Associate editor: S. Pestka The human cytomegalovirus Santo Landolfo a, * , Marisa Gariglio b , Giorgio Gribaudo a , David Lembo a a Department of Public Health and Microbiology, University of Turin, Via Santena 9, 10126 Turin, Italy b Department of Medical Sciences, University of Eastern Piedmont, Novara, Italy Abstract Human cytomegalovirus (HCMV), a betaherpesvirus, represents the major infectious cause of birth defects, as well as an important pathogen for immunocompromised individuals. The viral nucleocapsid containing a linear double-stranded DNA of 230 kb is surrounded by a proteinaceous tegument, which is itself enclosed by a loosely applied lipid bilayer. Expression of the HCMV genome is controlled by a cascade of transcriptional events that leads to the synthesis of three categories of viral proteins designated as immediate-early, early, and late. Clinical manifestations can be seen following primary infection, reinfection, or reactivation. About 10% of infants are infected by the age of 6 months following transmission from their mothers via the placenta, during delivery, or by breastfeeding. HCMV is a significant post-allograft pathogen and contributes to graft loss independently from graft rejection. Histopathologic examination of necropsy tissues demonstrates that the virus enters via the epithelium of the upper alimentary, respiratory, or genitourinary tracts. Hematogenous spreading is typically followed by infection of ductal epithelial cells. Infections are kept under control by the immune system. However, total HCMV clearance is rarely achieved, and the viral genome remains at selected sites in a latent state. Virological and molecular detection of HCMV, as well as serological demonstration of a specific immune response, are used for diagnosis. Treatment of HCMV infections is difficult because there are few options. The presently available drugs produced a significant clinical improvement, but suffer from poor oral bioavailability, low potency, development of resistance in clinical practice, and dose-limiting toxicities. D 2003 Elsevier Science Inc. All rights reserved. Keywords: HCMV; Gene expression; Epidemiology; Transplant; Diagnosis; Antiviral therapy Abbreviations: ab, antibody; AIDS, acquired immunodeficiency syndrome; AP, assembly protein; AP-1, activator protein-1; ATF, activating transcription factor; BMT, bone marrow transplant; CDV, cidofovir; CMV, cytomegalovirus; CPE, cytopathic effect; CREB, cyclic AMP-responsive element binding protein; crs, cis-repression signals; CTL, cytotoxic T lymphocyte; DB, dense bodies; E, early; ER, endoplasmic reticulum; GCV, ganciclovir; GPCR, G- protein-coupled receptor; HCMV, human cytomegalovirus; HIV, human immunodeficiency virus; IE, immediate-early; Ig, immunoglobulin; L, late; mCP, minor capsid protein; MCP, major capsid protein; MHC, major histocompatibility complex; MIE, major immediate-early; NF, nuclear factor; NK, natural killer; ORF, open reading frame; PBL, peripheral blood leukocytes; PCR, polymerase chain reaction; PFA, foscarnet; p.i., post-infection; Rb, Retinoblastoma; TAP, transporter associated with antigen processing; UL, unique long; US, unique short. Contents 1. Introduction ............................................ 270 2. The human cytomegalovirus ................................... 270 2.1. Virus structure ....................................... 270 2.2. Virus genome ....................................... 272 2.3. Virus growth cycle and viral gene expression ....................... 273 2.3.1. Range of cell permissivity and in vitro growth .................. 273 2.3.2. Virus binding and penetration .......................... 273 2.3.3. Regulation of viral gene expression ....................... 274 2.3.4. Characteristics and functions of the immediate-early proteins .......... 274 2.3.5. Characteristics and functions of the early proteins ................ 276 2.3.6. Viral DNA replication .............................. 277 0163-7258/03/$ – see front matter D 2003 Elsevier Science Inc. All rights reserved. doi:10.1016/S0163-7258(03)00034-2 * Corresponding author. Tel.: +39-11-6706604; fax: +39-11-6636436. E-mail address: [email protected] (S. Landolfo). www.elsevier.com/locate/pharmthera Pharmacology & Therapeutics 98 (2003) 269 – 297

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

www.elsevier.com/locate/pharmthera
Pharmacology & Therapeutics 98 (2003) 269–297
Associate editor: S. Pestka
The human cytomegalovirus
Santo Landolfoa,*, Marisa Garigliob, Giorgio Gribaudoa, David Lemboa
aDepartment of Public Health and Microbiology, University of Turin, Via Santena 9, 10126 Turin, ItalybDepartment of Medical Sciences, University of Eastern Piedmont, Novara, Italy
Abstract
Human cytomegalovirus (HCMV), a betaherpesvirus, represents the major infectious cause of birth defects, as well as an important
pathogen for immunocompromised individuals. The viral nucleocapsid containing a linear double-stranded DNA of 230 kb is surrounded by
a proteinaceous tegument, which is itself enclosed by a loosely applied lipid bilayer. Expression of the HCMV genome is controlled by a
cascade of transcriptional events that leads to the synthesis of three categories of viral proteins designated as immediate-early, early, and late.
Clinical manifestations can be seen following primary infection, reinfection, or reactivation. About 10% of infants are infected by the age of 6
months following transmission from their mothers via the placenta, during delivery, or by breastfeeding. HCMV is a significant post-allograft
pathogen and contributes to graft loss independently from graft rejection. Histopathologic examination of necropsy tissues demonstrates that
the virus enters via the epithelium of the upper alimentary, respiratory, or genitourinary tracts. Hematogenous spreading is typically followed
by infection of ductal epithelial cells. Infections are kept under control by the immune system. However, total HCMV clearance is rarely
achieved, and the viral genome remains at selected sites in a latent state. Virological and molecular detection of HCMV, as well as serological
demonstration of a specific immune response, are used for diagnosis. Treatment of HCMV infections is difficult because there are few
options. The presently available drugs produced a significant clinical improvement, but suffer from poor oral bioavailability, low potency,
development of resistance in clinical practice, and dose-limiting toxicities.
D 2003 Elsevier Science Inc. All rights reserved.
Keywords: HCMV; Gene expression; Epidemiology; Transplant; Diagnosis; Antiviral therapy
Abbreviations: ab, antibody; AIDS, acquired immunodeficiency syndrome; AP, assembly protein; AP-1, activator protein-1; ATF, activating transcription
factor; BMT, bone marrow transplant; CDV, cidofovir; CMV, cytomegalovirus; CPE, cytopathic effect; CREB, cyclic AMP-responsive element binding
protein; crs, cis-repression signals; CTL, cytotoxic T lymphocyte; DB, dense bodies; E, early; ER, endoplasmic reticulum; GCV, ganciclovir; GPCR, G-
protein-coupled receptor; HCMV, human cytomegalovirus; HIV, human immunodeficiency virus; IE, immediate-early; Ig, immunoglobulin; L, late; mCP,
minor capsid protein; MCP, major capsid protein; MHC, major histocompatibility complex; MIE, major immediate-early; NF, nuclear factor; NK, natural killer;
ORF, open reading frame; PBL, peripheral blood leukocytes; PCR, polymerase chain reaction; PFA, foscarnet; p.i., post-infection; Rb, Retinoblastoma; TAP,
transporter associated with antigen processing; UL, unique long; US, unique short.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 270
2. The human cytomegalovirus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 270
2.1. Virus structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 270
2.2. Virus genome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272
2.3. Virus growth cycle and viral gene expression . . . . . . . . . . . . . . . . . . . . . . . 273
2.3.1. Range of cell permissivity and in vitro growth . . . . . . . . . . . . . . . . . . 273
2.3.2. Virus binding and penetration . . . . . . . . . . . . . . . . . . . . . . . . . . 273
2.3.3. Regulation of viral gene expression . . . . . . . . . . . . . . . . . . . . . . . 274
2.3.4. Characteristics and functions of the immediate-early proteins . . . . . . . . . . 274
2.3.5. Characteristics and functions of the early proteins . . . . . . . . . . . . . . . . 276
2.3.6. Viral DNA replication . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 277
0163-7258/03/$ – see front matter D 2003 Elsevier Science Inc. All rights reserved.
doi:10.1016/S0163-7258(03)00034-2
* Corresponding author. Tel.: +39-11-6706604; fax: +39-11-6636436.
E-mail address: [email protected] (S. Landolfo).

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297270
2.3.7. Characteristics and functions of the late proteins . . . . . . . . . . . . . . . . . 278
2.3.8. Virion assembly, maturation, and egress . . . . . . . . . . . . . . . . . . . . . 278
3. Epidemiology of human cytomegalovirus infection . . . . . . . . . . . . . . . . . . . . . . . . 278
4. Infection routes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279
5. Pathogenesis and pathology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 280
6. Host defences . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
6.1. Cell-mediated immunity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
6.2. Humoral immunity. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 282
6.3. Immune evasion by human cytomegalovirus . . . . . . . . . . . . . . . . . . . . . . . 282
6.4. Persistence and release from the host . . . . . . . . . . . . . . . . . . . . . . . . . . . 283
7. Clinical features associated with human cytomegalovirus infection. . . . . . . . . . . . . . . . 284
7.1. Infection in normal hosts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
7.2. Congenital infection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
7.3. Cytomegalovirus infection in the immunocompromised host . . . . . . . . . . . . . . . 284
8. Diagnosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
8.1. Virus detection. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
8.1.1. Viremia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
8.1.2. Antigenemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
8.1.3. DNAemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
8.1.4. RNAemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
8.2. Detection of the immune response. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
9. Prevention of human cytomegalovirus infection and disease . . . . . . . . . . . . . . . . . . . 286
9.1. Human cytomegalovirus vaccines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
10. Antiviral treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 287
10.1. Currently available drugs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 287
10.2. Therapeutic approaches. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
10.2.1. Prophylaxis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
10.2.2. Pre-emptive treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
10.2.3. Treatment of an established disease . . . . . . . . . . . . . . . . . . . . . . . 288
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289
1. Introduction
The human cytomegalovirus (HCMV) is a widespread
pathogen responsible for generally asymptomatic and per-
sistent infections in healthy people. It may, however, cause
severe disease in the absence of an effective immune
response, as in immunologically immature and immunocom-
promised individuals. Its impact, therefore, has increased in
recent decades due to the rise in organ allografting, immu-
nosuppressive treatment, and human immunodeficiency
virus (HIV)-infected patients. Furthermore, it is the leading
infectious agent causing birth defects (Griffiths, 2000; Pass,
2001).
HCMV is a member of the betaherpesvirinae subfamily,
whose virion structure, kinetics of viral gene expression,
and persistence for the lifetime of their host are typical of
other herpesviruses. However, its strict species specificity,
salivary gland tropism, and slow growth in cell cultures
differentiate it as the prototype betaherpesvirus (Mocarski &
Courcelle, 2001).
This paper provides an overview of the general character-
istics of HCMV structure, growth, and replication, as well as
the pathogenesis, epidemiology, diagnosis, and management
of the infections it causes.
2. The human cytomegalovirus
2.1. Virus structure
The virion of HCMV consists of a 100-nm diameter
icosahedral nucleocapsid containing a 230-kbp, double-
stranded linear DNA genome surrounded by a proteinaceous
layer defined as the tegument or matrix, which, in turn, is
enclosed by a lipid bilayer containing a large number of
viral glycoproteins. The mature virion particle is 150–200
nm in diameter.
HCMV-infected cell cultures produce the infectious viri-
ons and another two types of morphological particles: non-
infectious enveloped particles and dense bodies (DB).
Noninfectious enveloped particles are defective viral par-
ticles composed of enveloped immature capsids (type B)
that lack DNA, but contain the viral scaffolding/assembly
protein (AP) normally absent from fully mature nucleocap-
sids (C-capsids). DB are enveloped particles that lack an
assembled nucleocapsid and viral DNA, but contain several
tegument proteins of which ppUL83 (pp65 or lower matrix
protein) is the most abundant. The relative amounts of the
three viral forms depends on the number of passages in cell
culture and the viral strain (Mocarski & Courcelle, 2001).
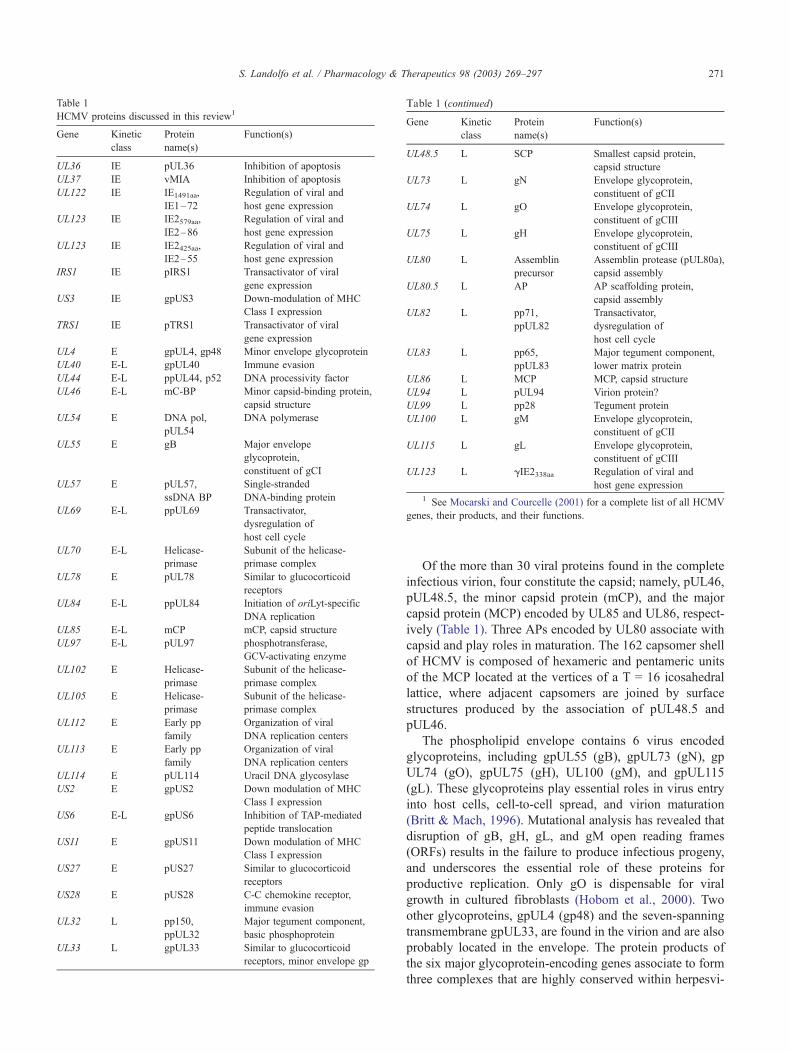
Table 1
HCMV proteins discussed in this review1
Gene Kinetic
class
Protein
name(s)
Function(s)
UL36 IE pUL36 Inhibition of apoptosis
UL37 IE vMIA Inhibition of apoptosis
UL122 IE IE1491aa,
IE1–72
Regulation of viral and
host gene expression
UL123 IE IE2579aa,
IE2–86
Regulation of viral and
host gene expression
UL123 IE IE2425aa,
IE2–55
Regulation of viral and
host gene expression
IRS1 IE pIRS1 Transactivator of viral
gene expression
US3 IE gpUS3 Down-modulation of MHC
Class I expression
TRS1 IE pTRS1 Transactivator of viral
gene expression
UL4 E gpUL4, gp48 Minor envelope glycoprotein
UL40 E-L gpUL40 Immune evasion
UL44 E-L ppUL44, p52 DNA processivity factor
UL46 E-L mC-BP Minor capsid-binding protein,
capsid structure
UL54 E DNA pol,
pUL54
DNA polymerase
UL55 E gB Major envelope
glycoprotein,
constituent of gCI
UL57 E pUL57,
ssDNA BP
Single-stranded
DNA-binding protein
UL69 E-L ppUL69 Transactivator,
dysregulation of
host cell cycle
UL70 E-L Helicase-
primase
Subunit of the helicase-
primase complex
UL78 E pUL78 Similar to glucocorticoid
receptors
UL84 E-L ppUL84 Initiation of oriLyt-specific
DNA replication
UL85 E-L mCP mCP, capsid structure
UL97 E-L pUL97 phosphotransferase,
GCV-activating enzyme
UL102 E Helicase-
primase
Subunit of the helicase-
primase complex
UL105 E Helicase-
primase
Subunit of the helicase-
primase complex
UL112 E Early pp
family
Organization of viral
DNA replication centers
UL113 E Early pp
family
Organization of viral
DNA replication centers
UL114 E pUL114 Uracil DNA glycosylase
US2 E gpUS2 Down modulation of MHC
Class I expression
US6 E-L gpUS6 Inhibition of TAP-mediated
peptide translocation
US11 E gpUS11 Down modulation of MHC
Class I expression
US27 E pUS27 Similar to glucocorticoid
receptors
US28 E pUS28 C-C chemokine receptor,
immune evasion
UL32 L pp150,
ppUL32
Major tegument component,
basic phosphoprotein
UL33 L gpUL33 Similar to glucocorticoid
receptors, minor envelope gp
Gene Kinetic
class
Protein
name(s)
Function(s)
UL48.5 L SCP Smallest capsid protein,
capsid structure
UL73 L gN Envelope glycoprotein,
constituent of gCII
UL74 L gO Envelope glycoprotein,
constituent of gCIII
UL75 L gH Envelope glycoprotein,
constituent of gCIII
UL80 L Assemblin
precursor
Assemblin protease (pUL80a),
capsid assembly
UL80.5 L AP AP scaffolding protein,
capsid assembly
UL82 L pp71,
ppUL82
Transactivator,
dysregulation of
host cell cycle
UL83 L pp65,
ppUL83
Major tegument component,
lower matrix protein
UL86 L MCP MCP, capsid structure
UL94 L pUL94 Virion protein?
UL99 L pp28 Tegument protein
UL100 L gM Envelope glycoprotein,
constituent of gCII
UL115 L gL Envelope glycoprotein,
constituent of gCIII
UL123 L gIE2338aa Regulation of viral and
host gene expression
1 See Mocarski and Courcelle (2001) for a complete list of all HCMV
genes, their products, and their functions.
Table 1 (continued)
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 271
Of the more than 30 viral proteins found in the complete
infectious virion, four constitute the capsid; namely, pUL46,
pUL48.5, the minor capsid protein (mCP), and the major
capsid protein (MCP) encoded by UL85 and UL86, respect-
ively (Table 1). Three APs encoded by UL80 associate with
capsid and play roles in maturation. The 162 capsomer shell
of HCMV is composed of hexameric and pentameric units
of the MCP located at the vertices of a T = 16 icosahedral
lattice, where adjacent capsomers are joined by surface
structures produced by the association of pUL48.5 and
pUL46.
The phospholipid envelope contains 6 virus encoded
glycoproteins, including gpUL55 (gB), gpUL73 (gN), gp
UL74 (gO), gpUL75 (gH), UL100 (gM), and gpUL115
(gL). These glycoproteins play essential roles in virus entry
into host cells, cell-to-cell spread, and virion maturation
(Britt & Mach, 1996). Mutational analysis has revealed that
disruption of gB, gH, gL, and gM open reading frames
(ORFs) results in the failure to produce infectious progeny,
and underscores the essential role of these proteins for
productive replication. Only gO is dispensable for viral
growth in cultured fibroblasts (Hobom et al., 2000). Two
other glycoproteins, gpUL4 (gp48) and the seven-spanning
transmembrane gpUL33, are found in the virion and are also
probably located in the envelope. The protein products of
the six major glycoprotein-encoding genes associate to form
three complexes that are highly conserved within herpesvi-

Fig. 1. Structure of the four HCMV genome isomers.
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297272
ruses and previously have been designated as gCI, gCII, and
gCIII (Gretch et al., 1988). At least two are required for viral
entry: gCI, composed of homodimeric gB molecules, and
gCIII, a heteroligomeric complex composed of gH, gL, and
gO (Theiler & Compton, 2001). gB is an essential gly-
coprotein that plays a crucial role in virus binding, since it is
the major cell surface, heparan sulfate proteoglycan-binding
glycoprotein. It also participates in viral entry, cell-to-cell
spread, and cell fusion. By virtue of its abundance and
ability to elicit neutralizing antibodies (abs), it has been
proposed as a molecule of choice for a recombinant subunit
vaccine (Gonczol & Plotkin, 2001). gCIII is necessary for
the final stage of virus entry via pH-independent fusion
between the viral envelope and the cell membrane (Huber &
Compton, 1998). gCII results from the association of gM
and gN (Kari et al., 1994). All three complexes induce
neutralizing abs. Because of their interaction with the
immune system, the genes encoding gB, gH, and gN have
been detected as highly polymorphic loci that can be
clustered in different genotypes in clinical isolates. The
occurrence of ab-escaping mutant viruses selected by the
pressure of the humoral immune response may be related to
cell tropism and virulence, and is thought to contribute to
the pathogenicity of HCMV.
The remaining 20–25 structural virion proteins probably
are located in the still poorly characterized amorphous layer
between the nucleocapsid and the envelope (Baldick &
Shenk, 1996). The tegument proteins may be involved in
the maturation of progeny virions, or may influence viral
and cellular events in the early stages of infection, such as
release of viral DNA from disassembling virus particles or
the regulation of viral and cellular promoters. Most tegu-
ment proteins are phosphorylated and are highly immuno-
genic. The most abundant are ppUL32 (pp150 or basic
phosphoprotein) and ppUL83. Due to its large amounts,
pp65 is the target antigen in antigenemia assays for rapid
diagnosis of HCMV-substained clinical infections. Tegu-
ment proteins such as ppUL69 and ppUL82 (pp71) may
play important regulatory roles in both viral and cellular
gene expression. They are, in fact, transactivators of viral
gene expression, and deeply impact on host cell physiology
by dysregulating cycle progression. UL69 stimulates
expression from the ie1/ie2 enhancer-promoter. It is required
for efficient viral replication, and arrests cells in G1 by an
unknown mechanism (Lu & Shenk, 1999; Hayashi et al.,
2000). UL82 is a transcriptional activator of activating
transcription factor (ATF)/cyclic AMP-response element
binding protein (CREB) or the activator protein-1 (AP-1)-
containing promoter and, thus, stimulates the activity of the
ie1/ie2 enhancer-promoter. It also increases the infectivity of
transfected viral DNA, is required for viral replication at low
multiplicities of infection, and accelerates the G1 to S
transition of quiescent cells (Liu & Stinski, 1992; Baldick
et al., 1997; Bresnahan & Shenk, 2000b). Two more trans-
activator proteins (pIRS1 and pTRS1) are associated with
virions and DB (Romanowski et al., 1997).
Five viral RNAs (designated as virion RNAs) have been
detected in preparations of highly purified, infectious
HCMV particles. These HCMV transcripts probably are
located in the tegument and are delivered to the host cell on
infection. However, the functional significance of their
presence in the virions and that of their protein products
remains to be defined (Bresnahan & Shenk, 2000a).
2.2. Virus genome
The HCMV genome is the largest of all herpesviruses and
has a high G+C content. Like that of herpes simplex virus-1,
it contains an arrangement of unique long (UL), unique short
(US), and repeat regions. Since each long and short region
can be oriented in either direction, four genome isomers are
produced in viral progeny (Class E structure) (Fig. 1). In
contrast, the genomes of animal CMV, as well as those of
other betaherpesviruses, are linear without repeat regions
(Class F genomes). Inversion of UL and US regions is
mediated by direct repeat sequences (a, b, c) at the genome
termini and by inverted repeat elements at the UL-US
junction (a0, b0, c0). The repeated a sequence that occurs as a
direct element at the termini and in the inverted orientation at
the UL/US junction promotes genome isomerization, since it
contains the cis-acting pac (packaging) elements needed for
DNA cleavage (Mocarski & Courcelle, 2001).
The AD169 laboratory strain is the only completely
sequenced HCMV. Analysis of its 230-kbp genome has
revealed that it encodes 225 ORFs of � 100 or more amino
acids (Chee et al., 1990a; Novotny et al., 2001). These
ORFs are designated sequentially according to their location
within the unique and repeated regions. Additional ORFs
have been identified in the Towne and Toledo laboratory
strains. In the latter, as well as in clinical isolates, the
inverted b0 repeat is deleted and replaced by an additional
UL region of � 15 kbp, containing 19 additional ORFs that
are absent in the AD169 genome (Cha et al., 1996). The
unique ORF UL1–154 and US1–36 blocks are separated
by duplicated IRL1–14 and J1I genes and the partially
repeated IRS1 gene. The UL region is flanked at the 50 end
by the duplicated TRL1–14 and J1L (identical to IRL1–14

Fig. 2. HCMV cytopathic effect on a human fibroblasts monolayer.
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 273
and J1I), whereas the US gene block is flanked at its 30 end
by the TRS1 gene and by the third copy of a J1 gene (J1S).
Comparison of the AD169 amino acid sequences with
those of other herpesvirus genomes has revealed that the
protein products of more than 40 ORFs share high similarity
to proteins encoded by alpha- and gammaherpesviruses
(Chee et al., 1990a; Karlin et al., 1994), and provided
further evidence of a common origin of the three subfam-
ilies. Of the herpesvirus-conserved ORFs, � 25% appear to
encode functions related to viral DNA metabolism and
replication, whereas the remaining 75% are thought to be
involved in the maturation and structural organization of
virions. The UL ORF-encoding functions involved in DNA
replication and repair, nucleotide metabolism, or virion
structure are grouped in seven conserved gene blocks (A–
G) also found in other herpesviruses, such as herpes simplex
virus-1 and Epstein-Barr virus, although in a different order
in the alpha- and gammaherpesviruses. The occurrence of
these conserved blocks in a characteristic order specific to
each subfamily suggests that the functions encoded are
probably conserved in all herpesviruses. The US ORF and
those located within the repeated regions of the HCMV
genome are less well conserved than in the other herpesvi-
ruses (Chee et al., 1990a).
Sequence homology searches and experimental biochem-
ical and/or genetic studies have assigned functional roles to
only some of the more than 200 HCMV ORFs (Novotny et
al., 2001). However, analysis of the phenotypes of spontan-
eous deletion mutants of the AD169 strain, as well as those
of virus-bearing deletions or inactivation at specific loci, has
indicated that the products of more than 50 HCMV ORFs
are dispensable for productive replication in fibroblast
cultures. These findings, along with observations that the
proteins responsible for functions common to all herpesvi-
ruses, such as basic DNA replication, virion organization,
and maturation, do not account for all the ORFs, indicate
that many ORFs still await functional characterization. It is
likely, therefore, that much of the coding capability has
evolved to optimize infection by influencing dissemination,
growth in target tissues and pathogenesis, and in counter-
acting host immune reactivity (Mocarski & Courcelle,
2001).
2.3. Virus growth cycle and viral gene expression
2.3.1. Range of cell permissivity and in vitro growth
Autopsy specimens show that HCMV infects a wide
range of epithelial tissues. The ductal epithelial cell is most
commonly infected, and develops a typical cytopathology
(Pass, 2001; Bissinger et al., 2002). However, during natural
infection, it is thought that HCMV replicates productively in
epithelial cells, endothelial cells, smooth muscle cells,
mesenchymal cells, hepatocytes, granulocytes, and mono-
cyte-derived macrophages (Plachter et al., 1996; Sinzger &
Jahn, 1996; Sinzger et al., 1996; Kahl et al., 2000; Bissinger
et al., 2002). Latent viral DNA, thus, can be detected in
macrophage-granulocyte progenitors in the bone marrow
and in peripheral monocytes (Kondo et al., 1994; Soder-
berg-Naucler et al., 1997). In contrast, in vitro, the only cells
fully permissive for replication of laboratory strains are
human skin or lung fibroblasts, whereas clinical isolates
replicate preferentially on endothelial cell cultures. Some
transformed cell lines derived from glioblastomas, such as
U373MG, as well as primary arterial smooth muscle cells,
support productive infection, although at lower levels than
in fibroblasts (Sinzger et al., 1995; Plachter et al., 1996). In
fibroblasts, the replication cycle is slow, with a typical
cytopathic effect (CPE) characterized by cell rounding and
enlargement with both intra- and perinuclear inclusions (Fig.
2). Only laboratory strains, such as AD169 or Towne,
produce a generalized CPE, with lysis of the entire cell
monolayer, and release high titers of virions starting from 3
to 4 days post-infection (p.i.). Recently, isolated strains
mainly spread on fibroblast monolayers by cell-to-cell
contact and produce a limited CPE, with dispersed foci
and poor virion yields (Pass, 2001).
2.3.2. Virus binding and penetration
Virus attachment and penetration are rapid and efficient
in both permissive and nonpermissive cell types. However,
since productive replication is observed in a very restricted
range of human cells, a post-penetration block to viral gene
expression is thought to restrict replication in nonpermissive
cells (Sinzger et al., 2000). The poorly characterized recep-
tor(s) for HCMV is widely distributed among host cell
types, and contributes to the broad viral tropism observed
during natural infections. Viral entry is the result of a
cascade of interactions between viral and cellular proteins
that culminate in fusion of the virion envelope with the
cellular plasma membrane by a pH-independent mechanism.
During the initial virus-cell interactions, as observed with
other herpesviruses, HCMV attaches to the cell surface by
low-affinity binding of gB to heparan sulfate proteoglycans
(Compton et al., 1993). The subsequent interaction of gB
with its nonheparin receptor then turns the weak adhesion of
the viral particle into a more stable binding or docking state.

S. Landolfo et al. / Pharmacology & T274
However, final fusion of the viral envelope with the cell
membrane to allow viral penetration is thought to require a
further priming event mediated by the heteroligomeric gH-
gL-gO complex with as yet unidentified receptors (Theiler
& Compton, 2001). Fusion of the virus and cell membranes
is followed by entry into the host cytoplasm of the nucle-
ocapsid and tegument proteins, and their rapid translocation
into the nucleus, where pp65 is detected < 1 hr p.i. Inter-
action of HCMV glycoproteins with their receptors is
enough to generate an intracellular signal transduction
pathway, leading to the alteration of cellular gene expres-
sion. Most changes in the profiles of host gene activity
resemble those induced by binding of interferons to their
receptors (Zhu et al., 1997, 1998; Browne et al., 2001). The
specific viral ligand triggering this response is gB, and its
interaction with its as yet unidentified receptor is thought to
be the main mechanism by which HCMV modifies host cell
gene expression in the very early phases of infection (Boyle
et al., 1999; Simmen et al., 2001). In addition, engagement
of gB and gH with their receptors is sufficient to induce the
activity of the cellular transcription factors nuclear factor-kB(NF-kB) and Sp1 (Yurochko et al., 1997).
2.3.3. Regulation of viral gene expression
During productive infection, the HCMV genome is
expressed in a temporally coordinated and regulated cascade
of transcriptional events that lead to the synthesis of three
categories of viral proteins described as immediate-early (IE
or a), early (E or b), and late (L or g) (Fig. 3). Failure in the
expression of early gene and subsequent viral DNA replica-
tion rather than attachment and/or penetration may be the
restricting event in nonpermissive cells. HCMV genes are
transcribed in the infected cell nucleus by RNA polymerase
II and the associated basal transcription machinery, with the
intervention of host-encoded transcription factors whose
activity may be stimulated by viral transactivators (Fortu-
nato & Spector, 1999; Mocarski & Courcelle, 2001).
Fig. 3. HCMV gene expression and viral gene pro
2.3.4. Characteristics and
functions of the immediate-early proteins
HCMV gene expression initiates from a few IE proteins
within l hr p.i. without de novo protein synthesis. The IE
genes include the major IE (MIE) UL122/123 genes (IE1
and IE2) and auxiliary genes, such as UL36–UL38,
UL115–UL119, IRS1/TRS1, and US3 (Table 1). The MIE
proteins, alone or in synergism, are required for subsequent
expression by acting as transactivators and autostimulators
of viral genes. In addition, these proteins have a deep impact
on host cell physiology since they regulate the expression of
a large number of host cell genes (Fortunato & Spector,
1999).
MIE proteins are encoded by the ie1/ie2 genes (UL122/
UL123), whose expression is regulated by a complex
enhancer-modulator element that functions in a tissue- and
cell-type-specific manner (Nelson et al., 1990; Meier &
Stinski, 1996), and exerts its strong transcriptional activity
through interactions with several host transcription factors
whose binding sites are closely distributed within the
regulatory element. An � 500-bp segment upstream from
the TATA box of the ie1/ie2 genes contains several repeat
elements with binding sites for NF-kB, AP-1, Sp1, and
CREB/ATF (Boshart et al., 1985). Since their cognate
DNA-binding activities are rapidly activated by HCMV
infection, their binding to the corresponding sites is thought
to contribute to the very strong activity of the MIE enhancer.
The CREB/ATF and AP-1 sites are also responsive to the
tegument transactivator ppUL82 (Liu & Stinski, 1992). In
addition, NF-1, serum response factor, Elk-1, CCAAT/
enhancer-binding protein, and YY1 sites have been iden-
tified within the enhancer segment. The cell type- and
differentiation state-specific enhancer activity is related to
the availability of appropriate transcription factors in a
specific cell type, and is thought to depend on the modulator
region. This element spans � 500 bp upstream from the
core of the MIE enhancer. In transient transfection assays,
herapeutics 98 (2003) 269–297
duct functions during productive infection.

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 275
the modulator region represses the transcriptional activity of
the enhancer in undifferentiated cells lines, although these
findings could not be reproduced with mutagenized viruses
in which the modulator element was removed (Nelson et al.,
1990; Meier & Stinski, 1997).
The MIE region produces a set of transcripts synthesized
from different start sites and differentially spliced and
polyadenylated to give rise to four protein products. The
most abundant in IE times of infection are IE1491aa and
IE2579aa. These share the same amino terminal 85 amino
acid long segment, but their differentially spliced exons are
4 (UL123) and 5 (UL122), respectively. In addition, a less
abundant IE2 isoform (IE2425aa) is encoded by a spliced
transcript bearing an in-frame deletion within UL122. The
fourth IE protein expressed from the MIE region is a true
late 40-kDa polypeptide (gIE2338aa) encoded by a polyade-
nylated transcript that originates late in infection from a start
site within UL122. Both IE1491aa and IE2579aa are expressed
throughout replication, albeit with slightly different kinetic
profiles. Their ability to regulate transcription of several
viral and cellular genes has been studied extensively (For-
tunato & Spector, 1999; Mocarski & Courcelle, 2001).
IE1479aa is a nuclear phosphoprotein of � 72 kDa (IE1-
72), whose functions are not needed for viral growth in
fibroblast cultures infected at high multiplicity of infection,
but are required after a low multiplicity of infection. The
presence of IE1-72, thus, is crucial when infection starts
from a single virion (Mocarski et al., 1996). Generation of
mutants lacking UL123 and, hence, IE1-72 expression has
confirmed in infected cells its functions, as predicted by
transfection assays. The first role of IE1-72 is consistent
with its ability to positively autoregulate expression of the
ie1/ie2 and US3 genes by activating the corresponding
enhancer elements through NF-kB-binding sites (Mocarski
et al., 1996). In addition, IE1-72 cooperates with IE2579aa to
regulate the expression of viral genes belonging to the
subsequent categories, as predicted by transient transfection
assays in which co-expression of the two IE proteins were
found to be required for maximal activity of the promoters
of E (UL44 and UL54), as well as L (UL83), genes
(Fortunato & Spector, 1999). IE1-72 is a weaker and less
promiscuous transactivator than IE2579aa. In these assays,
however, it stimulates the activity of several cellular pro-
moters, including those of dihydrofolate reductase, DNA
polymerase a, c-fos, c-myc, NF-kB p65 subunit, and thy-
midylate synthase genes (Hagemeier et al., 1992a; Wade et
al., 1992; Hayhurst et al., 1995; Yurochko et al., 1995;
Gribaudo et al., 2002). In addition, IE1 may affect the host
cell cycle regulatory pathway by targeting the Retinoblas-
toma (Rb) protein family and E2F-mediated transcriptional
mechanisms since it interacts with cellular p107, a member
of the Rb family, and activates an E2F-responsive promoter,
such as that of the DHFR gene, by alleviating p107-
mediated repression (Poma et al., 1996; Johnson et al.,
1999). Moreover, it interacts physically with E2F1 and
transactivates the DHFR promoter only if its E2F sites are
retained (Margolis et al., 1995). Lastly, IE1-72 is endowed
with a kinase activity that autophosphorylates and phos-
phorylates the p107 and p130, as well as E2F1, -2, and -3.
The putative catalytic domain of IE1-72 is also required for
the activation of E2F-dependent transcription (Pajovic et al.,
1997).
IE2579aa is an 86-kDa (IE2-86) nuclear phosphorylated
polypeptide whose activities are critical for viral replication.
A bacterial artificial chromosome clone of HCMV with a
deletion of exon 5 unique to IE2-86 (the UL122 ORF), in
fact, did not activate E gene expression nor yield progeny
virus upon transfection in permissive cells (Marchini et al.,
2001). Since it is difficult to generate mutant viruses lacking
IE2-86 and complementing long-term cell lines expressing
fully functional wild-type IE2-86, most studies have relied
on transient transfection experiments, many of which have
indeed established that IE2-86 behaves as a strong tran-
scriptional regulator by either stimulating or repressing both
HCMV and cellular genes (Fortunato & Spector, 1999;
Mocarski & Courcelle, 2001; Song & Stinski, 2002). During
productive infection, IE2-86 is the key protein regulating the
transition from the IE to the E, and very likely to the L
phases of HCMV gene expression. Characterization of its
function in transient transfection assays, in fact, has dem-
onstrated that alone or in combination with IE1, it trans-
activates several E and L promoters (Fortunato & Spector,
1999), and IE2-86-binding sites have been identified just
upstream from the TATA box of three E promoters. These
transactivating properties probably are attributable to the
interaction of IE2-86 with factors of the basal-transcription
machinery and a variety of promoter-specific transcription
factors. Recombinant IE2-86 produced in bacteria, in fact,
interacts with the TATA-binding protein, TFIIB, TAFII-130,
and TFIID (Hagemeier et al., 1992b; Caswell et al., 1993;
Jupp et al., 1993; Lukac et al., 1997). It also interacts in
vitro with other transcription factors, such as histone acetyl-
transferase, CREB, Sp1, c-Jun, and ATF-2, as well as with
cell-cycle regulatory factors, including p53, pRb, and p21
(Speir et al., 1993; Hagemeier et al., 1994; Sommer et al.,
1994; Fortunato et al., 1997). These interactions indicate
that IE2-86 may act as an adapter-like protein by stabilizing
basal and specific transcription factors on the promoter
region (Mocarski & Courcelle, 2001). Consistent with this
model, the occurrence of cis-acting regulatory elements;
namely, AP-1, CREB/ATF, and Sp1, upstream from some E
promoters contributes to the overall promoter responsive-
ness to IE2-86-mediated transactivation (Fortunato & Spec-
tor, 1999).
In addition to their regulation of the expression of E and
L genes, IE2-86 and gIE2338aa repress expression of ie1/ie2
and US3 (Fortunato & Spector, 1999; Mocarski & Cour-
celle, 2001). This down-regulation is mediated by IE2
binding to cis-repression signals (crs) sequences located
between the TATA box and the transcription initiation sites
of these genes (Pizzorno & Hayward, 1990; Cherrington et
al., 1991). By down-regulating transcription from its own

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297276
promoter, therefore, IE2-86 mediates autoregulation of its
own expression and contributes to the reduction of IE gene
expression in the late stages of infection. In vitro trans-
fection assays have established that binding of IE2-86 to crs
confers the IE2-dependent repression of homologous and
heterologous promoters, provided that the sequence element
is located near the transcription start site. Moreover, purified
bacterial IE2-86 binds to crs through its C-terminus and
alters RNA polymerase pre-initiation complex formation in
the in vitro repression of transcription assays.
IE2-86 also modulates several cell processes, including
cycle control and apoptosis (Zhu et al., 1995; Kalejta &
Shenk, 2002). In fact, expression of IE2-86 by either
transient transfection or replicative-defective adenovirus
arrests cells in an early S phase by mechanisms that are
not known yet (Wiebush & Hagemeier, 1999, 2001; Murphy
et al., 2000). Their limited DNA synthesis and high levels of
cyclin E-dependent kinase activity show that these cells are
blocked one step further than the G1 restriction point of the
cycle. IE2-86 stimulates quiescent, serum-starved cells into
the early S phase, but prevents them from progressing
through this phase, and cell division does not occur. Its
ability to arrest the cycle thus greatly contributes to the late
G1/early S phase synchronization and host DNA synthesis
inhibition observed in HCMV-infected cells (Jault et al.,
1995; Lu & Shenk, 1996; Bresnahan et al., 1996; Dittmer &
Mocarski, 1997; Salvant et al., 1998). These modifications
lead to a more favorable environment for viral replication,
since the precursors for DNA synthesis produced by the host
are available, but are not utilized by the DNA replication
machinery of the cell (Fortunato et al., 2000; Kalejta &
Shenk, 2002).
The functions of the other IE genes are heterogeneous.
TRS1 and IRS cooperate with IE1-72 and IE2-86 in the
transactivation of E promoters. The glycoproteins encoded
by the US3 gene are involved in the establishment of
immune evasion in infected cells, since these endoplasmic
reticulum (ER)-located proteins prevent the transport of the
assembled major histocompatibility complex (MHC) Class I
from ER to the Golgi and, thus, down-modulate MHC Class
I antigen presentation (Stasiak & Mocarski, 1992; Jones et
al., 1996). In contrast, the UL36–38 region encodes pro-
teins such as UL36 and UL37 that are endowed with anti-
apoptotic properties and that act at different steps in the
caspase cascade (Skaletskaya et al., 2001; Goldmacher,
2002). UL36, UL37, US3, and IRS1 are all dispensable
for replication in cell culture.
2.3.5. Characteristics and functions of the early proteins
Expression of E or b genes depends on the presence of
functional IE proteins and is unaffected by inhibitors of viral
DNA replication (Fortunato & Spector, 1999). They are
divided into two subclasses: b1 (E) and b2 (E-L) according
to their time of expression. b1 genes are transcribed within
4–8 hr p.i., b2 transcription 8–24 hr p.i.. The functional
data indicate that E genes encode mostly non-structural
proteins, including viral DNA replication factors, repair
enzymes, and proteins involved in immune evasion (Mocar-
ski & Courcelle, 2001).
The expression profiles of microarrays of viral DNA
recently have provided a temporal map of IE, E, and L
genes in the entire viral genome (Chambers et al., 1999).
Hybridization of such microarrays to cDNAs prepared from
HCMV-infected cells treated with ganciclovir (GCV) to
block viral DNA replication has revealed that 36% of the
more than 150 ORFs scored positive for expression were
unaffected by GCV and, therefore, classified as E. These E
genes are dispersed throughout the HCMV genome. Unlike
the genes in the UL region, most US genes show E class
expression properties. However, several E genes, such as
UL4, UL44, UL54, and UL112/113, are also transcribed late
in infection through several mechanisms, including activa-
tion of a promoter different and independent from that
transcriptionally active in the E times, initiation of tran-
scription from a new start site, and alteration of the splicing
pattern as infection proceeds (Fortunato & Spector, 1999;
Mocarski & Courcelle, 2001).
It is believed that transcription of E genes is stimulated by
IE2-86 alone or in cooperation with IE1-72 through trans-
activation of the corresponding promoters in a TATA box-
dependent manner that requires both the host basal transcrip-
tion initiation complex and sequence-specific transcription
factors, such as CREB/ATF and Sp1 (Fortunato & Spector,
1999). Both E and L transcripts may have a polycistro-
nic structure due to the relatively few polyadenylation sig-
nals in the genome that generate families of 30 co-terminal
transcripts. In addition, expression of several E genes
studied in some detail is regulated by both transcriptional
and post-transcriptional mechanisms (Mocarski & Cour-
celle, 2001).
Functional roles in viral DNA replication have been
identified for the UL112/113 family of DNA-binding pro-
teins that contribute to organization of the so-called replica-
tion centers within the nucleus of infected cells for the viral
DNA polymerase encoded by the UL54 ORF and the UL44
gene product that acts as a polymerase processivity factor
(Mocarski & Courcelle, 2001). Several other E proteins,
however, are involved in establishment of immune evasion
in the productively infected cell, such as the glycoproteins
encoded by US2 and US11, which bind the MHC Class I
heavy chains and transport them in a retrograde fashion
from the ER into the cytosol, where they are degraded by
the proteasome (Shamu et al., 1999; Story et al., 1999). The
E-expressed US27 and US28 ORFs have homology to the
CC chemokine receptors (Chee et al., 1990b). However,
US28 alone is a receptor for the CC chemokines RANTES
and monocyte chemoattractant peptide-1. It sequesters them
from the extracellular environment by internalization and,
thus, prevents elimination of HCMV-infected cells by che-
mokine-activated immune cells (Gao & Murphy, 1994;
Bodaghi et al., 1998). In addition, the E gene UL4 encodes
an early structural glycoprotein (gp48). This is a non-

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 277
essential component of the viral envelope, since a mutant
virus with disruption of the UL4 gene produces virus
progeny without impaired replication kinetics (Hobom et
al., 2000) (Table 1).
2.3.6. Viral DNA replication
HCMV genome replication, inversion, and packaging
occur in the nucleus of the infected cells. Viral DNA synthesis
begins later than 16 hr p.i. It requires the activities of essential
and specific viral proteins and the active contribution of
several cellular proteins (Mocarski & Courcelle, 2001).
Examination of CMV genome sequences has shown that,
unlike other herpesviruses, they do not encode deoxyribo-
nucleotide biosynthetic enzymes, such as thymidine kinase,
dihydrofolate reductase, thymidylate synthase, and an active
form of ribonucleotide reductase (Chee et al., 1990a; Raw-
linson et al., 1996). Thus, the virus must depend on the host
cell metabolism to ensure a sufficient supply of dNTPs for
its DNA replication. As a result, it does not shut off host
macromolecular synthesis, but stimulates cellular transcrip-
tion and translation. CMV evidently has developed strat-
egies to stimulate the biochemical pathways involved in the
biosynthesis of DNA precursors. Several reports have dem-
onstrated that the early consequences of CMV infection are
similar to those observed in serum-deprived cells exposed to
growth factors (Fortunato et al., 2000). These include cell
cycle functions, such as nuclear translocation of Cdk2;
induction of cyclin E and B; pRb hyperphosphorylation;
activation of E2F-dependent transcription; and activation of
c-myc, c-jun, and c-fos proto-oncogenes (Jault et al., 1995;
Margolis et al., 1995; Bresnahan et al., 1996, 1997; Salvant
et al., 1998). Moreover, there is a substantial increase in the
expression of cellular enzymes involved in nucleotide
metabolism, including thymidine kinase (Estes & Huang,
1977), ornithine decarboxylase (Isom, 1979) and topoiso-
merase II (Benson & Huang, 1990), dihydrofolate reductase
(Wade et al., 1992; Lembo et al., 1999; Song & Stinski,
2002), folylpolyglutamate synthetase (Cavallo et al., 2001),
thymidylate synthase (Gribaudo et al., 2000, 2002; Song &
Stinski, 2002), deoxycytidilate deaminase (Gribaudo et al.,
manuscript in preparation), and ribonucleotide reductase
(Lembo et al., 2000; Song & Stinski, 2002). Despite the
induction of an S phase-like state, CMV-infected cells, as
discussed in Section 2.3.4, fail to undergo cellular DNA
replication and division as a result of blocks in cell
cycle progression that prevent the host DNA replication
machinery from competing with the virus for access to
DNA precursors (Fortunato et al., 2000; Kalejta & Shenk,
2002). The ability of HCMV to stimulate the expression of
cellular enzymes for DNA precursor synthesis is crucial for
its productive replication in the quiescent or terminally
differentiated non-dividing cells it encounters during natural
infection, since their expression is stringently repressed in
these cells.
Six herpesvirus-conserved ORFs in the HCMV genome
provide the core replication proteins for viral DNA replica-
tion. Among them, the single-stranded DNA-binding pro-
tein ppUL57 prevents the reannealing of DNA strands
following unwinding by the helicase-primase complex.
This, in turn, is composed of three subunits encoded by
UL70, UL102, and UL105, the DNA polymerase encoded
by UL54 and the DNA polymerase processivity factor UL44
that prevents dissociation of UL54 from the template
(Griffiths, 2000; Mocarski & Courcelle, 2001). Replication
also requires other viral proteins to maximize DNA replica-
tion, such as UL84, UL112/113, and UL114. UL84 encodes
a 75-kDa phosphoprotein, which stably interacts with IE2-
86, functions as an origin-specific initiator factor, and
stimulates the viral origin (oriLyt)-dependent DNA syn-
thesis (Sarisky & Hayward, 1996). The phosphoproteins
encoded by the UL112/113 region regulate the establishment
of the so-called replication centers corresponding to sub-
nuclear sites of HCMV DNA synthesis. UL112/113 localize
to small intranuclear globular sites representing the early
precursors of the replication centers and recruit the core
replication proteins and enzymes (Penfold & Mocarski,
1997; Ahn et al., 1999). Finally, the protein encoded by
UL114 expresses a functional uracil DNA glycosylase
activity that appears to be required for efficient viral DNA
replication in post-mitotic cells, since a mutant virus with a
substitution mutation in UL114 showed a defect in transition
to high-level, late-phase DNA replication (Courcelle et al.,
2001). IE proteins, such as the transactivators encoded by
the ie1/ie2 and TRS1/IRS1 genes and those expressed by the
UL36-38 region, are also required for transient complemen-
tation of oriLyt-dependent DNA synthesis, although their
roles in this process are not known. HCMV DNA replica-
tion proceeds through initial circularization of the input
genome within 4 hr p.i., followed by DNA synthesis via a
bidirectional q mechanism from a single origin (oriLyt) of
replication that undergoes a switch to a late-phase rolling
circle form of DNA replication (Mocarski & Courcelle,
2001). The latter is responsible for most of the viral DNA
produced during the late stages of infection in the form of
large concatemeric replicating units lacking terminal frag-
ments that are subsequently cleaved into lengths that can be
encapsulated. The oriLyt of HCMV maps within the UL
region close to UL57 (Masse et al., 1992). It spans � 2000
bp, containing repeated nucleotide sequences, as well as
transcription factor-binding sites, and encodes a number of
short transcripts. The integrity of this region is needed for
efficient replication, since mutations targeting different
repeated sequence elements dramatically reduce the ini-
tiation of viral DNA synthesis (Mocarski & Courcelle,
2001).
During the late stages of viral DNA replication, newly
synthesized genomes mature through their inversion, cleav-
age, and packaging (McVoy & Adler, 1994). Inversion
occurs in concatemeric units and leads to the generation
of progeny genomes as a pool of four isomers that only
differ in the orientation of their L and S components (Fig.
1). The mechanism leading to inversion is thought to require

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297278
a recombination that needs the repeated a sequences at the
termini and the UL/US junction. Packaging of the genome
into preformed B capsids then follows its cleavage at the
essential cleavage/packaging signals pac1 and pac2. These
sequences are highly conserved among the herpesvirus
genomes and are contained within a 220-bp element located
in the S component of the HCMV DNA near the repeated a
sequence (Mocarski & Courcelle, 2001).
2.3.7. Characteristics and functions of the late proteins
The L proteins are the last class of gene products
expressed during HCMV replication (Table 1). Their tran-
scription begins more than 24 hr p.i. and requires prior viral
DNA replication (Fortunato & Spector, 1999). Late or
g gene expression leads to the synthesis of two subclasses
of L proteins (g1 and g2) in accordance with their time of
expression and sensitivity to viral DNA replication inhib-
itors. g1 (leaky late) transcription occurs 24–36 hr p.i., and
is reduced by such inhibitors. g2 (true late) transcription
occurs 24–48 hr p.i., and is strictly dependent on DNA
replication. The L proteins have mainly structural roles and
primarily contribute to the assembly and morphogenesis of
the virion (Mocarski & Courcelle, 2001).
Most HCMV genes belong to the L class. Examination of
the expression profiles of DNA microarrays from the whole
HCMV genome has shown that at 72 hr p.i., in the absence
of a viral DNA synthesis inhibitor, 26% and 32% of the
transcriptionally active genes are g1 and g2, respectively
(Chambers et al., 1999).
Little is known about the transcriptional regulation of L
genes and the requirement of viral and/or cellular factors for
their expression. Of the L genes studied in detail, the g1
UL83 gene promoter is transactivated by the combination of
IE2-86 and IE1-72, whereas deletion analysis of the g2
UL94 and UL99 promoters revealed that they require a
minimal promoter element containing little more then the
TATA box for full gene activation in late infection (For-
tunato & Spector, 1999; Mocarski & Courcelle, 2001).
2.3.8. Virion assembly, maturation, and egress
Formation of HCMV capsids and packaging of viral
DNA occur in the nucleus. Subsequently, nucleocapsids
acquire a primary envelopment by budding at the nuclear
membrane, and further mature through a de-envelopment/re-
envelopment process in the cytoplasm before leaving the
cell via an exocytotic-like pathway (Mocarski & Courcelle,
2001; Mettenleiter, 2002).
Nucleocapsid particles accumulate in inclusions that
confer the typical ‘‘owl’s eye’’ appearance of the infected
cell nucleus. The initial step is the interaction of pUL86 with
the scaffolding AP pUL80.5 (the AP precursor) in the
cytoplasm and subsequent translocation into the nucleus
(Gibson, 1996), where oligomerization of these complexes
catalyzed by AP leads to the formation of hexons and
pentons that interact with pUL85-pUL46 complexes to form
the B capsid precursor shell. The subsequent association of
capsid intermediates with pUL48.5 completes the formation
of B capsids that are now ready to package viral DNA and,
after insertion of viral genomes, remove AP (Butcher et al.,
1998). During capsid formation, a series of proteolytic
cleavages catalyzed by the assemblin protein (pUL80a)
leads to maturation of the UL80 precursor to assemblin/
APs and the dissociation of UL86 from AP (Gibson, 1996).
Capsids are initially enwrapped through budding at the
nuclear membrane, where they acquire a primary envelope
derived from its inner leaflet (Gibson, 1996). They then
cross the lumen, fuse with the outer leaflet of the nuclear
membrane or the ER membrane with which it is contiguous,
lose their primary envelope, and move into the cytoplasm.
Here, HCMV virion particles further mature by acquiring
their tegument. The tegumented capsids then receive their
definitive envelope by budding into vesicles of the Golgi
apparatus (Sanchez et al., 2000). Both tegumentation and re-
envelopment are driven by multiple specific protein-protein
interactions to secure the integrity of the viral particle
(Mettenleiter, 2002). These mature particles are retained
within the vesicles and transported to the cell surface via
the Golgi network, which is enlarged due to the accumula-
tion of nucleocapsids and DB. The Golgi alterations during
the late replication stages create inclusions around the
nucleus that result in its characteristic kidney-like appear-
ance (Pass, 2001). Progeny virus accumulates in the cyto-
plasm, and infectious virus is released into the extracellular
compartment beginning at 72 hr p.i. In the very late stages,
however, a substantial number of viral particles are still
associated with the cell.
3. Epidemiology of human cytomegalovirus infection
HCMV is one of the most successful parasites. It is found
in both the developed industrial societies and in isolated
aboriginal groups (Griffiths, 2000; Pass, 2001). Following
infection, it is excreted in body fluids (urine, saliva, tears,
semen, milk, and cervical secretions) for months to years.
Infection is usually mild and subclinical. The unsuspecting
host is thus able to spread the virus both vertically and
horizontally. Virus can appear following primary infection,
reinfection, or reactivation. About 10% of infants are
infected by the age of 6 months, following transmission
from their mothers via the placenta, during delivery, and by
breast feeding (Pass, 1985; Stagno et al., 1986). Trans-
placental infection can occur both in women infected for the
first time during pregnancy and those infected long before
conception (recurrent infection). Primary infection during
pregnancy as a source of fetal infection was suggested by
the observation that transplacental transmission ranges from
20% to 40% when primary infection varies from 0.7% to
4.1% (Pass & Boppana, 1999). In the case of recurrent
infections, high rates of congenital infections are observed
in populations with higher rates of maternal seropositivity,
suggesting that most congenital infections are caused by

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 279
reactivation of latent virus (Stagno et al., 1973; Yeager &
Martin, 1977). Stagno et al. (1977, 1982), for example,
found an incidence of � 2% of congenital infections among
births from mothers seropositive long before conception (as
documented by virus shedding and serum abs).
Infection during delivery is the outcome of shedding
from the vagina or cervix, followed by ingestion of infected
secretions by the offspring. Shedding close to the time of
delivery has been noted in from 2% to 28% of the mothers
(Reynolds et al., 1973). About 50% of the newborns may be
infected in this way, and they usually begin to shed the virus
themselves when they are 6 weeks old.
Breast-feeding is the most common route of transmis-
sion. Transmission is a function of its duration and the
relatively low virus load in the milk (Dworsky et al., 1983).
It has been shown, for example, that infants nursed for < 1
month do not become infected compared with almost 40%
of those nursed longer. Moreover, 69% of infants are
infected when the virus can be isolated from the milk,
whereas only 10% are infected when the mother is sero-
positive, but negative for virus in the milk. Polymerase
chain reaction (PCR) studies have also demonstrated a
strong relationship between the presence of viral DNA in
milk and transmission to the infant (Vochem et al., 1998).
The precise route of infection throughout childhood is not
known, although close contact is required. Infection increases
when people live in crowded, unhygienic conditions, and
hence is most common in socially disadvantaged countries.
Most children from populations with low socioeconomic
backgrounds, in fact, are infected at the onset of puberty,
whereas < 40% of adolescents from the industrial countries
are infected, followed by an increase of � 1% per year
(Griffiths, 2000; Pass, 2001). Infection is life long. Latent
CMV may reactivate and produce infectious virions that are
shed in the saliva, urine, and other body fluids. Reactivation
is usually asymptomatic, and enables the virus to spread
horizontally and vertically. The incidence of congenital
infection is highest in poor communities, since most women
are infected before puberty. Reactivation is thus a more
frequent cause of congenital infection than primary maternal
infection, although the latter presents a greater risk to the fetus
than recurrent infection. An epidemiological distinction is
often drawn between reinfection and reactivation as opposed
to latent infection. In clinical practice, however, the term
‘‘recurrent’’ is used to indicate both possibilities.
4. Infection routes
Primary infection does not usually result in a clinical
illness, and cannot be identified in pregnant women. Its
status as a risk factor for fetus infection is thus uncertain.
Intrauterine infection occurs in only one-third of pregnant
women with primary infection. The ways in which the fetus
escapes infection are unknown, although macrophages in the
placenta may constitute a barrier (Burton & Watson, 1997).
Ingestion of infected maternal in genital secretions or
breast milk is the main perinatal route of infection. ‘‘Copi-
ous’’ quantities of genital secretions containing high virus
titers as a result of recurrent maternal infection surround the
fetus during delivery, and upon ingestion, may result in
virus transmission to the newborn (Stagno et al., 1982).
Virus titers are usually low in breast milk, but long-term
feeding results in the build-up of an effective inoculum.
Newborns from women with infected breast milk who fed
with formula milk are not infected. Once ingested, the virus
infects the mucosa of the oropharynx, esophagus, or the
upper airways (Stagno et al., 1982).
The absence of symptoms following postnatal infection
makes it impossible to determine the transmission routes.
Molecular analysis of CMV isolates has greatly increased
our understanding of CMV epidemiology. PCR amplifica-
tion of selected segments of the genome and hybridization
of the variable junction region of the CMV genome require
only small amounts of virus for diagnosis and allow faster
sequencing of viral DNA. Chou and Dennison (1991)
compared envelope glycoprotein gene sequences and found
that clinical and laboratory strains can be grouped into
four gB genotypes, although epidemiologic and clinical
significance of the gB genotype distribution in immuno-
compromised patients remains unclear.
Primary infection with HCMV via blood transfusions was
noted in the mid-1960s, although epidemiologic studies have
demonstrated that this is an uncommon route. Attempts to
culture HCMV from fresh donor blood have rarely been
successful. It is thus assumed that the virus is latent in the
blood cells of healthy donors and is reactivated following
transfusion when they encounter an allogeneic stimulus. The
nature of the leukocytes carrying latent virus is unknown,
although attention is being increasingly focused on the
monocytes/macrophages (Plachter et al., 1996; Soderberg-
Naucler et al., 1997, 2001).
HCMV is a significant post-allograft pathogen. Several
studies have shown that seronegative recipients of an organ
from a seropositive donor are at risk of acquiring a primary
infection and that � 60%–80% develop a more severe
disease than seropositive recipients of seropositive organs,
suggesting that acquired immunity modulates infection
(Peterson et al., 1980; Pollard, 1988). The contribution
of immunity to resistance to HCMV disease, however,
decreases as the level of immunosuppression increases.
Variables that contribute to post-transplant infection include
donor and recipient serological status, type of immunosup-
pression, source of allograft, HLA matching of donor and
recipient, and type and amount of blood products used
(Rubin et al., 1985; Miller et al., 1991). Since the main
determinant of transmission via blood is the presence of
leukocytes, reduction of the cell contamination of blood
products has dramatically decreased the incidence of trans-
fusion-acquired infection in allograft recipients (Miller et al.,
1991). The effects of infection extend well beyond the
immediate post-transplant period. It appears that HCMV

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297280
contributes to graft loss independently of graft rejection.
Following kidney transplantation, a histologically distinct
glomerulopathy may result from infection of the graft and
may significantly alter its survival (Richardson et al., 1981;
Herrera et al., 1986). Following liver transplantation, a
distinct bile duct sclerosis syndrome has been related to
active HCMV infection. HCMV may perhaps increase the
expression of MHC antigens in the graft and may favor its
rejection (Donaldson et al., 1987; Vierling & Fennel, 1985),
although the way in which this could take place has not been
defined. The role of HCMV infection in rejection is most
convincing in the case of cardiac allografts. Accelerated
development of coronary atherosclerosis associated with
post-transplant HCMV infection has been reported and
found to be independent of reports of age, lipid levels, and
immunosuppression regimen (Loebe et al., 1990). Interac-
tions between HCMV IE proteins and p53 in vessel smooth
muscle cells have also been noted, and the virus may thus
contribute to the formation of atherosclerotic plaques in the
heart vessels (Streblow et al., 2001).
The incidence of HCMV infection following allogenic
bone marrow transplant (BMT) ranges from 32% to 70%,
with an average of 50%, regardless of the prior serological
status of the recipient and donor. Typing of strains has
shown that infection is due to a virus derived from the
recipient (Ruutu et al., 1990; Rubie et al., 1993; Winston et
al., 1993). Its incidence in seronegative recipients receiving
a seropositive marrow is lower than in seropositive recipi-
ents receiving a seropositive marrow, suggesting a transfer
of adoptive immunity from donor to recipient (Griffiths,
2000). Thus, the most critical event is reactivation of a latent
virus in seropositive BMT recipients, whereas in seroneg-
ative recipients, transmission mostly occurs through the
larger quantities of blood products that they receive com-
pared with solid organ transplant recipients. Increasing
evidence indicates that apart from the serological status of
donor and recipient, administration of blood products carries
a significant risk of exposure to HCMV infection following
BMT.
Another major risk factor is the immunosuppressive
regimen. Reactivation of infection ranges between 20%
with cyclosporine and 60% with OKT3 administration.
Hematologic, hepatic, and gastrointestinal abnormalities
are relatively common. Uncertainty, however, persists with
regard to both the role of virus replication in the onset of
graft versus host disease and the toxicity of the pretransplant
therapy. The most significant HCMV-associated syndrome
in BMT is viral pneumonia, with an incidence ranging
between 10 and 15%. Its pathogenesis is unknown, although
several mechanisms, such as uncontrolled replication lead-
ing to organ damage and respiratory failure or immunologic
destruction of the lungs triggered by HCMV, have been
suggested.
HCMV interaction during the induction and progression
of HIV infection has not been definitively established. Even
so, HCMV is a leading opportunistic agent in acquired
immunodeficiency syndrome (AIDS). Autopsies have
shown that 90% of patients develop active HCMV infection
and 40% display sight- and/or life-threatening diseases. The
increased survival of HIV-infected individuals following
more effective prophylaxis and treatment of bacterial and
protozoal infections, accompanied by virtual inactivity of
their immune system, has augmented the importance of
HCMV infection in these patients. Invasive HCMV infec-
tion, in fact, increases rapidly as CD4+ lymphocyte counts
fall (Gallant et al., 1992; Webster et al., 1992; Leach et al.,
1993). Three major organ systems are clinically affected: the
CNS, the lungs, and the gastrointestinal tract. Retinitis is the
most frequent CNS infection directly attributable to HCMV
replication, and the most sight-threatening (Vinters et al.,
1989; Sison et al., 1991; Pertel et al., 1992; Faber et al.,
1992). Autoptic immunohistochemical analysis has shown
HCMV presence in all layers of the retina, without under-
lying choroidal involvement. Absence of virus in the endo-
thelium of the retinal microvasculature indicates that its
hematogenous spread does not follow replication in these
cells, but that perhaps the virus breaks through the capillary
endothelium.
5. Pathogenesis and pathology
Histopathologic and immunohistochemical examination
of necropsy tissues has indicated that the virus initially
enters via the epithelium of the upper alimentary, respiratory,
or genitourinary tracts. However, since infection is readily
established by transfusion and transplantation, initial infec-
tion of epithelial cells does not seem essential (Sinzger &
Jahn, 1996). Cytotrophoblasts form a barrier between the
maternal and fetal circulation, but readily allow HCMV
replication in vitro, suggesting that the fetus is infected
hematogenously (Halwachs-Baumann et al., 1998; Hem-
mings et al., 1998). Leukocytes and vascular endothelial
cells aid the spread of HCMV. During acute infection,
HCMV can be isolated from buffy coat preparations of
leukocytes, but not from the plasma, and is presumably
carried by one or more cell subsets. Viral antigens are found
mainly in neutrophils and monocytes in acute infection
(Sinzger & Jahn, 1996) and transmission through blood
products from seropositive donors can be prevented by
removal of leukocytes (Pass, 2001). Moreover, immunohis-
tochemical double-labeling has shown that macrophages
infiltrating organ tissues support virus replication, whereas
granulocytes show no signs of viral late gene expression
(Sinzger & Jahn, 1996). Lastly, virus encoded chemokines
may facilitate dissemination of CMV from the initial
replication site by attracting neutrophils and monocytes
(Penfold et al., 1999). Saederup et al. (1999), in fact, have
demonstrated that mouse CMV-encoded b-chemokine pro-
motes monocyte-associated viremia, whereas viruses with
deletions in the gene for this b-chemokine have significantly
reduced levels of viremia. Endothelial cells detached from

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 281
the basal membrane as a result of HCMV infection are also
involved in hematogenous spreading of the virus. However,
the molecular mechanisms responsible for the interaction
between these cell populations and the microvascular sys-
tem, and subsequent transmission of the virus to the target
organs, have not been determined.
Hematogenous spreading is typically followed by infec-
tion of ductal epithelial cells. Fibroblasts are rarely in-
volved, despite the fact that the virus can replicate in such
cells in vitro. CMV is recognized histologically by its
characteristic ‘‘owl’s eye’’ intranuclear inclusions with a
surrounding halo and marginated chromatin. Typical cyto-
megalic cells are found in the salivary gland, bile duct,
bronchial and renal tubular epithelium, islet cells, epithelial
cells of the inner ear, the capillary endothelium, astrocytes,
and neurons (Griffiths, 2000; Pass, 2001). In cases of severe
disseminated disease, evidence of HCMV involvement can
be found in all organs. The present review, however, will be
confined to those most commonly involved (for further
details, see Ho, 1991 and Becroft, 1981). Salivary gland
involvement is probably chronic and probably the outcome
of subclinical congenital and perinatal infection, since it is
more frequent in infants and young children and diminishes
with age. In contrast, viruria is a constant feature in all age
groups as the result of replication in the genitourinary tract
(Plachter et al., 1996). In the kidneys, infected cells are
found in the proximal tubules, Henle’s loop, and the
collecting tubules, but seldom in the glomeruli. As already
mentioned in Section 4, infection is directly involved in
renal transplant dysfunction and rejection, whereas it rarely
leads to dysfunction in normal individuals. Elevated liver
enzyme levels indicative of subclinical hepatitis are fre-
quently associated with HCMV infection in immunocompe-
tent individuals. Cytomegalic cells are usually found in the
bile duct epithelium, less frequently in the capillary endo-
thelium, and rarely in the parenchyma. Together with
cytomegalic cells, areas of inflammatory infiltrates are a
dominant feature in histological sections, possibly suggest-
ing that liver dysfunction might result directly from virus
cytopathogenicity or indirectly from the inflammatory reac-
tion to HCMV infection (Phillips et al., 1977).
HCMV infection in HIV patients often affects other parts,
or even the entire digestive system (Rene et al., 1988). The
colon is the most common replication site, followed by the
esophagus, rectum, and small intestine. The direct effects of
infection range from punctate, superficial ulcers to deep
ulcerations, with extensive necrotizing involvement and gut
perforation.
HCMV pneumonia is infrequent in immunocompetent
individuals, but often is severe in immunosuppressed
patients, especially BMT and heart-lung recipients. Infected
cells are mainly observed in the alveolar and bronchial
epithelium, along with inflammatory infiltrates (Apperley
& Goldman, 1988; Wreghitt et al., 1988). The range of
clinical presentation, from severe pulmonary dysfunction
and limited focal infection, together with similar findings in
animal models, suggests that dysfunction may be due to
virus-associated dysregulation of the immune response.
CNS damage is a frequent feature of congenital infec-
tion (Fowler et al., 1992). Symptoms include mental
retardation, seizures, hypotonia, and hearing loss due to
various alterations in the form of many small, necrotic foci
surrounded by an inflammatory infiltrate. In histological
sections, inclusion-bearing cells positive for viral antigens
have been found in neurons, glia, ependyma, choroid
plexus, meninges, and vascular endothelium. These cells
are also widespread in the semicircular canals, vestibular
membrane, and cochleae. CNS involvement was once
regarded as infrequent, except in congenital infection.
Today, however, CNS tissues are the major target organs
in AIDS patients, where HCMV replicates efficiently in
various brain structures and in specialized organs, such as
the retina and cochlea. Histopathologic alterations include
microglial nodules, focal parenchymal necrosis, necrotizing
ventriculitis, hemorrhages, astrocytic proliferation, and
perivascular inflammation. Acute retinal infection fre-
quently leads to blindness, probably as the outcome of
direct lysis of the retinal pigment epithelial cells during
virus replication.
In conclusion, organ dysfunction seems to coincide with
HCMV replication, although the frequent association of an
inflammatory reaction indicates that other pathogenetic
mechanisms are also operative.
6. Host defences
HCMV infections are kept under control by the immune
system. Total HCMV clearance, however, is rarely achieved,
and the viral genome remains at selected sites in a latent
state (Reddehase et al., 1994). Initiation of productive
replication results in transient virus shedding and recrudes-
cent disease (Mocarski, 1996). Since severe infection is
usually restricted to individuals with impaired cell-mediated
immunity, it is evident that this arm of the immune response
provides the most protection. Even so, the supportive role of
the humoral system in keeping CMV loads below critical
thresholds must not be overlooked.
6.1. Cell-mediated immunity
Mouse models have been employed to determine the role
of specific components of the immune response and their
recognition of viral proteins (Koszinowski et al., 1990).
Early studies demonstrated that a CMV-specific cytotoxic T
lymphocyte (CTL) response was required for recovery from
CMV infection. Suppression of CTLs caused reactivation
and dissemination of natural infection. Further studies
showed that both natural killer (NK) cells and CD8+
cytotoxic T-cells are of primary importance in the preven-
tion of recurrence (Jonjic et al., 1990; Polic et al., 1996;
Hengel et al., 1998). Adoptive transfer of CD8+ cells

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297282
protected mice from lethal challenge independent of CD4+
T helper cells. CD8+ T-cell clones specific for both struc-
tural and nonstructural proteins were identified. However,
CD8+ depletion with abs or in mouse mutants deficient in
MHC Class I expression revealed additional antiviral activ-
ities mediated by CD4+-derived cytokines not seen when
CD8+ cells are present. Lastly, experiments with mice
deficient for perforin and granzyme- or Fas-mediated cyto-
toxicity have shown that replication is controlled by NK and
CD8+ cells via the perforin and granzyme-mediated path-
way, whereas the Fas-Fas ligand system is not critical (Riera
et al., 2000, 2001).
As already mentioned in Section 4, since infection (apart
from congenital forms) is most severe in patients with
dramatically impaired cell-mediated immunity, such as
BMT recipients, and those with AIDS, it is evident that this
arm of the immune response provides the most protection
(Pass et al., 1983; Zanghellini et al., 1999). Its investigation
in studies of lymphocyte proliferation in response to CMV
antigens has shown that in most seropositive immunocom-
petent adults, this usually takes the form of CD4+ cell
proliferation in response to envelope glycoproteins gB and
gH, the lower matrix protein pp65, and IE proteins, in
addition to other proteins (Liu et al., 1993; Davignon et
al., 1995; He et al., 1995; Beninga et al., 1995). The function
of proliferating CD4+ cells in resistance to HCMV infection
is not clear. Studies in mice point to several antiviral effector
functions, such as the production of cytokines and CD4+-
mediated cytotoxicity. The virus-encoded proteins that are
targets of CTLs include a collection of structural and non-
structural forms (Boppana & Britt, 1996; Kern et al., 1999).
Comparison of CTL precursor frequencies for envelope gB
or the 72-kDa IE1 protein revealed a low frequency of gB-
specific CTLs ( < 6%), whereas 18–58% of CTLs recog-
nized IE1-expressing target cells. The high frequencies for
pp65 and IE1 proteins found in subsequent studies show that
they are the chief targets of the CTL-mediated immune
response (Pass, 2001).
Although a direct role for virus-specific CTL responses
in the resolution of infection has yet to be defined, in vivo
evidence of the control of HCMV infection by CTLs has
been obtained through the adoptive transfer of virus-spe-
cific clones. Passive administration of autologous HCMV-
specific CD8+ CTLs at set intervals after BMT to sero-
negative recipients of marrow from seropositive donors
generated a vigorous CMV-specific CTL response without
onset of viremia or CMV disease (Walter et al., 1995).
These studies provided the first evidence for the role of
CTLs in controlling human CMV infection, and opened
new therapeutic perspectives for CMV treatment in organ
transplantation.
6.2. Humoral immunity
During primary infection, immunocompetent individuals
produce anti-HCMV immunoglobulin (Ig)M class abs that
persist for 3–4 months, followed a few weeks later by IgG
class abs that persist for life. Experimental and clinical
findings show that the humoral response is beneficial.
Mice immunized against murine CMV gB were protected
against a lethal challenge (Rapp et al., 1992), and immun-
ization of pregnant guinea pigs against guinea pig CMV
envelope glycoprotein protected their fetuses in the same
way (Harrison et al., 1995). Intrauterine infection is less
severe when transmitted by means of recurrent rather than
primary maternal infection (Griffiths, 2000). During prim-
ary infection, women who transmit the virus in utero have
high levels of total IgG characterized by low avidity and
neutralizing activity. Correlation of an enhanced ab
response with a poor fetal prognosis suggests that fetal
ab is responsible for immunopathology. Primary infection
is more frequent and more severe when the renal transplant
recipient is seronegative and the donor seropositive. Alle-
viation of this severity by preimmunization with the
attenuated Towne strain (Plotkin et al., 1984) or adminis-
tration of high-titer anti-CMV Igs (Snydman et al., 1987)
also shows that humoral immunity is beneficial. Many
CMV proteins are recognized by the humoral immune
system (Landini & Michelson, 1988; Landini et al.,
1988). The envelope glycoproteins (mainly gB and gH)
are the targets of virus-neutralizing abs in both human and
mouse models (Pass, 2001). The predominance of gB as a
target is best explained by its dominant immunogenicity
and abundance compared with other components of the
envelope. Anti-gH abs have a potent, complement-inde-
pendent, but minor neutralizing activity. Viral tegument
components, including pp28 (UL99), pp65 (UL83), and
pp150 (UL32), trigger an intense and long-lasting ab
response that provides an indirect measure of viral
replication and correlates with the clinical outcome. How-
ever, these abs are unable to react with the surface of virions
and infected cells, and are of limited importance in a
protective response.
6.3. Immune evasion by human cytomegalovirus
A characteristic feature of infection in the normal host
is persistence of the viral genome in a nonproductive form
at specific anatomical sites for months or even years
(Hengel et al., 1998). This ability to avoid elimination
by the immune system is the result of (1) induction of a
latent state of infection, (2) exploitation of immunologi-
cally privileged tissues for replication (i.e., epithelial cells
of the salivary glands expressing an insufficient number of
MHC Class I molecules to trigger clearance by CD8+ cell,
and (3) expression of genes that interfere with the immune
response (Mocarski, 2002). Table 2 summarizes the main
mechanisms exploited by HCMV to evade the immune
response. Escape from CD8+ cells is mediated by several
mechanisms, all of which block the expression of MHC
Class I molecules complexed with a potentially important
CTL target, the 72-kDa IE1 phosphoprotein, or which

Table 2
HCMV genes associated with immune evasion
Immune function Virus genes Mechanisms
NK cell activity UL 40 Up-regulation of HLA expression:
binding of inhibitory NK cell receptors
MHC Class I
antigen
US 2 Export of MHC Class I heavy
chains from the ER VIA Sec61
presentation/
processing
US 3 Retention of MHC Class I
complexes in the ER
US 6 Inhibition of TAP-mediated peptide
translocation into the ER
US 11 Dislocation of MHC Class I
heavy chains into cytosol
UL 83 Inhibition of 72-kDa IE1 antigen
presentation to CD+ T-cells
MHC Class II
antigen
presentation
US 2 Degradation of MHC Class II
antigen by down-regulating
antigen presentation
to CD4+ T-cells
Modulation of US 27/28 b-Chemokine-binding proteins
chemokine UL 78 Putative GPCR homologue
activity UL 144 Putative transferrin receptor
homologue
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 283
degrade MHC Class II molecule proteins, and, thus,
prevent presentation of viral antigen to CD4+ lympho-
cytes. HCMV deletion mutants have been used to map two
gene regions whose products are associated with the down-
regulation of MHC Class I complex formation; namely,
US11 and that spanning US2 and US5 (Machold et al.,
1997). Both US2- and US11-encoded proteins lead to the
accumulation of MHC Class I heavy chains in the cytosol
of infected cells, where they are degraded by the proteo-
some, thus shortening their half-life and preventing their
expression on the cell surface. Binding of peptides to
MHC Class I molecules depends on transport across the
ER membrane by a specific transporter complex desig-
nated TAP 1/2 (transporter associated with antigen pro-
cessing) (Sadasivan et al., 1996). Only after their
introduction into the ER can peptides form heterodimers
with MHC Class I molecules and b2-microglobulin and be
recognized by CD8+ CTLs. Despite a significant increase
in the expression of TAP molecules in infected cells, the
HCMV glycoprotein encoded by the US6 gene inactivates
the TAP system by interfering with formation of the TAP1-
TAP2-MHC Class I-b2-microglobulin complex. The find-
ing that lysis of HCMV-infected fibroblasts by IE-specific
CTL is usually low, but increases in fibroblasts infected
with a mutant HCMV lacking the UL83 encoding the
matrix pp65, suggests that phosphorylation of the 72-kDa
IE1 protein by pp65 interferes with its antigenic processing
and presentation to CD8+ cells (Wiertz et al., 1996; Gilbert
et al., 1996; Jones & Sun, 1997).
Experiments have demonstrated that NK cells are
involved in the control of CMV infection. Beige mice
(genetically deficient in functional NK cells) are more
susceptible to murine CMV (Shellam et al., 1981). In
humans, MHC Class I HLA–E molecules protect target
cells from NK cells by binding to inhibitory receptors on
their surface. A convincing mechanism for escape from NK
cell attack stems from the observation that the HCMV UL40
gene encodes for a glycoprotein containing a nine amino
acid sequence homologous to the MHC Class I sequence
that up-regulates the expression of Class I HLA-E molecules
(Tomasec et al., 2000).
Lastly, mechanisms interfering with chemokine-driven
inflammation are exploited by HCMV to evade the immune
response. HCMV-infected cells express viral G-protein-
coupled receptor (GPCR) homologues encoded by US27,
US28, UL33, and UL78, which are needed for virus
replication in vitro (Chee et al., 1990b; Bodaghi et al.,
1998). The US28-encoded GPCR binds to and sequesters
several b-chemokines and, thus, enables the virus to evade
the immune response.
6.4. Persistence and release from the host
Cells in bone marrow and peripheral blood are the chief
reservoirs for latent CMV infection. CMV DNA is found in
a small percentage of peripheral blood monocytes, and
gene expression is limited to the E genes. These findings
and the demonstration that tissue macrophages differenti-
ated from circulating monocytes also express E and L
genes have led to the formulation of the following model:
bone marrow precursors of blood monocytes are the site of
CMV latency and provide a means of dissemination upon
differentiation into circulating monocytes (Pass, 2001).
Differentiation of latently infected monocytes into macro-
phages leads to reactivation and productive infection.
Support for this model is provided by the reactivation of
CMV by allogeneic stimulation in vitro of peripheral blood
mononuclear cells from seropositive healthy donors (Soder-
berg-Naucler et al., 1997). Moreover, it has been found that
a small percentage (close to 0.01%) of CD33+/CD14+ or
CD33+/CD15+ bone marrow mononuclear cells from sero-
positive human donors express transcripts associated with
latency. In contrast, T lymphocytes, B lymphocytes, and
CD33- mature granulocytes and macrophages are negative
(Hahn et al., 1998).
Recurrent infection can be defined as indefinite, but
intermittent, excretion of the virus from single or multiple
sites. It should be distinguished from the prolonged excre-
tion typical of primary infection and also infection in the
immunocompromised host (Pass, 2001), where productive
infection, as measured by viral excretion, is markedly
increased. In allograft recipients and AIDS patients, the
incidence of virus excretion from multiple sites approaches
100%.
During primary, recurrent, or persistent infection, CMV
is shed in multiple body fluids, such as saliva, urine, tears,
cervicovaginal fluid, breast milk, and semen, probably due
to virus replication in glandular epithelial cells, accompan-
ied by virus release into excretions.

Table 3
Clinical syndromes associated with HCMV infection in the immunocom-
promised patient
Syndromes AIDS Solid transplant BMT
Esophagitis + + +
Gastritis + + +
Enterocolitis + + +
Hepatitis + + + +
Pancreatitis � + + �Pneumonitis � + ++
Retinitis + + + +
Encephalopathy + � �Polyradiculopathy + � �
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297284
7. Clinical features associated
with human cytomegalovirus infection
7.1. Infection in normal hosts
HCMV infection in normal immunocompetent hosts is
generally subclinical, but nonetheless, is thought to account
for 8% of all the cases of mononucleosis (Nesmith & Pass,
1995). Clinical manifestations of mononucleosis due to
HCMV are very similar to that induced by the more
common Epstein-Barr virus. Persistent fever, myalgia, head-
ache, cervical lymphadenopathy, splenomegaly, and non-
specific constitutional symptoms are common, and may
persist for weeks. Rash is also present in � 30% of patients.
Uncommon complications include pneumonia, myocarditis,
hemolytic anemia, retinitis, gastrointestinal ulceration, hep-
atitis, CNS involvement (Guillain-Barre syndrome), and
peripheral neuropathy. Common laboratory findings include
a negative heterophil ab response (Paul-Bunnel test), atyp-
ical lymphocytosis, and elevated liver aminotransferases.
Slight elevation of serum bilirubin and jaundice are occa-
sional findings.
7.2. Congenital infection
From 5% to 10% of congenitally infected newborns
display cytomegalic inclusion disease, whose symptoms
include intrauterine growth retardation, jaundice, hepatos-
plenomegaly, thrombocytopenia, petechiae, chorioretinitis,
and hepatitis, along with CNS involvement in the form of
microcephaly, encephalitis, seizures, and focal neurological
signs. Most of the non-CNS (liver and blood-forming
organs) manifestations are self-limiting and resolve without
therapy in the vast majority of cases. In contrast, the neuro-
logical damage is permanent and accounts for the long-term
morbidity and poor prognosis of cytomegalovirus inclusion
disease (Boppana et al., 1992). Long-term prospective
studies indicate that 80% of infants with symptomatic
congenital infection will display serious life-long neuro-
logical abnormalities. In 11–20%, damage caused by the
virus leads to such a severe life-threatening organ dysfunc-
tion that the patient dies during infancy.
Long-term follow-up shows that � 15% of the asympto-
matic infected infants at birth (90–95% are asymptomatic)
may develop hearing defects or impaired intellectual per-
formance. Differences in the reported incidence of hearing
defects are probably attributable to the use of different
evaluation methods.
7.3. Cytomegalovirus infection
in the immunocompromised host
HCMV is a significant opportunistic pathogen in immu-
nocompromised patients. Primary infection, reactivation of
latent virus, and reinfection are possible and are often
clinically silent. The onset of infection is marked by
spiking pyrexia, which may resolve in a few days (Pass,
2001). Its severity is roughly parallel with the level of
immunosuppression, and is greatest in BMT recipients and
AIDS patients with low CD4+ T-cell counts. Solid organ
transplant recipients, patients receiving immunosuppressive
chemotherapy, and subjects with congenital immunodefi-
ciencies may also be symptomatic. Table 3 summarizes the
major clinical diseases related to the type of immunocom-
promised host. Some patients may develop a CMV syn-
drome associated with pneumonitis. This has a poor
prognosis (up to 90% mortality). Retinitis may be present
alone following virus dissemination. Replication of CMV
in the gut may be asymptomatic or associated with
extensive ulceration due to erosion of the neighboring
blood vessels and hemorrhages. Replication in the CNS
of AIDS patients produces some of the symptoms
observed in congenital infection, and is often followed
by encephalopathy and/or peripheral polyradiculopathy. It
is not certain whether CMV infection and disease are
simply markers of the immune dysfunction that follows
HIV replication or whether CMV infection itself promotes
HIV progression, although it is clear that highly active
antiretroviral therapy has reduced HIV-1 viral load, along
with a reduction in CMV viremia and disease (Gerna et al.,
1998a; O’Sullivan et al., 1999). Clinical signs of HCMV
infection in transplant recipients may be absent or severe,
although severe infection is now less frequent as a result of
better prophylaxis. HCMV is initially localized in the
transplanted organ, i.e., hepatitis occurs generally in liver
transplant recipients, pancreatitis in pancreas transplant
recipients, but then spreads throughout the gastrointestinal
tract and to the retina, skin, endometrium, lungs, and CNS
(Rubin, 1994). HCMV disease is more difficult to treat in
BMT compared with solid organ transplant recipients, and
HCMV pneumonia has a high mortality rate, despite the
recent introduction of specific antiviral drugs (Reed et al.,
1988). Lastly, an immunosuppressive syndrome often
related to HCMV infection in the late post-transplant
period is characterized by superinfection with bacteria,
fungi, and protozoa, perhaps due to disturbance of both
the humoral and cellular immune response by HCMV.

Fig. 5. HCMV antigenemia assay. This picture was kindly provided by
Rossana Cavallo.
S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 285
8. Diagnosis
Virological and molecular detection of HCMV and sero-
logical demonstration of a specific immune response are
used for diagnosis, although acute infection is usually
diagnosed through detection of the virus in body fluids.
8.1. Virus detection
HCMV can be isolated from several body fluids, such as
urine, saliva, vaginal secretions, amniotic fluid, and blood,
following primary or recurrent infections (Gerna et al.,
1990; Revello et al., 1999). Moreover, HCMV-infected cells
can be identified in biopsy samples (e.g., liver, lungs),
owing to ‘‘owl’s eye’’ inclusions or, more efficiently, by
immunohistochemistry, in situ PCR, or nucleic acid hybrid-
ization. Electron microscopy is employed to detect the virus
in the urine from congenitally or perinatally infected infants
since, unlike other human herpesviruses, the HCMV virions
are present in high titers. However, it is generally agreed
that acute HCMV infection is best diagnosed by detection of
the virus in the blood since the level of viremia correlates
with the clinical picture and the prognosis. The most widely
used assays for detection and quantitation of HCMV load in
blood include tests for viremia, antigenemia, DNAemia, and
IE and L mRNAemia.
8.1.1. Viremia
Viremia is measured by an assay that allows the detec-
tion and the quantification of infectious viral particles in the
blood. Conventional methods are the inoculation of low
passage ( < 25) human embryonic lung or foreskin fibro-
blasts with peripheral blood leukocytes (PBL) and iden-
tification of the characteristic focal CPE (Fig. 2). This
usually develops very slowly, and the cultures must be
carefully observed for at least 21 days before being
reported as negative. This expensive and time-consuming
Fig. 4. HCMV viremia assay. This picture was kindly provided by Rossana
Cavallo.
technique has been replaced by the ‘‘shell vials’’ assay,
which retains the specificity and sensitivity of virus isola-
tion, but provides results within 24 hr (Gleaves et al., 1984;
Griffiths et al., 1984). A human fibroblast monolayer is
inoculated with 2� 105 PBL and fixed 18 hr later. The
nuclei of the infected fibroblasts are stained by immuno-
fluorescence using a monoclonal ab reactive with the MIE
protein p72 (Fig. 4). The assay is based on the assumption
that each stained cell was infected by a single PBL carrying
an infectious virus. Several studies indicate that trans-
planted patients with HCMV viremia in the range of 10–
100 infected fibroblasts following inoculation with 2� 105
PBL have a high risk of developing symptomatic HCMV
diseases (Gerna et al., 1995; Grossi et al., 1995) and should
be treated with antiviral drugs (Gerna et al., 1991). More-
over, viremia is a rapid way to monitor the efficacy of an
antiviral treatment since it decreases sharply 1 or 2 days
after the beginning of an effective regimen (Gerna et al.,
1993), whereas persistent or increasing viremia levels
predict the emergence of a drug-resistant HCMV strain
(Gerna et al., 1992a).
8.1.2. Antigenemia
Antigenemia is measured by the quantitation of leuko-
cyte nuclei positive, in an immunofluorescence assay, for
the HCMV lower matrix phosphoprotein pp65 in a cyto-
spin preparation of 2� 105 PBL (van der Bij et al., 1988a,
1988b; Revello et al., 1989; Gerna et al., 1992b) (Fig. 5).
This rapid assay provides a result in a few hours. Anti-
genemia is an indirect marker of disseminated infection,
since pp65 is transferred to uninfected leukocytes by
infected cells after transient fusion of plasma membranes
and exchange of cytoplasmic material (Gerna et al.,
2000b). By comparison with the test for infectious virus,
the test for viral antigen gives a positive result earlier after
the onset of infection and becomes negative later in the
late phase of a systemic infection. Since high levels of

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297286
antigen are frequently found in patients with HCMV
disease and low levels correlate with asymptomatic infec-
tions, the test for antigenemia is sensitive enough to
monitor infection and antiviral treatment in immunocom-
promised patients (van den Berg et al., 1989; Grossi et al.,
1995).
8.1.3. DNAemia
HCMV DNA in clinical samples is currently detected
and quantitated by qualitative or quantitative PCR. Qual-
itative PCR detects viral DNA in the amniotic fluid
(Revello et al., 1995) and the aqueous humor of AIDS
patients with HCMV retinits (Gerna et al., 1994a), whereas
quantitative PCR provides more clinically useful informa-
tion concerning the virus load in blood. PCR measurement
of DNA levels produces good prognostic information in
both transplant recipients and HIV-positive individuals
(Kidd et al., 1993; Gerna et al., 1994b, 1998b; Bowen et
al., 1997).
8.1.4. RNAemia
This assay is a good marker for monitoring active
HCMV replication and disease, since it detects mRNAs
transcribed from IE or L genes in PBL or whole blood
(Gozlan et al., 1996; Lam et al., 1998). Reverse transcrip-
tion-PCR was the first technique employed to detect viral
transcripts. Its major drawbacks, namely, instability of the
RNA and the difficulty of discriminating PCR products
from a DNA or RNA template when working with
unspliced viral transcripts, cause both false negatives and
false positives. However, these difficulties have now been
circumvented by nucleic acid sequence-based amplification,
which allows specific amplification of unspliced viral
mRNAs in a DNA background, and is a highly promising
technique for monitoring HCMV infection in transplant
recipients (Blok et al., 1998; Aono et al., 1998; Gerna et
al., 1999, 2000a) and for the early diagnosis of both
pregnant women with primary infection and congenitally
infected newborns (Revello et al., 2001).
8.2. Detection of the immune response
Serological determination of a past or recent HCMV
infection in immunocompetent individuals rests on detection
of virus-specific IgG or IgM abs. Occurrence of a primary
infection is conventionally deduced from seroconversion
from IgG ab negative to IgG ab positive in the interval
between two serological assays. A specific IgM response
may be serologic evidence of a recent primary infection.
Detection of IgM abs, however, is not reliable because false
positives may be induced by rheumatoid factor, antinuclear
abs, and other cross-reactive factors not yet identified.
Moreover, true-positive tests detect IgM persistence in
patients with a past HCMV infection. Interpretation of a
positive IgM assay in pregnant women is assisted by
determination of the avidity of HCMV-specific IgG, since
low-avidity IgG is produced early in infection and high-
avidity IgG is a marker of past or recurrent infection
(Blackburn et al., 1991; Grangeot-Keros et al., 1997;
Bodeus et al., 1998; Lazzarotto et al., 1999). Detection of
anti-HCMV IgG abs is a useful way of documenting a past
infection, and they are also a marker of potential infectivity,
since reactivation of a latent infection may occur. Serologic
assays are thus of great clinical importance in donor
selection, since HCMV can be transmitted by blood dona-
tion and organ transplantation.
9. Prevention of human
cytomegalovirus infection and disease
Steps to prevent infection in groups at risk have been
devised in light of what is known about the transmission
routes for HCMV. In solid organ transplant recipients, for
example, the risk of a primary infection could be reduced by
matching seronegative donors and recipients, although this
approach is hampered by the scarcity of donor organs
(Stratta et al., 1990). Prophylaxis with antiviral agents and
pre-emptive therapy (see Section 10) have proved useful in
transplant patients. Transmission to immunocompromised
individuals, pregnant women, and premature newborns
through donor blood can be avoided by using HCMV-
seronegative, filtered, or leukocyte-deprived blood products.
This method places a greater burden on blood transfusion
centers, but should be used whenever possible, since it
significantly reduces HCMV transmission (Gilbert et al.,
1989; Sayers et al., 1992). Administration of an HCMV
vaccine to secure pre-exposure immunization would be
another way of protecting groups at risk.
9.1. Human cytomegalovirus vaccines
Development of a vaccine to prevent congenital HCMV
infection is a priority concern, since the virus is a leading
cause of CNS damage in children (Pass, 1996). The poten-
tial benefit of vaccine-induced immunity has been estimated
at 40-fold reduction with respect to intrauterine infection
and 25- to 30-fold reduction with respect to decrease in CNS
damage (Britt, 1996). However, no vaccine is available and
live-virus vaccines have not obtained official approval (Elek
& Stern, 1974; Plotkin et al., 1994). Both humoral and cell-
mediated immunity are needed to prevent HCMV disease.
Many studies indicate that gB and pp65 are absolutely
necessary vaccine components to secure the induction of
neutralizing abs and CTL responses (Britt et al., 1990;
Gonczol et al., 1991; Riddell et al., 1991; Marshall et al.,
1992; Wills et al., 1996). Moreover, gH and gN for
neutralizing abs and IE1 and pp150 for CTL responses
could increase the protective immune response (Kern et al.,
1999; Gyulai et al., 2000). There are a variety of options for
the elaboration of a safe and effective vaccine: attenuated
live virus vaccines (Towne strain), recombinant live vac-

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 287
cines using chimeric viruses comprising genomic portions
of the virulent strain Toledo and the attenuated strain
Towne, canarypox-HCMV recombinants expressing gB
and pp65, vaccination with HCMV DB, subunit gB vaccine;
pp65 peptide-based vaccine, and DNA vaccines encoding
the gB and pp65 proteins. The attenuated Towne strain, the
recombinant gB, and the canarypox gB and pp65 vaccines
already have been tested in clinical trials.
The attenuated Towne vaccine has been tested both in
healthy volunteers and in transplant recipients (Plotkin et
al., 1975, 1976, 1989, 1991; Adler et al., 1995, 1998). It
induced neutralizing abs and an CTL response and reduced
HCMV disease in seronegative renal transplant recipients,
but did not prevent infection. However, there was no
evidence of its efficacy in a placebo-controlled trial in
seronegative mothers of HCMV-infected children. In con-
clusion, it is immunogenic, but the protection it offers is less
effective than that seen after natural infection. In addition, a
live virus is a potential risk factor in transplant recipients,
and all live attenuated herpesvirus vaccines are overshad-
owed by their possible oncogenicity. To circumvent these
risks, poxviruses with limited potential for replication in
humans (canarypox) have been tested as vehicles for the
expression of recombinant gB and pp65 (Adler et al., 1999;
Berencsi et al., 2001). A subunit vaccine composed of a
modified gB protein was evaluated after combination with a
new powerful adjuvant, MF59, based on an oil-in-water
emulsion of squalene. Results of a trial of 46 seronegative
adults established safety, immunogenicity, and optimal anti-
gen dose (Pass et al., 1999). Whether the neutralizing abs
elicited by this vaccine will be sufficient to prevent recur-
rence or primary HCMV infection is still an open question
(Drulak et al., 2000). Other promising preclinical ap-
proaches include vaccination with HCMV DB (Pepperl et
al., 2000) and administration of DNAvaccine encoding viral
immunogenic proteins to elicit a humoral and a CTL
response (Endresz et al., 1999, 2001).
10. Antiviral treatment
10.1. Currently available drugs
Treatment of HCMV infections is difficult because there
are few options. To date, the nucleoside analog GCV, the
nucleotide analog cidofovir [CDV; (S)-1-(3-hydroxy-2-
phosphonylmethoxypropyl) cytosine], and the pyrophos-
phate analog foscarnet (PFA) have been licensed for serious
or life-threatening HCMV infections in immunocompro-
mised individuals. These drugs have produced clinical
improvement in many patients, but suffer from poor
oral bioavailability, low potency, development of resist-
ance in clinical practice, and dose-limiting toxicities, and
hospitalization is sometimes required.
GCV is a competitive inhibitor of viral DNA polymerase
(UL54), and its antiviral activity requires monophosphor-
ylation in the infected cell by the phosphotransferase
encoded by the UL97 gene of HCMV (Littler et al.,
1992), followed by diphosphorylation by cellular kinases.
It is administered by intravenous infusion, and is usually the
front-line drug for the treatment of HCMV infections. An
oral formulation that is effective for prophylaxis in immu-
nocompromised patients has also been approved (Spector et
al., 1996). However, its side-effects include potentially
dose-limiting neutropenia and the emergence of GCV-
resistant strains due to UL97 and UL54 gene mutations,
mainly in AIDS patients during prolonged maintenance
therapy (Erice et al., 1989; Lurain et al., 1994; Chou et
al., 1995; Baldanti et al., 1995, 1998), and less frequently in
solid organ transplant recipients (Limaye et al., 2000, 2002).
PFA is a noncompetitive inhibitor of the pyrophosphate-
binding site of HCMV DNA polymerase, which does not
require prior activation by a virally encoded enzyme. It is
administered intravenously as an alternative to GCV in the
event of GCV resistance or severe side-effects, although its
application is limited by nephrotoxicity and disturbance of
the electrolyte balance (Vogel et al., 1998; Chrisp &
Clissold, 1991). Long-term exposure to PFA may lead to
the emergence of resistant strains due to UL54 mutations
(Knox et al., 1991; Baldanti et al., 1996).
CDV is a competitive inhibitor of the HCMV DNA
polymerase, and has been approved for the treatment of
HCMV retinitis (De Clercq, 1998). One of its advantages
compared with GCV and PFA is its long intracellular half-
life (Ho et al., 1992; Aduma et al., 1995). Its intermittent
intravenous administration is also possible. Toxicology
evaluations in animals have demonstrated that nephrotox-
icity is its major side-effect, although this can be reduced by
its less frequent administration, coupled with a high pro-
benecid dose and saline hydration (Cundy et al., 1994).
Cross-resistance between GCV and CDV could become a
concern since they share the same target, the product of the
UL54 gene. The emergence of GCV-resistant strains to
GCV through UL54 mutations in vitro or in AIDS patients
is well documented (Smith et al., 1997; Cherrington et al.,
1998). Double resistance to GCV and PFA in strains from
AIDS patients has been reported (Baldanti et al., 1996), and
high-level GCV-resistant strains with both UL97 and UL54
mutations were found to be cross-resistant to CDV (Smith et
al., 1997). At present, however, CDV is effective against
most of the GCV-resistant isolates studied, as well as PFA-
resistant viruses generated in vitro.
An antisense oligonucleotide against the HCMV IE2
mRNA (ISIS2922, fomivirsen) has also been developed
(Azad et al., 1993; Anderson et al., 1996) for intravitreal
application in patients with retinitis (De Smet et al., 1999)
who do not respond to conventional management.
A promising new class of anti-HCMV compounds is
formed of benzimidazole ribosides, which do not inhibit
the viral DNA polymerase. The benzimidazole analog
1263W94 has many characteristics that make it an attractive
candidate for development; namely, high potency in vitro,

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297288
selectivity, good oral bioavailability, and lower toxicity than
current therapies. Initial clinical trials have provided
encouraging results, including good tolerability and linear
pharmacokinetics over a wide dose range (Chulay et al.,
1999).
10.2. Therapeutic approaches
The antiviral agents described in the previous section are
used to treat and prevent HCMV infections in immunocom-
promised patients. In the immunocompetent host, infections
normally resolve spontaneously and antiviral therapy is not
indicated. Three major therapeutic approaches are currently
employed to manage HCMV infections and diseases: pro-
phylaxis, pre-emptive therapy, and treatment of an estab-
lished disease.
10.2.1. Prophylaxis
In prophylaxis, the antiviral drug is administered before
active HCMV infection is detected to prevent its occurrence.
The therapeutic value of an antiviral prophylaxis in AIDS
patients at high risk for HCMV disease is controversial. A
randomized clinical trial of oral GCV versus placebo found
that prophylaxis significantly reduced disseminated disease
and the incidence of retinitis (Spector et al., 1996). In
contrast, Brosgart et al. (1998) found no significant reduc-
tion in the occurrence of HCMV disease. Moreover, it is still
not clear whether oral GCV prophylaxis improves survival
and sight preservation compared with pre-emptive therapy
(Masur et al., 1996).
GCV prophylaxis after allogenic BMT in seropositive
recipients was significantly protective in two studies (Good-
rich et al., 1993; Boeckh & Bowden, 1995), but failed in a
third (Winston et al., 1993) and did not reduce overall
mortality (Goodrich et al., 1993; Winston et al., 1993;
Boeckh & Bowden, 1995), probably because drug-induced
neutropenia led to fatal bacterial and fungal infections.
In one study of heart transplant recipients, GCV prophy-
laxis exhibited a protective effect in seropositive patients, but
not in primary infection (Merigan et al., 1992), but the
opposite result was obtained in a different study (Macdonald
et al., 1995). For renal transplant recipients, prophylaxis is
recommended when the donor or recipient is seropositive
and if the recipient is subjected to an immunosuppressive
regimen, including an antilymphocyte treatment, or for a
seronegative recipient of a graft from a seropositive donor
(Jassal et al., 1998). In conclusion, antiviral prophylaxis may
be useful in some cases, but it is a high-cost strategy that
exposes all patients to drug toxicity (marrow suppression and
renal toxicity), the selection of antiviral resistance, and the
risk of late-onset HCMV infection and disease (Emery,
2001).
10.2.2. Pre-emptive treatment
Pre-emptive treatment was first described in the early
1990s (Rubin, 1991) as a means of reducing the incidence of
HCMV disease by withholding antiviral drugs until they
would be maximally effective. Antiviral treatment is initi-
ated when HCMV positivity is detected in the blood or
broncoalveolar lavage fluid using sensitive methods, such as
PCR, nucleic acid sequence-based amplification, and tests
for viral antigen. The advantages for the management of
HCMV infection include the targeting of antiviral therapy to
those most at risk for future disease, reducing the number of
patients exposed to antiviral toxicity, lowering the risk of
drug resistance, and maximizing the cost:benefit ratio. In the
BMT setting, pre-emptive GCV administration reduced
HCMV disease, and was literally life saving (Goodrich et
al., 1991; Einsele et al., 1995) in contrast to its lack of effect
on survival when used prophylactically (Goodrich et al.,
1993; Winston et al., 1993; Boeckh & Bowden, 1995). The
pre-emptive approach also prevented HCMV diseases in
liver transplant recipients (Singh et al., 1994). A prospective
study of the predictive value of PCR for CMV in asympto-
matic kidney transplant recipients indicated that while a
substantial number of recipients become positive for
HCMV-PCR after transplantation, only a minority would
develop CMV disease. Negative CMV-PCR assay is an
accurate negative predictor for CMV disease, but the value
of HCMV-PCR as a guide for pre-emptive anti-CMV
therapy in kidney transplant recipients is limited (Benedetti
et al., 1998).
10.2.3. Treatment of an established disease
It is generally agreed that it is better to prevent HCMV
diseases with pre-emptive approaches than to treat an
established disease, since an ongoing virus infection may
induce pathological phenomena unresponsive to antiviral
drugs and extensive tissue damage that may result in target
organ failure.
Clinical trials on AIDS patients with retinitis showed that
intravenous GCV, PFA, and CDV slow the progression of the
disease and lead to remission (Palestine et al., 1991; Spector
et al., 1993; Polis et al., 1995; Chavez-de la Paz et al., 1997;
Lalezari et al., 1998). In one study, PFA showed a higher
survival benefit than GCV (Anonymous, 1992). Retinitis can
also be treated with intravitreal injections of antiviral
compounds or by inserting an intraocular implant that
slowly releases the drug into the vitreous fluid (Masur et
al., 1996; Jacobson, 1997–1998). All antiviral compounds
suppress active replication of the virus, but do not eliminate
it. Unless immune reconstitution occurs as a consequence of
highly active antiretroviral therapy, chronic suppressive
maintenance anti-HCMV management is required, and will
be equally effective when administered intravenously, per os,
or locally (Drew et al., 1995). However, while oral and local
administration avoid the inconveniences and risks associated
with the intravenous route, they are less effective in prevent-
ing disease in the contralateral eye (Jacobson, 1997–1998;
Cheung & Teich, 1999). Maintenance therapy may result in
the appearance of resistant and refractory strains (Drew et al.,
1991; Cherrington et al., 1996; Jabs et al., 1998a, 1998b),

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 289
whose estimated emergence rate is � 25–33% after 9
months. These rates are similar for GCV, PFA, and CDV
(Jabs et al., 1998a, 1998b). The efficacy of the antisense
oligonucleotide fomivirsen in treating HCMV retinitis in
AIDS patients has been demonstrated recently (The Vitra-
vene Study Group, 2002a, 2002b).
GCVor PFA regimens have enjoyed some success in the
treatment of symptomatic gastrointestinal (esophagitis,
enterocolitis), respiratory (pneumonia), and neurologic
(encephalitis, peripheral neuropathy, polyradiculoneurop-
athy) HCMV diseases in AIDS (Dieterich et al., 1993;
Blanshard et al., 1995; Cheung & Teich, 1999).
No antiviral agents have been approved as yet for the
treatment of congenital HCMV infections.
Acknowledgments
The authors’ research is supported by grants from
MURST-CNR Biotechnology program L. 95/95, MURST
(40% and 60%), from the AIDS Research Project, and by
grants from the Ricerca Sanitaria Finalizzata (Regione
Piemonte).
References
Adler, S. P., Starr, S. E., Plotkin, S. A., Hempfling, S. H., Buis, J., Manning,
M. L., & Best, A. M. (1995). Immunity induced by primary human
cytomegalovirus infection protects against secondary infection among
women of childbearing age. J Infect Dis 171, 26–32.
Adler, S. P., Hempfling, S. H., Starr, S. E., Plotkin, S. A., & Riddell, S.
(1998). Safety and immunogenicity of the Towne strain cytomegalovi-
rus vaccine. Pediatr Infect Dis J 17, 200–206.
Adler, S. P., Plotkin, S. A., Gonczol, E., Cadoz, M., Meric, C., Wang, J. B.,
Dellamonica, P., Best, A. M., Zahradnik, J., Pincus, S., Berencsi, K.,
Cox, W. I., & Gyulai, Z. (1999). A canarypox vector expressing cyto-
megalovirus (CMV) glycoprotein B primes for antibody responses to a
live attenuated CMV vaccine (Towne). J Infect Dis 180, 843–846.
Aduma, P., Connelly, M. C., Srinivas, M. C., & Fridland, A. (1995). Meta-
bolic diversity and antiviral activities of acyclic nucleoside phospho-
nates. Mol Pharmacol 47, 816–822.
Ahn, J. H., Jang, W. J., & Hayward, G. S. (1999). The human cytomega-
lovirus IE2 and UL112-113 proteins accumulate in viral DNA replica-
tion compartments that initiate from the periphery of promyelocytic
leukemia protein associated nuclear bodies (PODs or ND10). J Virol
73, 10471–10548.
Anderson, K. P., Fox, M. C., Brown-Driver, V., Martin, M. J., & Azad, R. F.
(1996). Inhibition of human cytomegalovirus immediate-early gene ex-
pression by an antisense oligonucleotide complementary to immediate-
early RNA. Antimicrob Agents Chemother 40, 2004–2011.
Anonymous (1992). Mortality in patients with the acquired immunodefi-
ciency syndrome treated with either foscarnet or ganciclovir for cyto-
megalovirus retinitis. Studies of Ocular Complications of AIDS
Research Group, in collaboration with the AIDS Clinical Trials Group.
N Engl J Med 326, 213–220. Erratum: N Engl J Med 326, 1172
(1992).
Aono, T., Kondo, K., Miyoshi, H., Tanaka-Taya, K., Kondo, M., Osugi, Y.,
Hara, J., Okada, S., & Yamanishi, K. (1998). Monitoring of human
cytomegalovirus infections in pediatric bone marrow transplant recipi-
ents by nucleic acid sequence-based amplification. J Infect Dis 178,
1244–1249.
Apperley, J. F., & Goldman, J. M. (1988). Cytomegalovirus: biology, clin-
ical features and methods for diagnosis. Bone Marrow Transplant 3,
253–264.
Azad, R. F., Driver, V. B., Tanaka, K., Crooke, R. M., & Anderson, K. P.
(1993). Antiviral activity of a phosphorothioate oligonucleotide com-
plementary to RNA of the human cytomegalovirus major immediate-
early region. Antimicrobiol Agents Chemother 37, 1945–1954.
Baldanti, F., Silini, E., Sarasini, A., Talarico, C. L., Stanat, S. C., Biron, K. K.,
Furione, M., Bono, F., Palu, G., & Gerna, G. (1995). A three-nucleotide
deletion in the UL97 open reading frame is responsible for the ganci-
clovir resistance of a human cytomegalovirus clinical isolate. J Virol 69,
796–800.
Baldanti, F., Underwood, M. R., Stanat, S. C., Biron, K. K., Chou, S.,
Sarasini, A., Silini, E., & Gerna, G. (1996). Single amino acid
changes in the DNA polymerase confer foscarnet resistance and
slow-growth phenotype, while mutations in the UL97-encoded phos-
photransferase confer ganciclovir resistance in three double-resistant
human cytomegalovirus strains recovered from patients with AIDS.
J Virol 70, 1390–1395.
Baldanti, F., Simoncini, L., Talarico, C. L., Sarasini, A., Biron, K. K., &
Gerna, G. (1998). Emergence of a ganciclovir-resistant human cytome-
galovirus strain with a new UL97 mutation in an AIDS patient. AIDS
12, 816–818.
Baldick, C. J., & Shenk, T. (1996). Proteins associated with purified human
cytomegalovirus particles. J Virol 70, 6097–6105.
Baldick, C. J., Marchini, A., Patterson, C. E., & Shenk, T. (1997). Human
cytomegalovirus tegument protein pp71 (ppUL82) enhances the infec-
tivity of viral DNA and accelerates the infectious cycle. J Virol 71,
4400–4408.
Becroft, D. M. O. (1981). Prenatal cytomegalovirus infection: epidemiol-
ogy, pathology, and pathogenesis. In H. S. Rosenberg, & J. Bernstein
(Eds.), Perspective in Pediatric Pathology ( pp. 203–241). New York:
Masson Press.
Benedetti, E., Mihalov, M., Asolati, M., Kirby, J., Dunn, T., Raofi, V.,
Fontaine, M., & Pollak, R. (1998). A prospective study of the predictive
value of polymerase chain reaction assay for cytomegalovirus in asymp-
tomatic kidney transplant recipients. Clin Transplant 12, 391–395.
Beninga, J., Kropff, B., & Mach, M. (1995). Comparative analysis of four-
teen individual human cytomegalovirus proteins for helper T cell re-
sponse. J Gen Virol 76, 153–160.
Benson, J. D., & Huang, E.-S. (1990). Human cytomegalovirus induces
expression of cellular topoisomerase II. J Virol 64, 9–15.
Berencsi, K., Gyulai, Z., Gonczol, E., Pincus, S., Cox, W. I., Michelson, S.,
Kari, L., Meric, C., Cadoz, M., Zahradnik, J., Starr, S., & Plotkin, S.
(2001). A canarypox vector-expressing cytomegalovirus (CMV) phos-
phoprotein 65 induces long-lasting cytotoxic T cell responses in human
CMV-seronegative subjects. J Infect Dis 183, 1171–1179.
Bissinger, A. L., Singzer, C., Kaiserling, E., & Jahn, G. (2002). Human
cytomegalovirus as a direct pathogen: correlation of multiorgan in-
volvement and cell distribution with clinical and pathological findings
in a case of congenital inclusion disease. J Med Virol 67, 200–206.
Blackburn, N. K., Besselaar, T. G., Schoub, B. D., & O’Connell, K. F.
(1991). Differentiation of primary cytomegalovirus infection from re-
activation using the urea denaturation test for measuring antibody avid-
ity. J Med Virol 33, 6–9.
Blanshard, C., Benhamou, Y., Dohin, E., Lernestedt, J. O., Gazzard, B. G.,
& Katlama, C. (1995). Treatment of AIDS-associated gastrointestinal
cytomegalovirus infection with foscarnet and ganciclovir: a randomized
comparison. J Infect Dis 172, 622–628.
Blok, M. J., Goossens, V. J., Vanherle, S. J., Top, B., Tacken, N.,
Middeldorp, J. M., Christiaans, M. H., van Hooff, J. P., & Bruggeman,
C. A. (1998). Diagnostic value of monitoring human cytomegalovirus
late pp67 mRNA expression in renal-allograft recipients by nucleic acid
sequence-based amplification. J Clin Microbiol 36, 1341–1346.
Bodaghi, B., Jones, T. R., Zipeto, D., Vita, C., Sun, L., Laurent, L.,
Arenzana-Seisdedos, F., Virelizier, J. L., & Michelson, S. (1998).
Chemokine sequestration by viral chemoreceptors as a novel viral

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297290
escape strategy: withdrawal of chemokines from the environment of
cytomegalovirus-infected cells. J Exp Med 188, 855–866.
Bodeus, M., Feyder, S., & Goubau, P. (1998). Avidity of IgG antibodies
distinguishes primary from non-primary cytomegalovirus infection in
pregnant women. Clin Diagn Virol 9, 9–16.
Boeckh, M., & Bowden, R. (1995). Cytomegalovirus infection in marrow
transplantation. Cancer Treat Res 76, 97–136.
Boppana, S. B., & Britt, W. J. (1996). Recognition of human cytomegalo-
virus gene products by HCMV-specific cytotoxic T cells. Virology 222,
293–296.
Boppana, S. B., Pass, R. F., Britt, W. J., Stagno, S., & Alford, C. A. (1992).
Symptomatic congenital cytomegalovirus infection: neonatal morbidity
and mortality. Pediatr Infect Dis 11, 93–99.
Boshart, M., Weber, F., Jahn, G., Dorsch-Hasler, K., Fleckenstein, B., &
Schaffner, W. (1985). A very strong enhancer is located upstream of an
immediate early gene of human cytomegalovirus. Cell 41, 521–530.
Bowen, E. F., Sabin, C. A., Wilson, P., Griffiths, P. D., Davey, C. C.,
Johnson, M. A., & Emery, V. C. (1997). Cytomegalovirus (CMV) vir-
aemia detected by polymerase chain reaction identifies a group of HIV-
positive patients at high risk of CMV disease. AIDS 11, 889–893.
Boyle, K. A., Pietropaolo, R. L., & Compton, T. (1999). Engagement of the
cellular receptor for glycoprotein B of human cytomegalovirus activates
the interferon-responsive pathway. Mol Cell Biol 19, 3607–3613.
Bresnahan, W. A., & Shenk, T. (2000a). A subset of viral transcripts pack-
aged within human cytomegalovirus particles. Science 288, 2373–2376.
Bresnahan, W. A., & Shenk, T. (2000b). UL82 virion protein activates
expression of immediate early viral genes in human cytomegalovirus-
infected cells. Proc Natl Acad Sci USA 97, 14506–14511.
Bresnahan, W. A., Boldogh, I., Thompson, E. A., & Albrecht, T. (1996).
Human cytomegalovirus inhibits cellular DNA synthesis and arrests
productively infected cells in late G1. Virology 224, 150–160.
Bresnahan, W. A., Thompson, E. A., & Albrecht, T. (1997). Human cyto-
megalovirus infection results in altered Cdk2 subcellular localization.
J Gen Virol 78, 1993–1997.
Britt, W. J. (1996). Vaccines against human cytomegalovirus: time to test.
Trends Microbiol 4, 34–38.
Britt, W. J., & Mach, M. (1996). Human cytomegalovirus glycoproteins.
Intervirology 39, 401–412.
Britt, W. J., Vugler, L., Butfiloski, E. J., & Stephens, E. B. (1990). Cell
surface expression of human cytomegalovirus (HCMV) gp55-116 (gB):
use of HCMV-recombinant vaccinia virus-infected cells in analysis of
the human neutralizing antibody response. J Virol 64, 1079–1085.
Brosgart, C. L., Louis, T. A., Hillman, D. W., Craig, C. P., Alston, B.,
Fisher, E., Abrams, D. I., Luskin-Hawk, R. L., Sampson, J. H., Ward,
D. J., Thompson, M. A., & Torres, R. A. (1998). A randomized, pla-
cebo-controlled trial of the safety and efficacy of oral ganciclovir for
prophylaxis of cytomegalovirus disease in HIV-infected individuals.
Terry Beirn Community Programs for Clinical Research on AIDS. AIDS
12, 269–277.
Browne, E. P., Wing, B., Coleman, D., & Shenk, T. (2001). Altered cellular
mRNA levels in human cytomegalovirus infected fibroblasts: viral block
to the accumulation of antiviral mRNAs. J Virol 75, 12319–12330.
Burton, G. J., & Watson, A. L. (1997). The structure of the human placenta:
implications for initiating and defending against virus infections. Rev
Med Virol 7, 219–228.
Butcher, S. J., Aitken, J., Mitchell, J., Gowen, B., & Dargan, D. J. (1998).
Structure of the human cytomegalovirus B capsid by electron micro-
scopy and image reconstruction. J Struct Biol 124, 70–76.
Caswell, R., Hagemeier, C., Chiou, C.-J., Hayward, G., Kouzarides, T., &
Sinclair, J. H. (1993). The human cytomegalovirus 86K immediate
early (IE2) protein requires the basic region of the TATA-box binding
protein (TBP) for binding, and interacts with TBP and transcription
factor TFIIB via region of IE2 required for transcriptional regulation.
J Gen Virol 74, 2691–2698.
Cavallo, R., Lembo, D., Gribaudo, G., & Landolfo, S. (2001). Murine
cytomegalovirus infection induces cellular folylpolyglutamate synthe-
tase activity in quiescent cells. Intervirology 44, 224–226.
Cha, T. A., Tom, E., Kemble, G. W., Duke, G. M., Mocarski, E. S., &
Spaete, R. R. (1996). Human cytomegalovirus clinical isolates carry at
least 19 genes not found in laboratory strains. J Virol 70, 78–83.
Chambers, J., Angulo, A., Amaratunga, D., Guo, H., Jiang, Y., Wan, J. S.,
Bittner, A., Frueh, K., Jackson, M. R., Peterson, P. A., Erlander, M. G.,
& Ghazal, P. (1999). DNA microarrays of the complex human cytome-
galovirus genome: profiling kinetic class with drug sensitivity of viral
gene expression. J Virol 73, 5757–5766.
Chavez-de la Paz, E., Arevalo, J. F., Kirsch, L. S., Munguia, D.,
Rahhal, F. M., De Clercq, E., & Freeman, W. R. (1997). Anterior non-
granulomatous uveitis after intravitreal HPMPC (cidofovir) for the treat-
ment of cytomegalovirus retinitis. Analysis and prevention. Ophthal-
mology 104, 539–544. Erratum: Ophthalmology 104, 1063 (1997).
Chee, M. S., Bankier, A. T., Beck, S., Bohni, R., Brown, C. M., Cerny, R.,
Horsnell, T., Hutchison III, C. A., Kouzarides, T., Martignetti, J. A.,
Preddie, E., Satchwell, S. C., Tomlinson, P., Weston, K. M., & Barrell,
B. G. (1990a). Analysis of the protein-coding content of human cytome-
galovirus strain AD169. Curr Top Microbiol Immunol 154, 125–169.
Chee, M. S., Satchwell, S. C., Preddie, E., Weston, K. M., & Barrell, B. G.
(1990b). Human cytomegalovirus encodes three G protein-coupled re-
ceptor homologues. Nature 344, 774–777.
Cherrington, J. M., Khoury, E. L., & Mocarski, E. S. (1991). Human
cytomegalovirus IE2 negatively regulates a gene expression via a short
target sequence near the transcription start site. J Virol 65, 887–896.
Cherrington, J. M., Miner, R., Hitchcock, M. J., Lalezari, J. P., & Drew,
W. L. (1996). Susceptibility of human cytomegalovirus to cidofovir is
unchanged after limited in vivo exposure to various regimens of drug.
J Infect Dis 173, 987–992.
Cherrington, J. M., Allen, A. S. J. W., Mulato, S., Miner, R., Drew, W. L.,
& Chen, M. S. (1998). Sensitivities of human cytomegalovirus
(HCMV) clinical isolates to cidofovir. Antiviral Res 26, A319.
Cheung, T. W., & Teich, S. A. (1999). Cytomegalovirus infection in pa-
tients with HIV infection. Mt Sinai J Med 66, 113–124.
Chou, S. W., & Dennison, K. M. (1991). Analysis of interstrain variation
in cytomegalovirus glycoprotein B sequences encoding neutralization-
related epitopes. J Infect Dis 163, 1229–1234.
Chou, S., Guentzel, S., Michels, K. R., Miner, R. C., & Drew, W. L.
(1995). Frequency of UL97 phosphotransferase mutations related to
ganciclovir resistance in clinical cytomegalovirus isolates. J Infect Dis
172, 239–242.
Chrisp, P., & Clissold, S. P. (1991). Foscarnet: a review of its antiviral
activity, pharmacokinetic properties and therapeutic use in immunocom-
promised patients with cytomegalovirus retinitis. Drugs 41, 104–129.
Chulay, J., Biron, K., Wang, L., Underwood, M., Chamberlain, S., Frick,
L., Good, S., Davis, M., Harvey, R., Townsend, L., Drach, J., &
Koszalka, G. (1999). Development of novel benzimidazole riboside
compounds for treatment of cytomegalovirus disease. Adv Exp Med
Biol 458, 129–134.
Compton, T., Nowlin, D. M., & Cooper, N. R. (1993). Initiation of human
cytomegalovirus infection requires initial interaction with cell surface
heparan sulfate. Virology 193, 834–841.
Courcelle, C. T., Courcelle, J., Prichard, M. N., & Mocarski, E. S. (2001).
Requirement for uracil-DNA glycosylase during the transition to late-
phase cytomegalovirus DNA replication. J Virol 75, 7592–7601.
Cundy, K. C., Li, Z. H., & Lee, A. (1994). Effect of concomitant
probenecid on the tissue distribution and urinary excretion of HPMPC
in preclinical models. Pharm Res 11, S449.
Davignon, J. L., Clement, D., Alriquet, J., Michelson, S., & Davrinche, C.
(1995). Analysis of the proliferative T cell response to human cytome-
galovirus major immediate-early protein (IE1): phenotype, frequency
and variability. Scand J Immunol 41, 247–255.
De Clercq, E. (1998). Antiviral agents that are active against CMV: potential
of cidofovir for the treatment of CMV and other virus infections. In M.
Scholz, H. F. Rabenau, H. W. Doerr, & J. Cinatl, Jr. (Eds), CMV-Related
Immunopathology, Monographs in Virology, Vol. 21 (pp. 193–214).
Basel: Karger.
De Smet, M. D., Meenken, C. J., & van den Horn, G. J. (1999). Fomivirsen

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 291
a phosphorothioate oligonucleotide for the treatment of CMV retinitis.
Ocul Immunol Inflamm 7, 189–198.
Dieterich, D. T., Kotler, D. P., Busch, D. F., Crumpacker, C., Du Mond, C.,
Dearmand, B., & Buhles, W. (1993). Ganciclovir treatment of cytome-
galovirus colitis in AIDS: a randomized, double-blind, placebo-con-
trolled multicenter study. J Infect Dis 167, 278–282.
Dittmer, D., & Mocarski, E. S. (1997). Human cytomegalovirus infection
inhibits G1/S transition. J Virol 71, 1629–1634.
Donaldson, P. T., Alexander, G. J., O’Grady, J., Neuberger, J., Portmann,
B., Thick, M., Davis, H., Calne, R. Y., & Williams, R. (1987). Evidence
for an immune response to HLA class I antigens in the vanishing-bile
duct syndrome after liver transplantation. Lancet 1, 945–948.
Drew, W. L., Miner, R. C., Busch, D. F., Follansbee, S. E., Gullett, J., Me-
halko, S. G., Gordon, S. M., Owen, W. F., Jr., Matthews, T. R., & Buhles,
W. C. (1991). Prevalence of resistance in patients receiving ganciclovir
for serious cytomegalovirus infection. J Infect Dis 163, 716–719.
Drew, W. L., Ives, D., Lalezari, J. P., Crumpacker, C., Follansbee, S. E.,
Spector, S. A., Benson, C. A., Friedberg, D. N., Hubbard, L., Stempien,
M. J., Shadman, A., & Buhles, W. (1995). Oral ganciclovir as main-
tenance treatment for cytomegalovirus retinitis in patients with AIDS.
N Engl J Med 333, 615–620.
Drulak, M. W., Malinoski, F. J., Fuller, S. A., Stewart, S. S., Hoskin, S.,
Duliege, A. M., Sekulovich, R., Burke, R., & Winston, S. (2000).
Vaccination of seropositive subjects with CHIRON CMV gB subunit
vaccine combined with MF59 adjuvant for production of CMV immune
globulin. Viral Immunol 13, 49–56.
Dworsky, M., Yow, M., Stagno, S., Pass, R. F., & Alford, C. (1983).
Cytomegalovirus infection of breast milk and transmission in infancy.
Pediatrics 72, 295–299.
Einsele, H., Ehninger, G., Hebart, H., Wittkowski, K. M., Schuler, U., Jahn,
G., Mackes, P., Herter, M., Klingebiel, T., & Loffler, J. (1995). Poly-
merase chain reaction monitoring reduces the incidence of cytomega-
lovirus disease and the duration and side effects of antiviral therapy
after bone marrow transplantation. Blood 86, 2815–2820.
Elek, S. D., & Stern, H. (1974). Development of a vaccine against mental
retardation caused by cytomegalovirus infection in utero. Lancet i, 1–5.
Emery, V. C. (2001). Prophylaxis for CMV should not now replace pre-
emptive therapy in solid organ transplantation. RevMed Virol 11, 83–86.
Endresz, V., Kari, L., Berencsi, K., Kari, C., Gyulai, Z., Jeney, C., Pincus, S.,
Rodeck, U., Meric, C., Plotkin, S. A., & Gonczol, E. (1999). Induction of
human cytomegalovirus (HCMV)-glycoprotein B (gB)-specific neutral-
izing antibody and phosphoprotein 65 (pp65)-specific cytotoxic T lym-
phocyte responses by naked DNA immunization. Vaccine 17, 50–58.
Endresz, V., Burian, K., Berencsi, K., Gyulai, Z., Kari, L., Horton, H.,
Virok, D., Meric, C., Plotkin, S. A., & Gonczol, E. (2001). Optimization
of DNA immunization against human cytomegalovirus. Vaccine 19,
3972–3980.
Erice, A., Chou, S., Biron, K. K., Stanat, S. C., Balfour, H. H., Jr., &
Jordan, M. C. (1989). Progressive disease due to ganciclovir-resistant
cytomegalovirus in immunocompromised patients. N Engl J Med 320,
289–93.
Estes, J. E., & Huang, E. -S. (1977). Stimulation of cellular thymidine
kinase by human cytomegalovirus. J Virol 24, 13–21.
Faber, D. W., Wiley, C. A., Lynn, G. B., Gross, J. G., & Freeman, W. R.
(1992). Role of HIV and CMV in the pathogenesis of retinitis and
retinal vasculopathy in AIDS patients. Invest Ophthalmol Vis Sci 33,
2345–2353.
Fortunato, E. A., & Spector, D. H. (1999). Regulation of human cytome-
galovirus gene expression. Adv Virus Res 54, 61–128.
Fortunato, E. A., Sommer, M. H., Yoder, K., & Spector, D. H. (1997).
Identification of domains within the human cytomegalovirus major im-
mediate-early 86-kilodalton protein and the retinoblastoma protein re-
quired for physical and functional interaction with each other. J Virol
71, 8176–8185.
Fortunato, E. A., McElroy, A. K., Sanchez, V., & Spector, D. H. (2000).
Exploitation of cellular signalling and regulatory pathways by human
cytomegalovirus. Trends Microbiol 8, 111–119.
Fowler, K. B., Stagno, S., Pass, R. F., Britt, W. J., Boll, T. J., & Alford,
C. A. (1992). The outcome of congenital cytomegalovirus infection in
relation to maternal antibody status. N Engl J Med 326, 663–667.
Gallant, J. E., Moore, R. D., Richman, D. D., Keruly, J., & Chaisson, R. E.
(1992). Incidence and natural history of cytomegalovirus disease in
patients with advanced human immunodeficiency virus disease treated
with zidovudine. J Infect Dis 166, 1223–1227.
Gao, J. L., & Murphy, P. M. (1994). Human cytomegalovirus open reading
frame US28 encodes a functional b chemokine receptor. J Biol Chem
269, 28539–28542.
Gerna, G., Revello, M. G., Percivalle, E., Zavattoni, M., Marea, M., &
Battaglia, M. (1990). Quantification of human Cytomegalovirus viremia
by using monoclonal antibodies to different viral proteins. J Clin Mir-
obiol 28, 2681–2688.
Gerna, G., Zipeto, D., Parea, M., Revello, M. G., Silini, E., Percivalle, E.,
Zavattoni, M., Grossi, P., & Milanesi, G. (1991). Monitoring of human
cytomegalovirus infections and ganciclovir treatment in heart transplant
recipients by determination of viremia, antigenemia, and DNAemia.
J Infect Dis 164, 488–498.
Gerna, G., Baldanti, F., Zavattoni, M., Sarasini, A., Percivalle, E., & Rev-
ello, M. G. (1992a). Monitoring of ganciclovir sensitivity of multiple
human cytomegalovirus strains coinfecting blood of an AIDS patient by
an immediate-early antigen plaque assay. Antiviral Res 1, 333–345.
Gerna, G., Revello, M. G., Percivalle, E., & Morini, F. (1992b). Com-
parison of different immunostaining techniques and monoclonal anti-
bodies to the lower matrix phosphoprotein (pp65) for optimal
quantitation of human cytomegalovirus antigenemia. J Clin Microbiol
30, 1232–1237.
Gerna, G., Percivalle, E., Revello, M. G., & Morini, F. (1993). Correlation
of quantitative human cytomegalovirus pp65- p72- and p150-antigene-
mia, viremia and circulating endothelial giant cells with clinical symp-
toms and antiviral treatment in immunocompromised patients. Clin
Diagn Virol 1, 47–59.
Gerna, G., Baldanti, F., Sarasini, A., Furione, M., Percivalle, E., Revello,
M. G., Zipeto, D., & Zella, D. (1994a). Effect of foscarnet induction
treatment on quantitation of human cytomegalovirus (HCMV) DNA in
peripheral blood polymorphonuclear leukocytes and aqueous humor of
AIDS patients with HCMV retinitis. Antimicrob Agents Chemother 38,
38–44.
Gerna, G., Furione, M., Baldanti, F., & Sarasini, A. (1994b). Comparative
quantitation of human cytomegalovirus DNA in blood leukocytes and
plasma of transplant and AIDS patients. J Clin Microbiol 32, 709–717.
Gerna, G., Furione, M., Baldanti, F., Percivalle, E., Comoli, P., & Locatelli,
F. (1995). Quantitation of human cytomegalovirus DNA in bone mar-
row transplant recipients. Br J Haematol 91, 674–683.
Gerna, G., d’Arminio Monforte, A., Zavattoni, M., Sarasini, A., Testa, L.,
Revello, M. G., & Moroni, M. (1998a). Sharp drop in the prevalence of
human cytomegalovirus leuko-DNAemia in HIV-infected patients fol-
lowing highly active antiretroviral therapy. AIDS 12, 118–120.
Gerna, G., Zavattoni, M., Baldanti, F., Sarasini, A., Chezzi, L., Grossi, P., &
Revello, M. G. (1998b). Human cytomegalovirus (HCMV) leukodnae-
mia correlates more closely with clinical symptoms than antigenemia
and viremia in heart and heart-lung transplant recipients with primary
HCMV infection. Transplantation 65, 1378–1385.
Gerna, G., Baldanti, F., Middeldorp, J. M., Furione, M., Zavattoni, M.,
Lilleri, D., & Revello, M. G. (1999). Clinical significance of expression
of human cytomegalovirus pp67 late transcript in heart, lung, and bone
marrow transplant recipients as determined by nucleic acid sequence-
based amplification. J Clin Microbiol 37, 902–911.
Gerna, G., Baldanti, F., Lilleri, D., Parea, M., Alessandrino, E., Pagani, A.,
Locatelli, F., Middeldorp, J., & Revello, M. G. (2000a). Human cytome-
galovirus immediate-early mRNA detection by nucleic acid sequence-
based amplification as a new parameter for preemptive therapy in bone
marrow transplant recipients. J Clin Microbiol 38, 1845–1853.
Gerna, G., Percivalle, E., Baldanti, F., Sozzani, S., Lanzarini, P., Genini, E.,
Lilleri, D., & Revello, M. G. (2000b). Human cytomegalovirus repli-
cates abortively in polymorphonuclear leukocytes after transfer from

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297292
infected endothelial cells via transient microfusion events. J Virol 74,
5629–5638.
Gibson, W. (1996). Structure and assembly of the virion. Intervirology 39,
389–400.
Gilbert, G. L., Hayes, K., Hudson, I. L., & James, J. (1989). Prevention of
transfusion-acquired cytomegalovirus infection in infants by blood fil-
tration to remove leucocytes. Neonatal Cytomegalovirus Infection
Study Group. Lancet i, 1228–1231.
Gilbert, M. J., Riddell, S. R., Plachter, B., & Greenberg, P. D. (1996).
Cytomegalovirus selectively blocks antigen processing and presentation
of its immediate-early gene product. Nature 383, 720–722.
Gleaves, C. A., Smith, T. F., Shuster, E. A., & Pearson, G. R. (1984). Rapid
detection of cytomegalovirus in MRC-5 cells inoculated with urine
specimens by using low-speed centrifugation and monoclonal antibody
to an early antigen. J Clin Microbiol 19, 917–919.
Goldmacher, V. S. (2002). vMIA, a viral inhibitor of apoptosis targeting
mitochondria. Biochimie 84, 177–185.
Gonczol, E., & Plotkin, S. (2001). Development of a cytomegalovirus
vaccine: lessons from recent clinical trials. Expert Opin Biol Ther 1,
401–412.
Gonczol, E., deTaisne, C., Hirka, G., Berencsi, K., Lin, W. C., Paoletti, E., &
Plotkin, S. (1991). High expression of human cytomegalovirus (HCMV)-
gB protein in cells infected with a vaccinia-gB recombinant: the impor-
tance of the gB protein in HCMV immunity. Vaccine 9, 631–637.
Goodrich, J. M., Mori, M., Gleaves, C. A., Du Mond, C., Cays, M.,
Ebeling, D. F., Buhles, W. C., DeArmond, B., & Meyers, J. D.
(1991). Early treatment with ganciclovir to prevent cytomegalovirus
disease after allogeneic bone marrow transplantation. N Engl J Med
325, 1601–1607.
Goodrich, J. M., Bowden, R. A., Fisher, L., Keller, C., Schoch, G., &
Meyers, J. D. (1993). Ganciclovir prophylaxis to prevent cytomegalo-
virus disease after allogeneic marrow transplant. Ann Intern Med 118,
173–178.
Gozlan, J., Laporte, J. P., Lesage, S., Labopin, M., Najman, A., Gorin,
N. C., & Petit, J. C. (1996). Monitoring of cytomegalovirus infection
and disease in bone marrow recipients by reverse transcription-PCR and
comparison with PCR and blood and urine cultures. J Clin Microbiol
34, 2085–2088.
Grangeot-Keros, L., Mayaux, M. J., Lebon, P., Freymuth, F., Eugene, G.,
Stricker, R., & Dussaix, E. (1997). Value of cytomegalovirus (CMV)
IgG avidity index for the diagnosis of primary CMV infection in preg-
nant women. J Infect Dis 175, 944–946.
Gretch, D. R., Kari, B., Rasmussen, L., Gehrz, R. C., & Stinski, M. F.
(1988). Identification and characterization of three distinct families of
glycoprotein complexes in the envelopes of human cytomegalovirus.
J Virol 62, 875–881.
Gribaudo, G., Riera, L., Lembo, D., De Andrea, M., Gariglio, M.,
Rudge, T. L., Johnson, L. F., & Landolfo, S. (2000). Murine cyto-
megalovirus stimulates cellular thymidylate synthase gene expression
in quiescent cells and requires the enzyme for replication. J Virol 74,
4979–4987.
Gribaudo, G., Riera, L., Rudge, T. L., Caposio, P., Johnson, L. F., & Land-
olfo, S. (2002). Human cytomegalovirus infection induces cellular thy-
midylate synthase gene expression in quiescent fibroblasts. J Gen Virol
83, 2983–2993.
Griffiths, P. D. (2000). Cytomegalovirus. In A. J. Zuckerman, J. E. Banat-
vala, & J. R. Pattison (Eds.), Principles and Practice of Clinical Virol-
ogy ( pp. 79–116). London: John Wiley and Sons.
Griffiths, P. D., Panjwani, D. D., Stirk, P. R., Ball, M. G., Ganczakowski,
M., Blacklock, H. A., & Prentice, H. G. (1984). Rapid diagnosis of
cytomegalovirus infection in immunocompromised patients by detec-
tion of early antigen fluorescent foci. Lancet ii, 1242–1245.
Grossi, P., Minoli, L., Percivalle, E., Irish, W., Vigano, M., & Gerna, G.
(1995). Clinical and virological monitoring of human cytomegalovi-
rus infection in 294 heart transplant recipients. Transplantation 59,
847–851.
Gyulai, Z., Endresz, V., Burian, K., Pincus, S., Toldy, J., Cox, W. I., Meric,
C., Plotkin, S., Gonczol, E., & Berencsi, K. (2000). Cytotoxic T lym-
phocyte (CTL) responses to human cytomegalovirus pp65, IE1-Exon4,
gB, pp150, and pp28 in healthy individuals: reevaluation of prevalence
of IE1-specific CTLs. J Infect Dis 181, 1537–1546.
Hagemeier, C., Walker, S., Caswell, R., Kouzarides, T., & Sinclair, J. H.
(1992a). The 72K IE1 and 80K IE2 proteins of human cytomegalovirus
independently trans-activate the c-fos, c-myc and hsp70 promoter via
basal promoter elements. J Gen Virol 73, 2385–2393.
Hagemeier, C., Walker, S., Caswell, R., Kouzarides, T., & Sinclair, J. H.
(1992b). The human cytomegalovirus 80-kilodalton but not the 72-kilo-
dalton immediate-early protein transactivates heterologous promoters in
a TATA box-dependent mechanism and interacts directly with TFIID.
J Virol 66, 4452–4456.
Hagemeier, C., Caswell, R., Hayhurst, G., Sinclair, J. H., & Kouzarides, T.
(1994). Functional interaction between the HCMV IE2 transactivator
and the retinoblastoma protein. EMBO J 13, 2897–2903.
Hahn, G., Jores, R., & Mocarski, E. (1998). Cytomegalovirus remains latent
in a common precursor of dendritic and myeloid cells. Proc Natl Acad
Sci USA 95, 3937–3942.
Halwachs-Baumann, G., Wilders-Trucshnig, M., Desoye, G., Hahn, T.,
Kiesel, L., Klingel, K., Rieger, P., Jahn, G., & Sinzger, C. (1998).
Human trophoblast cells are permissive to the complete replicative
cycle of human cytomegalovirus. J Virol 72, 7598–7602.
Harrison, C. J., Britt, W. J., Chapan, N. M., Mullican, J., & Tracy, S.
(1995). Reduced congenital cytomegalovirus (CMV) infection after
maternal immunization with a guinea pig CMV glycoprotein before
gestational primary CMV infection in the guinea pig model. J Infect
Dis 172, 1212–1220.
Hayashi, M. L., Blankenship, C., & Shenk, T. (2000). Human cytomega-
lovirus UL69 protein is required for efficient accumulation of infected
cells in the G1 phase of the cell cycle. Proc Natl Acad Sci USA 97,
2692–2696.
Hayhurst, G. P., Bryant, L. A., Caswell, R. C., Walker, S. M., & Sinclair,
J. H. (1995). CCAAT box-dependent activation of the TATA-less hu-
man DNA polymerase a promoter by the human cytomegalovirus 72-
kilodalton major immediate-early protein. J Virol 69, 182–188.
He, H., Rinaldo, C. R., Jr., & Morel, P. A. (1995). T cell proliferative
responses for five human cytomegalovirus proteins in healthy seropos-
itive individuals: implications for vaccine development. J Gen Virol 76,
1603–1610.
Hemmings, D. G., Kilani, R., Nyukiforuk, C., Preiksaitis, J., & Guibert,
L. J. (1998). Permissive cytomegalovirus infection of primary villous
term and first trimester trophoblasts. J Virol 72, 4970–4979.
Hengel, H., Brune, W., & Koszinowski, U. H. (1998). Immune evasion by
cytomegalovirus-survival strategies of a highly adapted opportunist.
Trends Microbiol 6, 190–197.
Herrera, G. A., Alexander, R. W., Cooly, C. F., Luke, R. G., Kelly, D. R.,
Curtis, J. J., & Gockerman, J. P. (1986). Cytomegalovirus glomerulop-
athy: a controversial lesion. Kidney Int 29, 725–733.
Ho, M. (1991). Cytomegalovirus. In G. L. Mandell, J. E. Bennet, & R. Dolin
(Eds.),Principles and Practices of InfectiousDiseases ( pp. 1351–1364).
New York: Churchill Livingstone.
Ho, H.-T., Woods, K. L., Bronson, J. J., De Boeck, H., Martin, J. C., &
Hitchcock, M. J. (1992). Intracellular metabolism of the antiherpes
agent (S)-1-(3-hydroxy-2-(phosphonylmethoxy)propyl)-cytosine. Mol
Pharmacol 41, 97–202.
Hobom, U., Brune, W., Messerle, M., Hahn, G., & Koszinowski, U. H.
(2000). Fast screening procedures for random transposon libraries of
cloned herpesvirus genomes: mutational analysis of human cytomega-
lovirus envelope glycoproteins genes. J Virol 74, 7720–7729.
Huber, M. T., & Compton, T. (1998). The human cytomegalovirus UL74
gene encodes the third component of the glycoprotein H-glycoprotein
L-containing envelope complex. J Virol 72, 8191–8197.
Isom, H. C. (1979). Stimulation of ornithine decarboxylase by human
cytomegalovirus. J Gen Virol 42, 265–278.
Jabs, D. A., Enger, C., Dunn, J. P., & Forman, M., for the Cytomegalovirus
Retinitis and Viral Resistance Study Group (1998a). Cytomegalovirus

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 293
retinitis and viral resistance: 4. Ganciclovir resistance. J Infect Dis 177,
770–773.
Jabs, D. A., Enger, C., Forman, M., & Dunn, J. P., for the Cytomegalovirus
Retinitis and Viral Resistance Study Group (1998b). Incidence of
foscarnet resistance and cidofovir resistance in in patients treated
for cytomegalovirus retinitis. Antimicrob Agents Chemother 42,
2240–2244.
Jacobson, M. A. (1997–1998). Cytomegalovirus retinitis: new develop-
ments in prophylaxis and therapy. AIDS Clin Rev, 249–269.
Jassal, S. V., Roscoe, J. M., Zaltzman, J. S., Mazzulli, T., Krajden, M.,
Gadawski, M., Cattran, D. C., Cardella, C. J., Albert, S. E., & Cole,
E. H. (1998). Clinical practice guidelines: prevention of cytomegalovi-
rus disease after renal transplantation. J Am Soc Nephrol 9, 1697–1708.
Jault, F. M., Jault, J. M., Ruchti, F., Fortunato, E. A., Clark, C., Corbeil, J.,
Richman, D. D., & Spector, D. H. (1995). Cytomegalovirus infection
induces high levels of cyclins, phosphorylated Rb, and p53, leading to
cell cycle arrest. J Virol 69, 6697–6704.
Johnson, R. A., Yurochko, A. D., Poma, E. E., Zhu, L., & Huang, E.-S.
(1999). Domain mapping of the human cytomegalovirus IE1-72 and
cellular p107 protein-protein interaction and the possible functional
consequences. J Gen Virol 80, 1293–1303.
Jones, T. R., & Sun, L. (1997). Human cytomegalovirus US2 destabilizes
major histocompatibility complex class I heavy chains. J Virol 71,
2970–2979.
Jones, T. R., Wiertz, E. J., Sun, L., Fish, K. N., Nelson, J. A., &
Ploegh, H. L. (1996). Human cytomegalovirus US3 impairs transport
and maturation of major histocompatibility complex class I heavy
chains. Proc Natl Acad Sci USA 93, 11327–11333.
Jonjic, S., Pavic, I., Lucin, P., Rukavina, D., & Koszinowski, U. H. (1990).
Efficacious control of cytomegalovirus infection after long-term deple-
tion of CD8+ lymphocytes. J Virol 64, 5457–5464.
Jupp, R., Hoffmann, S., Stenberg, R. M., Nelson, J. A., & Ghazal, P.
(1993). Human cytomegalovirus IE86 protein interacts with promoter-
bound TATA-binding protein via a specific region distinct from the
autorepression domain. J Virol 67, 7539–7546.
Kahl, M., Siegel-Axel, D., Stenglein, S., Jahn, G., & Sinzger, C. (2000).
Efficient lytic infection of human arterial endothelial cells by human
cytomegalovirus strains. J Virol 74, 7628–7635.
Kalejta, R. F., & Shenk, T. (2002). Manipulation of the cell cycle by human
cytomegalovirus. Front Biosc 7, 295–306.
Kari, B., Li, W., Cooper, J., Goertz, R., & Radeke, B. (1994). The human
cytomegalovirus UL100 gene encodes the gC-II glycoproteins recog-
nized by group 2 monoclonal antibodies. J Gen Virol 75, 3081–3086.
Karlin, S., Mocarski, E. S., & Schachtel, G. A. (1994). Molecular evolution
of herpesviruses: genomic and protein sequence comparisons. J Virol
68, 1886–1902.
Kern, F., Surel, I. P., Faulhaber, N., Frommel, C., Schneider-Mergener, J.,
Schonemann, C., Reinke, P., & Volk, H. D. (1999). Target structures of
the CD8 + -T-cell response to human cytomegalovirus: the 72-kilodalton
major immediate-early protein revisited. J Virol 73, 8179–8184.
Kidd, I. M., Fox, J. C., Pillay, D., Charman, H., Griffiths, P. D., & Emery,
V. C. (1993). Provision of prognostic information in immunocompro-
mised patients by routine application of the polymerase chain reaction
for cytomegalovirus. Transplantation 56, 867–871.
Knox, K. K., Drobyski, W. R., & Carrigan, D. R. (1991). Cytomegalovirus
isolate resistant to ganciclovir and foscarnet from a marrow transplant
patient. Lancet ii, 1292–1293.
Kondo, K., Kaneshima, H., & Mocarski, E. S. (1994). Human cytomega-
lovirus latent infection of granulocyte-macrophage progenitors. Proc
Natl Acad Sci USA 91, 11879–11883.
Koszinowski, U. H., Del Val, M., & Reddehase, M. J. (1990). Cellular and
molecular basis of the protective immune response to cytomegalovirus
infection. Curr Top Microbiol Immunol 154, 189–215.
Lalezari, J. P., Holland, G. N., Kramer, F., McKinley, G. F., Kemper, C. A.,
Ives, D. V., Nelson, R., Hardy, W. D., Kuppermann, B. D., Northfelt,
D. W., Youle, M., Johnson, M., Lewis, R. A., Weinberg, D. V., Simon,
G. L., Wolitz, R. A., Ruby, A. E., Stagg, R. J., & Jaffe, H. S. (1998).
Randomized, controlled study of the safety and efficacy of intravenous
cidofovir for the treatment of relapsing cytomegalovirus retinitis in
patients with AIDS. J Acquir Immune Def Syndr Hum Retrovirol 17,
339–344.
Lam, K. M., Oldenburg, N., Khan, M. A., Gaylore, V., Mikhail, G. W.,
Strouhal, P. D., Middeldorp, J. M., Banner, N., & Yacoub, M. (1998).
Significance of reverse transcription polymerase chain reaction in the
detection of human cytomegalovirus gene transcripts in thoracic organ
transplant recipients. J Heart Lung Transplant 17, 555–565.
Landini, M. P., & Michelson, S. (1988). Human cytomegalovirus proteins.
In J. L. Melnick (Ed.), Progress in Medical Virology ( pp. 152–185).
Basel: Karger.
Landini, M. P., Rossier, E., & Schmitz, H. (1988). Antibodies to human
cytomegalovirus structural polypeptides during primary infection. J Vi-
rol Methods 22, 309–317.
Lazzarotto, T., Spezzacatena, P., Varani, S., Gabrielli, L., Pradelli, P., Guer-
ra, B., & Landini, M. P. (1999). Anticytomegalovirus (anti-CMV) im-
munoglobulin G avidity in identification of pregnant women at risk of
transmitting congenital CMV infection. Clin Diagn Lab Immunol 6,
127–129.
Leach, C. T., Cherry, J. D., English, P. A., Hennessey, K., Giorgi, J. V.,
Visscher, B. R., Dudley, J. P., & Detels, R. (1993). The relationship
between T-cells and CMV infection in asymptomatic HIV-1 antibody-
positive homosexual men. J Acquir Immune Def Syndr Hum Retrovirol
6, 407–413.
Lembo, D., Gribaudo, G., Cavallo, R., Riera, L., Angeretti, A., Hertel, L., &
Landolfo, S. (1999). Human cytomegalovirus stimulates cellular dihy-
drofolate reductase activity in quiescent cells. Intervirology 42, 30–36.
Lembo, D., Gribaudo, G., Hofer, A., Riera, L., Cornaglia, M., Mondo, A.,
Angeretti, A., Gariglio, M., Thelander, L., & Landolfo, S. (2000). Ex-
pression of an altered ribonucleotide reductase activity associated with
the replication of murine cytomegalovirus in quiescent fibroblasts.
J Virol 74, 11557–11565.
Limaye, A. P., Corey, L., Koelle, D. M., Davis, C. L., & Boeckh, M.
(2000). Emergence of ganciclovir-resistant cytomegalovirus disease
among recipients of solid-organ transplants. Lancet 356, 645–649.
Limaye, A. P., Raghu, G., Koelle, D. M., Ferrenberg, J., Huang, M. L., &
Boeckh, M. (2002). High incidence of ganciclovir-resistant cytomega-
lovirus infection among lung transplant recipients receiving preemptive
therapy. J Infect Dis 185, 20–27.
Littler, E., Stuart, A. D., & Chee, M. S. (1992). Human cytomegalovirus
UL97 open reading frame encodes a protein that phosphorylates the
antiviral nucleoside analogue ganciclovir. Nature 358, 160–162.
Liu, B., & Stinski, M. F. (1992). Human cytomegalovirus contains a tegu-
ment protein that enhances transcription from promoters with upstream
ATF and AP-1 cis-acting elements. J Virol 66, 4434–4444.
Liu, Y. N., Curtsinger, J., Donahue, P. R., Klaus, A., Optiz, G., Cooper, J.,
Karr, R. W., Bachs, F. H., & Gehrz, R. C. (1993). Molecular analysis of
the immune response to human cytomegalovirus glycoprotein B. I.
Mapping of HLA-restricted helper T cell epitopes on gp93. J Gen Virol
74, 2207–2214.
Loebe, M., Schuler, S., Zais, O., Warnecke, H., Fleck, E., & Hetzer, R.
(1990). Role of cytomegalovirus infection in the development of coro-
nary disease in the transplanted hearth. J Heart Transplant 9, 707–711.
Lu, M., & Shenk, T. (1996). Human cytomegalovirus infection inhibits cell
cycle progression at multiple points, including the transition from G1 to
S. J Virol 70, 8850–8857.
Lu, M., & Shenk, T. (1999). Human cytomegalovirus UL69 protein induces
cells to accumulate in G1 phase of the cell cycle. J Virol 73, 676–683.
Lukac, D. M., Harel, N. Y., Tanese, N., & Alwine, J. C. (1997). TAF-like
functions of human cytomegalovirus immediate-early proteins. J Virol
71, 7227–7239.
Lurain, N. S., Spafford, L. E., & Thompson, K. D. (1994). Mutation in the
UL97 open reading frame of human cytomegalovirus strains resistant to
ganciclovir. J Virol 68, 4427–4431.
Macdonald, P. S., Keogh, A. M., Marshman, D., Richens, D., Harvison, A.,
Kaan, A. M., & Spratt, P. M. (1995). A double-blind placebo-controlled

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297294
trial of low-dose ganciclovir to prevent cytomegalovirus disease after
heart transplantation. J Heart Lung Transplant 14, 32–38.
Machold, R. P., Wiertz, F. J., Jones, T. R., & Ploegh, H. L. (1997). The
HCMV gene products US11 and US2 differ in their ability to attack
allelic forms of murine major histocompatibility complex (MHC) class I
heavy chains. J Exp Med 185, 363–366.
Marchini, A., Liu, H., & Zhu, H. (2001). Human cytomegalovirus with
IE2 (UL122) deleted fails to express early lytic genes. J Virol 75,
1870–1878.
Margolis, M. J., Pajovic, S., Wong, E. L., Wade, M., Jupp, R., Nelson, J. A.,
& Clifford Azizkhan, J. (1995). Interaction of the 72-kilodalton human
cytomegalovirus IE1 gene product with E2F1 coincides with E2F-de-
pendent activation of dihydrofolate reductase transcription. J Virol 69,
7759–7767.
Marshall, G. S., Rabalais, G. P., Stout, G. G., & Waldeyer, S. L. (1992).
Antibodies to recombinant-derived glycoprotein B after natural human
cytomegalovirus infection correlate with neutralizing activity. J Infect
Dis 165, 381–384.
Masse, M. J., Karlin, S., Schachtel, G. A., & Mocarski, E. S. (1992).
Human cytomegalovirus origin of DNA replication (oriLyt) resides
within a highly complex repetitive region. Proc Natl Acad Sci USA
89, 5246–5250.
Masur, H., Whitcup, S. M., Cartwright, C., Polis, M., & Nussenblatt, R.
(1996). Advances in the management of AIDS-related cytomegalovirus
retinitis. Ann Intern Med 125, 126–136.
McVoy, M. A., & Adler, S. P. (1994). Human cytomegalovirus DNA rep-
licates after early circularization by concatemer formation, and inver-
sion occurs within concatemer. J Virol 68, 1040–1051.
Meier, J. L., & Stinski, M. F. (1996). Regulation of human cytomegalovirus
immediate-early gene expression. Intervirology 39, 331–342.
Meier, J. L., & Stinski, M. F. (1997). Effect of a modulator deletion on
transcription of the human cytomegalovirus major immediate-early
genes in infected undifferentiated and differentiated cells. J Virol 71,
1246–1255.
Merigan, T. C., Renlund, D. G., Keay, S., Bristow, M. R., Starnes, V.,
O’Connell, J. B., Resta, S., Dunn, D., Gamberg, P., & Ratkovec, R. M.
(1992). A controlled trial of ganciclovir to prevent cytomegalovirus
disease after heart transplantation. N Engl J Med 326, 1182–1186.
Mettenleiter, T. C. (2002). Herpesvirus assembly and egress. J Virol 76,
1537–1547.
Miller, W. J., McCullough, J., Balfour, H. H., Haake, R. J., Ramsay, N. K.,
Goldman, A., Bowman, R., & Kersey, J. (1991). Prevention of cytome-
galovirus infection following bonemarrow transplantation: a randomized
trial of blood product screening. Bone Marrow Transplant 7, 227–234.
Mocarski, E. S. (1996). Cytomegalovirus and their replication. In
B. N. Fields, D. M. Knipe, & P. M. Howley (Eds.), Virology
( pp. 2447–2492). Philadelphia: Lippincott-Raven.
Mocarski, E. S. (2002). Immunomodulation by cytomegaloviruses: manip-
ulative strategies beyond evasion. Trends Microbiol 10, 332–339.
Mocarski, E. S., & Courcelle, C. T. (2001). Cytomegalovirus and their
replication. In D. Knipe, & P. Howley (Eds.), Fields Virology ( pp.
2629–2673). Philadelphia: Lippincott, Williams and Wilkins.
Mocarski, E. S., Kemble, G. W., Lyle, J. M., & Greaves, R. F. (1996). A
deletion mutant in the human cytomegalovirus gene encoding IE1 (491
aa) is replication defective due to a failure in autoregulation. Proc Natl
Acad Sci USA 94, 11321–11326.
Murphy, E. A., Streblow, D. N., Nelson, J. A., & Stinski, M. F. (2000). The
human cytomegalovirus IE86 protein can block cell cycle progression
after inducing transition into the S phase of permissive cells. J Virol 74,
7108–7118.
Nelson, J. A., Gnann, J. W., & Ghazal, P. (1990). Regulation and tissue-
specific expression of human cytomegalovirus. Curr Top Microbiol
Immunol 154, 75–100.
Nesmith, J. D., & Pass, R. F. (1995). Cytomegalovirus infection in adoles-
cent. In G. D. Overturf, & R. F. Jacobs (Eds.), Adolescent Medicine:
State of the Art Reviews. Viral Infections in Adolescents ( pp. 79–90).
Philadelphia: Hanley and Belfus.
Novotny, J., Rigoutsos, I., Coleman, D., & Shenk, T. (2001). In silico
structural and functional analysis of the human cytomegalovirus
(HHV5) genome. J Mol Biol 310, 1151–1166.
O’Sullivan, C. E., Drew, W. L., McMullen, D. J., Miner, R., Lee, J. Y.,
Kaslow, R. A., Lazar, J. G., & Saag, M. S. (1999). Decrease of cyto-
megalovirus replication in human immunodeficiency virus-infected pa-
tients after treatment with highly active antiretroviral therapy. J Infect
Dis 180, 847–849.
Pajovic, S., Wong, E. L., Black, A. R., & Azizkhan, C. J. (1997). Identi-
fication of a viral kinase that phosphorylates specific E2Fs and pocket
proteins. Mol Cell Biol 17, 6459–6464.
Palestine, A. G., Polis, M. A., De Smet, M. D., Baird, B. F., Falloon, J.,
Kovacs, J. A., Davey, R. T., Zurlo, J. J., Zunich, K. M., Davis, M., et al.
(1991). A randomized, controlled trial of foscarnet in the treatment of
cytomegalovirus retinitis in patients with AIDS. Ann Intern Med 115,
665–673.
Pass, R. F. (1985). Epidemiology and transmission of cytomegalovirus
infection. J Infect Dis 152, 243–256.
Pass, R. F. (1996). Immunization strategy for prevention of congenital
cytomegalovirus infection. Infect Agents Dis 5, 240–244.
Pass, R. F. (2001). Cytomegalovirus. In D. Knipe, & P. Howley (Eds.),
Fields Virology ( pp. 2675–2705). Philadelphia: Lippincott, Williams
and Wilkins.
Pass, R. F., & Boppana, S. (1999). Cytomegalovirus. In D. J. Jeffries, &
C. N. Hudson (Eds.), Viral Infections in Obstetrics and Gynaecology
( pp. 35–56). New York: Arnold.
Pass, R. F., Stagno, S., & Britt, W. J. (1983). Specific cell-mediated im-
munity and the natural history of congenital infection with cytomega-
lovirus. J Infect Dis 148, 953–961.
Pass, R. F., Duliege, A. M., Boppana, S., Sekulovich, R., Percell, S., Britt,
W., & Burke, R. L. (1999). A subunit cytomegalovirus vaccine based on
recombinant envelope glycoprotein B and a new adjuvant. J Infect Dis
180, 970–975.
Penfold, M. E., & Mocarski, E. S. (1997). Formation of cytomegalovirus
DNA replication compartments defined by localization of viral proteins
and DNA synthesis. Virology 239, 46–61.
Penfold, M. E., Dairaghi, D. J., Duke, G. M., Saedurup, N., Mocarski,
E. S., Kemble, G. W., & Schall, T. J. (1999). Cytomegalovirus
encodes a potent alpha chemokine. Proc Natl Acad Sci USA 96,
9839–9844.
Pepperl, S., Munster, J., Mach, M., Harris, J. R., & Plachter, B. (2000).
Dense bodies of human cytomegalovirus induce both humoral and cel-
lular immune responses in the absence of viral gene expression. J Virol
74, 6132–6146.
Pertel, P., Hirschtick, R., Phair, J., Chmiel, J., Poggensee, L., & Murphy, R.
(1992). Risk of developing cytomegalovirus retinitis in persons infected
with the human immunodeficiency virus. J Acquir Immune Def Syndr
Hum Retrovirol 5, 1069–1074.
Peterson, P. K., Balfour Jr., H. H., Marker, S. C., Fryd, D. S., Howard, R. J.,
& Simmons, R. L. (1980). Cytomegalovirus disease in renal allograft
recipients: a prospective study of the clinical features, risk factors and
impact on renal transplantation. Medicine 59, 283–300.
Phillips, C. A., Fanning, W. L., & Gump, D. W. (1977). Cytomegalovirus in
immunologically normal adults. JAMA 238, 2299–2300.
Pizzorno, M. C., & Hayward, G. S. (1990). The IE2 gene products of
human cytomegalovirus specifically down-regulate expression from
the major immediate-early promoter through a target sequence located
near the cap site. J Virol 64, 6154–6165.
Plachter, B., Sinzger, C., & Jahn, G. (1996). Cell types involved in repli-
cation and distribution of human cytomegalovirus. Adv Virus Res 46,
195–261.
Plotkin, S. A., Furukawa, T., Zygraich, N., & Huygelen, C. (1975). Can-
didate cytomegalovirus strain for human vaccination. Infect Immun 12,
521–527.
Plotkin, S. A., Farquhar, J., & Horberger, E. (1976). Clinical trials of
immunization with the Towne 125 strain of human cytomegalovirus.
J Infect Dis 134, 470–475.

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 295
Plotkin, S. A., Smiley, M. L., Friedman, H. M., Starr, S. E., Fleisher, G. R.,
Wlodaver, C., Dafoe, D. C., Friedman, A. D., Grossman, R. A., &
Barker, C. F. (1984). Towne-vaccine induced prevention of cytomega-
lovirus disease after renal transplants. Lancet i, 528–530.
Plotkin, S. A., Starr, S. E., Friedman, H. M., Gonczol, E., & Weibel, R. E.
(1989). Protective effects of Towne cytomegalovirus vaccine against
low-passage cytomegalovirus administered as a challenge. J Infect
Dis 159, 860–865.
Plotkin, S. A., Starr, S. E., Friedman, H. M., Brayman, K., Harris, S.,
Jackson, S., Tustin, N. B., Grossman, R., Dafoe, D., & Barker, C.
(1991). Effect of Towne live virus vaccine on cytomegalovirus dis-
ease after renal transplant. A controlled trial. Ann Intern Med 114,
525–531.
Plotkin, S. A., Higgins, R., Kurtz, J. B., Morris, P. J., Campbell, D. A., Jr.,
Shope, T. C., Spector, S. A., & Dankner, W. M. (1994). Multicenter trial
of Towne strain attenuated virus vaccine in seronegative renal transplant
recipients. Transplantation 58, 1176–1178.
Polic, B., Jonjic, S., Pavic, I., Crnkovic, I., Zorica, I., Hengel, H., Lucin, P.,
& Koszinowski, U. H. (1996). Lack of MHC class I complex expression
has no effect on spread and control of cytomegalovirus infection in
vivo. J Gen Virol 77, 217–225.
Polis, M. A., Spooner, K. M., Baird, B. F., Manischewitz, J. F., Jaffe, H. S.,
Fisher, P. E., Falloon, J., Davey, R. T. J., Kovacs, J. A., Walker, R. E.,
Whitcup, S. M., Nussenblatt, R. B., Lane, H. C., & Masur, H. (1995).
Anticytomegaloviral activity and safety of cidofovir in patients with
human immunodeficiency virus infection and cytomegalovirus viruria.
Antimicrob Agents Chemother 39, 882–886.
Pollard, A. B. (1988). Cytomegalovirus infections in renal, heart, heart-lung
and liver transplantation. Pediatr Infect Dis J 7, 97–102.
Poma, E. E., Kowalik, T. F., Zhu, L., Sinclair, J. H., & Huang, E.-S. (1996).
The human cytomegalovirus IE1-72 protein interacts with the cellular
p107 protein and relieves p107-mediated transcriptional repression of
an E2F-responsive promoter. J Virol 70, 7867–7877.
Rapp, M., Messerle, M., Buhler, B., Tannheimer, M., Keil, G. M., &
Koszinowski, U. R. (1992). Identification of the murine cytomegalo-
virus glycoprotein B gene and its expression by recombinant vaccinia
virus. J Virol 66, 4399–4406.
Rawlinson, W. D., Farrell, H. E., & Barrell, B. G. (1996). Analysis of the
complete DNA sequence of murine cytomegalovirus. J Virol 70,
8833–8849.
Reddehase, M. J., Balthesen, M., Rapp, M., Jonjic, S., Pavic, I., & Koszi-
nowski, U. H. (1994). The conditions of primary infection define the
load of latent viral genome in organs and the risk of recurrent cytome-
galovirus disease. J Exp Med 179, 185–193.
Reed, E. C., Bowden, R. A., Dandliker, P. S., Lilleby, K. E., & Meyers,
J. D. (1988). Treatment of cytomegalovirus pneumonia with ganciclovir
and intravenous cytomegalovirus immunoglobulin in patients with bone
marrow transplants. Ann Intern Med 109, 783–788.
Rene, E., Marche, C., Chevalier, T., Rouzioux, C., Regnier, B., Saimot,
A. G., Negesse, Y., Matheron, S., Leport, C., & Wolff, B. (1988). Cy-
tomegalovirus colitis in patients with acquired immunodeficiency syn-
drome. Dig Dis Sci 33, 741–750.
Revello, M. G., Percivalle, E., Zavattoni, M., Parea, M., Grossi, P., &
Gerna, G. (1989). Detection of human cytomegalovirus immediate early
antigen in leukocytes as a marker of viremia in immunocompromised
patients. J Med Virol 29, 88–93.
Revello, M. G., Baldanti, F., Furione, M., Sarasini, A., Percivalle, E.,
Zavattoni, M., & Gerna, G. (1995). Polymerase chain reaction for pre-
natal diagnosis of congenital human cytomegalovirus infection. J Med
Virol 47, 462–466.
Revello, M. G., Zavattoni, M., Furione, M., Baldanti, F., & Gerna, G.
(1999). Quantification of human cytomegalovirus DNA in amniotic
fluid of mothers of congenitally infected fetuses. J Clin Mirobiol 37,
3350–3352.
Revello, M. G., Lilleri, D., Zavattoni, M., Stronati, M., Bollani, L., Mid-
deldorp, J. M., & Gerna, G. (2001). Human cytomegalovirus immedi-
ate-early messenger RNA in blood of pregnant women with primary
infection and of congenitally infected newborns. J Infect Dis 184,
1078–1081.
Reynolds, D. W., Stagno, S., Hosty, T. S., Tiller, M., & Alford, C. A., Jr.
(1973). Maternal cytomegalovirus excretion and perinatal infection. N
Engl J Med 289, 1–5.
Richardson, W. P., Colvin, R. B., Cheeseman, S. H., Tolkoff-Rubin, N. E.,
Herrin, J. T., Cosimi, A. B., Collins, A. B., Hirsch, M. S., McCluskey,
R. T., Russell, P. S., & Rubin, R. H. (1981). Glomerulopathy associated
with cytomegalovirus viremia in renal allografts. N Engl J Med 305,
57–63.
Riddell, S. R., Rabin, M., Geballe, A. P., Britt, W. J., & Greenberg, P. D.
(1991). Class I MHC-restricted cytotoxic T lymphocyte recognition of
cells infected with human cytomegalovirus does not require endogenous
viral gene expression. J Immunol 146, 2795–2804.
Riera, L., Gariglio, M., Valente, G., Mullbacher, A., Museteanu, C., Land-
olfo, S., & Simon, M. M. (2000). Murine cytomegalovirus replication in
salivary glands is controlled by both perforin and granzymes during
acute infection. Eur J Immunol 30, 1350–1355.
Riera, L., Gariglio, M., Pagano, M., Gaiola, O., Simon, M. M., & Landolfo,
S. (2001). Control of murine cytomegalovirus replication in salivary
glands during acute infection is independent of the Fas ligand/Fas sys-
tem. Microbiologica 24, 231–238.
Romanowski, M. J., Garrido-Guerrero, E., & Shenk, T. (1997). pIRS1 and
pTRS1 are present in human cytomegalovirus virions. J Virol 71,
5703–5705.
Rubie, H., Attal, M., Campardou, A. M., Gayet-Mengelle, C., Payen, C.,
Sanguignol, F., Calot, J. P., Charlet, J. P., Robert, A., & Huguet, F.
(1993). Risk factors for cytomegalovirus infection in BMT recipients
transfused exclusively with seronegative blood products. Bone Marrow
Transplant 11, 209–214.
Rubin, R. H. (1991). Preemptive therapy in immunocompromised hosts.
N Engl J Med 324, 1057–1059.
Rubin, R. H. (1994). Infection in the organ transplant recipient. In R. H.
Rubin, & L. S. Young (Eds.), Clinical Approach to Infection in the
Compromised Host (pp. 629–705). New York: Plenum Medical Book
Co.
Rubin, R. H., Tolkoff-Rubin, N. E., Oliver, D., Rota, T. R., Hamilton, J.,
Betts, R. F., Pass, R. F., Hillis, W., Szmuness,W., & Farrell, M. L. (1985).
Multicenter seroepidemiologic study of the impact of cytomegalovirus
infection on renal transplantation. Transplantation 40, 243–249.
Ruutu, P., Ruutu, T., Volin, L., Tukiainen, P., Ukkonen, P., & Hovi, T.
(1990). Cytomegalovirus is frequently isolated in bronchoalveolar lav-
age fluid of bone marrow transplant recipients without pneumonia. Ann
Intern Med 112, 913–916.
Sadasivan, B., Lehner, P. J., Ortmann, B., Spies, T., & Cresswell, P. (1996).
Role for clareticulin and a novel glycoprotein, tapasin, in the interaction
of MHC class I molecules with TAP. Immunity 5, 103–114.
Saederup, N., Lin, Y. C., Dairaghi, D. J., Schall, T. J., & Mocarski, E.
S. (1999). Cytomegalovirus-encoded b chemokine promotes mono-
cyte-associated viremia in the host. Proc Natl Acad Sci USA 96,
10881–10886.
Salvant, B. S., Fortunato, E. A., & Spector, D. H. (1998). Cell cycle
dysregulation by human cytomegalovirus: influence of the cell cycle
phase at the time of infection and effects on cyclin transcription. J Virol
72, 3729–3741.
Sanchez, V., Sztul, E., & Britt, W. (2000). Accumulation of virion tegument
and envelope proteins in a stable cytoplasmatic compartment during
human cytomegalovirus replication: characterization of a potential site
of virus assembly. J Virol 74, 975–986.
Sarisky, R. T., & Hayward, G. S. (1996). Evidence that the UL84 gene
product of human cytomegalovirus is essential for promoting oriLyt-
dependent DNA replication and formation of replication compartments
in cotransfection assays. J Virol 70, 7398–7413.
Sayers, M. H., Anderson, K. C., Goodnough, L. T., Kurtz, S. R., Lane,
T. A., Pisciotto, P., & Silberstein, L. E. (1992). Reducing the risk for
transfusion-transmitted cytomegalovirus infection. Ann Intern Med 116,
55–62.

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297296
Shamu, C. E., Story, C. M., Rapoport, T. A., & Ploegh, H. L. (1999). The
pathway of US11-dependent degradation of MHC class I heavy chains
involves a ubiquitin-conjugated intermediate. J Cell Biol 147, 45–58.
Shellam, G. R., Allen, J. E., Papdimitriou, J. M., & Bancroft, J. M. (1981).
Increased susceptibility to cytomegalovirus infection in beige mutant
mice. Proc Natl Acad Sci USA 78, 5104–5108.
Simmen, K. A., Singh, J., Luukkonen, B. G. M., Lopper, M., Bittner, A.,
Miller, N. E., Jackson, M. R., Compton, T., & Fruh, K. (2001). Global
modulation of cellular transcription by human cytomegalovirus is ini-
tiated by viral glycoprotein B. Proc Natl Acad Sci USA 98, 7140–7145.
Singh, N., Yu, V. L., Mieles, L., Wagener, M. M., Miner, R. C., & Gayowski,
T. (1994). High-dose acyclovir compared with short-course preemptive
ganciclovir therapy to prevent cytomegalovirus disease in liver transplant
recipients. A randomized trial. Ann Intern Med 120, 375–381.
Sinzger, C., & Jahn, G. (1996). Human cytomegalovirus cell tropism and
pathogenesis. Intervirology 39, 302–319.
Sinzger, C., Grefte, A., Plachter, B., Gouw, A. S., The, T. H., & Jahn, G.
(1995). Fibroblasts, epithelial cells, endothelial cells and smooth muscle
cells are major targets of human cytomegalovirus infection in lung and
gastrointestinal tissues. J Gen Virol 76, 741–750.
Sinzger, C., Plachter, B., Grefte, A., The, T. H., & Jahn, G. (1996). Tissue
macrophages are infected by human cytomegalovirus. J Infect Dis 173,
240–245.
Sinzger, C., Kahl, M., Laib, K., Klingel, K., Rieger, P., Plachter, B., & Jahn,
G. (2000). Tropism of human cytomegalovirus for endothelial cells is
determined by a post-entry step dependent on efficient translocation to
the nucleus. J Gen Virol 81, 3021–3035.
Sison, R. F., Holland, G. N., MacArthur, L. J., Wheeler, N. C., &
Gottlieb, M. S. (1991). Cytomegalovirus retinopathy as the initial
manifestation of the acquired immunodeficiency syndrome. Am J
Ophthalmol 112, 243–249.
Skaletskaya, A., Bartle, L. M., Chittenden, T., McCormick, A. L., Mocar-
ski, E. S., & Goldmacher, V. S. (2001). A cytomegalovirus-encoded
inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl
Acad Sci USA 98, 7829–7834.
Smith, I. L., Cherrington, J. M., Jiles, R. E., Fuller, M. D., Freeman, W. R.,
& Spector, S. A. (1997). High-level resistance of cytomegalovirus to
ganciclovir is associated with alterations in both the UL97 and DNA
polymerase genes. J Infect Dis 176, 69–77.
Snydman, D. R., Werner, B. G., Heinze-Lacey, B., Berardi, V. P., Tilney,
N. L., Kirkman, R. L., Milford, E. L., Cho, S. I., Bush Jr., H. L., &
Levey, A. S. (1987). Use of cytomegalovirus immune globulin to pre-
vent cytomegalovirus disease in renal transplant recipients. N Engl J
Med 317, 1049–1054.
Soderberg-Naucler, C., Fish, K. N., & Nelson, J. A. (1997). Reactivation of
latent human cytomegalovirus by allogeneic stimulation of blood cells
from healthy donors. Cell 91, 119–126.
Soderberg-Naucler, C., Streblow, D. N., Fish, K. N., Allan-Yorke, J., Smith,
P. P., & Nelson, J. A. (2001). Reactivation of latent human cytomega-
lovirus in CD14 + monocytes is differentiation dependent. J Virol 75,
7543–7554.
Sommer, M. H., Scully, A. L., & Spector, D. H. (1994). Transactivation by
the human cytomegalovirus IE2 86-kilodalton protein requires a domain
that binds to both the TATA box-binding protein and the retinoblastoma
protein. J Virol 68, 6223–6231.
Song, Y., & Stinski, M. F. (2002). Effect of the human cytomegalovirus
IE86 protein on expression of E2F-responsive genes: a DNA microarray
analysis. Proc Natl Acad Sci USA 99, 2836–2841.
Spector, S. A., Weingeist, T., Pollard, R. B., Dieterich, D. T., Samo, T.,
Benson, C. A., Busch, D. F., Freeman, W. R., Montague, P., Kaplan,
H. J., Keller, L., Crager, M., De Armond, B., Buhles, W., & Feinberg, J.
(1993). A randomized, controlled study of intravenous ganciclovir ther-
apy for cytomegalovirus peripheral retinitis in patients with AIDS.
AIDS Clinical Trials Group and Cytomegalovirus Cooperative Study
Group. J Infect Dis 168, 557–563.
Spector, S. A., McKinley, G. F., Lalezari, J. P., Samo, T., Andruczk, R.,
Follansbee, S., Sparti, P. D., Havlir, D. V., Simpson, G., Buhles, W.,
Wong, R., & Stempien, M. (1996). Oral ganciclovir for the prevention
of cytomegalovirus disease in persons with AIDS. Roche Cooperative
Oral Ganciclovir Study Group. N Engl J Med 334, 1491–1497.
Speir, E., Modali, R., Huang, E.-S., Leon, M. B., Shawl, F., Finkel, T., &
Epstein, S. E. (1993). Potential role of human cytomegalovirus and p53
interaction in coronary restenosis. Science 265, 391–394.
Stagno, S., Reynolds, D. W., Lakeman, A., Charamella, L. J., & Alford,
C. A. (1973). Congenital cytomegalovirus infection: consecutive occur-
rence due to viruses with similar antigenic compositions. Pediatrics 52,
788–794.
Stagno, S., Reynolds, D. W., Huang, E. S., Thames, S. D., Smith, R. J., &
Alford, C. A. (1977). Congenital cytomegalovirus infection: occurrence
in an immune population. N Engl J Med 296, 1254–1258.
Stagno, S., Pass, R. F., Dworsky, M. E., & Alford, C. A., Jr. (1982).
Maternal cytomegalovirus infection and perinatal transmission. Clin
Obstet Gynecol 25, 563–576.
Stagno, S., Pass, R. F., Cloud, G., Britt, W. J., Henderson, R. E., Walton,
P. D., Veren, D. A., Page, F., & Alford, C. A. (1986). Primary cytome-
galovirus infection in pregnancy. Incidence, transmission to fetus, and
clinical outcome. JAMA 256, 1904–1908.
Stasiak, P. C., & Mocarski, E. S. (1992). Transactivation of the human
cytomegalovirus ICP36 gene promoter requires the a gene product
TRS1 in addition to the IE1 and IE2. J Virol 66, 1050–1058.
Story, C. M., Furman, M. H., & Ploegh, H. L. (1999). The cytosolic tail of
class I MHC heavy chain is required for its dislocation by the human
cytomegalovirus US2 and US11 gene products. Proc Natl Acad Sci
USA 96, 8516–8521.
Stratta, R. J., Wood, R. P., Langnas, A. N., Duckworth, R. M., Shaefer,
M. S., Marujo, W., Pillen, T. J., Markin, R. S., & Shaw, B. W., Jr.
(1990). Donor selection for orthotopic liver transplantation: lack of an
effect of gender or cytomegalovirus (CMV) status. Transplant Proc 22,
410–413.
Streblow, D. N., Orloff, S. L., & Nelson, J. A. (2001). Do pathogens
accelerate atherosclerosis? J Nutr 13, 2798S–2804S.
Theiler, R. N., & Compton, T. (2001). Characterization of the signal peptide
processing and membrane association of human cytomegalovirus gly-
coprotein O. J Biol Chem 276, 39226–39231.
The Vitravene Study Group (2002a). A randomized controlled clinical trial
of intravitreous fomivirsen for treatment of newly diagnosed peripheral
cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 133,
467–474.
The Vitravene Study Group (2002b). Randomized dose-comparison studies
of intravitreous fomivirsen for treatment of cytomegalovirus retinitis
that has reactivated or is persistently active despite other therapies in
patients with AIDS. Am J Ophthalmol 133, 475–483.
Tomasec, P., Braud, V. M., Rickards, C., Powell, M. B., McSharry, B. P.,
Gadola, S., Cerundolo, V., Borysiewicz, L. K., McMichael, A. J., &
Wilkinson, G. W. (2000). Surface expression of HLA E an inhibitor of
natural killer cells, enhanced by human cytomegalovirus gpUL40. Sci-
ence 287, 1031–1033.
van den Berg, A. P., van der Bij, W., van Son, W. J., Anema, J., van der
Giessen, M., Schirm, J., Tegzess, A. M., & The, T. H. (1989). Cytome-
galovirus antigenemia as a useful marker of symptomatic cytomegalo-
virus infection after renal transplantation—a report of 130 consecutive
patients. Transplantation 48, 991–995.
van der Bij, W., Schirm, J., Torensma, R., van Son, W. J., Tegzess, A. M.,
& The, T. H. (1988a). Comparison between viremia and antigenemia
for detection of cytomegalovirus in blood. J Clin Microbiol 26,
2531–2535.
van der Bij, W., Torensma, R., van Son, W. J., Anema, J., Schirm, J.,
Tegzess, A. M., & The, T. H. (1988b). Rapid immunodiagnosis of
active cytomegalovirus infection by monoclonal antibody staining of
blood leucocytes. J Med Virol 25, 179–188.
Vierling, J. M., & Fennel, R. H. (1985). Histopathology of early and late
human hepatic allograft rejection: evidence of progressive destruction
of interlobular bile ducts. Hepatology 5, 1076–1082.
Vinters, H. V., Kwok, M. K., Ho, H. W., Anders, K. H., Tomiyasu, U.,

S. Landolfo et al. / Pharmacology & Therapeutics 98 (2003) 269–297 297
Wolfson, W. L., & Robert, F. (1989). Cytomegalovirus in the nervous
system of patients with the acquired immune deficiency syndrome.
Brain 112, 245–268.
Vochem, M., Hamprecht, K., Jahn, G., & Speer, C. P. (1998). Transmission
of cytomegalovirus to preterm infants through breast milk. Pediatr In-
fect Dis J 17, 53–58.
Vogel, J.-U., Scholz, M., & Cinatl, J., Jr. (1998). Treatment of CMV dis-
eases. Intervirology 40, 357–367.
Wade, M., Kowalik, T. F., Mudryj, M., Huang, E.-S., & Azizkhan, J. C.
(1992). E2F mediates dihydrofolate reducatse promoter activation and
multiprotein complex formation in human cytomegalovirus infection.
Mol Cell Biol 12, 4364–4374.
Walter, E. A., Greenberg, P. D., Gilbert, M. J., Finch, R. J., Watanabe, K. S.,
Thomas, E. D., & Riddell, S. R. (1995). Reconstitution of cellular
immunity against cytomegalovirus in recipients of allogeneic bone mar-
row by transfer of T-cell clones from the donor. N Engl J Med 333,
1038–1044.
Webster, A., Philips, A. N., Lee, C. A., Janossy, G., Kernoff, P. B., &
Griffiths, P. D. (1992). Cytomegalovirus (CMV) infection, CD4+ lym-
phocyte counts and the development of AIDS in HIV-1 infected hae-
mophiliac patients. Clin Exp Immunol 88, 6–9.
Wiebush, L., & Hagemeier, C. (1999). Human cytomegalovirus 86-kilo-
dalton IE2 protein blocks cell cycle progression in G1. J Virol 73,
9274–9283.
Wiebush, L., & Hagemeier, C. (2001). The human cytomegalovirus imme-
diate early 2 protein dissociates cellular DNA synthesis from cyclin-
dependent kinase activation. EMBO J 20, 1086–1098.
Wiertz, E. J., Jones, T. R., Sun, L., Bogyo, M., Geuze, H. J., & Ploegh,
H. L. (1996). The human cytomegalovirus US11 gene product dislo-
cates MHC class I heavy chains from the endoplasmic reticulum to the
cytosol. Cell 84, 769–779.
Wills, M. R., Carmichael, A. J., Mynard, K., Jin, X., Weekes, M. P.,
Plachter, B., & Sissons, J. G. (1996). The human cytotoxic T-lympho-
cyte (CTL) response to cytomegalovirus is dominated by structural
protein pp65: frequency, specificity, and T-cell receptor usage of
pp65-specific CTL. J Virol 70, 7569–7579.
Winston, D. J., Ho, W. G., Bartoni, K., Du Mond, C., Ebeling, D. F.,
Buhles, W. C., & Champlin, R. E. (1993). Ganciclovir prophylaxis of
cytomegalovirus infection and disease in allogeneic bone marrow trans-
plant recipients. Results of a placebo-controlled, double-blind trial. Ann
Intern Med 118, 179–184.
Wreghitt, T. G., Hakim, M., Gray, J. J., Kucia, S., Wallwork, J., & English,
T. A. (1988). Cytomegalovirus infections in heart and heart and lung
transplantation. J Clin Pathol 41, 660–667.
Yeager, A. S., & Martin, H. P. (1977). Congenital cytomegalovirus infec-
tion: outcome for the subsequent sibling. Clin Pediatr 16, 455–458.
Yurochko, A. D., Kowalik, T. F., Huong, S. M., & Huang, E.-S. (1995).
Human cytomegalovirus upregulates NF-kB activity by transactivating
the NF-kB p105/p50 and p65 promoter. J Virol 69, 5391–5400.
Yurochko, A. D., Mayo, M. W., Poma, E. E., Baldwin Jr., A. S., & Huang,
E.-S. (1997). Induction of the transcription factor Sp1 during human
cytomegalovirus infection mediates upregulation of the p65 and p105/
p50 NF-kB promoters. J Virol 71, 4638–4648.
Zanghellini, F., Boppana, S. B., Emery, V. C., Griffiths, P. D., & Pass, R. F.
(1999). Asymptomatic primary cytomegalovirus infection: virologic
and immunologic features. J Infect Dis 180, 702–707.
Zhu, H., Shen, Y., & Shenk, T. (1995). Human cytomegalovirus IE1 and
IE2 proteins block apoptosis. J Virol 69, 7960–7970.
Zhu, H., Cong, J. P., & Shenk, T. (1997). Use of differential display analysis
to assess the effect of human cytomegalovirus infection on the accu-
mulation of cellular RNAs: induction of the interferon-responsive
RNAs. Proc Natl Acad Sci USA 94, 13985–13990.
Zhu, H., Cong, J. P., Mamtora, G., Gingeras, T., & Shenk, T. (1998).
Cellular gene expression altered by human cytomegalovirus: global
monitoring with oligonucleotide arrays. Proc Natl Acad Sci USA 95,
14470–14475.
Related Documents











