The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Sciences THE EFFECTS OF VITAMIN A DEFICIENCY ON HOST DEFENSE DURING CITROBACTER RODENTIUM INFECTION A Dissertation in Pathobiology by Kaitlin L. McDaniel © 2015 Kaitlin L. McDaniel Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2015

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Pennsylvania State University
The Graduate School
Department of Veterinary and Biomedical Sciences
THE EFFECTS OF VITAMIN A DEFICIENCY ON HOST DEFENSE
DURING CITROBACTER RODENTIUM INFECTION
A Dissertation in
Pathobiology
by
Kaitlin L. McDaniel
© 2015 Kaitlin L. McDaniel
Submitted in Partial Fulfillment
of the Requirements for the Degree of
Doctor of Philosophy
December 2015

ii
The dissertation of Kaitlin L. McDaniel was reviewed and approved* by the following:
Margherita T. Cantorna Distinguished Professor of Nutrition and Immunology Dissertation Advisor Co-Chair of Committee
A. Catharine Ross Huck Chair and Professor of Nutrition Co-Chair of Committee
Na Xiong Associate Professor of Veterinary Science and Biomedical Sciences
Mary K. Kennett Professor of Veterinary Science and Biomedical Sciences
Connie J. Rogers Assistant Professor of Nutritional Science Anthony Schmitt Associate Professor of Molecular Virology Director, Pathobiology Graduate Program
*Signatures are on file in the Graduate School

iii
ABSTRACT
Vitamin A deficiency is still a prevalent problem in resource limited countries, affecting
more that 250 million preschool aged children. Vitamin A deficiency is associated with
increased severity of respiratory and diarrheal diseases, a major cause of childhood
mortality. Vitamin A supplementation is associated with increased vitamin A status and
decreased disease severity, indicating a link between vitamin A status and immune
function. Using the murine enteric pathogen, Citrobacter rodentium, I explored the
relationship between host vitamin A status, intestinal infection and immune response.
The first objective was to determine the effect of vitamin A deficiency on C. rodentium
infection. My study showed that vitamin A deficient (A-) hosts did not clear the infection
and 40% of the A- animals developed a lethal infection. Additionally, retinoic acid
treatment of A- hosts promoted infection clearance and survival. The findings from this
study suggest that A- hosts may harbor enteric pathogens but retinoic acid treatment was
effective during C. rodentium infection. The second objective was to determine how
retinoic acid promotes C. rodentium clearance in the A- host. My data shows that 1 dose
of retinoic acid in an A- host was effective at clearing infection. Two doses of retinoic
acid treatment promoted il17a expression in the colon of A- mice yet, retinoic acid
treatment of splenocytes in vitro inhibited IL-17 secretion. Vitamin A host status was
found to have no effect on the microbiota and B cell numbers in the gut, indicating that
other acquired immune cells may be targets of RA. To understand the effects of retinoid
signaling on T cells, T-dnRAR mice were generated. T-dnRAR mice had significantly
more CD8+ thymocytes but significantly fewer CD8+ T cells in the spleen and
mesenteric lymph node. Together, the work presented in this dissertation indicates that

iv
vitamin A deficient hosts may be asymptomatic carriers of enteric infections and that
vitamin A treatment during infection promotes a local IL-17a response that is associated
with clearance of C. rodentium.

v
TABLE OF CONTENTS
List of Figures……………………………………………………………………….
vi
List of Tables……………………………………………………………………..… Abbreviations……………………………………………………………………….
viii xi
Acknowledgements………………………………………………………………….
xii
Chapter 1: Introduction……………………………………………………………. Chapter 1 References……………………………………………………………….
1 17
Chapter 2: Vitamin A deficient hosts become non-symptomatic reservoirs of Escherichia coli-like enteric infections……………………………………………. Chapter 2 References……………………………………………………………….
27 45
Chapter 3: Short term retinoic acid treatment promote mucosal IL-17 expression leading to clearance of enteric infection……………………………… Chapter 3 References……………………………………………………………….
57 76
Chapter 4: Summary and conclusions …………………………………………… Chapter 4 References………………………………………………………………
91 97

vi
LIST OF FIGURES Figure 1-1: Vitamin A metabolism………………………………………………
26
Figure 2-1: A- mice are more susceptible to C. rodentium infection than A+ mice…………………………………………………………………………
47
Figure 2-2: Quantification of whole body imaging during C. rodentium infection…………………………………………………………………………..
48
Figure 2-3: T cells in the IEL of the colon from A+ and A- mice before and after infection…………………………………………………………………….
49
Figure 2-4: Colon histology from C. rodentium infected animals……………...
50
Figure 2-5: RA treatment of A- mice eliminates C. rodentium infection……..
51
Figure 2-6: Systemic C. rodentium in tissues…………………………………..
52
Figure 2-7: Systemic spread of C. rodentium following GI infection…………
53
Figure 2-8: The effect of vitamin A status on LPS response and i.v challenge with C. rodentium…………………………………………………………………
55
Figure 2-9: Natural transmission of C. rodentium is not affected by the host vitamin A status. ………………………………………………………………..
56
Figure 3-1: RA treatment starting at d7 results in similar clearance when RA is given at d0………………………………………………………………………
79
Figure 3-2: Short term RA treatment results in increased il17a mRNA…….. 80
Figure 3-3: The effects of RA treatment on mRNA expression in duodenum, ileum and colon at d11 post-infection and 2 doses of RA ……………………
82
Figure 3-4: The microbiota is not affected by host VA status……………….. 83
Figure 3-5: RA inhibition of IFN-γ and IL-17 in A- whole splenocytes and CD4+ T cell in vitro………………………………………………………………..
84
Figure 3-6: Serum antibody titers and GI B cell numbers are not affected by host vitamin A status …………………………………………………………...
85
Figure 3-7: RA treatment in vitro does not affect T-dnRAR splenocytes response to stimulation ………………………………………………………...
86

vii
Figure 3-8: RA signaling inhibition in T cells leads to increased CD8+ T cells in the thymus and decreased CD8+ T cells in the periphery…………………...
88
Figure 3-9: Gating strategy for flow cytometry analysis ………………….…... 89
Figure 5-1: Retinoic acid treatment during enteric infection promotes protective mucosal immune responses in the vitamin A deficient host……....
95

viii
LIST OF TABLES Table 2-1: Serum cytokines in A+ and A- mice………………………………...
54
Table 3-1: RT-PCR primer sequences…………………………………………..
78
Table 3-2: RA signaling inhibition in T cells leads to immune cell population imbalance in the periphery…………………………………………...…………
83

ix
ABBREVIATIONS
A-: vitamin A deficient hosts
A+: vitamin A sufficient hosts
CCL: chemokine ligand
CCR: CC-chemokine receptor
CD: cluster of differentiation
CFU: colony forming units
CTLA-4: cytotoxic T lymphocyte antigen 4
DBD: DNA binding domain
DC: dendritic cell
DN: double negative
dn-RAR: dominant negative retinoic acid receptor
DP: double positive
EtOH: ethanol
FOXP3: Forkhead box P3
GALT: gut associated lymphoid tissues
GI: gastrointestinal tract
IEL: intraepithelial lymphocytes
IFN: interferon
Ig: immunoglobulin
IL: interleukin
ip: intra-peritoneal

x
iv: intra-venous
KO: knock out
LBD: ligand binding domain
LCK: lymphocyte specific protein tyrosine kinase
LPL: lamina propria lymphocytes
LPS: lipopolysaccharide
MAdCAM-1: mucosal addressin cell adhesion molecule-1
MLN: mesenteric lymph nodes
NK: natural killer
NKT: natural killer T
NTD: NH2 terminal domain
PP: Peyer’s patches
RA: retinoic acid
RAR: retinoic acid receptor
RARE: retinoic acid response elements
RBP: retinol binding protein
ROR-γt: RAR orphan receptor γt
RXR: retinoid X receptor
SI: small intestine
STRA6: stimulated by retinoic acid gene 6
TCR: T cell receptor
T-dnRAR: T cell specific dn-RAR
TFG: transforming growth factor

xi
Th: T helper
TNF: tumor necrosis factor
Treg: regulatory T cell
TTR: transthyretin
VA: vitamin A
VAD: vitamin A deficiency
WT: wild type

xii
ACKNOWLEDGMENTS
First, I would like to thank my advisor, Dr. Margherita Cantorna for her help, guidance
and patience during my time at Penn State. Dr. Cantorna has given me invaluable advice
and life lessons that I will carry with me throughout my career. I am grateful for my
committee members: Dr. Catharine Ross, Dr. Na Xiong, Dr. Mary Kennett and Dr.
Connie Rogers for sharing their knowledge and their valuable support through my
studies. A special thanks to Dr. Rogers for her constant encouragement and mentoring
throughout my graduate school experience.
I would like to thank my lab members, Jamaal James and Yang-ding Lin for their support
and advice. Additionally, I have been fortunate enough to have three wonderful friends
in the lab. Veronika Weaver, Stephanie Bora and Lindsay Snyder have helped me with
countless experiments and have been friends in and out of lab. They truly made coming
to work in the lab enjoyable and helped me keep my sanity. I don’t know how I would
have survived graduate school without them. I would like to thank the past and present
members of the Cantorna lab, the Ross lab, the Veterinary and Biomedical Science
program, the members of the Flow Cytometry Core and the office staff for all of their
help during my time at Penn State. A special thanks to Dr. Ruth Nissly for her
encouragement and for letting me know that everything will work out in the end.
My family and friends deserved the largest and deepest thanks. I have been lucky
enough to have three families on my graduate school journey: my family in Wyoming,

xiii
my farm family on Rocky Top, and my CrossFit Nittany family. Not many people are
lucky enough to have experienced the love and fun that I have being so far from home.
My Wyoming family (Dad, Mom, and sister) always supported me no matter what, even
my insane decision to move across the country. They instilled qualities in me that have
helped me persevere to the end of graduate school. They have made me who I am today.
My Rocky Top family made me feel at home in a new place and I am forever grateful for
that. My CrossFit family gave me a place to be myself and learn and grow in ways I
didn’t know possible. I am a stronger, better person because of CrossFit Nittany and they
have saved my life. From the bottom of my heart, thank you to everybody who helped
me get to this point.

1
Chapter 1
Introduction

2
Vitamin A: Sources and metabolism
Vitamin A (VA) is a fat-soluble essential micronutrient that humans obtain
through their diet. VA is absorbed from the diet as previtamin A retinyl esters present
in dairy products and liver and provitamin A carotenoids (from leafy green vegetables
and bright colored fruits) (1). The most common form of dietary vitamin A is β-
carotene which is found in vegetables and fruits such as sweet potatoes, kale, carrots
and cantaloupe. Proper vitamin A intake is associated with eye health, cell
development and immune system health, making it an important nutrient.
Dietary vitamin A needs to be converted into an active form so that the body
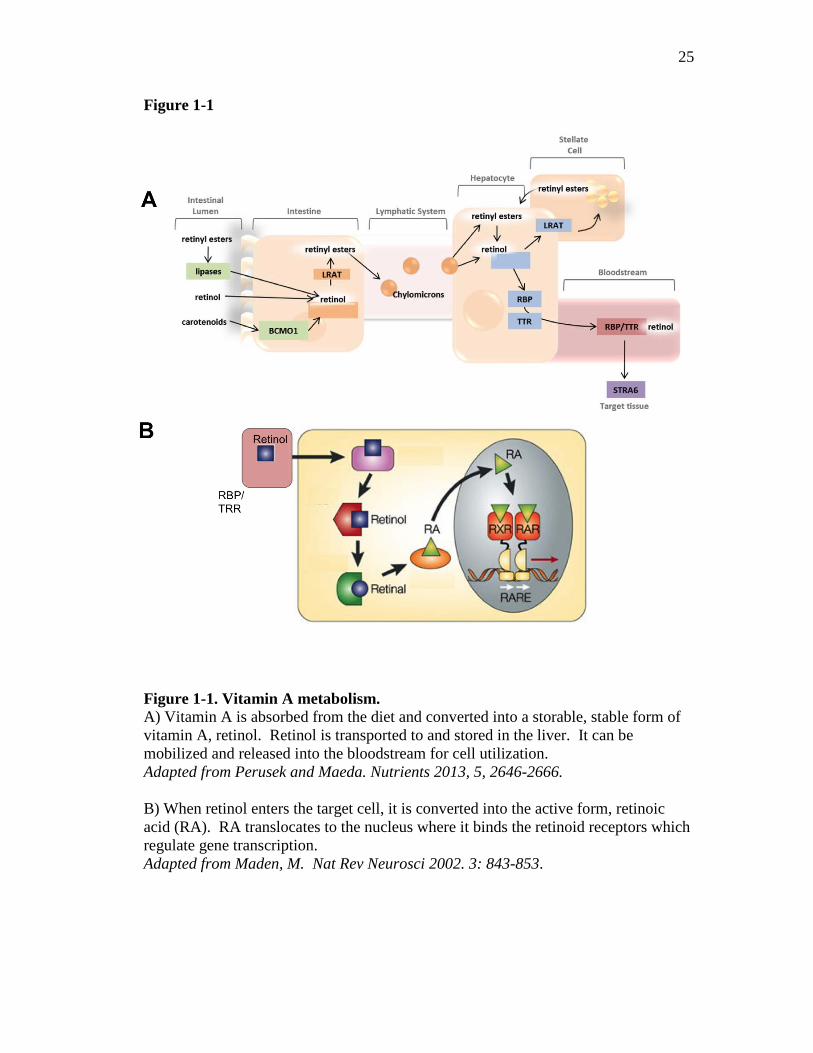
can use it. The metabolism of vitamin A has been summarized in Figure 1-1.
Briefly, once vitamin A has been absorbed into intestinal epithelial cells, the
associated proteins and fatty acids are removed. This frees the retinyl esters and
carotenoids so they can be processed by enzymes in the epithelial cells which convert
retinyl esters and carotenoids to retinol (2). Retinol becomes esterified into retinyl
palmitate and incorporated into chylomicrons where it enters the lymphatic system
and travels to the liver (3). The liver is the major storage organ of vitamin A and also
produces retinol binding protein (RBP) (4). Once the chylomicron enters the liver,
the retinyl palimitate is hydrolyzed into retinol where it is stored in the liver stellate
cells (5, 6). Retinol can associate with RBP which binds with transthyretin (TTR)
forming a traveling complex in the serum that can be transported to extrahepatic
tissues (1, 7). The RBP:TTR complex delivers retinol to the plasma membrane
receptor, STRA6 (stimulated by retinoic acid gene 6), on target cells. The binding to
the transport complex to STRA6 releases retinol from its binding proteins and retinol

3
enters the cell where it is converted to retinoic acid (RA) (Fig. 1-1A and 1-1B).
Dietary vitamin A must be converted into RA for the use of cells.
RA is the active metabolite of vitamin A. The effects of RA are exerted when
RA binds the retinoic acid receptor (RAR), which is found inside the nucleus (8).
The RAR is a member of the superfamily of nuclear receptors and has three isoforms,
RAR α, β and γ (8, 9). RARs are expressed in every cell of the body, but RARα is the
most common form (10). The RARs are divided into three domains that are
necessary for its function, including the COOH-terminal ligand binding domain
(LBD) which binds RA, the central DNA binding domain (DBD) which binds to
DNA sequences and the NH2 terminal domain (NTD) whose mechanism of action is
yet to be determined (11-13). The RAR heterodimerizes with the retinoid X receptor
(RXR) to form a complex that, in the presence of ligand, binds to retinoic acid
response elements (RARE) in the promoter regions of RA-inducible genes (11) (Fig.
1-1B). All-trans retinoic acid binds to the RAR, inducing conformational changes in
the ligand-binding domain which releases co-repressor proteins and recruits activator
proteins. The combination of RA, RAR:RXR heterodimer and activator proteins
make up a complex that induces gene transcription (11, 12). RAR signaling can also
inhibit gene transcription, but the mechanisms of inhibition are largely unknown. RA
largely regulates gene expression through nuclear receptor binding and co-activator
protein recruitment to gene promoter sites.
Vitamin A Deficiency
Vitamin A deficiency (14) remains a public health issue in resource-limited

4
countries, such as India and most of Africa (15). VAD is a critical concern in young
children and pregnant women (16, 17). It is estimated that over 20% of preschool
aged children are clinically vitamin A deficient (15, 17). Vitamin A deficiency in
infants occurs during breastfeeding because of the vitamin A deficient status of the
mothers. VAD causes xerophthalmia (night blindness) and is associated with
increased severity of infectious disease (18, 19). Vitamin A supplementation resulted
in a 26-30% reduction of mortality from respiratory and diarrheal diseases by
reducing disease severity in populations studied in Africa and India (18, 20). The
World Health Organization (WHO) reports vitamin A supplementation reduced
measles related mortality by 50% and diarrheal related mortality by 33% (21).
Additionally, studies showed that high dose vitamin A supplementation reduced
childhood mortality and morbidity in children without VAD (14, 22). Based on these
studies the WHO recommends that preschool aged children in counties where VAD is
common receive vitamin A supplementation (11, 15). The collective reports indicate
that there is a strong link between vitamin A status and resistance to infectious
disease.
Retinoic acid regulation of peripheral immune responses
The immune system is a network of cells, tissues and systems that protect the
host from disease. This defense system is composed of innate immune defenses
(barrier defenses and mechanisms that distinguish self from non-self) and adaptive
immune defenses (mechanisms that have immunological memory). The innate
immune response occurs quickly and initiates the adaptive immune response which

5
takes over once the initial innate response ends. Fully functional innate and adaptive
immunity are required for the host to fight infections.
Retinoic acid is a potent immune cell regulator. It has been shown to affect
both the innate and adaptive branches of the immune system. The effects of RA on
the innate immune system have been reviewed in detail elsewhere (23). The focus of
this dissertation is to elaborate on the effects of RA on the adaptive immune system,
mainly T cells.
T cells are a major part of the adaptive immune system. These cells mature in
the thymus and express T cell receptors, CD3 and either CD4 or CD8 on their
surface. CD4+ T cells are T helper (Th) cells that mediate immune responses
primarily though secretion of cytokines which regulate other immune cells such as B
cells and macrophages (24). Th cells are able to respond to a wide variety of
pathogens, regulate immune responses to control the magnitude of the immune
system’s response and protect against autoimmunity (24). Th cells are further
categorized based on the cytokines they secrete. Th1 cells are regulated by the
transcription factor, T-bet, and are potent cytokine producers, particularly interferon
(IFN) -γ that aids in the response to intracellular pathogens (25). Th2 cells secrete
interleukin (IL) -4, IL-5 and IL-10 and are regulated by the transcription factor,
GATA-3 (26). Th2 cells are important for host responses to helminthes and promote
B cell antibody production, the other half of the adaptive immune system (25). IFN-γ
secreted from Th1 cells inhibits Th2 cell function and IL-4 produced from Th2 cells
inhibit Th1 functions, indicating that Th1 and Th2 cells antagonize each other (27).

6
Another type of CD4+ T cells are Th17 cells. Th17 cells produce IL-17A,
IL17-F, IL-21 and IL-22 to control bacterial infections (28). Th17 cells are induced
in the presence of transforming growth factor (TFG)-β, IL-6 and IL-23 (29) and are
important for mucosal barrier function (24, 26, 28, 30). These cells are regulated by
the transcription factor, ROR-γt. The opposing cell lineage is regulatory T cells
(Tregs). Tregs express the transcription factor, FoxP3, and suppress the functions of
target T cells to inhibit immune responses (27). Naïve CD4+ T cells express both
FOXP3 and ROR-γt and when RA, TFG-β and low concentrations of IL-6 are
present, FOXP3 will inhibit Th17 cell development by inhibiting ROR-γt expression
(31, 32). Humans and mice with mutations in the foxp3 gene have severe immune
impairments indicating that Treg cells are vital to maintaining the balance between
inflammatory and anti-inflammatory responses (24, 25). Treg cells secrete anti-
inflammatory cytokines such as IL-10 and TGF-β to inhibit effector cell function
(27). They also express the surface marker CTLA-4 (cytotoxic T lymphocyte antigen
4) which competes for co-stimulatory receptors on effector cells (25). When CTLA-4
binds to effector cells, an inhibitory signal is sent to the effector cell and
inflammatory responses recede (25) . Additionally, IFN-γ and IL-17 has been
implicated in autoimmune disease pathology while IL-4 and IL-10 have been
associated with inflammation resolution (22). Maintaining the balance between
Th1/Th17 and Th2/Treg cells is required for inflammatory homeostasis in the host.
The balance between inflammatory and regulatory responses is mediated in
part by RA. RA promoted Th2 responses (IL-4, IL-5, IL-10) and inhibited Th1
responses (IFN- γ) in vitro and in vivo (33, 34). T cells treated in vitro with RA

7
produced less IFN-γ (34) while T cells from A- mice overproduced IFN-γ (35) and
under-produced regulatory cytokines (IL-4, IL-10) (36). Additionally, RA treatment
of A- T cells resulted in decreased IFN-γ transcription (36). However, a recent study
showed that RA signaling was needed for T-bet expression in memory CD4+ T cells
(37). Expression of a dominant negative RAR (dnRAR) in T cells (i.e. inhibition of
RA signaling in T cells) caused T cells to express high levels of IL-17 and express
both IL-17 and IFN-γ (37). Additionally, when dnRAR T cells were transferred into
Rag knockout (Rag KO) mice (mice that do not have T or B cells), the RagKO mice
that received the dnRAR T cells developed more severe colitis due to the dnRAR T
cells overproducing IL-17 and IL-17/IFN-γ (37). This new report is consistent with
RA inhibiting IL-17 and IFN-γ. Overall, the literature suggests a regulatory role for
RA in maintaining a balance between inflammatory and regulatory cytokine
production.
In addition to maintaining cytokine balance, RA can regulate the balance
between Th17 and Treg cells. RA, in combination with TGF-β, inhibited Th17 cell
induction from naïve CD4+ T cells in vitro and in vivo while promoting FoxP3
expression and IL-10 production from Treg cells (38, 39). Furthermore, RA inhibited
IL-6 mediated Th17 conversion, further promoting FoxP3+ Treg cell differentiation
(40). RA regulation of FoxP3 expression is largely mediated through RAR/RXR
binding at the FoxP3 promoter region (41). Maintaining balance between Th17 and
Treg cell responses is critical for ensuring that the immune system responds correctly
to stimuli and RA is a mediator of that balance (42-44). The literature suggests that
RA is an important mediator of immune response balance.

8
The mucosal immune system
The intestine is a complex environment that is constantly exposed to dietary
and bacterial antigens. The local immune system must distinguish between
commensals and pathogens by reacting effectively against pathogens but benignly to
commensals. The gut epithelial cells, microbiota and innate and adaptive immune
systems must work in conjunction to protect the host from breaches in the
gastrointestinal barrier.
The physical barrier created in the gut is the absolute first line of host defense
in the intestine. A thick layer of mucus secreted by goblet cells (specialized epithelial
cells) separates the microbiota and gut lumen from the host epithelial cells. Some
epithelial cells produce anti-microbial peptides, further bolstering the physical
defense between host and microbe. Furthermore, innate and innate-like immune cells
secrete cytokines that promote gut epithelial cell production of anti-microbial
peptides. Maintaining the physical barrier between host and intestinal lumen is vital
for host survival.
The immune compartments associated with intestinal immunity have been
termed gut associated lymphoid tissue (GALT). T cells in the gut reside in the
mesenteric lymph nodes (MLN), Peyers patches (45), and within the intraepithelial
lymphocyte (IEL) and lamina propria lymphocyte (LPL) compartments of the small
intestine (SI) and colon (46). Additionally, other immune cells such as dendritic
cells reside in the GALT. Dendritic cells (DCs) in the MLN, PP and LPL bridge the
gap between innate and adaptive immunity by initiating T cell responses (47). The
DC presents antigen to the T cell (47). In addition, the DC also provides co-

9
stimulation and cytokine signals that regulate T cell development into the effector T
cell types (47). The relationship between T cells and DCs is an important mediator of
immunity in the gut because these cells within the GALT are the first immune cells to
interact with pathogens that breach the host through the breaching the intestinal
epithelial barrier (48). The adaptive immune cells contained within these GALT
compartments are essential for maintaining a healthy host.
Retinoic acid and immune cell trafficking
One of the major functions of RA is the promotion of immune cell trafficking
to the mucosa. In a pivotal study, Iwata and colleagues determined that RA treatment
of T cells induced expression of the gut homing receptors, CCR9 and α4β7, on CD4+
T cells (44). CCR9 interacts with CCL25 on intestinal epithelial cells while α4β7
mediates cell trafficking to the intestinal endothelium though interactions with
mucosal addressin cell adhesion molecule-1 (MAdCAM-1) (11). Additionally, RA
induced CCR9 and α4β7 on other immune cells (49, 50). In support of the
observation that RA is important for immune homing to the gut, multiple groups have
reported that A- animals had fewer effector T cells in the gut and decreased gut
homing receptor expression (49, 51-53). Surprisingly, RA signaling was not needed
for homing since dnRAR T cells homed effectively to the gut of RagKO mice (37).
RA is an important metabolite for host defense.

10
Retinoic acid and barrier function
RA affects gut barrier function by supporting intestinal barrier integrity. VAD
in humans was associated with increased gut permeability (54) and vitamin A
supplementation was associated with improved gut integrity (55-57). Additionally,
studies done in animals models have shown that A- status resulted in increased gut
permeability and decreased goblet cell function (58-63). RA is important for
maintaining epithelial barrier in the GI tract.
When the epithelial barrier is breached, RA can promote epithelial repair.
During colitis, RA was bound to RAR/RXR and the IL-22 promoter inducing IL-22
production in innate lymphoid cells and γδ T cells (64). IL-22 is an important
cytokine for barrier function that largely targets epithelial cell to promotes tissue
regeneration (65) and is required for survival during enteric infection (66). These
studies suggest that RA plays a role in gut epithelial repair.
Once the epithelial barrier has been breached, immune cells respond. GALT
DCs are one of the first responders. These cells express CD103+ and have been
shown to express RA producing enzymes unlike their splenic counterparts (51, 67).
The RA produced by these DCs induced expression of the gut homing receptors,
CCR9 and α4β7, on T cells (51, 68, 69). Local production of RA, in conjunction with
the presence of TFG-β resulted in an environment that promoted Treg cell
development and IL-10 secretion (67, 70). Furthermore, DCs isolated from the
GALT of A- mice promoted the differentiation of Th17 cells in vitro (71). The SI has
a relatively high concentration of RA as determined by liquid chromatography–mass

11
spectrometry (11, 72). These findings support a role for RA in the promotion of
barrier functions.
Retinoic acid and the balance of mucosal Th17 and Treg cells
The balance between Th17 and Treg cells is highly important in the gut
because both are needed for host defense. If the Th17 response dominates over the
Treg response, excess inflammation and damage to the host can occur. However if
there is too much inhibition of inflammation, then pathogens can escape the gut and
become systemic, leading to sepsis. RA plays a role in maintaining the balance
between Th17 and Treg cells.
The effects of RA on Th17 cells are still not completely understood. Many in
vitro studies have shown that RA inhibited Th17 cell induction from naïve CD4+ T
cells (reviewed in (8)). Concentrations used in cell culture studies range from 10nM
to 1uM and result in suppressed Th17 cell induction and reduced IFN-γ and IL-17
secretion in vitro (11, 73, 74). Paradoxically, multiple groups have shown that A-
mice have almost no Th17 cells (73, 75, 76). In support of this observation, two
studies reported that 1-30nM RA in vitro is needed for IL-17 production from T cells
(73, 74) and upregulation of CCR9 and α4β7 expression on IL-17 producing T cells
(73), indicating that these concentrations of RA may promote Th17 cell induction.
Additionally, species of the gut microbiota can induce Th17 cell generation,
indicating that the intestinal environment may be a factor regarding the induction of
Th17 cells (77, 78). While there is no evidence that shows that RA directly affects
the microbiota (as bacteria do not have RARs or RXRs), the combination of local RA

12
in low concentrations (<30nM) and the presence of the microbiota may result is
different regulation of effector T cell differentiation in vivo. These data indicate that
the local environment and the concentration of RA may have different effects on
Th17 cells in vitro and in vivo.
Genetic mouse models have led to insight as to how RA signaling regulates
Th17 and Treg cells. Reports showed that Th17 cell differentiation is impaired in
RARα-deficient and A- CD4+ T cells, indicating that RA is needed for the
development of Th17 cells (75, 76). Conversely, when T cells expressed the dnRAR
(inhibited RA signaling in only T cells), they had a propensity to secrete more IL-17
than IFN-γ (37). dnRAR CD4+ T cells trafficked to the gut and caused more
intestinal inflammation when transferred into RagKO mice compared to WT cells
(37). More work is needed to fully understand how RA-RAR signaling affect Th17
cells in vivo.
T reg cell are critical to dampen the immune response so that the host is not
damaged by overactive inflammation. Treg cells have suppressive function, such as
inhibition of effector cell proliferation, secretion of regulatory cytokines and the
ability to traffic to the gut through CCR9 and α4β7 (79). CD103+ DCs can promote
the conversion of naïve CD4+ T cells to Treg cells in the gut (67). RA induced Tregs
are more suppressive during antigen specific CD8+ T cell driven acute intestinal
inflammation than Tregs induced without RA (80). However suppression following
CD4+ T cell transfer was no different between Treg cells induced with or without RA
in vitro (80). Interestingly, FoxP3+ T cell frequencies were not different in WT or
dnRAR T cells (37). Overall, RA promotes Treg cell induction in vitro however, the

13
role of RA in the promotion of Treg cell differentiation in vivo is less clear.
Currently, the effects of RA and RA signaling on Th17 and Treg cells are not
completely understood.
Vitamin A and disease outcomes
Due to its immune-regulatory functions, vitamin A is sometimes termed “the
anti-infective vitamin” (81, 82). In humans, VA supplementation was associated with
improved recovery from a wide variety of diseases, including malaria and measles
(83). Many studies looking at the interaction between VAD and human infections
have shown that VAD was associated with exacerbated disease (84). In HIV-1
infected humans, vitamin A supplementation decreased vertical transmission of HIV-
1 from mother to child (83, 85, 86) and decreased HIV-1 associated mortality in
children (83) (87). High dose vitamin A supplementation promoted recovery from
measles and decreased severity of malaria infection in young children (83, 88).
However, the effects of vitamin A supplementation in respiratory diseases were
conflicting as to whether vitamin A supplementation was beneficial, detrimental or
not different (83). Laboratory models of vitamin A deficiency indicated that multiple
immune responses were impaired in A- hosts. A- rodents had impaired antibody
responses and more Th1 cells and fewer Th2 cells (89-92). Conversely, A- mice had
decreased Th1 and Th17 responses following infection with Toxoplasma gondii (75).
RA treatment in vivo restored IFN-y production in A- T. gondii infected mice (75).
RA treatment in vitro was able to inhibit effector and memory T cell cytokine
production (93). The data suggest a role for vitamin A in host resistance to infection.

14
Vitamin A and gastrointestinal inflammation
Gastrointestinal infections are also regulated by vitamin A. A- mice
developed a more severe enterohemorrhagic E. coli infection compared to A+ mice
(94). Furthermore, RA treatment reduced colonic inflammation caused by dextran
sodium sulfate (DSS) and Citrobacter rodentium infection (64). RA treatment during
chemical and bacterial inflammation inhibited production of IL-17 and IFN-γ (64)
(76). In addition to inhibiting inflammatory cytokine production, RA treatment
increased anti-microbial peptide production and IL-22 production which
corresponded with decreased inflammation (64, 76). During C. rodentium infection,
A+ mice cleared the infection more quickly than A- mice (95, 96). VA
supplementation shortened the duration of enterotoxigenic E. coli infection in
children (97). These data suggest that vitamin A is a critical factor in limiting
intestinal inflammation.
Citrobacter rodentium: disease and pathogenesis
C. rodentium is a mouse pathogen that models human infections with
enteropathogenic Escherichia coli by causing attaching and effacing lesions of the
cecum and colon in mice (98). C. rodentium is transmitted via the fecal-oral route (as
mice are known to be coprophagic) and causes inflammation of the colon. The
severity of C. rodentium infection can range from a self-limiting colitis to severe
inflammation and death, usually due to dehydration (99-102). This mouse model of
infection is widely used to study host-pathogen interactions in the gastrointestinal
tract.

15
C. rodentium infection with laboratory grown bacteria is a highly reproducible
infection model. Following colonization, C. rodentium becomes more virulent,
adapting to the host and becoming more infectious to naïve animals (102, 103). This
adaptation allows C. rodentium to colonize the colon and rectum. By day 3 post
infection, the bacteria moves to the colon (100). Fecal shedding of C. rodentium
peaks between day 7 and 14 post-infection. Crypt hyperplasia develops and effector
immune cell infiltration occurs during this time period as well. During the peak of
infection, mice can exhibit weight loss and diarrhea (98, 100). Resistant mouse
strains clear the infection by day 30 post infection, while susceptible strains succumb
to persistent infections without clearing the pathogen (98). C. rodentium is a useful
model for studying host/pathogen interactions and mucosal immunity.
Immunity to Citrobacter rodentium
The innate immune system is required for early protection as demonstrated in
RagKO mice lacking T and B cells; these mice were unable to clear the bacteria and
eventually succumbed to the infection (104). Macrophages and innate lymphoid cells
produce IL-22 early during infection which promoted epithelial repair (100, 105,
106). IL-22 KO mice developed fatal infections within the first two weeks of C.
rodentium infection (66). Furthermore, IL-22 production induced an essential
adaptive Th17 responses, leading to clearance of C. rodentium (100, 105).
T cells and B cells are essential for resolution of C. rodentium infection since
mice without T cells (CD4 KO) or B cells (Igμ KO) developed fatal infections (100).
Furthermore, T cell dependent antibody, Th1 and Th17 responses were required for

16
bacterial clearance (99) (107). IFN-γ KO and IL-17 KO mice succumbed to lethal C.
rodentium infections (38, 108, 109). Together, the innate and adaptive immune
responses are required for protection from C. rodentium.
Objectives
In this dissertation, the effects of host vitamin A status and RA
supplementation on host defense to a bacterial infection with C. rodentium was
investigated. Some reports have shown that RA inhibits IFN-γ and IL-17 production
in vitro and in vivo from T cells however, other studies indicate that RA is critical for
Th1 and Th17 cell responses in vivo. It was unknown how the vitamin A deficient
host would respond to a C. rodentium infection, which requires Th1/Th17 immune
responses for clearance. Based on experiments that used A- mice and RA dosing in
models of gut inflammation, we hypothesized that the A- mice might be resistant to
C. rodentium infection. In Chapter 2, we tested the susceptibility of A- mice to C.
rodentium infection. In Chapter 3, we determined some of the RA targets in the gut
mucosa that would mediate host resistance to C. rodentium infection. The last
Chapter summarizes the major findings and puts the work in the context of the
growing literature on the role of vitamin A in regulating mucosal T cells in the gut.

17
REFERENCES:
1. Ross, A. C., R. Zolfaghari, and J. Weisz. 2001. Vitamin A: recent advances in the biotransformation, transport, and metabolism of retinoids. Curr Opin Gastroenterol 17: 184-192.
2. Harrison, E. H. 2005. Mechanisms of digestion and absorption of dietary vitamin A. Annu Rev Nutr 25: 87-103.
3. Seino, Y., T. Miki, H. Kiyonari, T. Abe, W. Fujimoto, K. Kimura, A. Takeuchi, Y. Takahashi, Y. Oiso, T. Iwanaga, and S. Seino. 2008. Isx participates in the maintenance of vitamin A metabolism by regulation of beta-carotene 15,15'-monooxygenase (Bcmo1) expression. J Biol Chem 283: 4905-4911.
4. Kanai, M., A. Raz, and D. S. Goodman. 1968. Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest 47: 2025-2044.
5. Blomhoff, R., K. R. Norum, and T. Berg. 1985. Hepatic uptake of [3H]retinol bound to the serum retinol binding protein involves both parenchymal and perisinusoidal stellate cells. J Biol Chem 260: 13571-13575.
6. Randolph, R. K., and A. C. Ross. 1991. Vitamin A status regulates hepatic lecithin: retinol acyltransferase activity in rats. J Biol Chem 266: 16453-16457.
7. Raz, A., and D. S. Goodman. 1969. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J Biol Chem 244: 3230-3237.
8. Mora, J. R., M. Iwata, and U. H. von Andrian. 2008. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 8: 685-698.
9. Evans, R. M. 1988. The steroid and thyroid hormone receptor superfamily. Science 240: 889-895.
10. Dolle, P. 2009. Developmental expression of retinoic acid receptors (RARs). Nucl Recept Signal 7: e006.
11. Guo, Y., C. Brown, C. Ortiz, and R. J. Noelle. 2015. Leukocyte homing, fate, and function are controlled by retinoic acid. Physiol Rev 95: 125-148.
12. Chambon, P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940-954.
13. Rochette-Egly, C., and P. Germain. 2009. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl Recept Signal 7: e005.
14. Coutsoudis, A., M. Broughton, and H. M. Coovadia. 1991. Vitamin A supplementation reduces measles morbidity in young African children: a randomized, placebo-controlled, double-blind trial. Am J Clin Nutr 54: 890-895.
15. WHO. 2009. Global prevalence of vitamin A deficiency in populations at risk 1995–2005: WHO global database on
vitamin A deficiency. In World Health Organization, Geneva. 16. Van, D. E., R. Kulier, A. M. Gulmezoglu, and J. Villar. 2002. Vitamin A
supplementation during pregnancy. Cochrane Database Syst Rev: CD001996.

18
17. West, K. P., Jr. 2002. Extent of vitamin A deficiency among preschool children and women of reproductive age. J Nutr 132: 2857S-2866S.
18. Glasziou, P. P., and D. E. Mackerras. 1993. Vitamin A supplementation in infectious diseases: a meta-analysis. BMJ 306: 366-370.
19. Sherwin, J. C., M. H. Reacher, W. H. Dean, and J. Ngondi. 2012. Epidemiology of vitamin A deficiency and xerophthalmia in at-risk populations. Trans R Soc Trop Med Hyg 106: 205-214.
20. Daulaire, N. M., E. S. Starbuck, R. M. Houston, M. S. Church, T. A. Stukel, and M. R. Pandey. 1992. Childhood mortality after a high dose of vitamin A in a high risk population. BMJ 304: 207-210.
21. WHO/UNICEF. 1998. Integration of vitamin A supplementation with immunization: Policy and programme implications. Report of Meeting.
22. Hussey, G. D., and M. Klein. 1990. A randomized, controlled trial of vitamin A in children with severe measles. N Engl J Med 323: 160-164.
23. Pino-Lagos, K., Y. Guo, and R. J. Noelle. 2010. Retinoic acid: a key player in immunity. Biofactors 36: 430-436.
24. Zhu, J., H. Yamane, and W. E. Paul. 2010. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28: 445-489.
25. Kuby, T. J. K. R. A. G. B. A. O. J. 2007. Kuby Immunology, 6th ed. W.H. Freeman, New York.
26. Brucklacher-Waldert, V., E. J. Carr, M. A. Linterman, and M. Veldhoen. 2014. Cellular Plasticity of CD4+ T Cells in the Intestine. Front Immunol 5: 488.
27. Corthay, A. 2009. How do regulatory T cells work? Scand J Immunol 70: 326-336.
28. Littman, D. R., and A. Y. Rudensky. 2010. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140: 845-858.
29. Bettelli, E., Y. Carrier, W. Gao, T. Korn, T. B. Strom, M. Oukka, H. L. Weiner, and V. K. Kuchroo. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235-238.
30. Garrido-Mesa, N., F. Algieri, A. Rodriguez Nogales, and J. Galvez. 2013. Functional plasticity of Th17 cells: implications in gastrointestinal tract function. Int Rev Immunol 32: 493-510.
31. Kimura, A., and T. Kishimoto. 2010. IL-6: regulator of Treg/Th17 balance. Eur J Immunol 40: 1830-1835.
32. Zhou, L., J. E. Lopes, M. M. Chong, Ivanov, II, R. Min, G. D. Victora, Y. Shen, J. Du, Y. P. Rubtsov, A. Y. Rudensky, S. F. Ziegler, and D. R. Littman. 2008. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453: 236-240.
33. Iwata, M., Y. Eshima, and H. Kagechika. 2003. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol 15: 1017-1025.
34. Cantorna, M. T., F. E. Nashold, and C. E. Hayes. 1994. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol 152: 1515-1522.

19
35. Carman, J. A., S. M. Smith, and C. E. Hayes. 1989. Characterization of a helper T lymphocyte defect in vitamin A-deficient mice. J Immunol 142: 388-393.
36. Cantorna, M. T., F. E. Nashold, and C. E. Hayes. 1995. Vitamin A deficiency results in a priming environment conducive for Th1 cell development. Eur J Immunol 25: 1673-1679.
37. Brown, C. C., D. Esterhazy, A. Sarde, M. London, V. Pullabhatla, I. Osma-Garcia, R. Al-Bader, C. Ortiz, R. Elgueta, M. Arno, E. de Rinaldis, D. Mucida, G. M. Lord, and R. J. Noelle. 2015. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42: 499-511.
38. Mangan, P. R., L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231-234.
39. Elias, K. M., A. Laurence, T. S. Davidson, G. Stephens, Y. Kanno, E. M. Shevach, and J. J. O'Shea. 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111: 1013-1020.
40. Mucida, D., Y. Park, G. Kim, O. Turovskaya, I. Scott, M. Kronenberg, and H. Cheroutre. 2007. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317: 256-260.
41. Xu, L., A. Kitani, C. Stuelten, G. McGrady, I. Fuss, and W. Strober. 2010. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 33: 313-325.
42. Benson, M. J., K. Pino-Lagos, M. Rosemblatt, and R. J. Noelle. 2007. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med 204: 1765-1774.
43. Xiao, S., H. Jin, T. Korn, S. M. Liu, M. Oukka, B. Lim, and V. K. Kuchroo. 2008. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 181: 2277-2284.
44. Zhou, X., N. Kong, J. Wang, H. Fan, H. Zou, D. Horwitz, D. Brand, Z. Liu, and S. G. Zheng. 2010. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol 185: 2675-2679.
45. Goverse, G., B. J. Olivier, R. Molenaar, M. Knippenberg, M. Greuter, T. Konijn, E. C. Cook, M. R. Beijer, D. M. Fedor, J. M. den Haan, J. L. Napoli, G. Bouma, and R. E. Mebius. 2015. Vitamin A metabolism and mucosal immune function are distinct between BALB/c and C57BL/6 mice. Eur J Immunol 45: 89-100.
46. Mowat, A. M., and W. W. Agace. 2014. Regional specialization within the intestinal immune system. Nat Rev Immunol 14: 667-685.

20
47. Gross, M., T. M. Salame, and S. Jung. 2015. Guardians of the Gut - Murine Intestinal Macrophages and Dendritic Cells. Front Immunol 6: 254.
48. Meresse, B., G. Malamut, and N. Cerf-Bensussan. 2012. Celiac disease: an immunological jigsaw. Immunity 36: 907-919.
49. Mora, J. R., M. Iwata, B. Eksteen, S. Y. Song, T. Junt, B. Senman, K. L. Otipoby, A. Yokota, H. Takeuchi, P. Ricciardi-Castagnoli, K. Rajewsky, D. H. Adams, and U. H. von Andrian. 2006. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314: 1157-1160.
50. Kim, M. H., E. J. Taparowsky, and C. H. Kim. 2015. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 43: 107-119.
51. Iwata, M., A. Hirakiyama, Y. Eshima, H. Kagechika, C. Kato, and S. Y. Song. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21: 527-538.
52. Bjersing, J. L., E. Telemo, U. Dahlgren, and L. A. Hanson. 2002. Loss of ileal IgA+ plasma cells and of CD4+ lymphocytes in ileal Peyer's patches of vitamin A deficient rats. Clin Exp Immunol 130: 404-408.
53. McDermott, M. R., D. A. Mark, A. D. Befus, B. S. Baliga, R. M. Suskind, and J. Bienenstock. 1982. Impaired intestinal localization of mesenteric lymphoblasts associated with vitamin A deficiency and protein-calorie malnutrition. Immunology 45: 1-5.
54. Quadro, L., M. V. Gamble, S. Vogel, A. A. Lima, R. Piantedosi, S. R. Moore, V. Colantuoni, M. E. Gottesman, R. L. Guerrant, and W. S. Blaner. 2000. Retinol and retinol-binding protein: gut integrity and circulating immunoglobulins. J Infect Dis 182 Suppl 1: S97-S102.
55. Thurnham, D. I., C. A. Northrop-Clewes, F. S. McCullough, B. S. Das, and P. G. Lunn. 2000. Innate immunity, gut integrity, and vitamin A in Gambian and Indian infants. J Infect Dis 182 Suppl 1: S23-28.
56. Filteau, S. M., N. C. Rollins, A. Coutsoudis, K. R. Sullivan, J. F. Willumsen, and A. M. Tomkins. 2001. The effect of antenatal vitamin A and beta-carotene supplementation on gut integrity of infants of HIV-infected South African women. J Pediatr Gastroenterol Nutr 32: 464-470.
57. Baltes, S., H. Nau, and A. Lampen. 2004. All-trans retinoic acid enhances differentiation and influences permeability of intestinal Caco-2 cells under serum-free conditions. Dev Growth Differ 46: 503-514.
58. Holland, R. E., C. J. Pfeiffer, N. J. Bruns, and K. E. Webb, Jr. 1993. Morphologic alterations in small intestinal epithelium of lambs fed vitamin A-depleted diet. Dig Dis Sci 38: 333-343.
59. Uni, Z., G. Zaiger, and R. Reifen. 1998. Vitamin A deficiency induces morphometric changes and decreased functionality in chicken small intestine. Br J Nutr 80: 401-407.
60. Olson, J. A., W. Rojanapo, and A. J. Lamb. 1981. The effect of vitamin A status on the differentiation and function of goblet cells in the rat intestine. Ann N Y Acad Sci 359: 181-191.

21
61. Rojanapo, W., A. J. Lamb, and J. A. Olson. 1980. The prevalence, metabolism and migration of goblet cells in rat intestine following the induction of rapid, synchronous vitamin A deficiency. J Nutr 110: 178-188.
62. Wiedermann, U., L. A. Hanson, T. Bremell, H. Kahu, and U. I. Dahlgren. 1995. Increased translocation of Escherichia coli and development of arthritis in vitamin A-deficient rats. Infect Immun 63: 3062-3068.
63. Shoda, R., D. Mahalanabis, M. A. Wahed, and M. J. Albert. 1995. Bacterial translocation in the rat model of lectin induced diarrhoea. Gut 36: 379-381.
64. Mielke, L. A., S. A. Jones, M. Raverdeau, R. Higgs, A. Stefanska, J. R. Groom, A. Misiak, L. S. Dungan, C. E. Sutton, G. Streubel, A. P. Bracken, and K. H. Mills. 2013. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210: 1117-1124.
65. Dudakov, J. A., A. M. Hanash, and M. R. van den Brink. 2015. Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33: 747-785.
66. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong, A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. de Sauvage, and W. Ouyang. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14: 282-289.
67. Coombes, J. L., K. R. Siddiqui, C. V. Arancibia-Carcamo, J. Hall, C. M. Sun, Y. Belkaid, and F. Powrie. 2007. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204: 1757-1764.
68. Kang, S. G., J. Park, J. Y. Cho, B. Ulrich, and C. H. Kim. 2011. Complementary roles of retinoic acid and TGF-beta1 in coordinated expression of mucosal integrins by T cells. Mucosal Immunol 4: 66-82.
69. Svensson, M., B. Johansson-Lindbom, F. Zapata, E. Jaensson, L. M. Austenaa, R. Blomhoff, and W. W. Agace. 2008. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol 1: 38-48.
70. Bakdash, G., L. T. Vogelpoel, T. M. van Capel, M. L. Kapsenberg, and E. C. de Jong. 2015. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol 8: 265-278.
71. Cha, H. R., S. Y. Chang, J. H. Chang, J. O. Kim, J. Y. Yang, C. H. Kim, and M. N. Kweon. 2010. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J Immunol 184: 6799-6806.
72. Villablanca, E. J., S. Wang, J. de Calisto, D. C. Gomes, M. A. Kane, J. L. Napoli, W. S. Blaner, H. Kagechika, R. Blomhoff, M. Rosemblatt, M. R. Bono, U. H. von Andrian, and J. R. Mora. 2011. MyD88 and retinoic acid signaling pathways interact to modulate gastrointestinal activities of dendritic cells. Gastroenterology 141: 176-185.
73. Wang, C., S. G. Kang, H. HogenEsch, P. E. Love, and C. H. Kim. 2010. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol 184: 5519-5526.

22
74. Uematsu, S., K. Fujimoto, M. H. Jang, B. G. Yang, Y. J. Jung, M. Nishiyama, S. Sato, T. Tsujimura, M. Yamamoto, Y. Yokota, H. Kiyono, M. Miyasaka, K. J. Ishii, and S. Akira. 2008. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol 9: 769-776.
75. Hall, J. A., J. L. Cannons, J. R. Grainger, L. M. Dos Santos, T. W. Hand, S. Naik, E. A. Wohlfert, D. B. Chou, G. Oldenhove, M. Robinson, M. E. Grigg, R. Kastenmayer, P. L. Schwartzberg, and Y. Belkaid. 2011. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34: 435-447.
76. Spencer, S. P., C. Wilhelm, Q. Yang, J. A. Hall, N. Bouladoux, A. Boyd, T. B. Nutman, J. F. Urban, Jr., J. Wang, T. R. Ramalingam, A. Bhandoola, T. A. Wynn, and Y. Belkaid. 2014. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343: 432-437.
77. Ivanov, II, K. Atarashi, N. Manel, E. L. Brodie, T. Shima, U. Karaoz, D. Wei, K. C. Goldfarb, C. A. Santee, S. V. Lynch, T. Tanoue, A. Imaoka, K. Itoh, K. Takeda, Y. Umesaki, K. Honda, and D. R. Littman. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485-498.
78. Zaph, C., Y. Du, S. A. Saenz, M. G. Nair, J. G. Perrigoue, B. C. Taylor, A. E. Troy, D. E. Kobuley, R. A. Kastelein, D. J. Cua, Y. Yu, and D. Artis. 2008. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med 205: 2191-2198.
79. Kang, S. G., H. W. Lim, O. M. Andrisani, H. E. Broxmeyer, and C. H. Kim. 2007. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol 179: 3724-3733.
80. Menning, A., C. Loddenkemper, A. M. Westendorf, B. Szilagyi, J. Buer, C. Siewert, A. Hamann, and J. Huehn. 2010. Retinoic acid-induced gut tropism improves the protective capacity of Treg in acute but not in chronic gut inflammation. Eur J Immunol 40: 2539-2548.
81. Semba, R. D. 1999. Vitamin A as "anti-infective" therapy, 1920-1940. J Nutr 129: 783-791.
82. Green, H. N., and E. Mellanby. 1928. Vitamin a as an Anti-Infective Agent. Br Med J 2: 691-696.
83. Stephensen, C. B. 2001. Vitamin A, infection, and immune function. Annu Rev Nutr 21: 167-192.
84. Ross, A. C., and C. B. Stephensen. 1996. Vitamin A and retinoids in antiviral responses. FASEB J 10: 979-985.
85. Semba, R. D., N. M. Graham, W. T. Caiaffa, J. B. Margolick, L. Clement, and D. Vlahov. 1993. Increased mortality associated with vitamin A deficiency during human immunodeficiency virus type 1 infection. Arch Intern Med 153: 2149-2154.
86. Greenberg, B. L., R. D. Semba, P. E. Vink, J. J. Farley, M. Sivapalasingam, R. W. Steketee, D. M. Thea, and E. E. Schoenbaum. 1997. Vitamin A deficiency and maternal-infant transmissions of HIV in two metropolitan areas in the United States. AIDS 11: 325-332.

23
87. Fawzi, W. W., R. L. Mbise, E. Hertzmark, M. R. Fataki, M. G. Herrera, G. Ndossi, and D. Spiegelman. 1999. A randomized trial of vitamin A supplements in relation to mortality among human immunodeficiency virus-infected and uninfected children in Tanzania. Pediatr Infect Dis J 18: 127-133.
88. West, C. E. 2000. Vitamin A and measles. Nutr Rev 58: S46-54. 89. Carman, J. A., L. Pond, F. Nashold, D. L. Wassom, and C. E. Hayes. 1992.
Immunity to Trichinella spiralis infection in vitamin A-deficient mice. J Exp Med 175: 111-120.
90. Stephensen, C. B., Z. Moldoveanu, and N. N. Gangopadhyay. 1996. Vitamin A deficiency diminishes the salivary immunoglobulin A response and enhances the serum immunoglobulin G response to influenza A virus infection in BALB/c mice. J Nutr 126: 94-102.
91. Ross, A. C. 1996. Vitamin A deficiency and retinoid repletion regulate the antibody response to bacterial antigens and the maintenance of natural killer cells. Clin Immunol Immunopathol 80: S63-72.
92. Wiedermann, U., L. A. Hanson, J. Holmgren, H. Kahu, and U. I. Dahlgren. 1993. Impaired mucosal antibody response to cholera toxin in vitamin A-deficient rats immunized with oral cholera vaccine. Infect Immun 61: 3952-3957.
93. Hill, J. A., J. A. Hall, C. M. Sun, Q. Cai, N. Ghyselinck, P. Chambon, Y. Belkaid, D. Mathis, and C. Benoist. 2008. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 29: 758-770.
94. Cabrera, G., R. J. Fernandez-Brando, M. J. Abrey-Recalde, A. Baschkier, A. Pinto, J. Goldstein, E. Zotta, R. Meiss, M. Rivas, and M. S. Palermo. 2014. Retinoid levels influence enterohemorrhagic Escherichia coli infection and Shiga toxin 2 susceptibility in mice. Infect Immun 82: 3948-3957.
95. Restori, K. H., K. L. McDaniel, A. E. Wray, M. T. Cantorna, and A. C. Ross. 2014. Streptococcus pneumoniae-induced pneumonia and Citrobacter rodentium-induced gut infection differentially alter vitamin A concentrations in the lung and liver of mice. J Nutr 144: 392-398.
96. McDaniel, K. L., K. H. Restori, J. W. Dodds, M. J. Kennett, A. C. Ross, and M. T. Cantorna. 2015. Vitamin A-Deficient Hosts Become Nonsymptomatic Reservoirs of Escherichia coli-Like Enteric Infections. Infect Immun 83: 2984-2991.
97. Long, K. Z., J. I. Santos, J. L. Rosado, T. Estrada-Garcia, M. Haas, A. Al Mamun, H. L. DuPont, and N. N. Nanthakumar. 2011. Vitamin A supplementation modifies the association between mucosal innate and adaptive immune responses and resolution of enteric pathogen infections. Am J Clin Nutr 93: 578-585.
98. Collins, J. W., K. M. Keeney, V. F. Crepin, V. A. Rathinam, K. A. Fitzgerald, B. B. Finlay, and G. Frankel. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12: 612-623.
99. Higgins, L. M., G. Frankel, G. Douce, G. Dougan, and T. T. MacDonald. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine

24
response and lesions similar to those in murine inflammatory bowel disease. Infect Immun 67: 3031-3039.
100. Mundy, R., T. T. MacDonald, G. Dougan, G. Frankel, and S. Wiles. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7: 1697-1706.
101. Borenshtein, D., K. A. Schlieper, B. H. Rickman, J. M. Chapman, C. W. Schweinfest, J. G. Fox, and D. B. Schauer. 2009. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect Immun 77: 3639-3650.
102. Wiles, S., K. M. Pickard, K. Peng, T. T. MacDonald, and G. Frankel. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun 74: 5391-5396.
103. Wiles, S., S. Clare, J. Harker, A. Huett, D. Young, G. Dougan, and G. Frankel. 2004. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol 6: 963-972.
104. Bry, L., and M. B. Brenner. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172: 433-441.
105. Sonnenberg, G. F., and D. Artis. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37: 601-610.
106. Peterson, L. W., and D. Artis. 2014. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141-153.
107. Vallance, B. A., W. Deng, L. A. Knodler, and B. B. Finlay. 2002. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun 70: 2070-2081.
108. Geddes, K., S. J. Rubino, J. G. Magalhaes, C. Streutker, L. Le Bourhis, J. H. Cho, S. J. Robertson, C. J. Kim, R. Kaul, D. J. Philpott, and S. E. Girardin. 2011. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17: 837-844.
109. Simmons, C. P., N. S. Goncalves, M. Ghaem-Maghami, M. Bajaj-Elliott, S. Clare, B. Neves, G. Frankel, G. Dougan, and T. T. MacDonald. 2002. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol 168: 1804-1812.
25
Figure 1-1
Figure 1-1. Vitamin A metabolism. A) Vitamin A is absorbed from the diet and converted into a storable, stable form of vitamin A, retinol. Retinol is transported to and stored in the liver. It can be mobilized and released into the bloodstream for cell utilization. Adapted from Perusek and Maeda. Nutrients 2013, 5, 2646-2666. B) When retinol enters the target cell, it is converted into the active form, retinoic acid (RA). RA translocates to the nucleus where it binds the retinoid receptors which regulate gene transcription. Adapted from Maden, M. Nat Rev Neurosci 2002. 3: 843-853.

27
Chapter 2
Vitamin A deficient hosts become non-symptomatic reservoirs of Escherichia coli-like enteric infections.
Chapter adapted from the manuscript entitled:
“Vitamin A deficient hosts become non-symptomatic reservoirs of Escherichia coli-like
enteric infections.”
Authors: Kaitlin L. McDaniel, Katherine H. Restori, Jeffery W. Dodds, Mary J.
Kennett1, A. Catharine Ross and Margherita T. Cantorna
Infect Immun. 2015 Jul;83(7):2984-91

28
ABSTACT
Vitamin A deficiency (A-) remains a public health concern in developing
countries and is associated with increased susceptibility to infection. Citrobacter
rodentium was used to model human Escherichia coli infections. A- mice developed a
severe and lethal (40%) infection. Vitamin A sufficient (A+) mice survived and cleared
the infection by d25. Retinoic acid treatment of A- mice at the peak of the infection
eliminated C. rodentium within 16d. Inflammation was not different in A+ and A- colons
although the A- mice were still infected at d37. Increased mortality of A- mice was not
due to systemic cytokine production, an inability to clear systemic C. rodentium or
increased virulence. Instead A- mice developed a severe gut infection with most of the A-
mice surviving and resolving inflammation but not eliminating the infection.
Improvements in vitamin A status might decrease susceptibility to enteric pathogens and
eliminate potential carriers from spreading infection to susceptible populations.

29
INTRODUCTION
Vitamin A deficiency is a significant problem in developing countries where
inadequate micronutrient intake remains a public health concern (1). The World Health
Organization estimates that over 20% of preschool aged children are clinically vitamin A
deficient (1, 2). Vitamin A deficiency contributes to the higher prevalence of respiratory
and diarrheal diseases as well as increased childhood mortality. Conversely, vitamin A
supplementation is practiced as a means to reduce mortality in preschool-aged children
by reducing severity of infectious diseases (3).
Vitamin A and its active metabolite, retinoic acid (RA) are important regulators of
T cell responses. RA inhibited IFN-γ production from T cells in vitro (4). In addition, T
cells from vitamin A deficient (A-) mice overproduced IFN-γ (5). RA also inhibited
Th17 cells in vitro and in vivo (6). RA increased the expression of the gut homing
receptors, α4β7 and CCR9, on T cells (7), which recruits T cells to the gut mucosa. In
vitro, RA inhibited IL-17 and induced expression of the transcription factor FoxP3,
associated with regulatory T cells, and IL-10 production (8). Vitamin A and RA are key
regulators of T cell cytokine production and gut homing.
Several lines of experimental evidence support a beneficial effect of vitamin A
and RA on the host response to infection (9, 10). In the gut, the mechanisms that account
for the anti-infective effects of vitamin A include support of B cell function and T cell-
dependent B-cell antibody responses (11). Gut infection of A- mice with Trichinella
spiralis resulted in T cells that produced IFN-γ but not IL-4, and as a result, reduced the
rate of parasite clearance (9). Furthermore, RA treatment reduced colonic inflammation
caused by dextran sodium sulfate and infection (12). The reduction in gastrointestinal

30
(GI) inflammation with RA treatment was attributed to the inhibition of IL-17 and IFN-γ
(12, 13). These data suggest that vitamin A and RA regulate T cell function to limit
inflammation following chemical and infectious injury in the gut.
Citrobacter rodentium is a mouse pathogen that models human infections with
enteropathogenic Escherichia coli and causes attaching and effacing lesions of the cecum
and colon in mice (14). The natural route of C. rodentium transmission is fecal-oral.
Resistant mouse strains including C57BL/6 mice are infected transiently with C.
rodentium and clear the infection within 2-3 weeks (14). The acquired immune system
was required for early protection from C. rodentium, as demonstrated in recombination-
activating gene (Rag) knockout (KO) mice lacking T and B cells that were unable to clear
the infection (15). Robust IL-22 production from innate lymphoid cells and macrophages
has been shown to induce protective Th17 responses that resulted in clearance (16, 17).
T cells and B cells were essential for resolution of C. rodentium infection since mice
without T cells (CD4 KO) or B cells (Igμ KO) developed fatal infections (17). Host
resistance to C. rodentium depends on IL-22 production from innate cells, T cells, B cells
and Th17 cells.
Here, we determined the effect of vitamin A deficiency on host resistance to C.
rodentium infection in C57BL/6 mice; a normally resistant mouse strain. Because of the
well-demonstrated inhibitory effects of RA on the differentiation of Th1 and Th17 cells,
we predicted that a bacterial infection that required Th17 cell responses for resistance
might be less severe in A- mice, and exacerbated in RA-treated mice. Interestingly and
contrary to expectations, A- mice developed a chronic infection with C. rodentium. The

31
C. rodentium infection was lethal for 40% of A- mice while none of the vitamin A
sufficient (A+) or RA treated A- mice died prematurely from infection.

32
MATERIALS AND METHODS
Mice. C57BL6 mice were originally from Jackson Laboratories (Bar Harbor, MN) and
bred at the Pennsylvania State University (University Park, PA) for experiments. Vitamin
A deficient (A-) and vitamin A sufficient (A+) mice generated as previously described (5,
18). Briefly, mice were fed a purified diet that did not contain any vitamin A (A-) or that
contained 25 µg of retinyl acetate (vitamin A) per day (A+). At weaning, mice were
continuously fed the A- or A+ diet until the end of the study. Serum retinol status was
determined by ultra pressure liquid chromatography at 6-7 weeks of age in pooled
samples. For some experiments, A- mice were treated with 37.5 µg of all-trans RA
(Sigma Aldrich, St Louis, MO) administered orally in 10 μl corn oil three times per week
(19) . For some experiments, mice were injected i.p. with E. coli O111:B4 LPS (6
mg/kg, Sigma-Aldrich). Experimental procedures were approved by the Office of
Research Protection Institutional Animal Care and Use Committee of the Pennsylvania
State University, University Park, PA.
C. rodentium infection. C. rodentium strain ICC169 (nalidixic acid resistant) and
bioluminescent strain ICC180 (kanamycin resistant) were kind gifts of Gad Frankel
(London School of Medicine and Dentistry, London UK). C. rodentium ICC169 was
cultured in Luria-Bertani (LB, EMD Chemicals, Inc., Gibbstown, NJ) broth containing 50
μg/ml nalidixic acid (Sigma-Aldrich) while, C. rodentium ICC180 was cultured in LB
broth containing 100 μg/ml kanamycin (Sigma-Aldrich). Mice 8-10 weeks of age were
infected by oral gavage with 5×109 CFU in 200 μl of C. rodentium strain unless
otherwise noted. For studies looking at in vivo infection kinetic, 5×109 CFU C.
rodentium ICC180 was used. Animals were imaged every other day using the IVIS50

33
small animal imaging system (Xenogen Corp., Alameda, CA, USA). Images were
analyzed by Living Image software (PerkinElmer, Waltham, MA). Additional groups of
mice were injected i.v. with 104-108 CFU of C. rodentium strain ICC169. Feces and
other tissues were collected, homogenized and plated in serial dilutions on LB agar plates
containing naladixic acid.
For most experiments mice were housed 1 per cage from the time of infection to prevent
transmission from mouse to mouse. Natural transmission experiments were performed as
previously described (20). Briefly, A-, RA d0, and A+ mice were infected via oral
gavage with 5×109 CFU. Three days post-infection, each infected “seed” mouse was co-
housed with two naive A+ mice.
Histology: Distal colon sections were fixed in 10% formalin, sectioned and stained with
hematoxylin and eosin (Pennsylvania State University Animal Diagnostic Laboratory,
University Park, PA). Specimens were coded and evaluated in a blinded fashion by a
board certified laboratory animal veterinarian with training in pathology. Crypt
measurements were taken at 100X using the cellSens software (Olympus Corp., Center
Valley, PA, USA). Sections were scored on a scale from 0 to 4 as follows: severity of
inflammation (0 = none, 1= minimal, 2= mild, 3= moderate, 4= extensive); epithelial
sloughing (0 = none, 1 = minimal, 2 = mild, 3= moderate, 4= extensive); distention of
muscularis (0 = none, 1 = minimal, 2 = mild, 3 = moderate, 4 = extensive); edema (0 =
none, 1 = minimal, 2= mild, 3 = moderate, 4 = extensive). Total histology scores were
generated by adding the scores for each category together, generating a value from 0-16
for each sample.
Flow cytometry: Colonic IELs were isolated as previously described and stained for

34
flow cytometry (21). Cells were counted and stained with PE-Cy5 TCRβ (BD
Pharmingen, San Jose, CA USA), FITC CD8β (eBioscience San Diego, CA, USA), PE-
Cy7 CD8α (BioLegend, San Diego, CA, USA) or PE Texas Red CD4 (Invitrogen,
Carlsbad, CA, USA). Cells were analyzed on a FC500 Bench top cytometer (Beckman
Coulter, Brea, CA, USA) and data was analyzed using FlowJo 7.6.1 software (Tree Star,
Ashland, OR, USA).
ELISA: Cytokine production in the serum was measured for TNF-α, IL-1β and IFN-γ
levels by ELISA and following the manufacturer’s instructions (BD Biosciences,
Minneapolis, MN).
Statistical Analysis: Statistical analyses were performed using GraphPad Prism
software (Graphpad, La Jolla, CA, USA). Two-tailed Student’s t-tests were used for
serum retinol analysis. One-way ANOVA with Tukey’s post-hoc test were used to
compare the systemic C. rodentium loads, bioluminescence quantification, and cell
population analysis. Two-way ANOVA with Bonferonni’s post-hoc test were used to
compare CFU, histology scores and crypt lengths. Log-rank tests were used for the
survival curves and ratios of serum cytokine producers. For all analyses, P<0.05 was
used as the limit for significance.

35
RESULTS
A- mice are more susceptible to C. rodentium infection than A+ mice. A+ and A-
mice were generated as previously described (5, 18). As expected (22), 6-7 week old A-
mice had significantly lower serum retinol than A+ mice (Fig. 2-1A). Bacterial fecal
shedding in A+ mice peaked around d10 and cleared within 25 days post-infection (Fig.
2-1B). The infection in A- mice followed the same kinetics as A+ mice until d14. A+
and A- mice had similar numbers of C. rodentium in the cecum and feces at d10 post-
infection (cecum data not shown and Fig. 2-1A). After d14 the A- mice continued to
shed high numbers of C. rodentium in their feces, whereas the A+ mice began to clear the
infection (Fig. 2-1B). All of the infected mice showed a small amount of weight loss (5%
of starting weight) within the first 2-4 days of infection but all of the A+ mice recovered
and no A+ mice died following C. rodentium infection (Fig. 1C). Conversely, some of
the A- mice failed to recover and instead lost significantly more of their initial body
weight (10-20%), which resulted in the premature lethality of 40% of the A- mice (Fig. 2-
1C). The A- mice that survived did not lose any more weight than the A+ mice, had
normal exploratory behaviors (data not shown) and otherwise were undistinguishable
from their A+ infected counterparts. A subset of A- mice die following weight loss and
the remaining mice resume normal behaviors although they have not cleared C.
rodentium.
To visualize the intestinal passage of C. rodentium in vivo, we made use of a
bioluminescent C. rodentium strain, and live animal imaging. Between d2 and d6 post-
infection, low levels of C. rodentium bioluminescence were detected in the upper parts of
the gastrointestinal (GI) tract of A+ mice (Fig. 2-1D). Just before the peak in fecal

36
shedding (Fig. 2-1B) the intensity of bioluminescence increased and moved into the
lower GI tract of A+ mice (Fig. 2-1D). After d10 of infection, the intensity decreased in
A+ mice and by d14, when fecal shedding of C. rodentium was on the decline in A+
mice, less bioluminescence was detected in the intact A+ animals (Fig. 2-1D). The
kinetics of C. rodentium transit in the A+ mice was similar to that reported previously in
wild-type mice (23). The bioluminescence in the A- mice matched that of the A+ mice at
d2 (Fig. 2-1D). However, as early as d4, the A- mice showed bioluminescence
throughout the GI tract, which persisted in intensity through d14 (Fig. 2-1D). Unlike the
A+ mice, the intensity of bioluminescence in A- mice between d4 and d14 was high
throughout the GI tract and significantly higher than in A+ mice (Fig. 2-1D and Fig. 2-2).
Thus, the transit of C. rodentium through the GI tract occurred more rapidly and persisted
for longer in A- than A+ mice and reflected the fecal shedding results.
T cell frequencies are lower in the vitamin A deficient gut. To determine if vitamin A
status causes differences in gut mucosal immune cell populations, the intraepithelial
lymphocyte (IEL) populations of the colon were characterized. IELs are in direct contact
with the intestinal epithelium and a source of T cells from the colon (24). In addition, the
T cells in the IEL are in close contact with the microbiota and enteric pathogens in the
colon. Fewer total IEL cells were isolated from the colons of uninfected A- versus A+
mice (Fig. 2-3A). C. rodentium infection resulted in a significant increase in the number
of cells in the colon of both A- and A+ mice (Fig. 2-3A). Interestingly, although the A+
mice resolved the infection by d37, as shown in Fig. 2-1B, the numbers of T cells
(TCRβ+) did not return to baseline by d37 pi (Fig. 2-3A and 2-3B). The total number of
IELs and TCRβ+ T cells in the colon of A- mice increased following infection but never

37
reached the levels present in infected A+ mice (Fig. 2-3A). The numbers of TCRβ+,
TCRβ+/CD8α+, and TCRβ+/CD8αβ+ T cells in the colon IEL compartment were lower
in A- than in A+ mice, both before and after infection (Fig. 2-3A-E). Therefore, although
the increases in total cell numbers and T cell subpopulations were similar in A+ and A-
mice, the A- mice consistently had fewer T cells in the colonic IEL compartment
compared to A+ mice. Differences in T cell numbers and populations between A+ and
A- mice were not associated with clearance of C. rodentium in A+ mice and persistence
of C. rodentium in A- mice.
Exacerbated inflammation and epithelial hyperplasia in A- mice following C.
rodentium infection. Histopathology sections of the colon were evaluated before and
after infection for signs of inflammation, tissue damage and hyperplasia, including
measurements of crypt length. Before infection, A+ and A- mice had low histopathology
scores that did not differ with vitamin A status (Fig. 2-3F and Fig. 2-4). Crypt lengths
also did not differ between uninfected A+ and A- mice (Fig. 2-3G). After infection, the
histopathology scores of A+ colons did not change significantly, either at peak infection
(d10) or after resolution of infection (d37) (Fig. 2-3F and 2-4). In A- mice,
histopathology scores were significantly higher at d10 post-infection, both compared with
baseline A- scores and scores for A+ mice at d10 (Fig. 2-3F and 2-4). By d37, the
histopathology scores of A+ and A- mice were the same in spite of the fact that A+ mice
had cleared the infection, while A- mice had not (Fig. 2-3F and 2-4). Crypt length in the
A+ mice was not affected by infection (Fig. 2-3G). Crypt length in the A- mice increased
significantly after infection and were significantly longer on both d10 and d37 than at
baseline and as compared to the A+ mice at all time points (Fig. 2-3G). Overall, although

38
A- mice exhibited more inflammation than A+ mice at d10 post-infection the effect was
not present at d37 even though A- mice continued to harbor C. rodentium and A+ mice
did not.
RA treatment of A- mice results in clearance of C. rodentium. To determine if RA
could rescue the severe infection in A- mice, A- mice were orally dosed with RA. RA
dosing began either 2 wks before infection (RA -d14), or on the day of infection (RA d0)
(Fig. 3). Once started, RA dosing continued 3 times weekly throughout the remainder of
the experiment. Treatment of A- mice with RA, either 2 wks before (RA -d14) or on the
day of infection (RA d0), resulted in fecal shedding of C. rodentium similar to that of A+
mice (Fig. 1B and Fig. 2-5). In a separate experiment, the RA d0 mice were also infected
with bioluminescent C. rodentium. The bioluminescence in the RA d0 mice was of a
high intensity and resembled that in A- rather than A+ mice (Fig. 2-1D and 2-2).
Interestingly, by d14 the intensity of bioluminescence in the RA d0 group was lower and
resembled the A+ rather than the A- mice (Fig. 2-1D and 2-2).
To determine whether RA could be used therapeutically in A- mice with an established
C. rodentium infection, the RA treatment of A- mice was started on d14 post-infection
(RA +d14). Day 14 of infection occurs just after the peak of fecal shedding and before
A+ mice show a rapid decline in C. rodentium shedding (Fig. 2-1B). Treatment of A-
mice with RA, three times weekly, beginning on d14 resulted in a gradual decline in fecal
shedding of C. rodentium (Fig. 2-5), which was significant as early as 4 days following
the start of RA treatment, as compared to untreated A- mice (d18 post-infection, Fig. 2-
5). As observed in earlier experiments, untreated A- mice maintained a persistent
infection while the RA +d14 treated mice cleared the infection 18d after the start of the

39
RA treatment (d32 post-infection, Fig. 2-5). Thus, RA treatment of A- mice cleared the
C. rodentium infection, which otherwise was persistent.
Increased C. rodentium CFU in the liver and spleen of A- mice. At d14 post-infection
mice that had been infected with the bioluminescence strain of C. rodentium were
euthanized to determine which organs harbored C. rodentium. In the A+ group,
bioluminescence was detected in the colon but not in other parts of the GI tract (Fig. 2-
6A). In addition, A+ mice did not have visible bioluminescence in their spleen or liver at
d14 post-infection (Fig. 2-6B). A- mice had high levels of bioluminescence in the upper
portions of the GI tract that were significantly higher than in A+ mice (Fig. 2-6C). A-
mice also had visible bioluminescence at d14 in both the spleen and liver (Fig. 2-7A and
Fig. 2-6B). The bioluminescence in the spleen and liver of RA d0 mice was more similar
to A+ mice than to A- mice at d14 post-infection (Fig. 2-7A and Fig. 2-6B).
Quantification of the bioluminescence showed that A- mice had higher bioluminescence
in the spleen, liver and SI than either the A+ or the RA d0 mice (Fig. 2-7A and Fig. 2-
6B).
Next we determined whether viable C. rodentium was detectable in the spleen and liver
of our mice following gastric infection with C. rodentium. Cultures of the spleen and
liver at d10 and d14 post-infection showed that A+ mice had detectable C. rodentium in
their internal organs (Fig. 2-7B, and d14 data not shown). A- mice had significantly
more C. rodentium in the spleen and liver than A+ mice (Fig. 2-7B), while the spleen and
liver of RA d0 mice did not differ from those of A+ mice but were significantly different
than those of A- mice (Fig. 2-7B). At d37 of infection the A- mice no longer had C.

40
rodentium present in the spleen and liver even though they continued to shed C.
rodentium in the feces (data not shown).
Mortality of A- mice following infection with C. rodentium is not due to over-
production of systemic cytokines. To determine whether A- mice died due to cytokine
over-production, serum cytokine levels were determined at before (d0) and at the peak
(d10) of infection in A+ and A- mice (Table 2-1). None of the A+ mice had detectable
levels of any cytokines before infection. At d0, several of the A- mice had detectable
levels of TNF-α, IL-1β or IFN-γ in their serum (Table 2-1). Infection resulted in
detectable TNF-α, IL-1β and IFN-γ in the serum of both A+ and A- mice. There was
significantly more A- mice with IL-1β in the serum (10 of 19) than A+ (1 of 13) mice at
d10 post-infection (Table 2-1). Serum IFN-γ was higher but not significantly different in
A- mice compared to A+ mice at d10 post-infection (P=0.0817). To measure the
capacity for cytokine production, LPS was injected ip into A- and A+ mice to measure
serum cytokine response. A- mice produced significantly more TNF-α than A+ mice
following LPS injection, while the IL-1β response was the same in A+ and A- mice (Fig.
2-8A and IL-1β data not shown). Overall A- mice were more likely to have IL-1β
detectable in the serum after C. rodentium infection and produced more TNF-α after LPS
than A+ mice.
We hypothesized that A- mice were dying following oral challenge with C. rodentium
because of systemic spread. We attempted to determine the LD50 of i.v. injected C.
rodentium in A+ mice. All of the A+ mice survived an i.v. dose of 108 C. rodentium and
readily cleared the bacteria. A- mice also survived an i.v. injection of 108 C. rodentium.
A+ mice had the highest number of organisms in the spleen and liver 1d post-i.v.

41
infection with 108 CFU, which declined at d3 and again at d5 and was completely
cleared by d10 post-infection (Fig. 2-8B and 2-8C). The group x time interaction showed
no differences in the clearance of i.v. injected C. rodentium between A+ and A- mice
(Fig. 2-8B and 2-8C). In addition, i.v. injection was not lethal and did not induce a
systemic cytokine response in either A+ or A- mice (data not shown).
Host vitamin A status does not alter C. rodentium infectivity. Previous reports have
demonstrated that natural infection of C. rodentium to co-housed uninfected mice occurs
with several fewer log units of bacteria than with laboratory grown C. rodentium (20).
We hypothesized that vitamin A and RA were attenuating the infectivity (virulence) of
C. rodentium and that C. rodentium that has been passaged through A- mice might be
more virulent. A+, RA d0, and A- seed mice were infected by gavage using laboratory
grown C. rodentium and 3d later each was co-housed with naïve A+ mice, housed in
groups of 2 with the seed mice. As described previously (20), all seed mice, despite their
vitamin A status, infected all co-housed naïve A+ mice within 3 days of exposure (data
not shown). Culturing of the feces, spleen and liver from the cage mates of A+, A- and
RA d0 seed mice showed that there were no differences in the CFU of C. rodentium
recovered in the spleen, liver and feces via natural transmission from either A+, A- or RA
d0 seed mice (Fig. 2-9). Thus the data do not show an effect of vitamin A status and or
RA treatment on C. rodentium infectivity.

42
DISCUSSION
A- mice were found to be significantly more susceptible to C. rodentium infection
compared to A+ mice. The increased susceptibility of A- mice included lethality of 40%
of the A- mice by d14. The kinetics of shedding of C. rodentium in A+ and A- mice were
not different before d14, after which the A+ mice proceeded to clear the infection while
the A- mice either died or developed a persistent infection. The data suggest that early
innate immune responses are adequate in A- mice during the initial phase of the infection,
when fecal bacterial counts are increasing, and instead suggest that A- mice failed to
generate an acquired immune response necessary to clear the infection. Our results
unexpectedly showed that A- mice became chronic carriers of C. rodentium, while no
longer showing symptoms (inflammation of the colon, diarrhea etc.). Correcting vitamin
A status or treating A- mice with RA effectively eliminated the infection and improved
survival of A- mice, suggesting an added benefit not previously recognized for vitamin A
and/or RA interventions.
The increased mortality of A- mice following GI infection was not due to the systemic
spread of C. rodentium, increased systemic cytokine production or the virulence of C.
rodentium. Others have reported mortality following gavage of C. rodentium in mice and
have attributed that mortality to systemic spread and poly-microbial sepsis (15, 25, 26).
Susceptible strains of mice (C3H/HeJ, C3H/HeOuJ and C3H/HeN) develop a severe
infection with 100% lethality due to bacterial translocation from the gut, cytokines in the
serum and crypt cell apoptosis (25). By comparison, C57BL/6 mice are relatively
resistant to C. rodentium (15, 17, 25). For the first time, our data clearly show that C.
rodentium does not grow following i.v. injection of large numbers of bacteria in either

43
A+ or A- mice, indicating that vitamin A deficiency does not affect the ability to survive
a systemic infection with C. rodentium. However, A- mice were more susceptible to oral
infection with C, rodentium. We necropsied one recently deceased and one moribund A-
mouse. Very little food was found in the stomach of either mouse, suggesting that the
mice had stopped eating. The necropsy of A- mice failed to identify evidence of systemic
infection (data not shown). It has been shown that LPS-induced proinflammatory
cytokines such as TNF-α and IL-1β can result in anorexia (27). Our data are consistent
with TNF-α and IL-1β induced anorexia, since the A- mice that died had lost significant
amounts of weight (10-20% of original body weight). In addition, A- mice had higher
TNF-α levels after LPS challenge and were more likely to have detectable IL-1β in their
serum after infection than A+ mice. Surviving A- mice may have been just below the
threshold of the response and therefore survived the infection-induced anorexia. Based
on the inability of C. rodentium to grow following an iv injection and the necropsies of
moribund and dead A- mice, we concluded that the mortality in our A- mice was not due
to sepsis and/or the systemic spread of the infection.
A- mice had reduced numbers of total colonic IELs and all IEL T cell subsets in the colon
before infection. Infection induced homing and expansion of T cells in the gut of both
A+ and A- mice. Although the A- mice were persistently infected with C. rodentium,
colonic inflammation as determined by histological score resolved and was not different
at d37 from A+ mice. This same phenomenon occurred in germfree mice mono-
associated with C. rodentium (28). In germfree mice C. rodentium infection was not
cleared but the inflammatory response in the colon was nevertheless resolved (28). It
therefore seems that vitamin A is not required to resolve inflammation in the colon and

44
that in the absence of vitamin A, T cells are able to arrive and respond to the infection.
However, the acquired immune response in the A- mice was ineffective at eliminating the
infection. The data suggest a requirement for vitamin A/RA to mount effective protective
immunity after the initial infection. Since RA treatments effectively reduced C.
rodentium numbers in already infected A- mice within 4 days of treatment, it will be of
interest to identify the targets of RA in the GI tract for C. rodentium clearance.
In our study, A- mice developed persistent and sometimes fatal enteric infections. The
cause of the premature lethality of A- mice was not due to sepsis and/or systemic spread
of C. rodentium. Instead it seems that a high load of C. rodentium in the gut of A- mice
results in infection-induced anorexia in 40% of the mice. When given early, vitamin A
and/or RA interventions protected A- mice from the lethality following C. rodentium
infection and when administered later RA cleared the persistent infections that occurred
in surviving A- mice. Our work has important implications for the developing world
where vitamin A deficiency is prevalent. In particular, vitamin A-deficient humans and
animals could be reservoirs for E. coli-like enteric pathogens. Vitamin A and RA
treatments might be useful interventions to decrease morbidity and mortality from enteric
infections. We show here a novel and unappreciated role for vitamin A/RA for
eliminating persistent enteric infections and show added benefits to improving vitamin A
status in the developing world.

45
REFERENCES: 1. WHO. 2009. Global prevalence of vitamin A deficiency in populations at risk
1995–2005: WHO global database on vitamin A deficiency. In World Health Organization, Geneva. 2. West, K. P., Jr. 2002. Extent of vitamin A deficiency among preschool children
and women of reproductive age. J Nutr 132: 2857S-2866S. 3. Fawzi, E. V. a. W. W. 2000. Vitamin A supplementation: implications for
morbidity and mortality in children. Journal of Infectious Diseases 2000: S112-133.
4. Cantorna, M. T., F. E. Nashold, and C. E. Hayes. 1994. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol 152: 1515-1522.
5. Carman, J. A., S. M. Smith, and C. E. Hayes. 1989. Characterization of a helper T lymphocyte defect in vitamin A-deficient mice. J Immunol 142: 388-393.
6. Mangan, P. R., L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231-234.
7. Iwata, M., A. Hirakiyama, Y. Eshima, H. Kagechika, C. Kato, and S. Y. Song. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21: 527-538.
8. Elias, K. M., A. Laurence, T. S. Davidson, G. Stephens, Y. Kanno, E. M. Shevach, and J. J. O'Shea. 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111: 1013-1020.
9. Carman, J. A., L. Pond, F. Nashold, D. L. Wassom, and C. E. Hayes. 1992. Immunity to Trichinella spiralis infection in vitamin A-deficient mice. J Exp Med 175: 111-120.
10. Ross, A. C., and C. B. Stephensen. 1996. Vitamin A and retinoids in antiviral responses. FASEB J 10: 979-985.
11. Mora, J. R., M. Iwata, and U. H. von Andrian. 2008. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 8: 685-698.
12. Mielke, L. A., S. A. Jones, M. Raverdeau, R. Higgs, A. Stefanska, J. R. Groom, A. Misiak, L. S. Dungan, C. E. Sutton, G. Streubel, A. P. Bracken, and K. H. Mills. 2013. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210: 1117-1124.
13. Spencer, S. P., C. Wilhelm, Q. Yang, J. A. Hall, N. Bouladoux, A. Boyd, T. B. Nutman, J. F. Urban, Jr., J. Wang, T. R. Ramalingam, A. Bhandoola, T. A. Wynn, and Y. Belkaid. 2014. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343: 432-437.
14. Collins, J. W., K. M. Keeney, V. F. Crepin, V. A. Rathinam, K. A. Fitzgerald, B. B. Finlay, and G. Frankel. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12: 612-623.

46
15. Bry, L., and M. B. Brenner. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172: 433-441.
16. Sonnenberg, G. F., and D. Artis. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37: 601-610.
17. Mundy, R., T. T. MacDonald, G. Dougan, G. Frankel, and S. Wiles. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7: 1697-1706.
18. Smith, S. M., N. S. Levy, and C. E. Hayes. 1987. Impaired immunity in vitamin A-deficient mice. J Nutr 117: 857-865.
19. Ma, Y., Q. Chen, and A. C. Ross. 2005. Retinoic acid and polyriboinosinic:polyribocytidylic acid stimulate robust anti-tetanus antibody production while differentially regulating type 1/type 2 cytokines and lymphocyte populations. J Immunol 174: 7961-7969.
20. Wiles, S., G. Dougan, and G. Frankel. 2005. Emergence of a 'hyperinfectious' bacterial state after passage of Citrobacter rodentium through the host gastrointestinal tract. Cell Microbiol 7: 1163-1172.
21. Lefrancois, L., and N. Lycke. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. Curr Protoc Immunol Chapter 3: Unit 3 19.
22. Restori, K. H., K. L. McDaniel, A. E. Wray, M. T. Cantorna, and A. C. Ross. 2014. Streptococcus pneumoniae-induced pneumonia and Citrobacter rodentium-induced gut infection differentially alter vitamin A concentrations in the lung and liver of mice. J Nutr 144: 392-398.
23. Basu, R., D. B. O'Quinn, D. J. Silberger, T. R. Schoeb, L. Fouser, W. Ouyang, R. D. Hatton, and C. T. Weaver. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37: 1061-1075.
24. Meresse, B., G. Malamut, and N. Cerf-Bensussan. 2012. Celiac disease: an immunological jigsaw. Immunity 36: 907-919.
25. Vallance, B. A., W. Deng, K. Jacobson, and B. B. Finlay. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect Immun 71: 3443-3453.
26. Ghosh, S., C. Dai, K. Brown, E. Rajendiran, S. Makarenko, J. Baker, C. Ma, S. Halder, M. Montero, V. A. Ionescu, A. Klegeris, B. A. Vallance, and D. L. Gibson. 2011. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol 301: G39-49.
27. Sarraf, P., R. C. Frederich, E. M. Turner, G. Ma, N. T. Jaskowiak, D. J. Rivet, 3rd, J. S. Flier, B. B. Lowell, D. L. Fraker, and H. R. Alexander. 1997. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 185: 171-175.
28. Kamada, N., Y. G. Kim, H. P. Sham, B. A. Vallance, J. L. Puente, E. C. Martens, and G. Nunez. 2012. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336: 1325-1329.
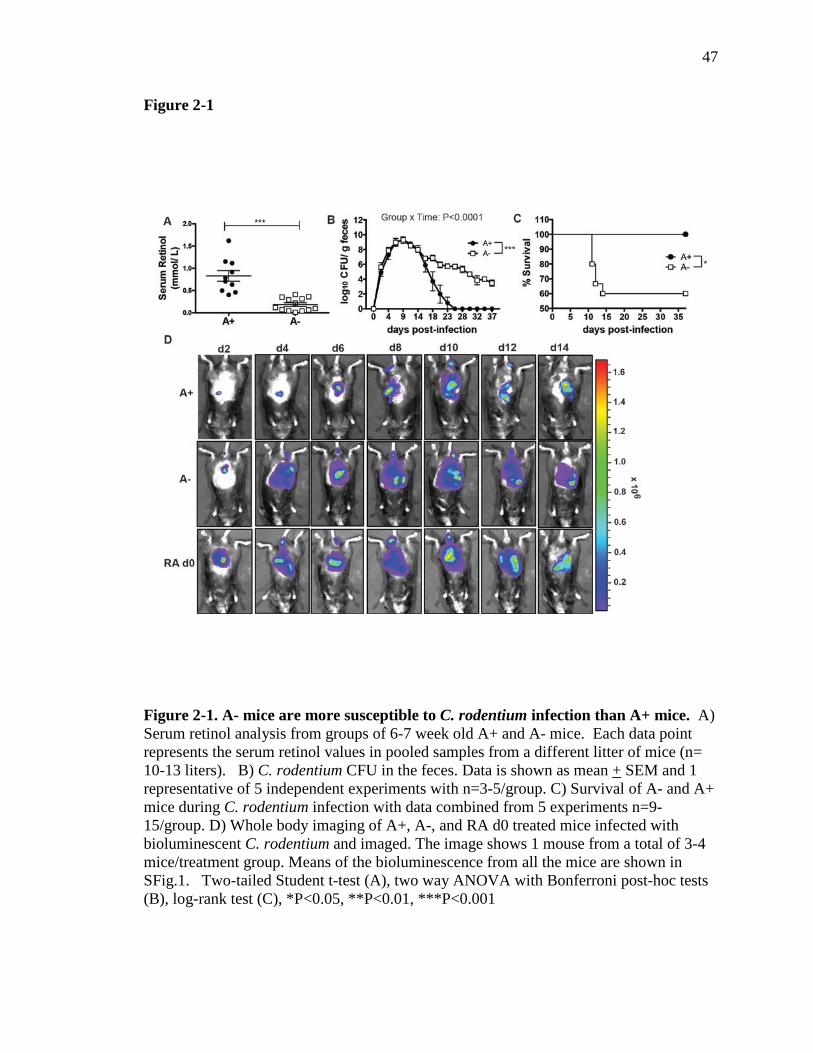
47
Figure 2-1
Figure 2-1. A- mice are more susceptible to C. rodentium infection than A+ mice. A) Serum retinol analysis from groups of 6-7 week old A+ and A- mice. Each data point represents the serum retinol values in pooled samples from a different litter of mice (n= 10-13 liters). B) C. rodentium CFU in the feces. Data is shown as mean + SEM and 1 representative of 5 independent experiments with n=3-5/group. C) Survival of A- and A+ mice during C. rodentium infection with data combined from 5 experiments n=9-15/group. D) Whole body imaging of A+, A-, and RA d0 treated mice infected with bioluminescent C. rodentium and imaged. The image shows 1 mouse from a total of 3-4 mice/treatment group. Means of the bioluminescence from all the mice are shown in SFig.1. Two-tailed Student t-test (A), two way ANOVA with Bonferroni post-hoc tests (B), log-rank test (C), *P<0.05, **P<0.01, ***P<0.001
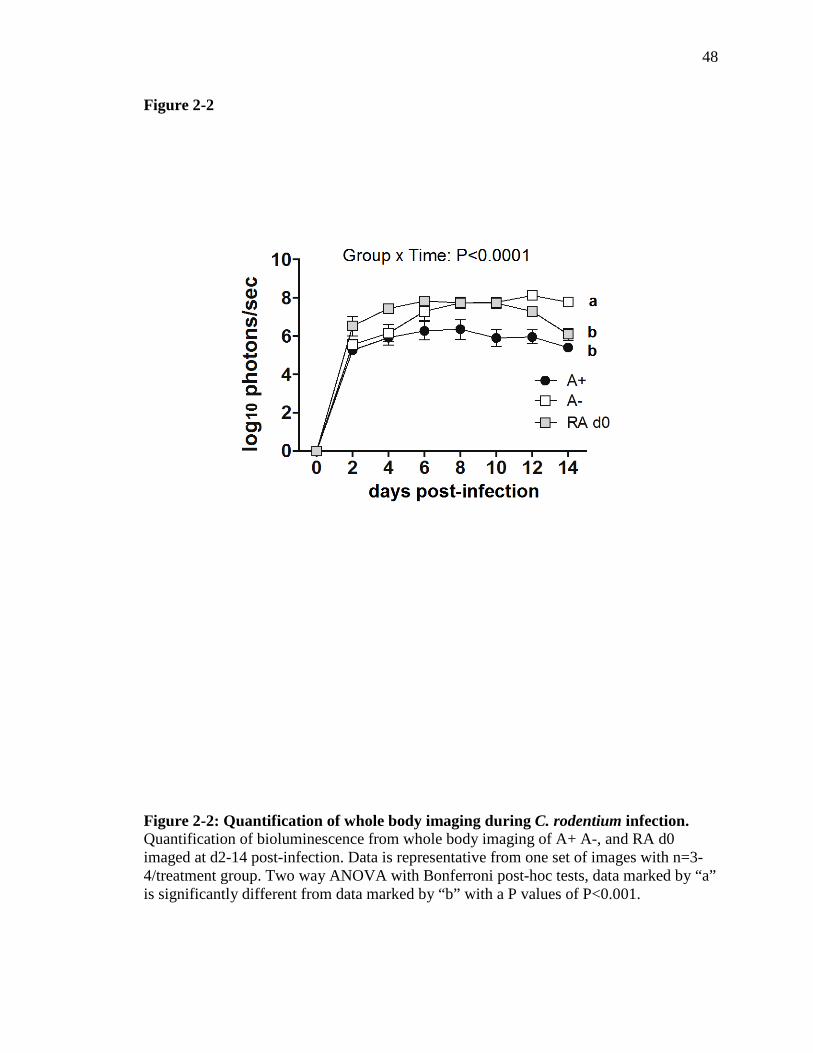
48
Figure 2-2 Figure 2-2: Quantification of whole body imaging during C. rodentium infection. Quantification of bioluminescence from whole body imaging of A+ A-, and RA d0 imaged at d2-14 post-infection. Data is representative from one set of images with n=3-4/treatment group. Two way ANOVA with Bonferroni post-hoc tests, data marked by “a” is significantly different from data marked by “b” with a P values of P<0.001.
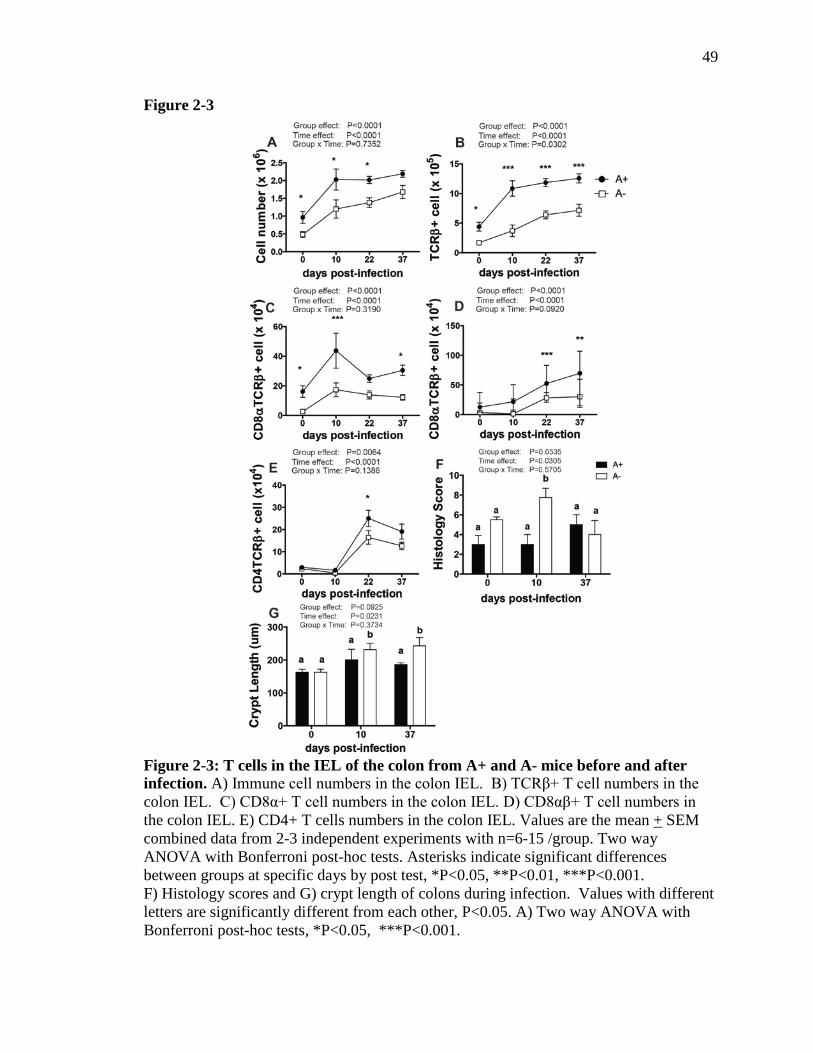
49
Figure 2-3
Figure 2-3: T cells in the IEL of the colon from A+ and A- mice before and after infection. A) Immune cell numbers in the colon IEL. B) TCRβ+ T cell numbers in the colon IEL. C) CD8α+ T cell numbers in the colon IEL. D) CD8αβ+ T cell numbers in the colon IEL. E) CD4+ T cells numbers in the colon IEL. Values are the mean + SEM combined data from 2-3 independent experiments with n=6-15 /group. Two way ANOVA with Bonferroni post-hoc tests. Asterisks indicate significant differences between groups at specific days by post test, *P<0.05, **P<0.01, ***P<0.001. F) Histology scores and G) crypt length of colons during infection. Values with different letters are significantly different from each other, P<0.05. A) Two way ANOVA with Bonferroni post-hoc tests, *P<0.05, ***P<0.001.
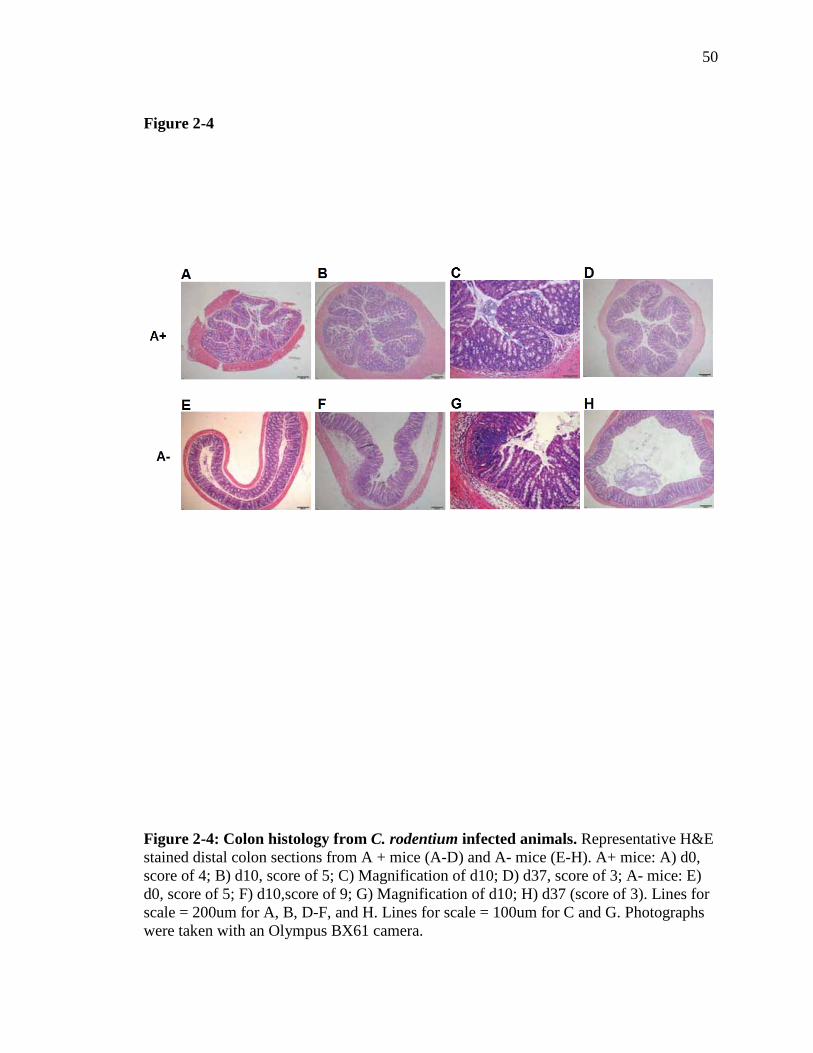
50
Figure 2-4
Figure 2-4: Colon histology from C. rodentium infected animals. Representative H&E stained distal colon sections from A + mice (A-D) and A- mice (E-H). A+ mice: A) d0, score of 4; B) d10, score of 5; C) Magnification of d10; D) d37, score of 3; A- mice: E) d0, score of 5; F) d10,score of 9; G) Magnification of d10; H) d37 (score of 3). Lines for scale = 200um for A, B, D-F, and H. Lines for scale = 100um for C and G. Photographs were taken with an Olympus BX61 camera.
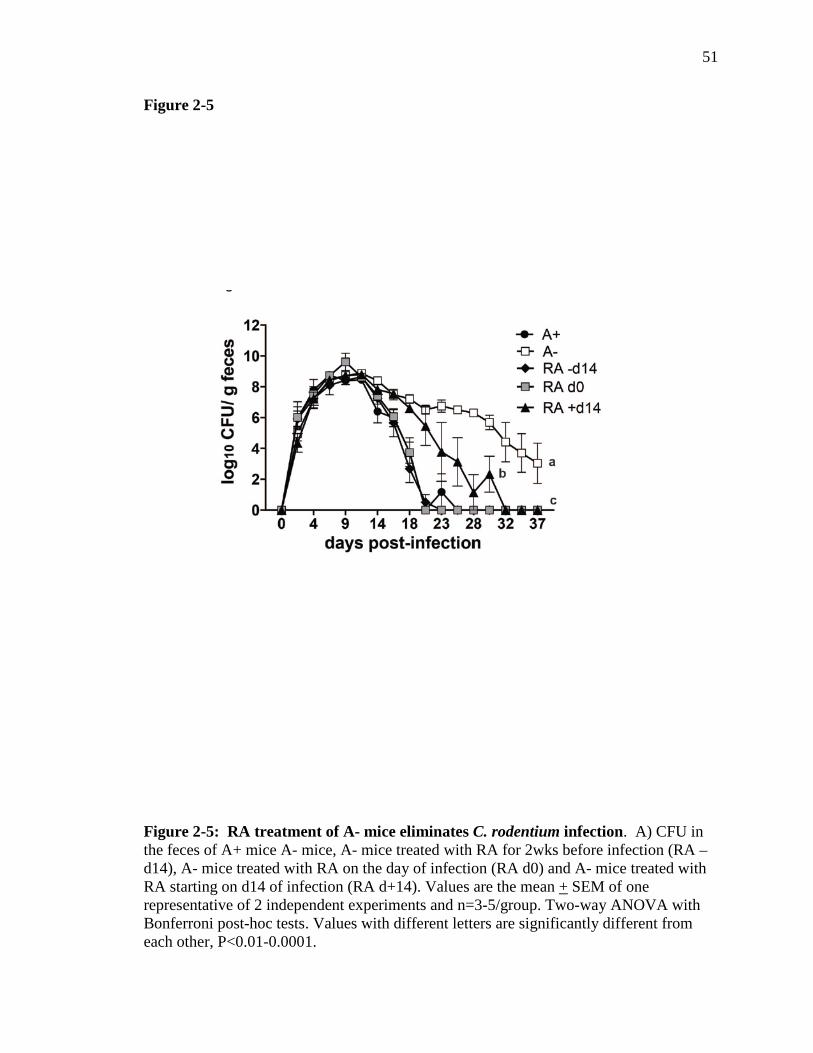
51
Figure 2-5
Figure 2-5: RA treatment of A- mice eliminates C. rodentium infection. A) CFU in the feces of A+ mice A- mice, A- mice treated with RA for 2wks before infection (RA –d14), A- mice treated with RA on the day of infection (RA d0) and A- mice treated with RA starting on d14 of infection (RA d+14). Values are the mean + SEM of one representative of 2 independent experiments and n=3-5/group. Two-way ANOVA with Bonferroni post-hoc tests. Values with different letters are significantly different from each other, P<0.01-0.0001.
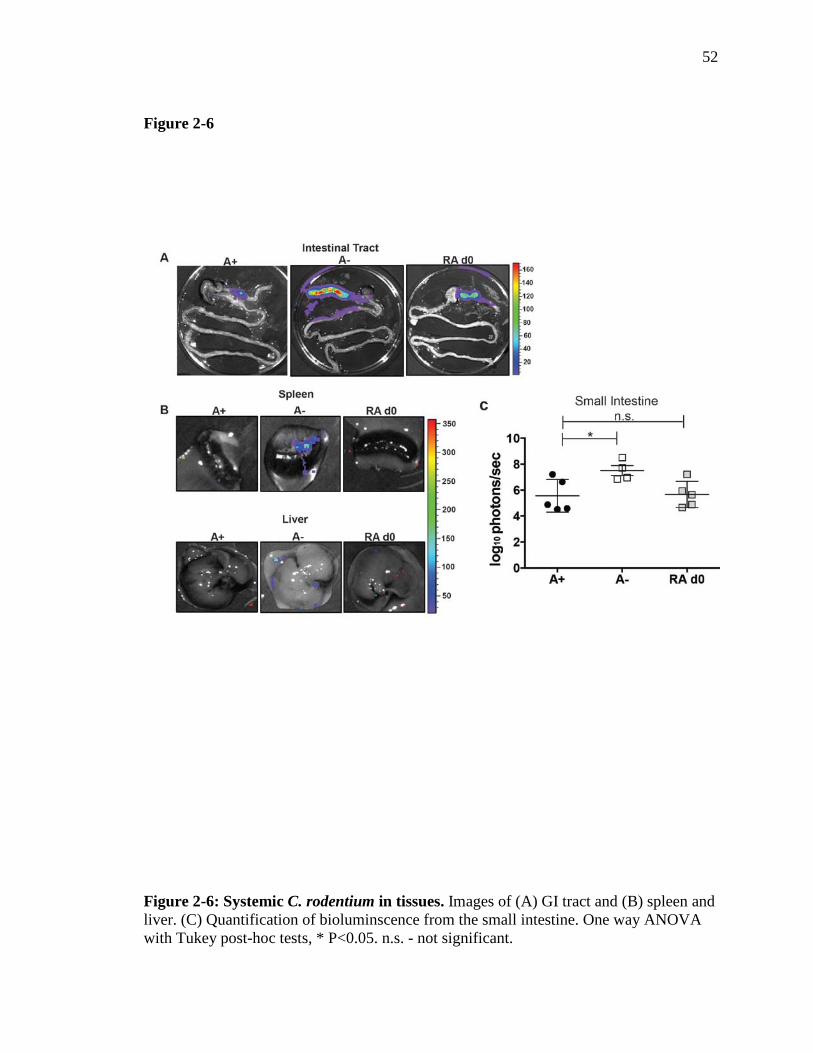
52
Figure 2-6
Figure 2-6: Systemic C. rodentium in tissues. Images of (A) GI tract and (B) spleen and liver. (C) Quantification of bioluminscence from the small intestine. One way ANOVA with Tukey post-hoc tests, * P<0.05. n.s. - not significant.
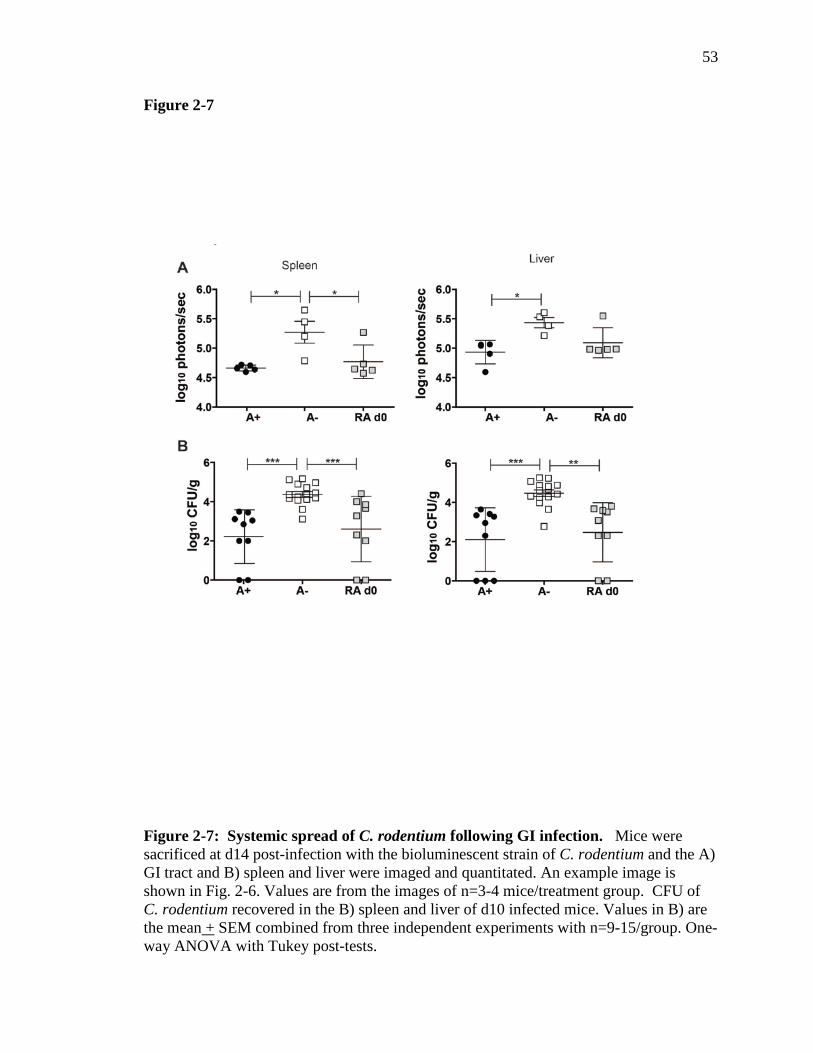
53
Figure 2-7
Figure 2-7: Systemic spread of C. rodentium following GI infection. Mice were sacrificed at d14 post-infection with the bioluminescent strain of C. rodentium and the A) GI tract and B) spleen and liver were imaged and quantitated. An example image is shown in Fig. 2-6. Values are from the images of n=3-4 mice/treatment group. CFU of C. rodentium recovered in the B) spleen and liver of d10 infected mice. Values in B) are the mean + SEM combined from three independent experiments with n=9-15/group. One-way ANOVA with Tukey post-tests.
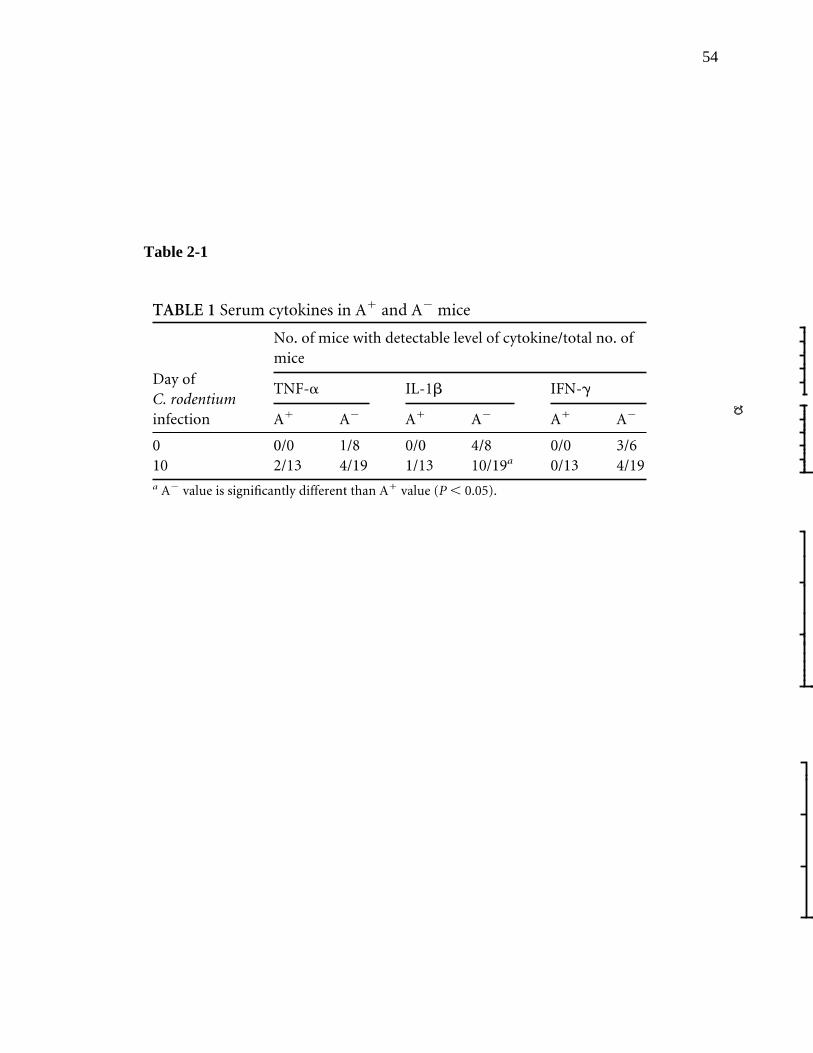
54
Table 2-1

55
Figure 2-8
Figure 2-8: The effect of vitamin A status on LPS response and i.v challenge with C. rodentium. A) Serum TNF-α in A- and A+ mice treated with LPS. CFU in the B) spleen and C) liver of mice following i.v. injection of C. rodentium. Values are the mean + SEM combined from two-three independent experiments with n=8-9/group/time-point. Two-tailed Student t-test (A), Two-way ANOVA with Bonferroni post-hoc tests (B and C). Asterisks indicate significant differences between groups at specific days by post test.; *P<0.05.
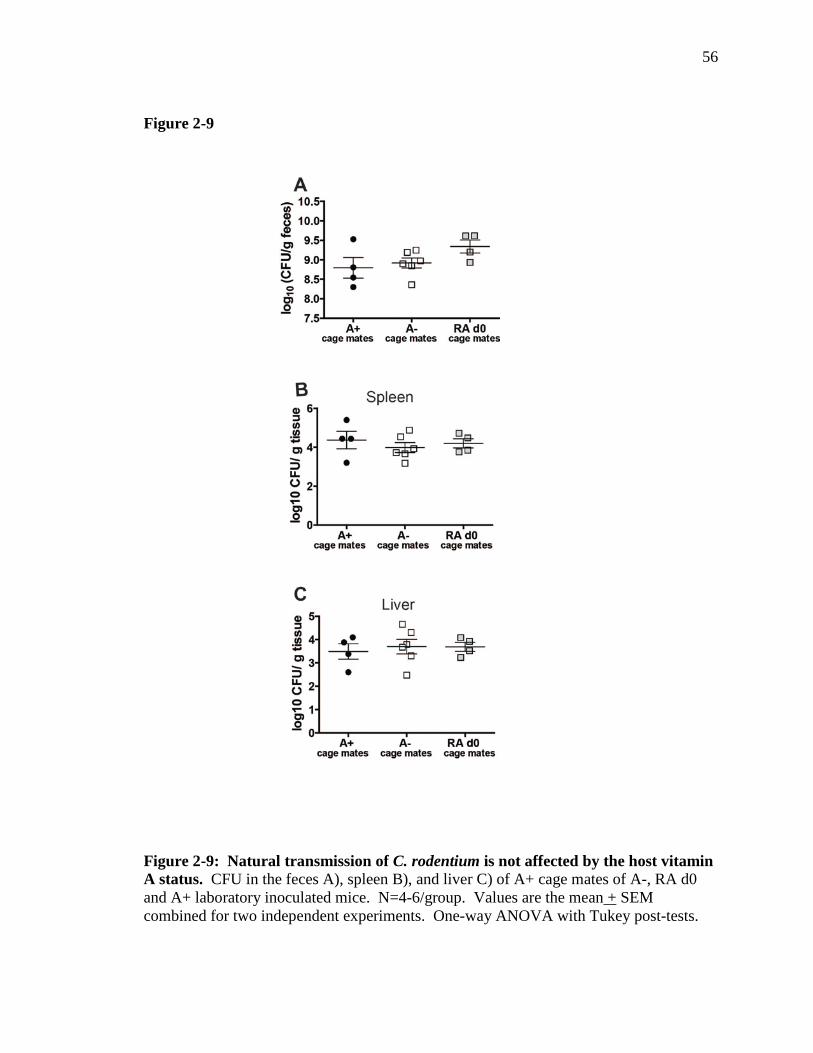
56
Figure 2-9
Figure 2-9: Natural transmission of C. rodentium is not affected by the host vitamin A status. CFU in the feces A), spleen B), and liver C) of A+ cage mates of A-, RA d0 and A+ laboratory inoculated mice. N=4-6/group. Values are the mean + SEM combined for two independent experiments. One-way ANOVA with Tukey post-tests.

57
Chapter 3
Short term retinoic acid treatment promotes mucosal IL-17 expression leading to clearance of enteric infection

58
ABSTRACT
Retinoic acid (RA), the active metabolite of vitamin A, is an important factor in
Citrobacter rodentium clearance. One dose of RA at d7 post infection was effective at
clearing C. rodentium infection in vitamin A deficient (A-) mice. The early effects of RA
included induction of il17a mRNA expression in the colon and regIIIy expression in the
GI tract that occurred after 4 days or 2 doses of RA treatment. One important source of
IL-17a in the gut are CD4+ T cells. Paradoxically, in vitro RA treatment of A- CD4+ T
cells inhibited IL-17a secretion. To study the specific effects of RA on T cells, mice
expressing a dominant negative form of the retinoic acid receptor (dnRAR) were crossed
with Lck-cre mice to generate T-dnRAR mice. T-dnRAR mice had significantly more
CD8+ single positive thymocytes and significantly fewer CD8+ T cells in the spleen and
mesenteric lymph nodes than WT mice. The targets of RA include T cells, IL-17a
production and CD8 T cell development and/or differentiation. In the gut induction of
mRNA for IL-17A was associated with the ability of RA treated mice to clear C.
rodentium.

59
INTRODUCTION
Vitamin A deficiency remains a significant problem in the human population,
particularly in resource limited countries where nutritional intake is a public health
concern (1). Vitamin A deficiency is suggested to contribute to higher prevalence of
diarrheal and respiratory diseases which leads to increased childhood mortality and
vitamin A supplementation effectively decreased mortality in young children by reducing
infection severity (2). Vitamin A deficient (A-) mice treated with retinoic acid (RA), the
active form of vitamin A, promoted clearance of an enteric bacterial infection (3).
Vitamin A is important for protection against infection but the mechanisms of protection
are unclear.
Vitamin A regulates immune cell development, phenotype, function and homing.
RA has a wide variety of effects on the immune system including upregulating gut
homing receptors, α4β7 and CCR9, on T and B cells (4-6) and balancing inflammatory
and regulatory immune responses (7). The balance between inflammatory and regulatory
responses are particularly important in the gut because the mucosal immune system has
to constantly discriminate pathogen from commensal and self from non-self. Dendritic
cells in the gut associated lymphoid tissue (GALT) have the capability to produce RA
(4). RA, in conjunction with transforming growth factor-β (TGF-β), promoted FoxP3+
expression in naïve CD4+ T cells (8, 9). Conversely, RA suppressed-6 and TGF-β
mediated Th17 cell induction (10, 11). These studies suggest that RA is an important
mediator of Th17 and Treg immune responses in vitro. Interestingly, in vivo A- mice had
fewer total T cells in the gut and decreased Th17 responses and increased Th1 responses

60
(12-16). How RA regulates T cells differently in vitro compared to in vivo is not
completely understood.
C. rodentium is a murine pathogen that models human enteropathogenic E. coli
infection and colonizes the intestinal epithelium of the colon through attaching and
effacing lesions (17). C. rodentium must compete with the host microbiota to first
establish colonization (17). Following colonization with C. rodentium, a protective
immune response is mounted, which requires IL-22 production from innate lymphoid
cells, macrophages and γδ T cells to promote intestinal epithelial barrier protection and
induce an adaptive Th17 cell response (18-22). Additionally, T cells and B cells are
required for clearance C. rodentium as mice deficient in T cells, B cells, or both develop
fatal infections within 20 days of inoculation (21, 23).
We have previously reported the effects of host vitamin A status on C. rodentium
infection. A+ mice cleared the bacteria but A- mice failed to clear the infection and
~40% of the A- mice died between d7 and d14 post-infection (3). RA treatment before,
at the time of infection and during infection promoted C. rodentium clearance in the A-
host (3). There was no difference in fecal shedding in A+, A-, or RA dosed animals
before day 10, which suggested that early innate immunity in the A- mice was adequate.
A- mice had significantly fewer T cells in the gut before infection, and although the
numbers of T cells increased with infection, A- mice had fewer T cells in the gut
throughout the course of infection (3). Other studies have shown that animals with
acquired immune defects succumb to C. rodentium infection between d7 and d14 post-
infection (23). Our studies indicate that A- mice are more susceptible to C. rodentium

61
infection, possibly due to an inability to mount an adequate mucosal acquired immune
response.
Our previous study suggested that inadequate acquired T cell responses in A-
mice might be the cause of increased susceptibility. Experiments were done to determine
the targets of RA that promote clearance of C. rodentium. RA treatment starting as early
as d7 post-infection was effective for clearance of C. rodentium. One dose, two doses
and three doses of RA starting at d7 post-infection induced bacterial clearance as well.
RA induced il17a mRNA expression in the colon within 4 days of treatment that was
associated with the successful clearance of C. rodentium. Two doses of RA also induced
regIIIy expression in the GI tract. Interestingly, RA treatment in vitro inhibited IFN-γ
and IL-17a secretion from A- CD4+ T cells. Expression of the dominant negative RAR
(dnRAR) in T cells using an Lck promoter resulted in increased CD8+ single positive
cells in the thymus but decreased CD8+ cells in the spleen and mesenteric lymph node
(MLN). Other possible targets of RA were B cells and the microbiota however no
differences in B cell function or microbiota were detected as a function of vitamin A
status and/or RA treatments. Our data indicates that in vivo RA treatment promoted IL-
17a production in the colon and RA signaling was important in CD8+ T cell development

62
MATERIALS AND METHODS
Mice: C57BL6 mice from Jackson Laboratories (Bar Harbor, MN) were bred at the
Pennsylvania State University (University Park, PA) for experiments. Vitamin A
deficient (A-) and vitamin A sufficient (A+) mice generated as previously described (15,
24). Briefly, mice were fed a purified diet that did not contain any vitamin A (A-) or that
contained 25 µg of retinyl acetate (vitamin A) per day (A+). At weaning, mice were
continuously fed the A- or A+ diet until the end of the study. Vitamin A status was
determined by using ultra pressure liquid chromatography to examine pooled serum
retinol levels in 6-7 week old mice. For some experiments, A- mice were treated with
37.5 µg of all-trans RA (Sigma Aldrich, St Louis, MO) administered orally in 10 μl corn
oil (25). Wild type (WT) C57Bl6 mice fed standard chow diets were used as controls for
microbiota studies.
T-dnRAR mice were generated by crossing Lck-cre mice from Jackson Laboratories (Bar
Harbor, MI) and dominant negative RARα mice, a kind gift from Dr. Randolph Noelle
(Dartmouth University, Lebanon, NH). Experimental procedures were approved by the
Office of Research Protection Institutional Animal Care and Use Committee of the
Pennsylvania State University, University Park, PA.
C. rodentium infection: C. rodentium strain ICC169 (nalidixic acid resistant) was a kind
gifts of Gad Frankel (London School of Medicine and Dentistry, London UK). C.
rodentium ICC169 was cultured in Luria-Bertani (LB, EMD Chemicals, Inc., Gibbstown,
NJ) broth containing 50 μg/ml nalidixic acid (Sigma-Aldrich) and mice 6-8 weeks of age
were infected by oral gavage with 5×109 CFU in 200 μl. Feces and other tissues were

63
collected, homogenized and plated in serial dilutions on LB agar plates containing
naladixic acid.
Cell isolation and culture: Spleens, thymus and MLNs were homogenized and lysed
with red blood cell lysis buffer to obtain single-cell suspensions. SI LPLs and colonic
LPLs were isolated at previously described and stained for flow cytometry (26). For
some experiments, CD4+ T cells from the spleen were purified using mouse CD4 cell
recovery column kits following the manufacturer’s instructions (Cedarlane Laboratories
Ltd, Burlington, NC). The purity of the CD4+ cells after enrichment was between 75-
82% CD4+ cells as determined by flow cytometry.
Cultures included the following concentrations of antibodies: 0.5 μg/ml anti-CD3 alone
or 0.5 μg/ml anti-CD3 with 5 μg/ml anti-CD28 (BD Pharmingen, San Diego, CA) in
RPMI 1640-C containing 10% FBS (Equitech-Bio, Inc, Kerrville, TX), 2 mM L-
glutamine, 5 mM β-mercaptoethanol (Invitrogen, Carlsbad, CA), and 10 μg/ml
gentamycin (Teknova, Hollister, CA). Some cultures contained 10nM of all-trans RA.
ELISAs: Supernatants were collected for cytokine detection and production of IFN-γ, IL-
4, IL-10 and IL-17 (BD Biosciences) were measured by ELISAs following the
manufacturer’s instructions. Serum was collected for antibody titer determination. Total
IgA production was measured by the Mouse IgA ELISA Quantitation Set (Bethyl
Laboratories, Inc, Montgomery, TX). C. rodentium specific antibody titers were
determined by coating ELISAs plates with 30ng/ml sonicated C. rodentium. Horseradish
peroxidase conjugated anti-mouse IgG1 and IgG2 (BD Pharmagen) were used as
detection antibodies and serially diluted pooled serum samples were used as standards to
determine relative values.

64
Flow cytometry: SI LPLs, colonic LPLs, splenocytes and thymocytes were collected and
processed according to standard protocol. Cells were counted and stained with FITC
B220, PE CD4, PE-CD19, PE-Cy5 TCRβ (BD Pharmingen, San Jose, CA USA) and
PE-Cy7 CD8α (BioLegend, San Diego, CA, USA). Cells were analyzed on a FC500
Bench top cytometer (Beckman Coulter, Brea, CA, USA) and data was analyzed using
FlowJo 7.6.1 software (Tree Star, Ashland, OR, USA).
RT-PCR: Whole tissues were snap frozen and RNA was isolated with TRiZOL
(Invitrogen). cDNA was synthesized using TaqMan reverse transcription reagents kit
(Applied Biosystems, Carlsbad, CA). RT- PCR was performed using SYBR green mix
(BioRad) and MyiQ Single-Color Real-Time PCR machine (BioRad). Serially diluted
standards for each gene of interest were run to determine relative copy number.
Expression levels were normalized to the expression of the average of HPRT and
GAPDH and values from A+ uninfected tissue samples were set at 1.
For bacterial DNA isolation, feces were collected from A- cagemates or A+ cagemates,
or from A- cagemates before and after two weeks of RA treatment (37.5 ug/10ul orally
three times a week). DNA was isolated from fecal samples using QIAamp DNA stool
minikit (Qiagen, Valcencia, CA). 10 ng/sample was amplified using SYBR green mix
(BioRad) and MyiQ Single-Color Real-Time PCR machine (BioRad). Data was analyzed
using the ΔΔCT method using 16S as the reference gene and expressed as relative
expression of specific bacterial phyla (27) compared to bacterial DNA isolated from mice
on standards chow diet set as 1. Primers for all RT-PCRs are in Table 3-1.
Statistical Analysis: Statistical analyses were performed using GraphPad Prism
software (Graphpad, La Jolla, CA, USA). Two-way ANOVA with Bonferonni’s post-

65
tests were used to compare CFUs, systemic bacterial loads and tissue RT-PCR across
different dietary groups. Student’s t-tests or nonparametric Mann Whitney tests were
used to compare bacterial DNA analysis, cytokine production and immune cell
populations between dietary groups or genotypes. For all analyses, P<0.05 was used as
the limit for significance.

66
RESULTS
RA treatment of A- mice at d7 promoted clearance.
A- mice failed to clear a C. rodentium infection (Figure 3-1A) and previously published
(3). RA treatment of A- mice at the time of infection (RA d0) resulted in bacterial
clearance (Figure 3-1A) (3). To determine whether RA treatment at the peak of infection
was as effective as at the day of infection, A- mice were treated continuously beginning
at day 7 post-infection (RA d7). RA d7 treatment was as effective as RA d0 treatment for
eliminating C. rodentium (Figure 3-1A). To determine how many RA doses were needed
to protect A- mice from C. rodentium, mice were infected for 7 days and received one
dose of RA (RA 1x) at d7, two doses of RA (RA 2x) at d7 and d9, or three doses of RA
(RA 3x) on d7, d9 and d11 post-infection. All three dosing schemes resulted in clearance
of C. rodentium from the A- host (Figure 3-1a and 31B). In one of 2 experiments, the
RA 3x treated A- mice cleared the infection by d23 and the 3X RA treatment was as
effective as continuous dosing with RA beginning at d0 or d7 (Figure 3-1A and 3-1B). In
the second experiment, the RA 3x group was not cleared more quickly than the other
groups (Figure 3-1C) and therefore the 1X, 2X and 3X RA treatment groups were equally
effective at clearing C. rodentium in A- mice. The average day of clearance from the
combined experiments was not significantly different between the RA 3x and the RA 1x
groups (d22 +1 vs. d26 +2, P=0.0892 by Mann-Whitney test). However, both
experiments showed that 1 dose of RA given at d7 post-infection effectively cleared C.
rodentium in the A- host.

67
RA treatment induced il17a expression in the colon.
To determine the short term effects of RA during C. rodentium infection, A- mice were
infected with C. rodentium for 7 days. At d7, A- mice received one oral dose of RA or
were left untreated. Half of the mice were sacrificed at d9 post-infection while the
remaining were dosed a second time with RA (RA 2X) or remained untreated and then
sacrificed at d11 post-infection (Figure 3-2A). The C. rodentium CFU in the spleen and
liver were not different at d9 or d11 post-infection in the RA 1x, RA 2x and A- mice
(Figure 3-2A). Short term RA treatment had no effect on the CFU in the spleen and liver.
To identify potential targets of RA in the colon, mRNA analysis of colon tissues were
performed. There were no differences in mRNA for il17a, il22, regIIIy, foxp3 or rorc in
the d9 and RA 1x treated mice compared to the A- d9 mRNA values (Figure 3-2B).
There were no differences in mRNA for il22, regIIIy, foxp3 or rorc mRNA expression in
the colons of d11 post-infection A- and 2x RA mice (Figure 3-2A). il17a mRNA
expression was significantly increased in the colon of RA 2x dosed mice compared to A-
mice at the same time point (P<0.01) (Figure 3-2A). Two doses of RA induced
expression of il-17a in the colon of A- mice. In addition, there was a significant
interaction effect on il17a between the VA groups and time (P=0.0105), suggesting that
the group effect differed over time. There was a significant time effect on il22 expression
(P=0.0344), indicating that IL-22 production increased during C. rodentium infection but
was not induced by oral RA treatment.
mRNA levels were measured in the duodenum and ileum of the small intestine (SI) and
colon after RA 2x and at d11 post C. rodentium infection. The duodenum contains the
highest relative concentration of host produced RA in the GI tract (28) and the ileum is

68
still colonized with C. rodentium at d10 post-infection (3). Expression of rorc, foxp3, il6,
il17a, il22 and regIIIy were significantly different based on location in the gut
(P<0.0001). We observed a significant increase in il17a expression in the colon of RA
treated mice (P<0.0286) and a significant interaction effect (P=0.0044) suggesting that
the group effect differs over time. Average expression was highest in the ileum and
lowest in the duodenum for all studied genes, indicating that gene expression can differ in
the GI tract based on location (Figure 3-3). A group effect was observed for regIIIy
(P=0.0169), indicating that RA treatment induced regIIIy expression throughout the GI
tract (Figure 3-3).
Vitamin A and the gut microbiota.
Previous reports have suggested that the microbiota regulated the ability to produce IL-17
(29). Additionally, one report showed that A- mice had fewer gut microbes in the SI and
feces as well as fewer mucosal Th17 cells (16). If the microbiota was different between
A+ and A-mice, this could explain the differences in IL-17 production and the inability of
the A- mice to clear C. rodentium. There were no differences in the bacterial 16S
ribosomal content of the feces from the mice as a consequence of vitamin A status
(Figure 3-4A). In addition, the feces from A+ and A- mice contained similar ratios of
Bacteriodies, Firmicutes, Actinobacter and γ-Proteobacter phyla DNA (Figure 3-4B).
Our results indicate that vitamin A status did not affect total bacteria in the feces or the
representation of 4 of the common phyla of bacteria found in mice.

69
Serum antibody levels and B cell numbers were not affected by vitamin A
deficiency.
Vitamin A regulates B cell responses (5, 7, 25). We hypothesized that A- mice failed to
mount a robust C. rodentium specific antibody response that might explain the inability to
clear an infection. There was no difference in total serum IgA or C. rodentium IgA levels
between A- and A+ mice but there was a significant interaction effect (P=0.0400) in total
IgA levels, indicating that the group effect differed over time (Figure 3-6A). C.
rodentium specific IgG1 and IgG2 titers were not affected by host vitamin A status
(Figure 3-6B). There was an observed time effect for C. rodentium specific IgA
(P=0.0005) and IgG2 (P=0.0025), indicating that antigen specific antibody levels
increased during infection. Additionally, there was no difference in B cell numbers in the
SI LPL (Figure 3-6C) or colonic LPL (Figure 3-6 D). B cell numbers and C. rodentium
specific antibody responses were not affected by vitamin A.
In vitro RA treatment inhibited Th1/Th17 cytokine production from T cells.
As previously published, 10nM RA inhibited IFN-γ secretion from A- splenocytes
(Figure 3-6A) (30). IL-17a production from A- and RA treated splenocytes was below
the limit of detection. RA treatment of enriched CD4+ T cells from mice significantly
inhibited IFN-γ and IL-17a production (Figure 3-6B). IL-4 and IL-10 levels were
undetectable in splenocyte and CD4+ T cell cultures (data not shown). Our data confirms
data in the literature demonstrating an inhibitory effect of RA on IFN-γ and IL-17
production from T cells in vitro.

70
Thymic development was impaired in mice that express a dnRAR receptor in T
cells.
T-dnRAR mice were generated to study the effects of retinoid signaling in T cells. To
confirm that the dnRAR was functioning in T cells, splenocytes were stimulated with
anti-CD3 antibody and treated with 10nM RA or ethanol as a control. RA treatment
significantly inhibited IFN-γ production in WT cells (Figure 3-7). RA treatment had no
effect on IFN-γ secretion from anti-CD3 stimulated T-dnRAR splenocytes (Figure 3-7).
There was no effect of RA on IL-17a in either the WT or T-dnRAR cultures for these
assays (Figure 3-7). These data indicated that T cells from the T-dnRAR mice were
unable to respond to RA treatment in vitro.
In the thymus, T cells develop through a double negative (DN) to a double positive (DP)
stage and then to single positive either CD4 or CD8 T cells. Frequencies of DN, DP,
CD4+ and CD8+ cells from the WT littermates were consistent with other reports (31).
WT mice had significantly more total thymocytess than T-dnRAR littermates (Table 3-2,
Figure 3-8 A). There were more Tcrβ+ T cells in the WT thymus compared to T-dnRAR
mice (Table 3-2). WT mice had more DP and less CD8 single positive thymocytes than
the T-dnRAR mice (Table 3-2, Figure 3-8A). In the periphery, WT mice had
significantly increased Tcrβ+ cells and CD8+ T cells in the spleen (Table 3-2, Figure 3-
8B). WT mice had more total cells and fewer CD8+ T cells in MLN than the T-dnRAR
mice (Table 3-2, Figure 3-8C). There were not differences in B cells (B220+) in the
spleen and MLN between WT and T-dnRAR mice (data not shown). Overall, the T-
dnRAR mice had fewer T cells in the thymus as well as in the periphery than their WT

71
counterparts, specifically decreased CD8+ T cells in the spleen and MLN. These data
indicate that RA signaling is important for CD8+ T cell development.

72
DISCUSSION
Here, for the first time, we show that short term treatment with RA during enteric
infection promoted bacterial clearance. Additionally, RA treatment of A- mice starting at
d7 post-infection was as effective as at the time of infection for clearance of C.
rodentium. Only 1 dose of RA was required for A- mice to clear a C. rodentium
infection. Induction of IL-17a mRNA expression in the colon of RA treated mice
corresponded with the elimination of C. rodentium in the RA treated A- mice. IL-17a
was induced in the colon following RA treatment. RA treatments of A- hosts could be
effective for eliminating carriers of Escherichia coli-like infections in the A- host.
The effect of RA in vitro and the effects of vitamin A in vivo on IL-17 and Th17 cells is
paradoxical. Our data confirms that RA treatment in vitro inhibited IL-17a production
from A- CD4+ T cells in vitro. Previously published in vitro studies have shown that RA
inhibited Th17 induction when IL-6 was absent (7, 32). Other in vitro experiments also
showed that 10nM to 1uM RA can inhibited Th17 cell induction and reduced IFN-γ and
IL-17a secretion (12, 33, 34). Two independent studies have shown that in vitro
treatment of naive T cells with 1nM RA was needed for IL-17a production (12, 34). All
referenced in vitro assays used naïve CD4+ T cells but used a variety of mouse strains
(C57BL/6 and BALB/c (12), C57BL6 OT-II (34), B6.SJL (13), or AKR/J (12)),
indicating that different mouse strains may respond to RA or metabolize vitamin A
differently (35). Our data show that RA in vivo induced IL-17a expression in the colon
of C57BL/6 mice. At this time, we do not know the source of the IL-17a mRNA in the
colon of RA treated mice. Several different populations of cells produce IL-17a

73
including T cells, innate lymphoid cells and monocytes (36). The effects of vitamin A in
vivo are not completely understood.
One explanation for the increased IL-17a expression as a result of RA treatment may be
the recruitment of Th17 cells from the periphery to the colon. We have observed that A-
mice had fewer colonic T cells than A+ mice (3) and it is known that RA upregulates gut
homing receptors on T cells in vitro (4). A- mice and retinoic acid receptor (RAR)α
deficient mice had very few Th17 cells in the gut (12-14, 16) . The decreased Th17 cells
in the gut of A- mice was also observed in a genetic mouse model of Crohn’s disease
(SAMP1/YP mouse model) however this seemed to have no effect on the model (37).
IL-17+ T cells generated in vitro in the presence of 1nM RA expressed high levels of
CCR9 and α4β7 (12). Furthermore, when these IL-17+ T cells were transferred into
RagKO mice, they trafficked to the gut and caused inflammation (12). If RA was
required to get Th17 cells to home to the gut during infection, this could account for the
observed increased expression of IL-17a in the RA treated animals compared to the
untreated mice.
To further determine possible targets of vitamin A in host resistance to C. rodentium, the
microbiota and B cells were examined. Because C. rodentium competes with the
commensal microbiota for colonization of the host, we measured 16S DNA and bacteria
phyla in the feces of A+ and A- mice. We found no differences in total bacterial DNA.
Additionally, we found no effect of vitamin A status on Bacteriodetes, Firmicutes,
Actinobacteria or γ-Proteobacteria. Others have reported that A- mice have fewer gut
bacteria in the ileum and feces by RT-PCR and bacterial cultures (16). A wide range of
factors affect gut microbiota composition (i.e. diet, animal facility, vendors) and these

74
factors along with experimental differences could account for the different results (38-
40). In addition, we found no differences in serum IgA, antigen specific IgA or antigen
specific IgG1 and IgG2 antibody levels in A- and A+ mice throughout infection or in B
cell numbers in the LP of the GI tract. Currently, our data does not support an effect of
vitamin A on the microbiota or B cells in the gut, however the relationship between RA
and gut microbiota or RA and mucosal B cells has not yet been thoroughly studied.
These data indicate that other acquired immune system components, like T cells, may be
a main target of RA during C. rodentium infection.
To examine the effects of RA on T cells, we generated T-dnRAR mice. Others have used
the dnRAR mouse to inhibit RA signaling in T cells using a CD4-cre mouse (31, 41).
Reports using the CD4 cre-dnRAR mice showed that, like in our study, the CD8+ single
positive thymocytes frequencies were increased compared to WT littermates (31).
However, there were no differences in the CD8+ T cell populations in the periphery
compared to WT mice in the CD4 cre-dnRAR mice and we showed that there were fewer
CD8+ in the periphery using the Lckcre-dnRAR mouse (31). Memory CD4+ T cell
numbers were not different between WT and CD4 cre-dnRAR mice although the CD4cre
dnRAR T cells produced more IL-17 and less IFN-γ than the WT T cells (41). By using
the Lck cre mouse, we have inhibited RA signaling at the DN stage of development while
the groups that use the CD4 cre mouse inhibited retinoid signaling at the DP stage (31,
41). Our data showed that when RA signaling is inhibited in T cells, double positive
(CD8+CD4+) thymocyte numbers were significantly decreased but CD8+ thymocyte
numbers were significantly increased. The Lck cre dnRAR CD8+ T cells may have a
defect in survival, maturation or proliferation. Defects in CD8+ T cells could result in

75
the inability of host to survive viral or intracellular bacterial infections. Thymic organ
culture and flow cytometry showed that in vitro RA treatment of human thymocytes
promoted CD4+ thymocyte development and inhibited CD8+ thymocyte development
from the DP stage, indicating that RA may affect CD8+ T cell development (42). The
data suggest that RA signaling is important for CD8+ T cell development and for
maintaining CD8+ T cells periphery.
From this study, we determined that RA treatment of A- mice at d7 post infection was
just as effective as RA treatment starting at d0 in clearing C. rodentium infection.
Additionally, we found that one dose of RA at d7 post infection eliminated C. rodentium
infection from the A- host. One target of RA in the colon that corresponded with C.
rodentium clearance was IL-17a mRNA levels. However, we do not know what cells
were making IL-17a. Determining which cells were producing IL-17 would be critical to
understanding the mechanisms underlying the effects of RA on host resistance in A-
mice.. The work shows that RA is a critical regulator of T cell development and the local
IL-17 response during C. rodentium infection.

76
REFERENCES: 1. WHO. 2009. Global prevalence of vitamin A deficiency in populations at risk
1995–2005: WHO global database on vitamin A deficiency. In World Health Organization, Geneva. 2. Fawzi, E. V. a. W. W. 2000. Vitamin A supplementation: implications for
morbidity and mortality in children. Journal of Infectious Diseases 2000: S112-133.
3. McDaniel, K. L., K. H. Restori, J. W. Dodds, M. J. Kennett, A. C. Ross, and M. T. Cantorna. 2015. Vitamin A-Deficient Hosts Become Nonsymptomatic Reservoirs of Escherichia coli-Like Enteric Infections. Infect Immun 83: 2984-2991.
4. Iwata, M., A. Hirakiyama, Y. Eshima, H. Kagechika, C. Kato, and S. Y. Song. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21: 527-538.
5. Mora, J. R., M. Iwata, B. Eksteen, S. Y. Song, T. Junt, B. Senman, K. L. Otipoby, A. Yokota, H. Takeuchi, P. Ricciardi-Castagnoli, K. Rajewsky, D. H. Adams, and U. H. von Andrian. 2006. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314: 1157-1160.
6. Iwata, M. 2009. Retinoic acid production by intestinal dendritic cells and its role in T-cell trafficking. Semin Immunol 21: 8-13.
7. Mora, J. R., M. Iwata, and U. H. von Andrian. 2008. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 8: 685-698.
8. Coombes, J. L., K. R. Siddiqui, C. V. Arancibia-Carcamo, J. Hall, C. M. Sun, Y. Belkaid, and F. Powrie. 2007. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204: 1757-1764.
9. Sun, C. M., J. A. Hall, R. B. Blank, N. Bouladoux, M. Oukka, J. R. Mora, and Y. Belkaid. 2007. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204: 1775-1785.
10. Mucida, D., Y. Park, G. Kim, O. Turovskaya, I. Scott, M. Kronenberg, and H. Cheroutre. 2007. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317: 256-260.
11. Elias, K. M., A. Laurence, T. S. Davidson, G. Stephens, Y. Kanno, E. M. Shevach, and J. J. O'Shea. 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111: 1013-1020.
12. Wang, C., S. G. Kang, H. HogenEsch, P. E. Love, and C. H. Kim. 2010. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol 184: 5519-5526.
13. Hall, J. A., J. L. Cannons, J. R. Grainger, L. M. Dos Santos, T. W. Hand, S. Naik, E. A. Wohlfert, D. B. Chou, G. Oldenhove, M. Robinson, M. E. Grigg, R. Kastenmayer, P. L. Schwartzberg, and Y. Belkaid. 2011. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34: 435-447.

77
14. Spencer, S. P., C. Wilhelm, Q. Yang, J. A. Hall, N. Bouladoux, A. Boyd, T. B. Nutman, J. F. Urban, Jr., J. Wang, T. R. Ramalingam, A. Bhandoola, T. A. Wynn, and Y. Belkaid. 2014. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343: 432-437.
15. Carman, J. A., S. M. Smith, and C. E. Hayes. 1989. Characterization of a helper T lymphocyte defect in vitamin A-deficient mice. J Immunol 142: 388-393.
16. Cha, H. R., S. Y. Chang, J. H. Chang, J. O. Kim, J. Y. Yang, C. H. Kim, and M. N. Kweon. 2010. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J Immunol 184: 6799-6806.
17. Collins, J. W., K. M. Keeney, V. F. Crepin, V. A. Rathinam, K. A. Fitzgerald, B. B. Finlay, and G. Frankel. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12: 612-623.
18. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong, A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. de Sauvage, and W. Ouyang. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14: 282-289.
19. Sonnenberg, G. F., and D. Artis. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37: 601-610.
20. Mielke, L. A., S. A. Jones, M. Raverdeau, R. Higgs, A. Stefanska, J. R. Groom, A. Misiak, L. S. Dungan, C. E. Sutton, G. Streubel, A. P. Bracken, and K. H. Mills. 2013. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210: 1117-1124.
21. Mundy, R., T. T. MacDonald, G. Dougan, G. Frankel, and S. Wiles. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7: 1697-1706.
22. Mangan, P. R., L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231-234.
23. Bry, L., and M. B. Brenner. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172: 433-441.
24. Smith, S. M., N. S. Levy, and C. E. Hayes. 1987. Impaired immunity in vitamin A-deficient mice. J Nutr 117: 857-865.
25. Ma, Y., Q. Chen, and A. C. Ross. 2005. Retinoic acid and polyriboinosinic:polyribocytidylic acid stimulate robust anti-tetanus antibody production while differentially regulating type 1/type 2 cytokines and lymphocyte populations. J Immunol 174: 7961-7969.
26. Lefrancois, L., and N. Lycke. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. Curr Protoc Immunol Chapter 3: Unit 3 19.
27. Bacchetti De Gregoris, T., N. Aldred, A. S. Clare, and J. G. Burgess. 2011. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J Microbiol Methods 86: 351-356.

78
28. Villablanca, E. J., S. Wang, J. de Calisto, D. C. Gomes, M. A. Kane, J. L. Napoli, W. S. Blaner, H. Kagechika, R. Blomhoff, M. Rosemblatt, M. R. Bono, U. H. von Andrian, and J. R. Mora. 2011. MyD88 and retinoic acid signaling pathways interact to modulate gastrointestinal activities of dendritic cells. Gastroenterology 141: 176-185.
29. Ivanov, II, K. Atarashi, N. Manel, E. L. Brodie, T. Shima, U. Karaoz, D. Wei, K. C. Goldfarb, C. A. Santee, S. V. Lynch, T. Tanoue, A. Imaoka, K. Itoh, K. Takeda, Y. Umesaki, K. Honda, and D. R. Littman. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485-498.
30. Cantorna, M. T., F. E. Nashold, and C. E. Hayes. 1994. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol 152: 1515-1522.
31. Pino-Lagos, K., Y. Guo, C. Brown, M. P. Alexander, R. Elgueta, K. A. Bennett, V. De Vries, E. Nowak, R. Blomhoff, S. Sockanathan, R. A. Chandraratna, E. Dmitrovsky, and R. J. Noelle. 2011. A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J Exp Med 208: 1767-1775.
32. Kimura, A., and T. Kishimoto. 2010. IL-6: regulator of Treg/Th17 balance. Eur J Immunol 40: 1830-1835.
33. Guo, Y., C. Brown, C. Ortiz, and R. J. Noelle. 2015. Leukocyte homing, fate, and function are controlled by retinoic acid. Physiol Rev 95: 125-148.
34. Uematsu, S., K. Fujimoto, M. H. Jang, B. G. Yang, Y. J. Jung, M. Nishiyama, S. Sato, T. Tsujimura, M. Yamamoto, Y. Yokota, H. Kiyono, M. Miyasaka, K. J. Ishii, and S. Akira. 2008. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol 9: 769-776.
35. Goverse, G., B. J. Olivier, R. Molenaar, M. Knippenberg, M. Greuter, T. Konijn, E. C. Cook, M. R. Beijer, D. M. Fedor, J. M. den Haan, J. L. Napoli, G. Bouma, and R. E. Mebius. 2015. Vitamin A metabolism and mucosal immune function are distinct between BALB/c and C57BL/6 mice. Eur J Immunol 45: 89-100.
36. Mills, K. H. 2008. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol 38: 2636-2649.
37. Kang, S. G., C. Wang, S. Matsumoto, and C. H. Kim. 2009. High and low vitamin A therapies induce distinct FoxP3+ T-cell subsets and effectively control intestinal inflammation. Gastroenterology 137: 1391-1402 e1391-1396.
38. Ericsson, A. C., J. W. Davis, W. Spollen, N. Bivens, S. Givan, C. E. Hagan, M. McIntosh, and C. L. Franklin. 2015. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS One 10: e0116704.
39. Hufeldt, M. R., D. S. Nielsen, F. K. Vogensen, T. Midtvedt, and A. K. Hansen. 2010. Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comp Med 60: 336-347.
40. Moschen, A. R., V. Wieser, and H. Tilg. 2012. Dietary Factors: Major Regulators of the Gut's Microbiota. Gut Liver 6: 411-416.
41. Brown, C. C., D. Esterhazy, A. Sarde, M. London, V. Pullabhatla, I. Osma-Garcia, R. Al-Bader, C. Ortiz, R. Elgueta, M. Arno, E. de Rinaldis, D. Mucida, G. M. Lord, and R. J. Noelle. 2015. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42: 499-511.

79
42. Zhou, X., W. Wang, and Y. Yang. 2008. The expression of retinoic acid receptors in thymus of young children and the effect of all-transretinoic acid on the development of T cells in thymus. J Clin Immunol 28: 85-91.
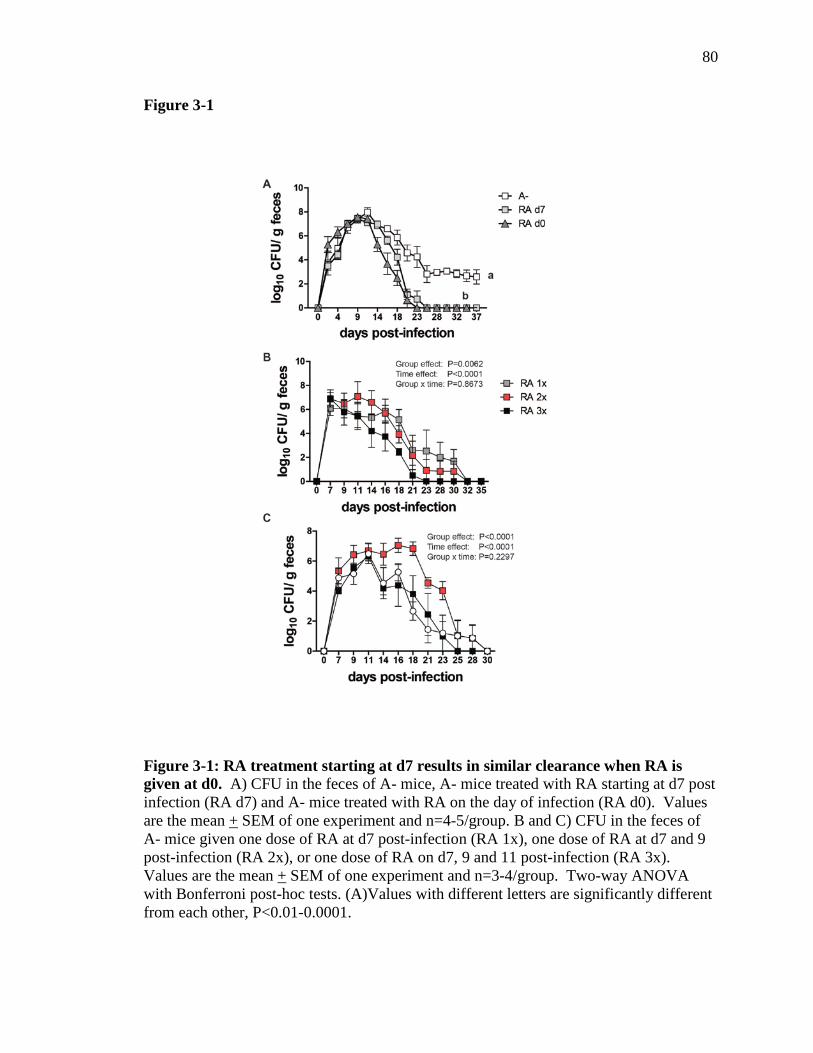
80
Figure 3-1 Figure 3-1: RA treatment starting at d7 results in similar clearance when RA is given at d0. A) CFU in the feces of A- mice, A- mice treated with RA starting at d7 post infection (RA d7) and A- mice treated with RA on the day of infection (RA d0). Values are the mean + SEM of one experiment and n=4-5/group. B and C) CFU in the feces of A- mice given one dose of RA at d7 post-infection (RA 1x), one dose of RA at d7 and 9 post-infection (RA 2x), or one dose of RA on d7, 9 and 11 post-infection (RA 3x). Values are the mean + SEM of one experiment and n=3-4/group. Two-way ANOVA with Bonferroni post-hoc tests. (A)Values with different letters are significantly different from each other, P<0.01-0.0001.
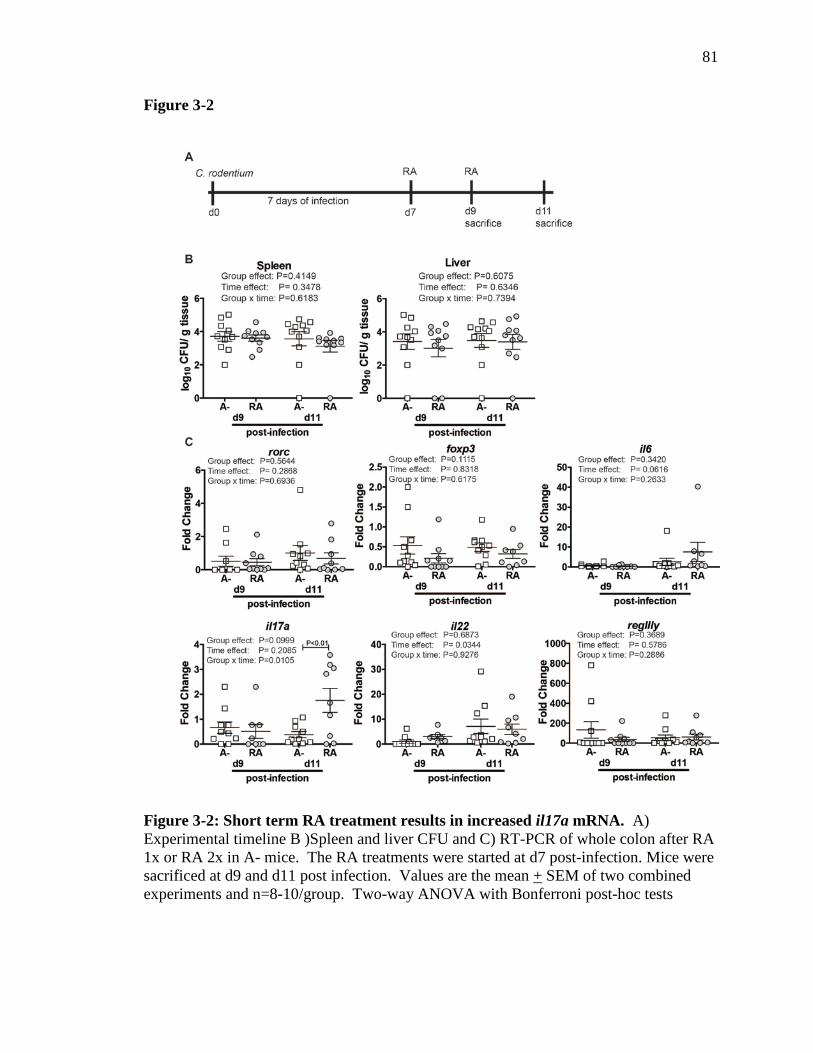
81
Figure 3-2
Figure 3-2: Short term RA treatment results in increased il17a mRNA. A) Experimental timeline B )Spleen and liver CFU and C) RT-PCR of whole colon after RA 1x or RA 2x in A- mice. The RA treatments were started at d7 post-infection. Mice were sacrificed at d9 and d11 post infection. Values are the mean + SEM of two combined experiments and n=8-10/group. Two-way ANOVA with Bonferroni post-hoc tests

82
Table 3-1: RT-PCR primer sequences HPRT: F: 3’-GTAATGTTATCGAGAAGTCAGAC-5’
R: 3’-CACCTTCTATATTAACTGTGACC-5’
GAPDH: F: 3’-TAGCTTCCACCTTCTACACCAAA;-5’ R: 3’-GATGTCGTTGTCCCACCACC-5’
FOXP3: F: 3’-GACGTATCGAGGGTCGAAGA-5’ R: 3’-CGAGAACGACGTAGCATCG-5’
RORC: F: 3’-AGGAGGGCACTTTTCTCCA-5’ R: 3’-GTTACACCCTCTACACCCT-5’
IL-17A: F: 3’-TGTGTCTACTTCGAGAGGGAC-5’ R: 3’-TAGTAGGGAGTTTCGACTCG-5’
IL-22: IL-6: REGIIIy
F: 3’-AGACCTACAAGACCAGCAGT-5’ R: 3’-CACCTCTCTAGTTCCGCTAA-5’ F: 3’-AGGACTAATATAGGTCAAACCCATCG-5’ R: 3’-AAGACCTCATGGTATCGATGGACC-5’ F: 3’-AAAACTAGTACCTCCTGTCCTT-5’ R: 3’-ACTTGGGTTGTCTCCACCTAC-5’
16S: F: 3’-AAACTCAAAKGAATTGACGG-5’ R: 3’-CTCACRRCACGAGCTGAC-5’
Bacteriodetes: F: 3’-CRAACAGGATTAGATACCCT-5’ R: 3’-GGTAAGGTTCCTCGCGTAT-5’
Firmicutes: F: 3’-TGAAACTYAAAGGAATTGACG-5’ R: 3’-ACCATGACCACCTGTC-5’
Actinobacteria: F: 3’-TACGGCCGCAAGGCTA-5’ R: 3’-TCRTCCCACCTTCCTCCG-5’
y-Proteobacteria F: 3’-TCGTCAGCTCGTGTYGTGA-5’ R: 3’-CGTAAGGGCCATGATG-5’
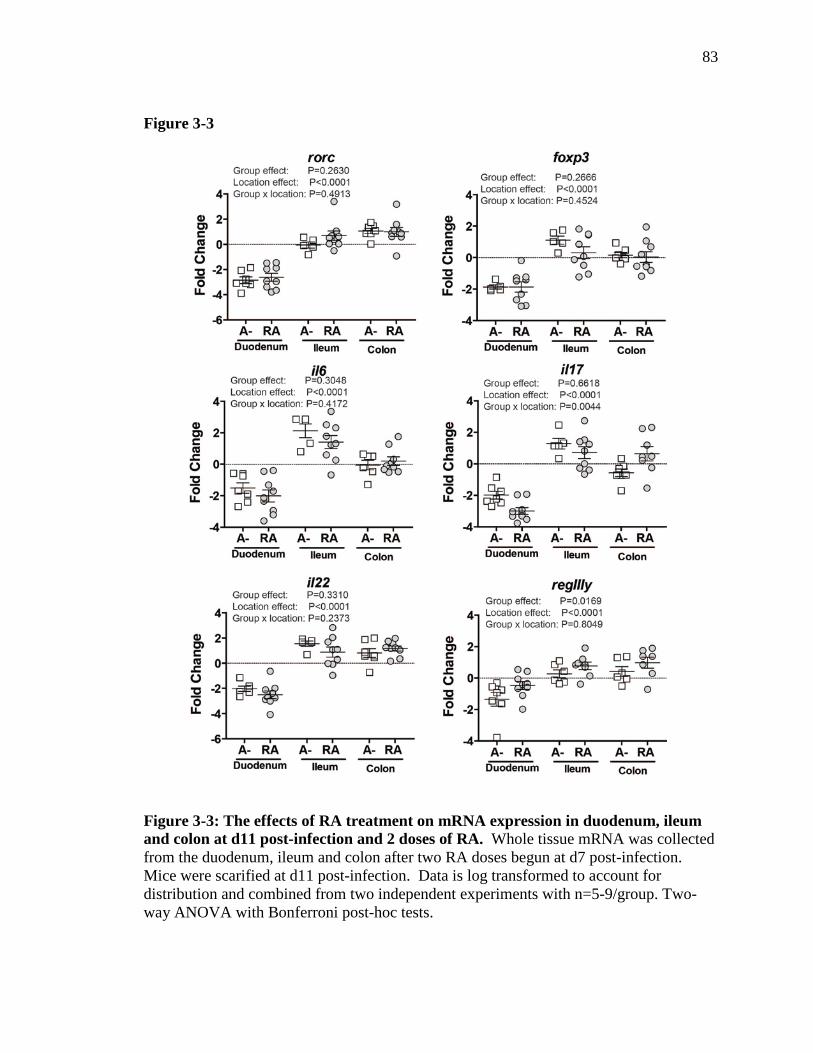
83
Figure 3-3 Figure 3-3: The effects of RA treatment on mRNA expression in duodenum, ileum and colon at d11 post-infection and 2 doses of RA. Whole tissue mRNA was collected from the duodenum, ileum and colon after two RA doses begun at d7 post-infection. Mice were scarified at d11 post-infection. Data is log transformed to account for distribution and combined from two independent experiments with n=5-9/group. Two-way ANOVA with Bonferroni post-hoc tests.
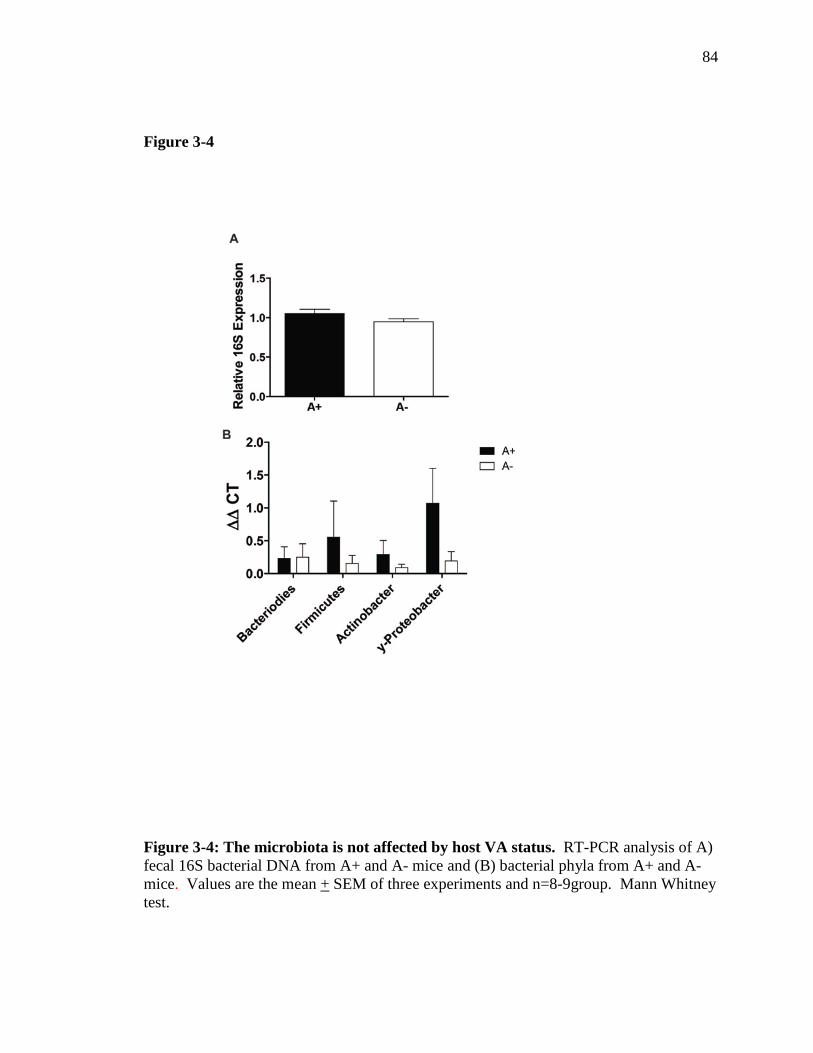
84
Figure 3-4
Figure 3-4: The microbiota is not affected by host VA status. RT-PCR analysis of A) fecal 16S bacterial DNA from A+ and A- mice and (B) bacterial phyla from A+ and A- mice. Values are the mean + SEM of three experiments and n=8-9group. Mann Whitney test.
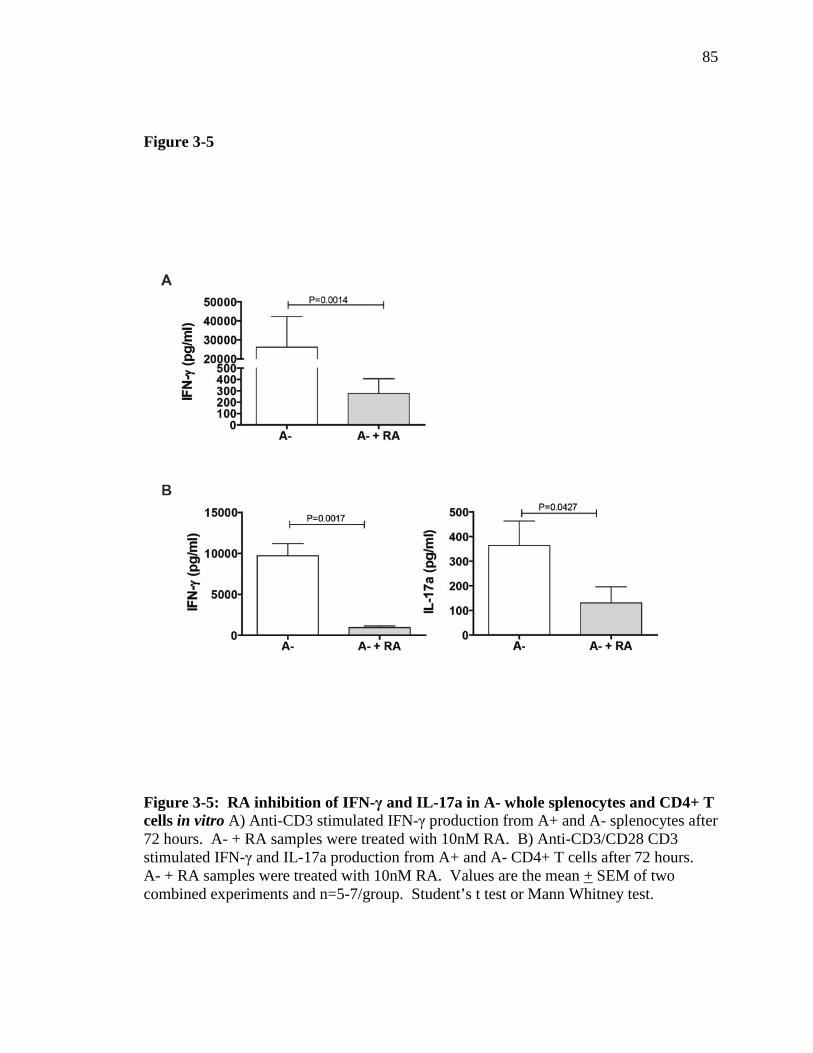
85
Figure 3-5
Figure 3-5: RA inhibition of IFN-γ and IL-17a in A- whole splenocytes and CD4+ T cells in vitro A) Anti-CD3 stimulated IFN-γ production from A+ and A- splenocytes after 72 hours. A- + RA samples were treated with 10nM RA. B) Anti-CD3/CD28 CD3 stimulated IFN-γ and IL-17a production from A+ and A- CD4+ T cells after 72 hours. A- + RA samples were treated with 10nM RA. Values are the mean + SEM of two combined experiments and n=5-7/group. Student’s t test or Mann Whitney test.
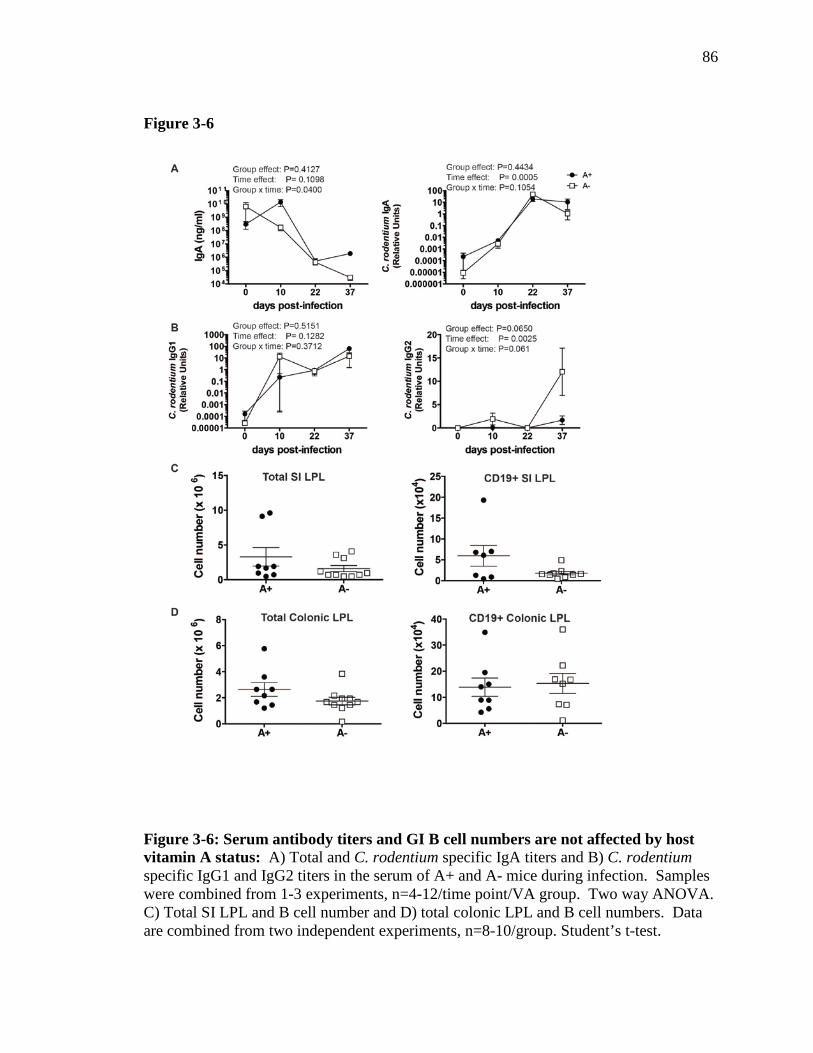
86
Figure 3-6
Figure 3-6: Serum antibody titers and GI B cell numbers are not affected by host vitamin A status: A) Total and C. rodentium specific IgA titers and B) C. rodentium specific IgG1 and IgG2 titers in the serum of A+ and A- mice during infection. Samples were combined from 1-3 experiments, n=4-12/time point/VA group. Two way ANOVA. C) Total SI LPL and B cell number and D) total colonic LPL and B cell numbers. Data are combined from two independent experiments, n=8-10/group. Student’s t-test.
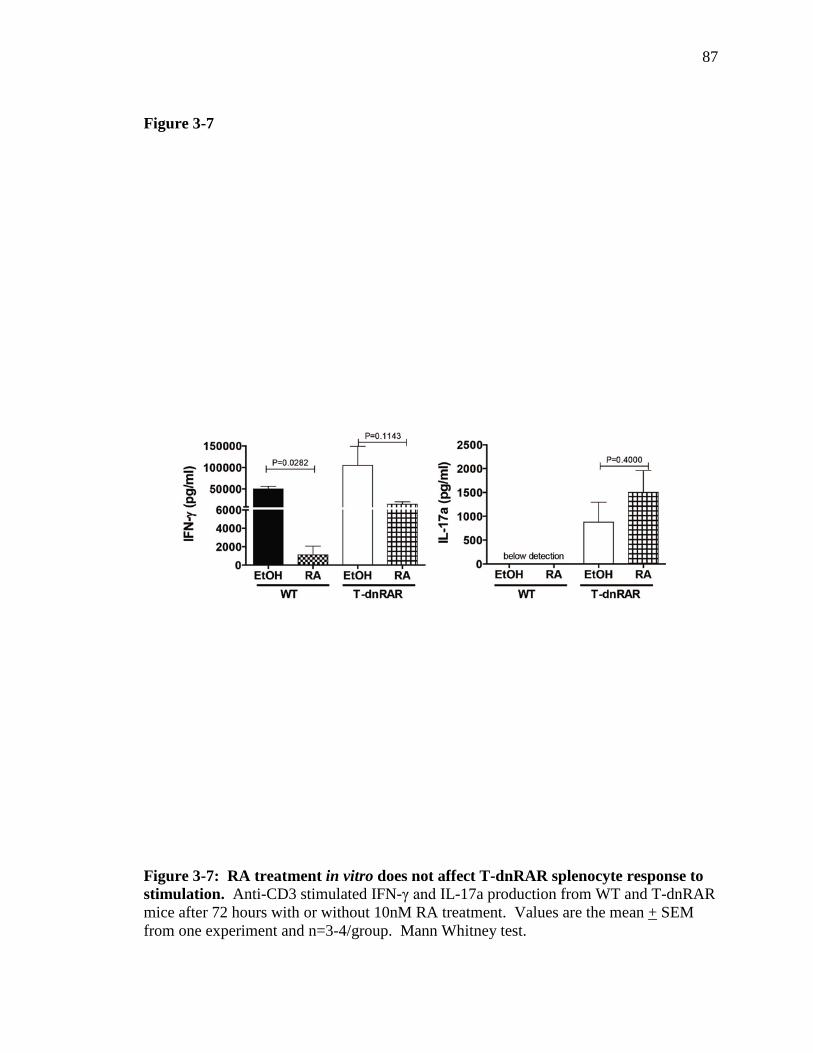
87
Figure 3-7
Figure 3-7: RA treatment in vitro does not affect T-dnRAR splenocyte response to stimulation. Anti-CD3 stimulated IFN-γ and IL-17a production from WT and T-dnRAR mice after 72 hours with or without 10nM RA treatment. Values are the mean + SEM from one experiment and n=3-4/group. Mann Whitney test.
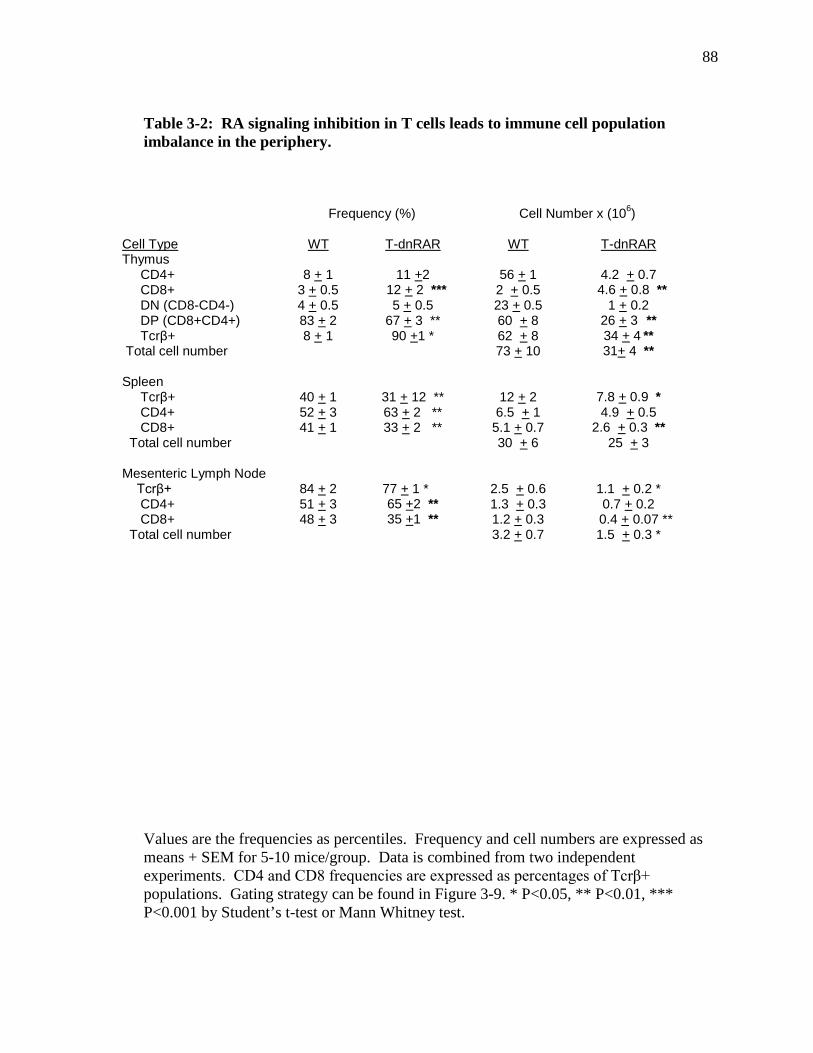
88
Table 3-2: RA signaling inhibition in T cells leads to immune cell population imbalance in the periphery.
Frequency (%)
Cell Number x (106)
Cell Type WT T-dnRAR WT T-dnRAR Thymus CD4+ 8 + 1 11 +2 56 + 1 4.2 + 0.7 CD8+ 3 + 0.5 12 + 2 *** 2 + 0.5 4.6 + 0.8 ** DN (CD8-CD4-) 4 + 0.5 5 + 0.5 23 + 0.5 1 + 0.2 DP (CD8+CD4+) 83 + 2 67 + 3 ** 60 + 8 26 + 3 ** Tcrβ+ 8 + 1 90 +1 * 62 + 8 34 + 4 ** Total cell number 73 + 10 31+ 4 ** Spleen Tcrβ+ 40 + 1 31 + 12 ** 12 + 2 7.8 + 0.9 * CD4+ 52 + 3 63 + 2 ** 6.5 + 1 4.9 + 0.5 CD8+ 41 + 1 33 + 2 ** 5.1 + 0.7 2.6 + 0.3 ** Total cell number 30 + 6 25 + 3 Mesenteric Lymph Node Tcrβ+ 84 + 2 77 + 1 * h 2.5 + 0.6 1.1 + 0.2 * CD4+ 51 + 3 65 +2 ** 1.3 + 0.3 0.7 + 0.2 CD8+ 48 + 3 35 +1 ** 1.2 + 0.3 0.4 + 0.07 ** Total cell number 3.2 + 0.7 1.5 + 0.3 *
Values are the frequencies as percentiles. Frequency and cell numbers are expressed as means + SEM for 5-10 mice/group. Data is combined from two independent experiments. CD4 and CD8 frequencies are expressed as percentages of Tcrβ+ populations. Gating strategy can be found in Figure 3-9. * P<0.05, ** P<0.01, *** P<0.001 by Student’s t-test or Mann Whitney test.
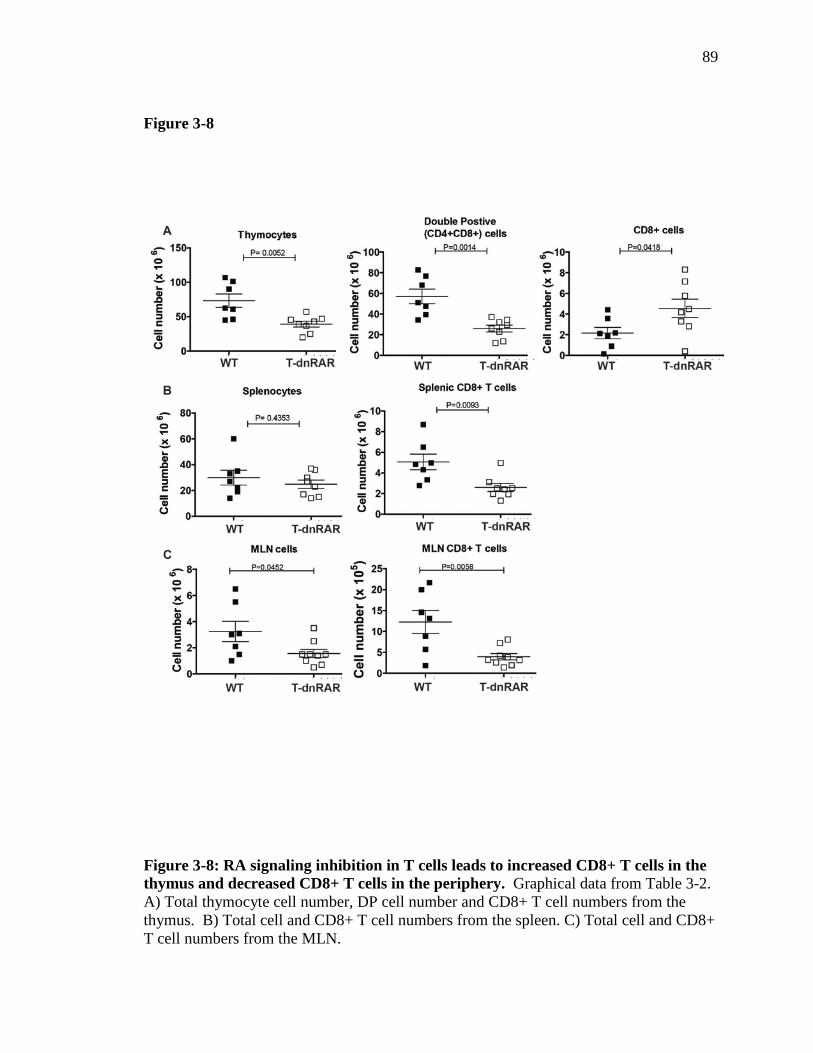
89
Figure 3-8
Figure 3-8: RA signaling inhibition in T cells leads to increased CD8+ T cells in the thymus and decreased CD8+ T cells in the periphery. Graphical data from Table 3-2. A) Total thymocyte cell number, DP cell number and CD8+ T cell numbers from the thymus. B) Total cell and CD8+ T cell numbers from the spleen. C) Total cell and CD8+ T cell numbers from the MLN.
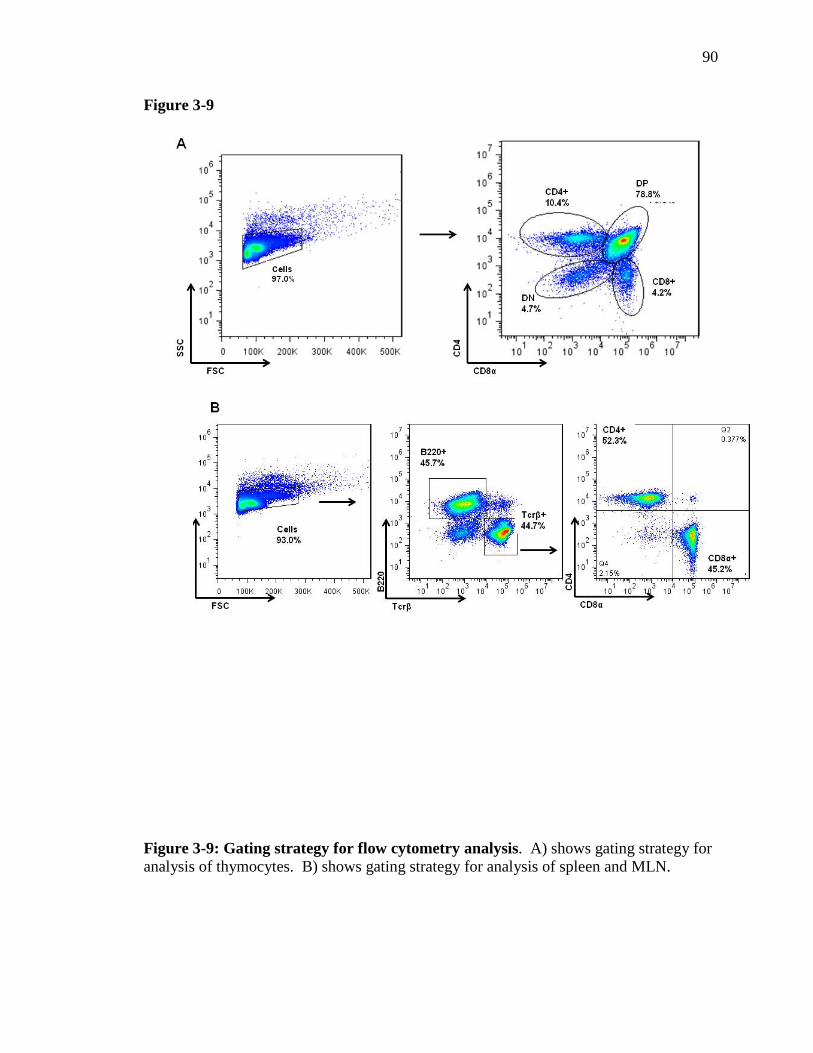
90
Figure 3-9
Figure 3-9: Gating strategy for flow cytometry analysis. A) shows gating strategy for analysis of thymocytes. B) shows gating strategy for analysis of spleen and MLN.

91
Chapter 4
Summary and conclusions

92
The role of vitamin A in enteric infection was investigated in this thesis. In
Chapter 2, the effect of host vitamin A status during Citrobacter rodentium infection was
evaluated. While it has been known that vitamin A deficiency is associated with
respiratory and diarrheal diseases in children (1, 2), the effects of vitamin A during C.
rodentium infection were unknown. I show here that A- mice failed to clear C.
rodentium infection. T cell numbers were decreased in the colon of A- mice compared to
A+ mice. Additionally, my work showed that RA treatment before infection, at the time
of infection and during infection eliminated the pathogen from the host. I observed 40%
mortality in A- mice following C. rodentium infection. Death following C. rodentium
infection had been attributed to sepsis (3-5), however I did not find elevated serum
cytokines in the A- mice compared to A+ mice. Furthermore, necropsies of moribund
mice did not show signs of sepsis but did find indicators of severe gut damage. Together
the data suggests that the C. rodentium related mortality in the A- host was not caused by
sepsis. However, this manuscript did not determine why the A- mice did not clear C.
rodentium or how RA treatment promoted infection clearance in A- mice. The major
conclusions from the first manuscript were that A- mice are susceptible to C. rodentium
infection and RA can be given before, at the time of or during infection to promote
survival and clearance of the infection. The work demonstrated a new role for vitamin A
for eliminating possible reservoirs of enteric bacteria in A- hosts (6).
The timing of RA treatment and the mechanisms by which RA regulated host
resistance to enteric infection were probed in Chapter 3. RA treatment starting at d7
post- infection was as effective as at the time of infection for C. rodentium clearance.

93
Furthermore, it was determined that one 37.5 ug dose of RA starting at d7 post infection
was enough to clear C. rodentium from the A- host. The data showed that RA treatment
during infection induced IL-17a production which was associated with clearance of C.
rodentium. Other reports indicated that A- mice have inadequate Th17 immune
responses and fewer Th17 cells in the gut (7) and our data showed that A- mice were
unable to clear an infection. Our previous study also showed that A- mice had fewer total
T cells in the colon compared to A+ mice (6) (7-10). However, in vitro RA inhibited IL-
17a production from A- CD4+ T cells. In addition to CD4+ T cells, IL-17a is produced
by many other cell types including CD8+ T cells, γδ T cells, innate lymphoid cells,
natural killer (NK) T cells, NK cells and neutrophils (11, 12). There are at least three
possibilities that could explain the induction of RA mRNA in the colon of A- mice
infected with C. rodentium and directly inhibiting IL-17a from CD4+ T cells: 1) RA
treatment promoted IL-17a production from other (CD8, γδT, ILC etc.) cells in the gut 2)
RA treatment promoted IL-17 producing immune cells to home to the gut or 3) RA
indirectly resulted in CD4+ T cells making more IL-17a in the gut via innate cell
production of IL-22. These three possibilities are not mutually exclusive. Further work
needs to be done to determine which of the mechanisms account for increased IL-17a in
the colon following RA treatment of C. rodentium infected mice.
Because the early shedding kinetics (before day 10-11) of C. rodentium were not
different in A-, A+ and RA treated mice, we hypothesized that the adaptive immune
response and not the early innate immune responses was the target of vitamin A in the
gut. In addition, it has been shown that both T cell and B cell responses are required for
the host to clear a C. rodentium infection (3). Our data showed that vitamin A status had

94
no effect on antibody responses during C. rodentium infection or B cell numbers in the
lamina propria of the SI and colon. T cells are well-described vitamin A targets. To
study the role of vitamin A in T cells we generated T-dnRAR mice. These mice
expressed the dnRAR in T cells under the Lck promoter and were therefore non-
responsive to RA treatment. Preliminary characterizations of the immune cell
populations in the T-dnRAR mice indicated that RA signaling was important for T cell
development and in particular the CD8+ T cells from the T-dnRAR mice were lower in
the periphery. The role of CD8+ T cells in host resistance is unclear since CD8 KO mice
survived and presumably cleared C. rodentium infection (3). It would be interesting to
see if IL-17a production from T-dnRAR CD8+ T cells is impaired compared to WT
CD8+ T cells.
The findings from this dissertation have implications in the treatment of enteric
pathogens, particularly in vitamin A deficient hosts. The work presented here highlights
a potential role for vitamin A supplements to eliminate reservoirs of enteric pathogens.
Vitamin A deficiency has been implicated as a factor important in the severity of
diarrheal disease and as a contributor to childhood mortality. I show that
supplementation with RA, the active form of vitamin A, reduced infection severity and
promoted survival of A- mice, indicating that vitamin A supplementation during enteric
infections in humans may be beneficial. Additionally, my work showed that RA
treatment may regulate IL-17a which is needed to clear C. rodentium (Figure 5-1).
Further experiments are needed to determine other targets of RA during C. rodentium
infection such as intestinal epithelial cells and other cytokines. It remains to be

95
determined whether vitamin A deficient humans become asymptomatic carriers of E. coli
or other enteric bacteria that have the potential to cause disease. A vitamin A supplement
that could clear bacterial infections and decrease carriers would be an effective way to
limit mortality and disease from enteric pathogens.
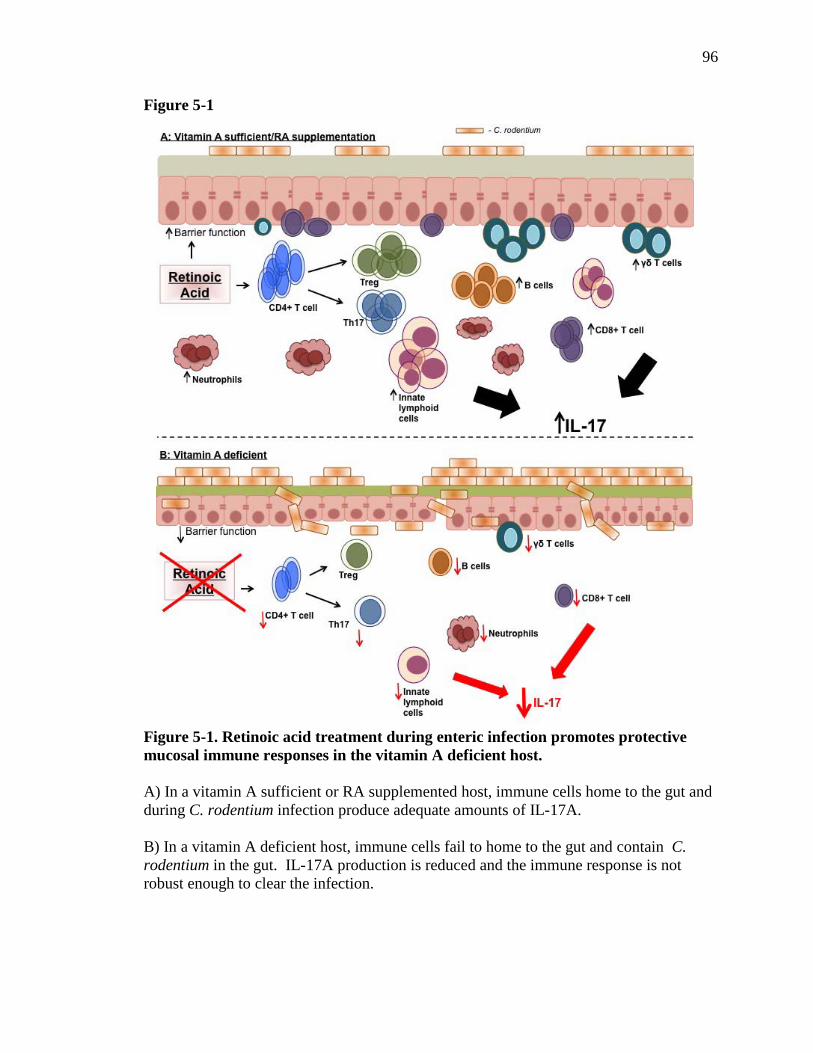
96
Figure 5-1
Figure 5-1. Retinoic acid treatment during enteric infection promotes protective mucosal immune responses in the vitamin A deficient host. A) In a vitamin A sufficient or RA supplemented host, immune cells home to the gut and during C. rodentium infection produce adequate amounts of IL-17A. B) In a vitamin A deficient host, immune cells fail to home to the gut and contain C. rodentium in the gut. IL-17A production is reduced and the immune response is not robust enough to clear the infection.

97
REFERENCES
1. WHO. 2009. Global prevalence of vitamin A deficiency in populations at risk 1995–2005: WHO global database on
vitamin A deficiency. In World Health Organization, Geneva. 2. West, K. P., Jr., G. R. Howard, and A. Sommer. 1989. Vitamin A and infection:
public health implications. Annu Rev Nutr 9: 63-86. 3. Bry, L., and M. B. Brenner. 2004. Critical role of T cell-dependent serum
antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172: 433-441.
4. Vallance, B. A., W. Deng, K. Jacobson, and B. B. Finlay. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect Immun 71: 3443-3453.
5. Ghosh, S., C. Dai, K. Brown, E. Rajendiran, S. Makarenko, J. Baker, C. Ma, S. Halder, M. Montero, V. A. Ionescu, A. Klegeris, B. A. Vallance, and D. L. Gibson. 2011. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol 301: G39-49.
6. McDaniel, K. L., K. H. Restori, J. W. Dodds, M. J. Kennett, A. C. Ross, and M. T. Cantorna. 2015. Vitamin A-Deficient Hosts Become Nonsymptomatic Reservoirs of Escherichia coli-Like Enteric Infections. Infect Immun 83: 2984-2991.
7. Cha, H. R., S. Y. Chang, J. H. Chang, J. O. Kim, J. Y. Yang, C. H. Kim, and M. N. Kweon. 2010. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J Immunol 184: 6799-6806.
8. Wang, C., S. G. Kang, H. HogenEsch, P. E. Love, and C. H. Kim. 2010. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol 184: 5519-5526.
9. Hall, J. A., J. L. Cannons, J. R. Grainger, L. M. Dos Santos, T. W. Hand, S. Naik, E. A. Wohlfert, D. B. Chou, G. Oldenhove, M. Robinson, M. E. Grigg, R. Kastenmayer, P. L. Schwartzberg, and Y. Belkaid. 2011. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34: 435-447.
10. Spencer, S. P., C. Wilhelm, Q. Yang, J. A. Hall, N. Bouladoux, A. Boyd, T. B. Nutman, J. F. Urban, Jr., J. Wang, T. R. Ramalingam, A. Bhandoola, T. A. Wynn, and Y. Belkaid. 2014. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343: 432-437.
11. Mills, K. H. 2008. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol 38: 2636-2649.
12. Jin, W., and C. Dong. 2013. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect 2: e60.

Vita Kaitlin L. McDaniel
Education 2010-present The Pennsylvania State University
Ph.D. in Pathobiology Advised by Dr. Margherita Cantorna, GPA: 3.8/4.0
2008-2010 University of Wyoming B.S. in Microbiology/B.S. in Molecular Biology Advised by Dr. E. Lee Belden, GPA 3.7/4.0
2006-2008 Northwest College A.S. in Biology, GPA 3.8/4.0
Publications
• McDaniel, K.L., K.H. Restori, J. Dodds, M.J. Kennett, A.C. Ross and M.T. Cantorna. 2015. Vitamin A deficient hosts become non-symptomatic reservoirs of Escherichia coli-like enteric infections. Infect Immun. 83:2984-91.
• Cantorna, M.T., K.L. McDaniel, S. Bora, J. Chen and J. James. 2014. Vitamin D, immune regulation, the microbiota and inflammatory bowel disease. Exp Biol Med (Maywood). 239: 1524-30.
• Ooi, J.H., K.L. McDaniel, V. Weaver and M.T. Cantorna. 2014. Murine CD8+ T cells but not macrophages express the vitamin D 1α-hydroxylase. J Nutr Biochem. 25: 58-65.
Professional Presentations
• Vitamin A deficient causes resistant Citrobacter rodentium intestinal infection and retinoic acid treatment mediates the local immune response. American Association of Immunologist Annual Meeting. New Orleans, LA. 2015. Poster
• Vitamin A deficient mice have a decreased ability to clear the enteric pathogen, Citrobacter rodentium and increased mortality during infection. American Association of Immunologist Annual Meeting. Pittsburgh, PA. 2014. Oral presentation and poster.
• Retinoic acid supplementation of vitamin A deficient mice promotes clearance of an infection with Citrobacter rodentium. 16th Annual Upstate New York Immunology Conference. Albany, NY. 2013. Poster
Awards and Honors
American Association of Immunologists Trainee Abstract Award 2014 PSU College of Agriculture Graduate Student Grant 2014 NIH Training Grant: Animal Models of Inflammation 2013 PSU College of Agriculture Mr. and Mrs. E. Bowsworth Grier Scholarship 2013 Pennsylvania State University Graduate School FEGR Scholarship Award 2010
Related Documents











