ARTICLE doi:10.1038/nature12782 Targeting Plasmodium PI(4)K to eliminate malaria Case W. McNamara 1 *, Marcus C. S. Lee 2 *, Chek Shik Lim 3 , Siau Hoi Lim 3 , Jason Roland 1 , Advait Nagle 1 , Oliver Simon 3 , Bryan K. S. Yeung 3 , Arnab K. Chatterjee 1 , Susan L. McCormack 1 , Micah J. Manary 4 , Anne-Marie Zeeman 5 , Koen J. Dechering 6 , T. R. Santha Kumar 2 , Philipp P. Henrich 2 , Kerstin Gagaring 1 , Maureen Ibanez 1 , Nobutaka Kato 1 , Kelli L. Kuhen 1 , Christoph Fischli 7 , Matthias Rottmann 7,8 , David M. Plouffe 1 , Badry Bursulaya 1 , Stephan Meister 4 , Lucia Rameh 9 , Joerg Trappe 10 , Dorothea Haasen 10 , Martijn Timmerman 6 , Robert W. Sauerwein 6,11 , Rossarin Suwanarusk 12 , Bruce Russell 12,13 , Laurent Renia 12 , Francois Nosten 14,15 , David C. Tully 1 , Clemens H. M. Kocken 5 , Richard J. Glynne 1 , Christophe Bodenreider 3 , David A. Fidock 2,16 , Thierry T. Diagana 3 & Elizabeth A. Winzeler 1,4 Achieving the goal of malaria elimination will depend on targeting Plasmodium pathways essential across all life stages. Here we identify a lipid kinase, phosphatidylinositol-4-OH kinase (PI(4)K), as the target of imidazopyrazines, a new antimalarial compound class that inhibits the intracellular development of multiple Plasmodium species at each stage of infection in the vertebrate host. Imidazopyrazines demonstrate potent preventive, therapeutic, and transmission-blocking activity in rodent malaria models, are active against blood-stage field isolates of the major human pathogens P. falciparum and P. vivax, and inhibit liver-stage hypnozoites in the simian parasite P. cynomolgi. We show that imidazopyrazines exert their effect through inhibitory interaction with the ATP-binding pocket of PI(4)K, altering the intracellular distribution of phosphatidylinositol-4-phosphate. Collectively, our data define PI(4)K as a key Plasmodium vulnerability, opening up new avenues of target-based discovery to identify drugs with an ideal activity profile for the prevention, treatment and elimination of malaria. To eliminate malaria, broadly acting medicines must be developed that cure the symptomatic asexual blood stage, clear the preceding liver- stage infection that can cause relapses and block parasite transmission to mosquitoes 1 . Relapse prevention is especially important for P. vivax, which forms intra-hepatic hypnozoites that can persist for years before reinitiating development and triggering blood-stage infection. Primaquine is the only licensed antimalarial capable of eliminating the hypnozoite reservoir and delivering a radical cure. However, side effects and weak activity against blood stages preclude widespread use of primaquine 2 . Because the target of primaquine and mechanism of action are not known, the search for radical cure drugs has been limited to related analogues, such as tafenoquine 3 . There is a clear need for druggable and chemically vali- dated targets that are essential in all life-cycle stages of the malaria parasite. Here we report that a parasite PI(4)K type III beta (PI(4)KIIIb), a ubiquitous eukaryotic enzyme that phosphorylates lipids to regulate intra- cellular signalling and trafficking, is inhibited by imidazopyrazines. In blood stages, imidazopyrazines block a late step in parasite development by disrupting plasma membrane ingression around developing daughter merozoites. This probably stems from altered phosphatidylinositol-4- phosphate (PI(4)P) pools and disrupted Rab11A-mediated membrane trafficking. To our knowledge, these findings are the first validation of a drug target that is required across all Plasmodium life-cycle stages. Imidazopyrazines target several life stages A cell-based screen against P. falciparum asexual blood-stage parasites 4 revealed a new class of antimalarials with an imidazopyrazine core (Fig. 1a). This compound was distinct from known antimalarials and was active against several drug-resistant strains (half-maximum inhibitory concentration (IC 50 ) 27–70 nM; Extended Data Table 1 and Extended Data Fig. 1a), suggesting a new mechanism of action. Synthetic deri- vatives with potency and pharmacokinetic properties suitable for test- ing in animal models were designed and showed activity in vivo (data shown for KDU691; Extended Data Table 2 and Extended Data Fig. 1b). We evaluated imidazopyrazines (Fig. 1a) against all parasite life-cycle stages in the vertebrate host. First, a cell-based assay showed potent inhibition of liver-stage development of the rodent parasite, P. yoelii, with IC 50 values less than 160nM and as low as 9 nM (Fig. 1b) for KDU691, KAI407 and KAI715. In vivo, KDU691 administered orally at the time of infection prophylactically protected mice from coloniza- tion with transgenic P. berghei sporozoites expressing luciferase. Just a single dose (7.5 mg kg 21 ; Fig. 1c and Extended Data Fig. 1c) prevented what would normally be a fatal outcome. To determine whether KDU691 could eliminate an existing infection, animals were dosed 24, 36 or 48 h after sporozoite inoculation, well after the onset of liver-stage develop- ment (which lasts 48 h in P. berghei). This delayed treatment rapidly eliminated the luciferase-expressing parasites with a single dose (Fig. 1c). KDU691 was also active at low concentrations (IC 50 , 196 nM; Fig. 1d) against in vitro cultured liver-resident hypnozoites of the simian para- site P. cynomolgi 5 . Collectively, these data demonstrate that KDU691 is active against liver stages in several Plasmodium species, including hypnozoites associated with malaria relapse. To assess imidazopyra- zine activity against a human parasite that forms hypnozoites, we tested 1 Genomics Institute of the Novartis Research Foundation, San Diego, California 92121, USA. 2 Department of Microbiology & Immunology, Columbia University Medical Center, New York, New York 10032, USA. 3 Novartis Institutes for Tropical Disease, 138670 Singapore. 4 Department of Pediatrics, School of Medicine, University of California, San Diego, La Jolla, California 92093, USA. 5 Department of Parasitology, Biomedical Primate Research Centre, PO Box 3306, 2280 GH Rijswijk, The Netherlands. 6 TropIQ Health Sciences, 6525 GA Nijmegen, The Netherlands. 7 Swiss Tropical and Public Health Institute, CH-4002 Basel, Switzerland. 8 University of Basel, CH-4003 Basel, Switzerland. 9 Department of Medicine, School of Medicine, Boston University, Boston, Massachusetts 02118, USA. 10 Novartis Institutes for BioMedical Research, CH-4002 Basel, Switzerland. 11 Department of Medical Microbiology, Radboud University, Nijmegen Medical Centre, PO Box 9101, 6500 HB Nijmegen, The Netherlands. 12 Laboratory of Malaria Immunobiology, Singapore Immunology Network, Agency for Science Technology and Research (A*STAR), Biopolis, 138648 Singapore. 13 Department of Microbiology, Yong Loo Lin School of Medicine, National University of Singapore, National University Health System, 117545 Singapore. 14 Centre for Tropical Medicine, Nuffield Department of Medicine, University of Oxford, Oxford OX3 7BN, UK. 15 Shoklo Malaria Research Unit, Mahidol-Oxford Tropical Medicine Research Unit, Faculty of Tropical Medicine, Mahidol University, Mae Sot 63110, Thailand. 16 Division of Infectious Diseases, Department of Medicine, Columbia University Medical Center, New York, New York 10032, USA. *These authors contributed equally to this work. 00 MONTH 2013 | VOL 000 | NATURE | 1 Macmillan Publishers Limited. All rights reserved ©2013

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ARTICLEdoi:10.1038/nature12782
Targeting Plasmodium PI(4)K toeliminate malariaCase W. McNamara1*, Marcus C. S. Lee2*, Chek Shik Lim3, Siau Hoi Lim3, Jason Roland1, Advait Nagle1, Oliver Simon3,Bryan K. S. Yeung3, Arnab K. Chatterjee1, Susan L. McCormack1, Micah J. Manary4, Anne-Marie Zeeman5, Koen J. Dechering6,T. R. Santha Kumar2, Philipp P. Henrich2, Kerstin Gagaring1, Maureen Ibanez1, Nobutaka Kato1, Kelli L. Kuhen1, Christoph Fischli7,Matthias Rottmann7,8, David M. Plouffe1, Badry Bursulaya1, Stephan Meister4, Lucia Rameh9, Joerg Trappe10, Dorothea Haasen10,Martijn Timmerman6, Robert W. Sauerwein6,11, Rossarin Suwanarusk12, Bruce Russell12,13, Laurent Renia12, Francois Nosten14,15,David C. Tully1, Clemens H. M. Kocken5, Richard J. Glynne1, Christophe Bodenreider3, David A. Fidock2,16, Thierry T. Diagana3
& Elizabeth A. Winzeler1,4
Achieving the goal of malaria elimination will depend on targeting Plasmodium pathways essential across all life stages.Here we identify a lipid kinase, phosphatidylinositol-4-OH kinase (PI(4)K), as the target of imidazopyrazines, a newantimalarial compound class that inhibits the intracellular development of multiple Plasmodium species at each stage ofinfection in the vertebrate host. Imidazopyrazines demonstrate potent preventive, therapeutic, and transmission-blockingactivity in rodent malaria models, are active against blood-stage field isolates of the major human pathogens P. falciparumand P. vivax, and inhibit liver-stage hypnozoites in the simian parasite P. cynomolgi. We show that imidazopyrazines exerttheir effect through inhibitory interaction with the ATP-binding pocket of PI(4)K, altering the intracellular distribution ofphosphatidylinositol-4-phosphate. Collectively, our data define PI(4)K as a key Plasmodium vulnerability, opening upnew avenues of target-based discovery to identify drugs with an ideal activity profile for the prevention, treatment andelimination of malaria.
To eliminate malaria, broadly acting medicines must be developed thatcure the symptomatic asexual blood stage, clear the preceding liver-stage infection that can cause relapses and block parasite transmissionto mosquitoes1. Relapse prevention is especially important for P. vivax,which forms intra-hepatic hypnozoites that can persist for years beforereinitiating development and triggering blood-stage infection. Primaquineis the only licensed antimalarial capable of eliminating the hypnozoitereservoir and delivering a radical cure. However, side effects and weakactivity against blood stages preclude widespread use of primaquine2.Because the target of primaquine and mechanism of action are not known,the search for radical cure drugs has been limited to related analogues, suchas tafenoquine3. There is a clear need for druggable and chemically vali-dated targets that are essential in all life-cycle stages of the malaria parasite.
Here we report that a parasite PI(4)K type III beta (PI(4)KIIIb), aubiquitous eukaryotic enzyme that phosphorylates lipids to regulate intra-cellular signalling and trafficking, is inhibited by imidazopyrazines. Inblood stages, imidazopyrazines block a late step in parasite developmentby disrupting plasma membrane ingression around developing daughtermerozoites. This probably stems from altered phosphatidylinositol-4-phosphate (PI(4)P) pools and disrupted Rab11A-mediated membranetrafficking. To our knowledge, these findings are the first validation ofa drug target that is required across all Plasmodium life-cycle stages.
Imidazopyrazines target several life stagesA cell-based screen against P. falciparum asexual blood-stage parasites4
revealed a new class of antimalarials with an imidazopyrazine core
(Fig. 1a). This compound was distinct from known antimalarials andwas active against several drug-resistant strains (half-maximum inhibitoryconcentration (IC50) 27–70 nM; Extended Data Table 1 and ExtendedData Fig. 1a), suggesting a new mechanism of action. Synthetic deri-vatives with potency and pharmacokinetic properties suitable for test-ing in animal models were designed and showed activity in vivo (datashown for KDU691; Extended Data Table 2 and Extended Data Fig. 1b).
We evaluated imidazopyrazines (Fig. 1a) against all parasite life-cyclestages in the vertebrate host. First, a cell-based assay showed potentinhibition of liver-stage development of the rodent parasite, P. yoelii,with IC50 values less than 160 nM and as low as 9 nM (Fig. 1b) forKDU691, KAI407 and KAI715. In vivo, KDU691 administered orallyat the time of infection prophylactically protected mice from coloniza-tion with transgenic P. berghei sporozoites expressing luciferase. Just asingle dose (7.5 mg kg21; Fig. 1c and Extended Data Fig. 1c) preventedwhat would normally be a fatal outcome. To determine whether KDU691could eliminate an existing infection, animals were dosed 24, 36 or 48 hafter sporozoite inoculation, well after the onset of liver-stage develop-ment (which lasts 48 h in P. berghei). This delayed treatment rapidlyeliminated the luciferase-expressing parasites with a single dose (Fig. 1c).KDU691 was also active at low concentrations (IC50 , 196 nM; Fig. 1d)against in vitro cultured liver-resident hypnozoites of the simian para-site P. cynomolgi5. Collectively, these data demonstrate that KDU691is active against liver stages in several Plasmodium species, includinghypnozoites associated with malaria relapse. To assess imidazopyra-zine activity against a human parasite that forms hypnozoites, we tested
1Genomics Institute of the Novartis Research Foundation, San Diego, California 92121, USA. 2Department of Microbiology & Immunology, Columbia University Medical Center, New York, New York 10032,USA. 3Novartis Institutes for Tropical Disease, 138670 Singapore. 4Department of Pediatrics, School of Medicine, University of California, San Diego, La Jolla, California 92093, USA. 5Department ofParasitology, Biomedical Primate Research Centre, PO Box 3306, 2280 GH Rijswijk, The Netherlands. 6TropIQ Health Sciences, 6525 GA Nijmegen, The Netherlands. 7Swiss Tropical and Public HealthInstitute, CH-4002 Basel, Switzerland. 8University of Basel, CH-4003 Basel, Switzerland. 9Department of Medicine, School of Medicine, Boston University, Boston, Massachusetts 02118, USA. 10NovartisInstitutes for BioMedical Research, CH-4002 Basel, Switzerland. 11Department of Medical Microbiology, Radboud University, Nijmegen Medical Centre, PO Box 9101, 6500 HB Nijmegen, The Netherlands.12Laboratory of Malaria Immunobiology, Singapore Immunology Network, Agency for Science Technology and Research (A*STAR), Biopolis, 138648 Singapore. 13Department of Microbiology, Yong LooLin School of Medicine, National University of Singapore, National University Health System, 117545 Singapore. 14Centre for Tropical Medicine, Nuffield Department of Medicine, University of Oxford,Oxford OX3 7BN, UK. 15Shoklo Malaria Research Unit, Mahidol-Oxford Tropical Medicine Research Unit, Faculty of Tropical Medicine, Mahidol University, Mae Sot 63110, Thailand. 16Division of InfectiousDiseases, Department of Medicine, Columbia University Medical Center, New York, New York 10032, USA.*These authors contributed equally to this work.
0 0 M O N T H 2 0 1 3 | V O L 0 0 0 | N A T U R E | 1
Macmillan Publishers Limited. All rights reserved©2013
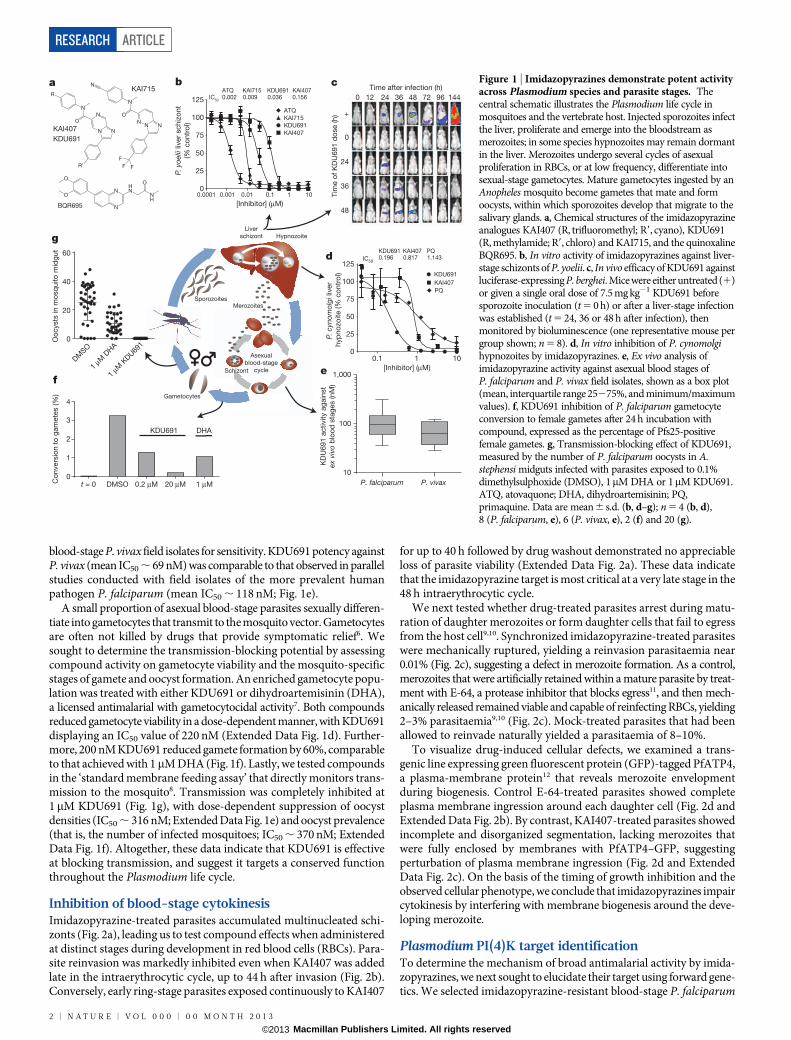
blood-stage P. vivax field isolates for sensitivity. KDU691 potency againstP. vivax (mean IC50 , 69 nM) was comparable to that observed in parallelstudies conducted with field isolates of the more prevalent humanpathogen P. falciparum (mean IC50 , 118 nM; Fig. 1e).
A small proportion of asexual blood-stage parasites sexually differen-tiate into gametocytes that transmit to the mosquito vector. Gametocytesare often not killed by drugs that provide symptomatic relief6. Wesought to determine the transmission-blocking potential by assessingcompound activity on gametocyte viability and the mosquito-specificstages of gamete and oocyst formation. An enriched gametocyte popu-lation was treated with either KDU691 or dihydroartemisinin (DHA),a licensed antimalarial with gametocytocidal activity7. Both compoundsreduced gametocyte viability in a dose-dependent manner, with KDU691displaying an IC50 value of 220 nM (Extended Data Fig. 1d). Further-more, 200 nM KDU691 reduced gamete formation by 60%, comparableto that achieved with 1mM DHA (Fig. 1f). Lastly, we tested compoundsin the ‘standard membrane feeding assay’ that directly monitors trans-mission to the mosquito8. Transmission was completely inhibited at1mM KDU691 (Fig. 1g), with dose-dependent suppression of oocystdensities (IC50 , 316 nM; Extended Data Fig. 1e) and oocyst prevalence(that is, the number of infected mosquitoes; IC50 , 370 nM; ExtendedData Fig. 1f). Altogether, these data indicate that KDU691 is effectiveat blocking transmission, and suggest it targets a conserved functionthroughout the Plasmodium life cycle.
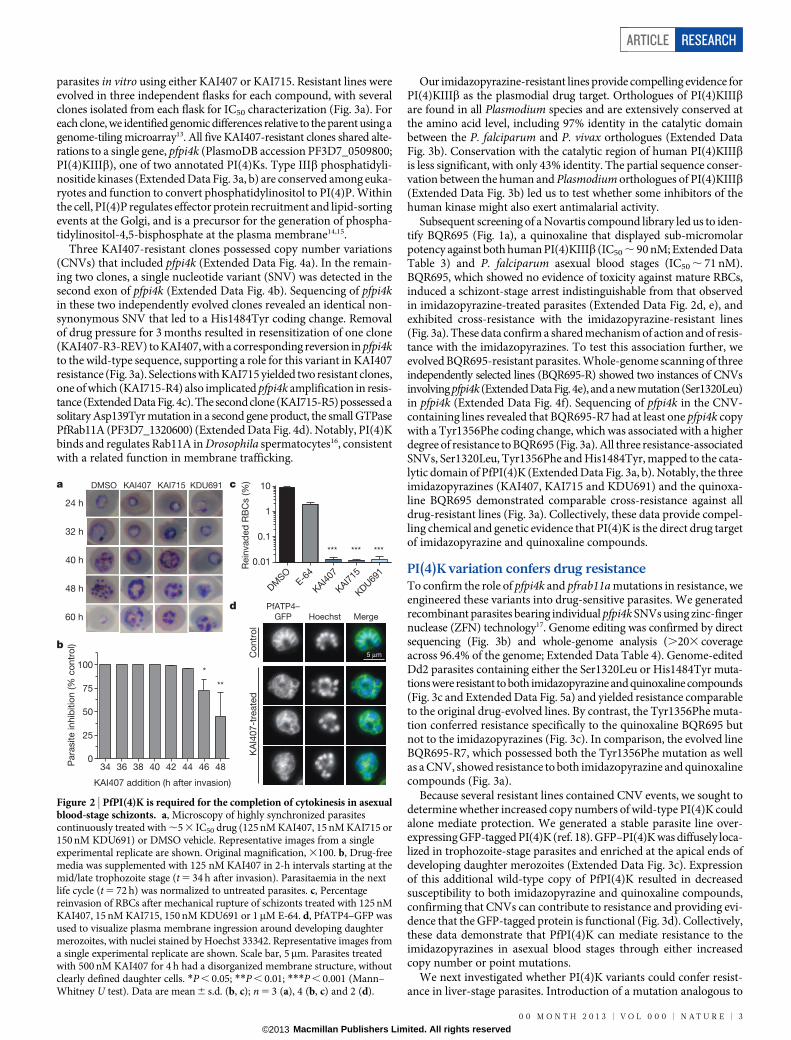
Inhibition of blood-stage cytokinesisImidazopyrazine-treated parasites accumulated multinucleated schi-zonts (Fig. 2a), leading us to test compound effects when administeredat distinct stages during development in red blood cells (RBCs). Para-site reinvasion was markedly inhibited even when KAI407 was addedlate in the intraerythrocytic cycle, up to 44 h after invasion (Fig. 2b).Conversely, early ring-stage parasites exposed continuously to KAI407
for up to 40 h followed by drug washout demonstrated no appreciableloss of parasite viability (Extended Data Fig. 2a). These data indicatethat the imidazopyrazine target is most critical at a very late stage in the48 h intraerythrocytic cycle.
We next tested whether drug-treated parasites arrest during matu-ration of daughter merozoites or form daughter cells that fail to egressfrom the host cell9,10. Synchronized imidazopyrazine-treated parasiteswere mechanically ruptured, yielding a reinvasion parasitaemia near0.01% (Fig. 2c), suggesting a defect in merozoite formation. As a control,merozoites that were artificially retained within a mature parasite by treat-ment with E-64, a protease inhibitor that blocks egress11, and then mech-anically released remained viable and capable of reinfecting RBCs, yielding2–3% parasitaemia9,10 (Fig. 2c). Mock-treated parasites that had beenallowed to reinvade naturally yielded a parasitaemia of 8–10%.
To visualize drug-induced cellular defects, we examined a trans-genic line expressing green fluorescent protein (GFP)-tagged PfATP4,a plasma-membrane protein12 that reveals merozoite envelopmentduring biogenesis. Control E-64-treated parasites showed completeplasma membrane ingression around each daughter cell (Fig. 2d andExtended Data Fig. 2b). By contrast, KAI407-treated parasites showedincomplete and disorganized segmentation, lacking merozoites thatwere fully enclosed by membranes with PfATP4–GFP, suggestingperturbation of plasma membrane ingression (Fig. 2d and ExtendedData Fig. 2c). On the basis of the timing of growth inhibition and theobserved cellular phenotype, we conclude that imidazopyrazines impaircytokinesis by interfering with membrane biogenesis around the deve-loping merozoite.
Plasmodium PI(4)K target identificationTo determine the mechanism of broad antimalarial activity by imida-zopyrazines, we next sought to elucidate their target using forward gene-tics. We selected imidazopyrazine-resistant blood-stage P. falciparum
KAI407
KDU691
BQR695
a cTime after infection (h)
0 12 24 36 48 72 96 144
+
0
24
36
48
b
0
25
50
75
100
125
[Inhibitor] (μM)
0.0001 0.001 0.01 10
KDU691
KAI407
KAI715
ATQ
ATQ0.002
KAI7150.009
KDU6910.036
KAI4070.156
100
25
50
75
100
125
PQ
KDU691
KAI407
10.1
KDU691 KAI407 PQ0.196 0.817 1.143d
P. falciparum P. vivax 10
100
1,000ef
g
1 μM
DHA
0
20
40
60
DM
SO
NH
OHN
N
N
O
O
R′
N
N
N
O
N
R
N
O
NN N
F
F F
NKAI715
0.1 1
1 μM
KDU69
1
Oo
cysts
in
mo
sq
uito
mid
gu
t
KD
U6
91
activity a
gain
st
ex v
ivo
blo
od
sta
ges (n
M)
P. c
ynom
olgi
liv
er
hyp
no
zo
ite (%
co
ntr
ol)
Tim
e o
f K
DU
69
1 d
ose (h
)
P. y
oelii
liv
er
sch
izo
nt
(% c
on
tro
l)
4
[Inhibitor] (μM)
Co
nvers
ion
to
gam
ete
s (%
)
0
1
2
3
KDU691 DHA
t = 0 DMSO 0.2 μM 20 μM 1 μM
Liver
schizont
Gametocytes
Hypnozoite
Asexual
blood-stage
cycle
MerozoitesSporozoites
Schizont
IC50
IC50
Figure 1 | Imidazopyrazines demonstrate potent activityacross Plasmodium species and parasite stages. Thecentral schematic illustrates the Plasmodium life cycle inmosquitoes and the vertebrate host. Injected sporozoites infectthe liver, proliferate and emerge into the bloodstream asmerozoites; in some species hypnozoites may remain dormantin the liver. Merozoites undergo several cycles of asexualproliferation in RBCs, or at low frequency, differentiate intosexual-stage gametocytes. Mature gametocytes ingested by anAnopheles mosquito become gametes that mate and formoocysts, within which sporozoites develop that migrate to thesalivary glands. a, Chemical structures of the imidazopyrazineanalogues KAI407 (R, trifluoromethyl; R9, cyano), KDU691(R, methylamide; R9, chloro) and KAI715, and the quinoxalineBQR695. b, In vitro activity of imidazopyrazines against liver-stage schizonts of P. yoelii. c, In vivo efficacy of KDU691 againstluciferase-expressing P. berghei. Mice were either untreated (1)or given a single oral dose of 7.5 mg kg21 KDU691 beforesporozoite inoculation (t 5 0 h) or after a liver-stage infectionwas established (t 5 24, 36 or 48 h after infection), thenmonitored by bioluminescence (one representative mouse pergroup shown; n 5 8). d, In vitro inhibition of P. cynomolgihypnozoites by imidazopyrazines. e, Ex vivo analysis ofimidazopyrazine activity against asexual blood stages ofP. falciparum and P. vivax field isolates, shown as a box plot(mean, interquartile range 25275%, and minimum/maximumvalues). f, KDU691 inhibition of P. falciparum gametocyteconversion to female gametes after 24 h incubation withcompound, expressed as the percentage of Pfs25-positivefemale gametes. g, Transmission-blocking effect of KDU691,measured by the number of P. falciparum oocysts in A.stephensi midguts infected with parasites exposed to 0.1%dimethylsulphoxide (DMSO), 1mM DHA or 1mM KDU691.ATQ, atovaquone; DHA, dihydroartemisinin; PQ,primaquine. Data are mean 6 s.d. (b, d–g); n 5 4 (b, d),8 (P. falciparum, e), 6 (P. vivax, e), 2 (f) and 20 (g).
RESEARCH ARTICLE
2 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013
parasites in vitro using either KAI407 or KAI715. Resistant lines wereevolved in three independent flasks for each compound, with severalclones isolated from each flask for IC50 characterization (Fig. 3a). Foreach clone, we identified genomic differences relative to the parent using agenome-tiling microarray13. All five KAI407-resistant clones shared alte-rations to a single gene, pfpi4k (PlasmoDB accession PF3D7_0509800;PI(4)KIIIb), one of two annotated PI(4)Ks. Type IIIb phosphatidyli-nositide kinases (Extended Data Fig. 3a, b) are conserved among euka-ryotes and function to convert phosphatidylinositol to PI(4)P. Withinthe cell, PI(4)P regulates effector protein recruitment and lipid-sortingevents at the Golgi, and is a precursor for the generation of phospha-tidylinositol-4,5-bisphosphate at the plasma membrane14,15.
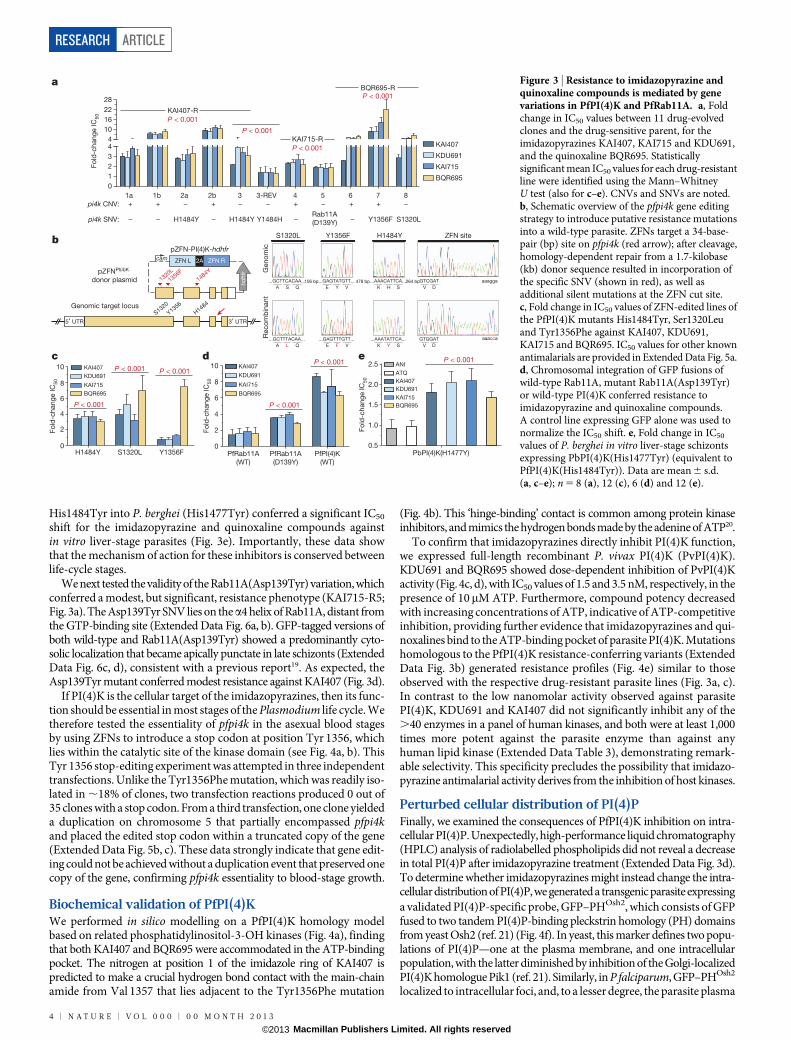
Three KAI407-resistant clones possessed copy number variations(CNVs) that included pfpi4k (Extended Data Fig. 4a). In the remain-ing two clones, a single nucleotide variant (SNV) was detected in thesecond exon of pfpi4k (Extended Data Fig. 4b). Sequencing of pfpi4kin these two independently evolved clones revealed an identical non-synonymous SNV that led to a His1484Tyr coding change. Removalof drug pressure for 3 months resulted in resensitization of one clone(KAI407-R3-REV) to KAI407, with a corresponding reversion in pfpi4kto the wild-type sequence, supporting a role for this variant in KAI407resistance (Fig. 3a). Selections with KAI715 yielded two resistant clones,one of which (KAI715-R4) also implicated pfpi4k amplification in resis-tance (Extended Data Fig. 4c). The second clone (KAI715-R5) possessed asolitary Asp139Tyr mutation in a second gene product, the small GTPasePfRab11A (PF3D7_1320600) (Extended Data Fig. 4d). Notably, PI(4)Kbinds and regulates Rab11A in Drosophila spermatocytes16, consistentwith a related function in membrane trafficking.
Our imidazopyrazine-resistant lines provide compelling evidence forPI(4)KIIIb as the plasmodial drug target. Orthologues of PI(4)KIIIbare found in all Plasmodium species and are extensively conserved atthe amino acid level, including 97% identity in the catalytic domainbetween the P. falciparum and P. vivax orthologues (Extended DataFig. 3b). Conservation with the catalytic region of human PI(4)KIIIbis less significant, with only 43% identity. The partial sequence conser-vation between the human and Plasmodium orthologues of PI(4)KIIIb(Extended Data Fig. 3b) led us to test whether some inhibitors of thehuman kinase might also exert antimalarial activity.
Subsequent screening of a Novartis compound library led us to iden-tify BQR695 (Fig. 1a), a quinoxaline that displayed sub-micromolarpotency against both human PI(4)KIIIb (IC50 , 90 nM; Extended DataTable 3) and P. falciparum asexual blood stages (IC50 , 71 nM).BQR695, which showed no evidence of toxicity against mature RBCs,induced a schizont-stage arrest indistinguishable from that observedin imidazopyrazine-treated parasites (Extended Data Fig. 2d, e), andexhibited cross-resistance with the imidazopyrazine-resistant lines(Fig. 3a). These data confirm a shared mechanism of action and of resis-tance with the imidazopyrazines. To test this association further, weevolved BQR695-resistant parasites. Whole-genome scanning of threeindependently selected lines (BQR695-R) showed two instances of CNVsinvolving pfpi4k (Extended Data Fig. 4e), and a new mutation (Ser1320Leu)in pfpi4k (Extended Data Fig. 4f). Sequencing of pfpi4k in the CNV-containing lines revealed that BQR695-R7 had at least one pfpi4k copywith a Tyr1356Phe coding change, which was associated with a higherdegree of resistance to BQR695 (Fig. 3a). All three resistance-associatedSNVs, Ser1320Leu, Tyr1356Phe and His1484Tyr, mapped to the cata-lytic domain of PfPI(4)K (Extended Data Fig. 3a, b). Notably, the threeimidazopyrazines (KAI407, KAI715 and KDU691) and the quinoxa-line BQR695 demonstrated comparable cross-resistance against alldrug-resistant lines (Fig. 3a). Collectively, these data provide compel-ling chemical and genetic evidence that PI(4)K is the direct drug targetof imidazopyrazine and quinoxaline compounds.
PI(4)K variation confers drug resistanceTo confirm the role of pfpi4k and pfrab11a mutations in resistance, weengineered these variants into drug-sensitive parasites. We generatedrecombinant parasites bearing individual pfpi4k SNVs using zinc-fingernuclease (ZFN) technology17. Genome editing was confirmed by directsequencing (Fig. 3b) and whole-genome analysis (.203 coverageacross 96.4% of the genome; Extended Data Table 4). Genome-editedDd2 parasites containing either the Ser1320Leu or His1484Tyr muta-tions were resistant to both imidazopyrazine and quinoxaline compounds(Fig. 3c and Extended Data Fig. 5a) and yielded resistance comparableto the original drug-evolved lines. By contrast, the Tyr1356Phe muta-tion conferred resistance specifically to the quinoxaline BQR695 butnot to the imidazopyrazines (Fig. 3c). In comparison, the evolved lineBQR695-R7, which possessed both the Tyr1356Phe mutation as wellas a CNV, showed resistance to both imidazopyrazine and quinoxalinecompounds (Fig. 3a).
Because several resistant lines contained CNV events, we sought todetermine whether increased copy numbers of wild-type PI(4)K couldalone mediate protection. We generated a stable parasite line over-expressing GFP-tagged PI(4)K (ref. 18). GFP–PI(4)K was diffusely loca-lized in trophozoite-stage parasites and enriched at the apical ends ofdeveloping daughter merozoites (Extended Data Fig. 3c). Expressionof this additional wild-type copy of PfPI(4)K resulted in decreasedsusceptibility to both imidazopyrazine and quinoxaline compounds,confirming that CNVs can contribute to resistance and providing evi-dence that the GFP-tagged protein is functional (Fig. 3d). Collectively,these data demonstrate that PfPI(4)K can mediate resistance to theimidazopyrazines in asexual blood stages through either increasedcopy number or point mutations.
We next investigated whether PI(4)K variants could confer resist-ance in liver-stage parasites. Introduction of a mutation analogous to
PfATP4–
GFP MergeHoechst
Co
ntr
ol
KA
I407-t
reate
d
5 μm
a
b
d
c
Para
site inhib
itio
n (%
co
ntr
ol)
Rein
vad
ed
RB
Cs (%
)
24 h
32 h
40 h
48 h
60 h
KDU691KAI407 KAI715DMSO
*** *** ***
**
*
KAI407 addition (h after invasion)
34 36 38 40 42 44 46 480
25
50
75
100
0.01
0.1
1
10
DM
SO
KAI715
KAI407
E-64
KDU69
1
Figure 2 | PfPI(4)K is required for the completion of cytokinesis in asexualblood-stage schizonts. a, Microscopy of highly synchronized parasitescontinuously treated with ,5 3 IC50 drug (125 nM KAI407, 15 nM KAI715 or150 nM KDU691) or DMSO vehicle. Representative images from a singleexperimental replicate are shown. Original magnification, 3100. b, Drug-freemedia was supplemented with 125 nM KAI407 in 2-h intervals starting at themid/late trophozoite stage (t 5 34 h after invasion). Parasitaemia in the nextlife cycle (t 5 72 h) was normalized to untreated parasites. c, Percentagereinvasion of RBCs after mechanical rupture of schizonts treated with 125 nMKAI407, 15 nM KAI715, 150 nM KDU691 or 1mM E-64. d, PfATP4–GFP wasused to visualize plasma membrane ingression around developing daughtermerozoites, with nuclei stained by Hoechst 33342. Representative images froma single experimental replicate are shown. Scale bar, 5mm. Parasites treatedwith 500 nM KAI407 for 4 h had a disorganized membrane structure, withoutclearly defined daughter cells. *P , 0.05; **P , 0.01; ***P , 0.001 (Mann–Whitney U test). Data are mean 6 s.d. (b, c); n 5 3 (a), 4 (b, c) and 2 (d).
ARTICLE RESEARCH
0 0 M O N T H 2 0 1 3 | V O L 0 0 0 | N A T U R E | 3
Macmillan Publishers Limited. All rights reserved©2013
His1484Tyr into P. berghei (His1477Tyr) conferred a significant IC50
shift for the imidazopyrazine and quinoxaline compounds againstin vitro liver-stage parasites (Fig. 3e). Importantly, these data showthat the mechanism of action for these inhibitors is conserved betweenlife-cycle stages.
We next tested the validity of the Rab11A(Asp139Tyr) variation, whichconferred a modest, but significant, resistance phenotype (KAI715-R5;Fig. 3a). The Asp139Tyr SNV lies on thea4 helix of Rab11A, distant fromthe GTP-binding site (Extended Data Fig. 6a, b). GFP-tagged versions ofboth wild-type and Rab11A(Asp139Tyr) showed a predominantly cyto-solic localization that became apically punctate in late schizonts (ExtendedData Fig. 6c, d), consistent with a previous report19. As expected, theAsp139Tyr mutant conferred modest resistance against KAI407 (Fig. 3d).
If PI(4)K is the cellular target of the imidazopyrazines, then its func-tion should be essential in most stages of the Plasmodium life cycle. Wetherefore tested the essentiality of pfpi4k in the asexual blood stagesby using ZFNs to introduce a stop codon at position Tyr 1356, whichlies within the catalytic site of the kinase domain (see Fig. 4a, b). ThisTyr 1356 stop-editing experiment was attempted in three independenttransfections. Unlike the Tyr1356Phe mutation, which was readily iso-lated in ,18% of clones, two transfection reactions produced 0 out of35 clones with a stop codon. From a third transfection, one clone yieldeda duplication on chromosome 5 that partially encompassed pfpi4kand placed the edited stop codon within a truncated copy of the gene(Extended Data Fig. 5b, c). These data strongly indicate that gene edit-ing could not be achieved without a duplication event that preserved onecopy of the gene, confirming pfpi4k essentiality to blood-stage growth.
Biochemical validation of PfPI(4)KWe performed in silico modelling on a PfPI(4)K homology modelbased on related phosphatidylinositol-3-OH kinases (Fig. 4a), findingthat both KAI407 and BQR695 were accommodated in the ATP-bindingpocket. The nitrogen at position 1 of the imidazole ring of KAI407 ispredicted to make a crucial hydrogen bond contact with the main-chainamide from Val 1357 that lies adjacent to the Tyr1356Phe mutation
(Fig. 4b). This ‘hinge-binding’ contact is common among protein kinaseinhibitors, and mimics the hydrogen bonds made by the adenine of ATP20.
To confirm that imidazopyrazines directly inhibit PI(4)K function,we expressed full-length recombinant P. vivax PI(4)K (PvPI(4)K).KDU691 and BQR695 showed dose-dependent inhibition of PvPI(4)Kactivity (Fig. 4c, d), with IC50 values of 1.5 and 3.5 nM, respectively, in thepresence of 10mM ATP. Furthermore, compound potency decreasedwith increasing concentrations of ATP, indicative of ATP-competitiveinhibition, providing further evidence that imidazopyrazines and qui-noxalines bind to the ATP-binding pocket of parasite PI(4)K. Mutationshomologous to the PfPI(4)K resistance-conferring variants (ExtendedData Fig. 3b) generated resistance profiles (Fig. 4e) similar to thoseobserved with the respective drug-resistant parasite lines (Fig. 3a, c).In contrast to the low nanomolar activity observed against parasitePI(4)K, KDU691 and KAI407 did not significantly inhibit any of the.40 enzymes in a panel of human kinases, and both were at least 1,000times more potent against the parasite enzyme than against anyhuman lipid kinase (Extended Data Table 3), demonstrating remark-able selectivity. This specificity precludes the possibility that imidazo-pyrazine antimalarial activity derives from the inhibition of host kinases.
Perturbed cellular distribution of PI(4)PFinally, we examined the consequences of PfPI(4)K inhibition on intra-cellular PI(4)P. Unexpectedly, high-performance liquid chromatography(HPLC) analysis of radiolabelled phospholipids did not reveal a decreasein total PI(4)P after imidazopyrazine treatment (Extended Data Fig. 3d).To determine whether imidazopyrazines might instead change the intra-cellular distribution of PI(4)P, we generated a transgenic parasite expressinga validated PI(4)P-specific probe, GFP–PHOsh2, which consists of GFPfused to two tandem PI(4)P-binding pleckstrin homology (PH) domainsfrom yeast Osh2 (ref. 21) (Fig. 4f). In yeast, this marker defines two popu-lations of PI(4)P—one at the plasma membrane, and one intracellularpopulation, with the latter diminished by inhibition of the Golgi-localizedPI(4)K homologue Pik1 (ref. 21). Similarly, in P falciparum, GFP–PHOsh2
localized to intracellular foci, and, to a lesser degree, the parasite plasma
Genomic target locus
pZFNPI(4)K
donor plasmid
pZFN-PI(4)K-hdhfrcam 2AZFN L ZFN R
hdhf
r
5′ UTR 3′ UTR
S1320
H1484
Y1356
1320L
1484Y
1356F
a
b
c d
S1320L ZFN siteH1484Y
Geno
mic
Y1356F
...GCTTCACAA...
A S Q
...GAGTATGTT...
E Y V
...AAACATTCA...
K H S
GTCGAT
V D
aaagga264 bp478 bp106 bp
Reco
mb
inant
...GCTTTACAA...
A L Q
...AAATATTCA...
K Y S
GTGGAT
V D
aaacca...GAGTTTGTT...
E F V
KAI407-R
KAI715-R
BQR695-RF
old
-chang
e IC
50
pi4k CNV:
pi4k SNV:
+
–
+
–
+
–
–
H1484Y
–
H1484Y
–
Y1484H
–
Rab11A
(D139Y)
+
–
+
–
–
S1320L
+
Y1356F
1a 1b 2a 2b 3 3-REV 4 6 7 8
KAI407
KDU691
KAI715
BQR695
5
0
1
2
3
44
10
16
22
28
P < 0.001
P < 0.001
P < 0.001
Fo
ld-c
hang
e IC
50
PfRab11A
(WT)
PfRab11A
(D139Y)
PfPI(4)K
(WT)
0
2
4
6
8
10 KAI407
KDU691
KAI715
BQR695
P < 0.001
P < 0.001
Fo
ld-c
hang
e IC
50
H1484Y S1320L Y1356F0
2
4
6
8
10 KAI407
KDU691
KAI715
BQR695
P < 0.001P < 0.001
P < 0.001
e
PbPI(4)K(H1477Y)0.5
1.0
1.5
2.0
2.5F
old
-chang
e IC
50
P < 0.001
KAI407
KDU691
KAI715
BQR695
ANI
ATQ
P < 0.001
Figure 3 | Resistance to imidazopyrazine andquinoxaline compounds is mediated by genevariations in PfPI(4)K and PfRab11A. a, Foldchange in IC50 values between 11 drug-evolvedclones and the drug-sensitive parent, for theimidazopyrazines KAI407, KAI715 and KDU691,and the quinoxaline BQR695. Statisticallysignificant mean IC50 values for each drug-resistantline were identified using the Mann–WhitneyU test (also for c–e). CNVs and SNVs are noted.b, Schematic overview of the pfpi4k gene editingstrategy to introduce putative resistance mutationsinto a wild-type parasite. ZFNs target a 34-base-pair (bp) site on pfpi4k (red arrow); after cleavage,homology-dependent repair from a 1.7-kilobase(kb) donor sequence resulted in incorporation ofthe specific SNV (shown in red), as well asadditional silent mutations at the ZFN cut site.c, Fold change in IC50 values of ZFN-edited lines ofthe PfPI(4)K mutants His1484Tyr, Ser1320Leuand Tyr1356Phe against KAI407, KDU691,KAI715 and BQR695. IC50 values for other knownantimalarials are provided in Extended Data Fig. 5a.d, Chromosomal integration of GFP fusions ofwild-type Rab11A, mutant Rab11A(Asp139Tyr)or wild-type PI(4)K conferred resistance toimidazopyrazine and quinoxaline compounds.A control line expressing GFP alone was used tonormalize the IC50 shift. e, Fold change in IC50
values of P. berghei in vitro liver-stage schizontsexpressing PbPI(4)K(His1477Tyr) (equivalent toPfPI(4)K(His1484Tyr)). Data are mean 6 s.d.(a, c–e); n 5 8 (a), 12 (c), 6 (d) and 12 (e).
RESEARCH ARTICLE
4 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013
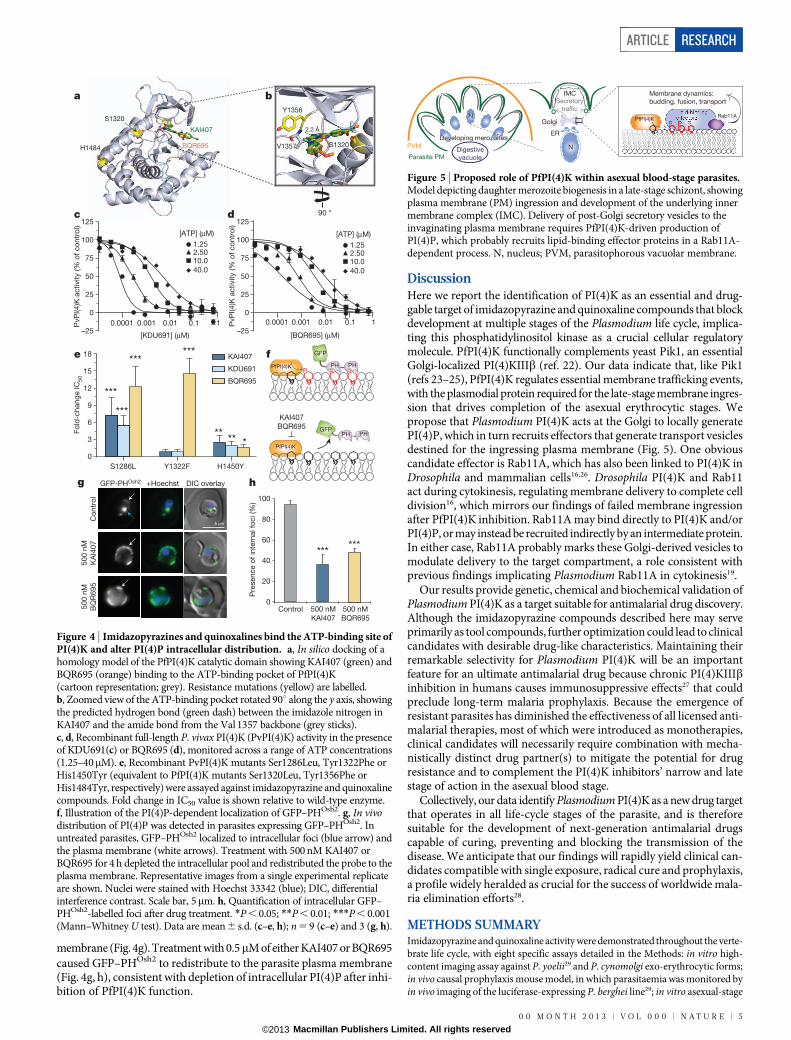
membrane (Fig. 4g). Treatment with 0.5mM of either KAI407 or BQR695caused GFP–PHOsh2 to redistribute to the parasite plasma membrane(Fig. 4g, h), consistent with depletion of intracellular PI(4)P after inhi-bition of PfPI(4)K function.
DiscussionHere we report the identification of PI(4)K as an essential and drug-gable target of imidazopyrazine and quinoxaline compounds that blockdevelopment at multiple stages of the Plasmodium life cycle, implica-ting this phosphatidylinositol kinase as a crucial cellular regulatorymolecule. PfPI(4)K functionally complements yeast Pik1, an essentialGolgi-localized PI(4)KIIIb (ref. 22). Our data indicate that, like Pik1(refs 23–25), PfPI(4)K regulates essential membrane trafficking events,with the plasmodial protein required for the late-stage membrane ingres-sion that drives completion of the asexual erythrocytic stages. Wepropose that Plasmodium PI(4)K acts at the Golgi to locally generatePI(4)P, which in turn recruits effectors that generate transport vesiclesdestined for the ingressing plasma membrane (Fig. 5). One obviouscandidate effector is Rab11A, which has also been linked to PI(4)K inDrosophila and mammalian cells16,26. Drosophila PI(4)K and Rab11act during cytokinesis, regulating membrane delivery to complete celldivision16, which mirrors our findings of failed membrane ingressionafter PfPI(4)K inhibition. Rab11A may bind directly to PI(4)K and/orPI(4)P, or may instead be recruited indirectly by an intermediate protein.In either case, Rab11A probably marks these Golgi-derived vesicles tomodulate delivery to the target compartment, a role consistent withprevious findings implicating Plasmodium Rab11A in cytokinesis19.
Our results provide genetic, chemical and biochemical validation ofPlasmodium PI(4)K as a target suitable for antimalarial drug discovery.Although the imidazopyrazine compounds described here may serveprimarily as tool compounds, further optimization could lead to clinicalcandidates with desirable drug-like characteristics. Maintaining theirremarkable selectivity for Plasmodium PI(4)K will be an importantfeature for an ultimate antimalarial drug because chronic PI(4)KIIIbinhibition in humans causes immunosuppressive effects27 that couldpreclude long-term malaria prophylaxis. Because the emergence ofresistant parasites has diminished the effectiveness of all licensed anti-malarial therapies, most of which were introduced as monotherapies,clinical candidates will necessarily require combination with mecha-nistically distinct drug partner(s) to mitigate the potential for drugresistance and to complement the PI(4)K inhibitors’ narrow and latestage of action in the asexual blood stage.
Collectively, our data identify Plasmodium PI(4)K as a new drug targetthat operates in all life-cycle stages of the parasite, and is thereforesuitable for the development of next-generation antimalarial drugscapable of curing, preventing and blocking the transmission of thedisease. We anticipate that our findings will rapidly yield clinical can-didates compatible with single exposure, radical cure and prophylaxis,a profile widely heralded as crucial for the success of worldwide mala-ria elimination efforts28.
METHODS SUMMARYImidazopyrazine and quinoxaline activity were demonstrated throughout the verte-brate life cycle, with eight specific assays detailed in the Methods: in vitro high-content imaging assay against P. yoelii29 and P. cynomolgi exo-erythrocytic forms;in vivo causal prophylaxis mouse model, in which parasitaemia was monitored byin vivo imaging of the luciferase-expressing P. berghei line29; in vitro asexual-stage
KAI407
BQR695H1484
S1320
S1320V1357
90 °
2.2 Å
Y1356
c d
e
h
f
g
P PPH PH
GFPPH PH
PfPI(4)KP
PfPI(4)K
KAI407
BQR695
GFP
DIC overlay+HoechstGFP-PHOsh2
Co
ntr
ol
5 μm
50
0 n
M
KA
I40
7
50
0 n
M
BQ
R695
a b
S1286L Y1322F H1450Y
0
3
6
9
12
15
18 KAI407
KDU691
BQR695
Fo
ld-c
han
ge IC
50
***
***
******
**** *
0.0001 0.001 0.01 0.1–25
0
25
50
75
100
125
[KDU691] (μM)
PvP
I(4)K
activity (%
of
co
ntr
ol)
[ATP] (μM)
1.252.5010.0
40.0
1 0.0001 0.001 0.01 0.1–25
0
25
50
75
100
125
[BQR695] (μM)
PvP
I(4)K
activity (%
of
co
ntr
ol)
[ATP] (μM)
1.252.5010.0
40.0
1
0
20
40
60
80
100
Control 500 nM
KAI407
500 nM
BQR695
Pre
sence o
f in
tern
al fo
ci (%
)
******
Figure 4 | Imidazopyrazines and quinoxalines bind the ATP-binding site ofPI(4)K and alter PI(4)P intracellular distribution. a, In silico docking of ahomology model of the PfPI(4)K catalytic domain showing KAI407 (green) andBQR695 (orange) binding to the ATP-binding pocket of PfPI(4)K(cartoon representation; grey). Resistance mutations (yellow) are labelled.b, Zoomed view of the ATP-binding pocket rotated 90u along the y axis, showingthe predicted hydrogen bond (green dash) between the imidazole nitrogen inKAI407 and the amide bond from the Val 1357 backbone (grey sticks).c, d, Recombinant full-length P. vivax PI(4)K (PvPI(4)K) activity in the presenceof KDU691(c) or BQR695 (d), monitored across a range of ATP concentrations(1.25–40mM). e, Recombinant PvPI(4)K mutants Ser1286Leu, Tyr1322Phe orHis1450Tyr (equivalent to PfPI(4)K mutants Ser1320Leu, Tyr1356Phe orHis1484Tyr, respectively) were assayed against imidazopyrazine and quinoxalinecompounds. Fold change in IC50 value is shown relative to wild-type enzyme.f, Illustration of the PI(4)P-dependent localization of GFP–PHOsh2. g, In vivodistribution of PI(4)P was detected in parasites expressing GFP–PHOsh2. Inuntreated parasites, GFP–PHOsh2 localized to intracellular foci (blue arrow) andthe plasma membrane (white arrows). Treatment with 500 nM KAI407 orBQR695 for 4 h depleted the intracellular pool and redistributed the probe to theplasma membrane. Representative images from a single experimental replicateare shown. Nuclei were stained with Hoechst 33342 (blue); DIC, differentialinterference contrast. Scale bar, 5mm. h, Quantification of intracellular GFP–PHOsh2-labelled foci after drug treatment. *P , 0.05; **P , 0.01; ***P , 0.001(Mann–Whitney U test). Data are mean 6 s.d. (c–e, h); n 5 9 (c–e) and 3 (g, h).
N
Digestive
vacuoleParasite PM
PVM
IMC
P P PPfPI(4)K
Rab11A
Membrane dynamics:
budding, fusion, transport
Developing merozoitesDeveloping merozoitesER
Golgi
N
Secretory
trafficLipid-bindingeffectors
Figure 5 | Proposed role of PfPI(4)K within asexual blood-stage parasites.Model depicting daughter merozoite biogenesis in a late-stage schizont, showingplasma membrane (PM) ingression and development of the underlying innermembrane complex (IMC). Delivery of post-Golgi secretory vesicles to theinvaginating plasma membrane requires PfPI(4)K-driven production ofPI(4)P, which probably recruits lipid-binding effector proteins in a Rab11A-dependent process. N, nucleus; PVM, parasitophorous vacuolar membrane.
ARTICLE RESEARCH
0 0 M O N T H 2 0 1 3 | V O L 0 0 0 | N A T U R E | 5
Macmillan Publishers Limited. All rights reserved©2013

activity against P. falciparum, measured using a modified SYBR Green cell proli-feration assay4; ex vivo asexual-stage assay against P. vivax and P. falciparum isolatescollected from Thailand30,31; gametocyte viability assays against P. falciparum NF54stage III2V gametocytes measured by a modified Plasmodium lactate dehydrogenaseassay32; in vitro gametogenesis assay against P. falciparum quantified by flow cytometry33;and P. falciparum transmission to Anopheles stephensi mosquitoes in a standardmembrane feeding assay34. Stage of action studies12 and a merozoite release assay9,10
were used to identify specific arrest of drug-treated asexual parasites in mid-schizogony.An established protocol13 was used for in vitro evolution and comparative genomicanalysis of KAI407-, KAI715- or BQR695-resistant lines. Resistance-conferringmutations were validated with ZFN approaches17 and standard transfection-basedmethods12. Genomic DNA libraries generated on an Illumina HiSeq 2000 wereanalysed with PlaTypUS, an internal WGS pipeline. A P. berghei line expressingPI(4)K(His1477Tyr) was generated by standard methods35, and resistance wasquantified against liver-stage parasites29. A homology model of PfPI(4)K was builtfrom atomic coordinates for human phosphatidylinositol-3-OH kinase gamma(PI(3)Kc; Protein Data Bank accession 3ENE) and used for flexible ligand dockingstudies within Glide (Schrodinger). Full-length PvPI(4)K was expressed in a SF9/baculovirus system and assayed for drug inhibition with the Transcreener ADP2
FP detection kit (BellBrook). PI(4)P pools were detected in asexual stage parasitesby expression of tandem Osh2 pleckstrin homology domains fused to GFP21.
Online Content Any additional Methods, Extended Data display items and SourceData are available in the online version of the paper; references unique to thesesections appear only in the online paper.
Received 23 July; accepted 15 October 2013.
Published online 27 November 2013.
1. Greenwood, B. M. et al. Malaria: progress, perils, and prospects for eradication.J. Clin. Invest. 118, 1266–1276 (2008).
2. Vale, N., Moreira, R. & Gomes, P. Primaquine revisited six decades after itsdiscovery. Eur. J. Med. Chem. 44, 937–953 (2009).
3. Wells, T. N., Burrows, J. N. & Baird, J. K. Targeting the hypnozoite reservoir ofPlasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 26,145–151 (2010).
4. Plouffe, D. et al. In silico activity profiling reveals the mechanism of action ofantimalarials discovered in a high-throughput screen. Proc. Natl Acad. Sci. USA105, 9059–9064 (2008).
5. Dembele, L. et al.Towardsan in vitro model of Plasmodiumhypnozoites suitable fordrug discovery. PLoS ONE 6, e18162 (2011).
6. Bousema, T. & Drakeley, C. Epidemiology and infectivity of Plasmodium falciparumand Plasmodium vivax gametocytes in relation to malaria control and elimination.Clin. Microbiol. Rev. 24, 377–410 (2011).
7. Adjalley, S. H. et al. Quantitative assessment of Plasmodium falciparum sexualdevelopment reveals potent transmission-blocking activity by methylene blue.Proc. Natl Acad. Sci. USA 108, E1214–E1223 (2011).
8. van Pelt-Koops, J. C. et al. The spiroindolone drug candidate NITD609 potentlyinhibits gametocytogenesis and blocks Plasmodium falciparum transmission toAnopheles mosquito vector. Antimicrob. Agents Chemother. 56, 3544–3548(2012).
9. Boyle, M. J. et al. Isolation of viable Plasmodium falciparum merozoites to defineerythrocyte invasion events and advance vaccine and drug development. Proc.Natl Acad. Sci. USA 107, 14378–14383 (2010).
10. Dvorin, J. D. et al. A plant-like kinase in Plasmodium falciparum regulates parasiteegress from erythrocytes. Science 328, 910–912 (2010).
11. Drew, M. E. et al. Plasmodium food vacuole plasmepsins are activated by falcipains.J. Biol. Chem. 283, 12870–12876 (2008).
12. Rottmann, M. et al. Spiroindolones, a potent compound class for the treatment ofmalaria. Science 329, 1175–1180 (2010).
13. Dharia, N. V. et al. Use of high-density tiling microarrays to identify mutationsglobally and elucidate mechanisms of drug resistance in Plasmodium falciparum.Genome Biol. 10, R21 (2009).
14. Balla, A. & Balla, T. Phosphatidylinositol 4-kinases: old enzymes with emergingfunctions. Trends Cell Biol. 16, 351–361 (2006).
15. Mayinger, P. Phosphoinositides and vesicular membrane traffic. Biochim. Biophys.Acta 1821, 1104–1113 (2012).
16. Polevoy, G. et al. Dual roles for the Drosophila PI 4-kinase four wheel drive inlocalizing Rab11 during cytokinesis. J. Cell Biol. 187, 847–858 (2009).
17. Straimer, J. et al. Site-specific genome editing in Plasmodium falciparum usingengineered zinc-finger nucleases. Nature Methods 9, 993–998 (2012).
18. Nkrumah, L. J. et al. Efficient site-specific integration in Plasmodium falciparumchromosomes mediated by mycobacteriophage Bxb1 integrase. Nature Methods3, 615–621 (2006).
19. Agop-Nersesian, C. et al. Rab11A-controlled assembly of the inner membranecomplex is required for completion of apicomplexan cytokinesis. PLoS Pathog. 5,e1000270 (2009).
20. Noble, M. E. M., Endicott, J. A. & Johnson, L. N. Protein kinase inhibitors: insightsinto drug design from structure. Science 303, 1800–1805 (2004).
21. Roy, A. & Levine, T. P. Multiple pools of phosphatidylinositol 4-phosphate detectedusing the pleckstrin homology domain of Osh2p. J. Biol. Chem. 279,44683–44689 (2004).
22. Kruger, T., Sanchez, C. P. & Lanzer, M. Complementation of Saccharomycescerevisiae pik1ts by a phosphatidylinositol 4-kinase from Plasmodium falciparum.Mol. Biochem. Parasitol. 172, 149–151 (2010).
23. Strahl, T., Hama, H., DeWald, D. B. & Thorner, J. Yeast phosphatidylinositol4-kinase, Pik1, has essential roles at the Golgi and in the nucleus. J. Cell Biol. 171,967–979 (2005).
24. Walch-Solimena, C. & Novick, P. The yeast phosphatidylinositol-4-OH kinase Pik1regulates secretion at the Golgi. Nature Cell Biol. 1, 523–525 (1999).
25. Hama, H., Schnieders, E. A., Thorner, J., Takemoto, J. Y. & DeWald, D. B. Directinvolvement of phosphatidylinositol 4-phosphate in secretion inthe yeast Saccharomyces cerevisiae. J. Biol. Chem. 274, 34294–34300 (1999).
26. de Graaf, P. et al. Phosphatidylinositol 4-kinaseb is critical for functionalassociationofRab11with theGolgi complex.Mol.Biol.Cell15,2038–2047 (2004).
27. LaMarche, M. J. et al. Anti-hepatitis C virus activity and toxicity of type IIIphosphatidylinositol-4-kinase beta inhibitors. Antimicrob. Agents Chemother. 56,5149–5156 (2012).
28. The malERA Consultative Group on Drugs. A research agenda for malariaeradication: drugs. PLoS Med. 8, e1000402 (2011).
29. Meister, S. et al. Imaging of Plasmodium liver stages to drive next-generationantimalarial drug discovery. Science 334, 1372–1377 (2011).
30. Russell, B. et al. Determinants of in vitro drug susceptibility testing of Plasmodiumvivax. Antimicrob. Agents Chemother. 52, 1040–1045 (2008).
31. Russell, B. M. et al. Simple in vitro assay for determining the sensitivity ofPlasmodium vivax isolates from fresh human blood to antimalarials in areaswhere P. vivax is endemic. Antimicrob. Agents Chemother. 47, 170–173 (2003).
32. D’Alessandro, S. et al. A Plasmodium falciparum screening assay foranti-gametocyte drugs based on parasite lactate dehydrogenase detection.J. Antimicrob. Chemother. 68, 2048–2058 (2013).
33. Dechering, K. J., Thompson, J., Dodemont, H. J., Eling, W. & Konings, R. N.Developmentally regulatedexpressionofpfs16, a marker for sexual differentiationof the human malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 89,235–244 (1997).
34. van der Kolk, M. et al. Evaluation of the standard membrane feeding assay (SMFA)for the determination of malaria transmission-reducing activity using empiricaldata. Parasitology 130, 13–22 (2005).
35. Janse, C. J., Ramesar, J. & Waters, A. P. High-efficiency transfection and drugselection of genetically transformed blood stages of the rodent malaria parasitePlasmodium berghei. Nature Protocols 1, 346–356 (2006).
Supplementary Information is available in the online version of the paper.
Acknowledgements We thank E. Miller for critical review of the manuscript and figuredesign. We also thank A. Rodriguez and the insectary core facility team at New YorkUniversity for reliable supplies of malaria-infected mosquitoes. We gratefullyacknowledge translational research grants (WT078285 and WT096157) from theWellcome Trust and funding from the Medicines for Malaria Venture (MMV) to theGenomics Institute of the Novartis Research Foundation, the Swiss Tropical and PublicHealth Institute, Columbia University, the Novartis Institute for Tropical Diseases, theSingapore Immunology Network and Horizontal Programme on Infectious Diseasesunder the Agency Science Technology and Research (A*STAR, Singapore), and theWellcome Trust (UK). Shoklo Malaria Research Unit is sponsored by The WellcomeTrust (UK), as part of the Oxford Tropical Medicine Research Programme of WellcomeTrust-Mahidol University. E.A.W. and D.A.F. are supported by grants from the Bill andMelinda Gates Foundation, MMV, and the National Institutes of Health (R01AI090141to E.A.W. and R01085584 and R01079709 to D.A.F.).
Author Contributions C.W.M., with assistance from S.L.M., M.I. and D.M.P., evolved andcharacterized drug-resistant parasite lines, analysed microarray data, and performedphenotypic studies. M.C.S.L. performed genome editing and other transgenic parasitestudies, as well as the fluorescence microscopy imaging. Additional experimentalcontributions were as follows: PvPI(4)K assay (C.S.L., S.H.L. and C.B.); imidazopyrazinechemistry (J.R., A.N., A.K.C. and D.C.T.); imidazopyrazine structure–activity studies (K.G.and K.L.K.); BQR695 re-synthesis (O.S.); quinoxaline development and human kinasepanels (J.T. and D.H.); mutant P. berghei strain generation (T.R.S.K. and P.P.H.);next-generation sequencing data analysis (M.J.M.); in vitro assay with P. cynomolgi(A.-M.Z. and C.H.M.K.); sexual-stage P. falciparum assays (M.T., K.J.D. andR.W.S.); ex vivoassays on P. falciparum and P. vivax clinical isolates (R.S., B.R., L.R. and F.N.); in vivoefficacy studies in the mouse model (blood stage, C.F. andM.R.; liver stage, N.K.); in vitroassay with P. yoelii (D.M.P., S.M., S.L.M. and K.G.); in silico docking studies (B.B.). L.R.quantified phosphatidylinositol phosphates. R.J.G. managed Genomics Institute of theNovartis Research Foundation (GNF) activities. B.K.S.Y. coordinated collaborativeefforts. C.W.M., M.C.S.L., C.B., D.A.F., T.T.D. and E.A.W. designed experiments andco-wrote the manuscript. C.W.M. and M.C.S.L. contributed equally to the study. Allauthors discussed the results and commented on the manuscript.
Author Information Reprints and permissions information is available atwww.nature.com/reprints. The authors declare competing financial interests: detailsare available in the online version of the paper. Readers are welcome to comment onthe online version of the paper. Correspondence and requests for materials should beaddressed to E.A.W ([email protected]) or T.T.D. ([email protected]).
RESEARCH ARTICLE
6 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013

METHODSStatistical analysis. Data are shown as mean 6 s.d., and represent at least two inde-pendent experiments performed in duplicate unless stated otherwise. Statisticalsignificance between groups was determined using an unpaired, nonparametricanalysis via the Mann–Whitney U test (with GraphPad Prism Software). TheMann–Whitney test does not require the assumption that the differences betweenthe two samples are normally distributed. P values were considered significant ifP , 0.05.P. berghei and P. yoelii sporozoite generation. The New York University Insec-tary provided A. stephensi mosquitoes infected with rodent malaria: Plasmodiumyoelii (17XNL), a transgenic P. berghei expressing luciferase (ANKA) or an engi-neered P. berghei ANKA line with H1477Y-PbPI(4)K (see below). Plasmodium-infected mosquitoes were dissected 22–25 days after an infective blood meal, andsporozoites purified as described previously29.P. yoelii liver schizont assay. A previously described29, in vitro high-contentimaging assay was used to measure the proliferation of P. yoelii sporozoites afterinvasion of a transgenic hepatoma line (HepG2-A16-CD81-EGFP; obtained fromthe laboratory of D. Mazier; tested monthly for mycoplasma contamination; usedfor up to 50 passages). Anti-PyHSP70-stained exoerythrocytic forms were quan-tified using the Opera Confocal High Content Screening System (PerkinElmer).Atovaquone and DMSO-treated wells were used as controls on each assay plate.IC50 values were obtained using mean parasite area and a custom curve fitting model,and a standard logistic regression model was applied for curve fitting.In vivo pharmacokinetic studies and causal prophylaxis efficacy. All animal-related procedures at GNF were non-blinded and conducted under an IACUC-approved protocol in compliance with Animal Welfare Act regulations and theGuide for the Care and Use of Laboratory Animals. To determine pharmacokineticproperties, non-randomized mice (n 5 3; naive male Balb/C; 7–9 weeks old; 23–25 g) were dosed with KDU691 (HCl salt; formulated in 75% polyethylene glycol300 (PEG300) and 25% of dextrose (5 mg ml21) in distilled water (D5W)). Samplesize and analysis protocol followed a standardized protocol36. The causal prophy-laxis model was carried out in two independent experiments with non-randomizedmice (n 5 4 per experimental group; female CD1; 7–9 weeks old; 25–32 g) inocu-lated with 1 3 105 P. berghei sporozoites (intravenous) and orally administered7.5 mg kg21 KDU691 (suspension formulation of 0.5% (w/v) methylcellulose and0.5% (v/v) Tween 80). Sample size was determined by a power analysis using binaryoutcome data (infected or cured mice), and showed that the use of four mice perexperimental group had 90% power to detect a difference of 75% (for example,three mice). The timing of drug administration occurred immediately after para-site inoculation (day 0), 24 h after inoculation (day 1), 36 h after inoculation, or48 h after inoculation (day 2). Parasitaemia was monitored by two methods. TheIn vivo Imaging System (IVIS 100, Xenogen; Calipar Life Science) was used toacquire the bioluminescence signal (5-min acquisitions) as previously described29.Alternatively, blood smear samples were obtained from each mouse on days 4, 5, 6,7, 10, 15, 21 and 30 after inoculation, stained with Giemsa, and viewed under amicroscope for visual detection of blood parasitaemia. Animals with parasitaemiaexceeding 20% were euthanized.P. cynomolgi liver assay: sporozoite generation. All Rhesus macaques (Macacamulatta) used in this study were bred in captivity for research purposes, and werehoused at the Biomedical Primate Research Centre (BPRC; AAALAC-certifiedinstitute) facilities under compliance with the Dutch law on animal experiments,European directive 86/609/EEC and with the ‘Standard for Humane Care and Useof Laboratory Animals by Foreign institutions’ identification number A5539-01,provided by the Department of Health and Human Services of the US NationalInstitutes of Health. The local independent ethical committee first approved allprotocols. Non-randomized rhesus macaques (male or female; 5214 years old; oneanimal per month) were infected with 1 3 106 P. cynomolgi M strain blood-stageparasites, and bled at peak parasitaemia. Approximately 300 female A. stephensimosquitoes (Sind-Kasur strain, Nijmegen University Medical Centre St Radboud)were fed with this blood as described previously37.P. cynomolgi liver assay: sporozoite infection of primary rhesus hepatocytes.Rhesus monkey hepatocytes were isolated from liver lobes as described by previously38.Sporozoite infections were performed within 3 days after hepatocyte isolation.Sporozoite inoculation of primary Rhesus hepatocytes was performed as describedpreviously5,39. On day 6, intracellular P. cynomolgi malaria parasites were fixed, stainedwith purified rabbit antiserum reactive against P. cynomolgi Hsp70.1 (ref. 5), andvisualized with FITC-labelled goat anti-rabbit IgG antibodies. Quantification ofsmall ‘hypnozoite’ exoerythrocytic forms (1 nucleus, a small round shape, maxi-mally 7mm in diameter) or large ‘developing parasite’ exoerythrocytic forms (morethan 1 nucleus, larger than 7mm and round or irregular shape) was determined foreach well using a high-content imaging system (Operetta, Perkin-Elmer).P. falciparum asexual blood-stage culture and assay. P. falciparum isolates weremaintained by standard methods40 in an atmosphere of 93% N2, 4% CO2, 3% O2
at 37 uC in complete culturing medium (10.4 g l21 RPMI 1640 (without phenolred, with 2.1 mM glutamine), 5.94 g l21 HEPES, 5 g l21 albumax II, 50 mg l21
hypoxanthine, 2.1 g l21 sodium bicarbonate, 10% human serum and 43 mg l21
gentamicin). Human erythrocytes served as host cells. In vitro antimalarial activitywas measured using a modified SYBR Green cell proliferation assay4.In vivo mouse therapeutic efficacy assay. All in vivo efficacy studies were approvedby the veterinary authorities of the Canton Basel-Stadt. In vivo antimalarial activitywas assessed in non-randomized, non-blinded mice (n 5 3 (once daily regimen) orn 5 5 (twice daily regimen) per experimental group; female NMRI; 6 weeks old;20–22 g) intravenously infected on day 0 with 2 3 107 erythrocytes infected withP. berghei GFP parasites (PbGFPCON, donated by A. P. Waters and C. J. Janse)40.Power analysis using a binary outcome data (infected or cured mice) showed thatthe use of three mice had .80% power to detect a difference of 67% (for example,two mice), whereas the power increased to .90% to detect a difference of 40% (forexample, two mice) when the sample size increased to five. Compounds formu-lated in 0.5% methylcellulose/0.5% Tween 80 were administered orally in a volumeof 10 ml kg21 as a single dose (24 h after infection), or as four consecutive doses (6,24, 48 and 72 h after infection). With the single-dose regimen at 96 h after infection,parasitaemia was measured using standard flow cytometry techniques or standardmicroscopy41. Activity was calculated as the difference between the mean percen-tage parasitaemia for the control and treated groups, expressed as a percentage ofthe control group. The survival time in days was also recorded up to 30 days afterinfection, at which time parasite-free animals were considered cured.Ex vivo schizont assay of P. falciparum and P. vivax isolates. Clinical isolates ofP. vivax and P. falciparum (eight samples each) were collected from the Mae Sotregion of Tak Province (Thailand) in 2011. All samples were collected from patientswith acute malaria (with a mono species parasitaemia of 5,000–8,000 parasites permicrolitre, .80% of the parasites at ring stage and no antimalarials or antibioticstaken by the patient in the past 30 days) attending the clinics of Shoklo MalariaResearch Unit. After obtaining written consent, the sensitivity of the parasite iso-lates was tested, as previously described30,31. The clinical samples were collected andtested in accordance to protocols approved by The Center for Clinical Vaccinologyand Tropical Medicine at University of Oxford (OXTREC 58-09), which had beenratified by the Ethics Committee of the Faculty of Tropical Medicine at MahidolUniversity.Gametocyte viability assay. P. falciparum NF54 stage III2V gametocytes wereisolated by discontinuous Percoll gradient centrifugation of parasite cultures treatedwith 50 mM N-acetyl-D-glucosamine for 3 days to kill asexual parasites. Game-tocytes were seeded in 96-well plates, and viability was measured by a modifiedPlasmodium lactate dehydrogenase assay using resazurin as a substrate followinga 72-h compound exposure32. Compounds were tested in a 9-point dose responseseries (0.1 nM–1mM in 0.5 log10 dilutions) in quadruplicate (two different plateswith two different parasite batches). Controls MIN (1mM DHA), MAX (vehicle)and full dose response of reference compound (DHA) were run in a standardizedplate format. Data were expressed as the percentage effect relative to the MIN andMAX wells of the assay plate. Data were analysed with GraphPad Prism 5 and IC50
values were calculated by applying a four-parameter nonlinear regression model.Gametogenesis assay. Cultures containing P. falciparum NF54 mature (stage V)gametocytes were pre-incubated with compound for 24 h. KDU691 was tested at0.2mM (1 3 IC50) and 20mM (100 3 IC50), and controls included 0.1% DMSO(vehicle) and 1mM DHA. Subsequently, gametogenesis was induced by addingxanthurenic acid to a final concentration of 50mM. An additional control includeda sample treated with 0.02% (w/v) NaN3 to block gametogenesis (sample t 5 0 h).Parasites were incubated for 8 h at 20 uC, and emerged gametes were immunostainedwith an Alexa-Fluor-488-labelled anti-Pfs25 antibody as previously described33.Gametes were quantified by flow cytometry, and data were expressed as the frac-tion of Pfs25-positive cells in the population of uninfected and infected RBCs.Standard membrane feeding assay. Cultures containing mature (stage V) P.falciparum NF54 gametocytes were pre-incubated with compound for 24 h. Cellswere spun down (10,000g, 20 s) and resuspended in human serum and RBCs to afinal haematocrit of 50%. Fresh compound was added and the blood meal was fedto A. stephensi mosquitoes. Seven days after the blood meal, the number of midgutoocysts was determined by microscopy34. Compounds were tested in a 9-point doseresponse series (0.1 nM–1mM in 0.5 log10 dilutions) in duplicate. Controls includedtwo vehicle controls (0.1% DMSO) and two positive controls (1mM DHA). Persample, a total of 20 mosquitoes were dissected to determine oocysts loads. Repre-sentative data are shown from one of two independent experiments and expressedas the percentage effect relative to vehicle control, and analysed with the GraphPadPrism 5 software package by applying a four-parameter nonlinear regression model.Parasite synchronization. In brief, a Dd2 culture was treated consecutively with5% sorbitol (6-h spacing), and 36 h after the first sorbitol treatment, the cultureswere captured on MACS LD columns (Miltenyi Biotec). Purified schizonts wereeluted with complete culturing media and combined into a fresh culture. The flask
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013

was incubated at 37 uC with gentle shaking (110 r.p.m.) for 2.5 h to promote opti-mal reinvasion. After this incubation, the culture was filtered over a fresh MACSLD column to re-capture any remaining schizonts, and to allow the uninfectedand newly parasitized RBCs with ring-stage parasites to flow through. The entir-ety of reinvasion occurred over a 3-h period, so the midpoint was used to establisht 5 0 h in the parasite life cycle.Onset of inhibition studies. A synchronized culture was divided into 100mlvolumes (,2% parasitaemia in 2.5% haematocrit) within a 96-well tissue cultureplate (BD Biosciences). Stage of action studies were carried out by immediatelyadding 3 3 IC50 inhibitor and parasites were visualized at t 5 24, 32, 40, 48 and60 h after synchronization. Giemsa-stained parasites were visualized (.1,000 cellsper sample) and recorded by a light microscope (Carl Zeiss Axiostar Plus micro-scope, 1003 objective lens) equipped with a PowerShot G6 digital camera (Canon).To test the effect of KAI407 exposure duration on parasite maturation throughout asingle (,48 h) asexual blood-stage cycle, 125 nM KAI407 was added either imme-diately after synchronization or every 2 h starting at life cycle t 5 34 h (trophozoitestage). Alternatively, 125 nM KAI407 was present in the media immediately aftersynchronization, and then washed out (3 3 1 ml culturing medium, then resus-pended to the original volume) at these same time points. In both experiments, theparasitaemia of ring-stage parasites was determined by microscopy 60 h after finalsynchronization. Drug-treated wells were compared to a DMSO-treated control todetermine the percentage inhibition.Physical disruption/merozoite release assay. Synchronized parasites were culturedto schizogony (t 5 42 h) and then treated with DMSO, 1mM E-64, or ,5 3 IC50
drug: 125 nM KAI407, 15 nM KAI715 or 150 nM KDU691. At t 5 51 h, parasiteswere sheared by 20 strokes through a 28.5-gauge needle as previously described10.The cultures were incubated for a further 8 h, and ring-stage parasitaemias deter-mined by microscopy (.1,000 cells counted per sample).Imaging of drug-treated PfATP4–GFP. Generation of the PfATP4–GFP lineand subsequent fluorescence imaging was as previously described12. The drug-arrested phenotype induced by KAI407 treatment of this line was investigatedwith sorbitol synchronized populations. Either 500 nM KAI407 or DMSO (control)was added to early schizonts (t 5 40 h) and incubated for 4 h. Cells were then imme-diately fixed with paraformaldehyde and glutaraldehyde for imaging. Images arerepresentative of two biological replicates.In vitro resistance selection and microarray analysis. A clonal population of P.falciparum Dd2 parasites was used to initiate two or three independent parasitecultures under the initial selection pressure of 12 nM KAI407, 1 nM KAI715 or40 nM BQR695. Stepwise drug evolution continued until the final concentrationwas at least threefold higher than the initial concentration (typically 802120 days).For each of the ten resistant strains, CNVs and SNVs were detected using a whole-genome tiling array and analysed with PfGenominator (freely available at http://winzeler.ucsd.edu/software.html)13. The susceptibility of each resistant strain toKAI407, KAI715, KDU691 and BQR695 was determined by the 72-h SYBR Greencell proliferation assay4, with four independent experiments assayed in duplicate.PI(4)K variation in a transgenic P. berghei line. Introduction of the PI(4)K-resistance-conferring mutation PbPI(4)K(His1477Tyr) (equivalent to His1484Tyrin PfPI(4)K) into a transgenic P. berghei ANKA line expressing luciferase (PbANKA;obtained from MR4, MRA-868) was generated using an established protocol35. Inbrief, an 1,857-bp fragment of PbPI(4)K (PBANKA_110940) was PCR-amplifiedusing primer pair p1/p2 (Supplementary Table 1) and cloned into a Topo TA vector(Invitrogen). Sequence fidelity was verified using primers p5 to p9. Subsequently,site-directed mutagenesis (Stratagene) was performed with primers p3/p4 to intro-duce the non-synonymous coding mutation C4519T (encodes Tyr 1477; mutationlocated 556 bp upstream of the stop codon). The KpnI/PspM01-digested fragmentwas cloned into pl0001-TgDHFR-KO (obtained from MR4, MRA-770), in whichthe pycrt 39 untranslated region (UTR) (terminating sequence; primers p12/p13)and PbPI(4)K 39 UTR (second homology site; primers p10/p11) were also included.In parallel, a second plasmid was constructed with the native pbpi4k gene fragmentto serve as recombinant control. After transfection, recombinant PbANKA para-sites were selected in vivo with non-randomized mice (female Swiss Webster mice,6 weeks old, ,25 g), and drinking water was supplemented with pyrimethamine.Animal protocols were approved and performed in accordance with rules and regu-lations of Institution Animal Care and Use Committee (IACUC) of ColumbiaUniversity. Presence of transfected parasites was verified with direct sequencingusing primers p14 to p20. Clonal parasite lines were isolated by in vivo limitingdilution, and the PI(4)K variation verified by direct sequencing. All primers forthis study are listed in Supplementary Table 1. Drug activity was measured in threeindependent experiments performed in quadruplicate using the in vitro P. yoeliiliver schizont assay.ZFN engineering. ZFNs targeting the genomic region encoding the kinase domainof pfpi4k (PF3D7_0509900) were designed by Sigma-Aldrich, and a ZFN pairrecognizing the sequence (59-CTGACAGTCGATGTGGTAAAAATAAAAGGA
AATA-39) at the exon-3/intron-3 boundary was selected based on activity in ayeast proxy assay42. The ZFN pair was subcloned into a pDC2-based expressionvector downstream of a calmodulin (PF14_0323) promoter essentially as described17,yielding the vector pZFNPI(4)K-hDHFR. A ,1.7-kb donor template, encompass-ing 1,217 bp of the pfpi4k coding sequence and 466 bp of the 39 UTR, was amp-lified with primers p3480 and p3498 and inserted into the BstAPI and ZraI sites ofpZFNPI(4)K-hDHFR to yield the editing plasmid pZFNPI(4)K-hDHFR. To preventrecleavage of an edited genome, three silent mutations were introduced by site-directed mutagenesis into the ZFN-binding site using primers p3496 and p3497(see Fig. 3b). Further modifications were overlaid on this donor sequence to gene-rate transfection plasmids bearing mutations at Ser1320Leu (pZFN-PI(4)KS1320L-hDHFR; primers p3506 and p3507), Tyr1356Phe (pZFN-PI(4)KY1356F-hDHFR;primers p3508 and p3509), Tyr1356stop (pZFN-PI(4)KY1356X-hDHFR; primersp3510 and p3511) and His1484Tyr (pZFN-PI(4)KH1484Y-hDHFR; primers p3512and p3513). The ZFN primers are listed in Supplementary Table 1.
A clonal Dd2 line was transfected with the pZFN-PI(4)K-hDHFR editingplasmids, and selected with 2.5 nM WR99210 the day after electroporation. Onday 6 after electroporation, WR99210 pressure was removed, and parasites wereobserved microscopically at day 14. Clonal parasites were obtained by limitingdilution, and successful editing was determined by direct sequencing. The suscep-tibility of each edited line to KAI407, KAI715, KDU691 and BQR695 was deter-mined by three independent experiments performed in triplicate using the 72-hSYBR Green cell proliferation assay4.Whole-genome sequencing analysis. Genomic DNA libraries were prepared forwhole-genome sequencing using the Illumina NexTera protocol of simultaneousfragmentation and adaptor ligation and indexed using the TruSeq Dual IndexSequencing primers (v. 2013, Illumina.). DNA libraries were clustered and run onan Illumina HiSeq 2000, according to manufacturer’s instructions. An internallydeveloped whole-genome sequencing pipeline, named PlaTypUS (M. Manary et al.,manuscript submitted), was used to align and analyse all whole-genome sequen-cing data. The program integrates many community-developed tools into its pro-cessing pipeline. To generate alignments, PlaTypUS follows a multi-step alignmentprocess to execute quality control measures on an alignment map. FASTQ filesobtained from sequencing were aligned to the Pf3D7 reference (PlasmoDB v9.1)for SNV and CNV analysis.Expression of GFP-tagged PfPI(4)K and PfRab11A. The full-length pfpi4k andpfrab11a genes were PCR amplified from genomic DNA using primers p3727/p3772and p3730/p3731, respectively, and inserted into a pDC2-based vector behind GFPto generate the expression plasmids pDC2-PbEF1alphapro-GFP-PfPI(4)K-attP-BSD and pDC2-PbEF1alphapro-GFP-PfRab11A-attP-BSD. The Asp139Tyr muta-tion was introduced by site-directed mutagenesis into the pfrab11a coding sequenceusing primers p3736/p3737, yielding the plasmid pDC2-PbEF1alphapro-GFP-PfRab11A-D139Y-attP-BSD. The expression plasmids were then transfected intothe Dd2-attB parasite line using attP 3 attB integrase-mediated recombination18.Drug activity against each transgenic line was determined by three independentexperiments performed in duplicate using the 72-h SYBR Green cell proliferationassay4.Human lipid kinase panel. The biochemical inhibitory activities of human lipidkinases were measured with either Kinase-Glo (Promega) for PI(4)KIIIb and thea and b isoforms of phosphatidylinositol-3-OH kinase (PI(3)K), or Adapta (Invi-trogen) for the d and c isoforms of PI(3)K. A detailed description of the Kinase-Glo assays is available from ref. 43. The Adapta assays were run as follows: 50 nl ofcompound dilutions were dispensed into white 384-well low-volume polystyreneplates (Greiner). Then, 5ml of purified recombinant PI(3)K and lipid substrate(phosphatidylinositol, or phosphatidylinositol-4,5-bisphosphate plus phosphati-dylserine) were dispensed and followed by a subsequent dispense of 5 ml ATP(final assay volume 10 ml). The enzymatic reaction was incubated at room tem-perature. The standard reaction buffer for the Adapta TR-FRET assay contained10 mM Tris–HCl, pH 7.5, 3 mM MgCl2, 50 mM NaCl, 1 mM dithiothreitol (DTT)and 0.05% CHAPS (v/v). Reactions were stopped with 5 ml of a mixture of EDTAcontaining the Eu31-labelled anti-ADP antibody and the Alexa-Fluor-647-labelledADP tracer in a proprietary TR-FRET dilution buffer. Plates were read 15–60 minlater in a Synergy2 reader using an integration time of 0.4 s and a delay of 0.05 s.Controls for 100% inhibition of the kinase reaction were performed by replacingPI(3)K with the equivalent volume of the standard reaction buffer. The controlsfor the 0% inhibition were performed with the solvent vehicle of the compounds(90% DMSO in H2O (v/v)). A reference compound was included in all assay platesand each compound was assayed in a 16-point, 1:3 dilution series.Human protein kinase panel. The human protein kinase selectivity profile forKAI407 and KDU691 was carried out against a panel of 40 recombinant kinases.Inhibition of kinase-dependent substrate phosphorylation was measured by theCaliper mobility shift assay technology using microfluidic capillary electroph-oresis as previously described44.
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013

Homology modelling. The P. falciparum PI(4)K model was built by homologymodelling using the X-ray crystallographic coordinates for human PI(3)Kc incomplex with an inhibitor (Protein Data Bank (PDB) accession 3ENE) as a tem-plate. The kinase domain of PfPI(4)K (amino acid boundaries: N1261–M1559)were aligned to that of PI(3)Kc (kinase domain: A557–A1102). Fully flexible liganddocking was performed with Glide (version 5.5; Schrodinger). The PyMOL Mole-cular Graphics System (v.1.2r2, Schrodinger) was used to render the models andprepare figures.Cloning and expression of PvPI(4)K. The full-length coding sequence of PvPI(4)K(PVX098_050) was codon-optimized for baculovirus expression, synthesized andcloned into pFastBac-HTa (Invitrogen) in frame with the amino-terminal poly-histidine tag using the EcoRI and HindIII restriction sites. Recombinant bacmidclones were generated by site-specific transposition in E. coli DH10Bac (Invitrogen).The pFastBacHTa-PvPI(4)K plasmid was used as a template for site-directed muta-genesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Theprimers used for mutagenesis are listed in Supplementary Table 1. All mutationsites, as well as absence of errors across the whole gene, were confirmed by directDNA sequencing. The subsequent steps for bacmid isolation, transfection and selec-tion of the recombinant viruses were performed according to the manufacturer’sprotocol (Bac-to-Bac system, Invitrogen).
SF9 cells, cultured in SF-900 III serum-free medium, were transfected withrecombinant baculovirus at 1/200 (v/v) and incubated at 27 uC for 72 h. The pelletswere collected after centrifugation and re-suspended in cell lysis buffer (20 mMTris-HCl, pH 7.5, 500 mM NaCl, 5% glycerol, 0.01% Triton X-100, 0.5 mM TCEPand 13 complete protease inhibitor cocktail without EDTA (Roche Diagnostics)).The cell suspension was lysed by sonication and the clarified supernatant wasloaded onto a 1 ml HisTrap affinity column (GE Healthcare) pre-equilibrated withbuffer A (20 mM Tris, pH 7.5, 500 mM NaCl, 5% glycerol, 0.5 mM TCEP, 20 mMimidazole and 13 complete protease inhibitor cocktail without EDTA). The proteinwas eluted with buffer B (buffer A with 300 mM imidazole), and fractions contain-ing PvPI(4)K were pooled and purified by a gel-filtration column (Hi-Load 16/60Superdex 200, GE Healthcare) equilibrated with 20 mM Tris, pH 7.5, 5% glycerol,500 mM NaCl, 5 mM DTT and 13 protease inhibitor cocktail without EDTA.Protein concentrations were determined by the calculated protein extinctioncoefficient (e280 nm 5 12,5875 M21 cm21). Aliquots were flash frozen in liquidnitrogen and immediately stored at 280 uC. All PvPI(4)K constructs were puri-fied with this same protocol.PI(4)K enzymatic assay. L-a-phosphatidylinositol (Avanti Polar Lipid), dis-solved in 3% n-octylglucoside (Roche Diagnostics), was used as the lipid substratefor the PI(4)K activity assay. PvPI(4)K was assayed using Transcreener ADP2 FPdetection kit (BellBrook) in a black, solid 384-well plate (Corning). The final assayvolume was 10 ml and contained 1 nM of the respective PvPI(4)K construct in10 mM Tris, pH 7.5, 1 mM DTT, 10 mM ATP, 5 mM Mn21, 0.2% Triton X-100and 11 mM phosphatidylinositol/octylglucoside. The enzyme reaction was per-formed for 1 h at room temperature and was stopped by adding 10ml of detectionmix containing 13 stop buffer, 4 nM AMP Alexa Fluor 633 tracer, and 20mg ml21
ADP antibody. Fluorescence polarization measurements were performed on theInfinite M1000 plate reader (Tecan) with lex 5 635 nm and lem 5 680 nm (20-nmbandwidth). IC50 values were calculated from three independent experimentsperformed in triplicate using Graphpad Prism software.In vivo detection of PI(4)P pools. To detect PI(4)P within the parasite, the PHdomain of yeast Osh2 was amplified from pREP41 (refs 21, 45) (provided by M. Ellisand F. Chang) using primers p2987b/p2988. The Osh2 PH domain was insertedas a dimer behind GFP to generate the expression vector pDC2-PbEF1alphapro-GFP-PHOsh2-attP-BSD, and transfected into the Dd2-attB parasite line using
attP 3 attB integrase-mediated recombination. Sorbitol synchronized parasites fol-lowed the above methodology to image drug-treated parasites expressing PfATP4–GFP. Parasites (100 cells per treatment) were imaged from three independentexperiments.Phosphatidylinositol phosphate quantification. The sorbitol/MACS methoddescribed above was used to generate a highly synchronized parasite culture (10%parasitaemia in 2.5% haematocrit). At t 5 41.5 h in the parasite life cycle, the culturewas washed three times with pre-warmed complete media prepared from phos-phate-free RPMI (US Biological; PF-CM). The final culture was resuspended in4 ml PF-CM and split into two equal volumes. One volume was treated with500 nM KAI407 and the remaining volume with DMSO and incubated at 37 uCfor 15 min. Phosphatidylinositol phosphate radiolabelling was initiated by theaddition of 800mCi ml21 Na2[32P]O4 (Perkin Elmer) and incubated for 1–4 h at37 uC (t 5 42–46 h in life cycle). The samples were then washed five times withchilled PBS plus 0.15% saponin. The final supernatant was aspirated, and to eachparasite pellet the following were successively added with mixing in between:0.4 ml 1 N HCl, 0.4 ml methanol and 0.4 ml CHCl3. Samples were vortexed vigo-rously and then centrifuged. The lower organic layer was transfer to a new tube andextracted once with 400ml (1.0:0.9 (v/v)) methanol:0.1 M EDTA, pH 8.0. The lowerorganic layer was transferred to a new tube and dried under nitrogen stream. Apreviously described method46 was used to quantify phosphatidylinositol phosphates.In brief, phosphatidylinositides were separated by anion-exchange high-performanceliquid chromatography (HPLC; Agilent) using an ammonium phosphate gradient,detected by a flow scintillation analyser (Perkin-Elmer), and quantified using ProFSAsoftware (Perkin-Elmer). The [32P]-labelled phosphatidylinositol phosphate peakswere identified using internal standards. The counts in each peak were normalizedagainst total phosphatidylinositol counts in each individual sample.
36. Li, C.et al. Amodern in vivopharmacokineticparadigm:combining snapshot, rapidand full PKapproaches to optimize andexpedite earlydrug discovery.DrugDiscov.Today 18, 71–78 (2013).
37. Ponnudurai, T. et al. Infectivity of cultured Plasmodium falciparum gametocytes tomosquitoes. Parasitology 98, 165–173 (1989).
38. Guguen-Guillouzo, C. et al. High yield preparation of isolated human adulthepatocytes by enzymatic perfusion of the liver. Cell Biol. Int. Rep. 6, 625–628(1982).
39. Mazier, D. et al. Complete development of hepatic stages ofPlasmodium falciparumin vitro. Science 227, 440–442 (1985).
40. Fidock, D. A., Nomura, T. & Wellems, T. E. Cycloguanil and its parent compoundproguanil demonstrate distinct activities against Plasmodium falciparum malariaparasites transformed with human dihydrofolate reductase. Mol. Pharmacol. 54,1140–1147 (1998).
41. Franke-Fayard, B. et al. A Plasmodium berghei reference line that constitutivelyexpresses GFP at a high level throughout the complete life cycle. Mol. Biochem.Parasitol. 137, 23–33 (2004).
42. Doyon, Y. et al. Heritable targeted gene disruption in zebrafish using designedzinc-finger nucleases. Nature Biotechnol. 26, 702–708 (2008).
43. Furet, P. et al. Discovery of NVP-BYL719 a potent and selectivephosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation.Bioorg. Med. Chem. Lett. 23, 3741–3748 (2013).
44. Manley, P. W. et al. Extended kinase profile and properties of the protein kinaseinhibitor nilotinib. Biochim. Biophys. Acta 1804, 445–453 (2010).
45. Onishi, M. et al. Role of septins in the orientation of forespore membraneextension during sporulation in fission yeast. Mol. Cell. Biol. 30, 2057–2074(2010).
46. Mandl, A., Sarkes, D., Carricaburu, V., Jung, V. & Rameh, L. Serumwithdrawal-induced accumulation of phosphoinositide 3-kinase lipids indifferentiating 3T3-L6 myoblasts: distinct roles for Ship2 and PTEN. Mol. Cell. Biol.27, 8098–8112 (2007).
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
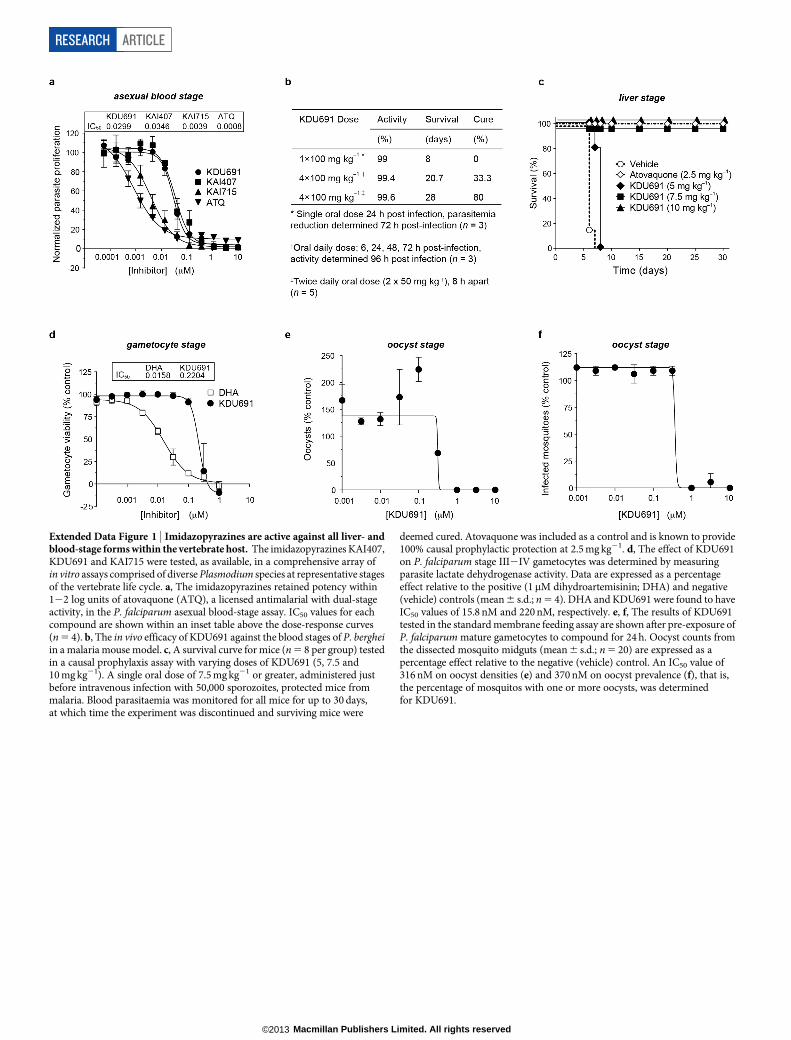
Extended Data Figure 1 | Imidazopyrazines are active against all liver- andblood-stage forms within the vertebrate host. The imidazopyrazines KAI407,KDU691 and KAI715 were tested, as available, in a comprehensive array ofin vitro assays comprised of diverse Plasmodium species at representative stagesof the vertebrate life cycle. a, The imidazopyrazines retained potency within122 log units of atovaquone (ATQ), a licensed antimalarial with dual-stageactivity, in the P. falciparum asexual blood-stage assay. IC50 values for eachcompound are shown within an inset table above the dose-response curves(n 5 4). b, The in vivo efficacy of KDU691 against the blood stages of P. bergheiin a malaria mouse model. c, A survival curve for mice (n 5 8 per group) testedin a causal prophylaxis assay with varying doses of KDU691 (5, 7.5 and10 mg kg21). A single oral dose of 7.5 mg kg21 or greater, administered justbefore intravenous infection with 50,000 sporozoites, protected mice frommalaria. Blood parasitaemia was monitored for all mice for up to 30 days,at which time the experiment was discontinued and surviving mice were
deemed cured. Atovaquone was included as a control and is known to provide100% causal prophylactic protection at 2.5 mg kg21. d, The effect of KDU691on P. falciparum stage III2IV gametocytes was determined by measuringparasite lactate dehydrogenase activity. Data are expressed as a percentageeffect relative to the positive (1mM dihydroartemisinin; DHA) and negative(vehicle) controls (mean 6 s.d.; n 5 4). DHA and KDU691 were found to haveIC50 values of 15.8 nM and 220 nM, respectively. e, f, The results of KDU691tested in the standard membrane feeding assay are shown after pre-exposure ofP. falciparum mature gametocytes to compound for 24 h. Oocyst counts fromthe dissected mosquito midguts (mean 6 s.d.; n 5 20) are expressed as apercentage effect relative to the negative (vehicle) control. An IC50 value of316 nM on oocyst densities (e) and 370 nM on oocyst prevalence (f), that is,the percentage of mosquitos with one or more oocysts, was determinedfor KDU691.
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013
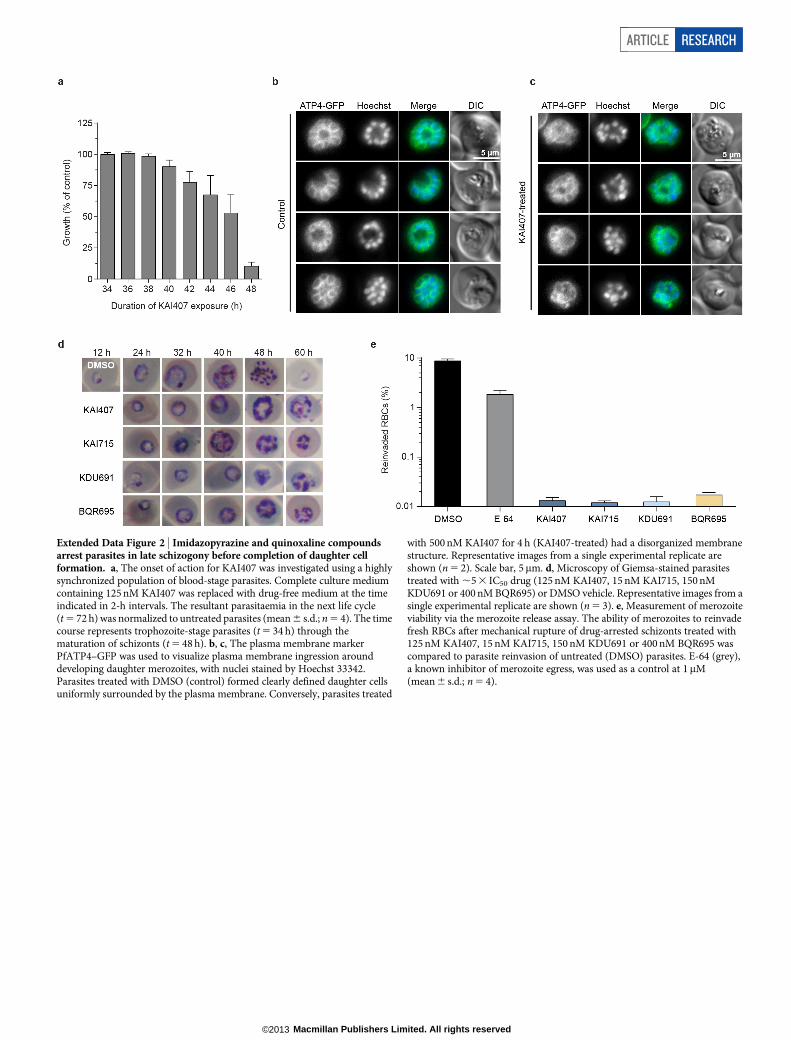
Extended Data Figure 2 | Imidazopyrazine and quinoxaline compoundsarrest parasites in late schizogony before completion of daughter cellformation. a, The onset of action for KAI407 was investigated using a highlysynchronized population of blood-stage parasites. Complete culture mediumcontaining 125 nM KAI407 was replaced with drug-free medium at the timeindicated in 2-h intervals. The resultant parasitaemia in the next life cycle(t 5 72 h) was normalized to untreated parasites (mean 6 s.d.; n 5 4). The timecourse represents trophozoite-stage parasites (t 5 34 h) through thematuration of schizonts (t 5 48 h). b, c, The plasma membrane markerPfATP4–GFP was used to visualize plasma membrane ingression arounddeveloping daughter merozoites, with nuclei stained by Hoechst 33342.Parasites treated with DMSO (control) formed clearly defined daughter cellsuniformly surrounded by the plasma membrane. Conversely, parasites treated
with 500 nM KAI407 for 4 h (KAI407-treated) had a disorganized membranestructure. Representative images from a single experimental replicate areshown (n 5 2). Scale bar, 5mm. d, Microscopy of Giemsa-stained parasitestreated with ,5 3 IC50 drug (125 nM KAI407, 15 nM KAI715, 150 nMKDU691 or 400 nM BQR695) or DMSO vehicle. Representative images from asingle experimental replicate are shown (n 5 3). e, Measurement of merozoiteviability via the merozoite release assay. The ability of merozoites to reinvadefresh RBCs after mechanical rupture of drug-arrested schizonts treated with125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 400 nM BQR695 wascompared to parasite reinvasion of untreated (DMSO) parasites. E-64 (grey),a known inhibitor of merozoite egress, was used as a control at 1mM(mean 6 s.d.; n 5 4).
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
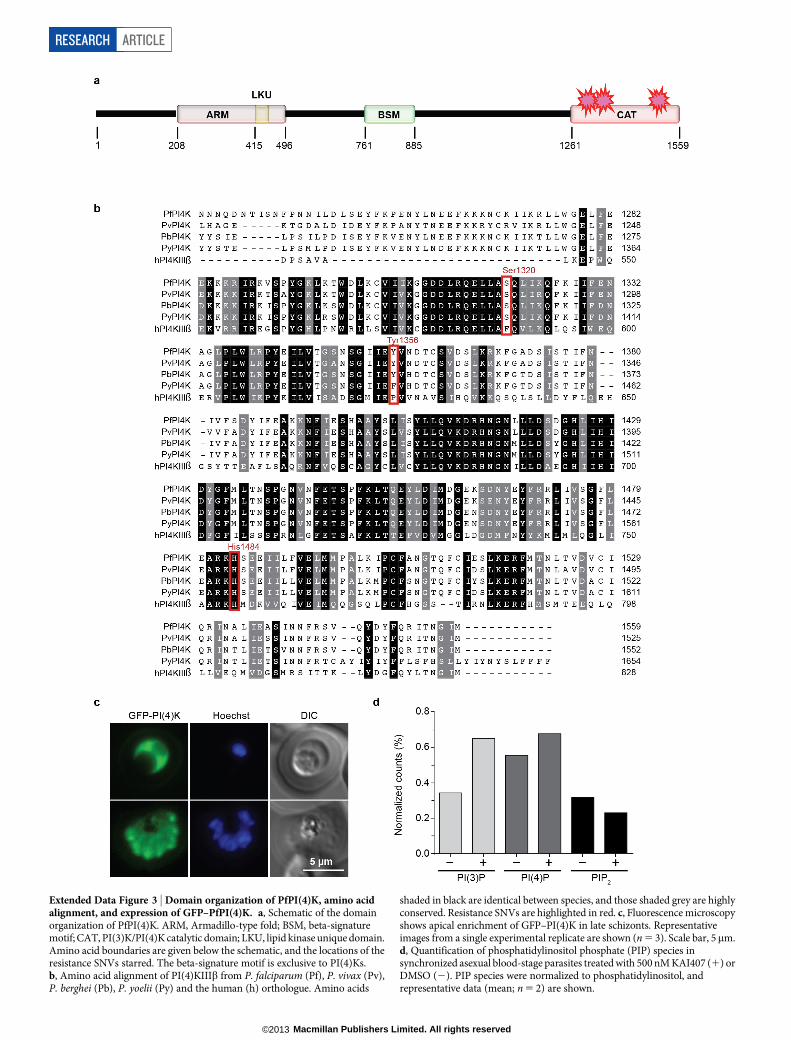
Extended Data Figure 3 | Domain organization of PfPI(4)K, amino acidalignment, and expression of GFP–PfPI(4)K. a, Schematic of the domainorganization of PfPI(4)K. ARM, Armadillo-type fold; BSM, beta-signaturemotif; CAT, PI(3)K/PI(4)K catalytic domain; LKU, lipid kinase unique domain.Amino acid boundaries are given below the schematic, and the locations of theresistance SNVs starred. The beta-signature motif is exclusive to PI(4)Ks.b, Amino acid alignment of PI(4)KIIIb from P. falciparum (Pf), P. vivax (Pv),P. berghei (Pb), P. yoelii (Py) and the human (h) orthologue. Amino acids
shaded in black are identical between species, and those shaded grey are highlyconserved. Resistance SNVs are highlighted in red. c, Fluorescence microscopyshows apical enrichment of GFP–PI(4)K in late schizonts. Representativeimages from a single experimental replicate are shown (n 5 3). Scale bar, 5mm.d, Quantification of phosphatidylinositol phosphate (PIP) species insynchronized asexual blood-stage parasites treated with 500 nM KAI407 (1) orDMSO (2). PIP species were normalized to phosphatidylinositol, andrepresentative data (mean; n 5 2) are shown.
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013
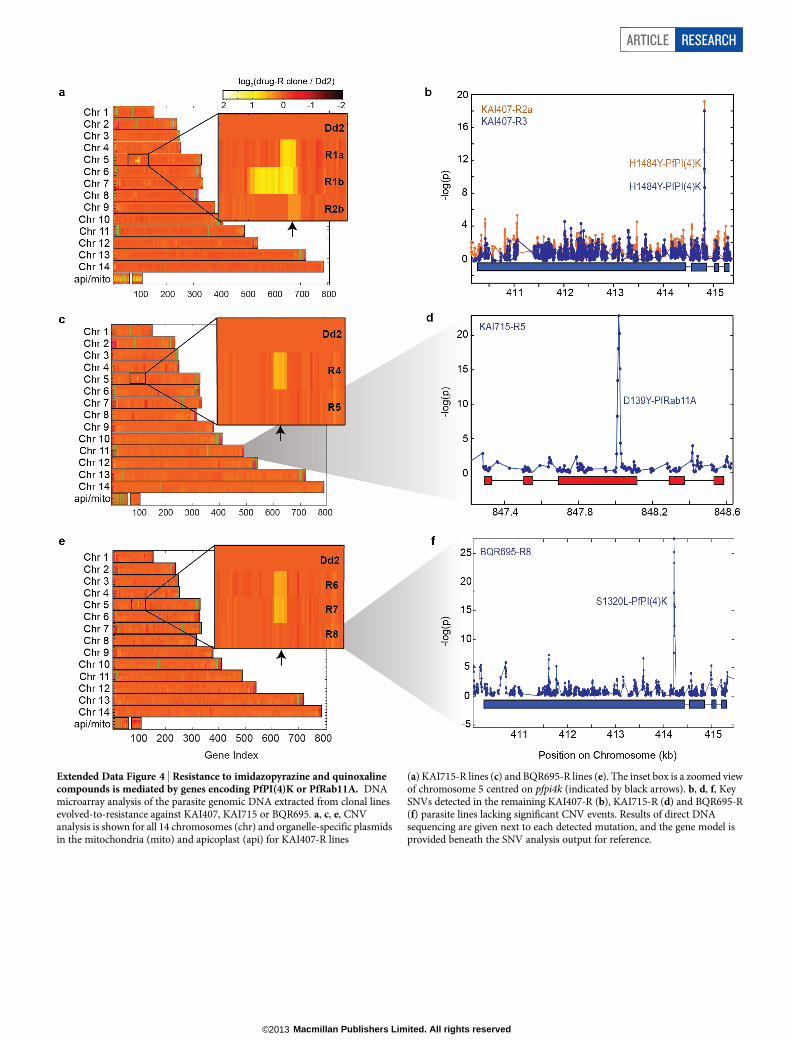
Extended Data Figure 4 | Resistance to imidazopyrazine and quinoxalinecompounds is mediated by genes encoding PfPI(4)K or PfRab11A. DNAmicroarray analysis of the parasite genomic DNA extracted from clonal linesevolved-to-resistance against KAI407, KAI715 or BQR695. a, c, e, CNVanalysis is shown for all 14 chromosomes (chr) and organelle-specific plasmidsin the mitochondria (mito) and apicoplast (api) for KAI407-R lines
(a) KAI715-R lines (c) and BQR695-R lines (e). The inset box is a zoomed viewof chromosome 5 centred on pfpi4k (indicated by black arrows). b, d, f, KeySNVs detected in the remaining KAI407-R (b), KAI715-R (d) and BQR695-R(f) parasite lines lacking significant CNV events. Results of direct DNAsequencing are given next to each detected mutation, and the gene model isprovided beneath the SNV analysis output for reference.
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
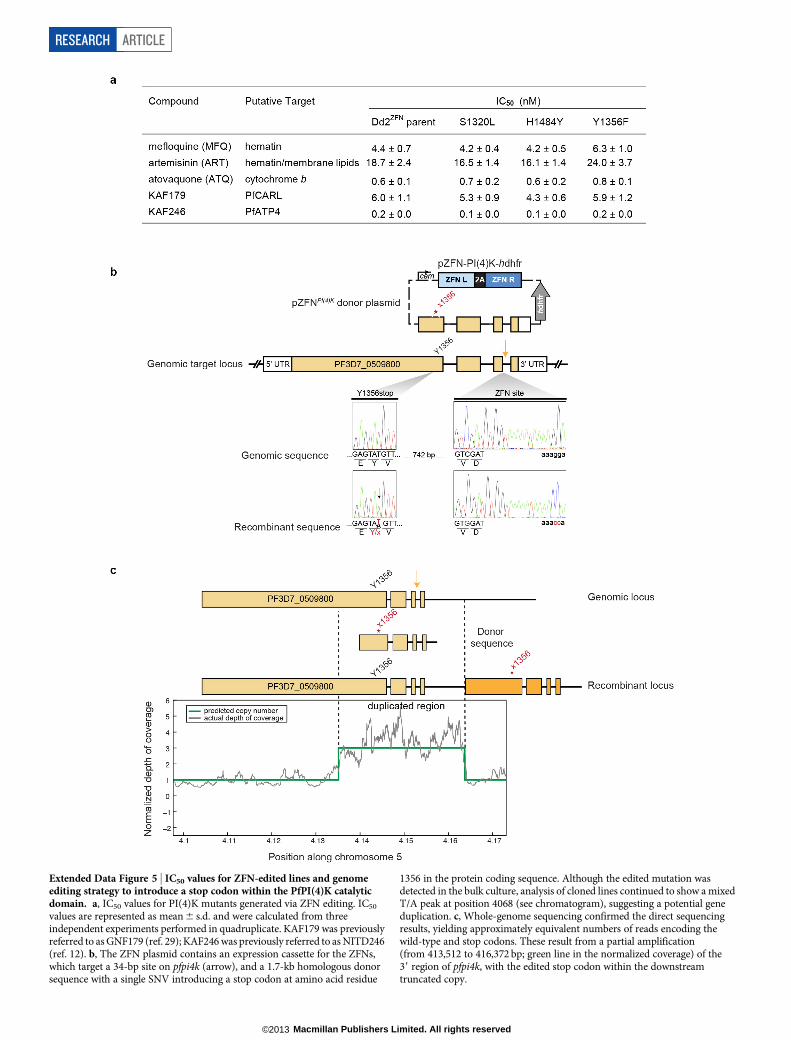
Extended Data Figure 5 | IC50 values for ZFN-edited lines and genomeediting strategy to introduce a stop codon within the PfPI(4)K catalyticdomain. a, IC50 values for PI(4)K mutants generated via ZFN editing. IC50
values are represented as mean 6 s.d. and were calculated from threeindependent experiments performed in quadruplicate. KAF179 was previouslyreferred to as GNF179 (ref. 29); KAF246 was previously referred to as NITD246(ref. 12). b, The ZFN plasmid contains an expression cassette for the ZFNs,which target a 34-bp site on pfpi4k (arrow), and a 1.7-kb homologous donorsequence with a single SNV introducing a stop codon at amino acid residue
1356 in the protein coding sequence. Although the edited mutation wasdetected in the bulk culture, analysis of cloned lines continued to show a mixedT/A peak at position 4068 (see chromatogram), suggesting a potential geneduplication. c, Whole-genome sequencing confirmed the direct sequencingresults, yielding approximately equivalent numbers of reads encoding thewild-type and stop codons. These result from a partial amplification(from 413,512 to 416,372 bp; green line in the normalized coverage) of the39 region of pfpi4k, with the edited stop codon within the downstreamtruncated copy.
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013
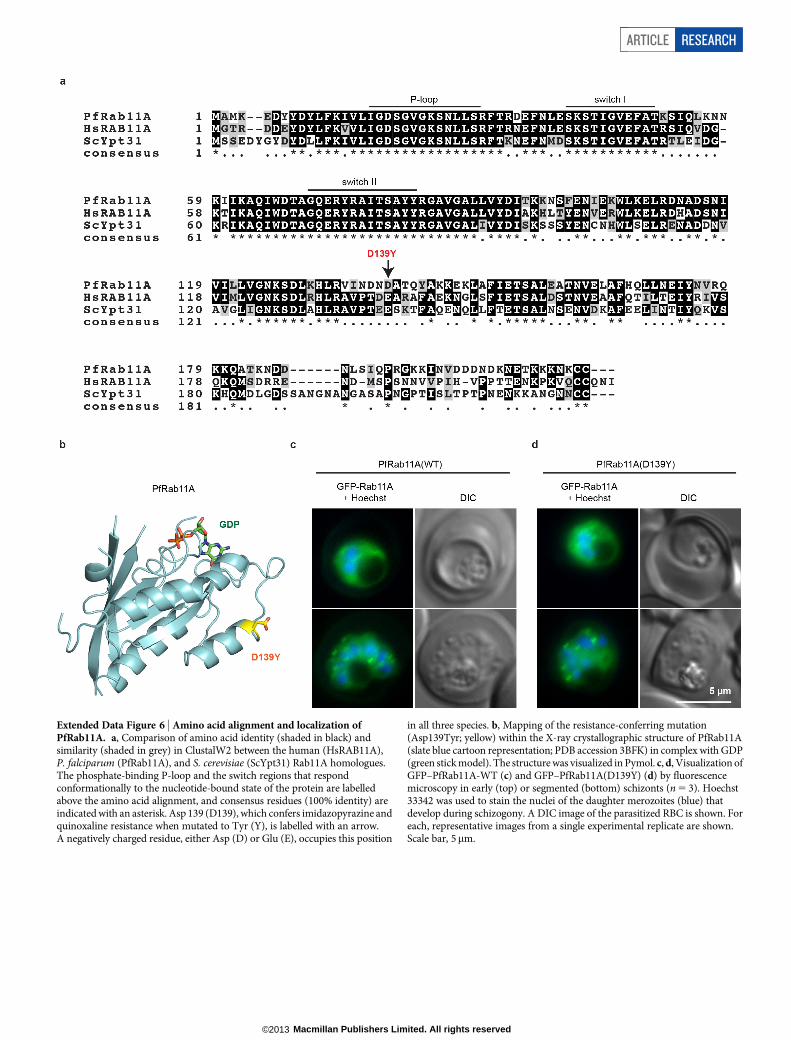
Extended Data Figure 6 | Amino acid alignment and localization ofPfRab11A. a, Comparison of amino acid identity (shaded in black) andsimilarity (shaded in grey) in ClustalW2 between the human (HsRAB11A),P. falciparum (PfRab11A), and S. cerevisiae (ScYpt31) Rab11A homologues.The phosphate-binding P-loop and the switch regions that respondconformationally to the nucleotide-bound state of the protein are labelledabove the amino acid alignment, and consensus residues (100% identity) areindicated with an asterisk. Asp 139 (D139), which confers imidazopyrazine andquinoxaline resistance when mutated to Tyr (Y), is labelled with an arrow.A negatively charged residue, either Asp (D) or Glu (E), occupies this position
in all three species. b, Mapping of the resistance-conferring mutation(Asp139Tyr; yellow) within the X-ray crystallographic structure of PfRab11A(slate blue cartoon representation; PDB accession 3BFK) in complex with GDP(green stick model). The structure was visualized in Pymol. c, d, Visualization ofGFP–PfRab11A-WT (c) and GFP–PfRab11A(D139Y) (d) by fluorescencemicroscopy in early (top) or segmented (bottom) schizonts (n 5 3). Hoechst33342 was used to stain the nuclei of the daughter merozoites (blue) thatdevelop during schizogony. A DIC image of the parasitized RBC is shown. Foreach, representative images from a single experimental replicate are shown.Scale bar, 5mm.
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
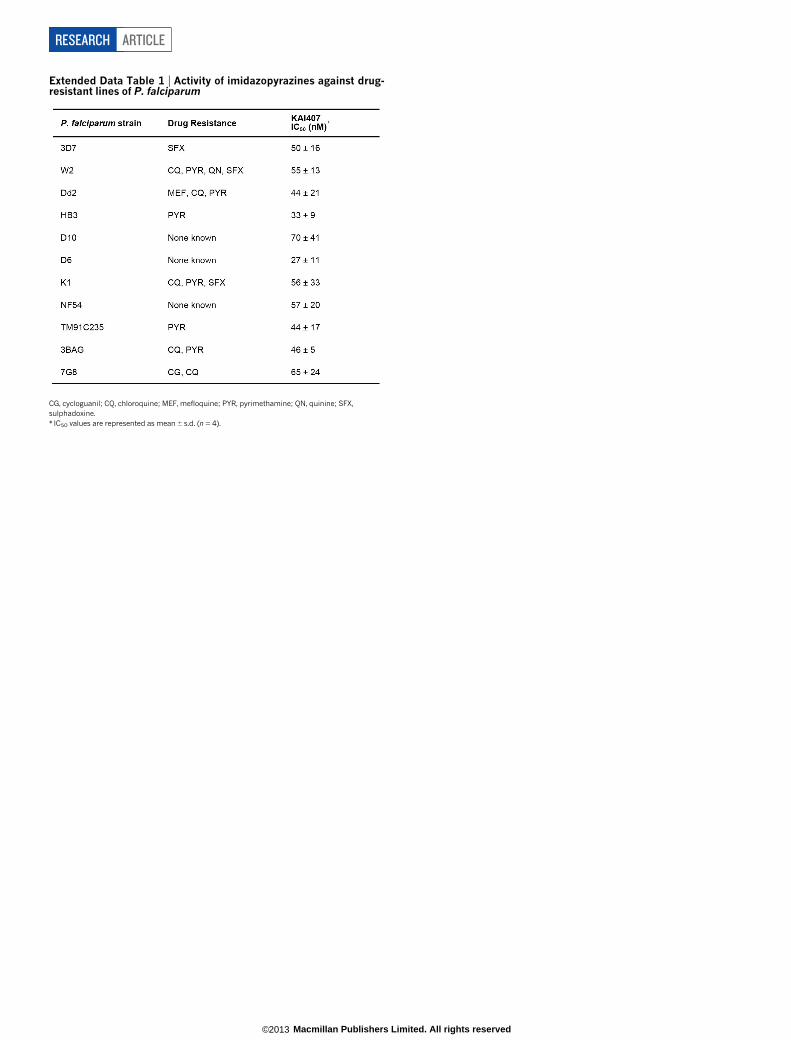
Extended Data Table 1 | Activity of imidazopyrazines against drug-resistant lines of P. falciparum
CG, cycloguanil; CQ, chloroquine; MEF, mefloquine; PYR, pyrimethamine; QN, quinine; SFX,sulphadoxine.* IC50 values are represented as mean 6 s.d. (n 5 4).
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013
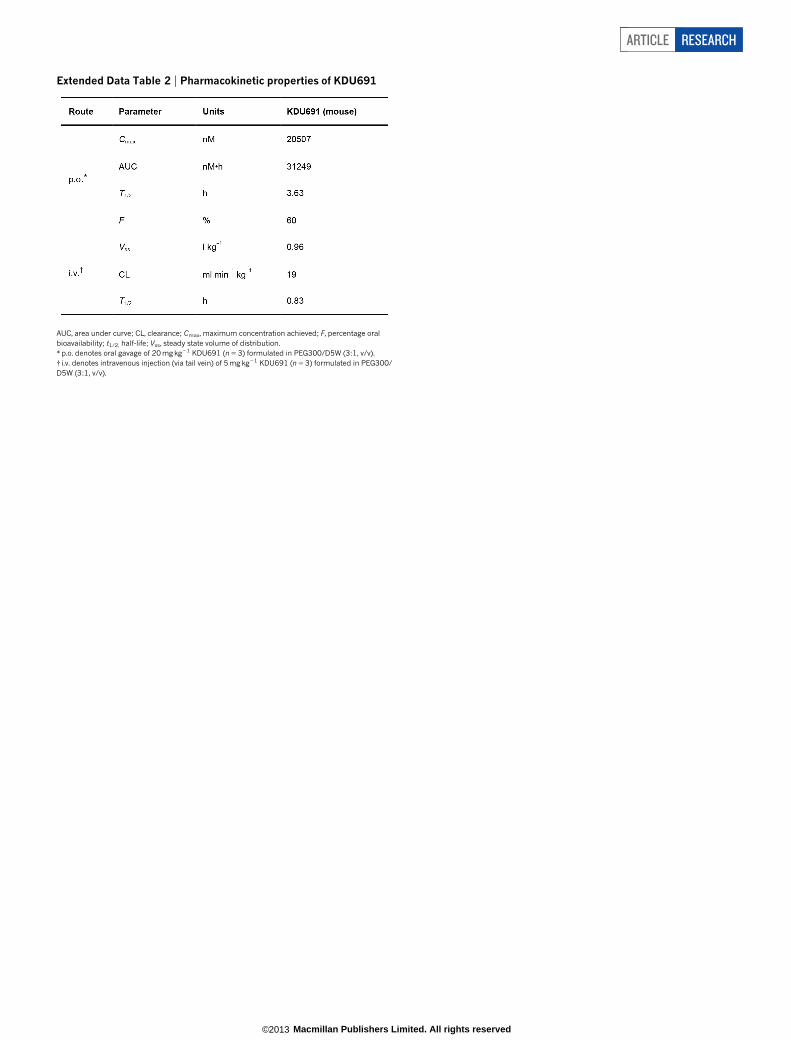
Extended Data Table 2 | Pharmacokinetic properties of KDU691
AUC, area under curve; CL, clearance; Cmax, maximum concentration achieved; F, percentage oralbioavailability; t1/2, half-life; Vss, steady state volume of distribution.*p.o. denotes oral gavage of 20 mg kg21 KDU691 (n 5 3) formulated in PEG300/D5W (3:1, v/v).{ i.v. denotes intravenous injection (via tail vein) of 5 mg kg21 KDU691 (n 5 3) formulated in PEG300/D5W (3:1, v/v).
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
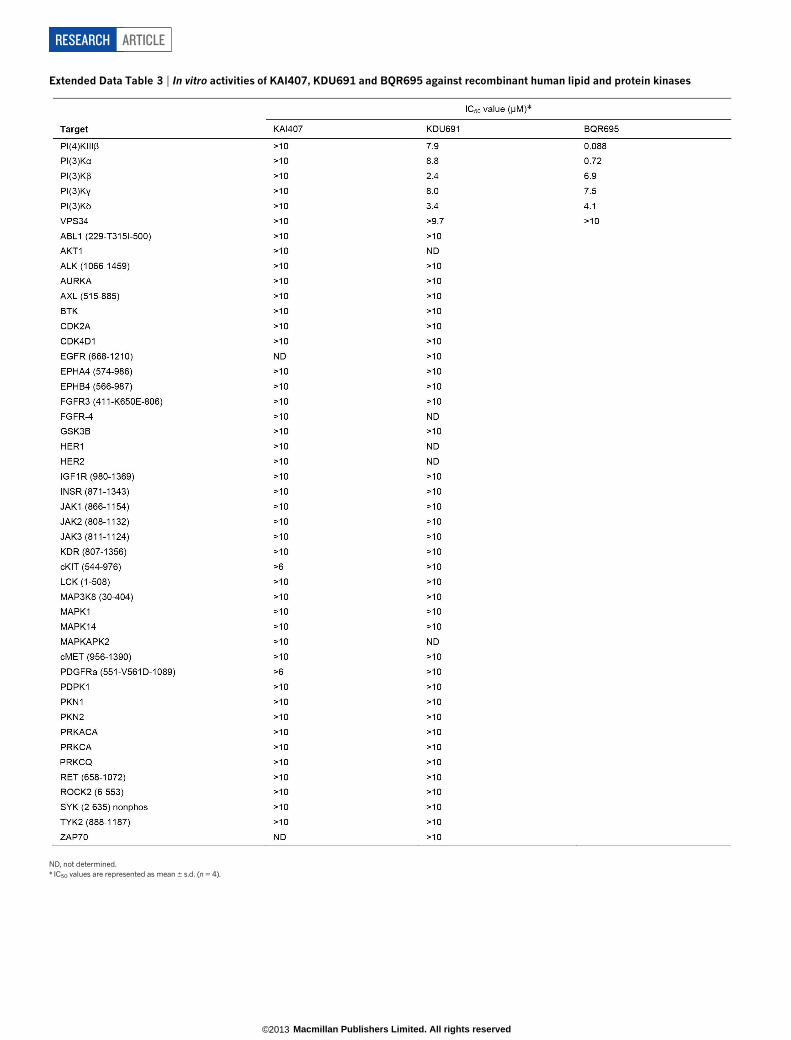
Extended Data Table 3 | In vitro activities of KAI407, KDU691 and BQR695 against recombinant human lipid and protein kinases
ND, not determined.* IC50 values are represented as mean 6 s.d. (n 5 4).
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013
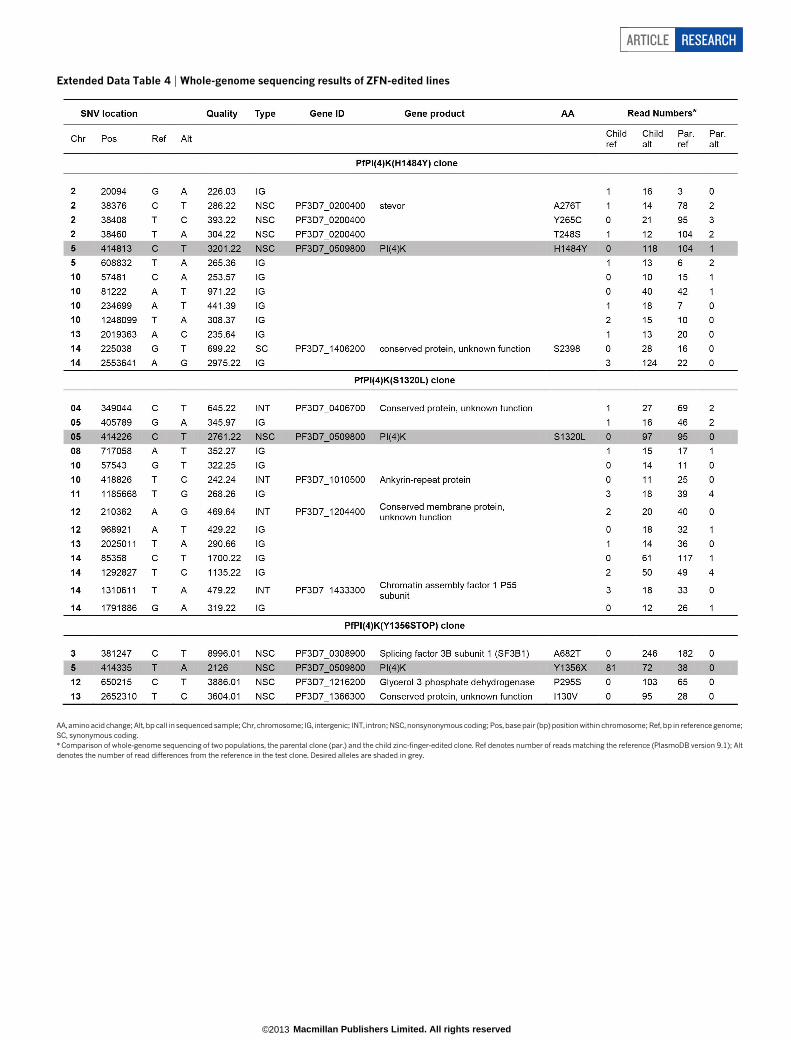
Extended Data Table 4 | Whole-genome sequencing results of ZFN-edited lines
AA, amino acid change; Alt, bp call in sequenced sample; Chr, chromosome; IG, intergenic; INT, intron; NSC, nonsynonymous coding; Pos, base pair (bp) position within chromosome; Ref, bp in reference genome;SC, synonymous coding.*Comparison of whole-genome sequencing of two populations, the parental clone (par.) and the child zinc-finger-edited clone. Ref denotes number of reads matching the reference (PlasmoDB version 9.1); Altdenotes the number of read differences from the reference in the test clone. Desired alleles are shaded in grey.
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013
Related Documents











