Original Article The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic–reperfused heart Raja B. Singh, Punam K. Chohan, Naranjan S. Dhalla, Thomas Netticadan * Institute of Cardiovascular Sciences, St. Boniface General Hospital Research Centre, and Department of Physiology, Faculty of Medicine, University of Manitoba, 351 Tache Avenue, Winnipeg, Man., Canada R2H 2A6 Received 17 February 2004; received in revised form 9 April 2004; accepted 19 April 2004 Available online 08 June 2004 Abstract Ca 2+ overload and free-radical injury are two mutually non-exclusive phenomena suggested to cause myocardial ischemia–reperfusion (IR)-induced contractile dysfunction; however, the mechanisms underlying their effects are not clear. One possible mechanism is the proteolytic modification of proteins by Ca 2+ -dependent proteases, such as calpains, which are activated during Ca 2+ overload that occurs in IR. The sarcoplasmic reticulum (SR) plays a central role in mediating cardiac contractility and therefore any impairment in SR function will induce cardiac contractile dysfunction. We therefore investigated the possibility whether SR proteins were the target for calpain action in IR. Langendorff-perfused rat hearts were subjected to IR in the presence and absence of leupeptin, a calpain inhibitor and the effects of calpain inhibition was examined on cardiac performance, SR function, and its regulation by protein phosphorylation as well as expression of SR Ca 2+ -cycling and -regulatory proteins. Our results show a depression in cardiac contractile function and activation of calpain during IR. Treatment with leupeptin recovered cardiac contractile function and attenuated calpain activity in IR hearts. The cardioprotection observed upon leupeptin treatment was associated with improved SR function and regulation. The recovery in SR function and regulation was consistent with prevention of IR-induced decrease in the expression of key SR Ca 2+ -handling and -regulatory proteins. Our results suggest that a downregulation of SR proteins by calpain may be a mechanism by which Ca 2+ overload causes cardiac contractile dysfunction during IR. © 2004 Elsevier Ltd. All rights reserved. Keywords: Sarcoplasmic reticulum; Ischemia reperfusion; Protease; Calpain 1. Introduction Early restoration of blood flow is critical for resuscitation of the ischemic myocardium; however, if reperfusion is not instituted within a certain time period of the ischemic insult it results in cardiac contractile dysfunction [1–4]. Post- ischemic myocardial dysfunction has been shown to occur as a consequence of reperfusion and not ischemia alone [1–4]. Ischemia–reperfusion (IR)-induced injury can vary from myocardial stunning to ventricular arrhythmias and cell death. Two decades of research has resulted in the emergence of the oxyradical and Ca 2+ -overload hypotheses as two sig- nificant mutually non-exclusive phenomena underlying IR injury [5]. However, the exact mechanisms by which ox- yradicals and Ca 2+ overload cause IR injury are not clear. In this regard, an increase in cytosolic-free Ca 2+ that occurs during IR [6] results in the activation of degradative en- zymes, such as Ca 2+ -activated proteases [7]. Calpain (EC 3.4.22.17) is a cysteine protease ubiquitously present in many tissues including the myocardium [8]. It causes limited proteolysis [9] and is activated by IR [10]. Calpains exist as two isoforms: I and II also called μ and m, respectively, due to their micro (μ) and milli (m) molar Ca 2+ requirements for activation [11,12]. Both isoforms are heterodimers made up of a large 80 kDa and a small 28–30 kDa subunit. Calpain has a Ca 2+ -binding domain similar to calmodulin and its activity is regulated by Ca 2+ concentration [11,12]. It has been impli- cated in the degradation of intracellular cytoskeletal proteins, such as fodrin [13], desmin, a-actinin, and spectrin [14], as well as a contractile protein troponin I [15], and could there- fore be a possible cause for IR-induced cardiac contractile dysfunction. Due to its ability to regulate intracellular Ca 2+ required for contraction and relaxation, the sarcoplasmic reticulum (SR) plays a central role in mediating cardiac contractility [16]. * Corresponding author. Tel.: +1-204-237-2691; fax: +1-204-233-6723. E-mail address: [email protected] (T. Netticadan). Journal of Molecular and Cellular Cardiology 37 (2004) 101–110 www.elsevier.com/locate/yjmcc © 2004 Elsevier Ltd. All rights reserved. doi:10.1016/j.yjmcc.2004.04.009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Original Article
The sarcoplasmic reticulum proteins are targets for calpain actionin the ischemic–reperfused heart
Raja B. Singh, Punam K. Chohan, Naranjan S. Dhalla, Thomas Netticadan *
Institute of Cardiovascular Sciences, St. Boniface General Hospital Research Centre, and Department of Physiology, Faculty of Medicine, University ofManitoba, 351 Tache Avenue, Winnipeg, Man., Canada R2H 2A6
Received 17 February 2004; received in revised form 9 April 2004; accepted 19 April 2004
Available online 08 June 2004
Abstract
Ca2+ overload and free-radical injury are two mutually non-exclusive phenomena suggested to cause myocardial ischemia–reperfusion(IR)-induced contractile dysfunction; however, the mechanisms underlying their effects are not clear. One possible mechanism is theproteolytic modification of proteins by Ca2+-dependent proteases, such as calpains, which are activated during Ca2+ overload that occurs in IR.The sarcoplasmic reticulum (SR) plays a central role in mediating cardiac contractility and therefore any impairment in SR function willinduce cardiac contractile dysfunction. We therefore investigated the possibility whether SR proteins were the target for calpain action in IR.Langendorff-perfused rat hearts were subjected to IR in the presence and absence of leupeptin, a calpain inhibitor and the effects of calpaininhibition was examined on cardiac performance, SR function, and its regulation by protein phosphorylation as well as expression of SRCa2+-cycling and -regulatory proteins. Our results show a depression in cardiac contractile function and activation of calpain during IR.Treatment with leupeptin recovered cardiac contractile function and attenuated calpain activity in IR hearts. The cardioprotection observedupon leupeptin treatment was associated with improved SR function and regulation. The recovery in SR function and regulation was consistentwith prevention of IR-induced decrease in the expression of key SR Ca2+-handling and -regulatory proteins. Our results suggest that adownregulation of SR proteins by calpain may be a mechanism by which Ca2+ overload causes cardiac contractile dysfunction during IR.© 2004 Elsevier Ltd. All rights reserved.
Keywords: Sarcoplasmic reticulum; Ischemia reperfusion; Protease; Calpain
1. Introduction
Early restoration of blood flow is critical for resuscitationof the ischemic myocardium; however, if reperfusion is notinstituted within a certain time period of the ischemic insult itresults in cardiac contractile dysfunction [1–4]. Post-ischemic myocardial dysfunction has been shown to occur asa consequence of reperfusion and not ischemia alone [1–4].Ischemia–reperfusion (IR)-induced injury can vary frommyocardial stunning to ventricular arrhythmias and celldeath. Two decades of research has resulted in the emergenceof the oxyradical and Ca2+-overload hypotheses as two sig-nificant mutually non-exclusive phenomena underlying IRinjury [5]. However, the exact mechanisms by which ox-yradicals and Ca2+ overload cause IR injury are not clear. Inthis regard, an increase in cytosolic-free Ca2+ that occurs
during IR [6] results in the activation of degradative en-zymes, such as Ca2+-activated proteases [7]. Calpain (EC3.4.22.17) is a cysteine protease ubiquitously present inmany tissues including the myocardium [8]. It causes limitedproteolysis [9] and is activated by IR [10]. Calpains exist astwo isoforms: I and II also called µ and m, respectively, due totheir micro (µ) and milli (m) molar Ca2+ requirements foractivation [11,12]. Both isoforms are heterodimers made upof a large 80 kDa and a small 28–30 kDa subunit. Calpain hasa Ca2+-binding domain similar to calmodulin and its activityis regulated by Ca2+ concentration [11,12]. It has been impli-cated in the degradation of intracellular cytoskeletal proteins,such as fodrin [13], desmin, a-actinin, and spectrin [14], aswell as a contractile protein troponin I [15], and could there-fore be a possible cause for IR-induced cardiac contractiledysfunction.
Due to its ability to regulate intracellular Ca2+ required forcontraction and relaxation, the sarcoplasmic reticulum (SR)plays a central role in mediating cardiac contractility [16].
* Corresponding author. Tel.: +1-204-237-2691; fax: +1-204-233-6723.E-mail address: [email protected] (T. Netticadan).
Journal of Molecular and Cellular Cardiology 37 (2004) 101–110
www.elsevier.com/locate/yjmcc
© 2004 Elsevier Ltd. All rights reserved.doi:10.1016/j.yjmcc.2004.04.009

Any impairment in SR function could therefore induce car-diac contractile dysfunction. Our earlier studies have shownthat the cardiac contractile function was reduced in IR heartsand this reduction was intimately associated with a depres-sion in SR function [17–20]. Decreased SR function was inturn caused by a downregulation of critical SR Ca2+-handlingproteins [17,19,20]. In view of the activation of calpains anda downregulation of key SR proteins in IR, we have investi-gated the possibility whether SR proteins were targets forcalpain action in the IR hearts.
2. Methods
All experimental protocols for animal studies were ap-proved by the Animal Care Committee of the University ofManitoba following guidelines set forth by the CanadianCouncil on Animal Care.
2.1. Heart perfusion and experimental protocol
Male Sprague–Dawley rats weighing 225–275 g wereanesthetized using a cocktail of ketamine and xylazine in adose of 1.0 ml/kg body weight and hearts rapidly excised,hung onto a Langendorff’s perfusion apparatus and perfusedwith Krebs–Henseleit solution (37 °C) gassed with a mixtureof 95% O2 and 5% CO2 at a pH of 7.4, containing (inmmol/l): 120 NaCl, 25 NaHCO3, 11 glucose, 4.7 KCl,1.2 KH2PO4, 1.2 MgSO4, and 1.25 CaCl2. The hearts wereelectrically stimulated at a rate of 300 beats/min (Harvard
6002 stimulator from Harvard Apparatus, Holliston, MA)and perfusion rate maintained at 10 ml/min. A water-filledlatex balloon was inserted in the left ventricle and connectedto a pressure transducer (Model 1050BP; BioPac system Inc.,Goleta, CA) to record left ventricular systolic and diastolicpressure measurements. Left ventricular developed pressure(LVDP), left ventricular end-diastolic pressure (LVEDP),rate of ventricular pressure development (+dP/dt), and therate of ventricular pressure decline/decay (–dP/dt) were cal-culated using the Acknowledge 3.5.3 software for Windows(BioPac system Inc., Goleta, CA).
All hearts were stabilized for a period of 20 min beforebeing assigned to different groups. Hearts were maintained ata constant temperature (37 °C) throughout the experiment.Control hearts were perfused for a period of 100 min after20 min of stabilization. Hearts were made globally ischemicby stopping the flow for 30 min. IR was induced by reperfus-ing the 30 min globally ischemic hearts for a period of60 min. In another group, IR hearts were treated with the bestdose of leupeptin 25 µmol/l, obtained after carrying out adose–response study. Leupeptin was infused for 10 min be-fore inducing ischemia and 20 min after ischemia beginningat the onset of reperfusion; control hearts were perfused for120 min. Another series of experiments was designed toobserve the effect of leupeptin pre-treatment (for 10 min) onhearts made globally ischemic for 30 min; control heartswere perfused for 60 min. The scheme for perfusion is shownin Fig. 1. At the end of the experiments hearts were freezeclamped and stored at –80 °C.
Fig. 1. Experimental protocol for perfusing isolated rat hearts under different conditions. In Panel A, 1 and 2 show control hearts treated with and withoutleupeptin. In Panel A, 3 and 4 show IR hearts treated with and without leupeptin. IR was induced by reperfusing the globally ischemic hearts for 60 min. Heartswere treated with leupeptin for 10 min before and 20 min after ischemia in the IR study. In Panel B, 1 and 2 show control hearts treated with and withoutleupeptin. In Panel B, 3 and 4 show ischemic hearts treated with and without leupeptin. Global ischemia was induced by stopping coronary flow for 30 min.Hearts were treated with leupeptin for 10 min before ischemia study. C1, control (2 h); C1T, control with leupeptin treatment; IR, 60 min reperfusion of heartsexposed to 30 min ischemia; IRT, IR hearts treated with leupeptin; C2, control (1 h); C2T, control with leupeptin treatment; I, hearts exposed to 30 min ischemia;IT, ischemic hearts treated with leupeptin.
102 R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

2.2. SR preparation
SR vesicles were obtained by a method described previ-ously [17–20]. Ventricular tissue was pulverized and homog-enized in a buffer containing (in mmol/l): 10 NaHCO3,5 NaN3, 15 Tris–HCl at pH 6.8 (10 ml/g tissue) with apolytron homogenizer (Brinkmann, Westbury, NY). The ho-mogenate was centrifuged for 20 min at 10,919 g. The result-ant pellet was discarded and the supernatant further centri-fuged for 45 min at 43,666 g (Beckman, JA 20). Thesupernatant obtained after the last step contained the cytoso-lic fraction and was aliquoted and frozen in liquid nitrogenprior to storage at –80 °C. The resultant pellet was resus-pended in a buffer containing 0.6 mol/l KCl and 20 mmol/lTris–HCl (pH 6.8) and centrifuged for 45 min at 43,666 g.The final pellet containing the SR fraction was suspended ina buffer containing 250 mM sucrose and 10 mM histidine(pH 7.0), aliquoted, and frozen in liquid nitrogen prior tostorage at –80 °C. All buffers used for isolation contained acocktail of protease inhibitors consisting of aprotinin, leu-peptin, AEBMSF, and 0.1% phenylmethylsulfonyl fluoride(PMSF) to prevent any degradation of proteins during theisolation procedure.
2.3. Calpain activity measurements
Calpain activity was measured in the cytosolic and SRfractions by a calpain assay kit from Calbiochem. The assayis based on the fluorometric detection of cleavage of calpainsubstrate Ac-LLY-AFC. Protein concentration of cytosolic/SR samples was normalized to 200 µg. Active calpain (1 µg/l)was added to the extraction buffer and used as a positivestandard. For the negative standard, a calpain inhibitor (Z-LLY-FMK) was added to the cytosol from control samples.The reaction buffer and substrate were added to all samplesincluding the standards. The reaction was carried out in a96-well plate that was protected from light and incubated for1 h at 37 °C with shaking. The samples were read in a Geminifluorescence microplate reader from Molecular Devices andresults expressed as relative fluorescent units (RFUs).
2.4. Measurement of Ca2+ uptake
SR Ca2+ uptake was measured by a procedure describedearlier [17,19,20]. The reaction mixture contained (inmmol/l): 50 Tris–maleate (pH 6.8), 5 NaN3, 5 ATP, 5 MgCl2,120 KCl, 5 potassium oxalate, 0.1 EGTA, 0.1 45CaCl2(20 mCi/l), and 25 µmol/l ruthenium red. Ruthenium red, ablocker of the SR Ca2+-release channel was added to inhibitSR Ca2+ release. The reaction was initiated by adding SRvesicles (20 µg protein) to the reaction mixture at 37 °C andterminated after 1 min by filtration. The filters were washed,dried, and counted in a beta scintillation counter (Beckman,USA). The Ca2+-uptake reaction was linear during 2 min ofthe incubation period.
2.5. Measurement of Ca2+-induced Ca2+ release
SR Ca2+ release was measured by a procedure describedearlier [17,19,20]. SR protein (0.5 mg/ml) was suspended ina loading buffer containing (in mmol/l): 100 KCl, 5 MgCl2,5 potassium oxalate, 5 NaN3, and 20 Tris–HCl (pH 6.8), andincubated with 10 µM 45CaCl2 (20 mCi/l) and 5 mM ATP for45 min at room temperature. Ca2+-induced Ca2+ release(CICR) was induced by adding 1 mmol/l EGTA plus1 mmol/l CaCl2 to the reaction mixture. The reaction wasterminated at 6 s by filtration. Filters were washed, dried, andcounted in a beta scintillation counter (Beckman, USA).CICR was completely prevented (95–97%) by the treatmentof SR preparations with 20 µmol/l ryanodine.
2.6. Measurement of Ca2+-calmodulin protein kinaseand protein kinase A activities
For protein kinase A (PKA) activity measurements, SRpreparations were isolated in the presence of a phosphataseinhibitor (1 mmol/l sodium pyrophosphate) to prevent anydephosphorylation occurring during the isolation procedure.Ca2+-calmodulin protein kinase (CaMK) and cyclic AMP-dependent PKA activities of the SR preparations were mea-sured by using Upstate Biotechnology (Lake Placid, NY)assay kits as described earlier [18,20]. The assay kit forCaMK activity is based on the phosphorylation of a specificsubstrate peptide, autocamtide by the transfer of thec-phosphate of [c-32P] ATP by CaMK-II. The assay kit forPKA activity measurement is based on the phosphorylationof a specific substrate, kemptide, by using the transfer of thec-phosphate of [c-32P] ATP by PKA. The activities of CaMKand PKA were calculated as the difference between valuesobtained in the presence and absence of the exogenous sub-strate. The reactions for CaMK and PKA were started byadding [c-32P] ATP to the reaction mixture containing therespective assay dilution buffers, substrates, and inhibitorcocktails (all provided in the kits) and incubated for 10 min at30 °C. The reaction was stopped by spotting the reactionmixture on phosphocellulose filter papers. Subsequently, fil-ters were washed thrice with phosphoric acid and once withacetone and counted in a beta scintillation counter (Beck-man, USA).
2.7. Western blot analysis
The protein content of SR Ca2+-cycling and -regulatoryproteins ryanodine receptor (RyR), sarcoendoplasmic reticu-lum Ca2+-ATPase (SERCA2a), phospholamban (PLB),calsequestrin (CQS), d-CaMK, a-PKA, Ser-16 PLB, Thr-17PLB as well as cytosolic calpain I, calpain II, and calpastatin(CS) were determined by western blot analysis as describedpreviously [17,19,20]. Protein samples (20–25 µg ofSR/cytosol) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and trans-ferred to polyvinylidene difluoride membranes for all except
103R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

RyR, which was transferred onto nitrocellulose membranes.Membranes were probed with monoclonal anti-SERCA2aand monoclonal anti-RyR antibodies obtained from AffinityBioreagents Inc., Golden, CO; monoclonal anti-PLB, poly-clonal anti-CQS, and polyclonal anti-a-PKA antibodies ob-tained from Upstate Biotechnology, Lake Placid, NY; poly-clonal anti-d-CaMK, polyclonal anti-calpain I, polyclonalanti-calpain II, polyclonal anti-CS, and polyclonal anti-Ser-16 PLB antibodies obtained from Santa Cruz Biotech-nology Inc., Santacruz, CA; polyclonal anti-Thr-17 PLB wasobtained from Badrilla, UK. Appropriate secondary antibod-ies were used and the antibody–antigen complexes in allmembranes were detected by the ECL (chemiluminescentdetection reagent, Amersham Life Science, Oakville, Ont.,Canada). An Imaging Densitometer model GS-800 (Bio-RadLtd., Hercules, CA) was used to scan the protein bands andquantified using the Quantity one 4.4.0 software from Bio-Rad.
Relative densities of proteins were scanned and the scanvalue for control in each group (comprising of control,leupeptin-treated control, IR, and leupeptin-treated IR) wastaken as 100% and accordingly others were expressed as %of control. Equal loading of SR and cytosolic protein sampleswas demonstrated by staining membranes subjected to west-ern blotting with Coomassie brilliant blue.
2.8. Statistical analysis
Results are expressed as mean ± S.E.M. and statisticallyevaluated by one-way analysis of variance (ANOVA) test. Alevel of P < 0.05 was considered the threshold for statisticalsignificance between the control and various experimentalgroups, and within the groups themselves.
3. Results
3.1. Calpain activity
Calpain activation has been proposed to occur duringischemia and IR [10]; we therefore examined this possibilityby measuring calpain activity in the cytosolic fraction ofcontrol, ischemic, and IR hearts. Calpain activity in ischemicand IR hearts was significantly higher than that observed inthe control hearts. Cytosolic calpain activity was seven timeshigher than that observed in the respective perfused controls(Fig. 2), while ischemic hearts showed almost twice as much
calpain activity as the controls. The cytosolic calpain activityin control hearts was negligible. Treatment of IR hearts withleupeptin significantly reduced IR-induced calpain activa-tion. We did not detect any calpain activity in the SR fractionsfrom all groups. We did not perform studies to identify whichisoform of calpain was being activated during ischemiaand/or IR, and our results reflect the total cytosolic calpainactivity. The protein contents of cytosolic calpain I and IIwere unaltered (Fig. 3A,B), whereas cytosolic CS proteincontent was decreased in IR hearts by about 30% of thecontrol values (Fig. 3C).
3.2. Cardiac performance
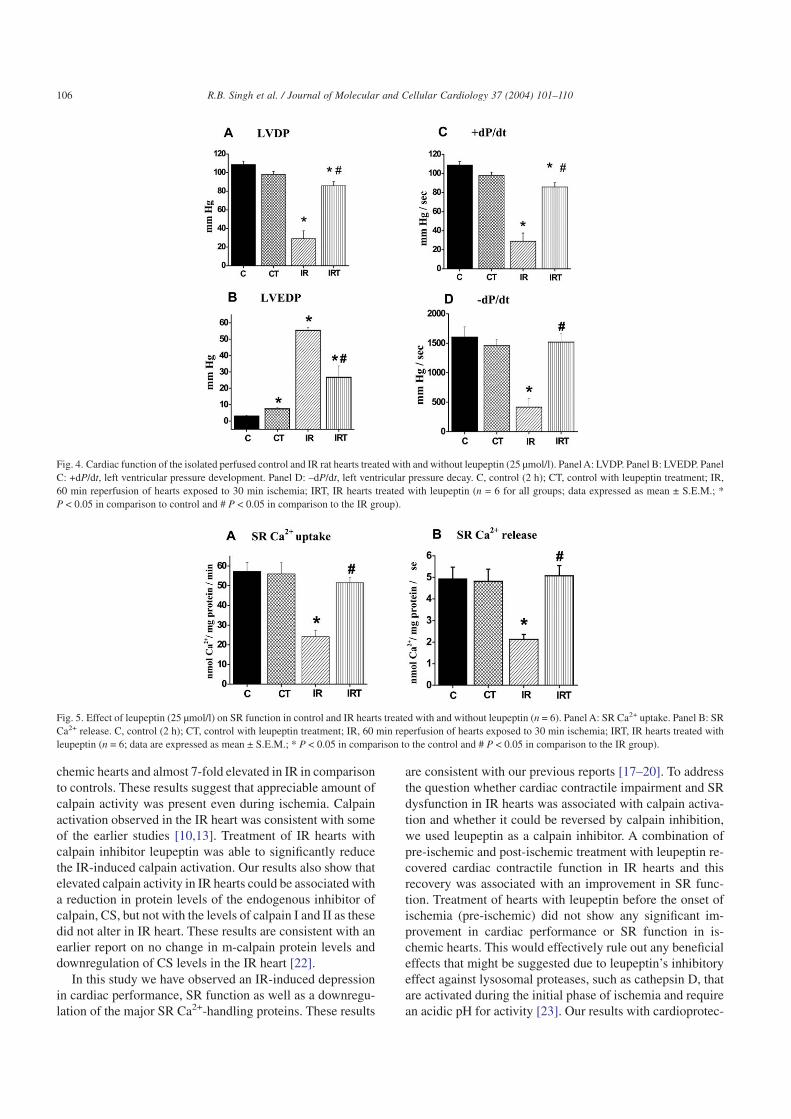
Calpain has been implicated in cardiac contractile dys-function due to IR [21]; we therefore examined whethercalpain inhibition using a calpain inhibitor, leupeptin, wouldrender cardioprotection in isolated perfused IR hearts. Heartssubjected to global ischemia for 30 min failed to generateLVDP, +dP/dt, and –dP/dt but showed a marked increase inLVEDP (Table 1). Pre-ischemic treatment with leupeptin didnot improve the cardiac function in ischemic hearts (Table 1).Reperfusion of the ischemic hearts for 60 min recovered thecontractile function, as represented by a 30–35% improve-ment in LVDP, +dP/dt, and –dP/dt of the respective pre-ischemic values but there was a marked increase in theLVEDP (Fig. 4, Panels A–D). Leupeptin (25 µmol/l) treat-ment markedly improved contractile recovery in IR hearts asreflected by an 80–85% recovery of LVDP, and 66–90%recovery in +dP/dt and –dP/dt, respectively, in comparison topre-ischemic values (Fig. 4, Panels A, C, and D). A marked
Table 1Cardiac function and SR Ca2+ uptake in isolated perfused controls and ischemic hearts treated with and without leupeptin. *P < 0.05 in comparison to control.C = Control, CT = Control + leupeptin treatment, I = Ischemia and IT = Ischemia + leupeptin treatment
Group LVDP LVEDP +dP/dt –dP/dt Ca2+ Uptabke(mm Hg) (mm Hg) (mm Hg/sec) (mm Hg/sec) (nmol Ca2+/mg protein/min
C 98.0 ± 4.7 6.5 ± 0.5 2196 ± 116 1408 ± 81 46.1 ± 2.9CT 98.3 ± 5.9 9.8 ± 4.0 2219 ± 154 1428 ± 69 43.2 ± 3.0I 1.4 ± 0.3* 43.1 ± 5.1* 17.1 ± 0.7* 17.1 ± 0.9* 18.9 ± 1.9*IT 1.0 ± 0.1* 39.5 ± 6.5* 12.4 ± 1.1* 12.4 ± 1.0* 23.7 ± 2.9*
Fig. 2. Cytosolic calpain activity of control, ischemic and IR hearts treatedwith and without leupeptin expressed as RFUs. CON, control (2 h); ISCH,30 min global no flow ischemia; IR, 60 min reperfusion of hearts exposed to30 min ischemia; IRT, IR hearts treated with leupeptin (25 µmol/l) (n = 6;data expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and# P < 0.05 in comparison to IR).
104 R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

reduction in LVEDP was observed with leupeptin treatmentin comparison to the IR hearts (Fig. 4, Panel B).
3.3. SR function
Since the SR plays a central role in determining the statusof cardiac contractility, its function was examined in control,ischemic, and IR hearts treated with and without leupeptin.SR Ca2+ uptake was significantly reduced by about 60% ofthe control values in the ischemic (Table 1) and IR hearts(Fig. 5, Panel A). While SR Ca2+ uptake in the ischemichearts did not show any significant recovery with leupeptintreatment (in comparison to control hearts) (Table 1), treat-ment improved SR Ca2+ uptake in IR hearts to about 90% ofthe control values (Fig. 5, Panel A). CICR was significantlydecreased in IR hearts by about 55% of the control values(Fig. 5, Panel B). Leupeptin treatment of IR hearts markedlyimproved SR Ca2+ release to almost control values (Fig. 5,Panel B).
3.4. SR protein content
As the SR function would be directly affected by changesin the protein content of SR Ca2+-cycling proteins, we exam-ined the protein levels of SERCA2a, RyR, CQS, and PLB incontrol and IR hearts treated with and without leupeptin.Reperfusion of the ischemic hearts decreased the proteincontent of SERCA2a (by 55%), PLB (by 40%), and RyR (by70%), in comparison to controls (Figs. 6, Panels A and B, and7, Panel A). Leupeptin treatment recovered the protein con-tent of SERCA2a, RyR, and PLB significantly. The CQSprotein content was slightly but not significantly higher in theIR group in comparison with controls (Fig. 7, Panel B).
3.5. PLB phosphorylation
SR Ca2+ uptake is dynamically regulated by PLB phos-phorylation mediated by SR-associated protein kinases, suchas CaMK and PKA; we therefore examined phosphorylationof PLB by using phospho-specific antibodies in control andIR hearts treated with and without leupeptin. PLB phospho-rylation at Thr-17 by CaMK was significantly decreased (by80%) during IR in comparison to controls (Fig. 8, Panel A).Phosphorylation of PLB at Ser-16 by PKA was also reduced(by 40%) in IR hearts in comparison to controls (Fig. 8, PanelB). Leupeptin treatment significantly improved PLB phos-phorylation at both Ser-16 and Thr-17 in the IR hearts (Fig. 8,Panels A and B).
3.6. CaMK and PKA activities
PLB phosphorylation at serine and threonine is dependenton the activities of the SR-associated CaMK and PKA; wetherefore determined these activities in control and IR groupstreated with and without leupeptin. SR-associated CaMKand PKA activities were significantly reduced by 54% and51%, respectively, in IR hearts in comparison to controls(Fig. 9, Panels A and C). Treatment of IR hearts with leupep-tin markedly improved the SR-associated CaMK and PKAactivities.
3.7. CaMK and PKA protein content
Changes in SR-associated CaMK and PKA activitiescould be due to their protein contents; therefore, the proteinlevels of the SR-associated d-CaMK and a-PKA were exam-ined in control and IR hearts treated with and without leupep-tin. Hearts subjected to IR showed a significant decrease inthe protein content of SR-associated d-CaMK (by 44%) anda-PKA (by 40%) in comparison to the controls (Fig. 9,Panels B and D). Treatment of IR hearts with leupeptinmarkedly improved the SR-associated d-CaMK and a-PKAprotein contents.
4. Discussion
Our results show that calpain activation occurred in theischemic and IR hearts; it was almost 2-fold higher in is-
Fig. 3. Cytosolic protein content of calpain I, II, and CS. Panel A: calpain I,(i) immunoreactive band of calpain I; (ii) western blot analysis of calpain I;(iii) Coomassie-stained membrane showing equivalent protein loading. Pa-nel B: calpain II, (i) immunoreactive band of calpain II; (ii) western blotanalysis of calpain II; (iii) Coomassie-stained membrane showing equiva-lent protein loading. Panel C: CS, (i) immunoreactive band of CS; (ii)western blot analysis of CS; (iii) Coomassie-stained membrane showingequivalent protein loading. C: control (2 h); IR: 60 min reperfusion of heartsexposed to 30 min ischemia (n = 6; data expressed as mean ± S.E.M.; *P < 0.05 in comparison to the control).
105R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110
chemic hearts and almost 7-fold elevated in IR in comparisonto controls. These results suggest that appreciable amount ofcalpain activity was present even during ischemia. Calpainactivation observed in the IR heart was consistent with someof the earlier studies [10,13]. Treatment of IR hearts withcalpain inhibitor leupeptin was able to significantly reducethe IR-induced calpain activation. Our results also show thatelevated calpain activity in IR hearts could be associated witha reduction in protein levels of the endogenous inhibitor ofcalpain, CS, but not with the levels of calpain I and II as thesedid not alter in IR heart. These results are consistent with anearlier report on no change in m-calpain protein levels anddownregulation of CS levels in the IR heart [22].
In this study we have observed an IR-induced depressionin cardiac performance, SR function as well as a downregu-lation of the major SR Ca2+-handling proteins. These results
are consistent with our previous reports [17–20]. To addressthe question whether cardiac contractile impairment and SRdysfunction in IR hearts was associated with calpain activa-tion and whether it could be reversed by calpain inhibition,we used leupeptin as a calpain inhibitor. A combination ofpre-ischemic and post-ischemic treatment with leupeptin re-covered cardiac contractile function in IR hearts and thisrecovery was associated with an improvement in SR func-tion. Treatment of hearts with leupeptin before the onset ofischemia (pre-ischemic) did not show any significant im-provement in cardiac performance or SR function in is-chemic hearts. This would effectively rule out any beneficialeffects that might be suggested due to leupeptin’s inhibitoryeffect against lysosomal proteases, such as cathepsin D, thatare activated during the initial phase of ischemia and requirean acidic pH for activity [23]. Our results with cardioprotec-
Fig. 4. Cardiac function of the isolated perfused control and IR rat hearts treated with and without leupeptin (25 µmol/l). Panel A: LVDP. Panel B: LVEDP. PanelC: +dP/dt, left ventricular pressure development. Panel D: –dP/dt, left ventricular pressure decay. C, control (2 h); CT, control with leupeptin treatment; IR,60 min reperfusion of hearts exposed to 30 min ischemia; IRT, IR hearts treated with leupeptin (n = 6 for all groups; data expressed as mean ± S.E.M.; *P < 0.05 in comparison to control and # P < 0.05 in comparison to the IR group).
Fig. 5. Effect of leupeptin (25 µmol/l) on SR function in control and IR hearts treated with and without leupeptin (n = 6). Panel A: SR Ca2+ uptake. Panel B: SRCa2+ release. C, control (2 h); CT, control with leupeptin treatment; IR, 60 min reperfusion of hearts exposed to 30 min ischemia; IRT, IR hearts treated withleupeptin (n = 6; data are expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and # P < 0.05 in comparison to the IR group).
106 R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110
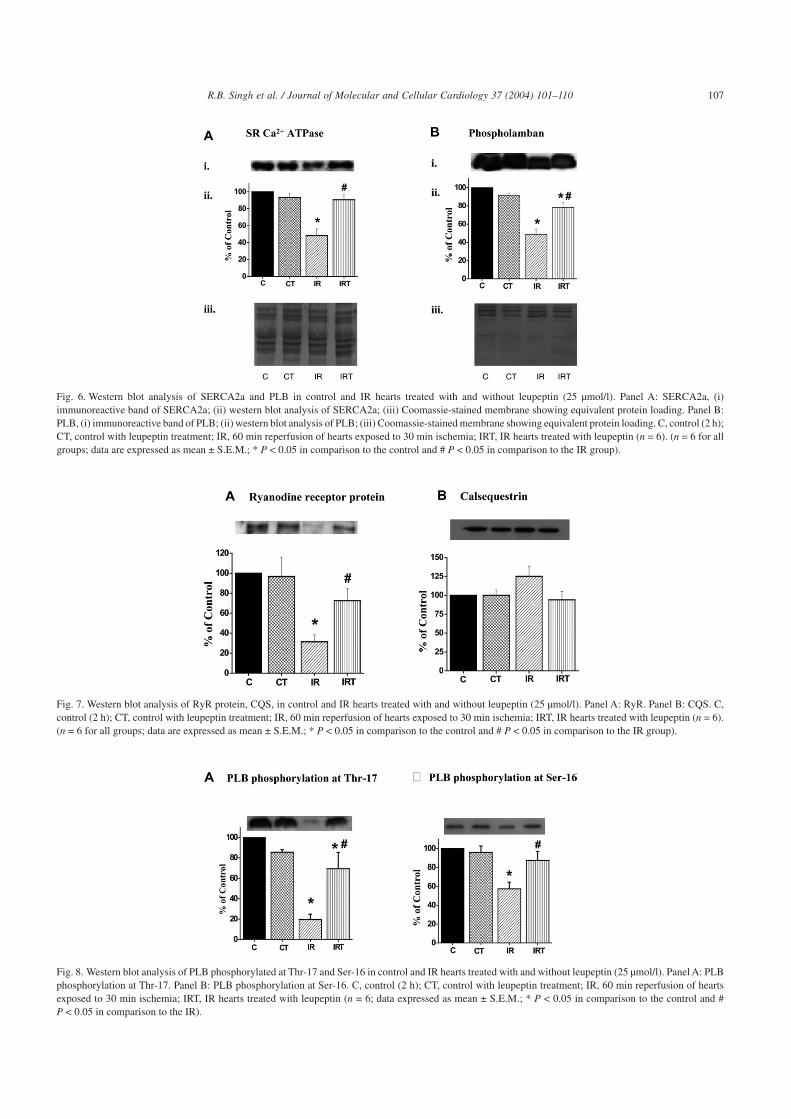
Fig. 6. Western blot analysis of SERCA2a and PLB in control and IR hearts treated with and without leupeptin (25 µmol/l). Panel A: SERCA2a, (i)immunoreactive band of SERCA2a; (ii) western blot analysis of SERCA2a; (iii) Coomassie-stained membrane showing equivalent protein loading. Panel B:PLB, (i) immunoreactive band of PLB; (ii) western blot analysis of PLB; (iii) Coomassie-stained membrane showing equivalent protein loading. C, control (2 h);CT, control with leupeptin treatment; IR, 60 min reperfusion of hearts exposed to 30 min ischemia; IRT, IR hearts treated with leupeptin (n = 6). (n = 6 for allgroups; data are expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and # P < 0.05 in comparison to the IR group).
Fig. 7. Western blot analysis of RyR protein, CQS, in control and IR hearts treated with and without leupeptin (25 µmol/l). Panel A: RyR. Panel B: CQS. C,control (2 h); CT, control with leupeptin treatment; IR, 60 min reperfusion of hearts exposed to 30 min ischemia; IRT, IR hearts treated with leupeptin (n = 6).(n = 6 for all groups; data are expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and # P < 0.05 in comparison to the IR group).
Fig. 8. Western blot analysis of PLB phosphorylated at Thr-17 and Ser-16 in control and IR hearts treated with and without leupeptin (25 µmol/l). Panel A: PLBphosphorylation at Thr-17. Panel B: PLB phosphorylation at Ser-16. C, control (2 h); CT, control with leupeptin treatment; IR, 60 min reperfusion of heartsexposed to 30 min ischemia; IRT, IR hearts treated with leupeptin (n = 6; data expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and #P < 0.05 in comparison to the IR).
107R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

tion rendered by leupeptin are consistent with earlier studies[14,21]; however, in these studies [14,21] leupeptin wasdissolved in dimethyl sulfoxide (DMSO), which has effectsof its own [24,25]. In fact, DMSO by itself has been shown toreduce calpain activity in IR hearts [10,13] and could there-fore exert beneficial effects against IR injury. Since we dis-solved leupeptin in distilled water, the cardioprotection ob-served in our study can be attributed solely to the effects ofleupeptin.
Improved SR function upon leupeptin treatment in the IRhearts could be partly attributed to a recovery in the proteincontent of major SR Ca2+-handling proteins, such as RyRand SERCA2a, and its regulator PLB in the IR hearts. Al-though PLB protein level was reduced in IR hearts, thedecrease in SERCA2a levels was comparatively more severe.Thus the ratio of PLB/SERCA2a protein levels was in-creased suggesting enhanced inhibition of SERCA2a by PLBin the IR hearts. By effectively recovering the protein levelsof PLB and SERCA2a, leupeptin treatment improved thisratio and in part relieved the inhibition of SERCA2a by PLBand improved SR Ca2+ uptake in IR hearts. Similarly, arecovery in RyR protein content upon leupeptin treatmentcould be responsible for the improvement in SR Ca2+ releasein IR hearts.
Leupeptin treatment also improved SR regulation by re-covering PLB phosphorylation in the IR heart. Phosphoryla-tion of PLB by CaMK and PKA are the key events thatde-inhibit SERCA2a and stimulate SR Ca2+ uptake [26].PLB phosphorylation by both kinases was downregulated inIR hearts suggesting increased inhibition of SERCA2a andreduced SR Ca2+ uptake. Our results are in slight variancewith those reported by Vittone et al. [27] who observed nochange in Ser-16 and Thr-17 phosphorylation of PLB in the
IR hearts. This difference could be due to the duration ofglobal ischemia and reperfusion in the two studies. Globalischemia was induced for 30 min and reperfusion maintainedfor 60 min in our study, whereas 20 min of global ischemiaand 30 min of reperfusion were used in the study by Vittoneet al. [27]. Treatment with leupeptin recovered PLB phos-phorylation by CaMK and PKA, and in part relieved theinhibition of SERCA2a by PLB leading to an improvementin SR Ca2+ uptake in IR hearts. The reduction in SR-associated CaMK and PKA activities was consistent with areduction of their respective protein contents in IR hearts.
Our study strongly suggests that IR-induced downregula-tion of the major SR Ca2+-cycling and -regulatory proteinsmay occur via proteolytic degradation/modification bycalpain. This downregulation may be responsible for de-pressed SR function and its regulation, which was partly orcompletely reversed by calpain inhibition. To our knowledgethere is only one report in literature that reported degradationof an SR protein, SERCA2a, by protease activation in the IRheart [28]. It must, however, be noted that the identificationof SERCA2a in that study [28] was not done by western blotanalysis and a band corresponding to 94 kDa was assumed tobe SERCA2a. Thus the identity of this protein as SERCA2ain that study [28] is questionable.
The proteolytic degradation/modification of SR proteinsby calpain may be a downstream mechanism of Ca2+ over-load mediated contractile dysfunction in IR hearts (Fig. 10).The effects of calpain on SR proteins may be in addition to itsdeleterious consequences on contractile proteins (such astroponin I) and cytoskeletal proteins (such as fodrin, desmin,and a-actinin) observed in some other studies [13–15]. It hasalso been reported that oxidative stress may render proteinssusceptible to proteolytic degradation [29]. In this regard, our
Fig. 9. SR-associated CaMK-II and cyclic AMP-dependent PKA activity and protein contents in control and IR hearts treated with and without leupeptin(25 µmol/l). Panel A: SR CaMK activity. Panel B: western blot analysis of SR d-CaMK protein content. Panel C: SR PKA activity. Panel D: western blot analysisof SR a-PKA (n = 6; data expressed as mean ± S.E.M.; * P < 0.05 in comparison to the control and # P < 0.05 in comparison to the IR group).
108 R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

earlier study [17] showed that although a combination ofpotent scavengers, such as superoxide dismutase plus cata-lase, improved cardiac function in IR hearts it did not recoverlevels of the SR Ca2+-cycling proteins. Thus it is possible thatthe downregulation observed in the present study could bedue to protease action on SR proteins that have been madesusceptible by IR-induced oxidative stress. Recent studieshave reported that leupeptin possesses free-radical-scavenging properties [30]. We cannot therefore rule out thecontribution of leupeptin’s antioxidant effects in renderingcardioprotection in IR hearts in addition to its calpain inhibi-tory effect as evident in the current study as well as reportedin other studies [14,21].
To the best of our knowledge this is the first study topresent clear evidence of the SR Ca2+-cycling and SR-regulatory proteins being the targets for calpain action and amajor cause for cardiac contractile dysfunction in IR hearts.
4.1. Limitation of the study
Although leupeptin inhibits calpain, it is not a very spe-cific inhibitor of calpains. We used leupeptin in this studysince the use of a highly specific peptide inhibitor to investi-gate all the parameters using the ex-vivo Langendorff heartmodel would be extremely expensive.
4.2. Clinical significance
Protease inhibitors are used under different clinical set-tings. Due to its effects on coagulation, fibrinolysis, andinflammation, aprotinin is used to reduce the loss of bloodand transfusion requirements that accompany cardiac sur-gery [31]. Tissue-type plasminogen activator has been usedclinically as thrombolytic agent for the management ofstroke and myocardial infarction [32]. Our study providesinsights into the mechanisms underlying the beneficial ef-
fects of calpain inhibition against IR injury. Prevention of SRprotein degradation/modification using protease inhibitorscould, therefore, be an additional strategy used to ameliorateIR injury in clinical settings.
Acknowledgements
The study was supported by a grant from the CanadianInstitute of Health Research (CIHR) Group in ExperimentalCardiology. N.S.D. holds CIHR/Pharmaceutical Researchand Development Chair in Cardiovascular Research sup-ported by Merck Frosst Canada.
References
[1] Piper HM, Garcia-Dorado D, Ovize M. A fresh look at reperfusioninjury. Cardiovasc Res 1998;38:291–300.
[2] Hearse DJ. Ischemia, reperfusion, and the determinants of tissueinjury. Cardiovasc Drug Ther 1990;4:767–76.
[3] Hearse DJ, Bolli R. Reperfusion induced injury: manifestations,mechanisms, and clinical relevance. Cardiovasc Res 1992;26:101–8.
[4] Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysi-ology, clinical manifestations and therapeutic options. Int J Cardiol1997;58:95–117.
[5] Gross GJ, Kersten JR, Warltier DC. Mechanisms of postischemiccontractile dysfunction. Ann Thorac Surg 1999;68:1898–904.
[6] Tani M, Neely JR. Role of intracellular Na+ in Ca2+ overload anddepressed recovery of ventricular function of reperfused ischemic rathearts. Possible involvement of H+–Na+ and Na+–Ca2+ exchange. CircRes 1989;65:1045–56.
[7] Barry WH. Mechanisms of myocardial cell injury during ischemiaand reperfusion. J Card Surg 1987;2:375–83.
[8] Ohsumi M, Hayashi M, Inomata M, Imahori K, Kawashima S. Iden-tity of calcium-activated neutral proteases from rabbit cardiac andskeletal muscle. Comp Biochem Physiol B 1984;79:643–6.
[9] Yajima Y, Kawashima S. Calpain function in differentiation of mes-enchymal cells. Biol Chem 2002;383:757–64.
[10] Yoshida K, Yamasaki Y, Kawashima S. Calpain activity alters in ratmyocardial subfractions after ischemia or reperfusion. Biochim Bio-phys Acta 1993;1182:215–20.
[11] Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system.Physiol Rev 2003;83:731–801.
[12] Carafoli E, Molinari M. Calpain: a protease in search of a function?Biochem Biophys Res Commun 1998;247:193–203.
[13] Yoshida K, Sorimachi Y, Fujiwara M, Hironaka K. Calpain is impli-cated in rat myocardial injury after ischemia or reperfusion. Jpn Circ J1995;59:40–8.
[14] Matsumura Y, Saeki E, Inoue M, Hori M, Kamada T, Kusuoka H.Inhomogeneous disappearance of myofilament-related cytoskeletalproteins in stunned myocardium of guinea pig. Circ Res 1996;79:447–54.
[15] Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role oftroponin I proteolysis in the pathogenesis of stunned myocardium.Circ Res 1997;80:393–9.
[16] Bers DM. Cardiac excitation–contraction coupling. Nature 2002;415:198–205.
[17] Temsah RM, Netticadan T, Chapman D, Takeda S, Mochizuki S,Dhalla NS. Alterations in sarcoplasmic reticulum function and geneexpression in ischemic–reperfused rat heart. Am J Physiol 1999;277:H584–94.
[18] Netticadan T, Temsah R, Osada M, Dhalla NS. Status ofCa2+/calmodulin protein kinase phosphorylation of cardiac SR pro-teins in ischemia–reperfusion. Am J Physiol 1999;277:C384–91.
Fig. 10. Proposed mechanisms underlying the development of contractiledysfunction during ischemia–reperfusion.
109R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110

[19] Temsah RM, Dyck C, Netticadan T, Chapman D, Elimban V,Dhalla NS. Effect of beta-adrenoceptor blockers on sarcoplasmicreticular function and gene expression in the ischemic–reperfusedheart. J Pharmacol Exp Ther 2000;293:15–23.
[20] Osada M, Netticadan T, Tamura K, Dhalla NS. Modification ofischemia–reperfusion-induced changes in cardiac sarcoplasmicreticulum by preconditioning. Am J Physiol 1998;274:H2025–34.
[21] Matsumura Y, Kusuoka H, Inoue M, Hori M, Kamada T. Protectiveeffect of the protease inhibitor leupeptin against myocardial stunning.J Cardiovasc Pharmacol 1993;22:135–42.
[22] Sorimachi Y, Harada K, Saido TC, Ono T, Kawashima S, Yoshida K.Downregulation of calpastatin in rat heart after brief ischemia andreperfusion. J Biochem (Tokyo) 1997;122:743–8.
[23] Wildenthal K, Decker RS, Poole AR, Griffin EE, Dingle JT. Sequen-tial lysosomal alterations during cardiac ischemia. I. Biochemical andimmunohistochemical changes. Lab Invest 1978;38:656–61.
[24] Ogura T, Kasamaki Y, McDonald TF. Force-relaxant actions of dim-ethyl sulfoxide on guinea-pig and rabbit papillary muscles. J Mol CellCardiol 1996;28:1777–88.
[25] Ogura T, Shuba LM, McDonald TF. Action potentials, ionic currentsand cell water in guinea pig ventricular preparations exposed todimethyl sulfoxide. J Pharmacol Exp Ther 1995;273:1273–86.
[26] MacLennan DH, Kranias EG. Phospholamban: a crucial regulator ofcardiac contractility. Nat Rev Mol Cell Biol 2003;4:566–77.
[27] Vittone L, Mundina-Weilenmann C, Said M, Ferrero P, Mattiazzi A.Time course and mechanisms of phosphorylation of phospholambanresidues in ischemia–reperfused rat hearts. Dissociation of phospho-lamban phosphorylation pathways. J Mol Cell Cardiol 2002;34:39–50.
[28] Yoshida Y, Shiga T, Imai S. Degradation of sarcoplasmic reticulumcalcium-pumping ATPase in ischemic–reperfused myocardium: roleof calcium-activated neutral protease. Basic Res Cardiol 1990;85:495–507.
[29] Zolotarjova N, Ho C, Mellgren RL, Askari A, Huang WH. Differentsensitivities of native and oxidized forms of Na+/K+-ATPase to intra-cellular proteinases. Biochim Biophys Acta 1994;1192:125–31.
[30] Perrin C, Vergely C, Zeller M, Rochette L. In vitro antioxidant prop-erties of calpain inhibitors: leupeptin and calpain inhibitor-1. Cell MolBiol 2002;48:OL267–70.
[31] Bull DA, Maurer J. Aprotinin and preservation of myocardial functionafter ischemia–reperfusion injury. Ann Thorac Surg 2003;75:S735–9.
[32] Rouf SA, Moo-Young M, ChistiY. Tissue-type plasminogen activator:characteristics, applications and production technology. BiotechnolAdv 1996;14:239–66.
110 R.B. Singh et al. / Journal of Molecular and Cellular Cardiology 37 (2004) 101–110
Related Documents











