Immunity Article The Transcription Factor IRF8 Activates Integrin-Mediated TGF- b Signaling and Promotes Neuroinflammation Yuko Yoshida, 1,8 Ryusuke Yoshimi, 1,8,9 Hiroaki Yoshii, 1 Daniel Kim, 1 Anup Dey, 1 Huabao Xiong, 2 Jeeva Munasinghe, 3 Itaru Yazawa, 4,10 Michael J. O’Donovan, 4 Olga A. Maximova, 5 Suveena Sharma, 6 Jinfang Zhu, 6 Hongsheng Wang, 7 Herbert C. Morse III, 7 and Keiko Ozato 1, * 1 Program in Genomics of Differentiation, NICHD, National Institutes of Health, Bethesda, MD 20892, USA 2 Immunology Institute, Department of Medicine, Mount Sinai School of Medicine, New York, NY 10029, USA 3 Laboratory of Functional and Molecular Imaging, NINDS, National Institutes of Health, Bethesda, MD 20892, USA 4 Laboratory of Neural Control, NINDS, National Institutes of Health, Bethesda, MD 20892, USA 5 Laboratory of Infectious Diseases, NIAID, National Institutes of Health, Bethesda, MD 20892, USA 6 Laboratory of Immunology, NIAID, National Institutes of Health, Bethesda, MD 20892, USA 7 Laboratory of Immunogenetics, NIAID, National Institutes of Health, Rockville, MD 20892, USA 8 These authors contributed equally to this work 9 Present address: Department of Internal Medicine and Clinical Immunology, Yokohama City University Graduate School of Medicine, Yokohama 236-0004, Japan 10 Present address: Department of Neurology, Showa University, Tokyo 142-8555, Japan *Correspondence: [email protected] http://dx.doi.org/10.1016/j.immuni.2013.11.022 SUMMARY Recent epidemiological studies have identified interferon regulatory factor 8 (IRF8) as a susceptibil- ity factor for multiple sclerosis (MS). However, how IRF8 influences the neuroinflammatory disease has remained unknown. By studying the role of IRF8 in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, we found that Irf8 / mice are resistant to EAE. Furthermore, expression of IRF8 in antigen-presenting cells (APCs, such as macrophages, dendritic cells, and microglia), but not in T cells, facilitated disease onset and pro- gression through multiple pathways. IRF8 enhanced avb8 integrin expression in APCs and activated TGF-b signaling leading to T helper 17 (Th17) cell differentiation. IRF8 induced a cytokine milieu that favored growth and maintenance of Th1 and Th17 cells, by stimulating interleukin-12 (IL-12) and IL-23 production, but inhibiting IL-27 during EAE. Finally, IRF8 activated microglia and exacerbated neuroinflammation. Together, this work provides mechanistic bases by which IRF8 contributes to the pathogenesis of MS. INTRODUCTION T helper 17 (Th17) cells promote inflammation and tissue injury and are associated with autoimmune diseases. However, they also play a role in pathogen resistance (Weaver et al., 2007). With the aid of transforming growth factor-b (TGF-b),inter- leukin-6 (IL-6), and other cytokines, antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, trigger development of Th17 cells (Bettelli et al., 2006). Recent studies revealed that TGF-b signaling is mediated by integrin molecules on APCs, which enables direct delivery of biologically active TGF-b into naive T cells (Acharya et al., 2010; Melton et al., 2010). Integrin-triggered TGF-b signaling results in activation of ROR family transcription factors that direct Th17 cell differentia- tion and production of the signature cytokine IL-17 (Ivanov et al., 2006; Yang et al., 2008). It also activates Treg generation (Travis et al., 2007). One of the best-studied autoimmune diseases causally asso- ciated with Th17 cells is experimental autoimmune encephalo- myelitis (EAE), a mouse model of multiple sclerosis (MS). MS is an inflammatory disease of the central nervous system (CNS) that involves demyelination and neuronal injury (Hauser and Oksenberg, 2006; Steinman and Zamvil, 2006). Mice lacking RORa, RORgt, and IL-17 are largely resistant to EAE (Ivanov et al., 2006; Komiyama et al., 2006). Correlating with results in the mouse, Th17 cells and IL-17 are present in the CNS of MS patients (Axtell et al., 2010). In addition to Th17 cells, Th1 cells play substantive roles in EAE, causing CNS lesions distinct from those by Th17 cells (Axtell et al., 2010; Kroenke et al., 2008; Stromnes et al., 2008). Involvement of Th1 cells in MS is also documented, adding to the similarity of EAE with MS (Lovett-Racke et al., 2011). Th17 cells, once developed, further proliferate in lymph nodes and then in the CNS, the processes dependent on IL-23, a cytokine of the IL-12 family, produced largely by macrophages and microglia (Becher et al., 2003; Chen et al., 2006; Cua et al., 2003). The classical IL-12p70 sup- ports the development and expansion of Th1 cells (Kroenke et al., 2008). Conversely, development of Th17 cells and EAE is suppressed by IL-27, another IL-12 family cytokine (Batten et al., 2006; Bettelli et al., 2006; Stumhofer et al., 2006). Although infiltrating T cells and APCs initiate CNS inflammation, activation of the resident microglia worsens the disease by increasing inflammation and neuronal damage (Sørensen et al., 1999; Star- ossom et al., 2012). Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 1 Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro- inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Immunity
Article
The Transcription Factor IRF8 ActivatesIntegrin-Mediated TGF-b Signalingand Promotes NeuroinflammationYuko Yoshida,1,8 Ryusuke Yoshimi,1,8,9 Hiroaki Yoshii,1 Daniel Kim,1 Anup Dey,1 Huabao Xiong,2 Jeeva Munasinghe,3
Itaru Yazawa,4,10 Michael J. O’Donovan,4 Olga A. Maximova,5 Suveena Sharma,6 Jinfang Zhu,6 Hongsheng Wang,7
Herbert C. Morse III,7 and Keiko Ozato1,*1Program in Genomics of Differentiation, NICHD, National Institutes of Health, Bethesda, MD 20892, USA2Immunology Institute, Department of Medicine, Mount Sinai School of Medicine, New York, NY 10029, USA3Laboratory of Functional and Molecular Imaging, NINDS, National Institutes of Health, Bethesda, MD 20892, USA4Laboratory of Neural Control, NINDS, National Institutes of Health, Bethesda, MD 20892, USA5Laboratory of Infectious Diseases, NIAID, National Institutes of Health, Bethesda, MD 20892, USA6Laboratory of Immunology, NIAID, National Institutes of Health, Bethesda, MD 20892, USA7Laboratory of Immunogenetics, NIAID, National Institutes of Health, Rockville, MD 20892, USA8These authors contributed equally to this work9Present address: Department of Internal Medicine and Clinical Immunology, Yokohama City University Graduate School of Medicine,Yokohama 236-0004, Japan10Present address: Department of Neurology, Showa University, Tokyo 142-8555, Japan
*Correspondence: [email protected]
http://dx.doi.org/10.1016/j.immuni.2013.11.022
SUMMARY
Recent epidemiological studies have identifiedinterferon regulatory factor 8 (IRF8) as a susceptibil-ity factor for multiple sclerosis (MS). However, howIRF8 influences the neuroinflammatory diseasehas remained unknown. By studying the role ofIRF8 in experimental autoimmune encephalomyelitis(EAE), a mouse model of MS, we found that Irf8�/�
mice are resistant to EAE. Furthermore, expressionof IRF8 in antigen-presenting cells (APCs, such asmacrophages, dendritic cells, and microglia), butnot in T cells, facilitated disease onset and pro-gression through multiple pathways. IRF8 enhancedavb8 integrin expression in APCs and activatedTGF-b signaling leading to T helper 17 (Th17) celldifferentiation. IRF8 induced a cytokine milieuthat favored growth and maintenance of Th1 andTh17 cells, by stimulating interleukin-12 (IL-12) andIL-23 production, but inhibiting IL-27 during EAE.Finally, IRF8 activated microglia and exacerbatedneuroinflammation. Together, this work providesmechanistic bases by which IRF8 contributes to thepathogenesis of MS.
INTRODUCTION
T helper 17 (Th17) cells promote inflammation and tissue injury
and are associated with autoimmune diseases. However, they
also play a role in pathogen resistance (Weaver et al., 2007).
With the aid of transforming growth factor-b (TGF-b),inter-
leukin-6 (IL-6), and other cytokines, antigen-presenting cells
(APCs), such as dendritic cells (DCs) and macrophages, trigger
development of Th17 cells (Bettelli et al., 2006). Recent studies
revealed that TGF-b signaling is mediated by integrin molecules
on APCs, which enables direct delivery of biologically active
TGF-b into naive T cells (Acharya et al., 2010; Melton et al.,
2010). Integrin-triggered TGF-b signaling results in activation of
ROR family transcription factors that direct Th17 cell differentia-
tion and production of the signature cytokine IL-17 (Ivanov et al.,
2006; Yang et al., 2008). It also activates Treg generation (Travis
et al., 2007).
One of the best-studied autoimmune diseases causally asso-
ciated with Th17 cells is experimental autoimmune encephalo-
myelitis (EAE), a mouse model of multiple sclerosis (MS). MS is
an inflammatory disease of the central nervous system (CNS)
that involves demyelination and neuronal injury (Hauser and
Oksenberg, 2006; Steinman and Zamvil, 2006). Mice lacking
RORa, RORgt, and IL-17 are largely resistant to EAE (Ivanov
et al., 2006; Komiyama et al., 2006). Correlating with results in
the mouse, Th17 cells and IL-17 are present in the CNS of MS
patients (Axtell et al., 2010). In addition to Th17 cells, Th1 cells
play substantive roles in EAE, causing CNS lesions distinct
from those by Th17 cells (Axtell et al., 2010; Kroenke et al.,
2008; Stromnes et al., 2008). Involvement of Th1 cells in MS is
also documented, adding to the similarity of EAE with MS
(Lovett-Racke et al., 2011). Th17 cells, once developed, further
proliferate in lymph nodes and then in the CNS, the processes
dependent on IL-23, a cytokine of the IL-12 family, produced
largely by macrophages and microglia (Becher et al., 2003;
Chen et al., 2006; Cua et al., 2003). The classical IL-12p70 sup-
ports the development and expansion of Th1 cells (Kroenke
et al., 2008). Conversely, development of Th17 cells and EAE is
suppressed by IL-27, another IL-12 family cytokine (Batten
et al., 2006; Bettelli et al., 2006; Stumhofer et al., 2006). Although
infiltrating T cells and APCs initiate CNS inflammation, activation
of the resident microglia worsens the disease by increasing
inflammation and neuronal damage (Sørensen et al., 1999; Star-
ossom et al., 2012).
Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 1
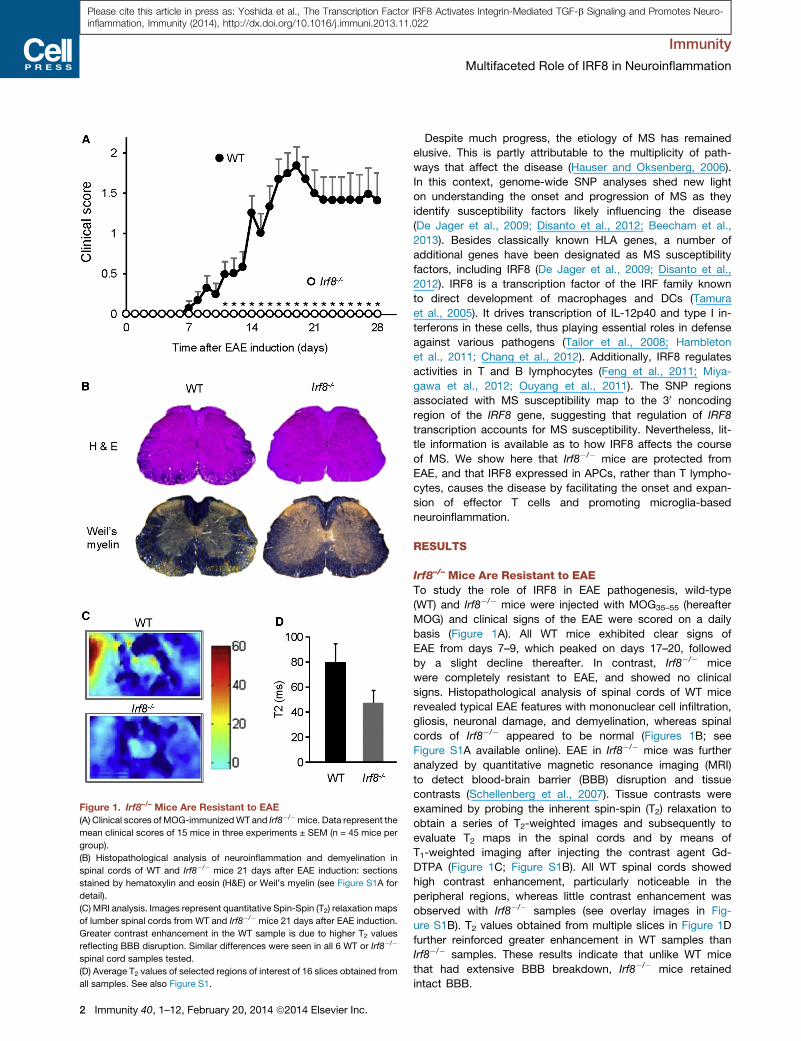
Figure 1. Irf8–/– Mice Are Resistant to EAE
(A) Clinical scores of MOG-immunizedWT and Irf8�/�mice. Data represent the
mean clinical scores of 15 mice in three experiments ± SEM (n = 45 mice per
group).
(B) Histopathological analysis of neuroinflammation and demyelination in
spinal cords of WT and Irf8�/� mice 21 days after EAE induction: sections
stained by hematoxylin and eosin (H&E) or Weil’s myelin (see Figure S1A for
detail).
(C) MRI analysis. Images represent quantitative Spin-Spin (T2) relaxation maps
of lumber spinal cords from WT and Irf8�/� mice 21 days after EAE induction.
Greater contrast enhancement in the WT sample is due to higher T2 values
reflecting BBB disruption. Similar differences were seen in all 6 WT or Irf8�/�
spinal cord samples tested.
(D) Average T2 values of selected regions of interest of 16 slices obtained from
all samples. See also Figure S1.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
2 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Despite much progress, the etiology of MS has remained
elusive. This is partly attributable to the multiplicity of path-
ways that affect the disease (Hauser and Oksenberg, 2006).
In this context, genome-wide SNP analyses shed new light
on understanding the onset and progression of MS as they
identify susceptibility factors likely influencing the disease
(De Jager et al., 2009; Disanto et al., 2012; Beecham et al.,
2013). Besides classically known HLA genes, a number of
additional genes have been designated as MS susceptibility
factors, including IRF8 (De Jager et al., 2009; Disanto et al.,
2012). IRF8 is a transcription factor of the IRF family known
to direct development of macrophages and DCs (Tamura
et al., 2005). It drives transcription of IL-12p40 and type I in-
terferons in these cells, thus playing essential roles in defense
against various pathogens (Tailor et al., 2008; Hambleton
et al., 2011; Chang et al., 2012). Additionally, IRF8 regulates
activities in T and B lymphocytes (Feng et al., 2011; Miya-
gawa et al., 2012; Ouyang et al., 2011). The SNP regions
associated with MS susceptibility map to the 30 noncoding
region of the IRF8 gene, suggesting that regulation of IRF8
transcription accounts for MS susceptibility. Nevertheless, lit-
tle information is available as to how IRF8 affects the course
of MS. We show here that Irf8�/� mice are protected from
EAE, and that IRF8 expressed in APCs, rather than T lympho-
cytes, causes the disease by facilitating the onset and expan-
sion of effector T cells and promoting microglia-based
neuroinflammation.
RESULTS
Irf8–/– Mice Are Resistant to EAETo study the role of IRF8 in EAE pathogenesis, wild-type
(WT) and Irf8�/� mice were injected with MOG35–55 (hereafter
MOG) and clinical signs of the EAE were scored on a daily
basis (Figure 1A). All WT mice exhibited clear signs of
EAE from days 7–9, which peaked on days 17–20, followed
by a slight decline thereafter. In contrast, Irf8�/� mice
were completely resistant to EAE, and showed no clinical
signs. Histopathological analysis of spinal cords of WT mice
revealed typical EAE features with mononuclear cell infiltration,
gliosis, neuronal damage, and demyelination, whereas spinal
cords of Irf8�/� appeared to be normal (Figures 1B; see
Figure S1A available online). EAE in Irf8�/� mice was further
analyzed by quantitative magnetic resonance imaging (MRI)
to detect blood-brain barrier (BBB) disruption and tissue
contrasts (Schellenberg et al., 2007). Tissue contrasts were
examined by probing the inherent spin-spin (T2) relaxation to
obtain a series of T2-weighted images and subsequently to
evaluate T2 maps in the spinal cords and by means of
T1-weighted imaging after injecting the contrast agent Gd-
DTPA (Figure 1C; Figure S1B). All WT spinal cords showed
high contrast enhancement, particularly noticeable in the
peripheral regions, whereas little contrast enhancement was
observed with Irf8�/� samples (see overlay images in Fig-
ure S1B). T2 values obtained from multiple slices in Figure 1D
further reinforced greater enhancement in WT samples than
Irf8�/� samples. These results indicate that unlike WT mice
that had extensive BBB breakdown, Irf8�/� mice retained
intact BBB.
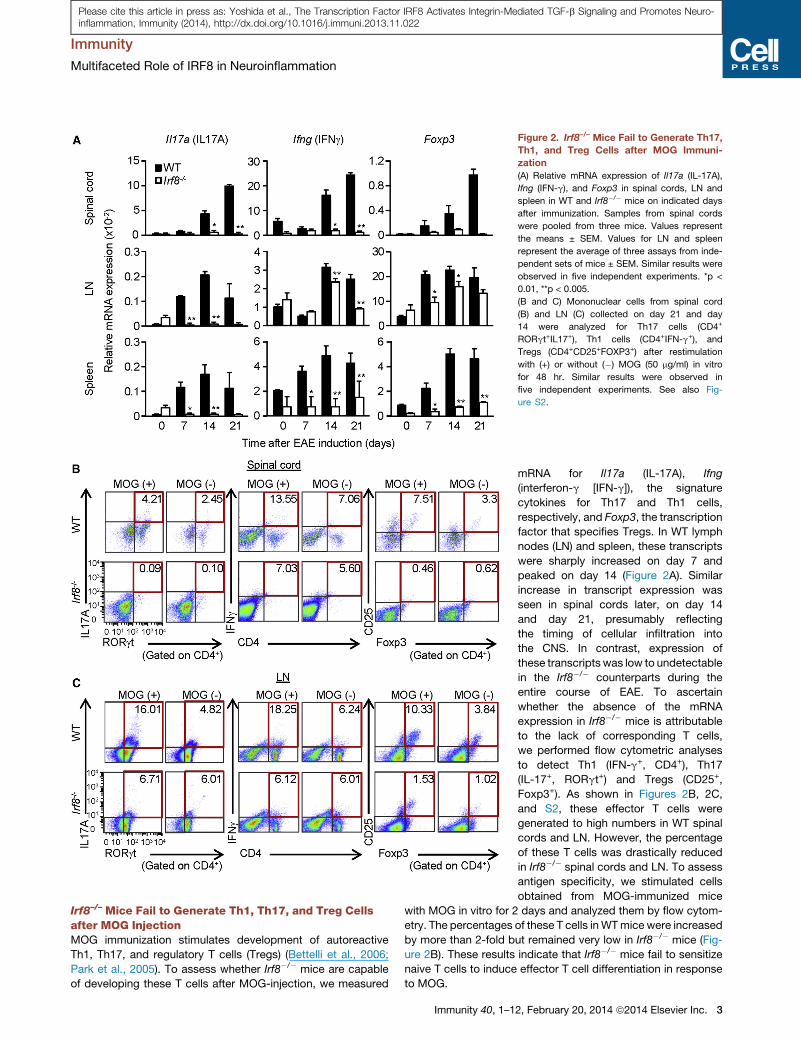
Figure 2. Irf8–/– Mice Fail to Generate Th17,
Th1, and Treg Cells after MOG Immuni-
zation
(A) Relative mRNA expression of Il17a (IL-17A),
Ifng (IFN-g), and Foxp3 in spinal cords, LN and
spleen in WT and Irf8�/� mice on indicated days
after immunization. Samples from spinal cords
were pooled from three mice. Values represent
the means ± SEM. Values for LN and spleen
represent the average of three assays from inde-
pendent sets of mice ± SEM. Similar results were
observed in five independent experiments. *p <
0.01, **p < 0.005.
(B and C) Mononuclear cells from spinal cord
(B) and LN (C) collected on day 21 and day
14 were analyzed for Th17 cells (CD4+
RORgt+IL17+), Th1 cells (CD4+IFN-g+), and
Tregs (CD4+CD25+FOXP3+) after restimulation
with (+) or without (�) MOG (50 mg/ml) in vitro
for 48 hr. Similar results were observed in
five independent experiments. See also Fig-
ure S2.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Irf8–/– Mice Fail to Generate Th1, Th17, and Treg Cellsafter MOG InjectionMOG immunization stimulates development of autoreactive
Th1, Th17, and regulatory T cells (Tregs) (Bettelli et al., 2006;
Park et al., 2005). To assess whether Irf8�/� mice are capable
of developing these T cells after MOG-injection, we measured
Immunity 40, 1–1
mRNA for Il17a (IL-17A), Ifng
(interferon-g [IFN-g]), the signature
cytokines for Th17 and Th1 cells,
respectively, and Foxp3, the transcription
factor that specifies Tregs. In WT lymph
nodes (LN) and spleen, these transcripts
were sharply increased on day 7 and
peaked on day 14 (Figure 2A). Similar
increase in transcript expression was
seen in spinal cords later, on day 14
and day 21, presumably reflecting
the timing of cellular infiltration into
the CNS. In contrast, expression of
these transcripts was low to undetectable
in the Irf8�/� counterparts during the
entire course of EAE. To ascertain
whether the absence of the mRNA
expression in Irf8�/� mice is attributable
to the lack of corresponding T cells,
we performed flow cytometric analyses
to detect Th1 (IFN-g+, CD4+), Th17
(IL-17+, RORgt+) and Tregs (CD25+,
Foxp3+). As shown in Figures 2B, 2C,
and S2, these effector T cells were
generated to high numbers in WT spinal
cords and LN. However, the percentage
of these T cells was drastically reduced
in Irf8�/� spinal cords and LN. To assess
antigen specificity, we stimulated cells
obtained from MOG-immunized mice
with MOG in vitro for 2 days and analyzed them by flow cytom-
etry. The percentages of these T cells inWTmice were increased
by more than 2-fold but remained very low in Irf8�/� mice (Fig-
ure 2B). These results indicate that Irf8�/� mice fail to sensitize
naive T cells to induce effector T cell differentiation in response
to MOG.
2, February 20, 2014 ª2014 Elsevier Inc. 3
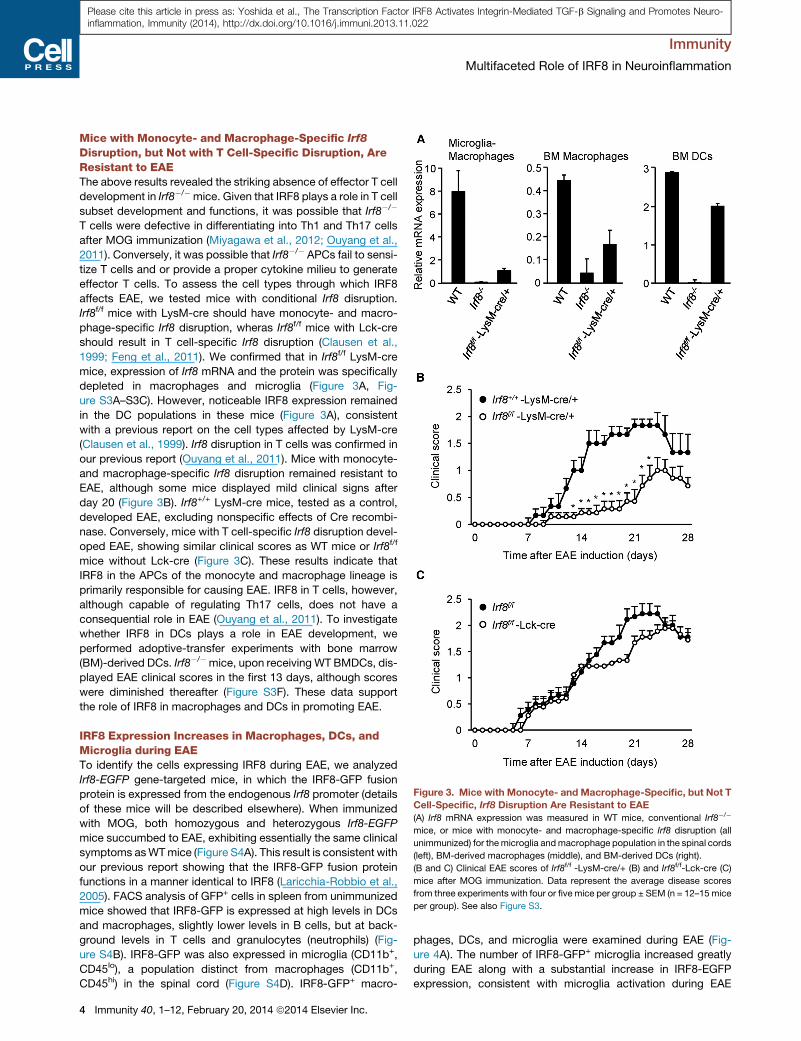
Figure 3. Mice with Monocyte- and Macrophage-Specific, but Not T
Cell-Specific, Irf8 Disruption Are Resistant to EAE
(A) Irf8 mRNA expression was measured in WT mice, conventional Irf8�/�
mice, or mice with monocyte- and macrophage-specific Irf8 disruption (all
unimmunized) for themicroglia andmacrophage population in the spinal cords
(left), BM-derived macrophages (middle), and BM-derived DCs (right).
(B and C) Clinical EAE scores of Irf8f/f -LysM-cre/+ (B) and Irf8f/f-Lck-cre (C)
mice after MOG immunization. Data represent the average disease scores
from three experiments with four or fivemice per group ± SEM (n = 12–15 mice
per group). See also Figure S3.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Mice with Monocyte- and Macrophage-Specific Irf8
Disruption, but Not with T Cell-Specific Disruption, AreResistant to EAEThe above results revealed the striking absence of effector T cell
development in Irf8�/�mice. Given that IRF8 plays a role in T cell
subset development and functions, it was possible that Irf8�/�
T cells were defective in differentiating into Th1 and Th17 cells
after MOG immunization (Miyagawa et al., 2012; Ouyang et al.,
2011). Conversely, it was possible that Irf8�/� APCs fail to sensi-
tize T cells and or provide a proper cytokine milieu to generate
effector T cells. To assess the cell types through which IRF8
affects EAE, we tested mice with conditional Irf8 disruption.
Irf8f/f mice with LysM-cre should have monocyte- and macro-
phage-specific Irf8 disruption, wheras Irf8f/f mice with Lck-cre
should result in T cell-specific Irf8 disruption (Clausen et al.,
1999; Feng et al., 2011). We confirmed that in Irf8f/f LysM-cre
mice, expression of Irf8 mRNA and the protein was specifically
depleted in macrophages and microglia (Figure 3A, Fig-
ure S3A–S3C). However, noticeable IRF8 expression remained
in the DC populations in these mice (Figure 3A), consistent
with a previous report on the cell types affected by LysM-cre
(Clausen et al., 1999). Irf8 disruption in T cells was confirmed in
our previous report (Ouyang et al., 2011). Mice with monocyte-
and macrophage-specific Irf8 disruption remained resistant to
EAE, although some mice displayed mild clinical signs after
day 20 (Figure 3B). Irf8+/+ LysM-cre mice, tested as a control,
developed EAE, excluding nonspecific effects of Cre recombi-
nase. Conversely, mice with T cell-specific Irf8 disruption devel-
oped EAE, showing similar clinical scores as WT mice or Irf8f/f
mice without Lck-cre (Figure 3C). These results indicate that
IRF8 in the APCs of the monocyte and macrophage lineage is
primarily responsible for causing EAE. IRF8 in T cells, however,
although capable of regulating Th17 cells, does not have a
consequential role in EAE (Ouyang et al., 2011). To investigate
whether IRF8 in DCs plays a role in EAE development, we
performed adoptive-transfer experiments with bone marrow
(BM)-derived DCs. Irf8�/�mice, upon receiving WT BMDCs, dis-
played EAE clinical scores in the first 13 days, although scores
were diminished thereafter (Figure S3F). These data support
the role of IRF8 in macrophages and DCs in promoting EAE.
IRF8 Expression Increases in Macrophages, DCs, andMicroglia during EAETo identify the cells expressing IRF8 during EAE, we analyzed
Irf8-EGFP gene-targeted mice, in which the IRF8-GFP fusion
protein is expressed from the endogenous Irf8 promoter (details
of these mice will be described elsewhere). When immunized
with MOG, both homozygous and heterozygous Irf8-EGFP
mice succumbed to EAE, exhibiting essentially the same clinical
symptoms asWTmice (Figure S4A). This result is consistent with
our previous report showing that the IRF8-GFP fusion protein
functions in a manner identical to IRF8 (Laricchia-Robbio et al.,
2005). FACS analysis of GFP+ cells in spleen from unimmunized
mice showed that IRF8-GFP is expressed at high levels in DCs
and macrophages, slightly lower levels in B cells, but at back-
ground levels in T cells and granulocytes (neutrophils) (Fig-
ure S4B). IRF8-GFP was also expressed in microglia (CD11b+,
CD45lo), a population distinct from macrophages (CD11b+,
CD45hi) in the spinal cord (Figure S4D). IRF8-GFP+ macro-
4 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
phages, DCs, and microglia were examined during EAE (Fig-
ure 4A). The number of IRF8-GFP+ microglia increased greatly
during EAE along with a substantial increase in IRF8-EGFP
expression, consistent with microglia activation during EAE
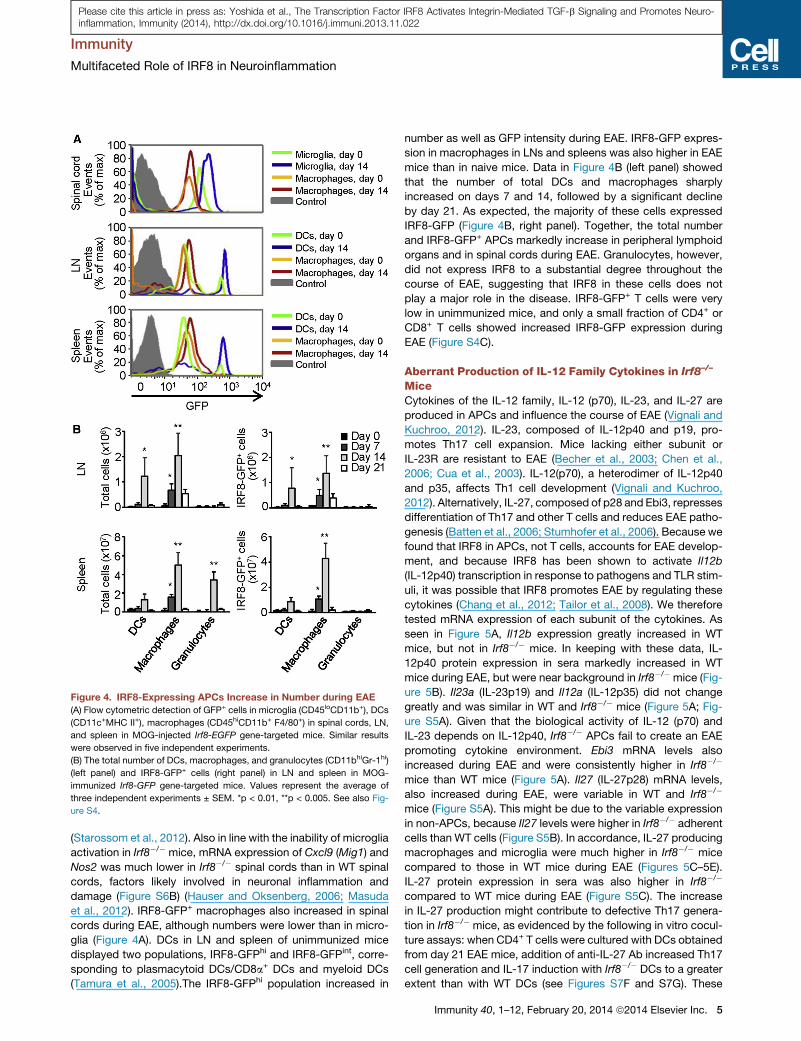
Figure 4. IRF8-Expressing APCs Increase in Number during EAE
(A) Flow cytometric detection of GFP+ cells in microglia (CD45loCD11b+), DCs
(CD11c+MHC II+), macrophages (CD45hiCD11b+ F4/80+) in spinal cords, LN,
and spleen in MOG-injected Irf8-EGFP gene-targeted mice. Similar results
were observed in five independent experiments.
(B) The total number of DCs, macrophages, and granulocytes (CD11bhiGr-1hi)
(left panel) and IRF8-GFP+ cells (right panel) in LN and spleen in MOG-
immunized Irf8-GFP gene-targeted mice. Values represent the average of
three independent experiments ± SEM. *p < 0.01, **p < 0.005. See also Fig-
ure S4.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
(Starossom et al., 2012). Also in line with the inability of microglia
activation in Irf8�/� mice, mRNA expression of Cxcl9 (Mig1) and
Nos2 was much lower in Irf8�/� spinal cords than in WT spinal
cords, factors likely involved in neuronal inflammation and
damage (Figure S6B) (Hauser and Oksenberg, 2006; Masuda
et al., 2012). IRF8-GFP+ macrophages also increased in spinal
cords during EAE, although numbers were lower than in micro-
glia (Figure 4A). DCs in LN and spleen of unimmunized mice
displayed two populations, IRF8-GFPhi and IRF8-GFPint, corre-
sponding to plasmacytoid DCs/CD8a+ DCs and myeloid DCs
(Tamura et al., 2005).The IRF8-GFPhi population increased in
number as well as GFP intensity during EAE. IRF8-GFP expres-
sion in macrophages in LNs and spleens was also higher in EAE
mice than in naive mice. Data in Figure 4B (left panel) showed
that the number of total DCs and macrophages sharply
increased on days 7 and 14, followed by a significant decline
by day 21. As expected, the majority of these cells expressed
IRF8-GFP (Figure 4B, right panel). Together, the total number
and IRF8-GFP+ APCs markedly increase in peripheral lymphoid
organs and in spinal cords during EAE. Granulocytes, however,
did not express IRF8 to a substantial degree throughout the
course of EAE, suggesting that IRF8 in these cells does not
play a major role in the disease. IRF8-GFP+ T cells were very
low in unimmunized mice, and only a small fraction of CD4+ or
CD8+ T cells showed increased IRF8-GFP expression during
EAE (Figure S4C).
Aberrant Production of IL-12 Family Cytokines in Irf8–/–
MiceCytokines of the IL-12 family, IL-12 (p70), IL-23, and IL-27 are
produced in APCs and influence the course of EAE (Vignali and
Kuchroo, 2012). IL-23, composed of IL-12p40 and p19, pro-
motes Th17 cell expansion. Mice lacking either subunit or
IL-23R are resistant to EAE (Becher et al., 2003; Chen et al.,
2006; Cua et al., 2003). IL-12(p70), a heterodimer of IL-12p40
and p35, affects Th1 cell development (Vignali and Kuchroo,
2012). Alternatively, IL-27, composed of p28 and Ebi3, represses
differentiation of Th17 and other T cells and reduces EAE patho-
genesis (Batten et al., 2006; Stumhofer et al., 2006). Because we
found that IRF8 in APCs, not T cells, accounts for EAE develop-
ment, and because IRF8 has been shown to activate Il12b
(IL-12p40) transcription in response to pathogens and TLR stim-
uli, it was possible that IRF8 promotes EAE by regulating these
cytokines (Chang et al., 2012; Tailor et al., 2008). We therefore
tested mRNA expression of each subunit of the cytokines. As
seen in Figure 5A, Il12b expression greatly increased in WT
mice, but not in Irf8�/� mice. In keeping with these data, IL-
12p40 protein expression in sera markedly increased in WT
mice during EAE, but were near background in Irf8�/� mice (Fig-
ure 5B). Il23a (IL-23p19) and Il12a (IL-12p35) did not change
greatly and was similar in WT and Irf8�/� mice (Figure 5A; Fig-
ure S5A). Given that the biological activity of IL-12 (p70) and
IL-23 depends on IL-12p40, Irf8�/� APCs fail to create an EAE
promoting cytokine environment. Ebi3 mRNA levels also
increased during EAE and were consistently higher in Irf8�/�
mice than WT mice (Figure 5A). Il27 (IL-27p28) mRNA levels,
also increased during EAE, were variable in WT and Irf8�/�
mice (Figure S5A). This might be due to the variable expression
in non-APCs, because Il27 levels were higher in Irf8�/� adherent
cells than WT cells (Figure S5B). In accordance, IL-27 producing
macrophages and microglia were much higher in Irf8�/� mice
compared to those in WT mice during EAE (Figures 5C–5E).
IL-27 protein expression in sera was also higher in Irf8�/�
compared to WT mice during EAE (Figure S5C). The increase
in IL-27 production might contribute to defective Th17 genera-
tion in Irf8�/� mice, as evidenced by the following in vitro cocul-
ture assays: when CD4+ T cells were cultured with DCs obtained
from day 21 EAE mice, addition of anti-IL-27 Ab increased Th17
cell generation and IL-17 induction with Irf8�/� DCs to a greater
extent than with WT DCs (see Figures S7F and S7G). These
Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 5
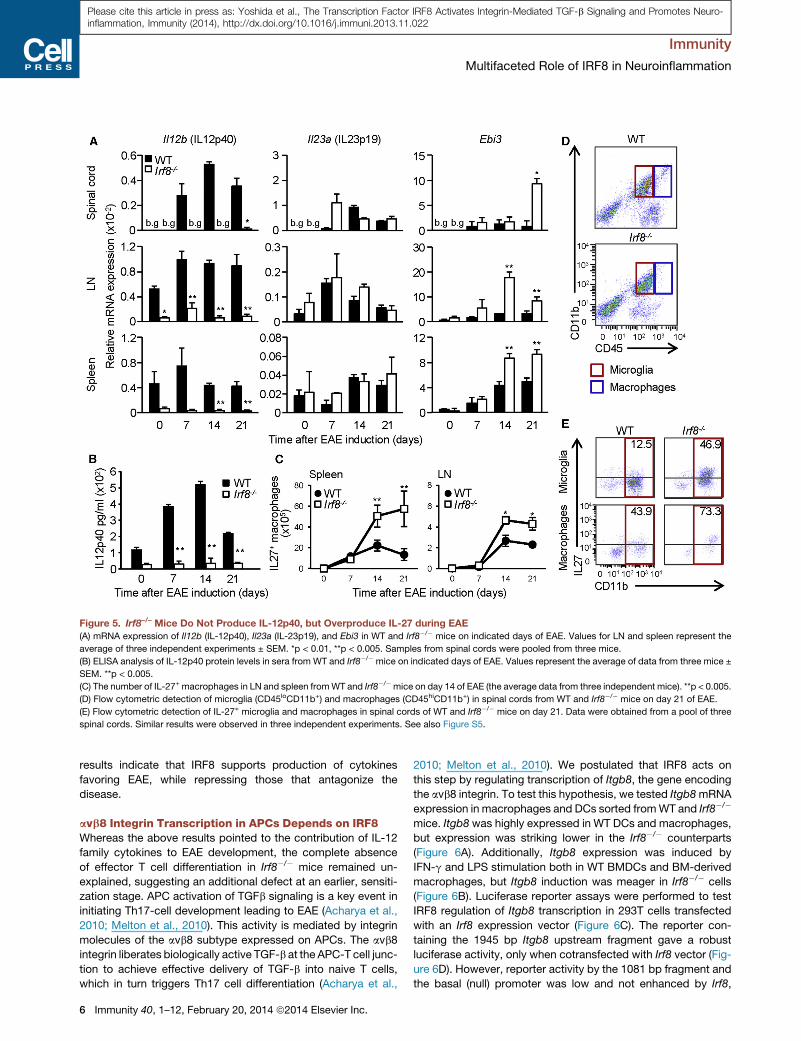
Figure 5. Irf8–/– Mice Do Not Produce IL-12p40, but Overproduce IL-27 during EAE
(A) mRNA expression of Il12b (IL-12p40), Il23a (IL-23p19), and Ebi3 in WT and Irf8�/� mice on indicated days of EAE. Values for LN and spleen represent the
average of three independent experiments ± SEM. *p < 0.01, **p < 0.005. Samples from spinal cords were pooled from three mice.
(B) ELISA analysis of IL-12p40 protein levels in sera from WT and Irf8�/� mice on indicated days of EAE. Values represent the average of data from three mice ±
SEM. **p < 0.005.
(C) The number of IL-27+ macrophages in LN and spleen fromWT and Irf8�/�mice on day 14 of EAE (the average data from three independent mice). **p < 0.005.
(D) Flow cytometric detection of microglia (CD45loCD11b+) and macrophages (CD45hiCD11b+) in spinal cords from WT and Irf8�/� mice on day 21 of EAE.
(E) Flow cytometric detection of IL-27+ microglia and macrophages in spinal cords of WT and Irf8�/� mice on day 21. Data were obtained from a pool of three
spinal cords. Similar results were observed in three independent experiments. See also Figure S5.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
results indicate that IRF8 supports production of cytokines
favoring EAE, while repressing those that antagonize the
disease.
avb8 Integrin Transcription in APCs Depends on IRF8Whereas the above results pointed to the contribution of IL-12
family cytokines to EAE development, the complete absence
of effector T cell differentiation in Irf8�/� mice remained un-
explained, suggesting an additional defect at an earlier, sensiti-
zation stage. APC activation of TGFb signaling is a key event in
initiating Th17-cell development leading to EAE (Acharya et al.,
2010; Melton et al., 2010). This activity is mediated by integrin
molecules of the avb8 subtype expressed on APCs. The avb8
integrin liberates biologically active TGF-b at the APC-T cell junc-
tion to achieve effective delivery of TGF-b into naive T cells,
which in turn triggers Th17 cell differentiation (Acharya et al.,
6 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
2010; Melton et al., 2010). We postulated that IRF8 acts on
this step by regulating transcription of Itgb8, the gene encoding
the avb8 integrin. To test this hypothesis, we tested Itgb8mRNA
expression in macrophages and DCs sorted fromWT and Irf8�/�
mice. Itgb8 was highly expressed in WT DCs and macrophages,
but expression was striking lower in the Irf8�/� counterparts
(Figure 6A). Additionally, Itgb8 expression was induced by
IFN-g and LPS stimulation both in WT BMDCs and BM-derived
macrophages, but Itgb8 induction was meager in Irf8�/� cells
(Figure 6B). Luciferase reporter assays were performed to test
IRF8 regulation of Itgb8 transcription in 293T cells transfected
with an Irf8 expression vector (Figure 6C). The reporter con-
taining the 1945 bp Itgb8 upstream fragment gave a robust
luciferase activity, only when cotransfected with Irf8 vector (Fig-
ure 6D). However, reporter activity by the 1081 bp fragment and
the basal (null) promoter was low and not enhanced by Irf8,
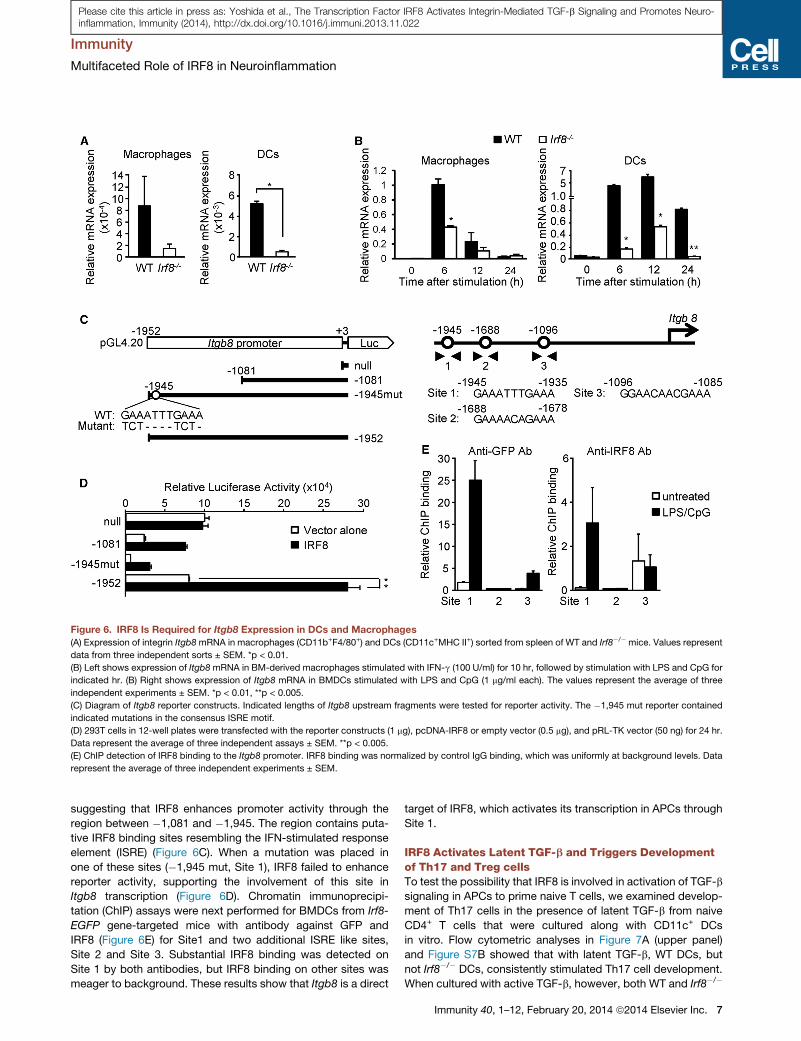
Figure 6. IRF8 Is Required for Itgb8 Expression in DCs and Macrophages
(A) Expression of integrin Itgb8mRNA in macrophages (CD11b+F4/80+) and DCs (CD11c+MHC II+) sorted from spleen of WT and Irf8�/� mice. Values represent
data from three independent sorts ± SEM. *p < 0.01.
(B) Left shows expression of Itgb8mRNA in BM-derived macrophages stimulated with IFN-g (100 U/ml) for 10 hr, followed by stimulation with LPS and CpG for
indicated hr. (B) Right shows expression of Itgb8 mRNA in BMDCs stimulated with LPS and CpG (1 mg/ml each). The values represent the average of three
independent experiments ± SEM. *p < 0.01, **p < 0.005.
(C) Diagram of Itgb8 reporter constructs. Indicated lengths of Itgb8 upstream fragments were tested for reporter activity. The �1,945 mut reporter contained
indicated mutations in the consensus ISRE motif.
(D) 293T cells in 12-well plates were transfected with the reporter constructs (1 mg), pcDNA-IRF8 or empty vector (0.5 mg), and pRL-TK vector (50 ng) for 24 hr.
Data represent the average of three independent assays ± SEM. **p < 0.005.
(E) ChIP detection of IRF8 binding to the Itgb8 promoter. IRF8 binding was normalized by control IgG binding, which was uniformly at background levels. Data
represent the average of three independent experiments ± SEM.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
suggesting that IRF8 enhances promoter activity through the
region between �1,081 and �1,945. The region contains puta-
tive IRF8 binding sites resembling the IFN-stimulated response
element (ISRE) (Figure 6C). When a mutation was placed in
one of these sites (�1,945 mut, Site 1), IRF8 failed to enhance
reporter activity, supporting the involvement of this site in
Itgb8 transcription (Figure 6D). Chromatin immunoprecipi-
tation (ChIP) assays were next performed for BMDCs from Irf8-
EGFP gene-targeted mice with antibody against GFP and
IRF8 (Figure 6E) for Site1 and two additional ISRE like sites,
Site 2 and Site 3. Substantial IRF8 binding was detected on
Site 1 by both antibodies, but IRF8 binding on other sites was
meager to background. These results show that Itgb8 is a direct
target of IRF8, which activates its transcription in APCs through
Site 1.
IRF8 Activates Latent TGF-b and Triggers Developmentof Th17 and Treg cellsTo test the possibility that IRF8 is involved in activation of TGF-b
signaling in APCs to prime naive T cells, we examined develop-
ment of Th17 cells in the presence of latent TGF-b from naive
CD4+ T cells that were cultured along with CD11c+ DCs
in vitro. Flow cytometric analyses in Figure 7A (upper panel)
and Figure S7B showed that with latent TGF-b, WT DCs, but
not Irf8�/� DCs, consistently stimulated Th17 cell development.
When cultured with active TGF-b, however, both WT and Irf8�/�
Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 7
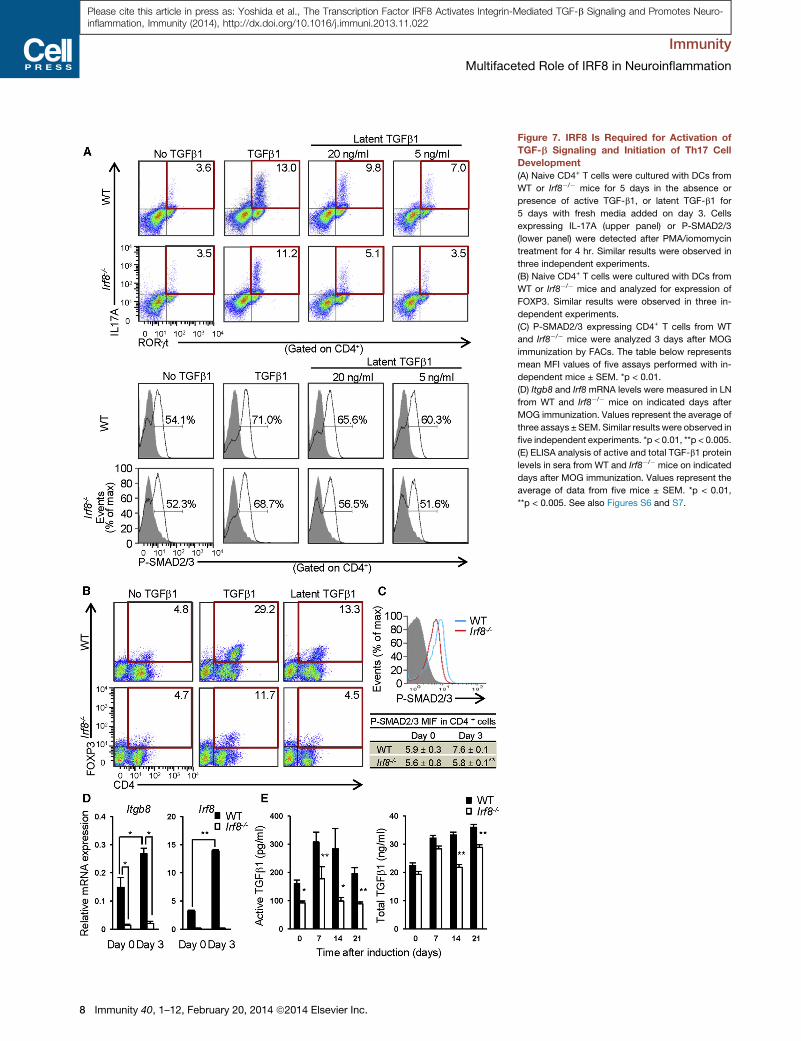
Figure 7. IRF8 Is Required for Activation of
TGF-b Signaling and Initiation of Th17 Cell
Development
(A) Naive CD4+ T cells were cultured with DCs from
WT or Irf8�/� mice for 5 days in the absence or
presence of active TGF-b1, or latent TGF-b1 for
5 days with fresh media added on day 3. Cells
expressing IL-17A (upper panel) or P-SMAD2/3
(lower panel) were detected after PMA/iomomycin
treatment for 4 hr. Similar results were observed in
three independent experiments.
(B) Naive CD4+ T cells were cultured with DCs from
WT or Irf8�/� mice and analyzed for expression of
FOXP3. Similar results were observed in three in-
dependent experiments.
(C) P-SMAD2/3 expressing CD4+ T cells from WT
and Irf8�/� mice were analyzed 3 days after MOG
immunization by FACs. The table below represents
mean MFI values of five assays performed with in-
dependent mice ± SEM. *p < 0.01.
(D) Itgb8 and Irf8 mRNA levels were measured in LN
from WT and Irf8�/� mice on indicated days after
MOG immunization. Values represent the average of
three assays ± SEM. Similar results were observed in
five independent experiments. *p < 0.01, **p < 0.005.
(E) ELISA analysis of active and total TGF-b1 protein
levels in sera from WT and Irf8�/� mice on indicated
days after MOG immunization. Values represent the
average of data from five mice ± SEM. *p < 0.01,
**p < 0.005. See also Figures S6 and S7.
Immunity
Multifaceted Role of IRF8 in Neuroinflammation
8 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022

Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
DCs supported Th17 cell generation, indicating defective TGF-b
activation in Irf8�/�DCs. As expected, no Th17 cells were gener-
ated without TGF-b or without DCs (Figure 7A; Figure S7A).
Consequently, the amount of IL-17 secreted in supernatants
was much lower when cultured with Irf8�/� DCs and latent
TGF-b, but comparable when cultured with active TGF-b
(Figure S7D). Moreover, Irf8�/� DCs did not stimulate Tregs
generation when cultured with latent TGF-b, while WT DCs did,
further supporting the idea that deficient TGF-b signaling
accounts for the priming defects in Irf8�/� DCs (Figure 7B). To
verify the role of IRF8 in TGF-b signaling, we tested SMAD
activation in APCs: among SMAD family of transcription factors
activated by TGF-b signaling, SMAD2 is shown to be critical for
Th17 cell activation (Malhotra et al., 2010). As shown in Figure 7A
(lower panel) and Figure S7C, WT DCs, but not Irf8�/� DCs led to
enhanced SMAD2 phosphorylation in T cells when incubated
with latent TGF-b. When incubated with active TGF-b, both WT
and Irf8�/� DCs enhanced SMAD2 phosphorylation. It is of
note that LPS induction of Il6 and Tnfa was comparable in WT
DCs and Irf8�/� DCs, verifying specificity of TGF-b signal
deficiency in Irf8�/� DCs (Figure S7E). Extending the above
in vitro observations to in vivo, we found that SMAD2 phosphor-
ylation was increased in CD4+ T cells upon EAE induction in WT
mice, but not Irf8�/�mice (Figure 7C). Moreover, Itgb8 and Irf8
transcript expression was enhanced in WT DCs during EAE,
but negligible in Irf8�/�DCs (Figure 7D).We also show that active
TGF-b protein levels in sera were lower in Irf8�/� mice than WT
mice during EAE, whereas total TGF-b protein levels varied,
although they tended to be lower in Irf8�/� mice (Figure 7E). In
addition, expression of Tgfb and Il6 mRNA was similar in WT
and Irf8�/� mice during EAE, indicating selective inability of
Irf8�/� mice in producing biologically active TGF-b (Figure S6A).
These results provide compelling evidence that IRF8 is essential
for TGF-b activation in APCs and hence sensitization of naive
CD4+ T to differentiate into Th17 cells and Tregs.
DISCUSSION
We show that Irf8�/� mice are resistant to EAE: Th17, Th1, and
Treg cells were not generated in Irf8�/� mice upon MOG
immunization. IRF8 in APCs, but not in T cells, promoted EAE
pathogenesis in a multifaceted manner. We show several key
mechanisms by which IRF8 promotes EAE, which likely con-
stitutes a basis of the MS risk factor (De Jager et al., 2009; Dis-
anto et al., 2012; Beecham et al., 2013).
IRF8 was required for TGF-b signaling that takes place at the
interface between APCs and naive T cells. The bulk of secreted
TGF-b is biologically inactive, as it is bound to the latency asso-
ciated protein (LAP). However, the avb8 integrin molecule on the
APC cell surface dissociates LAP to liberate functional TGF-b,
allowing direct delivery of TGF-b signal to T cells (Acharya
et al., 2010; Melton et al., 2010; Henderson and Sheppard,
2013). This step, coupled with TCR engagement drives T cells
to differentiate into Th17 cells (Acharya et al., 2010; Melton
et al., 2010). Our data show that IRF8 plays a role in this priming
step by enhancing Itgb8 transcription in APCs. The conclusion
that integrin-mediated TGF-b signaling depends on IRF8 is sup-
ported by the following observations: (1) Itgb8 transcript levels
were markedly lower in Irf8�/� APCs than WT cells, (2) IRF8
occupied the Itgb8 promoter and stimulated Itgb8 promoter
activity, (3) Irf8�/� DCs did not activate latent TGF-b and did
not initiate SMAD-based TGF-b signaling in T cells in vivo and
in vitro, and (4) Irf8�/� DCs failed to generate Th17 and Treg cells
in the presence of latent TGF-b. Consistent with these defects,
active TGF-b levels were lower in Irf8�/� mice during EAE.
Because Tregs are reported to promote Th17 cell development
under some conditions, defective Treg generation in Irf8�/�
mice might further contribute to the absence of EAE (Chen
et al., 2006; Pandiyan et al., 2011). Together, in view of the noted
requirement of TGF-b signaling for the EAE and for MS, IRF8 is
likely to exert a decisive influence on the onset of the disease
(Veldhoen et al., 2006).
Not only Th17 cells, but Th1 cells facilitate pathogenesis of
EAE and MS (Axtell et al., 2010; Kroenke et al., 2008; Lovett-
Racke et al., 2011). Th1 cells prompt macrophage infiltration
and NOS2 induction, whereas Th17 cells attract neutrophils
and stimulate GM-CSF production (Kroenke et al., 2008).
Although Th1 cell development might or might not depend on
TGF-b signaling, initial triggering of Th1 differentiation is likely
to be compromised in Irf8�/� mice as a result of the reduced
expression of MHC II and costimulatory molecules on Irf8�/�
APCs (Figure S6C).
Our results show that in addition to priming, IRF8 exerts a
major influence on subsequent stages of the disease, by facili-
tating expansion andmaintenance of Th17 and Th1 cells through
the regulation of IL-12 family cytokines. IL-12(p70) is critical for
development of Th1 cells in EAE, whereas IL-23 is necessary
for maintenance of Th17 cells (Becher et al., 2003; Cua et al.,
2003; Kroenke et al., 2008). IL-23 likely contributes not only to
EAE but to MS, because it is produced by microglia of MS
patients (Li et al., 2007). Due to the absence of the shared
IL-12p40 subunit, Irf8�/� mice are defective in producing both
IL-12p70 and IL-23, which would create an adverse environment
for the survival of Th1 and Th17 cells. Interestingly, Irf8�/� APCs
expressed greater levels of IL-27 thanWT cells, the cytokine that
negatively regulates development of Th17 and other T cells
(Stumhofer et al., 2006). Expression of Ebi3 mRNA was sig-
nificantly increased in Irf8�/� APCs relative to WT cells during
EAE. Irf8�/� mice had greater number of IL-27p28+ macro-
phages and microglia than WT mice. Together, IRF8 appears
to facilitate pro-Th1/Th17 cytokines and inhibits the anti-Th17
cytokine, generating a condition favoring inflammation and
exacerbation of EAE. The increase in IL-27 expression in Irf8�/�
mice was somewhat unexpected, considering the previous
report that IRF8 enhances IL-27p28 transcription in macro-
phages after TLR stimulation in vitro, while not affecting Ebi3
expression (Zhang et al., 2010). The different results obtained
in the two studies might be due to the differing cytokine environ-
ments in which IL-27 expression was examined, one in vitro after
TLR stimulation, the other in vivo after MOG immunization,
because it was previously found that IRF8 can act as a transcrip-
tional activator or a repressor in macrophages, depending on
stimulation (Chang et al., 2012). In addition, binding partners
for IRF8, such as IRF1, might also affect target gene regulation
(Horiuchi et al., 2012). In this respect, it is interesting to note
that Irf1�/� mice are partially resistant to EAE, suggesting the
possibility that IRF1 might cooperate with IRF8 to exacerbate
EAE (Tada et al., 1997).
Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 9

Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Microglia, the CNS-resident APCs, are activated in EAE and
play a role in disease progression (Starossom et al., 2012).
Analysis of Irf8-EGFP knockin mice found that the number of
IRF8-GFP+ microglia and levels of IRF8-GFP increased during
EAE. Although microglia are present in the CNS of Irf8�/� mice,
they express reduced levels of Iba1 and other microglia markers
and are deficient in IL-12p40 induction in vitro (Horiuchi et al.,
2012; Minten et al., 2012). Moreover, IRF8 is shown to be im-
portant for transforming quiescent microglia to a reactive
phenotype that produces proinflammatory cytokines after
peripheral nerve injury (Masuda et al., 2012). In agreement, our
results show that IRF8 plays a key role in microglial activation
during EAE by stimulating IL-12p40 expression, which in turn
increases intra-CNS amplification of Th1 and Th17 cells. We
also noted that Irf8�/� microglia produce more IL-27 than WT
mice. Thus, the ability to activate microglia might be another
attribute of IRF8 that boosts EAE disease progression. IRF8
might reinforce disease by enhancing expression of Ripk2 in
CNS-infiltrating DCs (Shaw et al., 2011). In light of the recent
report that IRF8 promotes Ly6C+ monocyte differentiation
(Kurotaki et al., 2013), which supports proinflammatory M1-
type macrophages, IRF8 appears to have a strong propensity
to steer APCs toward proinflammatory pathways and advance
EAE progression.
Lastly, it should be noted that IRF8 SNPs linked to the MS
susceptibility map to the 30 noncoding region of the IRF8 gene,
away from the transcription end sites (De Jager et al., 2009;
Disanto et al., 2012). This suggests that the SNPs are in a down-
stream regulatory sequence(s) yet to be fully characterized,
which influences IRF8 transcription in APCs. It will be of interest
to elucidate how this region affects IRF8 transcription and
whether drugs currently used or proposed for MS treatment
affect IRF8 expression.
In conclusion, IRF8, acting in APCs, activates TGF-b signaling
and primes naive T cells to initiate EAE. During the course of
disease, IRF8 fortifies inflammatory pathways to exacerbate
EAE pathogenesis. Together, this work highlights the multiplicity
of mechanisms by which IRF8 functions as a risk factor for MS.
EXPERIMENTAL PROCEDURES
Mice and EAE
Irf8�/�, Irf8-EGFP, and Irf8f/f mice of the C57BL/6 background and WT mice
were maintained in the NICHD animal facility (Feng et al., 2011) (H.W., data
not shown). LysM-cre (Lyz2tm1(cre)Ifo) and Lck-cre (Tg Lck-cre) mice (Jackson
Laboratories) were crossed with Irf8f/f mice to generate Irf8f/f-LysM-cre and
Irf8f/f Lck-cre mice (Clausen et al., 1999; Jakubzick et al., 2008). All animal
experiments were performed according to the animal study (ASP# 11-044)
approved by the Animal Care and Use Committees of NICHD, NIH. Female
mice (8–12 weeks old) were injected with 200 mg MOG35–55 in CFA containing
400 mg of killedM. tuberculosis subcutaneously at two sites and intraperitone-
ally with 100 ng of pertussis toxin, followed by injection of 100 ng pertussis
toxin 24 hr later (Hooke Laboratories). EAE symptoms were scored daily
according to the EAE scoring system.
Magnetic Resonance Imaging
MRI experiments were performed on a 7-T (Bruker Avance), 21 cm horizontal
scanner. A contiguous set of 1 mm thick, T1 weighted images (field-of-view
[FOV] = 25.6 3 12.8 mm, in-plane resolution of 100 mm, TE/TR = 6/100 ms),
encompassing the region between the Th9-L5 vertebrae (15 slices), were
acquired before and 5 min after intravenous administration of Gd-DTPA
(Berlex Laboratories). Quantitative T2 images (TE/TR = 20/2500 ms, in-plane
10 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
resolution of 200 mm, number of echoes = 16) were acquired with the same
number of slices and slice locations as the T1-weighted images. T2 data
were analyzed with software by MATLAB (Mathworks Inc). The resultant T2maps were used to evaluate T2 values for four regions of interests (area
0.016 cm2) per slice.
Cell Preparations, Culture, and Flow Cytometry
Microglia-macrophage populations were prepared from spinal cord single-cell
suspensions by a 37%/70% percoll density gradient centrifugation, followed
by incubation on tissue culture plates for 1 hr. CD11c+ DCs were isolated
from draining LN and spleen (90%–95%) as described Acharya et al., 2010;
Melton et al., 2010). Naive CD4+ T cells were isolated from LN of WT mice
with biotin-conjugated anti-CD8a, CD11b, CD11c, CD19, CD45R, NK1.1,
Gr-1 Abs (Biolegend), and streptavidin microbeads (Miltenyi Biotec). For
Th17 development, 2.5 3 105 T cells were cultured with 105 DCs for 5 days
with anti-IL4 (10 mg/ml, Bio X cell), anti-IL-12 (10 mg/ml, Bio X Cell), anti-
IFN-g (10 mg/ml, Bio X Cell), anti-CD3 (1 mg/ml, eBiosciences), IL-6
(10 ng/ml, Pepro Tec), IL-1b (10 ng/ml, Pepro Tec), and TGF-b1 (5 ng/ml,
R&D Systems) or latent TGF-b1 (5 or 20 ng/ml, R&D Systems), supplemented
with freshmedia on day 3. For Treg development, cells were cultured with anti-
IL-4 (10 mg/ml, Bio X cell), anti-IL12 (10 mg/ml, Bio X Cell), anti-IFN-g (10 mg/ml,
Bio X Cell), anti-CD3 (1 mg/ml, eBiosciences), anti-CD28 (3 mg/ml,
eBiosciences), IL-2 (100 U/ml, Pepro Tec), and TGF-b1 (5 ng/ml, R&D
Systems) or latent TGF-b1 (5 or 20 ng/ml, R&D Systems). Transcripts were
measured by quantitative RT-PCR and normalized by Hprt as described
(Chang et al., 2012). The concentrations of IL-12p40, IL-27, and TGF-b1 in
sera from MOG-immunized mice were measured by ELISA using commercial
kits (eBioscience and R&D Systems). Flow cytometry cells were blocked with
anti-mouse FcgR Ab (CD16/32, clone 2.4G2), stained with Abs below, and
analyzed on FACSCalibur (BD Biosciences). Data were analyzed by FlowJo
software. The following antibodies were used: PerCP-conjugated anti-CD4,
allophycocyanin-conjugated anti-CD25 Abs (BD Biosciences), allophycocya-
nin-conjugated anti-F4/80, PE-conjugated anti-CD11b and PerCP-conjugated
anti-CD45 Abs, BD Biosciences), PE-conjugated anti-IL17, FITC-conjugated
anti-IFN-g (BD Biosciences) and FITC-conjugated anti-IL27 Abs (R&D
Systems) with Cytyofix/Cytoperm kits (BD Biosciences), or allophycocyanin-
conjugated anti-RORgt, PE-conjugated anti-FOXP3 Abs (eBiosciences), and
PE-conjugated anti-Phospho SMAD3/2 Abs (BD Biosciences). To purify DCs
and macrophages, we treated spleen cell suspensions with the Fc receptor
blocker 2.4G2 mAb followed by staining with antibodies against Gr-1,
CD11c, MHC II, CD11b, and F4/80 and sorted with a FACSAria-sorter (BD
Biosciences). Dead cells were excluded by gating on 7AAD� cells. DCs
were sorted as MHC II+CD11c+ and macrophages as CD11b+F4/80+. Purity
of the sorted populations was greater than 95%.
Itgb8 Promoter and ChIP Assays
Itgb8 upstream fragments (�1,952 to +3, �1,081 to +3) were amplified from
mouse genomic DNA with Phusion high-fidelity DNA polymerase (New
England Biolab) with the forward primer containing KpnI, 50-CGCAGGTAC
CAGGAAGAAATTTGAAAAACC-30,or 50-CGCAGGTACCATCTGACAGTAGAA
TACATC-30 and the reverse primer containing BamHI, 50-TGTGGGATCCTA
GAAGGCCAAGG-3. The amplicons were inserted into the KpnI-BglII site of
pGL4.20 luciferase reporter (Promega). 293T cells were transfected with the
reporter, pcDNA-IRF8, and pRL-TK for 24 hr, and luciferase activity was
measured with Dual-Glo Luciferase System (Promega). Chromatin from 106
BMDCs was immunoprecipitated with 0.5 to 1 mg of normal immunoglobulin
G (IgG), affinity purified anti-IRF8 antibody, or anti-GFP antibody bound to
Dynabeads (Chang et al., 2012). The following primers were used for site 1:
50-GTGGCTCTAACCATATCTTC-30 (forward), and 50-TGAAGCTAAGGGGG
CAATCC-30 (reverse), site 2; 50-CAAATGATCCTCCTTAACAG-30 (forward),
and 50-GTAGGAGGTCCCACCAACCC-30 (reverse), site 3; 50-ATCTGCA
ACTTCTTGTTTCC-30 (forward), and 50-CAGACTTTGGAGATTTAGAC-30
(reverse). Input DNA (2%) was used for normalization.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this
article online at http://dx.doi.org/10.1016/j.immuni.2013.11.022.

Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
ACKNOWLEDGMENTS
We thank Alan Koretsky (NINDS) for his support and critical reading of the
manuscript, Takayuki Ito and Makoto Horiuchi (UC, Davis), Ulrich Siebenlist
(NIAID) for useful advice on experiments and reagents, and Mehrnoosh
Abshari for assistance in cell sorting and in vitro experiments. This work was
supported by the Intramural Program of NICHD, NIAID, and NINDS, National
Institutes of Health.
Received: March 14, 2013
Accepted: November 25, 2013
Published: January 30, 2014
REFERENCES
Acharya, M., Mukhopadhyay, S., Paıdassi, H., Jamil, T., Chow, C., Kissler, S.,
Stuart, L.M., Hynes, R.O., and Lacy-Hulbert, A. (2010). av Integrin expression
by DCs is required for Th17 cell differentiation and development of experi-
mental autoimmune encephalomyelitis in mice. J. Clin. Invest. 120, 4445–
4452.
Axtell, R.C., de Jong, B.A., Boniface, K., van der Voort, L.F., Bhat, R., De
Sarno, P., Naves, R., Han,M., Zhong, F., Castellanos, J.G., et al. (2010). T help-
er type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis
and experimental encephalomyelitis. Nat. Med. 16, 406–412.
Batten, M., Li, J., Yi, S., Kljavin, N.M., Danilenko, D.M., Lucas, S., Lee, J., de
Sauvage, F.J., and Ghilardi, N. (2006). Interleukin 27 limits autoimmune
encephalomyelitis by suppressing the development of interleukin 17-produc-
ing T cells. Nat. Immunol. 7, 929–936.
Becher, B., Durell, B.G., and Noelle, R.J. (2003). IL-23 produced by CNS-resi-
dent cells controls T cell encephalitogenicity during the effector phase of
experimental autoimmune encephalomyelitis. J. Clin. Invest. 112, 1186–1191.
Beecham, A.H., Patsopoulos, N.A., Xifara, D.K., Davis, M.F., Kemppinen, A.,
Cotsapas, C., Shah, T.S., Spencer, C., Booth, D., Goris, A., et al.;
International Multiple Sclerosis Genetics Consortium (IMSGC); Wellcome
Trust Case Control Consortium 2 (WTCCC2); International IBD Genetics
Consortium (IIBDGC) (2013). Analysis of immune-related loci identifies 48
new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360.
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T.B., Oukka, M., Weiner, H.L.,
and Kuchroo, V.K. (2006). Reciprocal developmental pathways for the gener-
ation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238.
Chang, T.H., Xu, S., Tailor, P., Kanno, T., and Ozato, K. (2012). The small ubiq-
uitin-like modifier-deconjugating enzyme sentrin-specific peptidase 1
switches IFN regulatory factor 8 from a repressor to an activator during macro-
phage activation. J. Immunol. 189, 3548–3556.
Chen, Y., Langrish, C.L., McKenzie, B., Joyce-Shaikh, B., Stumhofer, J.S.,
McClanahan, T., Blumenschein, W., Churakovsa, T., Low, J., Presta, L.,
et al. (2006). Anti-IL-23 therapy inhibits multiple inflammatory pathways and
ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 116, 1317–1326.
Clausen, B.E., Burkhardt, C., Reith, W., Renkawitz, R., and Forster, I. (1999).
Conditional gene targeting in macrophages and granulocytes using LysMcre
mice. Transgenic Res. 8, 265–277.
Cua, D.J., Sherlock, J., Chen, Y., Murphy, C.A., Joyce, B., Seymour, B.,
Lucian, L., To, W., Kwan, S., Churakova, T., et al. (2003). Interleukin-23 rather
than interleukin-12 is the critical cytokine for autoimmune inflammation of the
brain. Nature 421, 744–748.
De Jager, P.L., Jia, X., Wang, J., de Bakker, P.I.W., Ottoboni, L., Aggarwal,
N.T., Piccio, L., Raychaudhuri, S., Tran, D., Aubin, C., et al.; International MS
Genetics Consortium (2009). Meta-analysis of genome scans and replication
identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility
loci. Nat. Genet. 41, 776–782.
Disanto, G., Sandve, G.K., Berlanga-Taylor, A.J., Morahan, J.M., Dobson, R.,
Giovannoni, G., and Ramagopalan, S.V. (2012). Genomic regions associated
with multiple sclerosis are active in B cells. PLoS ONE 7, e32281.
Feng, J., Wang, H., Shin, D.M., Masiuk, M., Qi, C.F., and Morse, H.C., 3rd.
(2011). IFN regulatory factor 8 restricts the size of the marginal zone and follic-
ular B cell pools. J. Immunol. 186, 1458–1466.
Hambleton, S., Salem, S., Bustamante, J., Bigley, V., Boisson-Dupuis, S.,
Azevedo, J., Fortin, A., Haniffa, M., Ceron-Gutierrez, L., Bacon, C.M., et al.
(2011). IRF8 Mutations and Human Dendritic-Cell Immunodeficiency. N Engl
J Med.
Hauser, S.L., and Oksenberg, J.R. (2006). The neurobiology of multiple scle-
rosis: genes, inflammation, and neurodegeneration. Neuron 52, 61–76.
Henderson, N.C., and Sheppard, D. (2013). Integrin-mediated regulation of
TGFb in fibrosis. Biochim. Biophys. Acta 1832, 891–896.
Horiuchi, M., Wakayama, K., Itoh, A., Kawai, K., Pleasure, D., Ozato, K., and
Itoh, T. (2012). Interferon regulatory factor 8/interferon consensus sequence
binding protein is a critical transcription factor for the physiological phenotype
of microglia. J. Neuroinflammation 9, 227.
Ivanov, I.I., McKenzie, B.S., Zhou, L., Tadokoro, C.E., Lepelley, A., Lafaille,
J.J., Cua, D.J., and Littman, D.R. (2006). The orphan nuclear receptor
RORgammat directs the differentiation program of proinflammatory IL-17+ T
helper cells. Cell 126, 1121–1133.
Jakubzick, C., Bogunovic, M., Bonito, A.J., Kuan, E.L., Merad, M., and
Randolph, G.J. (2008). Lymph-migrating, tissue-derived dendritic cells are
minor constituents within steady-state lymph nodes. J. Exp. Med. 205,
2839–2850.
Komiyama, Y., Nakae, S., Matsuki, T., Nambu, A., Ishigame, H., Kakuta, S.,
Sudo, K., and Iwakura, Y. (2006). IL-17 plays an important role in the develop-
ment of experimental autoimmune encephalomyelitis. J. Immunol. 177,
566–573.
Kroenke, M.A., Carlson, T.J., Andjelkovic, A.V., and Segal, B.M. (2008). IL-12-
and IL-23-modulated T cells induce distinct types of EAE based on histology,
CNS chemokine profile, and response to cytokine inhibition. J. Exp. Med. 205,
1535–1541.
Kurotaki, D., Osato, N., Nishiyama, A., Yamamoto, M., Ban, T., Sato, H.,
Nakabayashi, J., Umehara, M., Miyake, N., Matsumoto, N., et al. (2013).
Essential role of the IRF8-KLF4 transcription factor cascade in murine mono-
cyte differentiation. Blood 121, 1839–1849.
Laricchia-Robbio, L., Tamura, T., Karpova, T., Sprague, B.L., McNally, J.G.,
and Ozato, K. (2005). Partner-regulated interaction of IFN regulatory factor 8
with chromatin visualized in live macrophages. Proc. Natl. Acad. Sci. USA
102, 14368–14373.
Li, Y., Chu, N., Hu, A., Gran, B., Rostami, A., and Zhang, G.-X. (2007).
Increased IL-23p19 expression in multiple sclerosis lesions and its induction
in microglia. Brain 130, 490–501.
Lovett-Racke, A.E., Yang, Y., and Racke, M.K. (2011). Th1 versus Th17: are
T cell cytokines relevant in multiple sclerosis? Biochim. Biophys. Acta 1812,
246–251.
Malhotra, N., Robertson, E., and Kang, J. (2010). SMAD2 is essential for TGF
beta-mediated Th17 cell generation. J. Biol. Chem. 285, 29044–29048.
Masuda, T., Tsuda, M., Yoshinaga, R., Tozaki-Saitoh, H., Ozato, K., Tamura,
T., and Inoue, K. (2012). IRF8 is a critical transcription factor for transforming
microglia into a reactive phenotype. Cell Rep 1, 334–340.
Melton, A.C., Bailey-Bucktrout, S.L., Travis, M.A., Fife, B.T., Bluestone, J.A.,
and Sheppard, D. (2010). Expression of avb8 integrin on dendritic cells regu-
lates Th17 cell development and experimental autoimmune encephalomyelitis
in mice. J. Clin. Invest. 120, 4436–4444.
Minten, C., Terry, R., Deffrasnes, C., King, N.J., and Campbell, I.L. (2012). IFN
regulatory factor 8 is a key constitutive determinant of the morphological and
molecular properties of microglia in the CNS. PLoS ONE 7, e49851.
Miyagawa, F., Zhang, H., Terunuma, A., Ozato, K., Tagaya, Y., and Katz, S.I.
(2012). Interferon regulatory factor 8 integrates T-cell receptor and cytokine-
signaling pathways and drives effector differentiation of CD8 T cells. Proc.
Natl. Acad. Sci. USA 109, 12123–12128.
Ouyang, X., Zhang, R., Yang, J., Li, Q., Qin, L., Zhu, C., Liu, J., Ning, H., Shin,
M.S., Gupta, M., et al. (2011). Transcription factor IRF8 directs a silencing
programme for TH17 cell differentiation. Nat Commun 2, 314.
Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc. 11

Immunity
Multifaceted Role of IRF8 in Neuroinflammation
Please cite this article in press as: Yoshida et al., The Transcription Factor IRF8 Activates Integrin-Mediated TGF-b Signaling and Promotes Neuro-inflammation, Immunity (2014), http://dx.doi.org/10.1016/j.immuni.2013.11.022
Pandiyan, P., Conti, H.R., Zheng, L., Peterson, A.C., Mathern, D.R.,
Hernandez-Santos, N., Edgerton, M., Gaffen, S.L., and Lenardo, M.J. (2011).
CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and
enhance host resistance in mouseCandida albicans Th17 cell infection model.
Immunity 34, 422–434.
Park, H., Li, Z., Yang, X.O., Chang, S.H., Nurieva, R., Wang, Y.H., Wang, Y.,
Hood, L., Zhu, Z., Tian, Q., and Dong, C. (2005). A distinct lineage of CD4
T cells regulates tissue inflammation by producing interleukin 17. Nat.
Immunol. 6, 1133–1141.
Schellenberg, A.E., Buist, R., Yong, V.W., Del Bigio, M.R., and Peeling, J.
(2007). Magnetic resonance imaging of blood-spinal cord barrier disruption
in mice with experimental autoimmune encephalomyelitis. Magn. Reson.
Med. 58, 298–305.
Shaw, P.J., Barr, M.J., Lukens, J.R., McGargill, M.A., Chi, H., Mak, T.W., and
Kanneganti, T.D. (2011). Signaling via the RIP2 adaptor protein in central
nervous system-infiltrating dendritic cells promotes inflammation and autoim-
munity. Immunity 34, 75–84.
Sørensen, T.L., Tani, M., Jensen, J., Pierce, V., Lucchinetti, C., Folcik, V.A.,
Qin, S., Rottman, J., Sellebjerg, F., Strieter, R.M., et al. (1999). Expression of
specific chemokines and chemokine receptors in the central nervous system
of multiple sclerosis patients. J. Clin. Invest. 103, 807–815.
Starossom, S.C., Mascanfroni, I.D., Imitola, J., Cao, L., Raddassi, K.,
Hernandez, S.F., Bassil, R., Croci, D.O., Cerliani, J.P., Delacour, D., et al.
(2012). Galectin-1 deactivates classically activated microglia and protects
from inflammation-induced neurodegeneration. Immunity 37, 249–263.
Steinman, L., and Zamvil, S.S. (2006). How to successfully apply animal
studies in experimental allergic encephalomyelitis to research onmultiple scle-
rosis. Ann. Neurol. 60, 12–21.
Stromnes, I.M., Cerretti, L.M., Liggitt, D., Harris, R.A., and Goverman, J.M.
(2008). Differential regulation of central nervous system autoimmunity by
T(H)1 and T(H)17 cells. Nat. Med. 14, 337–342.
Stumhofer, J.S., Laurence, A., Wilson, E.H., Huang, E., Tato, C.M., Johnson,
L.M., Villarino, A.V., Huang, Q., Yoshimura, A., Sehy, D., et al. (2006).
Interleukin 27 negatively regulates the development of interleukin 17-produc-
12 Immunity 40, 1–12, February 20, 2014 ª2014 Elsevier Inc.
ing T helper cells during chronic inflammation of the central nervous system.
Nat. Immunol. 7, 937–945.
Tada, Y., Ho, A., Matsuyama, T., and Mak, T.W. (1997). Reduced incidence
and severity of antigen-induced autoimmune diseases in mice lacking inter-
feron regulatory factor-1. J. Exp. Med. 185, 231–238.
Tailor, P., Tamura, T., Morse, H.C., 3rd, and Ozato, K. (2008). The BXH2
mutation in IRF8 differentially impairs dendritic cell subset development in
the mouse. Blood 111, 1942–1945.
Tamura, T., Tailor, P., Yamaoka, K., Kong, H.J., Tsujimura, H., O’Shea, J.J.,
Singh, H., and Ozato, K. (2005). IFN regulatory factor-4 and -8 govern dendritic
cell subset development and their functional diversity. J. Immunol. 174, 2573–
2581.
Travis, M.A., Reizis, B., Melton, A.C., Masteller, E., Tang, Q., Proctor, J.M.,
Wang, Y., Bernstein, X., Huang, X., Reichardt, L.F., et al. (2007). Loss of integ-
rin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice.
Nature 449, 361–365.
Veldhoen, M., Hocking, R.J., Flavell, R.A., and Stockinger, B. (2006). Signals
mediated by transforming growth factor-beta initiate autoimmune encephalo-
myelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol.
7, 1151–1156.
Vignali, D.A., and Kuchroo, V.K. (2012). IL-12 family cytokines: immunological
playmakers. Nat. Immunol. 13, 722–728.
Weaver, C.T., Hatton, R.D., Mangan, P.R., and Harrington, L.E. (2007). IL-17
family cytokines and the expanding diversity of effector T cell lineages.
Annu. Rev. Immunol. 25, 821–852.
Yang, X.O., Pappu, B.P., Nurieva, R., Akimzhanov, A., Kang, H.S., Chung, Y.,
Ma, L., Shah, B., Panopoulos, A.D., Schluns, K.S., et al. (2008). T helper 17
lineage differentiation is programmed by orphan nuclear receptors ROR alpha
and ROR gamma. Immunity 28, 29–39.
Zhang, J., Qian, X., Ning, H., Yang, J., Xiong, H., and Liu, J. (2010). Activation of
IL-27 p28 gene transcription by interferon regulatory factor 8 in cooperation
with interferon regulatory factor 1. J. Biol. Chem. 285, 21269–21281.
Related Documents