The prion protein binds thiamine Rolando Perez-Pineiro 1, *, Trent C. Bjorndahl 1, *, Mark V. Berjanskii 2 , David Hau 1 , Li Li 3 , Alan Hu- ang 3 , Rose Lee 3 , Ebrima Gibbs 3 , Carol Ladner 1 , Ying Wei Dong 1 , Ashenafi Abera 1 , Neil R. Cash- man 3 and David S. Wishart 1,2 1 Department of Biological Sciences, University of Alberta, Edmonton, AB, Canada 2 Department of Computing Science, University of Alberta, Edmonton, AB, Canada 3 Brain Research Centre, University of British Columbia, Vancouver, BC, Canada Keywords binding; in silico docking; NMR screening; prion protein; thiamine Correspondence D. S. Wishart, Departments of Biological Sciences and Computing Science, University of Alberta, Rm. 2-21, Athabasca Hall, University of Alberta, Edmonton, AB, Canada T6G 2E8 Fax: +1 780 492 1071 Tel: +1 780 492 0383 E-mail: [email protected] *These authors contributed equally to this study (Received 20 March 2011, revised 7 August 2011, accepted 11 August 2011) doi:10.1111/j.1742-4658.2011.08304.x Although highly conserved throughout evolution, the exact biological function of the prion protein is still unclear. In an effort to identify the potential biological functions of the prion protein we conducted a small- molecule screening assay using the Syrian hamster prion protein [shPrP(90– 232)]. The screen was performed using a library of 149 water-soluble metabolites that are known to pass through the blood–brain barrier. Using a combination of 1D NMR, fluorescence quenching and surface plasmon resonance we identified thiamine (vitamin B1) as a specific prion ligand with a binding constant of 60 lM. Subsequent studies showed that this interaction is evolutionarily conserved, with similar binding constants being seen for mouse, hamster and human prions. Various protein construct lengths, both with and without the unstructured N-terminal region in the presence and absence of copper, were examined. This indicates that the N-terminus has no influence on the protein’s ability to interact with thiamine. In addition to thiamine, the more biologically abundant forms of vitamin B1 (thiamine monophosphate and thiamine diphosphate) were also found to bind the prion protein with similar affinity. Heteronuclear NMR experiments were used to determine thiamine’s interaction site, which is located between helix 1 and the preceding loop. These data, in conjunction with computer-aided docking and molecular dynamics, were used to model the thiamine-binding pharmacophore and a comparison with other thia- mine binding proteins was performed to reveal the common features of interaction. Introduction Prion proteins (PrP) are endogenous, highly conserved membrane-anchored proteins that are particularly abundant in the neuronal cells of vertebrates. The mature form of the normal cellular isoform of the prion protein PrP c is a 200-residue glycoprotein that is tethered to the cell surface via a glycosyl-phosphati- dylinositol anchor at the C-terminus [1]. A b-rich, mis- folded isoform of PrP (generically designated PrP sc ) is the major macromolecule present in preparations of infectious prions. Prions are known to cause a variety of fatal, transmissible and incurable neurodegenerative diseases in both animals and humans. These include scrapie in sheep [2], bovine spongiform encephalopathy in cattle [3], chronic wasting disease in deer and elk [4], as well as Kuru, Creutzfeldt–Jacob disease and fatal familial insomnia in humans [3,5,6]. Prions cause Abbreviations huPrP, human prion protein; K A, association constant; K D, dissociation constant; moPrP, mouse prion protein; PrP c, cellular prion protein; PrP sc, scrapie prion protein; shPrP, Syrian hasmster prion protein. 4002 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The prion protein binds thiamineRolando Perez-Pineiro1,*, Trent C. Bjorndahl1,*, Mark V. Berjanskii2, David Hau1, Li Li3, Alan Hu-ang3, Rose Lee3, Ebrima Gibbs3, Carol Ladner1, Ying Wei Dong1, Ashenafi Abera1, Neil R. Cash-man3 and David S. Wishart1,2
1 Department of Biological Sciences, University of Alberta, Edmonton, AB, Canada
2 Department of Computing Science, University of Alberta, Edmonton, AB, Canada
3 Brain Research Centre, University of British Columbia, Vancouver, BC, Canada
Keywords
binding; in silico docking; NMR screening;
prion protein; thiamine
Correspondence
D. S. Wishart, Departments of Biological
Sciences and Computing Science, University
of Alberta, Rm. 2-21, Athabasca Hall,
University of Alberta, Edmonton, AB,
Canada T6G 2E8
Fax: +1 780 492 1071
Tel: +1 780 492 0383
E-mail: [email protected]
*These authors contributed equally to this
study
(Received 20 March 2011, revised 7 August
2011, accepted 11 August 2011)
doi:10.1111/j.1742-4658.2011.08304.x
Although highly conserved throughout evolution, the exact biological
function of the prion protein is still unclear. In an effort to identify the
potential biological functions of the prion protein we conducted a small-
molecule screening assay using the Syrian hamster prion protein [shPrP(90–
232)]. The screen was performed using a library of 149 water-soluble
metabolites that are known to pass through the blood–brain barrier. Using
a combination of 1D NMR, fluorescence quenching and surface plasmon
resonance we identified thiamine (vitamin B1) as a specific prion ligand
with a binding constant of � 60 lM. Subsequent studies showed that this
interaction is evolutionarily conserved, with similar binding constants being
seen for mouse, hamster and human prions. Various protein construct
lengths, both with and without the unstructured N-terminal region in
the presence and absence of copper, were examined. This indicates that the
N-terminus has no influence on the protein’s ability to interact with
thiamine. In addition to thiamine, the more biologically abundant forms of
vitamin B1 (thiamine monophosphate and thiamine diphosphate) were also
found to bind the prion protein with similar affinity. Heteronuclear NMR
experiments were used to determine thiamine’s interaction site, which is
located between helix 1 and the preceding loop. These data, in conjunction
with computer-aided docking and molecular dynamics, were used to model
the thiamine-binding pharmacophore and a comparison with other thia-
mine binding proteins was performed to reveal the common features of
interaction.
Introduction
Prion proteins (PrP) are endogenous, highly conserved
membrane-anchored proteins that are particularly
abundant in the neuronal cells of vertebrates. The
mature form of the normal cellular isoform of the
prion protein PrPc is a � 200-residue glycoprotein that
is tethered to the cell surface via a glycosyl-phosphati-
dylinositol anchor at the C-terminus [1]. A b-rich, mis-
folded isoform of PrP (generically designated PrPsc) is
the major macromolecule present in preparations of
infectious prions. Prions are known to cause a variety
of fatal, transmissible and incurable neurodegenerative
diseases in both animals and humans. These include
scrapie in sheep [2], bovine spongiform encephalopathy
in cattle [3], chronic wasting disease in deer and elk
[4], as well as Kuru, Creutzfeldt–Jacob disease and
fatal familial insomnia in humans [3,5,6]. Prions cause
Abbreviations
huPrP, human prion protein; KA, association constant; KD, dissociation constant; moPrP, mouse prion protein; PrPc, cellular prion protein;
PrPsc, scrapie prion protein; shPrP, Syrian hasmster prion protein.
4002 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

disease by converting from a soluble, helix-rich form
(PrPC) to an infectious b-rich form (PrPsc) that is both
insoluble and highly pathogenic [7].
After more than 30 years of study very little is
known about the physiological role of prion protein.
Most of the studies published to date have focused on
the identification of PrP-interacting partners such as
Cu2+, Ni2+, glycosaminoglycans, DNA, RNA and a
number of signaling proteins [8]. Based on these obser-
vations, the proposed physiological roles of PrPc range
from copper internalization and homeostasis to a vari-
ety of anti-apoptotic activities. Other potential func-
tions include protection against oxidative stress, cell
adhesion, cell signaling and the modulation of synaptic
structure and function [9,10]. More recent findings
suggest that PrPc may play a role in maintaining the
long-term integrity of peripheral myelin sheaths [11] or
even function as an antibacterial protein [12].
Aside from the identification of Cu2+ (and other
divalent metals such as Ni2+, Zn2+, Fe2+ and possibly
Mn2+) and hemin [13] as high-affinity PrP ligands,
there has been no published evidence that PrPc binds
any other endogenous small molecules. That is not to
say that PrP isoforms cannot, or do not, bind small
molecules. Indeed there are numerous studies that have
identified a variety of exogenous or xenobiotic ligands
that bind to either PrPc or PrPsc with relatively high
affinity. These include tetracyclines [14], quinacrines
[15], curcumin [16], simvastatin [17], Congo Red [18]
and others. However, these small molecules are not
endogenous molecules and they were identified through
chemical screens aimed at finding potential prion
therapies rather than potential PrP functions.
In an effort to identify potentially physiologically rel-
evant binding partners of the prion protein, we investi-
gated the binding of small molecules that would be
easily accessible to prion proteins. Taking into account
the extracellular location and the enrichment of PrPc in
the central nervous system, we chose to look at a collec-
tion of water-soluble metabolites that could easily pass
through the blood–brain barrier and which are found in
high abundance in central nervous system tissues or
biofluids. More specifically, we decided to screen recom-
binant prion proteins against a subset of 149 water-sol-
uble metabolites that were previously identified as being
enriched in human cerebrospinal fluid [19]. Using a
combination of 1D NMR, fluorescence quenching and
surface plasmon resonance, we found that thiamine
(vitamin B1) was the only ligand that exhibited clear
and specific binding to PrPc. The binding constant was
determined to be � 60 lm. Subsequent assessment of
thiamine binding showed that this interaction is evolu-
tionarily conserved, with similar binding constants
being seen for hamster, mouse and human prion pro-
teins. We also assessed the binding of other, more phys-
iologically abundant thiamine derivatives (thiamine
monophosphate and thiamine diphosphate) and deter-
mined the exact binding site for thiamine using a com-
bination of 2D heteronuclear NMR experiments and
computer-aided docking and molecular dynamics.
Results
1D NMR screening
One-dimensional NOESY NMR screening was per-
formed on a total of 149 water-soluble metabolites
(Table S1) in the presence and absence of recombinant
Syrian hamster prion protein [shPrP(90–232)]. Spectra
were compared for chemical shift, linewidth and ⁄orlineshape perturbations of the metabolite proton sig-
nals. The criteria for selecting potential binders
included the presence of new proton chemical shifts or
perturbations > 0.02 ppm, > 10% reduction in peak
intensity and ⁄or > 0.2 Hz broadening of the metabo-
lite signals. The parameters for the compound only
and compound + protein spectral collection were opti-
mized for detecting small molecules. Through this
NMR analysis, we identified two potential binders that
fit our criteria: thiamine and cytidine (Fig. 1A).
SPR and fluorescence studies of the
thiamine-prion complex
The binding of the two candidates identified from our
1D NMR screen to the prion protein, (Fig. 1A) were
also characterized using SPR as a second, independent
method. At ligand concentrations of 10 mm, binding is
clearly observed for thiamine (Fig. 1B). By contrast, the
affinity for cytidine to PrP as measured by SPR was
found to be very weak and we decided not pursue
further studies with this ligand. Caffeine, a negative con-
trol, exhibited nonspecific binding as manifested by the
negative binding response after dissociation. As shown
in Fig. 1C, the biosensor response arising from thiamine
binding is concentration dependent. Values of the asso-
ciation and dissociation constant (KD = 116 · 10)6m)
for this ligand were calculated using regression analysis
of the binding data.
Steady-state fluorescence quenching is a valuable
technique to study ligand–protein interactions if the
ligand binds near tyrosine or tryptophan residues [20].
Assessment of thiamine binding to PrP using
fluorescence quenching indicated a decrease in the flu-
orescence signal of shPrP at both 295 and 280 nm
after addition of thiamine (Fig. 2A) and the calculated
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4003

Stern–Volmer plot was linear for the selected concen-
tration range. Using the fluorescence data (see Materi-
als and methods) the estimated values for the
disassociation constants (KD = 65.36 · 10)6m) were
determined (Table 1, row 1). Interestingly, a similar
quenching profile was found for a longer version of
the Syrian hamster prion protein, shPrP(29–232) with
a histidine affinity tag (Table 1, row 2), the mouse
prion protein, moPrP(90–231) (Table 1, row 3) and
the full-length human prion protein, huPrP(23–230)
with histidine affinity tag (Table 1, row 4). These
results indicate that the binding of thiamine is con-
served across a wide range of mammalian species. Fur-
thermore, they also show that the binding is not
Fig. 1. (A) Chemical structures of thiamine and cytidine. (B) Biosen-
sor analysis of binding of thiamine, caffeine (negative control) and
cytidine to shPrP (90-232). (C) Biosensor response due to the
binding of thiamine (0 lM – light red, 50 lM – light magenta,
100 lM – yellow, 200 lM – cyan, 400 lM – blue, 800 lM – dark red,
1.6 M – dark magenta, 3.2 M – green) to the protein is concentra-
tion-dependent.
Fig. 2. (A) Fluorescence emission spectra of shPrP(90–232), 20 lM
in the presence of thiamine at different concentrations, (1) 0 M, (2)
10 lM (3) 20 lM (4) 40 lM (5) 80 lM (6) 160 lM (7) 250 lM (8)
320 lM (9) 510 lM (10) 700 lM; k = 295 nm. (B) The Stern–Volmer
plot of fluorescence quenching of shPrP(90–232) by thiamine.
Values for KD (65.36 · 10)6M), were obtained according to
equation (1).
PrP binds thiamine R. Perez-Pineiro et al.
4004 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS
affected by the presence of the His-tag or the unstruc-
tured, copper-binding N-terminus. Similar fluorescence
quenching of shPrP(29–232) was observed for the
mono and diphosphate analogs of thiamine and for
the moPrP(90–232) in the presence of a 3 m excess of
CuCl2. The calculated KA and KD values for these
constructs were in the same range of nonphosphorylat-
ed thiamine with the C-terminal core of the shPrP(90–
232) protein (Table 1, rows 5–9). The Stern–Volmer
plots are shown in Figs S1–S5.
Thiamine binding
Additional NMR studies were conducted to further
validate the PrP–thiamine binding and to identify the
site of interaction. A saturation transfer difference
TOCSY spectrum (STD-TOCSY) was collected to
identify the ligand protons that interacted with the
protein [21]. As observed from the reference spectrum
(Fig. S8B), the majority of thiamine protons, apart
from the methyl groups and the methylene signal at
5.3 p.p.m., were completely suppressed. Upon addition
of the shPrP(90–232), recovery of the remaining signals
was observed (Fig. S8C), whereas the methyl groups
and methylene proton displayed significant signal
Table 1. Binding affinity (KD) and correlation coefficients (C ) of
thiamine for different prion protein (PrP) constructs according to
equation (1).
Entry Ligand PrP construct KD (M) C
1 Thiamine shPrP(90–232) 65.36 · 10)6 0.9892
2 Thiamine shPrP(29–232) 66.66 · 10)6 0.9811
3 Thiamine moPrP(90–231)a 58.82 · 10)6 0.9679
4 Thiamine huPrP(23–230) 62.11 · 10)6 0.9693
5 Thiamine
(PO4)
shPrP(29–232) 72.72 · 10)6 0.99
6 Thiamine
(PO4)bshPrP(29–232) 69.52 · 10)6 0.9810
7 Thiamine +
CuCl2c
moPrP(90–232) 56.82 · 10)6 0.9991
8 Thiamine
(PO4) + CuCl2c
moPrP(90–232) 65.79 · 10)6 0.99
9 Thiamine(PO4)2 +
CuCl2c
moPrP(90–232) 59.88 · 10)6 0.99
10 Thiamine ShPrP(90–232)
pH 6.0
67.71 · 10)6 0.99
11 Thiamine ShPrP(90–232)
pH 8.0
68.90 · 10)6 0.99
a No His-tag. b Value calculated based on tyrosine quenching.c Three molar excess (3 ·) of CuCl2 in relation to the protein and
ligands were used.
10 9 8 7
H1 p.p.m.
130129128127126125124123122121120119118117116115114113112111110109108107
N15
p.p
.m.
10 9 8 7131
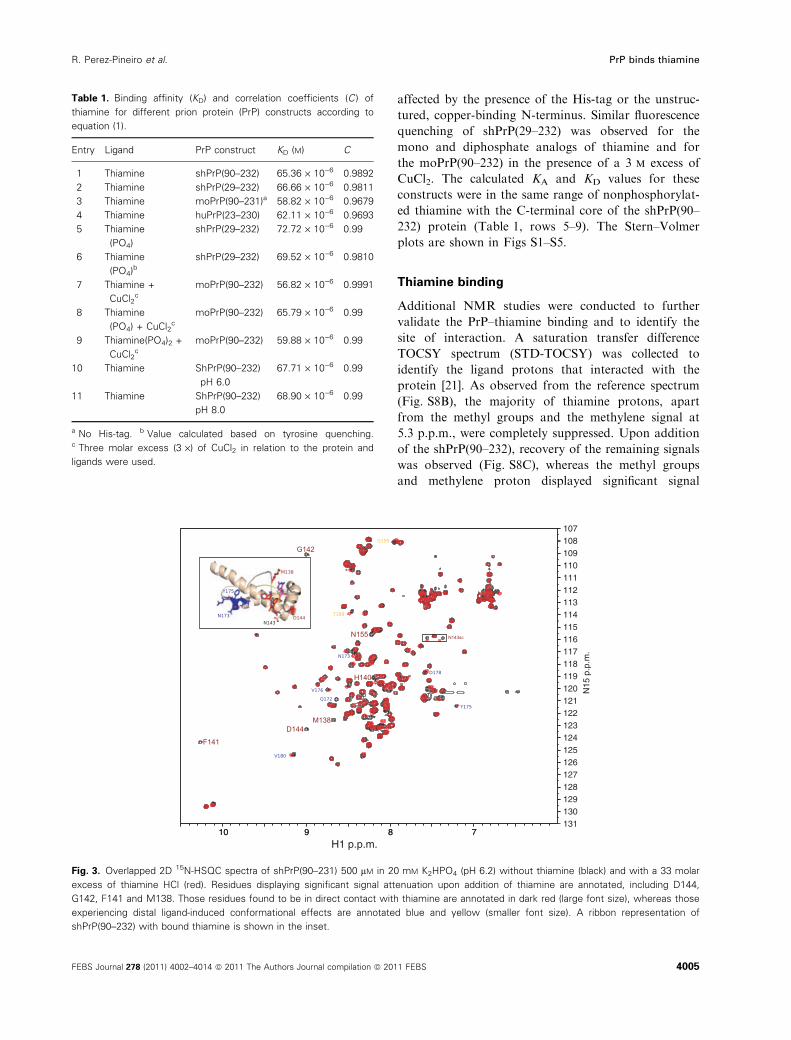
Fig. 3. Overlapped 2D 15N-HSQC spectra of shPrP(90–231) 500 lM in 20 mM K2HPO4 (pH 6.2) without thiamine (black) and with a 33 molar
excess of thiamine HCl (red). Residues displaying significant signal attenuation upon addition of thiamine are annotated, including D144,
G142, F141 and M138. Those residues found to be in direct contact with thiamine are annotated in dark red (large font size), whereas those
experiencing distal ligand-induced conformational effects are annotated blue and yellow (smaller font size). A ribbon representation of
shPrP(90–232) with bound thiamine is shown in the inset.
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4005

enhancement, indicating that all of thiamine’s protons
are involved in interaction with the prion protein.
The thiamine binding site was investigated using
heteronuclear NMR spectroscopy on a 15N-labeled
sample of the shPrP(90–232 with 6 · -His tag) protein
construct. Figure 3 shows the signal attenuation and
chemical shift changes in the 15N-HSQC spectrum of
the shPrP after addition of thiamine (33 : 1,
ligand ⁄protein molar ratio). The majority of attenu-
ated signals (M138, F141, G142 and D144) and those
with significant chemical shift perturbations (H140,
N143 side chain) cluster in an unstructured loop
adjacent to helix 1, which contains two tyrosines and a
tryptophan residue. These aromatic residues are within
the required 10 A of the interacting ligand to exhibit
fluorescence quenching at both 280 nm (Tyr, Trp exci-
tation) and 295 nm (Trp excitation). The perturbation
of the N143 side-chain amide is also noteworthy.
Although it is not in direct contact with thiamine, its
binding results in slight changes of the hydrogen bond
length between it and the E146 side-chain carboxylic
acid group. In the majority of the NMR structures
(PDB: 1B10), these side chains are involved with the
N-terminal capping of helix 1. In addition, a second
region of the protein exhibits conformation change,
which is likely a consequence of small changes in
hydrogen bond lengths or other minor conforma-
tional ⁄dynamic changes. However, no visible NOEs
could be observed between thiamine and this region,
which consists of the ‘amylome’ residues F169-
NNQNNY-175. Long-range effects between this region
and the distal residues in helix 3 have been noted in
previous studies [22]. Whether these distal effects on
the amylome region upon thiamine binding have any
significance or are an inconsequential artifact has yet
to be determined.
Although 15N-HSQC titration spectra can be useful
in identifying residues directly perturbed by ligand
binding, they are also sensitive to small perturbations in
hydrogen bond length, solvent structure and electric
field effects not directly related to ligand binding, result-
ing in a number of false-positive signals. Evidence of
this can be seen in the HSQC data presented in Fig. 3.
Also shown are a number of resonances that exhibited
some signal attenuation, likely due to ligand-induced
changes in the protein’s dynamics (i.e. intermediate
conformational exchange). In light of these hard-to-
interpret signal perturbations, we felt that additional
data were necessary to corroboate the initial findings.
Consequently, a NOESY experiment (Varian VNMRJ
v2.1b: tnnoesy) was performed on a sample containing
a 50 : 1 molar ratio of thiamine to the shPrP(90–232).
This experiment provided five unique NOE signals that
could be clearly assigned to protons within the putative
binding pocket (Fig. 4). Confirmation of the proton
chemical shift assignments was made from 15N TOC-
SY-HSQC and NOESY-HSQC data. Strip plots from
the residues providing NOE data are also provided in
the supporting information (Fig. S7). The attenuated
amide signal for G142 in the 15N-HSQC spectrum
tracks from 9.05 to 8.90 p.p.m. upon addition of
thiamine. This perturbation is also observed in the
TOCSY-HSQC data (Fig. S7), which dislays both free
and bound states in slow conformational exchange. The
Fig. 4. Regions of the tnnoesy spectrum collected on a 0.5 mM
shPrP(90–232) sample (20 mM KH2PO4, pH 7.0) with a 50-fold
molar excess of thiamine showing (A) the thiazolium proton
(9.38 p.p.m.) and the pyrimidine proton (8.00 p.p.m.) and (B) the
downfield shifted alkyl proton nearest to thiamine’s hydroxyl group.
The NOEs used to select the docking candiate shown in Fig. 5 are
annotated. Intrathiamine NOEs present a negative intensity (red),
while intermolecular shPrP to thiamine NOEs are positive (black).
Chemical shift values are shown and correspond to those identified
assigned in 15N-edited TOCSY-HSQC and NOESY-HSQC experi-
ments collected at pH 7.0 (Fig. S7A, B).
PrP binds thiamine R. Perez-Pineiro et al.
4006 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

tnnoesy spectum exhibits a very strong NOE between
thiamine’s aliphatic proton shift at 3.83 p.p.m. and
G142 amide proton (at 8.90 p.p.m.) in the bound state
(Fig. 4B). Other strong NOE data were observed
between thiamine’s pyrimidine and isoxazole ring pro-
tons and the protons for residues M138, M139, H140
and Y150 aromatic rings (Fig. 4A). From these NMR
data, the orientation of thiamine in complex with PrP
was modelled in-silico (Fig. 5). Analysis of the docking
result revealed that it was in good agreement with the
observed NMR and fluorescence quenching data. Spe-
cifically in this model, all NOEs were satisfied, the
ligand made good contact with residues displaying
NMR perturbations and the placement of the ligand is
within 10 A of W145, Y149 and Y150.
Discussion
By analyzing the fluorescence data, some additional
clues about the nature of PrP–thiamine interaction
could be obtained. The linearity of the Stern–Volmer
plot proves there is only one fluorogen that interacts
with the quencher (thiamine). However, these data alone
cannot distinguish between a dynamic or a static
quenching mechanism. To clarify this issue, we observed
only a slight decrease in the slope of the Stern–Volmer
plot when the experiments were conducted at higher
temperatures (40 �C, data not shown). This result is
characteristic of static quenching. By using equation (1):
Fo=F ¼ 1þ Kqs0½Q� andKA ¼ 1=KD ¼ Kqs0 ð1Þ
It is possible to calculate the apparent quenching con-
stants Kq of the two metabolites assuming the lifetime s0
of the biomolecules to be � 10 ns. It is known that the
maximal diffusion collision quenching constant of
quenchers is 2.0 · 1010 mÆs)1 [23]. In this case, the
apparent quenching constant calculated for thiamine
(1.53 · 1012 mÆs)1) is larger than the diffusion-con-
trolled quenching constant suggesting that the quench-
ing effect of thiamine on the prion protein is not due to
dynamic collisions. This result supports the fact that the
origin of the observed quenching effect is due to the for-
mation of a ground-state complex between the protein
and thiamine. We have also excluded a Forster energy
transfer mechanism as the other probable cause of static
quenching because in this case the fluorescence emission
spectrum of the prion protein does not overlap with the
UV absorption spectrum of the thiamine.
The results obtained from our SPR studies con-
firmed the thiamine affinity measurements made by flu-
orescence (� 65 lm). However, the KD value obtained
from SPR (� 116 lm) is about half that obtained by
our fluorescence experiments. The lower binding con-
stant as measured from SPR was not unexpected.
Indeed, given the fact that the prion protein was cova-
lently coupled (via lysine and arginine residues) to the
SPR chip, one would expect that this chemical cross-
linking would lead to a portion of the PrP molecules
having their binding sites shielded or inaccessible. It is
also notable that three arginine residues surround the
putative binding site (R136, R148 and R151). Further-
more, reduced binding constants are expected given
that some denaturation of the native structure is often
induced by the covalent modification [24].
As noted earlier, the prion protein does bind to other
endogenous ligands, including copper, zinc, manganese
and nickel. The binding sites for these metals are located
between residues 29 and 98, in the unstructured, N-ter-
minal octarepeat region [25,26], well away from where
thiamine binds. In most cases, the metal binding con-
stants are quite strong. The KD for copper is an aston-
ishingly tight 8 fm, the KD for nickel is � 20 nm, the KD
for zinc is � 100 nm, and the KD for manganese is rela-
tively weak at 200 lm [25]. The other endogenous ligand
that appears to bind to PrP is hemin, a naturally occur-
ring iron-binding porphyrin. Hemin appears to bind
exclusively in the unstructured N-terminus from resi-
dues 34 to 94, which corresponds to the octarepeat
region [13]. However, the binding affinity of hemin has
not yet been determined. Some non-natural porphyrin
analogs of have also been shown to bind to PrP with
low micromolar affinity [27] so it is likely that the KD
value for hemin would be in the same range. Among
nonendogenous or xenobiotic compounds that have
been shown to bind the cellular form of PrP, only quina-
crine has had both its binding site and binding affinity
Fig. 5. Thiamine docked onto a ribbon representation of the
shPrP(90–232). Residues displaying 15N-HSQC signal attenuation
are colored red. The NOEs obtained from the transferred NOE
experiment (supporting information) used to generate the model
are indicated with dashed yellow lines. The ribbon is colored yellow
at positions W145, Y149 and Y150, which are the residues respon-
sible for the fluorescence quenching results.
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4007

determined. In particular, quinacrine binds to the C-ter-
minal region of helix 3 with a rather low KD of 4.3 mm
[15]. Overall, these data suggest that the affinity of the
prion protein for thiamine is within the range of other
known PrP ligands. Furthermore, the thiamine binding
site appears to be unique and, so far as we know, does
not overlap with the binding site of other known
(endogenous or exogenous) prion ligands.
To assess whether the binding of thiamine to PrP
might share features with other thiamine binding pro-
teins, we compared our PrP–thiamine structure with
other known thiamine binding proteins including:
transketolase, thiamine pyrophosphokinase (from
mouse and Bacillus subtilus), thiamine phosphate syn-
thase and thiamine binding protein (Fig. 6). For all the
thiamine binding proteins, the amino substituted
pyrimidine ring occupies a hydrophobic pocket. Stabil-
ization and orientation of the ring is further provided
through hydrogen bond acceptor oxygen atoms. In the
case of pyrophosphokinase, salt bridges are also found
to the positively charged imidazolium nitrogen.
Finally, electrostatic and hydrogen bonds from the
backbone amides and side chain amide (Q, N) or
hydroxyl groups (S, T) and charged residues (D, E, R,
K and H) provide contacts for the thiamine hydroxyl
group or various phosphorylated forms. For the prion
protein, similar contacts are observed. Specifically, the
pyrimidine ring of thiamine sits in a hydrophobic
pocket created by Y150, Y141 and M138, and is also
stabilized electrostatically by the backbone carbonyl of
M139 and the Y150 hydroxyl group. In addition, the
positively charged imidazolium nitrogen forms a salt
bridge with the carboxylic acid side chain from D147,
and finally, the thiamine hydroxyl group is located
within a hydrogen-bond distance range (2.5 A) from
the backbone amide proton of G142. There are a num-
ber of additional residues (N143, D144, D147, R148)
in this region that can offer either electrostatic or
hydrogen bonding interactions to either the thiamine
hydroxyl group or its phosphate groups. Interestingly,
Fig. 6. Pharmacophore comparison between shPrP(90–232) and other thiamine binding proteins. The modeled structure of the shPrP(90–
232) protein with docked thiamine is shown in (A). (B) Thiamine phosphate synthase (PDB: 2TPS), (C) mouse thiamine pyrophosphokinase
(PDB: 1IG3), (D) Bacillus subtilis thiamine pyrophosphokinase (PDB: 3LM8), (E) thiamine binding protein (PDB: 2QRY) and (F) transketolase
(PDB: 3M34).
PrP binds thiamine R. Perez-Pineiro et al.
4008 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

it was noted that the H140 side chain played no direct
part in the interaction of thiamine. In order to validate
this observation, we performed additional binding
studies at pH 6 and 8, thereby titrating H140 through
its full range of pKa values [28], as well as most known
physiological pH ranges. The fluorescence quenching
data yielded no difference in the calculated KD con-
stants for these pH values, indicating that pH does not
influence thiamine binding (Table 1, Fig. S6). Other
prion protein constructs from nonmammalian species
(turtle and frog) show variability in the amino acid at
this position (N and Q respectively). These findings
suggest that the amino acid in this position plays no
significant role in the protein affinity for thiamine.
Potential biological consequences
Thiamine (vitamin B1) is an essential, water-soluble,
B vitamin that plays a critical role in carbohydrate
metabolism [29]. It is endogenously synthesized by bac-
teria and plants, but animals cannot synthesize it, so
thiamine must be obtained from the diet. Generally,
the unphosphorylated form is transported in the body,
whereas the phosphorylated forms of thiamine
(thiamine monophosphate, thiamine diphosphate and
thiamine triphosphate) are the active forms of the vita-
min. The human body keeps stores of 25–30 mg of
thiamine, with the greatest concentrations being in
metabolically active organs, such as skeletal muscle,
the brain, the heart, the kidneys and the liver [30].
Thiamine is known to bind to serum albumin [31], to a
hormonally regulated protein called thiamine binding
protein [32] and to a thiamine ⁄ folate transporter [33].
Thiamine binding protein and serum albumin have an
affinity for thiamine of � 1 lm [31,32]. Thiamine and
its phosphorylated derivatives have also been detected
in blood, cerebrospinal fluid, milk and several other
biofluids. The typical concentration of all forms of thi-
amine (free and phosphorylated) in human blood is
� 200–300 nm [30]. Thiamine is absorbed by active
transport and by passive diffusion.
Given the relatively low concentrations of thiamine
in the body, one might ask how a protein with a mod-
est (� 60 lm) affinity to thiamine could potentially
play a biologically meaningful role. One possibility is
that the prion protein functions to retain or concen-
trate thiamine in tissues more through avidity rather
than affinity. Many copies of a weak binding protein
on a cellular surface can create a ‘Velcro’ effect for
ligand binding. It has been estimated that prion pro-
teins are expressed at a level of between 2000 and 4000
copies per cell in peripheral tissues and organs and as
much as 50 000 copies per cell in cerebral tissue [34].
Clearly, if the role of PrP is to concentrate thiamine in
tissues, it would need to have a relatively weak binding
constant so that the thiamine could be released for
absorption and subsequent utilization by cells. It is
also important to note that the thiamine binding
constant reported here was determined for a soluble,
unglycosylated form of PrP rather than the native,
membrane-bound form. It may be that native, mem-
brane-bound PrP could exhibit higher affinities for thi-
amine because of the presence of the lipid bilayer or
other synergistic protein–protein interactions. It is
intriguing that prion proteins are highly expressed in
the brain, spinal cord, heart, kidney, lung, white blood
cells and lymphoid tissues [35]. These correlate well
with the tissues typically needing the highest levels of
thiamine in the body and the tissues with the highest
levels of carbohydrate metabolism.
In conclusion, we have identified, from an initial
screening of 149 water-soluble metabolites commonly
found in cerebrospinal fluid that the water-soluble vita-
min B1, thiamine, interacts with multiple constructs of
the prion protein. Three independent methods were
used to confirm the interaction. Two of them (SPR and
fluorescence quenching) led to binding constants in the
weak lm range, and NMR studies pinpointed the site
of interaction. Further docking studies were used to
ascertain the hydrogen bonds, electrostatic and lypo-
philic interactions that comprise the pharmacophore.
The residues involved with these interactions are
conserved across multiple mammalian species (Fig. 7).
Additional experiments with mouse, human and vari-
ous hamster prion constructs showed that this binding
was conserved across these mammalian species.
Materials and methods
Protein expression and purification
The expression and purification of recombinant shPrP(29–
232), shPrP(90–231) and shPrP(120–232), moPrP(90–231)
and huPrP(23–230) all followed a similar protocol.
Fig. 7. Structural sequence alignment for various prion protein con-
structs over the region containing the residues displaying 15N-HSQC
signal attenuation upon the addition of thiamine. The structure
alignment was performed using PYMOL (Warren Delano, ª 2004).
Residues showing phamacophore interactions with thiamine are
bolded italics. Hyrophobic residues are colored yellow, acid residues
are colored red, basic residues are colored blue.
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4009

Specifically, synthetic genes corresponding each construct
including a 22-residue N-terminal fusion tag containing
6 · His and a thrombin cleavage site (MGSSHHHHHH
SSGLVPRGSHML) were synthesized by DNA 2.0
(Menlo Park, CA, USA). The genes were cloned into a
pET15b expression vector between XhoI and EcoRI
restriction sites and heat shock transformed into Escheri-
chia coli strain BL21 (DE3). For expression, the trans-
formed cells were grown in 100 mL Luria–Bertani broth
plus 100 lgÆmL)1 ampicillin overnight to generate a star-
ter culture. Between 1% and 2% of this starter culture
was then used to inoculate 1 L of Luria–Bertani media
(giving a starting D600 of 0.1). The cells were allowed to
reach an D600 between 0.6 and 1.0 before induction with
1 mm isopropyl thiogalactoside. Twelve to eighteen hours
later, the cells were harvested by centrifugation at 1600 g
for 25 min at 4 �C. In addition, 15N-labeled shPrP(90–
232) was also expressed and purified from M9 media
(1.0 gÆL)1 15NH4Cl) for collection of heteronuclear NMR
data. The inclusion of the 6· His tag afforded a stan-
dardized nickel affinity purification strategy for all pro-
tein constructs. The details of the purification protocol
are described elsewhere [36].
NMR experiments
NMR spectra were acquired at 25 �C on a 500 MHz
Varian Unity INOVA spectrometer fitted with a 5 mm
HCN z-gradient pulsed-field gradient cryogenic probe
except for the STD-TOCSY, which was collected at 25 �Con a 800 MHz Varian Unity INOVA spectrometer fitted
with a 5 mm HCN xyz-gradient pulsed-field gradient cryo-
genic probe. All experiments were collected using Varian
BioPack pulse sequences (VNMRJ v2.1B). Spectra were
processed using nmrpipe [37] and analyzed with nmrpipe
and nmrviewj [38] unless stated otherwise.
Small molecule metabolites used in the ligand screening
were purchased from Sigma-Aldrich (St. Louis, MI, USA),
Fisher Scientific (Waltham, MA, USA), Acros Organics
(Geel, Belgium) and Alfa Aesar (Ward Hill, MA, USA)
and used without further purification. The final (cerebrospi-
nal fluid-compatible) metabolite library consisted of 149
different water-soluble chemicals. The complete list is
available in the Table S1. Seventeen screening sets contain-
ing between six and nine metabolites were manually
selected with the aid of Chemaxon’s jklustor v 5.0
(ChemAxon Kft, Budapest, Hungary) using the Ward algo-
rithm. Selection of each chemical set was made on the basis
of minimizing 1H NMR spectral overlap. Two sets of
1D NOESY spectra were collected on each set of metabo-
lites (with and without PrP protein). To collect the first
NOESY NMR spectrum, each metabolite set was dissolved
in 20 mm potassium phosphate buffer at pH 6.5 giving a
final concentration of 100 lm. Ten percent (v ⁄ v) D2O was
added to each sample to maintain a spectral lock, 1 mm of
2,2-dimethyl-2-silapentane-5-sulfonate was added for chemi-
cal shift referencing [39]. For each 1D NOESY spectrum,
48 000 points were averaged from 256 transients over a
sweep width of 6000 Hz. A recycle delay of 0.01 s and an
acquisition time of 4 s were used. The mixing time for the
screening experiments was 100 ms. Immediately following
collection of the reference compound spectra, the NMR
sample was used to reconstitute prealiquoted, lyophilized
shPrP(90–232) and a second spectrum was collected with
the same parameters. The molar ratio of protein to each
compound was 1 : 1. Matching spectra were superposed
and analyzed with the Chenomx NMR suite v6.0 to assess
chemical shift and linewidth perturbations of the metabolite
signals.
Saturation transfer difference TOCSY experiments were
collected on a 12.5 mm sample of thiamine (20 mm
K2HPO4, pH 7.5, 10% D2O) before and after the addition
of 500 lm shPrP(90–232). The spectra were collected using
4096 transients with a sweepwidth of 12000 Hz, a mixing
time of 10 ms, a recycle delay of 1 s and an acquisition
time of 2 s. On and off irradiation frequencies corre-
sponded to )0.73 and 24.5 p.p.m. respectively.
To collect the 2D 15N-HSQC titration spectra, a reference15N-HSQC spectrum of the shPrP(90–232), alone, was first
collected (300 lm, 350 lL, 20 mm KH2PO4, pH 7 and 6.2).
These 15N-HSQC reference spectrum was collected with
2048 complex points in the 1H dimension and 256 complex
points in the 15N dimension using a recycle delay of 1.5 s
(nt = 120, sw = 6000 Hz, sw1 = 1800 Hz). Thiamine HCl
was successively added to concentrations of 1, 2, 5 and
10 mm and the 15N-HSQC spectra recollected using identi-
cal acquisition parameters. The amide chemical shift data
for the prion-thiamine complex has been deposited into the
BioMagResBank (BMR17834).
A 2D tnnoesy experiment (Varian VNMRJ v2.1b) was
collected on a 500 lm shPrP(90–232) sample with 25 mm
thiamine in 20 mm KH2PO4, (pH 7.0, 350 lL, 10% D2O)
at 25 �C. The mixing time was 50 ms and a 1.5 s recycle
delay was used. Sixty-four transients were collected with
sweep widths of 6000 Hz in both the direct and indirect
detected dimensions (np = 1024, ni = 128). Three-dimen-
sional 15N edited TOCSY-HSQC and NOESY-HSQC [40]
experiments were acquired with a 500 lm shPrP(90–232)
sample prepared with 10 mm thiamine in 20 mm KH2PO4
(pH 7.0, 10% D2O, 350 lL). Sixteen transients were col-
lected for each experiment with sweepwidths of 6000 Hz
in the direct and first indirectly detected dimensions
(np = 1024, ni = 64). A sweep with of 1800 Hz was
used for the second indirectly detected dimension
(ni2 = 32). Mixing times of 50 and 100 ms were used for
the TOCSY and NOESY experiments respectively. A
recycle delay of 1.5 s was used and the experiments were
collected at 25 �C. All samples were transferred to
Shigemi (Shigemi Inc. Allison Park, PA, USA) microcell
NMR tubes (350 lL) prior to spectral acquisition.
PrP binds thiamine R. Perez-Pineiro et al.
4010 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

Steady-state fluorescence quenching
measurements
Fluorescence emission spectra were recorded on a PTI
MODEL-MP1 spectrofluorometer using a 1 cm fluores-
cence cell for all measurements. Two different excitation
wavelengths (295 and 280 nm) were used and the scan
range was 310–450 nm. Prior to collecting the fluorescence
spectra, the prion protein (20 lm) was dissolved in 100 lLof 20 mm potassium phosphate buffer at pH 6.0, 7.0 or 8.0
and incubated with increasing concentrations of the metab-
olites of interest (10–700 lm) for 30 min with shaking (800
rpm). To analyze the effect of copper on the binding of thi-
amine and thiamine-phosphate analogs to PrP, CuCl2 was
added to the mixture in a threefold excess previous to the
addition of the ligand. Data from these fluorescence experi-
ments were used to determine the apparent binding con-
stant according to equation (2):
Fo=F ¼ 1þ KA½Q� ð2Þ
Where KS = KA, is the formation constant of the donor–
acceptor (quencher–fluorogen) complex. The concentration
of the quencher [Q] after titration is taken to be its ratio to
protein concentration [Pt]: [Q] ⁄ [Pt]. From the slope of the
linear plot of Fo ⁄F versus [Q] ⁄ [Pt] the binding constant and
dissociation constant (1 ⁄KA) were estimated. The results are
expressed as mean values ± SD (n = 5–7).
SPR measurements
The SPR data was collected on a Biacore� system 3000
(BIAcore, Uppsala, Sweden) equipped with a CM5 sensor
chip. HBS-EP buffer (10 mm Hepes pH 7.5, 150 mm NaCl,
3.4 mm EDTA, 0.005% surfactant P20) was used for immo-
bilization of the protein. CM-dextran on A CM5 sensor chip
was activated by mixing equal volumes of 50 mm N-hydroxy
succinimide and 200 mm 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide followed by injection of the mixture over the
sensor chip surface for 7 min at a flow rate of 5 lLÆmin)1.
The shPrP(90–232) to be immobilized was injected over the
surface for 7 min. The unreacted sites on the sensor chip sur-
face were blocked by injection of 1 m ethanolamine, pH 8.5
for 7 min. Thiamine was diluted in HBS-EP-0.05% and
simultaneously injected over the PrP flow cell and the refer-
ence for 3 min at a flow rate of 30 lLÆmin)1. The dissocia-
tion phase was monitored for 2.5 min. The flow cell was
washed with glycine ⁄HCl (pH 1.7) at 60 lLÆmin)1 for 30 s
between each sample injection. Included in the assay were
positive controls (quinacrine HCl and congo red), com-
pounds known to bind shPrP(90–232), and a negative con-
trol, caffeine. The resultant sensorgrams, a plot of binding
response over time, were all double-referenced by first sub-
tracting the binding to the reference surface from the binding
to the active surface, and further subtracting out the binding
response of the sample diluents from all sensorgrams. The
dissociation constant (KD) for thiamine HCl was calculated
from data at doses ranging from 50 lm to 3.2 mm. Dilution
series sensorgrams were then evaluated using the steady-state
affinity analysis protocol of the biaevaluation 4.1.1 soft-
ware (GE HealthCare, Piscataway, NJ, USA), to obtain
affinity constants.
In silico docking protocol
The in silico docking protocol was initiated by screening
models of the shPrP NMR ensemble, PDB: 1B10 [41], for
structures having the propensity to bind thiamine in orien-
tations that are consistent with the NMR data. This step
was carried out using autodock v4.2 [42]. Coordinates of
the thiamine molecule in PDB format were obtained from
the Human Metabolome Database [18]. Hydrogen atoms
were added to the thiamine molecule using the openbabel
program [43]. Non-polar hydrogens in PrP and thiamine
were identified and merged with heavy atoms by auto-
dock tools 1.5.4 [44]. autodock tools were also
employed for calculating Gasteiger charges for both the
thiamine and PrP models. Six rotatable torsion angles in
thiamine molecule were identified by autodock tools and
allowed to freely rotate to perform flexible docking. The
protein model was treated as a rigid body during the
docking simulations. A large grid box (25 · 25 · 30 A)
centered in the vicinity of NMR-mapped thiamine binding
site (residues M138, M139, H140, F141, G142, D144 and
W145) was generated. This box also included helix 1, the
loop between b-sheet 1 and helix 1, the C-terminal half of
helix 2, the loop between helices 1 and 3, and the N-termi-
nal half of helix 3. Grid spacing was set to 0.375 A. The
atom-specific affinity map, electrostatic potential map, and
desolvation potential map were generated by the auto-
grid 4 program from the autodock package. The initial
dihedral offset of the ligand and the initial position of
ligand with respect to the protein model were selected ran-
domly for every docking run. A Lamarckian genetic algo-
rithm was selected to perform the docking simulations.
Two hundred and fifty docking runs were conducted using
the default autodock 4.2 parameters, except the following:
250 individuals in the ligand population, 2 500 000 energy
evaluations, 0.2 A translation step, 5� quaternion step, 5�torsion step.
During the second step, semiflexible docking was per-
formed with xplor-nih 2.27 [45] on the shPrP NMR
structure (PDB: 1B10 :model 13). This model showed
good agreement with the NOE data within the 6 A
upper-limit in our autodock simulations. Ligand and
protein side-chains were allowed to be flexible, while pro-
tein backbone was kept rigid. Thiamine topology and
parameter files for xplor were generated with the acpype
program (http://code.google.com/p/acpype/). After a
short initial energy minimization (50 steps), the thiamine
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4011

orientation was optimized with Cartesian dynamics for
6000 constant-temperature steps (T = 1000 K) and 6000
steps of simulated annealing to the final temperature of
300 K, followed by 400 minimization steps. A model that
had no NOE violations was selected as the final docking
pose. The coordinate data for this model has been depos-
ited into the Protein Data Bank (PDB accession no.
2LH8).
Acknowledgements
This project was funded by PrioNet Canada, the
Alberta Prion Research Institute and the National Insti-
tute for Nanotechnology (NINT). We would like to
thank the Canadian National High Field NMR Centre
(NANUC) for their assistance and use of the facilities.
The operation of NANUC is funded by the Canadian
Institutes of Health Research (CIHR), the Natural
Science and Engineering Research Council of Canada
(NSERC), and the University of Alberta.
References
1 Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95,
13363–13383.
2 Cosseddu GM, Agrimi U, Pinto J & Schudel AA (2007)
Advances in scrapie research. Rev Sci Techol 26, 657–
668.
3 Belay ED & Schonberger LB (2002) Variant Creutz-
feldt–Jakob disease and bovine spongiform encephalop-
athy. Clin Lab Med 22, 849–862.
4 Miller MW & Williams ES (2004) Chronic wasting dis-
ease of cervids. Curr Top Microbiol Immunol 284, 193–
214.
5 Collinge J, Whitfield J, McKintosh E, Beck J, Mead S,
Thomas DJ & Alpers M (2006) Kuru in the 21st cen-
tury – an acquired human prion disease with very long
incubation periods. Lancet 367, 2068–2074.
6 Almer G, Hainfellner JA, Brucke T, Jellinger K, Klein-
ert R, Bayer G, Windl O, Kretzschmar HA, Hill A,
Sidle K et al. (1999) Fatal familial insomnia: a new
Austrian family. Brain 122, 5–16.
7 Westergard L, Christensen HM & Harris DA (2007)
The cellular prion protein (PrP(C)): its physiological
function and role in disease. Biochim Biophys Acta
1772, 629–644.
8 Caughey B & Baron GS (2006) Prions and their part-
ners in crime. Nature 443, 803–810.
9 Watts JC & Westaway D (2007) The prion protein fam-
ily: diversity, rivalry, and dysfunction. Biochim Biophys
Acta 1772, 654–672.
10 Zomosa-Signoret V, Arnaud JD, Fontes P, Alvarez-
Martinez MT & Liautard JP (2008) Physiological role
of the cellular prion protein. Vet Res 39, 9.
11 Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H,
Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J
et al. (2010) Axonal prion protein is required for
peripheral myelin maintenance. Nat Neurosci 13,
310–318.
12 Pasupuleti M, Roupe M, Rydengard V, Surewicz K,
Surewicz WK, Chalupka A, Malmsten M, Sorensen OE
& Schmidtchen A (2009) Antimicrobial activity of
human prion protein is mediated by its N-terminal
region. PLoS ONE 4, 7358–7359.
13 Lee KS, Raymond LD, Schoen B, Raymond GL, Kett
L, Moore RA, Johnson LM, Kett L, Moore RA, John-
son LM et al. (2007) Hemin interactions and alterations
of the subcellular localization of prion protein. J Biol
Chem 282, 36525–36533.
14 Tagliavini F, Forloni G, Colombo L, Rossi G, Girola
L, Canciani B, Angeretti N, Giampaolo L, Peressini E,
Awan T et al. (2000) Tetracycline affects abnormal
properties of synthetic PrP peptides and PrPSc in vitro.
J Mol Biol 300, 1309–1322.
15 Vogtherr M, Grimme S, Elshorst B, Jacobs DM, Fiebig
K, Griesinger C & Zahn R (2003) Antimalarial drug
quinacrine binds to C-terminal helix of cellular prion
protein. J Med Chem 46, 3563–3564.
16 Hafner-Bratkovic I, Gaspersic J, Smid LM, Bresjanac
M & Jerala R (2008) Curcumin binds to the a-helicalintermediate and to the amyloid form of prion protein
– a new mechanism for the inhibition of PrPSc accumu-
lation. J Neurochem 104, 1553–1564.
17 Mok SWF, Thelen KM, Riemer C, Bamme T, Gultner
S, Lutjohann D & Baier M (2006) Simvastatin prolongs
survival times in prion infections of the central nervous
system. Biochem Biophys Res Commun 348, 697–
702.
18 Frid P, Anisimov SV & Popovic N (2007) Congo red
and protein aggregation in neurodegenerative diseases.
Brain Res Rev 53, 135–160.
19 Wishart DS, Knox C, Guo AC, Eisner R, Young N,
Gautam B, Hau DD, Psychogios N, Dong E, Bouatra
S et al. (2009) HMDB: a knowledge base for the human
metabolome. Nucleic Acids Res 37, D603–D610.
20 Lakowicz JR (2006) Principles of Fluorescence Spectros-
copy, 3rd edn. Springer, New York.
21 Mayer M & Meyer B (2001) Group epitope mapping by
saturation transfer difference NMR to identify segments
of a ligand in direct contact with a protein receptor.
J Am Chem Soc 123, 6108–6117.
22 Christen B, Hornemann S, Damberger FF & Wuthrich
K (2009) Prion protein NMR structure from Tammar
wallaby (Macropus euginii) shows that the B2–A2 loop
is modulated by long-range sequence effects. J Mol Biol
389, 833–845.
23 Yang M, Xi X & Yang P (2008) Thermodynamic analy-
sis of fluorescence enhancement and quenching theory
equations. Front Chem Chin 3, 254–261.
PrP binds thiamine R. Perez-Pineiro et al.
4012 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS

24 Matsunaga Y, Peretz D, Williamson A, Burton D, Me-
hlhorn I, Groth D, Cohen FE, Prusiner SB & Baldwin
MA (2001) Cryptic epitopes in N-terminally truncated
prion protein are exposed in the full-length molecule:
dependence of conformation on pH. Proteins 44, 110–
118.
25 Jackson GS, Murray I, Hosszu LLP, Gibbs N, Waltho
JP, Clarke AR & Collinge J (2001) Location and prop-
erties of metal-binding sites on the human prion pro-
tein. Proc Natl Acad Sci USA 98, 8531–8535.
26 Walter ED, Chattopadhyay M & Millhauser GL (2006)
The affinity of copper binding to the prion protein
octarepeat domain: evidence for negative cooperativity.
Biochemistry 45, 13083–13092.
27 Nicoll AJ, Trevitt CR, Tattum MH, Risse E, Quar-
terman E, Ibarra AA, Wright C, Jackson GS, Sessions
RB, Farrow M et al. (2010) Pharmacological chaperone
for the structured domain of human prion protein. Proc
Natl Acad Sci USA 107, 17610–17615.
28 Langella E, Impronta R, Crescenci O & Barone V
(2006) Assesing the acid–base and conformational
properties of histidine residues in human prion protein
(125–228) by means of pKa calculations and molecular
dynamics simulations. Protein Struct Funct Bioinform
64, 167–177.
29 Lonsdale D (2006) A review of the biochemistry,
metabolism and clinical benefits of thiamin(e) and its
derivatives. eCAM 3, 49–59.
30 Gangolf M, Czerniecki J, Radermecker M, Detry O,
Nisolle M, Jouan C, Martin D, Chantraine F, Lakaye
B, Wins P et al. (2010) Thiamine status in humans and
content of phosphorylated thiamine derivatives in biop-
sies and cultured cells. PLoS ONE 5, e13616.
31 Thom JY, Davis RE & Icke GC (1986) Protein binding
of thiamine in human plasma. Int J Vitam Nutr Res 56,
189–195.
32 Parkhomenko IM, Strokina AA, Pilipchuk SI, Step-
anenko SP, Chekhovskaia LI & Donchenko GV (2010)
Existence of two different active sites on thiamine
binding protein in plasma membranes of synaptosomes.
Ukr Biokhim Zh 82, 34–41.
33 Ganapathy V, Smith SB & Prasad PD (2004) SLC19:
the folate ⁄ thiamine transporter family. Pflugers Arch
447, 641–646.
34 Tichopad A, Pfaffl MW & Didier A (2003) Tissue-spe-
cific expression pattern of bovine prion gene: quantifi-
cation using real-time RT-PCR. Mol Cell Probe 17,
5–10.
35 Moudjou M, Frobert Y, Grassi J & La Bonnardiere C
(2001) Cellular prion protein status in sheep: tissue-
specific biochemical signatures. J Gen Virol 82, 2017–
2024.
36 Bjorndahl TC, Zhou GP, Liu XH, Perez-Pineiro R,
Semenchenko V, Saleem F, Acharya S, Bujold A,
Sobsey C & Wishart DS (2011) Detailed biophysical
characterization of the acid-induced PrPC to PrPB con-
version process. Biochemistry 50, 1162–1173.
37 Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J
& Bax A (1995) NMRPipe: a multidimensional spectral
processing system based on UNIX pipes. J Biomol
NMR 6, 277–293.
38 Johnson BA (2004) Using NMRView to visualize and
analyze the NMR spectra of macromolecules. Methods
Mol Biol (NY) 278, 313–352.
39 Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson
HJ, Oldfield E, Markley JL & Sykes BD (1995) 1H, 13C
and 15N chemical shift referencing in biomolecular
NMR. J Biomol NMR 6, 135–140.
40 Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A,
Gronenborn AM & Clore GM (1989) Overcoming the
overlap problem in the assignment of proton NMR
spectra of larger proteins by use of three-dimensional
heteronuclear proton–nitrogen-15 Hartmann–Hahn–
multiple quantum coherence and nuclear Overhauser–
multiple quantum coherence spectroscopy: application
to interleukin 1.beta. Biochemistry 28, 6150–
6156.
41 James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang
H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prus-
iner SB et al. (1997) Solution structure of a 142-residue
recombinant prion protein corresponding to the infec-
tious fragment of the scrapie isoform. Proc Natl Acad
Sci USA 94, 10086–10091.
42 Goodsell DS, Morris GM & Olson AJ (1996) Auto-
mated docking of flexible ligands: applications of auto-
dock. J Mol Recognit 9, 1–5.
43 Guha R, Howard MT, Hutchison GR, Murray-Rust P,
Rzepa H, Steinbeck C, Wegner J & Willighagen EL
(2006) The blue obelisks interoperability in chemical
informatics. J Chem Inf Model 46, 991–998.
44 Morris GM, Huey R, Lindstrom W, Sanner MF, Belew
RK, Goodsell DS & Olson AJ (2009) AutoDock4 and
AutoDockTools4: automated docking with selective
receptor flexibility. J Comput Chem 30, 2785–
2791.
45 Schwieters CD, Kuszewski JJ, Tjandra N & Clore GM
(2003) The Xplor-NIH NMR molecular structure deter-
mination package. J Magn Res 160, 66–74.
Supporting information
The following supplementary material is available:
Figs. S1–S6. Stern–Volmer plots for data presented in
Table 1.
Fig. S7. 15N edited TOCSY-HSQC and NOESY-
HSQC strip plots for selected residues in thiamine
binding pocket.
Fig. S8. NMR spectra of thiamine (free) and thiamine
bound to shPrP.
R. Perez-Pineiro et al. PrP binds thiamine
FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS 4013

Table S1. List of human metabolites used in the
screening process.
This supplementary material can be found in the
online version of this article.
Please note: As a service to our authors and read-
ers, this journal provides supporting information
supplied by the authors. Such materials are peer-
reviewed and may be re-organized for online deliv-
ery, but are not copy-edited or typeset. Technical
support issues arising from supporting information
(other than missing files) should be addressed to the
authors.
PrP binds thiamine R. Perez-Pineiro et al.
4014 FEBS Journal 278 (2011) 4002–4014 ª 2011 The Authors Journal compilation ª 2011 FEBS
Related Documents











