Nature Clinical Practice Cardiovascular Medicine (2007) 4, 558-569 Received 15 January 2007 | Accepted 8 June 2007 Mechanisms of Disease: the genetic basis of coronary heart disease Iftikhar J Kullo* and Keyue Ding Correspondence *Division of Cardiovascular Diseases, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA Email [email protected] SUMMARY Since completion of the human genome sequence, considerable progress has been made in determining the genetic basis of human diseases. Understanding the genet ic basis of coronary heart disease (CHD), the leading cause of mortality in developed countries, is a priority. Here we provide an update on the genetic basis of CHD, focusing mainly on the clinical manifestations rather than the risk factors, most of which are heritable and also influenced by genetic factors. The challenges faced when identifying clinically relevant genetic determinants of CHD include phenotypic and genetic heterogeneity, and gene– gene and gene–environment interactions. In addition, the etiologic spectrum includes common genetic variants with small effects, as well as rare genetic variants with large effec ts. Advances such as the catalogi ng of huma n geneti c varia tion, new statisti cal approaches for analyzing massive amounts of genetic data, and the development of high- thr oughput single-nucleotide polymorphism genotyping pla tfo rms, will increase the likelihood of success in the search for genetic determinants of CHD. Such knowledge could ref ine cardiovascular risk strati fic ation and fac ilitate the development of new therapies. Keywords: association, complex diseases, coronary heart disease, genetics, linkage Medscape Continuing Medical Education online Medscape, LLC is pleased to provide online continuing medical education (CME) for this journal article, allowing clinicians the opportunity to earn CME credit. Medscape, LLC is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide CME for physicians. Medscape, LLC designates this educational activity for a maximum of 1.0 AMA PRA Category 1 Credits ™ . Physicians should only claim credit commensurate with the extent of their participation in the activity. All other clinicians completing this activity will be issued a certificate of participation. To receive credit, please go to http://www.medscape.com/cme/ncp and complete the post-test. Learning objectives Upon completion of this activity, participants should be able to: 1. Desc ribe the epi demiol ogy of genet ic fact ors in coron ary hear t disea se (CHD). 2. Identi fy resul ts of li nkage studie s of genet ic fac tors in CHD. 3. Compare linkag e studies wit h associ atio n studies relating complex dise ases to genomic factors. 4. Specify limitations of studies li nking genet ic factors to compl ex diseas es. 5. Recogn ize possi ble contr ibuti ons of geneti c studies to the clinic al care of patie nts at risk for CHD.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 1/17
Nature Clinical Practice Cardiovascular Medicine (2007) 4, 558-569
Received 15 January 2007 | Accepted 8 June 2007
Mechanisms of Disease: the genetic basis of coronary heartdisease
Iftikhar J Kullo* and Keyue Ding
Correspondence *Division of Cardiovascular Diseases, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905,USA
Email [email protected]
SUMMARY
Since completion of the human genome sequence, considerable progress has been madein determining the genetic basis of human diseases. Understanding the genetic basis of
coronary heart disease (CHD), the leading cause of mortality in developed countries, is apriority. Here we provide an update on the genetic basis of CHD, focusing mainly on theclinical manifestations rather than the risk factors, most of which are heritable and also
influenced by genetic factors. The challenges faced when identifying clinically relevantgenetic determinants of CHD include phenotypic and genetic heterogeneity, and gene–
gene and gene–environment interactions. In addition, the etiologic spectrum includescommon genetic variants with small effects, as well as rare genetic variants with large
effects. Advances such as the cataloging of human genetic variation, new statisticalapproaches for analyzing massive amounts of genetic data, and the development of high-throughput single-nucleotide polymorphism genotyping platforms, will increase thelikelihood of success in the search for genetic determinants of CHD. Such knowledgecould refine cardiovascular risk stratification and facilitate the development of new
therapies.
Keywords:
association, complex diseases, coronary heart disease, genetics, linkage
Medscape Continuing Medical Education online
Medscape, LLC is pleased to provide online continuing medical education (CME) for this journal article, allowing clinicians the opportunity to earn CME credit. Medscape, LLC isaccredited by the Accreditation Council for Continuing Medical Education (ACCME) toprovide CME for physicians. Medscape, LLC designates this educational activity for a
maximum of 1.0 AMA PRA Category 1 Credits™
. Physicians should only claim creditcommensurate with the extent of their participation in the activity. All other clinicianscompleting this activity will be issued a certificate of participation. To receive credit,
please go to http://www.medscape.com/cme/ncp and complete the post-test.
Learning objectives
Upon completion of this activity, participants should be able to:
1. Describe the epidemiology of genetic factors in coronary heart disease (CHD).2. Identify results of linkage studies of genetic factors in CHD.3. Compare linkage studies with association studies relating complex diseases to
genomic factors.4. Specify limitations of studies linking genetic factors to complex diseases.
5. Recognize possible contributions of genetic studies to the clinical care of patientsat risk for CHD.

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 2/17
INTRODUCTION
In 2003 in the US alone, there were an estimated 1.2 million cases of coronary heartdisease (CHD), resulting in 479,000 deaths.1 Although recognition and treatment of established risk factors for CHD will reduce the disease burden considerably,
simultaneous efforts aimed at unraveling the genetic basis of CHD are important for thedevelopment of novel diagnostic and therapeutic methods.
Considerable progress has been made in determining the genetic basis of humandiseases since the human genome has been sequenced. The genetic determinants of
more than 1,600 Mendelian diseases are now known, and discovery of genomic regionsand genetic polymorphisms that influence susceptibility to common 'complex' diseases isaccelerating. In this Review, we discuss the challenges of elucidating the genetic basis of CHD, focusing mainly on its clinical manifestations rather than its risk factors, andsummarize the studies that have yielded insights into the genetic basis of this common,complex disease. We also discuss how recent advances in this field will increase thelikelihood of identifying genetic determinants of CHD.
THE CURRENT STATE OF KNOWLEDGE
Several challenges exist in identifying the genetic determinants of common, complexdiseases such as CHD (Table 1). These include phenotypic and genetic heterogeneity,gene–gene and gene–environment interactions, and the fact that the etiologic spectrum
ranges from common genetic variants with small effects to rare genetic variants withlarge effects. Below, we attempt to summarize the current state of knowledge about the
genetic basis of CHD. A glossary of some common genetic terms used in this Review canbe found in Box 1.
Table 1 Challenges in identifying genetic determinants of coronary heartdisease.
Challenge Comment
Abbreviation: CHD, coronary heart disease.
Phenotypicheterogeneity
CHD can manifest as several clinical phenotypes, including chronicstable angina, acute coronary syndrome, myocardial infarction,
sudden cardiac death, and history of coronary revascularization.Measures of coronary atherosclerotic burden such as coronary arterycalcium or angiographic coronary artery disease are objective andquantitative, unlike the dichotomous characterization (presence orabsence) of a history of a cardiovascular event. Atheroscleroticdisease burden and cardiovascular events such as myocardialinfarction are distinct phenotypes, however, the latter being related
more to plaque instability and rupture rather than plaque burden.
Geneticheterogeneity
Genetic heterogeneity is likely, given the multiple causal pathwaysthat lead to CHD. For example, low plasma levels of HDL cholesterolcan result from variation in genes from multiple metabolic pathways.Similarly, the transition from a stable coronary atherosclerotic plaqueto an unstable inflamed plaque could be the result of genetic variationin multiple genes that participate in the inflammatory cascade andmatrix degradation.
Small gene effects
A single genetic variant can constitute only a small proportion (e.g.
1– 2%) of the total genetic contribution towards complex diseasephenotypes, and an odds ratio of 1.1–1.5 is typical for a susceptibilityvariant of a candidate gene.86 Uncovering such small effects requireslarge sample sizes, and recognition of this fact has motivatedassembly of the so-called Biobanks in several European countries,including Iceland, England and Estonia.87

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 3/17
Challenge Comment
Gene–gene andgene–environmentinteractions
Identification of gene–gene and gene–environment interactions isessential for identification of genes responsible for complex diseases,and will require large sample sizes and adequate computationalresources.
Rare variants
causing complexdisease
Most current genetic epidemiology studies of complex diseases such
as CHD assume that common variants (i.e. minor allele frequency5%) account for much of the susceptibility to the disease. Bothcommon and rare variants, however, probably influence CHDsusceptibility. To uncover rare variants that influence susceptibility toCHD, resequencing or very large sample sizes will be required.
FAMILIAL CLUSTERING OF CORONARY HEART DISEASE
Twin and family studies have established that CHD aggregates in families and in fact
family history of early-onset CHD has long been considered a risk factor for the disease. 2
Although a contentious issue, the familial clustering of CHD could be partly explained byheritable quantitative variation in known CHD risk factors. Evidence suggests that familyhistory contributes to an increased risk of CHD independently of the known risk factors.3,
4 High-risk families make up a considerable proportion of early CHD cases in the generalpopulation. In one study, families with a history of early CHD represented only 14% of the general population but accounted for 72% of early CHD cases (men aged <55 years,women aged <65 years) and 48% of CHD at all ages.5 A history of early CHD in a first-
degree relative approximately doubles the risk of CHD, although the reported relative riskranges from 1.3–11.3.4, 6, 7, 8, 9, 10 The highest relative hazard of CHD-related death can be
seen in monozygotic twins, when one twin dies of early-onset CHD.4 Furthermore, siblinghistory of myocardial infarction seems to be a greater risk factor than parental history of early-onset CHD.11 A proposed family risk score for CHD evaluates the ratio of observed
CHD events to expected events in an individual's first-degree relatives, adjusted for ageand sex at the onset of the first event.12 A higher family risk score is associated withgreater CHD risk.
MENDELIAN DISORDERS ASSOCIATED WITH CORONARY HEART DISEASE
Mendelian disorders associated with CHD, such as familial hypercholesterolemia,
comprise single-gene traits that are transmitted in an autosomal dominant, recessive orX-linked manner. For example, mutations in the LDL receptor gene (LDLR), the ligand-
binding domain of apolipoprotein B100 ( APOB), and proprotein convertasesubtilisin/kexin type 9 gene (PCSK9) result in familial hypercholesterolemia transmitted
in an autosomal dominant manner. The examination of disease pathophysiology andgene function in such Mendelian disorders might increase our understanding of theetiology of complex traits.13 Additionally, common variation in genes implicated inMendelian disorders could be used to determine disease susceptibility in the generalpopulation. Several Mendelian disorders of lipid metabolism are associated with increasedCHD risk and have yielded novel insights into the mechanisms of CHD. Investigation of the molecular basis of the rare disorder familial homozygous hypercholesterolemia led tothe discovery of the pathways of LDL cholesterol metabolism and the subsequent
development of statins.14 Rare allelic variants of three candidate genes that influence HDLcholesterol metabolism ( ABCA1 [ATP-binding cassette A1], APOA1 [apolipoprotein A-1],
and LCAT [lecithin-cholesterol acyltransferase]) are associated with low HDL-cholesterollevel syndromes but are also found in individuals from the community with low HDLcholesterol levels.15, 16
LINKAGE STUDIES: IDENTIFICATION OF GENES RELATED TO CORONARY HEARTDISEASE

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 4/17
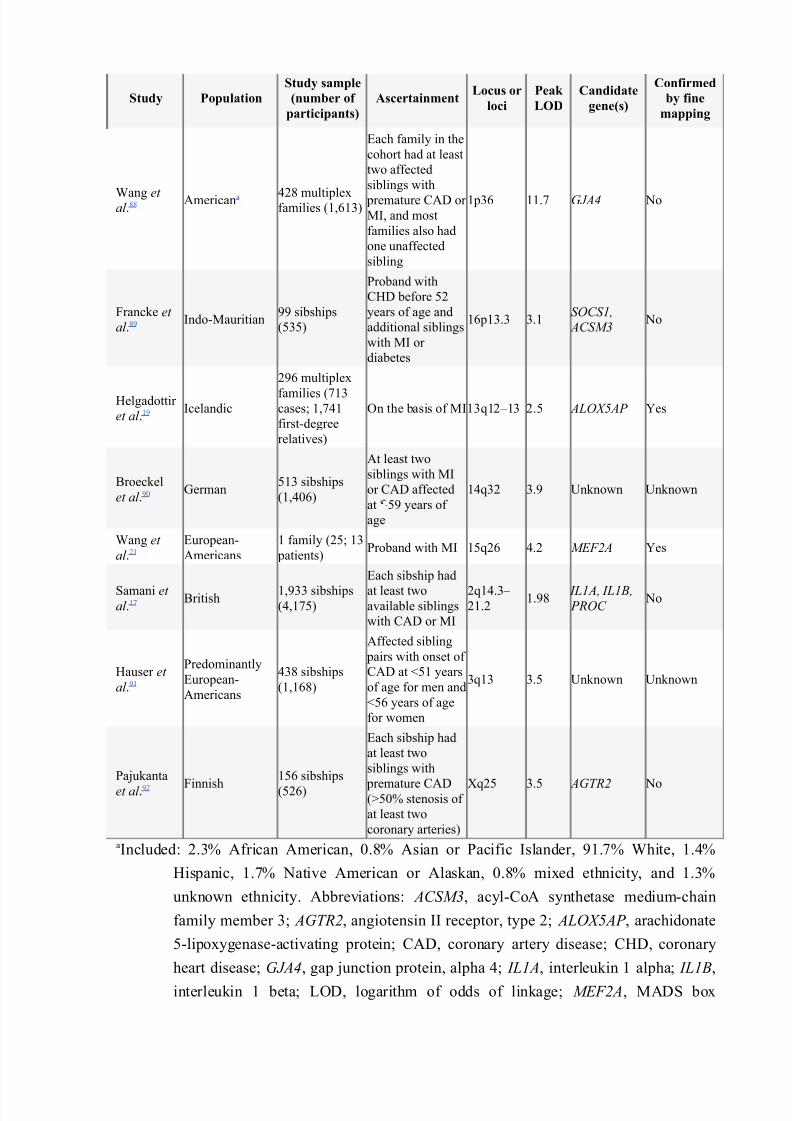
Linkage studies are performed by using polymorphic DNA markers (Box 2 and Figure 1).Microsatellite markers (short tandem repeat DNA sequences that are dispersedthroughout the human genome) are typically used in linkage studies, although single-nucleotide polymorphisms (SNPs) can also be used (Box 1). Several genome-widelinkage studies for myocardial infarction and coronary artery disease have been reported(Table 2). The largest study, the British Heart Foundation Family Heart Study, included4,175 individuals with CHD from 1,933 families recruited throughout the UK. 17 Despite
the large sample size, a statistically significant logarithmic odds of linkage (LOD) score(i.e. 3) was not obtained for any of the cardiovascular end points studied. For coronaryartery disease (verified by exercise stress test or angiography), the highest LOD score
was 2.70 (chromosome 2 at 149 cM) in families (n = 1,698) with age at onset of 56years or less. For myocardial infarction, an overlapping peak with a LOD score of 2.1 (at119.3 cM) in families (n = 801) with age of at onset 59 years or less, was observed.Genomic regions identified in the published linkage studies as being correlated with CHDare largely non-overlapping, suggesting genetic heterogeneity, although phenotypicheterogeneity could also have contributed to the non-replicability of results. Farrall et al .attempted to replicate a genomic locus for CHD by performing linkage analysis in twoindependent samples of European whites.18 The investigators found evidence of
replication for a locus on chromosome 17 (at 69 cM).
Figure 1. Different genetic markers used in linkage and association studies.
(A) A short tandem repeat consists of short sequences of DNA (normally 2–5 base pairs)that are repeated numerous times. A single-nucleotide polymorphism is a single
nucleotide change or variation that occurs in a DNA sequence. (B) A haplotype is thecombination of alleles (for different markers) that are located close together on the samechromosome and tend to be inherited together.
Table 2 Linkage studies of myocardial infarction and coronary artery disease.
8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 5/17
Study Population
Study sample
(number of
participants)
AscertainmentLocus or
loci
Peak
LOD
Candidate
gene(s)
Confirmed
by fine
mapping
Wang et al .88 Americana 428 multiplex
families (1,613)
Each family in the
cohort had at least
two affected
siblings with
premature CAD or MI, and most
families also had
one unaffected
sibling
1p36 11.7 GJA4 No
Francke et
al .89 Indo-Mauritian99 sibships
(535)
Proband with
CHD before 52
years of age and
additional siblings
with MI or
diabetes
16p13.3 3.1SOCS1,
ACSM3 No
Helgadottir
et al .19 Icelandic
296 multiplex
families (713cases; 1,741
first-degree
relatives)
On the basis of MI13q12–13 2.5 ALOX5AP Yes
Broeckel
et al .90 German513 sibships
(1,406)
At least two
siblings with MI
or CAD affected
at 59 years of
age
14q32 3.9 Unknown Unknown
Wang et
al .21
European-
Americans
1 family (25; 13
patients)Proband with MI 15q26 4.2 MEF2A Yes
Samani et al .17 British 1,933 sibships
(4,175)
Each sibship had
at least twoavailable siblings
with CAD or MI
2q14.3– 21.2
1.98 IL1A, IL1B, PROC
No
Hauser et
al .91
Predominantly
European-
Americans
438 sibships
(1,168)
Affected sibling
pairs with onset of
CAD at <51 years
of age for men and
<56 years of age
for women
3q13 3.5 Unknown Unknown
Pajukanta
et al .92 Finnish156 sibships
(526)
Each sibship had
at least two
siblings with
premature CAD(>50% stenosis of
at least two
coronary arteries)
Xq25 3.5 AGTR2 No
aIncluded: 2.3% African American, 0.8% Asian or Pacific Islander, 91.7% White, 1.4%
Hispanic, 1.7% Native American or Alaskan, 0.8% mixed ethnicity, and 1.3%
unknown ethnicity. Abbreviations: ACSM3, acyl-CoA synthetase medium-chain
family member 3; AGTR2, angiotensin II receptor, type 2; ALOX5AP , arachidonate
5-lipoxygenase-activating protein; CAD, coronary artery disease; CHD, coronary
heart disease; GJA4, gap junction protein, alpha 4; IL1A, interleukin 1 alpha; IL1B,
interleukin 1 beta; LOD, logarithm of odds of linkage; MEF2A, MADS box

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 6/17
transcription enhancer factor 2, polypeptide A; MI, myocardial infarction; PROC ,
protein C; SOCS1, suppressor of cytokine signaling 1.
BOX 2 LINKAGE STUDIES VERSUS ASSOCIATION STUDIES.
Linkage studies require enrollment of families with the disease or phenotype of
interest. Approximately 400 microsatellite markers throughout the genome are
genotyped, and statistical evidence of cotransmission of DNA markers with the disease or
phenotype of interest is sought. If significant evidence for cotransmission is found
between a marker and the disease, the marker is probably situated near the disease-
susceptibility gene of interest (within approximately 10 megabases). The figure (below)
shows the linkage analysis of a disease transmitted in a dominant fashion; the marker
allele A1 cosegregates with the disease. Once linkage is found, fine mapping using a
geographically narrower set of markers can identify the region linked to the disease more
precisely. Fine mapping can also be performed by analyses of haplotypes, in which
multiple DNA markers are analyzed together rather than individually. Alternatively,
polymorphisms in candidate genes can be tested to determine whether they associate
with disease, or the most likely candidate gene can be sequenced to identify a novelfunctional variant responsible for the disease phenotype.

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 7/17
Association studies compare allele frequencies in cases and controls to assess the
contribution of genetic variants to phenotypes. The figure (below) shows that the markerallele A6 is present more often in cases than controls and is, therefore, associated with
disease. Association studies use a case–control design and they do not require the study
of families. Family-based association studies using the transmission disequilibrium test
are, however, useful in reducing confounding caused by population stratification.107 Most
commonly, association studies test putative functional single-nucleotide polymorphisms
within candidate genes and regions. Alternatively, the entire set of markers in the gene
can be genotyped, which reduces bias, but entails considerable cost and adds complexity
to the statistical genetic analysis because of the large number of single-nucleotide
polymorphisms that can be present in a gene.
Helgadottir and colleagues showed the utility of linkage analysis in identifying new genesfor CHD.19 They performed linkage analysis with 1,068 microsatellite markers and found
a linkage signal (LOD 2.86) on chromosome 13 for 296 Icelandic families (713individuals) enrolled on the basis of a history of myocardial infarction. The investigators
then genotyped an additional 120 microsatellite markers in this interval in 802 cases of myocardial infarction and 837 controls, and found that a 4-SNP haplotype spanning the
ALOX5AP gene (encoding arachidonate 5-lipoxygenase-activating protein) was associatedwith a doubled risk of myocardial infarction. A subsequent study found that ALOX5AP wasassociated with CHD in an English population and associated with stroke in Icelandic andScottish populations.20 Another example of a novel gene identified by linkage analysis ina pedigree with several members affected by early-onset CHD is MEF2 (myocyteenhancer factor 2), a transcription factor expressed in coronary artery endothelium.21 The
results of these studies have not yet translated into specific genetic tests but may pointto novel drug targets; for example, an inhibitor of ALOX5AP pathway is being
investigated for clinical use.22
Genome-wide linkage studies for quantitative measures of atherosclerotic burden,including coronary artery calcium levels, carotid intima-media thickness and ankle–brachial index, have also been reported.23, 24, 25, 26 Although genomic regions with LODscores greater than 3 have been linked to some of these traits, specific genes responsiblefor the linkage signals have yet to be identified.

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 8/17
ASSOCIATION STUDIES: IDENTIFICATION OF GENES RELATED TO CORONARYHEART DISEASE
Association studies compare allele frequencies in cases and controls to assess thecontribution of genetic variants to phenotypes of interest (Box 2). In contrast to linkagestudies, association studies of complex diseases localize disease-related genomic regionsmore precisely and have greater statistical power for detecting small gene effects.27 A
major concern, however, is the considerable proportion of associations between geneticvariants and disease that are reported but not replicated.28 The difficulty encountered in
reproducing the results of genetic association studies could be attributable to severalissues common to epidemiologic risk factor studies,29, 30, 31, 32, 33 including faulty study
design, inaccurate phenotyping, bias introduced during ascertainment and analyses, andconfounding variables.34 Issues specific to genetic association studies are briefly
discussed below.
When disease-related alleles have only a small effect on the phenotype, the statisticalpower of association studies will be low, because such associations can be difficult toreproduce. Furthermore, gene effects are context-dependent and can be modified by thepresence of other genetic or environmental factors, which can vary within study
populations.35
Spurious associations result from the presence of genetically differentstrata in a study sample (population stratification), each strata has varying frequencies of disease and different allele frequencies at the marker locus. Another important cause of
irreproducible findings could be variation in linkage disequilibrium (Box 1).36 The geneticmarker used might be distinct from the polymorphism that affects disease but could be in
linkage disequilibrium with the polymorphism (i.e. they are inherited together in a unit).Notably, the degree of linkage disequilibrium between the polymorphism and the markercan also vary among study populations. Genetic heterogeneity, wherein the diseasephenotype results from multiple uncommon variations or variants with extremely lowfrequency37 (as posited by the 'common disease–rare variants' hypothesis),16, 38, 39 couldalso decrease the chances of replicating association study findings.
The results of several association studies for CHD have been validated by subsequentstudies or in independent samples (Table 3). An example is the external validation of the
role of ALOX5AP variants in several vascular disease phenotypes.19, 20 In another study,Ozaki et al . used two independent sample sets to validate that a functional SNP in the 5'-
untranslated regions of PSMA6 (proteasome subunit, alpha type 6) conferred anincreased risk for myocardial infarction.40 Connelly et al . identified the transcription factorGATA2 (GATA-binding protein 2), which regulates several endothelial-specific genes, as anovel susceptibility gene for CHD in two independent case–control samples.41 Anotherexample of association study 'replication' is the study by Shiffman et al .42 Theinvestigators genotyped 11,053 putative functional SNPs in 6,891 genes and used athree-step process to reduce the number of hypotheses tested, thus identifying variantsin four genes associated with myocardial infarction (PALLD, palladin, cytoskeletal
associated protein; ROS1, v-ros UR2 sarcoma virus oncogene homolog 1 (avian);
TAS2R50, taste receptor, type 2, member 50; and OR13G1, olfactory receptor, family13, subfamily G, member 1). Further investigation will be needed to assess the utility of these polymorphisms in assessing CHD risk or in identifying new targets for drug
therapy.
Table 3 Examples of single-nucleotide polymorphism or haplotype associationstudies of coronary heart diseasea.
Gene Gene nameSNPs or
haplotypeSNP class
Supportingstudy(ies)
ALOX5AP
Arachidonate 5-
lipoxgenase-activatingprotein Haplotype A NA Helgadottir et al .
20
APOE Apolipoprotein E 4 allele NonSyn Song et al .93 (a meta-

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 9/17
Gene Gene nameSNPs or
haplotypeSNP class
Supportingstudy(ies)
rs4986978 Intron analysis)
CX3CR1Chemokine (C-X3-C
motif) receptor 1rs3732378
NonSyn
(T280M)
Moatti et al .,94
McDermott et al .95
GJA4 Gap junction protein 1 rs1764391
NonSyn
(P319S)
Yamada et al .,96
Hirashiki et al .97
MMP3 Matrix metallopeptidase 3 rs3025058 Regulatory
Terashima et al .,98
Rauramaa et al .,99
Yamada et al .,96
Hirashiki et al .97
SERPINE
1
Serpin peptidase inhibitor,clade E member 1
rs1799889 RegulatoryMargaglione et al .,100
Yamada et al .96
PDE4DPhosphodiesterase 4D,cAMP-specific
haplotypes NA
Gretarsdottir et al .,101
Zee et al .,102 Brophy et al .103
PSMA6
Proteasome subunit type
6 rs1048990 5' UTR Ozaki et al .
40
TNFSF4Tumor necrosis factor(ligand) superfamily,
member 4
rs3850641 Intron Wang et al .104
LTA4H Leukotriene A4 hydrolase Haplotype K NA Helgadottir et al .105
USF1Upstream transcriptionfactor 1
Haplotypeacross USF1
NA Komulainen et al .106
GATA2 GATA binding protein 2rs2713604rs3803
Intron 5' UTR Connelly et al .41
aReplication implies in independent samples. Abbreviations: NA, not applicable; NonSyn,nonsynonymous; rs, reference SNP accession number; UTR, untranslated region.
NEW APPROACHES FOR IDENTIFYING GENETIC DETERMINANTS OF CHD
ACCURATE PHENOTYPING
Although considerable overlap exists among various CHD phenotypes (Table 1), theunderlying pathophysiology could vary considerably. Multiple risk factors and theirinteractions influence plaque stability and inflammation, platelet function, and thecoagulation cascade. Different combinations of these risk factors can, therefore,predispose individuals to the development of different phenotypes of CHD.43
Heterogeneity in the mechanisms underlying these phenotypes could explain why in
some cases there is no overlap between linked regions in related disease states—linkedregions that seem specific for myocardial infarction are not also specific for angiographic
coronary artery disease.18 At the outset of any genetic study, it is important to accuratelydefine the phenotype of cases and controls. Given the multiple possible presentations of CHD, careful characterization of the phenotype is especially important. Rigorous, uniformcriteria for cardiovascular events such as myocardial infarction or sudden cardiac deathshould be specified at the onset of the study. To avoid ascertainment bias, imagingstudies that provide information about both atherosclerotic burden and plaque activityshould be performed in community-based cohorts rather than in patients referred forsuch studies. To help diminish the likelihood of bias and reduce population stratification,
it is preferable that cases and control individuals are drawn from the same geographicregion and matched for age, sex and race. Furthermore, concomitant with efforts to
determine accurate phenotypes, further improvement is needed in the assessment andmeasurement of environmental factors relevant to CHD such as cigarette smoking,physical activity and dietary intake.

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 10/17
LINKAGE STUDIES
As shown by Helgadottir et al .19 and by Wang et al .,21 linkage analyses for complexdiseases have the potential to identify new candidate genes that previously would haveremained unsuspected on the basis of a priori knowledge of disease mechanisms. The
limitations associated with linkage studies of complex diseases include low statisticalpower and the inability to specify precise limits on the location of the causal gene ormutation. The statistical power of linkage studies could be improved by using larger
sample sizes and pedigrees, and disease susceptibility loci can be defined more preciselyby using a large number of markers across the genome. For example, John et al . used
11,245 SNPs in a genomic scan of families with rheumatoid arthritis and found that highSNP density localized disease susceptibility loci more precisely than the conventional 10-
cM microsatellite scan that used approximately 400 microsatellite markers.44
Linkage analysis in pedigrees is an unbiased approach for identifying genomic loci forquantitative disease phenotypes.45, 46 Quantitative traits have a simpler geneticarchitecture than the disease phenotype and could, therefore, be easier to map.
Quantitative traits related to atherosclerotic vascular disease and CHD that can bemeasured accurately, without bias in large population genetic studies include thefollowing: carotid intima-medial thickness, presence and quantity of coronary arterycalcium, coronary artery disease on angiography, coronary atherosclerotic burden on
intravascular ultrasonography, carotid or femoral artery plaque burden andcharacteristics on MRI, ankle–brachial index, and aortic pulse-wave velocity.
ADMIXTURE MAPPING
A new alternative to conventional linkage analysis is admixture mapping.47, 48, 49 This
technique can be applied in a population formed by relatively recent (e.g. 5 generations)admixture of two or more ancestral populations (e.g. African Americans who have WestAfrican and white European ancestry). For African Americans with a particular disease,genomic regions that have an unusually high proportion of ancestry from eitherEuropeans or Africans could harbor disease susceptibility variants. An example of the useof admixture mapping relevant to CHD is a study by Zhu et al .39 In this investigation, a
genomic admixture scan in 737 African Americans with hypertension and 573 controlsusing 269 microsatellite markers was performed. Evidence for association on
chromosomes 6q24 and 21q21 was found. Although confirmatory studies are needed,these results suggest that admixture mapping could be useful in identifying genome
regions that influence complex disease susceptibility.50
ASSOCIATION STUDIES
Developments include attempts to improve the replicability of association studies,
candidate-gene resequencing studies, and genome-wide association studies. To obtainrobust results from association studies the use of large samples (i.e. thousands of casesand controls, instead of hundreds) and stringent thresholds for statistical significancehave been proposed.51 Biologically plausible associations and risk alleles with functionaleffects are more likely to be 'true' associations, hence, should have replicable findings.
The replication of SNP disease associations in independent samples is crucial forvalidating results, and by genotyping 'neutral' markers throughout the genome,
potentially confounding population substructures can be excluded.52, 53 Alternatively,replication can be shown within a single study by dividing the study subjects into a 'test'
group and a 'validation' group,54 with both groups independently powered to detect anassociation. Association mapping could also be more successful with population isolatesin which genetic stratification is minimal, as shown by the success of the deCODE project
in Iceland.55
Candidate gene resequencing studies involve sequencing an entire candidate gene incases and controls and identifying the sequence variants that clearly differ in frequency

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 11/17
between the two groups. These studies are labor intensive and expensive but can identifyrare variants that influence complex diseases or traits. This approach was used toidentify rare variants of MC4R (the melanocortin 4 receptor gene) that were associatedwith severe early-onset obesity.56 Cohen et al . also successfully used this approach toidentify rare nonsynonymous SNPs that influence plasma levels of HDL cholesterol 16 andLDL cholesterol57 in the general population. Extension of this approach to a genomic scale('genome resequencing') with a large number of cases and controls would be the most
comprehensive means of identifying genetic variants underlying complex diseases.Although, genome resequencing in large case–control studies is not feasible at present, itcould soon become standard, as the costs for sequencing continue to drop.
In the interim, a genome-wide association approach, in which variants are tested forassociation with a trait or disease of interest, has become possible with data from the
HapMap project and high-throughput SNP-typing platforms.58 In 2002, the HapMapproject was undertaken to catalogue patterns of genetic variation as a means of identifying common genomic variants contributing to the cause of prevalent diseases.58, 59
The human genome seems to be organized into a series of haplotype blocks,60, 61, 62 eachhaplotype block shows low diversity and SNPs within a haplotype block show high linkagedisequilibrium. One strategy to reduce genotyping effort in association mapping of
complex diseases uses tag SNPs,63
which correlate with much of the common variation ina genomic region, and, therefore, could serve as a marker of this common variation.
Several studies have already reported convincing statistical evidence that links geneticpolymorphisms with CHD risk factors as well as with CHD phenotypes: polymorphisms in
INSIG264 (encoding insulin induced gene 2), and FTO65(fat mass and obesity associated)have been associated with obesity; polymorphisms in IFIH166 (interferon-induced helicaseC domain-containing protein 1) and IL2RA67 (interleukin-2 receptor alpha chain) havebeen associated with type 1 diabetes mellitus; polymorphisms in TCF7L268, 69
(transcription factor 7-like 2), SLC30A869, 70, 71, 72 (zinc transporter 8), a locus nearCDKN2A (cyclin-dependent kinase inhibitor 2A) and CDKN2B69, 71, 72 (cyclin-dependentkinase inhibitor 2B), IGF2BP269, 71, 72 (insulin-like growth factor 2 mRNA binding protein2), and in CDKAL169, 71, 72, 73 (CDK5 regulatory subunit associated protein 1-like 1) have
been associated with type 2 diabetes mellitus; and a polymorphism in a locus nearCDKN2A and CDKN2B on chromosome 9p21 has been associated with CHD in several
genome-wide association studies.74, 75, 76
Several different approaches fall under the rubric of genome-wide association studies(Figure 2). In broad terms, these approaches can be classified as 'map-based' (usinguniformly spaced SNPs or tag SNPs) or 'gene-based' (using putative functional SNPs).77
In the map-based approach, SNPs to be genotyped could be evenly spaced or tag SNPscould be used, with the presumption that linkage disequilibrium between such a tag SNPand the causal SNP would allow the detection of the causal SNP. The evenly spaced SNPscollection could provide sparse (e.g. 100,000 SNPs) or dense (e.g. 500,000–1,000,000
SNPs) coverage of the genome. The genotyping burden can be reduced markedly,however, by using tag SNPs across the genome. A collection of 250,000 tag SNPs, for
example, would cover approximately 85% of the genome.78 In the gene-based approach,putative functional SNPs throughout the genome are genotyped, including
nonsynonymous SNPs, regulatory SNPs and SNPs in splice sites.

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 12/17
Figure 2 Strategies for genome-wide association studies using SNPs.
These approaches can be classified as (A) 'map-based' (using uniformly spaced SNPs ortag SNPs) or (B) 'gene-based' (using putative functional SNPs).77 Under the 'map-based'approach, a subset of SNPs is selected for genotyping—these can be evenly spaced SNPsor tag SNPs. SNPs in strong linkage disequilibrium are likely to be inherited together, so
one can use a subset of 'tag' SNPs as proxies for the entire set. For the 'gene-based'approach, putatively functional SNPs located in regulatory regions and non-synonymous
SNPs are selected to be genotyped. Abbreviation: SNP, single-nucleotide polymorphism.
The development of appropriate statistical techniques to analyze the massive amount of genetic data gained from genome-wide association studies is crucial for the identificationof genes for complex diseases. As hundreds of thousands of potential statistical testsmight be computed in such studies, a major challenge is correction for the multiple
testing that must be performed.79 One approach is a multistage design that reduces thenumber of genotyped SNPs in each stage, achieving stepwise genome-wide
significance.28, 80 The Bonferroni correction is considered to be overly conservative, 81 andother approaches have been proposed, including estimation of the false discovery rate.82,
83
A CLINICAL PERSPECTIVE
We feel it is necessary to highlight three important considerations relevant to clinical
practice. First, as CHD clusters in families, obtaining detailed information on a patient'sfamily history is important. Family history has been described as a "... free, well-proven,personalized genomic tool that captures many of the genes and environmental
interactions and can serve as the cornerstone for individualized disease prevention."84
Second, although genetic testing is not part of current CHD risk stratification algorithms,it is likely that multilocus genotyping to assess CHD risk will become part of clinicalpractice in the future. Not unexpectedly, entrepreneurial zeal has overtaken carefulscientific validation, and several companies now market gene-based tests for assessing
cardiovascular risk to patients directly via the internet.85 Third, the immediate promise of identifying genetic determinants of CHD is greater in the therapeutic arena than in
refining risk prediction. This potential includes use of genetic tests to allow individualizedtreatment (pharmacogenetics) and to facilitate discovery of new molecular pathways of
CHD and drug targets. Of interest, the results of a drug trial that used the findings of agenetic study that identified a novel therapeutic target for CHD have already beenpublished.22

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 13/17
CONCLUSIONS
We have attempted to summarize the current state of knowledge about the genetic basisof CHD and the new approaches that might lead to further successes in elucidating thebasis of this disease. Increased knowledge of the genetic architecture of CHD will
improve risk prediction and facilitate the development of new therapies for patients withCHD. Although considerable challenges exist, advances such as high-throughput SNP
genotyping platforms and newer statistical and phenotyping methods show promise foraccelerating progress in this field. Well-designed studies are needed to define clinically
relevant phenotypes, identify genes and define environmental contributions to CHD.Given that CHD is a clinically heterogeneous chronic disease with multiple genetic andenvironmental contributions, identification of causal genes for this disease requires avigorous multidisciplinary approach that includes physician investigators and laboratoryscientists, and epidemiologists and statisticians with expertise in genetics. The task ischallenging, but the goals justify the effort and the expense.
KEY POINTS
•
Understanding the genetic basis of coronary heart disease (CHD) is a priority as itis projected to become the leading cause of mortality worldwide
• Challenges in identifying clinically relevant genetic determinants of complex
diseases such as CHD include phenotypic and genetic heterogeneity, gene–gene
and gene–environment interactions, and the fact that the etiologic spectrum
includes both common genetic variants with small effects as well as rare genetic
variants with large effects
• Linkage and association mapping are two conventional approaches in identifying
genetic determinants of CHD
• Advances such as cataloging of human genetic variation, the development of
high-throughput single-nucleotide polymorphisms genotyping platforms and
genome-wide association studies will increase the likelihood of success in the
search for genetic determinants of CHD al
REFERENCES
1. Thom T et al 2006 Heart disease and stroke statistics—2006 update: a report from the
American Heart Association Statistics Committee and Stroke StatisticsSubcommitteeCirculation113e85151 | Article | PubMed | ISI |
2. Scheuner MT2003 Genetic evaluation for coronary artery diseaseGenet
Med 5269285 | PubMed | ISI |
3. Murabito JM et al 2005 Sibling cardiovascular disease as a risk factor for cardiovascular
disease in middle-aged adults JAMA29431173123 | Article | PubMed | ISI | ChemPort |
4. Yusuf S et al 2004 Effect of potentially modifiable risk factors associated with myocardial
infarction in 52 countries (the INTERHEART study): case-controlstudyLancet 364937952 | Article | PubMed | ISI |
5. Williams RR et al 2001 Usefulness of cardiovascular family history data for population-based
preventive medicine and medical research (the Health Family Tree Study and the NHLBIFamily Heart Study) Am J Cardiol 87129135 | Article | PubMed | ISI | ChemPort |
6. Myers RH et al 1990 Parental history is an independent risk factor for coronary artery
disease: the Framingham Study Am Heart
J 120963969 | Article | PubMed | ISI | ChemPort |
7. Roncaglioni MC et al 1992 Role of family history in patients with myocardial infarction: an
Italian case-control study. GISSI-EFRIMInvestigatorsCirculation8520652072 | PubMed | ISI | ChemPort |
8. Friedlander Y et al 1998 Family history as a risk factor for primary cardiac
arrestCirculation97155160 | PubMed | ISI | ChemPort |
8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 14/17
9. Lloyd-Jones DM et al 2004 Parental cardiovascular disease as a risk factor for cardiovascular
disease in middle-aged adults: a prospective study of parents andoffspring JAMA29122042211 | Article | PubMed | ISI | ChemPort |
10. Kaikkonen KS et al 2006 Family history and the risk of sudden cardiac death as a
manifestation of an acute coronary eventCirculation11414621467 | Article | PubMed | ISI |
11. Nasir K et al 2004 Coronary artery calcification and family history of premature coronary
heart disease: sibling history is more strongly associated than parental
historyCirculation11021502156 | Article | PubMed | ISI |12. Li R et al 2000 Family risk score of coronary heart disease (CHD) as a predictor of CHD: the
Atherosclerosis Risk in Communities (ARIC) study and the NHLBI family heart studyGenet
Epidemiol 18236250 | Article | PubMed | ISI | ChemPort |
13. Antonarakis SEBeckmann JS2006 Mendelian disorders deserve more attentionNat Rev
Genet 7277282 | Article | PubMed | ISI | ChemPort |
14. Goldstein JLBrown MS1973 Familial hypercholesterolemia: identification of a defect in the
regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated withoverproduction of cholesterolProc Natl Acad Sci
USA7028042808 | Article | PubMed | ChemPort |
15. Frikke-Schmidt R et al 2004 Genetic variation in ABC transporter A1 contributes to HDL
cholesterol in the general population J Clin
Invest 11413431353 | Article | PubMed | ISI | ChemPort |
16. Cohen JC et al 2004 Multiple rare alleles contribute to low plasma levels of HDL
cholesterolScience305869872 | Article | PubMed | ISI | ChemPort |
17. 2005 A genomewide linkage study of 1,933 families affected by premature coronary artery
disease: The British Heart Foundation (BHF) Family Heart Study Am J HumGenet 7710111020 | Article | PubMed | ISI |
18. Farrall M et al 2006 Genome-wide mapping of susceptibility to coronary artery disease
identifies a novel replicated locus on chromosome 17PLoS
Genet 2e72 | Article | PubMed | ChemPort |
19. Helgadottir A et al 2004 The gene encoding 5-lipoxygenase activating protein confers risk of
myocardial infarction and strokeNat Genet 36233239 | Article | PubMed | ISI | ChemPort |
20. Helgadottir A et al 2005 Association between the gene encoding 5-lipoxygenase-activating
protein and stroke replicated in a Scottish population Am J Hum
Genet 76505509 | Article | PubMed | ISI | ChemPort |21. Wang L et al 2003 Mutation of MEF2A in an inherited disorder with features of coronary
artery diseaseScience30215781581 | Article | PubMed | ISI | ChemPort |
22. Hakonarson H et al 2005 Effects of a 5-lipoxygenase-activating protein inhibitor on
biomarkers associated with risk of myocardial infarction: a randomizedtrial JAMA29322452256 | Article | PubMed | ISI | ChemPort |
23. Lange LA et al 2002 Autosomal genome-wide scan for coronary artery calcification loci in
sibships at high risk for hypertension Arterioscler Thromb Vasc
Biol 22418423 | PubMed | ISI | ChemPort |
24. Fox CS et al 2004 Genomewide linkage analysis for internal carotid artery intimal medial
thickness: evidence for linkage to chromosome 12 Am J Hum
Genet 74253261 | Article | PubMed | ISI | ChemPort |
25. Wang D et al 2005 A genome-wide scan for carotid artery intima-media thickness: theMexican-American Coronary Artery Disease familystudyStroke36540545 | Article | PubMed | ISI | ChemPort |
26. Kullo IJ et al 2006 A genome-wide linkage scan for ankle-brachial index in African American
and non-Hispanic white subjects participating in the GENOAstudy Atherosclerosis187433438 | Article | PubMed | ISI | ChemPort |
27. Risch NMerikangas K1996 The future of genetic studies of complex human
diseasesScience27315161517 | Article | PubMed | ISI | ChemPort |
28. Hirschhorn JNDaly MJ2005 Genome-wide association studies for common diseases and
complex traitsNat Rev Genet 695108 | Article | PubMed | ISI | ChemPort |
29. Cardon LRPalmer LJ2003 Population stratification and spurious allelic
associationLancet 361598604 | Article | PubMed | ISI |
30. Colhoun HMet al
2003 Problems of reporting genetic associations with complexoutcomesLancet 361865872 | Article | PubMed | ISI |
31. Dahlman I et al 2002 Parameters for reliable results in genetic association studies in
common diseaseNat Genet 30149150 | Article | PubMed | ISI | ChemPort |

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 15/17
32. Freedman ML et al 2005 Systematic evaluation of genetic variation at the androgen receptor
locus and risk of prostate cancer in a multiethnic cohort study Am J Hum
Genet 768290 | Article | PubMed | ISI | ChemPort |
33. Romero R et al 2002 The design, execution, and interpretation of genetic association studies
to decipher complex diseases Am J Obstet Gynecol 18712991312 | Article | PubMed | ISI |
34. Ioannidis JP et al 2001 Replication validity of genetic association studiesNat
Genet 29306309 | Article | PubMed | ISI | ChemPort |
35. Sing CF et al 2003 Genes, environment, and cardiovascular disease Arterioscler ThrombVasc Biol 2311901196 | Article | PubMed | ISI | ChemPort |
36. Ardlie KG et al 2002 Patterns of linkage disequilibrium in the human genomeNat Rev
Genet 3299309 | Article | PubMed | ISI | ChemPort |
37. Pritchard JKCox NJ2002 The allelic architecture of human disease genes: common disease-
common variant...or not?Hum Mol Genet 1124172423 | Article | PubMed | ISI | ChemPort |
38. Liu PY et al 2005 A survey of haplotype variants at several disease candidate genes: the
importance of rare variants for complex diseases J Med
Genet 42221227 | Article | PubMed | ISI | ChemPort |
39. Zhu X et al 2005 Haplotypes produced from rare variants in the promoter and coding
regions of angiotensinogen contribute to variation in angiotensinogen levelsHum Mol
Genet 14639643 | Article | PubMed | ISI | ChemPort |
40. Ozaki K et al 2006 A functional SNP in PSMA6 confers risk of myocardial infarction in theJapanese populationNat Genet 38921925 | Article | PubMed | ISI | ChemPort |
41. Connelly JJ et al 2006 GATA2 is associated with familial early-onset coronary artery
diseasePLoS Genet 212651273 | ISI | ChemPort |
42. Shiffman D et al 2005 Identification of four gene variants associated with myocardial
infarction Am J Hum Genet 77596605 | Article | PubMed | ISI | ChemPort |
43. Hansson GK2005 Inflammation, atherosclerosis, and coronary artery diseaseN Engl J
Med 35216851695 | Article | PubMed | ISI | ChemPort |
44. John S et al 2004 Whole-genome scan, in a complex disease, using 11,245 single-
nucleotide polymorphisms: comparison with microsatellites Am J Hum
Genet 755464 | Article | PubMed | ISI | ChemPort |
45. Almasy LBlangero J2001 Endophenotypes as quantitative risk factors for psychiatric
disease: rationale and study design Am J Med Genet 1054244 | Article | PubMed | ISI | ChemPort |
46. Williams JTBlangero J1999 Power of variance component linkage analysis to detect
quantitative trait loci Ann Hum Genet 63545563 | Article | PubMed | ISI | ChemPort |
47. Patterson N et al 2004 Methods for high-density admixture mapping of disease genes Am J
Hum Genet 749791000 | Article | PubMed | ISI | ChemPort |
48. Smith MWO'Brien SJ2005 Mapping by admixture linkage disequilibrium: advances,
limitations and guidelinesNat Rev Genet 6623632 | Article | PubMed | ISI | ChemPort |
49. Reich DPatterson N2005 Will admixture mapping work to find disease genes?Philos Trans R
Soc Lond B Biol Sci 36016051607 | PubMed | ChemPort |
50. Darvasi AShifman S2005 The beauty of admixtureNat
Genet 37118119 | Article | PubMed | ISI | ChemPort |
51. Manly KF2005 Reliability of statistical associations between genes anddiseaseImmunogenetics57549558 | Article | PubMed | ISI |
52. Pritchard JKRosenberg NA1999 Use of unlinked genetic markers to detect population
stratification in association studies Am J Hum
Genet 65220228 | Article | PubMed | ISI | ChemPort |
53. Devlin BRoeder K1999 Genomic control for association
studiesBiometrics559971004 | Article | PubMed | ISI | ChemPort |
54. Ransohoff DF2004 Rules of evidence for cancer molecular-marker discovery and
validationNat Rev Cancer 4309314 | Article | PubMed | ISI | ChemPort |
55. Helgason A et al 2005 An Icelandic example of the impact of population structure on
association studiesNat Genet 379095 | Article | PubMed | ISI | ChemPort |
56. Vaisse C et al 2000 Melanocortin-4 receptor mutations are a frequent and heterogeneous
cause of morbid obesity J Clin Invest 106253262 | Article | PubMed | ISI | ChemPort |
57. Cohen J et al 2005 Low LDL cholesterol in individuals of African descent resulting from
frequent nonsense mutations in PCSK9Nat Genet 37161165 | Article | PubMed | ChemPort |

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 16/17
58. 2003 The International HapMap
ProjectNature426789796 | Article | PubMed | ISI | ChemPort |
59. Skelding KA et al 2007 The effect of HapMap on cardiovascular research and clinical
practiceNat Clin Pract Cardiovasc Med 4136142 | Article | PubMed | ISI | ChemPort |
60. Patil N et al 2001 Blocks of limited haplotype diversity revealed by high-resolution scanning
of human chromosome 21Science29417191723 | Article | PubMed | ISI | ChemPort |
61. Daly MJ et al 2001 High-resolution haplotype structure in the human genomeNat
Genet 29229232 | Article | PubMed | ISI | ChemPort |
62. Ding K et al 2005 The effect of haplotype block definitions on inference of haplotype block
structure and htSNPs selectionMol Biol Evol 22148159 | Article | PubMed | ISI | ChemPort |
63. Ding KKullo IJ2007 Methods for the selection of tagging SNPs: a comparison of tagging
efficiency and performanceEur J Hum Genet 15228236 | Article | PubMed | ISI | ChemPort |
64. Herbert A et al 2006 A common genetic variant is associated with adult and childhood
obesityScience312279283 | Article | PubMed | ISI | ChemPort |
65. Frayling TM et al 2007 A common variant in the FTO gene is associated with body mass
index and predisposes to childhood and adultobesityScience316889894 | Article | PubMed | ISI | ChemPort |
66. Smyth DJ et al 2006 A genome-wide association study of nonsynonymous SNPs identifies a
type 1 diabetes locus in the interferon-induced helicase (IFIH1) regionNat
Genet 38617619 | Article | PubMed | ISI | ChemPort |67. Vella A et al 2005 Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of
tag single-nucleotide polymorphisms Am J Hum
Genet 76773779 | Article | PubMed | ISI | ChemPort |
68. Grant SF et al 2006 Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of
type 2 diabetesNat Genet 38320323 | Article | PubMed | ISI | ChemPort |
69. Scott LJ et al 2007 A genome-wide association study of type 2 diabetes in Finns detects
multiple susceptibility variantsScience31613411345 | Article | PubMed | ISI | ChemPort |
70. Sladek R et al 2007 A genome-wide association study identifies novel risk loci for type 2
diabetesNature445881885 | Article | PubMed | ISI | ChemPort |
71. Saxena R et al 2007 Genome-wide association analysis identifies loci for type 2 diabetes
and triglyceride levelsScience31613311336 | Article | PubMed | ISI | ChemPort |
72. Zeggini E et al 2007 Replication of genome-wide association signals in UK samples revealsrisk loci for type 2 diabetesScience31613361341 | Article | PubMed | ISI | ChemPort |
73. Steinthorsdottir V et al 2007 A variant in CDKAL1 influences insulin response and risk of
type 2 diabetesNat Genet 39770775 | Article | PubMed | ISI | ChemPort |
74. McPherson R et al 2007 A common allele on chromosome 9 associated with coronary heart
diseaseScience31614881491 | Article | PubMed | ISI | ChemPort |
75. 2007 Genome-wide association study of 14,000 cases of seven common diseases and
3,000 shared controlsNature447661678 | Article | PubMed | ISI | ChemPort |
76. Helgadottir A et al 2007 A common variant on chromosome 9p21 affects the risk of
myocardial infarctionScience31614911493 | Article | PubMed | ISI | ChemPort |
77. Jorgenson EWitte JS2006 A gene-centric approach to genome-wide association studiesNat
Rev Genet 7885891 | Article | PubMed | ISI | ChemPort |
78. Altshuler D et al 2005 A haplotype map of the humangenomeNature43712991320 | Article | PubMed | ISI | ChemPort |
79. Thomas DC et al 2005 Recent developments in genomewide association scans: a workshop
summary and review Am J Hum Genet 77337345 | Article | PubMed | ISI | ChemPort |
80. Thomas D et al 2004 Two-stage sampling designs for gene association studiesGenet
Epidemiol 27401414 | Article | PubMed | ISI |
81. Perneger TV1998 What's wrong with Bonferroni
adjustmentsBMJ 31612361238 | PubMed | ChemPort |
82. Storey JDTibshirani R2003 Statistical significance for genomewide studiesProc Natl Acad Sci
USA10094409445 | Article | PubMed | ChemPort |
83. Churchill GADoerge RW1994 Empirical threshold values for quantitative trait
mappingGenetics138963971 | PubMed | ISI | ChemPort |
84. Guttmacher AE et al 2004 The family history—more important than everN Engl J Med 35123332336 | Article | PubMed | ISI | ChemPort |
85. Batchelder KMiller P2006 A change in the market—investing in diagnosticsNat
Biotechnol 24922926 | Article | PubMed | ISI | ChemPort |

8/6/2019 The Genetic Basis of Coronary Heart Disease
http://slidepdf.com/reader/full/the-genetic-basis-of-coronary-heart-disease 17/17
86. Risch NJ2000 Searching for genetic determinants in the new
millenniumNature405847856 | Article | PubMed | ISI | ChemPort |
87. Kaiser J2002 Biobanks: population databases boom, from Iceland to the
USScience29811581161 | Article | PubMed | ISI | ChemPort |
88. Wang Q et al 2004 Premature myocardial infarction novel susceptibility locus on
chromosome 1P34-36 identified by genomewide linkage analysis Am J Hum
Genet 74262271 | Article | PubMed | ISI | ChemPort |
89. Francke S et al 2001 A genome-wide scan for coronary heart disease suggests in Indo-Mauritians a susceptibility locus on chromosome 16p13 and replicates linkage with themetabolic syndrome on 3q27Hum Mol
Genet 1027512765 | Article | PubMed | ISI | ChemPort |
90. Broeckel U et al 2002 A comprehensive linkage analysis for myocardial infarction and its
related risk factorsNat Genet 30210214 | Article | PubMed | ISI | ChemPort |
91. Hauser ER et al 2004 A genomewide scan for early-onset coronary artery disease in 438
families: the GENECARD Study Am J Hum
Genet 75436447 | Article | PubMed | ISI | ChemPort |
92. Pajukanta P et al 2000 Two loci on chromosomes 2 and X for premature coronary heart
disease identified in early- and late-settlement populations of Finland Am J HumGenet 6714811493 | Article | PubMed | ISI | ChemPort |
93. Song Y et al 2004 Meta-analysis: apolipoprotein E genotypes and risk for coronary heartdisease Ann Intern Med 141137147 | PubMed | ISI |
94. Moatti D et al 2001 Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk
factor for coronary artery diseaseBlood 9719251928 | Article | PubMed | ISI | ChemPort |
95. McDermott DH et al 2003 Chemokine receptor mutant CX3CR1-M280 has impaired adhesive
function and correlates with protection from cardiovascular disease in humans J Clin
Invest 11112411250 | Article | PubMed | ISI | ChemPort |
96. Yamada Y et al 2002 Prediction of the risk of myocardial infarction from polymorphisms in
candidate genesN Engl J Med 34719161923 | Article | PubMed | ISI | ChemPort |
97. Hirashiki A et al 2003 Association of gene polymorphisms with coronary artery disease in
low- or high-risk subjects defined by conventional risk factors J Am Coll
Cardiol 4214291437 | Article | PubMed | ISI | ChemPort |
98. Terashima M et al 1999 Stromelysin promoter 5A/6A polymorphism is associated with acute
myocardial infarctionCirculation9927172719 | PubMed | ISI | ChemPort |
99. Rauramaa R et al 2000 Stromelysin-1 and interleukin-6 gene promoter polymorphisms are
determinants of asymptomatic carotid artery atherosclerosis Arterioscler Thromb Vasc Biol 2026572662 | PubMed | ISI | ChemPort |
100. Margaglione M et al 1998 The PAI-1 gene locus 4G/5G polymorphism is associated
with a family history of coronary artery disease Arterioscler Thromb Vasc
Biol 18152156 | PubMed | ISI | ChemPort |
101. Gretarsdottir S et al 2003 The gene encoding phosphodiesterase 4D confers risk of
ischemic strokeNat Genet 35131138 | Article | PubMed | ISI |
102. Zee RY et al 2006 Polymorphisms of the phosphodiesterase 4D, cAMP-specific
(PDE4D) gene and risk of ischemic stroke: a prospective, nested case-controlevaluationStroke3720122017 | Article | PubMed | ISI | ChemPort |
103. Brophy VH et al 2006 Association of phosphodiesterase 4D polymorphisms withischemic stroke in a US population stratified by hypertensionstatusStroke3713851390 | Article | PubMed | ISI | ChemPort |
104. Wang X et al 2005 Positional identification of TNFSF4, encoding OX40 ligand, as a
gene that influences atherosclerosis susceptibilityNat
Genet 37365372 | Article | PubMed | ISI | ChemPort |
105. Helgadottir A et al 2006 A variant of the gene encoding leukotriene A4 hydrolase
confers ethnicity-specific risk of myocardial infarctionNat
Genet 386874 | Article | PubMed | ISI | ChemPort |
106. Komulainen K et al 2006 Risk alleles of USF1 gene predict cardiovascular disease of
women in two prospective studiesPLoS Genet 2e69 | Article | PubMed | ChemPort |
107. Spielman RS et al 1993 Transmission test for linkage disequilibrium: the insulin
gene region and insulin-dependent diabetes mellitus (IDDM) Am J HumGenet 52506516 | PubMed | ISI | ChemPort |
Related Documents











