The mitochondrial genome of the water spider Argyroneta aquatica (Araneae: Cybaeidae) MINGXIN LIU,ZHISHENG ZHANG &ZUOGANG PENG Submitted: 28 June 2014 revised 23 September 2014; Accepted: 10 October 2014 doi:10.1111/zsc.12090 Liu, M., Zhang, Z., Peng, Z. (2015). The mitochondrial genome of the water spider Argyroneta aquatica (Araneae: Cybaeidae). —Zoologica Scripta, 44, 179–190. We sequenced nearly the entire mitochondrial genome of Argyroneta aquatica, a wholly underwater-living spider, thereby enhancing the available genomic information for Arachn- ida. The confirmed sequences contained the complete set of known genes present in other metazoan mitochondrial genomes. However, the mitochondrial gene order of A. aquatica was distinctly different from that of the most distant Chelicerata Limulus polyphemus (Xipho- sura), probably because of a series of gene translocations and/or inversions. Comparison of arachnid mitochondrial gene orders for the purpose of phylogenetic inference is only mini- mally useful, but provides a strong signal in closely related lineages. To test the basal rela- tionships and the evolutionary pattern of tRNA gene rearrangements among Arachnida, phylogenetic analyses using amino acid sequences of the 13 protein-coding genes were per- formed. An interesting feature, the five 135-bp tandem repeats and two 363-bp tandem repeats, was identified in the putative control region. Although control region tandem repeats have been reported in many other arachnid and metazoan species, this is the first time it has been described in spiders. Corresponding authors: Zhisheng Zhang and Zuogang Peng, Key Laboratory of Eco- environments in Three Gorges Reservoir Region (Ministry of Education), Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), School of Life Science, Southwest University, Chongqing 400715, China. E-mails: [email protected] and [email protected] Mingxin Liu, Zhisheng Zhang, and Zuogang Peng, Key Laboratory of Eco-environments in Three Gorges Reservoir Region (Ministry of Education), Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), School of Life Science, Southwest University, Chongqing, 400715, China. E-mail: [email protected] Background As of 2014, 44 906 spider species (Araneae) have been described and classified into 3 935 genera and 114 families (Platnick 2014). Among the 11 extant arachnid orders, Araneae (spiders) and Acari (mites and ticks) are by far the most diverse groups. Compared with the 53 published mitochondrial genomes of Acari, only 12 Araneae mito- chondrial genomes have been described (sourced from GenBank, June 2014). Typically, the metazoan mitochon- drial genome encodes 37 genes including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA) and two ribosomal RNA genes (rRNA), along with some non- coding sequences necessary for initiating and regulating transcription and replication (Clayton 1982; Zhang & Hewitt 1997; Boore 1999; Kolpakov et al. 2003). Mitochondrial genome evolution in many animal species has been studied for genome content and gene arrange- ments (Morrison et al. 2002; Murrell et al. 2003; Nardi et al. 2003; Fujita et al. 2007). Mitochondrial gene rearrangements are common among Chelicerata (Black & Roehrdanz 1998; Fahrein et al. 2007; Domes et al. 2008). Although molecular markers are commonly used for phylogenetic analysis, mitochondrial genome sequences and gene order have proven useful for phylogenetic reconstruc- tion among the Arthropoda (Arabi et al. 2012), including studies of Embioptera (Insecta) (Komoto et al. 2012), Hymenoptera (Insecta) (Dowton 1999), Parasitiformes (Arachnida: Acari) (Black & Roehrdanz 1998) and Ricinulei (Arachnida) (Fahrein et al. 2007). These studies have shown that whole mitochondrial genome sequencing allows comparisons between phylogenetically related groups to help determine the evolution of gene order. ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 179 Zoologica Scripta

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The mitochondrial genome of the water spider Argyronetaaquatica (Araneae: Cybaeidae)MINGXIN LIU, ZHISHENG ZHANG & ZUOGANG PENG
Submitted: 28 June 2014revised 23 September 2014; Accepted:10 October 2014doi:10.1111/zsc.12090
Liu, M., Zhang, Z., Peng, Z. (2015). The mitochondrial genome of the water spiderArgyroneta aquatica (Araneae: Cybaeidae). —Zoologica Scripta, 44, 179–190.We sequenced nearly the entire mitochondrial genome of Argyroneta aquatica, a whollyunderwater-living spider, thereby enhancing the available genomic information for Arachn-ida. The confirmed sequences contained the complete set of known genes present in othermetazoan mitochondrial genomes. However, the mitochondrial gene order of A. aquaticawas distinctly different from that of the most distant Chelicerata Limulus polyphemus (Xipho-sura), probably because of a series of gene translocations and/or inversions. Comparison ofarachnid mitochondrial gene orders for the purpose of phylogenetic inference is only mini-mally useful, but provides a strong signal in closely related lineages. To test the basal rela-tionships and the evolutionary pattern of tRNA gene rearrangements among Arachnida,phylogenetic analyses using amino acid sequences of the 13 protein-coding genes were per-formed. An interesting feature, the five 135-bp tandem repeats and two 363-bp tandemrepeats, was identified in the putative control region. Although control region tandemrepeats have been reported in many other arachnid and metazoan species, this is the firsttime it has been described in spiders.Corresponding authors: Zhisheng Zhang and Zuogang Peng, Key Laboratory of Eco-environments in Three Gorges Reservoir Region (Ministry of Education), Key Laboratory ofFreshwater Fish Reproduction and Development (Ministry of Education), School of Life Science,Southwest University, Chongqing 400715, China. E-mails: [email protected] [email protected] Liu, Zhisheng Zhang, and Zuogang Peng, Key Laboratory of Eco-environments in ThreeGorges Reservoir Region (Ministry of Education), Key Laboratory of Freshwater Fish Reproductionand Development (Ministry of Education), School of Life Science, Southwest University, Chongqing,400715, China. E-mail: [email protected]
BackgroundAs of 2014, 44 906 spider species (Araneae) have beendescribed and classified into 3 935 genera and 114 families(Platnick 2014). Among the 11 extant arachnid orders,Araneae (spiders) and Acari (mites and ticks) are by far themost diverse groups. Compared with the 53 publishedmitochondrial genomes of Acari, only 12 Araneae mito-chondrial genomes have been described (sourced fromGenBank, June 2014). Typically, the metazoan mitochon-drial genome encodes 37 genes including 13 protein-codinggenes (PCGs), 22 transfer RNA genes (tRNA) and tworibosomal RNA genes (rRNA), along with some non-coding sequences necessary for initiating and regulatingtranscription and replication (Clayton 1982; Zhang &Hewitt 1997; Boore 1999; Kolpakov et al. 2003).Mitochondrial genome evolution in many animal species
has been studied for genome content and gene arrange-ments (Morrison et al. 2002; Murrell et al. 2003; Nardiet al. 2003; Fujita et al. 2007). Mitochondrial generearrangements are common among Chelicerata (Black &Roehrdanz 1998; Fahrein et al. 2007; Domes et al. 2008).Although molecular markers are commonly used forphylogenetic analysis, mitochondrial genome sequences andgene order have proven useful for phylogenetic reconstruc-tion among the Arthropoda (Arabi et al. 2012), includingstudies of Embioptera (Insecta) (Komoto et al. 2012),Hymenoptera (Insecta) (Dowton 1999), Parasitiformes(Arachnida: Acari) (Black & Roehrdanz 1998) and Ricinulei(Arachnida) (Fahrein et al. 2007). These studies have shownthat whole mitochondrial genome sequencing allowscomparisons between phylogenetically related groups tohelp determine the evolution of gene order.
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 179
Zoologica Scripta

Fahrein et al. (2007) compared mitochondrial gene orderin Arachnida and revealed great variation, when comparedwith their most distant arthropod. Gene rearrangementshave been found in most arachnid orders. Arachnida is aspecies-rich class of Arthropoda, second only to Insecta(Masta 2010). Arachnida mitochondrial genomes haveyielded some surprising findings in their patterns of geneorganization and genome evolution (Davila et al. 2005;Shao et al. 2005). The water mite Unionicola foili and thedust mite Dermatophagoides pteronyssinus both possess novelgene arrangements with implications for phylogeneticinference (Dermauw et al. 2009; Ernsting et al. 2009).Therefore, Chelicerates represents a promising taxon forinvestigating the evolution of gene order. However, thelack of mitochondrial genome data for certain key taxa hashindered a thorough understanding of the evolution ofgene order.As with mitochondrial gene order, the secondary struc-
ture of tRNAs varies among some metazoan groups.Studies of molecular evolutionary patterns have allowedinferences about the secondary structure of tRNAs. Typi-cally, tRNAs have a highly conserved cloverleaf-shapedstructure (Dirheimer et al. 1995). Natural selection presum-ably maintains tRNA structure so that tRNA molecules caninteract effectively with other components of the transla-tion system (Masta & Boore 2008). Regions of evolution-arily conserved secondary structure may indicate theirimportance for correct functioning of the tRNA or protein.Masta & Boore (2008) classified the inferred tRNA struc-tures from 16 Arachnida mitochondrial genomes into fourdifferent types: (i) typical tRNA genes; (ii) tRNAs lacking aT arm; (iii) tRNAs lacking a D arm; (iv) tRNAs lacking awell-paired aminoacyl acceptor stem. Reduction in tRNAsize may lead to an inability to fold into the canonicalcloverleaf-shaped secondary structure and subsequentlyaffect protein synthesis. Mitochondrial tRNAs accumulatemore deleterious mutations relative to their nuclear tRNAcounterparts, as the mitochondrial genome is under greatermutational pressure (Lynch 1996). However, it is possiblethat an RNA editing mechanism plays a key role in post-transcriptional editing to modify atypical tRNAs (Masta &Boore 2004). Lavrov et al. (2000b) reported template-dependent RNA editing in an aminoacyl acceptor stem inLithobius forficatus (Myriapoda: Chilopoda: Lithobiomor-pha). In this system, the 30 end of the acceptor stem is usedas a template for 50 end synthesis, possibly by a RNA-dependent RNA polymerase.All published spider mitochondrial genomes contain
truncated or atypical tRNAs (Masta & Boore 2004; Qiuet al. 2005; Masta & Boore 2008). Although RNA editinghas been reported in many other groups (ranging fromprotozoa to metazoa), it has not been verified in the
mitochondrial tRNA of spiders. As there are differentatypical tRNA secondary structures in spiders, how RNAediting repairs tRNAs lacking a T arm, a D arm or awell-paired aminoacyl acceptor remains unknown. A cleardescription of RNA editing mechanisms and whether RNAediting is a phylogenetically useful tool remain unresolvedquestions in this field.Improvements in genomic technologies and interest in
mitochondrial genome organization and evolution haveled to pervasive whole mitochondrial genome sequencingof arthropods (Boore 2006). Despite an increasing under-standing of mitochondrial evolution within the Arthro-poda, the relationships within Arachnida have not beenwell resolved because of insufficient data or a lack ofimportant taxa. A Chelicerata phylogeny was constructedby analyzing cox1, and the 18S and 28S nuclear rRNAgenes (Arabi et al. 2012). However, despite having a dataset of 180 taxa, the study failed to elucidate the phylogenyof Arachnida. Several other studies describing the phylo-genetic relationships within Arachnida, based on mito-chondrial genomes, had either low bootstrap supportvalues for monophyly of Arachnida or poorly resolvedrelationships between orders (Masta et al. 2009; Arabiet al. 2012). Two main problems contribute to theremaining unresolved relationships between arachnidorders. First, incomplete taxa sampling may strongly affectthe phylogeny. Only Ovchinnikov & Masta (2012) used arelatively comprehensive data set of nine arachnid orders,other studies covered lower diversity of Arachnida. How-ever, reliable phylogenetic studies may be possible if aheavy sampling of Arachnida species is included. Second,heterogeneous substitution rates between Arachnida maylead to long-branch attraction.In this study, the mitochondrial genome of the water
spider Argyroneta aquatica was almost completelysequenced. All available arachnid taxa data, including nineof 11 orders, were then used to determine the phylogeneticrelationships between arachnid orders. Gene order evolu-tion was reconstructed by comparing all available arachnidtaxa to uncover possible evolutionary signals in gene rear-rangements. Finally, the highly repetitive sequences withinthe putative control region were investigated.
Materials and methodsSamples and DNA extraction
Argyroneta aquatica specimens were collected from lakeslocated in Wurih Tutala, Xilinhot, Inner Mongolia(China), in August 2011. All specimens were preserved in100% ethanol and stored at �20°C prior to DNA extrac-tion. Total genomic DNA was extracted using a DNeasyBlood and Tissue kit (Qiagen, Shanghai, China) followingthe manufacturer’s protocol.
180 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.

PCR and sequencing
Primers (Table S1) were designed based on the consensusnucleotide sequences of the mitochondrial genome of ninespider species (Calisoga longitarsis, Habronattus oregonensis,Heptathela hangzhouensis, Hypochilus thorelli, Liphistius era-wan, Nephila clavata, Ornithoctonus huwena, Pholcus phalan-gioides and Phyxioschema suthepium), together with theconserved primers for animal mitochondrial DNA(mtDNA) described by Simon et al. (2006). These primerswere used to amplify partial sequences of several mitochon-drial genes by polymerase chain reaction (PCR). Additionalspecies-specific primers (Table S1) were designed based onthe sequences obtained from these fragments and wereused for a second round PCR amplification. PCR mixturescontained 2.5 lL of 109 PCR buffer (Mg2+-free; Takara,Dalian, China), 1.5 lL of MgCl2 (25 mM), 2 lL of dNTPs(10 mM), 1 lL of each primer (10 mM), 0.25 lL of rTaqpolymerase (1 U; Takara), 3 lL of DNA template andsterile distilled H2O to a final volume of 30 lL. Amplifica-tion conditions were an initial denaturation at 94°C for2 min, followed by 30 cycles of 94°C for 30 s, 48–52°C for30 s and 72°C for 45–150 s. To generate long PCR frag-ments (>3 kb), reaction mixtures contained 5 lL of 109LA PCR buffer (Mg2+-plus; Takara), 8 lL of dNTPs(10 mM), 1 U of LA Taq polymerase (Takara), 3 lL ofDNA template and sterile distilled H2O to a final volumeof 50 lL. Amplification conditions consisted of an initialdenaturation at 94°C for 1 min, followed by 30 cycles of98°C for 10 s and 68°C for 4 min, then a final extension at72°C for 10 min. PCR products were visualized on 1.5%agarose gels stained with ethidium bromide and then puri-fied using a QIAquick Gel Extraction kit (Qiagen).Sequencing reactions were carried out on an ABI 3730sequencer by Beijing Genomics Institute (BGI, Beijing,China) with the same primers as those used for PCR. Forlong PCR fragments (>3 kb), walking primers for sequenc-ing were designed and supplied by BGI when needed.
Sequence assembly and gene annotation
Protein-coding and rRNA genes and gene boundaries wereidentified by BLAST search and by comparison with align-ments from other 12 spiders. tRNA genes were identifiedusing tRNAscan-SE (Lowe & Eddy 1997), using the fol-lowing parameters: Search Mode = ‘EufindtRNA-Cove’,Source = ‘Nematode Mito’, Genetic Code = ‘InvertebrateMito’ and Cove score cut-off = 0.1. Other tRNAs wereidentified by aligning all tRNA sequences and looking foranticodon arm motifs that were conserved among Araneae.As the control region often formed a potential stem-loopstructure, the Mfold Server (Zuker 2003) was used topredict the secondary structure of the putative controlregion. Base composition used to calculate the GC skew
was performed using MEGA version 5.0 (Tamura et al.2011). The mitochondrial genome map was drawn usingthe online tool mtviz (http://pacosy.informatik.uni-leipzig.de/mtviz) by arbitrarily filling the gaps, resulting in a totallength of 16 000 bp.
Phylogenetic analysis and gene rearrangements
To examine the phylogeny of Arachnida, we included allavailable 82 arachnid mitochondrial genome sequencesdeposited in GenBank (excluding Metaseiulus occidentalis,which lacks nad3 and nad6) along with the A. aquaticamitochondrial genome sequenced in this study. Sequencesfrom Daphnia pulex (Crustacea) (Crease 1999), Penaeus mon-odon (Crustacea) (Wilson et al. 2000), L. forficatus (Myria-poda) (Lavrov et al. 2000b) and Limulus polyphemus(Xiphosura) (Lavrov et al. 2000a) were used to root thephylogenetic tree. GenBank accession numbers are given inTable S2.All 13 PCGs were used in phylogenetic analyses. Each
of the PCGs was manually extracted from the mitochon-drial genome sequences of 87 taxa using the annotatedgene boundary information. Next, nucleotide sequenceswere translated into amino acid sequences using SEAVIEW
version 4 (Gouy et al. 2010), using the invertebrate mito-chondrial genetic code. Amino acid sequences were alignedin CLUSTAL X version 2.0 (Larkin et al. 2007) using theGonnet series protein matrix, with a gap opening penaltyof 10 and a gap extension penalty of 0.2. To ensure onlyhomologous amino acid sequences were used in phyloge-netic analyses, poorly conserved sites were subsequentlytrimmed using GBLOCKS version 0.91b (Talavera & Castre-sana 2007). Ambiguously aligned motifs were excludedusing the default settings, except that ‘data type’ was chan-ged to ‘protein’ and ‘allowed gap position’ was changed to‘with half’. The amino acid sequences of the 13 PCGs wereconcatenated into a single data set for subsequent phyloge-netic analysis.Maximum likelihood (ML) analysis was performed using
Treefinder (Jobb et al. 2004). Model selection was per-formed using PROTTEST version 3.0 (Darriba et al. 2011) foramino acid sequences. According to the Akaike informationcriterion, the mtArt + G + I model was optimal for MLanalysis of amino acid alignments. Bootstrapping was per-formed with 1 000 replicates to assess node support. MRBA-
YES version 3.1.2 (Huelsenbeck & Ronquist 2001) was usedto calculate Bayesian inference (BI). As the mtArt model isnot implemented in the current version of MRBAYES, themtRev + G + I model was used for phylogenetic analysis ofthe data set. The default parameters of MRBAYES wereused. Two runs of 1 000 000 generations were performed,and trees were sampled every 100 generations. Likelihoodvalues of the sampled trees were examined, and 2 500
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 181
M. Liu et al. � Mitochondrial genome of the water spider

samples were discarded as burn-in, with 7 500 sampledtrees used to calculate posterior probabilities.To explore genome rearrangements, we used the web-
based CREX software (Bernt et al. 2007) to conduct pair-wise comparisons of arachnid mitochondrial gene orders.We then mapped the determined genome rearrangementscenarios to corresponding branches. The rearrangementscenarios include transpositions (T), reverse transpositions(rT), reversals or inversion (I) and tandem-duplication-random-loss (TDRL) events. Species with uncommonmitochondrial gene content are excluded from ourcomparisons. Subsequently, Argiope amoena, A. bruennichi,Buthus occitanus, Centruroides limpidus, Leptotrombidiumpallidum, Mesobuthus martensii, Phalangium opilio andSteganacarus magnus were excluded because of geneduplication or deletion.
Results and discussionGenome content
The nearly complete mitochondrial genome of A. aquatica(GenBank accession number: KJ907736) was sequencedfrom overlapping PCR fragments, and the mitochondrialgenome map is shown in Fig. 1. The total length of all 13PCGs was 10 787 bp, with an overall G + C content of30.2%, ranging from 23.9% (atp8) to 33.8% (cox1). MostPCGs were predicted to use ATN as a start codon, whichis typical for metazoan mitochondria genes (Wolstenholme1992), while cox1, cox2, cox3, nad4 and nad6 were predictedto use a TTG start codon (Table S3). Only six PCGs hadincomplete stop codons (nad2, nad3, nad4, nad4L, nad5 andcytb), with only a single thymine residue. Similar incom-plete stop codons have been described in other Araneae, inwhich polyadenylation following transcription forms thecomplete TAA stop codon (Ojala et al. 1981). For theremaining PCGs, the standard TAA and TAG stop codonswere identified.We used Perna & Kocher’s (1995) method to determine
the nucleotide composition skew of PCGs, where GCskew = [G � C]/[G + C]. In A. aquatica, the GC skew waspositive in all majority strand-encoded genes and negativein all minority strand-encoded genes (Table S3). This find-ing agrees with a previous study that suggested that GCskew appears to be a good indicator of the strand-specificnucleotide frequency bias in mitochondrial genomes (Perna& Kocher 1995).We were unable to sequence the entire control region;
however, the remaining mitochondrial genome sequencecontained 13 PCGs, 22 tRNA genes and two rRNA genes.Genes were encoded on both strands and are shown inTable S3. The mitochondrial gene order of A. aquatica isshown in Fig. 2. While the protein-coding and rRNA genepositions are identical to those of the out-group species
L. polyphemus, a series of tRNA gene rearrangements haveoccurred in the evolutionary history of A. aquatica. A strik-ing feature is a tRNA translocation in which seven tRNAs(tRNAAsn, tRNASer(AGN), tRNAThr, tRNALeu(UUR),tRNAIle, tRNAGln, tRNATyr) have been rearranged into anew order (tRNALeu(UUR), tRNAAsn, tRNASer(AGN),tRNAIle, tRNAThr, tRNAGln, tRNATyr) (Fig. 2). Overlap-ping sequences were also common throughout the genome.A large non-coding region (352 bp in length) was identifiedbetween atp6 and cox3.
Transfer RNA genes
Our findings showed that all tRNAs in the A. aquaticamitochondrial genome lack a T arm, except for tRNASer1
and tRNASer2, in which the D arm is replaced by a loop(Data S1). Another feature observed in these tRNAs wasthe mismatched aminoacyl acceptor stem. Truncated tRNAgenes may be products of overlap with adjacent genes.While Masta & Boore (2004) disputed this possibility,Yamazaki et al. (1997) suggested that pressure to minimizethe size of the mitochondrial genome may lead to trunca-tion of these tRNAs, which is apparently deleterious, asatypical tRNA secondary structures cannot be functional inprotein synthesis. Thus, a putative RNA editing process
Fig. 1 Mitochondrial genome map of A. aquatica. PCGs, rRNAand tRNA genes: atp6 and atp8, ATP synthase subunits 6 and 8;cytb, cytochrome b; cox1–3, cytochrome c oxidase subunits 1–3;nad1–6 and nad4L stand for NADH dehydrogenase subunits 1–6and 4L in order; 12S, small ribosomal RNA; 16S, large ribosomalRNA; tRNA genes are labelled to the one letter amino acid code,where L1 = tRNALeu(CUN), L2 = tRNALeu(UUR), S1 = tRNASer
(AGN) and S2 = tRNASer(UCN); CR, putative control region.
182 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.

presumably exists to repair such deleterious mutations(Masta & Boore 2004). By this process, the mismatchesoccurring in the tRNA stem region could be corrected.Indeed, a variety of tRNA editing events have beenreported in mitochondrial tRNAs, ranging from protozoato plants and metazoan animals (Marechaldrouard et al.1993; Tomita et al. 1996; Laforest et al. 1997; Alfonzo et al.1999; Leigh & Lang 2004). However, although numeroustypes of RNA editing and thousands of editing sites havebeen described, the truncated tRNAs in spiders seem to beextremely unusual, as no existing RNA editing mechanismcan be applied to them. A study of inferred tRNA second-ary structures in eight arachnid mitochondrial genomesshowed that the majority of tRNA genes in four opisthot-hele spiders lacked sequences encoding a T arm (Masta &Boore 2008). However, all mitochondrial tRNAs in Pseudo-cellus pearsei (Arachnida: Ricinulei) can fold into the canoni-cal cloverleaf secondary structures (Fahrein et al. 2007). Inour study, we showed that 20 A. aquatica mitochondrialtRNAs lack a T arm, which is the greatest number of Tarm losses in any arachnid identified to date.Lavrov et al. (2000b) identified template-dependent RNA
editing in the centipede L. forficatus: the 30 end sequencesof the acceptor stem act as a template to synthesize thematching strand. This template-dependent mechanism cor-rected the mismatched aminoacyl acceptor stem in L. forfic-atus, but tRNAMet and tRNAAsn remained unedited andstill had a TV-replacement loop, while tRNASer1 had a Dloop. We can speculate that the mismatched aminoacylacceptor stems in A. aquatica mitochondrial tRNAs couldbe edited by template-dependent RNA editing, but there isno similar modification of T arm or D arm loss in spiders.Thus, we suggest that novel forms of RNA editing mayexist in Arachnida.Masta & Boore (2008) found that the tRNALeu(CUN)
mismatch aminoacyl acceptor appeared to be a shared char-acteristic among spiders, implying that some form of RNAediting is also a shared characteristic of this taxon. RNAediting is often confirmed by its specific mechanisms in
posttranscriptional modification. For example, A is modi-fied to I by adenosine deamination using tRNA-specificadenosine deaminases (Aphasizheva et al. 2011). The large-scale loss of sequences, resulting in TV-replacement loopsand D loops, suggests that arachnid mitochondrial genometRNAs are an ideal model for RNA editing research.However, both the molecular mechanisms and the enzymesinvolved remain undetermined.
Putative control region
The primer walking strategy used in the current studycould not successfully determine the sequence gap withinthe control region, and an inverse PCR method could notbe applied because no appropriate restriction sites in therepeated sequence were available. Highly repetitivesequences were identified within the sequenced portion ofthe control region, and we speculate that there are furtherrepetitive sequences in the undetermined part of thecontrol region. Although highly repetitive sequences havepreviously been reported in the mitochondrial genomes ofArachnida species, including Eremobates cf. palpisetulosus(Solifugae) (Masta & Boore 2008), M. martensii(Scorpiones) (Choi et al. 2007) and M. occidentalis (Acari)(Jeyaprakash & Hoy 2007), the repeat regions identified inA. aquatica are novel among Araneae. The putative controlregion tandem repeats are presented in Fig. 3.The first repeat region consists of five 135-bp tandem
repeats, while the second repeat region consists of two363-bp tandem repeats. Considering highly repetitivesequences (often A-T rich) in the control region inhibitDNA polymerase in insects and platyhelminths (Hu et al.2007), we may ascribe the failure of sequencing to thesetandem repeats.The control region in mitochondrial genomes initiates
and regulates transcription plus replication (Clayton 1982;Boore 1999). Part of this region can fold into a hairpin-likestructure. Masta et al. (2008) reported similar stem-loopstructures in the large non-coding regions of the Nothopugasp. and E. cf. palpisetulosus mitochondrial genomes. The
Fig. 2 Comparison of gene arrangement of Limulus polyphemus and Argyroneta aquatica. The circular genomes have been graphicallylinearized and arbitrarily chosen cox1 to show the gene rearrangement more clearly. The red, blue, grey and yellow box denote the PCGs,rRNAs, CR and tRNAs, respectively. Gene sizes are not drawn to scale.
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 183
M. Liu et al. � Mitochondrial genome of the water spider

predicted secondary structures of the control region repeatunits in A. aquatica are shown in Fig. S1. The two distinctrepeat units can both fold into several hairpin-like struc-tures not found in other spiders. And they are involved inthe control of replication and the initiation of transcription.Sumida et al. (2001) also showed similar stem-loop struc-tures could initiate replication and transcription in verte-brates. Unlike its terrestrial relatives, A. aquatica lives in alow dissolved-oxygen aquatic environment and may requirea more efficient oxidative respiratory chain to be able toperform normal biological activities (Schutz et al. 2007).The hairpins identified in the A. aquatica may increase thenumber of mitochondria and levels of specific enzymes toassist with survival in the aquatic environment. Although itis likely that this is an adaptive response of a terrestrial ani-mal to the colonization of an aquatic habitat, its molecularmechanisms remain to be elucidated.
Phylogenetic analysis
The phylogeny of arachnid orders has long been debated.Here, we carried out a phylogenetic analysis using a con-catenated amino acid data set of 13 PCGs from all availablearachnid mitochondrial genomes. Additionally, D. pulex(Crustacea), P. monodon (Crustacea), L. forficatus (Myria-poda) and L. polyphemus (Xiphosura) were chosen as out-groups to root the tree. To compare arachnid phylogeny,we summarized the hypothetical relationships proposed byother authors (Fig. 4). We could not include all extantarachnid orders, as samples from Schizomida and Palpigra-di have not yet to be sequenced for mitochondrial ge-nomes.Phylogenetic analysis of Chelicerata shows Pseudoscorpi-
ones is a monophyletic group only distantly related to
Solifugae (Fig. 5) and supports the sister group relationshipbetween Pseudoscorpiones and Acariformes, proposed byOvchinnikov & Masta (2012) and Regier et al. (2010).Studies on the structure of sperm and male genitalia inSolifugae also challenge the close relationship withPseudoscorpiones traditionally favoured (Alberti & Peretti2002). Morphological research has tended to place thePseudoscorpiones into a Opiliones + Scorpiones + Solifugaeclade (Shultz 1990; Wheeler & Hayashi 1998) (Fig. 4,A–E),while Regier et al. (2010) showed less than 50% bootstrapsupport for a sister relationship between these groups.Although the sister relationship between Solifugae andXiphosura was also recovered as Ovchinnikov & Masta(2012), in contrast, we find that sister group of other Cheli-cerates is Scorpiones. Scorpiones as sister of all other Cheli-cerates is also supported both by morphological data (Shultz2007) and molecular data (Masta 2010). Dunlop & Braddy(2001) have also argued for a close relationship betweenScorpiones and the extinct group Eurypterida.Acariformes was a sister group to Ricinulei in the analy-
ses of both Fahrein et al. (2007) and Domes et al. (2008)and is consistent with morphological hypotheses. WhenPseudoscorpiones taxa were added, Ricinulei + Acariformeswas split, in favour of the more traditional Ricinulei +Acari (including Pseudoscorpiones).Our results support the monophyly of each Arachnida
order, excluding the Acari. Currently, three major lineagesof Acari are recognized: Acariformes, Opilioacariformesand Parasitiformes (Walter et al. 1996; Dunlop & Alberti2008). While they are proposed to be closely related sistergroups, the monophyly of Acari is controversial. Ovchinni-kov & Masta (2012) reported that the Acari were split byPycnogonida and Pseudoscorpiones. Additionally, Fahrein
Fig. 3 The structural organization of the control region of Argyroneta aquatica. The flanking genes 12S, tRNAGln(Q), tRNAMet(M) and nad2are represented in coloured boxes. The gene sizes are not drawn to scale. The sequences of repeat units of both repeat region are alsoprovided.
184 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.

et al. (2007) and Domes et al. (2008) supported the para-phyly of Acari (Acariformes + Ricinulei). In this study, wealso find a polyphyletic relationship within the genusAmblyomma, as originally proposed by Burger et al. (2012)that A. elaphense and A. sphenodonti were not actually mem-bers of this genus.Although we did not find Arachnida to be monophyletic,
because of the placement of Xiphosura within Arachnida,the position of Xiphosura as a sister group to Arachnidahas been well supported in previous morphological andmolecular analyses (Shultz 2007; Regier et al. 2010).Monophyly of Arachnida is weakly supported in manymolecular phylogenies (Jones et al. 2007; Masta et al.2009). However, Masta (2010) proposed that Xiphosura iseither the sister group to Arachnida or diverged fromother ancient Arachnida lineages early in the diversificationof Chelicerata.
Gene rearrangements
The mitochondrial gene order of the water spider variesgreatly from that of L. polyphemus (Fig. 2) and from sevenother sequenced Araneae species. However, it shares thesame gene order as H. oregonensis and N. clavata, Telamoniavlijmi. These species, plus H. thorelli (Hypochilidae) andP. phalangioides (Pholcidae), represent the Araneomorphae
(Fig. 5). Our data suggest that some relationships amongthe Araneae can be distinguished by gene order synapo-morphies. The taxa in basal clades have the same geneorder as L. polyphemus, and basal taxa of each order alsoshare the same gene order. This further implies the useful-ness of gene order as a phylogenetic signal. Gene rear-rangements are very common in spiders and otherArachnida (Masta & Boore 2004; Qiu et al. 2005; Fahreinet al. 2007; Domes et al. 2008; Yuan et al. 2010). Addition-ally, Boore (1999) implied that gene rearrangements areuseful markers for resolving deep splits within phylogenies.Although the relative frequency of rearrangements variesbetween lineages, gene arrangements are becoming morewidely used as characters for resolving phylogenetic rela-tionships (Boore 1999; Dowton et al. 2002). Fahrein et al.(2007) compared the mitochondrial gene order from 23Arachnida species and found only eight species had thesame gene order as L. polyphemus. Masta (2010) addedOpiliones, represented by P. opilio, to a data set based onprevious studies to investigate the evolution of gene rear-rangement in Arachnida, and showed that gene order wasnot as robust a character as previously proposed for distin-guishing Arachnida phylogeny, but that it did provide aphylogenetic signal that resolved some Arachnida groups(Boore & Brown 1998; Masta et al. 2009; Masta 2010).
A B C
D E F
Fig. 4 Arachnida phylogeny of (A) Weygoldt & Paulus (1979), (B) Shultz (1990), (C) Wheeler & Hayashi (1998), (D) Giribet et al. (2002),(E) Shultz (2007) and (F) Regier et al. (2010).
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 185
M. Liu et al. � Mitochondrial genome of the water spider
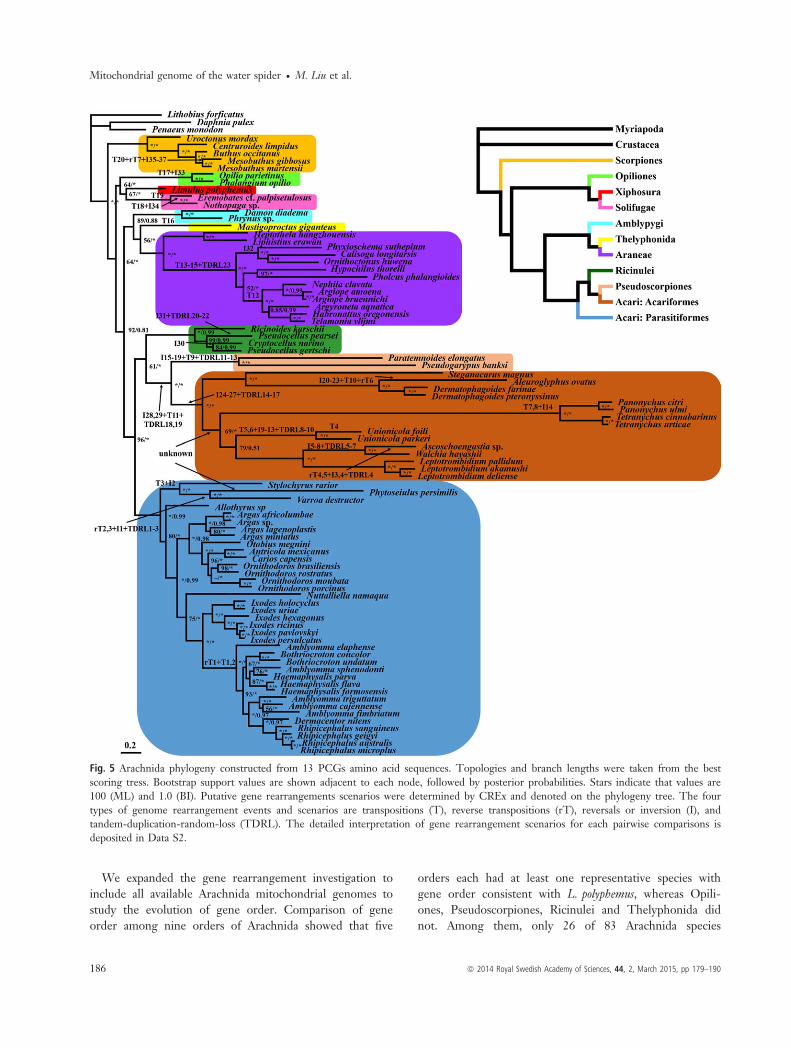
We expanded the gene rearrangement investigation toinclude all available Arachnida mitochondrial genomes tostudy the evolution of gene order. Comparison of geneorder among nine orders of Arachnida showed that five
orders each had at least one representative species withgene order consistent with L. polyphemus, whereas Opili-ones, Pseudoscorpiones, Ricinulei and Thelyphonida didnot. Among them, only 26 of 83 Arachnida species
Fig. 5 Arachnida phylogeny constructed from 13 PCGs amino acid sequences. Topologies and branch lengths were taken from the bestscoring tress. Bootstrap support values are shown adjacent to each node, followed by posterior probabilities. Stars indicate that values are100 (ML) and 1.0 (BI). Putative gene rearrangements scenarios were determined by CREx and denoted on the phylogeny tree. The fourtypes of genome rearrangement events and scenarios are transpositions (T), reverse transpositions (rT), reversals or inversion (I), andtandem-duplication-random-loss (TDRL). The detailed interpretation of gene rearrangement scenarios for each pairwise comparisons isdeposited in Data S2.
186 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.

retained the out-group pattern of L. polyphemus (Statonet al. 1997). Only one or two representative taxa for eachof the other orders were included in the current study,so gene order comparison was not comprehensive. Mostof the data we included were from Acari because of itsspecies richness in Arachnida (Bolland et al. 1998; Wal-ter & Proctor 1999; Harvey 2002). The gene orders ofAcari species display striking variation, which arose aftertheir divergence from a possible common ancestor. How-ever, in Parasitiformes, gene order is useful for dividingtaxa into three major groups, which we have termed theground pattern gene order group, the Amblyomma geneorder identical group and the gene order variation group.Several closely related genera from Parasitiformes sharethe same gene order, and experienced a series of translo-cations of tRNA, protein-coding or rRNA genes. Forexample, the gene orders of the genera Carios, Ixodes andOrnithodoros, belonging to the ground pattern gene ordergroup, are all consistent with that of L. polyphemus. Fur-thermore, they form a phylogenetically closely relatedclade that was well supported by our phylogenetic tree.Although the genera Amblyomma, Aponomma, Bothriocro-ton, Haemaphysalis and Rhipicephalus share the same geneorder, it differs from that of L. polyphemus. We suggestthat the gene order in this group was derived fromL. polyphemus after multiple rearrangements, which israrely reported, but widespread, in Acari. Despite geneorder being a relative weak signal for phylogenetic analy-sis, we were still able to summarize all the gene rear-rangement scenarios to provide a more comprehensivecomparison and gain further insight into mitochondrialgenome evolution among arachnids.Although many novel gene rearrangements were
described in Acari, data are still lacking from other orders,especially Schizomida and Palpigradi, for which no mito-chondrial genomes have been sequenced. Furthermore, asmitochondrial genomes have higher rates of evolution thanthe nuclear genome, saturation of the phylogenetic signalcan also be problematic in deep split phylogenies (Li1997). Novel mitochondrial genome features, such as com-plicated gene rearrangements or multiple control regions,are also vulnerable to long branches (Hassanin 2006). Clo-sely related lineages have implied strong signals for bothphylogenetic analysis and gene order evolution, whichallows us to investigate the mechanisms of gene order evo-lution in Arachnida. There are different mechanistic mod-els of how gene rearrangements occur. Boore et al. (1995)suggested a large number of possible gene rearrangementsmake it improbable that the same order would arise inde-pendently; shared-derived arrangements most likely indi-cate common ancestry. Gene rearrangements may betriggered by replication errors, such as inaccurate initiation
or termination, or strand slippage and mispairing, whichlead to partial duplication of mtDNA and loss of one copyof each repetitive gene (Li et al. 2009). Another mechanismof gene rearrangement is mtDNA duplication and non-ran-dom gene loss. According to this model, the transcriptionalpolarity and location of each gene copy in the repetitiveregion may be of significance in determining its destiny inthe genome (Lavrov et al. 2000b). Hitherto, no hypothesesof possible mechanisms of gene rearrangement have beenverified, and further experimental evidence is needed.
AcknowledgementsThe authors thank Luyu Wang, Zhong Li and Zongxu Li(Southwest University, China) for their support in speci-men sampling. We are also grateful to Wannes Dermauw(Ghent University, Belgium) for his help with tRNA sec-ondary structure drawing, anonymous reviewer and theeditor, Prof. Lutz Bachmann, for their helpful comments.This study was supported by the National Natural ScienceFoundation of China (31272267, 31471974) and the Fun-damental Research Funds for the Central Universities ofChina (XDJK2012C087) to Zhisheng Zhang and also bythe Program for New Century Excellent Talents in Uni-versity and the grant from the National Natural ScienceFoundation of China (31272283) to Zuogang Peng.
ReferencesAlberti, G. & Peretti, A. V. (2002). Fine structure of male genitalsystem and sperm in Solifugae does not support a sister-grouprelationship with Pseudoscorpiones (Arachnida). Journal ofArachnology, 30, 268–274.
Alfonzo, J. D., Blanc, V., Estevez, A. M., Rubio, M. A. T. &Simpson, L. (1999). C to U editing of the anticodon of importedmitochondrial tRNATrp allows decoding of the UGA stop codonin Leishmania tarentolae. EMBO Journal, 18, 7056–7062.
Aphasizheva, I., Maslov, D., Wang, X., Huang, L. & Aphasizhev,R. (2011). Pentatricopeptide repeat proteins stimulate mRNAadenylation/uridylation to activate mitochondrial translation intrypanosomes. Molecular Cell, 42, 106–117.
Arabi, J., Judson, M. L. I., Deharveng, L., Lourenco, W. R., Cru-aud, C. & Hassanin, A. (2012). Nucleotide composition of CO1sequences in Chelicerata (Arthropoda): detecting new mitoge-nomic rearrangements. Journal of Molecular Evolution, 74, 81–95.
Bernt, M., Merkle, D., Ramsch, K., Fritzsch, G., Perseke, M.,Bernhard, D., et al. (2007). CREx: inferring genomic rearrange-ments based on common intervals. Bioinformatics, 23, 2957–2958.
Black, W. C. & Roehrdanz, R. L. (1998). Mitochondrial geneorder is not conserved in Arthropods: Prostriate and Metastriatetick mitochondrial genomes. Molecular Biology and Evolution, 15,1772–1785.
Bolland, H. R., Gutierrez, J. & Flechtmann, C. H. W. (1998).World Catalogue of the Spider Mite Family (Acari: Tetranychidae).Leiden: Brill Academic Publishers.
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic AcidsResearch, 27, 1767–1780.
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 187
M. Liu et al. � Mitochondrial genome of the water spider

Boore, J. L. (2006). The use of genome-level characters for phylo-genetic reconstruction. Trends in Ecology & Evolution, 21, 439–446.
Boore, J. L. & Brown, W. M. (1998). Big trees from little ge-nomes: mitochondrial gene order as a phylogenetic tool. CurrentOpinion in Genetics & Development, 8, 668–674.
Boore, J. L., Collins, T. M., Stanton, D., Daehler, L. L. & Brown,W. M. (1995). Deducing the pattern of arthropod phylogenyfrom mitochondrial DNA rearrangements. Nature, 376, 163–165.
Burger, T. D., Shao, R., Beati, L., Miller, H. & Barker, S. C.(2012). Phylogenetic analysis of ticks (Acari: Ixodida) using mito-chondrial genomes and nuclear rRNA genes indicates that thegenus Amblyomma is polyphyletic. Molecular Phylogenetics andEvolution, 64, 45–55.
Choi, E. H., Park, S. J., Jang, K. H. & Hwang, W. (2007). Com-plete mitochondrial genome of a Chinese scorpion Mesobuthusmartensii (Chelicerata, Scorpiones, Buthidae). DNA Sequence, 18,459–471.
Clayton, D. A. (1982). Replication of animal mitochondrial DNA.Cell, 28, 693–705.
Crease, T. J. (1999). The complete sequence of the mitochondrialgenome of Daphnia pulex (Cladocera: Crustacea). Gene, 233, 89–99.
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. (2011).ProtTest 3: fast selection of best-fit models of protein evolution.Bioinformatics, 27, 1164–1165.
Davila, S., Pinero, D., Bustos, P., Cevallos, M. A. & Davila, G.(2005). The mitochondrial genome sequence of the scorpionCentruroides limpidus (Karsch 1879) (Chelicerata: Arachnida).Gene, 360, 92–102.
Dermauw, W., Van Leeuwen, T., Vanholme, B. & Tirry, L.(2009). The complete mitochondrial genome of the house dustmite Dermatophagoides pteronyssinus (Trouessart): a novel genearrangement among arthropods. BMC Genomics, 10, 107.
Dirheimer, G., Keith, G., Dumas, P. & Westhof, E. (1995). tRNAstructure, biosynthesis, and function. In D. S€oll & U. RajBhan-dary (Eds) Primary, Secondary and Tertiary Structures of TRNA(pp. 93–126). Washington, DC: American Society for Microbiol-ogy Press.
Domes, K., Maraun, M., Scheu, S. & Cameron, S. L. (2008). Thecomplete mitochondrial genome of the sexual oribatid mite Ste-ganacarus magnus: genome rearrangements and loss of tRNAs.BMC Genomics, 9, 532.
Dowton, M. (1999). Relationships among the cyclostome braconid(Hymenoptera: Braconidae) subfamilies inferred from a mito-chondrial tRNA gene rearrangement. Molecular Phylogenetics andEvolution, 11, 283–287.
Dowton, M., Castro, L. R. & Austin, A. D. (2002). Mitochondrialgene rearrangements as phylogenetic characters in the inverte-brates: the examination of genome ‘morphology’. InvertebrateSystematics, 16, 345–356.
Dunlop, J. A. & Alberti, G. (2008). The affinities of mites andticks: a review. Journal of Zoological Systematics and EvolutionaryResearch, 46, 1–18.
Dunlop, J. A. & Braddy, S. J. (2001). Scorpions and their sis-ter-group relationships. In V. Fet & P. A. Selden (Eds) Scorpions(pp. 1–24). London: British Arachnological Society.
Ernsting, B. R., Edwards, D. D., Aldred, K. J., Fites, J. S. & Neff,C. R. (2009). Mitochondrial genome sequence of Unionicola foili(Acari: Unionicolidae): a unique gene order with implications forphylogenetic inference. Experimental and Applied Acarology, 49,305–316.
Fahrein, K., Talarico, G., Braband, A. & Podsiadlowski, L. (2007).The complete mitochondrial genome of Pseudocellus pearsei(Chelicerata: Ricinulei) and a comparison of mitochondrial generearrangements in arachnida. BMC Genomics, 8, 36.
Fujita, M. K., Boore, J. L. & Moritz, C. (2007). Multiple originsand rapid evolution of duplicated mitochondrial genes in parthe-nogenetic geckos (Heteronotia binoei; Squamata, Gekkonidae).Molecular Biology and Evolution, 24, 2775–2786.
Giribet, G., Edgecombe, G. D., Wheeler, W. C. & Babbitt, C.(2002). Phylogeny and systematic position of Opiliones: a com-bined analysis of chelicerate relationships using morphologicaland molecular data. Cladistics, 18, 5–70.
Gouy, M., Guindon, S. & Gascuel, O. (2010). Seaview version 4: amultiplatform graphical user interface for sequence alignmentand phylogenetic tree building. Molecular Biology and Evolution,27, 221–224.
Harvey, M. S. (2002). The neglected cousins: what do we knowabout the smaller arachnid orders? Journal of Arachnology, 30,357–372.
Hassanin, A. (2006). Phylogeny of Arthropoda inferred from mito-chondrial sequences: strategies for limiting the misleading effectsof multiple changes in pattern and rates of substitution. Molecu-lar Phylogenetics and Evolution, 38, 100–116.
Hu, M., Jex, A. R., Campbell, B. E. & Gasser, R. B. (2007). LongPCR amplification of the entire mitochondrial genome fromindividual helminths for direct sequencing. Nature Protocols, 2,2339–2344.
Huelsenbeck, J. P. & Ronquist, F. (2001). Mrbayes: Bayesian infer-ence of phylogenetic trees. Bioinformatics, 17, 754–755.
Jeyaprakash, A. & Hoy, M. A. (2007). The mitochondrial genomeof the predatory mite Metaseiulus occidentalis (Arthropoda:Chelicerata: Acari: Phytoseiidae) is unexpectedly large and con-tains several novel features. Gene, 391, 264–274.
Jobb, G., von Haeseler, A. & Strimmer, K. (2004). Treefinder: apowerful graphical analysis environment for molecular phyloge-netics. BMC Evolutionary Biology, 4, 18.
Jones, M., Gantenbein, B., Fet, V. & Blaxter, M. (2007). Theeffect of model choice on phylogenetic inference using mito-chondrial sequence data: lessons from the scorpions. MolecularPhylogenetics and Evolution, 43, 583–595.
Kolpakov, R., Bana, G. & Kucherov, G. (2003). mreps: efficientand flexible detection of tandem repeats in DNA. Nucleic AcidsResearch, 31, 3672–3678.
Komoto, N., Yukuhiro, K. & Tomita, S. (2012). Novel gene rear-rangements in the mitochondrial genome of a webspinner, Apos-thonia japonica (Insecta: Embioptera). Genome, 55, 222–233.
Laforest, M. J., Roewer, I. & Lang, B. F. (1997). MitochondrialtRNAs in the lower fungus Spizellomyces punctatus: tRNA editingand UAG ‘stop’ codons recognized as leucine. Nucleic AcidsResearch, 25, 626–632.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R.,McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W andClustal X version 2.0. Bioinformatics, 23, 2947–2948.
188 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.

Lavrov, D. V., Boore, J. L. & Brown, W. M. (2000a). Thecomplete mitochondrial DNA sequence of the horseshoe crabLimulus polyphemus. Molecular Biology and Evolution, 17, 813–824.
Lavrov, D. V., Brown, W. M. & Boore, J. L. (2000b). A novel typeof RNA editing occurs in the mitochondrial tRNAs of thecentipede Lithobius forficatus. Proceedings of the National Academyof Sciences of the United States of America, 97, 13738–13742.
Leigh, J. & Lang, B. F. (2004). Mitochondrial 3’ tRNA editing inthe jakobid Seculamonas ecuadoriensis: a novel mechanism andimplications for tRNA processing. RNA, 10, 615–621.
Li, W. (1997). Molecular Evolution. Sunderland, MA: SinauerAssociates Incorporated.
Li, H., Gao, J., Liu, H. & Cai, W. (2009). Progress in theresearches on insect mitochondrial genome and analysis of geneorder. Science Foundation in China, 17, 39–45.
Lowe, T. M. & Eddy, S. R. (1997). tRNAscan-SE: a program forimproved detection of transfer RNA genes in genomic sequence.Nucleic Acids Research, 25, 955–964.
Lynch, M. (1996). Mutation accumulation in transfer RNAs:molecular evidence for Muller’s ratchet in mitochondrialgenomes. Molecular Biology and Evolution, 13, 209–220.
Marechaldrouard, L., Ramamonjisoa, D., Cosset, A., Weil, J. H. &Dietrich, A. (1993). Editing corrects mispairing in the acceptorstem of bean and potato mitochondrial phenylalanine transferRNAs. Nucleic Acids Research, 21, 4909–4914.
Masta, S. E. (2010). Mitochondrial rRNA secondary structures andgenome arrangements distinguish chelicerates: comparisons witha harvestman (Arachnida: Opiliones: Phalangium opilio). Gene,449, 9–21.
Masta, S. E. & Boore, J. L. (2004). The complete mitochondrialgenome sequence of the spider Habronattus oregonensis revealsrearranged and extremely truncated tRNAs. Molecular Biology andEvolution, 21, 893–902.
Masta, S. E. & Boore, J. L. (2008). Parallel evolution of truncatedtransfer RNA genes in arachnid mitochondrial genomes. Molecu-lar Biology and Evolution, 25, 949–959.
Masta, S. E., Klann, A. E. & Podsiadlowski, L. (2008). A compari-son of the mitochondrial genomes from two families of Solifugae(Arthropoda: Chelicerata): Eremobatidae and Ammotrechidae.Gene, 417, 35–42.
Masta, S. E., Longhorn, S. J. & Boore, J. L. (2009). Arachnid rela-tionships based on mitochondrial genomes: asymmetric nucleo-tide and amino acid bias affects phylogenetic analyses. MolecularPhylogenetics and Evolution, 50, 117–128.
Morrison, C. L., Harvey, A. W., Lavery, S., Tieu, K., Huang, Y.& Cunningham, C. W. (2002). Mitochondrial gene rearrange-ments confirm the parallel evolution of the crab-like form.Proceedings of the Royal Society B-Biological Sciences, 269, 345–350.
Murrell, A., Campbell, N. J. H. & Barker, S. C. (2003). The valueof idiosyncratic markers and changes to conserved tRNAsequences from the mitochondrial genome of hard ticks (Acari:Ixodida: Ixodidae) for phylogenetic inference. Systematic Biology,52, 296–310.
Nardi, F., Spinsanti, G., Boore, J. L., Carapelli, A., Dallai, R. &Frati, F. (2003). Hexapod origins: monophyletic or paraphyletic?Science, 299, 1887–1889.
Ojala, D., Montoya, J. & Attardi, G. (1981). tRNA punctuationmodel of RNA processing in human mitochondria. Nature, 290,470–474.
Ovchinnikov, S. & Masta, S. E. (2012). Pseudoscorpion mitochon-dria show rearranged genes and genome-wide reductions ofRNA gene sizes and inferred structures, yet typical nucleotidecomposition bias. BMC Evolutionary Biology, 12, 31.
Perna, N. T. & Kocher, T. D. (1995). Patterns of nucleotide com-position at fourfold degenerate sites of animal mitochondrial ge-nomes. Journal of Molecular Evolution, 41, 353–358.
Platnick, N. I. (2014). The world spider catalog, version 15. Avail-able via http://research.amnh.org/entomology/spiders/catalog/index.html
Qiu, Y., Song, D. X., Zhou, K. Y. & Sun, H. Y. (2005). The mito-chondrial sequences of Heptathela hangzhouensis and Ornithoctonushuwena reveal unique gene arrangements and atypical tRNAs.Journal of Molecular Evolution, 60, 57–71.
Regier, J. C., Shultz, J. W., Zwick, A., Hussey, A., Ball, B.,Wetzer, R., et al. (2010). Arthropod relationships revealed byphylogenomic analysis of nuclear protein-coding sequences.Nature, 463, 1079–1098.
Schutz, D., Taborsky, M. & Drapela, T. (2007). Air bells ofwater spiders are an extended phenotype modified in responseto gas composition. Journal of Experimental Zoology, 307A,549–555.
Shao, R. F., Mitani, H., Barker, S. C., Takahashi, M. & Fukunaga,M. (2005). Novel mitochondrial gene content and gene arrange-ment indicate illegitimate inter-mtDNA recombination in thechigger mite Leptotrombidium pallidum. Journal of MolecularEvolution, 60, 764–773.
Shultz, J. W. (1990). Evolutionary morphology and phylogeny ofArachnida. Cladistics, 6, 1–38.
Shultz, J. W. (2007). A phylogenetic analysis of the arachnidorders based on morphological characters. Zoological Journal ofthe Linnean Society, 150, 221–265.
Simon, C., Buckley, T. R., Frati, F., Stewart, J. B. & Beckenbach,A. T. (2006). Incorporating molecular evolution into phyloge-netic analysis, and a new compilation of conserved polymerasechain reaction primers for animal mitochondrial DNA. AnnualReview of Ecology Evolution and Systematics, 37, 545–579.
Staton, J. L., Daehler, L. L. & Brown, W. M. (1997). Mitochon-drial gene arrangement of the horseshoe crab Limulus polyphemusL.: conservation of major features among arthropod classes.Molecular Biology and Evolution, 14, 867–874.
Sumida, M., Kanamori, Y., Kaneda, H., Kato, Y., Nishioka, M.,Hasegawa, M., et al. (2001). Complete nucleotide sequence andgene rearrangement of the mitochondria genome of the Japanesepond frog Rana nigromaculata. Genes & Genetic Systems, 76,311–325.
Talavera, G. & Castresana, J. (2007). Improvement of phyloge-nies after removing divergent and ambiguously aligned blocksfrom protein sequence alignments. Systematic Biology, 56,564–577.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. &Kumar, S. (2011). MEGA5: molecular evolutionary geneticsanalysis using maximum likelihood, evolutionary distance, andmaximum parsimony methods. Molecular Biology and Evolution,28, 2731–2739.
Tomita, K., Ueda, T. & Watanabe, K. (1996). RNA editing in theacceptor stem of squid mitochondrial tRNATyr. Nucleic AcidsResearch, 24, 4987–4991.
ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190 189
M. Liu et al. � Mitochondrial genome of the water spider

Walter, D. E. & Proctor, H. C. (1999). Mites: Ecology, Evolutionand Behaviour. Sydney, NSW: University of New South WalesPress.
Walter, D. E., Krantz, G. & Lindquist, E. (1996) Acari. The mites.Version 13. Available via http://tolweb.org/Acari/2554/1996.12.13
Weygoldt, P. & Paulus, H. (1979). Unteruschungen zur morphol-ogie, taxonomie und phylogenie der chelicerata. Ii. Clado-gramme und die enfaltung der chelicerata. Zeitschrift f€urZoologische Systematik und Evolutionsforschung, 17, 117–200.
Wheeler, W. C. & Hayashi, C. Y. (1998). The phylogeny of theextant chelicerate orders. Cladistics, 14, 173–192.
Wilson, K., Cahill, V., Ballment, E. & Benzie, J. (2000). The com-plete sequence of the mitochondrial genome of the crustaceanPenaeus monodon: are malacostracan crustaceans more closelyrelated to insects than to branchiopods? Molecular Biology andEvolution, 17, 863–874.
Wolstenholme, D. R. (1992). Animal mitochondrial DNA:structure and evolution. International Review of Cytology, 141,173–216.
Yamazaki, N., Ueshima, R., Terrett, J. A., Yokobori, S., Kaifu, M.,Segawa, R., et al. (1997). Evolution of pulmonate gastropodmitochondrial genomes: comparisons of gene organizations ofEuhadra, Cepaea and Albinaria and implications of unusualtRNA secondary structures. Genetics, 145, 749–758.
Yuan, M. L., Wei, D. D., Wang, B. J., Dou, W. & Wang, J. J.(2010). The complete mitochondrial genome of the citrusred mite Panonychus citri (Acari: Tetranychidae): high genomerearrangement and extremely truncated tRNAs. BMC Genomics,11, 597.
Zhang, D. X. & Hewitt, G. M. (1997). Insect mitochondrialcontrol region: a review of its structure, evolution and usefulnessin evolutionary studies. Biochemical Systematics and Ecology, 25,99–120.
Zuker, M. (2003). Mfold web server for nucleic acid foldingand hybridization prediction. Nucleic Acids Research, 31,3406–3415.
Supporting InformationAdditional Supporting Information may be found in theonline version of this article:Fig. S1. Putative stem-loop structures of the 135 bp
repeat unit at the 50 end of control region; secondary struc-ture of the 363 bp repeat unit at the 30 end of controlregion was not given.Table S1. Primers used to amplify A. aquatica mitochon-
drial genome.Table S2. All available Arachnida taxa and outgroups
included in this study and their corresponding GenBankaccession numbers.Table S3. Mitochondrial genome organization, gene
lengths, start and stop codons of A. aquatica.Data S1. Inferred secondary structures of the mitochon-
drial 22 tRNAs from A. aquatica.Data S2. Summary of the detailed interpretation of all
CREx mitochondrial gene rearrangement scenarios.
190 ª 2014 Royal Swedish Academy of Sciences, 44, 2, March 2015, pp 179–190
Mitochondrial genome of the water spider � M. Liu et al.
Related Documents











