TGFb 1 represses proliferation of pancreatic carcinoma cells which correlates with Smad4-independent inhibition of ERK activation K Giehl 1 , B Seidel 2 , P Gierschik 1 , G Adler 2 and A Menke* ,2 1 Department of Pharmacology and Toxicology, University of Ulm, D-89070 Ulm, Germany; 2 Department of Internal Medicine I, University of Ulm, D-89070 Ulm, Germany Transforming growth factor beta (TGFb) is a tumor suppressor acting as inhibitor of cell cycle progression of epithelial cells. We show that treatment of the pancreatic carcinoma cell lines PANC-1 and BxPC-3 with TGFb 1 inhibits both growth factor-induced activation of the extracellular signal-regulated kinase 2 (ERK2) and translocation of the kinase to the nucleus. TGFb 1 causes a concentration-dependent reduction of cell proliferation in both cell lines. By measuring ERK activation, we can show that TGFb 1 is able to repress ERK activation induced by mitogenic stimuli such as EGF. This inhibitory eect of TGFb 1 is not mediated by suppres- sion of Ras or c-Raf-1 activation, but mediated by TGFb 1 -induced activation of a serine-threonine phospha- tase, as demonstrated by inhibition of phosphatases by treatment with okadaic acid. Results obtained in the Smad4-deficient pancreatic carcinoma cell line BxPC-3, demonstrate that TGFb 1 -induced growth inhibition is mediated by a Smad4-independent prevention of ERK2 activation. In contrast to the eects of TGFb 1 on epithelial cells, mesenchymal NIH3T3 fibroblasts exhibit elevated ERK2 activation and increased cell proliferation in response to TGFb 1 treatment. Smad4-independent phosphatase-mediated inhibition of mitogen-activated ERK2 represents a novel eector pathway contributing to suppression of epithelial pancreatic carcinoma cell proliferation by TGFb 1 , in addition to the well-known Smad-induced tumor suppressor activity of TGFb. Oncogene (2000) 19, 4531 – 4541. Keywords: TGFb; EGF; ERK2; proliferation control Introduction TGFb superfamily members are involved in multiple, mainly cell type-dependent cellular functions aecting dierentiation, cell survival, and cell proliferation (Massague, 1990). In epithelial cells, TGFbs cause a reversible cell growth arrest in G1 phase (Polyak, 1996; Sporn and Roberts, 1992). Growth inhibitory, im- munosuppressive, and apoptotic eects, characterize TGFb as a tumor suppressor. Naturally occurring inactivating mutations in TGFb receptors in human tumors support such a role for TGFb (Massague, 1998). TGFb 1 represents the prototype of the transforming growth factor b superfamily. The ligand binds to the type II receptor (TGFbRII), which heterodimerizes with a type I receptor. As a consequence, the type I protein is phosphorylated at serine/threonine residues by the constitutive active kinase-domain of the type II receptor, thereby inducing a number of intracellular signals (Attisano et al., 1993; Ebner et al., 1993; Franzen et al., 1993; recently reviewed in Piek et al., 1999). The best characterized signal pathway mediated by TGFb is the phosphorylation and activation of Smad2/3 proteins (Heldin et al., 1997; Heldin and Ostman, 1996). Activated Smad2/3 bind to Smad4, translocates into the nucleus, and regulates gene expression (Attisano and Wrana, 1998). Subsequently, expression of cyclic D1 and cdk4, which are essential for cell cycle progression, is downregulated (Ewen et al., 1993; Ko et al., 1998), whereas transcription of genes coding for collagen and other extracellular matrix proteins is enhanced (Roberts et al., 1990). Moreover, TGFbRII seems to cooperate directly with cell cycle proteins, such as cyclin B, representing an additional way to inhibit cell proliferation (Liu et al., 1999). In addition to directly aecting cyclin and cyclin- dependent kinases, TGFb receptors are binding partners for other proteins with signaling properties, such as the immunophilin FKBP-12 (Chen et al., 1997) or the a subunit of farnesylprotein transferase (FTa). Upon receptor activation FTa is phosphorylated by type I receptor. FTa is able to modify a series of molecules including the small GTPase Ras (Kawabata et al., 1995; Wang et al., 1996). The Ras-Raf-ERK/MAPK-cascade is one major signaling pathway regulating cell proliferation in many cell types (Lenormand et al., 1993; Pages et al., 1993). Binding of growth factors, like EGF, FGF, or HGF to their corresponding receptor tyrosine kinases results in association of the receptor with adaptor proteins Grb2 and Sos, followed by activation of the small GTPase Ras. GTP-bound Ras catalyzes the translocation of Raf to the plasma membrane, where the activated kinase phosphorylates the MAP kinase kinase MEK. MEK in turn phosphorylates the extracellular signal- regulated kinases ERK1 and ERK2 (Seger and Krebs, 1995). Activated ERKs translocate into the nucleus and stimulate gene expression by triggering transcrip- tion factors like ELK1 and ATF-2 (Brunet et al., 1999; Chen et al., 1992; Zheng and Guan, 1994). The ability of activation ERK kinases to phosphorylate Smads and thereby inactivating TGFb- or BMP-induced gene expression was recently described by Kretzschmar and coworkers (Kretzschmar et al., 1997, 1999). These results suggest a crosstalk between Ras/Raf/ERK- and TGFb-mediated proliferation control via Smad. Our results obtained in pancreatic carcinoma cell lines show that TGFb regulates cell cycle progression Oncogene (2000) 19, 4531 – 4541 ª 2000 Macmillan Publishers Ltd All rights reserved 0950 – 9232/00 $15.00 www.nature.com/onc *Correspondence: A Menke Received 20 January 2000; revised 14 July 2000; accepted 17 July 2000

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

TGFb1 represses proliferation of pancreatic carcinoma cells whichcorrelates with Smad4-independent inhibition of ERK activation
K Giehl1, B Seidel2, P Gierschik1, G Adler2 and A Menke*,2
1Department of Pharmacology and Toxicology, University of Ulm, D-89070 Ulm, Germany; 2Department of Internal Medicine I,University of Ulm, D-89070 Ulm, Germany
Transforming growth factor beta (TGFb) is a tumorsuppressor acting as inhibitor of cell cycle progression ofepithelial cells. We show that treatment of the pancreaticcarcinoma cell lines PANC-1 and BxPC-3 with TGFb1inhibits both growth factor-induced activation of theextracellular signal-regulated kinase 2 (ERK2) andtranslocation of the kinase to the nucleus. TGFb1 causesa concentration-dependent reduction of cell proliferationin both cell lines. By measuring ERK activation, we canshow that TGFb1 is able to repress ERK activationinduced by mitogenic stimuli such as EGF. Thisinhibitory e�ect of TGFb1 is not mediated by suppres-sion of Ras or c-Raf-1 activation, but mediated byTGFb1-induced activation of a serine-threonine phospha-tase, as demonstrated by inhibition of phosphatases bytreatment with okadaic acid. Results obtained in theSmad4-de®cient pancreatic carcinoma cell line BxPC-3,demonstrate that TGFb1-induced growth inhibition ismediated by a Smad4-independent prevention of ERK2activation. In contrast to the e�ects of TGFb1 onepithelial cells, mesenchymal NIH3T3 ®broblasts exhibitelevated ERK2 activation and increased cell proliferationin response to TGFb1 treatment. Smad4-independentphosphatase-mediated inhibition of mitogen-activatedERK2 represents a novel e�ector pathway contributingto suppression of epithelial pancreatic carcinoma cellproliferation by TGFb1, in addition to the well-knownSmad-induced tumor suppressor activity of TGFb.Oncogene (2000) 19, 4531 ± 4541.
Keywords: TGFb; EGF; ERK2; proliferation control
Introduction
TGFb superfamily members are involved in multiple,mainly cell type-dependent cellular functions a�ectingdi�erentiation, cell survival, and cell proliferation(Massague, 1990). In epithelial cells, TGFbs cause areversible cell growth arrest in G1 phase (Polyak, 1996;Sporn and Roberts, 1992). Growth inhibitory, im-munosuppressive, and apoptotic e�ects, characterizeTGFb as a tumor suppressor. Naturally occurringinactivating mutations in TGFb receptors in humantumors support such a role for TGFb (Massague,1998).
TGFb1 represents the prototype of the transforminggrowth factor b superfamily. The ligand binds to thetype II receptor (TGFbRII), which heterodimerizes
with a type I receptor. As a consequence, the type Iprotein is phosphorylated at serine/threonine residuesby the constitutive active kinase-domain of the type IIreceptor, thereby inducing a number of intracellularsignals (Attisano et al., 1993; Ebner et al., 1993;Franzen et al., 1993; recently reviewed in Piek et al.,1999). The best characterized signal pathway mediatedby TGFb is the phosphorylation and activation ofSmad2/3 proteins (Heldin et al., 1997; Heldin andOstman, 1996). Activated Smad2/3 bind to Smad4,translocates into the nucleus, and regulates geneexpression (Attisano and Wrana, 1998). Subsequently,expression of cyclic D1 and cdk4, which are essentialfor cell cycle progression, is downregulated (Ewen etal., 1993; Ko et al., 1998), whereas transcription ofgenes coding for collagen and other extracellularmatrix proteins is enhanced (Roberts et al., 1990).Moreover, TGFbRII seems to cooperate directly withcell cycle proteins, such as cyclin B, representing anadditional way to inhibit cell proliferation (Liu et al.,1999).
In addition to directly a�ecting cyclin and cyclin-dependent kinases, TGFb receptors are bindingpartners for other proteins with signaling properties,such as the immunophilin FKBP-12 (Chen et al., 1997)or the a subunit of farnesylprotein transferase (FTa).Upon receptor activation FTa is phosphorylated bytype I receptor. FTa is able to modify a series ofmolecules including the small GTPase Ras (Kawabataet al., 1995; Wang et al., 1996).
The Ras-Raf-ERK/MAPK-cascade is one majorsignaling pathway regulating cell proliferation in manycell types (Lenormand et al., 1993; Pages et al., 1993).Binding of growth factors, like EGF, FGF, or HGF totheir corresponding receptor tyrosine kinases results inassociation of the receptor with adaptor proteins Grb2and Sos, followed by activation of the small GTPaseRas. GTP-bound Ras catalyzes the translocation ofRaf to the plasma membrane, where the activatedkinase phosphorylates the MAP kinase kinase MEK.MEK in turn phosphorylates the extracellular signal-regulated kinases ERK1 and ERK2 (Seger and Krebs,1995). Activated ERKs translocate into the nucleusand stimulate gene expression by triggering transcrip-tion factors like ELK1 and ATF-2 (Brunet et al., 1999;Chen et al., 1992; Zheng and Guan, 1994). The abilityof activation ERK kinases to phosphorylate Smadsand thereby inactivating TGFb- or BMP-induced geneexpression was recently described by Kretzschmar andcoworkers (Kretzschmar et al., 1997, 1999). Theseresults suggest a crosstalk between Ras/Raf/ERK- andTGFb-mediated proliferation control via Smad.
Our results obtained in pancreatic carcinoma celllines show that TGFb regulates cell cycle progression
Oncogene (2000) 19, 4531 ± 4541ã 2000 Macmillan Publishers Ltd All rights reserved 0950 ± 9232/00 $15.00
www.nature.com/onc
*Correspondence: A MenkeReceived 20 January 2000; revised 14 July 2000; accepted 17 July2000

by a serine/threonine phosphatase-mediated inhibitionof growth factor-induced activation of ERK2. Thisinhibition results in reduction of serum-inducedproliferation of the epithelial pancreatic carcinoma celllines PANC-1 and BxPC-3. Since BxPC-3 cells arecharacterized by smad4-deletion, we conclude that inthese cells this inhibitory pathway represents amechanism independent of Smad4 activation, while inother cells it can operate in addition to Smad4-mediated growth inhibition. MIA PaCa-2 cells, whichlack a functional TGFb receptor type II, are notresponsive to TGFb1. Mesenchymal NIH3T3 ®bro-blasts react with increased proliferation and Ras/Raf/ERK activity to TGFb1 treatment.
Results
TGFb1-induced inhibition of epithelial cell proliferation
To analyse the in¯uence of TGFb1 on the proliferationof pancreatic carcinoma cell lines, DNA synthesis wasmeasured in the presence and absence of TGFb1.Treatment of PANC-1 and BxPC-3 cells with increas-ing amounts of TGFb1 (0.1 ± 10 ng/ml) resulted in aconcentration-dependent reduction of cell proliferationas measured by [3H]thymidine incorporation (Figure 1).Incubation of cells in growth medium containing 10%FCS and 10 ng/ml TGFb1 resulted in a 30% reductionof cell proliferation (quadruplicate values, n=5).PANC-1 cells were more sensitive to TGFb1 treatmentthan BxPC-3 cells, as demonstrated by the morepronounced e�ect of 0.1 and 1 ng/ml TGFb1 (Figure1). Quiescent PANC-1 cells (cultured for 36 h inDMEM+1% BSA) exhibited a more prominentgrowth inhibition in response to TGFb1 (45%, datanot shown). In contrast to PANC-1 and BxPC-3, MIAPaCa-2 cells, the third pancreatic carcinoma cell lineanalysed in this study, did not respond to TGFb1 evenwhen high concentrations of ligand were used.Proliferation of NIH3T3 ®broblasts, which were usedas a mesenchymal cell line, was not inhibited, butstimulated nearly twofold upon treatment with TGFb1(10 ng/ml). Addition of epidermal growth factor (EGF)(10 ng/ml) to serum-starved PANC-1 cells did notsigni®cantly change the thymidine incorporation, butstimulated the DNA synthesis more than ®vefold inNIH3T3 ®broblasts (data not shown).
Expression of TGFb1-associated signaling molecules
To characterize the signal transduction pathwayutilized by TGFb1, which accounts for the di�erente�ects in the analysed cell lines, we ®rst examined thepresence and abundance of the TGFb receptors type Iand II by Northern blot and immunoblot analyses.Receptor type speci®c Northern blot hybridizations(Figure 2a, left panel) revealed that both receptorproteins were expressed in PANC-1, BxPC-3 andNIH3T3 cells. The low signal intensity in NIH3T3cells may be due to the human speci®c cDNA probesused in these experiments. In contrast, MIA PaCa-2cells exhibited a distinct expression of TGFbRItranscript, but only a very low expression of TGFbRIImRNA with reduced molecular weight. These resultswere veri®ed on protein level by immunoblot analyses
of membrane fractions. For PANC-1, BxPC-3, andNIH3T3, TGFbRI and TGFbRII proteins were clearlydetectable. In contrast TGFbRI and TGFbRII werenot traceable in MIA PaCa-2 (Figure 2a, right panel).TGFbRII receptors were homogenously distributedover the cell membrane for PANC-1 and BxPC-3 cellsas revealed by immuno¯uorescence studies. There wasno signal for MIA PaCa-2 cells (data not shown).Thus, PANC-1, BxPC-3, and NIH3T3 cells areendowed with both receptor subtypes, while MIAPaCa-2 cells do not express the TGFbRII subtype,which explains the lack of responsiveness towardsTGFb1.
Smad proteins are substrates of TGFb type Ireceptor kinases and play a central role in transducingthe receptor signals to target genes in the nucleus.Because Smad4/DPC4 is deleted or inactivated in awide variety of pancreatic carcinomas, we analysed thecontent of Smad4 in the pancreatic carcinoma cell linesstudied here. PANC-1 and MIA PaCa-2 showedmarked Smad4 expression as analysed by RT±PCR,Northern blot (data not shown), and Western blotanalysis (Figure 2b). In contrast, BxPC-3 cellsexhibited no detectable Smad4, neither on mRNAnor on protein level, which is in agreement with datafrom the literature (Hahn et al., 1996; Schutte et al.,1996; Villanueva et al., 1998).
TGFb1-induced inhibition of ERK2
To characterize the possible mechanisms involved inTGFb1-mediated reduction of pancreatic carcinomacell proliferation, we analysed the in¯uence of TGFb1on the Ras-Raf-MEK-ERK/MAPK cascade, one ofthe best characterized signaling pathways involved in
Figure 1 In¯uence of TGFb1 on cell proliferation. Treatment ofPANC-1 and BxPC-3 cells with TGFb1 resulted in concentration-dependent inhibition of cell proliferation. MIA PaCa-2 cells,characterized by a lack of TGFb receptor type II, were unable torespond to TGFb. In contrast, NIH3T3 ®broblasts showed amarked increase in proliferation after treatment with TGFb1. Cellproliferation was measured by [3H]thymidine incorporationduring DNA synthesis. Untreated cells were de®ned as 100%,mean values+s.d. of ®ve independent assays are shown
TGFb inhibits ERK activationK Giehl et al
4532
Oncogene

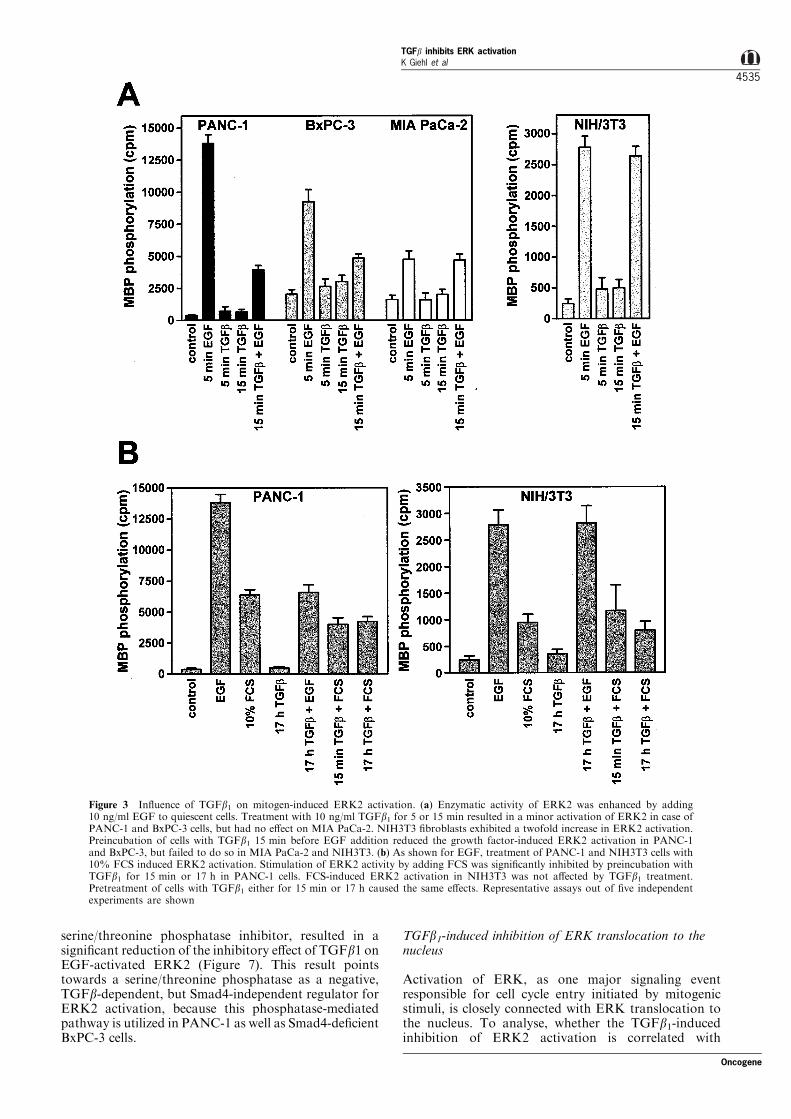
proliferation control. Activation of ERK1 and 2 isregulated by upstream protein kinases and RasGTPases, which receive signals from the plasmamembrane. As demonstrated in Figure 3a, treatmentof serum-starved PANC-1, BxPC-3, MIA PaCa-2, aswell as NIH3T3 cells with medium containing 10 ng/mlEGF led to a signi®cant stimulation of ERK2 activityin all cell lines. Only in NIH3T3 ®broblasts addition of
10 ng/ml TGFb1 for 15 min resulted in a signi®cantERK2 activation (2.2-fold), which is in line with theresults of TGFb1-mediated ERK activation in me-senchymal cells from other laboratories (Axmann et al.,1998; Reimann et al., 1997). Addition of TGFb110 min before mitogenic stimulation (5 min with TGFb1plus EGF), resulted in marked reduction of EGF-induced MBP phosphorylation of PANC-1 (71%,
Figure 2 Analyses of TGFb-associated signaling molecules. (a) TGFb receptor type I and II were detected in all cell lines onmRNA level (left panel) and as proteins in membrane fractions (right panel) except for MIA PaCa-2. The apparent molecularweight of TGFbR I is 55 kDa and of TGFbR II 70 kDa. (b) The intracellular TGFb e�ector protein Smad4 was not detectable inBxPC-3 cells, but clearly visible on mRNA level (left) and as protein in crude extracts (right) of PANC-1 and MIA PaCa-2carcinoma cell lines
Oncogene
TGFb inhibits ERK activationK Giehl et al
4533

n=5) and the Smad4-de®cient BxPC-3 cells (48%,n=5) cells, but had no in¯uence on ERK2 activationof MIA PaCa-2 and NIH3T3 cells.
To further characterize the in¯uence of TGFb1 ongrowth factor-induced ERK2 activation, serum-starvedPANC-1 and NIH3T3 cells were stimulated withserum. Treatment of cells for 5 min with mediumcontaining 10% FCS (Figure 3b) resulted in a 17-foldactivation of ERK2 for PANC-1 and fourfoldactivation for NIH3T3 ®broblasts, which is approxi-mately 50% of EGF-induced ERK activation. Pro-longed treatment with TGFb1 (17 h) had no signi®cante�ect on ERK2 activation for PANC-1. Long-termtreatment with TGFb1 exhibited the same inhibitorye�ect on EGF- or FCS-induced kinase stimulation aspretreatment for 10 min. To examine the speci®city ofthe inhibitory e�ect of TGFb1 for PANC-1, cells wereincubated with TGFb1 and a TGFb1-neutralizingantibody prior to mitogen treatment. Under theseconditions, ERK2 activation was maintained as incontrol cells not treated with TGFb1 (data not shown),con®rming the speci®city of the observed e�ects. Shortterm as well as prolonged TGFb1 treatment did notalter the FCS-induced activation of ERK2 in NIH3T3®broblasts.
Influence of TGFb1 on Ras and Raf activity
In order to analyse the signal transduction pathwayleading to reduced ERK2 activity and proliferation inthe pancreatic carcinoma cells in more detail, weexamined the activity of Ras and Raf proteins, whichare upstream activators of ERKs. The Ras-GTPcontent was estimated by pull-down experiments usinga recombinant GST-fusion protein of the Ras-bindingdomain (RBD) of Raf as an activation speci®c probefor endogenous Ras-GTP (de Rooij and Bos, 1997).Ras-GTP binds to RBD with an a�nity three orders ofmagnitude higher than that of Ras-GDP (Herrmann etal., 1995). The results of one representative assay outof four experiments are shown in Figure 4. EGFstimulation of PANC-1 and BxPC-3 cells resulted in anincreased amount of Ras-GTP which was about 3.5-times for PANC-1 and 6.8-times for BxPC-3 higher incomparison to quiescent control cells (Figure 4b).Neither 10 min nor 17 h pretreatment with 10 ng/mlTGFb1 was able to reduce this EGF-induced increasein Ras activation. Interestingly, TGFb1 treatment byitself resulted in an increased Ras activation asdetermined 5 and 15 min after ligand addition (Figure4). In control experiments, where a neutralizingantibody against TGFb1 was added together with theligand TGFb, Ras activation was not altered withregard to quiescent cells. In NIH3T3 cells, the GDP/GTP exchange on Ras was increased by EGF as wellas by TGFb1. Cotreatment of ®broblasts with EGFand TGFb1 did not result in additional Ras activationor in Ras inhibition as compared to treatment withEGF alone (Figure 4).
Ras-GTP interacts with the serine/threonine kinaseRaf, the ®rst protein kinase between Ras and ERK.Raf, which is activated by Ras, translocates to theplasma membrane. Since translocation from thecytoplasm to the inner surface of the plasma membraneis the ®rst indispensable step in Raf activation, wequanti®ed the amount of c-Raf-1 in cytosolic and
membrane fractions of PANC-1, BxPC-3, and NIH3T3cells by immunoblot analyses (Figure 5). Treatment ofPANC-1 and BxPC-3 cells with EGF caused a 3.5-foldincrease in membrane-associated c-Raf-1 in case ofPANC-1 and a 4.6-fold in case of BxPC-3, respectively.The corresponding cytoplasmic fraction showed aclearly reduced c-Raf-1 content. In contrast to Rasactivation, stimulation with TGFb1 did not result inincreased Raf activity. Pretreatment of PANC-1 andBxPC-3 cells with TGFb1 before EGF-stimulation didnot reduce the amount of c-Raf-1 at the plasmamembrane. Equal loading of both fractions wasveri®ed by reprobing the blots for b-catenin as amembrane and ERK2 as a cytoplasmic protein,respectively. NIH3T3 cells also reacted with enhancedRaf translocation to the membrane upon stimulationwith 10 ng/ml EGF, although the e�ect was lessdramatic compared to BxPC-3 cells. Preincubation ofthe cells with 10 ng/ml TGFb1 prior to EGF additiondid not result in decreased Raf activation. Further-more, TGFb1 itself induced Raf translocation to theplasma membrane. This is in line with observations,that TGFb1 is able to enhance proliferation and ERK2activity in ®broblasts. The TGFb1 e�ect on c-Raf-1was diminished by simultaneous addition of a neu-tralizing antibody against TGFb (Figure 5).
Influence of TGFb1 on ERK-dependent cell proliferation
To determine, if activated ERK2 is involved in TGFb1-mediated growth inhibition of pancreatic carcinomacell lines, we inhibited ERK activation by using theMEK-1 speci®c inhibitor PD98059. Cell proliferationwas quanti®ed by [3H]thymidine incorporation. Theinhibitor binds selectively to the inactive form ofMEK-1 and prevents activation by upstream activators(Dudley et al., 1995). As shown previously, 25 mMPD98059 is su�cient for complete ERK inhibition butdoes not cause cytotoxic e�ects (Giehl et al., 2000).Treatment of PANC-1, BxPC-3, and MIA PaCa-2cells, incubated in medium containing 10% FCS, withPD98059 resulted in a concentration-dependent reduc-tion of proliferation for all three cell lines. [3H]thymi-dine incorporation was reduced by nearly 20% using10 mM inhibitor, whereas 25 mM PD98059 resulted in30 ± 40% decrease (Figure 6). In PANC-1 cells,treatment with 25 mM PD98059 resulted in the sameproliferation reduction as growth inhibition afterTGFb1-treatment. Cotreatment of PANC-1 andBxPC-3 cells with 25 mM PD98059 and 10 ng/ml TGFb1resulted in the same growth inhibition as treatmentwith each inhibitor alone. MIA PaCA-2 cells were notresponsive to TGFb1 treatment, as shown above inFigure 1, but needed activated ERK for proliferation.
The serine/threonine phosphatase inhibitor okadaic acidsuppresses TGFb-induced ERK2 inhibition
To get further insights into the pathway utilized by TGFbto reduce growth factor-activated ERK2, we analysedthe signi®cance of phosphatases by using speci®cphosphatase inhibitors. Addition of the cell-permeabletyrosine phosphatase inhibitor sodium vanadate(2.5 mM) during incubation of cells with TGFb, had noa�ect on either EGF- or/and TGFb-mediated ERK2activity. In contrast, addition of okadaic acid (0.5 mM), a
TGFb inhibits ERK activationK Giehl et al
4534
Oncogene
serine/threonine phosphatase inhibitor, resulted in asigni®cant reduction of the inhibitory e�ect of TGFb1 onEGF-activated ERK2 (Figure 7). This result pointstowards a serine/threonine phosphatase as a negative,TGFb-dependent, but Smad4-independent regulator forERK2 activation, because this phosphatase-mediatedpathway is utilized in PANC-1 as well as Smad4-de®cientBxPC-3 cells.
TGFb1-induced inhibition of ERK translocation to thenucleus
Activation of ERK, as one major signaling eventresponsible for cell cycle entry initiated by mitogenicstimuli, is closely connected with ERK translocation tothe nucleus. To analyse, whether the TGFb1-inducedinhibition of ERK2 activation is correlated with
Figure 3 In¯uence of TGFb1 on mitogen-induced ERK2 activation. (a) Enzymatic activity of ERK2 was enhanced by adding10 ng/ml EGF to quiescent cells. Treatment with 10 ng/ml TGFb1 for 5 or 15 min resulted in a minor activation of ERK2 in case ofPANC-1 and BxPC-3 cells, but had no e�ect on MIA PaCa-2. NIH3T3 ®broblasts exhibited a twofold increase in ERK2 activation.Preincubation of cells with TGFb1 15 min before EGF addition reduced the growth factor-induced ERK2 activation in PANC-1and BxPC-3, but failed to do so in MIA PaCa-2 and NIH3T3. (b) As shown for EGF, treatment of PANC-1 and NIH3T3 cells with10% FCS induced ERK2 activation. Stimulation of ERK2 activity by adding FCS was signi®cantly inhibited by preincubation withTGFb1 for 15 min or 17 h in PANC-1 cells. FCS-induced ERK2 activation in NIH3T3 was not a�ected by TGFb1 treatment.Pretreatment of cells with TGFb1 either for 15 min or 17 h caused the same e�ects. Representative assays out of ®ve independentexperiments are shown
Oncogene
TGFb inhibits ERK activationK Giehl et al
4535

reduced ERK translocation to the nucleus, weexamined the subcellular localization of ERK2 afterEGF and/or TGFb1 treatment for 15 min. ERK2distribution in stimulated PANC-1 and BxPC-3 cellswas investigated by immunohistochemistry, using anantiserum against ERK2. As shown in Figure 8, EGFtreatment resulted in a rapid transport of ERK2 to thenucleus of both cell lines (Figure 8b,h). TGFb1 aloneinduced no enhanced nuclear staining in PANC-1(Figure 8c) and BxPC-3 cells (Figure 8i). Treatmentof the cells with TGFb1 in combination with EGF(Figure 8d,j) prevented the mitogen-induced kinasetranslocation in PANC-1 and BxPC-3 cells. Compar-able results were obtained using 10% FCS instead ofEGF (data not shown). To exclude a general inhibitionof nuclear transport activities, the ability of TGFb1 toinduce nuclear staining of Smad2/3 in PANC-1 cellswas analysed, as shown in Figure 8e,f. In summary,
these data con®rm that TGFb1-induced growth inhibi-tion of pancreatic carcinoma cells correlates withinactivation of ERK-dependent cell cycle progression.
Discussion
We have investigated the mechanism of TGFb1-mediated inhibition of growth factor-induced cellproliferation in epithelial pancreatic carcinoma celllines. Our results suggest that TGFb1 contributes toreduced cell proliferation by inhibiting mitogen-induced ERK activation independently of Smad4proteins at least in pancreatic carcinoma cell lines.Activation of the mitogen-activated protein kinasesERK1 and ERK2 by growth factors and othermitogenic proteins represents an essential step in cellcycle re-entry of most cell types (Seger and Krebs,
Figure 4 Stimulation of GDP/GTP exchange on Ras by TGFb1. Treatment of PANC-1, BxPC-3, and NIH3T3 cell with 10 ng/mlEGF induced a markedly elevated amount of Ras-GTP bound to GST-RBD. TGFb1 alone (10 ng/ml) was able to induce Rasactivation in all cell lines. Pretreatment with TGFb1 for 15 min followed by addition of 10 ng/ml EGF did not reduce EGF-inducedRas activation. Incubation of cells with TGFb1 and a neutralizing TGFb antibody (nAb) diminished TGFb-induced activation ofRas in PANC-1 cells. (a) (upper panel) Detection of GST-RBD-bound Ras-GTP using pan-Ras antibody. Immunodetection of Rasin cell lysates (30 mg) served as positive control; (lower panel) Detection of GST-RBD with anti-GST antibody to con®rm equalloading. One representative assay out of three is shown. (b) Quanti®cation of Ras activation by densitometric analyses of threeindependent experiments (mean values+s.e.mean)
TGFb inhibits ERK activationK Giehl et al
4536
Oncogene

1995). Addition of growth factors results in ERKphosphorylation and translocation into the nucleuswith subsequent induction of gene expression (Bron-dello et al., 1995; Brunet et al., 1999; Lenormand et al.,1993). Treatment of the pancreatic carcinoma cell linesPANC-1 and BxPC-3 with serum or EGF initiate amarked increase of ERK2 activity, which was clearlyreduced by TGFb1. The requirement of ERK activa-tion for the proliferation of the pancreatic carcinomacells is veri®ed by the use of the MEK inhibitorPD98059. It is likely that a unique target protein onthe level of MEK is inhibited, since this inhibitorcauses a similar reduction of cell proliferation as TGFb1
and both factors in combination are non-additive. Veryrecently, Dixon et al. (1999) reported that TGFb isable to inhibit p70 S6 kinase and ERK activation in rathepatocytes. Their results are in agreement with ourdata and document the signi®cance of the regulation ofERK by TGFb in epithelial cells. Other reports suggestthe importance of other members of the MAPK family,such as the Jun N-terminal kinase (JNK) for Smad-independent TGFb signaling processes (Hocevar et al.,1999).
The BxPC-3 cell line, which is characterized by ahomozygous deletion of smad4 gene (our data and(Villanueva et al., 1998)) responds to TGFb1 clearlybut less e�ective as Smad4-positive PANC-1 cells. Inaddition to the proposed model (Attisano and Wrana,1998; Heldin et al., 1997; Massague, 1998), that TGFbexerts growth inhibitory and transcriptional repression
Figure 5 Localization of c-Raf-1 after growth factor and TGFb1 treatment. Distribution of c-Raf-1 in membrane and cytosolicfractions of PANC-1, BxPC-3, and NIH3T3 was detected by Western blot analyses using c-Raf-1 antiserum. Stimulation ofquiescent cells with EGF (10 ng/ml) resulted in increased membrane localization in all cell lines. TGFb1 (10 ng/ml) had no e�ect onmembrane association of c-Raf-1 in PANC-1 and BxPC-3 cells, but caused a slight increase in membrane-bound c-Raf-1 in NIH3T3cells. Cotreatment with EGF and TGFb had the same e�ect as treatment with EGF alone. Addition of neutralizing TGFb antibodytogether with TGFb1 served as control. To con®rm equal protein loading, Western blots were reprobed with anti-b-catenin formembrane fractions and anti-ERK2 for cytosolic protein content. One representative assay out of ®ve is shown
Figure 6 Inhibition of cell proliferation by PD98059 and TGFb1.To compare the e�ects of MEK inhibitor PD98059- and TGFb1-reduced DNA synthesis of PANC-1, BxPC-3, and MIA PaCa-2,the cells were treated with 10 ng/ml TGFb1, 10 mM or 25 mMPD98059 as described in Materials and methods. Incubation withPD98059 resulted in concentration-dependent decrease (20 ± 40%)of DNA synthesis in all cell lines. TGFb and PD98059 reacted asnon-additive inhibitors. Mean values+s.e.mean of three indepen-dent experiments are expressed in per cent reduction in relation tountreated cells (100%)
Figure 7 Serine/threonine phosphatase inhibitor neutralizesTGFb1-inhibition. Treatment of cells with 0.5 mM okadaic acid(OA), an inhibitor of serine/threonine phosphatases, resulted insuppression of the TGFb-induced inhibition of activated ERK2 inPANC-1 as well as in BxPC-3 cells. In contrast, inhibition oftyrosine phosphatases using 2.5 mM sodium vanadate (OV) hadno e�ect on ERK activity. One representative assay out of three isshown
Oncogene
TGFb inhibits ERK activationK Giehl et al
4537

via Smad2/3 associated with Smad4, the pathwaypresented in this study suggest an additional anti-mitogenic e�ect independent of the latter protein.Very recently, Dai et al. (1999) reported Smad4/DPC4-independent, MEK1-dependent TGFb in¯u-ences on cell cycle progression, supporting theinterpretation of our ®ndings. Dissection of TGFb-
regulated cell proliferation and TGFb/Smad-depen-dent gene expression is also described by Rodeck et al.(1999). Melanoma cells, which were partially orcompletely resistant to TGFb-induced growth inhibi-tion, exhibited functional intact Smad-dependent geneexpression. These observations underscore the uncou-pling of the TGFb growth response from thetranscriptional response pathway.
The requirement of functional TGFb receptors forthe growth inhibitory e�ect of TGFb, as demonstratedin this study, is manifested by using TGFbRII-de®cientMIA PaCa-2 cells, as shown here and by Freeman etal. (1995). Stimulation of MIA PaCa-2 cells with TGFbdid not result in reduced proliferation or inhibition ofgrowth factor-induced ERK2 activation.
In contrast to the pathway suggested above,Massague and colleagues show an inverse pathwayin which TGFb signaling via Smads can be inhibitedby Ras-induced ERK activation (Calonge andMassague, 1999; Kretzschmar et al., 1999). Thesedivergent ®ndings implicate a ®ne tuned, eventuallycell type and signal-speci®c connection between theRas/ERK and TGFb/Smad pathways. The involve-ment of Ras in TGFb-mediated signal transductionwas also shown by Mulder and Morris (1992).TGFb-sensitive, non-transformed intestinal epithelialcells (IEC 4-1) showed a subtle activation of ERK1(p44MAPK) after addition of TGFb2 to proliferating,but serum-starved cells (Hartsough and Mulder,1995).
The inhibition of growth factor-induced ERK2stimulation in our system is not caused by reductionof c-Raf-1 and Ras activation due to mitogenexposure. The TGFb1-induced activation of Ras, butnot c-Raf-1 in pancreatic carcinoma cells might a�ectother, c-Raf-1 independent regulatory pathways, whichare currently unknown. The results presented heresuggest a serine/threonine phosphatase as a TGFb-dependent, but Ras/Raf-independent regulator ofERK, which directly or indirectly mediates ERK2inactivation. Although this phosphatase is not yetidenti®ed, results obtained by Griswold-Prenner et al.(1998) show that the regulatory subunit Ba of proteinphosphatase 2A (PP2A) enhances the growth inhibi-tory activity of TGFb in a receptor-dependent manner.This proposes PP2A as a likely candidate, because theenzyme is also characterized by its ability to dephos-phorylate activated MEK and ERK (Alessi et al.,1995).
In contrast to the epithelial cell system, mesenchymalcells respond di�erently to TGFb as shown here andby others (Axmann et al., 1998; Reimann et al., 1997).To support the speci®city of the observed e�ects inepithelial PANC-1 and BxPC-3 cells, we analysedNIH3T3 cells for comparison. TGFb1 increased the[3H]thymidine incorporation in mesenchymal cells andactivated Ras, c-Raf-1 and ERK2. Cotreatment of cellswith EGF and TGFb1 resulted in neither reducedmitogen-induced ERK2 activation nor prevention ofnuclear translocation.
In conclusion, TGFb1 can interfere with growthfactor-induced cell proliferation via ERK2 inactivationin epithelial pancreatic carcinoma cells. This pathwayoperates independently of Smad4 and can thereforefunction in addition to Smad4-dependent growthinhibition.
Figure 8 E�ect of TGFb1 on mitogen-induced ERK andSmad2/3 translocation. Localization of activated ERK2 inPANC-1 cells (a ± d) and BxPC-3 cells (g ± j) was determined byimmuno¯uorescence studies using anti-ERK2 antibody. Treat-ment of quiescent cells with 10 ng/ml EGF for 15 min resulted inintensive nuclear staining of PANC-1 as well as of BxPC-3 cells(b,f). TGFb1 (10 ng/ml) had no e�ect on ERK2 distribution inboth cell lines (c,g). Cotreatment of cells with EGF and TGFb1abrogates EGF-induced ERK2 translocation to the nucleus ofPANC-1 and BxPC-3 cells (d,h). Stimulation of cells with TGFb1resulted in e�cient Smad2/3 nuclear translocation in PANC-1cells (e,f), which excludes a general inhibitory mechanism ofnuclear transport by TGFb1. Scale bar represents 10 mm
TGFb inhibits ERK activationK Giehl et al
4538
Oncogene

Materials and methods
Antibodies and growth factors
Neutralizing TGFb antiserum (AB 101 NA) and anti-TGFbreceptor type II (AF 241 NA) were obtained from R&DSystems (Wiesbaden, Germany), antibodies against TGFbreceptor type I (T-19), ERK2 (C14), MEK-1 (C-18), Smad2/3and GST (Z-5) were from Santa Cruz Biotechnology (SantaCruz, CA, USA), anti-pan-Ras antibody (Ab-3) was fromOncogene Sciences (Cambridge, MA, USA), anti-b-cateninfrom Sigma (Deisenhofen, Germany), anti-Smad4 fromTransduction Laboratories (San Diego, CA, USA), andpolyclonal c-Raf-1 antiserum from Promega (Mannheim,Germany). The growth factors EGF and TGFb1 wereobtained from PeproTech (Rocky Hill, NJ, USA) and theMEK inhibitor PD98059 was purchased from NEB (BeverlyMA, USA). The phosphatase inhibitors okadaic acid andsodium vanadate were from Sigma (Deisenhofen, Germany).
Cell lines and culture conditions
The cell lines PANC-1 (ATCC, CRL 1469), BxPC-3 (ATCC,CRL 1687), MIA PaCa-2 (ATCC, CRL 1420) and NIH3T3(ATCC, CRL 1658) were obtained from American TypeCulture Collection (Rockville, MD, USA). Epithelial cellswere grown in DMEM complemented with 10% fetal calfserum (FCS) (Life Technologies, Karlsruhe, Germany), 1%non-essential amino acids and 1% L-glutamine. NIH3T3 cellswere cultured in DMEM containing 10% donor calf serum(Life Technologies) instead of FCS. Cells were incubated at378C in a humidi®ed atmosphere with 10% CO2. For Ras, c-Raf and ERK assays cells were stimulated with 10 ng/mlEGF or TGFb1. To analyse the TGFb in¯uence on EGF-induced enzymatic activity, the cells were stimulated with10 ng/ml EGF and 10 ng/ml TGFb1 for 5 min after apreincubation for 10 min with 10 ng/ml TGFb1. To inhibitprotein phosphatases, cells were incubated 15 min prior toaddition of growth factors with 2.5 mM sodium vanadate or0.5 mM okadaic acid.
Proliferation assay
DNA synthesis was determined by [methyl 3H]thymidineincorporation as described in (Menke et al., 1999). Brie¯y,cells were plated in 24-well plates (56104 cells/well) andallowed to attach for 24 h. Serum-starvation was performedby incubation with DMEM+1% bovine serum albumin(BSA) for 36 h or 12 h for NIH3T3, respectively, followed bystimulation with assay medium (DMEM+1% BSA+growthfactors and inhibitors) for additional 36 h. [3H]thymidine(1 mCi/well) was added for the last 24 h. Acid-insolubleradioactivity incorporated into protein was determined byliquid scintillation counting after solubilization in 0.1%NaOH, 1% sodium dodecyl sulfate (SDS).
ERK activity assay
ERK activity was quanti®ed in total RIPA cell lysates(50 mM Tris/HCl, pH 7.2, 150 mM NaCl, 1% Triton X-100,0.5% sodium deoxycholate, 0.1% SDS, 2 mM sodiumorthovanadate, 25 mM b-glycerophosphate, 10 mM sodiumpyrophosphate, 400 mM aprotinin) in an in vitro kinasereaction using a synthetic peptide (-APRTPGGRR-) derivedfrom myelin basic protein (MBP) as kinase substrate(Gardner et al., 1993). ERK2 was immunoprecipitated from0.5 mg of cellular protein for 2 h at 48C using 25 ml ofProtein-A agarose and 7 ml of ERK2 antibody. The kinasereaction was performed in kinase bu�er (15 mM Tris/HCl,pH 7.2, 15 mM MgCl2, 1 mM DTT), started by adding 2 mMATP, 30 mg of MBP peptide, and 2 mCi [g32P]ATP(5000 Ci mmol), and carried out for 10 min at 378C. Sampleswere spotted onto P81 phosphocellulose ®lter. Filters were
washed three times in 180 mM phosphoric acid and boundradioactivity was determined by Cerenkov radiation.
Ras-GTP binding assay
GTP-bound form of Ras was detected by using GST-RBD asan activation-speci®c probe for endogenous Ras-GTP (deRooij and Bos, 1997). Con¯uent cells were serum-starved for36 h (NIH3T3 for 12 h) in minimal medium (DMEM + 1%BSA) and stimulated with assay medium. Cells (1 ± 26107/100 mm cell culture dish) were washed with phosphatebu�ered saline (PBS) and lysed in 500 ml Ras-RIPA bu�er(50 mM Tris/HCl, pH 8.0, 150 mM NaCl, 0.5% sodiumdeoxycholate, 1% NP-40, 0.1% SDS, 1 mM leupeptin, 1 mM
phenylmethylsulfonyl ¯uoride). Protein concentration ofcleared cell lysate was determined by bicinchoninic acidassay (Pierce; Rockford, USA). Glutathione-Sepharose 4B(Pharmacia; Uppsala, Sweden) was preincubated with 150 mlGST-RBD extract at room temperature for 30 min. Recom-binant GST-RBD was prepared as described in (de Rooij andBos, 1997). Cell lysate (1 mg) was added to washed beadsand incubated at 48C for 2 h by end-over-end rotation.Proteins were separated on 12.5% SDS-polyacrylamide geland subsequently transferred to nitrocellulose membrane(Schleicher & SchuÈ ll, Dassel, Germany). Equal loading wascontrolled by immunostaining of GST-RBD using anti-GSTantibody. Precipitated endogenous Ras was detected by usingthe monoclonal pan-Ras (Ab-3) antibody, peroxidase-coupledgoat anti-mouse antiserum and ECL development (Pierce,Rockford, USA).
Raf assay
Raf activation was estimated by quantifying the amount of c-Raf-1 in cytosolic and membrane fractions. Cells were treatedwith assay medium as described for Ras assay and were thenhomogenized in KRH bu�er (100 mM KCl, 10 mM PIPES,pH 7.3, 3.5 mM MgCl2, 3 mM NaCl, 5 mM aprotinin, 1 mM
pefabloc, 10 mM leupeptin, 5 mM sodium vanadate and 5 mMsoybean trypsin inhibitor). Nuclei and unbroken cells wereremoved by centrifugation (200 g, 5 min). Cytosolic andmembrane fractions were separated by centrifugation at100 000 g and 48C for 30 min. Membranes were furtherpuri®ed by resuspending the pellet in KRH bu�er, followedby high speed centrifugation at 100 000 g and 48C for 1 h.Fractions of 30 mg protein were analysed for c-Raf-1 contentby Western blotting as described below. Equal loading wascontrolled by restaining the blots for b-catenin in case ofmembrane fractions and ERK2 of cytosolic samples.
Protein determination by Western blots
SDS-polyacrylamide gel electrophoresis (PAGE) was per-formed according to standard procedures (Laemmli, 1970).Sixty mg of lysate were separated for Smad4 detection and20 mg of the membrane fraction, produced as describedabove, for TGFb receptor analyses. Proteins were blotted bysemidry technique onto nitrocellulose membranes (Schleicher& SchuÈ ll, Dassel, Germany) and immunoreactive proteinswere visualized by using the enhanced chemiluminescence(ECL) Western blotting detection system (Pierce, Rockford,USA) or by using alkaline phosphatase-conjugated antibodies(Dianova, Hamburg, Germany) and nitroblue tetrazolium/bromo-chloro-indoyl phosphate staining.
RNA studies
RNA was extracted using the RNeasy Midi Kit from Qiagen(Hilden, Germany) according to manufacturer's instructions.Poly(A)+ RNA was isolated with Oligotex Kit (Qiagen). ForNorthern blot detection, 4 mg of poly(A)+ RNA wereseparated by electrophoresis and transferred on Hybond-N
Oncogene
TGFb inhibits ERK activationK Giehl et al
4539

membranes (Amersham, Braunschweig, Germany) as de-scribed in (Menke et al., 1997). Equal loading was veri®edby 18S rRNA hybridization, using a probe kindly providedby Dr T Gress (University of Ulm, Germany). Human-speci®c cDNA probes for TGFbRI and TGFbRII (ALK5)were kindly provided by Dr MG Brattain (Medical College ofOhio, Toledo, OH, USA), human-speci®c Smad4 cDNA wasa kind gift of Dr S Hahn (University of Bochum, Germany).Blots were hybridized with 32P-dCTP-labeled cDNA probesas described earlier (Menke et al., 1997). For RT±PCR,10 mg of total RNA were reversely transcribed using oligo-dTprimer and reverse transcriptase (Superscript, Life Technol-ogies, Karlsruhe, Germany). Complementary DNA wassynthesized for 60 min at 378C and stopped by incubationfor 15 min at 708C. PCR was performed with 2.5 mM MgCl2and 2.5 U Taq DNA Polymerase (Sigma). The ampli®cationprotocol was as follows: 35 cycles at 948C for 1 min, 528C for1 min, 728C for 2 min, followed by a ®nal extension time of10 min at 728C. The PCR primers were designed based onthe published cDNA sequence for Smad4 (Hahn et al., 1996)(sense 5'-ATGGACAATATGTCTATTA-3', antisense 5'-TCAGTCTAAAGGTTGTGG-3'). As a control, b-actinmRNA was ampli®ed from the used cDNAs with thefollowing ampli®cation protocol: 15 cycles at 948C for 45 s,608C for 1 min, 728C for 1.5 min, followed by a ®nalextension time of 10 min at 728C. The PCR primers were 5'-
GCGTACCTCATGAAGATCCT-3' for sense and 5'-GCG-GATGTCCACGTCACACT-3' for antisense.
Immunofluorescence analysis
Serum-starved PANC-1 and BxPC-3 cells (36 h inDMEM+1% BSA) were stimulated with 10 ng/ml EGF,10 ng/ml TGFb1 or 10 ng/ml TGFb1 plus 10 ng/ml EGF inminimal medium for 15 min. Cells were ®xed in 4%paraformaldehyde and permeabilized by treatment with0.1% Triton X-100 for 15 min. Unspeci®c binding wasblocked by treatment with PBS containing 3% BSA. Cellswere incubated for 1 h at 378C with anti-ERK2 or anti-Smad2/3. Protein localization was visualized by incubationwith a secondary Cy3-conjugated antibody (Biomol, Ham-burg Germany). Cells were examined by confocal laserscanning microscopy using a confocal microscope (TCS 4DLeica, Wetzlar, Germany).
AcknowledgmentsThe authors wish to thank A Mansard and S BraÈ g forexcellent technical assistance. This work was supported bythe SFB 518, DFG Kn200/4 and the BMBF (project 01-KJS-9605/2, IZKF Ulm).
References
Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM andCohen P. (1995). Curr. Biol., 5, 283 ± 295.
Attisano L, Carcamo J, Ventura F, Weis FM, Massague Jand Wrana JL. (1993). Cell, 75, 671 ± 680.
Attisano L and Wrana JL. (1998). Curr. Opin. Cell Biol., 10,188 ± 194.
Axmann A, Seidel D, Reimann T, Hempel U and WenzelKW. (1998). Biochem. Biophys. Res. Com., 249, 456 ± 460.
Brondello JM, McKenzie FR, Sun H, Tonks NK andPouysse gur J. (1995). Oncogene, 10, 1895 ± 1904.
Brunet A, Roux D, Lenormand P, Dowd S, Keyse S andPouysse gur J. (1999). EMBO J., 18, 664 ± 674.
Calonge MJ and Massague J. (1999). J. Biol. Chem., 274,33637 ± 33643.
Chen RH, Sarnecki C and Blenis J. (1992). Mol. Cell. Biol.,12, 915 ± 927.
Chen YG, Liu F and Massague J. (1997). EMBO J., 16,3866 ± 3876.
Dai JL, Schutte M, Bansal RK, Wilentz RE, Sugar AY andKern SE. (1999). Mol. Carcinog., 26, 37 ± 43.
de Rooij J and Bos JL. (1997). Oncogene, 14, 623 ± 625.Dixon M, Agius L, Yeaman SJ and Day CP. (1999).
Hepatology, 29, 1418 ± 1424.Dudley DT, Pang L, Decker SJ, Bridges AJ and Saltiel AR.
(1995). Proc. Natl. Acad. Sci. USA, 92, 7686 ± 7689.Ebner R, Chen RH, Lawler S, Zioncheck T and Derynck R.
(1993). Science, 262, 900 ± 902.Ewen ME, Sluss HK, Whitehouse LL and Livingston DM.
(1993). Cell, 74, 1009 ± 1020.Franzen P, ten Dijke P, Ichijo H, Yamashita H, Schulz P,
Heldin CH and Miyazono K. (1993). Cell, 75, 681 ± 692.Freeman JW, Mattingly CA and Strodel WE. (1995). J. Cell.
Physiol., 165, 155 ± 163.Gardner AM, Vaillancourt RR and Johnson GL. (1993). J.
Biol. Chem., 268, 17896 ± 17901.Giehl K, Skripczynski B, Mansard A, Menke A and
Gierschik P. (2000). Oncogene, 19, 2930 ± 2942.Griswold-Prenner I, Kamibayashi C, Maruoka EM, Mumby
MCandDerynckR. (1998).Mol.Cell.Biol.,18,6595 ± 6604.Hahn SA, SchutteM,HoqueATMS,Moskaluk CA, da Costa
LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ,Hruban RH and Kern SE. (1996). Science, 271, 350 ± 353.
Hartsough MT and Mulder KM. (1995). J. Biol. Chem., 270,7117 ± 7124.
Heldin CH, Miyazono K and ten Dijke P. (1997). Nature,390, 465 ± 471.
Heldin CH and Ostman A. (1996). Cytokine Growth Factor.Rev., 7, 3 ± 10.
Herrmann C, Martin GA and Wittinghofer A. (1995). J.Biol. Chem., 270, 2901 ± 2905.
Hocevar BA, Brown TL and Howe PH. (1999). EMBO J., 18,1345 ± 1356.
Kawabata M, Imamura T, Miyazono K, Engel ME andMoses HL. (1995). J. Biol. Chem., 270, 29628 ± 29631.
Ko TC, Yu W, Sakai T, Sheng H, Shao J, Beauchamp RDand Thompson EA. (1998). Oncogene, 16, 3445 ± 3454.
Kretzschmar M, Doody J and Massague J. (1997). Nature,389, 618 ± 622.
Kretzschmar M, Doody J, Timokhina I and Massague J.(1999). Genes Dev., 13, 804 ± 816.
Laemmli UK. (1970). Nature, 227, 680 ± 685.Lenormand P, Sardet C, Pages G, L'Allemain G, Brunet A
and Pouysse gur J. (1993). J. Cell Biol., 122, 1079 ± 1088.Liu JH, Wei S, Burnette PK, Gamero AM, Hutton M and
Djeu JY. (1999). Oncogene, 18, 269 ± 275.Massague J. (1990). Annu. Rev. Cell Biol., 6, 597 ± 641.Massague J. (1998). Annu. Rev. Biochem., 67, 753 ± 791.Menke A, Geerling I, Giehl K, Vogelmann R, Reinshagen M
and Adler G. (1999). Biochim. Biophys. Acta, 1449, 178 ±185.
Menke A, Yamaguchi H, Gress TM and Alder G. (1997).Gastroenterology, 113, 295 ± 303.
Mulder KM and Morris SL. (1992). J. Biol. Chem., 267,5029 ± 5031.
Pages G, Lenormand P, Allemain G, Chambard JC, MelocheS and Pouysse gur J. (1993). Proc. Natl. Acad. Sci. USA,90, 8319 ± 8323.
Piek E, Heldin CH and ten Dijke P. (1999). FASEB J., 13,2105 ± 2124.
Polyak K. (1996).Biochim. Biophys. Acta, 1242, 185 ± 199.Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R,
Seidel D andWenzel KW. (1997). FEBS Lett., 403, 57 ± 60.Roberts AB, Heine UI, Flanders KC and Sporn MB. (1990).
Ann. N.Y. Acad. Sci, 580, 225 ± 232.
TGFb inhibits ERK activationK Giehl et al
4540
Oncogene

Rodeck U, Nishiyama T andMauviel A. (1999). Cancer Res.,59, 547 ± 550.
Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM,Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H,Sidransky D, Casero Jr, RA, Meltzer PS, Hahn SA andKern SE. (1996). Cancer Res., 56, 2527 ± 2530.
Seger R and Krebs EG. (1995). FASEB J., 9, 726 ± 735.Sporn MB and Roberts AB. (1992). J. Cell Biol., 119, 1017 ±
1021.
Villanueva A, Garcia C, Paules AB, Vicente M, Megias M,Reyes G, de Villalonga P, Agell N, Lluis F, Bachs O andCapella G. (1998). Oncogene, 17, 1969 ± 1978.
Wang T, Danielson PD and Li B. (1996). Science, 271, 1120 ±1122.
Zheng CF and Guan KL. (1994). J. Biol. Chem., 269, 19947 ±19952.
Oncogene
TGFb inhibits ERK activationK Giehl et al
4541
Related Documents











