The Ministry of Health, Labour and Welfare Ministerial Notiˆcation No. 461 In accordance with the provisions of Article 41, Paragraph 1 of the Pharmaceutical AŠairs Law (Law No. 145, 1960), we hereby revise a part of the Japanese Phar- macopoeia (Ministerial Notiˆcation No. 111, 2001) as follows, and the revised Japanese Pharmacopoeia shall come into eŠect on January 1, 2005, [including dele- tion from O‹cial Monographs fro Part II in The Japanese Pharmacopoeia, Four- teenth Edition of the articles of Absorbent Cotton, Puriˆed Absorbent Cotton, Sterile Absorbent Cotton, Sterile Puriˆed Absorbent Cotton and Absorbent Gauze and Sterile Absorbent Gauze (hereinafter referred to as ``sanitary materials'')]. Provi- so: With respect to the drugs which are included in the Japanese Pharmacopoeia (hereinafter referred to as ``the old Japanese Pharmacopoeia'') [limited to those included in the Japanese Pharmacopoeia whose standards are changed with this notiˆcation published (hereinafter referred to as ``the new Japanese Phar- macopoeia'')] and those which are approved as of January 1, 2005 pursuant to the provisions of Article 14, Paragraph 1 of this Law (including cases where it shall apply mutatis mutandis under Article 23 of this Law; the same hereinafter) [including the drugs designated as those exempted from approval (hereinafter referred to as ``the drugs exempted from approval'') among the drugs etc. designated by the Minister of Health, Labour and Welfare as those exempted from manufacturing or import approval pursuant to the provisions of Article 14, Paragraph 1 of the Pharmaceutical AŠairs Law (Ministerial Notiˆcation No. 104, 1994), the standards established in the old Japanese Pharmacopoeia (limited to the standards for the relevant drugs) shall be recognized, up to June 30, 2006, as the standards established in the new Japanese Pharmacopoeia. With respect the drugs which are included in the new Japanese Pharmacopoeia (excluding those which are included in the old Japanese Phar- macopoeia) and those which are approved as of January 1, 2005 pursuant to the provisions of Article 14, Paragraph 1 of this Law (including the drugs exempted from approval), the drugs may be treated, up to June 30, 2006, as those which are not included in the new Japanese Pharmacopoeia. Further, sanitary materials may be treated, up to September 30, 2006, under the previous regulation. Hidehisa Otsuji The Minister of Health, Labour and Welfare December 28, 2004 (The texts referred to by the term ``as follows'' are omitted here. All of them are made available for public exhibition at the Evaluation and Licensing Division, Phar- maceutical and Medical Safety Bureau, Ministry of Health, Labour and Welfare, at each Regional Bureau of Health and Welfare and at each Prefectural O‹ce in Japan.) The term ``as follows'' here indicates the contents from Part I to Ultraviolet-visible Reference SpectraintheSupplementIItotheJapanesePharmacopoeiaFourteenthEdition(pp.1669 – 1866).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Ministry of Health, Labour andWelfare Ministerial Notiˆcation No. 461
In accordance with the provisions of Article 41, Paragraph 1 of the PharmaceuticalAŠairs Law (Law No. 145, 1960), we hereby revise a part of the Japanese Phar-macopoeia (Ministerial Notiˆcation No. 111, 2001) as follows, and the revisedJapanese Pharmacopoeia shall come into eŠect on January 1, 2005, [including dele-tion from O‹cial Monographs fro Part II in The Japanese Pharmacopoeia, Four-teenth Edition of the articles of Absorbent Cotton, Puriˆed Absorbent Cotton,Sterile Absorbent Cotton, Sterile Puriˆed Absorbent Cotton and Absorbent Gauzeand Sterile Absorbent Gauze (hereinafter referred to as ``sanitary materials'')]. Provi-so: With respect to the drugs which are included in the Japanese Pharmacopoeia(hereinafter referred to as ``the old Japanese Pharmacopoeia'') [limited to thoseincluded in the Japanese Pharmacopoeia whose standards are changed with thisnotiˆcation published (hereinafter referred to as ``the new Japanese Phar-macopoeia'')] and those which are approved as of January 1, 2005 pursuant to theprovisions of Article 14, Paragraph 1 of this Law (including cases where it shall applymutatis mutandis under Article 23 of this Law; the same hereinafter) [including thedrugs designated as those exempted from approval (hereinafter referred to as ``thedrugs exempted from approval'') among the drugs etc. designated by the Minister ofHealth, Labour and Welfare as those exempted from manufacturing or importapproval pursuant to the provisions of Article 14, Paragraph 1 of the PharmaceuticalAŠairs Law (Ministerial Notiˆcation No. 104, 1994), the standards established in theold Japanese Pharmacopoeia (limited to the standards for the relevant drugs) shall berecognized, up to June 30, 2006, as the standards established in the new JapanesePharmacopoeia. With respect the drugs which are included in the new JapanesePharmacopoeia (excluding those which are included in the old Japanese Phar-macopoeia) and those which are approved as of January 1, 2005 pursuant to theprovisions of Article 14, Paragraph 1 of this Law (including the drugs exempted fromapproval), the drugs may be treated, up to June 30, 2006, as those which are notincluded in the new Japanese Pharmacopoeia. Further, sanitary materials may betreated, up to September 30, 2006, under the previous regulation.
Hidehisa OtsujiThe Minister of Health, Labour and Welfare
December 28, 2004
(The texts referred to by the term ``as follows'' are omitted here. All of them aremade available for public exhibition at the Evaluation and Licensing Division, Phar-maceutical and Medical Safety Bureau, Ministry of Health, Labour and Welfare, ateach Regional Bureau of Health and Welfare and at each Prefectural O‹ce in Japan.)
The term ``as follows'' here indicates the contents from Part I to Ultraviolet-visible ReferenceSpectraintheSupplementIItotheJapanesePharmacopoeiaFourteenthEdition(pp.1669 – 1866).

CONTENTS
Preface ...................................................... iSupplement II to The Japanese Pharmacopoeia,Fourteenth Edition, Part I................. 1669–1754
General Tests, Processes and Apparatus ... 16696. Bacterial Endotoxins Test................. 1669
39. Nuclear Magnetic ResonanceSpectroscopy ................................. 1669
47. Pyrogen Test ................................. 167252. Residue on Ignition Test .................. 167253. Speciˆc Surface Area Determination... 167354. Sterility Test .................................. 167370. Reference Standards; Reagents, Test Solu-
tions; Standard Solutions for VolumetricAnalysis; Standard Solutions; MatchingFluids for Color; Optical Filters forWavelength and Transmission RateCalibration; and Measuring Instruments,Appliances .................................... 1677(1) Reference Standards ................. 1677(2) Reagents, Test Solutions............ 1677(3) Standard Solutions for Volumetric
Analysis ................................. 168574. Powder Particle Density
Determination................................ 1685O‹cial Monographs ............................. 1687
Supplement II to The Japanese Pharmacopoeia,Fourteenth Edition, Part II................ 1755–1778
General Rules for Crude Drugs ............... 1755O‹cial Monographs ............................. 1757
Infrared Reference Spectra ................ 1779–1789Part I ................................................ 1779Part II ............................................... 1789
Ultraviolet-visible Reference Spectra .... 1791–1799Part I ................................................ 1791
General Information5. International Harmonization Implemented
in the Japanese Pharmacopoeia FourteenthEdition ......................................... 1801
20. Amino Acid Analysis ...................... 181421. Capillary Electrophoresis.................. 182222. Isoelectric Focusing......................... 182823. Peptide Mapping ............................ 183024. Rapid Identiˆcation of Microorganisms
Based on Molecular BiologicalMethod ........................................ 1833
25. Solid and Particle Densities .............. 183526. Total Protein Assay ........................ 1836
Index.................................................... 1841Index in Japanese.................................... 1857

ii
Preface
The Fourteenth Edition of the Japanese Phar-macopoeia was promulgated on March 30, 2001 byMinisterial Notiˆcation No. 111 of the Ministry ofHealth, Labour and Welfare. To keep pace withprogress in medical and pharmaceutical sciences, inNovember 2001, the Council, at a meeting of theCommittee on Japanese Pharmacopoeia (JP), estab-lished the basic principles for the preparation of theJP Fifteenth Edition, setting out the characteristicsand roles of the JP, the deˆnite measures for therevision, the date of the revision, and the organizationof the Subcommittee on JP.
At the above meeting, the following ``ˆve pillars''were established as the basic principles of the JP:Making it more substantial by including all drugswhich are important from the viewpoint of health careand medical treatment; Making prompt partialrevision as necessary and facilitating smooth adminis-trative operation; Promoting international harmoni-zation; Ensuring transparency regarding the revisionand dissemination to the public of the JP; andPositively introducing contemporary analytical testsand developing reference standards. It was decided atthe meeting that each panel set up under the Subcom-mittee on JP should make eŠorts, on the basis of theseprinciples, to ensure that the JP is used more eŠective-ly in the ˆelds of health care and medical treatment bytaking appropriate measures, including getting theunderstanding and cooperation of other parties con-cerned.
The JP should comprise an o‹cial standard beingrequired to assure the quality of drugs in this countryin response to the progress in science and technologyand clinical demands at the time, it should deˆne thestandards for speciˆcations as well as the methods oftests to assure the overall quality of all drugs inprinciple, and it should have a role in clarifying thecriteria for quality of drugs which are recognized to beimportant from the viewpoint of medical treatment.
At the same time, it was agreed that the JP shouldbe prepared with the aid of the knowledge andexperience of many persons involved in the phar-maceuticals, that it should have the characteristics ofan o‹cial standard, which might be widely used by allparties concerned, that it should provide informationand understanding about the quality of drugs to thepublic, and that it should be conducive to smooth andeŠective government control of the quality of drugs,
and to securing and maintaining international con-sistency.
It was also agreed that JP articles should coverdrugs which are important from the viewpoint ofhealth care and medical treatment, clinical results andfrequency of use, as soon as possible after they reachthe market.
It was also decided to make a deˆnite rule for selec-tion of articles by clarifying the standards for selec-tion. The rule was shown in the verdict ``What thefuture Japanese Pharmacopoeia should be'' by thePharmaceutical AŠairs and Food Sanitation Council(PAFSC) on December 2002. The JP FifteenthEdition was decided to be slated for completion inApril 2006.
The panels on JP was reorganized into the followingeleven panels in accordance with the recommendationof the PAFSC: Panel on Planning and Revisions;Panel on Nomenclature for Pharmaceuticals; Panelon Excipients; Panel on Physico-chemical Tests; Panelon Medicinal Chemicals; Panel on BiologicallyDerived Drugs; Panel on Biological Tests; Panel onAntibiotics; Panel on Crude Drugs; Provisional Panelon Planning and Revisions and Panel on Phar-macopoeial Harmonization (PDG), followed by theestablishment of new panels: Panel on Water forPharmaceutical Preparations, Panel on JP ReferenceStandards, and three working groups under Panel onMedicinal Chemicals to expedite discussion of revisiondrafts of drug monographs.
In the Committee on Japanese Pharmacopoeia,Mitsuru Uchiyama took the role of chairman fromJanuary 2001 to December 2002, Tadao Terao fromJanuary to June 2003, and Takao Hayakawa fromJuly 2003 to December 2004.
It was decided that the JP will be revised not onlyevery ˆve years, in line with the basic principles for thepreparation of the JP Fifteenth Edition, but also asnecessary to take account of recent progress of scienceand in the interests of international harmonization.
In accordance with the revision principles, thepanels continued discussions on selection of articles,and revisions for General Notices, General Rules forPreparations, General Tests, and monographs ondrugs, and the supplement I to JP 14 was promulgatedon December 2002. After the promulgation of thesupplement I, the panels continued discussions to takeaccount of the progress of science and international

iiii Supplement II, JPXIVPreface
harmonization.Draft revisions covering subjects in General Rules
for Crude Drugs, General Tests, and monographs ondrugs, for which discussions were ˆnished betweenMarch 2002 and December 2003, were prepared for asupplement to the book. They were examined by theCommittee on Japanese Pharmacopoeia in September2004, followed by the PAFSC in December 2004, andthen submitted to the Minister of Health, Labour andWelfare.
Numbers of discussions in the panels to preparesupplement drafts were as follows: Panel on Planningand Revisions, 9 times; Panel on Nomenclature forPharmaceuticals, 10 times; Panel on Excipients, 11times; Panel on Physico-chemical Tests, 30 times;Panel on Medicinal Chemicals (including the workinggroups), 24 times; Panel on Biologically DerivedDrugs, 11 times; Panel on Biological Tests, 10 times;Panel on Antibiotics, 19 times; Panel on Crude Drugs,19 times; Provisional Panel on Planning and Revi-sions, 12 times; Panel on Pharmacopoeial Harmoniza-tion (PDG), 9 times; Panel on Water for Pharmaceuti-cal Preparations, 2 times; Panel on JP ReferenceStandards, 3 times.
It should be noted that in the preparation of thedrafts for the Supplement I, generous cooperation wasgiven by the Technical Committee of the Pharmaceuti-cal Manufacturer's Association of Tokyo and ofOsaka, the Crude Drugs Association of Tokyo, theJapan Pharmaceutical Excipients Council, theFederation of Crude Drugs Associations of Japan, theJapan Antibiotics Research Association, the JapanFlavor and Fragrance Manufacturer's Association,the Japan Medical Plants Federation, the JapanPharmaceutical Manufacturer's Association, theJapanese Society of Hospital Pharmacists, the JapanPharmaceutical Association, and the Japan OilseedProcessors Association.
In consequence of this revision, the JP FourteenthEdition carries 907 articles in Part I owing to theaddition of 27 articles and the deletion of 1 article; and484 articles in Part II owing to the addition of 12articles and the deletion of 9 articles.
The principles of description and the salient pointsof the revision in this volume are as follows:
1. The Supplement II to JP Fourteenth Editioncomprises the following items, in order: Notiˆcationof the Ministry of Health, Labour and Welfare;Contents; Preface; followed by General Tests, Proc-esses and Apparatus; monographs on drugs in Part I,and General Rules for Crude Drugs; monographs ondrugs in Part II, then followed by Ultraviolet-visual
Reference Spectra in Part I; Infrared ReferenceSpectra and Ultraviolet-visible Reference Spectra inPart II; General Information, and as an appendix, aCumulative Index containing references to the mainvolume, the supplement I and the supplement II.
2. The articles in General Tests, Processes andApparatus, Monographs on Drugs, Infrared Refer-ence Spectra and Ultraviolet-visible Reference Spectraare respectively placed in alphabetical order.
3. The following items in each monograph are putin the order shown below, except that unnecessaryitems are omitted depending on the nature of the drug:(1) English title(2) Commonly used name(s)(3) Latin title (only for Crude Drugs)(4) Title in Japanese(5) Structural formula or empirical formula(6) Molecular formula and molecular mass(7) Chemical name(8) Origin(9) Limits of the content of the ingredient(s)
and/or the unit of potency(10) Labeling requirements(11) Method of preparation(12) Description(13) Identiˆcation tests(14) Speciˆc physical and/or chemical values(15) Purity tests(16) Loss on drying, loss on ignition, and/or water(17) Residue on ignition, total ash, and/or acid-in-
soluble ash(18) Tests being required for pharmaceutical prepa-
rations and other special tests(19) Isomer ratio(20) Assay or the content of the ingredient(s)(21) Containers and storage(22) Expiration date(23) Others
4. In each monograph on a drug, the followingphysical and chemical values representing the proper-ties and quality of the drug are given in the order indi-cated below, except that unnecessary items are omitteddepending on the nature of the drug:(1) Alcohol number(2) Absorbance(3) Congealing point(4) Refractive index(5) Osmolarity(6) Optical rotation(7) Viscosity(8) pH(9) Speciˆc gravity
(10) Boiling point

iiiiiiSupplement II, JPXIV Preface
(11) Melting point(12) Acid value(13) Saponiˆcation value(14) Ester value(15) Hydroxyl value(16) Iodine value
5. Identiˆcation tests comprise the followingitems, which are generally put in the order given be-low:(1) Coloration reactions(2) Precipitation reactions(3) Decomposition reactions(4) Derivatives(5) Visible, ultraviolet or infrared spectra(6) Special reactions(7) Cations(8) Anions
6. Purity tests comprise the following items, whichare generally put in the order given below, except thatunnecessary items are omitted depending on thenature of the drug:(1) Color(2) Odor(3) Clarity and/or color of solution(4) Acidity or alkalinity(5) Acid(6) Alkali(7) Chloride(8) Sulfate(9) Sulˆte
(10) Nitrate(11) Nitrite(12) Carbonate(13) Bromide(14) Iodide(15) Soluble halide(16) Thiocyanide(17) Selenium(18) Cationic salts(19) Ammonium(20) Heavy metals(21) Iron(22) Manganese(23) Chromium(24) Bismuth(25) Tin(26) Aluminum(27) Zinc(28) Cadmium(29) Mercury(30) Copper(31) Lead
(32) Silver(33) Alkaline earth metals(34) Arsenic(35) Foreign matter(36) Related substances(37) Residual solvent(38) Other mixtures(39) Readily carbonizable substances
7. The following items of the General Tests, Proc-esses and Apparatus were partially revised:
Bacterial Endotoxins TestNuclear Magnetic Resonance Spectroscopy (1H)Pyrogen TestResidue on Ignition TestSpeciˆc Surface Area DeterminationSterility Test
8. The following test was added to the GeneralTests, Processes and Apparatus:
Powder Particle Density Determination
9. The following Reference Standard was deleted:Digitalis
10. The following Reference Standards wereadded:
AzithromycinCisplatinEtoposideFurosemideMethylprednisolone SuccinateNilvadipineThiamylalTranexamic AcidTrichlormethiazide
11. English and Latin titles of drugs are derived,in principle, from International NonproprietaryNames (INN) for Pharmaceutical Substances recom-mended by the World Health Organization. Japanesetitles are derived from the Japanese version of thisbook. The chemical names are based on the rules ofthe International Union of Pure and Applied Chemis-try (IUPAC).
12. Molecular formulas of organic compoundsbegin with C and then H, followed by other involvedelements in the alphabetical order of the symbols ofthe elements.
13. Structural formulas of drugs represent, as faras possible, steric conˆgurations.
14. Test procedures in monographs in Part I are,in principle, written in full even in correspondingmonographs in Part II, and vice versa. The test proce-

iviv Supplement II, JPXIVPreface
dures in monographs for preparations are also writtenin full even within the same part, except in the mono-graphs for preparations having a correspondingmonograph of their principal material substances.
15. The following articles were deleted fromO‹cial MonographPart I
Santonin Tablets
Part IIAbsorbent CottonPuriˆed Absorbent CottonSterile Absorbent CottonSterile Puriˆed Absorbent CottonAbsorbent GauzeSterile Absorbent GauzeAdhesive PlasterDigitalisPowdered Digitalis
16. The following articles were newly added too‹cial monographs:Part I
AlprostadilAzithromycin HydrateBenidipine HydrochlorideBenidipine Hydrochloride TabletsCefepime Dihydrochloride for InjectionCisplatinEperisone HydrochlorideEtoposideFlomoxef Sodium for InjectionFlopropione CapsulesFurosemide TabletsGlutathioneMethylprednisolone SuccinateMetoclopramide TabletsNicorandilNilvadipineNilvadipine TabletsOxytocinPirenzepine Hydrochloride HydratePiroxicamSerrapeptaseTiaramide Hydrochloride TabletsTizanidine HydrochlorideTranexamic Acid CapsulesTranexamic Acid InjectionTranexamic Acid TabletsTrichlormethiazide Tablets
Part IICnidium Monnieri FruitEpimedium Herb
Lindera RootLonicera Leaf and StemLycium BarkLycium FruitProcessed Aconite RootPowdered Processed Aconite RootProcessed GingerSappan WoodTermericTribulus Fruit
17. The following monographs were revised incommonly used name:Part I
Calcium Folinate
Part IICellulose Acetate Phthalate
18. The following monographs were revised instructural formula and in chemical name:Part I
Cefuroxime AxetilMepivacaine Hydrochloride
19. The part of origin of the following mono-graphs were deleted:Part II
Chorionic GonadotrophinSerum Gonadotrophin
20. The following monographs were revised inorigin:Part I
AcetohexamideAtropine Sulfate InjectionBenzylpenicillin BenzathineClarithromycinColchicineCytarabineDigoxin InjectionDigoxin TabletsDimorpholamineDimorpholamine InjectionEthionamideEtilefrine Hydrochloride TabletsFosfomycin CalciumFosfomycin SodiumFurosemidedl-Methylephedrine HydrochlorideMethyltestosteroneMethyltestosterone TabletsOxytocin InjectionPyridoxine HydrochloridePyridoxine Hydrochloride InjectionSodium Aurothiomalate

vvSupplement II, JPXIV Preface
Sodium ThiosulfateTestosterone Propionate InjectionThiamylal SodiumThiamylal Sodium for InjectionTinidazoleTranexamic AcidTrichlormethiazideVinblastine Sulfate for Injection
Part IIBenzyl AlcoholCellulose Acetate PhthalateCorn StarchOpium Alkaloids HydrochloridesPowdered AloePowdered Gardenia FruitPowdered Peony RootPowdered RhubarbPowdered Scutellaria RootRhubarbUncaria Thorn
21. The following monographs were revised byaddition or change in Method of preparation:Part I
Oxytocin Injection
Part IIChorionic GonadotrophinSerum Gonadotrophin
22. The following monographs were revised byaddition or change in Description and in test(s):Part I
AcetohexamideAnhydrous Citric acidAtropine Sulfate InjectionBenzylpenicillin BenzathineCefuroxime SodiumCiclosporinClarithromycinColchicineCytarabineDeferoxamine MesilateDigoxinDigoxin InjectionDigoxin TabletsDimorpholamineDimorpholamine InjectionEthionamideEtilefrine Hydrochloride TabletsFlavin Adenine Dinucleotide SodiumFosfomycin CalciumFosfomycin SodiumFurosemide
Heparin SodiumHeparin Sodium InjectionKallidinogenaseD-MannitolMeropenem Trihydratedl-Methylephedrine Hydrochloride10z dl-Methylephedrine Hydrochloride PowderMethyltestosteroneMethyltestosterone TabletsOxytocin InjectionPirenoxinePyrazinamidePyridoxine HydrochloridePyridoxine Hydrochloride InjectionSodium AurothiomalateSodium Polystyrene SulfonateSodium ThiosulfateTegafurTestosterone PropionateTestosterone Propionate InjectionThiamine HydrochlorideThiamylal SodiumThiamylal Sodium for InjectionTinidazoleTranexamic AcidTrichlormethiazideVasopressin InjectionVinblastine SulfateVinblastine Sulfate for Injection
Part IIBenzyl AlcoholCarmellose CalsiumCellulose Acetate PhthalateChorionic GonadotrophinChorionic Gonadotrophin for InjectionChrysanthemum FlowerCorn StarchCornus FruitGingerOpium Alkaloids HydrochloridesPotato StarchPowdered AloePowdered Gardenia FruitPowdered GingerPowdered Peony RootSerum GonadotrophinSerum Gonadotrophin for InjectionUncaria ThornWheat Starch
23. The following monographs were revised inIdentiˆcation by addition or change based on adop-tion of the Ultraviolet-visible Reference Spectra:

vivi Supplement II, JPXIVPreface
Part IAlprostadilBenidipine HydrochlorideCisplatinColchicineCytarabineEperisone HydrochlorideEthionamideEtoposideFurosemidedl-Methylephedrine HydrochlorideMethylprednisolone SuccinateMethyltestosteroneNilvadipineOxytocinPirenzepine Hydrochloride HydratePiroxicamPyridoxine HydrochlorideTestosterone PropionateThiamylal SodiumTizanidine HydrochlorideTrichlormethiazide
24. The following monographs were revised inIdentiˆcation by addition or change based on adop-tion of the Infrared Reference Spectra:Part I
AlprostadilAzithromycin HydrateBenidipine HydrochlorideCisplatinColchicineCytarabineDigoxinDimorpholamine
Eperisone HydrochlorideEthionamideEtoposideFurosemideGlutathionedl-Methylephedrine HydrochlorideMethylprednisolone SuccinateMethyltestosteroneNicorandilNilvadipinePirenzepine Hydrochloride HydratePiroxicamPyridoxine HydrochlorideTestosterone PropionateThiamylal SodiumTinidazoleTizanidine HydrochlorideTranexamic AcidTrichlormethiazideVinblastine Sulfate
Part IIBenzyl Alcohol
25. The following monographs were revised inContainers and storage:Part I
EthionamideVinblastine SulfateVinblastine Sulfate for Injection
Part IICorn StarchPotato StarchWheat Starch

viiviiSupplement II, JPXIV Preface
Those who were engaged in the preparation of theSupplement I to JP Fourteenth Edition are as follows:
Norio AimiMitsuo AokiKiichi AonukiNobuo AoyagiYoshichika ArakawaKeiko ArimotoKazuhide AshizawaShinichiro AsoYukio AsoHiroyuki FuchinoShigeyuki FujikuraGoro FunamotoAkihiro FurukawaYoshiharu GekiYukihiro GodaMorio HamashimaRuri HanajiriKouji HasegawaRyuichi HasegawaTakao Hayakawa*Masahiro HayashiKenji HiguchiKatsufuto HiramatsuFusayoshi HirayamaYukio HiyamaKunimoto HottaTakanori IchikawaNobukazu IgoshiKenichi InuiShigeru ItaiYuji ItoTakashi ItohShozo IwagamiAkemi KaiKazuaki KakehiTakemine KanaiNahoko KaniwaMotoko Kanke
Yoshiaki KatoNoriko KatoriNobuo KawaharaToru KawanishiToshiaki KawanishiNana KawasakiToshisuke KawasakiYoshiaki KawashimaKeiji KijimaTakao KiyoharaTakashi KobayashiToyohiko KobayashiMasayoshi KohaseShigeo KojimaHiroyasu KokuboEiichi KokueKatsuko KomatsuAkio KomuraToshifumi KondaSeizo KondoTakao KunisadaTakeshi KurataMasaaki KuriharaHaruo KuriyamaFumiyo KusuKumiko KusuyamaMasako MaedaMidori MakitaKeiichi MaruyamaToshio MasaokaToshihiko MatsubaraYoshihisa MatsudaNorio MatsukiShigeru MatsukiHayashi MatsukuraSatoshi MinobeHiroto MiyamotoNaoki Miyata
Michinao MizugakiTaiichi MizutaKaoru MorikawaOsamu MoritaTakashi MoritaToshimi MuraiHiroshi NakamuraTatsuya NakanoHiroyuki NakazawaMasaaki NaotsukaMasao NasuKoji NishijimaMotohiro NishijimaTatsumi NishiyamaTakashi NomotoYasuo OhnoYoshiro OhtaniMinoru OkadaSatoshi Okada**Kimiya OkazakiTsuneo OkuboHaruhiro OkudaMasami OtsukaEiji SakaiHideki SasakiTsuguo SasakiMotoyoshi SatakeAkihiro SatoMichiko SekiguchiSetsuko SekitaYasuo ShimadaKesamitsu ShimizuKyoko ShimuraFumitoshi ShinchoOsamu ShirotaKouichi ShudoHisashi SonobeShoko SueyoshiShinji Sugaya
Hisakazu SunadaHideyo SuzukiSenji SuzukiYoshikazu TakahashiTadahiro TakedaYasushi Takeda**Toyoshige TanabeHaruo TanakaToshihiro TanakaKenichi TanamotoTsuyoshi TanimotoSusumu TerabayashiTadao Terao*Reiko TeraokaHiroshi TokunagaKiyoshi TomiokaMotowo TomitaTatsuru TomizawaYosuke TsujiNobuchika TsumagariEriko UchidaMitsuru Uchiyama*Yoshimasa UeharaKazuichi UmemotoTakashi UnnoHaruo WatanabeMorimasa YagisawaTakehiko YajimaTeruhide YamaguchiKeiichi YamamotoKeiji YamamotoTosuke YamamotoKenichi YamazakiTakeshi YamazakiHikaru YodenChikako YomotaHitoo YoshidaKazumasa YoshikawaSumie Yoshioka
*Chairman, Committee on JP**Acting Chairman, Committee on JP

16691669
General Tests, Processesand Apparatus
Change the introduction to read:
General Tests, Processes and Apparatus includes commonmethods for tests and other articles related to them. Unlessotherwise speciˆed, the procedures for absorbance determi-nation, absorbance ratio determination, acid-neutralizingcapacity determination of gastrointestinal medicines,alcohol number determination, ammonium determination,arsenic determination, atomic absorption spectrophotomet-ry, test for bacterial endotoxins, boiling point determina-tion, distilling range determination, chloride determination,conductivity measurement, congealing point determination,test for content uniformity, determination of bulk andtapped densities, digestion test, disintegration test, dissolu-tion test, endpoint detection in titrimetry, ‰ame coloration,‰uorometry, foreign insoluble matter test for injections, gaschromatography, heavy metals determination, test for glasscontainers for injections, infrared spectrophotometry,insoluble particulate matter test for injections, insolubleparticulate matter test for ophthalmic solutions, iron deter-mination, liquid chromatography, loss on drying determina-tion, loss on ignition determination, mass variation test,melting point determination, test for metal particles inophthalmic ointments, methanol determination, methoxylassay, microbial assay for antibiotics, test for microbiallimit, test for microbial limit for crude drugs, mineral oildetermination, nitrogen determination, nuclear magneticresonance spectroscopy, optical rotation determination,osmolarity determination, oxygen ‰ask combustion method,paper chromatography, particle size distribution test forpreparations, pH determination, test for plastic containers,powder particle density determination, powder particle sizedetermination, test for pyrogen, qualitative test, test forreadily carbonizable substances, refractive index determina-tion, residual solvents test, residue on ignition determina-tion, test for rubber closure for aqueous infusions, speciˆcgravity and density determination, speciˆc surface areadetermination, test for sterility, sulfate determination,thermal analysis, thin-layer chromatography, test for totalorganic carbon, viscosity determination, vitamin A assay,test for volatile contaminants in ethanol, water determina-tion, and X-ray powder diŠraction are performed as directedin the corresponding articles under the General Tests, Proc-esses and Apparatus. The tests for melting point of fats,congealing point of fatty acids, speciˆc gravity, acid value,saponiˆcation value, ester value, hydroxyl value, unsaponiˆ-able matter and iodine value of fats and fatty oils areperformed as directed in the corresponding items under theFats and Fatty oils Test, and the tests for foreign matter and
loss on drying, total ash, acid-insoluble ash, extract content,essential oil content of crude drugs are performed as directedin the corresponding items under the Crude Drugs Test.
6. Bacterial Endotoxins TestChange the (ii) Interpretation of (2) Limit testunder Gel-clot techniques to read:
Gel-clot techniques(2) Limit test(ii) InterpretationThe test is valid when both replicates of solutions B and C
are positive and those of solution D are negative.The sample meets the endotoxin limit requirement of the
test when a negative result is found for both replicates ofsolution A.
Repeat the test in duplicate when the test results are posi-tive for one test but negative for the other one. The samplemeets the endotoxin limit requirement of the test when anegative result is found for both replicates of solution A inthe repeat test.
The sample does not meet the endotoxin limit requirementof the test when a positive result is found for both replicatesof the solution A at a dilution equal to the MVD. If the testis positive for the sample at a dilution less than the MVD,the test may be performed at a dilution not greater than theMVD.
39. Nuclear Magnetic ResonanceSpectroscopy (1H)
Change to read:
39. Nuclear Magnetic ResonanceSpectroscopy
Nuclear magnetic resonance (NMR) spectroscopy is basedon the phenomenon that speciˆc radio frequency radiation isabsorbed by magnetic nuclei in a sample placed in a magnet-ic ˆeld; target nuclei are 1H, 13C, 15N, 19F, 31P, etc. Thesenuclei have intrinsic spin angular momentum, of which themagnitude is given by I (I+1)/h/2p, where I is the spinquantum number and is integral or half-integral (I=1/2 for1H and 13C). When the magnetic nuclei are placed in a mag-netic ˆeld, they are oriented in 2I+1 possible orientations

16701670 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
corresponding to 2I+1 equally spaced energy levels (twoenergy levels for 1H and 13C). The transition between twosuccessive quantized energy levels corresponding to adjacentorientations can be induced by electromagnetic radiationwith a suitable frequency. The precise relation between theˆeld strength and the resonant frequency n is given by
n=g・H0
2pwhere H0 is the strength of the applied external magneticˆeld and g is the gyromagnetic ratio, a constant characteriz-ing a particular isotope. The absorption of radiation (NMRsignal) can occur only when the irradiating radio frequencysatisˆes the resonance condition. Since the absorptioncoe‹cient (the transition probability) does not depend onthe environment in which the nuclei are located, the intensityis basically proportional to the number of nuclei. The excessspins shifted to the higher energy levels by the transitionprocess return to the thermal equilibrium state at variousrates determined by a characteristic time constant (known asthe relaxation time).
A nucleus is shielded from the applied magnetic ˆeld bythe electrons belonging to its own atom and to the molecule.Therefore nuclei in diŠerent environments are shielded todiŠerent extents and resonate at diŠerent frequencies. ThediŠerence in resonance frequencies is deˆned as chemicalshift (d), which is independent of the strength of the magnet-ic ˆeld, and is given by
d=nS-nR
nR+dR
where,nS: The resonance frequency of the observed signal,nR: The resonance frequency of the reference signal,nR: The chemical shift of the reference signal (in the case
of the value not being 0).
The chemical shifts are normally expressed in ppm, adimensionless unit, by assuming the chemical shift of thereference compound as 0 ppm. When the chemical shift ofthe reference compound is not assumed to be 0 ppm, chemi-cal shifts of samples are corrected accordingly.
In addition to the shielding due to electrons, the nucleus issubjected to eŠects due to the spin orientations of othermagnetic nuclei through chemical bonds, resulting in anadditional splitting of the signal. The spacing between twoadjacent components of the signal is known as the spin-spincoupling constant (J). Coupling constants are measured inhertz and are independent of the strength of the externalmagnetic ˆeld. The increased number of interacting nucleiwill make the multiplet pattern more complex.
From the NMR spectrum the following four parameterscan be obtained: chemical shift, spin-spin coupling constant,resonance intensity (intensities of 1H are proportional to thenumber of nuclei and those of 13C and others are susceptibleto the nuclear Overhauser eŠect (NOE) and relaxation) andrelaxation time. These parameters are useful for structuraldetermination, identiˆcation and quantitative analysis ofmolecules. Spin decoupling, NOE, and two-dimensionalNMR techniques are also available for structural analysis.
SpectrometerThere are two types of spectrometers.(1) Fourier transform NMR (FT-NMR) spectrometers
(Figure 1)Target nuclei are simultaneously excited in all frequency
range of the nuclei by means of an intense radio frequencypulse. The FID (free induction decay) after the pulse isdetected, which is a time domain signal, is converted to afrequency domain spectrum by Fourier transformation.Number of data points suitable for the spectral range, ‰ipangle, acquisition time, delay time and number of scansshould be set appropriately.
Recently FT-NMR is commonly used because of its highsensitivity and various advanced applications.
Fig. 1 FT-NMR spectrometer
(2) Continuous wave NMR (CW-NMR) spectrometers(Figure 2)
In the case of the CW method, a spectrum is obtained bysweeping the radio frequency or magnetic ˆeld continuouslyover the frequency range of the nuclei being observed.
Fig. 2 CW-NMR spectrometer
MeasurementPrior to measurements, the sensitivity and resolution of
the instrument must be adjusted to the optimum levels usinga standard sample (ethylbenzene, 1,2-dichlorobenzene oracetaldehyde) dissolved in an appropriate NMR solvent.
(1) The sample dissolved in a suitable solvent is trans-

16711671Supplement II, JPXIV General Tests, Processes and Apparatus
ferred into an NMR tube. The reference compound can beadded directly to the sample solution (internal reference), ora sealed capillary tube containing the reference compoundcan be inserted into the NMR tube (external reference). Thesample solutions should be completely homogeneous. Inparticular, solid contaminants should be removed in order toobtain good spectra. Various deuterated NMR solvents arecommonly used for NMR measurement and the followingpoints should be considered in selecting an appropriatesolvent: (i) The solvent signals do not overlap with thesample signals. (ii) The sample must be soluble in the solventselected. (iii) The solvent does not react with the sample.Furthermore, it should be noted that chemical shifts candepend upon the solvent employed, sample concentrationand deuterium ion concentration, and that viscous solutionsusually give rather broad, poorly resolved spectra.
(2) For the reference standards use the reagents fornuclear magnetic resonance spectroscopy. For 1H and 13Cspectra, tetramethylsilane (TMS) is usually used as thereference compound for samples dissolved in organicsolvents. For samples dissolved in deuterium oxide, sodium2,2-dimethyl-2-silapentane-5-sulfonate (DSS) or sodium 3-(trimethylsilyl)propionate (TSP) is used. For other nuclei,nitromethane, trichloro‰uoromethane and phosphoric acidare used as reference compounds for 15N, 19F and 31P,respectively. Furthermore, chemical shifts of residual pro-tons in deuterated solvents and 13C in the solvent instead of areference compound can be used for 1H and 13C NMR.
Record of apparatus and measurement conditionsType of instrument, frequency, solvent, temperature,
sample concentration, reference compound, experimentaltechnique, etc. should be recorded to allow appropriatecomparison of spectra, because NMR spectra depend on themeasurement conditions.
IdentiˆcationThe sample solution is prepared and tested by the method
directed in each monograph. Usually in the case of 1H NMR,the sample is identiˆed by the following method.
(1) Identiˆcation by the use of chemical shift, multiplic-ity and relative intensity
When chemical shifts, multiplicities and relative intensi-ties of signals are deˆned, the sample can be identiˆed asbeing the same substance when all chemical shifts, multiplic-ities and relative intensities are the same as those prescribed.
(2) Identiˆcation by the use of a Reference StandardMeasurement conditions should be the same as those used
in the case of the Reference Standard. When the spectra of asample and the Reference Standard exhibit the samemultiplicities and relative intensities of signal at the samechemical shifts, the sample can be identiˆed as being thesame substance as the Reference Standard.
Experimental techniques of 1H and 13C NMR spectroscopyNMR spectroscopy includes one-, two- and multi-dimen-
sional techniques, which are used for various purposes.Spin decoupling, and NOE are available in one-dimen-
sional 1H spectroscopy. Spin decoupling can assign coupling
correlations. As NOE can observe correlations amongspatially proximate protons, the conˆguration and theconformation can be analyzed.
Broadband decoupling, INEPT and DEPT are usuallyapplied in one-dimensional 13C spectroscopy. The broad-band decoupling technique simpliˆes a spectrum andachieves enhancement of sensitivity. INEPT (insensitivenuclei enhanced by polarization transfer) and DEPT (distor-tionless enhancement of polarization transfer) enhance thesensitivity of 13C by means of polarization transfer fromdirectly bonded 1H with a large magnetic moment. They canbe applied to identify primary, secondary, tertiary orquarternary carbon.
Two-dimensional spectroscopy can observe all correlationpeaks between nuclei through spin-spin coupling or NOE ina single experiment, and there are many techniques forhomonuclear and heteronuclear measurements. Representa-tive techniques are described below.
COSY (2D correlation spectroscopy), HOHAHA(homonuclear Hartmann-Hahn spectroscopy) or TOCSY(total correlation spectroscopy): Correlation between pro-tons through scalar spin-spin coupling is obtained andintramolecular connectivities of hydrogen atoms arerevealed.
NOESY (2D nuclear Overhauser enhancement andexchange spectroscopy): NOE is measured by a two-dimen-sional technique. Approximate distances between spatiallyproximate hydrogen atoms are obtained to analyze the three-dimensional structure.
INADEQUATE (incredible natural abundance doublequantum transfer experiment): Although this technique isinsensitive because it involves double quantum transfer by13C-13C scalar coupling in a sample with natural isotopicabundance, the connectivities of all neighboring 13C nucleican be obtained to analyze the carbon skeleton.
HMQC (heteronuclear multiple quantum coherence): Thistechnique observes correlations between 1H and 13C withdirect spin-spin coupling using 1H detection and revealsintramolecular chemical bonds between hydrogen andcarbon atoms.
HMBC (heteronuclear multiple bond connectivity): Thistechnique observes correlations between 1H and 13C withlong range spin-spin coupling using 1H detection and revealsintramolecular connectivities of hydrogen and carbonatoms.
There are many other techniques such as DQF-COSY(double quantum ˆltered COSY) and HSQC (heteronuclearsingle quantum coherence). Furthermore, multidimensionalNMR techniques are used to analyze macromolecules.

16721672 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
47. Pyrogen TestChange to read:
The Pyrogen Test is a method to test the existence ofpyrogens by using rabbits.
Test animalsUse healthy mature rabbits, each weighing not less than
1.5 kg, which have not lost body mass when kept on aconstant diet for not less than one week. House the rabbitsindividually in an area free from disturbances likely to excitethem. Keep the temperature of the area constant between209C and 279C for at least 48 hours before and throughoutthe test. Before using a rabbit that has not previously beenused for a pyrogen test, condition it 1 to 3 days prior to thetest by conducting a sham test omitting the injection. Do notuse a rabbit for pyrogen tests more frequently than onceevery 48 hours, or after it has been given a test sample thatwas adjudged pyrogen-positive or that contained an antigenpresent commonly in the test sample to be examined.
Apparatus, instruments(1) Thermometer—Use a rectal thermometer or temper-
ature-measuring apparatus with an accuracy of ±0.19C orless.
(2) Syringe and injection needle—Depyrogenate thesyringes and needles in a hot-air oven using a validated proc-ess, usually by heating at 2509C for not less than 30 minutes.Sterilized syringes with needles are also available providedthat they have been validated to assure that they are free ofdetectable pyrogens and do not interfere with the test.
Test procedures(1) Quantity of injection—Unless otherwise speciˆed,
inject 10 mL of the sample per kg of body mass of eachrabbit.
(2) Procedure—Perform the test in a separate area at anenvironmental temperature similar to that of the roomwherein the animals were housed and free from disturbanceslikely to excite them. Withhold food from the rabbits forseveral hours before the ˆrst record of the temperature andthroughout the testing period. The test animals are usuallyrestrained with loosely ˆtting neck stocks that allow therabbits to assume a natural resting posture. Determine thetemperature of each rabbit by inserting the thermometer ortemperature-measuring probe into the rectum of the testanimal to a constant depth within the range of 60 mm to90 mm. The ``control temperature'' of each rabbit is themean of two temperature readings recorded for that rabbitat an interval of 30 min in the 40 min immediately precedingthe injection of the sample to be examined. Rabbits showinga temperature variation greater than 0.29C between the twosuccessive temperature readings or rabbits having an initialtemperature higher than 39.89C are withdrawn from thetest.
Warm the test solution to a temperature of 37±29Cbefore injection, and inject the solution slowly into themarginal vein of the ear of each rabbit over a period not
exceeding 10 min. Hypotonic test sample may be madeisotonic by the addition of pyrogen-free sodium chloride.Record the temperature of each rabbit during a period of3 hours after the injection, taking the measurements atintervals of not more than 30 min. The diŠerence betweenthe control temperature and the maximum temperature ofeach rabbit is taken to be the rise in body temperature.Consider any temperature decreases as zero rise.
Interpretation of resultsThe test is carried out on a group of three rabbits and the
result is judged on the basis of the sum of the three tempera-ture rises. Repeat if necessary on further groups of three rab-bits to a total of three groups, depending on the resultsobtained. If the summed response of the ˆrst group does notexceed 1.39C, the sample is judged to be pyrogen-negative.If the summed response exceeds 2.59C, the sample is judgedto be pyrogen-positive. If the summed response exceed1.39C but does not exceed 2.59C, repeat the test on anothergroup of three rabbits. If the summed response of the ˆrstand second group does not exceed 3.09C, the sample isjudged to be pyrogen-negative. If the summed response ofthe 6 rabbits exceeds 4.29C, the sample is judged to bepyrogen-positive. If the summed response exceeds 3.09C butdoes not exceed 4.29C, repeat the test on one more group ofthree rabbits. If the summed response of the 9 rabbits doesnot exceed 5.09C, the sample is judged to be pyrogen-negative. If the summed response exceeds 5.09C, the sampleis judged to be pyrogen-positive.
When the test sample is judged to be pyrogen-negative,the sample passes the pyrogen test.
52. Residue on Ignition TestChange the Procedure to read:
ProcedurePreviously ignite a suitable crucible (silica, platinum,
quartz or porcelain) at 600±509C for 30 minutes, and weighaccurately after cooling in a desiccator (silica gel or othersuitable dessicant).
Take the sample of the amount directed in the mono-graph, transfer into the ignited crucible, and weigh accurate-ly. When the quantity of the sample to be taken is indicatedin a volume, pipet exactly the amount directed in themonograph and transfer into the above crucible. Whendirected as ``after evaporating,'' heat properly to evaporatethe solution.
Moisten the sample with a small amount of sulfuric acid,usually 1 mL, then heat slowly at a temperature as low aspracticable until the sample is completely carbonized, andcool. Moisten again with a small amount (usually 1 mL) ofsulfuric acid, heat gently until white fumes are no longerevolved, and ignite at 600±509C until the residue is com-pletely incinerated. Ensure that ‰ames are not produced atany time during the procedure. Cool the crucible in a desic-cator (silica gel or other suitable dessicant), and reweighaccurately to calculate the percentage of residue.

16731673Supplement II, JPXIV General Tests, Processes and Apparatus
Unless otherwise speciˆed, if the amount of the residue soobtained exceeds the limit speciˆed in the individual mono-graph, repeat the moistening with sulfuric acid, heating andignition as before until constant mass is attained or until thepercentage of residue complies with the limit in the individ-ual monograph.
53. Speciˆc Surface AreaDetermination
Change to read the text up to the paragraphheaded ``Method 1 Dynamic Flow Method'' asfollows:
Speciˆc Surface Area Determination is a method to deter-mine the speciˆc surface area (the total surface area ofpowder per an unit mass) of a powdered pharmaceuticalpreparation by using the gas adsorption method. The gasadsorption method is a method for measuring the amount ofgas adsorbed on the surface of a powder sample as a func-tion of the pressure of the adsorbate gas, and the measure-ments are usually performed at the boiling point of liquidnitrogen (-1969C).
When the gas is physically adsorbed by the powdersample, the following relationship holds when P/P0 is in therange of 0.05 to 0.30 for pressure P of the adsorbate gas inequilibrium for the volume of gas adsorbed, Va.
1
VaØ P0
P-1»
=(C-1)VmC
×PP0+
1VmC
P: Partial vapor pressure of adsorbate gas in equilibrium(kPa)
P0: Saturated vapor pressure of the adsorbate gas at-1969C (kPa)
Va: Volume of gas adsorbed at equilibrium (mL)Vm: Volume of gas adsorbed in a monolayer (mL)C: Dimensionless constant relating to the enthalpy of
adsorption and condensation of the adsorbate gasThe speciˆc surface area, S, is determined from Vm, the
volume of gas adsorbed in a monolayer on the sample.
S=Vm×N×am×22400
S: Speciˆc surface area (m2/g)N: Avogadro constant 6.022×1023/mola: EŠective cross-sectional area of one adsorbate molecule
(m2)N20.162×10-18
Kr: 0.195×10-18
m: Mass of the test powder (g)Speciˆc surface area is generally expressed in unit of m2/g.Either of the methods described below can be used to
measure the gas adsorption.
54. Sterility TestChange to read:
Test for sterility is the method to establish the presence orabsence of viable microorganisms (bacteria and fungi) usingthe deˆned culturing method. Unless otherwise speciˆed, thetest is carried out by I. Membrane ˆltration method or II.Direct inoculation method. Water, reagents, test solutions,equipment, materials and all other requisites for the testshould be pre-sterilized. The test for sterility is carried outunder aseptic conditions. In order to achieve such condi-tions, the test environment has to be adapted to the way inwhich the sterility test is performed. The precautions takento avoid contamination are such that they do not aŠect anymicroorganisms which are to be revealed in the test. Theworking conditions in which the tests are performed aremonitored regularly by appropriate sampling of the workingarea and by carrying out appropriate controls.
Media and rinsing ‰uidsFluid thioglycolate medium, soybean-casein digest
medium are used, unless otherwise speciˆed. When it isdi‹cult to use ‰uid thioglycolate medium due to turbidity orviscosity of samples, alternative thioglycolate medium canbe used, provided it is heated on a water bath just prior touse and incubated under anaerobic conditions. Otherproducts of suitable quality yielding similar formulationsmay be used according to the indications on the label.
(1) Fluid thioglycolate mediumL-Cystine 0.5 gAgar 0.75 gSodium chloride 2.5 gGlucose, monohydrate/anhydrate 5.5/5.0 gYeast extract (water-soluble) 5.0 gPancreatic digest of casein 15.0 gSodium thioglycolate or 0.5 g
Thioglycolic acid 0.3 mLResazurin sodium solution (1 in 1000),
freshly prepared 1.0 mLWater 1000 mL
(pH after sterilization 7.1±0.2)Mix the L-cystine, agar, sodium chloride, glucose, water-
soluble yeast extract and pancreatic digest of casein with thewater, and heat until solution is eŠected. Dissolve thesodium thioglycolate or thioglycolic acid in the solution and,if necessary, add sodium hydroxide TS so that, after sterili-zation, the solution will have a pH of 7.1±0.2. If ˆltration isnecessary, heat the solution again without boiling and ˆlterwhile hot through moistened ˆlter paper. Add the resazurinsodium solution, mix and place the medium in suitablevessels which provide a ration of surface to depth of mediumsuch that not more than the upper half of the medium hasundergone a color change indicative of oxygen uptake at theend of the incubation period. Sterilize using a validatedprocess. Store the medium at a temperature between 2 –259C. If more than the upper one-third of the medium hasacquired a pink color, the medium may be restored once by

16741674 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
heating the containers in a water-bath or in free-‰owingsteam until the pink color disappears and cooling quickly,taking care to prevent the introduction of non-sterile air intothe container.
(2) Alternative thioglycolate mediumL-Cystine 0.5 gSodium chloride 2.5 gGlucose, monohydrate/anhydrate 5.5/5.0 gYeast extract (water-soluble) 5.0 gPancreatic digest of casein 15.0 gSodium thioglycolate or 0.5 g
Thioglycolic acid 0.3 mLWater 1000 mL
(pH after sterilization 7.1±0.2)The methods for preparation follow those of ‰uid
thioglycolate medium.(3) Soybean-casein digest medium
Casein peptone 17.0 gSoybean peptone 3.0 gSodium chloride 5.0 gDipotassium hydrogen phosphate 2.5 gGlucose, monohydrate/anhydrous 2.5/2.3 gWater 1000 mL
(pH after sterilization 7.3±0.2)Mix all the ingredients and heat until solution is eŠected.
If necessary, add sodium hydroxide TS so that, after sterili-zation, the solution will have a pH of 7.3±0.2. Filter, ifnecessary, to clarify, distribute into suitable vessels andsterilize using a validated process. Store at a temperaturebetween 2 – 259C in a sterile container.
(4) Rinsing ‰uidsMeat or casein peptone 1.0 gWater 1000 mL
(pH after sterilization 7.1±0.2)Dissolve animal tissue or casein peptone in water and
adjust the pH of the solution so that, after sterilization, itwill show 7.1±0.2. Filter, if necessary, to clarify, distributeinto suitable vessels and sterilize using a validated process.Store at a temperature between 2 – 259C in a sterilecontainer.
To rinsing ‰uid to be used for antibiotics or pharmaceuti-cal products containing an antimicrobial agent, a suitableneutralizer or inactive agent at concentration shown to beappropriate in the validation of the test can be added. Torinsing ‰uid to be used for oils, oily solutions, ointments orcreams, suitable emulsifying agent at a concentration shownto be appropriate in the validation of the test, for examplepolysorbate 80 at a concentration of 10 g/L can be added.
Suitability of mediaThe media used comply with the following tests, carried
out before or in parallel with the test on the product to beexamined.
(1) Sterility of mediaConˆrm the sterility of each sterilized batch of medium by
incubating a portion of the media at the speciˆed incubationtemperature for 14 days. No growth of microorganismsoccurs.
(2) Growth promotion testTest each batch of ready-prepared medium and each batch
(lot)of medium prepared either from dehydrated medium orfrom ingredients1. Inoculate a small number (not more than100 CFU) of microorganism listed in Table 1 or other strainsconsidered to be equivalent to these strains in containers ofeach medium. Each of the test organisms should showclearly visible growth in all inoculated media within 3 daysfor bacteria and within 5 days for fungi.
Table 1. Microorganisms for growth promotion test andthe validation test
Medium Test microorganisms Incubationconditions
Fluidthioglycolatemedium
Staphylococcus aureus(ATCC 6538, NBRC13276,CIP 4.83, NCTC 10788,NCIMB 9518)Pseudomonas aeruginosa(ATCC 9027, NBRC 13275,NCIMB 8626, CIP 82.1l8)Clostridium sporogenes(ATCC 19404, CIP 79.3,NCTC 532, or ATCC 11437,NBRC 14293)
Aerobic
Alternativethioglycolatemedium
Clostridium sporogenes(ATCC 19404, CIP 79.3,NCTC 532, or ATCC 11437,NBRC 14293)
Anaerobic
Soybean-caseindigest medium
Bacillus subtilis(ATCC 6633, NBRC 3134,CIP 52.62, NCIMB 8054)Candida albicans(ATCC 10231, NBRC 1594,IP 48.72, NCPF 3179)Aspergillus niger(ATCC 16404, NBRC 9455,IP 1431.83, IMI 149007)
Aerobic
Seed lot culture maintenance techniques (seed-lot systems)are used so that the viable microorganisms used for inocula-tion are not more than ˆve passages removed from theoriginal master seed-lot.
EŠective period of mediaIf prepared media are stored in unsealed containers, they
can be used for one month, provided that they are tested forgrowth promotion within two weeks of the time of use andthat color indicator requirements are met. If stored in tightcontainers, the media can be used for one year, providedthat they are tested for growth promotion within 3 monthsof the time of use and that the color indicator requirementsare met.
Validation testThe validation may be performed simultaneously with the
Test for sterility of the product to be examined in thefollowing cases.a) When the test for sterility has to be carried out on a new
product.

16751675Supplement II, JPXIV General Tests, Processes and Apparatus
b) Whenever there is a change in the experimental condi-tions of the test.
Carry out the test as described below under Test for sterili-ty of the product to be examined using exactly the samemethods except for the following modiˆcations.Membrane ˆltration After transferring the content of thecontainer or containers to be tested to the membrane add aninoculum of a small number of viable micro-organisms (notmore than 100 CFU) to the ˆnal portion of sterile diluentused to rinse the ˆlter.Direct inoculation After transferring the contents of thecontainer or containers to be tested to the culture mediumadd an inoculum of a small number of viable micro-organ-isms (not more than 100 CFU) to the medium.
In both cases use the same micro-organisms as thosedescribed above under Growth promotion test. Perform agrowth promotion test as a positive control. Incubate all thecontainers containing medium for not more than 5 days. Ifclearly visible growth of micro-organisms is obtained afterthe incubation, visually comparable to that in the controlvessel without product, either the product possesses noantimicrobial activity under the conditions of the test orsuch activity has been satisfactorily eliminated. The test forsterility may then be carried out without further modiˆca-tion. If clearly visible growth is not obtained in the presenceof the product to be tested, visually comparable to that inthe control vessels without product, the product possessesantimicrobial activity that has not been satisfactorily elimi-nated under the conditions of the test. Modify the conditionsin order to eliminate the antimicrobial activity and repeat thevalidation test.
In the membrane ˆltration, the antimicrobial activity shouldbe suppressed by suitable means such as replacement of themembrane ˆlters with less adsorptive ones, increase of theamount of rinsing ‰uid, or addition of a suitable inactivatingagent to the rinsing ‰uid. Do not exceed a washing cycle of5 times 100 mL per ˆlter, even if during validation it hasbeen demonstrated that such a cycle does not fully eliminatethe antimicrobial activity.
In the direct inoculation, use a suitable inactivating agentwhich does not aŠect the growth of microorganisms or in-crease the volume of medium irrespective of the prescriptionin II-2 so that no antimicrobial activity remains.
Test for sterility of the products to be examinedNumber of articles to be tested
Items to be used for the test are taken from the lot accord-ing to an appropriate sampling plan prepared by referring tothe numbers speciˆed in Table 2.
Table 2. Number of items to be taken from the lot
Number of items in the lot Minimum number of items tobe tested for each medium*
InjectionsNot more than 100 containers
More than 100 but not morethan 500 containersMore than 500 containers
For large-volume products(More than 100 mL)
10z or 4 containers, which-ever is greater
10 containers
2z or 20 containers, which-ever is less2z or 10 containers, which-ever is less
Ophthalmic and othernon-injectable products
Not more than 200 containers
More than 200 containersIf the product is presented inthe form of single-dose con-tainers, apply the schemeshown above for preparationsfor parenteral use
5z or 2 containers, which-ever is greater10 containers
Bulk solid productsUp to 4 containersMore than 5 containers butnot more than 50 containersMore than 50 containers
Each container20z or 4 containers, which-ever is greater2z or 10 containers, which-ever is greater
Antibiotic SolidsPharmacy bulk packages (º5 g)Pharmacy bulk packages (Æ5 g)
20 containers6 containers
*If the contents of one container are enough to inoculate thetwo media, this column gives the number of containers need-ed for both the media together.
Testing methodsThe test may be carried out using the technique of mem-
brane ˆltration or by direct inoculation of the culture mediawith the product to be examined. Appropriate negativecontrols are included. The technique of membrane ˆltrationis used whenever the nature of the product permits, that is,for ˆlterable aqueous preparations, for alcoholic or oilypreparations and for preparations miscible with or soluble inaqueous or oily solvents provided these solvents do not havean antimicrobial eŠect in the conditions of the test.
I. Membrane ˆltrationBy this method, a test article is ˆltered through a mem-
brane ˆlter, and the ˆlter is rinsed and incubated by beingtransferred to a medium or by adding a medium to the ˆltra-tion apparatus. Use membrane ˆlter made from suitablematerial having a nominal pore size of 0.45 mm or smaller.Use a ˆlter funnel sterilizable by the moist heat method orother methods and free from any leakage or back ‰ow whenˆltration is performed with the membrane in place. Thetechnique described below assumes that membranes about50 mm in diameter will be used. If ˆlters of a diŠerentdiameter are used the volumes of the dilutions and the

16761676 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
washings should be adjusted accordingly.I-1. Preparation of sample solution
a) Liquid medicine: Use as it is, as the sample solution.b) Solid medicine: In the case of a solid medicine, to be
administered after dissolving or suspending, the samplesolution is prepared with the provided solvent, isotonicsodium chloride solution or water to give the concentra-tion of use.
c) Oils and oily solutions: Oils and oily solutions ofsu‹ciently low viscosity may be ˆltered without dilutionthrough a dry membrane. Viscous oils may be diluted asnecessary with a suitable sterile diluent such as isopropylmyristate shown not to have antimicrobial activity in theconditions of the test.
d) Ointments and creams: Ointments in a fatty base andemulsions of the water-in-oil type may be diluted by usingsterile isopropyl myristate that has previously beenˆltered through a sterilizing membrane ˆlter or by usingother solvents not aŠecting the growth of microorgan-isms. Heat the sample preparation, if necessary, to notmore than 409C. In exceptional cases it may be necessaryto heat to not more than 449C.I-2. Quantities of sample solution to be testedUse for each medium not less than quantity of the product
prescribed in Table 3, unless otherwise speciˆed. If thecontents of one container are insu‹cient to inoculate thetwo media, twice or more containers shown in Table 2 areused. When using the technique of membrane ˆltration, use,whenever possible, the whole contents of the container, butnot less than the quantities indicated in Table 3, dilutingwhere necessary to about 100 mL with a suitable sterilerinsing ‰uid.
Table 3. Minimum quantity to be used for each medium
Quantity per container Minimum quantity to beused for each medium
LiquidsLess than 1 mL
1 – 40 mL
Greater than 40 mL andnot greater than 100 mLGreater than 100 mL
Antibiotic liquidsOther preparations solublein water or in isopropylmyristate
The whole contents of eachcontainerHalf the contents of eachcontainer but not less than1 mL20 mL
10z of the contents of thecontainer but not less than20 mL1 mLThe whole contents of eachcontainer to provide not lessthan 200 mg
Insoluble preparations, creamsand ointments to be suspendedor emulsiˆed
Use the contents of eachcontainer to provide not lessthan 200 mg
SolidsLess than 50 mg
50 mg or more but less than300 mg300 mg – 5 gGreater than 5 g
The whole contents of eachcontainerHalf the contents of each con-tainer but not less than 50 mg150 mg500 mg
I-3. ProceduresUsually complete the ˆltration of the sample solution with
one or two separate ˆlter funnels. Transfer the contents ofthe container or containers to be tested to the membrane ormembranes. If the sample solution is not readily ˆlterable, itmay be further diluted with rinsing ‰uid and thereafterˆltered. Rinse the membrane(s) with each 100-mL of rinsing‰uid per ˆlter for established cycles in the validation test.Provided the sample does not have antimicrobial activity,the rinsing procedure can be omitted. Employ either of thetwo methods described below for incubation of themembrane(s). Use the same volume of each medium as in thevalidation test.
(1) The processed membrane is aseptically transferredfrom the apparatus and cut into two equal parts, or half thevolume of sample solution is ˆltered into an entire mem-brane. Transfer each half of the cut membrane, or eachwhole membrane into the medium.
(2) After ˆltration of sample solution into the apparatusto which the membrane ˆlters are ˆtted, each medium isadded to the apparatus itself.
II. Direct inoculation of the culture mediumThis is the method by which the entire content or a portion
of the content of a sample container is transferred directly tothe culture medium and incubated. Usually, this method isapplied for medicines to which the membrane ˆltrationmethod cannot be applied or for which the application of thedirect transfer method, rather than the membrane ˆltration

16771677Supplement II, JPXIV General Tests, Processes and Apparatus
method, is rational.For products containing a mercurial preservative that
cannot be tested by the membrane-ˆltration method, ‰uidthioglycolate medium incubated at 20 – 259C may be usedinstead of Soybean-casein digest medium.
II-1. Preparation of sample solutionUsually, proceed as directed for the membrane ˆltration
method. In the case of an insoluble medicine, the product issuspended or crushed in a suitable manner and used as asample.a) Oily liquids. Use media to which have been added a suita-
ble emulsifying agent at a concentration shown to beappropriate in the validation of the test, for examplepolysorbate 80 at a concentration of 10 g/L.
b) Ointments and creams. Prepare by diluting to about 1 in10 by emulsifying with the chosen emulsifying agent in asuitable sterile diluent such as a 1 g/L neutral solution ofmeat or casein peptone. Transfer the diluted product to amedium not containing an emulsifying agent.II-2. Quantities of sample solution to be testedTransfer the quantity of the preparation to be examined
prescribed in Table 3, by using pipette, syringe or othersuitable inoculation devices, directly into the culturemedium so that the volume of the product is not more than10z of the volume of the medium, unless otherwiseprescribed. Shake cultures containing oily products gentlyeach observation day. However when thioglycolate mediumis used for the detection of anaerobic microorganisms keepshaking or mixing to a minimum in order to maintainanaerobic conditions.
Cultivation and observationFluid thioglycolate medium and Alternative thioglycolate
medium are to be incubated at 30 – 359C and Soybean-casein digest medium is to be incubated at 20 – 259C for notless than 14 days. Observe the cultures several times duringthe incubation period. If the material being tested rendersthe medium turbid so that the presence or absence ofmicrobial growth cannot be readily determined by visualexamination, 14 days after the beginning of incubationtransfer suitable portions of the medium to fresh vessels ofthe same medium and then incubate the original and transfervessels for not less than 4 days.
Observation and interpretation of resultsIf no evidence of microbial growth is found, the product
to be examined complies with the test for sterility. Ifevidence of microbial growth is found the product examineddoes not comply with the test for sterility, unless it can beclearly demonstrated that the test was invalid for causesunrelated to the product to be examined. If no evidence ofmicrobial growth is found in the repeat test the productcomplies with the Sterility Test. If microbial growth is foundin the repeat test the product does not comply with theSterility Test.
70. Reference Standards;Reagents, Test Solutions; StandardSolutions for Volumetric Analysis;
Standard Solutions; MatchingFluids for Color; Optical Filters forWavelength and Transmission Rate
Calibration; and MeasuringInstruments, Appliances
(1) Reference Standards
Add the following:
Azithromycin, Cisplatin, Etoposide, Furosemide, Methyl-prednisolone Succinate, Nilvadipine, Thiamylal, Tranexam-ic Acid, Trichlormethiazide
(2) Reagents, Test Solutions
Delete the following:
Cyclohexylamine for thin-layer chromatographyDicyclohexylurea for thin-layer chromatography
Change the following:
Calcium hydroxide for pH determination Calciumhydroxide prepared for pH determination.
[6]-Gingerol for thin-layer chromatography C17H26O4
A yellow-white to yellow, liquid or solid. Freely soluble inmethanol, in ethanol (99.5) and in diethyl ether, and practi-cally insoluble in water.
Purity Related substances—Dissolve 1.0 mg of [6]-gin-gerol for thin-layer chromatography in exactly 2 mL ofmethanol. Perform the test with 10 mL of this solution asdirected in the Identiˆcation under Ginger: any spot otherthan the principal spot at the Rf value of about 0.3 does notappear.
Piperidine hydrochloride C5H11N.HCl A white crys-talline powder. Dissolves in water and in methanol. The pHof the aqueous solution (1 in 20) is between 3.0 and 5.0.
Melting point: 247 – 2529CPurity Clarity and color of solution—Dissolve 1.0 g of
piperidine hydrochloride in 20 mL of water: the solution isclear and colorless.
Residue on ignition: not more than 0.10z (1 g).Content: not less than 99.0z. Assay—Dissolve about
0.25 g of piperidine hydrochloride, accurately weighed, in50 mL of water, add 5 mL of diluted nitric acid (1 in 3), andtitrate with 0.1 mol/L silver nitrate VS (potentiometric titra-tion). Perform a blank determination in the same manner,

16781678 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
and make any necessary correction.
Each mL of 0.1 mol/L silver nitrate VS=12.16 mg of C5H11N.HCl
Sodium 1-decanesulfonate C10H21NaO3S A white pow-der.
Purity Clarity and color of solution—Dissolve 1.0 g in20 mL of water: the solution is clear and colorless.
Loss on drying: not more than 3.0z (1 g, 1059C,3 hours).
Content: not less than 98.0z. Assay—Weigh accuratelyabout 0.45 g of sodium 1-decanesulfonate, dissolve in 50 mLof water, and pass through a column, about 1.2 cm in insidediameter and about 25 cm in length, packed with about20 mL of strongly acidic ion-exchange resin (0.3 to 1.0 mm,H type) at a ‰ow rate of about 4 mL per minute. Wash with150 mL of water at a ‰ow rate of about 4 mL per minute.Combine the washing and the elute, and titrate with0.1 mol/L sodium hydroxide VS (potentiometric titration).Perform a blank determination in the same manner, andmake any necessary correction.
Each mL of 0.1 mol/L sodium hydroxide VS=24.43 mg of C10H21NaO3S
Thioglycolate medium I for sterility test See ‰uidthioglycolate medium.
Thioglycolate medium II for sterility test See alternativethioglycolate medium.
Turmeric paper Macerate 20 g of powdered driedrhizome of Curcuma longa Linn áe with four 100 mL-portionsof cold water, decant the supernatant liquid each time, anddiscard it. Dry the residue at a temperature not over 1009C.Macerate the dried residue with 100 mL of ethanol (95) forseveral days, and ˆlter. Immerse ˆlter paper in this ethanoldecoction, and allow the ethanol (95) to evaporate spontane-ously in clean air.
Sensitivity—Dip a strip of turmeric paper, about 1.5 cmlength, in a solution of 1 mg of boric acid in a mixture of1 mL of hydrochloric acid and 4 mL of water, after 1 minuteremove the paper from the liquid, and allow it to dry spon-taneouly: the yellow color changes to brown. When the stripis moistened with ammonia TS, the color of the stripchanges to greenish black.
L-Tyrosine C9H11NO3 White, crystals or crystallinepowder. Odorless and tasteless. Freely soluble in formicacid, very slightly soluble in water, and practically insolublein ethanol (95) and in diethyl ether. It dissolves in dilutehydrochloric acid and in dilute nitric acid.
Optical rotation [a]D20: -10.5 –-12.59 (after drying,2.5 g, 1 mol/L hydrochloric acid TS, 50 mL, 100 mm).
Loss on drying: not more than 0.30z (1 g, 1059C,3 hours).
Content: not less than 99.0z. Assay—Weigh accuratelyabout 0.3 g of L-tyrosine, previously dried, dissolve in 6 mLof formic acid, add 50 mL of acetic acid (100), and titratewith 0.1 mol/L perchloric acid VS (potentiometric titra-tion). Perform a blank determination, and make any neces-
sary correction.
Each mL of 0.1 mol/L perchloric acid VS=18.12 mg of C9H11NO3
Add the following:
0.05 mol/L Acetic acid-sodium acetate buŠer solution,pH 4.6 Dissolve 6.6 g of sodium acetate trihydrate in900 mL of water, and add 3 mL of acetic acid and water tomake 1000 mL.
0.25 mol/L Acetic acid TS To 3 g of acetic acid (100)add water to make 200 mL.
Aconitine for purity C34H47NO11 White, crystals or crys-talline powder. Sparingly soluble in acetonitrile and inethanol (99.5), slightly soluble in diethyl ether, and practi-cally insoluble in water. Melting point: about 1859C (withdecomposition).
Identiˆcation—Determine the infrared absorption spec-trum of aconitine for purity as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry: it exhibits absorption at the wave numbers of about3500 cm-1, 1718 cm-1, 1278 cm-1, 1111 cm-1, 1097 cm-1
and 717 cm-1.Absorbance E1z
1 cm (230 nm): 211 – 243 [5 mg dried fornot less than 12 hours in a desiccator (reduced pressure notexceeding 0.67 kPa, phosphorus (V) oxide, 409C), ethanol(99.5), 200 mL].
Purity Related substances—(1) Dissolve 5.0 mg of aconitine for purity in 2 mL of
acetonitrile, and use as the sample solution. Pipet 1 mL ofthe sample solution, add acetonitrile to make exactly 50 mL,and use as the standard solution. Perform the test with thesesolutions as directed under the Thin-layer Chromatography.Spot 20 mL each of the sample solution and the standardsolution on a plate of silica gel for thin-layer chro-matography, and proceed the test as directed in the Identiˆ-cation in Processed Aconite Root: the spot other than theprincipal spot obtained with the sample solution is not moreintense than the spot with the standard solution.
(2) Dissolve 5.0 mg of aconitine for purity in 5 mL ofacetonitrile, and use as the sample solution. Pipet 1 mL ofthe sample solution, add acetonitrile to make exactly 50 mL,and use as the standard solution. Perform the test withexactly 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and determine eachpeak area by the automatic integration method: the totalarea of the peaks other than the peaks of aconitine and thesolvent obtained with the sample solution is not larger thanthe peak area of aconitine with the standard solution.Operating conditions
Detector, column, and column temperature: Proceed asdirected in the operating conditions in the Purity underProcessed Aconitine Root.
Mobile phase: A mixture of phosphate buŠer solution forprocessed aconite root and tetrahydrofuran (9:1).
Flow rate: Adjust the ‰ow rate so that the retention time

16791679Supplement II, JPXIV General Tests, Processes and Apparatus
of aconitine is about 26 minutes.Time span of measurement: About 3 times as long as the
retention time of aconitine.System suitability
Test for required detectability: Pipet 1 mL of the standardsolution, and add acetonitrile to make exactly 20 mL.Conˆrm that the peak area of aconitine obtained from 10 mLof this solution is equivalent to 3.5 to 6.5z of that obtainedfrom 10 mL of the standard solution.
System performance: Dissolve 1 mg each of aconitine forpurity, hypaconitine for purity and mesaconitine for purity,and 8 mg of jesaconitine for purity in 200 mL of acetonitrile.When the procedure is run with 10 mL of this solution underthe above operating conditions, mesaconitine, hypaconitine,aconitine and jesaconitine are eluted in this order, and eachresolution between these peaks is not less than 1.5, respec-tively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of aconitine is not more than 1.5z.
Water: not more than 1.0z [5 mg dried for not less than12 hours in a desiccator (reduced pressure not exceeding0.67 kPa, phosphorus (V) oxide, 409C), coulometric titra-tion].
Aconitum diester alkaloids standard solution for purityIt is a solution containing 10 mg of aconitine for purity,10 mg of jesaconitine for purity, 30 mg of hypaconitine forpurity and 20 mg of mesaconitine for purity in 1000 mL of amixture of phosphate buŠer solution for processed aconiteroot and acetonitrile (1:1). When proceed the test with 20 mLof this solution as directed in the Purity under ProcessedAconite Root, using the detection wavelength, 231 nm, thepeaks of aconitine, jesaconitine, hypaconitine andmesaconitine are observed, and the ratio of their peakheights is about 10:1:35:30. When proceed the test using thedetection wavelength, 254 nm, the peaks of aconitine,jesaconitine, hypaconitine and mesaconitine are observed,and the ratio of their peak heights is about 2:8:7:6.
Alternative thioglycolate medium See the Sterility Testunder the General Tests, Processes and Apparatus.
4-(Aminomethyl)benzoic acid C8H9NO2 A white pow-der.
Purity—Dissolve 10 mg of 4-(aminomethyl)benzoic acidin 100 mL of water, and use this as the sample solution.Pipet 1 mL of the sample solution, add water to makeexactly 20 mL, and use this solution as the standardsolution. Perform the test with exactly 20 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the operatingconditions as directed in the Purity (5) under TranexamicAcid, and determine each peak area by the automaticintegration method: each area of the peak other than 4-(aminomethyl)benzoic acid obtained from the sample solu-tion is not more than the peak area of 4-(aminomethyl)ben-zoic acid from the standard solution.
Ammonium amminetrichloroplatinate for liquid chro-matography Cl3H7N2Pt To 20 g of cisplatin add 600 mLof 6 mol/L hydrochloric acid TS, and heat under a re‰uxcondenser for 4 – 6 hours to boil while stirring. After cool-ing, evaporate the solvent, and dry the orange residue atroom temperature under reduced pressure. To the residue soobtained add 300 mL of methanol, and heat at about 509Cto dissolve. Filter, separate insoluble yellow solids, and washthe solids with 10 mL of methanol. Combine the ˆltrate andthe washing, heat at about 509C, and add slowly 100 mL ofethyl acetate while stirring. Cool the mixture to roomtemperature avoiding exposure to light, and allow to standat -109C for 1 hour. Filter the mixture to take oŠ theformed crystals, wash the crystals with 100 mL of acetone,combine the washing to the ˆltrate, and evaporate to drynessto obtain orange crystals. If necessary, repeat the puriˆca-tion procedure described above to take oŠ the insolublecrystals. To the orange crystals obtained add 300 to 500 mLof a mixture of acetone and methanol (5:1), and heat atabout 509C while stirring to dissolve. Filter while hot to takeoŠ the insoluble crystals, wash the crystals with the mixture,and combine the ˆltrate and washing. Repeat the procedureseveral times, and evaporate to dryness. Suspense thecrystals so obtained in 50 mL of acetone, ˆlter, wash thecrystals with 20 mL of acetone, and dry the crystals at roomtemperature under reduced pressure. It is a yellow-browncrystalline powder.
Identiˆcation—Determine the infrared absorption spec-trum of the substance to be examined, previously dried at809C for 3 hours, as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry: it exhibitsabsorption at the wave numbers of about 3480 cm-1,3220 cm-1, 1622 cm-1, 1408 cm-1 and 1321 cm-1.
Related substances—Cisplatin Conduct this procedureusing light-resistant vessels. Dissolve 10 mg in N,N-dimethylformamide to make exactly 10 mL, and use thissolution as the sample solution. Separately, dissolve 10 mgof Cisplatin in N,N-dimethylformamide to make exactly50 mL. Pipet 5 mL of this solution, add N,N-dimethylfor-mamide to make exactly 20 mL, and use this solution as thestandard solution. Perform the test with exactly 40 mL eachof the sample solution and the standard solution as directedunder the Liquid Chromatography according to the follow-ing conditions, and determine the peak area of cisplatin bythe automatic integration method: the peak area from thesample solution is not more than that from the standardsolution.Operating conditions
Proceed as directed in the operating conditions in theAssay under Cisplatin.System suitability
System performance: When the procedure is run with40 mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of cisplatin are not less than 2500 andnot more than 2.0, respectively.
System repeatability: When the test is repeated 6 timeswith 40 mL of the standard solution under the above

16801680 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
operating conditions, the relative standard deviation of thepeak area of cisplatin is not more than 5.0z.
Benidipine hydrochloride C28H31N3O6.HCl [Same asthe namesake monograph]
Benidipine hydrochloride for assay C28H31N3O6.HCl[Same as the monograph Benidipine Hydrochloride. Whendried, it contains not less than 99.5z of benidipinehydrochloride (C28H31N3O6.HCl)]
Benzoin C6H5CH(OH)COC6H5 White to pale yellow,crystals or powder.
Melting point: 132 – 1379C
N-Benzoyl-L-isoleucyl-L-glutamyl(g-OR)-glycyl-L-arginyl-p-nitroanilide hydrochloride An equal amount mixture oftwo components, R=H and R=CH3. A white powder.Slightly soluble in water.
Absorbance E1z1 cm (316 nm): 166 – 184 (10 mg, water,
300 mL).
Benzoylmesaconine hydrochloride for thin-layer chro-matography C31H43NO10.HCl.xH2O White, crystals orcrystalline powder. Soluble in water and in ethanol (99.5).Melting point: about 2509C (with decomposition).
Purity Related substances—Dissolve 1.0 mg of ben-zoylmesaconine hydrochloride for thin-layer chro-matography in exactly 10 mL of ethanol (99.5), and use thissolution as the sample solution. Perform the test with thissolution as directed under the Thin-layer Chromatography.Spot 10 mL of the sample solution on a plate of silica gel forthin-layer chromatography. Proceed the test as directed inthe Identiˆcation under Processed Aconite Root: no spotother than the principal spot at around Rf 0.4 appears.
4,4?-Bis(diethylamino)benzophenone(C2H5)2NC6H4]2CO Light yellow crystals.
Content: not less than 98z. Assay—Weigh accurately0.25 g of 4,4?-bis(diethylamino)benzophenone, dissolve in50 mL of acetic acid (100), and titrate with 0.1 mol/Lperchloric acid VS (potentiometric titration). Perform ablank titration in the same manner, and make any necessarycorrection.
Each mL of 0.1 mol/L perchloric acid VS=16.22 mg of C21H28N2O
Borate-hydrochloric acid buŠer solution, pH 9.0 Dis-solve 19.0 g of sodium borate in 900 mL of water, adjust thepH to exactly 9.0 with 1 mol/L hydrochloric acid TS, andadd water to make 1000 mL.
Bovine activated blood coagulation factor X A proteinobtained from bovine plasma. It has an activity to decom-pose prothrombin speciˆcally and limitedly and producethrombin. It does not contain thrombin and plasmin. Itcontains not less than 500 Units per mg protein. One unitindicates an amount of the factor X which hydrolyzes1 mmol of N-benzoyl-L-isoleucyl-L-glutamyl(g-OR)-glycyl-L-arginyl-p-nitroanilide in 1 minute at 259C.
Calibration ball for particle density measurement
Calibration ball with a known volume prepared for measure-ment of particle density. The volume of the calibration ballmust be accurately determined to the nearest 0.001 cm3.
Caprylic acid CH3(CH2)6COOH A clear and colorlessoily liquid, having a slight unpleasant odor. Freely soluble inethanol (95) and in chloroform, and very slightly soluble inwater.
Refractive index: nD20 1.426 – 1.430
Speciˆc gravity d420: 0.908 – 0.912
Distilling range 238 – 2429C, not less than 95 volz.
Chlorobutanol C4H7Cl3O [Same as the namesakemonograph in Part II]
Chlorogenic acid for thin-layer chromatographyC16H18O9.xH2O A white powder. Freely soluble inmethanol and in ethanol (99.5), and sparingly soluble inwater. Melting point: about 2059C (with decomposition).
Purity Related substances—Dissolve 1.0 mg of chloro-genic acid for thin-layer chromatography in 2 mL ofmethanol, and use this solution as the sample solution.Perform the test with this solution as directed under theThin-layer Chromatography. Spot 10 mL of the samplesolution on a plate of silica gel for thin-layer chro-matography, develop the plate with a mixture of ethylacetate, water and formic acid (6:1:1) to a distance of about10 cm, and air-dry the plate. Examine under ultraviolet light(main wavelength: 365 nm): no spot other than the principalspot at around Rf 0.5 appears.
Cisplatin [Same as the namesake monograph]
Cyclohexylamine C6H11NH2 A clear and colorlessliquid, having a characteristic amine-like odor. Miscible withwater, with N,N-dimethylformamide and with acetone.
Purity Related substances—Use cyclohexylamine as thesample solution. Separately, pipet 1 mL of cyclohexylamine,add hexane to make exactly 100 mL, and use this as the stan-dard solution. Perform the test as directed in the Thin-layerChromatography. Spot 5 mL each of the sample solution andthe standard solution on a plate of silica gel for thin-layerchromatography, develop the plate with a mixture of ethylacetate, methanol, ammonia water (28) and cyclohexane(6:2:1:1) to a distance of about 10 cm, and air-dry the plate.Allow the plate to stand in iodine vapor: the spot other thanthe principal spot obtained with the sample solution is notmore intense than the spot with the standard solution.
Cyclohexylmethanol C7H14O A liquid having slightcamphor odor. Soluble in ethanol (99.5).
Refractive index nD20: about 1.464
Bioling point: about 1859C
N,N?-Dibenzylethylenediamine diacetate A white toslightly pale yellow crystalline powder.
Identiˆcation—Determine the infrared absorption spec-trum of the substance to be examined as directed in thepotassium bromide disk method under the Infrared Spec-trophotometry: it exhibits absorption at the wave numbersof about 1530 cm-1, 1490 cm-1, 1460 cm-1, 1400 cm-1 and

16811681Supplement II, JPXIV General Tests, Processes and Apparatus
1290 cm-1.Content: not less than 99.0z. Assay—Weigh accurately
about 25 mg of N,N?-dibenzylethylenediamine diacetate,dissolve in 25 mL of methanol, and add a solution contain-ing 1.02 g of disodium hydrogen phosphate and 6.80 g ofpotassium dihydrogen phosphate in 1000 mL of water tomake exactly 50 mL. Pipet 5 mL of this solution, add amixture of the solution containing 1.02 g of disodium hydro-gen phosphate and 6.80 g of potassium dihydrogen phos-phate in 1000 mL of water and methanol (1:1) to makeexactly 20 mL, and use this solution as the sample solution.Separately, weigh accurately about 8 mg of acetic acid (100),add 25 mL of methanol, and add the solution containing1.02 g of disodium hydrogen phosphate and 6.80 g ofpotassium dihydrogen phosphate in 1000 mL of water tomake exactly 50 mL. Pipet 5 mL of this solution, add amixture of the solution containing 1.02 g of disodium hydro-gen phosphate and 6.80 g of potassium dihydrogen phos-phate in 1000 mL of water and methanol (1:1) to makeexactly 20 mL, and use this solution as the control solution.Perform the test with exactly 20 mL each of the sample solu-tion and the control solution as directed under the LiquidChromatography according to the following conditions, anddetermine each peak area by the automatic integrationmethod. After making correction for the peak areas basedon the valiance of the base-line and the peak of acetic acidon the chromatogram obtained with the sample solution,calculate the amount of N,N?-dibenzylethylenediamine bythe area percentage method.Operating conditions
Detector: An ultraviolet absorption photometer(wavelength: 220 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: A mixture of water, methanol and 0.25mol/L potassium dihydrogen phosphate TS, pH 3.5(11:7:2).
Flow rate: Adjust the ‰ow rate so that the retention timeof N,N?-dibenzylethylenediamine is about 4 minutes.
Time span of measurement: About 5 times as long as theretention time of N,N?-dibenzylethylenediamine.System suitability
System performance: Dissolve an amount of Benzyl-penicillin Benzathine, equivalent to about 85,000 Units, in25 mL of methanol, add a solution containing 1.02 g ofdisodium hydrogen phosphate and 6.80 g of potassiumdihydrogen phosphate in 1000 mL of water to make exactly50 mL. Pipet 5 mL of this solution, add a mixture of thesolution containing 1.02 g of disodium hydrogen phosphateand 6.80 g of potassium dihydrogen phosphate in 1000 mLof water and methanol (1:1) to make exactly 20 mL. Whenthe procedure is run with 20 mL of this solution under theabove operating conditions, N,N?-dibenzylethylenediamineand benzylpenicillin are eluted in this order with the resolu-
tion between these peaks being not less than 20.System repeatability: When the test is repeated 6 times
with 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of N,N?-dibenzylethylenediamine is not more than2.0z.
1,2-Dichlorobenzene C6H4Cl2 A colorless liquid.Speciˆc gravity d4
20: 1.306Boiling point: 180 – 1819C
2,6-Dichlorophenol C6H4Cl2O White to purplish whitecrystals.
Melting point: 65 – 679C
Dicyclohexyl C12H22
Speciˆc gravity d2020: about 0.864
Boiling point: about 2279CMelting point: about 49C
Dicyclohexylurea C6H11NHCONHC6H11 A white crys-talline powder, having no odor.
Purity Related substances—Dissolve 50 mg of dicyclo-hexylurea in methanol to make 100 mL. Pipet 10 mL of thissolution, and add methanol to make 100 mL. Pipet 20 mL ofthis solution, add 10 mL of 0.5 mol/L sodium hydroxideTS, shake, then add 5 mL of diluted hydrochloric acid (1 in10), and shake. Perform the test with 50 mL of this solutionas directed under the Liquid Chromatography according tothe following conditions, determine the area of each peak bythe automatic integration method, and calculate the amountby the area percentage method: the total amount of thepeaks other than dicyclohexylurea is not more than 3.0z.Operating conditions
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Purity (5) (ii) under Acetohexamide.
Time span of measurement: About 5 times as long as theretention time of dicyclohexylurea after the solvent peak.System suitability
Test for required detectability: To exactly 5 mL of thestandard solution add water to make exactly 200 mL.Conˆrm that the peak area of dicyclohexylurea obtainedwith 50 mL of this solution is equivalent to 1.8 to 3.3z ofthat with 50 mL of the standard solution.
System performance, and system repeatability: Proceed asdirected in the system suitability in the Purity (5) (ii) underAcetohexamide.
Digoxin C41H64O14 [Same as the namesake mono-graph]
0.04 mol/L Disodium dihydrogen ethylenediamine tetraa-cetate TS Dissolve 14.890 g of disodium dihydrogenethylenediamine tetraacetate dihydrate in water to make1000 mL.
Ethylbenzene C6H5C2H5 A colorless liquid. Freelysoluble in acetone and in ethanol (99.5), and practically in-soluble in water.
Speciˆc gravity d420: 0.862¿0.872

16821682 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
Boiling point: about 1359C
Flopropione [Same as the namesake monograph]
Flopropione for assay [Same as the monographFlopropione. It contains not less than 99.0z of ‰opropione(C9H10O4), calculated on the anhydrous basis.]
Fluid thioglycolate medium See the Sterility Test underthe General Tests, Processes and Apparatus.
85z Glycerin C3H8O3 [Same as the monographGlycerin]
Human antithrombin III Serine protease inhibitionfactor obtained from normal plasma of health human. It is aprotein, which inhibits the activities of thrombin andactivated blood coagulation factor X. It contains not lessthan 300 Units per mg protein. One unit indicates an amountof the antithrombin III which inhibits 1 unit of thrombin at259C under the existence of heparin.
Dried human normal plasma powder Freeze-driednormal plasma obtained from healthy human.
Hydrogen peroxide-sodium hydroxide TS To a mixtureof water and hydrogen peroxide (30) (9:1) add 3 drops ofbromophenol blue TS, and then add 0.01 mol/L sodiumhydroxide TS until a purple-blue color develops. Preparebefore use.
N-(2-Hydroxyethyl)isonicotinamide nitric esterC8H9N3O4 A white crystalline powder.
Identiˆcation—Determine the infrared absorption spec-trum of N-(2-hydroxyethyl)isonicotinamide nitric ester asdirected in the potassium bromide disk method under theInfrared Spectrophotometry: it exhibits absorption at thewave numbers of about 3270 cm-1, 1653 cm-1, 1546 cm-1
and 1283 cm-1.
Hypaconitine for purity C33H45NO10 White, crystals orcrystalline powder. Soluble in acetonitrile, sparingly solublein ethanol (99.5) and in diethyl ether, and practically insolu-ble in water. Melting point: about 1759C (with decomposi-tion).
Identiˆcation—Determine the infrared absorption spec-trum of hypaconitine for purity as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry: it exhibits absorption at the wave numbers of about3500 cm-1, 1728 cm-1, 1712 cm-1, 1278 cm-1, 1118 cm-1,1099 cm-1 and 714 cm-1.
Absorbance E1z1 cm (230 nm): 217 – 252 [5 mg dried for
not less than 12 hours in a desiccator (reduced pressure notexceeding 0.67 kPa, phosphorus (V) oxide, 409C), ethanol(99.5), 200 mL].
Purity Related substances—(1) Dissolve 5.0 mg ofhypaconitine for purity in 2 mL of acetonitrile, and use asthe sample solution. Pipet 1 mL of the sample solution, addacetonitrile to make exactly 50 mL, and use as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 20 mL each ofthe sample solution and the standard solution on a plate of
silica gel for thin-layer chromatography, and proceed thetest as directed in the Identiˆcation in Processed AconiteRoot: the spot other than the principal spot obtained withthe sample solution is not more intense than the spot withthe standard solution.
(2) Dissolve 5.0 mg of hypaconitine for purity in 5 mLof acetonitrile, and use as the sample solution. Pipet 1 mL ofthe sample solution, add acetonitrile to make exactly 50 mL,and use as the standard solution. Perform the test with ex-actly 10 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the total area ofthe peaks other than the peaks of hypaconitine and the sol-vent obtained with the sample solution is not larger than thepeak area of hypaconitine with the standard solution.Operating conditions
Detector, column, and column temperature: Proceed asdirected in the operating conditions in the Purity underProcessed Aconite Root.
Mobile phase: A mixture of phosphate buŠer solution forprocessed aconite root and tetrahydrofuran (9:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof hypaconitine is about 23 minutes.
Time span of measurement: About 3 times as long as theretention time of hypaconitine.System suitability
Test for required detectability: Pipet 1 mL of the standardsolution, and add acetonitrile to make exactly 20 mL.Conˆrm that the peak area of hypaconitine obtained from10 mL of this solution is equivalent to 3.5 to 6.5z of thatobtained from 10 mL of the standard solution.
System performance: Dissolve 1 mg each of aconitine forpurity, hypaconitine for purity and mesaconitine for purity,and 8 mg of jesaconitine for purity in 200 mL of acetonitrile.When the procedure is run with 10 mL of this solution underthe above operating conditions, mesaconitine, hypaconitine,aconitine and jesaconitine are eluted in this order, and eachresolution between these peaks is not less than 1.5, respec-tively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of hypaconitine is not more than 1.5z.
Water: not more than 1.0z [5 mg dried for not less than12 hours in a desiccator (reduced pressure not exceeding0.67 kPa, phosphorus (V) oxide, 409C), coulometric titra-tion].
Icariin for thin-layer chromatography C33H40O15 Lightyellow crystals. Very slightly soluble in methanol and inethanol (99.5), and practically insoluble in water. Meltingpoint: about 2349C (with decomposition).
Purity Related substances—Dissolve 1.0 mg of icariinfor thin-layer chromatography in 1 mL of methanol. Per-form the test with 10 mL of this solution as directed in theIdentiˆcation under Epimedium Herb: no spot other thanthe principal spot having Rf value about 0.4 appears.

16831683Supplement II, JPXIV General Tests, Processes and Apparatus
L-Isoleucine C6H13NO2 [Same as the namesake mono-graph]
Jesaconitine for purity C35H49NO12 A white powder.Freely soluble in acetonitrile, in ethanol (99.5) and in diethylether, and practically insoluble in water.
Identiˆcation—Determine the infrared absorption spec-trum of jesaconitine for purity as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry: it exhibits absorption at the wave numbers of about3500 cm-1, 1715 cm-1, 1607 cm-1, 1281 cm-1, 1259 cm-1,1099 cm-1 and 772 cm-1.
Absorbance E1z1 cm (258 nm): 270 – 291 [5 mg dried for
not less than 12 hours in a desiccator (reduced pressure notexceeding 0.67 kPa, phosphorus (V) oxide, 409C), ethanol(99.5), 200 mL].
Purity Related substances—(1) Dissolve 5.0 mg ofjesaconitine for purity in 2 mL of acetonitrile, and use as thesample solution. Pipet 1 mL of the sample solution, addacetonitrile to make exactly 50 mL, and use as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 20 mL each ofthe sample solution and the standard solution on a plate ofsilica gel for thin-layer chromatography, and proceed thetest as directed in the Identiˆcation in Processed AconiteRoot: the spot other than the principal spot is not more in-tense than the spot with the standard solution.
(2) Dissolve 5.0 mg of jesaconitine for purity in 5 mL ofacetonitrile, and use as the sample solution. Pipet 1 mL ofthe sample solution, add acetonitrile to make exactly 50 mL,and use as the standard solution. Perform the test with ex-actly 10 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the total area ofthe peaks other than the peaks of jesaconitine and the sol-vent is not larger than the peak area of jesaconitine with thestandard solution.Operating conditions
Detector, column, and column temperature: Proceed asdirected in the operating conditions in the Purity underProcessed Aconite Root.
Mobile phase: A mixture of phosphate buŠer solution forprocessed aconite root and tetrahydrofuran (9:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof jesaconitine is about 36 minutes.
Time span of measurement: About 3 times as long as theretention time of jesaconitine.System suitability
Test for required detectability: Pipet 1 mL of the standardsolution, and add acetonitrile to make exactly 20 mL.Conˆrm that the peak area of jesaconitine obtained from10 mL of this solution is equivalent to 3.5 to 6.5z of thatobtained from 10 mL of the standard solution.
System performance: Dissolve 5 mg each of aconitine forpurity, hypaconitine for purity and mesaconitine for purity,and 1 mg of jesaconitine for purity in 200 mL of acetonitrile.When the procedure is run with 10 mL of this solution under
the above operating conditions, mesaconitine, hypaconitine,aconitine and jesaconitine are eluted in this order, and eachresolution between these peaks is not less than 1.5, respec-tively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of jesaconitine is not more than 1.5z.
Water: not more than 1.0z [5 mg dried for not less than12 hours in a desiccator (reduced pressure not exceeding0.67 kPa, phosphorus (V) oxide, 409C), coulometrictitration].
Loganin for thin-layer chromatography C17H26O10
White, crystals or crystalline powder. Soluble in water,sparingly soluble in methanol, and very slightly soluble inethanol (99.5). Melting point: 221 – 2279C.
Purity Related substances—Dissolve 1.0 mg of loganinfor thin-layer chromatography in 2 mL of methanol. Per-form the test with 10 mL of this solution as directed in theIdentiˆcation under Cornus Fruit: any spot other than theprincipal spot at the Rf value of about 0.4 does not appear.
Mesaconitine for purity C33H45NO11 White, crystals orcrystalline powder. Slightly soluble in acetonitrile and inethanol (99.5), very slightly soluble in diethyl ether, andpractically insoluble in water. Melting point: about 1909C(with decomposition).
Identiˆcation—Determine the infrared absorption spec-trum of mesaconitine for purity as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry: it exhibits absorption at the wave numbers of about3510 cm-1, 1713 cm-1, 1277 cm-1, 1116 cm-1, 1098 cm-1
and 717 cm-1.Absorbance E1z
1 cm (230 nm): 211 – 247 [5 mg dried fornot less than 12 hours in a desiccator (reduced pressure notexceeding 0.67 kPa, phosphorus (V) oxide, 409C), ethanol(99.5), 200 mL].
Purity Related substances—(1) Dissolve 5.0 mg ofmesaconitine for purity in 2 mL of acetonitrile, and use asthe sample solution. Pipet 1 mL of the sample solution, addacetonitrile to make exactly 50 mL, and use as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 20 mL each ofthe sample solution and the standard solution on a plate ofsilica gel for thin-layer chromatography, and proceed thetest as directed in the Identiˆcation under Processed AconiteRoot: the spot other than the principal spot is not more in-tense than the spot with the standard solution.
(2) Dissolve 5.0 mg of mesaconitine for purity in 5 mLof acetonitrile, and use as the sample solution. Pipet 1 mL ofthe sample solution, add acetonitrile to make exactly 50 mL,and use as the standard solution. Perform the test with ex-actly 10 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the total area ofthe peaks other than the peaks of mesaconitine and the sol-vent is not larger than the peak area of mesaconitine with the

16841684 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
standard solution.Operating conditions
Detector, column, and column temperature: Proceed asdirected in the operating conditions in the Purity underProcessed Aconite Root.
Mobile phase: A mixture of phosphate buŠer solution forprocessed aconite root and tetrahydrofuran (9:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof mesaconitine is about 19 minutes.
Time span of measurement: About 3 times as long as theretention time of mesaconitine.System suitability
Test for required detectability: Pipet 1 mL of the standardsolution, and add acetonitrile to make exactly 20 mL.Conˆrm that the peak area of mesaconitine obtained from10 mL of this solution is equivalent to 3.5 to 6.5z of thatobtained from 10 mL of the standard solution.
System performance: Dissolve 1 mg each of aconitine forpurity, hypaconitine for purity and mesaconitine for purity,and 8 mg of jesaconitine for purity in 200 mL of acetonitrile.When the procedure is run with 10 mL of this solution underthe above operating conditions, mesaconitine, hypaconitine,aconitine and jesaconitine are eluted in this order, and eachresolution between these peaks is not less than 1.5, respec-tively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of mesaconitine is not more than 1.5z.
Water: not more than 1.0z [5 mg dried for not less than12 hours in a desiccator (reduced pressure not exceeding0.67 kPa, phosphorus (V) oxide, 409C), coulometric titra-tion].
Methanol for liquid chromatography CH3OH A clear,colorless liquid. Mixable with water.
Purity Ultraviolet-absorbing substances—Perform thetest as directed in the Ultraviolet-visible Spectrophotometryusing water as the blank: the absorbances at 210 nm, at220 nm, at 230 nm, at 240 nm and at 254 nm are not morethan 0.70, 0.30, 0.15, 0.07 and 0.02, respectively.
Metoclopramide for assay [Same as the monographMetoclopramide. When dried, it contains not less than99.0z of metoclopramide (C14H22ClN3O2).]
1,3-Naphthalenediol C10H8O2 Red-brown crystals orgray-brown powder. Freely soluble in water, in methanoland in ethanol (99.5). Melting point: about 1249C.
1,3-Naphthalenediol TS Dissolve 50 mg of 1,3-naphthalenediol in 25 mL of ethanol (99.5), and add 2.5 mLof phosphoric acid.
3-Nitrophenol C6H5NO3 A light yellow crystallinepowder.
Melting point: 96 – 999C
Osthole for thin-layer chromatography C15H16O3 Awhite crystalline powder, having no odor. Freely soluble inmethanol and in ethyl acetate, soluble in ethanol (99.5), and
practically insoluble in water. Melting point: 83 – 849C.Purity Related substances—Dissolve 1.0 mg of osthole
for thin-layer chromatography in 1 mL of methanol. Per-form the test with 10 mL of this solution as directed in theIdentiˆcation under Cnidium Monnieri Fruit: on spot ap-pears other than the principal spot at around Rf 0.3.
Oxytocin C43H66N12O12S2 [Same as the namesakemonograph]
D-Phenylglycine C8H9NO2 White, crystals or crystal-line powder. Slightly soluble in water.
Loss on drying: not more than 0.5z (1 g, 1059C,3 hours).
Content: not less than 98.5z. Assay—Weigh accuratelyabout 0.3 g of D-phenylglycine, previously dried, dissolve in3 mL of formic acid, add 50 mL of acetic acid (100), andtitrate with 0.1 mol/L perchloric acid VS (potentiometrictitration). Perform a blank determination in the samemanner, and make any necessary correction.
Each mL of 0.1 mol/L perchloric acid VS=15.12 mg of C8H9NO2
Phenylpiperazine hydrochloride C10H14N2.HCl Awhite powder.
Melting point: about 2479C (with decomposition).
Phenylsilanized silica gel for liquid chromatographyPrepared for liquid chromatography.
0.05 mol/L Phosphate buŠer solution, pH 3.5 To 1000mL of 0.05 mol/L potassium dihydrogen phosphate TS adda suitable amount of a solution of phosphoric acid (49 in10,000) to make a solution having pH 3.5.
Phosphate buŠer solution for processed aconite rootDissolve 19.3 g of disodium hydrogen phosphate 12-water in3660 mL of water, and add 12.7 g of phosphoric acid.
[6]-Shogaol for thin-layer chromatography C17H24O3
A pale yellow oil. Miscible with methanol, ethanol (99.5)and with diethyl ether, and practically insoluble in water.
Purity Related substances—Dissolve 1.0 mg of [6]-shogaol for thin-layer chromatography in 2 mL ofmethanol, and perform the test with this solution as directedunder the Thin-layer chromatography. Spot 10 mL on a plateof silica gel for thin-layer chromatography, develop the platewith a mixture of ethyl acetate and hexane (1:1) to a distanceof about 10 cm, and air-dry the plate. Spray evenly 4-dimethylaminobenzaldehyde TS on the plate, heat at 1059Cfor 5 minutes, and allow to cool: no spot other than theprincipal spot at around Rf 0.5 appears.
0.0375 mol/L Sodium 1-decanesulfonate TS Dissolve3.665 g of sodium 1-decanesulfonate in 400 mL of water.
Sodium dihydrogen phosphate NaH2PO4 A white,powder or crystalline powder. Freely soluble in water, andvery slightly soluble in ethanol (99.5). It has a hygroscopicproperty.
A solution is acidic.

16851685Supplement II, JPXIV General Tests, Processes and Apparatus
Soybean-casein digest medium See the Sterility Testunder the General Tests, Processes and Apparatus.
Thioacetamide C2H5NS A white crystalline powder orcolorless crystals. Freely soluble in water and in ethanol(99.5). Melting point: 112 – 1159C
Thioacetamide TS To 0.2 mL of a solution of thioa-cetamide (1 in 25) add 1 mL of a mixture of 15 mL ofsodium hydroxide TS, 5 mL of water and 20 mL of 85zglycerin, and heat in a water bath for 20 seconds. Preparebefore use.
Tiaramide hydrochloride for assay [Same as themonograph Tiaramide Hydrochloride. When dried, it con-tains not less than 99.0z of tiaramide hydrochloride(C15H18ClN3O3S.HCl).]
Trichloroacetic acid TS for serrapeptase Dissolve 1.80 gof trichloroacetic acid and 1.80 g of anhydrous sodiumacetate in 5.5 mL of 6 mol/L acetic acid TS and water tomake 100 mL.
Trichloro‰uoromethane CCl3F A colorless liquid orgas.
Speciˆc gravity d 417.2: 1.494
Boiling point: 23.79C
Vasopressin C46H65N15O12S2 A white powder.Constituent amino acids—Perform the test as directed in
the Constituent amino acids under Oxytocin, and calculatethe respective molar ratios with respect to glycine: 0.9 – 1.1for aspartic acid, 0.9 – 1.1 for glutamic acid, 0.9 – 1.1 forproline, 0.8 – 1.1 for tyrosine, 0.9 – 1.1 for phenylalanine,0.9 – 1.1 for arginine and 0.8 – 1.1 for cystine, and not morethan 0.03 for other amino acids.
0.04 mol/L Zinc chloride TS Dissolve 5.452 g of zincchloride in water to make 1000 mL.
(3) Standard Solutions forVolumetric Analysis
Change the following:
Iodine, 0.05 mol/L1000 mL of this solution contains 12.690 g of iodine
(I: 126.90).Preparation—Dissolve 13 g of iodine in 100 mL of a solu-
tion of potassium iodide (2 in 5), add 1 mL of dilutehydrochloric acid and water to make 1000 mL, and stan-dardize the solution as follows:
Standardization—Measure exactly 15 mL of the iodinesolution, and titrate with 0.1 mol/L sodium thiosulfate VS(Indicator method: starch TS; or potentiometric titration:platinum electrode). In the indicator method, when thesolution assumes a pale yellow color as the end point isapproached, add 3 mL of starch TS, and continue the titra-tion until the blue color disappears. Calculate the molarityfactor.
Note: Store protected from light. This solution, if storedfor a long period, should be restandardized before use.
Add the following:
Sodium Thiosulfate, 0.002 mol/L1000 mL of this solution contains 0.4964 g of sodium
thiosulfate pentahydrate (Na2S2O3.5H2O: 248.19).Preparation—Before use, dilute 0.1 mol/L sodium
thiosulfate VS with freshly boiled and cooled water to makeexactly 50 times the initial volume.
Add the following:
74. Powder Particle DensityDetermination
Powder Particle Density Determination is a method todetermine particle density of powdered pharmaceuticaldrugs or raw materials of drugs, and the gas displacementpycnometer is generally used. The powder density by thismethod is determined with an assumption that the volume ofthe gas displaced by the powder in a closed system is equal tothe volume of the powder. The bulk density at loose packingor the tapped density at tapping express the apparentdensities of the powder, since interparticulate void volumeof the powder is assumed to contribute a part of the volumeof the powder. On the contrary, the pycnometric particledensity expresses the powder density nearly equal to the crys-tal density, since the volume of the powder, that is deductedwith void volume of open pores accessible to gas, is counted.
Powder particle density is expressed in mass per unitvolume (kg/m3), and generally expressed in g/cm3.
ApparatusThe schematic diagram of particle density apparatus for
gas displacement pycnometric measurement is shown inFigure 1. The apparatus consists of a test cell in which thesample is placed, a reference cell and a manometer.
Generally, helium is used as the measurement gas. Theapparatus has to be equipped with a system capable ofpressuring the test cell to the deˆned pressure through themanometer.
Calibration of apparatus The volumes of the test cell(Vc) and the reference cell (Vr) must be accurately deter-mined to the nearest 0.001 cm3, and to assure accuracy of theresults of volume obtained, calibration of the apparatus iscarried out as follows using a calibration ball of knownvolume for particle density measurement. The ˆnal pressures(Pf) are determined for the initial empty test cell followed bythe test cell placed with the calibration ball for particledensity measurement in accordance with the procedures, andVc and Vr are calculated using the equation described in thesection of Procedure. Calculation can be made taking intoaccount that the sample volume (Vs) is zero in the ˆrst run.

16861686 Supplement II, JPXIVGeneral Tests, Processes and Apparatus
Vr: Reference cell volume (cm3)Vc: Test cell volume (cm3)Vs: Sample volume (cm3)M: Manometer
Figure 1. Schematic diagram of a gas displacement py-cnometer
ProcedureThe measurement of the particle density is carried out
between 15 and 309C, and temperature must not vary bymore than 29C during the course of measurement.
Firstly, weigh the mass of the test cell and record it. Afterweighing out the amount of the sample as described in theindividual monograph and placing it in the test cell, seal thecell in the pycnometer. Secondly, introduce the measure-ment gas (helium) into the test cell, and remove volatilecontaminants in the powder. If necessary, keep the samplepowder under reduced pressure to remove the volatilecontaminants in advance and use it as the test sample formeasurement.
Open the valve which connects the reference cell with thetest cell, conˆrm with the manometer that the pressure inside
the system is stable, and then read the system referencepressure (Pr). Secondly, close the valve that connects to thetwo cells, and introduce the measurement gas into the testcell to achieve positive pressure. Conˆrm with the manome-ter that the pressure inside the system is stable, and then readthe initial pressure (Pi). Open the valve to connect the testcell with the reference cell. After conˆrming that the indica-tor of the manometer is stable, read the ˆnal pressure (Pf),and calculate the sample volume (Vs) with the followingequation.
Vs=Vc-Vr
Pi-Pr
Pf-Pr-1
Vr: Reference cell volume (cm3)Vc: Test cell volume (cm3)Vs: Sample volume (cm3)Pi: Initial pressure (kPa)Pf: Final pressure (kPa)Pr: Reference pressure (kPa)
Repeat the measurement sequence for the same powdersample until consecutive measurements of the samplevolume agree to within 0.5z, and calculate the mean ofsample volumes (Vs). Finally, unload the test cell, weigh themass of test cell, and calculate the ˆnal sample mass bydeducting the empty cell mass from the test cell mass. Thepowder particle density r is calculated by the followingequation.
r= mVs
r: Powder particle density (g/cm3)m: Final sample mass (g)Vs: Sample volume (cm3)

16871687
O‹cial Monographsfor Part I
Acetohexamideアセトヘキサミド
Change the origin/limits of content to read:
Acetohexamide, when dried, contains not less than98.0z and not more than 101.0z of C15H20N2O4S.
Change the Description to read:
Description Acetohexamide occurs as a white to yellowishwhite powder.
It is freely soluble in N,N-dimethylformamide, sparinglysoluble in acetone, slightly soluble in methanol and inethanol (99.5), and practically insoluble in water.
Melting point: about 1859C (with decomposition).
Change the Purity (5) to read:
Purity(5) Related substances (i) Cyclohexylamine—Dissolve
exactly 1.0 g of Acetohexamide in exactly 30 mL of 0.5mol/L sodium hydroxide TS, add exactly 5 mL of hexane,shake vigorously for 60 minutes, allow to stand for 5minutes, and use the upper layer as the sample solution.Separately, dissolve exactly 50 mg of cyclohexylamine in 0.5mol/L sodium hydroxide TS to make exactly 50 mL. Pipet 2mL of this solution, and add 0.5 mol/L sodium hydroxideTS to make exactly 300 mL. Pipet 30 mL of this solution,add exactly 5 mL of hexane, shake vigorously for 60minutes, allow to stand for 5 minutes, and use the upper lay-er as the standard solution. Perform the test with exactly 2mL each of the sample solution and the standard solution asdirected under the Gas Chromatography according to thefollowing conditions, and determine the peak area of cyclo-hexylamine by the automatic integration method: the peakarea of cyclohexylamine is not more than that with the stan-dard solution.Operating conditions—
Detector: A hydrogen ‰ame-ionization detector.Column: A fused-silica column 0.53 mm in inside di-
ameter and 30 m in length, coated the inner surface withmethylsilicone polymer for gas chromatography 1.5 mm inthickness.
Column temperature: A constant temperature of about909C.
Injection port temperature: A constant temperature ofabout 1509C.
Detector temperature: A constant temperature of about2109C.
Carrier gas: HeliumFlow rate: Adjust the ‰ow rate so that the retention time
of cyclohexylamine is about 4 minutes.Split ratio: 1:1
System suitability—System performance: When the procedure is run with 2 mL
of the standard solution under the above operatingconditions, the number of theoretical plates of the peak ofcyclohexylamine is not less than 8000.
System repeatability: When the test is repeated 6 timeswith 2 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of cyclohexylamine is not more than 5z.
(ii) Dicyclohexylurea—Dissolve exactly 1.0 g of Aceto-hexamide in exactly 10 mL of 0.5 mol/L sodium hydroxideTS, add exactly 20 mL of methanol, shake, then add exactly5 mL of diluted hydrochloric acid (1 in 10), shake vigorouslyfor 15 minutes, and centrifuge. Filter 10 mL or more of thesupernatant liquid through a membrane ˆlter with pore sizeof not larger than 0.5 mm. Discard the ˆrst 5 mL of theˆltrate, and use the subsequent ˆltrate as the sample solu-tion. Separately, dissolve exactly 50 mg of dicyclohexylureain methanol to make exactly 100 mL. Pipet 2 mL of this so-lution, and add methanol to make exactly 100 mL. Pipet 20mL of this solution, add exactly 10 mL of 0.5 mol/L sodiumhydroxide TS, shake, then add exactly 5 mL of dilutedhydrochloric acid (1 in 10), shake, and use this solution asthe standard solution. Perform the test with exactly 50 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine the peak area of dicy-clohexylurea by the automatic integration method: the peakarea of dicyclohexylurea is not more than that with the stan-dard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 210 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 0.5 g of sodium hydroxide in 1000mL of 0.05 mol/L sodium dihydrogen phosphate TS, andadjust the pH to 6.5 with 0.5 mol/L sodium hydroxide TS.To 500 mL of this solution add 500 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention time

16881688 Supplement II, JPXIVO‹cial Monographs for Part I
of dicyclohexylurea is about 10 minutes.System suitability—
System performance: When the procedure is run with50 mL of the standard solution under the above operatingconditions, the number of theoretical plates of the peak ofdicyclohexylurea is not less than 10,000.
System repeatability: When the test is repeated 6 timeswith 50 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of dicyclohexylurea is not more than 2.0z.
(iii) Other related substances—Dissolve 0.10 g of Aceto-hexamide in 10 mL of acetone, and use this solution as thesample solution. Pipet 1 mL of the sample solution, and addacetone to make exactly 20 mL. Pipet two 1-mL portions ofthis solution, add acetone to make exactly 10 mL and25 mL, respectively, and use these solutions as the standardsolution (1) and the standard solution (2). Perform the testwith these solutions as directed under the Thin-layerChromatography. Spot 10 mL each of the sample solution,the standard solution (1) and the standard solution (2) on aplate of silica gel with ‰uorescent indicator for thin-layerchromatography. Develop the plate with a mixture of ethylacetate, methanol, ammonia water (28) and cyclohexane(6:2:1:1) to a distance of about 10 cm, and air-dry the plate.Examine under ultraviolet light (main wavelength: 254 nm):the spots other than the principal spot from the samplesolution are not more intense than spot from the standardsolution (1), and the number of them which are more intensethan the spot from the standard solution (2) is not morethan 4.
Add the following:
Alprostadilアルプロスタジル
Prostaglandin E1
C20H34O5: 354.487-s(1R,2R,3R)-3-Hydroxy-2-[(1E,3S)-3-hydroxyoct-1-en-1-yl]-5-oxocyclopentyltheptanoic acid [745-65-3]
Alprostadil, when dried, contains not less than97.0z and not more than 103.0z of C20H34O5.
Description Alprostadil occurs as white crystals or crystal-line powder.
It is freely soluble in ethanol (99.5) and in tetrahydrofu-ran, slightly soluble in acetonitrile, and practically insolublein water.
Identiˆcation (1) The absorption spectrum of a solutionof Alprostadil in ethanol (99.5) (1 in 100,000) determined asdirected under the Ultraviolet-visible Spectrophotometry
shows no absorption between 210 nm and 350 nm. Separate-ly, to 10 mL of this solution add 1 mL of potassium hydrox-ide-ethanol TS, allow to stand for 15 minutes, and determinethe absorption spectrum in the same way. Compare thespectrum so obtained with the Reference Spectrum or thespectrum of a solution of Alprostadil Reference Standardprepared in the same manner as the sample solution: bothspectra exhibit similar intensities of absorption at the samewavelengths.
(2) Determine the infrared absorption spectrum ofAlprostadil, previously dried, as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry, and compare the spectrum with the Reference Spectrumor the spectrum of previously dried Alprostadil ReferenceStandard: both spectra exhibit similar intensities of absorp-tion at the same wave numbers.
Optical rotation [a]D20: -53 –-619(after drying, 25 mg,tetrahydrofuran, 5 mL, 100 mm).
Melting point 114 – 1189C
Purity Related substances—Dissolve 4 mg of Alprostadilin 2 mL of a mixture of acetonitrile for liquid chro-matography and water (9:1), and use this solution as thesample solution. Pipet 0.5 mL of the sample solution, andadd the mixture of acetonitrile for liquid chromatographyand water (9:1) to make exactly 10 mL. Pipet 2 mL of thissolution, add the mixture of acetonitrile for liquid chro-matography and water (9:1) to make exactly 10 mL, and usethis solution as the standard solution. Perform the test withexactly 5 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the area of thepeaks, having the relative retention time of about 0.70 and1.26 with respect to alprostadil, is not larger than 1/2 timesthe peak area of alprostadil with the standard solution, thearea of the peaks, having the relative retention time of about0.88 and 1.18 with respect to alprostadil, is not larger thanthe peak area of alprostadil with the standard solution, thearea of the peaks other than alprostadil and the peaks men-tioned above is not larger than 1/10 times the peak area ofalprostadil with the standard solution and the total area ofthe peaks other than alprostadil is not larger than 2 times thepeak area of alprostadil with the standard solution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 5 times as long as theretention time of alprostadil after the solvent peak.System suitability—
Test for required detectability: Measure exactly 2 mL ofthe standard solution, add the mixture of acetonitrile forliquid chromatography and water (9:1) to make exactly20 mL. Conˆrm that the peak area of alprostadil obtainedwith 5 mL of this solution is equivalent to 7 to 13z of thatwith 5 mL of the standard solution.

16891689Supplement II, JPXIV O‹cial Monographs for Part I
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of alprostadil is not more than 1.5z.
Loss on drying Not more than 1.0z (0.1 g, in vacuum,phosphorus (V) oxide, 4 hours).
Assay Weigh accurately about 5 mg each of Alprostadiland Alprostadil Reference Standard, previously dried,dissolve in exactly 5 mL of the internal standard solution,add a mixture of acetonitrile for liquid chromatography andwater (9:1) to make 50 mL, and use these solutions as thesample solution and the standard solution, respectively.Perform the test with 5 mL each of the sample solution andthe standard solution as directed under the Liquid Chro-matography according to the following conditions, anddetermine the ratios, QT and QS, of the peak area ofalprostadil to that of the internal standard.
Amount (mg) of C20H34O5=WS×QT
QS
WS: Amount (mg) of Alprostadil Reference Standard
Internal standard solution—A solution of dimethyl phtha-late in the mixture of acetonitrile for liquid chromatographyand water (9:1) (1 in 10,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 196 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 9.07 g of potassium dihydrogenphosphate in water to make 1000 mL. Adjust the pH to 6.3with a solution prepared by dissolving 9.46 g of disodiumhydrogen phosphate in water to make 1000 mL, and diluteto 10 times its volume with water. To 360 mL of this solutionadd 110 mL of acetonitrile for liquid chromatography and30 mL of methanol for liquid chromatography.
Flow rate: Adjust the ‰ow rate so that the retention timeof alprostadil is about 10 minutes.System suitability—
System performance: When the procedure is run with 5 mLof the standard solution under the above operating condi-tions, alprostadil and the internal standard are eluted in thisorder with the resolution between these peaks being not lessthan 9.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of alprostadil to that of the internal standard isnot more than 1.0z.
Containers and storage Containers—Tight containers.Storage—Light-resistant, and at a temperature not
exceeding 59C.
Atropine Sulfate Injection硫酸アトロピン注射液
Change the origin/limits of content to read:
Atropine Sulfate Injection is an aqueous solutionfor injection.
It contains not less than 93.0z and not more than107.0z of the labeled amount of atropine sulfate[(C17H23NO3)2.H2SO4.H2O: 694.83].
Change the Identiˆcation (2) to read:
Identiˆcation(2) Evaporate an exactly measured volume of Atropine
Sulfate Injection, equivalent to 5 mg of Atropine Sulfateaccording to the labeled amount, on a water bath to dryness.After cooling, dissolve the residue in 1 mL of ethanol (95),and use this solution as the sample solution. If insolublesubstance remains, crush it, allow to stand, and use thesupernatant liquid as the sample solution. Separately,dissolve 10 mg of Atropine Sulfate Reference Standard in2 mL of ethanol (95), and use this solution as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 5 mL each ofthe sample solution and the standard solution on a plate ofsilica gel for thin-layer chromatography. Develop the platewith a mixture of acetone, water and ammonia water (28)(90:7:3) to a distance of about 10 cm, and dry the plate at809C for 10 minutes. After cooling, spray evenly Dragen-dorŠ's TS for spraying on the plate: the spots obtained fromthe sample solution and the standard solution show anorange color and the same Rf value.
Add the following next to Identiˆcation:
Bacterial endotoxins Less than 75 EU/mg.
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirement of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirement of the Insoluble Particu-late Matter Test for Injections.
Sterility Perform the test: it meets the requirement of theSterility Test.
Change the Assay to read:
Assay To an exactly measured volume of Atropine SulfateInjection, equivalent to about 5 mg of atropine sulfate[(C17H23NO3)2.H2SO4.H2O], add exactly 3 mL of the inter-nal standard solution and water to make 50 mL, and use this

16901690 Supplement II, JPXIVO‹cial Monographs for Part I
solution as the sample solution. Separately, weigh accuratelyabout 25 mg of Atropine Sulfate Reference Standard,separately determine its loss on drying in the same condi-tions as for Atropine Sulfate, and dissolve in water to makeexactly 50 mL. Pipet 10 mL of this solution, add exactly 3mL of the internal standard solution and water to make 50mL, and use this solution as the standard solution. Performthe test with 20 mL each of the sample solution and the stan-dard solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of atropine sulfate tothat of the internal standard.
Amount (mg) of atropine sulfate[(C17H23NO3)2.H2SO4.H2O]
=WS×QT
QS×
15×1.027
WS: Amount (mg) of Atropine Reference Standard, calcu-lated based on the dried basis
Internal standard solution—A solution of etilefrinehydrochloride (1 in 1000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 210 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: To 0.4 g of sodium lauryl sulfate add500 mL of diluted phosphoric acid (1 in 1000) to dissolve,and adjust the pH to 3.0 with sodium hydroxide TS. To240 mL of this solution add 70 mL of tetrahydrofuran, andmix.
Flow rate: Adjust the ‰ow rate so that the retention timeof atropine sulfate is about 16 minutes.System suitability—
System performance: When the procedure is run with 20mL of the standard solution under the above operatingconditions, the internal standard and atropine sulfate areeluted in this order with the resolution between these peaksbeing not less than 3.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of atropine sulfate to that of the internalstandard is not more than 1.5z.
Add the following:
Azithromycin Hydrateアジスロマイシン水和物
C38H72N2O12.2H2O: 785.02(2R,3S,4S,5R,6R,8R,11R,12R,13S,14R)-5-(3,4,6-Trideoxy-3-dimethylamino-b-D-xylo-hexopyranosyloxy)-3-(2,6-dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyloxy)-10-aza-6,12,13-trihydroxy-2,4,6,8,10,11,13-heptamethylhexadecan-14-olide dihydrate[117772-70-0]
Azithromycin Hydrate is the derivative oferythromycin.
It contains not less than 945 mg (potency) and notmore than 1030 mg (potency) per mg, calculated on theanhydrous basis. The potency of Azithromycin Hy-drate is expressed as mass (potency) of azithromycin(C38H72N2O12: 748.98).
Description Azithromycin Hydrate occurs as a whitecrystalline powder.
It is freely soluble in methanol and in ethanol (99.5), andpractically insoluble in water.
Identiˆcation Determine the infrared absorption spectrumof Azithromycin Hydrate as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry, and compare the spectrum with the Reference Spectrumor the spectrum of Azithromycin Reference Standard: bothspectra exhibit similar intensities of absorption at the samewave numbers.
Optical rotation [a]D20: -45 –-499(400 mg calculated onthe anhydrous basis, ethanol (99.5), 20 mL, 100 mm).
Purity (1) Heavy metals—Proceed with 1.0 g ofAzithromycin Hydrate according to Method 2, and performthe test. Prepare the control solution with 1.0 mL ofStandard Lead Solution (not more than 10 ppm).
(2) Related substances—Being speciˆed separately.(3) Residual solvent—Being speciˆed separately.
Water Not less than 4.0z and not more than 5.0z (0.4 g,volumetric titration, direct titration).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately an amount of Azithromycin

16911691Supplement II, JPXIV O‹cial Monographs for Part I
Hydrate and Azithromycin Reference Standard, equivalentto about 50 mg (potency), dissolve each in an adequateamount of a mixture of acetonitrile and water (3:2), addexactly 2 mL of the internal standard solution and themixture of acetonitrile and water (3:2) to make 50 mL, anduse these solutions as the sample solution and the standardsolution. Perform the test with 5 mL each of the samplesolution and the standard solution as directed under theLiquid Chromatography according to the following condi-tions, and determine the ratios, QT and QS, of the peak areaof azithromycin to that of the internal standard.
Amount [mg (potency)] of azithromycin (C38H72N2O12)
=WS×QR
QS×1000
WS: Amount [mg (potency)] of Azithromycin ReferenceStandard
Internal standard solution—A solution of 4,4?-bis(diethylamino)benzophenone in acetonitrile (3 in 4000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 215 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized polyvinyl alcohol gel polymer for liquid chro-matography (5 mm in particle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 6.97 g of dipotassium hydrogenphosphate in about 750 mL of water, adjust the pH to 11.0with potassium hydroxide TS, and add water to make 1000mL. To 400 mL of this solution add 600 mL of acetonitrilefor liquid chromatography.
Flow rate: Adjust the ‰ow rate so that the retention timeof azithromycin is about 10 minutes.System suitability—
System performance: When the procedure is run with 5 mLof the standard solution under the above operatingconditions, azithromycin and the internal standard areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratiosof the peak area of azithromycin to that of the internalstandard is not more than 1.0z.
Containers and storage Containers—Tight containers.
Add the following:
Benidipine Hydrochloride塩酸ベニジピン
C28H31N3O6.HCl: 542.023-[(3RS)-1-Benzylpiperidin-3-yl] 5-methyl (4RS)-1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate monohydrochloride [91599-74-5]
Benidipine Hydrochloride, when dried, contains notless than 99.0z and not more than 101.0z ofC28H31N3O6.HCl.
Description Benidipine Hydrochloride occurs as a yellowcrystalline powder.
It is very soluble in formic acid, soluble in methanol,sparingly soluble in ethanol (99.5), and practically insolublein water.
A solution of Benidipine Hydrochloride in methanol (1 in100) shows no optical rotation.
Melting point: about 2009C (with decomposition).
Identiˆcation (1) Determine the absorption spectrum ofa solution of Benidipine Hydrochloride in methanol (1 in100,000) as directed under the Ultraviolet-visible Spec-trophotometry, and compare the spectrum with theReference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofBenidipine Hydrochloride, previously dried, as directed inthe potassium chloride disk method under the Infrared Spec-trophotometry, and compare the spectrum with the Refer-ence Spectrum: both spectra exhibit similar intensities of ab-sorption at the same wave numbers.
(3) To 5 mL of a solution of Benidipine Hydrochloride(1 in 10) add 5 mL of ammonia TS, heat on a water bath for5 minutes, cool, and ˆlter. The ˆltrate, which is acidiˆedwith dilute nitric acid, responds to the Qualitative Tests (2)for chloride.
Purity (1) Heavy metals—Proceed with 1.0 g of Benidi-pine Hydrochloride according to Method 2, and perform thetest. Prepare the control solution with 2.0 mL of StandardLead Solution (not more than 20 ppm).
(2) Related substances—Dissolve 20 mg of BenidipineHydrochloride in 100 mL of a mixture of water andmethanol (1:1), and use this solution as the sample solution.Pipet 1 mL of the sample solution, add the mixture of waterand methanol (1:1) to make exactly 500 mL, and use thissolution as the standard solution. Perform the test with ex-actly 10 mL each of the sample solution and the standard so-

16921692 Supplement II, JPXIVO‹cial Monographs for Part I
lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the peak areas ofbisbenzylpiperidyl ester having the relative retention time ofabout 0.35 with respect to benidipine, dehydro derivativehaving the relative retention time of about 0.75 and otherrelated substances are not larger than 1/2 times the peakarea of benidipine with the standard solution, and the totalarea of the peaks other than benidipine is not larger than thepeak area of benidipine with the standard solution. For thiscalculation, use the peak areas of bisbenzylpiperidyl esterand dehydro derivative after multiplying by their responsefactors, 1.6, respectively.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 237 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 10 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (3 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of 0.05 mol/L potassium di-hydrogen phosphate TS, pH 3.0, methanol and tetra-hydrofuran (65:27:8).
Flow rate: Adjust the ‰ow rate so that the retention timeof benidipine is about 20 minutes.
Time span of measurement: About 2 times as long as theretention time of benidipine after the solvent peak.System suitability—
Test for required detectability: Measure exactly 5 mL ofthe standard solution, and add the mixture of water andmethanol (1:1) to make exactly 20 mL. Conˆrm that thepeak area of benidipine obtained with 10 mL of this solutionis equivalent to 18 to 32z of that with 10 mL of the standardsolution.
System performance: Dissolve 6 mg of BenidipineHydrochloride and 5 mg of benzoin in 200 mL of themixture of water and methanol (1:1). When the procedure isrun with 10 mL of this solution under the above operatingconditions, benzoin and benidipine are eluted in this orderwith the resolution between these peaks being not lessthan 8.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of benidipine is not more than 3.5z.
Loss on drying Not more than 0.5z (0.5 g, 1059C,2 hours).
Residue on ignition Not more than 0.10z (1 g).
Assay Weigh accurately about 0.7 g of BenidipineHydrochloride, previously dried, dissolve in 10 mL offormic acid, add 70 mL of acetic anhydride, and titrate with0.1 mol/L perchloric acid VS (potentiometric titration).Perform a blank determination in the same manner, andmake any necessary correction.
Each mL of 0.1 mol/L perchloric acid VS=54.20 mg of C28H31N3O6.HCl
Containers and storage Containers—Tight containers.
Add the following:
Benidipine Hydrochloride Tablets塩酸ベニジピン錠
Benidipine Hydrochloride Tablets contain notless than 95.0z and not more than 105.0z of thelabeled amount of benidipine hydrochloride(C28H31N3O6.HCl: 542.02).
Method of preparation Prepare as directed under Tablets,with Benidipine Hydrochloride.
Identiˆcation Shake well a quantity of powdered Benidi-pine Hydrochloride Tablets, equivalent to 10 mg of Benidi-pine Hydrochloride according to the labeled amount, with100 mL of methanol, and centrifuge. To 10 mL of the super-natant liquid add methanol to make 100 mL, and use this so-lution as the sample solution. Determine the absorptionspectrum of the sample solution as directed under theUltraviolet-visible Spectrophotometry: it exhibits maximabetween 235 nm and 239 nm, and between 350 nm and 360nm.
Purity Dehydro derivative—Weigh accurately the mass ofnot less than 20 Benidipine Hydrochloride Tablets, andpowder in an agate mortar. To an amount of the powder,equivalent to 20 mg of Benidipine Hydrochloride, add about80 mL of a mixture of diluted phosphoric acid (1 in 500) andmethanol (1:1), shake well, and add the mixture of dilutedphosphoric acid (1 in 500) and methanol (1:1) to makeexactly 100 mL. Filter through a membrane ˆlter with poresize of 0.45 mm, and use the ˆltrate as the sample solution.Separately, dissolve 20 mg of benidipine hydrochloride forassay in the mixture of diluted phosphoric acid (1 in 500) andmethanol (1:1) to make exactly 100 mL. Pipet 1 mL of thissolution, add the mixture of diluted phosphoric acid (1 in500) and methanol (1:1) to make exactly 100 mL, and usethis solution as the standard solution. Perform the test withexactly 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and determine eachpeak area by the automatic integration method: the peakarea of dehydro derivative having the relative retention timeof about 0.75 with respect to benidipine is not larger than1/2 times the peak area of benidipine with the standard solu-tion. For this calculation, use the peak area of dehydroderivative after multiplying by the relative response factor,1.6.Operating conditions—
Perform as directed in the operating conditions in theAssay.

16931693Supplement II, JPXIV O‹cial Monographs for Part I
System suitability—Test for required detectability: Measure exactly 2 mL of
the standard solution, and add the mixture of diluted phos-phoric acid (1 in 500) and methanol (1:1) to make exactly20 mL. Conˆrm that the peak area of benidipine obtainedwith 10 mL of this solution is equivalent to 7 to 13z of thatwith 10 mL of the standard solution.
System performance: Dissolve 6 mg of benidipinehydrochloride and 5 mg of benzoin in 200 mL of a mixtureof water and methanol (1:1). When the procedure is run with10 mL of this solution under the above operating conditions,benzoin and benidipine are eluted in this order with theresolution between these peaks being not less than 8.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of benidipine is not more than 2.0z.
Content uniformity Perform the test as directed in theAssay, and determine the content: it meets the requirementsof the Content Uniformity Test.
Dissolution Perform the test with 1 tablet of BenidipineHydrochloride Tablets at 50 revolutions per minuteaccording to Method 2 under the Dissolution Test using thesinker, using 900 mL of the 1st ‰uid under the Disintegra-tion Test as the dissolution medium. Withdraw 20 mL ormore of the dissolution medium 30 minutes after starting thetest for a 2-mg and a 4-mg tablet or 45 minutes after startingthe test for a 8-mg tablet, and ˆlter through a membraneˆlter with pore size of not more than 0.45 mm. Discard theˆrst 10 mL of the ˆltrate, pipet the subsequent V mL, andadd the 1st ‰uid to make exactly V? mL so that each mLcontains about 2.2 mg of benidipine hydrochloride(C28H31N3O6.HCl) according to the labeled amount. Pipet5 mL of this solution, add exactly 5 mL of the mobile phase,and use this solution as the sample solution. Separately,weigh accurately about 22 mg of benidipine hydrochloridefor assay, previously dried at 1059C for 2 hours, anddissolve in the mobile phase to make exactly 100 mL. Pipet2 mL of this solution, and add the mobile phase to makeexactly 50 mL. Pipet 5 mL of this solution, and add themobile phase to make exactly 20 mL. Pipet 5 mL of thissolution, add exactly 5 mL of the 1st ‰uid, and use this solu-tion as the standard solution. Perform the test with exactly50 mL each of the sample solution and the standard solutionas directed under the Liquid Chromatography according tothe following conditions, and determine the peak areas, AT
and AS, of benidipine: the dissolution rates for 2-mg or 4-mgtablet in 30 minutes and for 8-mg tablet in 45 minutes are notless than 80z and not less than 85z, respectively.
Dissolution rate (z) with respect to the labeled amount ofbenidipine hydrochloride(C28H31N3O6.HCl)
=WS×AT
AS×
V?V×
1C×9
WS: Amount (mg) of benidipine hydrochloride for assayC: Labeled amount (mg) of benidipine hydrochloride
(C28H31N3O6.HCl) in 1 tablet.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 237 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of 0.05 mol/L potassium di-hydrogen phosphate TS, pH 3.0 and acetonitrile (11:9).
Flow rate: Adjust the ‰ow rate so that the retention timeof benidipine is about 5 minutes.System suitability—
System performance: When the procedure is run with50 mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of benidipine are not less than 3000 andnot more than 2.0, respectively.
System repeatability: When the test is repeated 6 timeswith 50 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of thepeak area of benidipine is not more than 1.5z.
Assay Shake 1 tablet of Benidipine Hydrochloride Tabletswith 40 mL of a mixture of diluted phosphoric acid (1 in 500)and methanol (1:1) to disintegrate, add the mixture ofdiluted phosphoric acid (1 in 500) and methanol (1:1) tomake exactly V mL so that each mL contains 40 mg ofbenidipine hydrochloride (C28H31N3O6.HCl), and cen-trifuge. Pipet 20 mL of the supernatant liquid, add exactly10 mL of the internal standard solution and the mixture ofdiluted phosphoric acid (1 in 500) and methanol (1:1) tomake 50 mL, and use this solution as the sample solution.Separately, weigh accurately about 40 mg of benidipinehydrochloride for assay, previously dried at 1059C for 2hours, and dissolve in the mixture of diluted phosphoric acid(1 in 500) and methanol (1:1) to make exactly 100 mL. Pipet2 mL of this solution, add exactly 10 mL of the internal stan-dard solution and the mixture of diluted phosphoric acid (1in 500) and methanol (1:1) to make 50 mL, and use this solu-tion as the standard solution. Perform the test with 10 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine the ratios, QT and QS,of the peak area of benidipine to that of the internal stan-dard. Perform the test described above with not less than 10tablets, and calculate the average of these results.
Amount (mg) of benidipine hydrochloride(C28H31N3O6.HCl)
=WS×QT
QS×
V1000
WS: Amount (mg) of benidipine hydrochloride for assay
Internal standard solution—A solution of benzoin in amixture of water and methanol (1:1) (13 in 200,000).Operating conditions—

16941694 Supplement II, JPXIVO‹cial Monographs for Part I
Detector: An ultraviolet absorption photometer(wavelength: 237 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 10 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (3 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of 0.05 mol/L potassium di-hydrogen phosphate TS, pH 3.0, methanol and tetra-hydrofuran (65:27:8).
Flow rate: Adjust the ‰ow rate so that the retention timeof benidipine is about 20 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, the internal standard and benidipine are elutedin this order with the resolution between these peaks beingnot less than 8.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of benidipine to that of the internal standard isnot more than 1.0z.
Containers and storage Containers—Well-closed contain-ers.
Benzylpenicillin Benzathineベンジルペニシリンベンザチン
Change the origin/limits of content to read:
Benzylpenicillin Benzathine is the N,N?-diben-zylethylenediamine salt of a penicillin compoundhaving antibacterial activity produced by the growthof Penicillium species.
It contains not less than 1152 Units and not morethan 1272 Units per mg, calculated on the anhydrousbasis. The potency of Benzylpenicillin Benzathine isexpressed as unit calculated from the amount ofbenzylpenicillin sodium (C16H17N2NaO4S: 356.37).1 Unit of Benzylpenicillin Benzathine is equivalent to0.6 mg of benzylpenicillin sodium (C16H17N2NaO4S). Itcontains not less than 24.0z and not more than27.0z of N,N?-dibenzylethylenediamine (C16H20N2:240.34), calculated on the anhydrous basis.
Change the Purity (3) to read:
Purity(3) Related substances—Dissolve 70 mg of Benzyl-
penicillin Benzathine in 25 mL of methanol, add a solutionprepared by dissolving 1.02 g of disodium hydrogen phos-phate and 6.80 g of potassium dihydrogen phosphate inwater to make 1000 mL to make 50 mL, and use thissolution as the sample solution. Pipet 1 mL of the sample
solution, add the mobile phase A to make exactly 100 mL,and use this solution as the standard solution. Perform thetest with exactly 20 mL each of the sample solution and thestandard solution as directed under the Liquid Chro-matography according to the following conditions, and de-termine each peak area by the automatic integrationmethod: the area of the peak having the relative retentiontime of about 2.4 with respect to benzylpenicillin is not morethan 2 times the total area of the peaks of benzylpenicillinand N,N?-dibenzylethylenediamine obtained from the stan-dard solution, and the area of the peak other than benzyl-penicillin, N,N?-dibenzylethylenediamine and the peak hav-ing the relative retention time of about 2.4 is not more thanthe total area of the peaks of benzylpenicillin andN,N?-dibenzylethylenediamine obtained from the standardsolution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 220 nm).
Column: A stainless steel column 4.0 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase A: A mixture of water, methanol and0.25 mol/L potassium dihydrogen phosphate TS, pH 3.5(6:3:1).
Mobile phase B: A mixture of methanol, water and0.25 mol/L potassium dihydrogen phosphate TS, pH 3.5(6:3:1).
Flowing of the mobile phase: Control the gradient bymixing the mobile phases A and B as directed in thefollowing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
0 – 10 75 2510 – 20 75 ª 0 25 ª 10020 – 55 0 100
Flow rate: 1.0 mL/minTime span of measurement: About 3 times as long as the
retention time of benzylpenicillin after the solvent peak.System suitability—
Test for required detectability: To exactly 1 mL of thestandard solution add the mobile phase A to make exactly20 mL. Conˆrm that the peak area of benzylpenicillinobtained from 20 mL of this solution is equivalent to 3.5 to6.5z of that from the standard solution.
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, N,N?-dibenzylethylenediamine and benzyl-penicillin are eluted in this order with the resolution betweenthese peaks being not less than 25.
System repeatability: When the test is repeated 3 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peak

16951695Supplement II, JPXIV O‹cial Monographs for Part I
areas of benzylpenicillin is not more than 2.0z.Change the Assay to read:
Assay (1) Benzylpenicillin—Weigh accurately an amountof Benzylpenicillin Benzathine, equivalent to about 85,000Units, dissolve in 25 mL of methanol, and add a solutioncontaining 1.02 g of disodium hydrogen phosphate and6.80 g of potassium dihydrogen phosphate in 1000 mL ofwater to make exactly 50 mL. Pipet 5 mL of this solution,add a mixture of the solution containing 1.02 g of disodiumhydrogen phosphate and 6.80 g of potassium dihydrogenphosphate in 1000 mL of water and methanol (1:1) to makeexactly 20 mL, and use this solution as the sample solution.Separately, weigh accurately an amount of BenzylpenicillinPotassium Reference Standard, equivalent to about 85,000Units, and about 25 mg of N,N?-dibenzylethylenediaminediacetate, dissolve in 25 mL of methanol, and add thesolution containing 1.02 g of disodium hydrogen phosphateand 6.80 g of potassium dihydrogen phosphate in 1000 mLof water to make exactly 50 mL. Pipet 5 mL of this solution,add a mixture of the solution containing 1.02 g of disodiumhydrogen phosphate and 6.80 g of potassium dihydrogenphosphate in 1000 mL of water and methanol (1:1) to makeexactly 20 mL, and use this solution as the standardsolution. Perform the test with exactly 20 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and determine the peak areas, AT ands AS, ofbenzylpenicillin.
Amount (unit) of benzylpenicillin sodium (C16H17N2NaO4S)
=WS×AT
AS
WS: Amount (unit) of Benzylpenicillin Potassium Refer-ence Standard
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 220 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: A mixture of water, methanol and 0.25mol/L potassium dihydrogen phosphate TS, pH 3.5(11:7:2).
Flow rate: Adjust the ‰ow rate so that the retention timeof benzylpenicillin is about 18 minutes.System suitability—
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, N,N?-dibenzylethylenediamine and benzyl-penicillin are eluted in this order with the resolution betweenthese peaks being not less than 20.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviations of the peak
areas of N,N?-dibenzylethylenediamine and benzylpenicillinare not more than 2.0z, respectively.
(2) N,N?-Dibenzylethylenediamine—Determine theareas, AT and AS, of the peak corresponding to N,N?-diben-zylethylenediamine on the chromatograms obtained in (1)with the sample solution and the standard solution.
Amount (z) of N,N?-dibenzylethylenediamine (C16H20N2)
=WS
WT×
AT
AS×100×0.667
WS: Amount (mg) of N,N?-dibenzylethylenediaminediacetate
WT: Amount (mg) of the sample0.667: Conversion factor for the molecular mass
of N,N?-dibenzylethylenediamine diacetate(C16H20N2・2CH3COOH) to that of N,N?-diben-zylethylenediamine (benzathine, C16H20N2)
Add the following:
Cefepime Dihydrochloride forInjection注射用塩酸セフェピム
Cefepime Dihydrochloride for Injection is a prepa-ration for injection, which is dissolved before use.
It contains not less than 95.0z and not morethan 110.0z of the labeled amount of cefepime(C19H24N6O5S2: 480.56).
Method of preparation Prepare as directed under Injec-tions, with Cefepime Dihydrochloride.
Description Cefepime Dihydrochloride for Injectionoccurs as a white to pale yellow powder.
Identiˆcation (1) Dissolve 40 mg of Cefepime Di-hydrochloride in 2 mL of water, add 1 mL of a solution ofhydroxylammonium chloride (1 in 10) and 2 mL of sodiumhydroxide TS, allow to stand for 5 minutes, then add 3 mLof 1 mol/L hydrochloric acid TS and 3 drops of iron (III)chloride TS: a red-brown color develops.
(2) Determine the absorption spectrum of a solution ofCefepime Dihydrochloride for Injection (1 in 12,500) asdirected under the Ultraviolet-visible Spectrophotometry: itexhibits maxima between 233 nm and 237 nm and between255 nm and 259 nm.
pH The pH of a solution obtained by dissolving an amountof Cefepime Dihydrochloride for Injection, equivalent to0.5 g (potency) of Cefepime Dihydrochloride according tothe labeled amount, in 5 mL of water is between 4.0 and 6.0.
Purity (1) Clarity and color of solution—Dissolve anamount of Cefepime Dihydrochloride for Injection, equiva-lent to 0.5 g (potency) of Cefepime Dihydrochloride accord-ing to the labeled amount, in 5 mL of water: the solution isclear and colorless or light yellow. The color is not darker

16961696 Supplement II, JPXIVO‹cial Monographs for Part I
than Matching Fluid I.(2) N-Methylpyrrolidine—Weigh accurately an amount
of Cefepime Dihydrochloride for Injection, equivalent toabout 0.2 g (potency) of Cefepime Dihydrochloride accord-ing to the labeled amount, dissolve in diluted nitric acid (2 in625) to make exactly 20 mL, and use this solution as thesample solution. Separately, transfer 30 mL of water into a100-mL volumetric ‰ask, weigh accurately the mass of the‰ask, add about 0.125 g of N-methylpyrrolidine, then weighaccurately the mass, and add water to make exactly 100 mL.Pipet 4 mL of this solution, add diluted nitric acid (2 in3125) to make exactly 100 mL, and use this solution as thestandard solution. Perform the test with exactly 100 mL eachof the sample solution and the standard solution as directedunder the Liquid Chromatography according to the follow-ing conditions, and determine the peak areas of N-methyl-pyrrolidine, AT and AS, by the automatic integration methodwithin 20 minutes after the sample solution is prepared.Calculate the amount of N-methylpyrrolidine per mg (poten-cy) of Cefepime Dihydrochloride for Injection by thefollowing formula: not more than 1.0z.
Amount (z) of N-methylpyrrolidine
=WS×f
WT×
AT
AS×
1125
WS: Amount (mg) of N-methylpyrrolidineWT: Amount [mg (potency)] of cefepime in the sample
takenf: Purity (z) of N-methylpyrrolidine
Operating conditions—Proceed as directed in the operating conditions in the
Purity (3) under Cefepime Dihydrochloride.System suitability—
Proceed as directed in the system suitability in the Purity(3) under Cefepime Dihydrochloride.
Water Not more than 4.0z (Weigh accurately about50 mg of Cefepime Dihydrochloride for Injection, dissolvein exactly 2 mL of methanol for Karl Fischer method, andperform the test with exactly 0.5 mL of this solution. Coulo-metric titration).
Bacterial endotoxins Less than 0.06 EU/mg (potency).
Mass variation It meets the requirements of the MassVariation Test.
Foreign insoluble matter Perform the test according toMethod 2: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the InsolubleParticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Weigh accurately the mass of the contents of notless than 10 Cefepime Dihydrochloride for Injection. Weigh
accurately an amount of the content, equivalent to about60 mg (potency) of Cefepime Dihydrochloride according tothe labeled amount, dissolve in the mobile phase to makeexactly 50 mL, and use this solution as the sample solution.Separately, weigh accurately an amount of CefepimeDihydrochloride Reference Standard, equivalent to about60 mg (potency), dissolve in the mobile phase to makeexactly 50 mL, and use this solution as the standardsolution. Proceed as directed in the Assay under CefepimeDihydrochloride.
Amount [mg (potency)] of cefepime (C19H24N6O5S2)
=WS×AT
AS×1000
WS: Amount [mg (potency)] of Cefepime DihydrochlorideReference Standard
Containers and storage Containers—Hermetic containers.Storage—Light-resistant.
Cefuroxime Axetilセフロキシムアキセチル
Change the graphic formula to read:
Change the chemical name to read:
(1RS)-1-Acetoxyethyl (6R,7R)-3-carbamoyloxymethyl-7-[(Z)-2-furan-2-yl-2-methoxyiminoacetylamino]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate
Cefuroxime Sodiumセフロキシムナトリウム
Change the Purity (1) to read:
Purity (1) Clarity and color of solution—Dissolve 1.0 gof Cefuroxime Sodium in 10 mL of water: the solution isclear, and its absorbance at 450 nm is not more than 0.25.

16971697Supplement II, JPXIV O‹cial Monographs for Part I
Ciclosporinシクロスポリン
Change the Purity (3) to read:
Purity(3) Related substances—Use the sample solution ob-
tained in the Assay as the sample solution. Pipet 2 mL of thesample solution, add a mixture of water and acetonitrile(1:1) to make exactly 200 mL, and use this solution as thestandard solution. Perform the test with exactly 20 mL eachof the sample solution and the standard soluton as directedunder the Liquid Chromatography according to the follow-ing conditions. Determine each peak area of both solutionsby the automatic integration method: the area of the peakother than the ciclosporin is not more than 0.7 times of thepeak area of ciclosporin from the standard solution, and thetotal area of all peaks other than the ciclosporin is not morethan 1.5 times of the peak area of ciclosporin from the stan-dard solution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 2 times as long as theretention time of ciclosporin after the solvent peak.System suitability—
Test for required detection: To exactly 2 mL of the stan-dard solution add a mixture of water and acetonitrile (1:1) tomake exactly 20 mL. Conˆrm that the peak area of ciclospo-rin obtained from 20 mL of this solution is equivalent to 7 to13z of that of ciclosporin obtained from 20 mL of thestandard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of thepeak area of ciclosporin is not more than 3.0z.
Change the Assay to read:
Assay Weigh accurately about 30 mg each of Ciclosporinand Ciclosporin Reference Standard, previously determinedthe loss on drying as the same manner as Ciclosporin, anddissolve each in a mixture of water and acetonitrile (1:1) tomake exactly 25 mL, and use these solutions as the samplesolution and the standard solution. Perform the test with ex-actly 20 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine the peakareas, AT and AS, of ciclosporin.
Amount (mg) of C62H111N11O12
=WS×AS
AT
WS: Amount (mg) of Ciclosporin Reference Standard,
calculated on the dried basis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 210 nm).Column: A stainless steel column 4 mm in inside diameter
and 25 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).Connect the sample injection port and the column with astainless steel tube 0.3 mm in inside diameter and 1 m inlength.
Column temperature: A constant temperature of about809C (including the sample injection port and the connectingtube).
Mobile phase: A mixture of water, acetonitrile, tert-butylmethyl ether and phosphoric acid (520:430:50:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof ciclosporin is about 27 minutes.System suitability—
System performance: Dissolve 3 mg of Ciclosporin U in2.5 mL of a mixture of water and acetonitrile (1:1), and add2.5 mL of the standard solution. When the procedure is runwith 20 mL of this solution under the above operatingconditions, ciclosporin U and ciclosporin are eluted in thisorder with the resolution between these peaks being not lessthan 1.2.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of ciclosporin is not more than 1.0z.
Add the following:
Cisplatinシスプラチン
Cl2H6N2Pt: 300.05(SP-4-2)-Diamminedichloroplatinum [15663-27-1]
Cisplatin, when dried, contains not less than 98.0zand not more than 102.0z of Cl2H6N2Pt.
Description Cisplatin occurs as a yellow crystalline pow-der.
It is sparingly soluble in N,N-dimethylformamide, slightlysoluble in water, and practically insoluble in ethanol (99.5).
Identiˆcation (1) To 5 mL of a solution of Cisplatin (1 in2000) add 2 to 3 drops of a solution of tin (II) chloridedihydrate (1 in 100): a brown precipitate is formed.
(2) Determine the absorption spectrum of a solution ofCisplatin in a solution of sodium chloride in 0.01 mol/Lhydrochloric acid TS (9 in 1000) (1 in 2000) as directed underthe Ultraviolet-visible Spectrophotometry, and compare thespectrum with the Reference Spectrum or the spectrum of a

16981698 Supplement II, JPXIVO‹cial Monographs for Part I
solution of Cisplatin Reference Standard prepared in thesame manner as the sample solution: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
(3) Determine the infrared absorption spectrum ofCisplatin as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum or the spectrum ofCisplatin Reference Standard: both spectra exhibit similarintensities of absorption at the same wave numbers.
(4) A solution of Cisplatin (1 in 2000) responds to theQualitative Test (1) for chloride.
Purity Ammonium amminetrichloroplatinate—Conductthis procedure using light-resistant vessels. Dissolve 50 mgof Cisplatin in a solution of sodium chloride (9 in 1000) tomake exactly 100 mL, and use this solution as the samplesolution. Separately, dissolve 10 mg of ammonium am-minetrichloroplatinate for liquid chromatography, previous-ly dried at 809C for 3 hours, in the solution of sodiumchloride (9 in 1000) to make exactly 200 mL. Pipet 2 mL ofthis solution, add the solution of sodium chloride (9 in 1000)to make exactly 20 mL, and use this solution as the standardsolution. Perform the test with exactly 40 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and determine the peak area of ammoniumamminetrichloroplatinate by the automatic integrationmethod: the peak area from the sample solution is not morethan that from the standard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 209 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with silica gel forliquid chromatography having quaternary ammoniumgroups (10 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A solution of ammonium sulfate (1 in 800).Flow rate: Adjust the ‰ow rate so that the retention time
of ammonium amminetrichloroplatinate is about 8 minutes.System suitability—
System performance: When the procedure is run with40 mL of the standard solution under the above operatingconditions, the number of theoretical plates and thesymmetry factor of the peak of ammonium am-minetrichloroplatinate are not less than 1500 and not morethan 2.0, respectively.
System repeatability: When the test is repeated 6 timeswith 40 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of ammonium amminetrichloroplatinate is not morethan 3.0z.
Loss on drying Not more than 0.1z (1 g, 1059C, 4 hours).
Assay Conduct this procedure using light-resistant vessels.Weigh accurately about 25 mg each of Cisplatin andCisplatin Reference Standard, previously dried, dissolve in
N,N-dimethylformamide to make exactly 25 mL, and usethese solutions as the sample solution and the standardsolution. Perform the test with exactly 40 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and determine the peak areas, AT and AS, ofcisplatin by the automatic integration method.
Amount (mg) of Cl2H6N2Pt=WS×AT
AS
WS: Amount (mg) of Cisplatin Reference Standard
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 310 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 25 cm in length, packed with aminopropyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of ethyl acetate, methanol, waterand N,N-dimethylformamide (25:16:5:5).
Flow rate: Adjust the ‰ow rate so that the retention timeof cisplatin is about 4 minutes.System suitability—
System performance: When the procedure is run with40 mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of cisplatin are not less than 3000 andnot more than 2.0, respectively.
System repeatability: When the test is repeated 6 timeswith 40 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of cisplatin is not more than 1.0z.
Containers and storage Containers—Tight containers.
Anhydrous Citric Acid無水クエン酸
Change the Identiˆcation to read:
Identiˆcation Determine the infrared absorption spectrumof Anhydrous Citric Acid, previously dried at 1059C for2 hours, as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wave numbers.

16991699Supplement II, JPXIV O‹cial Monographs for Part I
Clarithromycinクラリスロマイシン
Change the origin/limits of content to read:
Clarithromycin is a derivative of erythromycin.It contains not less than 950 mg (potency) and not
more than 1050 mg (potency) per mg, calculated on theanhydrous basis. The potency of Clarithromycin isexpressed as mass (potency) of clarithromycin(C38H69NO13).
Change the Melting point to read:
Melting point 220 – 2279C
Change the Purity (2) and (3) to read:
Purity(2) Arsenic—Prepare the test solution with 1.0 g of
Clarithromycin according to Method 3, and perform the test(not more than 2 ppm).
(3) Related substances—Weigh accurately about 0.1 g ofClarithromycin, dissolve in the mobile phase to make exactly20 mL, and use this solution as the sample solution.Separately, weigh accurately about 10 mg of ClarithromycinReference Standard, dissolve in the mobile phase to makeexactly 20 mL, and use this solution as the standard solu-tion. Perform the test with exactly 10 mL each of the samplesolution and the standard solution as directed under the Liq-uid Chromatography according to the following conditions,and determine the each peak area by the automatic integra-tion method: the amount of each related substance calculat-ed on the anhydrous basis is not more than 2.0z, and thetotal of them is not more than 5.0z. Exclude any peak withan area of less than 0.05z.
Amount (z) of each related substance calculated on theanhydrous basis
=WS
WT×
AT
AS×100
Total amount (z) of the related substances calculated on theanhydrous basis
=WS
WT×
SAT
AS×100
WS: Amount (mg) of Clarithromycin Reference StandardWT: Amount (mg) of the sample, calculated on the anhy-
drous basisAS: Peak area of clarithromycin obtained with the stan-
dard solutionAT: Peak area of each related substance obtained with the
sample solutionSAT: Total area of the peaks other than clarithromycin
obtained with the sample solutionOperating conditions—
Detector, column, column temperature, mobile phase,
and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 5 times as long as theretention time of the main peak after 2 minutes of sampleinjection.System suitability—
Test for required detection: To exactly 2 mL of the stan-dard solution add the mobile phase to make exactly 10 mL,and use this solution as the solution for system suitabilitytest. Conˆrm that when the procedure is run with 10 mL ofthe solution for system suitability test, the peak area ofclarithromycin is equivalent to 14 – 26z of that obtainedfrom the standard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 10 mL of the solution for system suitability test underthe above operating conditions, the relative standarddeviation of the peak area of clarithromycin is not morethan 3.0z.
Colchicineコルヒチン
Change to read except the structural formulaand chemical name:
Colchicine contains not less than 97.0z and notmore than 102.0z of C22H25NO6, calculated on theanhydrous basis and corrected by the amount of ethylacetate.
Description Colchicine occurs as a yellowish whitepowder.
It is very soluble in methanol, freely soluble in N,N-dimethylformamide, in ethanol (95) and in acetic anhydride,and sparingly soluble in water.
It is colored by light.
Identiˆcation (1) Determine the absorption spectrum ofa solution of Colchicine in ethanol (95) (1 in 100,000) asdirected under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wavelengths.
(2) To 1 g of potassium bromide for infrared absorptionspectrum add 0.5 mL of a solution of Colchicine inmethanol (1 in 50), grind thoroughly, and dry in vacuum at809C for 1 hour. Determine the infrared absorption spec-trum of this powder as directed in the potassium bromidedisk method under the Infrared Spectrophotometry, andcompare the spectrum with the Reference Spectrum: bothspectra exhibit similar intensities of absorption at the samewave numbers.
Optical rotation [a]D20: -235 –-2509(0.1 g calculated onthe anhydrous basis and corrected by the amount of ethyl

17001700 Supplement II, JPXIVO‹cial Monographs for Part I
acetate, ethanol (95), 10 mL, 100 mm).
Purity (1) Colchiceine—Dissolve 0.10 g of Colchicine in10 mL of water, and to 5 mL of this solution add 2 drops ofiron (III) chloride TS: no deˆnite green color develops.
(2) Chloroform and ethyl acetate—Weigh accuratelyabout 0.60 g of Colchicine, dissolve in exactly 2 mL of theinternal standard solution, add N,N-dimethylformamide tomake 10 mL, and use this solution as the sample solution.Separately, weigh 0.30 g of chloroform using a 100-mLvolumetric ‰ask containing about 20 mL of N,N-dimethyl-formamide, and add N,N-dimethylformamide to make ex-actly 100 mL. Pipet 2 mL of this solution, add N,N-dimethylformamide to make exactly 200 mL, and use thissolution as the standard solution (1). Separately, weigh ac-curately about 1.8 g of ethyl acetate using a 100-mL volu-metric ‰ask containing about 20 mL of N,N-dimethylfor-mamide, and add N,N-dimethylformamide to make exactly100 mL. Pipet 2 mL of this solution, add exactly 2 mL of theinternal standard solution and N,N-dimethylformamide tomake 10 mL, and use this solution as the standard solution(2). Perform the test with 2 mL each of the sample solutionand the standard solutions (1) and (2) as directed under theGas Chromatography according to the following conditions:the peak area of chloroform is not more than that from thestandard solution (1). Determine the ratios of the peak areaof ethyl acetate to that of the internal standard, QT and QS,of the sample solution and the standard solution (2), and cal-culate the amount of ethyl acetate by the following formula:the amount of ethyl acetate is not more than 6.0z.
Amount (z) of ethyl acetate (C4H8O2)=WS
WT×
QT
QS×2
WS: Amount (g) of ethyl acetateWT: Amount (g) of the sample
Internal standard solution—A solution of 1-propanol inN,N-dimethylformamide (3 in 200)Operating conditions—
Detector: A hydrogen ‰ame-ionization detectorColumn: A fused silica column 0.53 mm in inside di-
ameter and 30 m in length, coated inside surface withpolyethylene glycol 20 M for gas chromatography 1.0 mm inthickness.
Column temperature: 609C for 7 minutes, then up to1009C at a rate of 409C per minute if necessary, and hold at1009C for 10 minutes.
Injection port temperature: A constant temperature ofabout 1309C
Detector temperature: A constant temperature of about2009C
Carrier gas: HeliumFlow rate: Adjust the ‰ow rate so that the retention time
of ethyl acetate is about 3 minutes.Split ratio: 1:20
System suitability—Test for required detectability: Pipet 2 mL of the standard
solution (2), and add N,N-dimethylformamide to makeexactly 25 mL. Pipet 1 mL of this solution, and add N,N-
dimethylformamide to make exactly 50 mL. Conˆrm thatthe peak area of ethyl acetate obtained from 2 mL of thissolution is equivalent to 0.11 to 0.21z of that obtained from2 mL of the standard solution (2).
System performance: To 1 mL of chloroform add N,N-dimethylformamide to make 10 mL. To 1 mL of this solu-tion add 2 mL of ethyl acetate and N,N-dimethylformamideto make 100 mL. To 2 mL of this solution add 2 mL of theinternal standard solution and N,N-dimethylformamide tomake 10 mL. When the procedure is run with 2 mL of thissolution under the above operating conditions, ethyl acetate,chloroform and the internal standard are eluted in this orderwith the resolution between the peaks of chloroform and theinternal standard being not less than 2.0.
System repeatability: When the test is repeated 3 timeswith 2 mL of the standard solution (2) under the aboveoperating conditions, the relative standard deviation of theratio of the peak area of ethyl acetate to that of the internalstandard is not more than 3.0z.
(3) Related substances Dissolve 60 mg of Colchicine in100 mL of diluted methanol (1 in 2). Pipet 1 mL of thissolution, add diluted methanol (1 in 2) to make exactly 100mL, and use this solution as the sample solution. Performthe test with 20 mL of the sample solution as directed underthe Liquid Chromatography according to the followingconditions, and determine each peak area by the automaticintegration method. Calculate the total amount of the peaksother than colchicine by the area percentage method: notmore than 5.0z.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4.6 mm in inside di-ameter and 25 cm in length, packed with octylsilanized silicagel for liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: To 450 mL of 0.05 mol/L potassium di-hydrogen phosphate TS add methanol to make 1000 mL.Adjust the pH to 5.5 with diluted phosphoric acid (7 in 200).
Flow rate: Adjust the ‰ow rate so that the retention timeof colchicine is about 7 minutes.
Time span of measurement: About 2 times as long as theretention time of colchicine after the solvent peak.System suitability—
Test for required detectability: Pipet 1 mL of the samplesolution, and add diluted methanol (1 in 2) to make exactly50 mL. Conˆrm that the peak area of colchicine obtainedfrom 20 mL of this solution is equivalent to 1.4 to 2.6z ofthat obtained from 20 mL of the sample solution.
System performance: When the procedure is run with20 mL of the sample solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of colchicine are not less than 6000 andnot more than 1.5, respectively.
System repeatability: When the test is repeated 6 timeswith 20 mL of the sample solution under the above operatingconditions, the relative standard deviation of the peak area

17011701Supplement II, JPXIV O‹cial Monographs for Part I
of colchicine is not more than 2.0z.
Water Not more than 2.0z (0.5 g, volumetric titration,back titration).
Assay Weigh accurately about 0.4 g of Colchicine, dissolvein 25 mL of acetic anhydride, and titrate with 0.05 mol/Lperchloric acid VS (potentiometric titration). Perform ablank determination in the same manner, and make anynecessary correction.
Each mL of 0.05 mol/L perchloric acid VS=19.97 mg of C22H25NO6
Containers and storage Containers—Tight containers.Storage—Light-resistant.
Cytarabineシタラビン
Change the origin/limits of content to read:
Cytarabine, when dried, contains not less than98.5z and not more than 101.0z of C9H13N3O5.
Change the Description to read:
Description Cytarabine occurs as white crystals or crystal-line powder.
It is freely soluble in water, soluble in acetic acid (100),and very slightly soluble in ethanol (99.5).
It dissolves in 0.1 mol/L hydrochloric acid TS.Melting point: about 2149C (with decomposition).
Change the Identiˆcation to read:
Identiˆcation (1) Determine the absorption spectrum ofa solution of Cytarabine in 0.1 mol/L hydrochloric acid TS(1 in 100,000) as directed under the Ultraviolet-visibleSpectrophotometry, and compare the spectrum with theReference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofCytarabine as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry, andcompare the spectrum with the Reference Spectrum: bothspectra exhibit similar intensities of absorption at the samewave numbers.
Delete the Absorbance:
Change the Purity (4) and (5) to read:
Purity(4) Related substances—Dissolve 0.10 g of Cytarabine in
10 mL of water, and use this solution as the sample solution.Pipet 1 mL of the sample solution, add water to makeexactly 200 mL, and use this solution as the standardsolution (1). Pipet 10 mL of the standard solution (1), add
water to make exactly 25 mL and use this solution as thestandard solution (2). Perform the test with these solutionsas directed under the Thin-layer Chromatography. Spot10 mL each of the sample solution and the standard solutions(1) and (2) on a plate of silica gel with ‰uorescent indicatorfor thin-layer chromatography. Develop the plate with1-butanol saturated with water to a distance of about 12 cm,and air-dry the plate. Examine under ultraviolet light (mainwavelength: 254 nm): the spots other than the principal spotfrom the sample solution are not more intense than the spotfrom the standard solution (1), and the number of themwhich are more intense than the spot from the standard solu-tion (2) is not more than two. Spray evenly acidic potassiumpermanganate TS on the plate: any spot other than the prin-cipal spot does not appear.
Deferoxamine Mesilateメシル酸デフェロキサミン
Change the Purity (6) to read:
Purity(6) Related substances—Dissolve 50 mg of Deferoxa-
mine Mesilata in 50 mL of the mobile phase, and use thissolution as the sample solution. Pipet 3 mL of the sample so-lution, add the mobile phase to make exactly 50 mL, and usethis solution as the standard solution. Perform the test withexactly 20 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions. Determine each peakarea of both solutions by the automatic integration method:the total area of all peaks other than the peak of deferoxa-mine is not larger than the peak area of deferoxamine fromthe standard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 230 nm).
Column: A stainless steel column 4 mm in inside diameterand 20 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (10 mm in particle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 1.32 g of diammonium hydrogenphosphate, 0.37 g of disodium dihydrogen ethylenediaminetetraacetate dihydrate and 1.08 g of sodium 1-heptanesul-fonate in 950 mL of water, and adjust the pH of thissolution to 2.8 with phosphoric acid. To 800 mL of thissolution add 100 mL of 2-propanol.
Flow rate: Adjust the ‰ow rate so that the retention timeof deferoxamine is about 15 minutes.
Time span of measurement: About two times as long asthe retention time of deferoxamine after the solvent peak.System suitability—
Test for required detection: To exactly 2 mL of the stan-dard solution add the mobile phase to make exactly 100 mL.Conˆrm that the peak area of deferoxamine obtained from

17021702 Supplement II, JPXIVO‹cial Monographs for Part I
20 mL of this solution is equivalent to 1.5 to 2.5z of that ofdeferoxamine obtained from 20 mL of the standard solution.
System performance: Dissolve 16 mg of DeferoxamineMesilate and 4 mg of methyl parahydroxybenzoate in 50 mLof the mobile phase. When the procedure is run with 20 mLof this solution under the above operating conditions,deferoxamine and methyl parahydroxybenzoate are eluted inthis order with the resolution between these peaks being notless than 4.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakareas of deferoxamine is not more than 3.0z.
Digoxinジゴキシン
Change the Description to read:
Description Digoxin occurs as colorless or white crystals ora white crystalline powder.
It is freely soluble in pyridine, slightly soluble in ethanol(95), very slightly soluble in acetic acid (100), and practicallyinsoluble in water.
Change the Identiˆcation (2) to read:
Identiˆcation(2) Determine the infrared absorption spectrum of
Digoxin, previously dried, as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry, and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wave numbers.
Change the Optical rotation to read:
Optical rotation [a]D20:+10.0 –+13.09(after drying, 0.20 g,dehydratead pyridine, 10 mL, 100 mm).
Change the Purity (2) to read:
Purity(2) Related substances—Dissolve exactly 25.0 mg of
Digoxin in 50 mL of warm ethanol (95), cool, and addethanol (95) to make exactly 100 mL. Pipet 10 mL of thissolution, add 10 mL of water and dilute ethanol to make 50mL, and use this solution as the sample solution. Separately,dissolve exactly 5.0 mg of Gitoxin Reference Standard,previously dried under reduced pressure at 1059C for 1 hour,in a mixture of acetonitrile and water (7:3) to make exactly200 mL. Pipet 2 mL of this solution, add dilute ethanol tomake 50 mL, and use this solution as the standard solution.Perform the test with exactly 10 mL each of the sample solu-tion and the standard solution as directed under the LiquidChromatography according to the following conditions, anddetermine the peak areas, AT and AS, of gitoxin: AT is notlarger than AS, and the total of the areas of the peaks other
than digitoxin and gitoxin, obtained by the area percentagemethod, is not more than 3z.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 4 times as long as theretention time of digoxin after the solvent peak.System suitability—
Test for required detectability: Pipet 1 mL of the samplesolution, add the mobile phase to make exactly 100 mL, anduse this solution as the solution for system suitability test.Pipet 1 mL of the solution, and add the mobile phase tomake exactly 10 mL. Conˆrm that the peak area of digoxinobtained from 10 mL of this solution is equivalent to 7 to13z of that from the solution for system suitability test.
System performance: Dissolve 25 mg of digoxin in 50 mLof warm ethanol (95), cool, and add ethanol (95) to makeexactly 100 mL. Pipet 10 mL of this solution, add exactly5 mL of a solution of propyl parahydroxybenzoate inethanol (95) (1 in 4000), 10 mL of water and dilute ethanol tomake 50 mL. When the procedure is run with 10 mL of thissolution under the above operating conditions, digoxin andpropyl parahydroxybenzoate are eluted in this order with theresolution between these peaks being not less than 5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of digoxin is not more than 1.0z.
Change the Assay to read:
Assay Weigh accurately about 25 mg each of Digoxin andDigoxin Reference Standard, previously dried, dissolve in50 mL of warm ethanol (95), cool, and add ethanol (95) tomake exactly 100 mL. Pipet 10 mL of these solutions, addexactly 5 mL of the internal standard solution, 10 mL ofwater and dilute ethanol to make 50 mL, and use thesesolutions as the sample solution and the standard solution.Perform the test with 10 mL each of the sample solution andthe standard solution as directed under the Liquid Chro-matography according to the following conditions, anddetermine the ratios, QT and QS, of the peak area of digoxinto that of the internal standard.
Amount (mg) of C41H64O14=WS×QT
QS
WS: Amount (mg) of Digoxin Reference Standard
Internal standard solution—A solution of propyl para-hydroxybenzoate in ethanol (95) (1 in 4000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 220 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about

17031703Supplement II, JPXIV O‹cial Monographs for Part I
309C.Mobile phase: A mixture of water and acetonitrile (7:3).Flow rate: Adjust the ‰ow rate so that the retention time
of digoxin is about 10 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, digoxin and the internal standard are eluted inthis order with the resolution between these peaks being notless than 5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of digoxin to that of the internal standard isnot more than 1.0z.
Change to read:
Digoxin Injectionジゴキシン注射液
Digoxin Injection is an aqueous solution forinjection.
It contains not less than 90.0z and not more than105.0z of the labeled amount of digoxin (C41H64O14:780.94).
Method of preparation Prepare as directed under Injec-tions, with a solution of Digoxin in 5 to 50 volz ethanol.
Description Digoxin Injection is a clear, colorless liquid.
Identiˆcation Dilute Digoxin Injection, if necessary, withmethanol so that each mL contains about 0.25 mg of Digox-in according to the labeled amount, and use this solution asthe sample solution. In case where ingredients are suspectedto aŠect the test, remove them by means of a solid-phase ex-traction. Separately, dissolve 0.5 mg of Digoxin ReferenceStandard in 2 mL of methanol, and use this solution as thestandard solution. Perform the test with these solutions asdirected under the Thin-layer Chromatography. Spot 10 mLeach of the sample solution and the standard solution on aplate of octadecylsilanized silica gel for thin-layer chro-matography. Develop the plate with a mixture of methanoland water (7:3) to a distance of about 10 cm, and air-dry theplate. Spray evenly a mixture of a solution of trichloroaceticacid in ethanol (99.5) (1 in 4) and a freshly prepared solutionof sodium toluenesulfonchloramide trihydrate (3 in 100)(4:1) on the plate, heat at 1109C for 10 minutes, andexamine under ultraviolet light (main wavelength: 366 nm):the Rf values of the principal spots with the sample solutionand the standard solution are not diŠerent each other.
Bacterial endotoxins Less than 200 EU/mg.
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign Insoluble
Matter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirement of the Insoluble Particu-late Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay To an exact volume of Digoxin Injection, equivalentto about 2.5 mg of digoxin (C41H64O14), add exactly 5 mL ofthe internal standard solution and dilute ethanol to make50 mL, and use this solution as the sample solution.Separately, weigh accurately about 25 mg of Digoxin Refer-ence Standard, previously dried under reduced pressure at1059C for 1 hour, dissolve in 50 mL of warm ethanol (95),cool, and add ethanol (95) to make exactly 100 mL. Pipet 10mL of this solution, add exactly 5 mL of the internal stan-dard solution, 10 mL of water and dilute ethanol to make 50mL, and use this solution as the standard solution. Performthe test with 10 mL each of the sample solution and the stan-dard solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of digoxin to that of theinternal standard.
Amount (mg) of digoxin (C41H64O14)=WS×QT
QS×
110
WS: Amount (mg) of Digoxin Reference Standard
Internal standard solution—A solution of propyl para-hydroxybenzoate in ethanol (95) (1 in 4000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 220 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about309C.
Mobile phase: A mixture of water and acetonitrile (7:3).Flow rate: Adjust the ‰ow rate so that the retention time
of digoxin is about 10 minutes.System suitability—
System performance: When the procedure is run with 10mL of the standard solution under the above operating con-ditions, digoxin and the internal standard are eluted in thisorder with the resolution between these peaks being not lessthan 5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of theratio of the peak area of digoxin to that of the internalstandard is not more than 1.0z.
Containers and storage Containers—Hermetic containers,and colored containers may be used.
Storage—Light-resistant.

17041704 Supplement II, JPXIVO‹cial Monographs for Part I
Change to read:
Digoxin Tabletsジゴキシン錠
Digoxin Tablets contain not less than 90.0z and notmore than 105.0z of the labeled amount of digoxin(C41H64O14: 780.94).
Method of preparation Prepare as directed under Tablets,with Digoxin.
Identiˆcation To an amount of pulverized DigoxinTablets, equivalent to 0.5 mg of Digoxin according to the la-beled amount, add 2 mL of methanol, shake for 10 minutes,ˆlter, and use the ˆltrate as the sample solution. Separately,dissolve 0.5 mg of Digoxin Reference Standard in 2 mL ofmethanol, and use this as the standard solution. Perform thetest with these solutions as directed under the Thin-layerChromatography. Spot 10 mL each of the sample solutionand the standard solution on a plate of octadecylsilanizedsilica gel for thin-layer chromatography. Develop the platewith a mixture of methanol and water (7:3) to a distance ofabout 10 cm, and air-dry the plate. Spray evenly a mixture ofa solution of trichloroacetic acid in ethanol (99.5) (1 in 4)and a freshly prepared solution of sodium toluenesulfon-chloramide trihydrate (3 in 100) (4:1) on the plate, heat at1109C for 10 minutes, and examine under ultraviolet light(main wavelength: 366 nm): the Rf values of the principalspots with the sample solution and the standard solution arenot diŠerent each other.
Content uniformity Perform the test according to the fol-lowing method: it meets the requirements of the ContentUniformity Test.
To 1 tablet of Digoxin Tablets add 0.5 mL of water todisintegrate, then add exactly 0.5 mL of the internal stan-dard solution, and add V mL of dilute ethanol so that eachmL contains about 21 mg of digoxin (C41H64O14). Exposurethis solution to ultrasonic waves for 20 minutes, shake for 5minutes, ˆlter, and use the ˆltrate as the sample solution.Separately, weigh accurately about 25 mg of Digoxin Refer-ence Standard, previously dried under reduced pressure at1059C for 1 hour, dissolve in 50 mL of warm ethanol (95),cool, and add ethanol (95) to make exactly 100 mL. Pipet10 mL of this solution, and add ethanol (95) to make exactly20 mL. Pipet 1 mL of this solution, add exactly 0.5 mL ofthe internal standard solution, then add 1.5 mL of water and(V – 2) mL of dilute ethanol, and use this as the standardsolution. Proceed with the sample solution and the standardsolution as directed in the Assay.
Amount (mg) of digoxin (C41H64O14)=WS×QT
QS×
1200
WS: Amount (mg) of Digoxin Reference Standard
Internal standard solution—A solution of propyl para-hydroxybenzoate in ethanol (95) (1 in 40,000/V).
Dissolution Perform the test with 1 tablet of DigoxinTablets, using 500 mL of diluted hydrochloric acid (3 in 500)at 100 revolutions per minute according to the Method 1 un-der the Dissolution Test as the dissolution medium.Withdraw 30 mL or more of the dissolved solution 60minutes after starting the test, and ˆlter through a mem-brane ˆlter (less than 0.8 mm in pore size). Discard the ˆrst10 mL of the ˆltrate, and use the subsequent ˆltrate as thesample solution. Separately, weigh accurately about 25 mgof Digoxin Reference Standard, previously dried in vacuumat 1059C for 1 hour, dissolve in a small portion of ethanol(95), and add a mixture of ethanol (95) and water (4:1) tomake exactly 500 mL. Pipet 5 mL of this solution, add thedissolution medium to make exactly 500 mL, and use this so-lution as the standard solution. Pipet 2 mL each of the sam-ple solution, the standard solution and the dissolution medi-um, and transfer to brown glass-stoppered test tubes. Addexactly 10 mL of 0.012 g/dL L-ascorbic acid-hydrochloricacid TS to these tubes, and shake. Immediately add exactly 1mL of dilute hydrogen peroxide TS, shake well, and allow tostand at a constant temperature between 259C and 309C for45 minutes. Determine the ‰uorescence intensities, FT, FS,and FB, of these solutions at 360 nm of the excitationwavelength and at 485 nm of the ‰uorescence wavelength asdirected under the Fluorometry, respectively: the dissolutionrate in 60 minutes is not less than 65z. No retest require-ment is applied to Digoxin Tablets.
Dissolution rate (z) with respect to the labeled amount ofdigoxin (C41H64O14)
=WS×FT-FB
FS-FB×
1C
WS: Amount (mg) of Digoxin Reference StandardC: The labeled amount (mg) of digoxin (C41H64O14) in 1
tablet
Assay Weigh accurately the mass of not less than 20Digoxin Tablets, and powder. Weigh accurately a portion ofthe powder, equivalent to about 2.5 mg of digoxin (C41H64O
14), add 30 mL of dilute ethanol, exposure to ultrasonicwaves for 20 minutes, and shake for 5 minutes. After cool-ing, add exactly 5 mL of the internal standard solution anddilute ethanol to make 50 mL, centrifuge, and use the super-natant liquid as the sample solution. Separately, weigh ac-curately about 25 mg of Digoxin Reference Standard, previ-ously dried under reduced pressure at 1059C for 1 hour , dis-solve in 50 mL of warm ethanol (95), cool, and add ethanol(95) to make exactly 100 mL. Pipet 10 mL of this solution,add exactly 5 mL of the internal standard solution, 10 mL ofwater and dilute ethanol to make 50 mL, and use this solu-tion as the standard solution. Perform the test with 10 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine the ratios, QT and QS,of the peak area of digoxin to that of the internal standard.
Amount (mg) of digoxin (C41H64O14)=WS×QT
QS×
110

17051705Supplement II, JPXIV O‹cial Monographs for Part I
WS: Amount (mg) of Digoxin Reference Standard
Internal standard solution—A solution of propyl para-hydroxybenzoate in ethanol (95) (1 in 4000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 220 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about309C.
Mobile phase: A mixture of water and acetonitrile (7:3).Flow rate: Adjust the ‰ow rate so that the retention time
of digoxin is about 10 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, digoxin and the internal standard are eluted inthis order with the resolution between these peaks being notless than 5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of digoxin to that of the internal standard isnot more than 1.0z.
Containers and storage Containers—Tight containers.Storage—Light-resistant.
Dimorpholamineジモルホラミン
Change the origin/limits of the content to read:
Dimorpholamine, when dried, contains not less than98.0z and not more than 101.0z of C20H38N4O4.
Change the Description to read:
Description Dimorpholamine is a white to light yellow,crystalline powder, masses or syrupy liquid.
It is very soluble in ethanol (99.5) and in acetic anhydride,and soluble in water.
The pH of a solution prepared by dissolving 1.0 g ofDimorpholamine in 10 mL of water is between 6.0 and 7.0.
It is hygroscopic.
Change the Identiˆcation to read:
Identiˆcation (1) Determine the absorption spectrum ofa solution of Dimorpholamine (1 in 50,000) as directedunder the Ultraviolet-visible Spectrophotometry, andcompare the spectrum with the Reference Spectrum: bothspectra exhibit similar intensities of absorption at the samewavelengths.
(2) Determine the infrared absorption spectrum of
Dimorpholamine, previously dried, as directed in the potas-sium bromide disk method under the Infrared Spectrophoto-metry, and compare the spectrum with the ReferenceSpectrum: both spectra exhibit similar intensities of absorp-tion at the same wave numbers.
Add the following next to Purity (4):
(5) Related substances—Dissolve 0.20 g of Dimorphola-mine in 10 mL of ethanol (99.5), and use this solution as thesample solution. Pipet 1 mL of the sample solution, addethanol (99.5) to make exactly 100 mL, and use this solutionas the standard solution. Perform the test with these solu-tions as directed under the Thin-layer Chromatography.Spot 10 mL each of the sample solution and the standardsolution on a plate of silica gel for thin-layer chro-matography. Develop the plate with a mixture of ethanol(99.5) and water (4:1) to a distance of about 10 cm, andair-dry the plate. Allow the plate to stand in iodine vapor for10 minutes: the spot other than the principal spot from thesample solution is not more intense than the spot from thestandard solution.
Change the Assay to read:
Assay Weigh accurately about 0.6 g of Dimorpholamine,previously dried, dissolve in 50 mL of acetic anhydride, andtitrate with 0.1 mol/L perchloric acid VS (potentiometrictitration). Perform a blank determination, and make anynecessary correction.
Each mL of 0.1 mol/L perchloric acid VS=39.85 mg of C20H38N4O4
Dimorpholamine Injectionジモルホラミン注射液
Change the origin/limits of content to read:
Dimorpholamine Injection is an aqueous solutionfor injection.
It contains not less than 95.0z and not more than105.0z of the labeled amount of dimorpholamine(C20H38N4O4: 398.54).
Change the Description to read:
Description Dimorpholamine Injection is a clear, colorlessliquid.
pH: 3.0 – 5.5
Change the Identiˆcation (2) to read:
Identiˆcation(2) To a volume of Dimorpholamine Injection, equiva-
lent to 50 mg of Dimorpholamine according to the labeledamount, add 1 mL of dilute hydrochloric acid, andevaporate on a water bath to dryness. Dissolve the residue in

17061706 Supplement II, JPXIVO‹cial Monographs for Part I
2 mL of hydrochloric acid, boil for 10 minutes under a re‰uxcondenser, and evaporate to dryness on a water bath.Dissolve the residue with 1 mL of water, neurtralize withsodium hydroxide TS, and add 0.2 mL of a solution ofacetaldehyde (1 in 20), 0.1 mL of sodium pentacyanonitrosylferrate (III) TS and 0.5 mL of sodium carbonate TS: a bluecolor develops.
Add the following next to Identiˆcation:
Bacterial endotoxins Less than 5.0 EU/mg. Perform thetest with the sample diluted to 0.15 w/vz with water forbacterial endotoxins test.
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Change the Assay to read:
Assay Measure exactly a volume of DimorpholamineInjection, equivalent to about 30 mg of dimorpholamine(C20H38N4O4), and add water to make exactly 200 mL. Pipet1 mL of this solution, shake with exactly 4 mL of the inernalstandard solution for 5 minutes, and use this solution as thesample solution. Separately, weigh accurately about 0.15 gof dimorpholamine for assay, previously dried in a desicca-tor (in vacuum, phosphorus (V) oxide) for 8 hours, anddissolve in water to make exactly 1000 mL. Pipet 1 mL ofthis solutionn, shake with exactly 4 mL of the inernalstandard solution for 5 minutes, and use this solution as thestandard solution. Perform the test with 10 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and calculate the ratios, QT and QS, of the peakarea of dimorpholamine to that of the inernal standard.
Amount (mg) of dimorpholamine (C20H38N4O4)
=WS×QT
QS×
15
WS: Amount (mg) of dimorpholamine for assay
Internal standard solution—A solution of butyl parahydrox-ybenzoate in acetonitrile (1 in 25,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 216 nm).
Column: A stainless steel column 4.6 mm in inside di-ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about409C.
Mobile phase: A mixture of water and acetonitrile (1:1).Flow rate: Adjust the ‰ow rate so that the retention time
of dimorpholamine is about 4 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, dimorpholamine and the internal standard areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of dimorpholamine to that of the internalstandard is not more than 1.0z.
Add the following:
Eperisone Hydrochloride塩酸エペリゾン
C17H25NO.HCl: 295.85(2RS)-1-(4-Ethylphenyl)-2-methyl-3-piperidin-1-ylpropan-1-one monohydrochloride [56839-43-1]
Eperisone Hydrochloride contains not less than98.5z and not more than 101.0z of C17H25NO.HCl,calculated on the anhydrous basis.
Description Eperisone Hydrochloride occurs as a whitecrystalline powder.
It is freely soluble in water, in methanol and in acetic acid(100), and soluble in ethanol (99.5).
Melting point: about 1679C (with decomposition).A solution of Eperisone Hydrochloride in methanol (1 in
100) shows no optical rotation.
Identiˆcation (1) Determine the absorption spectrum ofa solution of Eperisone Hydrochloride in methanol (1 in100,000) as directed under the Ultraviolet-visible Spec-trophotometry, and compare the spectrum with the Refer-ence Spectrum: both spectra exhibit similar intensities ofabsorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofEperisone Hydrochloride as directed in the potassium chlo-ride disk method under the Infrared Spectrophotometry,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wave numbers.
(3) A solution of Eperisone Hydrochloride (1 in 50)responds to the Qualitative Tests for chloride.
Purity (1) Heavy metals—Proceed with 1.0 g of Eperi-

17071707Supplement II, JPXIV O‹cial Monographs for Part I
sone Hydrochloride according to Method 1, and perform thetest. Prepare the control solution with 2.0 mL of StandardLead Solution (not more than 20 ppm).
(2) Piperidine hydrochloride—Dissolve 1.0 g of Eperi-sone Hydrochloride in 20 mL of water, add 2.0 mL ofdiluted hydrochloric acid (1 in 2), 2.0 mL of a solution ofcopper (II) sulfate pentahydrate (1 in 20) and 1.5 mL of am-monia solution (28), and use this solution as the sample solu-tion. Separately, to 2.0 mL of a solution of piperidinehydrochloride (1 in 1000) add 18 mL of water, 2.0 mL ofdiluted hydrochloric acid (1 in 2), 2.0 mL of a solution ofcopper (II) sulfate pentahydrate (1 in 20) and 1.5 mL ofammonia solution (28), and use this solution as the standardsolution. To each of the sample solution and the standardsolution add 10 mL of a mixture of isopropylether andcarbon disulˆde (3:1), shake for 30 seconds, allow them tostand for 2 minutes, and compare the color of the upperlayer: the color obtained from the sample solution is notmore darker than that from the standard solution.
(3) Related substances—Dissolve 0.1 g of EperisoneHydrochloride in 100 mL of the mobile phase, and use thissolution as the sample solution. Pipet 1 mL of the samplesolution, add the mobile phase to make exactly 100 mL, anduse this solution as the standard solution. Perform the testwith exactly 10 mL each of the sample solution and the stan-dard solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine eachpeak area by the automatic integration method: the totalarea of the peaks other than the peak of eperisone is notmore than 1/5 of the peak area of eperisone from the stan-dard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).Column: A stainless steel column 4.6 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about309C.
Mobile phase: A mixture of methanol, 0.0375 mol/L sodi-um 1-decanesulfonate TS and perchloric acid (600:400:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof eperisone is about 17 minutes.
Time span of measurement: About 2 times as long as theretention time of eperisone.System suitability—
Test for required detectability: Pipet 1 mL of the standardsolution, and add the mobile phase to make exactly 10 mL.Conˆrm that the peak area of eperisone obtained from 10mL of this solution is equivalent to 7 to 13z of that obtainedfrom 10 mL of the standard solution.
System performance: When the procedure is run with 10mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of eperisone are not less than 4000 stepsand not more than 2.0, respectively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-
ing conditions, the relative standard deviation of the peakarea of eperisone is not more than 3.0z.
Water Not more than 0.2z (0.1 g, coulometric titration).
Residue on ignition Not more than 0.2z (1 g).
Assay Weigh accurately about 0.6 g of EperisoneHydrochloride, dissolve in 20 mL of acetic acid (100), add80 mL of acetic anhydride, and titrate with 0.1 mol/L per-chloric acid VS (potentiometric titration). Perform a blankdetermination in the same manner, and make any necessarycorrection.
Each mL of 0.1 mol/L perchloric acid VS=29.58 mg of C17H25NO.HCl
Containers and storage Containers—Well-closed contain-ers.
Ethionamideエチオナミド
Change to read except the structural formulaand chemical name:
Ethionamide, when dried, contains not less than98.5z and not more than 101.0z of C8H10N2S.
Description Ethionamide occurs as yellow crystals or crys-talline powder, having a characteristic odor.
It is soluble in methanol and in acetic acid (100), sparinglysoluble in ethanol (99.5) and in acetone, and practically in-soluble in water.
Identiˆcation (1) Determine the absorption spectrum ofa solution of Ethionamide in methanol (3 in 160,000) asdirected under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wavelengths.
(2) Determine the infrared absorption spectrum ofEthionamide as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry, and com-pare the spectrum with the Reference Spectrum: both spec-tra exhibit similar intensities of absorption at the same wavenumbers.
Melting point 161 – 1659C
Purity(1) Acid—Dissolve 3.0 g of Ethionamide in 30 mL of
methanol by warming, add 90 mL of water, allow to stand inice water for 1 hour, and ˆlter. To 80 mL of the ˆltrate add0.8 mL of cresol red TS and 0.20 mL of 0.1 mol/L sodiumhydroxide VS: a red color develops.
(2) Heavy metals—Proceed with 1.0 g of Ethionamideaccording to Method 2, and perform the test. Prepare thecontrol solution with 2.0 mL of Standard Lead Solution (notmore than 20 ppm).

17081708 Supplement II, JPXIVO‹cial Monographs for Part I
(3) Arsenic—Prepare the test solution with 1.0 g ofEthionamide according to Method 3. Add 10 mL of a solu-tion of magnesium nitrate hexahydrate in ethanol (95) (1 in50), then add 1.5 mL of hydrogen peroxide (30), and ˆre toburn (not more than 2 ppm).
(4) Related substances—Conduct this procedure withoutexposure to light, using light-resistant vessels. Dissolve 0.20g of Ethionamide in 10 mL of acetone, and use this solutionas the sample solution. Pipet 0.5 mL of the sample solution,add acetone to make exactly 100 mL, and use this solution asthe standard solution (1). Separately, pipet exactly 0.2 mLof the sample solution, add acetone to make exactly 100 mL,and use this solution as the standard solution (2). Performthe test with these solutions as directed under the Thin-layerChromatography. Spot 10 mL each of the sample solutionand the standard solutions (1) and (2) on a plate of silica gelwith ‰uorescent indicator for thin-layer chromatography,develop with a mixture of ethyl acetate, hexane andmethanol (6:2:1) to a distance of about 15 cm, and air-drythe plate. Examine under ultraviolet light (main wavelength:254 nm): the spot other than the principal spot obtained withthe sample solution is not more intense than the spot withthe standard solution (1), and number of the spot other thanthe principal spot obtained with the sample solution which ismore intense than the spot with the standard solution (2) isnot more than one.
Loss on drying Not more than 0.5z (1 g, 1059C, 3 hours).
Residue on ignition Not more than 0.10z (1 g).
Assay Weigh accurately about 0.3 g of Ethionamide,previously dried, dissolve in 50 mL of acetic acid (100), andtitrate with 0.1 mol/L perchloric acid VS until the color ofthe solution changes from orange-red to dark orange-brown(indicator: 2 mL of p-naphtholbenzein TS). Perform a blankdetermination in the same manner, and make any necessarycorrection.
Each mL of 0.1 mol/L perchloric acid VS=16.62 mg of C8H10N2S
Containers and storage Containers—Well-closed contain-ers.
Etilefrine Hydrochloride Tablets塩酸エチレフリン錠
Change the origin/limits of content to read:
Etilefrine Hydrochloride Tablets contain not lessthan 93.0z and not more than 107.0z of the labeledamount of etilefrine hydrochloride (C10H15NO2.HCl:217.69).
Change the Identiˆcation to read:
Identiˆcation To a quantity of powdered EtilefrineHydrochloride Tablets, equivalent to 5 mg of EtilefrineHydrochloride according to the labeled amount, add 60 mLof diluted hydrochloric acid (1 in 1000), shake well, add40 mL of diluted hydrochloric acid (1 in 1000), and ˆlter.Determine the absorption spectrum of the ˆltrate as directedunder the Ultraviolet-visible Spectrophotometry, usingdiluted hydrochloric acid (1 in 1000) as the blank: it exhibitsa maximum between 271 nm and 275 nm.
Add the following next to Identiˆcation:
Content uniformity Perform the test according to the fol-lowing method: it meets the requirements of the ContentUniformity Test.
To 1 tablet of Etilefrine Hydrochloride Tablets add 60 mLof diluted hydrochloric acid (1 in 1000), and proceed asdirected in the Assay.
Amount (mg) of etilefrine hydrochloride (C10H15NO2.HCl)
=WS×AT
AS×
110
WS: Amount (mg) of etilefrine hydrochloride for assay
Change the Assay to read:
Assay Weigh accurately the mass of not less than 20Etilefrine Hydrochloride Tablets, and powder. Weigh ac-curately a portion of the powder, equivalent to about 5 mgof etilefrine hydrochloride (C10H15NO2.HCl), add 60 mL ofdiluted hydrochloric acid (1 in 1000), shake for 10 minutes,add diluted hydrochloric acid (1 in 1000) to make exactly 100mL, and ˆlter. Discard the ˆrst 20 mL of the ˆltrate, and usethe subsequent ˆltrate as the sample solution. Separately,weigh accurately about 50 mg of etilefrine hydrochloride forassay, previously dried at 1059C for 4 hours, and dissolve indiluted hydrochloric acid (1 in 1000) to make exactly 100mL. Pipet 10 mL of this solution, add diluted hydrochloricacid (1 in 1000) to make exactly 100 mL, and use this solu-tion as the standard solution. Perform the test with exactly20 mL each of the sample solution and the standard solutionas direct under the Liquid Chromatography according to thefollowing conditions, and determine the peak areas, AT andAS, of etilefrine.
Amount (mg) of etilefrine hydrochloride (C10H15NO2.HCl)
=WS×AT
AS×
110
WS: Amount (mg) of etilefrine hydrochloride for assay
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 220 nm).Column: A stainless steel column 4.6 mm in inside di-
ameter and 25 cm in length, packed with octylsilanized silicagel for liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about409C.

17091709Supplement II, JPXIV O‹cial Monographs for Part I
Mobile phase: Dissolve 5 g of sodium lauryl sulfate in940 mL of water and 500 mL of acetonitrile, and adjust thepH to 2.3 with phosphoric acid.
Flow rate: Adjust the ‰ow rate so that the retention timeof etilefrine is about 6 minutes.System suitability—
System performance: Dissolve 4 mg of bamethan sulfateand 4 mg of etilefrine hydrochloride in the mobile phase tomake 50 mL. When the procedure is run with 20 mL of thissolution under the above operating conditions, etilefrine andbamethan are eluted in this order with the resolutionbetween these peaks being not less than 5.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of etilefrine is not more than 1.0z.
Add the following:
Etoposideエトポシド
C29H32O13: 588.56(5R,5aR,8aR,9S)-9-s[4,6-O-(1R)-Ethylidene-b-D-glucopyranosyl]oxyt-5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)furo[3?,4?:6,7]naphtha[2,3-d]-1,3-dioxol-6(5aH)-one [33419-42-0]
Etoposide contains not less than 98.0z and notmore than 102.0z of C29H32O13, calculated on theanhydrous basis.
Description Etoposide occurs as white crystals or crystal-line powder.
It is sparingly soluble in methanol, slightly soluble inethanol (99.5), and very slightly soluble in water.
Melting point: about 2609C (with decomposition).
Identiˆcation (1) Determine the absorption spectrum ofa solution of Etoposide in methanol (1 in 10,000) as directedunder the Ultraviolet-visible Spectrophotometry, andcompare the spectrum with the Reference Spectrum or thespectrum of a solution of Etoposide Reference Standardprepared in the same manner as the sample solution: bothspectra exhibit similar intensities of absorption at the samewavelengths.
(2) Determine the infrared absorption spectrum of
Etoposide as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum or the spectrum ofEtoposide Reference Standard: both spectra exhibit similarintensities of absorption at the same wave numbers.
Optical rotation [a]D20: -100 –-1059(0.1 g calculated onthe anhydrous basis, methanol, 20 mL, 100 mm).
Purity (1) Heavy metals—Proceed with 2.0 g of Etopo-side according to Method 2, and perform the test. Preparethe control solution with 2.0 mL of Standard Lead Solution(not more than 10 ppm).
(2) Related substances—Dissolve 50 mg of Etoposide in10 mL of methanol, add the mobile phase to make 50 mL,and use this solution as the sample solution. Pipet 2 mL ofthe sample solution, add the mobile phase to make exactly200 mL, and use this solution as the standard solution. Per-form the test with exactly 50 mL each of the sample solutionand the standard solution as directed under the Liquid Chro-matography according to the following conditions, anddetermine each peak area by the automatic integrationmethod: the area of the peak other than etoposide is notlarger than 1/5 times the peak area of etoposide with thestandard solution, and the total area of the peaks other thanthe peak of etoposide with the sample solution is not largerthan 1/2 times the peak area of etoposide with the standardsolution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 3 times as long as theretention time of etoposide after the solvent peak.System suitability—
Test for required detectability: Measure exactly 1 mL ofthe standard solution, and add the mobile phase to makeexactly 10 mL. Conˆrm that the peak area of etoposideobtained with 50 mL of this solution is equivalent to 7 to13z of that with 50 mL of the standard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 50 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of etoposide is not more than 2.0z.
Water Not more than 4.0z (0.5 g, volumetric titration,direct titration).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 25 mg each of Etoposideand Etoposide Reference Standard, separately determinedthe water content, and dissolve separately in methanol tomake exactly 25 mL. Pipet 10 mL each of these solutions,add exactly 5 mL of the internal standard solution and themobile phase to make 50 mL, and use these solutions as thesample solution and the standard solution. Perform the testwith 50 mL each of the sample solution and the standard

17101710 Supplement II, JPXIVO‹cial Monographs for Part I
solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of etoposide to that ofthe internal standard.
Amount (mg) of C29H32O13=WS×QT
QS
WS: Amount (mg) of Etoposide Reference Standard,calculated on the anhydrous basis
Internal standard solution—A solution of 2,6-dichloro-phenol in methanol (3 in 2500).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 290 nm).
Column: A stainless steel column 3.9 mm in insidediameter and 30 cm in length, packed with phenylsilanizedsilica gel for liquid chromatography (10 mm in particlediameter).
Column temperature: A constant temperature of about359C.
Mobile phase: Dissolve 6.44 g of sodium sulfate decahy-drate in diluted acetic acid (100) (1 in 100) to make 1000 mL,and add 250 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof etoposide is about 20 minutes.System suitability—
System performance: Dissolve 10 mg of Etoposide in2 mL of methanol, add 8 mL of the mobile phase, and mixwell. Add 0.1 mL of diluted acetic acid (100) (1 in 25) and0.1 mL of phenolphthalein TS, and add sodium hydroxideTS until the color of the solution changes to faintly red. Af-ter allowing to stand for 15 minutes, add 0.1 mL of dilutedacetic acid (100) (1 in 25). When the procedure is run with 10mL of this solution under the above operating conditions, theresolution between the peak of etoposide and the peak hav-ing the relative retention time of about 1.3 with respect toetoposide is not less than 3.
System repeatability: When the test is repeated 6 timeswith 50 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of etoposide to that of the internal standard isnot more than 1.0z.
Containers and storage Containers—Tight containers.Storage—Light-resistant.
Flavin Adenine DinucleotideSodiumフラビンアデニンジヌクレオチドナトリウム
Change the Purity (5) to read:
Purity(5) Related substances—Dissolve 0.10 g of Flavin Ade-
nine Dinucleotide Sodium in 200 mL of the mobile phase,and use this solution as the sample solution. Perform the test
with 20 mL of the sample solution as directed under theLiquid Chromatography according to the following condi-tions. Determine the peak area, A, of ‰avin adeninedinucleotide and the total area, S, of peaks other than thepeak of ‰avin adenine dinucleotide by the automatic integra-tion method: S/(A+S) is not more than 0.10.Operating conditions—
Column, column temperature, mobile phase, ‰ow rate,and time span of measurement: Proceed as directed in theoperating conditions in the Procedure (ii) under Assay (1).
Detector: An ultraviolet absorption photometer(wavelength: 260 nm).System suitability—
Test for required detection: To exactly 2 mL of the samplesolution add the mobile phase to make exactly 20 mL, anduse this solution as the solution for system suitability test.Conˆrm that the peak area of ‰avin adenine dinucleotideobtained from 20 mL of this solution is equivalent to 8 to12z of that of ‰avin adenine dinucleotide obtained from20 mL of the sample solution.
System performance: Proceed as directed in the systemsuitability in the Procedure (ii) under Assay (1).
System repeatability: When the test is repeated 6 timeswith 20 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of ‰avin adenine dinucleotide is notmore than 1.0z.
Change the Assay (1) to read:
Assay (1) Procedure (i) Total ‰avin content—Conductthis procedure without exposure to daylight, using light-resistant vessels. Weigh accurately about 0.1 g of FlavinAdenine Dinucleotide Sodium, and dissolve in water tomake exactly 200 mL. Pipet 5 mL of this solution, add 5 mLof zinc chloride TS, and heat in a water bath for 30 minutes.After cooling, add water to make exactly 100 mL, and usethis solution as the sample solution. Separately, weighaccurately about 50 mg of Ribo‰avin Reference Standard,previously dried at 1059C for 2 hours, dissolve in 200 mL ofdiluted acetic acid (100) (1 in 100) by warming, cool, addwater to make exactly 500 mL. Pipet 10 mL of this solution,add water to make exactly 100 mL, and use this solution asthe standard solution. Determine the absorbances, AT and AS, of the sample solution and the standard solution at 450 nmas directed under the Ultraviolet-visible Spectrophotometry,using water as the blank.
Total amout (mg) of ‰avin
=WS×AT
AS×
45
WS: Amount (mg) of Ribo‰avin Reference Standard
(ii) Peak area ratio of ‰avin adenine dinucleotide—Dis-solve 0.1 g of Flavin Adenine Dinucleotide Sodium in 200mL of water, and use this solution as the sample solution.Perform the test with 5 mL of this solution as directed underthe Liquid Chromatography according to the followingconditions. Determine the peak area, A of ‰avin adenine

17111711Supplement II, JPXIV O‹cial Monographs for Part I
dinucleotide, and the total area, S, of the peaks other than‰avin adenine dinucleotide by the automatic integrationmethod.
Peak area ratio of ‰avin adenine dinucleotide
=1.08×A
1.08×A+S
Operating conditions—Detector: A visible spectrophotometer (wavelength: 450
nm).Column: A stainless steel column 4 mm in inside diameter
and 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about359C.
Mobile phase: A mixture of a solution of potassium di-hydrogen phosphate (1 in 500) and methanol (4:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof ‰avin adenine dinucleotide is about 10 minutes.
Time span of measurement: About 4.5 times as long as theretention time of ‰avin adenine dinucleotide.System suitability—
Test for required detection: To exactly 2 mL of the samplesolution add water to make exactly 20 mL, and use this solu-tion as the solution for system suitability test. Pipet 2 mL ofthe solution, and add water to make exactly 20 mL. Conˆrmthat the peak area of ‰avin adenine dinucleotide obtainedfrom 5 mL of this solution is equivalent to 8 to 12z of thatof ‰avin adenine dinucleotide obtained from 5 mL of thesolution for system suitability test.
System performance: Dissolve 20 mg each of FlavinAdenine Dinucleotide Sodium and ribo‰avin sodium phos-phate in 100 mL of water. When the procedure is run with5 mL of this solution under the above operating conditions,‰avin adenine dinucleotide and ribo‰avin phosphate areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 5 mL of the solution for system suitability test under theabove operating conditions, the relative standard deviationof the peak area of ‰avin adenine dinucleotide is not morethan 1.0z.
(2) Calculation
Amount (mg) of C27H31N9Na2O15P2
=fT×fR×2.2040
fT: Total amount (mg) of ‰avin in Flavin AdenineDinucleotide Sodium obtained from the procedure (i).
fR: Peak area ratio of ‰avin adenine dinucleotide in FlavinAdenine Dinucleotide Sodium obtained from theprocedure (ii).
Add the following:
Flomoxef Sodium for Injection注射用フロモキセフナトリウム
Flomoxef Sodium for Injection is a preparation forinjection which is dissolved before use.
It contains not less than 90.0z and not morethan 110.0z of the labeled amount of ‰omoxef(C15H18F2N6O7S2: 496.47).
Method of preparation Prepare as directed under Injec-tions, with Flomoxef Sodium.
Description Flomoxef Sodium for Injection occurs aswhite to light yellowish white, friable masses or powder.
Identiˆcation Proceed as directed in the Identiˆcation (3)under Flomoxef Sodium.
pH The pH of a solution obtained by dissolving an amountof Flomoxef Sodium for Injection, equivalent to 0.5 g(potency) of Flomoxef Sodium according to the labeledamount, in 5 mL of water is between 4.0 and 5.5.
Purity (1) Clarity and color of solution—Dissolve anamount of Flomoxef Sodium for Injection, equivalent to 1.0g (potency) of Flomoxef Sodium according to the labeledamount, in 10 mL of water: the solution is clear and color-less or pale yellow.
(2) 1-(2-Hydroxyethyl)-1H-tetrazol-5-thiol—Use thesample solution obtained in the Assay as the sample solu-tion. Weigh accurately about 20 mg of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol, and dissolve in water to make exactly100 mL. Pipet 5 mL of this solution, add exactly 25 mL ofthe internal standard solution and water to make 50 mL, anduse this solution as the standard solution. Perform the testwith 5 mL each of the sample solution and the standard solu-tion as directed under the Liquid Chromatography accord-ing to the following conditions, and determine the ratios, QT
and QS, of the peak area of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol to that of the internal standard. Calculate the amountof 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol per 1 g (potency)of Flomoxef Sodium for Injection by the following formula:not more than 10 mg.
Amount (mg) of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol(C3H6N4OS)
=WS×QT
QS×
110
WS: Amount (mg) of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol
Operating conditions—Proceed as directed in the Assay.
System suitability—Test for required detectability: Pipet 1 mL of the standard
solution, and add water to make exactly 20 mL. Conˆrmthat the peak area of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiolobtained from 5 mL of this solution is equivalent to 3.5 –6.5z of that obtained from 5 mL of the standard solution.

17121712 Supplement II, JPXIVO‹cial Monographs for Part I
System performance: When the procedure is run with 5 mLof the standard solution under the above operating condi-tions, 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol and the inter-nal standard are eluted in this order with the resolutionbetween these peaks being not less than 20.
System repeatability: When the test is repeated 3 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of 1-(2-hydroxyethyl)-1H-tetrazol-5-thiol tothat of the internal standard is not more than 1.0z.
Water Not more than 1.5z (0.5 g, volumetric titration,back titration).
Bacterial endotoxins Less than 0.025 EU/mg (potency).
Mass variation It meets the requirements of the MassVariation Test.
Foreign insoluble matter Perform the test according toMethod 2: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Weigh accurately the mass of the contents of notless than 10 Flomoxef Sodium for Injection, and calculatethe average mass of the content. Spread out thinly about 1 gof the content in a petri dish, allow the dish to stand in adesiccator containing a saturated solution of magnesiumbromide without light exposure to equilibrate the sample toconstant water content. Determine the water content,separately, with about 0.1 g of the sample according to themethod described in Water. Weigh accurately an amount ofthe sample, equivalent to about 50 mg (potency) of Flomox-ef Sodium according to the labeled amount, add exactly 50mL of the internal standard solution to dissolve, add waterto make 100 mL, and use this solution as the sample solu-tion. Separately weigh accurately about 50 mg (potency) ofFlomoxef Triethylammonium Reference Standard, addexactly 50 mL of the internal standard solution to dissolve,add water to make 100 mL, and use this solution as thestandard solution. Proceed as directed in the Assay underFlomoxef Sodium.
Amount [mg (potency)] of ‰omoxef (C15H18F2N6O7S2)
=WS×QT
QS×1000
WS: Amount [mg (potency)] of Flomoxef Triethylammo-nium Reference Standard
Internal standard solution—A solution of m-cresol (3 in1000).
Containers and storage Containers—Hermetic containers.Polyethylene or polypropylene containers for aqueous
injection may be used.
Add the following:
Flopropione Capsulesフロプロピオンカプセル
Flopropione Capsules contain not less than 93.0zand not more than 107.0z of the labeled amount of‰opropione (C9H10O4: 182.17).
Method of preparation Prepare as directed under theCapsules, with Flopropione.
Identiˆcation (1) Powder the contents of FlopropioneCapsules. To a portion of the powder, equivalent to 60 mgof Flopropione according to the labeled amount, add 40 mLof water, shake well, and ˆlter. To 5 mL of the ˆltrate add1 mL of iron (III) nitrate TS: a red-purple color appears.
(2) Powder the contents of Flopropione Capsules. To aportion of the powder, equivalent to 90 mg of Flopropioneaccording to the labeled amount, add 100 mL of ethanol(99.5), shake well, and ˆlter. To 5 mL of the ˆltrate addethanol (99.5) to make 50 mL. To 5 mL of this solution addethanol (99.5) to make 100 mL, and use this solution as thesample solution. Determine the absorption spectrum of thesample solution as directed under the Ultraviolet-visibleSpectrophotometry: it exhibits a maximum between 283 nmand 287 nm.
Content uniformity Perform the test as directed in theAssay, and determine the content: it meets the requirementsof the Content Uniformity Test.
Dissolution Being speciˆed separately.
Assay To 1 capsule of Flopropione Capsules add 43 mL ofa mixture of water and phosphoric acid (86:1), and disinte-grate the capsule at 509C in a water bath. After cooling, addacetonitrile to make exactly 100 mL, stir for 10 minutes, andcentrifuge a part of this solution for 5 minutes at 3000 rpm.Use the supernatant liquid as the sample solution. Separate-ly, weigh accurately about 40 mg of ‰opropione for assay,separately determine the water content in the same manneras Flopropione, add 70 mL of the mobile phase, and dis-solve by exposure for 10 minutes to ultrasonic vibration.Add the mobile phase to make exactly 100 mL, and use thissolution as the standard solution. Perform the test withexactly 5 mL each of the sample solution and the standardsolution as directed under the Liquid chromatography ac-cording to the following conditions, and determine the peakareas, AT and AS, of ‰opropione. Repeat the procedureabove with totally not less than 10 capsules, and calculatethe mean.
Amount (mg) of ‰opropione (C9H10O4)=WS×AT
AS
WS: Amount (mg) of ‰opropione for assay, calculated on

17131713Supplement II, JPXIV O‹cial Monographs for Part I
the anhydrous basis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 267 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about359C.
Mobile phase: A mixture of acetonitrile, water and phos-phoric acid (114:86:1)
Flow rate: Adjust the ‰ow rate so that the retention timeof ‰opropione is about 3 minutes.System suitability—
System performance: Dissolve 50 mg of ‰opropione in50 mL of the mobile phase. To 20 mL of the solution add25 mL of a solution prepared by dissolving 25 mg of ethylparahydroxybenzoate in 30 mL of acetonitrile and add waterto make 50 mL, and then add the mobile phase to make50 mL. When the procedure is run with 5 mL of this solutionunder the above operating conditions, ‰opropione and ethylparahydroxybenzoate are eluted in this order with the reso-lution between these peaks being not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of ‰opropione is not more than 1.0z.
Containers and storage Containers—Tight containers.
Fosfomycin Calciumホスホマイシンカルシウム
Change the origin/limits of content to read:
Fosfomycin Calcium is the calcium salt of a sub-stance having antibacterial actively produced by thegrowth of Streptomyces fradiae or by the chemicalsynthesis.
It contains not less than 725 mg (potency) and notmore than 805 mg (potency) per mg, calculated on theanhydrous basis. The potency of Fosfomycin Calciumis expressed as mass (potency) of fosfomycin(C3H7O4P: 138.06).
Change the Description to read:
Description Fosfomycin Calcium occurs as a white crystal-line powder.
It is slightly soluble in water, and practically insoluble inmethanol and in ethanol (99.5).
Change the Identiˆcation (2) to read:
Identiˆcation(2) Determine the spectrum of a solution of Fosfomycin
Calcium in heavy water for nuclear magnetic resonancespectroscopy (1 in 300), using sodium 3-trimethylsilyl-propanesulfonate for nuclear magnetic resonance spec-troscopy as an internal reference compound, as directedunder the Nuclear Magnetic Resonance Spectroscopy: itexhibits a double signal at around d 1.5 ppm, a duple doublesignal at around d 2.9 ppm, a multiple signal at around d 3.3ppm, and no signal at around d 1.4 ppm.
Change the Purity (1) to read:
Purity (1) Heavy metals—To 1.0 g of FosfomycinCalcium add 40 mL of 0.25 mol/L acetic acid TS and waterto make 50 mL. Proceed with this solution according toMethod 1, and perform the test. Prepare the control solutionwith 2.0 mL of Standard Lead Solution (not more than 20ppm).
Fosfomycin Sodiumホスホマイシンナトリウム
Change the origin/limits of content to read:
Fosfomycin Sodium is the sodium salt of a sub-stance having antibacterial activity produced by thegrowth of Streptomyces fradiae or by the chemicalsynthesis.
It contains not less than 725 mg (potency) and notmore than 770 mg (potency) per mg, calculated on theanhydrous basis. The potency of Fosfomycin Sodiumis expressed as mass (potency) of fosfomycin(C3H7O4P: 138.06).
Change the Description to read:
Description Fosfomycin Sodium occurs as a white crystal-line powder.
It is very soluble in water, sparingly soluble in methanol,and practically insoluble in ethanol (99.5).
Change the Identiˆcation (2) to read:
Identiˆcation(2) Determine the spectrum of a solution of Fosfomycin
Sodium in heavy water for nuclear magnetic resonancespectroscopy (1 in 300), using sodium 3-trimethylsilyl-propanesulfonate for nuclear magnetic resonance spec-troscopy as an internal reference compound, as directedunder the Nuclear Magnetic Resonance Spectroscopy: itexhibits a double signal at around d 1.5 ppm, a duple doublesignal at around d 2.8 ppm, a multiple signal at around d3.3 ppm, and no signal at around d 1.3 ppm.

17141714 Supplement II, JPXIVO‹cial Monographs for Part I
Furosemideフロセミド
Change the origin/limits of content to read:
Furosemide, when dried, contains not less than98.0z and not more than 101.0z of C12H11ClN2O5S.
Change the Description to read:
Description Furosemide occurs as white, crystals or crys-talline powder.
It is freely soluble in N,N-dimethylformamide, soluble inmethanol, sparingly soluble in ethanol (99.5), slightly solu-ble in acetonitrile and in acetic acid (100), and practicallyinsoluble in water.
It dissolves in dilute sodium hydroxide TS.It is gradually colored by light.Melting point: about 2059C (with decomposition).
Change the Identiˆcation to read:
Identiˆcation (1) Dissolve 25 mg of Furosemide in 10 mLof methanol. To 1 mL of this solution add 10 mL of 2mol/L hydrochloric acid TS. Heat the solution under are‰ux condenser on a water bath for 15 minutes, cool, andadd 18 mL of sodium hydroxide TS to make weakly acidic:the solution responds to the Qualitative Tests for primaryaromatic amines, producing a red to red-purple color.
(2) Determine the absorption spectrum of a solution ofFurosemide in dilute sodium hydroxide TS (1 in 125,000) asdirected under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum orthe spectrum of a solution of Furosemide ReferenceStandard prepared in the same manner as the sample solu-tion: both spectra exhibit similar intensities of absorption atthe same wavelengths.
(3) Determine the infrared absorption spectrum ofFurosemide as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry, and com-pare the spectrum with the Reference Spectrum or thespectrum of Furosemide Reference Standard: both spectraexhibit similar intensities of absorption at the same wavenumbers.
Change the Purity to read:
Purity (1) Clarity and color of solution—Dissolve 0.5 gof Furosemide in 10 mL of a solution of sodium hydroxide(1 in 50): the solution is clear and colorless.
(2) Chloride—Dissolve 2.6 g of Furosemide in 90 mL ofdilute sodium hydroxide TS, add 2 mL of nitric acid, andˆlter. To 25 mL of the ˆltrate add 6 mL of dilute nitric acidand water to make 50 mL, and perform the test using this so-lution as the test solution. Prepare the control solution asfollows: To 0.40 mL of 0.01 mol/L hydrochloric acid VSadd 6 mL of dilute nitric acid and water to make 50 mL (not
more than 0.020z).(3) Sulfate—To 20 mL of the ˆltrate obtained in (2) add
1 mL of dilute hydrochloric acid and water to make 50 mL,and perform the test using this solution as the test solution.Prepare the control solution as follows: To 0.35 mL of 0.005mol/L sulfuric acid VS add 1 mL of dilute hydrochloric acidand water to make 50 mL (not more than 0.030z).
(4) Heavy metals—Proceed with 2.0 g of Furosemideaccording to Method 2, and perform the test. Prepare thecontrol solution with 2.0 mL of Standard Lead Solution (notmore than 10 ppm).
(5) Related substances—Dissolve 25 mg of Furosemidein 25 mL of the dissolving solution, and use this solution asthe sample solution. Pipet 1 mL of the sample solution, addthe dissolving solution to make exactly 200 mL, and use thissolution as the standard solution. Perform the test with 20mL each of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine each peak area by theautomatic integration method: the area of each peakappeared ahead of the peak of furosemide is not more than2/5 times the peak area of furosemide from the standard so-lution, the area of each peak appeared behind the peak offurosemide is not more than 1/4 times the peak area offurosemide from the standard solution, and the total area ofthese peaks is not more than 2 times the peak area offurosemide from the standard solution.Dissolving solution—To 22 mL of acetic acid (100) add amixture of water and acetonitrile (1:1) to make 1000 mL.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 272 nm).
Column: A stainless steel column 4.6 mm in inside di-ameter and 25 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of water, tetrahydrofuran andacetic acid (100) (70:30:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof furosemide is about 18 minutes.
Time span of measurement: About 2.5 times as long as theretention time of furosemide after the solvent peak.System suitability—
Test for required detectability: Measure exactly 2 mL ofthe standard solution, and add the dissolving solution tomake exactly 50 mL. Conˆrm that the peak area offurosemide obtained from 20 mL of this solution is equiva-lent to 3.2 to 4.8z of that obtained from 20 mL of thestandard solution.
System performance: When the procedure is run with 20mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of furosemide is not less than 7000 andnot more than 1.5, respectively.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-

17151715Supplement II, JPXIV O‹cial Monographs for Part I
ing conditions, the relative standard deviation of the peakarea of furosemide is not more than 2.0z.
Change the Assay to read:
Assay Weigh accurately about 0.5 g of Furosemide, previ-ously dried, dissolve in 50 mL of N,N-dimethylformamide,and titrate with 0.1 mol/L sodium hydroxide VS until thecolor of the solution changes from yellow to blue (indicator:3 drops of bromothymol blue TS). Perform a blank determi-nation with a mixture of 50 mL of N,N-dimethylformamideand 15 mL of water, and make any necessary correction
Each mL of 0.1 mol/L sodium hydroxide VS=33.07 mg of C12H11ClN2O5S
Add the following:
Furosemide Tabletsフロセミド錠
Furosemide Tablets contain not less than 95.0z andnot more than 105.0z of the labeled amount offurosemide (C12H11ClN2O5S: 330.74).
Method of preparation Prepare as directed under Tablets,with Furosemide.
Identiˆcation (1) Shake well a quantity of powderedFurosemide Tablets, equivalent to 0.2 g of Furosemideaccording to the labeled amount, with 40 mL of acetone,and ˆlter. To 0.5 mL of the ˆltrate add 10 mL of 2 mol/Lhydrochloric acid TS, and heat under a re‰ux condenser on awater bath for 15 minutes. After cooling, add 18 mL ofsodium hydroxide TS to make the solution slightly acetic:the solution responds to the Qualitative Tests for primaryaromatic amines, producing a red to red-purple color.
(2) Determine the absorption spectrum of the samplesolution obtained in the Assay as directed under theUltraviolet-visible Spectrophotometry: it exhibits maximabetween 227 nm and 231 nm, between 269 nm and 273 nm,and between 330 nm and 336 nm.
Purity To a quantity of powdered Furosemide Tablets,equivalent to 40 mg of Furosemide according to the labeledamount, add about 30 mL of acetone, shake well, and addacetone to make exactly 50 mL. Centrifuge the solution, add3.0 mL of water to 1.0 mL of the supernatant liquid, cool inice, add 3.0 mL of dilute hydrochloric acid and 0.15 mL ofsodium nitrite TS, shake, and allow to stand for 1 minute.Add 1.0 mL of ammonium amidosulfate TS, shake well,allow to stand for 3 minutes, add 1.0 mL of N,N-diethyl-N?-1-naphthylethylenediamine oxalate TS, shake well, andallow to stand for 5 minutes. Perform the test with this solu-tion as directed under the Ultraviolet-visible Spectrophoto-metry, using a solution prepared in the same manner with1.0 mL of acetone as the blank: the absorbance at 530 nm isnot more than 0.10.
Content uniformity Perform the test according to the fol-lowing method: it meets the requirements of the ContentUniformity Test.
To 1 tablet of Furosemide Tablets add a suitable amountof 0.05 mol/L sodium hydroxide TS, shake to disintegrate,then add 0.05 mol/L sodium hydroxide TS to make exactlyV mL so that each mL contains about 0.4 mg of furosemide(C12H11ClN2O5S). Filter the solution, discard the ˆrst 10 mLof the ˆltrate, pipet the subsequent 2 mL of the ˆltrate, add0.05 mol/L sodium hydroxide TS to make exactly 100 mL,and use this solution as the sample solution. Proceed asdirected in the Assay.
Amount (mg) of furosemide (C12H11ClN2O5S)
=WS×AT
AS×
V100
WS: Amount (mg) of Furosemide Reference Standard
Dissolution Perform the test with 1 tablet of FurosemideTablets at 50 revolutions per minute according to Method 2under the Dissolution Test, using 900 mL of diluted phos-phate buŠer solution, pH 6.8 (1 in 2) as the dissolution medi-um. Withdraw 20 mL or more of the dissolution medium 15minutes after starting the test for a 20-mg tablet or 30minutes after for a 40-mg tablet, and ˆlter through a mem-brane ˆlter with pore size of not more than 0.45 mm. Discardthe ˆrst 10 mL of the ˆltrate, pipet V mL of the subsequentˆltrate, add diluted phosphate buŠer solution, pH 6.8 (1 in2) to make exactly V? mL so that each mL contains about 10mg of furosemide (C12H11ClN2O5S) according to the labeledamount, and use this solution as the sample solution.Separately, weigh accurately about 20 mg of FurosemideReference Standard, previously dried at 1059C for 4 hours,and dissolve in 5 mL of methanol, and add diluted phos-phate buŠer solution, pH 6.8 (1 in 2) to make exactly 100mL. Pipet 5 mL of this solution, add diluted phosphatebuŠer solution, pH 6.8 (1 in 2) to make exactly 100 mL, anduse this solution as the standard solution. Determine theabsorbances, AT and AS, of the sample solution and thestandard solution at 277 nm as directed under the Ultrav-iolet-visible Spectrophotometry: the dissolution rates for a20-mg tablet in 15 minutes and for a 40-mg tablet in 30minutes are not less than 80z, respectively.
Dissolution rate (z) with respect to the labeled amount offurosemide (C12H11ClN2O5S)
=WS×AT
AS×
V?V×
1C×45
WS: Amount (mg) of Furosemide Reference StandardC: Labeled amount (mg) of furosemide (C12H11ClN2O5S)
in 1 tablet
Assay Weigh accurately the mass of not less than 20Furosemide Tablets, and powder. Weigh accurately a por-tion of the powder, equivalent to about 40 mg of furosemide(C12H11ClN2O5S), add about 70 mL of 0.05 mol/L sodiumhydroxide TS, shake well, and add 0.05 mol/L sodiumhydroxide TS to make exactly 100 mL. Filter, discard theˆrst 10 mL of the ˆltrate, pipet 2 mL of the subsequent

17161716 Supplement II, JPXIVO‹cial Monographs for Part I
ˆltrate, add 0.05 mol/L sodium hydroxide TS to make ex-actly 100 mL, and use this solution as the sample solution.Separately, weigh accurately about 20 mg of FurosemideReference Standard, previously dried at 1059C for 4 hours,and dissolve in 0.05 mol/L sodium hydroxide TS to makeexactly 50 mL. Pipet 2 mL of this solution, add 0.05 mol/Lsodium hydroxide TS to make exactly 100 mL, and use thissolution as the standard solution. Determine the absor-bances, AT and AS, of the sample solution and the standardsolution at 271 nm as directed under the Ultraviolet-visibleSpectrophotometry.
Amount (mg) of furosemide (C12H11ClN2O5S)=WS×AT
AS
WS: Amount (mg) of Furosemide Reference Standard
Containers and storage Containers—Tight containers.Storage—Light-resistant.
Add the following:
Glutathioneグルタチオン
C10H17N3O6S: 307.32(2S)-2-Amino-4-[1-(carboxymethyl)carbamoyl-(2R)-2-sulfanylethylcarbamoyl]butanoic acid [70-18-8]
Glutathione, when dried, contains not less than98.0z and not more than 101.0z of C10H17N3O6S.
Description Glutathione occurs as a white crystallinepowder.
It is freely soluble in water, and practically insoluble inethanol (99.5).
Melting point: about 1859C (with decomposition).
Identiˆcation Determine the infrared absorption spectrumof Glutathione, previously dried, as directed in the potassi-um bromide disk method under the Infrared Spectrophoto-metry, and compare the spectrum with the Reference Spec-trum: both spectra exhibit similar intensities of absorption atthe same wave numbers.
Optical rotation [a]D20: -15.5 –-17.59(after drying, 2 g,water, 50 mL, 100 mm).
Purity (1) Clarity and color of solution—Dissolve 1.0 gof Glutathione in 10 mL of water: the solution is clear andcolorless.
(2) Heavy metals—Proceed with 2.0 g of Glutathioneaccording to Method 2, and perform the test. Prepare thecontrol solution with 2.0 mL of Standard Lead Solution (notmore than 10 ppm).
(3) Arsenic—Prepare the test solution with 1.0 g of
Glutathione according to Method 1, and perform the test(not more than 2 ppm).
(4) Related substances—Dissolve 50 mg of Glutathionein 100 mL of the mobile phase, and use this solution as thesample solution. Pipet 2 mL of the sample solution, add themobile phase to make exactly 100 mL, and use this solutionas the standard solution. Perform the test with exactly 10 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine each peak area by theautomatic integration method: the area of the peak havingthe relative retention time of about 4 with respect toglutathione is not more than 3/4 times the peak area ofglutathione from the standard solution, and the total area ofthe peaks other than the peak of glutathione is not morethan the peak area of glutathione from the standard solu-tion.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 210 nm).
Column: A stainless steel column 4.6 mm in inside di-ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about309C.
Mobile phase: Dissolve 6.8 g of potassium dihydrogenphosphate and 2.02 g of sodium 1-heptane sulfonate in 1000mL of water, and adjust the pH to 3.0 with phosphoric acid.To 970 mL of this solution add 30 mL of methanol.
Flow rate: Adjust the ‰ow rate so that the retention timeof glutathione is about 5 minutes.
Time span of measurement: About 6 times as long as theretention time of glutathione after the solvent peak.System suitability—
Test for required detectability: Pipet 10 mL of the stan-dard solution, and add the mobile phase to make exactly 100mL. Conˆrm that the peak area of glutathione obtainedfrom 10 mL of this solution is equivalent to 8 to 12z of thatobtained from 10 mL of the standard solution.
System performance: Dissolve 50 mg of glutathione,10 mg of D-phenylglycine and 50 mg of ascorbic acid in 100mL of water. When the procedure is run with 10 mL of thissolution under the above operating conditions, ascorbicacid, glutathione and D-phenylglycine are eluted in thisorder, and the resolutions between the peaks of ascorbic acidand glutathione and between the peaks of glutathione andD-phenylglycine are not less than 5, respectively.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of glutathione is not more than 1.5z.
Loss on drying Not more than 0.5z (1 g, 1059C, 3 hours).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 0.5 g of Glutathione,previously dried, dissolve in 50 mL of a solution of

17171717Supplement II, JPXIV O‹cial Monographs for Part I
metaphosphoric acid (1 in 50), and titrate with 0.05 mol/Liodine VS (indicator: 1 mL of starch TS). Perform a blankdetermination in the same manner, and make any necessarycorrection.
Each mL of 0.05 mol/L iodine VS=30.73 mg of C10H17N3O6S
Containers and storage Containers—Tight containers.
Heparin Sodiumヘパリンナトリウム
Change the Assay to read:
Assay (i) Substrate solution: Dissolve 15 mg of N-ben-zoyl-L-isoleucyl-L-glutamyl(g-OR)-glycyl-L-arginyl-p-nitroanilide hydrochloride in 20 mL of water.
(ii) Antithrombin III solution: Dissolve human an-tithrombin III in water to make a solution containing 1 Unitper mL.
(iii) Activated blood coagulation factor X solution: Dis-solve bovine activated blood coagulation factor X in waterto make a solution containing 0.426 Units per mL.
(iv) Human normal plasma: Dissolve an amount ofdried human normal plasma powder, equivalent to 1 mL ofhuman normal plasma, in 1 mL of water. Store at 2 – 109Cand use within a week.
(v) BuŠer solution: Dissolve 6.06 g of 2-amino-2-hydroxymethyl-1,3-propanediol in 750 mL of water, adjustthe pH to 8.4 with 1 mol/L hydrochloric acid TS, and addwater to make 1000 mL.
(vi) Reaction stop solution: To 20 mL of acetic acid(100) add water to make 40 mL.
(vii) Heparin standard solution: Dissolve Heparin Sodi-um Reference Standard in isotonic sodium chloride solutionto make a solution containing 10 Units per mL, and use asthe standard stock solution. To 100 mL of the standard stocksolution add the buŠer solution to make exactly 5 mL, anduse this solution as the standard solution. Prepare theheparin standard solutions (1), (2), (3), (4) and (5) byaddition of antithrombin III solution, human normalplasma and the buŠer solution to the standard solution asdirected in the following table.
Heparin standardsolution BuŠer
solution(mL)
AntithrombinIII solution
(mL)
Humannormalplasma(mL)
Standardsolution
(mL)No.Heparin
concentration(Unit/mL)
(1) 0 800 100 100 0
(2) 0.02 700 100 100 100
(3) 0.04 600 100 100 200
(4) 0.06 500 100 100 300
(5) 0.08 400 100 100 400
(viii) Sample solution: Weigh accurately an adequateamount of Heparin Sodium, dissolve in isotonic sodiumchloride solution so that each mL contains about 0.5 Unitsaccording to the labeled amount. To 100 mL of this solutionadd 100 mL of antithrombin III solution, 100 mL of humannormal plasma and 700 mL of the buŠer solution, and usethis solution as the sample solution.
(ix) Procedure: Transfer 400 mL of the sample solutionto a test tube, and warm at 379C for 4 minutes. Add 200 mLof the activated blood coagulation factor X solution, mixwell, warm at 379C for exactly 30 seconds, add 400 mL of thesubstrate solution, previously warmed at 379C, and mixwell. Allow the tube to stand at 379C for exactly 3 minutes,add 600 mL of the reaction stop solution, mix immediately,and determine the absorbance at 405 nm, using the blanksolution prepared by addition of 600 mL of the reaction stopsolution and 600 mL of water to 400 mL of the samplesolution. Proceed the same way with the heparin standardsolution (1), the heparin standard solution (2), the heparinstandard solution (3), the heparin standard solution (4) andthe heparin standard solution (5), and determine their absor-bances.
(x) Calculation: Plot the absorbances of the standard so-lutions on the vertical axis and their heparin concentrationson the horizontal axis to prepare a calibration curve. Deter-mine the heparin concentration, C, of the sample solutionfrom its absorbance by using the curve, and calculateheparin Units per mg of Heparin Sodium from the followingformula.
Units per mg of Heparin Sodium=C×10×ba
a: Amount of sample (mg)b: Total volume (mL) of isotonic sodium chloride solution
used to dissolve the sample to make the solutioncontaining about 0.5 Units per mL

17181718 Supplement II, JPXIVO‹cial Monographs for Part I
Heparin Sodium Injectionヘパリンナトリウム注射液
Add the following next to Bacterial endotoxins:
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Change the Assay to read:
Assay Proceed as directed in the Assay under HeparinSodium, replacing the sample solution indicated in (viii) andthe calculation in (x) with the following.
Sample solution: Measure exactly an adequate portion ofHeparin Sodium Injection according to the labeled Units,dilute it with isotonic sodium chloride solution so that eachmL contains about 0.5 Units. To 100 mL of this solution add100 mL of antithrombin III solution, 100 mL of human nor-mal plasma and 700 mL of the buŠer solution, and use thissolution as the sample solution.
Calculation: Plot the absorbances of the standard solu-tions on the vertical axis and their heparin concentrations onthe horizontal axis to prepare a calibration curve. Determinethe heparin concentration, C, of the sample solution from itsabsorbance by using the curve, and calculate heparin Unitsper mL of Heparin Sodium Injection from the followingformula.
Units per mL of Heparin Sodium Injection=C×10×ba
a: Amount of sample (mL)b: Total volume (mL) of isotonic sodium chloride solution
used to dilute the sample to make the solution contain-ing about 0.5 Units per mL
Kallidinogenaseカリジノゲナーゼ
Change the Assay to read:
Assay Weigh accurately an appropriate amount of Kal-lidinogenase according to the labeled Units, dissolve in 0.05mol/L phosphate buŠer solution, pH 7.0 to prepare a solu-tion containing about 10 Kallidinogenase Units per mL, anduse this solution as the sample stock solution. Pipet 4 mL ofthe sample stock solution, add exactly 1 mL of trypsin inhi-
bitor TS and 0.05 mol/L phosphate buŠer solution, pH 7.0to make exactly 10 mL, and use this solution as the samplesolution. Pipet 2.5 mL of substrate TS for kallidinogenaseassay (1), previously warmed at 30±0.59C for 5 minutes,place in a 1-cm cell, add exactly 0.5 mL of the sample solu-tion, warmed at 30±0.59C for 5 minutes, and start simul-taneously a chronograph. Perform the test at 30±0.59C asdirected under the Ultraviolet-visible Spectrophotometryusing water as the blank, and determine the absorbances at405 nm, AT2 and AT6, of this solution after allowing to standfor exactly 2 and 6 minutes. Separately, dissolve Kal-lidinogenase Reference Standard in 0.05 mol/L phosphatebuŠer solution, pH 7.0 to make a solutin so that each mLcontains exactly 10 Units, and use this solution as the stan-dard stock solution. Pipet 4 mL of the stock solution, addexactly 1 mL of trypsin inhibitor TS and 0.05 mol/L phos-phate buŠer solution, pH 7.0 to make exactly 10 mL, anduse this solution as the standard solution. Take exactly 0.5mL of the standard solution, perform the test in the samemanner as described for the sample solution, and determinethe absorbances, AS2 and AS6, of the solution after allowingto stand for exactly 2 and 6 minutes. Separately, take exactly1 mL of the trypsin inhibitor TS, and add 0.05 mol/L phos-phate buŠer solution, pH 7.0 to make exactly 10 mL. Pipet0.5 mL of this solution, perform the test in the same manneras described for the sample solution, and determine the ab-sorbances, AO2 and AO6, of the solution after allowing tostand for exactly 2 and 6 minutes.
Units per 1 mg of Kallidinogenase
=(AT6-AT2)-(AO6-AO2)(AS6-AS2)-(AO6-AO2)
×WS
a×
1b
WS: Amount (Units) of Kallidinogenase Reference Standarda: Volume (mL) of the standard stock solutionb: Amount (mg) of Kallidinogenase in 1 mL of the sample
stock solution
D-MannitolD-マンニトール
Change the Identiˆcation (2) to read:
Identiˆcation(2) Determine the infrared absorption spectrum of D-
Mannitol as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wave numbers.If any diŠerence appears between the spectra, dissolve 1 g ofD-Mannitol in 3 mL of warm water, then allow to stand at59C for 24 hours or until crystals appear, and ˆlter. Washthe crystals so obtained with a few amount of cold water,dry at 1059C for 4 hours, and perform the test with thecrystals.

17191719Supplement II, JPXIV O‹cial Monographs for Part I
Mepivacaine Hydrochloride塩酸メピバカイン
Change the graphic formula to read:
Change the chemical name to read:
(2RS)-N-(2,6-Dimethylphenyl)-1-methylpiperidine-2-carboxamide monohydrochloride
Meropenem Trihydrateメロペネム 三水和物
Change the water to read:
Water Not less than 11.4z and not more than 13.4z
(0.35 g, volumetric titration, direct titration).
dl-Methylephedrine Hydrochloridedl-塩酸メチルエフェドリン
Change the origin/limits of the content to read:
dl-Methylephedrine Hydrochloride, when dried,contains not less than 99.0z and not more than101.0z of C11H17NO.HCl.
Change the Description to read:
Description dl-Methylephedrine Hydrochloride occurs ascolorless crystals or a white, crystalline powder.
It is freely soluble in water, sparingly soluble in ethanol(99.5), slightly soluble in acetic acid (100), and practicallyinsoluble in acetic anhydride.
A solution of dl-Methylephedrine Hydrochloride (1 in 20)shows no optical rotation.
Change the Identiˆcation to read:
Identiˆcation (1) Determine the absorption spectrum ofa solution of dl-Methylephedrine Hydrochloride (1 in 2000)as directed under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wavelengths.
(2) Determine the infrared absorption spectrum of dl-Methylephedrine Hydrochloride, previously dried, as direct-ed in the potassium chloride disk method under the InfraredSpectrophotometry, and compare the spectrum with the
Reference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wave numbers.
(3) A solution of dl-Methylephedrine Hydrochloride(1 in 10) responds to the Qualitative Tests for chloride.
Add the following next to Identiˆcation:
pH The pH of a solution prepared by dissolving 1.0 g ofdl-Methylephedrine Hydrochloride in 20 mL of water is be-tween 4.5 and 6.0.
Change the Purity to read:
Purity (1) Clarity and color of solution—Dissolve 1.0 gof dl-Methylephedrine Hydrochloride in 10 mL of water: thesolution is clear and colorless.
(2) Heavy metals—Proceed with 1.0 g of dl-Methylephe-drine Hydrochloride according to Method 4, and performthe test. Prepare the control solution with 1.0 mL of Stan-dard Lead Solution (not more than 10 ppm).
(3) Related substances—Dissolve 50 mg of dl-Methylephedrine Hydrochloride in 20 mL of water, and usethis solution as the sample solution. Pipet 1 mL of thesample solution, add water to make exactly 100 mL, and usethis solution as the standard solution. Perform the test withexactly 20 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and determine eachpeak area by the automatic integration method: the totalarea of the peaks other than the peak of methylephedrine isnot more than the peak area of methylephedrine from thestandard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 257 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 13.6 g of potassium dihydrogenphosphate and 3 g of sodium 1-heptane sulfonate in 1000mL of water, and adjust the pH to 2.5 with phosphoric acid.To 900 mL of this solution add 200 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof methylephedrine is about 10 minutes.
Time span of measurement: About 2 times as long as theretention time of methylephedrine after the solvent peak.System suitability—
Test for required detectability: To exactly 2 mL of thestandard solution add water to make exactly 20 mL. Con-ˆrm that the peak area of methylephedrine obtained from 20mL of this solution is equivalent to 7 to 13z of that ofmethylephedrine obtained from 20 mL of the standard solu-tion.
System performance: Dissolve 50 mg of dl-Methylephe-drine Hydrochloride and 0.4 mg of methyl parahydroxyben-zoate in 50 mL of water. When the procedure is run with

17201720 Supplement II, JPXIVO‹cial Monographs for Part I
20 mL of this solution under the above operating conditions,methylephedrine and methyl parahydroxybenzoate areeluted in this order with the resolution between these peaksbeing not less than 3.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of methylephedrine is not more than 2.0z.
10z dl-MethylephedrineHydrochloride Powderdl-塩酸メチルエフェドリン散10z
Delete the Description:
Change the Identiˆcation to read:
Identiˆcation Determine the absorption spectrum of asolution of 10z dl-Methylephedrine Hydrochloride Powder(1 in 200) as directed under the Ultraviolet-visible Spec-trophotometry: it exhibits maxima between 250 nm and253 nm, between 255 nm and 259 nm, and between 261 nmand 264 nm.
Change the Assay to read:
Assay Weigh accurately about 0.5 g of 10z dl-Methylephedrine Hydrochloride Powder, add exactly 4 mLof the internal standard solution and 25 mL of water, shakevigorously for 20 minutes to dissolve, add water to make50 mL, ˆlter through a membrane ˆlter with pore size of0.45 mm, if necessary, discard the ˆrst 10 mL of the ˆltrate,and use the subsequent ˆltrate as the sample solution.Separately, weigh accurately about 50 mg of dl-methylephe-drine hydrochloride for assay, previously dried at 1059C for3 hours, add exactly 4 mL of the internal standard solutionand water to make 50 mL, and use this solution as thestandard solution. Perform the test with 20 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and determine the ratios of the peak area, QT
and QS, of methylephedrine to that of the internal standard.
Amount (mg) of dl-methylephedrine hydrochloride(C11H17NO.HCl)
=WS×QT
QS
WS: Amount (mg) of dl-methylephedrine hydrochloridefor assay
Internal standard solution—A solution of methyl para-hydroxybenzoate in acetonitrile (1 in 10,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 257 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-
silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 13.6 g of potassium dihydrogenphosphate and 3 g of sodium 1-heptane sulfonate in 1000mL of water, and adjust the pH to 2.5 with phosphoric acid.To 900 mL of this solution add 200 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof methylephedrine is about 10 minutes.System suitability—
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, methylephedrine and the internal standard areeluted in this order with the resolution between these peaksbeing not less than 3.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of methylephedrine to that of the internalstandard is not more than 1.0z.
Add the following:
Methylprednisolone Succinateコハク酸メチルプレドニゾロン
C26H34O8: 474.5411b,17,21-Trihydroxy-6a-methylpregna-1,4-diene-3,20-dione 21-(hydrogen succinate) [2921-57-5]
Methylprednisolone Succinate, when dried, containsnot less than 97.0z and not more than 103.0z ofC26H34O8.
Description Methylprednisolone Succinate occurs as awhite, crystals or crystalline powder.
It is soluble in methanol, sparingly soluble in ethanol (95),and practically insoluble in water.
Melting point: about 2359C (with decomposition).
Identiˆcation (1) Determine the absorption spectrum ofa solution of Methylprednisolone Succinate in methanol(1 in 50,000) as directed under the Ultraviolet-visibleSpectrophotometry, and compare the spectrum with theReference Spectrum or the spectrum of a solution of Methyl-prednisolone Succinate Reference Standard prepared in thesame manner as the sample solution: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum of

17211721Supplement II, JPXIV O‹cial Monographs for Part I
Methylprednisolone Succinate, previously dried, as directedin the potassium bromide disk method under the InfraredSpectrophotometry, and compare the spectrum with theReference Spectrum or the spectrum of previously driedMethylprednisolone Succinate Reference Standard: bothspectra exhibit similar intensities of absorption at the samewave numbers. In case when some diŠerences are found be-tween the spectra, repeat the test with residues obtained bydissolving these substances in ethanol (95), evaporating todryness, and drying.
Optical rotation [a]D25: +99 –+1039(after drying, 0.2 g,ethanol (95), 20 mL, 100 mm).
Purity (1) Heavy metals—Proceed with 1.0 g of Methyl-prednisolone Succinate according to Method 4, and performthe test. Prepare the control solution with 1.0 mL ofStandard Lead Solution (not more than 10 ppm).
(2) Arsenic—Prepare the test solution with 2.0 g ofMethylprednisolone Succinate according to Method 3, andperform the test (not more than 1 ppm).
(3) Related substances—Dissolve 15 mg of Methylpred-nisolone Succinate in 5 mL of methanol, add a mixture of0.05 mol/L phosphate buŠer solution, pH 3.5 and acetoni-trile (1:1) to make 50 mL, and use this solution as the samplesolution. Pipet 1 mL of the sample solution, add the mixtureof 0.05 mol/L phosphate buŠer solution, pH 3.5 andacetonitrile (1:1) to make exactly 100 mL, and use this solu-tion as the standard solution. Perform the test with exactly 5mL each of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine each peak area by theautomatic integration method: the area of the peaks otherthan the peak of methylprednisolone succinate is not morethan 1/2 of the peak area of methylprednisolone succinatefrom the standard solution, and the total area of the peaksother than the peak of methylprednisolone succinate is notmore than the peak area of methylprednisolone succinatefrom the standard solution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 3 times as long as theretention time of methylprednisolone succinate.System suitability—
Test for required detectability: Pipet 1 mL of the standardsolution, and add the mixture of 0.05 mol/L phosphatebuŠer solution, pH 3.5 and acetonitrile (1:1) to make exactly10 mL. Conˆrm that the peak area of methylprednisolonesuccinate obtained from 5 mL of this solution is equivalent to7 to 13z of that obtained from 5 mL of the standard solu-tion.
System performance: Proceed as directed in the Systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of methylprednisolone succinate is not more than
1.0z.
Loss on drying Not more than 1.0z (1 g, 1059C, 3 hours).
Residue on ignition Not more than 0.2z (0.5 g).
Assay Weigh accurately about 15 mg each of Methylpred-nisolone Succinate and Methylprednisolone SuccinateReference Standard, previously dried, dissolve separately in5 mL of methanol, and add the mixture of 0.05 mol/L phos-phate buŠer solution, pH 3.5 and acetonitrile (1:1) to makeexactly 50 mL. Pipet 5 mL each of these solutions, addexactly 5 mL of the internal standard solution, and use thesesolutions as the sample solution and the standard solution,respectively. Perform the test with 5 mL each of the samplesolution and the standard solution as directed under theLiquid Chromatography according to the following condi-tions, and determine the ratios, QT and QS, of the peak areaof methylprednisolone succinate to that of the internalstandard.
Amount (mg) of C26H34O8=WS×QT
QS
WS: Amount (mg) of Methylprednisolone SuccinateReference Standard
Internal standard solution—A solution of ethyl parahydrox-ybenzoate in a mixture of 0.05 mol/L phosphate buŠer solu-tion, pH 3.5 and acetonitrile (1:1) (3 in 20,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: To 1000 mL of 0.05 mol/L potassium di-hydrogen phosphate TS add a suitable amount of 0.05mol/L disodium hydrogen phosphate TS to make a solutionhaving pH 5.5. To 640 mL of this solution add 360 mL ofacetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof methylprednisolone succinate is about 6 minutes.System suitability—
System performance: When the procedure is run with 5 mLof the standard solution under the above operating condi-tions, methylprednisolone succinate and the internal stan-dard are eluted in this order with the resolution betweenthese peaks being not less than 6.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of methylprednisolone succinate to that of theinternal standard is not more than 1.0z.
Containers and storage Containers—Tight containers.

17221722 Supplement II, JPXIVO‹cial Monographs for Part I
Methyltestosteroneメチルテストステロン
Change the origin/limits of content to read:
Methyltestosterone, when dried, contains not lessthan 98.0z and not more than 102.0z of C20H30O2.
Change the Description to read:
Description Methyltestosterone occurs as white to paleyellow, crystals or crystalline powder.
It is freely soluble in methanol and in ethanol (95), andpractically insoluble in water.
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Methyltestosterone in ethanol (95) (1 in 100,000) as directedunder the Ultraviolet-visible Spectrophotometry, and com-pare the spectrum with the Reference Spectrum or the spec-trum of a solution of Methyltestosterone Reference Stan-dard prepared in the same manner as the sample solution:both spectra exhibit similar intensities of absorption at thesame wavelengths.
(2) Determine the infrared absorption spectrum ofMethyltestosterone, previously dried, as directed in thepotassium bromide disk method under the Infrared Spec-trophotometry, and compare the spectrum with the Refer-ence Spectrum or the spectrum of dried MethyltestosteroneReference Standard: both spectra exhibit similar intensitiesof absorption at the same wave numbers.
Change the Assay to read:
Assay Weigh accurately about 20 mg each ofMethyltestosterone and Methyltestosterone ReferenceStandard, previously dried in a desiccator (in vacuum, phos-phorus (V) oxide) for 10 hours, dissolve each in methanol tomake exactly 200 mL. Pipet 5 mL each of these solutions,add exactly 5 mL of the internal standard solution, addmethanol to make 50 mL, and use these solutions as thesample solution and the standard solution. Perform the testwith 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of methyltestosterone tothat of the internal standard.
Amount (mg) of C20H30O2
=WS×QT
QS
WS: Amount (mg) of Methyltestosterone ReferenceStandard
Internal standard solution—A solution of propyl para-hydroxybenzoate in methanol (1 in 10,000).
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 241 nm).Column: A stainless steel column 6 mm in inside diameter
and 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about359C.
Mobile phase: A mixture of acetonitrile and water (11:9).Flow rate: Adjust the ‰ow rate so that the retention time
of methyltestosterone is about 10 minutes.System suitability—
System performance: When the procedure is run with 10mL of the standard solution under the above operatingconditions, the internal standard and methyltestosterone areeluted in this order with the resolution between these peaksbeing not less than 9.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of theratios of the peak area of methyltestosterone to that of theinternal standard is not more than 1.0z.
Methyltestosterone Tabletsメチルテストステロン錠
Change the origin/limits of content to read:
Methyltestosterone Tablets contain not less than90.0z and not more than 110.0z of the labeledamount of methyltestosterone (C20H30O2: 302.45).
Change the Identiˆcation to read:
Identiˆcation To a portion of powdered Methyltestoster-one Tablets, equivalent to 0.01 g of Methyltestosteroneaccording to the labeled amount, add 50 mL of acetone,shake for 30 minutes, and ˆlter. Evaporate the ˆltrate todryness, dissolve the residue in 10 mL of acetone, and usethis solution as the sample solution. Separately, dissolve0.01 g of Methyltestosterone Reference Standard in 10 mLof acetone, and use this solution as the standard solution.Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 10 mL each of the samplesolution and the standard solution on a plate of silica gel forthin-layer chromatography. Develop the plate with a mix-ture of chloroform and ethanol (95) (9:1) to a distance ofabout 12 cm, and air-dry the plate. Spray evenly dilute sul-furic acid on the plate, and heat at 1109C for 10 minutes: thespot from the sample solution and the standard solutionshow the same Rf value.
Change the Assay to read:
Assay Weigh accurately the mass of not less than 20Methyltestosterone Tablets, and powder. Weigh accurately aportion of the powder, equivalent to about 25 mg of

17231723Supplement II, JPXIV O‹cial Monographs for Part I
methyltestosterone (C20H30O2), add about 70 mL ofmethanol, shake for 30 minutes, and add methanol to makeexactly 100 mL. Pipet 2 mL of this solution, add exactly 5mL of the internal standard solution and methanol to make50 mL, ˆlter through a membrane ˆlter (not exceeding 0.45mm in pore size), and use the ˆltrate as the sample solution.Separately, weigh accurately about 20 mg ofMethyltestosterone Reference Standard, previously dried ina desiccator (in vacuum, phosphorus (V) oxide) for 10hours, dissolve in methanol to make exactly 200 mL. Pipet 5mL of this solution, add exactly 5 mL of the internal stan-dard solution, add methanol to make 50 mL, and use this so-lution as the standard solution. Perform the test with 10 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine the ratios, QT and QS,of the peak area of methyltestosterone to that of the internalstandard.
Amount (mg) of methyltestosterone (C20H30O2)
=WS×QT
QS×
52
WS: Amount (mg) of Methyltestosterone ReferenceStandard
Internal standard solution—A solution of propyl para-hydroxybenzoate in methanol (1 in 10,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 241 nm).
Column: A stainless steel column 6 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about359C.
Mobile phase: A mixture of acetonitrile and water (11:9).Flow rate: Adjust the ‰ow rate so that the retention time
of methyltestosterone is about 10 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, the internal standard and methyltestosterone areeluted in this order with the resolution between these peaksbeing not less than 9.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of methyltestosterone to that of the internalstandard is not more than 1.0z.
Add the following:
Metoclopramide Tabletsメトクロプラミド錠
Metoclopramide Tablets contain not less than95.0z and not more than 105.0z of the labeledamount of metoclopramide (C14H22ClN3O2: 299.80).
Method of preparation Prepare as directed under Tablets,with Metoclopramide.
Identiˆcation (1) To a quantity of powderedMetoclopramide Tablets, equivalent to 50 mg ofMetoclopramide according to the labeled amount, add 15mL of 0.5 mol/L hydrochloric acid TS, and heat in a waterbath at 709C for 15 minutes while frequent shaking. Aftercooling, centrifuge for 10 minutes, and to 5 mL of the super-natant liquid add 1 mL of 4-dimethylaminobenzaldehyde-hydrochloric acid TS: a yellow color develops.
(2) Determine the absorption spectrum of the samplesolution obtained in the Assay as directed under theUltraviolet-visible Spectrophotometry: it exhibits maximabetween 270 nm and 274 nm, and between 306 nm and310 nm.
Content uniformity Perform the test according to thefollowing method: it meets the requirements of the ContentUniformity Test.
To 1 tablet of Metoclopramide Tablets add 10 mL of 0.1mol/L hydrochloric acid TS, disperse the particles with theaid of ultrasonic waves, then add 0.1 mol/L hydrochloricacid TS to make exactly 25 mL, and centrifuge for 10minutes. Pipet 4 mL of the supernatant liquid, add0.1 mol/L hydrochloric acid TS to make exactly V mL of asolution so that each mL contains about 12 mg ofmetoclopramide (C14H22ClN3O2), and use this solution as thesample solution. Separately, weigh accurately about 80 mgof metoclopramide for assay, previously dried at 1059C for3 hours, and dissolve in 0.1 mol/L hydrochloric acid TS tomake exactly 500 mL. Pipet 4 mL of this solution, add 0.1mol/L hydrochloric acid TS to make exactly 50 mL, and usethis solution as the standard solution. Determine the absor-bances, AT and AS, of the sample solution and the standardsolution at 308 nm as directed under the Ultraviolet-visibleSpectrophotometry.
Amount (mg) of metoclopramide (C14H22ClN3O2)
=WS×AT
AS×
V1000
WS: Amount (mg) of metoclopramide for assay
Dissolution Perform the test with 1 tablet ofMetoclopramide Tablets at 50 revolutions per minute ac-cording to Method 2 under the Dissolution Test, using 900mL of diluted phosphate buŠer solution, pH 6.8 (1 in 2) asthe dissolution medium. Withdraw 20 mL or more of thedissolution medium 45 minutes after starting the test for a3.84-mg tablet or 15 minutes after starting the test for a

17241724 Supplement II, JPXIVO‹cial Monographs for Part I
7.67-mg tablet, and ˆlter through a membrane ˆlter withpore size of not more than 0.5 mm. Discard the ˆrst 10 mL ofthe ˆltrate, pipet V mL of the subsequent ˆltrate, add dilut-ed phosphate buŠer solution, pH 6.8 (1 in 2) to make exactlyV? mL so that each mL contains about 4 mg ofmetoclopramide (C14H22ClN3O2) according to the labeledamount, and use this solution as the sample solution.Separately, weigh accurately about 20 mg ofmetoclopramide for assay, previously dried at 1059C for 3hours, and dissolve in diluted phosphate buŠer solution, pH6.8 (1 in 2) to make exactly 100 mL. Pipet 2 mL of this solu-tion, add diluted phosphate buŠer solution, pH 6.8 (1 in 2)to make exactly 100 mL, and use this solution as the stan-dard solution. Perform the test with exactly 50 mL each ofthe sample solution and the standard solution as directed un-der the Liquid Chromatography according to the followingconditions, and determine the peak areas, AT and AS, ofmetoclopramide. The dissolution rates for a 3.84-mg tabletin 45 minutes and for a 7.67-mg tablet in 15 minutes are notless than 80z and not less than 85z, respectively.
Dissolution rate (z) with respect to the labeled amount ofmetoclopramide (C14H22ClN3O2)
=WS×AT
AS×
V?V×
1C×18
WS: Amount (mg) of metoclopramide for assayC: Labeled amount (mg) of metoclopramide
(C14H22ClN3O2) in 1 tablet
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 275 nm).Column: A stainless steel column 4 mm in inside diameter
and 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 0.79 g of sodium lauryl sulfate in550 mL of water, and add 450 mL of acetonitrile and 0.3 mLof acetic acid (100).
Flow rate: Adjust the ‰ow rate so that the retention timeof metoclopramide is about 5 minutes.System suitability—
System performance: When the procedure is run with50 mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of metoclopramide are not less than3000 and not more than 1.5, respectively.
System repeatability: When the test is repeated 6 timeswith 50 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of metoclopramide is not more than 1.0z.
Assay Weigh accurately not less than 20 MetoclopramideTablets, and powder. Weigh accurately a portion of thepowder, equivalent to about 75 mg of metoclopramide(C14H22ClN3O2), add 300 mL of 0.1 mol/L hydrochloric acidTS, shake for 1 hour, and add 0.1 mol/L hydrochloric acidTS to make exactly 500 mL. Centrifuge for 10 minutes, pipet
4 mL of the supernatant liquid, add 0.1 mol/hydrochloricacid TS to make exactly 50 mL, and use this solution as thesample solution. Separately, weigh accurately about 80 mgof metoclopramide for assay, previously dried at 1059C for3 hours, and dissolve in 0.1 mol/L hydrochloric acid TS tomake exactly 500 mL. Pipet 4 mL of this solution, add 0.1mol/hydrochloric acid TS to make exactly 50 mL, and usethis solution as the standard solution. Determine the absor-bances, AT and AS, of the sample solution and the standardsolution at 308 nm as directed under the Ultraviolet-visibleSpectrophotometry.
Amount (mg) of metoclopramide (C14H22ClN3O2)
=WS×AT
AS
WS: Amount (mg) of metoclopramide for assay
Containers and storage Containers—Tight containers.
Add the following:
Nicorandilニコランジル
C8H9N3O4: 211.17N-[2-(Nitrooxy)ethyl]pyridine-3-carboxamide [65141-46-0]
Nicorandil contains not less than 98.5z and notmore than 101.0z of C8H9N3O4, calculated on theanhydrous basis.
Description Nicorandil occurs as white crystals.It is freely soluble in methanol, in ethanol (99.5) and in
acetic acid (100), soluble in acetic anhydride, and sparinglysoluble in water.
Melting point: about 929C (with decomposition).
Identiˆcation Determine the infrared absorption spectrumof Nicorandil as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry, and com-pare the spectrum with the Reference Spectrum: both spec-tra exhibit similar intensities of absorption at the same wavenumbers.
Purity (1) Sulfate—Dissolve 2.0 g of Nicorandil in 20 mLof dilute ethanol, add 1 mL of dilute hydrochloric acid andwater to make 50 mL, and perform the test using this solu-tion as the test solution. Prepare the control solution with0.40 mL of 0.005 mol/L sulfuric acid VS, 20 mL of diluteethanol and 1 mL of dilute hydrochloric acid, and dilutewith water to make 50 mL (not more than 0.010z).
(2) Heavy metals—Proceed with 2.0 g of Nicorandilaccording to Method 2, and perform the test. Prepare thecontrol solution with 2.0 mL of Standard Lead Solution (notmore than 10 ppm).

17251725Supplement II, JPXIV O‹cial Monographs for Part I
(3) Related substances—Dissolve 20 mg of Nicorandil in10 mL of the mobile phase, and use this solution as thesample solution. Perform the test with 10 mL of the samplesolution as directed under the Liquid Chromatographyaccording to the following conditions, and determine eachpeak area by the automatic integration method: the peakarea of N-(2-hydroxyethyl)isonicotinamide nitric ester,having the relative retention time of about 0.86 with respectto nicorandil, is not more than 0.5z of the peak area ofnicorandil, the area of all other peaks is less than 0.1z, andthe sum area of the peaks other than nicorandil and N-(2-hydroxyethyl)isonicotinamide nitric ester is not more than0.25z of the total peak area.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4 mm in inside diameterand 25 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of water, tetrahydrofuran,triethylamine and tri‰uoroacetic acid (982:10:5:3).
Flow rate: Adjust the ‰ow rate so that the retention timeof nicorandil is about 18 minutes.
Time span of measurement: About 3 times as long as theretention time of nicorandil after the solvent peak.System suitability—
Test for required detectability: Measure exactly 1 mL ofthe sample solution, add the mobile phase to make exactly500 mL, and use this solution as the solution for systemsuitability test. Pipet 1 mL of the solution for systemsuitability test, and add the mobile phase to make exactly20 mL. Conˆrm that the peak area of nicorandil obtainedwith 10 mL of this solution is equivalent to 2 to 8z of thatwith 10 mL of the solution for system suitability test.
System performance: Dissolve 10 mg of N-(2-hydrox-yethyl)isonicotinamide nitric ester in the mobile phase tomake 100 mL. To 1 mL of this solution add 10 mL of thesample solution. When the procedure is run with this solu-tion under the above operating conditions, N-(2-hydrox-yethyl)isonicotinamide nitric ester and nicorandil are elutedin this order with the resolution between these peaks beingnot less than 3.0.
System repeatability: When the test is repeated 6 timeswith 10 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of nicorandil is not more than 1.5z.
Water Not more than 0.1z (2 g, volumetric titration,direct titration).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 0.3 g of Nicorandil, dis-solve in 30 mL of a mixture of acetic anhydride and aceticacid (100) (7:3), and titrate with 0.1 mol/L perchloric acidVS (potentiometric titration). Perform a blank determina-tion in the same manner, and make any necessary correc-
tion.
Each mL of 0.1 mol/L perchloric acid VS=21.12 mg of C8H9N3O4
Containers and storage Containers—Tight containers.Storage—At a temperature between 29C and 89C.
Add the following:
Nilvadipineニルバジピン
C19H19N3O6: 385.375-Isopropyl 3-methyl (4RS)-2-cyano-1,4-dihydro-6-methyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate[75530-68-6]
Nilvadipine contains not less than 98.0z and notmore than 102.0z of C19H19N3O6.
Description Nilvadipine occurs as a yellow crystallinepowder.
It is freely soluble in acetonitrile, soluble in methanol,sparingly soluble in ethanol (99.5), and practically insolublein water.
A solution of Nilvadipine in acetonitrile (1 in 20) shows nooptical rotation.
Identiˆcation (1) Determine the absorption spectrum ofa solution of Nilvadipine in ethanol (99.5) (1 in 100,000) asdirected under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum orthe spectrum of a solution of Nilvadipine ReferenceStandard prepared in the same manner as the sample solu-tion: both spectra exhibit similar intensities of absorption atthe same wavelengths.
(2) Determine the infrared absorption spectrum ofNilvadipine as directed in the potassium bromide diskmethod under the Infrared Spectrophotometry, and com-pare the spectrum with the Reference Spectrum or thespectrum of Nilvadipine Reference Standard: both spectraexhibit similar intensities of absorption at the same wavenumbers.
Melting point 167 – 1719C
Purity (1) Heavy metals—Proceed with 2.0 g of Nilvadi-pine according to Method 2, and perform the test. Preparethe control solution with 2.0 mL of Standard Lead Solution(not more than 10 ppm).
(2) Related substances—Dissolve 20 mg of Nilvadipinein 20 mL of acetonitrile, and use this solution as the sample

17261726 Supplement II, JPXIVO‹cial Monographs for Part I
solution. Perform the test with 5 mL of the sample solutionas directed under the Liquid Chromatography according tothe following conditions. Determine each peak area by theautomatic integration method, and calculate the amount ofthem by the area percentage method: the amount of eachrelated substance is not more than 0.3z, and the total ofthem is not more than 0.5z.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 240 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of phosphate buŠer solution,pH 7.4, methanol and acetonitrile (32:27:18).
Flow rate: Adjust the ‰ow rate so that the retention timeof nilvadipine is about 12 minutes.
Time span of measurement: About 2.5 times as long as theretention time of nilvadipine after the solvent peak.System suitability—
Test for required detectability: Pipet 1 mL of the samplesolution, add acetonitrile to make exactly 100 mL, and usethis solution as the solution for system suitability test. Pipet1 mL of the solution for system suitability test, and addacetonitrile to make exactly 10 mL. Conˆrm that the peakarea of nilvadipine obtained from 5 mL of this solution isequivalent to 7 to 13z of that obtained from 5 mL of thesolution for system suitability test.
System performance: When the procedure is run with 5 mLof the solution for system suitability test under the aboveoperating conditions, the number of theoretical plates andthe symmetry factor of the peak of nilvadipine is not lessthan 3300 and not more than 1.3, respectively.
System repeatability: Pipet 1 mL of the solution forsystem suitability test, and add acetonitrile to make exactly10 mL. When the test is repeated 6 times with 5 mL of thissolution under the above operating conditions, the relativestandard deviation of the peak area of nilvadipine is notmore than 1.5z.
Loss on drying Not more than 0.1z (1 g, 1059C, 2 hours).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 25 mg each of Nilvadipineand Nilvadipine Reference Standard, dissolve in methanol tomake exactly 25 mL. Pipet 10 mL each of these solutions,add exactly 20 mL of the internal standard solution, 20 mLof water and methanol to make 100 mL, and use these solu-tions as the sample solution and the standard solution, re-spectively. Perform the test with 5 mL each of the sample so-lution and the standard solution as directed under the LiquidChromatography according to the following conditions, anddetermine the ratios, QT and QS, of the peak area of nilvadi-pine to that of the internal standard.
Amount (mg) of C19H19N3O6=WS×QT
QS
WS: Amount (mg) of Nilvadipine Reference Standard
Internal standard solution—A solution of acenaphthene inmethanol (1 in 200).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 2.5 g of diammonium hydrogenphosphate in 1000 mL of water, add 10 mL of tetrabutylam-monium hydoxide TS, adjust the pH to 7.0 with dilutedphosphoric acid (1 in 10), and add 900 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof nilvadipine is about 12 minutes.System suitability—
System performance: When the procedure is run with 5 mLof the standard solution under the above operating condi-tions, nilvadipine and the internal standard are eluted in thisorder with the resolution between these peaks being not lessthan 8.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of nilvadipine to that of the internal standardis not more than 1.0z.
Containers and storage Containers—Well-closed contain-ers.
Add the following:
Nilvadipine Tabletsニルバジピン錠
Nilvadipine Tablets contain not less than 93.0z andnot more than 107.0z of the labeled amount ofnilvadipine (C19H19N3O6: 385.37).
Method of preparation Prepare as directed under Tablets,with Nilvadipine.
Identiˆcation To a quantity of powdered NilvadipineTablets, equivalent to 1 mg of Nilvadipine according to thelabeled amount, add 100 mL of ethanol (99.5), shake for 10minutes, centrifuge, and use the supernatant liquid as thesample solution. Determine the absorption spectrum of thesample solution as directed under the Ultraviolet-visibleSpectrophotometry: it exhibits a maximum between 239 nmand 243 nm and a maximum having a broad-ranging absorp-tion between 371 nm and 381 nm.

17271727Supplement II, JPXIV O‹cial Monographs for Part I
Content uniformity Perform the test according to the fol-lowing method: it meets the requirements of the ContentUniformity Test.
To 1 tablet of Nilvadipine Tablets add V mL of a mixtureof acetonitrile and water (7:3) so that each mL of the solu-tion contains about 200 mg of nilvadipine (C19H19N3O6)according to the labeled amount, add exactly V mL of theinternal standard solution obtained in the Assay, and dis-perse the particles with the aid of ultrasonic waves. Cen-trifuge for 10 minutes, and use the supernatant liquid as thesample solution. Separately, weigh accurately about 20 mgof Nilvadipine Reference Standard, dissolve in the mixtureof acetonitrile and water (7:3) to make exactly 20 mL. Pipet5 mL of this solution, add exactly 25 mL of the internalstandard solution obtained in the Assay and the mixture ofacetonitrile and water (7:3) to make 50 mL, and use thissolution as the standard solution. Proceed as directed in theAssay.
Amount (mg) of nilvadipine (C19H19N3O6)
=WS×QT
QS×
V100
WS: Amount (mg) of Nilvadipine Reference Standard
Dissolution Perform the test with 1 tablet of NilvadipineTablets at 50 revolutions per minute according to Method 2under the Dissolution Test, using 900 mL of water as thedissolution medium. Withdraw 20 mL or more of the disso-lution medium 30 minutes after starting the test, and ˆlterthrough a membrane ˆlter with pore size of not more than0.5 mm. Discard the ˆrst 10 mL of the ˆltrate, pipet 10 mL ofthe subsequent ˆltrate, add exactly 1 mL of methanol, anduse this solution as the sample solution. Separately, weighaccurately an amount of Nilvadipine Reference Standard,equivalent to 10 times the labeled amount of NilvadipineTablets, and dissolve in methanol to make exactly 50 mL.Pipet 5 mL of this solution, and add methanol to makeexactly 100 mL. Pipet 1 mL of this solution, add exactly10 mL of water, and use this solution as the standardsolution. Perform the test with exactly 20 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the following con-ditions, and determine the peak areas, AT and AS, of nilvadi-pine: the dissolution rate in 30 minutes is not less than 85z.
Dissolution rate (z) with respect to the labeled amount ofnilvadipine (C19H19N3O6)
=WS×AT
AS×
1C×9
WS: Amount (mg) of Nilvadipine Reference StandardC: Labeled amount (mg) of nilvadipine (C19H19N3O6) in 1
tablet
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 242 nm).Column: A stainless steel column 4 mm in inside diameter
and 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of phosphate buŠer solution, pH7.4, methanol and acetonitrile (7:7:6)
Flow rate: Adjust the ‰ow rate so that the retention timeof nilvadipineis about 5 minutes.System suitability—
System performance: When the procedure is run with 20mL of the standard solution under the above operating con-ditions, the number of theoretical plates and the symmetryfactor of the peak of nilvadipine are not less than 2000 andnot more than 1.5, respectively.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of nilvadipine is not more than 1.5z.
Assay Weigh accurately not less than 20 NilvadipineTablets, and powder. Weigh accurately an amount of thepowder, equivalent to about 5 mg of nilvadipine(C19H19N3O6), add 10 mL of a mixture of acetonitrile andwater (7:3) and exactly 25 mL of the internal standard solu-tion, shake for 15 minutes, and add the mixture of acetoni-trile and water (7:3) to make 50 mL. Centrifuge, and use thesupernatant liquid as the sample solution. Separately, weighaccurately about 20 mg of Nilvadipine Reference Standard,dissolve in the mixture of acetonitrile and water (7:3) tomake exactly 20 mL. Pipet 5 mL of this solution, add exactly25 mL of the internal standard solution and the mixture ofacetonitrile and water (7:3) to make 50 mL, and use thissolution as the standard solution. Perform the test with 5 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine the ratios, QT and QS,of the peak area of nilvadipine to that of the internal stan-dard.
Amount (mg) of nilvadipine (C19H19N3O6)=WS×QT
QS×
14
WS: Amount (mg) of Nilvadipine Reference Standard
Internal standard solution—A solution of acenaphthene inacetonitrile (1 in 500).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 2.5 g of diammonium hydrogenphosphate in 1000 mL of water, add 10 mL of tetrabutylam-monium hydoxide TS, adjust the pH to 7.0 with dilutedphosphoric acid (1 in 10), and add 900 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof nilvadipine is about 12 minutes.System suitability—
System performance: When the procedure is run with 5 mL

17281728 Supplement II, JPXIVO‹cial Monographs for Part I
of the standard solution under the above operating condi-tions, nilvadipine and the internal standard are eluted in thisorder with the resolution between these peaks being not lessthan 8.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of nilvadipine to that of the internal standardis not more than 1.0z.
Containers and storage Containers—Well-closed contain-ers.
Add the following:
Oxytocinオキシトシン
C43H66N12O12S2: 1007.19[50-56-6]
Oxytocin is a synthetic peptide having the propertyof causing the contraction of uterine smooth muscle.
It contains not less than 540 oxytocin Units and notmore than 600 oxytocin Units per mg, calculated onthe dehydrated and de-acetic acid basis.
Description Oxytocin occurs as a white powder.It is very soluble in water, and freely soluble in ethanol
(99.5).It dissolves in hydrochloric acid TS.The pH of a solution prepared by dissolving 0.10 g of
Oxytocin in 10 mL of freshly boiled and cooled water is be-tween 4.0 and 6.0.
It is hygroscopic.
Identiˆcation Determine the absorption spectrum of asolution of Oxytocin (1 in 2000) as directed under theUltraviolet-visible Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
Constituent amino acids Put about 1 mg of Oxytocin in atest tube for hydrolysis, add 6 mol/L hydrochloric acid TSto dissolve, replace the air in the tube with Nitrogen, seal thetube under reduced pressure, and heat at 110 to 1159C for16 hours. After cooling, open the tube, evaporate thehydrolyzate to dryness under reduced pressure, add 2 mL of0.02 mol/L hydrochloric acid TS to dissolve the residue, anduse this solution as the sample solution. Separately, weighaccurately about 27 mg of L-aspartic acid, about 24 mg ofL-threonine, about 21 mg of L-serine, about 29 mg ofL-glutamic acid, about 23 mg of L-proline, about 15 mg ofglycine, about 18 mg of L-alanine, about 23 mg of L-valine,about 48 mg of L-cystine, about 30 mg of methionine, about26 mg of L-isoleucine, about 26 mg of L-leucine, about36 mg of L-tyrosine, about 33 mg of phenylalanine, about
37 mg of L-lysine hydrochloride, about 42 mg of L-histidinehydrochloride monohydrate and about 42 mg of L-argininehydrochloride, dissolve them in 10 mL of 1 mol/Lhydrochloric acid TS, and add water to make exactly100 mL. Pipet 5 mL of this solution, add water to make ex-actly 20 mL, and use this solution as the standard solution.Perform the test with exactly 20 mL each of the sample solu-tion and the standard solution as directed under the LiquidChromatography according to the following conditions, andcalculate the respective molar ratios with respect to leucine:0.95 – 1.05 for aspartic acid, 0.95 – 1.05 for glutamic acid,0.95 – 1.05 for proline, 0.95 – 1.05 for glycine, 0.80 – 1.10for isoleucine, 0.80 – 1.05 for tyrosine and 0.80 – 1.05 forcystine, and not more than 0.01 each for others.Operating conditions—
Detector: A visible spectrophotometer (wavelength: 440nm and 570 nm).Column: A stainless steel column 4.6 mm in inside diameterand 8 cm in length, packed with strongly acidic ion-exchangeresin for liquid chromatography (sodium type) composedwith a sulfonated polystyrene copolymer (3 mm in particlediameter).
Column temperature: A constant temperature of about579C.
Chemical reaction bath temperature: A constant tempera-ture of about 1309C.
Color developing time: About 1 minute.Mobile phase: Prepare mobile phases A, B and C accord-
ing to the following table.
Mobile phase A B C
Citric acid mono-hydrate 19.80 g 22.00 g 6.10 g
Trisodium citratedihydrate 6.19 g 7.74 g 26.67 g
Sodium chloride 5.66 g 7.07 g 54.35 gEthanol (99.5) 260.0 mL 20.0 mL —Benzyl alcohol — — 5.0 mLThiodiglycol 5.0 mL 5.0 mL —Lauromacrogolsolution (1 in 4) 4.0 mL 4.0 mL 4.0 mL
Capryric acid 0.1 mL 0.1 mL 0.1 mLWater a su‹cient
amounta su‹cient
amounta su‹cient
amount
Total amount 2000 mL 1000 mL 1000 mL
pH 3.3 3.2 4.9
Flowing of the mobile phase: Control the gradient by mix-ing the mobile phases A, B and C as directed in the followingtable.

17291729Supplement II, JPXIV O‹cial Monographs for Part I
Time afterinjection ofsample (min)
MobilephaseA (z)
MobilephaseB (z)
MobilephaseC (z)
0 – 9 100 0 09 – 25 0 100 0
25 – 61 0 100 ª 0 0 ª 10061 – 80 0 0 100
Reaction reagent: Mix 407 g of lithium acetate dihydrate,245 mL of acetic acid (100) and 801 mL of 1-methoxy-2-propanol, add water to make 2000 mL, stir for more than 10minutes while passing Nitrogen, and use this solution as So-lution A. Separately, to 1957 mL of 1-methoxy-2-propanoladd 77 g of ninhydrin and 0.134 g of sodium borohydride,stir for more than 30 minutes while passing Nitrogen, anduse this solution as Solution B. Mix Solution A and SolutionB before use.
Flow rate of mobile phase: About 0.26 mL per minute.Flow rate of reaction reagent: About 0.3 mL per minute.
System suitability—System performance: When the procedure is run with
20 mL of the standard solution under the above operatingconditions, aspartic acid, threonine, serine, glutamic acid,proline, glycine, alanine, valine, cystine, methionine, isoleu-cine, leucine, tyrosine, phenylalanine, lysine, histidine andarginine are eluted in this order with the resolutions betweenthe peaks of threonine and serine, glycine and alanine, andisoleucine and leucine being not less than 1.5, 1.4 and 1.2,respectively.
System repeatability: When the test is repeated 3 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviations of the peakarea of aspartic acid, proline, valine and arginine are notmore than 2.0z, respectively.
Purity (1) Acetic acid—Weigh accurately about 15 mg ofOxytocin, dissolve in the internal standard solution to makeexactly 10 mL, and use this solution as the sample solution.Separately, weigh accurately about 1 g of acetic acid (100),add the internal standard solution to make exactly 100 mL.Pipet 2 mL of this solution, add the internal standardsolution to make exactly 200 mL, and use this solution as thestandard solution. Perform the test with 10 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and calculate the ratios, QT and QS, of the peakarea of acetic acid to that of the internal standard: theamount of acetic acid is not less than 6.0z and not morethan 10.0z.
Amount (z) of acetic acid (C2H4O2)
=WS
WT×
QT
QS×
110
WS: Amount (mg) of acetic acid (100)WT: Amount (mg) of the sample
Internal standard solution—A solution of propionic acid inthe mobile phase (1 in 10,000).
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 210 nm).Column: A stainless steel column 4.6 mm in inside di-
ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about409C.
Mobile phase: To 0.7 mL of phosphoric acid add 900 mLof water, adjust the pH to 3.0 with 8 mol/L sodiumhydroxide TS, and add water to make 1000 mL. To 950 mLof this solution add 50 mL of methanol.
Flow rate: Adjust the ‰ow rate so that the retention timeof acetic acid is about 3 minutes.System suitability—
System performance: When the procedure is run with 10mL of the standard solution under the above operatingconditions, acetic acid and propionic acid are eluted in thisorder with the resolution between these peaks being not lessthan 14.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of acetic acid to that of the internal standard isnot more than 2.0z.
(2) Related substances—Dissolve 25 mg of Oxytocin in100 mL of the mobile phase A, and use this solution as thesample solution. Perform the test with 50 mL of the samplesolution as directed under the Liquid Chromatographyaccording to the following conditions, determine each peakarea by the automatic integration method, and calculate theamount of them by the area percentage method: the amountof each peak other than Oxytocin is not more than 1.5z,and the total of them is not more than 5.0z.Operating conditions—
Detector, column, column temperature, mobile phase,‰owing of mobile phase, and ‰ow rate: Proceed as directedin the operating conditions in the Assay.
Time span of measurement: About 2.5 times as long as theretention time of oxytocin.System suitability—
Test for required detectability: Measure exactly 1 mL ofthe sample solution, add the mobile phase A to make exactly100 mL, and use this solution as the solution for systemsuitability test. Pipet 1 mL of the solution for systemsuitability test, and add the mobile phase A to make exactly10 mL. Conˆrm that the peak area of oxytocin obtainedfrom 50 mL of this solution is equivalent to 5 to 15z of thatfrom 50 mL of the solution for system suitability test.
System performance: Dissolve an adequate amount ofoxytocin and vasopressin in the mobile phase A, so that eachmL contains about 0.1 mg each of them. When the proce-dure is run with 50 mL of this solution under the above oper-ating conditions, vasopressin and oxytocin are eluted in thisorder with the resolution between these peaks being not lessthan 14, and the symmetry factor of the peak of oxytocin isnot more than 1.5.

17301730 Supplement II, JPXIVO‹cial Monographs for Part I
System repeatability: When the test is repeated 6 timeswith 50 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of oxytocin is not more than 2.0z.
Water Not more than 5.0z (50 mg, coulometric titration).
Assay Weigh accurately an amount of Oxytocin, equiva-lent to about 13,000 Units, dissolve in the mobile phase A tomake exactly 100 mL, and use this solution as the samplesolution. Separately, dissolve oxytocin in 1 bottle of thePosterior Pituitary Reference Standard in the mobile phaseA to make a known concentration solution containing eachmL contains about 130 Units, and use this solution as thestandard solution. Perform the test with exactly 25 mL eachof the sample solution and the standard solution as directedunder the Liquid Chromatography according to the follow-ing conditions, and determine the peak areas, AT and AS, ofoxytocin.
Units per mg of Oxytocin, calculated on the dehydrated andde-acetic acid basis
=WS
WT×
AT
AS×100
WS: Units per mL of the standard solutionWT: Amount (mg) of the sample, calculated on the de-
hydrated and de-acetic acid basis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 220 nm).Column: A stainless steel column 4.6 mm in inside di-
ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about259C.
Mobile phase A: Dissolve 15.6 g of sodium dihydrogenphosphate dihydrate in 1000 mL of water.
Mobile phase B: A mixture of water and acetonitrile (1:1).Flowing of the mobile phase: Control the gradient by
mixing the mobile phases A and B as directed in thefollowing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
0 – 30 70 ª 40 30 ª 6030 – 30.1 40 ª 70 60 ª 3030.1 – 45 70 30
Flow rate: About 1.0 mL per minute.System suitability—
System performance: Dissolve an adequate amount ofoxytocin and vasopressin in the mobile phase A, so that eachmL contains about 0.1 mg each of them. When the proce-dure is run with 25 mL of this solution under the above oper-ating conditions, vasopressin and oxytocin are eluted in thisorder with the resolution between these peaks being not lessthan 14, and the symmetry factor of the peak of oxytocin is
not more than 1.5.System repeatability: When the test is repeated 6 times
with 25 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of oxytocin is not more than 1.0z.
Containers and storage Containers—Tight containers.Storage—At 2 to 89C.
Change to read:
Oxytocin Injectionオキシトシン注射液
Oxytocin Injection is an aqueous solution for injec-tion.
It contains not less than 90.0z and not more than110.0z of the labeled oxytocin Units.
Method of preparation Prepare as directed under Injec-tions, with Oxytocin.
Description Oxytocin Injection is a colorless, clear liquid.
pH 2.5 – 4.5
Bacterial endotoxins Less than 10 EU/oxytocin Unit.
Actual volume It meets the requirements of the Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Measure exactly a portion of Oxytocin Injectionaccording to the labeled Units, dilute with the diluent so thateach mL contains about 1 Unit, and use this solution as thesample solution. Separately, dissolve oxytocin in 1 bottle ofPosterior Pituitary Reference Standard in the mobile phaseA to make exactly 20 mL. Pipet a suitable volume of this so-lution, dilute with the diluent to make a known concentra-tion solution so that each mL contains about 1 Unit, and usethis solution as the standard solution. Perform the test withexactly 100 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and determine the peakareas, AT and AS, of oxytocin.
Units per mL of Oxytocin Injection=WS×AT
AS×
ba
WS: Units per mL of the standard solutiona: Volume (mL) of the sampleb: Total volume of the sample solution prepared by dilut-

17311731Supplement II, JPXIV O‹cial Monographs for Part I
ing with the diluentDiluent: Dissolve 5 g of chlorobutanol, 1.1 g of sodium
acetate trihydrate, 5 g of acetic acid (100) and 6mL of ethanol (99.5) in water to make 1000 mL.
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 220 nm).Column: A stainless steel column 4.6 mm in inside di-
ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about259C.
Mobile phase A: Dissolve 15.6 g of sodium dihydrogenphosphate dihydrate in 1000 mL of water.
Mobile phase B: A mixture of water and acetonitrile (1:1).Flowing of the mobile phase: Control the gradient by
mixing the mobile phases A and B as directed in the follow-ing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
0 – 30 70 ª 40 30 ª 6030 – 30.1 40 ª 70 60 ª 3030.1 – 45 70 30
Flow rate: About 1.0 mL per minute.System suitability—
System performance: Dissolve an adequate amount ofoxytocin and vasopressin in the mobile phase A, so that eachmL contains about 0.02 mg each of them. When the proce-dure is run with 100 mL of this solution under the aboveoperating conditions, vasopressin and oxytocin are eluted inthis order with the resolution between these peaks being notless than 14, and the symmetry factor of the peak of oxyto-cin is not more than 1.5.
System repeatability: When the test is repeated 6 timeswith 100 mL of the standard solution under the above oper-ating conditions, the relative standard deviation of the peakarea of oxytocin is not more than 2.0z.
Containers and storage Containers—Hermetic containers.Storage—In a cold place, and avoid freezing.
Pirenoxineピレノキシン
Change the Purity (2) to read:
Purity(2) Related substances—Dissolve 10 mg of Pirenoxine in
50 mL of the mobile phase, and use this solution as the sam-ple solution. Pipet 3 mL of the sample solution, add the mo-bile phase to make exactly 200 mL, and use this solution asthe standard solution. Perform the test with exactly 5 mLeach of the sample solution and the standard solution as
directed under the Liquid Chromatography according to thefollowing conditions. Determine each peak area of both so-lutions by the automatic integration method: the total areaof the peaks other than pirenoxine is not larger than the peakarea of pirenoxine from the standard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 230 nm).
Column: A stainless steel column 4 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about359C.
Mobile phase: Dissolve 1.39 g of tetra n-butylammoniumchloride and 4.5 g of disodium hydrogen phosphate 12-water in 1000 mL of water, and adjust the pH to 6.5 withphosphoric acid. To 700 mL of this solution add 200 mL ofacetonitrile and 30 mL of tetrahydrofuran, and mix.
Flow rate: Adjust the ‰ow rate so that the retention timeof pirenoxine is about 10 minutes.
Time span of measurement: About 3 times as long as theretention time of pirenoxine.System suitability—
Test for required detectability: To exactly 2 mL of thestandard solution add the mobile phase to make exactly 30mL. Conˆrm that the peak area of pirenoxine obtained from5 mL of this solution is equivalent to 5 to 8z of that ofpirenoxine obtained from 5 mL of the standard solution.
System performance: Dissolve 3 mg of Pirenoxine and16 mg of methyl parahydroxybenzoate in 100 mL of themobile phase. When the procedure is run with 5 mL of thissolution under the above operating conditions, pirenoxineand methyl parahydroxybenzoate are eluted in this orderwith the resolution between these peaks being not less than2.0.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of pirenoxine is not more than 1.0z.

17321732 Supplement II, JPXIVO‹cial Monographs for Part I
Add the following:
Pirenzepine Hydrochloride Hydrate塩酸ピレンゼピン水和物
C19H21N5O2.2HCl.H2O: 442.345,11-Dihydro-11-[(4-methylpiperazin-1-yl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one dihydrochloridemonohydrate [29868-97-1, anhydride]
Pirenzepine Hydrochloride Hydrate contains notless than 98.5z and not more than 101.0z of pirenze-pine hydrochloride (C19H21N5O2.2HCl: 424.32), calcu-lated on the anhydrous basis.
Description Pirenzepine Hydrochloride Hydrate occurs asa white to pale yellow crystalline powder.
It is freely soluble in water and in formic acid, slightlysoluble in methanol, and very slightly soluble in ethanol(99.5).
The pH of a solution by dissolving 1 g of PirenzepineHydrochloride Hydrate in 10 mL of water is between 1.0 and2.0.
Melting point: about 2459C (with decomposition).It is gradually colored by light.
Identiˆcation (1) Determine the absorption spectrum ofa solution of Pirenzepine Hydrochloride Hydrate (1 in40,000) as directed under the Ultraviolet-visible Spec-trophotometry, and compare the spectrum with theReference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofPirenzepine Hydrochloride Hydrate as directed in the potas-sium chloride disk method under the Infrared Spectrophoto-metry, and compare the spectrum with the ReferenceSpectrum: both spectra exhibit similar intensities of absorp-tion at the same wave numbers.
(3) A solution of Pirenzepine Hydrochloride Hydrate (1in 50) responds to the Qualitative Tests for chloride.
Purity (1) Clarity and color of solution—A solutionobtained by dissolving 1.0 g of Pirenzepine HydrochlorideHydrate in 10 mL of water is clear and not more color thanthat of the following control solution.
Control solution: To 1.2 mL of Matching ‰uid for color Fadd 8.8 mL of diluted hydrochloric acid (1 in 40).
(2) Heavy metals—Proceed with 2.0 g of PirenzepineHydrochloride Hydrate according to Method 2, and per-form the test. Prepare the control solution with 2.0 mL of
Standard Lead Solution (not more than 10 ppm).(3) Related substances—Dissolve 0.3 g of Pirenzepine
Hydrochloride Hydrate in 10 mL of water. To 1 mL of thissolution add 5 mL of methanol and the mobile phase A tomake 10 mL, and use this solution as the sample solution.Pipet 1 mL of the sample solution, and add 5 mL ofmethanol and the mobile phase A to make exactly 10 mL.Pipet 1 mL of this solution, add 5 mL of methanol and themobile phase A to make exactly 10 mL, and use this solutionas the standard solution. Perform the test with exactly 10 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine each peak area by theautomatic integration method: the area of the peak otherthan pirenzepine is not more than 3/10 times the peak areaof pirenzepine from the standard solution, and the total areaof the peaks other than pirenzepine is not more than 3/5times the peak area of pirenzepine from the standard solu-tion.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 283 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase A: Dissolve 2 g of sodium lauryl sulfate in900 mL of water, adjust the pH to 3.2 with acetic acid (100),and add water to make 1000 mL.
Mobile phase B: MethanolMobile phase C: AcetonitrileFlowing of the mobile phase: Control the gradient by
mixing the mobile phases A, B and C as directed in the fol-lowing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
Mobile phaseC (z)
0 – 15 55 ª 25 30 15 ª 4515 – 25 30 45
Flow rate: Adjust the ‰ow rate so that the retention timeof pirenzepine is about 8 minutes.
Time span of measurement: About 2 times as long as theretention time of pirenzepine after the solvent peak.System suitability—
Test for required detectability: Pipet 1 mL of the standardsolution, and add 5 mL of methanol and the mobile phase Ato make exactly 10 mL. Conˆrm that the peak area of piren-zepine obtained from 10 mL of this solution is equivalent to 7to 13z of that from 10 mL of the standard solution.
System performance: Dissolve 0.1 g of phenylpiperazinehydrochloride in 10 mL of methanol. Mix 1 mL of thissolution and 1 mL of the sample solution, and add 5 mL ofmethanol and the mobile phase A to make 10 mL. When theprocedure is run with 10 mL of this solution under the above

17331733Supplement II, JPXIV O‹cial Monographs for Part I
operating conditions, pirenzepine and phenylpiperazine areeluted in this order with the resolution between these peaksbeing not less than 5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of thepeak area of pirenzepine is not more than 2.0z.
Water Not less than 3.5z and not more than 5.0z (0.3 g,volumetric titration, direct titration).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 0.2 g of PirenzepineHydrochloride Hydrate, dissolve in 2 mL of formic acid,add 60 mL of acetic anhydride, and titrate with 0.1 mol/Lperchloric acid VS (potentiometric titration). Perform ablank determination in the same manner, and make anynecessary correction.
Each mL of 0.1 mol/L perchloric acid VS=14.14 mg of C19H21N5O2.2HCl
Containers and storage Containers—Well-closed contain-ers.
Storage—Light-resistant.
Add the following:
Piroxicamピロキシカム
C15H13N3O4S: 331.354-Hydroxy-2-methyl-N-(pyridin-2-yl)-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide [36322-90-4]
Piroxicam contains not less than 98.5z and notmore than 101.0z of C15H13N3O4S, calculated on thedried basis.
Description Piroxicam occurs as a white to pale yellowcrystalline powder.
It is sparingly soluble in acetic anhydride, slightly solublein acetonitrile, in methanol and in ethanol (99.5), very slight-ly soluble in acetic acid (100), and practically insoluble inwater.
Melting point: about 2009C (with decomposition).
Identiˆcation (1) Dissolve 0.1 g of Piroxicam in a mix-ture of methanol and 0.5 mol/L hydrochloric acid TS(490:1) to make 200 mL. To 1 mL of this solution add themixture of methanol and 0.5 mol/L hydrochloric acid TS(490:1) to make 100 mL. Determine the absorption spectrumof this solution as directed under the Ultraviolet-visibleSpectrophotometry, and compare the spectrum with the
Reference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofPiroxicam as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wave numbers.If any diŠerence appears between the spectra, dissolve thesample with dichloromethane, evaporate the solvent, dry theresidue on a water bath, and perform the test.
Purity (1) Heavy metals—Proceed with 1.0 g of Piroxic-am according to Method 2, and perform the test. Preparethe control solution with 2.0 mL of Standard Lead Solution(not more than 20 ppm).
(2) Related substances—Dissolve 75 mg of Piroxicam in50 mL of acetonitrile for liquid chromatography, and usethis solution as the sample solution. Pipet 1 mL of thesample solution, add acetonitrile for liquid chromatographyto make exactly 10 mL. Pipet 1 mL of this solution, addacetonitrile for liquid chromatography to make exactly 50mL, and use this solution as the standard solution. Performthe test with exactly 20 mL each of the sample solution andthe standard solution as directed under the Liquid Chro-matography according to the following conditions, anddetermine each peak area by the automatic integrationmethod: the area of the peak other than piroxicam is notlarger than the peak area of piroxicam with the standard so-lution, and the total area of the peaks other than piroxicamis not larger than 2 times the peak area of piroxicam with thestandard solution.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 230 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: A mixture of 0.05 mol/L potassium di-hydrogen phosphate TS, pH 3.0 and acetonitrile for liquidchromatography (3:2).
Flow rate: Adjust the ‰ow rate so that the retention timeof piroxicam is about 10 minutes.
Time span of measurement: About 5 times as long as theretention time of piroxicam after the solvent peak.System suitability—
Test for required detectability: To exactly 5 mL of thestandard solution add acetonitrile for liquid chro-matography to make exactly 20 mL. Conˆrm that the peakarea of piroxicam obtained with 20 mL of this solution isequivalent to 17.5 to 32.5z of that with 20 mL of thestandard solution.
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, the number of theoretical plates and the symmet-ry factor of the peak of piroxicam are not less than 6000 and

17341734 Supplement II, JPXIVO‹cial Monographs for Part I
not more than 1.5, respectively.System repeatability: When the test is repeated 6 times
with 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of piroxicam is not more than 2.0z.
Loss on drying Not more than 0.5z (1 g, 1059C, 3 hours).
Residue on ignition Not more than 0.2z (1 g).
Assay Weigh accurately about 0.25 g of Piroxicam, dis-solve in 60 mL of a mixture of acetic anhydride and aceticacid (100) (1:1), and titrate with 0.1 mol/L perchloric acidVS (potentiometric titration). Perform a blank determina-tion in the same manner, and make any necessary correc-tion.
Each mL of 0.1 mol/L perchloric acid VS=33.14 mg of C15H13N3O4S
Containers and storage Containers—Tight containers.
Pyrazinamideピラジナミド
Change the Description to read:
Description Pyrazinamide occurs as white crystals orcrystalline powder.
It is sparingly soluble in water and in methanol, andslightly soluble in ethanol (99.5) and in acetic anhydride.
Change the Purity to read:
Purity(1) Heavy metals—Proceed with 1.0 g of Pyrazinamide
according to Method 2, and perform the test. Prepare thecontrol solution with 2.0 mL of Standard Lead Solution (notmore than 20 ppm).
(2) Related substances—Dissolve 0.10 g of Pyrazin-amide in 10 mL of methanol, and use this solution as thesample solution. Pipet 1 mL of the sample solution, addmethanol to make exactly 200 mL, and use this solution asthe standard solution. Perform the test with these solutionsas directed under the Thin-layer Chromatography. Spot 20mL each of the sample solution and the standard solution ona plate of silica gel with ‰uorescent indicator for thin-layerchromatography. Develop the plate with a mixture of 1-butanol, water and acetic acid (100) (3:1:1) to a distance ofabout 10 cm, and air-dry the plate. Examine under ultravio-let light (main wavelength: 254 nm): the spot other than theprincipal spot obtained from the sample solution is not moreintense than the spot from the standard solution.
Change the Assay to read:
Assay Weigh accurately about 0.1 g of Pyrazinamide,previously dried, dissolve in 50 mL of acetic anhydride, andtitrate with 0.1 mol/L perchloric acid VS (potentiometrictitration). Perform a blank determination in the same man-
ner, and make any necessary correction.
Each mL of 0.1 mol/L perchloric acid VS=12.31 mg of C5H5N3O
Pyridoxine Hydrochloride塩酸ピリドキシン
Change the origin/limits of content to read:
Pyridoxine Hydrochloride, when dried, contains notless than 98.0z and not more than 101.0z ofC8H11NO3.HCl.
Change the Description to read:
Description Pyridoxine Hydrochloride occurs as a white topale yellow, crystalline powder.
It is freely soluble in water, slightly soluble in ethanol(99.5), and practically insoluble in acetic anhydride and inacetic acid (100).
It is gradually aŠected by light.Melting point: about 2069C (with decomposition).
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Pyridoxine Hydrochloride in 0.1 mol/L hydrochloric acidTS (1 in 100,000) as directed under the Ultraviolet-visibleSpectrophotometry, and compare the spectrum with theReference Spectrum or the spectrum of a solution ofPyridoxine Hydrochloride Reference Standard prepared inthe same manner as the sample solution: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofPyridoxine Hydrochloride as directed in the potassiumchloride disk method under the Infrared Spectrophotomet-ry, and compare the spectrum with the Reference Spectrumor the spectrum of Pyridoxine Hydrochloride ReferenceStandard: both spectra exhibit similar intensities of absorp-tion at the same wave numbers.
(3) A solution of Pyridoxine Hydrochloride (1 in 10)responds to the Qualitative Tests for chloride.
Add the following next to Identiˆcation:
pH The pH of a solution prepared by dissolving 1.0 g ofPyridoxine Hydrochloride in 50 mL of water is between 2.5and 3.5.
Add the following next to Purity (2):
(3) Related substances—Dissolve 1.0 g of PyridoxineHydrochloride in 10 mL of water, and use this solution asthe sample solution. Pipet 2.5 mL of the sample solution,and add water to make exactly 100 mL. Pipet 1 mL of thissolution, add water to make exactly 10 mL, and use this

17351735Supplement II, JPXIV O‹cial Monographs for Part I
solution as the standard solution. Perform the test with thesesolutions as directed under the Thin-layer Chromatography.Spot 2 mL each of the sample solution and the standardsolution on a plate of silica gel for thin-layer chro-matography, and air-dry the plate. Develop the plate with amixture of acetone, tetrahydrofuran, hexane and ammoniasolution (28) (65:13:13:9) to a distance of about 10 cm, andair-dry the plate. Spray evenly a solution of sodiumcarbonate in diluted ethanol (99.5) (3 in 10) (1 in 20) on theplate. After air-drying, spray evenly a solution of 2,6-dibromo-N-chloro-1,4-benzoquinone monoimine in ethanol(99.5) (1 in 1000) on the plate, and air-dry: the spot otherthan the principal spot obtained from the sample solution isnot more intense than the spot from the standard solution.
Pyridoxine Hydrochloride Injection塩酸ピリドキシン注射液
Change the origin/limits of content to read:
Pyridoxine Hydrochloride Injection is an aqueoussolution for injection.
It contains not less than 95.0z and not more than105.0z of the labeled amount of pyridoxinehydrochloride (C8H11NO3.HCl: 205.64).
Change the Identiˆcation to read:
Identiˆcation(1) To a volume of Pyridoxine Hydrochloride Injection,
equivalent to 50 mg of Pyridoxine Hydrochloride accordingto the labeled amount, add 0.1 mol/L hydrochloric acid TSto make 100 mL. To 2 mL of this solution add 0.1 mol/Lhydrochloric acid TS to make 100 mL. Determine theabsorption spectrum of this solution as directed under theUltraviolet-visible spectrophotometry: it exhibits a maxi-mum between 288 nm and 292 nm.
(2) To a volume of Pyridoxine Hydrochloride Injection,equivalent to 10 mg of Pyridoxine Hydrochloride accordingto the labeled amount, add water to make 10 mL, and usethis solution as the sample solution. Separately, dissolve 10mg of Pyridoxine Hydrochloride Reference Standard in 10mL of water, and use this solution as the standard solution.Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 2 mL each of the samplesolution and the standard solution on a plate of silica gel forthin-layer chromatography, and air-dry the plate. Developthe plate with a mixture of acetone, tetrahydrofuran, hexaneand ammonia solution (28) (65:13:13:9) to a distance ofabout 10 cm, and air-dry the plate. Spray evenly a solutionof sodium carbonate in diluted ethanol (99.5) (3 in 10) (1 in20) on the plate. After air-drying, spray evenly a solution of2,6-dibromo-N-chloro-1,4-benzoquinone monoimine inethanol (99.5) (1 in 1000) on the plate: the spots obtainedfrom the sample solution and the standard solution are bluein color and have the same Rf value.
Add the following next to Identiˆcation:
Endotoxins Less than 3.0 EU/mg.
Delete the following Monograph:
Santonin Tablets
Add the following:
Serrapeptaseセラペプターゼ
[95077-02-4]
Serrapeptase is the enzyme preparation having pro-teolytic activity, produced by the growth of Serratiaspecies. Usually, it is diluted with Lactose.
It contains not less than 2000 serrapeptase Units andnot more than 2600 serrapeptase Units per mg.
It is hygroscopic.
Description Serrapeptase occurs as a grayish white to lightbrown powder, having a slight characteristic odor.
Identiˆcation Dissolve 0.4 g of Serrapeptase in 100 mL ofacetic acid-sodium acetate buŠer solution, pH 5.0, transferexactly 1 mL each of this solution into three tubes, and referto them as A, B and C. To tube A add exactly 1 mL of water,to tubes B and C add exactly 1 mL of 0.04 mol/L disodiumdihydrogen ethylenediamine tetraacetate TS, mix gently,and allow them to stand in a water bath at 4±19C for about1 hour. Then, to the tube B add exactly 2 mL of 0.04 mol/Lzinc chloride TS, to the tubes A and C add exactly 2 mL ofwater, mix gently, and allow them to stand in a water bath at4±19C for about 1 hour. Pipet 1 mL each of these solu-tions, add borate-hydrochloric acid buŠer solution, pH 9.0to the solutions A and B to make exactly 200 mL, to the so-lution C to make exactly 50 mL, and use these solutions asthe sample solutions. Proceed with these sample solutions asdirected in the Assay: the activities of the solutions A and Bare almost the same, and the activity of the solution C is notmore than 5z of that of the solution A.
Activity of solutions A, B or C=AT
AS×
120×D×176
AS: Absorbance of the standard solutionAT: Absorbance of the sample solution20: Reaction time (minute)D: Dilution rate (200 for solution A and B, 50 for solution
C)176: Conversion factor (Total volume of enzyme reaction
solution/volume of ˆltrate taken×amount of tyrosine in 2mL of tyrosine standard solution)
Purity (1) Heavy metals—Put 1.0 g of Serrapeptase in a

17361736 Supplement II, JPXIVO‹cial Monographs for Part I
porcelain crucible, add 2 drops each of sulfuric acid andnitric acid, and incinerate by ignition. After cooling, to theresidue add 2 mL of hydrochloric acid, evaporate to drynesson a water bath, add 10 mL of a solution of hydroxylaminehydrochloride (3 in 100) and 2 mL of dilute acetic acid, andheat on a water bath for 5 minutes. After cooling, ˆlter ifnecessary, wash the ˆlter paper with 10 mL of water, put theˆltrate and washing in a Nessler tube, add water to make 50mL, and use this solution as the test solution. Prepare thecontrol solution as follows: Evaporate to dryness 2 dropseach of sulfuric acid and nitric acid on a sand bath, add 2mL of hydrochloric acid to the residue, evaporate to drynesson a water bath, add 2.0 mL of Standard Lead Solution, 10mL of a solution of hydroxylamine hydrochloride (3 in 100)and 2 mL of dilute acetic acid, and heat on a water bath for5 minutes. Proceed in the same manner as directed for thepreparation of the test solution, and add water to make 50mL (not more than 20 ppm).
(2) Arsenic—Prepare the test solution with 0.40 g ofSerrapeptase according to Method 3, excepting addition of 5mL of a solution of magnesium nitrate hexahydrate inethanol (95) (3 in 10) instead of a solution of magnesium ni-trate hexahydrate in ethanol (95) (1 in 50), evaporating todryness on a water bath, then incinerating with a small‰ame, and perform the test (not more than 5 ppm).
Loss on drying Not more than 7.0z (1 g, 1059C, 4 hours).
Residue on ignition Not more than 1.5z (1 g).
Assay (i) Sample solution: Dissolve exactly 0.100 g ofSerrapeptase in a solution of ammonium sulfate (1 in 20) tomake exactly 100 mL. Pipet 1 mL of this solution, addborate-hydrochloric acid buŠer solution, pH 9.0 to makeexactly 200 mL, and use this solution as the sample solution.
(ii) Tyrosine standard solution: Dissolve exactly 0.160 gof Tyrosine Reference Standard, previously dried at 1059Cfor 3 hours, in 0.2 mol/L hydrochloric acid TS to makeexactly 1000 mL. Pipet 10 mL of this solution, and add0.2 mol/L hydrochloric acid TS to make exactly 100 mL.Prepare before use.
(iii) Substrate solution: Previously determine the loss ondrying (609C, reduced pressure not exceeding 0.67 kPa, 3hours) of milk casein, previously dried. To exactly 1.20 g ofthe milk casein, calculated based on the loss on drying, add160 mL of a solution of sodium borate (19 in 1000), and heatin a water bath to dissolve. After cooling, adjust the pH toexactly 9.0 with 1 mol/L hydrochloric acid TS, and addborate-hydrochloric acid buŠer solution, pH 9.0 to make ex-actly 200 mL. Use after warming to 37±0.59C. Prepare be-fore use.
(iv) Precipitation reagent: Trichloroacetic acid TS forserrapeptase. Use after warming to 37±0.59C.
(v) Procedure: Pipet 1 mL of the sample solution, put ina glass-stoppered tube (15×130 mm), allow to stand at 37±0.59C for 5 minutes, add exactly 5 mL of the substratesolution, and mix well immediately. Allow to stand at 37±0.59C for exactly 20 minutes, add exactly 5 mL oftrichloroacetic acid TS for serrapeptase, mix, allow to stand
at 37±0.59C for 30 minutes, and ˆlter through a dried ˆlterpaper. Pipet 2 mL of the ˆltrate, add exactly 5 mL of asolution of anhydrous sodium carbonate (3 in 50), mix, addexactly 1 mL of diluted Folin's TS (1 in 3), mix well, andallow to stand at 37±0.59C for 30 minutes. Determine theabsorbance of this solution at 660 nm, A1, as directed underthe Ultraviolet-visible Spectrophotometry using water as theblank. Separately, pipet 1 mL of the sample solution, addexactly 5 mL of trichloroacetic acid TS for serrapeptase,mix, add exactly 5 mL of the substrate solution, allow tostand at 37±0.59C for 30 minutes, and proceed in the samemanner as directed above to determine the absorbance A2.Separately, pipet 2 mL of the tyrosine standard solution,add exactly 5 mL of a solution of anhydrous sodiumcarbonate (3 in 50), mix, add exactly 1 mL of diluted Folin'sTS (1 in 3), mix well, and proceed in the same manner asdirected above to determine the absorbance A3. Separately,pipet 2 mL of 0.2 mol/L hydrochloric acid TS, and proceedin the same manner as directed above to determine theabsorbance A4.
Serrapeptase Unit per mg of Serrapeptase
=A1-A2
A3-A4×
120×200×176
20: Reaction time (minute)200: Dilution rate176: Conversion factor (Total volume of enzyme reaction
solution/volume of ˆltrate taken×amount of tyro-sine in 2 mL of tyrosine standard solution)
One serrapeptase Unit corresponds to the amount ofserrapeptase which produces 5 mg of tyrosine per minutefrom 5 mL of the substrate solution under the above condi-tions.
Containers and storage Containers—Tight containers.
Sodium Aurothiomalate金チオリンゴ酸ナトリウム
Change the origin/limits of content to read:
Sodium Aurothiomalate contains not less than49.0z and not more than 52.5z of gold (Au: 196.97),calculated on the anhydrous basis and corrected by theamount of ethanol.
Change the Description to read:
Description Sodium Aurothiomalate occurs as white tolight yellow, powder or granules.
It is very soluble in water, and practically insoluble inethanol (99.5).
It is hygroscopic.It changes in color by light to greenish pale yellow.
Change the Identiˆcation (3) to read:Identiˆcation

17371737Supplement II, JPXIV O‹cial Monographs for Part I
(3) Place 2 mL of a solution of Sodium Aurothiomalate(1 in 10) in a porcelain crucible, add 1 mL of ammonia TSand 1 mL of hydrogen peroxide (30), evaporate to dryness,and ignite. Add 20 mL of water to the residue, and ˆlter: theresidue on the ˆlter paper occurs as a yellow or dark yellow,powder or granules.
Add the following next to Identiˆcation (3):
Identiˆcation(4) The ˆltrate obtained in (3) responds to the Qualita-
tive Tests for sodium salt.(5) The ˆltrate obtained in (3) responds to the Qualita-
tive Tests for sulfate.
Add the following next to Purity (3):
Purity(4) Ethanol—Weigh accurately about 0.2 g of Sodium
Aurothiomalate, add exactly 3 mL of the internal standardsolution and 2 mL of water to dissolve, and use this solutionas the sample solution. Separately, pipet 3 mL of ethanol(99.5), and add water to make exactly 1000 mL. Pipet 2 mLof this solution, add exactly 3 mL of the internal standardsolution, and use this solution as the standard solution.Perform the test with 2 mL each of the sample solution andthe standard solution as directed under the Gas Chro-matography according to the following conditions, anddetermine the ratios of the peak area of ethanol to that ofthe internal standard, QT and QS: the amount of ethanol isnot more than 3.0z.
Amount (mg) of ethanol=QT
QS×6×0.793
0.793: Density (g/mL) of ethanol (99.5) at 209C
Internal standard solution—A solution of 2-propanol (1 in500).Operating conditions—
Detector: Hydrogen ‰ame-ionization detector.Column: A column 3 mm in inside diameter and 3 m in
length, packed with porous styrene-divinylbenzenecopolymer for gas chromatography (particle diameter: 150 –180 mm) (average pore size: 0.0085 mm; 300 – 400 m2/g).
Column temperature: A constant temperature of about1809C.
Carrier gas: NitrogenFlow rate: Adjust the ‰ow rate so that the retention time
of the internal standard is about 7 minutes.System suitability—
System performance: When the procedure is run with 2 mLof the standard solution under the above operating condi-tions, ethanol and the internal standard are eluted in thisorder with the resolution between these peaks being not lessthan 4.
System repeatability: When the test is repeated 6 timeswith 2 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of ethanol to that of the internal standard is
not more than 2.0z.
Add the following next to Purity:
Water Not more than 5.0z (0.1 g, coulometric titration).Use a water vaporizer (heating temperature: 1059C; heatingtime: 30 minutes).
Delete the Loss on drying:
Change the Assay to read:
Assay Weigh accurately about 25 mg of SodiumAurothiomalate, and dissolve in 2 mL of aqua regia by heat-ing. After cooling, add water to make exactly 100 mL. Pipet2 mL of the solution, add water to make exactly 25 mL, anduse this solution as the sample solution. Separately, pipet 5mL, 10 mL and 15 mL of Standard Gold Solution for atom-ic absorption spectrophotometry, add water to make exactly25 mL, and use these solutions as the standard solutions (1),(2) and (3), respectively. Perform the test with the sample so-lution and the standard solutions (1), (2) and (3) as directedunder the Atomic Absorption Spectrophotometry under thefollowing conditions. Determine the amount of gold in thesample solution using the calibration curve obtained fromthe absorbances of the standard solutions.
Gas: Combustible gas—AcetyleneSupporting gas—Air
Lamp: Gold hollow-cathode lampWavelength: 242.8 nm
Sodium Polystyrene Sulfonateポリスチレンスルホン酸ナトリウム
Change the Purity (4) to read:
(4) Styrene—To 10.0 g of Sodium Polystyrene Sulfonateadd 10 mL of acetone, shake for 30 minutes, centrifuge, anduse the supernatant liquid as the sample solution. Separate-ly, dissolve 10 mg of styrene in acetone to make exactly 100mL. Pipet 1 mL of this solution, add acetone to makeexactly 100 mL, and use this solution as the standardsolution. Perform the test with 20 mL each of the sample so-lution and the standard solution as directed under the LiquidChromatography according to the following conditions, anddetermine peak areas, AT and AS, of styrene in each solu-tion: AT is not larger than AS.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4 mm in inside diameterand 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of water and acetonitrile (1:1).Flow rate: Adjust the ‰ow rate so that the retention time

17381738 Supplement II, JPXIVO‹cial Monographs for Part I
of styrene is about 8 minutes.System suitability—
System performance: Dissolve 20 mg each of styrene andbutyl parahydroxybenzoate in 100 mL of acetone. To 5 mLof this solution add acetone to make 100 mL. When theprocedure is run with 20 mL of this solution under the aboveoperating conditions, butyl parahydroxybenzoate and sty-rene are eluted in this order with the resolution betweenthese peaks being not less than 5.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of styrene is not more than 2.0z.
Sodium Thiosulfateチオ硫酸ナトリウム
Change the origin/limits of content to read:
Sodium Thiosulfate, when dried, contains not lessthan 99.0z and not more than 101.0z of sodiumthiosulfate (Na2S2O3: 158.11).
Change the Description to read:
Description Sodium Thiosulfate occurs as colorless, crys-tals or crystalline powder. It is odorless.
It is very soluble in water, and practically insoluble inethanol (99.5).
It eŒoresces in dry air, and is deliquescent in moist air.
Change the Identiˆcation to read:
Identiˆcation(1) A solution of Sodium Thiosulfate (1 in 10) responds
to the Qualitative Tests for thiosulfate.(2) A solution of Sodium Thiosulfate (1 in 10) responds
to the Qualitative Tests for sodium salt.
Change the Loss on drying to read:
Loss on drying 32.0 – 37.0z (1 g, in vacuum, 40 – 459C,16 hours).
Change the Assay to read:
Assay Weigh accurately about 0.4 g of Sodium Thiosul-fate, previously dried, dissolve in 30 mL of water, and titratewith 0.05 mol/L iodine VS (indicator: 1 mL of starch TS).
Each mL of 0.05 mol/L iodine VS=15.81 mg of Na2S2O3
Tegafurテガフール
Add the following next to Identiˆcation:
pH Dissolve 0.5 g of Tegafur in 50 mL of water: the pH ofthis solution is between 4.2 and 5.2.
Testosterone Propionateプロピオン酸テストステロン
Change the Description to read:
Description Testosterone Propionate occurs as white topale yellow crystals or crystalline powder.
It is freely soluble in methanol and in ethanol (95), andpractically insoluble in water.
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Testosterone Propionate in ethanol (95) (1 in 100,000) asdirected under the Ultraviolet-visible Spectrophotometry,and compare the spectrum with the Reference Spectrum orthe spectrum of a solution of Testosterone PropionateReference Standard prepared in the same manner as thesample solution: both spectra exhibit similar intensities ofabsorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofTestosterone Propionate, previously dried, as directed in thepotassium bromide disk method under the InfraredSpectrophotometry, and compare the spectrum with theReference Spectrum or the spectrum of TestosteronePropionate Reference Standard: both spectra exhibit similarintensities of absorption at the same wave numbers.
Change the Optical rotation to read:
Optical rotation [a]D20: +83 –+909 (after drying, 0.1 g,ethanol (95), 10 mL, 100 mm).
Change the Purity to read:
Purity Other steroids—Dissolve 40 mg of TestosteronePropionate in 2 mL of ethanol (95), and use this solution asthe sample solution. Pipet 1 mL of this solution, add ethanol(95) to make exactly 100 mL, and use this solution as thestandard solution. Perform the test with these solutions asdirected under the Thin-layer Chromatography. Spot 10 mLeach of the sample solution and the standard solution on aplate of silica gel with ‰uorescent indicator for thin-layerchromatography. Develop the plate with a mixture of chlo-roform and diethylamine (19:1) to a distance of about 15cm, and air-dry the plate. Examine under ultraviolet light(main wavelength: 254 nm): the spot other than the principal

17391739Supplement II, JPXIV O‹cial Monographs for Part I
spot from the sample solution is not more intense than thespot from the standard solution.
Change the Assay to read:
Assay Weigh accurately each about 10 mg of TestosteronePropionate and Testosterone Propionate ReferenceStandard, previously dried, and dissolve in methanol tomake exactly 100 mL. To exactly 5 mL of these solutionsadd exactly 5 mL of the internal standard solution andmethanol to make 20 mL, and use these solutions as thesample solution and the standard solution. Perform the testwith 5 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of testosteronepropionate to that of the internal standard.
Amount (mg) of C22H32O3
=WS×QT
QS
WS: Amount (mg) of Testosterone Propionate ReferenceStandard
Internal standard solution—A solution of Progesterone inmethanol (9 in 100,000).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 241 nm).
Column: A stainless steel column 4.6 mm in inside di-ameter and 15 cm in length, packed with octadecylsilanizedsilica gel for liquid chromatography (5 mm in particle di-ameter).
Column temperature: A constant temperature of about359C.
Mobile phase: A mixture of acetonitrile and water (7:3).Flow rate: Adjust the ‰ow rate so that the retention time
of testosterone propionate is about 10 minutes.System suitability—
System performance: When the procedure is run with 5 mLof the standard solution under the above operating condi-tions, the internal standard and testosterone propionate areeluted in this order with the resolution between these peaksbeing not less than 9.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of testosterone propionate to that of theinternal standard is not more than 1.0z.
Testosterone Propionate Injectionプロピオン酸テストステロン注射液
Change the origin/limits of content to read:
Testosterone Propionate Injection is an oily solutionfor injection.
It contains not less than 92.5z and not more than107.5z of the labeled amount of testosteronepropionate (C22H32O3: 344.49).
Change the Identiˆcation to read:
Identiˆcation Dissolve the residue obtained as directed inthe procedure in the Assay in exactly 20 mL of methanol,and use this solution as the sample solution. Separately,dissolve 1 mg of Testosterone Propionate ReferenceStandard in 10 mL of methanol, and use this solution as thestandard solution. Perform the test with these solutions asdirected under the Thin-layer Chromatography. Spot 10 mLeach of the sample solution and the standard solution on aplate of silica gel with ‰uorescent indicator for thin-layerchromatography. Develop the plate with a mixture ofchloroform and diethylamine (19:1) to a distance of about15 cm, and air-dry the plate. Examine under ultraviolet light(main wavelength: 254 nm): the Rf values of the principalspot with the sample solution and of the spot with the stan-dard solution are not diŠerent each other.
Add the following next to Identiˆcation:
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Change the Assay to read:
Assay(i) Chromatographic tube A glass tube about 1 cm in
inside diameter and about 18 cm in length, with a glass ˆlter(G3) at the lower end.
(ii) Chromatographic column To about 2 g of silica gelfor liquid chromatography add 5 mL of dichloromethane,and mix gently. Transfer and wash into the chromatographictube with the aid of dichloromethane, allow to elute thedichloromethane through the column, and put a ˆlter paperon the upper end of the silica gel.
(iii) Standard solution Weigh accurately about 10 mgof Testosterone Propionate Reference Standard, previouslydried at 1059C for 4 hours, and dissolve in methanol tomake exactly 100 mL. Pipet 5 mL of this solution, addexactly 5 mL of the internal standard solution and methanolto make exactly 20 mL.
(iv) Sample stock solution To exactly a volume ofTestosterone Propionate Injection, equivalent to about20 mg of testosterone propionate (C22H32O3), adddichloromethane to make exactly 20 mL.
(v) Procedure Transfer exactly 2 mL of the samplestock solution into the chromatographic column, andelute to the upper surface of the silica gel. Wash the innersurface of the chromatographic tube with 15 mL of

17401740 Supplement II, JPXIVO‹cial Monographs for Part I
dichloromethane, elute to the upper surface of the silica gel,and discard the eŒuent. Elute 15 mL of a mixture ofdichloromethane and methanol (39:1), discard the ˆrst5 mL of the eŒuent, and collect the subsequent eŒuent.Wash the lower part of the column with a few amount ofdichloromethane, combine the washings and the eŒuent,and evaporate the solvent under reduced pressure. Dissolvethe residue so obtained with methanol to make exactly 20mL. Pipet 5 mL of this solution, add exactly 5 mL of the in-ternal standard solution and methanol to make exactly 20mL, and use this solution as the sample solution. Performthe test with 5 mL each of the sample solution and the stan-dard solution as directed in the Assay under TestosteronePropionate.
Amount (mg) of testosterone propionate (C22H32O3)
=WS×QT
QS×2
WS: Amount (mg) of Testosterone Propionate ReferenceStandard
Thiamine Hydrochloride塩酸チアミン
Change the Purity (5) to read:
Purity(5) Related substances—Dissolve 0.10 g of Thiamine
Hydrochloride in 100 mL of the mobile phase, and use thissolution as the sample solution. Pipet 1 mL of the samplesolution, add the mobile phase to make exactly 100 mL, anduse this solution, as the standard solution. Perform the testwith exactly 10 mL each of the sample solution and the thestandard solution as directed under the Liquid Chro-matography according to the following conditions, and de-termine the area of each peak by the automatic integrationmethod: the total area of the peaks other than thiamine isnot larger than the peak area of thiamine from the standardsolution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 3 times as long as theretention time of thiamine.System suitability—
Test for required detectability: To exactly 5 mL of thestandard solution add water to make exactly 50 mL. Con-ˆrm that the peak area of thiamine obtained from 10 mL ofthis solution is equivalent to 7 to 13z of that of thiamineobtained from 10 mL of the standard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peak
area of thiamine is not more than 1.0z.
Change the Assay to read:
Assay Weigh accurately about 0.1 g each of ThiamineHydrochloride and Thiamine Hydrochloride ReferenceStandard (previously determine the water content), anddissolve them in the mobile phase to make exactly 50 mL. To10 mL each of the solutions, accurately measured, addexactly 5 mL each of the internal standard solution, add themobile phase to make 50 mL, and use these solutions as thesample solution and the standard solution. Perform the testwith 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and calculate the ratios,QT and QS, of the peak area of thiamine to that of the inter-nal standard.
Amount (mg) of C12H17ClN4OS.HCl
=WS×QT
QS
WS: Amount (mg) of Thiamine Hydrochloride ReferenceStandard, calculated on the anhydrous basis
Internal standard solution—A solution methyl benzoate inmethanol (1 in 50).Operating conditions—Detector: An ultraviolet absorption photometer(wavelength: 254 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 1.1 g of sodium 1-octanesulfonatein 1000 mL of diluted acetic acid (100) (1 in 100). To 600 mLof this solution add 400 mL of a mixture of methanol andacetonitrile (3:2).
Flow rate: Adjust the ‰ow rate so that the retention timeof thiamine is about 12 minutes.System suitability—
System performance: When the procedure is run with10 mL of the standard solution under the above operatingconditions, thiamine and the internal standard are eluted inthis order with the resolution between these peaks being notless than 6.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of thiamine to that of the internal standard isnot more than 1.0z.

17411741Supplement II, JPXIV O‹cial Monographs for Part I
Thiamylal Sodiumチアミラールナトリウム
Change the origin/limits of content to read:
Thiamylal Sodium contains not less than 97.5z andnot more than 101.0z of C12H17N2NaO2S, calculatedon the dried basis.
Change the Description to read:
Description Thiamylal Sodium occurs as light yellow crys-tals or powder.
It is very soluble in water, and freely soluble in ethanol(95).
The pH of a solution of Thiamylal Sodium (1 in 10) isbetween 10.0 and 11.0.
It is hygroscopic.It is gradually decomposed by light.Its solution in ethanol (95) (1 in 10) shows no optical
rotation.
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Thiamylal Sodium in ethanol (95) (7 in 1,000,000) as direct-ed under the Ultraviolet-visible Spectrophotometry, andcompare the spectrum with the Reference Spectrum: bothspectra exhibit similar intensities of absorption at the samewavelengths.
(2) Determine the infrared absorption spectrum ofThiamylal Sodium, previously dried, as directed in thepotassium bromide disk method under the InfraredSpectrophotometry, and compare the spectrum with theReference Spectrum: both spectra exhibit similar intensitiesof absorption at the same wave numbers.
(3) A solution of Thiamylal Sodium (1 in 10) responds tothe Qualitative Tests for sodium salt.
Change the Purity (3) to read:
Purity(3) Related substances—Dissolve 0.10 g of Thiamylal
Sodium in 10 mL of ethanol (95), and use this solution as thesample solution. Pipet 1 mL and 3 mL of the sample solu-tion, add ethanol (95) to make exactly 200 mL, and use thesesolutions as the standard solution (1) and the standard solu-tion (2). Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 10 mL each ofthe sample solution, the standard solution (1) and thestandard solution (2) on a plate of silica gel for thin-layerchromatography, develop with a mixture of toluene,methanol and ethyl acetate (40:7:3) to a distance of about12 cm, and air-dry the plate. Allow the plate to stand iniodine vapor for a night: the spot appeared around Rf 0.1obtained with the sample solution is not more intense than
the spot with the standard solution (2), and the spot otherthan the principal spot, the spot at origin and the spot men-tioned above obtained with the sample solution is not moreintense than the spot with the standard solution (1).
Change the Assay to read:
Assay Weigh accurately about 0.25 g of Thiamylal Sodi-um, dissolve in 50 mL of methanol and 5 mL of dilutehydrochloric acid, and add methanol to make exactly 100mL. Pipet 10 mL of this solution, and add methanol tomake exactly 100 mL. Pipet 5 mL of this solution, addexactly 10 mL of the internal standard solution and themobile phase to make 200 mL, and use this solution as thesample solution. Separately, weigh accurately about 23 mgof Thiamylal Reference Standard, previously dried at 1059Cfor 1 hour, dissolve in 50 mL of methanol and 0.5 mL ofdilute hydrochloric acid, and add methanol to make exactly100 mL. Pipet 5 mL of this solution, add exactly 10 mL ofthe internal standard solution and the mobile phase to make200 mL, and use this solution as the standard solution.Perform the test with 20 mL each of the sample solution andthe standard solution as directed under the Liquid Chro-matography according to the following conditions, anddetermine the ratios, QT and QS, of the peak area ofthiamylal to that of the internal standard.
Amount (mg) of C12H17N2NaO2S=WS×QT
QS×10×1.0864
WS: Amount (mg) of Thiamylal Reference Standard
Internal standard solution—A solution of phenyl benzoatein methanol (3 in 500).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 289 nm).
Column: A stainless steel column about 4 mm in insidediameter and about 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of methanol and 0.05 mol/Lacetic acid-sodium acetate buŠer solution, pH 4.6 (13:7).
Flow rate: Adjust the ‰ow rate so that the retention timeof thiamylal is about 6 minutes.System suitability—
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, thiamylal and the internal standard are eluted inthis order with the resolution between these peaks being notless than 12.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of thiamylal to that of the internal standard isnot more than 1.0z.

17421742 Supplement II, JPXIVO‹cial Monographs for Part I
Change to read:
Thiamylal Sodium for Injection注射用チアミラールナトリウム
Thiamylal Sodium for Injection is a preparation forinjection which is dissolved before use.
It contains not less than 93.0z and not more than107.0z of the labeled amount of thiamylal sodium(C12H17N2NaO2S: 276.33).
Method of preparation Prepare as directed under Injec-tions, with 100 parts of Thiamylal Sodium and 7 parts ofDried Sodium Carbonate in mass.
Description Thiamylal Sodium for Injection occurs as lightyellow crystals, powder or masses.
It is hygroscopic.It is gradually decomposed by light.
Identiˆcation (1) To 1.0 g of Thiamylal Sodium forInjection add 20 mL of ethanol (95), shake vigorously, andˆlter. Dissolve the precipitate so obtained in 1 mL of water,and add 1 mL of barium chloride TS: a white precipitate isproduced. Centrifuge this solution, take oŠ the supernatantliquid, and to the precipitate add dilute hydrochloric aciddropwise: the precipitate dissolves with eŠervescence.
(2) To 50 mg of Thiamylal Sodium for Injection add100 mL of ethanol (95), shake vigorously, and ˆlter. To 3mL of the ˆltrate add ethanol (95) to make 200 mL.Determine the absorption spectrum of this solution as direct-ed under the Ultraviolet-visible Spectrophotometry: itexhibits maxima between 236 nm and 240 nm, and between287 nm and 291 nm.
pH The pH of a solution obtained by dissolving 1.0 g ofThiamylal Sodium for Injection in 40 mL of water is be-tween 10.5 and 11.5.
Purity Related substances—To 0.10 g of Thiamylal Sodi-um for Injection add 10 mL of ethanol (95), shake vigorous-ly, ˆlter, and use the ˆltrate as the sample solution. Proceedas diected in the Purity (3) under Thiamylal Sodium.
Bacterial endotoxins Less than 1.0 EU/mg.
Mass variation It meets the requirements of the MassVariation Test.
Foreign insoluble matter Perform the test according toMethod 2: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Open carefully 10 containers of Thiamylal Sodiumfor Injection, dissolve the contents with water, wash out theinside of each container with water, combine them, and addwater to make exactly V mL so that each mL contains about5 mg of thiamylal sodium (C12H17N2NaO2S). Pipet 5 mL ofthis solution, and add 0.5 mL of dilute hydrochloric acidand methanol to make exactly 100 mL. Pipet 5 mL of thissolution, add exactly 10 mL of the internal standard solutionand the mobile phase to make 200 mL, and use this solutionas the sample solution. Proceed the test with the sample so-lution as directed in the Assay under Thiamylal Sodium.
Amount (mg) of thiamylal sodium (C12H17N2NaO2S) in 1container
=WS×QT
QS×
V50×1.0864
WS: Amount (mg) of Thiamylal Reference Standard
Internal standard solution—A solution of phenyl benzoatein methanol (3 in 500).
Containers and storage Containers—Hermetic containers.Storage—Light-resistant.
Add the following:
Tiaramide Hydrochloride Tablets塩酸チアラミド錠
Tiaramide Hydrochloride Tablets contain not lessthan 95.0z and not more than 105.0z of the labeledamount of tiaramide (C15H18ClN3O3S: 355.84).
Method of preparation Prepare as directed under Tablets,with Tiaramide Hydrochloride.
Identiˆcation (1) Determine the absorption spectrum ofthe sample solution obtained in the Assay as directed underthe Ultraviolet-visible Spectrophotometry: it exhibitsmaxima between 285 nm and 289 nm, and between 292 nmand 296 nm.
(2) To a quantity of powdered Tiaramide HydrochlorideTablets, equivalent to 0.1 g of tiaramide according to thelabeled amount, add 10 mL of diluted ethanol (99.5) (7 in10), shake well, ˆlter, and use the ˆltrate as the samplesolution. Separately, dissolve 0.11 g of tiaramide hydrochlo-ride for assay in 10 mL of diluted ethanol (99.5) (7 in 10),and use this solution as the standard solution. Perform thetest with these solutions as directed under the Thin-layerChromatography. Spot 20 mL each of the sample solutionand the standard solution on a plate of silica gel for thin-lay-er chromatography, develop with a mixture of 1-butamol,water and acetic acid (100) (4:2:1) to a distance of about 10cm, and dry the plate at 1009C for 30 minutes. Spray evenlyDragendorŠ's TS for spraying followed by diluted nitric acid(1 in 50) on the plate: the principal spot obtained with thesample solution and the spot with the standard solution areyellow-red in color and have the same Rf value.

17431743Supplement II, JPXIV O‹cial Monographs for Part I
Content uniformity Perform the test according to thefollowing method: it meets the requirements of the Contentuniformity Test.
To 1 tablet of Tiaramide Hydrochloride Tablets add avolume of 0.1 mol/L hydrochloric acid TS, equivalent to3/5 volume of V mL which makes a solution so that each mLcontains about 1 mg of tiaramide (C15H18ClN3O3S) accord-ing to the labeled amount, shake for 60 minutes, then add0.1 mol/L hydrochloric acid TS to make exactly V mL, andˆlter. Discard the ˆrst 20 mL of the ˆltrate, pipet 5 mL ofthe subsequent ˆltrate, add water to make exactly 100 mL,and use this solution as the sample solution. Separately,weigh accurately about 55 mg of tiaramide hydrochloridefor assay, previously dried at 1059C for 3 hours, and dis-solve in 0.1 mol/L hydrochloric acid TS to make exactly 50mL. Pipet 5 mL of this solution, add water to make exactly100 mL, and use this solution as the standard solution.Determine the absorbances, AT and AS, of the sample solu-tion and the standard solution at 294 nm as directed underthe Ultraviolet-visible Spectrophotometry.
Amount (mg) of tiaramide (C15H18ClN3O3S)
=WS×AT
AS×
V50×0.907
WS: Amount (mg) of tiaramide hydrochloride for assay
Dissolution Perform the test with 1 tablet of TiaramideHydrochloride Tablets at 50 revolutions per minute accord-ing to Method 2 under the Dissolution Test, using 900 mL ofwater as the dissolution medium. Withdraw 20 mL or moreof the dissolution medium 15 minutes after starting the testfor a 50-mg tablet or 30 minutes after starting the test for a100-mg tablet, and ˆlter through a membrane ˆlter with poresize of not more than 0.5 mm. Discard the ˆrst 10 mL of theˆltrate, pipet V mL of the subsequent ˆltrate, add water tomake exactly V? mL so that each mL contains about 56 mg oftiaramide (C15H18ClN3O3S) according to the labeled amount,and use this solution as the sample solution. Separately,weigh accurately about 15 mg of tiaramide hydrochloridefor assay, previously dried at 1059C for 3 hours, and dis-solve in water to make exactly 50 mL. Pipet 5 mL of thissolution, add water to make exactly 25 mL, and use this so-lution as the standard solution. Determine the absorbances,AT and AS, at 294 nm of the sample solution and the stan-dard solution as directed under the Ultraviolet-visibleSpectrophotometry: the dissolution rates for a 50-mg tabletin 15 minutes and a 100-mg tablet in 30 minutes are not lessthan 80z, respectively.
Dissolution rate (z) with respect to the labeled amount oftiaramide (C15H18ClN3O3S)
=WS×AT
AS×
V?V×
1C×360×0.907
WS: Amount (mg) of tiaramide hydrochloride for assayC: Labeled amount (mg) of tiaramide (C15H18ClN3O3S) in
1 tablet
Assay Weigh accurately the mass of more than 20Tiaramide Hydrochloride Tablets, and powder. Weigh ac-
curately an amount of the powder, equivalent to about 0.1 gof tiaramide (C15H18ClN3O3S), add 60 mL of 0.1 mol/Lhydrochloric acid TS, shake for 30 minutes, add 0.1 mol/Lhydrochloric acid TS to make exactly 100 mL, and ˆlter.Discard the ˆrst 20 mL of the ˆltrate, pipet 5 mL of the sub-sequent ˆltrate, add water to make exactly 100 mL, and usethis solution as the sample solution. Separately, weigh ac-curately about 0.11 g of tiaramide hydrochloride for assay,previously dried at 1059C for 3 hours, and dissolve in 0.1mol/L hydrochloric acid TS to make exactly 100 mL. Pipet5 mL of this solution, add water to make exactly 100 mL,and use this solution as the standard solution. Determine theabsorbances, AT and AS, of the sample solution and thestandard solution at 294 nm as directed under the Ultrav-iolet-visible Spectrophotometry.
Amount (mg) of tiaramide (C15H18ClN3O3S)
=WS×AT
AS×0.907
WS: Amount (mg) of tiaramide hydrochloride for assay
Containers and storage Containers—Tight containers.
Tinidazoleチニダゾール
Change the origin/limits of content to read:
Tinidazole, when dried, contains not less than98.5z and not more than 101.0z of C8H13N3O4S.
Change the Description to read:
Description Tinidazole occurs as a light yellow, crystallinepowder.
It is soluble in acetic anhydride and in acetone, sparinglysoluble in methanol, slightly soluble in ethanol (99.5), andvery slightly soluble in water.
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Tinidazole in methanol (1 in 50,000) as directed under theUltraviolet-visible Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofTinidazole as directed in the potassium bromide disk methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wave numbers.

17441744 Supplement II, JPXIVO‹cial Monographs for Part I
Add the following:
Tizanidine Hydrochloride塩酸チザニジン
C9H8ClN5S.HCl: 290.175-Chloro-4-[(4,5-dihydro-1H-imidazol-2-yl)amino]benzo[c][1,2,5]thiadiazole monohydrochloride[64461-82-1]
Tizanidine Hydrochloride, when dried, containsnot less than 99.0z and not more than 101.0z ofC9H8ClN5S.HCl.
Description Tizanidine Hydrochloride occurs as a white tolight yellowish white crystalline powder.
It is soluble in water, slightly soluble in ethanol (99.5), andpractically insoluble in acetic anhydride and in acetic acid(100).
Melting point: about 2909C (with decomposition).
Identiˆcation (1) Determine the absorption spectrum ofa solution of Tizanidine Hydrochloride in diluted 1 mol/Lammonia TS (1 in 10) (1 in 125,000) as directed under theUltraviolet-visible Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wavelengths.
(2) Determine the infrared absorption spectrum ofTizanidine Hydrochloride as directed in the potassium chlo-ride disk method under the Infrared Spectrophotometry,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar intensities of absorption at thesame wave numbers.
(3) A solution of Tizanidine Hydrochloride (1 in 50)responds to the Qualitative Tests for chloride.
Purity (1) Heavy metals—Proceed with 1.0 g of Tizani-dine Hydrochloride according to Method 3, and perform thetest. Prepare the control solution with 2.0 mL of StandardLead Solution (not more than 20 ppm).
(2) Related substances—Dissolve 60 mg of TizanidineHydrochloride in 10 mL of a mixture of water and acetoni-trile (17:3), and use this solution as the sample solution.Pipet 1 mL of the sample solution, add the mixture of waterand acetonitrile (17:3) to make exactly 200 mL, and use thissolution as the standard solution. Perform the test with ex-actly 10 mL of the sample solution and the standard solutionas directed under the Liquid Chromatography according tothe following conditions, and determine each peak area bythe automatic integration method: the area of the peak otherthan tizanidine is not larger than 1/5 times the peak area oftizanidine with the standard solution.Operating conditions—
Detector: An ultraviolet absorption photometer
(wavelength: 230 nm for about 3 minutes after sample injec-tion and 318 nm subsequently).
Column: A stainless steel column 4.6 mm in insidediameter and 12.5 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase A: A mixture of water and formic acid(200:1), adjusted to pH 8.5 with ammonia water (28).
Mobile phase B: A mixture of acetonitrile and the mobilephase A (4:1).
Flowing of the mobile phase: Control the gradient bymixing the mobile phases A and B as directed in the follow-ing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
0 – 10 81 ª 68 19 ª 3210 – 13 68 3213 – 26 68 ª 10 32 ª 9026 – 28 10 90
Flow rate: Adjust the ‰ow rate so that the retention timeof tizanidine is about 7 minute.
Time span of measurement: About 4 times as long as theretention time of tizanidine after the solvent peak.System suitability—
Test for required detectability: Measure exactly 2 mL ofthe standard solution, and add the mixture of water andacetonitrile (17:3) to make exactly 10 mL. Conˆrm that thepeak area of tizanidine obtained with 10 mL of this solutionis equivalent to 14 to 26z of that with 10 mL of the standardsolution.
System performance: Dissolve 2 mg each of TizanidineHydrochloride and p-toluenesulfonic acid monohydrate in100 mL of the mixture of water and acetonitrile (17:3).When the procedure is run with 10 mL of this solution underthe above operating conditions, p-toluenesulfonic acid andtizanidine are eluted in this order with the resolutionbetween these peaks being not less than 10.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tizanidine is not more than 2.0z.
Loss on drying Not more than 0.2z (1 g, 1059C, 3 hours).
Residue on ignition Not more than 0.1z (1 g).
Assay Weigh accurately about 0.2 g of TizanidineHydrochloride, previously dried, dissolve in 60 mL of amixture of acetic anhydride and acetic acid (100) (7:3) withthe aid of warming. After cooling, titrate with 0.1 mol/Lperchloric acid VS (potentiometric titration). Perform ablank determination in the same manner, and make anynecessary correction.

17451745Supplement II, JPXIV O‹cial Monographs for Part I
Each mL of 0.1 mol/L perchloric acid VS=29.02 mg of C9H8ClN5S.HCl
Containers and storage Containers—Well-closed contain-ers.
Tranexamic Acidトラネキサム酸
Change the origin/limits of content to read:
Tranexamic Acid, when dried, contains not less than98.0z and not more than 101.0z of C8H15NO2.
Change the Description to read:
Description Tranexamic Acid occurs as white crystals orcrystalline powder.
It is freely soluble in water, and practically insoluble inethanol (99.5).
Change the Identiˆcation to read:
Identiˆcation Determine the infrared absorption spectrumof Tranexamic Acid as directed in the potassium bromidedisk method under the Infrared Spectrophotometry, andcompare the spectrum with the Reference Spectrum or thespectrum of Tranexamic Acid Reference Standard: bothspectra exhibit similar intensities of absorption at the samewave numbers.
Add the following next to Identiˆcation:
pH The pH of a solution prepared by dissolving 1.0 g ofTranexamic Acid in 20 mL of water is between 7.0 and 8.0.
Change the Purity to read:
Purity (1) Clarity and color of solution—Dissolve 1.0 gof Tranexamic Acid in 10 mL of water: the solution is clearand colorless.
(2) Chloride—Perform the test with 1.0 g of TranexamicAcid. Prepare the control solution with 0.40 mL of 0.01mol/L hydrochloric acid VS (not more than 0.014z).
(3) Heavy metals—Dissolve 2.0 g of Tranexamic Acid inwater to make 20 mL, and use this solution as the samplestock solution. To 12 mL of the sample stock solution add2 mL of hydrochloric acid-ammonium acetate buŠer solu-tion, pH 3.5, mix, add 1.2 mL of thioacetamide TS , miximmediately, and use this solution as the sample solution.Separately, proceed in the same manner as above with amixture of 1 mL of Standard Lead Solution, 2 mL of thesample stock solution and 9 mL of water, and use the solu-tion so obtained as the standard solution. Separately, pro-ceed in the same manner with a mixture of 10 mL of waterand 2 mL of the sample stock solution, and use the solutionso obtained as the control solution. Conform that the colorof the standard solution is slightly darker than that of the
control solution. Compare the sample solution and the stan-dard solution 2 minutes after they are prepared: the color ofthe sample solution is not more intense than that of the stan-dard solution (not more than 10 ppm).
(4) Arsenic—Prepare the test solution by dissolving1.0 g of Tranexamic Acid in 10 mL of water, and performthe test (not more than 2 ppm).
(5) Related substances—Dissolve 0.20 g of TranexamicAcid in water to make exactly 20 mL, and use this solutionas the sample solution. Pipet 5 mL of the sample solution,and add water to make exactly 100 mL. Pipet 1 mL of thissolution, add water to make exactly 10 mL, and use thissolution as the standard solution. Perform the test with ex-actly 20 mL each of the sample solution and the standard so-lution as directed under the Liquid Chromatography accord-ing to the following conditions, and determine each peakarea by the automatic integration method: the area multi-plied by relative response factor 1.2 of the peak, having therelative retention time of about 1.5 with respect to tranexam-ic acid, is not more than 2/5 of the peak area of tranexamicacid from the standard solution, and the area of the peak,having the relative retention time of about 2.1 with respectto tranexamic acid, is not more than 1/5 of the peak area oftranexamic acid from the standard solution. The area ofeach peak other than tranexamic acid and other than thepeaks mentioned above is not more than 1/5 of the peakarea of tranexamic acid from the standard solution. For thiscomparison, use the area of the peaks, having the relativeretention time of about 1.1 and about 1.3, after multiplyingby their relative response factors 0.005 and 0.006, respec-tively. The total area of the peaks other than tranexamic acidis not more than the peak area of tranexamic acid from thestandard solution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 3 times as long as theretention time of tranexamic acid after the solvent peak.System suitability—
Test for required detectability: To exactly 5 mL of thestandard solution add water to make exactly 25 mL. Con-ˆrm that the peak area of tranexamic acid obtained from20 mL of this solution is equivalent to 14 to 26z of that from20 mL of the standard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tranexamic acid is not more than 7z.
Change the Assay to read:
Assay Weigh accurately about 50 mg each of TranexamicAcid and Tranexamic Acid Reference Standard, previouslydried, dissolve in water to make exactly 25 mL, and use thesesolutions as the sample solution and the standard solution.

17461746 Supplement II, JPXIVO‹cial Monographs for Part I
Perform the test with exactly 20 mL each of the samplesolution and the standard solution as directed under theLiquid Chromatography according to the following condi-tions, and determine the peak areas, AT and AS, of tranex-amic acid.
Amount (mg) of C8H15NO2=WS×AT
AS
WS: Amount (mg) of Tranexamic Acid ReferenceStandard
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 220 nm).Column: A stainless steel column 6.0 mm in inside
diameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: Dissolve 11.0 g of sodium dihydrogenphosphate in 500 mL of water, and add 5 mL of triethyla-mine and 1.4 g of sodium lauryl sulfate. Adjust the pH to2.5 with phosphoric acid or diluted phosphoric acid (1 in10), add water to make 600 mL, and add 400 mL ofmethanol.
Flow rate: Adjust the ‰ow rate so that the retention timeof tranexamic acid is about 20 minutes.System suitability—
System performance: To 5 mL of the standard solutionadd 1 mL of a solution of 4-(aminomethyl)benzoic acid (1 in10,000) and water to make 50 mL. When the procedure isrun with 20 mL of this solution under the above operatingconditions, tranexamic acid and 4-(aminomethyl)benzoicacid are eluted in this order with the resolution between thesepeaks being not less than 5.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tranexamic acid is not more than 0.6z.
Add the following:
Tranexamic Acid Capsulesトラネキサム酸カプセル
Tranexamic Acid Capsules contain not less than95.0z and not more than 105.0z of the labeledamount of tranexamic acid (C8H15NO2: 157.21).
Method of preparation Prepare as directed underCapsules, with Tranexamic Acid.
Identiˆcation Take an amount of powdered contents ofTranexamic Acid Capsules, equivalent to 0.5 g of Tranex-amic Acid according to the labeled amount, add 50 mL ofwater, shake well, and ˆlter. To 5 mL of the ˆltrate add1 mL of ninhydrin TS, and heat for 3 minutes: a dark purple
color develops.
Mass variation It meets the requirements of the MassVariation Test.
Dissolution Being speciˆed separately.
Assay Weigh accurately the mass of the contents of notless than 20 Tranexamic Acid Capsules, and powder. Weighaccurately an amount of the powder, equivalent to about 0.1g of tranexamic acid (C8H15NO2), add 30 mL of water, shakewell, and add water to make exactly 50 mL. Centrifuge, ˆlterthe supernatant liquid through a membrane ˆlter with poresize of not more than 0.45 mm, discard the ˆrst 10 mL of theˆltrate, and use the subsequent ˆltrate as the sample solu-tion. Separately, weigh accurately about 50 mg of Tranex-amic Acid Reference Standard, previously dried at 1059Cfor 2 hours, dissolve in water to make exactly 25 mL, anduse this solution as the standard solution. Perform the testwith exactly 30 mL each of the sample solution and the stan-dard solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine thepeak areas, AT and AS, of tranexamic acid.
Amount (mg) of tranexamic acid (C8H15NO2)
=WS×AT
AS×2
WS: Amount (mg) of Tranexamic Acid Reference Stan-dard
Operating conditions—Detector, column, and mobile phase: Proceed as directed
in the operating conditions in the Assay under TranexamicAcid.
Column temperature: A constant temperature of about359C.
Flow rate: Adjust the ‰ow rate so that the retention timeof tranexamic acid is about 16 minutes.System suitability—
System performance: To 5 mL of the standard solutionadd 1 mL of a solution of 4-(aminomethyl)benzoic acid (1 in10,000) and water to make 50 mL. When the procedure isrun with 30 mL of this solution under the above operatingconditions, tranexamic acid and 4-(aminomethyl)benzoicacid are eluted in this order with the resolution between thesepeaks being not less than 3.
System repeatability: When the test is repeated 6 timeswith 30 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tranexamic acid is not more than 1.0z.
Containers and storage Containers—Tight containers.

17471747Supplement II, JPXIV O‹cial Monographs for Part I
Add the following:
Tranexamic Acid Injectionトラネキサム酸注射液
Tranexamic Acid Injection is an aqueous injection.It contains not less than 95.0z and not more than
105.0z of the labeled amount of tranexamic acid(C8H15NO2: 157.21).
Method of preparation Prepare as directed underInjections, with Tranexamic Acid.
Description Tranexamic Acid Injection is a clear andcolorless liquid.
Identiˆcation To a volume of Tranexamic Acid Injection,equivalent to 50 mg of Tranexamic Acid according to thelabeled amount, add water to make 5 mL, add 1 mL ofninhydrin TS, and heat: a dark purple color develops.
pH 7.0 – 8.0
Bacterial endotoxins Not more than 0.12 EU/mg.
Actual volume It meets the requirements of Injections.
Foreign insoluble matter Perform the test according toMethod 1: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Take accurately a volume of Tranexamic AcidInjection, equivalent to about 0.1 g of tranexamic acid(C8H15NO2), add water to make exactly 50 mL, and use thissolution as the sample solution. Separately, weigh accuratelyabout 50 mg of Tranexamic Acid Reference Standard,previously dried at 1059C for 2 hours, dissolve in water tomake exactly 25 mL, and use this solution as the standardsolution. Perform the test with exactly 30 mL each of thesample solution and the standard solution as directed underthe Liquid Chromatography according to the followingconditions, and determine the peak areas, AT and AS, oftranexamic acid.
Amount (mg) of tranexamic acid (C8H15NO2)=WS×AT
AS
WS: Amount (mg) of Tranexamic Acid ReferenceStandard
Operating conditions—Detector, column, and mobile phase: Proceed as directed
in the operating conditions in the Assay under TranexamicAcid.
Column temperature: A constant temperature of about
359C.Flow rate: Adjust the ‰ow rate so that the retention time
of tranexamic acid is about 16 minutes.System suitability—
System performance: To 5 mL of the standard solutionadd 1 mL of a solution of 4-(aminomethyl)benzoic acid (1 in10,000) and water to make 50 mL. When the procedure isrun with 30 mL of this solution under the above operatingconditions, tranexamic acid and 4-(aminomethyl)benzoicacid are eluted in this order with the resolution between thesepeaks being not less than 3.
System repeatability: When the test is repeated 6 timeswith 30 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tranexamic acid is not more than 1.0z.
Containers and storage Containers—Hermetic containers.
Add the following:
Tranexamic Acid Tabletsトラネキサム酸錠
Tranexamic Acid Tablets contain not less than95.0z and not more than 105.0z of the labeledamount of tranexamic acid (C8H15NO2: 157.21).
Method of preparation Prepare as directed under Tablets,with Tranexamic Acid.
Identiˆcation To an amount of powdered TranexamicAcid Tablets, equivalent to 0.5 g of Tranexamic Acidaccording to the labeled amount, add 50 mL of water, shakewell, and ˆlter. To 5 mL of the ˆltrate add 1 mL ofninhydrin TS, and heat for 3 minutes: a dark purple colordevelops.
Mass variation It meets the requirements of the MassVariation Test.
Dissolution Being speciˆed separately.
Assay Weigh accurately the mass of not less than 20Tranexamic Acid Tablets, and powder. Weigh accurately aquantity of the powder, equivalent to about 5 g of tranexam-ic acid (C8H15NO2), add 150 mL of water, disintegrate thetablets completely with the aid of ultrasonic waves, and addwater to make exactly 200 mL. Centrifuge, pipet 4 mL of thesupernatant liquid, and add water to make exactly 50 mL.Filter through a membrane ˆlter with pore size of not morethan 0.45 mm, discard the ˆrst 10 mL of the ˆltrate, and usethe subsequent ˆltrate as the sample solution. Separately,weigh accurately about 50 mg of Tranexamic Acid ReferenceStandard, previously dried at 1059C for 2 hours, dissolve inwater to make exactly 25 mL, and use this solution as thestandard solution. Perform the test with exactly 30 mL eachof the sample solution and the standard solution as directedunder the Liquid Chromatography according to the follow-ing conditions, and determine the peak areas, AT and AS, of

17481748 Supplement II, JPXIVO‹cial Monographs for Part I
tranexamic acid.
Amount (mg) of tranexamic acid (C8H15NO2)
=WS×AT
AS×100
WS: Amount (mg) of Tranexamic Acid ReferenceStandard
Operating conditions—Detector, column, and mobile phase: Proceed as directed
in the operating conditions in the Assay under TranexamicAcid.
Column temperature: A constant temperature of about359C.
Flow rate: Adjust the ‰ow rate so that the retention timeof tranexamic acid is about 16 minutes.System suitability—
System performance: To 5 mL of the standard solutionadd 1 mL of a solution of 4-(aminomethyl)benzoic acid (1 in10,000) and water to make 50 mL. When the procedure isrun with 30 mL of this solution under the above operatingconditions, tranexamic acid and 4-(aminomethyl)benzoicacid are eluted in this order with the resolution between thesepeaks being not less than 3.
System repeatability: When the test is repeated 6 timeswith 30 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of tranexamic acid is not more than 1.0z.
Containers and storage Containers—Tight containers.
Trichlormethiazideトリクロルメチアジド
Change the origin/limits of the content to read:
Trichlormethiazide, when dried, contains not lessthan 97.5z and not more than 102.0z ofC8H8Cl3N3O4S2.
Change the Description to read:
Description Trichlormethiazide occurs as a white powder.It is freely soluble in N,N-dimethylformamide and in
acetone, slightly soluble in acetonitrile and in ethanol (95),and practically insoluble in water.
A solution of Trichlormethiazide in acetone (1 in 50)shows no optical rotation.
Melting point: about 2709C (with decomposition).
Change the Identiˆcation to read:
Identiˆcation(1) Determine the absorption spectrum of a solution of
Trichlormethiazide in ethanol (95) (3 in 250,000) as directedunder the Ultraviolet-visible Spectrophotometry, andcompare the spectrum with the Reference Spectrum or thespectrum of a solution of Trichlormethiazide Reference
Standard prepared in the same manner as the samplesolution: both spectra exhibit similar intensities of absorp-tion at the same wavelengths.
(2) Determine the infrared absorption spectrum ofTrichlormethiazide as directed in the potassium bromidedisk method under the Infrared Spectrophotometry, andcompare the spectrum with the Reference Spectrum or thespectrum of Trichlormethiazide Reference Standard: bothspectra exhibit similar intensities of absorption at the samewave numbers.
(3) Perform the test with Trichlormethiazide as directedunder the Flame Coloration Test (2): a green color appears.
Change the Purity (4) and (5) to read:
(4) Arsenic—Prepare the test solution with 0.6 g ofTrichlormethiazide according to Method 5, using 20 mL ofN,N-dimethylformamide, and perform the test (not morethan 3.3 ppm).
(5) Related substances―Dissolve 25 mg of Trichlor-methiazide in 50 mL of acetonitrile, and use the solution asthe sample solution. Perform the test with 10 mL of the sam-ple solution as directed under the Liquid Chromatographyaccording to the following conditions, determine each peakarea by the automatic integration method, and calculate theamount of related substances by the area percentagemethod: the amount of 4-amino-6-chlorobenzene-1,3-disul-fonamide, having the relative retention time of about 0.3with respect to trichlormethiazide, is not more than 2.0z,and the total amount of the related substances is not morethan 2.5z.Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 268 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with phenylsilanizedsilica gel for liquid chromatography (5 mm in particlediameter).
Column temperature: A constant temperature of about259C.
Mobile phase A: A mixture of diluted phosphoric acid(1 in 1000) and acetonitrile (3:1).
Mobile phase B: A mixture of acetonitrile and dilutedphosphoric acid (1 in 1000) (3:1).
Flowing of the mobile phase: Control the gradient bymixing the mobile phases A and B as directed in the follow-ing table.
Time after injectionof sample (min)
Mobile phaseA (volz)
Mobile phaseB (volz)
0 – 10 100 010 – 20 100 ª 0 0 ª 100
Flow rate: 1.5 mL per minuteTime span of measurement: About 2.5 times as long as the
retention time of trichlormethiazide after the solvent peak.System suitability—
Test for required detectability: To exactly 1 mL of the

17491749Supplement II, JPXIV O‹cial Monographs for Part I
sample solution add acetonitrile to make exactly 50 mL, anduse this solution as the solution for system suitability test.Pipet 1 mL of the solution, and add acetonitrile to make ex-actly 20 mL. Conˆrm that the peak area of trichlor-methiazide obtained from 10 mL of this solution is equiva-lent to 3.5 to 6.5z of that of trichlormethiazide obtainedfrom 10 mL of the solution for system suitability test.
System performance: To 5 mL of the solution for systemsuitability test add 5 mL of water, and warm in a water bathat 609C for 30 minutes. When the procedure is run with10 mL of this solution, after cooling, under the above operat-ing conditions, 4-amino-6-chlorobenzene-1,3-disulfonamideand trichlormethiazide are eluted in this order, the relativeretention time of 4-amino-6-chlorobenzene-1,3-disul-fonamide with respect to trichlormethiazide is about 0.3,and the number of theoretical plates and the symmetry fac-tor of the peak of trichlormethiazide are not less than 5000and not more than 1.2, respectively.
System repeatability: When the test is repeated 3 timeswith 10 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of trichlormethiazide is not more than2.0z.
Change the Assay to read:
Assay Weigh accurately about 25 mg of Trichlor-methiazide and Trichlormethiazide Reference Standard,previously dried, and dissolve separately in exactly 20 mL ofthe internal standard solution. To 1 mL of these solutionsadd acetonitrile to make 20 mL, and use these solutions asthe sample solution and the standard solution. Perform thetest with 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatographyaccording to the following conditions, and determine theratios, QT and QS, of the peak area of trichlormethiazide tothat of the internal standard.
Amount (mg) of C8H8Cl3N3O4S2=WS×QT
QS
WS: Amount (mg) of Trichlormethiazide Reference Stan-dard
Internal standard solution—A solution of 3-nitrophenol inacetonitrile (1 in 800).Operating conditions—
Detector: An ultraviolet absorption photometer(wavelength: 268 nm).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with phenylsilanizedsilica gel for liquid chromatography (5 mm in particlediameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of diluted phosphoric acid (1 in1000) and acetonitrile (3:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof trichlormethiazide is about 8 minutes.System suitability—
System performance: When the procedure is run with
10 mL of the standard solution under the above operatingconditions, the internal standard and trichlormethiazide areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of trichlormethiazide to that of the internalstandard is not more than 1.0z.
Add the following:
Trichlormethiazide Tabletsトリクロルメチアジド錠
Trichlormethiazide Tablets contain not less than93.0z and not more than 107.0z of the labeledamount of trichlormethiazide (C8H8Cl3N3O4S2:380.66).
Method of preparation Prepare as directed under Tablets,with Trichlormethiazide.
Identiˆcation To an amount of pulverized Trichlor-methiazide Tablets, equivalent to 4 mg of Trichlor-methiazide according to the labeled amount, add 10 mL ofacetone, shake vigorously for 5 minutes, centrifuge, and usethe supernatant liquid as the sample solution. Separately,dissolve 4 mg of Trichlormethiazide Reference Standard in10 mL of acetone, and use this solution as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 5 mL each ofthe sample solution and the standard solution on a plate ofsilica gel with ‰uorescent indicator for thin-layer chro-matography. Develop the plate with a mixture of ethylacetate, hexane and methanol (10:4:1) to a distance of about10 cm, and air-dry the plate. Examine under ultraviolet light(main wavelength: 254 nm): the principal spots from thesample solution and the standard solution show the same Rfvalue.
Purity Related substances—Pulverize a suitable amount ofTrichlormethiazide Tablets in an agate mortar. Take anamount of the powder, equivalent to 10 mg of Trichlor-methiazide according to the labeled amount, add 20 mL ofacetonitrile, shake vigorously for 15 minutes, centrifuge,and use the supernatant liquid as the sample solution. Per-form the test with 10 mL of the sample solution as directedunder the Liquid Chromatography according to the follow-ing conditions, determine each peak area by the automaticintegration method, and calculate the amount of eachrelated substance by the area percentage method: theamount of 4-amino-6-chlorobenzene-1,3-disulfoneamide,having the relative retention time of about 0.3 with respectto trichlormethiazide, is not more than 4.0z, and the totalamount of the peaks other than trichlormethiazide is notmore than 5.0z.

17501750 Supplement II, JPXIVO‹cial Monographs for Part I
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 268 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with phenylsilanizedsilica gel for liquid chromatography (5 mm in particlediameter).
Column temperature: A constant temperature of about259C.
Mobile phase A: A mixture of diluted phosphoric acid(1 in 1000) and acetonitrile (3:1).
Mobile phase B: A mixture of acetonitrile and dilutedphosphoric acid (1 in 1000) (3:1).
Flowing of the mobile phase: Control the gradient bymixing the mobile phases A and B as directed in the follow-ing table.
Time after injectionof sample (min)
Mobile phaseA (z)
Mobile phaseB (z)
0 – 10 100 010 – 20 100 ª 0 0 ª 100
Flow rate: 1.5 mL/minuteTime span of measurement: About 2.5 times as long as the
retention time of trichlormethiazide after the solvent peak.System suitability—
Test for required detectability: Dissolve 25 mg ofTrichlormethiazide in 50 mL of acetonitrile. Pipet 1 mL ofthis solution, add acetonitrile to make exactly 50 mL, anduse this solution as the solution for system suitability test.Pipet 1 mL of the solution for system suitability test, andadd acetonitrile to make exactly 20 mL. Conˆrm that thepeak area of trichlormethiazide obtained from 10 mL of thissolution is equivalent to 3.5 to 6.5z of that obtained from10 mL of the solution for system suitability test.
System performance: To 5 mL of the solution for systemsuitability test add 5 mL of water, and warm in a water bathof 609C for 30 minutes. When the procedure is run with 10mL of this solution, after cooling, under the above operatingconditions, 4-amino-6-chlorobenzene-1,3-disulfonamideand trichlormethiazide are eluted in this order, the relativeretention time of 4-amino-6-chlorobenzene-1,3-disul-fonamide with respect to trichlormethiazide is about 0.3,and the number of theoretical plates and the symmetry fac-tor of the peak of trichlormethiazide are not less than 5000and not more than 1.2, respectively.
System repeatability: When the test is repeated 3 timeswith 10 mL of the solution for system suitability test underthe above operating conditions, the relative standard devia-tion of the peak area of trichlormethiazide is not more than2.0z.
Content uniformity Perform the test according to the fol-lowing method: it meets the requirements of the ContentUniformity Test. To one tablet of TrichlormethiazideTablets add 5 mL of diluted phosphoric acid (1 in 50) to dis-integrate. Add exactly an amount of the internal standardsolution, equivalent to 10 mL per 2 mg of trichlormethiazide
(C8H8Cl3N3O4S2) according to the labeled amount, addacetonitrile to make 25 mL, shake vigorously for 15minutes, and centrifuge. To an amount of the supernatantliquid add the mobile phase to make a solution so that it con-tains about 40 mg of trichlormethiazide (C8H8Cl3N3O4S2) ineach mL, and use this solution as the sample solution.Separately, weigh accurately about 20 mg of Trichlor-methiazide Reference Standard, previously dried at 1059Cfor 3 hours, and dissolve in acetonitrile to make exactly 100mL. Pipet 5 mL of this solution, add exactly 5 mL of the in-ternal standard solution, add 10 mL of acetonitrile and 5 mLof diluted phosphoric acid (1 in 50), and use this solution asthe standard solution. Perform the test with 20 mL each ofthe sample solution and the standard solution as directed un-der the Liquid Chromatography according to the conditionsdescribed in Assay, and determine the ratios, QT and QS, ofthe peak area of trichlormethiazide to that of the internalstandard.
Amount (mg) of trichlormethiazide (C8H8Cl3N3O4S2)
=WS×QT
QS×C×
120
WS: Amount (mg) of Trichlormethiazide ReferenceStandard
C: Labeled amount (mg) of trichlormethiazide(C8H8Cl3N3O4S2) per tablet
Internal standard solution—A solution of 3-nitrophenol inacetonitrile (1 in 5000).
Dissolution Perform the test with 1 tablet of Trichlor-methiazide Tablets at 50 revolutions per minute according toMethod 2 under the Dissolution Test, using 900 mL of wateras the dissolution medium. Withdraw 20 mL or more of thedissolution medium 15 minutes after starting the test, andˆlter through a membrane ˆlter with pore size of not morethan 0.45 mm. Discard the ˆrst 10 mL of the ˆltrate, pipetthe subsequent V mL of the ˆltrate, add diluted phosphoricacid (1 in 50) to make exactly V? mL so that each mL con-tains about 1.1 mg of trichlormethiazide (C8H8Cl3N3O4S2)according to the labeled amount, and use this solution as thesample solution. Separately, weigh accurately about 22 mgof Trichlormethiazide Reference Standard, previously driedat 1059C for 3 hours, and dissolve in acetonitrile to make ex-actly 200 mL. Pipet 2 mL of this solution, add diluted phos-phoric acid (1 in 50) to make exactly 200 mL, and use this so-lution as the standard solution. Perform the test with exactly40 mL each of the sample solution and the standard solutionas directed under the Liquid Chromatography according tothe following conditions, and determine the peak areas, ATa
and ASa, of trichlormethiazide obtained with the sample so-lution and the standard solution, and the area, ATb, of thepeak, having the relative retention time of about 0.3 withrespect to trichlormethiazide, obtained with the sample solu-tion. The dissolution rate in 15 minutes is not less than 75z.
Dissolution rate (z) with respect to the labeled amount oftrichlormethiazide (C8H8Cl3N3O4S2)

17511751Supplement II, JPXIV O‹cial Monographs for Part I
=WS×ATa+ATb×0.95
ASa×
V?V×
1C×
92
WS: Amount (mg) of Trichlormethiazide Reference Stan-dard
C: Labeled amount (mg) of trichlormethiazide(C8H8Cl3N3O4S2) per tablet
Operating conditions—Proceed as directed in the operating conditions in the
Assay.System suitability—
System performance: Dissolve 25 mg of Trichlor-methiazide in 50 mL of acetonitrile. To 1 mL of this solutionadd acetonitrile to make 50 mL. To 5 mL of this solutionadd 5 mL of water, and heat at 609C in a water bath for 30minutes. After cooling, when the procedure is run with10 mL of this solution under the above operating conditions,4-amino-6-chlorobenzene-1,3-disulfonamide and trichlor-methiazide are eluted in this order, the relative retentiontime of 4-amino-6-chlorobenzene-1,3-disulfonamide withrespect to trichlormethiazide is about 0.3, and the number oftheoretical plates and the symmetry factor of the peak oftrichlormethiazide are not less than 5000 and not more than1.2, respectively.
System repeatability: When the test is repeated 6 timeswith 40 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of trichlormethiazide is not more than 2.0z.
Assay Weigh accurately the mass of not less than 20Trichlormethiazide Tablets, and pulverize the tablets in anagate mortar. Weigh accurately an amount of the powder,equivalent to about 2 mg of trichlormethiazide(C8H8Cl3N3O4S2) according to the labeled amount, add 5 mLof diluted phosphoric acid (1 in 50) and exactly 10 mL of theinternal standard solution, add 10 mL of acetonitrile, shakevigorously for 15 minutes, and centrifuge. To 2 mL of thesupernatant liquid add 2 mL of the mobile phase, and usethis solution as the sample solution. Separately, weigh ac-curately about 40 mg of Trichlormethiazide Reference Stan-dard, previously dried at 1059C for 3 hours, and dissolve inacetonitrile to make exactly 200 mL. Pipet 5 mL of this solu-tion, add exactly 5 mL of the internal standard solution, add10 mL of acetonitrile and 5 mL of diluted phosphoric acid (1in 50), and use this solution as the standard solution. Per-form the test with 20 mL each of the sample solution and thestandard solution as directed under the Liquid Chro-matography according to the following conditions, and de-termine the ratios, QT and QS, of the peak area of trichlor-methiazide to that of the internal standard.
Amount (mg) of trichlormethiazide (C8H8Cl3N3O4S2)
=WS×QT
QS×
120
WS: Amount (mg) of Trichlormethiazide ReferenceStandard
Internal standard solution—A solution of 3-nitrophenol inacetonitrile (1 in 5000).
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 268 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with phenylsilanizedsilica gel for liquid chromatography (5 mm in particlediameter).
Column temperature: A constant temperature of about259C.
Mobile phase: A mixture of diluted phosphoric acid (1 in1000) and acetonitrile (3:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof trichlormethiazide is about 8 minutes.System suitability—
System performance: When the procedure is run with20 mL of the standard solution under the above operatingconditions, the internal standard and trichlormethiazide areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the ratio ofthe peak area of trichlormethiazide to that of the internalstandard is not more than 1.0z.
Containers and storage Containers—Tight containers.
Vasopressin Injectionバソプレシン注射液
Change the paragraph (i) under Purity to read:
Purity(i) Standard stock solution: Dissolve 200 oxytocin Units
of Posterior Pituitary Reference Standard, according to thelabeled Units, in exactly 10 mL of diluted acetic acid (100)(1 in 400). Pipet 1 mL of this solution, and add diluted aceticacid (100) (1 in 400) to make exactly 10 mL. Store in a coldplace, avoiding freezing. Use within 6 months from the dateof preparation.
Change the paragraph (ii) under Assay to read:
Assay(ii) Standard stock solution: Dissolve 2000 vasopressin
Units of Posterior Pituitary Reference Standard, accordingto the labeled Units, in exactly 100 mL of diluted acetic acid(100) (1 in 400). Pipet 1 mL of this solution, and add dilutedacetic acid (100) (1 in 400) to make exactly 10 mL. Store in acold place, avoiding freezing. Use within 6 months from thedate of preparation.

17521752 Supplement II, JPXIVO‹cial Monographs for Part I
Vinblastine Sulfate硫酸ビンブラスチン
Change to read except the structural formula,chemical name and origin/limits of content:
Description Vinblastine Sulfate occurs as a white to paleyellow powder.
It is soluble in water, sparingly soluble in methanol, andpractically insoluble in ethanol (99.5).
It is hygroscopic.Optical rotation [a]D20: -28 –-359(0.20 g calculated on
the dried basis, methanol, 10 mL, 100 mm).
Identiˆcation (1) Determine the absorption spectrum ofa solution of Vinblastine Sulfate (1 in 50,000) as directedunder the Ultraviolet-visible Spectrophotometry, and com-pare the spectrum with the Reference Spectrum or thespectrum of a solution of Vinblastine Sulfate ReferenceStandard prepared in the same manner as the sample solu-tion: both spectra exhibit similar intensities of absorption atthe same wavelengths.
(2) Determine the infrared absorption spectrum ofVinblastine Sulfate as directed in the potassium bromidedisk method under the Infrared Spectrophotometry, andcompare the spectrum with the Reference Spectrum or thespectrum of Vinblastine Sulfate Reference Standard: bothspectra exhibit similar intensities of absorption at the samewave numbers.
(3) A solution of Vinblastine Sulfate (1 in 100) respondsto the Qualitative Tests for sulfate.
pH Dissolve 15 mg of Vinblastine Sulfate in 10 mL ofwater: the pH of this solution is between 3.5 and 5.0.
Purity (1) Clarity and color of solution—Dissolve 50 mgof Vinblastine Sulfate in 10 mL of water: the solution is clearand colorless.
(2) Related substances—Dissolve about 4 mg of Vin-blastine Sulfate in 10 mL of water, and use this solution asthe sample solution. Pipet 1 mL of the sample solution, addwater to make exactly 25 mL, and use this solution as thestandard solution. Perform the test with exactly 200 mL eachof the sample solution and the standard solution as directedunder the Liquid Chromatography according to the follow-ing conditions, and determine each peak area of these solu-tions by the automatic integration method: the area of peakother than the main peak is not larger than 1/4 of the peakarea of vinblastine from the standard solution, and the totalarea of the peaks other than the main peak is not larger than3/4 of the peak area of vinblastine from the standardsolution.Operating conditions—
Detector, column, column temperature, mobile phase,and ‰ow rate: Proceed as directed in the operating condi-tions in the Assay.
Time span of measurement: About 4 times as long as theretention time of vinblastine after the solvent peak.
System suitability—Test for required detectability: To exactly 2.5 mL of the
standard solution add water to make exactly 100 mL.Conˆrm that the peak area of vinblastine obtained from200 mL of this solution is equivalent to 1.7 to 3.3z of thatfrom 200 mL of the standard solution.
System performance: Proceed as directed in the systemsuitability in the Assay.
System repeatability: When the test is repeated 5 timeswith 200 mL of the standard solution under the aboveoperating conditions, the relative standard deviation of thepeak area of vinblastine is not more than 1.5z.
Loss on drying Perform the test with about 10 mg ofVinblastine Sulfate as directed in Method 2 under the Ther-mal Analysis according to the following conditions: notmore than 15.0z.Operating conditions—
Heating rate: 59C/minuteTemperature range: room temperature to 2009CAtmospheric gas: dried NitrogenFlow rate of atmospheric gas: 40 mL/minute
Assay Weigh accurately about 10 mg each of VinblastineSulfate and Vinblastine Sulfate Reference Standard(separately determine its loss on drying), dissolve in water tomake exactly 25 mL, and use these solutions as the samplesolution and the standard solution, respectively. Performthe test with exactly 20 mL each of the sample solution andthe standard solution as directed under the Liquid Chro-matography according to the following conditions, and de-termine the peak areas, AT and AS, of vinblastine.
Amount (mg) of C46H58N4O9.H2SO4=WS×AT
AS
WS: Amount (mg) of Vinblastine Sulfate ReferenceStandard, calculated on the dried basis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 262 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about259C.
Mobile phase: To 7 mL of diethylamine add water tomake 500 mL, and adjust the pH to 7.5 with phosphoricacid. To 380 mL of this solution add 620 mL of a mixture ofmethanol and acetonitrile (4:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof vinblastine is about 8 minutes.System suitability—
System performance: Dissolve 10 mg each of VinblastineSulfate and vincristine sulfate in 25 mL of water. When theprocedure is run with 20 mL of this solution under the aboveoperating conditions, vincristine and vinblastine are elutedin this order with the resolution between these peaks being

17531753Supplement II, JPXIV O‹cial Monographs for Part I
not less than 4.System repeatability: When the test is repeated 5 times
with 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of vinblastine is not more than 1.0z.
Containers and storage Containers—Tight containers.Storage—Light-resistant, at not exceeding -209C.
Change to read:
Vinblastine Sulfate for Injection注射用硫酸ビンブラスチン
Vinblastine Sulfate for Injection is a preparation forinjection, which is dissolved before use. It containsnot less than 90.0z and not more than 110.0z ofthe labeled amount of vinblastine sulfate(C46H58N4O9.H2SO4: 909.05).
Method of preparation Prepare as directed under Injec-tions, with Vinblastine Sulfate.
Description Vinblastine Sulfate for Injection occurs aswhite to pale yellow, light masses or powder.
It is freely soluble in water.The pH of a solution (1 in 1000) is 3.5 – 5.0.
Identiˆcation Proceed as directed in the Identiˆcation (1)under Vinblastine Sulfate.
Purity Related substances—Dissolve 4 mg of VinblastineSulfate for Injection in 10 mL of water, and use this solutionas the sample solution. Pipet 1 mL of the sample solution,add water to make exactly 25 mL, and use this solution asthe standard solution. Perform the test with exactly 200 mLeach of the sample solution and the standard solution asdirected under the Liquid Chromatography according to thefollowing conditions, and determine each peak area by theautomatic integration method: the area of the peak otherthan the main peak from the sample solution is not largerthan 1/2 of the peak area of vinblastine from the standardsolution, and the total area of the peaks other than the mainpeak is not larger than 2 times the peak area of vinblastinefrom the standard solution.Operating conditions—
Perform as directed in the operating conditions in Purity(2) under Vinblastine Sulfate.System suitability—
Perform as directed in the system suitability in Purity (2)under Vinblastine Sulfate.
Bacterial endotoxins Less than 10 EU/mg.
Content uniformity Perform the test according to the fol-
lowing method: it meets the requirements of the ContentUniformity Test. Open a container of Vinblastine Sulfatefor Injection, add an exact amount of water to make a solu-tion so that each mL contains about 0.4 mg of vinblastinesulfate (C46H58N4O9.H2SO4) according to the labeledamount, and use this solution as the sample solution.Separately, weigh accurately about 10 mg of VinblastineSulfate Reference Standard (separately determine its loss ondrying in the same manner as for Vinblastine Sulfate), dis-solve in water to make exactly 25 mL, and use this solutionas the standard solution. Proceed as directed in the Assayunder Vinblastine Sulfate.
Amount (mg) of vinblastine sulfate (C46H58N4O9.H2SO4)
=WS×AT
AS
WS: Amount (mg) of Vinblastine Sulfate ReferenceStandard, calculated on the dried basis
Foreign insoluble matter Perform the test according toMethod 2: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Assay Take an amount of Vinblastine Sulfate for Injec-tion, equivalent to 0.10 g of vinblastine sulfate(C46H58N4O9.H2SO4), dissolve each content with a suitableamount of water, transfer into a 100-mL volumetric ‰ask,wash each container with water, transfer the washings intothe volumetric ‰ask, and add water to make exactly 100 mL.Pipet 10 mL of this solution, add water to make exactly 25mL, and use this solution as the sample solution. Separately,weigh accurately about 10 mg of Vinblastine Sulfate Refer-ence Standard (separately determine its loss on drying in thesame manner as for Vinblastine Sulfate), dissolve in water tomake exactly 25 mL, and use this solution as the standardsolution. Proceed as directed in the Assay under VinblastineSulfate.
Amount (mg) of vinblastine sulfate (C46H58N4O9.H2SO4)
=WS×AT
AS
WS: Amount (mg) of Vinblastine Sulfate ReferenceStandard, calculated on the dried basis
Containers and storage Containers—Hermetic containers,and colored containers may be used.
Storage—Light-resistant, at 2 to 89C.

17551755
General Rulesfor Crude Drugs
Change the paragraph 1 to read:
1. Crude drugs in the monographs include medici-nal parts obtained from plants or animals, cell inclu-sions and secretes separated from the origins, theirextracts, and minerals. General Rules for Crude Drugsand the Crude Drugs in General Tests, Processes andApparatus are applicable to the followings:
Acacia, Achyranthes Root, Agar, Agar Powder,Akebia Stem, Alisma Rhizome, Aloe, AlpiniaO‹cinarum Rhizome, Amomum Seed, AnemarrhenaRhizome, Angelica Dahurica Root, Apricot Kernel,Areca, Artemisia Capillaris Flower, Asiasarum Root,Asparagus Tuber, Astragalus Root, AtractylodesLancea Rhizome, Atractylodes Rhizome, Bear Bile,Bearberry Leaf, Belladonna Root, Benzoin, BitterCardamon, Bitter Orange Peel, Bupleurum Root,Burdock Fruit, Calumba, Capsicum, Cardamon,Cassia Seed, Catalpa Fruit, Chrysanthemum Flower,Chuling, Cimicifuga Rhizome, Cinnamon Bark,Citrus Unshiu Peel, Clematis Root, Clove, CnidiumMonnieri Fruit, Cnidium Rhizome, Coix Seed, Con-durango, Coptis Rhizome, Cornus Fruit, CorydalisTuber, Cyperus Rhizome, Digenea, Dioscorea Rhi-zome, Ephedra Herb, Epimedium Herb, EucommiaBark, Evodia Fruit, Fennel, Forsythia Fruit, FritillariaBulb, Gambir, Gardenia Fruit, Gastrodia Tuber,Gentian, Geranium Herb, Ginger, Ginseng, GlehniaRoot, Glycyrrhiza, Gypsum, Hemp Fruit, Hoelen,Honey, Houttuynia Herb, Immature Orange,Imperata Rhizome, Ipecac, Japanese Angelica Root,Japanese Gentian, Japanese Valerian, Jujube, JujubeSeed, Lindera Root, Lithospermum Root, Longgu,Lonicera Leaf and Stem, Loquat Leaf, Lycium Bark,Lycium Fruit, Magnolia Bark, Magnolia Flower,Mallotus Bark, Mentha Herb, Moutan Bark,Mulberry Bark, Notopterygium Rhizome, NupharRhizome, Nux Vomica, Ophiopogon Tuber, OrientalBezoar, Oyster Shell, Panax Rhizome, Peach Kernel,Peony Root, Perilla Herb, Pharbitis Seed, Phelloden-
dron Bark, Picrasma Wood, Pinellia Tuber, PlantagoHerb, Plantago Seed, Platycodon Root, PolygalaRoot, Polygonum Root, Powdered Acacia, PowderedAlisma Rhizome, Powdered Aloe, PowderedAmomum Seed, Powdered Atractylodes LanceaRhizome, Powdered Atractylodes Rhizome, Pow-dered Calumba, Powdered Capsicum, PowderedCinnamon Bark, Powdered Clove, Powdered Cnidi-um Rhizome, Powdered Coix Seed, Powdered CoptisRhizome, Powdered Cyperus Rhizome, PowderedDioscorea Rhizome, Powdered Fennel, PowderedGambir, Powdered Gardenia Fruit, Powdered Genti-an, Powdered Geranium Herb, Powdered Ginger,Powdered Ginseng, Powdered Glycyrrhiza, PowderedHoelen, Powdered Ipecac, Powdered Japanese Angel-ica Root, Powdered Japanese Gentian, PowderedJapanese Valerian, Powdered Magnolia Bark, Pow-dered Moutan Bark, Powdered Oyster Shell, Pow-dered Panax Rhizome, Powdered Peach Kernel, Pow-dered Peony Root, Powdered Phellodendron Bark,Powdered Picrasma Wood, Powdered PlatycodonRoot, Powdered Polygala Root, Powdered Polypou-rus Sclerotium, Powdered Processed Aconite Root,Powdered Rhubarb, Powdered Rose Fruit, PowderedScutellaria Root, Powdered Senega, Powdered SennaLeaf, Powdered Smilax Rhizome, Powdered SophoraRoot, Powdered Sweet Hydrangea Leaf, PowderedSwertia Herb, Powdered Tragacanth, PowderedZanthoxylum Fruit, Processed Aconite Root, Proc-essed Ginger, Prunella Spike, Pueraria Root, RedGinseng, Rehmannia Root, Rhubarb, Rice Starch,Rose Fruit, Rosin, SaŒower, SaŠron, SaposhnikoviaRoot, Sappan Wood, Saussurea Root, SchisandraFruit, Schizonepeta Spike, Scopolia Rhizome, Scutel-laria Root, Senega, Senna Leaf, Sinomenium Stem,Smilax Rhizome, Sophora Root, Sweet HydrangeaLeaf, Swertia Herb, Termeric, Toad Venom,Tragacanth, Tribulus Fruit, Trichosanthes Root,Uncaria Thorn, Zanthoxylum Fruit, Zedoary.

17571757
O‹cial Monographsfor Part II
Delete the following Monographs:
Absorbent Cotton
Puriˆed Absorbent Cotton
Sterile Absorbent Cotton
Sterile Puriˆed Absorbent Cotton
Absorbent Gauze
Sterile Absorbent Gauze
Adhesive Plaster
Powdered Aloeアロエ末
Change the origin/limits of content to read:
Powdered Aloe is the powder of Aloe.It contains not less than 4.0z of barbaloin, calcu-
lated on the basis of dried material.
Change the Loss on drying to read:
Loss on drying Not more than 12.0z.
Add the following next to Extract content:
Component determination Weigh accurately about 0.1 gof Powdered Aloe, add 40 mL of methanol, and heat undera re‰ex condenser on a water bath for 30 minutes. Aftercooling, ˆlter, and add methanol to the ˆltrate to makeexactly 50 mL. Pipet 5 mL of the solution, add methanol tomake exactly 10 mL, and use this solution as the samplesolution. Separately, weigh accurately about 10 mg ofbarbaloin for component determination, previously dried ina desiccator (in vacuum, phosphorus (V) oxide) for 24hours, add 40 mg of oxalic acid dihydrate, and dissolve inmethanol to make exactly 100 mL. Pipet 5 mL of thesolution, add methanol to make exactly 10 mL, and use thissolution as the standard solution. Perform the test withexactly 5 mL each of the sample solution and the standard
solution as directed under the Liquid Chromatographyaccording to the following conditions, and determine thepeak areas of barbaloin, AT and AS, of both solutions.
Amount (mg) of barbaloin=WS×AT
AS×2
WS: Amount (mg) of barbaloin for component determina-tion
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 360 nm).Column: A stainless steel column about 6 mm in inside
diameter and about 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in parti-cle diameter).
Column temperature: A constant temperature of about309C.
Mobile phase: A mixture of water, acetonitrile and aceticacid (100) (74:26:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof barbaloin is about 12 minutes.System suitability—
System performance: To about 10 mg of barbaloin forcomponent determination add 40 mg of oxalic acid dihy-drate, and dissolve in methanol to make exactly 100 mL.Pipet 5 mL of the solution, add 1 mL of a solution of ethen-zamide in methanol (1 in 2000) and methanol to makeexactly 10 mL. When the procedure is run with 5 mL of thissolution under the above operating conditions except thewavelength of 300 nm, barbaloin and ethenzamide areeluted in this order with the resolution between these peaksbeing not less than 2.0.
System repeatability: When the test is repeated 6 timeswith 5 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of barbaloin is not more than 1.5z.
Benzyl Alcoholベンジルアルコール
Change to read except the structural formulaand chemical name:
Benzyl Alcohol contains not less than 98.0z andnot more than 100.5z of C7H8O.
The label states, where applicable, that it is suitablefor use in the manufacture of injection forms.

17581758 Supplement II, JPXIVO‹cial Monographs for Part II
Description Benzyl Alcohol is a clear, colorless oily liquid.It is miscible with ethanol (95), with fatty oils and with
volatile oils.It is soluble in water.Speciˆc gravity d 20
20: 1.043 – 1.049
Identiˆcation Determine the infrared absorption spectrumof Benzyl Alcohol as directed in the liquid ˆlm methodunder the Infrared Spectrophotometry, and compare thespectrum with the Reference Spectrum: both spectra exhibitsimilar intensities of absorption at the same wave numbers.
Refractive index nD20: 1.538 – 1.541
Purity (1) Clarity and color of solution—Dissolve 2.0mL of Benzyl Alcohol in 60 mL of water: the solution isclear and colorless.
(2) Acid—To 10 mL of Benzyl Alcohol add 10 mL ofneutralized ethanol, 2 drops of phenolphthalein TS and 1.0mL of 0.1 mol/L sodium hydroxide VS: the color of thesolution is red.
(3) Benzaldehyde and other related substances—UseBenzyl Alcohol as the sample solution. Separately, weighexactly 0.750 g of benzaldehyde and 0.500 g of cyclohex-ylmethanol, and add Benzyl Alcohol to make exactly 25 mL.Pipet 1 mL of this solution, add exactly 2 mL of ethylben-zene internal standard solution and exactly 3 mL of dicyclo-hexyl internal standard solution, then add Benzyl Alcohol tomake exactly 20 mL, and use this solution as the standardsolution (1). Perform the test with 0.1 mL each of the samplesolution and the standard solution (1) as directed under theGas Chromatography according to the following conditions:no peaks of ethylbenzene and dicyclohexyl appear on thechromatogram obtained with the sample solution. When0.1 mL of the standard solution (1) is injected, adjust thesensitivity of the detector so that the peak height of ethyl-benzene is not more than 30z of the full scale of the record-er. The peak area of benzaldehyde obtained with the samplesolution is not more than the deference between the peakareas of benzaldehyde of the sample solution and thestandard solution (1) (0.15z), and the peak area of cyclo-hexylmethanol with the sample solution is not more than thedeference between the peak areas of cyclohexylmethanol ofthe sample solution and the standard solution (1) (0.10z).The total area of the peaks having smaller retention timethan benzyl alcohol and other than benzaldehyde andcyclohexylmethanol obtained with the sample solution is notmore than 4 times the peak area of ethylbenzene with thestandard solution (1) (0.04z). The total area of the peakshaving larger retention time than benzaldehyde obtainedwith the sample solution is not more than the peak area ofcyclohexylmethanol with the standard solution (1) (0.3z).For these calculations the peak areas less than 1/100 timesthe peak area of ethylbenzene with the standard solution (1)are excluded.
Benzyl Alcohol labeled that it is suitable for use in themanufacture of injection forms meets the following require-ments.
Use Benzyl Alcohol as the sample solution. Separately,
weigh exactly 0.250 g of benzaldehyde and 0.500 g of cyclo-hexylmethanol, and add Benzyl Alcohol to make exactly25 mL. Pipet 1 mL of this solution, add exactly 2 mL of theethylbenzene internal standard solution and exactly 2 mL ofthe dicyclohexyl internal standard solution, then add BenzylAlcohol to make exactly 20 mL, and use this solution as thestandard solution (2). Perform the test with 0.1 mL each ofthe sample solution and the standard solution (2) as directedunder the Gas Chromatography according to the followingconditions: no peaks of ethylbenzene and dicyclohexylappear on the chromatogram obtained with the samplesolution. When 0.1 mL of the standard solution (2) is inject-ed, adjust the sensitivity of the detector so that the peakheight of ethylbenzene is not more than 30z of the full scaleof the recorder. The peak area of benzaldehyde of obtainedwith the sample solution is not more than the deferencebetween the peak areas of benzaldehyde of the samplesolution and the standard solution (2) (0.05z), and the peakarea of cyclohexylmethanol with the sample solution is notmore than the deference between the peak areas of cyclohex-ylmethanol of the sample solution and the standard solution(2) (0.10z). The total area of the peaks having smallerretention time than benzyl alcohol and other than benzalde-hyde and cyclohexylmethanol obtained with the samplesolution is not more than 2 times the peak area of ethylben-zene with the standard solution (2) (0.02z). The total areaof the peaks having larger retention time than benzaldehydeobtained with the sample solution is not more than the peakarea of cyclohexylmethanol with the standard solution (2)(0.2z). For these calculation the peak areas less than 1/100times the peak area of ethylbenzene with the standardsolution (2) are excluded.
Ethylbenzene internal standard solution: Dissolve exactly0.100 g of ethylbenzene in Benzyl Alcohol to make exactly10 mL. Pipet 2 mL of this solution, and add Benzyl Alcoholto make exactly 20 mL.
Dicyclohexyl internal standard solution: Dissolve exactly2.000 g of dicyclohexyl in Benzyl Alcohol to make exactly10 mL. Pipet 2 mL of this solution, and add Benzyl Alcoholto make exactly 20 mL.Operating conditions—
Detector: A hydrogen ‰ame-ionization detectorColumn: A fused silica column 0.33 mm in inside
diameter and 30 m in length, coated inside with polyethyleneglycol 20M for gas chromatography in 0.5 mm thickness.
Column temperature: Raise the temperature at a rate of59C per minutes from 509C to 2209C, and maintain at2209C for 35 minutes.
Temperature of injection port: A constant temperature ofabout 2009C.
Temperature of detector: A constant temperature ofabout 3109C.
Carrier gas: HeliumFlow rate: Adjust the ‰ow rate so that the retention time
of benzyl alcohol is between 24 and 28 minutes.Split ratio: Splitless
System suitability—System performance: When the procedure is run with the

17591759Supplement II, JPXIV O‹cial Monographs for Part II
standard solution (1) under the above operating conditions,the relative retention times of ethylbenzene, dicyclohexyl,benzaldehyde and cyclohexylmethanol with respect to benzylalcohol are about 0.28, about 0.59, about 0.68 and about0.71, respectively, and the resolution between the peaks ofbenzaldehyde and cyclohexylmethanol is not less than 3.0.In the case of Benzyl Alcohol labeled to use for injection,proceed with the standard solution (2) instead of thestandard solution (1).
(4) Peroxide value—Dissolve 5 g of Benzyl Alcohol in30 mL of a mixture of acetic acid (100) and chloroform (3:2)in a 250-mL glass-stoppered conical ‰ask. Add 0.5 mL ofpotassium iodide saturated solution, shake exactly for 1minute, add 30 mL of water, and titrate with 0.01 mol/Lsodium thiosulfate VS until the blue color of the solutiondisappears after addition of 10 mL of starch TS near the endpoint where the solution is a pale yellow color. Perform ablank determination in the same manner. Calculate theamount of peroxide by the following formula: not morethan 5.
Amount (mEq/kg) of peroxide=(V1-V0)
W×10
V1: Volume (mL) of 0.01 mol/L sodium thiosulfate VSconsumed in the test
V0: Volume (mL) of 0.01 mol/L sodium thiosulfate VSconsumed in the blank
W: Amount (g) of the sample
(5) Residue on evaporation—Perform the test afterconformation that the sample meets the requirement of theperoxide value. Evaporate to dryness 10.0 g of BenzylAlcohol on a water bath, dry the residue at 1059C for 1hour, and allow to cool in a desiccator: not more than 5 mg.
Assay Weigh accurately about 0.9 g of Benzyl Alcohol,add exactly 15.0 mL of a mixture of pyridine and aceticanhydride (7:1), and heat on a water bath under a re‰uxcondenser for 30 minutes. Cool, add 25 mL of water, andtitrate the excess acetic acid with 1 mol/L sodium hydroxideVS (indicator: 2 drops of phenolphthalein TS). Perform ablank determination.
Each mL of 1 mol/L sodium hydroxide VS=108.1 mg of C7H8O
Containers and storage Containers—Tight containers.Storage—Light-resistant.
Carmellose Calciumカルメロースカルシウム
Change the Description to read:
Description Carmellose Calcium occurs as a white toyellowish white powder.
It is practically insoluble in ethanol (95) and in diethylether.
It swells with water to form a suspension.The pH of a suspension, obtained by shaking 1.0 g of
Carmellose Calcium with 100 mL of water, is between 4.5and 6.0.
It is hygroscopic.
Change the Identiˆcation (1) and (4) to read:
Identiˆcation (1) Shake thoroughly 0.1 g of CarmelloseCalcium with 10 mL of water, followed by 2 mL of sodiumhydroxide TS, allow to stand for 10 minutes, and use thissolution as the sample solution. To 1 mL of the samplesolution add water to make 5 mL. To 1 drop of this solutionadd 0.5 mL of chromotropic acid TS, and heat in a waterbath for 10 minutes: a red-purple color develops.
(4) Ignite 1 g of Carmellose Calcium to ash, dissolve theresidue in 10 mL of water and 6 mL of acetic acid (31), andˆlter, if necessary. Boil the ˆltrate, cool, and neutralize withammonia TS: the solution responds to the Qualitative Tests(1) and (3) for calcium salt.
Change the Purity to read:
Purity (1) Alkali—Shake thoroughly 1.0 g of CarmelloseCalcium with 50 mL of freshly boiled and cooled water, andadd 2 drops of phenolphthalein TS: no red color develops.
(2) Chloride—Shake thoroughly 0.80 g of CarmelloseCalcium with 50 mL of water, add 10 mL of sodiumhydroxide TS to dissolved, add water to make 100 mL, anduse this solution as the sample solution. Heat 20 mL of thesample solution with 10 mL of 2 mol/L nitric acid on awater bath until a ‰occulent precipitate is produced. Aftercooling, centrifuge, and take out the supernatant liquid.Wash the precipitate with three 10-mL portions of water bycentrifuging each time, combine the supernatant and thewashings, and add water to make 100 mL. Take 25 mL ofthis solution, and add 1 mL of nitric acid and water to make50 mL. Perform the test using this solution as the testsolution. Prepare the control solution with 0.40 mL of 0.01mol/L hydrochloric acid VS (not more than 0.36z).
(3) Sulfate—Heat 10 mL of the sample solution ob-tained in (2) with 1 mL of hydrochloric acid in a water bathuntil a ‰occulent precipitate is produced. Cool, centrifuge,and take out the supernatant liquid. Wash the precipitatewith three 10-mL portions of water by centrifuging eachtime, combine the supernatant and the washings, and addwater to make 100 mL. Perform the test with 25 mL thissolution as the test solution. Prepare the control solutionwith 0.42 mL of 0.005 mol/L sulfuric acid VS. To the testsolution and the control solution add 1 mL of 3 mol/Lhydrochloric acid and 3 mL of barium chloride TS, then addwater to make 50 mL, and mix. Allow to stand for 10minutes, and compare the turbidity of these solutions: theturbidity obtained with the test solution is not more thanthat obtained with the control solution (not more than1.0z).
(4) Heavy metals—Proceed with 1.0 g of CarmelloseCalcium according to Method 2, and perform the test.Prepare the control solution with 2.0 mL of Standard Lead

17601760 Supplement II, JPXIVO‹cial Monographs for Part II
Solution (not more than 20 ppm).
Change the Residue on ignition to read:
Residue on ignition 10.0 – 20.0z (after drying, 1 g).
Cellulose Acetate Phthalate酢酸フタル酸セルロース
Add the following commonly used name:
Cellacefate
Change the origin/limits of the content to read:
Cellulose Acetate Phthalate is a reaction product ofphthalic anhydride and partially acetylated cellulose.
Cellulose Acetate Phthalate, calculated on theanhydrous and free acid-free basis, contains not lessthan 21.5z and not more than 26.0z of acetyl group(-COCH3: 43.04), and not less than 30.0z andnot more than 36.0z of carboxybenzoyl group(-COC6H4COOH: 149.12).
Change the Description to read:
Description Cellulose Acetate Phthalate occurs as a whitepowder or grain.
It is freely soluble in acetone, and practically insoluble inwater, in methanol and in ethanol (99.5).
Change the Identiˆcation to read:
Identiˆcation Determine the infrared absorption spectrumof Cellulose Acetate Phthalate as directed in the potassiumbromide disk method under the Infrared Spectrophotomet-ry, and compare the spectrum with the Reference Spectrumor spectrum of Cellulose Acetate Phthalate ReferenceStandard: both spectra exhibit similar intensities of absorp-tion at the same wave numbers.
Change the Water to read:
Water Not more than 5.0z (1 g, volumetric titration,direct titration, using a mixture of ethanol (99.5) anddichloromethane (3:2) instead of methanol for Karl Fischermethod).
Change the Assay (2) to read:
Assay(2) Acetyl group—Weigh accurately about 0.1 g of
Cellulose Acetate Phthalate, put in a glass-stoppered conical‰ask, add exactly 25 mL of 0.1 mol/L sodium hydroxideVS, and boil for 30 minutes under a re‰ux condenser. Aftercooling, add 5 drops of phenolphthalein TS, and titrate with0.1 mol/L hydrochloric acid VS. Perform a blank determi-nation.
Content (z) of free acids and bound acetyl group (C2H3O)
=AW×0.4304
A: Amount (mL) of 0.1 mol/L sodium hydroxide con-sumed, corrected by the blank determination
W: Amount (g) of the test sample, calculated on theanhydrous basis
Content (z) of acetyl group (C2H3O)
=P-0.5182×B
100-B×100-0.5772×C
B: Amount (z) of free acids obtained in Purity (2) Freeacids
C: Content (z) of carboxybenzoyl groupP: Content (z) of free acids and bound acetyl group
(C2H3O)
Chrysanthemum Flowerキクカ
Change the Extract content to read:
Extract content Dilute ethanol-extract: not less than30.0z.
Add the following:
Cnidium Monnieri Fruit
Cnidii Monnieris Fructus
ジャショウシ
Cnidium Monnieri Fruit is the fruit of Cnidiummonnieri Cusson (Umbelliferae).
Description Elliptical cremocarp, often each mericarpseparated; 2 – 3 mm in length, 1 – 2 mm in width; externallylight brown to brown, each mericarp usually with ˆvewinged longitudinal ridges; inner surface of mericarp almost‰at.
Odor, characteristic; it gives characteristic aroma, later aslight sensation of numbness on chewing.
Under a microscope, a transverse section reveals one oilcanal between longitudinal ridges, usually two oil canals inthe inner part of mericarp facing to gynophore; longitudinalridges composed of slightly ligniˆed parenchymatous cells,with vascular bundles in the base; epidermal cells andparenchymatous cells of longitudinal ridges contain solitarycrystals of calcium oxalate; parenchymatous cells ofalbumen contain oil drops and aleurone grains, and oc-casionally starch grains.
Identiˆcation To 1 g of pulverized Cnidium MonnieriFruit add 10 mL of ethyl acetate, shake for 10 minutes,ˆlter, and use the ˆltrate as the sample solution. Separately,

17611761Supplement II, JPXIV O‹cial Monographs for Part II
dissolve 1 mg of osthole for thin-layer chromatography in2 mL of methanol, and use this solution as the standardsolution. Perform the test with these solutions as directedunder the Thin-layer Chromatography. Spot 5 mL each ofthe sample solution and the standard solution on a plate ofsilica gel for thin-layer chromatography, develop the platewith a mixture of hexane and ethyl acetate (2:1) to a distanceof about 10 cm, and air-dry the plate. Examine under ultrav-iolet light (main wavelength: 365 nm): one of the spot amongthe several spots from the sample solution has the same colortone and the Rf value with the blue-white ‰uorescent spotfrom the standard solution.
Loss on drying Not more than 12.0z (6 hours).
Total ash Not more than 17.0z.
Acid-insoluble ash Not more than 6.0z.
Extract content Not less than 8.0z (dilute ethanol-solubleextract).
Change to read:
Corn Starch
Amylum Maydis
トウモロコシデンプン
Corn Starch consists of starch granules derived fromthe ripen seeds of Zea mays Linn áe (Gramineae).
Description Corn Starch occurs as white to pale yellowishwhite masses or powder.
It is practically insoluble in water and in ethanol (99.5).
Identiˆcation (1) Under a microscope, Corn Starch,preserved in a mixture of water and glycerin (1:1), appears asirregularly polygonal simple grains usually 2 – 23 mm indiameter, or irregularly orbicular or spherical simple grains25 – 35 mm in diameter; hilum appears as distinct cave or 2 –5 radial clefts; concentric striation absent; a black cross, itsintersection point on hilum, is observed when grains are putbetween two polarizing prisms ˆxed at right angle to eachother.
(2) To 1 g of Corn Starch add 50 mL of water, boil for 1minute, and allow to cool: a subtle white-turbid, pasty liquidis formed.
(3) To 10 mL of the pasty liquid obtained in (2) add0.04 mL of iodine TS: a orange-red to dark blue-purplecolor is formed, and the color disappears by heating.
pH Put 5.0 g of Corn Starch in a non-metal vessel, add25.0 mL of freshly boiled and cooled water, mix gently for 1minute, and allow to stand for 15 minutes: the pH of thesolution is between 4.0 and 7.0.
Purity (1) Iron—To 1.5 g of Corn Starch add 15 mL of 2mol/L hydrochloric acid TS, mix, ˆlter, and use the ˆltrateas the test solution. To 2.0 mL of Standard Iron Solution
add water to make 20 mL, and use as the control solution.Put 10 mL each of the test solution and the control solutionin test tubes, add 2 mL of a solution of citric acid (2 in 10)and 0.1 mL of mercapto acetic acid, and mix. Alkalize withammonia solution (28) to litmus paper, add water to make20 mL, and mix. Transfer 10 mL each of these solutions intotest tubes, allow to stand for 5 minutes, and compare thecolor of these solutions against a white background: thecolor of the test solution is not darker than that of thecontrol solution (not more than 10 ppm).
(2) Oxidizing substances—To 4.0 g of Corn Starch add50.0 mL of water, shake for 5 minutes, and centrifuge. To30.0 mL of the supernatant liquid add 1 mL of acetic acid(100) and 0.5 to 1.0 g of potassium iodide, shake, and allowto stand for 25 to 30 minutes at a dark place. Add 1 mL ofstarch TS, and titrate with 0.002 mol/L sodium thiosulfateVS until the color of the solution disappears. Perform ablank determination and make any necessary correction: thevolume of 0.002 mol/L sodium thiosulfate VS consumed isnot more than 1.4 mL (not more than 20 ppm, calculated ashydrogen peroxide).
(3) Sulfur dioxide—(i) Apparatus Use as shown in the following ˆgure.
The ˆgures are in mm.
A: Boiling ‰ask (500 mL)B: Funnel (100 mL)C: CondenserD: Test-tubeE: Tap
(ii) Procedure Introduce 150 mL of water into theboiling ‰ask, close the tap of the funnel, and pass carbondioxide through the whole system at a rate of 100±5 mL perminute. Pass cooling water through the condenser, and place10 mL of hydrogen peroxide-sodium hydroxide TS in thetest-tube. After 15 minutes, remove the funnel withoutinterrupting the stream of carbon dioxide, and introducethrough the opening into the ‰ask about 25 g of CornStarch, accurately weighed, with the aid of 100 mL of water.Apply tap grease to the outside of the connection part of the

17621762 Supplement II, JPXIVO‹cial Monographs for Part II
funnel, and load the funnel. Close the tap of the funnel,pour 80 mL of 2 mol/L hydrochloric acid TS into thefunnel, open the tap to introduce the hydrochloric acid intothe ‰ask, and close the tap while several mL of thehydrochloric acid remains, in order to avoid losing sulfurdioxide. Place the ‰ask in a water bath, and heat the mixturefor 1 hour. Transfer the contents of the test-tube with the aidof a little water to a wide-necked conical ‰ask. Heat in awater bath for 15 minutes, and cool. Add 0.1 mL ofbromophenol blue TS, and titrate with 0.1 mol/L sodiumhydroxide VS until the color changes from yellow to violet-blue lasting for at least 20 seconds. Perform a blank determi-nation and make any necessary correction. Calculate theamount of sulfur dioxide by applying the following formula:it is not more than 50 ppm.
Amount (ppm) of sulfur dioxide=VW×1000×3.203
W: Amount (g) of the sampleV: Amount (mL) of 0.1 mol/L sodium hydroxide VS
consumed
Loss on drying Not more than 15.0z (1 g, 1309C, 90minutes).
Reisdue on ignition Not more than 0.6z (1 g).
Containers and storage Containers—Well-closed contain-ers.
Cornus Fruitサンシュユ
Change the Identiˆcation to read:
Identiˆcation To 1 g of coarse cuttings of Cornus Fruitadd 10 mL of methanol, shake for 5 minutes, ˆlter, and usethe ˆltrate as the sample solution. Separately, dissolve 1 mgof loganin for thin-layer chromatography in 2 mL ofmethanol, and use this solution as the standard solution.Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 10 mL each of the samplesolution and the standard solution on a plate of silica gel forthin-layer chromatography. Develop with a mixture of ethylacetate, water and formic acid (6:1:1) to a distance of about10 cm, and air-dry the plate. Spray evenly 4-methoxybenzal-dehyde-sulfuric acid TS on the plate, and heat at 1059C for 5minutes: one of the spots from the sample solution is thesame with a red-purple spot from the standard solution incolor tone and Rf value.
Delete the following Monographs:
Digitalis
Powdered Digitalis
Add the following:
Epimedium Herb
Epimedii Herba
インヨウカク
Epimedium Herb is the terrestrial part of Epimedi-um pubescens Maximowicz, Epimedium brevicornumMaximowicz, Epimedium wushanense T. S. Ying,Epimedium sagittatum Maximowicz, Epimediumkoreanum Nakai, Epimedium grandi‰orum Morrenvar. thunbergianum Nakai or Epimedium sempervi-rens Nakai (Berberidaceae).
Description Epimedium Herb is composed of a stem and aternate to triternate compound leaf; lea‰et ovate to broadlyovate or ovate-lanceolate, 3 – 20 cm in length, 2 – 8 cm inwidth, petiolule 15 – 70 mm in length, apex of lea‰etacuminate, needle hair on margin 0.1 – 0.2 cm in length,base of lea‰et cordate to deeply cordate, lateral lea‰et asym-metry; upper surface green to greenish brown, sometimeslustrous, lower surface light green, often pilose, especiallyon vein densely pilose, papery or coriaceous; petiole andstem cylindrical, light yellowish brown to slightly purplishand light greenish brown, easily broken.
Odor, slight; taste, slightly bitter.Under a microscope, a transverse section of the leaf
reveals 3 – 6 vascular bundles in midvein; mesophyllcomposed of upper epidermis, single-layered palisade,spongy tissue and lower epidermis; leaf margins orbicular oroblong, sclerenchymatous; multi-cellular hairs on epidermis;8 – 20 vascular bundles in petiole and 6 – 15 vascular bundlesin petiolule. Under a microscope, a transverse section of thestem reveals a single to several-layered hypodermis, cortexof 4 – 10 layers of sclerenchymatous cells, vascular bundle13 – 30 in number, oblong to obovate.
Identiˆcation To 2 g of pulverized Epimedium Herb add20 mL of methanol, shake for 15 minutes, ˆlter, and use theˆltrate as the sample solution. Separately, dissolve 1 mg oficariin for thin-layer chromatography in 1 mL of methanol,and use this solution as the standard solution. Perform thetest with these solutions as directed under the Thin-layerChromatography. Spot 10 mL each of the sample solutionand the standard solution on a plate of silica gel with ‰uores-cent indicator for thin-layer chromatography. Develop theplate with a mixture of ethyl acetate, ethanol (99.5) andwater (8:2:1) to a distance of about 10 cm, and air-dry theplate. Examine under ultraviolet light (main wavelength: 254

17631763Supplement II, JPXIV O‹cial Monographs for Part II
nm): one spot among the spots from the sample solution anda dark purple spot from the standard solution show the samecolor tone and the same Rf value.
Loss on drying Not more than 12.5z (6 hours).
Total ash Not more than 8.5z.
Acid-insoluble ash Not more than 2.0z.
Extract content Not less than 17.0z (dilute ethanol-solu-ble extract).
Powdered Gardenia Fruitサンシシ末
Change the origin/limits of content to read:
Powdered Gardenia Fruit is the powder of GardeniaFruit.
It contains not less than 3.0z of geniposide, calcu-lated on the basis of dried material.
Change the Identiˆcation (1) to read:
Identiˆcation (1) To 1.0 g of Powdered Gardenia Fruit,previously dried in a desiccator (silica gel) for 24 hours, add100 mL of hot water, warm the mixture between 609C and709C for 30 minutes with frequent shaking, and ˆlter aftercooling. To 1.0 mL of the ˆltrate add water to make 10 mL:the color of the resulting solution is yellow and is not lighterthan that of the following control solution.
Control solution: Dissolve 9.8 mg of carbazochromesodium sulfonate in water to make exactly 10 mL. Pipet 1mL of this solution, and add water to make exactly 50 mL.
Add the following next to Identiˆcation:
Loss on drying Not more than 13.0z.
Add the following next to Total ash:
Component determination Weigh accurately about 0.5 gof Powdered Gardenia Fruit, transfer into a glass-stopperedcentrifuge tube, add 40 mL of diluted methanol (1 in 2),shake for 15 minutes, centrifuge, and take the supernatantliquid. To the residue add 40 mL of diluted methanol (1 in2), and perform as the same as above. Combine the extractsso obtained, and add diluted methanol (1 in 2) to makeexactly 100 mL. Pipet 5 mL of the solution, add methanol tomake exactly 20 mL, use this solution as the samplesolution. Separately, weigh accurately about 10 mg ofgeniposide for component determination, previously driedin a desiccator (in vacuum, phosphorus (V) oxide) for 24hours, and dissolve in methanol to make exactly 100 mL.Pipet 5 mL of the solution, add methanol to make exactly 10mL, and use this solution as the standard solution. Performthe test with exactly 10 mL each of the sample solution andthe standard solution as directed under the Liquid
Chromatography according to the following conditions, andmeasure the peak areas of geniposide, AT and AS, of bothsolutions.
Amount (mg) of geniposide=WS×AT
AS×2
WS: Amount (mg) of geniposide for component determi-nation
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 240 nm).Column: A stainless steel column 6 mm in inside diameter
and 15 cm in length, packed with octadecylsilanized silica gelfor liquid chromatography (5 mm in particle diameter).
Column temperature: A constant temperature of about309C.
Mobile phase: A mixture of water and acetonitrile (22:3).Flow rate: Adjust the ‰ow rate so that the retention time
of geniposide is about 15 minutes.System suitability—
System performance: Dissolve 1 mg each of geniposidefor component determination and caŠeine in methanol tomake 15 mL. When the procedure is run with 10 mL of thissolution under the above operating conditions, caŠeine andgeniposide are eluted in this order with the resolutionbetween these peaks being not less than 3.5.
System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of geniposide is not more than 1.5z.
Gingerショウキョウ
Change the Identiˆcation to read:
Identiˆcation To 2 g of pulverized Ginger add 5 mL ofdiethyl ether, shake for 10 minutes, ˆlter, and use the ˆltrateas the sample solution. Separately, dissolve 1 mg of [6]-gin-gerol for thin-layer chromatography in 2 mL of methanol,and use this solution as the standard solution. Perform thetest with these solutions as directed under the Thin-layerChromatography. Spot 10 mL of the sample solution and thestandard solution on a plate of silica gel for thin-layer chro-matography. Develop the plate with a mixture of ethylacetate and hexane (1:1) to a distance of about 10 cm, andair-dry the plate. Spray evenly 4-dimethylaminobenzalde-hyde TS on the plate, heat at 1059C for 5 minutes, and allowto cool: one of the spots from the sample solution and agreen spot from the standard solution show the same colortone and Rf value.

17641764 Supplement II, JPXIVO‹cial Monographs for Part II
Powdered Gingerショウキョウ末
Change the Identiˆcation to read:
Identiˆcation To 2 g of Powdered Ginger add 5 mL ofdiethyl ether, shake for 10 minutes, ˆlter, and use the ˆltrateas the sample solution. Separately, dissolve 1 mg of [6]-gin-gerol for thin-layer chromatography in 2 mL of methanol,and use this solution as the standard solution. Perform thetest with these solutions as directed under the Thin-layerChromatography. Spot 10 mL of the sample solution and thestandard solution on a plate of silica gel for thin-layer chro-matography. Develop the plate with a mixture of ethylacetate and hexane (1:1) to a distance of about 10 cm, andair-dry the plate. Spray evenly 4-dimethylaminobenzalde-hyde TS on the plate, heat at 1059C for 5 minutes, and allowto cool: one of the spots from the sample solution and agreen spot from the standard solution show the same colortone and Rf value.
Chorionic Gonadotrophin胎盤性性腺刺激ホルモン
Delete the origin/limits of content and add thefollowing:
Method of preparation Chorionic Gonadotrophin is adried preparation of gonad-stimulating hormone obtainedfrom the urine of healthy pregnant women after themanufacturing process to remove or inactivate the virus. Itcontains not less than 2500 chorionic gonadotrophin Unitsper mg, and contains not less than 3000 chorionicgonadotrophin Units per mg protein.
It contains not less than 80z and not more than 125z ofthe labeled Units of chorionic gonadotrophin.
Change the Description to read:
Description Chorionic Gonadotrophin occurs as a white tolight yellow-brown powder.
It is freely soluble in water.
Delete the Toxicity and Pyrogen, and add thefollowing:
Bacterial endotoxins Less than 0.03 EU/unit.
Abnormal toxicity Dilute Chorionic Gonadotrophin withisotonic sodium chloride solution so that each mL of thesolution contains 120 Units, and use this solution as thesample solution. Inject 5.0 mL of the sample solution intothe peritoneal cavity of each of 2 or more of well-nourished,healthy guinea pigs weighing about 350 g, and observe theconditions of the animals for more than 7 days: all theanimals exhibit no abnormalities.
Speciˆc activity When calculate from the results obtainedby the Assay and the following test, the speciˆc activity isnot less than 3000 chorionic gonadotrophin Units per mgprotein.
(i) Sample solution—To an exactly amount of ChorionicGonadotrophin add water to make a solution so that eachmL contains about 500 Units of chorionic gonadotrophinaccording to the labeled amount.
(ii) Standard solution—Weigh accurately about 10 mgof bovine serum albumin, and dissolve in water to makeexactly 20 mL. To a suitable volume of this solution addwater to make four solutions containing exactly 300, 200,100 and 50 mg of the albumin per mL, respectively.
(iii) Procedure—Pipet 0.5 mL each of the sample solu-tion and the standard solutions, put them in glass test tubesabout 18 mm in inside diameter and about 130 mm in length,add exactly 5 mL of alkaline copper TS, mix, and allow thetubes to stand in a water bath at 309C for 10 minutes. Thenadd exactly 0.5 mL of diluted Folin's TS (1 in 2), mix, andwarm in a water bath at 309C for 20 minutes. Determine theabsorbances of these solutions at 750 nm as directed underthe Ultraviolet-visible Spectrophotometry using a solutionobtained in the same manner with 0.5 mL of water as theblank.
Plot the absorbances of the standard solutions on thevertical axis and their protein concentrations on the horizon-tal axis to prepare a calibration curve, and determine theprotein content of the sample solution from its absorbanceby using this curve. Then calculate the amount of the proteinin the sample.
Change the Assay (i) to read:
Assay (i) Test animals—Select healthy female albino ratsweighing about 45 to 65 g.
Chorionic Gonadotrophin forInjection注射用胎盤性性腺刺激ホルモン
Change the Description to read:
Description Chorionic Gonadotrophin for Injectionoccurs as a white to light yellow-brown powder or masses.
Add the following next to Identiˆcation:
pH Prepare a solution so that each mL of isotonic sodiumchloride solution contains 2 mg of Chorionic Gonadotoro-phin for Injection: the pH of this solution is between 5.0 and7.0.
Delete the Pyrogaen and add the following:
Bacterial endotoxins Less than 0.03 EU/unit.
Mass variation When calculate the acceptance value usingthe mean of estimated contents of the units tested as M, it

17651765Supplement II, JPXIV O‹cial Monographs for Part II
meets the requirements of the Mass Variation Test.
Foreign insoluble matter Perform the test according toMethod 2: it meets the requirements of the Foreign InsolubleMatter Test for Injections.
Insoluble particulate matter Perform the test according toMethod 1: it meets the requirements of the Insoluble Par-ticulate Matter Test for Injections.
Sterility Perform the test according to the Membraneˆltration method: it meets the requirements of the SterilityTest.
Serum Gonadotrophin血清性性腺刺激ホルモン
Change the origin/limits of content to read:
Method of preparation Serum Gonadotrophin is a driedpreparation of gonad-stimulating hormone, obtained frompregnant mares' serum which has adequately inspectedviruses, and subjected to a suitable process for removal orinactivation of viruses. It contains not less than 2000 serumgonadotrophin Units per mg.
It contains not less than 80z and not more than 125z ofthe labeled Units of serum gonadotrophin.
Change the Description to read:
Description Serum Gonadotrophin occurs as a whitepowder.
It is freely soluble in water.
Add the following next to Purity:
Speciˆc activity When calculated from the results obtainedby the Assay and the following test, Serum Gonadotrophincontaines not less than 3000 serum gonadotrophin Units permg of protein.
(1) Standard solutions—Dissolve about 3 mg of bovineserum albumin in water to make a solution containing500 mg of the albumin in each mL. To this solution addwater to make four standard solutions so that each mLcontains exactly 200 mg, 150 mg, 100 mg and 50 mg of thealbumin, respectively.
(2) Sample solution—Dissolve about 1 mg of SerumGonadotrophin in water to make a solution containingexactly 180 mg in each mL.
(3) Sodium carbonate solution—Dissolve 2 g of sodiumcarbonate (standard reagent) in 0.1 mol/L sodiumhydroxide TS to make 100 mL.
(4) Sodium tartrate solution—Dissolve about 1 g ofsodium tartrate dihydrate in water to make 100 mL.
(5) Copper (II) sulfate solution—Dissolve 0.5 g ofcopper (II) sulfate pentahydrate in the sodium tartratesolution to make 100 mL.
(6) Alkaline copper solution—Mix 50 mL of the sodiumcarbonate solution and 1 mL of the copper (II) sulfate
solution. Prepare before use. Use within the day of prepara-tion.
(7) Procedure—Pipet 0.5 mL each of the standardsolutions and the sample solution in small test tubes, add3 mL of the alkaline copper solution to them, and mix.Allow them to stand at the room temperature for not lessthan 10 minutes, add 0.3 mL of diluted Folin's TS (1 in 2),mix immediately, and allow to stand for not less than 30minutes. Determine the absorbances of these solutions soobtained at 750 nm as directed under the Ultraviolet-visibleSpectrophotometry using a solution, prepared in the samemanner with 0.5 mL of water, as the blank. Plot the calibra-tion curve from the absorbances obtained with the standardsolutions, and determine the amount of protein in thesample solution from this curve.
Speciˆc activity (unit/mg protein)
=units per mg, obtained in the Assayamount (z) of protein in the sample
×100
Change the Toxicity to read:
Abnormal toxicity Dissolve Serum Gonadotrophin inisotonic sodium chloride solution so that each 5 mL of thesolution contains 4000 Units, and use this solution as thesample solution. Inject 5.0 mL of the sample solution intothe peritoneal cavity of each of 2 or more of well-nourished,healthy guinea pigs weighing about 350 g, and inject 0.5 mLof the sample solution into the peritoneal cavity of each of 2or more of well-nourished, healthy mice aged about 5 weeks.Observe the conditions of the animals for more than 7 days:all the animals exhibit no abnormalities.
Change the Pyrogen to read:
Bacterial endotoxins Less than 0.1 EU/unit.
Serum Gonadotrophin for Injection注射用血清性性腺刺激ホルモン
Change the Description to read:
Description Serum Gonadotrophin for Injection occurs aswhite powder or masses.
Add the following next to Identiˆcation:
pH Dissolve 30 mg of Serum Gonadotrophin for Injectionin 20 mL of isotonic sodium chloride solution: the pH of thissolution is between 5.0 and 7.0.
Delete the Pyrogen and add the following:
Bacterial endotoxins Less than 0.1 EU/unit.

17661766 Supplement II, JPXIVO‹cial Monographs for Part II
Add the following:
Lindera Root
Linderae Radix
ウヤク
Lindera Root is the root of Lindera strychnifoliaFernandez-Villar (Lauraceae).
Description Fusiform or rosary-like root, 10 – 15 cm inlength, 10 – 25 mm in diameter; externally yellowish brownto brown, with a few scars of rootlets; a transverse sectionreveals cortex brown, xylem light yellowish brown, concen-tric circles and radially arranged lines brown; dense and hardin texture.
Odor, camphor-like; taste, bitter.Under a microscope, a transverse section of the root with
periderm reveals a cork layer several cells thick, partiallyconsisting of cork stone cells; cortex parenchyma sometimescontains oil cells and ˆbers; in xylem, vessels-xylem ˆbersand rays are arranged alternately; parenchyatous cells ofcortex and xylem contain sandy and columnar crystals ofcalcium oxalate, simple starch grains 1 – 15 mm in diameter,and 2- to 4- compound starch grains.
Identiˆcation To 3 g of pulverized Lindera Root add40 mL of hexane, and warm under a re‰ux condenser on awater bath for 30 minutes. After cooling, ˆlter, to theresidue add 10 mL of ammonia TS and 30 mL of a mixtureof ethyl acetate and diethyl ether (1:1), shake vigorously for20 minutes, and centrifuge. Separate the supernatant liquid,add 10 g of anhydrous sodium sulfate, shake, and ˆlter.Evaporate the ˆltrate, dissolve the residue with 0.5 mL ofethanol (99.5), and use this solution as the sample solution.Perform the test with this solution as directed under theThin-layer Chromatography. Spot 20 mL of the samplesolution on a plate of silica gel for thin-layer chro-matography, develop the plate with a mixture of ethylacetate, methanol and ammonia water (28) (10:2:1) to adistance of about 10 cm, and air-dry the plate. Spray evenlyDragendorŠ's TS for spraying on the plate: a yellow-brownspot appears at around Rf 0.4.
Loss on drying Not more than 14.0z (6 hours).
Total ash Not more than 2.5z.
Extract content Not less than 6.0z (dilute ethanol-solubleextract).
Add the following:
Lonicera Leaf and Stem
Lonicerae Folium Cum Caulis
ニンドウ
Lonicera Leaf and Stem is the leaves and stems ofLonicera japonica Thunberg (Caprifoliaceae).
Description Leaves and opposite leaves on short stem;leaf, ovate and entire, with short petiole, 3 – 7 cm in length,1 – 3 cm in width; upper surface greenish brown, lowersurface light grayish green; under a magnifying glass, bothsurfaces pubescent. Stem, 1 – 4 mm in diameter; externallygrayish yellow-brown to purplish brown, a transversesection of stem, round and hollow.
Almost odorless; taste, slightly astringent, followed by alitter bitterness.
Under a microscope, a transverse section of leaf revealsthe outermost layer of upper and lower surfaces to be com-posed of a single-layered epidermis, uni-cellular non-glandu-lar hairs and multi-cellular glandular hairs on epidermis; inmidvein, several-layered collenchyma present beneath theepidermis and vascular bundles in the center; in mesophyll,palisade layer adjacent to upper epidermis, spongy tissue ad-jacent to lower epidermis; glandular hairs contain brownsecretion, parenchymatous cells contain aggregate crystalsof calcium oxalate, and occasionally starch grains.
Identiˆcation To 1 g of pulverized Lonicera Leaf and Stemadd 5 mL of methanol, shake for 5 minutes, centrifuge, anduse the supernatant liquid as the sample solution. Separate-ly, dissolve 1 mg of chlorogenic acid for thin-layer chro-matography in 2 mL of methanol, and use this solution asthe standard solution (1). Separately, dissolve 1 mg ofloganin for thin-layer chromatography in 2 mL of methanol,and use this solution as the standard solution (2). Performthe test with these solutions as directed under the Thin-layerChromatography. Spot 10 mL each of the sample solution,the standard solution (1) and the standard solution (2) on aplate of silica gel for thin-layer chromatography. Developthe plate with a mixture of ethyl acetate, water and formicacid (6:1:1) to a distance of about 10 cm, and air-dry theplate. Examine under ultraviolet light (main wavelength:365 nm): one of the spot among the several spots from thesample solution has the same color tone and Rf value withthe blue-white ‰uorescent spot from the standard solution(1). Spray evenly 4-methoxybenzaldehyde-sulfuric acid TSon the plate, and heat at 1059C for 5 minutes: one of thespot among the several spots from the sample solution hasthe same color tone and Rf value with the red-purple spotfrom the standard solution (2).
Purity Stem—Lonicera Leaf and Stem does not containsthe stems larger than 5 mm in diameter.
Loss on drying Not more than 12.0z (6 hours).

17671767Supplement II, JPXIV O‹cial Monographs for Part II
Total ash Not more than 9.0z.
Acid-insoluble ash Not more than 1.0z.
Extract content Not less than 12.0z (dilute ethanol-solu-ble extract).
Add the following:
Lycium Bark
Lycii Cortex
ジコッピ
Lycium Bark is the root bark of Lycium chinenseMiller or Lycium barbarum Linn áe (Solanaceae).
Description Tubular to semitubular bark, 1 – 6 mm inthickness; externally light brown to light yellowish brown,periderm peeled easily as scale; internally grayish brown,longitudinally striate; brittle in texture; fractured surface,grayish white, not ˆbrous.
Odor, weak and characteristic; taste, slightly sweet at ˆrst.Under a microscope, a transverse section reveals periderm
composed of a cork layer of several layers of thin walledcork cells; in cortex parenchymatous cells containing sandycrystals of calcium oxalate sparsely distributed, occasionallya few ˆbers observed; parenchymatous cells contain starchgrains, 1 – 10 mm in diameter; stone cells very rare.
Identiˆcation To 1.0 g of pulverized Lycium Bark add 10mL of methanol, shake for 15 minutes, ˆlter, and use theˆltrate as the sample solution. Perform the test with thissolution as directed under the Thin-layer Chromatography.Spot 10 mL of the sample solution on a plate of silica gel forthin-layer chromatography. Develop the plate with a mix-ture of 1-butanol, water, pyridine and acetic acid (100)(3:1:1:1) to a distance of about 10 cm, and air-dry the plate.Spray evenly DragendorŠ's TS for spraying on the plate,heat at 1059C for 3 minutes, then spray evenly sodium nitriteTS: a dark brown principal spot appears at around Rf 0.5.
Loss on drying Not more than 11.5z (6 hours).
Total ash Not more than 20.0z.
Acid-insoluble ash Not more than 3.0z.
Extract content Not less than 10.0z (dilute ethanol-solu-ble extract).
Add the following:
Lycium Fruit
Lycii Fructus
クコシ
Lycium Fruit is the fruit of Lycium chinense Milleror Lycium barbarum Linne (Solanaceae).
Description Fusiform fruit with acute apex, 6 – 20 mm inlength, 3 – 8 mm in diameter, pericarp red to dark red,externally roughly wrinkled; under a magnifying glass, atransverse section of fruit reveals two locules containingnumerous seeds; seed light brown to light yellowish brown,about 2 mm in a diameter, compressed reniform.
Odor, characteristic; taste, sweet, later slightly bitter.
Identiˆcation To 1.0 g of powdered Lycium Fruit add 5mL of ethyl acetate, shake for 15 minutes, ˆlter, and use theˆltrate as the sample solution. Perform the test with thissolution as directed under the Thin-layer Chromatography.Spot 20 mL of the sample solution on a plate of silica gel forthin-layer chromatography, develop the plate with a mixtureof hexane and ethyl acetate (10:1) to a distance of about10 cm, and air-dry the plate: a yellow principal spot appearsat around Rf 0.6.
Purity Foreign matter—It contains not more than 2.0z offoreign matter such as peduncle or others.
Total ash Not more than 8.0z.
Acid-insoluble ash Not more than 1.0z.
Extract content Not less than 35.0z (dilute ethanol-solu-ble extract).
Opium Alkaloids Hydrochlorides塩酸アヘンアルカロイド
Change the origin/limits of content to read:
Opium Alkaloids Hydrochlorides consists of thehydrochlorides of some of the main alkaloids obtainedfrom opium.
It contains not less than 47.0z and not more than52.0z of morphine (C17H19NO3: 285.34), and not lessthan 35.0z and not more than 41.0z of other opiumalkaloids.
Change the Description to read:
Description Opium Alkaloids Hydrochlorides occur as awhite to light brown powder.
It is soluble in water, and slightly soluble in ethanol (99.5).It is colored by light.

17681768 Supplement II, JPXIVO‹cial Monographs for Part II
Change the Identiˆcation (1) to read:
Identiˆcation (1) Dissolve 0.1 g of Opium AlkaloidsHydrochlorides in 10 mL of diluted ethanol (1 in 2), and usethis solution as the sample solution. Separately, dissolve60 mg of Morphine Hydrochloride, 40 mg of NoscapineHydrochloride, 10 mg of Codein Phosphate and 10 mg ofPapaverine Hydrochloride in 10 mL each of diluted ethanol(1 in 2), and use these solutions as the standard solutions (1),(2), (3) and (4), respectively. Perform the test with thesesolutions as directed under the Thin-layer Chromatography.Spot 20 mL each of the sample solution and the standardsolutions on a plate of silica gel with ‰uorescent indicatorfor thin-layer chromatography. Develop the plate with amixture of acetone, toluene, ethanol (99.5) and ammoniasolution (28) (20:20:3:1) to a distance of about 10 cm, andair-dry the plate. Examine under ultraviolet light (mainwavelength: 254 nm): each spot from the sample solution isthe same in color tone and Rf value with the correspondingspot from the standard solutions (1), (2), (3) and (4) (mor-phine, noscapine, codeine and papaverine).
Change the Assay to read:
Assay Weigh accurately about 0.1 g of Opium AlkaloidsHydrochlorides, and dissolve in water to make exactly 50mL, and use this solution as the sample solution. Separately,weigh accurately about 60 mg of morphine hydrochloridefor assay, dissolve in water to make exactly 50 mL, and usethis solution as the standard solution. Perform the test withexactly 20 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography ac-cording to the following conditions, and determine the peakareas of morphine, codeine, papaverine, thebaine, narceineand noscapine, AT1, AT2, AT3, AT4, AT5 and AT6, from thesample solution, and the peak area of morphine, AS, fromthe standard solution.
Amount (mg) of morphine (C17H19NO3)
=WS×AT1
AS×0.887
Amount (mg) of other opium alkaloids=WS
×AT2+AT3×0.29+AT4×0.20+AT5×0.19+AT6
AS×0.887
WS: Amount (mg) of morphine hydrochloride for assay,calculated on the anhydrous basis
The relative retention time of codine, papaverine, the-baine, narceine and noscapine with respect to morphineobtained under the following operating conditions are asfollows.
Component Relative retention timecodeine 1.1papaverine 1.9thebaine 2.5narceine 2.8
noscapine 3.6
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 285 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 1.0 g of sodium lauryl sulfate in500 mL of diluted phosphoric acid (1 in 1000), and adjustthe pH to 3.0 with sodium hydroxide TS. To 240 mL of thissolution add 70 mL of tetrahydrofuran, and mix.
Flow rate: Adjust the ‰ow rate so that the retention timeof morphine is about 10 minutes.System suitability—
System performance: Dissolve 60 mg of MorphineHydrochloride, 10 mg of Codeine Phosphate, 10 mg ofPapaverine Hydrochloride and 40 mg of NoscapineHydrochloride in water to make 50 mL. When the procedureis run with 20 mL of this solution under the above operatingconditions, morphine, codeine, papaverine and noscapineare eluted in this order with the complete separation betweenthese peaks and with the resolution between the peaks ofmorphine and codeine being not less than 1.5.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution under the above operat-ing conditions, the relative standard deviation of the peakarea of morphine is not more than 1.0z.
Powdered Peony Rootシャクヤク末
Change the origin/limits of content to read:
Powdered Peony Root is the powder of Peony Root.It contains not less than 2.0z of paeoni‰orin
(C23H28O11: 480.46), calculated on the dried basis.
Change the Assay to read:
Assay Weigh accurately about 0.5 g of Powdered PeonyRoot, add 50 mL of diluted methanol (1 in 2), heat under are‰ux condenser on a water bath for 30 minutes, cool, andˆlter. To the residue add 50 mL of diluted methanol (1 in 2),and proceed in the same manner. Combine the ˆltrates, adddiluted methanol (1 in 2) to make exactly 100 mL, and usethis solution as the sample solution. Separately, weigh ac-curately about 10 mg of Paeoni‰orin Reference Standard(separately detarmine the water content), dissolve in dilutedmethanol (1 in 2) to make exactly 100 mL, and use thissolution as the standard solution. Perform the test withexactly 10 mL each of the sample solution and the standardsolution as directed under the Liquid Chromatography

17691769Supplement II, JPXIV O‹cial Monographs for Part II
according to the following conditions. Determine the peakareas, AT and AS, of paeoni‰orin in each solution.
Amount (mg) of paeoni‰orin (C23H28O11)
=WS×AT
AS
WS: Amount (mg) of Paeoni‰orin Reference Standard,calculated on the anhydrous basis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 232 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about209C.
Mobile phase: A mixture of water, acetonitrile andphosphoric acid (850:150:1).
Flow rate: Adjust the ‰ow rate so that the retention timeof paeoni‰orin is about 10 minutes.System suitability—
System performance: Dissolve 1 mg each of Paeoni‰orinReference Standard and albi‰orin in diluted methanol (1 in2) to make 10 mL. When the procedure is run with 10 mL ofthis solution under the above operating conditions, albi‰orinand paeoni‰orin are eluted in this order with the resolutionbetween these peaks being not less than 2.5.
System repeatability: When the test is repeated 6 timeswith the standard solution under the above operatingconditions, the relative standard deviation of the peak areaof paeoni‰orin is not more than 1.5z.
Change to read:
Potato Starch
Amylum Solani
バレイショデンプン
Potato Starch consists of starch granules derivedfrom the tuber of Solanum tuberosum Linn áe(Solanaceae).
Description Potato Starch occurs as a white powder.It is practically insoluble in water and in ethanol (99.5).
Identiˆcation (1) Under a microscope, Potato Starch,preserved in a mixture of water and glycerin (1:1), appears asunevenly ovoid or pyriform simple grains usually 30 – 100mm, often more than 100 mm in diameter, or spherical simplegrains 10 – 35 mm in diameter, rarely 2- to 4-compoundgrains; ovoid or pyriform simple grains with eccentric hilum,spherical simple grains with non-centric or slightly eccentrichilum; striation distinct in all grains; a black cross, itsintersection point on hilum, is observed when grains are put
between two polarizing prisms ˆxed at right angle to eachother.
(2) To 1 g of Potato Starch add 50 mL of water, boil for1 minute, and allow to cool: a subtle white-turbid, pastyliquid is formed.
(3) To 1 mL of the pasty liquid obtained in (2) add 0.05mL of iodine TS: a orange-red to dark blue-purple color isformed, and the color disappears by heating.
pH Put 5.0 g of Potato Starch in a non-metal vessel, add25.0 mL of freshly boiled and cooled water, mix gently for 1minute, and allow to stand for 15 minutes: the pH of thesolution is between 5.0 and 8.0.
Purity (1) Iron—To 1.5 g of Potato Starch add 15 mL of2 mol/L hydrochloric acid TS, mix, ˆlter, and use the ˆltrateas the test solution. To 2.0 mL of Standard Iron Solutionadd water to make 20 mL, and use as the control solution.Put 10 mL each of the test solution and the control solutionin test tubes, add 2 mL of a solution of citric acid (2 in 10)and 0.1 mL of mercapto acetic acid, and mix. Alkalize withammonia solution (28) to litmus paper, add water to make20 mL, and mix. Transfer 10 mL each of these solutions intotest tubes, allow to stand for 5 minutes, and compare thecolor of these solutions against a white background: thecolor of the test solution is not darker than that of thecontrol solution (not more than 10 ppm).
(2) Oxidizing substances—To 4.0 g of Potato Starch add50.0 mL of water, shake for 5 minutes, and centrifuge. To30.0 mL of the supernatant liquid add 1 mL of acetic acid(100) and 0.5 to 1.0 g of potassium iodide, shake, and allowto stand for 25 to 30 minutes at a dark place. Add 1 mL ofstarch TS, and titrate with 0.002 mol/L sodium thiosulfateVS until the color of the solution disappears. Perform ablank determination and make any necessary correction: thevolume of 0.002 mol/L sodium thiosulfate VS consumed isnot more than 1.4 mL (not more than 20 ppm, calculated ashydrogen peroxide).
(3) Sulfur dioxide—(i) Apparatus Use as shown in the following ˆgure.

17701770 Supplement II, JPXIVO‹cial Monographs for Part II
The ˆgures are in mm.
A: Boiling ‰ask (500 mL)B: Funnel (100 mL)C: CondenserD: Test-tubeE: Tap
(ii) Procedure Introduce 150 mL of water into theboiling ‰ask, close the tap of the funnel, and pass carbondioxide through the whole system at a rate of 100±5 mL perminute. Pass cooling water through the condenser, and place10 mL of hydrogen peroxide-sodium hydroxide TS in thetest-tube. After 15 minutes, remove the funnel without inter-rupting the stream of carbon dioxide, and introduce throughthe opening into the ‰ask about 25 g of Potato Starch,accurately weighed, with the aid of 100 mL of water. Applytap grease to the outside of the connection part of thefunnel, and load the funnel. Close the tap of the funnel,pour 80 mL of 2 mol/L hydrochloric acid TS into thefunnel, open the tap to introduce the hydrochloric acid intothe ‰ask, and close the tap while several mL of thehydrochloric acid remains, in order to avoid losing sulfurdioxide. Place the ‰ask in a water bath, and heat the mixturefor 1 hour. Transfer the contents of the test-tube with the aidof a little water to a wide-necked conical ‰ask. Heat in awater bath for 15 minutes, and cool. Add 0.1 mL ofbromophenol blue TS, and titrate with 0.1 mol/L sodiumhydroxide VS until the color changes from yellow to violet-blue lasting for at least 20 seconds. Perform a blank determi-nation and make any necessary correction. Calculate theamount of sulfur dioxide by applying the following formula:it is not more than 50 ppm.
Amount (ppm) of sulfur dioxide=VW×1000×3.203
W: Amount (g) of the sampleV: Amount (mL) of 0.1 mol/L sodium hydroxide VS
consumed
Loss on drying Not more than 20.0z (1 g, 1309C, 90
minutes).
Residue on ignition Not more than 0.6z (1 g).
Containers and storage Containers—Well-closed contain-ers.
Add the following:
Processed Aconite Root
Processi Aconiti Radix
ブシ
Processed Aconite Root is the tuberous root ofAconitum carmichaeli Debeaux or Aconitumjaponicum Thunberg (Ranunculaceae) prepared bythe following processes.
Process 1: Autoclaving. [Processed Aconite Root 1]Process 2: Heating or autoclaving after rinsing in
the salt, rock salt or calcium chloride water. [Proc-essed Aconite Root 2]
Process 3: Treating with lime after rinsing in the saltwater. [Processed Aconite Root 3]
There are three kinds of Processed Aconite Rootbased on their preparing processes as shown above.
Processed Aconite Root 1, Processed Aconite Root2 and Processed Aconite Root 3 contain the totalalkaloid [as benzoylaconine (C32H45NO10: 603.70)] ofnot less than 0.7z and not more than 1.5z, not lessthan 0.1z and not more than 0.6z, and not less than0.5z and not more than 0.9z, calculated on the driedbases, respectively.
The label indicates the treating process.
DescriptionProcessed Aconite Root 1: Cut pieces irregularly polygonal,less than 10 mm in diameter; externally dark grayish brownto blackish brown; hard in texture; cut surface ‰at, lightbrown to dark brown, usually horny and lustrous.
Odor, weak and characteristic.Under a microscope, transverse and longitudinal sections
reveal pitted, scaraliform, reticulate and spiral vessels;starch grains in parenchymatous cells usually gelatinized butsometimes not gelatinized; starch grains, simple, spherical orellipsoid, 2 – 25 mm in diameter, or 2- to a dozen or so- com-pound, hilum of starch grain distinct.
Processed Aconite Root 2: Nearly obconical root, 15 – 30mm in length, 12 – 16 mm in diameter, slices cut longitudi-nally or transversely, 20 – 60 mm in length, 15 – 40 mm inwidth, and 200 – 700 mm in thickness, or cut pieces irregular-ly polygonal, less than 12 mm in diameter; externally lightbrown to dark brown or yellowish brown; hard in texture,usually without wrinkles; cut surface ‰at, light brown todark brown or yellowish white to light yellowish brown, usu-ally horny, semi-transparent and lustrous.
Odor, weak and characteristic.

17711771Supplement II, JPXIV O‹cial Monographs for Part II
Under a microscope, transverse and longitudinal sectionsreveal metaderm, primary cortex, endodermis, secondarycortex, cambium, and xylem; primary cortex containsoblong to oblong-square sclerenchymatous cells, 30 – 75 mmin short axis, 60 – 150 mm in long axis; endodermis singlelayered, endodermal cells elongated in tangential direction;cambium, star shaped or irregular polygons to orbicular; agroup of vessel in xylem v-shaped; sometimes isolated ringof cambium appears in secondary cortex or in pith; vessels,pitted, scaraliform, reticulate and spiral; starch grains inparenchymatous cells gelatinized.
Processed Aconite Root 3: Cut pieces irregularly polygonal,less than 5 mm in diameter; externally grayish brown; hardin texture; cut surface ‰at, light grayish brown to grayishwhite, not lustrous.
Odor, weak and characteristic.Under a microscope, transverse and longitudinal sections
reveal pitted, scaraliform, reticulate and spiral vessels;starch grains, simple, spherical or ellipsoid, 2 – 25 mm indiameter, or 2- to a dozen or so- compound, hilum of starchgrain distinct.
Identiˆcation To 3 g of pulverized Processed Aconite Rootadd 20 mL of diethyl ether and 2 mL of ammonia TS, shakefor 10 minutes, and centrifuge. Evaporate the ether layer todryness under reduced pressure, dissolve the residue in 1 mLof diethyl ether, and use this solution as the sample solution.Separately, dissolve 1 mg of benzoylmesaconine hydrochlo-ride for thin-layer chromatography in 10 mL of ethanol(99.5), and use this solution as the standard solution.Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 10 mL each of the samplesolution and the standard solution on a plate of silica gel forthin-layer chromatography, develop the plate with a mixtureof ethyl acetate, ethanol (99.5) and ammonia water (28)(40:3:2) to a distance of about 10 cm, and air-dry the plate.Spray evenly DragendorŠ's TS for spraying on the plate,air-dry the plate, and spray evenly sodium nitrite TS: one ofthe spot among the several spots from the sample solutionhas the same color tone and Rf value with the yellow-brownspot from the standard solution.
Purity Aconitum diester alkaloids (aconitine, jesaconitine,hypaconitine and mesaconitine)—Weigh accurately about0.5 g of pulverized Processed Aconite Root, put in a glass-stoppered centrifuge tube, suspend in 3.0 mL of water byshaking, and add 1.0 mL of ammonia TS and 20 mL ofdiethyl ether. Stopper tightly the tube, shake for 30 minutes,centrifuge, and separate the ether layer. To the residue add1.0 mL of ammonia TS and 20 mL of diethyl ether, andrepeat the above process two times. Combine all extracts,evaporate to dryness under reduced pressure at not morethan 409C, and dissolve the residue with exactly 10 mL of amixture of phosphate buŠer solution for aconite root andacetonitrile (1:1). Centrifuge this solution, and use thesupernatant liquid as the sample solution. Perform the testwith exactly 20 mL each of the sample solution and the aconi-tum diester alkaloids standard solution as directed under the
Liquid Chromatography according to the followingconditions, and determine the heights of the peaks corre-sponding to aconitine, jesaconitine, hypaconitine andmesaconitine, HTA and HSA, HTJ and HSJ, HTH and HSH, andHTM and HSM, respectively, and calculate the amounts ofthem by the following formulae: the amounts of aconitine,jesaconitine, hypaconitine and mesaconitine per g calculatedon the dried basis are not more than 60 mg, 60 mg, 280 mg and140 mg, respectively, and the total amount of them is notmore than 450 mg.
Amount (mg) of aconitine (C34H47NO11)
=CSA
W×
HTA
HSA×10
Amount (mg) of jesaconitine (C35H49NO12)
=CSJ
W×
HTJ
HSJ×10
Amount (mg) of hypaconitine (C33H45NO10)
=CSH
W×
HTH
HSH×10
Amount (mg) of mesaconitine (C33H45NO11)
=CSM
W×
HTM
HSM×10
CSA: Concentration (mg/mL) of aconitine for purity in theaconitum diester alkaloids standard solution forpurity
CSJ: Concentration (mg/mL) of jesaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
CSH: Concentration (mg/mL) of hypaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
CSM: Concentration (mg/mL) of mesaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
W: Amount (g) of the sample, calculated on the driedbasis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 231 nm for aconitine, hypaconitine andmesaconitine; 254 nm for jesaconitine).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: A mixture of phosphate buŠer solution foraconite root and tetrahydrofuran (183:17).
Flow rate: Adjust the ‰ow rate so that the retention timeof mesaconitine is about 31 minutes.System suitability—
System performance: When the procedure is run with20 mL of the aconitum diester alkaloids standard solutionfor purity under the above operating conditions, using 254

17721772 Supplement II, JPXIVO‹cial Monographs for Part II
nm, mesaconitine, hypaconitine, aconitine and jesaconitineare eluted in this order, and each resolution between theirpeaks is not less than 1.5, respectively.
System repeatability: To 1 mL of aconitum diesteralkaloids standard solution for purity add a mixture of phos-phate buŠer solution for aconite root and acetonitrile (1:1)to make 10 mL. When the test is repeated 6 times with 20 mLof this solution under the above operating conditions, using231 nm, the relative standard deviation of the peak height ofmesaconitine is not more than 1.5z.
Loss on drying Not more than 15.0z (6 hours).
Total ashProcessed Aconite Root 1: Not more than 4.0z.Processed Aconite Root 2: Not more than 12.0z.Processed Aconite Root 3: Not more than 19.0z.
Acid-insoluble ash Not more than 0.9z.
Assay Weigh accurately about 2 g of pulverized ProcessedAconite Root, put in a glass-stoppered centrifuge tube, andadd 1.6 mL of ammonia TS and 20 mL of diethyl ether.Stopper tightly the tube, shake for 30 minutes, centrifuge,and separate the ether layer. To the residue add 0.8 mL ofammonia TS and 20 mL of diethyl ether, and proceed asabove. Repeat this process more three times. Combine allextracts, evaporate to dryness under reduced pressure,dissolve the residue in 5 mL of ethanol (99.5), add 30 mL offreshly boiled and cooled water, and titrate with 0.01 mol/Lhydrochloric acid VS until the color of the solution changesfrom green to gray-blue through blue-green (indicator: 3drops of methyl red-methylene blue TS). Perform a blankdetermination and make any necessary correction.
Each mL of 0.01 mol/L hydrochloric acid VS=6.037 mg of total alkaloid [as benzoylaconine
(C32H45NO10: 603.70)]
Add the following:
Powdered Processed Aconite RootProcessi Aconiti Radix Pulverata
ブシ末
Powdered Processed Aconite Root is the powder ofProcessed Aconite Root prepared by Process 1 orProcess 2, or the powder of the tuberous root ofAconitum carmichaeli Debeaux or Aconitum japoni-cum Thunberg (Ranunculaceae) prepared by Process1.
Occasionally, it contains Corn Starch or Lactose.Process 1: Autoclaving. [Powdered Processed
Aconite Root 1]Process 2: Heating or autoclaving after rinsing in
the salt, rock salt or calcium chloride water. [Pow-dered Processed Aconite Root 2]
There are two kinds of Powdered Processed Aconite
Root based on their preparing processes as shownabove.
Powdered Processed Aconite Root 1 and PowderedProcessed Aconite Root 2 contain the total alkaloid[as benzoylaconine (C32H45NO10: 603.70)] of not lessthan 0.4z and not more than 1.2z, and not less than0.1z and not more than 0.3z, calculated on the driedbases, respectively.
The label indicates the treating process.
DescriptionPowdered Processed Aconite Root 1: Powdered ProcessedAconite Root 1 occurs as a light grayish brown powder. Ithas a characteristic odor.
Under a microscope, Powered Processed Aconite Root 1reveals gelatinized starch masses or starch grains and paren-chymatous cells containing them, fragments of reddishbrown metaderm, fragments of pitted, scaraliform, reticu-late and spiral vessels; also square to oblong-square scleren-chymatous cells, 30 – 150 mm in diameter, 100 – 250 mm inlength, cell wall of sclerenchymatous cells, 6 – 12 mm inthickness; starch grains of ``Processed Aconite Root'', sim-ple, spherical or ellipsoid, 2 – 25 mm in diameter, or 2- to adozen or so- compound, hilum of starch grain distinct.
Powdered Processed Aconite Root 2: Powdered ProcessedAconite Root 2 occurs as a light yellowish white powder. Ithas a characteristic odor.
Under a microscope, Powered Processed Aconite Root 2reveals gelatinized starch masses and parenchymatous cellscontaining them, fragments of reddish brown metaderm,fragments of pitted, scaraliform, reticulate and spiralvessels; also square to oblong-square sclerenchymatous cells,30 – 150 mm in diameter, 100 – 250 mm in length, cell wall ofsclerenchymatous cells, 6 – 12 mm in thickness.
Identiˆcation To 3 g of Powdered Processed Aconite Rootadd 2 mL of ammonia TS and 20 mL of diethyl ether, shakefor 10 minutes, and centrifuge. Evaporate the ether layer todryness under reduced pressure, dissolve the residue in 1 mLof diethyl ether, and use this solution as the sample solution.Separately, dissolve 1 mg of benzoylmesaconine hydrochlo-ride for thin-layer chromatography in 10 mL of ethanol(99.5), and use this solution as the standard solution.Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 10 mL each of the samplesolution and the standard solution on a plate of silica gel forthin-layer chromatography, develop the plate with a mixtureof ethyl acetate, ethanol (99.5) and ammonia water (28)(40:3:2) to a distance of about 10 cm, and air-dry the plate.Spray evenly DragendorŠ's TS for spraying on the plate,air-dry the plate, and spray evenly sodium nitrite TS: one ofthe spot among the several spots from the sample solutionhas the same color tone and Rf value with the yellow-brownspot from the standard solution.
Purity Aconitum diester alkaloids (aconitine, jesaconitine,hypaconitine and mesaconitine)—Weigh accurately about0.5 g of Powdered Processed Aconite Root, put in a glass-stoppered centrifuge tube, suspend in 3.0 mL of water by

17731773Supplement II, JPXIV O‹cial Monographs for Part II
shaking, and add 1.0 mL of ammonia TS and 20 mL ofdiethyl ether. Stopper tightly the tube, shake for 30 minutes,centrifuge, and separate the ether layer. To the residue add1.0 mL of ammonia TS and 20 mL of diethyl ether, andrepeat the above process two times. Combine all extracts,evaporate to dryness under reduced pressure at not morethan 409C, and dissolve the residue with exactly 10 mL of amixture of phosphate buŠer solution for aconite root andacetonitrile (1:1). Centrifuge this solution, and use thesupernatant liquid as the sample solution. Perform the testwith exactly 20 mL each of the sample solution and the aconi-tum diester alkaloids standard solution as directed under theLiquid Chromatography according to the followingconditions, and determine the heights of the peaks corre-sponding to aconitine, jesaconitine, hypaconitine andmesaconitine, HTA and HSA, HTJ and HSJ, HTH and HSH, andHTM and HSM, respectively, and calculate the amounts ofthem by the following formulae: the amounts of aconitine,jesaconitine, hypaconitine and mesaconitine per g calculatedon the dried basis are not more than 55 mg, 40 mg, 55 mg and120 mg, respectively, and the total amount of them is notmore than 230 mg.
Amount (mg) of aconitine (C34H47NO11)
=CSA
W×
HTA
HSA×10
Amount (mg) of jesaconitine (C35H49NO12)
=CSJ
W×
HTJ
HSJ×10
Amount (mg) of hypaconitine (C33H45NO10)
=CSH
W×
HTH
HSH×10
Amount (mg) of mesaconitine (C33H45NO11)
=CSM
W×
HTM
HSM×10
CSA: Concentration (mg/mL) of aconitine for purity in theaconitum diester alkaloids standard solution forpurity
CSJ: Concentration (mg/mL) of jesaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
CSH: Concentration (mg/mL) of hypaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
CSM: Concentration (mg/mL) of mesaconitine for purity inthe aconitum diester alkaloids standard solution forpurity
W: Amount (g) of the sample, calculated on the driedbasis
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 231 nm for aconitine, hypaconitine andmesaconitine; 254 nm for jesaconitine).
Column: A stainless steel column 4.6 mm in insidediameter and 15 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm in
particle diameter).Column temperature: A constant temperature of about
409C.Mobile phase: A mixture of phosphate buŠer solution for
aconite root and tetrahydrofuran (183:17).Flow rate: Adjust the ‰ow rate so that the retention time
of mesaconitine is about 31 minutes.System suitability—
System performance: When the procedure is run with 20mL of the aconitum diester alkaloids standard solution forpurity under the above operating conditions, using 254 nm,mesaconitine, hypaconitine, aconitine and jesaconitine areeluted in this order, and each resolution between their peaksis not less than 1.5, respectively.
System repeatability: To 1 mL of aconitum diesteralkaloids standard solution for purity add a mixture ofphosphate buŠer solution for aconite root and acetonitrile(1:1) to make 10 mL. When the test is repeated 6 times with20 mL of this solution under the above operating conditions,using 231 nm, the relative standard deviation of the peakheight of mesaconitine is not more than 1.5z.
Loss on drying Not more than 11.0z (6 hours).
Total ashPowdered Processed Aconite Root 1: Not more than
4.0z.Powdered Processed Aconite Root 2: Not more than
7.0z.
Acid-insoluble ash Not more than 0.7z.
Assay Weigh accurately about 2 g of Powdered ProcessedAconite Root, put in a glass-stoppered centrifuge tube, andadd 1.6 mL of ammonia TS and 20 mL of diethyl ether.Stopper tightly the tube, shake for 30 minutes, centrifuge,and separate the ether layer. To the residue add 0.8 mL ofammonia TS and 20 mL of diethyl ether, and proceed asabove. Repeat this process more three times. Combine allextracts, evaporate to dryness under reduced pressure,dissolve the residue in 5 mL of ethanol (99.5), add 30 mL offreshly boiled and cooled water, and titrate with 0.01 mol/Lhydrochloric acid VS until the color of the solution changesfrom green to gray-blue through blue-green (indicator: 3drops of methyl red-methylene blue TS). Perform a blankdetermination and make any necessary correction.
Each mL of 0.01 mol/L hydrochloric acid VS=6.037 mg of total alkaloid [as benzoylaconine
(C32H45NO10: 603.70)]

17741774 Supplement II, JPXIVO‹cial Monographs for Part II
Add the following:
Processed Ginger
Zingiberis Processum Rhizoma
カンキョウ
Processed Ginger is the rhizome of Zingibero‹cinale Roscoe (Zingiberaceae), after being passedthrough hot water or being steamed.
Description Irregularly compressed and often branchedmassive rhizome; branched parts slightly curved ovoid oroblong-ovoid, 2 – 4 cm in length, and 1 – 2 cm in diameter;external surface grayish yellow to grayish yellow-brown,with wrinkles and ring node; fractured surface brown todark brown, transparent and horny; under a magnifyingglass, a transverse section reveals cortex and stele distinctlydivided; vascular bundles scattered throughout the surface.
Odor, characteristic; taste, extremely pungent.Under a microscope, a transverse section reveals cork
layer, cortex and stele in this order from the outside; cortexand stele, divided by a single-layered endodermis, composedof parenchyma, vascular bundles scattered and surroundedby ˆber bundles; oil cells contain yellow oil-like substances,scattered in parenchyma; parenchymatous cells containsolitary crystals of calcium oxalate, and gelatinized starch.
Identiˆcation To 2 g of pulverized Processed Ginger add 5mL of diethyl ether, shake for 10 minutes, ˆlter, and use theˆltrate as the sample solution (1). To the residue add 5 mL ofmethanol, proceed in the same manner as above, and use soobtained solution as the sample solution (2). Separately,dissolve 1 mg of [6]-shogaol for thin-layer chromatographyin 2 mL of methanol, and use this solution as the standardsolution (1). Separately, dissolve 1 mg of Sucrose in 2 mL ofmethanol, and use this solution as the standard solution (2).Perform the test with these solutions as directed under theThin-layer Chromatography. Spot 10 mL each of the samplesolution (1) and the standard solution (1) on a plate of silicagel for thin-layer chromatography. Develop the plate with amixture of ethyl acetate and hexane (1:1) to a distance ofabout 10 cm, and air-dry the plate. Spray evenly 4-dimethylaminobenzaldehyde TS on the plate, heat at 1059Cfor 5 minutes, and allow to cool: one of the spot among theseveral spots from the sample solution (1) has the same colortone and Rf value with the green spot from the standardsolution (1). Spot 10 mL each of the sample solution (2) andthe standard solution (2) on a plate of silica gel for thin-layerchromatography, develop the plate with a mixture of 1-butanol, water and acetic acid (100) (8:5:3) to a distance ofabout 10 cm, and air-dry the plate. Spray evenly 1,3-naphthalenediol TS on the plate, and heat at 1059C for 5minutes: one of the spot among the several spots from thesample solution (2) has the same color tone and Rf valuewith the red-purple spot from the standard solution (2).
Loss on drying Not more than 15.0z (6 hours).
Total ash Not more than 6.5z.
Acid-insoluble ash Not more than 1.5z.
Extract content Not less than 8.0z (dilute ethanol-solubleextract).
Rhubarbダイオウ
Change the origin/limits of content to read:
Rhubarb is usually the rhizome of Rheum palmatumLinn áe, Rheum tanguticum Maximowicz, Rheumo‹cinale Baillon, Rheum coreanum Nakai or theirinterspeciˆc hybrids (Polygonaceae).
It contains not less than 0.25z of sennosides A(C42H38O20: 862.74), calculated on the basis of driedmaterial.
Powdered Rhubarbダイオウ末
Change the origin/limits of content to read:
Powdered Rhubarb is the powder of Rhubarb.It contains not less than 0.25z of sennoside A
(C42H38O20: 862.74), calculated on the basis of driedmaterials.
Add the following:
Sappan Wood
Sappan Lignum
ソボク
Sappan Wood is the duramen of Caesalpinia sappanLinn áe (Leguminosae).
Description Chips, slices or short pieces of wood; yellow-ish red to grayish yellow-brown, sometimes with light brownto grayish white splint woods; hard in texture; a transversesection shows a pattern like annual ring.
Almost odorless; almost tasteless.Under a microscope, a transverse section reveals ray
composed of 1 – 2 rows of slender and long cells; the areabetween rays ˆlled with ˆber cells, and large and oblongvessels scattered there; solitary crystals of calcium oxalate inparenchymatous cells of the innermost of xylem.
Identiˆcation To 0.5 g of pulverized Sappan Wood add 10mL of dilute ethanol, shake, and ˆlter. To 5 mL of theˆltrate add 2 to 3 drops of sodium hydroxide TS: a dark red

17751775Supplement II, JPXIV O‹cial Monographs for Part II
color develops.
Purity Put a small piece of Sappan Wood in calciumhydroxide TS: no purple-blue color develops.
Loss on drying Not more than 11.5z (6 hours).
Total ash Not more than 2.0z.
Extract content Not less than 7.0z (dilute ethanol-solubleextract).
Powdered Scutellaria Rootオウゴン末
Change the origin/limits of content to read:
Powdered Scutellaria Root is the powder of Scutel-laria Root.
It contains not less than 10.0z of baicalin(C21H18O11: 446.36), calculated on the basis of driedmaterial.
Add the following:
Termeric
Curcumae Rhizoma
ウコン
Termeric is the rhizome or being removed the corklayer from it of Curcuma longa Linn áe (Zingiberaceae),usually after being passed through hot water.
Description Termeric is a main rhizome or a lateral rhi-zome; main rhizome, nearly ovoid, about 3 cm in diameter,about 4 cm in length; lateral rhizome, cylindrical, withround tips, curved, about 1 cm in diameter, 2 - 6 cm inlength; both main and lateral rhizomes with cyclic nodes;rhizome with cork layer, yellowish brown, lustrous; rhizomewithout cork layer, dark yellowish red, with yellowish redpowders on surface; hard in texture, not easily broken;transversely cut surface yellowish brown to reddish brown,lustrous like wax.
Odor, characteristic; taste, slightly bitter and stimulant, itcolors a saliva yellow on chewing.
Under a microscope, a transverse section reveals theoutermost layer to be composed of a cork layer 4 – 10 cellsthick; sometimes a cork layer partly remains; cortex andstele, divided by a single-layered endodermis, composed ofparenchyma, vascular bundles scattered; oil cells scattered inparenchyma; parenchymatous cells contain yellow sub-stances, sandy and solitary crystals of calcium oxalate, andgelatinized starch.
Identiˆcation To 0.5 g of pulverized Termeric add 20 mLof methanol, shake for 15 minutes, ˆlter and use the ˆltrate
as the sample solution. Perform the test with this solution asdirected under the Thin-layer Chromatography. Spot 5 mLof the sample solution on a plate of silica gel for thin-layerchromatography. Develop the plate with a mixture of ethylacetate, hexane and acetic acid (100) (70:30:1) to a distanceof about 10 cm, and air-dry the plate: a yellow spot appearsat around Rf 0.4.
Loss on drying Not more than 17.0z (6 hours).
Total ash Not more than 7.5z.
Acid-insoluble ash Not more than 1.0z.
Extract content Not less than 9.0z (dilute ethanol-solubleextract).
Add the following:
Tribulus Fruit
Tribuli Fructus
シツリシ
Tribulus Fruit is the fruit of Tribulus terrestrisLinn áe (Zygophyllaceae).
Description Pentagonal star shaped fruit, composed of ˆvemericarps, 7 – 12 mm in diameter, often each mericarpseparated; externally grayish green to grayish brown; a pairof longer and shorter spines on surface of each mericarp, thelonger spine 3 – 7 mm in length, the shorter one 2 – 5 mm inlength, numerous small processes on midrib; pericarp hardin texture, cut surface light yellow; each mericarp contains 1– 3 seeds.
Almost odorless; taste, mild at ˆrst, followed by bitter-ness.
Under a microscope, a transverse section reveals epicarpcomposed of a single-layered epidermis; mesocarp com-posed of parenchyma and sclerenchyma layer; endocarpcomposed of several-layered ˆber cells; a single-layer of cellbetween mesocarp and endocarp contain solitary crystals ofcalcium oxalate; cotyledons of seed contain oil drops andaleurone grains, and occasionally starch grains.
Identiˆcation To 2 g of pulverized Tribulus Fruit add 5 mLof methanol, shake for 10 minutes, ˆlter, and use the ˆltrateas the sample solution. Perform the test with this solution asdirected under the Thin-layer Chromatography. Spot 10 mLof the sample solution on a plate of silica gel for thin-layerchromatography, develop the plate with a mixture of ethylacetate and water (40:1) to a distance of about 10 cm, andair-dry the plate. Spray evenly dilute sulfuric acid on theplate, heat at 1059C for 5 minutes, and examine underultraviolet light (main wavelength: 365 nm): a blue-white‰uorescent spot appears at around Rf 0.4.
Purity (1) Peduncle—Not more than 4.0z.(2) Foreign matters—Not more than 1.0z of foreign

17761776 Supplement II, JPXIVO‹cial Monographs for Part II
matters other than peduncle.
Loss on drying Not more than 11.0z (6 hours).
Total ash Not more than 13.0z.
Acid-insoluble ash Not more than 1.5z.
Extract content Not less than 8.5z (dilute ethanol-solubleextract).
Uncaria Thornチョウトウコウ
Change the origin/limits of the content to read:
Uncaria Thorn is, usually the prickle, of Uncariarhynchophylla Miquel, Uncaria sinensis Haviland orUncaria macrophylla Wallich (Rubiaceae).
Uncaria Thorn contains not less than 0.03z of totalalkaloids (rhynchophylline and hirstine), calculated onthe dried basis.
Change the Component determination to read:
Component determination Weigh accurately about 0.2 gof medium powdered Uncaria Thorn, transfer into a glass-stoppered centrifuge tube, add 30 mL of a mixture ofmethanol and dilute acetic acid (7:3), shake for 30 minutes,centrifuge, and separate the supernatant liquid. To theresidue add two 10-mL portions of a mixture of methanoland dilute acetic acid (7:3), proceed in the same manner, andcombine all of the supernatant liquid. To the combinedliquid add a mixture of methanol and dilute acetic acid (7:3)to make exactly 50 mL, and use this as the sample solution.Separately, weigh accurately about 5 mg of rhynchophyllinefor component determination, previously dried in a desicca-tor (silica gel) for 24 hours, and dissolve in a mixture ofmethanol and dilute acetic acid (7:3) to make exactly 100mL. Pipet 1 mL of this solution, add a mixture of methanoland dilute acetic acid (7:3) to make exactly 10 mL, and usethis solution as the standard solution (1). Separately, dis-solve 1 mg of hirsutine in 100 mL of a mixture of methanoland dilute acetic acid (7:3), and use this solution as the stan-dard solution (2). Perform the test with exactly 20 mL eachof the sample solution and the standard solution (1) and (2)as directed under the Liquid Chromatography according tothe following conditions, and determine the peak areas, ATa
and ATb, of rhynchophylline and hirsutine obtained from thesample solution, and the peak area, AS, of rhynchophyllinefrom the standard solution (1).
Amount (mg) of total alkaloids (rhynchophylline andhirstine)
=WS×ATa+1.405ATb
AS×
120
WS: Amount (mg) of rhynchophylline for componentdetermination
Operating conditions—Detector: An ultraviolet absorption photometer
(wavelength: 245 nm).Column: A stainless steel column 4.6 mm in inside
diameter and 25 cm in length, packed with octadecyl-silanized silica gel for liquid chromatography (5 mm inparticle diameter).
Column temperature: A constant temperature of about409C.
Mobile phase: Dissolve 3.85 g of ammonium acetate in200 mL of water, add 10 mL of acetic acid (100) and waterto make 1000 mL, and add 350 mL of acetonitrile.
Flow rate: Adjust the ‰ow rate so that the retention timeof rhynchophylline is about 17 minutes.System suitability—
System performance: Dissolve 5 mg of rhynchophyllinefor component determination in 100 mL of a mixture ofmethanol and dilute acetic acid (7:3). To 5 mL of this solu-tion add 1 mL of ammonia solution (28), and re‰ux for 10minutes or warm at about 509C for 2 hours. After cooling,to 1 mL of the solution so obtained add a mixture ofmethanol and dilute acetic acid (7:3) to make 5 mL. Whenthe procedure is run with 20 mL of this solution under theabove operating conditions, the peak of isorhynchophyllineis appears in addition to the peak of rhynchophylline, andthe resolution between these peaks is not less than 1.5.
System repeatability: When the test is repeated 6 timeswith 20 mL of the standard solution (1) under the aboveoperating conditions, the relative standard deviation of thepeak areas of rhynchophylline is not more than 1.5z.
Change to read:
Wheat Starch
Amylum Tritici
コムギデンプン
Wheat Starch consists of the starch granulesobtained from the seeds of Triticum aestivum Linn áe(Gramineae).
Description Wheat Starch occurs as white masses orpowder.
It is practically insoluble in water and in ethanol (99.5).
Identiˆcation (1) Under a microscope, Wheat Starch,preserved in a mixture of water and glycerin (1:1), appears aslarge and small sized simple grains, or quite rarely mediansized simple grains; usually, large sized grains usually 10 – 60mm in diameter, from upper view, disc like or quite rarelyreniform, centric hilum and striation indistinct or hardlydistinct, often cleft on marginal portion visible; from lateralview, narrowly ellipsoid or fusiform, hilum recognized as acleft along with long axis; small sized grains 2 – 10 mm indiameter, spherical or polygonal; a black cross, its intersec-tion point on hilum, is observed when grains are put between

17771777Supplement II, JPXIV O‹cial Monographs for Part II
two polarizing prisms ˆxed at right angle to each other.(2) To 1 g of Wheat Starch add 50 mL of water, boil for
1 minute, and allow to cool: a subtle white-turbid, pastyliquid is formed.
(3) To 1 mL of the pasty liquid obtained in (2) add 0.05mL of iodine TS: a dark blue-purple color is formed, and thecolor disappears by heating.
pH Put 5.0 g of Wheat Starch in a non-metal vessel, add25.0 mL of freshly boiled and cooled water, mix gently for 1minute, and allow to stand for 15 minutes: the pH of thesolution is between 4.5 and 7.0.
Purity (1) Iron—To 1.5 g of Wheat Starch add 15 mL of2 mol/L hydrochloric acid TS, mix, ˆlter, and use the ˆltrateas the test solution. To 2.0 mL of Standard Iron Solutionadd water to make 20 mL, and use as the control solution.Put 10 mL each of the test solution and the control solutionin test tubes, add 2 mL of a solution of citric acid (2 in 10)and 0.1 mL of mercapto acetic acid, and mix. Alkalize withammonia solution (28) to litmus paper, add water to make20 mL, and mix. Transfer 10 mL each of these solutions intotest tubes, allow to stand for 5 minutes, and compare thecolor of these solutions against a white background: thecolor of the test solution is not darker than that of thecontrol solution (not more than 10 ppm).
(2) Oxidizing substances—To 4.0 g of Wheat Starch add50.0 mL of water, shake for 5 minutes, and centrifuge. To30.0 mL of the supernatant liquid add 1 mL of acetic acid(100) and 0.5 to 1.0 g of potassium iodide, shake, and allowto stand for 25 to 30 minutes at a dark place. Add 1 mL ofstarch TS, and titrate with 0.002 mol/L sodium thiosulfateVS until the color of the solution disappears. Perform ablank determination and make any necessary correction: thevolume of 0.002 mol/L sodium thiosulfate VS consumed isnot more than 1.4 mL (not more than 20 ppm, calculated ashydrogen peroxide).
(3) Sulfur dioxide—(i) Apparatus Use as shown in the following ˆgure.
The ˆgures are in mm.
A: Boiling ‰ask (500 mL)B: Funnel (100 mL)C: CondenserD: Test-tubeE: Tap
(ii) Procedure Introduce 150 mL of water into the boil-ing ‰ask, close the tap of the funnel, and pass carbondioxide through the whole system at a rate of 100±5 mL perminute. Pass cooling water through the condenser, and place10 mL of hydrogen peroxide-sodium hydroxide TS in thetest-tube. After 15 minutes, remove the funnel without inter-rupting the stream of carbon dioxide, and introduce throughthe opening into the ‰ask about 25 g of Wheat Starch,accurately weighed, with the aid of 100 mL of water. Applytap grease to the outside of the connection part of thefunnel, and load the funnel. Close the tap of the funnel,pour 80 mL of 2 mol/L hydrochloric TS acid into thefunnel, open the tap to introduce the hydrochloric acid intothe ‰ask, and close the tap while several mL of thehydrochloric acid remains, in order to avoid losing sulfurdioxide. Place the ‰ask in a water bath, and heat the mixturefor 1 hour. Transfer the contents of the test-tube with the aidof a little water to a wide-necked conical ‰ask. Heat in awater bath for 15 minutes, and cool. Add 0.1 mL ofbromophenol blue TS, and titrate with 0.1 mol/L sodiumhydroxide VS until the color changes from yellow to violet-blue lasting for at least 20 seconds. Perform a blank determi-nation and make any necessary correction. Calculate theamount of sulfur dioxide by applying the following formula:it is not more than 50 ppm.
Amount (ppm) of sulfur dioxide= VW×1000×3.203
W: Amount (g) of the sampleV: Amount (mL) of 0.1 mol/L sodium hydroxide VS
consumed
Loss on drying Not more than 15.0z (1 g, 1309C, 90minutes).
Residue on ignition Not more than 0.6z (1 g).
Containers and storage Containers—Well-closed contain-ers.

17791779Supplement II, JPXIV Infrared Reference Spectra
Change to read:Part I

17801780 Supplement II, JPXIVInfrared Reference Spectra
Add the following:Part I

17811781Supplement II, JPXIV Infrared Reference Spectra

17821782 Supplement II, JPXIVInfrared Reference Spectra

17831783Supplement II, JPXIV Infrared Reference Spectra

17841784 Supplement II, JPXIVInfrared Reference Spectra

17851785Supplement II, JPXIV Infrared Reference Spectra
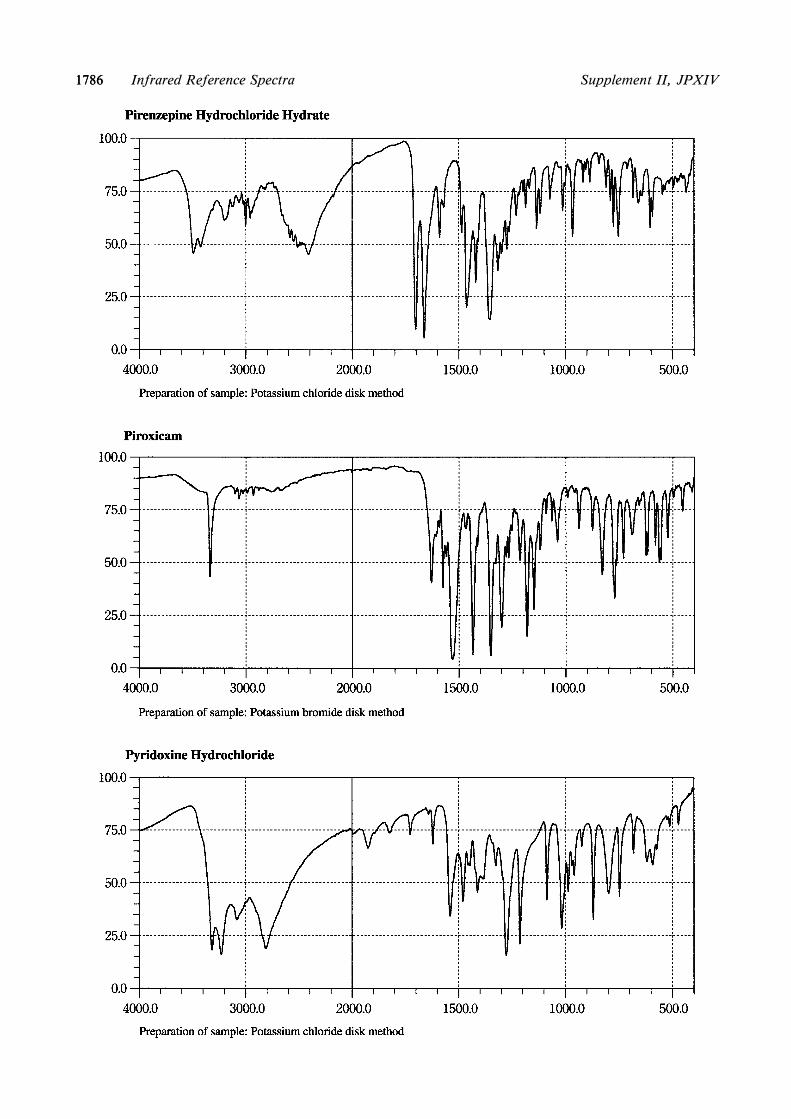
17861786 Supplement II, JPXIVInfrared Reference Spectra

17871787Supplement II, JPXIV Infrared Reference Spectra

17881788 Supplement II, JPXIVInfrared Reference Spectra

17891789Supplement II, JPXIV Infrared Reference Spectra
Add the following:Part II

17911791Supplement II, JPXIV Ultraviolet-visible Reference Spectra
Delete the following Ultraviolet-visible Reference Spectra:
Part I
Furosemide 1
Furosemide 2

17921792 Supplement II, JPXVUltraviolet-visible Reference Spectra
Change to read:Part I

17931793Supplement II, JPXV Ultraviolet-visible Reference Spectra
Add the following:Part I

17941794 Supplement II, JPXVUltraviolet-visible Reference Spectra

17951795Supplement II, JPXV Ultraviolet-visible Reference Spectra

17961796 Supplement II, JPXVUltraviolet-visible Reference Spectra

17971797Supplement II, JPXV Ultraviolet-visible Reference Spectra
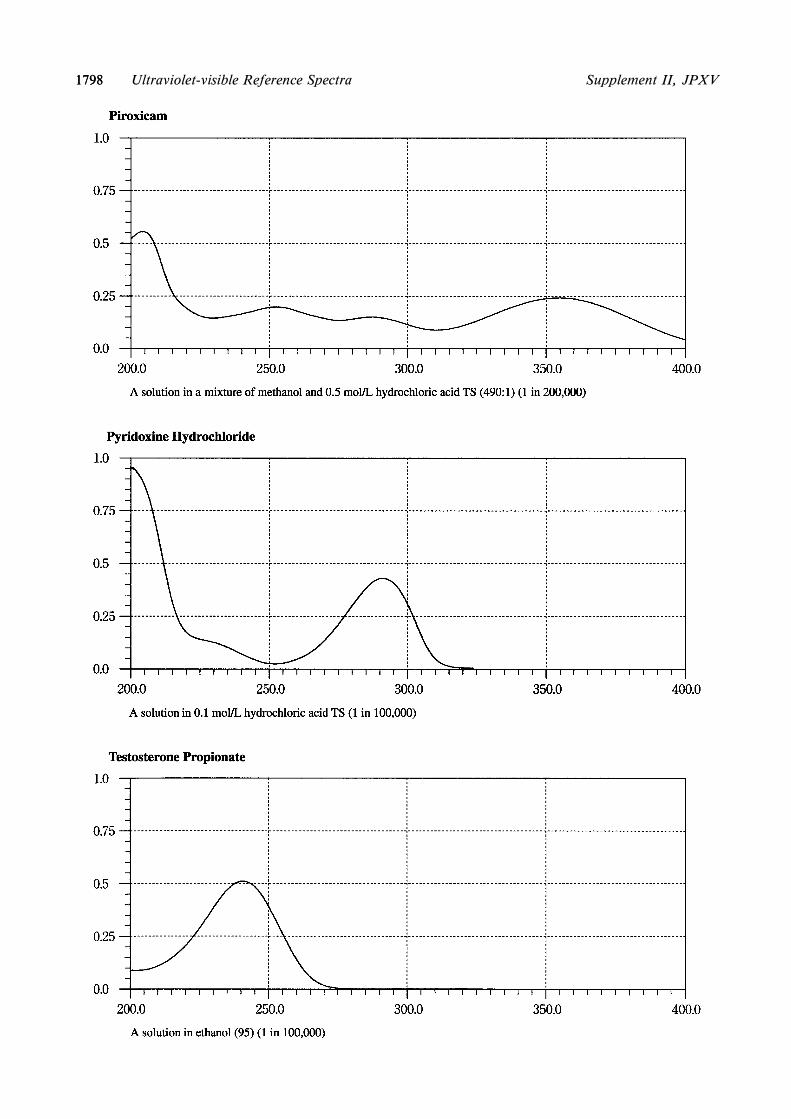
17981798 Supplement II, JPXVUltraviolet-visible Reference Spectra

17991799Supplement II, JPXV Ultraviolet-visible Reference Spectra

�������� ���� � �� ������
�������� ��������� ���
���������
��������� � �������� ��������
����� �������
��������� � �������� ��������
�������
��������� � ����� ��������
��������� � ������ ���� ��
�����
������� ������
��� ��������� ����!
�"� #�� ���
�$� ����
��������� �������
��� %��&����� ������
�"� '�����!�� ������
�$� ��������� ����!
�(� ����
������� ��� ���������
��&�� � � ���
# ��� %�
)��� �� &������ �������� ���
�*�%�
�������� ��������� ���
���������
��������� � �������� ��������
����� �������
��������� � �������� ��������
�������
��������� � ����� ��������
��������� � ������ ���� ��
�����
������� �������
��� ��������� ����!
�"� #�� ���
�$� ����
��������� �������
��� %��&����� ������
�"� '�����!�� ������
�$� ��������� ����!
�(� ����
������� ��� ���������
# ��� ��!��
# ��� %�
)��� �� &������ �������� ����*�%�
+��, %� ����� �� ����� � �� ��� �� &������ �������� -�� �!�� &�-� �� ��� ������������. &�� � ��
����� � #�� �� *������ ��������� ���� �� ����� /� ������� � 0� �(. �� ������ ����� ��� � ���������
&��� �� �� �1��! &�� ���� � �� ����� � 23 �!4
�53�
����� �����������
������ �� ���
! ������������ �������������
��"���� �� �# �"���
�#������"��� $������# ������
/��� �� -��� ������6���� ��� &� �!�� ����!
�� ������� �����������. �� 7��� ����� �����
������ ��� �� 0����� ����������� �� �������
� � �� 0����� ����������� 8������� ����� �0�
�(�4 %� �� ���-� � �� ��&�� &��-4 %� ������ ����
9�����6� ��� ���-� �� ������6� ��� -���� �
�� ������������ 9�����6���� �!���� �������.
��� �� ������ ���� 0� �( ���-� �� ��� �� �� �����
� 0� �(4 /� �� :����� ������. ���� �� �� �;����
&�-� 0� �( ��� �� �!���� �� ���-� �� �������
������4

�������� ���� � �� ������
���� �� ���������������� ���
���
������������
���� �� �������� ���
������������ � �������� �� ���� �������� � �����
���� � ��� ���������� ��� ������
���� ��� � ������ ����� ���� ����
�� �� � !���� � � ����!����� ���
���� ���� ��� ���� �� ��� ������ � ���
���� ���� �� �! ��� ! �� ��� ��"�
����� �� "� ������ � #�� �"��� !
�� ��� ��"����� � #�!��� �������$
��� !���� "$ ��� �������� ����"��
"���#� ��� ������� ��$�!�� ������
���� ��� ������ � ������ ����� "��!
���� ����� ��� ������� ���%�� �
��� ���� ��� &��� �� ��$�!�
�������� �������� � �������� �� ���� �������� � �����
���� ��� ��'�! ��� ������� (��� ���
)�����$ �� ��� ������ �� "� ��'�� � ��
����� � � *������ ���� � ���$ ���
������ ������ � ��� ����!���� ���
�������� �� ��� �"�*� ��"��� (���
������ �� ������� �*�������!��� ����
�������$ �� �*������� ��� ��������
�������� ���� � �� ������
�������� �������� ���
��������� �!���� ���"�� ���������� +������
+������ ���� ��� ��"���� ������������ ,��� ��� ����! -���
.��� ���!�$������ ����� .��� ���!�$����� ����� /��� 0 1
2�$��"��� ���� �!��� ����� 2�$"�������� �!��� �����
2�����$ 2�����$ �� ����
3��#�� �������� ���� �� ����"��� ������"��
��� ���!
3��#�� �������� ����
4������� ���� 4������� ���� /��� 5
,��"���� %������� ,��"���� %�������
6��� �������� 6��� ��������
���� ��� ������$ �� ��� ������ �� "� � ����� ���� ��� ������$ �� ��� ������� �� "� � �����
,��"���� %������� ,��"���� %�������
7)����� �������� �� &)�� ������ /��� 8
2���"�� ����� "� 2��� ������ +������ 9
:�� ��� ��$ �������� � :�� ��� ��$ ��������
:������� ��� ����� �� :������� ��� �����
6��� �������� �� ��� ������ ����� 6��� �������� �� ��� ������ �����
:�$ �)��� �� :�$ �)���
:������� ��� ����� "� :������� ��� �����
;�9 ���������� �� �������� ����������

������ ��� ��� ���� ��� ������ ��� ��� ����
�� ������ �
������� �� ��� �������� �� �� ������ ���� ��� �� ��� ������� ��� ������� �� ��� ����
����� �� �� ������
��� � � �
���� ��� �� �� � ��� �� �������� ������� ����
������ � ��� ��� ���� ������� ������� ���
� � �� �� ������ ! � � ��� ��� ��� � ��
������ "
#��� $%&%'%�'% (��� �� �� � ��� � ����������
��� �� ���� ��� �� � � )��!� *����� ��
#�� ��� � +�� ��� �� #��
#��� '% , �������� ��� ��� ���!� ������ ��
��� ��� � ��� ��� �� ���
#��� $%&%'%�$% , � ��� ���� �� �� � ��� ���
�� �� ��
#��� �% , � ��� ���� �� �� � ��� ��� ��
�� ��
#��� $%&%'%��% , � ��� ����� �� ��� �� �
����
#��� $% ����� �� ��� �� � ��-� ���� �
���
��� '. � ''
���/ '0 ������������ ��� �� �/ ��� �� � ���%
$0 1��� ������ �� ����/ ��� � �� ��� 2�%
�0 34�� � �� �� �� �� �/ 5�������� �� � ��� ����%
"0 34�� � �� �� �� �� � ����� � ��� � ����� ���/ 5���� ��� ��6 ��� �� ���%
70 ,� �� ��� ��� � �� ��� �� � �������� ����� � �� � ����� �� ������� �/ 8�� �������� ��� ������ � ���� ��
� �������2����� �� ����� �� 9� � � ��������� �� �� ������� �� $. � $7�� ����� �� ���������� �
� ��� �� ��%
&0 *� �� � ���!� ������ �� ��� ��� � �������� ��!�� �� ��/ ��� �� �� ��� ! � ����� �� �� �����
���� �� � � ������ ���� !������������ ���� � ���%
:0 ������ �� � � �� �� 9� � �� � � � � �������2����� �� ����/ '.. �; �� 2���%
�0 #������� �� ������ ���� ���� � �� �� �� ��� �� ��/ � �� ���� ������%
�0 < � ����� ��� � ���� �� � � ��� !� � � ��� �� � ���� �� ���!� � �����/ =� � � ���� �� ���!�
� ����� � ������� 6�� �� ��� ��� ������ ! � � ��� ��� ��� � ��% >�!��� ���� �� ��� ��� ��� �������
���?�� ������ � �� � ������ ���� ������ ��� � ��� ���� !�� ��� ���� � ��� ��� � �����% =� ��
� ��� �� � ���� �� ���!� � ����� � � ���� ��� � ������� ����� � ! � � (�� � �� #��% =� � ���� ��
���!� � ����� � � ���� ��� � ������� ��� ��� ������ ! � � (�� � �� #��%
'.0 # ���� �� @����� �� ��� �� � ��-� ���� � ���@/ (�� 2� �� � ���� �� � )���� #��%
''0 ����� �� ���������� �������� A��� ��� '.. �;0 �� � ��-� ���� � ���/ ,�6 ��� '. ����� ���%
������ '/ # ��� ������� ������ � � �������� ��%
������ $/ #� � � ������� � ''% =���� ��� A�0 ���� � )���� <��� ��� *������ ��� ��� BB5���� ���! �
��� 2�� =���� ��� �� � � � ����� �� � (�� � �� #��% =� � ��� �� ����� �� � � ������ ���� ���
����� ��� � ��� ! � � ����� �� ���� �� �� � ����� �� � ������� � � ������ ������%@@
������ �/ # ��� ��� ������ � C*%
������ "/ � ���� �� � � � � ���� � � )���� #��%
'�.����������� �� � ������� ����������

�������� ���� � �� ������
����� ������ ����� ������
��������� ���� �� ��� ������
������� ����� � ������� ����� ���
������� ����� � ������� ����� ���
� ����� �� ���������� ������ ��� � ����� �� ����������
������ ������ ��� ������
����� ������ ������ ��� ����� ������
������ ������ ��� ������
������ ��� !� ����
���!���� ������ �"� ���!����
#��!���� ������ �$� #������
������� ��� !� ����
����� ������ ��%� �����
���� ������ �&� ����
'�(���� ��� ��������)����� ���� ������ ���� '�(���� ��� ��������)
����� �������
������ ��� !� ����
*� �� �����( *� �� �����(
�� ������ ������+�� ��� !� ����
��� ���
�������� ���� � �� ������
������ ��� ��������� ������ ���
��������� ���� �� ��� ������
������� ����� ������� �����
�!!����� � �� ������� ������ ��� ,������ ��� ���� �� ���)
����
-������ ��.����.�� �.��� � ������ ��� -������ ��.���/�.�� �.)
��� �
0+��� � �� ������ �$� 0+�����
#��!���� ������ ��� #������
������� ��� !� ����
1���� 1����
#��!����� �� -����� �� �(������
�� ������ ������+�� ��� !� ����
��� ���
#����(� ,�������� ��� ����(�
�2%" ���������� �� �������� ����������

�������� ���� � �� ������
������ ���� �������� �������� ������ ���
��������� ���� �� ��� ������
������� ����� ������� �����
�������� � �� ������� ������ ��� ������� ��� ���� �� ����
����
������� ���������� ����� � ������ ��� ������� ����������� ���
��� �
!��� � �� ������ �"� !�����
#������� ������ �$� #������
������� ��� �� ����
%���� %����
#�������� �� ������ �� �&������
'� ������ ������!�� ��� �� ����
��� ���
#����&� ��������� ��� ����&�
�������� ���� � �� ����� ����������� ������
������������������� ������� �������� �������
��������� ���&��
������� ����� � ������� ����� ���
������� ����� ' ������� ����� �$�
������� ����� � ������� ����� �"�
������� ����� � ������� ����� �(�
��)������� ������ ��� ��)���
*� �� �����& *� �� �����&
������ �� �&������ ������ �� �&������
*��� �� ������� ������ �$� ��������
*��� �� ������ ������ �"� #������
�+,����������� �� � ������� ����������

�������� ���� � �� ����� ����������� ������
���� ������ ���� ������
��������� �����
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
�� ��
��� ������ ��� ���
��� � ������� ��� ��������
�������� �� �� ���� ������ ��� �����!�� �� �� ����
"���#�� ������� ������ ��� "��$�� �������
%��� �� ����� %��� �� �����
"���# ��� �# &������ �� �������
�������� ���� � �� ������
�������� ������� ����� ��������
��������� ��'��� �$ �#� �������
������� ���� ������� ����
(�������� (��������
%�'�� �$ $��� ��� ������ ��� )��� ����
* ��� * ���
&������ �� ������� &������ �� �������
�#�# ��� ������� ��� � ��� � � ��� ��!���
������� �$ ����� ��� � ��� ������ ����
� �+ �� �� ���� � ���� ����� �� ���� �
�������� ���� � �� ����� ����������� ������
���� ������ ���� ������
��������� �����
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
�� ��
%��� �� ����� %��� �� �����
&������ �� ������� &������ �� �������
%�'�� �$ ���� ������ ��� ���
%�'�� �$ �����!�� �� �� ���� ������ ��� �����!�� �� �� ����
"��$�� ������� �����'�� ���� ������ ��� "��$�� �������
�,-. ���������� �� �������� ����������

�������� ���� � �� ����� ����������� ������
������ ������ ������ ������
��������� �����
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
������� ���� � ������� ���� ���
�� ��
��� ������ ��� ���
�������� ����� ���� ������ ��� �������� ����� ����
�!�"�� ������� ������ ��� �!#�� �������
$��� �� ����� $��� �� �����
�!�" ��� �" %������ �� �������
�������� ���� � �� ������
����� ����� ������� �����
�������� �� �������������
�����!��"
����� �����������
���������������� �� �����������
���
�" � ���������� �# ��!� ���! &���
'�!�
�( �" � ���������� �# ��!� ���! &���
'�!�
��� ����� ��!� ���! &��� '�! )!��*
����"������
%������ ����������
+��*������� ����������
�( ��!� ���! &��� '�! )!������"���*
��� ����� ��� ����� ����������
�� %������ ����������
�� +��*������� ����������
�" � ���������� �# �������������
��,�� ����& '�! )!������"������
�( �" � ���������� �# �������������
��,�� ����& '�! )!������"������
���� ��� -����� ! �������������
��,�� � ��!� ���! &��� '�!�
����&�!�� �# �"� �! &��!��� � �*
�����
���� � ���� �# �"� �!
.( ���� ��� -����� ! �������������
��,�� � *��!� ���! &��� '�!�
�� ����&�!�� �# �"� �! &��!���
� ������
�� ���� � ���� �# �"� �!
/������ �"� �! �� �"� �!������"���*
��� �� � ��� �� �!������"������
��� � ����
�� /������ �"� �! �� �"� �!�����*
�"������ �� � ��� �� �!�����*
�"������ ��� � ����
��������� �# ������� �� '�!�
���& ���� �� ����
�!0�� �� ����
1( ��������� �# �������� �� '�!�
�� ���& ���� �� ����
�� �!0�� �� ����
����� �# � ���� � ��!�*
���! &��� '�!�
2( ����� �# � ���� � *
��!� ���! &��� '�!�
/�!���! �*/ �� �����&�� ���� 3( /�!���! �*/ �� �����&�� ����
- !�� ���� �# �"� 4��� 5( ��� ��!��� �# �"� 4���
6� ����� ���� �# &�������� 7( 6� ����� ���� �# &��������
%� ����8 4��� �!������ 4��� �!������
�!��9�� ��!����� �!��9�� 4
�5:3���������� �� � ������� ����������

��������� ����� ������� ��������� ����� �
������� ������� ������� �
��������� ������� ��������� �
���� ������� ���� �
������ ������ ����� ������ ������ � ��� ������ �����
�������������� ���� ����� �������������� ���� � ��� ���
���� � � ���������� �� �������
��
���� � � ���������� �� ������ ��
���� �� ���������� �� ������� ��
���� �� ���������� �� ������ ��
�������� ���� � �� ����� ����������� ������
����� ������� ����� �������
������� ������ �� ��� �����
���������� ����������
!�������� �� ������� �����" #�$ ������" �� ����� �� ����%
���
&��������� ���� &��������� ����
!�����" �����" #�$ !���
'�(�����"�� �� ����� �������
���������
�����" #)$ '�(�����"�� �� �����
������� ���������
�������� ����� �����" #*$ �������� �����
&������ � ���������� �����" #+$ &������ � ����������
!���" !���"
�������� ���� � �� ������
����� �����������
����� ��� �������� ����� ��� ��������
!�������� !��������
,����� ���������� ,����� ����������
&������� ������� -������� &������� ������� -�������
���������� �� ����������� ���������� �� �����������
&�����������" &�����������"
������ ���������� ������ ����������
����� �������� ����� ��������
������ ."����"��� ������ ."����"���
-����� � -����� �
."����"��� ������� ."����"��� �������
��������� ���������
-����� � -����� �
."����"��� ������� ."����"��� �������
/���� ����� ."����"��� /���� ����� ."����"���
-����� ) -����� )
�010 ���������� �� �������� ����������

���������� ������ ���������� ������
���� ����� ���������� ���� ����� ����������
������ � ������ �
��������� ������ ��������� ������
�������� ��������
������ � ������ �
���������� ������ ���������� ������
����� ����� ���������� ����� ����� ����������
������ � ������ �
���������� ������ ���������� ������
���� ����� ���������� ���� ����� ����������
������ � ������ �
������� ������ ������� ������
�������� ��������
������ � ������ �
��� ������� ��� �������
������� ������ ������� ������
�������� ��������
������ ! ������ !
��� ������� ��� �������
"��#���$���������� ������ "��#���$���������� ������
%&�� ������ %&�� ������
�������� ��������
������ '( ������ '(
������� ������ ������� ������
�������� ��������
������ '' ������ ''
������� ������� ������� �������
�������� ��������
������������� �) *$��� *��� *������� +������
����������
������������� �) *$��� *��� *������� +������
����������
������ ',�������$� -�������� .��������
+������ ���������
������ ',�������$� -�������� .��������
+������ ���������
������ /,�������$� ��* 0����$����� .����,
���� +������ ���������
������ /,�������$� ��* 0����$����� .����,
���� +������ ���������
������ 1,������$� �23" .���4���5�����
+������ ���������
������ 1,������$� �23" .���4���5�����
+������ ���������
������ �,������$� *6".���4���5����� +����,
�� ���������
������ �,������$� *6".���4���5����� +����,
�� ���������
������ �,������$� ��* .���4���5����� +����,
�� ���������
������ �,������$� ��* .���4���5����� +����,
�� ���������
������ �,������$� .*%,"� .���4���5�����
+������ ���������
������ �,������$� .*%,"� .���4���5�����
+������ ���������
������ �,������$� 0��","� .���4���5�����
+������ ���������
������ �,������$� 0��","� .���4���5�����
+������ ���������
'�(!���������� �� � ������� ����������

������ ������ �� ����� �������������
������ �������
������ ������ �� ����� �������������
������ �������
���� ���� ������ ��� �������� ���� ���� ������ ��� ��������
���� ������� ���� �������
����� ���� ���� ����� ����� ���� ���� �����
����� � ����� !������ ����� � ����� !������
"�� � ����� !������ "�� � ����� !������
�������� ���� � �� ������
����� �����������
��������� ������������� ��������� �������������
������ � ������ �
�������� #��� $������������
%�����������
&��� ������ ��������
'����(�
������
)������ �
��������
$���������� ��� ���� ��������
� *� ���� ��� ������������
� *� �+
%(���� ��������
��������� ,� ����� ����������
�������� #��� $������������
%�����������
&��� ������ ��������
'����(�
������
)������ �
��������
$���������� ��� ���� ��������
� *� ���� ��� ������������
� *� �+
%(���� ��������
��������� ,� ����� ����������
�������� ��� $������������
������������� �, ����
�������� ��� $������������
������������� �, ����
�������� &��������� ��� ���(
-�����( ����
������( �� ��� ����
��. ������ ������(
��� ���( ����
��/��������� ����
%�����������
'����(�
��������
!�� �����
�������� &��������� ��� ���(
-�����( ����
������( �� ��� ����
��. ������ ������(
��� ���( ����
��/��������� ����
%�����������
'����(�
��������
!�� �����
������� $������������ �������(���� 0�$"�1
%�����������
&��� ������ ��������
'����(�
)������ �
��������
$���������� ��� ���� ��������
������� $������������ �������(���� 0�$"�1
%�����������
&��� ������ ��������
'����(�
)������ �
��������
$���������� ��� ���� ��������
2�23 ���������� �� �������� ����������

���������� �� ��� � ��������� �
����� �
������� � ������
��������� � � ������ ������� ��
����� ���������
���������� �� ��� � ��������� �
����� �
������� � ������
��������� � � ������ ������� ��
����� ���������
����������� �
��������� ��
����������� �
��������� ��
����� ����������
������ ������ � ��� ������� �����
!�� ���� �
������ "��� �
������#� #� ��� !���
����� ����������
������ ������ � ��� ������� �����
!�� ���� �
������ "��� �
������#� #� ��� !���
�������� ���� � �� ������
����� �����������
����� ������ ����� ����� ������ �����
$��� � %
�������� � ���� �
���� � ���� �
� ������
&����#����������
��������� ��
$��� � %
�������� � ���� �
���� � ���� �
� ������
&����#����������
��������� ��
$��� � '
�������� � ���� ��
���� � ���� �
����(
!������� ��� � ���� ��
� �� ������� !������
�)� � ���� �
� ���� ��� *��� � ���� �
��(����� � �� !������
)������ " ���#�� ������+� ��� � !�����
� ������
��������� ��
,���������� ����������
� ���� )� *�� ���� !������
������ � ������ ���� !������
� ������
$��� � '
�������� � ���� ��
���� � ���� �
����(
!������� ��� � ���� ��
� �� ������� !������
-� �)� ��
� ���� ��� *��� � ���� �
��(����� � �� !������
)������ " ���+� ��
� ������
��������� ��
,���������� ����������
� ���� )� *�� ���� !������
������ � ������ ���� !������
� ������
.*����� � � �� �� //.*#
����0 ��� $������ !�#
1��������� � � �� � �����
� ����� ��� ����������
� � ���� � 2 ++ �� �����3
$��� � 4
�������� � ���� ��
���� � ���� �
����(
� ������ !������
$��� � 4
�������� � ���� ��
���� � ���� �
����(
� ������ !������
%5%%���������� �� � ������� ����������

���������
������ �
���������
������ �
������ �
�� ��� ������ �
���� ������
� �
���� ��
��� ���� �
������ ����� ���� �
���������� ���� �
���������
������ �
������ �
�� ��� ������ �
���� ������
� �
���� �� � ������ �
��� ���� �
������ ����� ���� �
���������� ���� �
���������
������ �
������ �
�� ��� ������ �
���� ������
� �
������ ���� �
���������
������ �
� ������� � ����� ���
����� ��
������ �
�� ��� ������ �
���� ������
� �
������ ���� �
���������
������ �
� ������� � ����� ���
����� ��
�� ��� �������� �� ������
���� �! �����"� � ������
#��$ ����� � ��������� ���
���� ���%���� � &���! ��
� ��� &���! � &��� � ��
������%'
������� �� ��� � �� � ��
�� ��� ������ � �� ���
���������'
������ (
�� ��� ������ �
���� ������
� �
���� ��
����� ��)��
����� *�� ���� �
*�� ���� �
���������
������ �
������ (
�� ��� ������ �
���� ������
� �
���� �� � ������ �
����� ��)��
����� *�� ���� �
*�� ���� �
���������
������ �
������ +
��������� �
��������� �
������ �
������ +
��������� �
��������� �
������ �
,-,. ���������� �� �������� ����������

�������� ���� � �� ������
����� �����������
��������� �������� ��������� ��������
��������� �� ���� ��������� �� ����
������ �� ���� ������ �� ����
� ���� � ����
���������� �������� �� ����������
���� ������ ���������
��� ����� �� ��� ���
�� ��� �� ���� ��������� ��
� ��� ����� �� ��� ����
���������� �������� �� ����������
���� ������ ���������
��� ����� �� ��� ���
���� ���������� ��
��� ����� �� ��� ����
!����� !�����
"������� �� ��� ������ ���������
#��$%��� �� &�������
"������� �� ��� ������ ���������
#'�$%��� �� "�������
"������ �� ���(������� �������� �����(
�����
"������ �� ���()������ �������� ���(
�������
' ���*�� "������ �� ��� ����� !����� ' ���*�� "������� �� ��� ����� !�����
����� �� +������� ������ �� +�������
������(!��� ������� !���
,������
��-��� ������� ��� ���������� �������� ��
��������� ��
,������ �� '�������
��-��� ������� ��� ���������� �������� ��
��������� ��
+������� ������� �'
��������� �������
+������� ������� �������
�� ��������� ������� ��
� ���*���
�������� ���� � �� ������
����� �����������
���� ������� ���� �������
��� ��� �� '�� � ��� ��� �� '�� �
��� �� ���� ! ��� �� ���� !
������� �� ����*����� ������� �� ����*�����
'�����&� +�&�� �� �� ���� .���� '�����&� +�&�� �� �� ���� .����
����������� �� '� � ����������� �� '� �
����������� �� ��� +�&�� ����� ����������� �� ��� +�&�� �����
����������� �� ��� ������� ����������� �� ��� �������
)��$������� �� / ��� ��������� +���������
0
��� ������
����
������ �� +�&�� �����
)��$������� �� / ��� ��������� +���������
0
��� ������
����
������ �� +�&�� �����
+�������� ��� '� �����
+�������� ��� +����
'�&���
+�������� ��� '� �����
+�������� ��� +����
'�&���
�1�2���������� �� � ������� ����������

������ ���
�� ���� ���������
������� ���������
����� �������
��� ����
������ ���
�� ���� ���������
������� ���������
����� �������
��� ����
����� � � ���������� �� ����� � ����� � � ���������� �� ����� �
���� �� ������ �� ��!"� �"����
���� #� ������$%� &� ��� ��� �������� ��
����� ��
���� �� ������ �� ��!"� �"���
���� #� ������$%� &� ��� ��� �������� ��
����� �
�'�( ���������� �� �������� ����������
��� ��� �������
��� ����� �� ���� ���
����� �� ���� ����� �� ��� ����� ���"� %� �� �)
������� ��� ���� �� ���������� �� ������� �� �������*
����� �* � ����� ������%���� ���������� �������
� ����� � �� ���������%�� �������" �� ��!������
��� � ���� �� ��� %� ��"��+� ����� ��������
��� �$%���� �� ��� ���� �� �� ������� �� ����� �
�������� ��� ��������� �� ��� �����%��� ������� ��
���� ��� ��"� �����%�� ��� �������� ���� ��� �
��%��%�� ,��� ������ �����������* ,���� ����� � ��
����� � �� ����� �� ���� ��, ���� �� � �����
�� ���� �� �� %� �� $%����� ������� � ����� �* ��
�������� ��� � ������ �� ������� �� ����� � �� �� �����
���� �� ����������* �� %����� ������� � ����� �
��%��%�� ����* �� �!�%�� ��"�������� ����"�� ���
����� � �����"* � �� ����� ������ ���� �� ���
��"�� �� ������ �� ������� �� ����� �� �� � ������ ��
�� ����+� �������-����� � �� �� �� �!� %� ���� ��
������%��� ������ ���� �� ����� .����,��" ���)
����-����� � �� �����* ��� ���� �� ���� ����� %��
�� �� ��� �� ��� ������� ��� ���� ���� �� ��
����� ������%���� ���������� ��� ���� �� ���)
���%��� �� ��� ��� ���� �� �������� ���!��+� ��� ��)
���
���������
����� %� ��� ���� �� ���� �� %%��� �� ��
�������"����� ������� �� ��� ���� �� ������ ��
��� ��� ����� %����� ������$%� �/� !��"� �� ���
%����� �������"����� ����%�������� ��"�� ���
������� ����� ���"��� �� ���� �� ���� ����%)
���� ,��� �������� �� ��,)���%�� �� ��"�)���%�� ��$%�
�������"��� ����� �� "�������" ������ ���
"� ���� ��� ����� ��� ���� �� ����� �� ����)
���"����� ���%��� ��� ����%���� �%� �!� ������%��
���!��+���� ��������* %��� ��� ���� � ���+� %��"
������%�� ���!��+����� ��� ������� � %%��� � %���!)
�����)!����� �� 0%�������� ������� ���� ��" �� ���
���!��+���� ����� %� � � ����� ��" �!��� 1��"�*
����"����2 � %� ��� ���������" ��� ���" �"�� ����
��� ������� � ��� $%��������� �� � �������� ���
����%�������� �� � ���� �����%���� ��� ���� ��
�����
������� ����������
3�/"��%� ����������� � �,� ������� ��� ���
���� �� ���������" ���� �� ����� 4�"� �%����
��"��� �� ������ 1��"�* ��, �%���� �� ��������� ��
�� �������%�� �� "������ �����������2� ��������
��"��� �� ���"� ��%������ �!��� ��, ,��/ %��" ����
��"�)���%�� ��$%� �������"���� 14�5 2 "� � ��)
!���� �������� �������� ����������� � �����"�
������ ��� ��"�� �� ������ �� ��� ��!��� �� �� %�� ��
�������" ��!��� ������ %�* /�����" ��!��� ����!���
��!��� * � ��� �����" ���� �� ���� ����%����)
���� �� ����� %���"���
5������� ������� �� �������� ��� $%���� �� ���
���� �� ����� ���� ��� ����%�������� �� ��,
��6� �� �� ��� ��������� 7��� ��� �������� �����
��� � ������� ����� ���� ��" �� ���������
��� %��� 7��� ����� ��� �� ��!��� ���8 ��� ���� ��
��� �� �� ����� ��� ,��� ����� �� � ��� ���� ��
,�� ��, ��)���� ���� �� �$%�!���� "��!�� 5���� ���
�%���� �� ���� ��� ���� !�� � ����� � ����
���%� %� �� �������%�� �� ���!�� ��!�� �� "������*
�����* � ������
� ,���)������� ����%���� � ������ ��� ��������
���� �� ���� ��%��� �� ��� ����%���� � %� ��
��%���� ��* �� � �� �� ����/� ��� ��� ��/* ������� �
��� �������* � ��� ������ �� ��� ���%�� �� ������
����%���� �� ��� �� �!� %� ���� �� � ��� �� ������ ��
����%���� ����� � ����� ��������� ���� �� ��%����
��� %���
�������� ������ ��������
��������� ���� �� �� � �� �����������
!����� ��� ���� �� ���� � �������� ����� �� �
$%��% ����%�� �� ���� �� � 9��� ���������" ����
�� ����������* ������� �� ����� � �� � �� ���+�
,��� ��� ��� ������ ������� �� �������� ��� ����"��)
�� �� ��� ������ ����� %��� 4�"��� �%���� ��!��� ��%�
��%��� � ���� %� ������� �� � ��� ���
�%�����
����������� �� ���������������
�������� �� ���� �� ���� ����%��������

�������������� �� � ������� ����������
������ ��� ��� ������ ��� ��� ��� ������� �����
� ������ � ������� � ��� ���� � ������ � � �����
���� ��� � ��������� ��� ���� ��� ��� � �� ���� �
����� � � ��� ��� ���� ��� � �������� � � ���
��� ��� �� ��� ������ �� �� ��� � ��� ������ �
�� ������� ��� ���� ������ ��� ��� ��� ������ �
����� ��!����� ���� ���� ������ ��� �������� ����
���� � ��� ��� ��� ����� ������"��� ����� ��������
� ��� � ��� ��!����� ���� ���� �� �� ������ #��
��� ������ � � ��� ��� ��� �� � ���� ������ ���
�� �� � �������� � � � ��� � ��� ��� ���� �� ���
������ ����� �� ����� ������ �� � ��� ���� �
��������� ��� ���� � ��� ��� � �������� �� ����� ���
��� �� � ����� ��� ��� �� � ��� ������ ����
������ � � ��� ��� ��� � �������� �� � �� ��� ����
��� ��� ���� ������ ��� ����� � � ��� ��� �����
� ��� ���� �� ������ ��� ����� ����� $����� ����
� ����� ����% � ��� ������"�� ��� ��� �� ���� � ����
������ �� ������� �������
& �� � ��� ��� ��� ������ ���� �� ����� �
��������� ���� ��� ��� � � � ��� ��� ���� ���
���� ��� ��� � �� ������ � ��� ����� ��� �� �
��� ������ ��� �� � ��� ��� ������� �� ��� �������
' ������ � (� � �������� � ��� ���� ��� ��� � � � ���
��� ��� �� ������� �� ���� � ����� ��� � �������
�� � � ��� ��� ��� ������� �� ��� ���� ����� ����
����� � ��� ��� �������� ��� ��� �� � ����� ����� �
����� ��� ��� �� ��� ���� ��� ��� � � � ��� ���
��� � ���� ��� �� � ��� ��� ���� & � � ����� ������
������� ��� ������ � �� �� ��)�����* � ������ ���
������ � (� �� ������ ���"����� �� ������ �� ���
����� � ����� � ��� � � ������ ��� ����������
������������
+ �������� ���� "���� ��� ��� ����� ������ �� �
� ����� � �� �� ��"���� ������ � � ��� ����������
� ��� ���� ,����� ����� � ��� ��� �� ������ �����
�� � � ��� ��� ���� � ����� ����������� ����� �� ����
�� �� ������� � ��� ��� �� ��� ��� � ����� �� �
��� ��� ����� ��� ��� ������ � ���� ���� �� ������
��� � ��� � ��� ��� ����� ������ ��� �� ������
��� �������� ��� ���� ���� � ������� � ���� �� ��������
��� ��� ��� � � "������� �� ' ����� ����������
����� � ��� ��� �������� ������ ��� ���
� ��� � �� ������ ��� �������� $����� ��� ����� �
� ��% � ��� ��� ������ � ��� �� ��� ������ ������
������ � $-.,% �� ���������� � � ��� ������� � ���� ��
��������� ��� �� � ��� ��� ���� '� ����� � �
��� ���������� �� ������� � ������ ������ ����
� ������� ��� ����� ��� �� ��!����� ������ /�����
���� ������ ��� ������� � � ������ ����� �� �� �
������� ������ � ��������� ��� ����� � ��� � ����
������� 0���� ��� ��� ��� � �� ���� � � ������
�� ���� $����� � ���� ����� �����% �� ����� � ��� �
��� ���������� ����� �� 1� ������� ��� �������
����� � $����� -.,%� ��� � �� �� �� ������� �����
����� � ������ ��� ��� ����� �� � ��� � �� �� ��
����� � ��� � � �� ������� � ������� ��� ���� ������
����� � ����� � ������ ��� ���� ������� '��� � � ��� �
� ��� ��� �������� � ��� ��� ��� ����� ������
���� ������� �� ���� ����� ��� ������ ������������
��� � "���� � ������� ��2 � � �� �� ���������
���������� ���� ����� �� ���������� �� ����� ��
����������� �� �� ���� ���� ����� �� ������ '
�������� ��� ��� �� ��� ����������� �� ��� �� �� � ���
����� � � ���
����� � ��� �����
'������ ������ �� � ��� ��� ����� ��"���� ����(��
�� ���� �� ������� ������ 1�!�� � �� ����� $����� ����
���� ����������% �� ��������� ���� ��� ��� ��� �����
�� �� ��� ��� �� � ��� ���� ��� �� ������ /��� ��
��� ������ � ��� ��� ���������� � � ��� ��� ���� ��
������ � � !����� �� ��!�� � �� ����� � ��� ������
���� ���� ���� ��� ���������� � ���� ��� � �� �������
� ���� ��� ������ � ���� ������� �� � ������
� ����� ����� ��� � ������� �� � ���� �� ����
��� ����� �� � ������ � �� + �� � ������"��� ���� �
��� �� ��!�� � �� ����� �� � �� ���� ����� ������ ���
� ���� ���� ��3 $�% ��4������ ��� �� ���� ���� ��
������������� 5#6+ ������� ������ ��� �� ���� ����
� ��� � ���� � ������� ��)����� ����� � �� �����
�� ������ ��� ���� �� ����� ����������* $7% ������
����� � ��� ��!�� � ����* $8% ��������� ���(���
�� � � � ��!�� ��������� ���� � ��� ��!�� � ����*
$9% ������������ ��� �� ���� �� � ��� ��!�� ����� � �����
� ���� $����� ��� ��%* �� $�% �� (���� ��
���� ��� ����� ��
� �� ��� ������� ��� � ������ ������ �� ���� �
� ��� � ������ �� ������ ���� �� ����� �� ������
��� ��� ������ '� ������� �� �� � ��� �
������ ������ �� �� ���� � �� ���� � ��� � ��� � �
���� ����� ��� ��� ���� � ��� ������ ������ ����� ���
����� ��� ���� � ��� ��� ���� � ��� �� ���� � ��� ��
&��� ��� ����� � ������ � � � ����� �� ��� ��� ��
� �� ������ ��� ��� ���� ������ ���� ���� ������
����� ���� � ����� � ��������� � �� ������ ������ ���
��� ��� � � ������ ������ � � ����� � � ���� ������
���� ���� �� ���� ������� ������� � �� �� �������� �
��� ���� � ��� ����� � �������� � ���� ������������ ���
������� ����� ������� �� � �� ���� � ��� �������
� ��� ���� ���� ���� ���� � � ����� � � ��!������� ��
���� ������ � �� ������ �� ������ ������� �� : �
����� ���� � ������ ������ �� � �������
�������� ������ ��� ��� ��� �� � ������� ����
�� ������������ � �� �� � �� ���� ������ ���� �����
��� ���� ��� ��� � �� �� �� ���� ���� � �������� �� ��
�� ����� � ���� ���� ���"�� ������� � ���� ���� ��
�������� ���� ��� ����� + �� �� ���� ��� ���
������� ������ � �������� ���� ��� ����� �� ����� ���
����� ����
� ����� �� �����
5��� ���� � �� ���� �� ������� ����� �� �������� � �
��� ��� ����� � ����� � ������ ��� ������ ����
� � ���� ���� ���� �� ���� ��� � � �� ��� �� �� �������

���� ���������� �� �������� ����������
����� ���� ��� ��������� �� ������� � ���� ���
��� � �������������� �� ����� ��� ������� � ���� � ����
���� ��� � ���� �� � ����� ������������ ���� �� �� ����
�� ������������ ������ ���� �� �� � ��!���� �� ������������
������������ ���� ��� ������������ ������ ���� "�#�$� %����
������� � ���� ��� ��� �� ��� ����&����� ���� �������
�� � ��� � ��� '(�% ����� ��������� ����� ��������� �� ��
����� ��� ����� ������� ����� � �� )������������� ����� �
�� ����� ��� ��� �� *++�% ��� , ���� ��� �� � �� � �� ��
��������� ������������� ���� ������� � ���� � )��-����
�� � ���� ���������� �������� ��� �� � �� � ���
)��� ������� � � ��� �� � ������ ������ ��� �������.&
��� � ������ ���� ������ ����� ���� ����� � � ��� ����
������� � ������-�� ��� ���������� �� ��� ��������� �� ���
����� � ��� �� ������� �� ������ �� �������� �� ������
����� ���� � �������� � �� ������/ ����� ��� ���������
��� �������� �� ������/ ���������� ����� ������� �!���&
����/ ��� �� ����� � �������� ��������� � �� ���� "���
�� ���� �������� � � ����� ��� ����� � �� ������ �� ����&
���� �� ��������� �� �� �����$� )�������� �� ���-����
������ "��� ���� 0++ �� �� ������� �� 0��1 (�$ ��
������������ �� �� ����� �� "�����$ �� ��� ���� ��� �� ���
�������� �� �� ��� ������ ��� ����� �� �!������� �� ���������
2� ����� ���� ��������� � �������� ��� ������ ��� �����
���� �� 2��&2��� 3��&3��� 2��&3��� ��� 3��&2�� ��� ��������
�������/ ��� � ������� ��� ��������� ��� �����������
�� ������ �� � ����� ���� ��� �������� ����� �� ���������
��� �� �� ��������� � �������� ��� ��������� ������ ��
���� ������� � ����� -����������� �� �1 ����� ���� � 4���
�� ��� ������� � ������-�� �� ������ ��� � �� �� �����
��� � ������� � 4��� �� ��� ������� � ������-�� �� ������
"������������ ����$ ��� ��� � ����������� �� ����� �����
���� � ���������� ��� ������ �� � ��� � ����� ������� �
������-�� ��� ������ ����� � ��� ������� ��� ��� ����&
��-�� ��� ��� �� ��� ���-������ ������ �������� � ������
����� ���� ���� ������� � �
) ����&���� � ���� "����� ����� ���� ����� � �� ����
������� � ���� �� 0,� ,�� ��� 10 ���� $ � ����� �������
�� �����.� ��� ������� ������������� �� ����� ���� ���� ���
�������� �� ������ �� �� �� ������� 5� ������� ���
�� ����� ������������� �� ������ ����� ���� "����� ����� ���
���������$ ��� � ������� � ����� ��� ���� ��� �� �!�������&
�� �� ��� ������ �� ��������� ��� ������� ������������� ��
��� � ����� ���� � ����&���� � ������� � ����� ��� �� �
� �� ��� ����� ���� ���� ��� �� �� ������ "����� � ����&
���� ��� ������$� 6����� ��� ������� � ���� ���� �� ���
����� � ��� �� ���� � ������ �� ��� � �� ���� � ��� ����� ��
��� ������ � �� �� � ��� �� ���� �������������� 2� ���
������� � ���� � ��� ����� ��� �� ���� ������������� �� ���
���� ��� ����� �� ������ �� ���������� �� �������� �� ���
������� � ��������� �
)� ��������� ����������� �� ��� ����&���� � ���� � ��
��7��� �� ����� ���� ����������� ������� �� ��� ���
������� � ��������� � ��� �� � ����� ��� ����� ���� ��
���� ���� ��� ��� ��������� ���� ��� ��� ���� �� �� ����&
���� �� ������ ����� ���� ����� � ����� �� ������ ������
��� ������� � � ��� � � ������� ���� ��� ����� ���� ����
��� �� �� ������ "����� 2��&3�� ���� $� '������ ���
������-�� ��� ���� ��� ����� � �� ������� ��� ��� �� ����
�� ��������� 8������� ���� ������� � �� ���� � �� ��� �
���� ��� ��-���� ����� �-������ � ��� � �����
��������� � ��� ������ ��������� ��� �������� �������&
� �� � �� ���� ������� ��� ���� ���������� ������������
����� ��� �������� ������� � ������-�� ��������
��-���� ���� � �� ������ � ��� ���� � ��������� �� ���
������ ��� ���� �����-���� �� ��� "����� ��������� ������&
� � �� �� �������� �� ������ ����� ���� $� %������ ������&
� � � � ��� � ��!���� �� ����� � � �� ���� � �� ��� ��� ��
����������� ��-���� ��� ���� ������� � ��� � ��������
���� �������� �� ����� ���� ������ �
9���# 6����� ������� ����� � �� �� �� ��� �������
�!������� ��� ������ ������� � ���� ��� ����������
��������� ��� ��������� �� ��������� ��� ������ �����&
���� �
������ �
)��� ������� � � ��� ������������ ���� ���������� �����
� ��� �� � ������ �������� � �� ��� ������������
������� � �������� ����� ���� ����� � � ��� �������� ��
����� �� ��� �������� ������ ��� ������������ �� ���� ����
����� � ����� �� � ����� ������������ ���� �������&
��� +��� �� ��+� �� ������
��������
������ ����� ���������� (���� ��� ������ �� �����
���� �� � ������� � ����� ��� ���� :9���# ��� ���� � �&
���� � ���� ���� �� ��� ���� ��� ��� ������ ��� ���� � ��
��� ��� ������� � �; )�� 0++ �� �� ���������� �������� ��
*++ �� �� �������.�� ������� <���.� ��� ���� ���� �� � ���
���&������� ����� ��� =��� ��� �� ������� 4���� ���
�������� �������.�� �� ��+�% ��� 0, ���� �� ������ ��
����� ���� ���� �� ������ �!�������� ������ ������� �
���� "����� ,� ��� 10 ���� $ ��� ���� ������� �� ����� � �
������� ���� ��� ������ � ��� ��������� �������.���
����� ����� ���������� ��� � ��� �� ��� �� � ���&
��� ���� ������� � �������� � ��� �� � �������� ���
���������� � ��� ���� ���� ������ �� ��� ���� ���
���������� %������������ �� ��� ���� ���� ��� ����
������� � �� � ������.�� �� � ��� ���� �� � ������� � �
(���� ���� ���������� ��� ����� ���� �� � �� �� ����
������� �� �������� ������ ������������ ��������� ���
���������� �������� ��� ��� ���� �� ������� ��� ��� �� �
����� )�� �� ����� ���� ���� �� ������ "� �� ����
0++ �� �� ������� �� 0��1 (�$ �� ��� ���� ��� �� ���
�� ��� ��� ���� �� ����� ��+�% ��� � 0,&���� ������� �
����� )��� ���� �������.� ��� ����� ����� )�� ������&
����� �� ��� ���� �� ��� ���� ���� � ������.��� )����
������� � � ��� ��� �� � ���� �� ������ �� ������ ���
�� ����� �����
������ �
�������� �!������� ������ ������� � � ������ �� ��
� ��� ������������� ������� ���� "8>4)$ � ��� ��������
�����
����� � ����� �� 0�* ����� 8>4) ��������
���� ����� ����� � )���� � �� �++ �� �� ���
������������ ����� �� � � ����� �� � ������� � ����� ���
������� � ���� � ����� �� � ������ ���� ��� ����� 0++ ��

�������������� �� � ������� ����������
�� ��� ���������� �������� �� �� �� ���� �� ������ ��
������ ������ �� �� �� �� �� � � �� ��� �� ���� � � ���
���������� �������� �� ��� ������ ���� �� ������ �� ����!
�� ����! �� ����� �"� ������� #��� ��� ������$ ���
��� ������ ���� �� � ��� �� ������ �� �� ������� �� �����
��� ������� ����
������ �
�������� �%������� �� ��� ��� ������ �� � ������� ��
����� ������������ ���� �&#� �� ��� ������� ����
����� � ����� �� # �������� ���������� � ���'(
��� ����� �� ����$ ��� �� � �)�� ������� ����$ "�� ��
������������ ����$ ��� �� �� ������
���� ����� ����� � #���� �� �� �� �� �� ���
� �����'������� ���� ���� �� � ��� �� � ������ ���� ��
������ ���� �� ������ �� � �� �� ���� *��� ����� "�� �( ��
��� ���������� �������� �� �� �� ���� �� ������ �� ������
������ �� �� �� �� �� � � �� ��� �� ���� � � ��� &#
�� ������ ���� �� ������ �� ����! �� ����� �� �� +�
������� #��� ��� ������$ ��� ������ ���� �� � ��� ��
������ �� � ������� �� ����� ��� ������� ���� ,����� �
�� � �������� �� ���� ������ ��� �� ��������� �� ���
������ �� ������ � �����
������ �
!�������-������� ��� ���������� �%������� �� �� �� ���
*��� �� �� ��� ���� ���� � ��� � ����� ��� ������
�� ��� �� ����� �� �� �� �� ��� ���� �� � ��� �� � ���
�� ��%��� �� ��� ���� ��� +� �� ���� ��� ���� �� �%���
�./��$ ��� ��������� �� ��� ����� ��� � �� � ���
������� �� � �����'������� ������ �� ��������� ��
"� �( �� �� ��� ����$ ��� ������ �� ���! �� � �������0 ����
��� �( �� ��� ��������� �������� �� ����� �� �%������� ��
����*�� �� � ����� �� �� �� +� ������� 1� ���� �������$
�������� �� ����� ��� �� ������� ���� ��� ���������� ��
����� ��� �� ���������� ������� �� �%���� ������ ��
������ � �� ��� ������ �� � ������ ���� ����� ���
������2�� ��� ����� ����3������� �� �� ����� ������� �� ���
� ������ �� ������� �� �%��� �� � ����� ��� ���� �� ����
��� ��� �� ����� ������ � � ������ �
������ �
!�������-������� �%������� �� ������������ �� ��� ���
��2��� ����� ��� ������ *��� ������ � ���
����� � ����� �� � ���'( ��� ����� �� ���� �������-
��� �"� �� ������$ �� *���� �� ����� ������ � ��� ��
������ � 3��� ������� ����� �� �"� �*'�� �� �����
������ � ������ ������������ �� �� �����
� �� � ����� ����� � �� � �����'������� ��� ������
�� ��������� �� ����� ����! �� "4 ��� � 5� ��� ���
��� ������$ ��� ��������-������� � ����� �� ��� ������ ��
����� ��� �� ������� ���� �� ��� ������ � ��� � ����� �� ���
���������� �������� ��� ������2�� ����*� ����� �� �����
����� � ���� ������ �$ ��� �� �� ��� 2����������� ��
���������� 6��������� �� ����� ��� �� � ��%�� � �� ���
�� ��� ���������� ��� ��� �*� �%������� � ������$ ������-
���� �����%��� ��� ���������� �������
������ �
!�������-������� �%������� �� ������������ *��� ��������
�����%��� �5678�
����� � ����� �� � ���'( ��� ����� �� ���� �������-
��� ��� �� ��� �� ������$ �� *���� 5678 �� ����� ��
������ � 3��� ������� ����� �� "� ��'��
���� ����� ����� � �� � �����'������� ��� ������
�� ��������� �� ����� ����! �� "4 ��� � 5� ��� ���
��� ������$ ��� ��������-������� � ����� �� ��� ������ ��
����� ��� �� ������� ���� �� ��� 5678 � ����� �� ���
���������� �������� #� �� ��� ���� �� ����� �� �������� ���
���������� �� �� ���� ���� ������$ �� �� ���������� ��
�������� ��� ������� ���� ����� � � �� �%������� ��� ������
�� ������ � � ������ ���������� � �� � ��� �� �������� ��
������� ����� � � �� � �����'������� ��� �������� � �
��������� ����� +�� ��*� ���� ����� �� ������ ��� ��
������� ���� ������ �� 9������ ���������$ ����������$
�� �����$ ��� � �������� � � ���� ����3��$ � ��������
������������� �������� �� ��� �������� *��� ���� ������2��
������ �
!�������-������� �������� ��� ��:������� �� ������������
�� � ���� ����� �� �������������� �������
����� �� ����� �� ����� �++ �( �� �� �����$ ���
�( �� 4-������� �����$ ��� �( �� � ���������������$ ���
�++ �( �� *��� �� � �������� �������� $ ��� ��%
������� #�� ��� � �����'������� ����*��� � ��� ���
��� �� � ��� ������ ����$ ��� ����� �� � �� �� ���� �����
��� ���!��" �������� �� ��� �� �� ����$ ���� �� ������
������ �� �� �� �� �� � � �� ���$ ��� �������� �� �����
����! �� � ������� ��� ����� ��� ���� ��� ������ ����$
��� � � �� �� � ������ ��������� �� �� ������� �� �����
������� ������� �� �� ������������� � �����'������� ���
���� �� ���� ��� ��� �� ����� � �������� ���� ����
� ����� �� �� �� �������������� ������� �� �� �� ���
�������������� *��� � � ����� ������ � ������ ���������� �
�� � ��� �� �������� �� ��� ��� ���� ��� �� ��� �� ���������-
�������� ����� � (���� ���������� ����� �� ���
�� �������������� ������� ��� ����� ����3������� �� ���
�-����� �� ����� � ��� ��� ��� �-����� � ��� �� ������ ��
��� � �����
������
!�������-������� �������� ��� ��:������� �� ������������
�� � ��2��� ����� �� �������������� �������
����! ����� ��� � ��� � ��� 3��� �� �� ���������/
� ���'( �� ��� ����� ��� ��; ��� ���������� 4 ����'(
������� �������� ����!# �������� $�$ � ���'( ���������
��� ����� ��� ����!# �������� %�$ ��� ��� �� "-�� ���-
��������� �� *��� ����!# �������� &�
����� �� ����� �� � ��� � � ��%�� � �� ���!# ��������
% ��� ���!# �������� $ �+/�� �� ������ � ��<� �� ��������
�� � ���'( ��������� ��� ����� ��� �� �"� ���'( ��
��� ����� ���
������� 5������� ����� �� �� �� ��� ���� ������ �� ��
�( �� ��� ���!��" ��������$ ��� ��� ����� "� �( �� ���!#
�������� & 7�� � ���� ��� ���� � � ��� �� " ��� � ��
��� ����� ��� � �� ��� �� : � ������� ��� �� ����������-
���� �������$ ��� ����� " �( �� 4-������� ����� �� ���
� ����� ��������$ ��� �������� �� �� ���������� " ��� � ��
��� ����� ��� � �� ��� �� : �� � �����'������� ��

���� ���������� �� �������� ����������
�������� ������� �� ��� ����� ������ �� ����� �� ���� �
�������������� ���� ������� ��� ��� ��������� ������ ���
� �� �� � � ������ ����� ���� ����� �� � ������ ��
������ �
���� ������ �� ������ �� ��� ������ �� � ������� ����
� � � � ����� ����!������� �� ����� ���
��� ����� ��� ������� �� � ������ ��� ������ ��
��������������� �� ����� �� ������� � ����� �� ����
�� � �� �"" �� �� ��������� �� ��� �� �� ������� #$%&�
����� ����� �� '�� ��� �������� ��������( �������� ��
� ������ ��� ������ ��
�������� ) ������ ��� ���� ������ � %" �� �� ��� �����
��������( ��� ��� ���� *�% �� �� ����� �������� �� +����
����� � ������ �� ����� ��� * ����� �� ���� ����������� �
��� ����� ,�� ��� ��� �!"����"������ �������� � � ��� �
��% ���� ��� ����� ������� ��� ������� �� �� ���( ��� ������
��� �� ��� � ���� -" � ����� �� ���� ����������� � ���
����� ./���0 1� ��� �� �� ������� �� ��� ����� � � �����2�(
���� ��� % �� �� �"" ������ ��������� �� ��� ���� *"
���� �� ����� � ��������3 ��� ����� �� � ������� ��� ��
�!���� �� *����������������� ��� ����� ������ �� � �������
�� ������� �� ��� ����� ������ �� ����� �� ���� � ������
��������� ���� ������� ��� ��� ��������� ������ ��� �
�� �� � � ������ ����� ���� ����� �� � ������ �� ���
+�����!�� ������������ �� ������ 2 �� � ��������� ��
+�����!��������� �� ��� �� �� � ������ ��
������ ��
���� ������ �� � ������� 2 �� � �� �� ����� � �� � ��
� �� �� ���� �� � �� � �� ������� � � !�� � ���4��� ./���0
��� ��� �� �� � �� �� ����� � �� � �� � �� �� ���� �� � �� �
������� �� ��� �� � ��� ������� �� �� ��� �� �� �� �
����� � �������3
���� �� ����� �� , ����� �� ����� � �� �" �� ��
� �� �� ����� � �� � #�� � �� �� ���� �� � �� �& ��� �� ��
"�* ����� ��� �� ����! ���
�������� �������� ���� *" �� �� ��� ���� ������ �� �
������ � ���( ��� ��� % �� �� ��� �������� ���������
,�� �" �� �� ������� �������( ��� ���� ������ ��� �� ���
������ � � � ������ ����� ����� ��� ��� ������ � ����
�����5�� �� �� ������ #� �� � ������ ��� ��� ���������
���� ����� �� �� �� � ��� ���� ��� ��� ��� ��� 5�� � ��
����� ���( ��� ��� ������ ���� ��� ���� �� � �������� �� ��
�� ������ ��
������ ��
,������ �� ��� ������ �� ��� ��������� �� ������ � �� �
��� ������ � �� �( ������� ���( ��� �� �� � ������ ��
,������ �� ��� ������ � �� � ��� ���� ��� ����� ���
����������� $%!( 2� �� ������ �� ��� ������ � �� �
��� ���� ��� ����� ��� ����������� ��!� ����� �������
� ��� ��� � ������� 2 �� �#�(���� 6���������!& ������
5��� #7�1& �� ������� ��� ������� �� ��� ������ �� ��� ����
�� � �� ������ �� � �� � ��� � �� ����� � �� � ��� ����(
������� ���( ���� �� � ������ �� ����� ������� ��� ����2
��� ������ �� ������ �� ��� ������� �� ��� ������ ��
������� �� � ����� ������ �� � ��� �������� �� ������ � �� �
��� ������ � �� � ��� �����
���� �� ����� ��� ������� ��� 4���� ����� ����� ���0 �
����� �� �� �" ������ �� 6�������� � �� � #�������� $&( �
����� �� �� % ����� ���� � �� ��������� �� ��� �"
������ �� 6�������� � �� � #�������� �&( ��� � ������
�������� ����� �� �� &(&�� ����������� �� ����� � ��
-8 �� �� 7�1 ��� �� #�������� �&�
�������� 1� � ����� ������ � ���( �������� ����
*"" �� �� ��� ���� ������( ��� ��� * �� �� �������� $ ��
�������� � ��� * �� �� �������� �� +��� ��� ������ � ���
� ������� ���� ��� ������ �� 8"�� ��� 9 ����� � ��� �����
��� ������ � ���� � ��5�� 2 �� 2���� �� ������ ��� �!����
��������� :!����� ��� � ��5�� ������ ����� � ��� 2 �� � ���
������� �� ����� �������( ��� ���� ���� � 5�� ��� ����� �
��� ���� � �� � �����5�� �� �� ���� ���� ����� ��
����������� ��� �(��� �� ������ �� � ��� �(��� �� ����
�� � �� � ��� ���� �� ��� �� ���� ������� ���� ��� �� ��
��� ���� ���� ����!������ ������������� ���� ��
�� �� �� � ����� �� ���������( 2��� �� �� ����!������ ��
��� ���� �� �� �� �� � ������� ��( ��� ������� �� ���
������ �� �������� ��� ��� ���� ��� �� � ;������ � ���
������ � �� � ��� ������ � �� � ������� ������ 2 ��
����� ��� 5�� ��� 7�1���� ��� 5�� �� � ������ �� ./���0
��� ������ ��( ���� �� ��( ���� ��( ���� ��( ��� � �� � ��
������ ������� ��� � ������� 7�1 ��� ��� 5�� ��< �
������ � 2 ����� 7�1 2 �� ���� �� � ��������� � ���
������ � ��������� � ��� ������ � �� �� ����� ����� �� ��
�� � ��� ���� �� ��� ����� ������ ���3
���������� �� �� �� �� � � ������ � ������ �� � !���
=�� �� �� �� � ����� � ����� ��� �! ��( ��� ��� ��� ��
�� �� ��� ����� �� ����� ������� �� ��� ���� � � �
�� � ��� ���� ��� ����� 1� �������( ���� �������� �� ���
�� �� �� � ����� � ����� ��� ������� ��� �� ���
������� �� �� ��� ���� �� �� �� �� ����!������
������������� �����2�� ���������� ��� ��� 5�� ��
#����( 2 �� � ���� � �� ���������������&� ����������
������ �� ����� ��� ��� � ���� 2 �� ������� ���� ����� �
����� ������� �� �;�� ����������( ���� �� ����� ���
����( ��� �������� �� � �� ��2��� % ��� �" �� �� ����� �
������ ��� ����� �� ��� ���� � �� �� �� �� � ����� ���
�� ���� ������ ��������� ��� ��� 5�� �� �� ��� ���� �� ��
�� �� #����( ����� ���� �������< 8��� �� � �����&�
����!���� � � �� �������� �� ���������������<
#� ������� ��&�5���5���������� ����� ��< $�
6������������������������< ���( >�6�����9�� �����5��
*��!���(-�� �5���& �����2�� �������������� �����
��������� ��� ��� 5�� �� ����� ��� ��� ��� ���� � �� ���
������ �� � �� ��2��� "�% ��� ��" �� �� ����� � ������ ���
����� � �� �� � �6������ �;�� ����� � ��� ��������
��������� ��� ��� 5�� �� ����� ��� �� ���� ������ �
���� ��� ��� ��� ��� �� � � ��� �� �� �� �( 2� �� ����� �
����� ��� ������ ���������� ��� ���������� ��� ��� 5�� ��
����� ��� ��� �������� �6������ ���� �����������
��� �� �� �� ��� ���� ���� ��������� ��� ��� 5�� �� �����
� ����
��� �����2 �� ������% �� � ���� ��� ���� ��� ��
�� �� �� � ����� �� 1���������� ��� �������� ��� �����
���������� ��� ��� ���� ������� ���� ?����������( ���

�������������� �� � ������� ����������
��������� �� ��� � ������������ ��� � ���� ��������
������ ��������� � ������ �������� � �����������
� ��� � ��� ����� ������� �� ��� ������ �� ���
��� ��������� �� �������� � ��� ��� � ������� ����
�����!� ���� ��� ��� ���� �� ��� � �������� ��
������� ��� ������ ������ � ��� "� �� �� ��� �
�������� ��� ���� ���� � �� ���!�� � ��� �� ��
��� ����� � ���� �� ��� ��# �� �� ��������� ���
���������� ����� � �
������ ������� �� ��������� ��������� �������
��������
"��$������ ����������� ���� �� ������ ���$
������ �������� � ��� �� ��� �� � ����� ������
�������� ��� ���������� ���� �� ��� � � % �����
&�$ �� ����$������ � ��� � �������� ��� ���
��� � �� ��� ���� ������ ��� ������� ���� � �� ���
� ��� '$ �� ����$������ � ��� � � �� ��� ��� ����
����� �� ���� �� ������� � ����� ���� �������
������� �� (������� �������� �) ���� �� ����$
���� *� �������� �� ��� ���� �� �� � ���$������
����� � ����� ��� ������� �� ������ �� ����
�� �+ �� ���� �������� % ���������� ������� � �����
�������� �� ����� ��������
,��� ��� ���� �� ��� ���� ���������� ��� �����
� ������ �� ������ �� ������ ����� %���� �� �
����� ����� ��� ���� ������ ����� �� ��� ��� ���$
��� ������� � -). ��� /�� ����� �� �� �������
���� ������ ����� �� ��� ��� ������ ������� �
00. ��� /�� �� ������ ������ ������ ��������� ��
���� �� ������ ���� ����� � ��������� � 00. ��
-). ��� �� ��� ��������� � ����� � � �� ��� ���
������������ �� ���� �� ���� ������
1������� ����� � �� ������ �� � �. ���� ��� �� � �� ���
���� �� ��������� � �� -. ���� ��� �������� 2� ��� �
�������� � � ����� �� ��� ���� �� 3. �� -.. ���� ����
��������� ��4���� �������� .����� /� � ��� ����
���� ����� ��� ���� ����� ��� � �� ����� �������$
� �� � � ����� ��� ��� ���� �� ��� � �� ���$
����5��������
������ ������� �� �� �� ��������� ���������
������� ��������
�$6����������� (76%* ��� ���� ������ ���� ��
��� ��� ��� �� ����� �������� �� ���� ������ 8���� ���
� ������� ������ � /�� ������ � �����!�� ��� ���
�� ������ �������!���� �� ��� � �� ���� �� �
���$������ ������������ /�� ���� �� ��� ������� �
��� �� ������ �� "� ������� �� ������ ��� ���
���� �� ���� �� ��� � �� ��� �� ����������
��� ��������� �� ��� ����������� ��� ��
%������� 76% ��� ��� ��� ���� ������ ����
(����� �� �� �������* �� ���� 8���� ��� � ��� �
��� �������� ���� ����� ����������� ���� ������
���� �� ��� ���� 76%� /�� �������� ������ �����$
�� ��� ����$������ ����� ��� ������� �� ����
���� �� �������� � �� ������ �������� ���� �����
����������� �� �� ������ �������!���� � ��� 76% ��
����� ������� �� �$����$&$� ����� �� 3$����$
��������� /�� �������!���� �� ������ ���� �� �� ���
����� �� ����� � ��� �������� ����� �� �����
������������
�������� �� ��� ���� �� �� � ���$������ �����
� ����� ��� ������� �� ������ �� ���� �� �+
�� ���� �������� %���� �� ������ �������!���� ��
������ ���� �� ���� 76%� ��� ����� � � �������
��� 8��������� �������� 9����� ��� ����� ��� �� 76%$
�������!�� ���� �� �� ��������� ���� � ��������
��������� �� :0� �� �� � ��� ��� ��������� ��
0-. ���
1������� ����� � �� ������ �� � ��� ��� �� �������
����� ��� �� � �� ��� ���� �� ��������� � 2� ��� �
�������� � � ����� �� ��� ���� �� ��� ������� ����� ��
��� ��� �� ������� ������ /� � ��� ���� ���� ������
��� ��� ������ ���� ������ ��� -.. �� �� ���� �����
������� � � � � ����� ��� ��� ���� �� ��� � ��
�������5��������
������ ������� �� ��� ���� ���!����� �������
��������
6������ ��������� (6"/;* ��� ���� ���� �� ��
���� ����������� ��� (6/;* ��������� ���� � �
������� ���� ���� �� ������� � 30- ��� /���������
�������� �������!���� �� ���� �� ���� 6"/;
�������� � ����� ��$�� � +6&; ������� ���� <= ��$
������ � � �� �� ���!� ��� ���� �� ���� ������
%���� ��� ������ � ������� ����� ����� ��� �����$
��!�� ���� �� � � ����� ��� �� ���!�� ��� �����
���# ���� �� ������� ���������� "� ��� ������� ��� ��$
>����� � #��� ���� �� ����� �� �� �� �����������
�� ��� � ��� ���� ����� �� �
�������� �� ��� 6/;$���� �� �� ����� ��$�� �
+6&; ���� 71� ����� � ����� ��� ������� �� �$
����� �� ���� �� ���������� �� ���������� ��
���� ���� �������� 6/;$���� �� ������ ���� �����
�� ��������� � 3-0 ���
1������� ����� � �� ������ �� � � ���� ��� �� � �� ���
���� �� ��������� � 2� ��� � �������� � � ����� �� ���
���� �� 3. �� -.. ���� ���� ��������� ��4���� �����$
��� .����� /� � ��� ���� ���� ������ ��� ����
����� ��� -.. �� �� �������5������� ����� ������� � �
� � ����� ��� ��� ���� �� ��� � �� ������� 5������� �
������ "������ �� �#� ���� ���!����� �������
��������
6������� �������!���� �� ���� �� ���� ?$����$
��������$�$������� ��������� � ��� (%@;* ��������
� ����� ��$�� � +6&; ������� ���� 8���������
�������� � � ���
?$%������������$�$������� ��������� � ���
(%@;* ��� ���� ���� �� �� ���� � ��� 8���� ���
�� ������� ��� ��������� (%@;$���� �� * ���� ��
������ ��� �� �� ��� � � ����� ��$�� � +6&;�
/��������� �������� �������!���� �� ���� �� ����
%@; �������� � ����� ��$�� � +6&; ������� � � ��
�� ���!� ��� ���� �� ���� ������
�������� �� ��� %@;$���� �� �� 71� ����� �
����� ��� ������� �� ������ �� ���� �� ����$

���� ���������� �� �������� ����������
������ � � �������� �� ����� ���� ���� ����� � � ���� �
�� � ��� ����������� ���� �� ����� ��������� �� ��� �
�� ������ ��������� �� ��� � ����� �� ��� ���� �
��� �� � ��� ��� �� ������� ���� ���� �� ��������
� � ��� ��� �! ���� ����� �� ������ "!�#��� �$
%����&������ '� ��� ������ �� ��#���! �!���!(�� )��*���� �� ��+ � !���� %����&�����$ ���!���!�� �
������ �� ��" ������$ �� ����� � ����� �������
��������(��� � ��,� #�� ��
-��, ����� �� ./0���� � ��� ��� ���������! ��
����� �� �� ����� � ���, �� �� ���#�������$ �� ���
����������� ���� ��� ��� ��1 ��� ���"����! � ���� ��
������� �������� ��������#�� ���!����
2��� �� ����� �� ������� � "� ����� ��� �� 3� �
��� ��� �� �� � ��� � ��$ �� �#� �� 0!�� 2��� ��
����� �� 0!� �� �##��������! ��� ���� 4��#�� �������!
�� "����� � ��� ���� � ��� � ��� ���*5 ���� ������
�� �1 ���� �� ����� ������ 6� �#������ ����
��� "� "����� ��� ��� ���!��� � ��������(�� #����
�!���!����� ����� �� ������ �� �� � � #����*#�#�����
������ ������ �� �� ������������� ������
�������
-�� ��� ��������(��� � ��� � ��� ���� ��#��������
���!�� )7-.+ ������ "! ���������#���� 8-50 ��#�����
���� �������� ���� �� �� ����� 9��� �� ��&�� ��� �
���� � ��� � ��� ���� ����� �� �� ���! ����� )����$
#����+�
��-����������!�� )7-.+ � �� �� ���� � ����
������ ��� �� ���� #�����! ���� ���#� � ��� �����!
����� �� ������ #��� ��� ��:�� �#������ � ������
�#�#�#�� � �� � "� ���� �� ��� ����� 7-. ������ ���
� ����� � �� ��&����! #��� �� ���������
#��,�� ; ������$ ��� ���"����! �� ���"����! � �&����
�����$ ��� ���� ��� ��#�� ,���� � �� ��� ��� ��$ ��,�
�� ����"�� � �������� ��������(��� �� ���!��� ����
� ������#��� � ��� ��� ���#�� ���� ��� �������
8�����$ �� , � ��� �����! ���� �� ���! ��� � ��� ���
"�� #������� ����"� ,� 9��� ����� ��� � ���� �
��� � ��� ���� ����� �� �� ���! ����� )����$ #����+�
9 �#����� �� ���� ����"� ,$ ���� �� ��&�� ��! "�
�"��� ���� ����� �� ��&�� ��� ��"�� � ������ � �
������ ��
-�� ��� ��������(��� � ��� � ��� ���� 7-. ��
������ "! � ���������#���� 8-50 ��#������ <� ���� �
��� ����"����! � ��� 7-.���� � �� ����������$ 8-50
��#����� �� ���!��� ��� #������� ����������! �����
�� ��������(���� 9�� ��&��� ��������#� �� �&��##��
���� � �������� ���� �� �� ��� ���� �� � ��������(��
��� � ���� =����� � � ������! � 7-.���������(��
��� � ��� �� ������ ���� � �� ����� ��������� �
�3� � �� � ������ ��������� � 3�� ��
2��� �� ������ �� �� �� �� ��� ��� ����� � � ����
"�� ��#����$ ������� ��� #�� �� �� ����� � ���!���
������ �� � #���
������ ������ �� ������� ������������� ������
�������
-�� ��� ��������(��� � ��� � ��� ����
)������!����+�("�(������!� ������ )2.<��0�+
������ "! ���������#���� 8-50 ��#����� ���� ����"��
����� ���� �� �� �����
)2�����!����+�("�(������!� ������ )2.<��0�+
�� � ���#��� ������ ��#�!�� �� ��� ��"���� �
��� � ���� .�� � ��� ��"���� ���� 2.<��0� )2.<��
��� � ���+ ��� �����! ���"�� �� ��� ��� �������
�"��#�� �� 3�% ��
2.<����� � ���$ ��� �� �������! ����� ��� � ���
�����������$ � "� ��#������ � 72� ��� � �
���������#���� 8-50 "! ��#�!�� ������� �!����� �
������ � � �������� �� �&���� "�>�� �������� ��#������
2.<����� � ��� ������ ��� ��� ��� ���� ��� ��
3�% � � ��� ����"�� �����
9��� ������ � ���!(� ��� ��� � ��� �� � �� #����
������� ���� ��� ��� � ��� �� ��� ���� ������ � ���������
�!$ 2.<��0� ��������(��� ����� #������ ��� ���������
�� &���� ��� � ��!#�#�� �������� "! #������
�!���!��� � ��� #����*#�#���� ���� ����� � ��� �� � ��
��� �#����������� � ��$ ������������ � �� �
����������� � �� ��� ��"�� ���������� � � ??-����
8!���!���@@� 9�� ���� � �����"��� ��������$ ��#������ ��
��������$ � ��� "� ���!��� "! #������ ����� ��
�����#�#�� � �� �� �����"��!�� � ��$ ���#� �
�����!$ "! �������� � #����*#�#���� ���� <9; ��� ��"��
���� ������ �� � ??-���� 8!���!���@@�
9�� �#������� ��� � ��$ ���� �� �� "�
���� �� ������ ������� � ���� �����$ �� ���� �#�� ��
������ � � ��������#�� ���� ����� ���� #��,� �
#�����! ��� � ���� A����!���� � "� ���� �� �
������ �������$ "� ���� �� �� ������ � � ��� �����
2��� �� ����� � 2.<����� � �� �� �"�� � #��� .�
������ �� � � � #�� � � ��������� 2.<����� � �� �
"� &�����������! ���!��� ���� �����"����!$ �� �! �� �
�� � � ��� ��"�!����� #���� �!���!���� �� ��&����� ��
�� � ���!����
������ ������ �� ������� ������������� ������
�������
-�� ��� ��������(��� � ��� � ��� ���� ��
����!�����!� ��������� )=:70�0�+ ������ "!
���������#���� 8-50 ��#����� ���� �������� ���� �
�� �� �����
��=����!�����!� ��������� )=:70�0�+ ��� �� ����
"�� #�����! �� �� ���! ��� � ��� � ��� �����!
����� �� #��� ��� 9�� ��� �� � =:70�0� ���� ���
� �� #� ���� ���� ���� ����� � �&���� ����� ��
�� �#����� � �� �� ��� 9�� ����������� ��� ���"��$ �!
��� �������� ���������� ����� �! "���,��� .������
=:70�0� �� ����� �� ������$ ��� ������ �� ��� ��
����� �� �����#��� �� � "� ��������� ������ ��� �
=:70���� � ����
=:70���� � ��� ��� ��#������ "! � ���������#����
8-50 ���� 72� ���� 9�� ��#����� �� ������ �� "!
������� ����� ������ ������! ��� � ������� � � ����
����� ������ �� � ��� � �� "�>�� )��B3�B��+ � � �������
� � �������� �� � ��� � �� "�>�� )��B��+$ �� �� ���
� �� ����������� ��� ��#������ � �� ������� '� � ����������

�������������� �� � ������� ����������
������ �� ����� �� �������� �� � �������� ���� ��
��� �� �� �� ������� ���������� � ��� �� ��� �� ��������
���������� � ��� ���
��� ���� ���� ����� �� �� ��� ��� ��� ����� � ��������
���� � ��� �� � ����!" �� �������� � ���� � ��� �����
� ����
������ ������ �� ����� ������������� ������
�������
#� ����� �������$����� � ����� � ��� ���� %&����&'&
�������$�&�&���&���&���$��� ()*+&,- ������� �� ������&
.���� /#"0 ��.������ ���� �������� ���� ���� �� �����
%&����&'&�������$�&�&���&���&���$��� ()*+&,- �� ��
���� ���� .���� ��� �� ����� ����� � ��� �� �� ������
����� ��� .��� ��� ����� � ��� �� �������$�� ����
)*+&, �� ������� �� ���0 � ��������
)*+&����� � �� ���������� �� ��.����� �� �� 1+2
����� � � ������&.���� /#"0 �� ��.������ �������
������� ������ ��������� � � ��������� ��� �3����� ��4�
������5 ��� �% ����� � �� ���������� �� ��.����� �� �
�������� �&����� �.�� � �� �� �� ���� �� �� �������
�������5 �� ���� �� �� ������ �� � ���� ��������.��
������ 6� � ��������� ������ �� ����� �� �������� ��
� �������� ���� �� ��� �� �� �� ������� ���������� �
'�� �� ��� �� �������� ���������� � �� ���
��� ����������� � ���� ������ �� ������ ��� ���� ��
� .� ����� 1#� �������$����� ������ (������ �-5
�� ������ .����� �� ��� � 1#� �� ��� �� ����5 ��� �����
�� ������������ � )*+&, ������� 1#�� ��� ���� ����
����� � �� � ����� � �� �� ����� �� ���� #�7�� ��������
��� � ������ � ����� �� �� � .����� ����������� �� ���
7��� .� ����� �������� �� ���� ������ � /#"0�
���� ���� ������ ��� ��������
8��� ���������� ��� ����� � �� ������ � � .�&
����!.�.���� ����������5 �� ������ �� ����� ���� ��� � ��
��������� ���. ������� ��.��.��� ��� �������� 2���� ���
�������� �� .������� �������� �� � �� ���������5 �����
������ ��� ��� ������ ������� ��� �� ���� .������� �������
9��������� �� ������ ��������� ����� � �� ���������5
��� ���� ����� � ��� (����5 ��� ��� ��� �����- �� �����
������������ �..�� ����� � ���3���� �� ��� (���� ����
���� ��� �� � �� �� � ���% #�- � ������ ���� � ����
��� (����- �� ��� �����.� � � ��� �� ���� ������ �����
��.� .���� ��������� �� ��� � ��� ����� � ���������
����� ����� ������5 ��� 3����������� ������ �������� �
�������5 ��.��.���5 ��������5 ������ ���5 ������5 ������&
����5 ��� ���5 ��� ����� �� � .�����!.�.���� ����������
��� �� ������� ��� ��� ����� ���� ������������� ���
������������
���� �������
����� ���� ���� ����� ���� �� ��� ����� � �.� �7
����� � �� ������� .� ��� ������� �� � .������ ���� �����
��� �� ����� � ���������� ����� � �� �������� ���� ����
��� ���� ��� ������ � ��� .����� ���� ������������� ��
��:����� ���� ��������� �� �� ���� �� ������� ���
�������� � � .�����!.�.���� ��� ��� ���� �..�� �������
0������ ������� ��� �������� ��� .��:� �������� �� ��� �&
�� � �� � ���������� 0�� ����� ��� ���� .� ��� � �� �
����� � �� .����� �� ��� ���� ���.�� �� ��� �����;
����<!�5
�� ��� � �< �� ��� .��: ��.����5 �� ����5 � ��� ����� � ��
���� ����= ��� � �� ��� ��� � .��: ��.�����5 �� ����5 �
��� ����� � ��� .����� �� ��� ���� ���.��� 0��.����� � ���
���� .� ��� � ��� ����� � ��� ���� ���� �� ���� ��
:���� .������ �� ���. ��������� � ������� ���
�������� � ��� ���.�� .������
�� ��!� ����� "������ ���� ���� �������� �� ���3��
�� �� ���� �� �������� ��� .����� �� �������� � ��
��:���� .����� ���.�� ����� ��� ����� � �� �������� �����
0�� ����� ��� ����5 �� ��5 � �� � � ����� ����� � �� ��
��� �����;
��8!����5
�� ��� � � �� ��� � ����� 3�������5 �� ����5 � ��� �����
� �� ���� ����= ��� �8 �� ��� ������ ���� ��� ������ �
���� ����� � ��5 �� ��� � ��� ������ � ��� ����
���� ��� ���� ��� ���������� ����� .�.���� ���� ���&
����� ��� ��� � ��� ������ � ��� � ����� ����� � ���
���� ���� �� �������� � ��� ����� ���� � ��� .����� �����$��
��� �..�.���� �� ���� � .������� ��� ��.������
�������� ����� � ���� > ��� ���� ��� ������ � ���
��:���� .����� �� ��������� (����5 �� 2+2&#�?6 ��������
� ���� �.� ��� �.�-5 ��� ����� � �� ��.������� � ���
��:���� .����� �� �� .��� ���� 0�� ����� ��� ����� �
������� � �� � ����� � �� �� ��� �����;
�!(�����!�8�-5
�� ��� � � �� ��� � ����� 3�������5 �� ����5 � ��� �����
� �� ���� ����= � �� ��� ����� ����5 �� ��5 � ��� .�����=
��� �8� �� ��� ���� ��� ������ � ��� ��:���� .������
#��!� ����� "������ ���� ���� �������� �� ���3��
�� �� ���� �� ����������� ��� ����� � �� ��.������� ���
.����� �� �������� � � .����� ���.�� � :���� ���� �&
�� ������ ��� ����� � �� ��.������� ����� ��� ����� � ��
�������� ����� 8��� ��� ��.������� � ��� .����� �����
�����$�� �� :����5 ��� �� ��.���� ��� � � ���� ���� �����
� ��� �� � ����� ����5 ����� ���� ����� � �� � ������
��� �� ��.������ �� ���� � ��.���� � .����� ����� &
���� (����5 ��.��.���5 �������5 ��������5 �����5 ������&
����-5 �� ��.���� ���� ������� (����5 � ������ ��� ���
������- ��� �� ����� � �� ������������ (����5 �� ��� ���
��� �����-�
*� ���� ����� ����� � ��� ���� �� � ����� ����
�.����� ��� .�����5 ����� ����� � ��� �� ����� ��
3������ ��� ������ � .������ 8���&� ����� ����� � ���
��5 ��.� ����5 ��.�����&��.������5 ���������&���������5
�������5 ��� ���5 .������������5 ������5 ��� �������� ����
���� �� �� ����7�� ����� �� ��.���� � ���� ���@� ���
�������� ������� +����� ��� 3�������5 �� ����5 � �� � � ���
����&� ����� ����� � ��� �� ��� ��.� ��� ����� �
������� � ���� ����� � �� �� ������ ��� .����� ������
����� �� �� � ����&� ����� ����� � ��� ������ ���
.����� ������ ������ �� ������� ��� .����� ������
��������� � �� � � ��� ����&� ����� ����� � ���
������ �� ������ ���������� ����� ��� ����� +�� ��
.����� ������ ������ � ����� ����� � ��� ���� ���� ��
��� �.����� ��������� �� ��� ����� ��.� ���� ������

���� ���������� �� �������� ����������
���� �� ������� �� ��� ���� �� ��������� ����������
���� ����������� ��� ���� ������ ������� �� ��� ������
��� ����� �� ������ ��� ������ ������� � ��� �������
����� ��� ������� � ���� ����� ���� �� ��� ����������
���� ������ ������� �� �������� ��� ����� ���� ��������
���� � ��� ������ �� ���������
��������� ��� ������ ������������� ��� �� ����������
�� ��� ������
� �!�"�
�� #���� � �� ��� �$����������� ��������� %�������� ��
���� �� ����� ���� ������� � ��� ����� ���� ���� ����&
��� �" �� ��� '��#� ������ ���� � ���� ����� ����� (��
����� ������ ������������� �� �� ��� ����� � ���
�������� ����� � ��� ������ ������������� ��� � ���
��������� ����� ������ ��������� �$������� ��������� ���
�������� �� ���� ������������ (�� ����� ������ ��������
������ �� ��� ����� �������� �� ������� �� ��� �������
��� � �������� �� �� ����� (�� �������� �� ��� �����
���� ����������� ���#��� ��� ������ ������ ��� ��� '��#�
����������� ��� �� ���� �� �������� ��� �������� ���
����� � ��� ������ �� ��� �������
��� ��� �������
��� ������� ��� ���������
������ ��� �����
�������� ������������� �� � �������� ������ � ��������
����� �� ��� ��������� ������ � ��������� � ������
�������� �������� �� �� ���������� ��������� ���� ���
��)����� � � ����������� ������� *����
(�� �������� ������� � �� ������� ���� �� ������� *���
� ��������� �� �� ��������� �� ��� ������������� ��������
� ��� ������� ��� ��� �������������� �������� � ��� ��+�
������ ��� ��������� (�� ������������� �������� � � ������
,���- ������� �� ��� ������������� � ��� ������ ,�������
������ �������� ��.� ��� �����- ��� ����� � ��� ��+� ��
#���� ��� �������� ��'�� ����� ,���� ��� ����� ������� �
��� ����������� �/� �������� ��� ��������-� (�� �����
��������� ������� ,���- � � ������� �������� � ��������
������ �� ���� �� ��� �%�������
���������� �
0����� �
��
�� �+����� ����� � ��� �������
�� �������� � ��� ���������� ���������
�� "��'�1� ����� � ��� �������
�� ������� �������
�� ����� ������ � ��� ���������
2��� �� ������� *��� �� ������� ������ ��� ��������
*���� #��� ��+�� � )�# � ������ �� �������� ������ ���
��������� ������ �������������� )�#� (�� ������� � ���
�������������� )�# ������� �� ��� �������������� �������
�� ,���- #���� �� ��� ������� �� ��� ����� ������� �� ���
�������� ������� #��� ��� ��� ��+� �������������� (��
�������������� ������� ,���- �� ���� �� ��� �%�������
���������� ��
��� �
��
�� ��������� �������� � ��� ��+��
�� .��� ��������� � ��� �������� �� ����
(�� ������� � ��� ������ ,�- �� ���� ���
���������
(�� ������������� �������� � ��� ������� ��� ���
�������������� �������� ��� ��� �� ��� ���� �������� � ��
�������� ���������� ��������� �� ��� ����� � ��� �������
3� ����� �������� �������������� ������ #��� ������ ��
��� �������� �������� �� ��� �������������� )�# ��� ����
��������� #��� �� ������ ���� ��� �������������� ��������
������� #��� ������ �� ��� ���� �������� �� ��� �������
������� )�# ��� ���� ��������� #��� �� ����� ���� ���
�������������� �������� 4��� ���������� �� #���� ���� ��
� ��� �������������� ������� #��� ������ �� ��� �����
��������� ������� � ��� �������� ���� ������� ��� ������
��� �� �������� �� ��� ���� ���
(�� ���� ,�- ��'�� �� ��� ������ �� ������ ��� �������� ,�-
�� ��� ��5������ ��� � ��� �������� �� ��� ��������� �����
,�������� �+����� ������- �� ���� �� ��� �$��������
���
��������
���
,�������-�
3� ������� �������� ����������� ���������� ���� �/ 6
��� ������� ����� ��� �� ����.�� ������� ����� �� ���
���� #���� �����%������� ��� �������������� )�# �� ��
����� �� �������� (�� �������������� )�# ���� �����
�������� �� �� �� �� � ���� ������������� �� �� ��
�������� �� ��� �������� ������� � ��� �������� 7� ����
������������� �� ��� �� �������� �� ����� � ������� ���
�������������� )�# �� ���� ���� ��� ���� #��� � ���
�������� � �� �������� ��� ������������� �����������
���!� �/ � ��� ��+� ���������
8 �� ��� ����������� � ��� ������ ���� ��� ���������
���� ������� ��� � ��� ������ ������� #����� ��� ���'�
����� ���������� �� �� ����������� .���� �������� �� ���
������������� ��������� 9��� ���������� ���� �� ��� ������
��� � ���� ������ ����� ������ �� ��+���� ����������
4��� ����� ���������� ��� ���� ����������� �� ��� �������
.��� ��������� �� �������� ��+����� � ��� ������ �����
��� �������� ,������������ ��+�����-� 3� ���� ����� ���� ���
�:������ � ��� .���� �$������ �� ��� ����� � ����������
������ ,�-� �� ���� ���
��,�������-����
�����
�� �������� ��+����� ���:����� � ��� ������ �� ���
��+��
3� �������� ���� ��������� ���� �� ���� ������������
������ ��������� ���� ��� �������� #���� ����������
����������� ���#��� ������ ��� ��+�� ������ � ��� ��5���
���� ����� ������� ���� ��.� ��� ��������� ��+� ������
��� ���� �����*������ ��������� �� ���� ����������
"�������� ���#��� �#� ����� ,�$������ �� ��� ������
����� �"- ��� �� �������� �� ���� ���� ��� �������������

�������������� �� � ������� ����������
������ �� � ������� � ������������ ������
������� �� � ������� ��� �� ���������� � �������� ���
� ���� �� ��� ������ ��������� � � ��������
���������������
� !��������
���� ��� ����� ������ ����� �������� �� � "� ������
���������
!���� ���� ������ ����� ������ �� � "� ������
!�����
������������#
���������
$� �������� ��� ������� ������ ������ �� ��������
���
%� �� �&����� �������� ������������ ��"�� �����
��
%"� ��'�� �����&����� �� � � ���� �&�� ���������
� ���������� ������ ��� �� ���� ��������
%"� ������� ��������� � � �� ��� ��� � �������
�������� �� � ��'�� �����&���� ��� �������� � �
��"�� ������
%� ��������� ������� ������ ���� �� ������������
" �� � " �� ���� "� ���� �����(� ���� �� ��������
�� �� ����� &��"��� "����" ������ "� � �����
��# ) � ���� �� � ������� ��� ����� �� � ��'��
�����&����# ) � ������� �� (�� "� � ������
���������� �� � �������� �
%� ������ ��*����� ������
%� ������ ��� � ������ � ����� �� ��������� ��
������ ������� ���� � ������ �� � ���������
������� � � ��&�� ���# + �� ����� ����� �� �������
��� ������� ������ �,- ��� &������ �� .�������
��� �� ������������� ����������� �� ���� �������
����� ������� ��� �� ����� ��� �����(� ����������#
+������ ������� �� �� ������&� �� �� ���� � ���
�� ����,-���������� ��� ����.�������� ����
�������
%� �������� ����� ��� � ������� � ������ ���
������� ������ � ������� �� ����������� � ����� �
���� ��������� ��������������
%� �������� ��� � ������ �������� �� � �������#
) � ��(����� �� � ��*����� ������� ��� �� ��������
��� ������ ��� ������� �������&� �������# /���� ��
��*����� ������ ���&��� �������� �� &����� ��*����� ���
�����0����� ��*�����# ) � ����� �� ��� �����
�������� ��������� �����0������� ������� �� ��
������ ����� ������� ������ � ������� �������������
����� �� ��*����� ����#
,�� � �������� � ��'�� �������� � ��������������
�� ��� � ����� ������ ��� � �������� ���������
���������� �� � �������� �� � ���������� ��������#
) � ������� �������� ������ �� (���� � ����&�
������� ��� �������� � �&��� ����� �������� � ����
�������� "� � ������� ����� �� ������� � �������
����� �� � ������� ������ � ��������� ���# $
�������� ������� ��������� � ��� �� ��&����� ��� ���
������� �� �� � �� ��&� ����������� �������� ����
�� � �����#
� ������� ���� ���������������
���������
+� ������� 1��� ������ ������� ������ ��� �������� ��
� ������� ��������� ��� ��'�� "� �� ��� ������&���&�
������# 2� �� �� ������ ��������� �0�� ���� �������
� ��'���� ��������� �� � ����� ������ �� �������
����� "� ��'���� &�������# ) � &����� �� ��� ����
������� �� � ������ ����� ������ �� � ���� ��� �
������������ .�" �� � ������� ���� 3����� 4���������#
5���� ��������� ��� �� ���� � �������� � ���������
������� �� ��� ��������� ��������� �� �����������
��������#
,���� �� ���� �� ������� ������ ������� � �������
�� �� ��� �����666� ��� ���� ������� ��666���
��66�666� ��� �� �������� ��# 7�� � � �� ��������
�� ��&�� �� ���� ������ ������� ������ ������� �������
��� �� ������� �&��� ��� ����� ��'������� �� ���
� ����������� ���� ��� �� �'����# ) �� ��������� ����
��� ��"� � ��������� �� � ��� ��������� �� �������
�� � ��� ������� � � ��������� ��'��#
������������
8����1���� �� � ��������� �� � �����9 ������� " ���
��&��� ��������� ��������� ��� ��� � ��*�� ���# ) �
���� ������ � �� ���������� �� � ��&������ �� �������
���� ��� ��������� ��� �������� ������ ���������#
������������ ����������
�������� $ :��� ����� �� �� ����� �� �����1��� �
������ &���� ��� ����� ���������# ��������� ��� ��
��&����� ���������� � ������ &����# ;�"�&��� ��
�������� �� � &���� ���� ��� ����� �9�����&� �� �������
���� ��&��� ���� � ��������� ���� �� � ���� ������
&������� �������� �� � ��'�� ������ � �������# ) ��
�'�� ������ ���� ���������� ��� ��������� ��������#
��������� <������ ������ ��� �� ����� ������ � �
��� ��� �� ��� � � ���� ��� � ������������ .�"
"� ��&� �"��� � �� ���# +� � ������� ������ ��
��&������ � ������������ .�" �� �"�� ���� � ���
��� ��� � ����� ������ "� ������������ ��������
������ �� � ������������ .�" "� ���� � � ���#
������������ ) � ���� �'�� �� ��������� �� ���
���&�� �� ��'�� &������� ��� ������� �������&��� ���
������� �� �������� &�����# +� ���� ������ �� ��������
�� ������� ��������� ��� ����� � ������������ � ����
�� �������� ��������� ��� �������� ��� ��� � ��������
�� � ���������#
���������� ) � ���������� �� � ������� ���� ���
������ �������� �������� � ������� ���� �������� ��
���������� ��� ��� �������# +��������� �� �'���&�
��� ��� �� ��� ��� �������� � ������ (���
�"��0��� � ������ &����� " �� ��������� ��������
���# =�� � ��&�� ��'�� ��� ������ (��� �� �����������
��� ���� ����� ���������������� ������ �� � ������
������� �� � �������# ) � ��� ��� �'��� � �������
���� ��������� �� � ����� &���� ��*���� ��� �
������� ����� �������#
����� � ��������� �� � ����� ��������� �� �
������� "� ���� ��������� �� ��� � �&��� ���
���������� � ��� �� ���������� �� � ��&������ �� �

���� ���������� �� �������� ����������
������� ������ � �� ������� ���� �� ���� �� ������
������� ���� ��� ������� � ����� ������� � ��
�������� ����� ���� �� ���� ������� ���� �� � ��� �!
� � ������� �� ��������� ������ ��"� ��������# � ��
�$��� ��������� �� �� ��"� ��������� � ����
���� ������� � � ��� �������� �� � � �� ���� ��
�� �������� �� ����� ��� � ������� ������ �� �� ��� �
�� ������� �� ���� � � ����� ����� �� ���� � � �
�� �������� ������ ������ ������� %� ��� �������
����&�&��� ���������� ��� ���� �� �� ���� � �� ����&
����������� ���� �� � � � �� �� ������� �� ����������
����������� ������ ��������
����� ���� ��� �������������' ������� ��"�� ��
�������� ������������ ���� � �������� ��"� �������
� �� �! � �� �� ������ � � ��� ������� � �� ���(�
��� �� ���� �
)���� � ��"�&�� ������� � ����� �������� ��� ���
��������� �� ����� �� �� ���(� � �� � ������ � *��
��� �� ������ ����� ���� �� ���� ����� � �������
� &����� ������ ������ �� ����� � ������ �������
�+��� �� � � ������� ������ �
, � ����� � ��"� �� �� ��� ��� � ���� �!#
�������� �����&������ -�� � � ����� ��������
����� ��' *�� �! �� �� ��"� �� �"�� ������� ��
������� � �� ����� �� �� � ���� � ��������� � � ��
��� �� � �� �����&������ -��� � ���� � � ������
������� � ��� �� � �� �! �� �� ��"� ��� ����� �
����� �� ��������� ��� ���# ��� ��� �� � ����� �� ��
����� ��� ������ � �������� , � ����� � �� ��"�
�! �� ����� � ������ �� �����&������ -���
������� ��������' .�� �� ������� ����� ��� ���� �&
���� ���# ��� �� ����� � �� �$����� ��"� � � ����� ��
��������� �� �� ����� � ��� �������� � �/� � �"�� ��
����� �� �� �(��� �� �� ������ ����� � �� *��
������ �� ���� ��� �� ������� � �� ��"� �� �����
������ � ������� � �� �����&������ -���
��������� ��� � ���� �����������' %� �� ������� ��
������ ������� � ����� ������ �� ����� � �� �������
��"�� *�� ��� ����� �� ���� ����� ������� ��
������� � �� �� ��� ����� �������������� � � ��&
�� � ��� ���� �� ����� �� �� ����� ���� ��� �� ���� ��
�� �� ��"�� � ����� � ����� �� ����� ������ � �
���� �� �� � � ������ �� ������� �������� �� ��
����� ������ �� ���� �� ������ � �� ��� �� �����
������ ����� � ��� ����� �� �� ��������� �� � ����
������� � ��� �� ������ � �� ������� � � ���� � �
��"�� ����� ��(� ��&� �&� � �&������� � # � �������
������� � � ��� ���� ������� ����� ���� ���0��� ���#
� �� �(���� ���� ������� ���� ������� ��������� ����
���# ������ 1�� ��� � ������� ������� � �� ����&�&
���� ������ � � �� ����� �� �������� �� ��
������� � � ��� �� �0� � � ����� �� �� � ����
� -�� �� �� ���������� .�� ����� �� ���� � �� �����&
�� � ����� ������� � �� �� �� ��� �� ����� �����&
�� ��������� � � �! �� �� ��"� � � �������� *��
��� �� ��� �� ��������� ���� �� ���� �� � ��� �� ����
������ �� ������� ���������
�� �� ����� ��� ������� ����
���� ��
� �������� ��� ������������� ������� �0�� �����
� ���� � �������� ����� ��� � ��� �� ��� �� � ��������
������ )�������� ��� ������ �����&�&���� ���� ��
������� ������ � � �������� ��(� �� �� ������ �����&
���� ���� ��� ����� ����� �� ���0 �� �� ��� � �
������ ����� ���� �� ���� ���������� 2�"��
���������� ������������� ��� � ������ ���� � � � 23,
����� �#� ����� ��� ���� ������ �����&�&���� �����
�� ��� �� ������� ������ � � ��� �������� ���� ��
�������� ��� �������������
����������� �� ���
*�� ���� �� ���� �� ���� � �������� ������������'
���� � �� ����� ���� � � �� �������� ����� ����� 4�&
�� � �� ����� ����� ���� �� ����&�� 0�� ��������������
�� ������ � ���� �� �������� �� �������(��� �� ��
�� ����� *��� �� ������� �� ��� � �� �����&������ ����
� � �� � �� ������ ����� ������ � �� ��������� ��
�� ���� �� ���� �� ���� � ������ � �� ����� � �� ��&
�� �� �� ������� ��"� ������� �� �� � ������ �������
������ � � �� ������� �� �� ���� �� ���� � � �� ��
�� ������ ������� ������ � � �&��������� �� �
���������� ����� � 5���� � 1�� � &����� � �� ��&
�� � �� ���� ��� � ������ � ������ �� � � �� � �����#�
�&��������� �� � � ���������� �� � ����� �����&
�� � ����&�� 0�� ���� �� �� �����(�� �� ������� � ��
������� ��"� ��� � ������ � �� ��� ������ ��������
������������ ����� # � � �� ���� � �� ��� ������
��� � �� ��� ������� � %� ����&�� 0�� �������������
����� �� ������ �� �� ������� �� ��� �� � �� �� �� &
��� �� ��������� � �/� �� ������ �� ����&�� 0��
,� � ���� � ������� � �� ������ �� �� ��� ����� � �
������� � �� ������� �� �� ������� 2�� � �� ������ ��
���� ����� � �� �����0� ��� � 5���� �� �� �����
2� �������� ����� ���� �� ���������� �������� ���� ��
�� �� �������������� ��������� ���������� �� � � ����
����� �� �� ��������� � �$����� ������� ��"�� ���� �
��� � � ������� ������ �� ���� ��� �� � ��������
������ *���� ������� ����� �� ����� � ����� ��
����&�� 0�� �������� *��� �� �� ������ � � ���� � �
����� �� ������ � � ����&����� �������� ���� � �����&
������ -��#� 6������ � �� ��� ����� ���� � 5����
�� ����� ������� �� ������� ������������� *��
������ �� �� ���� �� �� � ������ �� ��� � ������� ��
����� �������� ���� �� � ���� ������ �� �� ��� # �
�� ������� � �� ������ �� �� ��� ��� � ���� ����&
�� �������� ����#� , ������ � �� ��� ������ ����� �
� ������� � �� ������� �� �� ����� �� �� ���� ��"��
�� �� �� ��������� �� ���� ������� � �� ��"� �����
��� �������� ������ �� ��� ������ ���� � � �����0�&
��� � 5���� ��� �$��� �� �� �����
�� �� ����� ��������� ������
���� ��
� ��������� ������ �� �� ��������� ����� � �� ��
� -�� �� �� �� ������ ����� �� �� � �� ��� �� ������� �
� �! ����� �� ���� �� ��������� ���� � �� ������ � �

�������������� �� � ������� ����������
���� ��� ����������������� ������ ��������� �� ���
������� �����
�� ���� ���� ����� �! ���������� !������� � �������
!������� �� ������"�����
������� ��# �� ������� �� �� ��������#
$������ �� ��� ����# ��� ����� �� ����� ���� �����
����� �� ��������� ���� ��� ������ ����� �� ������
� �����%
$��&������ ������# ������ ����� ���� ��� �����
������ ���� ��� ����� ����� ���� ���������� ���
��������� ���� �� '���� ��� ��������� ���� �
��������� ���� ��� ������� �� ������ �! ��� �����
���� �� ���� ������ ��� �� ����!� ��� �( �������
�� ����� ��# )��� ��� ������ �� ������� ���������
����� ���� ��� ������ � ��� ����� ������� �� ����
��� ����� ���� ������ �( ������ !�� ���� ����
�(� �� ������ ����� �(�� *���� ���� ���� ��� ������
����� �� �� ������ ����� ����� ���� ��� �( ����
�������� �� ���� ���������� ����� �+� �� ��� ����� ����
�� ��� ��� ������
����������� ��# +! ������"���� �� �&���� !� ������
����� ��� ��� �! ��� !�������� �������� ��� ������� �
������#
$�� ��� '�� ������� ������"���� �� ����������� �����
��� !������� ���� ���� ��� ����� �! ��� ��������������
,��% ��� �������������� ,�� ���� �� ���� ������ ��
���� ��� !������� �! ��� ����������%
$�� ��� ������ ������� ������"���� �� ����������� ��
������� �������� ������ !�� ��� !������� ����%
$�� ��� ���� ������� ������"���� �� ������� !�� ���
!������� ���� �� ����� ���� �� ��� ������ ������ �
��� ���� ������ ��������� �� ��� �������� ������
!� ������"����� �� ��� �� ��� ��� �( �� ��� ������
� ���� ��� ������ �� ������� -� ��� �( �� �������
��� ������� �� ��������� � ������"�� �� ���
�������� �! ��� ������ ����� ������� ��� ���� ����
�� ��� ��� ��������
�� ������� �������� �������� � ��� ������� ��
��� �( ������ ���.���� ��� ����� �! ��������� ���
��� ������� �+ ������ ��� ������� �������� ���/�����
��� ��� ��������� �! ��� ������� '��� �� �� ��� ������ �!
��� ������������� �������� �! ��� ����� ���� ��� �(
���.����#
���0� ���.���
� ���.����
����������
�� ��� ������ �� �� ��������� �� ��� �����������
�! �������� �#
�����# 1����� ���������� !������� �������� ��� ����
������� '����� 022 3.�� �� �222 3.�� �� ��� !������� �����
���������# �� �������������� ,�� ���� �� ������ �
��������� ��������� �� ��� ������"���� ������ ���
������ 1���� �������� ���� �� ����� ��� ��������������
,���
��������# �� ���� ���� ������ �� '���� ����
�������� ���� �( ���� ��� ��� �+ �! ��� ���� �����
�������� �� ��� ������ ������ �� '���� ���� ��������
���� �( ����� ��� ��� �+ �! ��� ���� ���� ���������
4�������� ��� !� ��� ���� �� ������ �������� !� ���
������ � !�&������ �����
-������� �! ������� ���� � ���������������� �� ���
�������� �������� ����� �� ������� ���������� !���� �!
��� �� �������������� ,�� �� �������� ��� ����������
1������� ��������� � ������ ������� ��� �(
���� �� �� �� ����� �! ������� �� ����� � �������
�( ���� 5�� �( ���� � ���� �� ������� ��� ��������
��� ����� ����� ���� ���� � �������� �� ������
������ 1������� �� �� ���� �� �������� �������
���� ���� ���������� ����� !� ����� �! ������ �6���
*���� ��� !������� ���� ����������� �! ������� � ����
���������� ����� �� �� ��������� �! �������� ����� ����
�������� ���� � �������� ��!������ �� � "����������
������ (������ ��������� �� ��� ������������ ��
������� ��������
�� �� ���� �� ������ ������������� ��� �!
"��� ���
+� ������ ������6������ ����������� �������
�6�� ���� �� � ���������� �������� ����� ������� ���
!���� � ����������� ���� ��� ������ ������ �������
����� ����� �� ������ ��������� � ���������� �������
��� &����� ���� �� ��� �������������� ���� ����
����� �! ��������� ������� �� ��� ������� ���/����� �!
��� ������� �� ������&�� �� ����!�� �� ��������� �
����� �! ������������� �� ����������� +� �� �����
��&�� ��� �� �� ���� !� ��� ������� �! ���� ����� ��
����� �������� ��������� ��� �/������� ����� ��
���������� ���������� �! ������ �������������� 7�� �!
��� ���� ������ ���� ��!����� �� 89:1 �� ��� ������
��!���� ������ ������� ���!��� ������� ���� ���
!������ !� ������ ������� ��!����� ���� �
�������������������� ����� � ��� �����
�� ������� �������� �� � !������� -� ����� ��
�6���� �(� ����� �������������� ,�� �� ������� ��
����� ��� ������� ���� ���� �� ��� �������� �! ���
������� +! ������ ������� ���!�� �� �������� � ��� ���
!����� ��� ������������� ������� �! ��� ������ �������
�� �� ��� �������� ��������� ����� ��� ����� -� ������
��� ����� ������� ������� �������� �� ������ ���� ����
��� �� ��� ���6 ,�� �! ��� ����������� ��������� +� ��� ���
�! ����� �������� ����� ��� ����� �� ������� �������
��� ������� �� ��� &����� ����� �� �� �� �����
��������� ��������� ��� ����� ������� �������� ����
������ ���� �� ��� ������� ���/����� ������� ��� �������
�� ��� &����� ����� +� ��� ������������� ��� ��6�
����������� �� ��� ������� ������ � ���� �������
��� �! ��� �������������� ,�� �6� �� ��� �! ���
������� ��� ���� ������ ������� ����� ��� ��6� �� �����
��� ������� �������� ;� ���������� ����� �������� ���
������� �������� ������� �� ���� ��� ������� ���/�����
�! ��� ������ ������� ��� ������� �� ��� &����� ����� ��
�� ��� ������������� �������� �! ��� ������ �� ��� ������
�! ��������
<���� ��� �������� �� 89:1 �! ����� �� ��6��

���� ���������� �� �������� ����������
����� ��� �� �� ��������� ���������������� ��������� ��
�� ��� � �� ���� ���� ��� � ���������� �� ���� �� ��
������� ������ �� �� ��� � ����� ���� ���� �� �� ����
������ ���� ����� ����� ����� �� �� ����� �� �� � ��� ��
���� �� ��� � �� �� ����� �� ���� �� �� ����� �����
��� � � ���� ����� �� �� �� ���� ��
����!��"
�"��� �!���
���
�#
�$
�! ��������� ��� �� �� ��� ��
�" �������� ��� �� �� ������ ��� � ������ �� ��%
&����� �� �����%������� '�� ���(� ����� �� ���
��� �� ������ ��� ������� ���������
��� ����� ��������� ��� ���� � �� ��&����� �
����� ���(�� � �� �� # �� )))� ����� �������
���� ������ � ��� �������� �� �� �������
� ��������� ��*���� �� �� ��� ��
�# ��� � �� �� ������� �����
�$ ��� � �� �� ����� �����
+�(���� �� ���� ���� ���� ��� ������%���������
��� �� ��#� �� ���� ��
�#��
,�
���
�
����
������
��� �"���
���� �"
�������
� � ��� �� ��������� ����� ��� �� �� �� ��� ���
� ����������
��� �� ��� ������� ������� ��� ���� ��� ��� ���������
����������
#������� � � ��� �������� - ������ ��� �� �� �# ��� �
��� ���������� ����� ��� ���
�����������
.� ���� �������� �� � ������ �� �� �������
�� ���������� �� $/01 �� ����� ����� �� ����������
��� ���� ���������
��� ����� ����������
������� #�������� ��� �� ������� ������������ ��
����� ������� 2����� �� ������ �� ������ ��� �� �
3����� ��� ��� ����� ���� ���� ��� �� ������ �
������� �� ��������� ������� �� �� � 4� �� �� �����%
������ �� �� ���������� .��� 4�� ��� � �����5���� ����
���� ��� ������� � 4�� � �� �� ���� ���������� �������
6��� ��� ���������� �� �� ��� �������� �� �����
���� �����
����������� 7��������� �� ��������� ������ � �4��
�� ��������� ��*���� �� �� ��� � ���� �� � 4� ��
�� ������� �� �������� ������� ������������ �� ��
��������� �� �� � 4�� .�� �������� ������� � �� ��
��������� ��� �� �� ��� ��� .� � �� � ��� ������� ���%
�� ������� �� ���� �������� �� �� ��������� ��� ���
�� ��� ���
��������� 8� �� �� ��� ���� ��������� �������������
�� �������� �� �� ��������� ������ �� ������� �%
����� ������� � �� �������� ��� �� *����� �� �����%
������ )�������� ���� 4���� ����� �� ����� ����� ���
���� �� ������ 5�� ����(��� �� �������� ��������
������ ��������� ��� �� ������ �� ��������� *���%
��� .� ������� ����� �������� ��� ���������� ���� �
���� � 4� �� ������ 5�� �� ����- ���� �� �����
��� ���������
������������ ��� ��� ����������
���������� ���� ��� ������������� .� ��� �� � �%
�������� �� �� ��� ��� �� �� ���������� ���� �� ����%
����������� �4��� �� ���� ���� ���� �� ���5� �����%
���� ���������� 8���� �� ��� �� �� � � ���� ����� �
������� ������� ���� �� ������������ �� � �������� �� ��
����� ����� #��� ���� ���� �� $/01 ����� � ��3�%
� � ��� (� ��������� �� ��� �� ��9�"� �������� ��
������������ �� � �������� �� �� ����� ���� ������ ��
���� ���� �������
�!�� �" 8���� �� �2 �� ��� ����� �� ���������
��*���� �� ���%����� ��� ��� �� ��� ����� �� �����%
������� '�� �� ����� ����������� 8 ���� �� �� � 4�
�2 ����� �� �����%������� '�� �� ������
������� �� ���� ���� �� �� � ���� ��� �� �� $/01�
�� ����� �� � ����� �������� ����
#������ $��%���$ .� ������ $/01 ��������� ��
���������� ����� ��� ������� ���5�� ���������
��������� ����������� ���� ��� � � �� �� ����������
��� ����� .� ������ �� ��� ���5�� � ���� �����
��������� ��� �� �� ��������� �� �� ���������� #���
�� ������ �� ������� ���5�� �4��� �� �������� �������
������������� � ���� � �������� ������������ ��� � �
���� ������ � ������ ������� �� ������� ���5� ����
�� ������������ �� ������� �� ������ �4��� �� �����
�� �� ����� �� ������ ��� ������� �� �� ����� ��
���������� .� ����������� �� ������ �� �� ����� �� �
���� ������ �� ������� ������ �� ��� ������ ��� ����
�� ��������� ���� �� ����� � �������: �� ��� ���� ��
���������� ���������� ���� �� ����� � ��������
������ �� �� � ���� ��� �� ����� �����������
�����3� ���� ��� � ������ �����������������
&�����%�$ ��� �'���� $���������$ ��� �� ��������� ��
��������� ���� $/01� � ������ ������ �� ���� �� ��
������� ������ ���� ��������� �� � �� �� � �������� ��
� �� �� ������� ��������� ��������� $����� ����
��� � ����� ���� ������ ������������� �������� ����
����� �� �%�������%+%����� ����� ��� ������ ��� 1�����
���� ���� ��� ���� � ����� ���� ������ ��������������
� �� �� �����3������ � �� �� ���������� ��� �����
����� ������� ������� ������� � ����������
#�'�� ������%�$ #���� �������� ��� � ����� � � ��
����� ���������� �� ���� �������� �� �� � 4�� .�
������ �� ����� ���� �� �����3����� �� �� � 4� ���
���� � � �� � � �� ���������� �� ���������� ��� ��
���� �� ������ �� � ��������� �� ��������� ��� ���� ���
�� ����� ��
.� ������ �� � ������� ��� �� ����� ��� �%�����
����������� �� ��������� �� �� ������ �� � �� ������
�� ��������� �� �� ���������� �� $/01� .�� ������
��� � � ���� � �������� ������ �� ���%������ ����� ����
��� �� ��3 ������ �� ������� ������� ����� ������ ��
�� ����� �� ���� ��%��������� �����3� ���� ��

�������������� �� � ������� ����������
������
������������
� � � � ��� � ������ �� � ������������ ���� �
��� �� � ��� � ������ � �� ���� ��
������� ��� � ���� �� ���� ��� �� ���� ��� �
���� ��� ������� � � �� ��� �� � �������
������� ��� � �� �� ������� �� � ���
�������� !�� �� �� ���� ��� ����
"�� � ���� � � �� �� �� ���� ����� � �� � � ��
� ���� �� � � # ���� �� � ��� �� � �� �
���� � � �� ���
����������
$��� � � ��� �� ���� � ���� � ���� �� �
������� �� �������� ���� # ����� "��
��������� � ���� � ���� �� �� �� ��� ������
��� �� � � ��� � � # ���� �� � ���� � �� ���
������ � ������ � %�& �� � � �%�& � ���� � ��
� � � �� � ������ � � �� �� � ��� #������� ���
�� � ������ �� �� ��� � ��� %���� ��' ���
�������&� (� �� �� � ��� �� ���� ��� ����
%���� �� �� � �)������� �� ��������� ����& ��
���������
����� ����������
*� ���� � ���� � �� ���� �� � � ���� �� ��������
���� ����� ���� ��� ����� � � ��� � ���� (�
����� �� �� � � ��� ����� �� � ��� �� � ���� ��
����������� ���� (�� �� ����� � ��� %��& %����
��� ����� � ��������� ����� ��� ���&� �� ��
����� �� ����� � �� � %�&� ������ � ��� %�+& ��
�������� %�+&� *� ������� ������� � ����� � #����
����� ��� � �� �+ � � �� �������� �� ��� �� ��� �
)� ���� � ���! �� � � ��� � � � ���� � ����
� ���������� �� � ���� ���!�
�������� ������ � ���������� ������
(� �� �� ����� �� ����� � �� � %�& � � �
� ���� � ����� � #��������
��,�,-� �.��
��
�.� ���� ��� �� �� ��� �� ���� � � ���� ���� �
���� �� ��/���� � � ��������� � ������ ����
� � #���� �� � � � ������������ � � ������
���
��� !��� �� � � � � ��������
��������
(� �������� %.+& �!� � �� �� ����� � ���� ��
!� �������� � � � � ���� � ����� � #��������
�+�� ������.���.�&
��������
�.���.�
�.� �� �.�� ���� ��� ��� �� ��� ��� ���� � � ����
���� � ���� �� ��/���� � � ���������
� �� ������ ���� � � #�� �� !� �/ ��
� ���
��� �� ���� � � !���� � ��������
"�� ������� � � �������� � � � � ���� � ��
� ������ � ���� �� � � ��� %��& �!� !� � ���
������ � �� �� � �� �� ��� � ��� �� � ���� ��
� �� ��� � � %��& �� � ���� ��� � � ����� ���
� ���
�0����
��
������� �����
(� ������ � ��� %�+& �� � � � � � � ���� �
����� � #��������
�+��1�1,
��
�1�1,� !��� �� � � � ���!��� �� � � �
�����
�� ��� �� �!� � ��������� � ������ ���� �
� � � #���� �� � � ���� �� �� � � �
���!��� �� � � � �����
(�� ��� � �� ����� %� �� �� ��� ��� �� � � ��
�� � � 0���� ������ � ��& �� ��� ���� ��� ��
�� ����� %� �� �� ��� ��� �� ���� ��� ��& �
�������� � ��� ����� � � ���� 2��� ��� ��
�� ����� ������� � ��� � ��� ����� �� � � ���� �
�� ! ����� ��������� 3� ��� �� �� ��� � ���� �
� �� �� �� ����� �� � ���� ��� �� �� � �� ���� ���
�� �� �� � � ���� � � �� ���
3 � ��� � ���4� ��� �� � ���� �������� � �� ���
� �� �� ��� � ��� %�� � ����� ��� �� � ���� ��
)� ��4� ���& � � ��� � ����� ��� � ����� ��� ��
�� � ���� ����
������������ ����
(� ����� ���� �� )� ��4� ��� ���� ��������� �
���� �������� � ��� �� 5 �� �1 ��������� (� ���� ����
���� � �� %+06& �� � ���� � ����� � #��������
+06���
�
�� ���� �� � � � ������������ � � �������
�������� �� � ���������� � �� ��� !�� �
�������� ����� �������� � ���� ���� � � #����
�� � � � � � #� ��� � � ���� �� � ���� �
������ ��� ��� �� )� � � !�� ��� � !���
� ��������
�� � �� �� � � �������� �� � ���������� � ���
��� �� ��/���� �� �� ��� ������ ��� ��� ��
)� � � !�� ��� � !��� � � ������� �� � � �
�� � ���������� � �� ��� !�� � �������� ����
�� ������� ��� �� �������� ��� � )� ��� ����� �
�� � !�� ��� � � !���� � ������

���� ���������� �� �������� ����������
��� ��� �������
��� ��������� ��� ����
������ ���������
�������� ������ ����� �� � ����� � �������������
�� �������� ������� ������� � ���� �������� �����
��������� �� ������ �� �� � ���� � ������������� ��
������� ��� �� ������ � ������ � �������� ������
���� ������������ ��� ���!��� � �� ����� "���# ��
��������� ������ �� �� ��� � ���� � �$ �������� ��
���� ���� ���� �������� �� ��������%�� �$ �������# ����
����� �� ����������� &��' ���� ��� ����� � ����"
������� � �� ��� ��&��' ������ �� ���������� � �� ���#
��� ����� ��� �� ������� ������� ���� �� ��� �����
�� ��� � �$ �� �� �� ���� �� ���� �������� ���� ����#
���� ����� �� �������%�� ��� �������� ������ (�������
�� �� ���� �)�� )������ ������ � �$# ������� � ��
������ � ��������� ������
��������� ������
��� � ������ �� � �� ������� � �� �������� ����# �
��� �� �� ����� ��� ���� �� ��)�� �� � ��� ����� �� ��
����� "���� � ���# ��&�)��# ��)� ��� �� ������� ��
��*������ +�� �$ ������� ���� � ������ � ������ �� ��
�������� ���� �������# ��� ��������� �, ��� �����
����� �*� �� ����� -- ������..� ��������� �� �������
)����� �� ������� �� ������ ���� ����� �� �����)��
��������� � ������ +�� ������� )����� �� ������ �� ��
��� ��������# &��� ��� �� ���������� +�� ��� � ���
���� ��� �� �/��� ������ ���� �������� �� � ��������
� ������� ���)��� �� ������� � �� ��� &���� ����&���
����� ������� +�� ��������� �� ������� �� ����������
�� ������� �� ��*����� ����# &��� �� �������� �
������� � ����������� �����0
���1�����2���
�����2����
�0 3�*����� ��/��� � �� ������
���2��0 �$ �������
�0 ������� � �� ����� "���# �� )��� ��� �������
���2���0 4������� � �� ����� ������� &�� �� �$ ��
�� ������ ���� � �� ��
���� � ��� ���2��� �� � ��)�� ������ ���� ��
������# �� ��������� �� �� �����)�� �� ����� � �����&��
�$ ����� ��� �� ��������� �� ������� � �� ����� "����
5�������� ��&��� ������ ����� �� �� ��� ��� ��������
&�� ������ ��������� �� �� 6��� ����� �����)����� ��
��������� ��� �� ����)�� �� ����� ��������%�� �$
�������� &���� �� ��*����� ������# &��� ��� ���������
� ������ ���������# ��� ���������%�� &���� �� ���
������ 7������ ��������� ��� ��*����� �� �� ���� �� 8�8�
�$ ���� ��� �� �����)�� ����� � ��� �������� &�� ������
��������� &���� ��������%�� �$ �������� �� �����)�
������� ��*����� �� ������������ 8�88� �$ �����
������� ������
������ ������ ��� �� ���� � ������ ����������
���2�� ����������� $�)��� ��� �� �� ������ �� �� �����
����� ��� � �� ��� � ������� �� ������# � ��������# ��
������%�� &��� �� � ��� �� ���������# ����� �������� ��
��� "������ � ���������
+�� ��� ��6����� �� �������� � ������ �� ���������
������������� ���� �� ��������� �� ������ � ������
������ ����� ����������� � ��*���� �������� �� �� ���
��� �� ��� �� �������� �� ����� "���0 �� ����� ��� ��
������ &��� ��� ���������� ��)� �� ������� ���� ������
�� ���� ������� �� �������� � �� ������ �� ������
�� �� �� ��� ������� � �� �� ������ ����������
+�� ��� ��� �� �� ���� �� �� ������ �� &��� ��
�������� ����� �� �� ��� �� ������� � � �������
������� ���������� ��� ��� ��������� �������# �� ���
��� �� �� ���� �� � ���� �� &��� �� ������ � � ���� ��
��� �� ������� ���!��)��� &�� �� ������ � �����
��������� �� � ������� ����������# �� � �� �� ���� �� �
6������)� �� &��� �� ������ �� �������� ����� �
���������� �� ������� ������������ � �������� ��
�����)� ��������� � ������ �� �� ����� ���!� �
)���������
����� �
9� �������� �� ��� ������ � 0
:� ���������� �������� �� ����� �������# �����
��� ��&��� 7������� � �;88 4 ��)� ���� ���� ��� ���
��������� ������ ����� � ��)�� �� � ��������
��������� ������ � �� � 18 � ����� ��&�� ��
����������#
:� ����� ����� ��� ������ �� ������ � ����� ����#
� ������� �������# � ������ �� ���#
:� ����� �)�� &�� ������� �������� �� ��� ���
���� � �� ��� �� ����� � ����� &�'� � �������
&���# ����� ��� ��'����# ���������� &�� ��������
� ����� ��� ����� ����������
��������� ��� ���� �� ������������� ����� �������
����� �
+�� ����&��� ����� �� � ������� ��������� � �� ���
�������� �� ��' ������������� ���� ����# &��� �� ����
������ ����&��� ���� �� �� ����������
�������� �� �� ����
�� �� +�� ����� ���� ������� �� ������� � � �����
���� �9� �� &��� � �������� "�� �<� �� ����� � ������
�������� � �� ���# ��� �� ���� ������ �=�# � ����� �����
���� �3� ��� ����� � ���� �� ������ �������
������� >����

�������������� �� � ������� ����������
���� ���������� � ��� ������ ���� �� ���������
��� ��� �� ������������������������� �� ��� �� ��
������ �� ��� � ��� �� ���� �� ����� ��� ��� ��!� �� ��
��"������ �"���#�� �� ��� ���� ��"� ��� �� �� �� ��
� ��� ���� ������ $�! ����� � ��� �� �� ��� �� �����
��������� �� ��� ���� %��� ��� "������� #� �� ���
���� ��� "���� �""� ��� �"����� "��� ��� ������ ���
"��� ��� #� ��� ���"�� %��� &��� "����������� �
"��"���� ������ �� �� � �� ����� �������� ��� ��� ����
� ��� �� � �� ���� �� ������ � "��� ���� '� �� ��( ���
���� � ��� �� �����������������������������������
)��������� # ��� �"��� ������� ��� ��� "���� �� ���
�� � ���� ��� �� �����
�����
�������� ��� �� � ���� ��*�� �� �� ��� "�������
#�� �������� ��� � ���� ��� ����� � ""���� ������ ���� �
��� �������� �� � � ����� �+ ��� ��*�� ���� �� ����
������ ��� � ����� %��"��� ��� ���� �� ����� ��� ���������
�� ����� �� �"���#�� �� ��� ���� ��"�� %��� ����"� ��
"�"�� ��� ���"� �""�������� ��� � �� ���� �� �� ��,��
�� ��� � ��� ��"�� ���� ���� ���� ��� "��������� ��� ��
�� ��� ���� ��� ��������� �� ������ -�� �""� ��� "���������
+ ������ �� � �� ���� �� "������� ���� *���� ����������
"����� �� ". ���*��� �� �������� ��� �� )� ���� "�������
��� � ��� "������� ���� ����� � �� ���� �� ��� ���"� ��
�""��� ������� �� ��� ��"�� ����� "�"�� ����"�� / � �
����"� �� "�"�� �� ��� �� �� �� ��� � ��� ��"�� ���� ����
���� ��� ��������� �� �����0 ���� ��� ��� ����� ���
�*���� ��� ��� �������� ��� ���"�������� �� ��� ����� ���
������� �� ����� ��� ��� �� ��� ���� ��"�� -""� �����
"�"�� ���*� �� ���� ���� �� ��� � ����� ��������� ����
��� �� �� 1�� ��� ���� �� ���� ��� ��������� ��� �� �������
���� ��� ���*� '���"����� ��� ������ ��� �������� "���(�
%������ ���� ��� ���������� ��� ��� �� �""��� ��� �������
�� "��������� ��������� �� ��� ���� ��"�� 2����� �3 ���
� ����� ���� ��� �� ������ �� ��� ��!� �� �� ��������
"������� ��� ������,��� 4��� �����"�� ����� ��� ���"�
�""������� ����"� ��� ��� � �������� ���*�� )������ ��� �
�� #!�� �� ���� ��� ���������� ��� ��� �� "�����������
�� )�� ���� ���� ���� ���*�� �� ���� ���"���� �� ��� 5�
��� ���� ����� �3 ��� �� ���� ��� ��� ��� �� �� ��������
�� �� ����� )�� ���� ���� ���*�� ��� � �� �� ����� ���
�� ��� ��������� ������� �2� )�� ���� ��� 5� ��� ����
������� ��� � �� "����� ��3 ���� ���� ��������� �� ����
��� ��� ����� ��� �� �� ��,�� � ����� � ���� ���*�
�� ��� ������ ��� "������� ��� ��������� �� ��� ����� �� ���
������"���� ��� �� "��������� �� ��� ���� ��"��
��������� �� ��� ������� ��� �� ������� �� ���� ������
6���� ��������� �� ��� ����� ������ �� ����������
��� ��� �� ����� ��������� �� �������� � �� "����� ��
��� �� ���� � �7��� �� ��������� ����� ��� ��0
8��� �� �� ���������� ������ "������� �� ��� ��
��������� ������� ��� ��������� *����
8��� �� �� �������,�� ". ���������
8��� �� �� ��� ���
8��� �� �� � ��������� �� ��3����� ����������� ��� ���
�������� '��� ��( ���
8��������� �� ��� ���"� �""������� "����� ��� ��� ���
��3����� ���"� � ��� �� ��� �� �� ���"� �""�������
���*� �� ���*� ����� ���� "�"���
8��� �� �� �������� � ���� ����������� ��� ��� �����
����� �� ��� ������� #�� ��"����� �� � ���������� ���
�+ �"����� ��� ��� �� �� #!�� �� ������ ����� ������
���� � �7����� �����"�������� �� ���� ���������
8��� ��� ���� �� � "������ ��� ���"�
8��� �� �� � ������� ����� ����������
8��� �� �� � ����� ���
���� ����� �� ��� !���� "������ ��� ���
6���� ��������� ������� �� ��� ������� "����� �� ���
��"���� ���� � �� �� �������� ��� ������ �������� ���
�� ��� �� ������ ��� ��"�������0
8��������� �� � ����� ". ������� �� ������� ������������
����� �������� ��� �!��"� ��� ������ ". ���*��� ��
*���� ���������� "������
8���"������ ���� ��� ������"���� ��� "������ ���� ���
������� ��������� � ������� ��� ��� "��"������� �� ��
�!�������
8��� ����� �������� �������� �� "��������� �� ��� �����
��"��
����#� ��������� �� ��� $����� �����
9��������� �� ��� ����� ������ ��+ ���� ��� ��� �������
�� �"���#� � �������� ��� �� �"���#�� �� ����� �� �����
��"��� ����� ��� ��0
8��� �������� �� ��� �� ��� � '5 ��:� ������������� ��
����� ������������ �� *��" "������ �� �� ���� � � " �� �
��:� ��� �� ���(0 ���� "������� "����"����� �� �����
���������� "����� )� ���� ����� ��� �� ��� ��� �� ��� �
���� ����� �� *��" ��� "������ �� �� ����� )� ��� ��
���� ��� ����� �� ����� ��� � �� ��� �� "�����
������������ �� ��� "�������
8��� �� �� ��������� ������� ��������
8��� �� �� � �������� � �� �� ��������� ����� ���� '�� �
���� ������( �� ,����������� ����� ���� '�� �� /.-%2
�� /.-%2;(� ��� ��� �������� �� ��"����� �� ��� ����
"�� �� "����� "������� ���� � �� ���� �� "����"������ �
������ �� %���� �
2��"�� ��� �� �""��� �� ��� ���� �� ��� �� � � �� "���
���� ��� "������� ���� �!����� ". ����������� ���"��
��� � ��� �� �""��� ���� �� ������ ��������� � ���
������ ����"���� ��� ������ ��� ��� �""��� ��� "������
�� 5 "�������� �� ��� � '���� ����� ��� ���� ����(< ���
"������ �� � "������ �""��� �� �""����� ���� �� ��� � ���
��� �� ���������
- "��������� *���� �� �������� ������ ����� ��� ".
������� ������ ��� ����� ��� ��� � �� � � �� ��� ��� ���
�� � -��� � ��� �� ���������� �����������������
��� �����"���� �� ������ ���!��� ��� �� ������� ���� ��� ��
�������� ������ /������� ����� �� ������� �� ��� ��� "������
�� ����� �3 ��� ������� ��� �� ��� �� )������,�� ".
�������� ��� �� ��� �� ������� ���� "������
=>����� ����� '�""��!������ ?�/( �� ��� ��� ���� ���
� ��� �� � ��� ��� ��� �� ��"������� .� � #�� ����� ���
��� � ��� ���������� ��� ��� ��� ��� �� ��������� ���

���� ���������� �� �������� ����������
���� � ��� �� �� � ������� �� �
�������� ��� ��� ���
������ �������� ��� ����������� �������� �� ��������������
��� ����� �� �� � �� ���� ���� ��� �� ���� ������� ���
��� � �� ��� �������� ���� �� ���� � ���� ���� ���
����� � ��� � �� � ����� �� � � �� �� ���������
!�� ��� ! �� "� �� �� ��� �� �� � ��#��� �� ����$
����� ��� ����� ���� %���& %�'('�&$ ��� ) ���
����� � �� ���� � * ��#��� �� ����$ ����� ���
����� ���� %���& %�'('�&�
��� ��� �������
��� ���� �� ���� ��
������ ��� ���
+�,��� ��,,��� �� �� ������ �� ��� ,������$ ��,���� �
��� �!����� !� ���-* ���� ���� . ���� ��� � �����
�� �� ��/����� ������ �� � ,����� ���� ��� �� � ����
����� �� ,�,��� �������� �� ���� !� ��,������ ���
�����)����� �� � �������� �� � ��,������! � ������� .
�� � ,������ �� � �� ��,�! � �� ���������� ���� � �����
���� ������ ���� ��� ���� ����� ��� �� ������ �� �
������� �� ���, ������� �-* %��-*& ��������� �� ,���
�������� +�,��� ��,,��� �� � ���,������ ,��������
!������ � ���������� �!�����$ ���,���� � � ���������
������� �� ��������� ������ ���� �� � �����$ ���)��� �
,������ ������� �� � ,�����$ �� ��,�! � �� �������
���� � ������� �� ������� ��� ��������$ ��� ������
����� ,������ ���������� ��� ������ ��!� ��� 0�� ,���
��� ,������ ������ ������������ ��� ��� !� ��
��������� �� � � ������)� ��� ��� ���� �,,������
,���� �� ����� ���� �,��� �� � ,�,��� ��, � ,���
����� ��1���� �,���)����
2�� ��,�� ,������� ���� �� ��������� �� � �,, ����
��� �� ,�,��� ��,,��� ��� �� �� ������ � ��������/�
� ������� ,����� ,�����$ � ��� ��� � ��!� �� �� �
�#,������� ������� �� �� � ���� ��� �����!���� �-*
,������ ��� � ��� ��� � ���������� �� � ����� ,����
���$ � ������ ,����� ��!� �� �� �� �� � ������ � ������
� �� � ,����� ,�����$ �� � ���� � ,������� �� ,�����
�������
��� ���� �� ���
+�,��� ��,,��� �� �� � ������ ����$ !� ���� ���
���� �,��� �,���)� ��,� ��� ��� ������ ,������ * ���
� ���� ��� �� ��� ���� ��,�� �$ ��� ��� ������ �����
� ��� ������ � ����,��� 3�������� �� ��� ����� ��
!� ��������$ ��� �,,��,����$ �� �,���)� �������,��
* ,�,��� ��, ��� !� ������ �� � )����,��� �� � ,�����
��� �� � ��� ,����� �� ������ ������ ,�������� �
,������ � ���,�������� ������������ �� � ,����� !����
��� �/��� 4��� ��5�� ��,� ��� ��������� ��� � ���� �,�
��� �� � ,��������' ��� ���� ��� ,���)����� �� �
,�����$ �� � ,����� �� ,�� �� � ����� ����6 �� �����
� ������ �� � ,�,��� !����6 ���������,�� ��,������
�� � ,�,����6 ��� ��� ���� ��� �����)����� �� �
,�,����� * �� ���, � �� ������� ��� ������� �� ,��� �
�� � ��������� ������� �� � ��������� ������ � 7��, ��
� ������ �� ,�,��� !���� �� ���� ��� � � ����� ��� ���
/���� ��� �� ����,������� %����$ ��,���& ��� ����$ ������
�� ������ � ������ �������� * ��, ��� � ������
����� ,�,���� � !� ��������� � 8� � ��� ���$ ��
��� ��� �� ���� ��������$ � ��, ��� ��� ��
�,���)��� !������ ���� ,������ �� �� ��� � ����
,��) ���
����� � ��� ��� ���� �
.�� ���� ��� ,���)����� ��� ��������� ��� ��� ���� ��
!� � ����� �� ������ ����� ��������� ���������� �#��,����
��� ������� ,������ ���$ ��� ��������$ �� !� �,���)�� ��
� �������,� 9�������� �������� �� ,����� ���� �
������ ���� ��� � !� �� ������
����� �� �������� � ���� �� ���
2� �� ����� �� � �,,���� ���� ��� � � ������ ��
,�,��� !���� �� ��,��� �� � ,����� ����� ��� 2��
�� ����� ,������ ���� ��� ����������� �� � �,� ��
� ������ � !� ��, ���� :��/����� �� ������ : ��� �
�,� �� � ������ ���� ���� � ����� �������� ;�����
� ������ ����� ��� ��� �,���)��� ��� ���� �� 2�! � ��
2�� �� �� �� � ���� ����� ��� �� !� �#,����� �� ���
� ������ ����� ��� �����)���
��!�� "� 0#��, �� �� 7 ������ *����
�#�� $���� ��� �� �#
0�/����� 2��,��� %07 ��(� ��(& 7������� ���� ��
*�� ��� ���
7�����,���
%07 ��(� ���&
7������� ���� ��
����,�!��
�������� %����$
���$ <�$ * �$
��������&
+�,��� %07 ��(� ��� = & -���,���)� �����
���� ����,�,�����
%����7 0���,�,�����&
%07 ��(� ����&
7������� ���� ��
���
> ���� ����,�,�����
%���� �� ������ ����� 3�&
%07 ��(� ���?&
7������� ���� ��
> � ��� *�,
+�,��� �*�, ��� �
����,�,�����
%0���,�������� *�,�-&
%07 �� (���&
-������� ���� ��
*�,
7 ����,��� %07 ��(� ��& 7������� ���� ��
*��
7����� 7������� !������ 7������� ���� ��
<�
�-��������������!��/���
���� -������� ���� ��
7��
��.�����!��/��� ���� 7������� ���� ��
2�, ��� 2��
�� �� ���� *�, ��� +��
@-+;����� � 2�,

�������������� �� � ������� ����������
������������ � ����� ������� �� � � ��� �� � �
���������� �� � � ������� ������� ������� �� � � �
������������ �� ������� ��� �� ����� ��� ����������
�������� � � ���� ��� � � � ��� ��� ���� �� �� �������
�� ������ ������ !� ������ � ���� �� � ������� ���� ���
������� �� � ��������� ���� ������ � �� �""�""" ���
����� ������� ���� �� ��������� �� ������������� ��
����������# �� ������ ���� ������� ��� �� ���������
������������ � � � �������� ����� $����������� ��
������� ����� %��������� �������� �����% � � ��
��������� ��� ��������� �������� �� ������ ������������
�� � � ���� ��� �&������ ������ ���� �� � ������� ����
��� ��� �� �� ������� �� ����� ' � ���������� � ��
����� �� (����� �� �������� � ���������� )� �� ��� ����
��� �� ��������� �� ������ �� *$'+ �� ����������
�� ������ �� � �� �������� ��� ���� ������������ ����
� �� ���� � ����� ������ �� ������ � ���������
������������ � � � ������� ,���� ������ ��������� �
� � �� ��������� �� ����������� � � ������ �� �� ��������
� � ������ ���� ����� ���������� ��� ��������� ���� �
���������� �� � � �������� � � ��� �������� �� � �
����� ���������� $ ����� ���������� ���� ��� ��������
���� ��� ������ �������������� ������ � ��������� ��
��� ���� ������� )� �� �������������� ��� �� � �
������ �� � ������� ����� -���� ����. ��� �� ���� ��
������ � � ������ ���� �� ������ /� ����� � � ������ ��
��� ���� ������ �� ������� ���� ��� ����� ���� �������
�� � � ������� � � ����� ��������� �� ������ ��� ��(����� � �
������� ����� ���� �� �������
������ �� ������ ��� �������� ������� � � �
������ ��� ��� �� ��� �������� ������� ���� � �
������ �������� ��� �� ��������� �������� ������
���� ������� ����������� �&����� �� ��� ����
�������� �� �������� �� ���������� ����� ������� ����
� � ��������� �� ������� �� � � 0 ������� ��� �� �
������� ���� ������� ���(� ��� �� �������� �� ����
�������� �� ������� / �� �������� ������ �� � � ���� ��
������ �� ������� /� ���� ���� ��������� �������� ��
��������� ��� �� �������� �� � �* � �� � ��� ������ -����
�� �* 1 ��� ������.� � � ����� ���� � �� � � ������
����� ��� ������ ����� ���� ������ �� � � ���� ������
�������� ���� � ������� ��������� ���������� �������
� �� ����� � � ������������ ��� ����������� �� ������ ��
������� ��� � ��� � �� ����� ����� ��� � ����� �� �������
� ���������
�*2 / � �* �� � � ������ �&���� � ���������
��������� �� ������ � � ��������� �� � � ����������� ��
� � ��� ������� ����� ��� �&������ � �� ��� �������
������ �� � ������� ����� � �� ���� ����������
-���� �* 3� ����� ���. � ���������# ������� � �� ���
������ �� � ������� ����� � �� ��� ��(���� ����������
-�* �. � ������� 4� � ������ ����� � � �* �� � � �������
���� � ���� ��� ����� � � � ����� ������ �� � � ������
���� � � ������ ��� � ���� ��� � ��� ���� � �
������ �� � � ����������� ��������
�����������2 4 ����������� ������� 31�+ ��� �5�+ �
���6���� ��� ���� �������� / � ����������� ���� �
������� �� ����� � ����� ��� ��������� / � ���� ��
������ ����� ���� ��� ������ � � ����������� �� � � �������
����� ������� ���� ������� ��� ���� ���������� ��
����������� �� � � ����������� �� � � ������� ���������
��� �&������ ������ �� ���������� ����� ��������� �
��������� �� 7�+� ������� �� �� ������������ � ���
��������� ���� �������
����2 !� ��8���� ������ � ��������� � ��� ������
����� � ��������� � ����� �� �������� � � ������ ���
�� ����� � ����������� ��� ��� ���� ���������
������� /�� �� ������ ����� ���� 3 �� �" ����� / �
������� � ������� �� � � ������ �� �� ��� � � ���� ���
�������� � � � ������ ��� �� �� �������
������ �� �������� �����2 4�� �� �&������
������� �� ������� ���� ��� ���� �� �������� � �������
��� ���� ������ ��� -���� 9 �� 3" ����.� � � ������ ��
������� ���� � ������ �� ���� �� ���������� �� � �
� ��������� � ��� �������� 4 ������ �� �������� ����
������� 3"2� ��� 3""2� � �������� ����� !� � �����������
� �� � � ������� ���� ��� �� ����� � ��� �� ���� ����� ��
������ �������� 0���� ������ � � ���� ������� ������
������ ����� ���� �� �������� � � ��&� ���� � ������
����� %� � ��������� ����� /� ���� ��� ������
�������� � �� � � �� �������� �� � � �����6����
�������� � ����( ����������� � ���������� ��� � ���
��� ������� �� ��� � � �������� �&���� � � ���� �������
� ���������� �� ���������
:��� ��� �6��� ��� ���� �� �������� ������� ���
������ / � �������� �� � ��� �6�� ������� �� � � ���
��� ��� ������� /�� �6��� � �� ��� ���� ������������
���� ��� ��������� �� ������� ��� � ��� � /���� 3� !� � �
������� � ���� ����� ���� ������� � ��� * $����������
'6�� + ��������� � -;$ *$'+. ��� �� � ��������
�� ��� �� � � ���������� �� � ��������� � ����������
����� �� /�� �6��� ,��� ��� � � <�������� �� $������
;������ $ ��� * $���������� '6�� + ��������� �
-;$ *$'+.
!�� =&� ��� + ��������� � -!=+.
*����� ��� !��������� + ��������� � -*!+.
$������������ >�� =������� ����� -$4>=.� ������������
�
<���� ������� <������ $������������ >�� =������� �����
-<�< $4>=.
+������� =������� ����� -+=.
$���� + ��������� � * ?����� -$+*?.
* ?����� $���� =������� ����� -*?$=.
/ � ����� �� �������� ��� ����� � ���� � � ������
������ � *$'+ ���������� *$'+ ���� �������� ��� �����
� �� ��� ����������� ��������� ��� ����������� ��� ;$
*$'+� �������� ���� ������� � ������� � ������
������� � ��� � � �������� �� � � �� � � ������� ��� ��
���� � � �&���� � �� � � ���� �������� ?����� ������
��� ������ �� �������� ��� ����� ���� �� ������ �����
� ����������� @ �� ���� �������� � � � �������� ���
����� ��� � � *$'+ ������� � �� ��� ����� � � �����
�� ���� ������ �� ��� � � ��� �� � � � ��������� �

���� ���������� �� �������� ����������
����� �� � ���� �� ��� ���� �� �� �� �� ���� ����������
������������� ��� �� ��� ����� �� �� � ����� ��������� ���� �� ���������� �� ����� ��� ���� ��� �������� �� � ��� � �� ��� � ���� �� � �� � ������ ������ �� ��!� �� ��� ������ ��� "�� ������ ��� ����# �� �������� ��������� $���� � � ���� ������ ������ �� �����# � ��� � � ���� �� %&'( �� �� ����������� ���������$���� � ������ ������ �� ������ �������� �����# � � ��� � ���� �� %&�( ���� ���)��� ��� ��� �*��� �� �� $� �� ����� ��������� $���� � � ���� ������ ��������� �����# + � �� � � ���� �� %&�,( ���)���
������� ��� �� ����� ���� ���!� �� �� �� �� ���� �� ���� �� �� ������ ������ � ����� ���� �� ���� ��-������� �� ���� �� ������ .� ��������# ��� ���������������� �� ������� ������� � ����$��� � �� ����� ������ �# ���!���� �� �� ���� �� ���� � ����� ������� �� !������ � �� �� ����� ������� ����� ��/���� �$��� ������ �� ���� �����
��� � ��� ���� � ���!��� ��� -�0�$��� � � �� ����� �� ���1 ���� ���# ���� ���� � �� �1 � �� ��� � +�� ��������� �� ������ �� �� ��� ���� �� ���� ������ ��������%����# ��� ��� �� ����� �� �����(� 2���� �� �� ������������ ��# ���� ��� � �# ���������� ����# �� � �1$� ��� � �� ' %�� ������ ��� �������$���� ������ �( ��!����� $�� ���� �� � ��� �� ���� ������ �� 3�� �� ���� ��� ���� ��-������� �� ���� �� ���� 4�� � �� ��������� �������� 5����� � �� $� �����# ������#
�� ������ � �� ��� ���� 3 ������� ������ �� ��������� � ����� � ������ � �����0 �0 ����� 5����� � ����� �� �� � ���!��� ����� ������ �� �� �� �� �� ���)� �� ���� $���� 66��)��77 ���)� ��� �� �� ��������� �������� .����� �� 18&� ��� �� ���� � ���
��� �$��� ����� ��� ���� � �� $���� �� ���� ��!������� ��� �� ����!�� �� �� �� ��������� 9� ��� ������� �� �� � �$��� ����� � �$ �� ����� ������ �� �� �������) �� ��� ��� ��*��� � �� �$����� :�$��� ������ �������� ����� ������ � ����� �� ��� �� � �1 �������� �� �/�� �� � �� ��� �� ���)� � ��� ��� ��������� � $� ���� � ������ �� 18&� ��� �������� ���������� ������ ��� �� ��� �� �� ���� ��
������� �������� � �����!� ���� ���������$��� �� ��� -���� �� ��� �� �$��� ������ ���� ��� ��� � ��� & ����� �# �� �� �� �� �� �� ��� ���� �� �������� �� � �;< �� �� �� � ��� � ��� � 9 ��� � ���� �� �� �� ����!� $�� ���� %����# ��� ���� ����!� � � ��(# $� ������ � �� ��$�� �� !���� ��� �� ;< �� �� ����������� ���� ��� �� ���!���� � �0����� ��
��� ��� ������� �� �!����� ��������� �� �� �� � ���� ��� ����� ��� ��� ���� ��� ��� � ��� �$��� � ������ � �� ��� ���� �� �� ��� ���� �� ����� ��� �� �/�� �� � � ����� � �� �� ����� ���� ����� ��� ��������� ��� ��� ���� ��� ���� �� �� �� ��� ��� ����� ���� ��� ��� �������� 3 ����� �� �� �� ������� ����� ������� ��� �����!�� �� $� �� �������� �� � �=������� 2 �����# ����� �� ��� �� �0�� �� �� �� �� �������� �� � ��� ��� �� � �������� � ����� �� ��������� ����� � �������� �� � �� ��� �� ���� �� �� ��� ���� �
�� ��!����� �� �� �$����� �� ��� � ��� �$��� ���� �� . ���� �� � ������ ����� ���� ������ $�������� �� � �� =������� 2 ����� �� =������� :� �������� ���� ���� �������� ��������� 9 ��� ����� ��� �������� !����� ����� �� �� ��� �� �� ��� ��� ����$��� �# ���$���� �� � �� ��� ��# �� ������� �� �������� �� ������ ��������� ��� ���� ��� ��� ���� ��� � ��� �$��� ������ ��� ��� ��� ��� ������� ���� ����� � �� ��� ��������� �� ��� ��� ������ �� �� �� �� �� �� � �� �� �������� ��4����� ��
>�� ��� ��� ����� �� ���� �� � ��� ���� �� �� # ����� � ��� �$��� � ��4����� � ��� �� ��� ���� ��� ������!��� ����� �!� � �� ��������� . ��� ����# �� ���� �� ������ ���� �� �� !���� ��� �� �� ���# �� ��� ���� �� �� �� ������ � ��� ��� �� �� ��� ��� ����� � � �� ��� ����� ���!���� $� � � !������ �� �� �� )�� ������� ��� ��� �� �� ��� ���� �� �� ��� �� !���� � $� ��������� �� � �� ��� ��� �� �� �� �������� � ������?�������� � ����� ��� �� �������� ��� ��� ��� ��� ��� ����� �� �������� � ����� �� �������� � ����� ��� ���!� ��� �� � �� �� ���� �� �� ��� ��� ������ �� �� �� � ��� �� ������� "�� � ������� �� � !���� ��� ��# ������� ��� �� �0 ��� �� � !���� �� � �������� � ������� �������� � ����� �� $� ����# ���������� �� �� !���� ��� ��� �� ���� �� � � ����������!�� ����� �� �� ��� ������0 �� �� �� ����� ��� �� $� ����� �� �$�� ���@�� ��� ���� �� �� ��� 8�� ��� �� �� ����� ��� �� $�$�� ����� $� �� ������ �� �� ��� ��� ���)�� ����� ��������� ����� ��� A���� �� ���)� �����) ������ ��#�0�� ���) ��� �# ���) ����# ���) ����� ��� ���# ������ �*�����A �� $� ���� � ���� ��� ��� ������� ��� B������ � �� ��� �� ���� �� �� �� � ��� �������� �� ����# ����� ��� ��� �� �� ���� ��� ��� ������� �� ��4����� � �� $� ���������
��� ������� � ������� �� �� ����� �� �� ��������� ����� �� �������� � ����� ��� �� ��� �� ���� �� ������ ������� �� �������� �� 4�� � � �!� ����!����=���!��� �� �� ��� ���� ��� ���� �� �������� ����� ����$� �� ��� �� � ���� �� �0 ���� ��� ��� � ������� ����������� �� �0������� �� �� ���� �!� � ����� ��!�� ��%=2B(� B�/������ � �� ����!��� �� �������� �� ����� ���� ��� ���� ��� �0��� ��C ��������# �� ��� ���� �$��� � ��� � ���� ��!� � $� �� �$������ ��� $� � ������!��� �� �� �������� �� �� ��� ���� ��� ����� �������� � ��� ��4�� ��� � ��!� ��� �� �� ���� $� �������� � �� ���!����� ��������
<����� �������� �� �� ���� �!� �� � �� ���# �� ���)�������� % �� ���) ���� �� �� ���) ����� (# �� �$�� �����)�# �� �� �!����� ��� �� �� �� �� ����� �� �� ������. �� �� ������ �� �� ������ �� $� � ��� ����������� �� �� ���) ������� �� ��� �� $� �� ����� ��������� ������ �� � �D� %!?!( �0 ��� �� ����� ���������� � ����� �� �������� � ����� ����� � .� ��� ���)�� �� ����� ����� �� � �� �������� � ����� �� ��������� � ����� ����� ��!� �� ��� ���� �!� �� � �� ����� ���)� ������� �� ���# �� �� ��� � � �� �� ��������� �� �� �������
.� ���)� �� �� ����� ��� �� �� � ������� �� ��/���

�������������� �� � ������� ����������
������ �������� � �� ��� ���� ������� �� ����� ����� �
��� ��� ������ ��� ���� �������� ����� �� �� �������
�� ����� �������� ������� � �������� ����� ���
������� � ��� ��� ������ ��� ����� �� ������ �� ���
���� ������� �� ��� ������� � ���� ����� !� � ���� � ���
��� ����� � ���"������ ������� ���� ��� ������������
���� � ��� �� ��� ��� ��������� �������� �� ���������
������ ������ � �� ������ ��� �������� �� �������
�������� #�� ��� �� �� �����$���� ������� ���������
�������� ��� ��� ������� �� ������ ����� ���� ��� ����
�������� ��� ������� ��� ����� ������� �� ��� ������� ��
��� �� ����� �������� �������� �� ��� � � �� ������ ����
� ��� ���� ������� %���� ���� ���� ���������� ��� ����
���� ���� � ���� ���� ����� ��� ����� ������ ���
������� ���������� &��� ���������� ���� ��� ��� ���� ���
���� ���� ����"����� �� �� ������ �� !' ������������
��� ��� ��������� �� ����$����� () �������� ������� ���
����"����� �� �������� #���� ������ ��� � ������
��� �� ���� ���� ����������� ��$������ �� ���� ����� ��
�������� ���� �������� ��������� ������� ���������
�������� �� ��������� ������ ��� �� ��� ���� �����
#�� �� ����� �� ������ �� ��� �������� � �� ��� ����
����� �� ���� ������ ��� �� ���� ��� � �������� ����� ��
������� ����� ���� ��� ���� ��������� ����"�� � ���
������ ���� *��� ����� ��� �� ���������� ���� ��� ����
������ �������� � ��� ������ �� �� ������� ����������
������ � �� ���� ���� ���� ��������� � ������ ��
������� ������ ��� ����� �� �������� ����� � ���
�������� +����������� ��� ���������� �� ���� ������ ����
����� ������ �� ��� �� �� ��� ���� ������ ��� �� ��������$
�� ��� ��� �� ��� ����� ����� #�� ���������� � ����
�� ����� �� ���� �� ��� ������������ ���� �� ��� ���������
��������,��������� ������� #�� �������� �� ����$������$
��� �� ������ � ������� �� �������� � ����� ������
��� ���� �� ��� ������ �� ������� ���� � ����� ����$
��� � ������� ��� �������
#�� � � �� ��� ��� ��� ��� ���"����� �� ������
����� � �� ������� ���� ��������� ���� ������� �����
�������� �� � ���� �������� !� �������� ����� � ��� ��������$
�� �������� ���"����� �� ������ ����� ��� � ������ �
��-����� +� ��� ���������� ������� ������� ��� ��� ������
����������� �������� ���"������ �� ��� ���� ��� ������ �
������ ������� �� ��� ��������� ��������� �� �����
��������� ���� ��� ����� ��� ������ �� ������� � ���
������ ��� �� � ������ �� ��� ��� ����"�� �������
�������� ��� ����� ����� �� ������
#�� ������ ��� ������� �� ��� ��� �� ������ �����
����� ������ ��� � ������� �� ���������� �����������
#�� ��� �� � ������ �� �� � ������� ���� ���� ���
�� ��� ��� �� ����� ���������.���� �� ��� ������
������ ������ ������� ������� �� ������ ����� �
������� �� ���������� ���������� �� ���� ������� ���$
������.���� �� ���� �� ��� ������ ����� � ��� ������
��� /������ �� ���������.� ����� ����� ��� 0$��� ���
�� ������ �� ���� ���� �������� �� � �� ��� ������� ��
��� ��� �� ��� ������������ 1/&2�
3�� ���������.���� ��������� ���� 0$��� ��� �� ����$
�� ��� � �� ���� ������� ��� ����� ��� ��������� ������$
��� � ������ ��� &��� �����$�� ��� ����� ��� ��������� ��
������ ������ � � ���������� ���� � ����� ����� �� ������
���� ����� � �� ���� �� ��������� ��� �� �����$��� 4������
������������ �� ����"� ������ ����� ��� ����������
���� $������������� ��� ���� ���������� ����� �
���������� + �� ��� ������� �� ���� ���� �� �� � ���
�� ��� ������ �.�� !� ��� 0$��� ��� � �������� � �� ����
�� �� ������� ������ �� ������� 5$��� ��� �� ������ ��
������� � �� ������ ��� ��������������� ���
/+67!#%3$/& ��� ���� �� ���� ��� ���������.����
���������
#�� ��� �� /& ��� ���������.���� �� ������ ���� ���� �
�� ����� ������ �� ������� ������� �� �� ��� ��� �� ��$
��� 65$/& ��� ��������� �������� !� �������� � �������
������������ ��� ����$������� ����� ��������� ��.����
������� �� � �$��$8��� �����.�� 1/+67!#%32 �� ���� ��
���� ��� �� ���� ��� 13+92� #���� /& ��� ���� ����
���� �� �� ����� � ��"�� ������ ��� �� ����� �� ���
���� �� � �� ��� ��"����� ���� ��� ��������� #��
�� ������ �� ��� ������� �� ��� ������ ������ ��� �����
�������� ������ � ����� �� ����� ��� ����"�� ����� ��
��� ����� ����������$�������� ��������
!� ������ �� ��� �� ��� ��������� ��� ��� ������� �� ��$
������� �� ��� ������ ��� � ��� �� ��������� �� ������
� ��������� ������ ��� #�� ���� �� � ������� ����� ��
���������.���� �� � ������ ������� ������ ����� � ��
�������� ��� ������� ��� �� ����� :;� �� ��� ����������
�� ������ �� ��� ������ ����������
��� ��� �������
��� ����� ����� ����� ��
�� ����������� ���� ��
��� ���� ������� �� �����
#�� ������� �������� ��� ������ ��� ��� ����"�����
�� ��� ���� �� ��������� � 1������� ��� ����2� �����
� �$������� ������� ����� �� ��� ������� ����� �� ���� �����$
��� ��������� �� ��� ������ �� ����� ���� ����� �� ����
70+ �� ����� �� ������ #�� ����"����� �� �������
����� � ��� ������� ���� �� ������ ��������� ��� �� �������
��� ��������� ��� ������ �� ����� ������ 3������ ����
���� ���� �� ��������� � ����� � ��� ������� ����
��� ���� �������� ��������� ��������� ����� �� ����$
�������� ��������� ��� �� �� � ������ � �������
������� �� ������� ��� ����������� ����� �� ������
3�� ��� ����"����� �� ��������� �� ��������� �����$
�� � ����� ����� ����� �� ������������ ������������
��� ����� ��� �������� ��� ������� �� �� ��������
5� ����� ��� ����� �� ��������� � ��������� ��������
��� ���� ���� ��� ��� ����"����� �� ��������� �� ���
��� ��� ������ ��������� �� ��������� � ����� � ���
������� ���� ��� ���� �������� �������� ��� � ����$
����� ����� �� ���� �������� ��������� !� �������� ���

���� ���������� �� �������� ����������
�������� � � ���� �������� ����� � ���� ����
�������� ����� ������� �� ������ ��� ������� �� ���
���������� � �� � �������� �� �� �� ��� ���� ��� �� �
���� �������� �������� ��� ����� �� ��� ��!�� �� ����
��� � ��� "#$� ��"#$� % � �� �������� ��������� �
��� �������� � � ���� �������� �� ����� ����� ����
���� ����� � �� �������� � ���� ��&������� '��� ������
������� � ����� ��� � ������� � ������ ���� ����(
���� ����� � ������ ��&������ � �������� ���� �� � ��
�)* �"#$ ���� � � ������� ��� � �� ������� ����������
������ � ��'*� ���� � � ���� ������ ��* �"#$ ��� +��*
�"#$ � � �����% � �� ��� �� � ������ � � �� ��&������
��� � �� �� �� �������� ,�� �� ��������� �� ��� ������
� � ��� �� ����� � ����� ��� ��� �� � � �� �����(
��� �% ��� ��� �� � ���� ����� � �� �-������.�
�-��������% ��� � �� ��������� �&������ � ���������
/��� ���� ���� �� ������� � �� ���� ��� �� ��� ������
��� �� ���� �� ���� ������
�����������
�� 0#$ ��&������
1��� �� ���� � ��&������� ���� � ��� � ��� � ���������
��� �� �����
�2 0#$ �������
' ������� ���� 0#$ ��� ����� ������� �34"
�� ���� ��� ��&������� ��������
��������
'�� � �� ���� �� ������� ��� ��������� �� �� �-������
�� 3������� � � ������ 0#$
� �� ��� ��� ��� � ���� �������� �������� �
������ � � �������� �� �� �� ���� � � � �� �������%
� � ���� ��� ������ �� ��� � ������!�� ����� ��� �� ����
� �����% � ����� ������� � � � �� ������ �� ������ �� %
��� ��������� �� 5�� �6 � 0#$ ��������� � ��� � �� � ��+
�6 ��������� ���� �� �� ���� � ������ 7���% � 5�+ �6
� �� � � 7��� �� �� �� � ��+ �6 ��������� ��� ���
���������� � �5%555 ��� � � �5 ���� $��� ��� ��� � ��
���������% �� ����� �� ��������� �� 5�� �6 � 0#$
��������� � ��� �% ��� ��� ����� � �55�4 � � �5 ���� ��
�������% 34" ��� �� ��� � � ������� ��� ����� ����� ��
0#$ ��������� � ��� �� 8 � �����% 0#$ �-���� � ����
������ ��� � ��-�� � ����� ��� ������ � ��� ��
��������� ��� �� 34"�
2� 34"
$�� 2 �6 � ������ 0#$ �� 34" ����� � � ��� �� 9��
�58:�55" ������� � � ������� ��� �'*�8:�'*�" �������
� � �����% ��� ��� ���� �� �5 �������� � ������ � ;��4
� � �5 ���% ++�4 � � )5 ���% ��� <2�4 � � )5 ���� 0#$ ����(
���� ��� ������� �� � �55 �� �� �� ���� � ������� ���
�� � �+5 = �<5 �� ��������� � �� ����� �� �� ���� �
������ ������� � ������� � �� � ����� ������ � �� ��
� ��� � �� �� 34"�
�� 4 ����� � � 34" �� ����
,�- + �6 � 34" �� ��� ��� � �6 � � ����� ��>��
� ��� �% ����� � �� � ��+ �:�� ���� �� ��� ����% ��� �����
� ����� �� ����� ��� '$? ��>�� � ��� � ��(� �� � ����(
��� � � 4���� � �� ����� �� ����� ����� ���
���� ����� 0#$ ��!� �������� $��� �� ����� �� �����%
������ 34" �� ���� � � ����(�������� � ���2 �� ���
� ��� �� �������� � � ������ ���� � �� ������ ��!�� ��
������� ����� ��� �������% �� �� ������ ���� � � ��
���% ��� �-��� 0#$ �� ����� ���� ����� � ��������
0#$ �-���� � ���
�� 3������ � � 34" �� ����
"�� �� ����� �� ���� 34" ������� ��� �� -������ (
���� ���� ������ ��#'3 �� � 34" �� ���� �� �����
���� ����� ������� � ��� ���
+� @������� � � ������ 0#$
A��� ������ 0#$ �� �������� �� ����� �� ����%
�������� � /02)5 �� �� +5 ��:�6�
)� 6������� � 34" �� ���� ��� ��&������� �������
9�� �� ���� ����� 7� ��������(������� ��&�������
������ ������� � � �� ��������� 0#$ ��&������ � ��
�� ���� ��� ����� �� 34" �� ���� ��� ����� ��
������� �� �� ����� ��� �� �������
<� 3������ � � ��&������� ������(������� 34"
�� ����
'������� �� �� ��� �� <+ �6 � ������ ���� � �< �� �5
�� � ��+ �6 ��������� ���% ���� �� �� ��� ��� � � 25 ���%
��� ��������� � �+%555 ��� � � 25 ���� $��� ��� ��� �
���������% ��� 2+5 �6 � ������ ���� � �< �� �5 ��
��������� ��� ��������� � �+%555 ��� � � + ���� "�� ��
�� ��������� ��� ��� �� ����������
�� 0#$ � � � �� ��������
3���� ��&������� ������(������� 34" �� ���� �� ��
0#$ ��&������ ��� ���� �� ����� ��� ��&������ � ��
34" �� ����� 4 ����� �� ������ ����� ��� ��&�����
��� � �� �� �� B6$*' ��������
�������
�� ��&������� ��� �� � ��� ;5� ������ ��� � ��(
&����� �� �� �������% �� �������% ������� ��� �� ����
�� � �� ���
�� �� �� ���� � �������% � ����� �� � �55 ����� ����
������ � ��� �� +5 �+5 �� �� �� ��� ������ ���
�� �58 ������% ��� �� B6$*' �������� C�����
������ ������� ��� ������ �� ������� ������� � �� ����
������ ��������
2� �� �� ���� � �����% � ����� ��&������� ��� � � ��
�� ��� ������ ��� �� �'*�8 ������% ��� ��
B6$*' �������� C����� ������ ������� ��� ������ ��
������� ������� � �� ���� ������ ��������
��������� ���� �������
��� ��� ���� ������� �� !����� �� !�����������
������������ ��
0��� ��� ���) � � ��� ���� ������ ��� ��������������
�������� �������� �� ���� ���� �55 �6�
�"� � ���� ���� #�$�� ������� �% &��
0��� ��� 2��2 � � 2(���� (2(���� -������(�%�(
�� ������ � �� � ������� �� �� � ����% ����� �� �C
��5 ��� 5�2 � �:6 ���� ��� ��� ���� '*% ��� ��� ����
���� 255 �6�
�'� �( #�$�� ������
,�- ��5 �6 � � � �:6 ��� ��>�� � ��� �% �C ��5 ���
5�2 �6 � 5�+ � �:6 ��� ���� ������ ��� ��������������
�������� '*% ��� ��� ���� ���� �55 �6�

�������������� �� � ������� ����������
��� ��� ����� � ������
����� � � �� ��� ���� ���������� ����� ����� �� ��
���� ���� ����� ��� ��� ��� ���� ����� ���� �
��� ��� ������ ������
������� � �� ��� ����! � �"
�#$ ��%� �!! & �"
�� ����'" (�� )���� � �"
�� ����'" *������� )���� � �"
+����������� �#* )��,���� �� -'�"� � �"
.��� �/ �"
! 0��� ���)��� �� ��� ����'" 1�������1�2,���%�
,��2,���3��)��)������ 2,����2�����3 )+ � &3 ���
����'" )������ � �2�����3 1� ����'" ������ � �2���
��� ��� � � �'" ������
!! * ��� ���� ���������� 1 � ����'" ��2 �� �4$ ������
� 1����%,� ������ ������)2��)2���3 �*$ ����� � 1��
��%,������� ������)2��)2���3 �5$ ����� � 1����%�
,�,����� ������)2��)2��� ��� �$ ����� � 1����%�
,�2,����� ������)2��)2��� *�6 �� )��� ��� ����������
�2� ���)����� �� ������� ���� ��, � ��
��� ����� �� � ���� �
2� �� ���, 7���� �� �6 ����� ��2���3 � �2 �� �2
�,�)���� ��2�� ��� ������� �� )����3 �2 �,��������
��� ��2�� ��� ������� �� �#$ ��������� ��� �� �� -�
�� �))��)���� �6 ����� ����� 7�� ��� �2 �))���� � ���
)������ �� � ��
��� ������ �� �� ����� �� !�"�� ������
������� 1&1 � �� 1�������1�2,���%,��2,���3��
)��)������ �� �8 � �" �� ����� ���� ����� ��� ��� �" ��
� � ���'" ������ � ��2,����� �2,�������� ������
���� (3 ��� ��� 9��� �� ��7 ���� �"
�#� $���� �� �� ����� �� !�"�� ������
��� �� ������� ���������� *� � �� ��� ���� �� �� ���
)�)��� ���� � �� ����� �� �� ������ ����������
*� � �� ��� ����
�%� ������ ��
:�% � � � �� ������3 1 � �" �� ������� ����������
*� � �� ��� ����3 �� �" �� � ��� ���� �� �2��� �
������ ��3�������������2,��/�)2�,�)2����2������ �
������� �� �� ���� ��� ��� �" �� 9��� *��� ����������
�2 �������� �, 2�����3 ���� �2 ��� ���� �� ��� � /��53
��� )�)�� ���
�$�� &��� � !�"�� ������ ���'�� �� �� ������
������� � 1� � �� �����)2��� �� 3 � 1� � �� %,��
�,���� ;; ��� � /� � �� ������ � ��2,����� �2,������
��� ��������� ��2,���� �� �� �" �� 9���3 ��� ���
�� �" �� ��,���� ��� 9��� �� ��7 ��� �"
�$$� ��� (��)���
;�� $����
0������ ��;
���<
���44*554455*���
���*55*444*5**55���
; ��� =(�;
=(�<
���4**5**44�'5�554���
���5455*54*4���
��� ��� �������
*�+ ���� � ������ �� ������
�����, �� � ����� �� � )�9�� �� � ���� �� ����������
2�� ������ �>������� �)����� �� �2 9�, �� ���� ����
�� �2 ����)����� ��� ��� �����)����� ��� ����� �2�� %���
��9� �2 )������� �� ����� �2 )�9�� ������ >� ��
�� ������� �� ��2 ���3 ��� �2� �� ������ )��������
������� 4�����,3 �2� �� �2� ���� �� �>������� ��
�2 ����� �� )�9�� �����,
��� 5�,���� �����,? =� �� ��� �� �2�� �2 �,��� ��
2������ � 9��2 �� �����)����� ��� ���� 5�,���� �����,
�� ���� ����� �� �����,
�1� $������ �����,? 2 ���� )��� �� �2 %)�����
����, ������������ �)� )��� �� ���� ���� �� �� � )��� ��
�2 ��� �� �� �2 ����� �� �2 )�9��
��� 0 �7 �����,? 2 ����)����� ��� ���� ����� �� �2
)�9�� �� �� ���� ���� �� �� � )��� �� �2 ��� �� �� �2
����� �� �2 )�9�� 0 �7 �����, �� ���� ����� �))����
�����, 4�����,3 �2 )�9�� ������� �� ���� )��7���
��� �� ��))��� �� �>�� �� �2 � �7 �����, ��� �2 ��))�
�����,3 ��)�����,
4�����,3 �2 ������� �� ��6 �� �� ��� �� ����� ���,
�, ��)��� � ��� )��� �3 � � �2 ����� �� )�9�� �����,
�� ����� �, �2 ���� �� ���������� �� �2 ���� �� �� �2
)������� 2����3 �2 ����� �� )�9�� ������� ��� ����,
���, �)����� �� ��,���� ��� �� � �� ��,���������, �� �2
� ������ �������3 ��� ���� ����� �)����� �� �2
��2�� �� )�)������� �� 2������� �� �2 ���)� �� �����
)2� � ���� �� )�������, ����)2� � 5���6 ���,3 �� �� �
��� �2�� �9� ������ �� )�9��� �� �2������, ��������3 ��
��, � )������ �2�� �2 ������ >� �� �� �����, ��
������� �� �2�� ��,���� ��� �� �� �� ������ *� �2 �����
�� )�9�� )������ ������� �� ��)������ )2,����� )��)��
��� ��� �2 )�9��� )2����� ����� �� �� �� �2 )�9���
��9 �������� �� �� ��3 @�)��� $2������)��� �=A �)���
>� ��2 �����, ����������� �� BB$�9�� $������ �����,
�����������CC ��� �2 )������ �����, ��� �� BB��������
���� �� 0 �7 ��� �))� �������CC ��� �2 � �7 �����,
2 ����� �� )�9�� ������� �� %)���� �� ���� )�
��� ��� � �7�'���3 ��� ������, %)���� �� �'��� ��
�'�������� 7�'���
��,�� �� ���,
2 ��,���� �����, �� � � ������ �� �2 ����� ���� )�
��� ��� �3 %�� ��� �� ��� ����� �2�� �� ��� � � ������
��� )��� �� �2 ���� ��� )��7��� ��������� =� �� ��
��������� )��)��, ��������� �2 �)��>� ��,���� ��� �� � ��
�2 � ������3 ��� �� ��� ����� �, �2 ��2�� �� ������
������ 2 ��,���� �����, ��� � ������� ��2� �,
���� ������ �� �, ���)� ��� ����
* 2 ���� ���� ��,���� �����, �� ������� ����?
�� ;�� %��)�3 �2 ��,���������)2�� ���� ���� �
��� ���)������� �� �2 ��� ���� ������� �,
���%��� �2 )���� ��,���� ����, ���������� ����
���� ����� ��,���� �� �2 )�9�� ����, ����������
����

���� ���������� �� �������� ����������
�� �� ���� ���� � ��� ������� ��
�� ��� �������� ������ ������� �� ������� �� ��� ���� �
����� ���� ����� ��������� ��� ������ ������ ���� ���
������
�������� �����
��� ����� �� ������� �� �� � ��� ��� ��� ������ �������
��� ��� ���������� ����� ������ !������ ���"� �#�������$
����� ��$� ������� ��� ����� �� � ���� � ��� ����� ��
������ ��� ����� �� ������� ������� � ��� ����� � ���
����� ����������% ��� ��� ����� �� ���� ������� � ���
����� � ������������ &���� �� ������� �� �� ����������
������ �� ��� ������ ����� �� ������ � ��� ��� ����$
�����% ��� '������� &����� ���� ()* ��� �+�� ���
�� ������ �� ��� ,,&-��� &���� �� .������ .��������$
���//�
�� ��� �� ������ ������� �� ������� �� �������� ����
��� ����� � ��� ��� ������ ��% -�� � �� �������� -���
��� ��� ������ ����� �� ������% �� �0�������� � ����
� � �-� ���� � ��� �-���� )� �� ������ �������
������������% ��� ����� -��� ��� ��� ���� � ����$
��� � ��� ��� �� �� �� ����� �� � ���� � ����� � ���
�-���% ��� ��� ������ ���� � ���� ��� ������� � ���
��� �� �� ����� �� � ���� � ��� ����� � ��� �-����
.�� � ��� ���� ��1������� � ������ -�� � �� ����$
����� � ��� ��� ����% �� �� �� ��������� �� ���
����������� ��� � ����� �� �������� ��������% ���
�� ������ ����� �� ������� � � +���� ������ �-��� ��
��������� �� ���� ��1����� ��� ��� ������ ��������
2�� �% ��� ����� �� ������� �� ���� ����� �� ��� ����
�������� � ��� ���� ������� � �� ������� � �������$
�� ���������� ������% ��� �� �� -����� ���� ��
������ ������ ���� � ��� �� ����� ������ ���� ��
�-��� ��������
�� ��� ��� ��� ��������� ������� �� ��� ����� ��������
�������� ���� ����� ��� �� ����� ��� ������ ���� �� �
���� � ��� ������ � ��� ���� � ��� �-���% ���
�# ����� ��� ����� ��� ��� ��� ��� ���� ������
���� ��� ��3� ������ ���� ��� ��3� ����� � �������
� ��� �������� ������� � ��� ��#���� ��� ���
�������� �������� ������� ������ ��� ����������� ���
����� ����� �������� ��������% ��� ��� ��� ��� ��
��������� ��� +������ ���� � ������� � ������� 4�� �
���� ����� �� ������ � ��������� ��� ������� -�� �
�������� � ��� ��� ��3� ����� �� �� � ��� ���
�������� ��������% ��� ������ �������� ��������� �� ��
������� ��� �� �������
��� ����� �� ������ �����
��� ��� ������� � � �-��� �� ����� ��� ��������� �
���������� ����� ��� ����� �� � ���� � ��� ����� � ���
�-���� ��������% ��� ��� ������� ������� � ��� ���
�-��� ����� �� ������� ��� ��� ��� � ����������� �
����� ��� �� ��� �-�� ���� 5������% ��� � ��� ���������
��������� � � ��� ��� ��� ������ �� �������� � ��� ��� �
�����������% �� �� ���� ���� ��6 ��� � ��������� ��� ���
������� -��� �� ������ �������� ��������% �� �� ���������
� ��� ��� �- ��� ������������ -�� ���� ��� ��������
��� ��� ��������
'������� &����� ���� ()* ��� �+�� ,,.�����������
� ��� ��� ������ .��������//�
�� ��� ��� ������� �� ���������� �� ��������� ���
�������� ����� � � �-� ���� � �-��� ������
���� ��� ���� ������ ������ � � ���� �� � ���������
������� ! ������ ���� ������� 4���������% '�������
&����� ���� ()* ��� �+�� ��� ����� � ��������$
��� ��� ������� �� ��������� ��� ���� � �-��� �� �
������ ������ � �-� ����� ! ������ �����
�������
�� ��� ������ ������� �� ������� �� �� ���� ���� �������
� ��������� ������� �������� � �-��� ������� �����
����������� ��� ������� ��� �����% ���� �� �������
����� � +#�� ����������� ������ !������� ���� ���
��� �������% ��� ��� ����������� �� ������ ��
���������� ����� ��� ��� ����� �������� ������� ��
��� ����� �- ������������ �� -����� �� � �������
����� ! ������ ���� ������� 4���������% '�������
&����� ���� ()* ��� �+�� ��� ����� � ��������$
��� ��� ������ ������� �� ��������� ��� ���� � � +#��
����� � ��� ������ �-��� ! ������ �����
�������
��� ��� �������
��� ����� ������ �����
��� ���-��� �� ������ ��� ������� �� ������������ �
��� ������������ � ���� ������ ����� �� ������ ���$
�� ������������ 7���� �� ���0���% �� � �� 2&89% ��� ���
� ������� �� ���� ������ �� ���� �� ������������ ���
� ��� ���� ������ ����� ������ ��� ����� ���- �� ��
�������� �� �������� ����� ��� ��� ���� ��� ��� ���
:��; <���� -���� �� ��0�����% ��� ��������� -�����
������ �
&����� �� ������ ������ =* ����� �� � -��������� �
��> ��% ��� � ��� ������ � � ������ ���� � ���% ������
������� ��� ���������� ���� ������� �� ��� ����� � ����
������ &����� ������������ �� ��> �� �� ������ � ��� $
��� � ��� ������� ��� ��������� ����� � ��� ������� )�
��� ��1�� ���� � ������� ��� ������ ��� � ���� ������� �
�������� � ���� � -����% ����� �� �� ����������� ������� � ��
��� ��1��� ���� ���������� � �� �� ��������� �� -���
��� ��� ���������� �� ��?����� � 3�� ��1�� ������� ��
)� ��� ���������� � ������� �� � ����� ������� � ����
��������� ��� ����� � ����������� � ��� ��� ����������%
��� ������� ��� �� ��������� 5���������% �� �- �$
��������� ������ �� �� ������� �� ��� ������% �����$
�� ���� ��� ��� ����� �� ������� ���� �� �� ��������� ��
��������� ������� �� ������ � ��������� � �� ����� �
����� ��������� �� ��� �����������
:��; @��� ��� ���� 4�����% ��� 4������� 4�����% ���
��� ��1�� �� ��� ���� ����������� ������ ��������
������� ������� =����� ����-��� ��� �+�� �� ���
���������� �������% ������� � ������ � ��� ������� �
�������� � ������� � �������� �� ��� ������ ����� ���� ��

�� ������ �� ������ ��������� ��� ����������
�������� ��� ���������� ��������� �� ��
��� ���������� �� � ������� ����������
�� ��� !�"� ��� �� �� ��� ������������ �� �� #��
$�������%
���� ������ &������ � �����!� �������' �� �� ������
���� ��� �� �� ���������� !�"� �� �!���� � ��������
������ � ������������ �� (%) �� ) �� �� �*%
��� ����� +�����������' ������ �� �!���!���� ��
�� $������� $������� ��� �� #�� $������� �� �����, ���� ��
� -������� �� )�( ��. -��� � �����!� ��������������.
����� �� !�"� �� �� !���/% #� �!���� ������� ������. ��
������ ������ ! ����� �� �� ���� �� ������ ��������0
����� �� ! ����'�%
������ ������ #� �������' �� �� 12 ������������
����������� �� ������ ��� ! ������ !' �� ���������
�� ����� !' �� ��� ������% 3� �� ������� �� �������� ����
�� �������� �������!� �� ��, �� �� -������� �� ��
�������� ����� 4)5( �� �(( ���. ��������� �� �� ����� !��
������ �� �� ������� ������ �� �!���!��� �� �� ���
������% #� �������� �� �!���!��� �� )�( �� �� ��
�����0���������. ������ �� �!���!���� �� �� #��
$������� �� -�������� �� �)(. �)5. ��(. ��5. �6(. �65. ���
�5( ��% 1���� �� ����� �������� �����. ���� �� ��� ��
�� �!���� �!���!��� ����� �� ��� �� �� -�������.
��� ������ �� �������� ���� !�� 7����� �� ������
������% 8��� �� ����� �� �!�����. ��������� ��
�!���!��� ���� �� �� �����0��������� �� )�( ��% $�!�����
�� �!���!��� ���� �����0��������� ���� �� �����
�!���!��� �� )�( �� �� �!���� �� �!���!��� ���� �� ��
������ �� ��������% 8��������� -��� � 7��� ������ � (%)0��
�������' �� �����7������ !' ������������� ��' ! �������
�� ���� �� "�� �� �����0���������. �������' �� ��
�������� �� ������!�' ���!��%
��� ������ +������� �� ������������. �1. �� ������ ��
�� ��� ������ !' �� �������
�1��$ 4�19�$�.
�� -���� �$ �� �� ������������ �� �� $������� $�������:
��� �1 ��� �$ �� �� ������� �!���!���� �� �� #��
$������� ��� �� $������� $�������. ��������'%
������ �
#��� �����. �������' ����� �� �� �� *�-�' ����'. ��
!��� �� �� �������� !' ������ �� �� ����������'!���0
�������� ���� ���� �������� �� �� 8����0+�������;�
����� �����. �������� �� �� �!���!��� ������� ��
5( ��% #� 8����0+�������;� ����� ����� 48����;� #$�
����� ��������' -��� �'����� ������ �� �� ������. -����
��� ��� �� ��������� �� �� ������ �� �� ����' �� ��"���
�������% ����� �� ����� �� ������� �� ���������
��!������. � ������� ��� ������������ �� �� ������ ����
�� ��� ������ ��' ! ���% <�� ��������� �� ������0
��� ��!������ ���� �� ������ �� �� ��� ������ ��
������'. ����� �� ������ !��- ��� 3��������
$�!������ ����� �� ���������� �� �� #�� $�������% #�
"�� �� ��������� ��!������ ��� ! ������,� !' ��������
������� �� ������������ �� �� ������ ���� ��� ������
��=���� ��� ������� ��������% 2��������� �� ��
*�-�' ��� ���� �� �������� �� �������� ��������' ����0
������ ��� ! ��!������� ��� �� ����� �����!� !��-%
������� ������� 1���� ����-�� ����7� �� ��
���������� ���������. ������� �� ����� �������� ��
����� ������� ��� �� ������ ���� ��� �� �� !�"�
��� �� ����� �� #�� $�������% &���� �������� �� ����
�������� -��� �� ��� !�"� �� �!���� ��� �-� ���� 7�
$������� $�������� ������ ������������� !�-� 5 ���
�(( �� �� ������ �� �*. �� ������������� !��� ���'
�����%
���� ������ &������ � �����!� �������' �� �� ������
���� ��� �� �� ���������� !�"� �� �!���� � ��������
������ � ������������ -����� �� ���� �� �� ��������0
����� �� �� $������� $��������% >� ���������� !�"� -���
������ � �? �� �� ���� �� �( �� �(%5%
���� 1� �� !�"� ��� ��� �� #�� $������� ��� ��
$������� $��������%
������� �� ��������
������ ������� ������ &������ �(( �� �� ����� 433�
������ �����'���� ��� )(( �� �� ������ ������� ���'0
���� �� -���. ����� -��� -��� �� 5( �*. ��� ���% &������
�( � �� ���'����� ������ ���!���� �� -��� �� � 7���
����� �� 5( �*. ��� ���% $��-�' ���� �� ������ ���0
!���� �������� ���� �� ����� ������ �������� -��� ������%
����� ���� �������� ���� ����'%
�� ��� �� &������ 5 � �� ������ ����'� ������ ��
-���. ��� ����� -��� -��� �� �(( �*%
������ ������ ������ ����� � ������ �� 5� $&$
#$. +���� $����� �����. ��� $����� ?'������
$������� 46 �� �)5� 4)���% #��� ����� ��' ! ����� ��
���� �������� ��� �� �� ) -/�%
������ ��!� �� �� �( �* �� 8����;� #$ -��� 5( �*
�� -���% $��� �� �� ��!� !����. �� ���� ��������%
��� ����� #� � �* �� ��� $������� $�������. �� #��
$�������. ��� �� ����/. ��� � �* �� >�/���� +����
�����. ��� ���% >���- �� ����� �� ���� �������� ���
�( ������% >�� (%5 �* �� �� &����� 8����;� #$ �� ���
��������. ��� ��� ��� ��! ��������' ���� �� ��������.
��� ����- �� ����� �� ���� �������� ��� �( ������%
&����� �� �!���!���� �� �� ��������� ���� ��
$������� $�������� ��� �� #�� $������� �� �� -�������
�� ������� �!���!��� �� 5( ��. -��� � �����!� ���0
�����������. ����� �� �������� ���� �� ����/ �� �� ��
��������� �� ,��%
��� ������ @A�� #� ����������� �� �!���!��� ��
������ ������������ �� ��������: ��-��. �� �� ��������
���� ������������ ���� �� ��=�����' �����. �� -���
�������� �������'%B 1���� �� ����� �������� �����. ����
�� �!���!���� �� �� ��������� ���� �� $�������
$�������� ����� �� ������ �������������. ��� ������
�� �������� ���� !�� 7����� �� ������ ������% 8��� ��
�������� ���� �� �!����� ��� �� �!���!��� �� �� #��
$�������. ������ �� ������������ �� ������ �� �� #��
$�������%
"������� ��#��� �� 3� �� �����-��� �������. ���'0
������0�������������� ���� �� ���� �� � ��� ������ ��

�� ��� ���� � �� ��� � �� ������ ������� ��
���� ���������� �� �������� ����������
��� �� �������� ��������� �� ������ � � �� ���
��� �� ����� ��� ������� ��� ��� �� ���� � �����
��� �� ��� �� � � ���� � �� ��
������ ������ ��� ������� ������� � � �� � �
� ��� �� ��� ��� � !��� ����� � � ������ � �
�"# �� � �## �$�
����� ��������� ���� ������� ������� � � �� � �
���� � ���� ��� � !��� ����� � � ������ � � %� �
� �## �$�
��������� &�� #�� �$ � ' ��� �� ��� ��� (�����
� �$ � � � �� � � �� �� �� ����� ��� ) � �
� �� � ��* ��� ��� ! ���� � � � ��������� � � �#
������ &�� #�� �$ � ����� � ���� &�� (�����* ���
� � � � �� � ��� +������� � �###�� � � �#
�����* ����� �� ����* ��� ��� �� ��� ������� ����
!� � ���� (���� ��� �� �� �� ����� � � �$ �
&�,���� + ���� (������ �� ���� �� ������ � � ��� ' ���
�� -. �/ + � � ����� ���� ������� � �� ��� � �#
�# ����� ����� ����� � � � � ���������* ����
!��� ���� � � ������� � �� � � � �� ) � ��������
��������� ����� � � !�� � � � ����0 � !����* � �� �����
���� ����� � ���� ������� � � � �� & ��� ��� � �������
� ��� ����� � ������� � ��� 1������ �2����
�� �� ������ ��� ��� �2���� � � � ���� ��� ������*
�� ������� �� �� ��� �� �� �� �� ��� �� �� �����3
������ �
��� ��� �* � �� ��� �������� �� �� 1���� ��
�����* � ����� � �� ��� �� � ��� �� � 4%# ��
"5" �� ������� !��� �� ������ ���� 6��"# ��� ����
�� ��� ��� + ����� 1����� 1��� 6��"# ��� ���� � �
������ ������ ��� ����� ������� � �� �� ��* !��� ���
���� ���� � � �� ���� ��� � �� ����� �2����
�� ����
�������� �� ������ 7����� ���!�� ����8�� � ��
������� � � �����* ��� ��� �� ��������� ������� �
�� ��������� ������ � � �� �� �� ����� �� � �� ��2��
���� ������� �� ��� ' �� �� ���� � � �� � �� � �
�� � !� �� ���� ��2�� ��� � ��!�� ��� 8��
'������ ' �� �� ����� � ������ �� ��!��� �## ��
��� � �� � �� �� ��� �$* �� � ������ �� ���� ������
�������
���� �� ����� ��� ��� � ������ ����� � �� �� ��
����� �� � �� ���� ���� ��2�� ��� � � �� �
����� � � ������ � !�� �� ����� � �� � �������
�� � �� '������ ' �� ���
� ��� 7�� �� ��2�� ���� ������� �� ��� ' �� � ���
�� '������ ' �� ���
��������� ������� ��� ��� �## �� � ������ ����
6��"#�� � "# �$ � ���� � 95"�� -. �/ . ��� ���� ����
�� ���� ������ ���� 6 � ���* ��� �2���� �� ����
��� ��� �2���� �������3 &�� �## �$ � �� ��� �� ���*
���� !� !��� �### �$* ��� � � :��� �� � �� �
�� ��� 8��� ����� 9;����� . �� � ���������* ���
� �� �� 8����� ������ � �� ����� � �� � � � ������
����� -. �/ '� ! ������ � � �� ��� !�� ���� �����
� ���� � �� ������� :��� �� ������ ��� �� ����3
��������� &�� " �$ � �� + ����� (����� �## �$
� ���� '������ ' �� �* �� ��� ' �� �* ��� �� 1���,*
��� � �� ����� �� &� � � ����* !��� !�� ����
� � ���� ������� ������� �� ��� ������� � ��
� �� �� �� � �� '������ ' �� �� ��� �� ��� ' �� �
� "5" ��* !� � ������ ����� �� ����* ���� ��
1���, �� �� ������� <�� �
-. �/ � � ��� ����< 9����� ����� �� ���� �����/
�� ��� ���� �� ������� 1������ �2���� �� ��
������ ��� ��� �2���� � � � ���� ��� ������* ��
������� �� �� ��� �� �� �� �� ��� �� �� �����3 �����
��� �������� ��! �������� ���������* �� �������� ���
���� ���� � �� �� ������� �� ��� �� �� ���� =����
��,���� �������� ��� ������� !� �� ���� �������
�� �� ������ -. �/ ��� ���� ���� � ��� ������
�� �� � ������ � � � ������0 � !����* � �� �������
����� � ������ � ����� � ��>����� �����* !��
���� ��� �������3 7��� �� ����� ������� � ��� �* ��
�� ��� ������� � �� � �� �� �� � �� '������
' �� �� ������ �� �� �� � ������ ��* ��� �������
�� ������� ����� ��� 8�� �� �� �� � ��� :� � ��
������� ����� � ����� ��� �� ��� ������ � �� ���
' �� �* ������� �� � ������ � � �� �� � �� ���
' �� ��
������ �
��� ��� �* � �� ��� �������� �� �� ����� ���
��� � 1+& �����* � ����� � ����� � � �� �����
9+���� � ���� �� 9+��� � �� �� ��� ��� ����� �
��� ��� ������ � ���� ���� �� ���� �� �� ���
��� � ��� ��! �������� ���������� ;��� ��������
��������� ��� ������* ��� �2�� ��� �� ���<�� ��
��� �* �� ���� �� �� � ������ � � �� �� ��
����� �� ������ ��>��� � � ������� �����������
�������� �� ������ 7����� ���!�� ����8�� � ��
������� � � �����* ��� ��� �� ��������� ������� �
�� ��������� ������ � � �� �� �� ����� �� � �� ��2��
���� ������� �� ��� ' �� �� ���� � � �� � �� � �
�� � !� �� ���� ��2�� ��� � ��!�� ��� 8��
'������ ' �� �� ����� � ������ �� ��!��� �# ���
��## �� � �� �� ��� �$* �� � ������ �� ���� ������
�������
���� �� ����� ��� ��� � ������ ����� � �� �� ��
����� �� � �� ���� ���� ��2�� ��� � � �� �
����� � � ������ � !�� �� ����� � �� � �������
�� � �� '������ ' �� ���
� ��� 7�� �� ��2�� ���� ������� �� ��� ' �� � ���
�� '������ ' �� ���
�������� ��� �� �������
��� ������� ��� ��� �� � �# � � ����� ��� ���*
�# � � � ��� ���� ��� � � ������* ��? � � � ���
����� �������* 4 � � � ��� ���� ��* ��� 5�" � �
� ��� ���� ��� ���� ��� � !���� &�@��* � ���������*
!� � ��� ���� �� � � ��� ���� ��� ���� ��� �
�= � ����"� ���� !� !��� �### �$* ��� � �
������ �� ��� ������� ��� ��� �� � � � � � ����
9AA� ������ ���������� � !��� � 8��� � ���� � "# �$�

�������������� �� � ������� ����������
���������� ���� ��� � � � � �� �������
������� ��� �� � � ��� ��������
��������� ��� ��� � � ���� �������� ������ ��� !�"�
������ ��� ��� ����# $��� % � � ��� � ��&���
�������� '���(��� ��� "�����" �� �)�� �� �� ������" ���
��� ���� ��� ���$ � ��� � �� ��� �������� *����� +�
������" ���$��� ��� ����(���� ���� ��������� ��� �("�&
(����" � ��� "�����" ��� ��� �������� ������" ��� ���
!�"� ������ �� ,����- ����" �� �+% �� $��� � "����(��
" ���� ������� �"��� ��� ����# � "�� ��� ��"������� �
-��� ����� ��� "�����" ��� ���� � �� ��� �������
��� ��� �����"��. �������" � ������"� ��������.� '� "�(&
"�����" ���� $��� ���"� ������������ �� ��� ��"� ��� ��"���
����� �" �������� �� '���������� ��("�����" ����� �����
%� �����"� ��/����� ����� " ����" ��. ��0� ��/����� ���
��" �"� �����"����" ��� "������� ����� ��� ��"� �����
"���� (� ��� "����
����� ���� 12��3 !�� �������"�� � �("�(���� � �&
���� ����������� �" ��������4 �$�0�� �� ��� "�������
���0� ����������� ����� �" "�5������. "���� �� $���
� ���� ��������.�6 7"��� ��� ������ �����""�� ����� ��
��� �("�(����" � ��� "�����" ��� ��� ��������
������" 0��"�" ��� ����� �����������" ��� ���������
��� "������� ���0� (�"� 8����� ��� ����� ���"� 9�� ���
"������� ���0� " (������ ��� ��� �("�(���� � ��� !�"�
������ ��������� ��� ����������� � ����� �� ��� !�"�
�������
�� ��� �
!��" ����� �����. �������� � �" ��� ������ �""�. �"
(�"�� � ��� ���������� � �� ��� :��%�; �� $��� ����� ��
�� ��#����� "����� ��� ��� ��"������ ��0�� ���� � �("�&
(���� �� �<� ���
� ���� ���� ���� 7���"" ����$�"� " ���8�� �� ��� ��&
��0����� ����� � �� ��� � "����� � ��(���� =����
�� $���� ��� ����� ������ ��" (��� ��0��"�. �����&
����� (. ������� ����."�" :�"��� ��� �������&�& �����
��0��"�� ����� � +�%�; � � ��� ��������� "������� �
��������� �������� �� ��� ����� ����� ��"� �� "����
������� "����� :� �� ����;� >����� ����" � ���" "�����
$��� "���� ������� "����� :� �� ����; � (���� �� ��$��
���� ����� �������� ������" ��0��� �����������"
(��$��� ��� ��� �� �� �� � ��� �����������" (����
�0���. " ����� 12��3 $ ��" �"�" ��. (� ("��0�� �� ���
"�� �� ����� ��"� ��" "����8�����. ��/����� ��0�� � �����
���� ���� � ��(���� =����� � ��/����� "������� �����
��. (� �� �.�� �� "��� ��"�"�6
��� ���� ��� ?�� ��� � "����� � ��� ��"� ����� ��
"���� ������� "����� :� �� ����; ��0��� � �����������
$����� ��� ����� � ��� �����������" � ��� ��������
������"�
���� 7"� "���� ������� "����� :� �� ����;�
����� ���� >�""�0� �(�� ��<+ � � � �� :''; "������
�����.����� �� �� � � $���� $��� ������� �� ����""��.
��� ���$ � �� :������ �;� >�""�0� �(�� �<�+ � �
"���� ������� ���.����� ��� %��� � � ���.���" "����
���(���� �� �� � � $���� $��� ������� �� ����""��. ���
���$ � �� :������ �;� ��� ������" � ��� � ���
������ $��� $���� � %�� �� !��" ������ ������� �" "��(�� ��
�� ��� ������� �� + ����"� > �� �"� ��� ������� �� ��
��0�� " ���(����. � ������" ��. ���� ������
��������� ! �� 0���� � ��� �������� ������" ��� �
"����� � ��� !�"� ������ ��� �� �,��� 0���� � "����
�.������ "����� :+ �� ���; ��� ���� '���������. ��� �
0���� � ������ ������� �,��0����� � ��< 0���� � ���
!�"� ������ ��� ���� ���$ � "���� �� � ��� �������
(��$��� ���� ��� %��� �� �� ��"" ���� �� ������"� *�����
�� ������" ����� ��� ������� � ��� ������ ������� �����&
���� ��� �("�(����" � ��� �������� ������" ��� ���
"����� ��� ��� !�"� ������ �� ��� $�0������� � ����&
��� �("�(���� �� �<� �� $��� � "����(�� " ���� ���&
���� �"��� ��� ����# � "�� ��� ��"������� � -��� 12��3
��. "����� ���� ��0�� " ���(����. � � ���� ����� �" ��
���� ��(�� �� ���������� � ����� ������������6
����� ���� 7"��� ��� ���"�&",����" ������ �����""��
����� �� ��� �("�(����" � ��� �������� ������"
0��"�" ��� ����� �����������" ��� ��������� ��� "���&
���� ���0� (�"� 8����� ��� ����� ���" ��� ��������� ���
��������� ��5����� �� ��� ����� 12��3 *����� ��� ��0��
����� � ��� "�������" ��� �������"�� � �("�(���� �
����� ����������� �" � ��������. �������6 � "����(��
"."��� �" �� ���� .����" � ���� ��0��� � ���������
��5����� � �� ��"" ���� ����� 9�� ��� "������� ���0�
��� ��� �("�(���� � ��� !�"� ������ ��������� ���
����������� � ����� �� ��� ��"� " ������ ��#��� ��.
����""��. ���������
�� �������� ���� ���� ! ������-� ��� �/��� � ��������&
��� "�("�����" ��� ����� ��� (� ���� ������ ��� ���
������� ��"� " ������ �" ���$"� ��� ��� 0���� � ���
������������� ���� � � 0���� � � "����� � ��� ��"�
" ������ $������$ ��� "� �������� ��.�� ��� ��""�0� ���
���� ����� �� � "���� 0���� � ��� ��@ "����
�.������ !�� 7"� ��� "����� " (������ � �� ��� ���
!�"� �������
������ � !��" ��"� "�$" ������� ��/������ (��$���
�,��0����� '�A ��� ��(���� "�� ��"� ������� � ���
"���� �.������ ��� ��� ������ ������� �" � ��(����
������� ��"�5����� ������ ����� ��� ������� � ��� "����
�.������ � �� �������� ���� (��$��� ��� ������� � ���
"���� �.������ "����� ��� ��� ������� � ��� ������
������� $��� ��0� '�A "�� ��" � ������ ��" �"� ����
��(���� "�� ��"� !�� ������������� ���� ����� �"�� �
������-� ��� �/���" � ����������� "�("�����" ��" ��� (�
�"�� � ��������� ��� ����� ������ �� ��"� " ������" ��
�����������" (��$ ��� �� �� ��
�� ��� �
!��" B�������� ����� �" (�"�� � ��� ����0���-���� �
��� ����� $��� & ����������.�� :C?�; $���� �����" $���
��� �����. �����" � ��� ����� :���� 2=%&�������� ����
���� ��� ��� �&���� ��� � ��� �."��� ��"����";� !��
"��"���0��. � ��� ��"� ��� (� ������"�� (. �.���.-��� ���
����� (���� ��"����� =.���."�" ��#�" ��� �&���� ���
� ��� ��"������� ���� ����" � ��� ����� �0����(�� ��
������� $��� ��� C?� �������� !�� ����� ��,����" 0��.
"���� ,��������" � ��� ������

���� ���������� �� �������� ����������
����� ���� � ��� ��� �����������������
������ �� ���� ��� ����� � ���� ���� ��� �� �� �
�� ������ �� ������� ������ � ��!� ������������
���� ���� ���� ��� ���� "�� #���� ����� ������� ����
���� ���� ���� ��� �� �� �� ���� "�� � � �$ ������
�� %�������� �� ������&� ��� %�������� � ��%����
��� ����� �� %���� ��� �$ ��� �� �
�������� ������� '��� ������� � %���(�� �� ���
��������� ����!�%�� �� ���� ��� ��$������ ����� ��
��� ��$������ ������ $�� ��� %������ ����� �� � �� ��� �����
� �� �� %��%�� ��� "� � )������� *����� %������ �$ ��� ��
������ ���� ��� �� ����� �� ����� ��� $���� ��� (��
)����� )������� ����! ������������ ������� �� ��
+�� �! �$ %������ %�� �,� ��� ������������ ����! �����
%���
� �� ������ *� ���� ������ -����� �$ ��� %������
����� �� � �� ��� %%��%���� ����� �� ����� �������
����! ������������ ������ ��� ��!� �$ ��� ���������
���� �$ ��� )����� )�������
����� ' � ��� ����� � �� �� %��%�� ��� "� � )������� ��
��� )����� )�������
� �� ��� ��� ��������
����� �� � *� ���� ���� .� �/ ! �$ ����� ��� ��
����� �� �0� � ���� %�� ��� �������� �� %1 �$ �� �
*����� ���� ���� �� ���� �,� �� ���
����� ��� � �� �� *� ���� ���� �+� �! �$ ��
%���������� �� � 2 �, �$ �������� �� ��� �, �$
3���� 3����� �� ��� ��� � . �, �$ %����������� �+/�
���� ������ �� ��� "�� ������� � ���� � ���� ���%���
���� $�� � �� � / ���4
��� � �� �� "� 2 �, �$ )���4 ��� 5�!��� ��
�2 �, �$ +�����%�������� ���%�� � �� � /� ������
%���� �� � � "�� ��!��� � ���� $�� ��� �
���� �� ��0� � ��� �$ ��� )����� )������� �� ���
"� � )������� �� %1 ������� � � �� �� 2 6�� �� �, �$
��� "� � )������� �� ��� �$ ��� )����� )������� ����
��� �, �$ ��� 5�!���� �� ���� �� ��� � ���� ���%���
���� $�� �2 ������ ��� / �, �$ � 2 ���7, �����
�������� ")� �� ��� ' ��! ������ #����������
��������� ��� #���� ���� ����� ���� �$ ������� $��� ���
)����� )������� �� ��� "� � )������� � � ���������
������!�� �$ /�� �� �� � ��� ��� ������!�� �������
��� �� �22 �� 89���: "�� #���� ����� �$ � ���������
%������ � ��� ��� ���� ���� � ��������� ����� � ���
#���� ���� ����� �� ;
���������� "�� ������� ��% �$ #���� ����� �� %������
������������ � ����� ' ��! ��� ����� ��!�� ��� �������
%��� ��� #���� ���� ����� ���� �$ ��� ������� $��� ���
)����� )������� ��� � ��� %������ ������������ � ��
��������� ��� ����� ����� �� � (����! ��� %������ %����
<��� ��� ����� ����� � ������� �� ��� #���� ����
����� �� �$ ��� "� � )�������� ��������� ��� ������������
�$ %������ �� ��� �� � %������
� ���� �
"�� ������ � � �� �� �����!�� �� � ��� �$
%������ ������������ =����$������ �� �� � ��� %�� ����
�$ ����� �����!�����������! �� ���� �� ��� �� � %������
�� ���� ��� ������������ �$ %������ � ��� ������
9����!�� �� � ������-�� �� ��� ��� %������ ����� �� �
��� �� ��� ������� �� %������ %�� ������� �� � -����
�����������
���� �� � *�������� ��� �����!�� ������� �$ ��� %������
����� �� � �������� �� ������ �� ��� ������%���
>�������� �� ����������� � ������ $�� ��� ?0�����
�����!��
���� �� � >�������� �� ����������� � ������ $��
�����!�� �� � 6� � �����!�� �� � �� ������� � �
%��� � �� � � ����� ���� �$ ��� �%�� �� ��!�� �
���%������ %%������! �����>�� ����� %������ ������
����� �9�� �� ����� ����� �$ �����!�� �9��� $��� ���
�����!�� %�� ��� �� ��� �� � %������ )��� �� �������
������� ��� ������ ����� �� �����!�� ! � ����� � -����(��
���� ������ ����������� �������� ����� �� ������� ���
������ ����� �9�� ���� �&��� ��/� �� %������ ������� ������
!�� ������� �9�+ �� ����� ���� ��!�� ���� �� ��� �� ��
�� -����(�� ���� ����������� ����� �������� � %������
��$������ ����� �� ��$������ ������ ��� � ��������
%��� �� � ����� �� ���%� ����� �� ��� �� � %������ � � ��
�� �%����&� ��� ��0������ �� %��� � %������ �� ��
������ ��� � ���� �� ��� �� �
���������� "�� %������ ������������ � �������� �
�������! ��� �����!�� ������� �$ ��� �%�� � ��� 4����
�����!�� ������� �$ ��� %������ "�� 4���� �����!�� �������
�$ ��� %������ �� �� ���������� $��� ��� ������� ���%��
����� �$ ��� %������ �� � ���%�� �� ���� ��� �����!��
������� �$ ��� %%��%���� ��$������ ����� �� ��$������
������

18411841
INDEX
Page citations refer to the pages of the Supplement I, and to pages ofthe JP XIV main volume, including those where the text being revised inthe Supplement originally appeared. This Supplement II commenceswith page 1669 and succeeding Supplements will continue to be paged insequence.
1„1358 Main Volume of JP XIV1359„1668 Supplement I1669„1866 Supplement II
A
AbsorbentCotton, 851, 1757Cotton, Puriˆed, 851, 1757Cotton, Sterile, 852, 1757Cotton, Sterile, Puriˆed, 852, 1757Gauze, 853, 1757Gauze, Sterile, 854, 1757
Absorptive Ointment, 855Acacia, 855, 1543
Powdered, 855, 1543Acebutolol Hydrochloride, 219Aceglutamide Aluminum, 1377Acetaminophen, 219Acetazolamide, 220Acetic Acid, 856
Glacial, 856Acetohexamide, 221, 1687Acetylcholine Chloride for
Injection, 222Acetylkitasamycin, 223, 1378Acetylleucomycin, 223Acetylsalicylic Acid, 252
Tablets, 252Acetylspiramycin, 224, 1378Achyranthes Root, 857Aclarubicin Hydrochloride, 224, 1379Acrinol, 224
and Zinc Oxide Oil, 857and Zinc Oxide Ointment, 858
Actinomycin D, 225, 1381Adhesive Plaster, 858, 1757Adrenaline, 447
Hydrochloride Injection, 447Hydrochloride Solution, 448
AdsorbedDiphtheria-Puriˆed Pertussis-
Tetanus Combined Vaccine,1005
Diphtheria-Tetanus Combined
Toxoid, 913Diphtheria Toxoid for Adult Use,
912Habu-venom Toxoid, 938Hepatitis B Vaccine, 938Puriˆed Pertussis Vaccine, 1005Tetanus Toxoid, 1066
A‰oqualone, 225Agar, 859
Powdered, 859Ajmaline, 226
Tablets, 227Akebia Stem, 859Albumin Tannate, 227Alcohol, 914
Benzyl, 874Dehydrated, 914for Disinfection, 916Isopropyl, 556Stearyl, 1059
Aldioxa, 228Alimemazine Tartrate, 229Alisma Rhizome, 860
Powdered, 860Allopurinol, 229Aloe, 860, 1544
Powdered, 861, 1544, 1757Alprazolam, 230Alpinia O‹cinarum Rhizome, 1544Alprenolol Hydrochloride, 231Alprostadil, 1688
Alfadex, 231Alum, 862
Burnt, 863Solution, 861
AluminumAcetylsalicylate, 253Aspirin, 253Monostearate, 861Potassium Sulfate, 862Potassium Sulfate, Dried, 863Silicate, Natural, 233
Silicate, Synthetic, 235Sucrose Sulfate Ester, 766
Amantadine Hydrochloride, 235Ambenonium Chloride, 236Amidotrizoic Acid, 237Amikacin Sulfate, 238Aminoacetic Acid, 931Aminobenzylpenicillin, 246
Anhydrous, 246Sodium, 246
Aminophylline, 239Injection, 239
AmitriptylineHydrochloride, 240Hydrochloride Tablets, 240
Ammonia Water, 241Amobarbital, 242
Sodium for Injection, 242Amomum Seed, 863Amorphous Insulin Zinc Injection
(Aqueous Suspension), 542Amoxapine, 243Amoxicillin, 244, 1381Amphotericin B, 245Ampicillin, 246, 1382
Anhydrous, 246, 1384Ethoxycarbonyloxyethyl
Hydrochloride, 259Sodium, 246, 1385
AmpicillinphthalidylHydrochloride, 782
Amyl Nitrite, 246Anemarrhena Rhizome, 864Anesthamine, 465Anesthetic Ether, 462Angelica Dahurica Root, 864Anhydrous
Aminobenzylpenicillin, 246Ampicillin, 246CaŠeine, 294Citric Acid, 367, 1438, 1698Dibasic Calcium Phosphate, 879

18421842 Supplement II, JP XIVIndex
Lactose, 961Antiformin, Dental, 864Antipyrine, 247Apricot
Kernel, 864Kernel Water, 865
Arbekacin Sulfate, 247, 1386Areca, 866, 1545Arginine
Hydrochloride, 248Hydrochloride Injection, 249
L-ArginineHydrochloride, 248Hydrochloride Injection, 249
Aromatic Castor Oil, 889Arotinolol Hydrochloride, 249Arsenical Paste, 866Arsenic Trioxide, 250Arsenous Acid, 250Artemisia Capillaris Flower, 867Ascorbic Acid, 250
Injection, 251Powder, 251
Asiasarum Root, 867, 1545Asparagus Tuber, 1546Aspergillus Galactosidase, 921Aspirin, 252
Aluminum, 253Tablets, 252
Aspoxicillin, 254Astragalus Root, 867Astromicin Sulfate, 255, 1387Atractylodes
Lancea Rhizome, 869Rhizome, 868
AtropineSulfate, 255Sulfate Injection, 256, 1688
Azathioprine, 256Tablets, 256
Aztreonam, 258Azithromycin Hydrate, 1690
B
Bacampicillin Hydrochloride, 259,1389
Bacitracin, 1390Baclofen, 260
Tablets, 260Bamethan Sulfate, 262Barbital 263Barium Sulfate, 264Bark
Cinnamon, 896Cinnamon, Powdered, 897Magnolia, 970Magnolia, Powdered, 971Mallotus, 971Moutan, 979Moutan, Powdered, 980Mulberry, 981Phellodendron, 1007
Phellodendron, Powdered, 1008Bearberry Leaf, 870Bear Bile, 869Beclometasone Dipropionate, 264Beef Tallow, 870Beeswax
White, 871Yellow, 871
Bekanamycin Sulfate, 265, 1390Belladonna
Extract, 871Root, 872
Benidipine Hydrochloride, 1691Tablets, 1692
Benoxinate Hydrochloride, 661Benserazide Hydrochloride, 265Bentonite, 873Benzalkonium
Chloride, 266Chloride Solution, 266Chloride Solution 50,
Concentrated, 267Benzbromarone, 268, 1391Benzethonium
Chloride, 269Chloride Solution, 269
Benzocaine, 465Benzoic Acid, 270Benzoin, 873Benzyl
Alcohol, 874, 1758Benzoate, 874
BenzylpenicillinBenzathine, 1392, 1694Potassium, 270, 1393
BerberineChloride, 271Tannate, 272
Betahistine Mesilate, 273Betamethasone, 274
Dipropionate, 275Sodium Phosphate, 276, 1395Valerate, 276, 1395
Bethanechol Chloride, 278Bifonazole, 278Biperiden Hydrochloride, 279Bisacodyl, 280
Suppositories, 280, 1396Bismuth
Subgallate, 281Subnitrate, 282
BitterCardamon, 875Orange Peel, 875Tincture, 875
BleomycinHydrochloride, 283, 1396Sulfate, 283, 1398
Blood, Whole Human, 1080Boric Acid, 284Botulism Antitoxin, Equine, Freeze-
dried, 876Bromazepam, 284
Bromhexine Hydrochloride, 285Bromocriptine Mesilate, 286Bromovalerylurea, 287Bucumolol Hydrochloride, 287Bufetolol Hydrochloride, 288Bufexamac, 289
Cream, 289Ointment, 289
Bumetanide, 291Bunazosin Hydrochloride, 291Burdock Fruit, 1546Bupleurum Root, 876Bupranolol Hydrochloride, 292Burnt Alum, 863Busulfan, 293Butropium Bromide, 294Butyl Parahydroxybenzoate, 876Butyrate, Ribo‰avin, 734
C
Cacao Butter, 877CaŠeine, 294
and Sodium Benzoate, 296Anhydrous, 294
Calciferol, 449Calcium
Carbonate, Precipitated, 297Carboxymethylcellulose, 885Carmellose, 885Chloride, 298Chloride Injection, 298, 1399CMC, 885Folinate, 299Fosfomycin, 494Gluconate, 299Hydroxide, 877Lactate, 300Leucovorin, 299Oxide, 878Pantothenate, 301Para-aminosalicylate, 301Para-aminosalicylate Granules,
302Polystyrene Sulfonate, 303, 1399Stearate, 881
Calumba, 881Powdered, 881
Camellia Oil, 882Camostat Mesilate, 304Camphor, Synthetic, 306d-Camphor, 305, 1400dl-Camphor, 306, 1400Capsicum, 882, 1546
and Salicylic Acid Spirit, 884Powdered, 883, 1546Tincture, 883
Capsules, 885Cloˆbrate, 373Flurazepam, 488Indometacin, 535Sodium Iodide (123I), 755Sodium Iodide (131I), 755

18431843Supplement II, JP XIV Index
Sodium Iopodate, 757Vitamin A, 1075Vitamin A Oil, 1074
Captopril, 307Carbamazepine, 308Carbazochrome Sodium Sulfonate,
308, 1400Carbetapentane Citrate, 674Carbetaoebtene Citrete, 674Carbidopa, 309, 1401L-Carbocisteine, 310Carbolic Acid, 1010
for Disinfection, 1010Liqueˆed, 1011
Carbon Dioxide, 312Carboxymethylcellulose, 885
Calcium, 886Sodium, 887
Cardamon, 885Carmellose, 885
Calcium, 886, 1759Sodium, 887
Carmofur, 313Carnauba Wax, 888Carteolol Hydrochloride, 313Carumonam Sodium, 314, 1401Cassia Seed, 888Castor Oil, 888Catalpa Fruit, 889Cefaclor, 314, 1403Cefadroxil, 314Cefalexin, 315Cefaloridine, 317, 1405Cefalotin Sodium, 317, 1406Cefamandole Sodium, 317, 1407Cefapirin Sodium, 317Cefatrizine Propylene Glycolate, 318Cefazolin
Sodium, 319Sodium Hydrate, 321
Cefbuperazone Sodium, 322, 1409Cefcapene Pivoxil Hydrochloride,
322Cefdinir, 324Cefditoren Pivoxil, 325Cefepime Dihydrochloride, 326
for Injection, 1695Cefetamet Pivoxil Hydrochloride,
328Ceˆxime, 329Cefmenoxime Hydrochloride, 330,
1410Cefmetazole Sodium, 321Cefminox Sodium, 332Cefodizime Sodium, 1412Cefoperazone Sodium, 332Cefoselis Sulfate, 334Cefotaxime Sodium, 335, 1413Cefotetan, 335, 1415Cefotiam
Hexetil Hydrochloride, 335, 1417Hydrochloride, 335
Cefoxitin Sodium, 336, 1419
Cefozopran Hydrochloride, 337Cefpiramide Sodium, 338, 1421Cefpirome Sulfate, 338Cefpodoxime Proxetil, 1422Cefradine, 339Cefroxadine, 340, 1424Cefsulodin Sodium, 340Ceftazidime, 342Cefteram Pivoxil, 344, 1425Ceftibuten, 344, 1426Ceftizoxime Sodium, 345Ceftriaxone Sodium, 346, 1427Cefuroxime Axetil, 347, 1429, 1696Cefuroxime Sodium, 348, 1696Cellulose
Acetate Phthalate, 893, 1760Microcrystalline, 889Powdered, 892
Cetanol, 894Cetraxate Hydrochloride, 349, 1431Chloral Hydrate, 350Chloramphenicol, 351, 1432
Palmitate, 1432Sodium Succinate, 1434
Chlordiazepoxide, 351Powder, 351, 1434Tablets, 352, 1435
ChlorhexidineGluconate Solution, 353Hydrochloride, 354
Chlorinated Lime, 894Chlormadinone Acetate, 355, 1435Chlorobutanol, 894Chlorphenesin Carbamate, 356, 1436Chlorpheniramine
and Calcium Powder, 895Maleate, 357Maleate Injection, 357Maleate Powder, 358Maleate Tablets, 359
d-Chlorpheniramine Maleate, 359Chlorpromazine
Hydrochloride, 360Hydrochloride Injection, 361Hydrochloride Tablets, 361
Chlorpropamide, 362Tablets, 362
Cholecalciferol, 363Cholera Vaccine, 896Cholesterol, 896Chorionic
Gonadotrophin, 934, 1764Gonadotrophin for Injection, 936,
1764Chrysanthemum Flower, 1547, 1760Ciclacillin, 364, 1437Ciclosporin, 364, 1697
A, 364Cimetidine, 365Cimicifuga Rhizome, 896Cinchocaine Hydrochloride, 404Cinnamon
Bark, 896, 1547
Oil, 897Powdered, 1547
Cisplatin, 1697Citric Acid, 366, 1437
Anhydrous, 367, 1438, 1698Citrus Unshiu Peel, 898Clarithromycin, 367, 1699Clemastine Fumarate, 368Clematis Root, 1547Clindamycin
Hydrochloride, 1439Phosphate, 369, 1440
Clinoˆbrate, 369Clocapramine Hydrochloride, 370Clofedanol Hydrochloride, 371Cloˆbrate, 372
Capsules, 373Clomifene
Citrate, 373, 1441Citrate Tablets, 374
Clomipramine Hydrochloride, 375Clonazepam, 375Clonidine Hydrochloride, 376Cloperastine Hydrochloride, 377Clotiazepam, 378Clotrimazole, 379Clove, 898
Oil, 898Powdered, 898
Cloxacillin Sodium, 380, 1441Cloxazolam, 380CMC, 885
Calcium, 886Sodium, 887
CnidiumMonnieri Fruit, 1760Rhizome, 899
Cocaine Hydrochloride, 381Coconut Oil, 899Codeine
Phosphate, 382Phosphate Powder, 1, 382, 1442Phosphate Powder, 10, 383, 1442Phosphate Tablets, 384, 1443
Cod Liver Oil, 900, 1548Coix Seed, 900Colchicine, 384, 1699Colistin
Sodium Methanesulfonate, 385Sulfate, 1444
Colophonium, 1029Compound
Acrinol and Zinc Oxide Oil, 857Diastase and Sodium Bicarbonate
Powder, 907Hycodenone Injection, 995Iodine Glycerin, 950Methyl Salicylate Spirit, 977Oxycodone and Atropine
Injection, 996Oxycodone Injection, 995Phellodendron Powder for
Cataplasm, 1009, 1556

18441844 Supplement II, JP XIVIndex
Rhubarb and Senna Powder, 1028Salicylic Acid Spirit, 1033Scopolia Extract and Diastase
Powder, 1037Scopolia Extract and Tannic Acid
Ointment, 1039, 1560Scopolia Extract and Tannic Acid
Suppositories, 1041, 1560Thianthol and Salicylic Acid
Solution, 1067Vitamin B Powder, 1075
ConcentratedGlycerin, 504Glycerol, 504
Condurango, 900Fluidextract, 901
Coptis Rhizome, 901, 1548Powdered, 1548
CornOil, 903Starch, 903, 1761
Cornus Fruit, 903, 1762Cortisone
Acetate, 386Corydalis Tuber, 904Cotton, Absorbent, 851Cream, Bufexamac, 289Creosote, 904Cresol, 905
Solution, 905Croconazole Hydrochloride, 387Crude Glycyrrhiza Extract, 934Crystalline
Insulin Zinc Injection (AqueousSuspension), 543
Penicillin G Potassium, 270Crystal Violet, 617Cyanamide, 387Cyanocobalamin, 388
Injection, 389Cyclopentolate Hydrochloride, 389Cyclophosphamide, 390Cycloserine, 390Cyperus Rhizome, 906Cyproheptadine Hydrochloride, 391Cytarabine, 392, 1701
D
Dantrolene Sodium, 392Daunorubicin Hydrochloride, 1445Deferoxamine Mesilate, 393, 1701Dehydrated
Alcohol, 915Ethanol, 915
Dehydrocholate Sodium Injection,396
Dehydrocholic Acid, 394Injection, 396Puriˆed, 394
DemethylchlortetracyclineHydrochloride, 1446
Dental
Antiformin, 864Iodine Glycerin, 951Paraformaldehyde Paste, 1001Phenol with Camphor, 1011Sodium Hypochlorite Solution, 864Triozinc Paste, 1072
Dermatol, 281Deslanoside, 396
Injection, 397Dexamethasone, 397Dextran 906
40, 39840 Injection, 39870, 400Sulfate Sodium Sulfur 5, 401Sulfate Sodium Sulfur 18, 402
Dextrin, 906Dextromethorphan Hydrobromide,
402Diagnostic Sodium Citrate Solution,
754Diastase, 907
and Sodium Bicarbonate Powder,907
Diazepam, 403Dibasic
Calcium Phosphate, 878Sodium Phosphate, 907
Dibekacin Sulfate, 404, 1447Dibucaine Hydrochloride, 404Dichlorphenamide, 406
Tablets, 407Diclofenac Sodium, 405Diclofenamide, 406, 1448
Tablets, 407Dicloxacillin Sodium, 408Diethylcarbamazine
Citrate, 408Citrate Tablets, 409, 1449
DiethylstilbestrolDiphosphate, 493Diphosphate Tablets, 494
Difenidol Hydrochloride, 409Digenea, 908Digitalis, 908, 1762
Powdered, 910, 1762Digitoxin, 410
Tablets, 411Digoxin, 414, 1702
Injection, 415, 1703Tablets, 416, 1704
DihydrocodeinePhosphate, 417Phosphate Powder, 1, 417, 1449Phosphate Powder, 10, 418, 1450
Dihydroergotamine Mesilate, 418,1450
Dihydroergotoxine Mesilate, 412Dihydroxyaluminum Allantoinate,
228Dilazep Hydrochloride, 419Diltiazem Hydrochloride, 420, 1451Dilute
Hydrochloric Acid, 515Iodine Tincture, 950
Diluted Opium Powder, 987Dimemorfan Phosphate, 421Dimenhydrinate, 422Dimenhydrinate Tablets, 423Dimercaprol, 423
Injection, 424Dimorpholamine, 424, 1705
Injection, 425, 1705Dinoprost, 425Dionin, 467Dioscorea Rhizome, 911Diphenhydramine, 426
and Bromovalerylurea Powder,911
Hydrochloride, 427Phenol and Zinc Oxide Liniment,
912Tannate, 427
Diphenylhydantoin, 685Powder, 686Sodium for Injection, 686Tablets, 686
DiphtheriaAntitoxin, Equine, Freeze-dried,
912Toxoid, 912
Diphtheria-Tetanus CombinedToxoid, 912
Dipyridamole, 428, 1451Disodium
Edetate, 913Ethylenediaminetetraacetate, 913
Disopyramide, 429Distigmine
Bromide, 429Bromide Tablets, 430
Disulˆram, 430Dobutamine Hydrochloride, 431,
1452Dopamine
Hydrochloride, 432Hydrochloride Injection, 433, 1452
Dover's Powder, 993Doxapram Hydrochloride, 433Doxorubicin Hydrochloride, 434,
1453Doxycycline Hydrochloride, 434,
1454Dried
Aluminum Hydroxide Gel, 233Aluminum Hydroxide Gel Fine
Granules, 233Aluminum Potassium Sulfate, 863Sodium Carbonate, 1055Sodium Sulˆte, 1058Thyroid, 1069Yeast, 1083
DromostanolonePropionate, 435Propionate Injection, 436
Droperidol, 435, 1456

18451845Supplement II, JP XIV Index
DrostanolonePropionate, 435, 1456Propionate Injection, 436, 1456
Dydrogesterone, 437Tablets, 438
E
Ecarazine Hydrochloride, 811Ecothiopate Iodide, 438Edrophonium
Chloride, 439Chloride Injection, 440
EDTA Sodium, 913Elcatonin, 440En‰urane, 443, 1456Enoxacin, 444Enviomycin Sulfate, 1457Eperisone Hydrochloride, 1706Ephedra Herb, 914Ephedrine
Hydrochloride, 444Hydrochloride Injection, 445Hydrochloride Powder, 10z, 446Hydrochloride Tablets, 446
Epimedium Herb 1762Epinephrine, 447
Hydrochloride Injection, 447Hydrochloride Solution, 448Injection, 447Solution, 448
Epirenamine, 447Hydrochloride Injection, 447Hydrochloride Solution, 448
Epirizole, 448Epirubicin Hydrochloride, 1458Ergocalciferol, 449, 1460Ergometrine
Maleate, 450Maleate Injection, 451Maleate Tablets, 451
Ergotamine Tartrate, 452Erythromycin, 453, 1460
Ethylsuccinate, 453Lactobionate, 1462Stearate, 454
Estazolam, 454Estradiol
Benzoate, 455, 1462Benzoate Injection, 456Benzoate Injection (Aqueous
Suspension), 456Estriol, 457, 1463
Injection (Aqueous Suspension),458
Tablets, 458Etacrynic Acid, 459
Tablets, 460Ethacridine Lactate, 224Ethambutol Hydrochloride, 460Ethanol, 914
Dehydrated, 915for Disinfection, 916
Ethenzamide, 461Ether, 462
Anesthetic, 462Guaiacol Glyceryl, 505Polyoxyethylene Lauryl Alcohol,
964Ethinylestradiol, 463
Tablets, 463Ethionamide, 464, 1707Ethosuximide, 465Ethoxybenzamide, 461Ethyl
Aminobenzoate, 465Cysteine Hydrochloride, 466L-Cysteine Hydrochloride, 466Parahydroxybenzoate, 916
Ethylenediamine, 917Ethylmorphine Hydrochloride, 467Etilefrine
Hydrochloride, 468Hydrochloride Tablets, 468, 1708
Etizolam, 1463Etoposide, 1709Eucalyptus Oil, 917Eucommia Bark, 1548Evodia Fruit, 918, 1548Exsiccated Gypsum, 938Extract
Belladonna, 871Crude Glycyrrhiza, 934Glycyrrhiza, 933Nux Vomica, 984
F
Famotidine, 469for Injection, 469, 1464Powder, 469Tablets, 469
Faropenem Sodium, 472Fenbufen, 473Fennel, 918
Oil, 919Powdered, 918
Fentanyl Citrate, 473Ferrous Sulfate, 474Fine Granules, Aluminum Hydroxide
Gel, Dried, 233Flavin Adenine Dinucleotide
Sodium, 475, 1710Flavoxate Hydrochloride, 476Floctafenine, 477, 1465Flomoxef Sodium, 478, 1465
for Injection, 1711Flopropione, 478
Capsules, 1712Flower, Artemisia Capillaris, 867Flucytosine, 478Fludiazepam, 479Flunitrazepam, 480Fluocinolone Acetonide, 481Fluocinonide, 482Fluorescein Sodium, 483
Fluorometholone, 484Fluorouracil, 485Fluoxymesterone, 486Fluphenazine Enanthate, 487Flurazepam, 487
Capsules, 488Hydrochloride, 489
Flurbiprofen, 489, 1466Foeniculated Ammonia Spirit, 919Folic Acid, 490, 1466
Injection, 491Tablets, 491
Formalin, 919Water, 920
Formoterol Fumarate, 492Forsythia Fruit, 920Fosfestrol, 493
Tablets, 494Fosfomycin
Calcium, 494, 1713Sodium, 496, 1713
Fradiomycin Sulfate, 497, 1467Freeze-dried
BCG Vaccine (for PercutaneousUse), 869
Botulism Antitoxin, Equine, 876Diphtheria Antitoxin, Equine, 912Habu Antivenom, Equine, 938Inactivated Tissue Culture Rabies
Vaccine, 1024Japanese Encephalitis Vaccine,
957Live Attenuated Measles Vaccine,
972Live Attenuated Mumps Vaccine,
981Live Attenuated Rubella Vaccine,
1029Mamushi Antivenom, Equine, 972Smallpox Vaccine, 1053Smallpox Vaccine Prepared in Cell
Culture, 1053Tetanus Antitoxin, Equine, 1066
Fritillaria Bulb, 1549Fructose, 497
Injection, 498Fruit
Catalpa, 889Cornus, 903Evodia, 918Forsythia, 920Gardenia, 922Gardenia, Powdered, 923Rose, 1029Rose, Powdered, 1029Schisandra, 1035Zanthoxylum, 1083Zanthoxylum, Powdered, 1084
Furosemide, 498, 1714Tablets, 1715
Fursultiamine Hydrochloride, 499

18461846 Supplement II, JP XIVIndex
G
Gabexate Mesilate, 500b-Galactosidase (Aspergillus), 921b-Galactosidase (Penicillium), 921Gallium (67Ga) Citrate Injection, 501Gambir, 920
Powdered, 920Gardenia Fruit, 922, 1549
Powdered 1763Gas Gangrene Antitoxin, Equine, 923Gastrodia Tuber, 1550Gauze, Absorbent, 853Gel, Aluminum Hydroxide, Dried,
233Gelatin, 923
Puriˆed, 924Gentamicin Sulfate, 502, 1468Gentian, 925
and Sodium Bicarbonate Powder,926
Japanese, 957Powdered, 925Powdered, Japanese, 957
Geranium Herb, 926Ginger, 927, 1763, 1764
Powdered, 927Ginseng, 927
Powdered, 929Glacial Acetic Acid, 856Glehnia Root, 930Glibenclamide, 502Glucose, 503
Injection, 503Glutathione, 1716Glycerin, 504, 1469
and Potash Solution, 930Concentrated, 504, 1469
Glycerol, 504Concentrated, 504
Glyceryl Monostearate, 931Glycine, 931Glycyrrhiza, 932, 1550
Extract, 933Powdered, 933, 1550
Gramicidin, 1470Gonadotrophin
Chorionic, 934for Injection, Chorionic, 936for Injection, Serum, 937Serum, 936
Gramicidin 1470Granules
Calcium Para-aminosalicylate, 302Pas-calcium, 302
Griseofulvin, 505, 1471Guaiacol Glyceryl Ether, 505Guaifenesin, 505Guanabenz Acetate, 506Guanethidine Sulfate, 507Gypsum, 937
Exsiccated, 938
H
Habu Antivenom, Equine, Freeze-dried, 938
Haloperidol, 507Halothane, 508Haloxazolam, 509, 1472Hemp Fruit, 1551Heparin
Sodium, 510, 1717Sodium Injection, 511, 1718
HerbEphedra, 914Geranium, 926Houttuynia, 939Mentha, 974Perilla, 1004Plantago, 1014Powdered Geranium, 926Powdered Swertia, 1064Swertia, 1063
Homatropine Hydrobromide, 511Homochlorcyclizine Hydrochloride,
512, 1472Honey, 938Houttuynia Herb, 939Human Normal Immunoglobulin,
949Hycoato Injection, 996Hydralazine
Hydrochloride, 513Hydrochloride for Injection, 513Hydrochloride Powder, 513Hydrochloride Tablets, 514
Hydrochloric Acid, 514Dilute, 515Lemonade, 939
Hydrochlorothiazide, 515, 1473Hydrocortisone, 516
Acetate, 517, 1474and Diphenhydramine Ointment,
939Butyrate, 518Sodium Phosphate, 519, 1474Sodium Succinate, 520Succinate, 521
Hydrocotarnine Hydrochloride, 522Hydrogenated Oil, 940Hydrophilic
Ointment, 940Petrolatum, 1005
Hydrous Lanolin, 962Hydroxocobalamin Acetate, 523Hydroxypropylcellulose, 940
Low Substituted, 942Hydroxypropylmethylcellulose
Phthalate, 9472208, 9432906, 9442910, 945
HydroxyzineHydrochloride, 524Pamoate, 524
Hymecromone, 525
I
Ibuprofen, 526Ichthammol, 948Idarubicin Hydrochloride, 527, 1475Idoxuridine, 527
Ophthalmic Solution, 528, 1475Ifenprodil Tartrate, 529Imipenem, 530, 1476Imipramine
Hydrochloride, 530Hydrochloride Tablets, 531
Immature Orange, 948Immunoglobulin, Human Normal,
949Imperata Rhizome, 949, 1551Indenolol Hydrochloride, 532Indigocarmine, 533
Injection, 533Indium (111In) Chloride Injection, 534Indometacin, 534
Capsules, 535Suppositories, 536, 1477
In‰uenza HA Vaccine, 949Injection
Acetylcholine Chloride for, 222(Aqueous Suspension), Estradiol
Benzoate, 456(Aqueous Suspension), Estriol,
458(Aqueous Suspension), Insulin Zinc,
542(Aqueous Suspension), Insulin Zinc,
Amorphous, 542(Aqueous Suspension), Insulin Zinc,
Crystalline, 543(Aqueous Suspension), Insulin Zinc,
Protamine, 544(Aqueous Suspension), Isophane
Insulin, 541Adrenaline Hydrochloride, 447Aminophylline, 239Amobarbital Sodium for, 242Arginine Hydrochloride, 249L-Arginine Hydrochloride, 249Ascorbic Acid, 251Atropine Sulfate, 256Calcium Chloride, 298Chlorpheniramine Maleate, 357Chlorpromazine Hydrochloride,
361Cyanocobalamin, 389Dehydrocholate Sodium, 396Dehydrocholic Acid, 396Deslanoside, 397Dextran 40, 399Digoxin, 415Dimercaprol, 424Dimorpholamine, 425Diphenylhydantoin Sodium for,
686

18471847Supplement II, JP XIV Index
Dromostanolone Propionate, 436Drostanolone Propionate, 436Edrophonium Chloride, 440Ephedrine Hydrochloride, 445Epinephrine, 447Epinephrine Hydrochloride, 447Epirenamine Hydrochloride, 447Ergometrine Maleate, 451Estradiol Benzoate, 456Folic Acid, 491Fructose, 498Gallium (67Ga) Citrate, 501Glucose, 503Heparin Sodium, 511Human Serum Albumin, Iodinated
(131I), 546Hycoato, 996Hycodenone, Compound, 995Hydralazine Hydrochloride for,
513Indigocarmine, 533Indium (111In) Chloride, 534Insulin, 539Isoniazid, 554Levallorphan Tartrate, 570Lidocaine, 575Lidocaine Hydrochloride, 575Magnesium Sulfate, 586D-Mannite, 588D-Mannitol, 588Meglumine Amidotrizoate, 594Meglumine Iotalamate, 595Meglumine Sodium
Amidotrizote, 596Meglumine Sodium Iodamide, 597Metenolone Enanthate, 608Morphine and Atropine, 978Morphine Hydrochloride, 630Neostigmine Methylsulfate, 638Nicotinic Acid, 645Noradrenaline Hydrochloride, 650Norepinephrine, 650Norepirenamine Hydrochloride,
650Operidine, 679Opium Alkaloids and Atropine,
990Opium Alkaloids and
Scopolamine, 991Opium Alkaloids Hydrochlorides,
989Oxycodone and Atropine,
Compound, 996Oxycodone, Compound, 995Oxytocin, 665Paoaverine Hydrochloride, 671Pethidine Hydrochloride, 679Phenolsulfonphthalein, 682Phenytoin Sodium for, 686Prednisolone Sodium Succinate for,
705Procainamide Hydrochloride, 709Procaine Hydrochloride, 710
Progesterone, 714Protamine Sulfate, 719Pyridoxine Hydrochloride, 725Reserpine, 730Ribo‰avin Phosphate, 736Ribo‰avin Sodium Phosphate,
736Sodium Bicarbonate, 750Sodium Chloride, Isotonic, 752Sodium Chloride, 0.9, 752Sodium Chloride, 10, 752Sodium Chromate (51Cr), 752Sodium Iodohippurate (131I), 756Sodium Pertechnetate (99mTc), 758Sulfobromophthalein Sodium, 775Sulpyrine, 777Suxamethonium Chloride, 781Suxamethonium Chloride for, 781Testosterone Enanthate, 787Thallium (201Tl) Chloride, 791Thiamine Hydrochloride, 794Thiamylal Sodium for, 796Thiopental Sodium for, 798Tubocurarine Chloride, 831Tubocurarine Hydrochloride, 831Vasopressin, 837Vinblastine Sulfate for, 841Vitamin B12, 389Vitamin B1 Hydrochloride, 794Vitamin B2 Phosphate Ester, 736Vitamin B6, 725Vitamin C, 251Weak Opium Alkaloids and
Scopolamine, 992Xylitol, 845
Insulin, 536Insulin Human (GeneticalRecombination), 537
Injection, 539Zinc Injection (Aqueous
Suspension), 542Zinc Injection (Aqueous
Suspension), Amorphous, 542Zinc Injection (Aqueous
Suspension), Crystalline, 543Zinc Protamine Injection (Aqueous
Suspension), 544Iodamide, 545Iodinated (131I) Human Serum
Albumin Injection, 546Iodine, 546
Glycerin, Compound, 950Glycerin, Dental, 951Tincture, 949Tincture, Dilute, 950Salicylic Acid and Phenol Spirit,
952Iopamidol, 547Iopanoic Acid, 548, 1478
Tablets, 548, 1478Iotalamic Acid, 549Iotroxic Acid, 550Ipecac, 953
Powdered, 954Syrup, 955
Ipratropium Bromide, 550Iproveratril Hydrochloride, 839Isepamicin Sulfate, 552L-Isoleucine, 553Isoniazid, 553
Injection, 554, 1478Tablets, 554, 1478
Isophane Insulin Injection (AqueousSuspension), 541
l-Isoprenaline Hydrochloride, 555Isopropanol, 556Isopropyl Alcohol, 556Isopropylantipyrine, 556l-Isoproterenol Hydrochloride, 555Isosorbide, 557
Dinitrate, 558Dinitrate Tablets, 558
IsotonicSalt Solution, 752Sodium Chloride Injection, 752Sodium Chloride Solution, 752
J
JapaneseAngelica Root, 956Encephalitis Vaccine, 956Encephalitis Vaccine, Freeze-dried,
957Gentian, 957,Gentian, Powdered, 1551Valerian, 957
Josamycin, 559, 1479Propionate, 559, 1480
Jujube, 958Seed, 1551
K
Kainic Acid, 560and Santonin Powder, 958
Kallidinogenase, 560, 1718Kanamycin
Monosulfate, 1481Sulfate, 563, 1482
Kaolin, 959Kernel
Apricot, 864Peach, 1002Peach, Powdered, 1002
Ketamine Hydrochloride, 563Ketoprofen, 564Ketotifen Fumarate, 1483Kitasamycin, 565, 1484
Tartrate, 1485
L
Lactic Acid, 959Lactose, 960
Anhydrous, 961

18481848 Supplement II, JP XIVIndex
Lactulose, 566Lanatoside
C, 567C Tablets, 567
LanolinHydrous, 962Puriˆed, 963
Lard, 964Latamoxef Sodium, 569, 1486Lauromacrogol, 964Leaf
Bearberry, 870Senna, Powdered, 1046Sweet Hydrangea, Powdered, 1063Senna, 1044Sweet Hydrangea, 1063
Lenampicillin Hydrochloride, 1488L-Leucine, 569Leucomycin, 565Levallorphan
Tartrate, 570Tartrate Injection, 570
Levodopa, 571Levomepromazine Maleate, 572Levothyroxine
Sodium, 572Sodium Tablets, 573
Lidocaine, 574Hydrochloride Injection, 575Injection, 575
LightAnhydrous Silicic Acid, 1050Liquid Para‹n, 1001
LimeChlorinated, 894Quick, 878Slaked, 877
Lincomycin Hydrochloride, 576, 1490Lindera Root 1766Liothyronine
Sodium, 576Sodium Tablets, 577, 1490
LiqueˆedCarbolic Acid, 1011Phenol, 1011
Liquid Para‹n, 1000Lithium Carbonate, 578Lithospermum Root, 964Live
Attenuated Measles Vaccine, Freeze-dried, 972
Attenuated Mumps Vaccine, Freeze-dried, 981
Attenuated Rubella Vaccine, Freeze-dried, 1029
Oral Poliomyelitis Vaccine, 1015Longgu, 965Lonicera Leaf and Stem 1766Loquat Leaf, 1552Lorazepam, 579Low Substituted
Hydroxypropylcellulose, 942Loxoprofen Sodium, 580
LyciumBark, 1767Fruit, 1767
Lysine Hydrochloride, 581Lysozyme Hydrochloride, 1491L-Lysine Hydrochloride, 581
M
MacrogolOintment, 968400, 9651500, 9664000, 9676000, 96720000, 968
MagnesiumCarbonate, 582Oxide, 583Silicate, 584Stearate, 969Sulfate, 585Sulfate Injection, 586, 1492Sulfate Mixture, 970
Magnolia Bark, 970, 1552Flower, 1552Powdered, 1552
Mallotus Bark, 971Maltose, 586Mamushi Antivenom, Equine, Freeze-
dried, 972D-Mannite, 587
Injection, 588D-Mannitol, 587, 1718
Injection, 588, 1492Maprotiline Hydrochloride, 588Meclofenoxate Hydrochloride, 589Mecobalamin, 590Medazepam, 591Medicinal
Carbon, 972Soap, 973
Mefenamic Acid, 592Mefruside, 592
Tablets, 593Meglumine, 974
Amidotrizoate Injection, 594Iotalamate Injection, 595Sodium Amidotrizoate Injection,
596Sodium Iodamide Injection, 597
Melphalan, 597Menatetrenone, 598, 1492Mentha Herb, 974
Oil, 975Water, 975
dl-Menthol, 975l-Menthol, 976Mepenzolate Bromide, 599Mepirizole, 448Mepitiostane, 600, 1492Mepivacaine
Hydrochloride, 601, 1719
Hydrochloride Injection, 602Mequitazine, 602Merbromin, 604
Solution, 605Mercaptopurine, 603Mercurochrome, 604
Solution, 605Meropenem Trihydrate, 605, 1719Mestranol, 606Metenolone
Acetate, 607Enanthate, 607Enanthate Injection, 608
Methamphetamine Hydrochloride,609
L-Methionine, 609Methotrexate, 610, 1493Methoxsalen, 611Methylbenactyzium Bromide, 611Methylcellulose, 978Methylchlorophenylisoxazolylpenicil-
lin Sodium, 380Methyldichlorophenylisoxazolylpenicil-
lin Sodium, 408Methyldopa, 612
Tablets, 613dl-Methylephedrine
Hydrochloride, 613, 1719Hydrochloride Powder, 614Hydrochloride Powder, 10, 614,
1720Methylergometrine
Maleate, 615Maleate Tablets, 615
MethylParahydroxybenzoate, 976Salicylate, 618
Methylprednisolone, 616Succinate, 1720
Methylrosanilinium Chloride, 617Methyltestosterone, 618, 1722
Tablets, 619, 1722Meticrane, 619, 1493Metildigoxin, 620Metoclopramide, 621
Tablets, 1723Metronidazole, 622Metyrapone, 623Mexiletine Hydrochloride, 623Miconazole, 624
Nitrate, 625Microcrystalline Cellulose, 889Micronomicin Sulfate, 626, 1494Midecamycin, 626
Acetate, 627Migrenin, 627, 1495Minocycline Hydrochloride, 628,
1495Mitomycin C, 629, 1496Mixture, Magnesium Sulfate, 970Monobasic Calcium Phosphate, 880Monosodium
Trichloroethyl Phosphate, 823

18491849Supplement II, JP XIV Index
Trichloroethyl Phosphate Syrup,824
Morphineand Atropine Injection, 978, 1553Hydrochloride, 629, 1497Hydrochloride Injection, 630, 1497Hydrochloride Tablets, 631, 1498
Moutan Bark, 979Mulberry Bark, 980, 1554Mupirocin Calcium Hydrate, 631
N
Nadolol, 633Nalidixic Acid, 634Naloxone Hydrochloride, 634Naphazoline
and Chlorpheniramine Solution,981
Hydrochloride, 635Nitrate, 636
Naproxen, 636Narcotine, 654
Hydrochloride, 655Natural Aluminum Silicate, 233Neomycin Sulfate, 497Neostigmine
Methylsulfate, 637Methylsulfate Injection, 638
Netilmicin Sulfate, 638Nicardipine Hydrochloride, 639
Hydrochloride Injection, 640Niceritrol, 641Nicomol, 642
Tablets, 643Nicorandil 1724Nicotinamide, 643Nicotinic Acid, 644
Injection, 645Nifedipine, 645Nilvadipine, 1725
Tablets, 1726Nitrazepam, 646Nitrogen, 982Nitroglycerin Tablets, 647Nitrous Oxide, 648Noradrenaline, 649
Hydrochloride, 650Hydrochloride Injection,
Norepinephrine, 649, 1498Injection, 650, 1499
Norepirenamine, 649Hydrochloride Injection, 650
Norethisterone, 650Nor‰oxacin, 651Norgestrel, 652
and Ethinylestradiol Tablets, 652,1499
Nortriptyline Hydrochloride, 654Noscapine, 654
Hydrochloride, 655Notopterygium Rhizome, 1554Nuphar Rhizome, 983
NuxVomica, 983Vomica Extract, 984Vomica Extract Powder, 984Vomica Tincture, 985
Nystatin, 656
O
O‰oxacin, 1500Oil
Acrinol and Zinc Oxide, 857Acrinol and Zinc Oxide,
Compound, 857Aromatic Castor,Camellia, 889Castor, 888Cinnamon, 897Clove, 898Coconut, 899Cod Liver, 900Corn, 903Eucalyptus, 917Fennel, 919Hydrogenated, 940Mentha, 975Olive, 986Orange, 994Peanut, 1002Rape Seed, 1024Sesame, 1048Soybean, 1059Turpentine, 1073Vitamin A, 1074Zinc Oxide, 1085
OintmentAbsorptive, 855Acrinol and Zinc Oxide, 858Bufexamac, 290Hydrocortisone and
Diphenhydramine, 939Hydrophilic, 940Macrogol, 968Polyethylene Glycol, 968Scopolia Extract and Tannic Acid
Compound, 1039Simple, 1052Sulfur, Salicylic Acid and
Thianthol, 1062White, 1080Zinc Oxide, 1085
Olive Oil, 986Operidine, 678
Injection, 679Ophiopogon Tuber, 987Opium
Alkaloids and Atropine Injection,990
Alkaloids and ScopolamineInjection, 991
Alkaloids Hydrochlorides, 988,1555, 1767
Alkaloids Hydrochlorides
Injection, 989Ipecac Powder, 993Powdered, 987Tincture, 988
OrangeImmature, 948Oil, 994Peel Syrup, 994Peel Tincture, 995
Orciprenaline Sulfate, 656Oriental Bezoar, 995Oxapium Iodide, 657Oxaprozin, 658Oxazolam, 658Oxetacaine, 659Oxethazaine, 659Oxprenolol Hydrochloride, 660Oxybuprocaine Hydrochloride, 661Oxycodone Hydrochloride, 662Oxydol, 662Oxygen, 663Oxymetholone, 664Oxytetracycline Hydrochloride, 665,
1501Oxytocin, 1728
Injection, 665, 1730Oyster Shell, 997
P
Panax Rhizome, 998Powdered, 1555
Pancreatin, 999Pancuronium Bromide, 667Panipenem, 667Pantethine, 669Papaverine
Hydrochloride, 670Hydrochloride Injection, 671
Paracetamol, 219Para‹n, 999
Light Liquid, 1001Liquid, 1000
Paraformaldehyde, 671Paste
Arsenical, 866Paraformaldehyde, Dental, 1001Triozinc, Dental, 1072
Pas-calcium, 301Granules, 302
Peach Kernel, 1002Peanut Oil, 1002Peel
Bitter Orange, 875Citrus Unshiu, 898
Penbutolol Sulfate, 672Penicillin
G Potassium, 270Pentazocine, 672Pentobarbital Calcium, 673Pentoxyverine Citrate, 674Peony Root, 1003, 1555
Powdered, 1556, 1768

18501850 Supplement II, JP XIVIndex
Peplomycin Sulfate, 675, 1503Perilla Herb, 1004Perphenazine, 675
Maleate, 677Maleate Tablets, 678Tablets, 676
PethidineHydrochloride, 678, 1505Hydrochloride Injection, 679
PetrolatumWhite, 1005Yellow, 1006
Petroleum Benzin, 1006Pharbitis Seed, 1007Phellodendron
Bark, 1007, 1556Albumin Tannate and Bismuth
Subnitrate Powder, 1009, 1556Powdered, 1556Powder for Cataplasm, Compound,
1556Phenacetin, 680Phenazone, 247Phenethicillin Potassium, 1505Phenobarbital, 681
Powder, 681Powder, 10, 681
Phenol, 1010and Zinc Oxide Liniment, 1011for Disinfection, 1010Liqueˆed, 1011with Camphor, Dental, 1011
PhenolatedWater, 1012Water for Disinfection, 1012
Phenolsulfonphthalein, 682Injection, 682
Phenovalin and Magnesium OxidePowder, 1012, 1557
L-Phenylalanine, 683Phenylbutazone, 684Phenylephrine Hydrochloride, 684Phenytoin, 685
Powder, 686Sodium for Injection, 686Tablets, 686
Phytomenadione, 687Phytonadione, 687, 1507Picrasma Wood, 1013Pilocarpine Hydrochloride, 687Pimaricin, 1507Pindolol, 688Pinellia Tuber, 1013Pipemidic Acid Trihydrate, 689, 1508Piperacillin Sodium, 690, 1508Piperazine
Adipate, 691Phosphate, 692Phosphate Tablets, 692
Pirarubicin, 1509Pirenoxine, 693, 1731Pirenzepine Hydrochloride Hydrate,
1732
Piroxicam, 1733Pivmecillinam Hydrochloride, 693,
1510Plantago
Herb, 1014Seed, 1014
Plaster, Adhesive, 858Salicylic Acid, Adhesive, 1032
PlatycodonFluidextract, 1014Root, 1014Root, Powdered, 1015
PolyethyleneGlycol Ointment, 968Glycol 400, 965Glycol 1500, 966Glycol 4000, 967Glycol 6000, 967Glycol 20000, 968
PolygalaRoot, 1015Root, Powdered, 1016
Polygonum Root, 1557Polymixin B Sulfate, 694, 1511Polyoxyethylene Lauryl Alcohol
Ether, 964Polyoxyl 40 Stearate, 1016Polyporus Sclerotium, 1016Polysorbate 80, 1017Polyvidone, 1020Polyvinylpyrrolidone, 1020
K25, 1020K30, 1020K90, 1020
Poria Sclerotium, 1017Potash Soap, 1018Potassium
Benzylpenicillin, 270Bromide, 694Canrenoate, 694Carbonate, 1018Chloride, 695Clavulanate, 696, 1512Guaiacolsulfonate, 696Hydroxide, 1018Iodide, 697Penicillin G, 270Penicillin G, Crystalline, 270Permanganate, 698Sulfate, 1019
Potato Starch, 1019, 1769Povidone, 1020Povidone-Iodine, 698, 1513Powder
Ascorbic Acid, 251Chlordiazepoxide, 351Chlorpheniramine and Calcium,
895Chlorpheniramine Maleate, 358Codeine Phosphate, 1, 382Codeine Phosphate, 10, 383Diastase and Sodium Bicarbonate,
907
Diastase and Sodium Bicarbonate,Compound, 907
Dihydrocodeine Phosphate, 1,417
Dihydrocodeine Phosphate, 10,418
Diphenhydramine andBromovalerylurea, 911
Diphenylhydantoin, 686Dover's, 993Ephedrine Hydrochloride, 446Ephedrine Hydrochloride, 10,
446for Cataplasm, Phellodendron,
Compound, 1009Gentian and Sodium Bicarbonate,
926Hydralazine Hydrochloride, 513Kainic Acid and Santonin, 958dl-Methylephedrine
Hydrochloride, 614dl-Methylephedrine
Hydrochloride, 10, 614Nux Vomica Extract, 984Opium Ipecac, 993Opium, Diluted, 987Phellodendron, Albumin Tannate
and Bismuth Subnitrate, 1009Phenobarbital, 681Phenobarbital, 10, 681Phenovalin and Magnesium Oxide,
1012Phenytoin, 686Reserpine, 731Reserpine, 0.1, 731Rhubarb and Senna, Compound,
Ribo‰avin, 734Salicylated Alum, 1032Scopolia Extract, 1036Scopolia Extract and Carbon, 1037Scopolia Extract and Diastase,
Compound, 1037Scopolia Extract and Ethyl
Aminobenzoate, 1037Scopolia Extract, Papaverine and
Ethyl Aminobenzoate, 1038Swertia and Sodium Bicarbonate,
1065Thiamine Hydrochloride, 794Vitamin B, Compound,Vitamin B1 Hydrochloride, 794Vitamin B2, 734Vitamin C, 251Zinc Oxide Starch, 1086
PowderedAcacia, 855, 1543Agar, 859Alisma Rhizome, 860Aloe, 861, 1544, 1757Amomum Seed, 863Atractylodes Lancea Rhizome, 869Atractylodes Rhizome, 868Calumba, 881

18511851Supplement II, JP XIV Index
Capsicum, 883, 1546Cellulose, 892Cinnamon Bark, 897, 1547Clove, 898Cnidium Rhizome, 899Coix Seed, 900Coptis Rhizome, 902, 1548Cyperus Rhizome, 906Digitalis, 910, 1762Dioscorea Rhizome, 911Gambir, 920Gardenia Fruit, 923, 1763Gentian, 925Geranium Herb, 926Ginger, 927, 1764Ginseng, 929Glycyrrhiza, 933Ipecac, 954Japanese Angelica Root, 956Japanses Gentian, 957, 1551Japanese Valerian, 958Magnolia Bark, 971, 1552Moutan Bark, 980Opium, 987Oyster Shell, 998Panax Rhizome, 998, 1555Peach Kernel, 1002Peony Root, 1004, 1556, 1768Phellodendron Bark, 1008, 1556Picrasma Wood, 1013Platycodon Root, 1015Polygala Root, 1016Polyporus Sclerotium, 1016Poria Sclerotium, 1017Rhubarb, 1027, 1558, 1774Rose Fruit, 1029Scutellaria Root, 1043, 1560, 1775Senega, 1044Senna Leaf, 1046, 1561Smilax Rhizome, 1053Sophora Root, 1058Sweet Hydrangea Leaf, 1063Swertia Herb, 1064, 1562Tragacanth, 1072Zanthoxylum Fruit, 1084
Pranoprofen, 699Prazepam, 700
Tablets, 701Precipitated Calcium Carbonate, 297Prednisolone, 701
Acetate, 703, 1513Sodium Succinate for Injection,
705, 1513Succinate, 704Tablets, 702
Primidone, 706, 1514Probenecid, 706
Tablets, 707Procainamide
Hydrochloride, 708Hydrochloride Injection, 709Hydrochloride Tablets, 709
Procaine
Hydrochloride, 710Hydrochloride Injection, 710, 1514
Procarbazine Hydrochloride, 711,1515
Procaterol Hydrochloride, 712Processed Aconite Root, 1770
Powdered, 1772Processed Ginger, 1774Prochlorperazine
Maleate, 713Maleate Tablets, 713
Progesterone, 714Injection, 714
Proglumide, 715Promethazine Hydrochloride, 716Propantheline Bromide, 716Propranolol Hydrochloride, 717Propylene
Glycol, 1022Propyl Parahydroxybenzoate, 1021Propylthiouracil, 718
Tablets, 718Propyphenazone, 556Prostaglandin
E1, 1718E1 a-Cyclodextrin Clathrate
Compound, 231F2a, 425
ProtamineSulfate, 719Sulfate Injection, 719
Prothionamide, 720Protirelin, 720
Tartrate, 722Prunella Spike, 1023Pueraria Root, 1023, 1557Puriˆed
Absorbent Cotton, 851Dehydrocholic Acid, 395Gelatin, 924Lanolin, 963Shellac, 1049Water, 1079
Pyrantel Pamoate, 722Pyrazinamide, 723, 1515, 1734Pyridostigmine Bromide, 724Pyridoxine
Hydrochloride, 724, 1734Hydrochloride Injection, 725, 1735
Pyroxylin, 1023Pyrrolnitrin, 1516
Q
Quick Lime, 878Quinidine Sulfate, 725Quinine
Ethyl Carbonate, 726Hydrochloride, 727Sulfate, 728
R
Ranitidine Hydrochloride, 1517Rape Seed Oil, 1024Red Ginseng, 1024Rehmannia Root, 1025Reserpine, 729, 1518
Injection, 730Powder, 731Powder, 0.1, 731Tablets, 731
RetinolAcetate, 732, 1519Palmitate, 733, 1519
RhizomeAlisma, 860Alisma, Powdered, 860Anemarrhena, 864Atractylodes, 868Atractylodes Lancea, 869Atractylodes Lancea, Powdered,
869Atractylodes, Powdered, 868Cimicifuga, 896Cnidium, 899Cnidium, Powdered, 899Coptis, 901Coptis, Powdered, 902Cyperus, 906Cyperus, Powdered, 906Dioscorea, 911Dioscorea, Powdered, 911Imperate, 949Nuphar, 983Panax, 998Panax, Powdered, 998Scopolia, 1040
Rhubarb, 1026, 1558, 1774Powdered, 1027, 1558, 1774
Ribo‰avin, 733Butyrate, 734Phosphate, 735Phosphate Injection, 736Powder, 734Sodium Phosphate, 735Sodium Phosphate Injection, 736
Ribostamycin Sulfate, 737, 1520Rice Starch, 1028Rifampicin, 737, 1521Ringer's Solution, 1028Rokitamycin, 737Root
Achyranthes, 857Angelica Dahurica, 864Asiasarum, 867Astragalus, 867Belladonna, 872Bupleurum, 876Glehnia, 930Japanese Angelica, 956Japanese Angelica, Powdered, 956Lithospermum, 964Peony, 1003

18521852 Supplement II, JP XIVIndex
Peony, Powdered, 1004Platycodon, 1014Polygala, 1015Pueraria, 1023Rehmannia, 1025Saposhnikovia, 1034Saussurea, 1034Scutellaria, 1042Scutellaria, Powdered, 1043Sophora, 1058Sophora, Powdered, 1058Trichosanthes, 1072
Rose Fruit, 1029Rosin, 1029Roxithromycin, 738, 1522
S
Saccharated Pepsin, 1030Saccharin Sodium, 1030SaŒower, 1031SaŠron, 1031Salazosulfapyridine, 739Salbutamol Sulfate, 740Salicylated Alum Powder, 1032Salicylic Acid, 741
Plaster, Adhesive, 1032Spirit, 1033
Santonin, 742Tablets, 742, 1735
Saponated Cresol Solution, 905Saposhnikovia Root, 1034Sappan Wood, 1774Saussurea Root, 1034Schisandra Fruit, 1035Schizonepeta Spike, 1035Sclerotium
Polyporus, Powdered, 1016Poria, 1017Poria, Powdered, 1017
ScopolamineButylbromide, 743, 1522Hydrobromide, 744
ScopoliaExtract, 1035Extract and Carbon Powder, 1036Extract and Ethyl Aminobenzoate
Powder, 1037, 1559Extract and Tannic Acid
Ointment, Compound, 1560Suppositories, 1039Suppositories, Compound, 1560
Extract Powder, 1036Extract, Papaverine and Ethyl
Aminobenzoate Powder, 1038Rhizome, 1040
Scutellaria Root, 1042, 1560Powdered, 1560, 1775
Secretin, 744Seed
Amomum, 863Amomum, Powdered, 863Cassia, 888
Coix, 900Coix, Powdered, 900Pharbitis, 1007Plantago, 1014
Senega, 1043Powdered, 1044Syrup, 1044
Senna Leaf, 1044, 1560Powdered, 1046, 1561
Serrapeptase, 1735Serum
Gonadotrophin, 936, 1765Gonadotrophin for Injection, 937,
1765Sesame Oil, 1048Shellac
Puriˆed, 1049White, 1049
ShellOyster, 997Oyster, Powdered, 998
Siccaine, 1523Silicic Acid, Light Anhydrous, 1050Silver
Nitrate, 746Nitrate Ophthalmic Solution, 1041Protein, 1051Protein Solution, 1051
Simˆbrate, 746, 1524Simple
Ointment, 1052Syrup, 1052
Sinomenium Stem, 1052, 1562Sisomicin Sulfate, 747, 1524Slaked Lime, 877Smilax
Rhizome, 1053Rhizome, Powdered, 1053
SoapMedicinal, 973Potash, 1018
SodiumAcetate, 1053Aminobenzylpenicillin, 246Ampicillin, 246Aurothiomalate, 748, 1736Benzoate, 749Bicarbonate, 749Bicarbonate and Bitter Tincture
Mixture, 1054Bicarbonate Injection, 750, 1524Bisulˆte, 1054Borate, 750Bromide, 750Carbonate, 1055Carbonate, Dried,Carmellose, 887Cefalotin, 317Cefamandole, 317Cefapirin, 317Cefazolin, 319Cefbuperazone, 322Cefmetazole, 331
Cefoperazone, 332Cefotaxime, 335Cefoxitin, 336Cefpiramide, 338Cefsulodin, 340Ceftizoxime, 345Cefuroxime, 348Chloride, 751, 1524Chloride Injection, 0.9, 752Chloride Injection, 10, 752, 1525Chromate (51Cr) Injection, 752Citrate, 753Citrate Injection for Transfusion,
753Cloxacillin, 380CMC, 887Cromoglicate, 754Dantrolene, 392Diclofenac, 405Dicloxacillin, 408EDTA, 913Flavin Adenine Dinucleotide, 475Fluorescein, 483Fosfomycin, 496Fusidate, 1525Heparin, 510Hydrogen Sulˆte, 1054Hydroxide, 1056Iodide, 755Iodide (123I) Capsules, 755Iodide (131I) Capsules, 755Iodide (131I) Solution, 756Iodohippurate (131I) Injection, 756Iopodate, 756, 1526Iopodate Capsules, 757, 1526Iotalamate Injection, 757Latamoxef, 569Lauryl Sulfate, 1056Levothyroxine, 572Liothyronine, 576Loxoprofen, 580Metabisulˆte, 1057Methylchlorophenylisoxazolyl-
penicillin, 380Methyldichlorophenylisoxazolyl-
penicillin, 408Pertechnetate (99mTc) Injection, 758Phosphate, Dibasic, 907Picosulfate, 758Piperacillin, 690Polystyrene Sulfonate, 759, 1737Prasterone Sulfate, 760Pyrosulˆte, 1057Saccharin, 1030Salicylate, 761Sulˆte, Dried, 1058Thiosulfate, 762, 1738Thiosulfate Injection, 762Valproate, 762
SolutionAdrenaline Hydrochloride, 448Alum, 861Benzalkonium Chloride, 267

18531853Supplement II, JP XIV Index
Benzethonium Chloride, 269Chlorhexidine Gluconate, 353Cresol, 905Cresol, Saponated, 905Epinephrine, 448Epinephrine Hydrochloride, 448Epirenamine Hydrochloride, 448Glycerin and Potash, 930Idoxuridine Ophthalmic, 528Merbromin, 605Mercurochrome, 605Naphazoline and
Chlorpheniramine, 981Ringer's, 1028Salt, Isotonic, 752Silver Nitrate Ophthalmic, 1041Silver Protein, 1051Sodium Chloride, Isotonic, 752Sodium Hypochlorite, Dental, 864Sodium Iodide (131I), 756D-Sorbitol, 764Thianthol and Salicylic Acid,
Compound, 1067Tolnaftate, 816Zinc Sulfate Ophthalmic, 108650, Benzalkonium Chloride,
Concentrated, 267Sophora Root, 1058Sorbitan Sesquioleate, 1058D-Sorbitol, 763
Solution, 764Soybean Oil, 1059Spectinomycin Hydrochloride, 1526Spike
Prunella, 1023Schizonepeta, 1035
SpiritCapsicum and Salicylic Acid, 884Foeniculated Ammonia, 919Iodine, Salicylic Acid and Phenol,
952Methyl Salicylate, Compound, 977Salicylic Acid, 1033Salicylic Acid, Compound, 1033
Spironolactone, 765Starch
Corn, 903Potato, 1019Rice, 1028Wheat, 1080
Stearic Acid, 1059Stearyl Alcohol, 1059Stem
Akebia, 859Sinomenium, 1052
SterileAbsorbent Cotton, 852Absorbent Gauze, 853Puriˆed Absorbent Cotton, 852Puriˆed Water, 1079
Streptomycin Sulfate, 766, 1527Sucralfate, 766Sucrose, 1060
Sulbactam Sodium, 768Sulbenicillin Sodium, 769, 1528Sulfadiazine Silver, 769Sulfafurazole, 773Sulfamethizole, 770Sulfamethoxazole, 770Sulfamonomethoxine, 771Sulfasalazine, 739Sulˆnpyrazone, 772
Tablets, 773Sulˆsomezole, 770Sulˆsoxazole, 773Sulfobromophthalein
Sodium, 774Sodium Injection, 775
Sulfur, 775and Camphor Lotion, 1062Salicylic Acid and Thianthol
Ointment, 1062Sulpiride, 776Sulpyrine, 776
Injection, 777Sultamicillin Tosilate, 778Sultiame, 779Suppositorise
Bisacodyl, 280Indometacin, 536Scopolia Extract and Tannic Acid,
1039Scopolia Extract and Tannic Acid,
Compound, 1041Suxamethonium
Chloride, 780Chloride for Injection, 781Chloride Injection, 781
Sweet Hydrangea Leaf, 1063Swertia
and Sodium Bicarbonate Powder,1065, 1563
Herb, 1063, 1562Powdered, 1562
SyntheticAluminum Silicate, 235Camphor, 306
SyrupIpecac, 955Orange Peel, 994Senega, 1044Simple, 1052Trichloroethyl Phosphate,
Monosodium, 824Triclofos Sodium, 824
T
TabletsAcetylsalicylic Acid, 252Ajmaline, 227Amitriptyline Hydrochloride, 240Aspirin, 252Azathioprine, 258Baclofen, 260Chlordiazepoxide, 352
Chlorpheniramine Maleate, 359Chlorpromazine Hydrochloride,
361Chlorpropamide, 362Clomifene Citrate, 374Codeine Phosphate, 384Dichlorphenamide, 407Diclofenamide, 407Diethylcarbamazine Citrate, 409Diethylstilbestrol Diphosphate,
494Digitoxin, 411Digoxin, 416Dimenhydrinate, 423Diphenylhydantoin, 686Distigmine Bromide, 430Dydrogesterone, 438Ephedrine Hydrochloride, 446Ergometrine Maleate, 451Estriol, 458Etacrynic Acid, 460Ethinylestradiol, 463Etilefrine Hydrochloride, 468Folic Acid, 491Fosfestrol, 494Hydralazine Hydrochloride, 514Imipramine Hydrochloride, 531Iopanoic Acid, 548Isoniazid, 554Isosorbide Dinitrate, 558Lanatoside C, 567Levothyroxine Sodium, 573Liothyronine Sodium, 576Methyldopa, 613Methylergometrine Maleate, 615Methyltestosterone, 619Morphine Hydrochloride, 631Nitroglycerin, 647Norgestrel and Ethinylestradiol,
652Perphenazine, 676Perphenazine Maleate, 678Phenytoin, 686Piperazine Phosphate, 682Prazepam, 700Prednisolone, 702Probenecid, 707Procainamide Hydrochloride, 709Prochlorperazine Maleate, 713Propylthiouracil, 718Reserpine, 731Santonin, 742Sulˆnpyrazone, 773Thiamazole, 792Tipepidine Hibenzate, 806Tolbutamide, 814Trihexyphenidyl Hydrochloride,
825Trimethadione, 827Warfarin Potassium, 843
Talampicillin Hydrochloride, 782,1529
Talc, 1065

18541854 Supplement II, JP XIVIndex
Tannalbin, 227Tannic Acid, 782Tartaric Acid, 1066Tegafur, 782, 1530, 1738Teicoplanin, 783, 1530Terbutaline Sulfate, 786Termeric 1775Testosterone
Enanthate, 787Enanthate Injection, 787Propionate, 788, 1738Propionate Injection, 788, 1739
Tetanus Antitoxin, Equine, Freeze-dried, 1066
Tetracaine Hydrochloride, 789Tetracycline Hydrochloride, 789Thallium (201Tl) Chloride Injection,
791Theophylline, 791, 1531Thiamazole, 791
Tablets, 792Thiamine
Hydrochloride, 793, 1532, 1740Hydrochloride Injection, 794Hydrochloride Powder, 794Nitrate, 795, 1532
ThiamylalSodium, 796, 1741Sodium for Injection, 796, 1742
Thianthol, 1067Thiopental
Sodium, 797, 1532Sodium for Injection, 798
Thioridazine Hydrochloride, 798Thiotepa, 799L-Threonine, 799Thrombin, 1068Thymol, 1069Thyroid, Dried, 1069Tiaramide Hydrochloride, 800
Tablets, 1742Ticarcillin Sodium, 801Ticlopidine Hydrochloride, 802Timepidium Bromide, 803Tincture
Bitter, 875Capsicum, 883Iodine, 949Nux Vomica, 985Opium, 988Orange Peel, 995
Tinidazole, 804, 1743Tipepidine
Hibenzate, 804, 1533Hibenzate Tablets, 806
Titanium Oxide, 1070Tizanidine Hydrochloride, 1744Toad Venom, 1071, 1563Tobramycin, 807, 1533Tocopherol, 807, 1534
Acetate, 808, 1535Calcium Succinate, 809Nicotinate, 810
dl-a-Tocopherol, 807Acetate, 808Nicotinate, 810
Todralazine Hydrochloride, 811Toˆsopam, 812Tolazamide, 813Tolbutamide, 814
Tablets, 814Tolnaftate, 815
Solution, 816Tolperisone Hydrochloride, 816Toxoid
Diphtheria, 912Diphtheria-Tetanus, Combined,
913Diphtheria-Tetanus, Combined,
Adsorbed, 913for Adult Use, Diphtheria,
Adsorbed, 912Habu-venom, Adsorbed, 938Tetanus, Adsorbed, 1066
Tragacanth, 1072Powdered, 1072
Tranexamic Acid, 817, 1745Capsules, 1746Injection, 1747Tablets, 1747
Transfusion, Sodium Citrate Injectionfor, 753
Trapidil, 818Trepibutone, 819Tretoquinol Hydrochloride, 827Triamcinolone, 819, 1535
Acetonide, 820Triamterene, 821Tribulus Fruit, 1775Trichlormethiazide, 822, 1748
Tablets, 1749Trichloroethyl Phosphate,
Monosodium, 823Trichomycin, 823Trichosanthes Root, 1072Triclofos
Sodium, 823Sodium Syrup, 824
Trichomycin, 1536Trihexyphenidyl
Hydrochloride, 824Hydrochloride Tablets, 825
Trimebutine Maleate, 1537Trimetazidine Hydrochloride, 826Trimethadione, 827
Tablets, 827Trimetoquinol Hydrochloride, 827,1538Tropicamide, 828L-Tryptophan, 829Tuber
Corydalis, 904Ophiopogon, 987Pinellia, 1013
TubocurarineChloride, 830
Chloride Injection, 831Hydrochloride, 830Hydrochloride Injection, 831
Tulobuterol Hydrochloride, 831Turpentine Oil, 1073
U
Ubidecarenone, 832Ulinastatin, 833Uncaria Thorn, 1563, 1776Urea, 835Urokinase, 1073Ursodesoxycholic Acid, 835Uva Ursi Fluidextract, 1074
V
VaccineCholera, 896Diphtheria-Puriˆed Pertussis-
Tetanus Combined, Adsorbed,1005
Hepatitis B, Adsorbed, 938In‰uenza HA, 949Japanese Encephalitis, 956Japanese Encephalitis, Freeze-dried,
957Live Attenuated Measles, Freeze-d-
ried, 972Live Attenuated Mumps, Freeze-d-
ried, 981Live Attenuated Rubella, Freeze-d-
ried, 1029Live Oral Poliomyelitis, 1015Pertussis, Puriˆed, Adsorbed, 1005Rabies, Inactivated Tissue Culture,
Freeze-dried, 1024Smallpox, Freeze-dried, 1053Smallpox, Freeze-dried, Prepared in
Cell Culture, 1053Weil's Disease and Akiyami
Combined, 1080(for Percutaneous Use), BCG,
Freeze-dried, 869L-Valine, 836Vancomycin Hydrochloride, 837,
1539Vasopressin Injection, 837, 1751Verapamil Hydrochloride, 839Vinblastine
Sulfate, 840, 1752Sulfate for Injection, 841, 1753
Vincristine Sulfate, 842Vitamin
A Acetate, 732A Capsules, 1075A Oil, 1074, 1564A Oil Capsules, 1075, 1565A Palmitate, 733B1 Hydrochloride, 793B1 Hydrochloride Injection, 794B1 Hydrochloride Powder, 794

18551855Supplement II, JP XIV Index
B1 Nitrate, 795B2, 733B2 Phosphate Ester, 735B2 Phosphate Ester Injection, 736B2 Powder, 734B6, 724B6 Injection, 725B12, 388B12 Injection, 389C, 250C Injection, 251C Powder, 251D2, 449D3, 363E, 807E Acetate, 808E Calcium Succinate, 809E Nicotinate, 810K1, 687
W
WarfarinPotassium, 843Potassium Tablets, 843
Water, 1076
Ammonia, 241Apricot Kernel, 865for Disinfection, Phenolated, 1012for Injection, 1078Formalin, 920Mentha, 975Phenolated, 1012Puriˆed, 1079Sterile, Puriˆed, 1079
Wax, Carnauba, 888Weak Opium Alkaloids and
Scopolamine Injection, 992Weil's Disease and Akiyami CombinedVaccine, 1080Wheat Starch, 1080, 1776White
Beeswax, 871Ointment, 1080Petrolatum, 1005Shellac, 1049Soft Sugar, 1061
Whole Human Blood, 1080Wine, 1081Wood
Picrasma, 1013Picrasma, Powdered, 1013
X
Xylitol, 844Injection, 845, 1540
Y
Yeast, Dried, 1083Yellow
Beeswax, 871Petrolatum, 1006
Z
Zanthoxylum Fruit, 1083Zedoary, 1084Zinc
Chloride, 1084Oxide, 845Oxide Oil, 1085Oxide Ointment, 1085Oxide Starch Powder, 1086Sulfate, 846Sulfate Ophthalmic Solution, 1086
Zinostatin Stimalamer, 846

18571857
INDEX IN JAPANESE
ア
亜鉛華デンプン 1086亜鉛華軟膏 1085アカメガシワ 971アクチノマイシン D 225, 1381アクリノール 224アクリノール・亜鉛華軟膏 858アクリノール・チンク油 857アザチオプリン 256アザチオプリン錠 257亜酸化窒素 648アジスロマイシン水和物 1690アジピン酸ピペラジン 691アジマリン 226アジマリン錠 227亜硝酸アミル 246アスコルビン酸 250アスコルビン酸散 251アスコルビン酸注射液 251アズトレオナム 258アスピリン 252アスピリンアルミニウム 253アスピリン錠 252アスポキシシリン 254アセグルタミドアルミニウム 1377アセタゾラミド 220アセチルキタサマイシン 222, 1378アセチルスピラマイシン 224, 1378アセトアミノフェン 219アセトヘキサミド 221, 1687アセンヤク 920アセンヤク末 920亜ヒ酸パスタ 866アフロクアロン 225アヘンアルカロイド・アトロピン注射
液 990アヘンアルカロイド・スコポラミン注
射液 991アヘン散 987アヘンチンキ 988アヘン・トコン散 993アヘン末 987アマチャ 1063アマチャ末 1063アミドトリゾ酸 237アミドトリゾ酸ナトリウムメグルミン
注射液 596アミドトリゾ酸メグルミン注射液
594アミノ安息香酸エチル 465アミノフィリン 239アミノフィリン注射液 239アムホテリシン B 245
アモキサピン 243アモキシシリン 244, 1381アモバルビタール 242アラビアゴム 855, 1543アラビアゴム末 855, 1543亜硫酸水素ナトリウム 1054アルジオキサ 228アルプラゾラム 230アルプロスタジル 1688アルプロスタジルアルファデクス
231アロエ 860, 1544アロエ末 861, 1544, 1757アロプリノール 228アンソッコウ 873安息香酸 270安息香酸エストラジオール 455,
1462安息香酸エストラジオール水性懸濁注
射液 456安息香酸エストラジオール注射液
456安息香酸ナトリウム 749安息香酸ナトリウムカフェイン 296安息香酸ベンジル 874アンチピリン 247アンピシリン 246, 1382アンピシリンナトリウム 246, 1385アンモニア・ウイキョウ精 919アンモニア水 241
イ
イオウ 775イオウ・カンフルローション 1062イオウ・サリチル酸・チアントール軟膏
1062イオタラム酸 549イオタラム酸ナトリウム注射液 757イオタラム酸メグルミン注射液 595イオトロクス酸 550イオパノ酸 548, 1478イオパノ酸錠 548, 1478イオパミドール 547イオポダートナトリウム 756, 1526イオポダートナトリウムカプセル
757, 1526イクタモール 948イソソルビド 557イソニアジド 553イソニアジド錠 554, 1478イソニアジド注射液 554, 1478イソフェンインスリン水性懸濁注射液
541イソプロパノール 556
イソプロピルアンチピリン 556イドクスウリジン 527イドクスウリジン点眼液 528, 1475イブプロフェン 526イミペネム 530, 1476イレイセン 1547インジゴカルミン 533インジゴカルミン注射液 533インスリン 536インスリン亜鉛水性懸濁注射液 542インスリン注射液 539インチンコウ 867インドメタシン 534インドメタシンカプセル 535インドメタシン坐剤 536, 1477インフルエンザ HAワクチン 949インヨウカク 1762
ウ
ウイキョウ 918ウイキョウ末 918ウイキョウ油 918ウコン 1775ウヤク 1766ウリナスタチン 833ウルソデオキシコール酸 835ウロキナーゼ 1073ウワウルシ 870ウワウルシ流エキス 1074
エ
エイジツ 1029エイジツ末 1029液状フェノール 1011エスタゾラム 454エストリオール 457, 1463エストリオール錠 458エストリオール水性懸濁注射液 458エタクリン酸 459エタクリン酸錠 460エタノール 814エチオナミド 464, 1707エチゾラム 1463エチニルエストラジオール 463エチニルエストラジオール錠 463エチルコハク酸エリスロマイシン
453エチル炭酸キニーネ 727エチレンジアミン 917エデト酸ナトリウム 913エーテル 462エテンザミド 461エトスクシミド 465

18581858 Supplement II, JP XIVIndex in Japanese
エトポシド 1709エナント酸テストステロン 787エナント酸テストステロン注射液
787エナント酸フルフェナジン 487エナント酸メテノロン 607エナント酸メテノロン注射液 608エノキサシン 444エピネフリン 447エピネフリン液 447エピネフリン注射液 447エピリゾール 448エリスロマイシン 453, 1460L-イソロイシン 553エルカトニン 440L-カルボシステイン 310エルゴカルシフェロール 449, 1460L-トリプトファン 829L-トレオニン 799L-バリン 836L-フェニルアラニン 683L-メチオニン 609L-ロイシン 569塩化亜鉛 1084塩化アンベノニウム 236塩化インジウム (111In)注射液 534塩化エドロホニウム 439塩化エドロホニウム注射液 440塩化カリウム 695塩化カルシウム 298塩化カルシウム注射液 298, 1399塩化スキサメトニウム 781塩化スキサメトニウム注射液 781塩化タリウム (201Tl)注射液 791塩化ツボクラリン 830塩化ツボクラリン注射液 831塩化ナトリウム 751, 152410塩化ナトリウム注射液 752,
1525塩化ベタネコール 278塩化ベルベリン 271塩化ベンザルコニウム 266塩化ベンザルコニウム液 267塩化ベンゼトニウム 269塩化ベンゼトニウム液 269塩化メチルロザニリン 617塩化リゾチーム 1491エンゴサク 904塩酸 514塩酸アクラルビシン 224, 1379塩酸アセブトロール 219塩酸アヘンアルカロイド 988, 1555,
1767塩酸アヘンアルカロイド注射液 989塩酸アマンタジン 235塩酸アミトリプチリン 240塩酸アミトリプチリン錠 240塩酸アルプレノロール 231塩酸アロチノロール 249l-塩酸イソプレナリン 555塩酸イダルビシン 527, 1475塩酸イミプラミン 530塩酸イミプラミン錠 531
塩酸インデノロール 532塩酸エタンブトール 460塩酸エチルモルヒネ 467塩酸エチレフリン 468塩酸エチレフリン錠 468, 1708塩酸エピルビシン 1458塩酸エフェドリン 444塩酸エフェドリン散 10 446塩酸エフェドリン錠 446塩酸エフェドリン注射液 445塩酸エペリゾン 1706塩酸 L-アルギニン 248塩酸 L-アルギニン注射液 248塩酸 L-エチルシステイン 466塩酸 L-リジン 581塩酸オキシコドン 662塩酸オキシテトラサイクリン 665,
1501塩酸オキシブプロカイン 661塩酸オクスプレノロール 660塩酸カルテオロール 313塩酸キニーネ 727塩酸クリンダマイシン 1439塩酸クロカプラミン 370塩酸クロコナゾール 387塩酸クロニジン 376塩酸クロフェダノール 371塩酸クロペラスチン 377塩酸クロミプラミン 375塩酸クロルプロマジン 360塩酸クロルプロマジン錠 361塩酸クロルプロマジン注射液 361塩酸クロルヘキシジン 354塩酸ケタミン 563塩酸コカイン 381塩酸シクロペントラート 389塩酸ジフェニドール 409塩酸ジフェンヒドラミン 427塩酸ジブカイン 404塩酸シプロヘプタジン 391塩酸ジラゼプ 419塩酸ジルチアゼム 420, 1451塩酸スペクチノマイシン 1526塩酸セトラキサート 349, 1431塩酸セフェタメト ピボキシル 328塩酸セフェピム 326塩酸セフォゾプラン 337塩酸セフォチアム 335塩酸セフォチアムヘキセチル 335,
1417塩酸セフカペン ピボキシル 322塩酸セフメノキシム 330, 1410塩酸ダウノルビシン 1445塩酸タランピシリン 782, 1529塩酸チアミン 793, 1532, 1740塩酸チアミン散 794塩酸チアミン注射液 794塩酸チアラミド 800塩酸チアラミド錠 1742塩酸チオリダジン 798塩酸チクロピジン 802塩酸チザニジン 1744塩酸ツロブテロール 831
塩酸テトラカイン 789塩酸テトラサイクリン 789塩酸デメチルクロルテトラサイクリン
1446塩酸ドキサプラム 433塩酸ドキシサイクリン 434, 1454塩酸ドキソルビシン 434, 1453塩酸トドララジン 811塩酸ドパミン 432塩酸ドパミン注射液 433, 1452塩酸ドブタミン 431, 1452塩酸トリヘキシフェニジル 824塩酸トリヘキシフェニジル錠 825塩酸トリメタジジン 826塩酸トリメトキノール 827, 1538塩酸トルペリゾン 816塩酸ナファゾリン 635塩酸ナロキソン 634塩酸ニカルジピン 639塩酸ニカルジピン注射液 640塩酸ノスカピン 655塩酸ノルトリプチリン 654塩酸バカンピシリン 259, 1389塩酸パパベリン 670塩酸パパベリン注射液 671塩酸バンコマイシン 837, 1539塩酸ヒドララジン 513塩酸ヒドララジン散 513塩酸ヒドララジン錠 514塩酸ヒドロキシジン 524塩酸ヒドロコタルニン 522塩酸ピブメシリナム 693, 1510塩酸ビペリデン 279塩酸ピリドキシン 724, 1734塩酸ピリドキシン注射液 725, 1735塩酸ピレンゼピン水和物 1732塩酸ピロカルピン 687塩酸ベニジピン 1691塩酸ベニジピン錠 1692塩酸フェニレフリン 684塩酸ブクモロール 287塩酸ブナゾシン 291塩酸ブフェトロール 288塩酸ブプラノロール 292塩酸フラボキサート 476塩酸フルスルチアミン 499塩酸フルラゼパム 489塩酸ブレオマイシン 283, 1396塩酸プロカイン 710塩酸プロカインアミド 708塩酸プロカインアミド錠 709塩酸プロカインアミド注射液 709塩酸プロカイン注射液 710, 1514塩酸プロカテロール 712塩酸プロカルバジン 711, 1515塩酸プロプラノロール 717塩酸ブロムヘキシン 285塩酸プロメタジン 716塩酸ペチジン 678, 1505塩酸ペチジン注射液 679塩酸ベラパミル 839塩酸ベンセラジド 265塩酸ホモクロルシクリジン 512,

18591859Supplement II, JP XIV Index in Japanese
1472塩酸マプロチリン 588塩酸ミノサイクリン 628, 1495塩酸メキシレチン 623塩酸メクロフェノキサート 589塩酸メタンフェタミン 609dl-塩酸メチルエフェドリン 613,
1719dl-塩酸メチルエフェドリン散 10
614, 1720塩酸メピバカイン 601, 1719塩酸メピバカイン注射液 602塩酸モルヒネ 629, 1497塩酸モルヒネ錠 631, 1498塩酸モルヒネ注射液 630, 1497塩酸ラニチジン 1517塩酸リモナーデ 939塩酸リンコマイシン 576, 1490塩酸レナンピシリン 1488エンフルラン 443, 1456
オ
オウギ 867オウゴン 1042, 1560オウゴン末 1043, 1560, 1775黄色ワセリン 1006オウバク 1007, 1556オウバク・タンナルビン・ビスマス散
1009, 1556オウバク末 1008, 1556オウレン 901, 1548オウレン末 902, 1548オキサゾラム 658オキサプロジン 658オキシトシン 1728オキシトシン注射液 665, 1730オキシドール 662オキシメトロン 664オキセサゼイン 659オフロキサシン 1500オリブ油 986オレンジ油 994オンジ 1015オンジ末 1016
カ
カイニン酸 560カイニン酸・サントニン散 958カオリン 959カカオ脂 877加香ヒマシ油 889カゴソウ 1023カシュウ 1557ガジュツ 1084加水ラノリン 962ガスえそウマ抗毒素 923ガーゼ 853カッコン 1023, 1557過テクネチウム酸ナトリウム (99mTc)
注射液 758果糖 497
果糖注射液 498カノコソウ 957カノコソウ末 958カフェイン 294カプセル 885カプトプリル 307過マンガン酸カリウム 698b-ガラクトシダーゼ(アスペルギルス)
921b-ガラクトシダーゼ(ペニシリウム)
921カリジノゲナーゼ 560, 1718カリ石ケン 1018カルナウバロウ 888カルバゾクロムスルホン酸ナトリウム
308, 1400カルバマゼピン 308カルバミン酸クロルフェネシン 356,
1436カルビドパ 309, 1401カルメロース 885カルメロースカルシウム 886, 1759カルメロースナトリウム 887カルモナムナトリウム 314, 1401カルモフール 313カロコン 1072カンキョウ 1774カンゾウ 932, 1550カンゾウ末 1550乾燥亜硫酸ナトリウム 1058カンゾウエキス 933乾燥甲状腺 1069乾燥酵母 1083乾燥細胞培養痘そうワクチン 1053乾燥ジフテリアウマ抗毒素 912乾燥弱毒生おたふくかぜワクチン
981乾燥弱毒生風しんワクチン 1029乾燥弱毒生麻しんワクチン 972乾燥水酸化アルミニウムゲル 233乾燥水酸化アルミニウムゲル細粒
233カンゾウ粗エキス 934乾燥組織培養不活化狂犬病ワクチン
1024乾燥炭酸ナトリウム 1055乾燥痘そうワクチン 1053乾燥日本脳炎ワクチン 957乾燥破傷風ウマ抗毒素 1066乾燥はぶウマ抗毒素 938乾燥 BCGワクチン 869乾燥ボツリヌスウマ抗毒素 876カンゾウ末 933乾燥まむしウマ抗毒素 972乾燥硫酸アルミニウムカリウム 863カンテン 859カンテン末 859含糖ペプシン 1030d-カンフル 305, 1400dl-カンフル 306, 1400肝油 900, 1548カンレノ酸カリウム 694
キ
希塩酸 515キキョウ 1014キキョウ末 1015キキョウ流エキス 1014キクカ 1547, 1760キササゲ 889キジツ 948キシリトール 844キシリトール注射液 845, 1540吉草酸ベタメタゾン 277, 1395キタサマイシン 565, 1484牛脂 870吸水軟膏 855キョウカツ 1554キョウニン 864キョウニン水 865希ヨードチンキ 950金チオリンゴ酸ナトリウム 748,
1736
ク
グアイフェネシン 505グアヤコールスルホン酸カリウム
696クエン酸 366, 1437クエン酸ガリウム (67Ga)注射液 501クエン酸クロミフェン 373, 1441クエン酸クロミフェン錠 374クエン酸ジエチルカルバマジン 408クエン酸ジエチルカルバマジン錠
409, 1449クエン酸ナトリウム 753クエン酸フェンタニル 473クエン酸ペントキシベリン 674クコシ 1767クジン 1058クジン末 1058苦味重曹水 1054苦味チンキ 875クラブラン酸カリウム 696, 1512グラミシジン 1470クラリスロマイシン 367, 1699グリシン 931グリセオフルビン 505, 1471グリセリン 504, 1469グリセリンカリ液 930クリノフィブラート 369グリベンクラミド 502グルコン酸カルシウム 299グルコン酸クロルヘキシジン液 353グルタチオン 1716クレオソート 904クレゾール 905クレゾール水 905クレゾール石ケン液 905クロキサシリンナトリウム 380,
1441クロキサゾラム 380クロチアゼパム 378クロトリマゾール 379

18601860 Supplement II, JP XIVIndex in Japanese
クロナゼパム 375クロフィブラート 372クロフィブラートカプセル 373クロム酸ナトリウム (51Cr)注射液
752クロモグリク酸ナトリウム 754クロラムフェニコール 351, 1432クロルジアゼポキシド 351クロルジアゼポキシド散 351, 1434クロルジアゼポキシド錠 352, 1435クロルフェニラミン・カルシウム散
895クロルプロパミド 362クロルプロパミド錠 362クロロブタノール 894
ケ
ケイガイ 1035経口生ポリオワクチン 1015ケイ酸マグネシウム 584軽質無水ケイ酸 1050軽質流動パラフィン 1001ケイヒ 896, 1547ケイヒ末 897, 1547ケイヒ油 897結晶性インスリン亜鉛水性懸濁注射液
543結晶セルロース 889血清性性腺刺激ホルモン 936, 1765ケツメイシ 888ケトプロフェン 564ケンゴシ 1007ゲンチアナ 925ゲンチアナ・重曹散 926ゲンチアナ末 925ゲンノショウコ 926ゲンノショウコ末 926
コ
コウカ 1031硬化油 940コウジン 1024合成ケイ酸アルミニウム 235コウブシ 906コウブシ末 906コウボク 970, 1552コウボク末 970, 1552ゴオウ 995ゴシツ 857ゴシュユ 918, 1548コハク酸クロラムフェニコールナトリ
ウム 1434コハク酸トコフェロールカルシウム
809コハク酸ヒドロコルチゾン 521コハク酸ヒドロコルチゾンナトリウム
520コハク酸プレドニゾロン 704コハク酸メチルプレドニゾロン
1720ゴボウシ 1546
ゴマ油 1048ゴミシ 1035コムギデンプン 1080, 1776コメデンプン 1028コリスチンメタンスルホン酸ナトリウ
ム 385コルヒチン 384, 1699コレカルシフェロール 363コレステロール 896コレラワクチン 896コロンボ 881コロンボ末 881コンズランゴ 900コンズランゴ流エキス 901
サ
サイクロセリン 390サイコ 876サイシン 867, 1545酢酸 856酢酸グアナベンズ 506酢酸クロルマジノン 355, 1435酢酸コルチゾン 386酢酸トコフェロール 808, 1535酢酸ナトリウム 1053酢酸ヒドロキソコバラミン 523酢酸ヒドロコルチゾン 517, 1474酢酸フタル酸セルロース 893, 1760酢酸プレドニゾロン 703, 1513酢酸ミデカマイシン 627酢酸メテノロン 607酢酸レチノール 732, 1519サッカリンナトリウム 1030サフラン 1031サラシ粉 894サラシミツロウ 871サラゾスルファピリジン 739サリチル・ミョウバン散 1032サリチル酸 741サリチル酸精 1033サリチル酸ナトリウム 761サリチル酸絆創膏 1032サリチル酸メチル 618酸化亜鉛 845酸化カルシウム 878酸化チタン 1070酸化マグネシウム 583サンキライ 1053サンキライ末 1053三酸化ヒ素 250サンシシ 922, 1549サンシシ末 923, 1763サンシュユ 903, 1762サンショウ 1083サンショウ末 1084酸素 663サントニン 742サントニン錠 742サンヤク 911サンヤク末 911
シ
ジアスターゼ 907ジアスターゼ・重曹散 907ジアゼパム 403シアナミド 387シアノコバラミン 388シアノコバラミン注射液 389ジオウ 1025歯科用アンチホルミン 864歯科用トリオジンクパスタ 1072歯科用パラホルムパスタ 1001歯科用フェノール・カンフル 1011歯科用ヨード・グリセリン 951ジギタリス 908ジギタリス末 910ジギトキシン 410ジギトキシン錠 411シクラシリン 364, 1437ジクロキサシリンナトリウム 408シクロスポリン 364, 1697ジクロフェナクナトリウム 405ジクロフェナミド 406, 1448ジクロフェナミド錠 407シクロホスファミド 390ジゴキシン 414, 1702ジゴキシン錠 416, 1704ジゴキシン注射液 415, 1703ジコッピ 1767シコン 964次硝酸ビスマス 282シスプラチン 1697ジスルフィラム 430ジソピラミド 429シタラビン 392, 1701シッカニン 1523シツリシ 1775ジドロゲステロン 437ジドロゲステロン錠 438ジノスタチン スチマラマー 846ジノプロスト 425ジピリダモール 428, 1451ジフェンヒドラミン 426ジフェンヒドラミン・フェノール・亜鉛
華リニメント 912ジフェンヒドラミン・ワレリル尿素散
911ジフテリアトキソイド 912ジフテリア破傷風混合トキソイド
913ジプロピオン酸ベタメタゾン 275シメチジン 365ジメルカプロール 423ジメルカプロール注射液 424ジメンヒドリナート 422ジメンヒドリナート錠 423次没食子酸ビスマス 281ジモルホラミン 424, 1705ジモルホラミン注射液 425, 1705弱アヘンアルカロイド・スコポラミン
注射液 992シャクヤク 1003, 1555シャクヤク末 1004, 1556

18611861Supplement II, JP XIV Index in Japanese
ジャショウシ 1760シャゼンシ 1014シャゼンソウ 1014臭化イプラトロピウム 550臭化カリウム 694臭化ジスチグミン 429臭化ジスチグミン錠 430臭化水素酸スコポラミン 744臭化水素酸デキストロメトルファン
402臭化水素酸ホマトロピン 511臭化チメピジウム 803臭化ナトリウム 751臭化パンクロニウム 667臭化ピリドスチグミン 724臭化ブチルスコポラミン 743, 1522臭化ブトロピウム 294臭化プロパンテリン 716臭化メチルベナクチジウム 611臭化メペンゾラート 599ジュウヤク 939シュクシャ 863シュクシャ末 863, 1768酒石酸 1066酒石酸アリメマジン 229酒石酸イフェンプロジル 529酒石酸エルゴタミン 452酒石酸キタサマイシン 1485酒石酸プロチレリン 721酒石酸レバロルファン 570酒石酸レバロルファン注射液 570ショウキョウ 927, 1763ショウキョウ末 927, 1764硝酸イソソルビド 558硝酸イソソルビド錠 558硝酸銀 746硝酸銀点眼液 1041硝酸チアミン 795, 1532硝酸ナファゾリン 636硝酸ミコナゾール 625常水 1076ショウズク 885消毒用エタノール 916消毒用フェノール 1010消毒用フェノール水 1012ショウマ 896ジョサマイシン 559, 1478シンイ 1552親水軟膏 940親水ワセリン 1005診断用クエン酸ナトリウム液 754シンフィブラート 746, 1524
ス
水酸化カリウム 1018水酸化カルシウム 877水酸化ナトリウム 1056スクラルファート 766ステアリルアルコール 1059ステアリン酸 1059ステアリン酸エリスロマイシン 454ステアリン酸カルシウム 881
ステアリン酸ポリオキシル 40 1016ステアリン酸マグネシウム 969スピロノラクトン 765スルチアム 779スルバクタムナトリウム 768スルピリド 776スルピリン 776スルピリン注射液 777スルファジアジン銀 769スルファメチゾール 770スルファメトキサゾール 770スルファモノメトキシン 771スルフイソキサゾール 773スルフィンピラゾン 772スルフィンピラゾン錠 773スルベニシリンナトリウム 769,
1528スルホブロモフタレインナトリウム
774スルホブロモフタレインナトリウム注
射液 775
セ
成人用沈降ジフテリアトキソイド
912精製水 1079精製ゼラチン 924精製セラック 1049精製脱脂綿 851精製デヒドロコール酸 395精製白糖 1060精製ラノリン 963生理食塩液 752石油ベンジン 1006セクレチン 745セスキオレイン酸ソルビタン 1058セタノール 894セッコウ 937セネガ 1043セネガシロップ 1044セネガ末 1044セファクロル 314, 1403セファゾリンナトリウム 319セファゾリンナトリウム水和物 319セファトリジンプロピレングリコール
318セファドロキシル 314セファピリンナトリウム 317セファマンドールナトリウム 317,
1407セファレキシン 315セファロチンナトリウム 317, 1406セファロリジン 317, 1405セフィキシム 329セフォキシチンナトリウム 336,
1419セフォジジムナトリウム 1412セフォタキシムナトリウム 335,
1413セフォテタン 335, 1415セフォペラゾンナトリウム 332セフジトレン ピボキシル 325
セフジニル 324セフスロジンナトリウム 340セフタジジム 342セフチゾキシムナトリウム 345セフチブテン 344, 1426セフテラムピボキシル 344, 1425セフトリアキソンナトリウム 346,
1427セフピラミドナトリウム 338, 1421セフブペラゾンナトリウム 322,
1409セフポドキシムプロキセチル 1422セフミノクスナトリウム 332セフメタゾールナトリウム 331セフラジン 339セフロキサジン 340, 1424セフロキシムアキセチル 347, 1429,
1696セフロキシムナトリウム 348, 1696ゼラチン 923セラペプターゼ 1735センキュウ 899センキュウ末 899センコツ 983センソ 1071, 1563センナ 1044, 1560センナ末 1046, 1561センブリ 1063, 1562センブリ・重曹散 1065, 1563センブリ末 1064, 1562
ソ
ソウジュツ 869ソウジュツ末 869ソウハクヒ 981, 1554ソボク 1774ソヨウ 1004
タ
ダイオウ 1026, 1558, 1774ダイオウ末 1026, 1558, 1774ダイズ油 1059タイソウ 958胎盤性性腺刺激ホルモン 934, 1764タクシャ 860タクシャ末 860脱脂綿 851タルク 1065炭酸カリウム 1018炭酸水素ナトリウム 749炭酸水素ナトリウム注射液 750,
1524炭酸ナトリウム 1055炭酸マグネシウム 582炭酸リチウム 578単シロップ 1052ダントロレンナトリウム 392単軟膏 1052タンニン酸 782タンニン酸アルブミン 227タンニン酸ジフェンヒドラミン 427

18621862 Supplement II, JP XIVIndex in Japanese
タンニン酸ベルベリン 272
チ
チアマゾール 791チアマゾール錠 792チアミラールナトリウム 796, 1741チアントール 1067チオテパ 799チオペンタールナトリウム 797,
1532チオ硫酸ナトリウム 762, 1738チオ硫酸ナトリウム注射液 762チカルシリンナトリウム 801チクセツニンジン 998チクセツニンジン末 998, 1555窒素 982チニダゾール 804, 1743チモ 864チモール 1069注射用アモバルビタールナトリウム
242注射用塩化アセチルコリン 222注射用塩化スキサメトニウム 781注射用塩酸セフェピム 1695注射用塩酸ヒドララジン 513注射用血清性性腺刺激ホルモン 937,
1765注射用コハク酸プレドニゾロンナトリ
ウム 705, 1513注射用水 1078注射用胎盤性性腺刺激ホルモン 936,
1764注射用チアミラールナトリウム 796,
1742注射用チオペンタールナトリウム
798注射用ファモチジン 470, 1464注射用フェニトインナトリウム 686注射用フロモキセフナトリウム
1711注射用硫酸ビンブラスチン 841,
1753チョウジ 898チョウジ末 898チョウジ油 898チョウトウコウ 1563, 1776チョレイ 1061チョレイ末 1061チンク油 1085沈降ジフテリア破傷風混合トキソイド
913沈降精製百日せきジフテリア破傷風混
合ワクチン 1005沈降精製百日せきワクチン 1005沈降炭酸カルシウム 297沈降破傷風トキソイド 1066沈降はぶトキソイド 938沈降 B型肝炎ワクチン 938チンピ 898
ツ
ツバキ油 882
テ
テイコプラニン 783, 1530D-ソルビトール 763D-ソルビトール液 764D-マンニトール 587D-マンニトール注射液 588低置換度ヒドロキシプロピルセルロー
ス 942テオフィリン 791, 1531テガフール 782, 1530, 1738デキサメタゾン 397デキストラン硫酸ナトリウム イオウ
5 401デキストラン硫酸ナトリウム イオウ
18 402デキストラン 40 398デキストラン 40注射液 399デキストラン 70 400デキストリン 906デスラノシド 396デスラノシド注射液 397デヒドロコール酸 394デヒドロコール酸注射液 396テレビン油 1073天然ケイ酸アルミニウム 233テンマ 1550テンモンドウ 1546
ト
トウガラシ 882, 1546トウガラシ末 883, 1546トウガラシ・サリチル酸精 884トウガラシチンキ 883トウキ 956トウキ末 956トウニン 1002トウニン末 1002トウヒ 875トウヒシロップ 994トウヒチンキ 995トウモロコシデンプン 903, 1761トウモロコシ油 903トコフェロール 807, 1534トコン 953トコンシロップ 955トコン末 954トシル酸スルタミシリン 778トチュウ 1548トフィソパム 812トブラマイシン 807, 1533トラガント 1072トラガント末 1072トラザミド 813トラネキサム酸 817, 1745トラネキサム酸カプセル 1746トラネキサム酸錠 1747トラネキサム酸注射液 1747
トラピジル 818トリアムシノロン 819, 1535トリアムシノロンアセトニド 820トリアムテレン 821トリクロホスナトリウム 823トリクロホスナトリウムシロップ
824トリクロルメチアジド 822, 1748トリクロルメチアジド錠 1749トリコマイシン 823, 1536トリメタジオン 827トリメタジオン錠 827トルナフタート 815トルナフタート液 816トルブタミド 814トルブタミド錠 814トレピブトン 819トロピカミド 828ドロペリドール 435, 1456トロンビン 1068豚脂 964
ナ
ナイスタチン 656ナタネ油 1024ナドロール 633ナファゾリン・クロルフェニラミン液
981ナプロキセン 636ナリジクス酸 634
ニ
ニガキ 1013ニガキ末 1013ニコチン酸 644ニコチン酸アミド 643ニコチン酸注射液 645ニコチン酸トコフェロール 810ニコモール 642ニコモール錠 643ニコランジル 1724二酸化炭素 312ニセリトロール 641ニトラゼパム 646ニトログリセリン錠 647ニフェジピン 645日本脳炎ワクチン 956乳酸 959乳酸カルシウム 300乳糖 960尿素 835ニルバジピン 1725ニルバジピン錠 1726ニンジン 927ニンジン末 929ニンドウ 1766
ノ
濃塩化ベンザルコニウム液 50 267濃グリセリン 504, 1469

18631863Supplement II, JP XIV Index in Japanese
ノスカピン 654ノルエチステロン 650ノルエピネフリン 649, 1498ノルエピネフリン注射液 650, 1499ノルゲストレル 652ノルゲストレル・エチニルエストラジ
オール錠 652, 1499ノルフロキサシン 651
ハ
バイモ 1549白色セラック 1049白色軟膏 1080白色ワセリン 1005白糖 1061バクモンドウ 987バクロフェン 260バクロフェン錠 261バシトラシン 1390バソプレシン注射液 837, 1751ハチミツ 938ハッカ 974ハッカ水 975ハッカ油 975パップ用複方オウバク散 1009, 1556パニペネム 667ハマボウフウ 930パモ酸ヒドロキシジン 524パモ酸ピランテル 722パラアミノサリチル酸カルシウム
301パラアミノサリチル酸カルシウム顆粒
302パラオキシ安息香酸エチル 916パラオキシ安息香酸ブチル 876パラオキシ安息香酸プロピル 1021パラオキシ安息香酸メチル 976パラフィン 999パラホルムアルデヒド 671バルビタール 263バルプロ酸ナトリウム 762パルミチン酸クロラムフェニコール
1432パルミチン酸レチノール 733, 1519バレイショデンプン 1019, 1769ハロキサゾラム 509, 1472ハロタン 508ハロペリドール 507パンクレアチン 999ハンゲ 1013絆創膏 858パンテチン 669パントテン酸カルシウム 301
ヒ
ピコスルファートナトリウム 758ビサコジル 280ビサコジル坐剤 280, 1396ビタミン A油 1074, 1564ビタミン A油カプセル 1075, 1565ヒトインスリン(遺伝子組換え) 537
人全血液 1080人免疫グロブリン 949ヒドロキシプロピルセルロース 940ヒドロキシプロピルメチルセルロース
フタレート 947ヒドロキシプロピルメチルセルロース
2208 943ヒドロキシプロピルメチルセルロース
2906 944ヒドロキシプロピルメチルセルロース
2910 945ヒドロクロロチアジド 515, 1473ヒドロコルチゾン 516ヒドロコルチゾン・ジフェンヒドラミ
ン軟膏 939ピペミド酸三水和物 689, 1508ピペラシリンナトリウム 690, 1508ヒベンズ酸チペピジン 804, 1533ヒベンズ酸チペピジン錠 806ビホナゾール 278ヒマシ油 888ピマリシン 1507ヒメクロモン 525ビャクシ 864ビャクジュツ 868ビャクジュツ末 868氷酢酸 856ピラジナミド 723, 1515, 1734ピラルビシン 1509ピレノキシン 693, 1731ピロ亜硫酸ナトリウム 1057ピロキシカム 1733ピロキシリン 1023ピロールニトリン 1516ビワヨウ 1552ピンドロール 688ビンロウジ 866, 1545
フ
ファノバリン・マグネシア散 1557ファモチジン 469ファモチジン散 469ファモチジン錠 471ファロペネムナトリウム 472フィトナジオン 687, 1507フェナセチン 680フェニトイン 685フェニトイン散 686フェニトイン錠 686フェニルブタゾン 684フェネチシリンカリウム 1505フェノバリン・マグネシア散 1012フェノバルビタール 681フェノバルビタール散 10 681フェノール 1010フェノール・亜鉛華リニメント 1011フェノール水 1012フェノールスルホンフタレイン 682フェノールスルホンフタレイン注射液
682フェンブフェン 473複方アクリノール・チンク油 857
複方オキシコドン・アトロピン注射液996
複方オキシコドン注射液 995複方サリチル酸精 1033複方サリチル酸メチル精 977複方ジアスターゼ・重曹散 907複方ダイオウ・センナ散 1028複方チアントール・サリチル酸液
1067複方ビタミン B散 1075複方ヨード・グリセリン 950複方ロートエキス・ジアスターゼ散
1037複方ロートエキス・タンニン坐剤
1041, 1560複方ロートエキス・タンニン軟膏
1039, 1560ブクリョウ 1017ブクリョウ末 1017ブシ 1770ブシ末 1772フシジン酸ナトリウム 1525ブスルファン 293ブドウ酒 1081ブドウ糖 503ブドウ糖注射液 503ブフェキサマク 289ブフェキサマク軟膏 290ブフェキサマク乳剤性軟膏 289フマル酸クレマスチン 368フマル酸ケトチフェン 1483フマル酸ホルモテロール 492ブメタニド 291プラステロン硫酸ナトリウム 760プラゼパム 700プラゼパム錠 701プラノプロフェン 699フラビンアデニンジヌクレオチドナト
リウム 475, 1710プリミドン 706, 1514フルオキシメステロン 486フルオシノニド 482フルオシノロンアセトニド 481フルオレセインナトリウム 483フルオロウラシル 485フルオロメトロン 484フルジアゼパム 479フルシトシン 478フルニトラゼパム 480フルラゼパム 487フルラゼパムカプセル 488フルルビプロフェン 489, 1466プレドニゾロン 701プレドニゾロン錠 702フロクタフェニン 477, 1465プログルミド 715プロゲステロン 714プロゲステロン注射液 714フロセミド 498, 1714フロセミド錠 1715プロタミンインスリン亜鉛水性懸濁注
射液 544プロチオナミド 720

18641864 Supplement II, JP XIVIndex in Japanese
プロチレリン 720プロテイン銀 1051プロテイン銀液 1051プロピオン酸ジョサマイシン 559,
1480プロピオン酸テストステロン 788,
1738プロピオン酸テストステロン注射液
788, 1739プロピオン酸ドロスタノロン 435,
1456プロピオン酸ドロスタノロン注射液
436, 1456プロピオン酸ベクロメタゾン 264プロピルチオウラシル 718プロピルチオウラシル錠 718プロピレングリコール 1022フロプロピオン 478フロプロピオンカプセル 1712プロベネシド 706プロベネシド錠 707ブロマゼパム 284ブロムワレリル尿素 287フロモキセフナトリウム 478, 1465粉末セルロース 892
ヘ
ベタメタゾン 274ヘパリンナトリウム 510, 1717ヘパリンナトリウム注射液 511,
1718ベラドンナエキス 871ベラドンナコン 872ペルフェナジン 675ペルフェナジン錠 676ベンジルアルコール 874, 1758ベンジルペニシリンカリウム 270,
1393ベンジルペニシリンベンザチン
1392, 1694ベンズブロマロン 268, 1391ペンタゾシン 672ベントナイト 873ペントバルビタールカルシウム 673
ホ
ボウイ 1052, 1562ボウコン 949, 1551ホウ砂 750ホウ酸 284抱水クロラール 350ボウフウ 1034ホスフェストロール 493ホスフェストロール錠 494ホスホマイシンカルシウム 494,
1713ホスホマイシンナトリウム 496,
1713ボタンピ 979ボタンピ末 980ポビドン 1020
ポビドンヨード 699, 1513ホミカ 983ホミカエキス 984ホミカエキス散 984ホミカチンキ 985ポリスチレンスルホン酸カルシウム
303, 1399ポリスチレンスルホン酸ナトリウム
759, 1737ポリソルベート 80 1017ホリナートカルシウム 299ホルマリン 919ホルマリン水 920ボレイ 997ボレイ末 998
マ
マイトマイシン C 629, 1496マオウ 914マーキュロクロム 604マーキュロクロム液 605マクリ 908マクロゴール軟膏 968マクロゴール 400 965マクロゴール 1500 966マクロゴール 4000 967マクロゴール 6000 967マクロゴール 20000 968マシニン 1551麻酔用エーテル 462マルトース 586マレイン酸エルゴメトリン 450マレイン酸エルゴメトリン錠 451マレイン酸エルゴメトリン注射液
451マレイン酸クロルフェニラミン 357d-マレイン酸クロルフェニラミン
359マレイン酸クロルフェニラミン散
358マレイン酸クロルフェニラミン錠
359マレイン酸クロルフェニラミン注射液
357マレイン酸トリメブチン 1537マレイン酸プロクロルペラジン 713マレイン酸プロクロルペラジン錠
713マレイン酸ペルフェナジン 677マレイン酸ペルフェナジン錠 678マレイン酸メチルエルゴメトリン
615マレイン酸メチルエルゴメトリン錠
615マレイン酸レボメプロマジン 572D-マンニトール 587, 1718D-マンニトール 587, 1718D-マンニトール注射液 588, 1492
ミ
ミグレニン 627, 1495
ミコナゾール 624ミツロウ 871ミデカマイシン 626ミョウバン水 861
ム
無晶性インスリン亜鉛水性懸濁注射液
542無水アンピシリン 246, 1384無水エタノール 915無水カフェイン 295無水クエン酸 367, 1438, 1698無水乳糖 961無水リン酸水素カルシウム 879ムピロシンカルシウム 水和物 631
メ
メキタジン 602メグルミン 974メコバラミン 590メシル酸ガベキサート 500メシル酸カモスタット 304メシル酸ジヒドロエルゴタミン 418メシル酸ジヒドロエルゴトキシン
413, 1450メシル酸デフェロキサミン 393,
1701メシル酸ブロモクリプチン 286メシル酸ベタヒスチン 272メストラノール 606メダゼパム 591メチクラン 619, 1493メチラポン 623メチルジゴキシン 620メチルセルロース 978メチルテストステロン 618, 1722メチルテストステロン錠 619, 1722メチルドパ 612メチルドパ錠 613メチルプレドニゾロン 616メチル硫酸ネオスチグミン 637メチル硫酸ネオスチグミン注射液
638滅菌ガーゼ 854滅菌精製水 1079滅菌精製脱脂綿 852滅菌脱脂綿 852メトキサレン 611メトクロプラミド 621メトクロプラミド錠 1723メトトレキサート 610, 1493メトロニダゾール 622メナテトレノン 598, 1492メピチオスタン 600, 1492メフェナム酸 592メフルシド 592メフルシド錠 593メルカプトプリン 603メルファラン 597メロペネム 三水和物 605, 1719dl-メントール 975

18651865Supplement II, JP XIV Index in Japanese
l-メントール 976
モ
モクツウ 859モッコウ 1034モノステアリン酸アルミニウム 861モノステアリン酸グリセリン 931モルヒネ・アトロピン注射液 978,
1553
ヤ
焼セッコウ 938ヤクチ 875薬用石ケン 973薬用炭 972ヤシ油 899
ユ
ユウタン 869ユーカリ油 917輸血用クエン酸ナトリウム注射液
753ユビデカレノン 832
ヨ
ヨウ化エコチオパート 438ヨウ化オキサピウム 657ヨウ化カリウム 697ヨウ化人血清アルブミン (131I)注射液
546ヨウ化ナトリウム 755ヨウ化ナトリウム (123I)カプセル
755ヨウ化ナトリウム (131I)液 756ヨウ化ナトリウム (131I)カプセル
755ヨウ化ヒプル酸ナトリウム (131I)注射
液 756葉酸 490, 1466葉酸錠 491葉酸注射液 491ヨウ素 546ヨクイニン 900ヨクイニン末 900ヨーダミド 545ヨーダミドナトリウムメグルミン注射
液 597ヨード・サリチル酸・フェノール精
952ヨードチンキ 949ヨードホルム 546
ラ
ラウリル硫酸ナトリウム 1056ラウロマクロゴール 964酪酸ヒドロコルチゾン 518酪酸リボフラビン 734ラクツロース 566
ラクトビオン酸エリスロマイシン1462
ラタモキセフナトリウム 569, 1486ラッカセイ油 1002ラナトシド C 567ラナトシド C錠 567
リ
リオチロニンナトリウム 576リオチロニンナトリウム錠 577,
1490リドカイン 574リドカイン注射液 575リファンピシン 737, 1521リボフラビン 733リボフラビン散 734リュウコツ 965硫酸亜鉛 846硫酸亜鉛点眼液 1086硫酸アストロマイシン 255, 1387硫酸アトロピン 255硫酸アトロピン注射液 256, 1689硫酸アミカシン 238硫酸アルベカシン 247, 1386硫酸アルミニウムカリウム 862硫酸イセパマイシン 552硫酸エンビオマイシン 1457硫酸オルシプレナリン 656硫酸カナマイシン 563, 1482一硫酸カナマイシン 1481硫酸カリウム 1019硫酸キニジン 725硫酸キニーネ 728硫酸グアネチジン 507硫酸ゲンタマイシン 502, 1468硫酸コリスチン 1444硫酸サルブタモール 740硫酸シソマイシン 747, 1524硫酸ジベカシン 404, 1447硫酸ストレプトマイシン 766, 1527硫酸セフォセリス 334硫酸セフピロム 338硫酸鉄 474硫酸テルブタリン 786硫酸ネチルマイシン 630硫酸バメタン 262硫酸バリウム 264硫酸ビンクリスチン 842硫酸ビンブラスチン 840, 1752硫酸フラジオマイシン 497, 1467硫酸ブレオマイシン 283, 1398硫酸プロタミン 719硫酸プロタミン注射液 719硫酸ベカナマイシン 265, 1390硫酸ペプロマイシン 675, 1503硫酸ペンブトロール 672硫酸ポリミキシン B 694, 1511硫酸マグネシウム 585硫酸マグネシウム水 970硫酸マグネシウム注射液 586, 1492硫酸ミクロノマイシン 626, 1494硫酸リボスタマイシン 737, 1520
リュウタン 957リュウタン末 957, 1551流動パラフィン 1000リョウキョウ 1544リンゲル液 1028リン酸クリンダマイシン 369, 1440リン酸コデイン 382リン酸コデイン散 1 382, 1442リン酸コデイン散 10 383, 1442リン酸コデイン錠 384, 1443リン酸ジヒドロコデイン 417リン酸ジヒドロコデイン散 1 417,
1449リン酸ジヒドロコデイン散 10
418, 1450リン酸ジメモルファン 421リン酸水素カルシウム 878リン酸水素ナトリウム 907リン酸二水素カルシウム 880リン酸ヒドロコルチゾンナトリウム
519, 1474リン酸ピペラジン 692リン酸ピペラジン錠 692リン酸ベタメタゾンナトリウム 276,
1395リン酸リボフラビンナトリウム 735リン酸リボフラビンナトリウム注射液
736
レ
レセルピン 729, 1518レセルピン散 0.1 731レセルピン錠 731レセルピン注射液 730レボチロキシンナトリウム 572レボチロキシンナトリウム錠 573レボドパ 571レンギョウ 920
ロ
ロキシスロマイシン 738, 1522ロキソプロフェンナトリウム 580ロキタマイシン 737ロジン 1029ロートエキス 1035ロートエキス・アネスタミン散 1037,
1559ロートエキス・カーボン散 1037ロートエキス・タンニン坐剤 1039ロートエキス・パパベリン・アネスタミ
ン散 1038ロートエキス散 1036ロートコン 1040ロラゼパム 579
ワ
ワイル病秋やみ混合ワクチン 1080ワルファリンカリウム 843ワルファリンカリウム錠 843
Related Documents











