Coordination Chemistry Reviews 256 (2012) 328–351 Contents lists available at ScienceDirect Coordination Chemistry Reviews journal homepage: www.elsevier.com/locate/ccr Review Thermodynamics of lanthanide(III) complexation in non-aqueous solvents Plinio Di Bernardo a , Andrea Melchior b , Marilena Tolazzi b,∗ , Pier Luigi Zanonato a a Dipartimento di Scienze Chimiche, Università di Padova, Via Marzolo 1, 35131 Padova, Italy b Dipartimento di Scienze e Tecnologie Chimiche, Università di Udine, Via del Cotonificio 108, 33100 Udine, Italy Contents 1. Introduction ......................................................................................................................................... 329 2. Fundamental properties of Ln ions ................................................................................................................. 329 2.1. Relative stabilities of oxidation states ....................................................................................................... 329 2.2. Ionic radii .................................................................................................................................... 330 3. Lanthanide(III) solvation in non-aqueous solvents ................................................................................................. 332 3.1. Ln(III) ions preferential solvation ........................................................................................................... 332 3.2. Structural aspects of Ln(III) solvation ....................................................................................................... 332 4. Thermodynamics of complex formation with charged inorganic ligands .......................................................................... 333 5. Thermodynamics of complex formation with neutral organic ligands ............................................................................ 335 5.1. Coordination of O-donors ................................................................................................................... 335 5.1.1. Crown ethers ....................................................................................................................... 335 5.1.2. Complexation with diazamacrocycles and cryptands ............................................................................. 340 5.1.3. Solvent influence on crown ethers and cryptands complexation ................................................................. 343 5.1.4. Stabilization of lanthanide low oxidation states .................................................................................. 343 5.2. Coordination of N-donors ................................................................................................................... 343 5.2.1. Monodentate amines .............................................................................................................. 343 5.2.2. Polydentate amines ................................................................................................................ 344 5.2.3. Heterocyclic N-donor ligands ...................................................................................................... 346 6. Conclusions .......................................................................................................................................... 350 Acknowledgments .................................................................................................................................. 350 References ........................................................................................................................................... 350 article info Article history: Received 16 May 2011 Received in revised form 1 July 2011 Accepted 20 July 2011 Keywords: Lanthanides(III) Thermodynamics Complexation Solvation Non-aqueous solvents Neutral donors Charged inorganic ligands Coordination number Ionic radius abstract Lanthanide(III) coordination compounds are employed in several fundamental and applied research fields such as organic synthesis, bioinorganic chemistry, optical and magnetic imaging, catalysis, environment and geochemistry. All these applications have been favoured by the recent developments of a detailed knowledge of fundamental properties (electronic, spectroscopic, thermodynamic, magnetic, structural) of elements, ions and their compounds. Ln 3+ are hard acids and present strong affinity for charged ligands or neutral O- and N-donors, as indicated by a wide number of papers concerning formation of their complexes in solution. These studies allowed one to gain information on the complex stabilities, the metal-ion selectivity of a given ligand, the influence of the solvent on the nature and stability of the species in solution. Most of the above studies deal with aqueous solutions, while studies in non-aqueous media are less common. Despite more limited, investigations in aprotic solvents are particularly interesting as they allow one to extend the knowledge on the coordination chemistry of lanthanide(III), disclosing metal–ligand interactions not easily accessible in water due to ligand protonation equilibria, Ln(III) hydrolysis and strong hydration of the cations, which hampers interactions with neutral donors. This review analyzes a wide number of thermodynamic studies concerning formation of lanthanide(III) complexes with selected, simple neutral N-donors (amines, pyridines), O-donors (crown ethers, aza- crown ethers and cryptands) and charged inorganic ligands (halides, thiocyanate, nitrate, perchlorate, ∗ Corresponding author. Tel.: +39 0432 558852; fax: +39 0432 558803. E-mail address: [email protected] (M. Tolazzi). 0010-8545/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.ccr.2011.07.010

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

R
T
Pa
b
C
a
ARRA
KLTCSNNCCI
0d
Coordination Chemistry Reviews 256 (2012) 328–351
Contents lists available at ScienceDirect
Coordination Chemistry Reviews
journa l homepage: www.e lsev ier .com/ locate /ccr
eview
hermodynamics of lanthanide(III) complexation in non-aqueous solvents
linio Di Bernardoa, Andrea Melchiorb, Marilena Tolazzib,∗, Pier Luigi Zanonatoa
Dipartimento di Scienze Chimiche, Università di Padova, Via Marzolo 1, 35131 Padova, ItalyDipartimento di Scienze e Tecnologie Chimiche, Università di Udine, Via del Cotonificio 108, 33100 Udine, Italy
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3292. Fundamental properties of Ln ions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 329
2.1. Relative stabilities of oxidation states . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3292.2. Ionic radii . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 330
3. Lanthanide(III) solvation in non-aqueous solvents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3323.1. Ln(III) ions preferential solvation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3323.2. Structural aspects of Ln(III) solvation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 332
4. Thermodynamics of complex formation with charged inorganic ligands. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3335. Thermodynamics of complex formation with neutral organic ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335
5.1. Coordination of O-donors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3355.1.1. Crown ethers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3355.1.2. Complexation with diazamacrocycles and cryptands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3405.1.3. Solvent influence on crown ethers and cryptands complexation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3435.1.4. Stabilization of lanthanide low oxidation states . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 343
5.2. Coordination of N-donors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3435.2.1. Monodentate amines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3435.2.2. Polydentate amines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3445.2.3. Heterocyclic N-donor ligands. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 346
6. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 350Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 350References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 350
r t i c l e i n f o
rticle history:eceived 16 May 2011eceived in revised form 1 July 2011ccepted 20 July 2011
eywords:anthanides(III)hermodynamicsomplexationolvationon-aqueous solventseutral donors
a b s t r a c t
Lanthanide(III) coordination compounds are employed in several fundamental and applied research fieldssuch as organic synthesis, bioinorganic chemistry, optical and magnetic imaging, catalysis, environmentand geochemistry. All these applications have been favoured by the recent developments of a detailedknowledge of fundamental properties (electronic, spectroscopic, thermodynamic, magnetic, structural)of elements, ions and their compounds.
Ln3+ are hard acids and present strong affinity for charged ligands or neutral O- and N-donors, asindicated by a wide number of papers concerning formation of their complexes in solution. These studiesallowed one to gain information on the complex stabilities, the metal-ion selectivity of a given ligand,the influence of the solvent on the nature and stability of the species in solution. Most of the abovestudies deal with aqueous solutions, while studies in non-aqueous media are less common. Despite morelimited, investigations in aprotic solvents are particularly interesting as they allow one to extend the
harged inorganic ligandsoordination number
onic radius
knowledge on the coordination chemistry of lanthanide(III), disclosing metal–ligand interactions noteasily accessible in water due to ligand protonation equilibria, Ln(III) hydrolysis and strong hydration ofthe cations, which hampers interactions with neutral donors.
This review analyzes a wide number of thermodynamic studies concerning formation of lanthanide(III)complexes with selected, simple neutral N-donors (amines, pyridines), O-donors (crown ethers, aza-crown ethers and cryptands) and charged inorganic ligands (halides, thiocyanate, nitrate, perchlorate,
∗ Corresponding author. Tel.: +39 0432 558852; fax: +39 0432 558803.E-mail address: [email protected] (M. Tolazzi).
010-8545/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.ccr.2011.07.010

P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351 329
triflate) in non-aqueous solvents. The main aim of the review is to face the basic question of what arethe factors governing the complex stability and selectivity within the lanthanide series and how arethey influenced by different coordinating media. Fundamental properties of Ln ions, such as ionic radii,common oxidation states and structural aspects of their solvates are as well analyzed.Several points emerged from a critical analysis of the papers reviewed:
i) Ln3+ salts used in thermodynamic studies in poor coordinating solvents are often not completelydissociated and, in this case, the data obtained reflect multiple simultaneous equilibria in solution. Com-parisons between thermodynamic results in poor and high solvating media must be therefore regardedwith caution as they may refer to different reacting metal-species, hence, to different metal–ligandequilibria.
ii) High solvating aprotic media can be considered as ideal for thermodynamic studies since lan-thanide(III) is only present as Ln(solv)n
3+species. However, in this case, the strong solvation of Ln3+
ions hinders complex formations with weak or relatively weak donors.iii) Solvation of lanthanide(III) cations in non-aqueous solutions is generally a major factor in determining
the complex stabilities which, for the different kinds of ligands examined, follow the general trend:PC > AN > MeOH > DMF > DMSO.
1
daowa
odclMma(e
ttdnsoc
opwii
catpliswrc[
v
in aqueous solution [29,30]. The peculiar stability of the three Eu ,Yb2+ and Sm2+ ions can be explained by their electronic configura-tion in the f orbitals: (i) the 4f7 half-filled shell for Eu(II), ii) filledshell 4f14 for Yb(II), (iii) “nearly half-filled” 4f6 shell for Sm(II) [29].
1 Ln(III)/Ln(II) reduction potentials (at 298.15 K): −0.35, −1.1 and −1.5 V forLn = Eu, Yb and Sm respectively. Other values (V) are: −2.3 (Tm), −2.6 (Nd), −2.6 (Dy),−2.7 (Pr), −2.9 (Ho), −3.1 (La), −3.1 (Er), −3.2 (Ce), −3.7 (Tb), −3.9 (Gd) [30]. Slightly
. Introduction
Lanthanide compounds have been extensively used in the lastecades as luminescent chemosensors [1], for medical diagnosticsnd optical cell imaging [1–3], contrast reagents for magnetic res-nance imaging [4], shift reagents for NMR spectroscopy [5], asell as for applications in fundamental and applied science such
s organic synthesis, bioorganic chemistry, catalysis [6].These applications were favoured by the increased knowledge
f fundamental properties (electronic, spectroscopic, thermo-ynamic, magnetic, structural) of the elements, achieved as aonsequence of the rapid development of academic studies on theanthanide coordination chemistry during the last three decades.
ost of these studies mainly concerned compounds where theetal ions have the common +3 oxidation state (Ln3+) and behave
s hard acids with a strong affinity for hard bases like O-donorsneutral or negatively charged) or N-donors with which they formssentially non-directional bonds of a predominant ionic nature [7].
The ionic radii of Ln3+ cations decrease with the increase ofheir atomic number (lanthanide contraction) [7]. Their coordina-ion number (CN) and strength of solvation vary in solution withinifferent reaction media, thus strongly influencing the thermody-amic stability of the complexes. This is the reason why severaltudies have been carried out aimed at interpreting the changesf the thermodynamic properties of Ln3+ complexes in terms ofhanges of the CN along the series [8–10].
In addition, Ln3+ show chemical properties very similar to thosef actinides(III) (An3+). This chemical similitude is a challengingroblem in the separation of An3+ from excess of Ln3+ in the nuclearaste treatments [11,12] but it is also useful because the safer Ln3+
ons can be used as surrogates for the much more hazardous An3+
ons in the preparatory studies.A relatively limited number of papers reviewed specifically the
omplexation thermodynamics of Ln3+ ions in solution [13–19]nd some data can be found also in more general reviews on thehermodynamics of metal complex formation [20–22]. Most of theapers cited in these reviews concern aqueous solutions, while
ess frequently the complex formation in non-aqueous solventss specifically considered and discussed [15,16,19]. Interestingtructure–property relationships are discussed in other reviewshich, besides thermodynamic studies in non-aqueous media,
eport structural and spectroscopic data on the formation of heli-
ates [23], complexation of synthetic ionophores [24] and anions25].Despite more limited than in water, studies in aprotic sol-ents are particularly interesting as they allow one to disclose
© 2011 Elsevier B.V. All rights reserved.
metal–ligand interactions not easily accessible in aqueous solutiondue to ligand protonation equilibria, Ln(III) hydrolysis and stronghydration of the cations, which hampers interactions with neutraldonors.
The aim of this review is to focus the reader on the general fac-tors influencing the complexation reactions within the lanthanideseries and to face how stabilities, types of complexes formed andselectivity can be influenced by different coordinating media. Forthis reason the review covers the solution thermodynamics oflanthanide complexes with selected families of simple ligands:charged inorganic ligands, crown ethers, cryptands and aza-crownmacrocycles, amines, pyridines, azines. In order to keep the dis-cussion in line with the proposed aim, structurally more complexligands presenting supramolecular interactions [26–28] and form-ing fascinating molecular edifices are only mentioned here.
Particular attention is paid to the reacting species present inthe organic media. Most of the starting Ln(III) salts used in thethermodynamic studies here reviewed are in fact not completelydissociated in poor coordinating solvents: in these cases, in theabsence of a solid knowledge of the reacting species, this review willconsider the available thermodynamic data as “apparent” quanti-ties.
2. Fundamental properties of Ln ions
2.1. Relative stabilities of oxidation states
In general, the most common and stable oxidation state of lan-thanides is +3. The three elements showing stable +2 oxidationstate, Eu, Yb and Sm, have been widely studied in solution and arich organometallic chemistry has been developed [29]. The sta-bility of the +2 oxidation state for the three elements is evidentfrom the less negative Ln(III)/Ln(II) standard reduction potentials
1 2+
different reduction potentials values can be found in Ref. [31] with the exception ofLa for which also the value of −3.8 V is present. Data were obtained from thermo-chemical cycles [30] and refer to Ln(III) chlorides. Only for Eu the standard reductionpotential has been obtained by e.m.f. measurements [30].

330 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Nomenclature
List of acronyms and IUPAC names of the ligands12C4 1,4,7,10-tetraoxacyclododecane15C5 1,4,7,10,13-pentaoxacyclopentadecane18C6 1,4,7,10,13,16-hexaoxacyclooctadecaneA15C5 1,4,7,10-tetraoxa-13-azacyclopentadecaneA18C6 1,4,7,10-tetraoxa-13,16-diazacyclooctadecaneA618C6 1,4,7,10,13,16-hexaazacyclooctadecane21C7 1,4,7,10,13,16,19-heptaazacyclohenicosaneB15C5 2,3,5,6,8,9,11,12-octahydrobenzo[b][1,4,7,
10,13]pentaoxacyclopentadecineS15C5 (Z)-2,3-diphenyl-1,4,7,10,13-
pentaoxacyclopentadec-2-eneBisB15C5 1,2-bis(16-nitro-2,3,5,6,8,9,11,12-
octahydrobenzo[b][1,4,7,10,13]pentaoxacyclo-pentadecin-15-yl)disulfane
BzA15C5 13-benzyl-1,4,7,10-tetraoxa-13-azacyclopentadecane
BzA18C6 16-benzyl-1,4,7,10,13-pentaoxa-16-azacyclooctadecane
BzA215C7 19-benzyl-1,4,7,10,13,16-hexaoxa-19-azacyclohenicosane
16C5 1,4,7,10,13-pentaoxacyclohexadecaneMM16C5 15,15-dimethyl-1,4,7,10,13-
pentaoxacyclohexadecaneMeO(CH2)2OCH2: MR16C5 15-((2-methoxyethoxy)
methyl)-15-methyl-1,4,7,10,13-pentaoxacyclohexadecane
BzA16C5 7-benzyl-1,4,10,13-tetraoxa-7-azacyclohexadecane
R′A16C5 7-(2-methoxyethyl)-1,4,10,13-tetraoxa-7-azacyclohexadecane
12P4 2,5,8,11-tetraoxadodecane15P5 2,5,8,11,14-pentaoxapentadecane18P6 2,5,8,11,14,17-hexaoxaoctadecane21P7 2,5,8,11,14,17,20-heptaoxahenicosaneDC18C6 icosahydrodibenzo[b,k][1,4,7,10,
13,16]hexaoxacyclooctadecineDec18C6 2-decyl-1,4,7,10,13,16-hexaoxacyclooctadecaneDB18C6 6,7,9,10,17,18,20,21-octahydrodibenzo[b,k][1,4,7,
10,13,16]hexaoxacyclooctadecineDB30C10 6,7,9,10,12,13,15,16,23,24,26,27,29,30,32,33-
hexadecahydrodibenzo[b,q][1,4,7,10,13,16,19,22,25,28]decaoxacyclotriacontine
DBPY18C6 dibenzopyridino-18-Crown-6(2.1.1) 4,7,13,18-tetraoxa-1,10-diazabicyclo[8.5.5]icosane(2.2.1) 4,7,13,16,21-pentaoxa-1,10-
diazabicyclo[8.8.5]tricosane(2.2.2) 4,7,13,16,21,24-hexaoxa-1,10-
diazabicyclo[8.8.8]hexacosane(2.1) 1,4,10-trioxa-7,13-diazacyclopentadecane(2.2) 1,4,10,13-tetraoxa-7,16-diazacyclooctadecaneTButB15C5 15-tert-butyl-2,3,5,6,8,9,11,12-octahydro-
benzo[b][1,4,7,10,13]pentaoxacyclopentadecinePyO2[18]dieneN6 3,6,14,17,23,24-hexaaza-
tricyclo[17.3.1.1]tetracosa-1(23),8,10,12,(24),19,21-hexaene
DiTerDiB18C6 2,14-di-tert-butyl-6,7,9,10,17,18,20,21-octahydrodibenzo[b,k][1,4,7,10,13,16]hexaoxacy-clooctadecine
n-but n-butylamineen ethylenediaminedien diethylenetriamine
Trien triethylenetetramineTetren tetraethylenepentamineTren tris(2-aminoethyl)amineL1 2-methoxyethylamineL2 2-aminoethanolL3 2-methoxyethyletherL4 di(ethyleneglycol)L5 2,2-oxydiethylamineL6 1,5-diamino pentanethmd 2,2,6,6-tetramethyl-3,5-heptanedionatoFod 6,6,7,7,8,8,8-heptafluoro-2,2-dimethyloctane-3,5-
dionatotta thenoyltrifluoroacetonatoL7 2,2′;6′,2′′-terpyridineL8 (2,4,6-tri(pyridin-2yl)-1,3,5-triazineL9 2,6-bis(1,2,4-triazin-3-yl)pyridineL10 2,6-bis(5,6-dimethyl-1,2,4-triazin-3-yl)pyridineL11 2,6-bis(pyridin-2-yl)4-amino-1,3,5-triazineL12 2,6-bis(pyridin-2-yl)pyrimidineL13 2,6-bis(benzimidazol-2-yl)pyridineL14 (2,6-bis(5,6-dipropyl-1,2,4-triazin-3-yl)pyridineL15 6-(5,6-dipentyl-1,2,4-triazin-3-yl)-2,2′-bipyridineL16 (6,6′-bis(5,6-dipentyl-1,2,4-triazin-3-yl)-2,2′-
bipyridineL17 6,6′-bis(5,5,8,8-tetramethyl-5,6,7,8-
tetrahydrobenzo-1,2,4-triazin-3-yl)-2,2′-bipyridine
L18 tris(2-benzimidazolylmethyl)amineL19 tris[(2-pyridyl)methyl]amineL20 6,6′-bis[bis(2-pyridylmethyl)aminomethyl]-2,2′-
bipyridineL21 2,6-bis(benzimidazol-2-yl)pyridineL22 2,6-bis(1-methyl-benzimidazol-2-yl)pyridineL23 2,6-bis(1-3,5-dimethoxybenzyl-benzimidazol-2-
yl)pyridineL24 2,6-bis[1-(3,5-dimethoxybenzyl)benzimidazol-2-
yl)]-4-(diethylamino)phenyl-pyridineL25 6-bis[1-(3,5-dimethoxybenzyl)benzimidazol-2-
yl)]-4-nitrophenyl-pyridine.Bipy 2,2′-bipyridine
Phen 1,10-phenanthrolineAmong the divalent lanthanides Eu2+ is the most stable in aqueoussolutions.
Examples of solid salts of lanthanides in +2 oxidation state are2
LnCl2, LnBr2, LnI2, LnF2, LnS, LnSe and LnTe, LnSO4, LnCO3, LnD2and LnH2 and EuC2O4 [7].
The +4 oxidation state is relatively stable in the case of Ce, Prand Tb, being the Ce4+ chemistry the most extensive in this group[7]. Solid compounds containing tetrapositive Ce, Pr, Nd, Tb and Dyhave been characterized; some examples are LnF4 (Ln = Ce, Pr, Tb)and LnO2 (Ln = Ce, Pr, Tb) [7,32].
2.2. Ionic radii
Lanthanide(III) ions form compounds both in solution and in thesolid state with a variety of structural features which may not be
2 For LnCl2 (Ln = Nd, Sm, Eu, Dy, Tm and Yb); for LnBr2 (Ln = Sm, Yb, Eu); LnI2
(Ln = Nd, Sm, Eu, Dy, Tm, Yb, La, Ce, Pr, Gd); for LnF2 (Ln = Sm, Eu, Yb); for LnS, LnSeand LnTe (Ln = Sm, Eu, Tm and Yb); for LnSO4 (Ln = Sm, Eu); for LnCO3 (Ln = Sm, Euand Yb); for LnD2 and LnH2 (Ln = Eu, Yb).
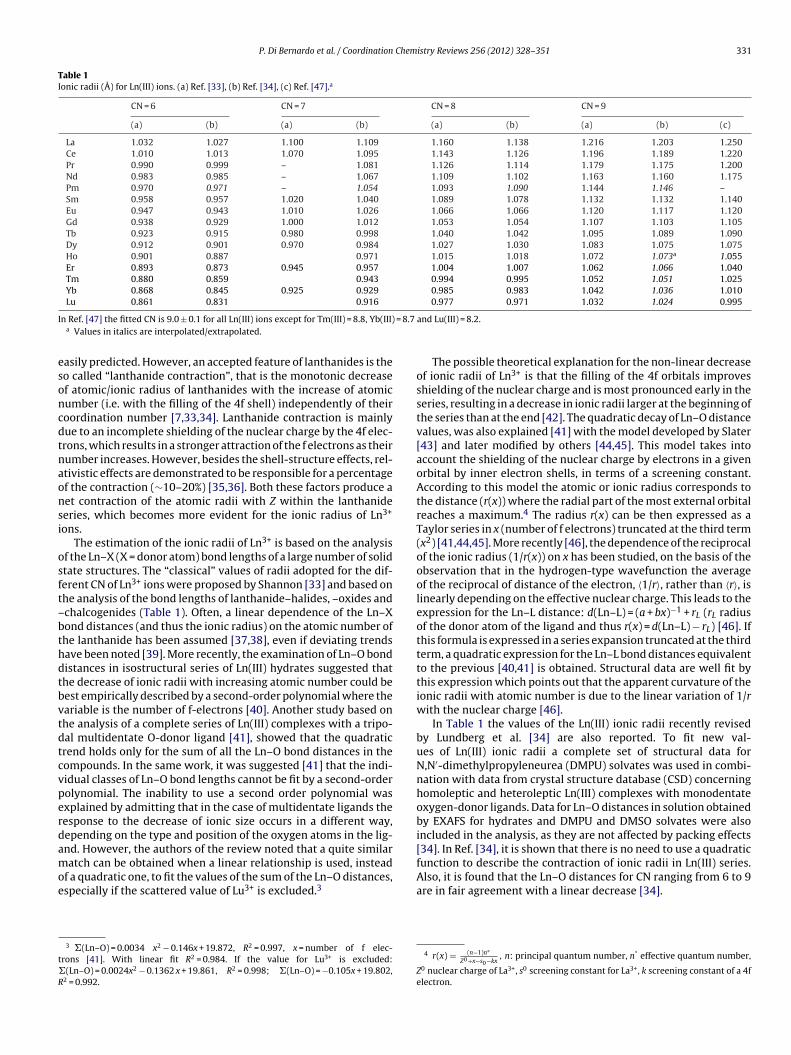
P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351 331
Table 1Ionic radii (Å) for Ln(III) ions. (a) Ref. [33], (b) Ref. [34], (c) Ref. [47].a
CN = 6 CN = 7 CN = 8 CN = 9
(a) (b) (a) (b) (a) (b) (a) (b) (c)
La 1.032 1.027 1.100 1.109 1.160 1.138 1.216 1.203 1.250Ce 1.010 1.013 1.070 1.095 1.143 1.126 1.196 1.189 1.220Pr 0.990 0.999 – 1.081 1.126 1.114 1.179 1.175 1.200Nd 0.983 0.985 – 1.067 1.109 1.102 1.163 1.160 1.175Pm 0.970 0.971 – 1.054 1.093 1.090 1.144 1.146 –Sm 0.958 0.957 1.020 1.040 1.089 1.078 1.132 1.132 1.140Eu 0.947 0.943 1.010 1.026 1.066 1.066 1.120 1.117 1.120Gd 0.938 0.929 1.000 1.012 1.053 1.054 1.107 1.103 1.105Tb 0.923 0.915 0.980 0.998 1.040 1.042 1.095 1.089 1.090Dy 0.912 0.901 0.970 0.984 1.027 1.030 1.083 1.075 1.075Ho 0.901 0.887 0.971 1.015 1.018 1.072 1.073a 1.055Er 0.893 0.873 0.945 0.957 1.004 1.007 1.062 1.066 1.040Tm 0.880 0.859 0.943 0.994 0.995 1.052 1.051 1.025Yb 0.868 0.845 0.925 0.929 0.985 0.983 1.042 1.036 1.010Lu 0.861 0.831 0.916 0.977 0.971 1.032 1.024 0.995
I = 8.7
esoncdtnaonsi
osft–bthdtbvtdtcvperdamoe
t�R
n Ref. [47] the fitted CN is 9.0 ± 0.1 for all Ln(III) ions except for Tm(III) = 8.8, Yb(III)a Values in italics are interpolated/extrapolated.
asily predicted. However, an accepted feature of lanthanides is theo called “lanthanide contraction”, that is the monotonic decreasef atomic/ionic radius of lanthanides with the increase of atomicumber (i.e. with the filling of the 4f shell) independently of theiroordination number [7,33,34]. Lanthanide contraction is mainlyue to an incomplete shielding of the nuclear charge by the 4f elec-rons, which results in a stronger attraction of the f electrons as theirumber increases. However, besides the shell-structure effects, rel-tivistic effects are demonstrated to be responsible for a percentagef the contraction (∼10–20%) [35,36]. Both these factors produce aet contraction of the atomic radii with Z within the lanthanideeries, which becomes more evident for the ionic radius of Ln3+
ons.The estimation of the ionic radii of Ln3+ is based on the analysis
f the Ln–X (X = donor atom) bond lengths of a large number of solidtate structures. The “classical” values of radii adopted for the dif-erent CN of Ln3+ ions were proposed by Shannon [33] and based onhe analysis of the bond lengths of lanthanide–halides, –oxides andchalcogenides (Table 1). Often, a linear dependence of the Ln–Xond distances (and thus the ionic radius) on the atomic number ofhe lanthanide has been assumed [37,38], even if deviating trendsave been noted [39]. More recently, the examination of Ln–O bondistances in isostructural series of Ln(III) hydrates suggested thathe decrease of ionic radii with increasing atomic number could beest empirically described by a second-order polynomial where theariable is the number of f-electrons [40]. Another study based onhe analysis of a complete series of Ln(III) complexes with a tripo-al multidentate O-donor ligand [41], showed that the quadraticrend holds only for the sum of all the Ln–O bond distances in theompounds. In the same work, it was suggested [41] that the indi-idual classes of Ln–O bond lengths cannot be fit by a second-orderolynomial. The inability to use a second order polynomial wasxplained by admitting that in the case of multidentate ligands theesponse to the decrease of ionic size occurs in a different way,epending on the type and position of the oxygen atoms in the lig-nd. However, the authors of the review noted that a quite similar
atch can be obtained when a linear relationship is used, insteadf a quadratic one, to fit the values of the sum of the Ln–O distances,specially if the scattered value of Lu3+ is excluded.3
3 �(Ln–O) = 0.0034 x2 − 0.146x + 19.872, R2 = 0.997, x = number of f elec-rons [41]. With linear fit R2 = 0.984. If the value for Lu3+ is excluded:(Ln–O) = 0.0024x2 − 0.1362 x + 19.861, R2 = 0.998; �(Ln–O) = −0.105x + 19.802,
2 = 0.992.
and Lu(III) = 8.2.
The possible theoretical explanation for the non-linear decreaseof ionic radii of Ln3+ is that the filling of the 4f orbitals improvesshielding of the nuclear charge and is most pronounced early in theseries, resulting in a decrease in ionic radii larger at the beginning ofthe series than at the end [42]. The quadratic decay of Ln–O distancevalues, was also explained [41] with the model developed by Slater[43] and later modified by others [44,45]. This model takes intoaccount the shielding of the nuclear charge by electrons in a givenorbital by inner electron shells, in terms of a screening constant.According to this model the atomic or ionic radius corresponds tothe distance (r(x)) where the radial part of the most external orbitalreaches a maximum.4 The radius r(x) can be then expressed as aTaylor series in x (number of f electrons) truncated at the third term(x2) [41,44,45]. More recently [46], the dependence of the reciprocalof the ionic radius (1/r(x)) on x has been studied, on the basis of theobservation that in the hydrogen-type wavefunction the averageof the reciprocal of distance of the electron, 〈1/r〉, rather than 〈r〉, islinearly depending on the effective nuclear charge. This leads to theexpression for the Ln–L distance: d(Ln–L) = (a + bx)−1 + rL (rL radiusof the donor atom of the ligand and thus r(x) = d(Ln–L) − rL) [46]. Ifthis formula is expressed in a series expansion truncated at the thirdterm, a quadratic expression for the Ln–L bond distances equivalentto the previous [40,41] is obtained. Structural data are well fit bythis expression which points out that the apparent curvature of theionic radii with atomic number is due to the linear variation of 1/rwith the nuclear charge [46].
In Table 1 the values of the Ln(III) ionic radii recently revisedby Lundberg et al. [34] are also reported. To fit new val-ues of Ln(III) ionic radii a complete set of structural data forN,N′-dimethylpropyleneurea (DMPU) solvates was used in combi-nation with data from crystal structure database (CSD) concerninghomoleptic and heteroleptic Ln(III) complexes with monodentateoxygen-donor ligands. Data for Ln–O distances in solution obtainedby EXAFS for hydrates and DMPU and DMSO solvates were alsoincluded in the analysis, as they are not affected by packing effects[34]. In Ref. [34], it is shown that there is no need to use a quadraticfunction to describe the contraction of ionic radii in Ln(III) series.
Also, it is found that the Ln–O distances for CN ranging from 6 to 9are in fair agreement with a linear decrease [34].4 r(x) = (n−1)n∗Z0+x−s0−kx
, n: principal quantum number, n* effective quantum number,
Z0 nuclear charge of La3+, s0 screening constant for La3+, k screening constant of a 4felectron.
3 Chemistry Reviews 256 (2012) 328–351
iE(9td
3
3
irsdm
pa(naeisatmssfwvseaiwscttpadp
[omalca
e[t
i
-110
-100
-90
-80
-70
-600.850 0.900 0.950 1.000 1.050
r-1 (Å-1)
ΔH
° tr /
kJ /
mol
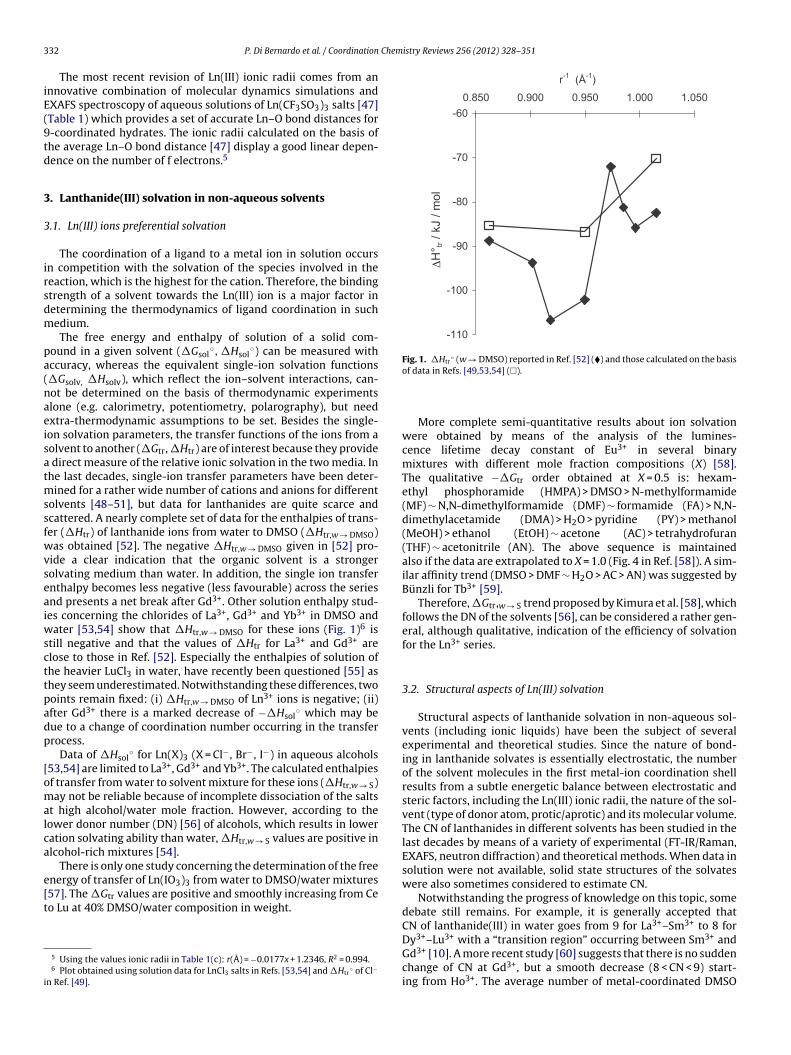
Fig. 1. �Htr◦ (w → DMSO) reported in Ref. [52] (�) and those calculated on the basis
32 P. Di Bernardo et al. / Coordination
The most recent revision of Ln(III) ionic radii comes from annnovative combination of molecular dynamics simulations andXAFS spectroscopy of aqueous solutions of Ln(CF3SO3)3 salts [47]Table 1) which provides a set of accurate Ln–O bond distances for-coordinated hydrates. The ionic radii calculated on the basis ofhe average Ln–O bond distance [47] display a good linear depen-ence on the number of f electrons.5
. Lanthanide(III) solvation in non-aqueous solvents
.1. Ln(III) ions preferential solvation
The coordination of a ligand to a metal ion in solution occursn competition with the solvation of the species involved in theeaction, which is the highest for the cation. Therefore, the bindingtrength of a solvent towards the Ln(III) ion is a major factor inetermining the thermodynamics of ligand coordination in suchedium.The free energy and enthalpy of solution of a solid com-
ound in a given solvent (�Gsol◦, �Hsol
◦) can be measured withccuracy, whereas the equivalent single-ion solvation functions�Gsolv, �Hsolv), which reflect the ion–solvent interactions, can-ot be determined on the basis of thermodynamic experimentslone (e.g. calorimetry, potentiometry, polarography), but needxtra-thermodynamic assumptions to be set. Besides the single-on solvation parameters, the transfer functions of the ions from aolvent to another (�Gtr, �Htr) are of interest because they providedirect measure of the relative ionic solvation in the two media. In
he last decades, single-ion transfer parameters have been deter-ined for a rather wide number of cations and anions for different
olvents [48–51], but data for lanthanides are quite scarce andcattered. A nearly complete set of data for the enthalpies of trans-er (�Htr) of lanthanide ions from water to DMSO (�Htr,w → DMSO)as obtained [52]. The negative �Htr,w → DMSO given in [52] pro-
ide a clear indication that the organic solvent is a strongerolvating medium than water. In addition, the single ion transfernthalpy becomes less negative (less favourable) across the seriesnd presents a net break after Gd3+. Other solution enthalpy stud-es concerning the chlorides of La3+, Gd3+ and Yb3+ in DMSO and
ater [53,54] show that �Htr,w → DMSO for these ions (Fig. 1)6 istill negative and that the values of �Htr for La3+ and Gd3+ arelose to those in Ref. [52]. Especially the enthalpies of solution ofhe heavier LuCl3 in water, have recently been questioned [55] ashey seem underestimated. Notwithstanding these differences, twooints remain fixed: (i) �Htr,w → DMSO of Ln3+ ions is negative; (ii)fter Gd3+ there is a marked decrease of −�Hsol
◦ which may beue to a change of coordination number occurring in the transferrocess.
Data of �Hsol◦ for Ln(X)3 (X = Cl−, Br−, I−) in aqueous alcohols
53,54] are limited to La3+, Gd3+ and Yb3+. The calculated enthalpiesf transfer from water to solvent mixture for these ions (�Htr,w → S)ay not be reliable because of incomplete dissociation of the salts
t high alcohol/water mole fraction. However, according to theower donor number (DN) [56] of alcohols, which results in loweration solvating ability than water, �Htr,w → S values are positive inlcohol-rich mixtures [54].
There is only one study concerning the determination of the free
nergy of transfer of Ln(IO3)3 from water to DMSO/water mixtures57]. The �Gtr values are positive and smoothly increasing from Ceo Lu at 40% DMSO/water composition in weight.5 Using the values ionic radii in Table 1(c): r(Å) = −0.0177x + 1.2346, R2 = 0.994.6 Plot obtained using solution data for LnCl3 salts in Refs. [53,54] and �Htr
◦ of Cl−
n Ref. [49].
of data in Refs. [49,53,54] (�).
More complete semi-quantitative results about ion solvationwere obtained by means of the analysis of the lumines-cence lifetime decay constant of Eu3+ in several binarymixtures with different mole fraction compositions (X) [58].The qualitative −�Gtr order obtained at X = 0.5 is: hexam-ethyl phosphoramide (HMPA) > DMSO > N-methylformamide(MF) ∼ N,N-dimethylformamide (DMF) ∼ formamide (FA) > N,N-dimethylacetamide (DMA) > H2O > pyridine (PY) > methanol(MeOH) > ethanol (EtOH) ∼ acetone (AC) > tetrahydrofuran(THF) ∼ acetonitrile (AN). The above sequence is maintainedalso if the data are extrapolated to X = 1.0 (Fig. 4 in Ref. [58]). A sim-ilar affinity trend (DMSO > DMF ∼ H2O > AC > AN) was suggested byBünzli for Tb3+ [59].
Therefore, �Gtr,w → S trend proposed by Kimura et al. [58], whichfollows the DN of the solvents [56], can be considered a rather gen-eral, although qualitative, indication of the efficiency of solvationfor the Ln3+ series.
3.2. Structural aspects of Ln(III) solvation
Structural aspects of lanthanide solvation in non-aqueous sol-vents (including ionic liquids) have been the subject of severalexperimental and theoretical studies. Since the nature of bond-ing in lanthanide solvates is essentially electrostatic, the numberof the solvent molecules in the first metal-ion coordination shellresults from a subtle energetic balance between electrostatic andsteric factors, including the Ln(III) ionic radii, the nature of the sol-vent (type of donor atom, protic/aprotic) and its molecular volume.The CN of lanthanides in different solvents has been studied in thelast decades by means of a variety of experimental (FT-IR/Raman,EXAFS, neutron diffraction) and theoretical methods. When data insolution were not available, solid state structures of the solvateswere also sometimes considered to estimate CN.
Notwithstanding the progress of knowledge on this topic, somedebate still remains. For example, it is generally accepted thatCN of lanthanide(III) in water goes from 9 for La3+–Sm3+ to 8 for
Dy3+–Lu3+ with a “transition region” occurring between Sm3+ andGd3+ [10]. A more recent study [60] suggests that there is no suddenchange of CN at Gd3+, but a smooth decrease (8 < CN < 9) start-ing from Ho3+. The average number of metal-coordinated DMSO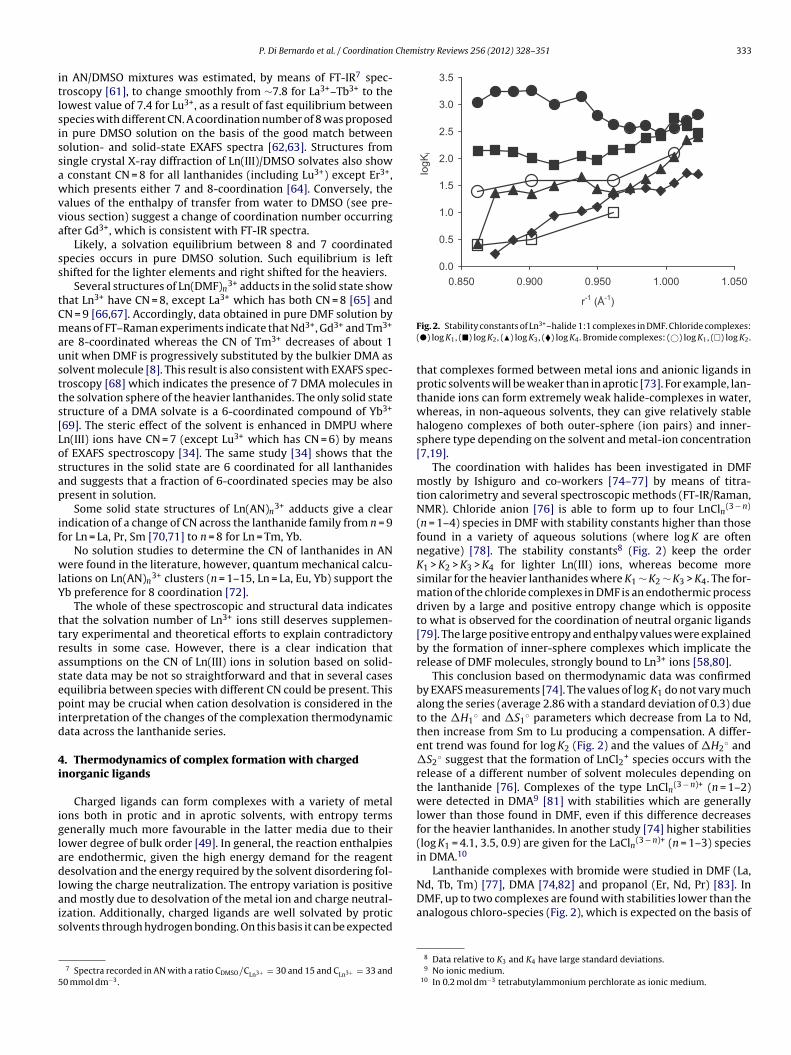
Chemistry Reviews 256 (2012) 328–351 333
itlsissawvva
ss
tCmaustts[Losap
if
wlY
ttrasepid
4i
igladlais
5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
0.850 0.900 1.0000.950 1.050
r-1 (Å-1)
logK
i
Nd, Tb, Tm) [77], DMA [74,82] and propanol (Er, Nd, Pr) [83]. In
P. Di Bernardo et al. / Coordination
n AN/DMSO mixtures was estimated, by means of FT-IR7 spec-roscopy [61], to change smoothly from ∼7.8 for La3+–Tb3+ to theowest value of 7.4 for Lu3+, as a result of fast equilibrium betweenpecies with different CN. A coordination number of 8 was proposedn pure DMSO solution on the basis of the good match betweenolution- and solid-state EXAFS spectra [62,63]. Structures fromingle crystal X-ray diffraction of Ln(III)/DMSO solvates also showconstant CN = 8 for all lanthanides (including Lu3+) except Er3+,hich presents either 7 and 8-coordination [64]. Conversely, the
alues of the enthalpy of transfer from water to DMSO (see pre-ious section) suggest a change of coordination number occurringfter Gd3+, which is consistent with FT-IR spectra.
Likely, a solvation equilibrium between 8 and 7 coordinatedpecies occurs in pure DMSO solution. Such equilibrium is lefthifted for the lighter elements and right shifted for the heaviers.
Several structures of Ln(DMF)n3+ adducts in the solid state show
hat Ln3+ have CN = 8, except La3+ which has both CN = 8 [65] andN = 9 [66,67]. Accordingly, data obtained in pure DMF solution byeans of FT–Raman experiments indicate that Nd3+, Gd3+ and Tm3+
re 8-coordinated whereas the CN of Tm3+ decreases of about 1nit when DMF is progressively substituted by the bulkier DMA asolvent molecule [8]. This result is also consistent with EXAFS spec-roscopy [68] which indicates the presence of 7 DMA molecules inhe solvation sphere of the heavier lanthanides. The only solid statetructure of a DMA solvate is a 6-coordinated compound of Yb3+
69]. The steric effect of the solvent is enhanced in DMPU wheren(III) ions have CN = 7 (except Lu3+ which has CN = 6) by meansf EXAFS spectroscopy [34]. The same study [34] shows that thetructures in the solid state are 6 coordinated for all lanthanidesnd suggests that a fraction of 6-coordinated species may be alsoresent in solution.
Some solid state structures of Ln(AN)n3+ adducts give a clear
ndication of a change of CN across the lanthanide family from n = 9or Ln = La, Pr, Sm [70,71] to n = 8 for Ln = Tm, Yb.
No solution studies to determine the CN of lanthanides in ANere found in the literature, however, quantum mechanical calcu-
ations on Ln(AN)n3+ clusters (n = 1–15, Ln = La, Eu, Yb) support the
b preference for 8 coordination [72].The whole of these spectroscopic and structural data indicates
hat the solvation number of Ln3+ ions still deserves supplemen-ary experimental and theoretical efforts to explain contradictoryesults in some case. However, there is a clear indication thatssumptions on the CN of Ln(III) ions in solution based on solid-tate data may be not so straightforward and that in several casesquilibria between species with different CN could be present. Thisoint may be crucial when cation desolvation is considered in the
nterpretation of the changes of the complexation thermodynamicata across the lanthanide series.
. Thermodynamics of complex formation with chargednorganic ligands
Charged ligands can form complexes with a variety of metalons both in protic and in aprotic solvents, with entropy termsenerally much more favourable in the latter media due to theirower degree of bulk order [49]. In general, the reaction enthalpiesre endothermic, given the high energy demand for the reagentesolvation and the energy required by the solvent disordering fol-
owing the charge neutralization. The entropy variation is positive
nd mostly due to desolvation of the metal ion and charge neutral-zation. Additionally, charged ligands are well solvated by proticolvents through hydrogen bonding. On this basis it can be expected7 Spectra recorded in AN with a ratio CDMSO/CLn3+ = 30 and 15 and CLn3+ = 33 and0 mmol dm−3.
Fig. 2. Stability constants of Ln3+–halide 1:1 complexes in DMF. Chloride complexes:(�) log K1, (�) log K2, (�) log K3, (�) log K4. Bromide complexes: (©) log K1, (�) log K2.
that complexes formed between metal ions and anionic ligands inprotic solvents will be weaker than in aprotic [73]. For example, lan-thanide ions can form extremely weak halide-complexes in water,whereas, in non-aqueous solvents, they can give relatively stablehalogeno complexes of both outer-sphere (ion pairs) and inner-sphere type depending on the solvent and metal-ion concentration[7,19].
The coordination with halides has been investigated in DMFmostly by Ishiguro and co-workers [74–77] by means of titra-tion calorimetry and several spectroscopic methods (FT-IR/Raman,NMR). Chloride anion [76] is able to form up to four LnCln(3 − n)
(n = 1–4) species in DMF with stability constants higher than thosefound in a variety of aqueous solutions (where log K are oftennegative) [78]. The stability constants8 (Fig. 2) keep the orderK1 > K2 > K3 > K4 for lighter Ln(III) ions, whereas become moresimilar for the heavier lanthanides where K1 ∼ K2 ∼ K3 > K4. The for-mation of the chloride complexes in DMF is an endothermic processdriven by a large and positive entropy change which is oppositeto what is observed for the coordination of neutral organic ligands[79]. The large positive entropy and enthalpy values were explainedby the formation of inner-sphere complexes which implicate therelease of DMF molecules, strongly bound to Ln3+ ions [58,80].
This conclusion based on thermodynamic data was confirmedby EXAFS measurements [74]. The values of log K1 do not vary muchalong the series (average 2.86 with a standard deviation of 0.3) dueto the �H1
◦ and �S1◦ parameters which decrease from La to Nd,
then increase from Sm to Lu producing a compensation. A differ-ent trend was found for log K2 (Fig. 2) and the values of �H2
◦ and�S2
◦ suggest that the formation of LnCl2+ species occurs with therelease of a different number of solvent molecules depending onthe lanthanide [76]. Complexes of the type LnCln(3 − n)+ (n = 1–2)were detected in DMA9 [81] with stabilities which are generallylower than those found in DMF, even if this difference decreasesfor the heavier lanthanides. In another study [74] higher stabilities(log K1 = 4.1, 3.5, 0.9) are given for the LaCln(3 − n)+ (n = 1–3) speciesin DMA.10
Lanthanide complexes with bromide were studied in DMF (La,
DMF, up to two complexes are found with stabilities lower than theanalogous chloro-species (Fig. 2), which is expected on the basis of
8 Data relative to K3 and K4 have large standard deviations.9 No ionic medium.
10 In 0.2 mol dm−3 tetrabutylammonium perchlorate as ionic medium.

3 Chem
tTfffics[atdf∼aottto
waflws
(bcen[tmwtut
aTnsrcttnsiRvr
dINIaEcdttN
Ln(trif)3 salts has been also shown in PC, while their salts arecompletely dissociated in strongly coordinating solvents such asDMF, DMA and DMSO [96,99,100].
11 Conductometry.12 Conductometric measurements on a 0.1 mol dm−3 solution of NEt4ClO4 in anhy-
drous PC at 298.15 K give a molar conductivity value �M = 39.5 �−1 cm2 mol−1 for this1:1 electrolyte. Values of �M of about 79 and 118 �−1 cm2 mol−1 at 298.15 K can beestimated for 1:2 and 1:3 electrolytes respectively. Data for 0.1 mol dm−3 PC solu-tions of La(III)-, Dy(III)- and Lu(III) triflates were found in the range of �M between
−1 2 −1
34 P. Di Bernardo et al. / Coordination
he HSAB principle, but �H1◦(Cl−) > �H1
◦(Br−) for all Ln(III) ions.hese values of enthalpy change are accompanied by a much lessavourable entropy variations suggesting that, contrarily to whatound for Cl− anion, no solvent molecules are released from therst solvation shell of the Ln(III) [77]. The outer-sphere nature of theomplexes formed by Ln(III) with Br− is supported by 89Y NMR mea-urements. On the other hand, in another article the same authors74], on the basis of 139La NMR, suggested an equilibrium betweenn inner and outer-sphere complex (even if shifted towards the lat-er) of the type [La(DMF)n
3+]Br− � [La(DMF)mBr]2+ + (n − m)DMF. Aifferent situation is present in DMA, where Ln(III) ions are able toorm up to three LaBrn
(3 − n) (n = 1–3) complexes with log K values1 unit higher than their counterparts in DMF. Also the enthalpynd entropy values significantly are higher than the correspondingnes in DMF indicating that inner-sphere complexes are formed inhis solvent. EXAFS measurements support this difference betweenhe Ln(III) bromo-complexes in DMF and DMA clearly indicating forhe Tm3+–Br− system an inner-sphere coordination in DMA anduter-sphere in DMF [74].
Formation of very weak lanthanide iodide complexes in DMFas also suggested [76], but log K1 is too uncertain to be reported
nd enthalpy of formation certainly negative [76]. The enthalpies oformation of the 1:1 lanthanide–halide complexes in DMF [76] fol-ow the order �H1
◦(Cl−) > �H1◦(Br−) > �H1
◦(I−), in agreementith a progressive change of nature of the complexes from inner
phere (Cl−) to outer-sphere (I−).Thiocyanate forms three successive complexes Ln(SCN)n
(3 − n)
n = 1–3 and Ln = La, Nd, Tb, Ho, Tm, Yb) in DMF [75] characterizedy positive values of �Hn
◦ and �Sn◦, as in the case of halides. Thio-
yanate coordinates to the Ln(III) ion through the N atom (which isxpected on the basis of the HSAB principle) and, while thermody-amic parameters are more similar to those of bromo-complexes77], rather than chloro-complexes [76], 89Y NMR suggests thathey have an inner-sphere nature. Thermodynamic parameters are
acroscopic indicators of the interactions occurring in solutionhereas NMR data are more focused to describe microscopic frame:
he deviation within the two different indications suggests that these of different techniques is fundamental before accounting forhe prevailing nature of the first coordination sphere.
There is little dependence of the thiocyanate log K1 on the Ln(III)s their values are scattered between 1.5 (La3+) and 1.8 (Nd3+,b3+, Yb3+). Also �H1
◦ (∼9 kJ mol−1) and �S1◦ (∼60 J mol−1 K−1) are
early constant along the series [75]. The second complex formed islightly less stable than the first while the third species suffers fromather large standard deviations to be considered for quantitativeomparisons [75]. The analysis of FT–Raman spectra also indicatehat SCN− anion is likely to displace only one solvent molecule andhat solvation equilibria are established between 9 and 8 coordi-ated species for La3+ complexes and between 8 and 7 coordinatedpecies for Tm3+, Yb3+ and Lu3+ [75]. The CN assumed for the Ln(III)n solution is consistent with solid state structures (see above) andaman spectra [8], while EXAFS in this case does not give a reliablealue since the coordination numbers found are scattered in theange 6–9 [19].
Lanthanide ions form only weak mono-nitrate and mono- andi-nitrate-complexes in propanol [83] and DMA [82] respectively.
n DMA the order of log K1,2 is NO3− < Br− < Cl− [82] for La3+ and
d3+, whereas the inverse order NO3− > Br− > Cl− holds for �H1
◦.n aqueous MeOH the interaction with neodymium, europiumnd erbium was investigated by UV–Vis spectroscopy [84,85]:u(III) ion forms [Eu(NO3)]2+, [Eu(NO3)2]+ and [Eu(NO3)3] solvatedomplexes. The magnitude of each of the equilibrium constantecreases with the increase of water mole fraction and in water, the
ris-complex is not present. Under the same experimental condi-ions, the stoichiometries of the nitrate complexes are different ford(III) and Er(III) compared with Eu(III) [84] and these differencesistry Reviews 256 (2012) 328–351
persist as the solvent composition is varied. At low water molefractions, Eu(III) and Er(III) form complexes up to the tris-complex,whereas Nd(III) only up to the di-nitrato compound.
Conductometric and spectroscopic experiments showed thatLn(III)–nitrates behave as non-electrolytes in anhydrous AN solu-tion [59,86,87]. From the structural point of view, FT-IR studies,carried out in AN/DMSO medium, indicate that nitrate bindslanthanide as bidentate [59,86]. Spectroscopy [88] also showedthat DMSO is more effective than water in displacing thenitrate in anhydrous AN resulting in a qualitative affinity order:NO3
− > DMSO > H2O > AN. In DMF solutions, conductimetric mea-surements showed that europium or terbium nitrates are 2:1(<0.02 M), or 1:1 (>0.02 M) electrolytes [89,90] and a inner-sphereLn(III)/nitrate interaction was concluded to exist [90].
Perchlorate is generally considered “non-reacting” anion andoften its salts are used to create ionic medium in thermody-namic studies. However, several solution studies demonstrate thatalso perchlorate can give complexes, whose nature is solvent-dependent [7,59]. The formation of moderately stable mono- andbi-dentate lanthanide complexes of perchlorate has been ascer-tained in anhydrous AN by means of FT-IR [91,92]). The apparentstability constants for the 1:1 complexes are affected by relativelylarge errors, nevertheless their values (for the heavier lanthanideslog K1 ranges from 1.8 to 2.7 [93]) suggest partial associationalready in relatively diluted solutions. However, no interactionbetween Ln3+ and ClO4
− ions was detected if a 7:1 excess of DMSOwith respect to the Ln(ClO4)3 concentration is added to the lan-thanide perchlorate solutions in anhydrous AN [91].
Trifluoromethanesulfonate anion (trif, triflate), which is oftenused in many preparations and experiments as substitute ofthe potentially explosive perchlorate, forms complexes with lan-thanides in anhydrous MeOH [94] and AN [95]. Until the studyin Ref. [95], the lanthanide triflates were considered strong elec-trolytes even in a poor coordinating solvent such as anhydrous AN[96]. The stepwise stability constants determined in anhydrous ANfor the equilibrium (1), where Ln = Gd–Lu:
Ln(trif)2+ + trif � Ln(trif)3 (K3) (1)
increase almost regularly from log K3 = 2.33 (Gd3+) to 2.58 (Lu3+)[95]. In the same study, it is also shown that perchlorates have anaffinity for Ln3+ ions about 10 times lower than triflates and that theaddition of an excess of water or DMSO to a solution of Lu(trif)3 inAN (Cw,DMSO/CLu(trif)3
> 20) favours the complete removal of oneand two anions from the undissociated salt for excess of waterand DMSO, respectively. In MeOH, the ratio K1(trif)/K1(ClO4
−) is∼1.15 indicating a moderate preference of lanthanides for thecoordination of triflate over ClO4
− [94]. Recently [97], it has beendemonstrated11 that Ln(trif)3 salts are completely dissociated atconcentrations lower than 5 × 10−5 M in anhydrous AN solutions.
The incomplete dissociation of Sm(trif)3 [98] and of some other12
80 and 85 � cm mol , thus indicating a partial association of triflate salts in PC.Similar results were obtained by comparison with other salts behaving as 1:1 elec-trolytes and using different, lower (down to 10−2 mol dm−3) molar concentrations(unpublished results by the authors of this review).

Chem
5o
letpf
mseimsvb(
L
cpt
L
ctslisil
tamcwfoaiclws
stmto�oa
erties of the benzo-derivative, whereas the opposite signs of the
P. Di Bernardo et al. / Coordination
. Thermodynamics of complex formation with neutralrganic ligands
When the formation of Ln(III) complexes with neutral organicigands is studied, the interpretation of the thermodynamic param-ters and of the solvent effect relies on a clear definition ofhe chemical nature of the reactants and products. This is are-requisite for comparing the thermodynamic parameters of dif-erent ions (or their variation along the Ln(III) series) and solvents.
An important aspect, which has been sometimes underesti-ated in the studies of lanthanide complexation in non-aqueous
olutions, is the incomplete dissociation of the lanthanide saltmployed to carry out the experiments (see Section 4), or the metal-on complexation by the anion of the salt used to form the ionic
edium. If this is the case, the reacting metal-species is not theimply solvated LnSs
3+ (S = solvent molecule; s = number of sol-ent molecules in the first coordination sphere of the metal ion)ut rather the complexes13 in equilibrium (2), where x ≤ s, m ≥ 1charges are omitted for simplicity).
nSxX(m − 1) + X � LnSyXm + (x − y)S (2)
This can very much complicate the complexation reaction whichan be described by the equilibrium (3), where, in the most com-licated case, z can be either x or y and, accordingly, n can be equalo (m − 1) or m.
nSzXn + pL � LnS(z-w)X(n-k)Lp + kX + wS (3)
In addition, since s, the number of solvent molecules in the firstoordination sphere of the metal ion, can vary for some of the ions inhe middle of the lanthanide series, it is clear that thermodynamictudies aimed at comparing the complexing strength of a givenigand towards Ln3+ ions must be carried out in solvents and inonic media which always assure the presence in solution of theame acceptor. The latter may be the simply solvated ion or one ofts complexes with X, which, however must be always the same toegitimate comparisons.
All these considerations indicate that a crucial step of thehermodynamic studies on the lanthanide complexation in non-queous solvents, is the choice of a starting salt, solvent, ionicedium which can assure the most accurate knowledge of the spe-
iation in the solution. Lanthanide perchlorate, nitrate or triflatesere used as metal cation sources for complexation studies in dif-
erent media such as AN, PC, DMSO and DMF (see Section 4). Mostf the data reported till now in the less coordinating among thebove solvents, refer to solutions where the inorganic anions of theonic medium (X− in Eqs. (2) and (3)) are in competition with theoncerned ligands for the coordination to the metal-ion. This canead to “thermodynamic data” obtained in uncontrolled conditions
hich have a quantitative value limited to the particular systemtudied and should not be used for general considerations.
This review covers all the literature data for the Ln(III)–ligandystems in the selected non-aqueous solvents. Some of the litera-ure data are discussed with the aim to enlighten general trends
ore than to compare the “apparent” thermodynamic values forhe complex formation, which not always refer to the real reactionccurring in solution. Accordingly, the authors will use the symbols
G, �H and �S, instead of �G◦, �H◦, �S◦ when, in their opinion,nly “apparent” thermodynamic parameters in the original studyre obtained.
13 X is a generic, non-innocent, negative monovalent ion present in solution.
istry Reviews 256 (2012) 328–351 335
5.1. Coordination of O-donors
Macrocyclic ligands define a hydrophilic cavity in which anionic substrate like a metal ion can be accommodated and shieldedfrom the environment. These synthetic macrocyclic receptors canbe designed to selectively bind a given metal ion on the basisof the general principles already mentioned and are discussed inseveral reviews [20–22]. The main aim of fundamental thermo-dynamic studies on Ln(III) complexation by macrocyclic donors innon-aqueous solutions in the last three decades has been to definethe influence of the ligand (structure, type and number of donoratoms) and the solvent on the stoichiometry and stability of thespecies formed with Ln(III) ions. This last information is of funda-mental importance in the development of new separation methods.
Lanthanide(III) ions are hard acids which form bonds with amarked ion–dipole character with neutral O- and N-donor ligands.Purely O-donors, like crown ethers are not able to form compoundsin water whereas stable complexes are formed in non-aqueoussolvents such as AN, MeOH and PC [16,101,102].
An aspect which has been carefully considered by many authorsis the study of the effect of ionic radius contraction on the stabilityof the species formed with a given macrocycle, that is to assess (andpossibly optimize) the selectivity for a given Ln(III) ion with respectto the rest of the series. However, the design of a macrocycle selec-tive for a given Ln(III) ion on the basis of the match between theionic radius and ring cavity is a difficult task, since the ionic radiusdifference between La(III) and Lu(III) is relatively small (∼0.18 A forCN = 9, Table 1), whereas the ionic radius of two consecutive triva-lent lanthanide ions decreases on average of ∼0.013 A (for CN = 9,Table 1). This change in ionic radius is small compared, for example,to the effect of the addition of a –CH2–CH2–O– unit to a crown etherwhich increases the cavity diameter by ∼0.5–0.7 A [7]. This meansthat selectivity may be only achieved through a delicate balance ofstructural and electronic factors.
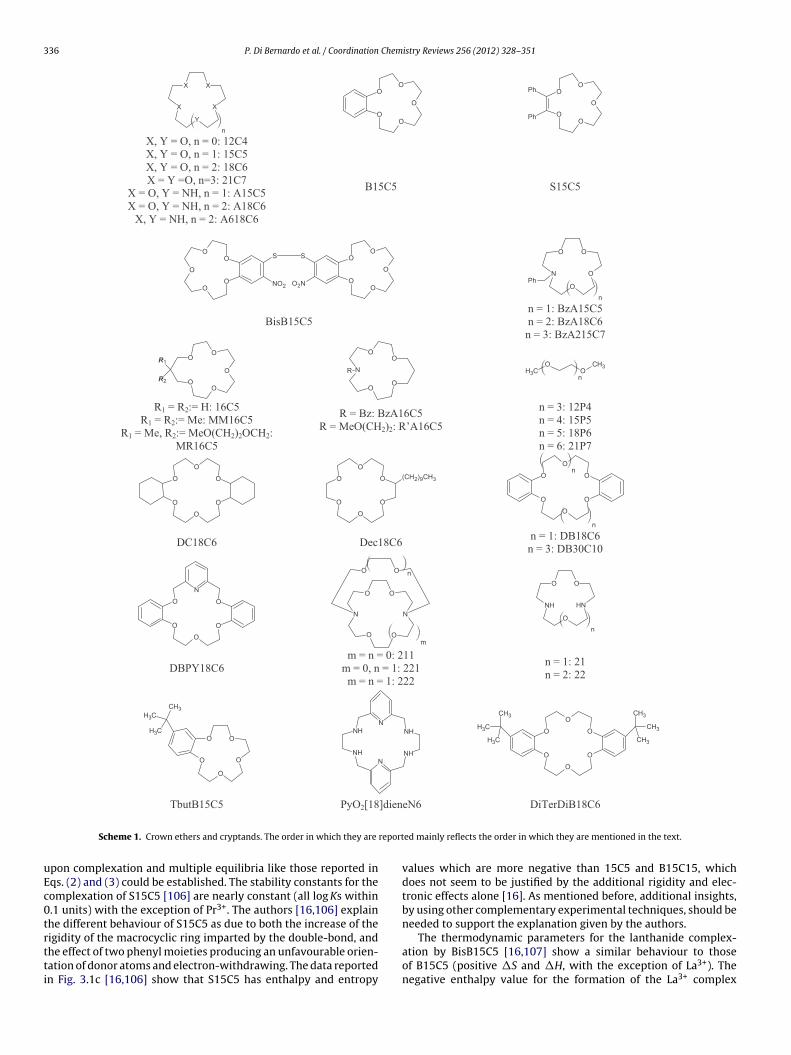
5.1.1. Crown ethersThe stability of the crown ethers lanthanide complexes (coro-
nates) mainly depends upon the relative size of the ligand cavityand the ionic diameter, the nature of the counterion and the sol-vent. A number of crown ethers (Scheme 1)14 form 1:1 complexesin AN with light lanthanide (La–Gd) nitrates [16] (Table 2), whichare not dissociated in that solvent [59,88,103].
The trends of the stability constants of the complexes for thevarious ligands are different across the Ln(III) series. The stability(log K) of 15C5 complexes decreases monotonously from La (5.17)to Gd (2.03) [104]. B15C5 shows a lower binding ability than 15C5(Table 2) for the same Ln3+ ions [16,105]; this was attributed to thehigher rigidity of the benzo-derivative and to the decreased elec-tron density on its donor atoms due to the withdrawing effect ofthe benzene ring. In addition, the stability of B15C5 lanthanide com-plexes decreases very steeply from La3+ to Pr3+ and then remainsalmost constant from Pr3+ to Gd3+. This result points out that evensmall changes in ionic radius could result in significant differencesin equilibrium constants, when the structural and electronic featureof the ligand are properly tuned. A peculiarity of these complexa-tion reactions is that they are enthalpy-driven for 15C5 (Fig. 3(1a))and entropy-driven for B15C5(Fig. 3(1b)). This behaviour for B15C5is assigned [105] to the modified electronic and structural prop-
reaction entropies to solvational effects which can be rather differ-ent for the two ligands. In addition, nitrate anions could be released
14 In Scheme 1 and in the successive ones the usual acronyms of the concernedligands are given, the complete IUPAC names of the ligands are reported at the endof the Review.
336 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Y
X X
X X
n
X, Y = O, n = 0: 12C4 X, Y = O, n = 1: 15C5 X, Y = O, n = 2: 18C6 X = Y =O, n= 3: 21C7
X = O, Y = NH, n = 1: A15C5 X = O, Y = NH, n = 2: A18C6
X, Y = NH, n = 2: A618C6
5C51S 5C51B
BisB15C 5 n = 1: BzA15C5 n = 2: BzA18C6 n = 3: BzA215C7
CH3O
OCH3
n
R1 = R2:= H: 16C 5 R1 = R2:= Me: MM16C5
R1 = Me , R2:= MeO(CH2)2OCH2:MR16C5
R = Bz: Bz A16C5 R = Me O(CH2)2: R’A16C5
n = 3: 12 P4 n = 4: 15 P5 n = 5: 18 P6 n = 6: 21 P7
O
O
O
O
O
On
n
6C81ceD 6C81CD n = 1: DB18C6 n = 3: DB3 0C10
DBPY18C6 m = n = 0: 211
m = 0, n = 1: 221 m = n = 1: 222
n = 1: 21 n = 2: 22
O
O
O
O
O
CH3CH3
CH3
OyP 5C51BtubT 2[18]dieneN6 DiTerDiB 18C6
O
O
O
O
OO
O
O
O
OPh
Ph
O
O
O
O
O
S
NO2
O
O
O
O
OS
O2N O
N O
O O
Ph
n
O
O
O
OOR1
R2
O
N
O
OO
R
O
O
O
O
O
O
O
O
O
O
O
O (CH2)9CH 3
O
O
O
O
N
O
O O
OO
N
O O
N
n
m
O
NH NH
O O
n
NHN
NH
NHN
NH O
O
O
O
O
O
CH3
CH3
CH3
CH3
CH3
CH3
report
uEc0trtti
Scheme 1. Crown ethers and cryptands. The order in which they are
pon complexation and multiple equilibria like those reported inqs. (2) and (3) could be established. The stability constants for theomplexation of S15C5 [106] are nearly constant (all log Ks within.1 units) with the exception of Pr3+. The authors [16,106] explainhe different behaviour of S15C5 as due to both the increase of the
igidity of the macrocyclic ring imparted by the double-bond, andhe effect of two phenyl moieties producing an unfavourable orien-ation of donor atoms and electron-withdrawing. The data reportedn Fig. 3.1c [16,106] show that S15C5 has enthalpy and entropyed mainly reflects the order in which they are mentioned in the text.
values which are more negative than 15C5 and B15C15, whichdoes not seem to be justified by the additional rigidity and elec-tronic effects alone [16]. As mentioned before, additional insights,by using other complementary experimental techniques, should beneeded to support the explanation given by the authors.
The thermodynamic parameters for the lanthanide complex-ation by BisB15C5 [16,107] show a similar behaviour to thoseof B15C5 (positive �S and �H, with the exception of La3+). Thenegative enthalpy value for the formation of the La3+ complex

P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351 337
Table 2Stability constants for complex formation of light lanthanide nitrates (unless otherwise specified) with crown ethers in AN at 298.15 K.
La Ce Pr Nd Sm Eu Gd
15C5 a 5.17 4.62 4.45 3.93 2.81 2.26 2.03B15C5 b 4.07 3.41 2.41 2.48 2.48 2.49 2.49S15C5 c 2.22 2.21 3.04 2.35 2.13 2.10 –BisB15C5 d 2.68 2.87 3.15 3.52 2.98 3.65 2.51BzA15C5 e 4.55 3.95 4.22 3.99 3.85 3.31 3.2416C5 a 2.54 2.49 2.76 3.81 4.14 – 3.66MM16C5 f 2.23 2.60 2.28 2.68 3.72 3.46 3.62MR16C5 a 3.76 3.18 3.07 3.04 3.01 – 3.10BzA16C5 f 2·52 2·43 3·27 2·48 2·46 2·36 2·37R′A16C5 g 2.82 3.01 3.47 4.41 3.50 2.71 2.8218C6 a 4.40 4.50 3.70 3.50 – 2.70 –18C6 h 3.29 3.57 2.63 2.44 2.03 1.84 1.32DB18C6 i 3.32 2.34 2.54 3.82 3.84 3.14 2.91DB18C6 l 5.54 4.61 3.34 2.96 2.23 2.10 2.57
3.62.7
a eOH; L
(toGCoec
d1mtlTwitwt
l1BemcIsfcintfsedA
a“efoga
show that the distance from the cation and the average plane of theoxygen atoms is 1.877 A in [La(12C4)(NO3)3H2O] [112], 1.462 A in[La(15C5)(NO3)3] [113] and 0.466 A in [La(18C6)(NO3)3] [114].
BzA18C6 m 3.77 3.46BzA21C7 n 3.01 2.66
– [104], b – [105], c –[106], d – [107], e – [108], f – [116], g – [115], h – [117] in M
extrapolated from temperature dependence of log K) is difficulto explain, especially if compared with the positive values for thether ions (e.g. �H = −40.3; +14.7; +58.4 kJ mol−1 for La3+, Ce3+,d3+, respectively, Fig. 3(1d)) [107]. The stability of the La3+ ande3+ complexes of BisB15C5 is lower than that of B15C5, thepposite holds for Pr3+–Gd3+. This was ascribed to the strongerlectron-withdrawing effect and to the cooperative binding of tworown-5 rings [16,107] in BisB15C5.
The stability (log K) of Ln3+ complexes with BzA15C5 [16,108].ecreases with increasing Z and, for La3+–Pr3+, is lower than with5C5 by ∼0.6 log units, whereas it is higher by 1–1.2 orders ofagnitude for Sm3+–Gd3+. The enthalpy terms are more negative
han for 15C5, however the complex formation of BzA15C5 is aittle more unfavoured by the slightly more negative �S values.he more negative �H and the higher stability of the complexesith increasing Z is attributed to the favourable effect of N-atom
nteraction which is prevailing with the heavier, more acidic, lan-hanides. In the case of lighter elements (La–Nd), the lariat effect,hich implies a critical balance between structural freezing and
he accompanying extensive desolvation, prevails [16].In summary, Fig. 3(1) shows that complex formation between
anthanide(III) ions and crown-ethers in AN is enthalpy-driven for5C5, S15C5 and BzA15C5; the opposite holds for BisB15C5 and15C5 (Fig. 3(1b) and (1d)). In addition, the negative formationnthalpies found for 15C5, S15C5 and BzA15C5 do not immediatelyean high complex stability, since a much or less large entropy
ompensation accompanies the reactions and define their �G [16].t has been noted [16] that the cation–ligand combination corre-ponding to smaller entropic loss leads to higher complex stabilityor this series of crown ethers and that one may conclude that theomplexation is enthalpy-driven in AN, but the cation selectivitys entropy-governed. However, from an analysis of the thermody-amic data in Fig. 3 and observation of Scheme 1, it emerges thathere is no systematic correlation between structural and electroniceatures of the ligands and thermodynamic parameters. Rather, iteems that more complicated equilibria (i.e. multiple species inquilibrium for the different ligand–lanthanide systems includingifferent solvational effects) can govern the complex formation inN solution.
The additional stability of the metal complexes with cyclic lig-nds with respect to the corresponding podates has been namedmacrocyclic effect”, generally explained on the basis of bothnthalpic and, in less extent, entropic factors [109]. The more
avourable enthalpy of solution of macrocyclic complex formationriginates mainly from the preorganization of the ligand and thereater basicity of the nitrogen/oxygen donors due to increasedlkylation. In particular, preorganization eliminates the energy cost2 4.05 4.33 4.35 4.592 2.28 2.67 2.59 2.35
nCl3, i – [120], l – [121]; Ln(ClO4)3 m – [16], n – [122].
needed to overcome the repulsive forces between the polar donorgroups in the reorientation of the ligand.
The macrocyclic effect has been studied by comparing the sta-bility constants of the complexes formed by Ln(III)–triflates andcyclic crown ethers containing from four to seven oxygen atomsand their open chain polyether analogues in PC [110]. The linearpolyethers considered in these studies were: 12P4, 15P5, 18P6 and21P7 [110] (Scheme 1). The conformation adopted in the solid stateby the podand in the [La(NO3)2(18P6)]+ is very similar to the onedisplayed by the corresponding macrocyclic ligand [111], exceptfor some helicity induced by the steric repulsion of the two methylgroups. Therefore, the difference between the stability constants ofthe 1:1 complexes of crown-ether and podand has been ascribed bythe authors [110] to the macrocyclic effect. The macrocyclic effectfor 12C4, 18C6 and 21C7 is approximately constant thorough thelanthanide series in PC: �log K = 1.0 ± 0.3 (12C4), 3.1 ± 0.2 (18C6),and 0.9 ± 0.2 (21C7) (�log K = log Kcrown − log Kpodand) [110]. For15C5 the effect is somewhat different because it increases 200 fold(Kcrown − Kpodand) from La to Tb and then stays approximately con-stant, with a magnitude five times larger than the macrocyclic effectfor 18C6 [110] (see also Fig. 4, where log K values in PC are reportedfor the complexes formed by 12C4, 15C5, 18C6 and 21C7 with La,Tb and Lu along with those for the corresponding podates). Therelative constancy of the macrocyclic effect along the 18C6 and21C7 series was assigned to the flexibility of the ligands, while inthe case of 12C4 the constancy was considered as due to the non-encapsulation of the metal ion [110]. The stability of the speciesformed by the Ln(III) ions and the podands has the tendency toincrease with the number of donor groups (Fig. 4). For the macro-cycles, there is a gradual shift of the maximum of stability from the18C6 coronates with lighter Ln(III) ions to the 15C5 coronates withheavier Ln(III) ions. The cause of this shift is attributed to a better fitbetween the cavities of 18C6 and 15C5 with lighter and heavier lan-thanide(III), respectively. In addition, also subtle solvation changesalong the lanthanide series and/or conformational variations of theligand and/or changes in the complex nature contribute to the shift.
Solid state structures show that the Ln(III) ion is located out ofthe average plane of the oxygen atoms in 12C4 and 15C5 while in18C6 the cation is nearly in-plane. For example, three structures ofLa3+–nitrate complexes with the above mentioned crown ethers15
15 CSD identifiers of structures used for the calculation POHDUL, CABLAS01 andLANITA.

338 P. Di Bernardo et al. / Coordination Chem
Fig. 3. Values of �G, �H, T�S for the complexation of lanthanide nitrates withcrown ethers at 298.15 K. 1: (a) 15C5, (b) B15C5,(c) S15C5, (d) BisB15C5, and (e)BzA15C5 in AN at 298.15 K. 2: (a) 16C5 (b) MM16C5 (c) MR16C5 (d) BzA16C5 (e)RlB
pe
Aiiabw
Mtctr
tonic decrease of stability with increasing Z for the 18C6 systemsis not only an ion size-related effect, but originated from a balancebetween ligand–cation binding and solvation. Once more, struc-
′A16C5 in AN at 298.15 K. 3: (a) 18C6 in AN (b) 18C6 in MeOH (c) DB18C6 withight lanthanide nitrates (d) DB18C6 with light lanthanide perchlorates in AN (e)zA18C6 in AN at 298.15 K.
Despite these data are confined to PC, it can be reasonably sup-osed that also in the poor coordinating AN similar macrocyclicffects occur.
The less-symmetrical 16C5 gives higher K values than 15C5 inN (Fig. 3(2b)) for the second part of the light lanthanide ions stud-
ed (Sm–Gd). This behaviour suggests [104] that the size-fit concepts likely to be more rigorous in the complexation of lanthanidesnd operates best only when very strict size-matching is realizedetween the ligand cavity and the cation diameter, as is the caseith 16C5 (1.8–1.9 A) and Sm–Gd (1.97–1.88 A) [104].
The introduction of methyl groups at the 15-position of 16C5 inM16C5 (Scheme 1) adds steric hindrance, increases the rigidity of
he macrocycle and lowers its binding constant. Also the enthalpyhanges for the formation of MM16C5 complexes are less nega-ive than for 15C5. However, these differences do not influence theelative cation selectivity of the macrocycles [104].
istry Reviews 256 (2012) 328–351
The crown ether MR16C5, which has donating atoms in a lariatarm, shows a flat profile (Table 2 and Fig. 3(2c)) [104]. The absenceof selectivity may be due to the fact that MR16C5 is able to form athree-dimensional cavity upon complexation which is more adapt-able to any Ln(III) ion. The complexation enthalpy for MR16C5 isnegative, while T�S, generally small (positive or negative), doesnot show a definite trend. This is unexpected, since the lariat armcan bind the Ln(III) ion causing a substantial desolvation, and can bejustified as arising from a more complex reality which involves bal-ance between the structural freezing and the extensive desolvationwhich accompanies complexation [16].
Due to its relatively flexible molecular structure as comparedwith MM16C5, the R′A16C5 crown ether (Scheme 1) gives the high-est K for Nd3+ and the lowest for La3+ and Eu3+ among the lightlanthanide ions, showing the highest selectivity for Nd3+ [115].This has been attributed [16] to the most positive entropy changein the Ln(III) investigated (T�S = +18.54 kJ mol−1 for Nd3+, with aminimum of −26.7 and a maximum of +7.32 kJ mol−1, for Gd3+
and Pr3+ respectively) (Fig. 3(2e)) which is maximized only whena very strict size-fit relationship is attained between the inducedthree-dimensional cavity of the N-pivot lariat ether and the ionicdiameter of the lanthanides. Therefore, the increased conforma-tional freedom of the lariat ether (R′A16C5) is essential for selectivecomplexation of the size-matched trivalent lanthanide ions [115].
When BzA16C5 [116] is compared with BzA15C5 it is evidentthat the ring enlargement lowers the log K values. The N-containingBzA16C5 [116] shows the most negative enthalpy and entropychanges than the rest of the 16C5 family (Fig. 3(3)), which theauthors considered as originated by a balance between enhancedion–dipole interactions and structural freezing upon complexation[16]. A peculiar feature of BzA16C5 is the highest stability found forPr3+ which could be assigned to an optimum size match betweenthis ion and the cavity of the ligand.
The K values of light Ln(NO3)3 with 18C6 in AN indicate a slightlyhigher selectivity for Ce3+ [16]. Note that the lanthanide diameteris smaller (2.43 for La to 2.24 A for Eu3+, for CN = 9, Table 1) than thecavity of the ligand (2.6–3.2 A) but the undissociated nitrates in ANare larger than the solvated trivalent lanthanides [16].
The stabilities of the complexes of lanthanide(III) with crown-ethers obtained in AN by using Ln(NO3)3 as “starting” salts, areabout one order of magnitude higher than those found in MeOHby titration calorimetry [117] using LnCl3 (Table 2). This trend isalso confirmed for the perchlorate salt of Ce(III) with the same lig-and [117]. In MeOH the profile of the complex stabilities is keptunchanged with respect to AN throughout the light lanthanideseries till Eu, from Gd no heat evolution was detected during titra-tions in MeOH. The similitude of the cation selectivity sequencein the two solvents is surprising as the thermodynamic parame-ters act in a completely opposite manner: in MeOH the reactionis entropy-driven while it is enthalpy-driven in AN (Fig. 3.2). Thisbehaviour has been rationalized [16] considering that in MeOHboth the dissociated trivalent lanthanide chlorides and the freeligand are heavily solvated16 and this explains the highly positivereaction entropy which results from extensive desolvation. On theother hand, lanthanide nitrates are not dissociated in AN and there-fore solvation of the salts is not as heavy as in MeOH. In addition,no strong solvation of free ligand is expected to occur in this sol-vent. As a consequence, complexation in AN is mainly driven byion–dipole interaction which gives highly negative �H. The mono-
16 Through ion–dipole and hydrogen-bonding interaction, respectively.
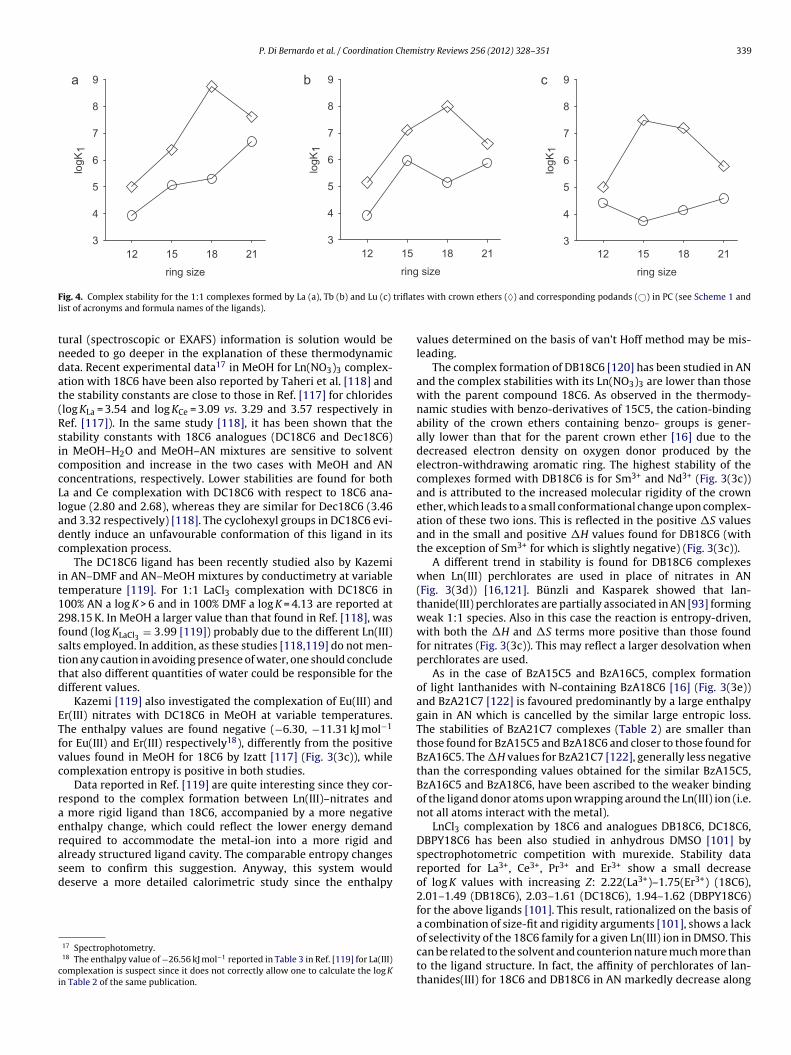
P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351 339
ring size
a b c
12 15 18 21
logK
1
3
4
5
6
7
8
9
ring size
12 15 18 21
logK
1
3
4
5
6
7
8
9
ring size
12 15 18 21
logK
1
3
4
5
6
7
8
9
F triflatel
tndat(RsiccLladc
it12fsttd
ETfvc
raerasd
ci
ig. 4. Complex stability for the 1:1 complexes formed by La (a), Tb (b) and Lu (c)ist of acronyms and formula names of the ligands).
ural (spectroscopic or EXAFS) information is solution would beeeded to go deeper in the explanation of these thermodynamicata. Recent experimental data17 in MeOH for Ln(NO3)3 complex-tion with 18C6 have been also reported by Taheri et al. [118] andhe stability constants are close to those in Ref. [117] for chlorideslog KLa = 3.54 and log KCe = 3.09 vs. 3.29 and 3.57 respectively inef. [117]). In the same study [118], it has been shown that thetability constants with 18C6 analogues (DC18C6 and Dec18C6)n MeOH–H2O and MeOH–AN mixtures are sensitive to solventomposition and increase in the two cases with MeOH and ANoncentrations, respectively. Lower stabilities are found for botha and Ce complexation with DC18C6 with respect to 18C6 ana-ogue (2.80 and 2.68), whereas they are similar for Dec18C6 (3.46nd 3.32 respectively) [118]. The cyclohexyl groups in DC18C6 evi-ently induce an unfavourable conformation of this ligand in itsomplexation process.
The DC18C6 ligand has been recently studied also by Kazemin AN–DMF and AN–MeOH mixtures by conductimetry at variableemperature [119]. For 1:1 LaCl3 complexation with DC18C6 in00% AN a log K > 6 and in 100% DMF a log K = 4.13 are reported at98.15 K. In MeOH a larger value than that found in Ref. [118], wasound (log KLaCl3 = 3.99 [119]) probably due to the different Ln(III)alts employed. In addition, as these studies [118,119] do not men-ion any caution in avoiding presence of water, one should concludehat also different quantities of water could be responsible for theifferent values.
Kazemi [119] also investigated the complexation of Eu(III) andr(III) nitrates with DC18C6 in MeOH at variable temperatures.he enthalpy values are found negative (−6.30, −11.31 kJ mol−1
or Eu(III) and Er(III) respectively18), differently from the positivealues found in MeOH for 18C6 by Izatt [117] (Fig. 3(3c)), whileomplexation entropy is positive in both studies.
Data reported in Ref. [119] are quite interesting since they cor-espond to the complex formation between Ln(III)–nitrates andmore rigid ligand than 18C6, accompanied by a more negative
nthalpy change, which could reflect the lower energy demandequired to accommodate the metal-ion into a more rigid andlready structured ligand cavity. The comparable entropy changes
eem to confirm this suggestion. Anyway, this system wouldeserve a more detailed calorimetric study since the enthalpy17 Spectrophotometry.18 The enthalpy value of −26.56 kJ mol−1 reported in Table 3 in Ref. [119] for La(III)omplexation is suspect since it does not correctly allow one to calculate the log Kn Table 2 of the same publication.
s with crown ethers (♦) and corresponding podands (©) in PC (see Scheme 1 and
values determined on the basis of van’t Hoff method may be mis-leading.
The complex formation of DB18C6 [120] has been studied in ANand the complex stabilities with its Ln(NO3)3 are lower than thosewith the parent compound 18C6. As observed in the thermody-namic studies with benzo-derivatives of 15C5, the cation-bindingability of the crown ethers containing benzo- groups is gener-ally lower than that for the parent crown ether [16] due to thedecreased electron density on oxygen donor produced by theelectron-withdrawing aromatic ring. The highest stability of thecomplexes formed with DB18C6 is for Sm3+ and Nd3+ (Fig. 3(3c))and is attributed to the increased molecular rigidity of the crownether, which leads to a small conformational change upon complex-ation of these two ions. This is reflected in the positive �S valuesand in the small and positive �H values found for DB18C6 (withthe exception of Sm3+ for which is slightly negative) (Fig. 3(3c)).
A different trend in stability is found for DB18C6 complexeswhen Ln(III) perchlorates are used in place of nitrates in AN(Fig. 3(3d)) [16,121]. Bünzli and Kasparek showed that lan-thanide(III) perchlorates are partially associated in AN [93] formingweak 1:1 species. Also in this case the reaction is entropy-driven,with both the �H and �S terms more positive than those foundfor nitrates (Fig. 3(3c)). This may reflect a larger desolvation whenperchlorates are used.
As in the case of BzA15C5 and BzA16C5, complex formationof light lanthanides with N-containing BzA18C6 [16] (Fig. 3(3e))and BzA21C7 [122] is favoured predominantly by a large enthalpygain in AN which is cancelled by the similar large entropic loss.The stabilities of BzA21C7 complexes (Table 2) are smaller thanthose found for BzA15C5 and BzA18C6 and closer to those found forBzA16C5. The �H values for BzA21C7 [122], generally less negativethan the corresponding values obtained for the similar BzA15C5,BzA16C5 and BzA18C6, have been ascribed to the weaker bindingof the ligand donor atoms upon wrapping around the Ln(III) ion (i.e.not all atoms interact with the metal).
LnCl3 complexation by 18C6 and analogues DB18C6, DC18C6,DBPY18C6 has been also studied in anhydrous DMSO [101] byspectrophotometric competition with murexide. Stability datareported for La3+, Ce3+, Pr3+ and Er3+ show a small decreaseof log K values with increasing Z: 2.22(La3+)–1.75(Er3+) (18C6),2.01–1.49 (DB18C6), 2.03–1.61 (DC18C6), 1.94–1.62 (DBPY18C6)for the above ligands [101]. This result, rationalized on the basis ofa combination of size-fit and rigidity arguments [101], shows a lack
of selectivity of the 18C6 family for a given Ln(III) ion in DMSO. Thiscan be related to the solvent and counterion nature much more thanto the ligand structure. In fact, the affinity of perchlorates of lan-thanides(III) for 18C6 and DB18C6 in AN markedly decrease along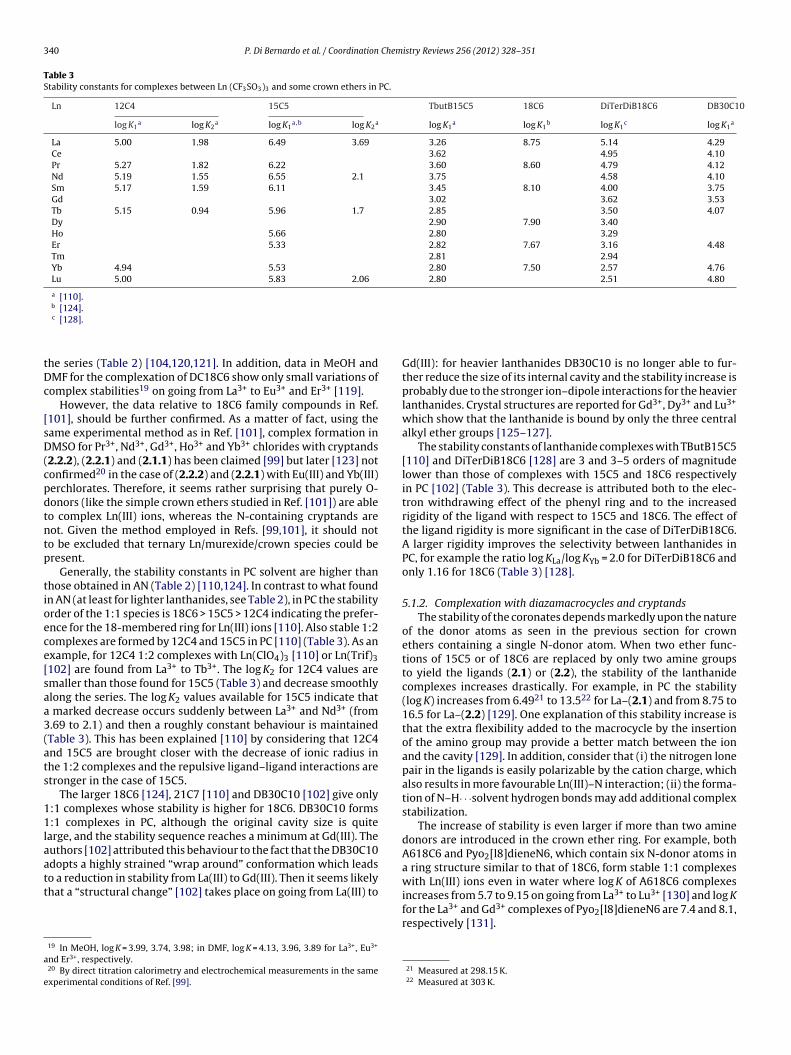
340 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Table 3Stability constants for complexes between Ln (CF3SO3)3 and some crown ethers in PC.
Ln 12C4 15C5 TbutB15C5 18C6 DiTerDiB18C6 DB30C10
log K1a log K2
a log K1a,b log K2
a log K1a log K1
b log K1c log K1
a
La 5.00 1.98 6.49 3.69 3.26 8.75 5.14 4.29Ce 3.62 4.95 4.10Pr 5.27 1.82 6.22 3.60 8.60 4.79 4.12Nd 5.19 1.55 6.55 2.1 3.75 4.58 4.10Sm 5.17 1.59 6.11 3.45 8.10 4.00 3.75Gd 3.02 3.62 3.53Tb 5.15 0.94 5.96 1.7 2.85 3.50 4.07Dy 2.90 7.90 3.40Ho 5.66 2.80 3.29Er 5.33 2.82 7.67 3.16 4.48Tm 2.81 2.94Yb 4.94 5.53 2.80 7.50 2.57 4.76Lu 5.00 5.83 2.06 2.80 2.51 4.80
a [110].
tDc
[sD(cpdtntp
tioece[saa3(ats
11laatt
a
e
b [124].c [128].
he series (Table 2) [104,120,121]. In addition, data in MeOH andMF for the complexation of DC18C6 show only small variations ofomplex stabilities19 on going from La3+ to Eu3+ and Er3+ [119].
However, the data relative to 18C6 family compounds in Ref.101], should be further confirmed. As a matter of fact, using theame experimental method as in Ref. [101], complex formation inMSO for Pr3+, Nd3+, Gd3+, Ho3+ and Yb3+ chlorides with cryptands
2.2.2), (2.2.1) and (2.1.1) has been claimed [99] but later [123] notonfirmed20 in the case of (2.2.2) and (2.2.1) with Eu(III) and Yb(III)erchlorates. Therefore, it seems rather surprising that purely O-onors (like the simple crown ethers studied in Ref. [101]) are ableo complex Ln(III) ions, whereas the N-containing cryptands areot. Given the method employed in Refs. [99,101], it should noto be excluded that ternary Ln/murexide/crown species could beresent.
Generally, the stability constants in PC solvent are higher thanhose obtained in AN (Table 2) [110,124]. In contrast to what foundn AN (at least for lighter lanthanides, see Table 2), in PC the stabilityrder of the 1:1 species is 18C6 > 15C5 > 12C4 indicating the prefer-nce for the 18-membered ring for Ln(III) ions [110]. Also stable 1:2omplexes are formed by 12C4 and 15C5 in PC [110] (Table 3). As anxample, for 12C4 1:2 complexes with Ln(ClO4)3 [110] or Ln(Trif)3102] are found from La3+ to Tb3+. The log K2 for 12C4 values aremaller than those found for 15C5 (Table 3) and decrease smoothlylong the series. The log K2 values available for 15C5 indicate thatmarked decrease occurs suddenly between La3+ and Nd3+ (from.69 to 2.1) and then a roughly constant behaviour is maintainedTable 3). This has been explained [110] by considering that 12C4nd 15C5 are brought closer with the decrease of ionic radius inhe 1:2 complexes and the repulsive ligand–ligand interactions aretronger in the case of 15C5.
The larger 18C6 [124], 21C7 [110] and DB30C10 [102] give only:1 complexes whose stability is higher for 18C6. DB30C10 forms:1 complexes in PC, although the original cavity size is quite
arge, and the stability sequence reaches a minimum at Gd(III). Theuthors [102] attributed this behaviour to the fact that the DB30C10
dopts a highly strained “wrap around” conformation which leadso a reduction in stability from La(III) to Gd(III). Then it seems likelyhat a “structural change” [102] takes place on going from La(III) to19 In MeOH, log K = 3.99, 3.74, 3.98; in DMF, log K = 4.13, 3.96, 3.89 for La3+, Eu3+
nd Er3+, respectively.20 By direct titration calorimetry and electrochemical measurements in the samexperimental conditions of Ref. [99].
Gd(III): for heavier lanthanides DB30C10 is no longer able to fur-ther reduce the size of its internal cavity and the stability increase isprobably due to the stronger ion–dipole interactions for the heavierlanthanides. Crystal structures are reported for Gd3+, Dy3+ and Lu3+
which show that the lanthanide is bound by only the three centralalkyl ether groups [125–127].
The stability constants of lanthanide complexes with TButB15C5[110] and DiTerDiB18C6 [128] are 3 and 3–5 orders of magnitudelower than those of complexes with 15C5 and 18C6 respectivelyin PC [102] (Table 3). This decrease is attributed both to the elec-tron withdrawing effect of the phenyl ring and to the increasedrigidity of the ligand with respect to 15C5 and 18C6. The effect ofthe ligand rigidity is more significant in the case of DiTerDiB18C6.A larger rigidity improves the selectivity between lanthanides inPC, for example the ratio log KLa/log KYb = 2.0 for DiTerDiB18C6 andonly 1.16 for 18C6 (Table 3) [128].
5.1.2. Complexation with diazamacrocycles and cryptandsThe stability of the coronates depends markedly upon the nature
of the donor atoms as seen in the previous section for crownethers containing a single N-donor atom. When two ether func-tions of 15C5 or of 18C6 are replaced by only two amine groupsto yield the ligands (2.1) or (2.2), the stability of the lanthanidecomplexes increases drastically. For example, in PC the stability(log K) increases from 6.4921 to 13.522 for La–(2.1) and from 8.75 to16.5 for La–(2.2) [129]. One explanation of this stability increase isthat the extra flexibility added to the macrocycle by the insertionof the amino group may provide a better match between the ionand the cavity [129]. In addition, consider that (i) the nitrogen lonepair in the ligands is easily polarizable by the cation charge, whichalso results in more favourable Ln(III)–N interaction; (ii) the forma-tion of N–H· · ·solvent hydrogen bonds may add additional complexstabilization.
The increase of stability is even larger if more than two aminedonors are introduced in the crown ether ring. For example, bothA618C6 and Pyo2[l8]dieneN6, which contain six N-donor atoms ina ring structure similar to that of 18C6, form stable 1:1 complexeswith Ln(III) ions even in water where log K of A618C6 complexes
increases from 5.7 to 9.15 on going from La3+ to Lu3+ [130] and log Kfor the La3+ and Gd3+ complexes of Pyo2[l8]dieneN6 are 7.4 and 8.1,respectively [131].21 Measured at 298.15 K.22 Measured at 303 K.
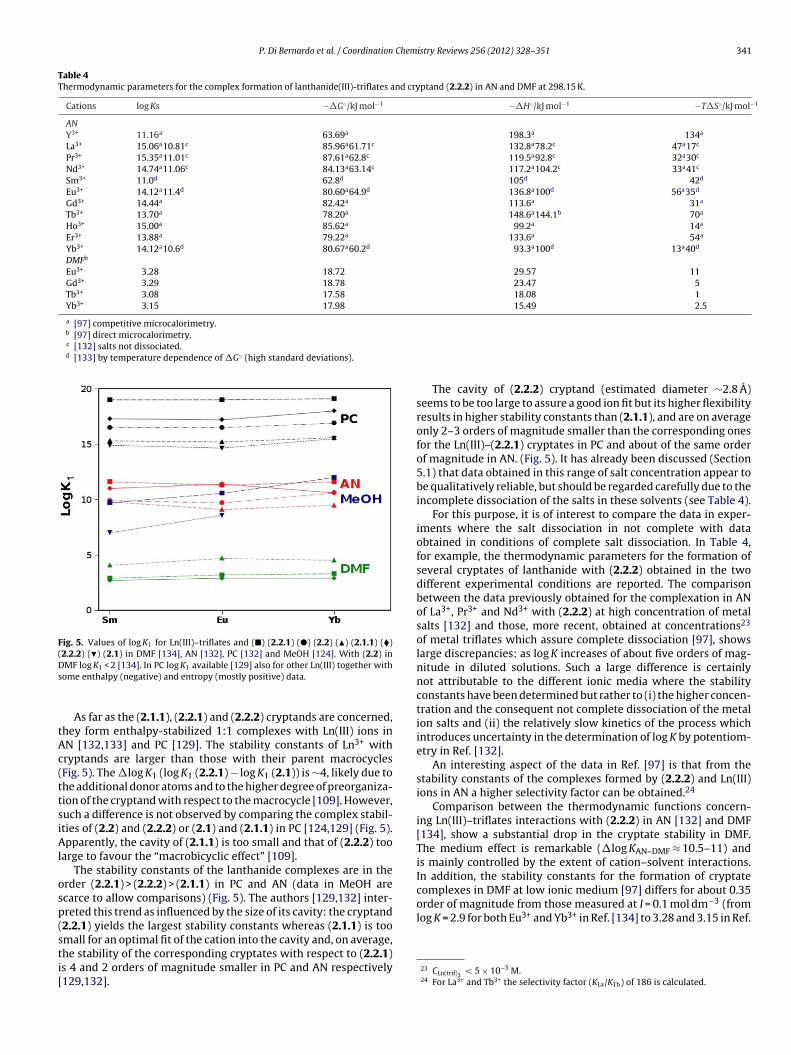
P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351 341
Table 4Thermodynamic parameters for the complex formation of lanthanide(III)-triflates and cryptand (2.2.2) in AN and DMF at 298.15 K.
Cations log Ks −�G◦/kJ mol−1 −�H◦/kJ mol−1 −T�S◦/kJ mol−1
ANY3+ 11.16a 63.69a 198.3a 134a
La3+ 15.06a10.81c 85.96a61.71c 132.8a78.2c 47a17c
Pr3+ 15.35a11.01c 87.61a62.8c 119.5a92.8c 32a30c
Nd3+ 14.74a11.06c 84.13a63.14c 117.2a104.2c 33a41c
Sm3+ 11.0d 62.8d 105d 42d
Eu3+ 14.12a11.4d 80.60a64.9d 136.8a100d 56a35d
Gd3+ 14.44a 82.42a 113.6a 31a
Tb3+ 13.70a 78.20a 148.6a144.1b 70a
Ho3+ 15.00a 85.62a 99.2a 14a
Er3+ 13.88a 79.22a 133.6a 54a
Yb3+ 14.12a10.6d 80.67a60.2d 93.3a100d 13a40d
DMFb
Eu3+ 3.28 18.72 29.57 11Gd3+ 3.29 18.78 23.47 5Tb3+ 3.08 17.58 18.08 1Yb3+ 3.15 17.98 15.49 2.5
a [97] competitive microcalorimetry.b [97] direct microcalorimetry.c [132] salts not dissociated.d [133] by temperature dependence of �G◦ (high standard deviations).
Fig. 5. Values of log K1 for Ln(III)–triflates and (�) (2.2.1) (�) (2.2) (�) (2.1.1) (�)(2.2.2) (�) (2.1) in DMF [134], AN [132], PC [132] and MeOH [124]. With (2.2) inDs
tAc(ttsiAl
osp(sti[
complexes in DMF at low ionic medium [97] differs for about 0.35order of magnitude from those measured at I = 0.1 mol dm−3 (fromlog K = 2.9 for both Eu3+ and Yb3+ in Ref. [134] to 3.28 and 3.15 in Ref.
MF log K1 < 2 [134]. In PC log K1 available [129] also for other Ln(III) together withome enthalpy (negative) and entropy (mostly positive) data.
As far as the (2.1.1), (2.2.1) and (2.2.2) cryptands are concerned,hey form enthalpy-stabilized 1:1 complexes with Ln(III) ions inN [132,133] and PC [129]. The stability constants of Ln3+ withryptands are larger than those with their parent macrocyclesFig. 5). The �log K1 (log K1 (2.2.1) − log K1 (2.1)) is ∼4, likely due tohe additional donor atoms and to the higher degree of preorganiza-ion of the cryptand with respect to the macrocycle [109]. However,uch a difference is not observed by comparing the complex stabil-ties of (2.2) and (2.2.2) or (2.1) and (2.1.1) in PC [124,129] (Fig. 5).pparently, the cavity of (2.1.1) is too small and that of (2.2.2) too
arge to favour the “macrobicyclic effect” [109].The stability constants of the lanthanide complexes are in the
rder (2.2.1) > (2.2.2) > (2.1.1) in PC and AN (data in MeOH arecarce to allow comparisons) (Fig. 5). The authors [129,132] inter-reted this trend as influenced by the size of its cavity: the cryptand2.2.1) yields the largest stability constants whereas (2.1.1) is toomall for an optimal fit of the cation into the cavity and, on average,
he stability of the corresponding cryptates with respect to (2.2.1)s 4 and 2 orders of magnitude smaller in PC and AN respectively129,132].The cavity of (2.2.2) cryptand (estimated diameter ∼2.8 A)seems to be too large to assure a good ion fit but its higher flexibilityresults in higher stability constants than (2.1.1), and are on averageonly 2–3 orders of magnitude smaller than the corresponding onesfor the Ln(III)–(2.2.1) cryptates in PC and about of the same orderof magnitude in AN. (Fig. 5). It has already been discussed (Section5.1) that data obtained in this range of salt concentration appear tobe qualitatively reliable, but should be regarded carefully due to theincomplete dissociation of the salts in these solvents (see Table 4).
For this purpose, it is of interest to compare the data in exper-iments where the salt dissociation in not complete with dataobtained in conditions of complete salt dissociation. In Table 4,for example, the thermodynamic parameters for the formation ofseveral cryptates of lanthanide with (2.2.2) obtained in the twodifferent experimental conditions are reported. The comparisonbetween the data previously obtained for the complexation in ANof La3+, Pr3+ and Nd3+ with (2.2.2) at high concentration of metalsalts [132] and those, more recent, obtained at concentrations23
of metal triflates which assure complete dissociation [97], showslarge discrepancies: as log K increases of about five orders of mag-nitude in diluted solutions. Such a large difference is certainlynot attributable to the different ionic media where the stabilityconstants have been determined but rather to (i) the higher concen-tration and the consequent not complete dissociation of the metalion salts and (ii) the relatively slow kinetics of the process whichintroduces uncertainty in the determination of log K by potentiom-etry in Ref. [132].
An interesting aspect of the data in Ref. [97] is that from thestability constants of the complexes formed by (2.2.2) and Ln(III)ions in AN a higher selectivity factor can be obtained.24
Comparison between the thermodynamic functions concern-ing Ln(III)–triflates interactions with (2.2.2) in AN [132] and DMF[134], show a substantial drop in the cryptate stability in DMF.The medium effect is remarkable (�log KAN–DMF ≈ 10.5–11) andis mainly controlled by the extent of cation–solvent interactions.In addition, the stability constants for the formation of cryptate
23 CLn(trif)3< 5 × 10−5 M.
24 For La3+ and Tb3+ the selectivity factor (KLa/KTb) of 186 is calculated.
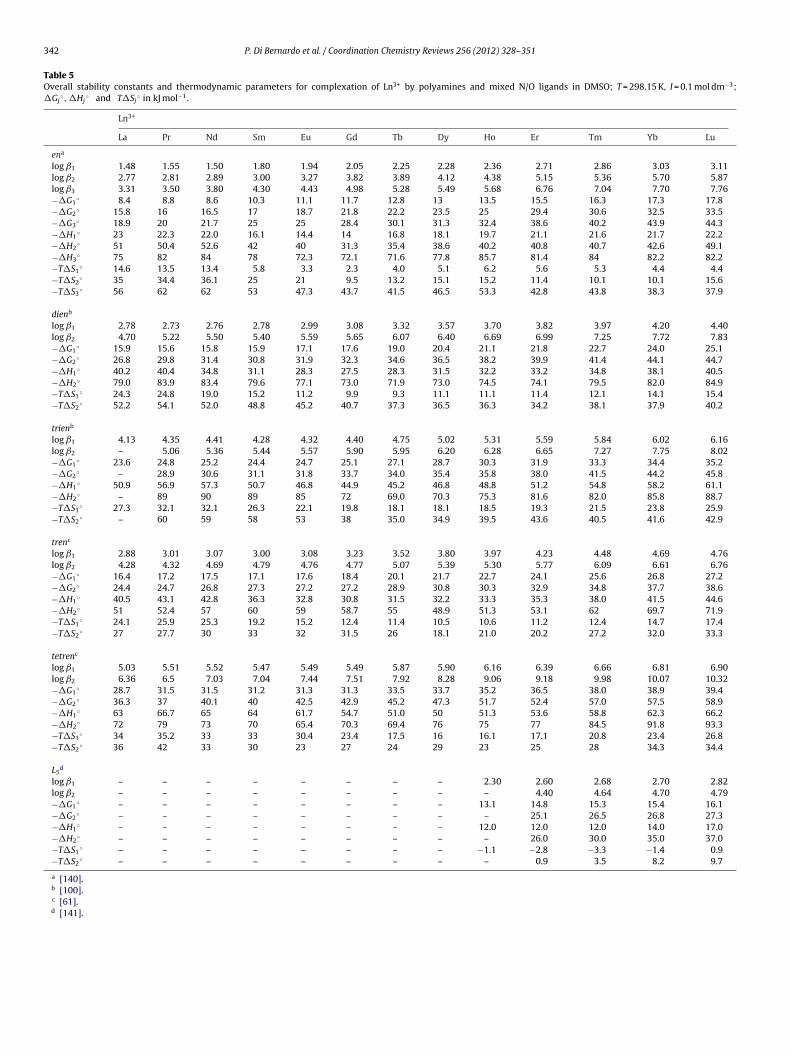
342 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Table 5Overall stability constants and thermodynamic parameters for complexation of Ln3+ by polyamines and mixed N/O ligands in DMSO; T = 298.15 K, I = 0.1 mol dm−3;�Gj
◦, �Hj◦ and T�Sj
◦ in kJ mol−1.
Ln3+
La Pr Nd Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
ena
log ˇ1 1.48 1.55 1.50 1.80 1.94 2.05 2.25 2.28 2.36 2.71 2.86 3.03 3.11log ˇ2 2.77 2.81 2.89 3.00 3.27 3.82 3.89 4.12 4.38 5.15 5.36 5.70 5.87log ˇ3 3.31 3.50 3.80 4.30 4.43 4.98 5.28 5.49 5.68 6.76 7.04 7.70 7.76−�G1
◦ 8.4 8.8 8.6 10.3 11.1 11.7 12.8 13 13.5 15.5 16.3 17.3 17.8−�G2
◦ 15.8 16 16.5 17 18.7 21.8 22.2 23.5 25 29.4 30.6 32.5 33.5−�G3
◦ 18.9 20 21.7 25 25 28.4 30.1 31.3 32.4 38.6 40.2 43.9 44.3−�H1
◦ 23 22.3 22.0 16.1 14.4 14 16.8 18.1 19.7 21.1 21.6 21.7 22.2−�H2
◦ 51 50.4 52.6 42 40 31.3 35.4 38.6 40.2 40.8 40.7 42.6 49.1−�H3
◦ 75 82 84 78 72.3 72.1 71.6 77.8 85.7 81.4 84 82.2 82.2−T�S1
◦ 14.6 13.5 13.4 5.8 3.3 2.3 4.0 5.1 6.2 5.6 5.3 4.4 4.4−T�S2
◦ 35 34.4 36.1 25 21 9.5 13.2 15.1 15.2 11.4 10.1 10.1 15.6−T�S3
◦ 56 62 62 53 47.3 43.7 41.5 46.5 53.3 42.8 43.8 38.3 37.9
dienb
log ˇ1 2.78 2.73 2.76 2.78 2.99 3.08 3.32 3.57 3.70 3.82 3.97 4.20 4.40log ˇ2 4.70 5.22 5.50 5.40 5.59 5.65 6.07 6.40 6.69 6.99 7.25 7.72 7.83−�G1
◦ 15.9 15.6 15.8 15.9 17.1 17.6 19.0 20.4 21.1 21.8 22.7 24.0 25.1−�G2
◦ 26.8 29.8 31.4 30.8 31.9 32.3 34.6 36.5 38.2 39.9 41.4 44.1 44.7−�H1
◦ 40.2 40.4 34.8 31.1 28.3 27.5 28.3 31.5 32.2 33.2 34.8 38.1 40.5−�H2
◦ 79.0 83.9 83.4 79.6 77.1 73.0 71.9 73.0 74.5 74.1 79.5 82.0 84.9−T�S1
◦ 24.3 24.8 19.0 15.2 11.2 9.9 9.3 11.1 11.1 11.4 12.1 14.1 15.4−T�S2
◦ 52.2 54.1 52.0 48.8 45.2 40.7 37.3 36.5 36.3 34.2 38.1 37.9 40.2
trienb
log ˇ1 4.13 4.35 4.41 4.28 4.32 4.40 4.75 5.02 5.31 5.59 5.84 6.02 6.16log ˇ2 – 5.06 5.36 5.44 5.57 5.90 5.95 6.20 6.28 6.65 7.27 7.75 8.02−�G1
◦ 23.6 24.8 25.2 24.4 24.7 25.1 27.1 28.7 30.3 31.9 33.3 34.4 35.2−�G2
◦ – 28.9 30.6 31.1 31.8 33.7 34.0 35.4 35.8 38.0 41.5 44.2 45.8−�H1
◦ 50.9 56.9 57.3 50.7 46.8 44.9 45.2 46.8 48.8 51.2 54.8 58.2 61.1−�H2
◦ – 89 90 89 85 72 69.0 70.3 75.3 81.6 82.0 85.8 88.7−T�S1
◦ 27.3 32.1 32.1 26.3 22.1 19.8 18.1 18.1 18.5 19.3 21.5 23.8 25.9−T�S2
◦ – 60 59 58 53 38 35.0 34.9 39.5 43.6 40.5 41.6 42.9
trenc
log ˇ1 2.88 3.01 3.07 3.00 3.08 3.23 3.52 3.80 3.97 4.23 4.48 4.69 4.76log ˇ2 4.28 4.32 4.69 4.79 4.76 4.77 5.07 5.39 5.30 5.77 6.09 6.61 6.76−�G1
◦ 16.4 17.2 17.5 17.1 17.6 18.4 20.1 21.7 22.7 24.1 25.6 26.8 27.2−�G2
◦ 24.4 24.7 26.8 27.3 27.2 27.2 28.9 30.8 30.3 32.9 34.8 37.7 38.6−�H1
◦ 40.5 43.1 42.8 36.3 32.8 30.8 31.5 32.2 33.3 35.3 38.0 41.5 44.6−�H2
◦ 51 52.4 57 60 59 58.7 55 48.9 51.3 53.1 62 69.7 71.9−T�S1
◦ 24.1 25.9 25.3 19.2 15.2 12.4 11.4 10.5 10.6 11.2 12.4 14.7 17.4−T�S2
◦ 27 27.7 30 33 32 31.5 26 18.1 21.0 20.2 27.2 32.0 33.3
tetrenc
log ˇ1 5.03 5.51 5.52 5.47 5.49 5.49 5.87 5.90 6.16 6.39 6.66 6.81 6.90log ˇ2 6.36 6.5 7.03 7.04 7.44 7.51 7.92 8.28 9.06 9.18 9.98 10.07 10.32−�G1
◦ 28.7 31.5 31.5 31.2 31.3 31.3 33.5 33.7 35.2 36.5 38.0 38.9 39.4−�G2
◦ 36.3 37 40.1 40 42.5 42.9 45.2 47.3 51.7 52.4 57.0 57.5 58.9−�H1
◦ 63 66.7 65 64 61.7 54.7 51.0 50 51.3 53.6 58.8 62.3 66.2−�H2
◦ 72 79 73 70 65.4 70.3 69.4 76 75 77 84.5 91.8 93.3−T�S1
◦ 34 35.2 33 33 30.4 23.4 17.5 16 16.1 17.1 20.8 23.4 26.8−T�S2
◦ 36 42 33 30 23 27 24 29 23 25 28 34.3 34.4
L5d
log ˇ1 – – – – – – – – 2.30 2.60 2.68 2.70 2.82log ˇ2 – – – – – – – – – 4.40 4.64 4.70 4.79−�G1
◦ – – – – – – – – 13.1 14.8 15.3 15.4 16.1−�G2
◦ – – – – – – – – – 25.1 26.5 26.8 27.3−�H1
◦ – – – – – – – – 12.0 12.0 12.0 14.0 17.0−�H2
◦ – – – – – – – – – 26.0 30.0 35.0 37.0−T�S1
◦ – – – – – – – – −1.1 −2.8 −3.3 −1.4 0.9−T�S2
◦ – – – – – – – – – 0.9 3.5 8.2 9.7
a [140].b [100].c [61].d [141].

Chem
[tdi
5c
isieti
mSano[hstiditp
tt(sctcaSb[
flotctmabi[
5
so
1
P. Di Bernardo et al. / Coordination
97]. This difference is in line with what expected on the basis ofhe different ionic medium. As mentioned (Section 5.1.1), thermo-ynamic data reported for (2.2.1) and (2.2.2) Ln(III)–ligand systems
n DMSO [99] should be revisited.
.1.3. Solvent influence on crown ethers and cryptandsomplexation
The influence of the solvent is dramatic and, generally speak-ng, the stability of lanthanide cryptates increases in the followingolvent sequence: DMSO < DMF < MeOH < AN < PC which is mostlyn agreement with the solvating properties of the solvents asxpressed by their Gutmann donor numbers [56] (with the excep-ion of PC) and with the trends of the preferential solvation obtainedn Refs. [58,59] (see Section 3.1).
The stability constants of cryptates of (2.2.1) and (2.2.2) areuch lower in AN than in PC: in the former solvent log K1 for
m3+, Eu3+ and Yb3+ is almost constant (Fig. 5, Ref. [133])25 andbout 7 orders of magnitude smaller than in PC [132]. Thermody-amic parameters are available in both media for the formationf (2.2.1) cryptates with other lanthanides(III), La3+, Pr3+ and Nd3+
132] and allow one to conclude that this large difference in stabilityas an entropic origin. In fact, in PC, cryptates are clearly enthalpytabilized,26 with favourable entropic contribution27 [132]. In AN,he enthalpic contribution is comparable with that in PC,28 but,n this case, the entropy is significantly unfavourable29 [132]. Theecrease of the reaction entropy with increasing atomic number
n both the solvents has been interpreted [132] as arising fromhe smaller solvation number for the heavier lanthanide ions and,ossibly, to an incomplete de-solvation.
Complex formation with cryptands has often been used forhe determination of single ion transfer parameters betweenwo solvents. The method is based on the approximation‘cryptate assumption’) that the inclusion complex (cryptate),hould interact in a similar way with the environment inomparison to uncomplexed cryptand with the consequencehat cryptate and cryptand transfer parameters should beomparable: �Gtr(cryptand) ≈ �Gtr(cryptate) [135]. Under thisssumption, single ion transfer free energy is calculated as �Gtr
1 → S2(Mn+) ≈ 2.3RT(log Kf(S1) − log Kf(S2)), where Kf is the sta-ility constant for the cryptate in each solvent (S1 and S2)135].
The single ion enthalpies of transfer from PC to AN determinedor the (2.2.1) and (2.2.2) Ln(III)–cryptates are independent of theigands but vary with the lanthanide ions [132]. Transfer enthalpyf the cryptates and of the Ln(III) ions is nearly equal indicating thathe cation recognition is performed by the solvent and not by theryptand [132]. Therefore, the complete shielding of the cation byhe cryptand, at the basis of the “cryptate assumption”, is not a good
odel for lanthanide cryptates. This may be due to the high chargend coordination numbers of lanthanide(III) ions, as suggestedy the crystal structures where the lanthanide is not embedded
n the cyptate, but it interacts with anions or solvent molecules136].
.1.4. Stabilization of lanthanide low oxidation statesIn solution, lanthanide ions usually exist in the +3 oxidation
tate, with a few exceptions (see Section 2.1). However their +2xidation state can be favoured by including the cations into lig-
25 Log K1 <fn0115>for (2.2.1): 11.6 (Sm3+), 11.3 (Eu3+) and 11.6 (Yb3+); for (2.2.2):1.0 (Sm3+), 11.4 (Eu3+) and 10.6 (Yb3+).26 �H = −77, −91 and −105 kJ mol−1 for La3+, Pr3+ and Nd3+, respectively.27 T�S = 29, 15 and 2 kJ mol−1, for La3+, Pr3+ and Nd3+, respectively.28 �H is between −75 and −107 kJ mol−1.29 T�S = −13, −28 and −40 kJ mol−1 for La3+, Pr3+ and Nd3+, respectively.
istry Reviews 256 (2012) 328–351 343
ands that can stabilize them. For example, since the beginning oftheir use as host for spherical metal ions, it has been realized thatcoronands and cryptands, may stabilize low oxidation states ofthe lanthanides because their somewhat flexible cavity can bet-ter adapt to the ionic radii of the Ln(II) ions which are usually0.16–0.20 A larger than those of Ln(III) ions [7].
In several solvents, including water, DMF, MeOH, the reductionof trivalent lanthanide coronates and cryptates always occurs ata higher potential than the reduction of the uncomplexed cations[133,134]. In some instances, e.g. in water, the cyclic voltammo-grams of the uncomplexed ions are irreversible, which is not thecase for the encapsulated cations. The difference in the formal redoxpotential �E between the complex and uncomplexed one-electronredox couples is related to the difference in Gibbs free energy forthese processes by the so-called Nernst–Peter equation30 [7].
Several effects influence the stabilization of Ln(II) over Ln(III).The stabilities of the Ln(III)–cryptates are inversely proportional toGutmann’s donor number of the solvent. This is not the case forthe Ln(II) coronates and cryptates, the stability of which is moreor less independent of the solvent. This leads to large variations inthe stabilization of Ln(II) over Ln(III) depending upon the solventand ligand. For example, studies on cryptates of lanthanides with(2.2.1) or (2.2.2) show that stabilization of +2 state complexes inPC or AN is small (log KII is at most two orders of magnitude largerthan log KIII) or even negative [137], while it is much more evidentin water where log KII is up to seven orders of magnitude higherthan log KIII [7].
In DMSO, the Eu(II) cryptates with (2.2.1) and (2.2.2) havesizeable stability [123], whereas Eu(III)- and, generally speaking,Ln(III)–cryptates do not form to an appreciable extent in this sol-vent for which also the use of the Nernst–Peter equation leadsto expected negative values for log KIII [7]. The thermodynamicstudy [123] shows that the Eu(II) cryptates of (2.2.1) and (2.2.2)are stabilized by a large enthalpic effect and de-stabilized by anunfavourable entropic contribution: log K1 = 5.80 and 5.33; �H1
◦ =−50.2 and −36.0 kJ mol−1; T�S1
◦ = −17.1 and −5.6 kJ mol−1 forEu(2.2.1)2+ and Eu(2.2.2)2+ systems, respectively [123].
Generally (2.2.2) forms the most stable Ln(II) complexes in allsolvents according with size considerations. In addition a markedpreorganization effect is present as the difference in stabilitybetween (2.2.2) and (2.2) complexes is 7.5 and 10 log units in AN[133] and DMF [134]. From all the above, it emerges that the stabil-ity of divalent lanthanide cryptates is controlled by size rather thanby solvent effects which dominate the cryptate stability of trivalentlanthanides.
5.2. Coordination of N-donors
The complexation reactions of trivalent lanthanide ions withpure nitrogen donors such as amines and polyamines are easilystudied in non-aqueous solution where difficulties due to hydroly-sis of the metal ions or ligand protonation are not present.
5.2.1. Monodentate aminesAlthough n-butylamine (n-but) has a larger DN than DMSO
(DNn-but = 42.0; DNDMSO = 29.8 [56]) no interaction with Ln(III)ions has been detected in this solvent [138] by means of bothpotentiometric and calorimetric measurements. Clearly, the high
concentration of the solvent compensates its lower donatingstrength and prevents the ligand–metal interaction.Strong interaction of n-but with heavier Ln(III)–triflates hasbeen indicated in AN [138] where the metal–ligand interactions
30 �E1/2 =(
RTF
)log
(KIIKIII
)= �GII
◦−�GIII◦
F .
344 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Scheme 2. Polyamines and mixed N/O donor ligands.
cTtatLmmc
mtaagrif
uttten
at
Fig. 6. Plot of log K1 for the formation of the lanthanide complexes of en (©), dien( ), tren (�), trien (�) and tetren (�) as a function of the ionic radii of the elements.Solvent DMSO, T = 298.15 K and I = 0.1 mol dm−3. The lines connecting symbols areeye guides.
Luminescence lifetime measurements on Eu3+ and Tb3+ com-plexes with en [139], show that each N–H vibrator unit of bonded
TO
D
an prevail over the metal–solvent interactions (DNAN = 14.1 [56]).his study was limited to Tb–Lu triflates as the lighter lanthanideriflates are sparingly soluble in AN. Conductometric, calorimetricnd FT-IR measurements show that the undissociated or par-ially dissociated species present in this medium (Ln(trif)3 andn(trif)2
+ (see Eq. (1), Section 4) form with n-but (L) strongononuclear successive complexes, Ln(trif)nLm
(3 − n)+; n = 2 or 3;= 1–4, whose stability is almost independent from the acceptor
harge.The bonding of n-but occurs with a preferential loss of solvent
olecules from the coordination sphere of the metal ions. Due tohe complexity of the system, only the apparent stability constantsnd the related thermodynamic values concerning the complexingbility of n-but for a system containing Ln(trif)3 and Ln(trif)2
+ areiven [138]. The stability of the four complexes increases almostegularly with the atomic number of metal ions. For example, log K1ncreases from 4.04 to 4.31 and log K4 from 10.81 to 11.46, on goingrom Tb3+ to Lu3+, respectively.
All the complexes were enthalpy stabilized and entropynfavoured. This pattern is typical for Ln3+ complexation by neu-ral ligands in aprotic solvent. The enthalpy change associated withhe complexation reactions is a measure of the difference betweenhe metal–ligand and metal–solvent bond energies. Thus, negativenthalpies reflect a larger interaction towards the Ln3+ ions of theeutral ligands with respect to the solvent molecules.
The desolvation of the cation upon coordination is rather smallnd consequently the negative entropy change observed for allhe n-but complexes emphasizes the predominance of the coor-
able 6verall stability constants and thermodynamic parameters for complexation of Ln3+ by d
Ln3+ Log ˇ1 �G1◦ �H1
◦ T�S1◦
La 5.29 −30.2 −44.9 −14.7Ce 5.58 −31.9 −54 −22.1Pr 5.76 −32.9 −59.7 −26.8Nd 5.90 −33.7 −61.6 −27.9Sm 5.74 −32.8 −54.8 −22.0Eu 5.66 −32.3 −52.3 −20.0Gd 5.69 −32.5 −46.8 −14.3Tb 5.73 −32.7 −47.6 −14.9Dy 6.03 −34.4 −47 −12.6Ho 6.19 −35.3 −47.7 −12.4Er 6.39 −36.5 −46.3 −9.8Tm 6.57 −37.5 −49.1 −11.6Yb 6.74 −38.5 −50.3 −11.8Lu 6.87 −39.2 −51.6 −12.4
ata from Ref. [79].
dination over the desolvation processes when neutral ligandscoordinate the lanthanide ions in non-aqueous solution.
In the absence of accurate FT-IR and conductivity measure-ments, the calorimetric and potentiometric results in Ref. [138]could have been wrongly interpreted as reflecting a simple free-metal-ion–amine interaction.
5.2.2. Polydentate aminesPolyamines, differently from monoamines, form relatively
strong complexes with Ln3 + ions in DMSO, where a variety of theseligands (Scheme 2) has been studied [61,100,139,140], to obtainstructure–stability relationships. The combination of complemen-tary experimental techniques (potentiometry, calorimetry, FT-IRand luminescence spectroscopy), provides an almost complete pic-ture of the behaviour of the polyamines towards Ln3+ ions in DMSO.
amine acts independently in the luminescence quenching pro-cesses, so that the observed decay constants (kobs) of the aminecomplexes of Eu3+ and Tb3+ can be linearly related to the average
ien in DMF; T = 298.15 K, I = 0.1 mol dm−3; �Gj◦, �Hj
◦ and T�Sj◦ in kJ mol−1.
Log ˇ2 �G2◦ �H2
◦ T�S2◦
8.36 −47.7 −89.7 −42.09.12 −52.1 −102.3 −50.29.48 −54.1 −107.1 −53.0
10.11 −57.7 −109.9 −52.210.31 −58.8 −113.6 −54.810.11 −57.7 −111.8 −54.110.25 −58.5 −111.4 −52.9
9.70 −55.4 −104.8 −49.410.08 −57.5 −102.2 −44.710.19 −58.2 −100.9 −42.710.43 −59.5 −94.5 −35.010.68 −61.0 −96.9 −35.910.82 −61.8 −98.2 −36.411.20 −63.9 −101.6 −37.7
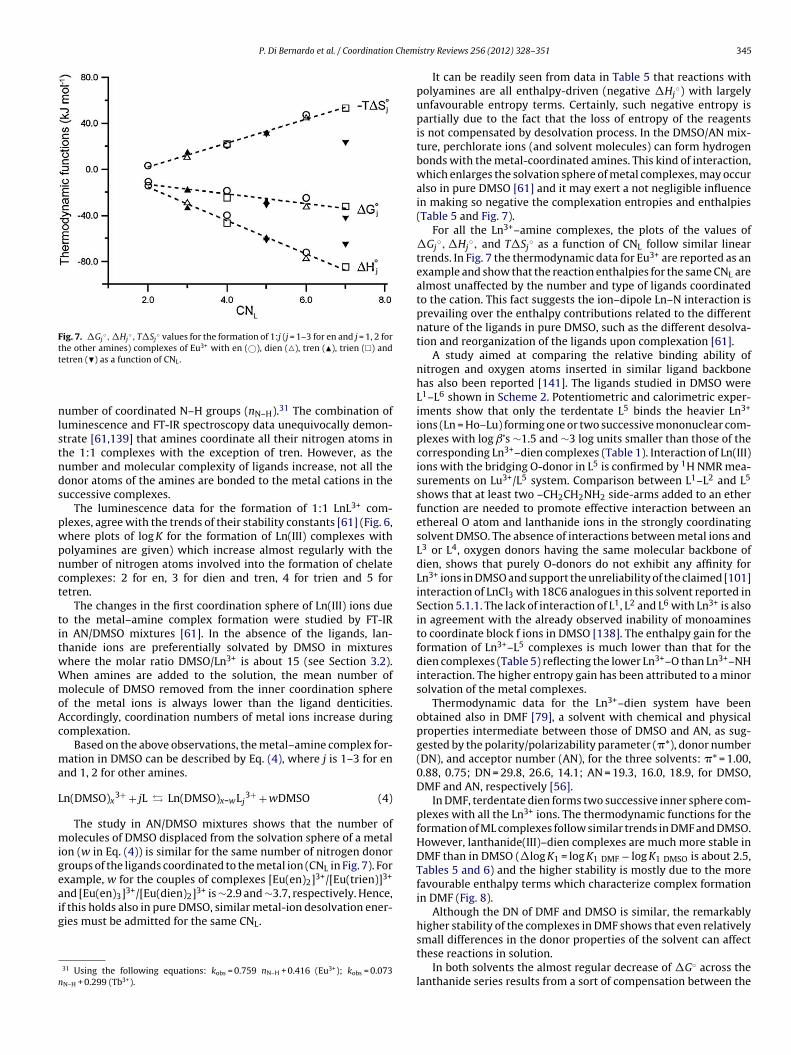
P. Di Bernardo et al. / Coordination Chem
Fig. 7. �Gj◦, �Hj
◦, T�Sj◦ values for the formation of 1:j (j = 1–3 for en and j = 1, 2 for
the other amines) complexes of Eu3+ with en (©), dien ( ), tren (�), trien (�) andt
nlstnds
pwpnct
titwWmoAc
ma
L
migeaig
n
etren (�) as a function of CNL.
umber of coordinated N–H groups (nN–H).31 The combination ofuminescence and FT-IR spectroscopy data unequivocally demon-trate [61,139] that amines coordinate all their nitrogen atoms inhe 1:1 complexes with the exception of tren. However, as theumber and molecular complexity of ligands increase, not all theonor atoms of the amines are bonded to the metal cations in theuccessive complexes.
The luminescence data for the formation of 1:1 LnL3+ com-lexes, agree with the trends of their stability constants [61] (Fig. 6,here plots of log K for the formation of Ln(III) complexes witholyamines are given) which increase almost regularly with theumber of nitrogen atoms involved into the formation of chelateomplexes: 2 for en, 3 for dien and tren, 4 for trien and 5 foretren.
The changes in the first coordination sphere of Ln(III) ions dueo the metal–amine complex formation were studied by FT-IRn AN/DMSO mixtures [61]. In the absence of the ligands, lan-hanide ions are preferentially solvated by DMSO in mixtureshere the molar ratio DMSO/Ln3+ is about 15 (see Section 3.2).hen amines are added to the solution, the mean number ofolecule of DMSO removed from the inner coordination sphere
f the metal ions is always lower than the ligand denticities.ccordingly, coordination numbers of metal ions increase duringomplexation.
Based on the above observations, the metal–amine complex for-ation in DMSO can be described by Eq. (4), where j is 1–3 for en
nd 1, 2 for other amines.
n(DMSO)x3+ + jL � Ln(DMSO)x-wLj
3+ + wDMSO (4)
The study in AN/DMSO mixtures shows that the number ofolecules of DMSO displaced from the solvation sphere of a metal
on (w in Eq. (4)) is similar for the same number of nitrogen donorroups of the ligands coordinated to the metal ion (CNL in Fig. 7). Forxample, w for the couples of complexes [Eu(en)2]3+/[Eu(trien)]3+
nd [Eu(en)3]3+/[Eu(dien)2]3+ is ∼2.9 and ∼3.7, respectively. Hence,f this holds also in pure DMSO, similar metal-ion desolvation ener-ies must be admitted for the same CNL.
31 Using the following equations: kobs = 0.759 nN–H + 0.416 (Eu3+); kobs = 0.073N–H + 0.299 (Tb3+).
istry Reviews 256 (2012) 328–351 345
It can be readily seen from data in Table 5 that reactions withpolyamines are all enthalpy-driven (negative �Hj
◦) with largelyunfavourable entropy terms. Certainly, such negative entropy ispartially due to the fact that the loss of entropy of the reagentsis not compensated by desolvation process. In the DMSO/AN mix-ture, perchlorate ions (and solvent molecules) can form hydrogenbonds with the metal-coordinated amines. This kind of interaction,which enlarges the solvation sphere of metal complexes, may occuralso in pure DMSO [61] and it may exert a not negligible influencein making so negative the complexation entropies and enthalpies(Table 5 and Fig. 7).
For all the Ln3+–amine complexes, the plots of the values of�Gj
◦, �Hj◦, and T�Sj
◦ as a function of CNL follow similar lineartrends. In Fig. 7 the thermodynamic data for Eu3+ are reported as anexample and show that the reaction enthalpies for the same CNL arealmost unaffected by the number and type of ligands coordinatedto the cation. This fact suggests the ion–dipole Ln–N interaction isprevailing over the enthalpy contributions related to the differentnature of the ligands in pure DMSO, such as the different desolva-tion and reorganization of the ligands upon complexation [61].
A study aimed at comparing the relative binding ability ofnitrogen and oxygen atoms inserted in similar ligand backbonehas also been reported [141]. The ligands studied in DMSO wereL1–L6 shown in Scheme 2. Potentiometric and calorimetric exper-iments show that only the terdentate L5 binds the heavier Ln3+
ions (Ln = Ho–Lu) forming one or two successive mononuclear com-plexes with log ˇ’s ∼1.5 and ∼3 log units smaller than those of thecorresponding Ln3+–dien complexes (Table 1). Interaction of Ln(III)ions with the bridging O-donor in L5 is confirmed by 1H NMR mea-surements on Lu3+/L5 system. Comparison between L1–L2 and L5
shows that at least two –CH2CH2NH2 side-arms added to an etherfunction are needed to promote effective interaction between anethereal O atom and lanthanide ions in the strongly coordinatingsolvent DMSO. The absence of interactions between metal ions andL3 or L4, oxygen donors having the same molecular backbone ofdien, shows that purely O-donors do not exhibit any affinity forLn3+ ions in DMSO and support the unreliability of the claimed [101]interaction of LnCl3 with 18C6 analogues in this solvent reported inSection 5.1.1. The lack of interaction of L1, L2 and L6 with Ln3+ is alsoin agreement with the already observed inability of monoaminesto coordinate block f ions in DMSO [138]. The enthalpy gain for theformation of Ln3+–L5 complexes is much lower than that for thedien complexes (Table 5) reflecting the lower Ln3+–O than Ln3+–NHinteraction. The higher entropy gain has been attributed to a minorsolvation of the metal complexes.
Thermodynamic data for the Ln3+–dien system have beenobtained also in DMF [79], a solvent with chemical and physicalproperties intermediate between those of DMSO and AN, as sug-gested by the polarity/polarizability parameter (�*), donor number(DN), and acceptor number (AN), for the three solvents: �* = 1.00,0.88, 0.75; DN = 29.8, 26.6, 14.1; AN = 19.3, 16.0, 18.9, for DMSO,DMF and AN, respectively [56].
In DMF, terdentate dien forms two successive inner sphere com-plexes with all the Ln3+ ions. The thermodynamic functions for theformation of ML complexes follow similar trends in DMF and DMSO.However, lanthanide(III)–dien complexes are much more stable inDMF than in DMSO (�log K1 = log K1 DMF − log K1 DMSO is about 2.5,Tables 5 and 6) and the higher stability is mostly due to the morefavourable enthalpy terms which characterize complex formationin DMF (Fig. 8).
Although the DN of DMF and DMSO is similar, the remarkablyhigher stability of the complexes in DMF shows that even relatively
small differences in the donor properties of the solvent can affectthese reactions in solution.In both solvents the almost regular decrease of �G◦ across thelanthanide series results from a sort of compensation between the
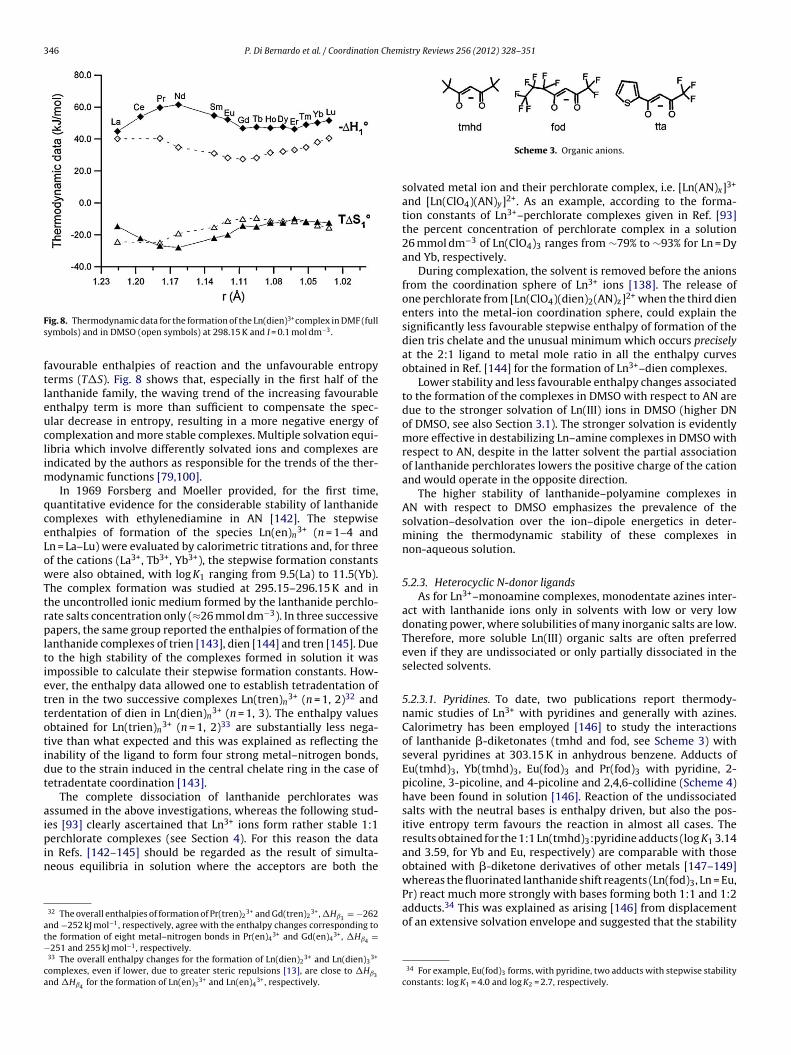
346 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
Fs
ftleuclim
qceLowTtrpltiettotidt
aipin
at−
ca
ig. 8. Thermodynamic data for the formation of the Ln(dien)3+complex in DMF (fullymbols) and in DMSO (open symbols) at 298.15 K and I = 0.1 mol dm−3.
avourable enthalpies of reaction and the unfavourable entropyerms (T�S). Fig. 8 shows that, especially in the first half of theanthanide family, the waving trend of the increasing favourablenthalpy term is more than sufficient to compensate the spec-lar decrease in entropy, resulting in a more negative energy ofomplexation and more stable complexes. Multiple solvation equi-ibria which involve differently solvated ions and complexes arendicated by the authors as responsible for the trends of the ther-
odynamic functions [79,100].In 1969 Forsberg and Moeller provided, for the first time,
uantitative evidence for the considerable stability of lanthanideomplexes with ethylenediamine in AN [142]. The stepwisenthalpies of formation of the species Ln(en)n
3+ (n = 1–4 andn = La–Lu) were evaluated by calorimetric titrations and, for threef the cations (La3+, Tb3+, Yb3+), the stepwise formation constantsere also obtained, with log K1 ranging from 9.5(La) to 11.5(Yb).
he complex formation was studied at 295.15–296.15 K and inhe uncontrolled ionic medium formed by the lanthanide perchlo-ate salts concentration only (≈26 mmol dm−3). In three successiveapers, the same group reported the enthalpies of formation of the
anthanide complexes of trien [143], dien [144] and tren [145]. Dueo the high stability of the complexes formed in solution it wasmpossible to calculate their stepwise formation constants. How-ver, the enthalpy data allowed one to establish tetradentation ofren in the two successive complexes Ln(tren)n
3+ (n = 1, 2)32 anderdentation of dien in Ln(dien)n
3+ (n = 1, 3). The enthalpy valuesbtained for Ln(trien)n
3+ (n = 1, 2)33 are substantially less nega-ive than what expected and this was explained as reflecting thenability of the ligand to form four strong metal–nitrogen bonds,ue to the strain induced in the central chelate ring in the case ofetradentate coordination [143].
The complete dissociation of lanthanide perchlorates wasssumed in the above investigations, whereas the following stud-es [93] clearly ascertained that Ln3+ ions form rather stable 1:1
erchlorate complexes (see Section 4). For this reason the datan Refs. [142–145] should be regarded as the result of simulta-eous equilibria in solution where the acceptors are both the
32 The overall enthalpies of formation of Pr(tren)23+ and Gd(tren)2
3+, �Hˇ3= −262
nd −252 kJ mol−1, respectively, agree with the enthalpy changes corresponding tohe formation of eight metal–nitrogen bonds in Pr(en)4
3+ and Gd(en)43+, �Hˇ4
=251 and 255 kJ mol−1, respectively.
33 The overall enthalpy changes for the formation of Ln(dien)23+ and Ln(dien)3
3+
omplexes, even if lower, due to greater steric repulsions [13], are close to �Hˇ3nd �Hˇ4
for the formation of Ln(en)33+ and Ln(en)4
3+, respectively.
Scheme 3. Organic anions.
solvated metal ion and their perchlorate complex, i.e. [Ln(AN)x]3+
and [Ln(ClO4)(AN)y]2+. As an example, according to the forma-tion constants of Ln3+–perchlorate complexes given in Ref. [93]the percent concentration of perchlorate complex in a solution26 mmol dm−3 of Ln(ClO4)3 ranges from ∼79% to ∼93% for Ln = Dyand Yb, respectively.
During complexation, the solvent is removed before the anionsfrom the coordination sphere of Ln3+ ions [138]. The release ofone perchlorate from [Ln(ClO4)(dien)2(AN)z]2+ when the third dienenters into the metal-ion coordination sphere, could explain thesignificantly less favourable stepwise enthalpy of formation of thedien tris chelate and the unusual minimum which occurs preciselyat the 2:1 ligand to metal mole ratio in all the enthalpy curvesobtained in Ref. [144] for the formation of Ln3+–dien complexes.
Lower stability and less favourable enthalpy changes associatedto the formation of the complexes in DMSO with respect to AN aredue to the stronger solvation of Ln(III) ions in DMSO (higher DNof DMSO, see also Section 3.1). The stronger solvation is evidentlymore effective in destabilizing Ln–amine complexes in DMSO withrespect to AN, despite in the latter solvent the partial associationof lanthanide perchlorates lowers the positive charge of the cationand would operate in the opposite direction.
The higher stability of lanthanide–polyamine complexes inAN with respect to DMSO emphasizes the prevalence of thesolvation–desolvation over the ion–dipole energetics in deter-mining the thermodynamic stability of these complexes innon-aqueous solution.
5.2.3. Heterocyclic N-donor ligandsAs for Ln3+–monoamine complexes, monodentate azines inter-
act with lanthanide ions only in solvents with low or very lowdonating power, where solubilities of many inorganic salts are low.Therefore, more soluble Ln(III) organic salts are often preferredeven if they are undissociated or only partially dissociated in theselected solvents.
5.2.3.1. Pyridines. To date, two publications report thermody-namic studies of Ln3+ with pyridines and generally with azines.Calorimetry has been employed [146] to study the interactionsof lanthanide �-diketonates (tmhd and fod, see Scheme 3) withseveral pyridines at 303.15 K in anhydrous benzene. Adducts ofEu(tmhd)3, Yb(tmhd)3, Eu(fod)3 and Pr(fod)3 with pyridine, 2-picoline, 3-picoline, and 4-picoline and 2,4,6-collidine (Scheme 4)have been found in solution [146]. Reaction of the undissociatedsalts with the neutral bases is enthalpy driven, but also the pos-itive entropy term favours the reaction in almost all cases. Theresults obtained for the 1:1 Ln(tmhd)3:pyridine adducts (log K1 3.14and 3.59, for Yb and Eu, respectively) are comparable with thoseobtained with �-diketone derivatives of other metals [147–149]whereas the fluorinated lanthanide shift reagents (Ln(fod)3, Ln = Eu,
Pr) react much more strongly with bases forming both 1:1 and 1:2adducts.34 This was explained as arising [146] from displacementof an extensive solvation envelope and suggested that the stability34 For example, Eu(fod)3 forms, with pyridine, two adducts with stepwise stabilityconstants: log K1 = 4.0 and log K2 = 2.7, respectively.

P. Di Bernardo et al. / Coordination Chem
oi
lSaewtfKpttLampttct[
5a(tlrt
LGbaaoltsib
distances become larger and the electron donor ability of Nc is
Scheme 4. Pyridines and azines.
f the fluorinated shift reagent adducts has as much to do with theirnteraction with the solvent and with pyridines.
Variable temperature 1H NMR was used [150] to study the equi-ibrium reaction of two trivalent cerocenes [Ce(C5H4R)3] (R = butyl,iMe3) with several azines (L) (Scheme 5) in toluene. 1:1 Lewis basedducts [Ce(C5H4R)3(L)] are formed in enthalpy-driven acid–basequilibria. The formation constants (KML) of the adducts increaseith the Lewis acidity of the metallocene and the Lewis basicity of
he azine. The values of KML are much greater35 for R = SiMe3 thanor R = butyl and a linear correlation is found between the log’s ofML and the hydrogen-bond basicity pKHB scale of the azines. Com-etition reactions of [Ce(C5H4R)3] and [U(C5H4R)3] with L showhat the stability constants of the uranocene adducts are greaterhan those of the cerocenes counterparts and that the selectivity ofin favour of U3+ increases with the � donor character of the met-llocene and is proportional to the � accepting ability of the azineolecule. This reflects the lower degree of covalence of the Ln com-
lexes with respect to those of actinides in agreement with recentheoretical indications which show that U(III) complexes withris[4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedione] have a covalentontribution in the form of back-bonding from filled orbitals cen-ered on uranium into empty �* orbitals on coordinated oxygen151].
.2.3.2. Oligopyridines–oligoazines. Several oligo-piridines and/or -zines complexes of more or less dissociated lanthanide(III) saltse.g. LnCl3, LnI3, Ln (diketonate)3) in different solvents were inves-igated (Scheme 5). Due to the rather inhomogeneous set of: (i)anthanide(III) salts, (ii) ligands and (iii) solvent media, a completeationalization of the whole data is not possible and a synthesis ofhese studies is reported below.
Thermodynamic parameters for the complexation of fourn(tta)3 (tta = thenoyltrifluoroacetonato (Scheme 3) and Ln = La, Nd,d and Lu) with bipy (Scheme 6) have been determined at 298.15 Ky direct titration calorimetry in chloroform in presence of moder-te amounts of water [152]. The neutral metal diketonates, whichre water solvated, coordinate up to two molecules of base with-ut replacement of the counterion. The adducts are stabilized byarge exothermic enthalpy changes due to relatively strain-free fit-ing of bipy with two potential nitrogen donor atoms. The complex
tabilities increase with Z notwithstanding the strongly contrast-ng entropy term, which decreases with Z. The large differencesetween �S1 and �S2 indicate that much of the water release35 About three orders of magnitude.
istry Reviews 256 (2012) 328–351 347
occurs in the formation of the first adduct M(tta)3bipy and/or thatthe second step involves a larger loss of rotational entropy for thebipyridyl molecule.
In anhydrous pyridine36 solution at 294 K, Ce(III) triiodide formsboth 1:1 (ML) and 1:2 (ML2) complexes with bipyridine (bipy = L),while Nd(III) triiodide formed only a 1:2 complex [153]. The 1:3(ML3) complexes were identified at low temperature with a largeexcess of L. In addition it has been observed that for Ce(III) andNd(III), MI2+ and MI3 present about the same affinity for L. Inter-estingly, a preference for the formation of ML2 complex was shownfor all the studied M(III) ions. In both cases, the driving force for thecomplex formation is always the enthalpy, however authors giveno any explanation for the positive values of the stepwise enthalpyand entropy which accompany formation of the second bipy adductof Ce.
Komiya et al. [154] studied the formation of binary complexesof phen (Scheme 6) with lanthanide perchlorates at 298.15 K inanhydrous DMF in which Ln(ClO4)3 are fully dissociated. Titrationcalorimety (Ln = La, Ce, Nd, Eu, Gd, Dy, Tm, Lu) and spectropho-tomety (Ln = Nd) were employed to obtain stability constantsand reaction enthalpies. The �H◦ and �S◦ for the formation ofLn(phen)3+ decrease in the order La > Ce > Nd, then increase in theorder Nd < Eu < Gd < Dy and again decrease in the order Dy > Tm > Lu(see Table 6).
The Ln(phen)3+ complex formed in DMF, is eight coordinate forLn = La, Ce, Nd, and seven coordinate for Ln = Dy, Tm, Lu; an equilib-rium between the seven and eight coordination is established for Euand Gd. The geometry equilibrium shifts to the lower coordinationnumber for the metal ion with a smaller ionic radius. Formation ofternary complexes of Ce3+, Nd3+ and Tm3+ with phen and chloride(LnCl(phen)2+, LnCl2(phen)+, and LnCl3(phen)) was also establishedin the same medium. The metal ion is seven coordinated in theternary TmCl(phen)(DMF)n
2+ complex whereas seven- and eight-coordinated species are in equilibrium for Ce3+ and Nd3+ complexes[154].
Ionova et al. with ab initio calculations and thermodynamicstudies [155], investigated the interaction in MeOH–water of Ln3+
ions with some polypyridines (L7–L13 Scheme 5).Ab initio calculations [155] show that the main factors affecting
the complex stabilities are: (i) the part of covalence of the lateralnitrogens of the ligand (Nl) in Ln–Nl bonds; (ii) the relative sizes ofthe cation and nitrogen cavity of the ligand; (iii) the electrostaticcapacity of the ligand and, (iv) the difference in the effective chargeson the lateral and central nitrogen atoms of the ligand (Nl and Nc,respectively).
The stability of the complexes is characterized by distinct cova-lence of the bonds between Ln3+ and the lateral coordinatingnitrogen atoms of the ligand. The lighter lanthanides (La3+, Ce3+,Pr3+ and Nd3+) do not fit into the rigid structure of the ligands:as a consequence the stability of complexes increases with theincrease of electron acceptor ability of Nl and with the increasingelectron donor ability of Nc. The central ring is negatively chargedin L11, but it has positive charge in L9. The strong donor–acceptorinteraction and the net covalence result in an enthalpy drivencomplexation of Ln3+ with L11, while entropy promotes the for-mation of the [Ln(L9)]3+ complex. Owing to the repulsion betweenthe cation and the central nitrogen atom in [Ln(L9)]3+, the cationis pushed away from the nitrogen cavity, the Ln–Nc interatomic
weak.
36 For Ce(III): log K1 = 0.6 and log ˇ2 = 2.5 with �HK1 = −20 kJ mol−1, �Hˇ2=
−10 kJ mol−1. For Nd(III): log ˇ2 = 2.9 and �Hˇ2= −30 kJ mol−1. Data available also
for complex formation of U(III) triiodide log K1 = 1.0, log ˇ2 = 3.1.
348 P. Di Bernardo et al. / Coordination Chemistry Reviews 256 (2012) 328–351
-heter
rnt2
wothemepotm
−
Scheme 5. Poly
A recent spectrophotometric and microcalorimetric study [156]eports the stability of the complexes formed by several Ln3+
itrates (Ln = La, Nd, Eu, Gd, Er and Yb) with selected tri- oretra-dentate triazine-pyridines (L14–L17 in Scheme 5) in MeOH at98.15 K.
Generally, mononuclear LnLj3+ complexes (j = 1, 2, 3) are formed,
ith different stoichiometries and stability constants dependingn the ligand and the cation. Due to their higher charge density,he heavier lanthanides (Er3+ and Yb3+) form complexes with theighest stability constants with all the ligands. Microcalorimetricxperiments were carried out only for Eu3+–L16 system. The for-ation of the 1:1 (Eu3+:L16) complex is both enthalpically and
ntropically driven (T�SK1 = 20 and �HK1 = −12 kJ mol−1); theositive entropy may reflect an increase of disorder due to the des-
lvation of the metal-ion and the exothermic �H can be relatedo the strength of the cation–ligand interaction. The stepwise for-ation of the 1:2 successive complex is characterized by �HK2 =16 kJ mol−1 which is slightly more favourable than T�SK2 =
Scheme 6. 2,2′-Bipyridyl and phenantroline.
ocyclic ligands.
12 kJ mol−1, which has been interpreted as the metal–ligand inter-action dominating desolvation effects.
The interaction of LaI3 with L18 and L19 (Scheme 5) has beenstudied in pyridine and two successive complexes (LaLI3 and(LaL2I3), L = L18, L19) were found [157]. Solution NMR studies showa clear difference in the behaviour of L18 and L19 towards complex-ation and a greater stability of the bis(ligand) complexes for L18
than for L19. The quotient K2/K1 between the stepwise formationconstants of the 1:2 and 1:1 La:L18 complexes is 35. The changeof anion in the La/L18 system from iodide to triflate increases theratio K2/K1 from 35 to 115 [158]. This observation is consistentwith the fact that the triflates are less strongly coordinating thaniodides and therefore are less competing with binding of the sec-ond ligand molecule. The uncommon preferential formation of thebis complex with respect to the mono complex, not observed inAN, is related to the presence in pyridine of strong �–� interactionbetween benzimidazole rings. The absence of such interaction inthe bis(L19) complex can explain its lower stability with respect tothe bis(L18) complex.
The N-donor ligand L20 in Scheme 5 forms in a MeOH solu-tion of EuCl3 a stable 1:1 complex [159–161]. The stability ofthe complex was determined by UV–Vis spectroscopy in MeOH
(log K = 7.09) and estimated in water (log K ∼ 5.4). In both mediathe average number of solvent molecules coordinated to the metalion is 0.5 [161]. The measure of the circularly polarized lumines-cence (CPL) of a solution of the Dy(L20)3+ complex, demonstrates
Chemistry Reviews 256 (2012) 328–351 349
ictfts
fw[ttsctcba1Ltp[c�Tbpastfarbiras
sbeaa+
mnip(narugt(gnt
A
P. Di Bernardo et al. / Coordination
ts chiral structure. No CPL was observed in the Eu(III) or Tb(III)omplex solutions because of efficient racemization. The varia-ion of the magnitude of the CPL as a function of temperatureor an aqueous solution of the dysprosium(III) complex is usedo characterize the solution equilibria between different chiralpecies[161].
Bünzli et al. studied in AN at 298.15 K [162] the complexesormed by Ln3+ ions with several derivatives of L21 (Scheme 5),hich are interesting for application as luminescent materials
163]. In particular, they determined37 the stability constants ofhe Ln3+ complexes with L22–L25 (Scheme 5). The planar aromaticridentate ligands, L22–L24 react with Ln3+ (Ln = La–Lu) to give theuccessive complexes [Ln(Li)n]3+ (i = 21–23; n = 1–3). The study ofomplex formation between L25 and Eu shows that L25 is not ableo form a 1:3 species. Stability constants of Ln–L22 systems indi-ate that 1:1 and 1:2 complexes display the usual thermodynamicehaviour associated with electrostatic effects (log ˇn smoothlynd uniformly increase with decreasing ionic radii) whereas the:3 complexes exhibit an unusual selectivity for the mid-rangen3+ ions (�log K3(Gd–Lu) = 4). A detailed investigation of the solu-ion structure of [Ln(L22)3]3+ (Ln = La–Dy) reveals that the closelyacked triple-helical structure found in the crystal structure ofEu(L22)3]3+ is retained in AN for the complete series. A sharpontrol of the coordination cavity results from the interstrand-stacking interactions which appear to be optimum for Gd3+.he binding of bulky substituents to the nitrogen atoms of theenzilimidazole side arms of L21 (i) severely affects the wrap-ing process, (ii) leads to less stable triple-helical building blocks,nd (iii) removes the size-discriminating effect. The exceedingtability of the 1:1 and 1:2 complexes, for L23 and L24, respec-ively, only allows calculation of the stepwise constants for theormation of [Ln(L23)2]3+, [Ln(L23)3]3+ and [Ln(L24)3]3+. Both L23
nd L24, analogously to L22, show maximum affinity for the mid-ange lanthanides [162]. The substitution of R2 with a relativelyulky neopentyl group in a benzylimidazol derivative of the fam-
ly L21–L25 [28] determines the loss of the �-stacking interactionsesulting in a destabilization of the 1:3 species in AN solutionnd causes a decrease of the molecular symmetry in the solidtate.
More complex ligands (L26–L29, Scheme 7) able to form triple-tranded homo- or hetero-polymetallic helicates of Ln(III) haveeen studied in AN solution and in the solid state [164–166]. Inter-stingly, cooperative effect in the assembly process is observednd explained by the high degree of pre-organization of the lig-nds which largely compensates the electrostatic repulsion of the3 charged ions [164].
As mentioned before, in this review the analysis of the ther-odynamics of formation of lanthanides with complex ligands is
ot a main aim, nevertheless some interesting innovations presentn these last studies should be highlighted (see Ref. [23] for a com-lete discussion). The polytopic nature of these helicating receptorsL26–L29) produces a variety of possible species, depending on theumber of sites in the ligand and on the Ln(III) ions, which arenalyzed by using statistical thermodynamic models [165]. Theelative abundance of these micro-species in solution is also mod-lated by the absolute binding affinity of each type of site for aiven Ln(III) ion, reflecting the specific free energy of complexa-ion [165,167]. Interactions between a pair of adjacent metal atomselectrostatic repulsion, mechanical coupling) is modelled by a sin-
le free energy term [165,167]. The results obtained indicate theon-equivalence of the central and terminal binding sites, sincehe model which implies identical absolute affinities for the two37 By means of spectrophotometry and potentiometric competitive titrations withg+ in 0.1 mol dm−3 Et4NClO4.
Scheme 7. Complex benzylimidazol derivatives.
types of sites fails in reproducing the experimental stability of thecomplexes formed [165]. The dependence of the absolute affinities(micro-constants) on the ionic radius provides size-discriminatingeffects that favour the formation of heterotrimetallic helicates inwhich the central site is occupied by the larger metal of the pair[165].
An extension of this “site-binding model”, has been then used tostudy the assembly of helical bi- tri- and tetra-nuclear supramolec-ular complexes in AN [23,166] and the formation of mononuclearspecies with the tridentate L7 and L21 (and their derivatives)[168]. This model is based on a statistical factor and five param-eters depending on: the site-specific affinity for the Ln(III) ion, anentropy-related term, one intermetallic and one interligand freeenergy term [166]. The statistical factor is calculated using bino-mial distributions or the symmetry number method [166] andgives good agreement with the experimental stability constants.Even if the symmetry number method implies assumptions on theCN of the Ln(III) (CN = 9 in AN) and on the complete dissociationof the salt, good predictions are obtained, despite a fraction ofLn(trif)n
(3 −n)+ species may be present in the experimental condi-tions [166]. The use of statistical factors adapted to self-assemblyprocesses combined with simple site-binding models is able topredicting stabilities of supramolecular complexes within a family[166]. This approach could be useful to go beyond chemical intu-
ition in the design of polytopic receptors able to selectively bindmultiple Ln(III) cations [23,169].
3 Chem
6
t
i
i
A
bE
R
50 P. Di Bernardo et al. / Coordination
. Conclusions
From the large number of thermodynamic data here examinedhe following main points emerge:
i) Due to the high charge of lanthanide ions, their salts canbe undissociated or only partially dissociated in poor coor-dinating solvents and, in this case, the data obtained reflectmultiple simultaneous equilibria established in solution. Com-parisons between thermodynamic results in poor and highsolvating media must be therefore regarded with caution asthey may refer to different reacting metal-species, hence, to dif-ferent metal–ligand equilibria. A main point emerging from thisreview is that only using several complementary experimen-tal techniques, a detailed comprehension of the real nature ofthe systems in solution can be obtained. Also, structural infor-mation and quantum chemical studies are highly desirable toproperly interpret, on the molecular level, thermodynamic data.As an example, in the absence of accurate FT-IR and conductivitymeasurements, calorimetric and potentiometric results alonecould be wrongly interpreted as due to interaction of n-butwith the solvated Ln(III) ions in AN rather than to the effect ofequilibria involving Ln(trif)3 and [Ln(trif)2]+ simultaneously. Inaddition, in the same solvent the thermodynamic data for Ln(III)complex formation with (2.2.2) cryptand drastically changeat low concentrations, where conductometric measurementsindicated the total dissociation of Ln(trif)3 salts, with respect todata referred to higher concentration range. Accordingly, greatattention must be paid to the range of Ln(III)-salts concentra-tions used in thermodynamic studies, to ensure that only thesolvated tervalent cation is the dominant species: in the absenceof a detailed knowledge of the nature of the metal receptor, thethermodynamic data should be considered as “apparent”.
ii) Lanthanide salts are dissociated in strong coordinating solvents(DMSO, DMF). Thus, these media are ideal for thermodynamicstudies since the lanthanide is always in the form of Ln(solv)n
3+.On the other hand, complexes with relatively poor donors,found in less solvating media (such as AN or PC) are often tooweak to be observed in DMSO or DMF.
ii) A convenient, selective, ligand for Ln(III) ions must have a largenumber of donor atoms in order to meet, in the absence of defi-nite steric requirements, their large coordination numbers. Inaddition, as clearly indicated by the thermodynamic data ofseveral crown ethers complexes, pre-organization of the coordi-nation sites and presence of additional donor arms in the ligandcan play an important role in achieving selectivity. For example,the introduction of the side arm in the lariat ether R′A16C5 isessential for acquiring a rather selective complexation of Nd(III)salts with respect to the other lanthanides(III).
v) Solvation of lanthanide(III) cations in non-aqueous solutions isgenerally a major factor in determining the complex stabili-ties which, for the different kinds of selected ligands, followthe trend: PC > AN > MeOH > DMF > DMSO.
cknowledgments
Financial support by University of Padova (project PRAT num-er CPDA08 5007/08) and “Regione Friuli Venezia Giulia” (projectCOMETA 2009) is acknowledged by the authors.
eferences
[1] J.C.G. Bünzli, C. Piguet, Chem. Soc. Rev. 34 (2005) 1048.[2] I. Hemmila, V. Laitala, J. Fluoresc. 15 (2005) 529.[3] E.J. New, D. Parker, D.G. Smith, J.W. Walton, Curr. Opin. Chem. Biol. 14 (2010)
238.[4] P. Caravan, Acc. Chem. Res. 42 (2009) 851.
istry Reviews 256 (2012) 328–351
[5] G. Otting, J. Biomol. NMR 42 (2008) 1.[6] M. Shibasaki, N. Yoshikawa, Chem. Rev. 102 (2002) 2187.[7] V.S. Sastri, J.C.G. Bünzli, V. Ramachandra Rao, G.V.S. Rayudu, J.R. Perumareddi,
Modern Aspects of Rare Earths and their Complexes, Elsevier B.V, Amsterdam,The Netherlands, 2003.
[8] Y. Umebayashi, K. Matsumoto, I. Mekata, S. Ishiguro, Phys. Chem. Chem. Phys.4 (2002) 5599.
[9] S. Ishiguro, Bull. Chem. Soc. Jpn. 70 (1997) 1465.[10] E.N. Rizkalla, G.R. Choppin, J. Alloys Compd. 180 (1992) 325.[11] Z. Kolarik, Chem. Rev. 108 (2008) 4208.[12] H.H. Dam, D.N. Reinhoudt, W. Verboom, Chem. Soc. Rev. 36 (2007) 367.[13] G.R. Choppin, D.R. Peterman, Coord. Chem. Rev. 174 (1998) 283.[14] A. Bianchi, L. Calabi, F. Corana, S. Fontana, P. Losi, A. Maiocchi, L. Paleari, B.
Valtancoli, Coord. Chem. Rev. 204 (2000) 309.[15] F. Arnaud-Neu, Chem. Soc. Rev. 23 (1994) 235.[16] Y. Liu, B.H. Han, Y.T. Chen, Coord. Chem. Rev. 200 (2000) 53.[17] R. Kumar, U.P. Singh, J. Rare Earth 23 (2005) 389.[18] C. Kremer, J. Torres, S. Dominguez, A. Mederos, Coord. Chem. Rev. 249 (2005)
567.[19] S. Ishiguro, Y. Umebayashi, M. Komiya, Coord. Chem. Rev. 226 (2002) 103.[20] R.M. Izatt, K. Pawlak, J.S. Bradshaw, R.L. Bruening, Chem. Rev. 95 (1995)
2529.[21] R.M. Izatt, K. Pawlak, J.S. Bradshaw, R.L. Bruening, Chem. Rev. 91 (1991) 1721.[22] R.M. Izatt, J.S. Bradshaw, S.A. Nielsen, J.D. Lamb, J.J. Christensen, D. Sen, Chem.
Rev. 85 (1985) 271.[23] C. Piguet, J.C.G. Bünzli, in: A.G. Karl (Ed.), Handbook on the Physics and Chem-
istry of Rare Earths, Elsevier, 2010 (Chapter 247).[24] J.C.G. Bünzli, in: A.G. Karl (Ed.), Handbook on the Physics and Chemistry of
Rare Earths, Elsevier, 1987 (Chapter 60).[25] J.C.G. Bünzli, A. Milicic-Tang, A.G. Karl (Eds.), Handbook on the Physics and
Chemistry of Rare Earths, Elsevier, 1995 (Chapter 145).[26] A.F.D. de Namor, K. Baron, S. Chahine, O. Jafou, J. Phys. Chem. A 108 (2004)
1082.[27] A.F.D. de Namor, O. Jafou, J. Phys. Chem. B 105 (2001) 8018.[28] G. Muller, J.C.G. Bünzli, K.J. Schenk, C. Piguet, G. Hopfgartner, Inorg. Chem. 40
(2001) 2642.[29] W.J. Evans, Coord. Chem. Rev. 206–207 (2000) 263.[30] L.R. Morss, Chem. Rev. 76 (1976) 827.[31] D.A. Johnson, J. Chem. Soc. Dalton Trans. (1974) 1671.[32] P.G. Eller, R.A. Penneman, J. Less-Common Met. 127 (1987) 19.[33] R.D. Shannon, Acta Crystallogr. A 32 (1976) 751.[34] D. Lundberg, I. Persson, L. Eriksson, P. D’Angelo, S. De Panfilis, Inorg. Chem. 49
(2010) 4420.[35] C. Clavaguera, J.P. Dognon, P. Pyykkö, Chem. Phys. Lett. 429 (2006) 8.[36] P. Pyykko, Chem. Rev. 88 (1988) 563.[37] A. Chatterjee, E.N. Maslen, K.J. Watson, Acta Crystallogr. B 44 (1988) 381.[38] D.A. Vander Griend, S. Malo, T.K. Wang, K.R. Poeppelmeier, J. Am. Chem. Soc.
122 (2000) 7308.[39] S. Siekierski, Inorg. Chim. Acta 109 (1985) 199.[40] E.A. Quadrelli, Inorg. Chem. 41 (2002) 167.[41] M. Seitz, A.G. Oliver, K.N. Raymond, J. Am. Chem. Soc. 129 (2007) 11153.[42] A.E. Clark, J. Chem. Theory Comput. 4 (2008) 708.[43] J.C. Slater, Phys. Rev. 36 (1930) 57.[44] E. Clementi, D.L. Raimondi, W.P. Reinhardt, J. Chem. Phys. 47 (1967) 1300.[45] E. Clementi, D.L. Raimondi, J. Chem. Phys. 38 (1963) 2686.[46] K.N. Raymond, D.L. Wellman, C. Sgarlata, A.P. Hill, C. R. Chim. 13 (2010) 849.[47] P. D’Angelo, A. Zitolo, V. Migliorati, G. Chillemi, M. Duvail, P. Vitorge, S. Abadie,
R. Spezia, Inorg. Chem. 50 (2011) 4572.[48] Y. Marcus, Ion Solvation, Wiley, New York, 1985.[49] S. Ahrland, Pure Appl. Chem. 62 (1990) 2077.[50] G. Gritzner, Electrochim. Acta 44 (1998) 73.[51] P. Smirnov, L.H. Weng, I. Persson, Phys. Chem. Chem. Phys. 3 (2001) 5248.[52] M.E. Clark, J.L. Bear, J. Inorg, Nucl. chem. 31 (1969) 2619.[53] M.J. Blandamer, J. Burgess, J. Kijowski, Inorg. Chim. Acta 58 (1982) 155.[54] J. Burgess, J. Kijowski, J. Inorg, Nucl. Chem. 43 (1981) 2389.[55] E.H.P. Cordfunke, R.J.M. Konings, Thermochim. Acta 375 (2001) 51.[56] Y. Marcus, The Properties of Solvents, John Wiley and Sons, Chichester, 1998.[57] H. Miyamoto, H. Iijima, M. Sugawara, Bull. Chem. Soc. Jpn. 59 (1986) 2973.[58] T. Kimura, R. Nagaishi, Y. Kato, Z. Yoshida, J. Alloys Compd. 323 (2001) 164.[59] J.C.G. Bünzli, M.M. Vuckovic, Inorg. Chim. Acta 73 (1983) 53.[60] I. Persson, P. D’Angelo, S. De Panfilis, M. Sandström, L. Eriksson, Chem. Eur. J.
14 (2008) 3056.[61] P. Di Bernardo, P.L. Zanonato, A. Melchior, R. Portanova, M. Tolazzi, G.R. Chop-
pin, Z. Wang, Inorg. Chem. 47 (2008) 1155.[62] I. Persson, E.D. Risberg, P. D’Angelo, S. De Panfilis, M. Sandström, A. Abbasi,
Inorg. Chem. 46 (2007) 7742.[63] A. Abbasi, E.D. Risberg, L. Eriksson, J. Mink, I. Persson, M. Sandström, Y.V.
Sidorov, M.Y. Skripkin, A.S. Ullstrom, Inorg. Chem. 46 (2007) 7731.[64] Q.X. Han, D.L. Zhang, J.R. Ma, J.P. Wang, J.Y. Niu, Chin. J. Struct. Chem. 26 (2007)
949.[65] S.S. Yarovoi, Y.V. Mironov, S.F. Solodovnikov, Z.A. Solodovnikova, D.Y. Nau-
mov, V.E. Fedorov, Russ. J. Coord. Chem. 32 (2006) 712.[66] S.M. Liu, C.E. Plecnik, E.A. Meyers, S.G. Shore, Inorg. Chem. 44 (2005) 282.[67] J.C. Berthet, P. Thuery, M. Ephritikhine, Polyhedron 25 (2006) 1700.[68] S. Ishiguro, Y. Umebayashi, K. Kato, R. Takahashi, K. Ozutsumi, J. Chem. Soc.
Faraday Trans. 94 (1998) 3607.

Chem
[
[
[[[[[
[[[[[
[[[
[
[[
[
[[
[[
P. Di Bernardo et al. / Coordination
[69] M.S. Grigor’ev, I.B. Shirokova, A.M. Fedoseev, C. Den Auwer, Crystallogr. Rep.48 (2003) 638.
[70] G.R. Willey, D.R. Aris, W. Errington, Inorg. Chim. Acta 318 (2001) 97.[71] G.B. Deacon, B. Gortler, P.C. Junk, E. Lork, R. Mews, J. Petersen, B. Zemva, J.
Chem. Soc. Dalton Trans. (1998) 3887.[72] M. Baaden, F. Berny, C. Madic, G. Wipff, J. Phys. Chem. A 104 (2000) 7659.[73] I. Persson, Pure Appl. Chem. 58 (1986) 1153.[74] S. Ishiguro, K. Kato, S. Nakasone, R. Takahashi, K. Ozutsumi, J. Chem. Soc.
Faraday Trans. 92 (1996) 1869.[75] R. Takahashi, S. Ishiguro, J. Chem. Soc. Faraday Trans. 88 (1992) 3165.[76] S. Ishiguro, R. Takahashi, Inorg. Chem. 30 (1991) 1854.[77] R. Takahashi, S. Ishiguro, J. Chem. Soc. Faraday Trans. 87 (1991) 3379.[78] Y. Hasegawa, K. Takashima, F. Watanabe, Bull. Chem. Soc. Jpn. 70 (1997) 1047.[79] C. Comuzzi, P. Di Bernardo, P. Polese, R. Portanova, M. Tolazzi, P.L. Zanonato,
Polyhedron 19 (2000) 2427.[80] J.C.G. Bünzli, C. Mabillard, J. Less-Common Met. 94 (1983) 317.[81] P.L.O. Volpe, A.P. Chagas, C. Airoldi, J. Inorg, Nucl. chem. 42 (1980) 1321.[82] C. Airoldi, P.L.O. Volpe, A.P. Chagas, Polyhedron 1 (1982) 49.[83] A.I. Krutous, I.M. Batyaev, Zh. Neorg. Khim. 19 (1974) 1234.[84] H.B. Silber, R. Bakhshandehfar, L.A. Contreras, F. Gaizer, M. Gonsalves, S. Ismail,
Inorg. Chem. 29 (1990) 4473.[85] H.B. Silber, M.S. Strozier, Inorg. Chim. Acta 128 (1987) 267.[86] J.C.G. Bünzli, A. Milicictang, C. Mabillard, Helv. Chim. Acta 76 (1993) 1292.[87] J.C.G. Bünzli, C. Mabillard, J.R. Yersin, Inorg. Chem. 21 (1982) 4214.[88] J.C.G. Bünzli, J.P. Metabanzoulou, P. Froidevaux, L.P. Jin, Inorg. Chem. 29 (1990)
3875.[89] J.C.G. Bünzli, J.R. Yersin, Helv. Chim. Acta 65 (1982) 2498.[90] J.C.G. Bünzli, M.M. Vuckovic, Inorg. Chim. Acta 95 (1984) 105.[91] J.C.G. Bünzli, C. Mabillard, Inorg. Chem. 25 (1986) 2750.[92] A. Milicictang, J.C.G. Bünzli, Inorg. Chim. Acta 192 (1992) 201.[93] J.C.G. Bünzli, V. Kasparek, Inorg. Chim. Acta 182 (1991) 101.[94] J.C.G. Bünzli, A.E. Merbach, R.M. Nielson, Inorg. Chim. Acta 139 (1987) 151.[95] P. Di Bernardo, G.R. Choppin, R. Portanova, P.L. Zanonato, Inorg. Chim. Acta
207 (1993) 85.[96] A. Seminara, E. Rizzarelli, Inorg. Chim. Acta 40 (1980) 249.[97] A.F.D. de Namor, S. Chahine, O. Jafou, K. Baron, J. Coord. Chem. 56 (2003) 1245.[98] F. Pilloud, J.C.G. Bünzli, Inorg. Chim. Acta 139 (1987) 153.[99] R. Pizer, R. Selzer, Inorg. Chem. 22 (1983) 1359.100] A. Cassol, G.R. Choppin, P. Di Bernardo, R. Portanova, M. Tolazzi, G. Tomat, P.L.
Zanonato, J. Chem. Soc. Dalton Trans. (1993) 1695.101] J. Zolgharnein, F. Riahii, S. Amani, J. Inclusion Phenom. Macrocycl. Chem. 45
(2003) 13.102] J. Massaux, J.F. Desreux, J. Am. Chem. Soc. 104 (1982) 2967.103] J.C.G. Bünzli, J.R. Yersin, C. Mabillard, Inorg. Chem. 21 (1982) 1471.104] Y. Liu, T. Lu, M. Tan, T. Hakushi, Y. Inoue, J. Phys. Chem. 97 (1993) 4548.105] D.Q. Liu, B.G. Jiang, J.Z. Yin, H.K. Yan, Acta Chim. Sin. 48 (1990) 452.106] Y. Liu, T.B. Lu, M.Y. Tan, I. Yoshihisa, H. Tadao, Acta Phys. Chim. Sin. 10 (1994)
336.107] Y. Liu, A.D. Qi, R.T. Chen, Y.M. Zhang, Acta Chim. Sin. 55 (1997) 1091.108] Y. Liu, T.B. Lu, M.Y. Tan, Y. Inoue, T. Hakushi, Acta Chim. Sin. 51 (1993) 874.109] A.E. Martell, R.D. Hancock, R.J. Motekaitis, Coord. Chem. Rev. 133 (1994) 39.110] J.C.G. Bünzli, F. Pilloud, Inorg. Chem. 28 (1989) 2638.111] J.C.G. Bünzli, J.M. Pfefferle, B. Ammann, G. Chapuis, F.J. Zuniga, Helv. Chim.
Acta 67 (1984) 1121.112] J.-G. Mao, Z.-S. Jin, F.-L. Yu, Chin. J. Struct. Chem. (1994) 276.113] R.D. Rogers, A.N. Rollins, J. Cryst. Spectrosc. 20 (1990) 389.114] J.D.J. Backerdirks, J.E. Cooke, A.M.R. Galas, J.S. Ghotra, C.J. Gray, F.A. Hart, M.B.
Hursthouse, J. Chem. Soc. Dalton Trans. (1980) 2191.115] Y. Liu, B.H. Han, Y.M. Li, R.T. Chen, M. Ouchi, Y. Inoue, J. Phys. Chem. 100 (1996)
17361.116] Y. Liu, X.P. Bai, Y. Inoue, M. Ouchi, J. Phys. Chem. B 102 (1998) 4871.117] R.M. Izatt, J.D. Lamb, J.J. Christensen, B.L. Haymore, J. Am. Chem. Soc. 99 (1977)
8344.118] K. Taheri, M. Chamsaz, G.H. Rounaghi, M.A. Fard, J. Inclusion Phenom. Macro-
cycl. Chem. 63 (2009) 43.119] M.S. Kazemi, J. Inclusion Phenom. Macrocycl. Chem. 68 (2010) 331.
120] Y. Liu, B.H. Han, Z.H. Zhang, J.H. Guo, Y.T. Chen, Thermochim. Acta 317 (1998)1.121] X.R. Huang, B.G. Jiang, J.Z. Yin, H.K. Yan, Acta Chim. Sin. 49 (1991) 359.122] Y. Liu, S.P. Dong, H. Zheng, Y. Inoue, M. Ouchi, Supramol. Chem. 11 (2000)
239.
istry Reviews 256 (2012) 328–351 351
[123] A. Cassol, P. Di Bernardo, G. Pilloni, M. Tolazzi, P.L. Zanonato, J. Chem. Soc.Dalton Trans. (1995) 2689.
[124] M.C. Almasio, F. Arnaud-Neu, M.J. Schwingweill, Helv. Chim. Acta 66 (1983)1296.
[125] T. Lu, X. Gan, M. Tan, K. Yu, Polyhedron 12 (1993) 2193.[126] Y. Shiping, J. Zonghui, L. Diazheng, W. Genglin, W. Ruji, W. Honggen, Y. Xinkan,
J. Inclusion Phenom. Macrocycl. Chem. 15 (1993) 159.[127] Y. Shiping, W. Genglin, W. Honggen, Y. Xinkan, Polyhedron 10 (1991) 1889.[128] J. Massaux, J.F. Desreux, C. Delchambre, G. Duyckaerts, Inorg. Chem. 19 (1980)
1893.[129] F. Arnaud-Neu, E.L. Loufouilou, M.J. Schwingweill, J. Chem. Soc. Dalton Trans.
(1986) 2629.[130] M. Kodama, T. Koike, A.B. Mahatma, E. Kimura, Inorg. Chem. 30 (1991) 1270.[131] G.L. Rothermel, L. Miao, A.L. Hill, S.C. Jackels, Inorg. Chem. 31 (1992) 4854.[132] A.F.D. Denamor, M.C. Ritt, M.J. Schwingweill, F. Arnaud-Neu, J. Chem. Soc.
Faraday Trans. 86 (1990) 89.[133] I. Marolleau, J.P. Gisselbrecht, M. Gross, F. Arnaud-Neu, M.J. Schwingweill, J.
Chem. Soc. Dalton Trans. (1990) 1285.[134] I. Marolleau, J.P. Gisselbrecht, M. Gross, F. Arnaud-Neu, M.J. Schwingweill, J.
Chem. Soc. Dalton Trans. (1989) 367.[135] A. Lewandowski, J. Chem. Soc. Faraday Trans. 1 (85) (1989) 4139.[136] J.G. Mao, Z.S. Jin, Polyhedron 13 (1994) 319.[137] E.L. Loufouilou, J.P. Gisselbrecht, Can. J. Chem. 66 (1988) 2172.[138] A. Cassol, P. Di Bernardo, R. Portanova, M. Tolazzi, P.L. Zanonato, Inorg. Chim.
Acta 262 (1997) 1.[139] Z.M. Wang, G.R. Choppin, P. Di Bernardo, P.L. Zanonato, R. Portanova, M.
Tolazzi, J. Chem. Soc. Dalton Trans. (1993) 2791.[140] A. Cassol, P. Di Bernardo, R. Portanova, M. Tolazzi, G. Tomat, P. Zanonato, J.
Chem. Soc. Dalton Trans. (1992) 469.[141] C. Comuzzi, P. Di Bernardo, R. Portanova, M. Tolazzi, P. Zanonato, Polyhedron
21 (2002) 1385.[142] J.H. Forsberg, T. Moeller, Inorg. Chem. 8 (1969) 889.[143] J.H. Forsberg, Coord. Chem. Rev. 10 (1973) 195.[144] J.H. Forsberg, C.A. Wathen, Inorg. Chem. 10 (1971) 1379.[145] J.H. Forsberg, T.M. Kubik, T. Moeller, K. Gucwa, Inorg. Chem. 10 (1971) 2656.[146] D.P. Graddon, L. Muir, J. Less-Common Met. (1981) 2434.[147] D.P. Graddon, T.T. Nyein, Aust. J. Chem. 27 (1974) 407.[148] D.R. Dakterni, D.P. Graddon, Aust. J. Chem. 26 (1973) 2537.[149] D.P. Graddon, K.B. Heng, Aust. J. Chem. 24 (1971) 1781.[150] T. Mehdoui, J.C. Berthet, P. Thuery, M. Ephritikhine, Dalton Trans. (2004) 579.[151] V. Vallet, A. Fischer, Z. Szabo, I. Grenthe, Dalton Trans. 39 (2010) 7666.[152] A.S. Kertes, E.F. Kassierer, Inorg. Chem. 11 (1972) 2108.[153] C. Riviere, M. Nierlich, M. Ephritikhine, C. Madic, Inorg. Chem. 40 (2001) 4428.[154] M. Komiya, Y. Nishikido, Y. Umebayashi, S. Ishiguro, J. Solution Chem. 31
(2002) 931.[155] G. Ionova, C. Rabbe, R. Guillaumont, S. Ionov, C. Madic, J.C. Krupa, D. Guilla-
neux, New J. Chem. 26 (2002) 234.[156] V. Hubscher-Bruder, J. Haddaoui, S. Bouhroum, F. Arnaud-Neu, Inorg. Chem.
49 (2010) 1363.[157] R. Wietzke, M. Mazzanti, J.M. Latour, J. Pecaut, J. Chem. Soc. Dalton Trans.
(2000) 4167.[158] R. Wietzke, M. Mazzanti, J.M. Latour, J. Pecaut, Chem Commun. (1999) 209.[159] P. Gawryszewska, J. Sokolnicki, A. Døssing, J.P. Riehl, G. Muller, J. Leg-
endziewicz, J. Phys. Chem. A 109 (2005) 3858.[160] A. Døssing, S.M. Kristensen, H. Toftlund, J.A. Wolny, Acta Chim. Scand. 53
(1999) 575.[161] A. Døssing, H. Toftlund, A. Hazell, J. Bourassa, P.C. Ford, J. Chem. Soc. Dalton
Trans. (1997) 335.[162] S. Petoud, J.C.G. Bünzli, F. Renaud, C. Piguet, K.J. Schenk, G. Hopfgartner, Inorg.
Chem. 36 (1997) 5750.[163] S.V. Eliseeva, J.C.G. Bünzli, Chem. Soc. Rev. 39 (2010) 189.[164] S. Floquet, N. Ouali, B. Bocquet, G. Bernardinelli, D. Imbert, J.C.G. Bünzli, G.
Hopfgartner, C. Piguet, Chem. Eur. J. 9 (2003) 1860.[165] S. Floquet, M. Borkovec, G. Bernardinelli, A. Pinto, L.A. Leuthold, G. Hopfgart-
ner, D. Imbert, J.C.G. Bünzli, C. Piguet, Chem. Eur. J. 10 (2004) 1091.[166] N. Dalla-Favera, J. Hamacek, M. Borkovec, D. Jeannerat, F. Gumy, J.C.G. Bünzli,
G. Ercolani, C. Piguet, Chem. Eur. J. 14 (2008) 2994.[167] G. Koper, M. Borkovec, J. Phys. Chem. B 105 (2001) 6666.[168] A. Escande, L. Guenee, K.L. Buchwalder, C. Piguet, Inorg. Chem. 48 (2009) 1132.[169] J.F. Lemonnier, L. Guénée, G. Bernardinelli, J.F. Vigier, B. Bocquet, C. Piguet,
Inorg. Chem. 49 (2010) 1252.
Related Documents











