The Application of Pericyclic, Photolytic, Chemoenzymatic and Cross-coupling Techniques to the Synthesis of Biologically Active Natural Products and Related Structures A thesis submitted for the Degree of Doctor of Philosophy of The Australian National University by Qiao Yan Research School of Chemistry Canberra, Australia April, 2017 © Copyright by Qiao Yan 2017 All Rights Reserved

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Application of Pericyclic, Photolytic, Chemoenzymatic
and Cross-coupling Techniques to the Synthesis of
Biologically Active Natural Products and Related Structures
A thesis submitted for the Degree of Doctor of Philosophy of
The Australian National University
by
Qiao Yan
Research School of Chemistry
Canberra, Australia
April, 2017
© Copyright by Qiao Yan 2017
All Rights Reserved


i
Declaration I declare that, to the best of my knowledge, the material presented in this thesis
represents the result of original work carried out by the author during the period 2012-
2017 and has not been presented for examination for any other degree. This thesis by
publication is comprised of four journal articles. Established methodologies have been
acknowledged, wherever possible, by citation of the original publications from which
they derive.
Qiao Yan
April, 2017


iii
Acknowledgements During my four and a half years of PhD life, I have been changed a lot by the
experience. In this long journey, I am lucky to have received plenty of help from a range
of people. Without them I would not have got to this point in a long but nevertheless
rewarding experience.
First, I would like to thank Professor Martin Banwell for his supervision,
encouragement and guidance. He gave me the opportunity to study in Banwell Group
and made me grow up in the past few years. His encouragement gave me the energy to
overcome the difficulties encountered in many of my experiments. In addition, when I
saw how diligent he is, working late almost every day, I told myself that I too must
work hard to become a successful scientist. All in all, both the Banwell chemistry and
habits have contributed so much to the entirety of my PhD studies. There is so much I
could say but put simply….many thanks Martin!
I am indebted to Dr Xinghua Ma. Although invariably busy working on his own
projects, Xinghua has always been so enthusiastic in discussions and provided many
effective solutions to the problems I encountered in my experiments. His careful
analysis and down-to-earth advice gave me the confidence to complete complex tasks.
Many thanks Xinghua and I wish you all the very best for your future.
I would also like to thank all the members of the Banwell Group, especially Nadia, Ping,
Shuxin and Benoit. In addition, I want to express my sincere thanks to my lab mates in
3.27, namely Xiang, Jeremy and Josh. I have spent almost every working day of the last
four and a half years with the three of you and am so grateful for your companionship
and advice.
I should also acknowledge the technical staff in Research School of Chemistry, in
particular Dr Anthony Wills, Dr Paul Carr, Mr Chris Blake and Ms Anithahini
Jeyasingham. Their patience and professionalism helped me acquire all the high quality
X-ray diffraction, nuclear magnetic resonance and mass spectrometric data sets required
for the completion of my research.

iv
Finally, I must give deep thanks to my parents, my husband and my sister. Although I
could not stay nearby my parents, they always understood me and supported my beliefs
and goals. Deepest thanks must also go to my husband Hao Zhang. He has always been
a patient listener and the provider of perfect advice. He is also my best friend with
whom I share both the happiness and sadness that comes with life. All my achievements
belong to both of us. I also feel exceptionally lucky to have an older sister who has
always taken care of me, as she has the rest of the family. All of them are the great
blessings of my life and I cherish our precious times together.

v
Publications and Presentations The following list details the publications and presentations that have resulted from the
author’s research work performed during her candidature for the Degree of Doctor of
Philosophy.
Publications:
1. Establishing the True Structure of the Sorbicillinoid-derived Isolate
Rezishanone C by Total Synthesis
Qiao Yan, Martin G. Banwell, Michelle L. Coote, Richmond Lee and Anthony C.
Willis
Chem. Asian J., 2017, 12, 1480.
2. Studies on the Photochemical Rearrangements of Enantiomerically Pure,
Polysubstituted and Variously Annulated Bicyclo[2.2.2]octenones
Qiao Yan, Benoit Bolte, Yuhua Bai, Martin G. Banwell, Anthony C. Willis and Paul
D. Carr
J. Org. Chem., 2017, 82, 8008.
3. A Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Route
to the Carbazole Natural Products 3-Methyl-9H-carbazole, Glycoborine,
Glycozoline, Clausazoline K, Mukonine and Karapinchamine A
Qiao Yan, Emma Gin, Malgorzata Wasinska-Kalwa, Martin G. Banwell and Paul D.
Carr
J. Org. Chem., 2017, 82, 4148.
4. A Unified Approach to the Isomeric α-, β-, γ- and δ-Carbolines via their
6,7,8,9-Tetrahydro Counterparts
Qiao Yan, Emma Gin, Martin G. Banwell, Anthony C. Willis and Paul D. Carr
J. Org. Chem., 2017, 82, 4328.

vi
Presentations:
1. Establishing the True Structure of a Sorbicillinoid-derived Isolate by
Chemoenzymatic Synthesis
Qiao Yan, Martin G. Banwell and Anthony C. Willis
Poster Presentation at The Royal Australian Chemical Institute Organic One-Day
Symposium, November 30th, 2016, Sydney, Australia.

vii
Commentary on the Contributions of Ms Qiao Yan to
the Four Papers Included in this PhD Thesis by
Publications
Publication 1
This is a communication detailing the author’s extensive experimental efforts directed
towards the total synthesis of ent-rezishanone C. The author carried out, single-
handedly, the entirety of the synthetic chemistry-based laboratory work reported in this
communication. Except for the reported computational chemistry studies, she wrote the
whole of the experimental section and conducted relevant literature surveys. In addition,
the author collated and formatted all of the reported spectral data presented in the
associated supporting information document. Dr Anthony C. Wills conducted the X-ray
crystallographic studies reported in this paper. Professor Martin Banwell wrote the body
of the paper.
Publication 2
This is a full paper detailing extensive experimental work concerned with the
photochemical rearrangements of bicyclo[2.2.2]octenones. The author carried out 90%
of the synthetic chemistry-based laboratory work reported in this article. She also wrote
95% of the experimental section and conducted relevant literature surveys. In addition,
the author collated and formatted all of the reported spectral data presented in the
supporting information document. Drs Anthony C. Wills and Paul D. Carr conducted
the X-ray crystallographic studies reported in this paper while Professor Martin Banwell
wrote the body of the paper.
Publication 3
This is a full paper detailing extensive experimental work directed towards the synthesis
of carbazole natural products 3-methyl-9H-carbazole, glycoborine, glycozoline,
clausazoline K, mukonine and karapinchamine A. The author carried out 70% of the
synthetic chemistry-based laboratory work reported in this article and also wrote the
whole of the experimental section as well as conducting relevant literature surveys. In

viii
addition, the author collated and formatted all the reported spectral data presented in the
supporting information document. Dr Paul D. Carr conducted the X-ray crystallographic
studies reported in this paper while Professor Martin Banwell wrote the body of the
paper.
Publication 4
This is a full paper detailing extensive experimental work directed towards a unified
approach to the isomeric α-, β-, γ- and δ-carbolines. The author carried out the entirety
of the synthetic chemistry-based laboratory work reported in this article, wrote the
whole of the experimental section and conducted relevant literature surveys. In addition,
she collated and formatted all of the reported spectral data presented in the supporting
information document. Drs Anthony C. Wills and Paul D. Carr conducted the X-ray
crystallographic studies reported in this paper. Professor Martin Banwell wrote the body
of the paper.

ix
Table of Contents
Declaration…………………………………………………………….i
Acknowledgements……………...…………………………………...iii
Publications and Presentations……..………………………………..v
Relative Contributions to Publications……..………………………vii
Table of Contents………...…………………………………………...ix
Abstract…………………...…………………………………………...1
Thesis Overview……………..…………………..…………………….3
Publication 1 ……………….……………………..………………….10
Publication 2 ….…………………………………….………………168
Publication 3 …………………………………………..……..……..301
Publication 4 ………………………………………………………..382
Appendices…….…………………………………………………….437


1
Abstract
The body of this thesis is comprised of four scientific articles and is preceded by an
overview that contextualises all of this submitted/published work.
The first major part of this thesis is comprised of Publication 1. This details work
concerned with establishing the true structure of the sorbicillinoid-derived isolate
rezishanone C by total synthesis. Specifically, the enantiomer, B, of what proved to be
the true structure, C, of the sorbicillinoid rezishanone C (sorbivinetone) was synthesized
from the homochiral cis-1,2-dihydrocatechol A that is itself generated through the
whole-cell biotransformation of toluene. These studies and dispersion-corrected DFT
calculations support the proposal that rezishanone C is an artefact of the isolation
process and arises through a Diels-Alder cycloaddition reaction between ethyl vinyl
ether and sorbicillinol (D).
The second major part of the thesis is comprised of Publication 2. This is concerned
with the synthesis and photochemical rearrangements of enantiomerically pure,
polysubstituted and, in some cases, variously annulated bicyclo[2.2.2]octenones.
Specifically, then, a series of bicyclo[2.2.2]octenones has been prepared by engaging
the enzymatically-derived and enantiomerically pure cis-1,2-dihydrocatechol A in either
inter- or intra-molecular Diels-Alder cycloaddition reactions with various dienophiles.
These polycyclic adducts or simple derivatives thereof were shown to readily participate
in both photochemically promoted 1,3-acyl migration and oxa-di-π-methane
rearrangement processes to give products such as E and F, respectively.
OH
OH
A
O
O
O
OOH
H
HO
O
OOH
O
O OH
O
OH
B C D
O
H
OO
HO
O
O
E F

2
The third major part of the thesis is comprised of Publication 3. This details the
establishment of a palladium-catalyzed Ullmann cross-coupling/reductive cyclization
route to the carbazole natural products 3-methyl-9H-carbazole, glycoborine,
glycozoline, clausazoline K, mukonine and karapinchamine A. These were prepared by
reductive cyclisation of the relevant 2-arylcyclohex-2-en-1-one (e.g. G) to the
corresponding tetrahydrocarbazole (e.g. H) and dehydrogenation of this to give the
target carbazole (e.g. I). Compounds such as G were themselves prepared using a
palladium-catalyzed Ullmann cross-coupling reaction that served to link the appropriate
2-iodocyclohex-2-en-1-one and o-halonitrobenzene.
The fourth and final part of the thesis is comprised of Publication 4. This details a
unified approach to the isomeric α-, β-, γ- and δ-carbolines via their 6,7,8,9-tetrahydro
counterparts. Specifically, then, a cross-coupling/reductive cyclisation protocol has been
employed in preparing all four carbolines. So, for example, the 2-nitropyridine (L),
which is readily generated through an efficient palladium-catalyzed Ullmann cross-
coupling reaction, is reductively cyclized under conventional conditions to give 6,7,8,9-
tetrahydro-α-carboline (M) that is itself readily aromatized to give α-carboline (N).
OO2N
NH N
H
OMe OMe OMe
G IH
O NO2NNN
H NNH
L M N

3
Thesis Overview
Publication 1: Establishing the True Structure of the Sorbicillinoid-derived Isolate
Rezishanone C by Total Synthesis
The sorbicillins are an ever-expanding class of polyketide-derived fungal metabolite1
that display significant structural diversity and (sometimes) unusual biological
activities.2 Rezishanone C3 is a representative member of the sub-type2c of sorbicillins
that arise through a Diels-Alder reaction between sorbicillinol and various non-
sorbicillinoid-derived compounds containing a dienophilic residue. In particular,
rezishanone C is thought to arise through reaction between ethyl vinyl ether (a common
contaminant in ethyl acetate, the solvent used to isolate this material) but precisely
which one of eight possible adducts represents the true structure of this artefact remains
unclear. Given the uncertainty regarding the structure of rezishanone C and informed by
theoretical calculations, the author developed a total synthesis of what proved to be ent-
rezishanone C.
Figure 1. Structures of rezishanone C, ent-rezishanone C and sorbicillinol
Thus, this publication details a twenty-three step total synthesis of ent-rezishanone C
(Scheme 1). Starting from cis-1,2-dihydrocatechol 1, and through the application of
intermolecular Diels-Alder and retro-aldol/aldol sequences, trans-diol 2 was formed.
Over nine steps this last compound was converted into ethyl ether 3 that was itself
subjected to cis-dihydroxylation under conditions defined by Bäckvall4 and so affording
diol 4. A Barton-McCombie deoxygenation reaction5 then a DMP (Dess-Martin
periodinane)-mediated oxidation followed and compound 4 was thereby transformed
into ketone 5. When the potassium enolate derived from compound 5 was treated with
(3E,5E)-2-oxo-3,5-heptadienenitrile6,7 then the desired C-acylated product 6 was formed.
Hydrolysis of the acetonide residue within compound 6 and oxidation of the resulting
sorbicillinol
7
O
O
O
OHO
H
ent-rezishanone C
O
O OH
O
OH
H
OHO
O
O
O
rezishanone C
HO
O OOH

4
diol then gave ent-rezishanone C. All the spectral data derived from this material
matched those reported for the isolate. Furthermore, the specific rotation of the
synthetic material was of the same magnitude but opposite sign to that reported for the
isolate and so definitively establishing the structure of rezishanone C as that shown in
Figure 1.
Scheme 1. Key steps involved in the synthesis of ent-rezishanone C
Publication 2: Studies on the Photochemical Rearrangements of Enantiomerically
Pure, Polysubstituted and Variously Annulated Bicyclo[2.2.2]octenones
Bicyclo[2.2.2]octenones including the parent system 7 (Scheme 2) are excellent
substrates for certain photochemically promoted rearrangement reactions.8 Specifically,
on irradiation in the presence of photosensitizers such as acetophenone they participate
in oxa-di-π-methane rearrangements and so affording, via a triplet pathway,
cyclopropannulated diquinanes such as 8. In contrast, on direct irradiation they engage,
now via a singlet pathway, in a 1,3-acyl migration reaction (Givens rearrangement) to
give bicyclo[4.2.0]oct-4-en-7-ones such as 9. Upon sustained irradiation photoproduct 9
and many of its derivatives can undergo decarbonylation to give the corresponding Δ2-
norcarene, e.g. 10.
OH
OH
1
OH
OOH O
OO
2 3
OO
O
4HOOH
6
O O
O
OH
O OO
O
5
O
4 steps 9 steps
KHMDS thenE,E-CH3(CH=CH)2COCN
THF–78 °C, 3 h
55% or 81% brsm
5% K2OsO4•H2Ocitric acid, NMO
CH3CN/H2O22 °C, 14 h
68%
6 steps
2 steps
O
O OH
O
OH
ent-rezishanone C

5
Scheme 2. Photochemical rearrangement reactions of bicyclo[2.2.2]octenone (7)
By way of example, then, Publication 2 describes methods for the synthesis of
enantiomerically pure bicyclo[2.2.2]octenones such as 11-14 (Figure 2)9 and the
engagement of these systems in the above-mentioned photochemical processes. As a
result a suite of novel diquinanes, bicyclo[4.2.0]octenones and/or bicyclo[4.1.0]octenes
was produced. A number of these photoproducts are potential precursors to a range of
terpenoid-type natural products.
Figure 2. Examples of enantiomerically pure bicyclo[2.2.2]octenones
Publication 3: A Palladium-Catalyzed Ullmann Cross-Coupling/Reductive
Cyclization Route to the Carbazole Natural Products 3-Methyl-9H-carbazole,
Glycoborine, Glycozoline, Clausazoline K, Mukonine and Karapinchamine A
9H-Carbazole and its various derivatives continue to fascinate organic chemists because
of their value in both medicine and materials science.10 Many biologically active natural
products embodying this framework have also been isolated, particularly from higher
plants.10d,10e,11 Accordingly, this publication details the development of a two-step
process leading to 1,2,3,4-tetrahydro-9H-carbazoles (formally 2,3,4,9-tetrahydro-1H-
carbazoles)12 that can then be oxidized (directly) to the corresponding carbazoles.13 A
relevant example leading to the natural product glycozoline is shown in Scheme 3.
O
H
H
O
7 8
9 10
oxa-di-π-methanerearrangement
1,3-acylmigration
decarbonylation
hνsens
hν(direct)
hν(direct)
H
HH
H
O
OOOHOO OOTBS
OOH OAcO
OAc
11 12 13 14

6
Scheme 3. The synthesis of glycozoline
By such means the author has been able to realize syntheses of the parent carbazole as
well as the natural products 3-methyl-9H-carbazole,14 glycoborine (a.k.a.
glycrophylamine),11a,15 glycozoline,11a,16 clauszoline K,17 mukonine18 and
karapinchamine A19 together with their mono-methoxylated congener 2-methoxy-9H-
carbazole (Figure 3).
Figure 3. Structures of certain carbazole-based natural products and a mono-
methoxylated congener
Publication 4: A Unified Approach to the Isomeric α-, β-, γ- and δ-Carbolines via
their 6,7,8,9-Tetrahydro Counterparts
The isomeric α-, β-, γ- and δ-carbolines (Figure 4) are important heterocyclic ring
systems20 encountered, albeit to varying extents, as key structural motifs in various
biologically active natural products.
Figure 4. The isomeric α-, β-, γ- and δ-carbolines (17-20, respectively)
OO2N
NH N
H15 16 glycozoline
OMe OMe OMeH2
Raney-Nickel 10 wt. % Pd/C
Ph2O, 210 °C0.33 h, 89%
MeOH, 22 °C16 h, 72%
Pd[0]-catalyzedUllmann cross-coupling
3-methyl-9H-carbazole glycozolineglycoborine
clauszoline K karapinchamine A
NH
NH
NH
OMe
NH
OHC
OMe
N OH
OMe
NH
OMe
CO2Me
mukonine
NH OMe
2-methoxy-9H-carbazole
17
NNH
18
NNH
19
N
NH
20
N
NH

7
This publication reports on the development of a unified approach (perhaps the first to
have been developed) to the carbolines involving palladium-catalyzed Ullmann cross-
coupling,21 reductive cyclization and dehydrogenation reactions as key steps, the last
being employed to convert tetrahydro-carbolines into their fully aromatic counterparts
(viz. the carbolines). The synthesis of α-carboline, as shown in Scheme 4, is illustrative
of the reaction sequence used.
Scheme 4. The synthesis of α-carboline
O N
H210 wt. % Pd/C 10 wt. % Pd/C
O2NNN
H NNH
Ph2O, 210 °C0.66 h, 97%
MeOH, 22 °C16 h, 75%
palladium-catalyzedUllmann cross-coupling
21 22 α-carboline

8
References
1. For examples of recently reported sorbicillins see: (a) El-Elimat, T.; Raja, H. A.;
Figueroa, M.; Swanson, S. M.; Falkinham III, J. O.; Lucas, D. M.; Grever, M. R.;
Wani, M. C.; Pearce, C. J.; Oberlies, N. H. J. Antibiot., 2015, 68, 191; (b) Zhai, M.-
M.; Qi, F.-M.; Li, J.; Jiang, C.-X.; Hou, Y.; Shi, Y.-P.; Di, D.-L.; Zhang, J.-W.; Wu,
Q.-X. J. Agric. Food Chem., 2016, 64, 2298.
2. For useful points-of entry into the literature on sorbicillinoids see: (a) Harned, A.
M.; Volp, K. A. Nat. Prod. Rep., 2011, 28, 1790; (b) Meng, J.; Wang, X.; Xu, D.;
Fu, X.; Zhang, X.; Lai, D.; Zhou, L.; Zhang, G. Molecules, 2016, 21, 715.
3. (a) Bringmann, G.; Lang, G.; Gulder, T. A. M.; Tsuruta, H.; Mühlbacher, J.;
Maksimenja, K.; Steffens, S.; Schaumann, K.; Stöhr, R.; Wiese, J.; Imhoff, J. F.;
Perovíc-Ottstadt, S.; Boreiko, O.; Müller, W. E. G. Tetrahedron, 2005, 61, 7252; (b)
Lee, D.; Lee, J. H.; Cai, X. F.; Shin, J. C.; Lee, K.; Hong, Y.-S.; Lee, J. J. J.
Antibiot., 2005, 58, 615; (c) Maskey, R. P.; Grün-Wollny, I.; Laatsch, H. J. Nat.
Prod., 2005, 68, 865; (d) Neumann, K.; Abdel-Lateff, A.; Wright, A. D.; Kehraus,
S.; Krick, A.; König, G. M. Eur. J. Org. Chem., 2007, 2268.
4. Ell, A.; Closson, A.; Adolfsson, H.; Bäckvall, J.-E. Adv. Synth. Catal., 2003, 345,
1012.
5. Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1, 1975, 16, 1574.
6. Volp, K. A.; Johnson, D. M.; Harned, A. M. Org. Lett., 2011, 13, 4486.
7. For a related conversion see Franck, G.; Brödner, K.; Helmchen, G. Org. Lett.,
2010, 12, 3886.
8. For a relevant and recent review see Banwell, M. G.; Bon, D. J.-Y. D. Applications
of the Di-π-Methane and Related Rearrangement Reactions in Chemical Synthesis.
In Molecular Rearrangements in Organic Synthesis: Rojas, C. M., Ed.; Wiley,
Hobokjen, NJ, 2015; Chapter 9, pp 261-288.
9. Juo, S.-Y.; Jang, Y.-J.; Liu, J.-Y.; Chu, C.-S.; Liao, C.-C.; Hung, S.-C. Angew.
Chem. Int. Ed. 2008, 47, 8082 and references cited therein.
10. Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis, 1994, 639.
11. See, for example, (a) Cheenpracha, S.; Laphookhieo, S. Phytochem. Lett. 2011, 4,
187; (b) Miller, C. M.; McCarthy, F. O. RSC Advances, 2012, 2, 8883; (c) Pieroni,
M.; Girmay, S.; Sun, D.; Sahu, R.; Tekwani, B. L.; Tan, G. T. Chem. Med. Chem.
2012, 7, 1895; (d) Russell, F.; Harmody, D.; McCarthy, P. J.; Pomponi, S. A.;

9
Wright, A. E. J. Nat. Prod. 2013, 76, 1989; (e) Kim, S.-H.; Ha, T.-K.-Q.; Oh, W.
K.; Shin, J.; Oh, D.-C. J. Nat. Prod. 2016, 79, 51; (f) Patel, O. P. S.; Mishra, A.;
Maurya, R.; Saini, D.; Pandey, J.; Taneja, I.; Raju, K. S. R.; Kanojiya, S.; Shukla,
S. K.; Srivastava, M. N.; Wahajuddin, M.; Tamrakar, A. K.; Srivastava, A. K.;
Yadav, P. P. J. Nat. Prod. 2016, 79, 1276.
12. Banwell, M. G.; Kelly, B. D.; Kokas, O. J.; Lupton, D. W. Org. Lett. 2003, 5, 2497.
13. Campaigne, E.; Lake, R. D. J. Org. Chem. 1959, 24, 478.
14. (a) Chakrabarty, M.; Nath, A. c.; Khasnobis, S.; Chakrabarty, M.; Konda, Y.;
Harigaya, Y.; Komiyama, K. Phytochem. 1997, 46, 751; (b) Cui, C.-B.; Yan, S.-Y.;
Cai, B.; Yao, X.-S. J. Asian. Nat. Prod. Res. 2002, 4, 233.
15. Chakravarty, A. K.; Sarkar, T.; Masuda, K.; Takey, T.; Doi, H.; Kotani, E.;
Shiojima, K. Indian J. Chem. Sec. B 2001, 40, 484.
16. (a) Chakraborty, D. P. Phytochem. 1969, 8, 769; (b) Li, W. S.; McChesney, J. D.;
El-Feraly, F. S. Phytochem. 1991, 30, 343.
17. (a) Birari, R.; Roy, S. K.; Singh, A.; Bhutani, K. Nat. Prod. Commun. 2009, 4,
1089; (b) Lin, W.; Wang, Y.; Lin, S.; Li, C.; Zhou, C.; Wang, S.; Huang, H.; Liu,
P.; Ye, G.; Shen, X. Eur. J. Med. Chem. 2012, 47, 214.
18. (a) Brenna, E.; Fuganti, C.; Serra, S. Tetrahedron, 1998, 54, 1585; (b)
Laphookhieo, S.; Sripisut, T.; Prawat, U.; Karalai, C. Heterocycles, 2009, 78, 2115.
19. Nakamura, S.; Nakashima, S.; Oda, Y.; Yokota, N.; Fujimoto, K.; Matsumoto, T.;
Ohta, T.; Ogawa, K.; Maeda, S.; Nishida, S.; Matsuda, H.; Yoshikawa, M. Bioorg.
Med. Chem. 2013, 21, 1043.
20. For useful points-of-entry into the substantial body of literature on these
compounds see (a) Smirnova, O. B.; Golovko, T. V.; Granik, V. G. Pharm. Chem.
J., 2011, 44, 654; (b) Hung, T. Q.; Dang, T. T.; Janke, J.; Villinger, A. Langer, P.
Org. Biomol. Chem., 2015, 13, 1375 and references cited therein.
21. Banwell, M. G.; Jones, M. T.; Reekie, T. A. Chem. New Zealand, 2011, 75, 122.


Publication One
Establishing the True Structure of the Sorbicillinoid-
derived Isolate Rezishanone C by Total Synthesis
Qiao Yan, Martin G. Banwell, Michelle L. Coote,
Richmond Lee and Anthony C. Willis
Chem. Asian J., 2017, 12, 1480.


Natural Product Synthesis
Establishing the True Structure of the Sorbicillinoid-DerivedIsolate Rezishanone C by Total Synthesis
Qiao Yan,[a] Martin G. Banwell,*[a] Michelle L. Coote,[a, b] Richmond Lee,[a, b] andAnthony C. Willis[a]
Abstract: The enantiomer, ent-4, of the true structure, 4,
of the sorbicillinoid rezishanone C (sorbivinetone) hasbeen synthesized from a homochiral cis-1,2-dihydrocate-chol that is itself generated through the whole-cell bio-transformation of toluene. These studies together withdispersion-corrected DFT calculations support the propos-
al that rezishanone C is an artefact of the isolation processand arises through a Diels–Alder reaction between ethyl
vinyl ether and sorbicillinol (3).
The sorbicillins are an ever-expanding class of polyketide-de-rived fungal metabolite[1] that display significant structural di-
versity and (sometimes) unusual biological activities.[2] Rezisha-
none C (sorbivinetone, Figure 1),[3] for which both structures1 and 2 have been suggested, can be classified as a member
of the so-called hybrid sub-type[2a] of sorbicillins that arisethrough a Diels–Alder reaction between sorbicillinol (3) and
various non-sorbicillinoid-derived compounds containing a di-enophilic residue. In the case of rezishanone C, it has been
suggested[3a] that ethyl vinyl ether is the dienophile involved.
Since this ether is a contaminant often found in ethyl acetate,the solvent used in extracting rezishanone C from various
fungal sources, it is quite possible that this compound is an ar-tefact of the isolation process rather than a true natural prod-
uct. Some support for this argument follows from the report[3a]
that the acetate of the racemic modification of compound 3(i.e. (:)-sorbicillinol acetate) reacts with ethyl vinyl ether under
ambient conditions to give a Diels–Alder adduct designated O-acetylrezishanone C. On this basis, Bringmann proposed[3a]
structure 1 for rezishanone C (a compound that would arisethrough a-face addition of ethyl vinyl ether to diene 3).
Others who have isolated what we now confirm to be thesame material have suggested that it may be one or other of
the C7 epimeric forms of compound 2 (that would arise if thesame dienophile added, depending on the C7 stereochemistry,
in either an exo or endo mode to the b face of diene 3).[3b–d] It
should be noted that each of the four research groups whohave reported isolating rezishanone C used a different solvent
to acquire their 1H and 13C NMR spectral data sets and thuspreventing direct comparisons between them.3
Rezishanone C, in any of its structurally proposed or plausi-ble alternate forms,[4] embodies a polyfunctionalized and ho-mochiral bicyclo[2.2.2]octane core bearing a bridgehead
methyl group, a structural motif that we have been able to as-semble by chemoenzymatic means during the course of ourstudies on the total synthesis of various terpenoids.[5] Accord-ingly, and given the absence of any prior studies[6] as well as
the ambiguities associated with its structure, we were attract-ed to rezishanone C as a synthetic target. As a prelude to un-
dertaking synthetic studies, we carried out dispersion-correct-ed DFT calculations to determine the transition state energiesassociated with the various possible modes of Diels–Alder cy-cloaddition that could take place between sorbicillinol (2) andethyl vinyl ether. These suggested that the reaction pathway
leading to compound 4 (rather than congener 1 or the 7R epi-meric form of 2) is the most energetically favorable one (see
the Supporting Information). As such, and given the nature ofthe homochiral starting material available to us, we pursuedthe total synthesis of ent-rezishanone C (ent-4, Figure 2), and
thereby established that the illustrated structure 4 is the cor-rect one for rezishanone C.
The opening stages of the synthesis are shown in Scheme 1and involved converting, under standard conditions, the readi-
Figure 1. The two possible structures, 1 and 2, originally proposed for re-zishanone C (sorbivinetone) and its likely biogenetic precursor sorbicillinol(3).
[a] Q. Yan, Prof. M. G. Banwell, Prof. M. L. Coote, Dr. R. Lee, Dr. A. C. WillisResearch School of ChemistryInstitute of Advanced StudiesThe Australian National UniversityCanberra, ACT 2601 (Australia)E-mail : [email protected]
[b] Prof. M. L. Coote, Dr. R. LeeARC Centre of Excellence for Electromaterials Science
Supporting information and the ORCID identification number(s) for the au-thor(s) of this article can be found under https ://doi.org/10.1002/asia.201700456.
This manuscript is part of a special issue celebrating the 100th anniversaryof the Royal Australian Chemical Institute (RACI). Click here to see the Tableof Contents of the special issue.
Chem. Asian J. 2017, 12, 1480 – 1484 T 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1480
CommunicationDOI: 10.1002/asia.201700456

ly available, enzymatically derived and homochiral cis-1,2-dihy-drocatechol 5[7] into the corresponding and previously report-
ed acetonide 6.[5, 8] Compound 6 was itself engaged in a ther-mally induced Diels–Alder cycloaddition reaction with a-chlor-
oacrylonitrile,[9] and the epimeric mixture of ortho adducts thusobtained were hydrolyzed using potassium hydroxide in tert-
butanol to give the known[5, 10] bicyclo[2.2.2]octenone 7 (53 %from 5). Cleavage of the acetonide moiety within the last com-pound was achieved using acidified AG-50W-X8 resin in hot
aqueous methanol, but this process was accompanied bya retro-aldol/aldol sequence that led to the production of the
trans-diol 8 (86 %). The structure of compound 8 was con-firmed via single-crystal X-ray analysis (see the Supporting In-
formation). The propensity for retro-aldol reactions to take
place within this framework was further emphasized on at-tempting to convert the ketone moiety within compound 8into the corresponding ethylene ketal. Thus, on treating thissubstrate with ethylene glycol in benzene under reflux, the un-
expected, ring-cleaved and crystalline cyclohexenone 9[10]
(48 %) was obtained.
Progress towards the bicyclo[2.2.2]octane core of target ent-4 could be made (Scheme 2) by selectively converting the hy-droxyl residue remote from the bridgehead methyl groupwithin compound 8 into the corresponding TBS ether 10(88 %). Reduction of the ketone moiety associated with com-pound 10 was achieved stereoselectively using DIBAL-H, and
so affording a chromatographically separable mixture of epi-mers 11[10] (15 %) and 12 (83 %). Two-fold acetylation of diol 12under standard conditions gave the diester 13 (98 %), and
treatment of the latter with tetra-n-butylammonium fluoride(TBAF) then gave alcohol 14[10] (91 %) that could be oxidized to
the corresponding ketone 15 (95 %) using Dess–Martin periodi-nane (DMP).[11] Reaction of compound 15 with methyl magne-
sium bromide at 0 8C for a brief period gave the cis diol 16[10]
(85 %) through selective si-face addition of the nucleophile to
the ketone carbonyl and as a result of the a-acetoxy group
hindering the corresponding re face. After saponification of theremaining acetate residue within compound 16, the triol 17[10]
(79 %) was converted, by standard methods, into the acetonide
Figure 2. The theoretically favored Diels–Alder adduct, 4, arising from the re-action of sorbicillinol (3) with ethyl vinyl ether and the structure of its enan-tiomer (ent-4) targeted for synthesis in the present study.
Scheme 1. Assembling the bicyclo[2.2.2]octane core of ent-rezishanone C(ent-4).
Scheme 2. Elaborating the bicyclo[2.2.2]octane core of ent-rezishanone C(ent-4).
Chem. Asian J. 2017, 12, 1480 – 1484 www.chemasianj.org T 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1481
Communication

18 (91 %). Finally, compound 18 was treated with sodium hy-dride and ethyl iodide and so generating the ethyl ether 19(86 %) that embodies a number of the key structural featuresof the core of ent-rezishanone C.
Our proposed conversion of compound 19 into target 1 re-quired that the double bond within the former compound be
manipulated such that a ketone carbonyl group is introducedadjacent to (rather than remote from) the bridgehead methyl
group. In one of a number of attempts to achieve such an out-
come alkene 19 (Scheme 3) was treated with meta-chloroper-
benzoic acid (mCPBA) and a single epoxide 20 (86 %) was ob-
tained. The high degree of selectivity associated with this elec-trophilic addition reaction may be the result of hydrogen
bonding between the epoxidizing agent and the oxygen ofthe pendant ethoxy group. Disappointingly, all efforts to effect
the reductive cleavage of this compound with the desired re-gioselectivity failed. So, for example, on reaction of com-
pound 20 with lithium triethylborohydride the undesired alco-
hol 21 (84 %) was obtained, perhaps as a result of electronic ef-fects exerted by the pendant ethoxy group and/or steric ones
arising from the methyl group at the junction between dioxo-lane and bicyclo[2.2.2]octane frameworks. The structure of
compound 21 (and, therefore, the precursor epoxide 20) fol-lowed from a single-crystal X-ray analysis of the crystalline triol
22[10] (59 % or 84 %, based on recovered starting material) pro-duced through hydrolysis of the acetonide group using acidi-fied AG-50W-X8 resin.
The above-mentioned difficulties were overcome by themeans outlined in Scheme 4. Thus, alkene 19 was subjected to
dihydroxylation under conditions defined by B-ckvall,[12] afford-ing a chromatographically separable mixture of products 23(22 %) and 24 (68 %). Presumably, the facial selectivity of this
oxidation process, which is essentially opposite to that ob-served in the conversion 19!20, is now dictated, to some
extent at least, by the steric demands of the pendant ethoxygroup. Treatment of the latter with para-methoxybenzalde-
hyde dimethylacetal (PMBDMA) in the presence of para-tolue-nesulfonic acid monohydrate (pTsOH·H2O) afforded the epimer-
ically pure acetal 25 (89 %) that was reductively cleaved with
diisobutylaluminum hydride (DIBAL-H) in CH2Cl2 and so givingthe diol mono-ether 26[10] in 95 % yield.[13] As a prelude to con-ducting a Barton–McCombie deoxygenation reaction,[14] com-
pound 26 was converted into the corresponding xanthate 27(96 %) under standard conditions and on treatment of this
with tris(trimethylsilyl)silane (TTMSS)[15] and azobisisobutyroni-trile (AIBN) reduction took place to give the anticipated ether
28 (79 %). 2,3-Dichloro-5,6-dicyanobenzoquinone (DDQ)[16] was
used to effect the oxidative cleavage of compound 28, afford-ing alcohol 29 (89 %) that was converted into the target
ketone 30[10, 17] (79 %) using DMP in pyridine. Disappointingly,when the enolate anion derived from ketone 30 [obtained by
treating it with potassium hexamethyl disilazide (KHMDS)] wasquenched with sorbyl chloride in THF at @78 8C, O-acylation
Scheme 3. An unwanted regiochemical outcome associated with manipulat-ing the olefinic residue in compound 19.
Scheme 4. Regiocontrolled manipulation of the olefinic residue within com-pound 19 and an O-acylation reaction.
Chem. Asian J. 2017, 12, 1480 – 1484 www.chemasianj.org T 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1482
Communication

rather than C-acylation took place, leading to the unstableenol ester 31 (84 %) as the exclusive product of reaction.
The difficulties detailed immediately above were overcomein the same manner as used by Harned and co-workers[6a]
during the course of their synthesis of sorbicillactone A. Thus,the potassium enolate derived from deprotonation of ketone
30 (Scheme 5) was treated with (3E,5E)-2-oxo-3,5-heptadieneni-
trile[6a, 18] in THF at @78 8C, and now the desired C-acylated
product 32 (55 %) was obtained as a crystalline solid, the struc-
ture of which was confirmed by single-crystal X-ray analysis.The completion of the target compound ent-4 from this point
involved hydrolysis[19] of the acetonide residue within com-pound 32 using acidified AG-50W-X8 resin then pyridinium
para-toluenesulfonate (PPTS).[20] Oxidation of the resulting diol33 (63 % or 82 %, based on recovered starting material) withthe Ley–Griffith reagent[21] then gave compound ent-4 (52 %).
All the spectral data acquired on compound ent-4 were incomplete accord with the assigned structure. In addition, thespecific rotation of this material was of similar magnitude butopposite sign to that reported for rezishanone C[22] while the1H and 13C NMR data recorded on the synthetic materialmatched those reported by Bringmann,[3a] Lee,[3b] Laatsch,[3c]
and Kçnig[3d] for the title isolate (see the Supporting Informa-tion for tabulated comparisons). The studies detailed aboveclearly indicate that rezishanone C has the illustrated structure
and not that originally depicted by Bringmann (structure 1).[3a]
Furthermore, the materials subsequently isolated by others are
one and the same compound (and not, for example, the 7R-epimeric form of 2). This outcome, when considered in con-
junction with the theoretical calculations described above,
adds weight to the hypothesis that rezishanone C is an artefactof the isolation process and arises as detailed above.[3a] The
present work also emphasizes the unique capacity of total syn-thesis to definitively establish/re-assign the structures of com-
pounds isolated from natural sources.[23]
Experimental Section
All computational methods, the derived data and the ensuing anal-yses are provided in the Supporting Information, as are the proce-dures for the preparation of all new compounds and the associat-ed spectroscopic and analytical data. Copies of the NMR spectra ofnew compounds, and X-ray plots and data for compounds 7, 8, 9,11, 14, 16, 17, 22, 26, 30 and 32 are also included in the SI.
Acknowledgements
We thank the Australian Research Council for financial support
through grants DP150101947 and CE140100012. QY is thegrateful recipient of PhD Scholarship provided by the CSC of
the People’s Republic of China. We are grateful to ProfessorBringmann (University of Werzburg) for kindly providing
copies of the various spectra recorded on sorbivinetone (re-zishanone C).
Conflict of interest
The authors declare no conflict of interest.
Keywords: density functional calculations · Diels–Alder ·natural products · sorbicillins · total synthesis
[1] For examples of recently reported sorbicillins see: a) T. El-Elimat, H. A.Raja, M. Figueroa, S. M. Swanson, J. O. Falkinham, III, D. M. Lucas, M. R.Grever, M. C. Wani, C. J. Pearce, N. H. Oberlies, J. Antibiot. 2015, 68, 191,b) M.-M. Zhai, F.-M. Qi, J. Li, C.-X. Jiang, Y. Hou, Y.-P. Shi, D.-L. Di, J.-W.Zhang, Q.-X. Wu, J. Agric. Food Chem. 2016, 64, 2298.
[2] For useful points-of entry into the literature on sorbicillinoids see:a) A. M. Harned, K. A. Volp, Nat. Prod. Rep. 2011, 28, 1790, b) J. Meng, X.Wang, D. Xu, X. Fu, X. Zhang, D. Lai, L. Zhou, G. Zhang, Molecules 2016,21, 715.
[3] a) G. Bringmann, G. Lang, T. A. M. Gulder, H. Tsuruta, J. Mehlbacher, K.Maksimenja, S. Steffens, K. Schaumann, R. Stçhr, J. Wiese, J. F. Imhoff, S.Perov&c-Ottstadt, O. Boreiko, W. E. G. Meller, Tetrahedron 2005, 61, 7252,b) D. Lee, J. H. Lee, X. F. Cai, J. C. Shin, K. Lee, Y.-S. Hong, J. J. Lee, J. Anti-biot. 2005, 58, 615, c) R. P. Maskey, I. Gren-Wollny, H. Laatsch, J. Nat.Prod. 2005, 68, 865, d) K. Neumann, A. Abdel-Lateff, A. D. Wright, S. Keh-raus, A. Krick, G. M. Kçnig, Eur. J. Org. Chem. 2007, 2268.
[4] In principle the dienophile ethyl vinyl ether could add to either the a-or b-face of the diene 3, in either an exo- or endo-mode and in eitheran ortho- or para-like manner.
[5] See D. J.-Y. D. Bon, M. G. Banwell, J. S. Ward, A. C. Willis, Tetrahedron2013, 69, 1363and references therein.
[6] For representative recent examples of studies on the synthesis of sorbi-cillinoids see: a) K. A. Volp, D. M. Johnson, A. M. Harned, Org. Lett. 2011,13, 4486, b) C. Qi, T. Qin, D. Suzuki, J. A. Porco, Jr. , J. Am. Chem. Soc.2014, 136, 3374.
[7] For reviews on cis-1,2-dihydrocatechols see: a) R. A. Johnson, Org. React.2004, 63, 117, b) T. Hudlicky, J. W. Reed, Synlett 2009, 685, c) S. E. Lewis,Chem. Commun. 2014, 50, 2821.
[8] T. Hudlicky, H. Luna, G. Barbieri, L. D. Kwart, J. Am. Chem. Soc. 1988, 110,4735.
[9] E. G. Mackay, C. G. Newton, Aust. J. Chem. 2016, 69, 1365.[10] X-ray analysis data for this compound are provided in the SI.[11] R. E. Ireland, L. Liu, J. Org. Chem. 1993, 58, 2899.[12] A. Pll, A. Closson, H. Adolfsson, J.-E. B-ckvall, Adv. Synth. Catal. 2003,
345, 1012.[13] For a related sequence see: M. G. Banwell, P. Darmos, M. D. McLeod,
D. C. R. Hockless, Synlett 1998, 897.
Scheme 5. A successful C-acylation reaction and completion of the synthesisof ent-rezishanone C (ent-4). [a] Based on recovered starting material.
Chem. Asian J. 2017, 12, 1480 – 1484 www.chemasianj.org T 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1483
Communication

[14] D. H. R. Barton, S. W. McCombie, J. Chem. Soc. Perkin Trans. 1 1975,1574.
[15] C. Chatgilialoglu, D. Griller, M. Lesage, J. Org. Chem. 1988, 53, 3641.[16] K. Horita, T. Yoshioka, T. Tanaka, Y. Oikawa, O. Yonemitsu, Tetrahedron
1986, 42, 3021.[17] Compound 23 can be converted into ketone 30 using a three-step re-
action sequence. See the Supporting Information for details.[18] For a related conversion see G. Franck, K. Brçdner, G. Helmchen, Org.
Lett. 2010, 12, 3886.[19] Only this two-stage hydrolysis process produced good yields of diol 33.[20] M. Miyashita, A. Yoshokoshi, P. A. Grieco, J. Org. Chem. 1977, 42, 3772.[21] S. V. Ley, J. Norman, W. P. Griffith, S. P. Marsden, Synthesis 1994, 639.
[22] The CD spectrum of compound ent-4 is provided in the Supporting In-formation and is essentially a “mirror image” to that of rezishanone C,kindly provided to us by Professor Bringmann.
[23] a) K. C. Nicolaou, S. A. Snyder, Angew. Chem. Int. Ed. 2005, 44, 1012;Angew. Chem. 2005, 117, 1036, b) M. E. Maier, Nat. Prod. Rep. 2009, 26,1105.
Manuscript received: March 26, 2017Revised manuscript received: April 28, 2017
Accepted manuscript online: April 28, 2017
Version of record online: June 5, 2017
Chem. Asian J. 2017, 12, 1480 – 1484 www.chemasianj.org T 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1484
Communication


Supporting Information
Establishing the True Structure of the Sorbicillinoid-DerivedIsolate Rezishanone C by Total Synthesis
Qiao Yan,[a] Martin G. Banwell,*[a] Michelle L. Coote,[a, b] Richmond Lee,[a, b] andAnthony C. Willis[a]
asia_201700456_sm_miscellaneous_information.pdf

! S1!
SUPPORTING INFORMATION FOR:
Establishing the True Structure of the Sorbicillinoid-derived Isolate Rezishanone C by Total Synthesis
Qiao Yan,a Martin G. Banwell,*, a Michelle L. Coote,a,b Richmond Lee a,b and Anthony C. Willis a
a Research School of Chemistry, Institute of Advanced Studies The Australian National University, Canberra, ACT 2601, Australia
b ARC Centre of Excellence for Electromaterials Science
Part A: Computational Studies S2
1. General Approach and Outcomes
2. Total Energies of Species
3. Gaussian Archive Entries for All Species
!Part B: Experimental Studies S8
4. General Experimental Procedures
5. Specific Chemical Transformations
6. Comparison of the 13C NMR Data Derived From Rezishanone C with
those Recorded on ent-Rezishanone C
7. CD Spectrum of Compound ent-4
8. Data Associated with Single-crystal X-ray Analyses of Compounds
7, 8, 9, 11, 14, 16, 17, 22, 26, 30 and 32
9. References
10. ORTEPs Derived from Single-crystal X-ray Analyses of Compounds
7, 8, 9, 11, 14, 16, 17, 22, 26, 30 and 32
11. 1H and 13C NMR Spectra of Compounds ent-4 and 7-35

! S2!
Part A: Computational Studies
1. General Approach and OutcomesAll quantum chemical calculations were carried out with Gaussian 09.1 The activationfree energy barriers in solution (ΔG‡) are reported relative to the individual freestarting materials: the diene and dienophile. The [4+2]-cycloaddition transition state(TS) electronic structures were optimized with the meta-hybrid functional M06-2X2
in conjunction with Grimme’s empirical dispersion correction D3 (zero damping),3herein termed M06-2X(D3), using Pople’s4 6-31G(d,p) basis set and the SMD5
polarizable continuum solvent model under ethyl acetate (EA) parameters. Theultrafine integral grid was selected during the optimization calculations to ensureaccuracy in the computed vibrational frequencies. Frequency calculations at 298Kbased on harmonic oscillator approximation were performed at the same level oftheory to verify that all TSs had one imaginary frequency and to compute thevibrational contributions to the entropies.Figure S1 shows the various possible transition structures for the [4+2]-cycloadditionreaction between sorbicillinol (3) and ethyl vinyl ether. We also endeavored to searchfor a step-wise [4+2] pathway but only the concerted one was found. From Figure S1it is seen that the lowest energy transition structure (designated here as TScDA-1aS)is that leading to ent-rezishanone C (ent-4). The next lowest transition structure issome 2.5 kcal mol–1 higher in energy and would thus not contribute significantly tothe reaction. As shown in Figure S1, the reason for this preference is predominantlysteric.
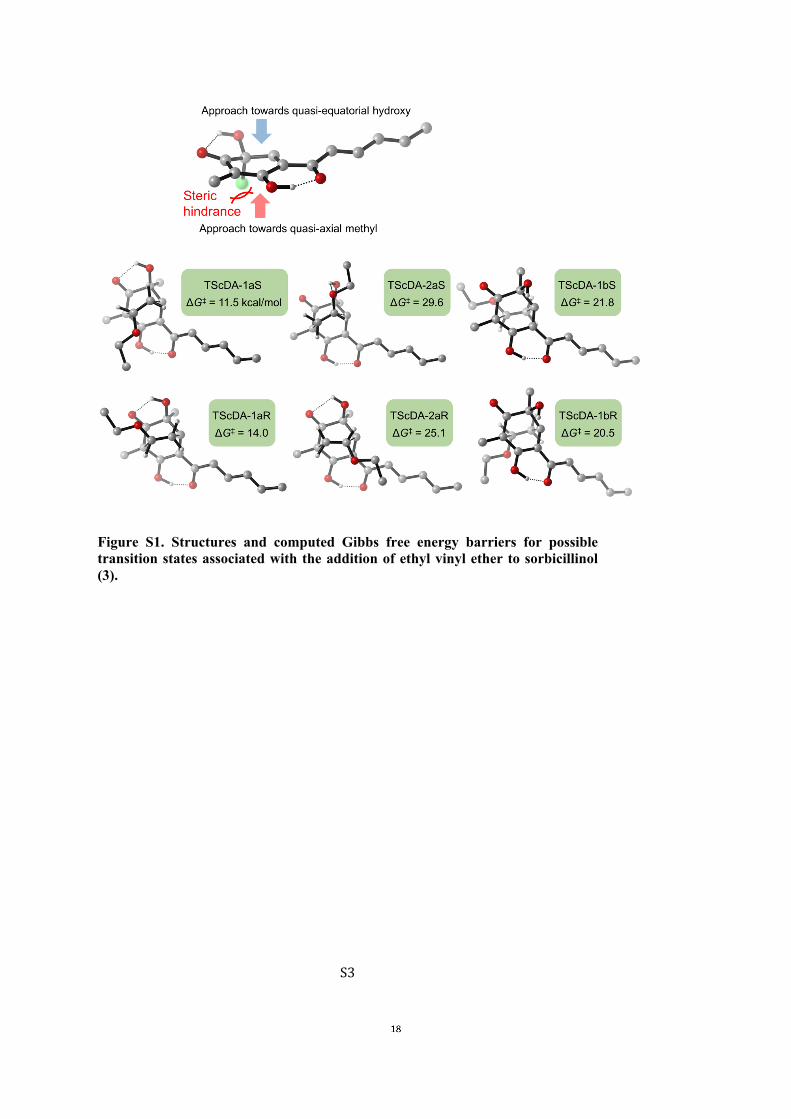
! S3!
Figure S1. Structures and computed Gibbs free energy barriers for possible transition states associated with the addition of ethyl vinyl ether to sorbicillinol (3).

! S4!
2. Total Energies of Species
Table S1. Energies and correctionsSpecies Ee Gcorrection Hcorrection ZPE G H Ee + ZPE Diene + dienophile -1075.98901 0.31310 0.41684 0.39008 -1075.67592 -1075.57217 -1075.59894
TScDA-1aS -1075.99632 0.33880 0.41802 0.39268 -1075.65751 -1075.57829 -1075.60363
TScDA-1aR -1075.99202 0.33845 0.41780 0.39233 -1075.65357 -1075.57422 -1075.59969
TScDA-2aS -1075.96614 0.33732 0.41765 0.39216 -1075.62882 -1075.54848 -1075.57398
TScDA-2aR -1075.97559 0.33967 0.41762 0.39249 -1075.63592 -1075.55797 -1075.58310
TScDA-1bS -1075.97955 0.33837 0.41799 0.39237 -1075.64118 -1075.56156 -1075.58718
TScDA-1bR -1075.98234 0.33910 0.41848 0.39305 -1075.64324 -1075.56386 -1075.58929All energy values are in hatrees. Ee is the electronic energy of the species. The energy corrections Gcorrection and Hcorrection and Zero Point Energy (ZPE) are added to Ee to constitute Gibb’s free energy (G), enthalpy (H) and the Ee + ZPE respectively.
3. Gaussian Archive Entries for All Speciesdiene 1\1\GINC-CA139\FOpt\RM062X\6-31G(d,p)\C14H16O4\ROOT\25-Nov-2015\0\\# m 062x/6-31G** opt=(maxcyc=200) scf=maxcyc=200 int=ultrafine freq # empi ricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate)\\Diene\\0,1\H,0. 9118471652,0.7607602863,1.7283129434\C,0.1551268325,0.1116187999,1.298 5048231\C,0.4098413776,-0.7043179035,0.2609841291\C,-0.6843127873,-1.5 701009665,-0.2593218829\C,-1.8943788889,-1.6900160766,0.362866934\C,1. 7561279323,-0.7650692082,-0.3825210329\C,2.905566831,-0.1254867215,0.2 649600274\H,2.7976888363,0.3088998433,1.2514618313\C,4.1004276879,-0.1 013309942,-0.3578786624\H,4.1782986604,-0.5525737361,-1.3464052082\C,5 .2976098793,0.4889509388,0.2007347235\H,5.229474379,0.9362420702,1.191 5679964\C,6.4637859718,0.4956193515,-0.4638072003\H,6.498005103,0.0395 895755,-1.4535924069\O,1.8887362015,-1.3599686844,-1.4605970159\C,-1.2 191827129,0.2671202915,1.86026615\C,-2.134988324,-0.9245279383,1.56506 37885\O,-3.0760467599,-1.1068489135,2.3323034454\O,-1.1434422387,0.476 8534779,3.2488645514\H,-1.9821446651,0.1243791629,3.5914649959\C,-1.86 69261517,1.4924337158,1.1722677981\H,-1.9374648635,1.3558912191,0.0902 926179\H,-2.871920573,1.621884268,1.5846551066\H,-1.2744295152,2.38401 02505,1.3903003518\O,-0.4522564725,-2.2990532726,-1.3471090501\H,0.467 6303633,-2.0846847507,-1.6565792445\C,-2.9195900933,-2.6763413524,-0.1 148552616\H,-3.8697935749,-2.49169118,0.3888882417\H,-3.065313454,-2.5 956258926,-1.1954655369\H,-2.6185656121,-3.7083702328,0.0950921827\C,7 .733296543,1.0848245403,0.0507555614\H,8.0961179432,1.8667433365,-0.62 55489385\H,7.6030492583,1.5154443712,1.0461209855\H,8.5191257616,0.322 7423047,0.0969522558\\Version=AS64L-G09RevD.01\State=1-A\HF=-843.66271 3\RMSD=4.198e-09\RMSF=2.771e-06\Dipole=2.8977284,1.4239484,-0.4058954\ Quadrupole=10.8581703,-2.6167901,-8.2413802,0.8479145,2.6773196,-0.781 7147\PG=C01 [X(C14H16O4)]\\@
dienophile 1\1\GINC-CA138\FOpt\RM062X\6-31G(d,p)\C4H8O1\ROOT\25-Nov-2015\0\\# m06 2x/6-31G** opt=(maxcyc=200) scf=maxcyc=200 int=ultrafine freq # empiri caldispersion=gd3 scrf=(smd,solvent=EthylEthanoate)\\Dienophile\\0,1\C ,-1.403718679,1.3926371956,-1.4945288669\C,-0.3521507403,1.6984085923, -0.7397477275\O,-1.3479173941,0.4578558643,-2.4731676641\C,-2.5684537044,0.2581938115,-3.1845290428\H,-3.3561482552,-0.0500943017,-2.4846590767\H,-2.8799109149,1.2010179362,-3.6529290445\C,-2.3302387688,-0.8078496772,-4.2290837283\H,-3.2476550637,-0.9873588261,-4.7954533259\H,-1.5478534861,-0.4953426323,-4.925270607\H,-2.0237957624,-1.7452903641,-3.7581671996\H,-0.4453472634,2.4529434227,0.0308176543\H,0.603549061,1.205139319,-0.8799616079\H,-2.3703590188,1.8807396697,-1.3643197731\\Version=AS64L-G09RevD.01\State=1-A\HF=-232.3263002\RMSD=5.907e-09\RMSF=4.863e-06\Dipole=-0.8227371,0.0521043,-0.2791994\Quadrupole=0.4241394,-0.363871,-0.0602684,-0.2959503,0.5605454,1.3506336\PG=C01 [X(C4H8O1)]\\@
TScDA-1aS 1\1\GINC-CA043\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\24-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=maxcyc=200 int=ultr afine freq # empiricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate)

! S5!
\\TS Diels-Alder concerted to product regio 1a (top) - S isomer\\0,1\H ,-0.3360018093,0.8635709343,1.0759069627\C,0.3938925775,0.2155934066,0 .5985407536\C,-0.0302969441,-1.0181731854,0.0757697514\C,0.9988445727, -1.9403480819,-0.3773111648\C,2.3525674673,-1.6506332187,-0.30483666\C ,-1.4138960889,-1.3000677958,-0.2562124002\C,-2.4535020226,-0.27950286 17,-0.029928811\H,-2.1789082757,0.6896542077,0.3720740718\C,-3.7386000 503,-0.5364427277,-0.3336660389\H,-3.9853950902,-1.5181452884,-0.73591 82707\C,-4.8233184446,0.4100083431,-0.1607059355\H,-4.577119261,1.3932 385886,0.2398286956\C,-6.0932857188,0.1139683683,-0.4741801392\H,-6.31 07825326,-0.877337855,-0.8728796928\O,-1.7465676254,-2.4062449777,-0.7 482651432\C,1.7499303742,0.2550906319,1.2606334497\C,2.7849324833,-0.4 873395465,0.4144272336\O,3.9499276272,-0.0799709649,0.462343303\O,2.19 09793816,1.5695963025,1.4857945363\H,3.1520872146,1.5183398354,1.33196 12769\C,1.6414725157,-0.4687950078,2.6207392488\H,1.31961288,-1.505997 9784,2.5097698247\H,0.9311303507,0.0654526961,3.2571071766\H,2.6251905 501,-0.4460030627,3.0982863918\O,0.6226342871,-3.0722984239,-0.9693841 267\H,-0.3899407908,-3.055988901,-0.9682497922\C,3.4015376009,-2.52430 18452,-0.9316563425\H,4.0188434557,-3.0137160319,-0.1696728965\H,4.081 5649454,-1.9272542069,-1.5478671207\H,2.9493113419,-3.2993322879,-1.55 20671276\C,-7.2492185457,1.0454423795,-0.3187862649\H,-7.7441581538,1. 2113575223,-1.2821760798\H,-6.9354354221,2.0118206408,0.0830407281\H,- 8.0039301011,0.6174711349,0.3504318918\C,1.2353286636,0.7503347251,-1. 9263227994\H,2.3083574265,0.6917451197,-2.1077327896\C,0.6597459832,1. 5533288143,-0.9758682852\O,0.4523494839,-0.064879729,-2.6032150334\C,1 .0817363568,-1.0116392029,-3.4920610542\H,1.7964137338,-1.6047701963,- 2.9120832566\H,1.6206483805,-0.4548574138,-4.2653637952\C,-0.014409887 2,-1.8714911824,-4.0707156754\H,0.4210562706,-2.6381152325,-4.71585566 23\H,-0.7097691065,-1.2700838291,-4.6611719414\H,-0.5664258695,-2.3630 492839,-3.2661015286\H,1.2661312121,2.305271334,-0.4886372151\H,-0.408 2653969,1.730495334,-1.061762253\\Version=AS64L-G09RevD.01\State=1-A\H F=-1075.9963159\RMSD=6.982e-09\RMSF=2.996e-06\Dipole=-1.8882506,1.2712 682,-1.1050708\Quadrupole=3.3414481,-6.9803872,3.6389391,-5.527101,-7. 4171643,-0.2596998\PG=C01 [X(C18H24O5)]\\@ TScDA-1aR 1\1\GINC-CA075\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\24-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=maxcyc=200 int=ultr afine freq # empiricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate) \\TS Diels-Alder concerted to product regio 1a (top) - R isomer\\0,1\H ,-0.2808218816,0.8374100752,0.7161396023\C,0.3629562437,0.0518649679,0 .3299677469\C,-0.1661440944,-1.2353008761,0.1515860946\C,0.7650918749, -2.2875177723,-0.2051919477\C,2.132700073,-2.0830851502,-0.3348617485\ C,-1.5921883209,-1.5034036526,0.0834644974\C,-2.5633616348,-0.40482929 6,0.2380700857\H,-2.2136868132,0.6042260823,0.4264809322\C,-3.88360922 24,-0.6435438637,0.1346435052\H,-4.2090702284,-1.6653847484,-0.0564565 338\C,-4.9090022107,0.3731204319,0.2634742715\H,-4.5865010347,1.395376 9065,0.4602248406\C,-6.2158433915,0.0919435032,0.1536531602\H,-6.50840 26487,-0.9400206716,-0.0426851032\O,-2.0149226526,-2.6655880801,-0.120 1286165\C,1.7899481358,0.1640155324,0.8032240334\C,2.7032452102,-0.841 1004296,0.0924966945\O,3.8956796206,-0.5283229877,-0.0112956045\O,2.28 97202989,1.463300294,0.6289455461\H,3.192358417,1.3296259233,0.2887736 006\C,1.814356583,-0.1802669627,2.3092648361\H,1.4424582254,-1.1875938 018,2.5079077284\H,1.2085048245,0.5454840458,2.8581833293\H,2.84982711 83,-0.104059475,2.6520878896\O,0.2943572623,-3.4972155654,-0.488335685 9\H,-0.7071510333,-3.4324578576,-0.3824197428\C,3.0176249211,-3.187075 0138,-0.840886778\H,4.0543731655,-2.8450869077,-0.8532723121\H,2.73846 00094,-3.5045033044,-1.8511335719\H,2.9522512998,-4.0738124709,-0.2013 591944\C,-7.317194298,1.0906908614,0.2824848953\H,-7.9163063488,1.1264 797116,-0.6342725816\H,-6.9299831593,2.0920804841,0.4851209509\H,-8.00 07502954,0.8097215858,1.0913599749\C,1.4364324964,0.3877920328,-2.2308 100552\C,0.4475393615,1.0262397564,-1.5269095603\O,2.6712568123,0.8553 901418,-2.2743281685\C,3.6296393969,0.0224989552,-2.9593464361\H,3.314 8714589,-0.071011439,-4.0047430794\H,3.6232836737,-0.9665153267,-2.489 7711368\C,4.9803788662,0.6812685738,-2.8372391453\H,5.7289906779,0.077 6707991,-3.3562879375\H,5.2598008612,0.7591360097,-1.784676422\H,4.967 6106593,1.6782597053,-3.2847079468\H,0.6574894312,2.0163438988,-1.1387 795272\H,-0.5764666662,0.7747136602,-1.7743612085\H,1.2421989559,-0.52 99582853,-2.7872941712\\Version=AS64L-G09RevD.01\State=1-A\HF=-1075.99 20184\RMSD=7.657e-09\RMSF=1.702e-06\Dipole=-1.65978,1.4327332,-1.07215 62\Quadrupole=10.922113,-12.7958623,1.8737493,-7.4666513,-8.841663,0.4 559919\PG=C01 [X(C18H24O5)]\\@ TScDA-2aS 1\1\GINC-CA043\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\24-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=maxcyc=200 int=ultr afine freq # empiricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate) \\TS Diels-Alder concerted to product regio 2a (top) - R isomer\\0,1\H

! S6!
,-0.2334157103,0.984924462,0.9043012491\C,0.4617064942,0.2140744917,0. 5836421072\C,-0.0237956745,-1.0603561504,0.256765379\C,0.9513705982,-2 .0163415054,-0.1500611421\C,2.2946418905,-1.6430314449,-0.2913488788\C ,-1.4449709658,-1.3666993089,0.0573863635\C,-2.4563781931,-0.309229151 ,0.2193882952\H,-2.1471283642,0.693266668,0.4927604145\C,-3.7605361133 ,-0.579852946,0.0249531255\H,-4.0404863313,-1.5969398597,-0.2472674486 \C,-4.8249603005,0.3936995115,0.1596881191\H,-4.5499864824,1.410906895 4,0.4372553821\C,-6.113407585,0.0775990238,-0.0395567645\H,-6.35794903 ,-0.948515975,-0.3158699645\O,-1.7982740502,-2.519559097,-0.2564522708 \C,1.8076410291,0.2914755529,1.2775913548\C,2.8376584666,-0.5323045893 ,0.5031129324\O,4.026930742,-0.2699600356,0.5737712011\O,2.2440106931, 1.6059661505,1.5051138394\H,2.5692902531,1.9445704665,0.6580757112\C,1 .6805890131,-0.3425213551,2.6774987344\H,1.3701430259,-1.387044887,2.6 294482273\H,0.9506091682,0.2243635089,3.2608181359\H,2.6557603494,-0.2 726933582,3.1656820331\O,0.5964548791,-3.221163262,-0.6067327839\H,-0. 3979106636,-3.2497957958,-0.5425318801\C,3.2564302843,-2.5684717373,-0 .9721970097\H,4.2287028812,-2.0831955028,-1.0683577055\H,2.8919597365, -2.8570124023,-1.9623716985\H,3.3873596396,-3.4901836394,-0.3948170609 \C,-7.2543513399,1.0298763453,0.0913279926\H,-7.8008775108,1.110854010 1,-0.8549315914\H,-6.9151149899,2.0260487239,0.3849013215\H,-7.9726582 998,0.6707150982,0.8366173075\H,2.8873572777,0.1717019714,-2.067858516 4\C,1.8312132541,0.0096936124,-1.8757463856\H,1.3431592576,-0.78491151 09,-2.4243688178\C,1.0690155919,1.043233894,-1.3589254951\H,0.01855610 88,1.1482023892,-1.6214774957\O,1.7244507577,2.2240381248,-1.082364266 3\C,0.8681924524,3.3012930383,-0.6999317985\H,0.3073454364,3.023988349 5,0.2011037351\H,0.1515757817,3.4909595323,-1.509393998\C,1.7328741804 ,4.5141253271,-0.4418950185\H,1.1038684593,5.3711833628,-0.1890844363\ H,2.3213034466,4.7624721966,-1.3283797674\H,2.4157124558,4.3342248069, 0.3933622333\\Version=AS64L-G09RevD.01\State=1-A\HF=-1075.9661372\RMSD =6.439e-09\RMSF=7.305e-07\Dipole=-3.1134839,1.7603694,-0.6959952\Quadr upole=3.1253256,-1.1532915,-1.9720342,-5.6372889,-4.5842957,-2.613618\ PG=C01 [X(C18H24O5)]\\@ TScDA-2aR 1\1\GINC-CA137\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\25-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=(novaracc,maxcyc=20 0) int=ultrafine freq # empiricaldispersion=gd3 scrf=(smd,solvent=Ethy lEthanoate) iop(1/8=1)\\TS Diels-Alder concerted to product regio 2a ( top) - R isomer\\0,1\H,-0.0180103034,-1.5682696255,0.564218462\C,0.778 0838345,-0.9332659451,0.1835552758\C,0.4685524271,0.0558675286,-0.7629 918252\C,1.5441448639,0.8957167772,-1.1758998934\C,2.8512051615,0.6654 349331,-0.7256978845\C,-0.9070874712,0.4477426406,-1.093546517\C,-2.03 13190961,-0.4280717448,-0.7174307424\H,-1.8275099462,-1.4273754948,-0. 3479185352\C,-3.3004414421,0.0062687658,-0.8291709962\H,-3.4690911787, 1.013464643,-1.2094381019\C,-4.4642846376,-0.7733984945,-0.4572436682\ H,-4.2971794632,-1.7830934807,-0.0830067768\C,-5.7122239374,-0.2900971 896,-0.5509094579\H,-5.8478908027,0.7236679927,-0.9286954238\O,-1.1309 77351,1.5013832054,-1.7198106878\C,2.0972685434,-1.6401992659,0.053742 547\C,3.2234812779,-0.659419549,-0.2640621421\O,4.3798904086,-1.036051 3916,-0.0889662642\O,2.409517802,-2.302326085,1.261689286\H,3.37181420 44,-2.4315282289,1.2299877623\C,2.0200428532,-2.6563317827,-1.09954498 86\H,1.8036023705,-2.1646279376,-2.0514700744\H,1.2416953873,-3.394226 0437,-0.8895913164\H,2.9845936731,-3.1668296321,-1.1776310047\O,1.3233 72414,1.9971003136,-1.8952288212\H,0.3304151036,2.0669257621,-1.980564 1925\C,3.9422397938,1.6530584377,-1.0043494189\H,4.3392819473,1.525946 9583,-2.0180976504\H,4.7674993411,1.5001402909,-0.3057190349\H,3.57336 46719,2.6771389724,-0.9193815518\C,-6.946799277,-1.0381877637,-0.17300 81827\H,-7.5021824247,-0.4975260643,0.6014334492\H,-6.7139199226,-2.03 87519193,0.1987567056\H,-7.6206855954,-1.132052987,-1.0317589386\H,2.4 690243224,1.8624639471,1.4918273068\C,2.3616235413,0.7953848817,1.6514 27164\H,3.2401646869,0.2363705924,1.9478127073\C,1.1067199935,0.259428 9233,1.8793619793\O,0.0623441918,1.1447307193,1.8642393377\C,-1.164030 8669,0.630426579,2.3706064223\H,-1.0445533031,0.3804807166,3.433822967 \H,-1.4325856451,-0.2917290732,1.8379644963\C,-2.2262277772,1.68789014 15,2.1733299993\H,-3.1990273622,1.3113245893,2.5002679909\H,-2.2951498 303,1.9606647489,1.116556026\H,-1.9871105178,2.5867617495,2.7471146332 \H,1.0057113368,-0.6263721111,2.5055675737\\Version=AS64L-G09RevD.01\S tate=1-A\HF=-1075.9755891\RMSD=9.470e-09\RMSF=2.574e-06\Dipole=-2.6951 235,-0.9435248,1.0622776\Quadrupole=7.7660361,-4.0201536,-3.7458825,4. 5393899,-0.174139,3.2361566\PG=C01 [X(C18H24O5)]\\@ TScDA-1bS 1\1\GINC-CA040\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\24-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=maxcyc=200 int=ultr afine freq # empiricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate) \\TS Diels-Alder concerted to product regio 1b (bottom) - S isomer\\0, 1\H,-0.3078639866,0.9212968229,-0.6614787015\C,0.346327319,0.129665527

! S7!
8,-0.3068050617\C,-0.1667809998,-1.1662038922,-0.179004716\C,0.7822571 492,-2.2197921041,0.146271712\C,2.1428342735,-2.0032191959,0.272067407 2\C,-1.5908352518,-1.4559943421,-0.1335403755\C,-2.5755158501,-0.36838 13315,-0.2799838837\H,-2.2390271304,0.6429325154,-0.4788120119\C,-3.89 2095816,-0.6200857982,-0.1614546254\H,-4.2056000855,-1.643923471,0.038 7719098\C,-4.9282655554,0.3862528834,-0.2850178562\H,-4.6171505354,1.4 101533764,-0.4913308099\C,-6.2313340251,0.094117881,-0.1601776528\H,-6 .5126585838,-0.9391941033,0.0452683836\O,-1.9997765032,-2.6271405933,0 .040749567\C,1.7794612898,0.2790415889,-0.799453148\C,2.7190383003,-0. 7341351494,-0.115990555\O,3.9145217434,-0.4681053299,-0.0123994427\O,1 .7369777076,-0.0380556199,-2.1965840837\H,1.4001214317,-0.9423564126,- 2.2824330102\C,2.3312880523,1.6887560741,-0.7267563719\H,2.5819050933, 1.9693034578,0.2958541803\H,3.2450130398,1.727120327,-1.3208678944\H,1 .6069484127,2.3957259506,-1.1417668994\O,0.323743201,-3.4455679812,0.3 894179514\H,-0.6764548547,-3.3897012087,0.2876353667\C,3.0412961135,-3 .1244485497,0.7133883517\H,4.0720394769,-2.7679282799,0.7556076849\H,2 .7578252221,-3.5101785409,1.6983676243\H,2.995387401,-3.9705760393,0.0 189788962\C,-7.3428042518,1.0822507333,-0.2842019741\H,-7.9332041825,1 .118373204,0.6381783833\H,-6.966582747,2.0857891165,-0.4967347502\H,-8 .0316139683,0.7902779777,-1.0846653462\C,1.4018607191,0.3160062581,2.2 894913703\C,0.4010543782,0.986820406,1.6317885072\O,2.6131679321,0.827 5330258,2.4088620558\C,3.6135730474,-0.0336608,2.9945546946\H,3.342666 7688,-0.2031916854,4.0424064808\H,3.603672339,-0.9876111238,2.45775450 17\C,4.945191205,0.6603784208,2.8587852385\H,5.7281085507,0.0376624046 ,3.2986751924\H,5.169605698,0.8165267009,1.8017691994\H,4.9355475738,1 .6230841456,3.3759934378\H,0.5598354566,2.027733703,1.3684201019\H,-0. 6171055261,0.677059521,1.8290744046\H,1.2371197578,-0.6636164103,2.738 6785169\\Version=AS64L-G09RevD.01\State=1-A\HF=-1075.9795462\RMSD=6.42 0e-09\RMSF=7.828e-06\Dipole=-2.1053653,1.6209969,1.5705496\Quadrupole= 8.2193915,-7.5404825,-0.6789089,-7.3337323,10.9378237,0.1989229\PG=C01 [X(C18H24O5)]\\@ TScDA-1bR 1\1\GINC-CA134\FTS\RM062X\6-31G(d,p)\C18H24O5\ROOT\25-Nov-2015\0\\# m0 62x/6-31G** opt=(ts,calcfc,noeigen,maxcyc=200) scf=maxcyc=200 int=ultr afine freq # empiricaldispersion=gd3 scrf=(smd,solvent=EthylEthanoate) \\TS Diels-Alder concerted to product regio 1b (bottom) - R isomer\\0, 1\H,0.648741236,0.9206698462,1.4398249904\C,-0.1227036707,0.3654075069 ,0.9127535266\C,0.246031723,-0.7823900162,0.2002493568\C,-0.8207376588 ,-1.673083453,-0.2412368707\C,-2.1591893075,-1.4476564118,0.012528868\ C,1.6064225369,-1.0348099093,-0.2403276845\C,2.6784819885,-0.069112784 1,0.067607522\H,2.4575165342,0.8152205469,0.6554994789\C,3.9277863214, -0.2710362697,-0.3892739531\H,4.1208742986,-1.1657189228,-0.9798240368 \C,5.040549773,0.6273951995,-0.150994881\H,4.8511204859,1.5214212801,0 .4427582557\C,6.269610412,0.3917206434,-0.6334495888\H,6.4291940197,-0 .5109414537,-1.2239606381\O,1.8893391324,-2.0645588213,-0.8967164803\C ,-1.4439230073,0.3385703645,1.6749836996\C,-2.5625631954,-0.3560315485 ,0.8682600844\O,-3.7325731761,-0.0339170681,1.0640176287\O,-1.18876925 99,-0.4549287528,2.8404929838\H,-0.9385331112,-1.3414612845,2.54021990 61\C,-1.905845668,1.6827008765,2.2036437851\H,-2.3247368131,2.30828222 77,1.4140637987\H,-2.692088494,1.5058930541,2.9379985775\H,-1.07571619 43,2.2068396816,2.6860748954\O,-0.4921393248,-2.7510123225,-0.95272221 76\H,0.5106146836,-2.7071292895,-1.0700425263\C,-3.2061501579,-2.35208 81419,-0.5746825774\H,-4.1813507729,-1.8651985264,-0.5113892264\H,-2.9 895317705,-2.5911360035,-1.6207194314\H,-3.2709639727,-3.3056079571,-0 .037350349\C,7.4522195496,1.2782745345,-0.4262046548\H,7.8563307482,1. 6146982944,-1.387478721\H,7.1960497283,2.1568485046,0.1707493697\H,8.2 587248625,0.7355768502,0.0792997611\C,-1.2887971706,1.4403472798,-1.26 44221993\C,-0.3153778657,1.9480525362,-0.4468198474\O,-0.9484935791,0. 6320533017,-2.2463998591\C,-2.036094532,0.0566459815,-3.003783507\H,-2 .8065607109,-0.275065665,-2.2998326527\H,-2.449422039,0.8416841198,-3. 645013324\C,-1.4844635054,-1.0941471397,-3.8081972453\H,-2.2901216279, -1.54443046,-4.3939518863\H,-0.7083222533,-0.749298436,-4.4960343522\H ,-1.0635495611,-1.8543233509,-3.1456622\H,-0.5712175715,2.7558274146,0 .2263330179\H,0.7141797497,1.8859123524,-0.7845953505\H,-2.3528518116, 1.6240415913,-1.108273245\\Version=AS64L-G09RevD.01\State=1-A\HF=-1075 .9823391\RMSD=7.808e-09\RMSF=2.441e-06\Dipole=2.0783348,2.2219918,-1.3 505933\Quadrupole=3.7475697,-0.4740664,-3.2735033,2.4713957,12.1393436 ,-0.610344\PG=C01 [X(C18H24O5)]\\@

! S8!
Part B: Experimental Studies
4. General Experimental Procedures
Unless otherwise specified, proton (1H) and carbon (13C) NMR spectra were recorded
at 18 °C in base-filtered CDCl3 on a spectrometer operating at 400 MHz for proton
and 100 MHz for carbon nuclei. 1H NMR data are recorded as follows: chemical shift
(δ) [multiplicity, coupling constant(s) J (Hz), relative integral] where multiplicity is
defined as s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet or
combinations of the above. In relevant cases, the signal due to residual CHCl3
appearing at δH 7.26 and the central resonance of the CDCl3 “triplet” appearing at δC
77.0 were used to reference 1H and 13C NMR spectra, respectively. Samples were
analyzed by infrared spectroscopy (νmax) as thin films on KBr plates. Optical rotations
were recorded using the sodium D-line (589 nm) in a cell with a path length of 1 dm,
at the concentrations indicated and in the specified solvent at 22 °C. Specific rotations
were then calculated in the usual manner. Low- and high-resolution electron impact
(EI) mass spectra were recorded on a double focusing, triple sector machine. Low-
and high-resolution ESI mass spectra were recorded on a triple-quadrupole mass
spectrometer operating in positive ion mode. Melting points are uncorrected.
Analytical thin layer chromatography (TLC) was performed on aluminum-backed 0.2
mm thick silica gel 60 F254 plates. Eluted plates were visualized using a 254 nm UV
lamp and/or by treatment with a suitable dip followed by heating. These dips included
phosphomolybdic acid/ceric sulfate/sulfuric acid (conc.)/water (37.5 g : 7.5 g : 37.5 g
: 720 mL), potassium permanganate/potassium carbonate/5% sodium hydroxide
aqueous solution/water (3 g : 20 g : 5 mL : 300 mL), and p-anisaldehyde or
vanillin/sulfuric acid (conc.)/ethanol (15 g : 2.5 mL : 250 mL). Flash chromatographic
separations were carried out following protocols defined by Still et al.6 with silica gel
60 (40-63 µm) as the stationary phase and using the AR- or HPLC-grade solvents
indicated. The melting points of solids purified by such means were recorded directly
(ie after they had crystallized from the concentrated chromatographic fractions).
Starting materials, reagents, drying agents, and other inorganic salts were generally
commercially available and were used as supplied. Tetrahydrofuran (THF), methanol
and dichloromethane were dried using a solvent purification system that is based upon
a technology originally described by Grubbs et al.7 Where necessary, reactions were
performed under a nitrogen atmosphere.

! S9!
5. Specific Chemical Transformations
(3aR,7aS)-2,2,4-Trimethyl-3a,7a-dihydrobenzo[d][1,3]dioxole (6)
A magnetically stirred solution of commercially available diol 5 (10.00 g, 79.3 mmol)
in 2,2-dimethoxypropane (320 mL) was cooled to −10 °C then treated with p-
toluenesulfonic acid monohydrate (1.60 g, 8.0 mmol). The ensuing mixture was
stirred at −10 °C for 0.40 h then treated with triethylamine (4.0 mL, 28.6 mmol)
before being concentrated under reduced pressure (no heating) to provide a brown oil.
This oil was dissolved in diethyl ether (400 mL) and the resulting solution washed
with NaOH (1 × 200 mL of a 1 M aqueous solution). The separated aqueous phase
was extracted with diethyl ether (1 × 200 mL) and the combined organic extracts
washed with water (2 × 200 mL) before being dried (MgSO4), filtered then
concentrated under reduced pressure to afford diene 6 (10.60 g, 80%) as a pale-yellow
oil. This material, the spectral data for which matched these reported previously,8 was
used immediately in the next step of the reaction sequence.
(3aS,4R,7S)-2,2,7-Trimethyl-3a,4,7,7a-tetrahydro-4,7-ethanobenzo[d][1,3]dioxol-
8-one (7)
Step i: A magnetically stirred solution of diene 6 (5.29 g, 31.8 mmol) and α-
chloroacrylonitrile (7.62 mL, 95.4 mmol) in benzene (62 mL) maintained under a
nitrogen atmosphere was heated under reflux for 18 h then cooled and concentrated
under reduced pressure. The residue thus obtained was subjected to flash column
chromatography (silica, 0:1 → 1:24 v/v ethyl acetate/40-60 petroleum ether gradient
O
O
6
OO
O
7

! S10!
elution) to give a 4:1 mixture of epimeric and previously reported Diels-Alder
adducts.9
Step ii: A magnetically stirred solution of the mixture of adducts (3.98 g, 15.7 mmol)
obtained from Step i in t-BuOH (120 mL) was treated with potassium hydroxide (3.52
g of pellets, 62.7 mmol) and the resulting mixture heated under reflux for 15 h before
being cooled then concentrated under reduced pressure. The residue thus obtained
was diluted with ethyl acetate (200 mL) and the ensuing mixture washed with water
(1 × 200 mL). The separated aqueous phase was treated with potassium carbonate
(1.47 g, 10.6 mmol) and extracted with ethyl acetate (2 × 100 mL) then the combined
organic phases were dried (MgSO4), filtered, and concentrated under reduced
pressure. The orange oil thus obtained was subjected to flash chromatography (silica,
0:1 → 1:9 v/v ethyl acetate/40-60 petroleum ether gradient elution) to afford, after
concentration of the appropriate fractions (Rf = 0.5 in 1:4 v/v ethyl acetate/40-60
petroleum ether), ketone 79 (2.91 g, 89%) as a colorless, crystalline solid, mp = 79-80
°C (lit.9 mp = 81-82 °C). 1H NMR (400 MHz, CDCl3) δ 6.34 (t, J = 7.1 Hz, 1H), 5.72 (d, J = 8.0 Hz, 1H), 4.49
(m, 1H), 4.05 (d, J = 7.1 Hz, 1H), 3.17 (m, 1H), 2.12 (dd, J = 18.7 and 3.9 Hz, 1H),
1.88 (d, J = 18.7 Hz, 1H), 1.37 (s, 3H), 1.30 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 210.5, 133.7, 131.6, 110.9, 79.9, 79.3, 54.8, 35.6,
35.3, 25.5, 25.1, 14.5.
IR (KBr) νmax 2977, 2935, 2887, 1731, 1374, 1208, 1093, 1070 cm–1.
MS (EI, 70 eV) m/z 208 (M+•, 30%), 193 [(M−CH3•)+, 40], 150 (60), 121 (70), 108
(100), 105 (72), 100 (70).
HRMS M+• calcd for C12H16O3 208.1099, found 208.1095.
Specific rotation [α]D = +313.5 (c = 0.8, CHCl3).
(1S,4R,7S,8S)-7,8-Dihydroxy-1-methylbicyclo[2.2.2]oct-5-en-2-one (8)
A magnetically stirred solution of compound 7 (2.40 g, 11.5 mmol) in methanol/water
(126 mL of a 105:21 v/v mixture) was treated with AG-50W-X8 resin (2.86 g of H+
OH
O
OH
8

! S11!
form). The ensuing mixture was heated at 65 °C for 16 h then cooled and filtered. The
retained solids were washed with methanol (40 mL) and the combined filtrates
concentrated under reduced pressure. The residue thus obtained was diluted with ethyl
acetate (40 mL) and the separated aqueous phase extracted with ethyl acetate (2 × 40
mL). The combined organic extracts were dried (MgSO4), filtered and concentrated
under reduced pressure and the residue thus obtained subjected to flash column
chromatography (silica, 1:1 → 1:0 v/v ethyl acetate/40-60 petroleum ether gradient
elution) to give, after concentration of the appropriate fractions (Rf = 0.2 in ethyl
acetate), diol 8 (1.67 g, 86%) as a colorless, crystalline solid, mp = 122-125 °C. 1H NMR (400 MHz, CD3OD) δ 6.45 (m, 1H), 5.82 (dd, J = 7.8 and 1.1 Hz, 1H), 3.91
(s, 1H), 3.45 (s, 1H), 2.93 (broad s, 1H), 2.07 (m, 2H), 1.23 (s, 3H) (signals due to
hydroxyl group protons not observed). 13C NMR (100 MHz, CD3OD) δ 213.1, 136.9, 132.6, 82.8, 80.0, 57.2, 40.5, 36.9,
14.8.
IR (KBr) νmax 3388, 1714, 1366, 1097, 1033, 744 cm–1.
MS (ESI, +ve) m/z 191 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C9H12NaO3 191.0684, found 191.0687.
Specific rotation [α]D = +487.8 (c = 0.6, CH3OH).
(5S)-5-((1,3-Dioxolan-2-yl)(hydroxy)methyl)-2-methylcyclohex-2-en-1-one (9)
A magnetically stirred solution of diol 8 (60 mg, 0.36 mmol) in benzene (5 mL) was
treated with ethylene glycol (240 µL, 4.30 mmol) and p-toluenesulfonic acid
monohydrate (12 mg, 0.06 mmol). The ensuing mixture was heated under reflux for
1.5 h then cooled and concentrated under reduced pressure to give a pale-yellow oil.
This oil was dissolved in ethyl acetate (50 mL) and the solution thus obtained washed
with NaHCO3 (1 × 50 mL of a saturated aqueous solution). The separated aqueous
phase was extracted with ethyl acetate (1 × 50 mL) and the combined organic phases
were then dried (MgSO4), filtered, and concentrated under reduced pressure. The
O
HO
O
O
H
9

! S12!
residue thus obtained was subjected to flash column chromatography (silica, 2:3 →
1:1 v/v ethyl acetate/40-60 petroleum ether elution) to give, after concentration of the
appropriate fractions (Rf = 0.5 in ethyl acetate), compound 9 (37 mg, 48%) as a
colorless, crystalline solid, mp = 112-114 °C. 1H NMR (400 MHz, CDCl3) δ 6.72 (m, 1H), 4.86 (d, J = 4.0 Hz, 1H), 4.10-3.85
(complex m, 4H), 3.53 (broad s, 1H), 2.70 (m, 1H), 2.45-2.32 (complex m, 4H), 1.93
(broad s, 1H), 1.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 199.8, 144.4, 135.7, 103.6, 73.9, 65.6, 65.4, 39.2,
37.7, 29.1, 15.9.
IR (KBr) νmax 3452, 2966, 2923, 2891, 1668, 1152, 1090, 1074, 963 cm–1.
MS (ESI, +ve) m/z 235 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C11H16NaO4 235.0946, found 235.0939.
Specific rotation [α]D = +34.0 (c = 0.5, CHCl3).
(1S,4R,7S,8S)-8-((tert-Butyldimethylsilyl)oxy)-7-hydroxy-1-methylbicyclo[2.2.2]-
oct-5-en-2-one (10)
A magnetically stirred solution of diol 8 (700 mg, 4.16 mmol) in dry dichloromethane
(45 mL) maintained under an atmosphere of nitrogen was treated with imidazole (1.13
g, 16.6 mmol), DMAP (51 mg, 0.42 mmol) and then TBSCl (1.88 g, 12.5 mmol). The
ensuing mixture was heated under reflux for 16 h then cooled to room temperature
before bring poured into NH4Cl (100 mL, a saturated aqueous solution) and extracted
with dichloromethane (3 × 100 mL). The combined organic phases were then dried
(MgSO4) filtered, and concentrated under reduced pressure. The residue thus obtained
was subjected to flash column chromatography (silica, 0:1 → 1:4 v/v ethyl acetate/40-
60 petroleum ether elution) to give, after concentration of the appropriate fractions (Rf
= 0.7 in ethyl acetate), compound 10 (1.04 g, 88%) as a colorless, crystalline solid,
mp = 71-73 °C.
OTBS
O
OH
10

! S13!
1H NMR (400 MHz, CDCl3) δ 6.42 (dd, J = 7.7 and 6.3 Hz, 1H), 5.77 (dd, J = 7.7 and
1.7 Hz, 1H), 3.96 (d, J = 3.2 Hz, 1H), 3.52 (d, J = 3.8 Hz, 1H), 2.86 (broad s, 1H)
2.14-2.03 (complex m, 2H), 1.96 (m, 1H), 1.27 (s, 3H), 0.89 (s, 9H), 0.11 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 210.3, 136.0, 130.6, 83.3, 79.8, 55.7, 39.8, 35.9, 25.9,
18.2, 14.4, −4.5(7), −4.6(1).
IR (KBr) νmax 3458, 2955, 2930, 2858, 1720, 1256, 1116, 1087, 838, 777 cm–1.
MS (ESI, +ve) m/z 305 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C15H26NaO3Si 305.1549, found 305.1549.
Specific rotation [α]D = +331.2 (c = 0.8, CHCl3).
(1R,2S,3S,4R,6S)-3-((tert-Butyldimethylsilyl)oxy)-1-methylbicyclo[2.2.2]oct-7-en-
e-2,6-diol (11) and (1R,2S,3S,4R,6R)-3-((tert-Butyldimethylsilyl)oxy)-1-
methylbicyclo[2.2.2]oct-7-ene-2,6-diol (12)
A magnetically stirred solution of ketone 10 (2.82 g, 10.0 mmol) in dry THF (140
mL) maintained under an atmosphere of nitrogen was cooled to −78 °C then treated,
dropwise over 1 h, with DIBAL-H (60 mL of a 1 M solution in THF, 60.0 mmol). The
ensuing mixture was stirred at −78 °C for 1 h before being warmed to 0 °C and
quenched by the slow addition of ice (CAUTION! EXOTHERMIC REACTION).
The ensuing mixture was poured into HCl (40 mL of a 1 M aqueous solution) and
extracted with ethyl acetate (3 × 200 mL). The combined organic phases were then
dried (MgSO4), filtered and concentrated under reduced pressure. The residue thus
obtained was subjected to flash column chromatography (silica, 1:19 → 3:7 v/v ethyl
acetate/40-60 petroleum ether gradient elution) to give two fractions, A and B.
Concentration of fraction A (Rf = 0.5 in 1:4 v/v ethyl acetate/40-60 petroleum
ether) afforded compound 11 (470 mg, 15%) as a colorless, crystalline solid, mp =
83-85 °C. 1H NMR (400 MHz, CDCl3) δ 6.14 (dd, J = 8.0 and 6.2 Hz, 1H), 5.75 (dd, J = 8.0 and
1.2 Hz, 1H), 3.85 (m, 1H), 3.63 (m, 1H), 3.32 (m, 1H), 3.06 (m, 1H), 2.62 (t, J = 6.8
OTBS
OH
OH
OTBS
OH
HO
11 12
+

! S14!
Hz, 1H), 2.48 (broads s, 1H), 2.10 (m, 1H), 1.55 (s, 1H), 1.39 (s, 3H), 0.88 (s, 9H),
0.10 (s, 3H), 0.09 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 134.3, 134.2, 85.8, 80.6, 74.2, 43.3, 38.3, 35.2, 26.0,
18.5, 18.3, −4.4(9), −4.5(4).
IR (KBr) νmax 3325, 2953, 2929, 2857, 1463, 1257, 1112, 1083, 1060, 836, 776 cm–1.
MS (ESI, +ve) m/z 307 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C15H28NaO3Si 307.1705, found 307.1705.
Specific rotation [α]D = +89.2 (c = 1.3, CHCl3).
Concentration of fraction B (Rf = 0.1 in 1:4 v/v ethyl acetate/40-60 petroleum
ether) afforded compound 12 (2.31 g, 83%) as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.33 (m, 1H), 5.76 (d, J = 8.1 Hz, 1H), 3.93 (d, J = 7.2
Hz, 1H), 3.54 (s, 1H), 3.25 (s, 1H), 2.49 (broad s, 1H), 2.15-2.03 (complex m, 1H),
1.30 (s, 3H), 1.22 (m, 1H), 0.86 (s, 9H), 0.05 (s, 6H) (signals due to hydroxyl group
protons not observed). 13C NMR (100 MHz, CDCl3) δ 134.8, 132.4, 83.2, 79.8, 68.4, 45.5, 39.2, 36.8, 26.0,
18.2, 18.0, −4.5, −4.6.
IR (KBr) νmax 3413, 2955, 2929, 2857, 1252, 1111, 1092, 1055, 934, 859, 836, 775,
718 cm–1.
MS (EI, 70 eV) m/z 284 (M+•, 3%), 227 (35), 165 (56), 135 (55), 117 (82), 75 (100).
HRMS M+• calcd for C15H28O3Si 284.1808, found 284.1807.
Specific rotation [α]D = +80.3 (c = 0.6, CHCl3).
(1R,2S,3S,4R,6R)-3-((tert-Butyldimethylsilyl)oxy)-1-methylbicyclo[2.2.2]oct-7-
ene-2,6-diyl Diacetate (13)
A magnetically stirred solution of compound 12 (1.70 g, 6.0 mmol) in dry
dichloromethane (212 mL) maintained at 0 °C under an atmosphere of nitrogen was
treated with DMAP (58 mg, 0.5 mmol), pyridine (10 mL, 123.6 mmol) and acetic
anhydride (5.5 mL, 58.6 mmol). After a further 1 h at 0 °C the reaction mixture was
warmed to 22 °C and then allowed to stir at this temperature for 16 h before being
OTBS
OAc
AcO
13

! S15!
poured into water (200 mL) and extracted with dichloromethane (3 × 200 mL). The
combined organic phases were dried (MgSO4), filtered and concentrated under
reduced pressure. The residue thus obtained was subjected to flash column
chromatography (silica, 0:1 → 1:9 v/v ethyl acetate/40-60 petroleum ether gradient
elution) to give, after concentration of the appropriate fractions (Rf = 0.5 in 3:7 v/v
ethyl acetate/40-60 petroleum ether), compound 13 (2.16 g, 98%) as a clear, colorless
oil. 1H NMR (400 MHz, CDCl3) δ 6.33 (m, 1H), 5.83 (d, J = 8.2 Hz, 1H), 4.97 (dd, J =
8.2 and 2.6 Hz, 1H), 4.60 (d, J = 1.6 Hz, 1H), 3.60 (m, 1H), 2.52 (m, 1H), 2.21 (m,
1H), 2.08 (s, 3H), 2.00 (s, 3H), 1.24 (m, 1H), 1.09 (s, 3H), 0.84 (s, 9H), 0.00 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 171.3, 170.3, 133.6, 132.2, 82.9, 77.3, 71.8, 42.6,
38.8, 34.4, 25.8, 21.3, 21.1, 18.1, 17.7, −4.8, −4.9.
IR (KBr) νmax 2955, 2858, 1743, 1372, 1228, 1114, 1096, 1061, 858, 838, 777 cm–1.
MS (EI, 70 eV) m/z 368 (M+•, <1%), 311 (16), 269 (22), 251 (24), 209 (30), 174 (33),
159 (32), 135 (65), 117 (100), 93 (60), 75 (53), 73 (45).
HRMS M+• calcd for C19H32O5Si 368.2019, found 368.2026.
Specific rotation [α]D = +22.1 (c = 0.4, CHCl3).
(1R,2S,3S,4R,6R)-3-Hydroxy-1-methylbicyclo[2.2.2]oct-7-ene-2,6-diyl Diacetate
(14)
A magnetically stirred solution of compound 13 (2.13 g, 5.8 mmol) in dry THF (187
mL) maintained at 0 °C under an atmosphere of nitrogen was treated, dropwise over
0.33 h, with TBAF (14.4 mL of a 1 M solution in THF, 14.4 mmol). After a further
0.66 h at 0 °C the reaction mixture was warmed to 22 °C and allowed to stir at this
temperature for 1 h then re-cooled to 0 °C and treated with NaHCO3 (200 mL of a
saturated aqueous solution). The ensuing mixture was extracted with ethyl acetate (3
× 200 mL) and the combined organic phases dried (MgSO4), filtered, and
concentrated under reduced pressure. The residue thus obtained was subjected to flash
column chromatography (silica, 3:7 v/v ethyl acetate/40-60 petroleum ether elution)
OH
OAc
AcO
14

! S16!
to give, after concentration of the appropriate fractions (Rf = 0.5 in ethyl acetate),
compound 14 (1.34 g, 91%) as a colorless, crystalline solid, mp = 82-86 °C. 1H NMR (400 MHz, CDCl3) δ 6.37 (m, 1H), 5.81 (d, J = 8.2 Hz, 1H), 4.98 (dd, J =
8.2 and 2.9 Hz, 1H), 3.90 (s, 1H), 3.48 (s, 1H), 3.18 (s, 1H), 2.66 (broad s, 1H), 2.18
(m, 1H), 2.09 (s, 3H), 1.98 (s, 3H), 1.23 (d, J = 14.2 Hz, 1H), 1.17 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.8, 171.1, 133.5, 132.3, 85.6, 76.9, 71.2, 41.4,
36.5, 34.5, 21.2, 21.0, 17.7.
IR (KBr) νmax 3493, 2975, 1733, 1374, 1249, 1055, 1028, 728 cm–1.
MS (EI, 70 eV) m/z 254 (M+•, 5%), 134 (32), 108 (100), 93 (60), 92 (43).
HRMS M+• calcd for C13H18O5 254.1154, found 254.1155.
Specific rotation [α]D = −99.2 (c = 0.3, CHCl3).
(1R,2S,4R,6R)-1-Methyl-3-oxobicyclo[2.2.2]oct-7-ene-2,6-diyl Diacetate (15)
A magnetically stirred solution of alcohol 14 (2.00 g, 7.9 mmol) in dry
dichloromethane (200 mL) maintained at 0 °C under an atmosphere of nitrogen was
treated with pyridine (4 mL, 49.5 mmol) then the Dess-Martin periodinane (6.00 g,
14.1 mmol). After a further 0.66 h at 0 °C the reaction mixture was warmed to 22 °C,
stirred at this temperature for 3 h then poured into water (200 mL) and extracted with
dichloromethane (3 × 200 mL). The combined organic phases were washed with
NaOH (100 mL of a 1 M aqueous solution) and HCl (100 mL of a 1 M aqueous
solution) before being dried (MgSO4), filtered, and concentrated under reduced
pressure. The residue thus obtained was subjected to flash column chromatography
(silica, 3:17 v/v ethyl acetate/40-60 petroleum ether elution) to give, after
concentration of the appropriate fractions (Rf = 0.3 in 3:7 v/v ethyl acetate/40-60
petroleum ether), ketone 15 (1.89 g, 95%) as a colorless, crystalline solid, mp = 101-
104 °C. 1H NMR (400 MHz, CDCl3) δ 6.38 (m, 1H), 6.07 (dd, J = 8.2 and 0.7 Hz, 1H), 5.15
(dd, J = 8.2 and 3.0 Hz, 1H), 4.90 (s, 1H), 3.14 (broad s, 1H), 2.52 (m, 1H), 2.13 (s,
3H), 2.02 (s, 3H), 1.59 (m, 1H), 1.20 (s, 3H).
OAc
AcOO
15

! S17!
13C NMR (100 MHz, CDCl3) δ 204.0, 170.8, 170.1, 136.6, 129.6, 72.1, 71.4, 46.8,
45.4, 35.5, 21.1, 20.7, 17.2.
IR (KBr) νmax 2917, 1737, 1372, 1223, 1037, 744 cm–1.
MS (ESI, +ve) m/z 275 [(M+Na)+, 45%], 249 (100), 231 (98), 133 (20).
HRMS m/z (M+Na)+ calcd for C13H16NaO5 275.0895, found 275.0895.
Specific rotation [α]D = +0.9 (c = 0.9, CHCl3).
(1S,2R,4R,7S,8R)-7,8-Dihydroxy-1,8-dimethylbicyclo[2.2.2]oct-5-en-2-yl Acetate
(16)
A magnetically stirred solution of ketone 15 (2.50 g, 9.9 mmol) in dry THF (200 mL)
maintained at 0 °C under an atmosphere of nitrogen was treated, in one portion, with
methylmagnesium bromide solution (10.7 mL of a 3 M solution in diethyl ether, 32.2
mmol). The ensuing mixture was stirred at 0 °C for 0.33 h then quenched by the slow
addition of ice (CAUTION! EXOTHERMIC REACTION). The ensuing mixture was
poured into NH4Cl (200 mL of a saturated aqueous solution) and extracted with ethyl
acetate (3 × 200 mL). The combined organic phases were then dried (MgSO4), filtered,
and concentrated under reduced pressure. The residue thus obtained was subjected to
flash column chromatography (silica, 1:4 v/v ethyl acetate/40-60 petroleum ether
elution) to give, after concentration of the appropriate fractions (Rf = 0.3 in 1:1 v/v
ethyl acetate/40-60 petroleum ether), compound 16 (1.90 mg, 85%) as a colorless,
crystalline solid, mp = 124-126 °C. 1H NMR (400 MHz, CDCl3) δ 6.32 (dd, J = 8.2 and 6.7 Hz, 1H), 5.74 (d, J = 8.2 Hz,
1H), 5.07 (dd, J = 8.2 and 3.5 Hz, 1H), 3.05 (s, 1H), 2.71 (m, 1H), 2.48 (m, 1H), 2.31
(broad s, 2H), 2.00 (s, 3H), 1.24 (s, 6H), 1.00 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 171.4, 134.5, 134.2, 77.4, 71.7, 69.7, 44.1, 43.0, 32.2,
30.6, 21.3, 18.0.
IR (KBr) νmax 3418, 2967, 1735, 1712, 1369, 1248, 1146, 1051, 1033, 937, 730 cm–1.
MS (ESI, +ve) m/z 475 [(2M+Na)+, 30%], 249 [(M+Na)+, 100].
HRMS (M+Na)+ calcd for C12H18NaO4 249.1103, found 249.1105.
OH
AcOOH
16

! S18!
Specific rotation [α]D = −60.3 (c = 0.6, CHCl3).
(1R,2R,3S,4R,5R)-2,4-Dimethylbicyclo[2.2.2]oct-7-ene-2,3,5-triol (17)
A magnetically stirred solution of diol 16 (960 mg, 4.2 mmol) in methanol (97 mL)
was treated with potassium carbonate (586 mg, 4.2 mmol). The ensuing mixture was
heated under reflux for 16 h then cooled and concentrated under reduced pressure.
The residue thus obtained was treated with water (30 mL) and extracted with ethyl
acetate (3 × 100 mL). The combined organic phases were then dried (MgSO4), filtered
and concentrated under reduced pressure and the residue thus obtained was subjected
to flash column chromatography (silica, 7:3 v/v ethyl acetate/40-60 petroleum ether
elution). Concentration of the appropriate fractions (Rf = 0.4 in ethyl acetate) then
gave triol 17 (620 mg, 79%) as a white, crystalline solid, mp = 180-183 °C. 1H NMR (400 MHz, CD3OD) δ 6.30 (m, 1H), 5.67 (d, J = 8.2 Hz, 1H), 3.91 (m, 1H),
2.94 (s, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 1.26 (s, 3H), 1.15 (s, 3H), 0.96 (dt, J = 13.2
and 3.2 Hz, 1H) (signals due to hydroxyl group protons not observed). 13C NMR (100 MHz, CD3OD) δ 135.6, 135.4, 78.6, 70.3, 68.2, 46.7, 44.6, 34.9, 30.7,
18.6.
IR (KBr) νmax 3362, 3246, 2955, 1372, 1286, 1140, 1064, 1030, 967, 919, 729 cm–1.
MS (ESI, +ve) m/z 265 (100%), 207 [(M+Na)+, 95].
HRMS (M+Na)+ calcd for C10H16NaO3 207.0997, found 207.0996.
Specific rotation [α]D = −10.8 (c = 0.1, CHCl3).
(3aS,4R,7R,9R)-2,2,4,7a-Tetramethyl-3a,4,7,7a-tetrahydro-4,7-ethanobenzo[d]-
[1,3]dioxol-9-ol (18)
OH
HOOH
17
O
HOO
18

! S19!
A magnetically stirred solution of triol 17 (1.40 g, 7.6 mmol) and p-toluenesulfonic
acid monohydrate (217 mg, 1.1 mmol) in dichloromethane (370 mL) maintained at 0
°C was treated with 2,2-dimethoxypropane (4.7 mL, 38.0 mmol). The ensuing
mixture was stirred at 0 °C for 2 h then warmed to 22 °C, stirred at this temperature
for 1 h then quenched with triethylamine (57 µL, 4.1 mmol). The ensuing mixture
was concentrated under reduced pressure and the residue thus obtained poured into
water (100 mL) and extracted with ethyl acetate (3 × 200 mL). The combined organic
phases were then dried (MgSO4), filtered, and concentrated under reduced pressure.
The residue thus obtained was subjected to flash column chromatography (silica, 1:4
v/v ethyl acetate/40-60 petroleum ether elution) to give, after concentration of the
appropriate fractions (Rf = 0.4 in 1:1 v/v ethyl acetate/40-60 petroleum ether), alcohol
18 (1.55 g, 91%) as a white, crystalline solid, mp = 122-124 °C. 1H NMR (400 MHz, CDCl3) δ 6.42 (t, J = 8.0 Hz, 1H), 5.68 (d, J = 8.0 Hz, 1H), 4.05
(d, J = 8.0 Hz, 1H), 3.60 (s, 1H), 2.63 (dd, J = 14.0 and 8.1 Hz, 1H), 2.51 (broad s,
1H), 1.45 (s, 3H), 1.39 (s, 3H), 1.38 (s, 3H), 1.35 (s, 3H), 1.26 (broad s, 1H), 1.01 (d,
J = 14.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 136.6, 133.2, 111.3, 87.3, 83.9, 69.6, 45.5, 42.8, 34.0,
28.9, 27.2(8), 27.2(5), 19.1.
IR (KBr) νmax 3388, 2967, 2945, 1368, 1207, 1121, 1040, 730 cm–1.
MS (EI) m/z 224 (M+•, <1%), 209 [(M−CH3•)+, 10], 166 (38), 123 (77), 114 (100),
107 (53), 95 (51).
HRMS (M−CH3•)+ calcd for C12H17O3 209.1178, found 209.1180.
Specific rotation [α]D = −3.4 (c = 0.4, CHCl3).
(3aS,4R,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethyl-3a,4,7,7a-tetrahydro-4,7-ethano-
benzo[d][1,3]dioxole (19)
Sodium hydride (936 mg of a 60% dispersion in mineral oil, 23.4 mmol) was added to
a magnetically stirred solution of alcohol 18 (350 mg, 1.6 mmol) in THF (18 mL)
O
OO
19

! S20!
maintained at room temperature under an atmosphere of nitrogen. The resulting
mixture was treated with iodoethane (753 µL, 9.4 mmol) then stirred at 22 °C for 18 h
before being cooled to 0 °C then quenched by the slow addition of ice (CAUTION!
EXOTHERMIC REACTION AND POSSIBILITY OF HYDROGEN EVOLUTION).
The ensuing mixture was poured into water (100 mL) and extracted with ethyl acetate
(3 × 100 mL). The combined organic phases were then dried (MgSO4), filtered, and
concentrated under reduced pressure. The residue thus obtained was subjected to flash
column chromatography (silica, 1:19 v/v ethyl acetate/40-60 petroleum ether elution)
to give, after concentration of the appropriate fractions (Rf = 0.6 in 3:7 v/v ethyl
acetate/40-60 petroleum ether), alcohol 19 (358 mg, 91%) as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.32 (m, 1H), 5.70 (d, J = 8.1 Hz, 1H), 3.72 (m, 1H),
3.54 (m, 2H), 3.34 (m, 1H), 2.51 (m, 1H), 2.44 (m, 1H), 1.46 (s, 3H), 1.39 (s, 3H),
1.34 (s, 3H), 1.33 (s, 3H), 1.17-1.10 (complex m, 4H). 13C NMR (100 MHz, CDCl3) δ 134.6, 134.1, 111.1, 87.5, 83.8, 77.0, 64.9, 44.7, 42.7,
31.1, 29.0, 27.5, 27.4, 19.1, 15.6.
IR (KBr) νmax 2975, 2933, 2874, 1372, 1243, 1207, 1120, 1095, 1054, 872, 715 cm–1.
MS (ESI, +ve) m/z 275 [(M+Na)+, 85%], 209 (100).
HRMS (M+Na)+ calcd for C15H24NaO3 275.1623, found 275.1624.
Specific rotation [α]D = −35.4 (c = 0.3, CHCl3).
(1aR,2S,2aS,6R,6aS,8R)-8-Ethoxy-2,4,4,5a-tetramethylhexahydro-2,6-ethano-
oxireno[2',3':4,5]benzo[1,2-d][1,3]dioxole (20)
A magnetically stirred solution of compound 19 (1.06 g, 4.2 mmol) in
dichloromethane (289 mL) maintained at 22 °C was treated with m-chloroperbenzoic
acid (6.20 g of approx. 77% - technical grade - material, 25.2 mmol). The resulting
mixture was stirred at 22 °C for 16 h then poured into water (200 mL) and extracted
with ethyl acetate (3 × 200 mL). The combined organic phases were washed with
NaOH (1 × 50 mL of a 1 M aqueous solution) before being dried (MgSO4), filtered
O
OOO
20

! S21!
and concentrated under reduced pressure. The residue thus obtained was subjected to
flash column chromatography (silica, 3:17 v/v ethyl acetate/40-60 petroleum ether
elution) to give, after concentration of the appropriate fractions (Rf = 0.3 in 3:7 v/v
ethyl acetate/40-60 petroleum ether), epoxide 20 (970 mg, 86%) as a white,
crystalline solid, mp = 57-59 °C. 1H NMR (400 MHz, CDCl3) δ 3.81 (s, 1H), 3.57 (m, 1H), 3.41 (m, 2H), 3.25 (m, 1H),
2.83 (m, 1H), 2.30 (m, 1H), 2.09 (m, 1H), 1.54 (m, 1H), 1.50 (s, 3H), 1.44 (s, 6H),
1.29 (s, 3H), 1.18 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 111.1, 86.6, 82.6, 74.8, 66.2, 54.6, 53.2, 42.7, 40.7,
28.2, 28.1, 27.3, 26.6, 19.2, 15.7.
IR (KBr) νmax 2977, 2936, 2871, 1375, 1208, 1121, 1100, 1069, 1019, 869 cm–1.
MS (ESI, +ve) m/z 559 [(2M+Na)+, 40%], 291 [(M+Na)+, 100].
HRMS (M+Na)+ calcd for C15H24NaO4 291.1572, found 291.1572.
Specific rotation [α]D = −76.7 (c = 0.6, CHCl3).
(3aS,4R,6R,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethylhexahydro-4,7-ethanobenzo-
[d][1,3]dioxol-6-ol (21)
A magnetically stirred solution of epoxide 20 (1.00 g, 3.7 mmol) in THF (48 mL)
maintained under nitrogen was cooled to 0 °C then treated, dropwise over 0.25 h, with
LiEt3BH (37 mL of a 1 M solution in THF, 37 mmol). The ensuing mixture was
stirred at 0 °C for 0.5 h before being warmed to 22 °C and stirred at this temperature
for 16 h. The resulting mixture was re-cooled to 0 °C then quenched by the slow
addition of ice (CAUTION! EXOTHERMIC REACTION AND POSSIBILITY OF
HYDROGEN EVOLUTION) before being diluted with ethyl acetate (20 mL). The
ensuing mixture was poured into water (200 mL) and extracted with ethyl acetate (3 ×
200 mL). The combined organic phases were then dried (MgSO4), filtered and
concentrated under reduced pressure and the ensuing light-yellow oil subjected to
flash chromatography (silica, 1:9 → 3:17 v/v ethyl acetate/40-60 petroleum ether
OOHOO
21

! S22!
gradient elution). Concentration of the appropriate fractions (Rf = 0.6 in 3:7 v/v ethyl
acetate/40-60 petroleum ether) then gave alcohol 21 (840 mg, 84%) as a clear,
colorless oil. 1H NMR (400 MHz, CDCl3) δ 3.84 (m, 1H), 3.69 (m, 1H), 3.51 (s, 1H), 3.43-3.34
(complex m, 2H), 2.14 (m, 1H), 1.95 (m, 1H), 1.75 (dd, J = 15.0 and 4.3 Hz, 1H),
1.61-1.57 (complex m, 2H), 1.48-1.42 (complex m, 4H), 1.39 (s, 3H), 1.37 (s, 3H),
1.20 (t, J = 7.0 Hz, 3H), 1.07 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 108.5, 86.8, 80.7, 75.6, 65.7, 64.9, 43.0, 38.1, 35.4,
26.8, 26.5, 26.3, 23.1, 22.0, 15.6.
IR (KBr) νmax 3501, 2975, 2871, 1378, 1206, 1056, 1011, 1081, 870 cm–1.
MS (ESI, +ve) m/z 293 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C15H26NaO4 293.1729, found 293.1725.
Specific rotation [α]D = +4.6 (c = 0.7, CHCl3).
(1S,2S,3R,4R,5R,7R)-7-Ethoxy-1,3-dimethylbicyclo[2.2.2]octane-2,3,5-triol (22)
A magnetically stirred solution of compound 21 (250 mg, 0.9 mmol) in
methanol/water (9.5 mL of an 4:1 v/v mixture) maintained at 22 °C was treated with
AG-50W-X8 resin (212 mg of H+ form) and the ensuing mixture heated at 65 °C for
40 h then cooled and filtered. The solids thus retained were washed with methanol (20
mL) and the combined filtrates concentrated under reduced pressure to give a light-
yellow oil. This material was subjected to flash column chromatography (silica,
1:1→1:0 v/v ethyl acetate/40-60 petroleum ether gradient elution) and thus affording
two fractions, A and B.
Concentration of the fraction A (Rf = 0.7 in 1:1 v/v ethyl acetate/40-60
petroleum ether) gave compound 21 (75 mg, 30% recovery) as a clear, colorless oil
that was identical, in all respects, with an authentic sample.
Concentration of the fraction B (Rf = 0.1 in 1:1 v/v ethyl acetate/40-60
petroleum ether) gave compound 22 (126 mg, 59% or 84% brsm) as a colorless,
OHOHHOO
22

! S23!
crystalline solid, mp = 87-90 °C. 1H NMR (400 MHz, CDCl3) δ 3.89 (broad s, 1H), 3.68 (m, 1H), 3.40 (m, 2H), 3.10
(m, 1H), 3.06 (s, 1H), 2.89 (d, J = 4.1 Hz, 1H), 2.57 (s, 1H), 2.14 (m, 1H), 1.81 (m,
2H), 1.63-1.50 (complex m, 2H), 1.31 (s, 3H), 1.19 (t, J = 7.0 Hz, 3H), 1.03 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 79.4, 74.9, 69.9, 65.6, 65.0, 44.6, 39.6, 36.3, 28.9,
23.0, 21.1, 15.6.
IR (KBr) νmax 3351, 2969, 2929, 1455, 1371, 1068, 1047, 1007, 973 cm–1.
MS (ESI, +ve) m/z 253 [(M+Na)+, 100%], 167 (20).
HRMS (M+Na)+ calcd for C12H22NaO4 253.1416, found 253.1412.
Specific rotation [α]D = −123.3 (c = 1.1, MeOH).
(3aS,4R,5R,6S,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethylhexahydro-4,7-ethanobenzo-
[d][1,3]dioxole-5,6-diol (23) and (3aS,4R,5S,6R,7R,9R)-9-Ethoxy-2,2,4,7a-tetra
methylhexahydro-4,7-ethanobenzo[d][1,3]dioxole-5,6-diol (24)
A magnetically stirred solution of compound 19 (1.10 g, 4.4 mmol) in
acetonitrile/water (44 mL of a 4:1 v/v mixture) maintained at 22 °C was treated with
4-methylmorpholine N-oxide (1.02 g, 8.7 mmol), citric acid (2.50 g, 13.0 mmol) and
potassium osmate dihydrate (77 mg, 0.2 mmol). The ensuing mixture was stirred at 22
°C for 14 h then quenched with Na2SO3 (10 mL of a saturated aqueous solution)
before being poured into water (50 mL) and extracted with ethyl acetate (3 × 50 mL).
The combined organic phases were then dried (MgSO4) filtered, and concentrated
under reduced pressure. The residue thus obtained was subjected to flash column
chromatography (silica, 1:9→3:7 v/v ethyl acetate/40-60 petroleum ether gradient
elution) to give two fractions, A and B.
Concentration of fraction A (Rf = 0.4 in 3:7 v/v ethyl acetate/petroleum ether)
afforded diol 24 (849 mg, 68%) as a white, crystalline solid, mp = 98-101 °C. 1H NMR (400 MHz, CDCl3) δ 4.20 (m, 1H), 4.06-4.02 (complex m, 2H), 3.54 (m,
1H), 3.38 (d, J = 8.3 Hz, 1H), 3.26 (m, 1H), 2.52 (m, 1H), 2.35 (complex m, 1H), 2.25
OOHOO
23
HO OO
O
24HOOH
+

! S24!
(m, 1H), 1.87 (s, 1H), 1.58 (s, 3H), 1.43 (s, 3H), 1.41 (s, 3H), 1.28 (s, 3H), 1.25 (m,
1H), 1.12 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 107.4, 82.5, 80.0, 75.3, 70.1, 68.6, 64.7, 44.2, 42.2,
29.6, 27.9, 26.8, 26.7, 18.8, 15.6.
IR (KBr) νmax 3406, 2975, 2936, 1379, 1206, 1183, 1097, 1051 cm–1.
MS (ESI, +ve) m/z 309 [(M+Na)+, 100%], 307 (60).
HRMS (M+Na)+ calcd for C15H26NaO5 309.1678, found 309.1679.
Specific rotation [α]D = −58.1 (c = 0.2, CHCl3).
Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/petroleum ether)
afforded diol 23 (270 mg, 22%) as a clear, pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.98 (m, 1H), 3.66 (m, 1H), 3.52 (d, J = 8.7 Hz, 1H),
3.48 (s, 1H), 3.33-3.26 (complex m, 2H), 2.14-2.02 (complex m, 2H), 1.87 (broad d, J
= 14.2 Hz, 1H), 1.42 (s, 3H), 1.38 (s, 6H), 1.20 (s, 3H), 1.16 (t, J = 7.0 Hz, 3H)
(signals due to hydroxyl group protons not observed). 13C NMR (100 MHz, CDCl3) δ 109.1, 85.9, 79.5, 76.9, 69.9, 65.3, 65.1, 42.2, 41.1,
26.8, 26.7, 26.6, 22.0, 18.3, 15.5.
IR (KBr) νmax 3471, 2980, 1451, 1378, 1244, 1209, 1177, 1075, 1044, 874 cm–1.
MS (ESI, +ve) m/z 309 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C15H26NaO5 309.1678, found 309.1678.
Specific rotation [α]D = −42.9 (c = 0.1, CHCl3).
(3aS,4S,4aS,6S,7aR,8R,10R)-10-Ethoxy-6-(4-methoxyphenyl)-2,2,4,8a-tetra-
methylhexahydro-4,8-ethanobenzo[1,2-d:4,5-d']bis([1,3]dioxole) (25)
A magnetically stirred solution of diol 24 (560 mg, 2.0 mmol) and p-
methoxybenzaldehyde dimethyl acetal (400 µL, 2.1 mmol) in THF (18 mL)
maintained under a nitrogen atmosphere was cooled to 0 °C then treated with p-
toluenesulfonic acid monohydrate (14 mg, 0.08 mmol). The ensuing mixture was
OO
O
OO
PMP
25

! S25!
stirred at 0 °C for 4 h then quenched with triethylamine (40 µL) before being
concentrated under reduced pressure. The residue thus obtained was dissolved in ethyl
acetate (50 mL) and the resulting solution washed with water (1 × 50 mL). The
separated aqueous phase was extracted with ethyl acetate (3 × 50 mL) and the
combined organic phases were then dried (MgSO4), filtered and concentrated under
reduced pressure to afford a light-yellow oil. Subjection of this material to flash
chromatography (silica, 1:19→1:9 v/v ethyl acetate/40-60 petroleum ether gradient
elution) and concentration of the appropriate fractions (Rf = 0.6 in 3:7 v/v ethyl
acetate/40-60 petroleum ether) gave compound 25 (704 mg, 89%) as a clear, colorless
oil. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 5.60
(s, 1H), 4.37-4.30 (complex m, 2H), 4.07 (s, 1H), 3.81 (s, 3H), 3.61-3.49 (complex m,
2H), 3.33 (m, 1H), 2.48 (m, 1H), 2.25 (m, 1H), 1.48 (s, 3H), 1.43 (s, 3H), 1.42 (s,
3H), 1.35 (s, 3H), 1.16 (t, J = 7.0 Hz, 3H), 1.10 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 160.2, 128.2, 128.0, 113.7, 107.8, 101.6, 82.6, 80.0,
77.9, 76.1, 74.2, 65.2, 55.4, 41.5, 41.4, 29.2, 28.0, 27.2, 17.6, 15.6 (one signal
obscured or overlapping).
IR (KBr) νmax 2975, 2933, 1616, 1518, 1248, 1172, 1151, 1054, 1033, 827 cm–1.
MS (ESI, +ve) m/z 427 [(M+Na)+, 100%], 405 [(M+H)+, 35], 309 (50), 177 (30), 61
(90).
HRMS (M+Na)+ calcd for C23H32NaO6 427.2097, found 427.2098.
Specific rotation [α]D = −110.3 (c = 1.9, CHCl3).
(3aS,4S,5S,6R,7R,9R)-9-Ethoxy-5-((4-methoxybenzyl)oxy)-2,2,4,7a-tetramethyl-
hexahydro-4,7-ethanobenzo[d][1,3]dioxol-6-ol (26)
!
A magnetically stirred solution of acetal 25 (1.60 g, 4.0 mmol) in dichloromethane
(42 mL) maintained under a nitrogen atmosphere was cooled to −78 °C then treated
OO
O
PMBOOH
26

! S26!
with DIBAL-H (29.7 mL of a 1 M solution in hexane, 29.7 mmol). The resulting
mixture was stirred at −78 °C for 1 h then at 0 °C for 1 h and after which time it was
quenched with iced-water (50 mL) (CAUTION! EXOTHERMIC REACTION AND
POSSIBILITY OF HYDROGEN EVOLUTION). The mixture thus obtained was
poured into HCl (50 mL of a 1 M aqueous solution) and extracted with ethyl acetate
(3 × 200 mL). The combined organic phases were then dried (MgSO4), filtered, and
concentrated under reduced pressure. The residue so produced was subjected to flash
column chromatography (silica, 0:1→1:9 v/v ethyl acetate/40-60 petroleum ether
gradient elution) and concentration of the appropriate fractions (Rf = 0.4 in 3:7 v/v
ethyl acetate/40-60 petroleum ether) gave compound 26 (1.53 g, 95%) as a colorless,
crystalline solid, mp = 110-113 °C. 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 4.58
(ABq, J = 10.8 Hz, 2H), 4.18 (m, 1H), 3.99 (s, 1H), 3.88 (d, J = 8.2 Hz, 1H), 3.81 (s,
3H), 3.56 (m, 1H), 3.37 (d, J = 8.2 Hz, 1H), 3.28 (m, 1H), 2.20 (m, 1H), 1.90 (s, 1H),
1.49 (s, 3H), 1.42 (s, 3H), 1.39 (s, 3H), 1.29 (s, 3H), 1.26 (m, 1H), 1.14 (t, J = 7.0 Hz,
3H) (signal due to hydroxyl group proton not observed). 13C NMR (100 MHz, CDCl3) δ 159.6, 130.2, 129.4, 114.1, 107.6, 82.9, 79.9, 79.2,
76.6, 75.2, 68.5, 64.6, 55.4, 44.3, 42.7, 29.4, 28.1, 26.8, 26.7, 19.1, 15.6.
IR (KBr) νmax 3494, 2974, 2935, 1613, 1515, 1379, 1247, 1206, 1182, 1094, 1054,
872 cm–1.
MS (ESI, +ve) m/z 429 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C23H34NaO6 429.2253, found 429.2259.
Specific rotation [α]D = −62.6 (c = 0.3, CHCl3).
O-((3aS,4S,5S,6R,7R,9R)-9-Ethoxy-5-((4-methoxybenzyl)oxy)-2,2,4,7a-tetrameth-
ylhexahydro-4,7-ethanobenzo[d][1,3]dioxol-6-yl) S-methyl Carbonodithioate (27)
!
A magnetically stirred solution of alcohol 26 (150 mg, 0.37 mmol) in dry THF (6 mL)
maintained under a nitrogen atmosphere at 22 °C was treated with sodium hydride (74
OO
O
PMBOOC(S)SMe
27

! S27!
mg of a 60% dispersion in mineral oil, 3.1 mmol). After 1.5 h carbon disulfide (330
µL, 5.5 mmol) was added to the reaction mixture that was then heated under reflux
for 2 h. The ensuing mixture was cooled to 22 °C then methyl iodide (120 µL, 1.9
mmol) added and stirring continued for 5 h. After this time, the reaction mixture was
cooled to 0 °C and quenched with iced water (12 mL) (CAUTION! EXOTHERMIC
REACTION AND POSSIBILITY OF HYDROGEN EVOLUTION). The ensuing
mixture was diluted with ethyl acetate (50 mL) then water (24 mL). The separated
aqueous phase was extracted with ethyl acetate (3 × 50 mL) and the combined organic
phases then dried (MgSO4), filtered, and concentrated under reduced pressure. The
residue thus obtained was subjected to flash column chromatography (silica, 1:19 v/v
ethyl acetate/40-60 petroleum ether elution) to give, after concentration of the
relevant fractions (Rf = 0.6), xanthate 27 (176 mg, 96%) as a clear, pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.2 Hz, 2H), 6.87 (d, J = 8.2 Hz, 2H), 5.99
(m, 1H), 4.59 (d, J = 10.5 Hz, 1H), 4.32 (d, J = 10.5 Hz, 1H), 4.07 (s, 1H), 4.00 (d, J
= 7.6 Hz, 1H), 3.81 (s, 3H), 360 (m, 1H), 3.41 (d, J = 8.0 Hz, 1H), 3.29 (m, 1H), 2.54
(s, 3H), 2.26 (m, 1H), 2.14 (broad s, 1H), 1.58 (s, 3H), 1.45-1.41 (complex m, 4H),
1.38 (s, 3H), 1.27 (s, 3H), 1.16 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 215.4, 159.2, 131.1, 129.4, 113.7, 107.6, 82.6, 80.4,
79.7, 77.1, 75.7, 74.7, 64.6, 55.4, 43.3, 41.8, 28.9, 27.1, 26.6, 26.5, 19.2, 18.7, 15.6.
IR (KBr) νmax 2972, 2935, 1515, 1248, 1206, 1062 cm–1.
MS (ESI, +ve) m/z 519 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C25H36NaO6S2 519.1851, found 519.1851.
Specific rotation [α]D = −9.0 (c = 0.6, CHCl3).
(3aS,4S,5R,7R,9R)-5-Ethoxy-9-((4-methoxybenzyl)oxy)-2,2,4,7a-tetramethyl-
hexahydro-4,7-ethanobenzo[d][1,3]dioxole (28)
!!
A magnetically stirred and de-oxygenated solution of xanthate 27 (468 mg, 0.94
OO
O
28
PMBO

! S28!
mmol) in toluene (84 mL) maintained under an atmosphere of nitrogen at 22 °C was
treated with AIBN (47 mg, 0.28 mmol) then (Me3Si)3SiH (840 µL, 2.72 mmol). The
ensuing mixture was heated at 100 °C for 1 h then cooled and concentrated under
reduced pressure. The residue thus obtained was subjected to flash column
chromatography (silica, 1:37.5:350 v/v ethyl acetate/dichloromethane/40-60
petroleum ether elution) to give, after concentration of the appropriate fractions (Rf =
0.6 twice in 1:2.5:8.5 v/v ethyl acetate/dichloromethane/40-60 petroleum ether),
compound 28 (291 mg, 79%) as a white, crystalline solid, mp = 80-84 °C. 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 4.50
(d, J = 11.2 Hz, 1H), 4.25 (d, J = 11.2 Hz, 1H), 4.00 (s, 1H), 3.81 (s, 3H), 3.68 (d, J =
8.3 Hz, 1H), 3.56 (complex m, 1H), 3.46 (d, J = 8.3 Hz, 1H), 3.30 (m, 1H), 2.23 (m,
1H), 1.88 (m, 1H), 1.80-1.73 (complex m, 2H), 1.50 (s, 3H), 1.43 (s, 3H), 1.38 (s,
3H), 1.24 (s, 3H), 1.21 (m, 1H), 1.14 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.0, 131.4, 129.0, 113.8, 107.0, 82.7, 80.7, 76.1,
75.2, 70.9, 64.7, 55.4, 42.1, 37.6, 30.2, 30.1, 26.7, 26.5, 25.9, 18.6, 15.6.
IR (KBr) νmax 2973, 2934, 1614, 1514, 1377, 1247, 1094, 1053, 821 cm–1.
MS (EI, 70 eV) m/z 390 (M+•, 10%), 375 [(M−CH3•)+, 75], 211 (75), 165 (90), 122
(95), 121 (100).
HRMS M+• calcd for C23H34O5 390.2406, found 390.2410.
Specific rotation [α]D = −84.1 (c = 0.4, CHCl3).
(3aS,4R,5R,7S,9R)-9-Ethoxy-2,2,4,7a-tetramethylhexahydro-4,7-ethanobenzo-
[d][1,3]dioxol-5-ol (29)
!!
!A magnetically stirred solution of ether 28 (109 mg, 0.28 mmol) in dichloromethane
(3.5 mL) maintained at 22 °C was treated with water (400 µL) then 2,3-dichloro-5,6-
dicyano-1,4-benzoquinone (95 mg, 0.42 mmol) and the resulting deep-green mixture
stirred for 0.25 h then quenched with NaHCO3 (20 mL of a saturated aqueous
solution). The mixture thus formed was extracted with dichloromethane (3 × 30 mL)
OO
O
29HO

! S29!
and the combined organic extracts were then dried (MgSO4), filtered and concentrated
under reduced pressure. The residue thus obtained was subjected to flash column
chromatography (silica, 1:9 v/v ethyl acetate/40-60 petroleum ether elution) to give,
after concentration of the appropriate fractions (Rf = 0.4 in 3:7 v/v ethyl acetate/40-60
petroleum ether), alcohol 29 (67 mg, 89%) as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.02 (d, J = 9.2 Hz, 1H), 3.98 (s, 1H), 3.52 (m, 1H),
3.46 (d, J = 8.6 Hz, 1H), 3.28 (m, 1H), 2.23 (m, 1H), 2.05 (m, 1H), 1.81 (broad s,
1H), 1.59 (m, 1H), 1.53 (s, 3H), 1.48 (s, 1H), 1.44 (s, 3H), 1.41 (s, 3H), 1.22 (s, 3H),
1.19 (m, 1H), 1.13 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 107.2, 82.6, 80.7, 75.3, 68.7, 64.7, 42.3, 37.8, 33.5,
30.1, 26.7, 26.6, 25.8, 18.3, 15.6.
IR (KBr) νmax 3493, 2973, 2938, 1377, 1247, 1207, 1096, 1052 cm–1.
MS (EI, 70 eV) m/z 255 [(M−CH3•)+, 100%], 195 (45), 194 (40), 151 (85), 149 (40),
123 (70).
HRMS (M−CH3•)+ calcd for C14H23O4 255.1596, found 255.1602.
Specific rotation [α]D = −74.5 (c = 0.4, CHCl3).
(3aS,4R,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethyltetrahydro-4,7-ethanobenzo-
[d][1,3]dioxol-5(4H)-one (30)
!
A magnetically stirred solution of alcohol 29 (120 mg, 0.44 mmol) in dry
dichloromethane (12 mL) maintained at 0 °C under an atmosphere of nitrogen was
treated with pyridine (240 µL, 2.97 mmol) then the Dess-Martin periodinane (DMP)
(339 mg, 0.80 mmol). After being maintained at 0 °C for a further 0.66 h the reaction
mixture was warmed to 22 °C, kept at this temperature for 0.5 h then poured into
water (30 mL) and extracted with dichloromethane (3 × 50 mL). The combined
organic phases were then dried (MgSO4), filtered and concentrated under reduced
pressure and the residue so formed subjected to flash column chromatography (silica,
1:9 v/v ethyl acetate/40-60 petroleum ether elution) to give, after concentration of the
OO
O
30
O

! S30!
appropriate fractions (Rf = 0.5 in 3:7 v/v ethyl acetate/40-60 petroleum ether),
compound 30 (94 mg, 79%) as a colorless, crystalline solid, mp = 62-64 °C. 1H NMR (400 MHz, CDCl3) δ 3.66 (d, J = 7.8 Hz, 1H), 3.62-3.58 (complex m, 1H),
3.56 (s, 1H), 3.29 (m, 1H), 2.49 (m, 1H), 2.35 (m, 1H), 2.21 (m, 2H), 1.60 (m, 1H),
1.50 (s, 3H), 1.39 (s, 3H), 1.34 (s, 3H), 1.22 (s, 3H), 1.09 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 212.0, 110.0, 84.6, 80.6, 75.4, 64.9, 54.5, 39.4, 38.4,
29.0, 26.5, 26.3, 25.7, 15.4, 14.8.
IR (KBr) νmax 2977, 2936, 2875, 1733, 1380, 1244, 1205, 1092, 1054, 890 cm–1.
MS (ESI, +ve) m/z 291 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C15H24NaO4 291.1572, found 291.1574.
Specific rotation [α]D = −27.2 (c = 0.3, CHCl3).
!(3aS,4R,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethyl-3a,4,7,7a-tetrahydro-4,7-ethano-
benzo[d][1,3]dioxol-5-yl (2E,4E)-hexa-2,4-dienoate (31)
!
A magnetically stirred solution of ketone 30 (49 mg, 0.18 mmol) in THF (5 mL)
maintained under a nitrogen atmosphere was cooled to −78 °C then treated with
KHMDS (540 µL of a 0.5 M solution in toluene, 0.27 mmol). The resulting mixture
was stirred at −78 °C for 1 h then sorbic chloride10 (27 µL, 0.22 mmol) was added
dropwise over 0.16 h. The ensuing mixture was stirred at −78 °C for 2 h then warmed
to 0 °C, quenched with NH4Cl (1 × 5 mL, a saturated aqueous solution) and extracted
with ethyl acetate (3 × 20 mL). The combined organic phases were washed with
NaHCO3 (1 × 20 mL of a saturated aqueous solution) before being dried (MgSO4),
filtered and concentrated under reduced pressure. The light-yellow oil thus obtained
was subjected to flash column chromatography (silica, 1:19→1:9 v/v ethyl acetate/40-
60 petroleum ether gradient elution) to give, after concentration of the appropriate
fractions (Rf = 0.6 in 3:7 v/v ethyl acetate/40-60 petroleum ether), the O-acylation
product 31 (55 mg, 84%) as a clear, colorless but unstable oil.
OO
O
31
OO

! S31!
1H NMR (400 MHz, CDCl3) δ 7.32 (m, 1H), 6.25-6.12 (complex m, 2H), 5.84 (m,
2H), 3.90 (s, 1H), 3.77 (d, J = 6.6 Hz, 1H), 3.57 (m, 1H), 3.39 (m, 1H), 2.57 (complex
m, 1H), 2,46 (m, 1H), 1.86 (d, J = 5.6 Hz, 3H), 1.48 (s, 3H), 1.46 (s, 3H), 1.40 (s,
3H), 1.24 (m, 1H), 1.22 (s, 3H), 1.14 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.7, 151.0, 146.8, 140.6, 129.9, 117.8, 116.8, 112.0,
86.5, 84.5, 77.5, 65.0, 48.0, 41.8, 30.3, 28.6, 27.3, 27.2, 18.8, 15.5, 14.5.
(3aS,4R,7R,9R,Z)-9-Ethoxy-6-((2E,4E)-1-hydroxyhexa-2,4-dien-1-ylidene)-
2,2,4,7a-tetramethyltetrahydro-4,7-ethanobenzo[d][1,3]dioxol-5(4H)-one (32)
!
A magnetically stirred solution of ketone 30 (89 mg, 0.33 mmol) in THF (20 mL)
maintained at −78 °C under a nitrogen atmosphere was treated, dropwise over 0.08 h,
with KHMDS (1.33 mL of a 0.5 M solution in toluene, 0.66 mmol). The resulting
mixture was stirred at −78 °C for 0.75 h then a solution of sorbyl cyanide10,11 (60 mg,
0.50 mmol) in THF (6 mL) was added dropwise over 0.16 h. The ensuing mixture
was stirred at −78 °C for 2 h, warmed to 0 °C, quenched at this temperature with
NH4Cl (30 mL of a saturated aqueous solution) and then extracted with ethyl acetate
(3 × 50 mL). The combined organic phases were then dried (MgSO4), filtered and
concentrated under reduced pressure to give a light-yellow oil that was subjected to
flash column chromatography (silica, 1:19 v/v ethyl acetate/40-60 petroleum ether
elution) and thereby affording two fractions, A and B.
Concentration of fraction A (Rf = 0.6, in 3:7 v/v ethyl acetate/40-60 petroleum
ether) gave compound 32 (66 mg, 55% or 81% brsm) as a pale-yellow, crystalline
solid, mp = 92-94 °C. 1H NMR (400 MHz, CDCl3) δ 14.12 (s, 1H), 7.21 (dd, J = 15.0 and 10.9 Hz, 1H),
6.24 (m, 1H), 6.10 (m, 2H), 3.74 (d, J = 6.9 Hz, 1H), 3.66 (s, 1H), 3.52 (m, 1H), 3.30
(m, 1H), 2.85 (broad s, 1H), 2.57 (m, 1H), 1.86 (d, J = 6.7 Hz, 3H), 1.50 (s, 3H), 1.44
(m, 1H), 1.41 (s, 3H), 1.30 (s, 6H), 1.09 (t, J = 7.0 Hz, 3H).
32
OO
O
OH
O

! S32!
13C NMR (100 MHz, CDCl3) δ 203.1, 165.9, 140.6, 138.1, 131.1, 118.7, 110.9, 109.9,
85.0, 82.2, 75.5, 65.0, 53.8, 40.3, 29.9, 27.1, 26.9(4), 26.8(6), 18.9, 15.3, 14.0.
IR (KBr) νmax 3463, 2977, 2934, 1737, 1607, 1569, 1446, 1378, 1239, 1096, 1053,
873 cm–1.
MS (ESI, +ve) m/z 385 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C21H30NaO5 385.1991, found 385.1994.
Specific rotation [α]D = +116.6 (c = 0.2, CHCl3).
Concentration of the fraction B (Rf = 0.2, in 3:7 v/v ethyl acetate/40-60
petroleum ether) gave compound 30 (29 mg, 33% recovery) as a clear, colorless solid
that was identical, in all respects, with an authentic sample.
!(1S,4R,5R,6S,7R,Z)-7-Ethoxy-5,6-dihydroxy-3-((2E,4E)-1-hydroxyhexa-2,4-dien-
1-ylidene)-1,5-dimethylbicyclo[2.2.2]octan-2-one (33)
!!
A magnetically stirred mixture of compound 32 (63 mg, 0.17 mmol) in acetic
acid/water (7.0 mL of a 4:1 v/v mixture) maintained at 22 °C was treated with AG-
50W-X8 resin (151 mg of H+ form) and the ensuing mixture was heated at 70 °C for
24 h before being cooled to 22 °C and treated with pyridinium p-toluenesulfonate (43
mg, 0.17 mmol). The ensuing mixture was stirred at 70 °C for 24 h then cooled and
filtered. The solid thus retained was washed with ethyl acetate (20 mL) and the
combined filtrates washed with water (1 x 20 mL). The separated aqueous phase was
extracted with ethyl acetate (1 x 50 mL) and the combined organic phases
concentrated under reduced pressure to give a light-yellow oil. Subjection of this
material to flash column chromatography (silica, 1:19 → 3:7 v/v ethyl acetate/40-60
petroleum ether elution) afforded two fractions, A and B.
Concentration of the fraction A (Rf = 0.6, in 3:7 v/v ethyl acetate/40-60
petroleum ether) gave compound 32 (15 mg, 24% recovery) as a light-yellow solid
that was identical, in all respects, with an authentic sample.
33
OOH
O
OH
OH

! S33!
Concentration of the fraction B (Rf = 0.2, in 3:7 v/v ethyl acetate/40-60
petroleum ether) gave diol 33 (35 mg, 63% or 82% brsm) as a white, amorphous
powder, mp = 51-54 °C. 1H NMR (600 MHz, CDCl3) δ 14.20 (s, 1H), 7.22 (dd, J = 14.9 and 11.0 Hz, 1H),
6.25 (m, 1H), 6.20-6.08 (complex m, 2H), 3.70 (m, 1H), 3.52 (m, 1H), 3.31 (m, 1H),
3.19 (s, 1H), 2.86 (m, 1H), 2.68 (m, 1H), 1.87 (d, J = 6.8 Hz, 3H), 1.38 (m, 1H), 1.27
(s, 3H), 1.22 (s, 3H), 1.10 (t, J = 7.0 Hz, 3H) (signals due to hydroxyl group protons
not observed). 13C NMR (150 MHz, CDCl3) δ 203.5, 165.9, 140.8, 138.3, 131.1, 118.7, 110.8, 76.1,
73.9, 70.8, 65.0, 54.9, 41.1, 31.0, 29.3, 18.9, 15.4, 12.9. 1H NMR (600 MHz, CD3OD) δ 7.20 (dd, J = 15.0 and 11.0 Hz, 1H), 6.43-6.26
(complex m, 2H), 6.14 (m, 1H), 3.75 (dd, J = 8.6 and 3.4 Hz, 1H), 3.53 (m, 1H), 3.29
(m, 1H), 3.05 (s, 1H), 2.88 (t, J = 2.0 Hz, 1H), 2.72 (m, 1H), 1.87 (dd, J = 6.9 and 1.5
Hz, 3H), 1.29 (m, 1H), 1.18 (s, 3H), 1.11 (s, 3H), 1.09 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CD3OD) δ 205.8, 166.3, 141.7, 138.8, 132.4, 119.9, 112.4, 77.0,
75.6, 71.0, 65.8, 56.2, 42.2, 32.0, 29.1, 18.8, 15.6, 13.4.
IR (KBr) νmax 3391, 2971, 2931, 1631, 1602, 1562, 1389, 1144, 1091, 991, 920, 732
cm–1.
MS (ESI, +ve) m/z 345 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C18H26NaO5 345.1678, found 345.1680.
Specific rotation [α]D = +16.0 (c = 0.8, CHCl3).
(1S,3R,4R,7R,Z)-7-Ethoxy-3-hydroxy-5-((2E,4E)-1-hydroxyhexa-2,4-dien-1-
ylidene)-1,3-dimethylbicyclo[2.2.2]octane-2,6-dione (ent-4)
A magnetically stirred solution of diol 33 (50 mg, 0.16 mmol) in dichloromethane (19
mL) was treated with 4-methylmorpholine N-oxide (49 mg, 0.418 mmol) and
molecular sieves (39 mg of powdered 4 Å material) then tetra-n-propylammonium
O
OOH
O
OH
ent-4

! S34!
perruthenate (11 mg, 0.031 mmol). The resulting mixture was stirred at 22 °C for 0.75
h then filtered through a pad of TLC-grade silica. The filtrate was concentrated under
reduced pressure to give a light-yellow oil and subjection of this material to flash
chromatography (silica, 1:19 → 1:9 v/v ethyl acetate/30-40 petroleum ether gradient
elution) afforded two fractions, A and B.
Concentration of the fraction A (Rf = 0.2 in 3:7 v/v ethyl acetate/40-60
petroleum ether) gave compound 33 (6 mg, 12% recovery) as a light-yellow,
amorphous powder that was identical with an authentic sample.
Concentration of the fraction B (Rf = 0.5 in 3:7 v/v ethyl acetate/40-60
petroleum ether) gave an oil that was subjected to flash chromatography (silica,
6:15:100 → 6:15:80 v/v ethyl acetate/dichloromethane/30-40 petroleum ether
gradient elution) to give, after concentration of the appropriate fractions (Rf = 0.5 in
3:7 v/v ethyl acetate/40-60 petroleum ether), compound ent-4 (26 mg, 52% or 59%
brsm) as a light-yellow, amorphous powder, mp = 83-85 °C. 1H NMR (600 MHz, CDCl3) δ 13.95 (s, 1H), 7.29 (dd, J = 14.9 and 10.9 Hz, 1H),
6.28 (m, 1H), 6.20-6.13 (complex m, 2H), 3.55 (m, 2H), 3.37 (m, 1H), 3.15 (m, 1H),
2.78 (m, 1H), 2.56 (broad s, 1H), 1.89 (dd, J = 6.8 and 1.3 Hz, 3H), 1.68 (m, 1H),
1.31 (s, 3H), 1.20 (s, 3H), 1.11 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 211.1, 196.5, 166.5, 141.9, 139.3, 131.0, 118.2, 110.5,
79.3, 74.7, 67.2, 65.8, 39.9, 30.8, 24.5, 19.0, 15.2, 9.1. 1H NMR (600 MHz, CD3OD) δ 7.27 (dd, J = 14.9 and 11.0 Hz, 1H), 6.38 (m, 2H),
6.18 (m, 1H), 3.61 (m, 1H), 3.58 (m, 1H), 3.34 (m, 1H), 3.16 (m, 1H), 2.83 (m, 1H),
1.87 (dd, J = 6.9 and 1.3 Hz, 3H), 1.63 (m, 1H), 1.20 (s, 3H), 1.14 (s, 3H), 1.10 (t, J =
7.0 Hz, 3H) (signals due to hydroxyl group protons not observed). 13C NMR (150 MHz, CD3OD) δ 210.3, 198.3, 167.2, 142.9, 139.8, 132.3, 119.5,
112.3, 79.9, 74.9, 68.7, 66.3, 41.7, 31.8, 24.0, 18.8, 15.5, 9.6. 1H NMR (600 MHz, C6D6) δ 14.65 (s, 1H), 7.33 (dd, J = 14.9 and 11.1 Hz, 1H), 5.97-
5.90 (complex m, 2H), 5.58 (m, 1H), 3.33 (m, 1H), 3.14 (m, 1H), 3.00 (m, 1H), 2.92
(m, 1H), 2.76 (m, 1H), 2.28 (s, 1H), 1.65 (s, 3H), 1.56 (m, 1H), 1.46 (dd, J = 6.9 and
1.4 Hz, 3H), 0.96 (s, 3H), 0.84 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, C6D6) δ 210.2, 197.1, 166.4, 141.7, 138.3, 131.2, 118.7, 111.0,
79.4, 74.6, 67.6, 65.5, 40.2, 31.0, 24.1, 18.6, 15.2, 9.7. 1H NMR [600 MHz, (CD3)2CO] δ 14.08 (s, 1H), 7.26 (dd, J = 14.9 and 11.1 Hz, 1H),
6.53 (d, J = 10.0 Hz, 1H), 6.41 (m, 1H), 6.23 (m, 1H), 4.81 (s, 1H), 3.64 (m, 1H),

! S35!
3.58 (m, 1H), 3.35 (m, 1H), 3.25 (m, 1H), 2.89 (m, 1H), 1.86 (d, J = 4.2 Hz, 3H), 1.64
(m, 1H), 1.20 (s, 3H), 1.16 (s, 3H), 1.07 (t, J = 7.0 Hz, 3H).
13C NMR [150 MHz, (CD3)2CO] δ 209.5, 198.0, 166.9, 142.0, 139.2, 132.0, 119.8,
112.2, 79.2, 74.3, 68.0, 65.6, 41.0, 31.5, 24.0, 18.8, 15.5, 9.5.
IR (KBr) νmax 3456, 2976, 2936, 2874, 1731, 1632, 1605, 1563, 1446, 1373, 1094,
1023, 998 cm–1.
MS (ESI, +ve) m/z 343 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C18H24NaO5 343.1521, found 343.1521.
Specific rotation [α]D = −234.0 (c = 0.1, MeOH) {lit.12 value (for 1) [α]D20 = +219 (c
0.1, methanol)}

! S36!
Scheme S1: Reaction Sequence Used for the Conversion of Diol 23 into Ketone 30
Experimental Protocols for the Conversion of Diol 23 into Ketone 30
(3aS,4R,5R,6S,7R,9R)-6-((tert-Butyldimethylsilyl)oxy)-9-ethoxy-2,2,4,7a-tetra
methylhexahydro-4,7-ethanobenzo[d][1,3]dioxol-5-ol (34)
Sodium hydride (370 mg of a 60% dispersion in mineral oil, 9.17 mmol) was added,
in one portion, to a magnetically stirred solution of diol 23 (530 mg, 1.85 mmol) in
THF (27 mL) maintained at 0 °C under an atmosphere of nitrogen. The ensuing
mixture was stirred at 0 °C for 0.5 h, warmed to 22 °C and then maintained at this
temperature for another 0.5 h before being re-cooled 0 °C then treated, dropwise over
0.16 h, with a solution of TBSCl (310 mg, 2.06 mmol) in THF (20 mL). The ensuing
mixture was stirred at 0 °C for 0.25 h then quenched with ice water (50 mL)
(CAUTION! EXOTHERMIC REACTION AND POSSIBILITY OF HYDROGEN
EVOLUTION) and extracted with ethyl acetate (3 × 100 mL). The combined organic
phases were then dried (MgSO4), filtered and concentrated under reduced pressure
and the residue thus obtained subjected to flash column chromatography (silica,
1:19→1:9 v/v ethyl acetate/40-60 petroleum ether gradient elution) to afford, after
OOHO O
23
HO OOTBSOO
34
HO OOTBSOO
35
O
OO
O
O
30
NaH thenTBSCl
THF0 - 22 oC
1.4 h, 61%
DMP, pyridine
CH2Cl20 - 22 °C
1.7 h, 89%
SmI2THF/MeOH
0 oC20 min, 93%
OOTBSOO
34
HO

! S37!
concentration of the appropriate fractions (Rf = 0.5 in 1:4 v/v ethyl acetate/40-60
petroleum ether), alcohol 34 (452 mg, 61%) as a colorless, crystalline solid, mp = 78-
80 °C. 1H NMR (800 MHz, CDCl3) δ 4.04 (dd, J = 7.5 and 4.3 Hz, 1H), 3.61 (m, 1H), 3.47
(s, 1H), 3.46 (s, 1H), 3.21-3.13 (complex m, 3H), 1.95 (m, 2H), 1.85 (s, 1H), 1.43 (s,
3H), 1.39 (s, 3H), 1.37 (s, 3H), 1.18 (s, 3H), 1.14 (t, J = 7.0 Hz, 3H), 0.91 (s, 9H),
0.08 (s, 3H), 0.06 (s, 3H). 13C NMR (200 MHz, CDCl3) δ 109.0, 86.3, 79.3, 76.1, 69.7, 66.6, 64.6, 43.9, 41.6,
27.1, 26.6, 25.9, 21.7, 18.4(9), 18.4(7), 15.6, −4.8, −5.1 (one signal obscured or
overlapping).
IR (KBr) νmax 3538, 2957, 2935, 2858, 1739, 1376, 1253, 1211, 1093, 1048, 895, 840,
778 cm–1.
MS (ESI, +ve) m/z 423 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C21H40NaO5Si 423.2543, found 423.2547.
Specific rotation [α]D = −16.2 (c = 0.4, CHCl3).
(3aS,4R,6S,7R,9R)-6-((tert-Butyldimethylsilyl)oxy)-9-ethoxy-2,2,4,7a-tetramethyl-
tetrahydro-4,7-ethanobenzo[d][1,3]dioxol-5(4H)-one (35)
A magnetically stirred solution of alcohol 34 (350 mg, 0.88 mmol) in dry
dichloromethane (45 mL) maintained at 0 °C under an atmosphere of nitrogen was
treated with pyridine (420 µL, 5.19 mmol) then the Dess-Martin periodinane (DMP)
(669 mg, 1.65 mmol). After stirring the reaction mixture at 0 °C for a further 0.66 h it
was warmed to 22 °C and maintained at this temperature for 1 h then poured into
water (100 mL) and extracted with dichloromethane (3 × 100 mL). The combined
organic phases were then dried (MgSO4), filtered and concentrated under reduced
pressure. The residue thus obtained was subjected to flash column chromatography
(silica, 1:19 v/v ethyl acetate/40-60 petroleum ether elution) and gave, after
concentration of the appropriate fractions (Rf = 0.7 in 3:7 v/v ethyl acetate/40-60
OOTBSOO
35
O

! S38!
petroleum ether), ketone 35 (311 mg, 89%) as a colorless, crystalline solid, mp = 61-
63 °C. 1H NMR (400 MHz, CDCl3) δ 3.72-3.69 (complex m, 2H), 3.60 (s, 1H), 3.55 (m, 1H),
3.23 (m, 1H), 2.26 (m, 1H), 2.15 (m, 1H), 2.03 (m, 1H), 1.50 (s, 3H), 1.42 (s, 3H),
1.40 (s, 3H), 1.18 (s, 3H), 1.08 (t, J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 210.6, 110.2, 84.5, 79.5, 75.2, 70.5, 64.3, 54.8, 46.4,
26.9, 26.7, 26.4, 25.8, 22.6, 18.3, 15.4, 14.4, −4.4, −5.1.
IR (KBr) νmax 2933, 2858, 1740, 1249, 1150, 1103, 1035, 837, 779 cm–1.
MS (ESI, +ve) m/z 421 [(M+Na)+, 100%].
HRMS (M+Na)+ calcd for C21H38NaO5Si 421.2386, found 421.2377.
Specific rotation [α]D = −60.0 (c = 0.4, CHCl3).
(3aS,4R,7R,9R)-9-Ethoxy-2,2,4,7a-tetramethyltetrahydro-4,7-ethanobenzo-
[d][1,3]dioxol-5(4H)-one (30)
A magnetically stirred solution of compound 35 (265 mg, 0.67 mmol) in
THF/methanol (40 mL of a 2:1 v/v mixture) maintained under an atmosphere of
nitrogen at 0 °C was treated, dropwise, with samarium diiodide (a 0.07-0.12 M
solution in THF, about 4.90 mmol) until the reaction mixture maintained a pale-
green/blue color for 0.16 h. At this point, the reaction mixture was poured into
NaHCO3 (100 mL of a saturated aqueous solution) and extracted with ethyl acetate (3
× 200 mL). The combined organic phases were then dried (MgSO4), filtered and
concentrated under reduced pressure. The residue thus obtained was subjected to flash
column chromatography (silica, 3:17 v/v ethyl acetate/40-60 petroleum ether elution)
and gave, after concentration of the appropriate fractions (Rf = 0.5 in 3:7 v/v ethyl
acetate/40-60 petroleum ether), ketone 30 (166 mg, 93%) as a colorless, crystalline
solid that was identical with an authentic sample.
!
OO
O
O
30
! S39!
Table S1. Comparison of the 13C NMR Spectral Data Sets Derived From Rezishanone C and ent-Rezishanone C in Various Solvents
CD3OD CDCl3 C6D6 (CD3)2CO
δca δcb △δ δcc δcb △δ δcd δcb △δ δce δcb △δ
210.4 210.3 −0.1 210.9 211.1 +0.2 210.5 210.2 −0.3 209.5 209.5 0
198.3 198.3 0 196.4 196.5 +0.1 197.0 197.1 +0.1 197.9 198.0 +0.1
167.3 167.2 −0.1 166.4 166.5 +0.1 166.4 166.4 0 166.9 16.9 0
142.9 142.9 0 141.8 141.9 +0.1 141.7 141.7 0 142.0 142.0 0
139.7 139.8 +0.1 139.2 139.3 +0.1 138.2 138.3 +0.1 139.2 139.2 0
132.3 132.3 0 130.9 131.0 +0.1 131.2 131.2 0 132.0 132.0 0
119.5 119.5 0 118.0 118.2 +0.2 118.6 118.7 +0.1 119.8 119.8 0
112.3 112.3 0 110.4 110.5 +0.1 111.0 111.0 0 112.1 122.2 +0.1
79.9 79.9 0 79.1 79.3 +0.2 79.3 79.4 +0.1 79.2 79.2 0
74.9 74.9 0 74.6 74.7 +0.1 74.3 74.6 +0.3 74.3 74.3 0
68.7 68.7 0 67.0 67.2 +0.2 67.6 67.6 0 68.0 68.0 0
66.3 66.3 0 65.6 65.8 +0.2 65.5 65.5 0 65.6 65.6 0
41.7 41.7 0 39.7 39.9 +0.2 40.2 40.2 0 40.9 41.0 +0.1
31.8 31.8 0 30.6 30.8 +0.2 30.9 31.0 +0.1 31.4 31.5 +0.1
24.0 24.0 0 24.3 24.5 +0.2 24.1 24.1 0 24.0 24.0 0
18.8 18.8 0 18.8 19.0 +0.2 18.6 18.6 0 18.8 18.8 0
15.4 15.5 +0.1 15.1 15.2 +0.1 15.2 15.2 0 15.5 15.5 0
9.6 9.6 0 8.9 9.1 +0.2 9.6 9.7 +0.1 9.5 9.5 0 aData obtained from reference 12 - recorded in CD3OD. bData obtained from present work-recorded at 150 MHz in CD3OD, CDCl3, C6D6, (CD3)2CO, respectively. cData obtained from reference 13 - recorded in CDCl3. dData obtained from reference 14 -recorded in C6D6 at 75 MHz. eData obtained from reference 15 - recorded in (CD3)2CO at 75 MHz.

! S40!
X-ray Crystallographic Studies
Crystallographic Data.
Compound 7. C12H16O3, M = 208.26, T = 150 K, trigonal, space group P3221, Z = 6, a
= 10.5437(1) Å, c = 17.0184(1) Å; V = 1638.46(2) Å3, Dx = 1.266 g cm–3, 2160
unique data (2θmax = 144.8°), R = 0.024 [for 2139 reflections with I > 2.0σ(I)]; Rw =
0.064 (all data), S = 1.01.
Compound 8. C9H12O3, M = 168.19, T = 150 K, orthorhombic, space group P212121, Z
= 4, a = 6.9885(1) Å, b = 7.9474(1) Å, c = 14.4684(1) Å; V = 803.58(2) Å3, Dx =
1.390 g cm–3, 1583 unique data (2θmax = 144.6°), R = 0.056 [for 1574 reflections with
I > 2.0σ(I)]; Rw = 0.056 (all data), S = 1.01.
!Compound 9. C11H16O4, M = 212.25, T = 200 K, orthorhombic, space group P212121,
Z = 4, a = 5.2471(3) Å, b = 10.1800(4) Å, c = 19.7165(9) Å; V = 1053.17(9) Å3, Dx =
1.339 g cm–3, 1121 unique data (2θmax = 50.0°), R = 0.47 [for 886 reflections with I >
2.0σ(I)]; Rw = 0.111 (all data), S = 0.95.
Compound 11. C15H28O3Si, M = 284.47, T = 150 K, monoclinic, space group C2, Z =
8, a = 31.7863(3) Å, b = 6.3670(1) Å, c = 18.8067(2) Å; β = 117.7194(12) °; V =
3369.35(8) Å3, Dx = 1.122 g cm–3, 5835 unique data (2θmax = 144.8°), R = 0.027 [for
5748 reflections with I > 2.0σ(I)]; Rw = 0.073 (all data), S = 1.00.
Compound 14. C13H18O5, M = 254.28, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 7.0640(1) Å, b = 8.5640(1) Å, c = 21.1355(2) Å; V = 1278.62(3) Å3, Dx =
1.321 g cm–3, 2534 unique data (2θmax = 144.8°), R = 0.025 [for 2492 reflections with
I > 2.0σ(I)]; Rw = 0.065 (all data), S = 1.00.
Compound 16. C12H18O4, M = 226.27, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 8.2169(1) Å, b = 10.8611(1) Å, c = 13.4792(1) Å; V = 1202.95(2) Å3, Dx =
1.249 g cm–3, 2380 unique data (2θmax = 144.8°), R = 0.026 [for 2350 reflections with
I > 2.0σ(I)]; Rw = 0.072 (all data), S = 1.00.

! S41!
Compound 17. C10H16O3, M = 184.24, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 6.4980(1) Å, b = 11.4064(1) Å, c = 12.4797(1) Å; V = 924.98(2) Å3, Dx =
1.323 g cm–3, 1733 unique data (2θmax = 139.6°), R = 0.025 [for 1723 reflections with
I > 2.0σ(I)]; Rw = 0.067 (all data), S = 1.01.
Compound 22. C12H22O4, M = 230.30, T = 150 K, orthorhombic, space group P212121,
Z = 12, a = 12.6340(1) Å, b = 13.4519(1) Å, c = 21.7470(1) Å; V = 3695.93(4) Å3, Dx
= 1.242 g cm–3, 7216 unique data (2θmax = 144.0°), R = 0.025 [for 7015 reflections
with I > 2.0σ(I)]; Rw = 0.062 (all data), S = 1.00.
Compound 26. C23H34O6, M = 406.52, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 7.9838(1) Å, b = 14.1145(1) Å, c = 18.8516(1) Å; V = 2124.34(3) Å3, Dx =
1.271 g cm–3, 4199 unique data (2θmax = 144.8°), R = 0.024 [for 4133 reflections with
I > 2.0σ(I)]; Rw = 0.061 (all data), S = 1.00.
Compound 30. C15H24O4, M = 268.35, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 7.6721(1) Å, b = 9.3354(2) Å, c = 20.5773(3) Å; V = 1473.79(4) Å3, Dx =
1.209 g cm–3, 2911 unique data (2θmax = 144.8°), R = 0.025 [for 2839 reflections with
I > 2.0σ(I)]; Rw = 0.063 (all data), S = 1.00.
Compound 32. C21H30O5, M = 362.47, T = 150 K, orthorhombic, space group P212121,
Z = 4, a = 7.3104(2) Å, b = 8.4083(2) Å, c = 33.0483(10) Å; V = 2031.41(10) Å3, Dx
= 1.185 g cm–3, 3805 unique data (2θmax = 144.6°), R = 0.036 [for 3624 reflections
with I > 2.0σ(I)]; Rw = 0.093 (all data), S = 1.01.
Structure Determination. The image for compound 9 was measured on a
diffractometer (Mo Kα, graphite monochromator, λ = 0.71073 Å) fitted with an area
detector and the data extracted using the DENZO/Scalepack package.16 Images for
compounds 7, 8, 11, 14, 16, 17, 22, 26, 30 and 32 were measured on a diffractometer
(Cu Kα, mirror monochromator, λ = 1.54184 Å) fitted with an area detector and the
data extracted using the CrysAlis package.17 The structure solutions for all eleven
compounds were solved by direct methods (SIR92)18 then refined using the
CRYSTALS program package.19 Atomic coordinates, bond lengths and angles, and

! S42!
displacement parameters have been deposited at the Cambridge Crystallographic Data
Centre (CCDC nos. 1526109-1526119). These data can be obtained free-of-charge via
www.ccdc.cam.ac.uk/data_request/cif, by emailing [email protected], or
by contacting The Cambridge Crystallographic Data Centre, 12 Union Road,
Cambridge CB2 1EZ, UK; fax: +44 1223 336033.

! S43!
References
1. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.;
Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.;
Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.;
Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.;
Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.;
Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd,
J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.;
Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi,
M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.;
Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin,
A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.;
Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.;
Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D.
J. Revision D.01 ed., Gaussian Inc., Wallingford CT, 2013.
2. Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215.
3. Goerigk, L.; Grimme, S. Phys. Chem. Chem. Phys. 2011, 13, 6670.
4. (a) Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724; (b)
Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257.
5. Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113,
6378.
6. Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.
7. Pangborn, A. B.; Giardello, M. A.; Grubbs, R. H.; Rosen, R. K.; Timmers, F. J.
Organometallics 1996, 15, 1518.
8. Banwell, M. G.; Dupuche, J. R.; Gable, R. W. Aust. J. Chem. 1996, 49, 639.
9. Banwell, M. G.; Darmos, P.; Hockless, D. C. R. Aust. J. Chem. 2004, 57, 41.
10. Volp, K. A.; Johnson, D. M.; Harned, A. M. Org. Lett. 2011, 13, 4486.
11. Santelli, M.; Abed, D. E.; Jellal, A. J. Org. Chem. 1986, 51, 1199.
12. Bringmann, G.; Lang, G.; Gulder, T. A. M.; Tsuruta, H.; Muhlbacher, J.;
Maksimenka, K.; Steffens, S.; Schaumann, K.; Stohr, R.; Wiese, J.; Imhoff, J.
F.; Perovic-Ottstadt, S.; Boreiko, O.; Muller, W. E. G. Tetrahedron 2005, 61,
7252.
13. Lee, D.; Lee, J. H.; Cai, X. F.; Shin, J. C.; Lee, K.; Hong, Y.; Lee, J. J. J.
Antibiot. 2005, 58, 615.

! S44!
14. Maskey, R. P.; Grun-Wollny, I.; Laatsch, H. J. Nat. Prod. 2005, 68, 865.
15. Neumann, K.; Abdel-Lateff, A.; Wright, A. D.; Kehraus, S.; Krick, A.; Konig,
G. M. Eur. J. Org. Chem. 2007, 2268.
16. DENZO–SMN. Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data
collected in oscillation mode. In Methods in Enzymology, Volume 276:
Macromolecular Crystallography, Part A; C. W. Carter Jr. and R. M. Sweet,
Eds.; Academic Press: New York, 1997; pp. 307–326.
17. CrysAlis PRO Version 1.171.37.35h (release 09-02-2015 CrysAlis171.NET)
(compiled Feb 9 2015,16:26:32) Agilent Technologies: Oxfordshire, UK.
18. SIR92. Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.
C.; Polidori, G.; Camalli, M. J. Appl. Crystallogr. 1994, 27, 435.
19. Betteridge, P. W.; Carruthers, J. R.; Cooper, R. I.; Prout, K.; Watkin, D. J. J.
Appl. Crystallogr. 2003, 36, 1487.
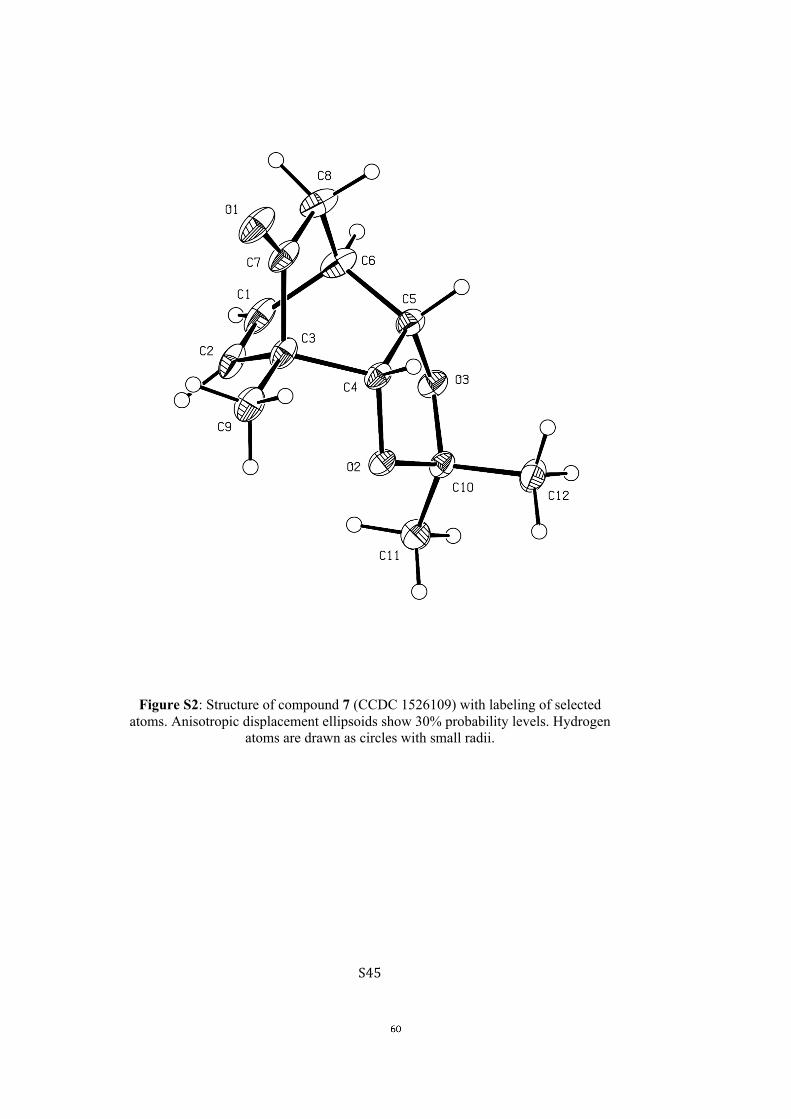
! S45!
Figure S2: Structure of compound 7 (CCDC 1526109) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.
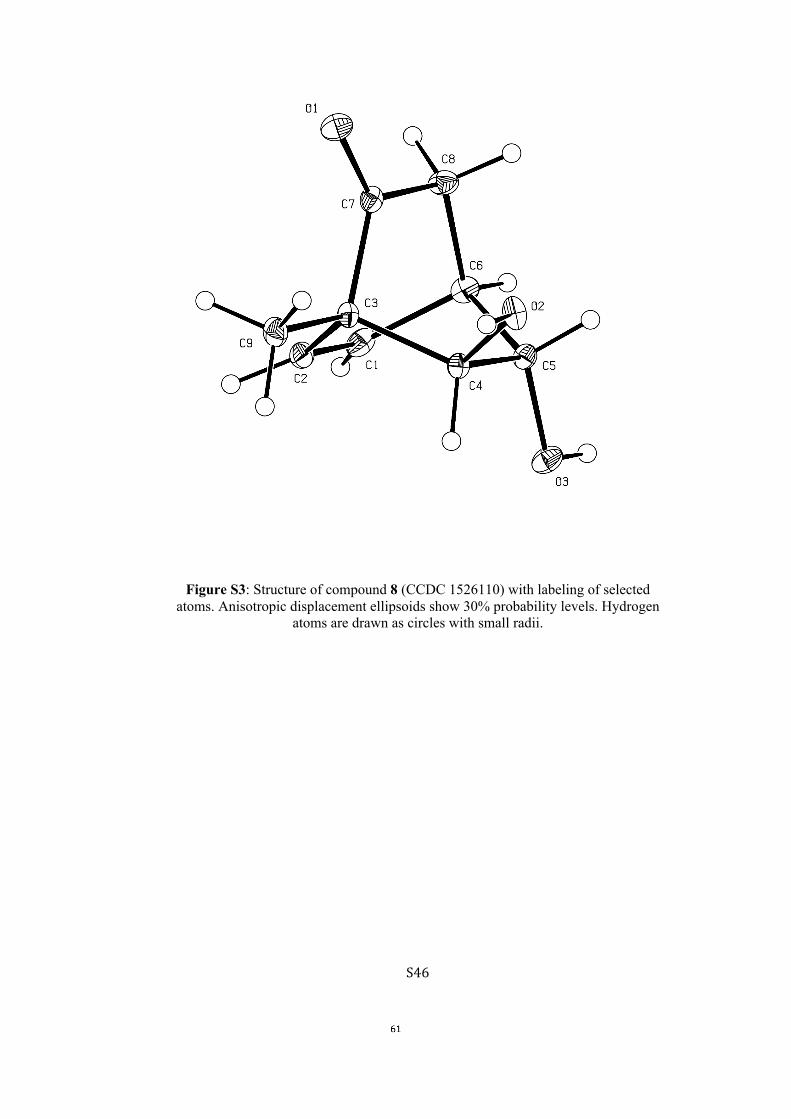
! S46!
Figure S3: Structure of compound 8 (CCDC 1526110) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.
! S47!
Figure S4: Structure of compound 9 (CCDC 1526111) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!!!!!!!!!
! S48!
Figure S5: Structure of compound 11 (CCDC 1526112) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!!!

! S49!
Figure S6: Structure of compound 14 (CCDC 1526113) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!!
! S50!
!
Figure S7: Structure of compound 16 (CCDC 1526114) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!! !
! S51!
Figure S8: Structure of compound 17 (CCDC 1526115) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!!!
! S52!
Figure S9: Structure of compound 22 (CCDC 1526116) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.
! S53!
Figure S10: Structure of compound 26 (CCDC 1526117) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.

! S54!
Figure S11: Structure of compound 30 (CCDC 1526118) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.

! S55!
Figure S12: Structure of compound 32 (CCDC 1526119) with labeling of selected atoms. Anisotropic displacement ellipsoids show 30% probability levels. Hydrogen
atoms are drawn as circles with small radii.!!!
!
S56!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
6.003.01
1.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
71
!
S57!
!5.6
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
6.5
f1 (
ppm
)
1.00
1.00
4.0
4.1
4.2
4.3
4.4
4.5
f1 (
ppm
)
1.00
1.00
1.9
2.0
2.1
f1 (
ppm
)
1.00
1.00
72
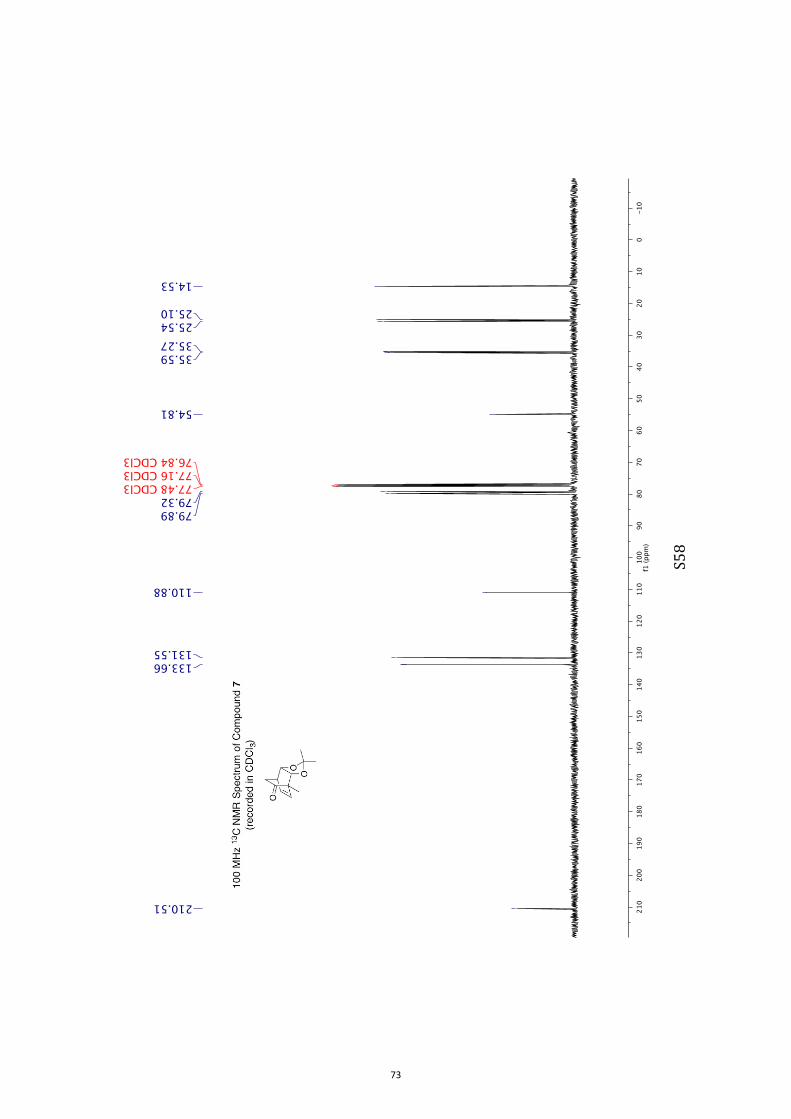
!
S58!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
14.53
25.1025.54
35.2735.59
54.81
76.84 CDCl377.16 CDCl377.48 CDCl379.3279.89
110.88
131.55133.66
210.51
73
!
S59!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.00
2.00
1.00
1.00
1.00
1.00
1.00
3.31 CD3OD_SPE
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
6.5
6.6
f1 (
ppm
)
1.00
1.00
74
!
S60!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
14.83
36.9240.4648.36 CD3OD_SPE48.57 CD3OD_SPE48.79 CD3OD_SPE49.00 CD3OD_SPE49.21 CD3OD_SPE49.42 CD3OD_SPE49.64 CD3OD_SPE57.17
79.9782.80
132.65136.88
213.10
75

!
S61!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.01
1.00
4.00
1.00
1.00
4.01
1.00
1.00
7.26 CDCl3
3.9
4.0
4.1
f1 (
ppm
)
4.01
76
!
S62!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.90
29.07
37.7039.20
65.4065.6373.9076.84 CDCl377.16 CDCl377.48 CDCl3
103.55
135.69
144.39
199.77
77

!
S63!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
6.03
9.04
3.01
1.002.00
1.01
1.00
1.00
1.00
1.00
7.26 CDCl3
78
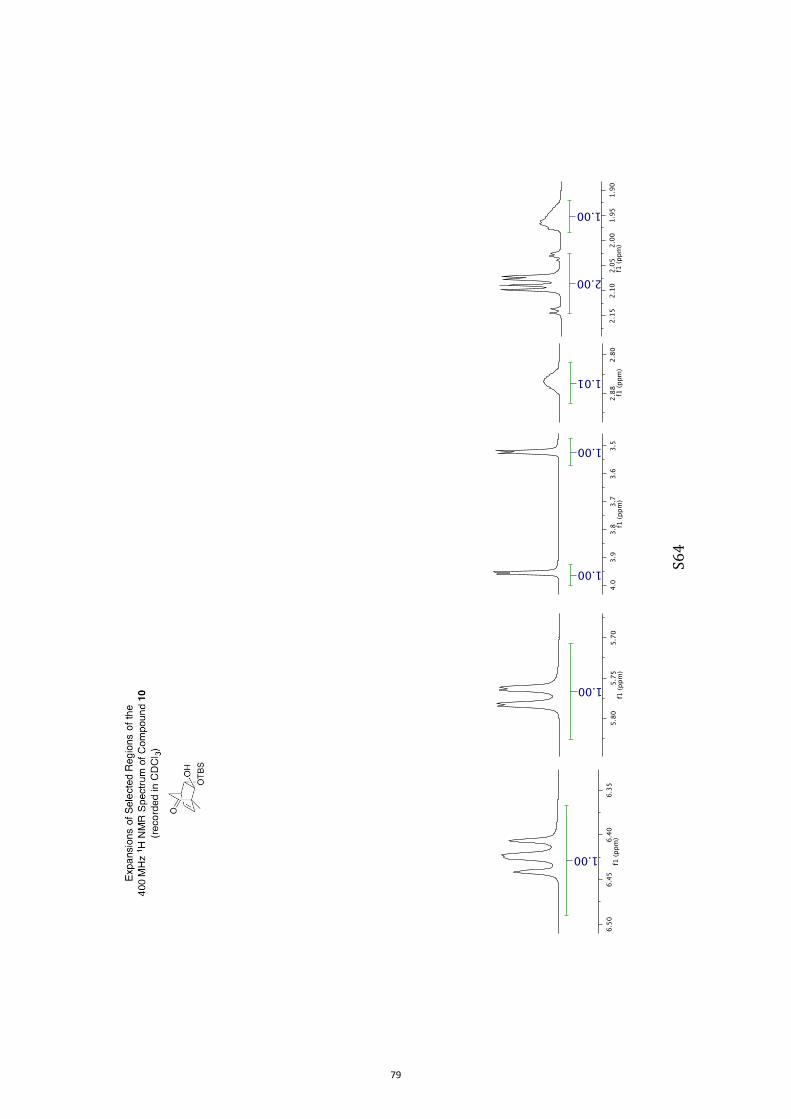
!
S64!
6.3
56
.40
6.4
56
.50
f1 (
ppm
)
1.00
5.7
05
.75
5.8
0f1
(ppm
)
1.00
3.5
3.6
3.7
3.8
3.9
4.0
f1 (
ppm
)
1.00
1.00
2.8
02
.88
f1 (
ppm
)
1.01
1.9
01
.95
2.0
02
.05
2.1
02
.15
f1 (
ppm
)
1.00
2.00
79
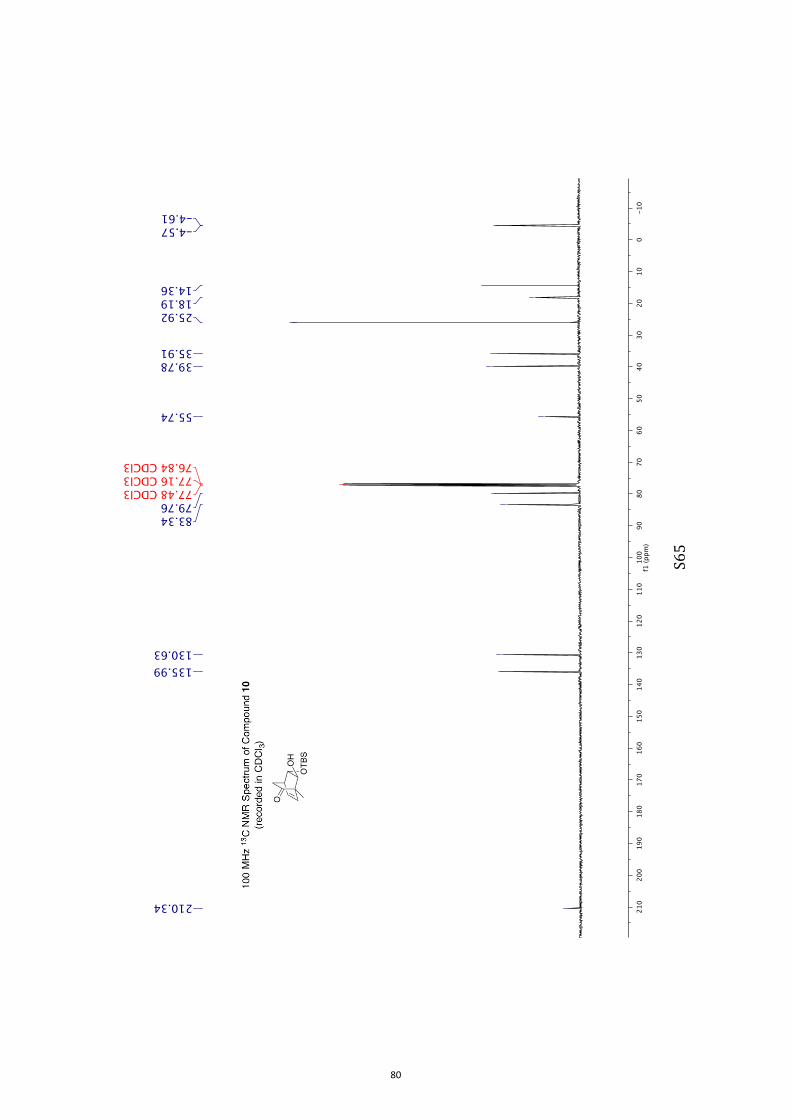
!
S65!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.61-4.57
14.3618.1925.92
35.9139.78
55.74
76.84 CDCl377.16 CDCl377.48 CDCl379.7683.34
130.63
135.99
210.34
80
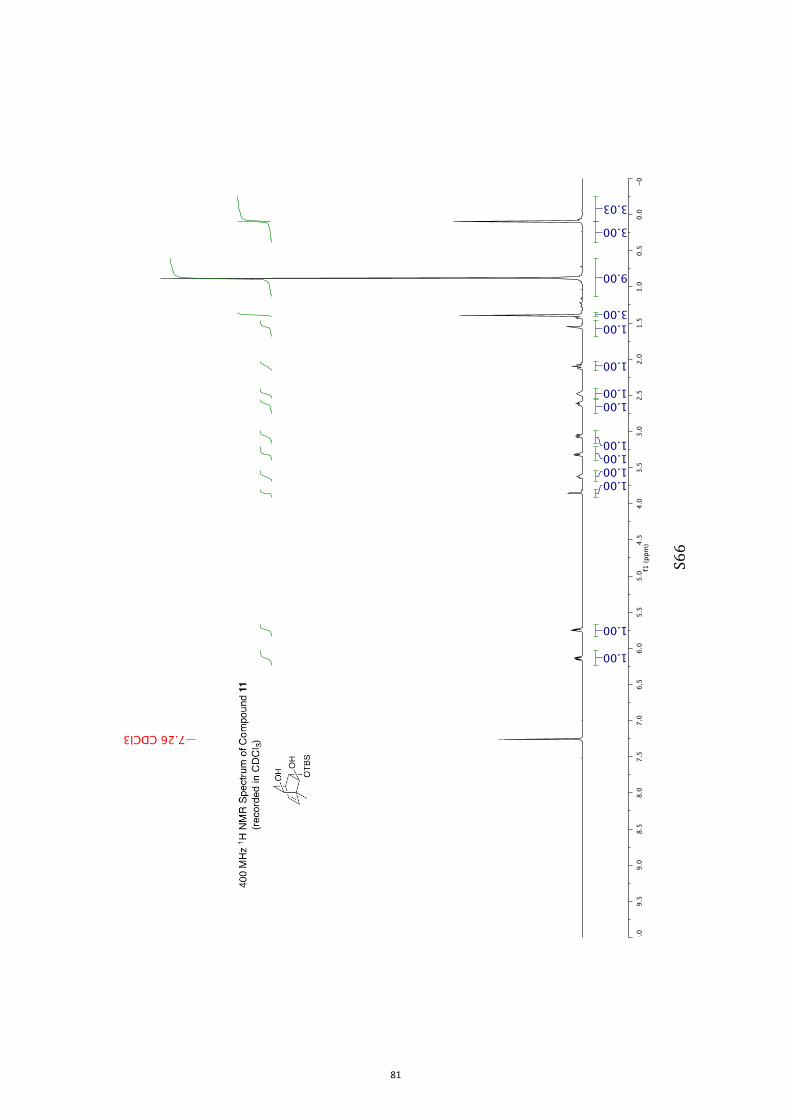
!
S66!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.03
3.00
9.00
3.00
1.00
1.00
1.001.00
1.001.001.001.00
1.00
1.00
7.26 CDCl3
81
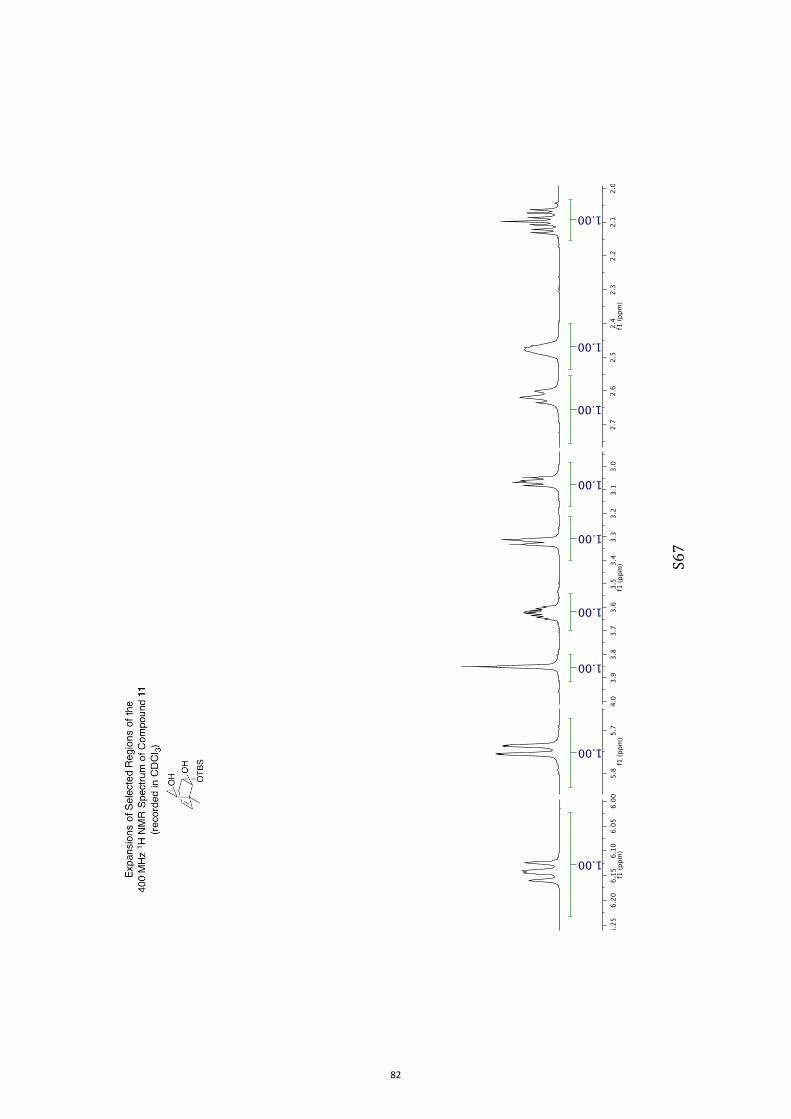
!
S67!
6.0
06.0
56.1
06.1
56.2
06.2
5f1
(ppm
)
1.00
5.7
5.8
f1 (
ppm
)
1.00
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
4.0
f1 (
ppm
)
1.00
1.00
1.00
1.00
2.0
2.1
2.2
2.3
2.4
2.5
2.6
2.7
f1 (
ppm
)
1.00
1.00
1.00
82
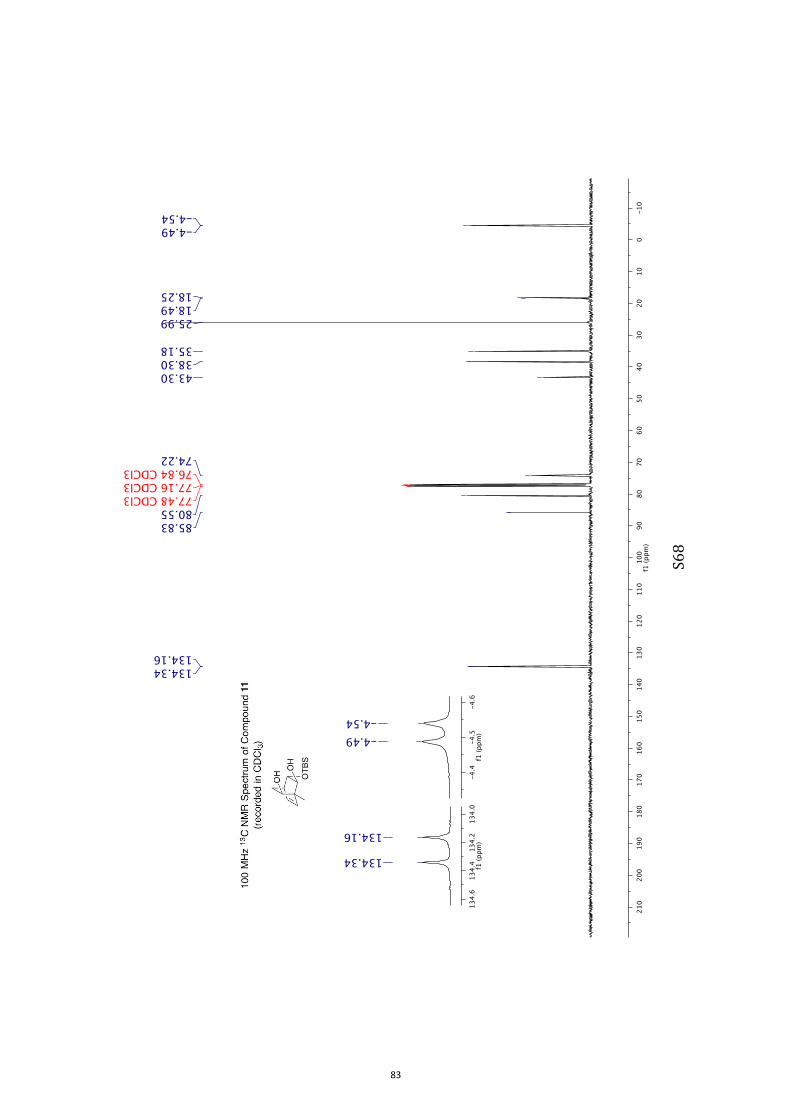
!
S68!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.54-4.49
18.2518.4925.99
35.1838.3043.30
74.2276.84 CDCl377.16 CDCl377.48 CDCl380.5585.83
134.16134.34
13
4.0
13
4.2
13
4.4
13
4.6
f1 (
ppm
)
134.16
134.34
-4
.6-4
.5-4
.4f1
(ppm
)
-4.54
-4.49
83
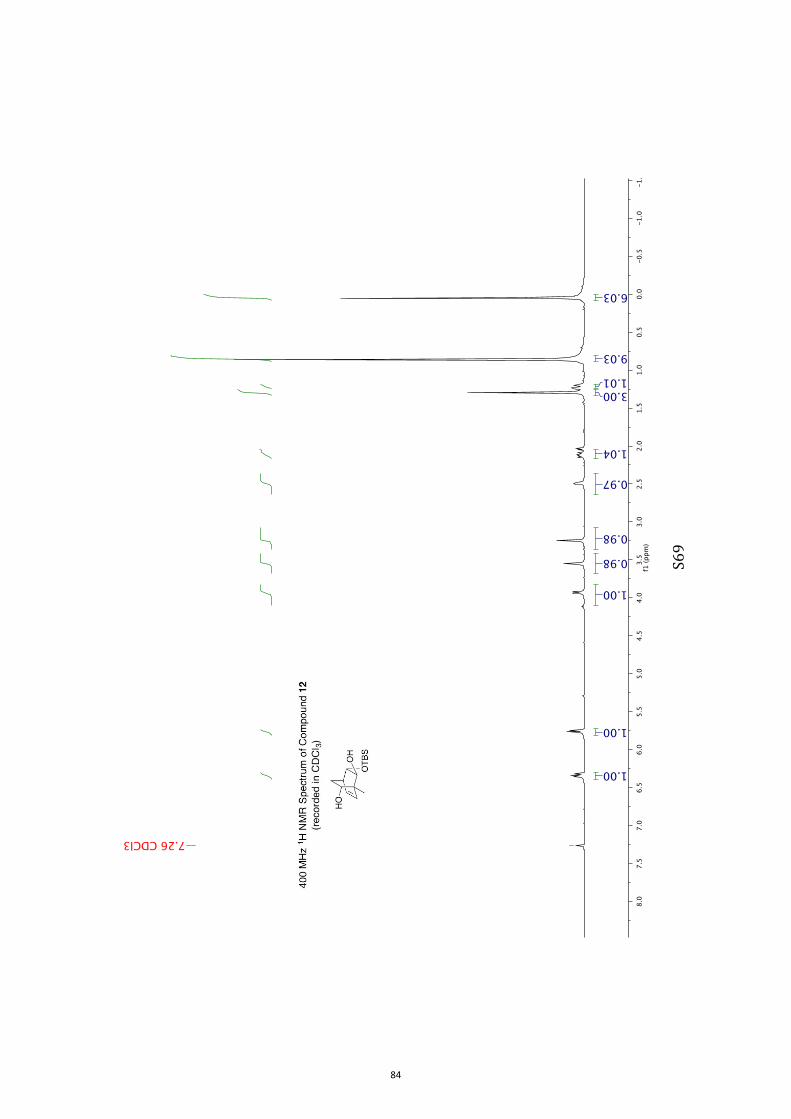
!
S69!
-1
.5-1
.0-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
6.03
9.03
1.013.00
1.04
0.97
0.98
0.98
1.00
1.00
1.00
7.26 CDCl3
84
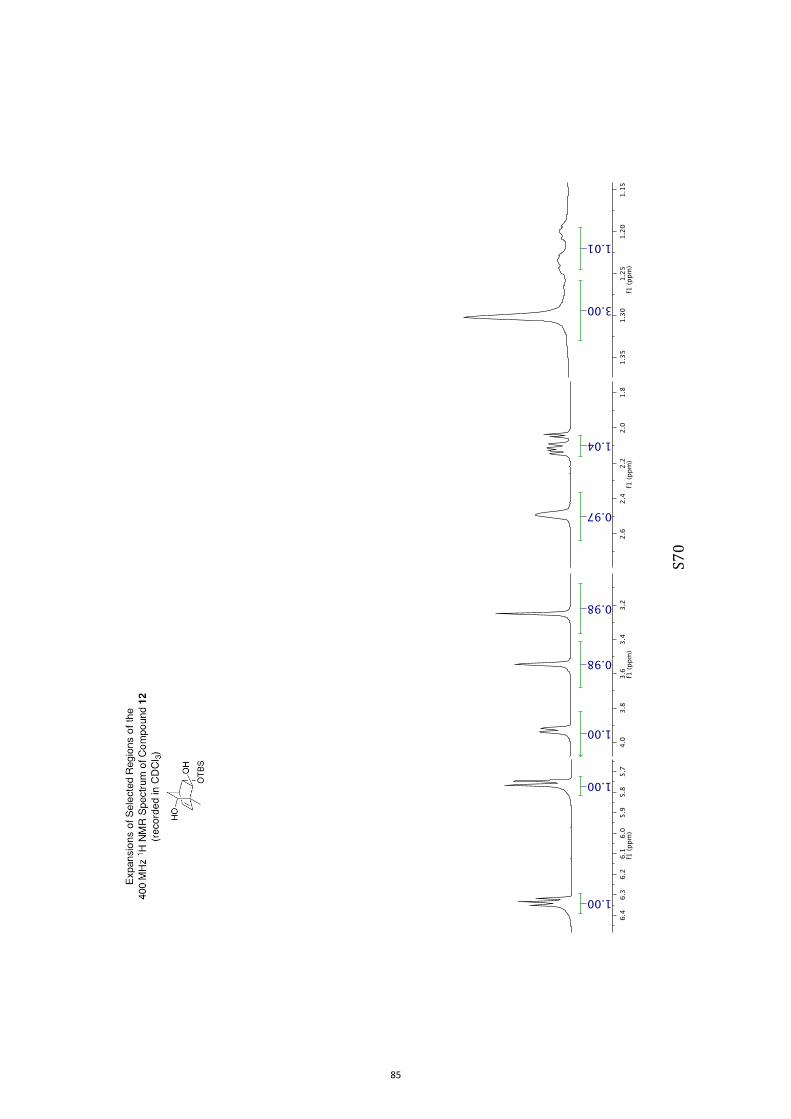
!
S70!
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
f1 (
ppm
)
1.00
1.00
3.2
3.4
3.6
3.8
4.0
f1 (
ppm
)
0.98
0.98
1.00
1.8
2.0
2.2
2.4
2.6
f1 (
ppm
)
1.04
0.97
1.1
51.2
01.2
51.3
01.3
5f1
(ppm
)
1.01
3.00
85
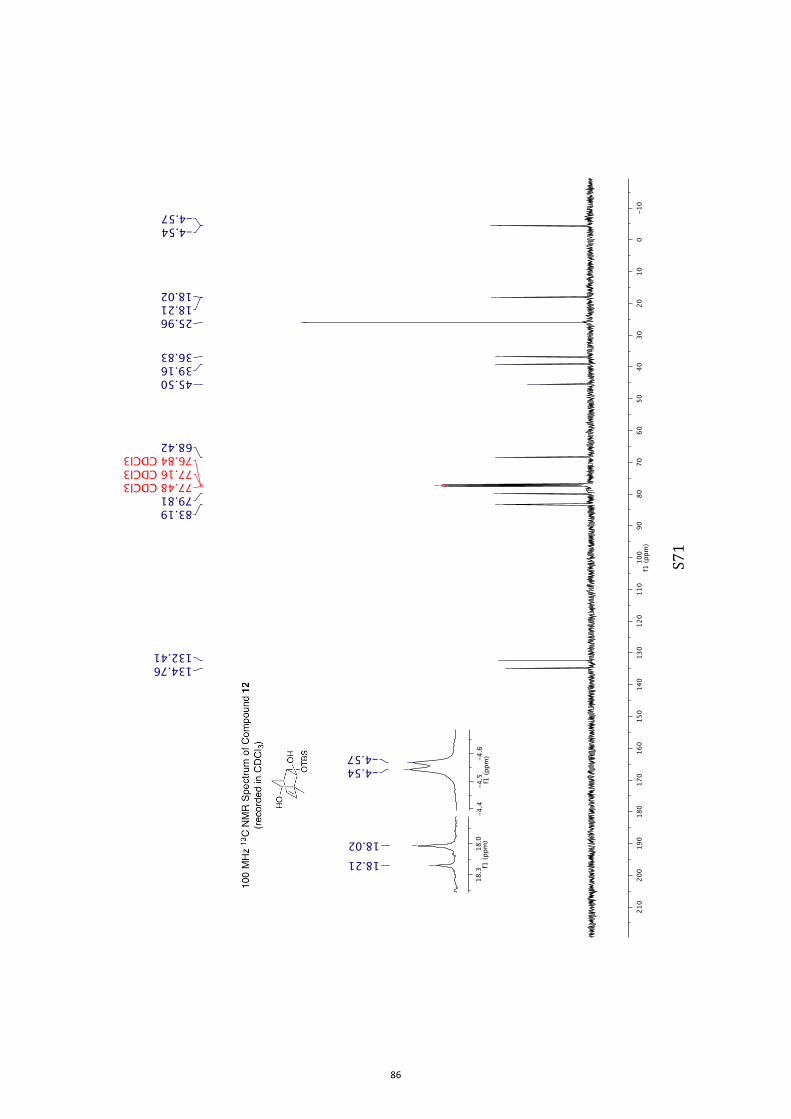
!
S71!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.57-4.54
18.0218.2125.96
36.8339.1645.50
68.4276.84 CDCl377.16 CDCl377.48 CDCl379.8183.19
132.41134.76
18
.01
8.3
f1 (
ppm
)18.02
18.21
-4
.6-4
.5-4
.4f1
(ppm
)-4.57-4.54
86

!
S72!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
5.98
9.003.001.00
3.003.001.001.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
87
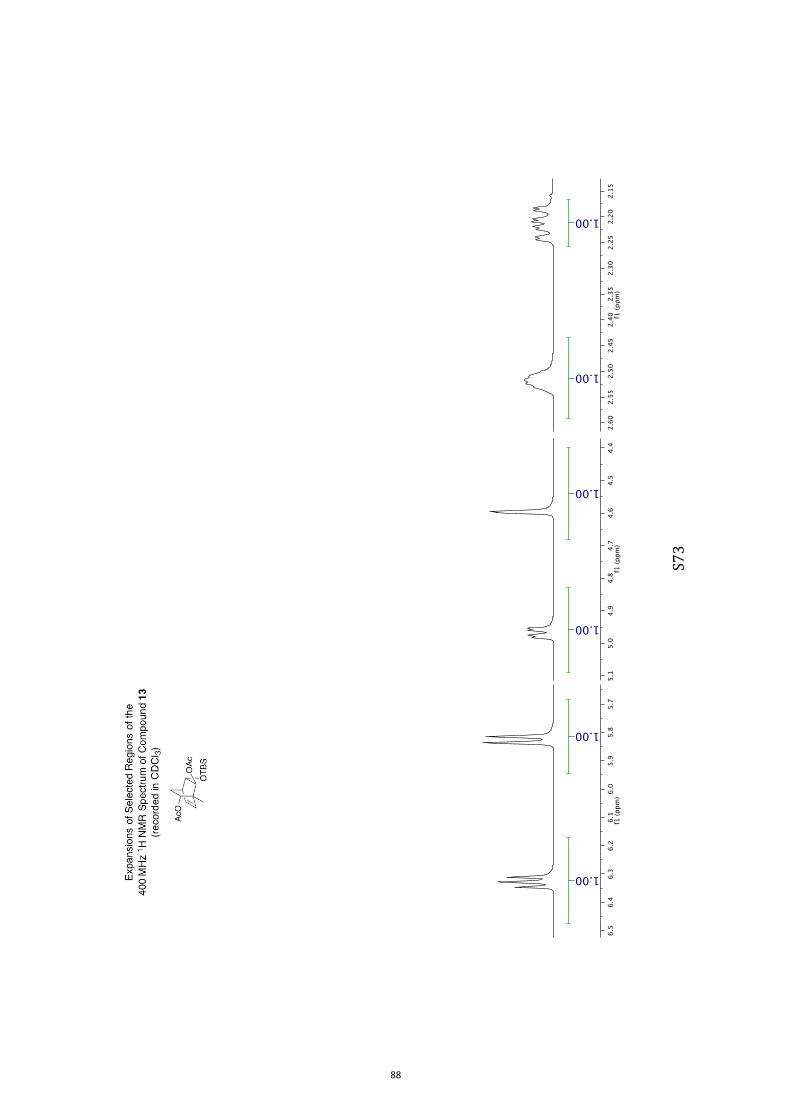
!
S73!
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
6.5
f1 (
ppm
)
1.00
1.00
4.4
4.5
4.6
4.7
4.8
4.9
5.0
5.1
f1 (
ppm
)1.00
1.00
2.1
52
.20
2.2
52
.30
2.3
52
.40
2.4
52
.50
2.5
52
.60
f1 (
ppm
)
1.00
1.00
88
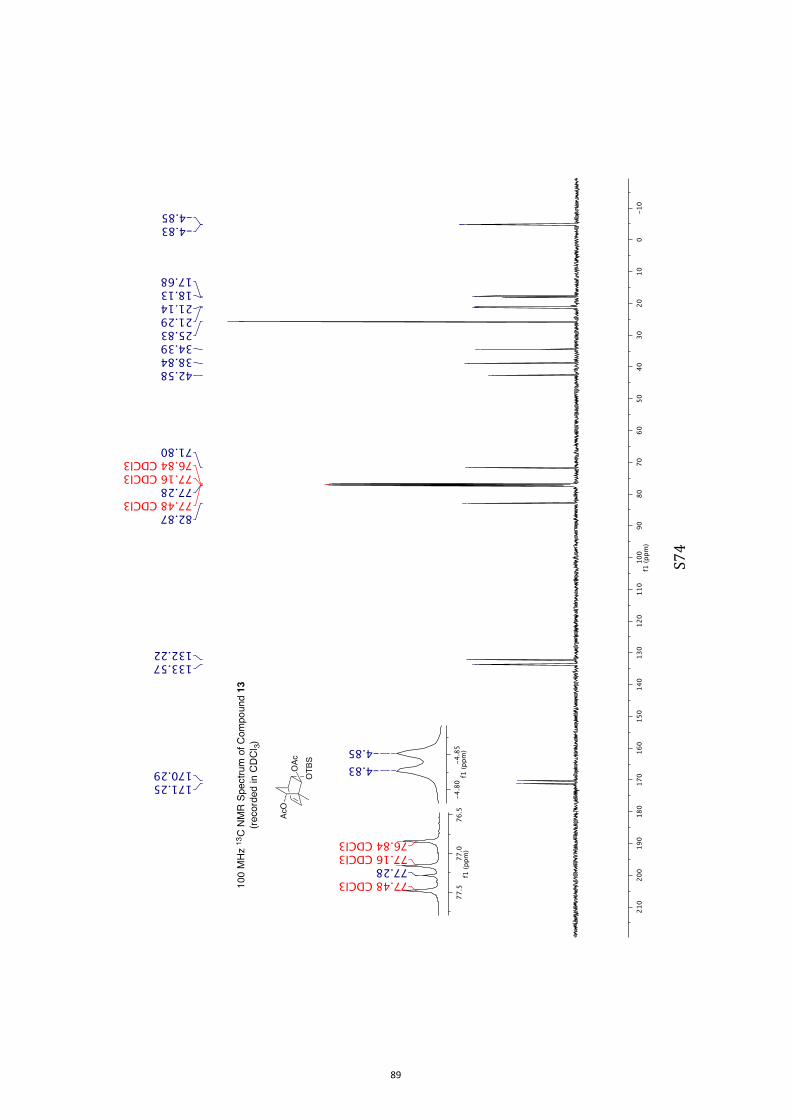
!
S74!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.85-4.83
17.6818.1321.1421.2925.8334.3938.8442.58
71.8076.84 CDCl377.16 CDCl377.2877.48 CDCl382.87
132.22133.57
170.29171.25
-4
.85
-4
.80
f1 (
ppm
)-4.85
-4.83
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl377.16 CDCl377.2877.48 CDCl3
89

!
S75!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.011.08
3.023.011.17
1.03
1.00
1.04
1.00
1.01
0.99
1.00
7.26 CDCl3
90
!
S76!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
17.6921.0321.16
34.5436.4941.37
71.2476.84 CDCl376.9377.16 CDCl377.48 CDCl385.56
132.27133.50
171.05172.82
77
.07
7.5
f1 (
ppm
)
76.84 CDCl376.9377.16 CDCl377.48 CDCl3
91
!
S77!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.02
1.00
3.003.01
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
92
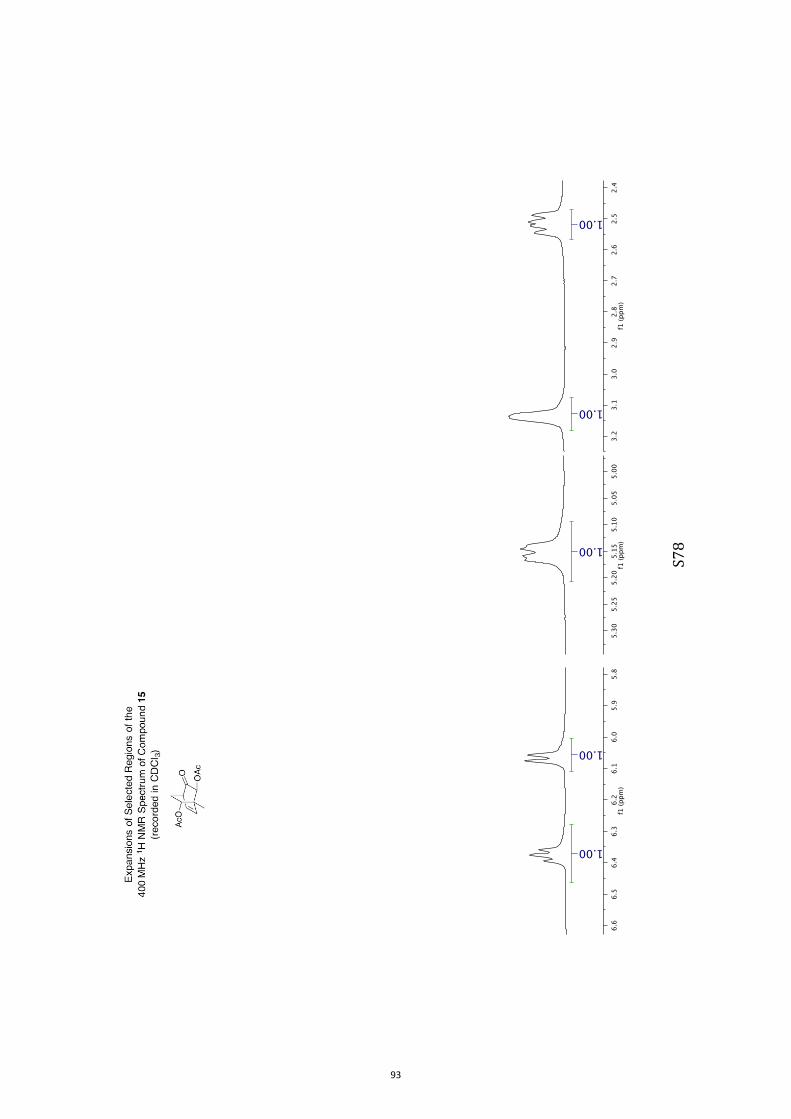
!
S78!
5.8
5.9
6.0
6.1
6.2
6.3
6.4
6.5
6.6
f1 (
ppm
)
1.00
1.00
5.0
05
.05
5.1
05
.15
5.2
05
.25
5.3
0f1
(ppm
)1.00
2.4
2.5
2.6
2.7
2.8
2.9
3.0
3.1
3.2
f1 (
ppm
)
1.00
1.00
93
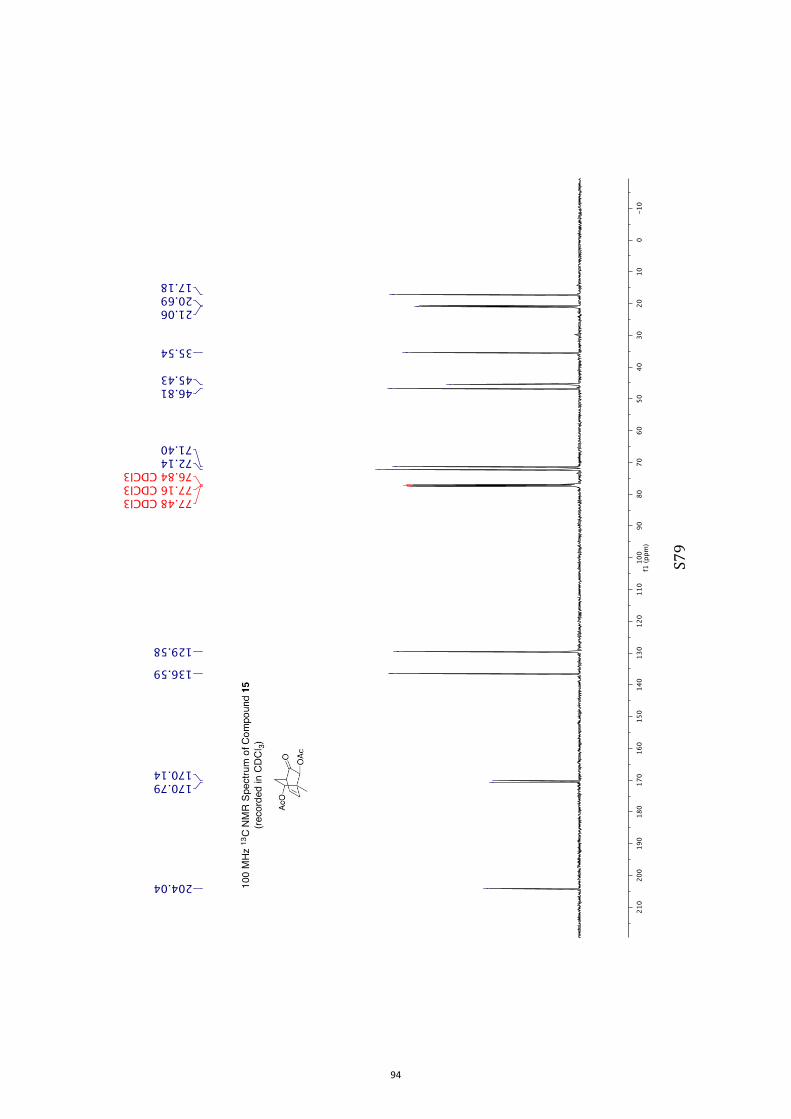
!
S79!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
17.1820.6921.06
35.54
45.4346.81
71.4072.1476.84 CDCl377.16 CDCl377.48 CDCl3
129.58
136.59
170.14170.79
204.04
94
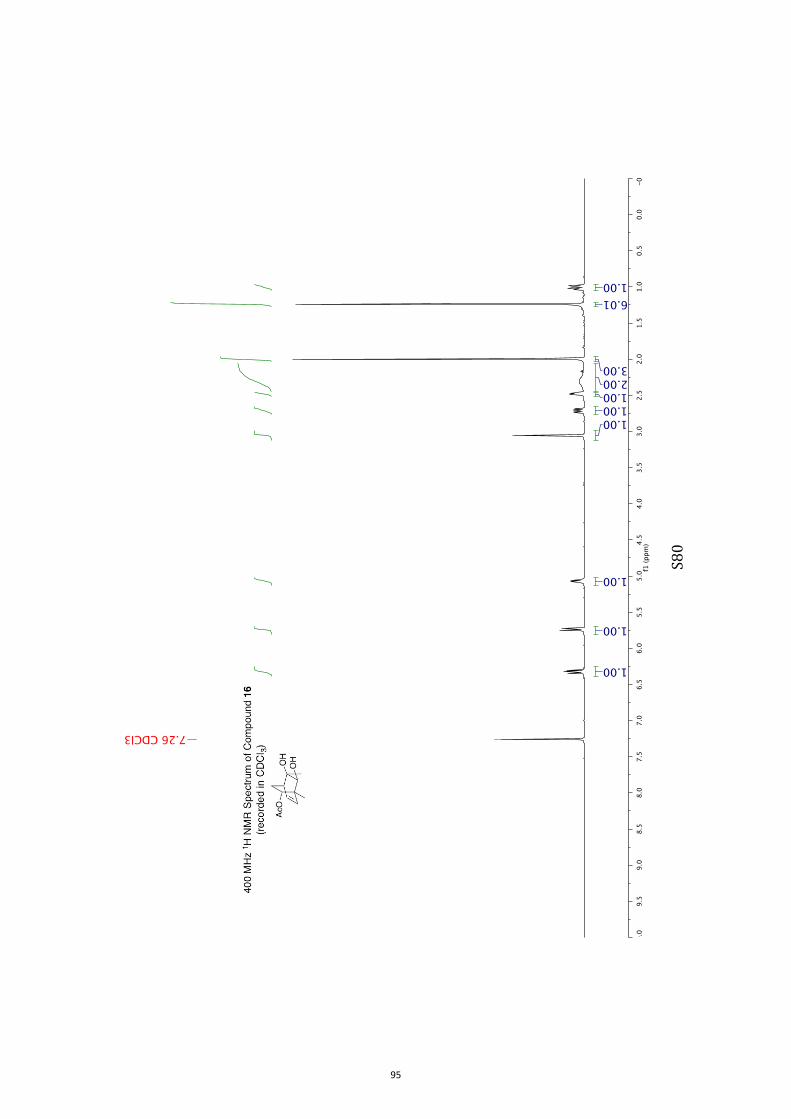
!
S80!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
1.00
6.01
3.002.001.001.001.00
1.00
1.00
1.00
7.26 CDCl3
95

!
S81!
5.6
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
f1 (
ppm
)
1.00
1.00
5.0
05
.05
5.1
05
.15
f1 (
ppm
)
1.00
2.2
2.3
2.4
2.5
2.6
2.7
2.8
f1 (
ppm
)
2.00
1.00
1.00
0.9
60
.98
1.0
01
.02
1.0
41
.06
f1 (
ppm
)
1.00
96
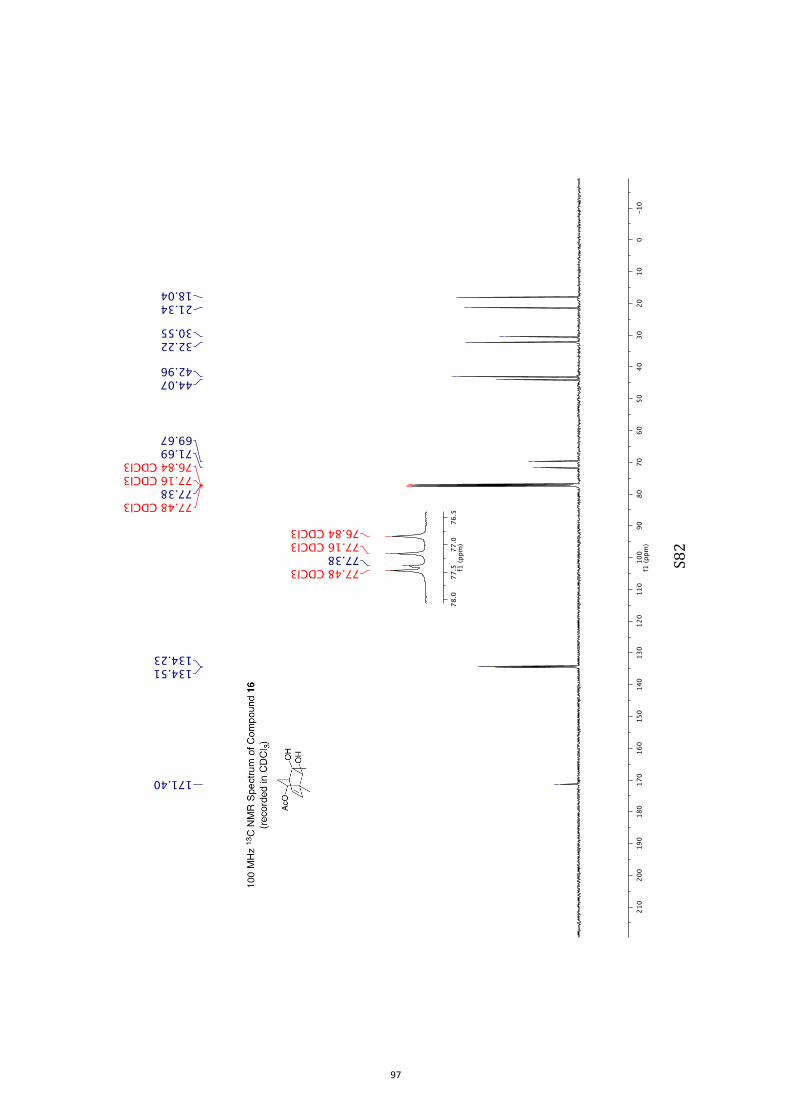
!
S82!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.0421.34
30.5532.22
42.9644.07
69.6771.6976.84 CDCl377.16 CDCl377.3877.48 CDCl3
134.23134.51
171.40
76
.57
7.0
77
.57
8.0
f1 (
ppm
)
76.84 CDCl377.16 CDCl377.3877.48 CDCl3
97
!
S83!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
1.003.003.00
1.001.001.00
1.00
1.00
1.00
3.31 CD3OD_SPE
98
!
S84!
5.5
5.6
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
6.5
f1 (
ppm
)
1.00
1.00
3.8
3.9
4.0
f1 (
ppm
)1.00
2.2
2.3
2.4
2.5
2.6
f1 (
ppm
)
1.00
1.000
.90
0.9
51
.00
f1 (
ppm
)
1.00
99
!
S85!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
18.64
30.7434.8544.5646.7448.36 CD3OD_SPE48.57 CD3OD_SPE48.79 CD3OD_SPE49.00 CD3OD_SPE49.21 CD3OD_SPE49.43 CD3OD_SPE49.64 CD3OD_SPE68.1970.2578.55
135.35135.62
100
!
S86!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
1.001.013.002.963.013.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
101
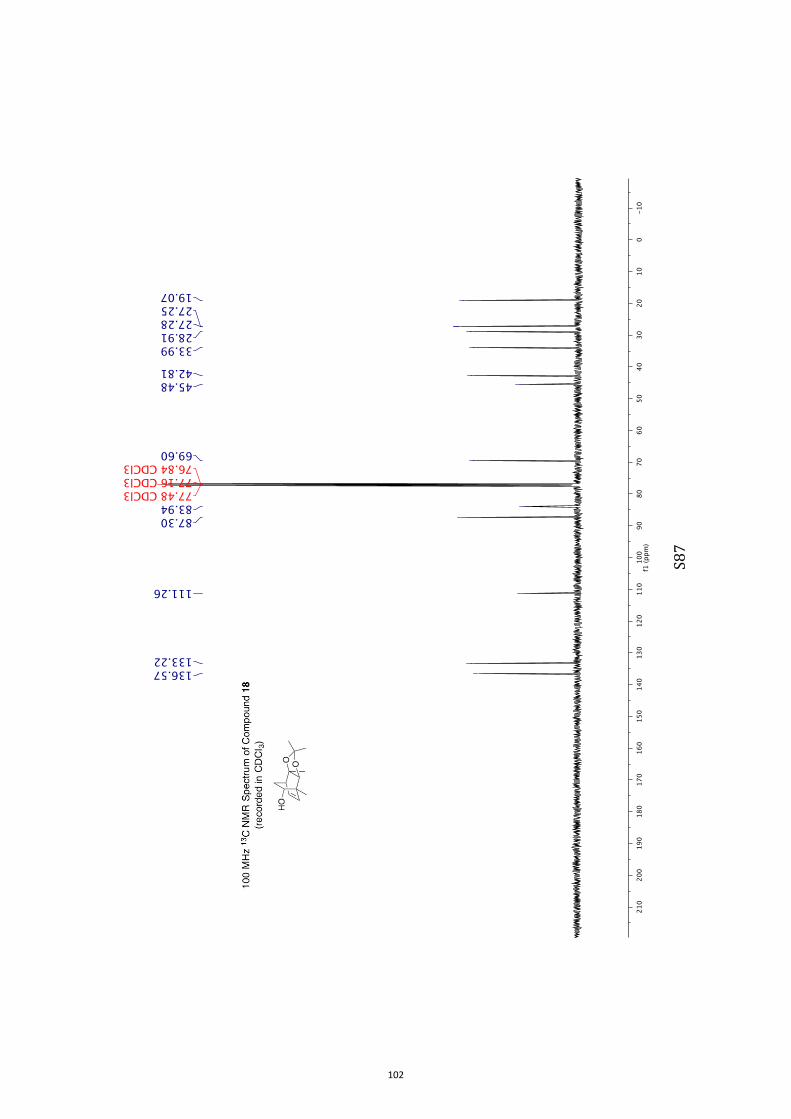
!
S87!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.0727.2527.2828.9133.99
42.8145.48
69.6076.84 CDCl377.16 CDCl377.48 CDCl383.9487.30
111.26
133.22136.57
102
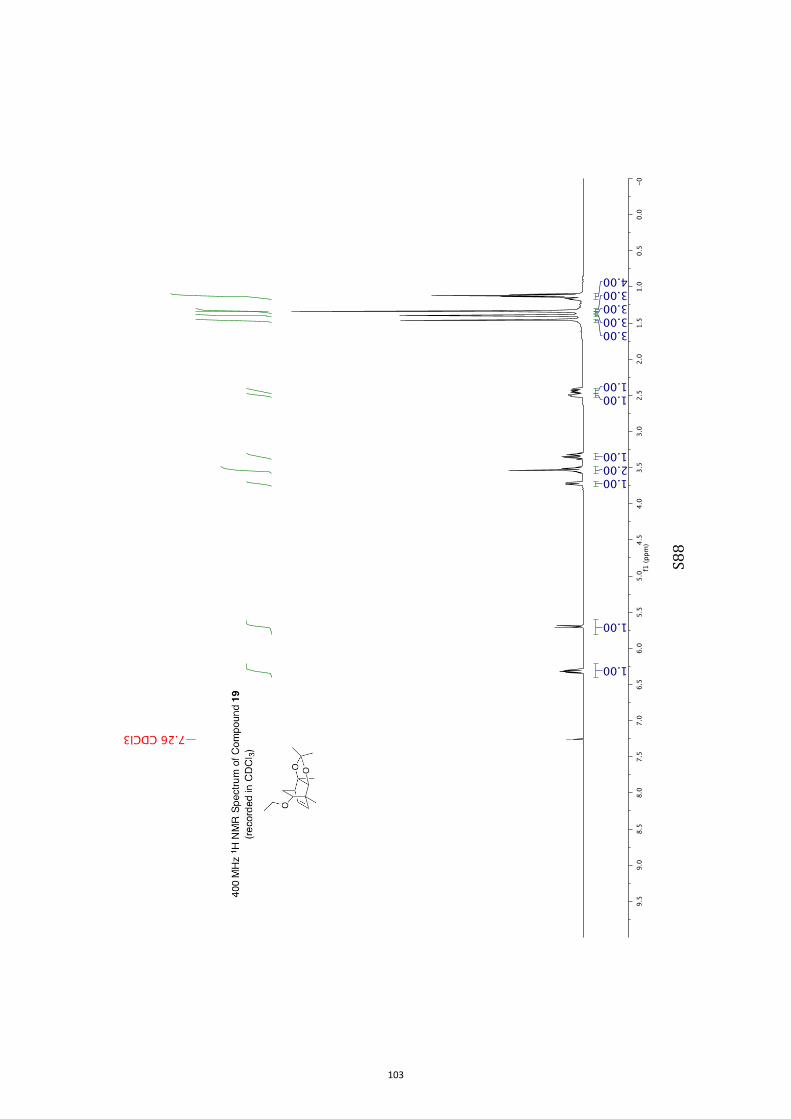
!
S88!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
4.003.003.003.003.00
1.001.00
1.002.001.00
1.00
1.00
7.26 CDCl3
103
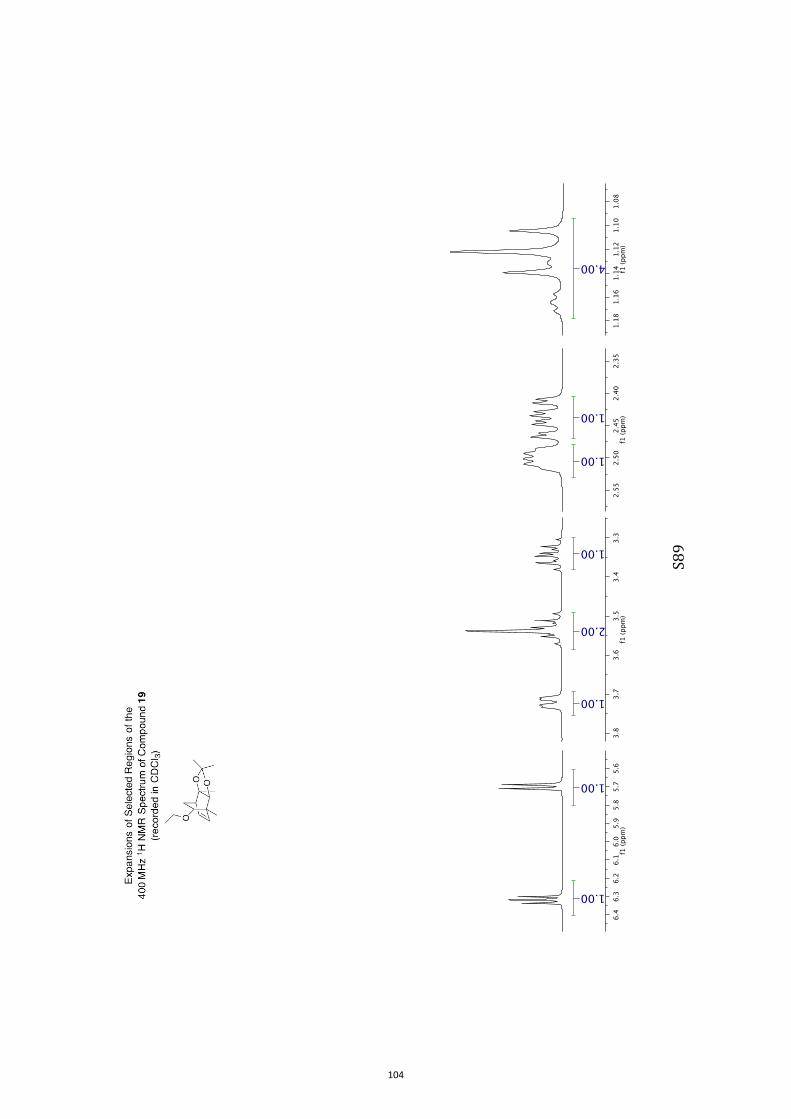
!
S89!
5.6
5.7
5.8
5.9
6.0
6.1
6.2
6.3
6.4
f1 (
ppm
)
1.00
1.00
3.3
3.4
3.5
3.6
3.7
3.8
f1 (
ppm
)
1.00
2.00
1.00
2.3
52
.40
2.4
52
.50
2.5
5f1
(ppm
)
1.00
1.00
1.0
81
.10
1.1
21
.14
1.1
61
.18
f1 (
ppm
)
4.00
104
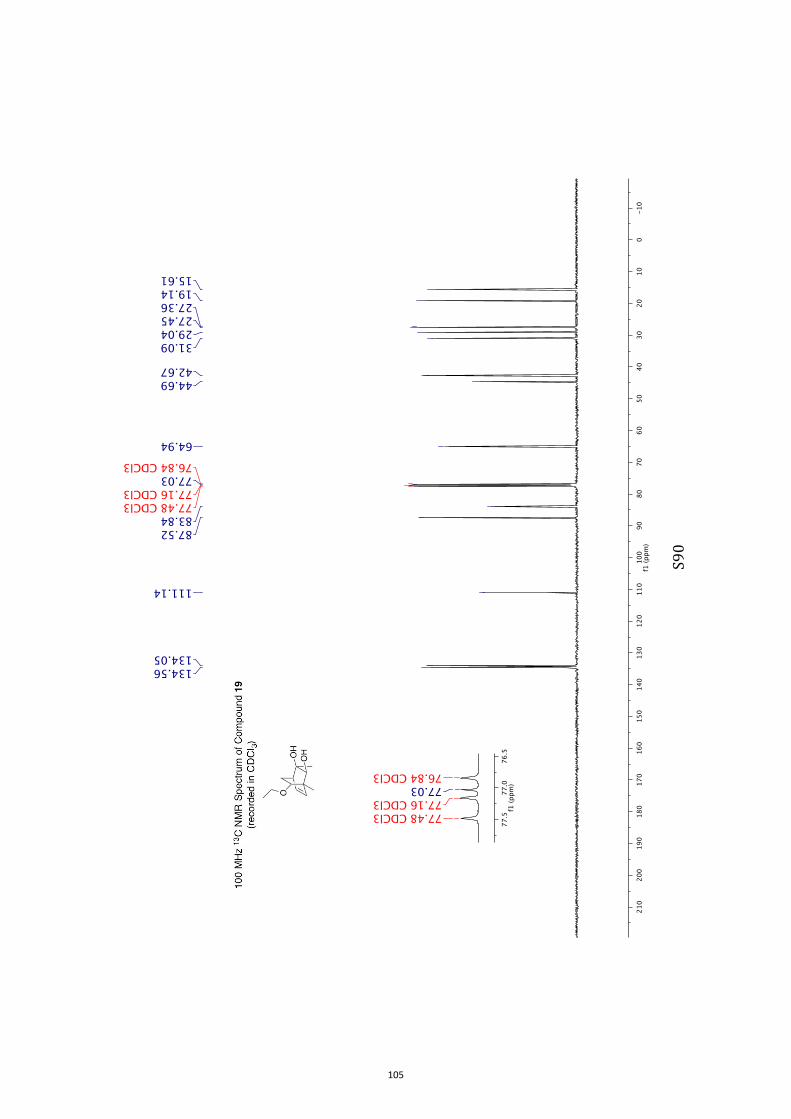
!
S90!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6119.1427.3627.4529.0431.09
42.6744.69
64.94
76.84 CDCl377.0377.16 CDCl377.48 CDCl383.8487.52
111.14
134.05134.56
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl377.0377.16 CDCl377.48 CDCl3
105

!
S91!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.006.003.001.00
1.00
1.00
1.00
1.002.001.001.00
7.26 CDCl3
106
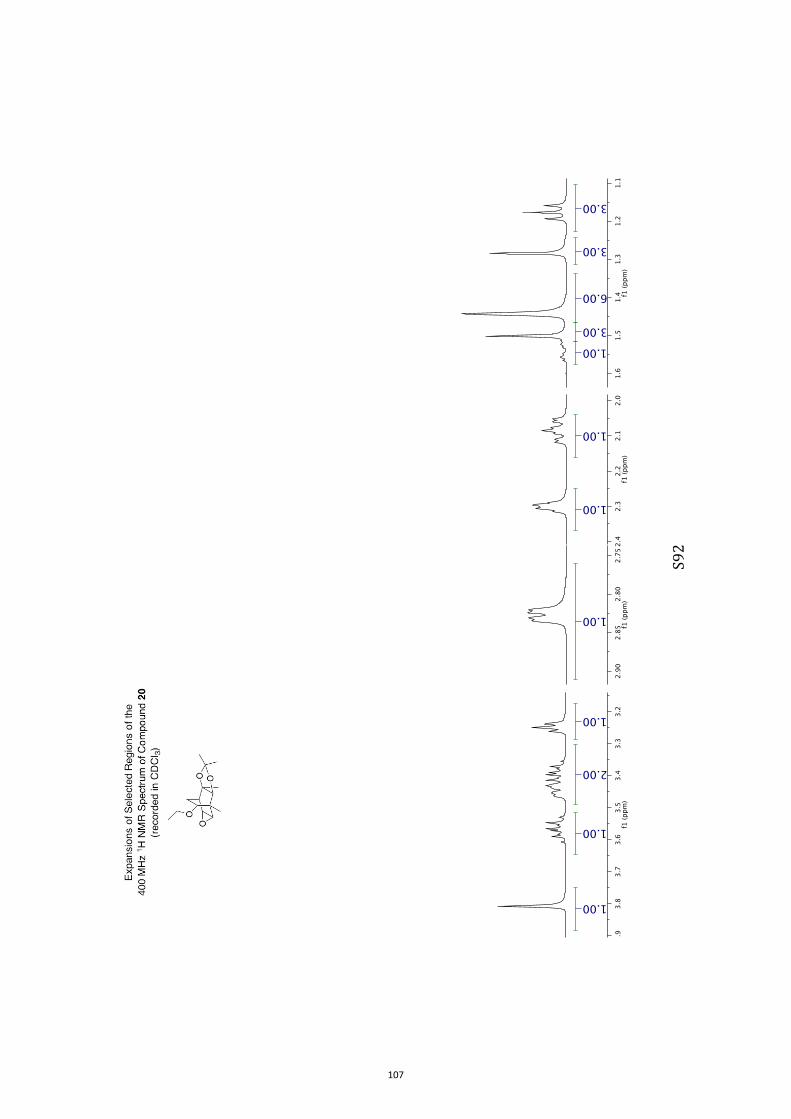
!
S92!
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
f1 (
ppm
)
1.00
2.00
1.00
1.00
2.7
52
.80
2.8
52
.90
f1 (
ppm
)
1.00
2.0
2.1
2.2
2.3
2.4
f1 (
ppm
)
1.00
1.001
.11
.21
.31
.41
.51
.6f1
(ppm
)
3.00
3.00
6.00
3.00
1.00
107

!
S93!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6919.1826.5927.3028.1228.18
40.6942.70
53.1754.61
66.1974.7676.84 CDCl377.16 CDCl377.48 CDCl382.6086.64
111.10
108
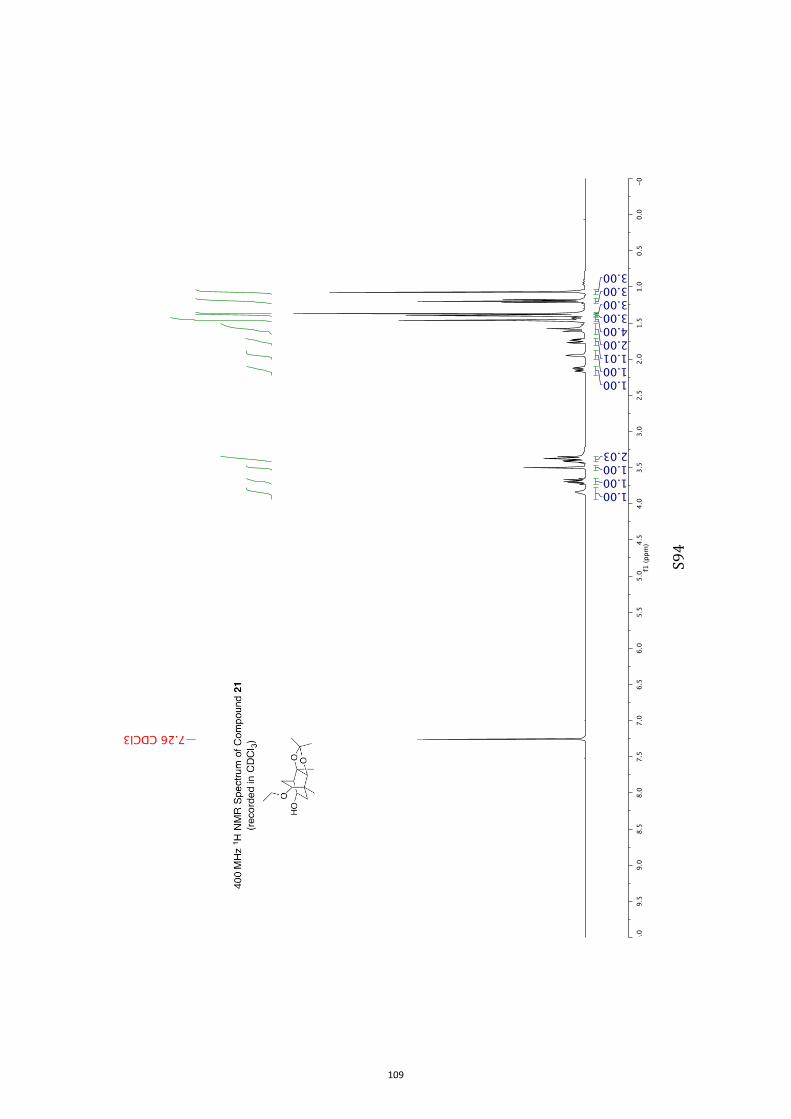
!
S94!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.003.003.004.002.001.011.001.00
2.031.001.001.00
7.26 CDCl3
109
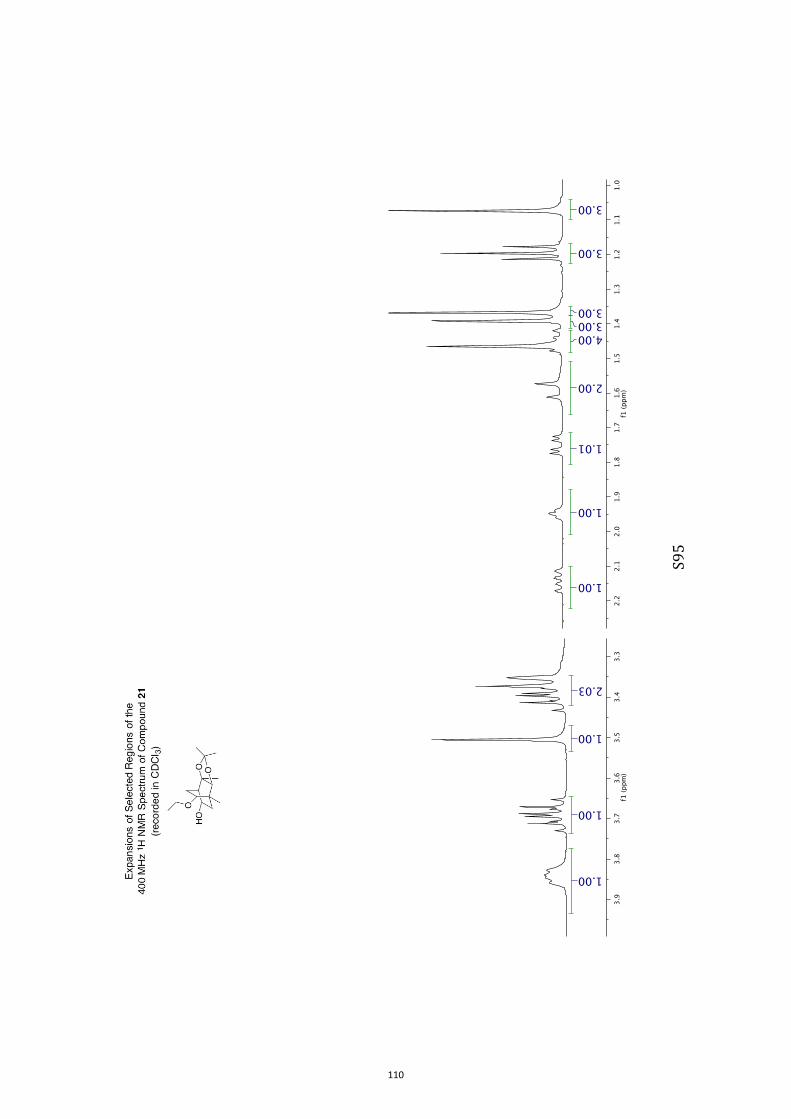
!
S95!
3.3
3.4
3.5
3.6
3.7
3.8
3.9
f1 (
ppm
)
2.03
1.00
1.00
1.00
1.0
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
f1 (
ppm
)
3.00
3.00
3.003.004.00
2.00
1.01
1.00
1.00
110

!
S96!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6022.0023.1226.2526.4826.8135.3938.1243.03
64.9365.6775.6276.84 CDCl377.16 CDCl377.48 CDCl380.7386.81
108.48
111

!
S97!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.003.002.022.001.00
1.001.001.001.002.001.001.00
7.26 CDCl3
112
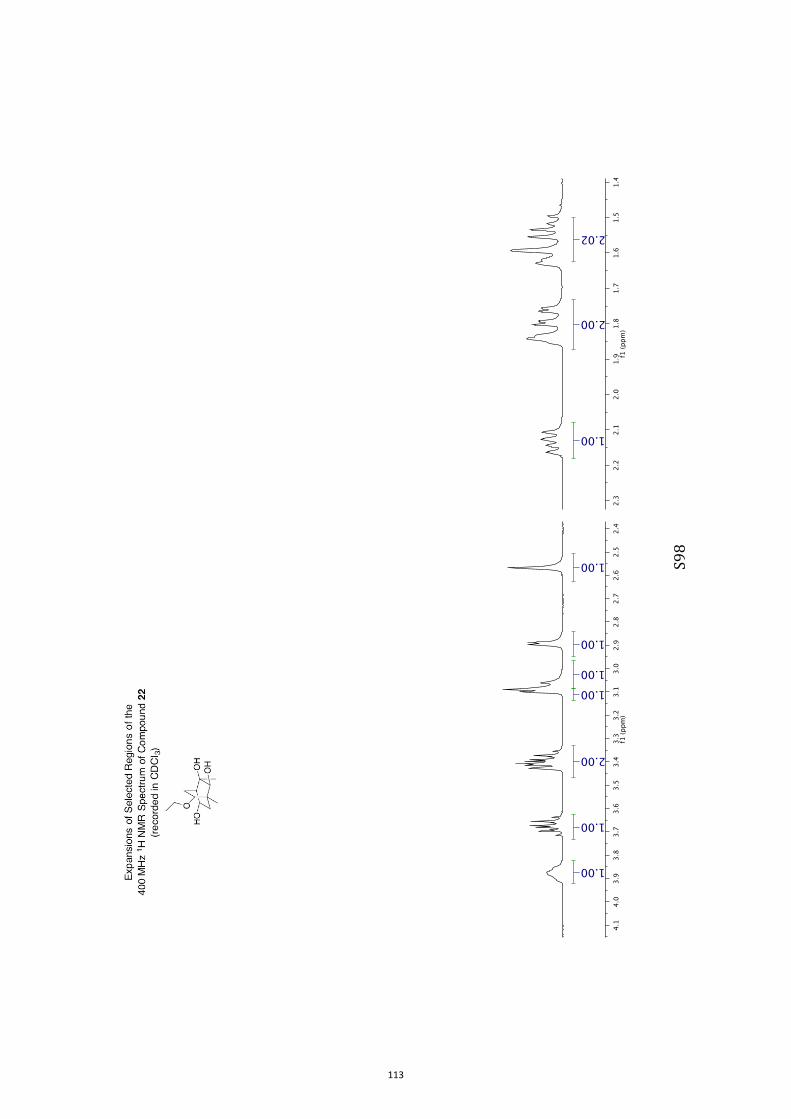
!
S98!
2.4
2.5
2.6
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
4.0
4.1
f1 (
ppm
)
1.00
1.00
1.00
1.00
2.00
1.00
1.00
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
2.3
f1 (
ppm
)
2.02
2.00
1.00
113
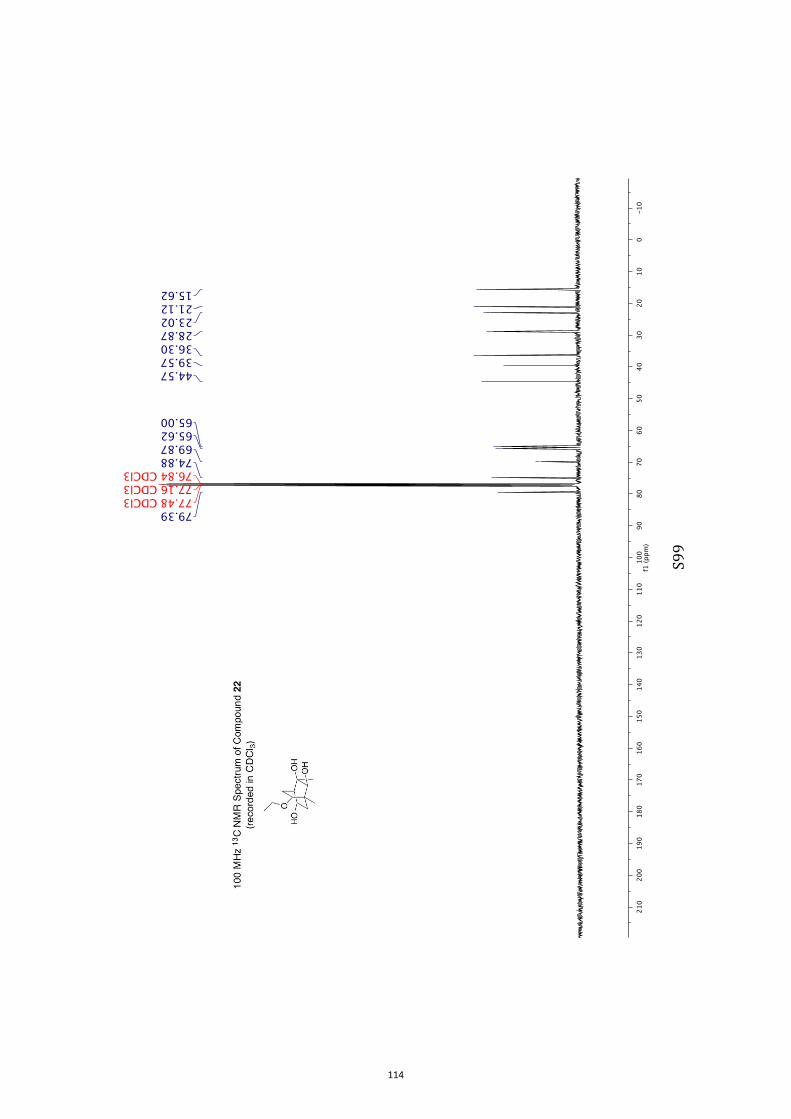
!
S99!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6221.1223.0228.8736.3039.5744.57
65.0065.6269.8774.8876.84 CDCl377.16 CDCl377.48 CDCl379.39
114

!
S100!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.003.006.013.00
1.00
2.00
2.011.001.001.00
1.00
7.26 CDCl3
115

!
S101!
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
4.0
4.1
f1 (
ppm
)
2.01
1.00
1.00
1.00
1.00
1.7
1.8
1.9
2.0
2.1
2.2
2.3
f1 (
ppm
)
1.00
2.00
0.9
51
.00
1.0
51
.10
1.1
51
.20
f1 (
ppm
)
3.00
3.00
116

!
S102!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.5418.3322.0426.6126.7126.83
41.0542.17
65.1265.2969.9376.84 CDCl376.8877.16 CDCl377.48 CDCl379.4785.89
109.15
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl376.8877.16 CDCl3
77.48 CDCl3
117
!
S103!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.001.012.882.872.953.021.001.000.981.00
1.001.001.002.001.00
7.26 CDCl3
118
!
S104!
3.9
4.0
4.1
4.2
4.3
f1 (
ppm
)
2.00
1.00
3.2
3.3
3.4
3.5
3.6
f1 (
ppm
)
1.00
1.00
1.00
2.2
02
.25
2.3
02
.35
2.4
02
.45
2.5
02
.55
f1 (
ppm
)
1.00
0.98
1.001
.05
1.1
01
.15
1.2
01
.25
1.3
0f1
(ppm
)
3.00
1.01
2.88
119

!
S105!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6218.7726.6726.7527.8629.60
42.1944.21
64.7168.6170.0975.2776.84 CDCl377.16 CDCl377.48 CDCl379.9982.54
107.43
120
!
S106!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
1.003.003.003.003.003.00
1.00
1.00
1.002.003.001.002.00
1.00
2.00
2.007.26 CDCl3
121
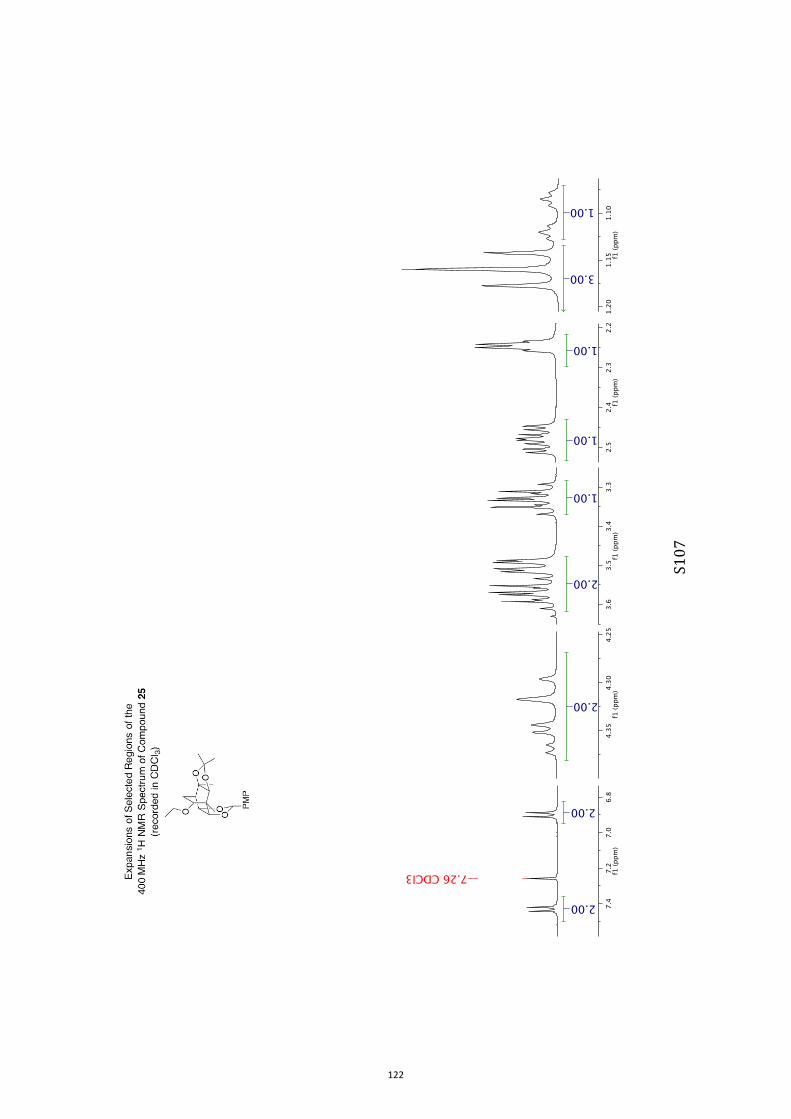
!
S107!
6.8
7.0
7.2
7.4
f1 (
ppm
)
2.00
2.00
7.26 CDCl3
4.2
54
.30
4.3
5f1
(ppm
)
2.00
3.3
3.4
3.5
3.6
f1 (
ppm
)1.00
2.00
2.2
2.3
2.4
2.5
f1 (
ppm
)
1.00
1.00
1.1
01
.15
1.2
0f1
(ppm
)
1.00
3.00
122
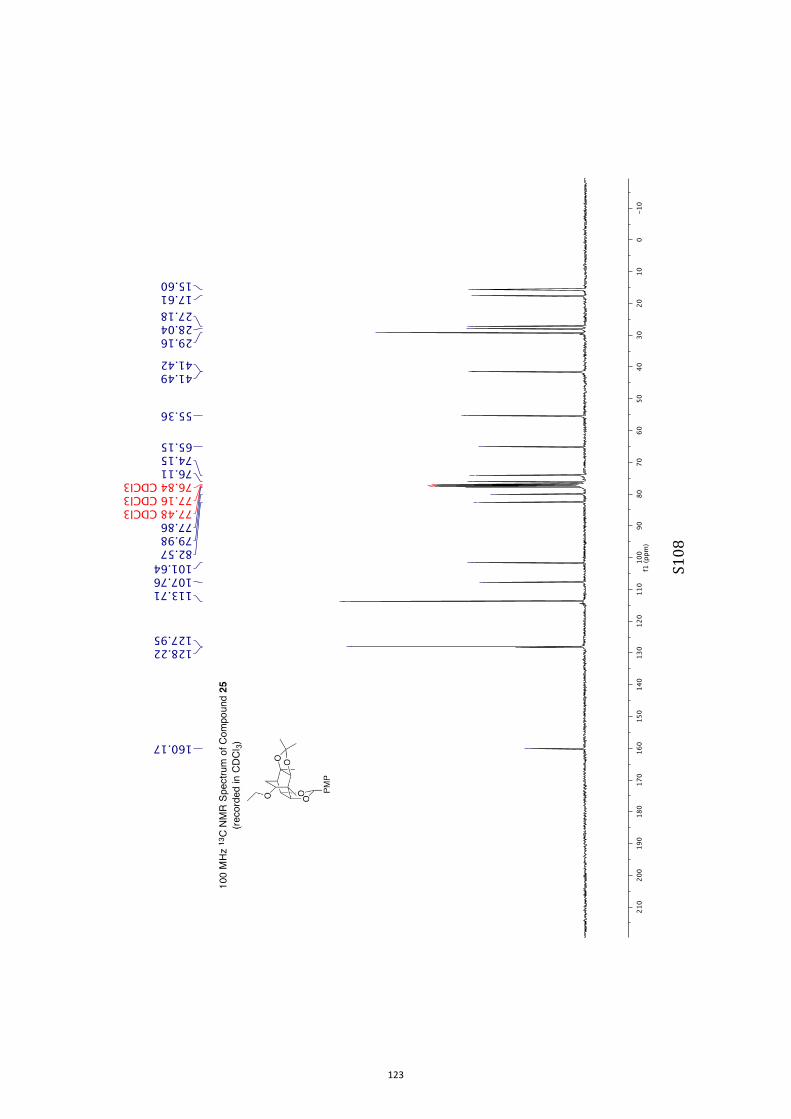
!
S108!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6017.61
27.1828.0429.16
41.4241.49
55.36
65.1574.1576.1176.84 CDCl377.16 CDCl377.48 CDCl377.8679.9882.57
101.64107.76113.71
127.95128.22
160.17
123
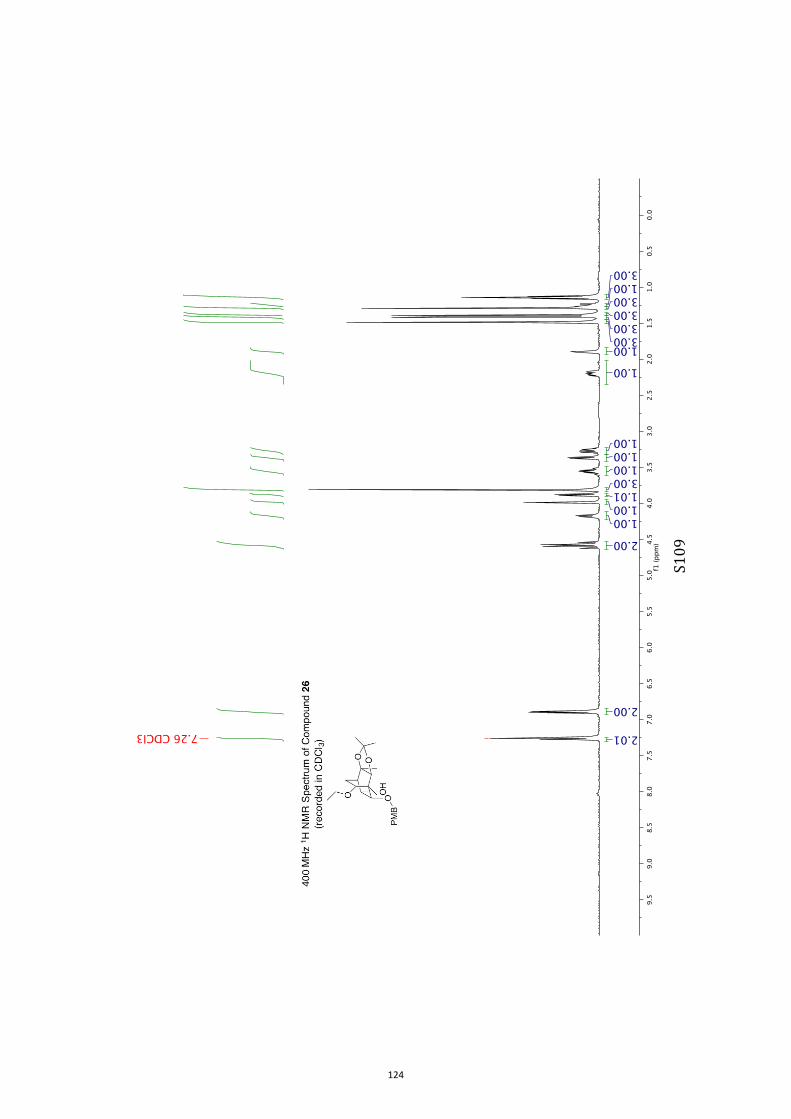
!
S109!
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
f1 (
ppm
)
3.001.003.003.003.003.001.00
1.00
1.001.001.003.001.011.001.00
2.00
2.00
2.017.26 CDCl3
124

!
S110!
6.9
7.0
7.1
7.2
7.3
f1 (
ppm
)
2.00
2.017.26 CDCl3
3.2
3.3
3.4
3.5
3.6
f1 (
ppm
)
1.00
1.00
1.00
2.0
52
.10
2.1
52
.20
2.2
52
.30
2.3
5f1
(ppm
)1.001
.05
1.1
01
.15
1.2
01
.25
1.3
0f1
(ppm
)
3.00
1.00
3.00
125
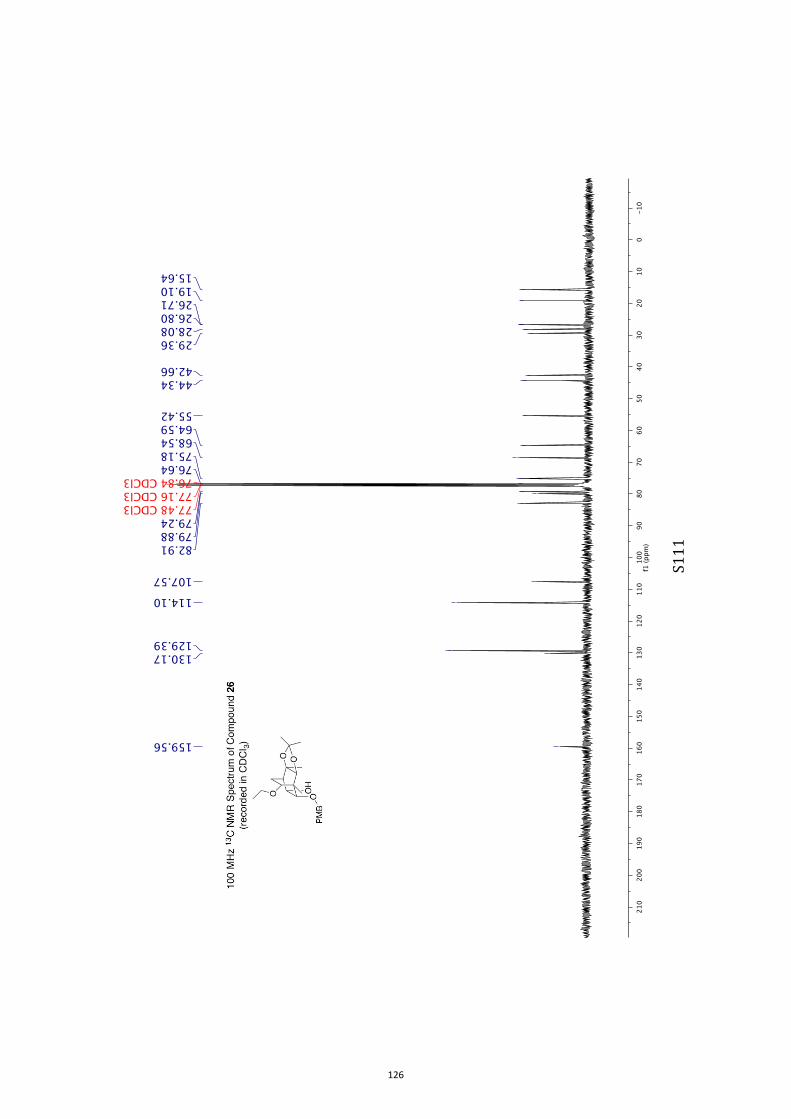
!
S111!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6419.1026.7126.8028.0829.36
42.6644.34
55.4264.5968.5475.1876.6476.84 CDCl377.16 CDCl377.48 CDCl379.2479.8882.91
107.57
114.10
129.39130.17
159.56
126
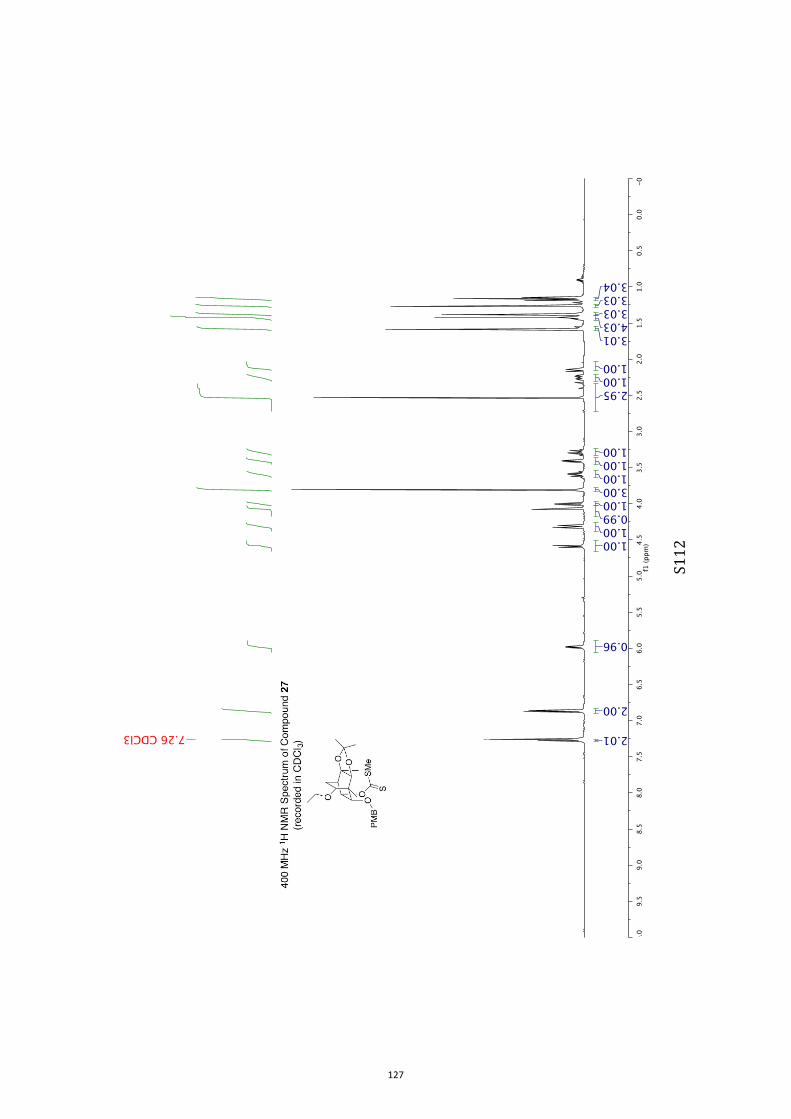
!
S112!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.043.033.034.033.01
1.001.002.95
1.001.001.003.001.000.991.001.00
0.96
2.00
2.017.26 CDCl3
127

!
S113!
5.9
05
.95
6.0
06
.05
f1 (
ppm
)0.96
4.0
4.1
4.2
4.3
4.4
4.5
4.6
f1 (
ppm
)
1.00
0.99
1.00
1.00
3.2
53
.30
3.3
53
.40
3.4
53
.50
3.5
53
.60
3.6
5f1
(ppm
)
1.00
1.00
1.00
1.4
01
.41
1.4
21
.43
1.4
41
.45
1.4
61
.47
1.4
8f1
(ppm
)
4.03
128
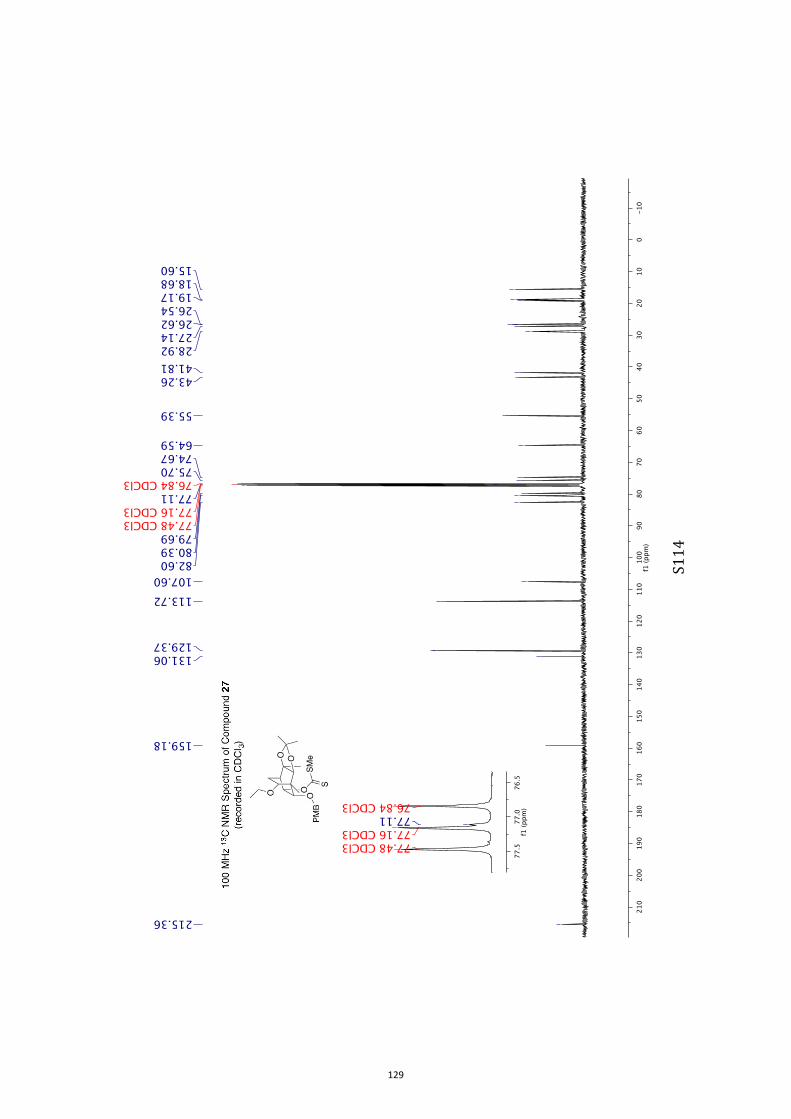
!
S114!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6018.6819.1726.5426.6227.1428.92
41.8143.26
55.39
64.5974.6775.7076.84 CDCl377.1177.16 CDCl377.48 CDCl379.6980.3982.60
107.60
113.72
129.37131.06
159.18
215.36
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl377.1177.16 CDCl377.48 CDCl3
129
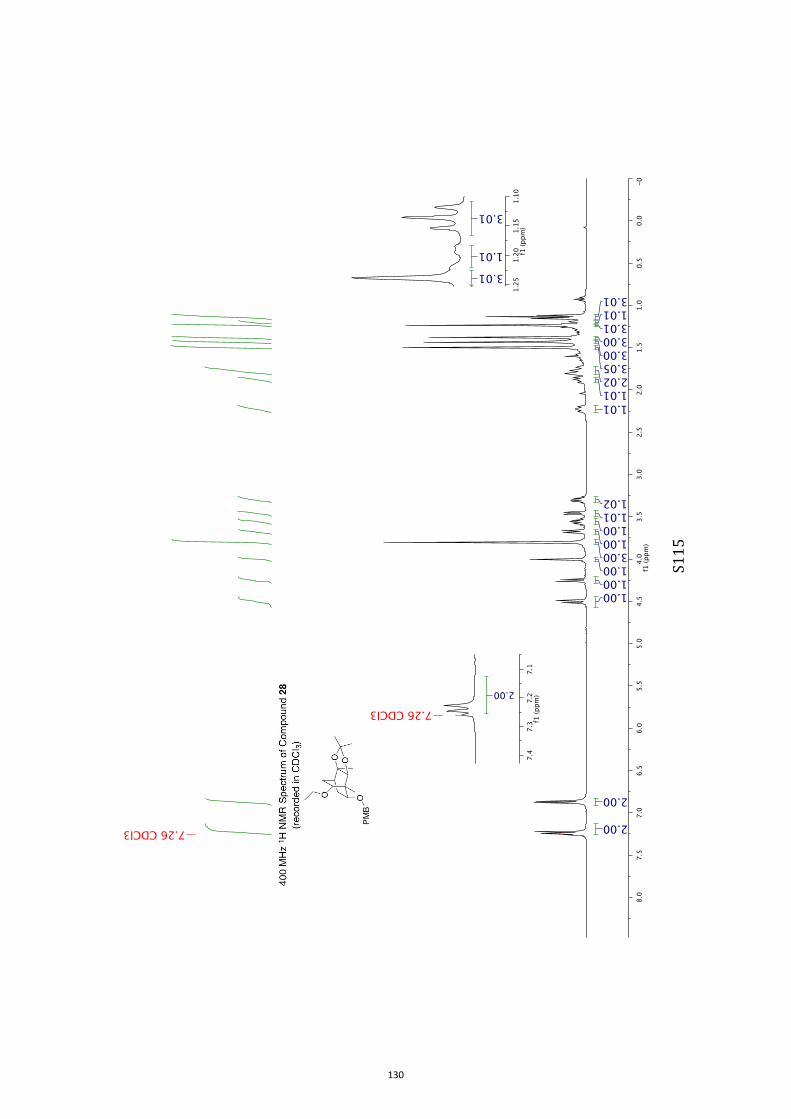
!
S115!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.011.013.013.003.003.052.021.01
1.01
1.021.011.001.003.001.001.001.00
2.00
2.007.26 CDCl3
7.1
7.2
7.3
7.4
f1 (
ppm
)
2.00
7.26 CDCl3
1.1
01
.15
1.2
01
.25
f1 (
ppm
)
3.01
1.01
3.01
130
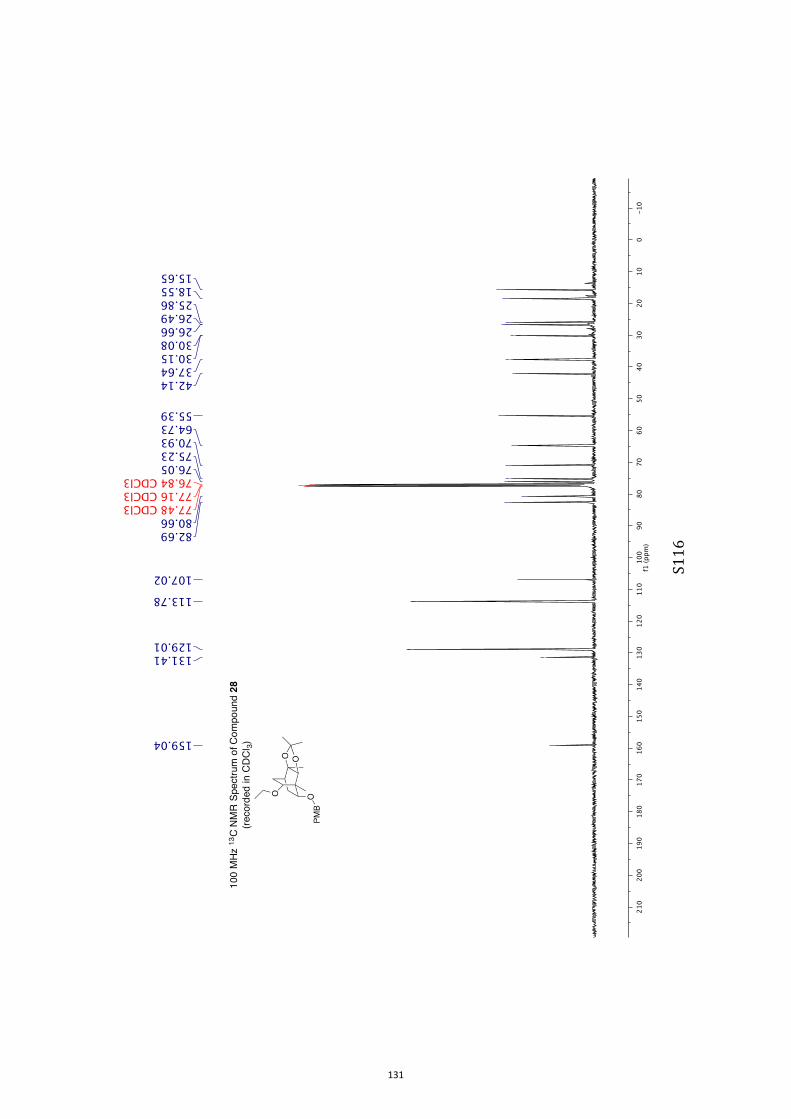
!
S116!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6518.5525.8626.4926.6630.0830.1537.6442.14
55.3964.7370.9375.2376.0576.84 CDCl377.16 CDCl377.48 CDCl380.6682.69
107.02
113.78
129.01131.41
159.04
131

!
S117!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.001.003.013.003.001.003.001.001.001.011.01
1.001.001.00
1.001.00
7.26 CDCl3
132

!
S118!
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
4.0
f1 (
ppm
)
1.00
1.00
1.00
1.00
1.00
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
2.3
f1 (
ppm
)
3.003.001.003.001.00
1.00
1.01
1.01
1.0
81
.10
1.1
21
.14
1.1
61
.18
1.2
01
.22
f1 (
ppm
)
3.00
1.00
3.01
133
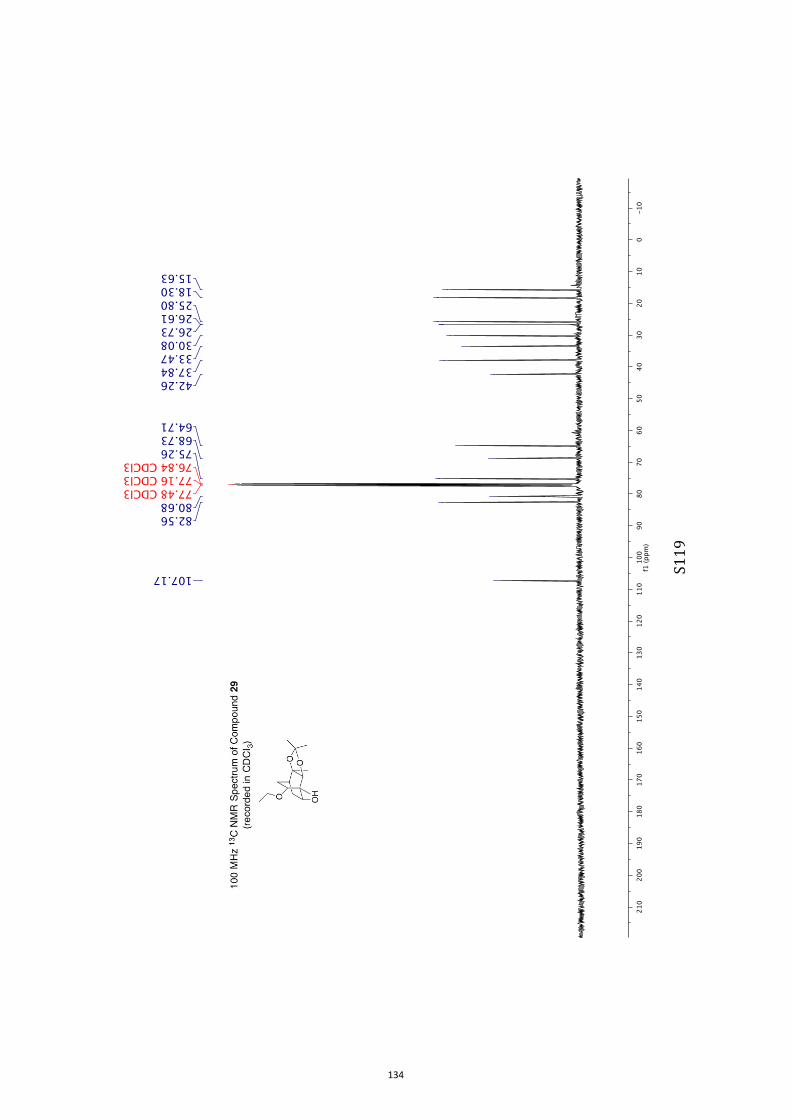
!
S119!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.6318.3025.8026.6126.7330.0833.4737.8442.26
64.7168.7375.2676.84 CDCl377.16 CDCl377.48 CDCl380.6882.56
107.17
134
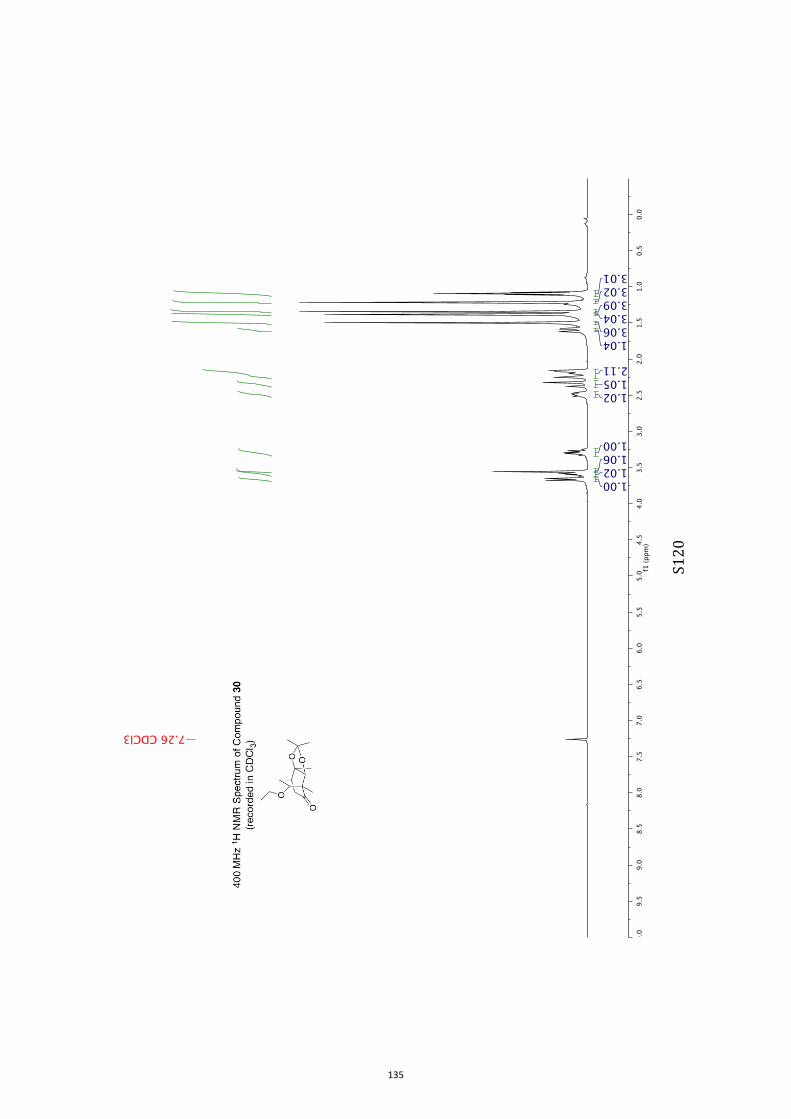
!
S120!
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
3.013.023.093.043.061.04
2.111.051.02
1.001.061.021.00
7.26 CDCl3
135
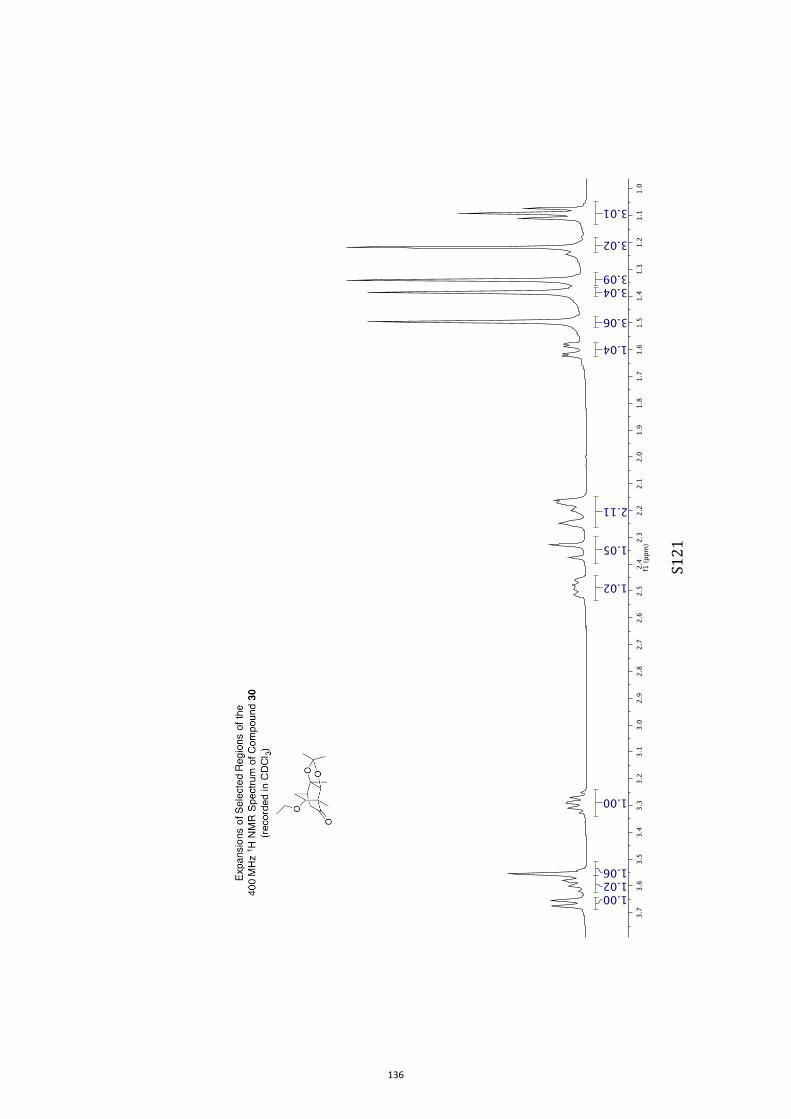
!
S121!
1.0
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
2.3
2.4
2.5
2.6
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
f1 (
ppm
)
3.01
3.02
3.093.04
3.06
1.04
2.11
1.05
1.02
1.00
1.061.021.00
136
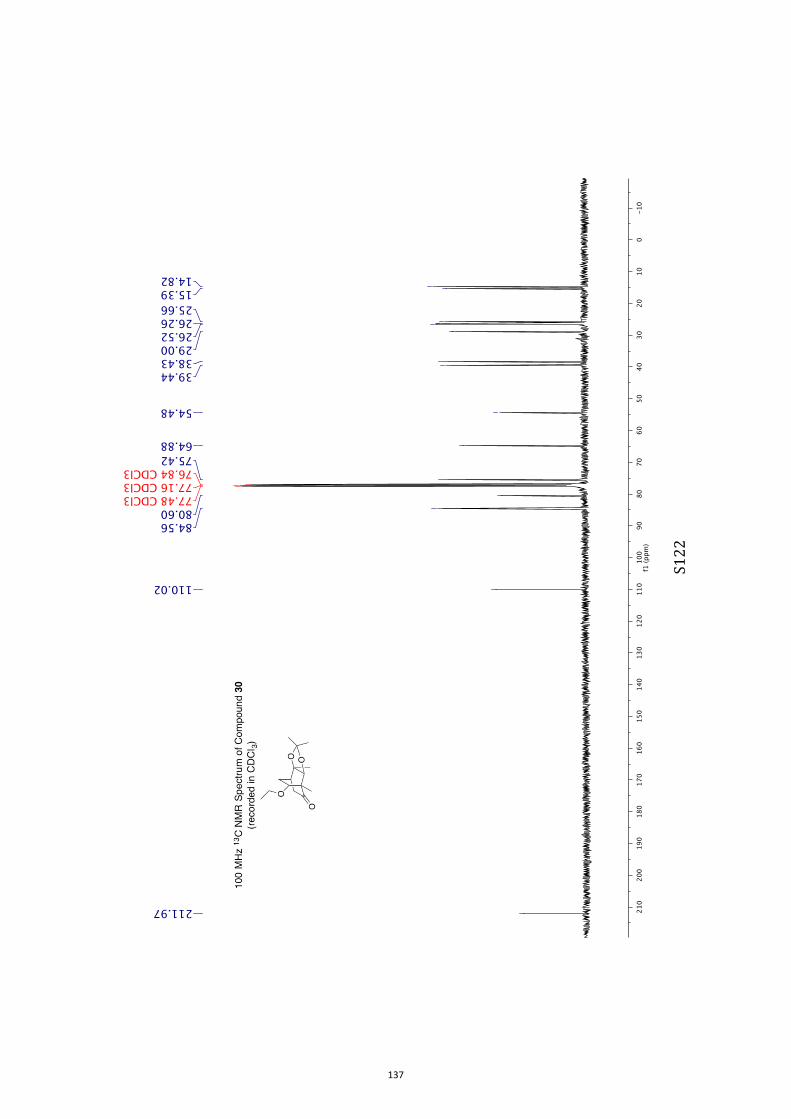
!
S122!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
14.8215.39
25.6626.2626.5229.0038.4339.44
54.48
64.88
75.4276.84 CDCl377.16 CDCl377.48 CDCl380.6084.56
110.02
211.97
137

!
S123!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.001.023.003.003.013.01
1.011.00
1.001.011.001.00
2.00
2.00
1.007.26 CDCl3
138
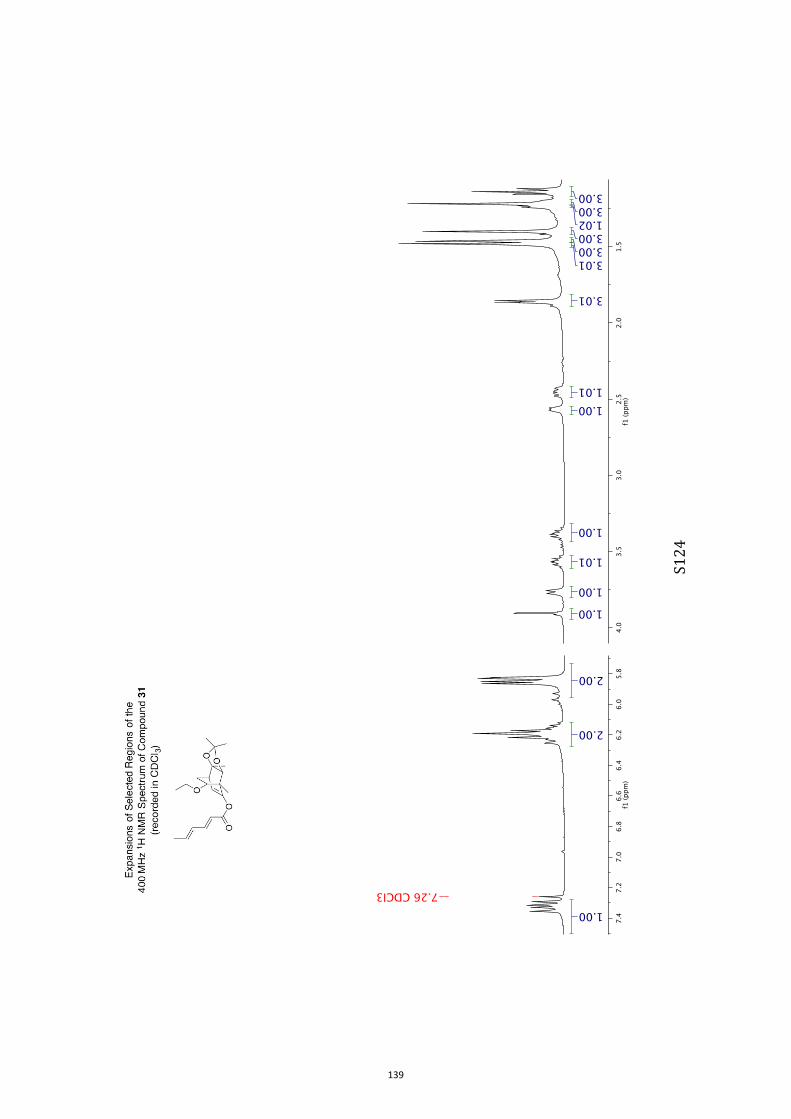
!
S124!
5.8
6.0
6.2
6.4
6.6
6.8
7.0
7.2
7.4
f1 (
ppm
)
2.00
2.00
1.00
7.26 CDCl3
1.5
2.0
2.5
3.0
3.5
4.0
f1 (
ppm
)
3.003.001.023.003.003.01
3.01
1.01
1.00
1.00
1.01
1.00
1.00
139
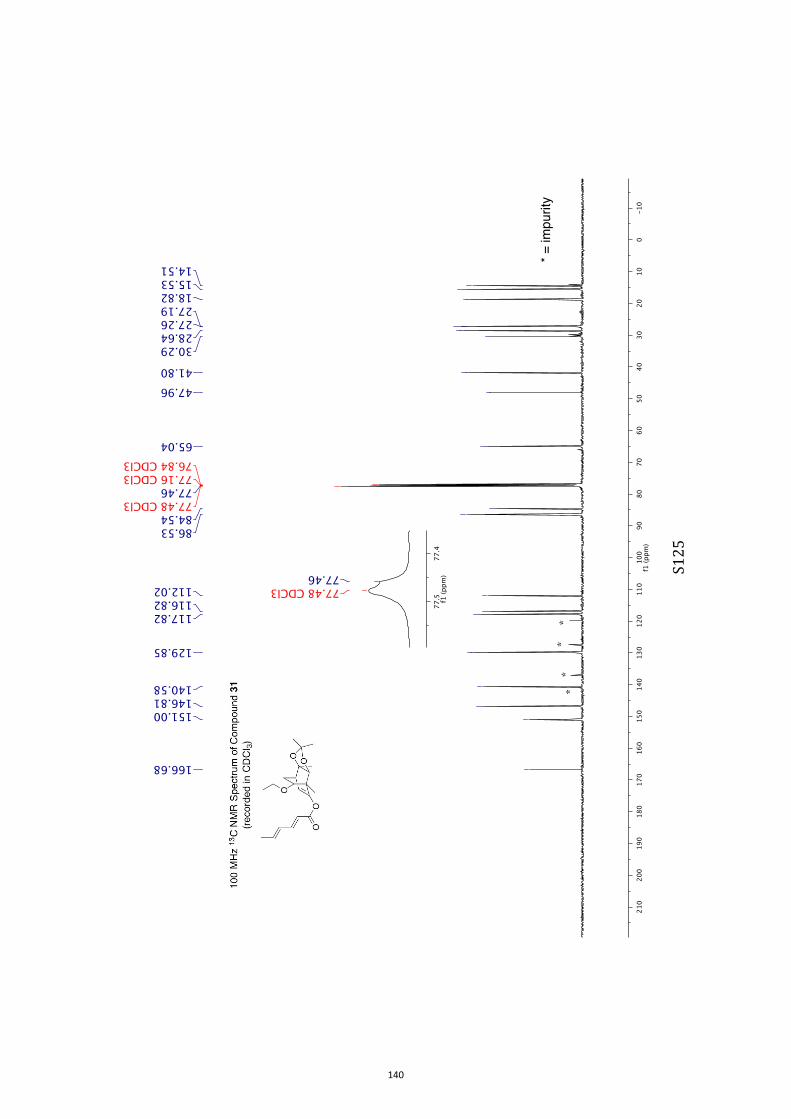
!
S125!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
14.5115.5318.8227.1927.2628.6430.29
41.80
47.96
65.04
76.84 CDCl377.16 CDCl377.4677.48 CDCl384.5486.53
112.02116.82117.82
129.85
140.58146.81151.00
166.68
**
**
77
.47
7.5 f1 (
ppm
)77.4677.48 CDCl3
*=
im
pu
rity
140

!
S126!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.51
5.0
f1 (
ppm
)
3.056.033.031.093.043.05
1.071.051.031.011.031.05
2.031.08
1.00
1.00
7.26 CDCl3
141
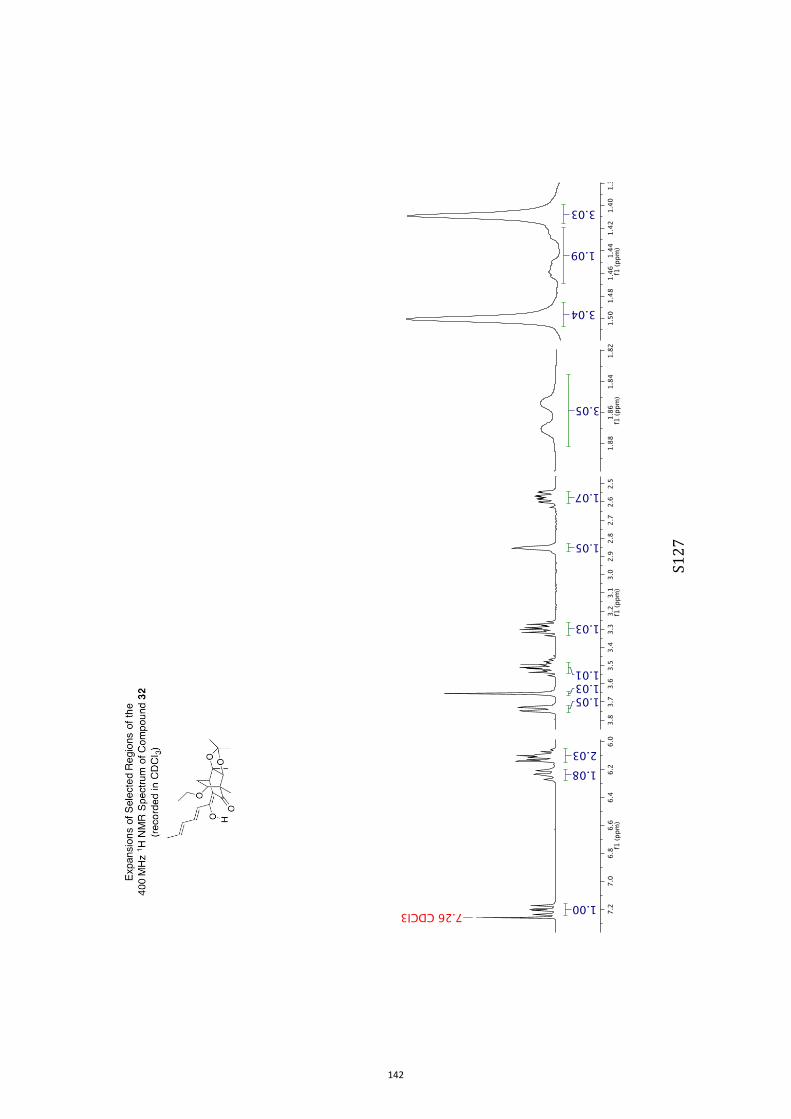
!
S127!
2.5
2.6
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
f1 (
ppm
)1.07
1.05
1.03
1.011.031.05
1.8
21.8
41.8
61.8
8f1
(ppm
)
3.05
1.3
81.4
01.4
21.4
41.4
61.4
81.5
0f1
(ppm
)
3.03
1.09
3.04
6.0
6.2
6.4
6.6
6.8
7.0
7.2
f1 (
ppm
)
2.03
1.08
1.007.26 CDCl3
142
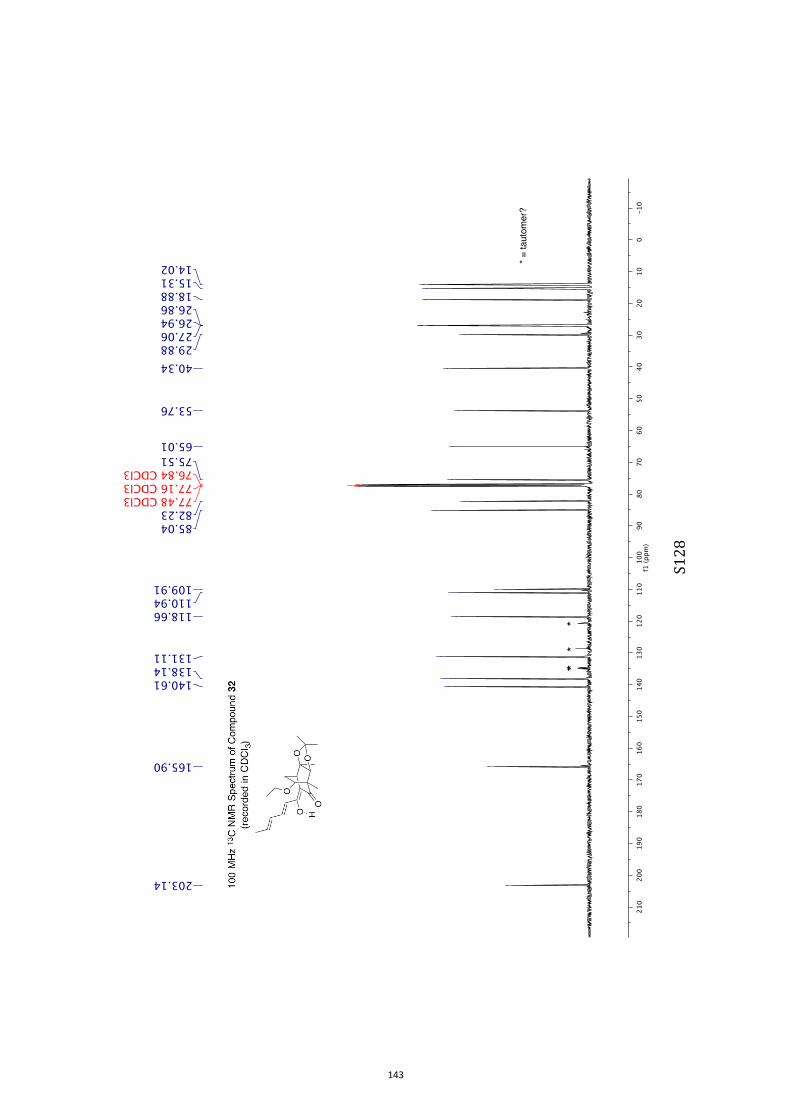
!
S128!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
14.0215.3118.8826.8626.9427.0629.88
40.34
53.76
65.01
75.5176.84 CDCl377.16 CDCl377.48 CDCl382.2385.04
109.91110.94118.66
131.11138.14140.61
165.90
203.14
***
*
* =
tauto
mer?
143
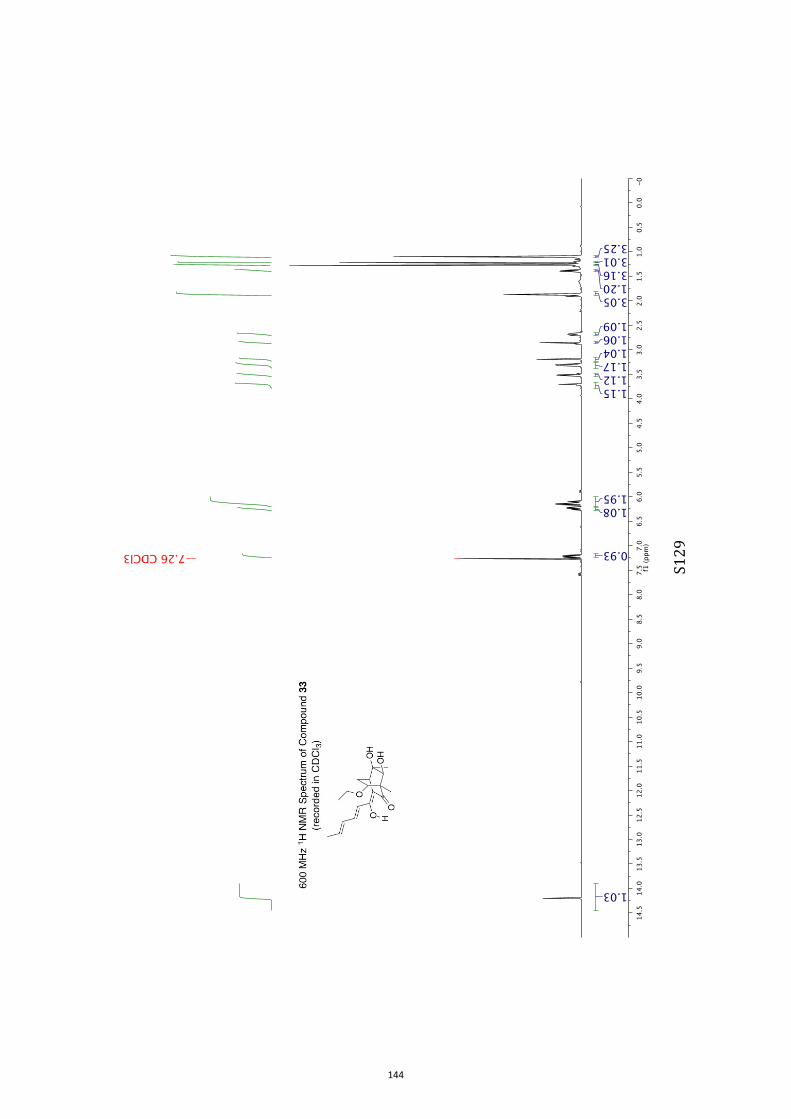
!
S129!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.5f1
(ppm
)
3.253.013.161.203.05
1.091.061.041.171.121.15
1.951.08
0.93
1.03
7.26 CDCl3
144
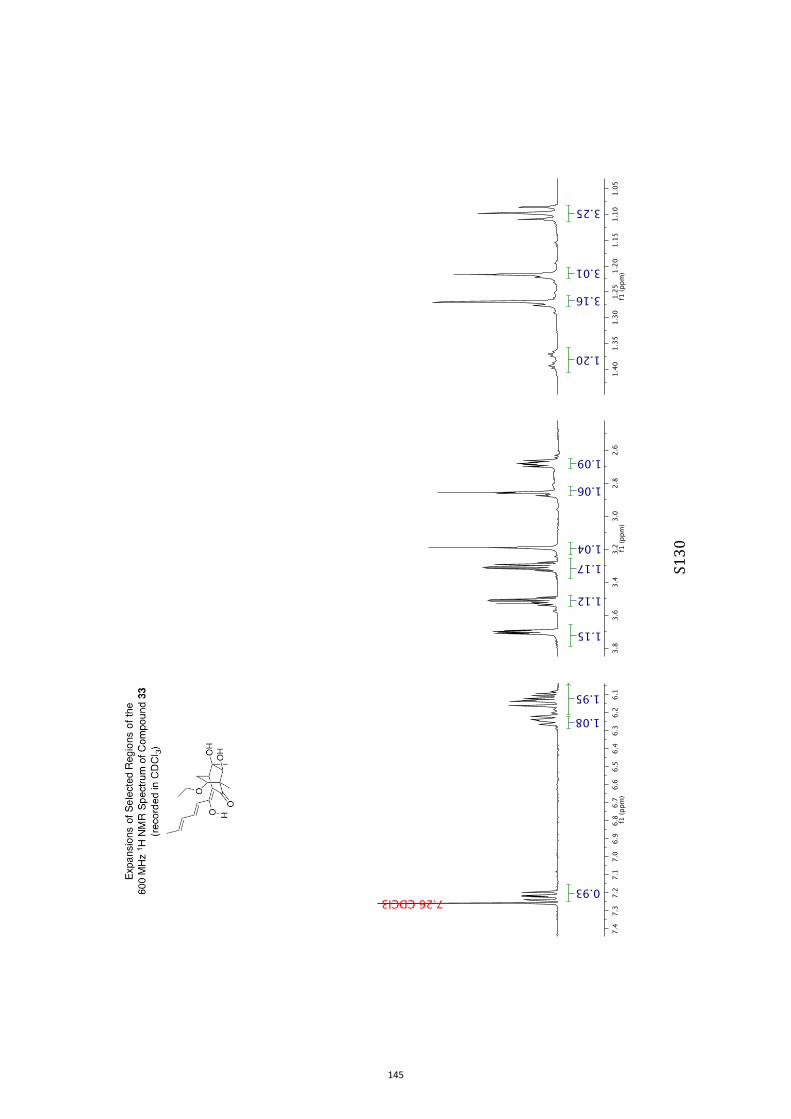
!
S130!
6.1
6.2
6.3
6.4
6.5
6.6
6.7
6.8
6.9
7.0
7.1
7.2
7.3
7.4
f1 (
ppm
)
1.95
1.08
0.937.26 CDCl3
2.6
2.8
3.0
3.2
3.4
3.6
3.8
f1 (
ppm
)
1.09
1.06
1.04
1.17
1.12
1.15
1.0
51
.10
1.1
51
.20
1.2
51
.30
1.3
51
.40
f1 (
ppm
)
3.25
3.01
3.16
1.20
145
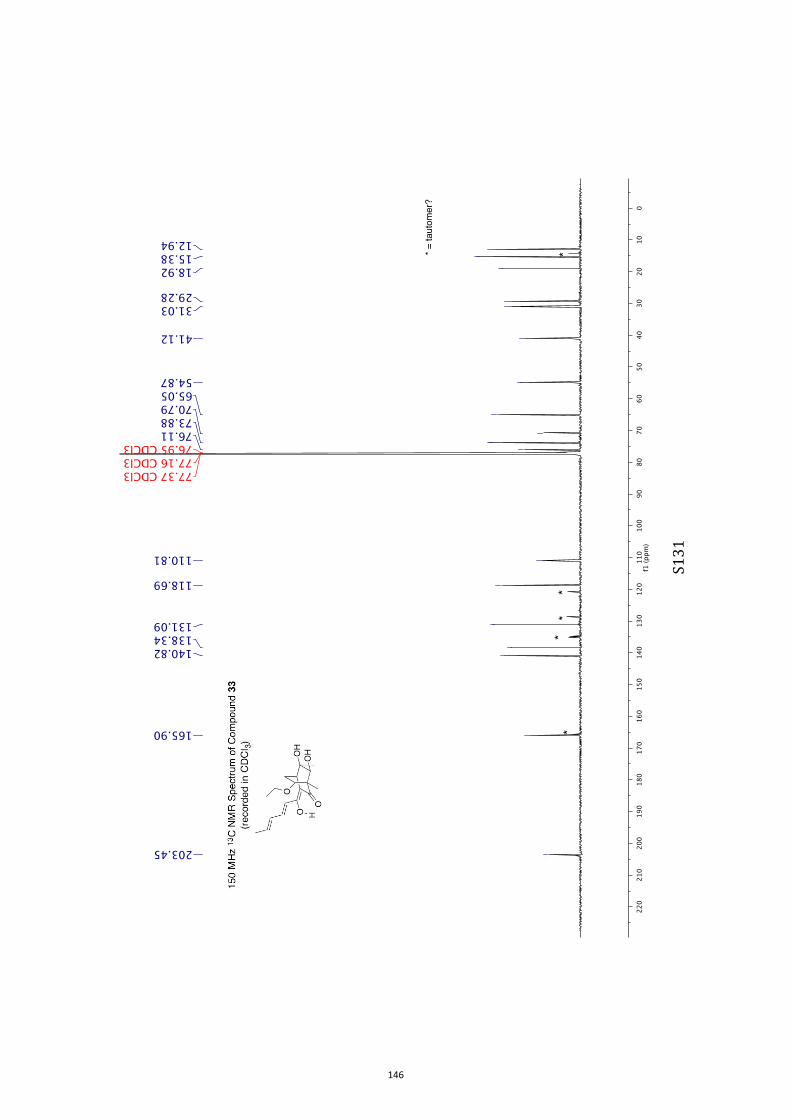
!
S131!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
f1 (
ppm
)
12.9415.3818.92
29.2831.03
41.12
54.8765.0570.7973.8876.1176.95 CDCl377.16 CDCl377.37 CDCl3
110.81
118.69
131.09138.34140.82
165.90
203.45
**
**
** =
tauto
mer?
146
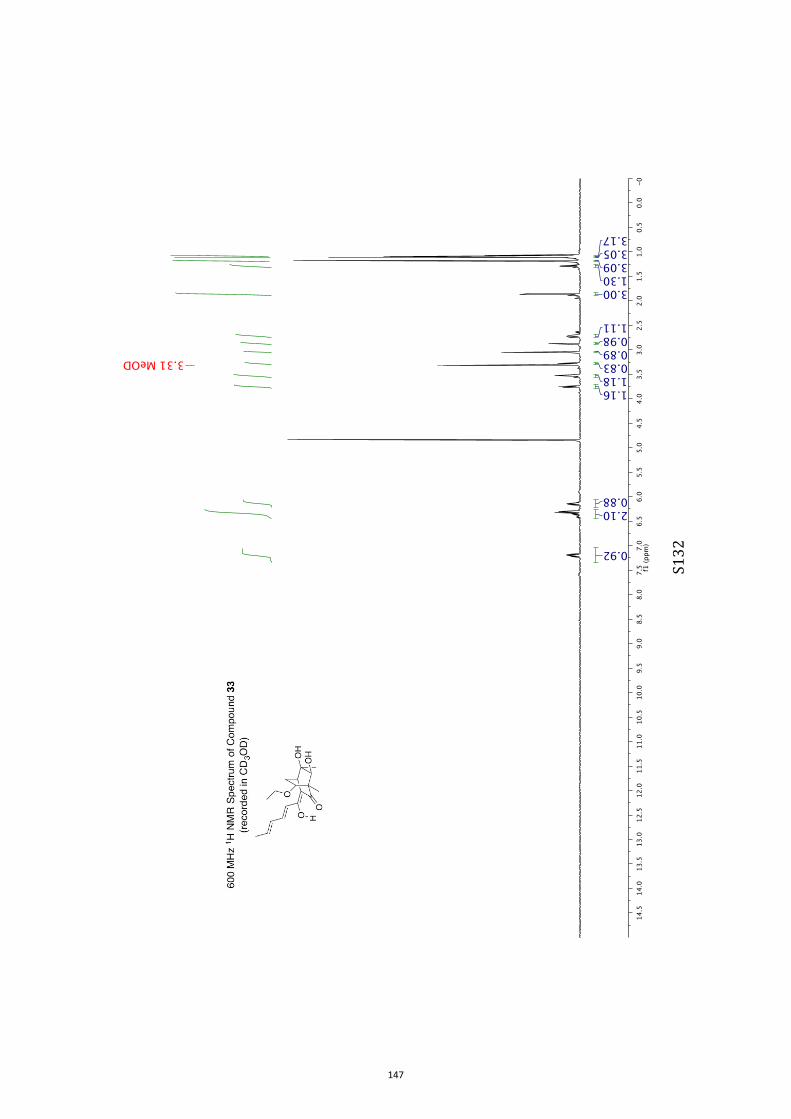
!
S132!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.5f1
(ppm
)
3.173.053.091.303.00
1.110.980.890.831.181.16
0.882.10
0.92
3.31 MeOD
147
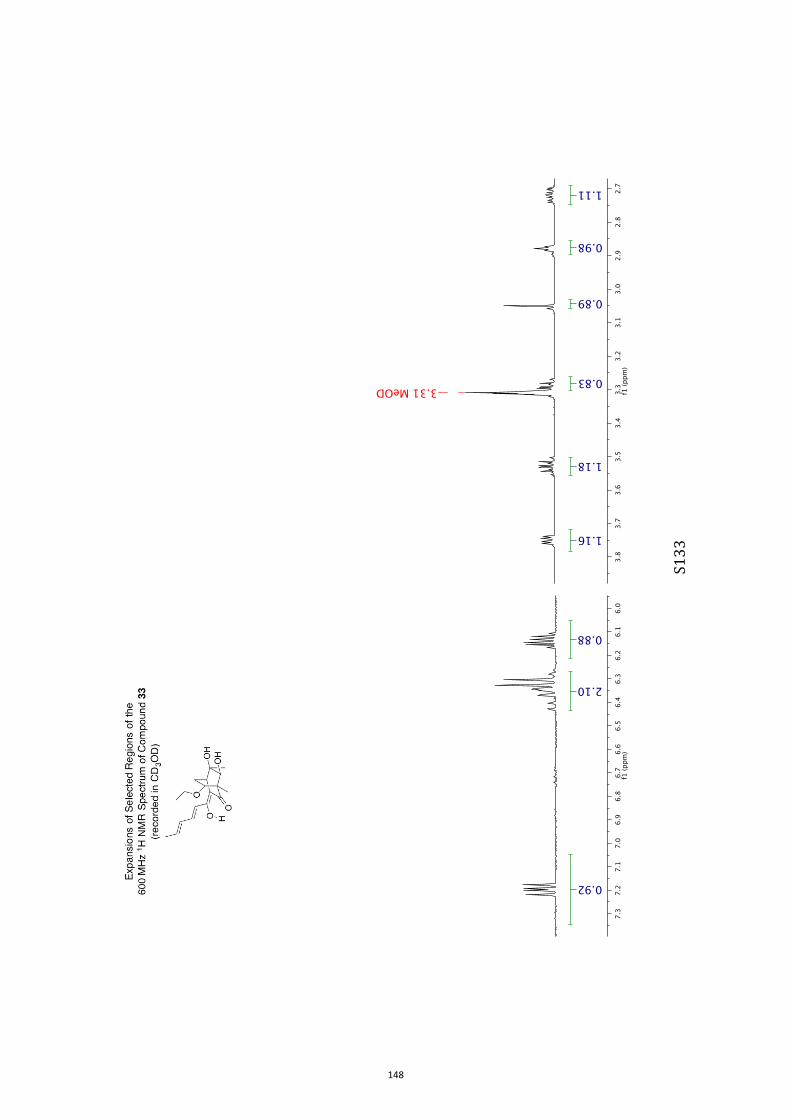
!
S133!
6.0
6.1
6.2
6.3
6.4
6.5
6.6
6.7
6.8
6.9
7.0
7.1
7.2
7.3
f1 (
ppm
)
0.88
2.10
0.92
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
f1 (
ppm
)
1.11
0.98
0.89
0.83
1.18
1.16
3.31 MeOD
148
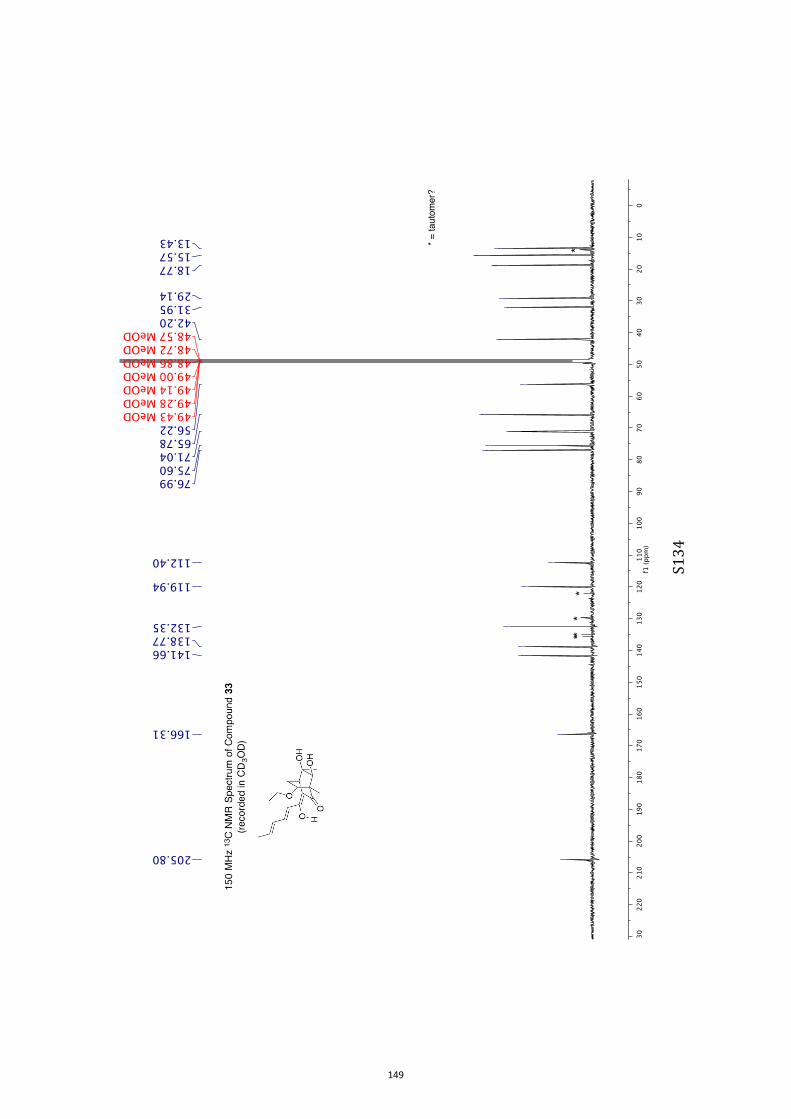
!
S134!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
23
0f1
(ppm
)
13.4315.5718.77
29.1431.9542.2048.57 MeOD48.72 MeOD48.86 MeOD49.00 MeOD49.14 MeOD49.28 MeOD49.43 MeOD56.2265.7871.0475.6076.99
112.40
119.94
132.35138.77141.66
166.31
205.80
***
**
* =
tauto
mer?
149

!
S135!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.003.00
9.003.003.003.003.003.00
1.002.00
3.001.020.941.00
1.00
7.26 CDCl3
150
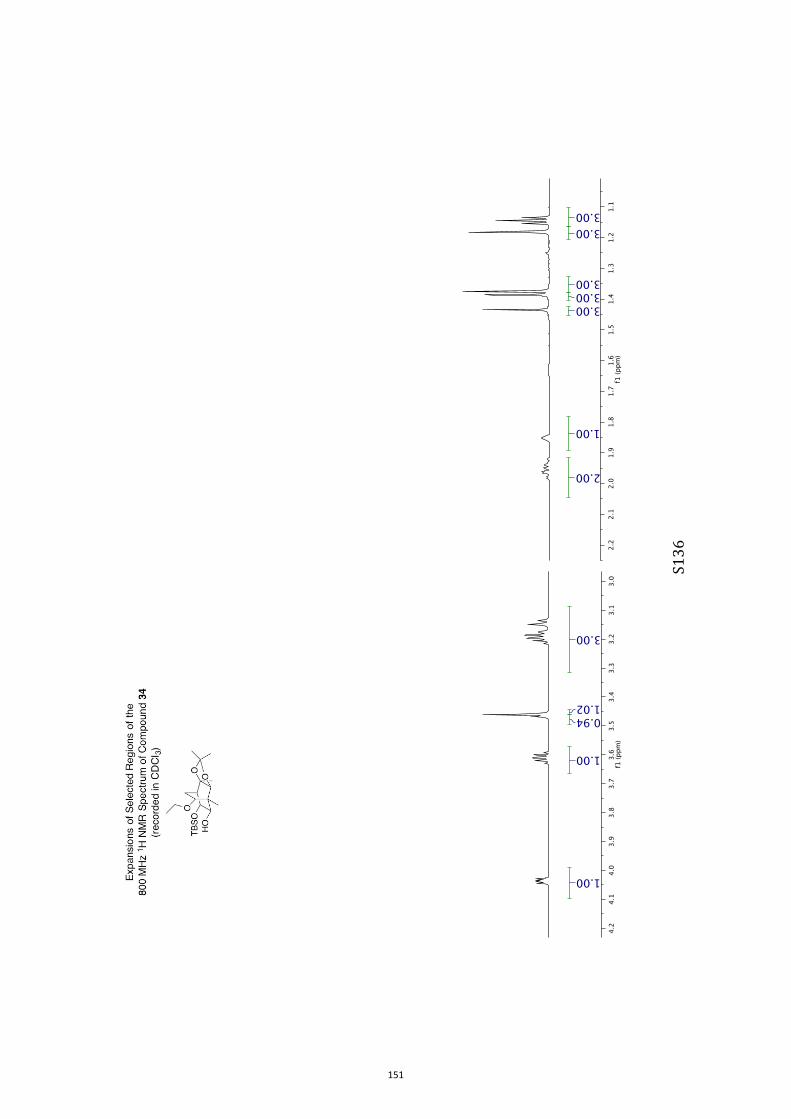
!
S136!
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
4.0
4.1
4.2
f1 (
ppm
)
3.00
1.020.94
1.00
1.00
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
f1 (
ppm
)
3.00
3.00
3.003.003.00
1.00
2.00
151
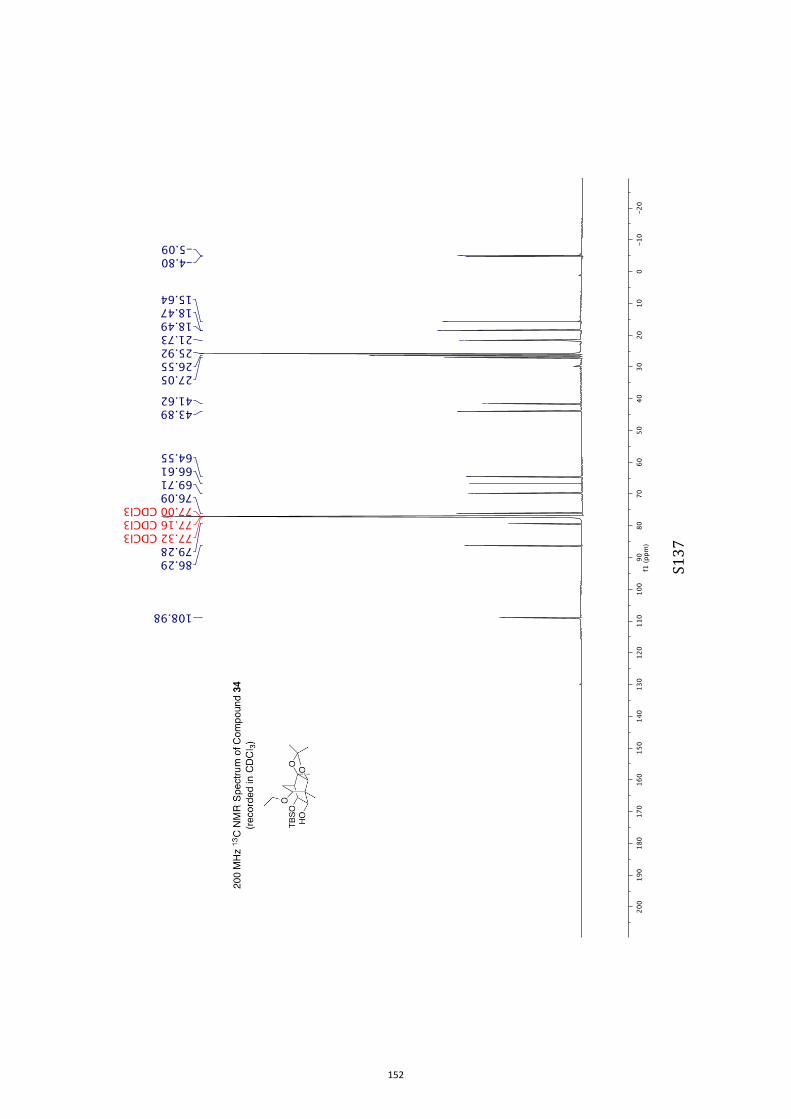
!
S137!
-2
0-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
0f1
(ppm
)
-5.09-4.80
15.6418.4718.4921.7325.9226.5527.05
41.6243.89
64.5566.6169.7176.0977.00 CDCl377.16 CDCl377.32 CDCl379.2886.29
108.98
152
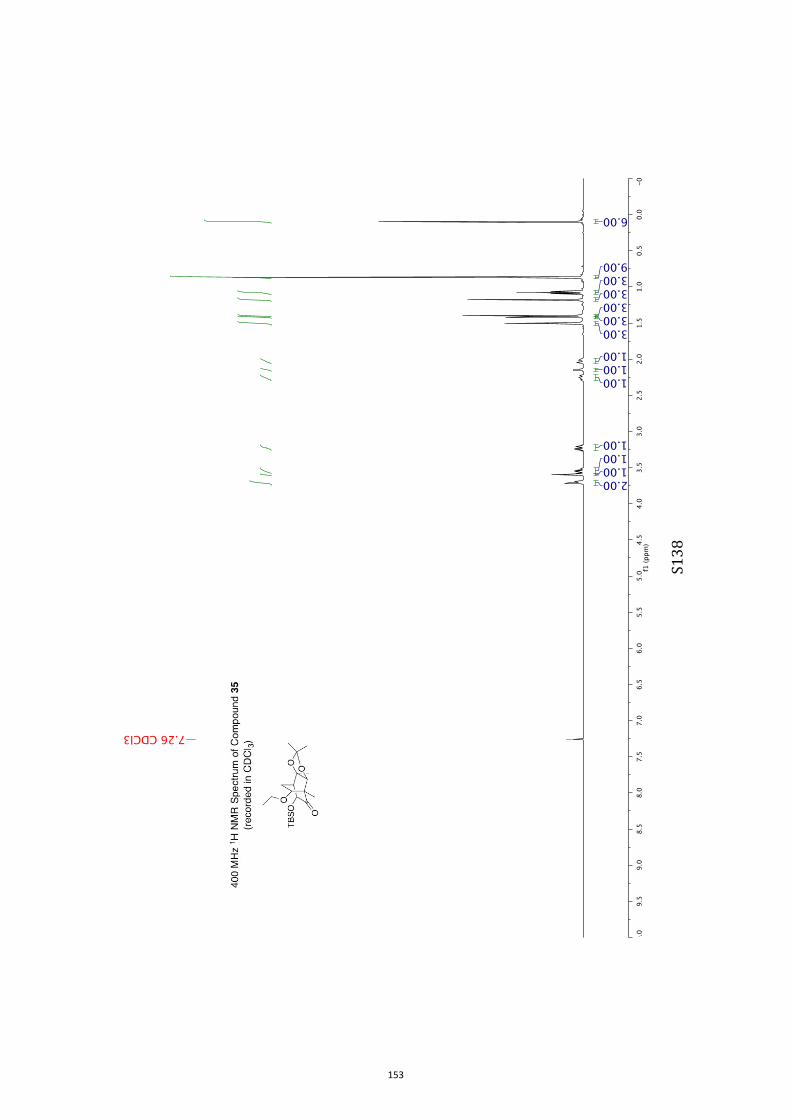
!
S138!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
6.00
9.003.003.003.003.003.00
1.001.001.00
1.001.001.002.00
7.26 CDCl3
153
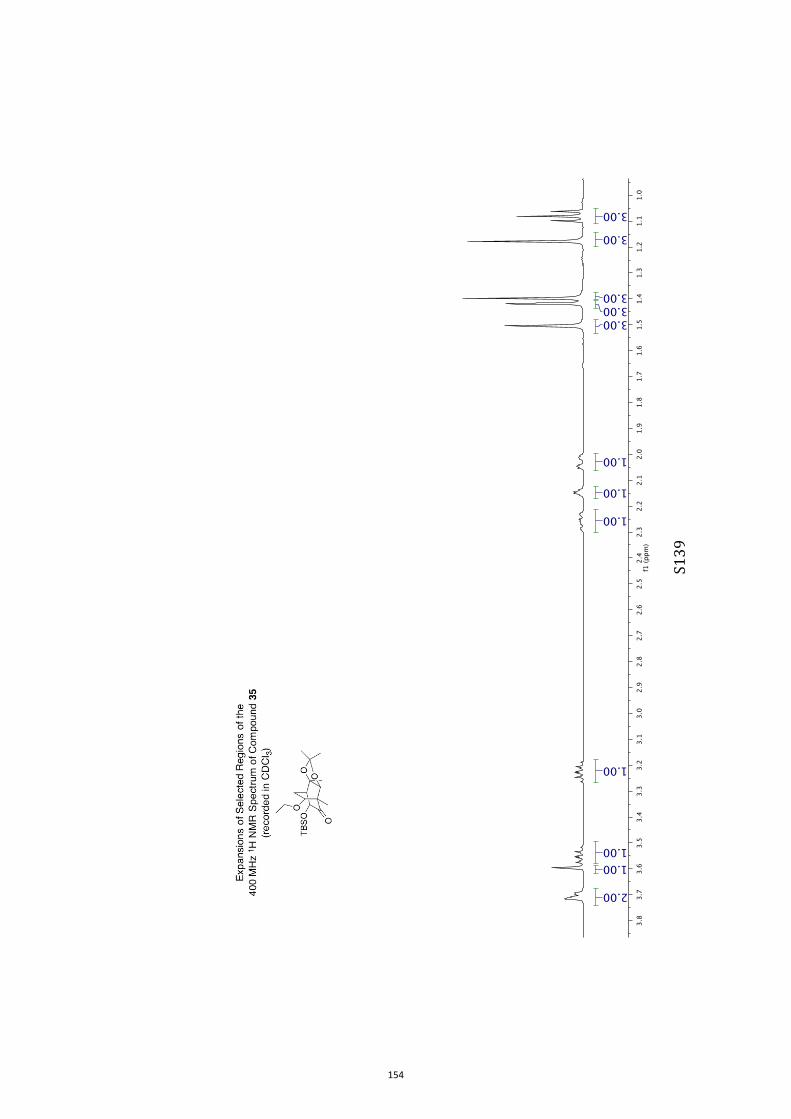
!
S139!
1.0
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2.0
2.1
2.2
2.3
2.4
2.5
2.6
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
f1 (
ppm
)
3.00
3.00
3.003.003.00
1.00
1.00
1.00
1.00
1.00
1.00
2.00
154
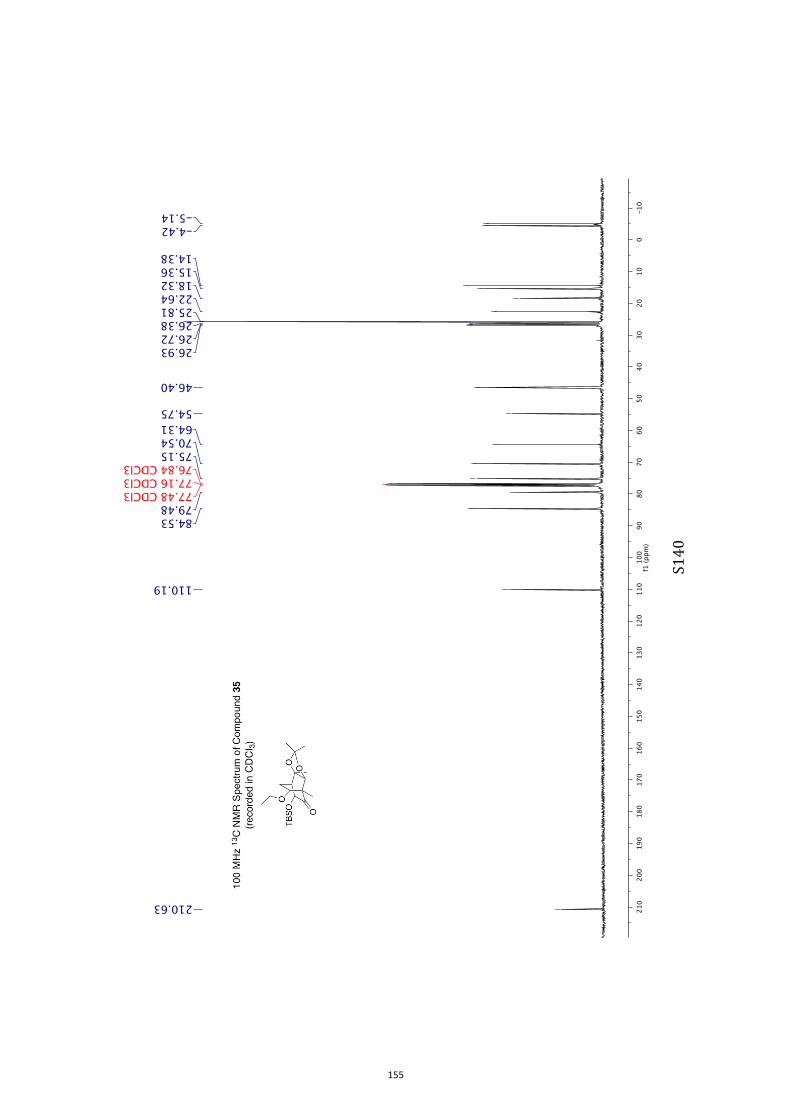
!
S140!
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-5.14-4.42
14.3815.3618.3222.6425.8126.3826.7226.93
46.40
54.75
64.3170.5475.1576.84 CDCl377.16 CDCl377.48 CDCl379.4884.53
110.19
210.63
155

!
S141!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.5f1
(ppm
)
3.083.073.001.173.07
1.001.081.071.082.02
2.041.16
1.00
1.01
7.26 CDCl3
156
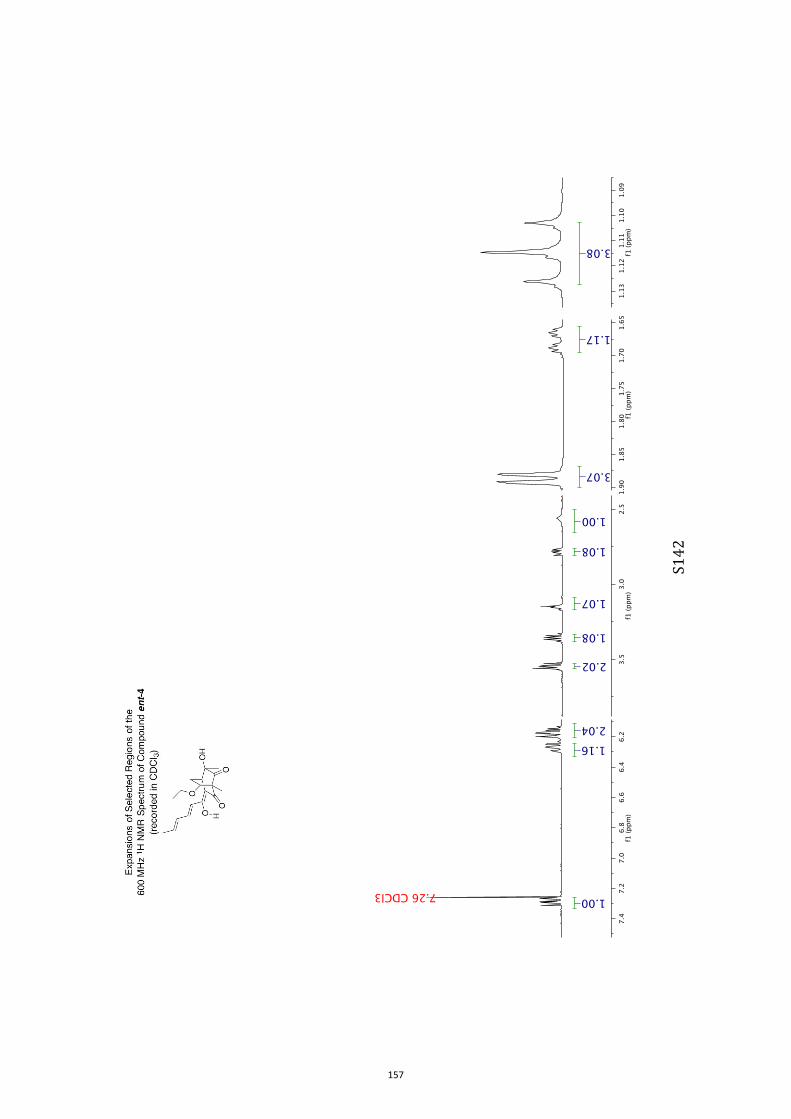
!
S142!
6.2
6.4
6.6
6.8
7.0
7.2
7.4
f1 (
ppm
)
2.04
1.16
1.007.26 CDCl3
2.5
3.0
3.5
f1 (
ppm
)
1.00
1.08
1.07
1.08
2.02
1.6
51.7
01.7
51.8
01.8
51.9
0f1
(ppm
)
1.17
3.07
1.0
91.1
01.1
11.1
21.1
3f1
(ppm
)
3.08
157
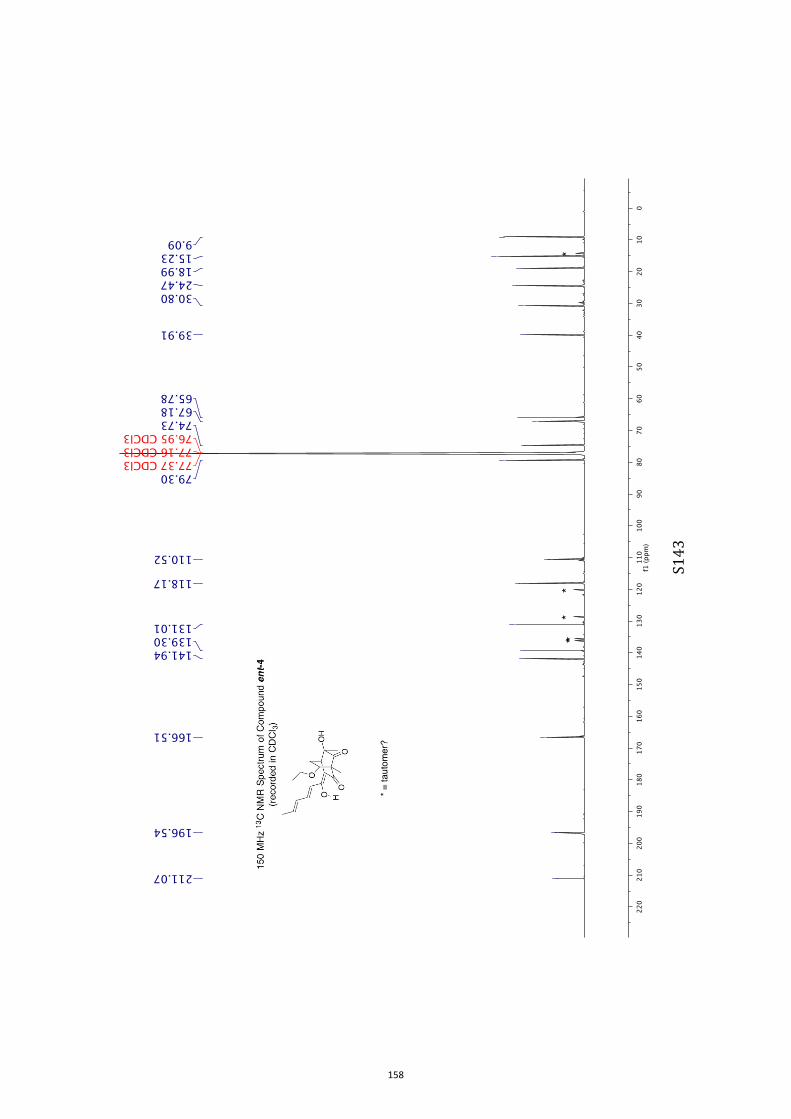
!
S143!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
f1 (
ppm
)
9.0915.2318.9924.4730.80
39.91
65.7867.1874.7376.95 CDCl377.16 CDCl377.37 CDCl379.30
110.52
118.17
131.01139.30141.94
166.51
196.54
211.07
**
**
*
* =
tauto
mer?
158

!
S144!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.5f1
(ppm
)
3.043.103.001.043.03
1.091.031.061.171.06
1.002.00
1.00
3.31 MeOD
159
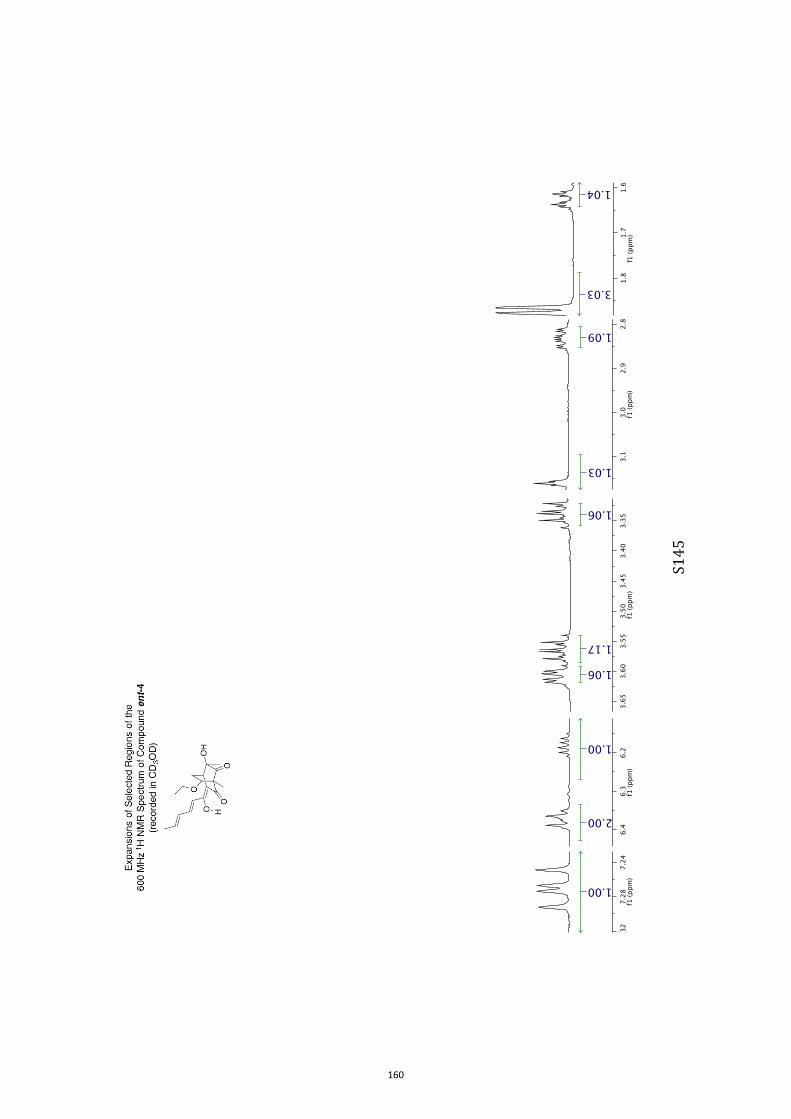
!
S145!
7.2
47.2
87.3
2f1
(ppm
)
1.00
6.2
6.3
6.4
f1 (
ppm
)
1.00
2.00
3.3
53.4
03.4
53.5
03.5
53.6
03.6
5f1
(ppm
)
1.06
1.17
1.06
2.8
2.9
3.0
3.1
f1 (
ppm
)
1.09
1.03
1.6
1.7
1.8
f1 (
ppm
)
1.04
3.03
160
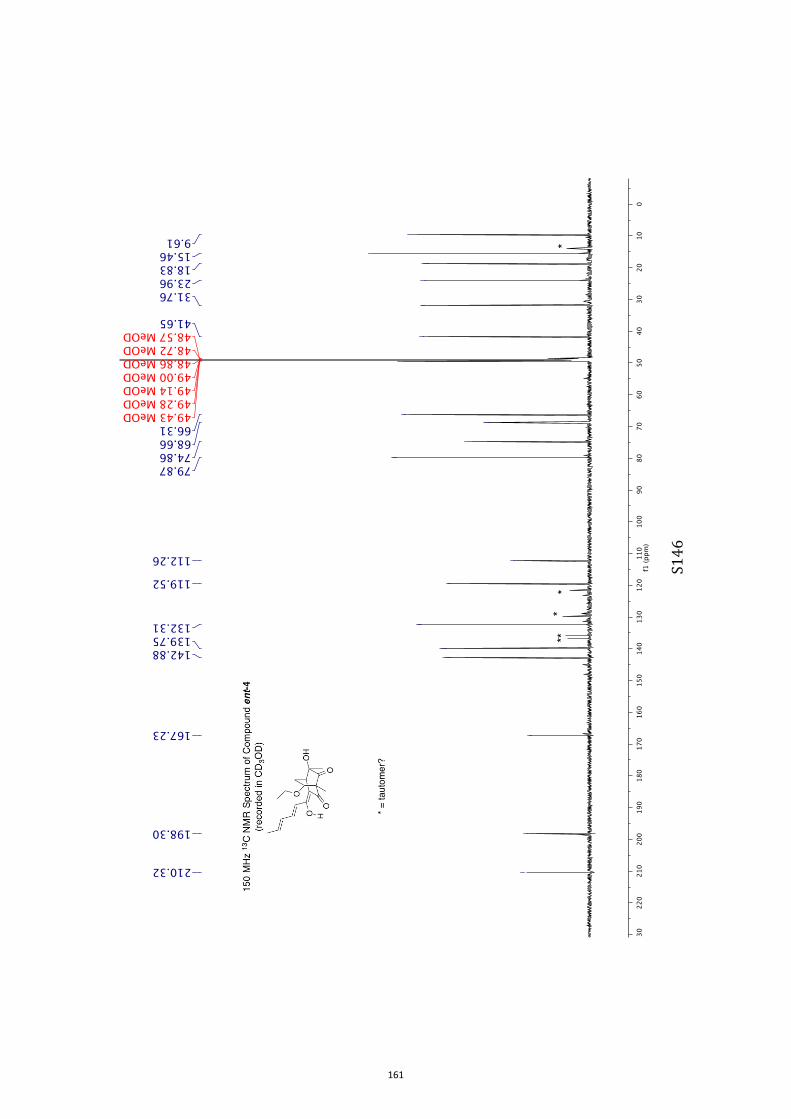
!
S146!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
23
0f1
(ppm
)
9.6115.4618.8323.9631.76
41.6548.57 MeOD48.72 MeOD48.86 MeOD49.00 MeOD49.14 MeOD49.28 MeOD49.43 MeOD66.3168.6674.8679.87
112.26
119.52
132.31139.75142.88
167.23
198.30
210.32
* **
**
* =
tauto
mer?
161
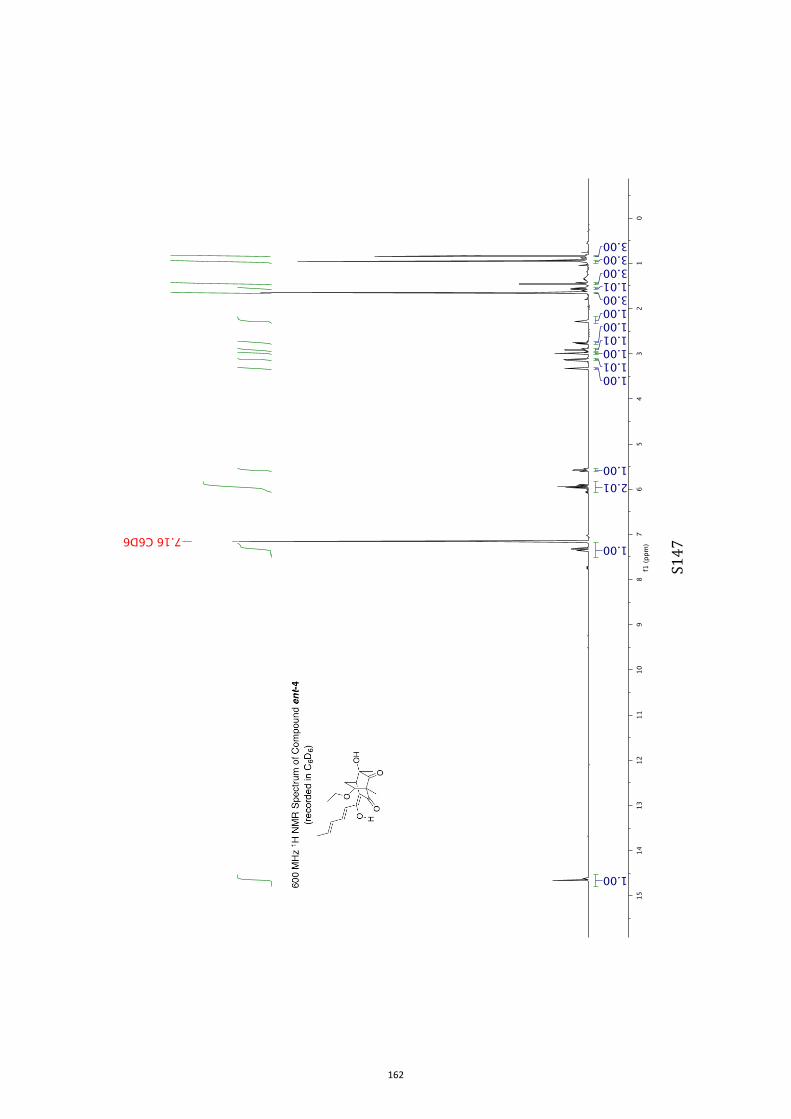
!
S147!
01
23
45
67
89
10
11
12
13
14
15
f1 (
ppm
)
3.003.003.001.013.001.001.001.011.001.011.00
1.00
2.01
1.00
1.00
7.16 C6D6
162

!
S148!
7.3
27
.36
f1 (
ppm
)
1.00
5.5
5.6
5.7
5.8
5.9
f1 (
ppm
)
1.00
2.01
2.8
2.9
3.0
3.1
3.2
3.3
f1 (
ppm
)
1.00
1.01
1.00
1.01
1.00
1.4
51
.50
1.5
5f1
(ppm
)
3.00
1.01
0.8
20
.83
0.8
40
.85
0.8
6f1
(ppm
)
3.00
163
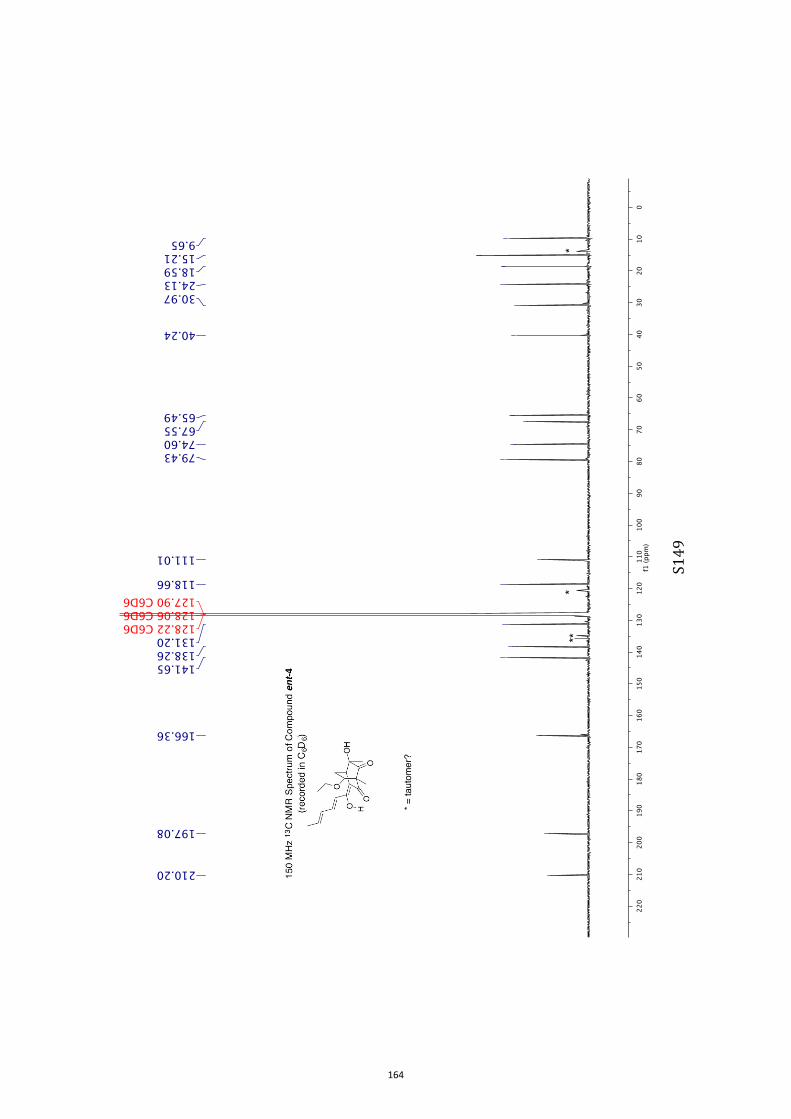
!
S149!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
f1 (
ppm
)
9.6515.2118.5924.1330.97
40.24
65.4967.5574.6079.43
111.01
118.66
127.90 C6D6128.06 C6D6128.22 C6D6131.20138.26141.65
166.36
197.08
210.20
* **
*
* =
tauto
mer?
164
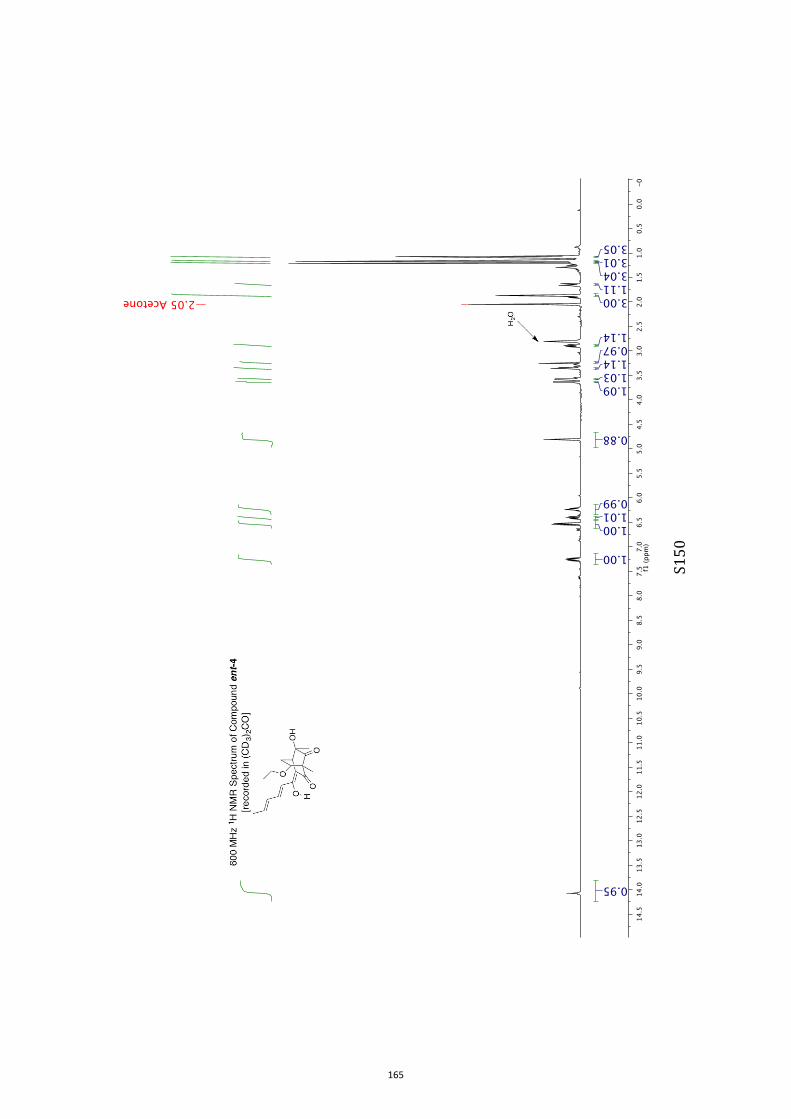
!
S150!
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.5f1
(ppm
)
3.053.013.041.113.00
1.140.971.141.031.09
0.88
0.991.011.00
1.00
0.95
2.05 Acetone
165

!
S151!
7.2
47
.26
7.2
87
.30
f1 (
ppm
)
1.00
6.2
6.3
6.4
6.5
f1 (
ppm
)
0.99
1.01
1.00
2.9
3.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
f1 (
ppm
)
1.14
0.97
1.14
1.03
1.09
1.6
51
.70
1.7
51
.80
1.8
5f1
(ppm
)
1.11
3.00
166
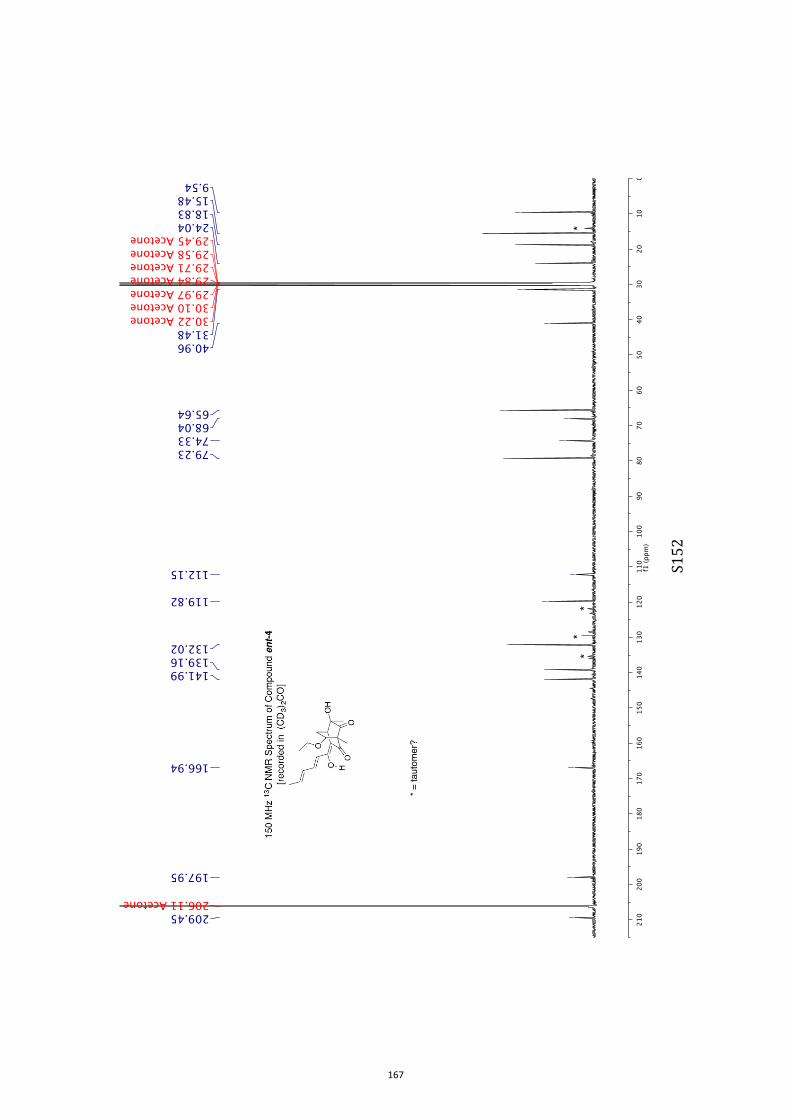
!
S152!
!!
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
0f1
(ppm
)
9.5415.4818.8324.0429.45 Acetone29.58 Acetone29.71 Acetone29.84 Acetone29.97 Acetone30.10 Acetone30.22 Acetone31.4840.96
65.6468.0474.3379.23
112.15
119.82
132.02139.16141.99
166.94
197.95
206.11 Acetone209.45
**
**
* =
tauto
mer?
167


168
Publication Two
Studies on the Photochemical Rearrangements of
Enantiomerically Pure, Polysubstituted and Variously
Annulated Bicyclo[2.2.2]octenones
Qiao Yan, Benoit Bolte, Yuhua Bai, Martin G. Banwell, Anthony C. Willis
and Paul D. Carr
J. Org. Chem., 2017, 82, 8008.


Studies on the Photochemical Rearrangements of EnantiomericallyPure, Polysubstituted, and Variously AnnulatedBicyclo[2.2.2]octenonesQiao Yan, Benoit Bolte, Yuhua Bai, Martin G. Banwell,* Anthony C. Willis, and Paul D. Carr
Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 2601, Australia
*S Supporting Information
ABSTRACT: A series of enantiomerically pure bicyclo[2.2.2]octenones, including the lactone-annulated system 26, has beenprepared by engaging derivatives of an enzymatically derived and homochiral cis-1,2-dihydrocatechol in inter- or intra-molecularDiels−Alder reactions. Systems such as 26 readily participate in photochemically promoted oxa-di-π-methane rearrangement or1,3-acyl migration processes to give products such as diquinane 34 or mixtures of cyclobutanone 36 and cyclopropane 38,respectively.
■ INTRODUCTION
Bicyclo[2.2.2]octenones including the parent system 1(Scheme 1) are excellent substrates for certain photochemicallypromoted rearrangement reactions.1 Specifically, on irradiationin the presence of photosensitizers such as acetophenone theyparticipate in oxa-di-π-methane rearrangements and, soaffording, via a triplet pathway, cyclopropannulated diquinanessuch as 2. In contrast, on direct irradiation they engage, now viaa singlet pathway, in a 1,3-acyl migration reaction (Givensrearrangement) to give bicyclo[4.2.0]oct-4-en-7-ones such as 3.Upon sustained irradiation, photoproduct 3 and many of its
derivatives can undergo decarbonylation to give the corre-sponding Δ2-norcarene, e.g. 4.The capacity to effectively exploit these valuable trans-
formations is contingent on having ready access tobicyclo[2.2.2]octenones, especially enantiomerically pureones. While various methods are available for the synthesis ofsuch systems,2 a particularly useful approach involves thefacially selective intermolecular cycloaddition of dienophiles toenantiomerically pure cis-1,2-dihydrocatechols such as 5(Figure 1) that are readily obtained through the whole-cell
biotransformation of the corresponding arene (toluene in thecase of compound 5).3 Indeed, we have exploited such adductsin the photochemically mediated total synthesis of a range ofsesquiterpenoid natural products including various triquinanes,4
protoilludanes,5 and the structure assigned to a sterpurene.6 Inseeking to extend such studies for the purposes of preparingnew molecular scaffolds, including those of use in drugdiscovery, we became interested in establishing the degree towhich the bicyclo[2.2.2]octenone framework could be sub-stituted and/or annulated and continue to engage in the above-
Received: May 21, 2017Published: July 3, 2017
Scheme 1. Oxa-di-π-methane, 1,3-Acyl Migration, andPhotodecarbonylation Reactions of the ParentBicyclo[2.2.2]octenone (1)
Figure 1. Homochiral cis-1,2-dihydrocatechol 5 and the derivedbicyclo[2.2.2]octenone-based photosubstrates 6 and 7.
Article
pubs.acs.org/joc
© 2017 American Chemical Society 8008 DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022

mentioned photochemical processes. Herein, we detail researchthat serves to emphasize the remarkable extent to which theseprocesses can be applied in a reliable fashion.In connection with our recently reported studies7 on the
synthesis of the sorbicillinoid-derived isolate rezishanone C, weprepared,7 using intermolecular Diels−Alder cycloadditionprocesses, the oxygenated bicyclo[2.2.2]octenones 6 and 7 ingram quantities from cis-1,2-dihydrocatechol 5. Accordingly,compounds 6 and 7 became the substrates used in the openingstages of the present work.
■ RESULTS AND DISCUSSIONIndependent irradiation (using a medium-pressure mercurylamp) of an acetone solution of each of compounds 6 and 7held in a round-bottomed flask made from borosilicate glassand containing ca. 1.4 molar equiv of acetophenone (Scheme2) afforded the anticipated oxa-di-π-methane rearrangement
products 8 (87% or 98% brsm) and 9 (90% or 99% brsm),respectively. All of the spectral data acquired on compounds 8and 9 were entirely consistent with the assigned structures, butfinal confirmation of these followed from single-crystal X-rayanalyses (see the Experimental Section and SupportingInformation for details).When dichloromethane solutions of substrates 6 and 7 were
subjected to direct irradiation for brief periods (less than 1 h)with the same type of lamp then the anticipated Givensrearrangement products 10 (70% or 84% brsm) and 11 (67%),respectively, were obtained. Upon sustained irradiation (5 h) ofthe same substrates, significant quantities of the correspondingdecarbonylated systems 12 (32%) and 13 (33%) were obtainedalong with reduced amounts of the corresponding andchromatographically separable cyclobutenones. Once again, allof the spectroscopic data acquired on compounds 10−13 werein accord with the assigned structures, but those of the first andthe last of these were confirmed by single-crystal X-ray analysis.Establishing the impact of various modes of ring-fusion on
the capacity of bicyclo[2.2.2]octenones to engage in oxa-di-π-
methane and 1,3-acyl migration reactions was another topic ofinterest. Therefore, building upon our earlier studies8 on theassembly of the relevant frameworks through the intramolecularDiels−Alder (IMDA) reactions of crotonate esters derivedfrom cis-1,2-dihydrocatechols, compound 5 was converted(Scheme 3) into the diesters 14 (81%) and 15 (74%) underpreviously established8 conditions. When each of compounds14 and 15 was heated in refluxing toluene they engaged in thetwo possible modes of cycloaddition and thus giving rise to thecorresponding pair of adducts, viz. lactones 16 (40% from 14),17 (41% from 15), 18 (26% from 14), and 19 (12% from 15),that could be separated from one another by conventionalchromatographic means. The remaining ester residues withineach of adducts 18 and 19 were cleaved by standard methodsand the resulting alcohols 20 (74%) and 21 (76%) oxidized tothe corresponding ketones 22 (86%) and 23 (82%),respectively, using the Dess−Martin periodinane.9 Similarly,esters 16 and 17 were cleaved using potassium carbonate inmethanol and the product alcohols 24 (79%) and 25 (94%),respectively, oxidized to the corresponding ketones, namely,compounds 26 (94%) and 27 (78%). Single-crystal X-rayanalyses were conducted on compounds 23 and 27, details ofwhich are presented in the Experimental Section and SI.The oxa-di-π-methane rearrangement of the lactone-annu-
lated bicyclo[2.2.2]octenones 22 and 23 took place readilywhen acetone solutions of each of these was irradiated in thepresence of acetophenone (Scheme 4). By such means thecyclopropane-annulated oxatriquinanes 28 (40%) and 29(43%) were obtained. In contrast, direct irradiation ofsubstrates 22 and 23 led to the corresponding mixtures ofcyclobutanones 30 (up to 75%) and 31 (up to 73%),respectively, as well as their decarbonylated counterparts 32(up to 37%) and 33 (up to 52%), respectively. The structuresof compounds 30 and 32 were confirmed by single-crystal X-ray analyses.Equivalent studies on the photochemical behaviors of the
isomeric lactone annulated bicyclo[2.2.2]octenones 26 and 27(Scheme 5) led to analogous outcomes. Specifically, theanticipated oxatriquinanes 34 (45%) and 35 (49%) wereobtained on photosensitized irradiation of these substrateswhile direct irradiation afforded mixtures of cyclobutanones 36(up to 70%) and 37 (up to 71%) as well as their decarbonylatedcongeners 38 (up to 35%) and 39 (up to 40%), respectively.The structures of compounds 34, 35, 36, and 38 were eachconfirmed by single-crystal X-ray analyses.It is worth noting that both the substrates and the
photoproducts shown in Scheme 5 are pseudoenantiomers oftheir counterparts shown in Scheme 4. Thus, for example, ifeach of compounds 30 (Scheme 4) and 36 (Scheme 5) lackedthe two methyl groups then they would be enantiomers.Accordingly, the protocols just described provide a means bywhich the single enantiomeric form of starting material 5 can beconverted into either enantiomeric form of a range of relativelycomplex molecular frameworks.In seeking to establish the effects of alternate modes of ring
fusion and other substituents on the capacity of bicyclo[2.2.2]-octenones to engage in photochemically promoted rearrange-ments, the behaviors of various polyhydro-4,7-ethanoindenonederivatives were explored. The routes shown in Scheme 6 wereused to prepare these cyclopentannulated systems. Thus, thepreviously described4d tricyclic system 40, prepared by areaction sequence involving an initial Diels−Alder cyclo-addition reaction between 2-cyclopenten-1-one and the
Scheme 2. Photochemical Behaviors ofBicyclo[2.2.2]octenones 6 and 7 under Either DirectIrradiation or Photosensitized Conditions
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8009

acetonide derived from cis-diol 5, was reduced with LiAlH4, andthe previously reported4d,5b alcohol 41 (39%) obtained in pureform after chromatographic separation from its coproducedepimer.4d,5b Compound 41, the structure of which wasconfirmed by single-crystal X-ray analysis, was acetylatedunder standard conditions to give ester 42 (97%), and theacetonide moiety associated with this last compound washydrolyzed by treating a methanol/water solution of it withacidified AG-50W-X8 resin. The product diol 43 (97%) wasselectively oxidized using the sterically demanding oxammo-
nium salt derived from p-TsOH·H2O-promoted disproportio-nation of 4-acetamido-TEMPO,10 and thus producing acyloin44 (97%) that was also subjected to single-crystal X-rayanalysis. Compound 44 was converted into derivatives 45(91%), 46 (77%), 47 (82%), and 48 (93%) by standardmethods, while samarium iodide mediated deoxygenation ofbenzoate 48 also allowed for the preparation of bicyclo[2.2.2]-octenone 49 (85%). Once again, the structures of compounds45, 47, and 48 were confirmed by single-crystal X-ray analyses.
Scheme 3. Synthesis of Lactone-Annulated Bicyclo[2.2.2]octenones 22, 23, 26, and 27
Scheme 4. Photochemical Behaviors ofBicyclo[2.2.2]octenones 22 and 23 under Either DirectIrradiation or Sensitized Conditions
Scheme 5. Photochemical Behaviors ofBicyclo[2.2.2]octenones 26 and 27 under Either DirectIrradiation or Sensitized Conditions
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8010

On direct irradiation of dichloromethane solutions of each ofcompounds 45−49 they engaged in 1,3-acyl migrationreactions to give the cyclobutanones 50 (43% or 99% brsm),51 (27% or 98% brsm), 52 (18% or 99% brsm), 53 (17% orquant brsm), and 54 (23% or 92% brsm), respectively.Irradiation of compounds 50 and 47 led to cyclopropanes 55(98%) and 56 (80%), respectively. The structures of all of these
photoproducts were determined through comprehensivespectroscopic analyses. In particular, the infrared spectrum ofeach of cyclobutanones 50−54 exhibited a characteristiccarbonyl absorption band in the range 1778−1792 cm−1 aswell as a carbonyl carbon resonance above δ 200 ppm in thecorresponding 13C NMR spectrum. The structure of compound53 was confirmed by single-crystal X-ray analysis.Given our extensive earlier studies4 on the oxa-di-π-methane
rearrangements of substrates very closely related to compounds45−49, we have not subjected these to photosensitizedirradiation but would fully expect them to behave in a similarmanner and thus leading to the corresponding cyclopropannu-lated triquinanes.In contrast to outcomes noted above (Scheme 6), when the
mesylate 57 (Scheme 7), which was readily prepared in 56%
yield from acyloin 44, was subjected (as a solution indichloromethane) to direct irradiation it engaged in an oxa-di-π-methane rearrangement reaction rather than a 1,3-acylmigration reaction and so affording the cyclopentannulated andstereochemically pure triquinane 58 in 48% yield (or 98% yieldbrsm). The configuration at the carbon bearing the mesyloxygroup is tentatively assigned as illustrated although the alternateone cannot be discounted as a result of the intervention of aphotoepimerization process.4b−d The precise origins of thisseemingly anomalous behavior of sulfonate ester 57 remainunclear at the present time and serve to highlight the verylimited understanding of the photochemical properties of suchsystems.11 These matters are the subject of ongoing studies inour laboratories.
■ CONCLUSIONSThe studies reported here have established that a wide range ofextensively substituted and variously annulated bicyclo[2.2.2]-octenones are capable of engaging in either oxa-di-π-methaneor 1,3-acyl migration reactions depending upon the mode ofphotoactivation (sensitized vs direct irradiation). The 1,3-acylmigration reactions leading to the isomeric cyclobutanones canbe followed by rapid photodecarbonylation processes to
Scheme 6. Synthesis of CyclopentannulatedBicyclo[2.2.2]octenones 45−49 and Their PhotochemicalBehaviors under Direct Irradiation Conditions
Scheme 7. Synthesis and Anomalous PhotochemicalBehavior of the Mesyloxy-SubstitutedBicyclo[2.2.2]octenone 57
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8011

coproduce the corresponding (but normally readily separable)cyclopropanes. A noteworthy feature of the photochemicalbehaviors of the annulated bicyclo[2.2.2]octenones studiedhere is that the mode of ring fusion (annulation) can have asignificant impact on the rates of both types of processes. Thus,for example, the lactone-annulated systems 22, 23, 26, and 27appear to engage in more rapid 1,3-acyl migration reactionsthan their cyclopentannulated counterparts 45−49. The samecould be said of the corresponding oxa-di-π-methane process,especially when the outcomes of our earlier studies4 are takeninto account. That is to say, the lactone annulated compoundsjust mentioned also appear to participate in more rapid triplet-sensitized rearrangement reactions than their cyclopentannu-lated counterparts. The origins of these variations are thesubject of ongoing studies, the outcomes of which will bereported in due course.
■ EXPERIMENTAL SECTIONGeneral Experimental Procedures. Unless otherwise specified,
proton (1H) and carbon (13C) NMR spectra were recorded at 18 °Cin base-filtered CDCl3 on a spectrometer operating at 400 MHz forproton and 100 MHz for carbon nuclei. 1H NMR data are recorded asfollows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz),relative integral] where multiplicity is defined as s = singlet; d =doublet; t = triplet; q = quartet; m = multiplet or combinations of theabove. In relevant cases, the signal due to residual CHCl3 appearing atδH 7.26 and the central resonance of the CDCl3 “triplet” appearing atδC 77.0 were used to reference 1H and 13C NMR spectra, respectively.Samples were analyzed by infrared spectroscopy (νmax) as thin films onKBr plates. Optical rotations were recorded using the sodium D-line(589 nm) in a cell with a path length of 1 dm, at the concentrationsindicated and in the specified solvent at 22 °C. Specific rotations werethen calculated in the usual manner. Low- and high-resolution electronimpact (EI) mass spectra were recorded on a double focusing, triplesector machine. Low- and high-resolution ESI mass spectra wererecorded on a triple-quadrupole mass spectrometer operating inpositive ion mode. Melting points are uncorrected. Analytical thinlayer chromatography (TLC) was performed on aluminum-backed 0.2mm thick silica gel 60 F254 plates. Eluted plates were visualized using a254 nm UV lamp and/or by treatment with a suitable dip followed byheating. These dips included phosphomolybdic acid/ceric sulfate/sulfuric acid (conc)/water (37.5 g: 7.5 g: 37.5 g: 720 mL), potassiumpermanganate/potassium carbonate/5% sodium hydroxide aqueoussolution/water (3 g: 20 g: 5 mL: 300 mL), and p-anisaldehyde orvanillin/sulfuric acid (conc)/ethanol (15 g: 2.5 mL: 250 mL). Flashchromatographic separations were carried out following protocolsdefined by Still et al.12 with silica gel 60 (40−63 μm) as the stationaryphase and using the AR- or HPLC-grade solvents indicated. Themelting points of solids purified by such means were recorded directly(ie after they had crystallized from the concentrated chromatographicfractions). Starting materials, reagents, drying agents, and otherinorganic salts were generally commercially available and were used assupplied. Tetrahydrofuran (THF), methanol, and dichloromethanewere dried using a solvent purification system that is based upon atechnology originally described by Grubbs et al.13 Where necessary,reactions were performed under a nitrogen atmosphere.Specific Chemical Transformations. (2a′R,3S,4S,4aR)-3,4-Dihy-
droxy-2b-methylhexahydrocyclopropa[cd]pentalen-2(1H)-one (8).A deoxygenated solution of compound 67 (94 mg, 0.56 mmol) andacetophenone (90 μL, 0.77 mmol) in acetone (100 mL) maintainedunder nitrogen was subjected to irradiation with a Hanovia 450 Wmedium-pressure mercury-vapor lamp for 2.5 h and then cooled andconcentrated under reduced pressure to give a yellow oil. This materialwas subjected to flash column chromatography (silica, 1:1 → 1:0 v/vethyl acetate/40−60 petroleum ether gradient elution) to afford twofractions, A and B.
Concentration of fraction A (Rf = 0.3 in ethyl acetate) afforded thestarting compound 6 (10 mg, 11% recovery) as a colorless, crystallinesolid that was identical, in all respects, with an authentic sample.
Concentration of fraction B (Rf = 0.1 in ethyl acetate) affordedcompound 8 (82 mg, 87% or 98% brsm) as a white, crystalline solid:mp = 169−171 °C; [α]D = +71.4 (c 1.0, methanol); 1H NMR (400MHz, CD3OD) δ 4.15 (d, J = 2.6 Hz, 1H), 3.74 (s, 1H), 2.81 (t, J =5.0 Hz, 1H), 2.72 (m, 1H), 2.42 (m, 1H), 2.08 (m, 1H), 1.82 (d, J =5.0 Hz, 1H), 1.37 (s, 3H) (signals due to O−H group protons notobserved); 13C NMR (100 MHz, CD3OD) δ 217.3, 88.2, 86.3, 47.8,47.0, 46.3(0), 46.2(8), 44.2, 21.5; IR νmax 3356, 2962, 2904, 1706,1693, 1290, 1040, 898 cm−1; MS (ESI, + ve) m/z 191 [(M + Na)+,100]; HRMS m/z (M + Na)+ calcd for C9H12O3Na 191.0684, found191.0685.
(2a′R,3S,4S,4aR)-4-((tert-Butyldimethylsilyl)oxy)-3-hydroxy-2b-methylhexahydrocyclopropa[cd]pentalen-2(1H)-one (9). A deoxy-genated solution of compound 77 (158 mg, 0.56 mmol) andacetophenone (90 μL, 0.77 mmol) in acetone (100 mL) maintainedunder nitrogen was subjected to irradiation with a Hanovia 450 Wmedium-pressure mercury-vapor lamp for 2.5 h. The cooled reactionmixture was concentrated under reduced pressure to give a clear,yellow oil, and this was subjected to flash column chromatography(silica, 1:9 → 3:7 v/v ethyl acetate/40−60 petroleum ether gradientelution) to afford two fractions, A and B.
Concentration of fraction A (Rf = 0.4 in 3:7 v/v ethyl acetate/40−60 petroleum ether) afforded compound 7 (13 mg, 8% recovery) as acolorless crystalline solid that was identical, in all respects, with anauthentic sample.
Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/40−60 petroleum ether) afforded compound 9 (143 mg, 90% or 99%brsm) as a white, crystalline solid: mp = 99−100 °C; [α]D = +41.8 (c1.0, CHCl3);
1H NMR (800 MHz, CDCl3) δ 4.20 (d, J = 2.5 Hz, 1H),3.81 (s, 1H), 2.76 (t, J = 5.1 Hz, 1H), 2.66 (m, 1H), 2.38 (m, 1H),2.13 (d, J = 17.9 Hz, 1H), 1.86 (d, J = 5.0 Hz, 1H), 1.79 (broad s, 1H),1.35 (s, 3H), 0.90 (s, 9H), 0.08 (s, 6H); 13C NMR (200 MHz, CDCl3)δ 213.3, 86.8, 85.8, 45.3, 45.0(4), 44.9(8), 44.4, 42.2, 24.9, 20.4, 17.2,− 5.6; IR νmax 3351, 2957, 2929, 2886, 2857, 1719, 1254, 1092, 867,836, 771 cm−1; MS (ESI, + ve) m/z 305 [(M + Na)+, 100], 283 [(M +H)+, 30]; HRMS m/z (M + Na)+ calcd for C15H26O3SiNa 305.1549,found 305.1548.
(1R,2S,3S,6R)-2,3-Dihydroxy-4-methylbicyclo[4.2.0]oct-4-en-7-one (10). A magnetically stirred and deoxygenated solution ofcompound 6 (100 mg, 0.59 mmol) in dichloromethane (100 mL)maintained under nitrogen was subjected to direct irradiation with aHanovia 450 W medium-pressure mercury-vapor lamp for 0.83 h. Thecooled reaction mixture was concentrated under reduced pressure togive a yellow solid that was subjected to flash column chromatography(silica, 2:3 → 1:0 v/v ethyl acetate/40−60 petroleum ether gradientelution) to afford two fractions, A and B.
Concentration of fraction A (Rf = 0.4 in ethyl acetate) affordedcompound 10 (70 mg, 70% or 84% brsm) as a white, crystalline solid:mp = 83−85 °C; [α]D = −234.4 (c 1.0, MeOH); 1H NMR (400 MHz,CD3OD) δ 5.40 (m, 1H), 3.96 (d, J = 6.9 Hz, 1H), 3.88 (m, 1H), 3.47(t, J = 7.5 Hz, 1H), 3.24 (m, 1H), 2.98 (m, 1H), 2.57 (m, 1H), 1.81 (s,3H) (signals due to O−H group protons not observed); 13C NMR(100 MHz, CD3OD) δ 209.0, 140.2, 116.7, 76.2, 73.6, 61.0, 50.1, 30.2,20.0; IR νmax 3391, 2882, 1769, 1066, 1047, 1014, 884 cm
−1; MS (ESI,+ ve) m/z 223 [(M + MeOH + Na)+, 40], 191 [(M + Na)+, 100];HRMS m/z (M + Na)+ calcd for C9H12O3Na 191.0684, found191.0684.
Concentration of fraction B (Rf = 0.3 in ethyl acetate) affordedcompound 6 (14 mg, 14% recovery) as a colorless, crystalline solidthat was identical, in all respects, with an authentic sample.
(1R,2S,3S,6R)-2-((tert-Butyldimethylsilyl)oxy)-3-hydroxy-4-methylbicyclo[4.2.0]oct-4-en-7-one (11). A magnetically stirred anddeoxygenated solution of compound 7 (100 mg, 0.35 mmol) indichloromethane (100 mL) maintained under nitrogen was subjectedto direct irradiation with a Hanovia 450 W medium-pressure mercury-vapor lamp for 0.67 h. The cooled reaction mixture was concentratedunder reduced pressure to give a pale-yellow oil. This was subjected to
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8012

flash column chromatography (silica, 1:9 v/v ethyl acetate/40−60petroleum ether elution) to give, after concentration of the appropriatefractions (Rf = 0.6 in 3:7 v/v ethyl acetate/40−60 petroleum ether),compound 11 (67 mg, 67%) as a white, crystalline solid: mp = 67−69°C; [α]D = −139.6 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ5.38 (m, 1H), 4.04 (d, J = 7.0 Hz, 1H), 3.86 (m, 1H), 3.57 (t, J = 7.7Hz, 1H), 3.24 (m, 1H), 2.94 (m, 1H), 2.57 (m, 1H), 2.18 (broad s,1H), 1.82 (s, 3H), 0.90 (s, 9H), 0.13 (s, 3H), 0.10 (s, 3H); 13C NMR(100 MHz, CDCl3) δ 207.0, 138.4, 115.1, 77.9, 73.4, 60.6, 50.2, 29.9,25.9, 19.1, 18.3, − 3.8, − 4.2; IR νmax 3499, 2954, 2930, 2857, 1779,1126, 1087, 836 cm−1; MS (ESI, + ve) m/z 337 [(M + MeOH + Na)+,30], 305 [(M + Na)+, 100]; HRMS m/z (M + Na)+ calcd forC15H26O3SiNa 305.1549, found 305.1554.(1R,2S,3S,6R)-4-Methylbicyclo[4.1.0]hept-4-ene-2,3-diol (12). A
magnetically stirred and deoxygenated solution of compound 6 (100mg, 0.59 mmol) in dichloromethane (100 mL) maintained under anitrogen atmosphere was subjected to direct irradiation with a Hanovia450 W medium-pressure mercury-vapor lamp for 5 h. The cooledreaction mixture was concentrated under reduced pressure to give abrown oil that was subjected to flash column chromatography (silica,1:4 → 1:1 v/v ethyl acetate/40−60 petroleum ether gradient elution)to afford two fractions, A and B.Concentration of fraction A (Rf = 0.6 in ethyl acetate) afforded
compound 12 (27 mg, 32%) as a pale-yellow oil: [α]D = +93.2 (c 0.8,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.00 (m, 1H), 4.04−3.99(complex m, 2H), 1.92 (broad s, 2H), 1.76 (s, 3H), 1.48−1.35(complex m, 2H), 1.03 (m, 1H), 0.64 (m, 1H); 13C NMR (100 MHz,CDCl3) δ 130.3, 127.8, 73.3, 70.1, 21.4, 19.6, 18.7, 11.0; IR νmax 3367,2920, 1445, 1260, 1048, 1001, 724 cm−1; MS (EI, 70 eV) m/z 140(M•+, 30), 122 [(M − H2O)
•+, 60], 93 (100), 79 (90), 77 (70), 70(60), 55 (60); HRMS m/z M•+ calcd for C8H12O2 140.0837, found140.0842.Concentration of fraction B (Rf = 0.4 in ethyl acetate) afforded
compound 10 (48 mg, 48%) as a white, crystalline solid that wasidentical, in all respects, with an authentic sample.(1R,2S,3S,6R)-2-((tert-Butyldimethylsilyl)oxy)-4-methylbicyclo-
[4.1.0]hept-4-en-3-ol (13). A magnetically stirred and deoxygenatedsolution of compound 7 (100 mg, 0.35 mmol) in dichloromethane(100 mL) maintained under a nitrogen atmosphere was subjected toirradiation with a Hanovia 450 W medium-pressure mercury-vaporlamp for 5 h. The cooled reaction mixture was concentrated underreduced pressure to give a brown oil that was subjected to flashcolumn chromatography (silica, 1:9 → 1:4 v/v ethyl acetate/40−60petroleum ether gradient elution) to afford two fractions, A and B.Concentration of fraction A (Rf = 0.7 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 13 (30 mg, 33%) as acolorless, crystalline solid: mp = 32−33 °C; [α]D = +35.3 (c 1.0,CHCl3).
1H NMR (400 MHz, CDCl3) δ 5.71 (m, 1H), 4.08 (d, J = 5.9Hz, 1H), 3.59 (m, 1H), 1.97 (broad s, 1H), 1.74 (s, 3H), 1.33−1.17(complex m, 2H), 0.99−0.94 (complex m, 1H), 0.92 (s, 9H), 0.32 (m,1H), 0.14 (s, 3H), 0.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ134.4, 124.7, 75.1, 74.0, 26.0, 19.6, 19.3, 18.7, 18.3, 12.0, − 4.1, − 4.6;IR νmax 3458, 2929, 2857, 1254, 1075, 1003, 835, 775 cm−1; MS (EI,70 eV) m/z 239 [(M − CH3
•)+, 30], 211 (35), 197 [(M − C4H9•)+,
35], 105 (60), 75 (100), 73 (95); HRMS m/z (M − CH3•)+ calcd for
C13H23O2Si 239.1467, found 239.1468.Concentration of fraction B (Rf = 0.6 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 11 (33 mg, 33%) as acolorless, crystalline solid that was identical, in all respects, with anauthentic sample.(1S,2R)-3-Methylcyclohexa-3,5-diene-1,2-diyl (2E,2′E)-Bis(but-2-
enoate) (14). A magnetically stirred solution of (1S,2R)-3-methyl-cyclohexa-3,5-diene-1,2-diol (5) (2.40 g, 19.0 mmol) in dry THF (284mL) was cooled to −78 °C and then treated, dropwise over 0.5 h, withn-BuLi (12.5 mL of a 1.6 M solution in hexane, 20.0 mmol). Theresulting solution was stirred at −78 °C for 0.17 h and then treatedwith crotonoyl chloride (1.80 mL, 19.02 mmol) over 0.33 h. Theresulting solution was stirred for 2 h at −78 °C and then treated,dropwise over 0.5 h, with n-BuLi (12.5 mL of a 1.6 M solution inhexane, 20.0 mmol). After 0.17 h, another portion of crotonoyl
chloride (1.80 mL, 19.02 mmol) was added over 0.33 h, and thenstirring of the recation mixture was continued at −78 °C for 1 h. Theensuing mixture was warmed to 22 °C and then quenched withNaHCO3 (96 mL of a saturated aqueous solution) before beingdiluted with diethyl ether (390 mL). The separated organic phase waswashed with NH4Cl (1 × 96 mL of a saturated aqueous solution) andthen dried (MgSO4), filtered, and concentrated under reducedpressure. The residue thus obtained was subjected to flash columnchromatography to give, after concentration of the appropriatefractions (Rf = 0.5 in 1:4 v/v ethyl acetate/40−60 petroleum ether),compound 14 (4.04 g, 81%) as a pale-yellow oil. The spectral datarecorded on this material matched those reported previously.8
(1S,2R)-3-Methylcyclohexa-3,5-diene-1,2-diyl (2E,2′E)-Bis(4-methylpent-2-enoate) (15). A magnetically stirred solution of(1S,2R)-3-methylcyclohexa-3,5-diene-1,2-diol (5) (2.40 g, 19.02mmol) in dry THF (284 mL) was cooled to −78 °C and thentreated, dropwise over 0.5 h, with n-BuLi (12.5 mL of a 1.6 M solutionin hexane, 20.0 mmol). The resulting solution was stirred at −78 °Cfor 0.17 h and then treated, dropwise over 0.33 h, with (E)-4-methylpent-2-enoyl chloride14 (2.50 mL, 19.02 mmol). The ensuingmixture was stirred at −78 °C for 2 h and again treated, dropwise over0.5 h, with n-BuLi (12.5 mL of a 1.6 M solution in hexane, 20.0mmol). After 0.17 h, further (E)-4-methylpent-2-enoyl chloride (1.80mL, 19.02 mmol) was added, again dropwise over 0.33 h, and stirringwas continued at −78 °C for 1 h. The reaction mixture was thenallowed to warm to 22 °C before being quenched with NaHCO3 (96mL of a saturated aqueous solution) and diluted with diethyl ether(390 mL). The separated organic phase was washed with NH4Cl (1 ×96 mL of a saturated aqueous solution), dried (MgSO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash column chromatography to give, after concentrationof the appropriate fractions (Rf = 0.6 in 1:4 v/v ethyl acetate/40−60petroleum ether), compound 15 (4.47 g, 74%) as a pale-yellow oil:[α]D = +71.6 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.98−6.88 (complex m, 2H), 6.06 (m, 1H), 5.88 (broadened d, J = 5.2 Hz,1H), 5.81−5.72 (complex m, 3H), 5.62 (s, 2H), 2.43 (m, 2H), 1.84 (s,3H), 1.05−1.02 (complex m, 12H); 13C NMR (100 MHz, CDCl3) δ166.5, 166.3, 156.5, 156.2, 134.7, 126.6, 123.0, 121.9, 118.3, 118.1,69.5, 68.0, 31.1, 31.0, 21.3, 21.2, 19.8 (two signals obscured oroverlapping); IR νmax 2963, 1717, 1651, 1297, 1260, 1157, 982, 859cm−1; MS (ESI, + ve) m/z 341 [(M + Na)+, 100]; HRMS m/z: (M +Na)+ calcd for C19H26O4Na 341.1729, found 341.1728.
3a,8-Dimethyl-2-oxo-2,3,3a,6,7,7a-hexahydro-3,6-methanoben-zofuran-7-yl (E)-But-2-enoate (16) and 6,8-Dimethyl-2-oxo-2,3,3a,6,7,7a-hexahydro-3,6-methanobenzofuran-7-yl (E)-But-2-enoate (18). A magnetically stirred solution of compound 14 (4.04g, 15.40 mmol) in toluene (97 mL) maintained under a nitrogenatmosphere was heated at reflux for 24 h then cooled to 22 °C andconcentrated under reduced pressure. The resulting yellow oil wassubjected to flash chromatography (silica, 1:19 v/v ethyl acetate/40−60 petroleum ether elution) to give two fractions, A and B.
Concentration of fraction A [Rf = 0.2(5) in 1:4 v/v ethyl acetate/40−60 petroleum ether] afforded compound 16 (1.62 g, 40%) as awhite, crystalline solid, mp = 45−47 °C. The spectral data recorded onthis material matched those reported previously.8
Concentration of fraction B (Rf = 0.2 in 1:4 v/v ethyl acetate/40−60 petroleum ether) afforded compound 18 (1.06 g, 26%) as a clear,colorless oil. The spectral data recorded on this material matchedthose reported previously.8
8-Isopropyl-3a-methyl-2-oxo-2,3,3a,6,7,7a-hexahydro-3,6-meth-anobenzofuran-7-yl (E)-4-Methylpent-2-enoate (17) and 8-Iso-propyl-6-methyl-2-oxo-2,3,3a,6,7,7a-hexahydro-3,6-methanoben-zofuran-7-yl (E)-4-Methylpent-2-enoate (19). A magnetically stirredsolution of compound 15 (4.47 g, 14.04 mmol) in toluene (88 mL)maintained under a nitrogen atmosphere was heated at reflux for 72 hthen cooled to 22 °C, and concentrated under reduced pressure. Theresulting yellow oil was subjected to flash chromatography (silica, 1:19v/v ethyl acetate/40−60 petroleum ether elution) to afford twofractions, A and B.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8013

Concentration of fraction A (Rf = 0.4 in 1:4 v/v ethyl acetate/40−60 petroleum ether) afforded compound 17 (1.83 g, 41%) as a white,crystalline solid: mp = 69−71 °C; [α]D = −115.4 (c 1.0, CHCl3);
1HNMR (400 MHz, CDCl3) δ 6.97 (dd, J = 15.7 and 6.5 Hz, 1H), 6.38(t, J = 7.6 Hz, 1H), 5.84 (m, 2H), 4.37 (dd, J = 6.7 and 2.2 Hz, 1H),4.24 (d, J = 6.7 Hz, 1H), 2.98 (m, 1H), 2.47 (m, 1H), 1.93−1.88(complex m, 2H), 1.40 (s, 3H), 1.27 (m, 1H), 1.08 (s, 3H), 1.06 (s,3H), 0.97 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H); 13C NMR(100 MHz, CDCl3) δ 179.3, 166.5, 156.8, 133.1, 132.2, 117.9, 77.3,70.6, 49.7, 46.9, 45.1, 38.8, 31.1, 30.5, 21.3, 21.2, 20.9, 20.8, 20.7; IRνmax 2962, 1783, 1717, 1264, 1160, 1038, 1009, 902 cm
−1; MS (EI, 70eV) m/z 318 (M•+, 5), 156 (15), 133 (10), 97 (100), 69 (10), 41 (20);HRMS m/z M•+ calcd for C19H26O4 318.1831, found 318.1834.Concentration of fraction B [Rf = 0.3(5) in 1:4 v/v ethyl acetate/
40−60 petroleum ether] afforded compound 19 (537 mg, 12%) as aclear, colorless oil: [α]D = +176.0 (c 1.0, CHCl3);
1H NMR (400MHz, CDCl3) δ 6.97 (dd, J = 15.7 and 6.5 Hz, 1H), 6.09 (d, J = 8.2Hz, 1H), 6.00 (dd, J = 8.2 and 6.6 Hz, 1H), 5.83 (dd, J = 15.7 and 1.5Hz, 1H), 4.57 (m, 1H), 4.05 (d, J = 6.7 Hz, 1H), 3.45 (m, 1H), 2.47(m, 1H), 2.30 (m, 1H), 2.16 (m, 1H), 2.01 (m, 1H), 1.24 (s, 3H), 1.08(s, 3H), 1.06 (s, 3H), 0.99 (d, J = 6.9 Hz, 3H), 0.61 (d, J = 6.8 Hz,3H); 13C NMR (100 MHz, CDCl3) δ 179.7, 166.7, 156.8, 140.3,124.6, 117.8, 74.4, 72.9, 48.0, 42.2, 40.6, 40.2, 31.1, 27.4, 22.8, 21.3,21.2, 19.9, 19.1; IR νmax 2961, 1786, 1718, 1264, 1158, 1027, 982, 907cm−1; MS (EI, 70 eV) m/z 318 (M•+, 5), 205 (5), 97 (100), 69 (15),41 (25); HRMS m/z M•+ calcd for C19H26O4 318.1831, found318.1832.7-Hydroxy-6,8-dimethyl-3a,6,7,7a-tetrahydro-3,6-methanoben-
zofuran-2(3H)-one (20). A magnetically stirred solution of compound18 (918 mg, 3.50 mmol) in methanol (55 mL) maintained at 0 °C wastreated, in one portion, with potassium carbonate (242 mg, 1.75mmol). The ensuing mixture was stirred at 0 °C for a further 0.5 h andthen warmed to 22 °C, stirred at this temperature for 1 h, and thenrecooled to 0 °C and treated with water (30 mL) before beingconcentrated under reduced pressure. The residue thus obtained wasextracted with ethyl acetate (3 × 100 mL), and the combined organicphases were then dried (MgSO4), filtered, and concentrated underreduced pressure. The resulting light-yellow oil was subjected to flashcolumn chromatography (silica, 3:7 → 1:1 v/v ethyl acetate/40−60petroleum ether gradient elution) to give, after concentration of theappropriate fractions (Rf = 0.1 in 3:7 v/v ethyl acetate/40−60petroleum ether), compound 20 (503 mg, 74%) as a white, crystallinesolid: mp = 114−116 °C; [α]D = +67.5 (c 1.0, CHCl3);
1H NMR (400MHz, CDCl3) δ 6.06−6.00 (complex m, 2H), 4.39 (t, J = 6.0 Hz, 1H),3.43 (m, 1H), 3.26 (t, J = 6.7 Hz, 1H), 2.23 (d, J = 6.8 Hz, 1H), 2.11(m, 1H), 2.01 (d, J = 4.5 Hz, 1H), 1.25 (s, 3H), 0.89 (d, J = 7.1 Hz,3H); 13C NMR (100 MHz, CDCl3) δ 178.9, 141.2, 124.2, 74.9, 71.9,46.9, 44.1, 40.4, 36.5, 18.8, 17.3; IR νmax 3417, 2968, 1760, 1750, 1184,1102, 1015, 713 cm−1; MS (ESI, + ve) m/z 217 [(M + Na)+, 100];HRMS m/z (M + Na)+ calcd for C11H14O3Na 217.0841, found217.0844.7-Hydroxy-8-isopropyl-6-methyl-3a,6,7,7a-tetrahydro-3,6-meth-
anobenzofuran-2(3H)-one (21). A magnetically stirred solution ofcompound 19 (1.11 g, 3.50 mmol) in methanol (55 mL) maintainedat 0 °C was treated, in one portion, with potassium carbonate (242mg, 1.75 mmol). After being kept for 0.5 h at 0 °C, the reactionmixture was warmed to 22 °C, maintained at this temperature for 1 h,and then recooled to 0 °C and treated with water (30 mL) beforebeing concentrated under reduced pressure. The residue thus obtainedwas extracted with ethyl acetate (3 × 100 mL), and the combinedorganic phases were then dried (MgSO4), filtered, and concentratedunder reduced pressure to give a light-yellow oil. This material wassubjected to flash column chromatography (silica, 3:17 → 3:7 v/vethyl acetate/40−60 petroleum ether gradient elution) and gave, afterconcentration of the appropriate fractions (Rf = 0.3 in 3:7 v/v ethylacetate/40−60 petroleum ether), compound 21 (590 mg, 76%) as awhite solid: mp = 120−122 °C; [α]D = +100.7 (c 1.0, CHCl3);
1HNMR (400 MHz, CDCl3) δ 6.09 (d, J = 8.1 Hz, 1H), 5.93 (m, 1H),4.35 (m, 1H), 3.43 (m, 1H), 3.12 (d, J = 6.9 Hz, 1H), 2.28 (m, 1H),2.03−1.95 (complex m, 2H), 1.26 (s, 3H), 0.97 (d, J = 6.7 Hz, 3H),
0.60 (d, J = 6.7 Hz, 3H) (signal due to O−H group proton notobserved); 13C NMR (100 MHz, CDCl3) δ 179.3, 141.8, 123.3, 74.8,72.9, 47.6, 44.0, 40.2 (3), 40.1 (7), 27.3, 22.7, 19.8, 18.8; IR νmax 3425,2958, 1765, 1170, 1100, 978, 723 cm−1; MS (ESI, + ve) m/z 467 [(2M+ Na)+, 30], 245 [(M + Na)+, 100], 223 [(M + H)+, 70]; HRMS m/z(M + Na)+ calcd for C13H18O3Na 245.1154, found 245.1156.
6,8-Dimethyl-3a,7a-dihydro-3,6-methanobenzofuran-2,7-(3H,6H)-dione (22). A magnetically stirred solution of compound 20(466 mg, 2.40 mmol) in dry dichloromethane (78 mL) maintained at0 °C under an atmosphere of nitrogen was treated with pyridine (1.2mL, 15.05 mmol) and then Dess−Martin periodinane (1.83 g, 4.32mmol). After a further 0.5 h, the reaction mixture was warmed to 22°C, maintained at this temperature for 3 h, and then poured into water(100 mL) and extracted with dichloromethane (3 × 100 mL). Thecombined organic phases were washed with NaOH (1 × 100 mL of a 1M aqueous solution) and then HCl (1 × 100 mL of a 1 M aqueoussolution) before being dried (MgSO4), filtered, and then concentratedunder reduced pressure. The residue thus obtained was subjected toflash column chromatography (silica, 1:9 → 3:17 v/v ethyl acetate/40−60 petroleum ether gradient elution) to give, after concentrationof the appropriate fractions (Rf = 0.3 in 3:7 v/v ethyl acetate/40−60petroleum ether), compound 22 (397 mg, 86%) as a white, crystallinesolid: mp = 121−124 °C; [α]D = +518.8 (c 1.0, CHCl3);
1H NMR(400 MHz, CDCl3) δ 6.32 (dd, J = 8.1 and 6.5 Hz, 1H), 6.02 (d, J =8.1 Hz, 1H), 4.15 (dd, J = 5.0 and 1.0 Hz, 1H), 3.66 (m, 1H), 2.37 (d,J = 5.0 Hz, 1H), 2.21 (m, 1H), 1.32 (s, 3H), 1.02 (d, J = 7.0 Hz, 3H);13C NMR (100 MHz, CDCl3) δ 201.5, 177.8, 136.7, 128.3, 73.9, 53.8,47.1, 41.3, 40.9, 16.1, 15.2; IR νmax 2976, 2937, 1785, 1736, 1159, 989,837, 713 cm−1; MS (ESI, + ve) m/z 247 [(M + MeOH + Na)+, 100],215 [(M + Na)+, 20]; HRMS m/z (M + MeOH + Na)+ calcd forC12H16O4Na 247.0946, found 247.0946.
8-Isopropyl-6-methyl-3a,7a-dihydro-3,6-methanobenzofuran-2,7(3H,6H)-dione (23). A magnetically stirred solution of compound21 (533 mg, 2.40 mmol) in dry dichloromethane (78 mL) maintainedat 0 °C under an atmosphere of nitrogen was treated with pyridine(1.2 mL, 15.05 mmol) and then the Dess−Martin periodinane (1.83 g,4.32 mmol). After a further 0.5 h the reaction mixture was warmed to22 °C, stirred at this temperature for 3 h, and then poured into water(100 mL) and extracted with dichloromethane (3 × 100 mL). Thecombined organic phases were washed with NaOH (1 × 100 mL of a 1M aqueous solution) then HCl (1 × 100 mL of a 1 M aqueoussolution) before being dried (MgSO4), filtered, and concentratedunder reduced pressure. The residue thus obtained was subjected toflash column chromatography (silica, 3:17 → 3:7 v/v ethyl acetate/40−60 petroleum ether gradient elution) and gave, after concentrationof the appropriate fractions (Rf = 0.3 in 3:7 v/v ethyl acetate/40−60petroleum ether), compound 23 (433 mg, 82%) as a white, crystallinesolid: mp = 114−116 °C; [α]D = +396.1 (c 0.8, CHCl3);
1H NMR(400 MHz, CDCl3) δ 6.19 (dd, J = 8.1 and 6.6 Hz, 1H), 6.04 (m, 1H),4.12 (dd, J = 5.0 and 1.1 Hz, 1H), 3.67 (m, 1H), 2.64 (m, 1H), 2.11(m, 1H), 1.99 (m, 1H), 1.36 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H), 0.72 (d,J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.7, 178.5, 136.8,127.3, 73.9, 53.6, 51.7, 40.9, 40.4, 28.2, 22.8, 20.0, 15.3; IR νmax 2961,1802, 1786, 1734, 1151, 1002, 981, 899 cm−1; MS (ESI, + ve) m/z 275[(M + MeOH + Na)+, 100], 243 [(M + Na)+, 40]; HRMS m/z (M +MeOH + Na)+ calcd for C14H20O3Na 275.1259, found 275.1251.
7-Hydroxy-3a,8-dimethyl-3a,6,7,7a-tetrahydro-3,6-methanoben-zofuran-2(3H)-one (24). A magnetically stirred solution of compound16 (918 mg, 3.50 mmol) in methanol (55 mL) maintained at 0 °Cunder an atmosphere of nitrogen was treated, in one portion, withpotassium carbonate (242 mg, 1.75 mmol). After a further 0.5 h, thereaction mixture was warmed to 22 °C, stirred at this temperature for1 h, and then recooled to 0 °C and treated with water (30 mL) beforebeing concentrated under reduced pressure. The residue thus obtainedwas extracted with ethyl acetate (3 × 100 mL), and the combinedorganic phases were then dried (MgSO4), filtered, and concentratedunder reduced pressure to give a light-yellow oil. This material wassubjected to flash column chromatography (silica, 1:4 → 3:7 v/v ethylacetate/40−60 petroleum ether gradient elution) and gave, afterconcentration of the appropriate fractions (Rf = 0.2 in 3:7 v/v ethyl
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8014

acetate/40−60 petroleum ether), compound 24 (538 mg, 79%) as awhite, crystalline solid: mp = 146−148 °C; [α]D = +15.8 (c 1.0,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.36 (t, J = 7.6 Hz, 1H), 5.81(dd, J = 7.6 and 1.0 Hz, 1H), 3.98 (d, J = 6.9 Hz, 1H), 3.57 (dd, J = 6.9and 2.2 Hz, 1H), 2.68 (m, 1H), 2.47 (m, 2H), 1.68 (s, 1H), 1.39 (s,3H), 0.93 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 179.0,135.4, 130.8, 79.0, 69.0, 52.2, 45.2, 45.0, 33.1, 21.1, 20.1; IR νmax 3402,2964, 1740, 1347, 1182, 1085, 992, 948, 713, 703 cm−1; MS (ESI, +ve) m/z 217 [(M + Na)+, 100%]; HRMS m/z (M + Na)+ calcd forC11H14O3Na 217.0841, found 217.0842.7-Hydroxy-8-isopropyl-3a-methyl-3a,6,7,7a-tetrahydro-3,6-
methanobenzofuran-2(3H)-one (25). A magnetically stirred solutionof compound 17 (1.11 g, 3.50 mmol) in methanol (55 mL)maintained at 0 °C under an atmosphere of nitrogen was treated, inone portion, with potassium carbonate (242 mg, 1.75 mmol). After afurther 0.5 h at 0 °C, the reaction mixture was warmed to 22 °C,stirred at this temperature for 1 h, and then recooled to 0 °C andtreated with water (30 mL) before being concentrated under reducedpressure. The ensuing mixture was extracted with ethyl acetate (3 ×100 mL), and the combined organic phases were dried (MgSO4),filtered, and concentrated under reduced pressure. The residue thusobtained was subjected to flash column chromatography (silica, 3:7 →1:1 v/v ethyl acetate/40−60 petroleum ether gradient elution) andgave, after concentration of the appropriate fractions (Rf = 0.2 in 3:7v/v ethyl acetate/40−60 petroleum ether), compound 25 (732 mg,94%) as a white, crystalline solid: mp = 60−61 °C; [α]D = +2.8 (c 0.9,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.35 (t, J = 8.3 Hz, 1H), 5.78(dd, J = 8.3 and 1.2 Hz, 1H), 3.99 (d, J = 6.9 Hz, 1H), 3.54 (dd, J = 6.9and 2.3 Hz, 1H), 2.89 (m, 1H), 2.48 (broad s, 1H), 1.85 (m, 2H), 1.38(s, 3H), 1.25 (m, 1H), 0.93 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.6 Hz,3H); 13C NMR (100 MHz, CDCl3) δ 179.1, 134.6, 131.1, 79.3, 68.9,49.7, 46.4, 44.9, 41.7, 30.7, 21.1, 20.8(0), 20.7(6); IR νmax 3452, 2962,1778, 1166, 1117, 993, 706 cm−1; MS (ESI, + ve) m/z 245 [(M +Na)+, 40], 223 [(M + H)+, 100], 205 (50), 159 (100), 109 (90), 90(70); HRMS m/z (M + H)+ calcd for C13H19O3 223.1334, found223.1331.3a,8-Dimethyl-3a,7a-dihydro-3,6-methanobenzofuran-2,7-
(3H,6H)-dione (26). A magnetically stirred solution of compound 24(466 mg, 2.40 mmol) in dry dichloromethane (78 mL) maintained at0 °C under an atmosphere of nitrogen was treated with pyridine (1.2mL, 15.05 mmol) and then the Dess−Martin periodinane (1.83 g, 4.32mmol). After being kept at 0 °C for a further 0.5 h, the reactionmixture was warmed to 22 °C and maintained at this temperature for 3h before being poured into water (100 mL) and extracted withdichloromethane (3 × 100 mL). The combined organic phases werewashed with NaOH (1 × 100 mL of a 1 M aqueous solution) and HCl(1 × 100 mL of a 1 M aqueous solution) and then dried (MgSO4),filtered, and concentrated under reduced pressure. The residue thusobtained was subjected to flash column chromatography (silica, 1:9 →3:17 v/v ethyl acetate/40−60 petroleum ether gradient elution) andgave, after concentration of the appropriate fractions (Rf = 0.3 in 3:7v/v ethyl acetate/40−60 petroleum ether), compound 26 (434 mg,94%) as a white, crystalline solid: mp = 135−136 °C; [α]D = −445.7(c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.37 (t, J = 7.5 Hz,1H), 6.13 (dd, J = 7.5 and 1.0 Hz, 1H), 3.75 (s, 1H), 3.27 (dd, J = 6.7and 3.1 Hz, 1H), 2.59 (m, 1H), 1.98 (s, 1H), 1.47 (s, 3H), 1.02 (d, J =7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.0, 177.7, 134.5,130.4, 78.5, 54.0, 51.9, 46.5, 37.2, 19.9, 18.4; IR νmax 2964, 1788, 1742,1312, 1161, 995, 875, 723 cm−1; MS (ESI, + ve) m/z 247 [(M +MeOH + Na)+, 100]; HRMS m/z (M + MeOH + Na)+ Calcd forC12H16O3Na 247.0946, found 247.0943.8-Isopropyl-3a-methyl-3a,7a-dihydro-3,6-methanobenzofuran-
2,7(3H,6H)-dione (27). A magnetically stirred solution of compound25 (533 mg, 2.40 mmol) in dry dichloromethane (78 mL) maintainedat 0 °C under an atmosphere of nitrogen was treated with pyridine(1.2 mL, 15.05 mmol) and then the Dess−Martin periodinane (1.83 g,4.32 mmol). After a further 0.5 h at 0 °C, the reaction mixture waswarmed to 22 °C and then stirred at this temperature for 3 h beforebeing poured into water (100 mL) and extracted with dichloro-methane (3 × 100 mL). The combined organic phases were washed
with NaOH (1 × 100 mL of a 1 M aqueous solution) and HCl (1 ×100 mL of a 1 M aqueous solution) then dried (MgSO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash column chromatography (silica, 1:9 → 3:17 v/vethyl acetate/40−60 petroleum ether gradient elution) and gave, afterconcentration of the appropriate fractions (Rf = 0.5 in 3:7 v/v ethylacetate/40−60 petroleum ether), compound 27 (413 mg, 78%) as awhite, crystalline solid: mp = 121−123 °C; [α]D = −332.5 (c 1.0,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.37 (t, J = 8.2 Hz, 1H), 6.09(dd, J = 8.2 and 1.1 Hz, 1H), 3.77 (s, 1H), 3.49 (dd, J = 6.7 and 3.1Hz, 1H), 2.19 (s, 1H), 1.93 (dd, J = 10.2 and 2.5 Hz, 1H), 1.47 (s,3H), 1.42 (m, 1H), 1.00 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H);13C NMR (100 MHz, CDCl3) δ 200.4, 177.7, 134.8, 129.8, 78.8, 51.1,50.2, 49.4, 46.2, 29.2, 20.6, 20.4, 19.9; IR νmax 2968, 17909, 1775, 1741,1160, 985, 713 cm−1; MS (ESI, + ve) m/z 275 [(M + MeOH + Na)+,100], 221 [(M + H)+, 10]; HRMS m/z (M + MeOH + Na)+ calcd forC14H20O3Na 275.1259, found 275.1260.
(1aS,1a′R,3aS,4S,4aR,4a′R)-4,4a-Dimethylhexahydro-1H-cyclopropa[3,4]pentaleno[1,6-bc]furan-1,3(1aH)-dione (28). A de-oxygenated and magnetically stirred solution of compound 22 (108mg, 0.56 mmol) and acetophenone (90 μL, 0.77 mmol) in acetone(100 mL) maintained under a nitrogen atmosphere was subjected toirradiation with a Hanovia 450 W medium-pressure mercury-vaporlamp for 0.83 h. The reaction mixture was then cooled andconcentrated under reduced pressure to give a brown oil that wassubjected to flash column chromatography (silica, 3:17 → 3:7 v/vethyl acetate/40−60 petroleum ether gradient elution). Concentrationof the appropriate fractions (Rf = 0.3 in 1:1 v/v ethyl acetate/40−60petroleum ether) then gave compound 28 (44 mg, 40%) as a pale-yellow oil: [α]D = +5.1 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3)δ 4.57 (dd, J = 9.0 and 2.2 Hz, 1H), 3.59 (m, 1H), 2.80 (t, J = 8.0 Hz,1H), 2.60 (m, 1H), 2.45 (m, 1H), 1.98 (m, 1H), 1.30 (d, J = 7.2 Hz,3H), 1.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.9, 176.8, 82.4,57.2, 46.1, 45.1, 45.0, 44.7, 38.1, 19.5, 18.4; IR νmax 2969, 2928, 1779,1726, 1192, 1152, 1054, 1031, 983 cm−1; MS (ESI, + ve) m/z 215 [(M+ Na)+, 100]; HRMS m/z (M + Na)+ calcd for C11H12O3Na 215.0684,found 215.0687.
(1aS,1a′R,3aS,4S,4aS,4a′R)-4-Isopropyl-4a-methylhexahydro-1H-cyclopropa[3,4]pentaleno[1,6-bc]furan-1,3(1aH)-dione (29). A de-oxygenated and magnetically stirred solution of compound 23 (123mg, 0.56 mmol) and acetophenone (90 μL, 0.77 mmol) in acetone(100 mL) maintained under a nitrogen atmosphere was subjected toirradiation with a Hanovia 450 W medium-pressure mercury-vaporlamp for 0.67 h. The reaction mixture thus formed was cooled andconcentrated under reduced pressure to give a yellow oil that wassubjected to flash column chromatography (silica, 3:17 → 1:3 v/vethyl acetate/40−60 petroleum ether gradient elution). Concentrationof the appropriate fractions (Rf = 0.4 in 1:1 v/v ethyl acetate/40−60petroleum ether) then gave compound 29 (53 mg, 43%) as a white,crystalline solid: mp = 107−109 °C; [α]D = −7.6 (c 1.0, CHCl3).
1HNMR (400 MHz, CDCl3) δ 4.60 (dd, J = 8.9 and 2.0 Hz, 1H), 3.61(m, 1H), 3.10 (dd, J = 8.5 and 6.5 Hz, 1H), 2.62 (m, 1H), 2.38 (m,1H), 2.15 (m, 1H), 1.88 (m, 1H), 1.27 (s, 3H), 1.05 (d, J = 6.9 Hz,3H), 0.97 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 206.9,177.4, 82.5, 55.0, 47.8, 46.6, 45.6, 44.0, 38.3, 29.2, 22.1, 19.5, 17.0; IRνmax 2963, 1780, 1728, 1196, 1150, 1049, 983 cm−1; MS (EI, 70 eV)m/z 220 (M•+, 25), 153 (40), 149 (90), 133 (40), 105 (60), 93 (100),91 (70), 84 (60), 77 (50), 59 (70); HRMS m/z M•+ calcd forC13H16O3 220.1099, found 220.1092.
(2aS,2a′R,3aR,6S,6aS)-5,6-Dimethyl-2a1,3a,6,6a-tetrahydro-1H-cyclobuta[cd]isobenzofuran-1,3(2aH)-dione (30). A deoxygenatedand magnetically stirred solution of compound 22 (100 mg, 0.52mmol) in dichloromethane (100 mL) maintained under a nitrogenatmosphere was subjected to irradiation with a Hanovia 450 Wmedium-pressure mercury-vapor lamp for 0.5 h. The reaction thusobtained was then cooled and concentrated under reduced pressure togive a yellow solid. Subjection of this material to flash columnchromatography (silica, 1:4 → 3:7 v/v ethyl acetate/40−60 petroleumether gradient elution) gave, after concentration of the appropriatefractions (Rf = 0.2 in 3:7 v/v ethyl acetate/40−60 petroleum ether),
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8015

compound 30 (75 mg, 75%) as a white, crystalline solid: mp = 161−163 °C; [α]D = −344.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3)δ 5.37 (m, 1H), 5.33 (m, 1H), 3.93 (m, 1H), 3.48 (m, 1H), 2.97 (dd, J= 10.0 and 2.1 Hz, 1H), 2.67 (m, 1H), 1.78 (s, 3H), 1.19 (d, J = 7.2Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.3, 177.5, 140.6, 112.9,89.8, 58.8, 43.3, 33.9, 26.8, 23.0, 19.8; IR νmax 2968, 1782, 1256, 1148,1141, 1130, 1004 cm−1; MS (ESI, + ve) m/z 247 [(M + MeOH +Na)+, 100]; HRMS m/z (M + MeOH + Na)+ calcd for C12H16O3Na247.0946, found 247.0945.(2aS,2a′R,3aR,6S,6aS)-6-Isopropyl-5-methyl-2a′,3a,6,6a-tetrahy-
dro-1H-cyclobuta[cd]isobenzofuran-1,3(2aH)-dione (31). A deoxy-genated and magnetically stirred solution of compound 23 (100 mg,0.45 mmol) in dichloromethane (100 mL) maintained under nitrogenwas subjected to irradiation with a Hanovia 450 W medium-pressuremercury-vapor lamp for 0.5 h. The reaction mixture thus obtained wascooled and concentrated under reduced pressure to give a yellow oil.Subjection of this material to flash column chromatography (silica, 1:4→ 3:7 v/v ethyl acetate/40−60 petroleum ether gradient elution)gave, after concentration of the appropriate fractions (Rf = 0.2 in 3:7v/v ethyl acetate/40−60 petroleum ether), compound 31 (73 mg,73%) as a clear, colorless oil: [α]D = −163.3 (c 0.7, CHCl3). 1H NMR(400 MHz, CDCl3) δ 5.47 (s, 1H), 5.27 (dd, J = 6.4 and 4.0 Hz, 1H),3.88 (m, 1H), 3.43 (m, 1H), 3.08 (dd, J = 10.1 and 1.4 Hz, 1H), 2.45(d, J = 3.9 Hz, 1H), 1.90 (m, 1H), 1.79 (s, 3H), 1.06 (d, J = 6.8 Hz,3H), 0.89 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.4,178.7, 138.9, 114.7, 89.1, 59.4, 45.9, 38.1, 30.8, 28.3, 24.5, 21.6, 19.6;IR νmax 2963, 1782, 1168, 1147, 1131, 1005, 838 cm
−1; MS (ESI, + ve)m/z 275 [(M + MeOH + Na)+, 100], 221 [(M + H)+, 25]; HRMS m/z (M + MeOH + Na)+ calcd for C14H20O3Na 275.1259, found275.1260.( 2 a S , 2 a ′R , 3 S , 5 aR , 5 bR ) - 3 , 4 -D ime t h y l - 2 a ′ , 3 , 5 a , 5 b -
tetrahydrocyclopropa[cd]isobenzofuran-2(2aH)-one (32). A deoxy-genated and magnetically stirred solution of compound 22 (100 mg,0.52 mmol) in dichloromethane (100 mL) maintained under anitrogen atmosphere was irradiated with a Hanovia 450 W medium-pressure mercury-vapor lamp for 5 h. The ensuing reaction mixturewas cooled to ca. 22 °C then concentrated under reduced pressure togive a brown oil. Subjection of this material to flash columnchromatography (silica, 1:9 → 3:7 v/v ethyl acetate/40−60 petroleumether gradient elution) gave two fractions, A and B.Concentration of fraction A (Rf = 0.5 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 32 (32 mg, 37%) as acolorless, crystalline solid: mp = 73−75 °C; [α]D = −102.8 (c 0.4,CHCl3);
1H NMR (400 MHz, CDCl3) δ 5.34 (s, 1H), 4.33 (dd, J =6.3 and 5.4 Hz, 1H), 2.89 (dd, J = 7.7 and 3.0 Hz, 1H), 2.23 (m, 1H),1.95 (m, 1H), 1.69 (s, 3H), 1.46 (m, 1H), 1.21 (d, J = 7.0 Hz, 3H);13C NMR (100 MHz, CDCl3) δ 180.3, 142.7, 112.7, 60.5, 44.7, 35.9,22.4, 18.7, 17.8, 11.6; IR νmax 2965, 1773, 1453, 1150, 1086, 1074, 922,794 cm−1; MS (ESI, + ve) m/z 187 [(M + Na)+, 100]; HRMS m/z (M+ Na)+ calcd for C10H12O2Na 187.0735, found 187.0735.Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 30 (28 mg, 28%) as a whitesolid that was identical with an authentic sample.(2aS,2a′R,3S,5aR,5bR)-3-Isopropyl-4-methyl-2a′,3,5a,5b-
tetrahydrocyclopropa[cd]isobenzofuran-2(2aH)-one (33). A deoxy-genated and magnetically stirred solution of compound 23 (100 mg,0.45 mmol) in dichloromethane (100 mL) maintained under anitrogen atmosphere was subjected to irradiation with a Hanovia 450W medium-pressure mercury-vapor lamp for 1.5 h. The ensuingmixture was cooled then concentrated under reduced pressure to givea brown oil. Subjection of this material to flash columnchromatography (silica, 1:9 → 3:7 v/v ethyl acetate/40−60 petroleumether gradient elution) afforded two fractions, A and B.Concentration of fraction A (Rf = 0.6 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) gave compound 33 (45 mg, 52%) as a clear,colorless oil: [α]D = −29.8 (c 1.0, CHCl3);
1H NMR (400 MHz,CDCl3) δ 5.45 (s, 1H), 4.33 (m, 1H), 3.03 (dd, J = 7.7 and 2.4 Hz,1H), 2.02−1.88 (complex m, 3H), 1.71 (s, 3H), 1.45 (m, 1H), 1.06 (d,J = 6.5 Hz, 3H), 0.98 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz,CDCl3) δ 181.1, 141.1, 114.8, 61.4, 47.6, 40.3, 30.7, 24.2, 21.7, 19.9,
18.0, 13.5; IR νmax 2960, 1769, 1149, 1127, 1087, 933, 781 cm−1; MS(ESI, + ve) m/z 215 [(M + Na)+, 100]; HRMS m/z (M + Na)+ calcdfor C12H16O2Na 215.1048, found 215.1048.
Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/40−60 petroleum ether) afforded compound 31 (25 mg, 25%) as a whitesolid that was identical with an authentic sample.
(1aR,1a ′S,3aR,4S,4a1R)-1a ′ ,4-Dimethylhexahydro-1H-cyclopropa[3,4]pentaleno[1,6-bc]furan-1,3(1aH)-dione (34). A de-oxygenated and magnetically stirred solution of compound 26 (108mg, 0.56 mmol) and acetophenone (90 μL, 0.77 mmol) in acetone(100 mL) maintained under nitrogen was subjected to irradiation witha Hanovia 450 W medium-pressure mercury-vapor lamp for 2.5 h. Theensuing reaction mixture was cooled then concentrated under reducedpressure to give a yellow oil. Subjection of this material to flash columnchromatography (silica, 3:17 → 3:7 v/v ethyl acetate/40−60petroleum ether gradient elution) gave, after concentration of theappropriate fractions (Rf = 0.3 in 1:1 v/v ethyl acetate/40−60petroleum ether), compound 34 (49 mg, 45%) as a white, crystallinesolid: mp = 154−156 °C, [α]D = +68.6 (c 1.0, CHCl3).
1H NMR (400MHz, CDCl3) δ 4.21 (s, 1H), 2.55−2.47 (complex m, 3H), 2.18 (m,1H), 1.97 (m, 1H), 1.54 (s, 3H), 1.38 (d, J = 6.8 Hz, 3H); 13C NMR(100 MHz, CDCl3) δ 206.9, 176.5, 87.7, 64.3, 55.0, 42.6, 40.3, 38.0,35.6, 22.6, 20.8; IR νmax 2970, 1781, 1726, 1238, 1163, 1033, 884, 874cm−1; MS (ESI, + ve) m/z 247 [(M + MeOH + Na)+, 20], 215 [(M +Na)+, 100], 193 (30); HRMS m/z (M + Na)+ calcd for C11H12O3Na215.0684, found 215.0676.
(1aR,1a′S,3aR,4S,4a′R)-4-Isopropyl-1a′-methylhexahydro-1H-cyclopropa[3,4]pentaleno[1,6-bc]furan-1,3(1aH)-dione (35). A de-oxygenated and magnetically stirred solution of compound 27 (123mg, 0.56 mmol) and acetophenone (90 μL, 0.77 mmol) in acetone(100 mL) maintained under a nitrogen atmosphere was irradiated witha Hanovia 450 W medium-pressure mercury-vapor lamp for 2.5 h. Thecooled reaction mixture was concentrated under reduced pressure togive a yellow oil and this subjected to flash column chromatography(silica, 3:17 → 1:3 v/v ethyl acetate/40−60 petroleum ether gradientelution). Concentration of the appropriate fractions (Rf = 0.5 in 1:1 v/v ethyl acetate/40−60 petroleum ether) then gave compound 35 (60mg, 49%) as a white, crystalline solid: mp = 102−103 °C; [α]D =+83.8 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ 4.22 (s, 1H),2.72 (d, J = 6.4 Hz, 1H), 2.41 (t, J = 4.0 Hz, 1H), 2.22−2.15 (complexm, 2H), 2.08 (m, 1H), 1.94 (m, 1H), 1.53 (s, 3H), 1.03 (d, J = 3.0 Hz,3H), 1.02 (d, J = 3.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 207.1,176.8, 87.6, 59.1, 54.6, 53.7, 37.4, 37.3, 35.5, 32.9, 20.6, 20.2, 19.4; IRνmax 2961, 1790, 1781, 1732, 1240, 1160, 1047, 889 cm
−1; MS (ESI, +ve) m/z 275 [(M + MeOH + Na)+, 20], 243 [(M + Na)+, 100];HRMS m/z (M + Na)+ calcd for C13H16O3Na 243.0997, found243.0993.
(2aR,2a′S,3aS,6S,6aR)-2a′,6-Dimethyl-2a′,3a,6,6a-tetrahydro-1H-cyclobuta[cd]isobenzofuran-1,3(2aH)-dione (36). A deoxygen-ated and magnetically stirred solution of compound 26 (100 mg, 0.52mmol) in dichloromethane (100 mL) maintained under a nitrogenatmosphere was irradiated with a Hanovia 450 W medium-pressuremercury-vapor lamp for 0.58 h then cooled and concentrated underreduced pressure to give a yellow solid. Subjection of this material toflash column chromatography (silica, 1:4 → 3:7 v/v ethyl acetate/40−60 petroleum ether gradient elution) then gave, after concentration ofthe appropriate fractions (Rf = 0.2 in 3:7 v/v ethyl acetate/40−60petroleum ether), compound 36 (70 mg, 70%) as a white, crystallinesolid: mp = 100−101 °C; [α]D = +220.5 (c 1.0, CHCl3).
1H NMR(400 MHz, CDCl3) δ 6.02 (m, 1H), 5.69 (dd, J = 10.0 and 4.0 Hz,1H), 4.82 (d, J = 4.0 Hz, 1H), 3.60 (m, 1H), 2.93 (m, 1H), 2.66 (s,1H), 1.70 (s, 3H), 1.23 (d, J = 7.3 Hz, 3H); 13C NMR (100 MHz,CDCl3) δ 200.1, 177.7, 132.6, 119.1, 93.5, 63.8, 48.5, 35.5, 30.1, 24.9,21.2; IR νmax 2965, 1779, 1191, 1096, 997, 744, 725 cm
−1; MS (ESI, +ve) m/z 247 [(M + MeOH + Na)+, 100%]; HRMS m/z (M + MeOH+ Na)+ calcd for C12H16O3Na 247.0946, found 247.0947.
(2aR,2a′S,3aS,6S,6aR)-6-Isopropyl-2a′-methyl-2a′,3a,6,6a-tetra-hydro-1H-cyclobuta[cd]isobenzofuran-1,3(2aH)-dione (37). A de-oxygenated and magnetically stirred solution of compound 27 (100mg, 0.45 mmol) in dichloromethane (100 mL) maintained under a
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8016

nitrogen atmosphere was irradiated with a Hanovia 450 W medium-pressure mercury-vapor lamp for 0.55 h then cooled and concentratedunder reduced pressure to give a yellow solid. Subjection of thismaterial to flash column chromatography (silica, 1:4 → 3:7 v/v ethylacetate/40−60 petroleum ether gradient elution) then gave, afterconcentration of the appropriate fractions (Rf = 0.2 in 3:7 v/v ethylacetate/40−60 petroleum ether), compound 37 (71 mg, 71%) as awhite, crystalline solid: mp = 96−98 °C; [α]D = +196.2 (c 1.0,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.11 (m, 1H), 5.73 (dd, J =10.1 and 3.6 Hz, 1H), 4.79 (d, J = 3.6 Hz, 1H), 3.56 (m, 1H), 2.95 (d,J = 1.7 Hz, 1H), 2.43 (t, J = 8.0 Hz, 1H), 1.68 (s, 3H), 1.66−1.60(complex m, 1H), 1.09 (d, J = 6.6 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H);13C NMR (100 MHz, CDCl3) δ 199.9, 178.5, 131.8, 120.0, 93.5, 64.2,44.8, 42.6, 35.9, 33.1, 24.1, 21.4, 21.3; IR νmax 2964, 1782, 1471, 1293,1193, 1000, 722 cm−1; MS (ESI, + ve) m/z 527 [(2M + 2MeOH +Na)+, 15], 275 [(M + MeOH + Na)+, 100], 221 [(M + H)+, 15];HRMS m/z (M + MeOH + Na)+ calcd for C14H20O3Na 275.1259,found 275.1257.( 2aR ,2a ′S ,3S , 5aS ,5bS ) -2a ′ , 3 -D imethy l - 2a 1 , 3 , 5a ,5b -
tetrahydrocyclopropa[cd]isobenzofuran-2(2aH)-one (38). A deoxy-genated and magnetically stirred solution of compound 26 (100 mg,0.52 mmol) in dichloromethane (100 mL) maintained under anitrogen atmosphere was irradiated with a Hanovia 450 W medium-pressure mercury-vapor lamp for 3.1 h then cooled and concentratedunder reduced pressure to give a brown oil. Subjection of this materialto flash column chromatography (silica, 1:9 → 3:7 v/v ethyl acetate/40−60 petroleum ether gradient elution) then gave two fractions, Aand B.Concentration of fraction A (Rf = 0.5 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 38 (30 mg, 35%) as acolorless, crystalline solid: mp = 40−42 °C; [α]D = +152.0 (c 0.5,CHCl3);
1H NMR (400 MHz, CDCl3) δ 5.95 (dd, J = 9.9 and 6.0 Hz,1H), 5.66 (dd, J = 9.9 and 4.5 Hz, 1H), 4.03 (d, J = 5.6 Hz, 1H), 2.65(s, 1H), 2.54 (m, 1H), 1.39 (s, 3H), 1.35 (m, 1H), 1.22 (d, J = 7.1 Hz,3H); 13C NMR (100 MHz, CDCl3) δ 180.7, 134.8, 119.4, 65.2, 49.3,31.7, 23.5, 20.6, 19.3, 18.9; IR νmax 2962, 2929, 1783, 1769, 1143,1090, 846, 750 cm−1; MS (EI, 70 eV) m/z 164 (M+•, 20%), 121 (30),107 (100), 91 (70), 77 (25), 65 (15); HRMS m/z M•+ calcd forC10H12O2 164.0837, found 164.0835.Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 36 (32 mg, 32%) as a white,crystalline solid that was identical, in all respects, with an authenticsample.(2aR,2a′S,3S,5aS,5bS)-3-Isopropyl-2a′-methyl-2a′,3,5a,5b-
tetrahydrocyclopropa[cd]isobenzofuran-2(2aH)-one (39). A deoxy-genated and magnetically stirred solution of compound 27 (100 mg,0.45 mmol) in dichloromethane (100 mL) maintained under anitrogen atmosphere was irradiated with a Hanovia 450 W medium-pressure mercury-vapor lamp for 3.1 h then cooled and concentratedunder reduced pressure to give a brown oil. Subjection of this materialto flash column chromatography (silica, 1:9 → 3:7 v/v ethyl acetate/40−60 petroleum ether elution) then gave two fractions, A and B.Concentration of fraction A (Rf = 0.6 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 39 (35 mg, 40%) as a yellowoil, [α]D = +166.7 (c 1.0, CHCl3).
1H NMR (400 MHz, CDCl3) δ 6.02(dd, J = 9.9 and 6.1 Hz, 1H), 5.70 (dd, J = 9.9 and 4.2 Hz, 1H), 4.02(d, J = 5.5 Hz, 1H), 2.92 (s, 1H), 2.02 (m, 1H), 1.79 (m, 1H), 1.36 (s,3H), 1.34 (m, 1H), 1.05 (d, J = 6.6 Hz, 3H), 0.99 (d, J = 6.6 Hz, 3H);13C NMR (100 MHz, CDCl3) δ 181.2, 134.0, 120.3, 65.5, 46.0, 43.8,32.3, 23.7, 21.8, 20.9, 20.1, 19.3; IR νmax 2966, 2872, 1778, 1148, 1092,834, 752 cm−1; MS (ESI, + ve) m/z 215 [(M + Na)+, 100%]; HRMSm/z (M + Na)+ calcd for C12H16O2Na 215.1048, found 215.1047.Concentration of fraction B (Rf = 0.2 in 3:7 v/v ethyl acetate/40−
60 petroleum ether) afforded compound 37 (34 mg, 34%) as a white,crystalline solid that was identical, in all respects, with an authenticsample.( 3aR ,4R ,4aR ,5S ,7aR ,8S ,8aS ) -2 ,2 ,4 ,6 ,6 -Pentamethy l -
3a,4a,5,6,7,7a,8,8a-octahydro-4H-4,8-ethenoindeno[5,6-d][1,3]-dioxol-5-yl Acetate (42). A magnetically stirred solution of compound414d,5b (5.00 g, 17.96 mmol), acetic anhydride (3.40 mL, 35.97 mmol)
and 4-(N,N-dimethylamino)pyridine (440 mg, 3.60 mmol) intriethylamine (7.50 mL, 53.8 mmol) was stirred at 0 °C for 2 h andthen warmed to 22 °C and stirred at this temperature for another 2 h.After this time, NH4Cl (50 mL of a saturated aqueous solution) wasadded to the reaction mixture, and the resulting suspension wasextracted with dichloromethane (3 × 50 mL). The combined organicfractions were washed with water (2 × 50 mL) and brine (1 × 50 mL)before being dried (MgSO4), filtered, and concentrated under reducedpressure. The residue thus obtained was subjected to flash columnchromatography (silica, 1:19 v/v ethyl acetate/40−60 petroleum etherelution) and gave, after concentration of the appropriate fractions (Rf= 0.3 in 1:9 v/v ethyl acetate/40−60 petroleum ether), compound 42(5.58 g, 97%) as a white, crystalline solid: mp = 135−137 °C; [α]D =+7.6 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ 5.86−5.80(complex m, 2H), 4.98 (d, J = 6.1 Hz, 1H), 4.22 (dd, J = 7.2 and 3.1Hz, 1H), 3.79 (d, J = 7.2 Hz, 1H), 2.75 (m, 1H), 2.22 (m, 1H), 2.07(m, 1H), 2.04 (s, 3H), 1.37 (m, 2H), 1.32 (s, 3H), 1.27 (s, 3H), 1.14(s, 3H), 0.95 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ170.4, 137.3, 125.7, 109.1, 84.1, 81.0, 80.1, 49.7, 44.7, 42.0, 40.8, 40.0,38.0, 25.6 (5), 25.6 (2), 25.2, 22.4, 21.6, 19.9; IR νmax 2961, 2934,1733, 1370, 1234, 1073, 723 cm−1; MS (ESI, + ve) m/z 343 [(M +Na)+, 100]; HRMS m/z (M + Na)+ calcd for C19H28O4Na 343.1885,found 343.1884.
(3S,3aR,4R,7S,7aR,8S,9R)-8,9-Dihydroxy-2,2,4-trimethyl-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-3-yl Acetate (43).AG-50W-X8 resin (3.32 g of H+ form) was added to a magneticallystirred solution of compound 42 in methanol/water (72 mL of a 5:1v/v mixture) and the resulting suspension heated under reflux for 67 hand then cooled to 22 °C before being filtered through a sintered glassfunnel. The filtrate was concentrated under reduced pressure and theresidue thus obtained subjected to flash column chromatography(silica, 3:7 v/v ethyl acetate/40−60 petroleum ether elution).Concentration of the appropriate fractions (Rf = 0.1 in 3:7 v/v ethylacetate/40−60 petroleum ether) gave compound 43 (1.80 g, 97%) asa white solid: mp = 73−74 °C; [α]D = −2.3 (c 1.1, CHCl3); 1H NMR(400 MHz, CDCl3) δ 5.99−5.91 (complex m, 2H), 4.97 (d, J = 6.1 Hz,1H), 3.88 (dd, J = 7.5 and 2.4 Hz, 1H), 3.40 (d, J = 7.5 Hz, 1H), 2.73(m, 1H), 2.52 (broad s, 2H), 2.27 (m, 1H), 2.13 (m, 1H), 2.03 (s,3H), 1.39 (m, 1H), 1.25 (t, J = 11.6 Hz, 1H), 1.15 (s, 3H), 0.93 (s,3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4, 138.8,126.8, 81.6, 75.4, 71.7, 50.4, 44.1, 42.0, 41.5, 41.3, 40.8, 25.5, 22.3,21.6, 19.6; IR νmax 3394, 2965, 2932, 1732, 1374, 1240, 1057 cm−1;MS m/z 583 [(2M + Na)+, 5], 303 [(M + Na)+, 100]; HRMS m/z (M+ Na)+ calcd for C16H24O4Na 303.1572, found 303.1577.
(3S,3aR,4R,7S,7aS,9R)-9-Hydroxy-2,2,4-trimethyl-8-oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-3-yl Acetate (44). Amagnetically stirred mixture of p-toluenesulfonic acid monohydrate(448 mg, 2.36 mmol) and 4-acetamido-TEMPO (502 mg, 2.35 mmol)in dichloromethane (10 mL) was maintained at 0 °C under a nitrogenatmosphere for 0.5 h and the resulting solution then added, dropwiseover 2 h, to a magnetically stirred solution of compound 43 (300 mg,1.07 mmol) in dichloromethane (10 mL) maintained at 0 °C. Theensuing mixture was stirred for a further 2 h at this temperature andthen quenched with NaHCO3 (50 mL of a saturated aqueoussolution). The resulting suspension was extracted with dichloro-methane (3 × 50 mL), and the combined organic phases were washedwith water (2 × 50 mL) and brine (1 × 50 mL) before being dried(Na2SO4), filtered, and concentrated under reduced pressure. Theresidue thus obtained was subjected to flash column chromatography(silica, 3:17 v/v ethyl acetate/40−60 petroleum ether elution) to give,after concentration of the appropriate fractions (Rf = 0.2 in 3:7 v/vethyl acetate/40−60 petroleum ether), compound 44 (289 mg, 97%)as a white, crystalline solid: mp = 147−149 °C; [α]D = +215.0 (c 1.1,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.08 (d, J = 7.9 Hz, 1H), 5.92(t, J = 7.9 Hz, 1H), 5.09 (d, J = 6.0 Hz, 1H), 3.34 (s, 1H), 3.15 (d, J =6.0 Hz, 1H), 2.72−2.49 (complex m, 3H), 2.06 (s, 3H), 1.52 (dd, J =12.2 and 7.4 Hz, 1H), 1.40 (t, J = 12.2 Hz, 1H), 1.22 (s, 3H), 1.00 (s,3H), 0.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 211.0, 170.2,142.2, 121.9, 81.1, 75.2, 51.8, 50.1, 44.8, 43.9, 41.7, 39.3, 25.6, 22.2,21.6, 18.1; IR νmax 3451, 3239, 2965, 1730, 1374, 1236, 1081, 1039
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8017

cm−1; MS (ESI, + ve) m/z 579 [(2M + Na)+, 10], 301 [(M + Na)+,100]; HRMS m/z (M + Na)+ calcd for C16H22O4Na 301.1416, found301.1411.(3S,3aR,4R,7S,7aS,9R)-2,2,4-Trimethyl-8-oxo-2,3,3a,4,7,7a-hexa-
hydro-1H-4,7-ethanoindene-3,9-diyl Diacetate (45). A magneticallystirred solution of compound 44 (239 mg, 0.86 mmol), 4-(N,N-dimethylamino)pyridine (11 mg, 0.09 mmol), and triethylamine (180μL, 1.29 mmol) in dichloromethane (10 mL) maintained at 0 °Cunder a nitrogen atmosphere was treated with acetic anhydride (97 μL,1.03 mmol). The ensuing mixture was kept at 0 °C for 2 h and then at22 °C for 16 h before being quenched with NH4Cl (10 mL of asaturated aqueous solution). The resulting mixture was extracted withdichloromethane (2 × 20 mL), and the combined organic phases werewashed with water (2 × 20 mL) and brine (1 × 20 mL) before beingdried (Na2SO4), filtered, and concentrated under reduced pressure.The residue thus obtained was subjected to flash columnchromatography (silica, 3:17 v/v ethyl acetate/40−60 petroleumether elution) and gave, after concentration of the appropriatefractions (Rf = 0.4 in 3:7 v/v ethyl acetate/40−60 petroleum ether),compound 45 (250 mg, 91%) as a white, crystalline solid: mp = 175−177 °C; [α]D = +200.0 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3)δ 6.09 (d, J = 8.1 Hz, 1H), 5.97 (t, J = 8.1 Hz, 1H), 5.09 (d, J = 6.0 Hz,1H), 4.85 (s, 1H), 3.17 (d, J = 6.0 Hz, 1H), 2.74 (m, 1H), 2.64 (dd, J= 10.6 and 6.0 Hz, 1H), 2.10 (s, 3H), 2.06 (s, 3H), 1.53 (dd, J = 12.2and 7.3 Hz, 1H), 1.39 (t, J = 12.2 Hz, 1H), 1.05 (s, 3H), 1.01 (s, 3H),0.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 205.2, 170.7, 170.1,141.4, 122.2, 81.1, 74.2, 51.5, 50.7, 44.9, 42.6, 41.7, 39.6, 25.5, 22.2,21.5, 20.7, 17.9; IR νmax 2979, 2965, 2932, 1721, 1374, 1230, 1053,1020 cm−1; MS (ESI, + ve) m/z 663 [(2M + Na)+, 20], 343 [(M +Na)+, 100], 321 [(M + H)+, 5]; HRMS m/z (M + Na)+ calcd forC18H24O5Na 343.1521, found 343.1505.(3S,3aR,4R,7S,7aS,9R)-9-(Methoxymethoxy)-2,2,4-trimethyl-8-
oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-3-yl Acetate(46). A magnetically stirred solution of compound 44 (280 mg, 1.00mmol), chloromethyl methyl ether (122 μL, 1.60 mmol), tetra-n-butylammonium iodide (37 mg, 0.10 mmol), and N,N-diisopropyle-thylamine (348 μL, 2.00 mmol) in dichloromethane (5 mL)maintained under a nitrogen atmosphere was stirred at 22 °C for 48h and then treated with NH4Cl (10 mL of a saturated aqueoussolution) and extracted with dichloromethane (2 × 20 mL). Thecombined organic phases were washed with water (2 × 20 mL) andbrine (1 × 20 mL) before being dried (Na2SO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash column chromatography (silica, 1:4 v/v ethylacetate/40−60 petroleum ether elution) and gave, after concentrationof the appropriate fractions (Rf = 0.3 in 1:4 v/v ethyl acetate/40−60petroleum ether), compound 46 (251 mg, 77%) as a pale-yellow oil:[α]D = +213.0 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.06 (d,J = 8.0 Hz, 1H), 5.90 (t, J = 8.0 Hz, 1H), 5.03 (d, J = 6.7 Hz, 2H), 4.59(d, J = 6.7 Hz, 1H), 3.38 (s, 3H), 3.31 (s, 1H), 3.04 (d, J = 6.3 Hz,1H), 2.61 (m, 1H), 2.46 (dd, J = 10.6 and 6.1 Hz, 1H), 2.03 (s, 3H),1.46 (dd, J = 12.1 and 7.4 Hz, 1H), 1.35 (t, J = 12.1 Hz, 1H), 1.14 (s,3H), 0.95 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ209.3, 170.1, 141.0, 121.5, 97.5, 81.1, 77.1, 56.3, 51.0, 50.7, 44.8, 43.2,41.5, 40.2, 25.3, 22.1, 21.5, 18.4; IR νmax 2966, 1731, 1372, 1239, 1150,1035, 710 cm−1; MS (ESI, + ve) m/z 667 [(2M + Na)+, 5], 345 [(M +Na)+, 100]; HRMS m/z (M + Na)+ calcd for C18H26O5Na 345.1678,found 345.1665.(3S,3aR,4R,7S,7aS,9R)-9-((tert-Butyldimethylsilyl)oxy)-2,2,4-tri-
methyl-8-oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-3-ylAcetate (47). A magnetically stirred solution of compound 44 (400mg, 1.44 mmol), tert-butyldimethylsilyl chloride (1.73 g, 11.48 mmol),and imidazole (980 mg, 14.39 mmol) in 1,2-dichloroethane (20 mL)kept under a nitrogen atmosphere was heated at reflux for 65 h andthen cooled, quenched with NH4Cl (10 mL of a saturated aqueoussolution), and extracted with dichloromethane (2 × 40 mL). Thecombined organic phases were washed with water (2 × 40 mL) andbrine (1 × 40 mL) before being dried (Na2SO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash column chromatography (silica, 2:5:100 v/v/v ethyl
acetate/dichloromethane/40−60 petroleum ether elution) and gave,after concentration of the appropriate fractions (Rf = 0.6 in 3:7 v/vethyl acetate/40−60 petroleum ether), compound 47 (465 mg, 82%)as a white, crystalline solid: mp = 71−72 °C; [α]D = +121.0 (c 1.0,CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.03 (d, J = 8.0 Hz, 1H), 5.87(t, J = 8.0 Hz, 1H), 5.06 (d, J = 6.0 Hz, 1H), 3.27 (s, 1H), 3.02 (dd, J =6.0 and 1.8 Hz, 1H), 2.62 (m, 1H), 2.42 (dd, J = 10.6 and 6.0 Hz, 1H),2.06 (s, 3H), 1.51−1.37 (complex m, 2H), 1.12 (s, 3H), 0.99 (s, 3H),0.86 (s, 12H), 0.10 (s, 3H), 0.09 (s, 3H); 13C NMR (100 MHz,CDCl3) δ 209.5, 170.3, 141.7, 120.9, 81.4, 76.0, 51.1, 50.5, 44.9, 44.4,41.7, 40.3, 26.0, 25.5, 22.3, 21.6, 19.0, 18.6, − 4.0, − 5.0; IR νmax 2959,2930, 2856, 1733, 1237, 1129, 1112, 1093, 858, 837, 778, 709 cm−1;MS m/z 415 [(M + Na)+, 100]; HRMS m/z (M + Na)+ calcd forC22H36O4SiNa 415.2281, found 415.2275.
(3S,3aR,4R,7S,7aS,9R)-3-Acetoxy-2,2,4-trimethyl-8-oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-9-yl Benzoate (48). Amagnetically stirred solution of compound 44 (2.90 g, 10.42 mmol), 4-(N,N-dimethylamino)pyridine (127 mg, 1.04 mmol), and triethyl-amine (2.9 mL, 21.00 mmol) in dichloromethane (50 mL) maintainedunder a nitrogen atmosphere at 0 °C was treated with benzoyl chloride(1.50 mL, 12.50 mmol). The ensuing mixture was allowed to warm toand then stirred at 22 °C for 16 h before being quenched with NH4Cl(50 mL of a saturated aqueous solution) and then extracted withdichloromethane (2 × 100 mL). The combined organic phases werewashed with water (2 × 100 mL) and then brine (1 × 100 mL) beforebeing dried (Na2SO4), filtered, and concentrated under reducedpressure. The residue thus obtained was subjected to flash columnchromatography (silica, 2:5:100 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether elution) and gave, after concentration of theappropriate fractions [Rf = 0.2(5) in 1:19 v/v ethyl acetate/40−60petroleum ether], compound 48 (3.69 g, 93%) as a white, crystallinesolid: mp = 134−136 °C; [α]D = +196.0 (c 1.1, CHCl3);
1H NMR(400 MHz, CDCl3) δ 7.99 (d, J = 7.3 Hz, 2H), 7.54 (t, J = 7.3 Hz,1H), 7.41 (t, J = 7.3 Hz, 2H), 6.20 (d, J = 8.1 Hz, 1H), 6.04 (t, J = 8.1Hz, 1H), 5.12−5.10 (complex m, 2H), 3.23 (dd, J = 6.6 and 1.6 Hz,1H), 2.80 (m, 1H), 2.72 (dd, J = 10.6 and 6.6 Hz, 1H), 2.07 (s, 3H),1.55 (dd, J = 12.1 and 7.3 Hz, 1H), 1.43 (m, 1H), 1.11 (s, 3H), 1.02 (s,3H), 0.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 205.0, 170.1,166.2, 141.3, 133.4, 130.0, 129.5, 128.5, 122.4, 81.1, 74.4, 51.3, 50.7,44.9, 42.9, 41.7, 39.7, 25.4, 22.2, 21.5, 18.0; IR νmax 2967, 1722, 1268,1238, 1108, 729, 706 cm−1; MS (ESI, + ve) m/z 787 [(2M + Na)+,100], 405 [(M + Na)+, 85]; HRMS m/z (M + Na)+ calcd forC23H26O5Na 405.1678, found 405.1682.
(1S,3aS,4S,7R,7aR)-2,2,7-Trimethyl-9-oxo-2,3,3a,4,7,7a-hexahy-dro-1H-4,7-ethanoinden-1-yl Acetate (49). A magnetically stirredsolution of compound 48 (3.30 g, 8.63 mmol) in deoxygenated THF/methanol (30 mL of a 9:1 v/v mixture) maintained at −78 °C underan argon atmosphere was treated, dropwise over 1 h, withsamarium(II) iodide (ca. 250 mL of a 0.086 M solution in THF) atwhich point a dark-blue color persisted for 0.25 h. The resultingmixture was then quenched with NH4Cl (100 mL of a saturatedaqueous solution) and the ensuing suspension extracted with diethylether (2 × 100 mL). The combined organic phases were washed withHCl (2 × 50 mL of a 1 M aqueous solution) and brine (1 × 100 mL)before being dried (Na2SO4), filtered, and concentrated under reducedpressure. The residue thus obtained was subjected to flash columnchromatography (silica, 1:9 v/v diethyl ether/n-hexane elution) andgave, after concentration of the appropriate fractions [Rf = 0.4(5) in1:9 v/v diethyl ether/n-hexane)], compound 49 (1.91 g, 85%) as aclear, colorless oil: [α]D = +161.0 (c 1.0, CHCl3);
1H NMR (400MHz, CDCl3) δ 6.10 (d, J = 8.1 Hz, 1H), 5.86 (t, J = 8.1 Hz, 1H), 4.93(d, J = 6.0 Hz, 1H), 2.96 (dd, J = 6.0 and 1.8 Hz, 1H), 2.60 (m, 1H),2.46 (m, 1H), 1.98 (s, 3H), 1.79 (s, 2H), 1.38 (m, 1H), 1.26 (t, J =11.5 Hz, 1H), 1.05 (s, 3H), 0.90 (s, 3H), 0.76 (s, 3H); 13C NMR (100MHz, CDCl3) δ 212.3, 170.0, 142.9, 123.0, 80.4, 53.6, 52.1, 47.7, 44.8,42.0, 41.2, 39.2, 25.4, 22.2, 22.0, 21.4; IR νmax 2965, 1726, 1373, 1239,1069, 707 cm−1; MS (ESI, + ve) m/z 547 [(2M + Na)+, 10], 285 [(M+ Na)+, 100], 263 [(M + H)+, 1]; HRMS m/z (M + H)+ calcd forC16H23O3 263.1647, found 263.1649.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8018

(1R,2aR,4aR,7S,7aR,7bS)-1-(Methoxymethoxy)-6,6,7b-trimethyl-2-oxo-2,2a,4a,5,6,7,7a,7b-octahydro-1H-cyclobuta[e]inden-7-yl Ac-etate (50). A magnetically stirred solution of compound 45 (100 mg,0.31 mmol) in dry, deoxygenated dichloromethane (30 mL)maintained under a nitrogen atmosphere was irradiated with aHanovia 100 W medium-pressure mercury-vapor lamp (maintained at5 °C) for 16 h. The reaction mixture was then cooled andconcentrated under reduced pressure and the residue thus obtainedsubjected to flash column chromatography (silica, 1:19 v/v ethylacetate/n-hexane elution) to afford two fractions, A and B.Concentration of fraction A [Rf = 0.3(5) in 1:19 v/v ethyl acetate/n-
hexane] gave compound 50 (43 mg, 43% or 99% brsm) as a clear,colorless oil: [α]D = −157.0 (c 0.7, CHCl3);
1H NMR (400 MHz,CDCl3) δ 5.76 (m, 1H), 5.73 (m, 1H), 5.54 (m, 1H), 4.97 (d, J = 4.2Hz, 1H), 3.02 (m, 1H), 2.96 (m, 1H), 2.87 (dd, J = 9.3 and 4.2 Hz,1H), 2.14 (s, 3H), 2.06 (m, 1H), 1.98 (s, 3H), 1.51 (dd, J = 13.3 and5.0 Hz, 1H), 1.09 (s, 3H), 1.03 (s, 3H), 0.92 (s, 3H); 13C NMR (100MHz, CDCl3) δ 201.3, 170.6, 169.9, 134.3, 117.2, 83.4, 83.2, 59.0,45.4, 43.5, 42.8, 34.8, 34.7, 28.8, 24.3, 21.3, 20.5, 18.4; IR νmax 2933,2873, 1792, 1735, 1373, 1224, 1087 cm−1; MS (ESI, + ve) m/z 375[(M + MeOH + Na)+, 20], 343 [(M + Na)+, 100]; HRMS m/z (M +Na)+ calcd for C18H24O5Na 343.1521, found 343.1520.Concentration of the fraction B [Rf = 0.2(5) in 1:19 v/v ethyl
acetate/n-hexane] gave compound 45 (56 mg, 56% recovery) as acolorless, crystalline solid that was identical, in all respects, with anauthentic sample.(1R,2aR,4aR,7S,7aR,7bS)-1-(Methoxymethoxy)-6,6,7b-trimethyl-
2-oxo-2,2a,4a,5,6,7,7a,7b-octahydro-1H-cyclobuta[e]inden-7-yl Ac-etate (51). A magnetically stirred solution of compound 46 (100 mg,0.31 mmol) in dry, deoxygenated dichloromethane (30 mL)maintained under a nitrogen atmosphere was irradiated with aHanovia 100 W medium-pressure mercury-vapor lamp (maintained at5 °C) for 16 h. The cooled reaction mixture was concentrated underreduced pressure and the residue thus obtained subjected to flashcolumn chromatography (silica, 1:9 v/v ethyl acetate/n-hexaneelution), thus affording two fractions, A and B.Concentration of the fraction A [Rf = 0.3(5) in 1:9 v/v ethyl
acetate/n-hexane] gave compound 51 (27 mg, 27% or 98% brsm) as aclear, colorless oil: [α]D = −324.0 (c 0.2, CHCl3);
1H NMR (400MHz, CDCl3) δ 5.70 (m, 1H), 5.55 (m, 1H), 5.00 (d, J = 4.1 Hz, 1H),4.67 (m, 3H), 3.42 (s, 3H), 2.90 (m, 1H), 2.85 (m, 1H), 2.76 (dd, J =9.4 and 4.1 Hz, 1H), 2.05 (m, 1H), 1.98 (s, 3H), 1.51 (dd, J = 13.2 and5.2 Hz, 1H), 1.11 (s, 3H), 1.06 (s, 3H), 0.93 (s, 3H); 13C NMR (100MHz, CDCl3) δ 204.5, 170.7, 133.4, 118.0, 97.3, 89.4, 83.2, 58.5, 56.1,45.4, 43.5, 43.2, 34.7, 34.4, 28.7, 24.2, 21.4, 18.5; IR νmax 2962, 2933,1787, 1733, 1373, 1240, 1110, 1067, 1012 cm−1; MS (ESI, + ve) m/z345 [(M + Na)+, 100]; HRMS m/z (M + Na)+ calcd for C18H26O5Na345.1678, found 345.1672.Concentration of the fraction B [Rf = 0.2(5) in 1:9 v/v ethyl
acetate/n-hexane] gave compound 46 (71 mg, 71% recovery) as aclear, colorless oil that was identical, in all respects, with an authenticsample.(3S,3aR,4R,7S,7aS,9R)-9-((tert-Butyldimethylsilyl)oxy)-2,2,4-tri-
methyl-8-oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoinden-3-ylAcetate (52). A magnetically stirred solution of compound 47 (100mg, 0.25 mmol) in dry, deoxygenated dichloromethane (30 mL)maintained under a nitrogen atmosphere was irradiated with a Hanovia100 W medium-pressure mercury-vapor lamp (maintained at 5 °C) for16 h. The cooled reaction mixture was concentrated under reducedpressure and the residue thus obtained subjected to flash columnchromatography (silica, 1:19 v/v ethyl acetate/n-hexane elution), thusthereby affording two fractions, A and B.Concentration of the fraction A (Rf = 0.4 in 1:19 v/v ethyl acetate/
n-hexane) gave compound 52 (18 mg, 18% or 99% brsm) as a clear,colorless oil: [α]D = −189.0 (c 2.3, CHCl3);
1H NMR (400 MHz,CDCl3) δ 5.64 (m, 1H), 5.55 (m, 1H), 4.98 (d, J = 4.1 Hz, 1H), 4.71(d, J = 2.8 Hz, 1H), 2.86−2.78 (complex m, 2H), 2.65 (dd, J = 9.3 and4.1 Hz, 1H), 2.03 (m, 1H), 1.97 (s, 3H), 1.50 (dd, J = 13.2 and 5.1 Hz,1H), 1.10 (s, 3H), 0.97 (s, 3H), 0.92 (s, 3H), 0.90 (s, 9H), 0.10 (s,3H), 0.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.1, 170.6,
132.8, 118.7, 85.0, 83.3, 58.0, 45.5, 43.5, 43.2, 34.8, 34.5, 28.8, 25.8,24.2, 21.3, 18.4, 18.2, − 4.6, − 4.9; IR νmax 2958, 2931, 2859, 1788,1736, 1239, 1108, 883, 838, 780 cm−1; MS (ESI, + ve) m/z 415 [(M +Na)+, 100], 393 [(M + H)+, 5]; HRMS m/z (M + Na)+ calcd forC22H36O4SiNa 415.2281, found 415.2284.
Concentration of the fraction B (Rf = 0.3 in 1:19 v/v ethyl acetate/n-hexane) gave compound 47 (81 mg, 81% recovery) as a colorless,crystalline solid that was identical, in all respects, with an authenticsample.
(1R,2aR,4aR,7S,7aR,7bS)-7-Acetoxy-6,6,7b-trimethyl-2-oxo-2,2a,4a,5,6,7,7a,7b-octahydro-1H-cyclobuta[e]inden-1-yl Benzoate(53). A magnetically stirred solution of compound 48 (100 mg, 0.26mmol) in dry, deoxygenated dichloromethane (30 mL) maintainedunder a nitrogen atmosphere was irradiated with a Hanovia 100 Wmedium-pressure mercury-vapor lamp (maintained at 5 °C) for 16 h.The reaction mixture was then cooled and concentrated under reducedpressure and the residue so obtained subjected to flash columnchromatography (silica, 1:19 v/v ethyl acetate/n-hexane elution), thusaffording two fractions, A and B.
Concentration of the fraction A [Rf = 0.3(5) in 1:19 v/v ethylacetate/n-hexane] gave compound 53 (17 mg, 17% or 100% brsm) asa colorless, crystalline solid: mp = 180−181 °C; [α]D = −672.0 (c 1.5,CHCl3);
1H NMR (400 MHz, CDCl3) δ 8.06 (m, 2H), 7.59 (t, J = 7.4Hz, 1H), 7.45 (t, J = 7.4 Hz, 2H), 5.97 (d, J = 2.8 Hz, 1H), 5.80 (m,1H), 5.59 (m, 1H), 4.98 (d, J = 4.2 Hz, 1H), 3.10 (m, 1H), 3.04 (m,1H), 2.97 (dd, J = 9.3 and 4.2 Hz, 1H), 2.08 (dd, J = 13.3 and 9.8 Hz,1H), 1.99 (s, 3H), 1.53 (dd, J = 13.3 and 5.0 Hz, 1H), 1.12 (s, 3H),1.10 (s, 3H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 201.1,170.6, 165.3, 134.3, 133.8, 130.1, 128.8, 128.7, 117.3, 83.6, 83.2, 59.2,45.4, 43.5, 42.9, 35.1, 34.8, 28.8, 24.3, 21.3, 18.5; IR νmax 2963, 2934,2872, 1790, 1724, 1372, 1237, 1104, 709 cm−1; MS (ESI, + ve) m/z787 [(2M + Na)+, 10], 437 [(M + MeOH + Na)+, 50], 405 [(M +Na)+, 100]; HRMS m/z (M + Na)+ calcd for C23H26O5Na 405.1678,found 405.1670.
Concentration of the fraction B [Rf = 0.2(5) in 1:19 v/v ethylacetate/n-hexane] gave compound 48 (83 mg, 83% recovery) as acolorless, crystalline solid that was identical, in all respects, with anauthentic sample.
( 2 a R , 4 a R , 7 S , 7 a R , 7 b S ) - 6 , 6 , 7 b - T r i m e t h y l - 2 - o x o -2,2a,4a,5,6,7,7a,7b-octahydro-1H-cyclobuta[e]inden-7-yl Acetate(54). A magnetically stirred solution of compound 49 (100 mg, 0.38mmol) in dry, deoxygenated dichloromethane (30 mL) maintainedunder a nitrogen atmosphere was irradiated with a Hanovia 100 Wmedium-pressure mercury-vapor lamp (maintained at 5 °C) for 16 h.The reaction mixture was then cooled and concentrated under reducedpressure and the ensuing residue subjected to flash columnchromatography (silica, 1:19 v/v ethyl acetate/n-hexane elution),thus affording two fractions, A and B.
Concentration of the fraction A [Rf = 0.3(5) in 1:19 v/v ethylacetate/n-hexane] gave compound 54 (23 mg, 23% or 92% brsm) as aclear, colorless oil, [α]D = −254.0 (c 3.4, CHCl3).
1H NMR (400MHz, CDCl3) δ 5.73 (m, 1H), 5.48 (m, 1H), 4.97 (d, J = 4.2 Hz, 1H),3.12 (dd, J = 16.2 and 2.1 Hz, 1H), 3.05 (m, 1H), 2.84 (m, 1H), 2.59(dd, J = 9.2 and 4.3 Hz, 1H), 2.51 (dd, J = 16.2 and 6.0 Hz, 1H), 2.01(dd, J = 13.2 and 9.7 Hz, 1H), 1.95 (s, 3H), 1.50 (dd, J = 13.2 and 4.8Hz, 1H), 1.19 (s, 3H), 1.08 (s, 3H), 0.90 (s, 3H); 13C NMR (100MHz, CDCl3) δ 206.4, 170.7, 133.7, 118.2, 83.5, 64.1, 56.7, 45.7, 44.3,43.4, 34.9, 29.0, 28.1, 25.5, 24.3, 21.3; IR νmax 3020, 2966, 2871, 1778,1731, 1373, 1239, 1020, 711 cm−1; MS (ESI, + ve) m/z 285 [(M +Na)+, 100]; HRMS m/z (M + Na)+ calcd for C16H22O3Na 285.1467,found 285.1472.
Concentration of the fraction B (Rf = 0.3 in 1:19 v/v ethyl acetate/n-hexane) gave compound 49 (69 mg, 69% recovery) as a colorless,crystalline solid that was identical, in all respects, with an authenticsample.
(1S,1aR,3aR,6S,6aR,6bR)-5,5,6b-Trimethyl-1,1a,3a,4,5,6,6a,6b-octahydrocyclopropa[e]indene-1,6-diyl Diacetate (55). A magneti-cally stirred solution of compound 50 (40 mg, 0.12 mmol) in dry,deoxygenated dichloromethane (10 mL) maintained under a nitrogenatmosphere was irradiated with a Hanovia 450 W medium-pressure
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8019

mercury-vapor lamp for 0.5 h. The reaction mixture was then cooledand concentrated under reduced pressure and the residue thusobtained subjected to flash column chromatography (silica, 1:9 v/vethyl acetate/n-hexane elution). Concentration of the appropriatefractions [Rf = 0.6(5) in 1:9 v/v ethyl acetate/n-hexane] gavecompound 55 (39 mg, 98%) as a clear, colorless oil: [α]D = +167.0 (c0.5, CHCl3);
1H NMR (400 MHz, CDCl3) δ 5.90 (m, 1H), 5.22 (dd, J= 10.0 and 1.9 Hz, 1H), 5.04 (d, J = 3.9 Hz, 1H), 3.77 (d, J = 2.3 Hz,1H), 2.82 (dd, J = 10.1 and 3.9 Hz, 1H), 2.64 (m, 1H), 2.05 (s, 3H),2.00 (s, 3H), 1.90 (dd, J = 13.1 and 9.6 Hz, 1H), 1.33 (dd, J = 13.1 and5.2 Hz, 1H), 1.22 (dd, J = 11.1 and 2.3 Hz, 1H), 1.06 (s, 6H), 0.90 (s,3H); 13C NMR (100 MHz, CDCl3) δ 171.8, 171.1, 129.9, 123.1, 84.0,63.4, 46.6, 42.8, 40.8, 35.4, 28.6, 24.3, 24.2, 22.1, 21.3, 20.9, 15.9; IRνmax 3026, 2966, 2933, 2872, 1733, 1371, 1227 cm−1; MS (ESI, + ve)m/z 347 [(M + MeOH + Na)+, 100%], 331 [(M + K)+, 20], 329 (50),315 [(M + Na)+, 60]; HRMS m/z (M + Na)+ calcd for C17H24O4Na315.1572, found 315.1570.(1S,1aR,3aR,6S,6aR,6bS)-1-((tert-Butyldimethylsilyl)oxy)-5,5,6b-
trimethyl-1,1a,-3a,4,5,6,6a,6b-octahydrocyclopropa[e]inden-6-ylAcetate (56). A magnetically stirred solution of compound 47 (100mg, 0.25 mmol) in dry, deoxygenated dichloromethane (30 mL)maintained under a nitrogen atmosphere was irradiated with a Hanovia450 W medium-pressure mercury-vapor lamp for 1.67 h. The reactionmixture was then cooled, concentrated under reduced pressure and theresidue thus obtained subjected to flash column chromatography(silica, 1:19 v/v ethyl acetate/40−60 petroleum ether) to afford twofractions, A and B.Concentration of the fraction A (Rf = 0.6 in 1:4 v/v ethyl acetate/
40−60 petroleum ether) gave compound 56 (74 mg, 80% or 87%brsm) as a clear, colorless oil: [α]D = −65.5 (c 1.0, CHCl3);
1H NMR[400 MHz, (CD3)2CO] δ 5.92 (m, 1H), 5.11 (dd, J = 10.0 and 2.1 Hz,1H), 5.01 (d, J = 4.1 Hz, 1H), 3.14 (d, J = 2.2 Hz, 1H), 2.78 (m, 1H),2.63 (m, 1H), 1.95 (s, 3H), 1.89 (m, 1H), 1.30 (dd, J = 13.0 and 5.2Hz, 1H), 1.09 (s, 3H), 1.06 (s, 3H), 0.97 (dd, J = 5.5 and 2.4 Hz, 1H),0.90 (s, 9H), 0.87 (s, 3H), 0.10(1) (s, 3H), 0.09(8) (s, 3H); 13C NMR[100 MHz, (CD3)2CO] δ 170.7, 128.9, 125.2, 84.4, 63.9, 47.3, 43.3,41.9, 36.2, 28.9, 27.7, 26.2, 24.7, 23.1, 21.1, 18.6, 15.9, − 4.9 (onesignal obscured or overlapping); IR νmax 3021, 2958, 2930, 2859, 1734,1240, 1153, 859, 835, 776 cm−1; MS (ESI, + ve) m/z 387 [(M + Na)+,100]; HRMS m/z (M + Na)+ calcd for C21H36O3SiNa 387.2331,found 387.2331.Concentration of the fraction B (Rf = 0.5 in 1:4 v/v ethyl acetate/
40−60 petroleum ether) gave compound 47 (8 mg, 8% recovery) as awhite, crystalline solid that was identical, in all respects, with anauthentic sample.(3S,3aR,4R,7S,7aS,9R)-2,2,4-Trimethyl-9-((methylsulfonyl)oxy)-8-
oxo-2,3,3a,4,-7,7a-hexahydro-1H-4,7-ethanoinden-3-yl Acetate(57). A magnetically stirred solution of compound 44 (280 mg, 1.01mmol), methanesulfonyl chloride (94 μL, 1.21 mmol) and triethyl-amine (212 μL, 1.52 mmol) in dichloromethane (5 mL) maintainedunder a nitrogen atmosphere was stirred at 22 °C for 48 h and thenquenched with NH4Cl (10 mL of a saturated aqueous solution) beforebeing extracted with dichloromethane (2 × 50 mL). The combinedorganic fractions were washed with water (2 × 50 mL) and brine (1 ×50 mL) and then dried (MgSO4), filtered, and concentrated underreduced pressure. The residue thus obtained was subjected to flashcolumn chromatography (silica, 1:9 v/v ethyl acetate/40−60petroleum ether elution) to give, after concentration of the appropriatefractions (Rf = 0.3 in 1:9 v/v ethyl acetate/40−60 petroleum ether),compound 57 (198 mg, 56%) as a light-yellow oil: [α]D = +114.0 (c4.3, CHCl3);
1H NMR (400 MHz, CDCl3) δ 6.08 (d, J = 8.1 Hz, 1H),5.95 (t, J = 8.1 Hz, 1H), 5.08 (d, J = 5.9 Hz, 1H), 4.31 (s, 1H), 3.19(m, 1H), 3.17 (s, 3H), 2.69 (m, 1H), 2.58 (dd, J = 10.6 and 6.0 Hz,1H), 2.05 (s, 3H), 1.53 (m, 1H), 1.39 (t, J = 12.0 Hz, 1H), 1.20 (s,3H), 0.98 (s, 3H), 0.85 (s, 3H); 13C NMR (100 MHz, CDCl3) δ204.1, 170.0, 140.9, 121.9, 81.3, 81.0, 50.7, 50.2, 44.9, 42.7, 41.6,39.5(1), 39.4(8), 25.3, 22.1, 21.5, 18.1; IR νmax 2968, 1730, 1358,1238, 1173, 965, 841 cm−1; MS (ESI, + ve) m/z 735 [(2M + Na)+,15], 379 [(M + Na)+, 100]; HRMS m/z (M + Na)+ calcd forC17H24O6SNa 379.1191, found 379.1193.
(1R,2a1S,2cR,5S,5aR,5bR)-4,4,5b-Trimethyl-1-((methylsulfonyl)-oxy)-2-oxodeca-hydro-1H-cyclopenta[a]cyclopropa[cd]pentalen-5-yl Acetate (58). A magnetically stirred solution of compound 57 (100mg, 0.28 mmol) in dry, deoxygenated dichloromethane (30 mL)maintained under a nitrogen atmosphere was irradiated with a Hanovia100 W medium-pressure mercury-vapor lamp (maintained at 5 °C) for12 h. The reaction mixture was then cooled and concentrated underreduced pressure and the residue thus obtained subjected to flashcolumn chromatography (silica, 1:9 v/v ethyl acetate/n-hexaneelution) to afford two fractions, A and B.
Concentration of the fraction A [Rf = 0.4(5) in 1:9 v/v ethylacetate/n-hexane] gave compound 58 (48 mg, 48% or 98% brsm) as aclear, colorless oil: [α]D = +161.0 (c 1.0, CHCl3).
1H NMR (400 MHz,CDCl3) δ 5.54 (s, 1H), 5.09 (d, J = 4.8 Hz, 1H), 3.05 (s, 3H), 2.85−2.72 (complex m, 2H), 2.50 (d, J = 4.8 Hz, 1H), 2.23 (s, 3H), 2.18 (s,1H), 1.71 (d, J = 4.8 Hz, 1H), 1.59 (dd, J = 8.7 and 2.3 Hz, 2H), 1.07(s, 3H), 0.97(2) (s, 3H), 0.96(7) (s, 3H); 13C NMR (100 MHz,CDCl3) δ 211.1, 170.2, 80.9, 78.8, 50.4, 45.2, 42.5, 42.4, 41.1, 39.3,37.6, 36.1, 32.9, 25.1, 22.7, 22.5, 21.2; IR νmax 2966, 2936, 2875, 1729,1356, 1236, 1172, 917, 842 cm−1; MS m/z 735 [(2M + Na)+, 5], 379[(M + Na)+, 100]; HRMS m/z (M + Na)+ calcd for C17H24O6SNa379.1191, found 379.1189.
Concentration of the fraction B (Rf = 0.3 in 1:9 v/v ethyl acetate/n-hexane) gave compound 57 (50 mg, 50% recovery) as a clear, colorlessoil that was identical, in all respects, with an authentic sample.
X-ray Crystallographic Studies. Crystallographic Data. Com-pound 8: C9H12O3, M = 168.19, T = 150 K, orthorhombic, spacegroup P212121, Z = 4, a = 7.3238(1) Å, b = 7.5553(1) Å, c =14.5389(2) Å; V = 804.49(2) Å3, Dx = 1.389 g cm−3, 1585 unique data(2θmax = 144.6°), R = 0.023 [for 1569 reflections with I > 2.0σ(I)]; Rw
= 0.057 (all data), S = 1.00.Compound 9: C15H26O3Si, M = 282.46, T = 150 K, monoclinic,
space group C2, Z = 4, a = 18.9377(5) Å, b = 6.2442(1) Å, c =14.6275(5) Å; β = 105.950(3)°; V = 1663.12(8) Å3, Dx = 1.128 gcm−3, 3042 unique data (2θmax = 144.8°), R = 0.032 [for 2889reflections with I > 2.0σ(I)]; Rw = 0.080 (all data), S = 0.99.
Compound 10: C9H12O3, M = 168.19, T = 150 K, orthorhombic,space group P212121, Z = 8, a = 8.23350(7) Å, b = 8.68949(8) Å, c =23.6396(2) Å; V = 1691.29(3) Å3, Dx = 1.321 g cm−3, 3334 uniquedata (2θmax = 144.6°), R = 0.04 [for 3257 reflections with I > 2.0σ(I)];Rw = 0.112 (all data), S = 1.01.
Compound 13: C14H26O2Si,M = 254.45, T = 150 K, orthorhombic,space group P21212, Z = 8, a = 18.3637(1) Å, b = 14.1093(1) Å, c =12.0122(1) Å; V = 3112.35(4) Å3, Dx = 1.086 g cm−3, 6159 uniquedata (2θmax = 145.0°), R = 0.025 [for 6101 reflections with I >2.0σ(I)]; Rw = 0.067 (all data), S = 1.01.
Compound 23: C13H16O3, M = 220.27, T = 150 K, orthorhombic,space group P212121, Z = 4, a = 7.27015(5) Å, b = 11.05109(8) Å, c =13.78627(9) Å; V = 1107.63(1) Å3, Dx = 1.321 g cm−3, 2186 uniquedata (2θmax = 144.8°), R = 0.022 [for 2152 reflections with I >2.0σ(I)]; Rw = 0.057 (all data), S = 1.01.Compound 27: C13H16O3, M = 220.27, T = 200 K, monoclinic,
space group P21, Z = 2, a = 7.2003(2) Å, b = 8.4682(2) Å, c =9.4258(3) Å; β = 91.1279(17)°; V = 574.61(3) Å3, Dx = 1.273 g cm−3,1778 unique data (2θmax = 60.0°), R = 0.048 [for 1655 reflections withI > 2.0σ(I)]; Rw = 0.156 (all data), S = 1.03.
Compound 30: C11H12O3, M = 192.21, T = 150 K, monoclinic,space group P21, Z = 2, a = 8.4949(4) Å, b = 5.9702(2) Å, c =9.3840(4) Å; β = 95.383(4)°; V = 473.82(3) Å3, Dx = 1.347 g cm−3,1032 unique data (2θmax = 144.8°), R = 0.045 [for 998 reflections withI > 2.0σ(I)]; Rw = 0.130 (all data), S = 1.03.
Compound 32: C10H12O2, M = 164.20, T = 150 K, orthorhombic,space group P212121, Z = 4, a = 7.8054(1) Å, b = 9.7920(1) Å, c =11.0580(1) Å; V = 845.17(2) Å3, Dx = 1.290 g cm−3, 1677 unique data(2θmax = 144.6°), R = 0.022 [for 1645 reflections with I > 2.0σ(I)]; Rw
= 0.057 (all data), S = 1.01.Compound 34: C11H12O3, M = 192.21, T = 150 K, monoclinic,
space group P21, Z = 2, a = 6.9303(2) Å, b = 7.7696(2) Å, c =8.6272(2) Å; β = 92.403(2)°; V = 464.13(2) Å3, Dx = 1.375 g cm−3,
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8020

993 unique data (2θmax = 145.2°), R = 0.037 [for 980 reflections with I> 2.0σ(I)]; Rw = 0.117 (all data), S = 1.04.Compound 35: C13H16O3, M = 220.27, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 7.3877(1) Å, b = 7.8178(1) Å, c =19.8494(2) Å; V = 1146.41(2) Å3, Dx = 1.276 g cm−3, 2267 uniquedata (2θmax = 144.8°), R = 0.030 [for 2238 reflections with I >2.0σ(I)]; Rw = 0.056 (all data), S = 1.00.Compound 36: C11H12O3, M = 192.21, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 7.4666(2) Å, b = 12.0831(5) Å, c =10.8645(4) Å; V = 980.19(6) Å3, Dx = 1.302 g cm−3, 1491 unique data(2θmax = 58.4°), R = 0.047 [for 1303 reflections with I > 2.0σ(I)]; Rw =0.108 (all data), S = 1.00.Compound 38: C10H12O2, M = 164.20, T = 150 K, hexagonal,
space group P65, Z = 6, a = 14.7787(4) Å, c = 7.6213(2) Å; V =1441.56(7) Å3, Dx = 1.135 g cm−3, 1379 unique data (2θmax = 58.8°),R = 0.045 [for 1283 reflections with I > 2.0σ(I)]; Rw = 0.111 (all data),S = 1.03.Compound 41: C17H26O3, M = 278.39, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 6.26451(4) Å, b = 12.09608(9) Å, c =20.26568(14) Å; V = 1535.65(2) Å3, Dx = 1.204 g cm−3, 3025 uniquedata (2θmax = 144.6°), R = 0.024 [for 2974 reflections with I >2.0σ(I)]; Rw = 0.062 (all data), S = 1.00.Compound 44: C16H22O4, M = 278.35, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 6.4307(1) Å, b = 8.7685(1) Å, c =26.1929(2) Å; V = 1476.95(3) Å3, Dx = 1.252 g cm−3, 2913 uniquedata (2θmax = 144.8°), R = 0.024 [for 2859 reflections with I >2.0σ(I)]; Rw = 0.063 (all data), S = 1.00.Compound 45: C18H24O5, M = 320.39, T = 150 K, monoclinic,
space group P21, Z = 2, a = 7.4300(3) Å, b = 9.7667(3) Å, c =12.1277(3) Å; β = 92.279(3)°; V = 879.37(5) Å3, Dx = 1.210 g cm−3,2377 unique data (2θmax = 58.6°), R = 0.038 [for 2242 reflections withI > 2.0σ(I)]; Rw = 0.093 (all data), S = 1.02.Compound 47: C22H36O4Si,M = 392.61, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 25.6494(7) Å, b = 14.1949(4) Å, c =6.2343(2) Å; V = 2269.85(11) Å3, Dx = 1.149 g cm−3, 5914 uniquedata (2θmax = 58.8°), R = 0.045 [for 5443 reflections with I > 2.0σ(I)];Rw = 0.102 (all data), S = 1.04.Compound 48: C23H26O5, M = 382.46, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 19.9195(6) Å, b = 11.6626(3) Å, c =8.7386(2) Å; V = 2030.09(9) Å3, Dx = 1.251 g cm−3, 2980 unique data(2θmax = 58.6°), R = 0.041 [for 2759 reflections with I > 2.0σ(I)]; Rw =0.101 (all data), S = 1.03.Compound 53: C23H26O5, M = 382.46, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 8.9344(2) Å, b = 15.4099(3) Å, c =14.9268(3) Å; V = 2055.09(7) Å3, Dx = 1.236 g cm−3, 3065 uniquedata (2θmax = 59.0°), R = 0.042 [for 2868 reflections with I > 2.0σ(I)];Rw = 0.112 (all data), S = 1.04.Structure Determination. Images for compound 27, 36, 38, 45, 47,
48, and 53 were measured on a diffractometer (Mo Kα, graphitemonochromator, λ = 0.71073 Å) fitted with an area detector and thedata extracted using the DENZO/Scalepack or CrysAlis package.15
Images for compounds 8, 9, 10, 13, 23, 30, 32, 34, 35, 41, and 44 weremeasured on a diffractometer (Cu Kα, mirror monochromator, λ=1.54184 Å) fitted with an area detector and the data extracted usingthe CrysAlis package.16 The structure solutions for all six compoundswere solved by direct methods (SIR92)17 then refined using theCRYSTALS program package.18 Atomic coordinates, bond lengthsand angles, and displacement parameters have been deposited at theCambridge Crystallographic Data Centre (CCDC nos. 1545223,154224, 1545225, 1545226, 1545227, 1545228, 1545229, 1545230,1545231, 1545232, 1545233, 1545234, 1545235, 1545236, 1545237,1545238, 1545239 and 1545240). These data can be obtained free ofcharge via www.ccdc.cam.ac.uk/data_request/cif, by emailing [email protected], or by contacting the Cambridge Crystallo-graphic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.joc.7b01243.
Anisotropic displacement ellipsoid plots from the single-crystal X-ray analyses of compounds 8−10, 13, 23, 27,30, 32, 34−36, 38, 41, 44, 45, 47, 48, and 53; 1H and13C NMR spectra of compounds 8−39 and 42−58(PDF)X-ray crystallographic data for compounds 8−10, 13, 23,27, 30, 32, and 34 (CIF)X-ray crystallographic data for compounds 35, 36, 38, 41,44, 45, 47, 48, and 53 (CIF)
■ AUTHOR INFORMATIONCorresponding Author*Tel: +61-2-6125-8202. Fax: +61-2-6125-8114. E-mail: [email protected] G. Banwell: 0000-0002-0582-475XNotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank the Australian Research Council and the Institute ofAdvanced Studies for financial support. Q.Y. is the gratefulrecipient of a CSC PhD Scholarship provided by theGovernment of the People’s Republic of China.
■ REFERENCES(1) For a relevant and recent review, see: Banwell, M. G.; Bon, D. J.-Y. D. Applications of the Di-π-Methane and Related RearrangementReactions in Chemical Synthesis. In Molecular Rearrangements inOrganic Synthesis; Rojas, C. M., Ed.; Wiley: Hoboken, NJ, 2015;Chapter 9, pp 261−288.(2) Luo, S.-Y.; Jang, Y.-J.; Liu, J.-Y.; Chu, C.-S.; Liao, C.-C.; Hung, S.-C. Angew. Chem., Int. Ed. 2008, 47, 8082 and references cited therein.(3) For reviews on cis-1,2-dihydrocatechols, see: (a) Johnson, R. A.Org. React. 2004, 63, 117. (b) Hudlicky, T.; Reed, J. W. Synlett 2009,2009, 685. (c) Lewis, S. E. Chem. Commun. 2014, 50, 2821.(d) Banwell, M. G.; Bolte, B.; Buckler, J. N.; Chang, E. L.; Lan, P.;Taher, E. S.; White, L. V.; Willis, A. C. J. Proc. R. Soc. New South Wales2016, 149, 34.(4) (a) Banwell, M. G.; Edwards, A. J.; Harfoot, G. J.; Jolliffe, K. A.Tetrahedron 2004, 60, 535. (b) Banwell, M. G.; Austin, K. A. B.; Willis,A. C. Tetrahedron 2007, 63, 6388. (c) Reekie, T. A.; Austin, K. A. B.;Banwell, M. G.; Willis, A. C. Aust. J. Chem. 2008, 61, 94. (d) Bon, D. J.-Y. D.; Banwell, M. G.; Willis, A. C. Tetrahedron 2010, 66, 7807.(e) Bon, D. J.-Y. D.; Banwell, M. G.; Cade, I. A.; Willis, A. C.Tetrahedron 2011, 67, 8348. (f) Bon, D. J.-Y. D.; Banwell, M. G.;Ward, J. S.; Willis, A. C. Tetrahedron 2013, 69, 1363.(5) (a) Schwartz, B. D.; Matousova, E.; White, R.; Banwell, M. G.;Willis, A. C. Org. Lett. 2013, 15, 1934. (b) Chang, E. L.; Bolte, B.; Lan,P.; Willis, A. C.; Banwell, M. G. J. Org. Chem. 2016, 81, 2078.(6) Lan, P.; Banwell, M. G.; Willis, A. C. Org. Lett. 2015, 17, 166.(7) Yan, Q.; Banwell, M. G.; Coote, M. L.; Lee, R.; Willis, A. C.Chem. - Asian J. 2017, 12, 1480.(8) Banwell, M. G.; Chun, C.; Edwards, A. J.; Vogtle, M. M. Aust. J.Chem. 2003, 56, 861.(9) Ireland, R. E.; Liu, L. J. Org. Chem. 1993, 58, 2899.(10) This protocol is based on one first described by Ma and Bobbitt:Ma, Z.; Bobbitt, J. M. J. Org. Chem. 1991, 56, 6110. For examples ofrelated oxidations see refs 4b−d.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8021

(11) Ravelli, D.; Fagnoni, M. Photochemistry of Phosphate andSulfonate Esters In CRC Handbook of Organic Photochemistry, 3rd ed.;Griesbeck, A., Oelgemoller, M., Ghetti, F., Ed.; CRC Press, BocaRaton, FL, 2012; Chapter 17, pp 393−417.(12) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.(13) Pangborn, A. B.; Giardello, M. A.; Grubbs, R. H.; Rosen, R. K.;Timmers, F. J. Organometallics 1996, 15, 1518.(14) Foley, D. A.; O’Leary, P.; Buckley, N. R.; Lawrence, S. E.;Maguire, A. R. Tetrahedron 2013, 69, 1778.(15) DENZO−SMN: Otwinowski, Z.; Minor, W. Processing of X-raydiffraction data collected in oscillation mode. In Methods inEnzymology; Carter, C. W., Jr., Sweet, R. M., Eds.; Academic Press:New York, 1997; Vol. 276: Macromolecular Crystallography, Part A,pp 307−326.(16) CrysAlis PRO, version 1.171.37.35h (release 09-02-2015CrysAlis171.NET) (compiled Feb 9 2015, 16:26:32) AgilentTechnologies: Oxfordshire, UK, 2015.(17) SIR92: Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi,A.; Burla, M. C.; Polidori, G.; Camalli, M. J. Appl. Crystallogr. 1994, 27,435.(18) Betteridge, P. W.; Carruthers, J. R.; Cooper, R. I.; Prout, K.;Watkin, D. J. J. Appl. Crystallogr. 2003, 36, 1487.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b01243J. Org. Chem. 2017, 82, 8008−8022
8022


S1
SUPP
ORT
ING
INFO
RMAT
ION
FO
R:
Stud
ies o
n th
e Ph
otoc
hem
ical
Rea
rran
gem
ents
of E
nant
iom
eric
ally
Pur
e, P
olys
ubst
itute
d an
d V
ario
usly
Ann
ulat
ed
Bic
yclo
[2.2
.2]o
cten
ones
Qia
o Y
an, B
enoi
t Bol
te, Y
uhua
Bai
, Mar
tin G
. Ban
wel
l* A
ntho
ny C
. Will
is a
nd P
aul D
. Car
r
Rese
arch
Sch
ool o
f Che
mis
try,
Inst
itute
of A
dvan
ced
Stud
ies,
The
Aust
ralia
n N
atio
nal U
nive
rsity
, Can
berr
a, A
CT
2601
, Aus
tral
ia
CO
NT
EN
TS
PA
GE
(i)A
niso
tropi
c D
ispl
acem
ent E
llips
oid
Plot
s fro
m th
e Si
ngle
-cry
stal
X-r
ay
Ana
lyse
s of C
ompo
unds
8, 9
, 10,
13,
23,
27,
30,
32,
34,
35,
36,
38,
41,
44, 4
5, 4
7, 4
8 an
d 53
S
2
(ii)
1 H a
nd 13
C N
MR
Spe
ctra
of C
ompo
unds
8-3
9 an
d 42
-58
S
20
184
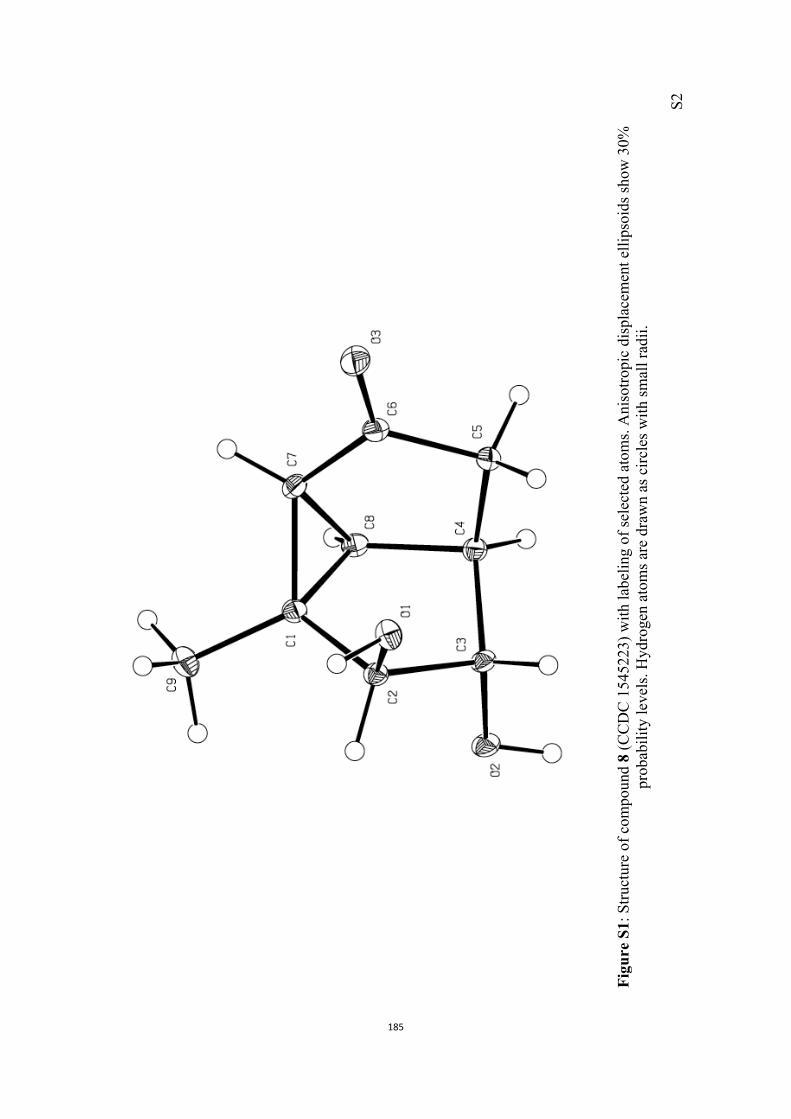
S2
Figu
re S
1: S
truct
ure
of c
ompo
und
8 (C
CD
C 1
5452
23) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
185
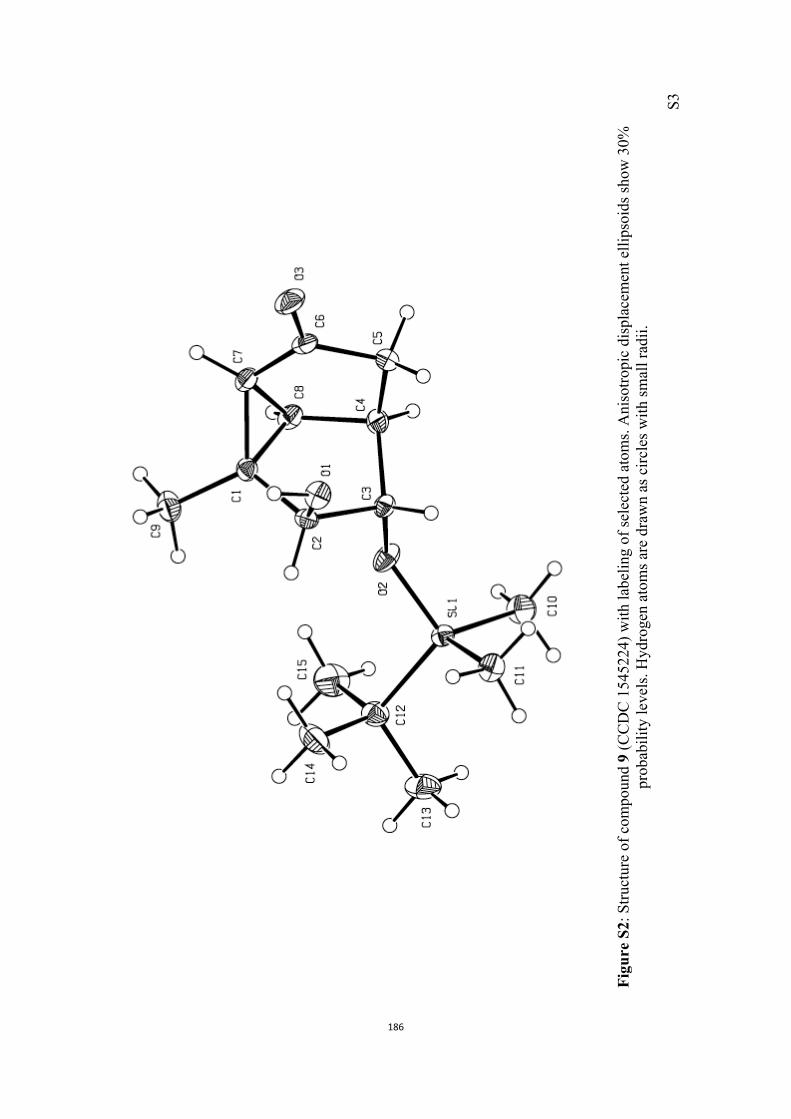
S3
Figu
re S
2: S
truct
ure
of c
ompo
und
9 (C
CD
C 1
5452
24) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
186
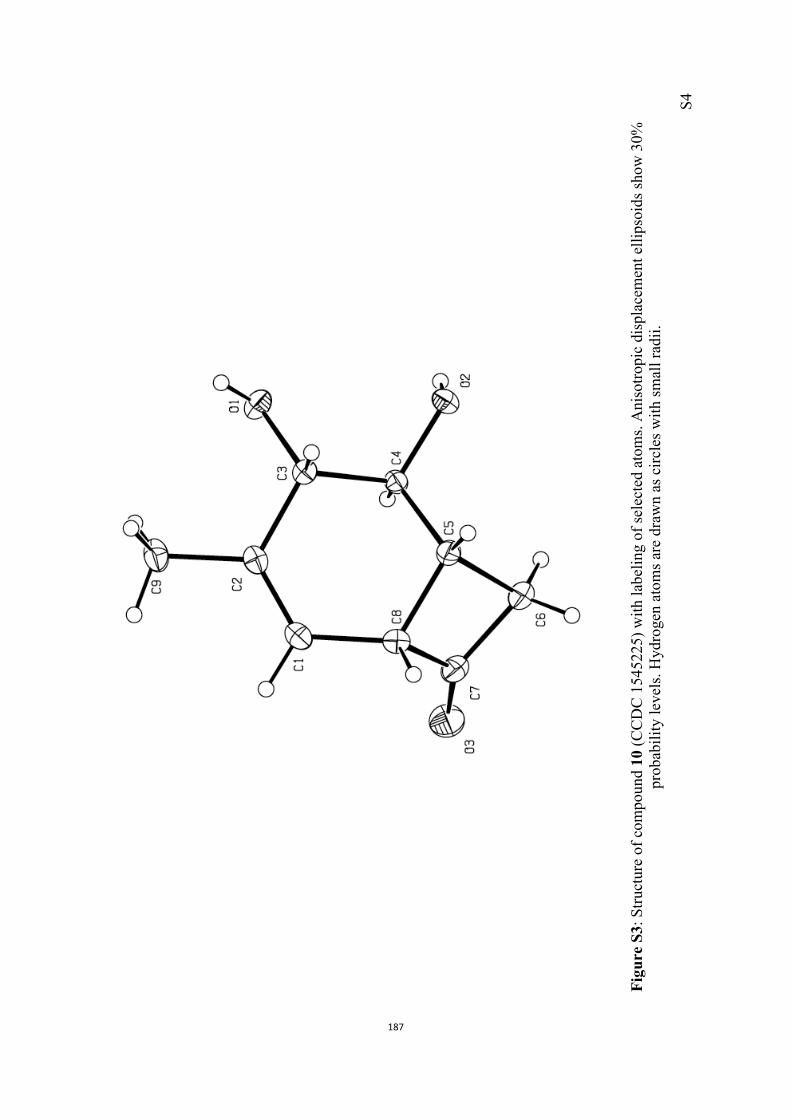
S4
Figu
re S
3: S
truct
ure
of c
ompo
und
10 (C
CD
C 1
5452
25) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
187
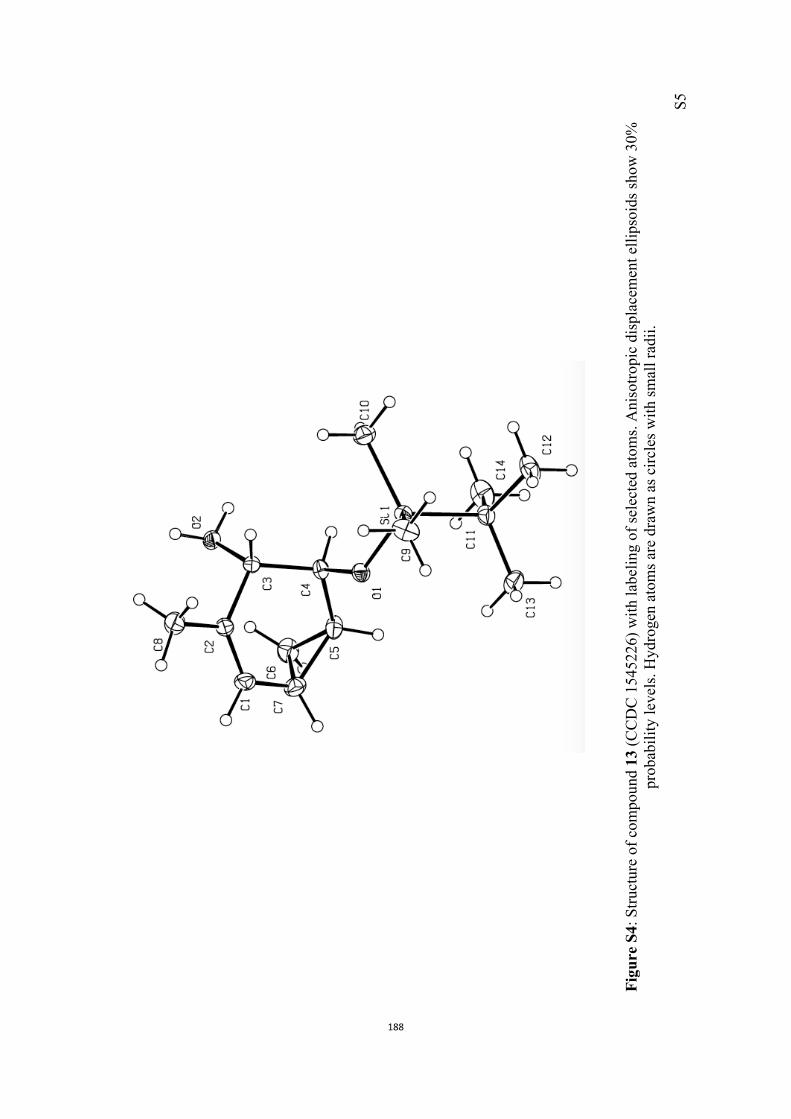
S5
Figu
re S
4: S
truct
ure
of c
ompo
und
13 (C
CD
C 1
5452
26) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
188

S6
Figu
re S
5: S
truct
ure
of c
ompo
und
23 (C
CD
C15
4522
7) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
189
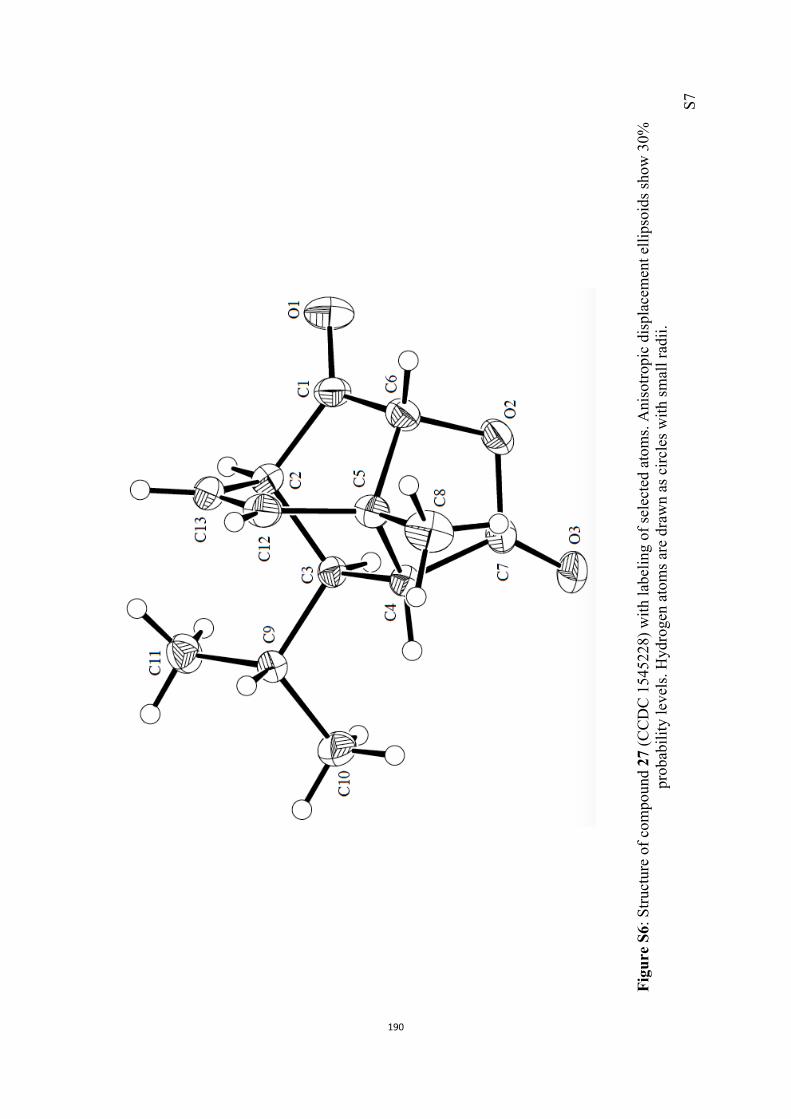
S7
Figu
re S
6: S
truct
ure
of c
ompo
und
27 (C
CD
C 1
5452
28) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
190
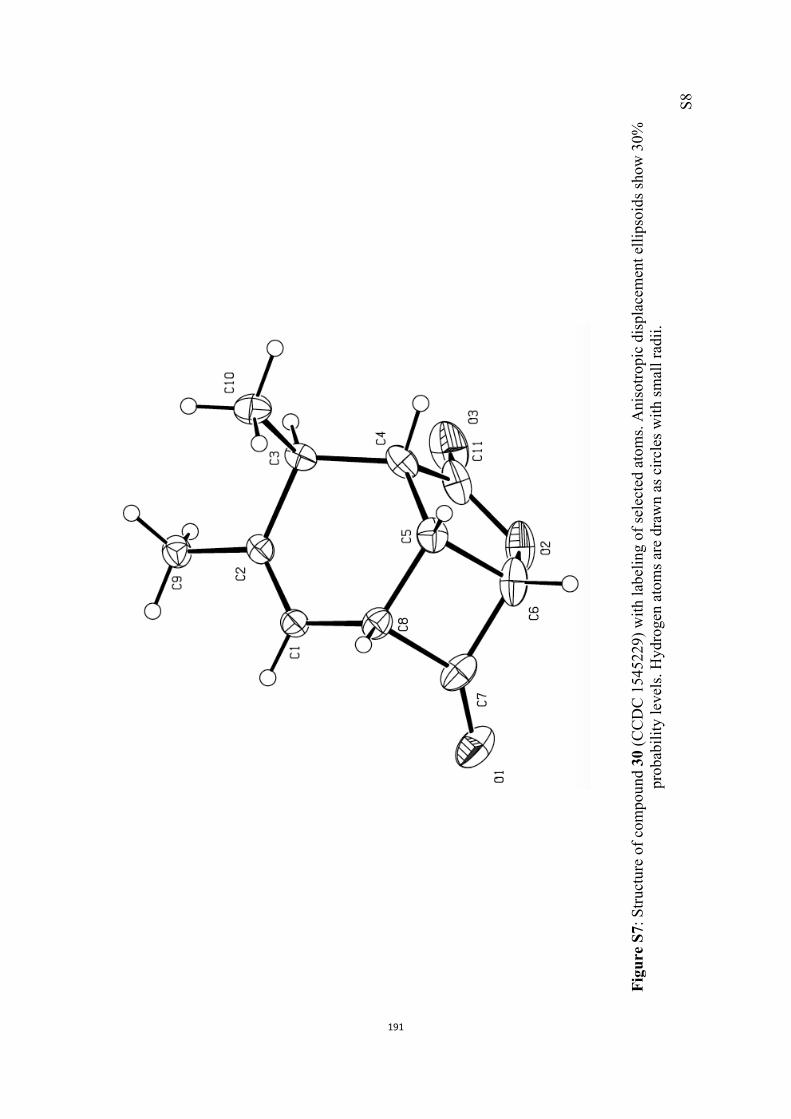
S8
Figu
re S
7: S
truct
ure
of c
ompo
und
30 (C
CD
C 1
5452
29) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
191
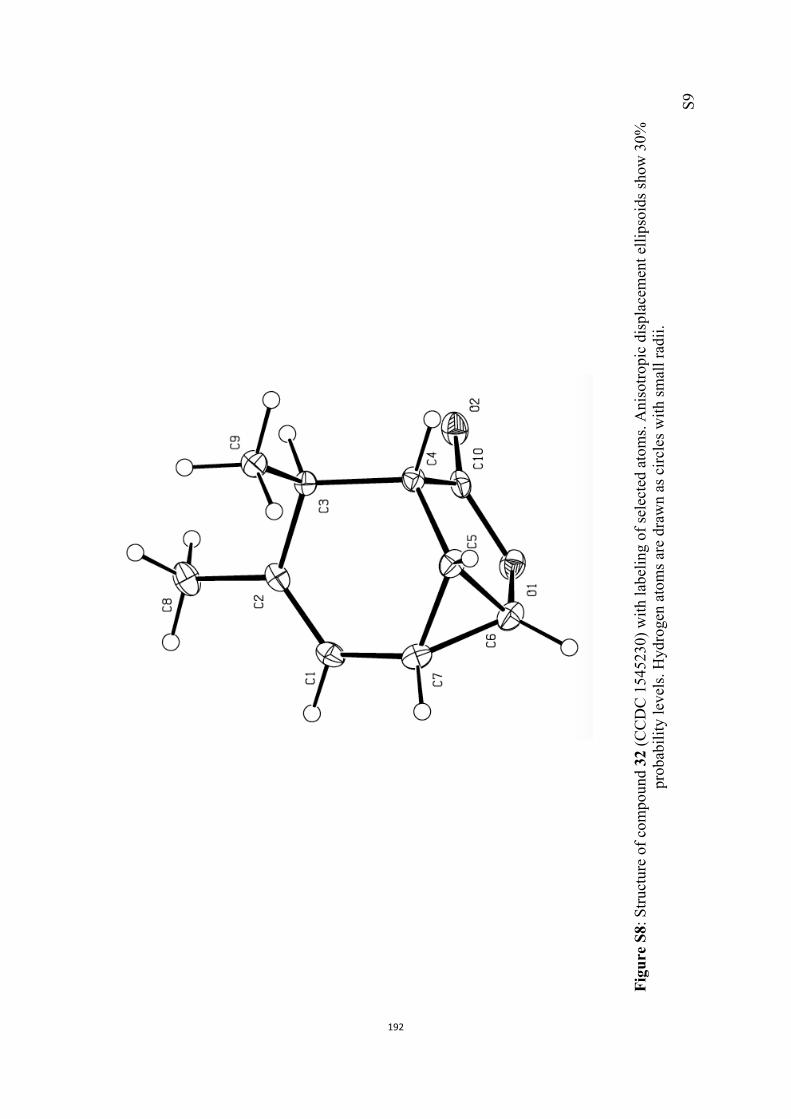
S9
Figu
re S
8: S
truct
ure
of c
ompo
und
32 (C
CD
C 1
5452
30) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
192
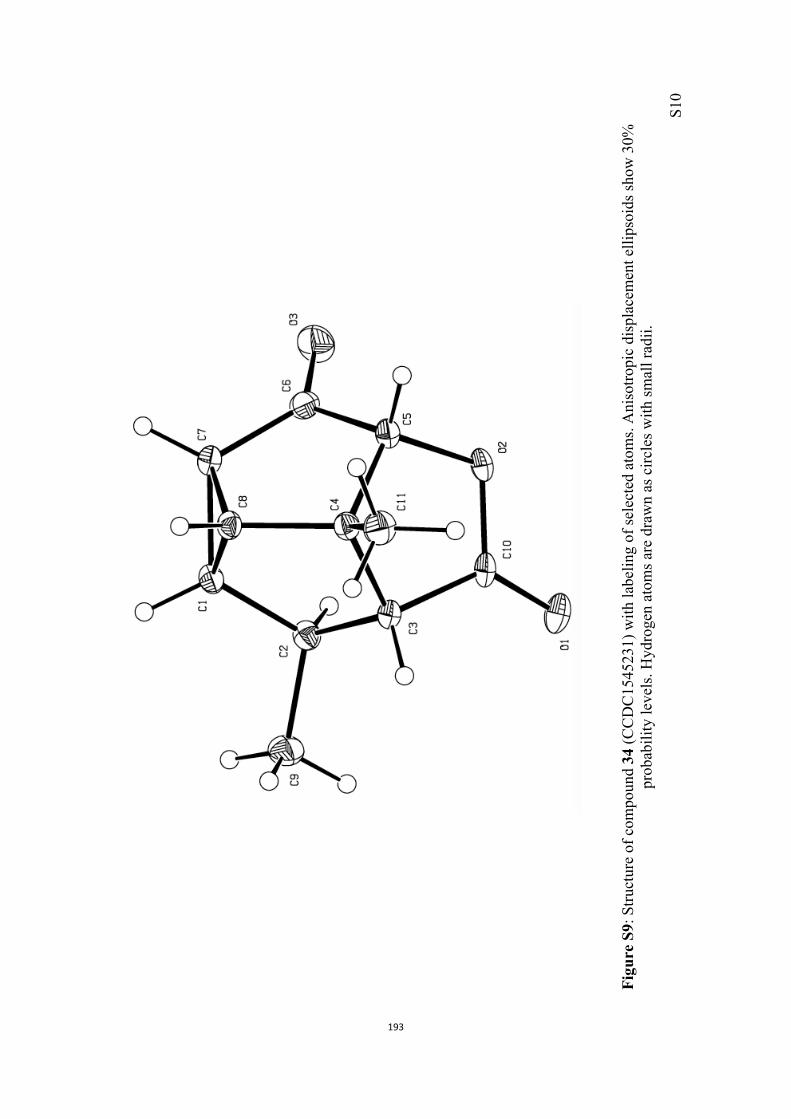
S10
Figu
re S
9: S
truct
ure
of c
ompo
und
34 (C
CD
C15
4523
1) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
193
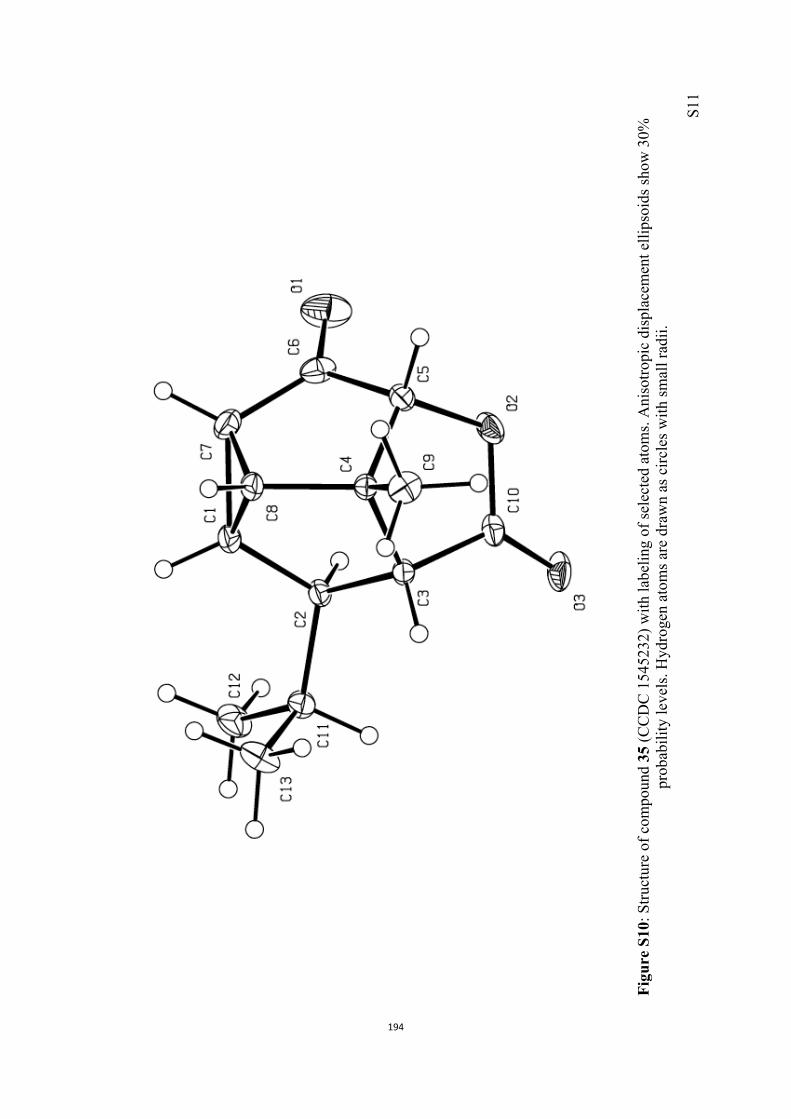
S11
Figu
re S
10: S
truct
ure
of c
ompo
und
35 (C
CD
C 1
5452
32) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
194
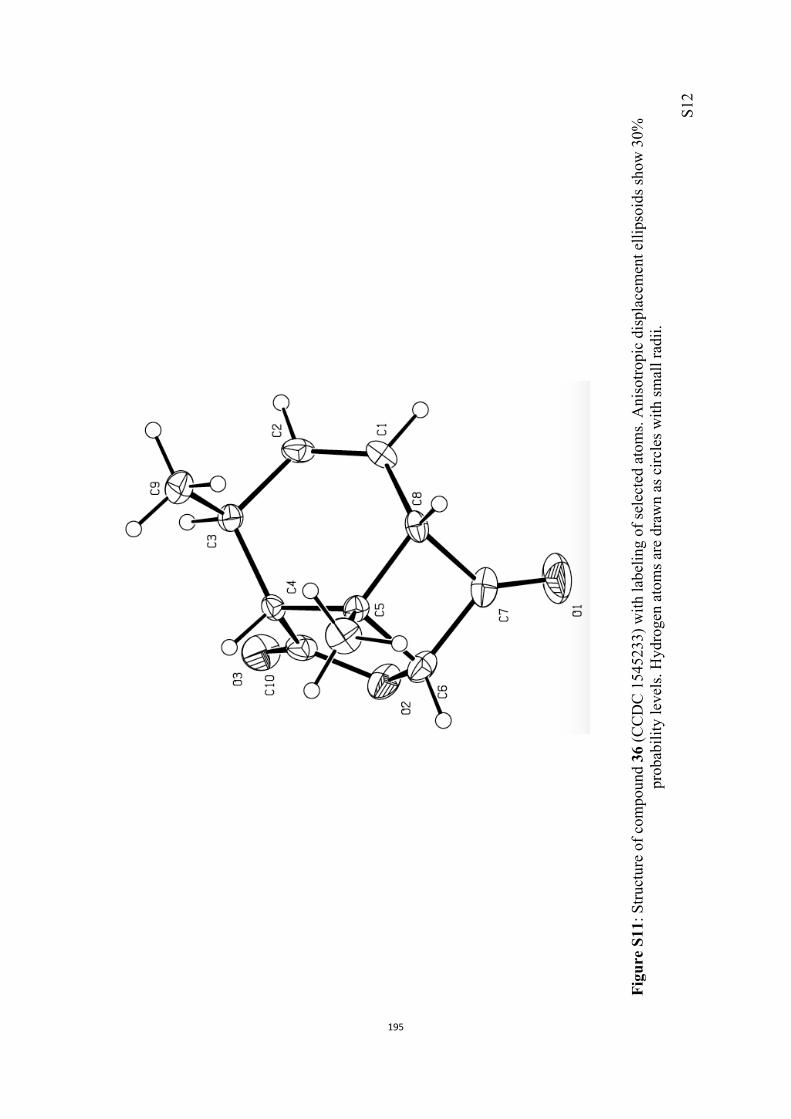
S12
Figu
re S
11: S
truct
ure
of c
ompo
und
36 (C
CD
C 1
5452
33) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
195
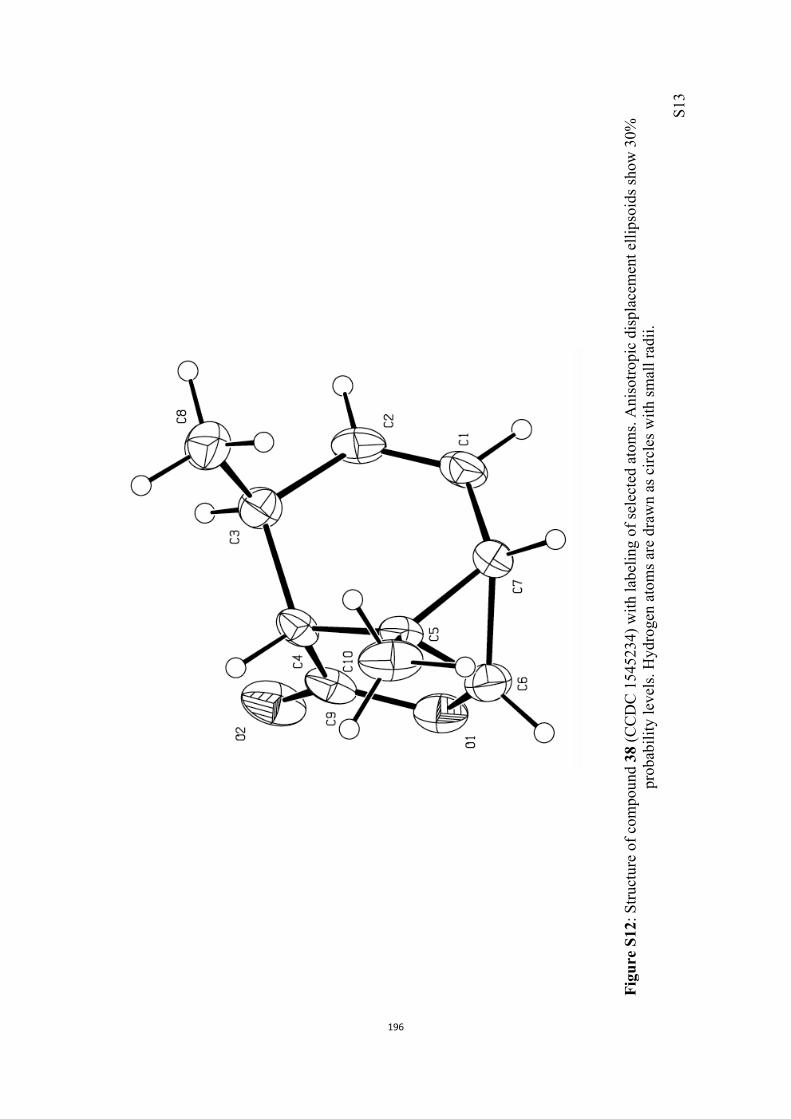
S13
Figu
re S
12: S
truct
ure
of c
ompo
und
38 (C
CD
C 1
5452
34) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
196
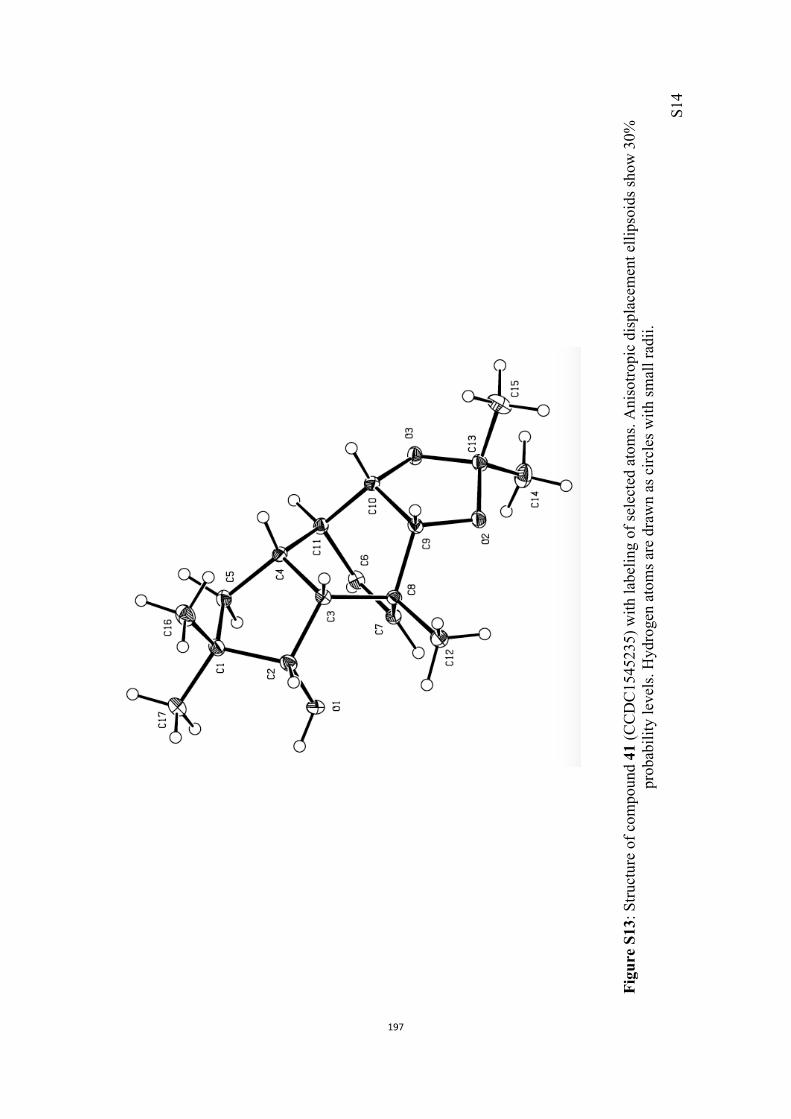
S14
Figu
re S
13: S
truct
ure
of c
ompo
und
41 (C
CD
C15
4523
5) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
197
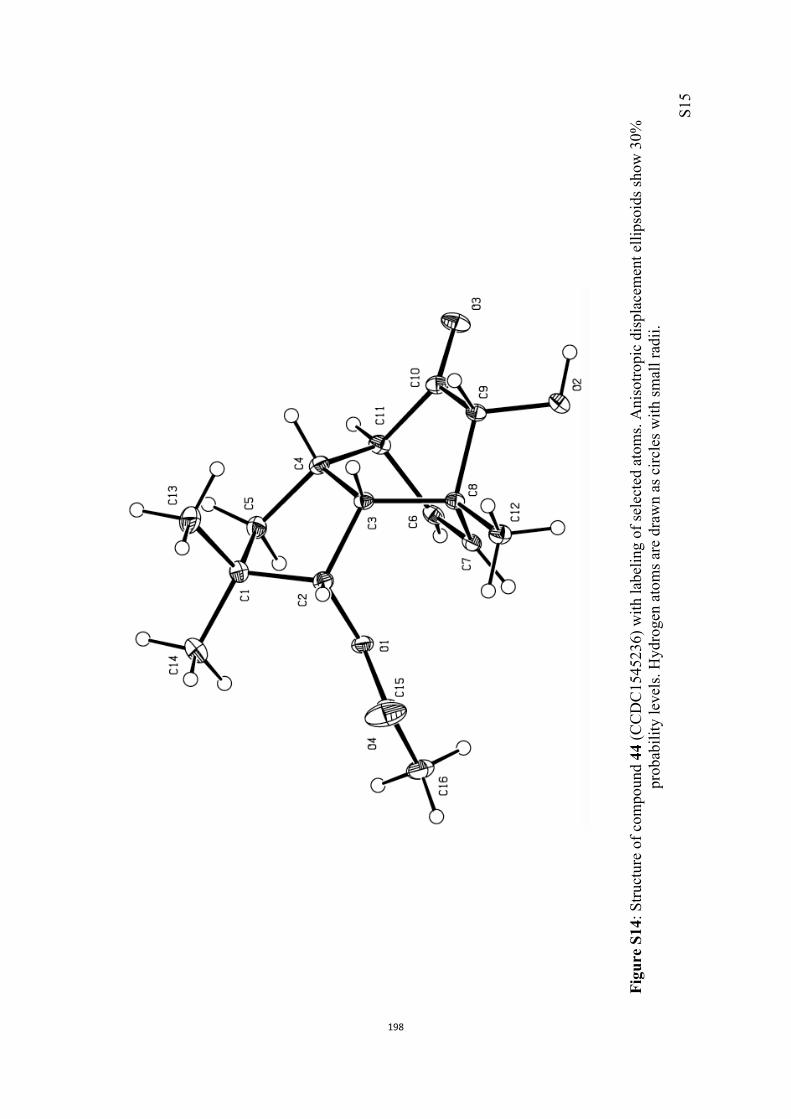
S15
Figu
re S
14: S
truct
ure
of c
ompo
und
44 (C
CD
C15
4523
6) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
198
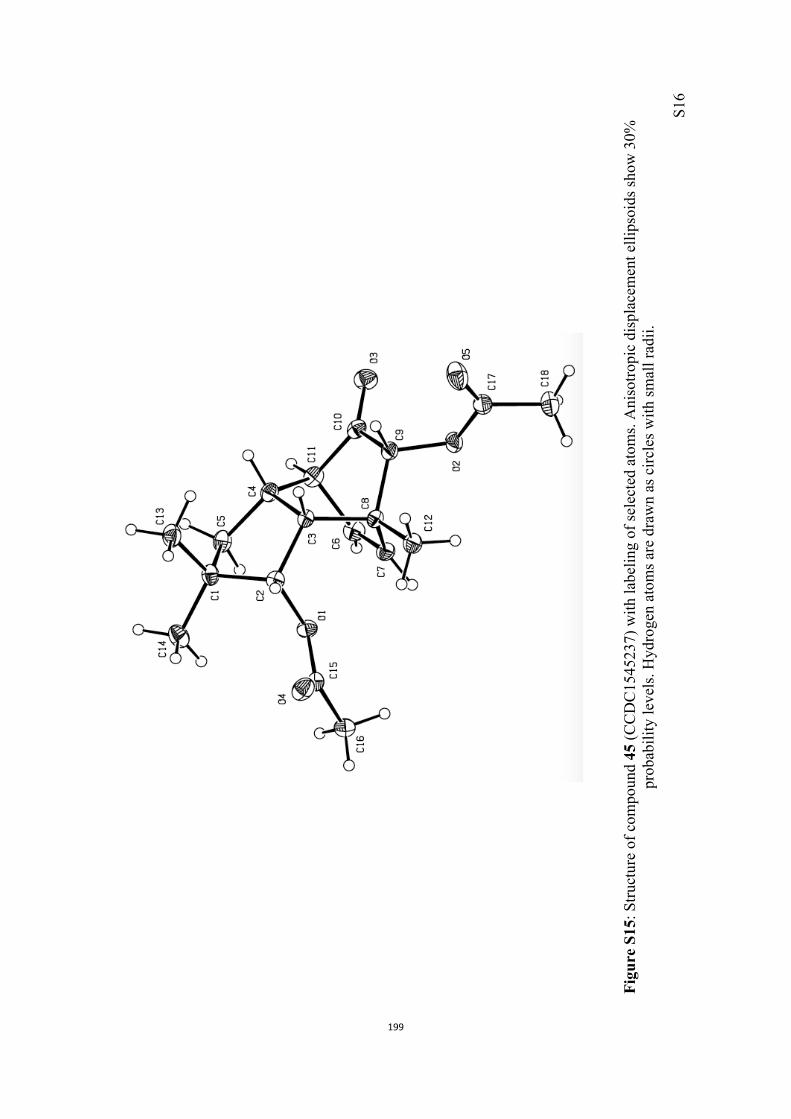
S16
Figu
re S
15: S
truct
ure
of c
ompo
und
45 (C
CD
C15
4523
7) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
199
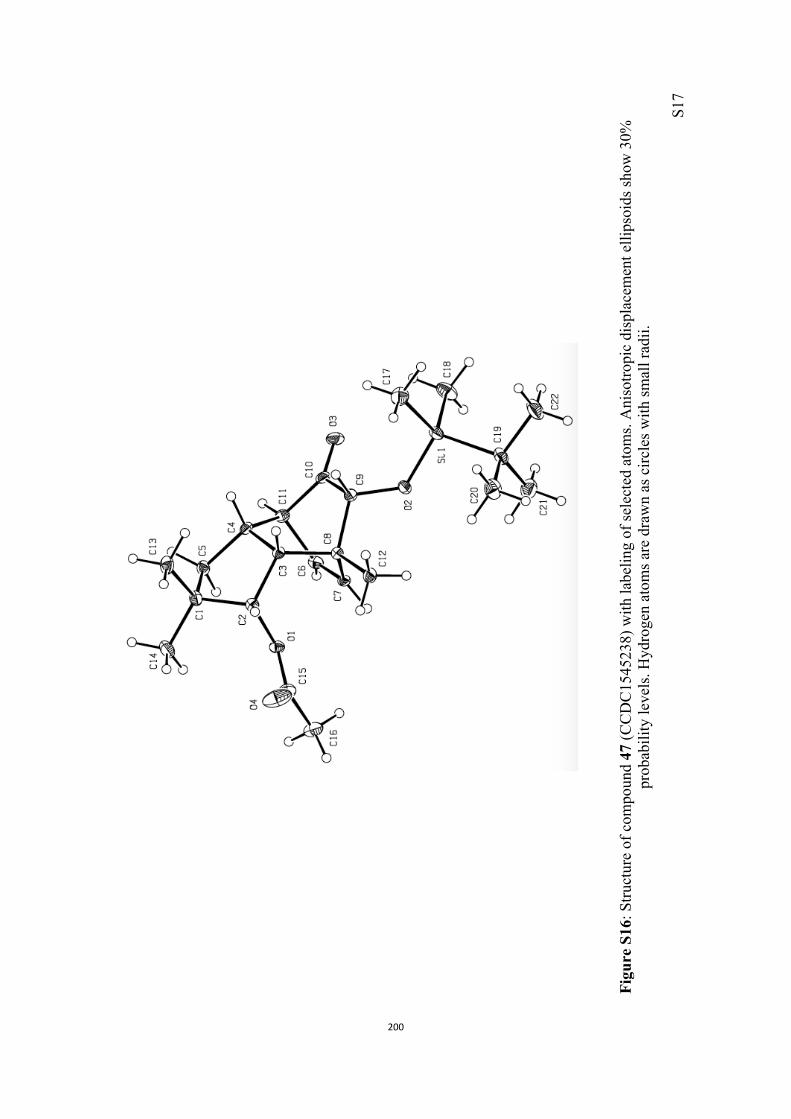
S17
Figu
re S
16: S
truct
ure
of c
ompo
und
47 (C
CD
C15
4523
8) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
200

S18
Figu
re S
17: S
truct
ure
of c
ompo
und
48 (C
CD
C 1
5452
39) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
201
S19
Figu
re S
18: S
truct
ure
of c
ompo
und
53 (C
CD
C15
4524
0) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
202
S20
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.02
1.001.031.001.001.00
1.00
1.00
3.31 CD3OD_SPE
203
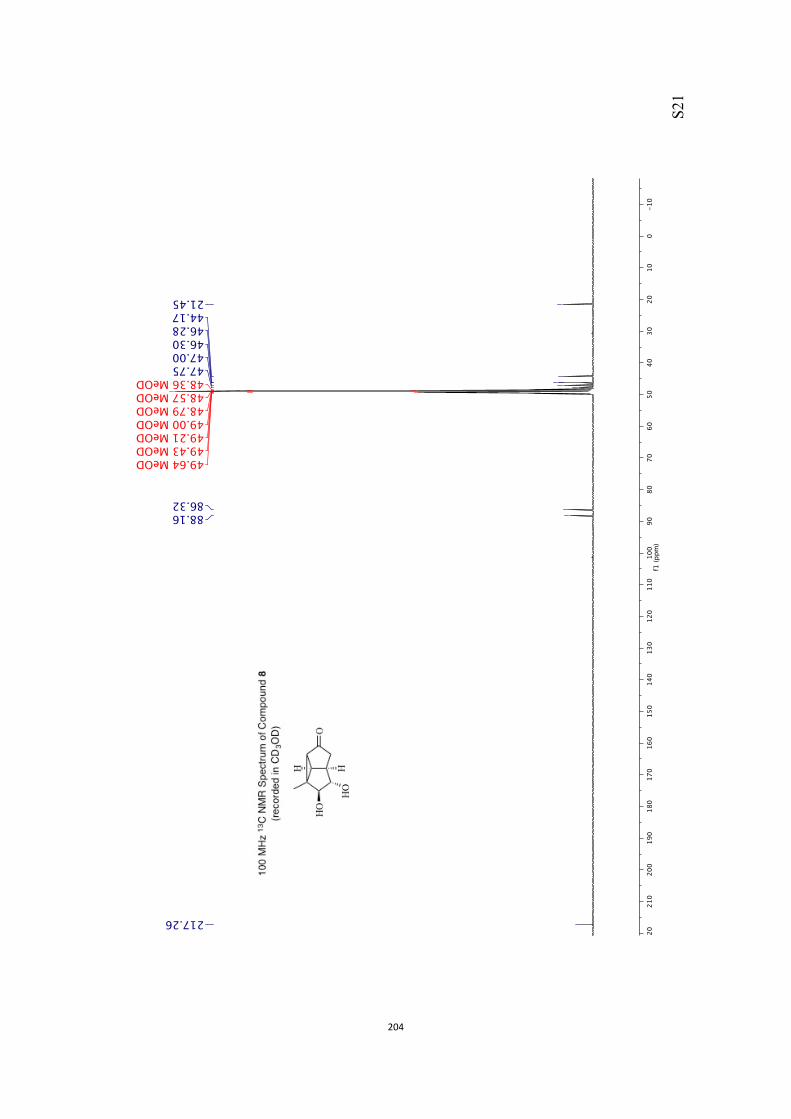
S21
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
f1 (
ppm
)
21.4544.1746.2846.3047.0047.7548.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.43 MeOD49.64 MeOD
86.3288.16
217.26
204
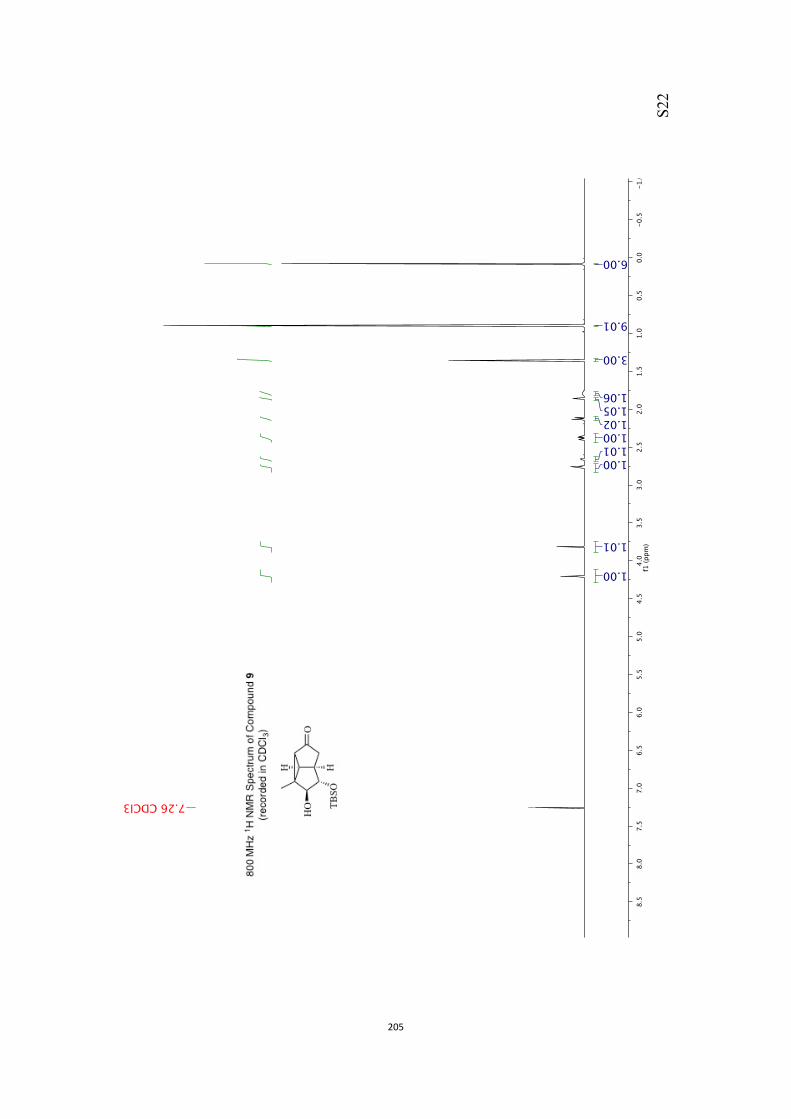
S22
-1
.0-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.5f1
(ppm
)
6.00
9.01
3.00
1.061.051.021.001.011.00
1.01
1.00
7.26 CDCl3
205
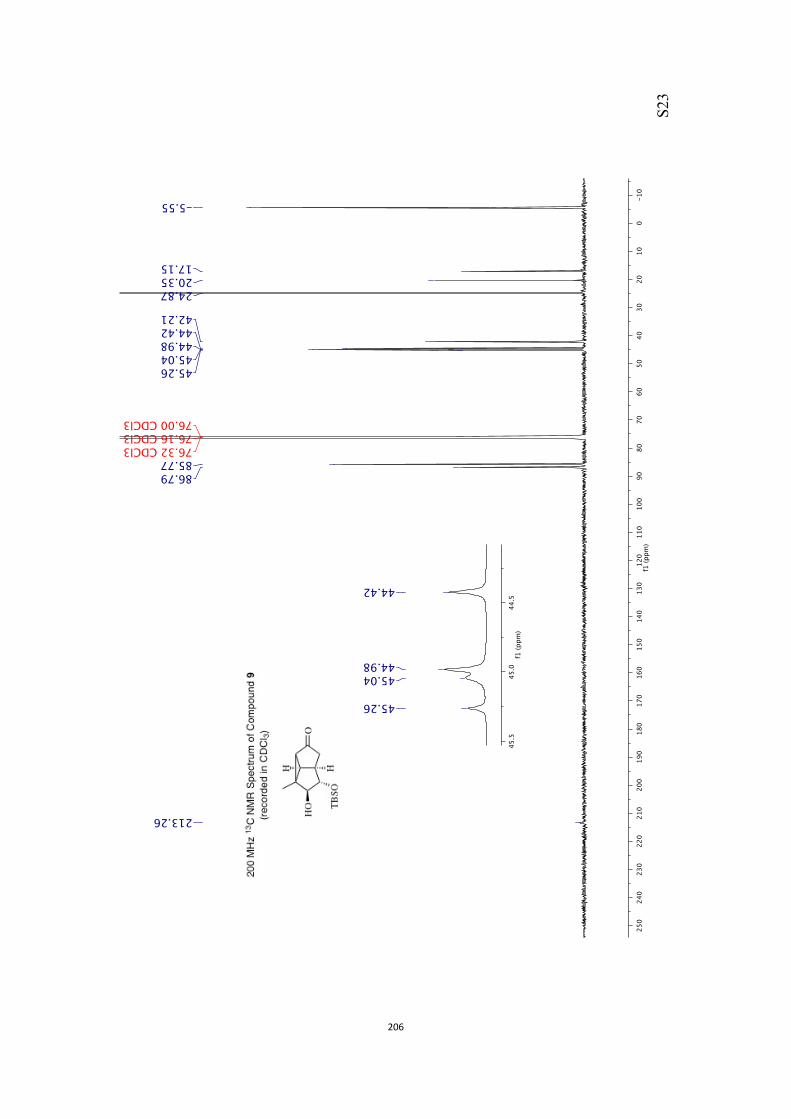
S23
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
02
30
24
02
50
f1 (
ppm
)
-5.55
17.1520.3524.87
42.2144.4244.9845.0445.26
76.00 CDCl376.16 CDCl376.32 CDCl385.7786.79
213.26
44
.54
5.0
45
.5f1
(ppm
)44.42
44.9845.04
45.26
206
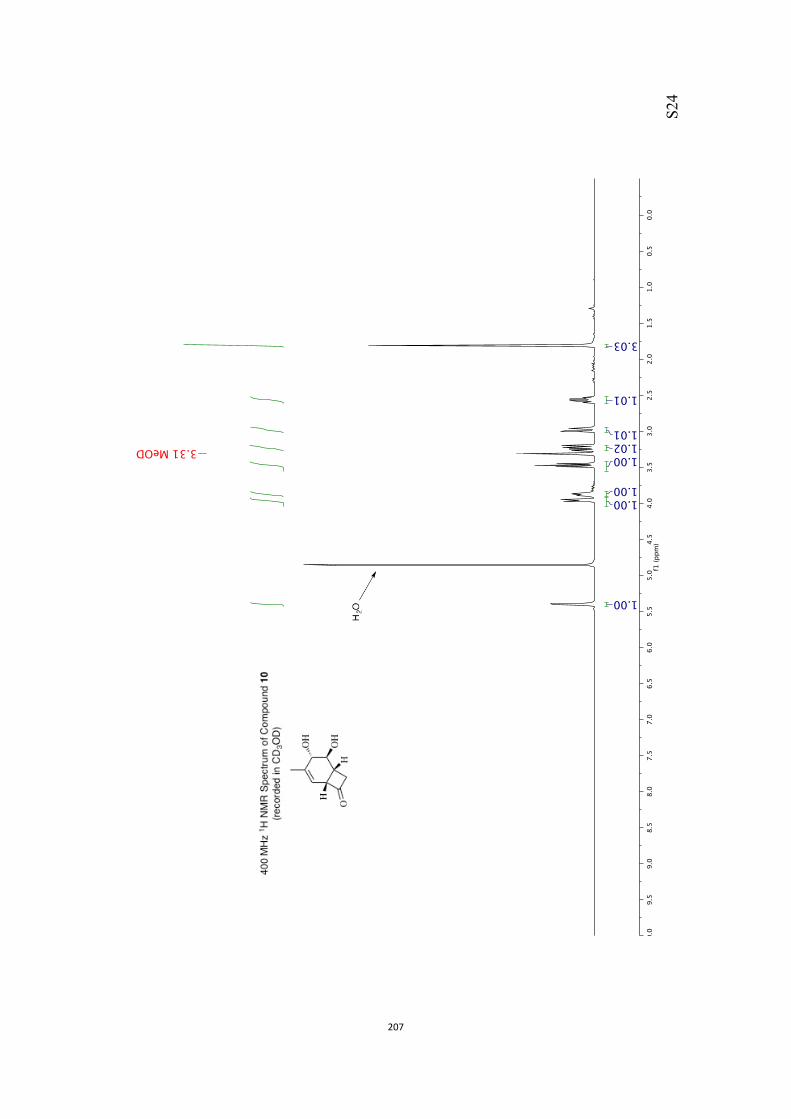
S24
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.03
1.01
1.011.021.00
1.001.00
1.00
3.31 MeOD
207
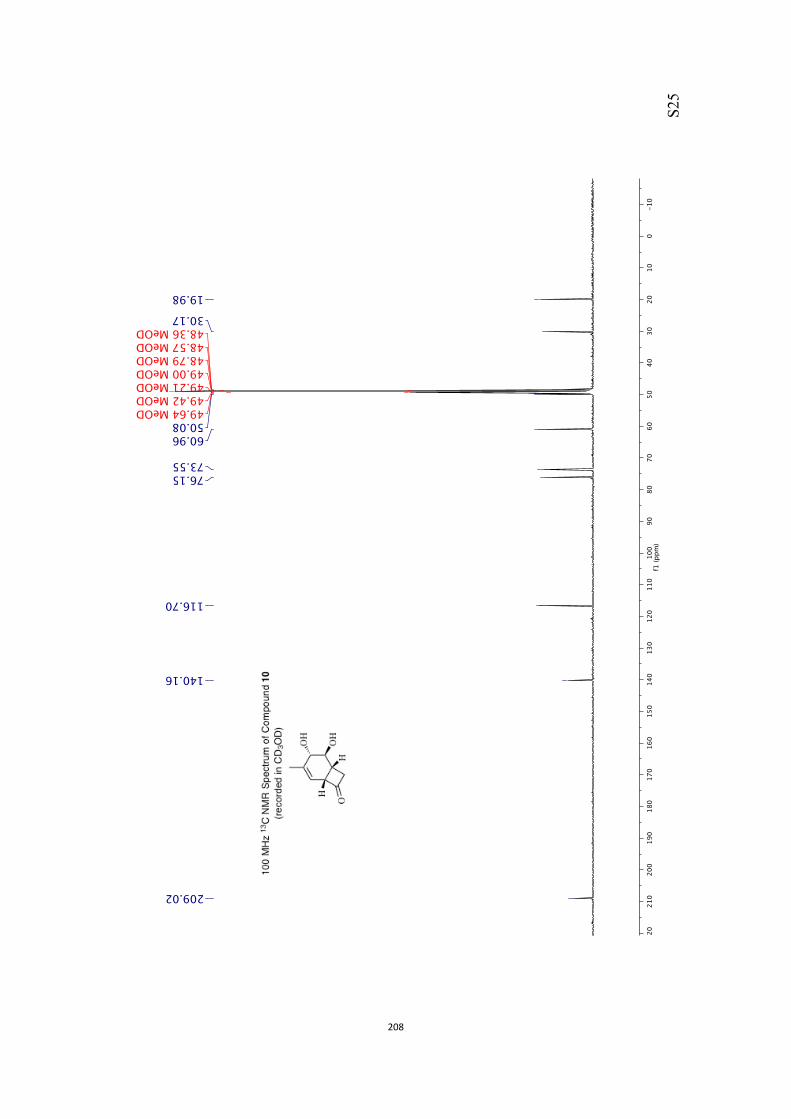
S25
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
f1 (
ppm
)
19.98
30.1748.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.42 MeOD49.64 MeOD50.0860.96
73.5576.15
116.70
140.16
209.02
208

S26
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.00
9.00
3.00
1.001.001.001.00
1.001.001.00
1.00
7.26 CDCl3
209
S27
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.22-3.82
18.2519.1325.9429.89
50.18
60.62
73.3676.84 CDCl377.16 CDCl377.48 CDCl377.86
115.06
138.35
207.02
-4
.5-4
.0-3
.5f1
(ppm
)
-4.22
-3.82
210
S28
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
1.00
1.00
2.003.012.00
2.01
1.00
7.26 CDCl3
211
S29
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
10.9518.7019.6021.35
70.1173.3376.84 CDCl377.16 CDCl377.48 CDCl3
127.84130.34
212
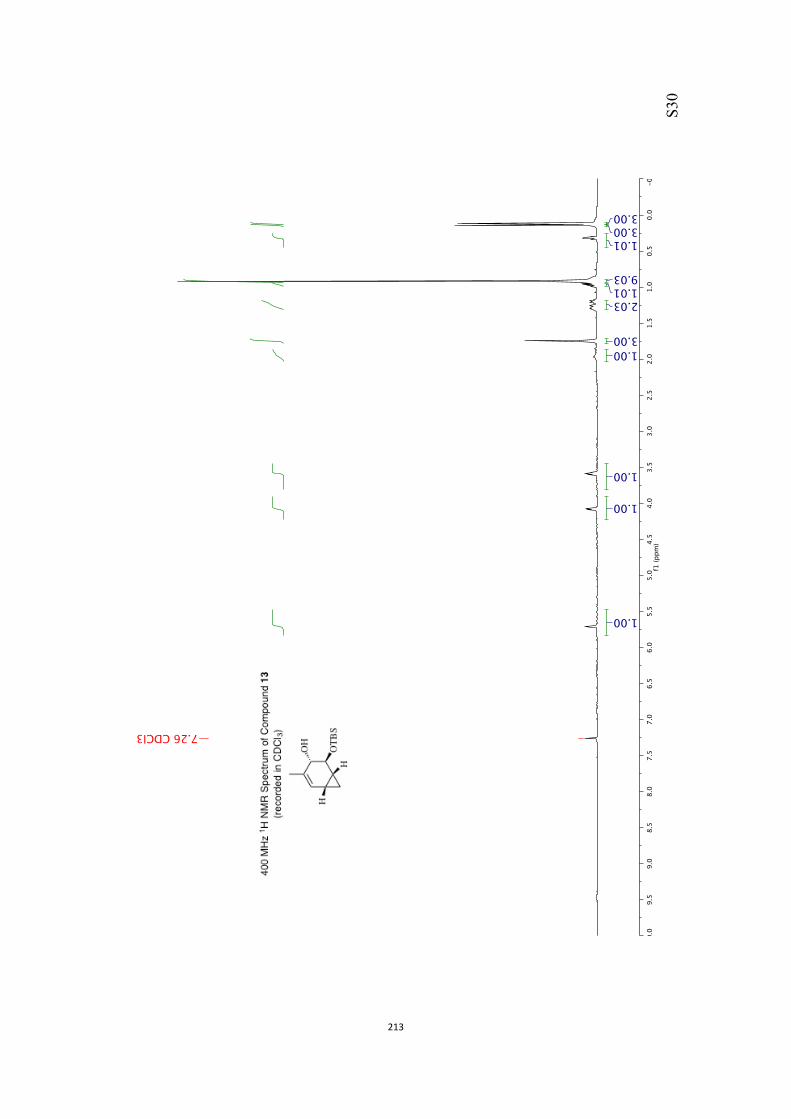
S30
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.003.001.01
9.031.012.03
3.00
1.00
1.00
1.00
1.00
7.26 CDCl3
213
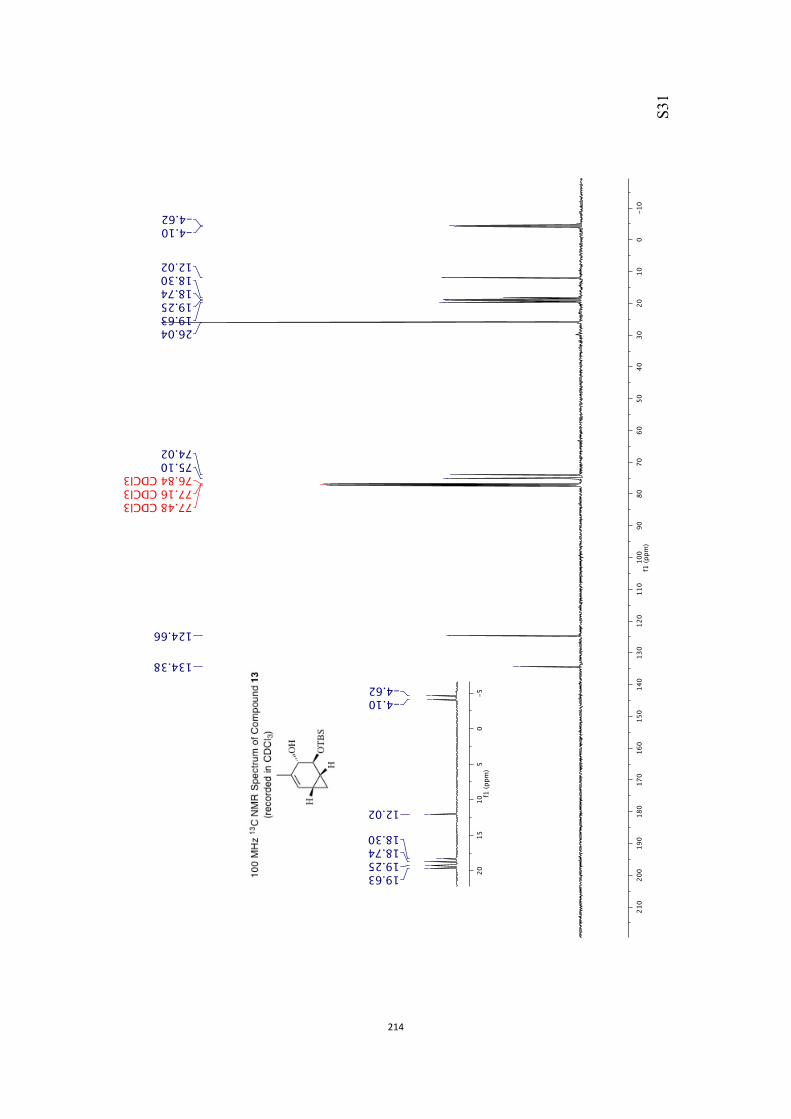
S31
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-4.62-4.10
12.0218.3018.7419.2519.6326.04
74.0275.1076.84 CDCl377.16 CDCl377.48 CDCl3
124.66
134.38
-5
05
10
15
20
f1 (
ppm
)
-4.62-4.10
12.02
18.3018.7419.2519.63
214
S32
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
9.09
2.004.001.03
2.01
7.26 CDCl3
215
S33
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.1718.1919.77
68.0369.7076.84 CDCl377.16 CDCl377.48 CDCl3
121.91122.40122.55123.01126.72134.79
145.37145.61
165.89166.09
**
*
* i
mpuri
ty
16
5.8
16
6.0
16
6.2
f1 (
ppm
)165.89
166.09
12
21
23
f1 (
ppm
)
121.91122.40122.55123.01
216
S34
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
12.01
3.08
2.05
2.013.011.001.00
2.00
7.26 CDCl3
217
S35
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.7721.2221.2631.0331.06
68.0369.5276.84 CDCl377.16 CDCl377.48 CDCl3
118.14118.32121.93123.01126.56134.65
156.22156.49
166.25166.50
31
.03
1.1 f1
(ppm
)31.0331.06
21
.12
1.2
21
.32
1.4
f1 (
ppm
)21.2221.26
16
01
65
f1 (
ppm
)
156.22156.49
166.25166.50
11
8.0
11
8.2
11
8.4 f1
(ppm
)
118.14
118.32
218
S36
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.001.003.00
1.00
1.00
1.00
1.00
2.00
1.00
1.00
7.26 CDCl3
219
S37
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.2419.8420.89
33.5141.8745.3552.17
70.5576.84 CDCl376.9977.16 CDCl377.48 CDCl3
122.08
131.86133.75
145.97
165.86
179.27
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl376.9977.16 CDCl3
77.48 CDCl3
220
S38
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.013.003.001.013.00
2.00
1.00
1.00
0.981.00
2.00
1.00
1.00
7.26 CDCl3
221
S39
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.7120.8020.9421.2021.2630.5231.1438.7745.1346.8849.67
70.5676.84 CDCl377.16 CDCl377.2777.48 CDCl3
117.87
132.19133.09
156.82
166.50
179.26
76
.57
7.0
77
.57
8.0
f1 (
ppm
)
76.84 CDCl377.16 CDCl377.2777.48 CDCl3
20
.52
1.0
21
.5f1
(ppm
)
20.7120.8020.9421.2021.26
222
S40
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
3.04
3.03
3.011.001.00
1.00
1.00
1.00
1.00
2.00
0.98
7.26 CDCl3
223
S41
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
17.1218.2319.03
37.0240.7642.1546.89
72.9573.4076.84 CDCl377.16 CDCl377.48 CDCl3
122.05125.47
139.53
145.84
165.99
179.27
224
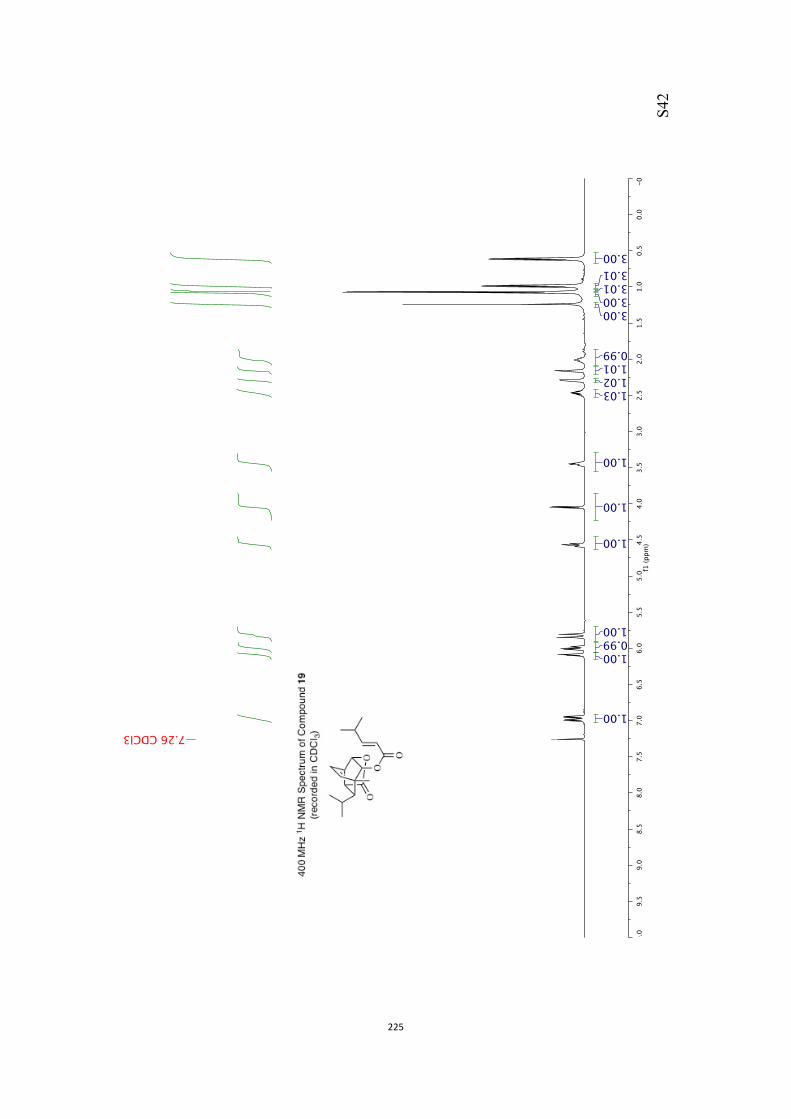
S42
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.00
3.013.013.003.00
0.991.011.021.03
1.00
1.00
1.00
1.000.991.00
1.00
7.26 CDCl3
225
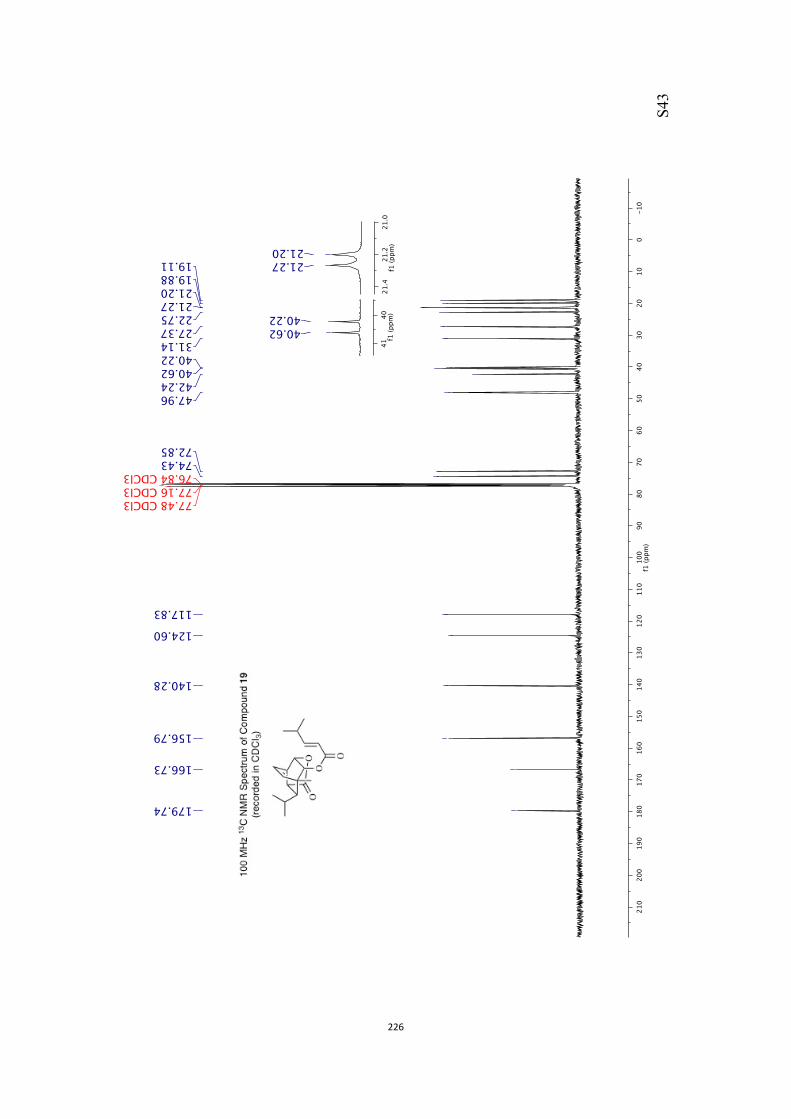
S43
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.1119.8821.2021.2722.7527.3731.1440.2240.6242.2447.96
72.8574.4376.84 CDCl377.16 CDCl377.48 CDCl3
117.83
124.60
140.28
156.79
166.73
179.74
40
41 f1
(ppm
)40.2240.62
21
.02
1.2
21
.4f1
(ppm
)21.2021.27
226
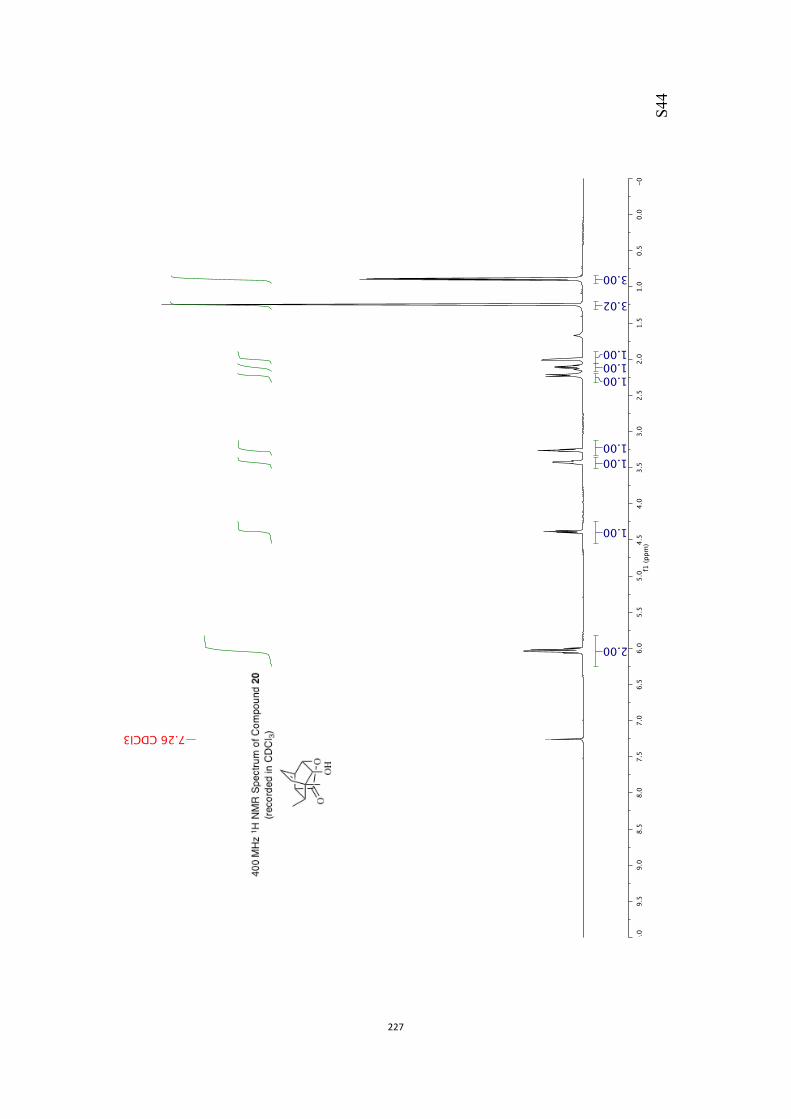
S44
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.00
3.02
1.001.001.00
1.00
1.00
1.00
2.00
7.26 CDCl3
227
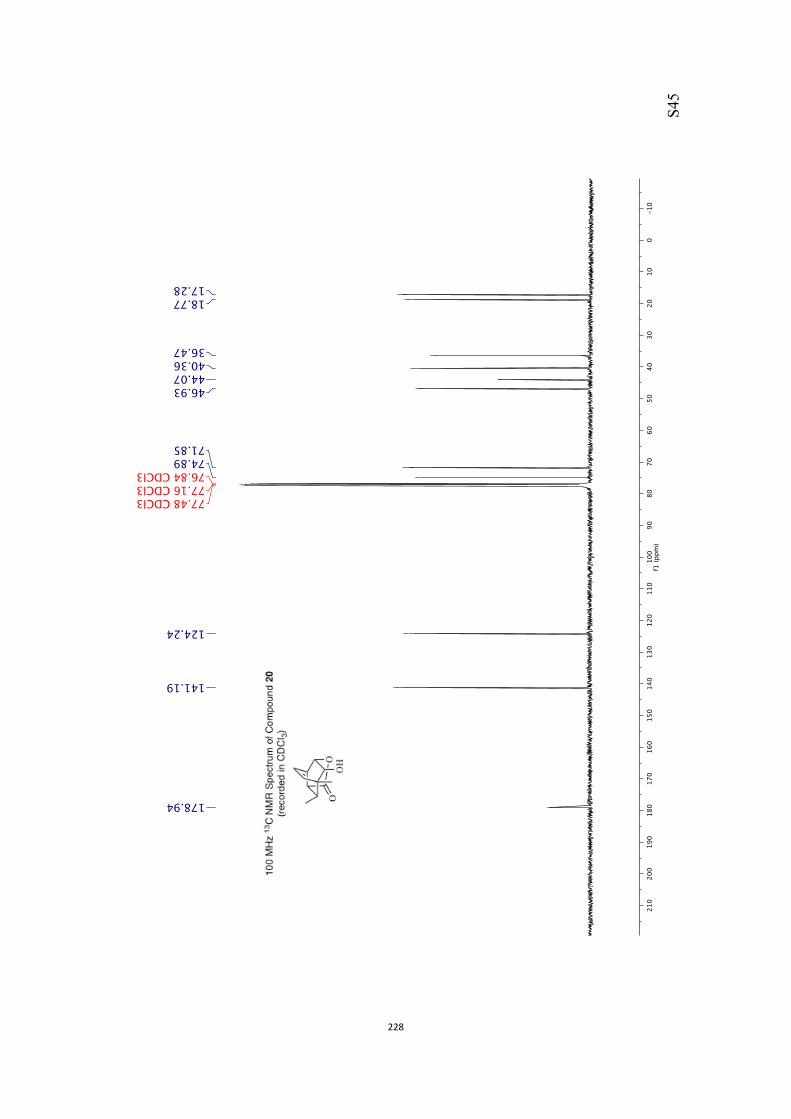
S45
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
17.2818.77
36.4740.3644.0746.93
71.8574.8976.84 CDCl377.16 CDCl377.48 CDCl3
124.24
141.19
178.94
228
S46
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.023.013.00
2.00
1.00
1.01
1.00
1.00
0.99
1.00
7.26 CDCl3
229
S47
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.8319.7722.7127.33
40.1740.2344.0347.58
72.9074.8476.84 CDCl377.16 CDCl377.48 CDCl3
123.32
141.84
179.28
40
.14
0.2
40
.3f1
(ppm
)
40.17
40.23
230
S48
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.05
3.03
1.00
1.00
1.01
0.97
1.00
1.00
7.26 CDCl3
231
S49
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.1616.14
40.9341.2547.1353.78
73.9476.84 CDCl377.16 CDCl377.48 CDCl3
128.31
136.74
177.81
201.54
40
.84
1.2
f1 (
ppm
)
40.93
41.25
232
S50
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.003.003.00
1.001.00
1.01
1.00
0.97
1.001.00
7.26 CDCl3
233
S51
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
15.3119.9622.7628.20
40.4440.89
51.7353.58
73.9076.84 CDCl377.16 CDCl377.48 CDCl3
127.28
136.75
178.45
200.65
234

S52
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.07
3.07
1.00
1.93
1.02
1.00
1.00
1.00
1.02
7.26 CDCl3
235
S53
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.0721.14
33.09
44.9845.1552.19
69.0276.84 CDCl377.16 CDCl377.48 CDCl379.01
130.77
135.35
178.98
44
.54
5.0
45
.5f1
(ppm
)
44.98
45.15
236
S54
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
f1 (
ppm
)
3.003.001.003.00
2.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
237
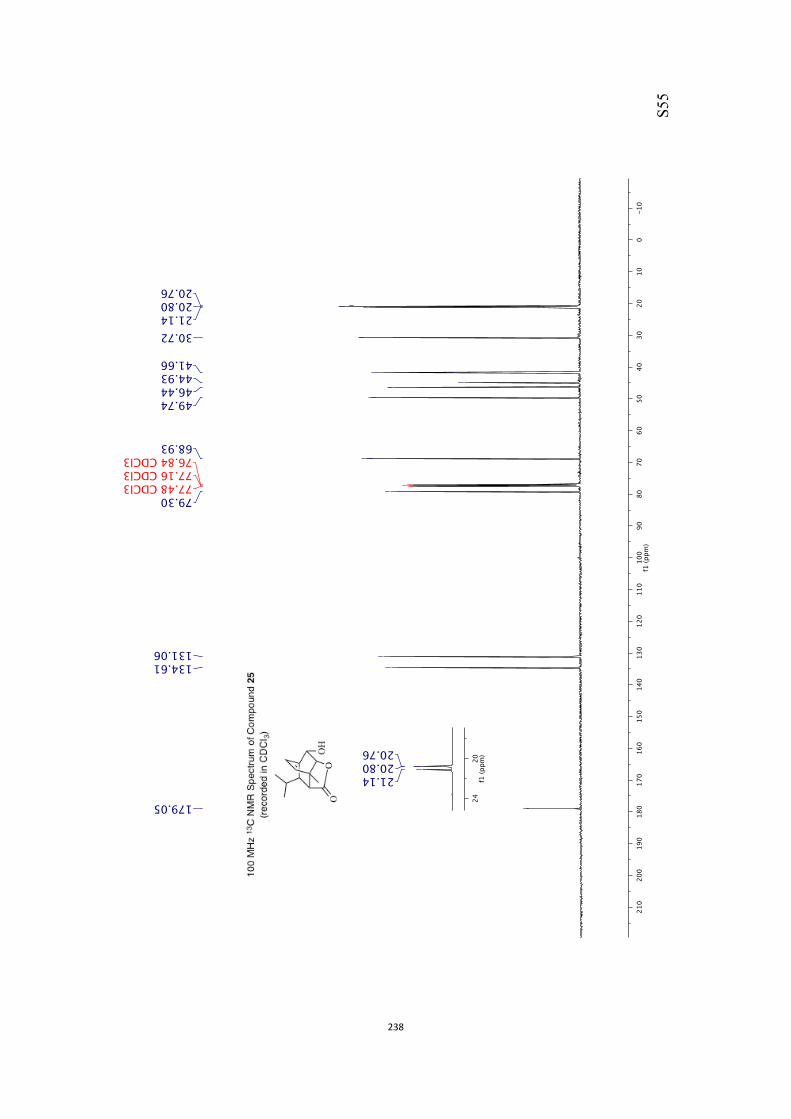
S55
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.7620.8021.14
30.72
41.6644.9346.4449.74
68.9376.84 CDCl377.16 CDCl377.48 CDCl379.30
131.06134.61
179.05
20
24
f1 (
ppm
)
20.7620.8021.14
238
S56
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.04
3.04
1.00
1.00
1.00
1.00
1.00
1.01
7.26 CDCl3
239
S57
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.3619.86
37.20
46.5051.9353.98
76.84 CDCl377.16 CDCl377.48 CDCl378.45
130.43134.54
177.67
199.95
240
S58
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.043.08
1.023.01
1.05
1.00
1.01
1.00
1.00
1.01
7.26 CDCl3
241
S59
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.9320.4420.63
29.21
46.2349.3850.2051.14
76.84 CDCl377.16 CDCl377.48 CDCl378.82
129.78
134.75
177.72
200.35
17
18
19
20
21
22
23
f1 (
ppm
)
19.9320.4420.63
242
S60
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.003.01
1.00
1.031.031.00
1.02
0.97
7.26 CDCl3
243
S61
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.3619.50
38.0644.7245.0045.1346.11
57.15
76.84 CDCl377.16 CDCl377.48 CDCl382.42
176.80
206.93
44
.54
5.0
45
.5f1
(ppm
)44.7245.0045.13
244
S62
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.023.013.00
1.021.011.001.00
1.01
1.02
1.00
7.26 CDCl3
245
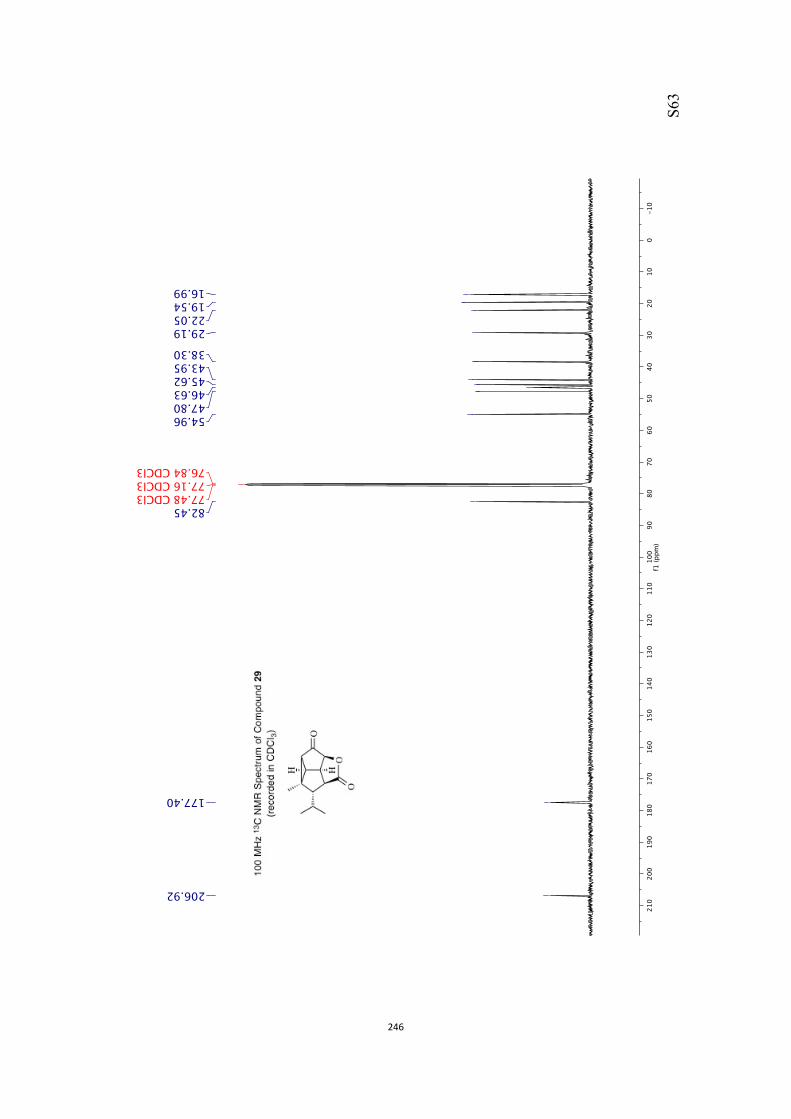
S63
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
16.9919.5422.0529.19
38.3043.9545.6246.6347.8054.96
76.84 CDCl377.16 CDCl377.48 CDCl382.45
177.40
206.92
246
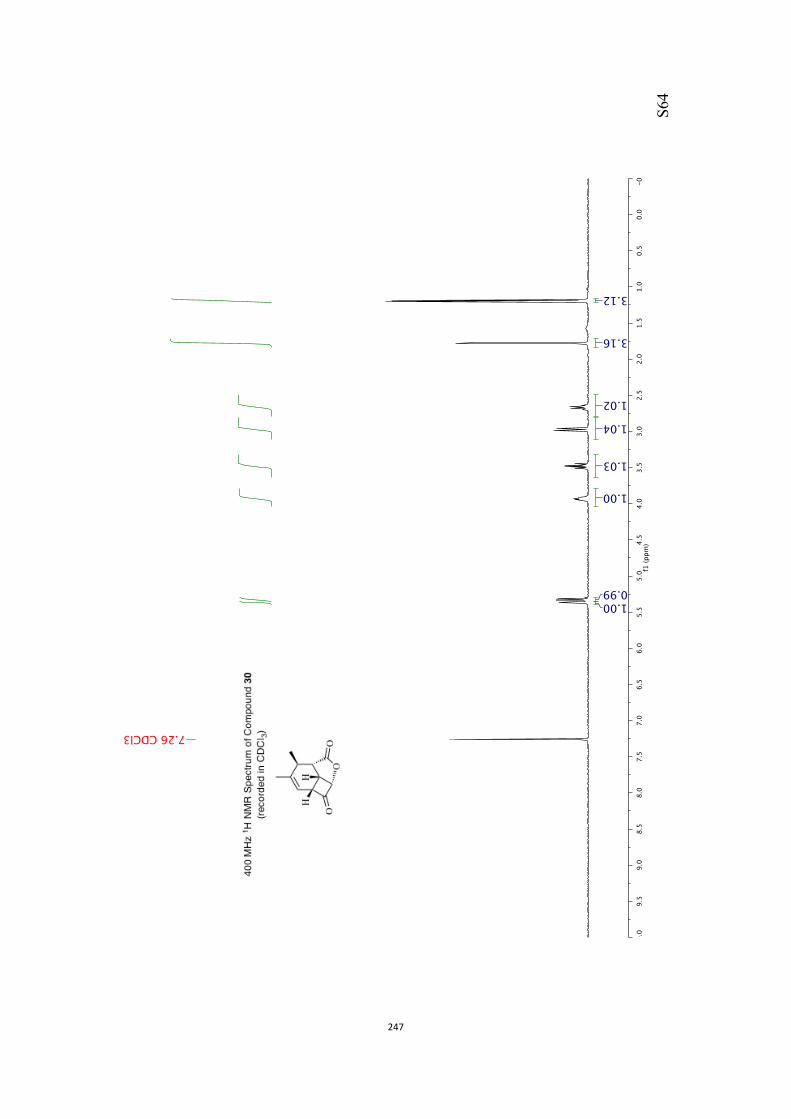
S64
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.12
3.16
1.02
1.04
1.03
1.00
0.991.00
7.26 CDCl3
247
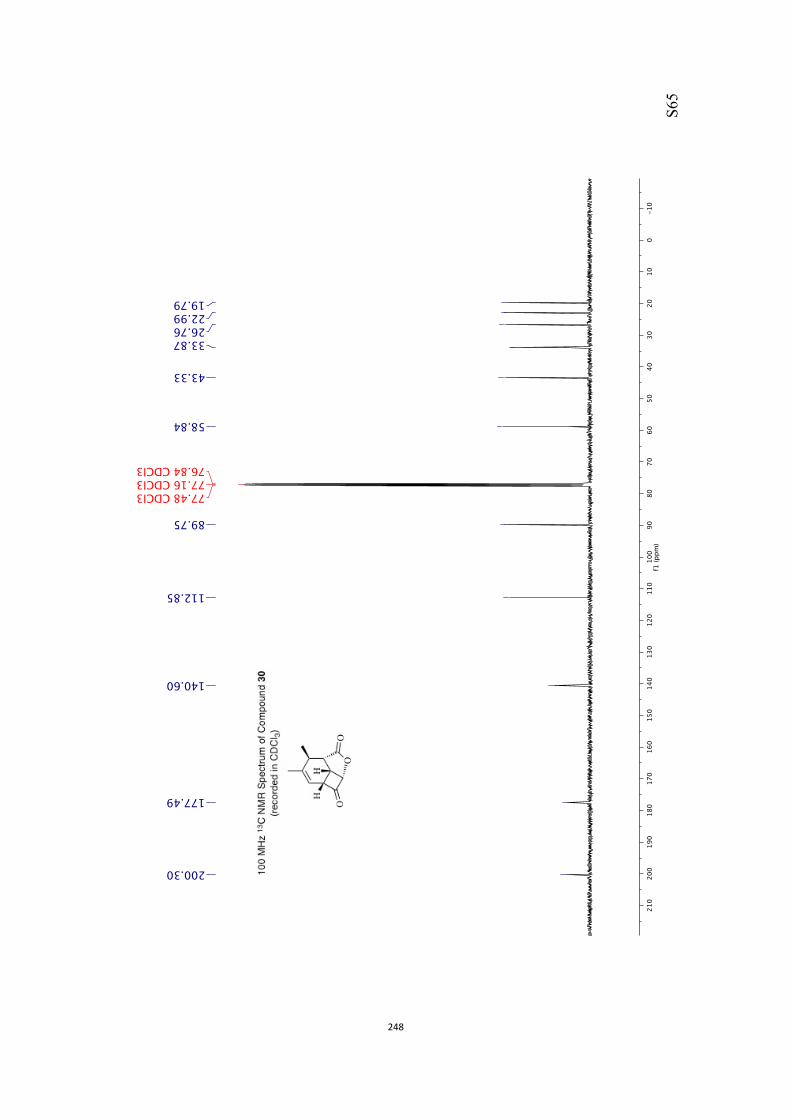
S65
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
19.7922.9926.7633.87
43.33
58.84
76.84 CDCl377.16 CDCl377.48 CDCl3
89.75
112.85
140.60
177.49
200.30
248
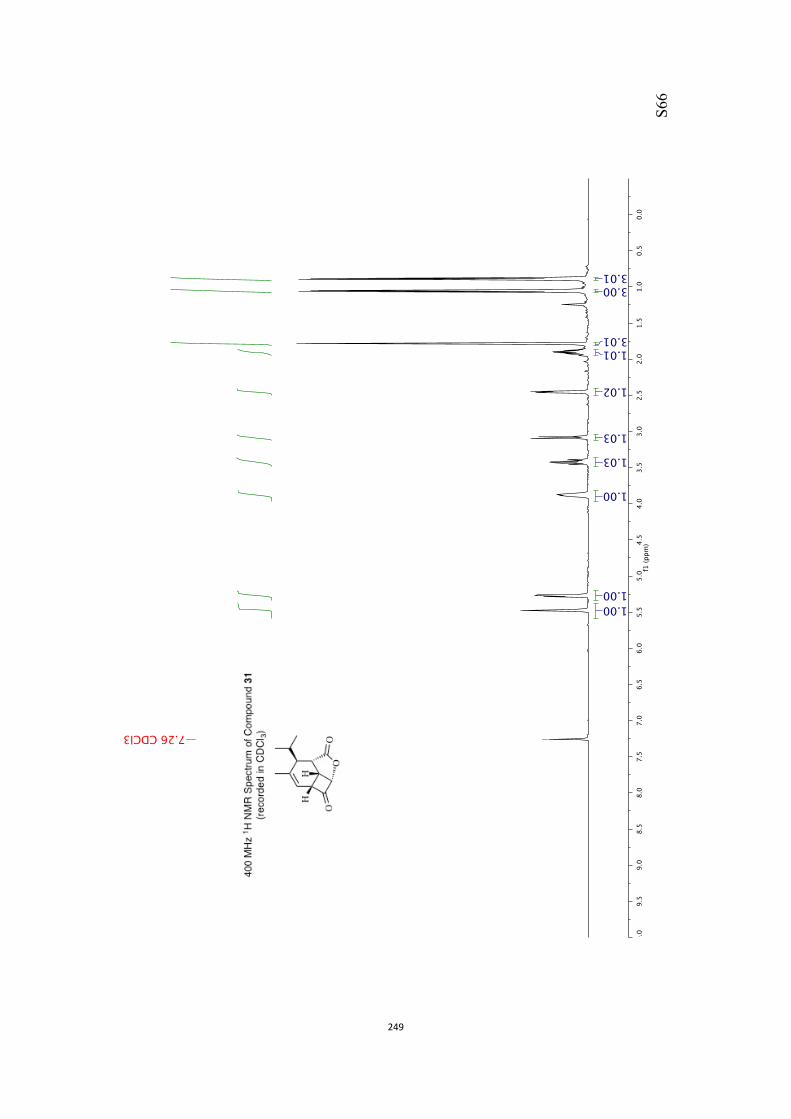
S66
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
3.013.00
3.011.01
1.02
1.03
1.03
1.00
1.00
1.00
7.26 CDCl3
249
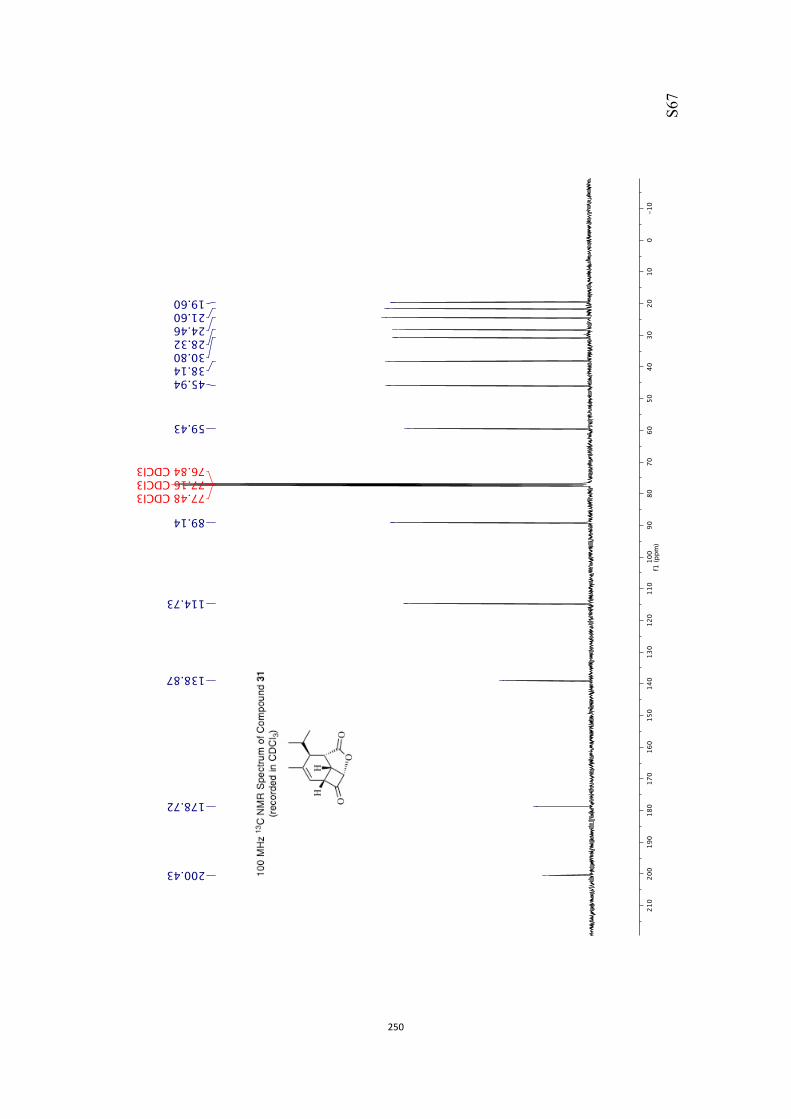
S67
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
19.6021.6024.4628.3230.8038.1445.94
59.43
76.84 CDCl377.16 CDCl377.48 CDCl3
89.14
114.73
138.87
178.72
200.43
250
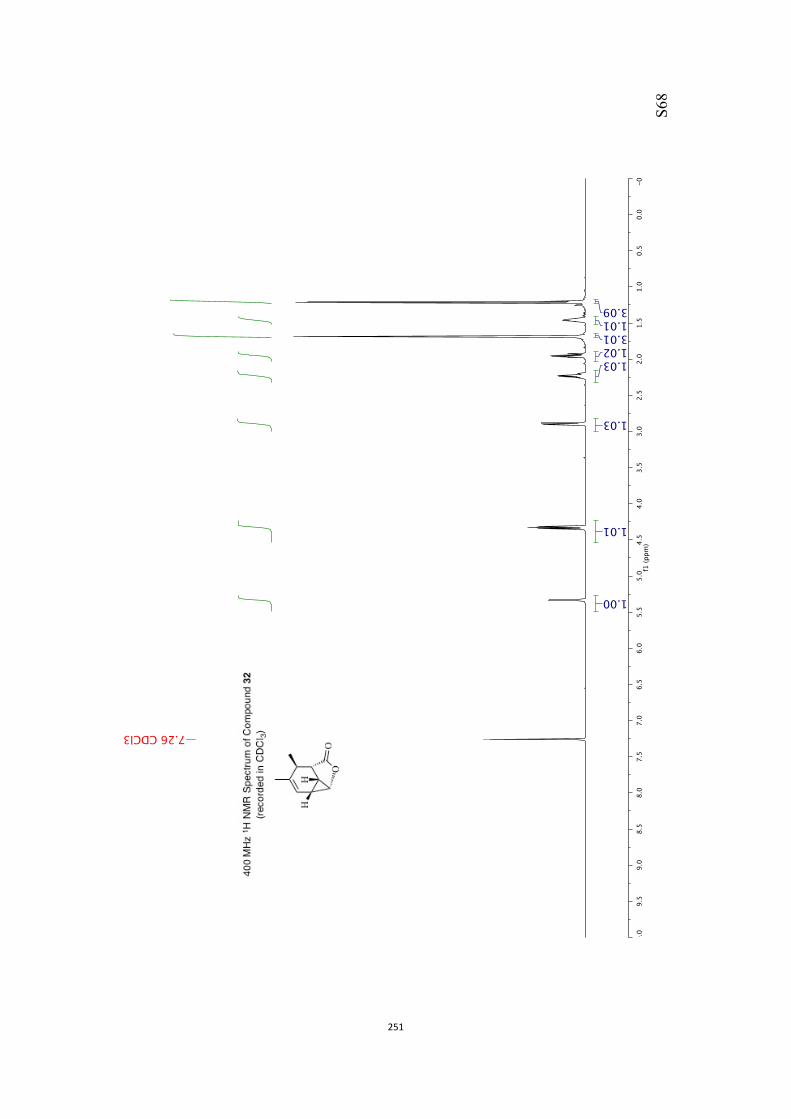
S68
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.091.013.011.021.03
1.03
1.01
1.00
7.26 CDCl3
251
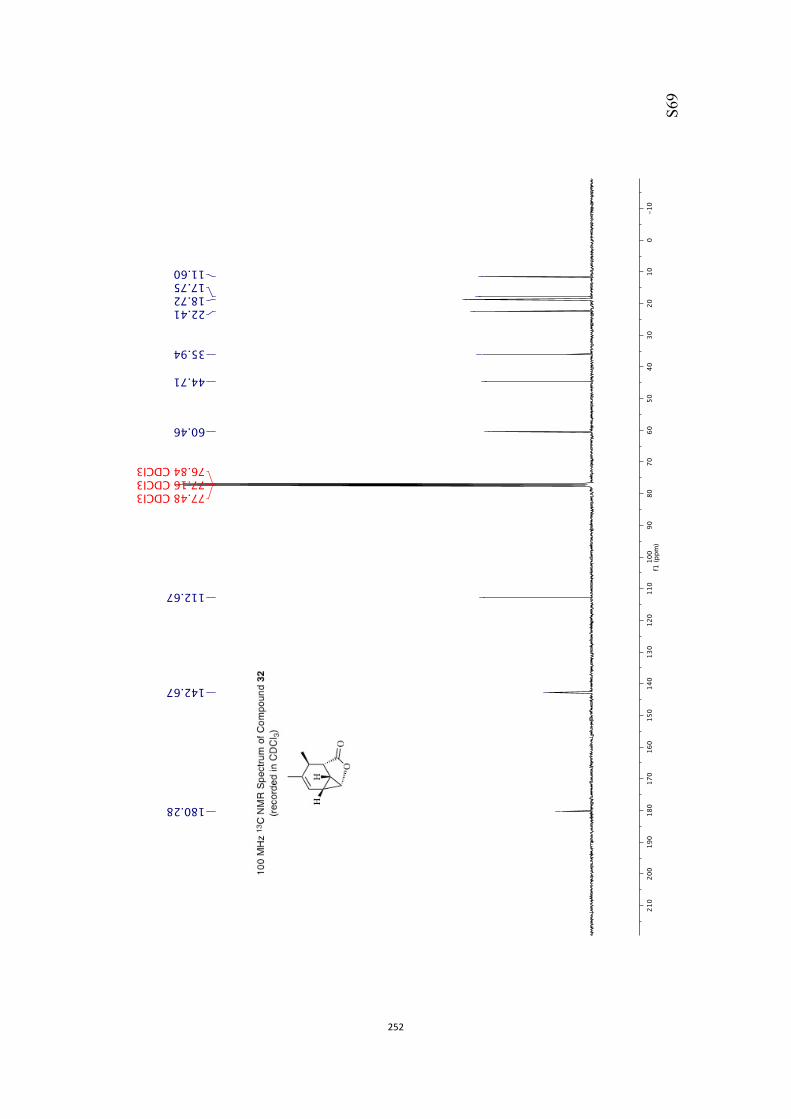
S69
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
11.6017.7518.7222.41
35.94
44.71
60.46
76.84 CDCl377.16 CDCl377.48 CDCl3
112.67
142.67
180.28
252
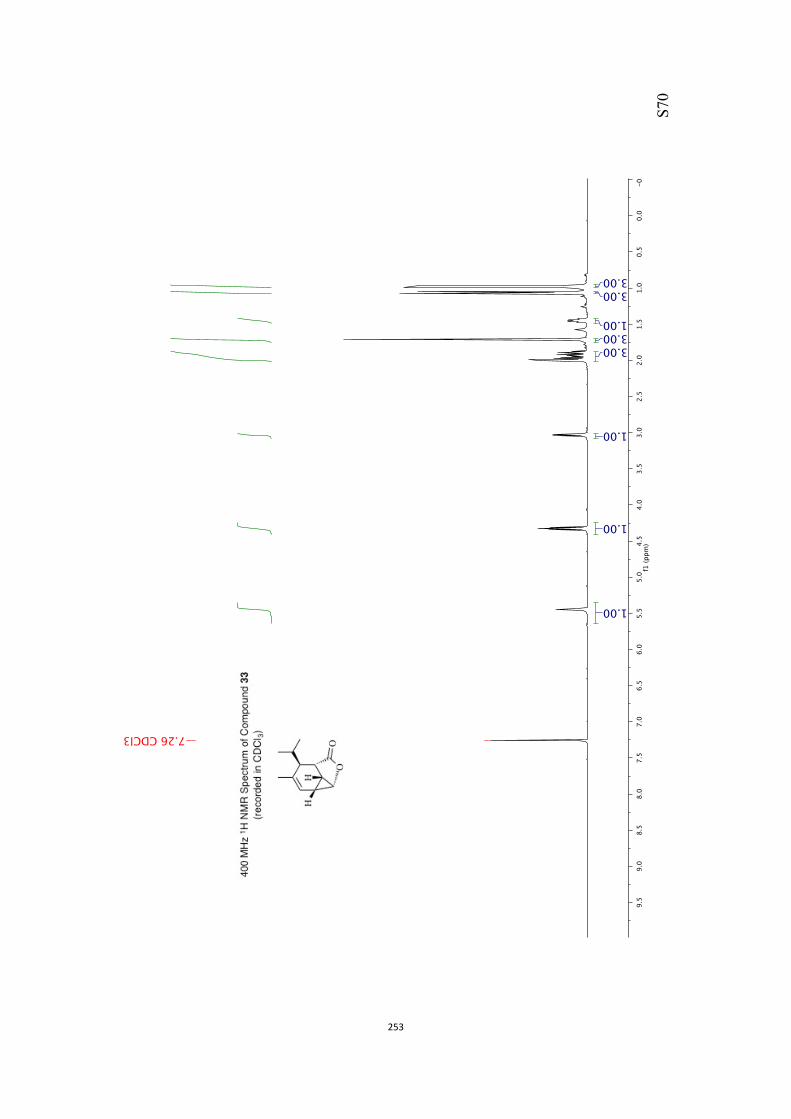
S70
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.003.00
1.003.003.00
1.00
1.00
1.00
7.26 CDCl3
253
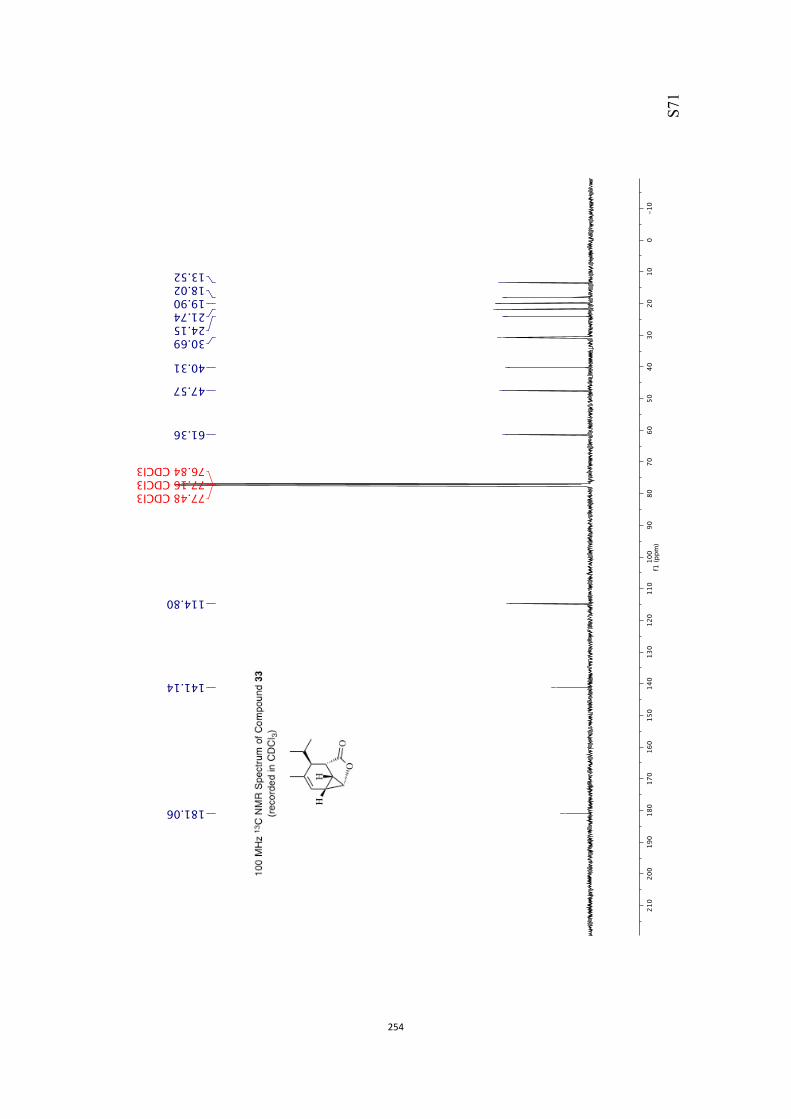
S71
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
13.5218.0219.9021.7424.1530.69
40.31
47.57
61.36
76.84 CDCl377.16 CDCl377.48 CDCl3
114.80
141.14
181.06
254
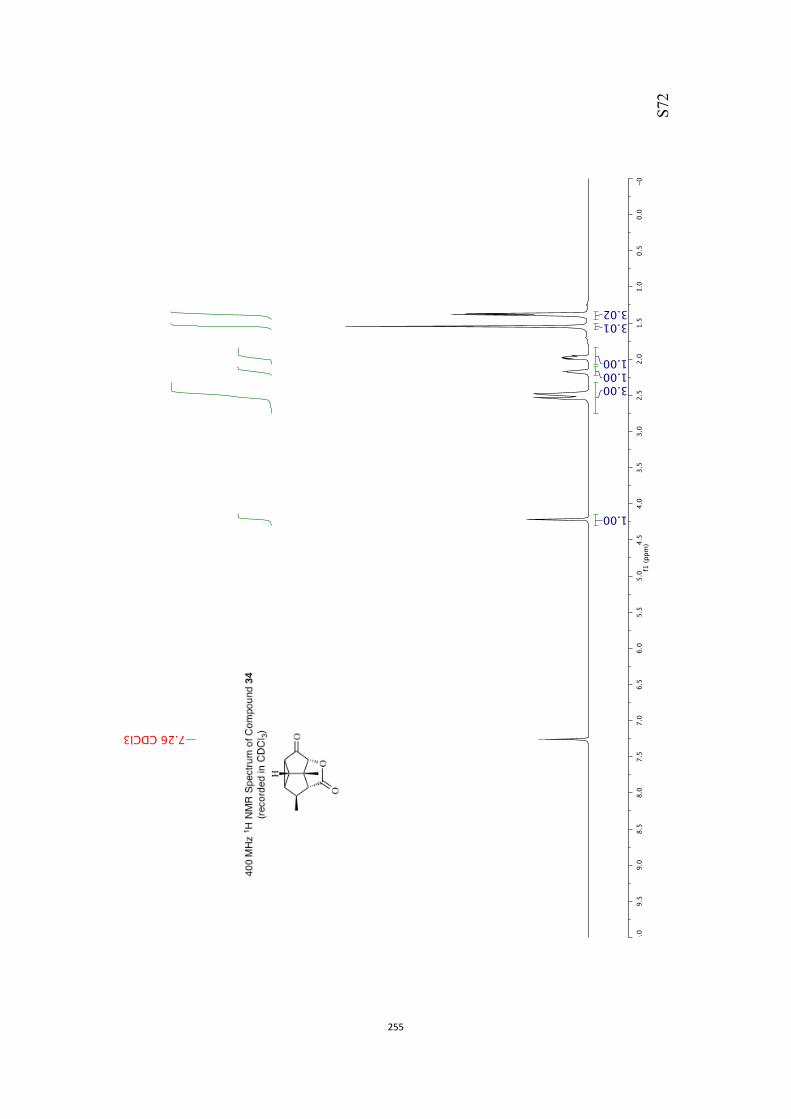
S72
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.023.01
1.001.003.00
1.00
7.26 CDCl3
255
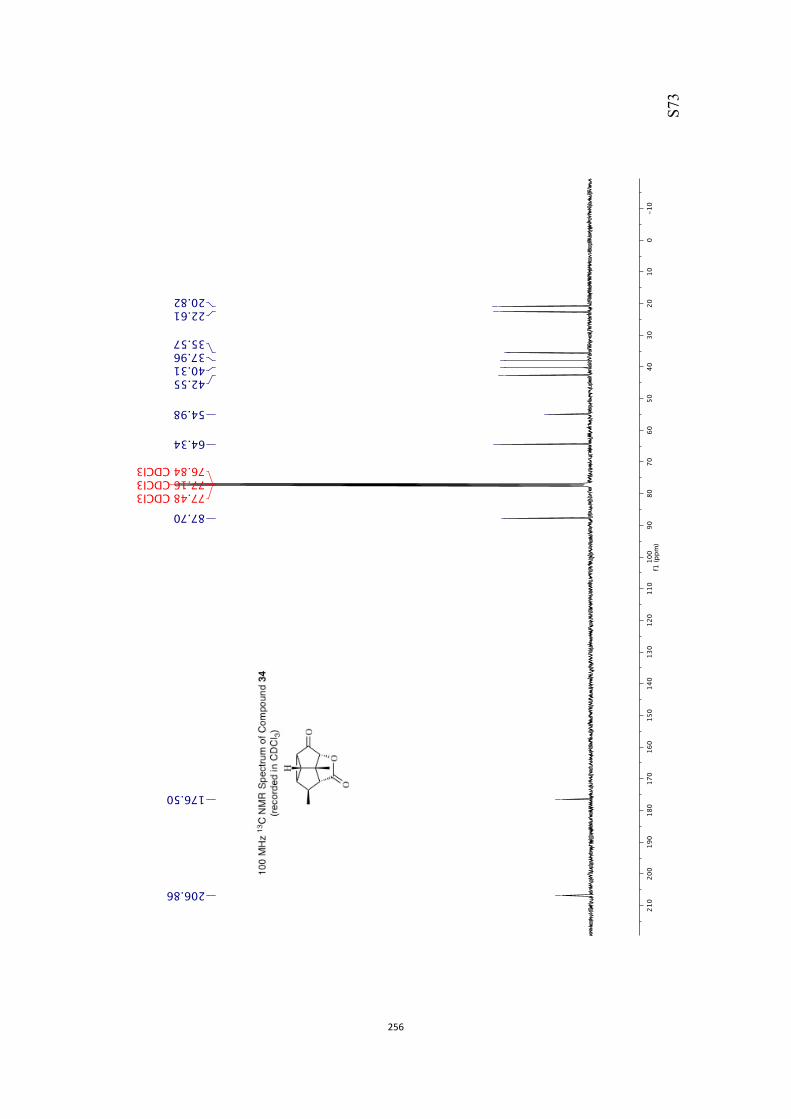
S73
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
20.8222.61
35.5737.9640.3142.55
54.98
64.34
76.84 CDCl377.16 CDCl377.48 CDCl3
87.70
176.50
206.86
256
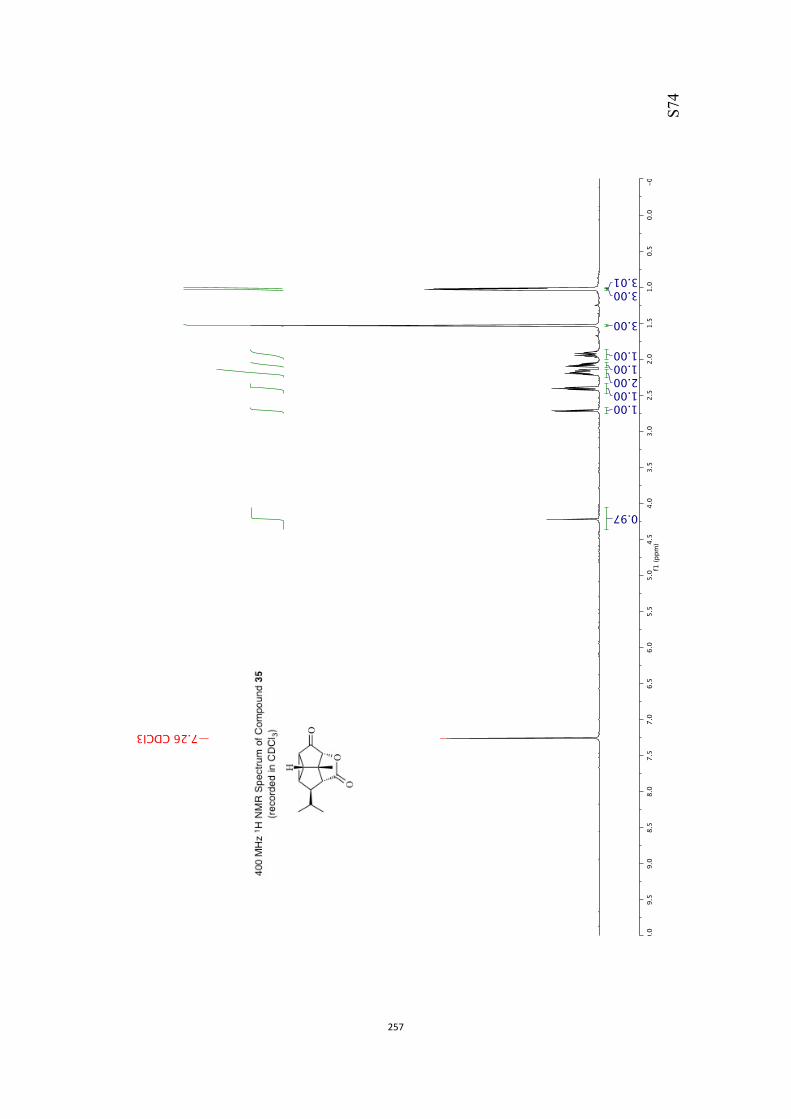
S74
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.013.00
3.00
1.001.002.001.001.00
0.97
7.26 CDCl3
257
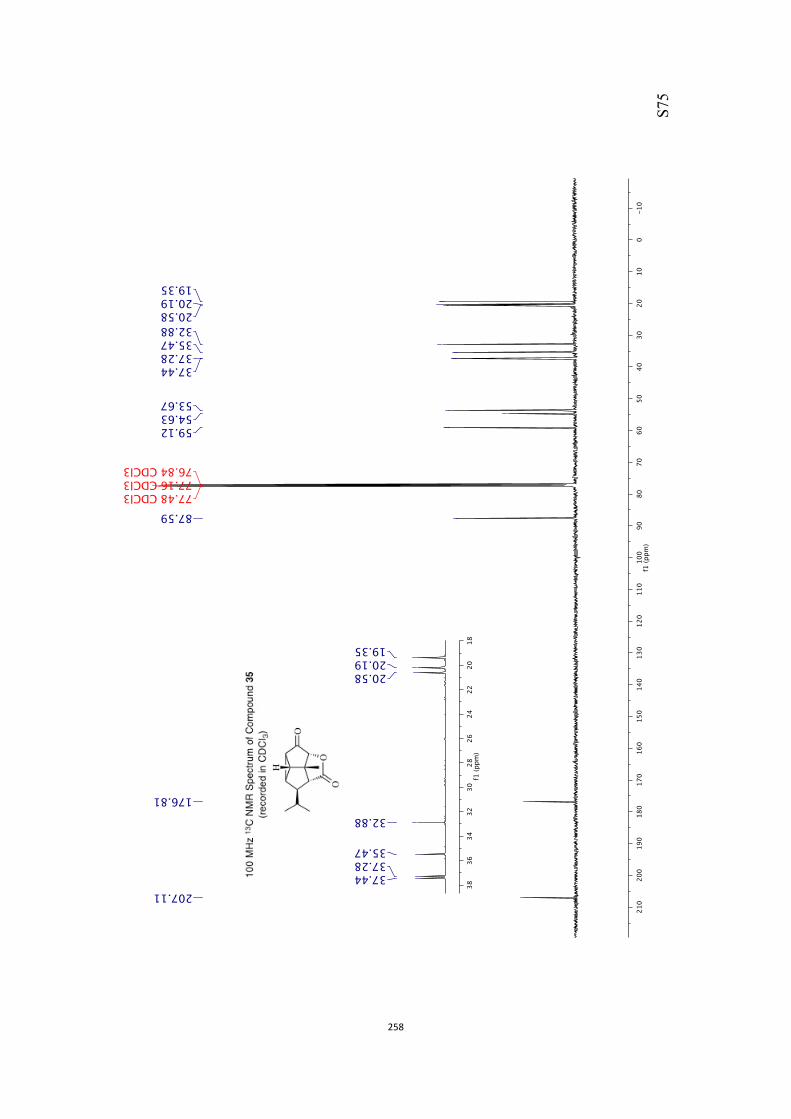
S75
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.3520.1920.58
32.8835.4737.2837.44
53.6754.6359.12
76.84 CDCl377.16 CDCl377.48 CDCl3
87.59
176.81
207.11
18
20
22
24
26
28
30
32
34
36
38
f1 (
ppm
)
19.3520.1920.58
32.88
35.4737.2837.44
258
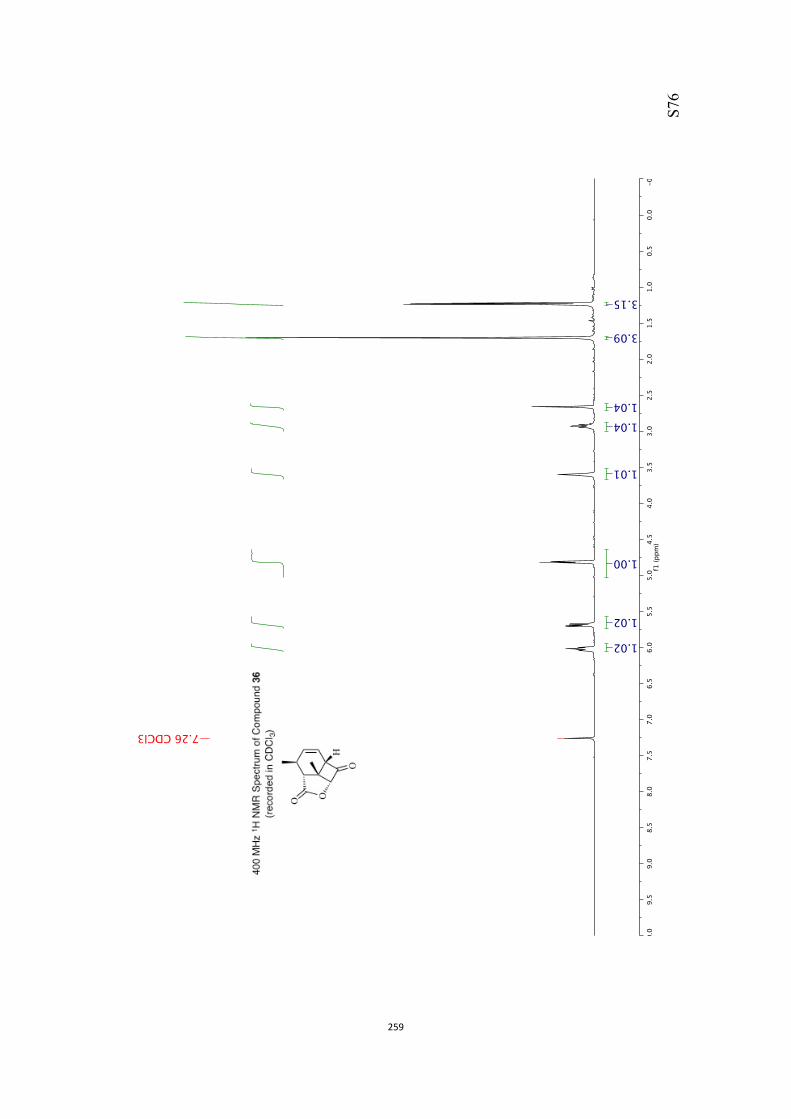
S76
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.15
3.09
1.04
1.04
1.01
1.00
1.02
1.02
7.26 CDCl3
259
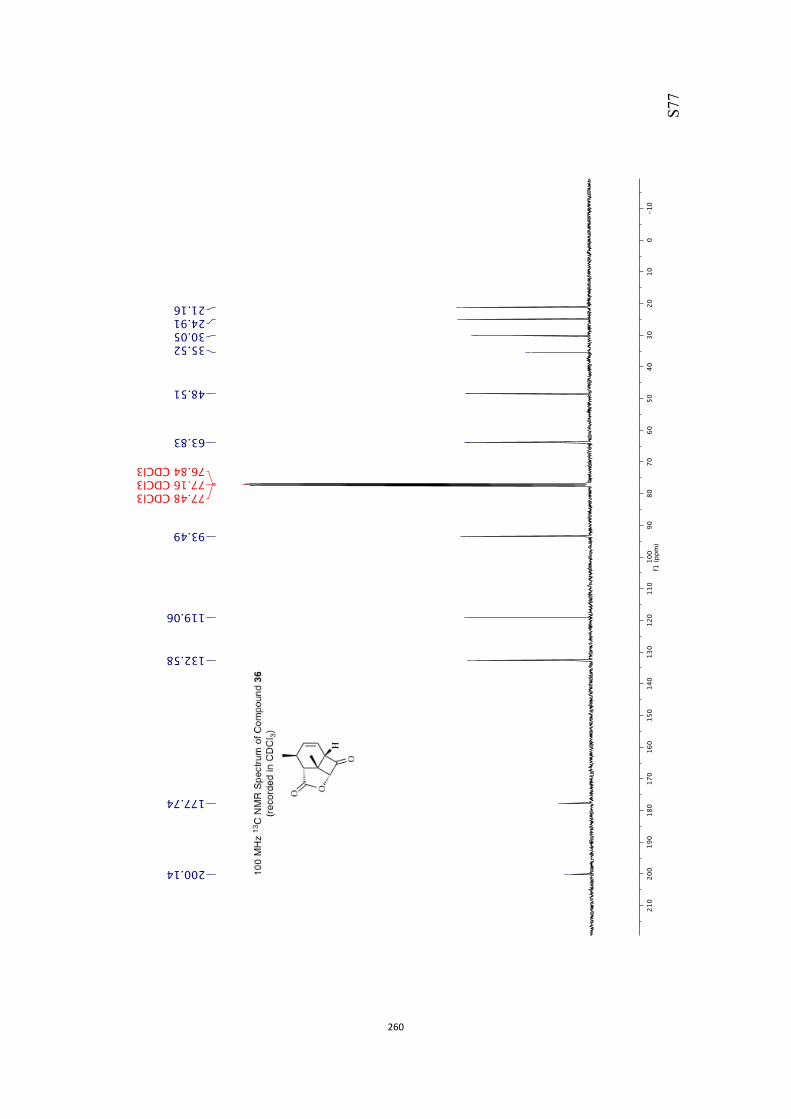
S77
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
21.1624.9130.0535.52
48.51
63.83
76.84 CDCl377.16 CDCl377.48 CDCl3
93.49
119.06
132.58
177.74
200.14
260
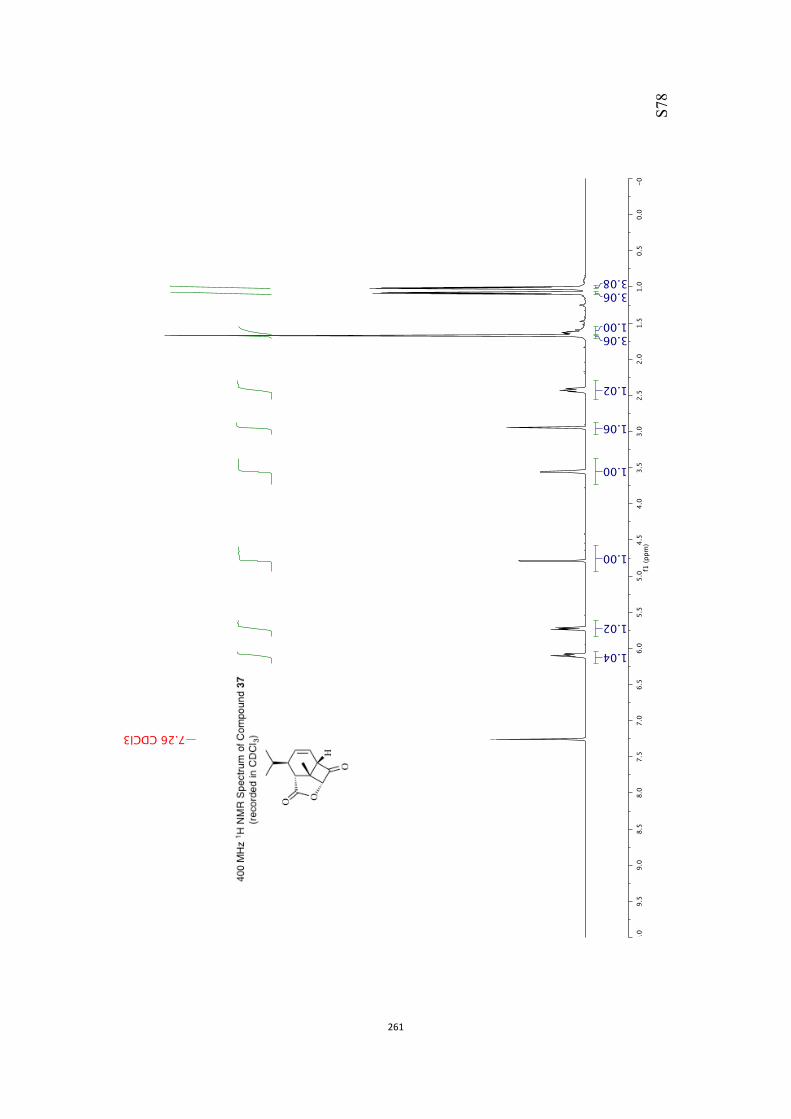
S78
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.083.06
1.003.06
1.02
1.06
1.00
1.00
1.02
1.04
7.26 CDCl3
261
S79
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
21.3421.4424.07
33.0735.8842.5644.83
64.15
76.84 CDCl377.16 CDCl377.48 CDCl3
93.52
120.01
131.79
178.52
199.87
21
.32
1.4
21
.5f1
(ppm
)
21.34
21.44
262
S80
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
3.001.003.00
1.001.00
1.00
1.00
1.00
7.26 CDCl3
263
S81
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
18.8719.3120.5923.5131.72
49.34
65.21
76.84 CDCl377.16 CDCl377.48 CDCl3
119.41
134.78
180.65
18
.51
9.0
19
.52
0.0
20
.52
1.0
f1 (
ppm
)
18.87
19.31
20.59
264
S82
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.003.001.003.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
265
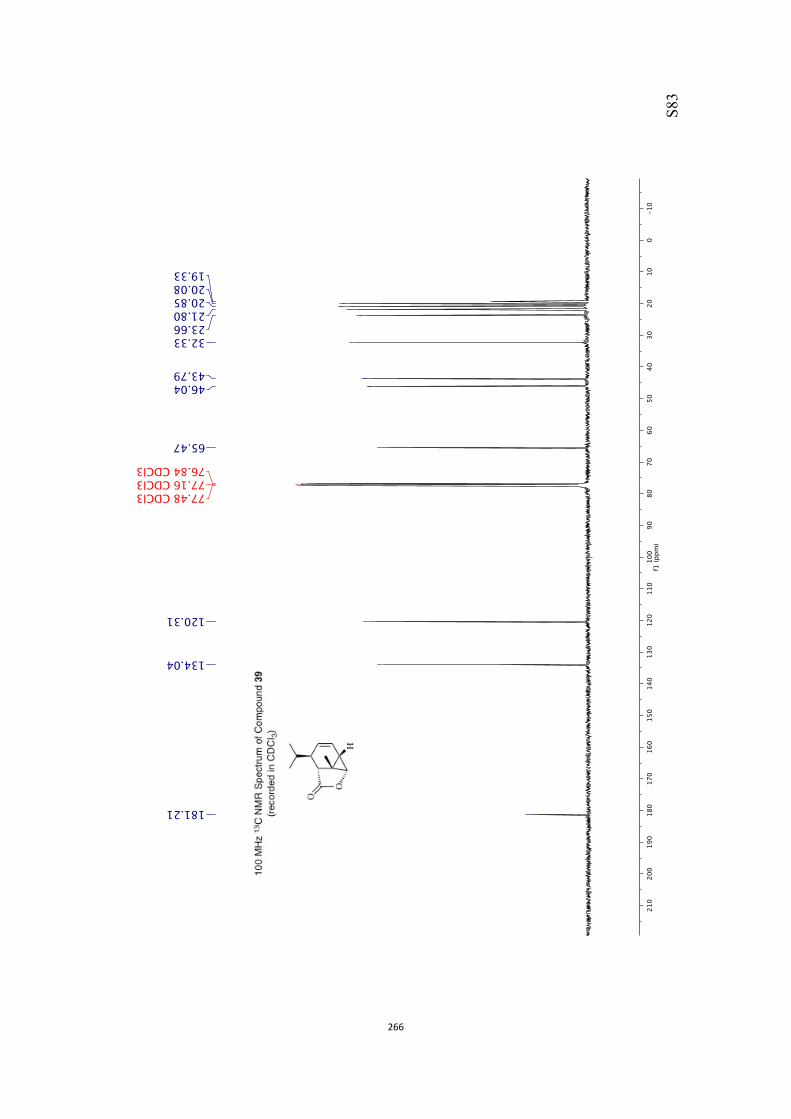
S83
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
19.3320.0820.8521.8023.6632.33
43.7946.04
65.47
76.84 CDCl377.16 CDCl377.48 CDCl3
120.31
134.04
181.21
266
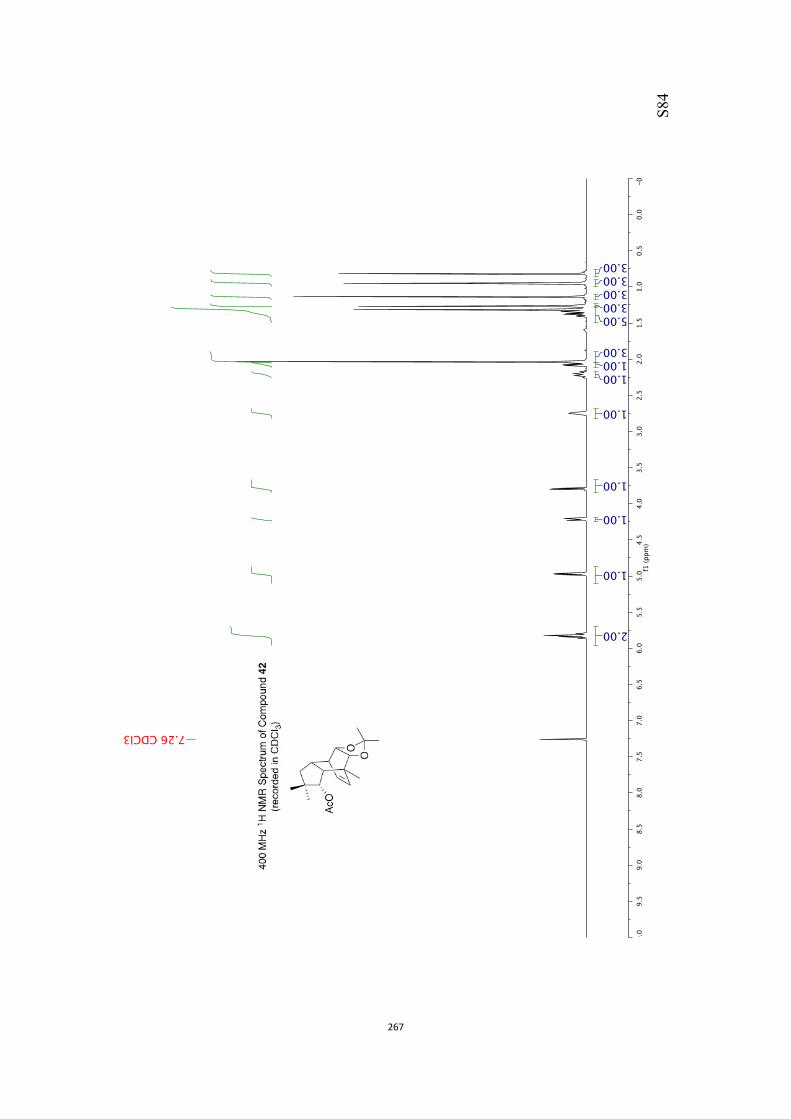
S84
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.003.003.003.005.00
3.001.001.00
1.00
1.00
1.00
1.00
2.00
7.26 CDCl3
267
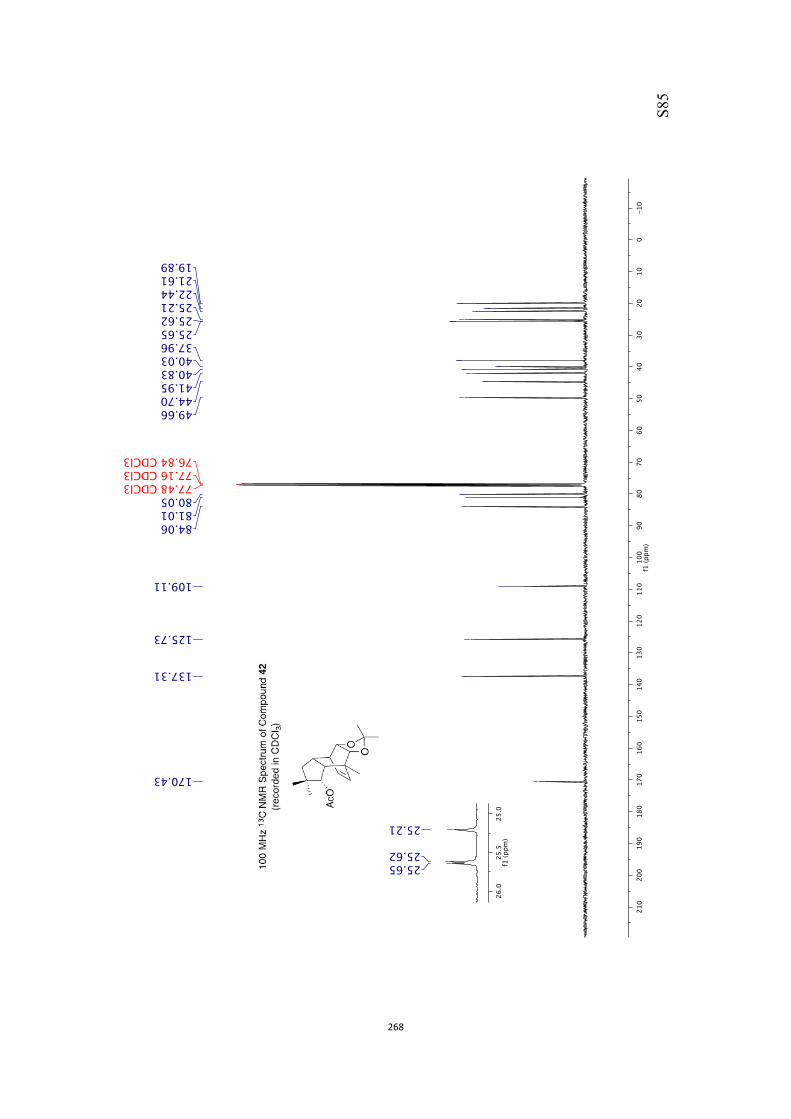
S85
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.8921.6122.4425.2125.6225.6537.9640.0340.8341.9544.7049.66
76.84 CDCl377.16 CDCl377.48 CDCl380.0581.0184.06
109.11
125.73
137.31
170.43
25
.02
5.5
26
.0f1
(ppm
)
25.21
25.6225.65
268
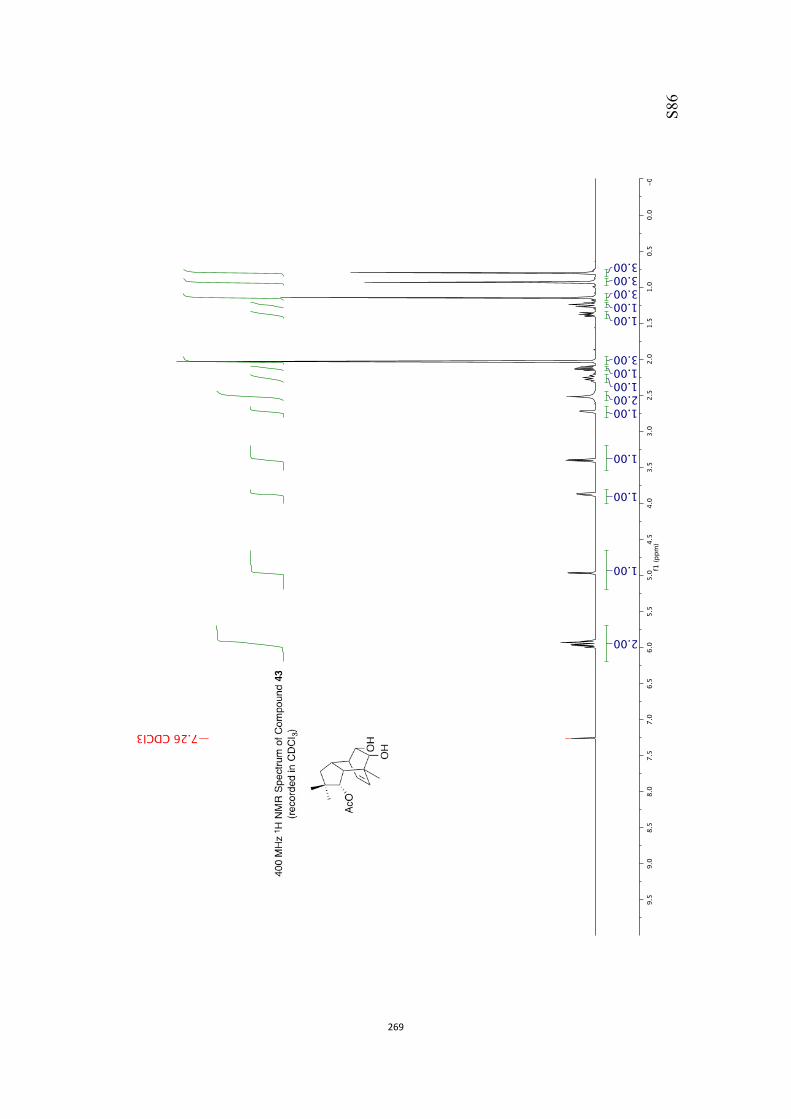
S86
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
f1 (
ppm
)
3.003.003.001.001.00
3.001.001.002.001.00
1.00
1.00
1.00
2.00
7.26 CDCl3
269
S87
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.5721.6122.3425.51
40.7841.3341.4842.0344.1450.44
71.7175.3976.84 CDCl377.16 CDCl377.48 CDCl381.61
126.82
138.75
170.43
40
41
42
43
f1 (
ppm
)
40.7841.3341.4842.03
270

S88
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.003.003.001.001.00
3.00
3.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
271
S89
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.08
21.56
22.23
25.56
39.33
41.68
43.87
44.82
50.10
51.81
75.16
76.84 CDCl3
77.16 CDCl3
77.48 CDCl3
81.11
121.90
142.16
170.19
210.99
272

S90
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.5f1
(ppm
)
3.003.003.001.001.003.003.001.001.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
273
S91
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
17.9220.7221.5322.2025.47
39.6241.6942.6044.9350.6551.45
74.1576.84 CDCl377.16 CDCl377.48 CDCl381.09
122.24
141.36
170.08170.67
205.16
274
S92
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.00
1.00
1.00
3.00
1.00
1.00
1.00
1.00
3.00
1.00
2.00
1.00
1.00
7.26 CDCl3
275
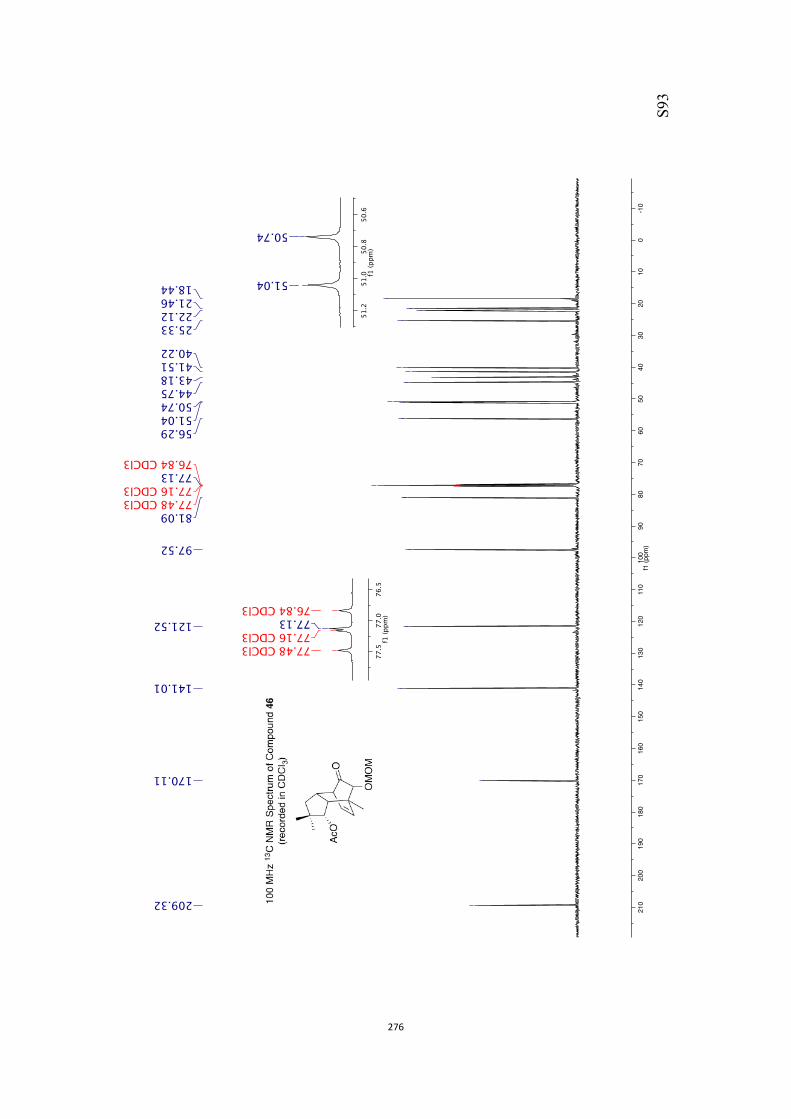
S93
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.4421.4622.1225.33
40.2241.5143.1844.7550.7451.0456.29
76.84 CDCl377.1377.16 CDCl377.48 CDCl381.09
97.52
121.52
141.01
170.11
209.32
76
.57
7.0
77
.5f1
(ppm
)
76.84 CDCl377.1377.16 CDCl377.48 CDCl3
50
.65
0.8
51
.05
1.2
f1 (
ppm
)
50.74
51.04
276
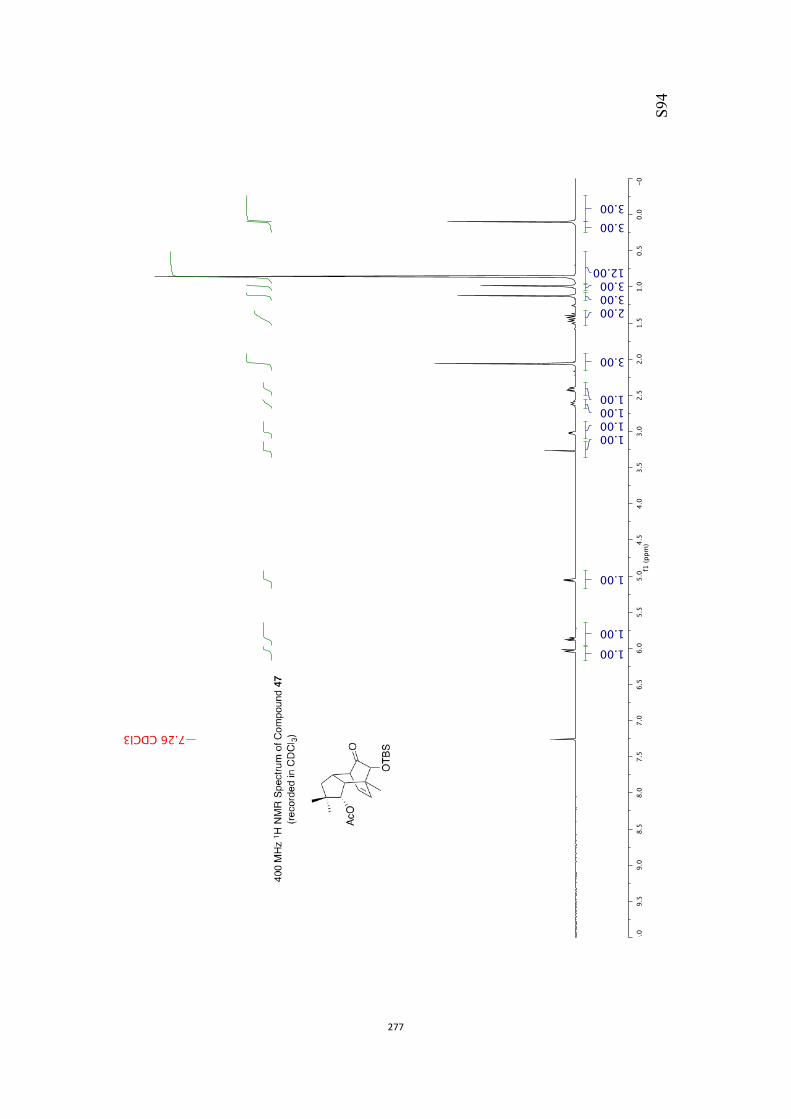
S94
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
f1 (
ppm
)
3.00
3.00
12.003.003.002.00
3.00
1.001.001.001.00
1.00
1.00
1.00
7.26 CDCl3
277
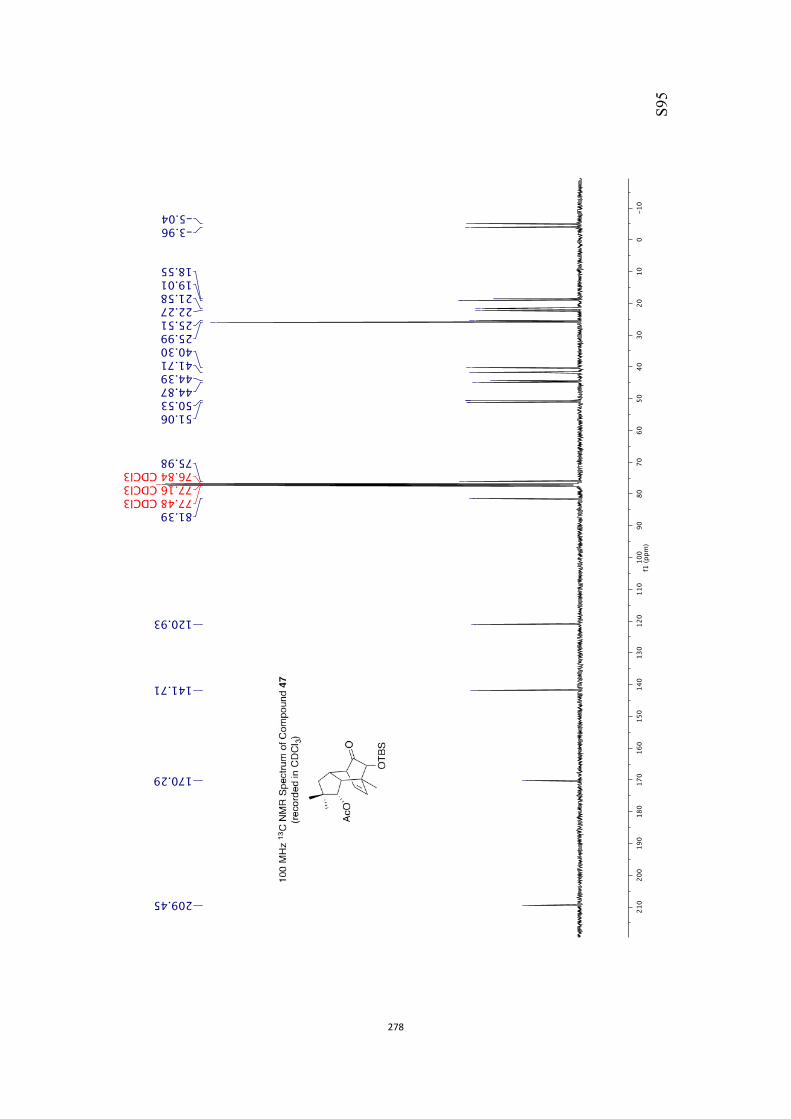
S95
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
-5.04-3.96
18.5519.0121.5822.2725.5125.9940.3041.7144.3944.8750.5351.06
75.9876.84 CDCl377.16 CDCl377.48 CDCl381.39
120.93
141.71
170.29
209.45
278
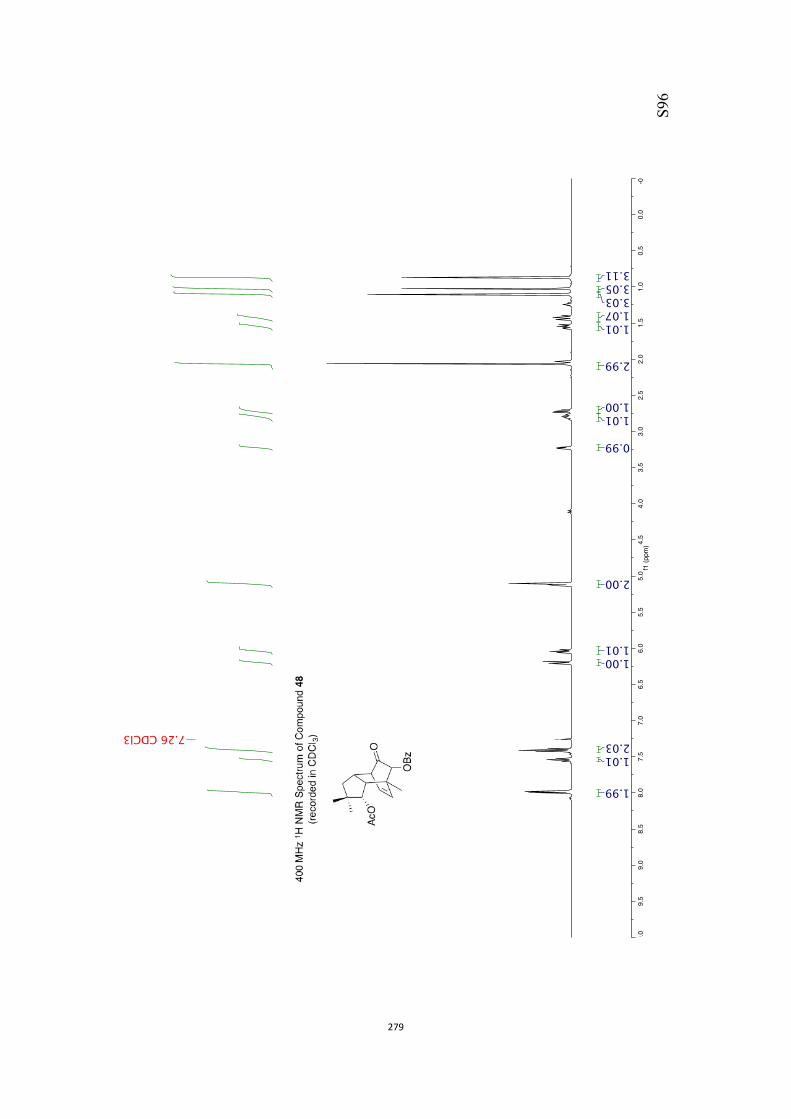
S96
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.11
3.05
3.03
1.07
1.01
2.99
1.00
1.01
0.99
2.00
1.01
1.00
2.03
1.01
1.99
7.26 CDCl3
279
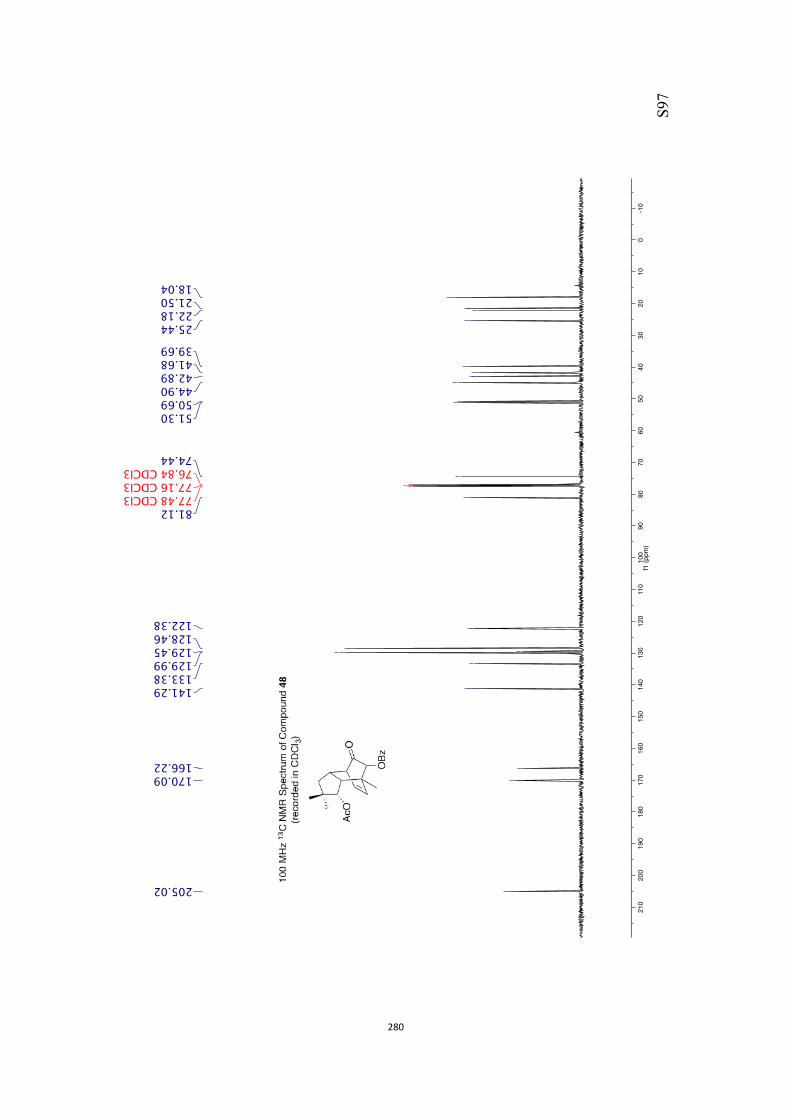
S97
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.04
21.50
22.18
25.44
39.69
41.68
42.89
44.90
50.69
51.30
74.44
76.84 CDCl3
77.16 CDCl3
77.48 CDCl3
81.12
122.38
128.46
129.45
129.99
133.38
141.29
166.22
170.09
205.02
280
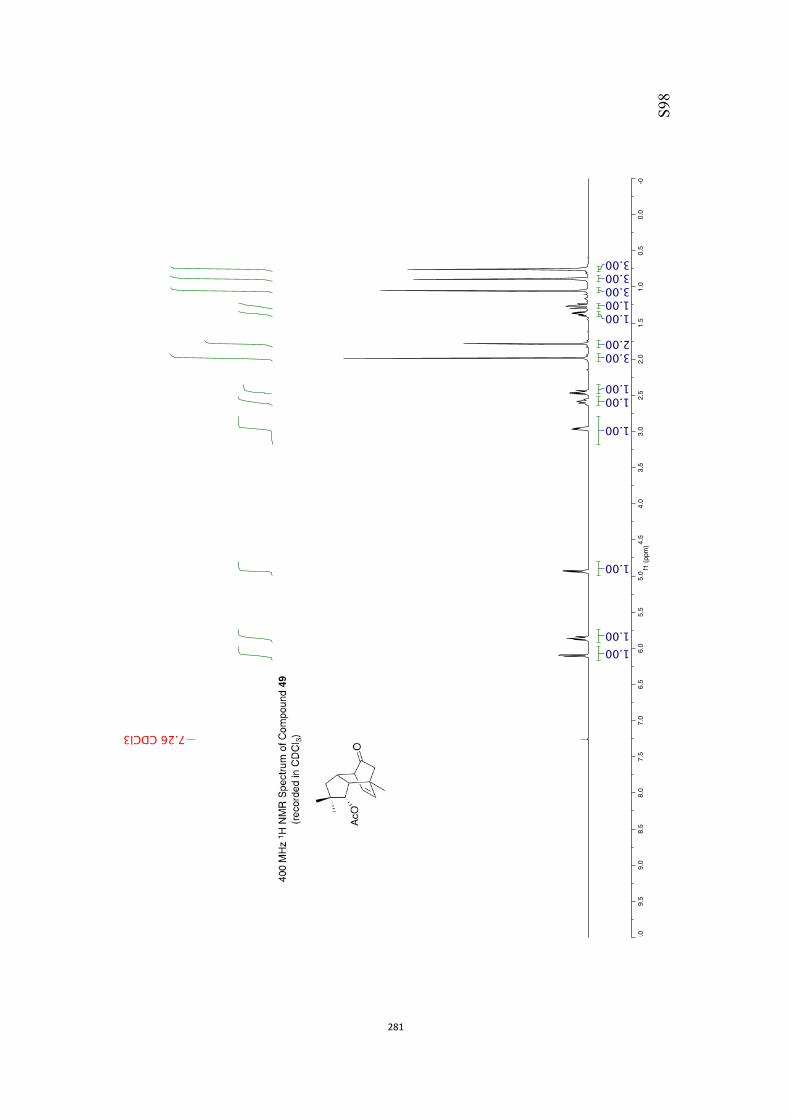
S98
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.00
1.00
1.00
2.00
3.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
281
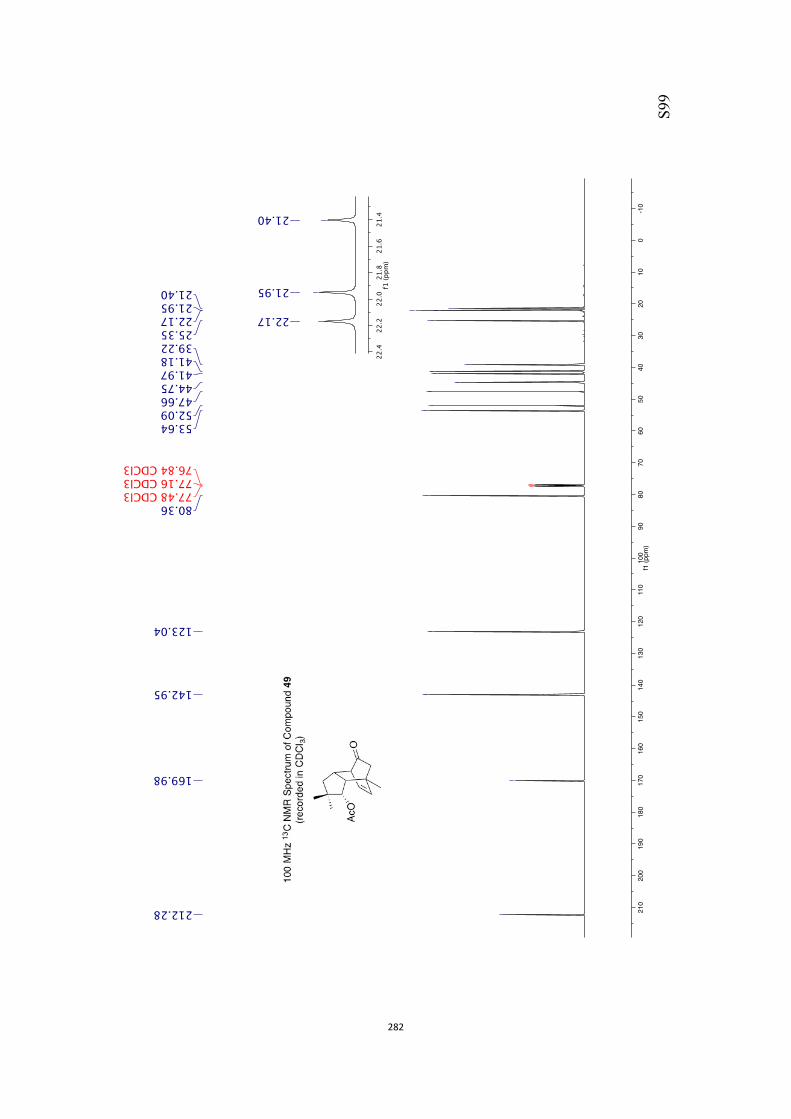
S99
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
21.4021.9522.1725.3539.2241.1841.9744.7547.6652.0953.64
76.84 CDCl377.16 CDCl377.48 CDCl380.36
123.04
142.95
169.98
212.28
21
.42
1.6
21
.82
2.0
22
.22
2.4
f1 (
ppm
)
21.40
21.95
22.17
282
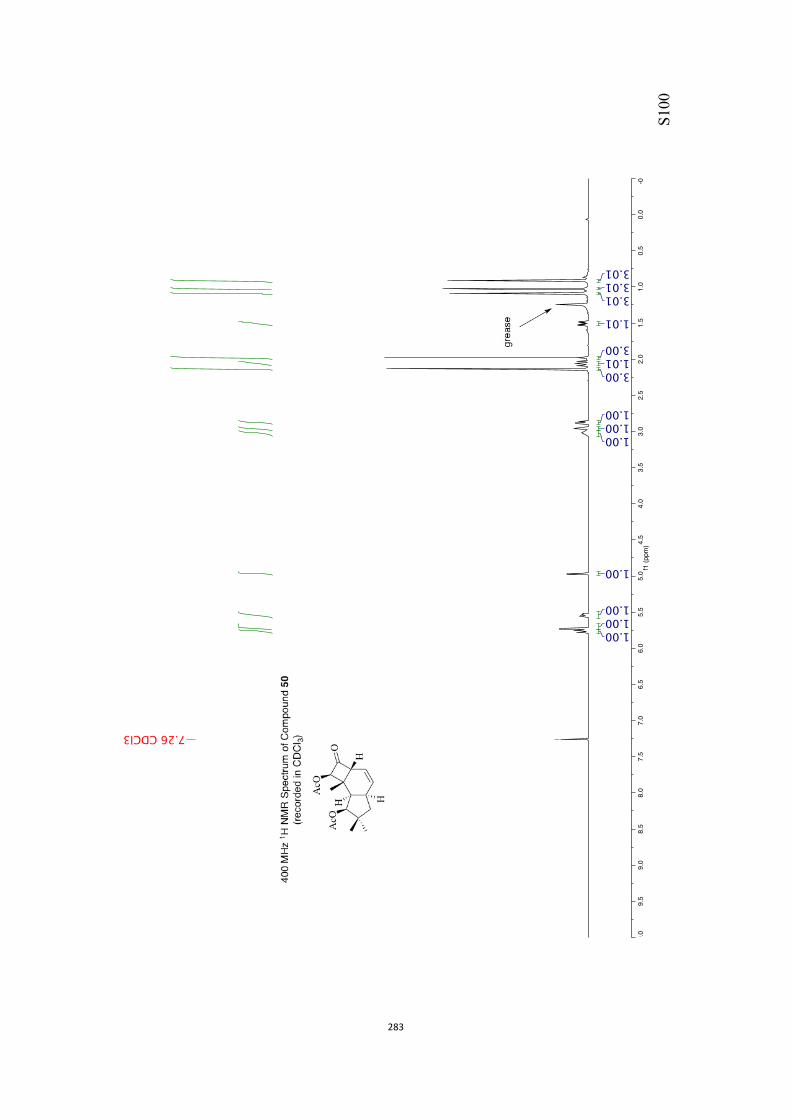
S100
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.01
3.01
3.01
1.01
3.00
1.01
3.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
283
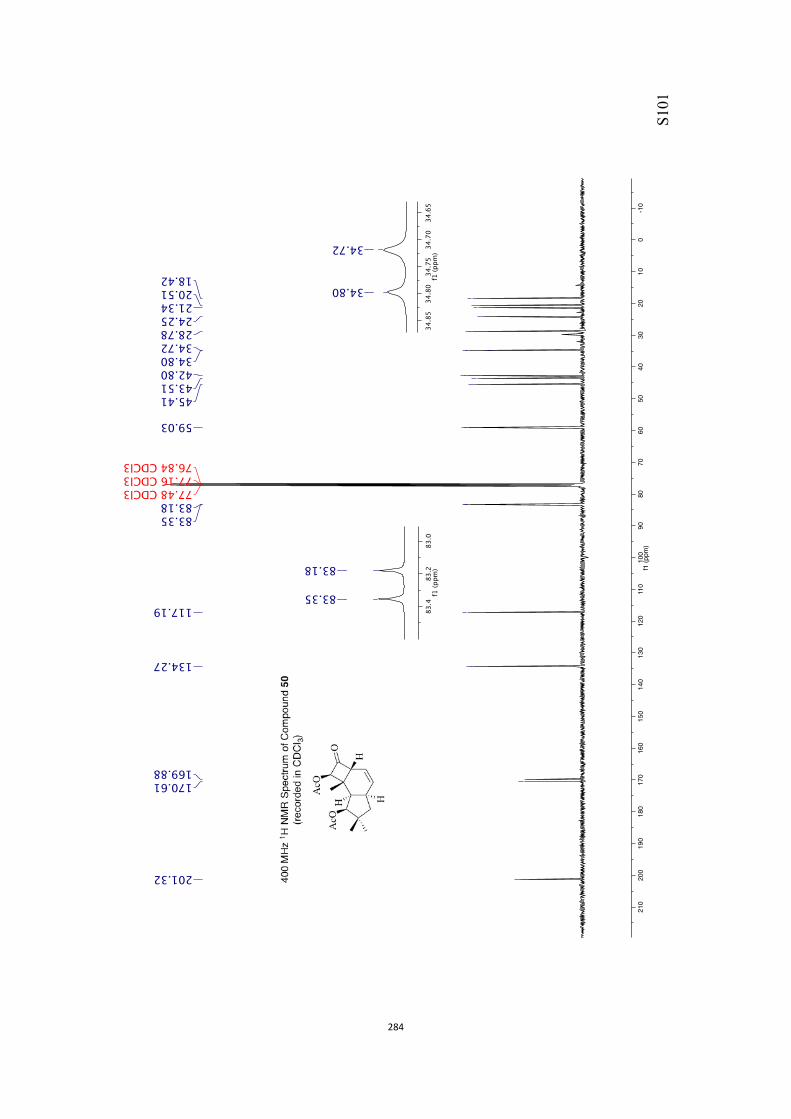
S101
-1
00
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.4220.5121.3424.2528.7834.7234.8042.8043.5145.41
59.03
76.84 CDCl377.16 CDCl377.48 CDCl383.1883.35
117.19
134.27
169.88170.61
201.32
83
.08
3.2
83
.4f1
(ppm
)
83.18
83.35
34
.65
34
.70
34
.75
34
.80
34
.85
f1 (
ppm
)
34.72
34.80
284
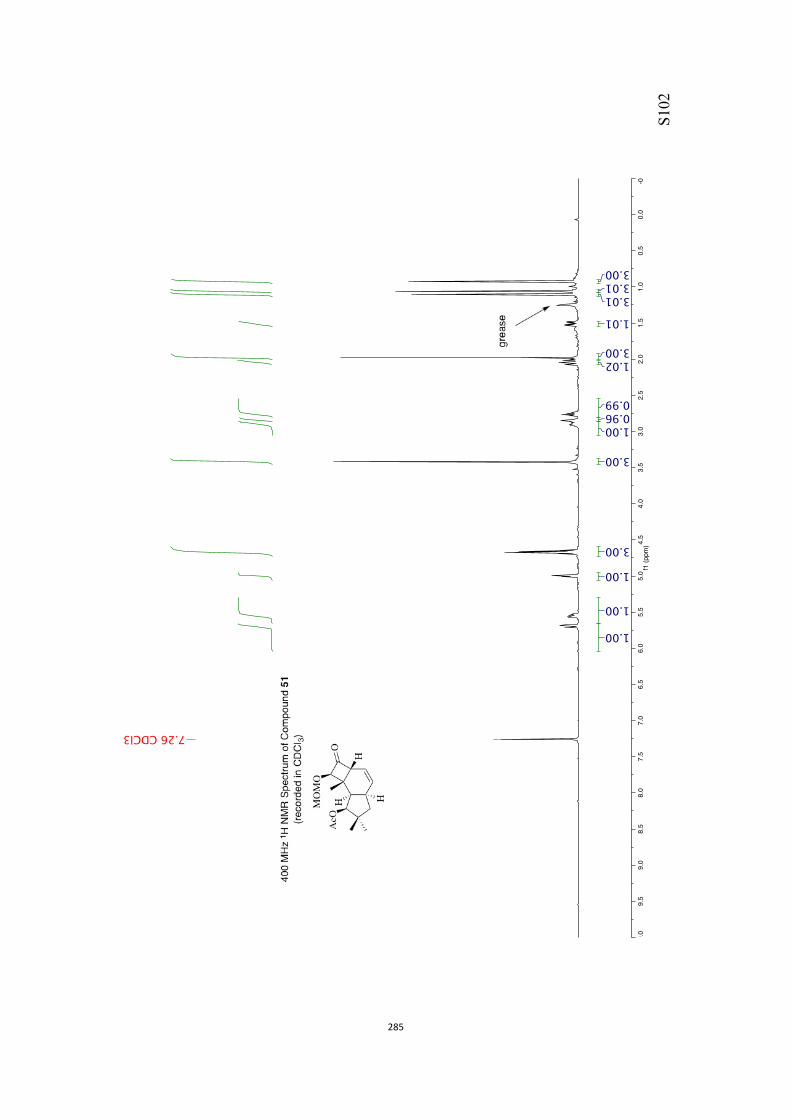
S102
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.01
3.01
1.01
3.00
1.02
0.99
0.96
1.00
3.00
3.00
1.00
1.00
1.00
7.26 CDCl3
285
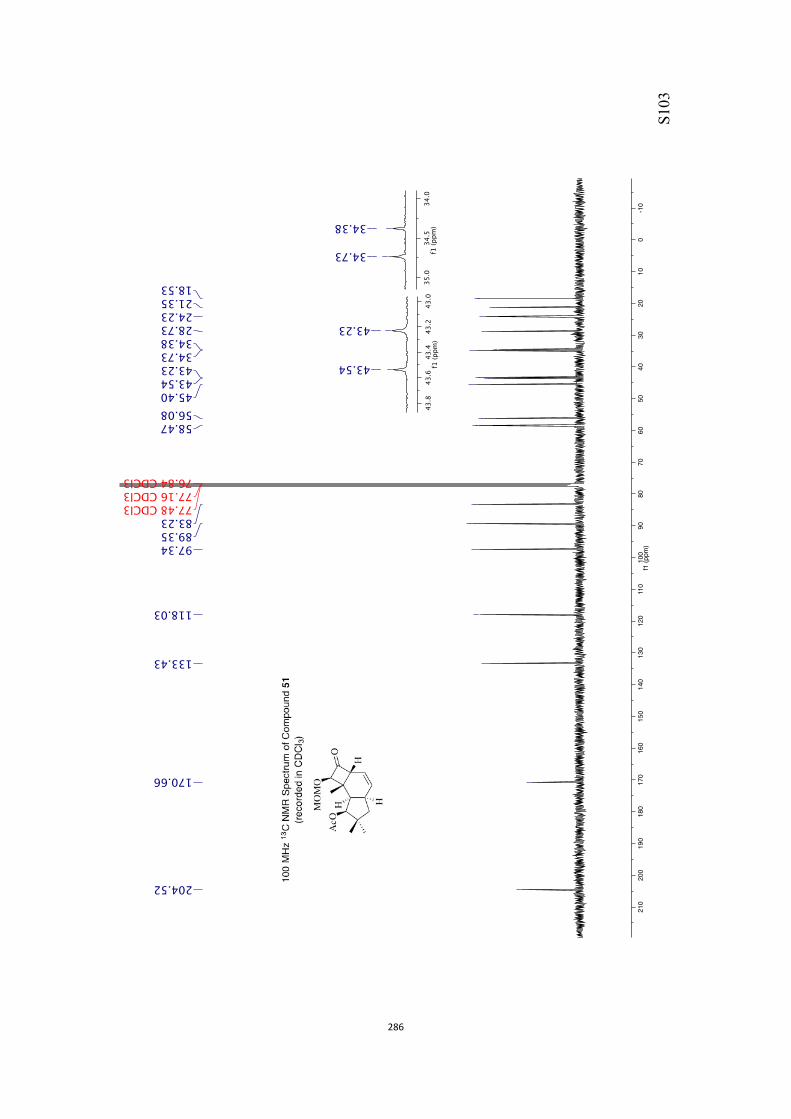
S103
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.5321.3524.2328.7334.3834.7343.2343.5445.40
56.0858.47
76.84 CDCl377.16 CDCl377.48 CDCl383.2389.3597.34
118.03
133.43
170.66
204.52
43
.04
3.2
43
.44
3.6
43
.8f1
(ppm
)
43.23
43.54
34
.03
4.5
35
.0f1
(ppm
)
34.38
34.73
286
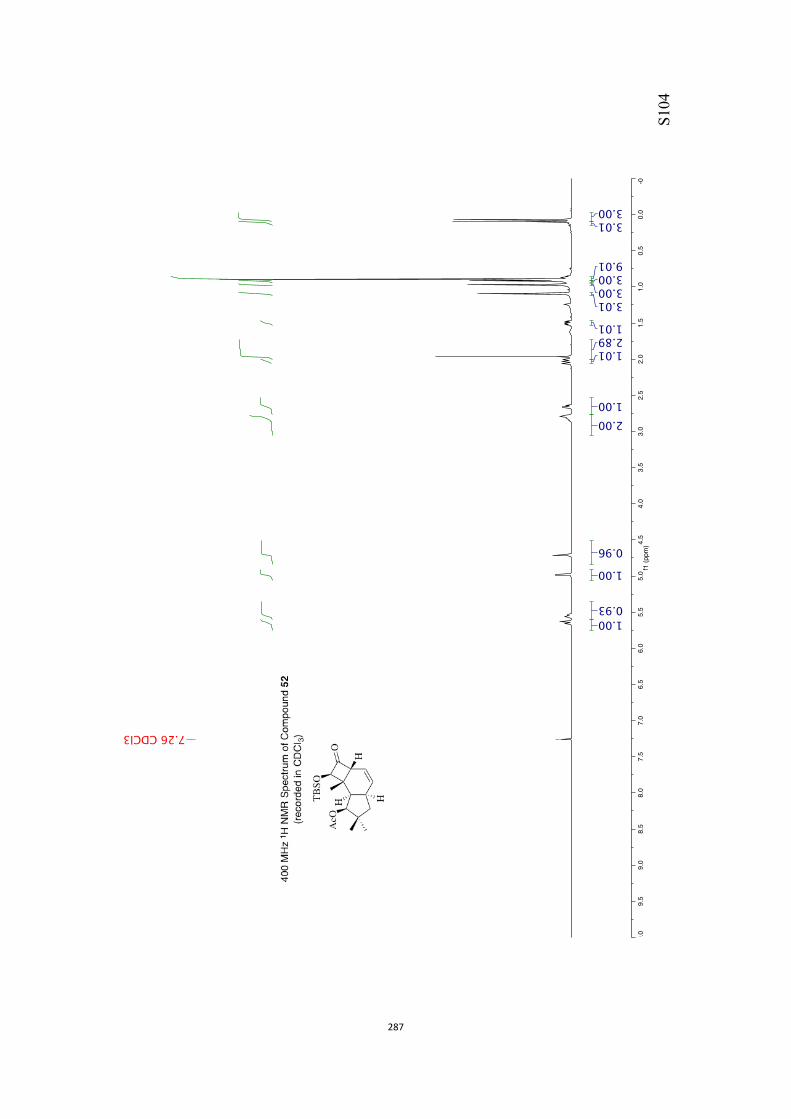
S104
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.01
9.01
3.00
3.00
3.01
1.01
2.89
1.01
1.00
2.00
0.96
1.00
0.93
1.00
7.26 CDCl3
287
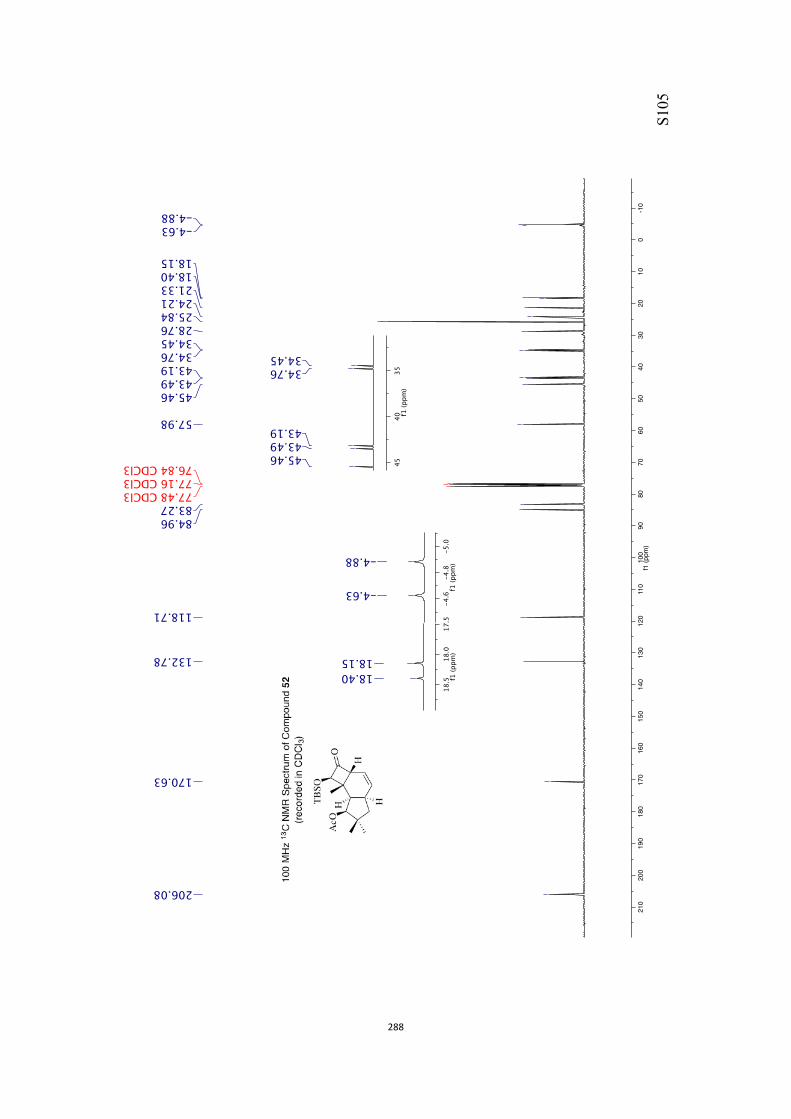
S105
-1
00
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
-4.88-4.63
18.1518.4021.3324.2125.8428.7634.4534.7643.1943.4945.46
57.98
76.84 CDCl377.16 CDCl377.48 CDCl383.2784.96
118.71
132.78
170.63
206.08
35
40
45
f1 (
ppm
)
34.4534.76
43.1943.4945.46
17
.51
8.0
18
.5 f1 (
ppm
)18.15
18.40-5
.0-4
.8-4
.6f1
(ppm
)
-4.88
-4.63
288
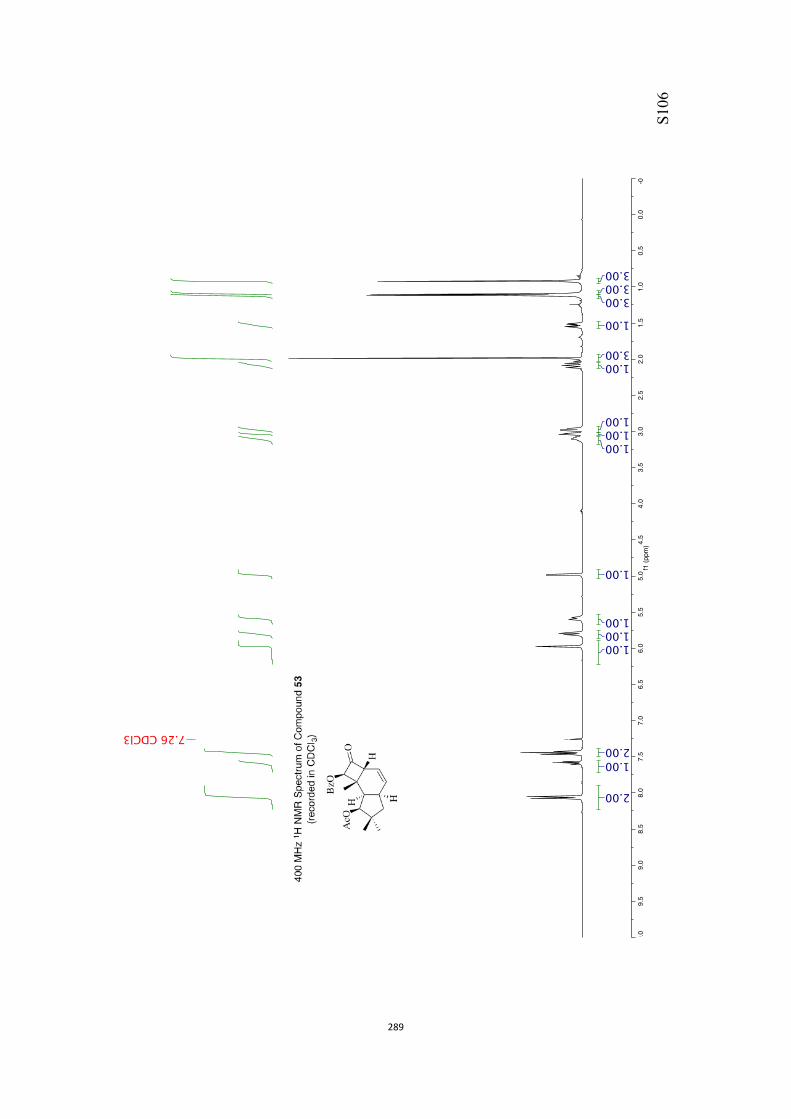
S106
-0
.50.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.00
1.00
3.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
2.00
1.00
2.00
7.26 CDCl3
289
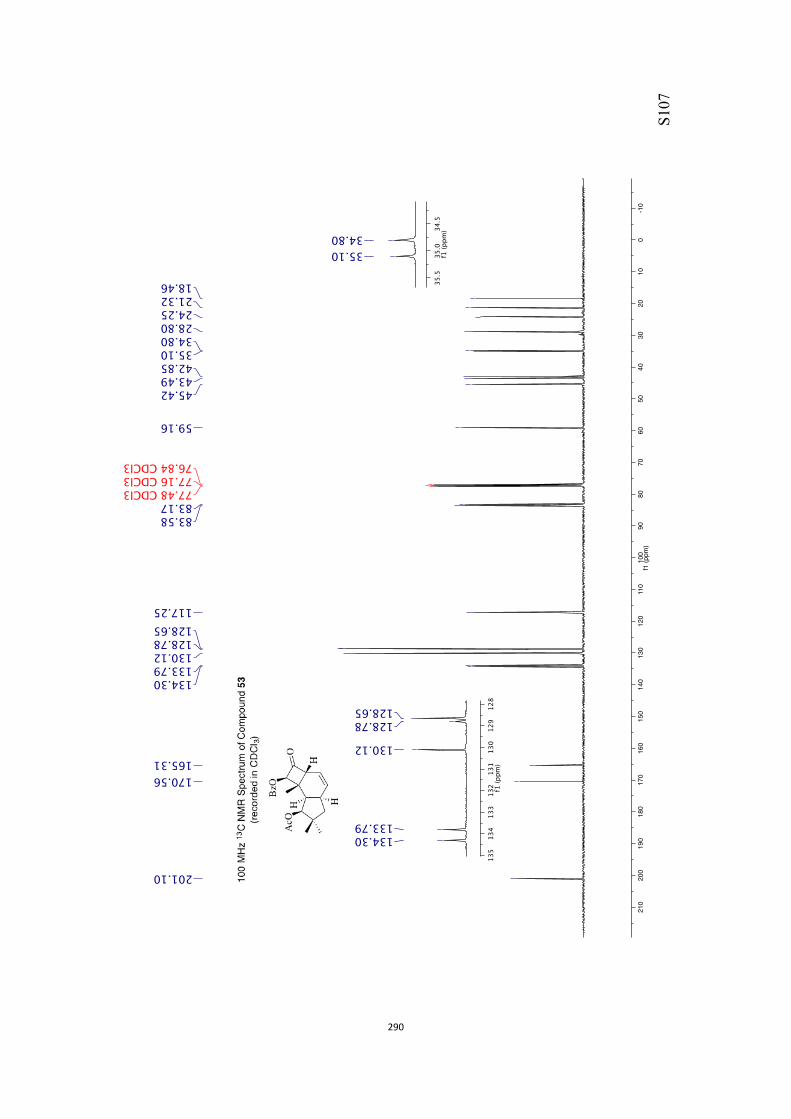
S107
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.4621.3224.2528.8034.8035.1042.8543.4945.42
59.16
76.84 CDCl377.16 CDCl377.48 CDCl383.1783.58
117.25
128.65128.78130.12133.79134.30
165.31
170.56
201.10
12
81
29
13
01
31
13
21
33
13
41
35
f1 (
ppm
)
128.65128.78
130.12
133.79134.30
34
.53
5.0
35
.5f1
(ppm
)34.80
35.10
290

S108
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.00
1.00
3.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
291
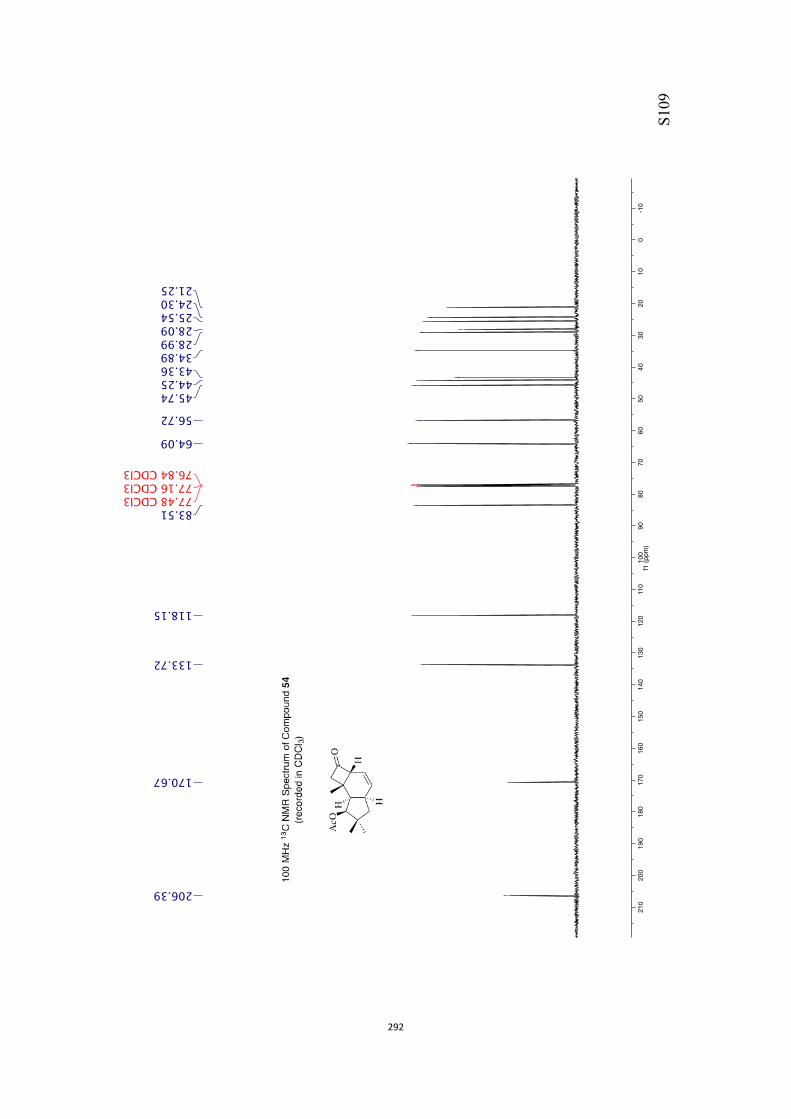
S109
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
21.25
24.30
25.54
28.09
28.99
34.89
43.36
44.25
45.74
56.72
64.09
76.84 CDCl3
77.16 CDCl3
77.48 CDCl3
83.51
118.15
133.72
170.67
206.39
292

S110
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.06
6.01
1.00
1.13
1.07
3.01
3.00
1.04
1.00
1.00
1.00
1.00
1.00
7.26 CDCl3
293
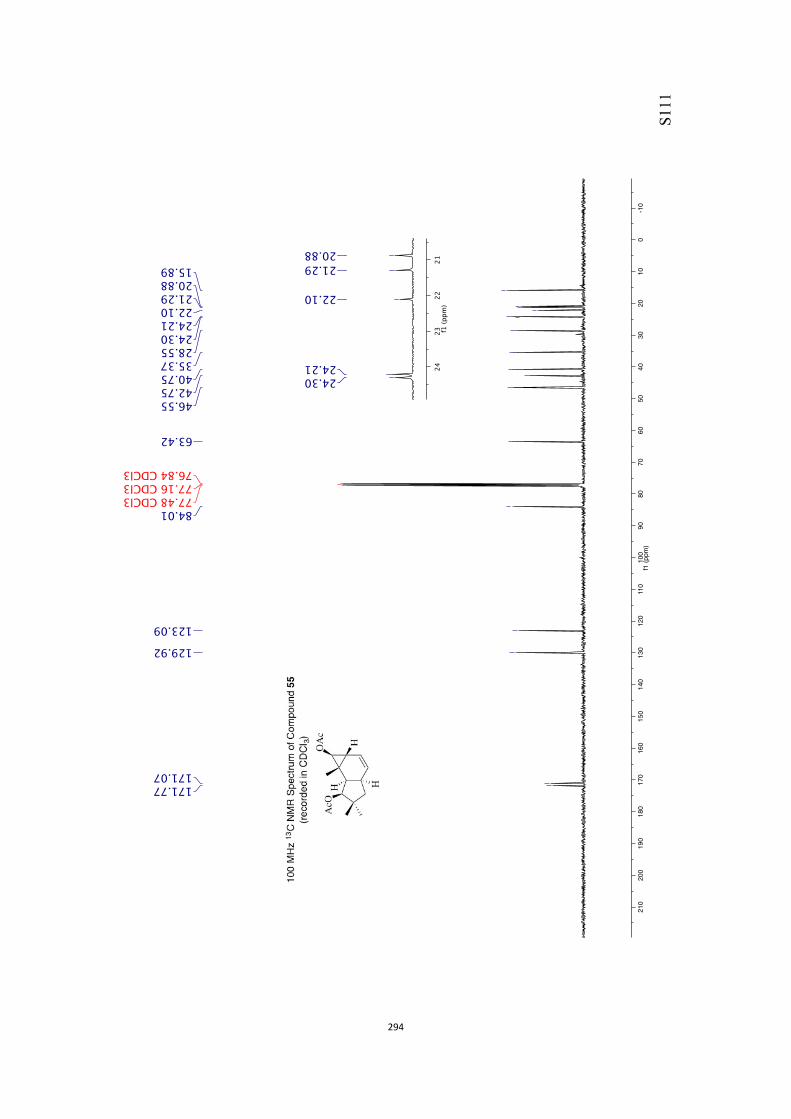
S111
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
15.8920.8821.2922.1024.2124.3028.5535.3740.7542.7546.55
63.42
76.84 CDCl377.16 CDCl377.48 CDCl384.01
123.09
129.92
171.07171.77
21
22
23
24
f1 (
ppm
)
20.88
21.29
22.10
24.2124.30
294

S112
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.0f1
(ppm
)
3.003.00
3.009.001.023.003.001.01
1.003.00
1.001.00
1.00
1.00
1.00
1.00
2.05 Acetone
295
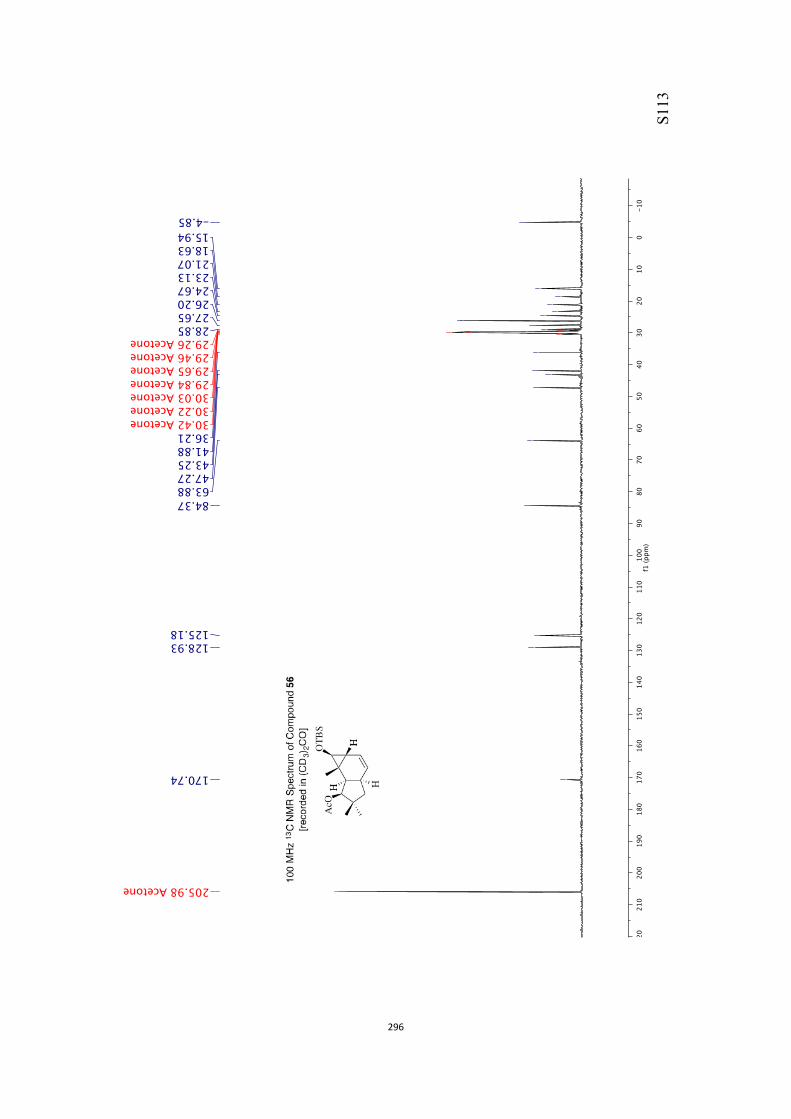
S113
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
-4.85
15.9418.6321.0723.1324.6726.2027.6528.8529.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone36.2141.8843.2547.2763.88
84.37
125.18128.93
170.74
205.98 Acetone
296
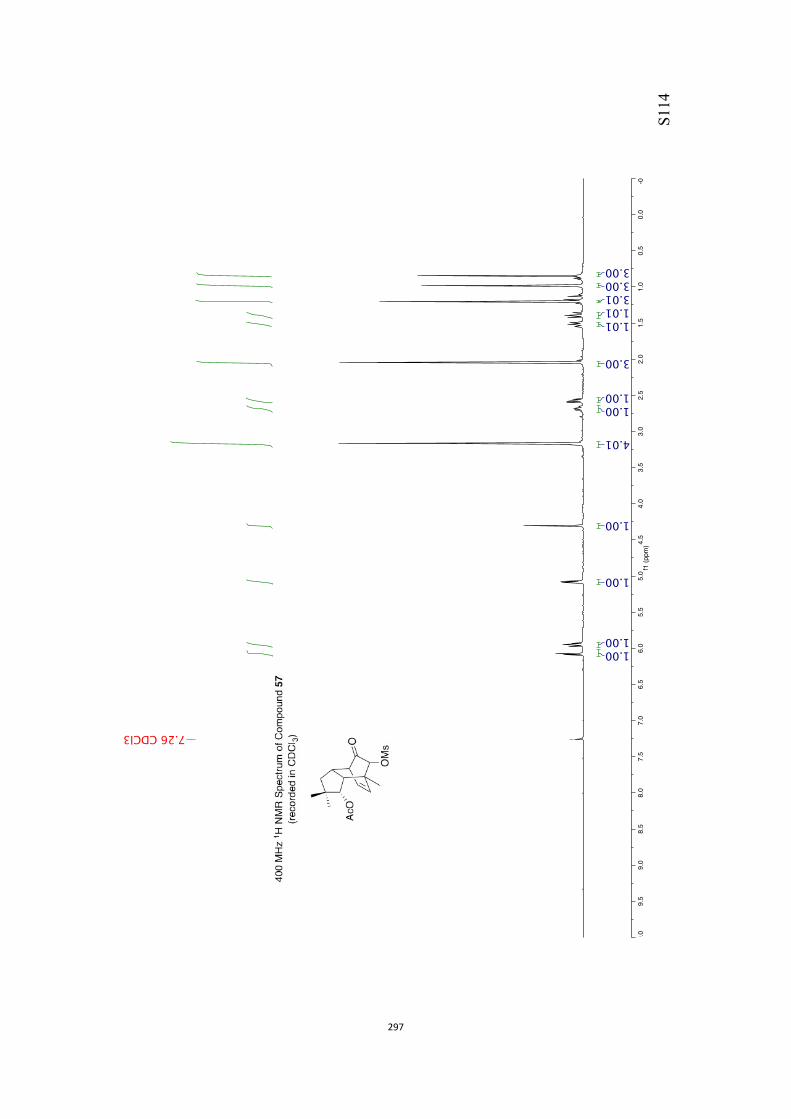
S114
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.01
1.01
1.01
3.00
1.00
1.00
4.01
1.00
1.00
1.00
1.00
7.26 CDCl3
297
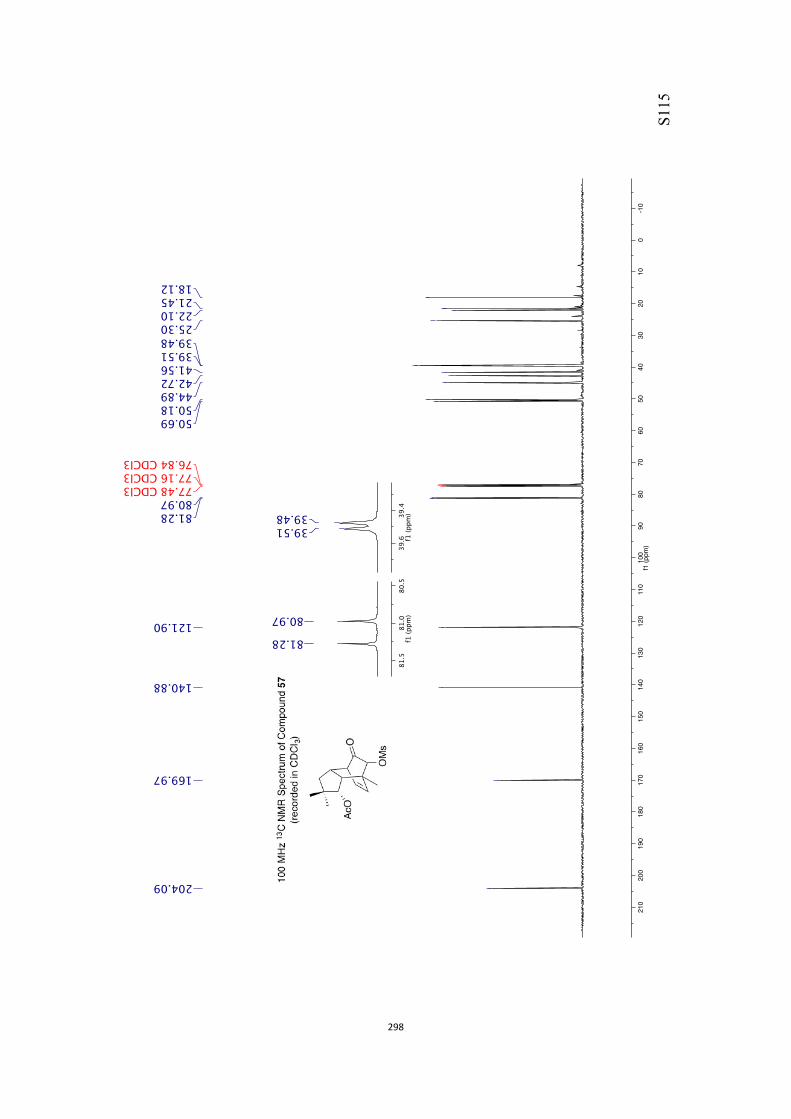
S115
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
18.1221.4522.1025.30
39.4839.5141.5642.7244.8950.1850.69
76.84 CDCl377.16 CDCl377.48 CDCl380.9781.28
121.90
140.88
169.97
204.09
80
.58
1.0
81
.5f1
(ppm
)80.97
81.28
39
.43
9.6 f1
(ppm
)39.4839.51
298
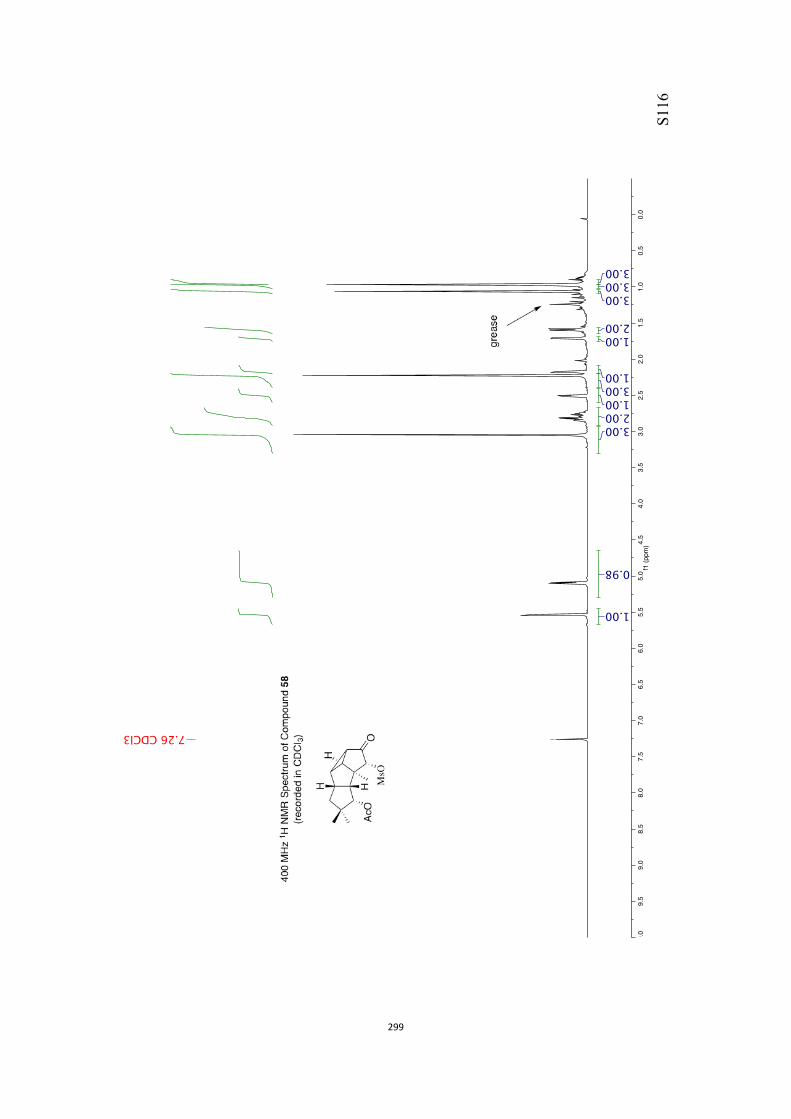
S116
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
f1 (
ppm
)
3.00
3.00
3.00
2.00
1.00
1.00
3.00
1.00
2.00
3.00
0.98
1.00
7.26 CDCl3
299
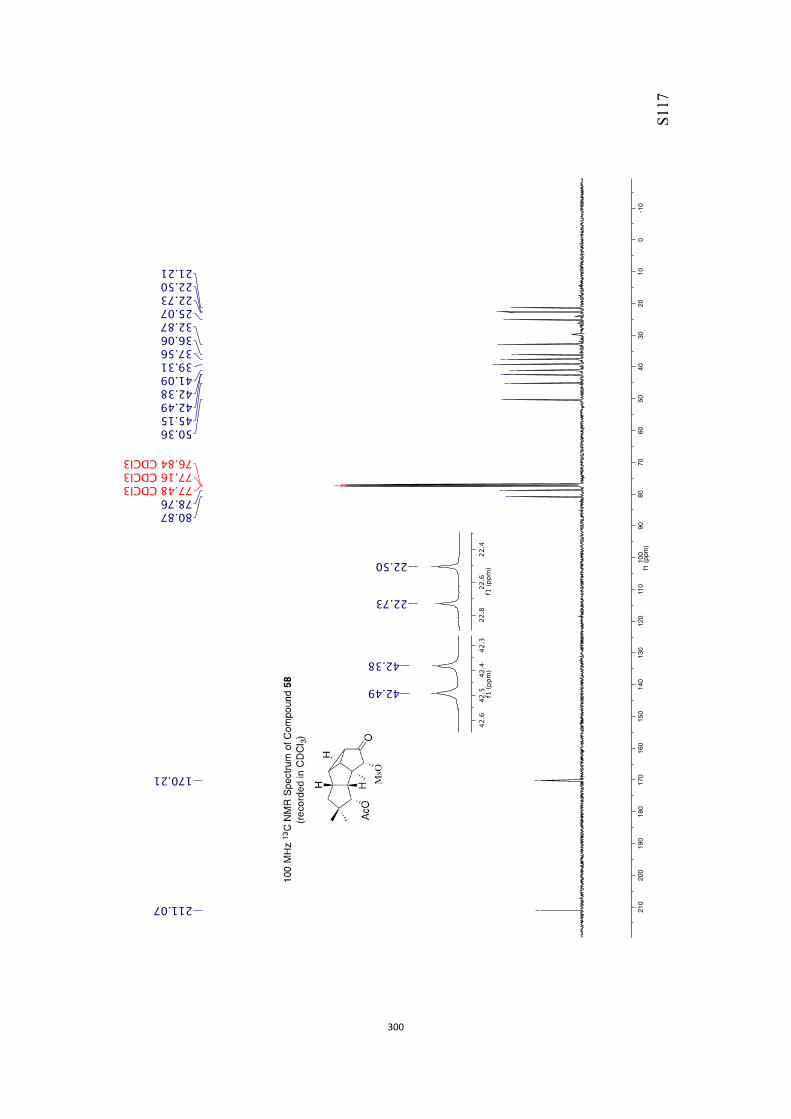
S117
-1
00
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
21.2122.5022.7325.0732.8736.0637.5639.3141.0942.3842.4945.1550.36
76.84 CDCl377.16 CDCl377.48 CDCl378.7680.87
170.21
211.07
42
.34
2.4
42
.54
2.6
f1 (
ppm
)
42.38
42.49
22
.42
2.6
22
.8f1
(ppm
)
22.50
22.73
300


301
Publication Three
A Palladium-Catalyzed Ullmann Cross-
Coupling/Reductive Cyclization Route to the
Carbazole Natural Products 3-Methyl-9H-carbazole,
Glycoborine, Glycozoline, Clauszoline K, Mukonine,
and Karapinchamine
Qiao Yan, Emma Gin, Malgorzata Wasinska-Kalwa,
Martin G. Banwell, and Paul D. Carr
J. Org. Chem., 2017, 82, 4148.


A Palladium-Catalyzed Ullmann Cross-Coupling/ReductiveCyclization Route to the Carbazole Natural Products3‑Methyl‑9H‑carbazole, Glycoborine, Glycozoline, Clauszoline K,Mukonine, and Karapinchamine AQiao Yan, Emma Gin, Malgorzata Wasinska-Kalwa, Martin G. Banwell,* and Paul D. Carr
Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, Australian CapitalTerritory 2601, Australia
*S Supporting Information
ABSTRACT: The title natural products 2−7 have been prepared byreductive cyclization of the relevant 2-arylcyclohex-2-en-1-one (e.g.20) to the corresponding tetrahydrocarbazole and dehydrogenation(aromatization) of this to give the target carbazole (e.g. 4).Compounds such as 20 were prepared using a palladium-catalyzedUllmann cross-coupling reaction between the appropriate 2-iodocyclohex-2-en-1-one and o-halonitrobenzene.
■ INTRODUCTION9H-Carbazole (1) (Figure 1) was first isolated from coal tarmore than 100 years ago,1 and since that time this aromatic
heterocycle and its various derivatives have fascinated organicchemists because of their value in both medicine and materialsscience.2 Many biologically active natural products embodyingthis framework have also been isolated, particularly from higherplants.2d,e,3 As such, the development of methods for thesynthesis of the carbazoles has been an ongoing field ofresearch. A suite of approaches to these compounds has beenreported, ranging from the classical Fischer−Borsche4 andGraebe−Ullmann5 routes to more contemporary ones such asthe cyclization of biarylnitrenes (Cadogan synthesis)6 orvariants thereof and the annulation of indoles,7 includingthrough electrocyclization processes. Despite the demonstratedutility of these and other approaches,8 perhaps the mosteffective route to carbazoles involves the cyclization ofdiarylamines, especially under oxidative conditions.9 Variationson this last approach have been used to great effect indeveloping total syntheses of a plethora of carbazole-containingnatural products, with particularly notable contributions havingbeing made, especially in recent times, by the Knolkergroup.2,9e−i,k,m
Sometime ago we reported10 that 1,2,3,4-tetrahydro-9H-carbazoles (formally 2,3,4,9-tetrahydro-1H-carbazoles) can beformed by a two-step process involving an initial the palladium-catalyzed Ullmann cross-coupling of 2-halocyclohex-2-en-1-ones with o-halonitrobenzenes and then subjecting the resulting2-arylcyclohex-2-en-1-ones to a reductive cyclization reaction.11
Since various methods are available for or could be applied tothe oxidation of tetrahydrocarbazoles to carbazoles,12 wesought to establish if the reaction sequence just mentionedcould provide a useful means for obtaining natural productsembodying the latter ring system. Herein we report theoutcomes of such studies and by which means we have beenable to realize syntheses of the parent carbazole (1) as well asthe natural products 3-methyl-9H-carbazole (2),13 glycoborine(3, aka glycrophylamine),3a,14 glycozoline (4),3a,15 clauszoline K(5),16 mukonine (6),17 and karapinchamine A (7)18 togetherwith their monomethoxylated congener 8 (Figure 2).
■ RESULTS AND DISCUSSIONOur initial studies focused on acquiring targets 1 and 2, and thisinvolved (Scheme 1) the initial palladium-catalyzed Ullmanncross-coupling of 2-iodocyclohex-2-en-1-one (9)19 or its C4-methylated counterpart 1011 with commercially available o-bromonitrobenzene (11) under conditions defined earlier,10
and thereby affording the anticipated and previously reported2-arylcyclohex-2-en-1-ones 1210,11 (85%) and 1311 (86%),respectively. A methanolic solution of each of compounds 12and 13 was then subjected to reaction with hydrogen in thepresence of commercially available W-2 Raney nickel at roomtemperature, and so affording the tetrahydrocarbazoles1410,11,12b (64%) and 1511,12b (58%), respectively. Various
Received: January 8, 2017Published: February 22, 2017
Figure 1. Structure of parent 9H-carbazole (1) and the associated ringlabeling and atom numbering.
Article
pubs.acs.org/joc
© 2017 American Chemical Society 4148 DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
302
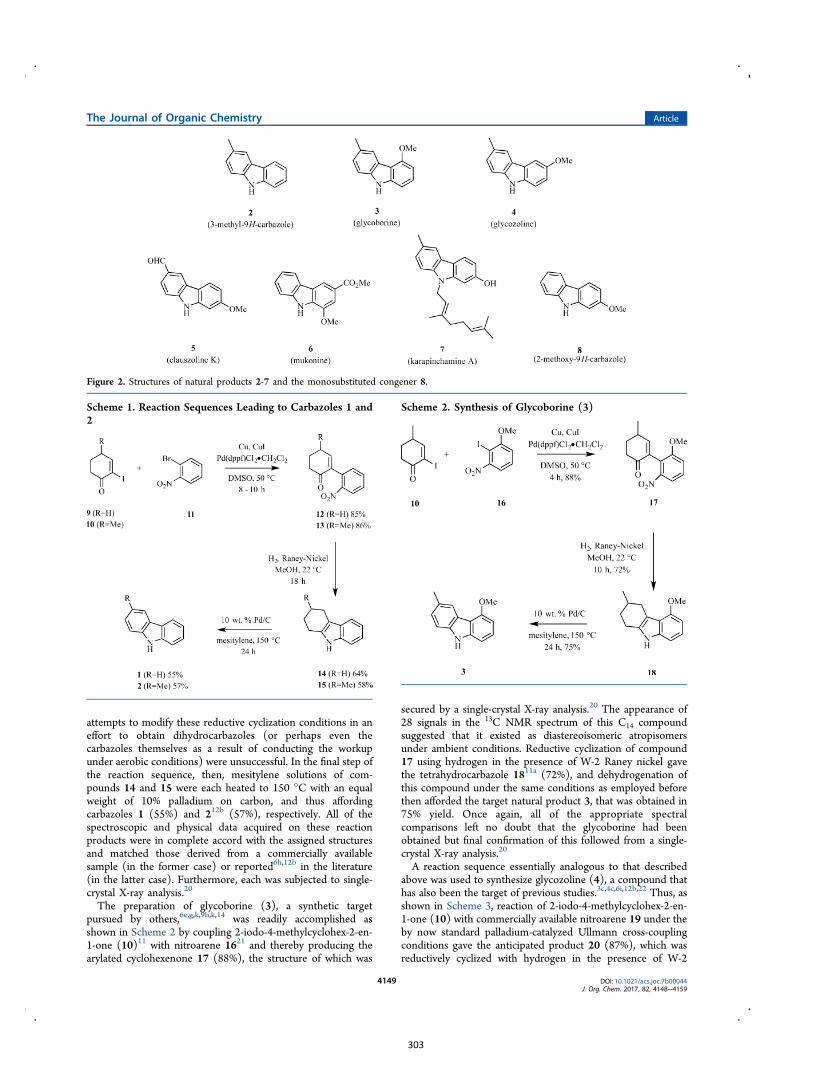
attempts to modify these reductive cyclization conditions in aneffort to obtain dihydrocarbazoles (or perhaps even thecarbazoles themselves as a result of conducting the workupunder aerobic conditions) were unsuccessful. In the final step ofthe reaction sequence, then, mesitylene solutions of com-pounds 14 and 15 were each heated to 150 °C with an equalweight of 10% palladium on carbon, and thus affordingcarbazoles 1 (55%) and 212b (57%), respectively. All of thespectroscopic and physical data acquired on these reactionproducts were in complete accord with the assigned structuresand matched those derived from a commercially availablesample (in the former case) or reported6h,12b in the literature(in the latter case). Furthermore, each was subjected to single-crystal X-ray analysis.20
The preparation of glycoborine (3), a synthetic targetpursued by others,6e,g,k,9h,k,14 was readily accomplished asshown in Scheme 2 by coupling 2-iodo-4-methylcyclohex-2-en-1-one (10)11 with nitroarene 1621 and thereby producing thearylated cyclohexenone 17 (88%), the structure of which was
secured by a single-crystal X-ray analysis.20 The appearance of28 signals in the 13C NMR spectrum of this C14 compoundsuggested that it existed as diastereoisomeric atropisomersunder ambient conditions. Reductive cyclization of compound17 using hydrogen in the presence of W-2 Raney nickel gavethe tetrahydrocarbazole 1811a (72%), and dehydrogenation ofthis compound under the same conditions as employed beforethen afforded the target natural product 3, that was obtained in75% yield. Once again, all of the appropriate spectralcomparisons left no doubt that the glycoborine had beenobtained but final confirmation of this followed from a single-crystal X-ray analysis.20
A reaction sequence essentially analogous to that describedabove was used to synthesize glycozoline (4), a compound thathas also been the target of previous studies.3c,4c,6i,12b,22 Thus, asshown in Scheme 3, reaction of 2-iodo-4-methylcyclohex-2-en-1-one (10) with commercially available nitroarene 19 under theby now standard palladium-catalyzed Ullmann cross-couplingconditions gave the anticipated product 20 (87%), which wasreductively cyclized with hydrogen in the presence of W-2
Figure 2. Structures of natural products 2-7 and the monosubstituted congener 8.
Scheme 1. Reaction Sequences Leading to Carbazoles 1 and2
Scheme 2. Synthesis of Glycoborine (3)
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4149
303
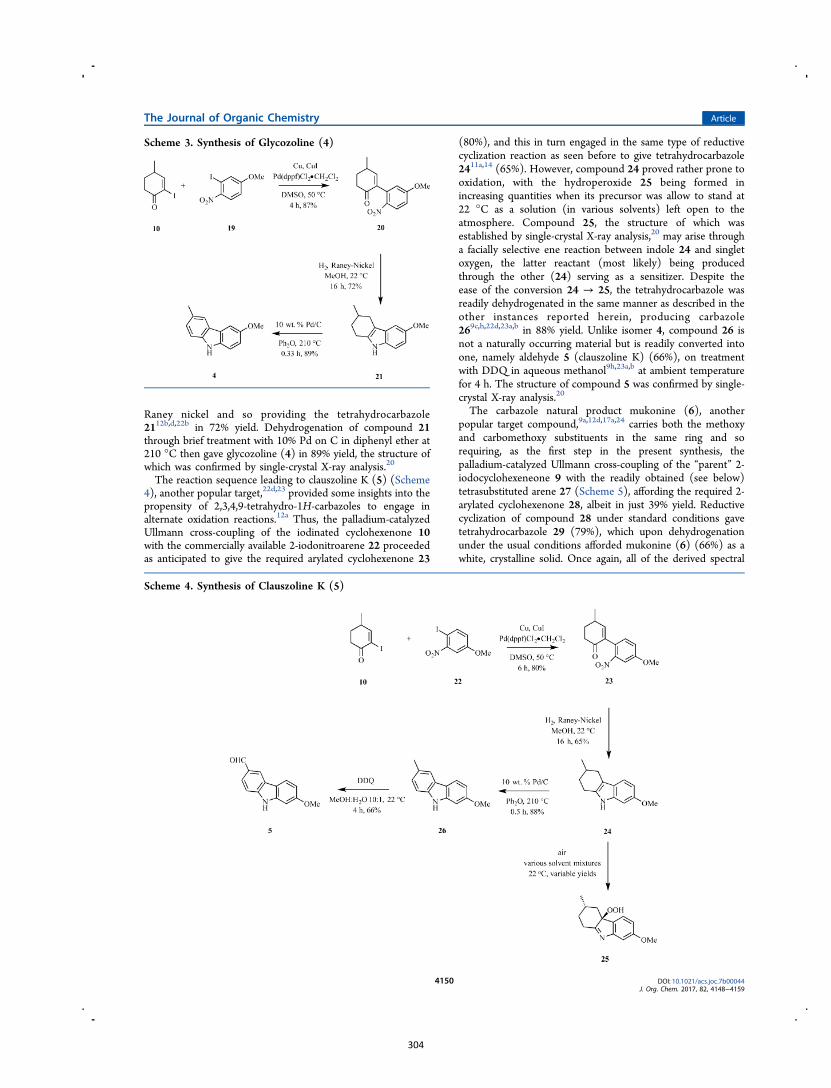
Raney nickel and so providing the tetrahydrocarbazole2112b,d,22b in 72% yield. Dehydrogenation of compound 21through brief treatment with 10% Pd on C in diphenyl ether at210 °C then gave glycozoline (4) in 89% yield, the structure ofwhich was confirmed by single-crystal X-ray analysis.20
The reaction sequence leading to clauszoline K (5) (Scheme4), another popular target,22d,23 provided some insights into thepropensity of 2,3,4,9-tetrahydro-1H-carbazoles to engage inalternate oxidation reactions.12a Thus, the palladium-catalyzedUllmann cross-coupling of the iodinated cyclohexenone 10with the commercially available 2-iodonitroarene 22 proceededas anticipated to give the required arylated cyclohexenone 23
(80%), and this in turn engaged in the same type of reductivecyclization reaction as seen before to give tetrahydrocarbazole2411a,14 (65%). However, compound 24 proved rather prone tooxidation, with the hydroperoxide 25 being formed inincreasing quantities when its precursor was allow to stand at22 °C as a solution (in various solvents) left open to theatmosphere. Compound 25, the structure of which wasestablished by single-crystal X-ray analysis,20 may arise througha facially selective ene reaction between indole 24 and singletoxygen, the latter reactant (most likely) being producedthrough the other (24) serving as a sensitizer. Despite theease of the conversion 24 → 25, the tetrahydrocarbazole wasreadily dehydrogenated in the same manner as described in theother instances reported herein, producing carbazole269c,h,22d,23a,b in 88% yield. Unlike isomer 4, compound 26 isnot a naturally occurring material but is readily converted intoone, namely aldehyde 5 (clauszoline K) (66%), on treatmentwith DDQ in aqueous methanol9h,23a,b at ambient temperaturefor 4 h. The structure of compound 5 was confirmed by single-crystal X-ray analysis.20
The carbazole natural product mukonine (6), anotherpopular target compound,9a,12d,17a,24 carries both the methoxyand carbomethoxy substituents in the same ring and sorequiring, as the first step in the present synthesis, thepalladium-catalyzed Ullmann cross-coupling of the “parent” 2-iodocyclohexeneone 9 with the readily obtained (see below)tetrasubstituted arene 27 (Scheme 5), affording the required 2-arylated cyclohexenone 28, albeit in just 39% yield. Reductivecyclization of compound 28 under standard conditions gavetetrahydrocarbazole 29 (79%), which upon dehydrogenationunder the usual conditions afforded mukonine (6) (66%) as awhite, crystalline solid. Once again, all of the derived spectral
Scheme 3. Synthesis of Glycozoline (4)
Scheme 4. Synthesis of Clauszoline K (5)
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4150
304
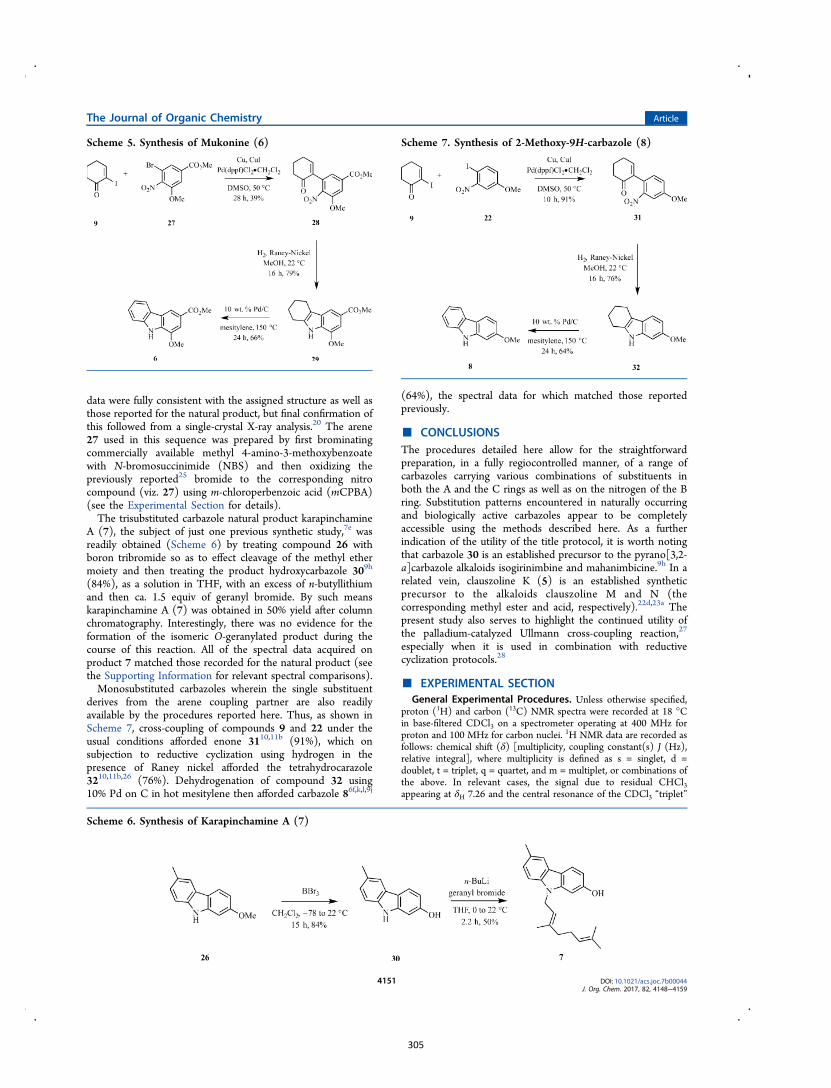
data were fully consistent with the assigned structure as well asthose reported for the natural product, but final confirmation ofthis followed from a single-crystal X-ray analysis.20 The arene27 used in this sequence was prepared by first brominatingcommercially available methyl 4-amino-3-methoxybenzoatewith N-bromosuccinimide (NBS) and then oxidizing thepreviously reported25 bromide to the corresponding nitrocompound (viz. 27) using m-chloroperbenzoic acid (mCPBA)(see the Experimental Section for details).The trisubstituted carbazole natural product karapinchamine
A (7), the subject of just one previous synthetic study,7e wasreadily obtained (Scheme 6) by treating compound 26 withboron tribromide so as to effect cleavage of the methyl ethermoiety and then treating the product hydroxycarbazole 309h
(84%), as a solution in THF, with an excess of n-butyllithiumand then ca. 1.5 equiv of geranyl bromide. By such meanskarapinchamine A (7) was obtained in 50% yield after columnchromatography. Interestingly, there was no evidence for theformation of the isomeric O-geranylated product during thecourse of this reaction. All of the spectral data acquired onproduct 7 matched those recorded for the natural product (seethe Supporting Information for relevant spectral comparisons).Monosubstituted carbazoles wherein the single substituent
derives from the arene coupling partner are also readilyavailable by the procedures reported here. Thus, as shown inScheme 7, cross-coupling of compounds 9 and 22 under theusual conditions afforded enone 3110,11b (91%), which onsubjection to reductive cyclization using hydrogen in thepresence of Raney nickel afforded the tetrahydrocarazole3210,11b,26 (76%). Dehydrogenation of compound 32 using10% Pd on C in hot mesitylene then afforded carbazole 86f,k,l,9j
(64%), the spectral data for which matched those reportedpreviously.
■ CONCLUSIONSThe procedures detailed here allow for the straightforwardpreparation, in a fully regiocontrolled manner, of a range ofcarbazoles carrying various combinations of substituents inboth the A and the C rings as well as on the nitrogen of the Bring. Substitution patterns encountered in naturally occurringand biologically active carbazoles appear to be completelyaccessible using the methods described here. As a furtherindication of the utility of the title protocol, it is worth notingthat carbazole 30 is an established precursor to the pyrano[3,2-a]carbazole alkaloids isogirinimbine and mahanimbicine.9h In arelated vein, clauszoline K (5) is an established syntheticprecursor to the alkaloids clauszoline M and N (thecorresponding methyl ester and acid, respectively).22d,23a Thepresent study also serves to highlight the continued utility ofthe palladium-catalyzed Ullmann cross-coupling reaction,27
especially when it is used in combination with reductivecyclization protocols.28
■ EXPERIMENTAL SECTIONGeneral Experimental Procedures. Unless otherwise specified,
proton (1H) and carbon (13C) NMR spectra were recorded at 18 °Cin base-filtered CDCl3 on a spectrometer operating at 400 MHz forproton and 100 MHz for carbon nuclei. 1H NMR data are recorded asfollows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz),relative integral], where multiplicity is defined as s = singlet, d =doublet, t = triplet, q = quartet, and m = multiplet, or combinations ofthe above. In relevant cases, the signal due to residual CHCl3appearing at δH 7.26 and the central resonance of the CDCl3 “triplet”
Scheme 5. Synthesis of Mukonine (6)
Scheme 6. Synthesis of Karapinchamine A (7)
Scheme 7. Synthesis of 2-Methoxy-9H-carbazole (8)
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4151
305

appearing at δC 77.0 were used to reference 1H and 13C NMR spectra,respectively. Samples were analyzed by infrared spectroscopy (νmax) asthin films on KBr plates. Low- and high-resolution electron impact(EI) mass spectra were recorded on a double-focusing, triple-sectormachine. Low- and high-resolution ESI mass spectra were recorded ona triple-quadrupole mass spectrometer operating in either positive ornegative ion mode. Melting points are uncorrected. Analytical thin-layer chromatography (TLC) was performed on aluminum-backed 0.2mm thick silica gel 60 F254 plates. Eluted plates were visualized using a254 nm UV lamp and/or by treatment with a suitable dip followed byheating. These dips included phosphomolybdic acid/ceric sulfate/sulfuric acid (concentrated)/water (37.5 g/7.5 g/37.5 g/720 mL),potassium permanganate/potassium carbonate/5% sodium hydroxideaqueous solution/water (3 g/20 g/5 mL/300 mL), and p-anisaldehydeor vanillin/sulfuric acid (concentrated)/ethanol (15 g/2.5 mL/250mL). Flash chromatographic separations were carried out followingprotocols defined by Still et al.29 with silica gel 60 (40−63 μm) as thestationary phase and using the AR- or HPLC-grade solvents indicated.The melting points of solids purified by such means were recordeddirectly (i.e. after they had crystallized from the concentratedchromatographic fractions). Starting materials, reagents, drying agents,and other inorganic salts were generally commercially available andwere used as supplied. The copper powder used in the palladium-catalyzed Ullmann cross-coupling reactions had a particle size of <75μm. Tetrahydrofuran (THF), methanol, and dichloromethane weredried using a solvent purification system that is based upon atechnology originally described by Grubbs et al.30 Where necessary,reactions were performed under a nitrogen atmosphere.Specific Chemical Transformations. 2-Iodocyclohex-2-en-1-
one (9). Following a procedure reported by Johnson et al.,31 amagnetically stirred solution of cyclohex-2-en-1-one (2.00 g, 20.8mmol) in chloroform/pyridine (80 mL of a 1/1 v/v mixture)maintained at 0 °C under a nitrogen atmosphere was treated, dropwiseover 2 h, with a solution of molecular iodine (18.5 g, 72.8 mmol) inchloroform/pyridine (80 mL of a 1/1 v/v mixture). The ensuingmixture was warmed to 22 °C, stirred at this temperature for 16 h thendiluted with diethyl ether (200 mL) and washed with water (1 × 100mL), Na2S2O3 (2 × 100 mL of a 20% w/v aqueous solution), HCl (2× 100 mL of a 1 M aqueous solution), water (2 × 100 mL) and brine(1 × 100 mL) before being dried (MgSO4), filtered, and concentratedunder reduced pressure to give a yellow oil. This was subjected to flashcolumn chromatography (silica, 2/5/70 v/v/v ethyl acetate/dichloro-methane/40−60 petroleum ether elution) to give, after concentrationof the appropriate fractions (Rf = 0.6 in 2/5/11 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether), 2-iodocyclohex-2-en-1-one919 (3.85 g, 83%) as a light yellow solid, mp 47−50 °C (lit.19 mp 47−49 °C): 1H NMR (400 MHz, CDCl3) δ 7.77 (t, J = 4.4 Hz, 1H), 2.67(m, 2H), 2.44 (m, 2H), 2.09 (m, 2H); 13C NMR (100 MHz, CDCl3) δ192.3, 159.6, 104.0, 37.4, 30.1, 23.0; IR νmax 2949, 2939, 1675, 1585,1421, 1314, 1120, 966, 914, 803, 703 cm−1; MS (EI, 70 eV) m/z 222(M+•, 100%), 194 (75), 67 (40); HRMS M+• calcd for C6H7
127IO221.9542, found 221.9540.2-Iodo-4-methylcyclohex-2-en-1-one (10). A magnetically stirred
solution of 4-methylcyclohex-2-en-1-one32 (2.29 g, 20.8 mmol) inchloroform/pyridine (80 mL of a 1/1 v/v mixture) maintained at 0 °Cunder a nitrogen atmosphere was treated, dropwise over 2 h, with asolution of molecular iodine (18.5 g, 72.8 mmol) in chloroform/pyridine (80 mL of a 1/1 v/v mixture). The ensuing mixture waswarmed to 22 °C, stirred at this temperature for 16 h, and then dilutedwith diethyl ether (200 mL) and washed with water (1 × 100 mL),Na2S2O3 (2 × 100 mL of a 20% w/v aqueous solution), HCl (2 × 100mL of a 1 M aqueous solution), water (2 × 100 mL), and brine (1 ×100 mL) before being dried (MgSO4), filtered, and concentratedunder reduced pressure to give a yellow oil. Subjection of this materialto flash column chromatography (silica, 1/19 v/v ethyl acetate/40−60petroleum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.5 in 1/5 v/v ethyl acetate/40−60 petroleum ether),compound 1011b (3.98 g, 81%) as a light yellow oil: 1H NMR (400MHz, CDCl3) δ 7.50 (d, J = 2.6 Hz, 1H), 2.66−2.56 (complex m, 2H),2.48−2.39 (complex m, 1H), 2.07 (m, 1H), 1.64 (m, 1H), 1.08 (d, J =
7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 191.8, 164.7, 102.9, 35.6,35.5, 30.6, 19.7; IR νmax 2959, 2930, 2870, 1686, 1585, 1454, 1318,1109, 790 cm−1; MS (EI, 70 eV) m/z 236 (M+•, 100%), 208 (25), 109(75), 81 (50), 53 (60); HRMS M+• calcd for C7H9
127IO 235.9698,found 235.9691.
2′-Nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-one (12). A magneti-cally stirred mixture of o-bromonitrobenzene (11) (941 mg, 4.7mmol), 2-iodocyclohex-2-en-1-one (9) (470 mg, 2.1 mmol), copperpowder (673 mg, 10.6 g atom), CuI (605 mg, 3.2 mmol), andPd(dppf)Cl2·CH2Cl2 (87 mg, 0.1 mmol) in deoxygenated DMSO (21mL) maintained under nitrogen was heated to 50 °C for 10 h. Thereaction mixture was then cooled to room temperature, quenched withwater (1 × 20 mL), diluted with ethyl acetate (1 × 40 mL), andfiltered through a pad of diatomaceous earth and silica gel. The padand the solids thus retained were washed with ethyl acetate (2 × 50mL), and the filtrate was washed with water (2 × 100 mL) and thenbrine (2 × 100 mL). The separated organic phase was dried (Na2SO4),filtered, and concentrated under reduced pressure to give a brown oil,and subjection of this material to flash chromatography (silica, 1/9 v/vethyl acetate/40−60 petroleum ether elution) gave, after concen-tration of the appropriate fractions (Rf = 0.4 in 1/1 v/v ethyl acetate/40−60 petroleum ether), compound 1210,11 (390 mg, 85%) as a lightyellow crystal, mp 96−99 °C (lit.10 mp 92−95 °C): 1H NMR (400MHz, CDCl3) δ 7.99 (m, 1H), 7.58 (m, 1H), 7.46 (m, 1H), 7.25 (m,1H), 7.00 (t, J = 4.2 Hz, 1H), 2.56 (m, 4H), 2.13 (m, 2H); 13C NMR(100 MHz, CDCl3) δ 196.6, 148.7, 146.8, 139.4, 133.4, 132.2, 131.8,128.8, 124.2, 38.4, 26.4, 22.7; IR νmax 2949, 2868, 1679, 1523, 1353,1158, 859, 788, 751, 709 cm−1; MS (ESI, +ve) m/z 240 [(M + Na)+,100%], 218 [(M + H)+, 10]; HRMS (M + H)+ calcd for C12H12NO3218.0817, found 218.0825.
5-Methyl-2′-nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-one (13). Amagnetically stirred mixture of o-bromonitrobenzene (11) (960 mg,4.75 mmol), 2-iodo-4-methylcyclohex-2-en-1-one (10) (510 mg, 2.16mmol), copper powder (687 mg, 10.80 g atom), CuI (617 mg, 3.24mmol), and Pd(dppf)Cl2·CH2Cl2 (88 mg, 0.11 mmol) indeoxygenated DMSO (22 mL) was heated to 50 °C under a nitrogenatmosphere for 8 h. The reaction mixture was then cooled to roomtemperature, quenched with water (20 mL), diluted with ethyl acetate(40 mL), and then filtered through a pad of diatomaceous earth andsilica gel. The pad and the solids thus retained were washed with ethylacetate (2 × 50 mL), and the organic phase associated with the filtratewas then washed with water (2 × 100 mL) and brine (2 × 100 mL)before being dried (Na2SO4), filtered, and concentrated under reducedpressure to give a brown oil. Subjection of this material to flashchromatography (silica, 1/9 v/v ethyl acetate/40−60 petroleum etherelution) gave, after concentration of the appropriate fractions (Rf = 0.5in 1/1 v/v ethyl acetate/40−60 petroleum ether), compound 1311
(430 mg, 86%) as a light yellow oil: 1H NMR (400 MHz, CDCl3) δ7.99 (m, 1H), 7.58 (m, 1H), 7.45 (m, 1H), 7.40 (d, J = 7.6 Hz, 1H),6.80 (d, J = 1.6 Hz, 1H), 2.79 (broad s, 1H), 2.65−2.47 (complex m,2H), 2.20 (m, 1H), 1.86−1.76 (complex m, 1H), 1.24 (m, 3H); 13CNMR (100 MHz, CDCl3) δ 196.6, 152.3, 148.7, 138.4, 133.4, 132.1,131.8, 128.9, 124.3, 37.2, 31.8, 30.7, 20.4; IR νmax 2960, 2872, 1681,1525, 1354, 1159, 788, 750 cm−1; MS (ESI, +ve) m/z 254 [(M +Na)+, 100%], 232 [(M + H)+, 10]; HRMS (M + Na)+ calcd forC13H13NNaO3 254.0793, found 254.0799.
2,3,4,9-Tetrahydro-1H-carbazole (14). A magnetically stirredmixture of compound 12 (100 mg, 0.46 mmol) and commerciallyavailable W-2 Raney nickel (200 mg, washed twice with absoluteethanol) in methanol (23 mL) was deoxygenated and then stirredunder an atmosphere of hydrogen at 22 °C for 18 h. After this timeand using an externally applied magnet to hold the solid associatedwith the reaction mixture within the flask, the supernatant liquid wasdecanted and the retained solid washed with methanol (2 × 30 mL)(Caution! Af ter these washings water should be added to the residual solidso as to prevent a f ire.). The methanolic solutions were combined andthen concentrated under reduced pressure to give a white solid.Subjection of this material to flash column chromatography (silica, 1/9v/v ethyl acetate/40−60 petroleum ether elution) gave, afterconcentration of the appropriate fractions (Rf = 0.6 in 1/5 v/v ethyl
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4152
306

acetate/40−60 petroleum ether), compound 1410,11,12b (50 mg, 64%)as a white solid, mp 104−106 °C (lit.12b mp 104−106 °C): 1H NMR[400 MHz, (CD3)2CO] δ 9.70 (broad s, 1H), 7.36 (d, J = 7.8 Hz, 1H),7.26 (d, J = 7.8 Hz, 1H), 7.02−6.92 (complex m, 2H), 2.73 (t, J = 5.9Hz, 2H), 2.68−2.65 (complex m, 2H), 1.92−1.81 (complex m, 4H);13C NMR [100 MHz, (CD3)2CO] δ 137.1, 135.1, 128.8, 121.1, 119.1,118.0, 111.3, 109.7, 24.2, 24.0, 23.8, 21.7; IR νmax 3396, 2926, 2849,1467, 1450, 1439, 1325, 1304, 1233, 736, 635 cm−1; MS (ESI, +ve) m/z 226 [(M + MeOH + Na)+, 100%], 188 (56), 172 [(M + H)+, 70];HRMS (M + H)+ calcd for C12H14N 172.1126, found 172.1121.3-Methyl-2,3,4,9-tetrahydro-1H-carbazole (15). A magnetically
stirred mixture of compound 13 (320 mg, 1.38 mmol) andcommercially available W-2 Raney nickel (640 mg, washed twicewith absolute ethanol) in methanol (69 mL) was deoxygenated andthen stirred under an atmosphere of hydrogen at 22 °C for 18 h. Afterthis time and using an externally applied magnet to hold the solidassociated with the reaction mixture within the flask, the supernatantliquid was decanted and the retained solid washed with methanol (2 ×30 mL) (Caution! Af ter these washings water should be added to theresidual solid so as to prevent a f ire.). The methanolic solutions werecombined and then concentrated under reduced pressure to give awhite solid. Subjection of this material to flash column chromatog-raphy (silica, 1/9 v/v ethyl acetate/40−60 petroleum ether elution) togive, after concentration of the appropriate fractions (Rf = 0.6 in 1/5v/v ethyl acetate/40−60 petroleum ether), compound 1511,12b (148mg, 58%) as a white solid, mp 98−100 °C (lit.12b mp 98−100 °C): 1HNMR [400 MHz, (CD3)2CO] δ 9.70 (broad s, 1H), 7.36 (d, J = 7.6Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.02−6.92 (complex m, 2H), 2.83−2.75 (complex m, 3H), 2.24 (m, 1H), 1.98−1.86 (complex m, 2H),1.53 (m, 1H), 1.13 (d, J = 6.6 Hz, 3H); 13C NMR [100 MHz,(CD3)2CO] δ 137.4, 134.9, 128.7, 121.1, 119.1, 118.0, 111.3, 109.7,32.3, 30.6, 30.2, 23.4, 22.1; IR νmax 3405, 2949, 2923, 1620, 1467,1326, 1232, 1009, 740 cm−1; MS (ESI, +ve) m/z 240 [(M + MeOH +Na)+, 100%], 202 (45), 186 [(M + H)+, 30]; HRMS (M + H)+ calcdfor C13H16N 186.1283, found 186.1286.9H-Carbazole (1). A magnetically stirred mixture of compound 14
(46 mg, 0.27 mmol) and 10 wt % Pd/C (46 mg) in mesitylene (5 mL)was stirred at 150 °C under an atmosphere of nitrogen for 24 h. Thecooled reaction mixture was filtered through filter paper, and the solidsthus retained were washed with ethyl acetate (2 × 15 mL). The filtratewas concentrated under reduced pressure, and the ensuing mixture ofproduct 1 and mesitylene was then subjected to flash columnchromatography (silica, 1/19 v/v ethyl acetate/40−60 petroleum etherelution). Concentration of the appropriate fractions (Rf = 0.6 in 1/5 v/v ethyl acetate/40−60 petroleum ether) then gave compound 1 (25mg, 55%) as a white, crystalline solid, mp 237−238 °C (lit.33 mp 245°C): 1H NMR [400 MHz, (CD3)2CO] δ 10.32 (broad s, 1H), 8.11 (d,J = 7.8 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.39 (m, 2H), 7.18 (m, 2H);13C NMR [100 MHz, (CD3)2CO] δ 141.0, 126.4, 124.0, 120.9, 119.6,111.7; IR νmax 3416, 3049, 1597, 1449, 1233, 928, 745, 722 cm−1; MS(ESI, − ve) m/z 166 [(M − H)−, 100%]; HRMS (M − H)− calcd forC12H8N 166.0657, found 166.0656.3-Methyl-9H-carbazole (2). A magnetically stirred mixture of
compound 15 (145 mg, 0.78 mmol) and 10 wt % Pd/C (145 mg) inmesitylene (15 mL) was stirred at 150 °C under an atmosphere ofnitrogen for 24 h. The cooled reaction mixture was filtered throughfilter paper, and the solids thus retained were washed with ethyl acetate(2 × 15 mL). The filtrate was concentrated under reduced pressure,and the ensuing mixture of product 2 and mesitylene was thensubjected to flash column chromatography (silica, 1/19 v/v ethylacetate/40−60 petroleum ether elution) to give, after concentration ofthe appropriate fractions (Rf = 0.6 in 1/3 v/v ethyl acetate/40−60petroleum ether), compound 212b (80 mg, 57%) as a white solid, mp200−202 °C (lit.12b mp 205−210 °C): 1H NMR [400 MHz,(CD3)2CO] δ 10.17 (broad s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.90(s, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.37−7.33(complex m, 1H), 7.22 (dd, J = 8.2 and 1.2 Hz, 1H), 7.15 (m, 1H),2.48 (s, 3H); 13C NMR [100 MHz, (CD3)2CO] δ 141.3, 139.2, 128.6,127.7, 126.2, 124.2, 123.8, 120.8, 120.7, 119.4, 111.6, 111.4, 21.5; IRνmax 3403, 3050, 2914, 2853, 1605, 1459, 1333, 1238, 805, 746, 726
cm−1; MS (ESI, −ve) m/z 180 [(M − H)−, 100%]; HRMS (M − H)−
calcd for C13H10N 180.0813, found 180.0804.2′-Methoxy-5-methyl-6′-nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-
one (17). A magnetically stirred mixture of 2-iodo-1-methoxy-3-nitrobenzene (16)21 (341 mg, 1.22 mmol), 2-iodo-4-methylcyclohex-2-en-1-one (10) (519 mg, 2.20 mmol), copper powder (311 mg, 4.89g atom), CuI (349 mg, 1.83 mmol), and Pd(dppf)Cl2·CH2Cl2 (100mg, 0.12 mmol) in deoxygenated DMSO (24 mL) was heated to 50°C under a nitrogen atmosphere for 4 h. The reaction mixture wasthen cooled to room temperature, quenched with water (10 mL),diluted with ethyl acetate (20 mL), and then filtered through a pad ofdiatomaceous earth and silica gel. The pad and the solids thus retainedwere washed with ethyl acetate (2 × 25 mL) and the organic phaseassociated with the filtrate then washed with water (2 × 50 mL) andbrine (2 × 50 mL) before being dried (Na2SO4), filtered, andconcentrated under reduced pressure to give a brown oil. Subjection ofthis material to flash chromatography (silica, 2/5/40 v/v/v ethylacetate/dichloromethane/40−60 petroleum ether elution) gave, afterconcentration of the appropriate fractions (Rf = 0.3 in 2/5/11 v/v/vethyl acetate/dichloromethane/40−60 petroleum ether), compound17 (281 mg, 88%) as a light yellow solid, mp 94−96 °C: 1H NMR(400 MHz, CDCl3) δ 7.53 (m, 1H), 7.40−7.10 (complex m, 2H), 6.63(m, 1H), 3.78 (m, 3H), 2.76−2.48 (complex m, 3H), 2.24−2.14(complex m, 1H), 1.90−1.76 (complex m, 1H), 1.20 (m, 3H); 13CNMR (100 MHz, CDCl3) δ 196.9, 196.7, 158.1, 158.0, 153.7, 153.5,150.3, 150.0, 133.0, 132.6, 129.1, 129.2, 121.5, 121.4, 116.1, 116.0,115.6, 115.4, 56.7, 56.6, 37.3, 37.1, 32.0, 31.6, 30.9, 30.5, 20.4, 20.1; IRνmax 2959, 1674, 1524, 1461, 1351, 1263, 1052, 794, 736 cm−1; MS(ESI, +ve) m/z 284 [(M + Na)+, 100%], 262 [(M + H)+, 15]; HRMS(M + Na)+ calcd for C14H15NNaO4 284.0899, found 284.0896.
5-Methoxy-3-methyl-2,3,4,9-tetrahydro-1H-carbazole (18). Amagnetically stirred mixture of compound 17 (211 mg, 0.81 mmol)and commercially available W-2 Raney nickel (422 mg, washed twicewith absolute ethanol) in methanol (40 mL) was deoxygenated andthen stirred under an atmosphere of hydrogen at 22 °C for 10 h. Afterthis time and using an externally applied magnet to hold the solidassociated with the reaction mixture within the flask, the supernatantliquid was decanted and the retained solid washed with methanol (2 ×15 mL) (Caution! Af ter these washings water should be added to theresidual solid so as to prevent a f ire.). The methanolic solutions werecombined and then concentrated under reduced pressure to give awhite solid. Subjection of this material to flash column chromatog-raphy (silica, 1/3/30 v/v/v acetone/dichloromethane/40−60 petro-leum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.5 in 1/3/6 v/v/v acetone/dichloromethane/40−60petroleum ether), compound 1811a (125 mg, 72%) as a white solid,mp 126−128 °C: 1H NMR [400 MHz, (CD3)2CO] δ 9.63 (broad s,1H), 6.91−6.85 (complex m, 2H), 6.41 (dd, J = 6.5 and 1.9 Hz, 1H),3.84 (s, 3H), 3.11 (dd, J = 16.0 and 4.7 Hz, 1H), 2.72 (m, 2H), 2.41(m, 1H), 1.92−1.79 (complex m, 2H), 1.48 (m, 1H), 1.10 (d, J = 6.6Hz, 3H); 13C NMR [100 MHz, (CD3)2CO] δ 155.1, 138.8, 132.8,121.8, 118.6, 109.6, 105.1, 99.7, 55.3, 32.7, 32.1, 30.8, 23.4, 22.2; IRνmax 3397, 2948, 2923, 2836, 1565, 1505, 1349, 1253, 1244, 1104, 775,729 cm−1; MS (ESI, +ve) m/z 232 (100%), 216 [(M + H)+, 90];HRMS (M + H)+ calcd for C14H18NO 216.1388, found 216.1393.
Glycoborine (3). A magnetically stirred mixture of compound 18(50 mg, 0.23 mmol) and 10 wt % Pd/C (50 mg) in mesitylene (5 mL)was stirred at 150 °C under an atmosphere of nitrogen for 24 h. Thecooled reaction mixture was filtered through filter paper, and the solidsthus retained were washed with ethyl acetate (2 × 10 mL). Thecombined filtrates were concentrated under reduced pressure, and theensuing mixture of product 3 and mesitylene was then subjected toflash column chromatography (silica, 2/5/62 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether elution) to give, afterconcentration of the appropriate fractions (Rf = 0.4(5) in 2/5/11 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether), com-pound 33a (35 mg, 75%) as a white solid, mp 130−132 °C (lit.3a mp132.0−134.6 °C): 1H NMR [400 MHz, (CD3)2CO] δ 10.17 (broad s,1H), 8.07 (s, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H),7.17 (dd, J = 8.2 and 1.2 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.68 (d, J =
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4153
307

8.0 Hz, 1H), 4.06 (s, 3H), 2.48 (s, 3H); 13C NMR [100 MHz,(CD3)2CO] δ 157.1, 142.7, 138.4, 128.5, 127.2, 126.7, 123.6, 123.5,113.0, 110.8, 104.7, 100.5, 55.6, 21.6; IR νmax 3402, 2917, 2838, 1608,1585, 1506, 1459, 1347, 1260, 1098, 750, 723 cm−1; MS (ESI, +ve) m/z 234 [(M + Na)+, 10%], 212 [(M + H)+, 100]; HRMS (M + H)+
calcd for C14H14NO 212.1075, found 212.1078.5′-Methoxy-5-methyl-2′-nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-
one (20). A magnetically stirred mixture of commercially available 2-iodo-4-methoxynitrobenzene (19) (150 mg, 0.54 mmol), 2-iodo-4-methylcyclohex-2-en-1-one (10) (229 mg, 0.97 mmol), copperpowder (137 mg, 2.15 g atom), CuI (154 mg, 0.81 mmol), andPd(dppf)Cl2·CH2Cl2 (44 mg, 0.05 mmol) in deoxygenated DMSO(12 mL) was heated to 50 °C under a nitrogen atmosphere for 4 h.The reaction mixture was then cooled to room temperature, quenchedwith water (5 mL), diluted with ethyl acetate (10 mL), and thenfiltered through a pad of diatomaceous earth and silica gel. The padand the solids thus retained were washed with ethyl acetate (2 × 15mL), and the separated organic phase associated with the filtrate wasthen washed with water (2 × 25 mL) and brine (2 × 25 mL) beforebeing dried (Na2SO4), filtered, and concentrated under reducedpressure to give a brown oil. Subjection of this material to flashchromatography (silica, 1/3/25 v/v/v acetone/dichloromethane/40−60 petroleum ether elution) gave, after concentration of theappropriate fractions [Rf = 0.5(5) in 1/3/6 v/v/v acetone/dichloro-methane/40−60 petroleum ether], compound 20 (125 mg, 87%) as alight yellow oil: 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 9.1 Hz,1H), 6.90 (dd, J = 9.1 and 2.6 Hz, 1H), 6.70 (m, 2H), 3.88 (s, 3H),2.76−2.51 (complex m, 3H), 2.21 (m, 1H), 1.84 (m, 1H), 1.25 (d, J =7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.8, 163.4, 151.2,141.7, 139.1, 135.2, 127.2, 117.2, 113.2, 56.1, 37.3, 31.8, 30.8, 20.5; IRνmax 2953, 1681, 1578, 1512, 1340, 1298, 1238, 1026, 844 cm−1; MS(ESI, +ve) m/z 284 [(M + Na)+, 100%], 262 [(M + H)+, 10]; HRMS(M + Na)+ calcd for C14H15NNaO4 284.0899, found 284.0890.6-Methoxy-3-methyl-2,3,4,9-tetrahydro-1H-carbazole (21). A
magnetically stirred mixture of compound 20 (100 mg, 0.38 mmol)and commercially available W-2 Raney nickel (200 mg, washed twicewith absolute ethanol) in methanol (19 mL) was deoxygenated andthen stirred under an atmosphere of hydrogen at 22 °C for 16 h. Afterthis time and using an externally applied magnet to hold the solidassociated with the reaction mixture within the flask, the supernatantliquid was decanted and the retained solid washed with methanol (2 ×10 mL) (Caution! Af ter these washings water should be added to theresidual solid so as to prevent a f ire.). The methanolic solutions werecombined and then concentrated under reduced pressure to give awhite solid. Subjection of this material to flash column chromatog-raphy (silica, 1/3/20 v/v/v acetone/dichloromethane/40−60 petro-leum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.6 in 1/3/6 v/v/v acetone/dichloromethane/40−60petroleum ether), compound 2112b,d,22b (50 mg, 61%) as a white solid,mp 108−109 °C: 1H NMR [400 MHz, (CD3)2CO] δ 9.52 (broad s,1H), 7.14 (d, J = 8.6 Hz, 1H), 6.88 (s, 1H), 6.65 (d, J = 8.6 Hz, 1H),3.78 (s, 3H), 2.80−2.73 (complex m, 3H), 2.20 (m, 1H), 1.95−1.85(complex m, 2H), 1.52 (m, 1H), 1.12 (d, J = 6.5 Hz, 3H); 13C NMR[100 MHz, (CD3)2CO] δ 154.6, 135.7, 132.5, 129.1, 111.8, 110.7,109.6, 100.6, 55.8, 32.4, 30.6, 30.4, 23.6, 22.1; IR νmax 3401, 2947,2909, 2830, 1591, 1481, 1454, 1431, 1210, 1137, 1030, 831, 796 cm−1;MS (ESI, −ve) m/z 214 [(M − H)−, 100%]; HRMS (M − H)− calcdfor C14H16NO 214.1232, found 214.1235.Glycozoline (4). A magnetically stirred mixture of compound 21
(44 mg, 0.20 mmol) and 10 wt % Pd/C (44 mg) in diphenyl ether (5mL) was stirred at 210 °C under an atmosphere of nitrogen for 0.33 h.The cooled reaction mixture was filtered through filter paper, and thesolids thus retained were washed with ethyl acetate (2 × 5 mL). Thecombined filtrates were concentrated under reduced pressure, and theensuing mixture of product 4 and diphenyl ether was then subjected toflash column chromatography (silica, 1/9 v/v ethyl acetate/40−60petroleum ether elution) to give, after concentration of the appropriatefractions (Rf = 0.4 in 3/7 v/v ethyl acetate/40−60 petroleum ether),compound 43a,15 (39 mg, 89%) as a white, crystalline solid, mp 180−181 °C (lit.22b mp. = 177−178 °C): 1H NMR [400 MHz, (CD3)2CO]
δ 9.96 (broad s, 1H), 7.87 (s, 1H), 7.63 (d, J = 2.2 Hz, 1H), 7.36 (t, J =8.5 Hz, 2H), 7.18 (d, J = 8.2 Hz, 1H), 7.00 (dd, J = 8.5 and 2.2 Hz,1H), 3.88 (s, 3H), 2.47 (s, 3H); 13C NMR [100 MHz, (CD3)2CO] δ154.5, 140.0, 136.1, 128.1, 127.7, 124.2(2), 124.2(0), 120.8, 115.6,112.3, 111.5, 103.5, 56.1, 21.5; IR νmax 3398, 2928, 1493, 1470, 1457,1208, 1148, 1033, 806 cm−1; MS (ESI, −ve) m/z 210 [(M − H)−,60%], 195 (100); HRMS (M − H)− calcd for C14H12NO 210.0919,found 210.0912.
4′-Methoxy-5-methyl-2′-nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-one (23). A magnetically stirred mixture of commercially available 1-iodo-4-methoxy-2-nitrobenzene (22) (341 mg, 1.22 mmol), 2-iodo-4-methylcyclohex-2-en-1-one (10) (519 mg, 2.20 mmol), copperpowder (311 mg, 4.89 g atom), CuI (349 mg, 1.83 mmol), andPd(dppf)Cl2·CH2Cl2 (100 mg, 0.12 mmol) in deoxygenated DMSO(24 mL) was heated to 50 °C under a nitrogen atmosphere for 6 h.The ensuing mixture was cooled to room temperature, quenched withwater (10 mL), diluted with ethyl acetate (20 mL), and then filteredthrough a pad of diatomaceous earth and silica gel. The pad and thesolids thus retained were washed with ethyl acetate (2 × 30 mL), andthe separated organic phase associated with the filtrate was washedwith water (2 × 50 mL) and brine (2 × 50 mL) before being dried(Na2SO4), filtered, and concentrated under reduced pressure to give abrown oil. Subjection of this material to flash column chromatography(silica, 2/5/64 v/v/v ethyl acetate/dichloromethane/40−60 petro-leum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.4 in 2/5/11 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether), compound 23 (256 mg, 80%) as a lightyellow oil: 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 2.3 Hz, 1H),7.15−7.09 (complex m, 2H), 6.75 (dd, J = 2.7 and 1.1 Hz, 1H), 3.85(s, 3H), 2.75 (m, 1H), 2.65−2.40 (complex m, 2H), 2.19 (m, 1H),1.80 (m, 1H), 1.24 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3)δ 196.9, 159.7, 151.8, 149.2, 138.1, 132.7, 124.2, 119.7, 109.3, 56.0,37.3, 31.8, 30.8, 20.5; IR νmax 2960, 1681, 1529, 1353, 1302, 1266,1236, 1032, 800 cm−1; MS (ESI, +ve) m/z 284 [(M + Na)+, 100%],262 [(M + H)+, 15]; HRMS (M + Na)+ calcd for C14H15NNaO4284.0899, found 284.0901.
7-Methoxy-3-methyl-2,3,4,9-tetrahydro-1H-carbazole (24) andtrans-4a-Hydroperoxy-7-methoxy-3-methyl-2,3,4,4a-tetrahydro-1H-carbazole (25). A magnetically stirred mixture of compound 23(202 mg, 0.77 mmol) and commercially available W-2 Raney nickel(404 mg, washed twice with absolute ethanol) in methanol (38 mL)was deoxygenated and then stirred under an atmosphere of hydrogenat 22 °C for 16 h. After this time and using an externally appliedmagnet to hold the solid associated with the reaction mixture withinthe flask, the supernatant liquid was decanted and the retained solidwashed with methanol (2 × 20 mL) (Caution! Af ter these washingswater should be added to the residual solid so as to prevent a f ire.). Themethanolic solutions were combined and then concentrated underreduced pressure to give a white solid. Subjection of this material toflash column chromatography (silica, 1/3/20 v/v/v acetone/dichloro-methane/40−60 petroleum ether elution) gave two fractions, A and B.
Concentration of fraction A (Rf = 0.5 in 1/3/6 v/v/v acetone/dichloromethane/40−60 petroleum ether) afforded compound2411a,14 (109 mg, 65%) as a white, crystalline solid, mp 130−132°C: 1H NMR [400 MHz, (CD3)2CO] δ 9.50 (broad s, 1H), 7.22 (d, J= 8.5 Hz, 1H), 6.83 (d, J = 2.2 Hz, 1H), 6.63 (dd, J = 8.5 and 2.2 Hz,1H), 3.76 (s, 3H), 2.83−2.71 (complex m, 3H), 2.19 (m, 1H), 1.90(m, 2H), 1.50 (m, 1H), 1.11 (d, J = 6.5 Hz, 3H); 13C NMR [100MHz, (CD3)2CO] δ 156.6, 138.2, 133.4, 123.2, 118.5, 109.5, 108.6,95.4, 55.7, 32.3, 30.6, 30.3, 23.5, 22.1; IR νmax 3390, 2909, 2835, 1630,1570, 1496, 1459, 1335, 1291, 1203, 1157, 1029, 826, 805 cm−1; MS(ESI, +ve) m/z 238 [(M + Na)+, 20%], 218 (100), 216 [(M + H)+,90]; HRMS (M + H)+ calcd for C14H18NO 216.1388, found 216.1386.
Concentration of fraction B (Rf = 0.1 in 1/3/6 v/v/v acetone/dichloromethane/40−60 petroleum ether) afforded hydroperoxide 25(15 mg, 8%) as a white, crystalline solid, mp 106−108 °C: 1H NMR[700 MHz, (CD3)2CO] δ δ 10.81 (s, 1H), 7.26 (d, J = 8.0 Hz, 1H),6.97 (d, J = 2.3 Hz, 1H), 6.71 (dd, J = 8.0 and 2.3 Hz, 1H), 3.82 (s,3H), 2.82−2.76 (complex m, 1H), 2.68 (m, 1H), 2.43 (m, 1H), 2.14(m, 2H), 1.13 (m, 1H), 0.95 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H); 13C
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4154
308

NMR [175 MHz, (CD3)2CO] δ 186.1, 162.2, 157.4, 131.5, 123.8,110.8, 107.4, 92.3, 55.8, 44.4, 37.4, 30.3, 28.1, 21.0; IR νmax 3100, 2954,2836, 1607, 1484, 1347, 1276, 1144, 1130, 1028, 845, 815 cm−1; MS(ESI, +ve) m/z 270 [(M + Na)+, 100%], 248 [(M + H)+, 10]; HRMS(ESI) (M + Na)+ calcd for C14H17NNaO3 270.1106, found 270.1107.2-Methoxy-6-methyl-9H-carbazole (26). A magnetically stirred
mixture of compound 24 (100 mg, 0.46 mmol) and 10 wt % Pd/C(100 mg) in diphenyl ether (10 mL) was stirred at 210 °C under anatmosphere of nitrogen for 0.5 h. The cooled reaction mixture wasfiltered through filter paper, and the solids thus retained were washedwith ethyl acetate (2 × 10 mL). The combined filtrates wereconcentrated under reduced pressure, and the ensuing mixture ofproduct 269c,h,22d,23a,b and diphenyl ether was then subjected to flashcolumn chromatography (silica, 2/5/60 v/v/v ethyl acetate/dichloro-methane/40−60 petroleum ether elution) to give, after concentrationof the appropriate fractions (Rf = 0.4 in 2/5/11 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether), compound 26 (87 mg,88%) as a white solid, mp 229−231 °C (lit.9c mp 228−229 °C): 1HNMR [400 MHz, (CD3)2CO] δ 10.03 (braod s, 1H), 7.92 (d, J = 8.6Hz, 1H), 7.78 (s, 1H), 7.32 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.2 Hz,1H), 6.99 (d, J = 2.2 Hz, 1H), 6.77 (dd, J = 8.6 and 2.2 Hz, 1H), 3.85(s, 3H), 2.46 (s, 3H); 13C NMR [100 MHz, (CD3)2CO] δ 160.0,142.7, 139.3, 128.6, 126.3, 124.4, 121.5, 120.0, 117.6, 111.1, 108.6,95.3, 55.7, 21.5; IR νmax 3391, 2962, 1615, 1460, 1310, 1261, 1164,1035, 811 cm−1; MS (ESI, +ve) m/z 212 [(M + H)+, 100%]; HRMS(M + H)+ calcd for C14H14NO 212.1075, found 212.1077.Clauszoline K (5). A magnetically stirred solution of 2-methoxy-6-
methyl-9H-carbazole (26) (50 mg, 0.24 mmol) in methanol/water (16mL of 10/1 v/v mixture) was treated with DDQ (226 mg, 0.99 mmol).The ensuing mixture was stirred at room temperature for 4 h and thendiluted with ethyl acetate (30 mL). The separated organic phase waswashed with NaHCO3 (2 × 15 mL of a saturated aqueous solution),water (1 × 30 mL), and brine (1 × 30 mL) before being dried(Na2SO4), filtered, and concentrated under reduced pressure. Theresidue thus obtained was subjected to flash chromatography (silica, 1/3 v/v ethyl acetate/40−60 petroleum ether elution) to give, afterconcentration of the appropriate fractions (Rf = 0.5 in 1/1 v/v ethylacetate/40−60 petroleum ether), compound 5 (35 mg, 66%) as ayellow solid, mp 170−171 °C (lit.22d mp 184−185 °C): 1H NMR[400 MHz, (CD3)2CO] δ 10.72 (broad s, 1H), 10.07 (s, 1H), 8.58 (s,1H), 8.12 (d, J = 8.6 Hz, 1H), 7.88 (broadened d, J = 8.4 Hz, 1H),7.59 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 2.3 Hz, 1H), 6.91 (dd, J = 8.6 and2.3 Hz, 1H), 3.89 (s, 3H); 13C NMR [100 MHz, (CD3)2CO] δ 192.0,160.8, 144.8, 143.1, 130.1, 126.3, 124.4, 123.5, 122.2, 117.6, 111.8,110.1, 96.0, 55.8; IR νmax 3306, 2923, 2849, 1667, 1604, 1569, 1456,1317, 1159, 817, 807 cm−1; MS (ESI, −ve) m/z 224 [(M − H)−,100%], 209 (54); HRMS (M − H)− calcd for C14H10NO2 224.0712,found 224.0705.Methyl 3-Bromo-5-methoxy-4-nitrobenzoate (27). Step i. A
magnetically stirred and ice-cold solution of methyl 4-amino-3-methoxybenzoate (3.00 mg, 16.56 mmol) in chloroform (33 mL)was treated, in portions over 1 h, with N-bromosuccinimide (NBS)(2.95, 16.6 mmol). The ensuing mixture was stirred at 5−10 °C for 2 hbefore being diluted with dichloromethane (1 × 200 mL) then washedwith Na2S2O3 (2 × 100 mL of a 20% w/v aqueous solution), water (2× 100 mL), and brine (1 × 100 mL). The separated organic phase wasthen dried (Na2SO4), filtered, and concentrated under reducedpressure. The residue thus obtained was subjected to flash columnchromatography (silica, 3/17 v/v ethyl acetate/40−60 petroleum etherelution) to give, after concentration of the appropriate fractions (Rf =0.6 in 1/3 v/v ethyl acetate/40−60 petroleum ether), methyl 4-amino-3-bromo-5-methoxybenzoate25 (3.70 g, 86%) as a yellow solid, mp90−91 °C: 1H NMR (CDCl3, 400 MHz) δ 7.81 (s, 1H), 7.38 (s, 1H),4.65 (broad s, 2H), 3.91 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz,CDCl3) δ 166.5, 146.3, 139.4, 127.1, 119.5, 109.9, 106.8, 56.2, 52.1; IRνmax 3503, 3399, 1692, 1603, 1566, 1502, 1435, 1270, 1209, 1041, 995,759 cm−1; MS (EI, 70 eV) m/z 261 and 259 (M+•, 98 and 100%,respectively), 246 and 244 (both 65 and 63, respectively), 230 and 228(both 54); HRMS M+• calcd for C9H10
79BrNO3 258.9844, found258.9846.
Step ii. A magnetically stirred solution of methyl 4-amino-3-bromo-5-methoxybenzoate (4.24 g, 16.31 mmol), obtained as describedimmediately above, in 1,2-dichloroethane (163 mL) was treated withm-chloroperbenzoic acid (mCPBA) (11.26 g of ca. 77% technicalgrade material, 65.3 mmol). The resulting mixture was heated to 70 °Cfor 14 h before being cooled, diluted with dichloromethane (150 mL),washed with NaOH (2 × 150 mL of a 1 M aqueous solution) followedby brine (1 × 100 mL), and then dried (MgSO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash column chromatography (silica, 1/8 v/v ethylacetate/40−60 petroleum ether elution) to give, after concentration ofthe appropriate fractions (Rf = 0.5 in 1/2 v/v ethyl acetate/40−60petroleum ether), methyl 3-bromo-5-methoxy-4-nitrobenzoate (27)(4.13 g, 87%) as a yellow solid, mp 103−104 °C: 1H NMR (400 MHz,CDCl3) δ 7.88 (d, J = 1.4 Hz, 1H), 7.65 (d, J = 1.4 Hz, 1H), 3.95(9)(s, 3H), 3.95(6) (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.4, 151.8,144.7, 133.1, 126.2, 113.8, 112.8, 57.1, 53.2; IR νmax 3097, 1730, 1540,1406, 1289, 1248, 1037, 984, 822, 764 cm−1; MS (EI, 70 eV) m/z 291and 289 (M+•, 98 and 100%, respectively) 259 and 257 (40 and 35,respectively) 199 and 197 (60 and 50, respectively); HRMS M+• calcdfor C9H8
79BrNO5 288.9586, found 288.9587.Methyl 5-Methoxy-6-nitro-2′-oxo-2′,3′,4′,5′-tetrahydro-[1,1′-bi-
phenyl]-3-carboxylate (28). A magnetically stirred mixture of methyl3-bromo-5-methoxy-4-nitrobenzoate (28) (2.56 g, 8.82 mmol), 2-iodocyclohex-2-en-1-one (9) (890 mg, 4.01 mmol), copper powder(1.27 g, 20.04 g atom), CuI (1.15 g, 6.01 mmol), and Pd(dppf)Cl2·CH2Cl2 (164 mg, 0.20 mmol) in deoxygenated DMSO (17 mL) washeated to 50 °C under a nitrogen atmosphere for 28 h. The reactionmixture was then cooled to room temperature, quenched with water(20 mL), diluted with ethyl acetate (40 mL) and filtered through a padof diatomaceous earth and silica gel. The pad and the solids thusretained were washed with ethyl acetate (2 × 60 mL), and the organicphase associated with the filtrate was washed with water (2 × 100 mL)and then brine (2 × 100 mL) before being dried (Na2SO4), filtered,and concentrated under reduced pressure to give a brown oil.Subjection of this material to flash column chromatography (silica, 1/9v/v ethyl acetate/40−60 petroleum ether elution) gave, afterconcentration of the appropriate fractions (Rf = 0.5 in 1/1 v/v ethylacetate/40−60 petroleum ether), compound 28 (478 mg, 39%) as ayellow oil: 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 1.3 Hz, 1H),7.51 (d, J = 1.3 Hz, 1H), 7.05 (t, J = 4.2 Hz, 1H), 3.96 (s, 3H), 3.94 (s,3H), 2.58−2.50 (complex m, 4H), 2.10 (m, 2H); 13C NMR (100MHz, CDCl3) δ 196.1, 165.4, 151.2, 150.2, 143.3, 136.3, 132.4, 131.6,124.2, 113.3, 57.0, 52.9, 38.3, 26.5, 22.7; IR νmax 2956, 2924, 1716,1683, 1535, 1360, 1245, 1022, 838, 766 cm−1; MS (ESI, +ve) m/z 328[(M + Na)+, 100%]; HRMS (M + Na)+ calcd for C15H15NNaO6
328.0797, found 328.0793.Methyl 8-Methoxy-2,3,4,9-tetrahydro-1H-carbazole-6-carboxy-
late (29). A magnetically stirred mixture of compound 28 (400 mg,1.31 mmol) and commercially available W-2 Raney nickel (800 mg,washed twice with absolute ethanol) in methanol (65 mL) wasdeoxygenated and then stirred under an atmosphere of hydrogen at 22°C for 16 h. After this time and using an externally applied magnet tohold the solid associated with the reaction mixture within the flask, thesupernatant liquid was decanted and the retained solid washed withmethanol (2 × 40 mL) (Caution! Af ter these washings water should beadded to the residual solid so as to prevent a f ire.). The methanolicsolutions were combined and then concentrated under reducedpressure to give a white solid. Subjection of this material to flashcolumn chromatography (silica, 3/17 v/v ethyl acetate/40−60petroleum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.6 in 1/2 v/v ethyl acetate/40−60 petroleum ether),compound 29 (268 mg, 79%) as a white solid, mp 181−182 °C: 1HNMR [400 MHz, (CD3)2CO] δ 10.15 (broad s, 1H), 7.81 (s, 1H),7.25 (s, 1H), 3.96 (s, 3H), 3.86 (s, 3H), 2.77 (m, 2H), 2.70 (m, 2H),1.92−1.82 (complex m, 4H); 13C NMR [100 MHz, (CD3)2CO] δ168.5, 146.2, 136.6, 129.7, 129.4, 122.1, 114.8, 111.7, 102.5, 55.7, 51.8,24.0, 23.9, 23.7, 21.6; IR νmax 3339, 2928, 2836, 1696, 1628, 1366,1326, 1231, 1188, 993, 765 cm−1; MS (ESI, +ve) m/z 541 (65%), 282
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4155
309

[(M + Na)+, 100]; HRMS (M + Na)+ calcd for C15H17NNaO3282.1106, found 282.1105.Mukonine (6). A magnetically stirred mixture of compound 29 (264
mg, 1.02 mmol) and 10 wt % Pd/C (264 mg) in mesitylene (20 mL)was stirred at 150 °C under an atmosphere of nitrogen for 24 h. Thecooled reaction mixture was filtered through filter paper, and the solidsthus retained were washed with ethyl acetate (2 × 20 mL). The filtratewas concentrated under reduced pressure and the ensuing mixture ofproduct 6 and mesitylene then subjected to flash columnchromatography (silica, 1/19 v/v ethyl acetate/40−60 petroleumether elution) to give, after concentration of the appropriate fractions(Rf = 0.5 in 1/3 v/v ethyl acetate/40−60 petroleum ether), compound617 (171 mg, 66%) as a white solid, mp 196−198 °C (lit.17a mp 193−195 °C): 1H NMR (400 MHz, CDCl3) δ 8.52 (broad s, 1H), 8.48 (s,1H), 8.10 (d, J = 7.8 Hz, 1H), 7.60 (s, 1H), 7.49−7.43 (complex m,2H), 7.28 (m, 1H), 4.05 (s, 3H), 3.98 (s, 3H); 13C NMR (100 MHz,CDCl3) δ 168.1, 145.2, 139.6, 133.0, 126.5, 123.9, 123.7, 122.0, 120.9,120.4, 116.4, 111.4, 106.8, 55.9, 52.2; IR νmax 3350, 2941, 1689, 1608,1585, 1433, 1338, 1255, 758, 732 cm−1; MS (ESI, +ve) m/z 533(28%), 278 [(M + Na)+, 100]; HRMS (M + Na)+ calcd forC15H13NNaO3 278.0793, found 278.0792.6-Methyl-9H-carbazol-2-ol (30). A magnetically stirred solution of
compound 26 (50 mg, 0.24 mmol) in dichloromethane (15 mL)maintained at −78 °C under an atmosphere of nitrogen was treated,dropwise, with BBr3 (470 μL of a 1 M solution in dichloromethane,0.47 mmol). The resulting mixture was stirred at −78 °C for another 2h and then warmed to −40 °C. After 2 h the reaction mixture wasrecooled to −78 °C and a further portion of BBr3 (520 μL of a 1 Msolution in dichloromethane, 0.52 mmol) added dropwise. Thereaction mixture was then warmed to 22 °C, stirred at this temperaturefor 11 h, cooled to 0 °C, and then quenched by the slow addition ofNaHCO3 (30 mL of a saturated aqueous solution). The resultingmixture was extracted with dichloromethane (1 × 20 mL), and thecombined organic phases were washed with water (1 × 20 mL) andbrine (1 × 20 mL) before being dried (Na2SO4), filtered, andconcentrated under reduced pressure. The residue thus obtained wassubjected to flash chromatography (silica, 3/17 v/v ethyl acetate/40−60 petroleum ether elution) to give, after concentration of theappropriate fractions [Rf = 0.2(5) in 3/7 v/v ethyl acetate/40−60petroleum ether], compound 309h (39 mg, 84%) as a white, crystallinesolid, mp 259−261 °C (lit.9h mp 230−233 °C): 1H NMR [400 MHz,(CD3)2CO] δ 9.91 (broad s, 1H), 8.25 (s, 1H), 7.84 (d, J = 8.4 Hz,1H), 7.74 (s, 1H), 7.28 (d, J = 8.2 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H),6.90 (d, J = 2.1 Hz, 1H), 6.71 (dd, J = 8.4 and 2.1 Hz, 1H), 2.45 (s,3H); 13C NMR (100 MHz, (CD3)2CO) δ 157.4, 143.0, 139.2, 128.4,126.0, 124.7, 121.5, 119.8, 117.1, 110.9, 109.1, 97.4, 21.5; IR νmax 3403,2922, 2853, 1633, 1616, 1490, 1459, 1296, 1026, 804 cm−1; MS (ESI,−ve) m/z 196 [(M − H)−, 100%]; HRMS (M − H)− calcd forC13H10NO 196.0762, found 196.0767.Karapinchamine A (7). A magnetically stirred solution of
compound 30 (27 mg, 0.14 mmol) in anhydrous THF (7 mL)maintained at 0 °C under a nitrogen atmosphere was treated,dropwise, with n-BuLi (340 μL of a 1.6 M solution in n-hexane, 0.55mmol). After 0.17 h, a solution of geranyl bromide (51 mg, 0.21mmol) in THF (7 mL) was added dropwise over 0.5 h. The resultingmixture was warmed to 22 °C, stirred at this temperature for 1.5 h,then cooled to 0 °C and quenched with NH4Cl (15 mL of a saturatedaqueous solution). The resulting mixture was extracted with ethylacetate (2 × 30 mL), and the combined organic phases were washedwith water (1 × 20 mL) and brine (1 × 20 mL) before being dried(Na2SO4), filtered, and concentrated under reduced pressure. Theresidue thus obtained was subjected to flash chromatography (silica, 1/9 v/v ethyl acetate/40−60 petroleum ether elution) to give, afterconcentration of the appropriate fractions (Rf = 0.6 in 3/7 v/v ethylacetate/40−60 petroleum ether), compound 77e,18 (23 mg, 50%) as awhite solid, mp 118−119 °C (lit.7e mp 124.3−125.6 °C): 1H NMR(400 MHz, CDCl3) δ 7.87 (d, J = 8.3 Hz, 1H), 7.78 (s, 1H), 7.23−7.18 (complex m, 2H), 6.78 (d, J = 1.9 Hz, 1H), 6.70 (dd, J = 8.3 and2.0 Hz, 1H), 5.24 (m, 1H), 5.02 (m, 1H), 4.88 (s, 1H), 4.79 (d, J = 6.1Hz, 2H), 2.52 (s, 3H), 2.09−1.98 (complex m, 4H), 1.90 (s, 3H), 1.62
(s, 3H), 1.55 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.6, 142.2,138.9, 138.7, 131.9, 128.4, 125.8, 123.9, 123.4, 121.2, 120.0, 119.6,117.1, 108.4, 107.7, 95.2, 41.3, 39.5, 26.4, 25.8, 21.5, 17.8, 16.8; IR νmax3278, 2922, 2855, 1635, 1610, 1487, 1471, 1348, 1171, 801 cm−1; MS(ESI, +ve) m/z 356 [(M + Na)+, 100%], 334 [(M + H)+, 20]; HRMS(M + Na)+ calcd for C23H27NNaO 356.1990, found 356.1994.
4′-Methoxy-2′-nitro-4,5-dihydro-[1,1′-biphenyl]-2(3H)-one (31).A magnetically stirred mixture of 1-iodo-4-methoxy-2-nitrobenzene(22) (912 mg, 3.27 mmol), 2-iodocyclohex-2-en-1-one (9) (330 mg,1.49 mmol), copper powder (472 mg, 7.43 g atom), CuI (425 mg, 2.23mmol), and Pd(dppf)Cl2·CH2Cl2 (61 mg, 0.07 mmol) indeoxygenated DMSO (15 mL) was heated to 50 °C under a nitrogenatmosphere for 10 h. The reaction mixture was then cooled to roomtemperature, quenched with water (15 mL), diluted with ethyl acetate(30 mL), and then filtered through a pad of diatomaceous earth andsilica gel. The pad and the solids thus retained were washed with ethylacetate (2 × 40 mL), and the separated organic phase associated withthe filtrate was washed with water (2 × 80 mL) and brine (2 × 80 mL)before being dried (Na2SO4), filtered, and concentrated under reducedpressure to give a brown oil. Subjection of this material to flash columnchromatography (silica, 2/5/44 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether elution) gave, after concentration of theappropriate fractions (Rf = 0.3 in 2/5/11 v/v/v ethyl acetate/dichloromethane/40−60 petroleum ether), compound 3110,11b (334mg, 91%) as a light yellow oil: 1H NMR [400 MHz, (CD3)2CO] δ7.51 (d, J = 2.2 Hz, 1H), 7.28 (m, 2H), 7.10 (m, 1H), 3.92 (s, 3H),2.58 (m, 2H), 2.46 (m, 2H), 2.09 (m, 2H); 13C NMR [100 MHz,(CD3)2CO] δ 196.5, 160.5, 150.4, 147.5, 139.5, 133.6, 125.1, 120.0,109.9, 56.4, 39.0, 27.0, 23.5; IR νmax 2945, 1678, 1525, 1496, 1353,1232, 1035, 913, 830, 798 cm−1; MS (ESI, +ve) m/z 270 [(M + Na)+,100%], 248 [(M + H)+, 10]; HRMS (M + Na)+ calcd forC13H13NNaO4 270.0742, found 270.0739.
7-Methoxy-2,3,4,9-tetrahydro-1H-carbazole (32). A magneticallystirred mixture of compound 31 (100 mg, 0.40 mmol) andcommercially available W-2 Raney nickel (200 mg, washed twicewith absolute ethanol) in methanol (23 mL) was deoxygenated andthen stirred under an atmosphere of hydrogen at 22 °C for 16 h. Afterthis time and using an externally applied magnet to hold the solidassociated with the reaction mixture within the flask, the supernatantliquid was decanted and the retained solid washed with methanol (2 ×20 mL) (Caution! Af ter these washings water should be added to theresidual solid so as to prevent a f ire.). The methanolic solutions werecombined and then concentrated under reduced pressure to give awhite solid. Subjection of this material to flash column chromatog-raphy (silica, 1/3/20 v/v/v acetone/dichloromethane/40−60 petro-leum ether elution) gave, after concentration of the appropriatefractions (Rf = 0.4(5) in 1/3/6 v/v/v acetone/dichloromethane/40−60 petroleum ether), compound 3210,11b,26 (62 mg, 76%) as a white,crystalline solid, mp 138−139 °C (lit.10 mp 136−142 °C): 1H NMR(400 MHz, CDCl3) δ 7.54 (broad s, 1H), 7.33 (d, J = 8.5 Hz, 1H),6.80 (d, J = 2.2 Hz, 1H), 6.75 (dd, J = 8.5 and 2.2 Hz, 1H), 3.84 (s,3H), 2.69 (m, 4H), 1.88 (m, 4H); 13C NMR (100 MHz, CDCl3) δ155.9, 136.5, 132.9, 122.5, 118.3, 110.1, 108.4, 95.0, 56.0, 23.4,23.3(5), 23.3(4), 21.1; IR νmax 3405, 2919, 2842, 1628, 1464, 1307,1213, 1156, 1029, 826, 803 cm−1; MS (ESI, +ve) m/z 202 [(M + H)+,100%]; HRMS (M + H)+ calcd for C13H16NO 202.1232, found202.1230.
2-Methoxy-9H-carbazole (8). A magnetically stirred mixture ofcompound 32 (40 mg, 0.20 mmol) and 10 wt % Pd/C (40 mg) inmesitylene (5 mL) was stirred at 150 °C under an atmosphere ofnitrogen for 24 h. The cooled reaction mixture was filtered throughfilter paper, and the solids thus retained were washed with ethyl acetate(2 × 10 mL). The combined filtrates were concentrated under reducedpressure, and the ensuing mixture of product 6 and mesitylene wasthen subjected to flash column chromatography (silica, 1/19 v/v ethylacetate/40−60 petroleum ether elution) to give, after concentration ofthe appropriate fractions (Rf = 0.3 in 1/5 v/v ethyl acetate/40−60petroleum ether), compound 86f,k,l,9j (25 mg, 64%) as a white,crystalline solid, mp 227−228 °C (lit.6f mp 233−235 °C): 1H NMR[400 MHz, (CD3)2CO] δ 10.18 (broad s, 1H), 7.97 (m, 2H), 7.44 (d,
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4156
310

J = 8.1 Hz, 1H), 7.28 (t, J = 7.9 Hz, 1H), 7.13 (m, 1H), 7.03 (broad s,1H), 6.80 (m, 1H), 3.86 (s, 3H); 13C NMR [100 MHz, (CD3)2CO] δ160.1, 142.4, 141.0, 125.0, 124.2, 121.6, 120.0, 119.7, 117.7, 111.4,108.8, 95.4, 55.7; IR νmax 3391, 2929, 1608, 1463, 1444, 1308, 1196,1163, 1033, 818, 727 cm−1; MS (ESI, −ve) m/z 196 [(M − H)−,100%]; HRMS (M − H)− calcd for C13H10NO 196.0762, found196.0753.X-ray Crystallographic Studies. Crystallographic Data. Com-
pound 1: C12H9N, Mr = 167.21, T = 150 K, orthorhombic, spacegroup Pnma, Z = 4, a = 7.6589(2) Å, b = 19.0353(5) Å, c = 5.6814(1)Å; V = 828.29(3) Å3, Dx = 1.341 g cm−3, 864 unique data (2θmax =147.4°), R = 0.035 [for 795 reflections with I > 2.0σ(I)]; Rw = 0.058(all data), S = 1.00.Compound 2: C13H11N, Mr = 181.24, T = 150 K, monoclinic, space
group P21/c, Z = 4, a = 20.3990(8) Å, b = 5.7846(2) Å, c = 7.8899(3)Å; β = 98.458(4)°; V = 920.88(6) Å3, Dx = 1.307 g cm−3, 1858 uniquedata (2θmax = 147.8°), R = 0.057 [for 1748 reflections with I >2.0σ(I)]; Rw = 0.096 (all data), S = 1.00.Compound 3: C14H13NO, Mr = 211.26, T = 150 K, monoclinic,
space group P21/n, Z = 4, a = 11.4714(17) Å, b = 5.6130(6) Å, c =16.915(2) Å; β = 95.648(12)°; V = 1083.9(2) Å3, Dx = 1.295 g cm−3,2160 unique data (2θmax = 151.8°), R = 0.058 [for 1763 reflectionswith I > 2.0σ(I)]; Rw = 0.164 (all data), S = 1.00.Compound 4: C14H13NO, Mr = 211.26, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 6.4420(1) Å, b = 7.5921(1) Å, c =21.9422(3) Å; V = 1073.16(3) Å3, Dx = 1.308 g cm−3, 2166 uniquedata (2θmax = 147.8°), R = 0.035 [for 2114 reflections with I >2.0σ(I)]; Rw = 0.064 (all data), S = 1.00.Compound 5: C14H11NO2, Mr = 225.25, T = 150 K, orthorhombic,
space group Pbca, Z = 8, a = 7.1367(1) Å, b = 13.2390(1) Å, c =22.5108(2) Å; V = 2126.88(4) Å3, Dx = 1.407 g cm−3, 2152 uniquedata (2θmax = 147.8°), R = 0.033 [for 2089 reflections with I >2.0σ(I)]; Rw = 0.058 (all data), S = 1.00.Compound 6: C15H13NO3, Mr = 255.27, T = 150 K, monoclinic,
space group C2/c, Z = 8, a = 15.7767(2) Å, b = 9.4629(1) Å, c =16.1402(2) Å; β = 91.7342(13)°; V = 2408.52(5) Å3, Dx = 1.408 gcm−3, 3284 unique data (2θmax = 59.6°), R = 0.039 [for 2869reflections with I > 2.0σ(I)]; Rw = 0.077 (all data), S = 1.00.Compound 17: C14H15NO4, Mr = 261.27, T = 150 K, monoclinic,
space group P21/c, Z = 8, a = 21.5986(4) Å, b = 6.89910(11) Å, c =17.6362(3) Å; β = 104.596(2)°; V = 2543.16(8) Å3, Dx = 1.365 gcm−3, 5147 unique data (2θmax = 147.6°), R = 0.084 [for 4826reflections with I > 2.0σ(I)]; Rw = 0.214 (all data), S = 1.12.Compound 25: C14H17NO3, Mr = 247.29, T = 150 K, monoclinic,
space group P21/c, Z = 4, a = 14.6135(2) Å, b = 5.8504(1) Å, c =14.6942 (2) Å; β = 96.2796(15)°; V = 1248.74(3) Å3, Dx = 1.315 gcm−3, 2542 unique data (2θmax = 147.6°), R = 0.036 [for 2433reflections with I > 2.0σ(I)]; Rw = 0.098 (all data), S = 1.00.Structure Determination. The image for compound 6 was
measured on a diffractometer (Mo Kα, graphite monochromator, λ= 0.71073 Å) fitted with an area detector, and the data were extractedusing the DENZO/Scalepack package.34 Images for compounds 1−5,17, and 25 were measured on a diffractometer (Cu Kα, mirrormonochromator, λ = 1.54184 Å) fitted with an area detector and thedata extracted using the CrysAlis package.35 The structure solutionsfor all eight compounds were solved by direct methods (SIR92)36 thenrefined using the CRYSTALS program package.37 Atomic coordinates,bond lengths and angles, and displacement parameters have beendeposited at the Cambridge Crystallographic Data Centre (CCDCnos. 1525166, 1525163, 1525164, 1525162, 1525167, 1525160,1525161, and 1525165 for compounds 1−6, 17, and 25, respectively).These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by e-mailing [email protected], or bycontacting The Cambridge Crystallographic Data Centre, 12 UnionRoad, Cambridge CB2 1EZ, U.K. (fax: + 44 1223 336033). Single-crystal X-ray analyses of compound 1 have been reported previously.38
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.joc.7b00044.
Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data (CIF)Crystallographic data and anisotropic displacementellipsoid plots derived from the single-crystal X-rayanalyses of compounds 1−6, 17, and 25, 1H and 13CNMR spectra of compounds 1−15, 17, 18, 20, 21, 23−32, and methyl 4-amino-3-bromo-5-methoxybenzoate(precursor to compound 27) (PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail for M.G.B.: [email protected] G. Banwell: 0000-0002-0582-475XNotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank the Australian Research Council and the Institute ofAdvanced Studies for financial support. Q.Y. is the gratefulrecipient of a PhD Scholarship provided by China ScholarshipCouncil of the People’s Republic of China. E.G. acknowledgesscholarship support from the ANU. M.W.-K. thanks theDepartment of Organic Chemistry, Faculty of Chemistry,Wroclaw University of Technology, Wroclaw, Poland for givingher leave to undertake research at the ANU.
■ REFERENCES(1) Graebe, C.; Glaser, C. Ber. Dtsch. Chem. Ges. 1872, 5, 12.(2) For useful points of entry into the relevant literature see:(a) Thevissen, K.; Marchand, A.; Chatlin, P.; Meert, E. M.; Cammue,B. P. Curr. Med. Chem. 2009, 16, 2205−2211. (b) Yaqub, G.; Huissain,E. A.; Rehman, M. A.; Mateen, B. Asian J. Chem. 2009, 21, 2485−2520.(c) Roy, J.; Jana, A. K.; Mal, D. Tetrahedron 2012, 68, 6099−6121.(d) Schmidt, A. W.; Reddy, K. R.; Knolker, H.-J. Chem. Rev. 2012, 112,3193−3328. (e) Gluszynska, A. Eur. J. Med. Chem. 2015, 94, 405−426.(f) Bashir, M.; Bano, A.; Ijaz, A. S.; Chaudhary, B. A. Molecules 2015,20, 13496−13517. (g) Sathiyan, G.; Sivakumar, E. K. T.;Ganesamoorthy, R.; Thangamuthu, R.; Sakthivel, P. Tetrahedron Lett.2016, 57, 243−252.(3) See, for example: (a) Cheenpracha, S.; Laphookhieo, S.Phytochem. Lett. 2011, 4, 187−189. (b) Miller, C. M.; McCarthy, F.O. RSC Adv. 2012, 2, 8883−8918. (c) Pieroni, M.; Girmay, S.; Sun, D.;Sahu, R.; Tekwani, B. L.; Tan, G. T. ChemMedChem 2012, 7, 1895−1900. (d) Russell, F.; Harmody, D.; McCarthy, P. J.; Pomponi, S. A.;Wright, A. E. J. Nat. Prod. 2013, 76, 1989−1992. (e) Kim, S.-H.; Ha,T.-K.-Q.; Oh, W. K.; Shin, J.; Oh, D.-C. J. Nat. Prod. 2016, 79, 51−58.(f) Patel, O. P. S.; Mishra, A.; Maurya, R.; Saini, D.; Pandey, J.; Taneja,I.; Raju, K. S. R.; Kanojiya, S.; Shukla, S. K.; Srivastava, M. N.;Wahajuddin, M.; Tamrakar, A. K.; Srivastava, A. K.; Yadav, P. P. J. Nat.Prod. 2016, 79, 1276−1284.(4) (a) Fischer, E.; Jourdan, F. Ber. Dtsch. Chem. Ges. 1883, 16,2241−2245. (b) Borsche, W.; Witte, A.; Bothe, W. Liebigs Ann. Chem.1908, 359, 49−80. For related and recent approaches see:
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4157
311

(c) Chakraborty, S.; Chattopadhyay, G.; Saha, C. J. Heterocyclic Chem.2013, 50, 91−98. (d) Wu, J.; Xie, Y.; Chen, X.; Deng, G.-J. Adv. Synth.Catal. 2016, 358, 3206−3211.(5) Graebe, C.; Ullmann, F. Liebigs Ann. Chem. 1896, 291, 16−17.(6) (a) Cadogan, J. I. G.; Cameron-Wood, M.; Mackie, R. K.; Searle,R. J. G. J. Chem. Soc. 1965, 4831−4837. For related and recentvariants see: (b) Smitrovich, J. H.; Davies, I. W. Org. Lett. 2004, 6,533−535. (c) Tsang, W. C. P.; Zheng, N.; Buchwald, S. L. J. Am.Chem. Soc. 2005, 127, 14560−14561. (d) Jordan-Hore, J. A.;Johansson, C. C. C.; Gulias, M.; Beck, E. M.; Gaunt, M. J. J. Am.Chem. Soc. 2008, 130, 16184−16186. (e) Kuethe, J. T.; Childers, K. G.Adv. Synth. Catal. 2008, 350, 1577−1586. (f) Stokes, B. J.; Jovanovic,H.; Dong, H.; Richert, K. J.; Riell, R. D.; Driver, T. G. J. Org. Chem.2009, 74, 3225−3228. (g) Gao, H.; Xu, Q.-L.; Yousufuddin, M.; Ess,D. H.; Kurti, L. Angew. Chem., Int. Ed. 2014, 53, 2701−2705.(h) Suzuki, C.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2015, 17,1597−1600. (i) Goo, D.-Y.; Woo, S. K. Org. Biomol. Chem. 2016, 14,122−130. (j) Yang, C.; Lin, K.; Huang, L.; Pan, W.-d.; Liu, S. BeilsteinJ. Org. Chem. 2016, 12, 2490−2494. (k) Yang, L.; Li, H.; Zhang, H.;Lu, H. Eur. J. Org. Chem. 2016, 2016, 5611−5615. (l) Alt, I. T.;Plietker, B. Angew. Chem., Int. Ed. 2016, 55, 1519−1522.(7) (a) Danheiser, R. L.; Brisbois, R. G.; Kowalczyk, J. J.; Miller, R. F.J. Am. Chem. Soc. 1990, 112, 3093−3100. (b) Praveen, C.; Perumal, P.T. Synlett 2011, 268−272. (c) Wang, J.; Zhu, H.-T.; Qiu, Y.-F.; Niu, Y.;Chen, S.; Li, Y.-X.; Liu, X.-Y.; Liang, Y.-M. Org. Lett. 2015, 17, 3186−3189. (d) James, M. J.; Clubley, R. E.; Palate, K. Y.; Procter, T. J.;Wyton, A. C.; O’Brien, P.; Taylor, R. J. K.; Unsworth, W. P. Org. Lett.2015, 17, 4372−4375. (e) Qiu, Y.; Zhou, J.; Fu, C.; Ma, S. Chem. - Eur.J. 2014, 20, 14589−14593. (f) Chen, S.; Li, Y.; Ni, P.; Huang, H.;Deng, G.-J. Org. Lett. 2016, 18, 5384−5387. (g) Huang, Y.-W.; Li, X.-Y.; Fu, L.-N.; Guo, Q.-X. Org. Lett. 2016, 18, 6200−6203. (h) Song,W.; Li, X.; Yang, K.; Zhao, X.-l.; Glazier, D. A.; Xi, B.-m.; Tang, W. J.Org. Chem. 2016, 81, 2930−2942. (i) Guo, B.; Huang, X.; Fu, C.; Ma,S. Chem. - Eur. J. 2016, 22, 18343−18348.(8) See, for example: (a) Knolker, H.-J.; Bauermeister, M.; Pannek, J.-B.; Wolpert, M. Synthesis 1995, 1995, 397−408. (b) Chakrabarty, S.;Chatterjee, I.; Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2013, 52,2968−2971.(9) See, for example: (a) Liegault, B.; Lee, D.; Huestis, M. P.; Stuart,D. R.; Fagnou, K. J. Org. Chem. 2008, 73, 5022−5028. (b) Ackermann,L.; Althammer, A.; Mayer, P. Synthesis 2009, 2009, 3493−3503.(c) Sridharan, V.; Martin, M. A.; Menendez, J. C. Eur. J. Org. Chem.2009, 2009, 4614−4621. (d) Hernandez-Perez, A. C.; Collins, S. K.Angew. Chem., Int. Ed. 2013, 52, 12696−12700. (e) Hesse, R.; Kataeva,O.; Schmidt, A. W.; Knolker, H.-J. Chem. - Eur. J. 2014, 20, 9504−9509. (f) Julich-Gruner, K. K.; Schmidt, A. W.; Knolker, H.-J. Synthesis2014, 46, 2651−2655. (g) Hesse, R.; Jager, A.; Schmidt, A. W.;Knolker, H.-J. Org. Biomol. Chem. 2014, 12, 3866−3876. (h) Gassner,C.; Hesse, R.; Schmidt, A. W.; Knolker, H.-J. Org. Biomol. Chem. 2014,12, 6490−6499. (i) Schuster, C.; Julich-Gruner, K. K.; Schnitzler, H.;Hesse, R.; Jager, A.; Schmidt, A. W.; Knolker, H.-J. J. Org. Chem. 2015,80, 5666−5673. (j) Wen, L.; Tang, L.; Yang, Y.; Zha, Z.; Wang, Z. Org.Lett. 2016, 18, 1278−1281. (k) Brutting, C.; Hesse, R.; Jager, A.;Kataeva, O.; Schmidt, A. W.; Knolker, H.-J. Chem. - Eur. J. 2016, 22,16897−16911. (l) Parisien-Collette, S.; Hernandez-Perez, A. C.;Collins, S. K. Org. Lett. 2016, 18, 4994−4997. (m) Kutz, S. K.;Schmidt, A. W.; Knolker, H.-J. Synthesis 2016, 49, 275−292.(10) Banwell, M. G.; Kelly, B. D.; Kokas, O. J.; Lupton, D. W. Org.Lett. 2003, 5, 2497−2500.(11) For a related approach involving Stille cross-coupling reactionssee: (a) Scott, T. L.; Yu, X.; Gorugantula, S. P.; Carrero-Martínez, G.;Soderberg, B. C. G. Tetrahedron 2006, 62, 10835−10842. (b) Scott, T.L.; Burke, N.; Carrero-Martinez, G.; Soderberg, B. C. G. Tetrahedron2007, 63, 1183−1190.(12) (a) Campaigne, E.; Lake, R. D. J. Org. Chem. 1959, 24, 478−487. (b) Humne, V.; Dangat, Y.; Vanka, K.; Lokhande, P. Org. Biomol.Chem. 2014, 12, 4832−4836. (c) Iosub, A. V.; Stahl, S. S. J. Am. Chem.Soc. 2015, 137, 3454−3457. (d) Humne, V. T.; Naykode, M. S.;Ghom, M. H.; Lokhande, P. D. Tetrahedron Lett. 2016, 57, 688−691.
(13) (a) Chakrabarty, M.; Nath, A. c.; Khasnobis, S.; Chakrabarty,M.; Konda, Y.; Harigaya, Y.; Komiyama, K. Phytochemistry 1997, 46,751−755. (b) Cui, C.-B.; Yan, S.-Y.; Cai, B.; Yao, X.-S. J. Asian Nat.Prod. Res. 2002, 4, 233−241.(14) Chakravarty, A. K.; Sarkar, T.; Masuda, K.; Takey, T.; Doi, H.;Kotani, E.; Shiojima, K. Indian J. Chem. Sec. B 2001, 40, 484−489.(15) (a) Chakraborty, D. P. Phytochemistry 1969, 8, 769−772. (b) Li,W. S.; McChesney, J. D.; El-Feraly, F. S. Phytochemistry 1991, 30,343−346.(16) (a) Birari, R.; Roy, S. K.; Singh, A.; Bhutani, K. Nat. Prod.Commun. 2009, 4, 1089−1092. (b) Lin, W.; Wang, Y.; Lin, S.; Li, C.;Zhou, C.; Wang, S.; Huang, H.; Liu, P.; Ye, G.; Shen, X. Eur. J. Med.Chem. 2012, 47, 214−220.(17) (a) Brenna, E.; Fuganti, C.; Serra, S. Tetrahedron 1998, 54,1585−1588. (b) Laphookhieo, S.; Sripisut, T.; Prawat, U.; Karalai, C.Heterocycles 2009, 78, 2115−2119.(18) Nakamura, S.; Nakashima, S.; Oda, Y.; Yokota, N.; Fujimoto, K.;Matsumoto, T.; Ohta, T.; Ogawa, K.; Maeda, S.; Nishida, S.; Matsuda,H.; Yoshikawa, M. Bioorg. Med. Chem. 2013, 21, 1043−1049.(19) Pandey, G.; Balakrishnan, M. J. Org. Chem. 2008, 73, 8128−8131.(20) Details of this X-ray analysis can be found in the ExperimentalSection and the Supporting Information.(21) Banwell, M. G.; Jones, M. T.; Loong, D. T. J.; Lupton, D. W.;Pinkerton, D. M.; Ray, J. K.; Willis, A. C. Tetrahedron 2010, 66, 9252−9262.(22) (a) Carruthers, W. Chem. Commun. 1966, 272. (b) Chakraborty,D. P.; Das, K. C.; Chowdhury, B. K. Phytochemistry 1969, 8, 773−776.(c) Forke, R.; Krahl, M. P.; Krause, T.; Schlechtingen, G.; Knolker, H.-J. Synlett 2007, 2007, 0268−0272. (d) Bautista, R.; Montoya, P. A.;Rebollar, A.; Burgueno, E.; Tamariz, J. Molecules 2013, 18, 10334−10351. (e) Naykode, M. S.; Humne, V. T.; Lokhande, P. D. J. Org.Chem. 2015, 80, 2392−2396. (f) Hesse, R.; Schmidt, A. W.; Knolker,H.-J. Tetrahedron 2015, 71, 3485−3490. (g) Momin, A. A.; Urmode,T. D.; Bhosale, S. M.; Kusurkar, R. S. Synth. Commun. 2016, 46, 1292−1298.(23) (a) Krahl, M. P.; Jager, A.; Krause, T.; Knolker, H.-J. Org.Biomol. Chem. 2006, 4, 3215−3219. (b) Mane, M. S.; Balaskar, R. S.;Gavade, S. N.; Pabrekar, P. N.; Shingare, M. S.; Mane, D. V. Chin.Chem. Lett. 2011, 22, 1039−1042. (c) Kong, W.; Fu, C.; Ma, S. Chem. -Eur. J. 2011, 17, 13134−13137.(24) (a) Bringmann, G.; Tasler, S.; Endress, H.; Peters, K.; Peters, E.-M. Synthesis 1998, 1998, 1501−1505. (b) Zempoalteca, A.; Tamariz, J.Heterocycles 2002, 57, 259−267. (c) Knolker, H.-J.; Wolpert, M.Tetrahedron 2003, 59, 5317−5322. (d) Kuwahara, A.; Nakano, K.;Nozaki, K. J. Org. Chem. 2005, 70, 413−419. (e) Liu, Z.; Larock, R. C.Tetrahedron 2007, 63, 347−355. (f) Tsang, W. C. P; Munday, R. H.;Brasche, G.; Zheng, N.; Buchwald, S. L. J. Org. Chem. 2008, 73, 7603−7610. (g) Hibino, S.; Tohyama, S.; Choshi, T.; Azuma, S.; Fujioka, H.Heterocycles 2009, 79, 955−965. (h) Borger, C.; Krahl, M. P.; Gruner,M.; Kataeva, O.; Knolker, H.-J. Org. Biomol. Chem. 2012, 10, 5189−5193. (i) Jana, A. K.; Pahari, P.; Mal, D. Synlett 2012, 23, 1769−1774.(j) Yu, J.; Wang, Y.; Zhang, P.; Wu, J. Synlett 2013, 24, 1448−1454.(25) Yang, W.; Zhou, J.; Wang, B.; Ren, H. Chem. - Eur. J. 2011, 17,13665−13669.(26) (a) Wender, P. A.; White, A. W. Tetrahedron 1983, 39, 3767−3776. (b) Johnson, P. D.; Sohn, J.-H.; Rawal, V. H. J. Org. Chem. 2006,71, 7899−7902.(27) Banwell, M. G.; Jones, M. T.; Reekie, T. A. Chem. New Zealand2011, 75, 122−127.(28) See, for example: (a) White, L. V.; Banwell, M. G. J. Org. Chem.2016, 81, 1617−1626. (b) Tang, F.; Banwell, M. G.; Willis, A. C. J.Org. Chem. 2016, 81, 2950−2957. (c) Tan, S. H.; Banwell, M. G.;Willis, A. C. J. Org. Chem. 2016, 81, 8022−8028. (d) Tang, F.;Banwell, M. G.; Willis, A. C. J. Org. Chem. 2016, 81, 10551−10557.(29) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923−2925.(30) Pangborn, A. B.; Giardello, M. A.; Grubbs, R. H.; Rosen, R. K.;Timmers, F. J. Organometallics 1996, 15, 1518−1520.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4158
312

(31) Johnson, C. R.; Adams, J. P.; Braun, M. P.; Senanayake, C. B.W.; Wovkulich, P. M.; Uskokovic, M. R. Tetrahedron Lett. 1992, 33,917−918.(32) Chong, B.-D.; Ji, Y. I.; Oh, S.-S.; Yang, J.-D.; Baik, W.; Koo, S. J.Org. Chem. 1997, 62, 9323−9325.(33) The Merck Index, 12th ed.; Merck and Co.: Kenilworth, NJ,1996; p 291.(34) DENZO-SMN: Otwinowski, Z.; Minor, W. Processing of X-raydiffraction data collected in oscillation mode. In Methods inEnzymology; Carter, C. W., Jr., Sweet, R. M., Eds.; Academic Press:New York, 1997; Vol. 276, Macromolecular Crystallography, Part A,pp 307−326.(35) CrysAlis PRO Version 1.171.37.35h (release 09-02-2015CrysAlis171.NET) (compiled Feb 9 2015,16:26:32); Agilent Tech-nologies, Oxfordshire, UK, 2015.(36) SIR92: Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi,A.; Burla, M. C.; Polidori, G.; Camalli, M. J. Appl. Crystallogr. 1994, 27,435−436.(37) Betteridge, P. W.; Carruthers, J. R.; Cooper, R. I.; Prout, K.;Watkin, D. J. J. Appl. Crystallogr. 2003, 36, 1487.(38) (a) Gerkin, R. E.; Reppart, W. J. Acta Crystallogr., Sect. C: Cryst.Struct. Commun. 1986, 42, 480−482. (b) Gajda, K.; Zarychta, B.;Kopka, K.; Daszkiewicz, Z.; Ejsmont, K. Acta Crystallogr., Sect. C:Struct. Chem. 2014, 70, 987−991.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00044J. Org. Chem. 2017, 82, 4148−4159
4159
313

S1
SUPP
ORT
ING
INFO
RMAT
ION
FO
R:
A P
alla
dium
-cat
alyz
ed U
llman
n C
ross
-cou
plin
g/R
educ
tive
Cyc
lisat
ion
Rou
te to
the
Car
bazo
le N
atur
al P
rodu
cts 3
-Met
hyl-9H
-car
bazo
le,
Gly
cobo
rine
, Gly
cozo
line,
Cla
uszo
line
K, M
ukon
ine
and
Kar
apin
cham
ine
A
Qia
o Y
an, E
mm
a G
in, M
algo
rzat
a W
asin
ska-
Kal
wa,
Mar
tin G
. Ban
wel
l* a
nd P
aul D
. Car
r
Rese
arch
Sch
ool o
f Che
mis
try,
Inst
itute
of A
dvan
ced
Stud
ies,
The
Aust
ralia
n N
atio
nal U
nive
rsity
, Can
berr
a, A
CT
2601
, Aus
tral
ia
CO
NT
EN
TS
PA
GE
(i)A
niso
tropi
c D
ispl
acem
ent E
llips
oid
Plot
s fro
m th
e Si
ngle
-cry
stal
X-r
ay
Ana
lyse
s of C
ompo
unds
1-6
, 17
and
25S2
(ii)
Tab
le S
1: C
ompa
rison
of t
he 13
C N
MR
Dat
a Re
cord
ed o
n Co
mpo
und 7
with
Lite
ratu
re V
alue
s for
Kar
apin
cham
ine
AS1
0
(iii)
1 H a
nd 13
C N
MR
Spe
ctra
of C
ompo
unds
1-1
5, 1
7, 1
8, 2
0, 2
1, 2
3-32
and
Met
hyl 4
-Am
ino-
3-br
omo-
5-m
etho
xybe
nzoa
te (p
recu
rsor
to c
ompo
und
27).
S11
314
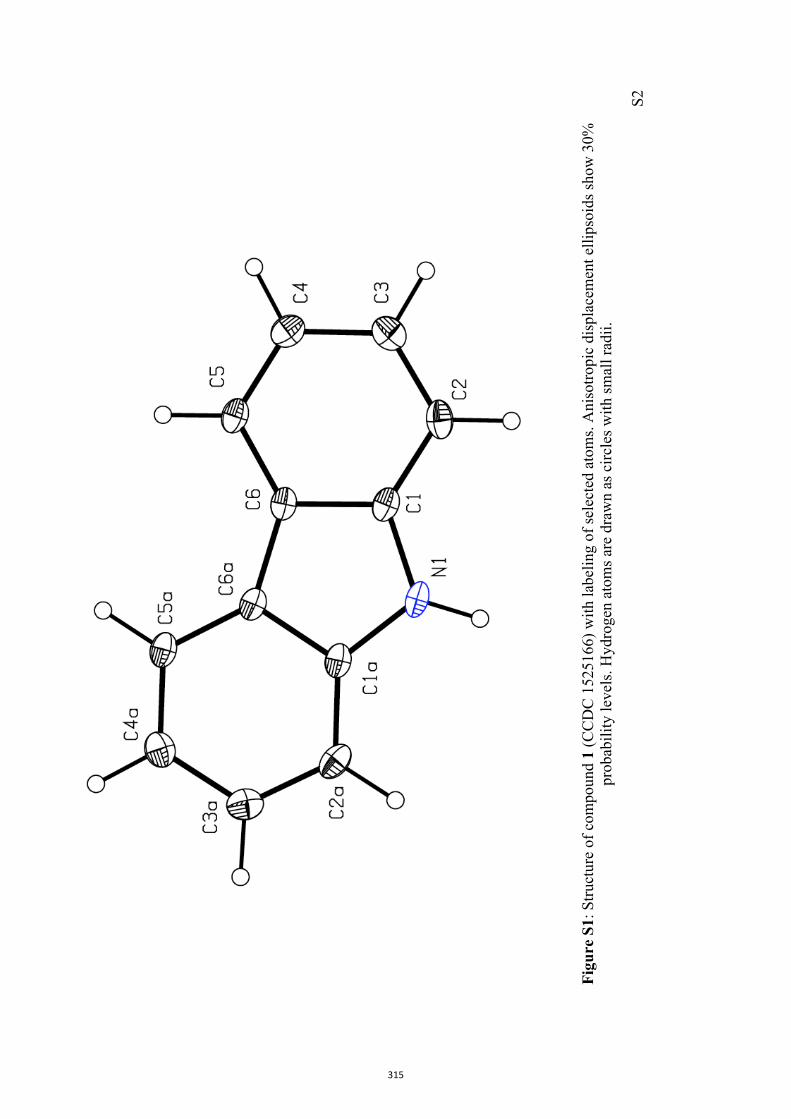
S2
Figu
re S
1: S
truct
ure
of c
ompo
und
1 (C
CD
C 1
5251
66) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
315
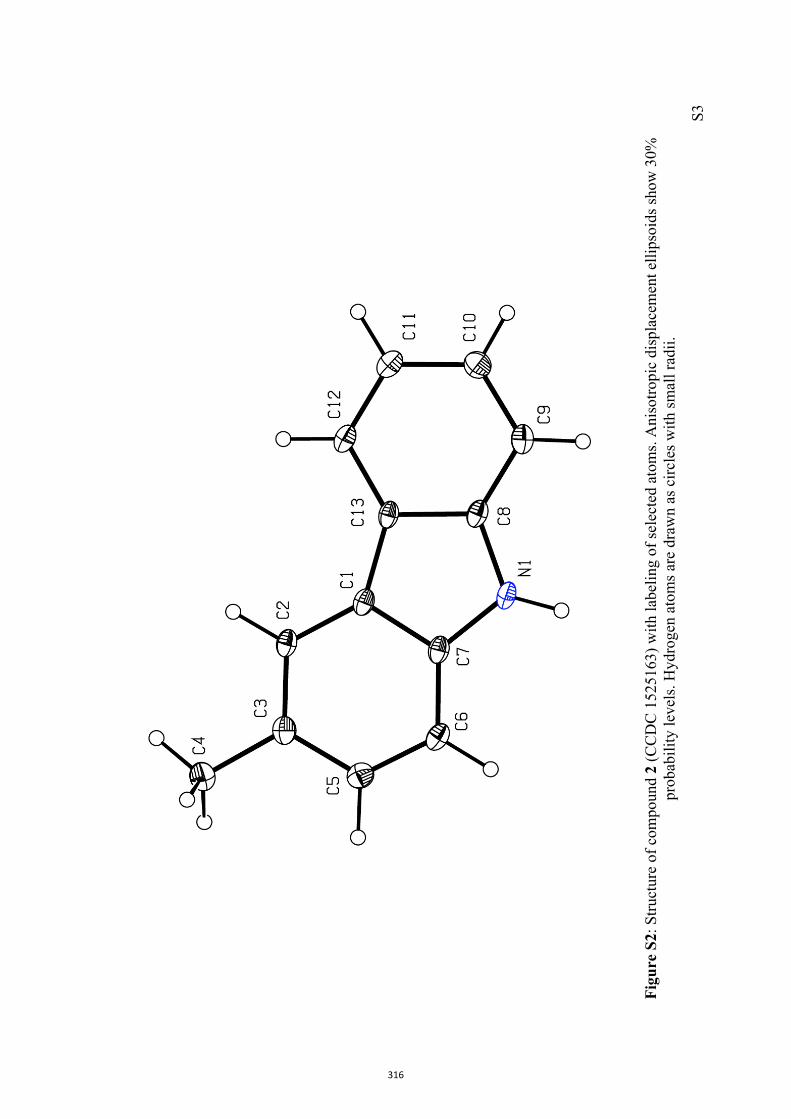
S3
Figu
re S
2: S
truct
ure
of c
ompo
und
2 (C
CD
C 1
5251
63) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
316
S4
Figu
re S
3: S
truct
ure
of c
ompo
und
3 (C
CD
C 1
5251
64) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
317
S5
Figu
re S
4: S
truct
ure
of c
ompo
und
4 (C
CD
C 1
5251
62) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
318
S6
Figu
re S
5: S
truct
ure
of c
ompo
und
5 (C
CD
C 1
5251
67) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
319
S7
Figu
re S
6: S
truct
ure
of c
ompo
und
6 (C
CD
C 1
5251
60) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
320
S8
Figu
re S
7: S
truct
ure
of c
ompo
und
17 (C
CD
C 1
5251
61) w
ith la
belin
g of
sele
cted
ato
ms.
Onl
y on
e of
the
two
mol
ecul
es p
rese
nt in
the
asym
met
ric u
nit i
s sho
wn.
The
re w
as so
me
diso
rder
ing
of a
tom
s C10
A a
nd C
11A
- on
ly th
e m
ajor
con
form
er is
show
n. T
his c
onfo
rmer
had
an
occu
panc
y of
81.
6%. A
niso
tropi
c di
spla
cem
ent e
llips
oids
show
30%
pro
babi
lity
leve
ls. H
ydro
gen
atom
s are
dra
wn
as c
ircle
s with
smal
l rad
ii.
321
S9
Figu
re S
8: S
truct
ure
of c
ompo
und
25 (C
CD
C 1
5251
65) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
322
S10
Tab
le S
1: C
ompa
riso
n of
the
13C
NM
R Sp
ectr
al D
ata
Repo
rted
by
Yosh
ikaw
a18 a
nd M
a7e fo
r Kar
apin
cham
ine
A w
ith th
e Eq
uiva
lent
Dat
a Re
cord
ed fo
r Com
poun
d 7
Prep
ared
by
the
Pres
ent R
oute
δ C
(e
x. Y
oshi
kaw
a)a
δ C
(ex.
Ma)
bδ C
(e
x. P
rese
nt R
oute
)cΔδ
δ C
(PR
d ) –δ C
(Y’k
awa)
15
4.5
154.
2 15
4.6
0.1
142.
0 14
1.9
142.
2 0.
2 13
8.8
138.
7 13
8.9
0.1
138.
5 13
8.4
138.
7 0.
2 13
1.8
131.
7 13
1.9
0.1
128.
3 12
8.1
128.
4 0.
1 12
5.6
125.
5 12
5.8
0.2
123.
7 12
3.7
123.
9 0.
2 12
3.3
123.
1 12
3.4
0.1
121.
0 12
1.0
121.
2 0.
2 11
9.8
119.
6 12
0.0
0.2
119.
5 11
9.4
119.
6 0.
1 11
7.0
116.
8 11
7.1
0.1
108.
3 10
8.3
108.
4 0.
1 10
7.5
107.
5 10
7.7
0.2
95.1
95
.1
95.2
0.
1 41
.2
40.9
41
.3
0.1
39.4
39
.3
39.5
0.
1 27
.2
26.2
26
.4
0.2
25.5
25
.5
25.8
0.
3 21
.4
21.3
21
.5
0.1
17.7
17
.6
17.8
0.
1 16
.6
16.4
16
.8
0.2
a Sp
ectru
m re
cord
ed in
CD
Cl 3
at 1
25 o
r 150
MH
z; b
Spec
trum
reco
rded
in C
DC
l 3 at
75
MH
z; c
Spec
trum
reco
rded
in
CD
Cl 3
at 1
00 M
Hz;
d PR
= p
rese
nt ro
ute.
323
S11
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
2.002.002.002.00
1.00
324

S12
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
f1 (
ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
111.69119.63120.86123.96126.40
140.99
206.16
325
S13
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.00
1.001.001.001.001.001.001.00
1.00
326
S14
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.4729.26 Acetone29.45 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
111.41111.64119.40120.74120.77123.83124.15126.21127.74128.60139.24141.30
206.15
11
1.2
11
1.4
11
1.6
11
1.8
f1 (
ppm
)111.41
111.64
12
0.0
12
0.5
12
1.0
12
1.5
f1 (
ppm
)120.74120.77
12
3.7
12
3.9
12
4.1
f1 (
ppm
)
123.83
124.15
327
S15
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.08
3.09
1.001.001.001.001.00
1.00
1.00
328

S16
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.5929.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.23 Acetone30.42 Acetone
55.63
100.45104.66110.77112.96
123.51123.56126.72127.18128.54138.44142.67
157.13
206.17
12
3.4
01
23
.45
12
3.5
01
23
.55
12
3.6
01
23
.65
12
3.7
0f1
(ppm
)
123.51
123.56
329
S17
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.00
3.00
1.001.012.001.001.00
1.00
330
S18
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.4629.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.23 Acetone30.42 Acetone
56.07
103.54111.50112.30115.59120.75124.20124.22127.66128.07136.14140.01
154.54
206.15
12
3.9
12
4.0
12
4.1
12
4.2
12
4.3
12
4.4
f1 (
ppm
)
124.20124.22
331
S19
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.14
1.001.001.001.001.001.00
1.00
1.00
332
S20
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
29.26 Acetone29.45 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
55.84
95.98
110.12111.78117.56122.20123.47124.43126.27130.12
143.13144.77
160.81
191.99
206.16
333
S21
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.003.00
1.011.951.001.001.001.00
7.26 CDCl3
334
S22
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
52.1855.87
76.84 CDCl377.16 CDCl377.48 CDCl3
106.81111.37116.36120.40120.87122.02123.73123.87126.47133.03139.64145.19
168.11
12
3.6
12
3.7
12
3.8
12
3.9
12
4.0
f1 (
ppm
)
123.73
123.87
335

S23
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.003.043.004.003.00
2.001.001.001.00
1.001.002.00
1.001.00
7.26 CDCl3
336
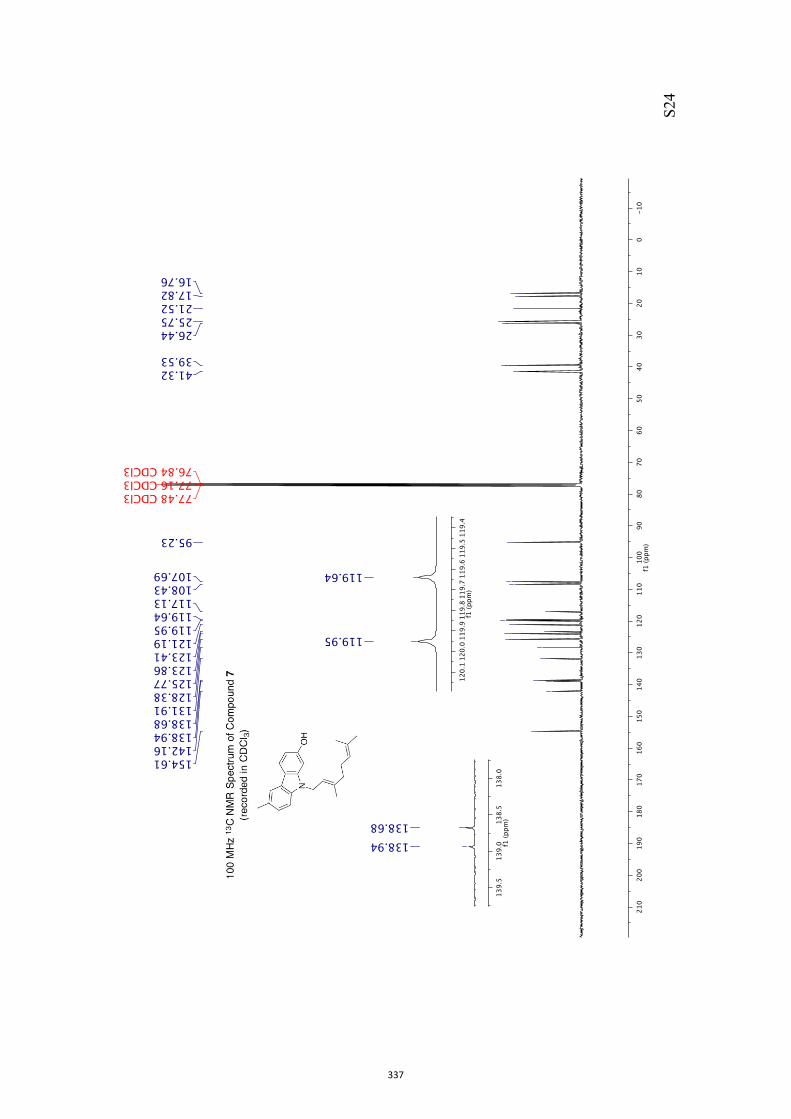
S24
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
16.7617.8221.5225.7526.44
39.5341.32
76.84 CDCl377.16 CDCl377.48 CDCl3
95.23
107.69108.43117.13119.64119.95121.19123.41123.86125.77128.38131.91138.68138.94142.16154.61
13
8.0
13
8.5
13
9.0
13
9.5
f1 (
ppm
)138.68
138.94
11
9.4
11
9.5
11
9.6
11
9.7
11
9.8
11
9.9
12
0.0
12
0.1
f1 (
ppm
)
119.64
119.95
337
S25
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.06
1.001.001.001.001.002.00
1.00
338

S26
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
55.72
95.39
108.78111.38117.74119.70119.98121.56124.24125.03
141.02142.36
160.09
206.15
11
9.2
11
9.4
11
9.6
11
9.8
12
0.0
12
0.2
f1 (
ppm
)119.70
119.98
339
S27
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
2.002.002.00
1.00
7.26 CDCl3
340
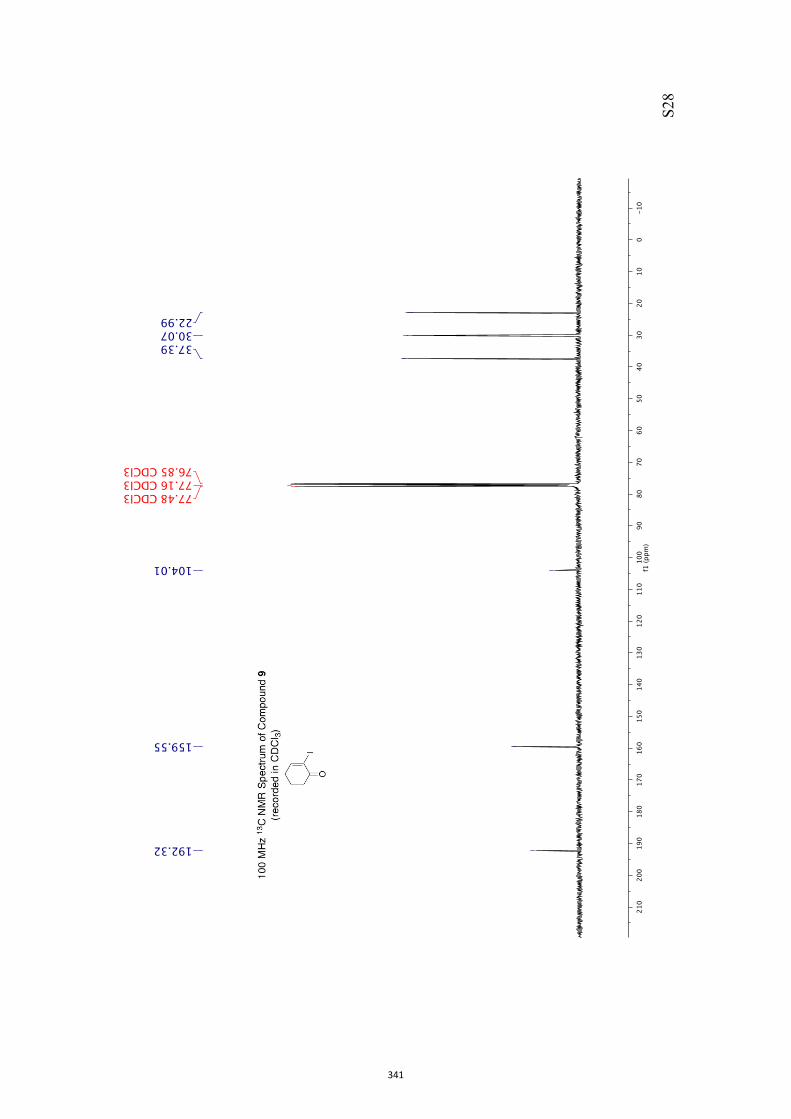
S28
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
22.9930.0737.39
76.85 CDCl377.16 CDCl377.48 CDCl3
104.01
159.55
192.32
341
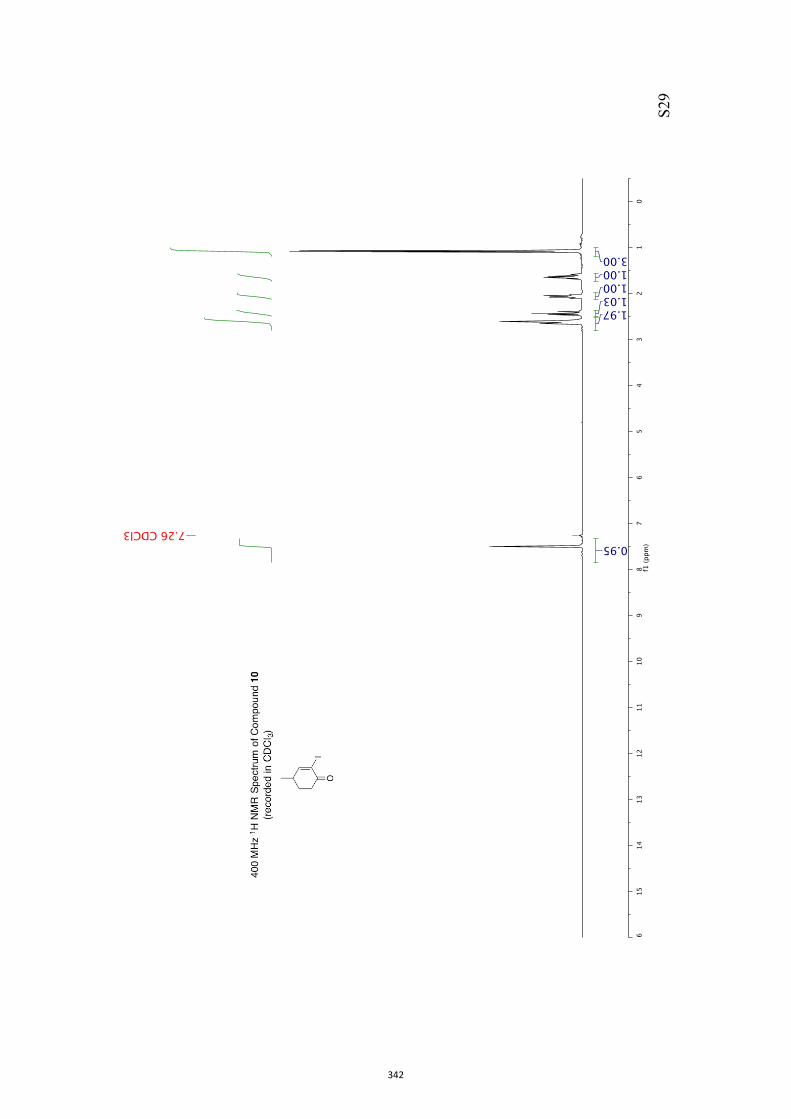
S29
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.001.001.001.031.97
0.95
7.26 CDCl3
342
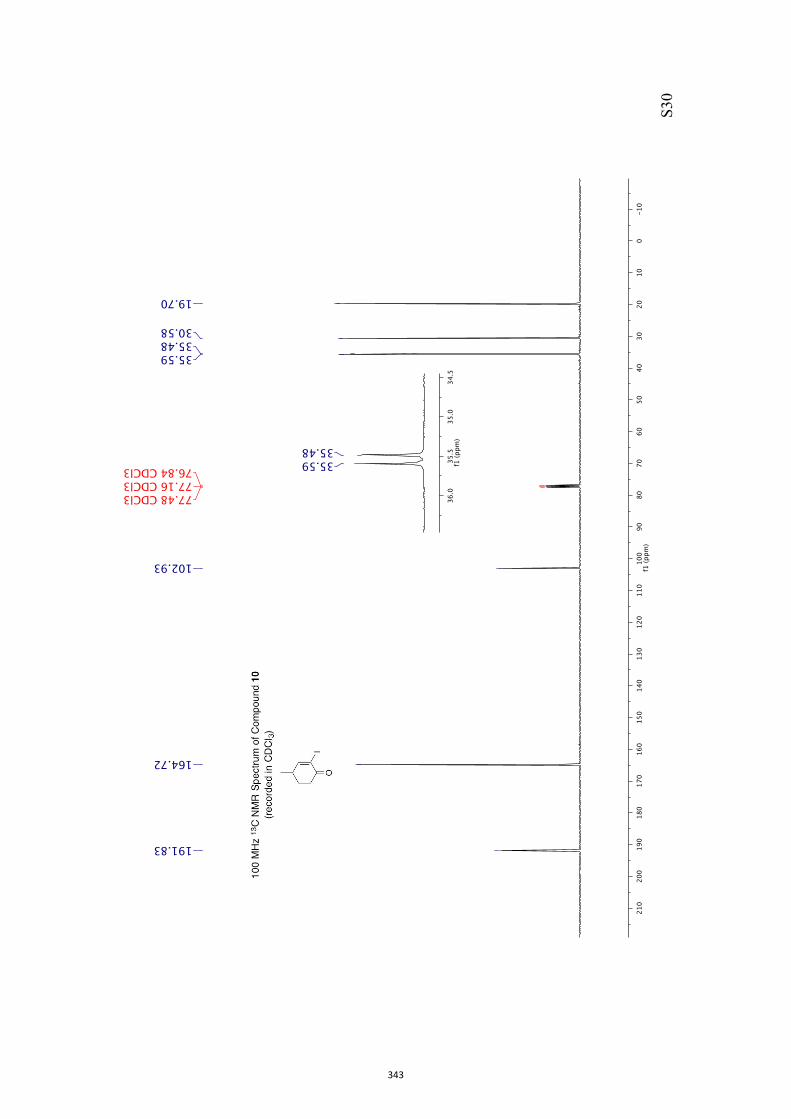
S30
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
19.70
30.5835.4835.59
76.84 CDCl377.16 CDCl377.48 CDCl3
102.93
164.72
191.83
34
.53
5.0
35
.53
6.0
f1 (
ppm
)
35.4835.59
343
S31
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
2.00
4.00
1.001.001.001.000.98
7.26 CDCl3
344
S32
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
22.6526.36
38.39
76.84 CDCl377.16 CDCl377.48 CDCl3
124.22128.83131.76132.17133.41139.44146.80148.65
196.58
345
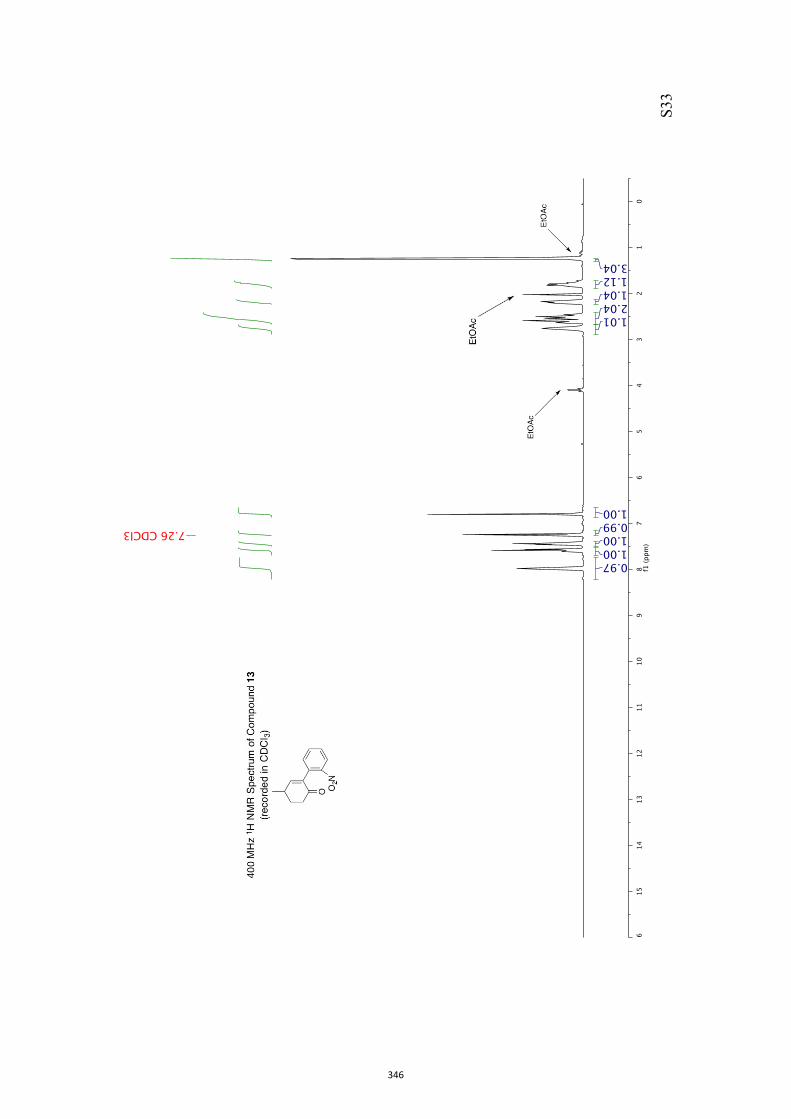
S33
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.041.121.042.041.01
1.000.991.001.000.97
7.26 CDCl3
346
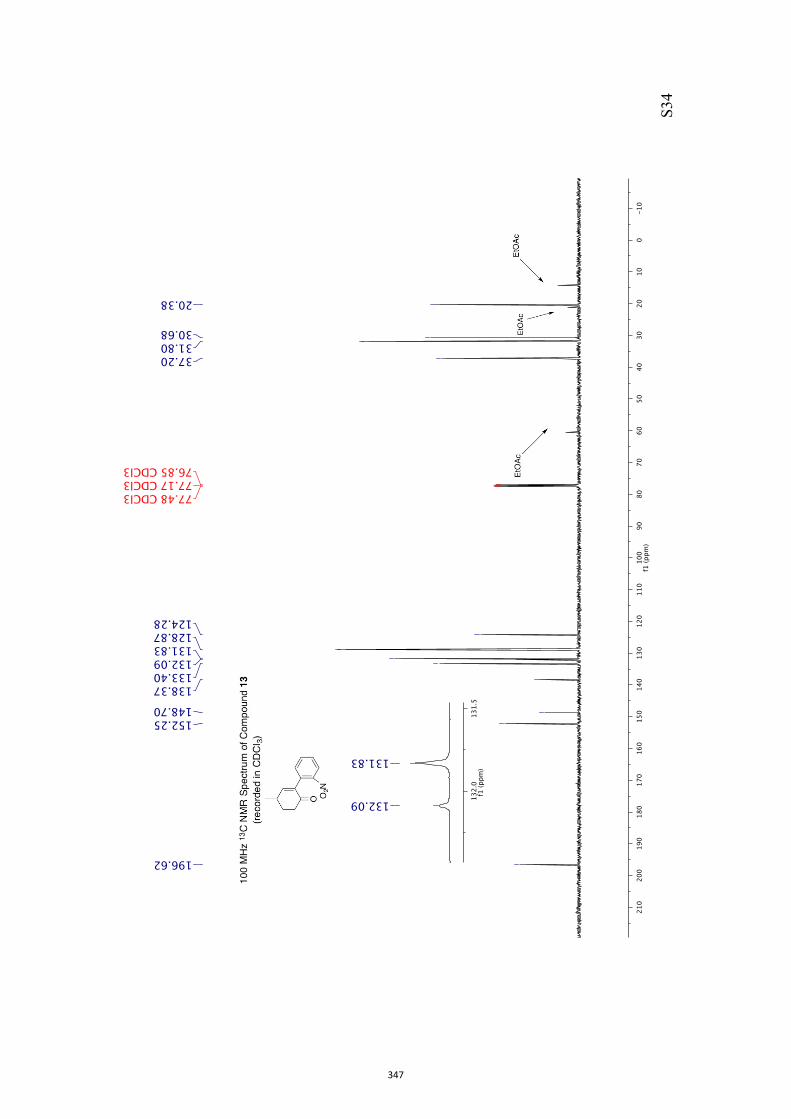
S34
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.38
30.6831.8037.20
76.85 CDCl377.17 CDCl377.48 CDCl3
124.28128.87131.83132.09133.40138.37
148.70152.25
196.62
13
1.5
13
2.0
f1 (
ppm
)
131.83
132.09
347
S35
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
4.12
2.002.01
2.001.001.00
1.00
348
S36
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.6523.7724.0324.1729.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.23 Acetone30.42 Acetone
109.69111.25118.04119.12121.11128.83135.10137.10
206.16
23
.62
3.8
24
.02
4.2
24
.4f1
(ppm
)
23.77
24.03
24.17
349
S37
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.121.082.101.043.25
2.051.001.00
0.98
350
S38
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
22.1223.4429.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.2230.42 Acetone30.5832.32
109.65111.27118.03119.11121.09128.73134.86137.43
206.15
28
29
30
31
f1 (
ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.2230.42 Acetone30.58
351
S39
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.05
1.121.103.17
3.08
0.981.001.001.00
7.26 CDCl3
352
S40
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.1020.4130.5430.8931.6031.9637.0737.33
56.6256.67
76.84 CDCl377.16 CDCl377.48 CDCl3
115.35115.55116.01116.08121.37121.47129.11129.15132.64133.00150.04150.33153.46153.73158.03158.10
196.69196.88
353
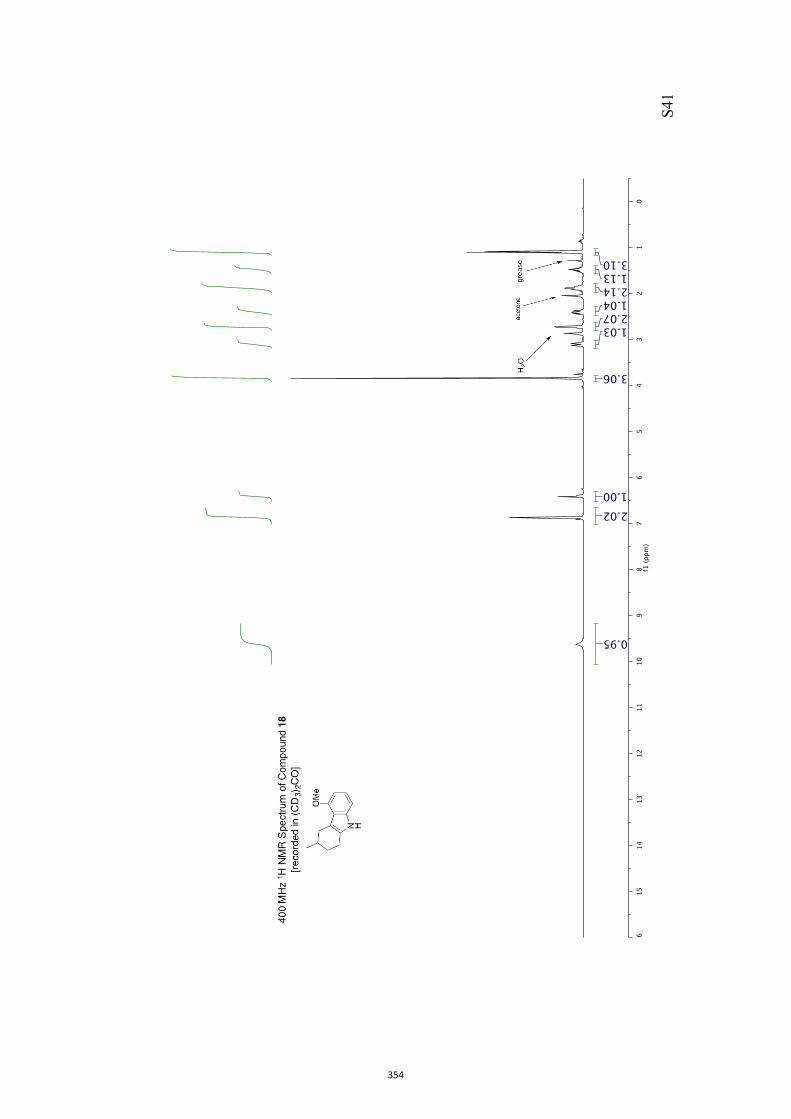
S41
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.101.132.141.042.071.03
3.06
1.00
2.02
0.95
354
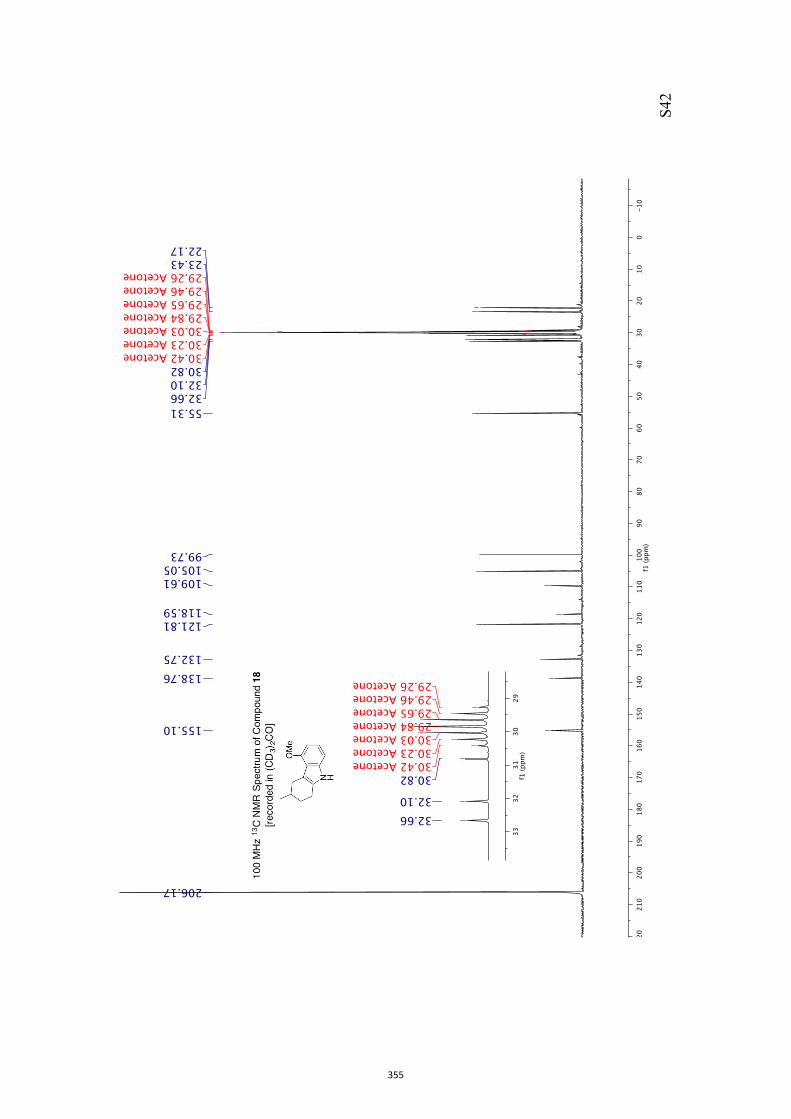
S42
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
22.1723.4329.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.23 Acetone30.42 Acetone30.8232.1032.66
55.31
99.73105.05109.61
118.59121.81
132.75
138.76
155.10
206.17
29
30
31
32
33
f1 (
ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.23 Acetone30.42 Acetone30.82
32.10
32.66
355
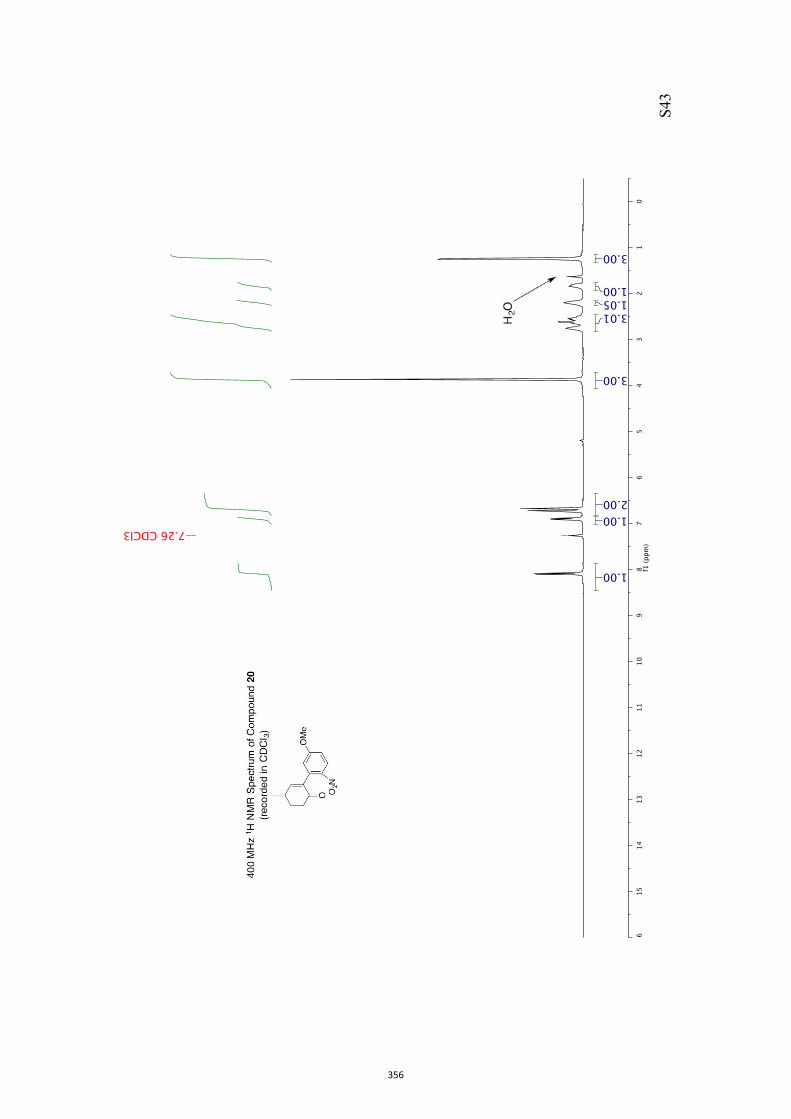
S43
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.00
1.001.053.01
3.00
2.00
1.00
1.00
7.26 CDCl3
356
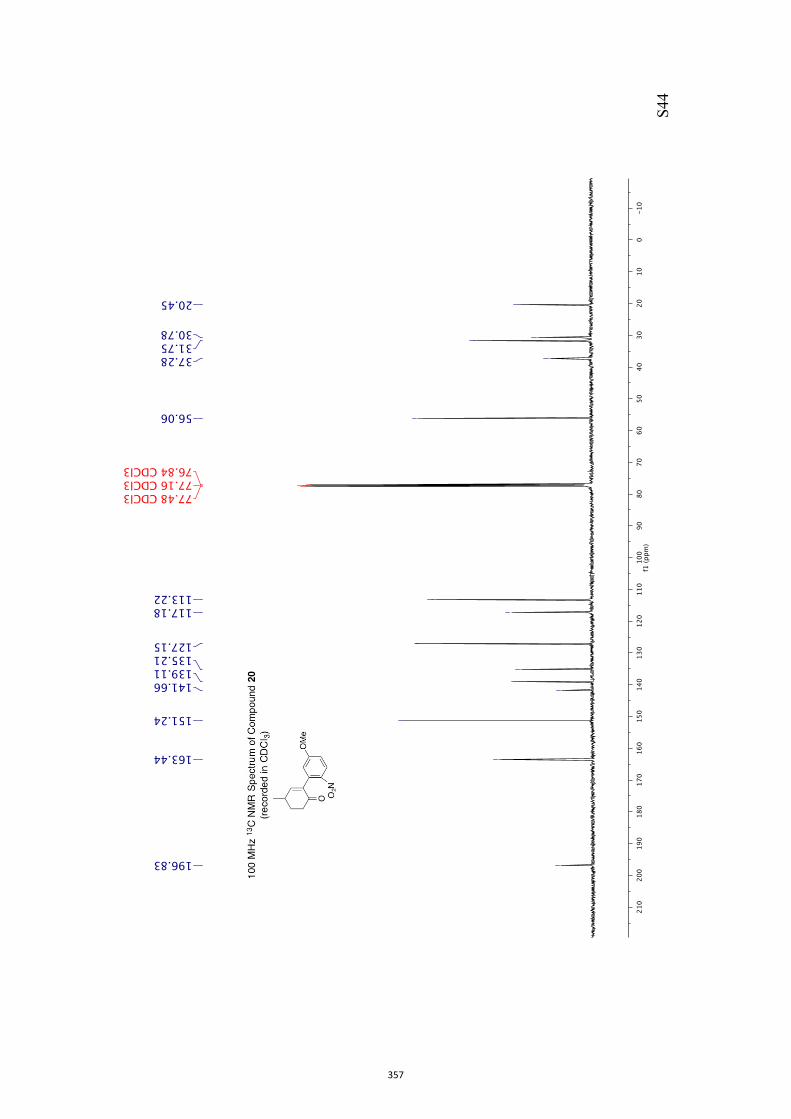
S44
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.45
30.7831.7537.28
56.06
76.84 CDCl377.16 CDCl377.48 CDCl3
113.22117.18
127.15135.21139.11141.66
151.24
163.44
196.83
357
S45
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.041.042.071.053.13
3.11
1.001.001.00
0.98
358
S46
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
22.1423.5629.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.3530.42 Acetone30.6332.36
55.84
100.59
109.60110.69111.78
129.10132.50135.71
154.57
206.12
29
.22
9.4
29
.62
9.8
30
.03
0.2
30
.43
0.6
f1 (
ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.3530.42 Acetone30.63
359
S47
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.101.031.041.041.071.06
3.08
1.002.001.00
7.26 CDCl3
360
S48
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.46
30.7631.8137.26
55.97
76.84 CDCl377.16 CDCl377.48 CDCl3
109.28
119.68124.24132.65138.05
149.21151.81159.66
196.93
361
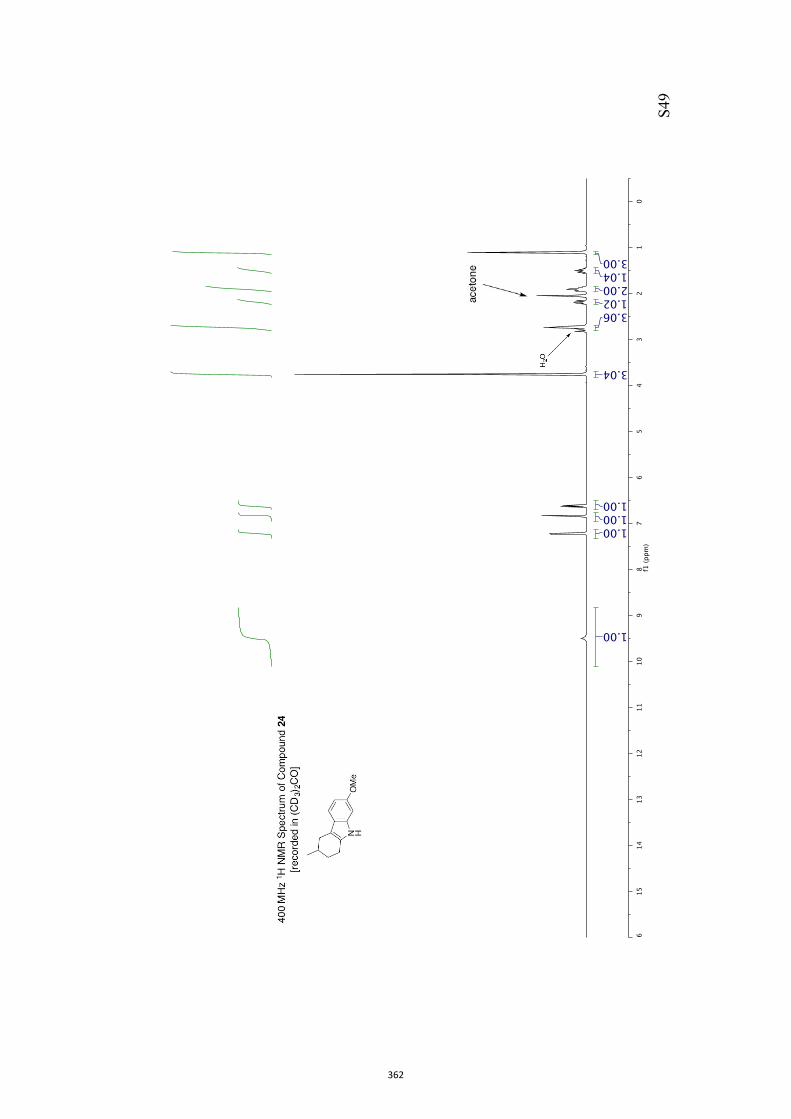
S49
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.001.042.001.023.06
3.04
1.001.001.00
1.00
362
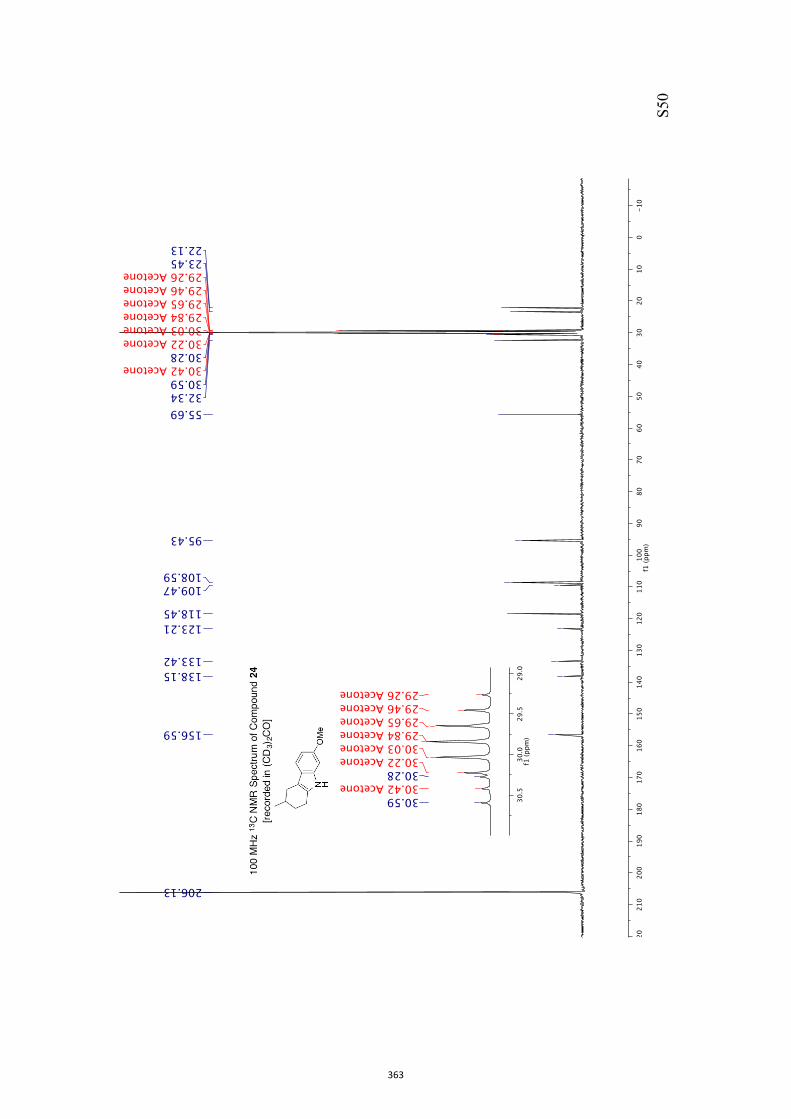
S50
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
22.1323.4529.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.2830.42 Acetone30.5932.34
55.69
95.43
108.59109.47
118.45
123.21
133.42
138.15
156.59
206.13
29
.02
9.5
30
.03
0.5
f1 (
ppm
)
29.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.2830.42 Acetone30.59
363
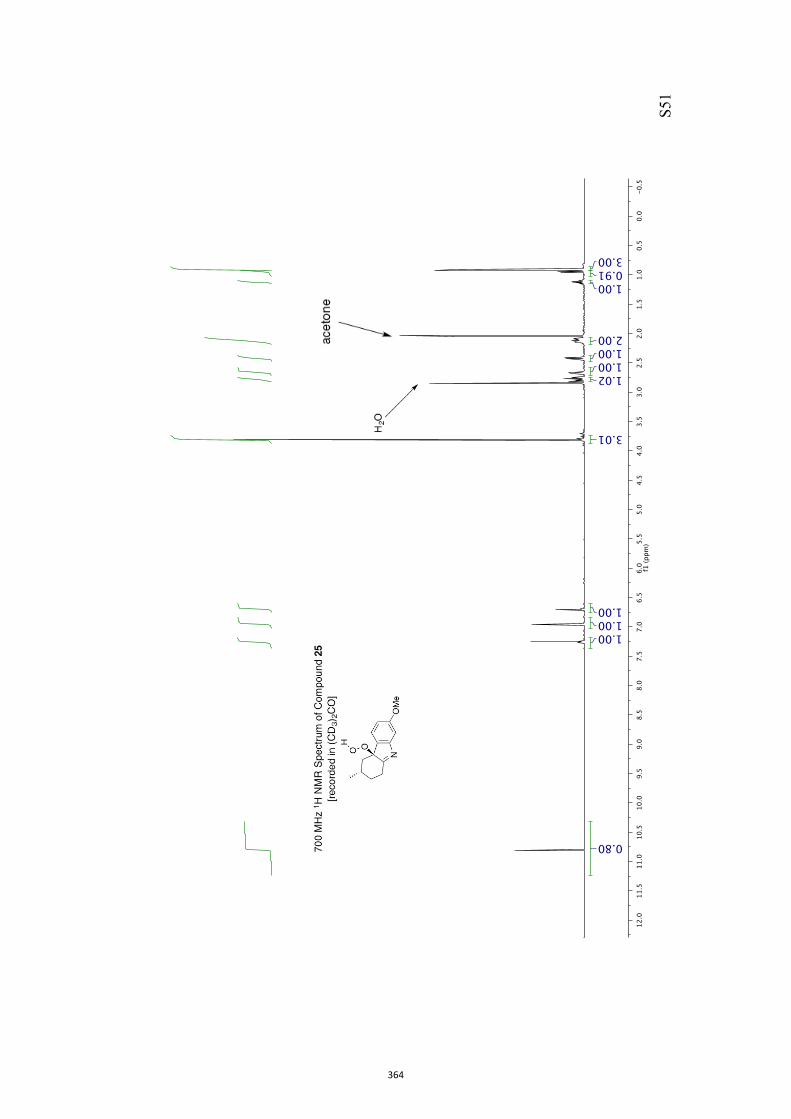
S51
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
3.000.911.00
2.001.001.001.02
3.01
1.001.001.00
0.80
364
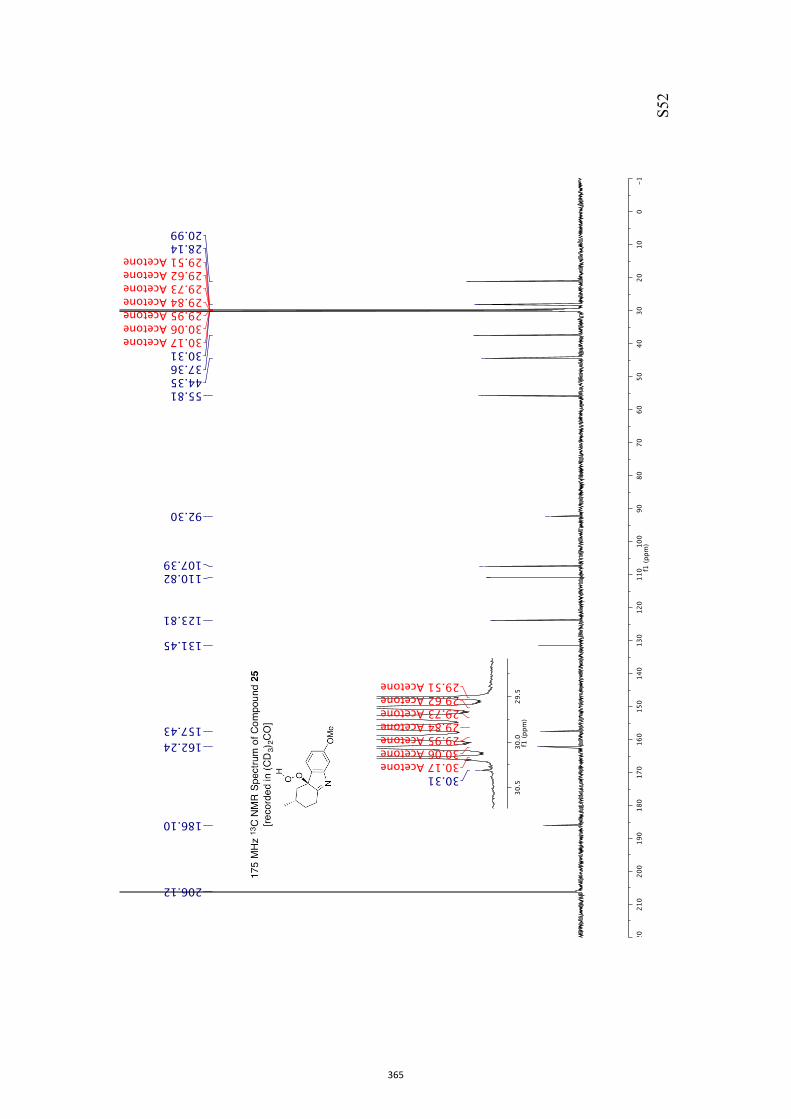
S52
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
20.9928.1429.51 Acetone29.62 Acetone29.73 Acetone29.84 Acetone29.95 Acetone30.06 Acetone30.17 Acetone30.3137.3644.3555.81
92.30
107.39110.82
123.81
131.45
157.43
162.24
186.10
206.12
29
.53
0.0
30
.5f1
(ppm
)
29.51 Acetone29.62 Acetone29.73 Acetone29.84 Acetone29.95 Acetone30.06 Acetone30.17 Acetone30.31
365
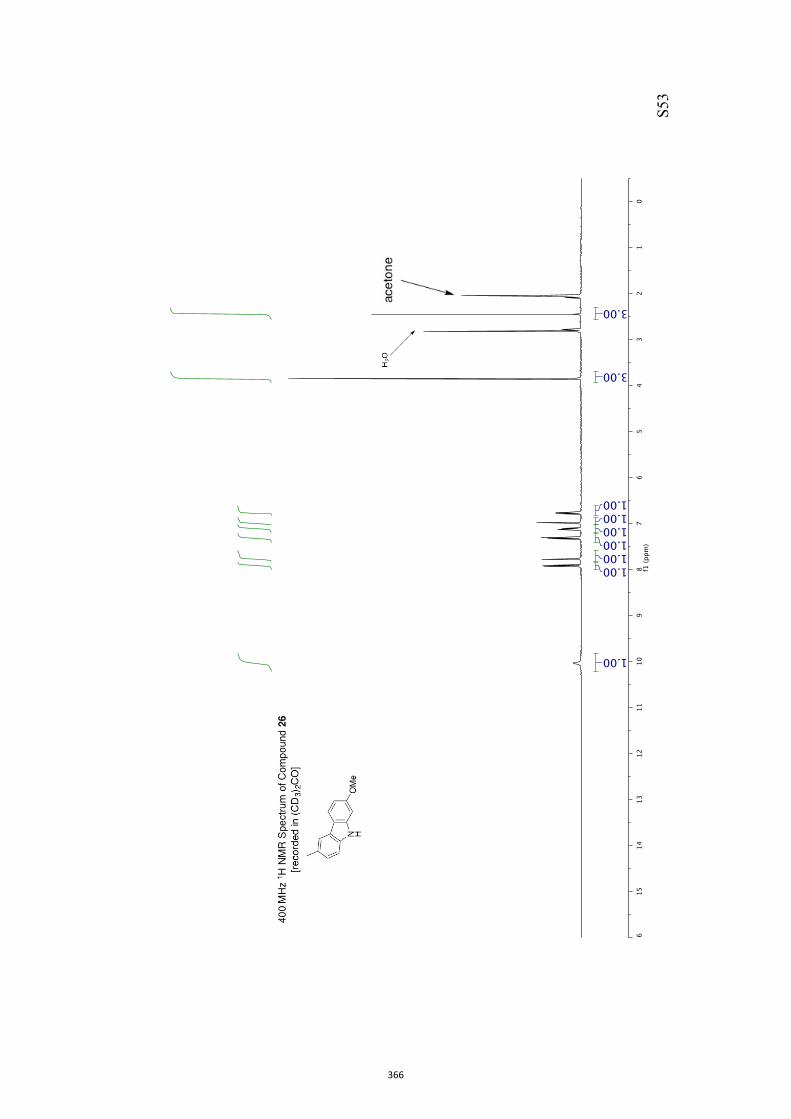
S53
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.00
3.00
1.001.001.001.001.001.00
1.00
366
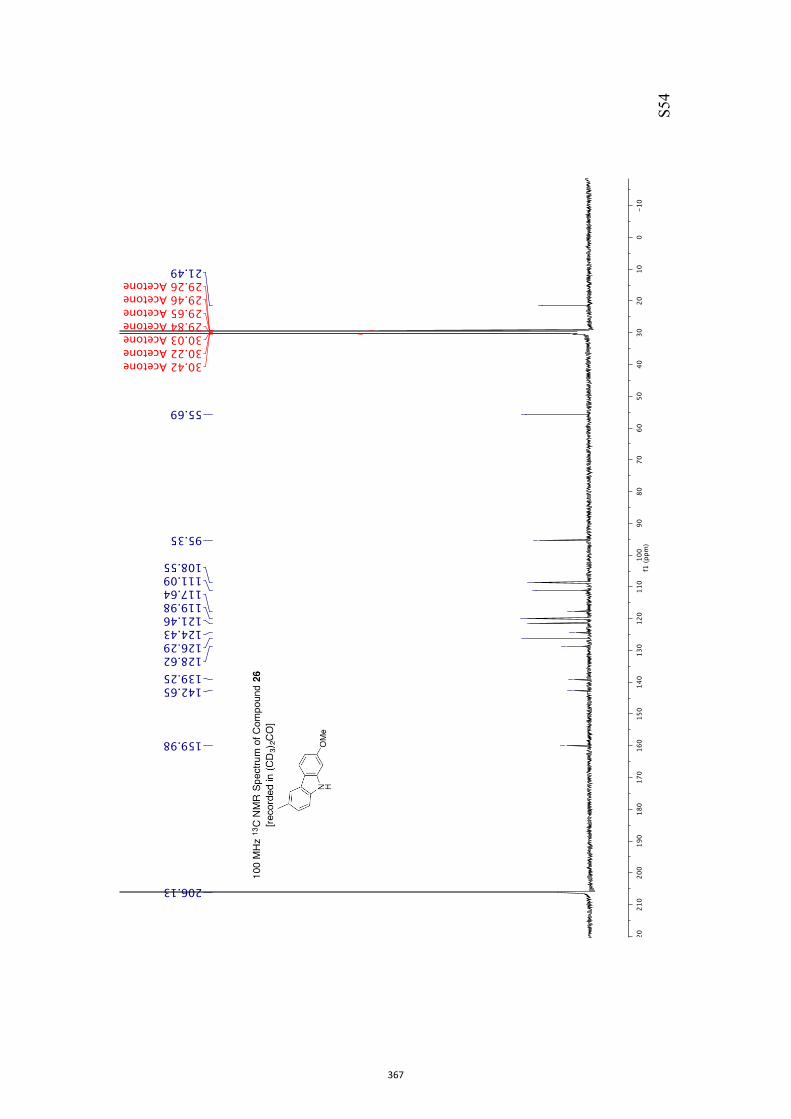
S54
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.4929.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
55.69
95.35
108.55111.09117.64119.98121.46124.43126.29128.62
139.25142.65
159.98
206.13
367
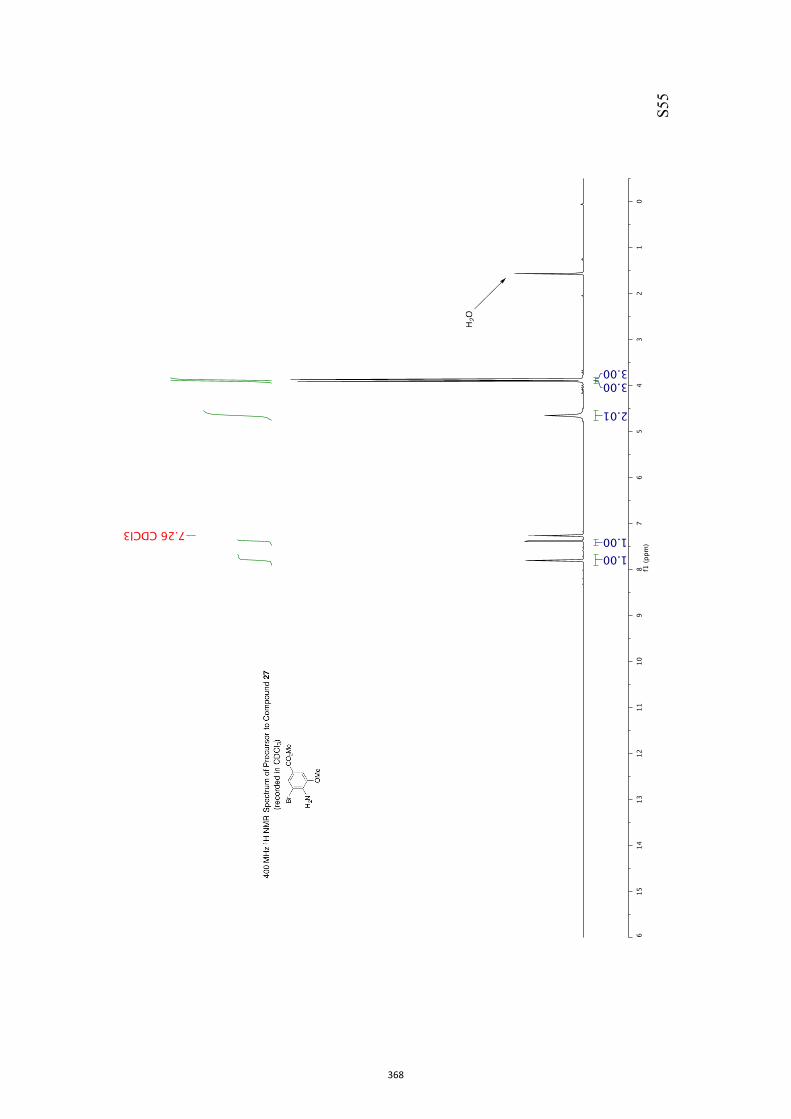
S55
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.003.00
2.01
1.00
1.00
7.26 CDCl3
368
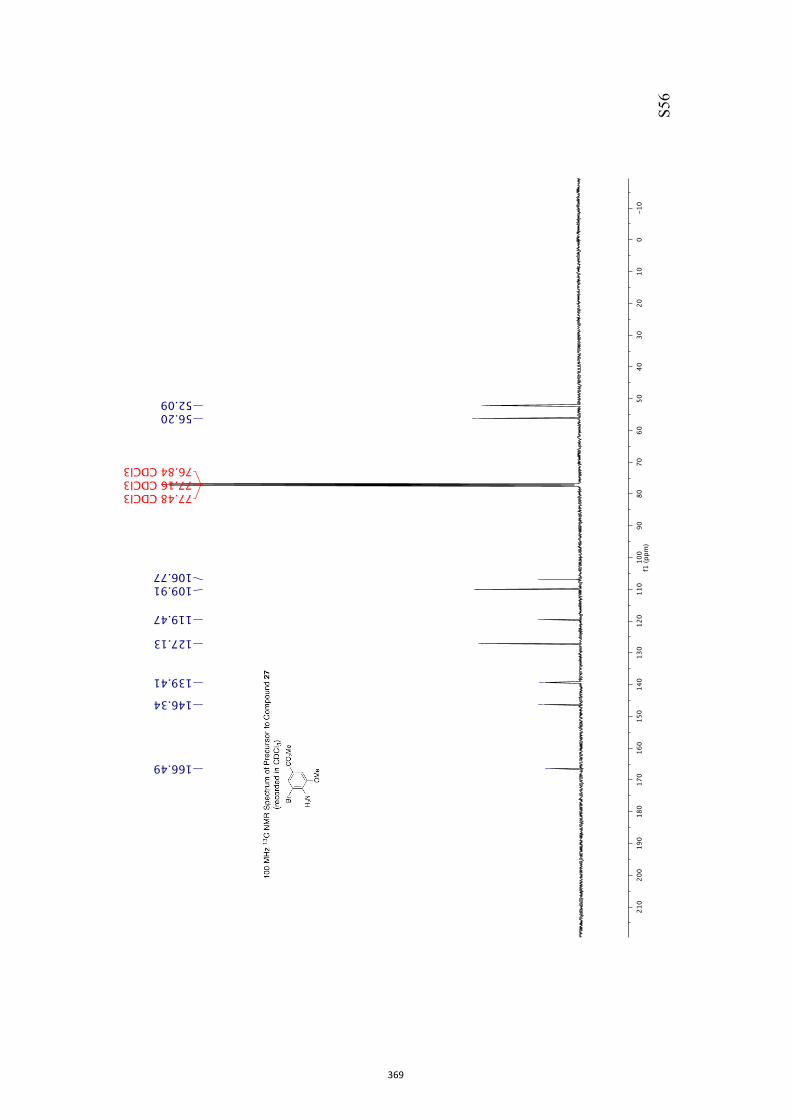
S56
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
52.0956.20
76.84 CDCl377.16 CDCl377.48 CDCl3
106.77109.91
119.47
127.13
139.41
146.34
166.49
369
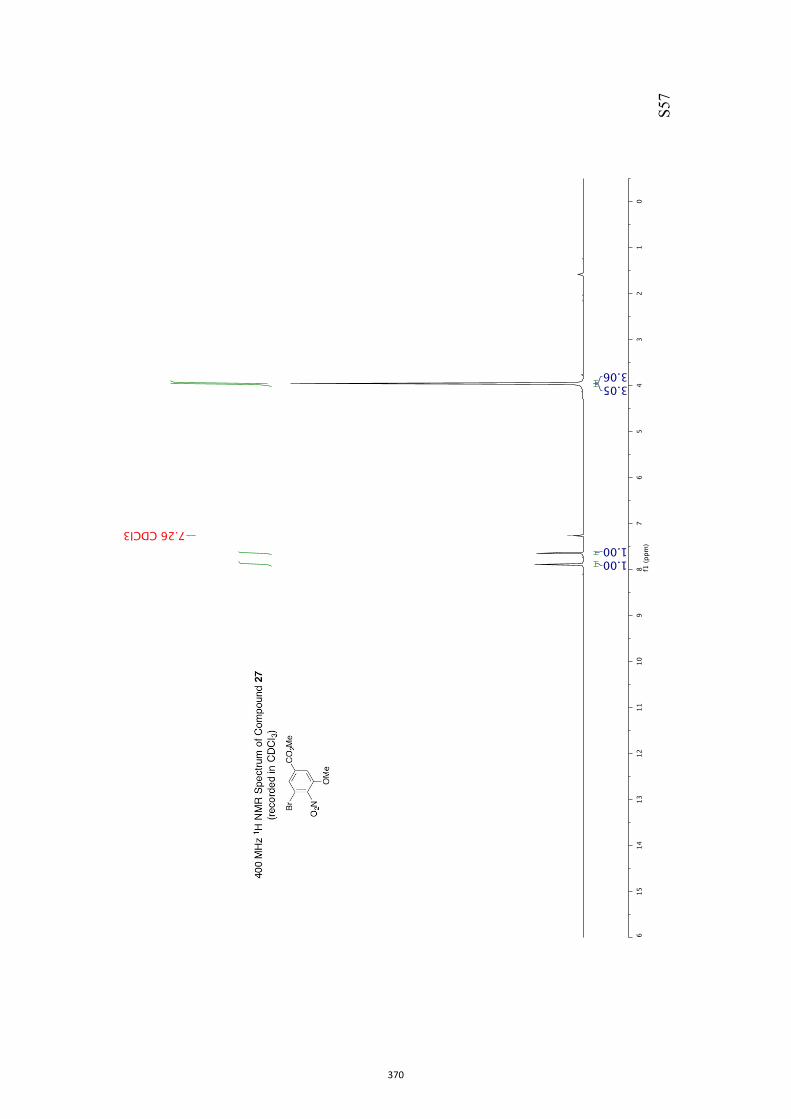
S57
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.063.05
1.001.00
7.26 CDCl3
370
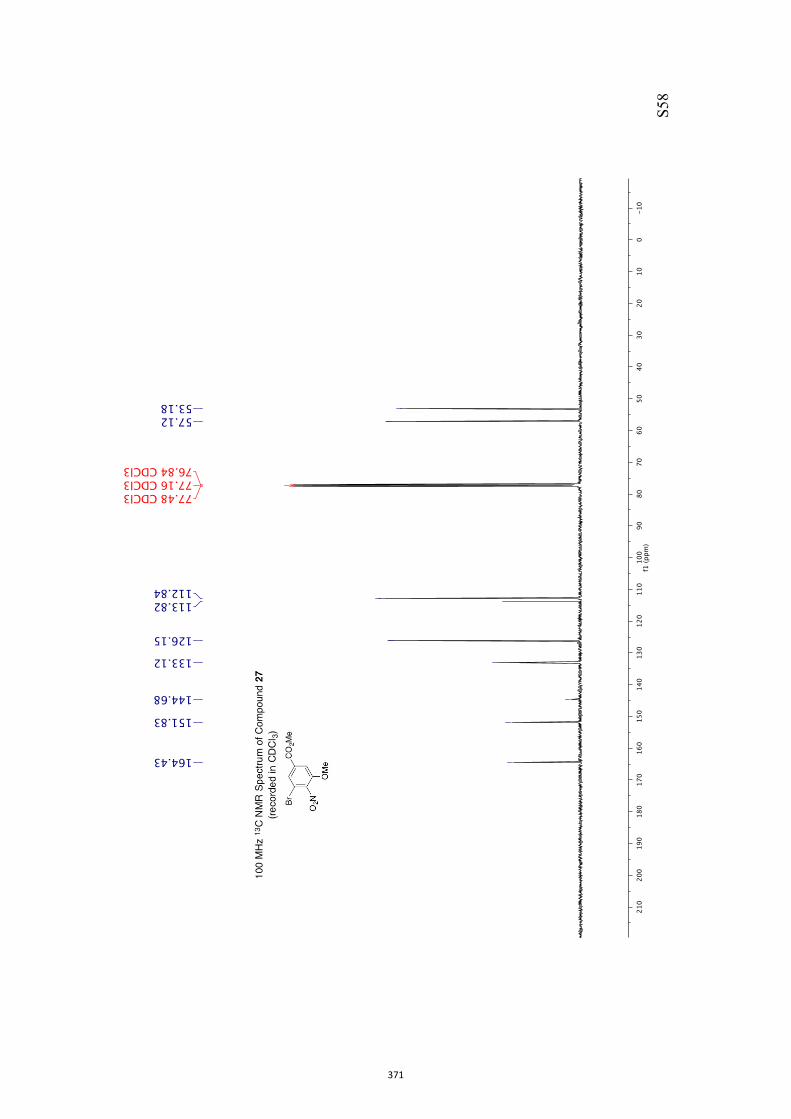
S58
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
53.1857.12
76.84 CDCl377.16 CDCl377.48 CDCl3
112.84113.82
126.15
133.12
144.68
151.83
164.43
371
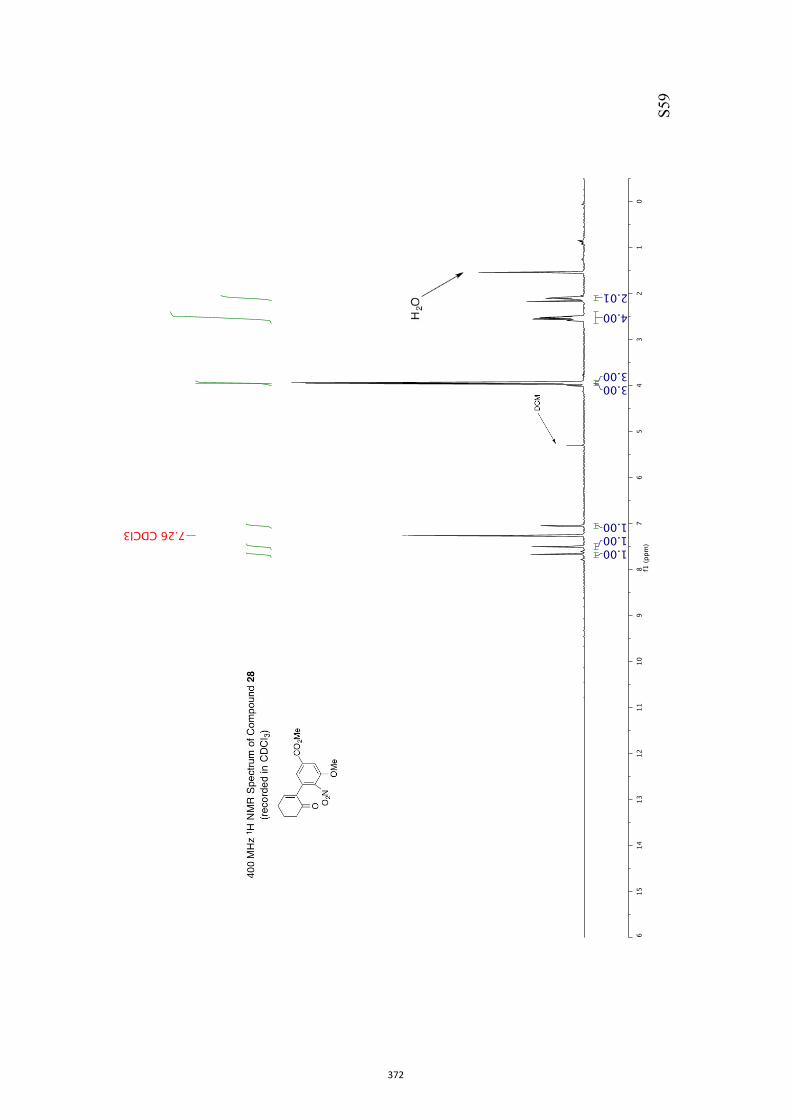
S59
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
2.01
4.00
3.003.00
1.001.001.00
7.26 CDCl3
372
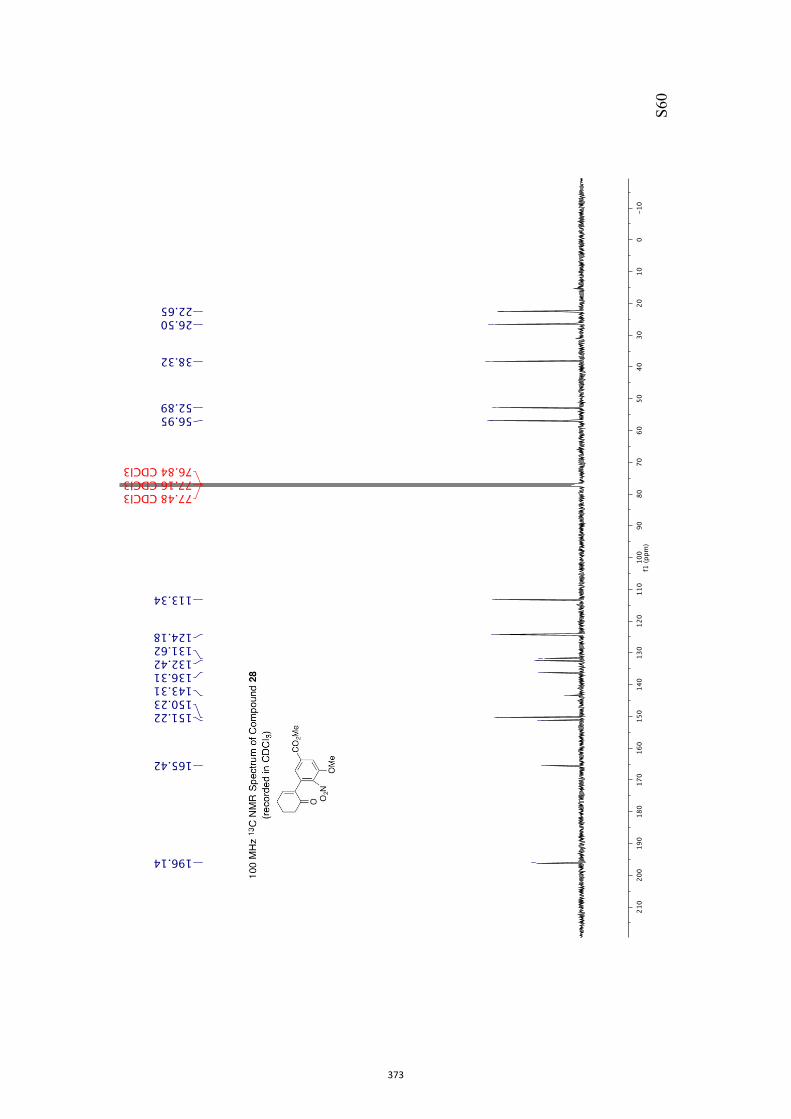
S60
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
22.6526.50
38.32
52.8956.95
76.84 CDCl377.16 CDCl377.48 CDCl3
113.34
124.18131.62132.42136.31143.31150.23151.22
165.42
196.14
373
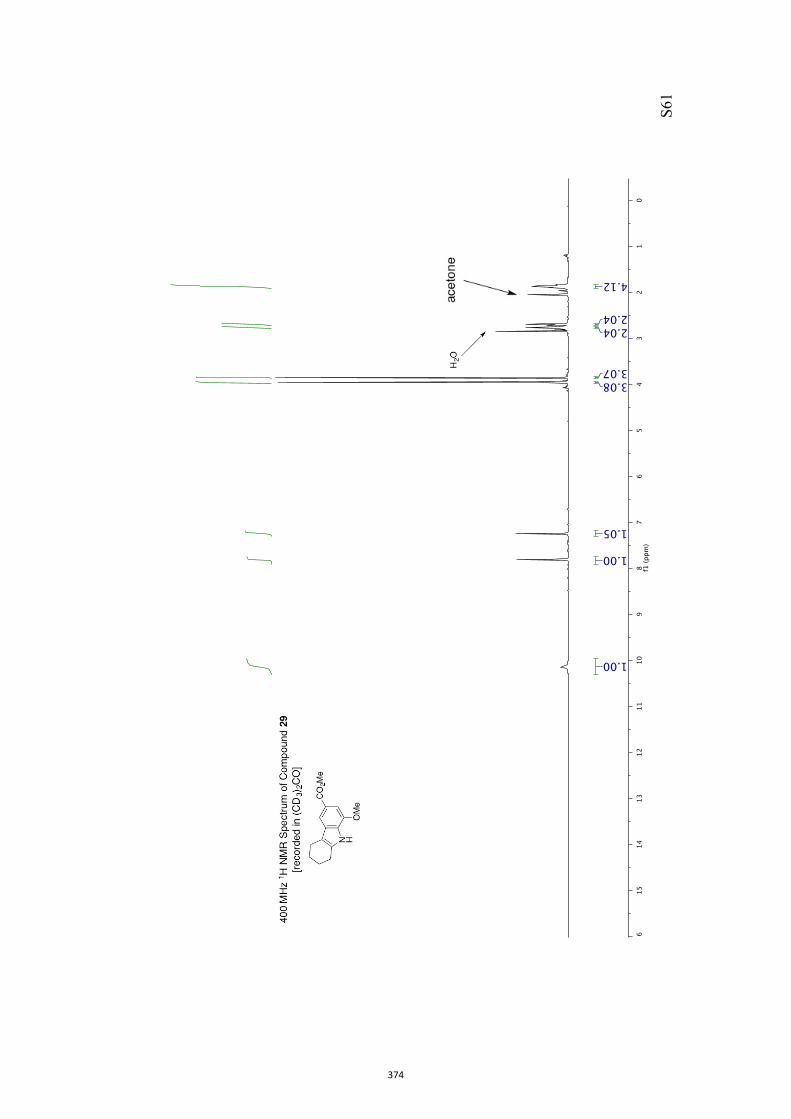
S61
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
4.12
2.042.04
3.073.08
1.05
1.00
1.00
374

S62
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.6223.6923.8623.9829.26 Acetone29.45 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
51.8155.72
102.53
111.73114.78122.08129.38129.71136.63
146.24
168.45
206.15
23
.42
3.6
23
.82
4.0
24
.2f1
(ppm
)
23.69
23.86
23.98
375
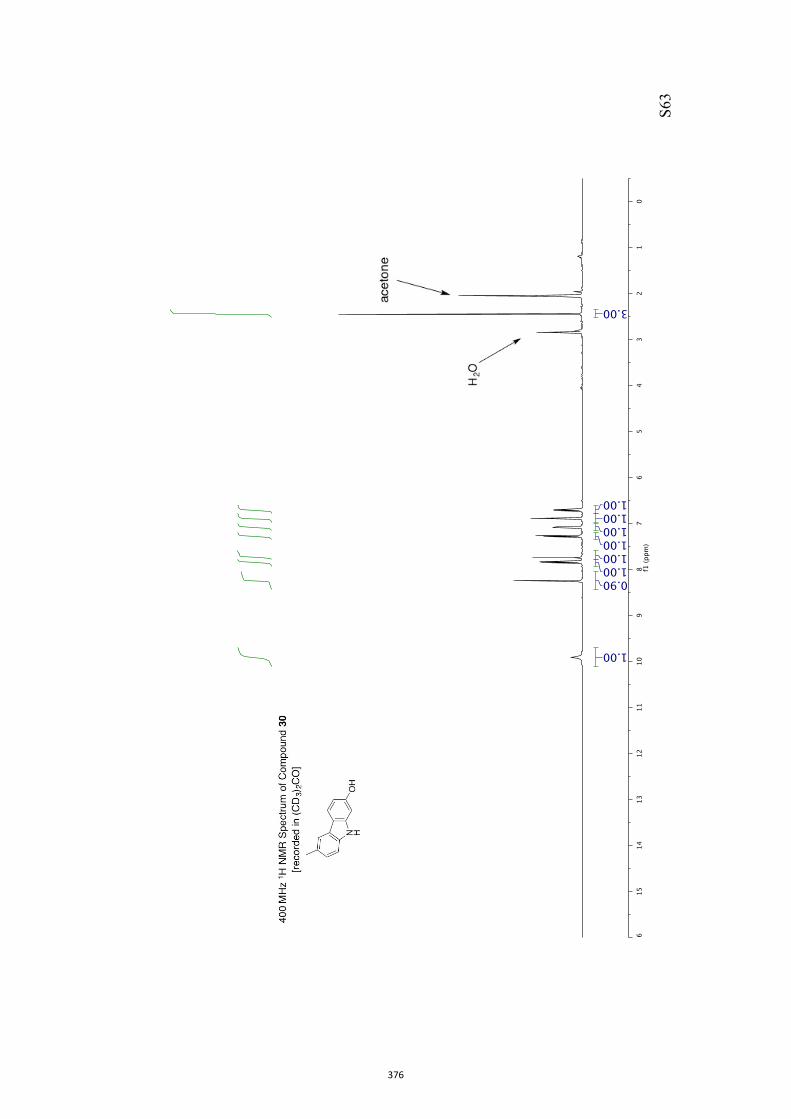
S63
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
3.00
1.001.001.001.001.001.000.90
1.00
376
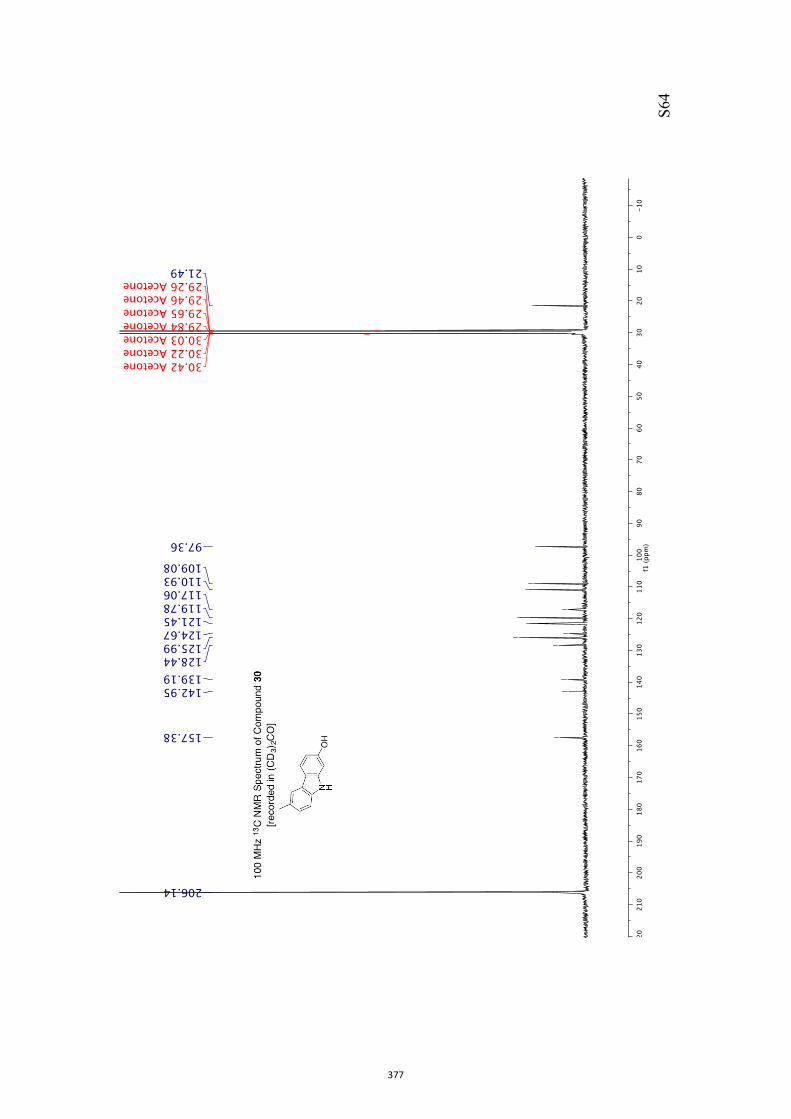
S64
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
21.4929.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone
97.36
109.08110.93117.06119.78121.45124.67125.99128.44
139.19142.95
157.38
206.14
377
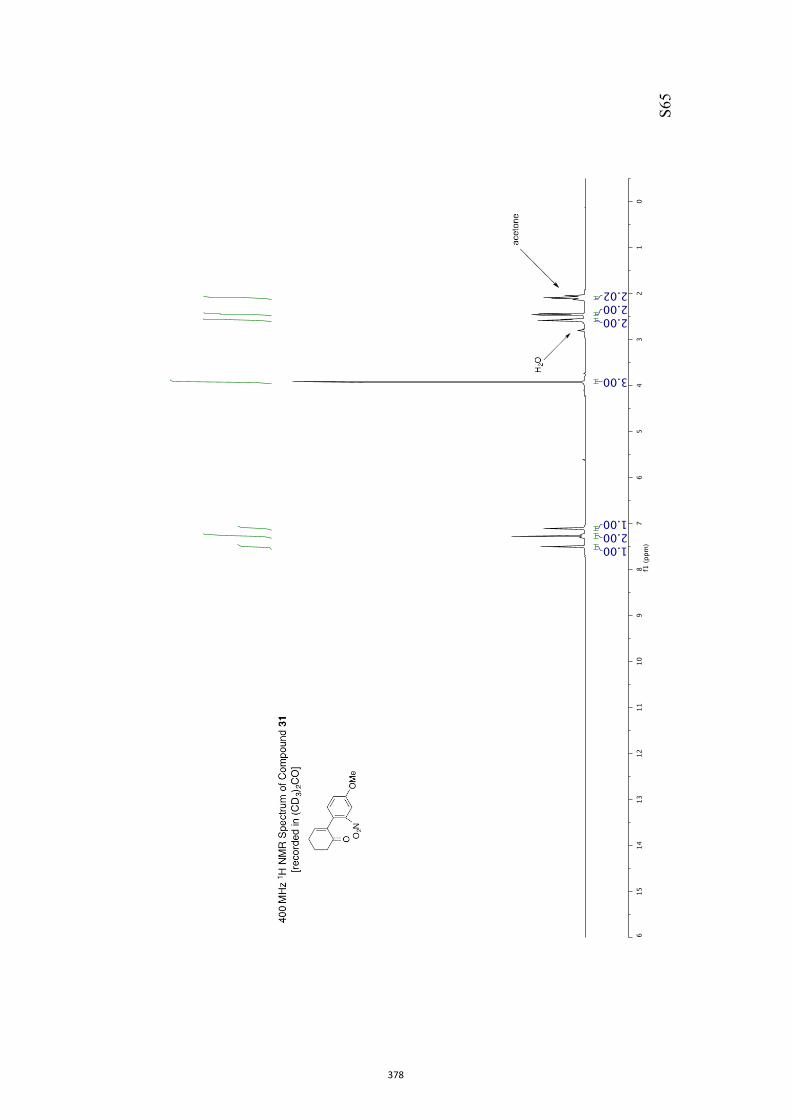
S65
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
2.022.002.00
3.00
1.002.001.00
378
S66
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
23.4826.9529.26 Acetone29.46 Acetone29.65 Acetone29.84 Acetone30.03 Acetone30.22 Acetone30.42 Acetone39.03
56.41
109.87
120.04
125.08
133.61139.48147.52150.42
160.54
196.47
206.10
379

S67
01
23
45
67
89
10
11
12
13
14
15
16
f1 (
ppm
)
4.10
4.00
3.16
1.001.001.001.00
7.26 CDCl3
380
S68
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
21.0823.3423.3523.44
55.97
76.84 CDCl377.16 CDCl377.48 CDCl3
95.00
108.40110.07118.31122.51
132.93136.49
155.94
23
.20
23
.25
23
.30
23
.35
23
.40
23
.45
23
.50
23
.55
f1 (
ppm
)
23.3423.35
23.44
381

382
Publication Four
A Unified Approach to the Isomericα-,β-,γ-,andδ-
Carbolines via their 6,7,8,9-Tetrahydro Counterparts
Qiao Yan, Emma Gin, Martin G. Banwell, Anthony C. Willis
and Paul D. Carr
J. Org. Chem., 2017, 82, 4328.


A Unified Approach to the Isomeric α‑, β‑, γ‑, and δ‑Carbolines viatheir 6,7,8,9-Tetrahydro CounterpartsQiao Yan, Emma Gin, Martin G. Banwell,* Anthony C. Willis, and Paul D. Carr
Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 2601, Australia
*S Supporting Information
ABSTRACT: A cross-coupling/reductive cyclization protocolhas been employed in a unified approach to all four carbolines.So, for example, the 2-nitropyridine 8, which is readily preparedthrough an efficient palladium-catalyzed Ullmann cross-couplingreaction, is reductively cyclized under conventional conditions togive 6,7,8,9-tetrahydro-α-carboline that is itself readily aromatizedto give α-carboline (1).
■ INTRODUCTIONThe isomeric α-, β-, γ-, and δ-carbolines (1−4, respectively, inFigure 1) are important heterocyclic rings systems.1 All are
found, albeit to varying extents, as key structural motifs innatural products. They also feature in a wide range ofmedicinally relevant compounds. The utility of their variousderivatives in materials science is a further focus of currentstudies.2 The α-carboline framework (1) is encountered in alimited number of naturally occurring anticancer agents and inthe neuro-protective alkaloid mescengricin.3 On the other hand,synthetically derived α-carbolines have shown anxiolytic, anti-inflammatory, central nervous system stimulating, and kinaseinhibitory properties.4 β-Carboline (2), itself a natural productisolated from both plants and micro-organisms, is the mostwell-known of the four systems and represents a keysubstructure associated with, for example, the eudistomineand manzamine classes of biologically active marine alkaloids.5
Many medicinal agents embodying this heterocyclic frameworkhave been identified.5,6 Derivatives of γ-carboline (3) have beenexplored extensively as anticancer and anti-Alzheimer agents,7
while those associated with δ-carboline (4) have been studied,inter alia, for their antibacterial and antitumor properties.1b,8 δ-Carboline-containing alkaloids have been isolated from, forexample, various West and Central African plants that areprized as sources of traditional medicines for treating malariaand certain infectious diseases.1b
A multitude of methods has been established for thesynthesis of the carbolines, including classical ones involvingGraebe−Ullmann, Fischer indolization, Bischler−Napieralski,and Pictet−Spengler reactions.9,10 Variations on the Cadogan
syntheses of carbazoles are also known,11 as are routesinvolving the annulation of pyridines onto indoles,9,10,12
including through Diels−Alder and electrocyclization pro-cesses. Generally speaking, though, “customized” approachesare required for the assembly of each of the α-, β-, γ-, and δ-carboline frameworks, thus prompting the search for moregeneral routes to them.13 There has been modest success in thisregard, with the most effective route involving the cyclization ofanilinopyridines.13g−j Recently, Driver13i and Ray13j have eachreported variations on such methods that allow access to threeof the four frameworks. It is against this background that wenow detail a distinct, operationally simple, and likely flexibleroute to all four of the isomeric carbolines and highlight theutility of this through the synthesis of the simple naturalproduct harman (5, Figure 2), a compound that displays anti-HIV and antibacterial properties.14
■ RESULTS AND DISCUSSIONThe pivotal steps associated with the unified approach to thecarbolines reported here are the palladium-catalyzed Ullmanncross-coupling15 of 2-iodocyclohex-2-en-1-one16 with therelevant halogenated nitropyridine and the reductive cyclizationof the ensuing 2-pyridylcyclohex-2-en-1-one to give thecorresponding 6,7,8,9-tetrahydrocarboline. Oxidation of thesetetrahydro compounds to their fully aromatic counterparts (viz.,the carbolines) was readily accomplished using 10 wt %palladium on carbon. This sequence mirrors that used in our
Received: February 10, 2017Published: March 17, 2017
Figure 1. Isomeric α-, β-, γ-, and δ-carbolines (1−4).
Figure 2. β-Carboline alkaloid harman (5).
Article
pubs.acs.org/joc
© 2017 American Chemical Society 4328 DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
383

recently reported syntheses of various carbazole-based naturalproducts, including glycoborine, glycozoline, clausazoline K,mukonine, and karapinchamine A.17
The synthesis of α-carboline (1), as shown in Scheme 1, isillustrative and starts with the palladium-catalyzed Ullmann
cross-coupling of the readily prepared17 2-iodocyclohex-2-en-1-one (6) with commercially available 3-bromo-2-nitropyridine(7), thus affording the 2-pyridylcyclohex-2-en-1-one (8) in 82%yield. In order to reduce the extent of homocoupling of thepyridine in this reaction, the iodo enone 6 was treated with acombination of copper metal, copper(I) iodide, and Pd(dppf)-Cl2·CH2Cl2 in DMSO at 50 °C for 0.75 h prior the addition ofcompound 7. Presumably this allows cupration of compound 6to take place prior to a palladium-catalyzed cross-couplingreaction with halide 7, thereby increasing the yields of product8. The reductive cyclization of compound 8 was effected usinghydrogen in the presence of catalytic amounts of 10 wt %palladium on carbon (Pd/C) in methanol at room temperaturefor 16 h, and this produced the 6,7,8,9-tetrahydrocarboline(9)18 in 75% yield. In our hands, the oxidation19 of compound9 to α-carboline (1)4a was best carried out by exposing theformer system to an equivalent mass of 10 wt % palladium oncarbon in diphenyl ether at 210 °C for 0.66 h. By such meanstarget 1 was obtained in 97% yield, and all of the spectral dataacquired on this material were in complete accord with theassigned structure and matched those reported in the literature.A single-crystal X-ray analysis was carried out on compound 1and details of this are provided in the Experimental Section andthe Supporting Information (SI).Given the use of 10 wt % palladium on carbon in both the
second and third steps of the reaction sequence, these could, inprinciple, be “telescoped” to establish a one-pot process. Todate, however, we have not been able to identify conditions thatallow for this to be conducted in both an operationally superiorway and with better outcomes.The synthesis of β-carboline (2) (Scheme 2) required 4-
iodo-3-nitropyridine (10)20 as a coupling partner, and this wasreadily obtained by reacting the commercially available chloroanalogue with sodium iodide in acetonitrile (see theExperimental Section for details). Cross-coupling of com-pounds 6 and 10 proceeded smoothly under essentially thesame conditions as employed for the conversion 6 + 7→ 8 andprovided the anticipated coupling product 11 in 86% yield.Reductive cyclization of compound 11 proceeded uneventfully
under the previously established conditions, and product 1221
(88%) was readily oxidized to target 2 (94%) upon briefexposure to an equal mass of 10 wt % palladium on carbon inhot diphenyl ether. Once again, all the spectral data acquiredfor β-carboline (2) matched those reported22 previously. Thestructure of compound 2 was also confirmed by single-crystalX-ray analysis.The analogous synthesis of γ-carboline (3) is shown in
Scheme 3, and in this instance the required pyridine (13) was a
commercially available material. Reductive cyclization of thecross-coupling product 14 (89%) produced from compound 6and 13 proceeded as anticipated to give the tetrahydrocarboline15,23 albeit in just 66% yield. Similarly, the oxidation of this lastcompound under the previously employed conditions was lessefficient than observed in the two previous cases, withcompound 324 being obtained in 77% yield. Once again, fullcharacterization of this product was undertaken, including bysingle-crystal X-ray analysis.The establishment of a unified approach to all the carbolines
followed from the successful synthesis of δ-carboline (4) by thepathway shown in Scheme 4. So, as before, the palladium-catalyzed Ullmann cross-coupling of iodo enone 6 with therequired and commercially available pyridine 16 proceededuneventfully to give product 17 (75%) that was reductivelycyclized in the usual manner to afford the tetrahydrocarboline1825 (65%). Oxidation of this last compound using an equal
Scheme 1. Synthesis of α-Carboline (1)
Scheme 2. Synthesis of β-Carboline (2)
Scheme 3. Synthesis of γ-Carboline (3)
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4329
384

mass of 10 wt % palladium on carbon in hot diphenyl etherthen gave δ-carboline (4)26 (92%) that was subject to the usualrange of spectroscopic analyses, including a single-crystal X-raystudy.In order to test the capacities of the above-mentioned
protocols to deliver substituted carbolines, the β-carboline-based natural product harman (5, Figure 2) was targeted forsynthesis. The required pyridine was prepared by the routeshown in Scheme 5. Thus, commercially available 2-
methylpyridin-4-amine (19) was subjected to a Sandmeyerreaction using water as the nucleophile, thus providing thepreviously reported nitric acid salt27 20 of 2-methylpyridin-4-ol.Aromatic nitration of this last compound could only beachieved under rather forcing conditions, thus providing a 1:3and inseparable mixture of pyridines 21 and 22 (70% combinedyield). Accordingly, this mixture was treated with POBr3 inrefluxing toluene, thereby affording what is presumed to be thecorresponding mixture of bromides 23 and 24 (92% combinedyield). These regioisomers could only be separated by HPLCtechniques but sufficient quantities of the pure form of thelatter could be accumulated by such means. The formerproduct (presumed to be compound 23) was not purified orsubject to any spectroscopic characterization.With compound 24 in hand, the synthesis of harman (5) was
completed by the now standard pathway shown in Scheme 6.Thus, palladium-catalyzed Ullmann cross-coupling of iodo
enone 6 with pyridine 24 delivered the required product 25 in84% yield. Reductive cyclization of the last compound underthe usual conditions gave tetrahydroharman 2628 (83%), whichcould be oxidized to the natural product 514,29 (91%) ontreatment with an equal mass of 10 wt % palladium on carbonin hot diphenyl ether. Once again, all the spectral data,including those derived from a single-crystal X-ray analysis,acquired for compound 5 confirmed the assigned structure, andappropriate comparisons with those reported14 for the naturalproduct were entirely favorable.
■ CONCLUSIONSThe reaction sequences reported here should allow for therational/logical design of pathways to a wide range of α-, β-, γ-,and δ-carbolines. This is all the more so given the increasinglyready availability of a wide range of polysubstituted pyridines30
and 2-iodocyclohex-2-en-1-ones. For similar reasons, theprotocols defined here should allow for ready access to awide range of azaindoles, compounds of considerable interestfrom a medicinal chemistry perspective.31 Studies exploitingsuch possibilities will be reported in due course.
■ EXPERIMENTAL SECTIONGeneral Experimental Procedures. Unless otherwise specified,
proton (1H) and carbon (13C) NMR spectra were recorded at 18 °Cin base-filtered CDCl3 on a spectrometer operating at 400 MHz forproton and 100 MHz for carbon nuclei. 1H NMR data are recorded asfollows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz),relative integral], where multiplicity is defined as s = singlet; d =doublet; t = triplet; q = quartet; m = multiplet or combinations of theabove. In relevant cases, the signal due to residual CHCl3 appearing atδH 7.26 and the central resonance of the CDCl3 “triplet” appearing atδC 77.0 were used to reference 1H and 13C NMR spectra, respectively.Samples were analyzed by infrared spectroscopy (νmax) as thin filmson KBr plates. Low- and high-resolution electron impact (EI) massspectra were recorded on a double-focusing, triple-sector machine.Low- and high-resolution ESI mass spectra were recorded on a triple-quadrupole mass spectrometer operating in positive ion mode. Meltingpoints are uncorrected. Analytical thin-layer chromatography (TLC)was performed on aluminum-backed 0.2 mm thick silica gel 60 F254plates. Eluted plates were visualized using a 254 nm UV lamp and/orby treatment with a suitable dip followed by heating. These dipsincluded phosphomolybdic acid/ceric sulfate/sulfuric acid (concd)/water (37.5 g/7.5 g/37.5 g/720 mL), potassium permanganate/potassium carbonate/5% sodium hydroxide aqueous solution/water (3g/20 g/5 mL/300 mL), and p-anisaldehyde or vanillin/sulfuric acid
Scheme 4. Synthesis of δ-Carboline (4)
Scheme 5. Route to the Trisubstituted Pyridine 24 Requiredfor the Synthesis of the β-Carboline Alkaloid Harman (5)
Scheme 6. Completion of the Synthesis of Harman (5)
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4330
385

(concd)/ethanol (15 g/2.5 mL/250 mL). Flash chromatographicseparations were carried out following protocols defined by Still et al.32
with silica gel 60 (40−63 μm) as the stationary phase and using theAR- or HPLC-grade solvents indicated. The melting points of solidspurified by such means were recorded directly (i.e., after they hadcrystallized from the concentrated chromatographic fractions). Startingmaterials, reagents, drying agents, and other inorganic salts weregenerally commercially available and were used as supplied. Thecopper powder used in the palladium-catalyzed Ullmann cross-coupling reactions had a particle size of <75 μm. Tetrahydrofuran(THF), methanol, and dichloromethane were dried using a solventpurification system that is based upon a technology originallydescribed by Grubbs et al.33 Where necessary, reactions wereperformed under a nitrogen atmosphere by subjecting the relevantsolution to reduced pressure for several minutes and then admittingnitrogen. This process was repeated three times.Specific Chemical Transformations. 2-(2-Nitropyridin-3-yl)-
cyclohex-2-en-1-one (8). A magnetically stirred mixture of 2-iodocyclohex-2-en-1-one (6)16 (2.63 g, 11.82 mmol), copper powder(1.50 g, 23.65 mmol), CuI (1.69 g, 8.87 mmol), and Pd(dppf)Cl2·CH2Cl2 (483 mg, 0.59 mmol) in degassed DMSO (118 mL) washeated at 50 °C under a nitrogen atmosphere for 0.75 h. After thistime, a solution of commercially available 3-bromo-2-nitropyridine (7)(1.20 g, 5.91 mmol) in degassed DMSO (30 mL) was added to thereaction mixture over 1.5 h. After a further 4 h, the reaction mixturewas cooled, quenched with water (30 mL), and then diluted with ethylacetate (50 mL). The ensuing mixture was filtered through a padcomprised of a mixture of diatomaceous earth and silica gel. The solidsthus retained were rinsed with ethyl acetate (2 × 50 mL), and theseparated organic phase associated with the combined filtrates waswashed with water (2 × 100 mL) and then brine (2 × 100 mL) beforebeing dried (Na2SO4), filtered, and concentrated under reducedpressure to give a brown oil. Subjection of this material to flash columnchromatography (silica, 1:3:11 v/v/v acetone/dichloromethane/40−60 petroleum ether elution) gave, after concentration of theappropriate fractions (Rf = 0.2 in 1:1 v/v ethyl acetate/40−60petroleum ether), compound 8 (1.06 g, 82%) as a light-brown solid:mp = 115−116 °C; 1H NMR (400 MHz, CDCl3) δ 8.52 (dd, J = 4.7and 1.7 Hz, 1H), 7.74 (dd, J = 7.6 and 1.7 Hz, 1H), 7.60 (dd, J = 7.6and 4.7 Hz, 1H), 7.08 (t, J = 4.2 Hz, 1H), 2.60 (m, 4H), 2.15 (m, 2H);13C NMR (100 MHz, CDCl3) δ 196.0, 157.0, 148.5, 147.8, 141.7,137.2, 128.1, 126.7, 38.2, 26.5, 22.6; IR νmax 2950, 1679, 1540, 1405,1366, 975, 864, 810, 707 cm−1; MS (ESI, +ve) m/z 241 [(M + Na)+,100%]; HRMS m/z (M + Na)+ calcd for C11H10N2NaO3 241.0589,found 241.0591.6,7,8,9-Tetrahydro-5H-pyrido[2,3-b]indole (9). A magnetically
stirred mixture of compound 8 (30 mg, 0.14 mmol) and 10 wt %Pd/C (12 mg) in degassed methanol (7 mL) was maintained under anatmosphere of hydrogen for 16 h at 22 °C and then filtered, and thesolids thus retained were washed with methanol (20 mL). Thecombined filtrates were concentrated under reduced pressure and thewhite solid thus obtained was subjected to flash column chromatog-raphy (silica, 1:4 v/v ethyl acetate/40−60 petroleum ether elution).Concentration of the appropriate fractions (Rf = 0.7 in ethyl acetate)then gave compound 918 (18 mg, 75%) as a white, crystalline solid: mp= 155−156 °C (lit.18 mp = 155−156 °C); 1H NMR (400 MHz,CDCl3) δ 10.65 (m, 1H), 8.19 (d, J = 4.3 Hz, 1H), 7.75 (dd, J = 7.7and 1.3 Hz, 1H), 7.01 (dd, J = 7.7 and 4.3 Hz, 1H), 2.84 (t, J = 6.0 Hz,2H), 2.70 (m, 2H), 1.98−1.86 (complex m, 4H); 13C NMR (100MHz, CDCl3) δ 148.9, 140.8, 135.4, 125.7, 120.8, 115.1, 108.4,23.3(4), 23.2(8), 23.1, 20.8; IR νmax 3149, 3075, 2921, 2846, 1587,1418, 1289, 786, 765, 677 cm−1; MS (ESI, +ve) m/z 173 [(M + H)+,100%]; HRMS m/z (M + H)+ calcd for C11H13N2 173.1079, found173.1078.9H-Pyrido[2,3-b]indole (α-Carboline, 1). A magnetically stirred
mixture of compound 9 (20 mg, 0.12 mmol) and 10 wt % Pd/C (20mg) in diphenyl ether (15 mL) maintained under a nitrogenatmosphere was heated at 210 °C for 0.66 h. The reaction mixturewas then cooled to room temperature and filtered (through filterpaper), and the solids so retained were washed with ethyl acetate (2 ×
15 mL). The combined filtrates were concentrated under reducedpressure, and the residue thus obtained was subjected to flash columnchromatography (silica, 0:1 → 1:4 v/v ethyl acetate/40−60 petroleumether gradient elution). Concentration of the appropriate fractions (Rf= 0.2 in 1:1 v/v ethyl acetate/40−60 petroleum ether) gave compound14a (19 mg, 97%) as a white, crystalline solid: mp = 200−202 °C (lit.4a
mp = 215−217 °C); 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 7.7and 1.5 Hz, 1H), 8.34 (dd, J = 4.9 and 1.2 Hz, 1H), 8.09 (d, J = 7.9 Hz,1H), 7.52 (d, J = 8.1 Hz, 1H), 7.48−7.44 (complex m, 1H), 7.26−7.19(complex m, 2H) (signal due to N−H group proton not observed);13C NMR (100 MHz, CD3OD) δ 152.9, 146.1, 140.6, 129.9, 128.0,122.0, 121.9, 121.0, 118.0, 116.1, 112.3; IR νmax 3048, 2984, 2906,1600, 1587, 1572, 1455, 1411, 1274, 998, 767, 736 cm−1; MS (ESI,+ve) m/z 169 [(M + H)+, 100%]; HRMS m/z (M + H)+ calcd forC11H9N2 169.0766, found 169.0768.
4-Iodo-3-nitropyridine (10). A magnetically stirred solution ofcommercially available 4-chloro-3-nitropyridine (1.00 g, 6.31 mmol) inacetonitrile (126 mL) maintained at ambient temperatures was treatedwith sodium iodide (17.02 g, 113.55 mmol). The ensuing mixture washeated under reflux for 2 h and then cooled to 22 °C and diluted withethyl acetate (200 mL). The resulting solution was washed withNa2CO3 (1 × 100 mL of a saturated aqueous solution), Na2SO3 (1 ×50 mL of a saturated solution), water (1 × 200 mL), and brine (1 ×100 mL) before being dried (Na2SO4,), filtered, and then concentratedunder reduced pressure. The residue thus obtained was subjected toflash column chromatography (silica, 1:19 v/v ethyl acetate/40−60petroleum ether elution) to give, after concentration of the appropriatefractions (Rf = 0.5 twice in 1:5 v/v ethyl acetate/40−60 petroleumether), 4-iodo-3-nitropyridine (10)20 (1.46 g, 92%) as a light-yellowsolid: mp = 80−82 °C; 1H NMR (400 MHz, CDCl3) δ 9.04 (broad s,1H), 8.35 (d, J = 5.1 Hz, 1H), 8.03 (d, J = 5.1 Hz, 1H); 13C NMR(100 MHz, CDCl3) δ 152.5, 149.6, 145.9, 136.5, 98.5; IR νmax 1572,1540, 1521, 1357, 1223, 1058, 835, 657 cm−1; MS (EI, 70 eV) m/z250 (M+•, 100%), 204 (70), 177 (60); HRMS m/z M+• calcd forC5H3
127IN2O2 249.9239, found 249.9236.2-(3-Nitropyridin-4-yl)cyclohex-2-en-1-one (11). A magnetically
stirred mixture of 2-iodocyclohex-2-en-1-one (6) (888 mg, 4.00mmol), copper powder (508 mg, 8.00 mmol), CuI (571 mg, 3.00mmol), and Pd(dppf)Cl2·CH2Cl2 (163 mg, 0.20 mmol) in degassedDMSO (40 mL) was heated at 50 °C under a nitrogen atmosphere for0.75 h. After this time a solution of compound 10 (500 mg, 2.00mmol) in degassed DMSO (10 mL) was added to the reaction mixtureover 1 h. After a further 0.75 h, the reaction mixture was cooled,quenched with water (10 mL), and then diluted with ethyl acetate (15mL). The ensuing mixture was filtered through a pad comprised of amixture of diatomaceous earth and silica gel. The solids thus retainedwere rinsed with ethyl acetate (2 × 15 mL), and the separated organicphase associated with the combined filtrates was washed with water (2× 30 mL) and then brine (2 × 30 mL) before being dried (Na2SO4),filtered, and concentrated under reduced pressure to give a brown oil.This was subjected to flash column chromatography (silica, 1:3:10 v/v/v acetone/dichloromethane/40−60 petroleum ether elution) andgave, after concentration of the appropriate fractions [Rf = 0.1(5) in1:1 v/v ethyl acetate/40−60 petroleum ether], compound 11 (374mg, 86%) as a light-yellow solid: mp = 105−107 °C; 1H NMR (400MHz, CDCl3) δ 9.18 (s, 1H), 8.78 (d, J = 4.9 Hz, 1H), 7.22 (d, J = 4.9Hz, 1H), 7.13 (t, J = 4.2 Hz, 1H), 2.63−2.57 (complex m, 4H), 2.19−2.13 (complex m, 2H); 13C NMR (100 MHz, CDCl3) δ 195.5, 153.9,148.7, 145.3, 145.0, 140.0, 137.5, 125.6, 38.2, 26.5, 22.5; IR νmax 2948,1679, 1600, 1542, 1523, 1357, 1217, 1159, 1121, 851, 716 cm−1; MS(ESI, +ve) m/z 219 [(M + H)+, 100%]; HRMS m/z (M + H)+ calcdfor C11H11N2O3 219.0770, found 219.0768.
6,7,8,9-Tetrahydro-5H-pyrido[3,4-b]indole (12). A magneticallystirred mixture of compound 11 (190 mg, 0.87 mmol) and 10 wt %Pd/C (76 mg) in degassed methanol (44 mL) was maintained underan atmosphere of hydrogen for 16 h at 22 °C and then filtered, and thesolids thus retained were washed with methanol (100 mL). Thecombined filtrates were concentrated under reduced pressure, and thewhite solid thus obtained was subjected to flash column chromatog-raphy (silica, 1:4 v/v methanol/dichloromethane elution) to give, after
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4331
386

concentration of the appropriate fractions [Rf = 0.3(5) in 1:1 v/vmethanol/dichloromethane], compound 1221 (132 mg, 88%) as awhite, crystalline solid: mp = 163−164 °C (lit.21 mp = 199−200 °C);1H NMR (700 MHz, CD3OD) δ 8.49 (broad s, 1H), 7.97 (broad s,1H), 7.36 (d, J = 4.9 Hz, 1H), 2.77 (t, J = 6.0 Hz, 2H), 2.65 (t, J = 6.0Hz, 2H), 1.92−1.83 (complex m, 4H) (signal due to N−H groupproton not observed); 13C NMR (175 MHz, CD3OD) δ 142.2, 136.9,134.5, 134.2, 132.6, 113.7, 110.4, 24.2(2), 24.2(0), 24.0, 21.6; IR νmax3143, 3040, 2926, 2850, 2839, 1569, 1471, 1442, 1359, 1142, 1030,808 cm−1; MS (ESI, +ve) m/z 173 [(M + H)+, 100%]; HRMS m/z(M + H)+ calcd for C11H13N2 173.1079, found 173.1078.9H-Pyrido[3,4-b]indole (β-Carboline, 2). A magnetically stirred
mixture of compound 12 (20 mg, 0.12 mmol) and 10 wt % Pd/C (20mg) in diphenyl ether (15 mL) maintained under a nitrogenatmosphere was heated at 210 °C for 0.66 h. The reaction mixturewas then cooled to room temperature and filtered (through filterpaper), and the solids so retained were washed with ethyl acetate (2 ×15 mL). The combined filtrates were concentrated under reducedpressure, and the residue thus obtained was subjected to flash columnchromatography (silica, 0:1 → 1:1 v/v ethyl acetate/40−60 petroleumether gradient elution) to give, after concentration of the appropriatefractions (Rf = 0.5 in 1:2 v/v methanol/dichloromethane), compound222 (18 mg, 94%) as a white, crystalline solid: mp = 210−211 °C (lit.22
mp = 199−201 °C); 1H NMR (400 MHz, CD3OD) δ 8.79 (s, 1H),8.27 (d, J = 5.3 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 5.3 Hz,1H), 7.56 (m, 2H), 7.29−7.23 (complex m, 1H) (signal due to N−Hgroup proton not observed); 13C NMR (100 MHz, CD3OD) δ 142.7,138.4, 137.8, 134.1, 130.4, 129.7, 122.7, 122.2, 120.8, 116.1, 112.8; IRνmax 3134, 3052, 2963, 2754, 1628, 1449, 1331, 1245, 746, 732 cm−1;MS (ESI, +ve) m/z 191 [(M + Na)+, 70%], 169 [(M + H)+, 100];HRMS m/z (M + H)+ calcd for C11H9N2 169.0766, found 169.0766.2-(4-Nitropyridin-3-yl)cyclohex-2-en-1-one (14). A magnetically
stirred mixture of 2-iodocyclohex-2-en-1-one (6) (2.19 g, 9.85 mmol),copper powder (1.26 g, 19.70 mmol), CuI (1.41 g, 7.39 mmol), andPd(dppf)Cl2·CH2Cl2 (400 mg, 0.49 mmol) in degassed DMSO (98mL) was heated at 50 °C under a nitrogen atmosphere for 0.75 h.After this time, a solution of compound 13 (1.00 g, 4.93 mmol) indegassed DMSO (25 mL) was added to the reaction mixture over 1.5h. After a further 4 h, the reaction mixture was cooled, quenched withwater (20 mL), and then diluted with ethyl acetate (1 × 30 mL). Theensuing mixture was filtered through a pad comprised of a mixture ofdiatomaceous earth and silica gel. The solids thus retained were rinsedwith ethyl acetate (2 × 30 mL), and the separated organic phaseassociated with the combined filtrates was washed with water (2 × 60mL) and then brine (2 × 60 mL) before being dried (Na2SO4),filtered, and concentrated under reduced pressure to give a brown oil.Subjection of this material to flash column chromatography (silica,1:3:11 v/v/v acetone/dichloromethane/40−60 petroleum etherelution) gave, after concentration of the appropriate fractions (Rf =0.2 in 1:3:6 v/v/v acetone/dichloromethane/40−60 petroleum ether),compound 14 (956 mg, 89%) as a light-brown solid: mp = 94−95 °C;1H NMR (400 MHz, CDCl3) δ 8.82 (d, J = 5.3 Hz, 1H), 8.61 (s, 1H),7.81 (d, J = 5.3 Hz, 1H), 7.17 (t, J = 4.2 Hz, 1H), 2.66−2.58 (complexm, 4H), 2.18 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 196.1, 154.1,152.8, 151.4, 148.6, 136.0, 125.4, 116.6, 38.2, 26.5, 22.6; IR νmax 2945,1677, 1557, 1529, 1401, 1358, 1222, 1159, 839, 705, 675 cm−1; MS(ESI, +ve) m/z 241 [(M + Na)+, 100%], 219 [(M + H)+, 15]; HRMSm/z (M + Na)+ calcd for C11H10N2NaO3 241.0589, found 241.0589.6,7,8,9-Tetrahydro-5H-pyrido[4,3-b]indole (15). A magnetically
stirred mixture of compound 14 (50 mg, 0.23 mmol) and 10 wt %Pd/C (20 mg) in degassed methanol (12 mL) was maintained underan atmosphere of hydrogen at 22 °C for 16 h and then filtered, and thesolids so retained were washed with methanol (25 mL). The combinedfiltrates were concentrated under reduced pressure, and the white solidthus obtained was subjected to flash column chromatography (silica,1:4 v/v methanol/dichloromethane elution) to give, after concen-tration of the appropriate fractions [Rf = 0.3(5) in 1:1 v/v methanol/dichloromethane], compound 1523 (26 mg, 66%) as a white,crystalline solid: mp = 223−224 °C (lit.23 mp = 269−271 °C); 1HNMR (700 MHz, CD3OD) δ 8.58 (broad s, 1H), 8.05 (broad s, 1H),
7.28 (d, J = 5.3 Hz, 1H), 2.74 (m, 4H), 1.95−1.87 (complex m, 4H)(signal due to N−H group proton not observed); 13C NMR (175MHz, CD3OD) δ 141.6, 139.9, 139.1, 138.1, 126.2, 110.3, 107.5, 24.2,24.1, 23.9, 21.6; IR νmax 2926, 2839, 2693, 1625, 1466, 1294, 1169,1142, 989, 802, 684 cm−1; MS (ESI, +ve) m/z 173 [(M + H)+, 100%];HRMS m/z (M + H)+ calcd for C11H13N2 173.1079, found 173.1078.
5H-Pyrido[4,3-b]indole (γ-Carboline, 3). A magnetically stirredmixture of compound 15 (20 mg, 0.12 mmol) and 10 wt % Pd/C (20mg) in diphenyl ether (15 mL) maintained under a nitrogenatmosphere was heated at 210 °C for 0.66 h. The reaction mixturewas then cooled to room temperature and filtered (through filterpaper), and the solids thus retained were washed with ethyl acetate (2× 15 mL). The combined filtrates were concentrated under reducedpressure, and the residue thus obtained was subjected to flash columnchromatography (silica, 40−60 petroleum ether elution → 1:3 v/vmethanol/dichloromethane gradient elution) to give, after concen-tration of the appropriate fractions [Rf = 0.2(5) in 1:3 v/v methanol/dichloromethane], compound 324 (15 mg, 77%) as a white, crystallinesolid: mp = 223−225 °C (lit.24 mp = 225−227 °C); 1H NMR [700MHz, (CD3)2SO] δ 11.69 (s, 1H), 9.33 (s, 1H), 8.43 (s, 1H), 8.22 (d,J = 7.8 Hz, 1H), 7.56 (m, 1H), 7.47 (m, 2H), 7.26 (t, J = 7.4 Hz, 1H);13C NMR [175 MHz, (CD3)2SO] δ 144.5, 143.5, 142.7, 139.5, 126.5,120.7, 120.6, 119.9, 119.4, 111.4, 106.4; 1H NMR (700 MHz,CD3OD) δ 9.20 (s, 1H), 8.36 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.53(d, J = 8.1 Hz, 1H), 7.49−7.46 (m, 2H), 7.28 (t, J = 7.4 Hz, 1H)(signal due to N−H group proton not observed); 13C NMR (175MHz, CD3OD) δ 145.8, 144.4, 142.7, 141.5, 128.2, 122.4, 121.6(4),121.6(0), 121.5, 112.4, 107.6; IR νmax 3062, 2956, 2806, 2679, 1607,1582, 1467, 1239, 999, 744 cm−1; MS (ESI, +ve) m/z 169 [(M + H)+,100%]; HRMS m/z (M + H)+ calcd for C11H9N2 169.0766, found169.0766.
2-(3-Nitropyridin-2-yl)cyclohex-2-en-1-one (17). A magneticallystirred mixture of 2-iodocyclohex-2-en-1-one (6) (1.09 g, 4.93 mmol),copper powder (630 mg, 9.85 mmol), CuI (704 mg, 3.69 mmol), andPd(dppf)Cl2·CH2Cl2 (200 mg, 0.25 mmol) in degassed DMSO (50mL) was heated at 50 °C under a nitrogen atmosphere for 0.75 h.After this time, a solution of compound 16 (500 mg, 4.93 mmol) indegassed DMSO (12 mL) was added to the reaction mixture over 1 h.After a further 2 h, the reaction mixture was cooled, quenched withwater (10 mL), diluted with ethyl acetate (15 mL), and then filteredthrough a pad comprised of a mixture of diatomaceous earth and silicagel. The solids thus retained were rinsed with ethyl acetate (2 × 15mL), and the separated organic phase associated with the combinedfiltrates was washed with water (2 × 30 mL) and then brine (2 × 30mL) before being dried (Na2SO4), filtered, and concentrated underreduced pressure to give a brown oil. Subjection of this material toflash column chromatography (silica, 1:3:12 v/v/v acetone/dichloro-methane/40−60 petroleum ether elution) gave, after concentration ofthe appropriate fractions [Rf = 0.1(5) in 1:3:6 v/v/v acetone/dichloromethane/40−60 petroleum ether], compound 17 (403 mg,75%) as a light-brown solid: mp = 104−106 °C; 1H NMR (400 MHz,CDCl3) δ 8.79 (dd, J = 4.7 and 1.3 Hz, 1H), 8.29 (dd, J = 8.2 and 1.3Hz, 1H), 7.49−7.43 (complex m, 2H), 2.65 (m, 2H), 2.58 (m, 2H),2.20−2.13 (complex m, 2H); 13C NMR (100 MHz, CDCl3) δ 196.2,152.9, 151.0, 149.4, 146.1, 139.2, 132.2, 123.4, 38.3, 26.6, 22.5; IR νmax2947, 1677, 1593, 1561, 1526, 1451, 1357, 765 cm−1; MS (ESI, +ve)m/z 241 [(M + Na)+, 100%], 219 [(M + H)+, 15]; HRMS m/z (M +Na)+ calcd for C11H10N2NaO3 241.0589, found 241.0589.
6,7,8,9-Tetrahydro-5H-pyrido[3,2-b]indole (18). A magneticallystirred mixture of compound 17 (213 mg, 0.98 mmol) and 10 wt %Pd/C (86 mg) in degassed methanol (49 mL) was maintained underan atmosphere of hydrogen for 16 h at 22 °C and then filtered, and thesolids so retained were washed with methanol (100 mL). Thecombined filtrates were concentrated under reduced pressure, and thewhite solid thus obtained was subjected to flash column chromatog-raphy (silica, 1:1 v/v ethyl acetate/40−60 petroleum ether elution) togive, after concentration of the appropriate fractions (Rf = 0.2 in 1:1 v/v ethyl acetate/40−60 petroleum ether), compound 1825 (109 mg,65%) as a white, crystalline solid: mp = 183−185 °C (lit.25 mp = 200−202 °C); 1H NMR (400 MHz, CDCl3) δ 8.86 (broad s, 1H), 8.38 (d, J
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4332
387

= 4.3 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.00 (m, 1H), 2.84 (m, 2H),2.76 (m, 2H), 1.95−1.84 (complex m, 4H); 13C NMR (100 MHz,CDCl3) δ 145.6, 141.9, 139.2, 129.1, 117.4, 115.8, 110.8, 23.8, 23.2,23.1, 20.1; IR νmax 3136, 3083, 3053, 2929, 2847, 1483, 1414, 1362,1286, 1144, 904, 767, 729 cm−1; MS (ESI, +ve) m/z 173 [(M + H)+,100%]; HRMS m/z (M + H)+ calcd for C11H13N2 173.1079, found173.1080.5H-Pyrido[3,2-b]indole (δ-Carboline, 4). A magnetically stirred
mixture of compound 18 (20 mg, 0.12 mmol) and 10 wt % Pd/C (20mg) in diphenyl ether (15 mL) maintained under a nitrogenatmosphere was heated at 210 °C for 0.66 h. The reaction mixturewas then cooled to room temperature and filtered (through filterpaper), and the solids thus retained were washed with ethyl acetate (2× 15 mL). The combined filtrates were concentrated under reducedpressure, and the residue thus obtained was subjected to flash columnchromatography (silica, 0:1 → 1:4 v/v ethyl acetate/40−60 petroleumether gradient elution) to give, after concentration of the appropriatefractions [Rf = 0.2(5) in 1:1 v/v ethyl acetate/40−60 petroleumether], compound 426 (18 mg, 92%) as a white, crystalline solid: mp =211−212 °C (lit.26 mp = 206−207 °C); 1H NMR (400 MHz,CD3OD) δ 8.39 (dd, J = 4.8 and 1.4 Hz, 1H), 8.28 (d, J = 7.9 Hz, 1H),7.88 (d, J = 8.2 Hz, 1H), 7.52 (m, 2H), 7.40 (m, 1H), 7.26 (m, 1H)(signal due to N−H group proton not observed); 13C NMR (100MHz, CD3OD) δ 142.4, 142.3, 141.5, 135.1, 129.0, 122.4, 121.4, 121.3,120.8, 119.9, 112.6; IR νmax 3057, 2979, 2919, 2848, 2760, 1629, 1460,1396, 1320, 1223, 741, 724 cm−1; MS (ESI, +ve) m/z 169 [(M + H)+,100%]; HRMS m/z (M + H)+ calcd for C11H9N2 169.0766, found169.0766.2-Methylpyridin-4-ol Nitrate (20). Commercially available 2-
methylpyridin-4-amine (8.00 g, 73.98 mmol) was dissolved in amixture of concentrated HNO3 (44.1 mL) and H2O (59.4 mL), andthe resulting solution was cooled, with vigorous magnetic stirring, to 0°C. A chilled solution of NaNO2 (7.40 g, 107.25 mmol) in water (21.7mL) was then added dropwise over 0.5 h, and the mixture thus formedstirred at 0 °C for 4 h before being allowed to warm to 22 °C and thenstirred at this temperature for another 10 h and was recooled to 0 °C.The resulting solid was removed by filtration, the filtrate wasconcentrated to about one-third of its original volume and thencooled to 0 °C, and a second crop of solid was removed by filtration.The combined solids were then air-dried to give the nitric acid salt27 ofcompound 20 (10.49 g, 82%) as a white, crystalline solid: mp = 162−164 °C (lit.27 mp = 164−165 °C); 1H NMR (400 MHz, CD3OD) δ8.34 (d, J = 6.5 Hz, 1H), 7.11−7.09 (complex m, 2H), 2.62 (s, 3H)(signals due to O−H and N−H group protons not observed); 13CNMR (100 MHz, CD3OD) δ 173.4, 156.0, 143.2, 114.7, 113.0, 19.3;IR νmax 3098, 2919, 2617, 1627, 1501, 1305, 1216, 830 cm
−1; MS (ESI,+ve) m/z 110 [(M + H)+, 100%]; HRMS m/z (M + H)+ calcd forC6H8NO 110.0606, found 110.0608.2-Methyl-4-nitropyridin-3-ol (21) and 2-Methyl-3-nitropyridin-4-
ol (22). A magnetically stirred solution of salt 20 (3.00 g, 17.43 mmol)in concentrated H2SO4 (6.9 mL) was cooled to 0 °C and then treated,dropwise, with fuming HNO3 (6.9 mL). The ensuing mixture wasstirred at 0 °C for 0.17 h, heated at 130 °C for 20 h, and then cooledto 0 °C and carefully neutralized with NaOH (25 mL of a 10 Maqueous solution). FC-grade silica gel (20 g) was added to the ensuingmixture, and this was then concentrated under reduced pressure. Thefree-flowing solid thus obtained was subjected to flash columnchromatography (silica, 1:19 v/v methanol/dichloromethane elution)to give, after concentration of the appropriate fractions (Rf = 0.6 in 1:1v/v methanol/dichloromethane), a 1:3 mixture of 2-methyl-5-nitropyridin-4-ol (21) and 2-methyl-3-nitropyridin-4-ol (22) (1.88 g,70% combined yield) as a white crystalline solid: mp = 148−150 °C;1H NMR [400 MHz, (CD3)2SO] δ (major product) 12.17 (broad s,1H), 7.72 (d, J = 7.5 Hz, 1H), 6.33 (d, J = 7.5 Hz, 1H), 2.29 (s, 3H);13C NMR [100 MHz, (CD3)2SO] δ (for mixture) 168.2, 151.2, 148.1,143.5, 142.2, 139.4, 137.8, 137.3, 120.7, 118.0, 18.2, 15.3; IR νmax 3037,2796, 1612, 1557, 1502, 1356, 1222, 836, 768 cm−1; MS (ESI, +ve) m/z 331 (42%), 177 [(M + Na)+, 100], 155 [(M + H)+, 10]; HRMS(ESI) m/z (M + Na)+ calcd for C6H6N2NaO3 177.0276, found177.0278.
4-Bromo-2-methyl-3-nitropyridine (24). A magnetically stirred 1:3mixture of 2-methyl-5-nitropyridin-4-ol (21) and 2-methyl-3-nitro-pyridin-4-ol (22) (1.12 g, 7.27 mmol), obtained as describedimmediately above, in toluene (8.0 mL) and maintained under anitrogen atmosphere was treated with POBr3 (2.19 g, 7.63 mmol). Theensuing mixture was heated under reflux for 10 h and then cooled to 0°C before being quenched with NaOH (30 mL of a 1 M aqueoussolution). The ensuing mixture was extracted with ethyl acetate (1 ×150 mL) and washed with H2O (2 × 150 mL). The separated organicphase was then dried (Na2SO4), filtered, and concentrated underreduced pressure. The solid so obtained was subjected to flash columnchromatography (silica, 1:3:20 v/v/v acetone/dichloromethane/40−60 petroleum ether elution) to give, after concentration of theappropriate fractions (Rf = 0.4 in 1:3:6 v/v/v acetone/dichloro-methane/40−60 petroleum ether), a 1:3 mixture of what is presumedto be 4-bromo-2-methyl-5-nitropyridine (23) and 4-bromo-2-methyl-3-nitropyridine (24) (1.46 g, 92% combined) as a white solid. A ca.800 mg portion of this material was subjected to semipreparativeHPLC (silica, normal phase, 17:83 v/v ethyl acetate/n-hexane elution,600 mL/h) to afford, after concentration of the relevant fractions (Rt =0.45 h), a pure sample of 4-bromo-2-methyl-3-nitropyridine (24) (ca.350 mg) as a white, crystalline solid: mp = 78−79 °C; 1H NMR (400MHz, CDCl3) δ 8.40 (d, J = 5.3 Hz, 1H), 7.50 (d, J = 5.3 Hz, 1H),2.60 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 151.8, 150.5, 148.5,126.3, 123.7, 20.9; IR νmax 3072, 1575, 1534, 1365, 1266, 846, 837, 714cm−1; MS (EI, 70 eV) m/z 218 and 216 (M+•, both 90%), 201 and199 (both 70), 172 and 170 (both 95), 93 and 91 (both 83), 62(100%); HRMS m/z M+• calcd for C6H5
79BrN2O2 215.9534, found.215.9533.
2-(2-Methyl-3-nitropyridin-4-yl)cyclohex-2-en-1-one (25). A mag-netically stirred mixture of 2-iodocyclohex-2-en-1-one (6) (205 mg,0.92 mmol), copper powder (117 mg, 1.84 mmol), CuI (132 mg, 0.69mmol), and Pd(dppf)Cl2·CH2Cl2 (38 mg, 0.05 mmol) in degassedDMSO (9 mL) was heated at 50 °C under a nitrogen atmosphere for0.75 h. After this time, a solution of compound 24 (100 mg, 0.46mmol) in degassed DMSO (3 mL) was added to the reaction mixtureover 0.66 h. After a further 6 h the reaction mixture was cooled,quenched with water (3 mL), and then diluted with ethyl acetate (5mL). The ensuing mixture was filtered through a pad comprised of amixture of diatomaceous earth and silica gel, and the solids so retainedwere rinsed with ethyl acetate (2 × 5 mL). The separated organicphase associated with the combined filtrates was washed with water (2× 10 mL) and then brine (2 × 10 mL) before being dried (Na2SO4),filtered, and concentrated under reduced pressure to give a brown oil.Subjection of this material to flash column chromatography (silica,3:9:40 v/v/v acetone/dichloromethane/40−60 petroleum etherelution) gave, after concentration of the appropriate fractions [Rf =0.1(5) in 1:3:6 v/v/v acetone/dichloromethane/40−60 petroleumether], compound 25 (90 mg, 84%) as a light-brown, crystalline solid:mp = 80−81 °C; 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 5.0 Hz,1H), 7.09−7.05 (complex m, 2H), 2.66 (s, 3H), 2.58−2.54 (complexm, 4H), 2.12 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 195.6, 151.4,150.8, 150.1, 146.6, 139.0, 136.3, 123.4, 38.2, 26.5, 22.5, 22.0; IR νmax2939, 1681, 1594, 1528, 1359, 1229, 860 cm−1; MS (ESI, +ve) m/z255 [(M + Na)+, 100%], 233 [(M + H)+, 10]; HRMS m/z (M + Na)+
calcd for C12H12N2NaO3 255.0746, found 255.0734.1-Methyl-6,7,8,9-tetrahydro-5H-pyrido[3,4-b]indole (26). A mag-
netically stirred mixture of compound 25 (103 mg, 0.44 mmol) and 10wt % Pd/C (42 mg) in degassed methanol (22 mL) was maintainedunder an atmosphere of hydrogen at 22 °C for 16 h and then filtered,and the solids thus retained were washed with methanol (50 mL). Thecombined filtrates were concentrated under reduced pressure, and thewhite solid thus obtained was subjected to flash column chromatog-raphy (silica, 1:4 v/v methanol/dichloromethane elution) to give, afterconcentration of the appropriate fractions (Rf = 0.2 in 1:1 v/vmethanol/dichloromethane), compound 2628 (69 mg, 83%) as awhite, crystalline solid: mp = 187−189 °C (lit.28 mp = 184−188 °C);1H NMR (400 MHz, CD3OD) δ 7.84 (d, J = 5.4 Hz, 1H), 7.19 (d, J =5.4 Hz, 1H), 2.78 (broad s, 2H), 2.63 (broad s, 5H), 1.91−1.84(complex m, 4H) (signal due to N−H group proton not observed);
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4333
388

13C NMR (100 MHz, CD3OD) δ 141.6, 141.2, 136.7, 133.7, 132.9,111.9, 110.6, 24.3, 24.2, 24.1, 21.7, 19.1; IR νmax 3045, 2930, 2846,1563, 1498, 1308, 1226, 809 cm−1; MS (ESI, +ve) m/z 187 [(M +H)+, 100%]; HRMS m/z (M + H)+ calcd for C12H15N2 187.1235,found 187.1232.1-Methyl-9H-pyrido[3,4-b]indole (Harman, 5). A magnetically
stirred mixture of compound 26 (36 mg, 0.19 mmol) and 10 wt %Pd/C (36 mg) in diphenyl ether (15 mL) maintained under a nitrogenatmosphere was heated at 210 °C for 0.66 h. The reaction mixture wasthen cooled to room temperature and filtered (through filter paper),and the solids so retained were washed with ethyl acetate (2 × 7 mL).The combined filtrates were concentrated under reduced pressure, andthe residue thus obtained was subjected to flash column chromatog-raphy (silica, 40−60 petroleum ether elution →1:9 v/v methanol/dichloromethane elution) to give, after concentration of theappropriate fractions (Rf = 0.5 in 1:3 v/v methanol/dichloromethane),compound 529 (32 mg, 91%) as a white, crystalline solid: mp = 233−235 °C (lit.29 mp = 235−238 °C); 1H NMR (400 MHz, CDCl3) δ8.91 (broad s, 1H), 8.38 (d, J = 5.4 Hz, 1H), 8.12 (d, J = 7.9 Hz, 1H),7.84 (d, J = 5.4 Hz, 1H), 7.56−7.51 (complex m, 2H), 7.29 (m, 1H),2.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.9, 140.3, 138.7,134.8, 128.5, 128.4, 122.2, 122.0, 120.2, 113.1, 111.7, 20.5; IR νmax
3135, 3068, 2925, 1626, 1568, 1504, 1450, 1323, 1251, 1237, 742cm−1; MS (ESI, +ve) m/z 183 [(M + H)+, 100%]; HRMS (ESI) m/z(M + H)+ calcd for C12H11N2 183.0922, found 183.0922.X-ray Crystallographic Studies. Crystallographic Data. Com-
pound 1. C11H8N2, M = 168.20, T = 150 K, monoclinic, space groupP21/n, Z = 4, a = 11.1814(2) Å, b = 5.53776(8) Å, c = 13.4546(2) Å, β= 96.2939(15)°, V = 828.09(2) Å3, Dx = 1.349 g cm−3, 1673 uniquereflections (2θmax = 147.6°), R = 0.035 [for 1520 reflections with I >2.0σ(I)], Rw = 0.089 (all data), S = 1.00.Compound 2. C11H8N2, M = 168.20, T = 150 K, orthorhombic,
space group P212121, Z = 4, a = 5.8440(1) Å, b = 9.8140(1) Å, c =14.4195(1) Å, V = 827.00(2) Å3, Dx = 1.351 g cm−3, 1004 uniquereflections (2θmax = 147.8°), R = 0.027 [for 998 reflections with I >2.0σ(I)], Rw = 0.073 (all data), S = 1.00.Compound 3. C11H8N2, M = 168.20, T = 150 K, orthorhombic,
space group Pna21, Z = 4, a = 17.1576(6) Å, b = 12.2955(6) Å, c =3.8213(2) Å, V = 806.15(6) Å3, Dx = 1.386 g cm−3, 944 uniquereflections (2θmax = 147.8°), R = 0.032 [for 906 reflections with I >2.0σ(I)], Rw = 0.062 (all data), S = 1.00.Compound 4. C11H8N2, M = 168.20, T = 150 K, monoclinic, space
group Ia, Z = 4, a = 12.1155(7) Å, b = 3.9036(2) Å, c = 18.1199(13)Å, β = 107.250(7)°, V = 818.42(9) Å3, Dx = 1.365 g cm−3, 820 uniquereflections (2θmax = 146.6°), R = 0.042 [for 807 reflections with I >2.0σ(I)], Rw = 0.107 (all data), S = 1.05.Compound 5. C12H10N2, M = 182.23, T = 150 K, orthorhombic,
space group P212121, Z = 8, a = 9.5865(2) Å, b = 13.2423(4) Å, c =15.1680(4) Å, V = 1925.54(9) Å3, Dx = 1.257 g cm−3, 1994 uniquereflections (2θmax = 52.0°), R = 0.032 [for 1805 reflections with I >2.0σ(I)], Rw = 0.073 (all data), S = 1.00.Structure Determination. Images for compound 5 were collected
on a diffractometer (Mo Kα, graphite monochromator, λ = 0.71073 Å)fitted with an area detector, and the data were extracted using CrysAlisPRO.34 Images for compounds 1, 2, 3, and 4 were measured on adiffractometer (Cu Kα, mirror monochromator, λ = 1.54184 Å) fittedwith an area detector, and the data were extracted using the CrysAlisPRO.34 The structure solutions for all 5 compounds were solved bydirect methods (SIR92)35 and then refined using the CRYSTALSprogram package.36 Atomic coordinates, bond lengths and angles, anddisplacement parameters have been deposited at the CambridgeCrystallographic Data Centre (CCDC nos. 1530002, 1530003,1530004, 1530005, 1530006). These data can be obtained free-of-charge via www.ccdc.cam.ac.uk/data_request/cif, by e-mailing [email protected], or by contacting The Cambridge Crystallo-graphic Data Centre (12 Union Road, Cambridge CB2 1EZ, UK; fax:+ 44 1223 336033).
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.joc.7b00323.
Crystallographic data for 1 in CIF format (CIF)Crystallographic data for 2 in CIF format (CIF)Crystallographic data for 3 in CIF format (CIF)Crystallographic data for 4 in CIF format (CIF)Crystallographic data for 5 in CIF format (CIF)ORTEPs derived from the single-crystal X-ray analyses ofcompounds 1−5 and 1H and 13C NMR spectra ofcompounds 1−5, 8−12, 14, 15, 17, 18, 20, 21/22, and24−26 (PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] G. Banwell: 0000-0002-0582-475XNotesThe authors declare no competing financial interest.Atomic coordinates, bond lengths and angles, and displacementparameters have been deposited at the Cambridge Crystallo-graphic Data Centre (CCDC nos. 1530002, 1530003, 1530004,1530005, 1530006). These data can be obtained free-of-chargevia www.ccdc.cam.ac.uk/data_request/cif, by emailing [email protected], or by contacting The CambridgeCrystallographic Data Centre at 12 Union Road, CambridgeCB2 1EZ, UK (fax: + 44 1223 336033).
■ ACKNOWLEDGMENTSWe thank the Australian Research Council and the Institute ofAdvanced Studies for financial support. Q.Y. is the gratefulrecipient of a PhD Scholarship provided by the ChinaScholarship Council of the People’s Republic of China.
■ REFERENCES(1) For useful points-of-entry into the substantial body of literatureon these compounds, see the following: (a) Smirnova, O. B.; Golovko,T. V.; Granik, V. G. Pharm. Chem. J. 2011, 44, 654. (b) Hung, T. Q.;Dang, T. T.; Janke, J.; Villinger, A.; Langer, P. Org. Biomol. Chem.2015, 13, 1375 and references cited therein.(2) See, for example, the following: Lee, C. W.; Lee, J. Y. Adv. Mater.2013, 25, 5450.(3) Basavaiah, D.; Reddy, D. M. Org. Biomol. Chem. 2012, 10, 8774and references cited therein.(4) (a) Laha, J. K.; Petrou, P.; Cuny, G. D. J. Org. Chem. 2009, 74,3152. (b) Ghahremanzadeh, R.; Ahadi, S.; Bazgir, A. Tetrahedron Lett.2009, 50, 7379. (c) Mineno, M.; Sera, M.; Ueda, T.; Mizuno, M.;Yamano, M.; Mizufune, H.; Zanka, A. Tetrahedron 2014, 70, 5550.(d) Mineno, M.; Sera, M.; Ueda, T.; Mizufune, H.; Zanka, A.; O’Bryan,C.; Brown, J.; Scorah, N. J. Org. Chem. 2015, 80, 1564 and referencescited therein.(5) (a) Zhu, Y.-P.; Liu, M.-C.; Cai, Q.; Jia, F.-C.; Wu, A.-X. Chem. -Eur. J. 2013, 19, 10132. (b) Dhiman, S.; Mishra, U. K.; Ramasastry, S.S. V. Angew. Chem. Int. Ed. 2016, 55, 7737. (c) Dighe, S. U.; Yadav, V.D.; Mahar, R.; Shukla, S. K.; Batra, S. Org. Lett. 2016, 18, 6010 andreferences cited therein.(6) See, for example, the following: (a) Kamlah, A.; Lirk, F.; Bracher,F. Tetrahedron 2016, 72, 837. (b) Du, H.; Gu, H.; Li, N.; Wang, J.MedChemComm 2016, 7, 636.(7) See, for example, the following: (a) Otto, R.; Penzis, R.; Gaube,F.; Winckler, T.; Appenroth, D.; Fleck, C.; Trankle, C.; Lehmann, J.;Enzensperger, C. Eur. J. Med. Chem. 2014, 87, 63. (b) Ran, X.; Zhao,
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4334
389

Y.; Liu, L.; Bai, L.; Yang, C.-Y.; Zhou, B.; Meagher, J. L.;Chinnaswamy, K.; Stuckey, J. A.; Wang, S. J. Med. Chem. 2015, 58,4927.(8) Cao, J.; Xu, Y.; Kong, Y.; Cui, Y.; Hu, Z.; Wang, G.; Deng, Y.; Lai,G. Org. Lett. 2012, 14, 38.(9) For some useful reviews see (a) Wadsworth, A. D.; Naysmith, B.J.; Brimble, M. A. Eur. J. Med. Chem. 2015, 97, 816. (b) Alekseyev, R.S.; Kurkin, A. V.; Yurovskaya, M. A. Chem. Heterocycl. Compd. 2009,45, 889. (c) Alekseyev, R. S.; Kurkin, A. V.; Yurovskaya, M. A. Chem.Heterocycl. Compd. 2010, 46, 777.(10) See also (a) Ishiyama, H.; Ohshita, K.; Abe, T.; Nakata, H.;Kobayashi, J. Bioorg. Med. Chem. 2008, 16, 3825. (b) Butin, A. V.;Pilipenko, A. S.; Milich, A. A.; Finko, A. V. Chem. Heterocycl. Compd.2009, 45, 613. (c) Dagar, A.; Biswas, S.; Samanta, S. RSC Adv. 2015, 5,52497. (d) Li, J.; Tang, Y.; Jin, H.-J.; Cui, Y.-D.; Zhang, L.-J.; Jiang, T.J. Asian Nat. Prod. Res. 2015, 17, 299. (e) Wang, G.; You, X.; Gan, Y.;Liu, Y. Org. Lett. 2017, 19, 110.(11) Peng, H.; Chen, X.; Chen, Y.; He, Q.; Xie, Y.; Yang, C.Tetrahedron 2011, 67, 5725.(12) (a) Zhang, H.; Larock, R. C. J. Org. Chem. 2002, 67, 9318.(b) Hostyn, S.; Van Baelen, G.; Lemiere, G. L. F.; Maes, B. U. W. Adv.Synth. Catal. 2008, 350, 2653. (c) Gupta, S.; Kumar, B.; Kundu, B. J.Org. Chem. 2011, 76, 10154. (d) Dassonneville, B.; Witulski, B.;Detert, H. Eur. J. Org. Chem. 2011, 2011, 2836. (e) Ma, Z.; Ni, F.;Woo, G. H. C.; Lo, S.-M.; Roveto, P. M.; Schaus, S. E.; Snyder, J. K.Beilstein J. Org. Chem. 2012, 8, 829. (f) Markey, S. J.; Lewis, W.;Moody, C. J. Org. Lett. 2013, 15, 6306. (g) Mineno, M.; Sera, M.;Ueda, T.; Mizuno, M.; Yamano, M.; Mizufune, H.; Zanka, A.Tetrahedron 2014, 70, 5550. (h) Pilipenko, A. S.; Uchuskin, M. G.;Trushkov, I. V.; Butin, A. V. Tetrahedron 2015, 71, 8786. (i) Dondas,H. A.; Hempshall, A.; Narramore, S.; Kilner, C.; Fishwick, C. W. G.;Grigg, R. Tetrahedron 2016, 72, 1316. (j) Hingane, D. G.; Parekh, N.P.; Khan, A.; Kusurkar, R. S. Synth. Commun. 2016, 46, 160. (k) He, L.;Allwein, S. P.; Dugan, B. J.; Knouse, K. W.; Ott, G. R.; Zificsak, C. A.Org. Synth. 2016, 93, 272.(13) (a) Clark, V. M.; Cox, A.; Herbert, E. J. J. Chem. Soc. C 1968,831. (b) Rocca, P.; Marsais, F.; Godard, A.; Queguiner, G. Tetrahedron1993, 49, 49. (c) Rocca, P.; Cochennec, C.; Marsais, F.; Thomas-dit-Dumont, L.; Mallet, M.; Godard, A.; Queguiner, G. J. Org. Chem. 1993,58, 7832. (d) Rocca, P.; Marsais, F.; Godard, A.; Queguiner, G.Tetrahedron 1993, 49, 3325. (e) Rocca, P.; Marsais, F.; Godard, A.;Queguiner, G. Tetrahedron Lett. 1993, 34, 7917. (f) Rocca, P.; Marsais,F.; Godard, A.; Queguiner, G.; Adams, L.; Alo, B. J. Heterocycl. Chem.1995, 32, 1171. (g) Iwaki, T.; Yasuhara, A.; Sakamoto, T. J. Chem. Soc.,Perkin Trans. 1 1999, 1505. (h) Laha, J. K.; Barolo, S. M.; Rossi, R. A.;Cuny, G. D. J. Org. Chem. 2011, 76, 6421. (i) Pumphrey, A. L.; Dong,H.; Driver, T. G. Angew. Chem. Int. Ed. 2012, 51, 5920. (j) Dhara, S.;Singha, R.; Ahmed, A.; Mandal, H.; Ghosh, M.; Nuree, Y.; Ray, J. K.RSC Adv. 2014, 4, 45163.(14) (a) Ishida, J.; Wang, H.-K.; Oyama, M.; Cosentino, M. L.; Hu,C.-Q.; Lee, K.-L. J. Nat. Prod. 2001, 64, 958. (b) Aassila, H.; Bourguet-Kondracki, M. L.; Rifai, S.; Fassouane, A.; Guyot, M. Mar. Biotechnol.2003, 5, 163. (c) Kusurkar, R. S.; Goswami, S. K.; Vyas, S. M.Tetrahedron Lett. 2003, 44, 4761.(15) Banwell, M. G.; Jones, M. T.; Reekie, T. A. Chem. New Zealand2011, 75, 122.(16) Pandey, G.; Balakrishnan, M. J. Org. Chem. 2008, 73, 8128.(17) Yan, Q.; Gin, E.; Wasinska-Kalwa, M.; Banwell, M. G.; Carr, P.D. J. Org. Chem. 2017, DOI: 10.1021/acs.joc.7b00044.(18) Okuda, S.; Robison, M. M. J. Am. Chem. Soc. 1959, 81, 740.(19) Various methods are available for the oxidation oftetrahydrocarbolines to their fully aromatic counterparts: (a) Panarese,J. D.; Waters, S. P. Org. Lett. 2010, 12, 4086. (b) Kamal, A.; Tangella,Y.; Manasa, K. L.; Sathish, M.; Srinivasulu, V.; Chetna, J.; Alarifi, A.Org. Biomol. Chem. 2015, 13, 8652. (c) Pakhare, D. S.; Kusurkar, R. S.Tetrahedron Lett. 2015, 56, 6012. (d) Hati, S.; Sen, S. Tetrahedron Lett.2016, 57, 1040.
(20) Burger, M.; Ding, Y.; Han, W.; Lindvall, M.; Nishiguchi, G. A.;Rico, A.; Smith, A.; Tanner, H.; Wan, L. PCT WO 2012/004217 A1,2012.(21) Abramovitch, R. A.; Adams, K. A. H. Can. J. Chem. 1962, 40,864.(22) Snyder, H. R.; Werber, F. X. J. Am. Chem. Soc. 1950, 72, 2962.(23) Mann, F. G.; Prior, A. F.; Willcox, T. J. J. Chem. Soc. 1959, 3830.(24) Smith, P. A. S.; Boyer, J. H. J. Am. Chem. Soc. 1951, 73, 2626.(25) Fontan, R.; Galvez, C.; Viladoms, P. Heterocycles 1981, 16, 1473.(26) Mazu, T. K.; Etukala, J. R.; Jacob, M. R.; Khan, S. I.; Walker, L.A.; Ablordeppey, S. Y. Eur. J. Med. Chem. 2011, 46, 2378.(27) Reich, M. F.; Fabio, P. F.; Lee, V. J.; Kuck, N. A.; Testa, R. T. J.Med. Chem. 1989, 32, 2474.(28) Remers, W. A.; Greenblatt, E. N.; Ellenbogen, L.; Weiss, M. J. J.Med. Chem. 1971, 14, 331.(29) Kusurkar, R. S.; Goswami, S. K. Tetrahedron 2004, 60, 5315.(30) See, for example, the following: (a) Hardegger, L. A.; Habegger,J.; Donohoe, T. J. Org. Lett. 2015, 17, 3222. (b) Sasaki, I. Synthesis2016, 48, 1974.(31) See, for example, the following: (a) Lachance, N.; April, M.;Joly, M.-C. Synthesis 2005, 2571. (b) Spergel, S. H.; Okoro, D. R.;Pitts, W. J. Org. Chem. 2010, 75, 5316.(32) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.(33) Pangborn, A. B.; Giardello, M. A.; Grubbs, R. H.; Rosen, R. K.;Timmers, F. J. Organometallics 1996, 15, 1518.(34) CrysAlis PRO Version 1.171.37.35h (released 02/09/2015CrysAlis171.NET); Agilent Technologies: Oxfordshire, UK.(35) SIR92: Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi,A.; Burla, M. C.; Polidori, G.; Camalli, M. J. Appl. Crystallogr. 1994, 27,435.(36) Betteridge, P. W.; Carruthers, J. R.; Cooper, R. I.; Prout, K.;Watkin, D. J. J. Appl. Crystallogr. 2003, 36, 1487.
The Journal of Organic Chemistry Article
DOI: 10.1021/acs.joc.7b00323J. Org. Chem. 2017, 82, 4328−4335
4335
390

S1
SUPP
ORT
ING
INFO
RMAT
ION
FO
R:
A U
nifie
d A
ppro
ach
to th
e Is
omer
ic α
-, β
-, γ-
and
δ-C
arbo
lines
via
thei
r 6,
7,8,
9-T
etra
hydr
o-co
unte
rpar
ts
Qia
o Y
an, E
mm
a G
in, M
artin
G. B
anw
ell*
Ant
hony
C. W
illis
and
Pau
l D. C
arr
Rese
arch
Sch
ool o
f Che
mis
try,
Inst
itute
of A
dvan
ced
Stud
ies,
The
Aust
ralia
n N
atio
nal U
nive
rsity
, Can
berr
a, A
CT
2601
, Aus
tral
ia
Emai
l: M
artin
.Ban
wel
l@an
u.ed
u.au
CO
NT
EN
TS
PA
GE
(i)A
niso
tropi
c D
ispl
acem
ent E
llips
oid
Plot
s fro
m th
e Si
ngle
-cry
stal
X-r
ay
Ana
lyse
s of C
ompo
unds
1-5
S2
(ii)
1 H a
nd 13
C N
MR
Spe
ctra
of C
ompo
unds
1-5
, 8, 9
, 10,
11,
12,
14,
15,
17,
18,
20,
21/2
2 an
d 24
-26
S7
391
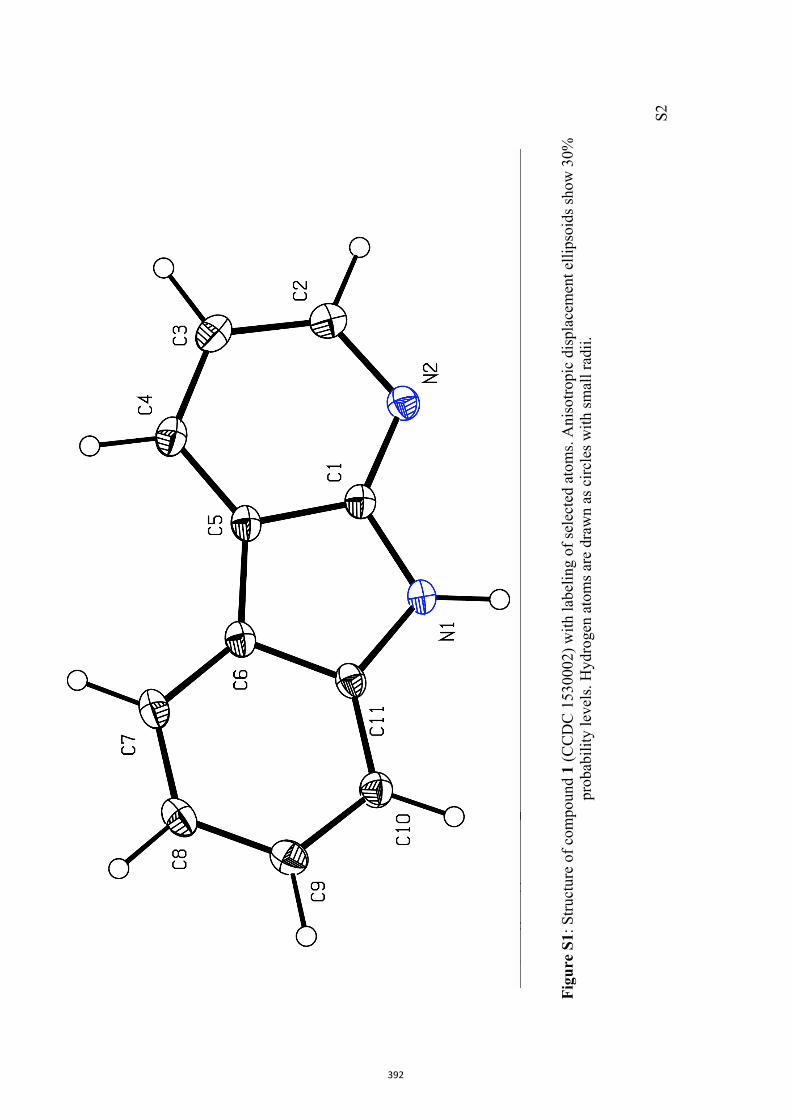
S2
Figu
re S
1: S
truct
ure
of c
ompo
und
1 (C
CD
C 1
5300
02) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
392
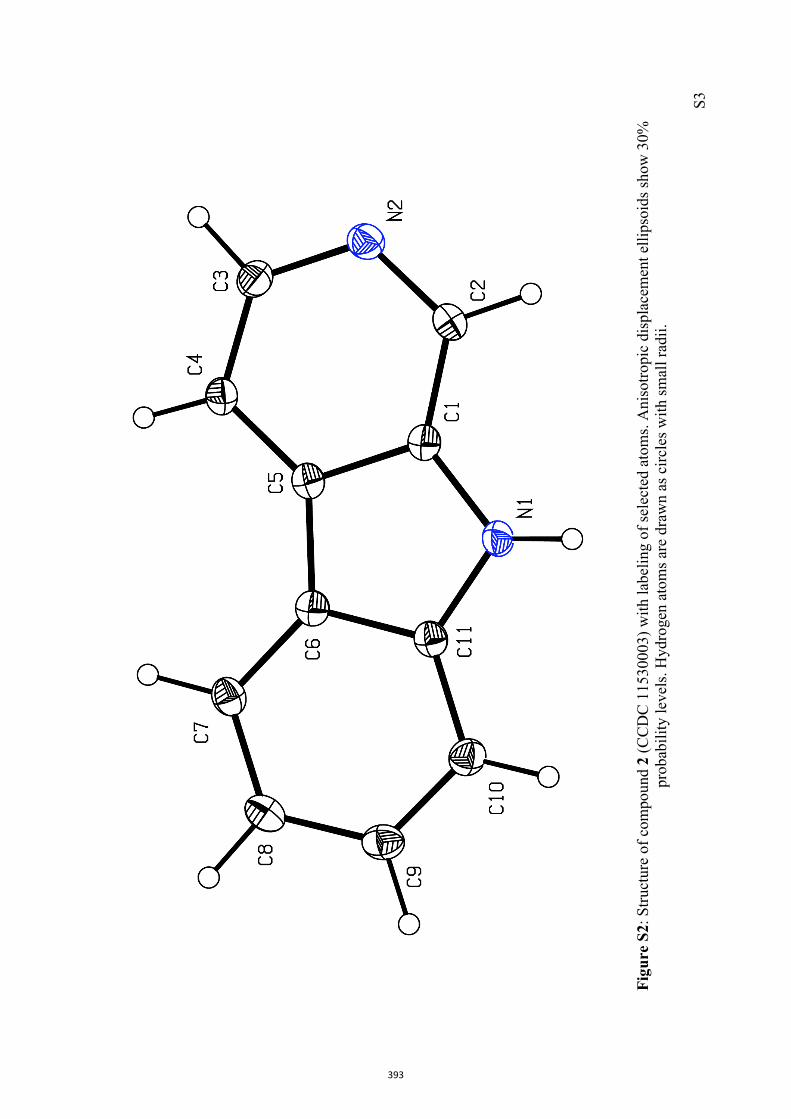
S3
Figu
re S
2: S
truct
ure
of c
ompo
und
2 (C
CD
C 1
1530
003)
with
labe
ling
of se
lect
ed a
tom
s. A
niso
tropi
c di
spla
cem
ent e
llips
oids
show
30%
pr
obab
ility
leve
ls. H
ydro
gen
atom
s are
dra
wn
as c
ircle
s with
smal
l rad
ii.
393
S4
Figu
re S
3: S
truct
ure
of c
ompo
und
3 (C
CD
C 1
5300
04) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
394
S5
Figu
re S
4: S
truct
ure
of c
ompo
und
4 (C
CD
C 1
5300
05) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
395
S6
Figu
re S
5: S
truct
ure
of c
ompo
und
5 (C
CD
C 1
5300
06) w
ith la
belin
g of
sele
cted
ato
ms.
Ani
sotro
pic
disp
lace
men
t elli
psoi
ds sh
ow 3
0%
prob
abili
ty le
vels
. Hyd
roge
n at
oms a
re d
raw
n as
circ
les w
ith sm
all r
adii.
396
S7
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
f1 (
ppm
)
2.001.001.00
1.001.001.00
3.31 MeOD
397
S8
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
48.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.43 MeOD49.64 MeOD
112.30116.10118.04121.02121.88122.04128.03129.94
140.57146.13152.90
12
1.8
12
2.0
12
2.2
f1 (
ppm
)
121.88
122.04
398
S9
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
1.00
2.00
1.001.001.00
1.00
3.31 MeOD
399
S10
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
48.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.42 MeOD49.64 MeOD
112.82116.07120.83122.18122.71129.74130.43134.06137.76138.36142.74
400
S11
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
12.0
f1 (
ppm
)
1.002.001.00
1.001.00
1.00
3.31 MeOD
401
S12
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
48.64 MeOD48.76 MeOD48.88 MeOD49.00 MeOD49.12 MeOD49.24 MeOD49.36 MeOD
107.62
112.42121.53121.60121.64122.42128.17141.50142.71144.40145.82
12
01
21
12
21
23
f1 (
ppm
)121.53121.60121.64
122.42
402
S13
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
12.0
12.5
13.0
f1 (
ppm
)
1.012.001.00
1.01
0.83
0.77
0.61
2.50 DMSO
403
S14
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
39.16 DMSO39.28 DMSO39.40 DMSO39.52 DMSO39.64 DMSO39.76 DMSO39.88 DMSO
106.37111.43119.43119.93120.56120.70126.54
139.50142.70143.47144.47
11
81
19
12
01
21
12
21
23
f1 (
ppm
)
119.43119.93120.56120.70
404
S15
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.5f1
(ppm
)
1.001.002.001.001.001.00
3.31 MeOD
405
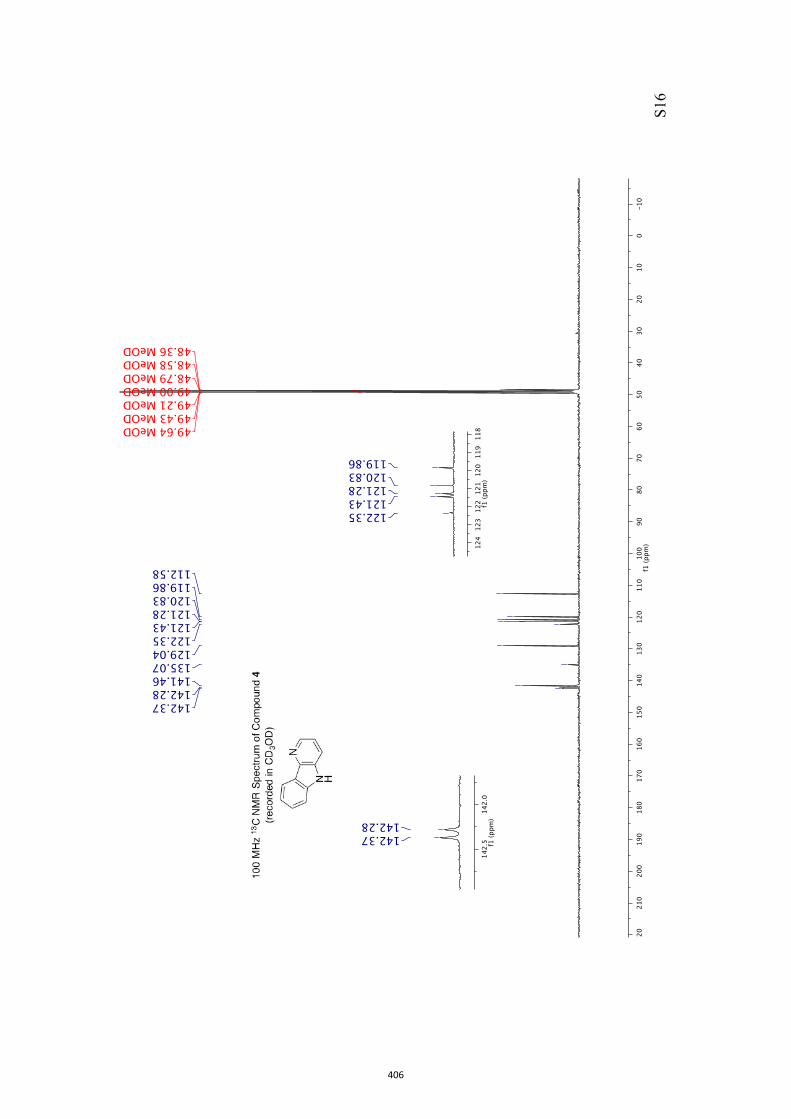
S16
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
48.36 MeOD48.58 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.43 MeOD49.64 MeOD
112.58119.86120.83121.28121.43122.35129.04135.07141.46142.28142.37
11
81
19
12
01
21
12
21
23
12
4f1
(ppm
)
119.86120.83121.28121.43122.35
14
2.0
14
2.5 f1
(ppm
)142.28142.37
406
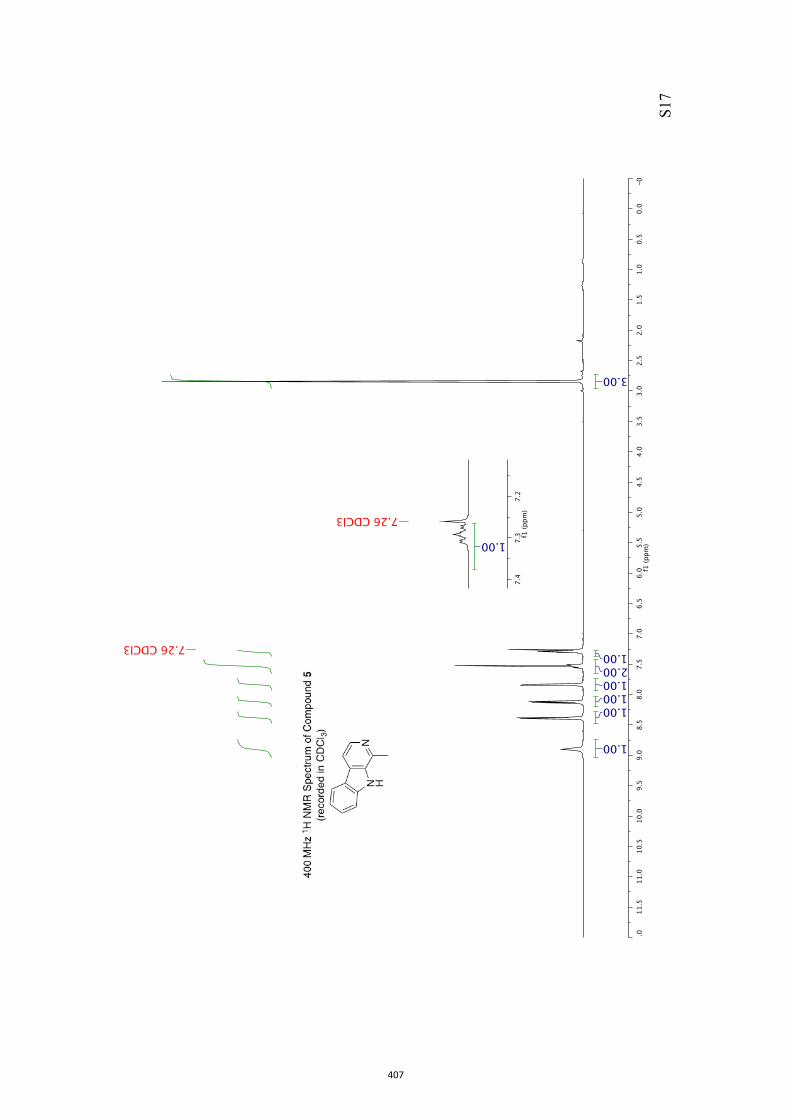
S17
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
3.00
1.002.001.001.001.00
1.00
7.26 CDCl3
7.2
7.3
7.4
f1 (
ppm
)
1.00
7.26 CDCl3
407
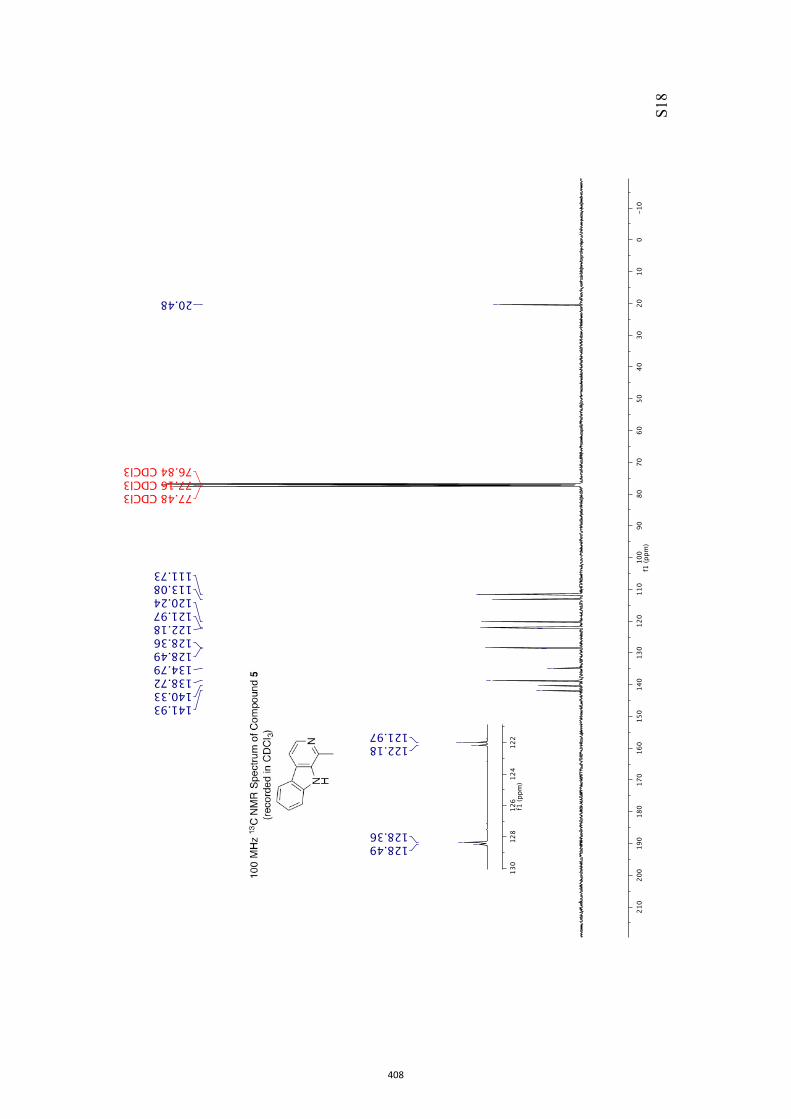
S18
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.48
76.84 CDCl377.16 CDCl377.48 CDCl3
111.73113.08120.24121.97122.18128.36128.49134.79138.72140.33141.93
12
21
24
12
61
28
13
0f1
(ppm
)
121.97122.18
128.36128.49
408
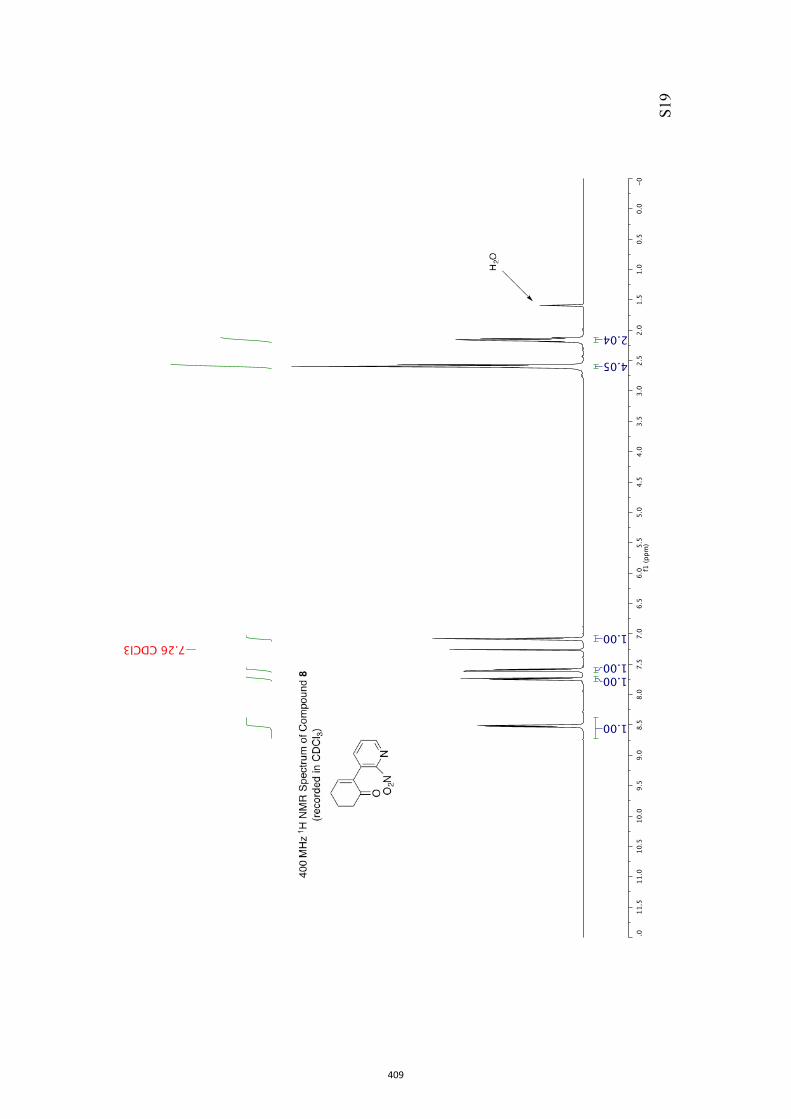
S19
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
2.04
4.05
1.00
1.001.00
1.00
7.26 CDCl3
409
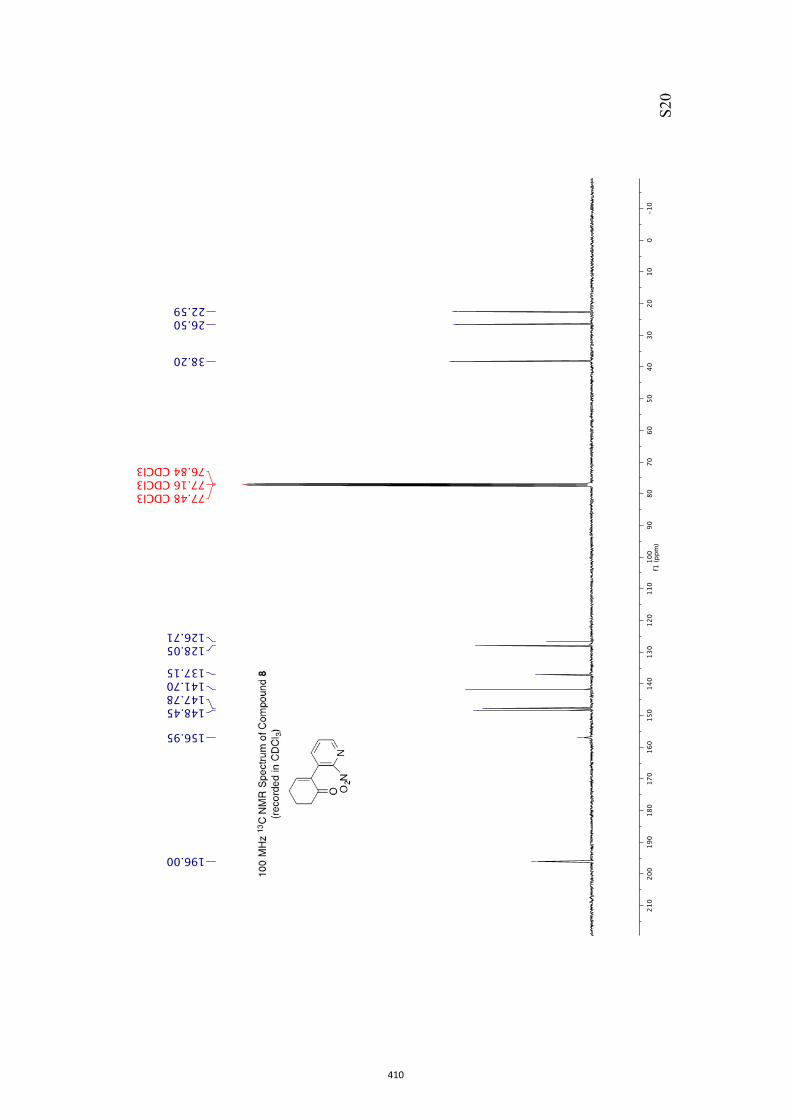
S20
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
22.5926.50
38.20
76.84 CDCl377.16 CDCl377.48 CDCl3
126.71128.05
137.15141.70147.78148.45
156.95
196.00
410
S21
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
4.10
2.062.03
1.00
1.00
1.00
0.98
7.26 CDCl3
411
S22
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.8123.1123.2823.34
76.84 CDCl377.16 CDCl377.48 CDCl3
108.38115.09120.83125.73
135.39140.80148.88
23
.02
3.1
23
.22
3.3
23
.42
3.5
f1 (
ppm
)
23.11
23.2823.34
412
S23
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
1.00
1.00
1.00
7.26 CDCl3
413
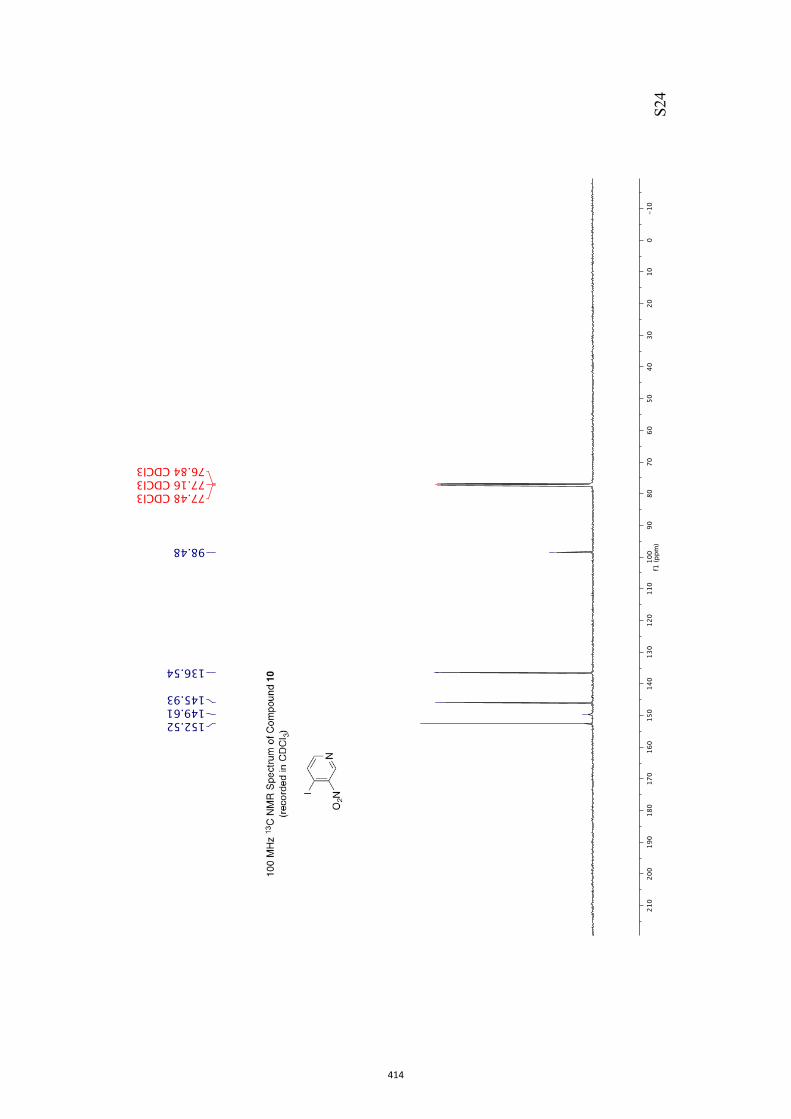
S24
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
76.84 CDCl377.16 CDCl377.48 CDCl3
98.48
136.54
145.93149.61152.52
414
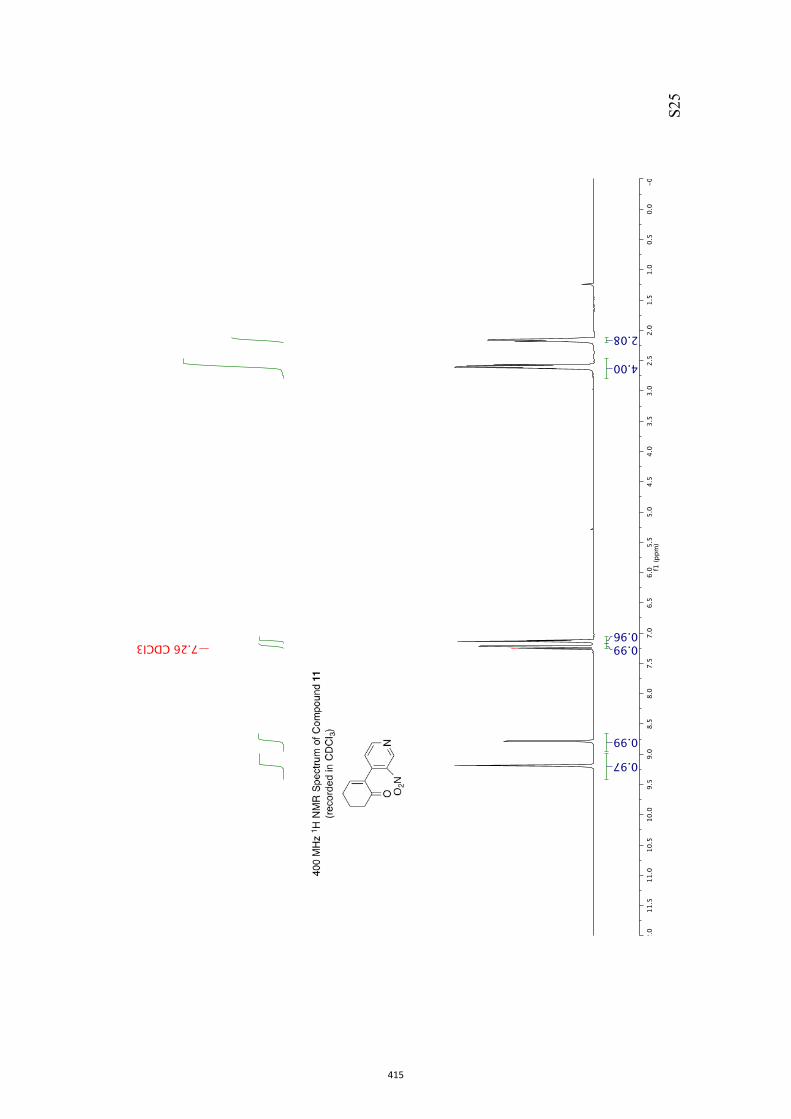
S25
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
12.0
f1 (
ppm
)
2.08
4.00
0.960.99
0.99
0.97
7.26 CDCl3
415
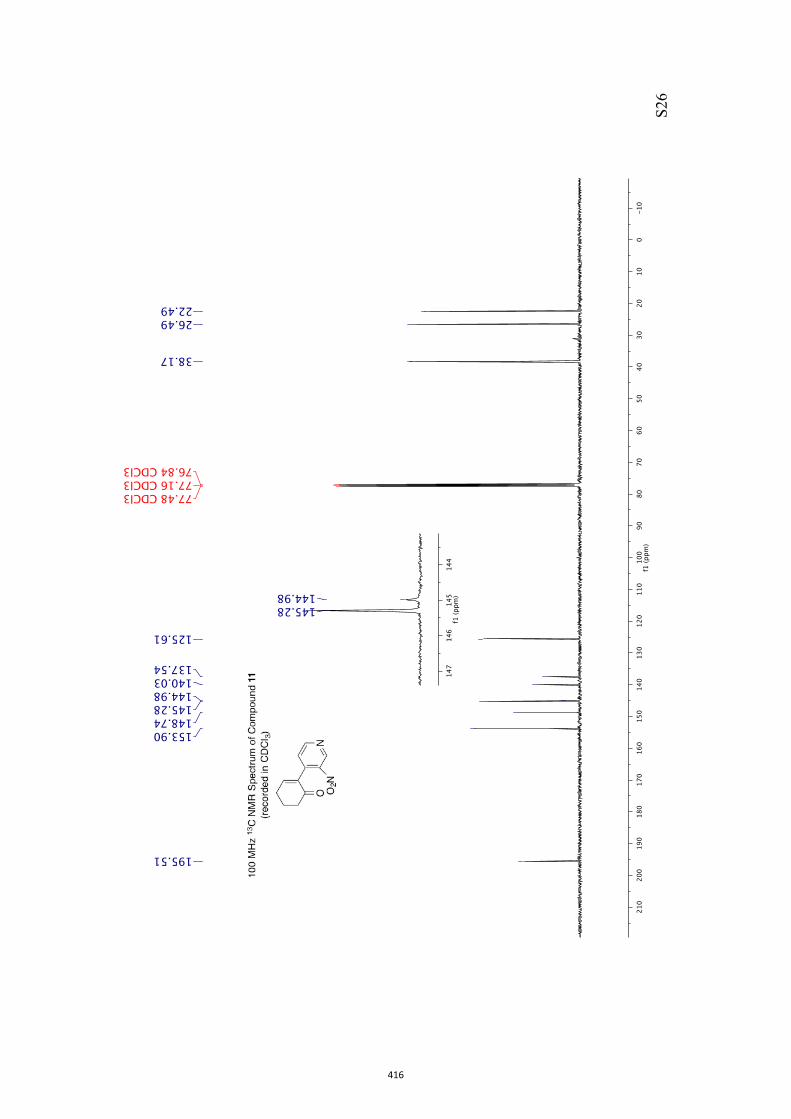
S26
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
22.4926.49
38.17
76.84 CDCl377.16 CDCl377.48 CDCl3
125.61
137.54140.03144.98145.28148.74153.90
195.51
14
41
45
14
61
47
f1 (
ppm
)144.98145.28
416
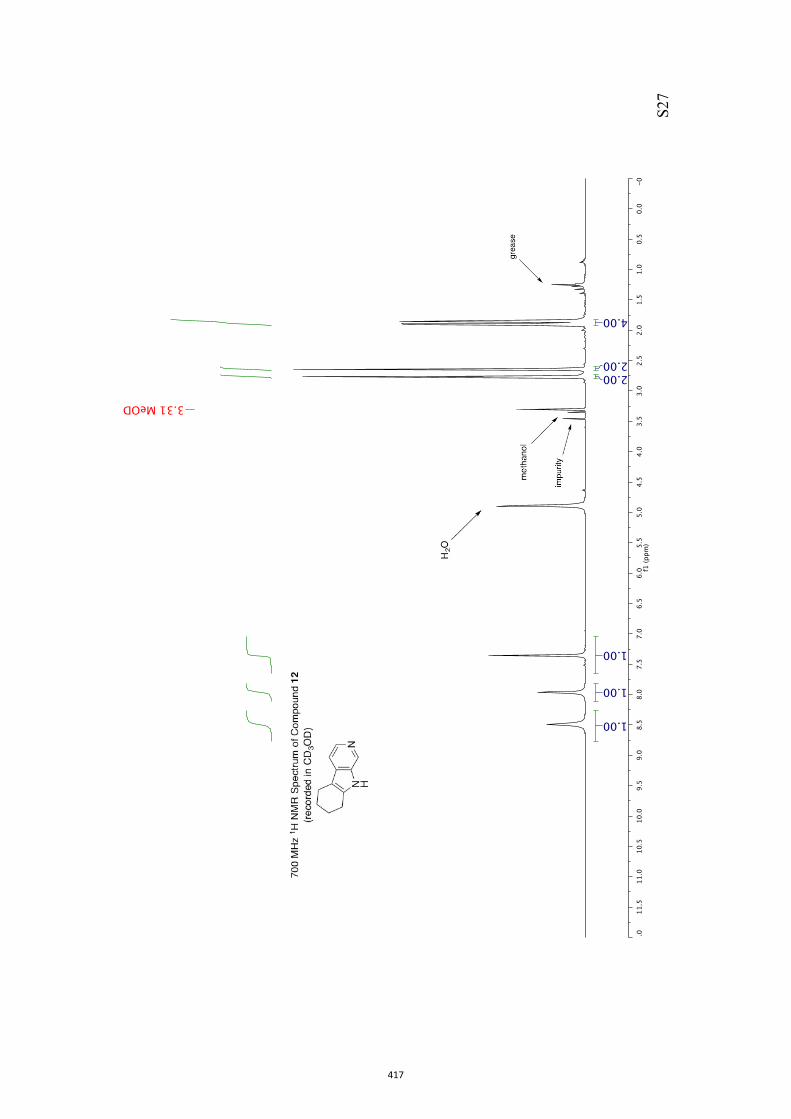
S27
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
4.00
2.002.00
1.00
1.00
1.00
3.31 MeOD
417
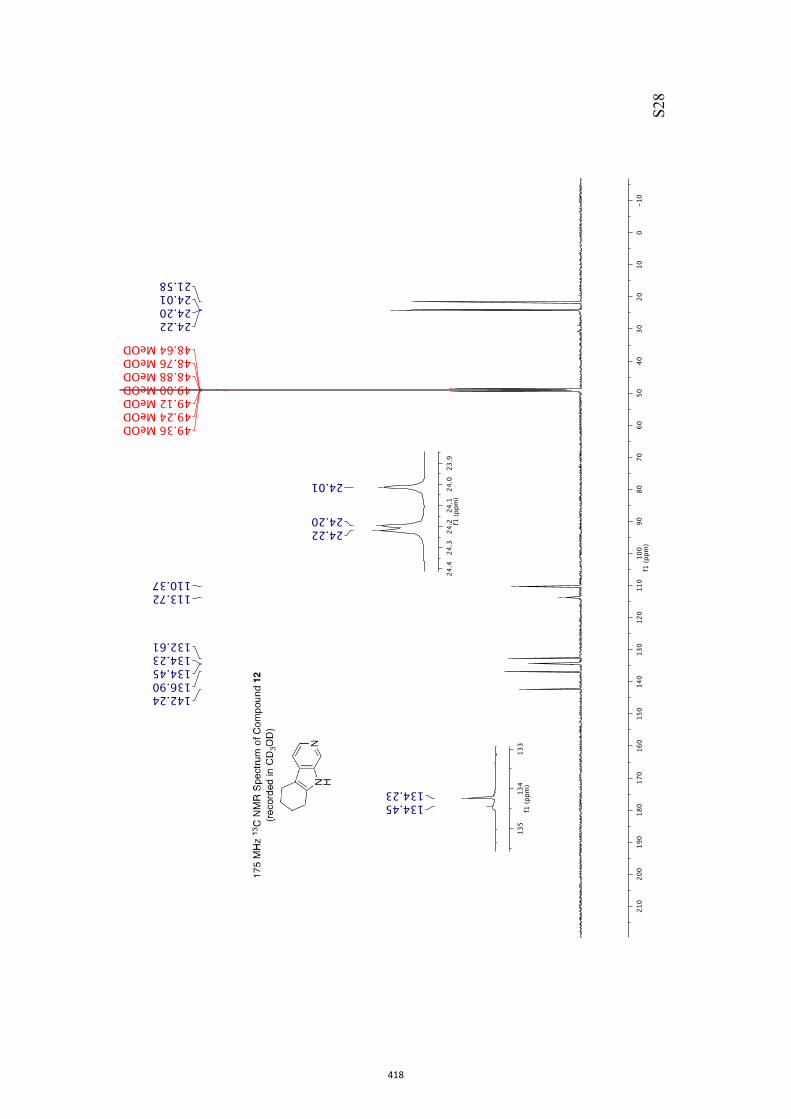
S28
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
21.5824.0124.2024.22
48.64 MeOD48.76 MeOD48.88 MeOD49.00 MeOD49.12 MeOD49.24 MeOD49.36 MeOD
110.37113.72
132.61134.23134.45136.90142.24
23
.92
4.0
24
.12
4.2
24
.32
4.4
f1 (
ppm
)
24.01
24.2024.22
13
31
34
13
5f1
(ppm
)134.23134.45
418
S29
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
2.00
4.00
1.00
1.00
1.00
1.00
7.26 CDCl3
419
S30
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
22.5926.54
38.19
76.84 CDCl377.16 CDCl377.48 CDCl3
116.59
125.42
135.97
148.57151.44152.77154.08
196.06
420
S31
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10
.0f1
(ppm
)
4.06
4.01
1.00
1.00
0.96
3.31 MeOD
421
S32
01
02
03
04
05
06
07
08
09
01
00
11
01
20
13
01
40
15
01
60
17
01
80
19
02
00
21
02
20
23
0f1
(ppm
)
21.6123.8724.0624.20
48.63 MeOD48.76 MeOD48.88 MeOD49.00 MeOD49.12 MeOD49.24 MeOD49.37 MeOD
107.54110.30
126.19
138.08139.08139.88141.57
23
24
25
f1 (
ppm
)23.8724.0624.20
422
S33
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
2.082.012.00
2.00
1.00
1.00
7.26 CDCl3
423
S34
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
f1 (
ppm
)
22.5026.58
38.25
76.84 CDCl377.16 CDCl377.48 CDCl3
123.36
132.15139.18146.14149.37151.04152.93
196.17
424

S35
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
12.0
f1 (
ppm
)
4.00
2.002.00
1.00
1.00
1.00
1.00
7.26 CDCl3
425
S36
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.1223.1023.1723.77
76.84 CDCl377.16 CDCl377.48 CDCl3
110.78115.82117.42
129.08
139.19141.91145.59
23
.02
3.5
24
.0f1
(ppm
)
23.1023.17
23.77
426
S37
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
9.0
9.5
10.0
10.5
11.0
11.5
12.0
f1 (
ppm
)
3.00
1.99
1.00
3.31 MeOD
427
S38
-10
010
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
f1 (
ppm
)
19.31
48.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.43 MeOD49.64 MeOD
113.04114.71
143.15
156.03
173.35
428
S39
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
12
.51
3.0
13
.51
4.0
14
.51
5.0
f1 (
ppm
)
0.92
3.00
1.33
1.00
0.33
1.33
2.50 DMSO
6.0
6.5
7.0
7.5
8.0
8.5
9.0
f1 (
ppm
)
1.33
1.00
0.33
429
S40
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
15.3018.20
38.90 DMSO39.10 DMSO39.31 DMSO39.52 DMSO39.73 DMSO39.94 DMSO40.15 DMSO
117.98120.72
137.25137.81139.43142.17143.45148.14151.15
168.15
430
S41
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
3.00
1.00
1.00
7.26 CDCl3
431
S42
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
20.88
76.84 CDCl377.16 CDCl377.48 CDCl3
123.73126.27
148.47150.48151.77
432
S43
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.51
2.0
f1 (
ppm
)
2.004.003.00
2.00
1.00
7.26 CDCl3
433
S44
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
f1 (
ppm
)
21.9722.5126.52
38.20
76.84 CDCl377.16 CDCl377.48 CDCl3
123.42
136.34138.95146.61150.09150.82151.38
195.61
434
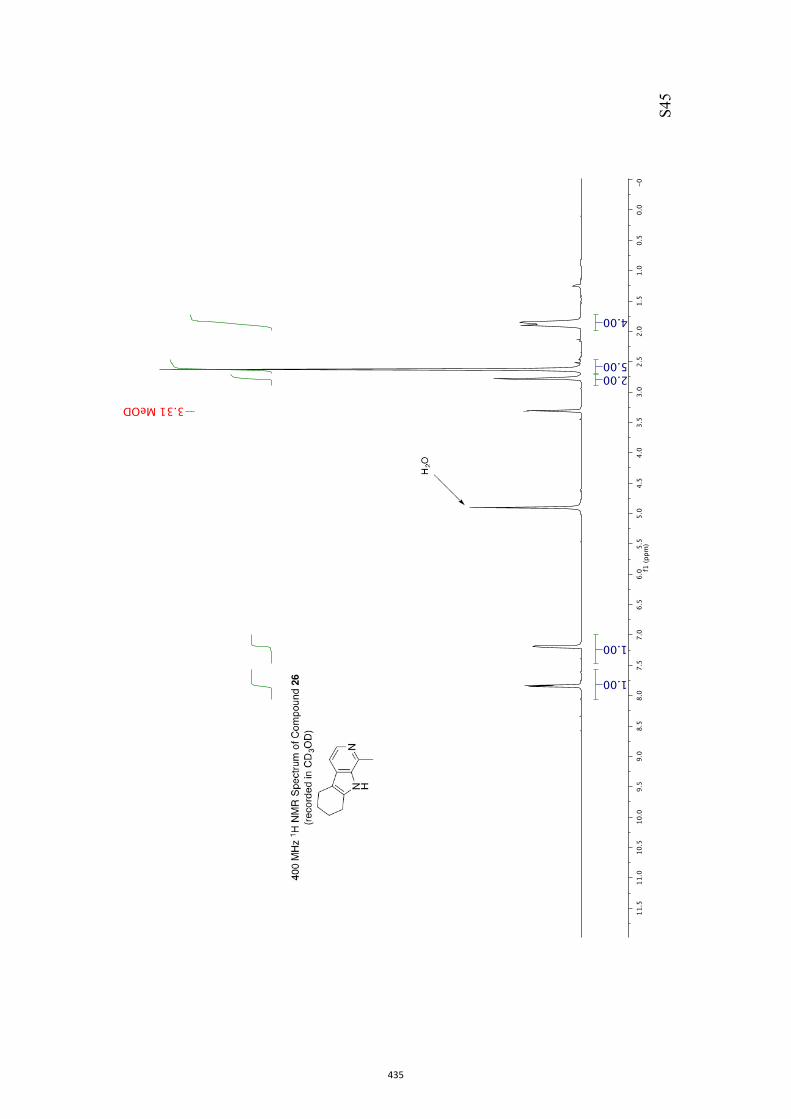
S45
-0
.50
.00
.51
.01
.52
.02
.53
.03
.54
.04
.55
.05
.56
.06
.57
.07
.58
.08
.59
.09
.51
0.0
10
.51
1.0
11
.5f1
(ppm
)
4.00
5.002.00
1.00
1.00
3.31 MeOD
435
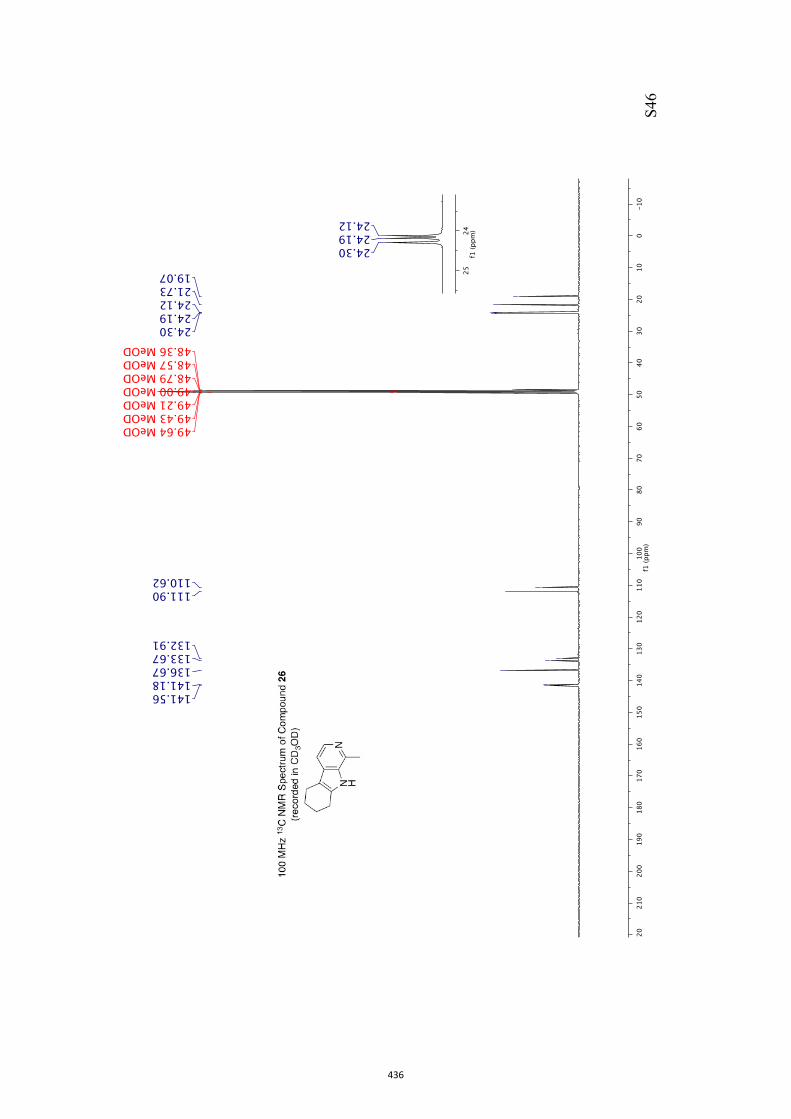
S46
-1
00
10
20
30
40
50
60
70
80
90
10
01
10
12
01
30
14
01
50
16
01
70
18
01
90
20
02
10
22
0f1
(ppm
)
19.0721.7324.1224.1924.30
48.36 MeOD48.57 MeOD48.79 MeOD49.00 MeOD49.21 MeOD49.43 MeOD49.64 MeOD
110.62111.90
132.91133.67136.67141.18141.56
24
25
f1 (
ppm
)
24.1224.1924.30
436
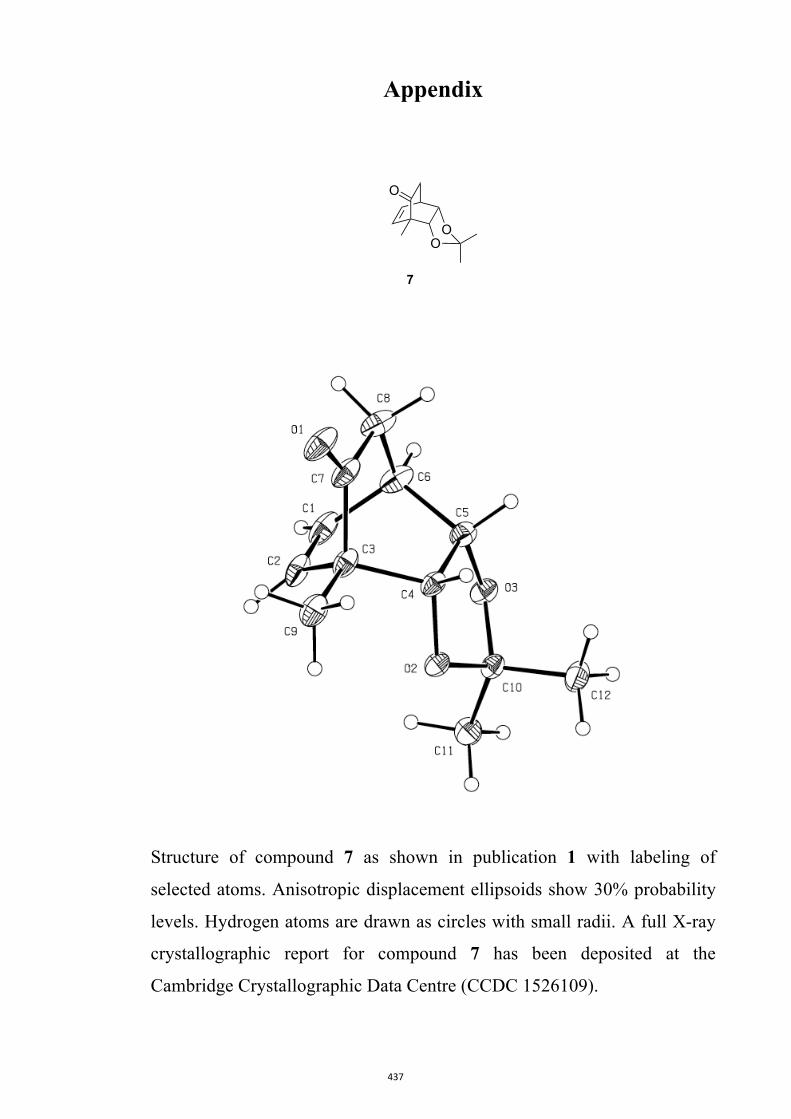
Appendix
Structure of compound 7 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 7 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526109).
OO
O
7
437
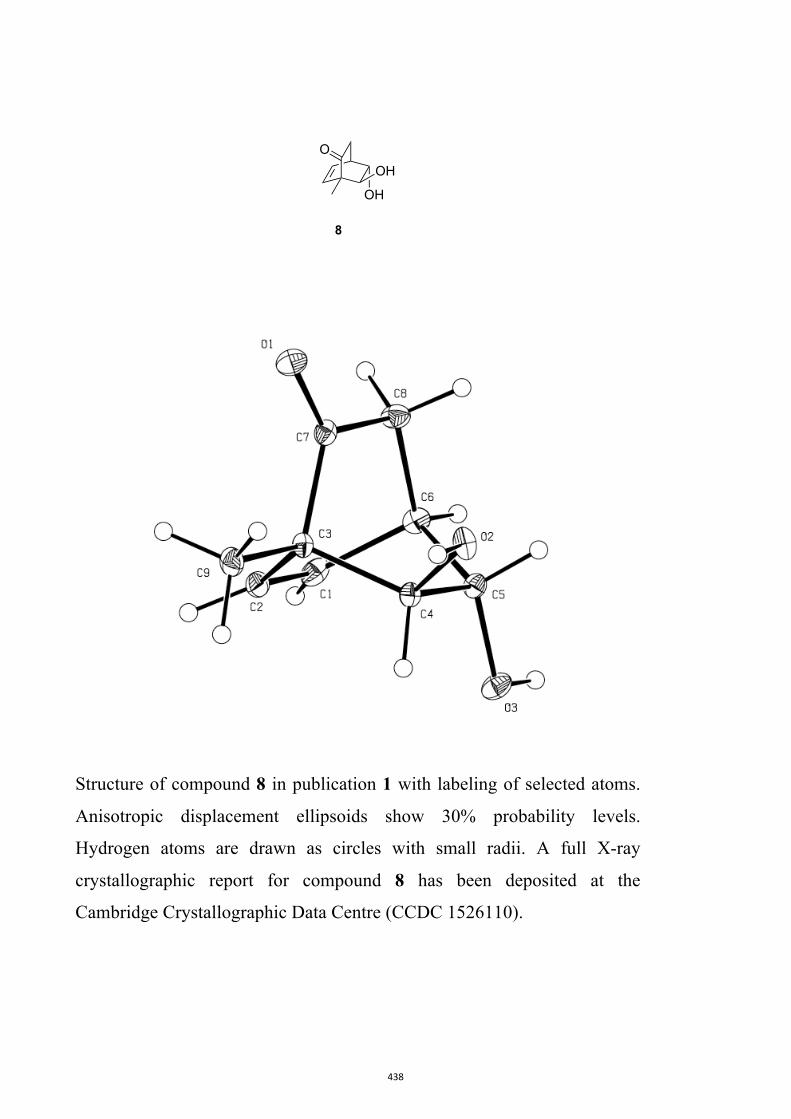
Structure of compound 8 in publication 1 with labeling of selected atoms.
Anisotropic displacement ellipsoids show 30% probability levels.
Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 8 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526110).
OH
OOH
8
438
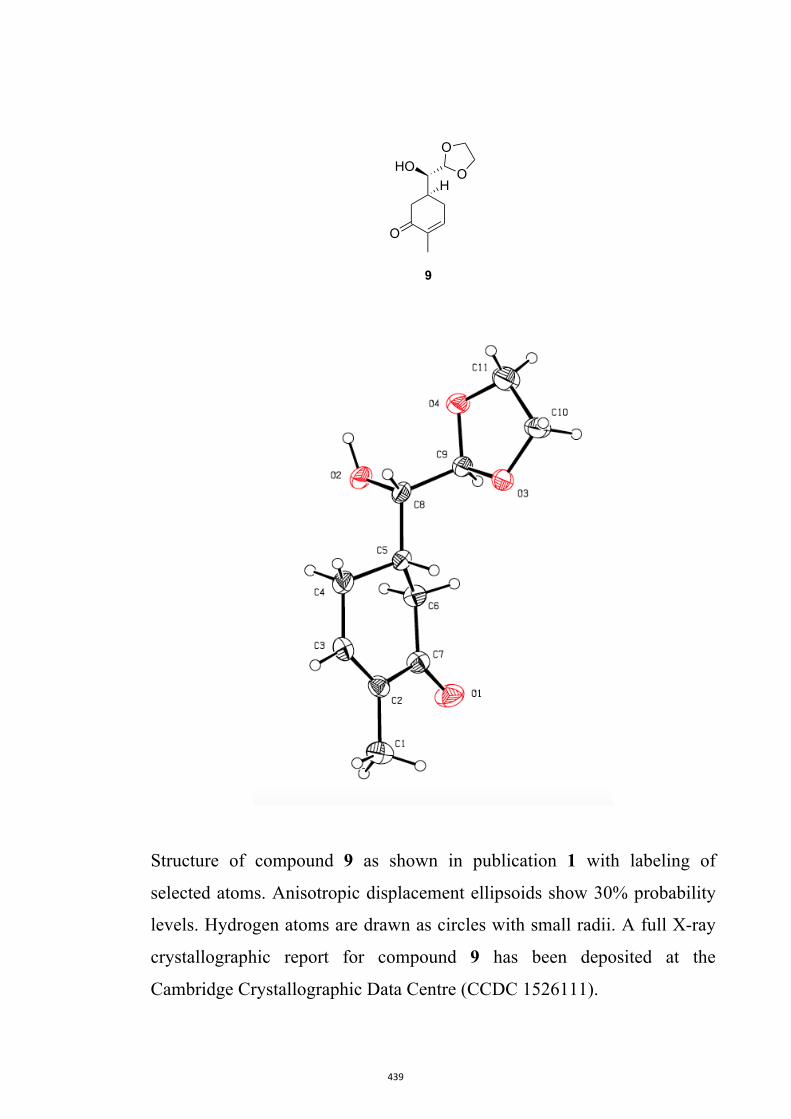
Structure of compound 9 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 9 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526111).
O
HO O
O
H
9
439
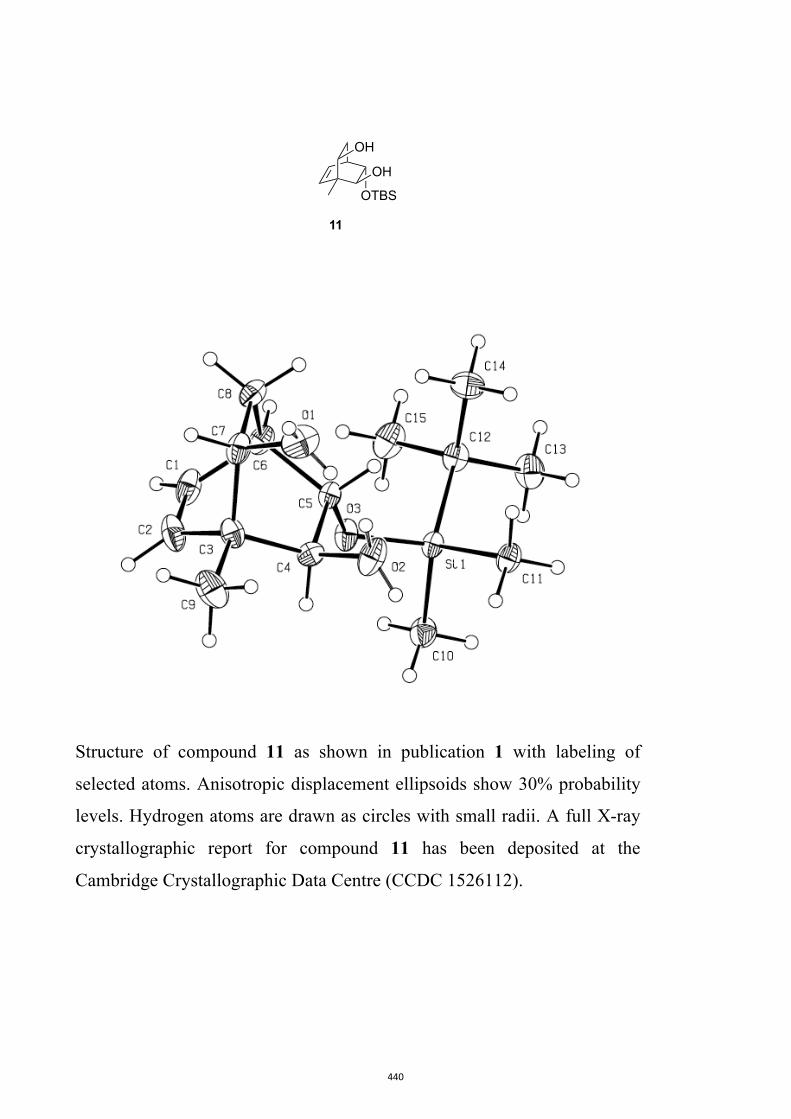
Structure of compound 11 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 11 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526112).
OTBS
OH
OH
11
440
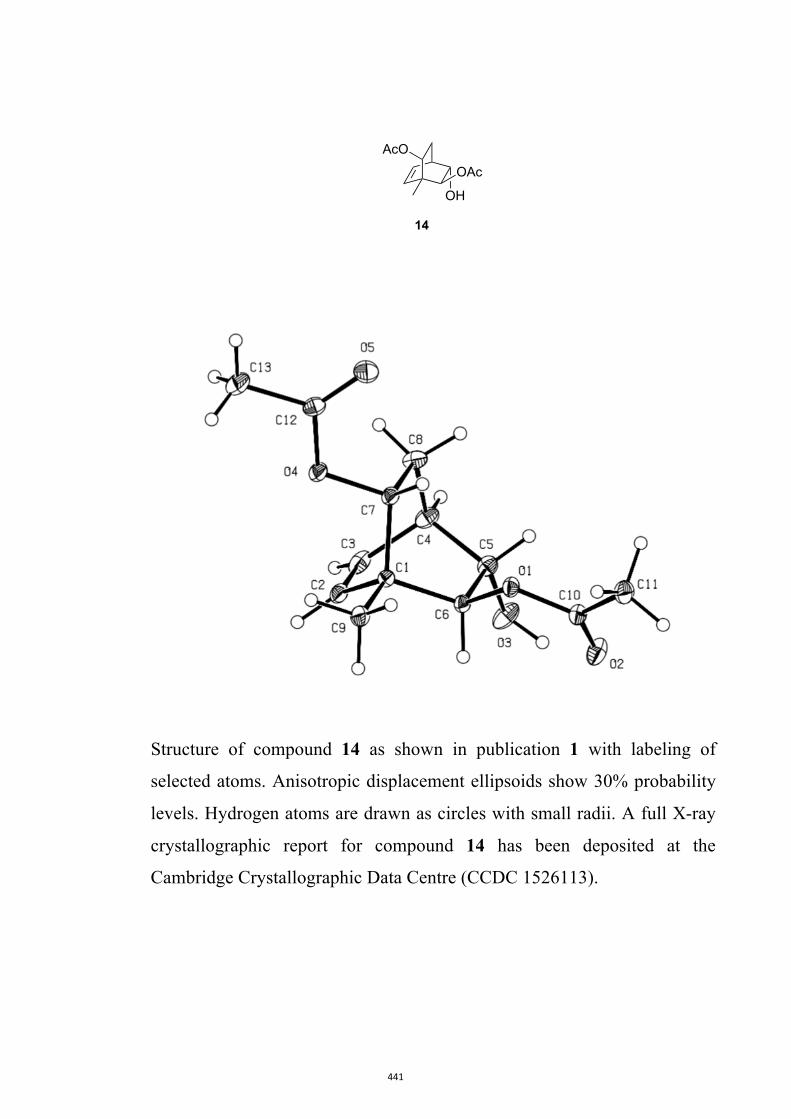
Structure of compound 14 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 14 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526113).
OH
OAc
AcO
14
441

Structure of compound 16 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 16 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526114).
OH
AcOOH
16
442
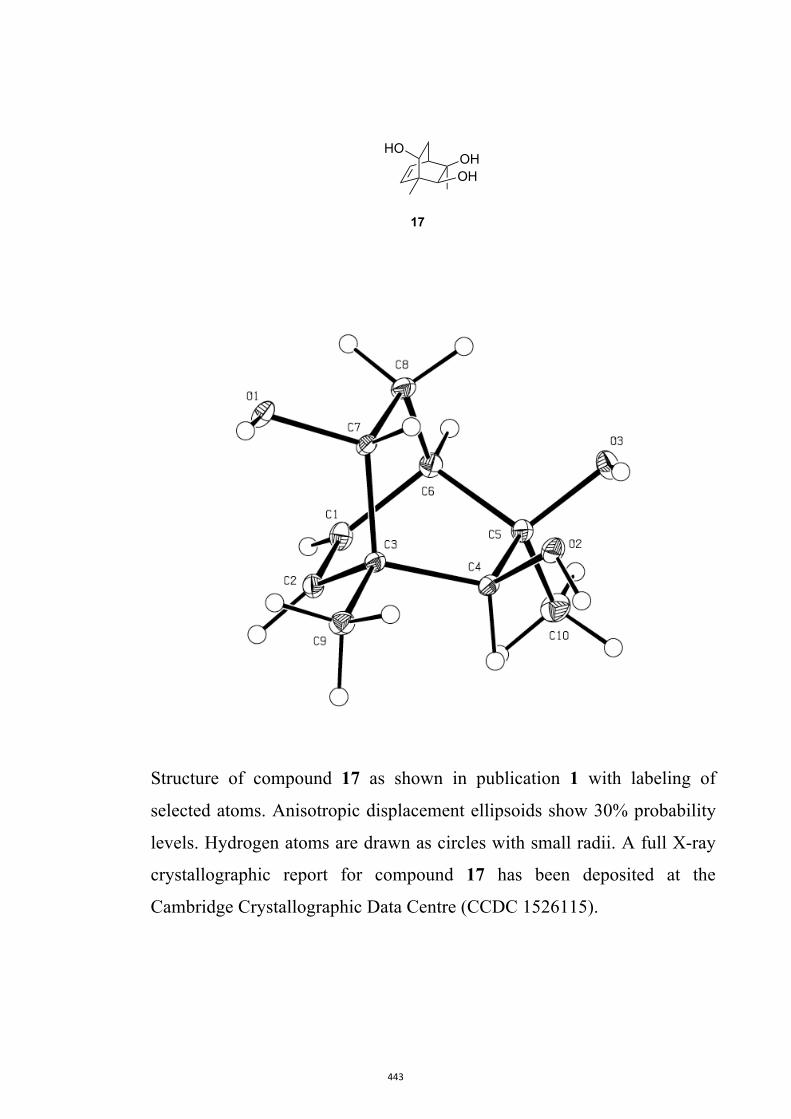
Structure of compound 17 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 17 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526115).
OH
HOOH
17
443
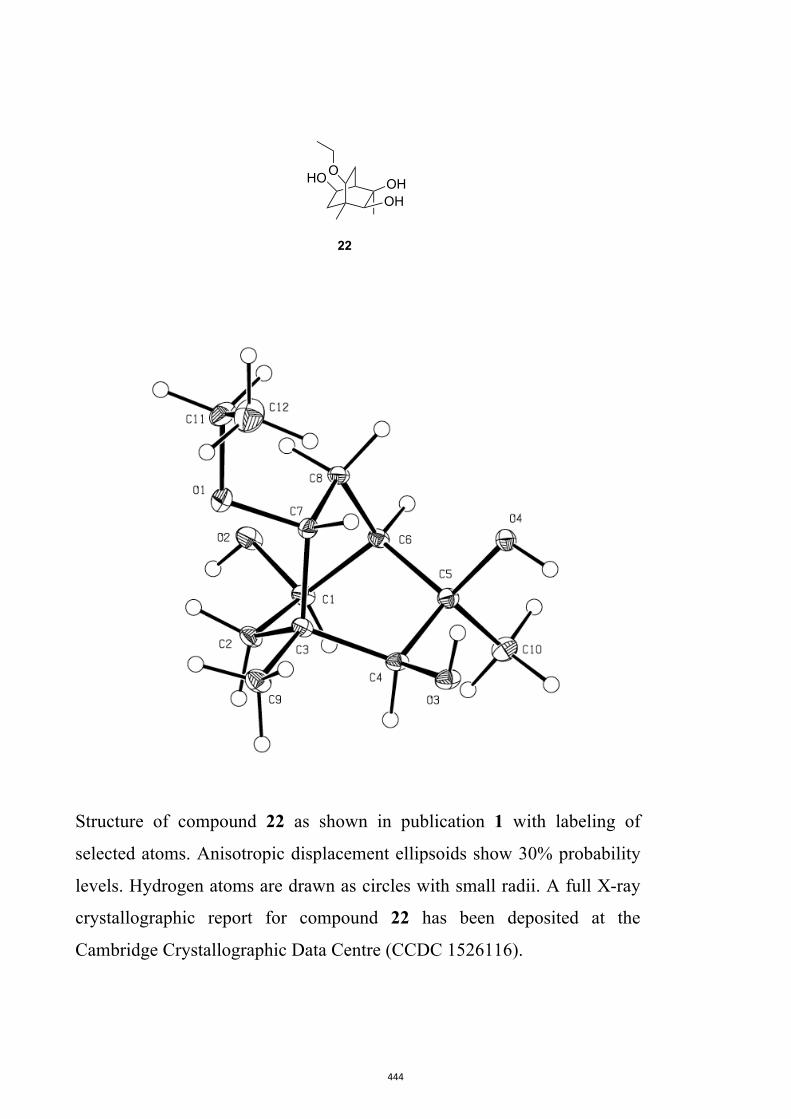
Structure of compound 22 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 22 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526116).
OHOHHOO
22
444
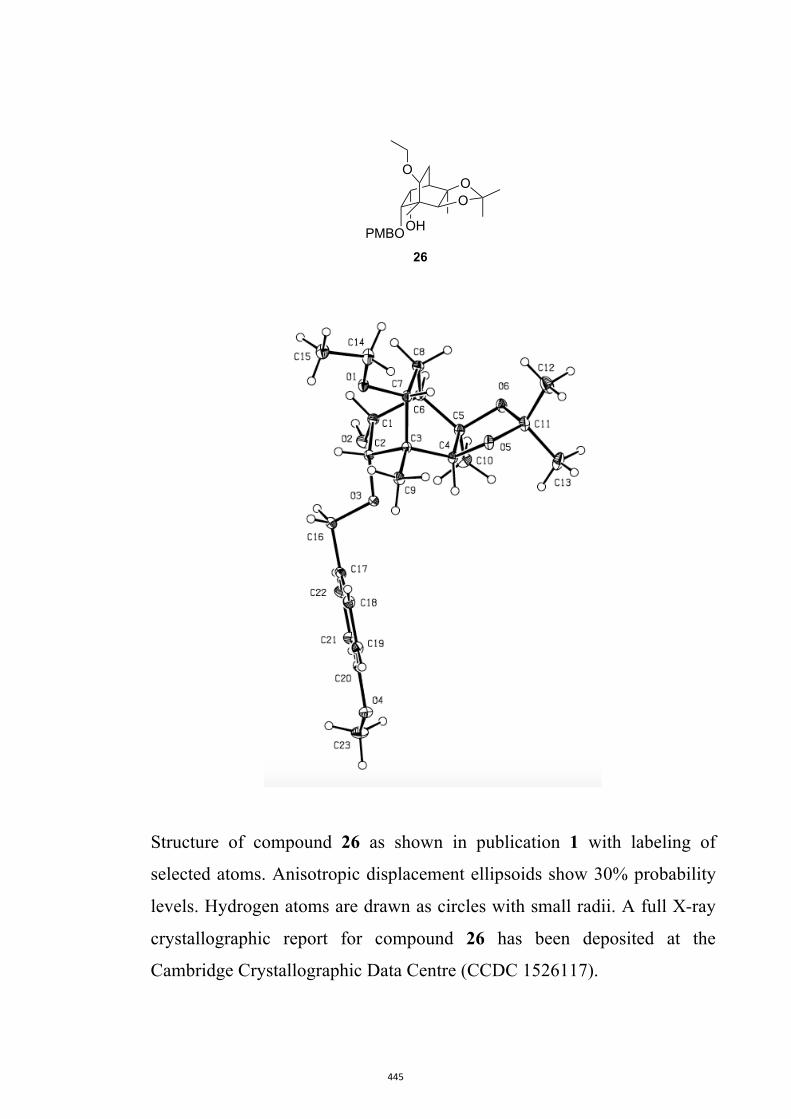
Structure of compound 26 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 26 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526117).
OO
O
PMBOOH
26
445
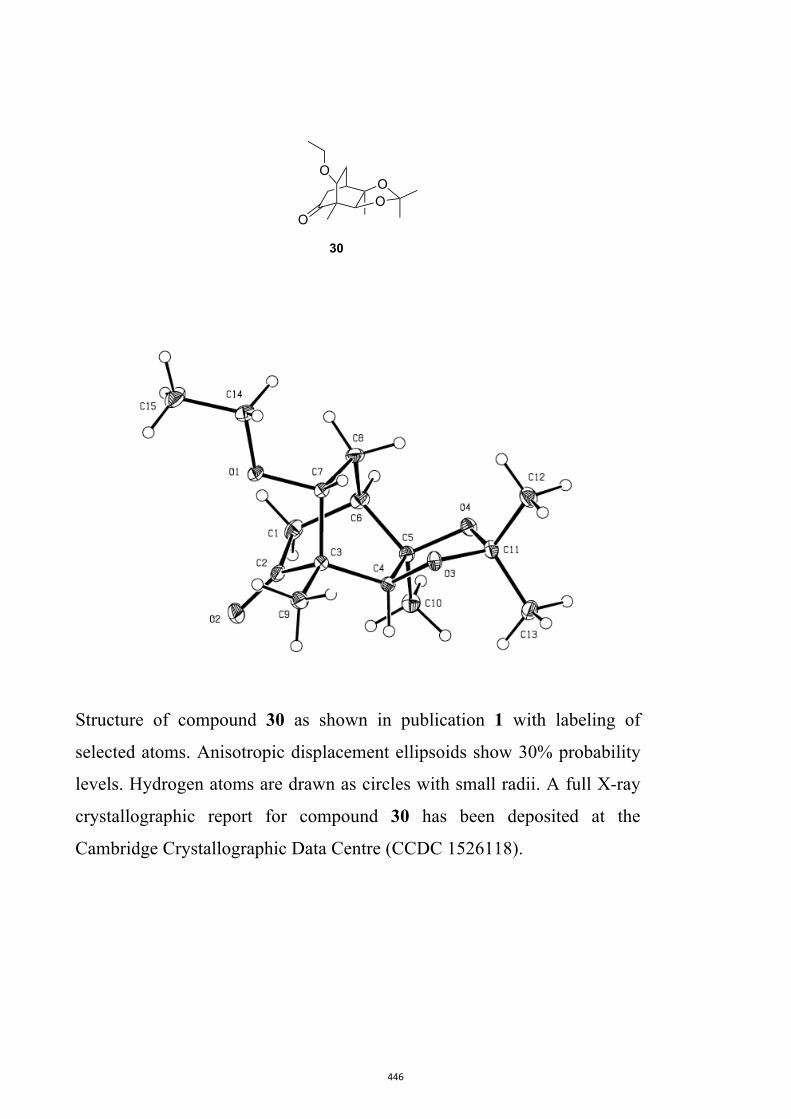
Structure of compound 30 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 30 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526118).
OO
O
30
O
446
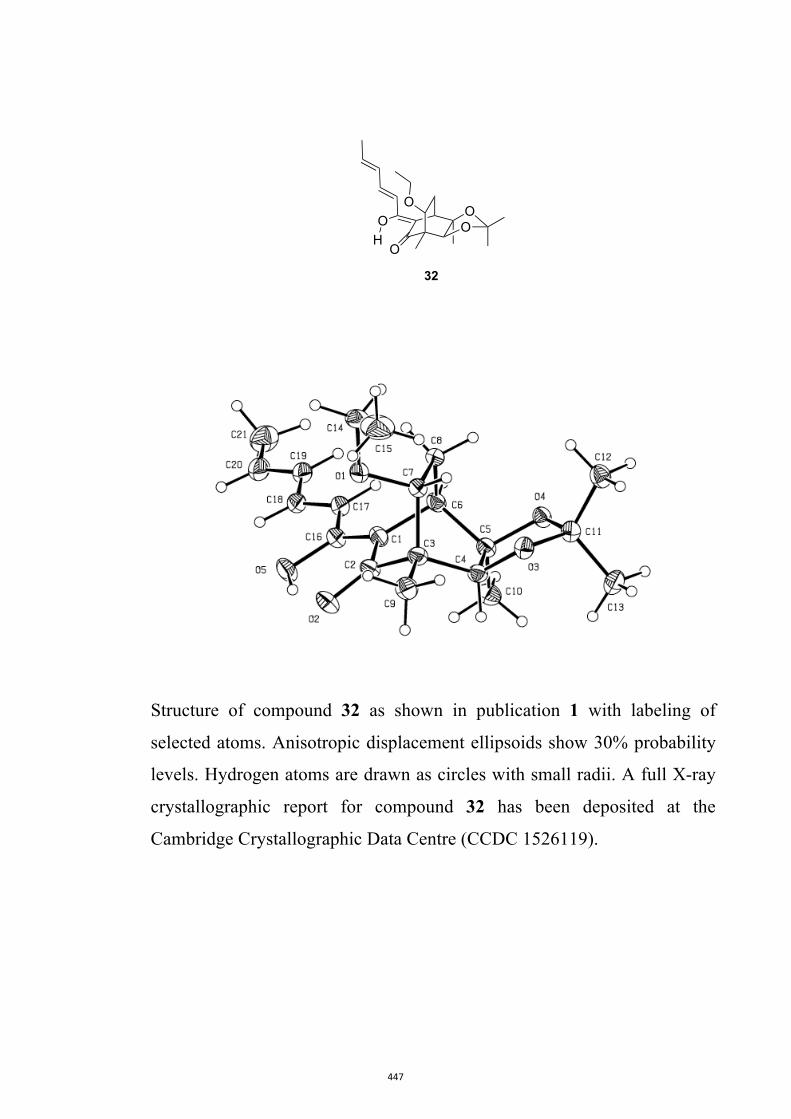
Structure of compound 32 as shown in publication 1 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 32 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1526119).
32
OO
O
OH
O
447
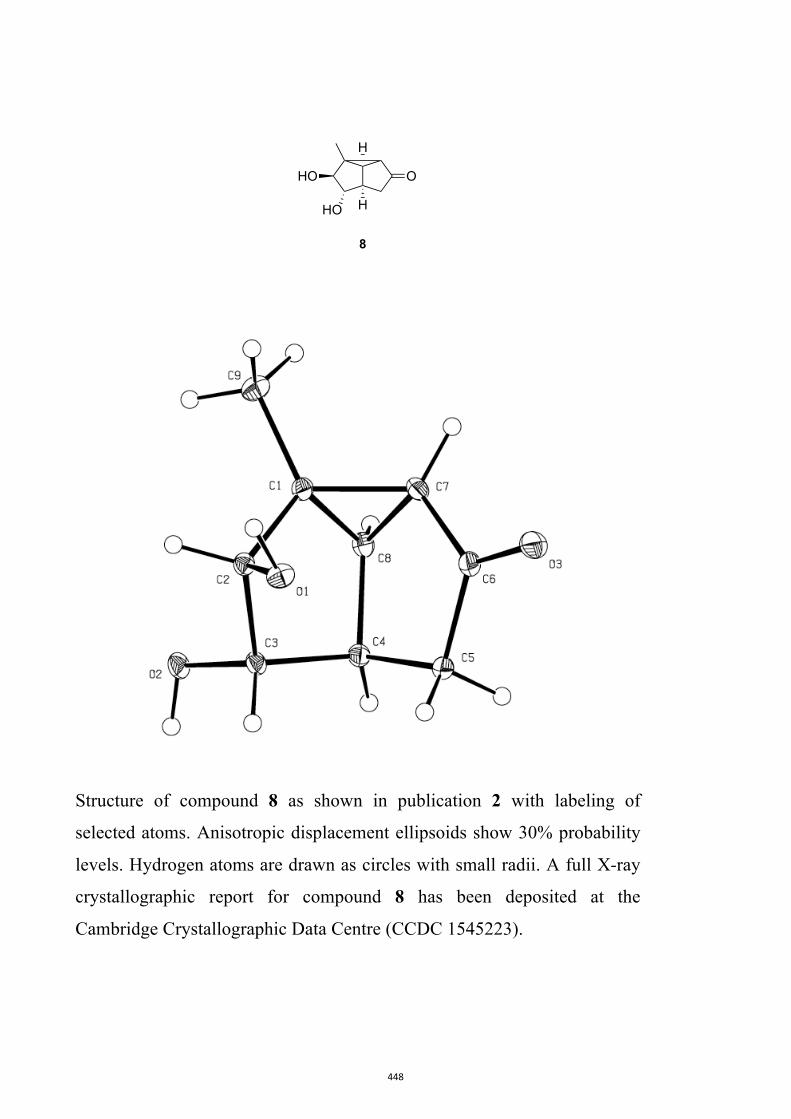
Structure of compound 8 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 8 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545223).
O
H
H
HO
HO
8
448
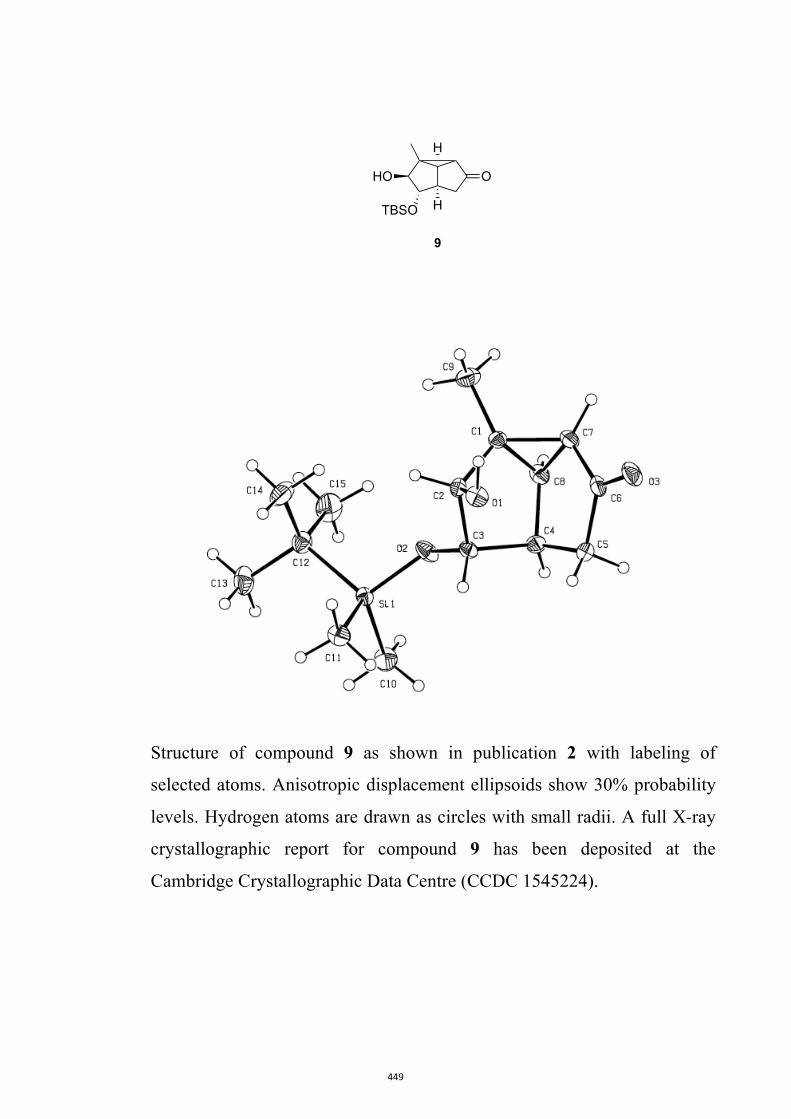
Structure of compound 9 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 9 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545224).
O
H
H
HO
TBSO
9
449
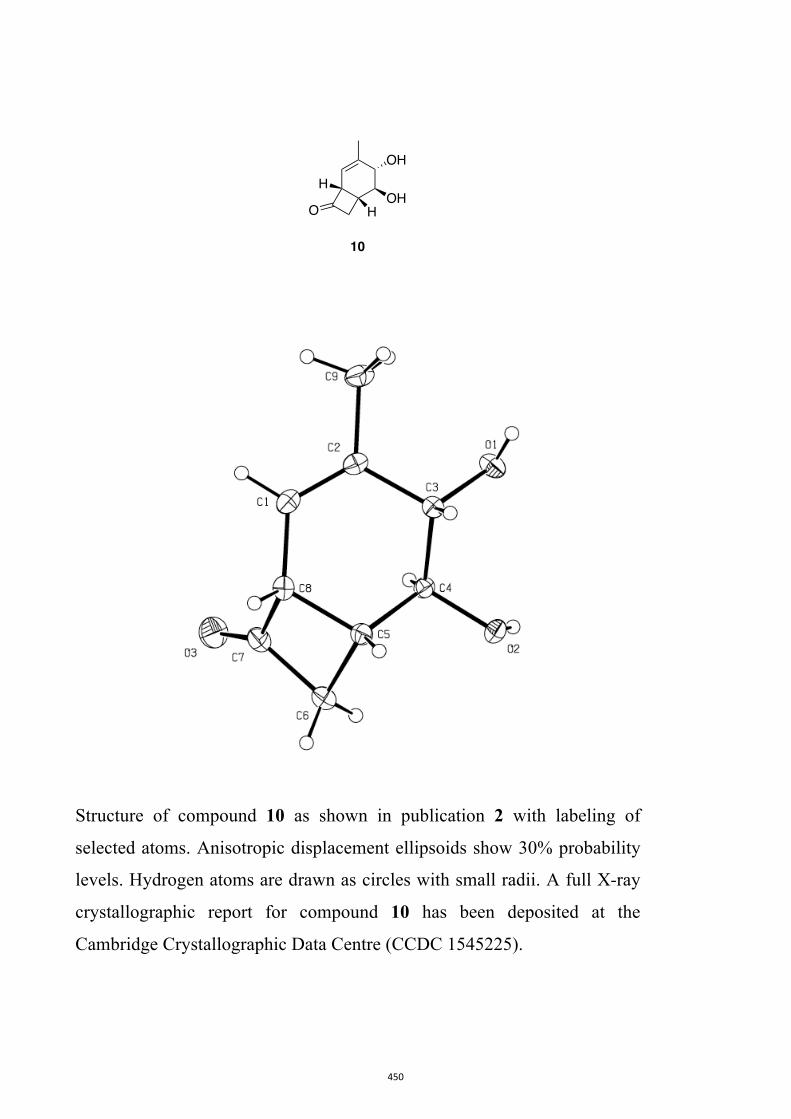
Structure of compound 10 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 10 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545225).
H
O H
10
OH
OH
450
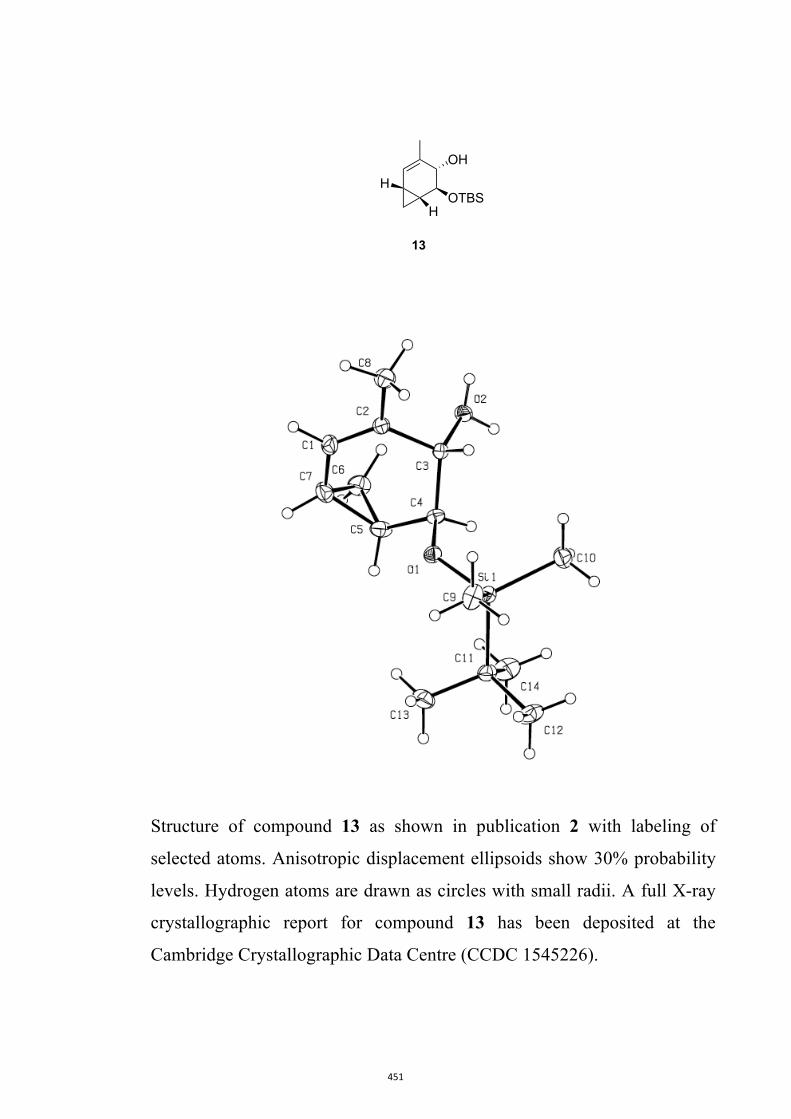
Structure of compound 13 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 13 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545226).
H
H
13
OH
OTBS
451
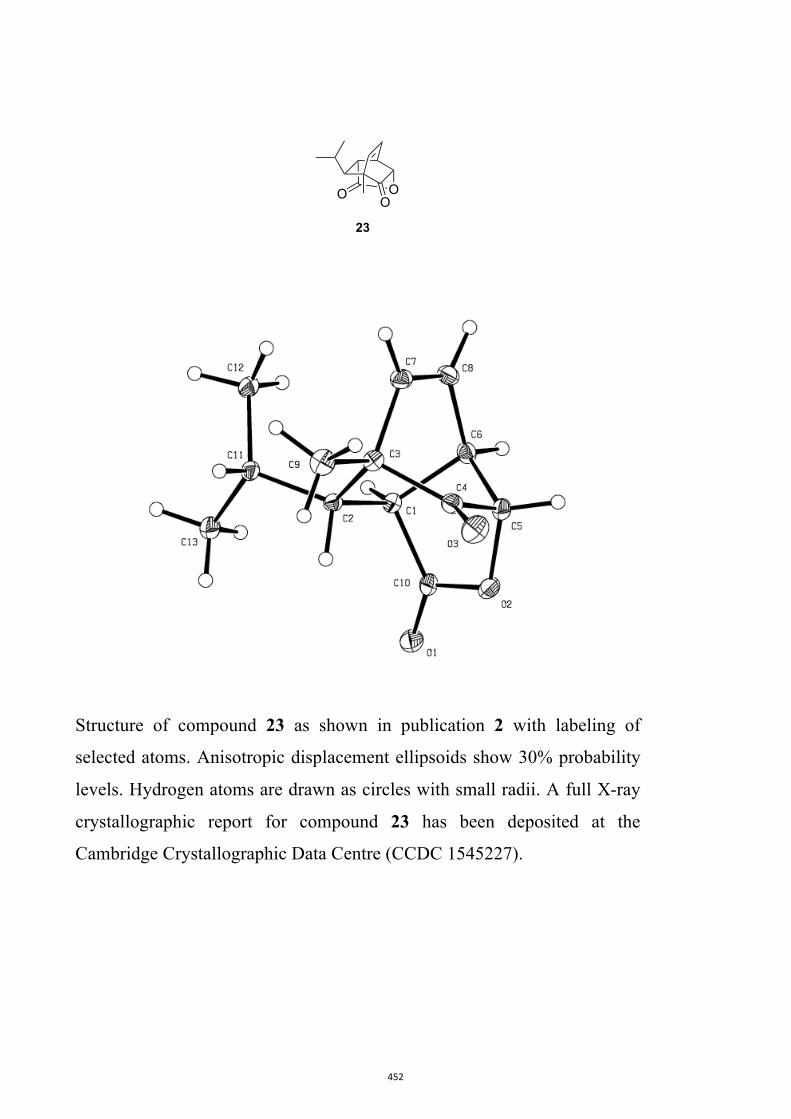
Structure of compound 23 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 23 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545227).
OO O
23
452
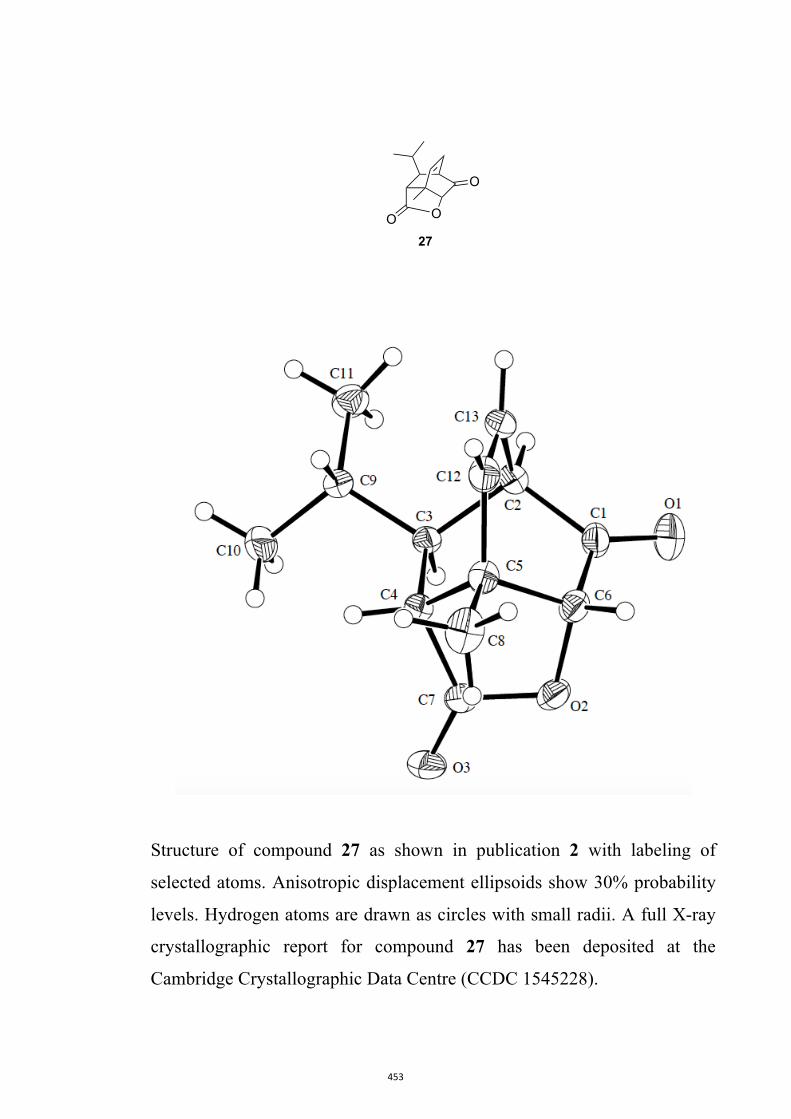
Structure of compound 27 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 27 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545228).
OO
O
27
453
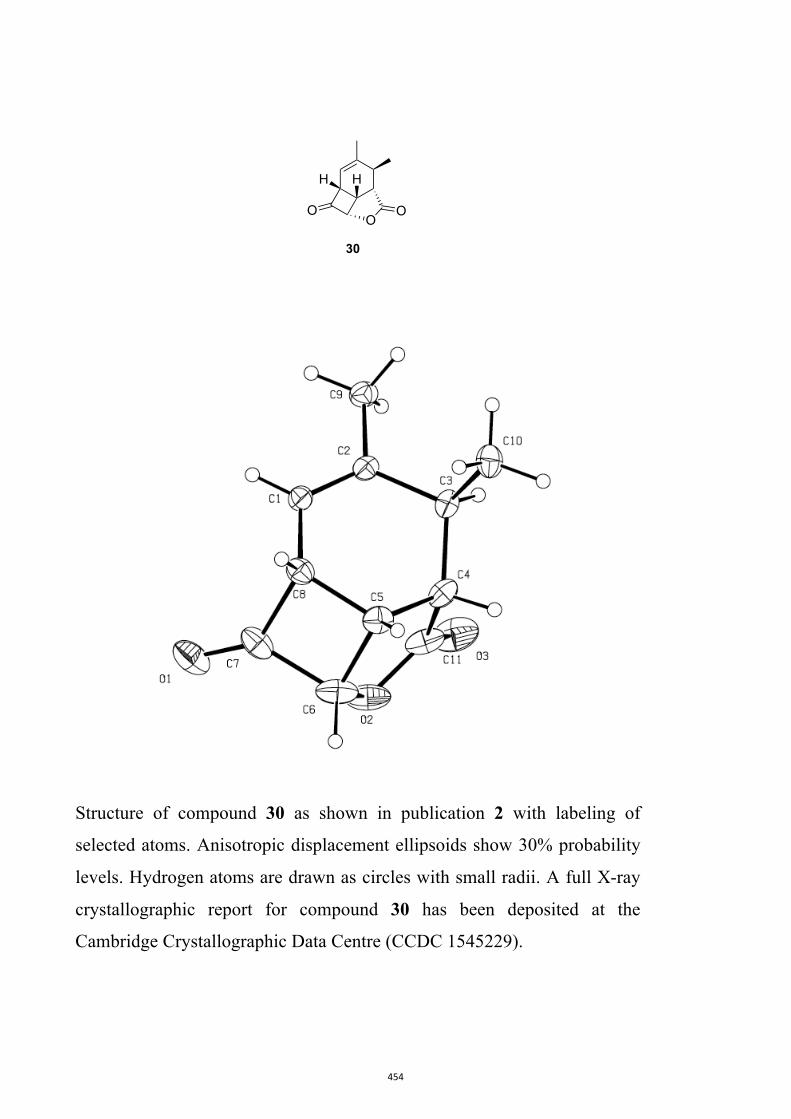
Structure of compound 30 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 30 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545229).
30
O
H H
OO
454
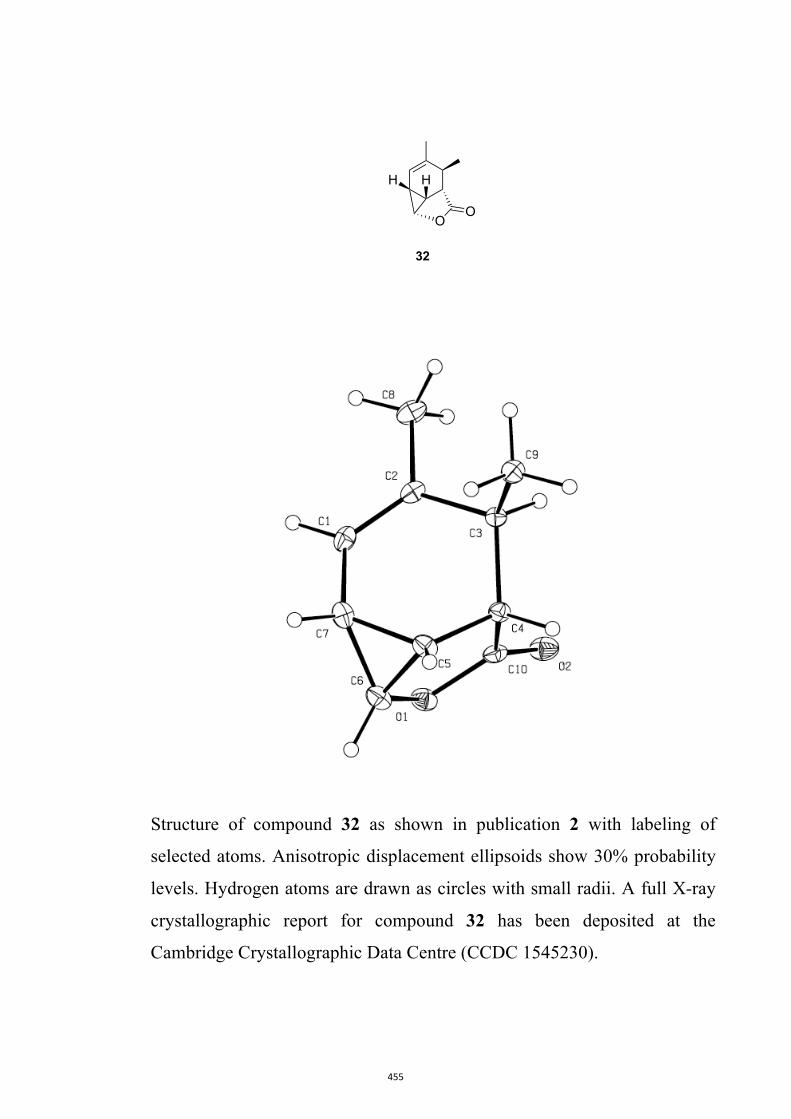
Structure of compound 32 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 32 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545230).
32
H H
OO
455
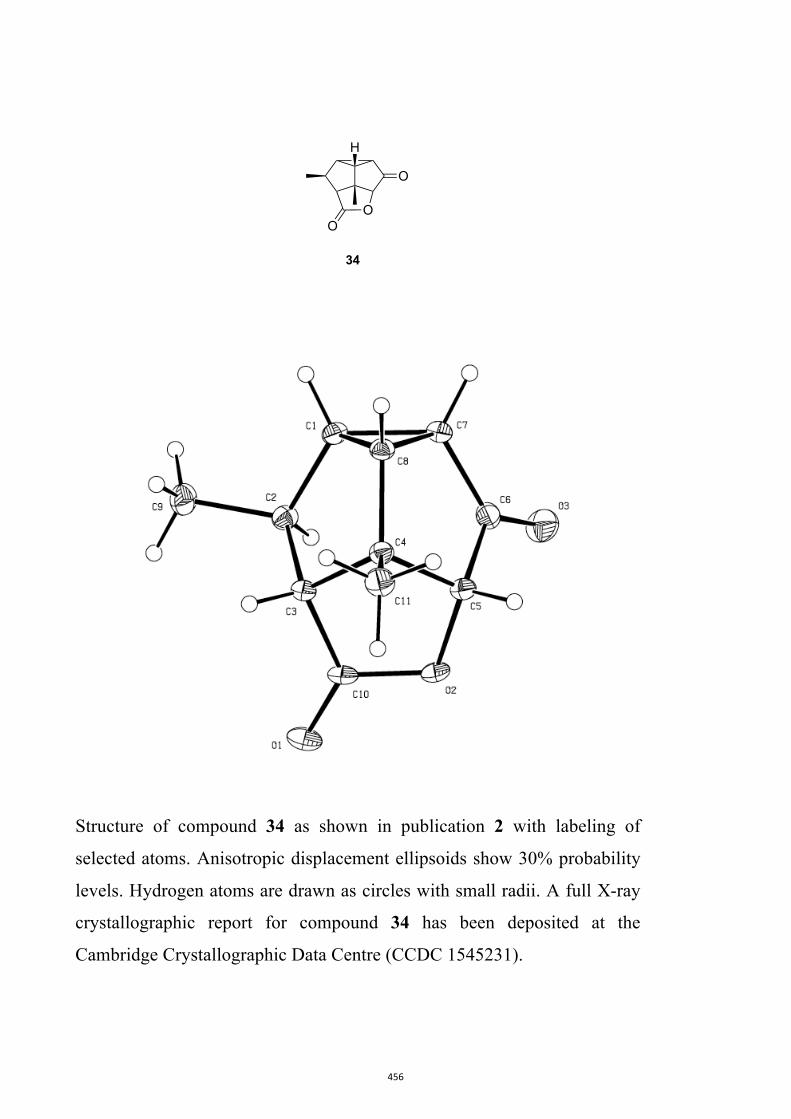
Structure of compound 34 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 34 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545231).
O
H
OO
34
456
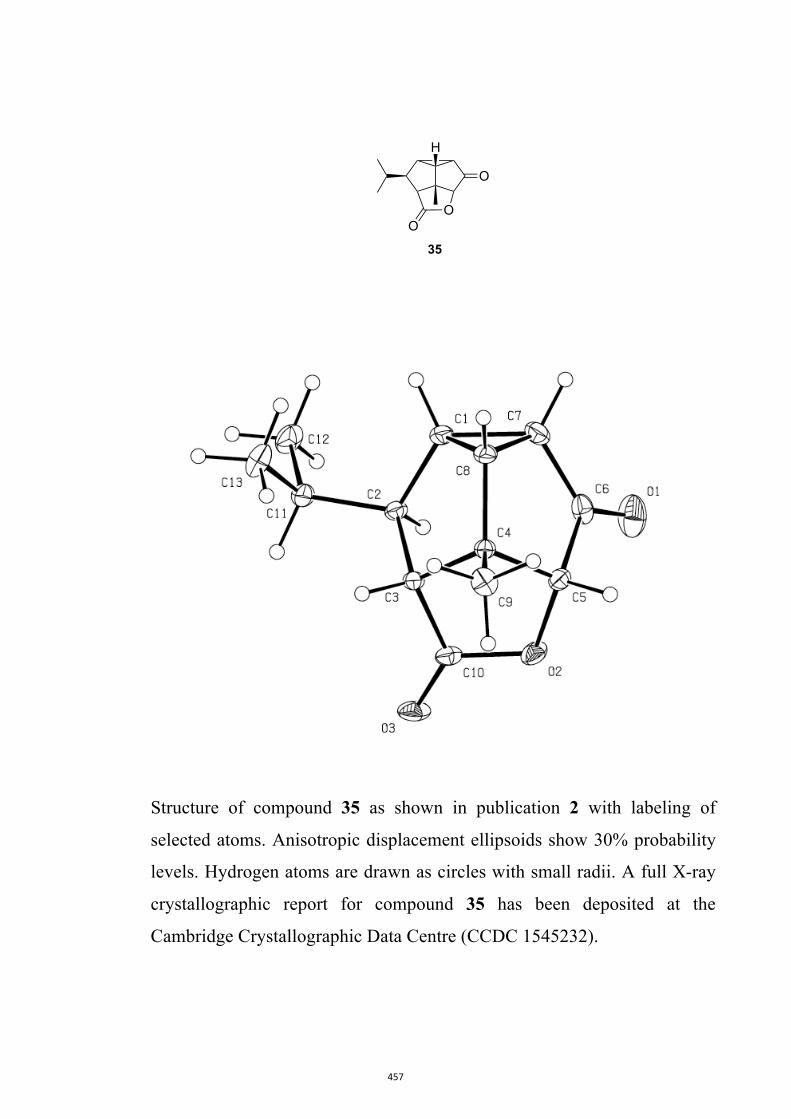
Structure of compound 35 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 35 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545232).
O
H
OO
35
457
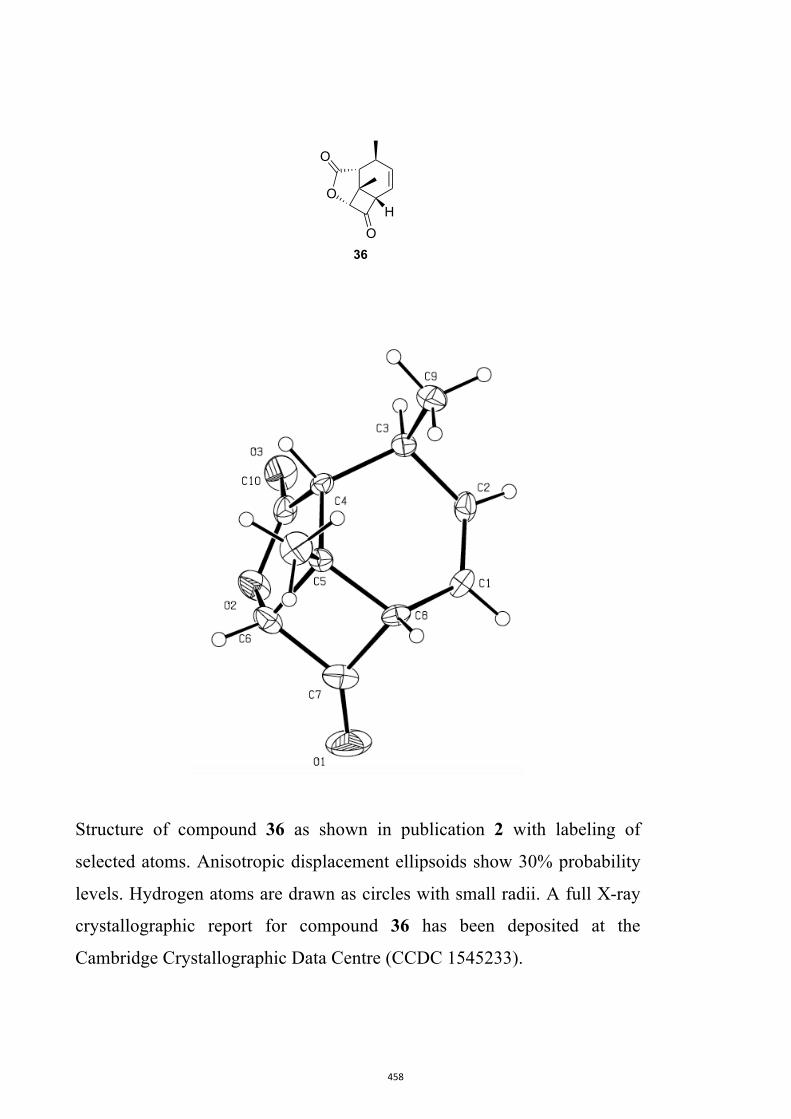
Structure of compound 36 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 36 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545233).
HO
O
O
36
458
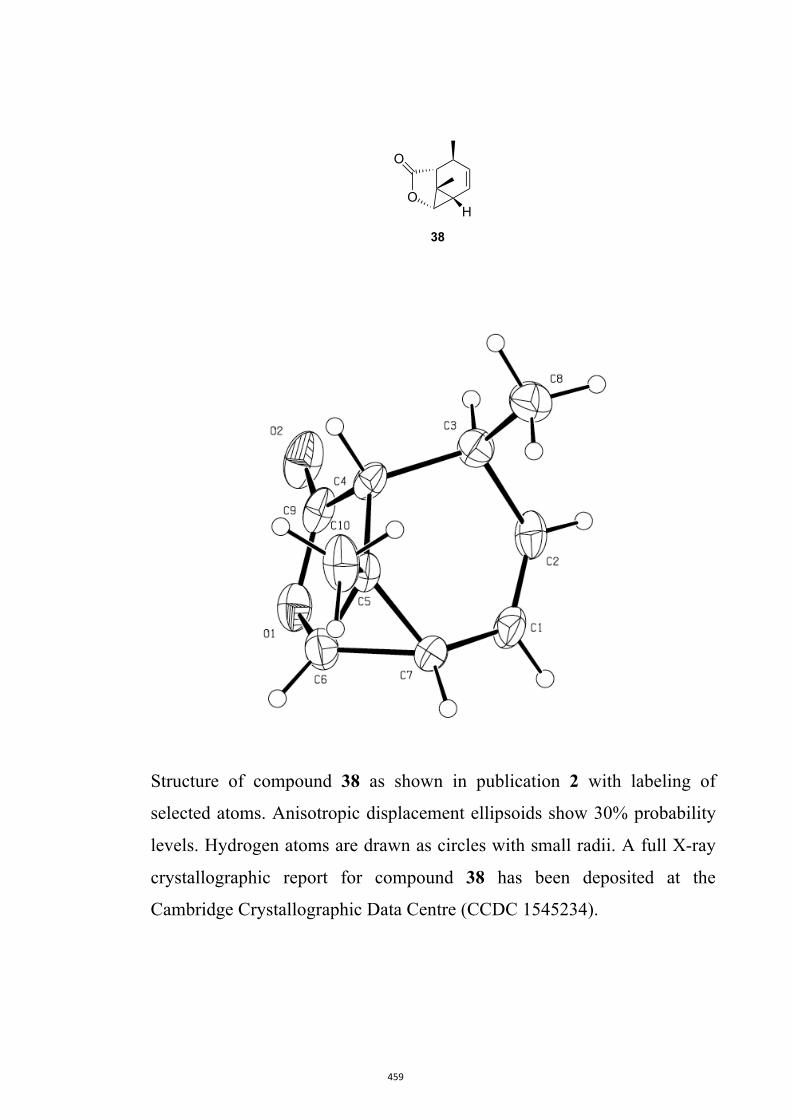
Structure of compound 38 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 38 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545234).
HO
O
38
459
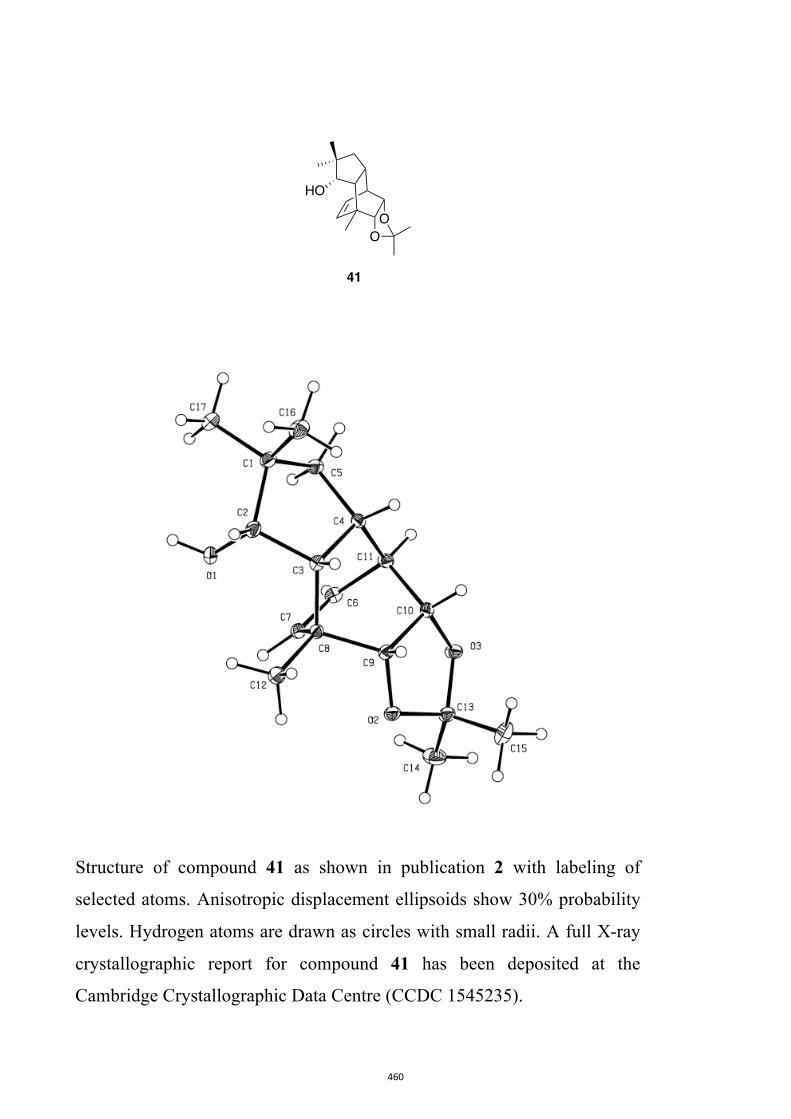
Structure of compound 41 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 41 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545235).
41
OO
HO
460
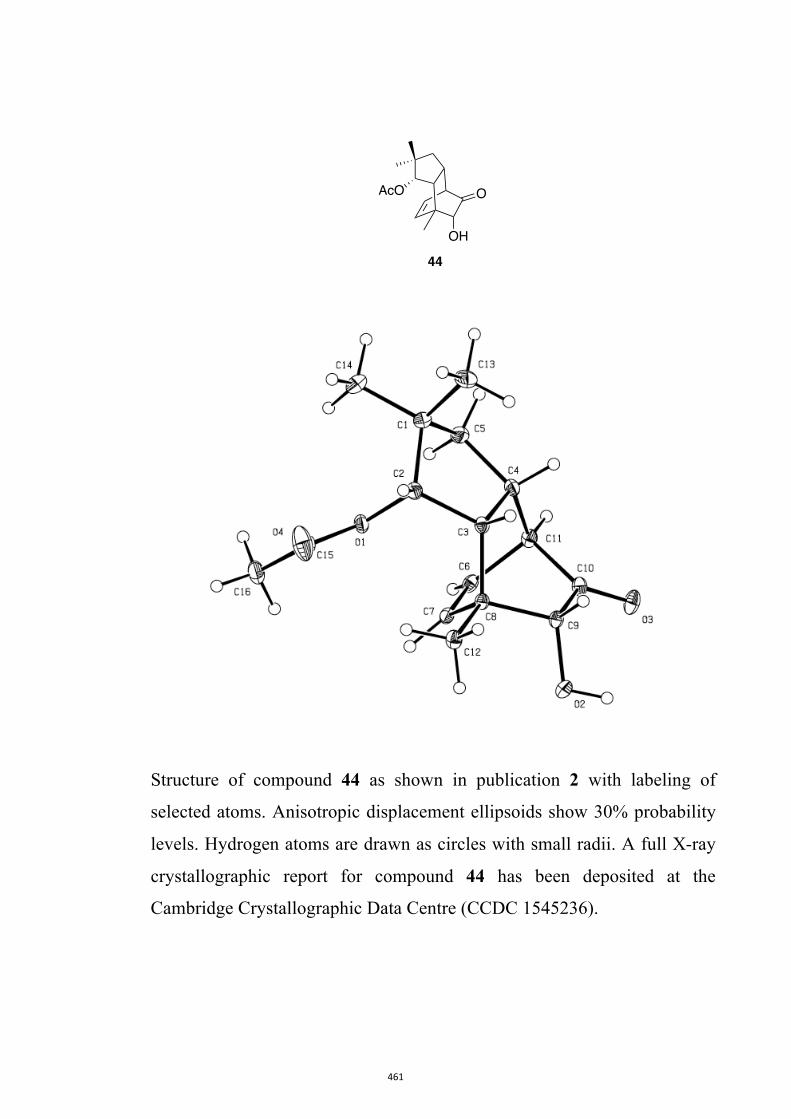
Structure of compound 44 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 44 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545236).
OAcO
OH
44
461
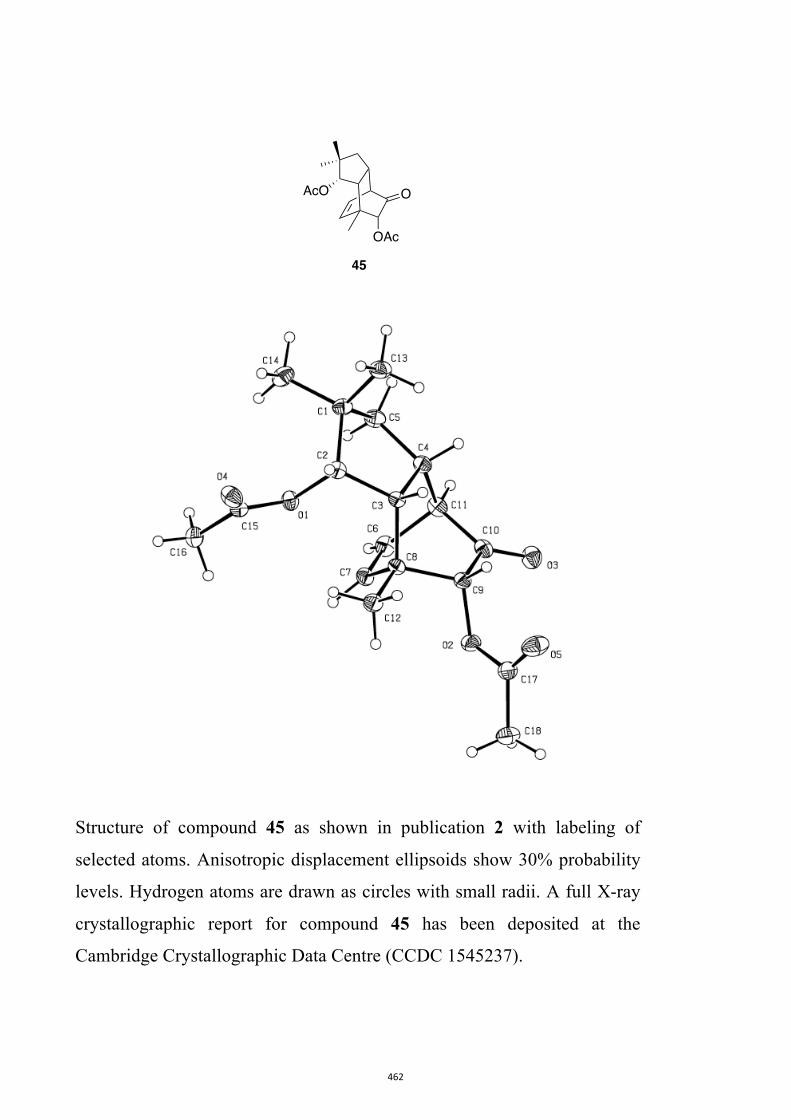
Structure of compound 45 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 45 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545237).
OAcO
OAc
45
462
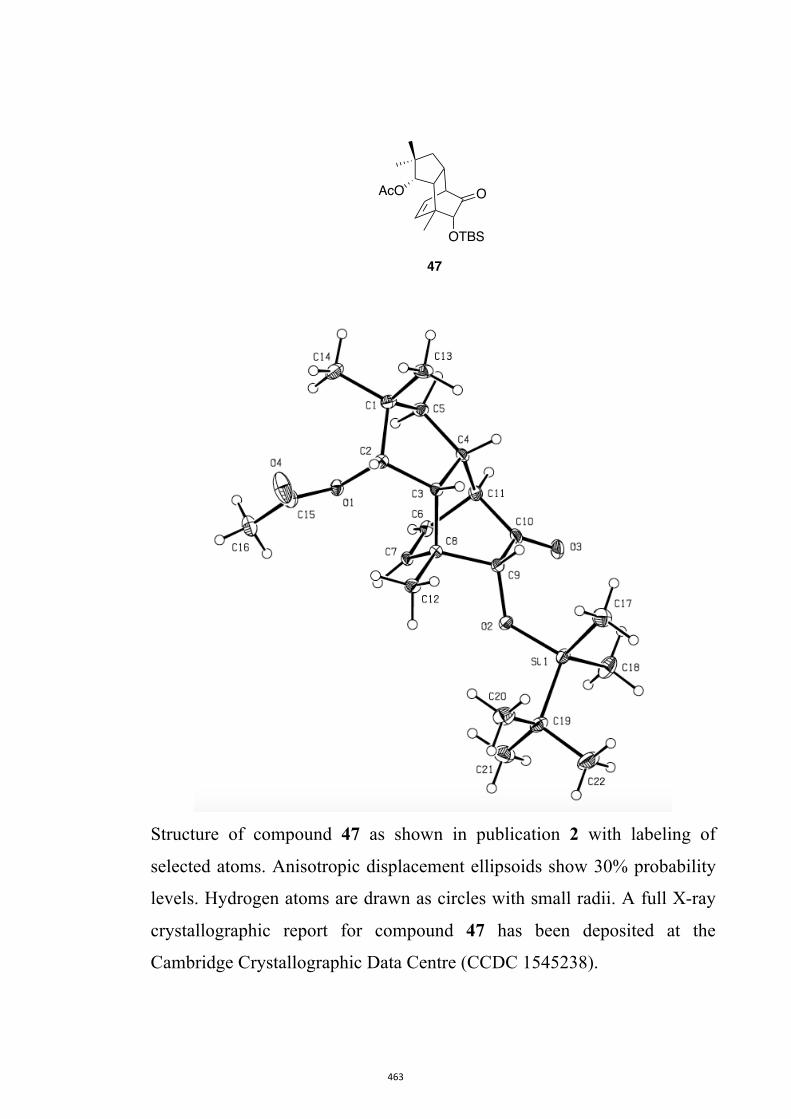
Structure of compound 47 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 47 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545238).
OAcO
OTBS
47
463
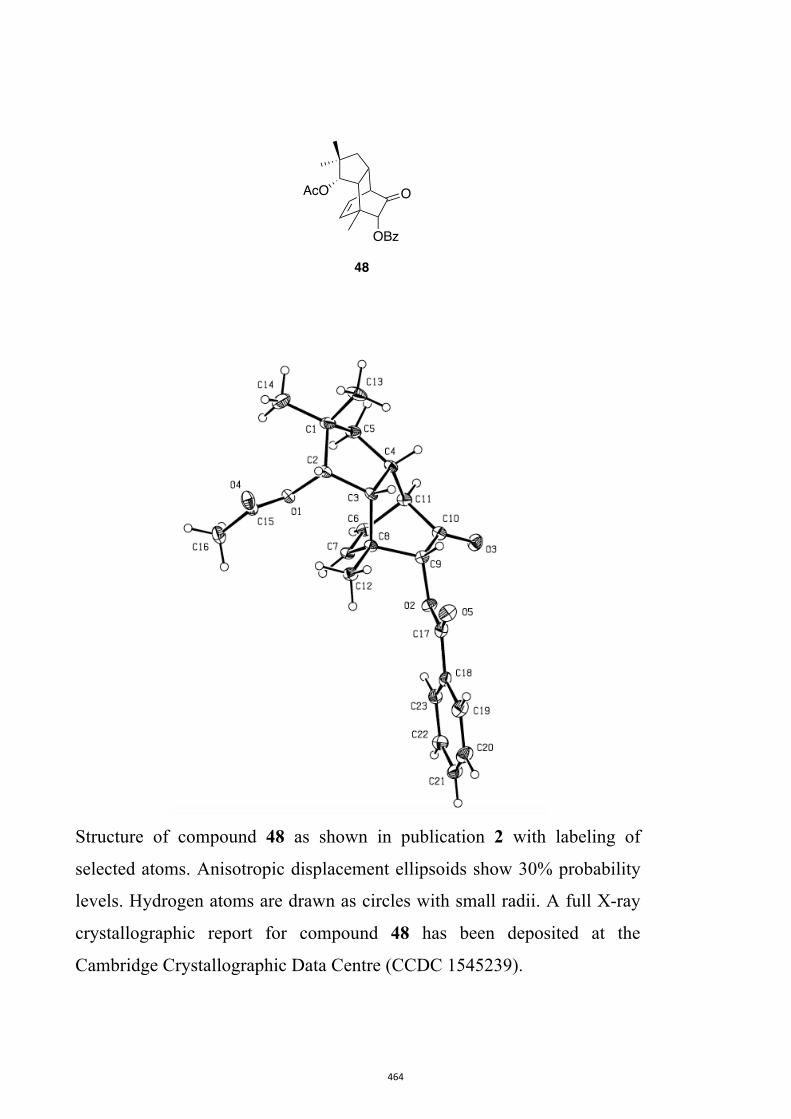
Structure of compound 48 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 48 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545239).
OAcO
OBz
48
464
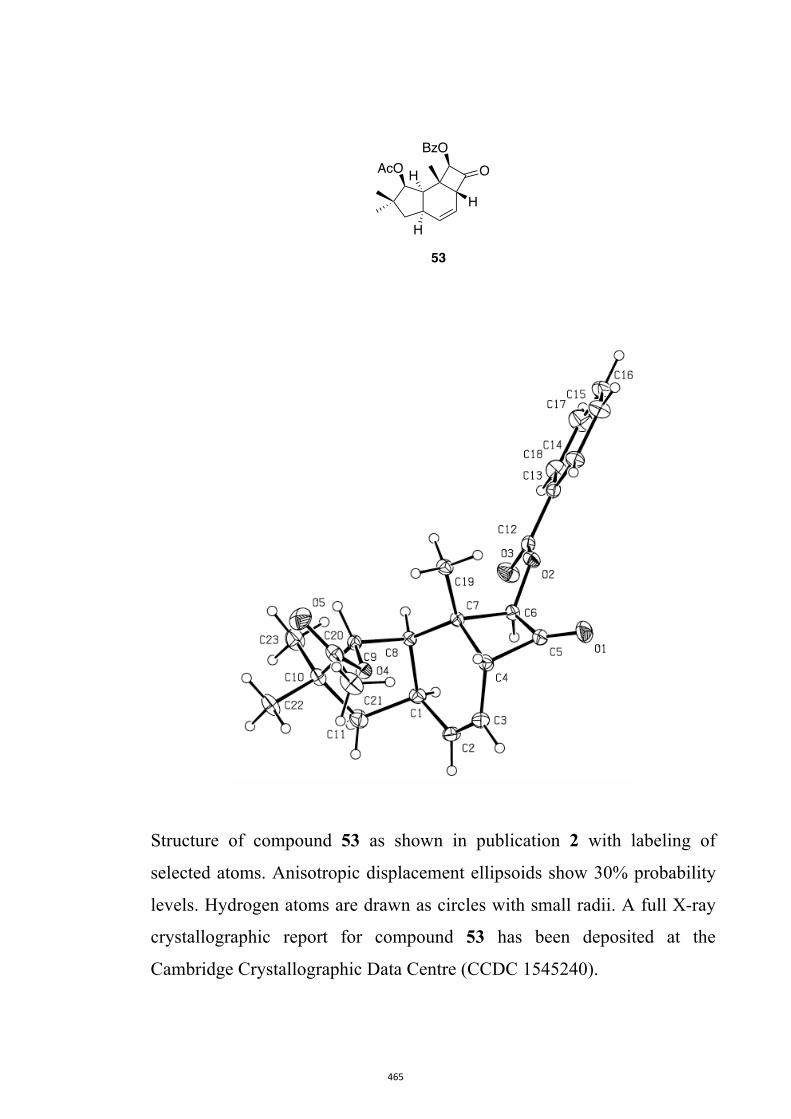
Structure of compound 53 as shown in publication 2 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 53 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1545240).
H
H
H
OBzO
AcO
53
465
Structure of compound 1 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 1 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525166).
NH
1
466
Structure of compound 2 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 2 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525163).
NH
2
467
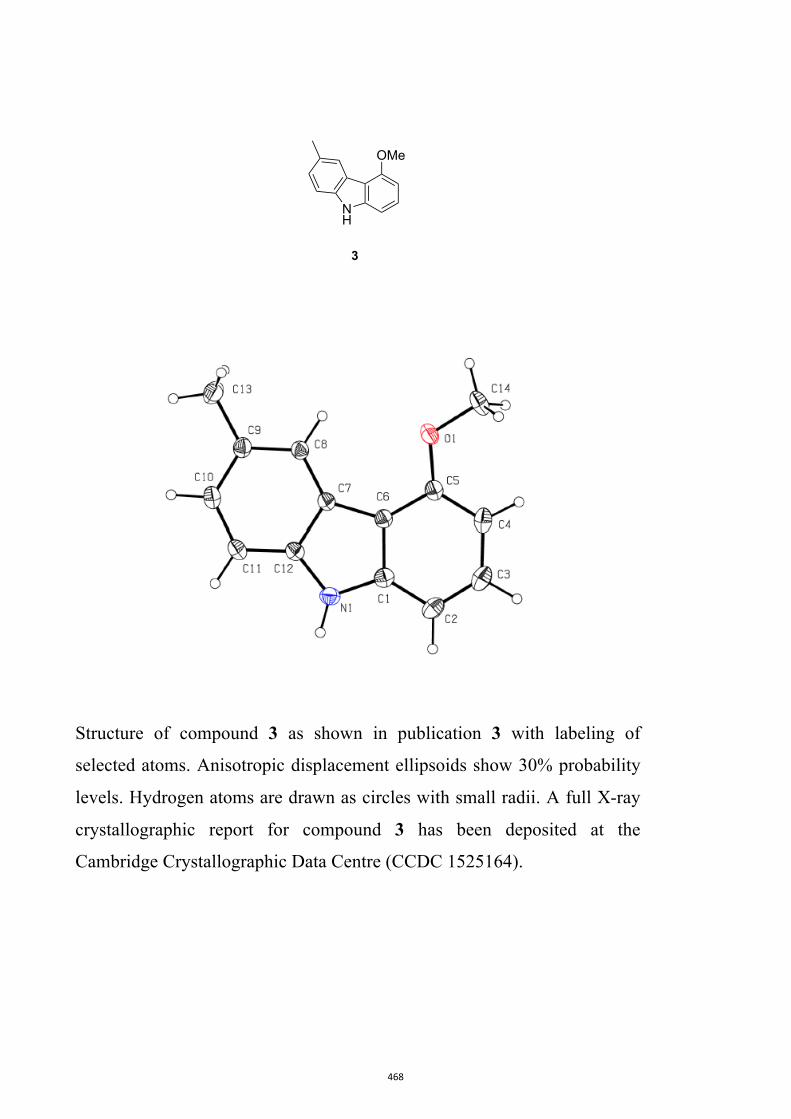
Structure of compound 3 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 3 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525164).
NH
3
OMe
468
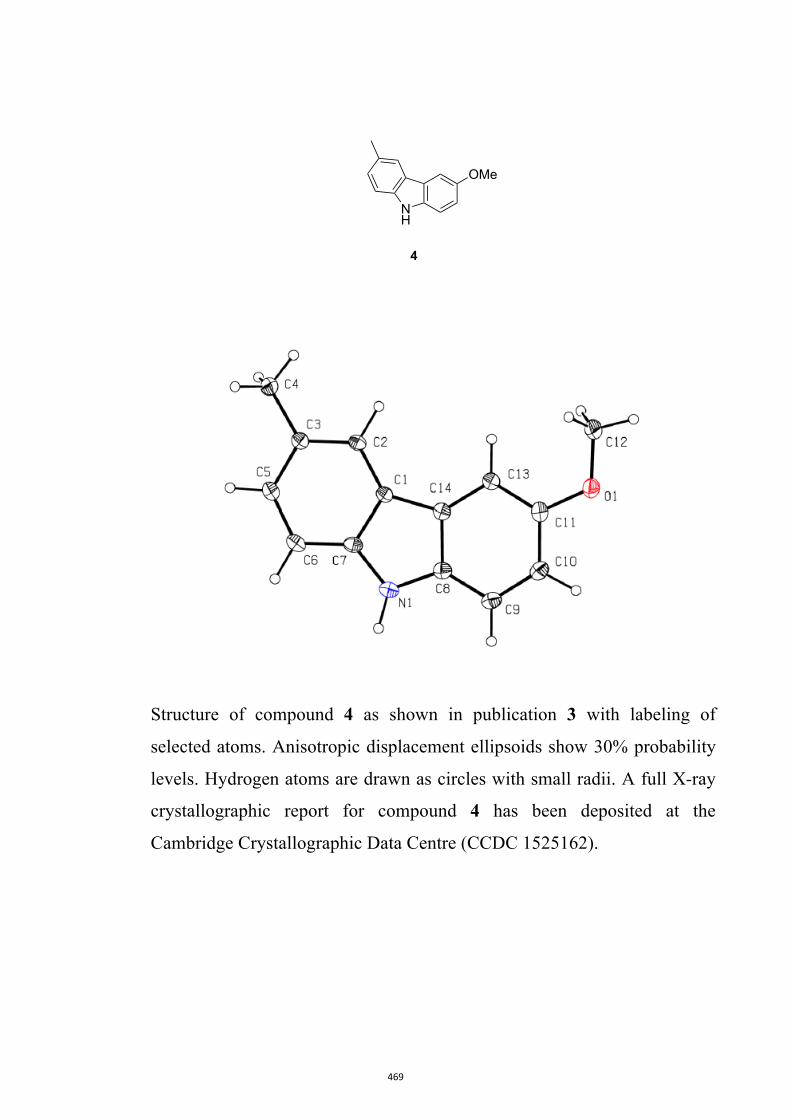
Structure of compound 4 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 4 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525162).
NH
4
OMe
469
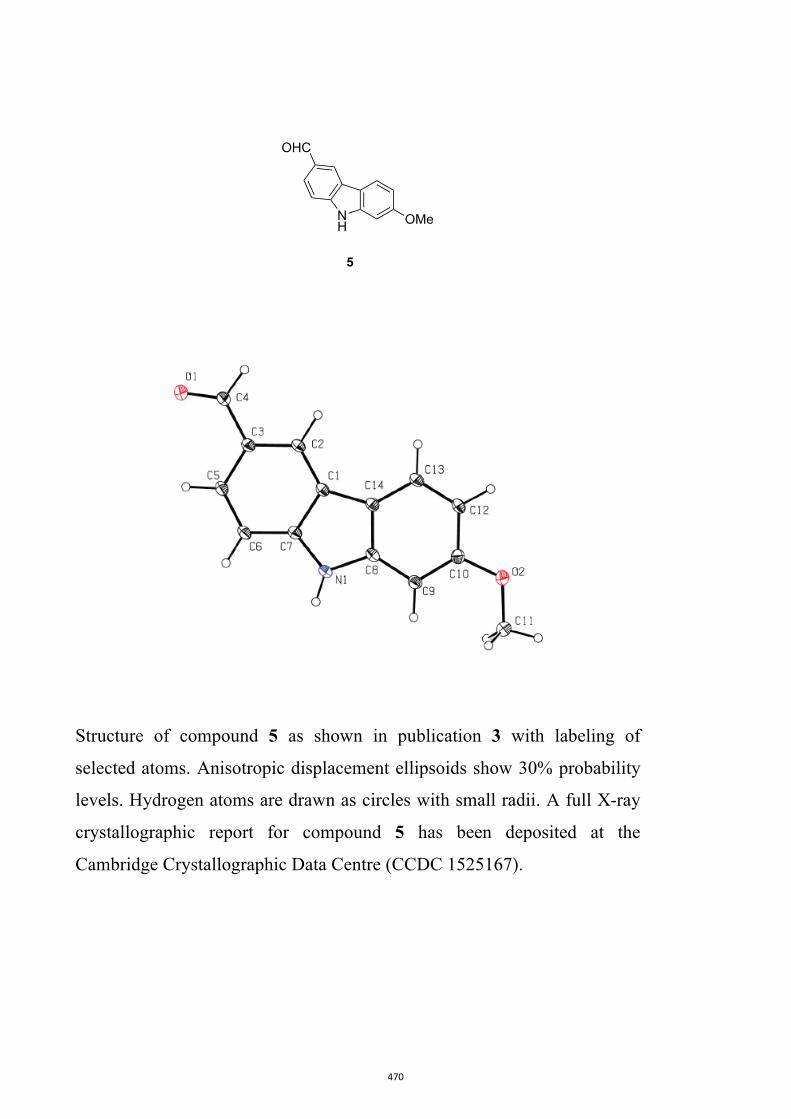
Structure of compound 5 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 5 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525167).
NH
5
OHC
OMe
470
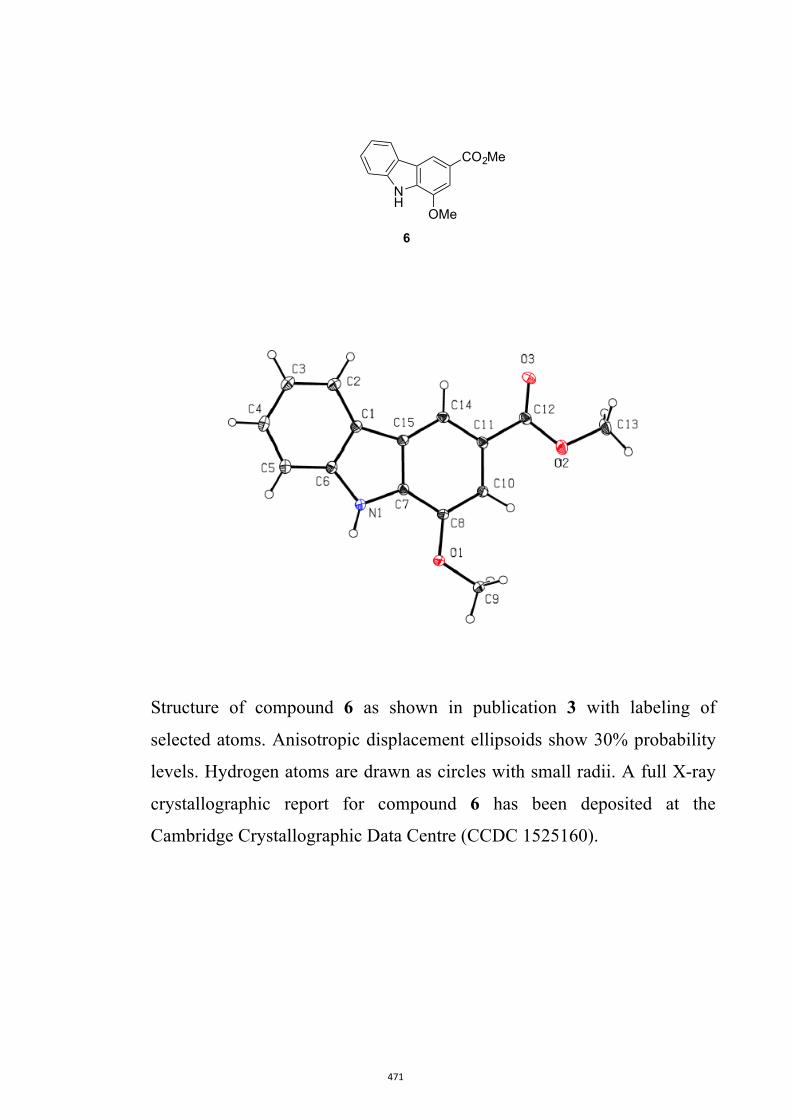
Structure of compound 6 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 6 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525160).
NH
6
CO2Me
OMe
471
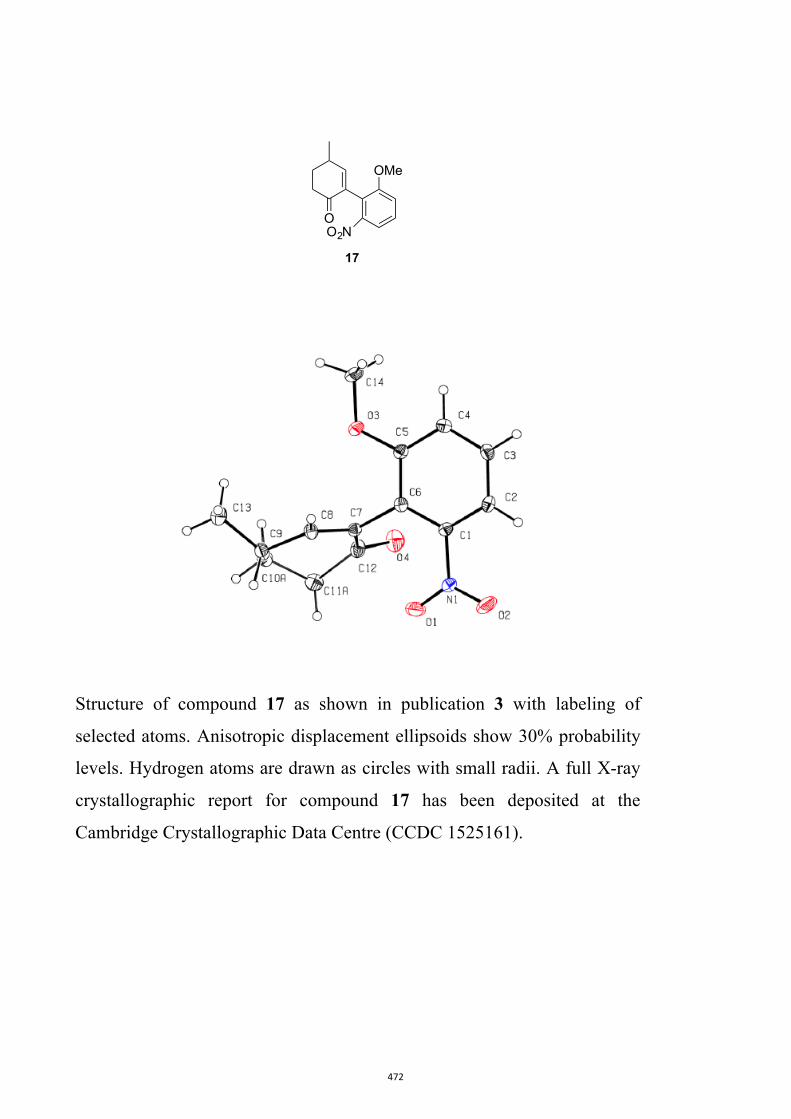
Structure of compound 17 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 17 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525161).
OO2N
17
OMe
472
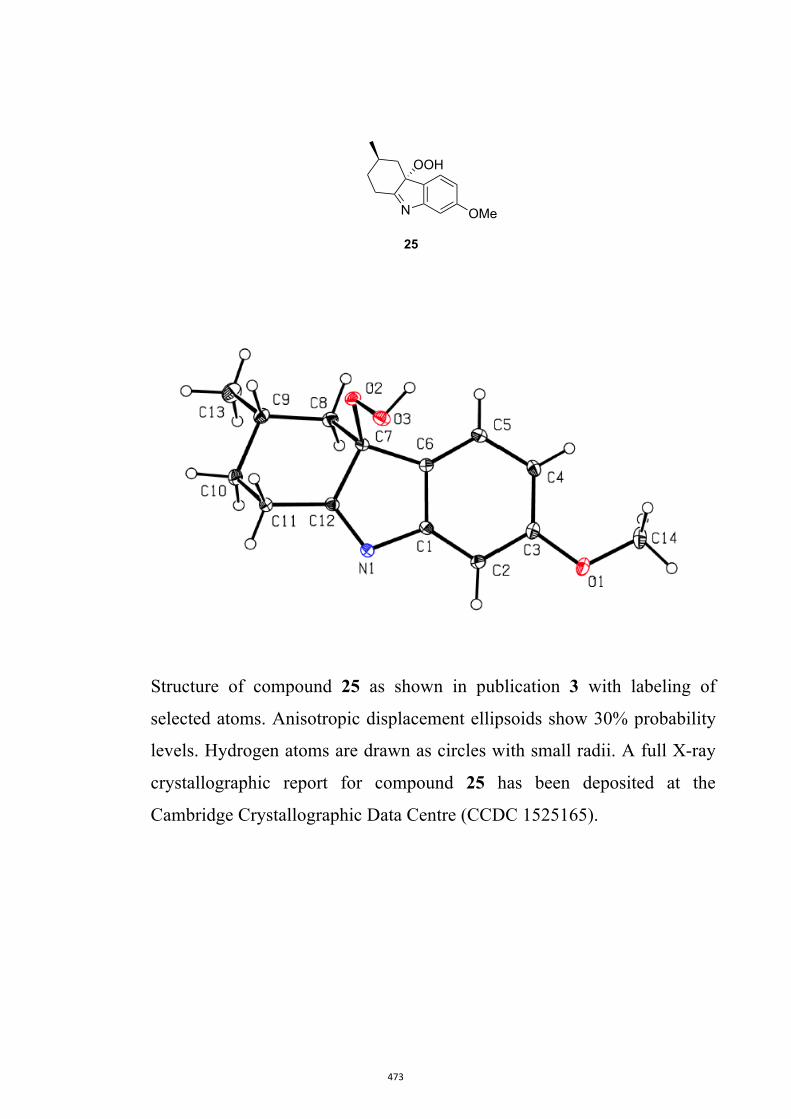
Structure of compound 25 as shown in publication 3 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 25 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1525165).
N
25
OMe
OOH
473
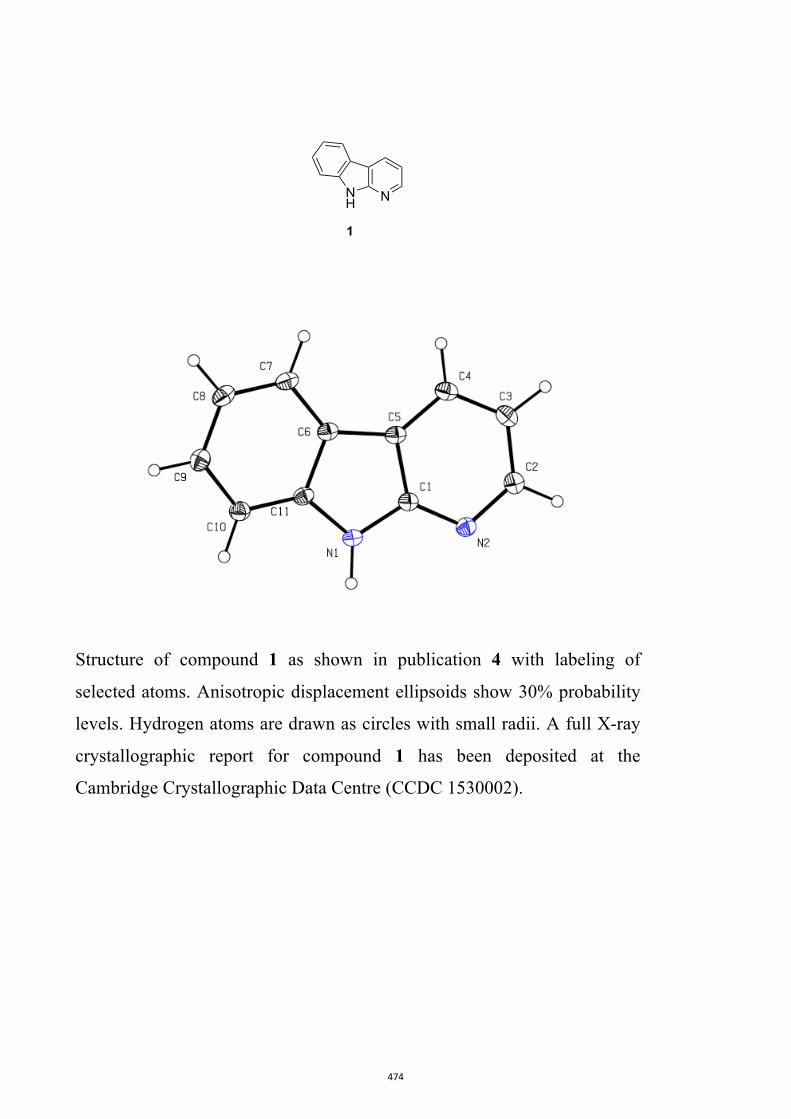
Structure of compound 1 as shown in publication 4 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 1 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1530002).
1
NNH
474
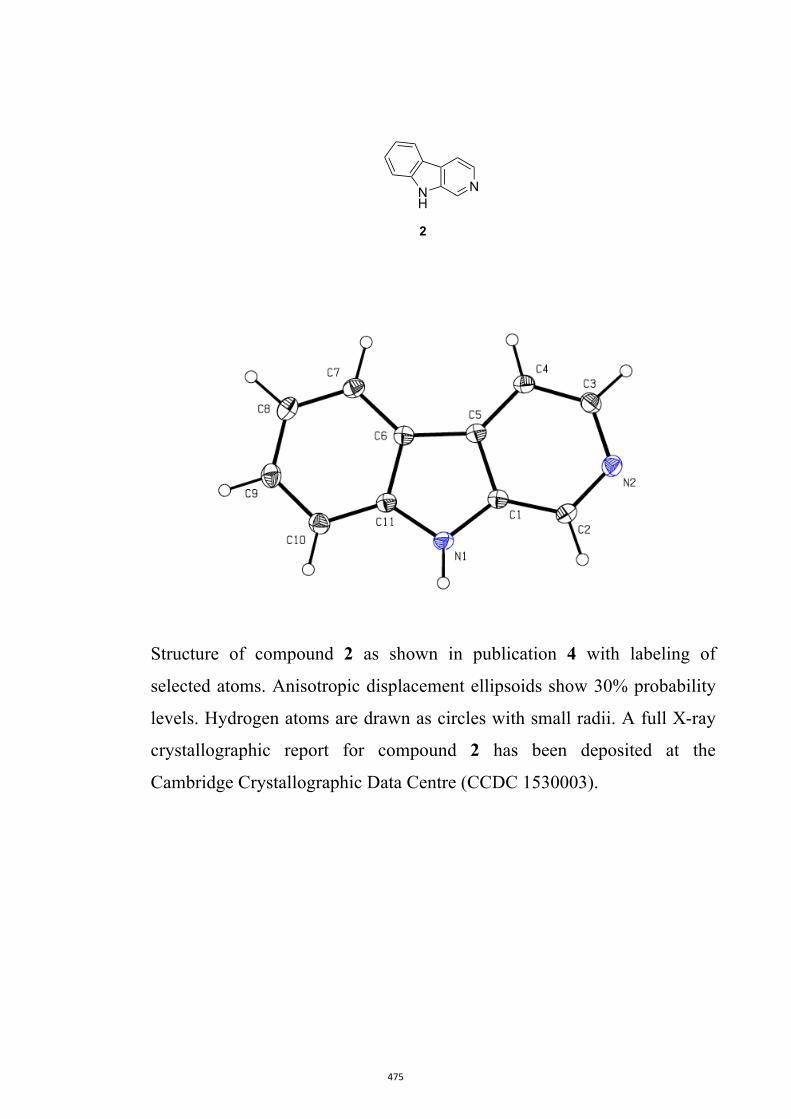
Structure of compound 2 as shown in publication 4 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 2 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1530003).
2
NNH
475
Structure of compound 3 as shown in publication 4 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 3 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1530004).
3
N
NH
476
Structure of compound 4 as shown in publication 4 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 4 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1530005).
4
N
NH
477
Structure of compound 5 as shown in publication 4 with labeling of
selected atoms. Anisotropic displacement ellipsoids show 30% probability
levels. Hydrogen atoms are drawn as circles with small radii. A full X-ray
crystallographic report for compound 5 has been deposited at the
Cambridge Crystallographic Data Centre (CCDC 1530006).
5
NNH
478
Related Documents











