The three-dimensional structure of an eukaryotic glutamine synthetase: Functional implications of its oligomeric structure Oscar Llorca c , Marco Betti b , Jose ´ M. Gonza ´lez a , Alfonso Valencia a , Antonio J. Ma ´rquez b , Jose ´ M. Valpuesta a, * a Centro Nacional de Biotecnologı ´a, C.S.I.C. Campus de la Universidad Auto ´ noma de Madrid, Darwin 3, 28049 Madrid, Spain b Departamento de Bioquı ´mica Vegetal y Biologı ´a Molecular, Facultad de Quı ´mica, Universidad de Sevilla, Spain c Centro de Investigaciones Biolo ´ gicas, C.S.I.C. Ramiro de Maeztu 9, 28040 Madrid, Spain Received 28 March 2006; received in revised form 5 June 2006; accepted 8 June 2006 Available online 21 June 2006 Abstract The structure of the prokaryotic glutamine synthetases type I (GS-I), key enzymes in nitrogen metabolism, was determined several years ago by X-ray diffraction, and consists of a double hexameric ring. The structure of the eukaryotic GS from the plant Phaseolus vulgaris (Glutamine synthetase type II; GS-II) has now been determined at low-resolution using electron microscopy and image process- ing, and consists of an octamer composed of two tetramers placed back-to-back and rotated 90° with respect to each other. The olig- omeric structure possesses a twofold symmetry, very suggestive of each tetramer being composed of two dimers. This is reinforced by the fact that dimers are isolated as a stable albeit non-functional species during the purification procedure. Given the fact that the active site of all types of GS is formed by highly conserved residues located in the interface of two interacting monomers, the geometry of the reconstructed tetramer suggests that it only contains two functional active sites, i.e., an active site per dimer. This is supported by bio- chemical data, which reveal that while the octamer binds eight ATP molecules, it only binds four molecules of the transition state ana- logue and GS inhibitor methionine-(S)-sulfoximine-P (MetSox-P). All this suggests for the GS-II enzyme an oligomeric structure containing four active sites and four possible regulatory sites, which might point to a complex regulatory behavior. Ó 2006 Elsevier Inc. All rights reserved. Keywords: Glutamine synthetase; Electron microscopy; Oligomerization; Domain swapping 1. Introduction Glutamine synthetase (GS; EC 6.3.1.2) 1 is one of the key enzymes in nitrogen metabolism since it catalyses the synthesis of glutamine from glutamate according to the following reaction: glutamate + NH 4 + + ATP fi gluta- mine + ADP + P i +H + (for a more comprehensive review see Stadtman and Ginsburg, 1974). GS is an ubiquitous enzyme found in all organisms through three different types of proteins: GS-I, which is found mostly in prokaryotes; GS-II, which is located mostly in eukaryotes, and GS-III, which is also found in prokaryotes. GS-I is by far the best characterized of all GS types. The structure of GS-I from several organisms has been determined at atomic resolution (Almassy et al., 1986; Gill and Eisenberg, 2001; Gill et al., 2002) and it has been found to be a dodecamer built up by two back-to-back hexameric rings. The active site of GS-I, whose residues are conserved in all types of GS, is located between adjacent, intra-ring monomers (Almassy et al., 1986) so that the oligomer possesses 12 active sites contain- ing each one two metal ions (Mg 2+ or Mn 2+ ) that are crucial to the enzymatic activity. Each monomer, with an average length of 470 residues (Fig. 1), is divided in 1047-8477/$ - see front matter Ó 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.jsb.2006.06.003 * Corresponding author. Fax: +34 915854506. E-mail address: [email protected] (J.M. Valpuesta). 1 Abbreviations used: MetSox, L-methionine-(S)-sulfoximine; MetSox-P, phosphorylated L-methionine-(S)-sulfoximine; GS II, type II glutamine synthetase; GS I, type I glutamine synthetase; IPTG, isopropyl-b-D- thiogalactopyranoside. www.elsevier.com/locate/yjsbi Journal of Structural Biology 156 (2006) 469–479 Journal of Structural Biology

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of
www.elsevier.com/locate/yjsbi
Journal of Structural Biology 156 (2006) 469–479
StructuralBiology
The three-dimensional structure of an eukaryotic glutaminesynthetase: Functional implications of its oligomeric structure
Oscar Llorca c, Marco Betti b, Jose M. Gonzalez a, Alfonso Valencia a,Antonio J. Marquez b, Jose M. Valpuesta a,*
a Centro Nacional de Biotecnologıa, C.S.I.C. Campus de la Universidad Autonoma de Madrid, Darwin 3, 28049 Madrid, Spainb Departamento de Bioquımica Vegetal y Biologıa Molecular, Facultad de Quımica, Universidad de Sevilla, Spain
c Centro de Investigaciones Biologicas, C.S.I.C. Ramiro de Maeztu 9, 28040 Madrid, Spain
Received 28 March 2006; received in revised form 5 June 2006; accepted 8 June 2006Available online 21 June 2006
Abstract
The structure of the prokaryotic glutamine synthetases type I (GS-I), key enzymes in nitrogen metabolism, was determined severalyears ago by X-ray diffraction, and consists of a double hexameric ring. The structure of the eukaryotic GS from the plant Phaseolus
vulgaris (Glutamine synthetase type II; GS-II) has now been determined at low-resolution using electron microscopy and image process-ing, and consists of an octamer composed of two tetramers placed back-to-back and rotated 90� with respect to each other. The olig-omeric structure possesses a twofold symmetry, very suggestive of each tetramer being composed of two dimers. This is reinforced bythe fact that dimers are isolated as a stable albeit non-functional species during the purification procedure. Given the fact that the activesite of all types of GS is formed by highly conserved residues located in the interface of two interacting monomers, the geometry of thereconstructed tetramer suggests that it only contains two functional active sites, i.e., an active site per dimer. This is supported by bio-chemical data, which reveal that while the octamer binds eight ATP molecules, it only binds four molecules of the transition state ana-logue and GS inhibitor methionine-(S)-sulfoximine-P (MetSox-P). All this suggests for the GS-II enzyme an oligomeric structurecontaining four active sites and four possible regulatory sites, which might point to a complex regulatory behavior.� 2006 Elsevier Inc. All rights reserved.
Keywords: Glutamine synthetase; Electron microscopy; Oligomerization; Domain swapping
1. Introduction
Glutamine synthetase (GS; EC 6.3.1.2)1 is one of the keyenzymes in nitrogen metabolism since it catalyses thesynthesis of glutamine from glutamate according to thefollowing reaction: glutamate + NH4
+ + ATP fi gluta-mine + ADP + Pi + H+ (for a more comprehensive reviewsee Stadtman and Ginsburg, 1974). GS is an ubiquitous
1047-8477/$ - see front matter � 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.jsb.2006.06.003
* Corresponding author. Fax: +34 915854506.E-mail address: [email protected] (J.M. Valpuesta).
1 Abbreviations used: MetSox, L-methionine-(S)-sulfoximine; MetSox-P,phosphorylated L-methionine-(S)-sulfoximine; GS II, type II glutaminesynthetase; GS I, type I glutamine synthetase; IPTG, isopropyl-b-D-thiogalactopyranoside.
enzyme found in all organisms through three different typesof proteins: GS-I, which is found mostly in prokaryotes;GS-II, which is located mostly in eukaryotes, and GS-III,which is also found in prokaryotes. GS-I is by far the bestcharacterized of all GS types. The structure of GS-I fromseveral organisms has been determined at atomic resolution(Almassy et al., 1986; Gill and Eisenberg, 2001; Gill et al.,2002) and it has been found to be a dodecamer built up bytwo back-to-back hexameric rings. The active site of GS-I,whose residues are conserved in all types of GS, is locatedbetween adjacent, intra-ring monomers (Almassy et al.,1986) so that the oligomer possesses 12 active sites contain-ing each one two metal ions (Mg2+ or Mn2+) that arecrucial to the enzymatic activity. Each monomer, with anaverage length of �470 residues (Fig. 1), is divided in

Fig. 1. Sequence comparison and secondary structure prediction of GS-II from P. vulgaris. Alignment of Salmonella typhimurium GS-I (GLNA_SALTY)and Phaseolus vulgaris GS-II (GLNA2_PHAVU) sequences. Residues are coloured according to conservation: identities in black background andsimilarities in grey background. The secondary structure of S. typhimurium GS-I was determined with DSSP (Kabsch and Sander, 1983) from the PDB file1f1h, while secondary structure of P. vulgaris GS-II was predicted with PSIPRED (McGuffin et al., 2000). Helices are depicted as green zigzag lines, andstrands as red arrows. Residues forming part of the active site are marked with an asterisk. The regions in the S. typhimurium GS-I sequence involved ininter-ring contacts are marked with a blue line (see Fig. 6A for a different visualization of these two domains). Residues of the N-terminal domain are inbold letters (Almassy et al., 1986).
470 O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479
two domains, each contributing to the active site of adja-cent monomers. The smaller, N-terminal domain, containsmostly a sheet made by six antiparallel b-strands of whichtwo form part of the active site, whereas the larger C-termi-nal domain, which is mainly a-helical, contains sixb-strands which hold most of the residues building theactive site. The dodecamer is maintained mainly by hydro-phobic interactions between the two hexameric rings.
The GS-II type, a smaller protein than GS-I (�370residues average length; Fig. 1), has been less studiedthan its prokaryotic counterpart both in functional andstructural terms. Nevertheless, the oligomeric state ofGS-II has been the subject of study for more than twodecades and several models have been generated, basedon electron microscopy and biochemical studies, whichpoint to GS-II as an octamer made by two superimposedrings formed in turn by four identically placed subunits(McParland et al., 1976; Pushkin et al., 1981, 1985;Tsuprun et al., 1987; Boksha et al., 2002). Some of thesestudies suggest a shift between the two tetrameric ringsof 40� (Pushkin et al., 1981, 1985; Tsuprun et al.,1987). More recent electron microscopy results point toGS-II as a double heptamer (Kiang, 2001). In this work,we have determined the three-dimensional reconstructionof GS-II from the plant Phaseolus vulgaris using electronmicroscopy and image processing. Our structural results,which are confirmed by biochemical and biophysicaldata, reveal that GS-II is an octamer built by two tetra-mers which are placed back-to-back and rotated 90� withrespect to each other. The basic symmetry found for thethree-dimensional structure (c2) and some biochemicaldata strongly suggest that the tetramers are formed bythe interaction of two preformed dimers so that thereare only two active sites per tetramer. The functionalimplications of this finding are discussed.
2. Materials and methods
2.1. Overexpression and purification of GS-II fromP. vulgaris
Phaseolus vulgaris Glna cDNA was obtained by PCRamplification using the plasmid pcGS-18a (Clemente andMarquez, 1999) as template. The primers used added twonew restriction sites: SphI at the 5 0end (5 0GCGCGCATGCATCAACCTCAACCTC-3 0) and SalI at the 3 0end (5 0-GCGCGTCGACTCATGGTTTCCAGAG-3 0) to allow fur-ther cloning. The resulting 1.070 bp PCR product was clonedinto the pCR-Script Amp SK(+) plasmid (Stratagene). Theinsert was then isolated by digestion with SphI and SalIand cloned into SphI–SalI cutted pQE30 plasmid (Qiagen).The recombinant plasmid so obtained (pQE30GSa,4.53 kb in size) contained the GSa cDNA fused in-framewith a N-terminal 6-histidine tag. The coding region ofpQE30GSa was sequenced in order to exclude mutationalevents that may have occurred during the cloning process.The fusion construct was transformed into Escherichia coli
strain SG13009. The recombinant cells were cultured at37 �C overnight, after which the expression of recombinantGS was induced by adding IPTG 0.5 mM. Cells were furthercultured at 20 �C for 72 h and subsequently pelleted.
Pellets of E. coli cells were resuspended in 20 ml extrac-tion buffer (10 mM Tris–HCl, pH 7.5, containing 10 mMMgCl2; 0.05% Triton X-100; PMSF 100 lg ml�1; leupeptin2 lg ml�1; pepstatin A 2 lg ml�1) per gram of fresh weight.The cells were broken by sonication, and the correspondinghomogenate was clarified by centrifugation for 30 min at4 �C and 27 000g and diluted with an equal volume of(10 mM Tris–HCl, pH 7.5, containing 10 mM MgCl2;0.05% Triton X-100; 0.6 M NaCl; 20% glycerol; 20 mMimidazol–HCl, pH 8.0; PMSF 100 lg ml�1; leupeptin

O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479 471
2 lg ml�1; pepstatin A 2 lg ml�1). The crude extract wasloaded onto a column packed with 6 ml of Ni–NTA aga-rose (Qiagen), and the column was washed first with40 ml of 10 mM TrisÆHCl pH 7.5 containing 10 mM MgCl2and 10 mM imidazolÆHCl pH 8.0, followed by a secondwash with 40 ml of 10 mM TrisÆHCl pH 7.5 containing10 mM MgCl2 and 20 mM imidazolÆHCl pH 8.0. Recombi-nant GS was eluted with 10 mM TrisÆHCl, pH 7.5, contain-ing 10 mM MgCl2 and 200 mM imidazolÆHCl, pH 8.0, andthe fractions with best GS specific activity were pooled andconcentrated with a Millipore Ultrafree-4 centrifugal filterunit with a molecular weight cut-off of 10 kDa. All purifi-cation procedures were carried out at 0–4 �C. Homogeneitypurification of GS was confirmed by SDS–PAGE andimmunoblot analysis (not shown).
2.2. Enzymatic analysis
The GS protein concentration was determined using theBradford microassay (Bio-Rad). GS biosynthetic andtransferase activities were assayed as described (Montaniniet al., 2003). The functional active sites of GS-II from P.vulgaris were titrated with MetSox-P by incubating theenzyme in the presence of MetSox, [32c-P]ATP and anexcess of cold ATP. 0.5 mg of purified GS were incubatedfor 1 h at 30 �C in 100 mM TrisÆHCl, pH 7.50, containing100 mM MgCl2; 1 mM ATP; 10 mM MetSox; and from30 · 106 to 50 · 106 total dpm of [32c-P]ATP (3000 Ci/mmol, 10 mCi ml�1, supplied by Amersham–Biosciences).The formation of MetSox-P was confirmed by the irrevers-ible inactivation of about 95% of the total GS activity.Excess labelled nucleotides were removed with 2–3 desalt-ing steps on Sephadex G25. The protein-containing frac-tions were assayed for total radioactivity on a BeckmannLS 6000IC liquid scintillation counter and total proteinusing the Bio-Rad protein assay (that was preferred toOD280 as the formation of MetSox-P-ADP in the GS IIactive site caused a relevant change of the proteinspectrum).
The transferase GS activity was stained in situ in the gelaccording to the following protocol: after the electrophore-sis the gel was immersed for 30 min at 37 �C in 75 ml of asolution containing 6.75 mmol L-glutamine, 9 mmolMOPS, pH 7.0, 0.18 mmol MnCl2, 9 mmol NH2OHÆHCl,4.5 mmol NaOH, 3.75 lmol ADP and 3.75 mmolNa2HASO4. After the incubation, the solution containingthe substrates was discarded and c-glutamylhydroxamaterevealed by incubating the gel for 10 min at room temper-ature in 100 ml of 0.22% (w/v) HCl containing 10 mmol ofFeCl3Æ6H2O and 6 mmol of trichloroacetic acid. After abrief wash with deionized water, the gel was stained withCoomassie blue.
2.3. Hydrodynamic analysis
Separation of Phaseolus GS-II oligomers was carriedout by gel-filtration chromatography in a column of
Sephacryl S-300 HR (1.6 · 70 cm; Amersham–Biosciences).Purified protein samples from metal-affinity chromatogra-phy were concentrated to a final volume of 2 ml using aUltrafree-4 column (10 kDa molecular mass cut-off) andloaded onto the column. The chromatography was carriedout at a flow rate of 10 ml h�1 with 10 mM TrisÆHCl, pH7.50, containing 10 mM MgCl2 and 0.1 M KCl. The frac-tions were assayed for GS transferase activity and OD280.
Analytical ultracentrifugation was carried out using aBeckman Optima XL-A ultracentrifuge. Sedimentation datawere analyzed using the SVEDBERG program (Philo,1997). The sedimentation coefficients obtained were correct-ed for the temperature and the buffer composition using theprogram SEDNTERP (Laue et al., 1992) to calculate thesedimentation coefficient at 20 �C and water (s20,w).
2.4. Electron microscopy and image processing
For EM visualization, 5 ll-aliquots of GS-II wereapplied to glow-discharged carbon grids for 1 min and thenstained for 1 min with 2% uranyl acetate. Images wererecorded at 0�-tilt in a JEOL 1200EX-II electron micro-scope operated at 100 KV and recorded at 60000· nominalmagnification under low dose conditions.
Micrographs were digitized in a Zeiss SCAI scannerwith a sampling window corresponding to 3.5 A/pixel.The three-dimensional reconstruction of GS-II complexwas generated from 5578 negatively stained, randomly ori-ented particles, using the EMAN package for single-parti-cle three-dimensional reconstruction (Ludtke et al., 1999).The initial volume was generated by the common lines pro-cedure included in the EMAN package, using the averageclasses obtained after multivariate statistical analysis. Dur-ing the initial set of refinements, no symmetry was imposedto the reconstruction procedure. However, once it was evi-dent the existence of a twofold symmetry in the structure, ac2 symmetry was imposed in the subsequent refinements.Symmetrization was applied to the volumes generated aftereach round of refinement. Two possible twofold axis ofsymmetry were scrutinized (along each of the two viewsobserved in the EM fields) and it was chosen the one thatpreserved the structural features of the non-symmetrizedreconstruction. To further verify the correctness of thesymmetrization, the raw particles were confronted withstructures symmetrized along the two possible axis usingthe ‘‘multirefine’’ command in EMAN, and the greatmajority of particles were classified within the chosen sym-metry. In the last step of the refinement, the whole data setwas sorted into 250 representative classes containingbetween 5 and 30 images per class depending on the abun-dance of images of the corresponding view. Only someselected projections of the final volume and their corre-sponding averages are shown in Fig. 4. The final resolutionwas estimated to be 25 A with the 0.5 criterion for the Fou-rier shell correlation coefficient between two independentreconstructions. Visualization of the volumes was carriedout using AMIRA (http://amira.zeb.de). Volumes were
472 O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479
rendered with the handedness that better matched theatomic model (see below).
2.5. Modeling of GS-II
The homology model of a GS-II isozyme alpha mono-mer from P. vulgaris (UniProt Accession No. P04771)was built by homology modeling based on the structureof Salmonella typhimurium GS-I (PDB accession number1f1h) as a template. A pairwise target–template alignmentwas extracted from a multiple sequence alignment of repre-sentative GS-I and GS-II sequences with T-COFFEE(Notredame et al., 2000). The secondary structure of GS-II sequence, predicted with PSIPRED (Jones, 1999), wasused to refine the sequence–structure alignment, whichserved as input for the modeling by the MODELLER6v2 program (Sali and Blundell, 1993) with default param-eters. The quality of the model was assessed with What-Check (Hooft et al., 1996) and Prosa II (Sippl, 1993).
To build a model of the GS-II dimer which conservedthe geometry of the GS-I active site, two single copies ofthe homology model of the GS-II monomer were fitted intoa GS-I intra-ring dimeric structure (pdb 1f1h, chains A andB) with Swiss PDB Viewer (Guex and Peitsch, 1997).
Finally, the homology model thus generated was manu-ally fitted into the three-dimensional reconstruction of theGS-II from P. vulgaris.
3. Results
3.1. Biochemical characterization of GS-II
The fractions containing pure recombinant homopoly-meric GSa from the plant Phaseolus vulgaris (GS-II from
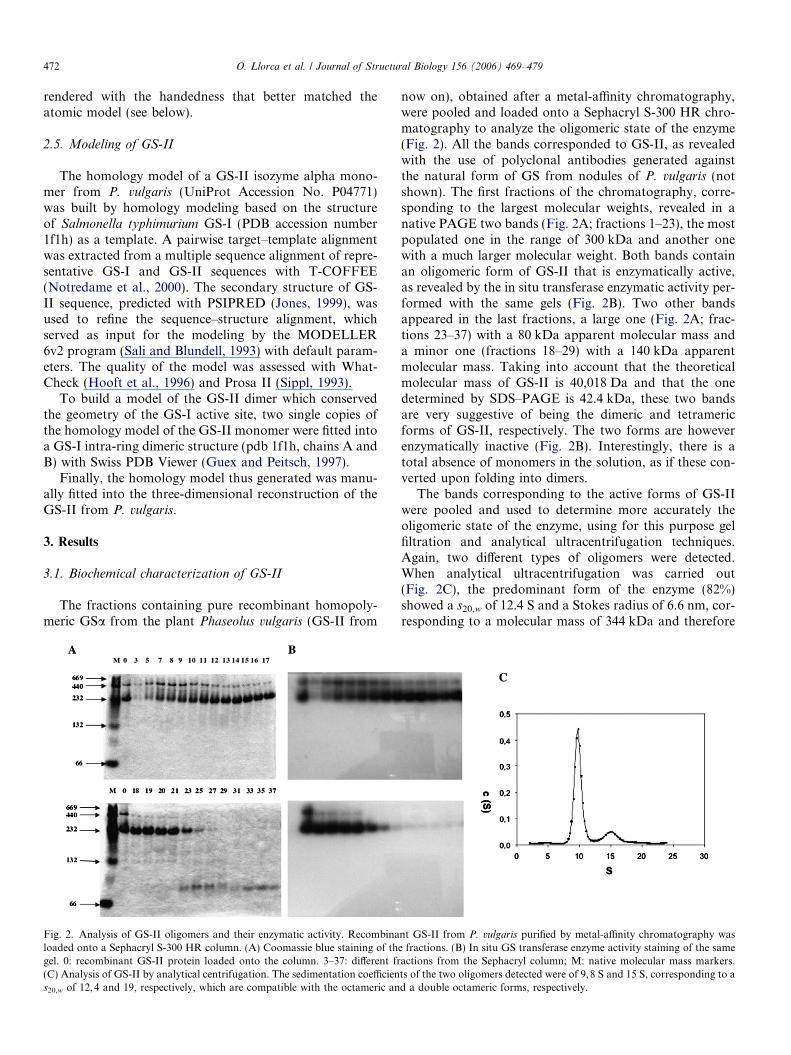
Fig. 2. Analysis of GS-II oligomers and their enzymatic activity. Recombinaloaded onto a Sephacryl S-300 HR column. (A) Coomassie blue staining of thgel. 0: recombinant GS-II protein loaded onto the column. 3–37: different fr(C) Analysis of GS-II by analytical centrifugation. The sedimentation coefficiens20,w of 12,4 and 19, respectively, which are compatible with the octameric an
now on), obtained after a metal-affinity chromatography,were pooled and loaded onto a Sephacryl S-300 HR chro-matography to analyze the oligomeric state of the enzyme(Fig. 2). All the bands corresponded to GS-II, as revealedwith the use of polyclonal antibodies generated againstthe natural form of GS from nodules of P. vulgaris (notshown). The first fractions of the chromatography, corre-sponding to the largest molecular weights, revealed in anative PAGE two bands (Fig. 2A; fractions 1–23), the mostpopulated one in the range of 300 kDa and another onewith a much larger molecular weight. Both bands containan oligomeric form of GS-II that is enzymatically active,as revealed by the in situ transferase enzymatic activity per-formed with the same gels (Fig. 2B). Two other bandsappeared in the last fractions, a large one (Fig. 2A; frac-tions 23–37) with a 80 kDa apparent molecular mass anda minor one (fractions 18–29) with a 140 kDa apparentmolecular mass. Taking into account that the theoreticalmolecular mass of GS-II is 40,018 Da and that the onedetermined by SDS–PAGE is 42.4 kDa, these two bandsare very suggestive of being the dimeric and tetramericforms of GS-II, respectively. The two forms are howeverenzymatically inactive (Fig. 2B). Interestingly, there is atotal absence of monomers in the solution, as if these con-verted upon folding into dimers.
The bands corresponding to the active forms of GS-IIwere pooled and used to determine more accurately theoligomeric state of the enzyme, using for this purpose gelfiltration and analytical ultracentrifugation techniques.Again, two different types of oligomers were detected.When analytical ultracentrifugation was carried out(Fig. 2C), the predominant form of the enzyme (82%)showed a s20,w of 12.4 S and a Stokes radius of 6.6 nm, cor-responding to a molecular mass of 344 kDa and therefore
nt GS-II from P. vulgaris purified by metal-affinity chromatography wase fractions. (B) In situ GS transferase enzyme activity staining of the sameactions from the Sephacryl column; M: native molecular mass markers.ts of the two oligomers detected were of 9,8 S and 15 S, corresponding to ad a double octameric forms, respectively.

Table 1Main physicochemical parameters of purified type II GS from P. vulgaris
Sedimentation coefficient at 10.5 �C in buffer 9.8Sedimentation coefficient at 20 �C in water (s20,w) 12.4Stokes’ radius (nm) 6.64Subunit Mr
Theoretic 40 018From SDS–PAGE 42 415
Native Mra 3 44 257
Calculated subunit numberFor subunit Mr = 40 018 (theoretic) 8.6For subunit Mr = 42 415 (SDS) 8.1
Transferase activity (U mg�1) 243MetSox-P bound per octamerb 4.25 ± 1.30b
a Determined from s20,w and Stokes radius according to the Siegel andMonty equation (Siegel and Monty, 1966). The Stokes radius wasdetermined by gel filtration and sedimentation coefficients by analyticalultracentrifugation.
b Mean ± SD of three independent experiments. Other details in Section 2.
Fig. 3. Electron microscopy of negatively stained GS-II from P. vulgaris.(A) An electron micrograph field of GS-II. Arrowheads point to the mostpopulated views (side views). Arrows point to the less populated views(end-on views). Bar indicates 100 nm. (B) A gallery of GS-II particlesselected for three-dimensional reconstruction.
O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479 473
to an octamer (Table 1), as it has been previously describedfor other GS-II enzymes (Eisenberg et al., 2000). A smallproportion (18%) of the recombinant GS-II enzyme waspresent as a higher molecular weight form with a s20,w of19 S and a Stokes radius of 9.7 nm, corresponding to amolecular mass of 772 kDa, compatible with a double octa-mer. In situ GS enzymatic activity staining in non-denatur-ing gels revealed again that both types of oligomersobtained were catalytically active (Fig. 2B). In summary,our results point to the generation of a functional octamerthrough the formation of transient dimeric and tetramericforms. A proportion of a hexadecameric form exists, prob-ably through the formation of a double octamer.
3.2. Three-dimensional structure of GS-II
Although several electron microscopy studies have beenperformed with GS-II enzymes (Pushkin et al., 1981, 1985;Tsuprun et al., 1987; Boksha et al., 2002), which in somecases have resulted in a model of the oligomeric state ofthe enzyme, no three-dimensional reconstruction of anyGS-II has to our knowledge been so far reported. Recently,a three-dimensional structure of human GS-II was generat-ed that depicts GS-II as a double heptamer (Kiang, 2001).The structure is however reminiscent to that of the chap-eronin GroEL, a common impurity in the purification pro-cess of GS. We wanted to determine the three-dimensionalstructure of GS-II from P. vulgaris and as a first step in thereconstruction process, GS-II from P. vulgaris was nega-tively stained in 2% uranyl acetate and observed by elec-tron microscopy (Fig. 3A). Only a single type of particlewith roughly two main views can be observed. The mostcommon one corresponds to those particles having twoparallel stain-excluding masses linked by others with lessdensity, which we called side view (Fig. 3A, arrowheads),and the less frequent corresponds to those particles withsimilar stain-excluding masses in the four corners of theparticle, which we termed end-on view (Fig. 3A, arrows).Still, each of these two types of views would actually
enclose a collection of different views where the particlehas rotated along its axes, such as the rocking of the parti-cle due to rotation along the longitudinal axis. To carry outthe three-dimensional reconstruction of GS-II, 5578 parti-cles were selected (Fig. 3B) and processed as described inSection 2 without any symmetry assumption (Fig. 4A).After several rounds of refinement, the three-dimensionalstructure obtained revealed a clear twofold symmetry alongone of the axes. Fig. 4B shows the two top views, whichreveal in both cases an apparent twofold rotational symme-try. The three-dimensional reconstruction shows two dis-tinct side views, each alternating every 90�, one of themdisplaying no density in the central area (Fig. 4C, bottompanel), while the other revealing a stain-excluding mass inthe center (Fig. 4C, bottom panel). Consequently, a two-fold rotational symmetry was imposed throughout the restof the reconstruction procedure.
The final twofold symmetrized three-dimensional recon-struction (Fig. 5A–F), obtained after several further refine-ment iterations, shows an almost cubic structure of �100 Aheight formed by two back-to-back square rings (see topand bottom masses in Fig. 5E and F). This two-ringarrangement is very similar to that found for GS-I, exceptthat the prokaryotic enzyme each ring is 143 A wide and isformed by six monomers (Fig. 5G). The four-subunitarrangement within each ring follows a clear twofold sym-metry (imposed for a better reconstruction but neverthelesspresent in the non-symmetrized particle; see Figs. 4A and5C and D), which strongly suggests that each tetramericring is not built by four monomers but rather by the inter-action of two dimers. This is a surprising finding since most

Fig. 4. The three-dimensional reconstruction of GS-II without symmetry imposition. (A) A gallery of representative projections (left) and theircorresponding averages (right) of the three-dimensional reconstruction without any symmetry imposition (Section 2). (B) The two end-on views of thenon-symmetrized volume showing a clear two-fold symmetry. (C) Two of the side views of the same three-dimensional reconstruction.
Fig. 5. Three-dimensional reconstruction of GS-II. (A and F) Three different views of the three-dimensional reconstruction. (A and B) The two end-onviews of the particle. (C and D) The same two views in transparent fashion showing in the interior and as a solid representation the 43% of the totalvolume. (E and F) The two side views of the particle, revealing the two face-to-face interacting rings. (G and H) The two end-on and side views,respectively, of the hexameric structure of GS-I from S. typhimurium (pdb file 1f1h) with the resolution limited at 25 A. The asterisk points to the locationof one of the GS-I active sites.
474 O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479
of the literature concerning the structure of GS-II hasdescribed the oligomer as possessing a quaternary symme-try. However, our finding is in accordance with the bio-chemical data revealing the existence of a stablepopulation of dimers (Fig. 2). Another interesting pointof the structure obtained is the arrangement between thetwo rings, rotated 90� with respect to each other (comparethe two end-on views in Fig. 5A–D). This is a surprising
arrangement, which might have important functionalimplications (see below). Four domains, located at everycorner of the interface between the two rings, are involvedin inter-ring interaction.
In summary, the three-dimensional structure of GS-IIgenerated from electron microscopy and image processingis compatible with an octamer built by two tetramers, eachone built by two dimers. The two tetramers are placed

O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479 475
back-to-back and rotated 90� with respect to each other.The structure determined suggests, since no other type ofparticle was observed in the micrographs, that the minor-itary, hexadecameric form of GS-II determined by gel fil-tration and ultracentrifugation techniques is a doubleoctamer that has been separated into octamers, probablyduring the preparation of the sample for EM observation.
3.3. A model for the arrangement of the GS-II octamer
To characterize in more detail some of the points raisedin the previous section, we performed a docking analysis ofthe three-dimensional structure of GS-II obtained. Sincethere is no high-resolution information available on anyGS-II protein, we reasoned that the best solution was togenerate an homology model of GS-II based on the atomicstructure of GS-I from S. typhimurium (pdb file 1f1h; Gilland Eisenberg, 2001). Although the sequence homologybetween the two GS types is low (21% average sequenceidentity; Fig. 1), two facts prompted us to use the atomicmodel of GS-II. First, the sequence homology is very highwith respect to the residues forming part of the active site,which are strictly conserved (Fig. 1). This clearly points toa similar architecture of the region forming the active sitein both types of GS, a region that is located between adja-cent intra-ring monomers (Almassy et al., 1986). Second, astructure prediction for the GS-II sequences (Fig. 1) sug-gests a secondary structure pattern similar to the GS-Istructures known. Taking all this into account and the factthat the GS-II tetramer seems to be built by dimers insteadof monomers, we generated an homology model of a GS-IIdimer using the atomic structure of a dimer of S. typhimu-
rium GS-I as a template (Fig. 6A), to maintain the structur-al arrangement of the active site between the twomonomers (Fig. 6B). Docking of this homology model intothe GS-II three-dimensional reconstruction is reasonablewith respect to the main mass of the dimer (Fig. 6C andD), and serves to reinforce the notion of the GS-II ringbeing formed by a double dimer, but also reveals regionsof the three-dimensional reconstruction that are not filledby the atomic model. This clearly suggests that even ifGS-II shares a similar secondary structure to that of GS-I, there must be large reorientations of the two GS-II sub-domains to adequately fill the three-dimensional structureobtained by electron microscopy. Nevertheless, some ofthe differences observed in the docking analysis have todo with the low-resolution of the EM reconstruction. Thisis the case for the funnel described for the active site in theGS-I atomic structure (Eisenberg et al., 2000), which is notrevealed in the three-dimensional structure of GS-II(Fig. 6C), but the same can be observed when the resolu-tion of the atomic structure of GS-I is limited to 25 A(see Fig. 5G and H).
The most important difference in the docking analysislies however in the inter-ring domain of the three-dimensional reconstruction, whose density is not filled bythe homology model (Fig. 6D). This may be explained
by the fact that the two most important domains involvedin the inter-ring interaction in GS-I (residues encompassedby a blue line in Fig. 1) are absent in the GS-II sequences:an a-helical region encompassing residues 435–468 in the S.
typhimurium sequence (arrow in Fig. 6A) and a hairpinconstituted by two b-strands and located between residues137–152 of the same sequence (arrowhead in Fig. 6A).Therefore, the interaction between the two tetramers inGS-II must follow a completely different pattern to thatof GS-I, probably involving movements of the regions clos-er to the opposed monomer, which include the loopsencompassed by residues 134–153, 235–245 and 275–300in the P. vulgaris GS-II sequence. Interestingly, the inter-ring domain is variable even within the GS-I proteins, sincethe hairpin described above is only present in the GS-Ifound in Gram-negative bacteria. The apparent less tightinteraction between the two rings of GS-II with respectto their GS-I counterparts is consistent with the tendencyof eukaryotic GS to be naturally present as a mixture ofoctamers and tetramers (Eisenberg et al., 2000), while thedissociation of GS-I requires the removal of the metalcofactors with EDTA and the modification of sulfhydrilgroups (Maurizi and Ginsburg, 1985).
In summary, docking of the homology model of a GS-IIdimer obtained from the atomic structure of a prokaryoticcounterpart into the EM volume, reinforces the notion ofeach of the GS-II tetramers built by the interaction oftwo dimers. These, according to our biochemical results,are present during the purification procedure and mightbe the basic structure from which the octamer is built.There are differences however, especially in the inter-ringdomain, which can only be explained by large molecularrearrangements of the GS-II structure as compared to thatof GS-I.
4. Discussion
4.1. Structural and functional considerations
From the geometry of the three-dimensional reconstruc-tion it clearly follows that unlike in the case of the GS-Ienzyme, which possesses one active site per subunit(although located between two adjacent monomers), GS-II must contain one active site per dimer (i.e., half activesite per monomer). This notion was first proposed byEisenberg (1987) who, based on the high conservationamong the GS sequences of the residues involved in theenzyme functionality, reasoned that for the geometry ofthe active site present in an hexameric form to be main-tained in a tetrameric one, the GS-II enzyme had to bearranged so that the tetrameric ring be formed by pairsof dimers. This packing reduces the number of active sitesto the ones already included in the dimer and therefore tohalf the potential ones (‘‘Half-of-the-sites reactivity’’;Eisenberg, 1987). This hypothesis was against the structur-al information obtained at that time for the GS-II oligo-mers, which pointed to an octamer built up by two
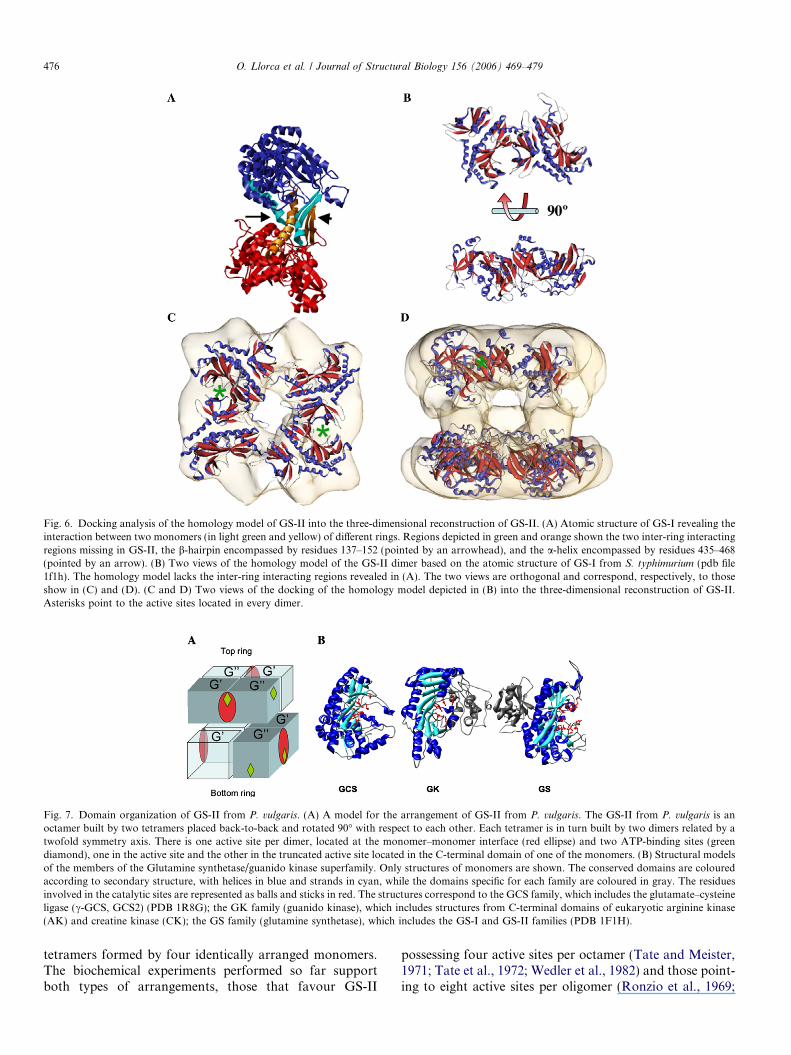
Fig. 6. Docking analysis of the homology model of GS-II into the three-dimensional reconstruction of GS-II. (A) Atomic structure of GS-I revealing theinteraction between two monomers (in light green and yellow) of different rings. Regions depicted in green and orange shown the two inter-ring interactingregions missing in GS-II, the b-hairpin encompassed by residues 137–152 (pointed by an arrowhead), and the a-helix encompassed by residues 435–468(pointed by an arrow). (B) Two views of the homology model of the GS-II dimer based on the atomic structure of GS-I from S. typhimurium (pdb file1f1h). The homology model lacks the inter-ring interacting regions revealed in (A). The two views are orthogonal and correspond, respectively, to thoseshow in (C) and (D). (C and D) Two views of the docking of the homology model depicted in (B) into the three-dimensional reconstruction of GS-II.Asterisks point to the active sites located in every dimer.
Fig. 7. Domain organization of GS-II from P. vulgaris. (A) A model for the arrangement of GS-II from P. vulgaris. The GS-II from P. vulgaris is anoctamer built by two tetramers placed back-to-back and rotated 90� with respect to each other. Each tetramer is in turn built by two dimers related by atwofold symmetry axis. There is one active site per dimer, located at the monomer–monomer interface (red ellipse) and two ATP-binding sites (greendiamond), one in the active site and the other in the truncated active site located in the C-terminal domain of one of the monomers. (B) Structural modelsof the members of the Glutamine synthetase/guanido kinase superfamily. Only structures of monomers are shown. The conserved domains are colouredaccording to secondary structure, with helices in blue and strands in cyan, while the domains specific for each family are coloured in gray. The residuesinvolved in the catalytic sites are represented as balls and sticks in red. The structures correspond to the GCS family, which includes the glutamate–cysteineligase (c-GCS, GCS2) (PDB 1R8G); the GK family (guanido kinase), which includes structures from C-terminal domains of eukaryotic arginine kinase(AK) and creatine kinase (CK); the GS family (glutamine synthetase), which includes the GS-I and GS-II families (PDB 1F1H).
476 O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479
tetramers formed by four identically arranged monomers.The biochemical experiments performed so far supportboth types of arrangements, those that favour GS-II
possessing four active sites per octamer (Tate and Meister,1971; Tate et al., 1972; Wedler et al., 1982) and those point-ing to eight active sites per oligomer (Ronzio et al., 1969;

O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479 477
Ericson and Brunn, 1985; Maurizi et al., 1986; Hopfneret al., 1988). Our own results with GS-II from P. vulgaris
are compatible with the ‘‘half-of-the-sites reactivity’’hypothesis not only in structural terms but also biochemi-cally (Table 1), since active site titration experiments car-ried out with the transition state analogue and GSirreversible inhibitor MetSox-P imply the presence of fourbinding sites per octamer. This ‘‘half-of-the sites reactivity’’could be due to pre-existing half-sites reactivity or to anallosteric behaviour of the enzyme. Interestingly, bindingexperiments with ATP reveal that every GS-II subunitbinds one ATP molecule (Betti et al., 2002), which indicatesthat the dimer maintains two ATP-binding sites, located inthe C-terminal domain of each monomer.
Our three-dimensional reconstruction together with thedocking analysis and the biochemical results obtained sug-gests that each dimer contains one active site, whichaccording to what is known for GS-I and GS-II enzymesmust be located in the inter-monomer region, and twoATP-binding sites, one of them in the active site and theother in the C-terminal domain of the other monomer. Thisarrangement and the fact that the two tetrameric rings arerotated 90� with respect to each other, allows one activesite and one free ATP-binding site, which might have a reg-ulatory role, in each of the four ‘‘side faces’’ of the octamer(Fig. 7A). The tetrameric ring is formed by a dimer ofdimers, and for for this to happen the two monomers with-in the dimer must have at least a slightly different structure(G 0 and G00, see Fig. 7A). The arrangement of the two typesof GS-II subunits (G 0 and G00) in the octamer is such thatintra-ring interaction must occur between the two differenttypes of subunits (G 0 with G00) but the inter-ring ones onlywith the same type of subunits (G 0 with G 0 or G00 with G00;Fig. 7A), generating in the latter case two different kinds ofinter-ring interactions that are clearly visible in the three-dimensional reconstruction of GS-II (see Fig. 4E and F).In any case, the GS-II inter-ring domain must have a differ-ent set of interaction to that of GS-I since the residuesinvolved in the latter are missing in the former.
The biochemical results obtained indicate that for GS-IIto be active the octameric, double ring must be generated,since neither the dimer nor the tetramer are functional.This strongly suggests that GS-II is highly cooperative,something which has already been reported with regardto different GS substrates and cofactors like L-glutamate(Montanini et al., 2003) and Mg2+/Mn2+ (Denman andWedler, 1984; Clemente and Marquez, 1999; Sakakibaraet al., 1996; Hirel and Gadal, 1980). The GS-II studied inthis work has revealed positive cooperativity to metal cat-ions and glutamine (data not shown), thus strenghteningthis hypothesis. Different types of metabolic effectors andallosteric sites have been also reported for mammalianGS-II (Eisenberg et al., 2000). In addition, several GS-IIenzymes have also been reported to be regulated in vitroby association–dissociation of subunits, such in humans(Denman and Wedler, 1984), plants (Mack, 1998) and fungi(Mora et al., 1980).
4.2. Structural context of GS enzymes
GS enzymes belong to a large family of structures witha peculiar distribution of the active sites, located in theinter-domain interfaces. It is interesting to realize thatall these proteins carry out their catalytic activity eitherby interacting with additional domains or with auxiliaryproteins. The GS-I monomer (and the predicted second-ary structure for the P. vulgaris GS-II sequence suggeststhe same for the GS-II enzyme; see Fig. 1) is formed bytwo clearly separated domains, a small N-terminaldomain and a larger C-terminal one. The two domainscontribute to the formation of the active sites, each inan adjacent monomer. The two GS domains belong totwo different protein families (as classified in SCOP, Mur-zin et al., 1995). SCOP classifies proteins with knownstructure using a hierarchy with four categories: ‘‘class’’,‘‘fold’’, ‘‘superfamily’’ and ‘‘family’’, according to increas-ing homology. Families within the same superfamily arethought to possess not only a structural similarity but alsoa probable evolutionary relationship. The GS N-terminaldomain forms a separated superfamily (‘‘Glutamine syn-thetase N-terminal domain’’) within the beta-Grasp fold,meaning that no remote sequence homology to other pro-teins with solved structures has been found. However, theC-terminal domain belongs to the ‘‘Glutamine synthetase/guanido kinase’’ superfamily within the fold of the samename. This superfamily contains the families ‘‘Glutaminesynthetase catalytic domain’’ (GS), ‘‘Guanido kinase cat-alytic domain’’ (GK), and ‘‘Glutamate-cysteine ligasedomain’’ (GCS) (Fig. 7B). The GK family includes struc-tures from C-terminal domains of eukaryotic argininekinase and creatine kinase, which are found in differentquaternary structures: octamers, dimers and monomers.Proteins in this family also present a N-terminal domainwith the same size as beta-Grasp domain in GS, but witha different fold (‘‘Guanido kinase N-terminal domain’’ inSCOP). The active site is formed by residues on both N-and C-terminal domains from the same monomer. TheGCS family contains structures such as the prokaryoticcarboxylate-amine ligase YbdK, which can be found asa monomer or a dimer. The active site of this enzyme islocated within the sequence of each monomer and is strik-ingly similar to the C-terminal part of the GS active site(Abbott et al., 2001; Lehmann et al., 2004), with whichit shares some substrates (glutamate and ATP). Thesethree families present a detectable similarity at structural(Fig. 7B) and sequence levels, strongly suggesting thatthey probably arose from a common ancestor having anequivalent structure to the C-terminal domain of GS. Itis quite likely that during their divergence, the GS andGK families acquired an additional domain, which helpedthem to specialize by adopting new substrate specificity.
With this information it is possible to propose a firstmodel for the evolution of the Glutamine synthetase/guanido kinase superfamily (see Supplementary Material).The future availability of new structures from a wider

478 O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479
range of organisms will help to sharpen the details of theevolution of this interesting protein family.
Acknowledgments
The authors acknowledge the help of Drs. G. Rivasand C. Alfonso with the ultracentrifugation analyses.This work was supported by grant BFU2004-00232 toJ.M.V, BFU2004-02753 and BFU2005-03120 to A.J.Mand SAF2002-01715 to O.L. A.J.M and M.B. alsoacknowledge the financial support given by the Juntade Andalucıa (CVI-163). A.V. and J.M.G work wassupported by grants LSHG-CT-2004-503567 andBIO2004-00875.
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.jsb.2006.06.003.
References
Abbott, J.J., Pei, J., Ford, J.L., Qi, Y., Grishin, V.N., Pitcher, L.A.,Phillips, M.A., Grishin, N.V., 2001. Structure prediction and active siteanalysis of the metal binding determinants in gamma-glutamylcysteinesynthetase. J. Biol. Chem. 276, 42099–42107.
Almassy, R.J., Janson, C.A., Hamlin, R., Huong, N.H., Eisenberg, D.,1986. Novel subunitsubunit interactions in the structure of glutaminesynthetase. Nature 323, 304–309.
Betti, M., Marquez, A.J., Yanes, C., Maestre, A., 2002. ATPbinding to purified homopolymeric plant glutamine synthetasestudied by isothermal titration calorimetry. Thermochim. Acta394, 63–71.
Boksha, I.S., Schonfeld, H.J., Langen, H., Muller, F., Tereshkina, E.B.,Burbaeba, G.S., 2002. Glutamine synthetase isolated from humanbrain: octameric structure and homology of partial primary structurewith human liver glutamine synthetase. Biochemistry (Moscow) 67,1012–1020.
Clemente, M.T., Marquez, A.J., 1999. Functional importance of Asp56from the a-polypeptide of Phaseolus vulgaris glutamine synthetase. Anessential residue for transferase but not for biosynthetic enzymeactivity. Eur. J. Biochem. 264, 453–460.
Denman, R.B., Wedler, F.C., 1984. Association-dissociation of mamma-lian brain glutamine synthetase: effects of metal ions and other ligands.Arch. Biochem. Biophys. 232, 427–440.
Eisenberg, D., 1987. Some evolutionary relationships of the primarybiological catalysts glutamine synthetase and RuBisCo. Cold SpringHarbor Quant. Biol. 52, 483–490.
Eisenberg, D., Gill, H.S., Pfluegl, G.M.U., Rotstein, S.H., 2000. Struc-ture–function relationships of glutamine synthetases. Biochim. Bio-phys. Acta 1477, 122–145.
Ericson, M.C., Brunn, S.A., 1985. Cysteine residues at the active site ofglutamine synthetase from spinach leaves. Biochim. Biophys. Res.Commun. 133, 527–531.
Gill, H.S., Eisenberg, D., 2001. The crystal structure of phosphinothricinin the active site of glutamine synthetase illuminates the mechanism ofenzymatic inhibition. Biochemistry 40, 1903–1912.
Gill, H.S., Pfluegl, G.M.U., Eisenberg, D., 2002. Multicopy crystal-lographic refinement of a relaxed glutamine synthetase fromMycobacterium tuberculosis highlights flexible loops in theenzymatic mechanism and its regulation. Biochemistry 41, 9863–9872.
Guex, N., Peitsch, M.C., 1997. SWISS_MODEL and the Swiss-PdbView-er: an environment for comparative protein modeling. Electrophoresis18, 2714–2723.
Hirel, B., Gadal, P., 1980. Glutamine synthetase in rice: a comparativestudy of the enzymes from root and leaves. Plant Physiol. 66, 619–623.
Hooft, R.W.W., Vriend, G., Sander, C., Abola, E.E., 1996. Errors inprotein structures. Nature 381, 272.
Hopfner, M., Reifferscheid, G., Wild, A., 1988. Molecular composition ofglutamine synthetase of Sinapsis alba L. Z. Naturforsch. 43c, 194–198.
Jones, D.T., 1999. Protein secondary structure prediction based onposition-specific scoring matrices. J. Mol. Biol. 292, 195–202.
Kabsch, W., Sander, C., 1983. Dictionary of protein secondary structure:pattern recognition of hydrogen-bonded and geometrical features.Biopolymers 22, 2577–2637.
Kiang, C.-H., 2001. Single-particle study of protein assembly. Phys. Rev.E 64, 041911 1–3.
Laue, T.M., Shall, B.D., Ridgeway, T.M., Pelletier, S.L., 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In:Harding, S.E., Rowe, A.J., Horton, J.C. (Eds.), Analytical ultracen-trifugation in biochemistry and polymer science. The Royal Society ofChemistry Cambridge, pp. 90–125.
Lehmann, C., Doseeva, V., Pullalarevu, S., Krajewski, W., Howard, A.,Herzberg, O., 2004. YbdK is a carboxylate–amine ligase with agamma-glutamyl:cysteine ligase activity: crystal structure and enzy-matic assays. Proteins 56, 376–383.
Liu, Y., Eisenberg, D., 2002. 3D domain swapping: as domains continueto swap. Protein Sci. 11, 1285–1299.
Ludtke, S.J., Baldwin, P.R., Chiu, W., 1999. EMAN: semi automatedsoftware for high-resolution single-particle reconstructions. J. Struct.Biol. 128, 82–97.
Mack, G., 1998. Glutamine synthetase isoenzymes, oligomers andsubunits from hairy roots of Beta vulgaris L. var. lutea. Planta 205,113–120.
Maurizi, M.R., Ginsburg, A., 1985. Active-site ligand binding and subunitinteractions in glutamine synthetase from Escherichia coli. Curr. Top.Cell. Reg. 26, 191–206.
Maurizi, M.R., Pinfofsky, H.B., McFarland, P.J., Ginsburg, A., 1986.Mg2+ is bound to glutamine synthetase extracted from bovine orovine in the presence of L-methionine–S-sulphoximine phosphate.Arch. Biohem. Biophys. 256, 494–500.
McGuffin, L.J., Bryson, K., Jones, D.T., 2000. The PSIPRED proteinstructure prediction server. Bioinformatics 16, 404–405.
McParland, R.H., Guevara, J.G., Becker, R.R., Evans, H.J., 1976. Thepurification and properties of the glutamine synthetase from thecytosol of soya-bean root nodules. Biochem. J. 153, 597–606.
Montanini, B., Betti, M., Marquez, A.J., Balestrini, R., Bonfante, P.,Ottonello, S., 2003. Distinctive properties and expression profiles ofglutamine synthetase from a plant symbiotic fungus. Biochem. J. 373,357–368.
Mora, J., Davila, G., Espın, G., Gonzales, A., Guzman, J., Hernandez,G., Hummelt, G., Lara, M., Martınez, E., Mora, Y., Romero, D.,1980. Glutamine metabolism in Neurospora crassa. In: Glutamine:Metabolism, Enzymology and Regulation. Academic Press, NewYork, pp. 185–211.
Murzin, A.G., Brenner, S.E., Hubbard, T., Chothia, C., 1995. SCOP: astructural classification of proteins database for the investigation ofsequences and structures. J. Mol. Biol. 247, 536–540.
Notredame, C., Higgins, D.G., Heringa, J., 2000. T-Coffee: a novelmethod for fast and accurate multiple sequence alignment. J. Mol.Biol. 302, 205–217.
Philo, J.S., 1997. An improved function for fitting sedimentation velocitydata for low molecular-weight solutes. Biophys. J. 72, 435–444.
Pushkin, A.V., Tsuprun, V.L., Dzhokharidze, T.Z., Evstingneeva, Z.G.,Kretovich, W.L., 1981. Glutamine synthetase from the pumpkin leafcytosol. Biochim. Biophys. Acta 662, 160–162.
Pushkin, A.V., Antoniuk, L., Solovieva, N.A., Shubin, V.V., Evstingn-eeva, Z.G., Kretovich, W.L., Cherednikova, T.V., Tsuprun, V.L.,Zograf, O.N., Kiselev, N.A., 1985. Glutamine synthetase of pea leaf

O. Llorca et al. / Journal of Structural Biology 156 (2006) 469–479 479
and seed cytosol. Structure and properties. Biochim. Biophys. Acta828, 336–350.
Ronzio, R.A., Rowe, W.B., Meister, A., 1969. Studies on the mechanismof inhibition of glutamine synthetase by methionine sulfoximine.Biochemistry 8, 1066–1075.
Sakakibara, H., Shimizu, H., Hase, T., Yamazaki, Y., Takao, T.,Shimonishi, Y., Sugiyama, T., 1996. Molecular identification andcharacterization of cytosolic isoforms of glutamine synthetase in maizeroots. J. Biol. Chem. 271, 29561–29568.
Sali, A., Blundell, T.L., 1993. Comparative protein modelling bysatisfaction of spatial restraints. J. Mol. Biol. 234, 779–815.
Siegel, L.M., Monty, K.J., 1966. Determination of molecular weights andfrictional ratios of proteins in impure systems by use of gel filtrationand density gradient centrifugation. Application to crude preparationsof sulphite and hydroxylamine reductases. Biochim. Biophys. Acta112, 346–362.
Sippl, M.J., 1993. Recognition of errors in three-dimensional structures ofproteins. Proteins 17, 355–362.
Stadtman, E.R., Ginsburg, A., 1974. The glutamine synthetase ofEscherichia coli: structure and control. In: Boyer, P.D. (Ed.), TheEnzymes, vol. 10. Academic Press, New York, pp. 755–807.
Tsoka, S., Ouzounis, C.A., 2000. Prediction of protein interactions:metabolic enzymes are frequently involved in gene fusion. Nat. Genet.26, 141–142.
Tsuprun, V.L., Zograf, O.N., Orlova, E.V., Kiselev, N.A., Pushkin, A.V.,Shiffelova, G.E., Solovieva, N.A., Evstingneeva, Z.G., Kretovich,W.L., 1987. Electron microscopy of multiple forms of glutaminesynthetase from bacteriods and the cytosol of yellow lupin rootnodules. Biochim. Biophys. Acta 913, 368–376.
Tate, S.S., Meister, A., 1971. Regulation of rat liver glutaminesynthetases: activation by a-ketoglutarate and inhibition by glycine,alanine and carbamyl phosphate. Proc. Natl. Acad. Sci. USA 68,781–785.
Tate, S.S., Leu, F.Y., Meister, A., 1972. Rat liver glutamine synthetase.Preparation, properties and mechanism of inhibition by carbamylphosphate. J. Biol. Chem. 247, 5312–5321.
Vogel, C., Bashton, M., Kerrison, N.D., Chothia, C., Teichmann, S.A.,2004. Structure, function and evolution of multidomain proteins. Curr.Opin. Struct. Biol. 14, 208–216.
Wedler, F.C., Denman, R.B., Roby, W.G., 1982. Glutamine synthetasefrom ovine brain is a manganese (II) enzyme. Biochemistry 21, 6389–6396.
Related Documents











