The Bile Acid Sensor Farnesoid X Receptor Is a Modulator of Liver Immunity in a Rodent Model of Acute Hepatitis 1 Andrea Mencarelli,* Barbara Renga,* Marco Migliorati,* Sabrina Cipriani,* Eleonora Distrutti, † Luca Santucci, † and Stefano Fiorucci 2 * Immune-mediated liver diseases including autoimmune and viral hepatitis are a major health problem worldwide. In this study, we report that activation of the farnesoid X receptor (FXR), a member of the ligand-activated nuclear receptor superfamily and bile sensor highly expressed in the liver, attenuates liver injury in a model of autoimmune hepatitis induced by Con A. We found that FXR gene ablation results in a time-dependent increase of liver expression (up to 20-fold in a 9-mo-old mouse) of osteopontin, a NKT cell-derived extracellular matrix protein and immunoregulatory cytokine. In comparison to wild-type, FXR / mice are more susceptible to Con A-induced hepatitis and react to Con A administration by an unregulated production of osteopontin. Administering wild-type mice with a synthetic FXR agonist attenuated Con A-induced liver damage and liver expression of the osteopontin gene. By in vitro studies, we found that FXR is expressed by primarily isolated NKT cells and its ablation favors ostepontin production in response to Con A. Chromatin immunoprecipitation assay and coimmunoprecipitation experiments demonstrate that the short heterodimer partner (SHP), a nuclear receptor and FXR target, was expressed by NKT cell hybridomas and increased in response to FXR activation. FXR activates SHP that interacts with and inhibits c-Jun binding to the osteopontin promoter. These data indicate that in NKT cells, FXR activation causes a SHP-mediated inhibition of osteopontin production. These data support the notion that the bile acid sensor FXR regulates the activation of liver NKT cells. The Journal of Immu- nology, 2009, 183: 6657– 6666. T cell-mediated liver diseases, including autoimmune and viral hepatitis, are associated with significant morbidity and mortality worldwide and represent a serious concern in the clinical setting (1, 2). The Con A-induced hepatitis in mice is a model of T cell-mediated liver diseases (3). Following Con A administration, liver histology shows massive granulocyte accu- mulation, CD4 T cell infiltration, and hepatocyte necrosis/apop- tosis (3, 4). The induction of this hepatitis depends on CD1d-re- stricted invariant NKT cells, that express an invariant V14J18 TCR (5, 6). Thus, J18 / and CD1d / mice that lack NKT cells are resistant to Con A-induced hepatic injury (5, 6), whereas the adoptive transfer of NKT cells isolated from wild-type (WT) 3 mice into healthy CD1d / or J18 / mice restores hepatic injury mediated by Con A. Previous studies have shown that Con A ac- tivates hepatic resident NKT cells to produce IL-4 (5, 6); IL-4, in turn, acts on NKT cells in an autocrine fashion to induce Fas ligand (FasL) expression on these cells, which subsequently promotes hepatocyte cell death (i.e., cytotoxicity), possibly by interacting with Fas-expressing hepatocytes. In agreement with this concept is the observation that Fas, FasL, and IL-4 exert proinflammatory effects during Con A-induced hepatitis since mice deficient in any of the aforementioned mediators are resistant to Con A-induced liver damage (7–9). In addition to these cytokines, hepatic NKT cells release os- teopontin (OPN), an extracellular matrix glycoprotein known for its role in supporting adhesion and migration of inflammatory cells (10, 11). OPN plays a nondispensable role in the development of Con A-induced hepatitis in mice (12–14), as demonstrated by the fact that neutralizing OPN function by OPN gene deletion or by using OPN small-interfering RNA protects against Con A-induced liver injury. In addition, OPN overexpression occurs in other mod- els of liver injury (15–17) and OPN dysregulation facilitates in- flammation-associated fibrosis in several organs including lung, heart, and liver (18 –20). The farnesoid X receptor (FXR; NR1H4) is a member of the nuclear receptor superfamily of ligand-activated regulatory factors (21–23). Nuclear receptors have been shown to regulate the es- sential aspect of innate immunity (24 –26) and production of OPN in several experimental settings (27–29), but this information lacks for FXR. FXR is highly expressed in the liver, intestine, kidney, and adrenal gland where it function as a bile acid sensor (30 –32). Thus, there is circumstantial evidence to link the activity of FXR to the regulation of bile acid synthesis from cholesterol, providing a link between nutrient absorption and regulation of key steps in the intermediate metabolism. In addition, because bile acids might be toxic for the liver, activation of FXR by natural and synthetic ligands has been shown to prevent bile acid-induced liver toxicity in a variety of rodent models (33–39). The homeostatic function of FXR in the liver is highlighted by the demonstration that mice harboring a disrupted FXR (FXR / ) develop spontaneously an *Dipartimento di Medicina Clinica e Sperimentale, University of Perugia, Perugia, Italy; and † Azienda Ospedaliera di Perugia, Perugia, Italy Received for publication April 30, 2009. Accepted for publication August 31, 2009. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1 A.M. has carried out animal studies, cytokine measurement, and flow cytometry and wrote the manuscript. S.C. has performed the immunohistochemistry. B.R. and M.M. carried out RT-PCR, Western blot, ChIP, and coimmunoprecipitation experiments. S.F., E.D., and L.S. designed the study and wrote the manuscript. 2 Address correspondence and reprint requests to Dr. Stefano Fiorucci, University of Perugia, Dipartimento di Medicina Clinica e Sperimentale, Via E. Dal Pozzo; 06122 Perugia, Italy. E-mail address: fi[email protected] 3 Abbreviations used in this paper: WT, wild type; 6E-CDCA, 6-ethylchenodeoxy- cholic acid; ChIP, chromatin immunoprecipitation; FXR, farnesoid X receptor; MPO, myeloperoxidase; OPN, osteopontin; SHP, short heterodimer partner; FasL, Fas li- gand; AST, aspartate aminotransferase; C T , cycle threshold; PMN, polymorphonu- clear neutrophil; HVEM, herpes virus entry mediator. Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00 The Journal of Immunology www.jimmunol.org/cgi/doi/10.4049/jimmunol.0901347

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Bile Acid Sensor Farnesoid X Receptor Is a Modulator ofLiver Immunity in a Rodent Model of Acute Hepatitis1
Andrea Mencarelli,* Barbara Renga,* Marco Migliorati,* Sabrina Cipriani,*Eleonora Distrutti,† Luca Santucci,† and Stefano Fiorucci2*
Immune-mediated liver diseases including autoimmune and viral hepatitis are a major health problem worldwide. In this study,we report that activation of the farnesoid X receptor (FXR), a member of the ligand-activated nuclear receptor superfamily andbile sensor highly expressed in the liver, attenuates liver injury in a model of autoimmune hepatitis induced by Con A. We foundthat FXR gene ablation results in a time-dependent increase of liver expression (up to 20-fold in a 9-mo-old mouse) of osteopontin,a NKT cell-derived extracellular matrix protein and immunoregulatory cytokine. In comparison to wild-type, FXR�/� mice aremore susceptible to Con A-induced hepatitis and react to Con A administration by an unregulated production of osteopontin.Administering wild-type mice with a synthetic FXR agonist attenuated Con A-induced liver damage and liver expression of theosteopontin gene. By in vitro studies, we found that FXR is expressed by primarily isolated NKT cells and its ablation favorsostepontin production in response to Con A. Chromatin immunoprecipitation assay and coimmunoprecipitation experimentsdemonstrate that the short heterodimer partner (SHP), a nuclear receptor and FXR target, was expressed by NKT cell hybridomasand increased in response to FXR activation. FXR activates SHP that interacts with and inhibits c-Jun binding to the osteopontinpromoter. These data indicate that in NKT cells, FXR activation causes a SHP-mediated inhibition of osteopontin production.These data support the notion that the bile acid sensor FXR regulates the activation of liver NKT cells. The Journal of Immu-nology, 2009, 183: 6657–6666.
T cell-mediated liver diseases, including autoimmune andviral hepatitis, are associated with significant morbidityand mortality worldwide and represent a serious concern
in the clinical setting (1, 2). The Con A-induced hepatitis in miceis a model of T cell-mediated liver diseases (3). Following Con Aadministration, liver histology shows massive granulocyte accu-mulation, CD4� T cell infiltration, and hepatocyte necrosis/apop-tosis (3, 4). The induction of this hepatitis depends on CD1d-re-stricted invariant NKT cells, that express an invariant V�14J�18TCR (5, 6). Thus, J�18�/� and CD1d�/� mice that lack NKT cellsare resistant to Con A-induced hepatic injury (5, 6), whereas theadoptive transfer of NKT cells isolated from wild-type (WT)3 miceinto healthy CD1d�/� or J�18�/� mice restores hepatic injurymediated by Con A. Previous studies have shown that Con A ac-tivates hepatic resident NKT cells to produce IL-4 (5, 6); IL-4, inturn, acts on NKT cells in an autocrine fashion to induce Fas ligand
(FasL) expression on these cells, which subsequently promoteshepatocyte cell death (i.e., cytotoxicity), possibly by interactingwith Fas-expressing hepatocytes. In agreement with this concept isthe observation that Fas, FasL, and IL-4 exert proinflammatoryeffects during Con A-induced hepatitis since mice deficient in anyof the aforementioned mediators are resistant to Con A-inducedliver damage (7–9).
In addition to these cytokines, hepatic NKT cells release os-teopontin (OPN), an extracellular matrix glycoprotein known forits role in supporting adhesion and migration of inflammatory cells(10, 11). OPN plays a nondispensable role in the development ofCon A-induced hepatitis in mice (12–14), as demonstrated by thefact that neutralizing OPN function by OPN gene deletion or byusing OPN small-interfering RNA protects against Con A-inducedliver injury. In addition, OPN overexpression occurs in other mod-els of liver injury (15–17) and OPN dysregulation facilitates in-flammation-associated fibrosis in several organs including lung,heart, and liver (18–20).
The farnesoid X receptor (FXR; NR1H4) is a member of thenuclear receptor superfamily of ligand-activated regulatory factors(21–23). Nuclear receptors have been shown to regulate the es-sential aspect of innate immunity (24–26) and production of OPNin several experimental settings (27–29), but this information lacksfor FXR. FXR is highly expressed in the liver, intestine, kidney,and adrenal gland where it function as a bile acid sensor (30–32).Thus, there is circumstantial evidence to link the activity of FXRto the regulation of bile acid synthesis from cholesterol, providinga link between nutrient absorption and regulation of key steps inthe intermediate metabolism. In addition, because bile acids mightbe toxic for the liver, activation of FXR by natural and syntheticligands has been shown to prevent bile acid-induced liver toxicityin a variety of rodent models (33–39). The homeostatic function ofFXR in the liver is highlighted by the demonstration that miceharboring a disrupted FXR (FXR�/�) develop spontaneously an
*Dipartimento di Medicina Clinica e Sperimentale, University of Perugia, Perugia,Italy; and †Azienda Ospedaliera di Perugia, Perugia, Italy
Received for publication April 30, 2009. Accepted for publication August 31, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 A.M. has carried out animal studies, cytokine measurement, and flow cytometry andwrote the manuscript. S.C. has performed the immunohistochemistry. B.R. and M.M.carried out RT-PCR, Western blot, ChIP, and coimmunoprecipitation experiments.S.F., E.D., and L.S. designed the study and wrote the manuscript.2 Address correspondence and reprint requests to Dr. Stefano Fiorucci, University ofPerugia, Dipartimento di Medicina Clinica e Sperimentale, Via E. Dal Pozzo; 06122Perugia, Italy. E-mail address: [email protected] Abbreviations used in this paper: WT, wild type; 6E-CDCA, 6-ethylchenodeoxy-cholic acid; ChIP, chromatin immunoprecipitation; FXR, farnesoid X receptor; MPO,myeloperoxidase; OPN, osteopontin; SHP, short heterodimer partner; FasL, Fas li-gand; AST, aspartate aminotransferase; CT, cycle threshold; PMN, polymorphonu-clear neutrophil; HVEM, herpes virus entry mediator.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0901347

array of hepatocellular abnormalities including adenomas and car-cinomas. This pattern associates with an increased liver epithelialcell proliferation and expression of proinflammatory cytokines andoncogenes (40, 41).
In the present study, we demonstrate that FXR�/� mice spon-taneously develop hepatic damage and liver immune dysregulationand show enhanced susceptibly to T cell-mediated hepatitis. Inaddition, we provide evidence that NKT cells express a functionalFXR and that FXR ligands activate counterregulatory signals inNKT cells that prevent hepatitis by inhibiting Con A-induced OPNsynthesis.
Materials and MethodsMice
Specific pathogen-free C57BL/6 mice (5–7 wk old) were purchased fromCharles River Laboratories. Homozygous FXR�/� mice (C57BL/6BJ6 ge-netic background) were originally provided by F. J. Gonzalez (NationalInstitutes of Health, Bethesda, MD). Animals were maintained with a 12-hlight/12-h dark cycle with free access to food and water. All procedureswere approved by the Animal Care and Ethics Committees of the Univer-sity of Perugia.
Cell lines and culture media
The HepG2 cell line was cultured in MEM supplemented with 10% FBS,100 U/ml penicillin, and 100 mg/ml streptomycin. DN32.D3 cells, a mu-rine NKT cell hybridoma cell line (42), provided by Dr. A. Bendelac(Princeton University, Princeton, NJ), were cultured in DMEM supple-mented with 5% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Treatments
FXR WT and FXR�/� mice were injected i.v. with a freshly prepared ConA, type IV, in a pyrogen-free saline (Sigma-Aldrich). In a first set of ex-periments, we have investigated whether FXR gene ablation sensitizesmice to Con A-induced liver injury. To this end, FXR�/� and WT micewere administered with a lethal dose of Con A (25 mg/kg), with or withoutpretreatment with 6�-ethyl-chenodeoxycholic acid (6E-CDCA; 10 mg/kgi.p., 30 min before Con A), a semisynthetic FXR ligand. Mortality wasassessed over the next 2 days. In a second set of experiments, FXR�/� andWT mice were administered with a sublethal dose of Con A (10 mg/kg),with or without pretreatment with the same agent as described above. Micewere then sacrificed under pentothal anesthesia at 4, 8, 24, and 48 h afterCon A administration and blood and livers were collected. Plasma aspartateaminotransferase (AST) was used as a marker of hepatic injury.
OPN immunohistochemistry
Cryostat liver sections, 5-�m thickness, were washed in PBS, pretreatedwith H2O2 for 10 min, and then incubated in a blocking solution containing10% goat serum and 0.1% Triton X-100 in PBS. After overnight incubationat 4°C with an 1/200 primary anti-OPN Ab (Sigma-Aldrich), slides werewashed three times in PBS and incubated for 2 h at room temperature witha biotinylated secondary Ab (Vector Laboratories) diluted 1/250 in PBS.Finally, slides were incubated with the avidin-biotin complex (Vectastain;Vector Laboratories) and visualized with diaminobenzidine.
Isolation of liver-infiltrating mononuclear cells and flowcytometric analysis
Mice were sacrificed 2 h after Con A administration. Liver were perfusedthrough the portal vein with 5 ml of PBS. The livers were harvested, dis-persed through a stainless steel mesh (100-�m pore size), and washed withHBSS containing 10% bovine calf serum. The dispersed cells were mixedwith a 33% Percoll solution (Amersham Biosciences) containing 100 U/mlheparin (Sigma-Aldrich) and centrifuged for 20 min (800 � g) at roomtemperature. Next, the cell pellet was suspended in 2 ml of ammoniumchloride solution for 2 min for RBC lysis. The isolated mononuclear cellswere then suspended in HBSS and kept on ice until stained with anti-CD3,anti-CD4, anti-Gr1, anti-FasL, anti-CD11b, anti-CD14, and anti-NK1.1Abs (BD Biosciences) for flow cytometry analysis (Epics XL System;Beckman Coulter).
Isolation of liver NKT cells
Livers were treated as described above. After RBC lysis, cell number andviability were determined by trypan blue dye exclusion. Subsequently, B
cells were removed by adherence on plates coated with affinity-purifiedgoat anti-mouse IgG� IgM Abs twice. T cells, NKT cells, and NK cellswere isolated using anti-CD5 mAb and anti-DX5 mAb to avoid the stim-ulation of these cells by cross-linking with mAbs. CD5-positive cells, in-cluding both T and NKT cells, were positively selected on anti-CD5 mAb-coated plates (BD Pharmingen). Then, DX5�CD5� NKT cells werepositively separated from DX5�CD5� T cells by anti-NK (DX5) Micro-Beads (Miltenyi Biotec). Purity of cell fractions was verified by two-colorflow cytometry analysis using FITC-conjugated anti-mouse CD3 and PE-conjugated anti-NK1.1 mAbs (BD Pharmingen). The NKT cell fractioncontained �90% CD3�NK1.1� NKT cells and �10% CD3�NK1.1� Tcells or CD3�NK1.1� NK cells. NKT-purified cells, obtained from WTmice, were used to evaluate FXR expression by quantitative real-time RT-PCR (qRT-PCR) and compared with HepG2 cells. In an additional set ofexperiments, NKT liver cells obtained from WT and FXR�/� mice, 5 moold (n � 15), were incubated with 5 �g/ml Con A for 16 h. At the end ofincubation, the cells were collected and OPN expression was assessed byqRT-PCR.
Evaluation of FXR activity in ND32.D3 cells
Immunoprecipitation. DN32.D3 cells (42), a NKT hybridoma, were serumstarved for 24 h and then incubated for 18 h with 10 �M 6E-CDCA and 1�g/ml Con A at 37°C. 6E-CDCA was added 30 min before Con A/LPStreatments. Cellular lysates were first diluted with E1A lysis buffer con-taining phosphatase and protease inhibitors, sonicated, and than immuno-precipitated with specific Ab anti-SHP (Q-14, sc-15283) from Santa CruzBiotechnology and with protein A beads (Amersham Biosciences). Proteinsamples were denatured by heating at 90°C in SDS sample reducing bufferand resolved by electrophoresis on 10% SDS-polyacrylamide gels. Afterprotein transfer to the nitrocellulose membranes, the filters were probedwith anti-c-Jun Ab (N, sc-45; Santa Cruz Biotechnology). Proteins werethen visualized by chemiluminescence using Super Signal West FEMTOreagent (Pierce).Western blotting. Total lysates from DN32.D3 cells were prepared by sol-ubilization in NuPAGE sample buffer containing sample reducing agent.Proteins were separated by PAGE, transferred to nitrocellulose membranes(Bio-Rad), and then probed with primary anti-FXR Ab (Abcam). The anti-Ig G rabbit Ab (Bio-Rad) was used as a secondary Ab, and specific proteinbands were visualized by chemiluminescence using Super Signal WestFEMTO reagent (Pierce, Rockford, IL).Chromatin immunoprecipitation (ChIP) assay. Cross-linking and ChIPassays were performed according to the manufacturer’s protocols (Abcam),with minor modifications. Briefly, DN32.D3 cells were cross-linked with1% formaldehyde at room temperature and then the reaction was termi-nated by the addition of glycine at a final concentration of 0.125 M. Cellswere washed in ice-cold PBS and lysed with SDS lysis buffer (1% SDS, 10mM EDTA, and 50 mM Tris-HCl, pH 8). Cellular lysates were diluted withChIP dilution buffer, sonicated, and immunoprecipitated with specific Abs:anti-SHP and anti-c-Jun from (Santa Cruz Biotechnology). Immunopre-cipitates were collected with protein A beads (Amersham Biosciences) andwashed sequentially first with a low-salt wash buffer and then with a high-salt wash buffer using the manufacturer’s recommended procedures. DNAwas eluted by addition of 1% SDS and 0.1 M NaHCO3, and the cross-linking reactions were reversed by heating the mixture to 65°C overnight.The DNA was recovered from immunoprecipitated material by proteinaseK treatment at 65°C for 1 h followed by phenol:chloroform (1:1) extrac-tion, ethanol precipitation, and dissolved into 50 �l of water. For real-timePCR, 5 �l of ChIP products from DN32.D3 cells were used to performamplification of the mouse OPN gene promoter using the following prim-ers against the AP-1 binding site: AGGAGGAAGTGTAGGAGCAGGTand GGCTCAGACCTCCCAGAATTTA.Real-time PCR. Quantization of the expression of selected genes was per-formed by real-time qRT-PCR. Total RNA were obtained from livers andcells and isolated with TRIzol reagent (Invitrogen), incubated with DNaseI, and reverse-transcribed with Superscript II (Invitrogen) according to themanufacturer’s specifications. For real-time PCR, 100 ng of template wasused in a 25-�l reaction containing a 0.3 �M concentration of each primerand 12.5 �l of 2� SYBR Green PCR Master Mix (Bio-Rad). All reactionswere performed in triplicate using the following cycling conditions: 2 minat 95°C, followed by 50 cycles of 95°C for 10 s and 60°C for 30 s usingan iCycler iQ instrument (Bio-Rad). The mean value of the replicates foreach sample was calculated and expressed as cycle threshold (CT). Theamount of gene expression was then calculated as the difference (�CT)between the CT value of the sample for the target gene and the mean CT
value of that sample for the endogenous control (GAPDH). Relative ex-pression was calculated as the difference (��CT) between the �CT valuesof the test and control samples for each target gene. The relative level of
6658 FXR AND LIVER NKT CELLS

expression was measured as 2 � ��CT. All PCR primers were designed withthe software PRIMER3-OUTPUT using published sequence data obtainedfrom the National Center for Biotechnology Information database.
Mouse primers were as follows: FXR, TGTGAGGGCTGCAAAGGTTand ACATCCCCATCTTGGAC; IL-4, CCTCACAGCAACGAAGAACAand ATCGAAAAGCCCGAAAGAGT; IFN-�, GCGTCATTGAATCACACCTG and GACCTGTGGGGTTGACCT; OPN, CTGTTGCCCAGCTTCTAGG and GGCTTTGGAACTTGCTTGAC; TNF-�, ACGGCATGGATCTCAAAGAC and GTGGGTGAGGAGCACGTAGT; SHP,AAGGGCTTGCTGGACAGTTA and TCTCTTCTTCCGCCCTATCA;18S, ACCGCAGCTAGGAATAATGGA and GCCTCAGTTCCGAAAACC; and GAPDH, CTGAGTATGTCGTGGAGTCTAC and GTTGGTGGTGCAGGATGCATTG. Human primers were as follows: FXR,TACATGCGAAGAAAGTGTCAAGA and ACTGTCTTCATTCACGGTCTGAT; GAPDH, GAAGGTGAAGGTCGGAGT and CATGGGTGGAATCATATTGGAA; and 18S, CGGCTACCACATCCAAGCAA andGCTGGAATTACCGCGGCT.Cytokine measurements. Plasma cytokine concentrations were determinedusing Bio-Plex Cytokine Assays (Mouse 23-Plex Panel 171-FII241; Bio-Rad) according to the manufacturer’s directions. Plasma samples were in-cubated for 2 h at room temperature and cytokine concentrations weremeasured on a single plate with a Bio-Plex 200 system and then analyzedwith Bio-Plex Manager 5.0 software.Myeloperoxidase (MPO) activity. Neutrophil infiltration in the liver wasmeasured by assessing MPO activity. Liver lysates were prepared by ho-mogenizing 100 mg of tissue in 1 ml of T-PER buffer. Liver homogenateswere then centrifuged at 15,000 rpm for 15 min and supernatants werecollected for MPO activity measurement by using a spectrophotometricassay with tetramethylbenzidine as a substrate. Standard curve was per-formed using purified MPO (Sigma-Aldrich).Data analysis. All values are expressed as mean � SE of mice per ex-periments. The variation between data sets were tested with the unpaireddata and ANOVA.
ResultsFXR�/� mice spontaneously develop a progressive liver injuryassociated with liver immunity dysregulation
We first evaluated hepatic histopathology and expression of reg-ulatory cytokines and chemokines at 2, 5, and 9 mo of age in FXRnull and WT mice. Histopathological evaluation demonstrates thatFXR deficiency results in a progressive liver injury characterized byhepatocytes vacuolization, lipid deposit, and focal necrosis (data notshown). As shown in Fig. 1, these changes associate with liver im-
munity dysregulation, as demonstrated by the robust induction ofIFN-� (9-fold increase vs WT mice) and OPN (20-fold increasevs WT mice) in 9-mo-old mice ( p � 0.05 vs WT; n � 6). Thesechanges associated with a strong reduction in the expression of liverSHP, a FXR-regulated gene ( p � 0.05 vs WT mice; n � 6).
FXR gene ablation exacerbates, while FXR activation protects,against Con A-induced liver injury
To explore whether FXR gene ablation predisposes to hepatitisinduced by Con A, we injected 8-wk-old FXR�/� and WT micewith a lethal dose of Con A (25 mg/kg), with or without pretreat-ment with 6E-CDCA (10 mg/kg). As shown in Fig. 2A, this treat-ment resulted in a 24-h mortality of 80% in WT mice. This figurewas further exacerbated by FXR gene ablation, with an earlyhigher mortality (50% at 4 h; p � 0.05 vs WT) and the 100%mortality at 24 h. Administering FXR WT mice with 6E-CDCAreduced lethality induced by Con A administration, highlightingthe protective role of the receptor in this model of liver injury ( p �0.05 vs Con A alone).
In the next series of experiments, we measured AST plasmalevels and assessed liver histology in mice exposed to a sublethaldose of Con A (10 mg/kg). As shown in Fig. 2B, administering WTmice with Con A resulted in a time-dependent increase in ASTplasma levels. In comparison to WT mice, Con A administration toFXR�/� mice resulted in significantly higher AST levels at 24 h( p � 0.05, vs WT mice; n � 8). Interestingly, pretreatment with6E-CDCA attenuated liver injury caused by Con A administration,as measured by AST plasma levels ( p � 0.05; n � 8). Moreover,liver MPO activity (Fig. 2C), a measure of neutrophil accumula-tion into the liver, was significantly elevated in all groups within4 h but remained markedly elevated at 24 h only in Con A-treatedFXR�/� mice. Pretreatment with 6E-CDCA resulted in a signifi-cant reduction in Con A-induced liver MPO activity ( p � 0.05 vssaline-treated mice, n � 8). In comparison to WT mice, the se-verity of liver injury caused by Con A was markedly exacerbatedby FXR gene ablation, as demonstrated by the histopathologicalanalysis (Fig. 2D). In aggregates, these data establish that FXR
FIGURE 1. Liver expression ofregulatory cytokines and SHP, a FXR-regulated gene, in WT and FXR�/�
mice at 2, 5, and 9 mo of age. �, p �0.05 vs WT mice n � 6.
6659The Journal of Immunology

ablation exacerbates, while FXR activation protects against ConA-induced hepatitis.
FXR regulates liver NKT cell activation
To gain information on the mechanisms involved in liver protec-tion exerted by FXR activation, we then examined the immunephenotype of liver-infiltrating cells in WT mice administered ConA in the presence of a FXR ligand. In addition, we also investi-gated whether Con A administration to FXR�/� mice results in adifferent phenotype of liver-infiltrating leukocytes. We found thatCon A administration resulted in an 3-fold increase in the abso-lute number of leukocytes infiltrating the liver at 2 h (data notshown). The phenotypic characterization of these liver-infiltratingcells demonstrated that Con A administration associates with arobust increase in the percentage of CD14� cells (macrophages)and Gr1� cells (polymorphonuclear neutrophil (PMN)) while thepercentage of CD3� cells was slightly reduced (Fig. 3). This effectwas attenuated by treating mice with 6E-CDCA at a dose of 10mg/kg (Fig. 3). Moreover, flow cytometry analysis revealed thatliver-infiltrating CD14� and Gr1� cells were significantly moreactivated than cells isolated from naive animals as assessed bymeasuring the expression of CD11b on both cell types. Again,
treating the mice with a FXR agonist reduced this pattern. Admin-istering mice with 6E-CDCA was also effective in reducing theexpression of FasL on CD3� and NKT cells (Fig. 3). Hepatic NKTcells, but not T cells, constitutively express FasL mRNA. There-fore, the rapid appearance of FasL on the surface of Con A-stim-ulated NKT cells may be predominantly mediated by the deliveryof intracellularly stored FasL to the cell surface. A precedent work(6) indicated that NKT Fas/FasL cascade is involved in the earlyphase of Con A-induced hepatitis. Consistent with this finding,down-regulation of FasL on NKT cells in mice administered 6E-CDCA associated with a reduction of AST levels at 4–8 h afterCon A administration. Although these data suggest that FXR ac-tivation limits the infiltration of activated leukocytes in this model,challenging FXR�/�mice with Con A did not generate a distinc-tive signature either in the number or the relative percentage ofinfiltrating cells in comparison to FXR WT mice (Fig. 3). Thissuggests that FXR agonism attenuates activation of NKT in themodel of Con A-induced hepatitis.
FXR regulates OPN liver expression
We then investigated whether FXR gene ablation modulates therelease of cytokines and chemokines known to play a pathogenic
FIGURE 2. FXR gene ablation exacerbates while FXR activation protects against Con A-induced liver injury. A, Effect of FXR gene ablation on ConA-induced mortality. FXR�/� and WT mice (8 wk old) were injected with a lethal dose of Con A (25 mg/kg). In addition, WT mice were administeredi.p. with 6E-CDCA (10 mg/kg), a semisynthetic FXR ligand, 30 min before Con A injection. �, p � 0.05 vs WT Con A alone group; n � 4 experiments.B, Effect of FXR gene ablation and FXR activation on hepatitis induced by a sublethal dose of Con A. Time course of AST plasma levels in FXR�/� andWT mice administrated Con A (10 mg/kg) � 6E-CDCA (10 mg/kg). �, p � 0.05 vs WT Con A alone group; n � 8 for group and n � 3 experiments. C,Time course of MPO liver activity in FXR�/� and WT mice treated with Con A (10 mg/kg) � 6E-CDCA (10 mg/kg). �, p � 0.05 vs WT Con A alonegroup; n � 8 for group and n � 3 experiments. D, Liver histopathology. Liver sections from naive FXR�/� and WT mice and mice treated with Con A.Paraffin sections were stained with H&E. Original magnifications, �200.
6660 FXR AND LIVER NKT CELLS

role in the model of Con A-induced hepatitis. As shown in Fig. 4,Con A administration resulted in a robust induction of liver ex-pression of several cytokines. This effect was insensitive to thepresence of FXR, with the exception of OPN. Thus, FXR�/� micereact to Con A administration with a dramatic up-regulation ofOPN mRNA in the liver (25-fold increase in comparison to WTmice; Fig. 4D). These findings were further corroborated by themeasurement of OPN protein (ELISA and immunohistochemistry)in the liver (Fig. 5). Immunohistochemistry analysis (Fig. 5B)demonstrated a focal increase in the OPN expression. OPN ex-pression was concentrated in areas of liver damage, suggesting arole for this glycoprotein in favoring emigration of inflammatorycells into necrotic tissues. FXR activation with 6E-CDCA in WTmice reduced mRNA liver expression of OPN and other proin-flammatory cytokines measured 24 h after Con A injection (Fig. 4;p � 0.05 vs saline-treated mice, n � 8). The inhibitory effects of
6E-CDCA were lost in mice harboring a disrupted FXR gene. Con-firming mRNA data, administering mice with 6E-CDCA caused arobust reduction in the liver expression of OPN protein content asmeasured by ELISA and immunohistochemistry ( p � 0.05 vs sa-line-treated mice, n � 8; Fig. 5). Finally, 6E-CDCA administrationto Con A-treated WT mice effectively reduced the plasma levels ofseveral cytokines and chemokines (KC, MIP-1�, IFN-�, andTNF-�; supplemental Fig. 14).
FXR/SHP directly regulates OPN gene expression on activatedNKT cells
Because these data suggest that FXR regulates the liver expressionof OPN, we have then designed a series of studies to investigatewhether FXR regulates OPN expression in liver-derived NKTcells. The experiment shown in Fig. 6 demonstrates that liver-derived NKT cells express FXR (Fig. 6, A and B). CD3�NK1.1�
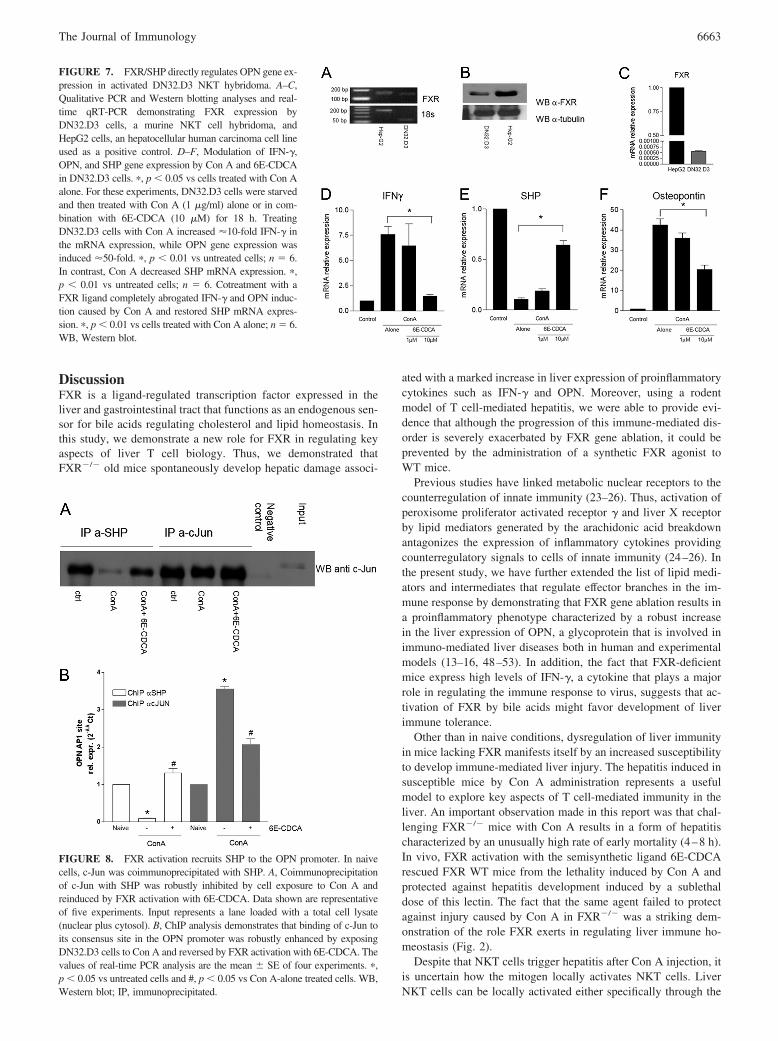
cells (90% purity; n � 4) express FXR (Fig. 6, A–C). By usingliver-derived NKT cells isolated from WT and FXR�/� mice, wefound that FXR exerts a direct regulatory role on OPN generation.Thus, as shown in Fig. 6D, not only OPN expression was 3-foldhigher in FXR�/� NKT cells in comparison to WT cells, but,while exposure of WT NKT cells to Con A resulted in a robustinduction of OPN mRNA expression ( p � 0.05 vs untreated),OPN expression was boosted up to 7-fold in FXR�/� NKT cells( p � 0.01 vs WT cells treated with Con A). To gain insight on themolecular mechanisms involved in OPN regulation by FXR inNKT cells, additional experiments were conducted using a NKTcell hybridoma (the murine V�14invt TCR� CD1d-specific T-Thybridoma DN32.D3 (39). By qualitative RT-PCR, Western blotanalysis and real-time qRT-PCR, we found that DN32.D3 cells,nonactivated by Con A, express FXR along with the FXR-regu-lated gene SHP (Fig. 7). This finding is in line with the observationthat adult peripheral blood-derived CD56 NK cells express FXR,as reported by the gene atlas (http://symatlas.gnf.org). TreatingDN32.D3 with Con A for 18 h resulted in a 10-fold increase inIFN-� mRNA expression, while expression of the OPN gene wasinduced by 50-fold ( p � 0.01 vs untreated, n � 6; Fig. 7, D andE). Induction of cytokine expression by Con A was associated witha significant reduction in SHP mRNA expression (Fig. 7F). Thesechanges were reversed by treating DN32.D3 cells with the FXR
4 The online version of this article contains supplemental material.
FIGURE 3. Phenotypic character-ization of liver-infiltrating cells inFXR�/� and WT mice administeredCon A. FXR�/� and WT mice wereadministrated Con A (10 mg/kg). Inaddition, WT mice were administeredi.p. with 6E-CDCA a semisyntheticFXR ligand (10 mg/kg) 30 min beforeCon A injection. Two hours after drugtreatment, mice were sacrificed andliver-infiltrating cells were collectedfor the flow cytometric analysis (seeMaterials and Methods). Cells werestained with anti-CD3, anti-CD4, anti-Gr1, anti-FasL, anti-CD11b, anti-CD14, and anti-NK1.1 The values in-dicated are the mean � SE. �, p �0.05 vs WT naive mice group and #,p � 0.05 vs WT Con A alone group.MFI, Mean fluorescence intensity.
FIGURE 4. Effect of Con A administration on liver mRNA expressionof proinflammatory cytokines. FXR�/� and WT mice were administeredCon A (10 mg/kg) alone or in combination with 6E-CDCA (10 mg/kg).Liver samples were collected 24 h after Con A administration. �, p � 0.05vs WT naive mice group; n � 5 and #, p � 0.05 vs WT Con A alone group;n � 5. A, IFN-� mRNA expression; B, TNF� mRNA expression; C, IL-4mRNA expression; D, osteopontin mRNA expression.
6661The Journal of Immunology

ligand 6E-CDCA (Fig. 7, D–F). Thus, exposure of DN32.D3 cellsto 10 �M of the FXR ligand completely abrogated IFN-� and OPNinduction caused by Con A and restored SHP mRNA expression( p � 0.05; n � 6), highlighting the negative correlation existingbetween cytokines/chemokines and SHP gene expression in NKTcell hybridomas.
Previous studies have shown a regulatory function for the AP-1consensus site located at �76 bp in the transcriptional regulation ofthe OPN gene (43–45). The importance of this AP-1 site for thetranscriptional activity of the OPN promoter has been highlighted bya complete loss of basal and inducible OPN promoter activity aftersite-directed mutagenesis (43–45). We have previously shown thatFXR activation induces a SHP-dependent inhibition of JunD andc-Jun binding to the AP-1 binding site (46, 47). We have there-fore performed a ChIP assay and PCR amplification using
primer pairs that cover the AP-1 consensus site at �76 in theOPN promoter. Results from these experiments demonstratedthat this promoter region coimmunoprecipitated with c-Jun inCon A-treated DN32.D3 cells (Fig. 8B) and that this effect wasreduced in cells treated with 6E-CDCA. In addition, immunopre-cipitation experiments demonstrated that exposure to 6E-CDCAinduces SHP association with c-Jun in Con A-activated cells (Fig.8A). Furthermore, by ChIP assay, we found that SHP associateswith the OPN promoter in untreated cells (Fig. 8B). Although SHPbinding to the OPN promoter was markedly reduced by exposureof DN32.D3 cells to Con A (Fig. 8B), this interaction was robustlyreinduced by coexposing DN32.D3 cells treated with Con A to6E-CDCA. In concert, these findings indicate that Con A-in-duced c-Jun binding to the AP-1 consensus site at �76 of theproximal OPN promoter is inhibited by FXR/SHP activation.
FIGURE 5. FXR gene ablation re-sulted in an unregulated generation ofOPN. OPN liver expression was as-sessed in FXR�/� and WT mice 24after the mice had been treated withCon A (10 mg/kg). In addition, WTmice were administered 6E-CDCA(10 mg/kg). A, Quantification of OPNprotein levels. The results were nor-malized with the protein sample con-centrations (�, p � 0.05 vs WT ConA alone group. n � 8). B, OPN im-munohistochemistry. B1, Naive WTmice; B2, WT mice administered ConA; B3, WT mice administered Con Aand 6E-CDCA; B4, naive FXR�/�
mice; and B5, FXR�/� mice admin-istered Con A.
FIGURE 6. FXR expression and OPN regulation inliver-derived NKT cells. A and B, Isolation and charac-terization of liver NKT cells (CD�NK1.1� cells) fromWT and FXR�/� mice. Purity of cell fractions was ver-ified by two-color flow cytometry analysis using FITC-conjugated anti-mouse CD3 and PE-conjugated anti-NK1.1 mAbs. An example of liver mononuclear cellsbefore extraction protocol is shown in A, 10% of cellsare CD� and NK1.�1. B, A cell population after thepurification protocol, see Materials and Methods, with90% of cells staining positively with anti-CD3 andanti-NK1.1 (n � 4). C and D, FXR expression in liverNKT cells as assessed by qualitative and quantitativeRT-PCR. HepG2 cells, an hepatocellular human carci-noma cell line, was used as a positive control. E, OPNgene expression in liver NKT cells obtained from WTand FXR null mice, 5 mo old mice. Cells were incu-bated alone or in combination with 1 �g/ml Con A for16 h. �, p � 0.05 vs WT cells not treated and ��, p �0.05 vs WT cells treated with Con A alone (n � 4).
6662 FXR AND LIVER NKT CELLS
DiscussionFXR is a ligand-regulated transcription factor expressed in theliver and gastrointestinal tract that functions as an endogenous sen-sor for bile acids regulating cholesterol and lipid homeostasis. Inthis study, we demonstrate a new role for FXR in regulating keyaspects of liver T cell biology. Thus, we demonstrated thatFXR�/� old mice spontaneously develop hepatic damage associ-
ated with a marked increase in liver expression of proinflammatorycytokines such as IFN-� and OPN. Moreover, using a rodentmodel of T cell-mediated hepatitis, we were able to provide evi-dence that although the progression of this immune-mediated dis-order is severely exacerbated by FXR gene ablation, it could beprevented by the administration of a synthetic FXR agonist toWT mice.
Previous studies have linked metabolic nuclear receptors to thecounterregulation of innate immunity (23–26). Thus, activation ofperoxisome proliferator activated receptor � and liver X receptorby lipid mediators generated by the arachidonic acid breakdownantagonizes the expression of inflammatory cytokines providingcounterregulatory signals to cells of innate immunity (24–26). Inthe present study, we have further extended the list of lipid medi-ators and intermediates that regulate effector branches in the im-mune response by demonstrating that FXR gene ablation results ina proinflammatory phenotype characterized by a robust increasein the liver expression of OPN, a glycoprotein that is involved inimmuno-mediated liver diseases both in human and experimentalmodels (13–16, 48–53). In addition, the fact that FXR-deficientmice express high levels of IFN-�, a cytokine that plays a majorrole in regulating the immune response to virus, suggests that ac-tivation of FXR by bile acids might favor development of liverimmune tolerance.
Other than in naive conditions, dysregulation of liver immunityin mice lacking FXR manifests itself by an increased susceptibilityto develop immune-mediated liver injury. The hepatitis induced insusceptible mice by Con A administration represents a usefulmodel to explore key aspects of T cell-mediated immunity in theliver. An important observation made in this report was that chal-lenging FXR�/� mice with Con A results in a form of hepatitischaracterized by an unusually high rate of early mortality (4–8 h).In vivo, FXR activation with the semisynthetic ligand 6E-CDCArescued FXR WT mice from the lethality induced by Con A andprotected against hepatitis development induced by a sublethaldose of this lectin. The fact that the same agent failed to protectagainst injury caused by Con A in FXR�/� was a striking dem-onstration of the role FXR exerts in regulating liver immune ho-meostasis (Fig. 2).
Despite that NKT cells trigger hepatitis after Con A injection, itis uncertain how the mitogen locally activates NKT cells. LiverNKT cells can be locally activated either specifically through the
FIGURE 7. FXR/SHP directly regulates OPN gene ex-pression in activated DN32.D3 NKT hybridoma. A–C,Qualitative PCR and Western blotting analyses and real-time qRT-PCR demonstrating FXR expression byDN32.D3 cells, a murine NKT cell hybridoma, andHepG2 cells, an hepatocellular human carcinoma cell lineused as a positive control. D–F, Modulation of IFN-�,OPN, and SHP gene expression by Con A and 6E-CDCAin DN32.D3 cells. �, p � 0.05 vs cells treated with Con Aalone. For these experiments, DN32.D3 cells were starvedand then treated with Con A (1 �g/ml) alone or in com-bination with 6E-CDCA (10 �M) for 18 h. TreatingDN32.D3 cells with Con A increased 10-fold IFN-� inthe mRNA expression, while OPN gene expression wasinduced 50-fold. �, p � 0.01 vs untreated cells; n � 6.In contrast, Con A decreased SHP mRNA expression. �,p � 0.01 vs untreated cells; n � 6. Cotreatment with aFXR ligand completely abrogated IFN-� and OPN induc-tion caused by Con A and restored SHP mRNA expres-sion. �, p � 0.01 vs cells treated with Con A alone; n � 6.WB, Western blot.
FIGURE 8. FXR activation recruits SHP to the OPN promoter. In naivecells, c-Jun was coimmunoprecipitated with SHP. A, Coimmunoprecipitationof c-Jun with SHP was robustly inhibited by cell exposure to Con A andreinduced by FXR activation with 6E-CDCA. Data shown are representativeof five experiments. Input represents a lane loaded with a total cell lysate(nuclear plus cytosol). B, ChIP analysis demonstrates that binding of c-Jun toits consensus site in the OPN promoter was robustly enhanced by exposingDN32.D3 cells to Con A and reversed by FXR activation with 6E-CDCA. Thevalues of real-time PCR analysis are the mean � SE of four experiments. �,p � 0.05 vs untreated cells and #, p � 0.05 vs Con A-alone treated cells. WB,Western blot; IP, immunoprecipitated.
6663The Journal of Immunology

TCR (e.g., by glycolipid-presenting cells) or nonspecificallythrough cytokines such as IL-12 or IL-18 (54). In vivo, activationof T cells is an APC-dependent process, but the APC type thatpresents the lectin in the liver in this model is not defined. Usingherpes virus entry mediator (HVEM)-deficient mice, Wahl et al.(54) have shown that nonparenchymal APC are essential for acti-vating NKT cells in the Con A model. Because HVEM is ex-pressed by most cells of the immune system (T cells, B cells, NKcells, dendritic cells, and monocytes) and by some nonimmunecells such as hepatocytes, the specific cell subtypes that function asAPC in this model were not strictly defined. However, becauseintrahepatic dendritic cells express HVEM on the surface and NKTmaturation in the Con A model requires IL-12, these professionalAPC are an attractive candidate (54). In addition to professionalAPC, it is suggested that liver sinusoidal cells could play a role inNKT cell activation. NKT cells in the liver do not transmigrateoutside the vascular system and remain within the sinusoids. Gei-ssmann et al. (55) have elegantly shown that NKT cells patrolwithin hepatic sinusoids at 10–20 �m/min and stop upon TCR-mediated activation, thus providing a local, intravascular immunesurveillance (56). Liver NKT cells are CXCR6 positive (55).CXCR6 is a chemokine receptor that can serve in conjunction withCD4 as a coreceptor for infection with some human and mostsimian immunodeficiency viruses (HIV-1, HIV-2, and SIV) andsimilarly to CCR5 and CXCR3 has an expression pattern restrictedto memory/effector T cells such as NKT cells. CXCR6 has oneknown ligand, CXCL16, a transmembrane chemokine highly ex-pressed by liver sinusoidal cells (55). CXCR-deficient mice have adecreased susceptibility to hepatitis induced by Con A. Becausesinusoidal cells are the main site of lymphocyte adhesion/transmi-gration and adhesion molecule expression in the hepatic venoussystem during Con A-induced hepatitis (57), it is has been sug-gested that these cells can function as nonprofessional APC forNKT cells. Interestingly, endothelial cells (58) and liver sinusoidalcells express a functionally active FXR that regulates multiple ef-fector functions in these cells (59).
Activation of NKT cells in response to Con A can be also par-tially modulated in an autocrine fashion. Previous studies havedemonstrated that OPN derived from activated NKT cells is es-sential in the development of hepatitis induced by Con A (12–14).Thus, Con A administration results in activation of liver NKT cellsthat secrete OPN. The interaction of NKT cells with the thrombin-cleaved form of OPN through �9�1 and �4�1 integrins furtheractivates NKT cells in an autocrine fashion(12). However, it isintriguingly that in contrast to the cleaved form (that is mostlyacting through integrin receptors), the intact isoform of OPN bindsto CD44 (60). Because CD44 directly activates NKT cells with adifferent mechanism in comparison to conventional T cells (61,62), this interaction could further strengthen the pathogenic role ofthe OPN-NKT cell pathway in the model of Con A-induced liverinjury.
There is circumstantial evidence to support the concept that ac-tivated NKT cells expressing FasL contribute to Con A-inducedliver cell injury (12). On the other hand, OPN, along with MIP-2,recruits neutrophils into the liver (12). Upon interaction of OPNwith its receptors on neutrophils, the latter cells became activated,secreted MPO, and contributed to additional liver cell damage(12). Confirming this scenario, we demonstrated that FXR activa-tion exerts protective activity in Con A-induced hepatitis by coun-teracting OPN expression. This view is confirmed by the followingobservations: 1) FXR�/� old mice spontaneously develop liverdamage associated with a strong (20-fold) increase of liver OPNmRNA expression, 2) induction of OPN liver expression by Con Aadministration is amplified in FXR�/� mice, and 3) FXR activa-
tion in WT mice reduces liver OPN gene expression. In aggre-gates, these data support the notion that attenuation of liver ex-pression of OPN could be the pivotal mechanism through whichFXR exerts its counterregulatory role in this model. Thus, atten-uation of OPN synthesis observed in mice treated with the FXRagonist could manifest its effects by a reduction of liver-activatedNKT cells harboring FasL on their surface. Because these cells areinstrumental to the liver damage caused by Con A, this effect islikely to play an important role in the protective effect we observedin response to FXR activation (6). In addition, because of the es-sential role OPN plays in recruiting PMN into the liver in thismodel, inhibition of OPN synthesis by the FXR agonist is alsolikely to contribute to the protective effect we measured on thenumber of liver-infiltrating PMN, another cell type that contributesto liver injury in this model (12, 62).
One important observation we made was that treating WT micewith a FXR agonist attenuates changes in blood plasma levels ofKC (63) and MIP-1� (62) (supplemental Fig. 1). Because MIP-1�levels are elevated in the serum of patients with T cell-mediatedliver diseases and this chemokine appears to be involved in re-cruitment of CD4� cells in the Con A-induced hepatitis (64), it istempting to speculate that MIP-1� represents an additional targetfor FXR.
Previous studies have shown that SHP mediates some of theregulatory activities exerted by FXR (33, 36, 46, 47). SHP is anatypical nuclear receptor, lacking a ligand-binding domain. SHPactivation by FXR ligands in hepatocytes leads to the repression ofcholesterol 7�-hydroxylase expression, the rate-limiting enzyme inthe neutral pathway that leads to bile acid production from cho-lesterol (33). SHP is also a known target for other nuclear recep-tors, including the estrogen receptors and PPAR� (65, 66), and isessential in the regulation of the expression/activity of hepatocyteNF4 and retinoid X receptor (66). A body of evidence suggest thatSHP represents an important mediator of FXR activity and acts asa corepressor of FXR target genes in different physiological con-texts (33, 36, 46, 47). To investigate the molecular mechanism(s)by which FXR regulates OPN production, we conducted in vitroexperiments using a NKT cell hybridoma. The results of theseexperiments demonstrate that DN32.D3 cells express FXR andSHP and that FXR activation leads to induction of SHP expression.Because the analysis of the OPN promoter failed to demonstrateda FXR-responsive element and OPN is positively regulated by in-teraction of AP-1 elements to its promoter (43–45), we have in-vestigated whether FXR activation inhibits the interaction of AP-1with the OPN promoter. The results of these studies demonstratethat activation of FXR leads to a displacement of c-Jun from itsconsensus site in the OPN promoter. The mechanism throughwhich FXR mediates this effect requires SHP, because FXR acti-vation leads to a direct interaction of SHP with c-Jun, an AP-1constituent, and the formation of a SHP-c-Jun protein-protein com-plex. Coimmunoprecipitation experiments and ChIP analysis dem-onstrated that SHP binds to the OPN promoter in untreated cells,but is released in cells exposed to Con A. This interaction is re-stored by treating the cells with the FXR ligand. In concert, thesefindings indicate that Con A-induced c-Jun binding to the AP-1consensus site at �76 of the proximal OPN promoter is inhibitedby FXR/SHP activation.
In summary, ablation of the bile acid sensor FXR results in adysregulated generation of OPN in the liver and exacerbation ofthe liver injury caused by Con A. Activation of FXR by a syntheticligand rescues from liver damage caused by Con A by a mecha-nism that involves inhibition of NKT cell activation and OPN pro-duction. Regulation of OPN production by NKT cells represents a
6664 FXR AND LIVER NKT CELLS

new mechanism for regulation of liver-immune homeostasis by thebile acid sensor FXR.
DisclosuresThe authors have no financial conflict of interest.
References1. Diao, J., R. Garces, and C. D. Richardson. 2001. X protein of hepatitis B virus
modulates cytokine and growth factor related signal transduction pathways dur-ing the course of viral infections and hepatocarcinogenesis. Cytokine GrowthFactor Rev. 12: 189–205.
2. McFarlane, I. G. 1999. Pathogenesis of autoimmune hepatitis. Biomed. Pharma-cother. 53: 255–263.
3. Tiegs, G., J. Hentschel, and A. Wendel. 1992. A T cell-dependent. experimentalliver injury in mice inducible by concanavalin A. J. Clin. Invest. 90: 196–203.
4. Chen, D., R. J. McKallip, A. Zeytun, Y. Do, C. Lombard, J. L. Robertson,T. W. Mak, P. S. Nagarkatti, and M. Nagarkatti. 2001. CD44-deficient miceexhibit enhanced hepatitis after concanavalin A injection: evidence for involve-ment of CD44 in activation-induced cell death. J. Immunol. 166: 5889–5897.
5. Kaneko, Y., M. Harada, T. Kawano, M. Yamashita, Y. Shibata, F. Gejyo,T. Nakayama, and M. Taniguchi. 2000. Augmentation of V�14 NKT cell-me-diated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in thedevelopment of concanavalin A-induced hepatitis. J. Exp. Med. 191: 105–114.
6. Takeda, K., Y. Hayakawa, L. Van Kaer, H. Matsuda, H. Yagita, and K. Okumura.2000. Critical contribution of liver natural killer T cells to a murine model ofhepatitis. Proc. Natl. Acad. Sci. USA 97: 5498–5503.
7. Burdin, N., L. Brossay, Y. Koezuka, S. T. Smiley, M. J. Grusby, M. Gui,M. Taniguchi, K. Hayakawa, and M. Kronenberg. 1998. Selective ability ofmouse CD1 to present glycolipids: �-galactosylceramide specifically stimulatesV�14-NK T lymphocytes. J. Immunol. 161: 3271–3281.
8. Mizuhara, H., E. O’Neill, N. Seki, T. Ogawa, C. Kusunoki, K. Otsuka, S. Satoh,M. Niwa, H. Senoha, and H. Fujiwara. 1994. T cell activation-associated hepaticinjury: mediation by tumor necrosis factor and protection by Interleukin 6. J. Exp.Med. 79: 1529–1537.
9. Louis, H., A. Le Moine, V. Flamand, N. Nagy, E. Quertinmont, F. Paulart,D. Abramowicz, O. Le Moine, M. Goldman, and J. Deviere. 2002. Critical roleof interleukin 5 and eosinophils in concanavalin A- induced hepatitis in mice.Gastroenterology 122: 2001–2010.
10. Ashkar, S., G. F. Weber, V. Panoutsakopoulou, M. Sanchirico Jansson,S. Zawaideh, S. R. Rittling, D. T. Denhardt, M. J. Glimcher, and H. Cantor. 2000.Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Sci-ence 287: 860–864.
11. O’Regan, A. W., G. J. Nau, G. L. Chupp, and J. S. Berman. 2000. Osteopontin(Eta-1) in cell-mediated immunity: teaching an old dog new tricks. Immunol.Today 21: 475–478.
12. Diao, H., S. Kon, K. Iwabuchi, C. Kimura, J. Morimoto, D. Ito, T. Segawa,M. Maeda, J. Hamuro, T. Nakayama, et al. 2004. Osteopontin as a mediator ofNKT cell function in T cell-mediated liver diseases. Immunity 21: 539–550.
13. Saito, Y., S. Kon, Y. Fujiwara, Y. Nakayama, D. Kurotaki, N. Fukuda,C. Kimura, M. Kanayama, K. Ito, H. Diao, et al. 2007. Osteopontin small inter-fering RNA protects mice from fulminant hepatitis. Hum. Gene Ther. 18:1205–1214.
14. Kon, S., M. Ikesue, C. Kimura, M. Aoki, Y. Nakayama, Y. Saito, D. Kurotaki,H. Diao, Y. Matsui, T. Segawa, et al. 2008. Syndecan-4 protects against os-teopontin-mediated acute hepatic injury by masking functional domains of os-teopontin. J. Exp. Med. 205: 25–33.
15. Morimoto, J., M. Inobe, C. Kimura, S. Kon, H. Diao, T. Aoki Miyazaki,D. T. Denhardt, S. Rittling, and T. Uede. 2004. Osteopontin affects the persis-tence of �-glucan-induced hepatic granuloma formation and tissue injury throughtwo distinct mechanisms. Int. Immunol. 16: 477–488.
16. Tanaka, K., J. Morimoto, S. Kon, C. Kimura, M. Inobe, H. Diao, G. Hirschfeld,J. M. Weiss, and T. Uede. 2004. Effect of osteopontin alleles on �-glucan-inducedgranuloma formation in the mouse liver. Am. J. Pathol. 164: 567–575.
17. Bertola, A., V. Deveaux, S. Bonnafous, D. Rousseau, R. Anty, A. Wakkach,M. Dahman, J. Tordjman, K. Clement, S. E. McQuaid, et al. 2009. Elevatedexpression of osteopontin may be related to adipose tissue macrophage accumu-lation and liver steatosis in morbid obesity. Diabetes 58: 125–133.
18. O’Regan, A. 2003. The role of osteopontin in lung disease. Cytokine GrowthFactor Rev. 14: 479–488.
19. Sahai, A., P. Malladi, H. Melin-Aldana, R. M. Green, and P. F. Whitington. 2004.Upregulation of osteopontin expression is involved in the development of non-alcoholic steatohepatitis in a dietary murine model. Am. J. Physiol. 287:G264–G273.
20. Okamoto, H. 2007. Osteopontin and cardiovascular system. Mol. Cell Biochem.300: 1–7.
21. Fiorucci, S., G. Rizzo, A. Donini, E. Distrutti, and L. Santucci. 2007. Targetingfarnesoid X receptor for liver and metabolic disorders. Trends Mol. Med. 13:298–309.
22. Renga, B., M. Migliorati, A. Mencarelli, and S. Fiorucci. 2009. Reciprocal reg-ulation of the bile acid-activated receptor FXR and the interferon-�-STAT-1pathway in macrophages. Biochim. Biophys. Acta 1792: 564–573.
23. Mangelsdorf, D. J., C. Thummel, M. Beato, P. Herrlich, G. Schutz, K. Umesono,B. Blumberg, P. Kastner, M. Mark, P. Chambon, and R. M. Evans. 1995. Thenuclear receptor superfamily: the second decade. Cell 83: 835–839.
24. Glass, C. K., and S. Ogawa. 2006. Combinatorial roles of nuclear receptors ininflammation and immunity. Nat. Rev. Immunol. 6: 44–55.
25. Glass, C. K., and M. G. Rosenfeld. 2000. The coregulator exchange in transcrip-tional functions of nuclear receptors. Genes Dev. 14: 121–141.
26. Castrillo, A., and P. Tontonoz. 2004. Nuclear receptors in macrophage biology:at the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev.Biol. 20: 455–480.
27. Nakamachi, T., T. Nomiyama, F. Gizard, E. B. Heywood, K. L. Jones, Y. Zhao,L. Fuentes, K. Takebayashi, Y. Aso, B. Staels, et al. 2007. PPAR� agonist sup-press osteopontin expression in macrophages and decrease plasma levels in pa-tients with type 2 diabetes. Diabetes 56: 1662–1670.
28. Oyama, Y., N. Akuzawa, R. Nagai, and M. Kurabayashi. 2002. PPAR� ligandinhibits osteopontin gene expression through interference with binding of nuclearfactor to A/T-rich sequence in THP-1 cells. Circ. Res. 90: 348–355.
29. Ogawa, D., J. F. Stone, Y. Takata, F. Blaschke, V. H. Chu, D. A. Towler,R. E. Law, W. A. Hsueh, and D. Bruemmer. 2005. Liver X receptor agonistinhibit cytokine-induced osteopontin expression in macrophages through inter-ference with activator protein-1 signaling pathways. Circ. Res. 95: 59–67.
30. Parks, D. J., S. G. Blanchard, R. K. Bledsoe, G. Chandra, T. G. Consler,S. A. Kliewer, J. B. Stimmel, T. M. Willson, A. M. Zavacki, D. D. Moore, andJ. M. Lehmann. 1999. Bile acids: natural ligands for an orphan nuclear receptor.Science 284: 1365–1368.
31. Makishima, M., A. Y. Okamot, J. J. Repa, H. Tu, R. M. Learned, A. Luk,M. V. Hull, K. D. Lustig, D. J. Mangelsdorf, and B. Shan. 1999. Identification ofa nuclear receptor for bile acids. Science 284: 1362–1365.
32. Wang, H., J. Chen, K. Hollister, L. C. Sowers, and B. M. Forman. 1999. En-dogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3:543–553.
33. Goodwin, B., S. A. Jones, R. R. Price, M. A. Watson, D. D. McKee, L. B. Moore,C. Galardi, J. G. Wilson, M. C. Lewis, M. E. Roth, et al. 2000. A regulatorycascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acidbiosynthesis. Mol. Cell 6: 517–526.
34. Lu, T. T., M. Makishima, J. J. Repa, K. Schoonjans, T. A. Kerr, J. Auwerx, andD. J. Mangelsdorf. 2000. Molecular basis or feedback regulation of bile acidsynthesis by nuclear receptors. Mol. Cell. 6: 507–515.
35. Kalaany, N. Y., and D. J. Mangelsdorf. 2006. LXRS and FXR: the yin and yangof cholesterol and fat metabolism. Annu. Rev. Physiol. 68: 159–191.
36. Watanabe, M., S. M. Houten, L. Wang, A. Moschetta, D. J. Mangelsdorf,R. A. Heyman, D. D. Moore, and J. Auwerx. 2004. Bile acids lower triglyceridelevels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113:1408–1418.
37. Zhang, Y., F. Y. Lee, G. Barrera, H. Lee, C. Vales, F. J. Gonzalez, T. M. Willson,and P. A. Edwards. 2006. Activation of the nuclear receptor FXR improves hy-perglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA103: 1006–1011.
38. Ma, K., P. K. Saha, L. Chan, and D. D. Moore. 2006. Farnesoid X receptor isessential for normal glucose homeostasis. J. Clin. Invest. 116: 1102–1109.
39. Cariou, B., K. van Harmelen, K. D. Duran-Sandoval, T. H. van Dijk,A. Grefhorst, M. Abdelkarim, S. Caron, G. Torpier, J. C. Fruchart, F. J. Gonzalez,et al. 2006. The farnesoid X receptor modulates adiposity and peripheral insulinsensitivity in mice. J. Biol. Chem. 281: 11039–11049.
40. Insook, K., K. Morimura, Y. Shah, Q. Yang, J. M. Ward, and J. F. Gonzalez.2007. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Car-cinogenesis 28: 940–946.
41. Yang, F., X. Huang, T. Yi, Y. Yen, D. D. Moore, and W. Huang. 2007. Spon-taneous development of liver tumors in the absence of the bile acid receptorfarnesoid X receptor. Cancer Res. 67: 863–867.
42. Bendelac, A., O. Lantz, M. E. Quimby, J. W. Yewdell, J. R. Bennink, andR. R. Brutkiewicz. 1995. CD1 recognition by mouse NK1� T lymphocytes. Sci-ence 268: 863–865.
43. Renault, M. A., S. Jalvy, I. Belloc, S. Pasquet, S. Sena, M. Olive, C. Desgranges,and A. P. Gadeau. 2003. AP-1 is involved in UTP-induced osteopontin expres-sion in arterial smooth muscle cells. Circ. Res. 93: 674–681.
44. Bidder, M., J. S. Shao, N. Charlton-Kachigian, A. P. Loewy, C. F. Semenkovich,and D. A. Towler. 2002. Osteopontin transcription in aortic vascular smoothmuscle cells is controlled by glucose-regulated upstream stimulatory factor andactivator protein-1 activities. J. Biol. Chem. 277: 44485–44496.
45. Ogawa, D., J. F. Stone, Y. Takata, F. Blaschke, Van H. Chu, D. A. Towler,R. E. Law, W. A. Hsueh, and D. Bruemmer. 2005. Liver X receptor agonistsinhibit cytokine-induced osteopontin expression in macrophages through inter-ference with activator protein-1 signaling pathways. Circ. Res. 96: 59–67.
46. Fiorucci, S., G. Rizzo, E. Antonelli, B. Renga, A. Mencarelli, L. Riccardi,S. Orlandi, M. Pruzanski, A. Morelli, and R. Pellicciari. 2005. A farnesoid Xreceptor-small heterodimer partner regulatory cascade modulates tissue metallo-proteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellatecells and promotes resolution of liver fibrosis. J. Pharmacol. Exp. Ther. 314:584–595.
47. Fiorucci, S., E. Antonelli, G. Rizzo, B. Renga, A. Mencarelli, L. Riccardi,S. Orlandi, R. Pellicciari, and A. Morelli. 2004. The nuclear receptor SHP me-diates inhibition of hepatic stellate cells by FXR and protects against liver fibro-sis. Gastroenterology 127: 1497–1512.
48. Giachelli, C. M., D. Lombardi, R. J. Johnson, C. E. Murry, and M. Almeida.1998. Evidence for a role of osteopontin in macrophage infiltration in response topathological stimuli in vivo. Am. J. Pathol. 152: 353–358.
49. Ashkar, S., G. F. Weber, V. Panoutsakopoulou, M. E. Sanchirico, M. Jansson,S. Zawaideh, S. R. Rittling, D. T. Denhardt, M. J. Glimcher, and H. Cantor. 2000.
6665The Journal of Immunology

Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Sci-ence 287: 860–864.
50. Weber, G. F., S. Zawaideh, S. Hikita, V. A. Kumar, H. Cantor, and S. Ashkar.2002. Phosphorylation- dependent interaction of osteopontin with its receptorsregulates macrophage migration and activation. J. Leukocyte Biol. 72: 752–761.
51. O’ Regan, A. W., G. L. Chupp, J. A. Lowry, M. Goetschkes, N. Mulligan, andJ. S. Berman. 1999. Osteopontin is associated with T cells in sarcoid granulomasand has T cell adhesive and cytokine-like properties in vitro. J. Immunol. 162:1024–1031.
52. O’Regan, A., and J. S. Berman. 2000. Osteopontin: a key cytokine in cell-me-diated and granulomatous inflammation. Int. J. Exp. Pathol. 81: 373–890.
53. Nau, G. J., G. L. Chupp, J. F. Emile, E. Jouanguy, J. S. Berman, J. L. Casanova,and R. A. Young. 2000. Osteopontin expression correlates with clinical outcomein patients with mycobacterial infection. Am. J. Pathol. 157: 37–42.
54. Wahl, C., U. A. Wegenka, F. Leithauser, R. Schirmbeck, and J. Reimann. 2009.IL-22-dependent attenuation of T cell-dependent (cona) hepatitis in herpes virusentry mediator deficiency. J. Immunol. 182: 4521–4528
55. Geissmann, F., T. O. Cameron, S. Sidobre, N. Manlongat, M. Kronenberg,M. J. Briskin, M. L. Dustin, and D. R. Littman. 2005. Intravascular immunesurveillance by CXCR6� NKT cells patrolling liver sinusoids. PLoS Biol.3: e113.
56. Commentary. 2005. Killers on patrol: liver lymphocytes remain in the bloodvessels. PLoS Biol. 3: e154.
57. Morikawa, H., K. Hachiya, H. Mizuhara, H. Fujiwara, S. Nishiguchi, S. Shiomi,T. Kuroki, and K. Kaneda. 2004. Sublobular veins as the main site of lymphocyteadhesion/transmigration and adhesion molecule expression in the porto-sinusoi-dal-hepatic venous system during concanavalin A-induced hepatitis in mice.Hepatology 31: 83–94.
58. Li, J., A. Wilson, R. Kuruba, Q. Zhang, X. Gao, F. He, L. M. Zhang, B. R. Pitt,W. Xie, and S. Li. 2008. FXR-mediated regulation of eNOS expression in vas-cular endothelial cells. Cardiovasc. Res. 77: 169–177.
59. Das, A., M. E. Fernandez-Zapico, S. Cao, J. Yao, S. Fiorucci, R. P. Hebbel,R. Urrutia, and V. H. Shah. 2006. Disruption of an SP2/KLF6 repression complexby SHP is required for farnesoid X receptor-induced endothelial cell migration.J. Biol. Chem. 281: 39105–39113.
60. Weber, G. F., S. Ashkar, M. J. Glimcher, and H. Cantor. 1996. Receptor-ligandinteraction between CD44 and osteopontin (Eta-1). Science 271: 509–512.
61. Larkin, J., G. J. Renukaradhya, V. Sriram, W. Du, J. Gervay-Hague, andR. R. Brutkiewicz. 2006. CD44 differentially activates mouse NK T cells andconventional T cells. J. Immunol. 177: 268–279.
62. Bonder, C. S., M. N. Ajuebor, L. D. Zbytnuik, P. Kubes, and M. G. Swain. 2004.Essential role for neutrophil recruitment to the liver in concanavalin A-inducedhepatitis. J. Immunol. 172: 45–53.
63. Stefanovic, L., D. A. Brenner, and B. Stefanovic. 2005. Direct hepatotoxic effectof KC chemokine in the liver without infiltration of neutrophils. Exp. Biol. Med.230: 573–586.
64. Hsieh, C. H., M. Frink, Y. C. Hsieh, W. H. Kan, J. T. Hsu, M. G. Schwacha,M. A. Choudhry, and I. H. Chaudry. 2008. The role of MIP-1� in the develop-ment of systemic inflammatory response and organ injury following trauma hem-orrhage. J. Immunol. 181: 2806–2812.
65. Lai, K., D. C. Harnish, and M. J. Evans. 2003. Estrogen receptor � regulatesexpression of the orphan receptor small heterodimer partner. J. Biol. Chem. 278:36418–36429.
66. Lee, Y. K., H. Dell, D. H. Dowhan, M. Hadzopoulou-Cladaras, and D. D. Moore.2000. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 andretinoid X receptor transactivation: two mechanisms for repression. Mol. CellBiol. 20: 187–195.
6666 FXR AND LIVER NKT CELLS
Related Documents











