This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
The structure of horseradish peroxidase C characterized as a molten globule stateafter Ca2+ depletion
Krisztián Szigeti a,b, László Smeller b, Szabolcs Osváth b, Zsuzsanna Majer c, Judit Fidy a,b,⁎a Research Group for Membrane Biology, Hungarian Academy of Sciences, 1113 Budapest, Hungaryb Department of Biophysics and Radiation Biology, Semmelweis University, 1088 Budapest, Hungaryc Institute of Chemistry, Lorand Eotvos University, 1117 Budapest, Hungary
a b s t r a c ta r t i c l e i n f o
Article history:Received 6 May 2008Received in revised form 25 July 2008Accepted 13 August 2008Available online 5 September 2008
Keywords:Horseradish peroxidaseCa2+ depletionpH effectSecondary structureCircular dichroismConformational dynamicsHigh pressureH/D exchangeFTIR
The structure and activity of native horseradish peroxidase C (HRP) is stabilized by two bound Ca2+ ions.Earlier studies suggested a critical role of one of the bound Ca2+ ions but with conflicting conclusionsconcerning their respective importance. In this work we compare the native and totally Ca2+-depleted formsof the enzyme using pH-, pressure-, viscosity- and temperature-dependent UV absorption, CD, H/Dexchange-FTIR spectroscopy and by binding the substrate benzohydroxamic acid (BHA). We report that Ca2+-depletion does not change the alpha helical content of the protein, but strongly modifies the tertiarystructure and dynamics to yield a homogeneously loosened molten globule-like structure. We relateobserved tertiary changes in the heme pocket to changes in the dipole orientation and coordination of adistal water molecule. Deprotonation of distal His42, linked to Asp43, itself coordinated to the distal Ca2+,perturbs a H-bonding network connecting this Ca2+ to the heme crevice that involves the distal water. Themeasured effects of Ca2+ depletion can be interpreted as supporting a structural role for the distal Ca2+ andfor its enhanced significance in finetuning the protein structure to optimize enzyme activity.
© 2008 Elsevier B.V. All rights reserved.
1. Introduction
Horseradish peroxidase (HRP) is a typical, broadly studiedrepresentative of class III heme peroxidases, in which the activesite and the Ca2+ binding sites are evolutionary conserved [1]. It is amonomeric glycoprotein with a molecular mass of 44 kDa. There is abroad biotechnological interest concerning this enzyme because ofits unusually high stability under a variety of conditions [2–4].Essential structural elements include a Fe3+ protoporphyrin IX (btype heme) with a histidine (His170) as fifth ligand to the Fe3+ andan extended hydrogen bonding network, four disulfide bonds andtwo heptacoordinated Ca2+ ions, one at the distal and one at theproximal side of the heme. The analysis of the B-factors in X-raycrystallography shows that the distal Ca2+ binding site is more buriedin a rigid protein core [5].
The importance of the enzyme reaction of HRP prompted a greatnumber of works in prominent scientific laboratories related to the
structural conditions of enzymatic activity [6–9], long before thecrystal structure of the enzyme came out. Reports have also beenpublished concerning the structural role of bound Ca2+ ions in theenzyme activity of HRP. Haschke and Friedhoff were the first todescribe a clearly verified protocol for the removal of both boundCa2+ ions [10]. Their protocol had been used in a controlled way byMorishima's group [11–13] who reported results of NMR, EPR andabsorption spectroscopy. They proposed that the two Ca2+ bindingsites influence the heme environment, the rate constants of thecatalytic cycle, and the spin state of the heme, but by differentmechanisms, and that only one of the binding sites has importantrole in the regulation of enzyme activity. Just before the long awaitedcrystal structure of HRP became available [14], the same groupreported on extended mutation studies aiming to reveal thestructural role of amino acids and details of the earlier suggestedH-bonding network around the heme [15–19]. These studiesconcluded on the determining role of the bound Ca2+ ion at thedistal side of the heme. Besides this group, several authors followedthe same protocol or used different approaches — such as simplyadding EDTA to remove Ca2+ [20,21]. Some of these studies werewrong assuming complete Ca2+ depletion, as EDTA only removes theproximal Ca2+ [5].
Besides the early NMR and EPR works, optical absorption andresonance Raman spectroscopy – methods also yielding informationabout the environment and conformation of the heme as well as about
Biochimica et Biophysica Acta 1784 (2008) 1965–1974
Abbreviations: HRP, Horseradish peroxidase isoenzyme C; disHis, His42, histidine,not coordinated to Fe3+ of the heme; proHis, His170, histidine coordinated to Fe3+ of theheme; CD, circular dichroism; ANS, anilinonaphtalene sulphonic acid; CT band, chargetransfer band; HS, high spin (S=5/2; LS, low spin (S=3/2; QS, quantum mechanicallymixed HS and LS states; LT, low temperature; RT, room temperature⁎ Corresponding author. 1088 Puskin u. 9, Budapest, Hungary. Tel.: +36 1 267 62 61.
E-mail address: [email protected] (J. Fidy).URL: http://www.biofiz.sote.hu (J. Fidy).
1570-9639/$ – see front matter © 2008 Elsevier B.V. All rights reserved.doi:10.1016/j.bbapap.2008.08.015
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbapap

Author's personal copy
the spin state of the Fe3+ – were also applied to study HRP. Smulevichand coworkers reported on the structure of the heme environment innative HRP C and its isoenzyme A2 in both their ferrous and ferricstates in a series of papers, as well as on the effect of bound Ca2+
[11,22–29]. Comparing the native and partially Ca2+-depleted HRP (i.e.one Ca2+/molecule) by using room temperature (RT) optical andresonance Raman spectroscopy, a significant structural effect wasattributed to the proximal Ca2+ [28]. This conclusion does not agreewith the early studies of Morishima's group, who attributedimportance to the distal Ca2+ [8,13] and with results of authors whoreported only minor structural effects when Ca2+ was removed simplyby a treatment with EDTA [20,21], that is, when supposedly only theproximal Ca2+ was removed.
Our group addressed the role of Ca2+ ions also by using computersimulations. We performed 500 ps [30,31] and 2 ns [32] moleculardynamics calculations on the native HRP structure and thoseproduced by computer simulation techniques after partial and totalCa2+ depletion. By normal coordinate structural decompositionanalysis of the heme group we found that the deformation of theheme group from D4h symmetry was significant in the native form[33], but total Ca2+ depletion changed both the in-plane and out-of-plane distortions and diminishes nonplanarity [31,33]. Normal modeanalysis showed a lack of coupled dynamics of the heme and proteinmotions [32] after Ca2+ removal. Experimental studies in collabora-tion with Schweitzer-Stenner's group by polarized resonance Ramanspectroscopy on partially Ca2+-depleted samples [5] suggested moresignificant structural effects than those reported by Smulevich et al.[28] and somewhat different changes in the spin state of the heme.We attribute this to differences in the sample preparation techni-ques, even if they seemed to be similar. Preliminary low temperature(LT) absorption spectroscopy measurements on totally Ca2+-depletedsamples yielded, however, encouraging results. The Ca2+-depletioneffect was found significant in such measurements, and thesample preparation method proved to be reliable and reproducible[31]. Thus we decided to perform a detailed LT optical spectroscopystudy including other biophysical methods. By comparing native HRPwith the totally Ca2+-depleted condition we wanted to obtain aclearer conclusion about the structural role of binding (both) Ca2+
ions and suggest which of the two are more important in enzymeactivity.
Here we show that Ca2+ removal does not significantly affect thesecondary structural units of the protein but significantly influencesthe tertiary structure and overall conformational dynamics trans-forming the protein into a molten globule-like state. We identifystructural rearrangements at the heme distal side connected to thedisHis residue and a water molecule as a reason for decreasedenzyme activity. We conclude that binding Ca2+ at the distal sidehas a dramatic effect on the tertiary structure of the whole proteinand also finetunes the tertiary structure of the active site forbiochemical activity.
2. Materials and methods
Horseradish peroxidase isoenzyme C (HRPC) was purchased fromSigma and further purified by chromatography. After purification, theRZ (Reinheitszahl) was 3.2 and the percentage of izoenzyme C washigher than 95%. Ca2+ depletion was based on a modified protocol ofHaschke et al. [10]. The enzyme dissolved in 50 mM Tris/HCl (pH 8.4)buffer was incubated for 4 h in 6 M guanidine hydrochloride and10 mM EDTA at room temperature (RT). After this treatment, adialysis against 10 mM EDTA and 50 mM Tris/HCl (pH 7.4) wasstarted with frequent buffer changes, and than continued withdialysis against the same buffer for 24 h, then against distilled waterfor another 24 h. The final pH was adjusted by dialysis against50 mM Tris/HCl (pH 7.6). The sample was concentrated using aMillipore 8010 Stirred Cell with Millipore YM 10 filter. Special care
had to be taken to avoid contamination by even trace amounts ofCa2+. For this reason chromatography could not be applied to controlthe purity and homogeneity of the Ca2+-depleted sample due tocontamination from the glassware. The presence of denaturatedproteins in the final sample could be excluded based on the RZ valueand low level of light scattering. Dynamic light scattering measure-ments (data not shown) showed a single population of particles inthe solution which overlapped with the population found in thenative state.
Total reflection X-ray fluorescence (TXRF Extra IIA, AtomicaInstruments, Germany) spectroscopy was used to determine theCa2+ content [34] of the samples relative to the Fe3+ of the heme. Thismeasurement is performed on a dry film deposited onto a quartzplate. A samplewas considered Ca2+-depleted if themolar ratio of Ca2+
and protein was less than 0.4±0.1 mol Ca2+ compared to 2 mol of Ca2+
for one mole of protein in the native enzyme. Our experience was thatwithout disrupting the rigid structure around the distal Ca2+ in thenative state by partial denaturation it is impossible to remove thedistal Ca2+ by incubation with EDTA.
For UV-VIS optical spectroscopy (absorption, fluorescence, CD), theprotein concentration was set to 10 μM. In the 6.0–8.7 pH range, thepH was adjusted by mixing TRIS and BES buffers to 100 mM of finalbuffer concentration. At every pH, the sample was incubated for 4–5 hbefore the experiments to reach equilibrium. The pH of both the nativeand the Ca-depleted HRP could be changed reversibly withoutdenaturation.
Absorption spectra at RT were recorded in a 1 ml quartz cuvette(10 mm optical path) with a Varian Cary 4 spectrophotometer using0.5 nm step, 0.5 s integration time and 1.5 nm bandwidth.
Circular dichroism experiments were performed on a Jasco J 710spectropolarimeter at 25 °C in a 1 ml quartz cuvette. Circulardichroism in the UV region (190–260 nm) was monitored in acylindrical quartz cell of 0.2 mm pathlength, while in the visiblerange (260–500 nm) it was monitored using a glass cuvette of10 mm pathlength. Five spectra were averaged with a scanningspeed of 50 nm/min and 0.2 nm step size. ANS binding was followedwith fluorescence emission intensity measurements at 460 nm(λexc=380 nm) with a Jobin Yvon Fluorolog spectrophotometer using1 s integration time and 1.0 nm bandwidth at 25 °C in a 1 ml quartzcuvette.
Absorption spectroscopy under increased hydrostatic pressurewasperformed by a diamond anvil cell (Diacell, Leicester, UK) placed in theVarian Cary 4 spectrophotometer using 0.5 nm step, 0.5 s integrationtime and 1.5 nm bandwidth. The pressure was varied from atmo-spheric up to 12 kbar at 25 °C.
Absorption spectra at LT were recorded in 50% (V/V) glycerol/buffer, which ensures good optical quality at cryogenic temperatures.The samples were placed in a sample holder with 0.5 mm path length,and the measurements were performed using a CTI-Cryogenics Model22 refrigeration unit (Cryophysics SA, Geneva) inserted in the cavity ofa double beam Varian Cary 4 UV-VIS spectrophotometer.
Infrared spectra were recorded on a Bruker Vertex 80v FTIRspectrometer at 25 °C in an Attenuated Total Reflection (ATR)cuvette equipped with a liquid nitrogen – cooled MCT solid statedetector. 4 interferograms were normally combined after registra-tion at a resolution of 1 cm−1. The IR spectra were used tocharacterize the flexibility and conformational dynamics of thestudied proteins based on H/D exchange kinetics. Lyophilizedsamples were dissolved within a few seconds in deuterated100 mM TRIS (pD 8.2) or 100 mM BES (pD 6.8) buffer (reaching aconcentration of about 1.3 mM), and IR spectra were measured infunction of time to follow the kinetics of H/D exchange. We usedthe method of data collection and evaluation introduced by Hvidt etal. [35,36]. The peak absorbances of amide I and amide II bandswere evaluated from the spectra at 1650 and 1547.5 cm−1
respectively, and the fraction of unexchangeable hydrogens (X) at
1966 K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
each registration time was calculated from the ratio of bandintensities corrected for the background. It was assumed that thefluctuations by which the NH bonds become exposed to H/Dexchange are the rate limiting factor in the exchange (EX2 or non-cooperative exchange mechanism):
NHi;buried²k1
k−1NHi;exposed Y
kexchNDi ð1Þ
In this mechanism, kexch is a uniform rate constant for theexchange of totally exposed amide hydrogens, denoted by k0. ThepH- and temperature-dependence of this “chemical” exchange rateconstant has been studied and described as [35]:
k0 ¼ 10−pH þ 10pH −6� �
100:05 T −25ð Þs−1 ð2Þ
where T is the temperature in centigrades. Under dilute conditions forthe proteins, the exchange can be described as the result of a sum ofindependent first order reactions for the NH groups of variousexchange conditions:
X ¼ n−1 ∑n
i¼1e−pik0t ð3Þ
where n is the number of NH groups per protein molecule, and pik0 isthe exchange rate constant of the i'th amino acid, pi being theprobability of solvent exposure of the i'th amide group.
From the data, X versus log(k0t) curves – called relaxation spectra –were determined and compared with the functions corresponding tothose of polymers with uniformly accessible groups determined forgiven probabilities of exposure.
3. Results
3.1. Sample preparation
We considered critical to verify the amount of remaining boundCa2+ after Ca2+ depletion by an independent method. Fig. 1 showsthe total reflection X-ray fluorescence spectrum of Ca2+-depletedHRP. The Ca2+ content of the samples relative to the Fe3+ of theheme can be determined from the area below the peaks aftercalibration. The data that we show correspond to an average Ca2+
content of 0.4 mol of Fe3+ compared to the 2 mol of Ca2+ for onemole of protein in the native enzyme. Smulevich et al. reported [28]similar UV-VIS absorption spectra for the native state of HRP and forthe state of partial Ca2+ depletion (supposedly the lack of proximalCa2+). We show below that the absorption spectrum of HRPsubjected to our Ca2+ depletion procedure is very different fromthat of the native state.
3.2. Optical absorption measurements at room temperature,and pH effects
Literature data concerning pH-dependent effects support the roleof the protonation state of the disHis in maintaining enzyme activity[9,37]. It was also suggested that this residue can be characterized bya pK of slightly above 7 in HRP. Thus, a pH-dependent study wasimplemented in the range of 6.0 to 8.7 with the hope that aprotonation change at disHis would unravel the role of the bounddistal Ca2+ in maintaining the bonding network around the heme. Inoptical absorption measurements at RT the native sample did notshow a pH-dependent behavior, however, the spectrum of the Ca2+-depleted sample was definitely pH sensitive in the studied range. Fig.2 shows characteristic optical absorption spectra of native HRPrecorded at pH 6.8 (it is unchanged in the whole studied pH range)and that of Ca2+-depleted HRP, recorded at pH 6.8, and pH 8.2 at25 °C. In the latter case the spectra are very different: The Soret bandhas a significant shoulder at the short wavelength side at pH 8.2. Thedecrease of the extinction coefficient of both the Soret maximumand the Q band as well as the shift of the peaks report a significantstructural change in the vicinity of the heme in Ca2+-depleted HRPupon changing the pH to be more basic. The maximum positions ofthe main absorption bands and the extinction coefficients of theSoret maxima at RT for the three different samples are summarizedin Table 1.
Fig. 3 shows the position of the Soret band (around 400 nm) andof the charge transfer (CT) band (around 640 nm) in the spectrum ofCa2+-depleted HRP as function of pH. The data for the Soret and CTmaxima can be well fitted with a Henderson–Hassebalch curvedescribing the protonation-deprotonation reaction of a single proton-able residue. The pKa-s obtained from the two bands coincide withinexperimental error (7.4±0.1, and 7.3±0.2 for the Soret and the CTmaximum respectively). These measurements indicate that in Ca2+-
Fig. 1. Total reflection X-ray fluorescence (TXRF) spectrum of Ca2+-depleted HRP. Thepeaks corresponding to Fe and Ca are labeled.
Fig. 2. Absorption spectra of native and Ca2+-depleted HRP. Absorption spectra of 10mMnative HRP at pH 6.8 (continuous line), Ca2+-depleted HRP at pH 6.8 (dotted line) andCa2+-depleted HRP at pH 8.2 (dashed line). Spectra were recorded at 25 °C in 20 mMTRIS/BES buffer. The inset shows the long wavelength range of the spectra.
Table 1Soret-, Q-, and charge transfer (CT) band maximum positions (λ (nm)) at atmosphericand 10 kbar pressure in the absorption spectra and molar extinction coefficient for theSoret band
Sample Soret (nm) Q (nm) CT (nm)
λ (nm) λ (nm) at10 kbar
HRP 402.5 425.0 103 500; 540 644Ca-depleted HRP pH 6.8 403 410.4 81±7 499; 540 641Ca-depleted HRP pH 8.2 390 – 54±6 480, 500,
540, 560627
The error of the measurement is 1 nm.
ɛ (mM−1 cm−1)
1967K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974
Author's personal copy
depleted HRP there is a characteristic structural change in theenvironment of the heme that influences its electronic states, andwhich corresponds to the deprotonation of a single residue with a pKa
of about 7.4. This corresponds to our hypothesis that the deprotona-tion of the disHis residue affects the heme environment in the absenceof Ca2+.
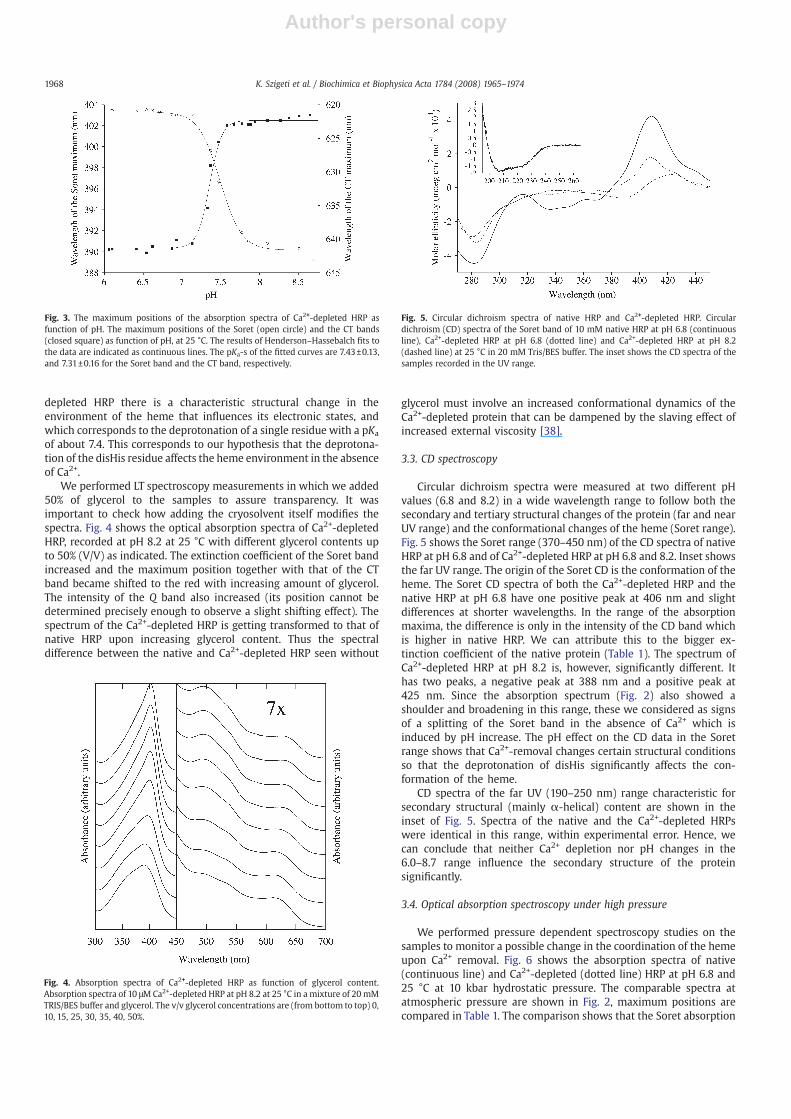
We performed LT spectroscopy measurements in which we added50% of glycerol to the samples to assure transparency. It wasimportant to check how adding the cryosolvent itself modifies thespectra. Fig. 4 shows the optical absorption spectra of Ca2+-depletedHRP, recorded at pH 8.2 at 25 °C with different glycerol contents upto 50% (V/V) as indicated. The extinction coefficient of the Soret bandincreased and the maximum position together with that of the CTband became shifted to the red with increasing amount of glycerol.The intensity of the Q band also increased (its position cannot bedetermined precisely enough to observe a slight shifting effect). Thespectrum of the Ca2+-depleted HRP is getting transformed to that ofnative HRP upon increasing glycerol content. Thus the spectraldifference between the native and Ca2+-depleted HRP seen without
glycerol must involve an increased conformational dynamics of theCa2+-depleted protein that can be dampened by the slaving effect ofincreased external viscosity [38].
3.3. CD spectroscopy
Circular dichroism spectra were measured at two different pHvalues (6.8 and 8.2) in a wide wavelength range to follow both thesecondary and tertiary structural changes of the protein (far and nearUV range) and the conformational changes of the heme (Soret range).Fig. 5 shows the Soret range (370–450 nm) of the CD spectra of nativeHRP at pH 6.8 and of Ca2+-depleted HRP at pH 6.8 and 8.2. Inset showsthe far UV range. The origin of the Soret CD is the conformation of theheme. The Soret CD spectra of both the Ca2+-depleted HRP and thenative HRP at pH 6.8 have one positive peak at 406 nm and slightdifferences at shorter wavelengths. In the range of the absorptionmaxima, the difference is only in the intensity of the CD band whichis higher in native HRP. We can attribute this to the bigger ex-tinction coefficient of the native protein (Table 1). The spectrum ofCa2+-depleted HRP at pH 8.2 is, however, significantly different. Ithas two peaks, a negative peak at 388 nm and a positive peak at425 nm. Since the absorption spectrum (Fig. 2) also showed ashoulder and broadening in this range, these we considered as signsof a splitting of the Soret band in the absence of Ca2+ which isinduced by pH increase. The pH effect on the CD data in the Soretrange shows that Ca2+-removal changes certain structural conditionsso that the deprotonation of disHis significantly affects the con-formation of the heme.
CD spectra of the far UV (190–250 nm) range characteristic forsecondary structural (mainly α-helical) content are shown in theinset of Fig. 5. Spectra of the native and the Ca2+-depleted HRPswere identical in this range, within experimental error. Hence, wecan conclude that neither Ca2+ depletion nor pH changes in the6.0–8.7 range influence the secondary structure of the proteinsignificantly.
3.4. Optical absorption spectroscopy under high pressure
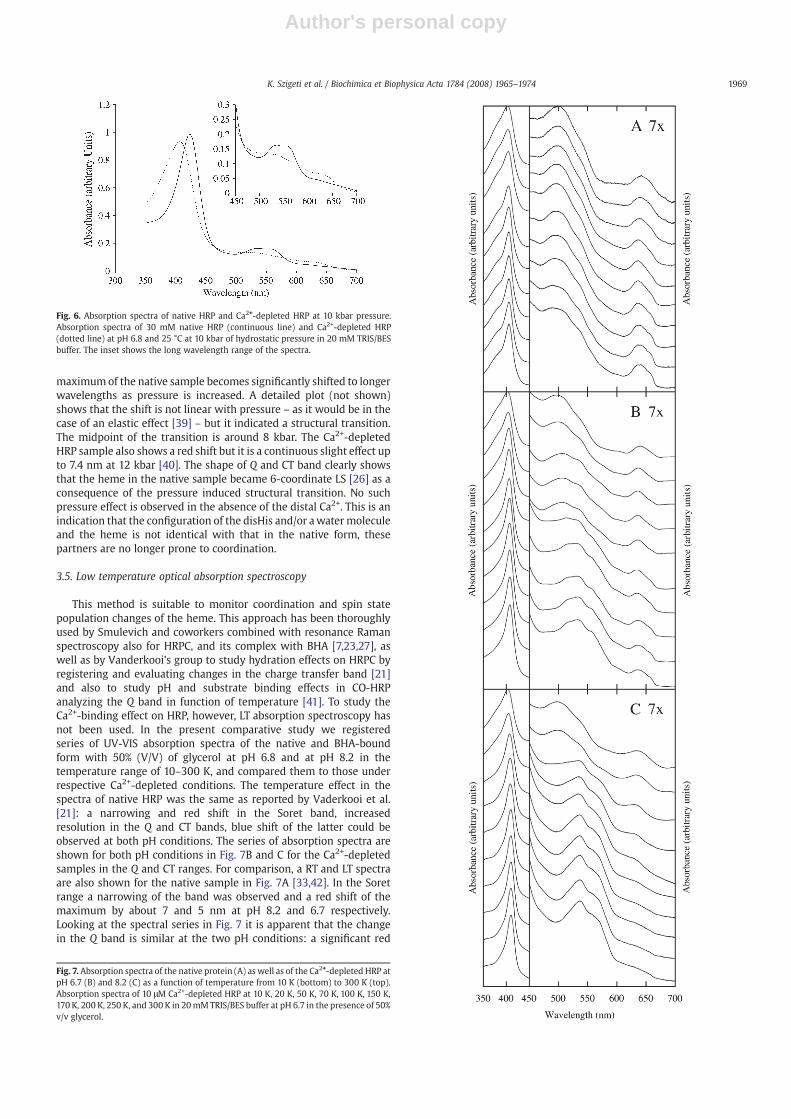
We performed pressure dependent spectroscopy studies on thesamples to monitor a possible change in the coordination of the hemeupon Ca2+ removal. Fig. 6 shows the absorption spectra of native(continuous line) and Ca2+-depleted (dotted line) HRP at pH 6.8 and25 °C at 10 kbar hydrostatic pressure. The comparable spectra atatmospheric pressure are shown in Fig. 2, maximum positions arecompared in Table 1. The comparison shows that the Soret absorption
Fig. 3. The maximum positions of the absorption spectra of Ca2+-depleted HRP asfunction of pH. The maximum positions of the Soret (open circle) and the CT bands(closed square) as function of pH, at 25 °C. The results of Henderson–Hassebalch fits tothe data are indicated as continuous lines. The pKa-s of the fitted curves are 7.43±0.13,and 7.31±0.16 for the Soret band and the CT band, respectively.
Fig. 4. Absorption spectra of Ca2+-depleted HRP as function of glycerol content.Absorption spectra of 10 μMCa2+-depleted HRP at pH 8.2 at 25 °C in a mixture of 20 mMTRIS/BES buffer and glycerol. The v/v glycerol concentrations are (from bottom to top) 0,10, 15, 25, 30, 35, 40, 50%.
Fig. 5. Circular dichroism spectra of native HRP and Ca2+-depleted HRP. Circulardichroism (CD) spectra of the Soret band of 10 mM native HRP at pH 6.8 (continuousline), Ca2+-depleted HRP at pH 6.8 (dotted line) and Ca2+-depleted HRP at pH 8.2(dashed line) at 25 °C in 20 mM Tris/BES buffer. The inset shows the CD spectra of thesamples recorded in the UV range.
1968 K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974
Author's personal copy
maximumof the native sample becomes significantly shifted to longerwavelengths as pressure is increased. A detailed plot (not shown)shows that the shift is not linear with pressure – as it would be in thecase of an elastic effect [39] – but it indicated a structural transition.The midpoint of the transition is around 8 kbar. The Ca2+-depletedHRP sample also shows a red shift but it is a continuous slight effect upto 7.4 nm at 12 kbar [40]. The shape of Q and CT band clearly showsthat the heme in the native sample became 6-coordinate LS [26] as aconsequence of the pressure induced structural transition. No suchpressure effect is observed in the absence of the distal Ca2+. This is anindication that the configuration of the disHis and/or awatermoleculeand the heme is not identical with that in the native form, thesepartners are no longer prone to coordination.
3.5. Low temperature optical absorption spectroscopy
This method is suitable to monitor coordination and spin statepopulation changes of the heme. This approach has been thoroughlyused by Smulevich and coworkers combined with resonance Ramanspectroscopy also for HRPC, and its complex with BHA [7,23,27], aswell as by Vanderkooi's group to study hydration effects on HRPC byregistering and evaluating changes in the charge transfer band [21]and also to study pH and substrate binding effects in CO-HRPanalyzing the Q band in function of temperature [41]. To study theCa2+-binding effect on HRP, however, LT absorption spectroscopy hasnot been used. In the present comparative study we registeredseries of UV-VIS absorption spectra of the native and BHA-boundform with 50% (V/V) of glycerol at pH 6.8 and at pH 8.2 in thetemperature range of 10–300 K, and compared them to those underrespective Ca2+-depleted conditions. The temperature effect in thespectra of native HRP was the same as reported by Vaderkooi et al.[21]: a narrowing and red shift in the Soret band, increasedresolution in the Q and CT bands, blue shift of the latter could beobserved at both pH conditions. The series of absorption spectra areshown for both pH conditions in Fig. 7B and C for the Ca2+-depletedsamples in the Q and CT ranges. For comparison, a RT and LT spectraare also shown for the native sample in Fig. 7A [33,42]. In the Soretrange a narrowing of the band was observed and a red shift of themaximum by about 7 and 5 nm at pH 8.2 and 6.7 respectively.Looking at the spectral series in Fig. 7 it is apparent that the changein the Q band is similar at the two pH conditions: a significant red
Fig. 7. Absorption spectra of the native protein (A) as well as of the Ca2+-depleted HRP atpH 6.7 (B) and 8.2 (C) as a function of temperature from 10 K (bottom) to 300 K (top).Absorption spectra of 10 μM Ca2+-depleted HRP at 10 K, 20 K, 50 K, 70 K, 100 K, 150 K,170 K, 200 K, 250 K, and 300 K in 20mMTRIS/BES buffer at pH 6.7 in the presence of 50%v/v glycerol.
Fig. 6. Absorption spectra of native HRP and Ca2+-depleted HRP at 10 kbar pressure.Absorption spectra of 30 mM native HRP (continuous line) and Ca2+-depleted HRP(dotted line) at pH 6.8 and 25 °C at 10 kbar of hydrostatic pressure in 20 mM TRIS/BESbuffer. The inset shows the long wavelength range of the spectra.
1969K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
shift and increased resolution develop while lowering the tempera-ture. The change in the CT band, however, is totally different. At pH8.2 (Fig. 7C), the relative intensity of this band is decreased relativeto that at pH 6.7 already at RT, and it totally disappears around theglass transition temperature and below. In Fig. 8 we show results ofthe same experiment on the native HRP at pH 6.7 (Fig. 8A) and theCa2+-depleted sample (Fig. 8B), both with BHA bound. The latterspectra were not sensitive to a change in the pH from 6 to 9. In theSoret range, basically only a narrowing (and a slight red shift) canbe observed while lowering the temperature. When comparing theCT band of the native and Ca2+-depleted samples, a similar changecan be concluded: the position of the band does not change but anincreased resolution – may be, a splitting even, especially in thecase of the native sample – is becoming evident toward LT. The Qband behaves differently in the two samples: the change in thenative sample is like a splitting at LT, while in the Ca2+-depletedsample a definite red shift with increased resolution and/or splittingis seen toward LT.
3.6. H/D exchange followed by Fourier Transform Infrared spectroscopy
We were interested in looking at tertiary structural changespossibly induced by Ca2+ removal. We applied the FTIR H/D exchangemethod to unravel such effects. Fig 9. shows the summarized data ofthe relaxation spectra–semilogarithmic plots of the fraction ofunexchangeable amide hydrogens as a function of time [35]. Thespectra of native and Ca2+-depleted HRP were recorded at pD 6.8 in100 mM BES buffer at 25 °C. The results show that the kinetics of H/Dexchange is dramatically different in the native and in the Ca2+-depletedHRP. The kinetics in the native form is highly inhomogeneous,it covers a broad range in solvent accessibility. In the Ca2+-depletedsample, the plot is almost identical with that of a polypeptide withuniformly accessible residues. It is noteworthy that this uniformaccessibility is definitely higher than that of the most accessibleresidues in the native form. The loss of tertiary contacts withoutsignificant change in the secondary structural elements characterizesa transition to a molten globule-like state of the structure [43]. Wethus performed anilinonaphtalene sulphonic acid (ANS) bindingstudies. The fluorescence quantum yield of ANS is known to increasewhen it binds to hydrophobic sites of the protein structure [44], thusit can be used to test the presence of the molten globule state [45].We found a dramatically increased ANS binding to the Ca2+-depleted
Fig. 10. Fluorescence emission spectra of ANS in the presence of native and Ca2+-depleted HRP Titration of 10 μM native HRP at pH 6.8 (open square), Ca2+-depleted HRPat pH 6.8 (open triangle) and Ca2+-depleted HRP at pH 8.2 (open circle) at 25 °C in100 mM Tris/BES buffer with ANS, followed with fluorescence emission intensitymeasurements at 460 nm (λexc=380 nm).
Fig. 9. Results of H/D exchange-FTIR experiment for native HRP and Ca2+-depleted HRP.Function of unexchangeable hydrogens (X) as a function of time. The sample contained:200mM native (open square) or Ca2+-depleted HRP (open triangle) at pD 6.8 in 100mMTRIS pD 6.8 at 25 °C. k0 is the chemical exchange rate constant for amide hydrogenstotally exposed to solvent, k0=(10pH+10pH −6)100.05(T −25). The dotted curves are X-log(k0t) functions corresponding to polymers with amide hydrogens of identicalprobability of exposure as indicated at the curve.
Fig. 8. Absorption spectra of the native (A) and Ca2+-depleted HRP (B) with BHA asfunction of temperature from 10 K (bottom) to 300 K (top) at pH 6.7. Absorption spectraof 10 μM native HRP Ca2+-depleted HRP with 2.06 mM BHA added at 10 K, 20 K, 50 K,70 K, 100 K, 150 K, 170 K, 200 K, 250 K, and 300 K in mixture of 50% 20 mM TRIS/BESbuffer and 50% v/v glycerol.
1970 K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
form of the protein (Fig. 10). At saturation, the fluorescence signalincreased by about a factor of ten at pH 6.8 and by about a factor of20 at pH 8.2. These data confirm the conclusion from the H/Dexchange experiment that total Ca2+ depletion induces a transition inthe tertiary structure of the protein toward a molten globule-like,loosened state, with amide hydrogens more exposed to H/Dexchange. The pH effect on this loosening could not be studied byFTIR, since the kinetics was already so fast at pH 6.8 that it was at theedge of being measurable.
4. Discussion
4.1. Removal of both the proximal and distal Ca2+ ions does not affect thealpha helical content of the protein
The effect of Ca2+-removal by treatment with EDTA on thesecondary structure of HRP has been studied by IR spectroscopy inour group [20] and also in collaborationwith Vanderkooi's group [21].Very little effect was detected in the amide I/I′ band region afterdeconvolution [20] indicating negligible change in the alpha helicalcontent – approximately 2–3% – in both studies. We now believe thattreatment with EDTA alone removes primarily the loosely boundproximal Ca2+. Thus one conclusion from these studies was thatremoval of the loosely bound Ca2+ does not influence the secondarystructure of the protein. When the treatment with EDTA wascombined with high hydrostatic pressure, we observed that therigidly bound Ca2+ ions can be exposed to the chelator at about 2 kbar,and the protein becomes depleted from both Ca2+ ions under suchconditions. Analysis of the spectral series at various pressuresconfirmed that no significant change in the secondary structureoccurred up to the range of pressure denaturation at 10 kbar. Ourearlier molecular dynamics simulation studies [30] showed only anegligible change in the radius of gyration after the removal of bothCa2+ ions. Tanaka et al. reported [9,17,18] that mutations leading to theloss of the distal Ca2+ binding ability did not produce observablechanges in the protein structure. These results are in accordance withthe present CD spectroscopy data showing that even after the removalof both Ca ions there is no change in the alpha helical content in thepH range of 6.0–8.7.
In type II peroxidases (lignin-, mangenase-, versatile peroxidase)the role of bound Ca2+ has also been investigated. The experimentalresults showed that upon Ca2+ removal, the enzyme activity was lost(by 98%) and the secondary structure also changed in these proteins[46–48]. This behavior is totally different from that of HRP. Inspectionof X-ray crystallography data concerning these other peroxidasesshows that the location of the disulfide bridges and the Ca2+ bindingsites are farther away in these proteins while in HRP one of the fourdisulfide bridges is near the distal Ca2+ binding site. This structuralfeature may be the reason why HRP remains stable even after Ca2+
depletion.
4.2. The tertiary structure after Ca2+ depletion
The relaxation spectra (Fig. 9) showed significantly faster H/Dexchange kinetics in the Ca2+-depleted sample than in native HRP. Thekinetics of the Ca2+-free sample at the higher pH was immeasurablyfast, while in the native structure, the pH influenced the exchangesolely by its chemical effect (see “Materials and methods” for details)— data not shown. Accessibility of the amide hydrogens is very diversein native HRP – reflecting the variety of native tertiary contacts –, butthe structural inhomogeneity disappeared in the Ca2+-depletedprotein (Fig. 9). The relaxation spectrum of this condition is close toa case of uniform accessibility (dashed lines in Fig. 9). The solventexposed fraction for this Ca2+-free condition can be estimated fromthe figure as ∼10−n (where 4bnb5). This extent of burial of amidehydrogens is still significant and confirms that the conformation is still
ordered (secondary structure is maintained). It is possible, that at thehigher pH condition, the relaxation curve – if measurable – wouldcorrespond to an even higher degree of exposure of the amidehydrogens, but according to the CD data, we have to suppose that thesecondary structural order does not become lost even at the basic pH.Zavodszky and coworkers found that the Zn-containing Carbonic-Anhydrase B in the apo-form behaved like a denatured protein withtotally exposed amide hydrogens [36]. Denaturation causes uniformaccessibility with a probability of 1. In another study concerning theregulatory domain of Physarum myosin, the removal of bound Ca2+
ions – although somewhat increased the overall flexibility but – didnot influence the inhomogeneity of the tertiary structure as revealedin a H/D exchange-FTIR study [49]. The structure of Ca2+-depleted HRP,as revealed by our H/D exchange study, represents an interesting casewhere the protein maintains its secondary structure with uniformlyloose tertiary contacts after the removal of bound metal ions. If weconsider the X-ray data of native HRP, one can easily reason for theloosening upon the removal of the distal Ca2+. As we discussed in [33],the analysis of the B-factors of X-ray crystallography shows a rigid corein the native protein, and the distal Ca2+ binding site is in the center ofthis rigid region. One could suppose that the fact that Ca2+ depletioncauses only loosening and not the total loss of structural order isrelated to the presence of the four disulfide bridges of the structure.This idea, however, must be reconciled with our earlier pressure-stability studies [20] that have shown an outstanding stability of thisprotein even under reduced conditions. In this respect one has to takeinto account another circumstance that may alter the conclusion fromthe experimental data at high pressure. One has to remember thateight glycan chains are attached to the surface of HRP [3], and underhigh pressure, these sugar chains may be pushed onto the surface ofthe molecule leading to an enhancement of structural stability. Underthe conditions of atmospheric pressure, they might not be enforced tolay onto the surface.We believe that both the disulfide bridges and theglycans may contribute to structural stability even in the absence ofCa2+ ions.
ANS binding studies confirmed the H/D exchange results: thedramatically increased ANS binding capability after Ca2+ depletion isan indication for – at least temporary – exposure of hydrophobicbinding sites that were buried in the native structure. Enhancement ofANS binding without the loss of secondary structure is characteristicfor a molten globule-like structure [45]. The observed ensembleaveraged structure at a given pH we consider as an equilibriumbetween the native andmolten globule-like regions in the structure ofthe molecules. The pH increase enhances the proportion of moltenglobule structures thus the population is more capable of ANS bindingat pH 8.2. Thus we identify the effect of Ca2+-removal as a native tomolten globule transition in the structure.
The narrowing effect of glycerol on the absorption spectrum in thecase of Ca2+ depletion (Fig. 4) is also in accordance with the result thatthe removal of Ca2+ ions causes a general loosening of the structure.Thus the increase of external viscosity (reducing conformationaldynamics) and/or the penetration of glycerol molecules into theprotein may yield a more defined structural distribution of the hemecrevice that manifests itself in a narrowed inhomogeneous broad-ening of the spectral bands [50,51]. Glycerol has no such effect on thenative form of HRP.
4.3. Structural effects of Ca2+ depletion at the heme crevice
Results of LT spectroscopy allow to elaborate a structural model forthe consequence of Ca2+ depletion on the immediate neighborhood ofthe heme. It has been shown in a series of optical absorption andresonance Raman studies on peroxidases that at LT, the porphyrin ringshrinks and a strengthening of the packing forces around the hemecan be supposed [26,27]. At LT, weak coordination and orientationeffects around the heme at RT may be enhanced and thus detected.
1971K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
Howes et al. reported very little change in optical absorption at RTupon Ca2+ depletion, a slight blue shift of the bands, with an averagelack of Ca2+ as 1 mol/1 mol protein [28]. They did not observe changesof the extinction in the Soret band. Under our Ca2+-depletioncondition, we have observed a significant reduction of the Soretextinction coefficient in the Ca2+-depleted form (Table 1.) and the LToptical absorption study clearly indicated a difference between thenative and Ca2+-depleted forms as well (Figs. 7 and 8) (Thiscorresponds to the fact that the extent of Ca2+ depletion was clearlydifferent in the cited study.). An increased resolution and somenarrowing of the Soret band can be seen also in the case of the nativeform at LT. The difference between the two spectra, however, becomesdramatically observable in the Q and CT ranges at basic pH. In theCa2+-depleted sample, at pH 8.2, a component present in smallproportion within the red side of the broad Q band at RT is gainingintensity at LT, and the CT band disappears [31]. The change in the Qrange is not very different at pH 6.7, but the CT band shows adifference. Based on these effects we propose that in the Ca2+-depleted sample awatermolecule –mobile at RT in the loosely packedstructure – in the distal side of the pocket gets oriented by its electricdipole to the heme. The consequence of this is the lowering ofelectronic energies and a red shift in the spectrum [52]. Such arearrangement certainly would affect the whole bonding networkaround the heme. At acidic pH, the water molecule may not besterically allowed to be coordinated to the Fe3+ of the heme. When thedisHis gets deprotonated, however, the coordination becomesallowed. The CD results show that the degeneracy of the Q bandpromoted by the 6-coordinated state of the heme, becomes resolvedat pH 8.2 by splitting into two energetically and also structurallydifferent states (Jahn-Teller effect — [53]). Both of these states areclose to be planar, and 6c, but to manifest this coordination, thesterical enforcement achieved by LT is necessary. This is verified by theresult of the high pressure study: at RT, high pressure was not able toinduce the 6c state in the Ca-depleted sample, thus it confirms thepresence of a distribution of loosely coordinated structures at pH 7.The pressure dependent study also shows that in the native structure,a steric hindrance – a consequence of the ordered structure of thecrevice – opposes the coordination of the sixth ligand (water) that canonly be overcome close to the denaturation pressure, at ∼10 kbar.
In the studies, we made use of the known structural effect ofbinding BHA in the distal side of the heme, that has also been usedin other studies to test structural perturbance on the distal side [28].The idea is based on the published crystal structure of the HRP–BHAcomplex [54]. The non-physiological substrate binds at the distalside of the heme, almost parallel with the heme plane, but too far todirectly interact with it. A water molecule has been shown to bealso present in the distal cavity that was suggested to becoordinated to the Fe3+ ion. In fact, the crystal structure showsthat when BHA binds, the water molecule is shifted closer to theFe3+ of the heme. With BHA bound, the RT absorption spectra arevery similar for the native and the Ca2+-depleted form at bothcompared pH values (Fig. 8A and B). Howes et al. also used theeffect of BHA binding on the spectra and found at RT that there wasonly a slight blue shift of the CT band in the case of the Ca2+-depleted sample as the effect of binding BHA — similarly to ourobservation. The authors used this experience as a strong argumentfor an important role of the proximal Ca2+ [28]. Going to LT yield,however, different effects under our conditions of Ca2+ depletion,especially in the Q range. In the CT range, the cease of bandintensity observed without BHA at pH 8.2 and LT was not observed,but an increased resolution – may be splitting even – becomesvisible instead, just like in the native form with BHA. A red shift inthe Q band upon lowering the temperature is also seen in the caseof the Ca2+-depleted sample with bound BHA (Fig. 8B). In this case,however, the Soret band position does not show a red shift, it onlynarrows with a slightly increased resolution to show a second
component. Thus, the reason cannot be a dipole interaction broughtabout by the shrinking of the heme pocket dimensions toward LT,since in that case, the Soret band should also shift. Also, thecomplex shape of the red shifted Q band suggests a different reasonfor the change in position and resolution. We suggest to see theconsequence of a change in the population, and possibly also in theseparation of two split energy levels in the Q range. A similarsplitting effect of binding a similar substrate, napthohydroxamicacid to Mg-porphyrin-substituted HRP has been reported earlier[55]. The increased resolution at LT in the CT band can indicate alsoa splitting effect. We now similarly suggest – in agreement withHowes et al. [56] – that the heme becomes 6c involving the distalwater molecule when BHA is bound. We propose that at RT, there isnot enough space for the water on the distal side to freely adjustitself (rotate) for dipole orientation [12], thus a red shift is not seenat LT. The 6c state, however, increases the planarity of the heme,which together with an altered symmetry of the electric chargedistribution in the heme crevice promotes the splitting of thedegenerate level responsible for the Q transition [57].
From the comparison of data shown in Fig. 8A and B one can clearlyconclude that binding BHA does not totally shield the heme from thestructural effect of Ca2+ depletion. The spectra of the BHA-bound Ca2+-depleted protein being insensitive to the pH is an indication that theremaining effect is not related to the distal His42. When BHA is notthere, and the Ca2+ ions are removed, the pH effect on the LT spectrashows that the distal histidine is gaining significance in establishingthe environment of the heme. The Soret band of the Ca2+-free samplebecomes significantly narrower at RT when BHA is bound, thus thisbinding may restore somewhat the loosening effect of Ca2+ depletion,but the interaction with the immediate partners of the heme will notbe restored. In the discussion above, we strongly relied on the effect ofa water molecule in the distal side of the heme crevice [12]. The pHeffects in the spectral studies can be clearly related to the role of thedistal His42 [9]. Molecular dynamics simulations suggest that total Ca2+ depletion decreases the distance between Fe3+ and His42 [30,58].The role of the disHis alone, however, would not be able to account forthe effects.
We conclude that coordination and dipole orientation effects onthe distal side may be directly related to a water molecule, but it mustbe connected to the distal His42. In the spectral changes, the role ofaltered position and orientation between the heme and other
Fig. 11. Structural model structure of the heme and the distal Ca2+ binding site of HRP.The distal Ca2+ is coordinated, to one of the oxygen atoms of the Asp43 carboxylategroup to one oxygen of the backbone carbonyl group and to one oxygen of thecarboxylate group of the Glu64 via a water molecule. Glu64 is connected to Asn70 via ahydrogen bond and Asn70 and disHis are connected with hydrogen bond as well. ThedisHis and Arg 38 are connected to Fe3+ via a water molecule. The proximal Ca2+ islinked to the proHis (His170) via Thr171 which is coordinated to the Fe3+ of the heme.The Ca2+ and Fe3+ ions are indicated as yellow and purple spacefill spheres. The figurewas created from the 1atj.pdb coordinates [14].
1972 K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
neighboring aromatic amino acids in the heme pocket (Phe, Tyr) mustalso be considered.
4.4. Structural aspects of the pH effect on the protein structure in theabsence of Ca2+ ions
The pH effect confirms earlier suggestions for the structuralconnection of the distal Ca2+ and the distal His42. Based on theavailable structural data on native HRP [14], and on suggestedmodels for bonding networks around the heme we suggest astructural model to interpret the pH-dependent Ca2+-depletioneffects. We show a sketch of this structural model in Fig. 11. Basedon EPR, NMR and resonance Raman investigations of Asn70 mutants[37] it was established that a H-bond must exist between Asn70 andHis42, which was later confirmed by all crystal structures [14]. Theenzyme activity of Glu64, and the Asn70 mutants, was shown to bedecreased dramatically compared to the wild type enzyme [16,17].Mutation of Glu64 to Gly, Ser or Pro causes the dissociation of thedistal Ca2+ [18]. These results together with the structural data leadto the conclusion that Glu64 interacts with the distal Ca2+ through awater molecule and thus strengthens the stability of the distal Ca2+
binding site. Asn70 is connected to disHis42 and to the distal Ca2+ viaGlu64 and water. The neighboring residue of His42 is Asp43 which iscoordinated to the distal Ca2+. His42 is connected to the Fe3+ of theheme via water and also connects to the Ca2+ via Asp43. Thisstructure explains our experimental findings. Deprotonation ofdisHis42 eliminates the hydrogen bond between the disHis andAsn70 above pH 7.4, but the distal Ca2+ can still stabilize theadequate position of it. Ca2+ depletion combined with basic pHeliminates the two stabilizing bonds which keep His42 in theorientation necessary for the enzyme function. Both stabilizingbranch become affected in the Ca2+-depleted protein, lacking thehydrogen bridge between Asn70 and His42 above pH 7.4.
4.5. Conclusions
The effect of deleting both Ca2+ ions of HRPC – that are vital for theenzyme activity – does not change the alpha helicity of the protein,but it changes the whole tertiary structure to a loosened, moltenglobule-like conformation. The Ca2+ depletion also introduces tertiarystructural changes in the immediate vicinity of the heme. The RT andLT absorption spectroscopy data can be interpreted by a significantrearrangement of the structure in the distal side, involving themobility, role in the H-bonding network and coordination of a watermolecule defining also the position of the distal histidine that isindirectly bound to the distal Ca2+ if present. We did not observeeffects that needed to involve the role of the proximal Ca2+. Thesignificant structural effects in the overall protein structure and also inthe heme pocket reported in this study indicate that the binding of thedistal Ca2+ is of essential importance.
Acknowledgements
The authors acknowledge support from Hungarian Grants, OTKAT049213 (SL), TÉT A-4/2005 (JF, SzO, KSz), and ETT 512/2006 (JF, SzO),and from the PhD program (no. 3/1) of the Semmelweis UniversityBudapest.
References
[1] A.T. Smith, N.C. Veitch, Structural versatility of heme peroxidases for substratebinding and catalysis, Curr. Opin. Chem. Biol. 2 (1998) 269–278.
[2] O. Ryan, M.R. Smyth, C. Fagain, Horseradish peroxidase: the analyst's friend, z 28(1994) 129–146.
[3] N.C. Veitch, A.T. Smith, Horseradish peroxidase, Adv. Inorg. Chem. 51 (2001)107–162.
[4] H.B. Dunford, Heme Peroxidases, Wiley-VCH, New York, 1999.
[5] Q. Huang, M. Laberge, K. Szigeti, J. Fidy, R. Schweitzer-Stenner, Resonance Ramanspectroscopy study of change of iron spin state in horseradish peroxidase Cinduced by removal of calcium, Biopolymers 72 (2003) 241–248.
[6] G.N. La Mar, J.S. de Ropp, K.M. Smith, K.C. Langry, Proton nuclear magneticresonance study of the electronic and molecular structure of the heme crevice inhorseradish peroxidase, J. Biol. Chem. 255 (1980) 6646–6652.
[7] G. Smulevich, A.M. English, A.R. Mantini, M.P. Marzocchi, Resonance Ramaninvestigation of ferric iron in horseradish peroxidase and its aromatic donorcomplexes at room and low temperatures, Biochemistry 30 (1991) 772–779.
[8] I. Morishima, S. Ogawa, Proton nuclear magnetic resonance spectra of compoundsI and II of horseradish peroxidase, Biochemistry 17 (1978) 4384–4388.
[9] J. Teraoka, T. Kitagawa, Structural implication of the heme-linked ionization ofhorseradish peroxidase probed by the Fe-Histidine stretching Raman line, J. Biol.Chem. 256 (1981) 3969–3977.
[10] R. Haschke, J.M. Friedhoff, Calcium-related properties of horseradish peroxidase,Biochem. Biophys. Res. Commun. 80 (1978) 1039–1042.
[11] S. Ogawa, Y. Shiro, I. Morishima, Calcium binding by horseradish peroxidase C andthe heme environmental structure, Biochem. Biophys. Res. Commun. 90 (1979)674–678.
[12] Y. Shiro, M. Kurono, I. Morishima, Presence of Endogenous calcium ion and itsfunctional and structural regulation in horseradish peroxidase, J. Biol. Chem. 261(1986) 9382–9390.
[13] I. Morishima, M. Kurono, Y. Shiro, Presence of endogenous calcium ion inhorseradish peroxidase, J. Biol. Chem. 261 (1986) 9391–9399.
[14] M. Gajhede, D.J. Schuller, A. Henriksen, A.T. Smith, T.L. Poulos, Crystal structure ofhorseradish peroxidase C at 2.15 A resolution, Nat. Struct. Mol. Biol. 4 (1997)1032–1038.
[15] S. Nagano, M. Tanaka, K. Ishimori, Y. Watanabe, I. Morishima, Catalytic roles of thedistal site asparagine-histidine couple in peroxidases, Biochemistry 35 (1996)14251–14258.
[16] M. Tanaka, S. Nagano, K. Ishimori, I. Morishima, Hydrogen bond network in thedistal site of peroxidases: spectroscopic properties of Asn70-Asp horseradishperoxidase mutant, Biochemistry 36 (1997) 9791–9798.
[17] M. Tanaka, A. Morimoto, K. Ishimori, I. Morishima, Structure–activity relation ofhorseradish peroxidases as studied with mutations at heme distal and proximalsites, Pure Appl. Chem. 70 (1998) 911–916.
[18] M. Tanaka, K. Ishimori, M. Mukai, T. Kitagawa, I. Morishima, Catalytic activities andstructural properties of horseradish peroxidase distal His42→Glu or Gln mutant,Biochemistry 36 (1997) 9889–9898.
[19] M. Tanaka, K. Ishimori, I. Morishima, Structural roles of the highly conserved Gluresidue in heme distal site of peroxidase, Biochemitry 37 (1998) 2629–2638.
[20] L. Smeller, F. Meersman, J. Fidy, K. Heremans, High-pressure FTIR Study of thestability of horseradish peroxidase. Effect of heme substitution, ligand binding,Ca removal, and reduction of the disulfide bonds, Biochemistry 42 (2003)553–561.
[21] A.D. Kaposi, J. Fidy, E.S. Manas, J.M. Vanderkooi, W.W. Wright, Horseradishperoxidase monitored by infrared spectroscopy: effect of temperature, substrateand calcium, Biochim. Biophys. Acta 1435 (1999) 41–50.
[22] A. Feis, M.P. Marzocchi, M. Paoli, G. Smulevich, Spin state and axial ligand bondingin the hydroxide complexes of metmyoglobin, methemoglobin, and horseradishperoxidase at room and low temperatures, Biochemistry 33 (1994) 4577–4583.
[23] B.D. Howes, J.N. Rodrigez-Lopez, A.T. Smith, G. Smulevich, Mutation of distalresidues of horseradish peroxidase: influence on substrate binding and cavityproperties, Biochemistry 36 (1997) 1532–1543.
[24] A. Feis, B.D. Howes, C. Indiani, G. Smulevich, Resonance Raman and electronicabsorption spectra of horseradish peroxidase isozyme A2: evidence for aquantum-mixed spin species, J. Raman Spectrosc. 29 (1998) 933–938.
[25] G. Smulevich, A. Feis, C. Indiani, M. Becucci, M.P. Marzocchi, Peroxidase-benzhydroxamic acid complexes: spectroscopic evidence that a Fe-H2O distanceof 2.6 angstrom can correspond to hexa-coordinate high-spin heme, J. Biol. Inorg.Chem. 4 (1999) 39–47.
[26] B.D. Howes, A. Feiss, C. Indiani, M.P. Marzocchi, G. Smulevich, Formation of twotype of low-spin heme in horseradish peroxidase isoenzyme A2 at lowtemperature, J. Biol. Inorg. Chem. 5 (2000) 227–235.
[27] C. Indiani, A. Feis, B.D. Howes, M.P. Marzocchi, G. Smulevich, Benzohydroxamicacid-peroxidase complexes: spectroscopic characterization of a novel heme spinspecies, JACS 122 (2000) 7368–7376.
[28] B.D. Howes, A. Feiss, L. Raimondi, C. Indiani, G. Smulevich, The critical role of theproximal calcium ion in the structural properties of horseradish peroxidase, J. Biol.Chem. 276 (2001) 40704–40711.
[29] H.A. Heering, A.T. Smith, G. Smulevich, Spectroscopic characterization ofmutations at the Phe41 position in the distal haem pocket of horseradishperoxidase C: structural and functional consequences, Biochem. J. 363 (2002)571–579.
[30] M. Laberge, Q. Huang, R. Schweitzer-Stenner, J. Fidy, The endogenous calcium ionsof horseradish peroxidase C are required to maintain the functional nonplanarityof the heme, Biophys. J. 84 (2003) 2542–2552.
[31] M. Laberge, K. Szigeti, J. Fidy, The charge transfer band in horseradish peroxidasecorrelates with heme in-plane distortions induced by calcium removal, Biopoly-mers 74 (2004) 41–45.
[32] M. Laberge, I. Kovesi, T. Yonetani, J. Fidy, Normal mode analysis of the horseradishperoxidase collective motions: correlationwith spectroscopicalliy observed hemedistortions, Biopolymers 82 (2006) 425–429.
[33] Q. Huang, K. Szigeti, J. Fidy, R. Schweitzer-Stenner, Structural disorder of nativehorseradish peroxidase C probed by resonance Raman and low-temperatureoptical absorption spectroscopy, J. Phys. Chem., B 107 (2003) 2822–2830.
1973K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974

Author's personal copy
[34] A. Prange, H. Boddeker, W. Michaelis, Multielement determination of traceelements in whole blood and blood serum by TXRF, Fresenius Z. Anal. Chem. 335(1989) 914–918.
[35] A. Hvidt, K. Wallevik, Conformational changes in human serum albumin asrevealed by hydrogen-deuterium exchange studies, J. Biol. Chem. 247 (1971)1530–1535.
[36] P. Zavodszky, J.T. Johansen, A. Hvidt, Hydrogen-exchange study of the conforma-tional stability of human carbonic anhydrase B and its metallocomplexes, Eur. J.Biochem. 56 (1975) 67–72.
[37] M. Mukai, S. Nagano, M. Tanaka, K. Ishimori, I. Morishima, T. Ogura, Y.Watanabe, T.Kitagawa, Effects of concerted hydrogen bonding of distal histidine on active sitestructures of horseradish peroxidase. Resonance Raman studies with Asn70Mutants, J. Am. Chem. Soc. 119 (1997) 1758–1766.
[38] H. Frauenfelder, P.W. Fenimore, B.H. McMahon, Hydration, slaving and proteinfunction, Biophys. Chem. 98 (2002) 35–48.
[39] B.B. Laird, J. Skinner, Microscopic theory of reversible pressure broadening inhole-burning spectra of impurities in glasses, Chem. Phys. 90 (1989)3274–3281.
[40] L. Smeller, F. Meersman, J. Fidy, K. Heremans, High-pressure FTIR Study of thestability of horseradish peroxidase. Effect of heme substitution, ligand binding,Ca removal, and reduction of the disulfide bonds, Biochemistry 42 (2003)553–561.
[41] A.D. Kaposi, W.W. Wright, J. Fidy, S.S. Stavrov, J.M. Vanderkooi, I. Rasnik,Carbonmonoxy horseradish peroxidase as a function of pH and substrate:influence of local electric fields on the optical and infrared spectra, Biochemistry40 (2001) 3483–3491.
[42] B. Zelent, A.D. Kaposi, N.V. Nucci, K.A. Sharp, S.D. Dalosto, W.W. Wright, J.M.Vanderkooi, Water channel of horseradish peroxidase studied by the charge-transfer absorption band of Ferric heme, J. Phys. Chem., B 108 (2004) 10317–10324.
[43] M. Ohgushi, A. Wada, “Molten-globule state”: a compact form of globular proteinswith mobile side-chains, FEBS. Lett. 164 (1983) 14–21.
[44] L. Stryer, The interaction of a naphthalene dye with apomyoglobin andapohemoglobin. A fluorescent probe of non-polar binding sites, J. Mol. Biol. 13(1965) 482–495.
[45] O.B. Ptitsyn, R.H. Pain, G.V. Semisotnov, E. Zerovnik, O.I. Razgulyaev, Evidence for amolten globule state as a general intermediate in protein folding, FEBS Lett. 262(1990) 20–24.
[46] G.R.J. Sutherland, L.S. Zapanta, M. Tien, S.D. Aust, Role of calcium in maintainingthe heme environment of manganese peroxidase, Biochemistry 36 (1997)3654–3662.
[47] G. Nie, S.D. Aust, Spectal changes of lignin peroxidase during reversibleinactivation, Biochemistry 36 (1997) 5113–5119.
[48] J. Verdin, R. Pogni, A. Baeza, C.M. Barato, R. Basosi, R. Vasquez-Duhalt, Mechanismof versatile peroxidase inactivation by Ca2+ depletion, Biophys. Chem. 121 (2006)163–170.
[49] J.E. Debreczeni, L. Farkas, V. Harmat, C.s. Hetenyi, I. Hajdu, P. Zavodszky, K. Kohama,L. Nyitrai, Structural evidence for non-canonical binding of Ca2+myosin, J. Biol.Chem. 280 (2005) 41458–41464.
[50] J. Schlichter, J. Friedrich, L. Herenyi, J. Fidy, Trehalose effect on low temperatureprotein dynamics: fluctuation and relaxation phenomena, Biophys. J. 80 (2001)2011–2017.
[51] H. Frauenfelder, P.W. Fenimore, B.H. McMahon, Hydration, slaving and proteinfunction, Biophys. Chem. 98 (2002) 35–48.
[52] W.C. Galley, R.M. Purkey, Role of heterogeneity of the solvation site in electronicspectra in solution, Proc. Natl. Acad. Sci. 67 (1970) 1116–1121.
[53] I.B. Bersuker, S.S. Stavrov, Structure and properties of metalloporphyrins andhemoproteins: the vibronic approach, Coord. Chem. Rev. 88 (1988) 1–68.
[54] A. Henriksen, D.J. Schuller, K. Meno, K.G. Welinder, A.T. Smith, M. Gajhede,Structural interactions between horseradish peroxidase C and the substratebenzhydroxamic acid determined by X-ray crystallography, Biochemistry 37(1998) 8054–8060.
[55] E. Balog, K. Kis-Petik, J. Fidy, M. Köhler, J. Friedrich, Interpretation of multiple Q(0,0) bands in the absorption spectrum of Mg-mesoporphyrin embedded inhorseradish peroxidase, Biophys. J. 73 (1997) 397–405.
[56] G. Smulevich, A. Feis, C. Indiani, M. Becucci, M.P. Marzocchi. Peroxidase-benzhydroxamic acid complexes: spectroscopic evidence that a Fe-H2O distanceof 2.6 angstrom can correspond to hexa-coordinate high-spin heme. J. Biol. Inorg.Chem. 4 (99) (1999) 39–47.
[57] J. Fidy, K.G. Paul, J.M. Vanderkooi, Differences in the binding of aromatic substratesto horseradish peroxidase revealed by fluorescence line narrowing, Biochemistry28 (1989) 531–7541.
[58] L. Banci, P. Carloni, A. Diaz, G.G. Savellini, Molecular dynamics calculation onperoxidases: the effect of calcium ions on protein structure, J. Biol. Inorg. Chem. 1(1996) 264–272.
1974 K. Szigeti et al. / Biochimica et Biophysica Acta 1784 (2008) 1965–1974
Related Documents











