Endocrine Pharmacology The calcimimetic AMG 641 abrogates parathyroid hyperplasia, bone and vascular calcification abnormalities in uremic rats Charles Henley a, ⁎, James Davis a , Gerald Miller a , Edward Shatzen a , Russ Cattley b , Xiaodong Li a , David Martin a, 1 , Wei Yao c , Nancy Lane c , Victoria Shalhoub a a Department of Metabolic Disorders, Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320, USA b Department of Pathology, Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320, USA c Department of Medicine, University of California at Davis, Medical School, Sacramento, California 95817, USA abstract article info Article history: Received 18 December 2008 Received in revised form 27 April 2009 Accepted 14 May 2009 Available online 24 May 2009 Keywords: Bone Calcimimetic Calcium-sensing receptor Parathyroid gland hyperplasia Secondary hyperparathyroidism Vascular calcification Vitamin D sterol Calcimimetics and vitamin D sterols reduce serum parathyroid hormone (PTH) in patients with secondary hyperparathyroidism receiving dialysis, a disease state associated with parathyroid hyperplasia, vascular calcification, bone disease, and increased mortality. The aim of this study was to determine the effects of the research calcimimetic AMG 641 (Amgen, Inc., Thousand Oaks, CA) or calcitriol (Sigma Aldrich Corporation, St. Louis, MO) on vascular calcification in a rodent model of progressive uremia with accompanying secondary hyperparathyroidism induced by dietary adenine. Treatment effects on parathyroid gland hyperplasia and bone loss were also investigated. Rats were treated daily with vehicle, calcitriol (10 ng), AMG 641 (3 mg/kg), or no treatment during the 4 week period the animals were fed adenine. The uremia-induced increases in serum PTH levels were significantly attenuated by both AMG 641 (N 90%) and calcitriol (~50%). AMG 641 significantly reduced calcium–phosphorus product (Ca × P) and significantly attenuated the development of both parathyroid hyperplasia and vascular calcification. In addition, AMG 641 prevented the defects in trabecular bone volume, trabecular number, and bone mineralization, as well as increases in trabecular spacing in this rodent model of secondary hyperparathyroidism. Calcitriol (10 ng/rat) decreased osteoid surface/bone surface, but had no effects on other bone parameters, or parathyroid hyperplasia (likely due to the lower PTH suppressive effect of calcitriol at the dose used in this study). However, this dose of calcitriol significantly exacerbated vascular calcification. These results suggest that calcimimetics can reduce the development of vascular calcification, parathyroid hyperplasia and bone abnormalities associated with secondary hyperparathyroidism. © 2009 Elsevier B.V. All rights reserved. 1. Introduction Patients with chronic kidney disease receiving dialysis often develop secondary hyperparathyroidism characterized by increased serum parathyroid hormone (PTH), increased serum phosphorous, decreased serum calcium and calcitriol (Goodman and Quarles, 2008; Drueke et al., 2007). Secondary hyperparathyroidism is accompanied by parathyroid hyperplasia and excessive synthesis and secretion of PTH, which can result in disproportionate bone resorption and other bone disorders, soft tissue and vascular calcification, and significant risk for cardiovascular morbidity and mortality (Block et al., 2004; de Francisco, 2004; Hebert, 2006; Kalantar-Zadeh et al., 2006; Young et al., 2005). Evidence suggests that reductions in PTH and serum phosphorus may slow or prevent secondary hyperparathyroidism-associated parathyroid hyperplasia, vascular calcification, and renal osteodystro- phy. Traditional therapeutic approaches rely on the actions of vitamin D sterols, which, while able to decrease PTH levels, have also been associated with hypercalcemia and vascular calcification in preclinical studies (Henley et al., 2005; Lopez et al., 2006). Calcimimetics (e.g., cinacalcet HCl), pharmacologic agents that act directly at the calcium- sensing receptor in the parathyroid gland to reduce PTH secretion, represent a relatively new therapeutic approach. Evidence suggests that calcimimetics may slow or prevent parathyroid hyperplasia in uremic animals (Colloton et al., 2005; Wada et al., 1997) without inducing vascular calcification (Henley et al., 2005; Lopez et al., 2006). Moreover, preclinical (Wada et al., 1998) and clinical (Lien et al., 2005; Cunningham et al., 2005) evidence suggests that calcimimetics improve bone health, including reducing the incidence of fractures. Some clinical data show that calcitriol may influence bone remodeling and ameliorate osteitis fibrosa (Slatopolsky et al., 1984; Andress et al., European Journal of Pharmacology 616 (2009) 306–313 ⁎ Corresponding author. Department of Metabolic Disorders, MS-15-2-A, Amgen Inc, One Amgen Center Drive, Thousand Oaks, CA 91320, USA. Tel.: +1 805 447 7023. E-mail address: [email protected] (C. Henley). 1 Current address: Department of Biology, Nektar Therapeutics, San Carlos, Ca USA. 0014-2999/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.ejphar.2009.05.013 Contents lists available at ScienceDirect European Journal of Pharmacology journal homepage: www.elsevier.com/locate/ejphar

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

European Journal of Pharmacology 616 (2009) 306–313
Contents lists available at ScienceDirect
European Journal of Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /e jphar
Endocrine Pharmacology
The calcimimetic AMG 641 abrogates parathyroid hyperplasia, bone and vascularcalcification abnormalities in uremic rats
Charles Henley a,⁎, James Davis a, Gerald Miller a, Edward Shatzen a, Russ Cattley b, Xiaodong Li a,David Martin a,1, Wei Yao c, Nancy Lane c, Victoria Shalhoub a
a Department of Metabolic Disorders, Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320, USAb Department of Pathology, Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320, USAc Department of Medicine, University of California at Davis, Medical School, Sacramento, California 95817, USA
⁎ Corresponding author. Department of Metabolic DisOne Amgen Center Drive, Thousand Oaks, CA 91320, US
E-mail address: [email protected] (C. Henley).1 Current address: Department of Biology, Nektar The
0014-2999/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.ejphar.2009.05.013
a b s t r a c t
a r t i c l e i n f oArticle history:Received 18 December 2008Received in revised form 27 April 2009Accepted 14 May 2009Available online 24 May 2009
Keywords:BoneCalcimimeticCalcium-sensing receptorParathyroid gland hyperplasiaSecondary hyperparathyroidismVascular calcificationVitamin D sterol
Calcimimetics and vitamin D sterols reduce serum parathyroid hormone (PTH) in patients with secondaryhyperparathyroidism receiving dialysis, a disease state associated with parathyroid hyperplasia, vascularcalcification, bone disease, and increased mortality. The aim of this study was to determine the effects of theresearch calcimimetic AMG 641 (Amgen, Inc., Thousand Oaks, CA) or calcitriol (Sigma Aldrich Corporation, St.Louis, MO) on vascular calcification in a rodent model of progressive uremia with accompanying secondaryhyperparathyroidism induced by dietary adenine. Treatment effects on parathyroid gland hyperplasia andbone loss were also investigated. Rats were treated daily with vehicle, calcitriol (10 ng), AMG 641 (3 mg/kg),or no treatment during the 4 week period the animals were fed adenine. The uremia-induced increases inserum PTH levels were significantly attenuated by both AMG 641 (N90%) and calcitriol (~50%). AMG 641significantly reduced calcium–phosphorus product (Ca×P) and significantly attenuated the development ofboth parathyroid hyperplasia and vascular calcification. In addition, AMG 641 prevented the defects intrabecular bone volume, trabecular number, and bone mineralization, as well as increases in trabecularspacing in this rodent model of secondary hyperparathyroidism. Calcitriol (10 ng/rat) decreased osteoidsurface/bone surface, but had no effects on other bone parameters, or parathyroid hyperplasia (likely due tothe lower PTH suppressive effect of calcitriol at the dose used in this study). However, this dose of calcitriolsignificantly exacerbated vascular calcification. These results suggest that calcimimetics can reduce thedevelopment of vascular calcification, parathyroid hyperplasia and bone abnormalities associated withsecondary hyperparathyroidism.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
Patientswith chronic kidney disease receiving dialysis often developsecondary hyperparathyroidism characterized by increased serumparathyroid hormone (PTH), increased serum phosphorous, decreasedserumcalciumand calcitriol (GoodmanandQuarles, 2008;Drueke et al.,2007). Secondary hyperparathyroidism is accompanied by parathyroidhyperplasia and excessive synthesis and secretion of PTH, which canresult in disproportionate bone resorptionandother bonedisorders, softtissue and vascular calcification, and significant risk for cardiovascularmorbidity and mortality (Block et al., 2004; de Francisco, 2004; Hebert,2006; Kalantar-Zadeh et al., 2006; Young et al., 2005).
orders, MS-15-2-A, Amgen Inc,A. Tel.: +1 805 447 7023.
rapeutics, San Carlos, Ca USA.
ll rights reserved.
Evidence suggests that reductions in PTH and serum phosphorusmay slow or prevent secondary hyperparathyroidism-associatedparathyroid hyperplasia, vascular calcification, and renal osteodystro-phy. Traditional therapeutic approaches rely on the actions of vitaminD sterols, which, while able to decrease PTH levels, have also beenassociated with hypercalcemia and vascular calcification in preclinicalstudies (Henley et al., 2005; Lopez et al., 2006). Calcimimetics (e.g.,cinacalcet HCl), pharmacologic agents that act directly at the calcium-sensing receptor in the parathyroid gland to reduce PTH secretion,represent a relatively new therapeutic approach. Evidence suggeststhat calcimimetics may slow or prevent parathyroid hyperplasia inuremic animals (Colloton et al., 2005; Wada et al., 1997) withoutinducing vascular calcification (Henley et al., 2005; Lopez et al., 2006).Moreover, preclinical (Wada et al., 1998) and clinical (Lien et al., 2005;Cunningham et al., 2005) evidence suggests that calcimimeticsimprove bone health, including reducing the incidence of fractures.Some clinical data show that calcitriol may influence bone remodelingand ameliorate osteitis fibrosa (Slatopolsky et al., 1984; Andress et al.,
307C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
1989), although other studies have shown either no or detrimentaleffects on bone remodeling (Costa et al., 2003; Pahl et al., 1995).
There are currently no reports of calcimimetic effects on the totalityof biochemical (PTH, calcium, phosphorus, Ca×P) and pathologicalparameters of secondary hyperparathyroidism (parathyroid hyperplasia,vascular calcification and renal osteodystrophy) in a rodent model ofuremia with secondary hyperparathyroidism. Herein, we investigatedwhether the research calcimimetic AMG 641 (chemical name: (1R)-N-((6-(methyloxy)-4′-(trifluoromethyl)-3-biphenylyl)methyl)-1-pheny-lethanamine) could abrogate these characteristic biochemical andpathological changes without exacerbating vascular calcificationobserved in the adenine-treated uremic rat. AMG 641 is an arylalk-ylamine with a molecular weight of approximately 400 g/mol, is morepotent than cinacalcet, and has approximately a 3-fold longer half-lifeand a larger volume of distribution. We also investigated the effectsof calcitriol on the above parameters utilizing a dose that would signif-icantly lower PTH while avoiding the potentially deleterious effects ofhypercalcemia.
2. Materials and methods
Male Sprague–Dawley rats (350–390 g) were purchased fromHarlan (Indianapolis, IN). Rats were pair-housed under a 12 h/12 hlight/dark cycle and given ad libitum access to standard rat chow (1.2%calcium, 0.9% phosphorus) and water. Experiments were performedunder protocols approved by Amgen's Internal Animal Care and UseCommittee.
2.1. Adenine-induced uremia and drug administration
Rats were randomly assigned into treatment groups based on thenormal distribution of baseline body weights, then fed a diet contain-ing 0.75% adenine (Adenine freebase A8626, Sigma Aldrich, St. Louis,MO) or a control diet with equivalent amounts of calcium (1.1%) andphosphorus (0.9%) (Dyets, Inc, Bethlehem, PA) for 4weeks. Concurrentwith adenine exposure, rats were divided into the following dailytreatment groups: normal, non-adenine control (n=8), adeninecontrol (no treatment; n=9), vehicle for AMG 641 (12% captisol inwater p.o.; n=9), AMG641 (3mg/kg, p.o.; n=9), vehicle for calcitriol(0.19% ethanol in phosphate buffered saline, s.c.; n=10), or calcitriol(1α, 25-dihydroxycholecalciferol, Sigma Aldrich Corp, St. Louis, MO,10 ng/rat [~0.025–0.028 μg/kg], s.c.; n=10). The dose of calcitriol waschosen since higher doses resulted in significant vascular calcificationin subtotal nephrectomized rats (Henley et al., 2005), and we wantedto reduce serum PTH significantly while attempting to avoid over-whelming vascular calcification in the rat uremia adenine model.
2.2. Aorta histopathology
Twenty-four hours following the last dose, rats were carbondioxide-euthanized. The thoracic aorta was then excised and fixed inaqueous buffered zinc formalin (Z-fix; Anatech Ltd; Battle Creek, MI),paraffin embedded, sectioned longitudinally, and von Kossa stained. A
Table 1Biochemical markers.
Treatment No adenine Adenine Adenine+AMG 641 vehicle Adenine+
Creatinine mg/dL 0.3±0.01 3.5±0.2a 3.5±0.3a 2.8±0.1a
BUN mg/dL 18±1 160±12a 146±9a 125±5a
iPTH pg/ml 35±3 565±91a 656±76a 13±3c
Ca mg/dL 10.6±0.1 10.7±0.1 10.8±0.2 9.4±0.3a
P mg/dL 7.2±0.2 23.0±2.0a 21.1±0.9a 21.8±1.0a
Measurements were taken 24 h after treatment administration on treatment day 28. Valuea Pb0.05 significantly different from standard chow (no adenine, no treatment).b Pb0.05 significantly different from adenine + calcitriol vehicle treated group.c Pb0.05 significantly different from adenine + AMG 641 vehicle.
board-certified pathologist blinded to treatment groups evaluated andscored the stained sections within a single session (to avoid drift inscoring outcome). One section per animal was scored on a 0–5 scale:0=no calcification,1=minimal, 2=mild, 3=moderate, 4=marked,and 5=severe calcification.
2.3. Blood biochemistries
PTH, total serum calcium and phosphorus, ionized calcium, bloodurea nitrogen [BUN], and creatinine were determined from bloodcollected from the retro-orbital sinuses of isoflurane anesthetized rats.Ionized calcium was measured using a Ciba-Corning 634 ISE Ca++/pHAnalyzer (Ciba-Corning Diagnostics Corp, Medfield, MA) immediatelyafter collection into heparinized capillary tubes. Blood for the remainingparameters was collected into SST (clot activator) brand blood tubes.Serumwas removed and stored at−70 °C. A blood chemistry analyzer(AU 400; Olympus, Melville, NY) was used to determine calcium,phosphorus, BUN, and creatinine, and rat PTH(1–34) immunoradiometricassay kits (Immutopics, San Clemente, CA) were used to determine PTHlevels.
2.4. Parathyroid hyperplasia
After sacrifice, the laryngo-tracheal complex was removed, storedin zinc-buffered formalin (2 to 3 days), transferred to 70% alcohol andtrimmed. The thyroid and parathyroid glands were dissected fromeach other, and parathyroid glands were blotted dry on a lint-free Kimwipe (KimberlyClark Corp., Roswell, GA, USA), weighed, and paraffinembedded. Sections (5 μm) were placed onto charged slides (VWRScientific, West Chester, PA, USA). Increases in the number of pro-liferating parathyroid cells were determined by immunostainingusing a proliferating cell nuclear antigen (PCNA) staining kit (ZymedLaboratories, Inc., South San Francisco, CA, USA). Parathyroid area wasdetermined as previously described (Colloton et al., 2005) using anarea-measurement graticle with 0.01 mm2 grids. Data is expressed asthe number of PCNA-positive cells/mm2.
Total gland weight is expressed as parathyroid gland weight/bodyweight.
2.5. Bone histopathology and histomorphometry
Approximately 1/3 of the distal femur and 1/3 midshaft femur wasprepared using slow speed saw. Undecalcified segmentswere processedthroughdefattingand infiltration andembedded inmethylmathacrylate.Frontal sections of the distal femurwere obtained near themiddle of thebone using a Leica/Jung 2255 microtome set at 4 μm. Distal femursections were counter-stained with Von Kossa and tetrachrome bonestains. Histomorphometric evaluations (Bioquant Image Analysis Cor-poration,Nashville, TN)of the stained slideswereperformed in ablindedmanner with the nomenclature and calculations based on standardizedterms and formulae (Parfitt et al., 1987), such as percent bone volumeper tissue volume (%BV/TV), percentosteoblast surface per bone surface(%ObS/BS), percent osteoclast surface per bone surface (%OcS/BS),
AMG 641 (3 mg/kg) Adenine+calcitriol vehicle Adenine+calcitriol (10 ng/rat)
3.6±0.4a 3.3±0.4a
159±10a 139±5a
801±70a 396±57a,b,c 10.8±0.2 11.0±0.2
20.1±1.7a 18.7±0.7a
s are mean±S.E.M. (n=8–10).

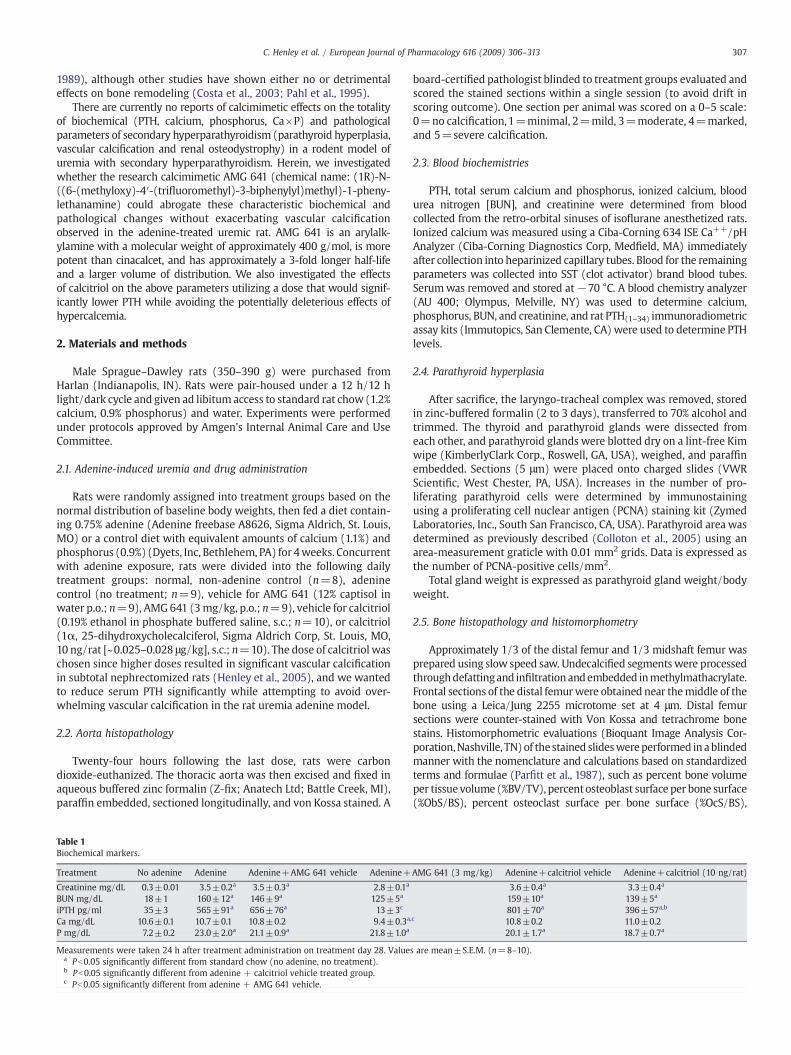
Fig. 1. Von Kossa-stained sections of aortas showing mineralization from animals fed adenine (0.75%) for 4 weeks and treated for 4 weeks with either (A) AMG-641 (3 mg/kg, p.o.);(B) 10% captisol-vehicle for AMG 641 (1 ml/rat, p.o.); (C) calcitriol (10 ng/rat, s.c.) or (D) PBS-vehicle for calcitriol (0.2 ml/rat s.c.).
Table 2Parathyroid PCNA-positive cells from adenine-induced uremic rats.
PCNA-positive cells (cells/mm2)
Weeks Treatments
Noadenine
Adenine Adenine+AMG 641vehicle
Adenine+AMG 641
Adenine+calcitriolvehicle
Adenine+calcitriol
1 ND 111±14a 93±9a 72±12 106±18a 97±15a
2 ND 116±11a 120±18a 50±10b 108±8a 107±12a
3 ND 144±16a 140±16a 52±9c 135±16a 141±13a
4 33±6 167±18a 157±22a 40±5c 147±19a 142±19a
Parathyroid PCNA-positive cell numbers, values are mean±S.E.M. (n=16–20).a Pb0.01 significantly different from standard chow (no adenine, no treatment values
measured at week 4).b Pb0.01.c Pb0.001 significantly different from adenine+AMG 641 vehicle.
308 C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
trabecular spacing (TbSp, measured in μm), trabecular number (TbN)/millimeter, and percent osteoid surface/bone surface (%OS/BS).
Analyses were performed in the secondary spongiosa of the distalfemurs that included the trabecular area between 0.1 mm and 1.1 mmdistal to the lowest point of the growth plate. Structural evaluation ofthe trabecular bone at the metaphysis included measurement of bonearea, tissue area, and bone perimeter; measurement of cellular levelactivities included osteoid, osteoclastic, and osteoblastic perimeters.
2.6. Statistical analyses
The mean and standard error of the mean was determined andstatistical analyses were used to compare differences between thetreatment groups in body weight, serum chemistry and biochemicalmarkers of bone turnover, and parathyroid gland weights. Analysis ofvariance (ANOVA; α=0.05) was used to test for overall treatmenteffects and post-hoc analysis (Fisher's Protected Least Squares Differ-ence tests) was used for between group comparisons (α=0.05). Pairedt-test (α=0.05) were used to test for differences between predose andpostdose values for a given treatment condition and day.
3. Results
3.1. Effects of AMG 641 or calcitriol on blood biochemical parameters
Adenine-induced uremia was confirmed by significant increases inserum creatinine and BUN compared with untreated controls (Pb0.05;Table 1), and the uremic state was unaffected by any of the treatments(Table 1). While a slight decrease in creatinine and BUN levels wereobserved in theAMG641group (P=0.045 comparedwithvehicle), theyremained grossly elevated (6- to 10-fold) over the normal values. It isnoteworthy that Piecha et al., have reported that the calcimimetic R-568or calcitriol have beneficial effects on progression of renal damage insubtotal nephrectomized rats (Piecha et al., 2008). Since we did notassess the myriad of parameters that were assessed in their study wecannot definitively say that AMG641was nephroprotective in our study.Secondary hyperparathyroidism was confirmed by a significant rise inserum PTH in rats receiving dietary adenine compared with untreatedcontrols (Pb0.05). Uremic rats (on adenine diet) lost an average of~32.6% body weight over the course of the study. The rats receivingstandard chow were weight stable; gaining 20 g over the time course.
Body weight in the uremic animals was unaffected by any treatment(i.e., AMG 641, calcitriol or their vehicles).
Both calcitriol and AMG 641 significantly reduced serum PTH(Table 1). Rats treated with AMG 641 exhibited greater than 90%reduction in PTH comparedwith animals treatedwith its vehicle (13±3vs 656±76 pg/mL respectively, Pb0.001). Rats in the calcitriol-treatment group exhibited a significant reduction in PTH comparedwithvehicle-treated controls (396±57vs801±70pg/mL, respectively,Pb0.01). AMG641preventedadenine-induced increase in PTHsuch thatPTH levels were not statistically different than those observed in non-adenine controls. It is important to note that the magnitude of PTHsuppression by calcitriol was 30-fold less than was observed for AMG641. The calcitriol dose (10 ng/rat) was limited to avoid significanthypercalcemia and moderate to severe vascular calcification ob-served with higher doses in uremic rats (Henley et al., 2005; Milleret al., 2006a,b). Serum PTH levels in the corresponding vehicle-treatedgroups were not significantly different from the adenine-induceduremic rats receiving no treatment (Table 1).
Total serum calcium was unchanged in any treatment groupcompared with non-adenine controls, with the exception of AMG641-treated rats (Table 1), which exhibited significantly reduced totalserum calcium compared with all other groups (Pb0.05, Table 1).Serum phosphorus was significantly increased in adenine-fed ratscomparedwith rats receiving the adenine-free diet (Pb0.05), regardlessof treatment group.
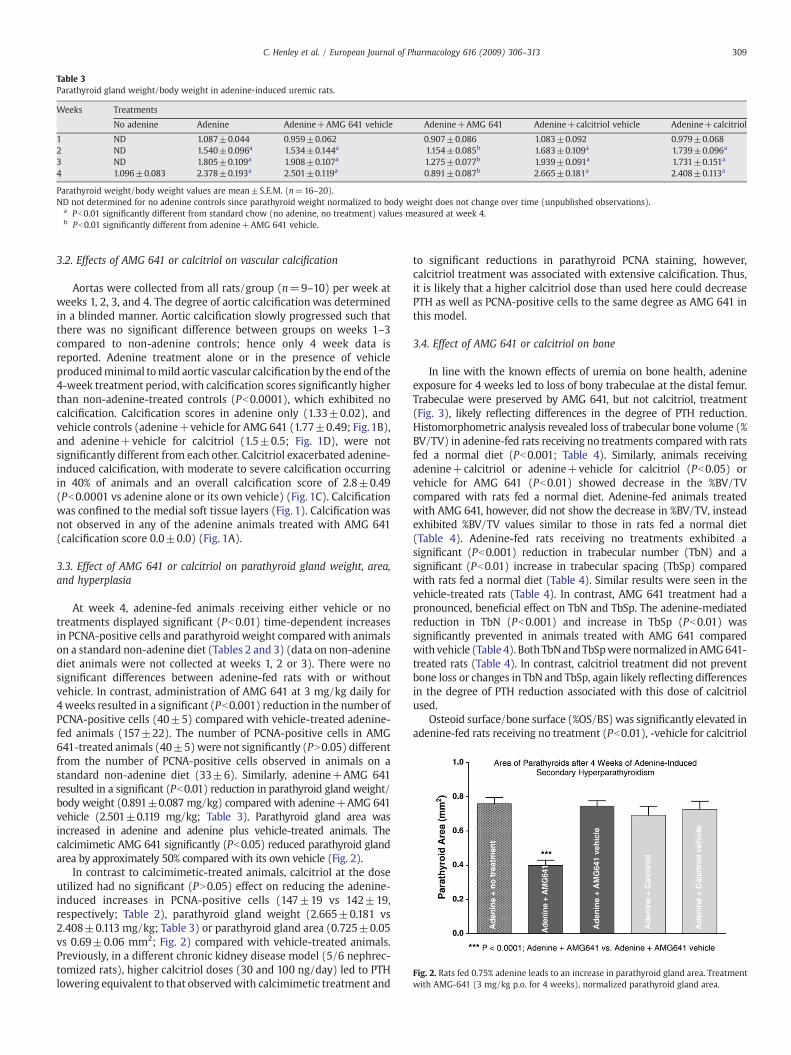
Table 3Parathyroid gland weight/body weight in adenine-induced uremic rats.
Weeks Treatments
No adenine Adenine Adenine+AMG 641 vehicle Adenine+AMG 641 Adenine+calcitriol vehicle Adenine+calcitriol
1 ND 1.087±0.044 0.959±0.062 0.907±0.086 1.083±0.092 0.979±0.0682 ND 1.540±0.096a 1.534±0.144a 1.154±0.085b 1.683±0.109a 1.739±0.096a
3 ND 1.805±0.109a 1.908±0.107a 1.275±0.077b 1.939±0.091a 1.731±0.151a
4 1.096±0.083 2.378±0.193a 2.501±0.119a 0.891±0.087b 2.665±0.181a 2.408±0.113a
Parathyroid weight/body weight values are mean±S.E.M. (n=16–20).ND not determined for no adenine controls since parathyroid weight normalized to body weight does not change over time (unpublished observations).
a Pb0.01 significantly different from standard chow (no adenine, no treatment) values measured at week 4.b Pb0.01 significantly different from adenine+AMG 641 vehicle.
Fig. 2. Rats fed 0.75% adenine leads to an increase in parathyroid gland area. Treatmentwith AMG-641 (3 mg/kg p.o. for 4 weeks), normalized parathyroid gland area.
309C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
3.2. Effects of AMG 641 or calcitriol on vascular calcification
Aortas were collected from all rats/group (n=9–10) per week atweeks 1, 2, 3, and 4. The degree of aortic calcification was determinedin a blinded manner. Aortic calcification slowly progressed such thatthere was no significant difference between groups on weeks 1–3compared to non-adenine controls; hence only 4 week data isreported. Adenine treatment alone or in the presence of vehicleproducedminimal tomild aortic vascular calcification by the end of the4-week treatment period, with calcification scores significantly higherthan non-adenine-treated controls (Pb0.0001), which exhibited nocalcification. Calcification scores in adenine only (1.33±0.02), andvehicle controls (adenine+vehicle for AMG 641 (1.77±0.49; Fig. 1B),and adenine+vehicle for calcitriol (1.5±0.5; Fig. 1D), were notsignificantly different from each other. Calcitriol exacerbated adenine-induced calcification, with moderate to severe calcification occurringin 40% of animals and an overall calcification score of 2.8±0.49(Pb0.0001 vs adenine alone or its own vehicle) (Fig. 1C). Calcificationwas confined to the medial soft tissue layers (Fig. 1). Calcification wasnot observed in any of the adenine animals treated with AMG 641(calcification score 0.0±0.0) (Fig. 1A).
3.3. Effect of AMG 641 or calcitriol on parathyroid gland weight, area,and hyperplasia
At week 4, adenine-fed animals receiving either vehicle or notreatments displayed significant (Pb0.01) time-dependent increasesin PCNA-positive cells and parathyroid weight compared with animalson a standard non-adenine diet (Tables 2 and 3) (data on non-adeninediet animals were not collected at weeks 1, 2 or 3). There were nosignificant differences between adenine-fed rats with or withoutvehicle. In contrast, administration of AMG 641 at 3 mg/kg daily for4weeks resulted in a significant (Pb0.001) reduction in the number ofPCNA-positive cells (40±5) compared with vehicle-treated adenine-fed animals (157±22). The number of PCNA-positive cells in AMG641-treated animals (40±5)were not significantly (PN0.05) differentfrom the number of PCNA-positive cells observed in animals on astandard non-adenine diet (33±6). Similarly, adenine+AMG 641resulted in a significant (Pb0.01) reduction in parathyroid gland weight/body weight (0.891±0.087 mg/kg) compared with adenine+AMG 641vehicle (2.501±0.119 mg/kg; Table 3). Parathyroid gland area wasincreased in adenine and adenine plus vehicle-treated animals. Thecalcimimetic AMG 641 significantly (Pb0.05) reduced parathyroid glandarea by approximately 50% compared with its own vehicle (Fig. 2).
In contrast to calcimimetic-treated animals, calcitriol at the doseutilized had no significant (PN0.05) effect on reducing the adenine-induced increases in PCNA-positive cells (147±19 vs 142±19,respectively; Table 2), parathyroid gland weight (2.665±0.181 vs2.408±0.113 mg/kg; Table 3) or parathyroid gland area (0.725±0.05vs 0.69±0.06 mm2; Fig. 2) compared with vehicle-treated animals.Previously, in a different chronic kidney disease model (5/6 nephrec-tomized rats), higher calcitriol doses (30 and 100 ng/day) led to PTHlowering equivalent to that observedwith calcimimetic treatment and
to significant reductions in parathyroid PCNA staining, however,calcitriol treatment was associated with extensive calcification. Thus,it is likely that a higher calcitriol dose than used here could decreasePTH as well as PCNA-positive cells to the same degree as AMG 641 inthis model.
3.4. Effect of AMG 641 or calcitriol on bone
In line with the known effects of uremia on bone health, adenineexposure for 4 weeks led to loss of bony trabeculae at the distal femur.Trabeculae were preserved by AMG 641, but not calcitriol, treatment(Fig. 3), likely reflecting differences in the degree of PTH reduction.Histomorphometric analysis revealed loss of trabecular bone volume (%BV/TV) in adenine-fed rats receiving no treatments compared with ratsfed a normal diet (Pb0.001; Table 4). Similarly, animals receivingadenine+calcitriol or adenine+vehicle for calcitriol (Pb0.05) orvehicle for AMG 641 (Pb0.01) showed decrease in the %BV/TVcompared with rats fed a normal diet. Adenine-fed animals treatedwith AMG 641, however, did not show the decrease in %BV/TV, insteadexhibited %BV/TV values similar to those in rats fed a normal diet(Table 4). Adenine-fed rats receiving no treatments exhibited asignificant (Pb0.001) reduction in trabecular number (TbN) and asignificant (Pb0.01) increase in trabecular spacing (TbSp) comparedwith rats fed a normal diet (Table 4). Similar results were seen in thevehicle-treated rats (Table 4). In contrast, AMG 641 treatment had apronounced, beneficial effect on TbN and TbSp. The adenine-mediatedreduction in TbN (Pb0.001) and increase in TbSp (Pb0.01) wassignificantly prevented in animals treated with AMG 641 comparedwith vehicle (Table 4). Both TbNandTbSpwere normalized inAMG641-treated rats (Table 4). In contrast, calcitriol treatment did not preventbone loss or changes in TbN and TbSp, again likely reflecting differencesin the degree of PTH reduction associated with this dose of calcitriolused.
Osteoid surface/bone surface (%OS/BS) was significantly elevated inadenine-fed rats receiving no treatment (Pb0.01), -vehicle for calcitriol
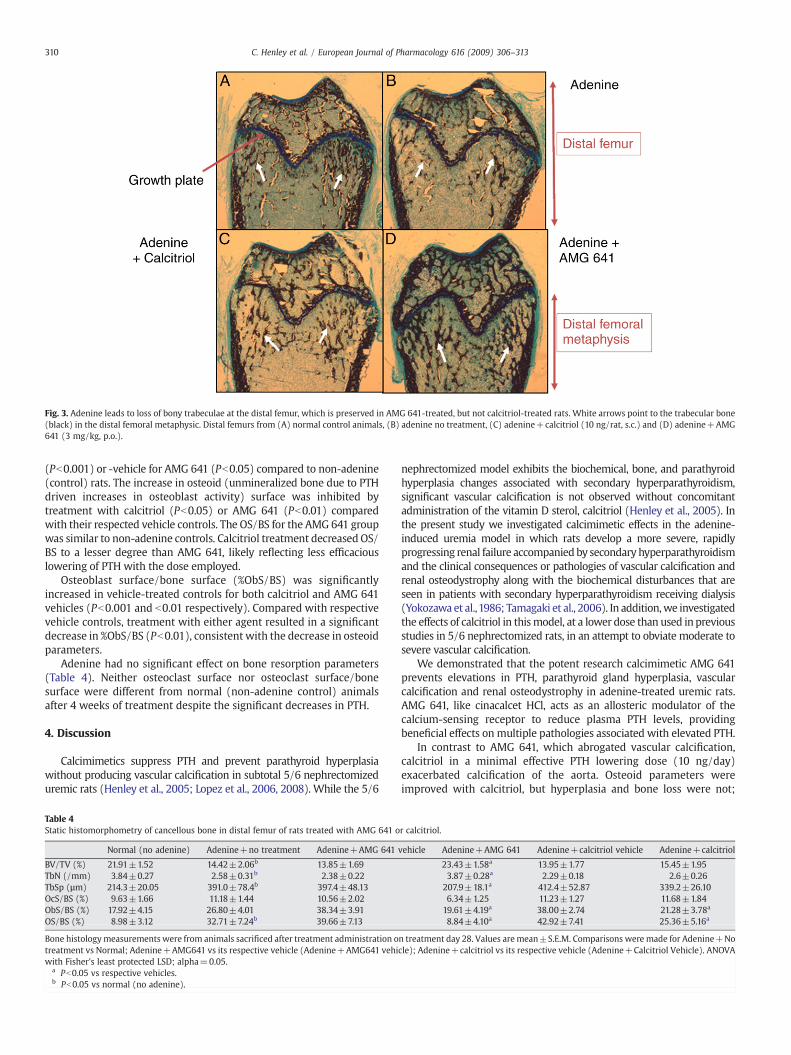
Fig. 3. Adenine leads to loss of bony trabeculae at the distal femur, which is preserved in AMG 641-treated, but not calcitriol-treated rats. White arrows point to the trabecular bone(black) in the distal femoral metaphysic. Distal femurs from (A) normal control animals, (B) adenine no treatment, (C) adenine+calcitriol (10 ng/rat, s.c.) and (D) adenine+AMG641 (3 mg/kg, p.o.).
310 C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
(Pb0.001) or -vehicle for AMG 641 (Pb0.05) compared to non-adenine(control) rats. The increase in osteoid (unmineralized bone due to PTHdriven increases in osteoblast activity) surface was inhibited bytreatment with calcitriol (Pb0.05) or AMG 641 (Pb0.01) comparedwith their respected vehicle controls. The OS/BS for the AMG 641 groupwas similar to non-adenine controls. Calcitriol treatment decreased OS/BS to a lesser degree than AMG 641, likely reflecting less efficaciouslowering of PTH with the dose employed.
Osteoblast surface/bone surface (%ObS/BS) was significantlyincreased in vehicle-treated controls for both calcitriol and AMG 641vehicles (Pb0.001 and b0.01 respectively). Compared with respectivevehicle controls, treatment with either agent resulted in a significantdecrease in %ObS/BS (Pb0.01), consistent with the decrease in osteoidparameters.
Adenine had no significant effect on bone resorption parameters(Table 4). Neither osteoclast surface nor osteoclast surface/bonesurface were different from normal (non-adenine control) animalsafter 4 weeks of treatment despite the significant decreases in PTH.
4. Discussion
Calcimimetics suppress PTH and prevent parathyroid hyperplasiawithout producing vascular calcification in subtotal 5/6 nephrectomizeduremic rats (Henley et al., 2005; Lopez et al., 2006, 2008). While the 5/6
Table 4Static histomorphometry of cancellous bone in distal femur of rats treated with AMG 641 o
Normal (no adenine) Adenine+no treatment Adenine+AMG 641
BV/TV (%) 21.91±1.52 14.42±2.06b 13.85±1.69TbN (/mm) 3.84±0.27 2.58±0.31b 2.38±0.22TbSp (µm) 214.3±20.05 391.0±78.4b 397.4±48.13OcS/BS (%) 9.63±1.66 11.18±1.44 10.56±2.02ObS/BS (%) 17.92±4.15 26.80±4.01 38.34±3.91OS/BS (%) 8.98±3.12 32.71±7.24b 39.66±7.13
Bone histologymeasurements were from animals sacrificed after treatment administration otreatment vs Normal; Adenine+AMG641 vs its respective vehicle (Adenine+AMG641 vehiwith Fisher's least protected LSD; alpha=0.05.
a Pb0.05 vs respective vehicles.b Pb0.05 vs normal (no adenine).
nephrectomized model exhibits the biochemical, bone, and parathyroidhyperplasia changes associated with secondary hyperparathyroidism,significant vascular calcification is not observed without concomitantadministration of the vitamin D sterol, calcitriol (Henley et al., 2005). Inthe present study we investigated calcimimetic effects in the adenine-induced uremia model in which rats develop a more severe, rapidlyprogressing renal failure accompanied by secondary hyperparathyroidismand the clinical consequences or pathologies of vascular calcification andrenal osteodystrophy along with the biochemical disturbances that areseen in patients with secondary hyperparathyroidism receiving dialysis(Yokozawa et al.,1986; Tamagaki et al., 2006). In addition,we investigatedthe effects of calcitriol in thismodel, at a lower dose than used in previousstudies in 5/6 nephrectomized rats, in an attempt to obviate moderate tosevere vascular calcification.
We demonstrated that the potent research calcimimetic AMG 641prevents elevations in PTH, parathyroid gland hyperplasia, vascularcalcification and renal osteodystrophy in adenine-treated uremic rats.AMG 641, like cinacalcet HCl, acts as an allosteric modulator of thecalcium-sensing receptor to reduce plasma PTH levels, providingbeneficial effects on multiple pathologies associated with elevated PTH.
In contrast to AMG 641, which abrogated vascular calcification,calcitriol in a minimal effective PTH lowering dose (10 ng/day)exacerbated calcification of the aorta. Osteoid parameters wereimproved with calcitriol, but hyperplasia and bone loss were not;
r calcitriol.
vehicle Adenine+AMG 641 Adenine+calcitriol vehicle Adenine+calcitriol
23.43±1.58a 13.95±1.77 15.45±1.953.87±0.28a 2.29±0.18 2.6±0.26207.9±18.1a 412.4±52.87 339.2±26.106.34±1.25 11.23±1.27 11.68±1.8419.61±4.19a 38.00±2.74 21.28±3.78a
8.84±4.10a 42.92±7.41 25.36±5.16a
n treatment day 28. Values aremean±S.E.M. Comparisons were made for Adenine+Nocle); Adenine+calcitriol vs its respective vehicle (Adenine+Calcitriol Vehicle). ANOVA

311C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
while AMG 641 prevented parathyroid cell proliferation, andwas boneprotective. Prevention of parathyroid hyperplasia and bone disease byAMG 641 may be secondary to serum lowering of PTH. The use ofsufficiently high doses of calcitriol to reduce PTH to levels observedwith AMG 641 would likely result in similar effects on bone andhyperplasia (Cozzolino et al., 2001), but at the expense of increasedvascular calcification (Henley et al., 2005; Lopez et al., 2006, 2008).Vitamin D sterols exhibit varied effects, including increasing serumcalcium and phosphorus levels (Tentori et al., 2006) and mediatingexpression of bone-associated genes in vascular smooth muscle cells(Shalhoub et al., 2006a,b).
An important question regarding calcimimetic or vitamin D sterol usein secondary hyperparathyroidism is whether treatment modifies para-thyroid gland hyperplasia and reduces glandmass.Wepreviously showedthat cinacalcet prevented acute parathyroid hyperplasia in uremic rats(Colloton et al., 2005). The present study with AMG 641 confirms thatcalcimimetics attenuate progression of parathyroid hyperplasia andcorroborates several reports indicating marked reduction in numbers ofS-phase, PCNA-positive and overall parathyroid cell numbers in calcimi-metic-treated, uremic rats (Wada et al., 2000; Chin et al., 2000;Mizobuchiet al., 2004; Colloton et al., 2005). Of note, AMG 641 was able to preventparathyroid gland hyperplasia even in the presence of extremely highserum levels of phosphorus, a known direct stimulator of parathyroidhyperplasia (Almaden et al., 1996; Slatopolsky et al., 1996).
Several studies support the notion that calcium-sensing receptorsignaling is a major determinant in controlling hyperplasia and diseaseprogression (Brown and MacLeod, 2001; Drueke et al., 2007).Parathyroid hyperplasia is observed in patients with neonatal severehyperparathyroidism, a disease attributed to calcium-sensing receptorloss of function mutations (Chattopadhyay et al., 1996). Furthermore,parathyroid hyperplasia is observed in calcium-sensing receptor nullmice, further demonstrating that the absence of the calcium-sensingreceptor can influence chief cell proliferation innon-uremic animals (Hoet al., 1995). Interestingly, vitamin D receptor knockout mice developparathyroid hyperplasia that ismitigated by a high calciumdiet (Li et al.,1998). As previously noted we did not use higher doses of calcitriol thatwould result in increased serum calcium to prevent hyperplasia in thecurrent study because of excessive aortic calcification.
Studies have demonstrated that calcitriol can prevent parathyroidcell proliferation in uremic rats when given at the very start of chronicrenal failure (Szabo et al., 1989). A decline in both calcium-sensingreceptor and vitamin D receptor expression is characteristic ofhyperplastic parathyroid cells (see review Drueke et al., 2007). Onceparathyroid cell proliferation advances, traditional therapies such ascalcium and calcitriol become ineffective (i.e., refractory to subse-quent treatment). Recent studies have shown that the down-regulation of both vitamin D receptor and calcium-sensing receptormRNA and protein in uremic animals can be reversed by calcimimetics(Mizobuchi et al., 2004; Rodríguez et al., 2007) and some, but not all,evidence suggests that vitamin D sterols may also increase calcium-sensing receptor expression in the parathyroid gland and kidney of therat (Brown et al., 1996; Rogers et al., 1995). Vitamin D-elicitedregulation of the parathyroid gland calcium-sensing receptor wouldtend to facilitate inhibition of parathyroid function in the uremic stateby increasing sensitivity to serum calcium. Furthermore, vitamin D-induced increases in serum calcium could secondarily decrease PTHsecretion via increased parathyroid calcium-sensing receptor expres-sion and subsequent inhibition of parathyroid hyperplasia (seeRodriguez et al., 2005). Thus, upregulation of the calcium-sensingreceptor may lead to reduced hyperplasia.
The precise molecular mechanism for decreased parathyroid cellproliferation by calcimimetics is unclear. Miller et al. (2006)demonstrated that cinacalcet reduced hyperplasia in long-termuremic rats with established secondary hyperparathyroidism,whereas discontinuation of cinacalcet resulted in a gradual return ofparathyroid hyperplasia. The reduction in cellular proliferation as
measured by PCNA staining was associated with a concomitant rise inthe number of cells expressing the cyclin-dependent kinase inhibitorp21. The increase in p21 was not sustained upon discontinuation oftreatment. These findings suggest that the calcimimetic-mediatedincrease in p21 in uremic rats may mediate, at least in part, the effectson hyperplasia. The direct mechanism(s) by which calcimimeticsregulate hyperplasia has not been adequately investigated and is thefocus of further study.
Increasing evidence suggests that disordered mineral metabolismand bone disease, common complications of chronic kidney disease,are associated with increased risk for cardiovascular calcification(Chertow et al., 2002; Block et al., 2005; Braun et al., 2004), morbidity,and mortality (Block and Cunningham, 2006; Block et al., 2007;Matsuoka et al., 2004). The underlying mechanisms for this linkageare not completely understood, but are likely related to vascularcalcification leading to changes in cardiovascular structure andfunction (Ketteler et al., 2005; London et al., 2005).
The present data, coupled with previous findings (Tamagaki et al.,2006), demonstrate that rats fed a 0.75% adenine diet for four weeksexhibit vascular calcification. In agreement with Tamagaki et al.(2006), medial calcification, one of the characteristics of vascularcalcification in hemodialysis patients, was confirmed in these studies.This type of calcification is independent of lipids and seems to berelated to the expression of numerous bone-associated proteins (Jonoet al., 2000; Tyson et al., 2003). It is generally accepted that elevatedCa×P predisposes patients with secondary hyperparathyroidism tovascular and tissue calcification and increased cardiovascular mortal-ity risk (Goodman et al., 2004; Block et al., 1998). O'Neill (2007)recently downplayed the assumption that Ca×P product drivesectopic calcification and highlighted the fact that calcification is acomplex biological process not governed completely by CaHPO4
precipitation (O'Neill, 2007). In this study, serum calcium levels werereduced by AMG 641 without a pronounced effect on serumphosphorus. Reduction in blood calcium mediated by calcimimeticshas been observed in both hemodialysis patients as well as acutelynephrectomized animals, which suggests that this phenomenon maybe mediated in part through non-renal mechanisms (Fox et al., 1999).Calcimimetics in rats cause a transient increase in serum calcitonin,which may contribute to the rate of onset of the observed decrease inserum calcium levels (Nemeth et al., 2004; Fox et al., 1999).
The importance of serum calcium in giving rise to vascularcalcification independent of phosphorus levels was demonstrated bythe ability of AMG 641 to prevent the development of vascularcalcification despite the highly elevated phosphorus levels, whichhave been associated with the rapid development of vascularcalcification (Tamagaki et al., 2006). The ability of calcimimetics toprevent vascular calcification is consistent with the effect of thecalcimimetics R-568, AMG 641 or cinacalcet in 5/6 nephrectomizedrats (Lopez et al., 2006, 2008; Kawata et al., 2008), and of R-568 inattenuating calcium-induced mineral disposition in cultured vascularsmooth muscle cells (Alam et al., 2009). These authors suggested thatthe loss of functional vascular smooth muscle cell calcium-sensingreceptor induced by disturbances in serum calcium and phosphorus instage 5 chronic kidney disease exacerbates mineral deposition.Calcimimetics may prevent calcium-sensing receptor loss and/orimprove calcium-sensing receptor functionality to attenuate thecalcification process; similar to the calcimimetic-induced increase inparathyroid gland calcium-sensing receptor expression from rats withsecondary hyperparathyroidism (Mizobuchi et al., 2004) and in theintima of uremic rats (Koleganova et al., 2009). Recently resolution ofsoft tissue calcification was observed in a hemodialysis patientfollowing six months of treatment with cinacalcet (Zerbi et al., 2008).
Persistently elevated levels of PTH, as seen in secondary hyperpar-athyroidism, lead to high-turnover bone disease, the severity of whichis directly proportional to the magnitude of the disease and over-production of PTH. The calcimimetic R-568 reversed the development

312 C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
of osteitis fibrosa and restored cortical bone strength throughnormalization of serum PTH in uremic rats (Wada et al., 1998).Consistent with this observation, preclinical and clinical data havesuggested that cinacalcet may reduce bone turnover, tissue fibrosis,bone loss and fractures (Lien et al., 2005; Cunningham et al., 2005).Likewise, some evidence suggests that calcitriol can ameliorateosteitis fibrosa (Slatopolsky et al., 1984; Andress et al., 1989), whileother clinical studies show either no or detrimental effects on boneremodeling (Costa et al., 2003; Pahl et al., 1995).
Consistent with a previous study (Tamagaki et al., 2006), adenine-fed rats presented severe bone lesions within 4 weeks. AMG 641prevented adenine-induced loss in bone volume and trabecularnumber, increase in trabecular spacing, and defects in bone miner-alization. Although PTH levels were low in the AMG 641-treatedanimals (to 13 pg/ml), they were not significantly lower than normalnon-uremic controls, and did not result in adynamic bone disease orsignificant alterations in bone resorption parameters.
In conclusion, the present study indicates that the calcimimeticAMG 641 reduced circulating PTH levels, and prevented parathyroidhyperplasia, vascular calcification and bone abnormalities associatedwith secondary hyperparathyroidism. The ability of calcimimetics tomodify disease progression and morbidities related to secondaryhyperparathyroidism as observed in preclinical models is currentlybeing tested clinically in stage 5 chronic kidney disease patients withsecondary hyperparathyroidism.
Acknowledgements
Funding for this study and the preparation of this manuscript wasprovided by Amgen, Inc. Writing support was provided by WilliamStark, Jr. and Holly Tomlin (both are employees of, and stockholders in,Amgen, Inc).
References
Alam, M.U., Kirton, J.P., Wilkinson, F.L., Towers, E., Sinha, S., Rouhi, M., Vizard, T.N., Sage,A.P., Martin, D., Ward, D.T., Alexander, M.Y., Riccardi, D., Canfield, A.E., 2009.Calcification is associated with loss of functional calcium-sensing receptor invascular smooth muscle cells. Cardiovasc. Res. 81, 260–268.
Almaden, Y., Canalejo, A., Hernandez, A., Ballesteros, E., Garcia-Navarro, S., Torres, A.,Rodríguez, M., 1996. Direct effect of phosphorus on PTH secretion from whole ratparathyroid tissue in vitro. J. Bone Miner. Res. 11, 970–976.
Andress, D.L., Norris, K.C., Coburn, J.W., Slatopolsky, E.A., Sherrard, D.J.,1989. Intravenouscalcitriol in the treatment of refractory osteitis fibrosa of chronic renal failure. NewEng. J. Medicine. 321, 274–279.
Block, G.A., Cunningham, J., 2006. Morbidity and mortality associated with abnormal-ities in bone and mineral metabolism in CKD. In: Olgaard, K. (Ed.), Clinical Guide tothe Basics of Bone and Mineral Metabolismin CKD. InNational Kidney Foundation,New York, NY, pp. 77–92. Chapter 4.
Block, G.A., Hulbert, S., Levin, N.W., Port, F.K., 1998. Association of serum phosphorusand calcium×phosphorus product with mortality risk in chronic hemodialysispatients: a national study. Am. J. Kidney Dis. 31, 607–617.
Block, G.A.,Martin, K.J., De Francisco, A.L.M., Turner, S.A., Avram,M.M., Suranyi,M.G.,Hercz,G., Cunningham, J., Abu-Alfa, A.K., Messa, P., Coyne, D.W., Locatelli, F., Cohen, R.M.,Evanepoel, P., Moe, S.M., Fournier, A., Braun, J., McCary, L.C., Zani, V.J., Olson, K.A.,Drueke, T.B., Goodman, W.G., 2004. Cinacalcet for secondary hyperparathyroidism inpatients receiving hemodialysis. N. Engl. J. Med. 350, 1516–1525.
Block, G.A., Raggi, P., Bellasi, A., Kooienga, L., Spiegel, D.M., 2007.Mortalityeffectof coronarycalcification and phosphate binder choice in incident hemodialysis patients. KidneyInt. 71, 438–441.
Block, G.A., Spiegal, D.M., Ehrlich, J., Mehta, R., Lindbergh, J., Dreisbach, A., Raggi, P.,2005. Effects of sevelamer and calcium on coronary artery calcification in patientsnew to hemodialysis. Kidney Int. 68, 1815–1824.
Braun, J., Asmus, H.G., Holzer, H., Brunkhorst, R., Krause, R., Schulz, W., Neumayer, H.H.,Raggi, P., Bommer, J., 2004. Long-term comparison of a calcium-free phosphatebinder and calcium carbonate–phosphorus metabolism and cardiovascular calci-fication. Clin. Nephrol. 62, 104–115.
Brown, E.M., MacLeod, R.J., 2001. Extracellular calcium sensing and extracellularcalcium signaling. Physiol. Rev. 81, 239–297.
Brown, A.J., Zhong, M., Finch, J., Ritter, C., McCracken, R., Morrissey, J., Slatopolsky, E.,1996. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium.Am. J. Physiol. 270, F454–F460.
Chattopadhyay, N., Mithal, A., Brown, E.M., 1996. The calcium-sensing receptor: awindow into the physiology and pathophysiology of mineral ion metabolism.Endocr. Rev. 17, 289–307.
Chertow, G.M., Burke, S.K., Raggi, P., Treat to Goal Working Group, 2002. Sevelamerattenuates the progression of coronary and aortic calcification in hemodialysispatients. Kidney Int. 62, 245–252.
Chin, J., Miller, S.C., Wada, M., Nagano, N., Nemeth, E.F., Fox, J., 2000. Activation of thecalcium receptor by a calcimimetic compound halts the progression of secondaryhyperparathyroidism in uremic rats. J. Am. Soc. Nephrol. 11, 903–911.
Colloton, M., Shatzen, E., Miller, G., Stehman-Breen, C., Wada, M., Lacey, D., Martin, D.,2005. Cinacalcet HCl attenuates parathyroid hyperplasia in a ratmodel of secondaryhyperparathyroidism. Kidney Int. 67, 467–476.
Costa, A.F., dos Reis, L.M., Riberio, M.C., Moyses, R.M., Jorgetti, V., 2003. Effects ofcalcitriol on parathyroid function and on bone remodeling in secondaryhyperparathyroidism. Nephrol. Dial. Transplant. 18, 743–749.
Cozzolino, M., Lu, Y., Finch, J., Slatopolsky, E., Dusso, A.S., 2001. p21WAF1 and TGF-mediate parathyroid growth arrest by vitamin D and high calcium. Kidney Int. 60,2109–2117.
Cunningham, J., Danese, M., Olson, K., Klassen, P., Chertow, G.M., 2005. Effects of thecalcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-relatedquality of life in secondary hyperparathyroidism. Kidney Int. 68, 1793–1800.
de Francisco, A.L., 2004. Secondary hyperparathyroidism: review of the disease and itstreatment. Clin. Ther. 26, 1976–1993.
Drueke, T., Martin, D., Rodriguez, M., 2007. Can calcimimetics inhibit parathyroidhyperplasia? Evidence from preclinical studies. Nephrol. Dial. Transplant. 22,1828–1839.
Fox, J., Lowe, S.H., Conklin, R.L., Petty, B.A., Nemeth, E.F., 1999. Calcimimetic compoundNPS R-568 stimulates calcitonin secretion but selectively targets parathyroid glandCa2+ receptor in rats. J. Pharmacol. Exp. Ther. 290, 480–486.
Goodman, W.G., London, G., Amann, K., Block, G.A., Giachelli, C., Hruska, K.A., Ketteler,M., Levin, A., Massy, Z., McCarron, D.A., Raggi, P., Shanahan, C.M., Yorioka, N.,Vascular Calcification Work Group, 2004. Vascular calcification in chronic kidneydisease. Am. J. Kidney Dis. 43, 572–579.
Goodman, W.G., Quarles, L.D., 2008. Development and progression of secondaryhyperparathyroidism in chronic kidney disease: lessons from molecular genetics.Kidney Int. 74, 276–288.
Hebert, S.C., 2006. Therapeutic use of calcimimetics. Annu. Rev. Med. 57, 349–364.Henley, C., Colloton, M., Cattley, R.C., Shatzen, E., Towler, D.A., Lacey, D., Martin, D., 2005.
1,25-Dihydroxyvitamin D3 but not cinacalcet HCl treatment mediates aorticmineralization in a rat model of secondary hyperparathyroidism. Nephrol. Dial.Transplant. 20, 1370–1377.
Ho, C., Conner, D.A., Pollak, M.R., Ladd, D.J., Kifor, O., Warren, H.B., Brown, E.M., Seidman,J.G., Seidman, C.E., 1995. A mouse model of human familial hypocalciurichypercalcemia and neonatal severe hyperparathyroidism. Nat.Genet. 11, 389–394.
Jono, S., McKee, M.D., Murry, C.E., Shiol, A., Nishizawa, Y., Mori, K., Morii, H., Giachelli, C.M.,2000. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 87,E10–17.
Kalantar-Zadeh, K., Kuwae, N., Regidor, D.L., Kovesdy, C.P., Kilpatrick, R.D., Shinaberger, C.S.,McAllister, C.J., Budoff, M.J., Salusky, J.B., Kopple, J.D., 2006. Survival predictability oftime-varying indicators of bonedisease inmaintenancehemodialysis patients. KidneyInt. 70, 771–780.
Kawata, T., Nagano, N., Obi, M., Miyata, S., Koyama, C., Kobayahsi, N., Wakita, S., Wada,M., 2008. Cinacalcet suppresses calcification of the aorta and heart in uremic rats.Kidney Int. 74, 1270–1277.
Ketteler, M., Gross, M.L., Ritz, E., 2005. Calcification and cardiovascular problems in renalfailure. Kidney Int. 94 (Suppl), S120–S127.
Koleganova, N., Piecha, G., Ritz, E., Schmitt, C.P., Gross, M.L., 2009. A calcimimetic (R-568),but not calcitriol, prevents vascular remodeling in uremia. Kidney Int. 75, 60–71.
Li, Y., Amling, M., Pirro, A.E., Priemel, M., Meuse, J., Baron, R., Delling, G., Demay, M.B.,1998. Normalization of mineral ion homeostasis by dietary means preventshyperparathyroidism, rickets and osteomalacia, but not alopecia in vitamin Dreceptor-ablated mice. Endocrinology. 139, 4391–4396.
Lien, Y.H., Silva, A.L., Whittman, D., 2005. Effects of cinacalcet on bone mineral density inpatients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 20, 132–137.
London, G.M., Marchais, S.J., Guerin, A.P., Metivier, F., 2005. Arteriosclerosis, vascularcalcifications and cardiovascular disease in uremia. Curr. Opin. Nephrol. Hypertens.14, 525–531.
Lopez, I., Aguilera-Tejero, E., Mendoza, F.J., Almaden, Y., Perez, J., Martin, D., Rodrigues,M., 2006. Calcimimetic R-568 decreases extraosseous calcifications in uremic ratstreated with calcitriol. J. Am. Soc. Nephrol. 17, 795–804.
Lopez, I., Mendoza, F.J., Aguilera-Tejero, E., Perez, J., Guerrero, F., Martin, D., Rodriguez,M., 2008. The effect of calcitriol, paricalcitol and a calcimimetic on extraosseouscalcifications in uremic rats. Kidney Int. 73, 300–307.
Matsuoka, M., Iseki, K., Tamashiro, M., Fujimoto, N., Higa, N., Touma, T., Takishita, S.,2004. Impact of high coronary artery calcification score (CACS) on survival inpatients on chronic hemodialysis. Clin. Exp. Nephrol. 8, 54–58.
Miller, G., Davis, J., Shatzen, E., Lott, F., Haas, K., Cattley, R., Lacey, D., Tasker, A., Henley, C.,Martin, D., 2006a. Calcimimetic reduces aortic calcification in calcitriol-treateduremic rats. Am. Soc. Nephrol. 17, 698A, Abstr. SAPO584.
Miller, J., Davis, J., Van, G., Shatzen, E., Henley, C., Martin, D., 2006b. Inhibition ofparathyroid gland hyperplasia and increased expression of p21 in the parathyroidare reversed upon discontinuation of cinacalcet HCl treatment. Nephrol, Dial.Transplant. 21 (4), iv30.
Mizobuchi, M., Hatamura, I., Ogata, H., Saji, F., Uda, S., Shiizaki, K., Sakaguchi, T., Negi, S.,Kinugasa, E., Koshikawa, S., Akizawa, T., 2004. Calcimimetic compound upregulatesdecreased calcium-sensing receptor expression level in parathyroid glands of ratswith chronic renal insufficiency. J. Am. Soc. Nephrol. 15, 2579–2587.
Nemeth, E.F., Heaton, W.H., Miller, M., Fox, J., Balandrin, M.F., Van Wagenen, B.C.,Colloton, M., Karbon, W., Scherrer, J., Shatzen, E., Rishton, G., Scully, S., Oi, M., Harris,

313C. Henley et al. / European Journal of Pharmacology 616 (2009) 306–313
R., Lacey, D., Martin, D., 2004. Pharmacodynamics of the type II calcimimeticscompound, cinacalcet HCl. J. Pharmacol. Exp. Ther. 308, 627–635.
O'Neill, W.C., 2007. The fallacy of the calcium–phosphorous product. Kidney Int. 72,792–796.
Pahl, M., Jara, A., Bover, J., Felsenfeld, A.J., 1995. Studies in a hemodialysis patientindicating that calcitriol may have a direct suppressive effect on bone. Nephron 71,228–232.
Parfitt, A.M., Drezner, M.K., Glorieux, F.H., Kanis, J.A., Malluche, H., Meunier, P.J., Ott, S.M.,Recker, R.R., 1987. Bone histomorphometry: standardization of nomenclature,symbols, and units. Report of the ASBMR Histomorphometry NomenclatureCommittee. J. Bone Miner. Res. 2, 595–610.
Piecha, G., Kokeny, G., Nakagawa, K., Koleganova, N., Geldvvev, A., Berger, I., Ritz, E.,Schmitt, C.P., Gross, M.L., 2008. Calcimimetic R-568 or calcitriol: equally beneficialon progression of renal damage in subtotally nephrectomized rats. Am. J. Physiol.Renal Physiol. 294, F748–757.
Rodríguez,M.E., Almaden, Y., Canadillas, S., Canalejo, A., Siendones, E., Lopez, I., Aguilera-Tejero, E., Martin, D., Rodríguez, M., 2007. The calcimimetic R-568 increases vitaminD receptor expression in rat parathyroid glands. Am. J. Physiol. Renal Physiol. 292,F1390–F1395.
Rodriguez,M., Nemeth, E.,Martin, D., 2005. The calcium-sensing receptor: a key factor inthe pathogenesis of secondary hyperparathyroidism. Am. J. Physiol. Renal Physiol.288, F253–F264.
Rogers, K.V., Dunn, C.K., Herbert, S.C., Brown, E.M., Nemeth, E.F., 1995. Pharmacologicalcomparison of bovine parathyroid, human parathyroid, and rat kidney calciumreceptors expressed in HEK 293 cells. J. Bone Miner. Res. 10, S483.
Shalhoub, V., Shatzen, E., Henley, C., Boedigheimer, M., McNinch, J., Manuoukian, R.,Damore, M., Fitzpatrick, D., Haas, K., Twomey, B., Kiaei, P., Lacey, D.L., Martin, D.,2006a. Calcification inhibitors and Wnt signaling proteins are implicated in bovineartery smooth muscle cell calcification in the presence of phosphate and vitamin Dsterols. Calcif. Tissue Int. 79, 431–442.
Shalhoub, V., Shatzen, E., Ward, S.C., Young, J., Boedigheimer, M., Twehues, L., Damore,M., Kiaei, P., Fitzpatrick, D., Henley, C., Haas, K., Lacey, D., Martin, D., 2006b. Effects ofCalcitriol and Paricalcitol on Gene Expression Patterns in Human Coronary ArterySmooth Muscle Cells. Abstract at XLIII ERA-EDTA Congress — July 15–18.
Slatopolsky, E., Finch, J., Denda, M., Ritter, C., Zhong, M., Dusso, A., MacDonald, P.N.,Brown, A.J., 1996. Phosphorus restriction prevents parathyroid gland growth. Highphosphorus directly stimulates PTH secretion in vitro. J. Clin. Invest. 97, 2534–2540.
Slatopolsky, E., Weerts, C., Thielan, J., Horst, R., Harter, H., Martin, K.J., 1984. Markedsuppression of secondary hyperparathyroidism by intravenous administration of1,25-dihydroxy-cholecalciferol in uremic patients. J. Clin. Invest. 74, 2136–2143.
Szabo, A., Merke, J., Beier, E., Mall, G., Ritz, E., 1989. 1,25(OH)2 vitamin D3 inhibitsparathyroid cell proliferation in experimental uremia. Kidney Int. 35, 1049–1056.
Tamagaki, K., Yuan, Q., Ohkawa, H., Imazeki, I., Moriouchi, Y., Imai, N., Sasaki, S., Takeda, K.,Fukagawa, M., 2006. Severe hyperparathyroidism with bone abnormalities andmetastatic calcification in ratswithadenine-induceduraemia.Nephrol.Dial. Transplant.21, 651–659.
Tentori, F., Hunt,W.C., Stidley, C.A., Rohrscheib,M.R., Bedrick, E.J.,Meyer, K.B., Johnson, H.K.,Zager, P.G., Medical Directors of Dialysis Clinic Inc., 2006. Mortality risk amonghemodialysispatients receivingdifferent vitaminDanalogs. Kidney Int. 70,1858–1865.
Tyson,K.L., Reynolds, J.L.,McNair, R., Zhang,O.,Weissberg, P.L., Shanahan,C.M., 2003.Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns ofexpression inhumanarterial calcification.Arterioscler. Thromb.Vasc.Biol. 23, 489–494.
Wada, M., Furuya, Y., Sakiyama, J., Kobayashi, N., Miyata, S., Ishii, H., Pagano, N.,1997. Thecalcimimetic compound NPS R-568 suppresses parathyroid cell proliferation in ratswith renal insufficiency. Control of parathyroid cell growth via a calcium receptor.J. Clin. Invest. 15, 2977–2983.
Wada, M., Ishii, H., Furuya, Y., Fox, J., Nemeth, E.F., Nagano, N., 1998. NPS R-568 halts orreverses osteitis fibrosa in uremic rats. Kidney Int. 53, 448–453.
Wada, M., Nagano, N., Furuya, Y., Chin, J., Nemeth, E.F., Fox, J., 2000. Calcimimetic NPS R-568 prevents parathyroid hyperplasia in rats with severe secondary hyperpara-thyroidism. Kidney Int. 57, 50–58.
Yokozawa, T., Zheng, P.D., Oura, H., Koizumi, F., 1986. Animal model of adenine-inducedchronic renal failure in rats. Nephron 44, 230–234.
Young, E.W., Albert, J.M., Satayathum, S., Goodkin, D.A., Pisoni, R.L., Akiba, T., Akizawa, T.,Kurokawa, K., Bommer, J., Piera, L., Port, F.K., 2005. Predictors and consequences ofaltered mineral metabolism: The Dialysis Outcomes and Practice Patterns Study.Kidney Int. 67 (3), 1179–1187.
Zerbi, S., Ruggiero, P., Pedrini, L.A., 2008. Massive soft tissue calcification and cinacalcet.J. Clin. Endo. Metab. 93, 1121–1122.
Related Documents











