The use of the generator coordinate method for designing basis set. Application to oxo-diperoxo molybdenum complexes Fabrı ´cio R. Sensato a, * , Roge ´rio Custodio a , Quezia B. Cass b , Elson Longo b , Marcelo Z. Hernandes c , Ricardo L. Longo c , Juan Andre ´s d a Instituto de Quı ´mica, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas, SP, Brazil b Departamento de Quı ´mica, Universidade Federal de Sa ˜o Carlos, C.P. 676, 13565-905 Sao Carlos, SP, Brazil c Departamento de Quı ´mica Fundamental, Universidade Federal de Pernambuco, 50540-760 Recife, PE, Brazil d Departament de Cie `ncies Experimentals, Universitat Jaume I, Apartat 224, 12080 Castello, Spain Received 10 December 2001; accepted 22 April 2002 Abstract The molecular and electronic structures of MoO(O 2 ) 2 (1), MoO(O 2 ) 2 (OPy) (2) and MoO(O 2 ) 2 (OPy)(H 2 O) (3) complexes were investigated at the Hartree – Fock and density functional method (B3LYP) calculation levels. The generator coordinate method (GCM) has been used to design basis sets that properly represent the electronic density on the Mo and O atoms for all electron calculations, while a variant of the GCM method has been employed to design a valence basis set for the Mo atom for the pseudopotential calculations. Compound 1 adopts a distorted tetragonal pyramid structure, where the four peroxo oxygen atoms are located in the same plane, which is perpendicular to the axis defined by the Mo and oxo oxygen atoms. An analysis based upon the geometrical and electronic parameters and the vibrational frequencies renders that 1 can be described as two peroxide fragments bonded to the MoO moiety. 2 and 3 complexes are bipyramidal pentagonal structures, with the OPy ligand occupying a quasi-equatorial position in the same plane as the two peroxo triangles while the H 2 O ligand is situated trans to the oxo group in 3. A comparison between theoretical and experimental results for the geometry and vibrational frequencies of 3 complex shows good agreement. The relationship between the reactivity of 1, 2 and 3 complexes and their coordination number has been established by analyzing the values of the vibrational frequencies, frontier molecular orbitals and the values of electron affinities. q 2002 Elsevier Science B.V. All rights reserved. Keywords: Generator coordinate method; Basis sets; Oxo-diperoxo molybdenum complexes; Oxygen transfer reaction; Electronic structure calculations 1. Introduction Oxo-diperoxo complexes of transition metals are an important class of compounds used in the oxidation of a variety of organic substrates [1–4]. In the 1960s, oxo-diperoxo transition metal compounds with general formula MO(O 2 ) 2 L 1 L 2 (M ¼ Mo or W and donor ligands L 1 , L 2 ¼ pyridine, dimethylformamide or hexamethylpho- sphoric triamide), were synthesized by Mimoun [1, 2,4]. Since then, several oxo-diperoxo-molyb- denum complexes, known as Mimoun complexes, have been prepared [5–9] and several reviews on this topic have been published by Jorgensen [10], Dickman and Pope [11] and Morris [12]. 0166-1280/02/$ - see front matter q 2002 Elsevier Science B.V. All rights reserved. PII: S0166-1280(02)00202-6 Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 www.elsevier.com/locate/theochem * Corresponding author. E-mail address: [email protected] (F.R. Sensato).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The use of the generator coordinate method for designing basis set.
Application to oxo-diperoxo molybdenum complexes
Fabrıcio R. Sensatoa,*, Rogerio Custodioa, Quezia B. Cassb, Elson Longob,Marcelo Z. Hernandesc, Ricardo L. Longoc, Juan Andresd
aInstituto de Quımica, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas, SP, BrazilbDepartamento de Quımica, Universidade Federal de Sao Carlos, C.P. 676, 13565-905 Sao Carlos, SP, BrazilcDepartamento de Quımica Fundamental, Universidade Federal de Pernambuco, 50540-760 Recife, PE, Brazil
dDepartament de Ciencies Experimentals, Universitat Jaume I, Apartat 224, 12080 Castello, Spain
Received 10 December 2001; accepted 22 April 2002
Abstract
The molecular and electronic structures of MoO(O2)2 (1), MoO(O2)2(OPy) (2) and MoO(O2)2(OPy)(H2O) (3) complexes
were investigated at the Hartree–Fock and density functional method (B3LYP) calculation levels. The generator coordinate
method (GCM) has been used to design basis sets that properly represent the electronic density on the Mo and O atoms for all
electron calculations, while a variant of the GCM method has been employed to design a valence basis set for the Mo atom for
the pseudopotential calculations. Compound 1 adopts a distorted tetragonal pyramid structure, where the four peroxo oxygen
atoms are located in the same plane, which is perpendicular to the axis defined by the Mo and oxo oxygen atoms. An analysis
based upon the geometrical and electronic parameters and the vibrational frequencies renders that 1 can be described as two
peroxide fragments bonded to the MoO moiety. 2 and 3 complexes are bipyramidal pentagonal structures, with the OPy ligand
occupying a quasi-equatorial position in the same plane as the two peroxo triangles while the H2O ligand is situated trans to the
oxo group in 3. A comparison between theoretical and experimental results for the geometry and vibrational frequencies of 3
complex shows good agreement. The relationship between the reactivity of 1, 2 and 3 complexes and their coordination number
has been established by analyzing the values of the vibrational frequencies, frontier molecular orbitals and the values of electron
affinities. q 2002 Elsevier Science B.V. All rights reserved.
Keywords: Generator coordinate method; Basis sets; Oxo-diperoxo molybdenum complexes; Oxygen transfer reaction; Electronic structure
calculations
1. Introduction
Oxo-diperoxo complexes of transition metals
are an important class of compounds used in the
oxidation of a variety of organic substrates [1–4].
In the 1960s, oxo-diperoxo transition metal
compounds with general formula MO(O2)2L1L2
(M ¼ Mo or W and donor ligands L1, L2 ¼
pyridine, dimethylformamide or hexamethylpho-
sphoric triamide), were synthesized by Mimoun [1,
2,4]. Since then, several oxo-diperoxo-molyb-
denum complexes, known as Mimoun complexes,
have been prepared [5–9] and several reviews on
this topic have been published by Jorgensen [10],
Dickman and Pope [11] and Morris [12].
0166-1280/02/$ - see front matter q 2002 Elsevier Science B.V. All rights reserved.
PII: S0 16 6 -1 28 0 (0 2) 00 2 02 -6
Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264
www.elsevier.com/locate/theochem
* Corresponding author.
E-mail address: [email protected] (F.R. Sensato).

The complex MoO(O2)2(OPy), was first syn-
thesized and characterized via infrared spectroscopy
by Mimoun et al. [1]. The relative stability and
chemical reactivity of several diperoxo complexes
MO(O2)2L1L2 (M ¼ Mo and W, L1 ¼ amine oxide,
tertiary phosphine oxide, or tertiary arsine oxide and
L2 ¼ L1 or H2O) have been established by Westland
et al. [13]. The pyridine oxide complexes (L1 ¼ OPy)
were found to be the most unstable, exhibiting the
lowest activation energy for the loss of an oxygen
molecule. However, as far as we know, there is no
study concerning the molecular structure and the
reactivity of MoO(O2)2 (1), MoO(O2)2(OPy) (2) and
MoO(O2)2(OPy)(H2O) (3) (Fig. 1). Only recently, 3
compound has been synthesized and structurally
characterized by our research group [14].
Mononuclear molybdenum complexes containing
dioxygen ligands have been the subject of several
theoretical studies [15–24]. The catalytic properties
of oxo-bisperoxo-molybdenum complexes are depen-
dent on both number and type of ligand attached to the
Mo ion. Therefore, the knowledge of the electronic
and molecular structures of these complexes can
provide an understanding and rationalization of their
catalytic behavior.
The main concern of the present work is to
determine the electronic and molecular structures of
1, 2 and 3 complexes, using Hartree–Fock (HF) and
density functional (DFT; B3LYP) methods. The goal
is therefore to establish a relationship between their
electronic and molecular structures with their reactiv-
ity and catalytic activity.
The layout of this paper is presented as it follows.
Section 2 is devoted to the description of the
computational procedure together with a detailed
formulation of the generator coordinate method
(GCM). This method was employed to obtain accurate
all-electron basis sets in order to represent adequately
the Mo and O atoms and a valence basis set for Mo
atom adapted to be used with the effective core
potentials (ECPs). In Section 3, the results are
reported and discussed. Section 4 closes this paper.
2. Computational procedure and generator
coordinate method
All calculations were carried out with theFig. 1. Optimized molecular structure of (A) MoO(O2)2 (1); (B)
MoO(O2)2(OPy) (2) and (C) MoO(O2)2(OPy)(H2O) (3) complexes.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264252

GAUSSIAN 98 program [25] without any symmetry
constraint. The HF and DFT with the B3LYP
functional were then selected [26–29]. The optimized
structure have been identified as energy minima by
calculation of the eigenvalues of the hessian matrices
associated with the corresponding stationary point in
the potential surface [30]. Supplementary vibrational
and spectral analyses were carried out with the
GAUSSVIEW 2.0 program [31] and the ORTEP-3
program [32] was used to visualize and to draw the
molecular structures.
Uncontracted basis sets for representing Mo
(17s11p8d) [33] and O (9s4p) [34] have been used,
while the standard 6-31G(d,p) basis sets have been
employed for the C, N and H atoms [35]. The
adequacy and necessary corrections for the basis
functions in the valence region of the Mo and O
atoms were carried out by the application of the
Fig. 2. Weight functions for the p (solid line) and d (dotted line) atomic orbitals of molybdenum obtained from all-electron calculations at the
ROHF level using (a) the preprocessed Huzinaga’s original (18s11p8d) basis set and (b) the (18s12p9d) improved basis set. Both calculations
have been performed at the equilibrium geometry of the [MoO(O2)2(OPy)(H2O)]2 species.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 253

GCM [36,37], which has been used to design basis
sets for atoms and molecules (see Refs. [38–46] and
references therein). The basic assumption of this
method is to describe one-electron function as an
integral transform:
cið1Þ ¼ð1
0fið1;aÞfiðaÞda; with i ¼ 1;…; n ð1Þ
where fi and fi are the generator and weight functions,
respectively, and a is the generator coordinate. The
mathematical and computational foundations of this
method are described elsewhere [47–50]. The most
significant aspect is that the use of numerical
techniques recovers the Hartree–Fock–Roothaan
equations and allows the use of conventional ab initio
molecular orbital programs. Therefore, the linear
combination of the uncontracted basis function
coefficients can be associated with discretized weight
functions. In practice, the analysis of the adequacy of
a basis function is considerably simple and requires
only a careful observation of the approximated weight
functions through the behavior of the LCAO coeffi-
cients at the SCF level with an uncontracted basis set.
A convenient way to visualize the tendency of the
approximated weight functions is to define a logar-
ithmic set of exponents, ViðkÞ ¼ lnðaiÞ; and create
plots of the LCAO coefficients with respect to the
corresponding Vi. In the relabeled space the largest V
represent the innermost primitives and negative V the
primitives in the valence region.
Fig. 2 shows, as an example, the p and d
approximated weight functions for Mo in the outer
molecular orbitals of the [MoO(O2)2(OPy)(H2O)]2
(32) anionic complex obtained with a (18s11p8d)
uncontracted geometric basis set for Mo which has
previously been preprocessed into a geometric basis
set represented by
aiðkÞ ¼ exp½V0ðkÞ þ ði 2 1ÞDVðkÞ�;
with i ¼ 1;…; nk
ð2Þ
where nk is the number of primitives in the mesh
of symmetry k, V0 and DV are the initial exponent
and a constant distance between the exponents in
the logarithmic space, respectively. The anionic
species was chosen to provide elements that
characterized the adequacy of the basis functions
because we are particularly interested in calculating
electron affinities, which are evaluated in the
present study as a difference between the total
energies of the anion radical and the neutral
compound. The need for diffuse functions in
order to adequately describe the electronic distri-
bution in the anionic system emerges naturally
from the analysis of the weight functions, which
provides a precise indication of the number of
primitives to be included in the basis set [42]. The
s weight function, not presented in Fig. 2, correctly
goes to zero at the boundaries of ln(a ), indicating
a satisfactory basis set. However, the p (solid line)
and d (dotted line) weight functions show the need
for enlargement of the respective basis sets in the
valence region (negative Vi ¼ lnðaiÞ), since both
functions present a significant gap between the last
coefficient of each symmetry and the expected zero
of the weight function for all molecular orbitals.
The improvement of the weight functions is
accomplished by enlarging the basis set in the
deficient region until they converge to zero. More
diffuse functions can be added to the basis function
preserving the geometric tendency shown in Eq. (2)
Table 1
Improved (18s12p9d1f) basis set for the Mo atom
Molybdenum basis set
s p d f
0.0305417a 0.1319095a 0.0716065a 0.5018503
0.0817269a 0.3420828a 0.1895673a
0.2186936a 0.8871281a 0.5018503a
0.5852040a 2.3006017a 1.3285717a
1.5659521a 5.9661829a 3.5171896a
4.1903435a 15.4721863a 9.3112195a
11.2129734a 40.1242390 24.6500243a
30.0048843a 104.0547550 65.2571551
80.2903073 269.8466640 172.7583000
214.8494686 699.7971604
574.9173927 1814.7938477
1538.4259995 4706.3304856
4116.6863033
11015.8734476
29477.4629092
78878.9762064
211072.8764730
564811.5800334
a Exponents that must to be used along with the Hay and Wadt
ECP.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264254

using the equation
aiþ1ðkÞ ¼a2
i ðkÞ
ai21ðkÞð3Þ
where aiðkÞ is the lowest exponent of symmetry
species k, ai21ðkÞ is the next lowest exponent and
aiþ1ðkÞ the exponent of the diffuse function to be
added to the basis set. In this study, the weight
functions of 32 were analyzed for each basis
function symmetry in each molecular orbital for
Mo and O. Corrections of the basis set for the Mo
atom were achieved by increasing the (17s11p8d)
basis in the valence region up to a (18s12p9d)
geometric basis set and for the O atom from (9s4p)
up to (11s5p) (Tables 1 and 2).
Calculations involving transition metal are com-
putationally demanding, which can be considerably
reduced by replacing the inner (core) electrons by
ECP. All-electron basis sets should generally not be
used together with ECPs [51]. The modifications of
the basis set for Mo using pseudopotential was also
carried out using GCM and are described in detail
elsewhere [43,44,46]. Eq. (1) shows that the complete
space of the a exponent should comprise a range from
0 to 1. Although in practice the a space is limited to a
narrower region, a large number of functions must be
included either in atomic or molecular calculations to
describe appropriately the regions near the nuclei.
However, the limits of integration in Eq. (1) need to
be changed from (0,1) to (0,acut) when the core
electrons are replaced by a pseudopotential, which is
equivalent to saying that the basis set has no effect on
the wavefunction in a range between (acut,1) when
the pseudopotential starts to take effect. Mathemat-
ically the use of ECP changes Eq. (1) to
cið1Þ ¼ðacut
0fið1;aÞfiðaÞda; with i ¼ 1;…; n
ð4Þ
In order to evaluate the effect of the pseudopotential
on the wavefunction, Fig. 3 presents the Mo s weight
functions for the 5s atomic orbital calculated with the
previously improved (18s12p9d) basis set at the HF
level of theory with all electron (dashed line) and the
same basis set in conjunction with the Hay and Wadt
(HW) quasi-relativistic pseudopotential [52] (dotted
line). The two weight functions are practically
equivalent in the valence region, more specifically,
for V larger than 3.4, however, they are quite different
in the inner region. This value of V (<3.4) also
indicates where the pseudopotential starts to take
effect, i.e. the value (acut) at which the original basis
set should be terminated. This analysis suggests that
some primitive functions can be removed from the
inner region for an ECP calculation. Numerical tests
have shown that basis sets can be removed after the
first gaussian exponent where the weight function
presents essentially no change [43], i.e. for V larger
than 3.4 in Fig. 3. The Mo weight functions from Fig.
3 show that the first eight s gaussian functions must be
retained in the calculation using pseudopotential. The
cutoff in the inner region for p, d and other meshes can
be taken as the same for the s mesh. Consequently the
(18s12p9d) basis set can be drastically reduced to a
(8s6p7d) basis function when HW pseudopotential is
used. The discretization parameters (Eq. (2)) defining
the uncontracted (18s12p9d) all-electron basis set for
Mo atom are s mesh: V0 ¼ 23.489, DV ¼ 0.984,
N ¼ 18; p mesh: V0 ¼ 22.026, DV ¼ 0.953,
N ¼ 12; d mesh: V0 ¼ 22.637, DV ¼ 0.974,
N ¼ 9. The same values are used for the ECP basis
set, except that N ¼ 8; 6 and 7, for the s, p and d
meshes, respectively. The optimized geometric par-
ameters for the O atom are for s mesh, V0 ¼ 23.821,
DV ¼ 1.310, N ¼ 11 whereas for p mesh,
V0 ¼ 22.675, DV ¼ 1.388, N ¼ 5: It is important
to note that Walch et al. [53] has also presented
supplemental basis functions to the Huzinaga’s basis
set for the Mo atom, namely, two additional diffuse
Table 2
Improved (11s5p2d) basis set for the O atom
Oxygen basis set
s p d
0.0219125 0.0689206 1.1056131
0.0811708 0.2760426 0.2760462
0.3006826 1.1056131
1.1138250 4.4282302
4.1259653 17.7360626
15.2839002
56.6164738
209.7255980
776.8909554
2877.8535497
10660.4935937
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 255

functions were required for the p mesh:
a1(p) ¼ 0.1339 (Va1(p) ¼ 22.011) and
a2(p) ¼ 0.0462 (Va2(p) ¼ 23.076) while for the d
mesh an additional exponent a1(d) ¼ 0.0562
(Va1(d) ¼ 22.80) was suggested. The value of
Va1(p) for the p mesh is very similar to the value
obtained by us, V0(p) ¼ 22.026 (Table 1) for the
most diffuse exponent. A comparison of the exponent
values for the d mesh obtained by us,
V0(d) ¼ 22.637 and Walch et al. [53],
Va1(d) ¼ 22.80, also shows good agreement.
In addition to these diffuse functions, Walch et al.
[53] also recommended a set of 4f functions for the
basis set of the Mo atom. The Hellmann–Feynman
force analysis and total energy convergence were used
as criteria to include polarization functions on Mo and
O centers. Such a procedure is based on the magnitude
of the electric field on the nuclei in the molecular
system [41,44] and suggests the need of one f valence
polarization function (0.502) for the Mo center and
two d functions (0.276 and 1.106) for the O atoms.
It is important to note that we obtained similar
exponents for our basis function to those obtained by
Walch et al. [53] who used a much more elaborate and
demanding procedure than the GMC.
Optimized exponents for each mesh of Mo and O
Fig. 3. Molybdenum s weight function for the 5s atomic orbital calculated at the HF level using the improved (18s12p9d) basis set. The dashed
line is the s weight function from all-electron calculations; the dotted line is the weight function using the same basis set and the pseudopotential
by Hay and Wadt; the solid line is the s weight function using only improved outermost primitives and pseudopotential.
Table 3
Selected computing methods (level of calculation þ basis set)
Computing methods A B C D
Level of theory HF HF DFT–B3LYP DFT–B3LYP
Mo (8s6p7d1f) þ ECP (18s12p9d1f) (8s6p7d1f) þ ECP (18s12p9d1f)
O (11s5p2d) (11s5p2d) (11s5p2d) (11s5p2d)
C, N, H 6-31G(d,p) 6-31G(d,p) 6-31G(d,p) 6-31G(d,p)
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264256
atoms are listed in Tables 1 and 2. As a result, all-
electron basis sets (18s12p9d1f) and (11s5p2d) for
Mo and O atoms, respectively, have been designed,
while a valence basis set of (8s6p7d1f) has been
designed to be used with the HW-ECP for the Mo
atom. Although it seems natural to transfer the
concept of ‘atomic core’ into Kohn-Sham density
functional calculations, there are some problems
associated with this procedure. The performance of
ECPs adjusted at the HF level is well documented but
their application in density functional calculations has
been the subject of some research [51,54]. Therefore,
we have defined four computing methods, which are
summarized in Table 3.
3. Results and discussion
Optimized molecular structures of the compounds
1, 2 and 3 with A, B, C and D computing methods are
presented in Tables 4–6, respectively. The results
show that 1 has a pyramidal arrangement with the
peroxo oxygen atoms located in the same plane (Fig.
1(a)). This plane is perpendicular to the axis formed
by the Mo and the oxo ligand, Mo–O(1), and a value
around 458 is found for the bond angles associated
with the dioxygen ligands, O(3)–Mo–O(4) and
O(5)–Mo–O(6). Tilting of peroxo ligands yields
that these groups are asymmetrically bound to the
molybdenum center. The oxygen centers of peroxo
group are not equivalent, more specifically, the Mo–
O(3) and Mo–O(5) bond distances (back-side) are
approximately 0.04 A shorter than the Mo–O(4) and
Mo–O(6) ones, for all computational methods used.
These relative differences decrease on going from 1 to
2 and 3. For the sake of comparison we have
calculated the difference in total energy between the
conformation of 1 belonging to the groups Cs and C2v
(symmetric disposal of the back-side and front-side
oxygen atoms with respect to the Mo center). Cs
conformation is proven to be more stable by about
70.0 kcal/mol than the C2v structure.
Factors controlling this asymmetry can be found by
means of an analysis of the atomic orbital contri-
butions of representative molecular orbitals. A
detailed analysis of the valence molecular orbitals
calculated at C computing method reveals that the
HOMO (28.2 eV) and HOMO-1 (28.8 eV) of 1 are
localized on the peroxo oxygen atoms and are
associated to the O–O antibonding orbital pzp(O–O)
Table 4
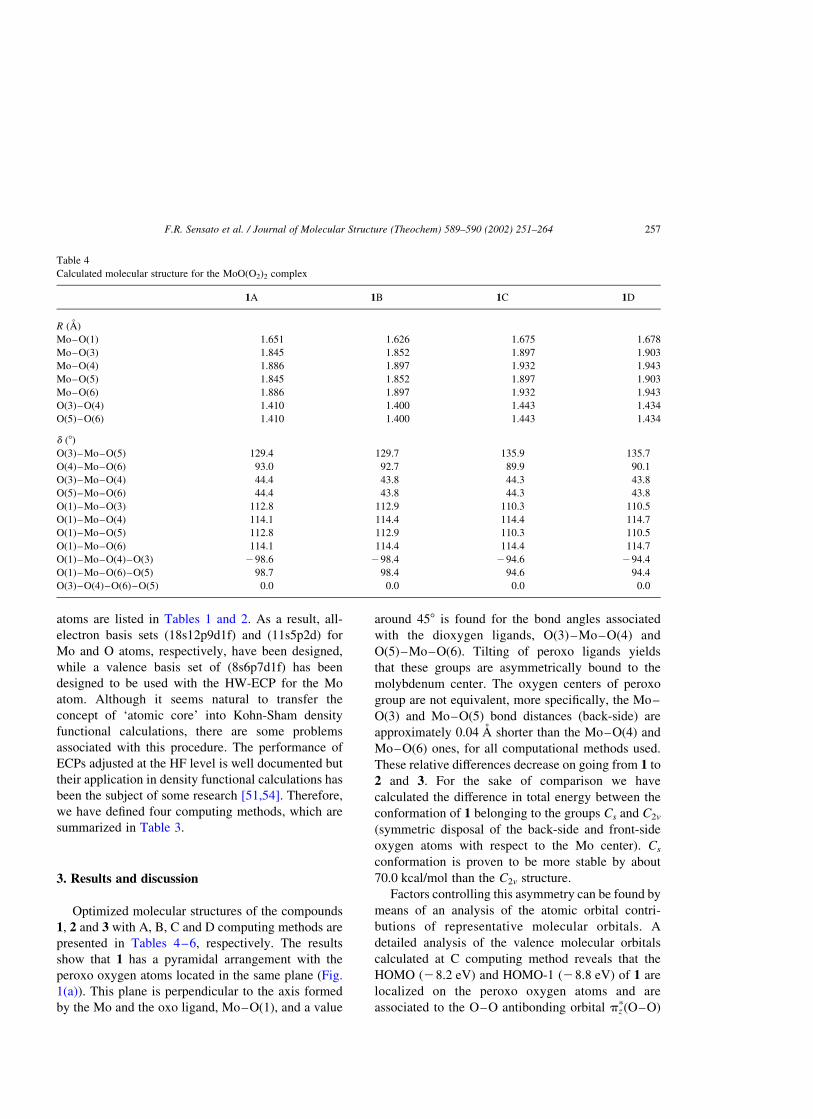
Calculated molecular structure for the MoO(O2)2 complex
1A 1B 1C 1D
R (A)
Mo–O(1) 1.651 1.626 1.675 1.678
Mo–O(3) 1.845 1.852 1.897 1.903
Mo–O(4) 1.886 1.897 1.932 1.943
Mo–O(5) 1.845 1.852 1.897 1.903
Mo–O(6) 1.886 1.897 1.932 1.943
O(3)–O(4) 1.410 1.400 1.443 1.434
O(5)–O(6) 1.410 1.400 1.443 1.434
d (8)
O(3)–Mo–O(5) 129.4 129.7 135.9 135.7
O(4)–Mo–O(6) 93.0 92.7 89.9 90.1
O(3)–Mo–O(4) 44.4 43.8 44.3 43.8
O(5)–Mo–O(6) 44.4 43.8 44.3 43.8
O(1)–Mo–O(3) 112.8 112.9 110.3 110.5
O(1)–Mo–O(4) 114.1 114.4 114.4 114.7
O(1)–Mo–O(5) 112.8 112.9 110.3 110.5
O(1)–Mo–O(6) 114.1 114.4 114.4 114.7
O(1)–Mo–O(4)–O(3) 298.6 298.4 294.6 294.4
O(1)–Mo–O(6)–O(5) 98.7 98.4 94.6 94.4
O(3)–O(4)–O(6)–O(5) 0.0 0.0 0.0 0.0
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 257

(see Fig. 4(a) and (b), respectively); HOMO-2
(210.3 eV; see Fig. 4(c)) is dominated by the py
orbitals of the oxygen atoms belonging to the peroxo
group,pyp(O–O), and the dxy and dx 2 2 dy
2 orbitals of the
Mo atom. Thus, the disposal of each peroxo group with
respect to the molybdenum center is dictated by the
angle between the lobes of the dxy and dx 2 2 dy2
orbitals of molybdenum. HOMO-3 (210.8 eV) is
illustrated in Fig. 4(d) and the dominant contributions
are associated to the py orbitals of both oxo oxygen
(O1) and Mo atoms being associated to p(Mo–O1)
bond. In addition, a very strong overlap between the
py orbital of the Mo center and back-side oxygen is
observed. As a result, these features of HOMO-2 and
HOMO-3 are responsible for the asymmetry exhibited
by 1 and related complexes. HOMO-4 (210.9 eV)
has a strong contribution of dxy orbital of the Mo atom,
which overlaps with px and py orbitals of the back- and
front-side oxygen atoms, respectively. HOMO-5
(211.6 eV) is related to the bonding orbital pz(O–
O) of the peroxo groups and the bonding interaction
between the Mo and oxo oxygen atoms. Since the
HOMO-4 and HOMO-5 orbitals do not contribute to
the observed asymmetry they are not shown in Fig. 4.
Although there are no experimental data for the
bis-peroxo structure 1; it might be formed as a result
of the ligand(s) dissociation from the Mo ion of 2 or 3
during the oxidation reaction as it has been suggested
by Mimoun [2]. The introduction of a pyridine oxide
ligand (OPy) between the back-side oxygen atoms in
1 leads to 2 and causes an increase in the Mo–O(3)
and Mo–O(5) bond lengths and a smaller increase in
the Mo–O(4) and Mo–O(6) bond distances. This
leads to a smaller asymmetry of the peroxo groups of
0.02 A compared to 0.04 A of 1. However, HF and
B3LYP computing methods yielded an opposite trend
for O–O; the bond distances in the peroxo groups,
namely, the HF method predicts a small decrease
(0.01 A) in the O(3)–O(4) and O(5)–O(6) bond
distances whereas a increase of the same magnitude is
Table 5
Calculated molecular structure for the MoO(O2)2(OPy) complex
2A 2B 2C 2D
R (A)
Mo–O(1) 1.629 1.630 1.681 1.684
Mo–O(3) 1.884 1.889 1.925 1.931
Mo–O(4) 1.901 1.909 1.942 1.952
Mo–O(5) 1.884 1.889 1.925 1.931
Mo–O(6) 1.901 1.909 1.942 1.952
Mo–O(7) 2.098 2.110 2.127 2.140
O(3)–O(4) 1.400 1.394 1.453 1.447
O(5)–O(6) 1.400 1.394 1.453 1.447
O(7)–N(8) 1.326 1.326 1.337 1.336
d (8)
O(1)–Mo–O(7) 96.2 96.6 95.7 96.0
O(3)–Mo–O(5) 130.8 130.7 132.7 132.4
O(4)–Mo–O(6) 89.1 89.0 88.2 88.2
O(3)–Mo–O(4) 43.4 43.1 44.1 43.8
O(5)–Mo–O(6) 43.4 43.1 44.1 43.8
O(1)–Mo–O(3) 114.1 114.2 113.0 113.2
O(1)–Mo–O(4) 110.1 110.5 110.5 110.9
O(1)–Mo–O(5) 114.1 114.2 113.0 113.2
O(1)–Mo–O(6) 110.1 110.5 110.5 110.9
Mo–O(7)–N(8) 118.4 118.4 116.7 116.6
O(1)–Mo–O(4)–O(3) 2104.2 2103.9 2102.3 2102.2
O(1)–Mo–O(6)–O(5) 104.2 103.9 102.4 102.2
O(3)–O(4)–O(6)–O(5) 0.0 0.0 0.0 0.0
O(1)–Mo–O(7)–N(8) 180.0 180.0 180.0 179.9
Mo–O(7)–N(8)–C(9) 91.0 91.0 90.7 90.7
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264258

obtained with B3LYP. The Mo-centered bond angles
between the oxo oxygen O(1) and the back-side
oxygen atoms (O(3) and O(5)) increase approximately
38, whereas the bond angles formed with the front-side
oxygen atoms (O(4) and O(6)) decrease nearly 48. All
four computing methods show that the oxo oxygen,
the back-side oxygen and Mo atoms lie onto the same
plane, which leads to a 08 dihedral angle O(3)–Mo–
O(1)–O(5) for all 1, 2 and 3 compounds.
The most noticeable feature induced by adding a
water molecule into 2 to yield 3 is the increase of the
O(3)–Mo–O(5) bond angle from approximately 1308
to around 1408. The Mo-centered bond angles formed
between the oxo and the back- and front-side oxygen
atoms decrease 58 and 38, respectively.
The X-ray structure of 3 has recently been
determined [14], and a comparison between the
theoretical and experimental data shows a very good
agreement, except for the bond distance between the
metal ion and the oxygen atom of the water ligand,
Mo(2)–O(19), that has been calculated to be 2.80 A,
which compares poorly with the experimental data
2.30 A. A similar result was reported by Rosch et al.
[55] who have compared experimental and theoretical
results for an analogue complex ReO(O2)2(CH3)(H2-
O), namely, the experimental data is 2.25 A, whereas
the calculated is 2.48 A, whose discrepancy has been
rationalized in terms of a co-crystallized ether
molecule. More recently, upon investigating the olefin
epoxidation by peroxo complexes with structures
(NH3)(L)M(O)22n(h 2-O2)1þn (n ¼ 0, 1; L ¼ none,
NH3; M ¼ Cr, Mo, W) Rosch et al. [20] inferred that
the corresponding part of the potential energy surfaces
is rather flat so that small perturbations may cause
Table 6
Calculated and experimental molecular structure for the MoO(O2)2(OPy)(H2O) complex
3A 3B 3C 3D Exp. [14]
R (A)
Mo–O(1) 1.622 1.624 1.676 1.679 1.670
Mo–O(3) 1.899 1.904 1.940 1.946 1.955
Mo–O(4) 1.905 1.913 1.949 1.958 1.919
Mo–O(5) 1.899 1.904 1.940 1.946 1.955
Mo–O(6) 1.905 1.913 1.949 1.958 1.919
Mo–O(7) 2.090 2.102 2.117 2.130 2.076
O(3)–O(4) 1.400 1.395 1.452 1.446 1.470
O(5)–O(6) 1.400 1.395 1.452 1.446 1.470
O(7)–N(8) 1.325 1.324 1.338 1.337 1.354
Mo–O(19) 2.820 2.844 2.811 2.838 2.295
Hwat–O(3)back-side 2.522 2.523 2.489 2.491 2.718
Hwat–O(4)front-side 2.353 2.347 2.249 2.240 2.832
d (8)
O(1)–Mo–O(7) 96.8 97.2 96.3 96.7 89.2
O(3)–Mo–O(5) 141.4 140.8 143.0 142.5 156.7
O(4)–Mo–O(6) 90.2 90.2 89.5 89.6 88.9
O(3)–Mo–O(4) 43.2 42.9 43.8 43.5 44.6
O(5)–Mo–O(6) 43.2 42.9 43.8 43.5 44.6
O(1)–Mo–O(3) 109.0 109.3 108.1 108.4 101.6
O(1)–Mo–O(4) 107.7 108.0 107.9 108.3 102.8
O(1)–Mo–O(5) 109.0 109.3 108.1 108.4 101.6
O(1)–Mo–O(6) 107.7 108.0 107.9 108.3 102.8
Mo–O(7)–N(8) 120.5 120.4 118.1 117.0 123.0
O(1)–Mo–O(19) 178.3 178.1 177.8 177.6 170.8
O(1)–Mo–O(4)–O(3) 299.2 299.2 297.7 297.6 293.5
O(1)–Mo–O(6)–O(5) 99.2 99.2 97.7 97.7 93.5
O(3)–O(4)–O(6)–O(5) 0.0 0.0 0.0 0.0 0.0
O(1)–Mo–O(7)–N(8) 180.0 180.0 180.0 180.0 180.0
Mo–O(7)–N(8)–C(9) 90.7 90.7 90.4 90.4 90.8
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 259

large structural effect. In addition, Frenking et al. [21]
have studied the equilibrium geometries of the
molybdenum oxo and peroxo compounds MoOn
(O2)32n and the related complexes [MoOn(O2)32n
(OPH3)] and [MoOn(O2)32n(OPH3)(H2O)] (n ¼ 0–3)
by DFT–B3LYP calculations and found a similar
discrepancy, which was partly attributed to inherent
solid-state effects. Thiel et al. [23] have also found
that the calculated distance between the axial ligand
and the molybdenum atom is overestimated with
respect to the corresponding distance for analogous
molybdenum peroxo complexes. It is important to
note that even in the experimental structure determi-
nation, the Mo–O(19) (2.295 A) distance is reported
to be larger than the Mo–O(7) distance (2.064 A).
The difference between the axial and equatorial bond
lengths has been reported to occur in analogue
complexes which has been supposed to be trans
influence of O(1) [6–9]. However, the considerable
discrepancy between our theoretical and experimental
results for Mo–O(19) distance cannot be regarded as
only resulting from such an effect. In addition, we
have characterized experimentally that the water
ligand forms intermolecular hydrogen bonds with
the peroxo groups of two other adjacent complexes in
the crystal [14]. Our calculation has not taken into
account these intermolecular interactions and, as a
result, a spurious and strong intramolecular inter-
action between the hydrogen atom of water ligand and
the peroxo group is favored (see Table 6 for the
calculated bond distance between the Hwat and front-
side oxygen atoms). Such an intramolecular inter-
action of the hydrogen atom towards the peroxo group
causes a displacement of the oxygen of the water in an
opposite direction leading to a very large value for the
Mo–O(19) bond. When the environment effect is
modeled by two methanol molecules that form
hydrogen bonds with the coordinated water, such a
bond distance decrease to a value of 2.3 A. A detailed
analysis along with the experimental data is presented
elsewhere [14].
An analysis of the relativistic effects can be
performed by comparing the results obtained with
the A and B, and the C and D computing methods. A
shorter Mo – oxygen bond distances have been
obtained when relativistic ECP was used (Tables
4–6). These findings have also been observed by van
Wullen for a similar analysis of the Mo(CO)6 complex
[51].
Once the molecular structure has been obtained,
the vibrational frequencies were calculated. Several
recent studies have shown that the vibrational
frequencies calculated with DFT methods agree well
with the experimental data (see Ref. [56] and
references therein). In addition, vibrational frequen-
cies for a large number of inorganic molecules have
been calculated using B3LYP functional in conjunc-
tion with the HW-ECP, which yielded a reasonable
agreement with the experiment [57]. Therefore, the C
and D methods were used and the assignments of the
more significant infrared bands are compared to
experimental results in Table 7. Calculated frequency
values are known to be overestimated by 10–15%,
partly due to the neglect of the anharmonic effects and
the inherent limitations of the theory. In order to
address this systematic difference, it has been
proposed that scaling factors be obtained by compar-
ing calculated values with large experimental data sets
[56–59]. We have determined the scaling factors for
the C and D computing methods by comparing
experimental and theoretical results for the main
vibrational modes of compound 3. Optimum scaling
factors were obtained in the same way as that
described in the literature [56–59] and the values
Fig. 4. Dominant orbital contribution for some occupied molecular
orbitals of the MoO(O2)2 complex calculated with the C method: (a)
HOMO; (b) HOMO-1; (c) HOMO-2 and (d) HOMO-3.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264260

0.9056 and 0.9097 were obtained for the C and D
methods, respectively. The calculated vibrational
frequencies of the 1, 2 and 3 complexes after scaling
are listed in Table 7 along with the experimental
values. The corresponding vibrational modes, from i
to ix, are also presented in Table 7. An analysis of the
results points out that a good agreement between the
theoretical values calculated with 3C and 3D and
experimental data is obtained. The overall root mean
square errors for the fundamental frequencies for 3C
and 3D are 22 and 23 cm21, respectively, affording
unambiguous assignment of the observed vibrational
frequencies for the coordination polyhedron around
Mo atom.
On going from 1 to 2 for either C or D methods, the
most prominent feature is the displacement by
43 cm21 of the vibrational mode iv to a lower
frequency value caused by the introduction of the
OPy ligand which elongates the bond distance
between the Mo atom and the back-side oxygens
(see Tables 4 and 5 for corresponding bond distances).
The calculated values for the vibrational frequencies
of 1, 2 and 3 suggest that the O–O fragment is
associated with a peroxo group (e.g. for H2O2, the
distance O–O and the corresponding vibrational
frequency are 1.453 A and 882 cm21, respectively
[60]). In addition, the stretching movement associated
with the Mo–OPy and Mo–OH2 bonds of 2 and 3
complexes present very low values (around 360 and
115 cm21, respectively), characterizing the flatness of
the potential curves associated with these ligands.
The metal-mediated epoxidation of olefins mech-
anism has been subjected to a great deal of
experimental and theoretical studies. Two main
mechanisms concerning stoichiometric olefin epox-
idation by MoO(O2)2(L1,L2) have generated a long
Table 7
Selected calculated and experimental vibrational frequencies (cm21) for compounds 1, 2 and 3
1C 2C 3C 1D 2D 3D Exp. [1] Exp. [13] Exp. [14]
(i) 945 933 937 942 930 934 960 970 969
(ii) 881 880 876 892 887 883 875 873
(iii) 872 871 868 881 878 876 860 859
(iv) 638 595 594 637 594 592 580 577 582
(v) 588 578 562 585 574 558 540 537
(vi) 502 504 495 493 499 490
(vii) 515 526 505 509 519 503
(viii) Mo–OPy 361 358 359 356
(ix) Mo–OH2 116 113
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 261
controversy, namely, Mimoun [2] proposed a coordi-
nation of the olefin at the metal and a subsequent
cycloinsertion into a metal–peroxo bond forming a
five-membered dioxymetallocycle that decomposes
into an epoxide and the corresponding monoperoxo
complex, while Sharpless [61] suggested an alterna-
tive mechanism involving transfer of one of the
peroxo oxygens to the olefin through a three-
membered ring transition state. Recently, two theor-
etical studies have corroborated the mechanism
suggested by Sharpless [20,22].
Still, an important point to consider is how the
reactivity of the oxo-peroxo complexes depends upon
the coordination around the metal atom. We have
observed experimentally that the oxo-peroxo molyb-
denum complex in its dehydrate form (compounds
related to 1 or 2) is more reactive towards olefin and
sulfide oxidation that the hydrated ones (compounds
related to 3) [62]. The question of whether either 1 or
2 is the ultimate moiety remains unanswered. The
approach of an olefin molecule towards the oxo-
peroxo complex may be able to induce the displace-
ment of the OPy ligand (substitution mode ) so that the
olefin attacks directly 1. Otherwise, the ligand may
remain bonded to metal atom during the formation of
an olefin-coordinated specie (addition mode ).
Olefin epoxidation by d0 metal–peroxo complexes
closely resemble epoxidation by purely organic
oxidants. This provides support for the conclusion
that oxygen transfer by metal catalyzed reactions
presents also an electrophilic character. Using an
NDDO approach, Filatov et al. [17] has compared the
electron affinities of MoO(O2)2L1 (L1 ¼ H2O or NH3)
and MoO(O2)2L1L2 (L1 and L2 ¼ H2O or NH3), and
inferred that the lower electron affinity exhibited by
the seven-coordinated species is the reason for these
complexes being much less reactive towards epoxida-
tion of olefins than the six-coordinated ones. In light
of this, we have calculated the electron affinities of 1,
2 and 3 complexes at the C level, which were
computed as the difference between the total energies
of the radical anion and the neutral compound by two
approaches, namely, with the former being calculated
at the geometry for the latter (vertical electron affinity,
VEA), and the anionic specie being allowed to relax
(adiabatic electron affinity, AEA). The corresponding
values of VEA and AEA are summarized in Table 8.
As expected, the calculated AEA values are larger
than the corresponding VEA ones; however, both
approaches yielded the same qualitative and quanti-
tative trend. Compound 1 is the most electrophilic
whereas 2 and 3 should exhibit similar electrophili-
city. The electrophilic properties of 1, 2 and 3 are also
reflected by the pertinent charge distribution. Table 9
presents the NBO population analysis of 1, 2 and 3
calculated with the C method. The smaller electron
affinities of 2 and 3 is caused by the electron donation
of approximately 0.3 electrons from the OPy ligand to
the MoO(O2)2 moiety. As a result, electron affinity
considerations render that compound 1 could be
expected to be the ultimate specie toward the
epoxidation of olefins.
Olefin epoxidation by a d0 metal peroxo is also
generally considered as an electrophilic attack of the
peroxo oxygen on the olefin. Based on a charge
decomposition analysis (CDA) scheme [63] on the
transition state related to the Sharpless mechanism,
Frenking et al. [22] suggested that the epoxidation
with metallo peroxides should be considered as a
nucleophilic attack of ethylene toward the sp orbital
of the O–O bond. Rosch et al. [20] have also
established a linear correlation between the energy
of the molecular orbital with dominant contribution of
sp(O–O) orbital of the complex and the height of the
activation barrier for the interaction between the
Table 8
The calculated vertical (VEA) and adiabatic (AEA) electron
affinities (eV) for compounds 1, 2 and 3 with the C method
1C 2C 3C
VEA 2.9 1.2 1.1
AEA 3.3 1.4 1.4
Table 9
Calculated atomic charges based on the NBO population analysis
for compounds 1, 2 and 3 with the C method
1C 2C 3C
O(1) 20.51 20.52 20.48
Mo 1.84 1.79 1.77
O(3), O(5) 20.35 20.41 20.41
O(4), O(6) 20.31 20.36 20.39
O(7) 20.56 20.56
N(8) 0.05 0.06
O(19) 21.02
H(20), H(21) 0.53
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264262

olefin molecule and the oxo-peroxo complex of Mo.
That is, for a fixed electron donor, the activation
barrier increases if the energy of the molecular orbital
with dominant contribution of sp(O–O) level also
increases, even if it is not the LUMO of the complex.
An analysis of the molecular orbital structure for
compound 1 calculated at C method reveals that the
energy associated to the molecular orbital with
dominant contribution of sp(O – O) level is
20.7 eV, while a value of approximately 1.0 eV
was found for compounds 2 and 3 indicating that the
compound 1 is the most reactive towards nucleophilic
attack. The calculated LUMO energy values also
support this trend. The LUMO energy of 1 is 25.1 eV
while the values of 23.1 and 22.7 eV were found for
2 and 3, respectively.
The Mo–H2O bond energy of 3 as well as the Mo–
OPy bond energy of 2 have also been calculated with
the C method, and they are 29.7 and 244.6 kcal/mol,
respectively. As a result, there might be two
competitive effects: the electrophilic properties as
well as the value of the energy associated to the
molecular orbital with dominant contribution of the
sp(O–O) level for 1, 2 and 3 favor the substitution
approach of the olefin molecule, while the Mo–OPy
bond energy supports the addition process.
4. Conclusions
The electronic and molecular structures of oxo-
diperoxo complexes: MoO(O2)2, MoO(O2)2OPy and
MoO(O2)2(OPy)(H2O), where OPy ¼ pyridine N-
oxide, have been investigated using ab initio HF and
DFT (B3LYP) methods. The GCM has been used to
design all-electrons basis sets for the oxygen and
molybdenum atoms as well as a valence basis set for
the Mo atom for pseudopotential calculations. The
GCM approach has proven to be quite versatile,
efficient, and simple to be used and implemented. The
results obtained with the designed valence basis set
for Mo atom adapted to be used with the HW-ECP
compared very well with the all-electron ones. The
atomic orbital contributions of the oxygen and Mo
atoms to the HOMO-2 and HOMO-3 molecular
orbitals are responsible for generating an asymmetric
(Cs) structure for all complexes investigated. The
MoO(O2)2 compound has proven to be the most
reactive towards oxygen transfer based upon the
energy of the sp(O–O) molecular orbital and the
values of the electron affinities of each complex
studied. The reliability of these results is strengthened
by the very good agreement between the calculated
and experimental molecular structures as well as for
the vibrational frequencies.
Acknowledgments
This work was supported by the Brazilian funding
agencies: FAPESP/CEPID and by the ‘Programa de
Cooperacao Internacional’ supported by CAPES
(Brazil) and Ministerio de Educacion y Cultura del
Gobierno Espanol (Spain). The authors also wish to
thank the computer centers CENAPAD-NAR-UFS-
Car, CENAPAD-SP, CESUP-RG (Brazil), Servei
d’Informatica de la Universitat Jaume I-Castellon-
Spain for making their computational facilities
available.
References
[1] H. Mimoun, I.S. Roch, L. Sajus, Bull. Soc. Chim. Fr. 5 (1969)
1481.
[2] H. Mimoun, I.S. Roch, L. Sajus, Tetrahedron 26 (1970) 37.
[3] S.A. Matlin, P.G. Sammes, R. Upton, J. Chem. Soc. Perkin
Trans. 1 (1979) 2481.
[4] H. Mimoun, Angew. Chem., Int. Ed. Engl. 21 (1982) 734.
[5] J.M. Le Carpentier, R. Schlupp, R. Weiss, Acta Crystallogr., B
28 (1972) 1278.
[6] E.O. Schlemper, G.N. Schrauzer, L.A. Hughes, Polyhedron 3
(1984) 377.
[7] W.P. Griffith, A.M.Z. Slawin, K.M. Thompson, D.J. Williams,
Chem. Soc. Chem. Commun. (1994) 569.
[8] P. Martın-Zarza, P. Gili, F.V. Rodrıguez-Romero, C. Ruiz-
Perez, X. Solans, Inorg. Chim. Acta 223 (1994) 173.
[9] W.R. Thiel, T. Priermeier, Angew. Chem., Int. Ed. Engl. 34
(1995) 1737.
[10] K.A. Jørgensen, Chem. Rev. 89 (1989) 431.
[11] M.H. Dickman, M.T. Pope, Chem. Rev. 94 (1994) 569.
[12] M.J. Morris, Coord. Chem. Rev. 152 (1996) 309.
[13] A.D. Westland, F. Haque, J. Bouchard, Inorg. Chem. 19
(1980) 2255.
[14] F.R. Sensato, Q.B. Cass, E. Longo, J. Zukerman-Schpector, R.
Custodio, J. Andres, M.Z. Hernandes, R.L. Longo, Inorg.
Chem. 40 (2001) 6022.
[15] K.F. Purcell, Organometallics 4 (1985) 509.
[16] K.A. Jørgensen, R. Hoffmann, Acta Chem. Scand. B 40 (1986)
411.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264 263

[17] M.J. Filatov, K.V. Shalyaev, E.P. Talsi, J. Mol. Catal. 87
(1994) L5.
[18] L. Salles, J.-Y. Piquemal, R. Thouvenot, C. Minot, J.-M.
Bregeault, J. Mol. Catal. A 117 (1997) 375.
[19] I.V. Yudanov, C. Di Valentin, P. Gisdakis, N. Rosch, J. Mol.
Catal. A 158 (2000) 189.
[20] C. Di Valentin, P. Gisdakis, I.V. Yudanov, N. Rosch, J. Org.
Chem. 65 (2000) 2996.
[21] D.V. Deubel, J. Sundermeyer, G. Frenking, Inorg. Chem. 39
(2000) 2314.
[22] D.V. Deubel, J. Sundermeyer, G. Frenking, J. Am. Chem. Soc.
122 (2000) 10101.
[23] A. Hroch, G. Gemmecker, W.R. Thiel, Eur. J. Inorg. Chem. 5
(2000) 1107.
[24] D.V. Deubel, J. Sundermeyer, G. Frenking, Org. Lett. 3 (2001)
329.
[25] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A.
Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery,
Jr., R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam,
A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi,
V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.
Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,
Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck,
K. Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz,
B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi,
R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham,
C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe,
P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L. Andres,
C. Gonzalez, M. Head-Gordon, E.S. Replogle, J.A. Pople,
Gaussian, Inc., Pittsburgh PA, 1998.
[26] S.H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58 (1980) 1200.
[27] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[28] A.D. Becke, Phys. Rev. A 38 (1988) 3098.
[29] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[30] J.B. Foresman, A. Frish, Exploring Chemistry with Electronic
Structure Methods, Gaussian Inc, Pittsburgh, 1993.
[31] GAUSSVIEW (2.0), Gaussian Inc., Pittsburgh, PA, 1998.
[32] L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565.
[33] S. Huzinaga, J. Chem. Phys. 66 (1977) 4245.
[34] F.B. Duijneveldt, IBM Res. J. (1971) 945.
[35] R. Ditchfield, W.J. Hehre, J.A. Pople, J. Chem. Phys. 52
(1970) 5001.
[36] D.L. Hill, J.A. Wheeler, Phys. Rev. 89 (1953) 1102.
[37] J.J. Griffin, J.A. Wheeler, Phys. Rev. 108 (1957) 311.
[38] R.O. Vianna, R. Custodio, H. Chacham, J.R. Mohallem, Int.
J. Quantum Chem. Symp. 26 (1992) 311.
[39] R. Custodio, J.D. Goddard, J. Mol. Struct. (Theochem) 277
(1992) 263.
[40] R. Custodio, M. Giordan, N.H. Morgon, J.D. Goddard, Int.
J. Quantum Chem. 42 (1992) 411.
[41] R. Custodio, J.D. Goddard, M. Giordan, N.H. Morgon, Can.
J. Chem. 70 (1992) 580.
[42] R. Custodio, J.D. Goddard, J. Mol. Struct. (Theochem) 281
(1993) 75.
[43] R. Custodio, W.M. Davis, J.D. Goddard, J. Mol. Struct.
(Theochem) 315 (1994) 163.
[44] N.H. Morgon, R. Custodio, J.M. Riveros, Chem. Phys. Lett.
235 (1995) 436.
[45] N.H. Morgon, R. Custodio, J.G.R. Tostes, C.A. Taft, J. Mol.
Struct. (Theochem) 335 (1995) 11.
[46] M. Giordan, R. Custodio, N.H. Morgon, Chem. Phys. Lett. 279
(1997) 396.
[47] J.R. Mohallem, R.M. Dreizler, M. Trsic, Int. J. Quantum
Chem. Symp. 20 (1986) 45.
[48] J.R. Mohallem, Z. Phys. D 3 (1986) 339.
[49] J.R. Mohallem, M. Trsic, J. Chem. Phys. 86 (1987) 5043.
[50] H.F.M. Costa, M. Trsic, J.R. Mohallem, Mol. Phys. 62 (1987)
91.
[51] C.V. Wullen, Int. J. Quantum Chem. 58 (1996) 147.
[52] P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 299.
[53] S.P. Walch, C.W. Bauschlicher Jr, C.J. Nelin, J. Chem. Phys.
79 (1983) 3600.
[54] T.V. Russo, R.L. Martin, P.J. Hay, J. Phys. Chem. 99 (1995)
17085.
[55] P. Gisdakis, S. Antonczak, S. Kostlmeier, W.A. Herrmann, N.
Rosch, Angew. Chem., Int. Ed. Engl. 37 (1998) 2211.
[56] M.W. Wong, Chem. Phys. Lett. 26 (1996) 391.
[57] I. Bytheway, M.W. Wong, Chem. Phys. Lett. 282 (1998) 219.
[58] J.A. Pople, H.B. Schlegel, R. Krishnan, D.J. Defrees, J.S.
Binkley, M.J. Frisch, R.A. Whiteside, R.F. Hout, W.J. Hehre,
Int. J. Quantum Chem. Symp. 15 (1981) 269.
[59] J.A. Pople, A.P. Scott, M.W. Wong, L. Radom, Isr. J. Chem.
33 (1993) 345.
[60] N.N. Greenwood, A. Earnshaw, Chemistry of the Elements,
Pergamon Press, Oxford, 1984.
[61] K.B. Sharpless, J.M. Townsend, D.R. Williams, J. Am. Chem.
Soc. 94 (1972) 295.
[62] M.Z. Hernandes, Thesis, Universidade Federal de Sao Carlos,
Sao Carlos, 1998.
[63] S. Dapprich, G. Frenking, J. Phys. Chem. 99 (1995) 9352.
F.R. Sensato et al. / Journal of Molecular Structure (Theochem) 589–590 (2002) 251–264264
Related Documents











