# WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim Reprint Ribosome Structure and Translation The Ribosome through the Looking Glass One for all : Ribosomes are the translation machines of the cell which convert genetic information into protein structure in the same manner in all animals. Recent crystallographic and cryo-electron micro- scopic reconstructions of ribosomes have revolutionized our understanding of the translation process. The superposed il- lustration shows the impressive structural similarity of prokaryotic and eukar- yotic ribosomes. D. N. Wilson, K. H. Nierhaus* 3464 – 3486 Keywords: amino acids · proteins · ribosomes · RNA · translation 2003 – 42/30

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

� WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Reprint
Ribosome Structure and Translation
The Ribosome through the Looking Glass
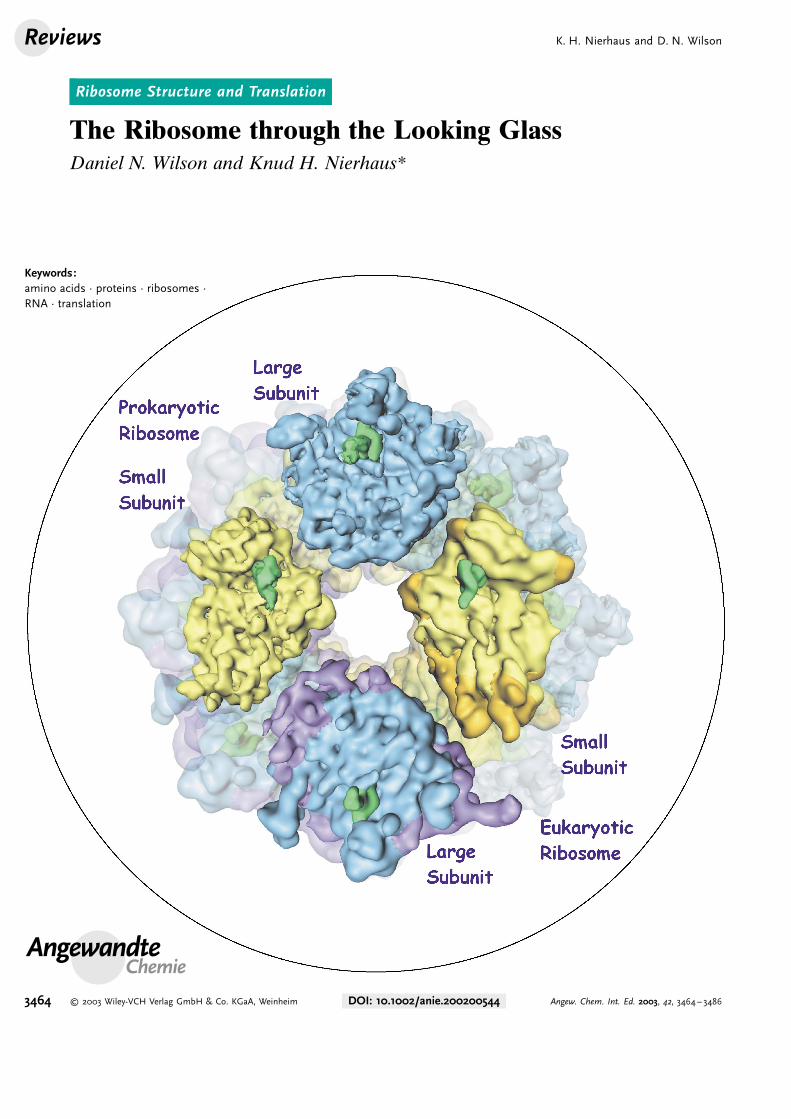
One for all : Ribosomes are the translationmachines of the cell which convert geneticinformation into protein structure in thesame manner in all animals. Recentcrystallographic and cryo-electron micro-scopic reconstructions of ribosomes haverevolutionized our understanding of thetranslation process. The superposed il-lustration shows the impressive structuralsimilarity of prokaryotic and eukar-yotic ribosomes.
D. N. Wilson,K. H. Nierhaus* 3464 – 3486
Keywords: amino acids · proteins ·ribosomes · RNA · translation
2003 – 42/30
Ribosome Structure and Translation
The Ribosome through the Looking GlassDaniel N. Wilson and Knud H. Nierhaus*
AngewandteChemie
Keywords:amino acids · proteins · ribosomes ·RNA · translation
K. H. Nierhaus and D. N. WilsonReviews
3464 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim DOI: 10.1002/anie.200200544 Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

1. The Wonders of the Translational World
The ribosome is a translator. It uses the informationcontained in messenger RNA (mRNA) to produce thecorresponding sequence of amino acids, thus linking theworlds of nucleic acids (DNA and RNA) and proteins. It doesthis by providing the platform on which each codon of themRNA is matched with the amino acid it encodes. Thephysical link between the worlds of RNA and protein is thepool of transfer RNAs (tRNAs). One end of each tRNAspecies, the anticodon, is complementary to the codon of themRNA while the other end, termed the CCA end, iscovalently attached to the amino acid specific for thatcodon. The correct aminoacylation of tRNAs with theappropriate amino acid is equally important for ensuringthe fidelity of translation. This task is performed by synthe-tases, such that for each of the 20 amino acids there is acorreponding synthetase, which recognizes on the one handthe amino acids and on the other hand all tRNAs whichdecode this amino acid. The job of every ribosome is to ensurethat the mRNA is read in the correct frame and that eachtRNA faithfully follows the code. The ribosome performs thisprocess with amazing accuracy and at high speed: 10–20 amino acids are incorporated per second into the growingnascent chain, with only one error in every 3000 codonsdeciphered. To understand how the ribosome achieves thisfeat, an understanding of the overall structure of the ribosomewill be necessary.
1.1. Common Structural Features of theRibosome
All ribosomes are composed of twosubunits of unequal size. Bacterialribosomes have a relative sedimenta-tion rate of 70S and can be separatedinto a large 50S subunit and a small 30Ssubunit. Eukaryotic ribosomes arelarger: Saccharomyces cerevisiae(yeast) ribosomes, for example, sedi-ment at 80S and are separable into 60Sand 40S subunits. Each subunit of aribosome is a ribonucleoprotein par-ticle. In the eubacteria Escherichia coli
one third of the mass of a ribosome consists of protein and theother two thirds of ribosomal RNA (rRNA): The 50S subunitcontains both a 5S (120 nucleotides) and a 23S rRNA (about2900 nucleotides), while the 30S subunit contains a single16S rRNA (approximately 1500 nucleotides). The proteinfraction consists of 21 different proteins in the small subunitand 33 proteins in the large subunit. Eukaryotic ribosomeshave longer rRNAs (because of the insertion of additionalsequences at specific regions termed expansion sequences(ESs)), an additional rRNA, and 20–30 extra ribosomalproteins, which together account for the 30% increase in sizerelative to E. coli ribosomes.
The overall three-dimensional shapes of the 70S ribosomeand its component subunits have been characterized by avariety of electronmicroscopy techniques since the 1980s. Thesmall subunit was described anthropomorphically with ahead, connected by a neck to a body with a shoulder and aplatform (Figure 1a). The large subunit presents a morecompact structure consisting of a rounded base with threeprotuberances, termed the L1, central protuberance, and theL7/L12 stalk (Figure 1b). A vast improvement in the reso-lution was achieved by the introduction of single-particle
[*] Prof. Dr. K. H. Nierhaus, Dr. D. N. WilsonMax-Planck-Institut f%r molekulare GenetikIhnestrasse 73, 14195 Berlin (Germany)Fax: (+49)30-8413-1594E-mail: [email protected]
For almost 20 years crystallographers have sought to solve thestructure of the ribosome, the largest and most complicated RNA–protein complex in the cell. All ribosomes are composed of a large andsmall subunit which for the humble bacterial ribosome comprise morethan 4000 ribonucleotides, 54 different proteins, and have a molecularmass totaling over 2.5 million Daltons. The past few years have seenthe resolution of structures at the atomic level for both large and smallsubunits and of the complete 70S ribosome from Thermus thermo-philus at a resolution of 5.5-+. Soaking of small ligands (such asantibiotics, substrate analogues, and small translational factors) intothe crystals of the subunits has revolutionized our understanding of thecentral functions of the ribosome. Coupled with the power of cryo-electron microscopic studies of translation complexes, a collection ofsnap-shots is accumulating, which can be assembled to create a likelymotion picture of the bacterial ribosome during translation. Recentanalyses show yeast ribosomes have a remarkable structural similarityto bacterial ribosomes. This Review aims to follow the bacterialribosome through each sequential “frame” of the translation cycle,emphasizing at each point the features that are found in all organisms.
From the Contents
1. The Wonders of theTranslational World 3465
2. The Path of the tRNA throughthe Ribosome 3468
3. Summary and Outlook 3483
Ribosome Structure and TranslationAngewandte
Chemie
3465Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 DOI: 10.1002/anie.200200544 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

reconstruction of cryo-electron microscopy images.[1] Thegeneral structural features of the ribosome remained athigher resolution, but more detailed structural featuresappeared, such as the beak and toe or spur on the 30S subunit.
More recently, cryo-EM analysis has been used toexamine eukaryotic ribosomes and subunits.[2–5] These recon-structions show that despite the extra size, yeast andmammalian ribosomes show extensive structural similaritywith their bacterial counterparts (Figure 1c and d). Thedifferences, which stem principally from additional rRNA andproteins in the eukaryote ribosome (shaded darker inFigure 1c and d), are located predominantly in the surfacesof the subunits exposed to the cytoplasm and not to the
intersubunit surface where the subunits interact with oneanother. Another feature of all ribosomes is a tunnel thattraverses the large subunit; it starts at the peptidyltransferase(PTF) center at the interface and exits at the base orcytoplasm side of the large subunit. The growing polypeptidechain is believed to travel through the tunnel before exitinginto the cytosol of the cell. The tunnel has a length ofapproximately 100 @ and can house between 30 and 50 aminoacid residues of the growing polypeptide chain.
In the case of proteins that are targeted to organelles andcell compartments, such as the endoplasmic reticulum,chloroplasts, or mitochondria, the ribosome must dock witha pore or protein-conducting channel on the surface of the
Knud H. Nierhaus studied medicine andcompleted his thesis with Prof. Klaus Betkein T�bingen. In 1968 he joined the MPI f�rMolekulare Genetik in Berlin, where he cur-rently leads a research group studying differ-ent aspects of translation. He is“außerplanm-ßiger Professor” at the TU,Berlin and “adjunct Professor” at theMoscow State University. His main achieve-ments include: the development of amethod to degrade the large subunit fromE. coli ribosomes and the detection of athird tRNA binding site on the ribosome.
Daniel N. Wilson studied Biochemistry andMolecular Biology at Victoria University,Wellington, New Zealand. He carried out hisPhD in the laboratory of Prof. Warren Tatein the Biochemistry Department at the Uni-versity of Otago, Dunedin, New Zealand. Inhis thesis he focused on the mechanisms oftranslational termination and recodingevents. Following completion of his studiesin 1999, he was awarded an Alexander von -Humboldt and currently works in the labora-tory of Prof. Nierhaus at the MPI f�r Mole-kulare Genetik in Berlin.
Figure 1. Comparison of the small and large ribosomal subunit from bacteria with those of a lower eukaryote. Cryo-electron microscopic recon-structions of the small (a) and large subunit (b) of the bacteria Escherichia coli are compared with the small (c) and large subunit (d) of the yeastSaccharomyces cerevisiae and the recent high-resolution crystal structures for the small subunit from the bacteria Thermus thermophilus (e) andlarge subunit from the archeon Haloarcula marismortui (f). All subunits are viewed from the interface side with the P-tRNA present in the cryo-EMreconstructions (green). Additional masses within the yeast 80S ribosome compared with that of a bacterial ribosome are shown in dark yellow(40S subunit) and purple (60S subunit). Landmarks of the small subunits include: b, body; bk, beak; h, head; lf, left foot; rf, right foot; pt, plat-form; sh, shoulder; and sp, spur. Landmarks for the large subunit: CP, central protuberance; L1, L1 protuberance; SB, stalk base; St, L7/L12stalk; H34, helix 34; H38, helix 38; and SRL, sarcin–ricin loop. Cryo-EM images adapted from Spahn et al.[3]
K. H. Nierhaus and D. N. WilsonReviews
3466 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

organellar membrane, through which the nascent chain iscotranslationally exported. Such a complex from yeast,namely the 80S ribosomes of S. cerevisiae bound to theSec61 pore protein, a protein involved in protein transportinto the endoplasmic reticulum, has been analyzed by cryo-EM. The results of these studies show that the funnel-shapedpore sits directly over the exit of the tunnel,[4–6] thussupporting the role of the tunnel as the conduit for thenascent chain.
The presence of the tunnel in ribosomes of all organismsimplies its importance, yet its function still remains specula-tive. The tunnel has been proposed to provide an environmentsuitable for the early stages of protein folding or to simplyprotect the nascent chain from proteases until sufficientamino acids have been synthesized to enable secondarystructure formation. Recently, a more active role for thetunnel has been proposed, based on the observation ofsequence-specific recognition by ribosomal components ofnascent oligopeptides within the tunnel, which were shown toinfluence both protein elongation and translation termination(reviewed by Tenson and Ehrenberg).[7]
1.2. The Ribosome up Close
The last few years have seen the arrival of high-resolutioncrystal structures of the small subunit from the thermophilicbacterium Thermus thermophilus (3 @; Figure 1e),[8,9] thelarge subunit from the archaebacterium Haloarcula maris-mortui (2.4 @; Figure 1 f),[10] and also more recently the largesubunit from the mesophilic eubacterium Deinococcus radio-durans (3.1 @).[11] A number of excellent reviews followedshortly after, in which the implications of these structureswere discussed in regard to our extensive knowledge of thefunctional aspects of protein synthesis.[12, 13]
With the arrival of high-resolution subunit structures, itwas now possible to correlate the low-resolution features withparticular rRNA helices and/or ribosomal proteins. Forexample, the beak of the small subunit consists exclusivelyof helix h33, the spur of h6, and the central protuberance ismade up of the 5S rRNA, part of 23S rRNA, as well asribosomal proteins L5, L18, L25, and L33. A more detailedanalysis of the subunit structures reveals an immediatelydiscernible difference in the overall assignment of thedomains of the rRNA secondary structure relative to thetertiary domains: In the 30S subunit, the rRNA can be simplydivided into tertiary domains, with each domain correspond-ing with a structural landmark of the 30S subunit; forexample, the 5’-domain of the 16S RNA forms the bodyfrom the toe to the shoulder, the middle domain forms theplatform, and the large 3’-domain the head, and finally thesmall 3’-domain runs down the intersubunit surface of the 30Ssubunit. In contrast, rRNA in the 50S subunit has a muchmore compact interwoven secondary structure, perhapssuggesting that the 50S subunit is older in evolutionaryterms, and has had more time to evolve such a complexorganization of the domain[14] and/or that the 30S subunitrequires more flexibility to perform its function.
With the crystal structures of the ribosomal subunits came20 new and complete structures for both small[8] and large[10]
subunits of ribosomal proteins. A special feature of many ofthese ribosomal proteins is the presence of a globular domain,which is usually bound to the surface of the subunit, as well asa long filamentous extension, which penetrates deep into thecenter of the ribosome, the ribosomal RNA core. Ribosomalproteins are commonly found bound to junctions betweenrRNA helices, thereby often connecting different domains. Acomprehensive analysis of the protein–rRNA interactions inthe small (30S) subunit revealed that the globular proteinstend to bind early in the assembly process whereas theproteins with long extensions assemble later.[15]
Perhaps one of the biggest surprises from the crystalstructures was that, despite a tenfold increase in the amountof RNA structure known, almost all of the secondarystructure motifs had been seen before, which suggests thatthe number of motifs is limited. A common feature of theribosome is the highly helical nature of the rRNA. Regionsthat were predicted to be single-stranded loop regionsactually appear as slightly irregular double-stranded exten-sions of neighboring helices in the crystal structure. Further-more, helical regions stack end-to-end to form long quasi-continuous helical structures. The proportion of adenineresidues is significantly underrepresented within these helicalor paired regions (Table 1).[16] The significance, both func-
tional and structural, of these residues is evident from theparticipation of adenine residues in the so-called “A-minor”motif, a recurring feature of the ribosome which is importantin the stabilization of the rRNA tertiary structure.[17] Ingeneral, the A-minor motif constitutes an interaction betweenan adenine residue and the minor groove of an rRNA helixand is not limited to ribosome structures, and have beenobserved previously in Tetrahymena and hepatitis delta virusribozymes.[18] Four variations in this motif have been identi-fied in the ribosome,[17] two of which, type I and II, play acrucial role in ribosome function by being involved in bothdecoding and formation of a peptide bond. Since the adenineresidues involved at both of these sites are universallyconserved, the implication is that the mechanisms of decodingand formation of a peptide bond are also conserved betweenprokaryotes and eukaryotes. The details of these interactionsare discussed more thoroughly in Sections 2.2.2 and 2.3.2,respectively.
Table 1: Frequency and distribution for each ribonucleotide withinbacterial 16S and 23S rRNAs secondary structure models.[a]
Nucleotide G C A U
overall frequency 31.4 22.4 25.7 20.5distribution withinhelical regions
36.6 14.5
frequency withinunpaired regions
12.5 42.6
distribution withinunpaired regions
30.1 22.3 66.2 41.5
unpaired/paired ratio 0.43/1 0.29/1 1.96/1 0.71/1
[a] Data sourced from Gutell et al.[16]
Ribosome Structure and TranslationAngewandte
Chemie
3467Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

2. The Path of the tRNA through the Ribosome
The “decoding site” is located on the small subunit. This isthe site where the codons of the mRNA are recognized anddeciphered by the complementary anticodon of the tRNA.There are three sites on the ribosome that tRNAs can occupy(A, P, and E; Figure 2). The A site is where the aminoacyltRNA (aa-tRNA) binds, according to the codon displayed atthis site. It is the aa-tRNA that brings in the new amino acid toextend the growing polypeptide chain. The P site is where thepeptidyl-tRNA is bound before formation of the peptidebond. This is the tRNA carrying the nascent polypeptidechain. The E site is the exit site for the deacylated oruncharged tRNA. The tRNAs move through each of the sitessequentially during translation, starting at the A site andpassing through the P site to the E site, before leaving the
ribosome. The exception is the binding of the very first tRNA(the initiator tRNA), which binds directly to the P site(Figure 2a).
Initiator tRNAs decode the start codon, usually AUG, andcarry the amino acid formylmethionine in bacteria ormethionine in eukaryotes (including archaea). The codonfollowing the start codon is displayed at the A site anddictates which aa-tRNA will now bind (Figure 2b). The aa-tRNAs are delivered to the A site in the form of a ternarycomplex consisting of an elongation factor (EF-Tu in bacteriaand EF1a in eukaryotes), GTP, and the aa-tRNA. After GTPhydrolysis, EF-Tu·GDP is released from the ribosome and theaa-tRNA docks into the A site (Figure 2c). The formation ofa peptide bond involves the transfer of the peptidyl moiety ofthe P-tRNA to the aminoacyl moiety of the A-tRNA: It isnoteworthy that the whole polypeptide chain is added to thenew amino acid rather than the addition of the new aminoacid to the chain. Formation of peptide bonds occurs on thelarge subunit at the PTF center. The formation of the peptidebond had no significant change in the positions of the twotRNAs (Figure 2d), although the P site now contains anuncharged tRNA and the A site contains a peptidyl-tRNA.
Transfer of the A- and P-tRNAs to the P and E sites istermed translocation and is mediated by a second elongationfactor (EF-G in bacteria (Figure 2e) and EF2 in eukaryotes).In simple terms, the role of the elongation factors is toaccelerate the elongation cycle to the rate of 50 msec perelongation cycle in vivo. The rate without elongation factors isabout four orders of magnitude slower[20] because of the highenergy barrier (120 kJmol�1) that separates the pre- andposttranslocational states (Figure 2c and f, respectively) inthe E. coli ribosomes.[21] Translocation places the deacylatedtRNA at the E site and peptidyl-tRNA at the P site, thusfreeing the A site for the binding of the next aa-tRNA(Figure 2 f). Binding of the next A-tRNA releases the E-tRNA (Figure 2g) and so the cycle repeats (that is, back toFigure 2c) until a stop codon appears in the A site. At thispoint protein termination factors release the completedpolypeptide and dissociate the ribosome into subunits inpreparation for the next round of translation.
2.1. Initiation of Protein Synthesis: Subunit Association andIntersubunit Bridges
The translation of functionally active protein requires thatmRNA be positioned on the 30S subunit such that the startcodon will be read first and in the correct frame. Initiation at acodon before or after the start codon would produce either anextended or truncated protein, either of which may beinactive. The placement of the start codon must also beprecise: As codons are composed of three nucleotides, thisopens the possibility of initiating translation in an incorrectframe through selection of the incorrect first nucleotide of thecodon. Thus, the precision and specificity of the initiationphase is crucial for cell viability.
How does the ribosome select the correct start codon andensure the specificity of the interaction with the initiatingtRNA? In fact, there is no single answer to this question, since
Figure 2. Overview of the translation cycle. Multiple cryo-electronmicroscopic studies have determined the binding positions of thetRNA and elongation factor on the 70S ribosome during differentstages of the elongation cycle (see Ref. [19] and references therein).The small 30S subunit is in yellow, the 50S large subunit in blue. Thepositions of the ribosomal elongations factors have been overlaid ontoa 3D map of the ribosome at a resolution of 11.5 F to generate a sche-matic overview of the elongation cycle, the details of which are pro-vided in the text. Adapted from Agrawal et al.[19]
K. H. Nierhaus and D. N. WilsonReviews
3468 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

there are so many exceptions to the rule. In general, however,the positioning of the mRNA utilizes the untranslated region(UTR) upstream of the start codon, which directs placementof the mRNA on the small subunit. In bacteria, themechanism is relatively simple and involves a stretch ofnucleotides (the Shine–Dalgarno (SD) sequence) that inter-acts with a complementary sequence within the 3’ end of the16S rRNA (the anti-SD sequence). This interaction wasdirectly visualized recently in the Fourier difference mapsbetween empty 70S ribosomes and those carrying mRNAs.[22]
The situation is much more complex in eukaryotesbecause of the increased regulatory mechanisms operatingon translation. In most cases, the 5’-end of eukaryotic mRNAsare modified with a guanine “cap” structure. This capstructure is recognized by specific initiation factors, whichdirect binding of both the mRNA and the initiator Met-tRNAto the 40S subunit. The mRNA is then “scanned” downstream(in the 3’ direction) until the first AUG start codon is found.The large 60S subunit can now bind and protein synthesiscommences. In certain cases, the UTRs contain complexsecondary structures, which are recognized and bound bylarge heteromeric protein complexes. Some of these proteinfactors interact directly with the 40S subunit to mediatemRNA positioning (reviewed recently by Pestova andet al.).[23]
Despite these differences between translation initiation inbacteria and eukaryotes, a number of distinct similarities arealso emerging.[24] For example, formation of initiation com-plexes in both prokaryotes and eukaryotes involves thebinding of the mRNA and initiator tRNA to the smallsubunit, such that the initiator tRNA is present at the P site ofthe small subunit. This is a unique situation as all subsequentaa-tRNAs that participate in translation will enter theribosome through the A site. As discussed in Section 2.2.2,it is the codon–anticodon interactions at the A site that aremonitored carefully by the ribosomes to ensure translationalfidelity. Initiation, by allowing direct P-site binding, bypassesthis important “monitoring” step.
How then does the ribosome ensure that the correcttRNA binds the start codon and that the start codon is placedexactly at the P site? A number of specialist initiation factorshave evolved to ensure the fidelity of P-site binding duringinitiation. In bacteria, this process is mediated by threeinitiation factors, IF1, IF2, and IF3 (reviewed by Gualerzi andet al.).[25] The only ribosome complexes with protein trans-lation factors that have been solved so far are those withinitiation factors IF1 and a domain of IF3.[26,27]
IF3, which may be the first initiation factor to bind to the30S subunit, displays dual functions. Firstly, by acting as ananti-association factor, it prevents formation of the 70Sribosome by prohibiting association of the 30S subunit with50S subunits. Secondly, it plays an important role in codon–anticodon discrimination in the P site. IF3 is composed of twodomains separated by a long lysine-rich linker. The C-terminal domain (CTD) is sufficient for ribosome bindingand fulfills the first function of the factor, while the N-terminal domain (NTD) has been implicated in the secondfunction. The crystal structure of the CTD on the 30S subunithas been solved, which allowed the NTD to be docked into
the structure.[27] The binding site of the CTD of IF3 suggeststhat the anti-association function of this domain operatesthrough conformational changes, not through direct stericobstruction as previously thought. Furthermore, the NTD isproposed to monitor correct codon–anticodon interactions atthe P site, not through a direct interaction, but through spacerestrictions, such that only correct binding orientations arepossible.[27]
Recently, footprinting experiments using IF3 led to acontradictory model for IF3 binding on the 30S subunit whichsuggested the CTD of IF3 does in fact prevent subunitassociation directly.[28] This result raises the question ofwhether IF3 has two binding sites on the 30S subunit: perhapsone associated with initiation, thus checking the codon–anti-codon interaction, and the other during ribosome disassem-bly.
Despite the single unequivocal binding site of IF1 on theribosome, it's exact function still remains a mystery. The smallsize of IF1 (less than 10 kDa) allowed the factor to be soakedinto crystals of the 30S subunit.[26] IF1 was found to bind in theA site which suggested that it helps prevent prematurebinding of the A site to the initiator tRNA. It was howevershown that factor-free 30S subunits bind tRNA exclusively atthe P site in the presence of a suitable mRNA,[29] so thisadditional verification seems unnecessary. The binding of IF1induces long-range conformational changes within the inter-subunit surface, particularly within the helix h44 of the 30Ssubunit, which together with IF3 may promote association ofthe subunit.[26] Bacterial IF2 binds specifically to initiatortRNAs and directs them to the 30S subunit. Delivery of theinitiator tRNA by IF2 is enhanced by IF1. Interestingly, thecentral region of eukaryotic translation initiation factoreIF1A forms a so-called “OB fold”, as seen for IF1, andthus may bind in an analogous fashion to the A site of the40S subunit.[30] Homologues to IF2 have been found ineukaryotes and archaea, eIF5B and aIF5B, respectively, andhave been shown to interact directly with eIF1A (see thereview by Pestova et al.[23]). The complex containing the40S subunit, the initiator tRNA, initiation factors, and themRNAwith the AUG codon at the P site is called the 43S pre-initiation complex. Association of this pre-initiation complexwith the 60S subunit is stimulated by eIF5B. It is likely thatthe association of the small and large subunit results inconformational change that stimulates hydrolysis of GTP byIF2 or eIF5B and subsequently their release from theribosome.
A number of contact points between the small and largesubunit have been identified in the bacterial 70S ribosome,which are termed intersubunit bridges (Figure 3a and b).[31,32]
The functional importance of these intersubunit bridges isemphasized by identification of corresponding bridges in theeukaryotic 80S ribosome (yeast; Figure 3c and d).[3] As wellas being important for association of the ribosomal subunits,these bridges probably play an important role in movement ofthe tRNAs through the ribosome (see Section 2.4) and insignaling between the decoding center on the small subunitand the PTF center on the large subunit. Closer inspection ofthe bridges within bacterial and yeast ribosomes reveals thatmany of the bridges, namely B2a, B3, B5a, and B5b, involve
Ribosome Structure and TranslationAngewandte
Chemie
3469Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

contacts with h44, a helix of the 30S subunit intimatelyinvolved in decoding.
2.2. The A Site: Decoding, Mimicry, and Antibiotic Interaction
With the exception of initiation, the ribosomal A site isthe entry point for charged aa-tRNAs. Here the ribosomemust determine whether the incoming tRNA is correct withrespect to the codon of the A site or not, that is, whether itshould be accepted into the A site or rejected. The moleculardetails of this “decision” have been recently revealed andconfirm a 20-year-old hypothesis.[33a] The A site also contrib-utes to the binding site of the elongation factors. During eachstage of protein synthesis—initiation, elongation, and termi-nation—translation factors interact with the ribosomal A site.Structures for a number of these translation factors have beendetermined. Regions of these structures display an overallsimilarity with one another, but more specifically they exhibita remarkable similarity with the dimensions of a tRNA, the“true” substrate of the A site. This phenomenon, in which aprotein factor mimics the A-tRNA substrate, is an example ofmolecular mimicry—a reoccurring theme in the translation(see the review by Nissen et al.[34]) that may well have been
overextrapolated. As yet there are no ribosome crystalcomplexes with elongation or termination factors. Thereason for this is probably that the large size of the translationfactors (40–80 kDa) prohibits the use of simple soakingexperiments and instead requires more complex cocrystalli-zation experiments. Thus, most of the structural informationfor these complexes comes from cryo-EM studies. In contrast,the soaking of small ligands, such as antibiotics, into theribosome crystals has been very successful. The decoding siteis the target for a number of potent translation inhibitors. Thestructures of antibiotics, such as tetracycline, paromomycin,streptomycin, hygromycin, and spectinomycin, have beensolved to atomic resolution in a complex with the 30S subunit.
2.2.1. The Universal Problem of Aminoacyl-tRNA Selection
Following dissociation of the initiation factors, the ribo-some is now primed with an aa-tRNA bound at the P site. Thecodon displayed in the vacant A site is specific for a singlespecies of tRNA that has a perfectly complementary anti-codon, the cognate tRNA. However, there are many othertRNA competitors that can interfere with this selectionprocess: 41 tRNAs with different anticodons exist in thebacterium Escherichia coli and even more in the eukaryoticcell. To complicate matters further, three to five or six of thesetRNAs (near-cognate tRNAs) will have an anticodon similarto the cognate tRNA. The remaining 90% have a dissimilaranticodon and are termed noncognate tRNAs. The problem iscompounded further when one considers that the aa-tRNAsare delivered in the form of a ternary complex, that is, in acomplex with the elongation factor EF-Tu and GTP. Theribosome must therefore discriminate between relativelylarge ternary complexes (72 kDa), which present multiplepotential interaction sites to the ribosome, on the basis of asmall discrimination area, the anticodon (1 kDa).
The ratio of the large surface area and the smalldiscrimination surface defines the corresponding energyratio: binding is dominated by a relatively large free energy,with only a tiny fraction corresponding to the discriminationenergy. The unusual molecular selection problem of theribosome consists of the fact that a large part of the bindingenergy of the 41 different ternary complexes (E. coli) isidentical, and thus the discrimination is based on the tinyfraction corresponding to the discrimination energy. Thediscrimination potential of the discrimination energy can onlybe reached under equilibrium conditions. In this case wherethe binding energy is relatively large, the equilibrium can onlybe reached after long time periods—in other words, thisprocess must be slow to be accurate.
Since we know that protein synthesis is a relatively fastand accurate process, the ribosome must overcome thishurdle. But how? A model has been proposed which over-comes this problem by simply dividing the occupation atsite A into two distinct events: a decoding step followed by anaccommodation step (see the review in Ref. [35]). During theinitial decoding step, the A site is in a low affinity state, whichreduces interaction of the ternary complex to codon–anti-codon interactions, thus excluding general contacts of thetRNA and elongation factor. By restricting the binding
Figure 3. Comparison of bridging positions between the subunits ofthe bacteria and yeast ribosomes. a, b) The 30S (blue) and 50S (gray)ribosomal subunits of Thermus thermophilus are shown from theirintersubunit sides. The bridges are marked in red and are annotatedaccording to the nomenclature from Gabashvili et al.[31] Bridges B1, B2,and B3–5 are labeled in blue, green, and orange, respectively. Thefigure was adapted from Cate et al.[32] c,d): The 40S (yellow) and 60S(blue) ribosomal subunits of Saccharomyces cerevisiae are shown fromthe interface side. The bridges are labeled in red. Those common toT. thermophilus are labeled in blue (B1), green (B2), and orange (B3–B5), while additional intersubunit connections in the yeast ribosomeare labeled eB8–eB11 in red. Note that bridges B6 and B7 are notincluded for simplicity. Adapted from Spahn et al.[3]
K. H. Nierhaus and D. N. WilsonReviews
3470 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

surface of the ternary complex to the discriminating feature,that is, the anticodon, the binding energy is both small andapproximately equivalent to the discrimination energy. Sincethe binding energy is small, equilibrium can be rapidlyattained, thus ensuring that the efficiency of the reaction isretained. The second step, accommodation in the A site,requires release of the aa-tRNA from the ternary complex.This step utilizes the nondiscriminatory binding energy todock the tRNA precisely into the A site and the attachedaminoacyl residue into the PTF center on the 50S subunit inpreparation for formation of a peptide bond. As we will seelater, accommodation of the aa-tRNA in the A site isaccompanied by release of the E-tRNA. Evidently, thissecond step of A-site binding involves gross conformationalchanges within the ribosome[21] and thus can be thought of as arelatively slow process relative to the decoding step. A-sitebinding occurs through a coupled reaction system consistingof a fast initial or decoding step and a slow accommodationstep. This has the important consequence that the initialreaction operates at equilibrium even when under steady-state conditions. The complete process—the fast decodingstep with a subsequent slower accommodation step—resultsin the discriminatory potential of codon–anticodon interac-tions being efficient and the rate of proton synthesis high.
Recently, the first step of A-site binding (low affinityA site) was visualized by cryo-EM analysis of ternary com-plexes stalled the A site with the antibiotic kirromycin.[36,37]
Although kirromycin allows GTP hydrolysis of EF-Tu, itinhibits the associated conformational changes in EF-Tu thatare necessary for dissociation from the ribosome. The cryo-EM reconstructions suggest that the anticodon stem loop(ASL) of the tRNA is kinked to allow codon–anticodoninteraction, and thus overcomes the unfavorable incomingangle of the tRNA to the A site as dictated by the ternarycomplex (see Figure 2b).[36]
As already indicated, the accommodation of an aa-tRNAinto the A site involves the dissociation of EF-Tu·GDP fromthe ribosome, a process which is coupled with the hydrolysisof GTP. It is interesting to note that in E. coli up to two GTPsare hydrolyzed during the incorporation of cognate-tRNAsand up to six GTPs during the incorporation of near-cognate-tRNAs, whereas noncognate-tRNAs do not trigger EF-Tu-dependent GTP hydrolysis at all.[38] This observation addsfurther weight to the argument that the tRNA discriminationis governed predominantly by anticodon–codon interactionsduring the initial binding step. The next question is: How arethe cognate and near-cognate tRNAs discriminated? This is aquestion that can now be answered at the molecular level, asis discussed in the next section.
2.2.2. Decoding of Aminoacyl-tRNAs
Amodel for the discrimination between cognate and near-cognate aa-tRNAs was proposed by Potapov about 20 yearsago.[33a] According to this model, the decoding center of theribosome recognizes the anticodon–codon duplex, in partic-ular, sensing the stereochemical correctness of the partialWatson–Crick base pairing and the positioning of thephosphate–sugar backbone within this structure. A test of
this hypothesis was performed with an mRNA that carried aDNA codon at one of the three ribosomal sites.[33b] If thestability of the base pairs, that is, the hydrogen bonds betweencodon–anticodon bases of the Watson–Crick base pairs, is thesole requirement for the recognition step, then a 2’-deoxybase in the codon should not affect the decoding process. If,however, the stereochemical correctness of the base pairing istested, that is, including the positioning of the sugar group,then a 2’-deoxy base should impair the decoding process. Itwas found that a deoxycodon at the A site was disastrous fortRNA binding at this site, whereas a deoxycodon at the P sitehad no effect on tRNA binding to the P site. This result was inagreement with earlier data showing that DNA could notperform the same function as an mRNA (see Potapovet al.[33b] and references therein).
Recently, the components of the ribosome directlyinvolved in decoding were identified by crystallography at aresolution of 3.1 @.[39] The crystal packing of the 30S subunitof Thermus thermophilus showed that the spur (h6) of onesubunit was placed fortuitously into the P site of another, thusmimicking the anticodon stem loop of a P-tRNA. Anothersurprise was that the base-pairing partner to the P-tRNAmimic was the 3’-end of the 16S rRNA, which extended intothe decoding center by folding back upon itself. This situation,with the P site filled, enabled Ramakrishnan and co-workersto then soak an ASL fragment (ASL-tRNA) and a comple-mentary mRNA fragment into these crystals to study aa-tRNA decoding.[39]
The binding of mRNA and cognate aa-tRNA induces twomajor rearrangements within the ribosomal decoding center:the universally conserved residues A1492 and A1493 flip outof the internal loop of h44, while the universally conservedbase G530 switches from a syn to an anti conformation.Through this process A1493 recognizes the minor groove ofthe first base pair of the codon–anticodon helix in the A site.The first base pair between ASL-tRNA and the mRNAconsists of position A36 andU1 in Figure 4a, respectively, andrecognition takes place through a type I A-minor motif. Threehydrogen bonds are formed between A1493 and the firstposition of the codon–anticodon duplex (two with the 2’-OHgroups of A36 and U1 and another with the O2 of U1). It isnoteworthy that the third hydrogen bond is not sequence-specific as might be expected, since the O2 position of thepyrimidines and the N3 position of purines occupy equivalentpositions in the minor groove of a double helix and both arehydrogen-bond acceptors.
The second base pair (A35-U2) is also monitored by 2’-OH interactions, but this time by two bases, namely A1492and G530 (this type II A-minor interaction is seen inFigure 4b). A1492 and G530 are locked in position bysecondary interactions with protein S12 (serine 50) andanother universally conserved residue C518. Thus, it seemsthat the monitoring of the middle base pair of a codon–anticodon duplex is more rigid than the first base pair. Thisfinding fits well with the observation that the middle base pairplays the most important role in coding an amino acid,followed by the first base pair. In contrast, the third position isless rigorously monitored and plays no role in the decoding ofmRNA information. This less rigorous checking of the third
Ribosome Structure and TranslationAngewandte
Chemie
3471Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

position allows latitude for wobble interactions (Figure 4c).This is evident in the third base pair (G34-U3) where theminor groove remains exposed, despite direct interactionswith C1054 and G530 and indirect metal-mediated interac-tions with C518 and proline 48 of the ribosomal protein S12.Taken together, these results clearly confirm the Potapovhypothesis and explain how decoding operates through therecognition of the correct stereochemistry of the A-formcodon–anticodon duplex. Furthermore, since the ribosomalcomponents involved are universally conserved, this suggeststhat the mechanism of decoding is likely to be similar for allribosomes.
Prior to the Potapov hypothesis, it had been proposed thatthe ribosome utilized a “proofreading mechanism” toimprove the accuracy of translation.[40,41] This mechanismwas suggested to operate by re-selection of the correctsubstrate during a so-called “discarding step”, after the initialbinding of the A-tRNA. Since re-selection is dependent uponrelease of the tRNA from EF-Tu and is accompanied by GTPcleavage, the GTP consumption for the incorporation of acognate and near-cognate amino acid provides a measure ofthe power of proofreading. Insofar as the crystal structure ofEF-Tu and the ribosome are concerned, the proofreadingmechanism does not have its own active center; instead it canbe described in terms of a kinetic effect that occurs after therelease of the binary complex EF-Tu·GDP.[42] Thus, a simplemodel for kinetic proofreading is the following: The bindingenergy during the decoding step (first step of A-site binding,see Section 2.2.1) is lower for the near-cognate aa-tRNA thanfor the cognate one, therefore, the probability of triggeringthe gross-conformational change required for A-site accom-
modation of the aa-tRNA (second step of A-site binding) islower than for the near-cognate. This in turn prolongs theresting time of the near-cognate aa-tRNA at the low-affinityA site and provides an additional chance for the near-cognateaa-tRNA to fall off the low-affinity A site.[43] Re-binding ofthis near-cognate aa-tRNA is unlikely in the presence ofcompeting ternary complexes that have an affinity two tothree orders of magnitude higher for the A site than thenaked aa-tRNA.[44]
The importance of the proofreading step can be quanti-tatively determined by taking advantage of the fact that theproofreading mechanism requires EF-Tu-dependent GTPhydrolysis. The accuracy of aa-tRNA selection in the presenceof EF-Tu and a noncleavable GTP analogue was determinedto be 1:1000,[45] an accuracy only a factor of 3 lower than thatseen in vivo (1:3000). The same threefold difference was alsodetermined for the GTP consumption per incorporation ofcognate versus near-cognate amino acids.[38] Thus, it is clearthat the significant contribution to the accuracy of translation(1000-fold) lies within the stereochemical monitoring of thecodon–anticodon duplex by the ribosome—as predicted byPotapov—and that the “proofreading mechanism” plays onlya minor role in improving the accuracy. This view wasqualitatively confirmed by direct measurement of the dis-crimination power of the initial binding without proofreading,where the binding of cognate and near-cognate ASL-tRNAfragments to the A site of 70S ribosomes were compared. Theaccuracy was found to be between 1:350 to 1:500, furtheremphasizing that the “lion's share” of the ribosomal accuracyis carried by the initial binding.[46]
2.2.3. Mimicry at the Ribosomal A Site
The A site is not restricted to binding tRNAs exclusively.During the various stages of the elongation cycle a number oftranslational factors interact at the A site. The first structuresdetermined for these translational factors were those of thetranslational factors EF-G[47,48] and EF-Tu.[49, 50] Interestingly,the structure of the latter, in the form of a ternary complexEF-Tu·GTP·tRNA,[51] had a striking similarity to that of EF-G·GDP, such that domains 3–5 of EF-G closely mimic thetRNA in the ternary complex (Figure 5a and b; see the reviewby Nissen et al.[34]). This observation suggested that thebinding pocket of the A site constrains the translationalfactors binding there to conform to a tRNA-like shape. In thepast few years, structures for various termination factors, alsothought to interact with the A site, have generally supportedthis concept. The recent structures of the ribosome recyclingfactor (RRF) perhaps presented the most convincing tRNAmimic, exhibiting a similar “L” shape and dimensions as atRNA (Figure 5c).[53] Although the structures of the bacterialRF2 factor (Figure 5d)[56] and eukaryotic human eRF1[57]
factor deviate significantly from the simple tRNA structure,they do reveal overall domain arrangements that wereproposed to span the ribosome in an analogous fashion totRNA: One domain extends into the decoding site of thesmall subunit and another reaches towards the PTC on thelarge subunit. However, a recent study has contradicted theseresults: Cryo-EM microscopy analyses of the termination
Figure 4. The principles of decoding in the A site of the ribosome.a) The first base pair of a codon–anticodon interaction (position 1)exemplifies a Type I A-minor motif: A1493 binds to the minor grooveof the A36-U1 base pair through H bonds (dotted lines). b) Position 2illustrates a type II A-minor motif: A1492 and G530 act in tandem torecognize the stereochemical correctness of the A35-U2 base pairusing H bonds. c) The third (or wobble) base pair (G34-U3) is lessrigorously monitored. C1054 stacks against G34 while U3 interactsdirectly with G530 and indirectly with C518 and proline 48 of S12through a magnesium ion (magenta). All nucleotides involved inmonitoring positions 1 and 2 are universally conserved. Adaptedfrom Ogle et al.[39]
K. H. Nierhaus and D. N. WilsonReviews
3472 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

complex of RF2 bound to the ribosome revealed that RF2undergoes dramatic rearrangement upon ribosome binding(Figure 5e).[58, 59] This conformational change was not totallyunexpected since two regions within the protein factor, the socalled “tripeptide anticodon” and the GGQ motif were only23 @ apart in the solution structure of RF2 (Figure 5d). Sincethese regions had been associated with decoding of the stopcodon and hydrolysis at the PTF center, respectively, theywould need to be separated by about 70 @ to make theappropriate interactions (Figure 5 f).[58,59]
The structural mimicry of a tRNA by RRF was alsobrought into doubt when hydroxyl radical probing experi-ments suggested that the RRF is oriented upside down on theribosome when the structural similarities with a tRNA wereconsidered.[60] Confirmation of this orientation, however,awaits cryo-EM analysis of the ribosomal RRF complexes.Whether RRF undergoes similar structural rearrangements asseen for RF2 seems unlikely, but it is clear that functional,rather than structural mimicry, is a more appropriate term inthis case: After release of the polypeptide chain by thetermination release factors, the RRF binds the ribosome andis involved in dissociating the ribosome into subunits, thusrecycling them for the next round of translation. This process
is mediated by EF-G, which has been proposed to translocateRRF from the A to the P site, thus simulating its tRNAtranslocative role during elongation.[61]
Mimicry of RNA by a protein may be a more commonfeature in ribosomes than first realized. Organellar ribosomesgenerally have shorter rRNA components than E. coli.Recent analyses of the chloroplast and mitochondrial ribo-some components suggests that these rRNA losses arecompensated for by both increases in size of the ribosomalprotein homologues and the presence of additional organelle-specific ribosomal proteins.[62] Mitochondria represent anextreme example in that the protein component of theribosomes represents two-thirds of the mass (instead of one-third as in E. coli ribosomes). The rRNA that remains consistspredominantly of universally conserved residues that arelocated at the active centers of the ribosome, that is, thedecoding center on the 30S subunit and the PTF center on thelarge subunit,[63] thus reinforcing the importance of theseregions.
2.2.4. Antibiotic Antagonists of A Site Decoding
To date, the structures of seven antibiotics, namelytetracycline, paromomycin, spectinomycin, streptomycin,pactamycin, hygromycin B, and edeine, have been solved incomplexes with the 30S subunit.[27, 39,64,65] Although the pri-mary binding sites of these antibiotic are distinct from oneanother, they all target functionally important regions of the30S subunit, mainly rRNA-rich regions associated with tRNAinteraction or movement through the ribosome. Here we willfocus our attention on tetracycline and paromomycin, bothbeing antibiotics that bind within the decoding site.
Independent studies identified two common tetracyclinebinding sites on the 30S subunit (Figure 6a).[27,65] In both
Figure 5. Molecular mimicry of tRNAs by translation factors. Compari-son of the crystal structures for a) EF-G·GDP with domains 3–5 in gold(pdb1fmn),[52] b) EF-Tu·GTP·tRNA (pdb1ttt),[51] c) (pdb1eh1),[54] andd) RF2 (pdb1gqe).[56] e) Cryo-EM reconstruction of RF2 (red) bound tothe 70S ribosome of E. coli (30S in yellow and 50S in blue). DC: decod-ing center, GAC: GTPase-associated center, P: P-site tRNA, PTC: pepti-dyltransferase center. f) Modeling of the RF2 crystal structure into theelectron density of RF2 seen in (e). In (d) and (f) the Roman numeralsindicate the RF2 domains which are colored accordingly, and the GGQand SPF motifs are indicated in gray and pink, respectively. Thedashed white line delineates the RF2 electron density from the ribo-some electron density. The pictures of the crystal structures weregenerated with Swisspdb viewer[55] and rendered with POVRAY.Cryo-EM data adapted from Rawat et al.[59]
Figure 6. Binding of tetracycline (Tet) to the 30S subunit of T thermo-philus. a) Overview of the primary and secondary Tet binding sites(red). b) Close-up view of the primary binding site, with the position ofthe A-site tRNA (red) and mRNA (yellow). Helix 18 (brown), h31(green), h34 (blue), and h44 (cyan) are represented in ribbon format.c) The secondary binding site includes h11 (purple) and h27 (yellow).The switch described in the text involves interconversion between thebase-pair configuration from red to green. Figure (a) was generatedfrom pdb file 1HNW using Swisspdb viewer[55] and rendered withPOVRAY. Figures (b) and (c) are adapted from Brodersen et al.[65]
Ribosome Structure and TranslationAngewandte
Chemie
3473Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

cases, the site with the highest occupancy, which is located in acrevice between the head and shoulder of the 30S subunit,was taken to be the primary binding site (Figure 6b). In thisposition, tetracyclin, which has a system of four fused rings,interact predominately with the 16S rRNA through theoxygen atoms located along one side of the molecule. Theoxygen atoms form hydrogen bonds with the exposed sugarphosphate backbone of helix h34. Thus, the hydrophilic sideof the tetracycline interacts with the 16S RNA while thehydrophobic side is sited in the lumen of the A site. This issurprising since interaction between two molecules is usuallythrough their hydrophobic regions. The primary binding siteof tetracyclin can be expected to overlap with the position ofthe A site tRNA, and thus the mechanism of action oftetracycline most probably results from a direct inhibition ofaa-tRNA during the accommodation step of A-site binding.For two reasons it seems unlikely that the initial binding of theaa-tRNA would be affected: 1) the tetracyline binding site islocated on the opposite side of the tRNA from the site ofinitial anticodon–codon interaction, 2) during the delivery ofaa-tRNA to the A site, the anticodon- stem loop of the aa-tRNA of EF-Tu was kinked and presented at an angle,[36,37]
which would be predicted to initially avoid contact with thebound tetracycline.[27,65] This is in line with the observationthat tetracycline does not inhibit the EF-Tu-dependentGTPase hydrolysis,[66] a step that occurs after the initialbinding of the ternary complex. The rRNA bases of theprimary binding site of tetracycline are poorly conservedbetween prokaryotes and eukaryotes, and its universal modeof action can be explained since the interaction of tetracyclinis almost exclusively made with the sugar–phosphate back-bone. In contrast, the binding sites of pactamycin, edeine, andhygromycin B are highly conserved, and in these cases themain interactions are with the conserved bases.
The secondary binding site of tetracyclin is sandwichedbetween h11 and h27 in the body of the 30S subunit(Figure 6c). Although h27 has been proposed as a conforma-tion switch modulating the translational fidelity of base pairsin E. coli,[67] it seems unlikely that this secondary site plays arole in tetracyclin inhibition. The protein Tet(O) removestetracycline from the ribosome and thus mediates resistanceagainst this drug. Probing experiments with dimethyl sulfate(DMS) in the presence of Tet(O) demonstrated that tetracy-cline was removed from the primary rather than from thesecondary binding site.[68]
The aminoglycoside paromomycin is well known forincreasing the rate of translational misreading. The bindingsite for paromomycin, determined at a resolution of 3 @, wasobserved to involve contacts exclusively with h44 of the16S rRNA.[39,64] Paromomycin binding induces the universallyconserved residues A1492 and A1493 to flip out of h44 in afashion reminiscent of that observed during the binding of aa-tRNA to the A site. This conformational change is broughtabout by the insertion of one of the four rings (ring I) ofparomomycin into h44. In this position, ring I mimics anucleotide base: it stacks with G1491 and hydrogen bondswith A1408. The stability of this conformation is furtherreinforced by hydrogen-bonding interactions between ring Iand the backbone of the flipped out A1493. Significantly,
rings I and II of paromomycin are found in a number of otheraminogylcosides, such as the antibiotics neomycin, gentamy-cin, and kanamycin families, which suggests that misreadingby these antibiotics operates through a similar mechanism.
As we have seen in Section 2.2.2, the formation ofappropriate codon–anticodon interactions is monitored bythe formation of A-minor interactions between A1492 andA1493 with the codon–anticodon helix. Presumably theenergy required to flip out A1492 and A1493 during decodingis compensated for by interactions established with thecodon–anticodon helix, thus stabilizing this conformation. Inthe presence of near-cognate tRNA, the prediction is thatthese compensatory interactions are not sufficient to stabilizethe flipping out of A1492 and A1493, and thus accommoda-tion of the A site does not occur. However, in the presence ofparomomycin, the normally uncompensated losses of energyare absorbed by the paromomycin that has already inducedA1492 and A1493 to flip out and stabilized them in this openconformation. The outcome is that a near-cognate tRNAbecomes fully accommodated into the A site in the presenceof paromomycin and thus results in mis-incorporation of anamino acid.
2.3. The P Site and the Peptidyltransferase Center
Historically, much controversy has surrounded the ques-tion regarding the catalytic “heart” of the ribosome, thepeptidyltransferase (PTF) center. Specifically, the questionsposed related to whether this active site was predominantlyprotein or rRNA. The answers to these questions arrived withthe X-ray structure of the 50S subunit, in particular the50S subunit complexed with a transition intermediate of thePTF reaction.[69] The location of the transition intermediateimmediately suggested that, in fact, the catalytic center of theribosome is exclusively composed of RNA. Furthermore,analyses of the residues within proximity to the CCA endanalogues of the tRNA led to the proposal of an acid–basecatalysis mechanism for the PTF reaction involving theuniversally conserved A2451—a proposal that came underimmediate attack from a number of research groups whopresented biochemical and genetic data to the contrary.[70–72]
The ongoing debate as to the exact catalytic contribution ofthe ribosome and the residues involved is the topic of a recentreview.[73] Thus, within a short space of time the debate hadmoved from whether or not RNA or a protein constituted thecatalytic domain, onto more detailed mechanistic questions.
In Section 2.3.1 we analyze the interactions of thepeptidyl-tRNA at the P site—certainly the tRNA bindingsite on which most information from different species hasbeen gathered—and illustrate the universal features. Themechanism of PTF is examined in Section 2.3.2, with partic-ular focus on how the ribosome assists or catalyzes thetransfer of the growing polypeptide chain from the P-tRNA toA-tRNA. The extreme conservation of residues within thePTF center suggests that the mechanism of PTF is universallyconserved. Finally, the PTF center and its immediate vicinityare the target for a number of antibiotics, the structures ofsome clinically important examples of which, such as chlor-
K. H. Nierhaus and D. N. WilsonReviews
3474 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

amphenicol, clindamycin, and a number of macrolides, havebeen solved in complexes with the 50S subunit.[11,74] Thesestructures provide not only an insight into the mechanism ofantibiotic inhibition and ribosome function, but may pave theway for the design of specific antibiotics to combat theincrease in antibiotic-resistant bacteria.[75]
2.3.1. Peptidyl-tRNA Contacts at the P Site of the Ribosome
In the past, the tRNA positions on the ribosome werestudied using a large number of biochemical approaches thathave identified a number of rRNA nucleotides that areassociated with each tRNA site (see Table 3 in Ref. [13]).After a crystal structure of the Thermus thermophilus 70Sribosome in complex with three tRNAs was solved with aresolution of 5.5 @,[76] it became clear that the identifiedresidues either directly contact the tRNA or their alteredmodification pattern could be explained by conformationalchanges within binding regions. An excellent example illus-trating this correlation is the analysis of cleavage patternsfrom ribosome-bound phosphorothioated tRNAs.[77] Thecontact patterns suggested that only 15% of the P-tRNA(nucleotides 29–43, which comprise theanticodon loop and two adjacent stembase pairs) interact with the 30S subunitwhile the remaining 85% is in contact withthe 50S subunit (Figure 7). The fact that theprotection patterns cover the entire tRNAagain dispels the earlier assumptions thattRNAs bind the ribosome using only theirextremities, that is, through anticodon–codon interactions in the small subunit andthe CCA end in the large subunit. Further-more, the ribosome contacts all threetRNAs at universally conserved parts oftheir structure.[76, 77] Phosphorothioate pro-tection studies have also suggested a con-formational change of the P-tRNA uponbinding to the ribosome.[79] Such a change isevident for the P-tRNA in the 70S/tRNA3
crystal structure, where the P-tRNA isslightly kinked around the junction of theD loop and anticodon stem.[76] A similarconformational change was necessary todock the crystal structure for the yeasttRNAPhe into the P-site electron densityduring the cryo-EM reconstruction of acomplex of P-tRNA bound to the 80Ssubunit of a yeast.[3]
We have chosen to analyze P-tRNAbinding interactions with the yeast ribo-some[3] in detail because of the availabilityof data, with a view to highlight theconservation in the interaction. A comparison of the contactsbetween rRNA and the P-tRNA in both bacteria and yeast(Table 2) illustrates the similarity in the contacts.[3, 32] The P-tRNA is fixed very tightly on the bacterial 30S subunitthrough approximately six interactions with the rRNA (a–f inFigure 8a). The codon–anticodon duplex in the P site is
positioned in the major groove of the noncanonical helicalstructure at the base of h44 and is fixed with a number of“ribosomal fingers” mainly to the sugar–phosphate back-bone.[32] A similar position for the P-tRNA and arrangementof contacts is also observed for the yeast ribosome (Fig-ure 8b). Two of the rRNA interactions with the P-tRNA in
Figure 7. P-tRNA contacts with the 70S ribosome. The region of the P-tRNA that makes contact with the 30S subunit (15%) and the 50S sub-unit (85%) are shown in blue and red, respectively.[77] The ribosomalcomponents within a 10-F radius of the P-tRNA are indicated for the30S subunit (rRNA: green, proteins: cyan), and for the 50S subunit(rRNA: yellow, proteins: orange). H: helices of the tRNA, S and L: pro-teins of the small and large subunits, respectively. The figure was gen-erated from pdb files 1GIX/1GIY[76] using the program RASMOL[78] andrendered with POVRAY.
Table 2: Comparison of ribosomal intersubunit bridge components between the bacteria Thermusthermophilus[32] and the yeast Saccharomyces cerevisiae.[3]
Subunit tRNA posi-tion[a]
Ribosomal com-ponent(T. thermophilus)
rRNA contact(T. thermophilus)
Ribosomal component(S. cerevisiae)
rRNA con-tact(S. cerevisiae)
small 28 16S h30 122929/30 16S h30 1229 18S h30 1229/123032 RpS16 Tyr141/
Arg14234 16S h31 966 18S h31 96635 S9 Arg12836 S13 116–12038 (39) 16S h24 790 (18S h24) (790)(40) 41 (16S h42) (1339) 18S h43 1338(41) 42 (16S h42) (1338) 18S h43 1339
large 2 RpL10 Lys1013 23S H80 2255–2256 25S H80 22854 25S H80 228611–13 (12–13)
(23S H69) (1908–1909) 25S H69 1908–1910
14 25S H69 192425–26 23S H69 1922–192351/52/63 RpL10 Arg2456 (56–57) (RpL5) (55–66) RpL11 Tyr5171–72 25S H93 259473 (75) 23S H93 (2602) 25S H93 260274/76 23S P loop/L90–
932252/2585
[a] tRNA positions from yeast cryo-EM analysis are approximate because of limitations in the resolution.
Ribosome Structure and TranslationAngewandte
Chemie
3475Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

Thermus thermophilus are strengthened by interaction withC-terminal ends of ribosomal proteins S9 and S13. The C-terminal end of S9 is highly conserved and contains auniversally conserved arginine residue that appears to contactthe phosphate group of position 35 in the anticodon loop ofthe P-tRNA. Again, a similar interaction is seen in yeast withrpS16, the homologue of bacterial S9. In contrast, rpS18, thehomologue of S13, does not have a corresponding C-terminalsequence and, on the basis of its position in the 80S ribosome,is probably not involved in P-tRNA fixation. Interactionbetween the large subunit and the P-tRNA also involvescontacts between H69 and the D loop, while the T loopcontacts rpL11 in eukaryotes and the homologue L5 inbacteria (Table 2 and Figure 8c and d). Interestingly, boththese ribosomal components are involved in the formation ofbridges between subunits, which suggests they may play adynamic role in translation, for example, translocation of thetRNAs (see Section 2.4.2). Although the single-strandedCCA end of the P-tRNA is not resolved in the 80S complex,its position in the crystal structure suggests there is interactionwithin the PTF center. This is evident from the proximity of
the CCA end to base 2602 in the yeast ribosome complex,which in the bacterial ribosome complex contacts position 75,the penultimate base of the CCA end of the P-tRNA.
It is clear from the multiple tRNA contact points with theribosome that positioning of the tRNA involves a complexnetwork of interactions. The distinct similarity in the arrange-ment and make-up of the ribosomal components that interactwith a P-tRNA, despite the billions of years separating yeastsand eubacteria, suggests an important role for these compo-nents during translation. Accurate positioning of tRNAs isessential for ribosomal function. As seen in Section 2.2, theprocess of decoding is governed by the stereochemicalarrangement of the tRNAs relative to the mRNA and theribosome. Many of the contact points are components ofintersubunit bridges, which suggests that these interactionsnot only “lock” the tRNA in position but may be involved intransporting it through the ribosome (see Section 2.4).Furthermore, tight fixation of both A- and P-tRNAs may bethe prerequisite for efficient formation of the peptide bond, aswill be described in the following section.
2.3.2. The Ribosome is a Ribozyme
The PTF reaction is the central enzymatic activity of thelarge subunit. It occurs when a pretranslocational (PRE) stateis reached, that is, when a peptidyl-tRNA is located in theP site and an aa-tRNA is in the A site. The two L-shapedtRNAs at the P and A sites form an angle of about 408,[19, 76,80]
while the acceptor stems of both tRNAs are parallel to eachother, such that they can move in a translational movementrelative to each other. In contrast, the CCA ends of bothtRNAs at the PTF center have rotation symmetry, beingarranged at an angle of approximately 1808 to each other. Thetwist needed to accomplish this rotation occurs almostentirely between nucleotides 72 and 74 of the tRNA.[81]
Recently, this rotational symmetry of the A- and P-tRNACCA ends was shown to be complemented by two sets of PTFnucleotides surrounding each of the CCA ends that are alsorelated by a rotational symmetry.[82] This ribosomal structuremight play an essential role in guiding the CCA ends duringtranslocation from the A and P sites to the P and E sites,respectively.
During PTF the a-amino group of the A-tRNA attacksthe carbonyl group of the peptidyl residue of the P-tRNA,which is linked through an ester bond to the tRNA moiety.This results in the formation of a tetrahedral intermediate,which leads to formation of a peptidyl bond. As a result, theaa-tRNA becomes a peptidyl-tRNA extended by one amino-acyl residue, and the former peptidyl-tRNA is stripped of thepeptidyl residue to become a deacylated tRNA (Figure 9).
A long-standing debate within the field of translationconcerned whether, or not, the PTF reaction was catalyzed byproteins or rRNA. This question was answered with theidentification of the PTF center. A putative transition stateanalogue of the PTF reaction was soaked into crystals of the50S subunit from Haloarcula marismortui.[69] This analogue(the so-called Yarus inhibitor) is a mimic of the CCA end of aP-tRNA attached to puromycin in the A site (Figure 9), and isa strong competitive inhibitor of the A site substrate.[84] The
Figure 8. Details of P-tRNA interactions with the small and large subu-nit of Thermus thermophilus and Saccharomyces cerevisiae ribosomes.The P-tRNA (red) contacts made with the 30S (a) and 50S (c) subunitsfrom T. thermophilus are similar to those made by the P-tRNA (green)with the 40S (b) and 60S (d) subunits from S. cerevisiae. The smallsubunit rRNA is colored cyan (a) or yellow (b), while the ribosomalproteins are colored either purple (a) or red (b). The large subunitrRNA is colored gray (c) or blue (d), while the ribosomal proteins arecolored magenta (c) or red (d). The spheres in (a) and (c) representrRNA bases that are protected from chemical probes upon tRNA bind-ing. In (a) the six ribosomal fingers that hold the anti-codon stem–loop (ASL) in place are indicated by a–f. The figure is adapted fromYusupov et al.[76] and Spahn et al.[3]
K. H. Nierhaus and D. N. WilsonReviews
3476 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

region bound by the inhibitor is densely packed with highlyconserved bases of the 23S rRNA, mainly derived from theso-called PTF ring of domain V. Although there are 15 pro-teins that interact with domain V of the 23S rRNA, only theextensions of proteins L2, L3, L4, and L10e come within 20 @of the active site (Figure 10). The fact that the active center ofthe ribosome is made exclusively from RNA means that theribosome is a true ribozyme.
The debate has now turned to whether the PTF reactionsimply utilizes a physical principle or whether an additionalchemical principle also applies, that is, whether besides theaccurate stereochemical arrangement of the substrate (phys-ical principle), a chemical principal, such as a general acid–base catalysis, is also involved.
The universally conserved residue A2451 of domain V isthe nearest base to the transition analogue (Figure9). It was
thought to be a good candidate for a general acid–basecatalyst, since its N3 atom is about 3 @ from the oxygen atomand 4 @ from the nitrogen atom of the phosphoamide in theYarus inhibitor (Figure 11a). This proposal was strengthenedwhen the pKa value of the A2451 residue was found to beabnormally high at neutral pH (pKa= 7), which is six pH unitshigher than expected.[71,85] This property is essential foracid–base catalysis, as it allows for easy donation andwithdrawal of a proton from the a-amino group of the aa-tRNA at the A site. According to the same model,[69]
protonation of A2451 would also allow formation of ahydrogen bond with the carbonyl oxyanion of the tetrahedraltransition state analogue (Figure 9). However, modificationof position 2451 with dimethyl sulfate showed that pH de-pendence was only displayed by inactive ribosomes, not bythe active ribosomes.[86] Several research groups reported
Figure 9. The peptidyltransferase reaction. The picture bottom right shows the Yarus inhibitor CCdAp-puromycin (CCdApPmn) that was used toidentify the PTF center of the ribosome. The interactions of the Yarus inhibitor with the rRNA were deduced from the 50S crystal structure ofH. marismortui ribosomes after soaking the inhibitor into the crystals. It was concluded that the protonated N3 atom of A2451 makes a H bridgeto the O2 atom that was thought to mark the position of the oxyanion of the tetrahedral intermediate formed during peptide-bond formation[69]
(see the schematic representation in step b). The figure shows the four possible steps of peptide-bond formation according to recent crystallo-graphic and biochemical data.[69, 73,81,85] The essential features are: a) C74 and C75 of the P-site tRNA (green) form a Watson–Crick base pair withG2252 and G2251, respectively, of the P loop (blue). Likewise, C75 from the A-site substrate (red) forms a Watson–Crick base pair with G2553(A loop). The a-amino function of the A-site aa-tRNA is an ammonium ion at pH 7.[83] b) Deprotonation of the ammonium ion triggers the nucleo-philic attack of the a-amino function on the carbonyl group of the P site substrate, which results in the tetrahedral intermediate T� . The secondaryNH2 group forms a hydrogen bond with the N3 atom of A2451 and a second with either the 2’OH group of the A76 ribose at the P site (shownhere) or alternatively with the 2’OH group of A2451. The oxyanion of the tetrahedral intermediate points away from the N3-A2451[81] and thuscannot, in contrast to the previous proposal, form a H bridge.[69] c) Further deprotonation of the secondary a-NH2 group leads to the tetrahedralintermediate T� and the PTF reaction is completed by an elimination step (d). The peptidyl residue is linked to the aminoacyl-tRNA at the A sitethrough a peptide bond.
Ribosome Structure and TranslationAngewandte
Chemie
3477Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

shortly after that A2451 was not essential for formation of thepeptide bond, since ribosomes bearing mutations at posi-tion 2451 exhibited only modest (2- to 14-fold) decreases inthe rate of peptidyl transfer[72] and were instead shown to bedefective in substrate binding.[70]
The next step in the elucidation of the PTF reaction wasidentifying the position of the products and was obtained bysoaking A- and P-site substrates into enzymatically activeH. marismortui 50S crystals.[87] The structure determined to aresolution of 2.4–3.0 @ was that after formation of the peptidebond but before translocation, and showed that the deacy-lated CCA bound to the P site had its 3’OH group inproximity to the N3 atom of A2451 (Figure 11b).[87]
Evidence followed, however, that A2451 was not involvedin stabilizing the transition-state analogue: If the oxyanion ofthe tetrahedral intermediate is hydrogen bonded to theN3 atom of A2451, then this N3 atom must be protonated ataround pH 7 and therefore should loose its proton at pH>
7.3. In this case, one would expect the affinity of the Yarusinhibitor to be strongly pH-dependent, since the hydrogenbond would contribute significantly (up to three orders ofmagnitude) to the affinity. To test this hypothesis, Strobel andco-workers determined the affinity of the Yarus inhibitor forthe 50S subunit between pH 5 and 8.5, and found that itremained unchanged.[85] This result is inconsistent with theidea that the oxyanion is stabilized by a hydrogen bond to theN3 atom of A2451. The same conclusion was drawn bysubsequent crystallographic studies showing that the oxy-anion of the tetrahedral intermediate points away from theN3 atom of A2451, thus excluding the possibility of theformation of a hydrogen bond between these two atoms.[81]
Furthermore, the Yarus inhibitor is not an honest mimic ofthe transition state. The distance between the O2 atom of theYarus inhibitor and the C2’ atom of the deoxy-A76 (dA76)ribose at the P site is only 2.8 @ (arrowed in Figure 11a). A
physiologically P-site substrate contains a 2’OH group at thisC2’ atom which is essential for formation of a peptide bond[88]
and would sterically clash with the O2 atom of the Yarusinhibitor positioned as observed in the crystal. The essentialnature of this 2’-OH group might be explained by theobservation that the a-NH2 group of the A site possiblyforms a hydrogen bond with this 2’-OH group (illustrated inFigure 9b).
The general base catalysis debate flared up again whenRodnina and co-workers presented evidence that formationof a peptide bond depends on two ionizable groups, one with a
Figure 10. The A- and P-site products (red and green, respectively),bound at the peptidyltransferase center of the 50S subunit. The pro-teins that reach within about 20 F of the PTF center include proteinsL2 (purple), L3 (blue), L4 (red), and L10e (cyan). This figure was gen-erated from pdb file 1KQS[87] using Swisspdb viewer[55] and renderedwith POVRAY.
Figure 11. Tight fixation of the CCA ends of the P- (green) and A-tRNAs (red) observed in the 50S subunit from H. marismortui in acomplex with a) the Yarus inhibitor and b) the products following for-mation of the peptide bond. The N3 atom of A2451 (dark blue) is3.4 F from the O2 atom of the Yarus inhibitor (see also Figure 9). Thesame O2 atom is only 2.8 F from the 2’-deoxy position of A76(arrowed). Selected rRNA residues of domain V of the 23S rRNA arecolored light blue, including the A- and P-loop bases that participate infixation of the CCA ends of the A and P sites (E. coli numbering). In(b) C74 and C75 of the P site have been omitted for clarity. Dashesindicate hydrogen bonding and rRNA nucleotides use the followingcolor scheme: O red, P yellow, N blue, C dark blue. Figures (a) and (b)were generated from pdb files 1FFZ[69] and 1KQS,[87] respectively, usingSwisspdb viewer[55] and rendered with POVRAY.
K. H. Nierhaus and D. N. WilsonReviews
3478 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

pKa value of 6.9 and the other with a pKa value of 7.5.[89] Theformer value was shown to be associated with the a-NH2
group of puromycin used in the kinetic experiments, while thelatter value seemed to be associated with the ribosome. Theionizable group seemed to belong to A2451, since theformation of a peptide bond was about 130 times slowerthan normal when catalyzed by a ribosome bearing anA2451U mutation, and had lost the pH dependence. How-ever, an alternative explanation suggested by the authors wasthat the protonated group is part of the A2450:C2063 basepair lying directly behind A2451 (shown in Figure 11b). Inthis case, the ionizable group would be the N1 atom of A2450.Although a distance of 7 @ from the N1 atom to the a-NH2
group is too long for hydrogen transfer, a postulatedconformational change of the PTF center might bringA2450 within range.[89] The assumption of conformationalchanges again increases the number of possible candidatesthat might play a role in the kind of chemical catalysis that isadvocated here. We note that evidence was presented thatHis229 of protein L2 might also be involved in this catal-ysis,[90] although current atom maps place this residue morethan 20 @ away from the tetrahedral intermediate of thetransition state.
At the moment it can be said that a direct role of A2451 ina general acid–base catalysis can hardly be reconciled with theobservation that A2451 in active ribosomes, in contrast toinactive ribosomes, does not contain a titratable group at thispKa value.[86]
In fact, the ribosome need not use any direct chemicalinvolvement in the catalysis of the PTF reaction, such as theformation of a transient covalent interaction between thesubstrate (tRNAs) and the enzyme (the ribosome, or morespecifically in this case the rRNA). The template modelpredicts that tight stereochemical arrangement of substratesrelative to one another would be sufficient to provide thedramatic acceleration of the reaction rate needed for theformation of a peptide bond (see the review in Ref. [91]). Inthis case the role of A2451 would be to remove a proton fromthe free nucleophilic a-NH2 group of the A-site substrate orform a hydrogen bond with the a-NH2 group, thus promotingformation of a peptide bond through proper positioning of theNH2 group. The reaction scheme would be something like thatpresented in Figure 9 and described in more detail in thecorresponding legend.
Tight fixation of the CCA ends of the P- and A-tRNAs isexactly what is observed both in the analogue-soaked 50Scrystal structure (Figure 11a) and the products containing apeptide bond following soaking of the A- and P-site substrates(Figure 11b). The CCA end is locked into position in theP site by formation of two Watson–Crick base pairs (C74 andC75 with G2252 and G2251, respectively). A76 stacks on theribose of A2451 (shown clearly in Figure 11b). The CCA endof the aa-tRNA in the A site is fixed by: 1) Watson–Crickbase pairing between C75 and G2553, 2) a type-I A-minormotif between A76 and the G2583-U2506 base pair, and 3) anadditional hydrogen-bonding interaction between the 2’-OHgroup of A76 and U2585. Tight fixation of the CCA ends ofboth A- and P-tRNAs at the PTF center underlines theimportance of the (purely physical) template model in
formation of a peptide bond, namely, that precise stereo-chemical fixation is predominantly responsible for theenormous acceleration of the reaction. The rate of formationof a peptide bond on the ribosome at about 50 s�1 wasestimated to be approximately a factor of 105 faster than theuncatalyzed reaction (the rate in the absence of ribosomes).[91]
The ribosomal PTF reaction without chemical catalysis (thatis, when the ionizable group of the ribosome is protonated atpH< 7) occurs with a rate of about 0.5 s�1,[89] which is stillmore than 1000 times faster than the uncatalyzed reaction. Ifthis estimation is correct then the purely physical mode ofpeptide-bond formation (the exact stereochemical arrange-ment of the reactants) represents approximately 90% of thereaction rate, with the catalytic component making up theremaining 10%.
2.3.3. Antibiotic Action on the 50S Subunit
The functional importance of rRNA at the ribosomeactive centers is reiterated by the sites of interaction of avariety of clinically relevant antibiotics, such as chloramphe-nicol (Cam) and clindamycin, which inhibit PTF activity.[11]
Cam is well-known as an specific bacterial inhibitor of theCCA-aa end of an A-site tRNA[92] but does not inhibit theCCA fixation of a P-site tRNA.[93] The binding site of Camwas determined to a resolution of 3.5 @ by soaking theantibiotic into crystals of the 50S subunit of D. radioduransand shown to involve interaction with seven nucleotideswithin the PTF center.[11] A number of these interactions areindirect as they are mediated through putative Mg2+ ions.Since moieties important for the antibiotic action of Camconstitute these interactions, this result suggests that thepresence of the ions are of extreme importance for antibioticbinding and PTF inhibition. The position of the “tail” of Camwithin the PTF center is such that it reaches towards, and mayeven displace, the CCA end of the A-site tRNA. Thisobservation is in agreement with the result that althoughtRNAs can bind in the presence of Cam, they cannot undergoformation of a peptide bond, probably because the CCA-aaend of the A-site tRNA is displaced from the correct position.Cam inhibition in vitro is dependent on the nature of thepeptidyl residue and the A-site substrate. In particular, Cam isa less effective inhibitor against aromatic amino acids such asphenylalanine. This leads to the conclusion that these aminoacids can actually displace Cam during formation of thepeptide bond by competing with the phenyl group of the drugfor binding. The major overlap between Cam and an aminoacid in the A-site tRNA lends credence to the idea that Camoperates predominantly by displacing the aminoacyl residueof the A-site tRNA, which probably indirectly displaces theCCA end.
In contrast with chloramphenicol, the binding site deter-mined for the lincosamide clindamycin spans between boththe A and P sites at the PTF center.[11] The majority of theinteractions involve hydrogen bonds between hydroxy groupson the sugar moiety and bases within the PTF center; thesecontacts are in agreement with most of the available mutationresistance data. The proline group of clindamycin overlapswith the position of the phenyl group of Cam, which is in line
Ribosome Structure and TranslationAngewandte
Chemie
3479Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

with the A-site nature of clindamycin inhibition. TheC8’ atom on the proline moiety of clindamycin comes within2.5 @ of the N3 atom of C2452 and is thus in proximity to a P-site-bound tRNA. Thus, the binding position of clindamycintraverses both the A and P sites, and would be expected todisturb the positioning of amino acid moieties at both sites.
The positions of no less than seven distinct members ofthe macrolide family of antibiotics have been solved incomplexes with the 50S subunit.[11,74] This large family ofantibiotics can be divided into three classes on the basis of thesize of the lactone ring. Erythromycin and two erythromycinderivatives (clarithromycin and roxithromycin) are represen-tatives of the 14-membered-ring class, all of which have beensolved in complexes with the 50S subunit of D. radiodurans.[11]
From the two larger 15- and 16-membered classes, the bindingsites of azithromycin, spiramycin, tylosin, and carbomycin Ahave been solved in complexes with the 50S subunit fromH. marismortui.[74]
The binding positions determined for these macrolides aregenerally in good agreement with one another, located withinthe polypeptide tunnel in proximity to the PTF center. In thisposition, the macrolides would be expected to block thetunnel, thus preventing passage of the nascent polypeptidechain (Figure 12). This proposal can be reconciled with theobservation that macrolides cannot inhibit actively translatingribosomes, since the presence of the polypeptide chain in thetunnel precludes the macrolide from binding. Ribosomesprebound with macrolides are able to synthesize oligopep-tides up to five amino acids in length, depending on themacrolide present. Since the lactone ring of the boundmacrolides are positioned in the tunnel such that the sugarextensions from the C5 position extend towards the PTF
center, the length of the side chain dictates the number ofpeptide bonds that can be formed; for example, tri- ortetrapeptides can be formed in the presence of erythromycin,which has a monosaccharide at the C5 position, whereas onlydipeptides are formed in the presence of tylosin andspiramycin, which bear C5-disaccharides. Carbomycin A,which has an additional isobutyrate group on the disacchar-ide, even prevents formation of the first peptide bond. Theisobutyrate group of carbomycin A reaches so far into thePTF center that it occupies the position that the amino acidmoiety of the A-site substrate would normally occupy.[74] Thedirect interaction of the macrolides with position 2058 makesit easy to understand how modifications and mutations at thisposition in E. coli provide resistance to this class of anti-biotics. In contrast, erythromycin (or its derivatives) andazithromycin make no direct contact with ribosomal pro-teins L4 or L22, which suggests that the mutations withinthese proteins that confer resistance must do so indirectlythrough conformational changes of the 23S rRNA.
A further interesting discovery was the formation of areversible covalent bond between the acetaldehyde substitu-ent at position C6 of the 16-membered ring macrolides andthe N6 atom of A2062 (E. coli).[74] The formation of thiscarbinolamine is specific for the 16-membered group, sincethe smaller macrolides do not contain a correspondingaldehyde functional group. Lastly, it is important to notethat although the overall binding sites for each macrolide arein general agreement between the two studies discussed here,there are some significant differences in the orientation andconformation of the lactone ring and cladinose sugar moietyof the macrolides bound on the ribosome. In one studyinteraction with the tunnel wall is made through the hydro-phobic face of the lactone ring, whereas in the otherinteraction is made through a series of hydrogen bonds.Whether these discrepancies arise because of differences ininterpretation, differential binding to ribosomes from partic-ular species, or actual differences in the binding of themacrolides themselves is unclear. The last possibility wouldbe surprising since azithromycin and erythromycin differ onlyby the absence of a ketone oxygen atom and the addition ofmethyl nitrogen atom. Re-analysis of these structures and theduplication of identical antibiotics for each ribosome speciesshould resolve this problem.
2.4. P- and A-tRNA Translocation within the Ribosome
The position of the tRNAs remains unchanged afterformation of the peptide bond. This has been demonstratedby cryo-EM analyses of E. coli ribosome complexes[19] andmost strikingly by soaking A- and P-site substrates into active50S H. marismortui crystals and solving the structure of thereaction products.[87] After formation of the peptide bond theribosome must transfer the products—the peptidyl-tRNA inthe A site and deacylated tRNA in the P site to the P andE sites, respectively–-thus, shifting the ribosome from the pre-to the posttranslational state (Figure 2c and 2 f respectively).This process is termed translocation. It must be extremelyaccurate at both ends of the tRNA molecule: The anticodon–
Figure 12. The macrolide carbomycin A bound in the tunnel of theH. marismortui 50S subunit. The rRNA and proteins are represented asviolet ribbons and carbomycin A as a red space-filling model. The 50Ssubunit is viewed from the cytoplasmic side (backside) looking up thetunnel to the PTF center. This figure was created from pdb file 1K8A[74]
using Swiss-pdb viewer[55] and rendered with POVRAY.
K. H. Nierhaus and D. N. WilsonReviews
3480 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

codon complex must be moved exactly 10 @ (the length ofone codon), longer or shorter movements would result in theribosome losing the reading frame. At the other end of the A-site peptidyl-tRNA, the CCA end must also be movedprecisely into the P site so as to set up the next PTF reactionwith the incoming aa-tRNA. Incorrect placement of thepeptidyl-tRNA at the P site could be disastrous for theformation of a peptide bond and result in the abortion oftranslation.
Ribosomes have an innate translocase activity, but it ismore than one order of magnitude slower than that of the EF-G-catalyzed reaction.[94] This observation implies that thestructures necessary to move the tRNAs reside in theribosome and that the role of EF-G/EF2 is to reduce theactivation energy barrier that separates the two sets of tRNApositions (A plus P and P plus E). Important questionsremaining unanswered are: How does EF-G/EF2 mediatetranslocation and which ribosomal components are involvedin the transfer of the tRNAs?
2.4.1. Conservation in the Binding Site of Elongation Factor-G
The crystal structure of EF-G has been solved in theabsence of the nucleotide[48] and in the complex with GDP(Figure 5a).[47] The GTP form is the active one, which binds tothe ribosome and triggers translocation. Hydrolysis of GTPinactivates EF-G and dissociates it from the ribosome (see thereview by Kaziro).[95] EF-G belongs to the same subfamily ofG proteins as IF2, RF3, and EF-Tu, the latter of which hasbeen crystallized in both GTP (active) and GDP (inactive)forms which exhibit large domain shifts relative to oneanother.[50]
Cryo-EM reconstructions of EF-G bound to bacterial70S ribosomes at a resolution of 17.5–20 @[96–98] and EF2 toeukaryotic 80S ribosomes at 17.5 A resolution[99] show similarbinding sites for both factors (Figure 13a and b). Antibiotics
were used to trap the elongation factor on the ribosome inthese complexes. EF-G was trapped on the 70S ribosomeusing the antibiotic fusidic acid. This fixation allows trans-location and GTP hydrolysis, but blocks the switch into theGDP conformer of the factor, thus preventing dissociationfrom the ribosome. The eukaryotic eEF2 was locked onto theyeast ribosome using the antifungal sordarin, which is thoughtto function analogously to fusidic acid.[99] The complex wasformed with a typical pretranslational state, that is, A- and P-tRNAs were present. As expected, the tRNAs were trans-located to the P and E sites, but of special interest is that thetip, domain IV, of EF-G was shown to occupy the position ofthe A site. Similarly, EF2 in the EF2–ribosome complex alsooccupied the A site and came very close to the position of theP-tRNA (Figure 13c and d). EF-G-mediated translocation isalso possible in the presence of nonhydrolyzable GTPanalogues, such as GDPNP, which suggests that binding ofEF-G alone is sufficient for translocation and that hydrolysisis necessary for the conformational change and release of EF-G·GDP.[95]
For classical G proteins, a GTPase-activating protein(GAP) stimulates the G-protein-mediated hydrolysis ofGTP. In the case of EF-G/EF2, the GAP is provided bycomponents of the ribosome. There are certainly grosschanges visible upon the binding of each elongation factorto the ribosome. One of the most striking changes is seenwithin the “stalk” region; there is no electron density for thisregion in the empty 70S and 80S ribosomes but becomesordered upon EF-G/EF2 binding,[96,97,99] which supports itsuniversal role in factor binding. Perhaps the best candidatesfor the GAP role are a region of the 23S rRNA termed thesarcin–ricin loop (SRL) and the pentameric stalk complex ofthe ribosomal proteins L10·(L7/L12)4. The SRL is so namedbecause cleavage after G2661 of the bacterial 23S rRNAwithin this region by a-sarcin inhibits all activities dependenton the elongation factor.[100] Similar effects are seen afterremoving the neighboring base A2660 (E. coli nomenclature)in the 26S rRNA of yeast by the N-glycosidase activity of thericin A-chain.[101] Furthermore, this region contains thelongest (12 nucleotides) universally conserved stretch ofrRNA, which underlines its functional importance.
Recently, hybrid ribosomes were constructed in which theproteins at the GTPase center from E. coli (L7/L12 and L10)were replaced with their eukaryotic counterparts from rat P1/P2 and P0, respectively.[102] Both the in vitro translation andGTPase activity of the resultant hybrid ribosomes was strictlydependent on the presence of the eukaryotic elongationfactors, EF2 and EF1a. This result reflects not only thespecificity of the interaction between the stalk proteins andthe elongation factors from each species, but also theimportance of the stalk proteins in mediating elongationfactor GTPase activity.
The ribosomal protein L11 (and associated L11 bindingsite on the 23S rRNA) is often considered as a candidate fortaking over the function of the GAP. This is becausemutations in both L11 and its binding site on the 23S rRNAcan confer resistance against the antibiotic thiostrepton, apotent inhibitor of EF-G- and EF-Tu-dependent GTPaseactivities.[103] However, the direct involvement of L11 in the
Figure 13. Comparison of cryo-electron microscopic analyses of theEF-G·70S complex from E. coli (a) and the EF2·80S complex fromS. cerevisiae (b); the small subunit is on the left side and the large sub-unit on the right. The same orientation is seen on the left of an empty80S ribosome (yellow: 40S, blue 60S). The relative arrangements ofEF-G and P-tRNA (c) and EF2 (red) and P-tRNA (d) are illustrated. Theabbreviations of the features are as in Figure 1, while roman numeralson EF-G and EF2 refer to the domains of these factors. The figure wasmodified from Gomez-Lorenzo et al.[99]
Ribosome Structure and TranslationAngewandte
Chemie
3481Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

factor-dependent GTPase is not very likely, since mutantslacking L11 are viable, although extremely compromised,[104]
and the IF-2-dependent GTPase is stimulated rather thanblocked by thiostrepton.[105] Furthermore, replacement ofeither the L10·(L7/L12)4 complex or L11 with the equivalentrat protein showed that the P0·(P1·P2)2 complex, but not theeukaryotic counterpart to L11 (RL12), was responsible forfactor specificity and associated GTPase dependence,although addition of L11 or RL12 did stimulate proteinsynthesis significantly.[102] Thus, the role of L11 is unclear. L11is in any case in proximity to the elongation factors: Cryo-EManalyses of EF-G bound to 70S ribosomes revealed that theN-terminal domain of L11 is shifted upon binding of EF-G soas to form an arclike connection with the G domain of EF-G.[106] This arclike connection is also observed in the EF2–80Scomplex although it is broader and fused to a greaterextent.[99] Thus it seems likely that EF-G binding stimulatesconformational changes in the ribosome, probably throughthe L10·(L7/L12)4 complex, which triggers translocation ofthe A- and P-tRNAs. The question remaining is how are thetRNAs actually moved?
2.4.2. Dynamics within the Ribosome
In the a-e model for translocation a moveable domainwithin the ribosome is hypothesized to carry the A- and P-tRNAs during translocation (see the review in Ref. [107]).Evidence for this model comes from testing the accessibilityof phosphate groups on the tRNAs in the pre- and post-translational states. The essential observation was that theprotection patterns of A- and P-tRNAs differ from oneanother, but the corresponding tRNAs exhibit the sameprotection patterns in the pre- and the posttranslationalstates. This observation suggests that distinct ribosomalcomponents are involved in carrying the tRNAs from thepre- to the posttranslational state. There are a number ofcandidates that may play a role in the translocation of thetRNAs or even constitute portions of the moveable domains.
Distinct regions within the crystal structures are disor-dered, which most likely reflects the flexibility of thesecomponents. A classic example is the stalk region, which, asalready mentioned, only becomes ordered upon binding ofthe elongation factor. Another is the L1 region, the flexibilityof which may regulate release of E-tRNA (see Section 2.5).There are also certain structures that become either orderedor rearranged upon association of the subunit. Most of theseelements are constituents of the bridges between the subunits.One striking example is the universally conserved helix H69in domain IV of the larger RNA subunit. H69 is the majorelement of bridge B2a, the largest inter-subunit bridge (seeFigure 3), and is disordered in the structure of the 50S subunitof H. marismortui, but ordered in the 50S subunit of D. radio-durans. Comparison of the latter structure with the 70Sstructure of T. thermophilus shows that H69 swings outtowards h44, another very flexible element, upon association.In this extended conformation H69 would be predicted tomake contact with both A and P tRNAs.[108] Another elementthat is not fully resolved in either of the 50S structures is H38of domain II, a constituent of bridge B1a and often called the
“A-site finger” because it contacts the A site of tRNA. Aspreviously mentioned, both B1a and B2a bridge elementshave corresponding counterparts within the 80S yeast ribo-some, thus strengthening their candidacy for a role intranslocation.
2.5. The E Site and Translational Fidelity2.5.1. Discovery of a Third Universal tRNA Binding Site on
Ribosomes
Despite identification of the E site in bacterial ribosomesin the early 1980s,[109,110] it is only in recent years that it hasappeared in some textbooks. The E site has also beendiscovered in archea[111] and in eukaryotes, both yeast[112]
and mammals,[113] which suggests that it is a universal featureof ribosomes. The E site is specific for deacylated tRNA andwill not bind peptidyl- or aminoacyl-tRNAs. This result is inaccordance with the idea that the E site only accepts thedeacylated tRNA from the P site following transfer of thenascent chain to the A-tRNA. At least part of the controversyover the existence of the E site stems from its instability undercertain buffer conditions (see the review in Ref. [107]). Stablebinding in the E site has been shown to be dependent onphysiological buffer conditions, such as in the presence of alow magnesium concentration (3–6 mm) and polyamines.Nonphysiological buffer conditions dissociate the E-tRNA,as nicely illustrated by the visualization of tRNAs inposttranslational complexes under different buffer conditionsusing cryo-EM.[114, 115] Under nonphysiological conditions, theE-tRNA electron density was lost from the E site itself andextra electron density appeared contacting the L1 stalk (theE2 site). It was thought that this position might reflect thepath of a tRNA that has left the E site as it dissociates fromthe ribosome and that the very flexible L1 stalk might play arole in dissociating the E-tRNA from the ribosome. This ideahas gained recent support from a comparison of the crystalstructure of the D. radiodurans 50S subunit with the T. ther-mophilus 70S structure, where the position of the L1 armdiffers by 308 (Figure 14). In the latter structure, the L1 arm isclosed and contacts the elbow of the E-tRNA, which preventsits release from the ribosome. In contrast, the L1 arm in theD. radiodurans structure is open, which may serve to releasethe E-tRNA during translation.[108] In fact, this mechanismmay be universal. In yeast, there is an enormous 70 @difference in the position of the L1 stalk in the P-tRNA-bound 80S ribosome (pseudo-pretranslational state) and astalled translating ribosome (artificially induced posttransla-tional state).[3,4] In light of the conformational flexibility of theL1 stalk (see below), the E2 electron density may not evenrepresent a tRNA, which may have totally dissociated fromthe ribosome; instead it may merely reflect a buffer-inducedconformational change within the L1 region.
The existence of the E site was also confirmed by thepresence of three tRNAs in the crystal structure of the70S subunit of T. thermophilus.[76] The E-tRNA results pre-dominantly from the presence of endogenous E-tRNA thatco-purified with the ribosomes, thus illustrating the stability ofthe binding despite the lengthy purification procedure. The
K. H. Nierhaus and D. N. WilsonReviews
3482 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

existence of the E site is finally being accepted, but itsimportance has yet to be fully recognized.
2.5.2. Importance of the E-Site: Fidelity and Reading FrameMaintenance
The fact that the E site has been discovered in theribosomes of both prokaryotes and eukaryotes suggests that itshould play an important role during translation. Thepresence of a deacylated tRNA at the E site has beenshown to modulate aa-tRNA selection at the A site (see thereview in Ref. [116]). In particular, an occupied E site inducesa low affinity A site, such that interaction of the ternarycomplex is dictated by anticodon interactions. This allowsonly cognate and near-cognate tRNAs to bind, thus eliminat-ing 90% of the competing noncognate tRNAs (the impor-tance of this was discussed in Section 2.2.1). In contrast, whenthe E-tRNA is absent, the A site has high affinity, whichpermits interaction of all tRNAs, including the erroneousincorporation of noncognate tRNAs. These two situationshave been demonstrated in vitro.[117] Simply, in the presenceof P-tRNA alone, both noncognate Asp-tRNA and cognatePhe-tRNA could bind to an A site UUU codon (Figure 15a),but in the presence of an E-tRNA (and P-tRNA), only thecognate Phe-tRNA could bind the A site (Figure 15b and c).Binding of the A-tRNA releases the E-tRNA from the E site,so that at any one time during translation, except during thefirst decoding step, there are never more than two tRNAspresent on the ribosome. Furthermore, the E-tRNA must becognate to the E site codon, as a near-cognate E-tRNA couldnot prevent erroneous incorporation of the Asp-tRNA.[117] Itis significant that a cognate tRNA must be present at theE site; this observation indicates that codon–anticodon inter-
actions at the E site is the signal that tells the ribosome toadopt the posttranslational state and display a low affinityA site. Further evidence for anticodon–codon interactions atthe E site comes from the gag-pol recoding site of HIV-1.Ribosomes recode this site using a posttranslocationalslippage mechanism, that is, slippage into the �1 readingframe occurs after translocation, where both P- and E-tRNAsslip simultaneously in the �1 direction.[118] Another recodingsite illustrating the importance of the E site is the + 1frameshifting site of the termination factor RF2. At codonposition 26 within the RF2 mRNA a + 1 frameshift isrequired to avoid an internal UGA stop. This internal stopcodon is specifically recognized by the RF2 protein and thusprovides an autoregulatory mechanism, such that when RF2protein levels are high in the cell, RF2 termination at theframeshift is favored and a truncated and inactive RF2 pro-tein is produced which is rapidly degraded. When RF2 levelsare low in the cell, termination loses out to frameshifting andthe full-length active RF2 protein is produced. One importantfeature that stimulates frameshifting in this case is anupstream Shine–Dalgarno (SD) type sequence that interactswith a complementary anti-SD sequence within the16S rRNA of the 30S subunit. Interestingly, the SD-anti-SDinteraction includes the first position of the E-site codon.Recently it could be demonstrated that formation of the SD-anti-SD duplex displaces the E-tRNA from the ribosome andby doing so promotes frameshifting.[119] Thus the implicationis that the presence of the E-tRNA at the E site is necessaryfor maintaining the correct reading frame during translation.
3. Summary and Outlook
Although we have taken a huge leap forward during thepast years in our understanding of ribosome structure and
Figure 14. Movement of the L1 arm. A region of the crystal structure ofthe D. radiodurans 50S subunit (D50S) showing the 23S rRNA (gray)with the L1 arm highlighted (yellow). The position of the L1 arm in thecrystal structure of the T. thermophilus 70S subunit (T50S) is superim-posed (green) with the putative pivot point for the L1 arm and ismarked with a red dot. The positions of the A- (cyan), P- (blue), and E-tRNAs (pink) are included, with the anticodon arms pointing out ofthe plane of the paper. Adapted from Harms et al.[108]
Figure 15. The role of the E site in the accuracy of decoding. Ribosomecomplexes were prepared using poly(U)-mRNA where the P site wasoccupied with AcPhe-tRNA. A-site binding of cognate Phe-tRNA(codon UUU) and noncognate Asp-tRNA (codon GAC/U) was per-formed in the absence (a) or presence (b) of an E-tRNA. Binding ofthe Phe-tRNA or Asp-tRNAs to the A site was monitored by measuringthe formation of the dipeptides AcPhe-Phe or AcPhe-Asp, respectively(c). Both AcPhe-Phe and AcPhe-Asp were formed in the absence of E-tRNA, which indicates that erroneous incorporation of Asp-tRNA at theA site occurred, while normal amounts of AcPhe-Phe with backgroundlevels of AcPhe-Asp were formed in the presence of E-tRNA, thus indi-cating that the cognate Phe-tRNA predominantly bound the A site.Data taken from Geigenm%ller et al.[117]
Ribosome Structure and TranslationAngewandte
Chemie
3483Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

function, there is still much that eludes researchers. Whilesome of these questions remain fundamental to the process oftranslation, for example, those pertaining to the mechanism ofpeptidyltransferase, translocation, or the role of the tunnel,there are many questions that are perhaps more peripheral,being associated with translational regulation. This is partic-ularly true in the case of eukaryotic translation, with itsmultitude of factors, additional ribosomal proteins, and rRNAexpansion elements. Some of the answers to these questionswill arrive with higher resolution structures of ribosomalcomplexes and translational intermediates. As high-resolu-tion structures are dependent on the production of diffractingcrystals it is hard to predict with any certainty the time scalefor the preparation of eukaryotic ribosome crystals orprokaryotic ribosome complexes, such as pre- and posttrans-locational states, or with large translational factors bound.Unfortunately, these are static structures but the ribosome is ahighly dynamic machine, therefore, dissection of its mecha-nism will require a concerted approach from many differentangles, both biochemical and biophysical. Given that proteinsynthesis is fundamental to all life forms and provides suchinsight into general molecular interactions, surely the pursuitfor further knowledge must be one worth striving for!
Abbreviations
A site aminoacyl(aa)-tRNA siteA-tRNA tRNA in the A siteCTD carboxy-terminal domainE site, exit site, specific fordeacylated tRNAE-tRNA tRNA at the E siteGAP GTPase activating proteinNTD amino-terminal domainP site peptidyl-tRNA site before
peptide-bond formationP-tRNA tRNA at the P sitePOST posttranslocational statePRE pretranslocational statePTF peptidyltransferaseSD Shine–DalgarnoSRL sarcin–ricin loopUTR untranslated region of an
mRNA
We would like to thank Christian Spahn for supplying high-resolution images of Figure 1a–d, 3b, 8b and d, 8zlem Tastanfor kindly providing Figure 7, and Meredith Ross, OliverVesper, and Sean Connell for critical reading of the Manu-script. D.N.W. would like to thank the Alexander von Hum-boldt foundation for support. K.H.N. acknowledges thesupport of the Deutsche Forschungsgemeinschaft (Ni174/8-2and Ni174/-9-2).
Received: July 23, 2002 [A544]
[1] J. Frank, Bioessays 2001, 23, 725 – 732.[2] P. Dube, M. Wieske, H. Stark, M. Schatz, J. Stahl, F. Zemlin, G.
Lutsch, M. van Heel, Structure 1998, 6, 389 – 399.[3] C. M. Spahn, R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G.
Blobel, J. Frank, Cell 2001, 107, 373 – 386.[4] R. Beckmann, C. M. Spahn, N. Eswar, J. Helmers, P. A.
Penczek, A. Sali, J. Frank, G. Blobel, Cell 2001, 107, 361 – 372.[5] a) D. G. Morgan, J. F. Menetret, A. Neuhof, T. A. Rapoport,
C. W. Akey, J. Mol. Biol. 2002, 324, 871 – 886; b) J. F. Menetret,A. Neuhof, D. G. Morgan, K. Plath, M. Radermacher, T. A.Rapoport, C. W. Akey, Mol. Cell 2000, 6, 1219 – 1232.
[6] R. Beckmann, D. Bubeck, R. Grassucci, P. Penczek, A.Verschoor, G. Blobel, J. Frank, Science 1997, 278, 2123 – 2126.
[7] T. Tenson, M. Ehrenberg, Cell 2002, 108, 591 – 594.[8] B. T. Wimberly, D. E. Brodersen,W. M. Clemons, R. J. Morgan-
Warren, A. P. Carter, C. Vonrhein, T. Hartsch, V. Ramak-rishnan, Nature 2000, 407, 327 – 339.
[9] F. Schluenzen, A. Tocilj, R. Zarivach, J. Harms, M. Gluehmann,D. Janell, A. Bashan, H. Bartels, I. Agmon, F. Franceschi, A.Yonath, Cell 2000, 102, 615 – 623.
[10] N. Ban, P. Nissen, J. Hansen, P. B. Moore, T. A. Steitz, Science2000, 289, 905 – 920.
[11] F. SchlSnzen, R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R.Albrecht, A. Yonath, F. Franceschi, Nature 2001, 413, 814 – 821.
[12] a) V. Ramakrishnan, Cell 2002, 108, 557 – 572; b) P. B. Moore,T. A. Steitz, Nature 2002, 418, 229 – 235; c) A. Yonath, Annu.Rev. Biophys. Biomol. Struct. 2002, 31, 257 – 273.
[13] D. N. Wilson, G. Blaha, S. R. Connell, P. V. Ivanov, H. Jenke, U.Stelzl, Y. Teraoka, K. H. Nierhaus, Curr. Protein Pept. Sci. 2002,3, 1 – 53.
[14] P. Schimmel, B. Henderson, Proc. Natl. Acad. Sci. USA 1994,91, 11283 – 11286.
[15] D. E. Brodersen, W. M. Clemons, Jr., A. P. Carter, B. T. Wim-berly, V. Ramakrishnan, J. Mol. Biol. 2002, 316, 725 – 768.
[16] R. R. Gutell, J. J. Cannone, Z. Shang, Y. Du, M. J. Serra, J. Mol.Biol. 2000, 304, 335 – 354.
[17] P. Nissen, J. A. Ippolito, N. Ban, P. B. Moore, T. A. Steitz, Proc.Natl. Acad. Sci. USA 2001, 98, 4899 – 4903.
[18] a) J. H. Cate, A. R. Gooding, E. Podell, K. H. Zhou, B. L.Golden, C. E. Kundrot, T. R. Cech, J. A. Doudna, Science 1996,273, 1678 – 1685; b) A. R. Ferre-D'Amare, K. Zhou, J. A.Doudna, Nature 1998, 395, 567 – 574.
[19] R. K. Agrawal, C. M. T. Spahn, P. Penczek, R. A. Grassucci,K. H. Nierhaus, J. Frank, J. Cell Biol. 2000, 150, 447 – 459.
[20] L. P. Gavrilova, O. E. Kostiashkina, V. E. Koteliansky, N. M.Rutkevitch, A. S. Spirin, J. Mol. Biol. 1976, 101, 537 – 552.
[21] S. Schilling-Bartetzko, A. Bartetzko, K. H. Nierhaus, J. Biol.Chem. 1992, 267, 4703 – 4712.
[22] G. Z. Yusupova, M. M. Yusupov, J. H. Cate, H. F. Noller, Cell2001, 106, 233 – 241.
[23] T. V. Pestova, V. G. Kolupaeva, I. B. Lomakin, E. V. Pilipenko,I. N. Shatsky, V. I. Agol, C. U. T. Hellen, Proc. Natl. Acad. Sci.USA 2001, 98, 7029 – 7036.
[24] A. Roll-Mecak, B. S. Shin, T. E. Dever, S. K. Burley, TrendsBiochem. Sci. 2001, 26, 705 – 709.
[25] C. Gualerzi, L. Brandi, E. Caserta, A. La teana, R. Spurio, J.Tomsic, C. Pon in The ribosome. structure, function, antibiotics,and cellular interactions (Eds.: R. A. Garrett, S. R. Douthwaite,A. Liljas, A. T. Matheson, P. B. Moore, H. F. Noller), AmericanSociety for Microbiology, Washington, DC, 2000, pp. 477 – 494.
[26] A. P. Carter, W. M. Clemons, Jr., D. E. Brodersen, R. J.Morgan-Warren, T. Hartsch, B. T. Wimberly, V. Ramakrishnan,Science 2001, 291, 498 – 501.
[27] M. Pioletti, F. Schlunzen, J. Harms, R. Zarivach, M. Gluhmann,H. Avila, A. Bashan, H. Bartels, T. Auerbach, C. Jacobi, T.
K. H. Nierhaus and D. N. WilsonReviews
3484 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486

Hartsch, A. Yonath, F. Franceschi, EMBO J. 2001, 20, 1829 –1839.
[28] A. Dallas, H. F. Noller, Mol. Cell 2001, 8, 855 – 864.[29] A. Gnirke, K. H. Nierhaus, J. Biol. Chem. 1986, 261, 14506 –
14514.[30] A. Roll-Mecak, C. Cao, T. E. Dever, S. K. Burley, Cell 2000,
103, 781 – 792.[31] I. S. Gabashvili, R. K. Agrawal, C. M. T. Spahn, R. A. Grass-
ucci, D. I. Svergun, J. Frank, P. Penczek, Cell 2000, 100, 537 –549.
[32] J. H. Cate, M. M. Yusupov, G. Z. Yusupova, T. N. Earnest, H. F.Noller, Science 1999, 285, 2095 – 2104.
[33] a) A. P. Potapov, FEBS Lett. 1982, 146, 28 – 33; b) A. P.Potapov, F. J. Triana-Alonso, K. H. Nierhaus, J. Biol. Chem.1995, 270, 17680 – 17684.
[34] P. Nissen, M. Kjeldgaard, J. Nyborg, EMBO J. 2000, 19, 489 –495.
[35] K. H. Nierhaus, Mol. Microbiol. 1993, 9, 661 – 669.[36] M. Valle, J. Sengupta, N. K. Swami, R. A. Grassucci, N.
Burkhardt, K. H. Nierhaus, R. K. Agrawal, J. Frank, EMBOJ. 2002, 21, 3557 – 3567.
[37] H. Stark, M. V. Rodnina, H. J. Wieden, F. Zemlin, W. Winter-meyer, M. Van Heel, Nat. Struct. Biol. 2002, 15, 15 – 20.
[38] A. Weijland, A. Parmeggiani, Science 1993, 259, 1311 – 1314.[39] J. M. Ogle, D. E. Brodersen, W. M. Clemons, Jr, M. J. Tarry,
A. P. Carter, V. Ramakrishnan, Science 2001, 292, 897 – 902.[40] J. J. Hopfield, Proc. Natl. Acad. Sci. USA 1974, 71, 4135 – 4139.[41] J. Ninio, Biochimie 1975, 57, 587 – 595.[42] M. Ehrenberg, D. Andersson, K. Mohman, P. Jelenc, T.
Ruusala, C. G. Kurland in Structure, function and genetics ofribosomes (Eds.: B. Hardesty, G. Kramer), Springer, New York,1986, pp. 573 – 585.
[43] M. V. Rodnina, T. Daviter, K. Gromadski, W. Wintermeyer,Biochimie 2002, 84, 745 – 754.
[44] S. Schilling-Bartetzko, F. Franceschi, H. Sternbach, K. H.Nierhaus, J. Biol. Chem. 1992, 267, 4693 – 4702.
[45] A. M. Karim, R. C. Thompson, J. Biol. Chem. 1986, 261, 3238 –3243.
[46] J. M. Ogle, F. V. Murphy, M. J. Tarry, V. Ramakrishnan, Cell2002, 111, 721 – 732.
[47] J. Czworkowski, J. Wang, T. A. Seitz, P. B. Moore, EMBO J.1994, 13, 3661 – 3668.
[48] A. Aevarsson, E. Brazhnikov, M. Garber, J. Zheltonosova, Y.Chirgadze, S. Alkaradaghi, L. A. Svensson, A. Liljas, EMBO J.1994, 13, 3669 – 3677.
[49] M. Kjeldgaard, J. Nyborg, J. Mol. Biol. 1992, 223, 721 – 742.[50] H. Berchtold, L. Reshetnikova, C. O. A. Reiser, N. K.
Schirmer, M. Sprinzl, R. Hilgenfeld, Nature 1993, 365, 126 –132.
[51] P. Nissen, M. Kjeldgaard, S. Thirup, G. Polekhina, L. Reshetni-kova, B. F. C. Clark, J. Nyborg, Science 1995, 270, 1464 – 1472.
[52] M. Laurberg, O. Kristensen, K. Martemyanov, A. T. Gudkov, I.Nagaev, D. Hughes, A. Liljas, J. Mol. Biol. 2000, 303, 593 – 603.
[53] a) M. Selmer, S. Al-Karadaghi, G. Hirakawa, A. Kaji, A. Liljas,Science 1999, 286, 2349 – 2352; b) K. K. Kim, K. Min, S. W. Suh,EMBO J. 2000, 19, 2362 – 2370; c) T. Yoshida, S. Uchiyama, H.Nakano, H. Kashimori, H. Kijima, T. Ohshima, Y. Saihara, T.Ishino, H. Shimahara, K. Yokose, T. Ohkubo, A. Kaji, Y.Kobayashi, Biochemistry 2001, 40, 2387 – 2396.
[54] T. Toyoda, O. F. Tin, K. Ito, T. Fujiwara, T. Kumasaka, M.Yamamoto, M. B. Garber, Y. Nakamura, RNA 2000, 6, 1432 –1444.
[55] N. Guex, M. C. Peitsch, Electrophoresis 1997, 18, 2714 – 2723.[56] B. Vestergaard, L. Van, G. Andersen, J. Nyborg, R. Bucking-
ham, M. Kjeldgaard, Mol. Cell 2001, 8, 1375 – 1382.[57] H. Song, P. Mugnier, A. Das, H. Webb, D. Evans, M. Tuite, B.
Hemmings, D. Barford, Cell 2000, 100, 311 – 321.
[58] B. P. Klaholz, T. Pape, A. V. Zavialov, A. G. Myasnikov, E. V.Orlova, B. Vestergaard, M. Ehrenberg, M. Van Heel, Nature2003, 421, 90 – 94.
[59] U. B. Rawat, A. V. Zavialov, J. Sengupta, M. Valle, R. A.Grassucci, J. Linde, B. Vestergaard, M. Ehrenberg, J. Frank,Nature 2003, 421, 87 – 90.
[60] L. Lancaster, M. C. Kiel, A. Kaji, H. F. Noller, Cell 2002, 111,129 – 140.
[61] G. Hirokawa, M. C. Kiel, A. Muto, M. Selmer, V. S. Raj, A.Liljas, K. Igarashi, H. Kaji, A. Kaji, EMBO J. 2002, 21, 2272 –2281.
[62] a) K. Yamaguchi, K. von Knoblauch, A. R. Subramanian, J.Biol. Chem. 2000, 275, 28455 – 28465; b) K. Yamaguchi, A. R.Subramanian, J. Biol. Chem. 2000, 275, 28466 – 28482; c) T.Suzuki, M. Terasaki, C. Takemoto-Hori, T. Hanada, T. Ueda, A.Wada, K. Watanabe, J. Biol. Chem. 2001, 276, 33181 – 33195;d) E. C. Koc, W. Burkhart, K. Blackburn, M. B. Moyer, D. M.Schlatzer, A. Moseley, L. Spremulli, J. Biol. Chem. 2001, 276,43958 – 43969; e) E. C. Koc, W. Burkhart, K. Blackburn, A.Moseley, L. Spremulli, J. Biol. Chem. 2001, 276, 19363 – 19374.
[63] J. A. Mears, J. J. Cannone, S. M. Stagg, R. R. Gutell, R. K.Agrawal, S. C. Harvey, J. Mol. Biol. 2002, 321, 215 – 234.
[64] A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly, V. Ramakrishnan, Nature 2000, 407,340 – 348.
[65] D. E. Brodersen, W. M. Clemons, A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly, V. Ramakrishnan, Cell 2000, 103,1143 – 1154.
[66] J. Gordon, J. Biol. Chem. 1969, 244, 5680 – 5686.[67] J. S. Lodmell, A. E. Dahlberg, Science 1997, 277, 1262 – 1267.[68] S. R. Connell, C. A. Trieber, U. Stelzl, E. Einfeldt, D. E. Taylor,
K. H. Nierhaus, Mol. Microbiol. 2002, 45, 1463 – 1472.[69] P. Nissen, J. Hansen, N. Ban, P. B. Moore, T. A. Steitz, Science
2000, 289, 920 – 930.[70] N. Polacek, M. Gaynor, A. Yassin, A. S. Mankin, Nature 2001,
411, 498 – 501.[71] G. W. Muth, L. Ortoleva-Donnelly, S. A. Strobel, Science 2000,
289, 947 – 950.[72] J. Thompson, D. F. Kim, M. O'Connor, K. R. Lieberman, M. A.
Bayfield, S. T. Gregory, R. Green, H. F. Noller, A. E. Dahlberg,Proc. Natl. Acad. Sci. USA 2001, 98, 9002 – 9007.
[73] R. Green, J. R. Lorsch, Cell 2002, 110, 665 – 668.[74] J. L. Hansen, J. A. Ippolito, N. Ban, P. Nissen, P. B. Moore, T. A.
Steitz, Mol. Cell 2002, 10, 117 – 128.[75] D. Knowles, N. Foloppe, N. Matassova, A. Murchie, Curr. Opin.
Pharmacol. 2002, 2, 501.[76] M. M. Yusupov, G. Z. Yusupova, A. Baucom, K. Lieberman,
T. N. Earnest, J. H. Cate, H. F. Noller, Science 2001, 292, 883 –896.
[77] M. A. SchTfer, A. O. Tastan, S. Patzke, G. Blaha, C. M. Spahn,D. N. Wilson, K. H. Nierhaus, J. Biol. Chem. 2002, 277, 19095 –19105.
[78] R. A. Sayle, E. J. Milner-White, Trends Biochem. Sci. 1995, 20,374.
[79] M. Dabrowski, C. M. T. Spahn, K. H. Nierhaus, EMBO J. 1995,14, 4872 – 4882.
[80] K. H. Nierhaus, J. Wadzack, N. Burkhardt, R. JSnemann, W.Meerwinck, R. Willumeit, H. B. Stuhrmann, Proc. Natl. Acad.Sci. USA 1998, 95, 945 – 950.
[81] J. L. Hansen, T. M. Schmeing, P. B. Moore, T. A. Steitz, Proc.Natl. Acad. Sci. USA 2002, 99, 11670 – 11675.
[82] A. Bashan, I. Agmon, R. Zarivach, F. Schluenzen, J. Harms, R.Berisio, H. Bartels, F. Franceschi, T. Auerbach, H. A. Hansen,E. Kossoy, M. Kessler, A. Yonath, Mol. Cell 2003, 11, 91 – 102.
[83] J. M. Berg, J. R. Lorsch, Science 2001, 291, 203.[84] M. Welch, J. Chastang, M. Yarus, Biochemistry 1995, 34, 385 –
390.
Ribosome Structure and TranslationAngewandte
Chemie
3485Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486 www.angewandte.org � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

[85] K. M. Parnell, A. Seila, S. A. Strobel, Proc. Natl. Acad. Sci.USA 2002, 99, 11658 – 11663.
[86] M. A. Bayfield, A. E. Dahlberg, U. Schulmeister, S. Dorner, A.Barta, Proc. Natl. Acad. Sci. USA 2001, 98, 10096 – 10101.
[87] T. M. Schmeing, A. C. Seila, J. L. Hansen, B. Freeborn, J. K.Soukup, S. A. Scaringe, S. A. Strobel, P. B. Moore, T. A. Steitz,Nat. Struct. Biol. 2002, 9, 225 – 230.
[88] K. Quiggle, G. Kumar, T. W. Ott, E. K. Ryu, S. Chladek,Biochemistry 1981, 20, 3480 – 3485.
[89] V. I. Katunin, G. W. Muth, S. A. Strobel, W. Wintermeyer,M. V. Rodnina, Mol. Cell 2002, 10, 339 – 346.
[90] G. Diedrich, C. M. T. Spahn, U. Stelzl, M. A. SchTfer, T.Wooten, D. E. Bochariov, B. S. Cooperman, R. R. Traut,K. H. Nierhaus, EMBO J. 2000, 19, 5241 – 5250.
[91] K. H. Nierhaus, H. Schulze, B. S. Cooperman, Biochem. Int.1980, 1, 185 – 192.
[92] M. L. Celma, R. E. Monro, D. Vazquez, FEBS Lett. 1971, 13,247 – 251.
[93] B. Ulbrich, G. Mertens, K. H. Nierhaus, Arch. Biochem.Biophys. 1978, 190, 149 – 154.
[94] K. Bergemann, K. H. Nierhaus, J. Biol. Chem. 1983, 258,15105 – 15113.
[95] Y. Kaziro, Biochim. Biophys. Acta 1978, 505, 95 – 127.[96] R. Agrawal, P. Penczek, R. Grassucci, J. Frank, Proc. Natl.
Acad. Sci. USA 1998, 95, 6134 – 6138.[97] R. K. Agrawal, A. B. Heagle, P. Penczek, R. A. Grassucci, J.
Frank, Nat. Struct. Biol. 1999, 6, 643 – 647.[98] H. Stark, M. V. Rodnina, H. J. Wieden, M. van Heel, W.
Wintermeyer, Cell 2000, 100, 301 – 309.[99] M. G. Gomez-Lorenzo, C. M. T. Spahn, R. K. Agrawal, R. A.
Grassucci, P. Penczek, K. Chakraburtty, J. P. G. Ballesta, J. L.Lavandera, J. F. Garcia-Bustos, J. Frank, EMBO J. 2000, 19,2710 – 2718.
[100] T. P. Hausner, J. Atmadja, K. H. Nierhaus, Biochimie 1987, 69,911 – 923.
[101] Y. Endo, K. Tsurugi, J. Biol. Chem. 1987, 262, 8128 – 8130.[102] T. Uchiumi, S. Honma, T. Nomura, E. R. Dabbs, A. Hachimori,
J. Biol. Chem. 2002, 277, 3857 – 3862.[103] E. Cundliffe in The ribosome: structure, function and evolution
(Eds.: W. E. Hill, A. Dahlberg, R. A. Garrett, P. B. Moore, D.
Schlessinger, J. R. Warners), American Society for Microbiol-ogy, Washington, DC, 1990, pp. 479 – 490.
[104] G. StUffler, E. Cundliffe, M. StUffle-Meilicke, E. R. Dabbs, J.Biol. Chem. 1980, 255, 10517 – 10522.
[105] D. M. Cameron, J. Thompson, P. E. March, A. E. Dahlberg, J.Mol. Biol. 2002, 319, 27 – 35.
[106] R. K. Agrawal, J. Linde, J. Sengupta, K. H. Nierhaus, J. Frank, J.Mol. Biol. 2001, 311, 777 – 787.
[107] K. H. Nierhaus, C. M. T. Spahn, N. Burkhardt, M. Dabrowski,G. Diedrich, E. Einfeldt, D. Kamp, V. Marquez, S. Patzke,M. A. SchTfer, U. Stelzl, G. Blaha, R. Willumeit, H. B.Stuhrmann in The ribosome. structure, function, antibiotics,and cellular interactions (Eds.: R. A. Garrett, S. R. Douthwaite,A. Liljas, A. T. Matheson, P. B. Moore, H. F. Noller), AmericanSociety for Microbiology, Washington, DC, 2000, pp. 319 – 335.
[108] J. Harms, F. Schluenzen, R. Zarivach, A. Bashan, S. Gat, I.Agmon, H. Bartels, F. Franceschi, A. Yonath, Cell 2001, 107,679 – 688.
[109] H.-J. Rheinberger, K. H. Nierhaus, Biochem. Int. 1980, 1, 297 –303.
[110] H.-J. Rheinberger, H. Sternbach, K. H. Nierhaus, Proc. Natl.Acad. Sci. USA 1981, 78, 5310 – 5314.
[111] H. Saruyama, K. H. Nierhaus, Mol. Gen. Genet. 1986, 204, 221 –228.
[112] F. Triana, K. Nierhaus, K. Chakraburtty, Biochem. Mol. Biol.Int. 1994, 33, 909 – 915.
[113] A. V. Elskaya, G. V. Ovcharenko, S. S. Palchevskii, Z. M.Petrushenko, F. J. Triana-Alonso, K. H. Nierhaus, Biochemistry1997, 36, 10492 – 10497.
[114] R. K. Agrawal, P. Penczek, R. A. Grassucci, N. Burkhardt,K. H. Nierhaus, J. Frank, J. Biol. Chem. 1999, 274, 8723 – 8729.
[115] G. Blaha, U. Stelzl, C. M. T. Spahn, R. K. Agrawal, J. Frank,K. H. Nierhaus, Methods Enzymol. 2000, 317, 292 – 309.
[116] K. H. Nierhaus, Biochemistry 1990, 29, 4997 – 5008.[117] U. GeigenmSller, K. H. Nierhaus, EMBO J. 1990, 9, 4527 –
4533.[118] J. A. Horsfield, D. N. Wilson, S. A. Mannering, F. M. Adamski,
W. P. Tate, Nucleic Acids Res. 1995, 23, 1487 – 1494.[119] V. Marquez, D. N. Wilson, F. Triana-Alonso, W. Tate, K. H.
Nierhaus, unpublished results
K. H. Nierhaus and D. N. WilsonReviews
3486 � 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org Angew. Chem. Int. Ed. 2003, 42, 3464 – 3486
Related Documents











![Through the looking [Google] Glass](https://static.cupdf.com/doc/110x72/58ed70341a28abdb228b46c9/through-the-looking-google-glass.jpg)