Published: October 18, 2011 r2011 American Chemical Society 9649 dx.doi.org/10.1021/es2026533 | Environ. Sci. Technol. 2011, 45, 9649–9657 ARTICLE pubs.acs.org/est The Atmospheric Photolysis of o-Tolualdehyde Grainne M. Clifford, † Aur elie Hadj-Aïssa, † Robert M. Healy, † Abdelwahid Mellouki, ‡ Amalia Mu~ noz, § Klaus Wirtz, § Montserrat Martín Reviejo, § Esther Borr as, § and John C. Wenger †, * † Department of Chemistry and Environmental Research Institute, University College Cork, Cork, Ireland ‡ CNRS ICARE, F-45071 Orl eans 2, France § Instituto Universitario UMH-CEAM, C/Charles R. Darwin 14, Parque Tecnol ogico, 46980 Paterna, Valencia, Spain b S Supporting Information ’ INTRODUCTION o-Tolualdehyde is an aromatic aldehyde regularly detected in ambient air. 13 It is emitted into the atmosphere as a primary pollutant from automobile exhausts 4,5 and can also be formed in situ from the hydroxyl radical (OH) initiated oxidation of o-xylene. 3,6,7 The subsequent degradation of o-tolualdehyde in the atmosphere may contribute to the formation of secondary species such as ozone, nitrates, and organic aerosol, which are major constituents of polluted air in the troposphere. 8 A detailed knowledge of the kinetics and mechanisms of these atmospheric degradation processes is therefore required to fully understand the environmental impact of o-tolualdehyde and its parent compound, o-xylene. The potential gas-phase removal processes for o-tolualdehyde are reaction with OH, the nitrate radical (NO 3 ), ozone, chlorine atoms, and direct photolysis by sunlight. 8 Laboratory kinetic studies indicate that the reactions with ozone and Cl atoms are of negligible importance and that reaction with NO 3 is a minor pathway compared to OH-initiated oxidation. 9,10 The gas-phase absorption cross section of o-tolualdehyde is relatively strong in the actinic region, indicating that photolysis may also be an important loss process if the quantum yield is close to unity. 11 Some preliminary studies of the gas-phase photolysis of o-tolualdehyde have been performed at the outdoor European Photoreactor (EUPHORE) in Valencia, Spain; 12,13 however, the reported value for the photolysis rate coefficient is somewhat uncertain and no mechanistic information was obtained. In this work a more detailed investigation of the photolysis of o- tolualdehyde by natural sunlight has been performed at EU- PHORE. The photolysis rate coefficient has been determined, reaction products identified and secondary organic aerosol formation observed. Supplementary experiments have also been performed in an indoor simulation chamber using an artificial light source. The results provide new information on the atmo- spheric degradation of o-tolualdehyde and its potential impact on the environment. ’ EXPERIMENTAL SECTION EUPHORE Chamber. The sunlight photolysis of o-tolualde- hyde was investigated in Chamber B at the EUPHORE facility which consists of a 204.5 m 3 hemispherical reactor made of FEP Teflon foil. Technical details concerning the facility and its Received: August 1, 2011 Accepted: October 18, 2011 Revised: October 17, 2011 ABSTRACT: The photolysis of o-tolualdehyde by natural sunlight has been investigated at the large outdoor European Photoreactor (EUPHORE) in Valencia, Spain. The photolysis rate coefficient was measured directly under different solar flux levels, with values in the range j(o-tolualdehyde) = (1.622.15) 10 4 s 1 observed, yielding an average value of j(o-tolualdehyde)/j(NO 2 ) = (2.53 ( 0.25) 10 2 . The estimated photolysis lifetime is 12 h, confirming that direct photolysis by sunlight is the major atmo- spheric degradation pathway for o-tolualdehyde. Published UV absorption cross-section data were used to derive an effective quantum yield (290400 nm) close to unity, within experimental error. Possible reaction pathways for the formation of the major photolysis products, benzocyclobutenol (tentatively identified) and o-phthalaldehyde, are proposed. Appreciable yields (513%) of secondary organic aerosol (SOA) were observed at EUPHORE and also during supplementary experiments performed in an indoor chamber using an artificial light source. Off-line analysis by gas chromatographymass spectrometry allowed identification of o-phthalaldehyde, phthalide, phthalic anhydride, o-toluic acid, and phthalaldehydic acid in the particle phase.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Published: October 18, 2011
r 2011 American Chemical Society 9649 dx.doi.org/10.1021/es2026533 | Environ. Sci. Technol. 2011, 45, 9649–9657
ARTICLE
pubs.acs.org/est
The Atmospheric Photolysis of o-TolualdehydeGrainne M. Clifford,† Aur�elie Hadj-Aïssa,† Robert M. Healy,† Abdelwahid Mellouki,‡ Amalia Mu~noz,§
Klaus Wirtz,§ Montserrat Martín Reviejo,§ Esther Borr�as,§ and John C. Wenger†,*†Department of Chemistry and Environmental Research Institute, University College Cork, Cork, Ireland‡CNRS ICARE, F-45071 Orl�eans 2, France§Instituto Universitario UMH-CEAM, C/Charles R. Darwin 14, Parque Tecnol�ogico, 46980 Paterna, Valencia, Spain
bS Supporting Information
’ INTRODUCTION
o-Tolualdehyde is an aromatic aldehyde regularly detectedin ambient air.1�3 It is emitted into the atmosphere as a primarypollutant from automobile exhausts 4,5 and can also be formedin situ from the hydroxyl radical (OH) initiated oxidation ofo-xylene.3,6,7 The subsequent degradation of o-tolualdehyde inthe atmosphere may contribute to the formation of secondaryspecies such as ozone, nitrates, and organic aerosol, which aremajor constituents of polluted air in the troposphere.8 A detailedknowledge of the kinetics and mechanisms of these atmosphericdegradation processes is therefore required to fully understandthe environmental impact of o-tolualdehyde and its parentcompound, o-xylene.
The potential gas-phase removal processes for o-tolualdehydeare reaction with OH, the nitrate radical (NO3), ozone, chlorineatoms, and direct photolysis by sunlight.8 Laboratory kineticstudies indicate that the reactions with ozone and Cl atoms are ofnegligible importance and that reaction with NO3 is a minorpathway compared to OH-initiated oxidation.9,10 The gas-phaseabsorption cross section of o-tolualdehyde is relatively strong inthe actinic region, indicating that photolysis may also be animportant loss process if the quantum yield is close to unity.11
Some preliminary studies of the gas-phase photolysis ofo-tolualdehyde have been performed at the outdoor European
Photoreactor (EUPHORE) in Valencia, Spain;12,13 however, thereported value for the photolysis rate coefficient is somewhatuncertain and no mechanistic information was obtained. In thiswork a more detailed investigation of the photolysis of o-tolualdehyde by natural sunlight has been performed at EU-PHORE. The photolysis rate coefficient has been determined,reaction products identified and secondary organic aerosolformation observed. Supplementary experiments have also beenperformed in an indoor simulation chamber using an artificiallight source. The results provide new information on the atmo-spheric degradation of o-tolualdehyde and its potential impact onthe environment.
’EXPERIMENTAL SECTION
EUPHORE Chamber. The sunlight photolysis of o-tolualde-hyde was investigated in Chamber B at the EUPHORE facilitywhich consists of a 204.5 m3 hemispherical reactor made of FEPTeflon foil. Technical details concerning the facility and its
Received: August 1, 2011Accepted: October 18, 2011Revised: October 17, 2011
ABSTRACT: The photolysis of o-tolualdehyde by natural sunlighthas been investigated at the large outdoor European Photoreactor(EUPHORE) in Valencia, Spain. The photolysis rate coefficientwasmeasured directly under different solar flux levels, with values inthe range j(o-tolualdehyde) = (1.62�2.15) � 10�4 s�1 observed,yielding an average value of j(o-tolualdehyde)/j(NO2) = (2.53 (0.25) � 10�2. The estimated photolysis lifetime is 1�2 h,confirming that direct photolysis by sunlight is the major atmo-spheric degradation pathway for o-tolualdehyde. Published UVabsorption cross-section data were used to derive an effectivequantum yield (290�400 nm) close to unity, within experimentalerror. Possible reaction pathways for the formation of the majorphotolysis products, benzocyclobutenol (tentatively identified)and o-phthalaldehyde, are proposed. Appreciable yields (5�13%) of secondary organic aerosol (SOA)were observed at EUPHOREand also during supplementary experiments performed in an indoor chamber using an artificial light source. Off-line analysis by gaschromatography�mass spectrometry allowed identification of o-phthalaldehyde, phthalide, phthalic anhydride, o-toluic acid, andphthalaldehydic acid in the particle phase.

9650 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
application for photolysis experiments have been previouslyreported in the literature.14�18
The chamber was cleaned by flushing with purified air over-night and filled to atmospheric pressure. o-Tolualdehyde wasintroduced into the chamber and allowed to mix for approxi-mately one hour while its loss to the chamber walls wasmeasured. Photolysis was initiated by opening the protectivehousing to expose the contents of the chamber to sunlight for2�3 h. The temperature inside the chamber increased slightly asthe experiments progressed but was always within the range296�308 K. The solar actinic flux over the range 290�520 nmwas measured using a calibrated spectroradiometer (BenthamDM300). A full spectral scan took 420�430 s.Chemical analysis was performed by in situ FTIR spectro-
scopy and gas chromatography (GC). The FTIR spectrometer(Nicolet Magna 550) was operated at 1 cm�1 resolution andspectra over the range 600�4000 cm�1 were derived from thecoaddition of 270 scans collected over 5 min. The GC (Fisons8160), equipped with flame ionization and photoionizationdetectors (FID and PID), was operated using a 30 m DB-624fused silica capillary column (J&W Scientific, 0.32 mm i.d.,1.8 μm film). Air was sampled from the chamber into a 3 cm3
sampling loop and injected onto the column operated at 150 �C.The reactants and products were quantified using calibratedFTIR spectra and GC sensitivity factors obtained by introducingknown volumes of pure materials into the chamber. The leak ratewas determined by adding about 20 ppbV of SF6 to the chamberand measuring its loss by FTIR spectroscopy.Additional chemical analysis was afforded by gas chromato-
graphy�mass spectrometry (GC�MS) using a Varian GC 3400interfaced to a Saturn 2000 ion trap mass spectrometer. TheGC�MS incorporated a cryogenic sample preconcentration trap(SPT) containing glass beads cooled to �160 �C. The SPT wasoperated for 5 min at a flow of 40 cm3 min�1. Desorption wasperformed at 270 �C onto a 30 m HP Innowax column(0.25 mm id, 0.25 μm film thickness). The column was heldat�20 �C for 7 min and increased to 250 at 10 �C min�1. TheGC�MS was operated in electron ionization (EI) mode overthe m/z range 46�250. Products were identified by compari-son with chromatographic retention times and mass spectra ofauthentic standards.A scanning mobility particle sizer (SMPS), comprising of a
condensation particle counter (TSI 3022A) and differentialmobility analyzer (TSI 3081), was used to measure particlesize distribution, number and volume concentrations with atime resolution of ca. 5 min. Aerosol mass concentrations weremeasured using a tapered element oscillating microbalance(TEOM, Rupprecht and Patashnick 1400a) fitted with a PM1
inlet and operated at a total flow of 16.7 L min�1. For stableoperation of the TEOM system, the sampling line and thesensor unit were held at 27 �C. The cabinet temperature ofthe SMPS was 25 �C. Both the TEOM and SMPS systems werestarted prior to the introduction of reactants to confirm that noparticles were present in the chamber before photolysis wasinitiated. The instruments were also operated for several hoursafter the chamber was closed to measure the loss of particles tothe chamber walls. Off-line chemical analysis of the particleswas performed by GC�MS using a method reported pre-viously.19 Particles were collected onto a 47 mm quartz fiberfilter at a flow rate of 80 L min�1 for 1 h and subsequentlyextracted under sonication in 5 mL of a CH2Cl2/CH3CN(1:1) mixture. The extract was derivatized with O-(2,3,4,5,6-
pentafluorobenzyl)-hydroxylamine (PFBHA) and N-methyl-N-trimethylsilyltrifluoro-acetamide (MSTFA) to react withcarbonyls and hydroxyl-containing compounds respectively.One μL was injected into the GC�MS (TRACE-DSQ II,Thermo Fisher Scientific Co., Waltham, MA) fitted with anRTX-5MS column (30 m � 0.25 mm i.d. � 0.25 μm filmthickness, Thermo Fisher Scientific Co).Indoor Chamber. A number of experiments were also con-
ducted in the 3.91 m3 FEP Teflon indoor chamber at UniversityCollege Cork, described in detail elsewhere.20 The chamber wassurrounded by 18 lamps (Philips TL05, 40 W; 320 nm < λ >480 nm; λmax = 360 nm) which deliver a light intensity equivalentto j(NO2) = 1.56 � 10�3 s�1. Photolysis of o-tolualdehyde wasinitiated by turning on the lamps for 2�3 h. Quantitative analysisof gas-phase species was performed by in situ FTIR spectroscopyusing the procedures outlined above. A SMPS, consisting of acondensation particle counter (TSI 3010) and a differentialmobility analyzer (TSI 3080), was used to measure particleproperties at ca. 5 min intervals. Additional chemical analysiswas provided by GC�MS. The contents of the chamber weresampled using a 1 cm3 gastight syringe (Hamilton) and injecteddirectly into the GC�MS instrument (Varian GC 3800 inter-faced to a MS Saturn 2000). Typically five samples were takenper experiment. The GC�MS was operated in electron ioniza-tion (EI) mode over the m/z range 46�250. Chromatographicseparation was achieved using a CP-Sil8 fused silica capillarycolumn (Varian, 30 m, 0.25 mm i.d, 0.25 μm film thickness)operated at 60 �C for 1min and then increased to 290 �C at a rateof 10 �C per minute. A flow rate of 5 cm3min�1 was used and theinjector temperature was 250 �C. Products were identified andquantified by comparison with chromatographic retention timesand mass spectra of authentic standards. It should be notedhowever, that reliable quantitative information could not beobtained for all products due to sampling losses associated withuse of the syringe. Further off-line detection of carbonyl reactionproducts in the gas and particle phase was performed using adenuder-filter sampling method coupled with PFBHA derivati-zation and GC�MS analysis.21,22
Materials.All organic compounds were obtained fromAldrichChemical Co. at the highest purity available (stated purities>97%) and used without further purification. Sulfur hexafluoride(99.9%) was obtained from Messer Griesheim, Germany.
’RESULTS AND DISCUSSION
Photolysis Rate Coefficient. Photolysis of o-tolualdehydefollows first order kinetics:
o-tolualdehyde þ hνfjproducts ðIÞ
where j is the photolysis rate coefficient. Assuming photolysis isthe only loss process, j can be determined from a simple firstorder kinetic plot:
ln½o-tolualdehyde�t=½o-tolualdehyde�0 ¼ � jt ðIIÞwhere the subscripts 0 and t refer to the concentrations at initialtime 0 and elapsed time t, respectively. Test experiments carriedout in the presence and absence of excess amounts (10�25ppmV) ofOH radical scavenger (isopropanol and cyclohexane inthe indoor and outdoor chambers, respectively) produced verysimilar decay rates, indicating that loss of o-tolualdehyde dueto reaction with OH in the chambers was negligible. However,

9651 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
o-tolualdehyde was found to undergo a small amount of depositionto the walls of the reactor. The rate coefficient for this process(kwall) was determined by measuring the first order decay of thecompound in the dark for about one hour prior to photolysis.This value was incorporated into the overall decay as follows:
ln½o-tolualdehyde�t=½o-tolualdehyde�0 � kwallt ¼ � jt ðIIIÞ
Although dilution was also observed during the EUPHOREexperiments, the rate determined by measuring the loss of SF6from the chamber, kSF6, was lower than the wall loss and istherefore already incorporated into kwall. Thus a plot in the formof eq III should yield a straight line with gradient �j.
Concentration�time profiles and kinetic plots in the form ofeq III were generated for all experiments. The concentration�time profile for the EUPHORE experiment conducted onthird July is presented in Figure 1 and clearly shows the rapiddecay of o-tolualdehyde following exposure of the chamber tosunlight. The light intensity is represented by the photolysis ratecoefficient for NO2, j(NO2), which was calculated from the solarflux measurements of the spectroradiometer and recommendedvalues for the absorption cross-section and quantum yield.23 Thecorresponding kinetic plot used to calculate j from these FTIRspectroscopic measurements is shown in Figure S1 (SupportingInformation) and exhibits good linearity and a near-zero inter-cept. The value for j(o-tolualdehyde) is listed in Table 1 along
Table 1. Experimental Details for the Photolysis of o-Tolualdehyde in the Outdoor (EUPHORE) and Indoor SimulationChambersa
EUPHORE indoor chamber
19 September 2002 3 July 2003 4 July 2003 four experiments
initial concentration (ppbV) 525 508 256 364, 518, 712, 1354
irradiation period 11.59 � 14:24 09:44�12:27 11:36�13:53 1�3 h
j(NO2)average (s�1) (6.41 ( 0.64) � 10�3 (8.13 ( 0.81) � 10�3 (8.44 ( 0.84) � 10�3 1.56 � 10�3
kwall (s�1) (7.80 ( 0.47) � 10�6 (1.03 ( 0.47) � 10�5 (1.68 ( 0.50) � 10�5 (7.03 ( 0.90) � 10�6
kSF6 (s�1) (6.57 ( 0.72) � 10�6 (5.48 ( 0.90) � 10�6
j(o-tolualdehyde)FTIR (s�1) (1.97 ( 0.05) � 10�4 (4.16 ( 0.23) � 10�5
j(o-tolualdehyde)GC‑PID (s�1) (1.62 ( 0.05) � 10�4 (2.12 ( 0.05) � 10�4 (2.15 ( 0.06) � 10�4
j(o-tolualdehyde)average (s�1) (1.62 ( 0.05) � 10�4 (2.05 ( 0.05) � 10�4 (2.15 ( 0.06) � 10�4 (4.16 ( 0.23) � 10�5
j(o-tolualdehyde)/j(NO2) (2.53 ( 0.25) � 10�2 (2.52 ( 0.25) � 10�2 (2.55 ( 0.26) � 10�2 (2.66 ( 0.23) � 10�2
maximum theoretical loss rate (s�1) (1.33 ( 0.13) � 10�4 (1.85 ( 0.19) � 10�4 (1.90 ( 0.19) � 10�4
effective quantum yield 1.22 ( 0.03 1.11 ( 0.03 1.13 ( 0.05
molar yield of benzocyclobutenol 0.77 ( 0.04 0.71 ( 0.06
molar yield of o-phthalaldehyde 0.21 ( 0.02 0.22 ( 0.02
yield of aerosol 0.104 0.049, 0.059, 0.080, 0.133a Except for j(NO2), quoted errors are twice the standard deviation arising from the least squares fit of the data and include the uncertainty in calibrationand response factors. For j(NO2) and the maximum theoretical loss rate, the estimated error is 10%. The molar yield of benzocyclobutenol is based onthe use of 1-indanol as a surrogate compound.
Figure 1. Concentration�time profile and j(NO2) during the photolysis of o-tolualdehyde at EUPHORE on 3 July 2003. The vertical dotted linesindicate the time the chamber was opened (09:44) and closed (12:27).
9652 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
with a summary of the reaction conditions and results obtainedfor all of the photolysis experiments.As shown in Table 1, there is very good agreement between
the j values determined using FTIR spectroscopy and GC-PID inthe experiment performed on third July 2003. In the experimentperformed on fourth July 2003, o-tolualdehyde was amongseveral aromatic aldehydes subjected to photolysis in the cham-ber and FTIR spectroscopy could not be used for analysis due tooverlapping absorption bands. The value for j(o-tolualdehyde)obtained by GC-PID in this experiment is very similar to thatobtained on third July 2003, indicating that the presence ofthe other compounds (2,3-dimethylbenzaldehyde and 2,6-di-methylbenzaldehyde) did not affect the photolysis rate. Theaverage values for j(NO2) and hence j(o-tolualdehyde)/j(NO2)in these two EUPHORE experiments were also very similar,reflecting the fact that they were performed under almostidentical, cloud-free conditions. As expected, the sunlight inten-sity was somewhat lower for the experiment performed on 19thSeptember 2002 and although a slightly lower value was observedfor j(o-tolualdehyde), the j(o-tolualdehyde)/j(NO2) ratio wasvirtually the same. This indicates that the average value ofj(o-tolualdehyde)/j(NO2) = (2.53 ( 0.25) � 10�2 is a usefulparameter for calculating the photolysis rate of o-tolualdehydeunder different light conditions in chambers or in the realatmosphere.The effective quantum yield for photolysis of o-tolualdehyde,
jeff was determined using the following expression;
jeff ¼ jexp=jmax ðIVÞwhere jexp and jmax are the experimentally observed and max-imum theoretical values of the photolysis rate coefficient respec-tively. The latter term was calculated using the solar flux intensitymeasurements of the spectroradiometer, the absorption crosssection data reported by Thiault et al.,11 and assuming a quantumyield of unity over the atmospheric absorption range of thecompound. The values obtained for jeff during the EUPHOREexperiments are in reasonable agreement, yielding an average ofjeff = (1.15 ( 0.05). However, the calculated value of jmax doesnot include the uncertainty in the reported absorption cross
sections, which is estimated to be 15% below 340 nm and 20% inthe range 340�363 nm.11 The results therefore suggest that,within experimental error, the effective quantum yield forphotolysis of o-tolualdehyde by natural sunlight is unity.The results of this work can be compared to those obtained in
the preliminary studies also performed at EUPHORE. A value ofj(o-tolualdehyde) = (2.00 ( 0.10) � 10�4 s�1 was obtained byVolkamer et al.12 in one experiment during February, where thesolar zenith angle was 50� and the UV flux reduced by around afactor of 2 compared to midsummer. In contrast, Thiault et al.13
obtained a value of j(o-tolualdehyde) = (1.10( 0.20)� 10�4 s�1
during an experiment performed in April. Values ofj(o-tolualdehyde)/j(NO2) = 0.032 and jeff = 0.6 were alsoreported, but no information was provided on whether the wallloss of o-tolualdehyde was taken into account during data analysis.It is interesting note that in these preliminary studies,12,13 thephotolysis of benzaldehyde, m- and p-tolualdehyde was found tobe negligible, suggesting that the presence of the methyl group inthe ortho position is a key factor in determining the photolysisefficiency of o-tolualdehyde.The photolysis rate of o-tolualdehyde in the indoor chamber
was around a factor of 5 slower than in the outdoor chamber.This result was expected since the intensity and wavelengthdistribution of UV light produced from the TL05 lamps is quitedifferent from natural sunlight. However, a useful comparisonbetween the two chambers can be made by examining the valuesdetermined for j(o-tolualdehyde)/j(NO2), shown in Table 1.The values are in very close agreement, indicating that the TL05lamps used in the indoor chamber studies provide reasonablyrealistic light conditions for studies of the atmospheric photolysisof the aromatic aldehydes.Photolysis Products. Gas-phase products arising from the
photolysis of o-tolualdehyde were determined by FTIR spectro-scopy and GC�MS. The FTIR spectra obtained in both chambersenabled identification and quantification of o-phthalaldehydeand carbon monoxide as reaction products. Phthalide and formicacid were also detected during the later stages of the experiments,but below the limits of quantification. The FTIR product spectraalso contain significant absorption features around 1050 cm�1
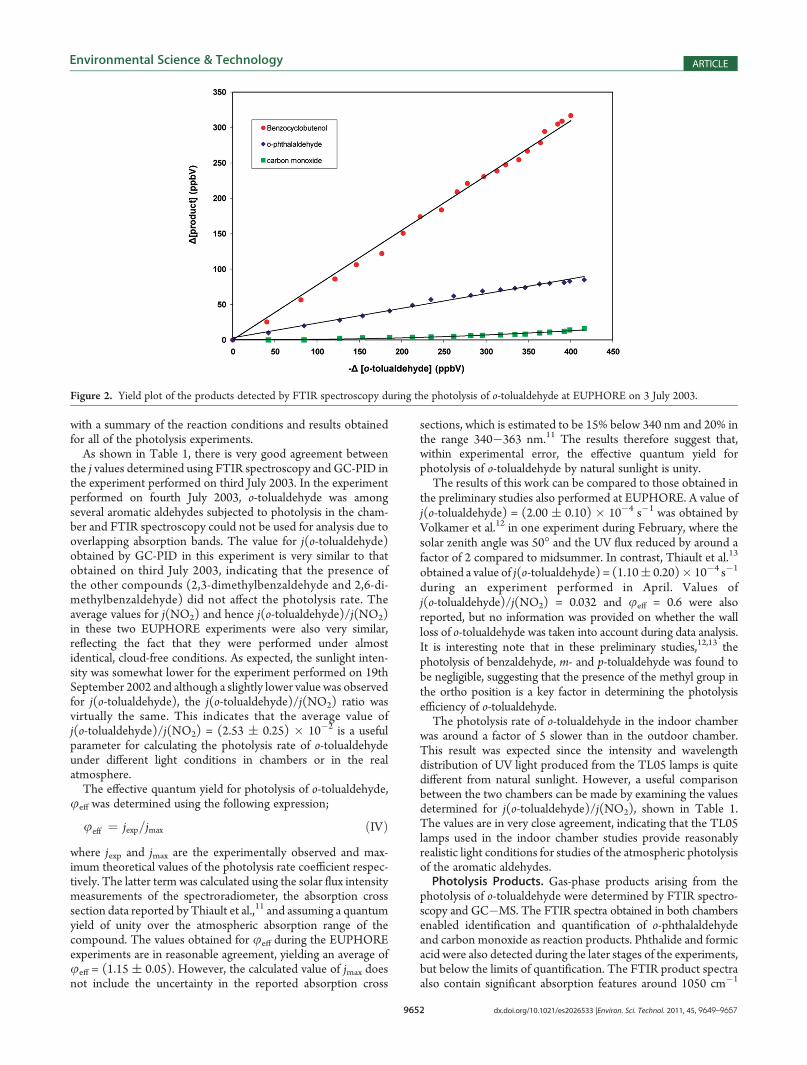
Figure 2. Yield plot of the products detected by FTIR spectroscopy during the photolysis of o-tolualdehyde at EUPHORE on 3 July 2003.

9653 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
and just below 1400 cm�1 and 750 cm�1 (Figure S2, SupportingInformation). The peak at 1050 cm�1 is characteristic of theO�H bending vibration in an alcohol, and inspection of theliterature reveals that UV photolysis of o-alkyl aromatic carbonylcompounds in solution and in the condensed phase leads tothe efficient formation of the corresponding dienols and ben-zocyclobutenols.24,25 The dienol and benzocyclobutenol ex-pected from photolysis of o-tolualdehyde are not commerciallyavailable, however, their IR absorption spectra (not quantified)in a low temperaturematrix have been reported previously.26 Themain absorption bands of benzocyclobutenol in the fingerprintregion, located at 1055 cm�1 (OH bend), 1212 cm�1 and1401 cm�1, are all apparent in the product spectra obtainedhere, while there is no evidence for the presence of the dienol (at1256 cm�1 (OH bend) and 1102 cm�1 (CO stretch)). It istherefore proposed that benzocyclobutenol is a product of thegas-phase photolysis of o-tolualdehyde. Since benzocyclobutenolis not commercially available, 1-indanol, the similarly structuredaromatic cyclic alcohol, was used as a surrogate compound:
The main absorption features of 1-indanol are virtuallyidentical to those observed in the product spectra (SupportingInformation Figure S2) thus providing further evidence tosupport the formation of benzocyclobutenol as a reactionproduct from photolysis of o-tolualdehyde. Calibrated infraredspectra of 1-indanol were subsequently used for quantificationof benzocyclobutenol and the concentration�time profile inFigure 1 shows that the aromatic cyclic alcohol is in fact the majorreaction product. The corresponding product yield plots forbenzocyclobutenol and o-phthalaldehyde displayed in Figure 2are linear, indicating that these compounds are primary productsof the reaction and are not removed to any significant extentduring the time scale of the experiments. Only trace amounts ofcarbon monoxide were observed and the yield plot in Figure 2 iscurved, indicating that it is most likely a secondary product.Similar results were also obtained from analysis of the FTIRspectra obtained in the indoor chamber experiments, Table 1.A number of gas-phase photolysis products were also detected
using GC�MS. During the indoor chamber experiments, o-phthalaldehyde, phthalide, phthalic anhydride, o-toluic acid, ando-cresol were identified by comparison of the retention times andmass spectra with those of standards (Figure S3 and Table S1,Supporting Information). The molar yields of o-phthalaldehydeand phthalide were (0.25 ( 0.04) and (0.02 ( 0.01), in goodagreement with the results obtained by FTIR spectroscopy.Analysis of the denuder extracts showed the formation of o-phthalaldehyde and very small amounts of glyoxal. Quantifica-tion of the other products identified by direct injection GC�MSproved difficult due to losses during the sampling procedure.However, there was no evidence for the presence of benzocy-clobutenol in the mass spectra of the products. Similar resultswere obtained at EUPHORE, where o-phthalaldehyde andphthalide were also detected as the major products, along witho-cresol and 2-hydroxymethylbenzaldehyde in trace amounts. Asmall peak, possibly due to benzocyclobutenol was also observedin the GC�MS product spectra, but could not be confirmed dueto the lack of a standard. The benzocyclobutenols are known toundergo thermal decomposition above 80 �C24 and it therefore
seems very likely that benzocyclobutenol decomposed during theGC�MS analysis, either in the heated injector or on the columnitself. In fact, degradation of the aromatic cyclic alcohol could beresponsible for generating some of the aromatic products(phthalide, phthalic anhydride, o-cresol) which were detectedby GC�MS but not by in situ FTIR spectroscopy.The information obtained from these product studies can be
used to propose a mechanism for the atmospheric photolysis ofo-tolualdehyde, shown in Scheme 1. The initial step in themechanism is photoexcitation of the aldehyde through thenfπ* electronic transition followed by intramolecular hydrogenabstraction (Norrish Type II process) to produce a 1,4-biradicalspecies.24,25 Based on the distribution of identified reactionproducts, two possible reactions for the 1,4-biradical are pro-posed, cyclization to form benzocyclobutenol and reaction withO2 to produce o-phthalaldehyde. The cyclization reaction wasinitially proposed by Yang27 to explain the formation of cyclicalcohols from photolysis of aliphatic ketones in solution. How-ever, more recent studies on o-alkyl aromatic carbonyl com-pounds, in solution and in the solid phase, indicate that directcyclization of the 1,4-biradical is unlikely, and that formation ofthe dienol, in both (Z)- and (E)-configurations is preferred.24,25
The (Z)-dienol is very short-lived and reverts to the startingaldehyde via a rapid 1,5-H atom shift, whereas the (E)-dienolundergoes thermal conrotatory ring closure to form benzocyclo-butenol. As indicated above, there is no evidence for theformation of the dienol in these experiments, although its lifetimein the gas-phase may be too short to be detected. Nevertheless,the possibility that formation of benzocyclobutenol proceeds viathe (E)-dienol cannot be ruled out.The formation of o-phthalaldehyde is postulated to occur via
reaction of the 1,4-biradical with O2. It is possible that thispathway may involve a number of concurrent or subsequentsteps; H-atom abstraction from theOH group and addition of O2
to the methylene unit to form a peroxy radical. In the absence ofNO, the peroxy radicals react together, with the major reactionpathway resulting in oxy radicals which also react withO2 to formo-phthalaldehyde. The minor reaction pathway involves combi-nation of the peroxy radicals to produce two molecular productsin one step; o-phthalaldehyde and 2-hydroxymethylbenzalde-hyde. A very small amount of the latter species was detected byGC�MS in the EUPHORE experiment. However, for the sake ofsimplicity, this mechanistic detail is omitted from Scheme 1.
Scheme 1

9654 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
Additional reaction pathways are required to explain theformation of the minor products detected by GC�MS and FTIRspectroscopy. Phthalide and phthalic anhydride are known to begenerated from the UV photolysis of the primary product, o-phthalaldehyde.28 The formation of the other aromatic com-pounds, o-cresol and o-toluic acid, is more difficult to explain andsince these products were only detected in very small amounts,speculative mechanisms for their formation will not be consid-ered here. As indicated above, carbon monoxide appears to beformed as a secondary product. Benzocylcobutenol is notexpected to undergo rapid photolysis under the conditionsemployed in the chambers, suggesting that photolysis of o-phthalaldehyde is the most likely source of carbon monoxide.However, preliminary experiments on the photolysis of o-phthal-aldehyde in both chambers did not generate detectable levels ofcarbon monoxide. The origin of the secondary carbon monoxidetherefore remains unknown.Secondary Organic Aerosol Formation. Secondary Organic
Aerosol (SOA) formation was observed in all experimentsperformed in both chambers. The evolution of the aerosolproduced from photolysis of o-tolualdehyde in the EUPHOREchamber is shown in Figure 3. A large number of particles with amean diameter of around 20 nm were observed within 5 min ofthe start of photolysis. This initial “burst” of particles alsocorresponded to the greatest number present during the reac-tion. As photolysis continued, there was a gradual reduction inparticle number and a corresponding increase in average particlediameter due to coagulation. Particles with a mean diameter ofaround 70 nm were present at the end of the experiment. Verysimilar results were obtained in the indoor chamber, althoughthe greatest number of particles was typically observed around10 min later when the mean diameter was 25�35 nm. This slightdifference in the particle-time profile is probably due to the factthat the photolysis process, and hence particle formation, wasslower in the indoor chamber.
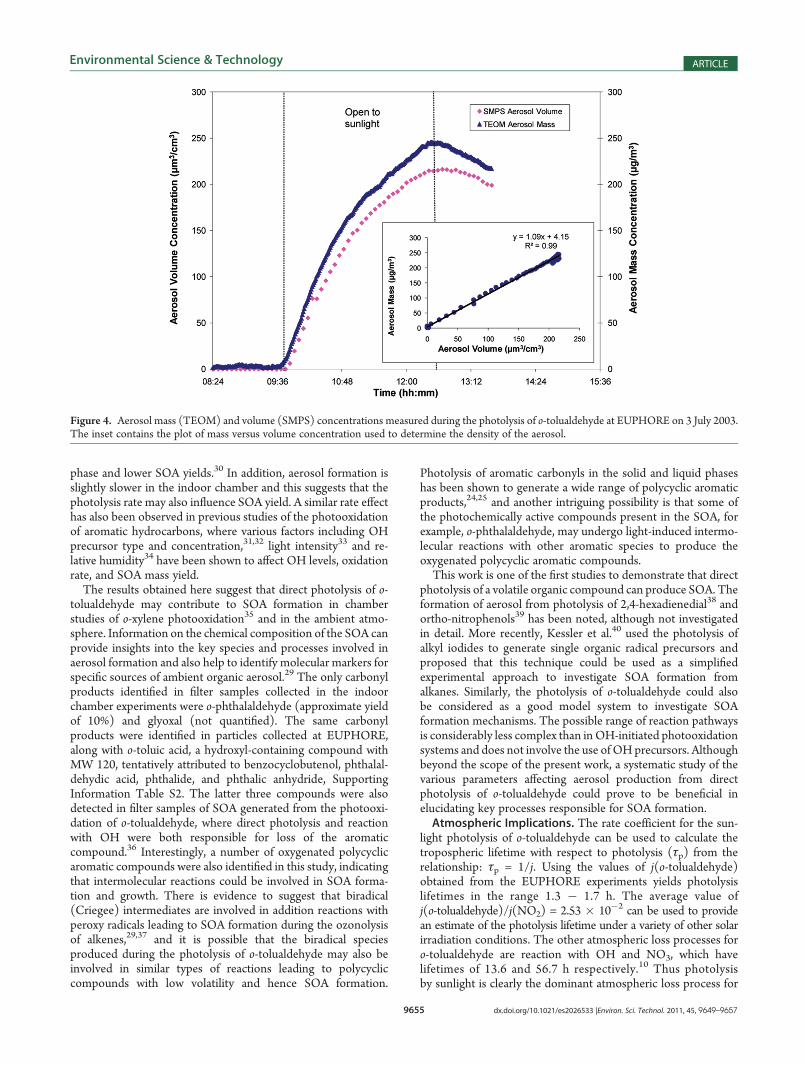
The mass and volume concentration of SOA generated in theEUPHORE experiment on third July 2003 is shown in Figure 4.The density of SOA formed in the experiment was determinedto be (1.09 ( 0.01) g cm�3 by plotting the measured mass(TEOM) versus the volume (SMPS), Figure 4 (inset). Thecalculated density is lower than the value of 1.35 g cm�3
determined for SOA generated from photooxidation of benzenein the EUPHORE chamber using the same method,15 and alsoat the lower end of the range of effective densities (1.06 �1.45 g cm�3) for laboratory-generated SOA from anthropogenicprecursors.29 The maximum aerosol concentration was observedat the end of the experiment when all of the o-tolualdehyde hadreacted. After closing the chamber, the aerosol was found toundergo a first order decay, k = (1.46( 0.10)� 10�5 s�1, due todeposition at the walls of the reactor. This wall loss factor wasused to provide a corrected value of 258 μg m�3 for themaximum aerosol mass concentration, which when divided bythe mass of o-tolualdehyde reacted (2491 μg m�3) gives anaerosol yield of 0.104.SOA yields were also obtained from four different experiments
performed in the indoor chamber by converting the measuredvolume concentrations to mass concentrations using the densityof 1.09 g cm�3. As shown in Table 1, the SOA yields were foundto increase with starting concentration of o-tolualdehyde. Thiseffect has been observed for many other SOA precursors and isconsistent with gas/particle partitioning theory.29 The partition-ing of semivolatile reaction products to the aerosol phaseincreases with the amount of available aerosol mass, resultingin higher yields of SOA when greater precursor concentrationsare used. It is interesting to note that the yield obtained for theexperiment with an initial concentration of 518 ppbV is con-siderably lower than that determined in the equivalent experi-ment at EUPHORE. The semivolatile reaction products wouldbe expected to undergo a higher degree of wall loss in the smallerindoor chamber, resulting in less partitioning to the particle
Figure 3. Number and size distribution of particles formed during the photolysis of o-tolualdehyde at EUPHORE on 3 July 2003. The numbers in thelegend refer to time during the experiment.
9655 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
phase and lower SOA yields.30 In addition, aerosol formation isslightly slower in the indoor chamber and this suggests that thephotolysis rate may also influence SOA yield. A similar rate effecthas also been observed in previous studies of the photooxidationof aromatic hydrocarbons, where various factors including OHprecursor type and concentration,31,32 light intensity33 and re-lative humidity34 have been shown to affect OH levels, oxidationrate, and SOA mass yield.The results obtained here suggest that direct photolysis of o-
tolualdehyde may contribute to SOA formation in chamberstudies of o-xylene photooxidation35 and in the ambient atmo-sphere. Information on the chemical composition of the SOA canprovide insights into the key species and processes involved inaerosol formation and also help to identify molecular markers forspecific sources of ambient organic aerosol.29 The only carbonylproducts identified in filter samples collected in the indoorchamber experiments were o-phthalaldehyde (approximate yieldof 10%) and glyoxal (not quantified). The same carbonylproducts were identified in particles collected at EUPHORE,along with o-toluic acid, a hydroxyl-containing compound withMW 120, tentatively attributed to benzocyclobutenol, phthalal-dehydic acid, phthalide, and phthalic anhydride, SupportingInformation Table S2. The latter three compounds were alsodetected in filter samples of SOA generated from the photooxi-dation of o-tolualdehyde, where direct photolysis and reactionwith OH were both responsible for loss of the aromaticcompound.36 Interestingly, a number of oxygenated polycyclicaromatic compounds were also identified in this study, indicatingthat intermolecular reactions could be involved in SOA forma-tion and growth. There is evidence to suggest that biradical(Criegee) intermediates are involved in addition reactions withperoxy radicals leading to SOA formation during the ozonolysisof alkenes,29,37 and it is possible that the biradical speciesproduced during the photolysis of o-tolualdehyde may also beinvolved in similar types of reactions leading to polycycliccompounds with low volatility and hence SOA formation.
Photolysis of aromatic carbonyls in the solid and liquid phaseshas been shown to generate a wide range of polycyclic aromaticproducts,24,25 and another intriguing possibility is that some ofthe photochemically active compounds present in the SOA, forexample, o-phthalaldehyde, may undergo light-induced intermo-lecular reactions with other aromatic species to produce theoxygenated polycyclic aromatic compounds.This work is one of the first studies to demonstrate that direct
photolysis of a volatile organic compound can produce SOA. Theformation of aerosol from photolysis of 2,4-hexadienedial38 andortho-nitrophenols39 has been noted, although not investigatedin detail. More recently, Kessler et al.40 used the photolysis ofalkyl iodides to generate single organic radical precursors andproposed that this technique could be used as a simplifiedexperimental approach to investigate SOA formation fromalkanes. Similarly, the photolysis of o-tolualdehyde could alsobe considered as a good model system to investigate SOAformation mechanisms. The possible range of reaction pathwaysis considerably less complex than inOH-initiated photooxidationsystems and does not involve the use of OHprecursors. Althoughbeyond the scope of the present work, a systematic study of thevarious parameters affecting aerosol production from directphotolysis of o-tolualdehyde could prove to be beneficial inelucidating key processes responsible for SOA formation.Atmospheric Implications. The rate coefficient for the sun-
light photolysis of o-tolualdehyde can be used to calculate thetropospheric lifetime with respect to photolysis (τp) from therelationship: τp = 1/j. Using the values of j(o-tolualdehyde)obtained from the EUPHORE experiments yields photolysislifetimes in the range 1.3 � 1.7 h. The average value ofj(o-tolualdehyde)/j(NO2) = 2.53 � 10�2 can be used to providean estimate of the photolysis lifetime under a variety of other solarirradiation conditions. The other atmospheric loss processes foro-tolualdehyde are reaction with OH and NO3, which havelifetimes of 13.6 and 56.7 h respectively.10 Thus photolysisby sunlight is clearly the dominant atmospheric loss process for
Figure 4. Aerosol mass (TEOM) and volume (SMPS) concentrations measured during the photolysis of o-tolualdehyde at EUPHORE on 3 July 2003.The inset contains the plot of mass versus volume concentration used to determine the density of the aerosol.

9656 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
o-tolualdehyde. The short photolysis lifetime indicates that ifreleased or formed in the atmosphere, it will readily photolyze inthe troposphere and could contribute to the production of ozoneand secondary organic aerosol. Atmospheric degradation of thereaction products could also contribute to the formation ofozone and other oxidants. The major atmospheric fate ofbenzocyclobutenol is likely to be gas-phase reaction with OHradicals, with an estimated lifetime of around 24 h,41 whereas o-phthalaldehyde can undergo both photolysis and reaction withOH radicals.28 Finally, the kinetic and mechanistic informationobtained for the photolysis of o-tolualdehyde could be includedin photochemical degradation models, such as the MasterChemical Mechanism,42 that are used to predict secondarypollutant formation. It is envisaged that the inclusion of thischemistry will help to improve the prediction capability of themodels.
’ASSOCIATED CONTENT
bS Supporting Information. Photolytic loss plot, FTIRspectra, GC�MS data. This material is available free of chargevia the Internet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*Phone: +353 21 4902454; fax: +353 21 4903014; e-mail:[email protected].
’ACKNOWLEDGMENT
This work was supported by the Higher Education Authorityin Ireland and the European Commission through the researchproject EUROCHAMP 2 (contract number 228335). TheInstituto Universitario CEAM-UMH is partly supported byGeneralitat Valenciana, Fundaci�on Bancaja, and the projectsGRACCIE (Consolider-Ingenio 2010) and FEEDBACKS(Prometeo - Generalitat Valenciana).
’REFERENCES
(1) Feng, Y.; Wen, S.; Chen, Y.; Wang, X.; L€u, H.; Bi, X.; Sheng, G.;Fu, J. Ambient levels of carbonyl compounds and their sources inGuangzhou, China. Atmos. Environ. 2005, 39, 1789–1695.(2) L€u, H.; Cai, Q.-Y.; Wen, S.; Chi, Y.; Guo, S.; Sheng, G.; Fu, J.
Seasonal and diurnal variations of carbonyl compounds in the urbanatmosphere of Guangzhou, China. Sci. Total Environ. 2010, 408,3523–3529.(3) Obermeyer, G.; Aschmann, S. M.; Atkinson, R.; Arey, J. Carbo-
nyl atmospheric reaction products of aromatic hydrocarbons in ambientair. Atmos. Environ. 2009, 43, 3736–3744.(4) Kean, A. J.; Grosjean, E; Grosjean, D.; Harley, R. On-road
measurement of carbonyls in California light-duty vehicle emissions.Environ. Sci. Technol. 2001, 35, 4198–4204.(5) Jakober, C. A.; Robert, M. A.; Riddle, S. G.; Destaillats, H.;
Charles, M. J.; Green, P. G.; Kleeman, M. J. Carbonyl emissions fromgasoline and diesel motor vehicles. Environ. Sci. Technol. 2008, 42,4697–4703.(6) Calvert, J. G. A.; Becker, K. H.; Kamens, R. M.; Seinfeld, J. H.;
Wallington, T. J.; Yarwood, G.,TheMechanisms of Atmospheric Oxidationof Aromatic Hydrocarbons; Oxford University Press: Oxford, UK, 2002.(7) Atkinson, R; Aschmann, S. M.; Arey, J. Formation of ring-
retaining products from the OH radical-initiated reactions of o-, m-,and p-xylene. Int. J. Chem. Kinet. 1991, 23, 77–97.
(8) Atkinson, R.; Arey, J. Atmospheric degradation of volatileorganic compounds. Chem. Rev. 2003, 103 (12), 4605–4638.
(9) Thiault, G; Mellouki, A.; Le Bras, G. Kinetics of gas phasereactions of OH and Cl with aromatic aldehydes. Phys. Chem. Chem.Phys. 2002, 4, 2194–2199.
(10) Clifford, G. M.; Th€uner, L. P.; Wenger, J. C.; Shallcross, D. E.Kinetics of the gas-phase reactions of OH and NO3 radicals witharomatic aldehydes. J. Photochem. Photobiol., A 2005, 176, 172–182.
(11) Thiault, G.; Mellouki, A.; Le Bras, G.; Chakir, A.; Sokolowski-Gomez, N.; Daumont, D. UV-absorption cross sections of benzalde-hyde, ortho-, meta-, and para-tolualdehyde. J. Photochem. Photobiol., A2004, 162, 273–281.
(12) Volkamer, R.; Platt, U.; Wirtz, K. The European Photoreactor(EUPHORE), 3rd Annual Report 2000; Barnes, I., Sidebottom, H., Eds.;Bergische Universit€at Wuppertal: Wuppertal, Germany, 2001, 1.
(13) Thiault, G.; Mellouki, A.; Le Bras, G.; Wirtz, K. The EuropeanPhotoreactor (EUPHORE), 4th Annual Report 2001; Barnes, I., Ed.;Bergische Universit€at Wuppertal: Wuppertal, Germany, 2003, 29.
(14) Klotz, B.; Sørensen, S.; Barnes, I.; Becker, K. H.; Etzkorn, T.;Volkammer, R.; Platt, U.; Wirtz, K.; Martín-Reviejo, M. Atmosphericoxidation of toluene in a large-volume outdoor photoreactor: In situdetermination of ring-retaining product yields. J. Phys. Chem. 1998, 102,10289–10299.
(15) Martín-Reviejo, M; Wirtz, K. Is benzene a precursor forsecondary organic aerosol?. Environ. Sci. Technol. 2005, 39, 1045–1054.
(16) Wenger, J. C.; Le Calv�e, S.; Sidebottom, H. W.; Wirtz, K.;Martín-Reviejo, M; Franklin, J. A. Photolysis of chloral under atmo-spheric conditions. Environ. Sci. Technol. 2004, 38, 831–837.
(17) Sellev�ag, S. R.; Kelly, T.; Sidebottom, H.; Nielsen, C. J. A studyof the IR and UV-Vis absorption cross-sections, photolysis and OH-initiated oxidation of CF3CHO and CF3CH2CHO. Phys. Chem. Chem.Phys. 2004, 6, 1243–1252.
(18) O’Connor, M. P.; Wenger, J. C.; Mellouki, A.; Wirtz, K.;Mu~noz, A. The atmospheric photolysis of E-2-hexenal, Z-3-hexenaland E,E-2,4-hexadienal. Phys. Chem. Chem. Phys. 2006, 8, 5236–5246.
(19) Borr�as, E.; Tortajada-Genaro, L. A. Determination of oxygenatedcompounds in secondary organic aerosol from isoprene and toluene smogchamber experiments. Int. J. Environ. Anal. Chem. 2011in press.
(20) Th€uner, L. P.; Bardini, P.; Rea, G. J.; Wenger, J. C. Kinetics ofthe gas-phase reactions of OH and NO3 radicals with dimethylphenols.J. Phys. Chem. A. 2004, 108, 11019–11025.
(21) Temime, B.; Healy, R. M.; Wenger, J. C. A denuder-filtersampling technique for the detection of gas and particle phase carbonylcompounds. Environ. Sci. Technol. 2007, 41, 6514–6520.
(22) Healy, R. M.; Wenger, J. C.; Metzger, A.; Duplissy, J.; Kalberer,M.; Dommen, J. Gas/particle partitioning of carbonyls in the photo-oxidation of isoprene and 1,3,5-trimethylbenzene. Atmos. Chem. Phys.2008, 8, 3215–3220.
(23) DeMore, W. B.; Sander, S. P.; Golden, D. M.; Hampson, R. F.;Kurylo, M. J.; Howard, C. J.; Ravishankara, A. R.; Kolb, C. E.; Molina,M. J. In Chemical Kinetics and Photochemical Data for use in StratosphericModelling, JPL Publication 97-4; Jet Propulsion Laboratory, Pasadena,CA, 1997.
(24) Wagner, P. J.; Subrahmanyam, D.; Park, B.-S. Mechanism forthe photocyclization of o -alkyl ketones to cyclobutenols. J. Am. Chem.Soc. 1991, 113, 709–710.
(25) Moorthy, J. N.;Mal, P.; Natarajan, R.; Venugopalan, P. Efficientphotocyclization of o-alkylbenzaldehydes in the solid state: Directobservation of E-xylylenols en route to benzocyclobutenols. J. Org.Chem. 2001, 66, 7013–7019.
(26) Gebicki, J.; Krantz, A. Trapping of photoenols from o-tolualde-hydes in gasmatrices. Dependence of photoenol formation on the nature ofthe carbonyl function. J. Chem. Soc. Perkin Trans. 2 1984, 1623–1627.
(27) Yang, N. C.; Yang, D-D. H. Photochemical reactions of ketonesin solution. J. Am. Chem. Soc. 1958, 80, 2913–2914.
(28) Wang, L.; Arey, J.; Atkinson, R. Kinetics and products ofphotolysis and reaction with OH radicals of a series of aromatic carbonylcompounds. Environ. Sci. Technol. 2006, 40, 5465–5471.

9657 dx.doi.org/10.1021/es2026533 |Environ. Sci. Technol. 2011, 45, 9649–9657
Environmental Science & Technology ARTICLE
(29) Hallquist, M.; Wenger, J. C.; Baltensperger, U.; Rudich, Y.;Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N. M.; George, C.;Goldstein, A. H.; Hamilton, J. F.; Herrmann, H.; Hoffmann, T.; Iinuma,Y.; Jang, M.; Jenkin, M. E.; Jimenez, J. L.; Kiendler-Scharr, A.; Maenhaut,W.; McFiggans, G.; Mentel, T. F.; Monod, A.; Prevot, A. S. H.; Seinfeld,J. H.; Surratt, J. D.; Szmigielski, R.; Wildt, J. The formation, propertiesand impact of secondary organic aerosol: Current and emerging issues.Atmos. Chem. Phys. 2009, 9, 5155–5236.(30) Matsunaga, A; Ziemann, P. J. Gas-wall partitioning of organic
compounds in a Teflon film chamber and potential effects on reactionproduct and aerosol yield measurements. Aerosol Sci. Technol. 2010, 44,881–892.(31) Ng, N. L.; Kroll, J. H.; Chan, A. W. H.; Chhabra, P.; Flagan,
R. C.; Seinfeld, J. H. Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos. Chem. Phys. 2007, 7, 3909–3922.(32) Song, C.; Na, K. S.; Warren, B.; Malloy, Q.; Cocker, D. R.
Secondary organic aerosol formation from m-xylene in the absence ofNOx. Environ. Sci. Technol. 2007, 41, 7409–7416.(33) Warren, B.; Song, C.; Cocker, D. R. Light intensity and light
source influence on secondary organic aerosol formation for the m-xylene/NOx photooxidation system. Environ. Sci. Technol. 2008,42, 5461–5466.(34) Healy, R. M.; Temime, B.; Kuprovskyte, K.; Wenger, J. C. The
effect of relative humidity on gas/particle partitioning and aerosol massyield in the photooxidation of p-xylene. Environ. Sci. Technol. 2009, 43,1884–1889.(35) Song, C.; Na, K.; Warren, B.; Malloy, Q.; Cocker, D. R.
Secondary organic aerosol formation from the photooxidation of p-and o-xylene. Environ. Sci. Technol. 2007, 41, 7403–7408.(36) Webb, P. J.; Hamilton, J. F.; Lewis, A. C.;Wirtz, K. Formation of
oxygenated-polycyclic aromatic compounds in aerosol from the photo-oxidation of o-tolualdehyde. Polycyclic Aromat. Compd. 2006, 26,236–252.(37) Sadezky, A.; Winterhalter, R.; Kanawati, B.; R€ompp, A.; Spengler,
B.; Mellouki, A.; Le Bras, G.; Chaimbault, P.; Moortgat, G. K. Oligomerformation during gas-phase ozonolysis of small alkenes and enol ethers:New evidence for the central role of the Criegee Intermediate asoligomer chain unit. Atmos. Chem. Phys. 2008, 8, 2667–2699.(38) Klotz, B.; Barnes, I.; Becker, K. H. Kinetic study of the gas-phase
photolysis and OH radical reaction of E,Z- and E,E-2,4-hexadienedial.Int. J. Chem. Kinet. 1999, 31, 689–697.(39) Bejan, I.; Abd El Aal, Y.; Barnes, I.; Benter, T.; Bohn, B.;
Wiesen, P.; Kleffmann, J. The photolysis of ortho-nitrophenols: A newgas phase source of HONO. Phys. Chem. Chem. Phys. 2006, 8,2028–2035.(40) Kessler, S. H.; Nah, T.; Carrasquillo, A. J.; Jayne, J. T.;Worsnop,
D. R.; Wilson, K. R.; Kroll, J. H. Formation of secondary organic aerosolfrom the direct photolytic generation of organic radicals. J. Phys. Chem.Lett. 2011, 2, 1295–1300.(41) US Environmental Protection Agency. Estimation Programs
Interface (EPI) Suite v.3.20, 2007, http://www.epa.gov/oppt/expo-sure/pubs/episuitedl.htm (accessed October 7, 2011).(42) Bloss, C.; Wagner, V.; Jenkin, M. E.; Volkamer, R.; Bloss, W. J.;
Lee, J. D.; Heard, D. E.; Wirtz, K.; Martin-Reviejo, M.; Rea, G.; Wenger,J. C.; Pilling, M. J. Development of a detailed chemical mechanism(MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons.Atmos. Chem. Phys. 2005, 5, 641–664.
Related Documents











