 7, 7, 14, 10) 99.09 6.8 (6, 7, 7, 7, 7) OZ277 25.2 (18, 30, 18, 30, 30) 99.98 10.2 (8, 10, 8, 10, 15)
No Drug 4.0 (4, 4, 4, 4, 4) 0 3.0 (3, 3, 3, 3, 3) nd = not determined Table S6: In vivo phamacokinetic analysis - impact of infection on exposure profile.
Cmax (ng/mL)
Tmax (h)
AUCtot (h)*(ng/mL)
T 1/2 (h )
Clearance (L/h)
Vz (L)
Vss (L)
1 Control 80.48 1 291.34 1.85446 80.3186 214.885 257.938 2 Control 173 1 299.317 1.33499 78.1779 150.57 204.841 3 Control 64.85 1 147.257 1.44923 158.906 332.241 393.892
mean 106.11 1 245.97 1.54 105.33 232.5 285.55
1 Berg 76.66 2 364.987 2.38413 64.112 220.518 306.167 2 Berg 58.73 1 244.207 2.72154 95.8204 376.224 442.388 3 Berg 95.02 2 262.468 1.25088 89.1536 160.89 250.662 Mean 76.80 1.66 290.55 2.11 83.02 252.53 333.07
Mice dosed orally with 0.75 mg RKA182 (salt) in water (16 mg/kg) at time 0 Blood sample centrifuged and plasma sample collected and frozen Control = RKA mice received drug only Berg = RKA mice were infected with P.berghei at approximately 4% parasitemia Supplementary Figures Figure S1: Main metabolic pathways in the amide tetraoxanes
168

9
Supplementay Data X-Ray Data for 15 (RKA182) CCDC 757528. C28H45N3O5, M = 503.67, colourless prism, 0.33 x 0.27 x 0.15 mm3, monoclinic, P21/c (No. 14), a = 20.387(4) b = 6.1810(1), c = 20.945(4) Å, β= 106.617(4)°, V = 2587.4(9) Å3, Z = 4, Dc = 1.293 g/cm3, T = 100(2)K, 17914 reflections collected, 6876 unique, Rint = 0.0435, Final GooF = 0.984, R1 = 0.0467, R2 = 0.1099,328 parameters, 0 restraints, µ = 0.088 mm-1 Supplementary Materials and Methods Chemistry Preparation of ethyl 2-(4-oxocyclohexyl)acetate 8 A solution of ethyl 2-(1,4-dioxaspiro[4.5]decan-8-yl)acetate (13 mmol) in 1M HCl (30 ml) was stirred at room temperature for 2-4 hr. The aqueous solution was neutralised with sodium bicarbonate and extracted with dichloromethane (3 × 15 ml). The combined organic extracts were washed with brine (10 ml), dried over MgSO4, filtered and concentrated under reduced pressure to give a colourless liquid. Purification by flash column chromatography gave 8 in 94%. 1HNMR (400MHz, CDCl3) δH, 1.28 (t, 3H, J = 7.1 Hz, CH3), 1.43-1.56 (m, 2H, CH2), 2.06-2.13 (m, 2H, CH2), 2.32 (s, 2H, CH2CO), 2.36-2.42 ( m, 5H, CH2/CH), 4.15(q, 2H, J = 7.1 Hz, OCH2), 13CNMR (100MHz, CDCl3), δC 14.6, 32.7, 33.4, 40.5, 40.9, 60.7, 172.8, 211.7 MS (CI), [M + NH4] + (100), 202.1. Preparation of ethyl 2-(4,4-dihydroperoxycyclohexyl)acetate 9 50% aq. Hydrogen peroxide (4 ml) was added to a stirring solution of 8 (6.31 mmol) in formic acid (8 ml) and acetonitrile (8 ml) at 0 °C. The solution was allowed to warm to room temperature and stirred for 15 min. The solution was diluted with dichloromethane (30 ml) and washed with water (10 ml), saturated aq. NaHCO3 (10 ml) and brine (10 ml). The organic phase was dried over MgSO4, filtered and concentrated to give 9 1HNMR (400MHz, CDCl3) δH, 1.26(t, 3H, J = 7.15Hz, CH3), 1.62(m, 2H, cyclohexyl),1.78(m, 4H, cyclohexyl), 1.92(m, 2H, cyclohexyl), 2.22 (d, 2H, J = 13.51Hz, CH2CO), 2.4 (m, 1H, CH), 4.14 (q, 2H, J = 7.15Hz, OCH2), 8.55 (bs, 2H, OH). 13CNMR (100MHz, CDCl3), δC 14.20, 24.78, 28.19, 41.76, 60.67, 109.58. MS (ES+), [M + Na] + (100), 389.1 [2M + Na] + 257.3 Preparation of ethyl dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-ylacetate 10 A solution of 9 in DCM (10mL) was added to a stirring solution of 2-adamantanone (9.46 mmol) and rhenium (VII) oxide (0.13 mmol) in DCM (10 mL) at 0 °C. The solution was allowed to warm to room temperature and stirred for 45 min before being filtered through a plug of silica and the filtrate was concentrated under reduced pressure to give a white solid. Purification by flash chromatography on silica gel (10/90 ethyl acetate/n-hexane) gave 10 as a white solid in 48% yield. Mpt. 60-62oC Vmax (CHCl3)/cm-1 1446.8, 1718.5, 2858.9, 2922.3, 3003.8 1HNMR (400MHz, CDCl3) δ H, 1.25(t, 3H, J = 7.31Hz, CH3), 1.28-1.37(m, 2H, CH2), 1.48-1.79(m, 10H, CH2), 1.87(bs, 2H, CH2), 1.91-2.20(m, 9H, CH2/CH), 2.23(d, 2H, J = 6.83Hz, CH2CO), 4.13(q, 2H, J = 7.21Hz, CH2)13CNMR (100MHz, CDCl3), δC 14.62, 27.48, 27.87, 34.10, 36.72, 37.37, 39.65, 41.12, 47.38, 60.62, 107.99, 110.78, 173.00 MS (ES+), [M + Na] + (100), 389.1 [2M + Na] + 755.2 HRMS calculated for 389.1940 C20H30O6Na found, 389.1954. Preparation of dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-ylacetic acid 11 A solution of 10 (3.06 mmol) in 10% w/v potassium hydroxide/methanol (10 ml) was stirred at reflux for 90 min. The solution was allowed to cool to room temperature and concentrated under reduced pressure. The resulting residue was taken up in water (15 ml) and washed with diethyl ether (3 × 10 ml). The aqueous layer was acidified with concentrated hydrochloric acid and a white precipitate formed. Diethyl ether (15 ml) was added to dissolve the precipitate and the aqueous phase extracted with diethyl ether (2 × 10 ml). The combined organic phases were washed with brine (10 ml), dried over Na2SO4, filtered and concentrated under reduced pressure to give a white solid. Recrystallisation from ethanol gave 11 as a white solid in 94% yield. Vmax (CHCl3)/cm-1 991.8, 1057.5, 1446.7, 1694.3, 2844.0, 2924.8, 3005.7 3355.7. 1HNMR (400MHz, CDCl3) δH, 1.22-1.46(m, 2H, CH2), 1.50-1.90(m, 12H, CH2), 1.01-2.05(m, 4H, CH2), 2.06-2.15(m, 5H, CH), 2.29(d, 2H, J = 6.83Hz, CH2CO). 13CNMR (100MHz, CDCl3), δ C 27.47, 27.84, 33.52, 33.86, 36.69, 37.35, 39.65, 40.75, 47.34, 108.89, 110.79, 178.23. MS (ES+), [M - H] + (100), 337.2 HRMS calculated for 337.1651 C18H25O6 found, 337.1663. Preparation of amide tetraoxanes 12-18
General procedure
169

10
To a solution of the acid 11 (0.89 mmol) in dry DCM (20 mL), with added triethylamine (1.34mmol, 1.5 eq) and ethylchloroformate (0.89mmol, 1.0 eq). The reaction was stirred for 60 minutes at 0oC. (0.89 mmol, 1.0 eq) of the required amine was added, and after 30 minutes of stirring the reaction mixture was warmed to room temperature. After 90 minutes, it was diluted with water and extracted with DCM. The organic extracts were washed with brine, dried over anhydrous Na2SO4. The crude products were purified by flash column chromatography.
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-yl)-1-(4-
isobutylpiperazin-1-yl)ethanone 12
The general procedure above was followed to give 12 as a white solid in 87% yield. 1H NMR (400MHz, CDCl3) δH, 0.89 (m, 6H, J = 6.46Hz, CH3), 1.17-1.36 (m, 2H, adamantylidene), 1.50-1.84 (m, 14H, adamantylidene/CH2), 1.86 bs, 2H, CH2), 1.90-2.04 (m, 6H, CH), 2.08 (d, 2H, J = 7.40Hz, CH2CO), 2.20-2.27 (m, 2H, NCH2), 2.35 (t, 4H, CH2N), 3.45(t, 2H, J = 4.74Hz, NCH2), 3.62(t, 2H, J = 4.74Hz, NCH2). 13C NMR (100MHz, CDCl3), δC 21.2, 25.8, 27.4, 33.5, 34.4, 37.3, 39.4, 42.1, 46.3, 53.7, 54.2, 67.2, 108.2, 110.8, 170.7. MS (ES+), [M + Na] + (100), 463.3 HRMS calculated for 463.3172 C26H43O5N2Na, found 4632.3187. Elemental analysis C: 67.62, H: 9.18, N: 6.09 (required values C: 67.50, H: 9.15, N: 6.06).
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan] -4-yl)-1-(morpholin-4-yl)ethanone 13
The general procedure above was followed to give 13 as a white solid in 81% yield. Mpt. 139-140 oC υmax (CHCl3)/cm-1 1442.3, 1632.5, 2858.9, 2913.2, 3003.8 1H NMR (400MHz, CDCl3) δH, 1.11-1.38 (m, 2H, CH2), 1.50-1.82 (m, 12H, CH2), 1.85 (bs, 2H, CH2), 1.90-2.18 (m, 5H, CH), 2.30 (d, 2H, J = 7.0 Hz, CH2CO), 3.46 (t, 2H, J = 4.6 Hz, NCH2), 3.60-3.69 (m, 6H, NCH2/CH2O) 13C NMR (100MHz, CDCl3), δC 27.5, 28.9, 33.5, 33.6, 34.3, 37.4, 39.2, 42.4, 46.6, 67.3, 108.1, 110.8, 170.9 MS (ES+), [M + Na] + (100), 430.2 [2M + Na] +, 837.4 HRMS calculated for 430.2206 C22H33O6NNa, found 430.2213. Elemental analysis C: 65.10, H: 8.18, N: 3.39 (required values C: 64.84, H: 8.16, N: 3.44)
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-yl)-1-[4-(morpholin-4-
yl)piperidin-1-yl]ethanone 14
The general procedure above was followed to give 14 as a white solid in 88% yield. 1H NMR (400MHz, CDCl3) δH 1.20-2.03 (m, 20H, CH2), 2.24 (bs, 2H, CH2), 2.40 (m, 1H, CH), 2.56 (bs, 6H, CH2), 2.78 (m, 1H, CH), 3.01 (t, 2H, J = 11.5 Hz, CH2), 3.72 (t, 6H, J = 4.6 Hz, CH2), 3.91 (d, 2H, J = 13.6 Hz, CH2), 4.65 (d, 2H, J = 11.4 Hz, CH2) 13C NMR (100MHz, CDCl3), δC 27.5, 28.5, 29.4, 33.5, 34.4, 37.4, 39.4, 41.3, 45.4, 50.2, 62.3, 67.5, 108.2, 110.8, 170.6 MS (ES+), [M + H ] + (100) 491.3 HRMS calculated for 491.3121C28H42O6N2, found 491.3127. Elemental analysis C: 65.96, H: 8.55, N: 5.67 (required values C: 66.10, H: 8.63, N: 5.71).
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-yl)-1-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]ethanone 15
The general procedure above was followed to give 15 as a white solid in 86% yield. Mpt. 128-130 oC. 1H NMR (400MHz, CDCl3) δH 1.20-2.07 (m, 27H, CH/CH2), 2.23 (bs, 2H, CH2), 2.28 (s, 3H, NCH3), 2.36-2.68 (m, 7H, CH/CH2), 3.00 (t, 2H, J = 11.1 Hz, CH2), 3.90 (d, 2H, J = 11.0 Hz, CH2), 4.65 (d, 2H, J = 13.4 Hz, CH2). 13C NMR (100MHz, CDCl3), δC 27.5, 28.6, 29.6, 33.5, 34.4, 37.3, 39.4, 41.5, 45.5, 46.4, 49.5, 55.7, 62.1, 108.2, 110.8, 170.5. MS (ES+), [M + H ] + (100) 504.4 HRMS calculated for 504.3437 C28H46O5N3, found 504.3426. Elemental analysis C: 66.44, H: 9.06, N: 8.34 (required values C: 66.77, H: 9.04, N: 8.30).
170

11
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-yl)-1-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]ethanone 16 The general procedure above was followed to give 16 as a white solid in 88% yield. 1H NMR (400MHz, CDCl3) δH 1.26 (bs, 2H, adamantylidene), 1.52-2.07 (m, 27H, CH/CH2), 2.26 (bs, 6H, CH/CH2), 2.56 (t, 4H, J = 4.5Hz, CH2), 2.90 (d, 2H, J = 11.9 Hz, CH2), 3.46 ( t, 2H, J = 3.8 Hz, CH2), 3.62 (t, 2H, J = 3.8 Hz, CH2) 13C NMR (100MHz, CDCl3), δC 27.5, 28.5, 33.5, 34.3, 37.3, 39.3, 42.3, 46.5, 49.4, 49.7, 55.7, 61.9, 108.2, 110.8, 170.6 MS (ES+), [M +H ] + (100) 504.3 HRMS calculated for 504.3437 C28H46O5, found 504.3462. Elemental analysis C: 66.68, H: 9.03, N: 8.33 (required values C: 66.77, H: 9.04, N: 8.30).
Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]
-4-yl)-N-[2-(pyrrolidin-1-yl)ethyl]acetamide 17
The general procedure above was followed to give 17 as a white solid in 76% yield. Mpt. 142-144oC. Vmax (CHCl3)/cm-1
1446.7, 1559.9, 1641.1, 2859.1, 2931.2, 2937.7, 3260.8 1HNMR (400MHz, CDCl3) δH 1.19-1.35(m, 2H, CH2), 1.50-1.83(m, 14H, CH2), 1.83-1.89(m, 4H, CH2), 1.90-2.04(m, 5H, CH), 2.12(d, 2H, J = 7.02Hz, CH2CO), 2.50-2.67(m, 6H, NCH2/CH2N), 3.31(q, 4H, J = 5.50Hz, CH2), 6.55(bs, 1H, NH). 13CNMR (100MHz, CDCl3), δ C 23.81, 23.83, 27.47, 27.85, 28.61, 33.52, 33.53, 34.45, 36.69, 37.35, 37.95, 39.64, 39.80, 43.51, 47.36, 50.89, 54.29, 55.33, 55.57, 61.06, 108.12, 110.74, 172.53. MS (ES+), [M + Na] + (100), 457.2 [2M + Na] +, 891.3 HRMS calculated for 457.2678 C24H38O5N2Na found, 457.2680. Elemental analysis C: 66.29, H: 8.80, N: 6.44 (required values C: 66.33, H: 8.81, N: 6.45). Preparation of 2-(dispiro[cyclohexane-1,3'-[1,2,4,5]tetroxane-6',2''-tricyclo[3.3.1.13,7]decan]-4-yl)-1-[4-(pyrrolidin-1-yl)piperidin-1-yl]ethanone 18 The general procedure above was followed to give 18 as a white solid in 82% yield. 1H NMR (400MHz, CDCl3) δH 1.20-1.36 (m, 4H, adamantylidene), 1.42-1.80 (m, 14H, admantylidene/CH2), 1.81-1.89 (m, 8H, CH2), 1.90-2.04 (m, 5H, adamantylidene/CH), 2.24 (d, 2H, J = 7.22 Hz, CH2CO), 2.62-2.73 (m, 4H, NCH2), 3.05 (t, 1H, J = 11.6 Hz, NH), 3.85 (t, 2H, J = 13.5 Hz, NCH2), 4.56 (t, 2H, J = 13.5 Hz, NCH2). 13C NMR (100MHz, CDCl3), δC 23.7, 27.5, 31.0, 32.0, 33.6, 34.4, 37.4, 39.4, 40.7, 44.8, 51.7, 62.1, 108.2, 110.8, 170.5. MS (ES+), [M + Na] + (100), 475.3 HRMS calculated for 475.3172 C27H43O5N2Na, found 475.3163. Elemental analysis C: 68.22, H: 8.90, N: 5.91 (required values C: 68.32, H: 8.92, N: 5.90). General procedure for the reaction of tetraoxanes with heme: Hemin stock solution (10 mM in 0.1 M NaOH) and acetonitrile were freshly degassed prior to use. Hemin solution (2.0 mL, 0.02 mmol) was added to a suspension of tetraoxane RKA099 (10.0 mg, 0.02 mmol, 1 equiv.) and dithionite (41 mg, 0.20 mmol, 10 equiv.) in acetonitrile (2.0 mL) under argon. The mixture was stirred at room temperature. TLC monitoring showed that all the starting peroxide was consumed within 20 min. the crude mixture was analyzed by RP-HPLC. HPLC analysis conditions: Column: C18-Reversed phase preparative column; Gradient: From 70% MeOH/0.1% TFA to 100% MeOH/0.1% TFA over 60 min; Flow rate: 0.5 mL/min ; Wavelength: 430 nm. In Vitro Antimalarial Screening Cultures were grown in flasks containing human erythrocytes (2-5%) with parasitemia in the range of 1% to 10% suspended in RPMI 1640 medium supplemented with 25 mM HEPES and 32 mM NaHCO3, and 10% human serum (complete medium). Cultures were gassed with a mixture of 3% O2, 4% CO2 and 93% N2. Antimalarial activity was assessed with an adaption of the 48-h sensitivity assay of Desjardins et al. using [H]-hypoxanthine incorporation as an assessment of parasite growth.1 Stock drug solutions were prepared in 100% dimethylsulphoxide (DMSO) and diluted to the appropriate concentration using complete medium. Assays were performed in sterile 96-well microtitre plates, each plate contained 200 µl of parasite culture (2% parasitemia, 0.5% haematocrit) with or without 10 µl drug dilutions. Each drug was tested in triplicate and parasite growth
compared to control wells (which consituted 100 % parasite growth). After 24-h incubation at 37oC, 0.5 µCi hypoxanthine was added to each well. Cultures were incubated for a further 24 h before they were harvested onto filter-mats, dried for 1 h at
55oC and counted using a Wallac 1450 Microbeta Trilux Liquid scintillation and luminescence counter. IC50 values were calculated by interpolation of the probit transformation of the log dose - response curve. In vivo Antimalarial Screening In vivo data was determined using 30 mg/kg oral (po) and subcutaneous (sc) doses in a 4–days Peter’s test.2 For subcutaneous administration, compounds were dissolved in 10% dimethylsulfoxide (DMSO) 0.05% Tween 80 (Sigma, Dorset, UK) in distilled water. For oral admistration, compounds were dissolved in standard suspending formula (SSV) [0.5% sodium carboxymethylcellulose, 0.5% benzyl alcohol, 0.4% Tween 80, 0.9% NaCl (all Sigma)]. Subcutaneous (s.c) or oral (p.o) treatment was done with 0.2ml of a solution of the test compound two hours (day 0) and on days 1, 2, and 3 post infections. Parasitaemia was determined by microscopic examination of Giemsa stained blood films taken on day 4. Microscopic counts
171

12
of blood films from each mouse were processed using MICROSOFT@EXCELL spreadsheet (Microsoft Corp.) and expressed as percentage of inhibition from the arithmetic mean parasitaemias of each group in relation to the untreated group. Experiments described in Table S5 were generated at the Swiss Tropical and Public Health Institute with a slightly modified methodology. NMRI mice were infected intravenously on day 0 with the Plasmodium berghei GFP ANKA malaria strain (2 × 107 parasitized erythrocytes, donation from AP Waters and CJ Janse, Leiden University). The mice were treated orally with either three doses (24, 48, and 72 h post-infection) or a single dose (24 h post-infection) of compound with a volume of 10 mL/kg. Blood parasitemia levels were determined by FACS analysis. In vivo pharmacokinetic methods Dimethylsulphoxide (DMSO) was obtained from Fisher (UK). Tetraglycol was obtained from Sigma-Aldrich (UK). Ethanol was obtained from Hayman (UK) and sterile saline (0.9%, w/v) was obtained from Baxter (UK).All other chemicals used on this study were reagent grade or higher and were obtained from standard commercial suppliers. Seventy-eight male Sprague Dawley rats, aged 8-12 weeks, were supplied by Charles River (UK) Limited and surgically prepared with an indwelling femoral vein cannula. After a post-surgery recovery period of at least 5 days and prior to dose administration, a pre-study health evaluation was conducted on these animals and the results were found to be satisfactory.A standard laboratory diet and mains tap water were available ad libitum to the animals.Clinical signs were monitored at regular intervals throughout the study in order to assess any reaction to treatment.
Intravenous Dose Formulation and Administration
For intravenous administration, the test items were formulated as solutions in 5% DMSO, 5% ethanol, 50% tetraglycol and 40% sterile saline (v/v/v/v) to achieve the intravenous target concentrations of 1 mg/mL. The weight of each formulation was recorded in order to determine the theoretical dose concentration. Each intravenous formulation was administered to a group of three animals as a slow bolus over ca 30s via a tail vein at a target dose volume of 1 mL/kg to achieve a target dose level of 1 mg/kg. The dose volume administered was calculated according to the body weight of the animals on the day prior to administration. Three aliquots of each formulation (100 µL) were taken on completion of dose preparation and stored at ca -80°C.
Oral Dose Formulation and Administration
For oral administration, the test items were formulated as solutions in 5% DMSO, 5% ethanol and 90% tetraglycol (v/v/v) to achieve final target concentrations of 10 mg/mL. The weight of each formulation was recorded in order to determine the theoretical dose concentration.Each oral formulation was administered to a group of three animals via gastric gavage at a target dose volume of 1 mL/kg to achieve a target dose level of 10 mg/kg. The dose volume administered was calculated according to the body weight of the animals on the day prior to administration. Three aliquots of each formulation (100 µL) were taken on completion of dose preparation and stored at ca -80°C. All blood samples (ca 0.5 mL) were collected into glass tubes containing lithium heparin as an anti-coagulant from each animal via the femoral vein cannula at the following sampling times post dose: 0.25, 0.5, 1, 2 and 6 h post intravenous dose and 0.5, 1, 2, 4 and 6 h post oral dose. As soon as practically possible, each sample was centrifuged at 3000 r.p.m. for 10 min at ca 4°C. Plasma was separated and transferred into glass tubes. Plasma samples were stored frozen at ca -80°C prior to analysis.For each analysis batch, a calibration series of samples containing the test item (analyte) of interest were prepared using 50 µL of control plasma to give a range of concentrations generally 1 to 1000 ng/mL. The standards were prepared and analysed in batches along with freshly prepared blank samples. Plasma (50 µL) samples and standards were analysed and prepared for each analyte using a method based upon protein precipitation with acetonitrile containing and internal standard, Nifedipine (added at a matrix concentration of 100 ng/mL). An aliquot of the supernatant was analysed by reverse phase LC-MS/MS using a heat assisted electrospray interface in negative ion mode. The intravenous and oral pharmacokinetics of each test item in male rats were characterised using mean plasma concentration vs time data. The pharmacokinetic parameters C0, Cmax(obs), Tmax(obs), AUC(0-t), AUC(0-∞), T½, CL, CL/F and F were estimated using WinNonLin v4.1 pharmacokinetic software. Speed of action Swiss outbred 18-20g male CD-1 mice (Charles Rivers, UK) were kept in specific pathogen-free conditions and fed ad libitum. Mice were infected intravenously with 4 x 106 P. berghei NY infected red cells obtained from donor mice, randomized and divided in groups of five mice (day 0). RKA182 or artesunate (ASN) were dissolved in standard suspending formula (SSV) [0.5% sodium carboxymethylcellulose, 0.5% benzyl alcohol, 0.4% Tween 80, 0.9% NaCl (all Sigma)] and administered by oral gavage in a single oral dose on day two post-infection. Parasitaemias were determined by microscopic examination of Giemsa stained thin blood films at 0, 0.5h, 1h, 2h, 2.5h, 6h, 8h, 24h, 48h post-treatment and the microscopic counts for each mouse processed using MICROSOFT ® EXCEL, and plotted as the arithmetic mean parasitaemia versus time. Additional Information
172

13
A full toxicological profile has been carried out on tetraoxane 15. The compound recorded no positive responses in the Ames test showing no mutagenicity in strains of Salmonella typhimurium and Escherichai coli, was found not to be clastogenic when tested with Chinese hamster ovary cells in vitro and demonstrated no potential to induce forward mutation in mouse lymphoma cells. Single dose oral toxicity experiments in Sprague-Dawley rats gave a maximum tolerated dose of 400 mg/kg. The cardiac toxicity (QT interval prolongation) was also evaluated by the conventional whole-cell patch-clamp technique by recording outward potassium currents [human Ether-a-Go-go related gene (hERG)] from single cells of HEK-293. An IC50 of greater than 10 uM was observed and testing of 15 against the multidrug resistant cancer cell line KB gave an IC50 of greater than 21 mM giving the compound a therapeutic index (TI) of greater than 14,000 (See Table S2). Compound 15 also did not inhibit any of the major human cytochrome P450’s with all IC50s greater than 20 mM and most above 100 mM. References 1. Desjardins, R. E.; Canfield, C. J.; Haynes, J. D.; Chulay, J. D.Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. 2. Peters, W.; Robinson, B. L. Malaria; Academic Press: San Diego, 1999.
173

14
Entry for the Table of Contents
Antimalarial Agents
((Author(s), Corresponding Author(s)*)) __________ Page – Page
Identification of the First 1,2,4,5-Antimalarial Tetraoxane Drug-Development Candidate (RKA 182) with Superior Properties to the Semi-Synthetic Artemisinins
From a library of over 150 1,2,4,5-tetraoxanes we have candidate selected a molecule, RKA182, for full formal preclinical development. RKA182 has outstanding in vitro activity against sensitive and resistant strains of P.falciparum (IC50=1.1 nM (K1), 0.8 nM (3D7)) and retains this level of activity against South East Asian isolates that failed ACT combination chemotherapy. The drug candidate has a fast rate of parasite kill, superior in vivo activity to artesunate (ED50=1.33 mg/kg and ED90=4.18 mg/kg) in rodent models of malaria and improved DMPK characteristics and stability in the presence of infected red blood cells when compared to synthetic ozonides or semi-synthetic artemisinins.
174


Author: Fatima Bousejra‐El Garah
Title: Role of metals in the mechanism of action of antimalarial peroxides
Thesis supervisor: Dr Anne Robert
Abstract: The 1,2,4‐trioxane core structure of artemisinin is essential for its activity. It was shown
that iron(II) catalyses the reductive cleavage of the peroxide bond of the drug, leading to
the formation of C‐centered radicals. These radicals are able to alkylate heme resulting
from host cell hemoglobin digestion, and parasitic proteins. These alkylation processes
are believed to induce parasite death.
In the present work, we studied the two main mechanisms proposed in the literature for
artemisinin, namely heme alkylation and PfATP6 inhibition. In addition, we also explored
the possible bio‐activation of artemisinin by copper enzymes.
We report our investigation into the reactivity of metal salts and complexes, such as
heme, toward highly active antimalarial peroxide‐containing drugs, namely artemisone,
trioxaquines, trioxolanes, and tetraoxanes.
Overall, our results with heme confirmed that the alkylating properties of artemisinin, in
particular in malaria‐infected mice, are not limited to this natural compound, but are
shared with other potent peroxide‐containing drugs. It is likely that heme alkylation plays
a very important role in their anti‐plasmodial mechanism of action.
In this work, we also considered an alternative mechanism of action for artemisinin,
based on the inhibition of PfATP6. The main result is that the predicted binding affinity of
the tested compounds does not correlate with their in vitro antimalarial activity.
Key words: Malaria / Treatment / Antimalarial peroxides / Artemisinin / Trioxaquines / Tetraoxanes / Heme / Alkylation / PfATP6 / Modelling
Laboratoire de Chimie de Coordination du CNRS 205 route de Narbonne, 31077 Toulouse, Cedex 4

Auteur : Fatima Bousejra‐El Garah
Titre : Role of metals ism of action of antimalarial peroxides in the mechan
Directeur de thèse : Dr Anne Robert
Lieu et date de soutenance : Toulouse, le 18 mars 2010
Résumé : Le cycle 1,2,4‐trioxane de l’artémisinine est essentiel pour son activité antipaludique. Le
fer(II) peut catalyser la réaction de réduction de la liaison peroxyde de l’artémisinine et
conduire à la formation de radicaux. Ces radicaux alkylent l’hème libéré lors de la
dégradation de l’hémoglobine par Plasmodium pour former des produits de couplage
appelés adduits hème‐artémisinine. Ce processus d’alkylation pourrait être directement
ou indirectement responsable de la mort du parasite. La connaissance du mécanisme
d’action de l’artémisinine est le point de départ de ce projet de thèse qui se divise en deux
parties distinctes. La première partie consiste à étudier le rôle de complexes métalliques,
l’hème en particulier, dans le mécanisme d’action in vitro et in vivo de nouveaux
peroxydes synthétiques ou hémi‐synthétiques (artémisone, trioxaquines, trioxolanes et
tétraoxanes). Nous avons caractérisé des adduits hème‐drogue dans tous les cas, et
econfirmé l’importance de l’hème dans l’activité antipaludique d ces molécules.
Une étude de docking a également été réalisée entre ces molécules et la protéine
parasitaire PfATP6 présentée dans la littérature comme une autre cible de l’artémisinine.
Les principaux résultats de notre étude montrent qu’il n’y a pas de corrélation entre
l’affinité pour la protéine PfATP6 et l’activité in vitro des molécules testées.
Mots clés : Paludisme / Traitement / Peroxydes antipaludiques / Artémisinine / Trioxaquine Alkylation / PfATP6 / Modélisation s / Tétraoxanes / Hème /
Spécialité : Chimie – Biologie – Santé
Laboratoire de Chimie de Coordination du CNRS 205 route de Narbonne, 31077 Toulouse, Cedex 4
Related Documents











