Editor-in-Chief, James O. Armitage, MD | ASCOPost.com Charting a New Course: From Clinical Investigator to University President A Conversation With Fadlo R. Khuri, MD, FACP By Jo Cavallo W hat first intrigued Fadlo R. Khuri, MD, FACP, about the prospect of becoming the 16th Pres- ident of the American University of Beirut (AUB) in Lebanon was the chance to give back to an institution and a country that had given him so much. Born in Boston, Massachuses, in 1963, Dr. Khuri was raised in Beirut and followed his great grandfathers, paternal grandfather, father—Raja N. Khuri, MD, later served as Dean of the AUB Medical School—and mother in aending AUB, but he leſt the university in 1982, dur- ing the country’s raging civil war, to study at Yale Uni- versity in New Haven, Connecticut. “I didn’t leave because of the war,” said Dr. Khuri. “I leſt because I was aract- ed to explore aspects of Yale in terms of exposure to a broader liberal arts education while preparing for medical school, which would have been more dif- ficult at AUB at that time.” Aſter graduating from Yale, Dr. Khuri earned his Journal Spotlight A few weeks ago, I read an op-ed 1 in e New York Times wrien by Stan Collender, a patient with Merkel cell carcinoma, a rare and aggressive type of skin cancer. In his article, he described his participation in a clinical trial for a new drug he is hoping will stem progression of his cancer and the need for more patients to enroll in clinical studies so effective new medi- cations can be approved and quickly get into the hands of patients who need them. Without clinical trials, wrote Mr. Collender, promising new drugs won’t be tested, and progress against cancer will be slowed. Our Patients Are the True Heroes of Cancer Research By Julie M. Vose, MD, MBA, FASCO Oncology Meetings Coverage Debates and Didactics in Hematology and Oncology�������������������� 3–6 Best of ASCO �������������������� 14–17, 20–22 World Congress of Psycho-oncology ������������������������������ 24–27 National Cancer Policy Forum�������� 35–36 New Orleans Summer Cancer Meeting ��������������� 40–43, 45–46 S. Vincent Rajkumar, MD, on Myeloma ����38 Oncology Worldwide������������������������ 47, 67 Direct From ASCO �������������������������� 55–58 Women in Oncology: Carolyn D. Runowicz, MD, FASCO���������������������������� 76 MORE IN THIS ISSUE continued on page 65 Oncology Worldwide Adding Elotuzumab to Lenalidomide Plus Dexamethasone Improves Progression-Free Survival in Refractory Multiple Myeloma By Mahew Stenger I n an interim analysis of the phase III ELO- QUENT-2 trial reported in e New England Jour- nal of Medicine, Sagar Lonial, MD, of Emory Univer- sity School of Medicine, Meletios Dimopoulos, MD, of National and Kapodistrian University of Athens, and colleagues found that the addition of elotuzumab to lenalidomide (Revlimid)/dexamethasone signifi- cantly increased progression-free survival in patients with relapsed or refractory multiple myeloma. 1 Elotuzumab is an immunostimulatory monoclo- nal antibody targeting signaling lymphocytic activa- tion molecule F7 (also known as SLAM7, SLAMF7, and CS1). Bone marrow myeloma cells (approxi- mately 95%) express SLAM7, as do natural killer cells. Elotuzumab directly activates natural killer cells and mediates antibody-dependent cell-mediat- ed cytotoxicity via the CD16 pathway. Study Details In this open-label trial conducted at 168 sites worldwide, 648 patients were randomly assigned between June 2011 and November 2012 to receive elotuzumab plus lenalido- mide/dexamethasone (n = 321) or lenalidomide/ dexamethasone alone (n = 325). Elotuzumab was given intravenously at 10 mg/kg on days 1, 8, 15, and 22 during 28-day cycles, along with lenalidomide at 25 mg/d on days 1 to 21 and dexamethasone orally at 40 mg during the week without elotuzumab and intra- venously at 8 mg plus 28 mg orally on the day of elo- tuzumab administration. Patients in the control group Sagar Lonial, MD Myeloma, Lymphoma, Leukemia 1, 5, 37–39, 60, 61 | Breast Cancer 17, 40, 87, 88, 104 | Coriolus Versicolor 92 VOLUME 6, ISSUE 16 SEPTEMBER 10, 2015 continued on page 37 continued on page 121 There is no question that the American University of Beirut casts a very large shadow and has a major impact on the political discourse in the region, the development of modern engineering and knowledge, and medical technology and research. —Fadlo R. Khuri, MD, FACP Dr. Vose is President of ASCO, the Neuman M. and Mil- dred E. Harris Professional Chair and Chief of the Oncol- ogy/Hematology Division in the Department of Internal Medicine at the University of Nebraska Medical Center, and the Associate Director of Clinical Research at the Fred & Pamela Buffe Cancer Center in Omaha. A Harborside Press® Publication Send your comments to [email protected]

Tap Vol 6 Issue 16
Jul 23, 2016
In an interim analysis of the phase III ELOQUENT-2 trial reported in The New England Journal of Medicine, Sagar Lonial, MD, of Emory University School of Medicine, Meletios Dimopoulos, MD, of National and Kapodistrian University of Athens, and colleagues found that the addition of elotuzumab to lenalidomide (Revlimid)/dexamethasone significantly increased progression-free survival in patients with relapsed or refractory multiple myeloma.
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Editor-in-Chief, James O. Armitage, MD | ASCOPost.com
Charting a New Course: From Clinical Investigator to University PresidentA Conversation With Fadlo R. Khuri, MD, FACPBy Jo Cavallo
What first intrigued Fadlo R. Khuri, MD, FACP, about the prospect of becoming the 16th Pres-
ident of the American University of Beirut (AUB) in Lebanon was the chance to give back to an institution and a country that had given him so much. Born in Boston, Massachusetts, in 1963, Dr. Khuri was raised
in Beirut and followed his great grandfathers, paternal grandfather, father—Raja N. Khuri, MD, later served as Dean of the AUB Medical School—and mother in attending AUB, but he left the university in 1982, dur-ing the country’s raging civil war, to study at Yale Uni-versity in New Haven, Connecticut.
“I didn’t leave because of the war,” said Dr. Khuri. “I left because I was attract-ed to explore aspects of Yale in terms of exposure to a broader liberal arts education while preparing for medical school, which would have been more dif-ficult at AUB at that time.”
After graduating from Yale, Dr. Khuri earned his
Journal Spotlight
A few weeks ago, I read an op-ed1 in The New York Times written by Stan Collender, a
patient with Merkel cell carcinoma, a rare and aggressive type of skin cancer. In his article, he described his participation in a clinical trial for a new drug he is hoping will stem progression of his cancer and the need for more patients to enroll in clinical studies so effective new medi-cations can be approved and quickly get into the hands of patients who need them. Without clinical trials, wrote Mr. Collender, promising new drugs won’t be tested, and progress against cancer will be slowed.
Our Patients Are the True Heroes of Cancer Research
By Julie M. Vose, MD, MBA, FASCO
Oncology Meetings CoverageDebates and Didactics in Hematology and Oncology ��������������������3–6Best of ASCO �������������������� 14–17, 20–22World Congress of Psycho-oncology ������������������������������ 24–27National Cancer Policy Forum �������� 35–36New Orleans Summer Cancer Meeting ��������������� 40–43, 45–46
S. Vincent Rajkumar, MD, on Myeloma ����38Oncology Worldwide ������������������������ 47, 67Direct From ASCO �������������������������� 55–58Women in Oncology: Carolyn D. Runowicz, MD, FASCO����������������������������76
MORE IN THIS ISSUE
continued on page 65
Oncology Worldwide
Adding Elotuzumab to Lenalidomide Plus Dexamethasone Improves Progression-Free Survival in Refractory Multiple Myeloma By Matthew Stenger
In an interim analysis of the phase III ELO-QUENT-2 trial reported in The New England Jour-
nal of Medicine, Sagar Lonial, MD, of Emory Univer-sity School of Medicine, Meletios Dimopoulos, MD, of National and Kapodistrian University of Athens, and colleagues found that the addition of elotuzumab to lenalidomide (Revlimid)/dexamethasone signifi-cantly increased progression-free survival in patients with relapsed or refractory multiple myeloma.1
Elotuzumab is an immunostimulatory monoclo-nal antibody targeting signaling lymphocytic activa-tion molecule F7 (also known as SLAM7, SLAMF7, and CS1). Bone marrow myeloma cells (approxi-mately 95%) express SLAM7, as do natural killer cells. Elotuzumab directly activates natural killer cells and mediates antibody-dependent cell-mediat-ed cytotoxicity via the CD16 pathway.
Study DetailsIn this open-label trial
conducted at 168 sites worldwide, 648 patients were randomly assigned between June 2011 and November 2012 to receive elotuzumab plus lenalido-mide/dexamethasone (n = 321) or lenalidomide/dexamethasone alone (n = 325). Elotuzumab was given intravenously at 10 mg/kg on days 1, 8, 15, and 22 during 28-day cycles, along with lenalidomide at 25 mg/d on days 1 to 21 and dexamethasone orally at 40 mg during the week without elotuzumab and intra-venously at 8 mg plus 28 mg orally on the day of elo-tuzumab administration. Patients in the control group
Sagar Lonial, MD
Myeloma, Lymphoma, Leukemia 1, 5, 37–39, 60, 61 | Breast Cancer 17, 40, 87, 88, 104 | Coriolus Versicolor 92 VOLUME 6, ISSUE 16SEPTEMBER 10, 2015
continued on page 37
continued on page 121
There is no question that the American University of Beirut casts a very large shadow and has a major impact on the political discourse in the region, the development of modern engineering and knowledge, and medical technology and research.
—Fadlo R. Khuri, MD, FACP
Dr. Vose is President of ASCO, the Neuman M. and Mil-dred E. Harris Professional Chair and Chief of the Oncol-ogy/Hematology Division in the Department of Internal Medicine at the University of Nebraska Medical Center, and the Associate Director of Clinical Research at the Fred & Pamela Buffett Cancer Center in Omaha.
A Harborside Press® PublicationSend your comments to [email protected]

PAGE 2 The ASCO Post | SEPTEMBER 10, 2015
Disclaimer: The ideas and opinions expressed in The ASCO Post™ do not necessarily reflect those of Harborside Press®, LLC, HSP News Service, LLC, or the American Society of Clinical Oncology, Inc. (ASCO®). The mention of any product, service, or therapy in this publication should not be construed as an endorsement of the products mentioned. It is the responsibility of the treating physician or other health-care provider, relying on independent experience and knowledge of the patient, to determine the appropri-ate treatment for the patient. Readers are advised to check the appropriate medical literature and the product information currently provided by the manufacturer of each product or therapy to be administered to verify the dosage, method, and duration of administration, or contraindications. Readers are also encouraged to contact the manufacturer with questions about the features or limitations of any products. Harborside Press®, HSP News Service, LLC, and ASCO® assume no responsibility for any injury or damage to persons or property arising out of or related to any use of material contained in this publication or to any errors or omissions.
James O. Armitage, MD Editor-in-Chief
Elizabeth Reed, MD Deputy Editor University of Nebraska Medical Center
Associate EditorsJame Abraham, MD Cleveland Clinic
Syed A. Abutalib, MD Cancer Treatment Centers of America
Manmeet Ahluwalia, MD, FACP Cleveland Clinic
Chandrakanth Are, MD University of Nebraska Medical Center
Joseph S. Bailes, MD Texas Oncology
Laurence H. Baker, DO University of Michigan Health System
Richard R. Barakat, MD Memorial Sloan Kettering Cancer Center
Charles L. Bennett, MD, PhD, MPP University of South Carolina, Columbia
Douglas W. Blayney, MD Stanford University Medical Center
Philip D. Bonomi, MD Rush University Medical Center
Richard Boxer, MD University of Wisconsin School of Medicine
Harold J. Burstein, MD Dana-Farber Cancer Institute
Robert W. Carlson, MD National Comprehensive Cancer Network
Barrie R. Cassileth, PhD Memorial Sloan Kettering Cancer Center
Jay S. Cooper, MD Maimonides Medical Center
John Cox, DO Texas Oncology
E. David Crawford, MD University of Colorado
Nancy E. Davidson, MD University of Pittsburgh Cancer Institute
George D. Demetri, MD Dana-Farber Cancer Institute
Paul F. Engstrom, MD Fox Chase Cancer Center
David S. Ettinger, MD Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Bishoy Morris Faltas, MD Weill Cornell Medical College
John A. Fracchia, MD New York Urological Associates
Alison Freifeld, MD University of Nebraska Medical Center
Louis B. Harrison, MD Moffitt Cancer Center
Jimmie C. Holland, MD Memorial Sloan Kettering Cancer Center
Clifford A. Hudis, MD, FACP Memorial Sloan Kettering Cancer Center
Nora Janjan, MD, MPSA, MBA National Center for Policy Analysis
Hagop M. Kantarjian, MD MD Anderson Cancer Center
Mario E. Lacouture, MD Memorial Sloan Kettering Cancer Center
Theodore S. Lawrence, MD, PhD University of Michigan Comprehensive Cancer Center
Stephen J. Lemon, MD, MPH Oncology Associates, PC, Omaha
Stuart Lichtman, MD Memorial Sloan Kettering Cancer Center Commack, New York
Michael P. Link, MD Stanford University Medical Center
John L. Marshall, MD Ruesch Center for the Cure of GI Cancer at Georgetown University
Mary S. McCabe, RN, MA Memorial Sloan Kettering Cancer Center
William T. McGivney, PhD Philadelphia, Pennsylvania
James L. Mulshine, MD Rush University Medical Center
Derek Raghavan, MD, PhD Levine Cancer Institute Carolinas HealthCare System
Steven T. Rosen, MD City of Hope National Medical Center
Lee S. Schwartzberg, MD University of Tennessee Health Science Center
Andrew D. Seidman, MD Memorial Sloan Kettering Cancer Center
Samuel Silver, MD, PhD University of Michigan Health System
George W. Sledge, MD Stanford University School of Medicine
Thomas J. Smith, MD Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Jamie Von Roenn, MD American Society of Clinical Oncology
Lynn D. Wilson, MD Yale University School of Medicine
Stanley H. Winokur, MD Singer Island, Florida
William C. Wood, MD Winship Cancer Institute, Emory University
International EditorsClement Adebamowo, BM, ChB (Hons), ScD University of Ibadan, Nigeria
Eduardo Cazap, MD, PhD International Union Against Cancer (UICC) Buenos Aires, Argentina
Rakesh Chopra, MD Artemis Healthsciences Institute Gurgaon, Haryana, India
Nagi El-Saghir, MD American University of Beirut, Lebanon
Mary Gospodarowicz, MD Princess Margaret Hospital Toronto, Ontario, Canada
Jacek Jassem, MD Medical University of Gdansk, Poland
David Khayat, MD Pitie-Salpetriere Hospital, Paris, France
Tony Mok, MD The Chinese University of Hong Kong Shatin, Hong Kong
Eliezer Robinson, MD National Council for Oncology Israeli Cancer Association, Haifa, Israel
Nagahiro Saijo, MD, PhD Kinki University School of Medicine Osaka, Japan
John F. Smyth, MD University of Edinburgh Edinburgh, Scotland
Daniel A. Vorobiof, MD Sandton Oncology Centre Johannesburg, South Africa
Editorial Board
The ASCO Post (ISSN 2154-3283), USPS Publicaton Number 6885, is published semi-monthly, except monthly in January by Harborside Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724, under a license arrangement with the American Society of Clinical Oncology, Inc. (ASCO®). Periodicals Postage paid at Cold Spring Harbor, NY, and additional mailing offices.
Change of Address: Postmaster send address changes to The ASCO Post, c/o Harborside Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724. ASCO Members: If you would like to cancel your subscription to The ASCO Post or need to update your mailing address, please visit your personalized page on ASCO.org. For personal-ized service, please contact ASCO Member Services at (888) 282-2552, (703) 299-0158, or via email at [email protected]. Non ASCO Members: To initiate or cancel a subscription or to update your mailing address, please email [email protected] or fax (631) 692-0805.
Copyright ©2015 by Harborside Press®, LLC. All rights reserved. Reproduction in whole or in part, in any form, without prior written permission of the publisher is pro-
hibited. For permission inquiries, contact [email protected].
Editorial Mission: The ASCO Post communicates timely in-formation to a broad audience of oncology specialists, help-ing to advance the highest quality multidisciplinary cancer care. The ASCO Post publishes highly validated coverage of cancer research and policy news, patient care and clinical practice issues, and thoughtful commentary from leaders in the field and others with an interest in clinical oncology.
Circulation: The ASCO Post is sent free of charge to ap-proximately 27,000 physicians and nurses, including all US-based ASCO members. Medical, surgical, pediatric, and gynecologic oncologists, hematologists, and he-matologist/oncologists in the United States who are not members of ASCO will be eligible for a complimentary subscription. ASCO members outside of the United States receive complimentary access to The ASCO Post online at www.ASCOPost.com.
Paid subscriptions to The ASCO Post are available for all other interested individuals. Individual Domestic: $300;
Canada: $436; Individual International: $575; Institutional Domestic: $370; Canada: $507; Institutional International: $645. Single Copy Domestic: $57; Canada: $65; Interna-tional: $72. Contact [email protected].
Correspondence: Address general inquiries to Harbor-side Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724. Phone: 631.692.0800; Fax: 631.692.0805. Address editorial correspondence to James O. Armitage, MD, Edi-tor-in-Chief, c/o Cara Glynn, phone: 631.935.7654; e-mail: [email protected].
Advertising: For information on advertising rates, re-prints, or supplements, contact Leslie Dubin, phone: 631.935.7660; e-mail: [email protected].
Notice to Advertisers: Advertiser and advertising agency recognize and accept that the following language ap-pears within the publication: “All statements, including product claims, are those of the person or organization making the statement or claim. Neither the publisher nor ASCO adopts any such statement or claim as its own, and any such statement or claim does not necessarily reflect the opinion of the publisher or ASCO.”
Advertiser and advertising agency accept and assume li-ability for all content (including text, representations, il-lustrations, opinions, and facts) of advertisements print-ed, and also assume responsibility for any claims made against the publisher or ASCO arising from or related to such advertisements. In the event that legal action or a claim is made against the publisher or ASCO arising from or related to such advertisements, advertiser and advertising agency agree to fully defend, indemnify, and hold harmless the publisher and ASCO, and to pay any judgment, expenses, and legal fees incurred by the publisher and by ASCO as a result of said legal action or claim. The publisher reserves the right to reject any advertising that it believes is not in keeping with the publication’s standards.
The publisher is not liable for delays in delivery and/or non-delivery in the event of Act of God, action by any government or quasi-governmental entity, fire, flood, in-surrection, riot, explosion, embargo, strikes (whether legal or illegal), labor or material shortage, transportation inter-ruption of any kind, work slow-down, or any condition be-yond the control of the publisher affecting production or delivery in any manner.
Harborside Press® Publishing Staff Conor Lynch, Executive Editor [email protected]
Cara H. Glynn, Director of Editorial [email protected]
Andrew Nash, Associate Director of Editorial [email protected]
Jo Cavallo, Senior Editor and Correspondent [email protected]
Randi Londer Gould and Susan Reckling, Senior Editors [email protected] [email protected]
Sarah McGullam, Web Editor [email protected]
Michael Buckley, Art Director [email protected]
Regine M. Lombardo, Senior Graphic Designer [email protected]
Terri Caivano and Brittany Bordonaro, Layout Artists [email protected] [email protected]
Gail van Koot, Editorial Coordinator [email protected]
Elizabeth Janetschek, Editorial Assistant [email protected]
Norman Virtue, Production Manager [email protected]
Shannon Meserve, Circulation Manager [email protected]
Jeannine Coronna, Vice President, Director of Operations [email protected]
Frank Buchner, Chief Technology Officer [email protected]
Leslie Dubin, Vice-President, Director of Sales [email protected]
Anthony Cutrone, President [email protected]
John A. Gentile, Jr, Chairman [email protected]
Contributing Writers:Meg Barbor, Charlotte Bath, Chase Doyle, Kirsten Boyd Goldberg, Margot Fromer, Alice Goodman, Caroline Helwick, Beth Howard, Susan London, Caroline McNeil, Eileen O’Gara-Kurtis, Ronald Piana, Matthew Stenger
Contributing Artists: Portraits by Keith Witmer, Keith Witmer Illustrations.
Disclosure information available at ASCOPost.com.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 3
Debates and Didactics in Hematology and Oncology
Positioning Targeted and Immunotherapy-Based Approaches in Lung CancerBy Caroline Helwick
W ith immunotherapy changing the face of lung cancer, is there
still a place for targeted therapy? Two experts from Emory University debated this issue at the 2015 Debates and Di-dactics in Hematology and Oncology Conference held in Sea Island, Georgia. Fadlo Khuri, MD, was recently named President of the American University of Beirut in Lebanon, and Suresh S. Ramalingam, MD, is Professor and Di-rector of Medical Oncology at Emory.
Targeting Driver Mutations“In my view,” Dr. Khuri maintained,
“when you have highly characterized genomic drivers, you are going to go with genomically targeted therapy.”
Lung cancer exhibits at least three targetable mutations, at least one of which is harbored by almost two-thirds of patients with adenocarcinomas. Half or more of these patients will respond to targeted agents, with a duration of benefit that can be “quite striking,” Dr. Khuri said.
Numerous benefits remain unique to targeted agents, he said, including the opportunity to target specific drivers of oncogene-addicted tumors, high re-sponse rates, reliability and availability of biomarkers in genomic testing, a fa-vorable cost-benefit ratio, and an under-standing of mechanisms of resistance.
The prognosis of non–small cell lung cancer (NSCLC) dramatically changed with the discovery of mutations in the epidermal growth factor (EGFR). The ability to target such tumors with ty-rosine kinase inhibitors led to a “sea change” in the management of EGFR-mutated patients. EGFR inhibitors be-came and remain the first-line standard of care, he said.
The emergence of resistance has been the Achilles heel of targeted ther-
apy, but drugs in development are tack-ling that problem, especially by over-coming the hallmark T790M mutation. About half the patients whose disease progresses demonstrate this alteration.
“We now have impressive data show-ing that we understand this mutation, its biology, and its prognosis, and we have developed treatments that target T790M,” he said.
Patients with the T790M mutation almost invariably respond to the third-generation EGFR inhibitor AZD9291. In a study of 138 mutation-positive pa-tients, the disease control rate was 95%.1 Similar agents are in development, in-cluding rociletinib (CO-1686), which has produced responses in almost 60% of T790M-mutated patients.2
The ALK fusion gene represents the other “success story” of targeted management. Showing striking ac-tivity (doubling of progression-free survival) in the 5% of patients with NSCLC who have this alteration, the ALK inhibitor crizotinib (Xalkori) was rapidly approved.3
Resistance to crizotinib is also be-ing tackled as more is learned about the mechanisms of resistance to ALK inhibitors, which differ from those ob-served with EGFR inhibitors. Third-generation ALK inhibitors in develop-
ment, including ceritinib (Zykadia) and alectinib, are effective in more than 50% of crizotinib-treated patients. The dual ALK/EGFR inhibitor AP26113 is also active in patients with secondary resis-tance to ALK, he noted.
BRAF, the third driver mutation that can be targeted, is identified in 1% to 2% of patients. Monotherapy with dabrafenib (Tafinlar) yielded a disease
control rate of 56% at 12 weeks and a median duration of response of 11.8 months in previously treated patients.4 It may be even more effective when combined with trametinib (Mekinist), Dr. Khuri said.
A Place for ImmunotherapySince targeted therapy is limited to
mutation subsets, there is a place for im-munotherapy, especially as second-line therapy, Dr. Khuri acknowledged. “If you know you have a driver mutation, you go with targeted therapy. But with-out a known mutation, in the second-line setting, you go with nivolumab [Opdivo], which is dramatically supe-rior to docetaxel,” he said.
“No question, there is no more ex-citing science than cancer immuno-therapy. Especially with combination immunotherapy, we may see dramatic, durable responses,” he acknowledged.
However, Dr. Khuri pointed out that the benefits may be accompanied by more toxicity (especially with combina-tions) than is seen with targeted thera-py. “One of the most evident toxicities is to the bank account!” he added.
For a median progression-free sur-vival of 6.9 months, nivolumab mono-therapy costs more than $100,000. For a median progression-free survival of 11.4 months, the cost approaches $300,000, according to some calculations.
“In my view, this is probably more than the market can bear,” Dr. Khuri commented. Coupled with a 20% copay for patients, “this will be unsustainable.”
Dr. Ramalingam pointed out that as good as targeted therapy is, this ap-proach has never cured a patient with lung cancer. “Immunotherapy is going to, by far, have a stronger impact on lung cancer than targeted treatment has had so far,” he maintained. “Immunothera-py improves survival, and this cannot be said for targeted agents.”
In addition, he added, immunother-apy can be applied to all-comers, and that unlike targeted therapy, its effect is not preferential to persons who have a little to no smoking history (ie, not the average patient). Immunotherapy ad-dresses the “garden variety lung cancer,” which is smoking-derived tumors with high mutational burdens, he indicated.
Dr. Ramalingam further reminded listeners that although 65% of patients have a driver mutation, 25% of them will be in KRAS, “which still begs for a treatment option.” Targeted treatments, he pointed out, “are therefore used in a small percentage of our patients.”
Additional obstacles to the use of targeted therapies are the emergence of resistance, for which current options are limited, and their failure to be effective
Thoracic Oncology
continued on page 4
If you know you have a driver mutation, you go with targeted therapy. But without a known mutation, in the second-line setting, you go with nivolumab, which is dramatically superior to docetaxel.
—Fadlo Khuri, MD, FACP
Learn more at jadprolive.com For conference exhibitor or sponsorship opportunities, contact David Horowitz
at 631-935-7658 or email at [email protected]
Learn more at Learn more at Learn more at Learn more at jadprolive.comLearn more at jadprolive.comjadprolive.comLearn more at jadprolive.comjadprolive.comjadprolive.comLearn more at Learn more at
November 5-8, 2015JW Marriott Phoenix Desert Ridge
Save the Date
A CE/CME Conference for Advanced Practitioners in Oncology

PAGE 4 The ASCO Post | SEPTEMBER 10, 2015
Debates and Didactics in Hematology and Oncology
earlier in the disease course. However, targeted agents do have the edge over immunotherapy in terms of response rates, he acknowledged, but added that when patients do respond to immuno-therapy, their benefits can be durable.
Survival Benefits With Immunotherapy
“We also now have overall survival data showing immunotherapy to be su-perior,” said Dr. Ramalingam.
In a phase I study of nivolumab in refractory NSCLC,5 2-year survival was 24%, and at 3 years, 18% of heavily pretreated patients remained free from progression, he noted. “To be alive 3 years later is very exciting for patients who have run out of options,” added Dr. Ramalingam.
In the phase III CheckMate 017 trial in previously treated squamous cell carci-noma, nivolumab significantly improved overall survival over docetaxel in the second-line setting.6 The hazard ratio of 0.59 compares favorably with that seen with targeted agents vs chemotherapy in the front line, “and this is second line,” he added. “First-line data will be out in a year or so, and if this is a sign of what’s to come, we are in for an exciting time.”
The possibility of long-term survival is further illustrated by a study led by Dr. Ramalingam of nivolumab in refrac-tory patients with squamous cell carci-noma.7 Although the response rate was only 14%, almost all responders had ongoing responses, whether or not they continued on nivolumab.
“That tells us the power of immuno-therapy,” he commented. “With target-ed therapy, the median progression-free survival is 10 to 13 months. With these patients, we haven’t reached the median
duration of response.”Activity in nonsquamous histol-
ogy also has shown that the anti–PD-1 (programmed cell death protein 1) an-tibodies are not “one-trick ponies,” he continued. Although it yielded a slight-ly lower hazard ratio (0.73), nivolumab improved survival over docetaxel as second-line treatment of nonsquamous patients in the phase III CheckMate 057 study.8 “These two trials show that immunotherapy trumps targeted ther-apy in the second line,” he maintained.
Beyond NivolumabStrong data are also emerging for
anti–PD-1/PD-L1 (its ligand) agents other than nivolumab. In the phase II POPLAR trial, atezolizumab significant-ly improved overall survival vs docetaxel in patients with strong PD-L1 expres-sion; the response rate was 38%, and me-dian overall survival was not reached.9
Although such findings suggest that PD-L1 expression may help select patients, noted Dr. Ramalingam, “These drugs work even in PD-L1–negative patients. Their outcomes are at least as good as with che-motherapy, and we need to bear this in mind in terms of selecting patients.”
Impressive outcomes were also achieved among PD-L1–positive pa-tients receiving pembrolizumab (Key-truda) in KEYNOTE-001.10 In updated
results, high expressers (≥ 50% stain-ing) had a response rate of 52%, a me-dian progression-free survival of 12.5 months, and a median overall survival that had not been reached.
The anti–PD-1/PD-L1 agents may be even more effective in combination with other agents. In early-phase studies, pembrolizumab plus chemotherapy has yielded a disease control rate of 100%, with relatively good tolerability, he added.
In conclusion, Dr. Ramalingam maintained that immunotherapy offers
the best chance of long-term survival in advanced lung cancer and a means of changing the biology of this disease. n
Disclosure: Dr. Khuri reported no potential conflicts of interest. Dr. Ramalingam has served on advisory boards for and has received honoraria from AstraZeneca, Bristol-Myers Squibb, Merck, and Genentech.
References1. Jänne PA, Yang JC, Kim DW, et al:
AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 372:1689-1699, 2015.
2. Sequist LV, Soria JC, Goldman JW, et al: Rociletinib in EGFR-mutated non-small cell lung cancer. N Engl J Med 372:1700-1709, 2015.
3. Shaw AT, Kim DW, Nakagawa K, et al: Crizotinib versus chemotherapy in ad-
vanced ALK-positive lung cancer. N Engl J Med 368:2385-2394, 2013.
4. Planchard D, Kim TM, Mazières J, et al: Dabrafenib in patients with BRAF V600E-mutant advanced non-small cell lung cancer: A multicenter, open-label phase II trial (BRF113928). 2014 ESMO Congress. Abstract LBA38_PR. Presented September 29, 2014.
5. Brahmer J, Horn L, Antonia S: Nivolumab in patients with non-small cell lung cancer: Overall survival and long-term safety in phase 1 trial. 2013 IASLC 15th World Conference on Lung Cancer. Ab-stract MO 18.03.
6. Spigel DR, Reckamp KL, Rizvi NA, et al: A phase III study (CheckMate 017) of nivolumab vs docetaxel in previously treated advanced or metastatic squamous cell non-small cell lung cancer. 2015 ASCO Annual Meeting. Abstract 8009. Presented May 29, 2015.
7. Rizvi NA, Mazières J, Planchard D, et al: Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for pa-tients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol 16:257-265, 2015.
8. Paz-Ares, L, Horn L, Borghaei H, et al: Phase III, randomized trial (CheckMate 057) of nivolumab versus docetaxel in ad-vanced non-squamous cell non-small cell lung cancer. 2015 ASCO Annual Meeting. Abstract LBA109. Presented May 29, 2015.
9. Spira AI, Park K, Mazières J, et al: Efficacy, safety and predictive biomarker results from a randomized phase II study comparing MPDL3280A vs docetaxel in 2L/3L NSCLC (POPLAR). 2015 ASCO Annual Meeting. Abstract 8010. Presented May 29, 2015.
10. Garon EB, Rizvi NA, Hui R et al: Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372:2018-2028, 2015.
Approaches to Lung Cancercontinued from page 3
Immunotherapy is going to have a stronger impact on lung cancer than targeted treatment has had so far. Immunotherapy improves survival, and this cannot be said for targeted agents.
—Suresh S. Ramalingam, MD
Delivered to your inbox every weekday evening. Visit ASCOPost.com to learn more.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 5
Debates and Didactics in Hematology and Oncology
Indolent Lymphoma: A More Complex Malignancy Than Once ThoughtBy Caroline Helwick
With a growing number of options for follicular lymphoma, clini-
cians may wonder whether there is one best regimen. James O. Armitage, MD, FACP, FRCP, Professor of Medicine at the University of Nebraska, Omaha—and Editor-in-Chief of The ASCO Post—tackled this question and offered recom-mendations at the 2015 Debates and Didactics in Hematology and Oncology in Sea Island, Georgia, an annual confer-ence sponsored by Emory University.
Approach to TreatmentFollicular lymphoma is actually a
more complex malignancy than is often believed, according to Dr. Armitage. “It’s becoming increasingly clear that follicu-lar lymphoma is complex and that the relationship between tumor cells and surrounding cells is important.”
Of note, some patients with grade 3 follicular lymphoma have a disease course more like diffuse large B-cell lymphoma. This is important to recog-nize, because it should dictate a more aggressive treatment, he said. “If you want to cure these patients, you’d bet-ter treat them with a [diffuse large B-cell lymphoma]-type regimen,” he rec-ommended, adding that he typically treats all grade 3 follicular lymphomas this way.
The other important question is how aggressively to treat stage I (ie, local-ized) follicular lymphoma. This early disease will respond well to radiothera-py alone. Studies have uniformly shown that almost half of patients treated with radiotherapy alone are continuously free of disease at 10 years. “Based on this, we usually treat localized follicular lymphoma with involved-field radio-therapy alone,” he said.
A Role for ‘Watch and Wait’? Given the excellent long-term out-
comes with rituximab (Rituxan), is there a place for observation in indolent disease?
The Nebraska Lymphoma Study Group documented the 10-year over-all survival rate for patients treated be-tween 1982 and 2000 (ie, before the ap-proval of rituximab) as approximately 50%. This rose to about 75% after the year 2000, he noted.
“Since our current treatment is so good, is it appropriate to watch and wait? Some think not, because of the impact of rituximab on the natural his-tory of lymphoma,” Dr. Armitage noted.
He maintained that observation can be appropriate for patients who are asymptomatic and either do not want therapy or are elderly and frail. The ca-veat is that close monitoring is manda-tory; delaying treatment may decrease survival, though this remains unproven, he said.
“You must be willing to observe these patients closely. Seeing them every 6 months is not enough. I see patients monthly at first, then every 3 months at most. If you are going to watch and wait, you cannot ignore these patients!”
CHOP-R vs CVP-RThere are many active regimens for
patients with disseminated follicular lymphoma.
“There are patients for whom ritux-imab alone is reasonable, but for ritux-imab-containing combinations, which is best?” Dr. Armitage asked.
He discussed as options CHOP-R (cyclophosphamide, doxorubicin, vin-cristine, prednisone, and rituximab), CVP-R (cyclophosphamide, vincris-tine, prednisone, and rituximab), B-R (bendamustine [Treanda] and ritux-imab), fludarabine-based regimens, and lenalidomide [Revlimid] plus rituximab.
An Italian study compared three regimens and found that complete re-mission rates were higher for CHOP-R (74%) than for CVP-R (67%) and fludarabine/mitoxantrone/rituximab (72%).1 Progression-free survival at 3.5 years was also longer with CHOP-R (68% vs 52% and 63%, respectively). The PRIMA study showed that CHOP-R produced higher complete remission rates than CVP-R and progression-free survival similar to that associated with FCM-R (fludarabine, cyclophospha-
mide, mitoxantrone, and rituximab).2 CHOP-R, therefore, is more active
than CVP-R, and because of toxicity, fludarabine-based regimens are rarely used in the United States. The bigger question is whether to use CHOP-R or B-R, he said.
CHOP-R vs B-RThe German NHL1 trial appeared
to show superiority of B-R over CHOP-R, based on a longer progression-free survival (P = .0281).3 But the results of NHL1 were not substantiated by the U.S.-based BRIGHT trial, which compared B-R to CHOP-R/CVP-R.4 Among the follicular/indolent lympho-ma cohort, the complete remission rate with B-R was much lower (27%) than it was in NHL1 (44%).
“When the U.S. group tried to re-produce the NHL1 study, they found no difference in the regimens,” he said.
The toxicity profiles of the two regi-mens, however, do differ in ways that may be important to patients. There is more vomiting and hypersensitiv-ity reaction with bendamustine but more alopecia and neuropathy with CHOP-R.5 The lack of alopecia with bendamustine has popularized its use, he added.
Comparability between the two regimens was further demonstrated in a matched-pair analysis reported from The University of Texas MD Anderson Cancer Center.6 After six cycles, complete remission rates were 91% with CHOP-R and 93% with B-R; 2-year progression-free sur-vival was 82% and 85%, respectively; and 2-year overall survival was 99% and 94%, respectively. Although the CHOP-R group contained more pa-tients with high-risk criteria, patient
outcomes were no worse, he noted. “I think both of these regimens work,
and one regimen is not better than the other. Both are better than CVP-R, and fludarabine is too toxic,” Dr. Armitage concluded.
The New Option: R2 The newest option is ritux-
imab/lenalidomide, the so-called R-squared or R2 regimen. R2 pro-duced a very high complete remis-sion rate (87%) in an MD Anderson series; 93% of evaluable patients be-came positron-emission tomography (PET)-negative, and the 2-year pro-gression-free survival rate was 89%.7 In a multi-institutional study, the complete remission rate was lower (72%), but 2-year progression-free survival remained high (89%).8 In a 154-patient study, European investi-gators also reported a lower complete remission rate (36%).9
“As more patients are treated, the results are sometimes not as good as the original report, and this is com-mon,” Dr. Armitage commented. “There has been no comparative trial, but this regimen might offer a similar likelihood to CHOP-R and B-R for keeping low-grade follicular lympho-ma patients well.”
Deciding on a RegimenAll things considered, Dr. Armitage
weighs the treatment options by asking the following questions: Is the patient fit? How old is the patient? Is the pa-tient symptomatic? What side effects is the patient willing to accept?
Patients who are not generally healthy are more likely to receive rituximab alone. Patients who are ill from cancer are most likely to benefit from CHOP-R or B-R. Elderly pa-tients are more likely to receive B-R than CHOP-R. Side effects should be weighed and patient preferences considered.
Optimal Treatment Duration?Dr. Armitage emphasized that the
aim of treatment is to achieve a com-plete remission. “It’s not okay to just treat the patient for a while and make him feel better. We know there is a huge survival advantage to achieving a com-plete remission,” he said.
A recent pooled analysis of three trials of patients receiving at least six
Hematology
For most patients, relapse is a horrible thing, so I offer maintenance therapy because I know this will increase the time before we need to treat again, if ever. Maintenance keeps patients in remission longer, but we don’t know if it keeps them alive longer.
—James O. Armitage, MD, FACP, FRCP
continued on page 6

PAGE 6 The ASCO Post | SEPTEMBER 10, 2015
Debates and Didactics in Hematology and Oncology
cycles of chemotherapy plus rituximab confirmed the value of achieving a negative PET/computed tomography (CT) scan after induction.10 The hazard ratios associated with having a positive PET/CT were 3.9 for progression-free survival (P < .0001) and 6.7 for overall survival (P = .0002).
More uncertain is the value of maintenance rituximab in this set-ting. The PRIMA study “convincingly showed” that maintenance will pro-long remission (hazard ratio = 0.50, P < .0001), but it did not demonstrate improved survival.2 The U.S.-based RESORT study, on the other hand, showed no difference in “time to treatment failure” between patients on the maintenance arm and those being observed only. This was defined as progression within 6 months of the last rituximab dose, no response to rituximab retreatment, initiation of alternative therapy, or inability to complete protocol therapy.11
The investigators concluded that in-termittent treatment was as effective as maintenance rituximab. In Dr. Armit-age’s opinion, the study’s median fol-low-up of 3.8 years may be too short to determine true differences in outcomes.
“You have to decide how important it is to the patient to be continuously well,” he commented. “For most pa-tients, relapse is a horrible thing, so I offer maintenance therapy because I know this will increase the time before we need to treat again, if ever. Mainte-nance keeps patients in remission lon-ger, but we don’t know if it keeps them alive longer.” n
Disclosure: Dr. Armitage reported no potential conflicts of interest.
References1. Federico M, Luminari S, Dondi A, et
al: R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage follicular lymphoma: Re-sults of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol 31:1506-1513, 2013.
2. Salles G, Seymour JF, Offner F, et al: Rituximab maintenance for 2 years in pa-tients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): A phase 3, ran-domized controlled trial. Lancet 377:42-51, 2011.
3. Rummel MJ, Niederle N, Masch-meyer G, et al: Bendamustine plus ritux-imab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-
label, multicenter, randomized, phase 3 non-inferiority trial. Lancet 381:1203-1210, 2013.
4. Flinn I, van der Jagt R, Kahl B, et al: An open-label, randomized study of bendamustine and rituximab compared with rituximab, cyclophosphamide, vin-cristine, and prednisone (R-CVP) or rituximab, cyclophosphamide, doxo-rubicin, vincristine, and prednisone in first-line treatment of advanced indolent non-Hodgkin’s lymphoma or mantle cell lymphoma: The BRIGHT study. 2012 ASH Annual Meeting. Abstract 902. Pre-sented December 11, 2012.
5. Macdonald D, van der Jagt R, Burke JM, et al: Different safety profiles of first-line bendamustine-rituximab, R-CHOP, and R-CVP in an open-label, random-ized study of indolent non-Hodgkin lymphoma and mantle cell lymphoma: The BRIGHT study. 2013 ASCO Annual Meeting. Abstract 8565. Presented May 31, 2013.
6. Phansalkar K, Ahmed M, Fowler N, et al: R-bendamustine vs R-CHOP as initial treatment for advanced stage low grade fol-licular lymphoma: A matched-pair analysis. 2014 ASH Annual Meeting. Abstract 3048. Presented December 7, 2014.
7. Fowler NH, Neelapu SS, Hagemeis-ter FB, et al: Lenalidomide and rituximab for untreated indolent lymphoma: Final
results of a phase II study. 2012 ASH An-nual Meeting. Abstract 901. Presented De-cember 11, 2012.
8. Martin P, Jung S, Johnson J, et al: CALGB 50803 (Alliance): A phase II trial of lenalidomide plus rituximab in patients with previously untreated follicular lym-phoma. 2014 ASCO Annual Meeting. Ab-stract 8521. Presented May 30, 2014.
9. Kimby E, Martinelli G, Ostenstad B, et al: Rituximab plus lenalidomide improves the complete remission rate in comparison with rituximab monotherapy in untreated follicular lymphoma patients in need of therapy: Primary endpoint analysis of the randomized phase 2 trial SAKK 35/10. 2014 ASH Annual Meeting. Abstract 799. Presented December 9, 2014.
10. Trotman J, Luminari J, Boussetta S, et al: Prognostic value of PET-CT af-ter first-line therapy in patients with fol-licular lymphoma: A pooled analysis of central scan review in three multicenter studies. Lancet Haematol 1(1):e17-e27, October 2014.
11. Kahl B, Hong F, Williams ME, et al: Results of Eastern Cooperative Oncol-ogy Group protocol E4420 (RESORT): A randomized phase III study comparing two different rituximab dosing strategies for low tumor burden follicular lymphoma. 2011 ASH Annual Meeting. Abstract LBA-6. Pre-sented December 13, 2011.
Indolent Lymphomacontinued from page 5
a
Don’t Miss These Important Reports in This Issue of The ASCO Post
Laura J. Esserman, MD, MBA, on Postmastectomy Pain see page 104
Pasquale W. Benedetto, MD, on High Cure Rates in Germ Cell Tumors see page 44
Charles Prober, MD, on Online Medical Education see page 72
Patricia Ganz, MD, and others on Mobile App for Breast Cancer Survivors see page 88
Georgina V. Long, MD, on Dabrafenib Plus Trametinib in BRAF V600-Mutant Melanoma see page 60
Peter Agre, MD, on a New Lung Cancer Vaccine Collaboration see page 47
Mark Pegram, MD, on Tumor Complexity and Technology see page 41
Louise Morrell, MD, on Genetic Testing in Breast Cancer see page 40
Carolyn D. Runowicz, MD, FASCO, on Her Career in Gynecologic Cancer Treatment see page 76
Visit The ASCO Post online at ASCOPost.com


(nivolumab) ,.,
Expect More. Do More.
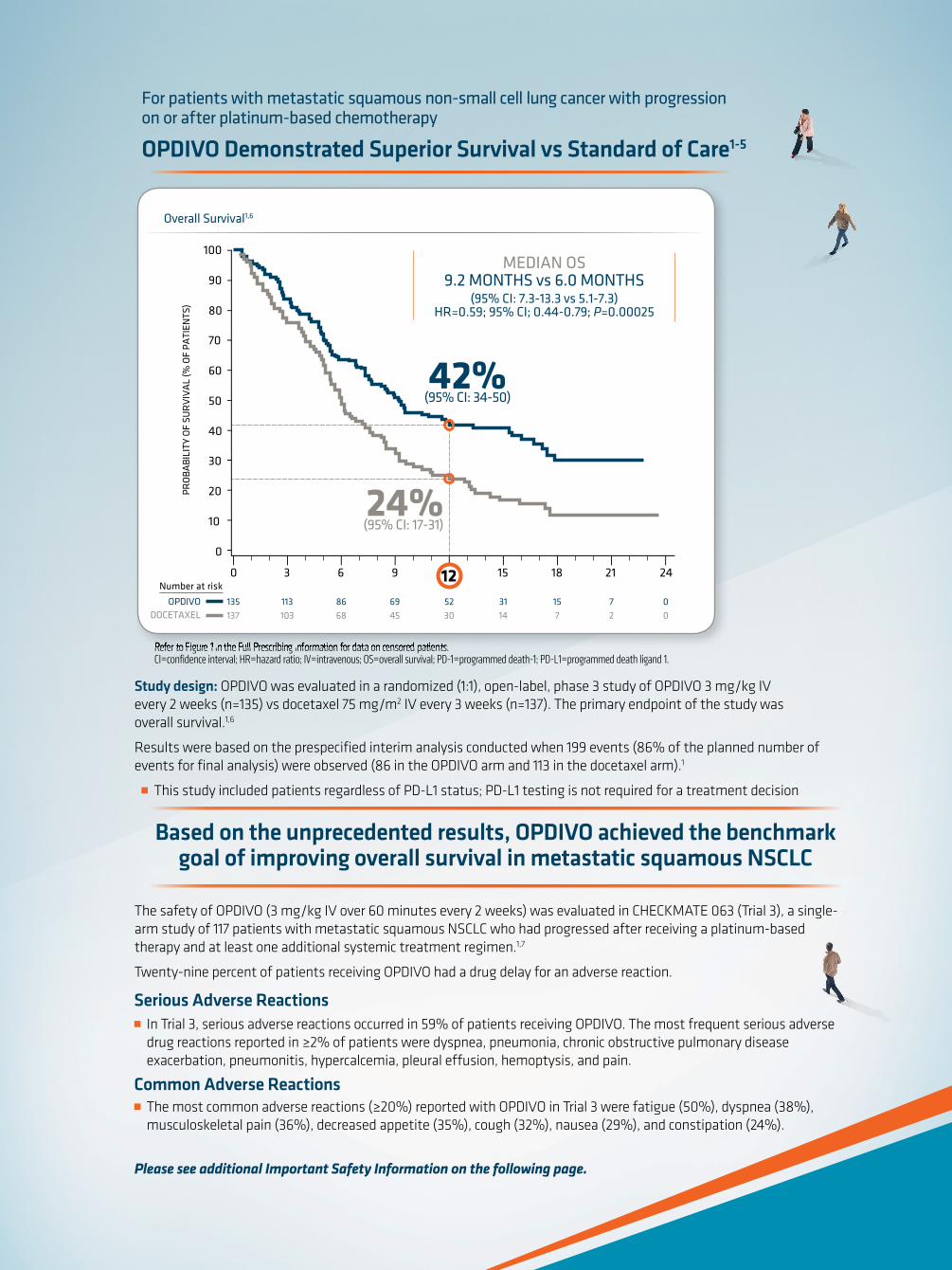
Proven Superior Survival With the Only lmmuno-Oncology
Therapy in Previously Treated Metastatic Squamous NSCLC
INDICATION
OPDIVO® (nivolumab) is indicated for the treatment of patients with metastatic squamous non-small cell lung cancer
(NSCLC) with progression on or after platinum-based chemotherapy.
SELECT IMPORTANT SAFETY INFORMATION
OPDIVO is associated with the following Warnings and Precautions including immune-mediated: pneumonitis,
colitis, hepatitis, nephritis and renal dysfunction, hypothyroidism, hyperthyroidism, other adverse reactions;
and embryofetal toxicity.
@ Please visit www.OPDIVO.com/hcp for more information





PAGE 14 The ASCO Post | SEPTEMBER 10, 2015
Best of ASCO®
Colorectal Liver Metastases: Thumbs Up for Radiofrequency Ablation, Jury Still Out for Selective Internal RadiotherapyBy Alice Goodman
Two “firsts” in studies of colorectal liver metastases were highlighted
at the 2015 ASCO Annual Meeting: the first prospective randomized trial to evaluate radiofrequency ablation plus chemotherapy1 and the first large randomized phase III trial to study liv-er-directed selective internal radiation therapy.2 Radiofrequency ablation plus chemotherapy appeared to have clear benefit in this setting, whereas the re-sults for selective internal radiotherapy in first-line therapy are currently less conclusive. Daniel G. Haller, MD, of Abramson Cancer Center at the Uni-versity of Pennsylvania, discussed both trials at Best of ASCO® 2015 in Boston.
Radiofrequency Ablation Plus Chemotherapy
Unresectable liver metastases treated with radiofrequency ablation plus che-motherapy improved long-term overall and progression-free survival compared with chemotherapy alone in patients with metastatic colorectal cancer, according to the results of the randomized phase II study of the EORTC NCRI CCSG-ALM Intergroup 40004 (CLOCC).
“This is the first study to prospec-tively evaluate radiofrequency ablation plus chemotherapy. This study showed that radiofrequency plus chemo-therapy is clearly superior to systemic [chemo] therapy in terms of progres-sion-free survival and overall survival in colorectal cancer patients with inoper-able liver metastases. This is a no-brain-er, and this study is unlikely to ever be repeated,” said Dr. Haller, who reviewed CLOCC and other important studies in gastrointestinal (colorectal) cancer se-lected for the Best of ASCO 2015.
Between 2002 and 2007, the study
enrolled patients with colorectal cancer who had up to nine liver metastases and no extrahepatic disease. A total of 119 patients were randomized to receive chemotherapy alone or radiofrequency ablation plus chemotherapy. In both arms, chemotherapy consisted of 6 months of FOLFOX (leucovorin, fluo-rouracil, oxaliplatin), and bevacizumab (Avastin) was added in 2005. In both arms, resection was allowed if chemo-therapy converted unresectable metas-tases to resectable.
In the radiofrequency ablation arm, 56 patients were treated with radiofre-quency ablation, and 27 patients (45%) underwent hepatic resection. Of the 51 patients in the chemotherapy arm, 6 eventually underwent hepatic resection.
As reported in 2010, 30-month overall survival (primary endpoint) was 61.7% for radiofrequency abla-tion plus chemotherapy and 57.6% for chemotherapy alone, which was higher than expected.
At a median follow-up of 9.2 years, the difference in overall survival sig-nificantly favored the radiofrequency ablation arm (P = .01). Median overall survival was 45.6 months in the radio-
frequency ablation arm vs 40.5 months in the chemotherapy-alone arm. Eight-year overall survival was 35.9% vs 8.9%, respectively (P = .010). A total of 92 deaths were reported: 39 in the radiofrequency ablation arm and 53 in the chemotherapy-alone arm. Progres-sive disease accounted for 34 and 48 deaths, respectively.
Progression-free survival was signifi-cantly superior in the radiofrequency ablation arm. With long-term follow-up, median progression-free survival
was 16.8 months for radiofrequency ablation vs 9.9 months for chemother-apy alone (P = .025). Eight-year pro-gression-free survival was 22% vs 2%, respectively (P = .005).
SIRFLOX StudyIn the randomized, phase III
SIRFLOX study, first-line therapy for patients with unresectable liver me-tastases with selective internal radio-therapy added to modified FOLFOX6 plus or minus bevacizumab failed to im-prove overall progression-free survival compared with modified FOLFOX6 plus or minus bevacizumab (which was not part of the randomization scheme) did better than those who did not.
Selective internal radiotherapy em-ploys yttrium-90–labelled resin micro-spheres or glass beads as a liver-directed therapy, and it has been approved by the U.S. Food and Drug Administration for the treatment of hepatic cell carcinoma, neuroendocrine tumors, and metastatic colorectal cancer.
SIRFLOX randomized 530 patients with nonresectable liver-only or liver-dominant metastatic colorectal cancer with no prior therapy in a 1:1 ratio to receive treatment with modified FOLFOX6 plus or minus bevacizumab or modified FOLFOX6 plus selective
internal radiotherapy plus or minus bevacizumab. Patients were prestrati-fied according to extrahepatic metas-tasis, degree of liver involvement, and intended use of bevacizumab.
Inclusion criteria allowed extrahe-patic metastases as follows: up to five lung metastases smaller than 1 cm and lymph nodes in a single anatomic re-gion. Less than 40% of patients had extrahepatic disease. About 50% of pa-tients received bevacizumab.
For the primary endpoint of overall progression-free survival, there was no significant difference between the two treatment arms. “However, progres-sion-free survival in the liver (a second-ary endpoint) had interesting results,” Dr. Haller noted.
Median progression-free survival in the liver was 12.6 months for chemother-apy vs 20.5 months for selective internal radiotherapy (P = .002), representing a 7.9-month improvement and a 31% risk reduction for disease progression in the liver. Complete response in the liver was achieved in 2% of the chemotherapy arm vs 6% in the selective internal radiother-apy arm (P = .020).
“Selective internal radiotherapy did not enable more liver resections and does not convert patients or downstage them,” Dr. Haller said.
More adverse events occurred with patients treated with selective internal radiotherapy. The rate of any adverse event was 73.4% for chemotherapy and 85.4% for selective internal radiotherapy. Grade 3 or higher adverse events follow: neutropenia, 28.5% for chemotherapy vs 40.7% for selective internal radiothera-py; febrile neutropenia, 1.9% vs 6.1%; thrombocytopenia, 2.6% vs 9.8%.
Future analysis of these data will include liver-only vs liver-dominant outcomes, depth of response, quality of life, cost-effectiveness of a single treatment of selective internal radio-therapy vs systemic treatment, and overall survival.
“Selective internal radiotherapy achieved an 8-month improvement in progression-free survival in the liver, but it is not clear what this means for the lifespan of the patient. Selective in-ternal radiotherapy did not add much to the overall response rate, but it was superior in the liver,” he continued. “However, toxicities are noteworthy.”
Nonsurgical Treatments for Colorectal Liver Metastases
■ Recent studies have helped to refine the usefulness of two nonsurgical treatments of colorectal liver metastases: radiofrequency ablation plus chemotherapy and selective internal radiotherapy.
■ Radiofrequency ablation plus chemotherapy appeared to achieve superior overall survival and progression-free survival vs chemotherapy alone.
■ Selective internal radiotherapy plus chemotherapy did not improve progression-free survival overall but did reduce the incidence of progression in the liver.
■ Overall survival data for selective internal radiotherapy will be analyzed across three large randomized trials, and results are expected in about 2 years.
continued on page 15
Gastrointestinal Oncology
The goal of these treatments is to allow patients to be resected, delay biliary obstruction, [and] maintain liver function, so patients can get later lines of therapy. The timing is questionable, but I believe in the middle of chemotherapy is optimal.
—Daniel G. Haller, MD

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 15
Best of ASCO®
SIRFLOX is the first of three stud-ies to look at a pooled analysis of overall survival; the other two ongoing trials are FOXFIRE and FOXFIRE global. Overall survival results of all three tri-als, for a total of 2,017 patients, should be available in about 2 years.
Additional Discussion“There are multiple options for treat-
ment of metastatic disease to the liver. They include systemic therapy, surgery, neoadjuvant therapy, liver-directed therapy, selective internal radiotherapy [SIRFLOX], and radiofrequency abla-
tion [CLOCC],” Dr. Haller said. “The goal of these latter treatments is to al-low patients to be resected, delay biliary obstruction, [and] maintain liver func-tion, so patients can get later lines of therapy. The timing is questionable, but I believe in the middle of chemotherapy is optimal,” he said.
“[The phase II Intergroup 40004] study included patients with unresect-able liver metastasis. Patients were allowed chemotherapy prior to ran-domization. The study had a huge bias in that there were more patients with a single liver metastasis in the radio-frequency ablation arm. In addition, about 50% of patients treated with ra-
diofrequency ablation had resection, making it difficult to distinguish which patients were helped by radiofrequen-cy ablation. Also, there were small numbers of patients, and they were highly selected,” Dr. Haller continued.
“Selective internal radiotherapy bought patients an extra 8 months of progression-free survival in the liver, but this therapy has toxicities. Analysis of overall survival and the impact of the initial large increase in liver progression-free survival on further lines of therapy will add more data on this modality,” concluded Dr. Haller. n
Disclosure: Dr. Haller is on the advisory board of Sirtex.
References1. Ruers T, Punt CJA, van Coevorden F, et
al: Radiofrequency ablation combined with chemotherapy for unresectable colorectal liver metastases: Long-term survival results of a randomized phase II study of the EORTC-NCRI CCSG-ALM Intergroup 40004 (CLOCC). 2015 ASCO Annual Meeting. Abstract 3501. Presented May 29, 2015.
2. Gibbs P, Heinemann V, Sharma NK, et al: SIRFLOX: Randomized phase III trial comparing first-line mFOLFOX6 ± beva-cizumab versus mFOLFOX6 ± selective internal radiation therapy ± bevacizumab in patients with metastatic colorectal can-cer. 2015 ASCO Annual Meeting. Abstract 3502. Presented May 29, 2015.
Colorectal Liver Metastasescontinued from page 14
Secondary Prevention in Metastatic Colorectal Cancer: Benefits of Vitamin D and Aspirin ExploredBy Alice Goodman
Two low-cost, low-tech options may lead to a survival benefit in
metastatic colorectal cancer, according to separate retrospective studies select-ed for the Best of ASCO® 2015. The first study suggested that vitamin D supple-mentation is worthy of investigation in this regard,1 and the second study added to existing evidence that regular use of aspirin may improve survival in colorectal cancer.2
“Various standard therapies can improve survival in metastatic colorectal cancer. What else can you do to improve survival in this disease? The take-home message from these studies is that vitamin D levels and as-pirin are lifestyle factors most consis-tently associated with improved out-comes among patients with colorectal cancer. There is strong supportive evidence for both of these interven-tions, and if confirmed, these low-cost options could have a substantial impact for the treatment of colorectal cancer,” said Daniel G. Haller, MD, of Abramson Cancer Center, Univer-sity of Pennsylvania, Philadelphia, discussant of these two studies at the Best of ASCO® 2015.
Vitamin D LevelsHigher plasma concentrations of
plasma 25-hydroxyvitamin D [25(OH)D, vitamin D] are associated with sig-nificantly improved survival in patients with metastatic colorectal cancer treat-ed with chemotherapy and biologics, according to a prospective pretherapy assessment of patients enrolled in CALGB/SWOG 80404, a random-
ized phase III trial of chemotherapy plus bevacizumab (Avastin), cetuximab (Erbitux), or both, prior to the KRAS-wild type amendment for this study.
The study included 2,334 patients randomized as follows: chemotherapy plus bevacizumab (n = 899 patients), cetuximab (902 patients), or both (533 patients). Plasma vitamin D was mea-sured prior to receiving chemotherapy in 1,043 patients. The primary study re-sults showed no significant difference in overall survival among the arms.
Significantly lower plasma vitamin D levels were found in male and black patients; those living in the northeast; and those with lower dietary and sup-plemental vitamin D intake, an Eastern Cooperative Oncology Group perfor-mance status of 1 (vs 0), tumoral RAS mutation, higher body mass index, low-er levels of physical activity, and a blood draw during the winter and spring.
Patients in the highest quintile had significantly improved overall survival compared with those in the two low-est quintiles, after adjusting for patho-logic and clinical prognostic factors: median of 32.6 months vs 24.5 months (P trend = .001).
Improved progression-free survival was also significantly associated with higher plasma concentrations of vita-min D: median of 12.2 months vs 10.1 months, respectively (P trend = .01). Re-sults were consistent across subgroups.
“These results showed a 35% im-provement in overall survival and a 21% improvement in median progres-sion-free survival in the highest quintile compared to the lowest. According to a
Forest plot, every subgroup did better with higher levels vs lower levels,” Dr. Haller noted.
Strengths of the study are that vita-min D levels were measured after di-agnosis but before treatment and that there is detailed information on other prognostic factors. Patients received protocol-based therapy, and follow-up was standard.
“Limitations [of the study] include lack of data on vitamin D level before the cancer diagnosis. It is also not clear if a sin-gle measure of a single form of vitamin D is the best measure,” Dr. Haller continued.
“There is a preponderance of evi-dence from several countries that pa-tients with higher vitamin D levels have improved outcomes. This is strong, con-sistent evidence. It is not clear, however, whether correcting vitamin D levels will lead to improved outcomes,” he noted.
Remaining questions are whether vitamin D supplementation should be given in adjuvant settings, whether vitamin D supplementation improves outcomes only in vitamin D–defi-cient patients, and what are the best formulations of vitamin D supple-
ments. Two randomized trials are looking at these questions.
“If these findings are confirmed, vita-min D supplementation will represent a low-cost option to further improve metastatic colorectal cancer outcomes,” he stated.
Aspirin UseAspirin has been validated in pri-
mary prevention of colorectal cancer. A population-based, retrospective, cohort study conducted in Norway between 2004 and 2011 in an unselected co-hort of 25,644 patients with colorectal cancer found that aspirin as secondary prevention was associated with im-proved survival. Twenty-five percent of patients (n = 6,109) were exposed to aspirin, as defined by a prescription of aspirin for more than 6 months after diagnosis of colorectal cancer.
At a median follow-up of 2.2 years, overall survival and colorectal can-cer–specific survival were significantly improved in patients who took aspi-rin. Among those exposed to aspirin, 2,088 (34.2%) deaths were recorded,
Gastrointestinal Cancer
Vitamin D Levels and Aspirin Use in Colorectal Cancer
■ Higher levels of plasma vitamin D are associated with improved survival in patients with metastatic colorectal cancer.
■ It is not known at present whether vitamin D supplementation can improve survival in patients with low levels of plasma vitamin D.
■ A large population-based study found that aspirin exposure improves overall survival and progression-free survival in patients with metastatic colorectal cancer.
continued on page 16

PAGE 16 The ASCO Post | SEPTEMBER 10, 2015
Best of ASCO®
of which 1,172 (19.2%) were attrib-uted to colorectal cancer. Among nonexposed patients, 7,595 deaths (38.9%) were recorded, of which 6,356 (33.5%) were colorectal can-cer–specific. An adjusted multivariate analysis found that aspirin exposure after diagnosis of colorectal cancer was an independent risk factor associated with improved colorectal cancer–spe-cific and overall survival (P < .001 for both comparisons).
Dr. Haller cited the following
strengths of this study: a large popu-lation, unselected patients, high-qual-ity validated data, complete follow-up data, no recall bias, and no lead-time bias. Study limitations are that the dose and duration of aspirin treatment are not known, and there are no data on toxicities.
Remaining questions on the use of aspirin are its optimal dose and dura-tion and identification of molecular markers for which patients will benefit.
“The majority of studies show a posi-tive effect on colorectal cancer–specific survival and overall survival. I think the
answer to whether aspirin should be used is a resounding yes,” he said.
Results of two ongoing trials are awaited: CLEAR (Alliance 80702 trial) in stage III colorectal cancer, which has a 2 × 2 factorial design: 3 vs 6 months of adjuvant FOLFOX (leu-covorin, fluorouracil, and oxaliplatin) with or without celecoxib for 3 years; and the British Add-Aspirin trial, a randomized, double-blind, placebo-controlled trial after survival in non-metastatic colorectal cancer. n
Disclosure: Dr. Haller reported no potential conflicts of interest.
References1. Ng K , Venook AP, Sato K , et
al: Vitamin D status and survival of metastatic colorectal cancer patients: Results from CALGB/SWOG 80405 (Alliance). 2015 ASCO Annual Meet-ing. Abstract 3503. Presented May 29, 2015.
2. Bains S, Mahic M, Cvancarova M, et al: Impact of aspirin as secondary prevention in an unselected cohort of 25,644 patients with colorectal cancer: A population-based study. 2015 ASCO Annual Meeting. Abstract 3504. Pre-sented May 29, 2015.
Vitamin D and Aspirincontinued from page 15
Optimal Timing of Hormonal Therapy for Biochemically Recurrent Prostate Cancer Remains UnclearBy Alice Goodman
There is no consensus as to whether it is better to treat immediately or
to delay androgen-deprivation therapy in patients with a rising prostate-spe-cific antigen (PSA) level (“biochemical relapse”) after curative therapy for pros-tate cancer. A phase III study, selected for the Best of ASCO® 2015, found that immediate androgen-deprivation ther-apy improved overall survival and time to clinical disease progression, but the results were not definitive.
This trial has several issues that make it difficult to walk away with a firm conclusion, according to Terence W. Friedlander, MD, of the University of California, San Francisco, who re-viewed this trial and its implications at the Best of ASCO 2015.
Study DetailsAbout 20% to 50% of patients will
relapse following definitive therapy for prostate cancer. The Timing of Androgen-Deprivation Therapy in Prostate Cancer Patients With a Rising PSA (TOAD) study sought to address the optimal tim-ing of androgen-deprivation therapy in this setting. Men were eligible for the trial if they had a PSA doubling time of less than 12 months following curative thera-
py or metastasis or symptoms.Only one-third of the planned accru-
al occurred, with 293 patients enrolled from September 2004 to July 2012. Median follow-up was 5 years. Most patients had slowly progressing disease, and about two-thirds of each arm went on intermittent therapy.
Patients were randomized in a 1:1 ratio to receive immediate intervention with androgen-deprivation therapy (arm B) or delayed therapy (arm A). About one-third of patients in arm A initiated androgen-deprivation therapy within 2 years, whereas 49% started therapy later than 4 years on trial or had not yet begun therapy.
The mean age was about 70 years at trial initiation. About two-thirds of
patients had curative radiation therapy, and about one-third underwent surgery plus radiation as curative therapy. The relapse-free interval was less than 10 months in about one-third of patients and 10 months or longer in two-thirds.
Survival Data and ToxicityAt 5 years, overall survival was about
10% higher if androgen-deprivation therapy was started immediately: 86%
vs 76%, respectively; 6-year overall sur-vival was 81% vs 65.4%, respectively (P = .05). Nonsignificant reductions were also observed in prostate cancer–specific deaths and deaths due to all causes.
Overall, 46 deaths were reported, in-cluding 30 deaths in the delayed-therapy arm and 16 deaths in the immediate-ther-apy arm. “There were only 18 prostate cancer deaths in this study—6 of these deaths were in the immediate [androgen-deprivation therapy] arm and 10 were in the delayed arm. It is difficult to interpret these data to know if this is a real differ-ence,” Dr. Friedlander commented.
Progression-free survival (both dis-tant and local) was improved with im-mediate therapy (P = .001) compared with delayed therapy. However, time to castration resistance did not differ be-tween the two study arms.
A subgroup analysis suggested that intermittent androgen-deprivation ther-apy might be superior to continuous androgen-deprivation therapy. However, due to poor accrual, the study was un-derpowered to detect a difference in this and other secondary endpoints.
Adverse events related to androgen-deprivation therapy were more common
in the immediate-therapy arm than in the delayed-therapy arm: 80% vs 50%.
‘Not Practice-Changing’“This was a challenging trial to con-
duct, and accrual was disappointing. It appears that overall survival may be im-proved by immediate vs delayed initia-tion of androgen-deprivation therapy, but the benefits are likely to be modest and have to be balanced with the con-siderable side effects of hormonal treat-ment,” stated Dr. Friedlander.
“This study is not practice-changing. When faced with a patient with a ris-ing PSA following curative therapy, the physician should discuss therapeu-tic options incorporating the risks of treatment,” he said. The way forward will depend on finding better ways to identify which patients need androgen-deprivation therapy upfront and which ones can safely delay it. n
Disclosure: Dr. Friedlander reported no potential conflicts of interest.
Reference1. Duchesne et al: 2015 ASCO Annual
Meeting. Abstract 5007. Presented May 29, 2015.
This study is not practice-changing. When faced with a patient with a rising PSA following curative therapy, the physician should discuss therapeutic options incorporating the risks of treatment.
—Terence W. Friedlander, MD
Genitourinary Cancer
Androgen-Deprivation Therapy for Recurrent Prostate Cancer
■ Immediate initiation of androgen-deprivation therapy may have a survival advantage over delayed therapy in the setting of biochemical-only recurrence following curative radiation therapy, surgery, or both, according to the results of a large collaborative trial.
■ However, these study findings are not definitive due to poor accrual and a heterogeneous patient base.
■ The decision as to when to initiate androgen-deprivation therapy should be individualized, based on a discussion with the patient of risks vs benefits.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 17
Best of ASCO®
Role of Surgery and Adjuvant Radiotherapy in the Treatment of Breast Cancer ExploredBy Alice Goodman
Two separate retrospective studies have further refined our understand-
ing of the respective contributions of sur-gery and radiotherapy in the treatment of breast cancer. However, these studies are not definitive, and “gold standard” trials are needed to arrive at definitive recom-mendations. Both abstracts were present-ed at the 2015 ASCO Annual Meeting and were discussed at the Best of ASCO® 2015 by Steven Isakoff, MD, of Massa-chusetts General Hospital, Boston.
A retrospective review of a large data-base suggested that surgery improves sur-vival in intermediate- and high-grade duc-tal carcinoma in situ but does not affect survival in low-grade ductal carcinoma in situ. It may be possible to forgo surgery in low-grade ductal carcinoma in situ, but further prospective study is needed.
A retrospective meta-analysis found that adding adjuvant radiation therapy to neoadjuvant treatment improves out-comes in women with operable and nonoperable breast cancer, regardless of whether they achieve pathologic complete response to neoadjuvant therapy. In that study, women with and without patholog-ic complete response who did not receive radiation therapy had worse outcomes.
Surgical Management of Ductal Carcinoma in Situ
Widespread use of mammography has led to increased detection of duc-tal carcinoma in situ. Currently, about 60,000 new cases of ductal carcinoma in situ are reported each year in the United States. An estimated 40% to 85% of these cases are low grade.
Current management of ductal carci-noma in situ is lumpectomy with radia-tion or mastectomy. The magnitude of the survival benefit of surgery in ductal carcinoma in situ is not well known. Yasuaki Sagara, MD, Research Fellow in Surgery at Brigham and Women’s Hos-pital in Boston, and coauthors designed a study to explore whether surgical man-agement improves survival in women
with low-grade ductal carcinoma in situ.The study was based on more than
96,000 cases of ductal carcinoma in situ entered in the Surveillance, Epidemi-ology, and End Results (SEER) data-base between 1988 and 2011. Of them, 57,000 cases met the inclusion criteria; 56,053 cases (98%) were managed sur-gically, and, for various reasons, 1,169 (2%) did not undergo surgery. These two cohorts of patients were compared.
The authors found that nonsurgically managed patients had a higher proportion of low-grade ductal carcinoma in situ and did not receive radiation therapy. Those treated with surgery were more likely to have high-grade ductal carcinoma in situ and to receive radiation therapy.
At a median follow-up of 72 months, 576 breast cancer–specific deaths (1%) were reported; 3,600 deaths from other causes (6.4%) were also reported.
Overall, surgically managed patients had significantly improved 10-year breast cancer–specific survival compared with the nonsurgical group: 98% vs 93%, re-spectively (P < .001). Overall survival was 89% vs 83%, respectively. Nuclear grade was associated with a survival benefit in the surgery group (P = .0003).
“The main message of this study is that there was no difference in 10-year breast cancer–specific survival for low-grade duc-tal carcinoma in situ patients who did or did not have surgery. These are relatively small numbers, but there is no discernible difference. But there is a difference in breast
cancer–specific survival in the high-grade group favoring surgery,” said Dr. Isakoff.
“We perform surgery in ductal carci-noma in situ to exclude the presence of invasive cancer and to reduce the likeli-hood of developing invasive cancer. At the time of surgery, about 20% to 25% of patients with ductal carcinoma in situ are upstaged. Radiotherapy after surgery reduces the risk of invasive recurrence in low-grade ductal carcinoma in situ
from 30% to 12%; 7.5% of recurrences in low- or intermediate-stage patients are actually invasive cancer. There is a linear path from ductal carcinoma in situ to in-creased risk of invasive cancer and then increased risk of mortality,” he continued.
An ongoing, prospective, random-ized trial is currently addressing the issue of whether low-risk ductal carcinoma in situ can be safely treated without surgery. Called LORIS, the study is comparing surgery vs active monitoring in patients with low-risk ductal carcinoma in situ.
Radiotherapy Post Neoadjuvant Therapy
It is controversial whether to incor-porate response to neoadjuvant therapy into decision-making regarding the addi-tion of adjuvant radiotherapy for patients with breast cancer. A pooled analysis of three GEPAR trials (GeparTrio, Gepar-Quattro, and GeparQuinto) was under-taken to address this question.
“Local recurrence rates after neoad-juvant and adjuvant chemotherapy have been shown to be similar if adequate lo-cal therapy is performed. Treatment re-sponse to neoadjuvant therapy is consid-ered prognostic,” Dr. Isakoff told listeners.
The study, which was reported at the 2015 ASCO Annual Meeting by David Krug, MD, of the University Hos-pital, Heidelberg, Germany, suggests that adjuvant radiotherapy should be added regardless of whether patients achieve
pathologic complete response or not.The pooled analysis included 3,370
patients with operable and nonoper-able breast cancer for whom there were available data on the use of radiotherapy (57% of a total of 5,780 patients). Lo-coregional recurrence was defined as recurrence in the breast, chest wall, or regional lymph nodes.
At a median follow-up of 50.3 months, the overall recurrence rate was 8.8%. Five-year locoregional recurrence-free survival was 89.6% with radiother-apy vs 81.8% without (P < .004). Five-year disease-free survival was 74.5% vs 66.7%, respectively (P = .087).
“The top-line result is the general ben-efit of radiotherapy in locoregional recur-rence and disease-free survival,” Dr. Isakoff said. “It gets more interesting when you look at subgroup results. Patients with pathologic complete response still had im-provements with radiotherapy, and there was also an improvement with radiothera-py in patients who did not achieve patho-logic complete response. The greatest benefit was observed in patients who had pathologic complete response and were node-negative after neoadjuvant therapy.”
For those who achieved pathologic complete response, 5-year locoregional recurrence-free survival was 94.7% with radiotherapy vs 85.8% without (P = .043); 5-year disease-free survival was 86.0% vs 60.3%, respectively (P = .004).
For those without pathologic com-plete response, 5-year locoregional re-currence-free survival was 88.4% with radiotherapy vs 81.1% without radio-therapy (P = .030); 5-year disease-free survival was 71.7% vs 67.4%, respective-ly (P = .519).
Multivariate analysis adjusted for baseline parameters, tumor stage, and pathologic complete response showed that radiotherapy was an independent risk factor for locoregional recurrence-free survival (P = .038) but not for dis-ease-free survival (P = .728).
“Radiotherapy is an independent prognostic factor for locoregional re-currence and disease-free survival after neoadjuvant therapy. We still don’t know which patients can avoid radiotherapy. We have to take these data with a grain of salt. Information on radiotherapy was available for only 57% of patients in these three trials. The three studies had differ-ent characteristics, making cross-study
Breast Cancer
There is a linear path from ductal carcinoma in situ to increased risk of invasive cancer and then increased risk of mortality.
—Steven Isakoff, MD
continued on page 20
Potential Benefits of Surgery and Adjuvant Radiotherapy
■ Surgery appears to improve survival in high- and intermediate-risk ductal carcinoma in situ, but outcomes are similar for low-risk ductal carcinoma in situ with or without surgery.
■ Adjuvant radiotherapy appears to improve locoregional control and disease-free survival regardless of whether pathologic complete response is achieved with neoadjuvant therapy.


• Infections: In Study 1, 1% of XTANDI patients compared to
0.3% of placebo patients died from infections or sepsis. In
Study 2, 1 patient in each treatment group (0.1%) had an
infection resulting in death.
• Falls (including fall-related injuries), occurred in 9% of XTANDI
patients and 4% of placebo patients. Falls were not associated
with loss of consciousness or seizure. Fall-related injuries were
more severe in XTANDI patients, and included non-pathologic
fractures, joint injuries, and hematomas.
• Hypertension occurred in 11% of XTANDI patients and 4% of
placebo patients. No patients experienced hypertensive crisis.
Medical history of hypertension was balanced between arms.
Hypertension led to study discontinuation in< 1% of all patients.
Drug Interactions Effect of Other Drugs on XTANDI Avoid strong CYP2C8 inhibitors,
as they can increase the plasma exposure to XTANDI. If co
administration cannot be avoided, reduce the dose of XTANDI.
Avoid strong or moderate CYP3A4 or CYP2C8 inducers as they
can alter the plasma exposure to XTANDI.
-,, astellas 1:v� JVf E DIV-\TION
© 2015 Astellas Pharma US, Inc. All rights reserved. Printed in USA. 076-1003-PM 8/15 XTANDI, Astellas, and the flying star logo are trademarks of Astellas Pharma Inc.
Effect of XTANDI on Other Drugs Avoid CYP3A4, CYP2C9, and
CYP2C19 substrates with a narrow therapeutic index, as XTANDI
may decrease the plasma exposures of these drugs. If XTANDI
is co-administered with warfarin (CYP2C9 substrate), conduct
additional INR monitoring.
Please see adjacent pages for Brief Summary of Full Prescribing Information.
·i-As seen in the PREVAIL trial (Study 2): a multinational, double-blind, randomized, phase 3 trial that enrolled 1717 patients with metastatic CRPC that progressedon GnRH therapy or after bilateral orchiectomy, and who had not received priorcytotoxic chemotherapy. All patients continued on GnRH therapy.1•2
§Results from this analysis were consistent with those from the prespecified interim analysis. llln the PREVAIL trial, 27% of patients in the XTANDI arm and 30% of patientsin the placebo arm received glucocorticoids for varying reasons. In the AFFIRMtrial (Study 1), 48% of patients in the XTANDI arm and 4 6% of patients inthe placebo arm received glucocorticoids. AFFIRM was a phase 3, multicenter,placebo-controlled, randomized trial that enrolled 1199 patients with metastaticCRPC who had previously received docetaxel.1
References: 1. XTANDI [package insert]. Northbrook, IL: Astellas Pharma US, Inc. 2. Beer TM, Armstrong AJ, Rathkopf DE, et al, for the PREVAIL Investigators. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med.2014;371:424-433. 3. Data on file, Medivation, Inc.

PAGE 20 The ASCO Post | SEPTEMBER 10, 2015
Best of ASCO®
comparisons difficult,” Dr. Isakoff stated.Additionally, the no-radiation group
in the mastectomy patients included only 151 patients. A multivariate analy-sis found that radiotherapy also pre-dicted locoregional recurrence in these patients but not disease-free survival.
An ongoing NSABP/RTOG study should shed more light on this issue. That study includes patients with posi-tive lymph nodes before treatment who undergo randomization to receive post-mastectomy radiotherapy or not or—in the case of breast-conserving surgery—to whole-breast irradiation with or without regional nodal irradiation.
“This study should help decide which patients should be treated with radiotherapy and whether radiotherapy can be avoided if the patient achieves pathologic complete response after neo-adjuvant therapy,” he said.
“The data from the GEPAR meta-anal-ysis suggest that at the moment, treatment should be based on prechemotherapy
characteristics,” Dr. Isakoff commented. nDisclosure: Drs. Sagara and Isakoff reported
no potential conflicts of interest. Dr. Krug has received travel expenses from Accuray.
References1. Sagara Y, et al: 2015 ASCO Annual Meet-
ing. Abstract 1006. Presented May 29, 2015.2. Krug D, et al: 2015 ASCO Annual Meet-
ing. Abstract 1008. Presented May 29, 2015.
Breast Cancer Treatmentcontinued from page 17
XTANDI® (enzalutamide) capsules for oral use Initial U.S. Approval: 2012BRIEF SUMMARY OF PRESCRIBING INFORMATION
The following is a brief summary. Please see the package insert for full prescribing information.
INDICATIONS AND USAGE
XTANDI is indicated for the treatment of patients with metastatic castration-resistant prostate cancer (CRPC).
CONTRAINDICATIONS
Pregnancy XTANDI can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. XTANDI is not indicated for use in women. XTANDI is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss.
WARNINGS AND PRECAUTIONS
Seizure In Study 1, which enrolled patients who previously received docetaxel, 7 of 800 (0.9%) patients treated with XTANDI experienced a seizure and no patients treated with placebo experienced a seizure. Seizure occurred from 31 to 603 days after initiation of XTANDI. In Study 2, 1 of 871 (0.1%) chemotherapy-naive patients treated with XTANDI and 1 of 844 (0.1%) patients treated with placebo experienced a seizure. Patients experiencing seizure were permanently discontinued from therapy and all seizure events resolved. There is no clinical trial experience re-administering XTANDI to patients who experienced seizure.Limited safety data are available in patients with predisposing factors for seizure because these patients were generally excluded from the trials. These exclusion criteria included a history of seizure, underlying brain injury with loss of consciousness, transient ischemic attack within the past 12 months, cerebral vascular accident, brain metastases, and brain arteriovenous malformation. Study 1 excluded the use of concomitant medications that may lower the seizure threshold, whereas Study 2 permitted the use of these medications.
Because of the risk of seizure associated with XTANDI use, patients should be advised of the risk of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others. Permanently discontinue XTANDI in patients who develop a seizure during treatment.Posterior Reversible Encephalopathy Syndrome (PRES)There have been reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving XTANDI [see Adverse Reactions (6.2)]. PRES is a neurological disorder which can present with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual and neurological disturbances, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). Discontinue XTANDI in patients who develop PRES.
ADVERSE REACTIONS
Clinical Trial Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Two randomized clinical trials enrolled patients with metastatic prostate cancer that has progressed on androgen deprivation therapy (GnRH therapy or bilateral orchiectomy), a disease setting that is also defined as metastatic CRPC. In both studies, patients received XTANDI 160 mg orally once daily in the active treatment arm or placebo in the control arm. All patients continued androgen deprivation therapy. Patients were allowed, but not required, to take glucocorticoids.The most common adverse reactions (≥ 10%) that occurred more commonly (≥ 2% over placebo) in the
XTANDI-treated patients from the two randomized clinical trials were asthenia/fatigue, back pain, decreased appetite, constipation, arthralgia, diarrhea, hot flush, upper respiratory tract infection, peripheral edema, dyspnea, musculoskeletal pain, weight decreased, headache, hypertension, and dizziness/vertigo.
Study 1: Metastatic Castration-Resistant Prostate Cancer Following Chemotherapy
Study 1 enrolled 1199 patients with metastatic CRPC who had previously received docetaxel. The median duration of treatment was 8.3 months with XTANDI and 3.0 months with placebo. During the trial, 48% of patients on the XTANDI arm and 46% of patients on the placebo arm received glucocorticoids.Grade 3 and higher adverse reactions were reported among 47% of XTANDI-treated patients and 53% of placebo-treated patients. Discontinuations due to adverse events were reported for 16% of XTANDI-treated patients and 18% of placebo-treated patients. The most common adverse reaction leading to treatment discontinuation was seizure, which occurred in 0.9% of the XTANDI-treated patients compared to none (0%) of the placebo-treated patients. Table 1 shows adverse reactions reported in Study 1 that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
Table 1. Adverse Reactions in Study 1 XTANDIN = 800
PlaceboN = 399
Grade 1-4a
(%)
Grade 3-4(%)
Grade 1-4(%)
Grade 3-4(%)
General DisordersAsthenic Conditionsb 50.6 9.0 44.4 9.3
Peripheral Edema 15.4 1.0 13.3 0.8
Musculoskeletal And Connective Tissue DisordersBack Pain 26.4 5.3 24.3 4.0Arthralgia 20.5 2.5 17.3 1.8Musculoskeletal Pain 15.0 1.3 11.5 0.3
Muscular Weakness 9.8 1.5 6.8 1.8
Musculoskeletal Stiffness 2.6 0.3 0.3 0.0
Gastrointestinal DisordersDiarrhea 21.8 1.1 17.5 0.3Vascular DisordersHot Flush 20.3 0.0 10.3 0.0Hypertension 6.4 2.1 2.8 1.3Nervous System DisordersHeadache 12.1 0.9 5.5 0.0Dizzinessc 9.5 0.5 7.5 0.5Spinal Cord Compression and Cauda Equina Syndrome
7.4 6.6 4.5 3.8
Paresthesia 6.6 0.0 4.5 0.0Mental Impairment Disordersd
4.3 0.3 1.8 0.0
Hypoesthesia 4.0 0.3 1.8 0.0Infections And InfestationsUpper Respiratory Tract Infectione
10.9 0.0 6.5 0.3
Lower Respiratory Tract And Lung Infectionf
8.5 2.4 4.8 1.3
Psychiatric DisordersInsomnia 8.8 0.0 6.0 0.5Anxiety 6.5 0.3 4.0 0.0Renal And Urinary DisordersHematuria 6.9 1.8 4.5 1.0Pollakiuria 4.8 0.0 2.5 0.0Injury, Poisoning And Procedural ComplicationsFall 4.6 0.3 1.3 0.0Non-pathologic Fractures 4.0 1.4 0.8 0.3
Skin And Subcutaneous Tissue DisordersPruritus 3.8 0.0 1.3 0.0Dry Skin 3.5 0.0 1.3 0.0
Table 1. Adverse Reactions in Study 1 Respiratory DisordersEpistaxis 3.3 0.1 1.3 0.3a CTCAE v4b Includes asthenia and fatigue.c Includes dizziness and vertigo.d Includes amnesia, memory impairment, cognitive disorder,
and disturbance in attention.e Includes nasopharyngitis, upper respiratory tract infection,
sinusitis, rhinitis, pharyngitis, and laryngitis.f Includes pneumonia, lower respiratory tract infection,
bronchitis, and lung infection.
Study 2: Chemotherapy-naive Metastatic Castration-Resistant Prostate Cancer
Study 2 enrolled 1717 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy, of whom 1715 received at least one dose of study drug. The median duration of treatment was 17.5 months with XTANDI and 4.6 months with placebo. Grade 3-4 adverse reactions were reported in 44% of XTANDI-treated patients and 37% of placebo-treated patients. Discontinuations due to adverse events were reported for 6% of XTANDI-treated patients and 6% of placebo-treated patients. The most common adverse reaction leading to treatment discontinuation was fatigue/asthenia, which occurred in 1% of patients on each treatment arm. Table 2 includes adverse reactions reported in Study 2 that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
Table 2. Adverse Reactions in Study 2XTANDIN = 871
PlaceboN = 844
Grade 1-4a
(%)
Grade 3-4(%)
Grade 1-4(%)
Grade 3-4(%)
General DisordersAsthenic Conditionsb 46.9 3.4 33.0 2.8Peripheral Edema 11.5 0.2 8.2 0.4
Musculoskeletal And Connective Tissue DisordersBack Pain 28.6 2.5 22.4 3.0Arthralgia 21.4 1.6 16.1 1.1Gastrointestinal DisordersConstipation 23.2 0.7 17.3 0.4Diarrhea 16.8 0.3 14.3 0.4Vascular DisordersHot Flush 18.0 0.1 7.8 0.0Hypertension 14.2 7.2 4.1 2.3Nervous System DisordersDizzinessc 11.3 0.3 7.1 0.0Headache 11.0 0.2 7.0 0.4Dysgeusia 7.6 0.1 3.7 0.0Mental Impairment Disordersd
5.7 0.0 1.3 0.1
Restless Legs Syndrome 2.1 0.1 0.4 0.0
Respiratory DisordersDyspneae 11.0 0.6 8.5 0.6Infections And InfestationsUpper Respiratory Tract Infectionf
16.4 0.0 10.5 0.0
Lower Respiratory Tract And Lung Infectiong
7.9 1.5 4.7 1.1
Psychiatric DisordersInsomnia 8.2 0.1 5.7 0.0Renal And Urinary DisordersHematuria 8.8 1.3 5.8 1.3Injury, Poisoning And Procedural ComplicationsFall 12.7 1.6 5.3 0.7Non-Pathological Fracture 8.8 2.1 3.0 1.1
Metabolism and Nutrition DisordersDecreased Appetite 18.9 0.3 16.4 0.7
InvestigationsWeight Decreased 12.4 0.8 8.5 0.2
Reproductive System and Breast DisordersGynecomastia 3.4 0.0 1.4 0.0
(cont.)
K
Cosmos Communications 1
1js
31376a 08.20.15 133
QC
Table 2. Adverse Reactions in Study 2a CTCAE v4b Includes asthenia and fatigue. c Includes dizziness and vertigo.d Includes amnesia, memory impairment, cognitive disorder,
and disturbance in attention.e Includes dyspnea, exertional dyspnea, and dyspnea at rest.f Includes nasopharyngitis, upper respiratory tract infection,
sinusitis, rhinitis, pharyngitis, and laryngitis.g Includes pneumonia, lower respiratory tract infection,
bronchitis, and lung infection.
Laboratory AbnormalitiesIn the two randomized clinical trials, Grade 1-4 neutropenia occurred in 15% of patients treated with XTANDI (1% Grade 3-4) and in 6% of patients treated with placebo (0.5% Grade 3-4). The incidence of Grade 1-4 thrombocytopenia was 6% of patients treated with XTANDI (0.3% Grade 3-4) and 5% of patients treated with placebo (0.5% Grade 3-4). Grade 1-4 elevations in ALT occurred in 10% of patients treated with XTANDI (0.2% Grade 3-4) and 16% of patients treated with placebo (0.2% Grade 3-4). Grade 1-4 elevations in bilirubin occurred in 3% of patients treated with XTANDI (0.1% Grade 3-4) and 2% of patients treated with placebo (no Grade 3-4).InfectionsIn Study 1, 1% of patients treated with XTANDI compared to 0.3% of patients treated with placebo died from infections or sepsis. In Study 2, 1 patient in each treatment group (0.1%) had an infection resulting in death.Falls and Fall-related InjuriesIn the two randomized clinical trials, falls including fall- related injuries, occurred in 9% of patients treated with XTANDI compared to 4% of patients treated with placebo. Falls were not associated with loss of consciousness or seizure. Fall-related injuries were more severe in patients treated with XTANDI and included non-pathologic fractures, joint injuries, and hematomas.HypertensionIn the two randomized trials, hypertension was reported in 11% of patients receiving XTANDI and 4% of patients receiving placebo. No patients experienced hypertensive crisis. Medical history of hypertension was balanced between arms. Hypertension led to study discontinuation in < 1% of patients in each arm.Post-Marketing ExperienceThe following additional adverse reactions have been identified during post approval use of XTANDI. Because these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.Neurological Disorders: posterior reversible encephalopathy syndrome (PRES)
DRUG INTERACTIONS
Drugs that Inhibit or Induce CYP2C8Co-administration of a strong CYP2C8 inhibitor (gemfibrozil) increased the composite area under the plasma concentration-time curve (AUC) of enzalutamide plus N-desmethyl enzalutamide by 2.2-fold in healthy volunteers. Co-administration of XTANDI with strong CYP2C8 inhibitors should be avoided if possible. If co-administration of XTANDI with a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of XTANDI.The effects of CYP2C8 inducers on the pharmacokinetics of enzalutamide have not been evaluated in vivo. Co- administration of XTANDI with strong or moderate CYP2C8 inducers (e.g., rifampin) may alter the plasma exposure of XTANDI and should be avoided if possible. Selection of a concomitant medication with no or minimal CYP2C8 induction potential is recommended.Drugs that Inhibit or Induce CYP3A4Co-administration of a strong CYP3A4 inhibitor (itraconazole) increased the composite AUC of enzalutamide plus N-desmethyl enzalutamide by 1.3-fold in healthy volunteers.The effects of CYP3A4 inducers on the pharmacokinetics of enzalutamide have not been evaluated in vivo. Co- administration of XTANDI with strong CYP3A4 inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifabutin, rifampin, rifapentine) may decrease the plasma exposure of XTANDI and should be avoided if possible. Selection of a concomitant medication with no or minimal CYP3A4 induction potential is recommended. Moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin) and St. John’s Wort may also reduce the plasma
exposure of XTANDI and should be avoided if possible.Effect of XTANDI on Drug Metabolizing EnzymesEnzalutamide is a strong CYP3A4 inducer and a moderate CYP2C9 and CYP2C19 inducer in humans. At steady state, XTANDI reduced the plasma exposure to midazolam (CYP3A4 substrate), warfarin (CYP2C9 substrate), and omeprazole (CYP2C19 substrate). Concomitant use of XTANDI with narrow therapeutic index drugs that are metabolized by CYP3A4 (e.g., alfentanil, cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus and tacrolimus), CYP2C9 (e.g., phenytoin, warfarin) and CYP2C19 (e.g., S-mephenytoin) should be avoided, as enzalutamide may decrease their exposure. If co-administration with warfarin cannot be avoided, conduct additional INR monitoring.
USE IN SPECIFIC POPULATIONS
Pregnancy- Pregnancy Category X.Risk SummaryXTANDI can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. While there are no human data on the use of XTANDI in pregnancy and XTANDI is not indicated for use in women, it is important to know that maternal use of an androgen receptor inhibitor could affect development of the fetus. Enzalutamide caused embryo-fetal toxicity in mice at exposures that were lower than in patients receiving the recommended dose. XTANDI is contraindicated in women who are or may become pregnant while receiving the drug. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss. Advise females of reproductive potential to avoid becoming pregnant during treatment with XTANDI.Animal DataIn an embryo-fetal developmental toxicity study in mice, enzalutamide caused developmental toxicity when administered at oral doses of 10 or 30 mg/kg/day throughout the period of organogenesis (gestational days 6-15). Findings included embryo-fetal lethality (increased post-implantation loss and resorptions) and decreased anogenital distance at ≥ 10 mg/kg/day, and cleft palate and absent palatine bone at 30 mg/kg/day. Doses of 30 mg/kg/day caused maternal toxicity. The doses tested in mice (1, 10 and 30 mg/kg/day) resulted in systemic exposures (AUC) approximately 0.04, 0.4 and 1.1 times, respectively, the exposures in patients. Enzalutamide did not cause developmental toxicity in rabbits when administered throughout the period of organogenesis (gestational days 6-18) at dose levels up to 10 mg/kg/day (approximately 0.4 times the exposures in patients based on AUC).Nursing MothersXTANDI is not indicated for use in women. It is not known if enzalutamide is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from XTANDI, a decision should be made to either discontinue nursing, or discontinue the drug taking into account the importance of the drug to the mother.Pediatric UseSafety and effectiveness of XTANDI in pediatric patients have not been established.Geriatric UseOf 1671 patients who received XTANDI in the two randomized clinical trials, 75% were 65 and over, while 31% were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.Patients with Renal ImpairmentA dedicated renal impairment trial for XTANDI has not been conducted. Based on the population pharmacokinetic analysis using data from clinical trials in patients with metastatic CRPC and healthy volunteers, no significant difference in enzalutamide clearance was observed in patients with pre-existing mild to moderate renal impairment (30 mL/min ≤ creatinine clearance [CrCL] ≤ 89 mL/min) compared to patients and volunteers with baseline normal renal function (CrCL ≥ 90 mL/min). No initial dosage adjustment is necessary for patients with mild to moderate renal impairment. Severe renal impairment (CrCL < 30 mL/min) and end-stage renal
disease have not been assessed.Patients with Hepatic ImpairmentA dedicated hepatic impairment trial compared the composite systemic exposure of enzalutamide plus N-desmethyl enzalutamide in volunteers with baseline mild or moderate hepatic impairment (Child-Pugh Class A and B, respectively) versus healthy controls with normal hepatic function. The composite AUC of enzalutamide plus N-desmethyl enzalutamide was similar in volunteers with mild or moderate baseline hepatic impairment compared to volunteers with normal hepatic function. No initial dosage adjustment is necessary for patients with baseline mild or moderate hepatic impairment. Baseline severe hepatic impairment (Child-Pugh Class C) has not been assessed.
OVERDOSAGE
In the event of an overdose, stop treatment with XTANDI and initiate general supportive measures taking into consideration the half-life of 5.8 days. In a dose escalation study, no seizures were reported at ≤ 240 mg daily, whereas 3 seizures were reported, 1 each at 360 mg, 480 mg, and 600 mg daily. Patients may be at increased risk of seizure following an overdose.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of FertilityLong-term animal studies have not been conducted to evaluate the carcinogenic potential of enzalutamide.Enzalutamide did not induce mutations in the bacterial reverse mutation (Ames) assay and was not genotoxic in either the in vitro mouse lymphoma thymidine kinase (Tk) gene mutation assay or the in vivo mouse micronucleus assay.Based on nonclinical findings in repeat-dose toxicology studies, which were consistent with the pharmacological activity of enzalutamide, male fertility may be impaired by treatment with XTANDI. In a 26-week study in rats, atrophy of the prostate and seminal vesicles was observed at ≥ 30 mg/kg/day (equal to the human exposure based on AUC). In 4-, 13-, and 39-week studies in dogs, hypospermatogenesis and atrophy of the prostate and epididymides were observed at ≥ 4 mg/kg/day (0.3 times the human exposure based on AUC).
Manufactured by: Catalent Pharma Solutions, LLC, St. Petersburg, FL 33716
Manufactured for and Distributed by: Astellas Pharma US, Inc., Northbrook, IL 60062
Marketed by:Astellas Pharma US, Inc., Northbrook, IL 60062Medivation, Inc., San Francisco, CA 94105
Revised: August 2015
14L082-XTA
Rx Only
© 2015 Astellas Pharma US, Inc.
XTANDI® is a registered trademark of Astellas Pharma Inc.
076-1119-PM
(cont.)
K
Cosmos Communications 1
1js
31376a 08.20.15 133
QC

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 21
Best of ASCO®
HPV16 DNA in Post-Treatment Oral Rinses Signals Poor Prognosis in Oropharyngeal CancerBy Alice Goodman
Detectable oral HPV16 DNA in oral rinses post treatment for
oropharyngeal cancer appears to be
a harbinger of poor prognosis and can predict recurrence. Oral HPV16 DNA rinses are a potential tool for
long-term tumor surveillance, ac-cording to a study selected for the Best of ASCO® 2015.1
“The goal is to identify progressors early enough to intervene therapeuti-cally. Persistent human papillomavi-rus (HPV) DNA is infrequent in oral rinses post treatment, but when it is present, it signals reinfection and is a useful marker for recurrence as well as survival,” said Robert L. Ferris, MD, PhD, University of Pittsburgh Cancer Institute, who discussed this study at the Best of ASCO® 2015.
There are two distinct types of head and neck cancer: carcinogen-exposed and HPV-associated cancer. The incidence of HPV head and neck cancer has increased dramatically over the past few decades, and it is now the cause of the vast majority of oropharyngeal cancers.
HPV oropharyngeal cancers have distinct clinical and biologic character-istics. “The prognosis is uniquely good, with about an 80% long-term survival. About 10% to 25% progress within 2 years of first-line treatment. HPV oro-pharyngeal cancers have a favorable re-sponse to all types of salvage therapy,” he said.
HPV16 DNA (the type of HPV most responsible for 95% of oropha-ryngeal cancers) is detectable in oral rinses but only in up to two-thirds of patients.
Head/Neck Cancer
Persistent human papillomavirus DNA is
infrequent in oral rinses post treatment, but when it is
present, it signals reinfection and is a useful marker
for recurrence as well as survival.
—Robert L. Ferris, MD, PhD
continued on page 22
XTANDI® (enzalutamide) capsules for oral use Initial U.S. Approval: 2012BRIEF SUMMARY OF PRESCRIBING INFORMATION
The following is a brief summary. Please see the package insert for full prescribing information.
INDICATIONS AND USAGE
XTANDI is indicated for the treatment of patients with metastatic castration-resistant prostate cancer (CRPC).
CONTRAINDICATIONS
Pregnancy XTANDI can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. XTANDI is not indicated for use in women. XTANDI is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss.
WARNINGS AND PRECAUTIONS
Seizure In Study 1, which enrolled patients who previously received docetaxel, 7 of 800 (0.9%) patients treated with XTANDI experienced a seizure and no patients treated with placebo experienced a seizure. Seizure occurred from 31 to 603 days after initiation of XTANDI. In Study 2, 1 of 871 (0.1%) chemotherapy-naive patients treated with XTANDI and 1 of 844 (0.1%) patients treated with placebo experienced a seizure. Patients experiencing seizure were permanently discontinued from therapy and all seizure events resolved. There is no clinical trial experience re-administering XTANDI to patients who experienced seizure.Limited safety data are available in patients with predisposing factors for seizure because these patients were generally excluded from the trials. These exclusion criteria included a history of seizure, underlying brain injury with loss of consciousness, transient ischemic attack within the past 12 months, cerebral vascular accident, brain metastases, and brain arteriovenous malformation. Study 1 excluded the use of concomitant medications that may lower the seizure threshold, whereas Study 2 permitted the use of these medications.
Because of the risk of seizure associated with XTANDI use, patients should be advised of the risk of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others. Permanently discontinue XTANDI in patients who develop a seizure during treatment.Posterior Reversible Encephalopathy Syndrome (PRES)There have been reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving XTANDI [see Adverse Reactions (6.2)]. PRES is a neurological disorder which can present with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual and neurological disturbances, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). Discontinue XTANDI in patients who develop PRES.
ADVERSE REACTIONS
Clinical Trial Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Two randomized clinical trials enrolled patients with metastatic prostate cancer that has progressed on androgen deprivation therapy (GnRH therapy or bilateral orchiectomy), a disease setting that is also defined as metastatic CRPC. In both studies, patients received XTANDI 160 mg orally once daily in the active treatment arm or placebo in the control arm. All patients continued androgen deprivation therapy. Patients were allowed, but not required, to take glucocorticoids.The most common adverse reactions (≥ 10%) that occurred more commonly (≥ 2% over placebo) in the
XTANDI-treated patients from the two randomized clinical trials were asthenia/fatigue, back pain, decreased appetite, constipation, arthralgia, diarrhea, hot flush, upper respiratory tract infection, peripheral edema, dyspnea, musculoskeletal pain, weight decreased, headache, hypertension, and dizziness/vertigo.
Study 1: Metastatic Castration-Resistant Prostate Cancer Following Chemotherapy
Study 1 enrolled 1199 patients with metastatic CRPC who had previously received docetaxel. The median duration of treatment was 8.3 months with XTANDI and 3.0 months with placebo. During the trial, 48% of patients on the XTANDI arm and 46% of patients on the placebo arm received glucocorticoids.Grade 3 and higher adverse reactions were reported among 47% of XTANDI-treated patients and 53% of placebo-treated patients. Discontinuations due to adverse events were reported for 16% of XTANDI-treated patients and 18% of placebo-treated patients. The most common adverse reaction leading to treatment discontinuation was seizure, which occurred in 0.9% of the XTANDI-treated patients compared to none (0%) of the placebo-treated patients. Table 1 shows adverse reactions reported in Study 1 that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
Table 1. Adverse Reactions in Study 1 XTANDIN = 800
PlaceboN = 399
Grade 1-4a
(%)
Grade 3-4(%)
Grade 1-4(%)
Grade 3-4(%)
General DisordersAsthenic Conditionsb 50.6 9.0 44.4 9.3
Peripheral Edema 15.4 1.0 13.3 0.8
Musculoskeletal And Connective Tissue DisordersBack Pain 26.4 5.3 24.3 4.0Arthralgia 20.5 2.5 17.3 1.8Musculoskeletal Pain 15.0 1.3 11.5 0.3
Muscular Weakness 9.8 1.5 6.8 1.8
Musculoskeletal Stiffness 2.6 0.3 0.3 0.0
Gastrointestinal DisordersDiarrhea 21.8 1.1 17.5 0.3Vascular DisordersHot Flush 20.3 0.0 10.3 0.0Hypertension 6.4 2.1 2.8 1.3Nervous System DisordersHeadache 12.1 0.9 5.5 0.0Dizzinessc 9.5 0.5 7.5 0.5Spinal Cord Compression and Cauda Equina Syndrome
7.4 6.6 4.5 3.8
Paresthesia 6.6 0.0 4.5 0.0Mental Impairment Disordersd
4.3 0.3 1.8 0.0
Hypoesthesia 4.0 0.3 1.8 0.0Infections And InfestationsUpper Respiratory Tract Infectione
10.9 0.0 6.5 0.3
Lower Respiratory Tract And Lung Infectionf
8.5 2.4 4.8 1.3
Psychiatric DisordersInsomnia 8.8 0.0 6.0 0.5Anxiety 6.5 0.3 4.0 0.0Renal And Urinary DisordersHematuria 6.9 1.8 4.5 1.0Pollakiuria 4.8 0.0 2.5 0.0Injury, Poisoning And Procedural ComplicationsFall 4.6 0.3 1.3 0.0Non-pathologic Fractures 4.0 1.4 0.8 0.3
Skin And Subcutaneous Tissue DisordersPruritus 3.8 0.0 1.3 0.0Dry Skin 3.5 0.0 1.3 0.0
Table 1. Adverse Reactions in Study 1 Respiratory DisordersEpistaxis 3.3 0.1 1.3 0.3a CTCAE v4b Includes asthenia and fatigue.c Includes dizziness and vertigo.d Includes amnesia, memory impairment, cognitive disorder,
and disturbance in attention.e Includes nasopharyngitis, upper respiratory tract infection,
sinusitis, rhinitis, pharyngitis, and laryngitis.f Includes pneumonia, lower respiratory tract infection,
bronchitis, and lung infection.
Study 2: Chemotherapy-naive Metastatic Castration-Resistant Prostate Cancer
Study 2 enrolled 1717 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy, of whom 1715 received at least one dose of study drug. The median duration of treatment was 17.5 months with XTANDI and 4.6 months with placebo. Grade 3-4 adverse reactions were reported in 44% of XTANDI-treated patients and 37% of placebo-treated patients. Discontinuations due to adverse events were reported for 6% of XTANDI-treated patients and 6% of placebo-treated patients. The most common adverse reaction leading to treatment discontinuation was fatigue/asthenia, which occurred in 1% of patients on each treatment arm. Table 2 includes adverse reactions reported in Study 2 that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
Table 2. Adverse Reactions in Study 2XTANDIN = 871
PlaceboN = 844
Grade 1-4a
(%)
Grade 3-4(%)
Grade 1-4(%)
Grade 3-4(%)
General DisordersAsthenic Conditionsb 46.9 3.4 33.0 2.8Peripheral Edema 11.5 0.2 8.2 0.4
Musculoskeletal And Connective Tissue DisordersBack Pain 28.6 2.5 22.4 3.0Arthralgia 21.4 1.6 16.1 1.1Gastrointestinal DisordersConstipation 23.2 0.7 17.3 0.4Diarrhea 16.8 0.3 14.3 0.4Vascular DisordersHot Flush 18.0 0.1 7.8 0.0Hypertension 14.2 7.2 4.1 2.3Nervous System DisordersDizzinessc 11.3 0.3 7.1 0.0Headache 11.0 0.2 7.0 0.4Dysgeusia 7.6 0.1 3.7 0.0Mental Impairment Disordersd
5.7 0.0 1.3 0.1
Restless Legs Syndrome 2.1 0.1 0.4 0.0
Respiratory DisordersDyspneae 11.0 0.6 8.5 0.6Infections And InfestationsUpper Respiratory Tract Infectionf
16.4 0.0 10.5 0.0
Lower Respiratory Tract And Lung Infectiong
7.9 1.5 4.7 1.1
Psychiatric DisordersInsomnia 8.2 0.1 5.7 0.0Renal And Urinary DisordersHematuria 8.8 1.3 5.8 1.3Injury, Poisoning And Procedural ComplicationsFall 12.7 1.6 5.3 0.7Non-Pathological Fracture 8.8 2.1 3.0 1.1
Metabolism and Nutrition DisordersDecreased Appetite 18.9 0.3 16.4 0.7
InvestigationsWeight Decreased 12.4 0.8 8.5 0.2
Reproductive System and Breast DisordersGynecomastia 3.4 0.0 1.4 0.0
(cont.)
K
Cosmos Communications 1
1js
31376a 08.20.15 133
QC
Table 2. Adverse Reactions in Study 2a CTCAE v4b Includes asthenia and fatigue. c Includes dizziness and vertigo.d Includes amnesia, memory impairment, cognitive disorder,
and disturbance in attention.e Includes dyspnea, exertional dyspnea, and dyspnea at rest.f Includes nasopharyngitis, upper respiratory tract infection,
sinusitis, rhinitis, pharyngitis, and laryngitis.g Includes pneumonia, lower respiratory tract infection,
bronchitis, and lung infection.
Laboratory AbnormalitiesIn the two randomized clinical trials, Grade 1-4 neutropenia occurred in 15% of patients treated with XTANDI (1% Grade 3-4) and in 6% of patients treated with placebo (0.5% Grade 3-4). The incidence of Grade 1-4 thrombocytopenia was 6% of patients treated with XTANDI (0.3% Grade 3-4) and 5% of patients treated with placebo (0.5% Grade 3-4). Grade 1-4 elevations in ALT occurred in 10% of patients treated with XTANDI (0.2% Grade 3-4) and 16% of patients treated with placebo (0.2% Grade 3-4). Grade 1-4 elevations in bilirubin occurred in 3% of patients treated with XTANDI (0.1% Grade 3-4) and 2% of patients treated with placebo (no Grade 3-4).InfectionsIn Study 1, 1% of patients treated with XTANDI compared to 0.3% of patients treated with placebo died from infections or sepsis. In Study 2, 1 patient in each treatment group (0.1%) had an infection resulting in death.Falls and Fall-related InjuriesIn the two randomized clinical trials, falls including fall- related injuries, occurred in 9% of patients treated with XTANDI compared to 4% of patients treated with placebo. Falls were not associated with loss of consciousness or seizure. Fall-related injuries were more severe in patients treated with XTANDI and included non-pathologic fractures, joint injuries, and hematomas.HypertensionIn the two randomized trials, hypertension was reported in 11% of patients receiving XTANDI and 4% of patients receiving placebo. No patients experienced hypertensive crisis. Medical history of hypertension was balanced between arms. Hypertension led to study discontinuation in < 1% of patients in each arm.Post-Marketing ExperienceThe following additional adverse reactions have been identified during post approval use of XTANDI. Because these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.Neurological Disorders: posterior reversible encephalopathy syndrome (PRES)
DRUG INTERACTIONS
Drugs that Inhibit or Induce CYP2C8Co-administration of a strong CYP2C8 inhibitor (gemfibrozil) increased the composite area under the plasma concentration-time curve (AUC) of enzalutamide plus N-desmethyl enzalutamide by 2.2-fold in healthy volunteers. Co-administration of XTANDI with strong CYP2C8 inhibitors should be avoided if possible. If co-administration of XTANDI with a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of XTANDI.The effects of CYP2C8 inducers on the pharmacokinetics of enzalutamide have not been evaluated in vivo. Co- administration of XTANDI with strong or moderate CYP2C8 inducers (e.g., rifampin) may alter the plasma exposure of XTANDI and should be avoided if possible. Selection of a concomitant medication with no or minimal CYP2C8 induction potential is recommended.Drugs that Inhibit or Induce CYP3A4Co-administration of a strong CYP3A4 inhibitor (itraconazole) increased the composite AUC of enzalutamide plus N-desmethyl enzalutamide by 1.3-fold in healthy volunteers.The effects of CYP3A4 inducers on the pharmacokinetics of enzalutamide have not been evaluated in vivo. Co- administration of XTANDI with strong CYP3A4 inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifabutin, rifampin, rifapentine) may decrease the plasma exposure of XTANDI and should be avoided if possible. Selection of a concomitant medication with no or minimal CYP3A4 induction potential is recommended. Moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin) and St. John’s Wort may also reduce the plasma
exposure of XTANDI and should be avoided if possible.Effect of XTANDI on Drug Metabolizing EnzymesEnzalutamide is a strong CYP3A4 inducer and a moderate CYP2C9 and CYP2C19 inducer in humans. At steady state, XTANDI reduced the plasma exposure to midazolam (CYP3A4 substrate), warfarin (CYP2C9 substrate), and omeprazole (CYP2C19 substrate). Concomitant use of XTANDI with narrow therapeutic index drugs that are metabolized by CYP3A4 (e.g., alfentanil, cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus and tacrolimus), CYP2C9 (e.g., phenytoin, warfarin) and CYP2C19 (e.g., S-mephenytoin) should be avoided, as enzalutamide may decrease their exposure. If co-administration with warfarin cannot be avoided, conduct additional INR monitoring.
USE IN SPECIFIC POPULATIONS
Pregnancy- Pregnancy Category X.Risk SummaryXTANDI can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. While there are no human data on the use of XTANDI in pregnancy and XTANDI is not indicated for use in women, it is important to know that maternal use of an androgen receptor inhibitor could affect development of the fetus. Enzalutamide caused embryo-fetal toxicity in mice at exposures that were lower than in patients receiving the recommended dose. XTANDI is contraindicated in women who are or may become pregnant while receiving the drug. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss. Advise females of reproductive potential to avoid becoming pregnant during treatment with XTANDI.Animal DataIn an embryo-fetal developmental toxicity study in mice, enzalutamide caused developmental toxicity when administered at oral doses of 10 or 30 mg/kg/day throughout the period of organogenesis (gestational days 6-15). Findings included embryo-fetal lethality (increased post-implantation loss and resorptions) and decreased anogenital distance at ≥ 10 mg/kg/day, and cleft palate and absent palatine bone at 30 mg/kg/day. Doses of 30 mg/kg/day caused maternal toxicity. The doses tested in mice (1, 10 and 30 mg/kg/day) resulted in systemic exposures (AUC) approximately 0.04, 0.4 and 1.1 times, respectively, the exposures in patients. Enzalutamide did not cause developmental toxicity in rabbits when administered throughout the period of organogenesis (gestational days 6-18) at dose levels up to 10 mg/kg/day (approximately 0.4 times the exposures in patients based on AUC).Nursing MothersXTANDI is not indicated for use in women. It is not known if enzalutamide is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from XTANDI, a decision should be made to either discontinue nursing, or discontinue the drug taking into account the importance of the drug to the mother.Pediatric UseSafety and effectiveness of XTANDI in pediatric patients have not been established.Geriatric UseOf 1671 patients who received XTANDI in the two randomized clinical trials, 75% were 65 and over, while 31% were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.Patients with Renal ImpairmentA dedicated renal impairment trial for XTANDI has not been conducted. Based on the population pharmacokinetic analysis using data from clinical trials in patients with metastatic CRPC and healthy volunteers, no significant difference in enzalutamide clearance was observed in patients with pre-existing mild to moderate renal impairment (30 mL/min ≤ creatinine clearance [CrCL] ≤ 89 mL/min) compared to patients and volunteers with baseline normal renal function (CrCL ≥ 90 mL/min). No initial dosage adjustment is necessary for patients with mild to moderate renal impairment. Severe renal impairment (CrCL < 30 mL/min) and end-stage renal
disease have not been assessed.Patients with Hepatic ImpairmentA dedicated hepatic impairment trial compared the composite systemic exposure of enzalutamide plus N-desmethyl enzalutamide in volunteers with baseline mild or moderate hepatic impairment (Child-Pugh Class A and B, respectively) versus healthy controls with normal hepatic function. The composite AUC of enzalutamide plus N-desmethyl enzalutamide was similar in volunteers with mild or moderate baseline hepatic impairment compared to volunteers with normal hepatic function. No initial dosage adjustment is necessary for patients with baseline mild or moderate hepatic impairment. Baseline severe hepatic impairment (Child-Pugh Class C) has not been assessed.
OVERDOSAGE
In the event of an overdose, stop treatment with XTANDI and initiate general supportive measures taking into consideration the half-life of 5.8 days. In a dose escalation study, no seizures were reported at ≤ 240 mg daily, whereas 3 seizures were reported, 1 each at 360 mg, 480 mg, and 600 mg daily. Patients may be at increased risk of seizure following an overdose.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of FertilityLong-term animal studies have not been conducted to evaluate the carcinogenic potential of enzalutamide.Enzalutamide did not induce mutations in the bacterial reverse mutation (Ames) assay and was not genotoxic in either the in vitro mouse lymphoma thymidine kinase (Tk) gene mutation assay or the in vivo mouse micronucleus assay.Based on nonclinical findings in repeat-dose toxicology studies, which were consistent with the pharmacological activity of enzalutamide, male fertility may be impaired by treatment with XTANDI. In a 26-week study in rats, atrophy of the prostate and seminal vesicles was observed at ≥ 30 mg/kg/day (equal to the human exposure based on AUC). In 4-, 13-, and 39-week studies in dogs, hypospermatogenesis and atrophy of the prostate and epididymides were observed at ≥ 4 mg/kg/day (0.3 times the human exposure based on AUC).
Manufactured by: Catalent Pharma Solutions, LLC, St. Petersburg, FL 33716
Manufactured for and Distributed by: Astellas Pharma US, Inc., Northbrook, IL 60062
Marketed by:Astellas Pharma US, Inc., Northbrook, IL 60062Medivation, Inc., San Francisco, CA 94105
Revised: August 2015
14L082-XTA
Rx Only
© 2015 Astellas Pharma US, Inc.
XTANDI® is a registered trademark of Astellas Pharma Inc.
076-1119-PM
(cont.)
K
Cosmos Communications 1
1js
31376a 08.20.15 133
QC

PAGE 22 The ASCO Post | SEPTEMBER 10, 2015
Best of ASCO®
Study DetailsThe study enrolled 124 patients
with HPV-associated oropharyngeal cancer treated with curative intent at four different centers from October 2009 to May 2013. Oral rinses were collected at baseline after diagnosis and at 9, 12, 18, and 24 months post treatment.
Ninety percent of the patients were male, 58% were never smokers, 85% were married at diagnosis, 65% had small tumors, and 70% had ad-vanced nodal status.
At baseline, 67 patients (54%) had evidence of HPV16 in their oral rinses. Post treatment, persistent HPV16 was detected in five patients (4%) and newly detected HPV16 was found in one patient (1%). Ninety-three percent of patients were cleared of oral HPV16.
HPV typically recurs within 4
years, Dr. Ferris explained. At 33 months of follow-up, there were 14 recurrences and 6 deaths; 2-year disease-free survival was 92%, and 2-year overall survival was 98%.
Prognostic ImplicationsThe presence of HPV16 DNA
post treatment was strongly associ-ated with survival. All five patients with persistent HPV16 DNA died.
The five patients with persistent HPV16 all had recurrences; three of them died of oropharyngeal cancer. Three patients had local recurrence, one patient had distant recurrence alone, and one patient had local and distant recurrences. The median time to first post-treatment recurrence was 7 months.
The specificity of detectable HPV16 DNA in oral rinses was 100%. “When patients had it, they recurred. The specificity for the test was 43%. Some recurrences were not associ-
ated with detectable HPV16 DNA,” Dr. Ferris told listeners.
In this study, post-treatment speci-mens were not collected during the first 9 months after diagnosis. Dr. Fer-ris questioned whether the presence of HPV16 DNA in oral rinses reflected persistent infection or reinfection.
In addition, other issues are that reproducible results were not con-firmed before treatment and the rea-
sons for new infection are unclear. nDisclosure: Dr. Ferris reported no potential
conflicts of interest.
Reference1. Rettig EM, Wentz A, Posner MR,
et al: Prognostic implication of persistent HPV16 DNA detection in oral rinses for HPV-positive oropharyngeal carcinoma. 2015 ASCO Annual Meeting. Abstract 6005. Presented May 29, 2015.
HPV16 DNA and Recurrent Oropharyngeal Cancer
■ The persistence of HPV16 DNA in oral rinses post treatment is infrequent but strongly predictive for recurrence of oropharyngeal cancer, particularly local recurrence.
■ Persistent oral HPV16 DNA (from diagnosis to post treatment) in oral rinses predicts disease-free survival and overall survival.
■ Pretreatment oral HPV16 DNA does not appear to be a useful marker for recurrence.
■ Detectable HPV16 DNA in post-treatment oral rinses may be useful for long-term monitoring of patients with HPV-positive oropharyngeal cancer.
Oropharyngeal Cancercontinued from page 21
Announcements
IPOS Announces 2015 Award Winners at the World Congress of Psycho-Oncology
The International Psycho-Oncol-ogy Society (IPOS) announced
four award winners at the 2015 World Congress of Psycho-Oncology, held July 28 to August 1 in Washington, DC.
Arthur M. Sutherland Award: William H. Redd, PhD
Dr. Redd is Professor of Oncological Sciences at the Mount Sinai Hospital. He is credited with introducing behav-ioral psychology and behavioral medi-cine to oncology; he identified the role of respondent and operant conditioning in the development of cancer treatment side effects, recognizing the severe con-ditioned aversions patients can develop as they undergo their treatments. He then went on to develop distraction and other intervention techniques to help pa-tients counter these conditioned effects.
Bernard Fox Memorial Award: Susanne O. Dalton, MD, PhD
Dr. Dalton is a Senior Researcher in Survivorship Unit of the Danish Can-cer Research Council, and conducts research primarily focusing on social inequality in cancer and on physical, psychological, and socioeconomic consequences of cancer. Since 2011,
she has been head of the Research Group on Social Inequality in Sur-vivorship. She leads a series of stud-ies exploring issues of cancer among patients with mental illness, and in another series, delineated the roles of sociodemographic factors in cancer survivorship. Her work has contrib-uted significantly to the evidence base highlighting social inequalities that can exist in cancer care and recovery.
Noemi Fishman Award for Lifetime Clinical Excellence: Andrew J. Roth, MD
Since 2007, Dr. Roth has been the Training Program Director for the clinical psycho- oncology fellowship training program at Memorial Sloan Kettering Cancer Center. He has pio-
neered the field of prostate cancer psycho- oncology, being among the first psychiatrist in the world special-izing primarily in the psychiatric and psychosocial problems of men with prostate cancer. Notably, Dr. Roth was in the team that developed the “Dis-tress Thermometer,” now widely used as a tool to identify distress in patients with cancer. He has supervised and helped train over 100 clinical fellows in psycho-oncology.
Hiroomi Kawano New Investigator Award: Katriina Whitaker, PhD, CPsychol
Dr. Whittaker is currently Senior Lecturer at the University of Surrey. Her doctoral thesis involved a novel application of theoretical models from
the study of anxiety disorders to inves-tigate anxiety in cancer patients, estab-lishing the importance of involuntary thoughts, memories, and images in explaining mood disorder in these pa-tients, and identifying an additional role for negative appraisals by patients of these experiences. She demonstrat-ed the clinical utility of this research by using a psychological intervention specifically targeted on these main-taining processes, reducing associated distress substantially. n
Katriina Whitaker, PhD, CPsychol
Susanne O. Dalton, MD, PhD Andrew J. Roth, MDWilliam H. Redd, PhD
See pages 24 to 27 for more on the World
Congress of Psycho-Oncology.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 23
Announcements
Three New Officers Elected to ASTRO’s Board of Directors
The American Society for Radia-tion Oncology (ASTRO) has
elected three new officers to the Board of Directors and three members to serve on the Nominating Committee. Terms for all positions begin on Oc-tober 20, 2015 at ASTRO’s 57th An-nual Meeting. The new officers to the Board of Directors are:
President-Elect: Brian D. Kavanagh, MD, MPH, FASTRO
Dr. Kavanagh is Professor and Interim Chair of the Department of Radiation Oncology at the University of Colorado School of Medicine, Denver, and an at-tending physician at the University of Colorado Hospital. His service to ASTRO includes his current roles as Co-Chair of the Health Policy Committee and as the ASTRO Advisor to the American Medi-cal Association/Specialty Society RVS Update Committee. He is currently Co-Director of the ASTRO-AANS Stereo-tactic Radiosurgery Registry, and he has served ASTRO as a member of the Board of Directors (2011–2015) and in multiple other leadership positions. His additional service has included the Center for Medi-care and Medicaid Services’ Hospital Out-patient Payment Advisory Panel and the Ambulatory Payment Classification Advi-sory Panel, as well as a member of the Na-tional Comprehensive Cancer Network’s (NCCN) Policy Advisory Group.
Health Policy Council Vice-Chair: Michael R. Kuettel, MD, PhD, MBA, FASTRO
Dr. Kuettel is Professor and Chair of the Department of Radiation Medicine and The Barbara C. and George H. Hyde Chair in Radiation Medicine at Roswell Park Cancer Institute and State Univer-sity of New York at Buffalo; he is Profes-sor, Department of Molecular and Cel-lular Biophysics at Roswell Park Cancer Institute. Dr. Kuettel is Clinical Direc-tor of Radiation Oncology at Kaleida Health System. His service to ASTRO includes his current roles as Co-Chair of the Health Policy Committee and as the ASTRO Advisor to the American Medi-cal Association/Specialty Society RVS Update Committee.
Science Council Vice-Chair: Daniel Low, PhD
Dr. Low is Professor in Radiation On-cology and Vice-Chair of Medical Physics in the Department of Radiation Oncol-ogy at the University of California, Los Angeles. He currently serves ASTRO as a member of the Funding Advocacy
and Clinical Tri-als Committee, the Best Practices Sub-committee, and the Multidisciplinary Quality Assurance Subcommittee.
Nominating CommitteeThe new Nominating Committee
members are Quynh-Thu Le, MD, FASTRO, Stanford University; John W. Rieke, MD, MultiCare Regional Cancer Center; and Indrin J. Chetty, PhD, MS, Henry Ford Health System. nBrian D. Kavanagh,MD Michael R. Kuettel, MD Daniel Low, PhD
NEARLY 2 OUT OF 3
CASES ARE RELATED TO T790M
( )
0%
T790M Is the Most Common Mechanism
of Acquired Resistance to First-Generation
EGFR TKI Therapy1
Study of 155 patients with radiographic progression following
a response or durable stable disease with frst-generation
EGFR TKI therapy.
Other rare mechanisms of acquired resistance may include
BRAF, FGFR, and PIK3CA mutations, and transformation to
small-cell histology.10,11
10% 20% 30% 40% 50% 60% 70%
HER2
amplifcation
(3/24)
13%(95% CI, 3%–32%)
METamplifcation
(4/75)
5%(95% CI, 1%–13%)
T790M(98/155)
63% 95% CI, 55%–70%
In EGFRm+ advanced NSCLC,
NEARLY 2 OUT OF 3 CASES OF PROGRESSION WITH FIRST-
GENERATION EGFR TKIs ARE RELATED TO THE T790M MUTATION1,2
©2015 AstraZeneca. All rights reserved. 3140404 6/15
Lung cancer is the leading cause of cancer-related deaths both in the US and worldwide.3,4
For NSCLC EGFRm+ patients, the recommended f rst-line treatment is EGFR tyrosine kinase inhibitors (TKIs).5
The majority of tumors will acquire EGFR TKI–resistance mutations
Despite initial high response rates with f rst-generation EGFR TKIs, many tumors
will develop new mutations and become resistant.6,7 A major barrier to disease
control is resistance to treatment. Resistance to f rst-generation therapy will
develop in most patients with EGFRm+ advanced NSCLC on a currently approved
EGFR TKI.7
After disease progression, clinical guidelines recommend subsequent
treatments including either continuing with an EGFR TKI therapy or beginning
platinum-based chemotherapy.5
Nearly 2 out of 3 cases of progressionwith fi rst-generation EGFR TKIs are related to the T790M mutation
In patients with NSCLC who are EGFRm+, T790M
is an acquired mutation and has been identif ed as
the most common mechanism of acquired resistance
in nearly 2 out of 3 patients.1,2 Development of T790M
mutation may confer resistance through several
potential mechanisms, which may include8,9:
- Steric hindrance, which reduces receptor binding of
reversible EGFR TKIs
- Increased binding aff nity of EGFR for ATP, resulting in
reduced TKI potency
Discovering the cause of resistance
Patients should be monitored for radiologic or clinical progression. Tumors can also be assessed for molecular progression to uncover
additional acquired mutations.1,12-16 When patients with EGFRm+ status progress, prior to changing therapy, a biopsy is reasonable to identify
mechanisms of acquired resistance, as stated in NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®).5
AstraZeneca is a leader in lung cancer research
AstraZeneca is conducting ongoing research to understand the science of the T790M mutation as a driver of resistance.
References: 1. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240-2247. 2. Arcila ME, Oxnard GR,
Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169-1180. 3. American Cancer Society.
Cancer Facts & Figures 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed March 17, 2015. 4. GLOBOCAN 2012. http://globocan.iarc.fr. Accessed February 9, 2015. 5. Referenced
with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.7.2015. ©National Comprehensive Cancer Network, Inc. 2015. All rights reserved. Accessed June 12, 2015. To view
the most recent and complete version of the guideline, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive
Cancer Network, Inc. 6. Mok TS, Wu YL, Thongprasert S, et al. Gef tinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947-957. 7. Sequist LV, Yang JCH, Yamamoto N, et al. Phase III study of afatinib or cisplatin
plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327-3334. 8. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non–small-cell lung cancer to gef tinib. N
Engl J Med. 2005;352:786-792. 9. Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affnity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070-2075. 10. Cheng L, Alexander
RE, MacLennan GT, et al. Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol. 2012;25:347-369. 11. Ware KE, Marshall ME, Heasley LY, et al. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell
lines through de-repression of FGFR2 and FGFR3 expression. PLoS One. 2010;5:e14117. doi:10.1371/journal.pone.0014117.12. Johnson KR, Ringland C, Stokes BJ, et al. Response rate or time to progression as predictors of survival in trials of
metastatic colorectal cancer or non-small-cell lung cancer: a meta-analysis. Lancet. 2006;7:741-746. 13. Lussier YA, Khodarev NN, Regan K, et al. Oligo- and polymetastatic progression in lung metastasis(es) patients is associated with specif c
microRNAs. PLoS One. 2012;7:e50141. doi:10.1371/journal.pone.0050141. 14. Jackman DM, Miller VA, Cioffredi, et al. Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non–small cell
lung cancer patients: results of an online tumor registry of clinical trials. Clin Cancer Res. 2009;15:5267-5273. 15. Noronha V, Joshi A, Gokarn A, et al. The importance of brain metastasis in EGFR mutation positive NSCLC patients. Chemother Res
Pract. doi:10.1155/2014/856156. 16. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-247.
Find out more at EGFRevolution.com.

PAGE 24 The ASCO Post | SEPTEMBER 10, 2015PAGE 24 The ASCO Post | SEPTEMBER 10, 2015
World Congress of Psycho-oncology
International Psycho-Oncology Society and American Psychosocial Oncology Society Meet to Foster Psychosocial Oncology WorldwideBy Margot J. Fromer
The International Psycho-Oncology Society (IPOS) has partnered with
the American Psychosocial Oncology Society (APOS) for the 17th World Con-gress of Psycho-Oncology, held in late July 2015 in Washington, DC. Its theme, “From National to Global: Implementing the Standard of Psychosocial Care in On-cology,” described the awareness that psy-chosocial care has become a recognized subspecialty of cancer care in the United States and many other Western coun-tries—but regrettably this is not the case in many countries in the rest of the world, notably Africa and South America.
IPOS recently celebrated its 30th an-niversary, and its more then 7,000 pro-fessional federated members work in over 60 countries to “foster the science and practice of psychosocial oncology to improve the care of people affected by cancer worldwide,” according to Luzia Travado, PhD, President of IPOS.
APOS has 450 practitioners and sci-entists in psychiatry, psychology, social work, nursing, clergy, and patient advo-cacy in this country, said its President, Michelle Corove Fingeret, PhD.
The Lisbon DeclarationThe IPOS Human Rights Task Force
has been working since 2008 to raise awareness and support for the relevance of psychosocial care as a human right. To that end, in November 2014, at the World Congress of Psycho-Oncology in Lisbon, the IPOS Board endorsed the Lisbon Declaration: Psychosocial Cancer Care
as a Human Right. Furthermore, in 2015, IPOS added this declaration to its Inter-national Standards of Quality in Cancer Care, which now include: (1) Psychoso-cial cancer care should be recognized as a universal human right; (2) Quality cancer care must integrate the psychosocial do-main into routine care; and (3) Distress should be measured as the sixth vital sign after temperature, blood pressure, pulse, respiratory rate, and pain. This standard has been endorsed by 75 organizations worldwide and, in 2013, was codified in a revision of the World Cancer Declaration.
Writing and approving a declaration are important, but it does not necessarily translate into easing the lives of actual pa-tients with cancer, and the vast majority of them still suffer without significant help. Dr. Travado, Head of Psycho-oncology at Champalimaud Cancer Center, Lisbon, acknowledged that the bulk of the work lies ahead. “We have worked hard to influ-ence national cancer policies to include psychosocial care and have succeeded in many countries, but we need to continue to pursue this goal and take further steps to turn this standard into international global clinical practice worldwide.”
William Breitbart, MD, Chairman, Department of Psychiatry and Behav-ioral Sciences, Memorial Sloan Ketter-ing Cancer Center, New York, said that at least half of all patients with cancer experience psychosocial distress at some time during the course of their ill-ness, and it must be addressed.
The Lisbon Declaration, which follows on the heels of the World Health Organi-zation’s declaration of the Right to Health, is considered a landmark document to ad-vance the human rights of all patients with cancer, especially those in underserved areas. (The human Right to Health states that everyone has the right to the highest attainable standard of physical and mental health, which includes access to all medi-cal services, sanitation, adequate food, de-cent housing, healthy working conditions,
and a clean environment.) The Lisbon Declaration stresses the need to:• Strengthen health systems to increase
effective cancer control• Reduce the stigma of cancer and dispel
the many myths surrounding the disease• Establish universal access to screen-
ing and early detection• Improve access to services across the
cancer care continuum for all patients and all family members, who also suffer
• Ensure universal availability of pain control and distress management
• Improve the education and training of health-care professionals“These commitments oblige govern-
ments to provide access, by means of relevant legislation, to psychosocial care for all cancer patients,” said Dr. Breitbart.
Even more, policymakers, in the health and political arenas, must be made to understand that palliative care is to be included as a critical compo-nent of cancer care—as important as screening, early detection, and active treatment. And beyond understanding of and agreement with the fundamental right to psychosocial care is the neces-sity to make sufficient funds available to carry out necessary implementation.
Children With CancerAndrea Patenaude, PhD, Director
of Psychology Research and Clinical Services, Center for Cancer Genetics and Prevention, Dana-Farber Cancer Institute, and Associate Professor of Psychology at Harvard Medical School, described the human rights challenges in Africa, especially those facing chil-dren with cancer and their families.
“In many parts of the world, it is stan-dard to deny children with cancer the right to know their diagnosis, the treat-ments they will receive, their current health status, and the likely outcome of their disease. They are desperately in need of concurrent psychosocial care provided by a caring, trusted adult with whom young patients can discuss their feelings and fears about the disease, its treatment, and possible separation from family,” stated Dr. Patenaude.
Basic human rights to psychosocial care, palliative care, and pain and symptom con-trol do not exist in many parts of the world. Even worse are the countless examples of ethical mistreatment and downright cru-elty. For example, Dr. Patenaude described what has happened at the Moi Hospital
in the Rift Valley Province of Kenya. “We have reports that approximately half of the children of families that do not have health insurance [78% of parents of children with cancer in a recent study did not] are de-tained in the hospital—usually without the presence of their parents or much attention from hospital staff—after they have com-pleted treatment until the parents pay the bill. This can last for 10 days to 2 weeks. If the child has died, the corpse is held hos-tage until the hospital bill is paid.”
This is a shocking and extreme denial of rights, said Dr. Patenaude, but there are many less dramatic yet equally im-portant ethical issues in the psychoso-cial care of children with cancer. Gaps in the definition and understanding of minimal essential services that should be available to all children are wide and deep. As defined by the U.N. Con-vention on the Rights of the Child, the following items should be addressed: interventions to help children cope with the disease and its ramifications; the presence of parents during the child’s hospitalization; freedom from pain; age-appropriate explanations of what is and will be happening to the child; and interaction with peers and the opportu-nity to play and express emotions.
Cultural Beliefs, Unmet NeedsThe Middle East has among the high-
est population growth rates in the world, which brings constant dynamic chal-lenges. More than nine in ten people (93%) in the Middle East–North Africa region are Muslim. Lea Baider, PhD,
Psychosocial Care
Luzia Travado, PhD
Michelle Corove Fingeret, PhD
William Breitbart, MD
Andrea Patenaude, PhD
Lea Baider, PhD

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 25ASCOPost.com | SEPTEMBER 10, 2015 PAGE 25
World Congress of Psycho-oncology
Professor of Health Psychology and Director, Psychosocial Department, As-suta Medical Center, Tel Aviv, described what happens to many Muslim women in the Middle East when they get cancer.
“The most intimate events of life draw us into the health-care system and expose our own fundamental attitudes, moral values, and beliefs, as well as the beliefs and behaviors of those who love us and take care of us. Religion and belief systems affect psychological and physi-cal health because they influence coping strategies, social and family behavior, and attitudes about health. They can—and do, among many Muslims—lead to fear, mistrust, silence in the face of ill-ness, and the belief that cancer is a pun-ishment visited upon women by Allah.”
Dr. Baider explained that in Muslim countries, belief systems and health ritu-als appear unscientific and contradictory when compared to evidence-based West-ern medical care. A pervasive culture of collective silence influences the way women perceive their bodies and their health. Nevertheless, to pursue any pos-sible health change, it is vital to listen carefully to the traditional and religious values of women in Muslim societies. Then we could create not conflicts but in-tegration and cooperation in health care.
Daisuke Fujisawa, MD, PhD, of the Department of Neuropsychiatry and Palliative Care Center, Keio University School of Medicine, Tokyo, who spoke for Luigi Grassi, MD, Professor and Chair of Psychiatry, University of Ferrara (Italy) Section of Psychiatry, who could not at-tend the meeting, said that psychosocial oncology services are available in many parts of the world—but still too few.
For example, in a country in Africa, there are only 10 oncologists for every 40 million people, and patients often need to travel 600 kilometers or more to a cancer center where there are only rudimentary services. He described what these services can and should do: treat all patients with cancer; ensure the right to enjoy the highest attainable standard of physical and mental health; and provide psychosocial services and interventions in the event of illness.
From Principles to Practical CareStephen Connor, PhD, Senior Fel-
low, Worldwide Hospice Palliative Care Alliance (WHPCA) and member of the World Health Organization Ad Hoc Advisory Group on Palliative and Long-Term Care, said that human rights ad-vocacy must move from statements of principles and goals to practical clinical implementation. Efforts to advance psy-chosocial cancer care must be seen as a
human rights issue and therefore acted upon rather than just talked about.
Over a million people die every week around the world, he said, and 80% of them have highly restricted access to or no pain relief. An estimated 40 million people worldwide are in need of pallia-tive care at any one time, but less than 10% receive it. More than 75% of all countries provide little or no palliative care to patients with cancer.
“The need for palliative care has nev-er been greater and is increasing rapidly because the world’s population is aging, and the incidence of cancer and other noncommunicable diseases is increas-ing. This is especially true outside North America, Europe, and Australia. The un-met need is enormous,” said Dr. Connor.
“WHPCA’s advocacy began with the Seoul Declaration in 2005, which called for access to palliative care as a human right. The group emerged from the Seoul meeting of Asian hospice networks as an alliance of national and regional hospice and palliative care associations. A series of global summits and further declara-tions on palliative care were held and cre-ated a strong international association.”
WHPCA believes that no one with a life-limiting condition should live and die with unnecessary pain and distress due to lack of access to quality care. Its objectives include being a strong, sustainable, and efficient global membership organization exclusively focused on hospice and pal-liative care; being an effective and power-ful global voice and advocate for hospice and palliative care; providing integration of palliative care into country health sys-tems, drawing on and supporting local technical expertise; and assisting national associations to integrate palliative care into national undergraduate and postgraduate health science curricula and health and so-cial care workers’ training programs.
Goals of Palliative CareCokie Roberts, a journalist for
National Public Radio and a survi-vor of breast cancer, introduced the plenary session panelists and asked them to describe their ideas about
what is important with regard to pal-liative care and how it can be furthered. Jamie Von Roenn, MD, ASCO Senior Director of Education, Science, and Professional Development, said, “We need to educate people, we need to talk about death and dying, and we need to develop a model of integrated care.”
Small cancer centers and community
oncology practices provide almost no pal-liative care at all—and that’s where 80% of cancer is treated in the United States. Moreover, because do-not-resuscitate orders and end-of-life care are regulated by states, there is no national uniformity about how such care is to be provided—and how much choice patients have about the way they approach death.
Joanne Wolfe, MD, Chief, Divi-sion of Pediatric Palliative Care, De-partment of Psychosocial Oncology and Palliative Care, Dana-Farber Can-cer Institute, and Director of Palliative Care at Boston Children’s Hospital, described the Pediatric Advanced Care Team (PACT) that helps children and their parents through serious illness. “All our young patients receive inter-disciplinary care (primary palliative care) early in their treatment, which is critically important when a child is di-agnosed with cancer. The care team is ‘wrapped around’ the child and his or her family, so they feel protected and know they have knowledgeable and caring clinicians to whom to turn. Pal-liative care subspecialty consultation is reserved for more complicated cases such as the need for intensive symp-tom management.”
The Dana-Farber PACT team consists of clinicians who specialize in palliative care, symptom management, complemen-
tary medicine, spirituality-religion, social work, and community resources. These professionals consult with the patient, his or her parents, siblings, and the pri-mary care team to ease pain and manage symptoms and side effects; foster commu-nication among families and health-care providers; and coordinate inpatient, out-patient, and home-care services.
Dr. Wolfe said that caregivers at Dana-Farber have found social media very use-ful for children with cancer. “This is how they are used to communicating with one another, and we can use these me-dia to educate people and to disseminate whatever messages we feel will help.”
Stein Kaasa, MD, Professor of Medi-cine, Institute of Cancer Research and Molecular Medicine, University Hospital of Trondheim (Norway), said that pro-viders should be encouraging patients to think about palliative care as early in their treatment as possible, rather than shying away from discussions of death, dying, and hospice care. “And there’s no reason to use euphemisms or call palliation anything other than it is. It is a good thing, so let’s not change its name,” Dr. Kaasa added.
In Norway, palliative care has its own diagnosis-related group and is integrated into the billing system in most institu-tions. In this country, said Dr. Wolfe, Medicare and most private insurers do not reimburse for advanced planning conversations, but they soon will. “It’s on the horizon,” he noted. In the United Kingdom, spending on palliative care is “hugely varied,” said Irene J. Higginson, OBE, Director, Cicely Saunders In-stitute, Professor, King’s College Lon-don—the world’s first purpose-built in-stitute for palliative care.
Dr. Higginson said that the purpose of palliative care is first to determine what patients and families need. “There are 13 or 14 common physical symptoms that we can help with, as well as innumerable psychosocial ones. This requires critical evaluation, and we must do it early. Even though we start it earlier in the UK than you do here, people still come to pallia-tive care far too late in the course of their illness,” she explained.
Medical students at her institution spent time at a hospice, which made them more likely to recommend palliative care when they began practice. “In the United States, palliative care is being integrated into the medical curriculum, but it’s very slow,” concluded Dr. Von Roenn. She did note, however, that as it appears in medical schools, it follows in clinical practice. n
Disclosure: Drs. Travado, Breitbart, Patenaude, Baider, Fujisawa, Grassi, Connor, Von Roenn, Wolfe, Kaasa, and Higginson reported no potential conflicts of interest.
Jamie Von Roenn, MD
The need for palliative care has never been greater and is increasing rapidly because the world’s population is aging, and the incidence of cancer and other noncommunicable diseases is increasing.
—Stephen Connor, PhD

PAGE 26 The ASCO Post | SEPTEMBER 10, 2015PAGE 26 The ASCO Post | SEPTEMBER 10, 2015
World Congress of Psycho-oncology
Suicide After Cancer: Understanding the Challenges Across the Treatment Trajectory By Margot J. Fromer
Suicidal thoughts and impulses are among the most challenging symp-
toms in patients with cancer, and they may occur both during and after treatment. It has long been known that a cancer diag-nosis carries an increased risk for suicide, but the problem is not widely addressed. Suicide is one of the few remaining taboo topics of conversation among the general public, and even health professionals are unprepared to understand and respond to their patients’ suicidal thoughts. The topic of suicide after cancer was featured at the 2015 World Congress of Psycho-Oncol-ogy, held in late July in Washington, DC.
Caring for suicidal patients is de-manding and requires risk assessment, possibly urgent mental health care, knowledge of ways to manage symp-toms, clarity about personal and pro-fessional ethics, willingness to grant patient autonomy, and recognition of legal liability. Moreover, most suicide research has focused on people with psychiatric rather than physical condi-tions, making it difficult to know how well it fits with patients with cancer.
Patients With Cancer May Think About It
Andrew Roth, MD, a psychiatrist at Memorial Sloan Kettering Cancer Center, New York, said that he has seen many patients with cancer who, in a va-riety of ways, express the thought: “If I’m going to die anyway, what difference does it make if I kill myself?” It is best to help patients derive as much meaning and pleasure from life despite the can-cer, which may alleviate ideas and plans for suicide, he explained.
The risk for suicide is definitely el-evated in patients with cancer, said Dr. Roth. The more comorbidities a patient has, the higher the risk, often double that of the general population.
“We need to integrate principles and practices from the psychiatric setting, es-pecially suicide screening—and especial-ly early after initial cancer diagnosis, when the rate is higher. We want to know if a patient has a history of suicide attempts, poorly controlled pain, a poor prognosis, and physical impairment and/or loss of mobility. Is the patient elderly? Has there been a history of depression?”
Suicide ideation is not necessarily a precursor to the act itself, but it should be talked about. Dr. Roth also said it is impor-tant to ask patients with cancer whether
they have been or are now thinking about killing themselves and, if so, whether they have a plan in mind. “Asking about sui-cidal thoughts does not put new ideas into
someone’s mind, having never been there before. Many cancer patients think about whether it would be better not to be alive at some time or another after diagnosis.”
Ways to Look at Suicidal Thoughts
Keith Wilson, PhD, Associate Scien-tist, Ottawa Hospital Research Institute and Staff Psychologist, The Ottawa Hospi-tal Rehabilitation Centre, Canada, report-ed that palliative care studies have looked at suicidal concerns in three ways: a desire for death, suicidal ideation, and interest in receiving physician-assisted suicide.
All three were addressed in the Ca-nadian National Palliative Care Survey of 381 patients with advanced cancer. Participants were asked their thoughts about suicide, and they were interviewed to determine the presence of common mental disorders. Thirty percent ac-knowledged a transient desire for death, but only 12% had a genuine desire to die. Some degree of suicidal ideation was re-ported by 16%, but only 4% thought of it often or knew how they would do it. Close to 6% would have requested as-sisted suicide at the time of the interview.
“There was overlap across the catego-ries but also differences,” said Dr. Wilson. The prevalence of depression and/or
anxiety among participants with a desire for death, serious suicidal ideation, and an interest in receiving assistance with suicide was 52.2%, 53.3%, and 40.1%, respectively.
The study concluded that occasional wishes for death are common among patients with advanced cancer, but more serious concerns are often asso-ciated with depression and/or anxiety. Therefore, said Dr. Wilson, “Expression of a desire for death or suicide by a ter-minally ill patient should raise a suspi-cion about mental health problems but is not in itself clearly indicative of them.”
Identifying Survivors at RiskSuicide remains a risk for survivors,
sometimes for years afterward, even if cancer has been cured or is in remission. How can this be? Shouldn’t making it through a fatal disease confer a love of life, a sense of profound gratitude?
Apparently not, said Christopher Recklitis, PhD, Associate Professor of Pediatrics, Dana-Farber Cancer Institute and Harvard Medical School. He looked at data from cohort studies of long-term cancer survivors, and compared with the general population, he found a significant increase in suicide ideation and completed suicide. Although the risk decreases as time passes, it depends largely on the site of the cancer, gender, age, and permanent or long-term handicaps and health problems.
Newer studies have examined the rela-tionship of survivor health outcomes with suicidality and revealed that risk is associ-
ated with physical function and the late effects of treatment, the latter of which are particularly prevalent in survivors of child-hood cancer. Suicidal ideation is driven by physical as well as emotional health, but it is interesting to note that 30% to 45% of survivors with suicidal ideation report no significant symptoms of depression, so screening for emotional distress alone will not always identify suicide risk.
Even if you do not have cancer any longer, if you feel bad much of the time or have major handicapping after-effects, life may not be enjoyable enough to keep going. In fact, said Dr. Recklitis, “Suicide ideation increases with a diminution of self-assessment of overall health, as well as an objective assessment of such. The risk factor is 30% to 50%, and more than half of those people do not have classic symp-toms of clinical depression.” Therefore, he said, it is imperative to develop ways to identify survivors at high risk.
Donald Rosenstein, MD, Professor of Psychiatry and Director, Comprehen-sive Cancer Support Program, Univer-sity of North Carolina, Chapel Hill, said that even though many patients with cancer and survivors are not clinically de-pressed as they think about suicide, the Patient Health Questionnaire (PHQ-9) can be a useful assessment tool.
It is a nine-item scale that measures the presence and severity of symptoms of depression. The tool is short and can be administered in person, over the phone, or self-administered. Available in 30 languages, it is well validated and docu-mented. The first eight questions are the ones usually asked of people thought to be depressed, and the last question is about suicide ideation. According to Dr. Rosen-stein, however, that question “has been shown to result in fairly high false-positive rates, and so, is not a good choice for broad screening efforts within oncology.” n
Disclosure: Drs. Roth, Wilson, Recklitis, and Rosenstein reported no potential conflicts of interest.
Supportive Care
Asking about suicidal thoughts does not put new ideas into someone’s mind, having never been there before. Many cancer patients think about whether it would be better not to be alive at some time or another after diagnosis.
—Andrew Roth, MD
Keith Wilson, PhD
Christopher Recklitis, PhD
Donald Rosenstein, MD

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 5 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

The Merck Access Program for KEYTRUDA for Injection
Visitmerckaccessprogram-keytruda.com
Call855-257-3932
• Speak with a dedicated representative Monday to Friday between 8 AM and 8 PM ET.
• Ask to be contacted by a field reimbursement associate.
OR
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14
32946_BUILD.indd 4 12/9/14 4:21 PM32946_01_p.indd 3 12/10/14 12:57 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 27ASCOPost.com | SEPTEMBER 10, 2015 PAGE 27
World Congress of Psycho-oncology
Coping With Aging and Cancer: Psychosocial Factors and Geriatric-Specific InterventionsBy Margot J. Fromer
“You can’t control the wind, but you can adjust your sails,” said
Mindy Greenstein, PhD, consulting psychologist and author, to begin her talk at the 2015 World Congress of Psycho-Oncology, held in July in Wash-ington, DC. The sense of this proverb pervaded the entire session on cancer and aging.
Elders are the fastest-growing popula-tion of cancer patients with cancer, said Dr. Greenstein, and their needs are both similar to and different from younger ones. Older patients with cancer are in the midst of a “double whammy” in that they face a life-threatening illness within the context of life’s shortening.
Cancer and Aging: Similar yet Different
Similarities between cancer and the negative aspects of aging include coping with the sword of Damocles while liv-ing a normal life; uncertainty and vul-nerability; learning to make the most of the present; focusing on what is most meaningful in life; and reinforcing one’s most adaptive character strengths and virtues. The last aspect, said Dr. Green-stein, includes transcendence, temper-ance, wisdom, humanity, courage, and a sense of social justice. “These are char-acteristics that can help one get through the cancer crisis, but they also can serve as a bridge to the younger generation.”
There are two major differences be-tween cancer and aging. First, young pa-tients with cancer know how different their experience is from that of others their age, whereas older patients with cancer see others like them all around. Second, older patients have had long life experiences dealing with crises of one sort or another and can use what they have learned to deal with cancer. By and large, younger patients do not.
In many ways, said Dr. Greenstein,
coping with cancer is a lot like coping with aging, so clinicians can teach pa-tients to use skills derived from the lat-ter to get them through the former.
Silver Tsunami‘Silver tsunami’ is the term being
used to describe the crisis in cancer care, said Jimmie Holland, MD, Wayne E. Chapman Chair in Psychiatric Oncol-ogy, Memorial Sloan Kettering Cancer Center (Memorial), New York. It refers to people with the highest incidence of cancer (59% of all patients) and for whom there are often the fewest psy-chosocial resources.
Dr. Holland described two psycho-social resources she developed: Can-cer and Aging Reflections of Elders (CARE) and a reading group known as the Vintage Readers Book Club.
The latter began as a group of two—Dr. Holland and her granddaughter—who decided to start a book club reading the Harvard Classics. When her grand-daughter went off to college, she suggest-ed continuing the club with some older patients with cancer, most of whom were home alone, isolated, lonely, and bored. It began as an outgrowth of the Aging and Cancer Group at Memorial but quickly became much more popular.
“We meet once a month over lunch to read and discuss the world’s classic literature, with special emphasis on the issues and problems of older people. For example, we read Ben Franklin’s thoughts on happiness and Cicero’s essay, Old Age. One of the best things about these discussions is the recogni-tion of the universality of human ex-perience and how people learn to see themselves in a broader context.”
Dr. Holland continued: “Many types of cancer are now treated as a chronic illness, and for older people, once treat-ment is finished and they accept that they’re going to survive—at least for a while—it’s just another one of the dis-tressing vagaries of age.”
Older adults in general are often de-pressed (10% to 20% of them), and 5% to 6% suffer from anxiety disorders. Older patients with cancer report depression at a rate of 17% to 25% and anxiety at a rate of 21% to 28%. Although these numbers are only estimates, it is clear that getting older and having cancer at the same time is not much fun.
Help Is AvailableThere are, however, interventions
that can help to improve quality of life, including physical therapy, cognitive behavioral training, education about problems such as symptoms and side effects, spiritual guidance, and assis-tance with financial resources. And there is CARE, a five-session tele-phone psychotherapy program that helps people cope with the double whammy of aging and cancer.
The pilot study consisted of 61 pa-tients with cancer (prostate, breast, lym-phoma, lung, or gynecologic) who were
recruited by letter. They had to be 70 years of age or older, 6 months post diagnosis, and either in active treatment or 6 months past treatment. They also were required to have high distress and/or anxiety levels. Master’s-level counselors conducted the telephone sessions, detailed as follows:• Session 1 is an introduction and over-
view in which the therapist intro-duces the program and listens to the patient’s story.
• Session 2 deals with coping with loss-es and facing the unknowns of cancer and aging, as well as with the com-bined problems of illness and aging, present and future fears, concerns, and worries.
• Session 3 addresses loneliness, the di-minished socializing that arises from both aging and cancer, and the stigma attached to aging in our society.
• Session 4 attempts to make peace with one’s life and acquire wisdom by coming to terms with one’s chang-ing sense of self, contributing to the greater good, and passing on what one has learned.
• Session 5 consists of reflection and review.After completing the pilot, people’s
depression and anxiety decreased, as did loneliness. The ability to cope and plan ahead improved, as did finding meaning in life. Dr. Holland concluded: “Now that the program is up and run-ning, we may add intervention sessions, and we certainly need to do a larger and longer study, but the CARE program has a definitive role in improving the lives of older people with cancer.” n
Disclosure: Drs. Greenstein and Holland reported no potential conflicts of interest.
More on Psychosocial Oncology and Aging
For more on Aging, don’t miss the captivating new book,
Lighter as We Go: Virtues, Charac-ter Strengths, and Aging, by Mindy Greenstein, PhD, and Jimmie Holland, MD, both of Memorial Sloan Kettering Cancer Center in New York. Available on Amazon and at other booksellers.
For more from the World Con-gress, don’t miss The ASCO Post Newsreels, video interviews with faculty recorded during the July 2015 World Congress of Psycho-oncology in Washington, DC.
Visit http://video.ascopost.com/ n
Geriatric Oncology
Now that the program is up and running, we may add intervention sessions, and we certainly need to do a larger and longer study, but the CARE program has a definitive role in improving the lives of older people with cancer.
—Jimmie Holland, MD
Mindy Greenstein, PhD

PAGE 28 The ASCO Post | SEPTEMBER 10, 2015
In the Clinic
Brentuximab Vedotin for Consolidation Therapy in High-Risk Hodgkin LymphomaBy Matthew Stenger
On August 17, 2015, brentuximab vedotin (Adcetris) was approved
for consolidation treatment post au-tologous hematopoietic stem cell transplantation in patients with classic Hodgkin lymphoma who are at high risk of relapse or progression.1,2
Supporting Efficacy DataApproval was based on a double-blind
phase III trial (AETHERA) showing prolonged progression-free survival with brentuximab vedotin vs placebo on inde-pendent review.2,3 A total of 327 evaluable patients defined as high risk (response to front-line therapy defined as refractory, re-lapsed disease occurring within 12 months or relapsed disease with extranodal in-volvement) were randomized to receive brentuximab vedotin 1.8 mg/kg given IV over 30 minutes (n = 167) or placebo (n = 160) every 3 weeks for up to 16 cycles. All patients received best supportive care.
Patients were required to have had complete or partial response or stable dis-ease during the most recent salvage thera-py. Patients had a median age of 32 years (range = 18–76 years), most were male (53%) and white (94%), and the median number of prior systemic therapies (ex-cluding autologous hematopoietic stem cell transplantation) was 2 (range = 2–8).
Median follow-up for progression-free survival was 22 months (range = 0–49 months). Median progression-free sur-vival was 42.9+ months (95% confidence interval [CI] = 30.4–42.9+ months) in the brentuximab vedotin group vs 24.1 months (95% CI = 11.5 to not estimable) in the placebo group (stratified hazard ratio = 0.57, P = .001). At the time of the progression-free survival analysis, interim
analysis of overall survival showed no sig-nificant difference between the groups. Patients in the placebo group could re-ceive brentuximab vedotin in a separate trial after disease progression.
How It WorksBrentuximab vedotin is an antibody-
drug conjugate consisting of a chimeric IgG1 antibody directed against CD30 and the small-molecule microtubule-disrupting agent MMAE (monomethyl auristatin E), which is covalently at-tached to the antibody via a linker. Pre-clinical findings indicated that binding of the conjugate to CD30-expressing cells is followed by internalization of the conjugate-CD30 complex and release of MMAE via proteolytic cleavage. Binding of MMAE to tubulin disrupts the micro-tubule network within the cell, inducing cell-cycle arrest and apoptosis.
How It Is GivenThe recommended dosage for bren-
tuximab vedotin as consolidation thera-py post autologous hematopoietic stem cell transplantation is 1.8 mg/kg up to 180 mg given IV over 30 minutes every 3 weeks. Treatment should be initiated within 4 to 6 weeks after autologous hematopoietic stem cell transplanta-tion or upon recovery from transplan-tation and should be continued until a maximum of 16 cycles, disease progres-sion, or unacceptable toxicity. Infusion should be interrupted for infusion reac-tion and discontinued for anaphylaxis.
The starting dose of brentuximab does not need to be changed in patients with mild or moderate renal impair-ment, but the drug should be avoided in patients with severe renal impair-ment. The recommended starting dose is 1.2 mg/kg up to 120 mg in patients with mild hepatic impairment; the drug should be avoided in patients with moderate or severe hepatic impairment.
Treatment should be held for new or worsening grade 2 or 3 neuropathy un-til recovery to grade 1 or baseline and then restarted at 1.2 mg/kg; treatment should be discontinued for grade 4 pe-ripheral neuropathy. Treatment should be held for grade 3 or 4 neutropenia until resolution to baseline or grade ≤ 2. Granulocyte colony-stimulating factor (G-CSF) prophylaxis should be con-sidered for subsequent cycles. If grade 4 neutropenia recurs despite G-CSF
prophylaxis, treatment discontinuation or dose reduction to 1.2 mg/kg should be considered.
MMAE is primarily metabolized by CYP3A. Coadministration of brentux-imab vedotin with the strong CYP3A4 inhibitor ketoconazole increased exposure to MMAE by approximately 34%; patients who are receiving strong CYP3A4 inhibi-tors (eg, ketoconazole, clarithromycin, in-dinavir [Crixivan], ritonavir [Norvir], bo-ceprevir [Victrelis]) concomitantly with brentuximab vedotin should be closely monitored for adverse reactions.
Coadministration of brentuximab with the strong CYP3A4 inducer ri-fampin reduced exposure to MMAE by approximately 46%. Coadministra-tion with P-glycoprotein inhibitors (eg, ketoconazole, cyclosporin A, ritonavir, verapamil) may increase exposure to MMAE, and patients receiving con-comitant treatment should be closely monitored for adverse reactions.
Safety ProfileIn the phase III trial, the most com-
mon adverse events of any grade in the brentuximab vedotin group were neutro-penia (78% vs 34% in the placebo group), peripheral sensory neuropathy (56% vs 16%), thrombocytopenia (41% vs 20%), anemia (27% vs 19%), and upper respi-ratory tract infection (26% vs 23%). The most common grade 3 or 4 adverse events were neutropenia (39% vs 10%), periph-eral sensory neuropathy (10% vs 1%), and peripheral motor neuropathy (6% vs 1%). Infusion-related reactions occurred in 15% (grade 3 in 1.7%) vs 2% of patients. Pulmo-nary toxicity occurred in 5% vs 3%.
Overall, 67% of patients receiving bren-tuximab vedotin had neuropathy of any grade. The median time to first onset was 14 weeks for any grade neuropathy, 27 weeks for grade 2 neuropathy, and 34 weeks for grade 3 neuropathy. The median time from onset to resolution or improvement was 23 weeks for any grade, 24 weeks for grade 2, and 25 weeks for grade 3. Complete reso-
lution occurred in 59% of patients; of 41% with residual neuropathy, 26% had partial improvement at last evaluation.
Serious adverse events occurred in 25% of the brentuximab vedotin group, with the most common being pneumo-nia (4%), pyrexia (4%), vomiting (3%), nausea (2%), hepatotoxicity (2%), and peripheral sensory neuropathy (2%). The most common adverse events lead-ing to dose delays were neutropenia (22%), peripheral sensory neuropathy (16%), upper respiratory tract infection (6%), and peripheral motor neuropathy (6%). Adverse events led to treatment discontinuation in 32% of patients, with the most common causes being periph-eral sensory neuropathy (14%), pe-ripheral motor neuropathy (7%), acute respiratory distress syndrome (1%), par-esthesia (1%), and vomiting (1%).
Brentuximab vedotin carries a boxed warning for progressive multifocal leu-koencephalopathy, including fatality. It also carries warnings/precautions for peripheral neuropathy, anaphylaxis and infusion reactions, hematologic toxici-ties, serious infections and opportunistic infections, tumor lysis syndrome, hepa-totoxicity, pulmonary toxicity, serious dermatologic reactions, and embryo-fe-tal toxicity. Complete blood cell counts should be monitored before each dose. Liver enzymes and bilirubin should be routinely monitored. The drug is contra-indicated with concomitant bleomycin due to pulmonary toxicity. n
References1. U.S. Food and Drug Administration:
Brentuximab vedotin. Available at www.fda.gov. Accessed August 27, 2015.
2. Adcetris (brentuximab vedotin) for injection prescribing information, Seat-tle Genetics, Inc, August 2015. Available at http://www.accessdata.fda.gov/drug-satfda_docs/label/2015/125388s080lbl.pdf. Accessed August 27, 2015.
3. Moskowitz CH, et al: Lancet 385:1853-1862, 2015.
Brentuximab Vedotin in High-Risk Hodgkin Lymphoma
■ Brentuximab vedotin (Adcetris) has been approved for consolidation treatment post autologous hematopoietic stem cell transplantation in patients with classic Hodgkin lymphoma who are at high risk of relapse or progression.
■ The recommended dosage for brentuximab vedotin as consolidation therapy post autologous hematopoietic stem cell transplantation is 1.8 mg/kg up to 180 mg given IV over 30 minutes every 3 weeks.
Hematology
In the Clinic provides overviews of novel oncology agents, addressing indications, mechanisms, admin-istration recommendations, safety profiles, and other essential infor-mation needed for the appropriate clinical use of these drugs.
OF NOTEBrentuximab vedotin carries a boxed warning for progressive mul-tifocal leukoencephalopathy, in-cluding fatality.

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 35
National Cancer Policy Forum
New Techniques in Oncologic Surgery and Radiology: Some Worth the Expense, Some Not So MuchBy Margot J. Fromer
In recent years, patients with cancer have had the benefit of much high
technology: proton-beam radiotherapy, intensity-modulated radiation therapy, various minimally invasive surgery techniques, and robots in the operating room. They all receive hype in the pro-fessional and public press, and patients and physicians demand their use. They are all more expensive—many wildly so—than the techniques they replace. In light of this trend, a recent National Cancer Policy Forum workshop titled “Appropriate Use of Advanced Tech-nologies for Radiation Therapy and Surgery in Oncology” sought to exam-ine the clinical benefits and compara-tive effectiveness of emerging advanced technologies for cancer treatment in ra-diotherapy and surgery.
“The increased cost of novel treat-ments without adequate assessment on patient outcomes warrants further dis-cussion,” said Ralph R. Weichselbaum, MD, Chair of Radiation and Cellular Oncology at the University of Chicago Medicine and Planning Committee Co-Chair of the workshop. “We need a criti-cal assessment of the clinical [efficacy] and cost-effectiveness of emerging tech-nologies in radiation and surgery.”
In addition, new technology of-ten dominates discussions of value in health care, said Ya-Chen Tina Shih, PhD, Planning Committee Co-Chair. The goal, she said, is to find the best treatments that patients and the health-care system can afford. However, evalu-
ating the clinical benefit of new devices is more difficult than testing new drugs due to the difference in regulatory over-sights. Unlike new drugs that require clinical trials for U.S. Food and Drug Administration approval, devices often proliferate the market without evidence established in clinical trials.
Advances in RadiotherapyRadiation technology has been bur-
geoning of late, with intensity-modu-lated radiation therapy, various forms of stereotactic radiotherapy, particle beam therapy, and proton-beam radio-therapy. They all have the potential for better clinical outcomes and reduced treatment-related morbidity, but they are all significantly more expensive than older techniques. Moreover, some do not necessarily confer extra benefit.
Some of the “sexy” new machinery may be better than its older counter-parts, and although there is some evi-dence to that effect, there is insufficient comparative research. This is especially troubling, because about half of all pa-tients with cancer receive radiotherapy.
“There are three basic principles un-derlying radiotherapy,” said Anthony Zietman, MD, Associate Director, Har-vard Radiation Oncology and Director, Genitourinary Service, Massachusetts General Hospital: direct radiation-relat-ed complications do not occur in nonir-radiated tissue; irradiating normal tissue does not benefit patients; and the thera-peutic ratio can be optimized by maxi-
mizing the radiation dose to the tumor and minimizing the normal tissue dose.
Although these principles appear to clearly obvious, until recently, radiation oncologists have not been able to fully protect normal tissue, even as they did a good job irradiating tumors. Now there is proton-beam radiotherapy, the “point of the arrow,” as Dr. Zietman called it.
Traditional x-rays (photons) attenu-ate progressively with the depth of tissue, but they continue to deposit radiation beyond the tumor target, thus bathing normal tissue in unwanted radiation. However, proton therapy has a precisely defined range with no radiation deliv-ered beyond a specified point. Hence, there is no damage to normal tissue.
Although using proton-beam radio-therapy may seem like a no-brainer in theory, it may not necessarily be the case in practice. The technology is compli-cated, the machinery is enormous and tremendously heavy (requiring its own real estate), and the cost ranges in the hundreds of millions for its initial instal-lation and many millions each year for its use and maintenance. Very few cancer centers have the required resources.
Proton beams are generated by a cyclotron, which accelerates hydrogen protons to two-thirds the speed of light. The hydrogen protons are then shot through and focused by magnets toward a gantry that can rotate 360° around the patient. Inside the gantry is a nozzle that weighs 21,000 pounds. This nozzle guides the beam to the patient who, in schematic depictions, appears no bigger than a bug amid the gigantic machinery.
There are currently 14 of these be-hemoths in the United States, 11 are under construction, and more are in the offing.
Proton-beam radiotherapy has clini-cal advantages, said Dr. Zietman. The integral dose of radiation is smaller, and there is no exit dose. Therefore, it im-proves acute treatment tolerance, allows
delivery of higher doses of radiation, and can be integrated with systemic chemo-therapy. It also reduces late effects.
It is thought that children have the most to gain from proton therapy be-cause of the profoundly negative effects of any type of radiation on growth and development as well as a substantial risk of radiation-induced cancers over the remainder of their lives. There is, however, no classic comparative effec-tiveness research to prove this claims because most physicians would be uncomfortable randomizing their pe-diatric patients is such a trial in which the conventional radiation arm is very likely to prove inferior.
Adult indications for protons in-clude tumors of the skull base, eyes, spine, and sacrum. “It’s not so much that the evidence is strong for these in-dications but rather that the alternatives are unacceptable,” said Dr. Zietman.
Randomized controlled trials of proton-beam radiotherapy in both children and adults are problematic. There are ethical objections; trials need to be very large and very long, and the advantages are small and ar-rive late; and the initial investment for the technology is huge—perhaps too big for a treatment that may have limited advantages. Despite these con-siderations, Dr. Zietman said proton-beam radiotherapy is here to stay for a number of reasons. The treatment can be accurate, effective, and perhaps the best choice for pediatric solid tu-mors and some adult tumors. It rides the current minimally invasive popu-larity wave. Its technical and biologic advances create a “personalized” dose. New centers are quickly being estab-lished around the world.
Money Still MattersEven if clinical research indicated
that proton-beam therapy is an un-
Technology
Ralph R. Weichselbaum, MD
Anthony Zietman, MD
continued on page 36
Visit The ASCO Post website at ASCOPost.com

PAGE 36 The ASCO Post | SEPTEMBER 10, 2015
National Cancer Policy Forum
equivocally good thing, cost remains a problem. James Yu, MD, Associate Professor of Therapeutic Radiology, Yale University, said that cost has always been problematic, just not as dramatic as it is now. He noted that in 1979, the National Research Council reported expenditures of $4 billion for new tech-nology, and no one knew whether it did any good. That sum now seems down-right paltry.
The personal “financial toxicity” of health care is staggering now, he said, and the burden affects the quality of life of patients with cancer, so even though a treatment is physically safe, it can have an ultimately negative af-fect. “Therefore, rejecting low value or overly expensive treatments should be a top priority.”
What about Medicare, which pays for most of it? Reimbursement for in-tensity-modulated radiation therapy and proton-beam radiotherapy has surpassed older technologies, and the cost is passed on to taxpayers and patients. For example, in prostate cancer in 2005, three-dimensional ra-
diotherapy costs $20,588; intensity-modulated radiation therapy costs $31,574; and in 2008 the price tag for proton-beam radiotherapy is $13,753 more than intensity-modulated ra-diation therapy. In 2005, intensity-modulated radiation therapy cost Medicare $282 million more than three-dimensional radiotherapy for prostate cancer, though reimburse-ment has steadily declined over the past decade. In its place, proton-beam radiotherapy has the potential to cost Medicare hundreds of mil-lions of dollars more than intensity-modulated radiation therapy —for prostate cancer alone.
Dr. Yu said that although about 50% of all patients with cancer require radiotherapy, only 1.6% of all National Institutes of Health cancer funding went to radiation research in 2013. This amount should be significantly increased, he added. Moreover, insur-ance coverage with development of ev-idence is not applied evenly; therefore, all patients undergoing treatment with a new technology should be enrolled in a study.
Deciding on coverage of any de-vice or technology requires review of high-quality studies that provide direct evidence of positive outcomes. The problem is that many, probably most, have not benefited from such stud-ies. Although there is intense public interest in covering high-tech devices, published evidence has been sugges-tive of benefit but insufficient. In addi-tion, clinical trials usually do not have enough subjects representative of the Medicare population.
Advances in SurgeryThe challenge in surgery, said David
C. Miller, MD, Associate Professor of Urology, University of Michigan Medical School, Ann Arbor, is to enhance acces-sibility to minimally invasive procedures.
One such avenue is the popular, but not necessarily always better, da Vinci Surgical System for robotic-assisted laparoscopic surgery, cleared by the U.S. Food and Drug Administration in 2000. It carries a number of advantages:• Better visualization of the operative
field: three-dimensional, high-defini-tion, and 10x magnification.
• Greater flexibility of the robot arm’s “wrist” than a natural wrist.
• More precise movements and damp-ening of hand tremors.
• Improved ergonomics.• Facilitation of laparoscopic surgery,
which carries the advantages of small-er incisions, shorter hospital stay, and easier short-term recovery.The da Vinci Surgical System en-
joyed widespread and almost immedi-ate success. For example, of the 111 surgeons in Michigan who performed 3,730 prostatectomies between March 2012 and June 2015, 93% used this sys-tem over open surgery. The robot is also used for cancer of the bladder, kidneys,
uterus, cervix, ovaries, colon, pancreas, thyroid, lung, esophagus, tonsil, and tongue base.
It’s popular, but is it really better? No one knows for sure because al-though there are observational studies about its advantages, except for cys-tectomy, rectal excision, and radical prostatectomy, there are few random-ized controlled trials. And there is no obvious reason to believe it is better for cancer control or that it provides better functional outcomes such as uri-nary control and erectile function, said Dr. Miller. In addition, current data are somewhat mixed regarding the ben-efits of robotic surgery for functional outcomes such as urinary control and erectile function.
Like proton-beam radiotherapy, the da Vinci Surgical System is often more expensive than open surgery. Each unit costs more than $1 million; annual service costs at least $150,000; the instruments are disposable and cost about $2,000 per case. At least one study estimated that this adds up to an average of $3,200 more per case than open surgery.
Is it worth it? “We don’t know yet,” said Dr. Miller. “The da Vinci has defi-nite benefits and some unintended ad-verse consequences, including higher cost. Comparative clinical [efficacy] and cost-effectiveness has not been fully defined, so we need to make a greater effort to improve its perfor-mance and outcomes.” n
Disclosure: Drs. Weichselbaum, Shih, Zietman, and Miller reported no potential conflicts of interest. Dr. Yu has received research funding from 21st Century Oncology LLC.
David C. Miller, MD
James Yu, MD
New Techniques in Surgery and Radiologycontinued from page 35
How Do You Speak With Your Patients About Their Level of Distress?
The ASCO Post Newsreels is pleased to present Jimmie C. Holland, MD, of Memorial Sloan Kettering Cancer Center, and Tammy A. Schuler, PhD, of the Association for Behavioral and Cognitive Therapies. Drs. Holland and Schuler demonstrate a dialogue between a clinician and a recently diagnosed cancer patient whose distress was discovered by the Distress Thermometer.
Watch this short video recorded during the 2015 World Congress on Psycho-oncology, July 2015, Washington, DC.
Visit http://bit.ly/1UOBUUp for this demonstration and more on The ASCO Post Newsreels.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 37
Journal Spotlight
received lenalidomide at 25 mg on days 1 to 21 and oral dexamethasone at 40 mg on days 1, 8, 15, and 22.
The coprimary endpoints were progression-free survival and objective response rate. The primary analysis of progression-free survival used indepen-dent review committee assessment of tumor response and censoring of data for patients who received subsequent myeloma therapy or missed assess-ments. Intention-to-treat analysis was performed as a supportive evaluation.
The elotuzumab and control groups were generally balanced for age (me-dian, 67 and 66 years), sex (60% and 59% male), Eastern Cooperative On-cology Group performance status (0 for 50% and 45%, 1 for 43% and 45%, 2 for 8% and 11%), race/ethnicity (82% and 86% white, 10% Asian in both), presence of del(17p) (32% in both), presence of t(4:14) (9% and 10%), and stage (I in 44% and 42%, II in 32% in both, III in 21% in both).
The study groups were also balanced for number of previous treatments (me-dian of 2 in both, ≥ 3 in 16% in both), previous stem cell transplantation (52% and 57%), and previous therapies (bort-ezomib [Velcade] in 68% and 71%, mel-
phalan in 69% and 61%, thalidomide [Thalomid] in 48% in both, lenalido-mide in 5% and 6%). Response to the most recent line of therapy was refrac-tory for 35% in both groups, including 22% and 21% refractory to bortezomib and 9% and 11% refractory to thalido-mide, and relapse in 65% in both.
Progression-Free SurvivalAfter a median follow-up of 24.5
months, median progression-free sur-vival in the primary analysis was 19.4 months (95% confidence interval [CI] = 16.6–22.2 months) in the elotuzu-mab group vs 14.9 months (95% CI = 12.1–17.2 months) in the control group (hazard ratio [HR] = 0.70, P < .001). The outcome was similar in the inten-
tion-to-treat analysis (HR = 0.68, 95% CI = 0.56–0.83). Rates of progression-free survival were 68% vs 57% at 1 year and 41% vs 27% at 2 years in the elotu-zumab and control groups, respectively.
The benefit of elotuzumab was con-sistent across subgroups, including pa-tients aged 65 years and older and those with resistance to the most recent line of therapy, stage III disease, previous expo-sure to bortezomib or immunomodu-latory drugs, previous stem cell trans-plantation, and those with the del(17p) variant. Multivariate analysis suggested
an increased benefit of elotuzumab among patients diagnosed at least 3.5 years before study entry (median, 26.0 vs 17.3 months, HR = 0.55, P < .001).
Overall response rates were 79% vs 66% (odds ratio = 1.9, P < .001), with a complete response in 4% vs 7%. At the time of progression-free survival analysis, when 49% of prespecified
deaths for final overall survival analysis had occurred, 30% of the elotuzumab group and 37% of the control group had died.
Adverse EventsThe most common grade 3 or 4 he-
matologic adverse events in the elotu-zumab group were lymphocytopenia (77% vs 49% in control group), neu-tropenia (34% vs 44%), anemia (19% vs 21%), and thrombocytopenia (19% vs 20%). The most common grade 3 or 4 nonhematologic adverse events were
fatigue (8% vs 8%) and diarrhea (5% vs 4%). Grade 3 or 4 cardiac disorders oc-curred in 4% and 6%, and grade 3 or 4 renal disorders occurred in 4% in both groups. Serious adverse events were re-ported in 65% and 57% of patients.
Infections were reported in 81% vs 74% of patients, with infection rates being identical in the two groups af-ter adjustment for drug exposure (197 events per 100 patient-years). Infusion reactions were reported in 33 elotu-zumab patients (10%, grade 1 or 2 in 29 and grade 3 in 4), with 70% of reactions occurring during the first dose. Infusion was interrupted in 5% of patients. Reactions resolved in all patients except 2 (1%), with both dis-continuing treatment.
The investigators concluded: “Pa-tients with relapsed or refractory mul-tiple myeloma who received a combi-nation of elotuzumab, lenalidomide, and dexamethasone had a significant relative reduction of 30% in the risk of disease progression or death.” n
Disclosure: The study was funded by Bristol-Myers Squibb and AbbVie Biotherapeutics. For full disclosures of the study authors, visit www.nejm.org.
Reference1. Lonial S, Dimopoulos M, Palumbo A,
et al: Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 373:621-631, 2015.
Hematology
Role of Elotuzumab in Myeloma
■ The addition of elotuzumab to lenalidomide-dexamethasone significantly increased progression-free survival in patients with relapsed/refractory multiple myeloma.
■ Benefit was observed across subgroups, including patients with resistance to their most recent therapy and those with the del(17p) variant.
Refractory Multiple Myelomacontinued from page 1
Patients with relapsed or refractory multiple myeloma who received a combination of elotuzumab, lenalidomide,
and dexamethasone had a significant relative reduction of 30% in the risk of disease progression or death.
—Sagar Lonial, MD, Meletios Dimopoulos, MD, and colleagues
Meletios Dimopoulos, MD
Elotuzumab Ushers in a New Era in Myeloma TherapyBy S. Vincent Rajkumar, MD
The long wait for monoclonal anti-bodies for the treatment of multiple
myeloma is over. In the landmark ELO-QUENT-2 study, reviewed in this issue of The ASCO Post, Lonial and colleagues convincingly demonstrate the effective-ness of elotuzumab, a monoclonal anti-body directed against SLAMF7, in the treatment of relapsed multiple myeloma.1
The key findings of this randomized trial are straightforward: An absolute
improvement in response rate of 15%, a median improvement in progression-free survival of 4.5 months, and no sig-nificant added toxicity were noted with the addition of elotuzumab to standard therapy. The results are clear and com-pelling. I believe that elotuzumab will be approved for the treatment of myeloma within the next several months and will usher in a new era in myeloma therapy.
Key Points From ELOQUENT-2There are some key points from
ELOQUENT-2 worthy of highlight-ing. First, elotuzumab does not pos-
sess significant single-agent activity in myeloma but yet appears to provide a meaningful therapeutic benefit in this trial when combined with lenalido-mide [Revlimid]/low-dose dexameth-asone. This finding raises the blinds on the increasing complexity of develop-ing myeloma treatments. The demon-stration of the clinical activity of drugs like elotuzumab, which need partner drugs to deliver synergistic benefit, re-quires large phase III trials and consid-erable risk of failure. This is in contrast to drugs like carfilzomib (Kyprolis), which have clear single-agent activity
that can garner accelerated approval based on single-arm phase II trials.
Second, the trial illustrates the limitations of using median time-to-event estimates to assess the value of a new drug in the relapsed/refractory myeloma setting. Although the me-dian improvement in progression-free survival is modest, the true benefit of elotuzumab is best appreciated in the proportion of patients who are progression free at 2 years, where the difference is much more striking: 27% (control) vs 41% (elotuzumab).
continued on page 38
Dr. Rajkumar is Professor of Medicine, Division of Hematology, Mayo Clinic, Rochester, Minnesota.

PAGE 38 The ASCO Post | SEPTEMBER 10, 2015
Perspective
Third, I applaud the study team for designing a control arm (lenalido-mide and low-dose dexamethasone) that is more in line with actual clinical practice rather than simply playing it
safe and using a control arm that is un-necessarily more toxic. Thus, although the full approval of lenalidomide in relapsed myeloma was in combina-tion with high-dose dexamethasone, the elotuzumab trial used low-dose dexamethasone based on data from a nonregulatory cooperative group trial showing that lowering the dose of dexamethasone reduced morbidity and mortality.
This approach is also in contrast to the trial supporting approval of pano-binostat (Farydak), in which use of the approved dose of bortezomib (Vel-cade) rather than the less-toxic once-weekly schedule resulted in grade 3 or
4 neuropathy in approximately 25% of patients. The moral of the story is that the control arm for a regulatory study does not need to be an approved regi-men: It just needs to be one that is con-sidered by investigators in the field to be a reasonable standard.
Study CaveatsThere are a few important caveats
in interpreting this study. The trial was not blinded, which raises some uncertainty in the progression-free survival estimates. There was no im-provement in quality of life. Merely demonstrating that a drug did not worsen quality of life is simply not enough; after all, we are hoping that delaying disease progression is as-sociated with at least some improve-ment in patients’ perceived quality of life. Finally, there is no evidence that patients lived longer as a result of the therapy, since no overall survival benefit has been shown so far; sur-
vival estimates are “immature”—we shall wait and see.
So, what then is the value of a new drug that appears to have benefit based solely on surrogate endpoints, with no real clinical benefit having been shown yet in terms of either quality of life or overall survival? It de-pends on the setting. In this case, we have a new drug that is well tolerated in the treatment of relapsed myeloma. In this setting, progression-free sur-vival has been consistently shown to be an excellent surrogate of clinical benefit in myeloma.
Therefore, I have no reservation in stating that the results of this trial are sufficient proof of the effectiveness of elotuzumab in relapsed myeloma. Had this study been in the front-line or maintenance setting with an approved drug, we would need to see evidence of true clinical benefit to be enthusiastic. Thus, the front-line elotuzumab study will be subject to greater scrutiny.
Real-World ImplicationsWhat are the real-world implica-
tions of this study once the drug is approved? Strangely, how well elotu-zumab is used in the clinic may de-pend largely on how another mono-clonal antibody, daratumumab, fares in the approval process.
Daratumumab targets CD38 and, unlike elotuzumab, has substantial single-agent activity (approximately
30% in heavily pretreated patients). This means it may secure accelerated approval based on phase II data and may be on the market at nearly the same time as elotuzumab. Time will tell. I expect that there will be com-petition between the two drugs. Since myeloma is incurable, I would try elo-tuzumab if daratumumab does not work and vice versa. It is also likely that these antibodies will be added not just to the regimens that were used in regulatory trials but to all manner of myeloma treatments, analogous to the way rituximab (Rituxan) is used in lymphoma. However, the complicat-ing twist in myeloma is that we may have to adjudicate the clinical value of two antibodies simultaneously.
I do expect new monoclonal anti-bodies in myeloma to be pricey, similar to other new myeloma drugs. The cost will pose an enormous financial dilem-ma in the United States and elsewhere in the world, especially if it comes to pass that we wind up combining two monoclonal antibodies with carfilzo-mib (Kyprolis), pomalidomide (Pom-alyst), and dexamethasone. n
Disclosure: Dr. Rajkumar reported no potential conflicts of interest.
Reference1. Lonial S, Dimopoulos M, Palumbo
A, et al: Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 373:621-631, 2015.
Strangely, how well elotuzumab is used in the clinic may depend largely on how another monoclonal antibody, daratumumab, fares in the approval process.
—S. Vincent Rajkumar, MD
S. Vincent Rajkumar, MDcontinued from page 38
Announcements
NCI and Sage Bionetworks Present: Up for A Challenge? Award up to $50,000
The National Cancer Institute (NCI) Division of Cancer Con-
trol and Population Sciences is partner-ing with Sage Bionetworks for “Up for a Challenge (U4C): Stimulating Innova-tion in Breast Cancer Genetic Epidemi-ology.” This prize competition aims to explore the genomic basis of breast can-cer in diverse populations. Researchers who may be interested in this compe-tition would be scientists involved in genetic epidemiology, bioinformatics, and data science.
Studies suggest that genetic factors play a role in determining who is at in-
creased risk of developing breast can-cer, as well as what type of breast cancer they develop. The goal of U4C is to en-courage innovative approaches to more fully explain the genetic basis of breast cancer.
Specifically, U4C aims to use in-novative approaches to identify novel pathways—including new genes or combinations of genes, genetic variants, or sets of genomic features—involved in breast cancer risk in order to gener-ate new biologic hypotheses. Through U4C, NCI aims to increase the number and diversity of researchers address-
ing a difficult problem by encouraging broader collaborations. Ideally, this will result in new insights that could help improve breast cancer prevention, screening, and/or treatment.
Newly Accessible DataU4C participants will be able to
apply for controlled access to genetic epidemiologic data from thousands of breast cancer cases and controls from ethnically diverse populations. Some of these data sets are being made available to researchers for the first time for this competition.
NCI will award up to $50,000 and other prizes based on the identification of novel findings, replication of find-ings, innovation of approach, evidence of novel biologic hypotheses, and col-laboration. U4C offers participants an opportunity to advance scientific knowledge of the causes of breast can-cer, the second most common cancer in the world.
Visit the U4C website (https://www.synapse.org/upforachallenge) for more information about the goals of U4C, applications, due dates, rules, and other frequently asked questions. n

Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 2 left hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
More than 1,000 days.And every day tells a story.More than 1,000 days.More than 1,000 days.
For men with mCRPC who progressed on ADT
In a clinical trial, patients had a median overall survival on ZYTIGA® (abiraterone acetate) of…*
Please see brief summary of full Prescribing Information on subsequent pages.
Janssen Biotech, Inc.© Janssen Biotech, Inc. 2014 6/14 016819-140612
5.235.3
Co-primary end point—overall survival: hazard ratio (HR)=0.792; 95% CI: 0.655, 0.956; P=0.0151; prespecifi ed value for statistical signifi cance not reached.
Co-primary end point—radiographic progression-free survival: median not reached for ZYTIGA® plus prednisone vs a median of 8.28 months for placebo plus prednisone. HR=0.425; 95% CI: 0.347, 0.522; P<0.0001.
MONTHS IMPROVEMENT IN MEDIAN OVERALL SURVIVALcompared with placebo plus prednisone.
MONTHS MEDIAN OVERALL SURVIVAL FOR ZYTIGA® plus prednisone†vs 30.1 MONTHS with placebo plus prednisone (active compound).‡
IMPORTANT SAFETY INFORMATION (cont)Increased ZYTIGA® Exposures With Food—ZYTIGA® must be taken on an empty stomach. No food should be eaten for at least two hours before the dose of ZYTIGA® is taken and for at least one hour after the dose of ZYTIGA® is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state.Hepatotoxicity—Monitor liver function and modify, withhold, or discontinue ZYTIGA® dosing as recommended (see Prescribing Information for more information). Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA®, every two weeks for the fi rst three months of treatment, and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above fi ve times the upper limit of normal (ULN) or the bilirubin rises above three times the ULN, interrupt ZYTIGA® treatment and closely monitor liver function.* Study Design: ZYTIGA®, in combination with prednisone, was evaluated in a phase 3, randomized, double-blind, placebo-controlled, multicenter trial in patients with mCRPC who had not received prior chemotherapy (N=1,088). Patients were using a luteinizing hormone-releasing hormone (LHRH) agonist or were previously treated with orchiectomy. In the ZYTIGA® arm, patients received ZYTIGA® 1,000 mg orally once daily + prednisone 5 mg orally twice daily. In the placebo arm, patients received placebo orally once daily + prednisone 5 mg orally twice daily. In this study, the co-primary effi cacy end points were overall survival (OS) and radiographic progression-free survival.
ADT=androgen-deprivation therapy.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in
Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 3 right hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
Learn more today at www.zytigahcp.com. Every day tells a story.
0033
07-1
3092
4
Drug Interactions—Based on in vitro data, ZYTIGA® is a substrate of CYP3A4. In a drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA® treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA® dosing frequency only during the co-administration period [see Dosage and Administration (2.3)]. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful eff ect on the pharmacokinetics of abiraterone.ZYTIGA® is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6. Avoid co-administration with CYP2D6 substrates with a narrow therapeutic index. If alternative treatments cannot be used, exercise caution and consider a dose reduction of the CYP2D6 substrate drug. In vitro, ZYTIGA® inhibits CYP2C8. There are no clinical data on the use of ZYTIGA® with drugs that are substrates of CYP2C8. Patients should be monitored closely for signs of toxicity related to the CYP2C8 substrate if used concomitantly with abiraterone acetate.Use in Specifi c Populations—Do not use ZYTIGA® in patients with baseline severe hepatic impairment (Child-Pugh Class C).† At a prespecifi ed interim analysis for OS, 37% (200/546) of patients treated with ZYTIGA® plus prednisone compared with 43% (234/542) of patients treated with placebo plus prednisone had died.
‡Prednisone, as a single agent, is not approved for the treatment of prostate cancer.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in

Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 2 left hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
More than 1,000 days.And every day tells a story.More than 1,000 days.More than 1,000 days.
For men with mCRPC who progressed on ADT
In a clinical trial, patients had a median overall survival on ZYTIGA® (abiraterone acetate) of…*
Please see brief summary of full Prescribing Information on subsequent pages.
Janssen Biotech, Inc.© Janssen Biotech, Inc. 2014 6/14 016819-140612
5.235.3
Co-primary end point—overall survival: hazard ratio (HR)=0.792; 95% CI: 0.655, 0.956; P=0.0151; prespecifi ed value for statistical signifi cance not reached.
Co-primary end point—radiographic progression-free survival: median not reached for ZYTIGA® plus prednisone vs a median of 8.28 months for placebo plus prednisone. HR=0.425; 95% CI: 0.347, 0.522; P<0.0001.
MONTHS IMPROVEMENT IN MEDIAN OVERALL SURVIVALcompared with placebo plus prednisone.
MONTHS MEDIAN OVERALL SURVIVAL FOR ZYTIGA® plus prednisone†vs 30.1 MONTHS with placebo plus prednisone (active compound).‡
IMPORTANT SAFETY INFORMATION (cont)Increased ZYTIGA® Exposures With Food—ZYTIGA® must be taken on an empty stomach. No food should be eaten for at least two hours before the dose of ZYTIGA® is taken and for at least one hour after the dose of ZYTIGA® is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state.Hepatotoxicity—Monitor liver function and modify, withhold, or discontinue ZYTIGA® dosing as recommended (see Prescribing Information for more information). Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA®, every two weeks for the fi rst three months of treatment, and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above fi ve times the upper limit of normal (ULN) or the bilirubin rises above three times the ULN, interrupt ZYTIGA® treatment and closely monitor liver function.* Study Design: ZYTIGA®, in combination with prednisone, was evaluated in a phase 3, randomized, double-blind, placebo-controlled, multicenter trial in patients with mCRPC who had not received prior chemotherapy (N=1,088). Patients were using a luteinizing hormone-releasing hormone (LHRH) agonist or were previously treated with orchiectomy. In the ZYTIGA® arm, patients received ZYTIGA® 1,000 mg orally once daily + prednisone 5 mg orally twice daily. In the placebo arm, patients received placebo orally once daily + prednisone 5 mg orally twice daily. In this study, the co-primary effi cacy end points were overall survival (OS) and radiographic progression-free survival.
ADT=androgen-deprivation therapy.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in
Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 3 right hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
Learn more today at www.zytigahcp.com. Every day tells a story.
0033
07-1
3092
4
Drug Interactions—Based on in vitro data, ZYTIGA® is a substrate of CYP3A4. In a drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA® treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA® dosing frequency only during the co-administration period [see Dosage and Administration (2.3)]. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful eff ect on the pharmacokinetics of abiraterone.ZYTIGA® is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6. Avoid co-administration with CYP2D6 substrates with a narrow therapeutic index. If alternative treatments cannot be used, exercise caution and consider a dose reduction of the CYP2D6 substrate drug. In vitro, ZYTIGA® inhibits CYP2C8. There are no clinical data on the use of ZYTIGA® with drugs that are substrates of CYP2C8. Patients should be monitored closely for signs of toxicity related to the CYP2C8 substrate if used concomitantly with abiraterone acetate.Use in Specifi c Populations—Do not use ZYTIGA® in patients with baseline severe hepatic impairment (Child-Pugh Class C).† At a prespecifi ed interim analysis for OS, 37% (200/546) of patients treated with ZYTIGA® plus prednisone compared with 43% (234/542) of patients treated with placebo plus prednisone had died.
‡Prednisone, as a single agent, is not approved for the treatment of prostate cancer.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in

Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 2 left hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
More than 1,000 days.And every day tells a story.More than 1,000 days.More than 1,000 days.
For men with mCRPC who progressed on ADT
In a clinical trial, patients had a median overall survival on ZYTIGA® (abiraterone acetate) of…*
Please see brief summary of full Prescribing Information on subsequent pages.
Janssen Biotech, Inc.© Janssen Biotech, Inc. 2014 6/14 016819-140612
5.235.3
Co-primary end point—overall survival: hazard ratio (HR)=0.792; 95% CI: 0.655, 0.956; P=0.0151; prespecifi ed value for statistical signifi cance not reached.
Co-primary end point—radiographic progression-free survival: median not reached for ZYTIGA® plus prednisone vs a median of 8.28 months for placebo plus prednisone. HR=0.425; 95% CI: 0.347, 0.522; P<0.0001.
MONTHS IMPROVEMENT IN MEDIAN OVERALL SURVIVALcompared with placebo plus prednisone.
MONTHS MEDIAN OVERALL SURVIVAL FOR ZYTIGA® plus prednisone†vs 30.1 MONTHS with placebo plus prednisone (active compound).‡
IMPORTANT SAFETY INFORMATION (cont)Increased ZYTIGA® Exposures With Food—ZYTIGA® must be taken on an empty stomach. No food should be eaten for at least two hours before the dose of ZYTIGA® is taken and for at least one hour after the dose of ZYTIGA® is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state.Hepatotoxicity—Monitor liver function and modify, withhold, or discontinue ZYTIGA® dosing as recommended (see Prescribing Information for more information). Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA®, every two weeks for the fi rst three months of treatment, and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above fi ve times the upper limit of normal (ULN) or the bilirubin rises above three times the ULN, interrupt ZYTIGA® treatment and closely monitor liver function.* Study Design: ZYTIGA®, in combination with prednisone, was evaluated in a phase 3, randomized, double-blind, placebo-controlled, multicenter trial in patients with mCRPC who had not received prior chemotherapy (N=1,088). Patients were using a luteinizing hormone-releasing hormone (LHRH) agonist or were previously treated with orchiectomy. In the ZYTIGA® arm, patients received ZYTIGA® 1,000 mg orally once daily + prednisone 5 mg orally twice daily. In the placebo arm, patients received placebo orally once daily + prednisone 5 mg orally twice daily. In this study, the co-primary effi cacy end points were overall survival (OS) and radiographic progression-free survival.
ADT=androgen-deprivation therapy.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in
Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 3 right hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
Learn more today at www.zytigahcp.com. Every day tells a story.
0033
07-1
3092
4
Drug Interactions—Based on in vitro data, ZYTIGA® is a substrate of CYP3A4. In a drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA® treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA® dosing frequency only during the co-administration period [see Dosage and Administration (2.3)]. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful eff ect on the pharmacokinetics of abiraterone.ZYTIGA® is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6. Avoid co-administration with CYP2D6 substrates with a narrow therapeutic index. If alternative treatments cannot be used, exercise caution and consider a dose reduction of the CYP2D6 substrate drug. In vitro, ZYTIGA® inhibits CYP2C8. There are no clinical data on the use of ZYTIGA® with drugs that are substrates of CYP2C8. Patients should be monitored closely for signs of toxicity related to the CYP2C8 substrate if used concomitantly with abiraterone acetate.Use in Specifi c Populations—Do not use ZYTIGA® in patients with baseline severe hepatic impairment (Child-Pugh Class C).† At a prespecifi ed interim analysis for OS, 37% (200/546) of patients treated with ZYTIGA® plus prednisone compared with 43% (234/542) of patients treated with placebo plus prednisone had died.
‡Prednisone, as a single agent, is not approved for the treatment of prostate cancer.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in
Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 2 left hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
More than 1,000 days.And every day tells a story.More than 1,000 days.More than 1,000 days.
For men with mCRPC who progressed on ADT
In a clinical trial, patients had a median overall survival on ZYTIGA® (abiraterone acetate) of…*
Please see brief summary of full Prescribing Information on subsequent pages.
Janssen Biotech, Inc.© Janssen Biotech, Inc. 2014 6/14 016819-140612
5.235.3
Co-primary end point—overall survival: hazard ratio (HR)=0.792; 95% CI: 0.655, 0.956; P=0.0151; prespecifi ed value for statistical signifi cance not reached.
Co-primary end point—radiographic progression-free survival: median not reached for ZYTIGA® plus prednisone vs a median of 8.28 months for placebo plus prednisone. HR=0.425; 95% CI: 0.347, 0.522; P<0.0001.
MONTHS IMPROVEMENT IN MEDIAN OVERALL SURVIVALcompared with placebo plus prednisone.
MONTHS MEDIAN OVERALL SURVIVAL FOR ZYTIGA® plus prednisone†vs 30.1 MONTHS with placebo plus prednisone (active compound).‡
IMPORTANT SAFETY INFORMATION (cont)Increased ZYTIGA® Exposures With Food—ZYTIGA® must be taken on an empty stomach. No food should be eaten for at least two hours before the dose of ZYTIGA® is taken and for at least one hour after the dose of ZYTIGA® is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state.Hepatotoxicity—Monitor liver function and modify, withhold, or discontinue ZYTIGA® dosing as recommended (see Prescribing Information for more information). Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA®, every two weeks for the fi rst three months of treatment, and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above fi ve times the upper limit of normal (ULN) or the bilirubin rises above three times the ULN, interrupt ZYTIGA® treatment and closely monitor liver function.* Study Design: ZYTIGA®, in combination with prednisone, was evaluated in a phase 3, randomized, double-blind, placebo-controlled, multicenter trial in patients with mCRPC who had not received prior chemotherapy (N=1,088). Patients were using a luteinizing hormone-releasing hormone (LHRH) agonist or were previously treated with orchiectomy. In the ZYTIGA® arm, patients received ZYTIGA® 1,000 mg orally once daily + prednisone 5 mg orally twice daily. In the placebo arm, patients received placebo orally once daily + prednisone 5 mg orally twice daily. In this study, the co-primary effi cacy end points were overall survival (OS) and radiographic progression-free survival.
ADT=androgen-deprivation therapy.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in
Date: 12/29/14 Customer Code: 016819-140612 Group 360 Job #: 719920File Name: 016819-140612_719920_v1 (pg 3 right hand) Brand: ZytigaSize: 7.625" x 10.5" Colors: CMYK Description: Is there more to the story?Pub: ASCO POST - Digital Issue (2/10/15 Issue)
K P G75 M50 K75 Y50 GN M25 B C75 M75 K25 Y C50 M G25 C Y75 K50 C25 G50 Y25 R
Learn more today at www.zytigahcp.com. Every day tells a story.
0033
07-1
3092
4
Drug Interactions—Based on in vitro data, ZYTIGA® is a substrate of CYP3A4. In a drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA® treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA® dosing frequency only during the co-administration period [see Dosage and Administration (2.3)]. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful eff ect on the pharmacokinetics of abiraterone.ZYTIGA® is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6. Avoid co-administration with CYP2D6 substrates with a narrow therapeutic index. If alternative treatments cannot be used, exercise caution and consider a dose reduction of the CYP2D6 substrate drug. In vitro, ZYTIGA® inhibits CYP2C8. There are no clinical data on the use of ZYTIGA® with drugs that are substrates of CYP2C8. Patients should be monitored closely for signs of toxicity related to the CYP2C8 substrate if used concomitantly with abiraterone acetate.Use in Specifi c Populations—Do not use ZYTIGA® in patients with baseline severe hepatic impairment (Child-Pugh Class C).† At a prespecifi ed interim analysis for OS, 37% (200/546) of patients treated with ZYTIGA® plus prednisone compared with 43% (234/542) of patients treated with placebo plus prednisone had died.
‡Prednisone, as a single agent, is not approved for the treatment of prostate cancer.
B:10.75 in
B:7.875 in
T:10.5 in
T:7.625 in
S:10 in
S:6.625 in

ZYTIGA® (abiraterone acetate) TabletsBrief Summary of Prescribing Information.INDICATIONS AND USAGE ZYTIGA is a CYP17 inhibitor indicated in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer.CONTRAINDICATIONSPregnancy: ZYTIGA can cause fetal harm when administered to a pregnant woman. ZYTIGA is not indicated for use in women. ZYTIGA is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss [see Use in Specific Populations].WARNINGS AND PRECAUTIONSHypertension, Hypokalemia and Fluid Retention Due to Mineralocorticoid Excess: ZYTIGA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1) in full Prescribing Information]. In the two randomized clinical trials, grade 3 to 4 hypertension occurred in 2% of patients, grade 3 to 4 hypokalemia in 4% of patients, and grade 3 to 4 edema in 1% of patients treated with ZYTIGA [see Adverse Reactions]. Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in the incidence and severity of these adverse reactions. Use caution when treating patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, e.g., those with heart failure, recent myocardial infarction or ventricular arrhythmia. Use ZYTIGA with caution in patients with a history of cardiovascular disease. The safety of ZYTIGA in patients with left ventricular ejection fraction <50% or New York Heart Association (NYHA) Class III or IV heart failure (in Study 1) or NYHA Class II to IV heart failure (in Study 2) was not established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14) in full Prescribing Information]. Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with ZYTIGA.Adrenocortical Insufficiency: Adrenal insufficiency occurred in the two randomized clinical studies in 0.5% of patients taking ZYTIGA and in 0.2% of patients taking placebo. Adrenocortical insufficiency was reported in patients receiving ZYTIGA in combination with prednisone, following interruption of daily steroids and/or with concurrent infection or stress. Use caution and monitor for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from prednisone, have prednisone dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with ZYTIGA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions].Hepatotoxicity: In the two randomized clinical trials, grade 3 or 4 ALT or AST increases (at least 5X ULN) were reported in 4% of patients who received ZYTIGA, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to liver enzyme increases occurred in 1% of patients taking ZYTIGA. No deaths clearly related to ZYTIGA were reported due to hepatotoxicity events. Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced ZYTIGA dose of 250 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt ZYTIGA treatment and closely monitor liver function.Re-treatment with ZYTIGA at a reduced dose level may take place only after return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN [see Dosage and Administration (2.2) in full Prescribing Information].The safety of ZYTIGA re-treatment of patients who develop AST or ALT greater than or equal to 20X ULN and/or bilirubin greater than or equal to 10X ULN is unknown.Increased ZYTIGA Exposures with Food: ZYTIGA must be taken on an empty stomach. No food should be consumed for at least two hours before the dose of ZYTIGA is taken and for at least one hour after the dose of ZYTIGA
is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state. The safety of these increased exposures when multiple doses of abiraterone acetate are taken with food has not been assessed [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3) in full Prescribing Information].ADVERSE REACTIONSThe following are discussed in more detail in other sections of the labeling:• Hypertension, Hypokalemia, and Fluid Retention due to Mineralocorti-
coid Excess [see Warnings and Precautions].• Adrenocortical Insufficiency [see Warnings and Precautions].• Hepatotoxicity [see Warnings and Precautions].• Increased ZYTIGA Exposures with Food [see Warnings and
Precautions].Clinical Trial Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.Two randomized placebo-controlled, multicenter clinical trials enrolled patients who had metastatic castration-resistant prostate cancer who were using a gonadotropin-releasing hormone (GnRH) agonist or were previously treated with orchiectomy. In both Study 1 and Study 2 ZYTIGA was administered at a dose of 1,000 mg daily in combination with prednisone 5 mg twice daily in the active treatment arms. Placebo plus prednisone 5 mg twice daily was given to control patients. The most common adverse drug reactions (≥10%) reported in the two randomized clinical trials that occurred more commonly (>2%) in the abiraterone acetate arm were fatigue, joint swelling or discomfort, edema, hot flush, diarrhea, vomiting, cough, hypertension, dyspnea, urinary tract infection and contusion. The most common laboratory abnormalities (>20%) reported in the two randomized clinical trials that occurred more commonly (≥2%) in the abiraterone acetate arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, elevated AST, hypo phosphatemia, elevated ALT and hypokalemia.Study 1: Metastatic CRPC Following Chemotherapy: Study 1 enrolled 1195 patients with metastatic CRPC who had received prior docetaxel chemotherapy. Patients were not eligible if AST and/or ALT ≥2.5X ULN in the absence of liver metastases. Patients with liver metastases were excluded if AST and/or ALT >5X ULN.Table 1 shows adverse reactions on the ZYTIGA arm in Study 1 that occurred with a ≥2% absolute increase in frequency compared to placebo or were events of special interest. The median duration of treatment with ZYTIGA was 8 months.Table 1: Adverse Reactions due to ZYTIGA in Study 1
ZYTIGA with Prednisone (N=791)
Placebo with Prednisone (N=394)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort2 29.5 4.2 23.4 4.1Muscle discomfort3 26.2 3.0 23.1 2.3
General disordersEdema4 26.7 1.9 18.3 0.8
Vascular disordersHot flush 19.0 0.3 16.8 0.3Hypertension 8.5 1.3 6.9 0.3
Gastrointestinal disorders
Diarrhea 17.6 0.6 13.5 1.3Dyspepsia 6.1 0 3.3 0
Infections and infestations
Urinary tract infection
11.52.1 7.1 0.5
Upper respiratory tract infection 5.4 0 2.5 0
Respiratory, thoracic and mediastinal disorders
Cough 10.6 0 7.6 0Renal and urinary disorders
Urinary frequency 7.2 0.3 5.1 0.3Nocturia 6.2 0 4.1 0
ZYTIGA® (abiraterone acetate) Tablets
Table 1: Adverse Reactions due to ZYTIGA in Study 1 (continued) ZYTIGA with
Prednisone (N=791)Placebo with
Prednisone (N=394)System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4
Adverse reaction % % % %Injury, poisoning and procedural complications
Fractures5 5.9 1.4 2.3 0Cardiac disorders
Arrhythmia6 7.2 1.1 4.6 1.0Chest pain or chest discomfort7 3.8 0.5 2.8 0Cardiac failure8 2.3 1.9 1.0 0.3
1 Adverse events graded according to CTCAE version 3.02 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness 3 Includes terms Muscle spasms, Musculoskeletal pain, Myalgia,
Musculoskeletal discomfort, and Musculoskeletal stiffness4 Includes terms Edema, Edema peripheral, Pitting edema, and Generalized
edema5 Includes all fractures with the exception of pathological fracture6 Includes terms Arrhythmia, Tachycardia, Atrial fibrillation,
Supraventricular tachycardia, Atrial tachycardia, Ventricular tachycardia, Atrial flutter, Bradycardia, Atrioventricular block complete, Conduction disorder, and Bradyarrhythmia
7 Includes terms Angina pectoris, Chest pain, and Angina unstable. Myocardial infarction or ischemia occurred more commonly in the placebo arm than in the ZYTIGA arm (1.3% vs. 1.1% respectively).
8 Includes terms Cardiac failure, Cardiac failure congestive, Left ventricular dysfunction, Cardiogenic shock, Cardiomegaly, Cardiomyopathy, and Ejection fraction decreased
Table 2 shows laboratory abnormalities of interest from Study 1. Grade 3-4 low serum phosphorus (7%) and low potassium (5%) occurred at a greater than or equal to 5% rate in the ZYTIGA arm.
Table 2: Laboratory Abnormalities of Interest in Study 1Abiraterone (N=791) Placebo (N=394)
Laboratory Abnormality
All Grades (%)
Grade 3-4 (%)
All Grades (%)
Grade 3-4 (%)
Hypertriglyceridemia 62.5 0.4 53.0 0High AST 30.6 2.1 36.3 1.5Hypokalemia 28.3 5.3 19.8 1.0Hypophosphatemia 23.8 7.2 15.7 5.8High ALT 11.1 1.4 10.4 0.8High Total Bilirubin 6.6 0.1 4.6 0
Study 2: Metastatic CRPC Prior to Chemotherapy: Study 2 enrolled 1088 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy. Patients were ineligible if AST and/or ALT ≥2.5X ULN and patients were excluded if they had liver metastases.Table 3 shows adverse reactions on the ZYTIGA arm in Study 2 that occurred with a ≥2% absolute increase in frequency compared to placebo. The median duration of treatment with ZYTIGA was 13.8 months.
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
General disordersFatigue 39.1 2.2 34.3 1.7Edema2 25.1 0.4 20.7 1.1Pyrexia 8.7 0.6 5.9 0.2
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort3 30.3 2.0 25.2 2.0Groin pain 6.6 0.4 4.1 0.7
Gastrointestinal disorders
Constipation 23.1 0.4 19.1 0.6Diarrhea 21.6 0.9 17.8 0.9Dyspepsia 11.1 0.0 5.0 0.2
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2 (continued)
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Vascular disordersHot flush 22.3 0.2 18.1 0.0Hypertension 21.6 3.9 13.1 3.0
Respiratory, thoracic and mediastinal disorders
Cough 17.3 0.0 13.5 0.2Dyspnea 11.8 2.4 9.6 0.9
Psychiatric disordersInsomnia 13.5 0.2 11.3 0.0
Injury, poisoning and procedural complications
Contusion 13.3 0.0 9.1 0.0Falls 5.9 0.0 3.3 0.0
Infections and infestations
Upper respiratory tract infection 12.7 0.0 8.0 0.0Nasopharyngitis 10.7 0.0 8.1 0.0
Renal and urinary disorders
Hematuria 10.3 1.3 5.6 0.6Skin and subcutaneous tissue disorders
Rash 8.1 0.0 3.7 0.01 Adverse events graded according to CTCAE version 3.02 Includes terms Edema peripheral, Pitting edema, and Generalized
edema3 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness
Table 4 shows laboratory abnormalities that occurred in greater than 15% of patients, and more frequently (>5%) in the ZYTIGA arm compared to placebo in Study 2. Grade 3-4 lymphopenia (9%), hyperglycemia (7%) and high alanine aminotransferase (6%) occurred at a greater than 5% rate in the ZYTIGA arm. Table 4: Laboratory Abnormalities in >15% of Patients in the ZYTIGA
Arm of Study 2Abiraterone (N=542) Placebo (N=540)
Laboratory Abnormality
Grade 1-4%
Grade 3-4%
Grade 1-4%
Grade 3-4%
HematologyLymphopenia 38.2 8.7 31.7 7.4
ChemistryHyperglycemia1 56.6 6.5 50.9 5.2High ALT 41.9 6.1 29.1 0.7High AST 37.3 3.1 28.7 1.1Hypernatremia 32.8 0.4 25.0 0.2Hypokalemia 17.2 2.8 10.2 1.7
1Based on non-fasting blood drawsCardiovascular Adverse Reactions: In the combined data for studies 1 and 2, cardiac failure occurred more commonly in patients treated with ZYTIGA compared to patients on the placebo arm (2.1% versus 0.7%). Grade 3-4 cardiac failure occurred in 1.6% of patients taking ZYTIGA and led to 5 treatment discontinuations and 2 deaths. Grade 3-4 cardiac failure occurred in 0.2% of patients taking placebo. There were no treatment discontinuations and one death due to cardiac failure in the placebo group. In Study 1 and 2, the majority of arrhythmias were grade 1 or 2. There was one death associated with arrhythmia and one patient with sudden death in the ZYTIGA arms and no deaths in the placebo arms. There were 7 (0.5%) deaths due to cardiorespiratory arrest in the ZYTIGA arms and 3 (0.3%) deaths in the placebo arms. Myocardial ischemia or myocardial infarction led to death in 3 patients in the placebo arms and 2 deaths in the ZYTIGA arms. Post Marketing ExperienceThe following additional adverse reactions have been identified during post approval use of ZYTIGA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.Respiratory, Thoracic and Mediastinal Disorders: non-infectious pneumonitis.
ZYTIGA® (abiraterone acetate) Tablets ZYTIGA® (abiraterone acetate) Tablets
ZYTIGA® (abiraterone acetate) TabletsBrief Summary of Prescribing Information.INDICATIONS AND USAGE ZYTIGA is a CYP17 inhibitor indicated in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer.CONTRAINDICATIONSPregnancy: ZYTIGA can cause fetal harm when administered to a pregnant woman. ZYTIGA is not indicated for use in women. ZYTIGA is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss [see Use in Specific Populations].WARNINGS AND PRECAUTIONSHypertension, Hypokalemia and Fluid Retention Due to Mineralocorticoid Excess: ZYTIGA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1) in full Prescribing Information]. In the two randomized clinical trials, grade 3 to 4 hypertension occurred in 2% of patients, grade 3 to 4 hypokalemia in 4% of patients, and grade 3 to 4 edema in 1% of patients treated with ZYTIGA [see Adverse Reactions]. Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in the incidence and severity of these adverse reactions. Use caution when treating patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, e.g., those with heart failure, recent myocardial infarction or ventricular arrhythmia. Use ZYTIGA with caution in patients with a history of cardiovascular disease. The safety of ZYTIGA in patients with left ventricular ejection fraction <50% or New York Heart Association (NYHA) Class III or IV heart failure (in Study 1) or NYHA Class II to IV heart failure (in Study 2) was not established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14) in full Prescribing Information]. Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with ZYTIGA.Adrenocortical Insufficiency: Adrenal insufficiency occurred in the two randomized clinical studies in 0.5% of patients taking ZYTIGA and in 0.2% of patients taking placebo. Adrenocortical insufficiency was reported in patients receiving ZYTIGA in combination with prednisone, following interruption of daily steroids and/or with concurrent infection or stress. Use caution and monitor for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from prednisone, have prednisone dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with ZYTIGA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions].Hepatotoxicity: In the two randomized clinical trials, grade 3 or 4 ALT or AST increases (at least 5X ULN) were reported in 4% of patients who received ZYTIGA, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to liver enzyme increases occurred in 1% of patients taking ZYTIGA. No deaths clearly related to ZYTIGA were reported due to hepatotoxicity events. Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced ZYTIGA dose of 250 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt ZYTIGA treatment and closely monitor liver function.Re-treatment with ZYTIGA at a reduced dose level may take place only after return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN [see Dosage and Administration (2.2) in full Prescribing Information].The safety of ZYTIGA re-treatment of patients who develop AST or ALT greater than or equal to 20X ULN and/or bilirubin greater than or equal to 10X ULN is unknown.Increased ZYTIGA Exposures with Food: ZYTIGA must be taken on an empty stomach. No food should be consumed for at least two hours before the dose of ZYTIGA is taken and for at least one hour after the dose of ZYTIGA
is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state. The safety of these increased exposures when multiple doses of abiraterone acetate are taken with food has not been assessed [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3) in full Prescribing Information].ADVERSE REACTIONSThe following are discussed in more detail in other sections of the labeling:• Hypertension, Hypokalemia, and Fluid Retention due to Mineralocorti-
coid Excess [see Warnings and Precautions].• Adrenocortical Insufficiency [see Warnings and Precautions].• Hepatotoxicity [see Warnings and Precautions].• Increased ZYTIGA Exposures with Food [see Warnings and
Precautions].Clinical Trial Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.Two randomized placebo-controlled, multicenter clinical trials enrolled patients who had metastatic castration-resistant prostate cancer who were using a gonadotropin-releasing hormone (GnRH) agonist or were previously treated with orchiectomy. In both Study 1 and Study 2 ZYTIGA was administered at a dose of 1,000 mg daily in combination with prednisone 5 mg twice daily in the active treatment arms. Placebo plus prednisone 5 mg twice daily was given to control patients. The most common adverse drug reactions (≥10%) reported in the two randomized clinical trials that occurred more commonly (>2%) in the abiraterone acetate arm were fatigue, joint swelling or discomfort, edema, hot flush, diarrhea, vomiting, cough, hypertension, dyspnea, urinary tract infection and contusion. The most common laboratory abnormalities (>20%) reported in the two randomized clinical trials that occurred more commonly (≥2%) in the abiraterone acetate arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, elevated AST, hypo phosphatemia, elevated ALT and hypokalemia.Study 1: Metastatic CRPC Following Chemotherapy: Study 1 enrolled 1195 patients with metastatic CRPC who had received prior docetaxel chemotherapy. Patients were not eligible if AST and/or ALT ≥2.5X ULN in the absence of liver metastases. Patients with liver metastases were excluded if AST and/or ALT >5X ULN.Table 1 shows adverse reactions on the ZYTIGA arm in Study 1 that occurred with a ≥2% absolute increase in frequency compared to placebo or were events of special interest. The median duration of treatment with ZYTIGA was 8 months.Table 1: Adverse Reactions due to ZYTIGA in Study 1
ZYTIGA with Prednisone (N=791)
Placebo with Prednisone (N=394)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort2 29.5 4.2 23.4 4.1Muscle discomfort3 26.2 3.0 23.1 2.3
General disordersEdema4 26.7 1.9 18.3 0.8
Vascular disordersHot flush 19.0 0.3 16.8 0.3Hypertension 8.5 1.3 6.9 0.3
Gastrointestinal disorders
Diarrhea 17.6 0.6 13.5 1.3Dyspepsia 6.1 0 3.3 0
Infections and infestations
Urinary tract infection
11.52.1 7.1 0.5
Upper respiratory tract infection 5.4 0 2.5 0
Respiratory, thoracic and mediastinal disorders
Cough 10.6 0 7.6 0Renal and urinary disorders
Urinary frequency 7.2 0.3 5.1 0.3Nocturia 6.2 0 4.1 0
ZYTIGA® (abiraterone acetate) Tablets
Table 1: Adverse Reactions due to ZYTIGA in Study 1 (continued) ZYTIGA with
Prednisone (N=791)Placebo with
Prednisone (N=394)System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4
Adverse reaction % % % %Injury, poisoning and procedural complications
Fractures5 5.9 1.4 2.3 0Cardiac disorders
Arrhythmia6 7.2 1.1 4.6 1.0Chest pain or chest discomfort7 3.8 0.5 2.8 0Cardiac failure8 2.3 1.9 1.0 0.3
1 Adverse events graded according to CTCAE version 3.02 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness 3 Includes terms Muscle spasms, Musculoskeletal pain, Myalgia,
Musculoskeletal discomfort, and Musculoskeletal stiffness4 Includes terms Edema, Edema peripheral, Pitting edema, and Generalized
edema5 Includes all fractures with the exception of pathological fracture6 Includes terms Arrhythmia, Tachycardia, Atrial fibrillation,
Supraventricular tachycardia, Atrial tachycardia, Ventricular tachycardia, Atrial flutter, Bradycardia, Atrioventricular block complete, Conduction disorder, and Bradyarrhythmia
7 Includes terms Angina pectoris, Chest pain, and Angina unstable. Myocardial infarction or ischemia occurred more commonly in the placebo arm than in the ZYTIGA arm (1.3% vs. 1.1% respectively).
8 Includes terms Cardiac failure, Cardiac failure congestive, Left ventricular dysfunction, Cardiogenic shock, Cardiomegaly, Cardiomyopathy, and Ejection fraction decreased
Table 2 shows laboratory abnormalities of interest from Study 1. Grade 3-4 low serum phosphorus (7%) and low potassium (5%) occurred at a greater than or equal to 5% rate in the ZYTIGA arm.
Table 2: Laboratory Abnormalities of Interest in Study 1Abiraterone (N=791) Placebo (N=394)
Laboratory Abnormality
All Grades (%)
Grade 3-4 (%)
All Grades (%)
Grade 3-4 (%)
Hypertriglyceridemia 62.5 0.4 53.0 0High AST 30.6 2.1 36.3 1.5Hypokalemia 28.3 5.3 19.8 1.0Hypophosphatemia 23.8 7.2 15.7 5.8High ALT 11.1 1.4 10.4 0.8High Total Bilirubin 6.6 0.1 4.6 0
Study 2: Metastatic CRPC Prior to Chemotherapy: Study 2 enrolled 1088 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy. Patients were ineligible if AST and/or ALT ≥2.5X ULN and patients were excluded if they had liver metastases.Table 3 shows adverse reactions on the ZYTIGA arm in Study 2 that occurred with a ≥2% absolute increase in frequency compared to placebo. The median duration of treatment with ZYTIGA was 13.8 months.
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
General disordersFatigue 39.1 2.2 34.3 1.7Edema2 25.1 0.4 20.7 1.1Pyrexia 8.7 0.6 5.9 0.2
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort3 30.3 2.0 25.2 2.0Groin pain 6.6 0.4 4.1 0.7
Gastrointestinal disorders
Constipation 23.1 0.4 19.1 0.6Diarrhea 21.6 0.9 17.8 0.9Dyspepsia 11.1 0.0 5.0 0.2
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2 (continued)
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Vascular disordersHot flush 22.3 0.2 18.1 0.0Hypertension 21.6 3.9 13.1 3.0
Respiratory, thoracic and mediastinal disorders
Cough 17.3 0.0 13.5 0.2Dyspnea 11.8 2.4 9.6 0.9
Psychiatric disordersInsomnia 13.5 0.2 11.3 0.0
Injury, poisoning and procedural complications
Contusion 13.3 0.0 9.1 0.0Falls 5.9 0.0 3.3 0.0
Infections and infestations
Upper respiratory tract infection 12.7 0.0 8.0 0.0Nasopharyngitis 10.7 0.0 8.1 0.0
Renal and urinary disorders
Hematuria 10.3 1.3 5.6 0.6Skin and subcutaneous tissue disorders
Rash 8.1 0.0 3.7 0.01 Adverse events graded according to CTCAE version 3.02 Includes terms Edema peripheral, Pitting edema, and Generalized
edema3 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness
Table 4 shows laboratory abnormalities that occurred in greater than 15% of patients, and more frequently (>5%) in the ZYTIGA arm compared to placebo in Study 2. Grade 3-4 lymphopenia (9%), hyperglycemia (7%) and high alanine aminotransferase (6%) occurred at a greater than 5% rate in the ZYTIGA arm. Table 4: Laboratory Abnormalities in >15% of Patients in the ZYTIGA
Arm of Study 2Abiraterone (N=542) Placebo (N=540)
Laboratory Abnormality
Grade 1-4%
Grade 3-4%
Grade 1-4%
Grade 3-4%
HematologyLymphopenia 38.2 8.7 31.7 7.4
ChemistryHyperglycemia1 56.6 6.5 50.9 5.2High ALT 41.9 6.1 29.1 0.7High AST 37.3 3.1 28.7 1.1Hypernatremia 32.8 0.4 25.0 0.2Hypokalemia 17.2 2.8 10.2 1.7
1Based on non-fasting blood drawsCardiovascular Adverse Reactions: In the combined data for studies 1 and 2, cardiac failure occurred more commonly in patients treated with ZYTIGA compared to patients on the placebo arm (2.1% versus 0.7%). Grade 3-4 cardiac failure occurred in 1.6% of patients taking ZYTIGA and led to 5 treatment discontinuations and 2 deaths. Grade 3-4 cardiac failure occurred in 0.2% of patients taking placebo. There were no treatment discontinuations and one death due to cardiac failure in the placebo group. In Study 1 and 2, the majority of arrhythmias were grade 1 or 2. There was one death associated with arrhythmia and one patient with sudden death in the ZYTIGA arms and no deaths in the placebo arms. There were 7 (0.5%) deaths due to cardiorespiratory arrest in the ZYTIGA arms and 3 (0.3%) deaths in the placebo arms. Myocardial ischemia or myocardial infarction led to death in 3 patients in the placebo arms and 2 deaths in the ZYTIGA arms. Post Marketing ExperienceThe following additional adverse reactions have been identified during post approval use of ZYTIGA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.Respiratory, Thoracic and Mediastinal Disorders: non-infectious pneumonitis.
ZYTIGA® (abiraterone acetate) Tablets ZYTIGA® (abiraterone acetate) TabletsZYTIGA® (abiraterone acetate) TabletsBrief Summary of Prescribing Information.INDICATIONS AND USAGE ZYTIGA is a CYP17 inhibitor indicated in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer.CONTRAINDICATIONSPregnancy: ZYTIGA can cause fetal harm when administered to a pregnant woman. ZYTIGA is not indicated for use in women. ZYTIGA is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss [see Use in Specific Populations].WARNINGS AND PRECAUTIONSHypertension, Hypokalemia and Fluid Retention Due to Mineralocorticoid Excess: ZYTIGA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1) in full Prescribing Information]. In the two randomized clinical trials, grade 3 to 4 hypertension occurred in 2% of patients, grade 3 to 4 hypokalemia in 4% of patients, and grade 3 to 4 edema in 1% of patients treated with ZYTIGA [see Adverse Reactions]. Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in the incidence and severity of these adverse reactions. Use caution when treating patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, e.g., those with heart failure, recent myocardial infarction or ventricular arrhythmia. Use ZYTIGA with caution in patients with a history of cardiovascular disease. The safety of ZYTIGA in patients with left ventricular ejection fraction <50% or New York Heart Association (NYHA) Class III or IV heart failure (in Study 1) or NYHA Class II to IV heart failure (in Study 2) was not established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14) in full Prescribing Information]. Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with ZYTIGA.Adrenocortical Insufficiency: Adrenal insufficiency occurred in the two randomized clinical studies in 0.5% of patients taking ZYTIGA and in 0.2% of patients taking placebo. Adrenocortical insufficiency was reported in patients receiving ZYTIGA in combination with prednisone, following interruption of daily steroids and/or with concurrent infection or stress. Use caution and monitor for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from prednisone, have prednisone dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with ZYTIGA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions].Hepatotoxicity: In the two randomized clinical trials, grade 3 or 4 ALT or AST increases (at least 5X ULN) were reported in 4% of patients who received ZYTIGA, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to liver enzyme increases occurred in 1% of patients taking ZYTIGA. No deaths clearly related to ZYTIGA were reported due to hepatotoxicity events. Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced ZYTIGA dose of 250 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient’s baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt ZYTIGA treatment and closely monitor liver function.Re-treatment with ZYTIGA at a reduced dose level may take place only after return of liver function tests to the patient’s baseline or to AST and ALT less than or equal to 2.5X ULN and total bilirubin less than or equal to 1.5X ULN [see Dosage and Administration (2.2) in full Prescribing Information].The safety of ZYTIGA re-treatment of patients who develop AST or ALT greater than or equal to 20X ULN and/or bilirubin greater than or equal to 10X ULN is unknown.Increased ZYTIGA Exposures with Food: ZYTIGA must be taken on an empty stomach. No food should be consumed for at least two hours before the dose of ZYTIGA is taken and for at least one hour after the dose of ZYTIGA
is taken. Abiraterone Cmax and AUC0-∞ (exposure) were increased up to 17- and 10-fold higher, respectively, when a single dose of abiraterone acetate was administered with a meal compared to a fasted state. The safety of these increased exposures when multiple doses of abiraterone acetate are taken with food has not been assessed [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3) in full Prescribing Information].ADVERSE REACTIONSThe following are discussed in more detail in other sections of the labeling:• Hypertension, Hypokalemia, and Fluid Retention due to Mineralocorti-
coid Excess [see Warnings and Precautions].• Adrenocortical Insufficiency [see Warnings and Precautions].• Hepatotoxicity [see Warnings and Precautions].• Increased ZYTIGA Exposures with Food [see Warnings and
Precautions].Clinical Trial Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.Two randomized placebo-controlled, multicenter clinical trials enrolled patients who had metastatic castration-resistant prostate cancer who were using a gonadotropin-releasing hormone (GnRH) agonist or were previously treated with orchiectomy. In both Study 1 and Study 2 ZYTIGA was administered at a dose of 1,000 mg daily in combination with prednisone 5 mg twice daily in the active treatment arms. Placebo plus prednisone 5 mg twice daily was given to control patients. The most common adverse drug reactions (≥10%) reported in the two randomized clinical trials that occurred more commonly (>2%) in the abiraterone acetate arm were fatigue, joint swelling or discomfort, edema, hot flush, diarrhea, vomiting, cough, hypertension, dyspnea, urinary tract infection and contusion. The most common laboratory abnormalities (>20%) reported in the two randomized clinical trials that occurred more commonly (≥2%) in the abiraterone acetate arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, elevated AST, hypo phosphatemia, elevated ALT and hypokalemia.Study 1: Metastatic CRPC Following Chemotherapy: Study 1 enrolled 1195 patients with metastatic CRPC who had received prior docetaxel chemotherapy. Patients were not eligible if AST and/or ALT ≥2.5X ULN in the absence of liver metastases. Patients with liver metastases were excluded if AST and/or ALT >5X ULN.Table 1 shows adverse reactions on the ZYTIGA arm in Study 1 that occurred with a ≥2% absolute increase in frequency compared to placebo or were events of special interest. The median duration of treatment with ZYTIGA was 8 months.Table 1: Adverse Reactions due to ZYTIGA in Study 1
ZYTIGA with Prednisone (N=791)
Placebo with Prednisone (N=394)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort2 29.5 4.2 23.4 4.1Muscle discomfort3 26.2 3.0 23.1 2.3
General disordersEdema4 26.7 1.9 18.3 0.8
Vascular disordersHot flush 19.0 0.3 16.8 0.3Hypertension 8.5 1.3 6.9 0.3
Gastrointestinal disorders
Diarrhea 17.6 0.6 13.5 1.3Dyspepsia 6.1 0 3.3 0
Infections and infestations
Urinary tract infection
11.52.1 7.1 0.5
Upper respiratory tract infection 5.4 0 2.5 0
Respiratory, thoracic and mediastinal disorders
Cough 10.6 0 7.6 0Renal and urinary disorders
Urinary frequency 7.2 0.3 5.1 0.3Nocturia 6.2 0 4.1 0
ZYTIGA® (abiraterone acetate) Tablets
Table 1: Adverse Reactions due to ZYTIGA in Study 1 (continued) ZYTIGA with
Prednisone (N=791)Placebo with
Prednisone (N=394)System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4
Adverse reaction % % % %Injury, poisoning and procedural complications
Fractures5 5.9 1.4 2.3 0Cardiac disorders
Arrhythmia6 7.2 1.1 4.6 1.0Chest pain or chest discomfort7 3.8 0.5 2.8 0Cardiac failure8 2.3 1.9 1.0 0.3
1 Adverse events graded according to CTCAE version 3.02 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness 3 Includes terms Muscle spasms, Musculoskeletal pain, Myalgia,
Musculoskeletal discomfort, and Musculoskeletal stiffness4 Includes terms Edema, Edema peripheral, Pitting edema, and Generalized
edema5 Includes all fractures with the exception of pathological fracture6 Includes terms Arrhythmia, Tachycardia, Atrial fibrillation,
Supraventricular tachycardia, Atrial tachycardia, Ventricular tachycardia, Atrial flutter, Bradycardia, Atrioventricular block complete, Conduction disorder, and Bradyarrhythmia
7 Includes terms Angina pectoris, Chest pain, and Angina unstable. Myocardial infarction or ischemia occurred more commonly in the placebo arm than in the ZYTIGA arm (1.3% vs. 1.1% respectively).
8 Includes terms Cardiac failure, Cardiac failure congestive, Left ventricular dysfunction, Cardiogenic shock, Cardiomegaly, Cardiomyopathy, and Ejection fraction decreased
Table 2 shows laboratory abnormalities of interest from Study 1. Grade 3-4 low serum phosphorus (7%) and low potassium (5%) occurred at a greater than or equal to 5% rate in the ZYTIGA arm.
Table 2: Laboratory Abnormalities of Interest in Study 1Abiraterone (N=791) Placebo (N=394)
Laboratory Abnormality
All Grades (%)
Grade 3-4 (%)
All Grades (%)
Grade 3-4 (%)
Hypertriglyceridemia 62.5 0.4 53.0 0High AST 30.6 2.1 36.3 1.5Hypokalemia 28.3 5.3 19.8 1.0Hypophosphatemia 23.8 7.2 15.7 5.8High ALT 11.1 1.4 10.4 0.8High Total Bilirubin 6.6 0.1 4.6 0
Study 2: Metastatic CRPC Prior to Chemotherapy: Study 2 enrolled 1088 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy. Patients were ineligible if AST and/or ALT ≥2.5X ULN and patients were excluded if they had liver metastases.Table 3 shows adverse reactions on the ZYTIGA arm in Study 2 that occurred with a ≥2% absolute increase in frequency compared to placebo. The median duration of treatment with ZYTIGA was 13.8 months.
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
General disordersFatigue 39.1 2.2 34.3 1.7Edema2 25.1 0.4 20.7 1.1Pyrexia 8.7 0.6 5.9 0.2
Musculoskeletal and connective tissue disorders
Joint swelling/discomfort3 30.3 2.0 25.2 2.0Groin pain 6.6 0.4 4.1 0.7
Gastrointestinal disorders
Constipation 23.1 0.4 19.1 0.6Diarrhea 21.6 0.9 17.8 0.9Dyspepsia 11.1 0.0 5.0 0.2
Table 3: Adverse Reactions in ≥5% of Patients on the ZYTIGA Arm in Study 2 (continued)
ZYTIGA with Prednisone (N=542)
Placebo with Prednisone (N=540)
System/Organ Class All Grades1 Grade 3-4 All Grades Grade 3-4Adverse reaction % % % %
Vascular disordersHot flush 22.3 0.2 18.1 0.0Hypertension 21.6 3.9 13.1 3.0
Respiratory, thoracic and mediastinal disorders
Cough 17.3 0.0 13.5 0.2Dyspnea 11.8 2.4 9.6 0.9
Psychiatric disordersInsomnia 13.5 0.2 11.3 0.0
Injury, poisoning and procedural complications
Contusion 13.3 0.0 9.1 0.0Falls 5.9 0.0 3.3 0.0
Infections and infestations
Upper respiratory tract infection 12.7 0.0 8.0 0.0Nasopharyngitis 10.7 0.0 8.1 0.0
Renal and urinary disorders
Hematuria 10.3 1.3 5.6 0.6Skin and subcutaneous tissue disorders
Rash 8.1 0.0 3.7 0.01 Adverse events graded according to CTCAE version 3.02 Includes terms Edema peripheral, Pitting edema, and Generalized
edema3 Includes terms Arthritis, Arthralgia, Joint swelling, and Joint stiffness
Table 4 shows laboratory abnormalities that occurred in greater than 15% of patients, and more frequently (>5%) in the ZYTIGA arm compared to placebo in Study 2. Grade 3-4 lymphopenia (9%), hyperglycemia (7%) and high alanine aminotransferase (6%) occurred at a greater than 5% rate in the ZYTIGA arm. Table 4: Laboratory Abnormalities in >15% of Patients in the ZYTIGA
Arm of Study 2Abiraterone (N=542) Placebo (N=540)
Laboratory Abnormality
Grade 1-4%
Grade 3-4%
Grade 1-4%
Grade 3-4%
HematologyLymphopenia 38.2 8.7 31.7 7.4
ChemistryHyperglycemia1 56.6 6.5 50.9 5.2High ALT 41.9 6.1 29.1 0.7High AST 37.3 3.1 28.7 1.1Hypernatremia 32.8 0.4 25.0 0.2Hypokalemia 17.2 2.8 10.2 1.7
1Based on non-fasting blood drawsCardiovascular Adverse Reactions: In the combined data for studies 1 and 2, cardiac failure occurred more commonly in patients treated with ZYTIGA compared to patients on the placebo arm (2.1% versus 0.7%). Grade 3-4 cardiac failure occurred in 1.6% of patients taking ZYTIGA and led to 5 treatment discontinuations and 2 deaths. Grade 3-4 cardiac failure occurred in 0.2% of patients taking placebo. There were no treatment discontinuations and one death due to cardiac failure in the placebo group. In Study 1 and 2, the majority of arrhythmias were grade 1 or 2. There was one death associated with arrhythmia and one patient with sudden death in the ZYTIGA arms and no deaths in the placebo arms. There were 7 (0.5%) deaths due to cardiorespiratory arrest in the ZYTIGA arms and 3 (0.3%) deaths in the placebo arms. Myocardial ischemia or myocardial infarction led to death in 3 patients in the placebo arms and 2 deaths in the ZYTIGA arms. Post Marketing ExperienceThe following additional adverse reactions have been identified during post approval use of ZYTIGA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.Respiratory, Thoracic and Mediastinal Disorders: non-infectious pneumonitis.
ZYTIGA® (abiraterone acetate) Tablets ZYTIGA® (abiraterone acetate) Tablets

DRUG INTERACTIONSDrugs that Inhibit or Induce CYP3A4 Enzymes: Based on in vitro data, ZYTIGA is a substrate of CYP3A4. In a dedicated drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3) in full Prescribing Information]. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone [see Clinical Pharmacology (12.3) in full Prescribing Information].Effects of Abiraterone on Drug Metabolizing Enzymes: ZYTIGA is an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6. In a CYP2D6 drug-drug interaction trial, the Cmax and AUC of dextromethorphan (CYP2D6 substrate) were increased 2.8- and 2.9-fold, respectively, when dextromethorphan was given with abiraterone acetate 1,000 mg daily and prednisone 5 mg twice daily. Avoid co-administration of abiraterone acetate with substrates of CYP2D6 with a narrow therapeutic index (e.g., thioridazine). If alternative treatments cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate drug [see Clinical Pharmacology (12.3) in full Prescribing Information].In vitro, ZYTIGA inhibits CYP2C8. There are no clinical data on the use of ZYTIGA with drugs that are substrates of CYP2C8. However, patients should be monitored closely for signs of toxicity related to the CYP2C8 substrate if used concomitantly with abiraterone acetate. USE IN SPECIFIC POPULATIONSPregnancy: Pregnancy Category X [see Contraindications].: ZYTIGA can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. While there are no adequate and well-controlled studies with ZYTIGA in pregnant women and ZYTIGA is not indicated for use in women, it is important to know that maternal use of a CYP17 inhibitor could affect development of the fetus. Abiraterone acetate caused developmental toxicity in pregnant rats at exposures that were lower than in patients receiving the recommended dose. ZYTIGA is contraindicated in women who are or may become pregnant while receiving the drug. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for pregnancy loss. Advise females of reproductive potential to avoid becoming pregnant during treatment with ZYTIGA.In an embryo-fetal developmental toxicity study in rats, abiraterone acetate caused developmental toxicity when administered at oral doses of 10, 30 or 100 mg/kg/day throughout the period of organogenesis (gestational days 6-17). Findings included embryo-fetal lethality (increased post implantation loss and resorptions and decreased number of live fetuses), fetal developmental delay (skeletal effects) and urogenital effects (bilateral ureter dilation) at doses ≥10 mg/kg/day, decreased fetal ano-genital distance at ≥30 mg/kg/day, and decreased fetal body weight at 100 mg/kg/day. Doses ≥10 mg/kg/day caused maternal toxicity. The doses tested in rats resulted in systemic exposures (AUC) approximately 0.03, 0.1 and 0.3 times, respectively, the AUC in patients.Nursing Mothers: ZYTIGA is not indicated for use in women. It is not known if abiraterone acetate is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from ZYTIGA, a decision should be made to either discontinue nursing, or discontinue the drug taking into account the importance of the drug to the mother.Pediatric Use: Safety and effectiveness of ZYTIGA in pediatric patients have not been established.Geriatric Use: Of the total number of patients receiving ZYTIGA in phase 3 trials, 73% of patients were 65 years and over and 30% were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.Patients with Hepatic Impairment: The pharmacokinetics of abiraterone were examined in subjects with baseline mild (n=8) or moderate (n=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone after a single oral 1,000 mg dose of ZYTIGA increased by approximately 1.1-fold and 3.6-fold in subjects with mild and moderate baseline hepatic impairment, respectively compared to subjects with normal hepatic function.In another trial, the pharmacokinetics of abiraterone were examined in subjects with baseline severe (n=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone increased by approximately 7-fold and the fraction of free drug increased 2-fold in subjects with severe baseline hepatic impairment compared to subjects with normal hepatic function.
No dosage adjustment is necessary for patients with baseline mild hepatic impairment. In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of ZYTIGA to 250 mg once daily. Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C). If elevations in ALT or AST >5X ULN or total bilirubin >3X ULN occur in patients with baseline moderate hepatic impairment, discontinue ZYTIGA treatment [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3) in full Prescribing Information].For patients who develop hepatotoxicity during treatment, interruption of treatment and dosage adjustment may be required [see Dosage and Administration (2.2) in full Prescribing Information, Warnings and Precautions, and Clinical Pharmacology (12.3)] in full Prescribing Information.Patients with Renal Impairment: In a dedicated renal impairment trial, the mean PK parameters were comparable between healthy subjects with normal renal function (N=8) and those with end stage renal disease (ESRD) on hemodialysis (N=8) after a single oral 1,000 mg dose of ZYTIGA. No dosage adjustment is necessary for patients with renal impairment [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3) in full Prescribing Information].OVERDOSAGEHuman experience of overdose with ZYTIGA is limited.There is no specific antidote. In the event of an overdose, stop ZYTIGA, undertake general supportive measures, including monitoring for arrhythmias and cardiac failure and assess liver function.Storage and Handling: Store at 20°C to 25°C (68°F to 77°F); excursions permitted in the range from 15°C to 30°C (59°F to 86°F) [see USP controlled room temperature].Based on its mechanism of action, ZYTIGA may harm a developing fetus. Therefore, women who are pregnant or women who may be pregnant should not handle ZYTIGA without protection, e.g., gloves [see Use in Specific Populations].PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information)• Patients should be informed that ZYTIGA and prednisone are used
together and that they should not interrupt or stop either of these medications without consulting their physician.
• Patients receiving GnRH agonists should be informed that they need to maintain this treatment during the course of treatment with ZYTIGA and prednisone.
• Patients should be informed that ZYTIGA must not be taken with food and that no food should be consumed for at least two hours before the dose of ZYTIGA is taken and for at least one hour after the dose of ZYTIGA is taken. They should be informed that the tablets should be swallowed whole with water without crushing or chewing. Patients should be informed that taking ZYTIGA with food causes increased exposure and this may result in adverse reactions.
• Patients should be informed that ZYTIGA is taken once daily and prednisone is taken twice daily according to their physician’s instructions.
• Patients should be informed that in the event of a missed daily dose of ZYTIGA or prednisone, they should take their normal dose the following day. If more than one daily dose is skipped, patients should be told to inform their physician.
• Patients should be apprised of the common side effects associated with ZYTIGA, including peripheral edema, hypokalemia, hypertension, elevated liver function tests, and urinary tract infection. Direct the patient to a complete list of adverse drug reactions in PATIENT INFORMATION.
• Patients should be advised that their liver function will be monitored using blood tests.
• Patients should be informed that ZYTIGA may harm a developing fetus; thus, women who are pregnant or women who may be pregnant should not handle ZYTIGA without protection, e.g., gloves. Patients should also be informed that it is not known whether abiraterone or its metabolites are present in semen and they should use a condom if having sex with a pregnant woman. The patient should use a condom and another effective method of birth control if he is having sex with a woman of child-bearing potential. These measures are required during and for one week after treatment with ZYTIGA.
Manufactured by:Patheon Inc.Mississauga, CanadaManufactured for:Janssen Biotech, Inc.Horsham, PA 19044© Janssen Biotech, Inc. 2012 Revised: May 2014 015924-140528
ZYTIGA® (abiraterone acetate) Tablets ZYTIGA® (abiraterone acetate) Tablets

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 39
Journal Spotlight
Novel Combination Increases Progression-Free Survival in CLL Patients Who Are Not Candidates for FludarabineBy Matthew Stenger
In the phase III COMPLEMENT 1 trial reported in The Lancet, Peter
Hillmen, MB, ChB, of St. James’s University Hospital, Leeds, and col-leagues found that the addition of the anti-CD20 antibody ofatumumab (Arzerra) to chlorambucil (Leukeran) increased progression-free survival among patients with chronic lympho-cytic leukemia (CLL) who were not considered candidates for fludarabine due to older age or comorbidities.1
Study DetailsIn this open-label trial, 447 patients
from 16 countries were randomly as-signed between December 2008 and May 2011 to receive chlorambucil at 10 mg/m2 on days 1 to 7 with (n = 221) or without (n = 226) ofatumum-ab every 28 days for 3 to 12 cycles. Ofatumumab was given at 300 mg on day 1 and 1,000 mg on day 8 of cycle 1, followed by 1,000 mg on day 1 of sub-sequent cycles.
Patients could not be considered candidates for fludarabine-based treatment. The primary endpoint was progression-free survival on indepen-dent review in the intention-to-treat population.
The combination and chlorambucil groups were generally balanced for age (median, 69 and 70 years, 69% in both ≥ 65 years), sex (64% and 62% men),
Binet stage (A in 35% and 31%, B in 33% and 38%, C in 32% and 31%), Eastern Cooperative Oncology Group performance status (0 for 39% and 38%, 1 for 53% and 54%), B symptoms (53% in both), and all other baseline characteristics.
Progression-Free SurvivalBoth groups received a median of
six treatment cycles. Median follow-up was 28.9 months. Median progression-
free survival was 22.4 months (95% confidence interval [CI] = 19.0–25.2 months) in the chlorambucil/ofatu-mumab group vs 13.1 months (95% CI = 10.6–13.8 months) in the chloram-bucil group (hazard ratio [HR] = 0.57, P < .0001).
Hazard ratios favored combination treatment in all subgroups. Combina-tion treatment was associated with consistent benefit in patients aged < 65 years (HR = 0.54, 95% CI = 0.34–0.85), ≥ 65 years (HR = 0.57, 95% CI = 0.43–0.76), and ≥ 75 years (HR = 0.56, 95% CI = 0.35–0.89).
Time to Next TherapyThe combination group had
significantly prolonged time to next therapy (median, 39.8 vs 24.7 months, HR = 0.49, P < .0001). After a median of 6 months of treatment in
both groups, the median treatment-free period was 34 vs 19 months. Sal-vage therapy was used in 29% vs 44% of patients, with the most common treatment being rituximab (Rituxan) alone or in combination (17% and 31%).
Objective response rates were 82% vs 69% (odds ratio = 2.16, P = .001), with complete response in 14% vs 1%. No difference in overall survival was observed at the time of analysis, with death occurring in 15% of the combination group and 18% of the chlorambucil group (HR = 0.91, P = .666). Two-year survival was 89% vs 87% and 3-year survival was 85% vs 83%.
Adverse EventsAdverse events ≥ grade 3 occurred
in 50% of the combination group and 43% of the chlorambucil group, in-cluding 59% and 51% of patients aged ≥ 65 years.
Among grade ≥ 3 adverse events, neutropenia occurred in 26% vs 14% (30% vs 15% of patients aged ≥ 65 years), thrombocytopenia in 5% vs 10% (7% vs 10%), anemia in 5% vs 5% (5% vs 6%), and infection in 9% vs 12% (13% vs 15%). Grade ≥ 3 infu-sion-related reactions occurred in 10% of the combination group.
Adverse events led to discontinu-ation of study treatment in 13% of both groups and in 15% of patients aged ≥ 65 years in both groups. Death considered related to study treatment occurred during the treatment period in three patients in the chlorambucil-ofatumumab group and two in the chlorambucil group.
Study ImplicationsThe investigators concluded:
“Addition of ofatumumab to chlo-rambucil led to clinically important
improvements with a manageable side-effect profile in treatment-naive patients with chronic lymphocytic leukaemia who were elderly or had comorbidities. Chlorambucil plus ofatumumab is therefore an impor-tant treatment option for these pa-tients who cannot tolerate more in-tensive therapy.”
They noted: “More research is warranted to establish whether and how ofatumumab and obinutuzum-ab [Gazyva] differentiate from each other and from rituximab; to estab-lish if one antibody is more suit-able for a specific patient subgroup; and to explore which chlorambucil backbone regimen is the most com-plementary to anti-CD20 antibody therapy.” n
Disclosure: The study was funded by GlaxoSmithKline and Genmab A/S. For full disclosures of the study authors, visit www.thelancet.com.
Reference1. Hillmen P, Robak T, Janssens A,
et al: Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lympho-cytic leukaemia (COMPLEMENT 1): A randomised, multicentre, open-label phase 3 trial. Lancet 385:1873-1883, 2015.
Hematology
Addition of ofatumumab to chlorambucil led to clinically important improvements with a manageable side-effect profile in treatment-naive patients with chronic lymphocytic leukaemia who were elderly or had comorbidities.
—Peter Hillmen, MB, ChB
Improving Therapy for Chronic Lymphocytic
Leukemia
■ The addition of ofatumumab to chlorambucil significantly prolonged progression-free survival.
■ Similar degree of benefit was observed in older and younger patients.
Childhood Cancer Awareness Month

PAGE 40 The ASCO Post | SEPTEMBER 10, 2015
New Orleans Summer Cancer Meeting
Genetic Testing in Breast Cancer Offers Much Information but Poses Challenges in InterpretationBy Caroline Helwick
For breast cancer patients with ro-bust family histories, medical on-
cologists should be testing not only for BRCA1/2 mutations, but also for large duplications and deletions as well as for PALB2 mutations.
“These [findings] have proven util-ity in testing breast cancer patients,” said Louise E. Morrell, MD, Medical Director of the Lynn Cancer Institute, Boca Raton, Florida, in an update on germline testing for breast cancer at the 2015 New Orleans Summer Can-cer Meeting.1
Large Duplications and Deletions in BRCA
Comprehensive BRCA testing includes complete sequencing of the BRCA1 and BRCA2 genes and an additional procedure to identify five common large rearrangements in the BRCA1 gene. BART—the BRACAnalysis Large Rearrangement Test—was introduced in 2006 and detects additional rare large genomic rearrangements in both BRCA1 and BRCA2. About 1% to 2% of individu-als who meet the family history criteria for BRCA gene testing will have a mu-tation detected by the test. The guide-lines of the National Comprehensive Cancer Network (NCCN) now rec-ommend large rearrangement deletion and duplication testing to be included in BRCA gene testing.
“The original BRCA analysis [rou-tine Sanger sequencing] looked for sequence errors, which is like going through sentences and finding errors. But if you cut out a whole paragraph, sequencing doesn’t find the error. This is what large-deletion analysis finds,” she explained.
In very high-risk families, 9% of all mutations are due to large duplications and deletions. This additional testing identifies an additional 2.3% of muta-tions in such families, though far less among patients with more modest risk (those with triple-negative disease or only one affected family member).
“This means that for patients with a [considerable] family history who are looking for an explanation for the many cancers [in their families], there is a 2.3% chance you will find a muta-tion testing for large rearrangement delection duplication when tests were negative before,” she said.
Beyond BRCA“Today, oncologists will be testing
for many abnormalities beyond BRCA, one being PALB2,” she said. While 10 times less frequent than BRCA, it is a genetically important factor.
PALB2 is a partner and localizer of BRCA2 and is part of the homol-ogous DNA recombination-repair pathway. PALB2 accounts for 2.4% of familial aggregates of breast cancer and renders a 33% lifetime risk in car-riers. This risk increases to 58% if the person has more than two affected first-degree relatives.
“We have learned that family his-tory matters a lot with regard to risk associated with this mutation,” Dr. Morrelll said. “We now know that these genes and those we discover in the future should not be viewed in isolation. They need an accompa-nying familial phenotype to be most damaging.”
Information from 17,000 BRCA mu-tation carriers has been evaluated by the Consortium of Investigators of Modifi-ers of BRCA1/2 (CIMBA). This has led to the identification of risk modifiers for these mutation carriers, which enhanc-es the accuracy of risk prediction for the individual patient.
“We are realizing that not all pa-tients are the same,” she said. “This may have added value to the patient who has the mutation but doesn’t want her ovaries out yet.”
Understanding Panel Testing Panel testing with next-generation
sequencing is now offered by more than a dozen laboratories. Data from panel testing are being published, confirming the challenges with es-timates of cancer risk and the prob-lematic issues of variants of unknown significance and incidental findings.
These observations appear relatively consistent across laboratories and in-stitutions, she said.
Panels should always include the high-risk and intermediate-risk genes. High-risk alleles are rare-to–very rare and impose a very high relative risk (up to 10-fold) for breast cancer. In this category are TP53, BRCA1, BRCA2, PTEN, CDH1, and STK11. Intermediate-risk alleles are rare with essentially a two- to four-fold increase in the risk of cancer. They include BRIP1, ATM, PALB2, and CHEK2.
Panel testing reports “actionable findings,” which are findings that could “plausibly lead to a specific medical recommendation or inter-vention,” she said. It does not mean that taking such action has demon-strated clinical utility, such as im-provement in survival.
Panels evaluate 10 times the num-ber of genes as single-gene testing, but they also increase the number of incidental findings and greatly multiply the number of variants of unknown significance. A variant of unknown significance is an altera-tion that reflects only a minor change along a gene that may be 10,000 to 15,000 nucleotides long. For exam-ple, in the sentence, “The big red dog ran out,” a variant of unknown sig-nificance might cause it to read, “The big ned dog ran out,” she explained.
“Based on this minor change, one would not necessarily expect a vari-ant of unknown significance to have significant health consequences, but the variants of unknown significance classification means that we do not yet know,” she said. “Determining the significance of variants is proving to be a major challenge to interpreting multiplex panel testing.”
The clinician can try to make sense of a variant of unknown signifi-cance by looking at its location with-in a gene and its protein functional-ity, cosegregation with disease in affected families, prevalence in a con-trol population, and other aspects. Still, confusion reigns with variants of unknown significance, “and this is where we must dedicate consider-able time, expertise, and resources in interpreting tests for our patients and families: the interpretaton is very challenging,” she remarked.
Other issues with panel testing create challenges for clinicians: lack of publication of the results of valida-tion studies, lack of standardization for validation among labs, and un-known accuracy of panel tests. Labs can also differ in other ways: how they report results, their “transpar-ency,” depth of coverage with next-generation sequencing, detection of large-deletion duplication, methods of defining variants of unknown sig-nificance, genes tested, and payment approaches.
Differences in interpretation may be the most confusing to clinicians and patients. “A patient can have a test at one lab, and a gene is called a mutation. Her sister can use a differ-ent lab, get the same finding, and it’s not called a mutation. We are not al-ways getting the same interpretation of the same finding,” Dr. Morrell said.
When to Consider Panel Testing
“When should we test for a panel of cancer genes? That’s a question that is not yet answered,” she indicated.
Dr. Morrell said she opts for panel testing when the results can help fami-lies understand their strong family his-tory and when the results are likely to be actionable. “Mostly, panel testing helps future generations,” she added.
She noted that 10% of breast cancer patients with a strong family history who undergo panel testing will have a deleterious mutation, of which about 6% will be due to BRCA1 or BRCA2 and 4% due to genes other than BRCA1/2. The panel test should include PALB2, which carries a lifetime risk of about 33% to 58%. For estrogen receptor–positive breast cancer patients, CHEK2,
Breast Cancer
You will gain information if you do a panel test, but not all findings will be actionable. Be prepared for variants of unknown significance, which average one or two per test.
—Louise E. Morrell, MD
continued on page 41

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 41
New Orleans Summer Cancer Meeting
Emerging Technology Will Help Tackle Tumor Complexity By Caroline Helwick
Emerging laboratory technology will be “moving the bar forward” in
terms of molecular markers, genomics, and gene-expression profiling, with the potential for huge payoffs to oncologists and patients, according to Mark Pegram, MD, the Susy Yuan-Huey Hung Profes-sor of Medicine at Stanford School of Medicine, Director of the Breast Cancer Oncology Program at Stanford Women’s Cancer Center, and Co-Director of Stan-ford’s Translational Oncology Program in Palo Alto, California.
At the 10th Annual New Orleans Summer Cancer Meeting, Dr. Pegram described novel approaches as they might be applied in breast cancer, show-ing enthusiasm for their possibilities, tempered with a note of caution.1
“These are technologies emerging in the lab, and what the future may hold for their clinical application, we don’t yet know,” he said. “Hopefully, in the end, these will be useful for clinical de-cision-making and result in improved patient outcomes.”
Dr. Pegram also described the chal-lenge of developing targeted therapies for rare mutations, even with these so-phisticated tools.
End of the Pathologist?“At Stanford, surgical oncology has
conspired to try to eliminate the need for the pathologist,” Dr. Pegram quipped.
Virtually all the information use-ful to oncologists comes by way of the pathologist’s review—an established practice that might be disrupted by cutting-edge technology called selec-tive negative ion mode desorption elec-trospray ionization mass spectrometry, developed at Stanford, he said. Desorp-tion electrospray ionization mass spec-trometry is an ionization technique that can image biologic samples without the need for extensive sample preparation, investigating the distribution of diag-nostic lipids and metabolites directly from tiny tissue sections.
As the developers explained in a published investigation,2 “Samples are bombarded with microdroplets that dissolve hundreds of lipids and metab-olites. The splash forms secondary mi-crodroplets that enter a mass spectrom-eter, providing a detailed chemical map of the distribution of molecules within the sample surface.”
Dr. Pegram added, “On a thin sam-ple of tumor tissue you can measure, based on their molecular weight, all the biologic elements that splash out of that tumor sample. You find lipids, phos-pholipids, proteins, products of metab-olism—everything that’s in there.”
Molecular masses of these multiple types of molecules from the tumor are compared to normal tissue and used to construct patterns that differentiate the tumor from the epithelium and stroma. The technology may someday provide a means of interrogating tumor tissue without the need for a large surgical sam-ple or reading by pathologist, he said.
Where Next-Generation Sequencing Falls Short
Outside of a handful of breast can-cer–associated genes with frequencies greater than 10%, “the sobering reality” is that most other mutations are “one-offs” that are unique to individuals or small groups of individuals, Dr. Pegram noted. Next-generation sequencing may be able to identify these new tar-gets, but developing drugs to target them will be difficult.
The HER2 kinase mutation is an example. This mutation is found in pa-tients with HER2-negative breast can-cer, especially of the lobular subtype. It occurs in the absence of gene amplifica-tion and has a frequency of only 1.6% (3% in lobular tumors).
“Because these mutations tend to occur in the kinase domain, it sounds reasonable to treat with lapatinib [Tykerb]; however, this doesn’t work,” he indicated. “These recombinant con-
structs of mutants do not bind to lapa-tinib anymore, but they do bind to the tyrosine kinase inhibitor neratinib.”
In a collaboration spearheaded by the group at Washington University in St. Louis, Dr. Pegram and colleagues are studying neratinib in a small cohort of patients with HER2 mutations. Taking into account the fraction of patients who harbor a HER2 kinase mutation, meet all eligibility criteria, and are willing to
participate in a clinical trial, the research-ers calculated the “number needed to study” neratinib in this population, con-cluding that in the best-case scenario, 125 patients must be screened to yield 1 enrollee. Nevertheless, the study did launch, with a low double-digit number of patients currently enrolled.
“One response to neratinib, a drug we thought would have exquisite activ-ity,” Dr. Pegram said. This is “sobering” if it foreshadows the payoff for such en-deavors, he commented.
Tackling Heterogeneity“Another conundrum in tumor
genome sequencing campaigns is the degree of heterogeneity in breast cancer,” he continued. Ge-nomes sequenced for more than 100-fold coverage will identify separate clones within the same primary tumor that have different genotypes—and this can be observed early, at diagnosis.
One of the first examples of this
complex heterogeneity was a dis-covery made in renal cell carcinoma. Gerlinger and colleagues evaluated spatially separated samples from the primary tumor and associated metas-tases using exome sequencing.3 A sin-gle biopsy showed about 70 mutations that accounted for only half of all the mutations identified across multiple biopsies. Only 34% were contained in all regions.
“Breast cancer is the same,” Dr. Pegram indicated. “We are wrestling with how to accommodate this heterogeneity.”
Harnessing the Dynamic Nature of Mutations
Moreover, these sequences are ever-changing as a consequence of selection pressure after treatment. It may be pos-sible, however, to turn this characteris-tic into a positive feature.
Vanderbilt investigators studied the molecular landscape of tumors after neoadjuvant chemotherapy in patients with triple-negative breast cancer, fo-cusing on the molecular underpinnings of patients lacking a complete patholog-ic response (a possible marker of resis-tance). By next-generation sequencing and digital RNA expression analysis, they identified diverse molecular le-sions and pathway activation in drug-resistant tumor cells, compared to pre-treatment biopsies; they hypothesized
ATM, and TP53 should be included in the panel.
“You will gain information if you do a panel test, but not all findings will be actionable. Be prepared for variants of unknown significance, which can average one or two per test,” she said. Despite these issues,
she added, “There’s value in collect-ing these data…. We should be doing all we can to gather this information.”
Dr. Morrell suggested that phy-sicians consult the NCCN Clinical Practice Guidelines for Genetic/Familial High-Risk Assessment2 and the National Institutes of Health GeneReviews3 websites for informa-tion about unfamiliar genes that are
reported in test results. n Disclosure: Dr. Morrell reported no
potential conflicts of interest.
References1. Morrell L: Next generation sequencing
and inherited risk for cancer: BRCA, Lynch and beyond. 2015 New Orleans Summer Cancer Meeting. Dr. Peter A. Cassileth Me-morial Lecture. Presented July 18, 2015.
2. National Comprehensive Cancer Network: Genetic/Familial High-Risk Assessment, Version 2.2015. Available at www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed August 14, 2015.
3. Pagon RA, Adam MP, Ardinger HH, et al (eds): GeneReviews. Available at www.ncbi.nlm.nih.gov/books/NBK1116. Ac-cessed August 14, 2015.
Genetic Testingcontinued from page 40
Technology
These are technologies emerging in the lab, and what the future may hold for their clinical application, we don’t yet know. Hopefully, in the end, these will be useful for clinical decision-making and result in improved patient outcomes.
—Mark Pegram, MD
continued on page 42

PAGE 42 The ASCO Post | SEPTEMBER 10, 2015
New Orleans Summer Cancer Meeting
Tumor Complexitycontinued from page 41that these findings may mirror micro-metastasis destined to recur clinically. Ninety percent of the tumors contained a genetic alteration potentially treatable with available targeted drugs.4
“Tumors were enriched for muta-tions along pathways we all know—those related to DNA repair, growth factor receptor amplification, PI3K/AKT, Ras/RAF/ERK, and cell cycle,” Dr. Pegram noted. “This suggests that with this type of analysis we may have the opportunity for personal-ized adjuvant treatment.”
Mutations of the estrogen recep-tor are further examples of the dy-namic nature of mutations. Estrogen receptor mutations occur in up to 20% of metastatic tumors (vs about 1% in primary tumors). The frequen-cy appears to be enriched with prior endocrine treatment, and there is a clear trend toward rising mutation frequency from primary disease to early metastatic disease to late meta-static disease.
“These mutations are also quite interesting,” he observed. “They cluster in the ligand-binding domain, which begs the question of whether antiestrogen therapeutics will hit them…. This could prove to be an opportunity, now that we understand that estrogen receptor mutations are not uncommon.”
‘Composite’ Look at Heterogeneity
Could circulating tumor cells provide a snapshot of the tumor’s global picture? A new cell separation method that employs a microfluidic device yields live tumor cells that can not only be enumerated but analyzed for their components, such as steroid receptor, HER2, mutations, and pro-teins.
“In theory, this could be power-ful,” Dr. Pegram commented. “We could measure the percentage of cells that are [estrogen receptor]–positive in the circulation, and if it reflects the global tumor burden, we can estimate the degree of heterogeneity of that marker and perhaps correlate that with the expectation of response to an antiestrogen, for example.”
It is actually now possible to iso-late single cells and observe their makeup as well. In a collaboration with Amy Herr, PhD, of the Depart-ment of Bioengineering, University of California at Berkeley, who has pioneered a new technology, Dr. Pe-
gram’s lab is doing this, focusing on the protein content of circulating tu-mor cells. Using a single-cell Western blot analysis platform, researchers place individual cells into microw-ells, where they can be lysed and elec-trophoresed, producing a readout of all proteins within that cell.5
Single cells can also be subjected
to whole-exome sequencing. This may better capture “what’s in the gen-eral mix” of the tumor, as compared to a biopsy of just one site. This ap-proach captures cell-to-cell hetero-geneity; provides objective, quanti-tative, reproducible, digital data for archiving; and “spares precious pa-tient tissue,” Dr. Pegram said. “Maybe
this is a path forward toward getting a handle on the heterogeneity of circu-lating tumor cells.”
Moreover, cells may not be needed at all to produce a global snapshot, as tumor-specific DNA can now be iso-lated from the circulation. This cell-free DNA approach can correlate the amount of circulating DNA with radio-
Advertisement not displayed in digital edition at advertiser’s request

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 43
New Orleans Summer Cancer Meeting
graphic findings, can quantitate second-site mutations, and can be applied to any tumor type. “This could be very in-teresting in terms of following patients with metastatic disease,” he said.
Dr. Pegram concluded by acknowl-edging that the path to the develop-ment of new biomarkers is long and arduous but could ultimately lead to
better treatments and outcomes. nDisclosure: Dr. Pegram reported no potential
conflicts of interest.
References1. Pegram M: Novel molecular markers,
genomics, and gene expression profile: Mov-ing forward the bar for breast cancer therapy development. 2015 New Orleans Summer
Cancer Meeting. Presented July 18, 2015.2. Eberlin LS, Gabay M, Fan AC, et al:
Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc Natl Acad Sci USA 111:10450-10455, 2014.
3. Gerlinger M, Rowan AJ, Horswell S, et al: Intratumor heterogeneity and branched evolution revealed by multiregion sequenc-ing. N Engl J Med 366:883-892, 2012.
4. Balko JM, Giltnane JM, Wang K, et al: Molecular profiling of the residual disease of triple-negative breast cancer after neoad-juvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 4:232-245, 2014.
5. Hughes AJ, Spelke DP, Xu Z, et al: Single-cell western blotting. Nat Methods 11:749-755, 2014.
Advertisement not displayed in digital edition at advertiser’s request

PAGE 44 The ASCO Post | SEPTEMBER 10, 2015
New Orleans Summer Cancer Meeting
State-of-the-Art Management of Germ Cell Tumors Produces High Cure RatesBy Caroline Helwick
Pasquale W. Benedetto, MD, the Leonard M. Miller Professor of
Medicine at the University of Miami/Sylvester Comprehensive Cancer Cen-ter, recently spoke at the 2015 New
Orleans Summer Cancer Meeting about his approach to diagnosing and treating germ cell tumors in men.1 The ASCO Post was there to capture Dr. Benedetto’s insights.
The Basics Germ cell tumors derive from an
ovum or spermatozoon, or precursor cells thereof. While germ cell tumors are primarily gonadal (95%), they can arise
in the mediastinum, retroperitoneum, or central nervous system, or constitute mid-line germ cell syndrome.
“These are curable malignancies in more than 90% of cases. Patients diag-
Advertisement not displayed in digital edition at advertiser’s request

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 45
New Orleans Summer Cancer MeetingGerm Cell Tumors
continued on page 46
nosed with localized disease can be cured with surgery, but nonlocalized disease can also be cured,” Dr. Benedetto emphasized. “It is incumbent upon medical oncologists to identify these tumors, especially when the presentation is not typical, and to treat intensively to achieve complete remission.”
In men, germ cell tumors are broadly divided into seminoma and nonsemino-
matous disease. The most primitive form of the latter is embryonal carcinoma. Dif-ferentiation along embryonic lines gives rise to any of the three germ layers (endo-, ecto-, or mesoderm), which in aggregate is called teratoma. Differentiation along extraembryonic lines gives rise to chorio-carcinoma or yolk sac tumor.
A distinction between seminoma and
nonseminomatous lesions is important. Seminomas are biologically indolent, whereas nonseminomas can be aggres-sive and more likely to metastasize.
Signs Pointing to Germ Cell Tumor
“Any patient between the ages of 15 and 40 with an intratesticular mass has
germ cell tumor until proven otherwise,” he said. Other possible presentations in-clude anterior superior mediastinal mass and localized retroperitoneal mass.
In addition, oncologists should sus-pect a germ cell tumor in any male pa-tient with a midline tumor described as “poorly differentiated carcinoma” without an obvious primary (ie, mid-line germ cell syndrome); with elevated alpha-fetoprotein or beta–human cho-rionic gonadotropin (hCG); or with a tumor of unknown origin with isochro-mosome 12p, a molecular feature char-acteristic of germ cell neoplasms.
Importantly, occult disease, including recurrences, can be documented by these serum markers. Both alpha-fetoprotein and beta-hCG can be elevated in non-seminomatous lesions. In seminomas, alpha-fetoprotein is never elevated, and beta-hCG may or may not be elevated.
Staging of Germ Cell CancersGerm cell tumors are limited to
stage I disease in 85% of seminomas but in only 35% of nonseminomatous lesions. Fortunately, this distinction does not determine outcomes, he said.
“We can cure both these popula-tions to the same degree. It’s simply the mechanism by which we get there or the relative need for any other treat-ment after the primary surgery,” Dr. Benedetto explained.
Tumors are best staged using the In-ternational Germ Cell Cancer Consensus Group classification system, which relies heavily on tumor marker levels and takes into account the presence and sites of me-tastases and magnitude of tumor marker elevations, risk-stratifying patients into good-, intermediate-, and poor-risk sub-sets. With seminomas, there is no cat-egory for “poor risk,” as all these patients “should be cured,” he added.
Understanding Tumor MarkersWhile tumor markers are vital com-
ponents of assessment, in the case of seminoma they do not define the disease stage. “There is no such thing as a semi-noma with [alpha-fetoprotein] elevation,” he said. Patients labeled as having “semi-noma” who have elevated alpha-fetopro-tein, without an alternative explanation, undoubtedly have a nonseminomatous component to their disease and should be treated according to the algorithm for nonseminomatous disease.
A marker can only be interpreted with an understanding of its half-life, which is 5 to 7 days for alpha-fetopro-tein and 2 to 3 days for beta-hCG. This information helps monitor the patient
Advertisement not displayed in digital edition at advertiser’s request

PAGE 46 The ASCO Post | SEPTEMBER 10, 2015
New Orleans Summer Cancer Meeting
without the use of imaging.“A single assay is uninterpretable,”
he said. “If a marker is elevated a few weeks after treatment, you don’t know what it means unless you have a previ-ous report. You need at least two points to determine whether the difference between those points is within the pre-dicted range, according to the half-life of the marker.”
A persistently elevated tumor marker after surgery or chemotherapy is “un-equivocal evidence” of disease, suggesting that the patient has not achieved a com-plete remission. A rising level indicates persistent, recurrent, or new disease.
Milestones That Inform Current Treatment
Dr. Benedetto described several discoveries and milestones in the treat-ment of these tumors that have in-formed the current standard of care: • The efficacy of cisplatin is critical to
inducing complete remission in dis-seminated testis cancer; carboplatin should not be substituted.
• Intensity of treatment, not length of treatment, matters.
• Complete remissions are durable, and maintenance therapy is unnecessary.
• Risk-assessment criteria are based on the Indiana staging system, classify-ing disseminated disease as minimal-, moderate-, or high-risk.
• Bleomycin/etoposide/cisplatin is
equivalent to cisplatin/vinblastine/bleomycin, is less toxic, and remains the standard of care.
• For good-risk patients, three cycles of bleomycin/etoposide/cisplatin are equivalent to four cycles, and for poor-risk patients, four cycles of bleomycin/etoposide/cisplatin are better than four cycles of cisplatin/vinblastine/bleomycin.
• Etoposide/ifosfamide/cisplatin or upfront high-dose chemotherapy and stem cell transplantation is not better than bleomycin/etoposide/cisplatin as first-line therapy for poor-risk patients.“Everything we do is to maximize the
patient achieving a complete response,” he elaborated. “With 12 weeks of che-motherapy, we cure most patients.”
Practical Treatment TipsDr. Benedetto offered a number of
clinical pearls for treating germ cell tumors:• Treat good-risk patients with 9 weeks
of bleomycin/etoposide/cisplatin; treat poor-risk patients for 12 weeks.
• Maintain treatment intensity over the 21-day cycles. Do not get off sched-ule; do not initiate on a Wednesday unless you can treat the patient over the weekend.
• Do not hold any dose of chemo-therapy for a low absolute neutrophil count, as these patients typically do not develop febrile neutropenia.
• Do not give growth factors to the av-erage patient.
• Do not repeat computed tomography (CT) imaging during chemotherapy; simply repeat tumor marker assess-ments at the beginning of each cycle. If markers are decreasing predictably, do not change treatment.
Treatment of Stage I DiseaseSince most patients with stage I dis-
ease are cured by the primary surgery, adjuvant chemotherapy is not war-ranted for most, if not all, patients. Af-ter orchiectomy, 70% of patients with nonseminomatous disease are cured. Most of the 30% who relapse will do so within a few months, and their recur-
rences occur with good-risk features. Therefore, they are highly, essentially universally, curable.
“There is nothing to be lost with surveillance and nothing to be gained with intervention,” Dr. Benedetto maintained. “It’s better to wait for the few patients who will relapse, and then treat them for cure with chemo-therapy and avoid overtreatment of many who are destined never to have a recurrence.”
The only patient for whom adjuvant treatment might be considered is one
who may not be compliant with surveil-lance. His surveillance strategy involves CT scans of the abdomen only (not the lungs or pelvis) and chest x-rays.
Imaging, Residual MassesDr. Benedetto emphasized, “CT
scans in the middle of chemotherapy will only confuse you…. All you really need to know is whether tumor markers are going down with treatment.”
Postchemotherapy retroperitoneal exploration may be the key to achieving long-term remission in patients with a residual mass > 1 cm or after salvage therapy, he said.
Residual masses in nonseminoma-tous patients will be viable germ cell tumor in only 8%, teratoma in 50%, and fibrosis in 43%. Teratomas must be re-moved, as they can dedifferentiate and become resistant tumors.
For nonseminomas, positron-emis-sion tomography (PET) scanning is not necessary for staging or for evaluating outcomes, as PET cannot distinguish between teratoma and cancer and can-not predict for relapse, he said. For seminoma, however, PET can predict for residual disease > 3 cm with 100% accuracy; therefore, it is useful for gaug-ing response but not for initial staging.
Predicting, Treating Recurrences“You don’t want to need salvage ther-
apy, since we don’t cure many patients that way. Do things right the first time,” Dr. Benedetto exhorted attendees.
While first-line chemotherapy is al-most 100% curative, the cure rate drops to less than 40% in the second-line set-ting. Salvage chemotherapy should be ifosfamide-based, preferably including paclitaxel, ifosfamide, and platinum. The use of transplant can be discussed with patients. n
Disclosure: Dr. Benedetto reported no potential conflicts of interest.
Reference1. Benedetto PW: How do I treat a pa-
tient diagnosed with a seminoma and a non-seminoma testicular cancer: State-of-the-art management to obtain a high cure rate. 2015 New Orleans Summer Cancer Meeting. Presented July 18, 2015.
Everything we do is to maximize the patient achieving a complete response. With 12 weeks of
chemotherapy, we cure most patients. —Pasquale W. Benedetto, MD
Germ Cell Tumorscontinued from page 45
Resilient New Orleans: 10 Years After KatrinaBy Caroline Helwick
I t’s been 10 years since floodwa-ters washed away lives in New
Orleans—for most, figuratively or temporarily, but for more than 1,800, literally. We who call this place home all lost something—homes, posses-sions, jobs, pets, loved ones, our san-ity. Mold and muck marked our days for a long time, but now, we seem somehow better for it.
Ten years later we are 10 times stronger, with sturdier levees and homes, better public schools and public housing, state- ‐of- ‐the- ‐art hospi-tals, greater commerce (more shops and restaurants!), and an influx of young entrepreneurs that has fresh-ened our outlook.
We have hosted a Super Bowl (and
even won one!) and welcomed a grow-ing number of conventions (including the 2013 American Society of Hema-tology Annual Meeting). NOLA has been called the #1 Brainpower City (Forbes), the #1 Most Improved Metro Area (Wall Street Journal), the #1 Most Inspiring U.S. City (Good Magazine, 2014), and more. Katrina was a defin-
ing moment for us. We’ve rebuilt—but upon our centuries- ‐old traditions for which we are known and loved.
In fact, there has been so much to cheer about that it’s easy to forget that not everything, and not everyone, has “come back.” They may never. And we are still plagued by problems common to other great American Cities. We still mourn. We are still in recovery But we are “irrepressible,” to quote President Obama. We have perhaps an unwar-ranted confidence in our future.
We will keep moving forward—but keeping an eye to the sky, of course. n
Caroline Helwick is a reporter for The ASCO Post and lives in New Orleans.
Caroline Helwick

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 47
In MemoriamOncology Worldwide
Roswell Park Cancer Institute Partners With Cuban Scientists to Develop Lung Cancer VaccineA thaw in 5 decades of enmity between the United States and Cuba could lead to scientific progress in both countries.By Jo Cavallo
J ust 4 months after President Barack Obama’s announcement in Decem-
ber 2014 that there would be an easing of the trade embargo between the Unit-ed States and Cuba, a deal was struck between Roswell Park Cancer Institute in Buffalo, New York, and the Center for Molecular Immunology (CIM) in Havana, Cuba, to test a immunotherapy vaccine for non–small cell lung cancer (NSCLC) in the United States.
The vaccine, known as CimaVax EGF, was developed by CIM and has been approved for the treatment of late-stage NSCLC in Cuba and Peru. Although not a cure, CimaVax has been shown in phase III clinical trials in stage III and IV NSCLC to extend survival between 4 and 6 months, curtail symp-toms like coughing and breathlessness, and improve overall quality of life. The vaccine is now being tested in Japan and several European countries.
The agreement between Roswell Park Cancer Institute and CMI to de-velop CimaVax in the United States was made during the New York State Trade Mission to Cuba in April 2015, but a partnership between the two institu-tions has been in place since 2011, when investigators from Roswell Park and
CIM began exchanging academic and preclinical research information. Soon after, Roswell Park received a licensing agreement from the Government Cen-ter for Quality Control of Medicines, the Cuban regulatory authority, to test CimaVax in the laboratory.
Long-Term ObjectivesIf the U.S. Food and Drug Adminis-
tration (FDA) approves Roswell Park’s investigational new drug application, which is expected by early 2016, the cancer center will launch a phase I study of the vaccine to investigate toxicities, followed by a phase I study of the drug to test efficacy in patients with early-stage NSCLC.
“Our hope for this vaccine longer term, beyond those early studies, is to see how effective it is in patients that don’t have as much tumor bur-den as stage III or IV lung cancer patients,” said Candace S. Johnson, PhD, President and CEO, Wallace Family Chair in Translational Re-search, and Professor of Oncology at Roswell Park Cancer Institute.
“After we finish our toxicity phase I approach, which won’t take long to complete since the drug has already
been tested in early clinical studies in Cuba, we would like to study the vaccine in patients presenting with a single malignant nodule, because even though those patients are good candi-dates for surgery, they are at high risk for disease recurrence. We want to see if we can elicit a more long-term re-sponse in early-stage patients.” If the studies prove successful in the treat-ment of lung cancer, according to Dr. Johnson, Roswell Park proposes to test CimaVax in other cancer types with epidermal growth factor receptor (EGFR) mutations, including prostate, breast, and colon cancers, and for the prevention of epithelial cancers.
CimaVax EGF works by depriving
tumors of epidermal growth factor by making the protein immunogenic and inducing an immune response that re-duces the amount of circulating EGF protein in patients, stunting tumor pro-gression. Targeting EGF protein, said Dr. Johnson, provides a totally different mechanism for prohibiting cancer cell growth than the tyrosine kinase inhibi-tors approved by the FDA in the treat-ment of advanced-stage NSCLC, in-cluding erlotinib, gefitinib (Iressa), and afatinib (Gilotrif), and so may produce fewer side effects.
“Because CimaVax immunizes pa-tients to make antibodies against the EGF protein, it is inherently probably not going to have the side effects of a drug like erlotinib, where you are trying to achieve some inhibition of receptor activity,” said Dr. Johnson. “Inducing an immune response with CimaVax could have side effects like fever and chills, but actually the reports from the Cuban sci-entists show very little toxicity from the vaccine. We will have to sort this all out ourselves once we begin clinical studies.”
In addition to CimaVax, Cuba’s CIM has developed a second vaccine for NSCLC called racotumomab (Vaxira) and the monoclonal antibody nimotu-zumab for the treatment of glioblasto-ma, which is being studied in both adult and pediatric patients.
Therapy for Diabetic Foot UlcersInnovative oncology drugs are not
the only Cuban-made medications at-tracting attention outside their shores. Heberprot-P, a novel therapy for ad-vanced diabetic foot ulcers, was devel-oped 9 years ago by the Center for Ge-netic Engineering and Biotechnology
Thoracic Cancer
In Cuba: Doing More With Less
■ $4 million: Science ministry’s research and development budget in 2015
■ $2 million: Gross domestic product per peer-reviewed scientific paper, about 3% of the U.S. amount
■ 6,000: Approximate number of scientists in Cuba
■ 559: Approximate number of Cubans who earned a BS in natural sciences or mathematics in 2007–2008
■ $36: How much most scientists in Cuba earn each monthSource: Stone R: In from the cold. Science 348:746-751, 2015.
continued on page 48
Roswell Park Cancer Institute/Center for Molecular Immunology, Havana
Members of Roswell Park Cancer Institute, including Mary Reid, PhD, Director of Cancer Screening and Survivorship (right, front), and the Center for Molecular Immunology, including Kalet León, Director of Research and Development (left, front), gather with colleagues outside the center in Havana, Cuba, after a meeting to discuss development of CimaVax EGF in November 2014.

PAGE 48 The ASCO Post | SEPTEMBER 10, 2015
In MemoriamOncology Worldwide
in Havana. The drug, which contains recombinant human epidermal growth factor, has been shown in clinical stud-ies to accelerate healing of deep and complex ulcers and reduce diabetes-re-lated amputations,1 according to some reports by between 75% and 80%. Al-though not available for clinical testing in the United States because of the trade embargo, Heberprot-P is being used in 26 other countries and is reported to have treated as many as 165,000 pa-tients with diabetic foot ulcers.2
Coming in From the ColdScientific collaborations between
the United States and Cuba are not new and date as far back as the mid-19th century, when Joseph Henry, the first Secretary of the Smithsonian Institu-tion, and naturalist Spencer Fullerton Baird, the Smithsonian’s Assistant Sec-retary, began exchanging letters, docu-ments, and scientific literature with Felipe Poey, a professor of zoology and comparative anatomy at the University
of Havana and the founder of Havana’s Museum of Natural History. Other scientific partnerships followed, most notably between American physician Jesse Lazear, MD, and Cuban scientist Carlos Finlay, MD, whose ground-breaking investigations in 1900 proved that mosquitoes carry yellow fever.
By 1961, however, diplomatic ties between the two countries were sev-ered as the Cold War heated up, and a full economic embargo, which includ-ed prohibitions on the importation of equipment and supplies and stringent travel restrictions, was imposed by the United States on Cuba. The Cuban Mis-sile Crisis the following year cemented U.S. economic and diplomatic isolation of Cuba for the next 53 years.
Despite the embargo’s crippling eco-nomic sanctions on Cuba, which has stymied the import of computer technol-ogy and modern medical research equip-ment—CMI has only one flow cytome-ter to study cancer cells, according to Dr. Johnson—the country has maintained a thriving scientific community. This com-munity includes 6,000 scientists3 and a universal health-care system that has re-duced the infant mortality rate to 4.7 per 1,000 live births in 2014, compared with 6.17 in the United States,4 and at a frac-tion of the cost. According to the World Bank, in 2013, expenditure on health care per capita in the United States was $9,146, compared with $603 in Cuba.5
“The Cubans have had to do more with less [see sidebar] and have had to use their innovative minds to create so-lutions to achieve what they’ve wanted to,” said Dr. Johnson. “They may not have the fancy equipment that we do, but their infant mortality rate is better than ours, and if you look at the mean survival rate of their citizens, 79 years, it’s not different from ours.”
Forging New Scientific Collaborations
Although the economic embargo has stifled scientific collaborations be-tween the United States and Cuba, it has not completely eliminated them, and U.S. scientists have been traveling to Cuba in recent years, financing the trips with private or foundation funds. “I have had the opportunity to make scientific visits to Cuba, and it is obvi-ous that while our governments have had difficulty communicating, we have not,” said Peter Agre, MD, Bloom-berg Distinguished Professor and Di-rector of the Johns Hopkins Medical Research Institute, Department of Molecular Microbiology and Immu-nology, Bloomberg School of Public
Health; Past President of the Ameri-can Association for the Advancement of Science; and a co-recipient of the 2003 Nobel Prize in Chemistry.
“While Cuba is desperately poor be-cause of the embargo, the scientists there are doing remarkable work and have had some formidable success in cancer and other research, and we need to know about it,” continued Dr. Agre. “It is in our
best interest to have the opportunity to meet our counterparts face-to-face, not just in occasional e-mails, and share our knowledge. Cancer knows no socioeco-nomic boundaries, and the advances that are likely to occur from the collabo-ration between the scientists at Roswell Park Cancer Institute and CMI could help all Americans and people in other countries as well. I have no doubt that the collaboration will lead to productive developments in research and maybe breakthroughs, which will translate into improved clinical care for patients with lung cancer and other diseases.”
A New Era in DétenteSince the announcement in Decem-
ber 2014 of a policy shift toward Cuba, the United States and Cuba have re-opened their embassies in Havana and Washington, DC, reestablishing diplo-matic ties between the two countries. In addition, Cuba was removed from the list of nations that sponsor terrorism, signaling a new era in détente. However, until the American trade embargo is lift-ed, which will take an act of Congress and is unlikely to happen soon, the abil-ity to fully engage in collaborative scien-tific research between the two countries will remain limited and opportunities for progress slowed.
“After more than half a century with-out diplomatic relations, a variety of is-sues will need to be resolved between the two countries, including visa obsta-cles and overcoming obstructed sharing of data, resources, and knowledge, and many would argue against a warmer re-
lationship until certain steps are taken first. But science deserves a chance to move forward now,” said Sergio Jorge Pastrana, Foreign Secretary and Execu-tive Director of the Academy of Scienc-es of Cuba in Havana.
“Joint research in almost any field can only work for the best aims and needs of both countries and should be favored without any prerequisites. The evidence
strongly suggests that joint scientific re-search between Cuba and the United States can provide opportunities for progress and capacity building in both countries and elsewhere,” he added.
Mr. Pastrana continued: “There is a lot of misunderstanding between our two countries because of the policy that has been in place for more than 50 years. I only wish to assure all Ameri-cans that if they come to Cuba, they will find it is very different from the idea that has been created by misun-derstanding for too long.” n
Disclosure: Drs. Agre and Pastrana reported no potential conflicts of interest.
References1. Berlanga J, Fernández JI, López, E, et
al: Heberprot-P: A novel product for treat-ing advanced diabetic foot ulcer. MEDICC Rev 15:11-15, 2013.
2. Reed G: Renewed U.S.-Cuba relations: Saving American lives and limbs? Huffing-ton Post, posted January 24, 2015; updated March 26, 2015. Available at http://www.huffingtonpost.com/gail-reed/renewed-us-cuba-relations-_b_6537518.html. Accessed August 18, 2015.
3. Stone R: In from the cold. Science 348:746-751, 2015.
4. Central Intelligence Agency: The World Factbook. Available at https://www.cia.gov/library/publications/the-world-factbook/rankorder/2091rank.html. Ac-cessed August 18, 2015.
5. The World Bank: Health expenditure per capita (current US$). Available at data.worldbank.org/indicator/sh.xpd.pcap. Ac-cessed August 18, 2015.
Getting There From Here: What You Need to Know About Traveling to Cuba
In January 2015, the U.S. De-partment of the Treasury and
the U.S. Department of Com-merce announced changes re-lated to the easing of sanctions on Cuba, which loosened some restrictions on travel for Ameri-cans, although some rules still ap-ply. There are 12 legal categories of travel to Cuba, including for professional research and profes-sional meetings. All authorized travelers to Cuba need a valid passport and a visa to enter the country, but it is no longer nec-essary to apply for a license. For information regarding the type of visa required to travel to Cuba, visit the Cuban Embassy web-site at www.cubadiplomatica.cu/sicw/EN/ConsularServices.aspx.
For additional information on travel to Cuba, visit the U.S. De-partment of Treasury website at http://www.treasury.gov/press-center/press-releases/Pages/jl9740.aspx. n
U.S.-Cuban Partnershipcontinued from page 47
Cancer knows no socioeconomic boundaries, and the advances that are likely to occur from the collaboration between the scientists at Roswell Park Cancer Institute and the Cuban Center for Molecular Immunology could help all Americans and people in other countries as well.
—Peter Agre, MD

If she has ovarian cancer
TEST FOR BRCA
If indicated*
TREAT WITH LYNPARZA
Please see the following pages for additional Safety Information
and Brief Summary of the full Prescribing Information.
INDICATION
LYNPARZA is indicated as monotherapy in patients with deleterious or suspected deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.
The indication is approved under accelerated approval based on objective response rate and duration of response. Continued approval for this indication may be contingent upon verifi cation and
description of clinical benefi t in confi rmatory trials.
SELECT SAFETY INFORMATION
Myelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confi rmed in 2% of patients enrolled in both a single arm monotherapy trial (6 out of 298) and a randomized placebo controlled trial (3 out of 136). Overall, MDS/AML were reported in <1% of patients (22 out of 2,618) treated with LYNPARZA. The majority of MDS/AML cases were fatal (17 out of 22) and the duration of therapy in patients who developed secondary MDS/AML varied from <6 months to >2 years.
Monitor complete blood count testing at baseline and monthly thereafter. Do not start LYNPARZA until patients have recovered from hematological toxicity caused by previous chemotherapy (≤CTCAE Grade 1). For prolonged hematological toxicities, interrupt LYNPARZA and monitor blood counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confi rmed, discontinue LYNPARZA.
*
Help her continue the fi ght withthe fi rst approved PARP inhibitor1
Trim: 10.5 x 14

LYNPARZA demonstrated an objective response
rate of 34% in patients with BRCA-mutated
advanced ovarian cancer who had been treated
with 3 or more lines of chemotherapy1
The effi cacy of LYNPARZA was investigated in a single-arm study of patients with deleterious or
suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancer. A total of 137 patients
with measurable gBRCAm-associated ovarian cancer treated with three or more prior lines of
chemotherapy were enrolled. Effi cacy was based on objective response rate and duration of response.1
Objective response rate was defi ned as a ≥30% reduction in target lesion size, according to
RECIST criteria, as measured by CT or MRI and confi rmed at least 4 weeks later.2
• The rate of partial response was 32% and the rate of complete response was 2%1
Please see the following pages for additional Safety Information and Brief Summary of the full Prescribing Information.
0 2010 30
PERCENTAGE OF PATIENTS WHO RESPONDED TO THERAPY
34%
OBJECTIVE RESPONSE RATE(95% CI: 26, 42)
MEDIAN DURATION OF RESPONSE7.9MONTHS
(95% CI: 5.6, 9.6)
Trim: 10.5 x 14
Warnings and Precautions
Myelodysplastic syndrome/Acute Myeloid Leukemia
Myelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confi rmed in 6 out
of 298 (2%) patients enrolled in a single arm trial of LYNPARZA monotherapy, in patients with
deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a
randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with
advanced ovarian cancer treated with LYNPARZA. Overall, MDS/AML were reported in 22 of
2,618 (<1%) patients treated with LYNPARZA. The majority of MDS/AML cases (17 of 22 cases)
were fatal, and the duration of therapy with LYNPARZA in patients who developed secondary
MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous
chemotherapy with platinum agents and/or other DNA damaging agents.
Monitor complete blood count testing at baseline and monthly thereafter. Do not start LYNPARZA
until patients have recovered from hematological toxicity caused by previous chemotherapy
(≤CTCAE Grade 1). For prolonged hematological toxicities, interrupt LYNPARZA and monitor blood
counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks,
refer the patient to a hematologist for further investigations, including bone marrow analysis and
blood sample for cytogenetics. If MDS/AML is confi rmed, discontinue LYNPARZA.
Pneumonitis
Pneumonitis, including fatal cases, occurred in <1% of patients treated with LYNPARZA. If patients
present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a
radiological abnormality occurs, interrupt treatment with LYNPARZA and initiate prompt investigation.
If pneumonitis is confi rmed, discontinue LYNPARZA.
Embryo-Fetal Toxicity
LYNPARZA can cause fetal harm when administered to a pregnant woman based on its mechanism of
action and fi ndings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at
exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the
patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Advise females of reproductive potential to avoid becoming pregnant while taking LYNPARZA.
If contraceptive methods are being considered, use effective contraception during treatment and
for at least one month after receiving the last dose of LYNPARZA.
Use in Nursing Mothers Nursing Mothers
It is not known whether olaparib is excreted in human milk. Because many drugs are excreted
in human milk and because of the potential for serious adverse reactions in nursing infants from
olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug,
taking into account the importance of the drug to the mother.
Trim: 10.5 x 14

LYNPARZA demonstrated an objective response
rate of 34% in patients with BRCA-mutated
advanced ovarian cancer who had been treated
with 3 or more lines of chemotherapy1
The effi cacy of LYNPARZA was investigated in a single-arm study of patients with deleterious or
suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancer. A total of 137 patients
with measurable gBRCAm-associated ovarian cancer treated with three or more prior lines of
chemotherapy were enrolled. Effi cacy was based on objective response rate and duration of response.1
Objective response rate was defi ned as a ≥30% reduction in target lesion size, according to
RECIST criteria, as measured by CT or MRI and confi rmed at least 4 weeks later.2
• The rate of partial response was 32% and the rate of complete response was 2%1
Please see the following pages for additional Safety Information and Brief Summary of the full Prescribing Information.
0 2010 30
PERCENTAGE OF PATIENTS WHO RESPONDED TO THERAPY
34%
OBJECTIVE RESPONSE RATE(95% CI: 26, 42)
MEDIAN DURATION OF RESPONSE7.9MONTHS
(95% CI: 5.6, 9.6)
Trim: 10.5 x 14
Warnings and Precautions
Myelodysplastic syndrome/Acute Myeloid Leukemia
Myelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confi rmed in 6 out
of 298 (2%) patients enrolled in a single arm trial of LYNPARZA monotherapy, in patients with
deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a
randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with
advanced ovarian cancer treated with LYNPARZA. Overall, MDS/AML were reported in 22 of
2,618 (<1%) patients treated with LYNPARZA. The majority of MDS/AML cases (17 of 22 cases)
were fatal, and the duration of therapy with LYNPARZA in patients who developed secondary
MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous
chemotherapy with platinum agents and/or other DNA damaging agents.
Monitor complete blood count testing at baseline and monthly thereafter. Do not start LYNPARZA
until patients have recovered from hematological toxicity caused by previous chemotherapy
(≤CTCAE Grade 1). For prolonged hematological toxicities, interrupt LYNPARZA and monitor blood
counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks,
refer the patient to a hematologist for further investigations, including bone marrow analysis and
blood sample for cytogenetics. If MDS/AML is confi rmed, discontinue LYNPARZA.
Pneumonitis
Pneumonitis, including fatal cases, occurred in <1% of patients treated with LYNPARZA. If patients
present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a
radiological abnormality occurs, interrupt treatment with LYNPARZA and initiate prompt investigation.
If pneumonitis is confi rmed, discontinue LYNPARZA.
Embryo-Fetal Toxicity
LYNPARZA can cause fetal harm when administered to a pregnant woman based on its mechanism of
action and fi ndings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at
exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the
patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Advise females of reproductive potential to avoid becoming pregnant while taking LYNPARZA.
If contraceptive methods are being considered, use effective contraception during treatment and
for at least one month after receiving the last dose of LYNPARZA.
Use in Nursing Mothers Nursing Mothers
It is not known whether olaparib is excreted in human milk. Because many drugs are excreted
in human milk and because of the potential for serious adverse reactions in nursing infants from
olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug,
taking into account the importance of the drug to the mother.
Trim: 10.5 x 14
Safety and tolerability of LYNPARZA • LYNPARZA 400 mg twice daily was evaluated as monotherapy in 223 patients with BRCA-mutated
advanced ovarian cancer who had 3 or more prior lines of chemotherapy in 6 clinical trials1
Please see accompanying Brief Summary of Full Prescribing Information.
LYNPARZA is a trademark of the AstraZeneca group of companies.©2015 AstraZeneca. All rights reserved. 3118712 Last Updated 4/15
References: 1. LYNPARZA [package insert]. Wilmington, DE: AstraZeneca; 2014. 2. Eisenhauer EA, Therasse P, Bogaerts J, et al.
New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247.
BLOOD AND LYMPHATIC DISORDERS
GASTROINTESTINAL DISORDERS
GENERAL DISORDERS
INFECTIONS AND INFESTATIONS
MUSCULOSKELETAL AND CONNECTIVE
TISSUE DISORDERS
Anemia 34 18
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
Abdominal pain/discomfort 43 8
Fatigue/asthenia
Nasopharyngitis/URI
66 8
26 0
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
CTCAE GRADES 1-4 (%) CTCAE GRADES 3-4 (%)
LYNPARZA 400 MG TWICE DAILY n=223Adverse Reactions Reported
in ≥20% of Patients1
Trim: 10.5 x 14
The safety and tolerability of LYNPARZA were also evaluated
in a randomized, placebo-controlled study1
• LYNPARZA 400 mg twice daily was evaluated as maintenance monotherapy in a randomized, placebo-controlled
clinical trial of 96 patients with germline BRCA-mutated platinum-sensitive ovarian cancer who had received
2 or more lines of platinum-containing chemotherapy1
• Frequently occurring adverse reactions and lab abnormalities were consistent with those seen in the 6 clinical
trials, with the addition of back pain, headache, cough, rash, and dysgeusia1
aPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
To learn more, including how to order LYNPARZA,
please visit www.lynparza.com
Decrease in absolute neutrophil count (neutropenia) 25 7
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatininea 30 2
Decrease in hemoglobin (anemia) 90 15
LYNPARZA 400 MG TWICE DAILY
n=223
CTCAE GRADES 1-4 (%) CTCAE GRADES 3-4 (%)
Laboratory Abnormalities1
Trim: 10.5 x 14

Safety and tolerability of LYNPARZA • LYNPARZA 400 mg twice daily was evaluated as monotherapy in 223 patients with BRCA-mutated
advanced ovarian cancer who had 3 or more prior lines of chemotherapy in 6 clinical trials1
Please see accompanying Brief Summary of Full Prescribing Information.
LYNPARZA is a trademark of the AstraZeneca group of companies.©2015 AstraZeneca. All rights reserved. 3118712 Last Updated 4/15
References: 1. LYNPARZA [package insert]. Wilmington, DE: AstraZeneca; 2014. 2. Eisenhauer EA, Therasse P, Bogaerts J, et al.
New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247.
BLOOD AND LYMPHATIC DISORDERS
GASTROINTESTINAL DISORDERS
GENERAL DISORDERS
INFECTIONS AND INFESTATIONS
MUSCULOSKELETAL AND CONNECTIVE
TISSUE DISORDERS
Anemia 34 18
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
Abdominal pain/discomfort 43 8
Fatigue/asthenia
Nasopharyngitis/URI
66 8
26 0
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
CTCAE GRADES 1-4 (%) CTCAE GRADES 3-4 (%)
LYNPARZA 400 MG TWICE DAILY n=223Adverse Reactions Reported
in ≥20% of Patients1
Trim: 10.5 x 14
The safety and tolerability of LYNPARZA were also evaluated
in a randomized, placebo-controlled study1
• LYNPARZA 400 mg twice daily was evaluated as maintenance monotherapy in a randomized, placebo-controlled
clinical trial of 96 patients with germline BRCA-mutated platinum-sensitive ovarian cancer who had received
2 or more lines of platinum-containing chemotherapy1
• Frequently occurring adverse reactions and lab abnormalities were consistent with those seen in the 6 clinical
trials, with the addition of back pain, headache, cough, rash, and dysgeusia1
aPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
To learn more, including how to order LYNPARZA,
please visit www.lynparza.com
Decrease in absolute neutrophil count (neutropenia) 25 7
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatininea 30 2
Decrease in hemoglobin (anemia) 90 15
LYNPARZA 400 MG TWICE DAILY
n=223
CTCAE GRADES 1-4 (%) CTCAE GRADES 3-4 (%)
Laboratory Abnormalities1
Trim: 10.5 x 14

PAGE 54 The ASCO Post | SEPTEMBER 10, 2015
Announcements
St. Jude Names Keith Perry, MBA, as Chief Information Officer
S t. Jude Children’s Research Hospi-tal has named Keith Perry, MBA,
as Chief Information Officer (CIO) to provide strategic counsel and leader-ship for the hospital’s information tech-nology initiatives.
“Our internal information tech-
nology systems and operations play a critical role in the success of our insti-tution,” said James R. Downing, MD, St. Jude President and Chief Executive Officer. “Keith’s extensive knowledge will support the utilization of new technologies and advancements to
provide the highest quality of care for our patients and families.”
Mr. Perry joins St. Jude from the University of Texas MD Anderson Can-cer Center, where he served as Associ-ate Vice President and Deputy Chief Information Officer. nKeith Perry, MBA
LYNPARZA™ (olaparib) capsules, for oral use
Brief Summary of Prescribing Information. For complete prescribing information consult official package insert.
INDICATIONS AND USAGETreatment of gBRCA-mutated advanced ovarian cancerLynparza is indicated as monotherapy in patients with deleterious or suspected deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.The indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14) in the full Prescribing Information]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATIONPatient SelectionSelect patients for the treatment of advanced ovarian cancer with Lynparza based on the presence of deleterious or suspected deleterious germline BRCA-mutations [see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information]. Information on FDA-approved test for the detection of BRCA-mutations is available at http://www.fda.gov/companiondiagnostics.Recommended DosingThe recommended dose of Lynparza is 400 mg (eight 50 mg capsules) taken twice daily, for a total daily dose of 800 mg. Continue treatment until disease progression or unacceptable toxicity.If a patient misses a dose of Lynparza, instruct patients to take their next dose at its scheduled time.Swallow capsule whole. Do not chew, dissolve, or open capsule. Do not take capsules which appear deformed or show evidence of leakage [see How Supplied/Storage and Handling (16.2) in the full Prescribing Information].Dose Adjustments for Adverse ReactionsTo manage adverse reactions, consider dose interruption of treatment or dose reduction.The recommended dose reduction is to 200 mg (four 50 mg capsules) taken twice daily, for a total daily dose of 400 mg.If a further final dose reduction is required, then reduce to 100 mg (two 50 mg capsules) taken twice daily, for a total daily dose of 200 mg.Dose Modifications for Use with CYP3A InhibitorsAvoid concomitant use of strong and moderate CYP3A inhibitors and consider alternative agents with less CYP3A inhibition. If the inhibitor cannot be avoided, reduce the Lynparza dose to 150 mg (three 50 mg capsules) taken twice daily for a strong CYP3A inhibitor or 200 mg (four 50 mg capsules) taken twice daily for a moderate CYP3A inhibitor [see Drug Interactions (7.2) in the full Prescribing Information].
CONTRAINDICATIONSNone
WARNINGS AND PRECAUTIONSMyelodysplastic syndrome/Acute Myeloid LeukemiaMyelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confirmed in 6 out of 298 (2%) patients enrolled in a single arm trial of Lynparza monotherapy, in patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with advanced ovarian cancer treated with Lynparza. Overall, MDS/AML were reported in 22 of 2,618 (<1%) patients treated with Lynparza. The majority of MDS/AML cases (17 of 22 cases) were fatal, and the duration of therapy with Lynparza in patients who developed secondary MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous chemotherapy with platinum agents and/or other DNA damaging agents.Monitor complete blood count testing at baseline and monthly thereafter. Do not start Lynparza until patients have recovered from hematological toxicity caused by previous chemotherapy (�CTCAE Grade 1). For prolonged hematological toxicities, interrupt Lynparza and monitor blood counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Lynparza.PneumonitisPneumonitis, including fatal cases, occurred in <1% of patients treated with Lynparza. If patients present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a radiological abnormality occurs, interrupt treatment with Lynparza and initiate prompt investigation. If pneumonitis is confirmed, discontinue Lynparza.Embryo-Fetal ToxicityLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus [see Use in Specific Populations (8.1) in the full Prescribing Information].Advise females of reproductive potential to avoid becoming pregnant while taking Lynparza. If contraceptive methods are being considered, use effective contraception during treatment and for at least one month after receiving the last dose of Lynparza [see Use in Specific Populations (8.6) in the full Prescribing Information].
ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the labeling:• Myelodysplastic syndrome/Acute Myeloid Leukemia [see Warnings and Precautions
(5.1) in the full Prescribing Information]• Pneumonitis [see Warnings and Precautions (5.2) in the full Prescribing Information]
Clinical Trial ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Lynparza 400 mg twice daily as monotherapy, has been studied in 300 patients with gBRCA-mutated advanced ovarian cancer, and 223 of these patients had received 3 or more prior lines of chemotherapy.In the 223 patients with gBRCA-mutated ovarian cancer who received 3 or more prior lines of chemotherapy (including 137 patients in Study 1 with measureable disease) [see Clinical Studies (14) in the full Prescribing Information] adverse reactions led to dose interruption in 40% of patients, dose reduction in 4%, and discontinuation in 7%. There were 8 (4%) patients with adverse reactions leading to death, two were attributed to acute leukemia, and one each was attributed to COPD, cerebrovascular accident, intestinal perforation, pulmonary embolism, sepsis, and suture rupture. Table 1 presents the frequency of adverse reactions reported in �20% of 223 patients (in 6 studies) with gBRCA-mutated advanced ovarian cancer who had received 3 or more prior lines of chemotherapy who were treated with Lynparza 400 mg twice daily. The median exposure to Lynparza in these patients was 158 days.
Table 1 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
3 or more lines of prior chemotherapy
Adverse Reaction Grades 1-4 Grades 3-4 N=223 N=223 % % Blood and Lymphatic disorders
Anemia 34 18
Gastrointestinal disorders
Abdominal pain/discomfort 43 8
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
General disorders
Fatigue/asthenia 66 8
Infections and infestations
Nasopharyngitis/URI 26 0
Musculoskeletal and Connective Tissue disorders
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
Table 2 presents the frequency of abnormal laboratory findings in the 223 patients with gBRCA-mutated advanced ovarian cancer who had received three or more prior lines of chemotherapy receiving Lynparza 400 mg twice daily.
Table 2 Laboratory Abnormalities Reported in Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
Laboratory Parameter* 3 or more lines of prior chemotherapy
Grades 1-4 Grades 3-4 N=223 N=223 % %
Decrease in hemoglobin (anemia) 90 15
Decrease in absolute neutrophil count 25 7 (neutropenia)
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatinine* 30 2* Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
The following adverse reactions and laboratory abnormalities have been identified in �10 to <20% of the 223 patients receiving Lynparza and not included in the table: cough, constipation, dysgeusia, peripheral edema, back pain, dizziness, headache, urinary tract infection, dyspnea, and rash. The following adverse reactions and laboratory abnormalities have been identified in �1 to <10% of the 223 patients receiving Lynparza and not included in the table: leukopenia, stomatitis, peripheral neuropathy, pyrexia, hypomagnesemia, hyperglycemia, anxiety, depression, insomnia, dysuria, urinary incontinence, vulvovaginal disorder, dry skin/ eczema, pruritis, hypertension, venous thrombosis (including pulmonary embolism), and hot flush.Table 3 presents adverse reactions reported in �20% of patients from a randomized trial of Lynparza 400 mg twice daily as maintenance monotherapy compared to placebo in patients with platinum sensitive, relapsed, high-grade serous ovarian cancer following treatment with 2 or more platinum-containing regimens. Table 4 presents the laboratory abnormalities in patients from this randomized trial. Of the 96 patients with gBRCA- mutation, 53 received Lynparza, and 43 received placebo. The median duration on treatment with Lynparza was 11.1 months for patients with a gBRCA mutation compared to 4.4 months for patients with gBRCA mutation on placebo.Adverse reactions led to dose interruptions in 26% of those receiving Lynparza and 7% of those receiving placebo; dose reductions in 15% of Lynparza and 5% of placebo patients; and discontinuation in 9% of Lynparza and 0% in placebo patients. One (2%) patient on Lynparza died as a result of an adverse reaction.
Trim: 7.625 x 10.5
LYNPARZATM (olaparib) capsules 2
Table 3 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Adverse Reactions Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Blood and Lymphatic disorders Anemia 25 4 7 2 Gastrointestinal disorders Abdominal pain/discomfort 47 0 58 2 Decreased appetite 25 0 14 0 Nausea 75 2 37 0 Vomiting 32 4 9 0 Diarrhea 28 4 21 2 Dyspepsia 25 0 14 0 Dysgeusia 21 0 9 0 General disorders Fatigue (including asthenia, lethargy) 68 6 53 2 Infections and infestations Nasopharyngitis/Pharyngitis/URI 43 0 16 0 Musculoskeletal and Connective tissue disorders Arthralgia/Musculoskeletal pain 32 4 21 0 Myalgia 25 2 12 0 Back pain 25 6 21 0 Nervous system disorder Headache 25 0 19 2 Respiratory, Thoracic, Mediastinal disorders Cough 21 0 14 0 Skin and Subcutaneous Tissue Dermatitis/Rash 25 0 14 0
Table 4 Laboratory Abnormalities in Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Laboratory parameter* Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Decrease in hemoglobin 85 8 58 2 Decrease in absolute neutrophil count 32 8 23 0 Decrease in platelets 26 6 19 0 Mean corpuscular volume elevation 85 - 44 - Increase in creatinine* 26 0 5 0 * Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
DRUG INTERACTIONSOlaparib is primarily metabolized by CYP3A.Anticancer AgentsClinical studies of Lynparza in combination with other myelosuppressive anticancer agents, including DNA damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.Drugs that may Increase Olaparib Plasma ConcentrationsIn patients (N=57), co-administration of itraconazole, a strong CYP3A inhibitor, increased AUC of olaparib by 2.7-fold. A moderate CYP3A inhibitor, fluconazole, is predicted to increase the AUC of olaparib by 2-fold.Avoid concomitant use of strong CYP3A inhibitors (e.g., itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritinovir, lopinavir/ ritinovir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir) and moderate CYP3A inhibitors (e.g., amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil). If the strong or moderate CYP3A inhibitors must be co-administered, reduce the dose of Lynparza [see Dosage and Administration (2.4) in the full Prescribing Information].Avoid grapefruit and Seville oranges during Lynparza treatment [see Dosage and Adminis-tration (2.4) and Clinical Pharmacology (12.3) in the full Prescribing Information].Drugs that may Decrease Olaparib Plasma ConcentrationsIn patients (N=22), co-administration of rifampicin, a strong CYP3A inducer, decreased AUC of olaparib by 87%. A moderate CYP3A inducer, efavirenz, is predicted to decrease the AUC of olaparib by 50-60%.Avoid concomitant use of strong CYP3A inducers (e.g., phenytoin, rifampicin, carbamazepine, St. John’s Wort) and moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin). If a moderate CYP3A inducer cannot be avoided, be aware of a potential for decreased efficacy of Lynparza [see Clinical Pharmacology (12.3) in the full Prescribing Information].USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions (5.3) in the full Prescribing Information] Risk summaryLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If this drug is used during pregnancy, or if a patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for loss of the pregnancy.Animal DataIn a fertility and early embryonic development study in female rats, olaparib was administered orally for 14 days before mating through to day 6 of pregnancy, which
resulted in increased post-implantation loss at a dose level of 15 mg/kg/day (with maternal systemic exposures approximately 11% of the human exposure (AUC0-24h) at the recommended dose).In an embryo-fetal development study, pregnant rats received oral doses of 0.05 and 0.5 mg/kg/day olaparib during the period of organogenesis. A dose of 0.5 mg/kg/day (with maternal systemic exposures approximately 0.3% of human exposure (AUC0-24h) at the recommended dose) caused embryo-fetal toxicities including increased post-implantation loss and major malformations of the eyes (anophthalmia, microphthalmia), vertebrae/ribs (extra rib or ossification center; fused or absent neural arches, ribs, and sternebrae), skull (fused exoccipital) and diaphragm (hernia). Additional abnormalities or variants included incomplete or absent ossification (vertebrae/sternebrae, ribs, limbs) and other findings in the vertebrae/sternebrae, pelvic girdle, lung, thymus, liver, ureter and umbilical artery. Some findings noted above in the eyes, ribs and ureter were observed at a dose of 0.05 mg/kg/day olaparib at lower incidence.Nursing MothersIt is not known whether olaparib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric UseThe safety and efficacy of Lynparza has not been established in pediatric patients.Geriatric UseIn clinical studies of Lynparza enrolling 735 patients with advanced solid tumors [the majority (69%) of whom had ovarian cancer] who received Lynparza 400 mg twice daily as monotherapy, 148 (20%) of patients were aged �65 years. The safety profile was similar irrespective of age with the exception of AEs of CTCAE �3 which were reported more frequently in patients aged �65 years (53.4%) than those <65 years (43.4%). No individual adverse event or System Organ Class accounted for this observed difference.Females of Reproductive PotentialLynparza can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise female patients of reproductive potential to avoid pregnancy while taking Lynparza. If contraceptive methods are being considered, use highly effective contraception during treatment with Lynparza and for at least one month following the last dose of Lynparza. Instruct patients to contact their health-care provider if they become pregnant, or if pregnancy is suspected, while taking Lynparza.Hepatic ImpairmentThe effect of hepatic impairment on exposure to Lynparza has not been studied. Patients with bilirubin >1.5 X ULN and AST/ALT �2.5 X ULN (�5 X ULN in the presence of liver metastases) were excluded from Lynparza clinical trials. There are no data in patients with baseline hepatic impairment (serum bilirubin >1.5 X ULN) [see Clinical Pharmacology (12.3) in the full Prescribing Information].Renal ImpairmentBased on preliminary data, a 1.5 fold increase in mean exposure (AUC) was observed in patients with mild renal impairment (CLcr = 50-80 mL/min) compared to patients with normal renal function (CLcr >80 mL/min). No dose adjustment to the starting dose is required in patients with CLcr of 50 to 80 mL/min, but patients should be monitored closely for toxicity. There are no data in patients with moderate or severe renal impairment (CLcr <50 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in the full Prescribing Information].OVERDOSAGEThere is no specific treatment in the event of Lynparza overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should follow general supportive measures and should treat symptomatically.17 PATIENT COUNSELING INFORMATIONSEE FDA-APPROVED PATIENT LABELING (MEDICATION GUIDE)• Dosing Instructions: Inform patients on how to take Lynparza [see Dosage and
Administration (2.1) in the full Prescribing Information]. Lynparza should be taken twice daily. Instruct patients that if they miss a dose of Lynparza, not to take an extra dose to make up for the one that they missed. They should take their next normal dose at the usual time. Each capsule should be swallowed whole. Do not chew, dissolve, or open the capsule. Patient should not take Lynparza with grapefruit or Seville oranges.
• MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called ‘myelodysplastic syndrome’ (MDS) or ‘acute myeloid leukemia’ (AML) which have been reported in patients treated with Lynparza [see Warnings and Precautions (5.1) in the full Prescribing Information].
• Pneumonitis: Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including shortness of breath, fever, cough, or wheezing [see Warnings and Precautions (5.2) in the full Prescribing Information].
• Pregnancy and Females of Reproductive Potential: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for at least one month after receiving the last dose of Lynparza [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.6) in the full Prescribing Information].
• Nursing Mothers: Advise patients not to breastfeed while taking Lynparza [see Use in Special Populations (8.3) in the full Prescribing Information].
• Nausea/vomiting: Advise patients that mild or moderate nausea and/or vomiting is very common in patients receiving Lynparza and that they should contact their healthcare provider who will advise on available antiemetic treatment options.
Distributed by:AstraZeneca Pharmaceuticals LP, Wilmington, DE 198503079901 12/14 Issued: 12/2014
Trim: 7.625 x 10.5

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 55
Direct From ASCO
ASCO Guidelines: A Collaborative Effort
The ASCO Guidelines Program has worked with other professional
societies and guideline development organizations in an effort to expand the ASCO guideline portfolio and harmonize recommended care options across promi-nent guideline development groups.
For ASCO, this effort began with a sys-tematic and proactive approach in identi-fying guideline topics for development. ASCO then reached out to partnering or-ganizations to explore the development of joint guidelines where none currently ex-ist, or to consider endorsement or adap-
tation of recommendations from recently developed evidence-based guidelines.
Guideline Advisory GroupsThe recent formation of Guideline
Advisory Groups within ASCO is criti-cal to the model. Advisory Groups were
created based on a recommendation by the guidelines team of the ASCO Lead-ership Development Program to the ASCO Board of Directors.
Advisory groups comprise con-tent experts that advise ASCO’s Clini-cal Practice Guidelines Committee (CPGC) on the highest priority topics to consider for guideline development within a given area. There are currently seven Advisory Groups, with four fo-cusing on specific disease sites—breast, gastrointestinal, genitourinary, and tho-racic cancers—and three dedicated to topics within survivorship, supportive care, and resource stratification.
“Guideline advisory groups have been one of the most innovative and produc-tive efforts to come out of the ASCO Guideline Program in the last couple of years,” said Gary H. Lyman, MD, MPH,
Order ASCO Answers Guides to Cancer for Your PracticeASCO Answers Guides to breast,
colorectal, lung, and prostate can-cers are designed to help newly diag-nosed patients understand their disease and treatment options. These com-prehensive, patient-friendly booklets contain trusted information about the diagnosis, treatment, side effects, and psychosocial effects of cancer. These guides also provide space for recording details about diagnosis and treatment plans, a feature that allows patients to easily go back and find specific informa-tion when needed. Printed copies can be purchased from the ASCO Univer-sity Bookstore, in packs of 50, at cancer.net/eStore. Shipping is free, and ASCO members save 20%. n
© 2015. American Society of Clinical Oncology. All rights reserved.
continued on page 58
Gary H. Lyman, MD, MPH, FASCO
LYNPARZA™ (olaparib) capsules, for oral use
Brief Summary of Prescribing Information. For complete prescribing information consult official package insert.
INDICATIONS AND USAGETreatment of gBRCA-mutated advanced ovarian cancerLynparza is indicated as monotherapy in patients with deleterious or suspected deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.The indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14) in the full Prescribing Information]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATIONPatient SelectionSelect patients for the treatment of advanced ovarian cancer with Lynparza based on the presence of deleterious or suspected deleterious germline BRCA-mutations [see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information]. Information on FDA-approved test for the detection of BRCA-mutations is available at http://www.fda.gov/companiondiagnostics.Recommended DosingThe recommended dose of Lynparza is 400 mg (eight 50 mg capsules) taken twice daily, for a total daily dose of 800 mg. Continue treatment until disease progression or unacceptable toxicity.If a patient misses a dose of Lynparza, instruct patients to take their next dose at its scheduled time.Swallow capsule whole. Do not chew, dissolve, or open capsule. Do not take capsules which appear deformed or show evidence of leakage [see How Supplied/Storage and Handling (16.2) in the full Prescribing Information].Dose Adjustments for Adverse ReactionsTo manage adverse reactions, consider dose interruption of treatment or dose reduction.The recommended dose reduction is to 200 mg (four 50 mg capsules) taken twice daily, for a total daily dose of 400 mg.If a further final dose reduction is required, then reduce to 100 mg (two 50 mg capsules) taken twice daily, for a total daily dose of 200 mg.Dose Modifications for Use with CYP3A InhibitorsAvoid concomitant use of strong and moderate CYP3A inhibitors and consider alternative agents with less CYP3A inhibition. If the inhibitor cannot be avoided, reduce the Lynparza dose to 150 mg (three 50 mg capsules) taken twice daily for a strong CYP3A inhibitor or 200 mg (four 50 mg capsules) taken twice daily for a moderate CYP3A inhibitor [see Drug Interactions (7.2) in the full Prescribing Information].
CONTRAINDICATIONSNone
WARNINGS AND PRECAUTIONSMyelodysplastic syndrome/Acute Myeloid LeukemiaMyelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confirmed in 6 out of 298 (2%) patients enrolled in a single arm trial of Lynparza monotherapy, in patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with advanced ovarian cancer treated with Lynparza. Overall, MDS/AML were reported in 22 of 2,618 (<1%) patients treated with Lynparza. The majority of MDS/AML cases (17 of 22 cases) were fatal, and the duration of therapy with Lynparza in patients who developed secondary MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous chemotherapy with platinum agents and/or other DNA damaging agents.Monitor complete blood count testing at baseline and monthly thereafter. Do not start Lynparza until patients have recovered from hematological toxicity caused by previous chemotherapy (�CTCAE Grade 1). For prolonged hematological toxicities, interrupt Lynparza and monitor blood counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Lynparza.PneumonitisPneumonitis, including fatal cases, occurred in <1% of patients treated with Lynparza. If patients present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a radiological abnormality occurs, interrupt treatment with Lynparza and initiate prompt investigation. If pneumonitis is confirmed, discontinue Lynparza.Embryo-Fetal ToxicityLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus [see Use in Specific Populations (8.1) in the full Prescribing Information].Advise females of reproductive potential to avoid becoming pregnant while taking Lynparza. If contraceptive methods are being considered, use effective contraception during treatment and for at least one month after receiving the last dose of Lynparza [see Use in Specific Populations (8.6) in the full Prescribing Information].
ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the labeling:• Myelodysplastic syndrome/Acute Myeloid Leukemia [see Warnings and Precautions
(5.1) in the full Prescribing Information]• Pneumonitis [see Warnings and Precautions (5.2) in the full Prescribing Information]
Clinical Trial ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Lynparza 400 mg twice daily as monotherapy, has been studied in 300 patients with gBRCA-mutated advanced ovarian cancer, and 223 of these patients had received 3 or more prior lines of chemotherapy.In the 223 patients with gBRCA-mutated ovarian cancer who received 3 or more prior lines of chemotherapy (including 137 patients in Study 1 with measureable disease) [see Clinical Studies (14) in the full Prescribing Information] adverse reactions led to dose interruption in 40% of patients, dose reduction in 4%, and discontinuation in 7%. There were 8 (4%) patients with adverse reactions leading to death, two were attributed to acute leukemia, and one each was attributed to COPD, cerebrovascular accident, intestinal perforation, pulmonary embolism, sepsis, and suture rupture. Table 1 presents the frequency of adverse reactions reported in �20% of 223 patients (in 6 studies) with gBRCA-mutated advanced ovarian cancer who had received 3 or more prior lines of chemotherapy who were treated with Lynparza 400 mg twice daily. The median exposure to Lynparza in these patients was 158 days.
Table 1 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
3 or more lines of prior chemotherapy
Adverse Reaction Grades 1-4 Grades 3-4 N=223 N=223 % % Blood and Lymphatic disorders
Anemia 34 18
Gastrointestinal disorders
Abdominal pain/discomfort 43 8
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
General disorders
Fatigue/asthenia 66 8
Infections and infestations
Nasopharyngitis/URI 26 0
Musculoskeletal and Connective Tissue disorders
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
Table 2 presents the frequency of abnormal laboratory findings in the 223 patients with gBRCA-mutated advanced ovarian cancer who had received three or more prior lines of chemotherapy receiving Lynparza 400 mg twice daily.
Table 2 Laboratory Abnormalities Reported in Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
Laboratory Parameter* 3 or more lines of prior chemotherapy
Grades 1-4 Grades 3-4 N=223 N=223 % %
Decrease in hemoglobin (anemia) 90 15
Decrease in absolute neutrophil count 25 7 (neutropenia)
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatinine* 30 2* Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
The following adverse reactions and laboratory abnormalities have been identified in �10 to <20% of the 223 patients receiving Lynparza and not included in the table: cough, constipation, dysgeusia, peripheral edema, back pain, dizziness, headache, urinary tract infection, dyspnea, and rash. The following adverse reactions and laboratory abnormalities have been identified in �1 to <10% of the 223 patients receiving Lynparza and not included in the table: leukopenia, stomatitis, peripheral neuropathy, pyrexia, hypomagnesemia, hyperglycemia, anxiety, depression, insomnia, dysuria, urinary incontinence, vulvovaginal disorder, dry skin/ eczema, pruritis, hypertension, venous thrombosis (including pulmonary embolism), and hot flush.Table 3 presents adverse reactions reported in �20% of patients from a randomized trial of Lynparza 400 mg twice daily as maintenance monotherapy compared to placebo in patients with platinum sensitive, relapsed, high-grade serous ovarian cancer following treatment with 2 or more platinum-containing regimens. Table 4 presents the laboratory abnormalities in patients from this randomized trial. Of the 96 patients with gBRCA- mutation, 53 received Lynparza, and 43 received placebo. The median duration on treatment with Lynparza was 11.1 months for patients with a gBRCA mutation compared to 4.4 months for patients with gBRCA mutation on placebo.Adverse reactions led to dose interruptions in 26% of those receiving Lynparza and 7% of those receiving placebo; dose reductions in 15% of Lynparza and 5% of placebo patients; and discontinuation in 9% of Lynparza and 0% in placebo patients. One (2%) patient on Lynparza died as a result of an adverse reaction.
Trim: 7.625 x 10.5
LYNPARZATM (olaparib) capsules 2
Table 3 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Adverse Reactions Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Blood and Lymphatic disorders Anemia 25 4 7 2 Gastrointestinal disorders Abdominal pain/discomfort 47 0 58 2 Decreased appetite 25 0 14 0 Nausea 75 2 37 0 Vomiting 32 4 9 0 Diarrhea 28 4 21 2 Dyspepsia 25 0 14 0 Dysgeusia 21 0 9 0 General disorders Fatigue (including asthenia, lethargy) 68 6 53 2 Infections and infestations Nasopharyngitis/Pharyngitis/URI 43 0 16 0 Musculoskeletal and Connective tissue disorders Arthralgia/Musculoskeletal pain 32 4 21 0 Myalgia 25 2 12 0 Back pain 25 6 21 0 Nervous system disorder Headache 25 0 19 2 Respiratory, Thoracic, Mediastinal disorders Cough 21 0 14 0 Skin and Subcutaneous Tissue Dermatitis/Rash 25 0 14 0
Table 4 Laboratory Abnormalities in Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Laboratory parameter* Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Decrease in hemoglobin 85 8 58 2 Decrease in absolute neutrophil count 32 8 23 0 Decrease in platelets 26 6 19 0 Mean corpuscular volume elevation 85 - 44 - Increase in creatinine* 26 0 5 0 * Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
DRUG INTERACTIONSOlaparib is primarily metabolized by CYP3A.Anticancer AgentsClinical studies of Lynparza in combination with other myelosuppressive anticancer agents, including DNA damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.Drugs that may Increase Olaparib Plasma ConcentrationsIn patients (N=57), co-administration of itraconazole, a strong CYP3A inhibitor, increased AUC of olaparib by 2.7-fold. A moderate CYP3A inhibitor, fluconazole, is predicted to increase the AUC of olaparib by 2-fold.Avoid concomitant use of strong CYP3A inhibitors (e.g., itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritinovir, lopinavir/ ritinovir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir) and moderate CYP3A inhibitors (e.g., amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil). If the strong or moderate CYP3A inhibitors must be co-administered, reduce the dose of Lynparza [see Dosage and Administration (2.4) in the full Prescribing Information].Avoid grapefruit and Seville oranges during Lynparza treatment [see Dosage and Adminis-tration (2.4) and Clinical Pharmacology (12.3) in the full Prescribing Information].Drugs that may Decrease Olaparib Plasma ConcentrationsIn patients (N=22), co-administration of rifampicin, a strong CYP3A inducer, decreased AUC of olaparib by 87%. A moderate CYP3A inducer, efavirenz, is predicted to decrease the AUC of olaparib by 50-60%.Avoid concomitant use of strong CYP3A inducers (e.g., phenytoin, rifampicin, carbamazepine, St. John’s Wort) and moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin). If a moderate CYP3A inducer cannot be avoided, be aware of a potential for decreased efficacy of Lynparza [see Clinical Pharmacology (12.3) in the full Prescribing Information].USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions (5.3) in the full Prescribing Information] Risk summaryLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If this drug is used during pregnancy, or if a patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for loss of the pregnancy.Animal DataIn a fertility and early embryonic development study in female rats, olaparib was administered orally for 14 days before mating through to day 6 of pregnancy, which
resulted in increased post-implantation loss at a dose level of 15 mg/kg/day (with maternal systemic exposures approximately 11% of the human exposure (AUC0-24h) at the recommended dose).In an embryo-fetal development study, pregnant rats received oral doses of 0.05 and 0.5 mg/kg/day olaparib during the period of organogenesis. A dose of 0.5 mg/kg/day (with maternal systemic exposures approximately 0.3% of human exposure (AUC0-24h) at the recommended dose) caused embryo-fetal toxicities including increased post-implantation loss and major malformations of the eyes (anophthalmia, microphthalmia), vertebrae/ribs (extra rib or ossification center; fused or absent neural arches, ribs, and sternebrae), skull (fused exoccipital) and diaphragm (hernia). Additional abnormalities or variants included incomplete or absent ossification (vertebrae/sternebrae, ribs, limbs) and other findings in the vertebrae/sternebrae, pelvic girdle, lung, thymus, liver, ureter and umbilical artery. Some findings noted above in the eyes, ribs and ureter were observed at a dose of 0.05 mg/kg/day olaparib at lower incidence.Nursing MothersIt is not known whether olaparib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric UseThe safety and efficacy of Lynparza has not been established in pediatric patients.Geriatric UseIn clinical studies of Lynparza enrolling 735 patients with advanced solid tumors [the majority (69%) of whom had ovarian cancer] who received Lynparza 400 mg twice daily as monotherapy, 148 (20%) of patients were aged �65 years. The safety profile was similar irrespective of age with the exception of AEs of CTCAE �3 which were reported more frequently in patients aged �65 years (53.4%) than those <65 years (43.4%). No individual adverse event or System Organ Class accounted for this observed difference.Females of Reproductive PotentialLynparza can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise female patients of reproductive potential to avoid pregnancy while taking Lynparza. If contraceptive methods are being considered, use highly effective contraception during treatment with Lynparza and for at least one month following the last dose of Lynparza. Instruct patients to contact their health-care provider if they become pregnant, or if pregnancy is suspected, while taking Lynparza.Hepatic ImpairmentThe effect of hepatic impairment on exposure to Lynparza has not been studied. Patients with bilirubin >1.5 X ULN and AST/ALT �2.5 X ULN (�5 X ULN in the presence of liver metastases) were excluded from Lynparza clinical trials. There are no data in patients with baseline hepatic impairment (serum bilirubin >1.5 X ULN) [see Clinical Pharmacology (12.3) in the full Prescribing Information].Renal ImpairmentBased on preliminary data, a 1.5 fold increase in mean exposure (AUC) was observed in patients with mild renal impairment (CLcr = 50-80 mL/min) compared to patients with normal renal function (CLcr >80 mL/min). No dose adjustment to the starting dose is required in patients with CLcr of 50 to 80 mL/min, but patients should be monitored closely for toxicity. There are no data in patients with moderate or severe renal impairment (CLcr <50 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in the full Prescribing Information].OVERDOSAGEThere is no specific treatment in the event of Lynparza overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should follow general supportive measures and should treat symptomatically.17 PATIENT COUNSELING INFORMATIONSEE FDA-APPROVED PATIENT LABELING (MEDICATION GUIDE)• Dosing Instructions: Inform patients on how to take Lynparza [see Dosage and
Administration (2.1) in the full Prescribing Information]. Lynparza should be taken twice daily. Instruct patients that if they miss a dose of Lynparza, not to take an extra dose to make up for the one that they missed. They should take their next normal dose at the usual time. Each capsule should be swallowed whole. Do not chew, dissolve, or open the capsule. Patient should not take Lynparza with grapefruit or Seville oranges.
• MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called ‘myelodysplastic syndrome’ (MDS) or ‘acute myeloid leukemia’ (AML) which have been reported in patients treated with Lynparza [see Warnings and Precautions (5.1) in the full Prescribing Information].
• Pneumonitis: Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including shortness of breath, fever, cough, or wheezing [see Warnings and Precautions (5.2) in the full Prescribing Information].
• Pregnancy and Females of Reproductive Potential: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for at least one month after receiving the last dose of Lynparza [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.6) in the full Prescribing Information].
• Nursing Mothers: Advise patients not to breastfeed while taking Lynparza [see Use in Special Populations (8.3) in the full Prescribing Information].
• Nausea/vomiting: Advise patients that mild or moderate nausea and/or vomiting is very common in patients receiving Lynparza and that they should contact their healthcare provider who will advise on available antiemetic treatment options.
Distributed by:AstraZeneca Pharmaceuticals LP, Wilmington, DE 198503079901 12/14 Issued: 12/2014
Trim: 7.625 x 10.5

PAGE 56 The ASCO Post | SEPTEMBER 10, 2015
Direct From ASCO
Conquer Cancer Foundation Young Investigator Award Applications Now Open
The Conquer Cancer Foundation’s Young Investigator Award (YIA)
provides research funding to promis-ing physicians to support the transition from final years of training to faculty appointment and to encourage and pro-mote quality research in clinical oncol-ogy. The online application for 2016 YIAs is open now through September 24, 2015.
The YIA is a 1-year research grant totaling $50,000, paid in two incre-ments to the awardee’s institution. The applicant must be a physician (MD, DO, or international equiva-lent) who is currently within the last 2 years of final subspecialty training at an academic medical institution at the time of grant submission. Ap-plicants should be planning an inves-tigative career in clinical oncology.
Areas of interest include (but are not limited to) breast cancer, cholangio-carcinoma, fibrolamellar cancer, kid-ney cancer, pediatrics, sarcoma, sup-portive care, cancer prevention, and health disparities.
To learn more about how YIAs have launched careers and led to breakthroughs in cancer research, and to submit your application, visit conquercancerfoundation.org/yia. n
© 2015. American Society of Clinical Oncology. All rights reserved.
Journal of Clinical Oncology Special Series Issues Provide Updates on Important Topics in Oncology Practice
The Journal of Clinical Oncology ( JCO) annually publishes sev-
eral Special Series of reviews—disease-specific issues dedicated to providing readers with concise, authoritative up-dates on important topics in oncology practice. Each issue of the Special Series explores one specific area of cancer care through a collection of articles written by leaders in the field and an introduc-tory overview authored by the issue’s Associate and Guest Editors.
For 2015, the line-up of Special Se-ries includes:
Gastrointestinal Cancers: Where We Are and Where We Are Going• Associate Editors: Alan P. Venook,
MD, and Joel E. Tepper, MD, FASCO
• Guest Editor: Jeffrey A. Meyerhardt, MD, MPH
Topics will include immunothera-py, radiation, diet and lifestyle, genetic screening, gastric and esophageal can-cers, hepatocellular carcinoma, and more.
Advances in Head and Neck Cancer• Associate Editor: Danny Rischin,
MD• Guest Editors: Quynh-Thu Le, MD,
and Robert L. Ferris, MD, PhD, FACS
Topics will include human papillo-mavirus–negative pharyngeal cancer, advances in surgery and radiotherapy, survivorship, quality of life and surveil-lance, nasopharyngeal cancer, nonmela-noma cutaneous head and neck cancer, and more.
Pediatric Cancer: Progress Through Collaboration• Associate Editor: Melissa M.
Hudson, MD• Guest Editors: Ching-Hon Pui,
MD, and William H. Meyer, MD
Topics will include acute lympho-blastic leukemia, Hodgkin lymphoma, non-Hodgkin lymphoma, central ner-vous system tumors, osteosarcoma, Ew-ing sarcoma, and more.
Global Cancer Medicine• Associate Editor: Tony Mok, MD• Guest Editor: Lawrence N.
Shulman, MD
Topics will include structural bar-riers to diagnosis and treatment, re-source-constrained settings, global pe-diatric oncology, infections and cancer, cancer genomics, and more.
The JCO Special Series on Gastroin-testinal Cancers is available now at JCO.org, and the remaining three issues will be published later this year.
JCO Editor-in-Chief Stephen A. Cannistra, MD, FASCO, explained why the journal focused specifically on these four topics in cancer care.
“Gastrointestinal cancers, pediatric oncology, head and neck tumors, and global oncology were chosen for this year’s collection because each of these areas has witnessed important changes over the past several years, from novel diagnostics and treatment approaches to greater insight into disease pathogen-esis,” Dr. Cannistra said. “We felt that issues on these topics would be of value
to our broad readership, which includes clinicians, clinical investigators, and pa-tients. There should be something of in-terest in this year’s collection for every reader of JCO.”
“The Special Series represents JCO’s ongoing effort to provide readers with a rapid, self-contained, and efficient way of updating their knowledge in a variety of diseases seen in everyday practice,” Dr. Cannistra said. n
Originally printed in ASCO Daily News. © American Society of Clinical Oncology. “Journal of Clinical Oncology to Feature Four Special Series Issues in 2015.” am.asco.org , June 1, 2015. All rights reserved.
Stephen A. Cannistra, MD, FASCO
Genitourinary Cancers SymposiumJanuary 7–9, 2016
Moscone West Building
San Francisco, California
Inaugural Cancer Survivorship Symposium: Advancing Care and Research
January 15–16, 2016
San Francisco Marriott Marquis
San Francisco, California
Gastrointestinal Cancers SymposiumJanuary 21–23, 2016
Moscone West Building
San Francisco, California
Save the Date

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 57
Direct From ASCO
This September, Triple Your Impact
At the 2015 ASCO Annual Meeting, the Conquer Cancer
Foundation (CCF) launched The Campaign to Conquer Cancer—a comprehensive, $150 million fund-raising campaign supporting break-through research and sharing vital knowledge with physicians, patients, and families. In support of The Campaign, CCF Board Member Raj Mantena, RPh, announced his inten-tion to match individual donations to CCF dollar-for-dollar, up to $1 mil-lion, for the coming year.
This September, Mr. Mantena is raising the stakes: He will match all donations two-to-one, tripling the impact of gifts made between Sep-tember 1 and September 30. In light of this, CCF has set a goal to raise
$50,000 by September 30. As with regular gifts, matching
gift funds will support the Founda-tion mission area each donor selects. Options include the CCF Grants and
Awards Program, Cancer.Net and ASCO’s patient information pro-gram, CancerLinQ™ (ASCO’s rapid learning system), and more. If a do-
nor does not have a particular mis-sion area in mind, he or she can al-ways give to CCF’s “Area of Greatest Need,” supplying triple the support where it is needed most.
No matter which mission area a donor selects, all gifts count toward The Campaign to Conquer Cancer’s $150 million goal. While Mr. Man-tena’s challenge means each donation goes three times as far, it also means that each gift brings the Foundation three times closer to realizing its vi-sion: a world free from cancer.
“We cannot thank Raj Mantena enough for his tremendous demon-stration of support at this landmark moment in the Foundation’s history,” said CCF Executive Director and Chief Philanthropic Officer Nancy R . Daly, MS, MPH. “His generosity has a powerful impact on programs and resources advancing cancer pre-vention, treatment, and cures. We sincerely hope his matching gift chal-lenge will inspire others to come to-gether and give in the name of con-quering cancer.”
History has shown we can—and have—made progress: The can-cer death rate has dropped 20% in the past 20 years thanks, in part, to the hard work of dedicated oncol-ogy professionals. But it is critical we continue this momentum, which can-not be maintained without the sup-port of generous donors. Thanks to Mr. Mantena, your gift this Septem-ber will triple the impact advancing progress against cancer.
Donors can participate in this matching gift challenge at conquercancerfoundation.org/match, or by mailing a check to the Conquer Cancer Foundation at P.O. Box 896076, Charlotte, NC, 28289-6076. Mail dona-tions must be postmarked by Septem-ber 30, 2015 to qualify for the two-to-one match period.
Will you step up to the challenge? n
Originally printed in ASCO Connection. © American Society of Clinical Oncology. “This September: Triple Your Impact.” ASCO Connection, September 2015. All rights reserved.
Nancy R. Daly, MS, MPH
Raj Mantena, RPh, and ASCO Past President Peter Paul Yu, MD, FASCO, at the ASCO Annual Meeting.
Top 10 most-accessed articles published in 2011 in Journal of Clinical Oncology
Top 5 articles recently published in
Journal of Oncology Practice
What’s Hot in
JOPFeasibility and Effectiveness of a Pilot Program to Facilitate
Quality Improvement Learning in Oncology: Experience of the
American Society of Clinical Oncology Quality Training Program
by Arif H. Kamal, et al
Health-Related Quality of Life of Food-Insecure Ethnic Minority
Patients With Cancer
by Francesca Gany, et al
Are Patients With Cancer Less Willing to Share Their Health
Information? Privacy, Sensitivity, and Social Purpose
by David Grande, et al
Impact of the National Cancer Institute Community Cancer
Centers Program on Clinical Trial and Related Activities at a
Community Cancer Center in Rural Nebraska
by Mehmet Sitki Copur, et al
Complications Associated With Use of Long-Term Central Venous
Catheters Among Commercially Insured Women With Breast
Cancer
by Allison Lipitz-Snyderman, et al
JOP.ascopubs.org
Volume 7, Issue 3 May 2011
The Authoritative Resource for Oncology Practices
Report on the ASCO 2010 Provider-Payer Initiative Meeting By Michael N. Neuss, MD, et al
Subspecialization in Community Oncology: Option or Necessity? By Dean H. Gesme, MD, et al
Current Hepatitis B Screening Practices and Clinical Experience of Reactivation in Patients Undergoing Chemotherapy for Solid Tumors: A Nationwide Survey of Medical Oncologists By Fiona L. Day, FRACP, et al
Barriers to Recruitment of Rural Patients in Cancer Clinical Trials By Shamsuddin Virani, MB, BS, et al
Partners and Partnerships: Trends in Private Oncology Practice By Thomas A. Paivanas, MHSA
Journal of oncology Practice
www.jop.ascopubs.org

PAGE 58 The ASCO Post | SEPTEMBER 10, 2015
Direct From ASCO
FASCO, of the Fred Hutchinson Cancer Research Cen- ter and the University of Washington. Dr. Lyman is a member of ASCO’s Board of Directors and the Chair of the Methodology Subcommittee of the CPGC.
Prior to these advisory groups, guide-line development typically relied on ad hoc proposals from ASCO members. “While still encouraging member sugges-tions, this new organization allows for a more systematic and comprehensive ap-proach to guideline development with a broad scope of coverage, and better priori-tization,” Dr. Lyman said.
A Collaborative ApproachProviding a list of prioritized guideline
topics each year, along with the order in which guidelines will be developed, will facilitate collaborative efforts with other guideline development groups. Such col-laborative efforts can include joint guide-line development, guideline endorsement, or guideline adaptation.
The prioritized list of topics allows ASCO staff to contact other guideline de-velopment groups about possible collabo-ration in creating new guidelines. Joint guideline development occurs when the topics and scope of effort of two or more guideline development groups align, and there is an agreed-upon process for guide-line development. Development of a joint guideline allows for a more efficient use of resources and facilitates increased guide-line standardization, credibility, and dis-semination.
ASCO collaborates with organizations striving to meet the standards of the In-stitute of Medicine (IOM) and Council of Medical Specialty Societies (CMSS). This includes managing conflicts of inter-est among guideline development group members, establishing evidence-based recommendations, and presenting rec-
ommendations in a clear and transparent manner.1
ASCO does not mandate how collabo-rators should create and present guide-lines, as long as they primarily adhere to the IOM and CMSS standards. An orga-nization can contract out its guideline de-velopment; produce it in-house; or have a unique development, quality appraisal, and approval process. Documents do not have to contain specific sections, nor must data be summarized in a certain format. Collaborators typically follow the format used to publish their guideline develop-ment efforts, and the components are gen-erally consistent across the collaborating guideline development groups.
Collaborations between ASCO and other guideline development groups can take several forms. In the traditional joint collaboration model, organizations typically share the human and/or financial resources equally, with a similar number of represen-tatives on the guideline development panel dividing the writing responsibility.
In a more recent collaborative guideline development, one organization takes the lead, whereas others play more of a sup-porting role. If ASCO takes the lead, the Society provides the majority of the re-sources, whereas collaborators send official representatives to sit on the expert panel and approve the final guideline. If both organizations approve of the guideline, it is published as a joint effort and placed on the organizations’ websites. In this manner, guideline productivity can increase. Guide-lines can reach a wider audience and have a greater impact with dual publication to their respective memberships.
“ASCO is very flexible in making col-laborations work, either from having one representative on the panel to com-pletely coproducing or endorsing other organizations’ guidelines,” said Sharon H. Giordano, MD, MPH, of the University of Texas MD Anderson Cancer Center, and Chair of ASCO’s CPGC.
CAP PartnershipsASCO has created two joint guidelines
with the College of American Patholo-gists (CAP) since the organizations began working together in 2008, and they have followed the traditional model of equal responsibility.
The guideline development process can take longer together than when created by either organization alone. However, the result is a document with more strength, credibility, and reach. “Even if there is ad-ditional time, it is incredibly valuable to work with clinicians on the guideline,” said Elizabeth A. Wagar, MD, of The University of Texas MD Anderson Cancer Center, and Chair of CAP’s Pathology and Laboratory Quality Center Committee. Whereas oncologists help CAP members understand the therapeutic options based on biomarker testing, Dr. Wagar said, pa-thologists help the oncologists understand issues with obtaining high-quality data and clear results.
Expansion and Shortened Production Time
Guideline endorsements and adap-tations allow ASCO to rapidly expand its guideline portfolio, an action en-couraged by the ASCO membership that will facilitate guideline integration into ASCO’s new CancerLinQ™ initia-tive. Once Guideline Advisory Groups receive confirmation of prioritized topics from the CPGC, literature is re-viewed to see if any guidelines already
exist on each topic from other organi-zations. If the guidelines are evidence-based and up-to-date—and the Guide-line Advisory Group agrees with the recommendations—the group con-siders endorsing the guideline. If the Guideline Advisory Group agrees with most, but not all, of the recommenda-tions of an existing guideline, it may opt to modify the guideline and produce an adapted version.
ASCO has published several endorse-ments and adaptations on a variety of topics over the past year, and several more endorse-ments are currently in press. An independent panel at ASCO adapted a pan-Canadian practice guideline concerning fatigue in adult cancer survivors in 2014.2 The adaptation process took a few months, but a de novo guideline likely would have taken at least 1 year, Dr. Lyman said.
With more than 50 completed guide-lines, endorsements, and adaptations, and more than 20 topics currently in devel-opment, the ASCO Guidelines Program has worked to increase its portfolio of ev-idence-based clinical practice guidelines to better meet the needs of its membership and the patients they serve. n
References1. Institute of Medicine: Standards for
developing trustworthy clinical practice guidelines. Available at www. iom.edu/Re-ports/2011/Clinical-Practice-Guidelines-We-Can-Trust/Standards.aspx. Accessed May 13, 2015.
2. Bower JE, Bak K, Berger A, et al: Screening, assessment, and management of fatigue in adult survivors of cancer: An American Society of Clinical Oncology clinical practice guideline adaptation. J Clin Oncol 32:1840-1850, 2014.
Originally printed in ASCO Daily News. © American Society of Clinical Oncology. “ASCO Guidelines: A Collaborative Effort” am.asco.org, May 29, 2015. All rights reserved.
Join Us for the Community Research Forum Annual Meeting This year’s Community Research
Forum Annual Meeting is right around the corner. There is an exciting lineup scheduled for the meeting, with participants and presenters representing a range of practices and types of research programs. Participants will engage with one another in breakout sessions to ad-dress topics related to conducting clinical research in a community-based practice,
including coverage analyses of clinical trials, contract negotiations, exemplary attributes of trial sites, and practical considerations for precision medicine. The meeting will include presentations by internationally recognized speakers on personalized medicine and the chal-lenges of molecular profiling. There will also be discussions about clinical trial opportunities.
The Community Research Forum has also developed valuable tools for community researchers, including the ASCO Research Program Quality As-sessment Tool, the ASCO Clinical Trial Workload Assessment Tool, and an online repository of resources. Please join fellow colleagues from community research sites for this great opportunity to advance clinical research. The meeting will
take place September 20 to 21, 2015, in Alexandria, Virginia.
Visit the Community Research Forum webpage at www.asco.org/ communityresearchforum to register for Meeting, and for additional information about the Forum and its resources. n
© 2015. American Society of Clinical Oncology. All rights reserved.
ASCO Guidelinescontinued from page 55
Sharon H. Giordano, MD, MPH

ASCO ® 2 e s m o 3 St. Gallen consensus4
N I C Ed i a g n o s t i c S
g u i d a n c e 5
N C C NGUIDELINES®1
GENB-24117 Invasive Ad_6-6_ASCO-POST.indd 1 4/16/15 1:44 PM

PAGE 60 The ASCO Post | SEPTEMBER 10, 2015
Journal Spotlight
Dabrafenib Plus Trametinib Improves Overall Survival vs Dabrafenib in BRAF V600–Mutant MelanomaBy Matthew Stenger
O verall survival results of the phase III COMBI-d trial reported
in The Lancet by Georgina V. Long, MD, and colleagues showed that the combination of the BRAF inhibitor dabrafenib (Tafinlar) with the MEK inhibitor trametinib (Mekinist) re-sulted in significantly prolonged overall survival vs dabrafenib alone in BRAF V600–mutant melanoma.1 The primary analysis of the trial had shown that the combination significantly prolonged progression-free survival.2
Study DetailsIn this double-blind trial, 423 pre-
viously untreated patients from 14 countries with BRAF V600E/K mu-tation–positive unresectable stage IIIC or IV melanoma were randomly assigned between May 2012 and No-vember 2012 to receive oral dab-rafenib at 150 mg twice daily and oral trametinib at 2 mg once daily (n = 211) or dabrafenib and placebo (n = 212). The primary endpoint was pro-gression-free survival; overall survival was a secondary endpoint.
The combination and dabrafenib groups were generally balanced for age (median, 55 and 56.5 years), sex (53% and 54% male), Eastern Cooperative Oncology Group performance status (0 for 73% and 71%, 1 for 26% and 29%), BRAF mutation (V600E in 85% in both, V600K in 15% and 14%), stage (IVM1c in 67% and 65%; IIIc, IVM1a-b in 33%
and 34%), lactate dehydrogenase level (greater than the upper limit of normal in 36% and 33%), visceral disease (78% and 68%), and number of disease sites (at least three in 48% and 43%).
Improved Overall SurvivalAt the final data cutoff ( January
2015), after 222 patients had died, me-dian overall survival was 25.1 months (95% confidence interval [CI] = 19.2 months to not reached) in the combi-nation group vs 18.7 months (95% CI
= 15.2–23.7 months) in the dabrafenib group (hazard ratio [HR] = 0.71, P = .0107). Overall survival was 74% vs 68% at 1 year and 51% vs 42% at 2 years.
Consistent benefit of the combi-nation was observed across all sub-groups, with hazard ratios significantly favoring the combination among pa-tients with Val600Glu mutation, stage IVM1c disease, baseline lactate de-hydrogenase above the upper limit of normal, visceral disease, and at least three disease sites.
At the current analysis, after an additional 17 months of follow-up since the primary analysis, median progression-free survival was 11.0 months (95% CI = 8.0–13.9 months) in the combination group vs 8.8 months (95% CI = 5.9–9.3 months) in the dabrafenib group (HR = 0.67, P = .0004, unadjusted for multiple testing). Consistent benefit of the
combination was observed across all subgroups.
In the primary analysis, at a median follow-up of 9 months, median progres-sion-free survival had been 9.3 vs 8.8 months (HR = 0.75, P = .0348). Over-all response rates were 69% (complete response in 16%) vs 53% (complete re-sponse in 13%, P = .0014).
Subsequent TherapyA total of 29% of the combination
group and 31% of the dabrafenib group continued study treatment for at least 15 days after progression. Subsequent therapy, excluding study therapy, was used in 33% and 51% of patients, in the combination and dabrafenib groups, respectively, with the most common treatments being ipilimumab (Yervoy, 18% and 28%), dacarbazine (8% and 11%), and vemurafenib (Zelboraf, 8% and 11%); anti–programmed cell death protein 1 (PD-1) treatment was re-ceived by 3% and 7%.
Adverse EventsThe most common treatment-relat-
ed adverse events of any grade in the combination group were pyrexia (52% vs 25%), chills (28% vs 14%), fatigue (27% vs 28%), and rash (24% vs 20%), whereas those in the dabrafenib group were hyperkeratosis (33% vs 6%), fa-tigue (28% vs 27%), hand-foot syn-drome (27% vs 6%), and alopecia (26% vs 5%). The combination group also had lower rates of cutaneous squamous cell carcinoma (3% vs 9%), and skin papilloma (1% vs 18%).
Treatment-related grade 3 adverse events occurred in 32% vs 30% of pa-tients (grade 4 events in one combina-tion recipient and three dabrafenib re-cipients), with the most common being pyrexia (7% vs 2%) in the combination group and cutaneous squamous cell car-cinoma (9% vs 3%) in the dabrafenib group. New primary malignant melano-mas occurred in < 1% and 2% of patients and noncutaneous treatment-emergent cancers were reported in 1% and 2%.
Treatment was discontinued due to adverse events in 11% of the combi-nation group vs 7% of the dabrafenib group, with the most common reasons being pyrexia (2% vs 1%) and decreased ejection fraction (1% vs 1%).
The investigators concluded: “The improvement in overall survival estab-lishes the combination of dabrafenib and trametinib as the standard targeted treat-ment for BRAF Val600 mutation–posi-tive melanoma. Studies assessing dab-rafenib and trametinib in combination with immunotherapies are ongoing.” n
Disclosure: The study was funded by GlaxoSmithKline. For full disclosures of the study authors, visit www.thelancet.com.
References1. Long GV, Stroyakovskiy D, Gogas H,
et al: Dabrafenib and trametinib versus dab-rafenib and placebo for Val600 BRAF-mu-tant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386:444-451, 2015.
2. Long GV, Stroyakovskiy D, Gogas H, et al: Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371:1877-1888, 2014.
Dermatologic Oncology
The improvement in overall survival establishes the combination of dabrafenib and trametinib as the standard targeted treatment for BRAF Val600 mutation–positive melanoma.
—Georgina V. Long, MD, and colleagues
Dabrafenib/Trametinib for Melanoma
■ Dabrafenib plus trametinib significantly prolonged overall survival and progression-free survival in melanoma patients.
■ Combination treatment was associated with lower rates of cutaneous squamous cell carcinoma, hyperkeratoses, skin papilloma, alopecia, and hand-foot syndrome.
COMBI-d Trial and the Need to Guide Progress in Melanoma TreatmentMichael S. Sabel, MD, FACS, of the University of Michigan Health Systems, Ann Arbor, offers his perspective on the COMBI-d trial discussed above. See page 61.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 61
Perspective
COMBI-d Trial and the Need to Guide Progress in Melanoma Treatment By Michael S. Sabel, MD, FACS
A s reviewed in this issue of The ASCO Post, Long et al1 have
reported the final overall survival analysis of the COMBI-d phase III trial comparing combination therapy with the BRAF inhibitor dabrafenib (Tafinlar) and the MEK inhibitor tra-metinib (Mekinist) to monotherapy with dabrafenib alone, confirming the superiority of combination therapy in patients with advanced melanoma harboring the BRAF V600 mutation. As witnessed before, such combina-tion therapy not only improves surviv-al, but also reduces many of the com-plications associated with paradoxical activation of the MAPK pathway.
Further InsightWhile the COMBI-d study find-
ings serve primarily to support the idea that combination therapy should be the standard targeted therapy for patients with BRAF V600 mutation–positive melanoma, the more mature survival analysis provides further in-sight into the rapidly changing man-agement of advanced melanoma. It has been stated before, but cannot be overemphasized, that this is a unique and challenging period for clinicians treating patients with melanoma. Af-ter years of limited options, the rapid introduction and development of two distinct classes of systemic agents for melanoma have been extremely excit-ing but uniquely perplexing, as each new clinical trial raises additional questions regarding both clinical ap-plication and basic science.
In their discussion, the authors compare the progression-free and overall survival data with those from trials of single-agent immunothera-pies and find that they compare fa-vorably. This shines a light on one of the challenges facing clinicians—the most appropriate choice of therapy in patients with a BRAF V600 muta-
tion. And, while it may be tempting to compare 1-year survival data between trials, the patient populations being treated are not the same, as immuno-therapy trials are often restricted to, or dominated by, patients with BRAF wild-type melanoma.
In addition, these are moving targets. At the same time Long et al are providing additional support for combination targeted therapy, new data are emerging support-ing combination immunotherapy. These classes of drugs are also sub-stantially different in terms of ease of delivery, cost, expected toxicities, and the time course of response, all of which must be considered when translating clinical trial results to in-dividual patients.
Persistent Holes in Our Knowledge
These questions highlight many of the issues we have to address moving forward, particularly as ongoing tri-als begin to explore the combination of these two classes. With new agents being developed, many of the clini-cal trials are designed with a focus on comparing efficacy and establishing position. We must be diligent in assur-ing that ongoing research continues to address persistent holes in our knowl-
edge—not only the most appropriate clinical use of current drugs, but also gaining additional insight into the ba-sic science that dictates their successes (and failures).
The triumphs of the checkpoint inhibitors ipilimumab (Yervoy), pem-
brolizumab (Keytruda), and nivolu-mab (Opdivo) have been a tremen-dous boon to patients and have altered the landscape of melanoma manage-ment, but have also challenged our understanding of the interactions be-tween the immune system and cancer. Likewise, data such as those presented by Long et al regarding dabrafenib and trametinib highlight ongoing ques-tions regarding the molecular basis of melanoma.
These questions will only be-come more apparent as we continue to explore future combinations and shift to the adjuvant setting, where the goals of therapy and the biol-ogy of the disease can be consider-ably different. Our clinical research must strive not only to document moderate improvements in out-come, but to identify predictive fac-tors that optimize patient selection and address gaps in our basic sci-ence knowledge. These might then allow us to optimize the timing and sequence of combination therapies
in concordance with how these agents function and interact and identify new potential targets for intervention. The “bench to bed-side” model should more appropri-ately be visualized as a continuous “back and forth” between the bench and bedside.
Curbing OverenthusiasmWith each newly reported success
of both targeted therapies and immu-notherapies, it is sometimes hard to prevent our enthusiasm and excite-ment from overshadowing some of the gaps in knowledge we still have. We must also be aware that this same phenomenon is present in our pa-tients, who are also witness to the trumpeted successes of the new sys-temic agents in the media.
In surgical oncology, we have seen an increasing, and concerning, trend for patients refusing surgery or seeking to greatly limit surgery, sec-ondary to their enthusiasm for “new drugs” they read about. This includes patients declining completion node dissections as well as those refusing surgery for clinically evident stage III disease or isolated, resectable M1a disease.
While it is important that our re-search efforts moving forward address these clinical questions regarding the multidisciplinary management of melanoma, we also must spend time educating patients, in both the office and the media, about the limitations of these therapies and the many ques-tions that still exist regarding their op-timal use. n
Disclosure: Dr. Sabel reported no potential conflicts of interest.
Reference1. Long GV, Stroyakovskiy D, Gogas
H, et al: Dabrafenib and trametinib ver-sus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised con-trolled trial. Lancet. May 29, 2015 (early release online).
While it is important that our research efforts moving forward address these clinical questions regarding the multidisciplinary management of melanoma, we also must spend time educating patients, in both the office and the media, about the limitations of these therapies and the questions that still exist regarding their optimal use.
—Michael S. Sabel, MD, FACS
Dr. Sabel is Chief, Division of Surgical Oncology, University of Michigan Health Systems, Ann Arbor.
Visit The ASCO Post website at ASCOPost.com

PAGE 62 The ASCO Post | SEPTEMBER 10, 2015
Announcements
Seventeen ASTRO Members Awarded Fellows Designation
The American Society for Radia-tion Oncology (ASTRO) has se-
lected 17 distinguished members to receive the ASTRO Fellows designa-tion. The 2015 class of Fellows will receive the recognition at the Awards Ceremony at ASTRO’s 57th Annual Meeting on Tuesday, October 20 in San Antonio, Texas.
ASTRO Fellows designation, or FASTRO, honors those individuals who have been active or emeritus members of ASTRO for at least 15 years, given the equivalent of 10 years of service to ASTRO, and significant-ly added to the field of radiation on-cology in the areas of research, educa-tion, patient care, and/or service and leadership.
Including the 2015 class of Fel-lows, 259 ASTRO members have received the FASTRO designation since the inception of the award in 2006.
2015 Fellows ClassThe members of the 2015 Fellows
class are:May Abdel-Wahab, MD, PhD, Di-
rector of the Division of Human Health in the Department of Nuclear Sciences and Applications, International Atomic Energy Agency
Kaled M. Alektiar, MD, Attending Radiation Oncologist, Memorial Sloan Kettering Cancer Center
Manjeet Chadha, MD, Attend-ing Physician and Associate Chair of the Charles and Bernice Blitman Department of Radiation Oncology, Mount Sinai Beth Israel
A. Bapsi Chakravarthy, MD, Pro-fessor and Residency Program Director in the Department of Radiation Oncol-ogy, Vanderbilt University
Eric L. Chang, MD, Professor and Chair of the Department of Radiation Oncology, Keck School of Medicine of University of Southern California
Joel M. Cherlow, MD, PhD, Radia-tion Oncologist, Long Beach Radiation Oncology Medical Group and Orange County CyberKnife and Radiation On-cology Center, and Medical Director, Department of Radiation Oncology, Saddleback Memorial Hospital
Martin Colman, MD, Professor and Chair of the Department of Radia-tion Oncology and John Sealy Distin-guished Centennial Chair, The Univer-sity of Texas Medical Branch, Galveston
Carol A. Hahn, MD, Associate Pro-fessor, Department of Radiation Oncol-ogy, Duke Cancer Institute
James M. Larner, MD, Professor of Radiation Oncology and Chair, Department of Radiation Oncology, and Professor of Medicine, Depart-ment of Internal Medicine/ He-matology Oncology, University of Virginia
Robert C. Miller, MD, MBA, Pro-fessor of Radiation Oncology, Mayo Clinic Florida, and Medical Director, Mayo Clinic Operations
Arnold dela Cruz Paulino, MD, Professor and Pediatric Radiation On-cology Fellowship Director, Depart-
ment of Radiation Oncology, The Uni-versity of Texas MD Anderson Cancer Center
Rachel Rabinovitch, MD, Profes-sor, Departments of Radiation Oncol-ogy and Medicine, University of Colo-rado School of Medicine

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 63
Announcements
C. Leland Rogers, MD, Clinical Pro-fessor, Department of Radiation Oncol-ogy, Virginia Commonwealth University
David I. Rosenthal, MD, Section Chief and Director of Translational Re-search, Head and Neck Division of Radia-tion Oncology, and Professor, Department of Radiation Oncology, The University of Texas MD Anderson Cancer Center
Richard Kenneth Valicenti, MD, MA, Professor and Chair of the De-partment of Radiation Oncology, Uni-versity of California, Davis School of Medicine
Julia White, MD, Professor, Depart-ment of Radiation Oncology, The Ohio State University Comprehensive Can-cer Center
Yan Yu, PhD, MBA, Professor, Vice-Chair, and Director of Medical Physics, Department of Radiation Oncology, Thomas Jefferson University
“The 17 Fellows chosen this year have demonstrated their dedication to the field of radiation oncology. Their work has impacted and benefited pa-tients and the field of radiation oncology
in a variety of ways, including patient care, research, education, service, and leadership,” said Bruce G. Haffty, MD, FASTRO, Chair of ASTRO’s Board of Directors and Professor and Chairman of the Department of Radiation Oncol-ogy at Rutgers–Robert Wood Johnson Medical School and Associate Director at the Cancer Institute of New Jersey. n

PAGE 64 The ASCO Post | SEPTEMBER 10, 2015
Announcements
Massimo Cristofanilli, MD, Named Associate Director for Precision Medicine and Translational Research at Lurie Cancer Center
B reast cancer expert Massimo Cristofanilli, MD, has been ap-
pointed Associate Director for Precision Medicine and Translational Research at the Robert H. Lurie Comprehensive
Cancer Center of Northwestern Uni-versity and Director of Northwestern Onco-SET (Sequence, Evaluate, Treat). Dr. Cristofanilli will also be Professor of Medicine in the Division of Hematolo-
gy-Oncology at Northwestern Univer-sity Feinberg School of Medicine.
An expert in translational research and treatment of patients with inflam-matory breast cancer, Dr. Cristofanilli
will be an integral part of the medical oncology team providing breast cancer patients with the most effective treat-ment options available at the Maggie Daley Center for Women’s Cancer Care in Prentice Women’s Hospital.
As Associate Director for Precision Medicine and Director of Northwest-ern Onco-SET, Dr. Cristofanilli will oversee the development of Onco-SET and related clinical and research opera-tions. The program personalizes cancer care for patients by sequencing the in-dividual genetic profile of their tumors and evaluating the results to provide the treatments or clinical trials that will of-fer the greatest benefit.
Dr. Cristofanilli comes to North-western from Thomas Jefferson Uni-versity and Hospitals, where he served as Director of Jefferson Breast Care Center and Deputy Director of Transla-tional Research at the Kimmel Cancer Center. Previously, Dr. Cristofanilli was Chair of the Department of Medical Oncology at Fox Chase Cancer Center and Executive Director of the Morgan Welch Inflammatory Breast Cancer Program and Clinic at The University of Texas MD Anderson Cancer Center.
“We have the unprecedented op-portunity to design a patient-centered, biology-driven model of cancer care combining sophisticated tissue and blood-based molecular diagnostic tech-nologies and innovative treatments,” said Dr. Cristofanilli. “We plan to con-tinue the establishment of strategic partnerships and to involve our patients in an educational and informative jour-ney to advance their understanding of their disease and treatments.” n
Massimo Cristofanilli, MD
Visit The ASCO Post
website at ASCOPost.com

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 65
Expert’s Corner
Oncology Worldwide
medical degree from Columbia Univer-sity College of Physicians and Surgeons in New York and completed his medical residency in internal medicine at Bos-ton City Hospital, followed by a fellow-ship in hematology and medical oncol-ogy at Tufts University/New England Medical Center. Dr. Khuri then began a remarkable 20-year career as one of the leading translational clinical investiga-tors and physicians in lung and aerodi-gestive medical oncology, first at MD Anderson Cancer Center in Houston and for the past 13 years at Emory Uni-versity School of Medicine in Atlanta.
Before starting his new position at AUB on September 1, 2015, Dr. Khuri was Deputy Director of Emory’s Win-ship Cancer Institute, Professor and Chair of the Department of Hematol-ogy and Medical Oncology, Executive Associate Dean for Research, and the Roberto C. Goizueta Distinguished Chair for Cancer Research at Emory. During his tenure at Emory, Dr. Khuri was instrumental in securing a Nation-al Cancer Institute (NCI) designation for Winship Cancer Institute, the only cancer institute in Georgia to achieve an NCI designation, and was co-head of the $12.5 million NCI-funded Specialized Program of Research Ex-cellence (SPORE) in head and neck cancer.
Dr. Khuri is the recipient of numer-ous awards, including the American Association for Cancer Research Rich-ard and Hinda Rosenthal Memorial Award and the Nagi Sahyoun Award of the Middle East Medical Assembly for his groundbreaking research in lung and aerodigestive cancers. He has been named one of America’s Top Cancer Doctors by Castle Connolly Medical for the past 8 years. Dr. Khuri is a long-time member of ASCO and has served as a member of ASCO’s Cancer Preven-tion Committee, the Conquer Cancer Foundation Grants Selection Commit-tee, the Journal of Clinical Oncology Edi-torial Board, and the Scientific Program Committee.
The ASCO Post talked with Dr. Khuri about his decision to leave Emo-ry University; the new responsibilities and challenges he faces; and his goal of making AUB the premier liberal arts institution in the Middle East.
Deep Roots in Family HistoryPlease talk about the decision-mak-
ing that went into changing the course of your career from leading clinical investi-gator and physician in the field of lung
and aerodigestive medical oncology to university president.
I was elected to the Board of Trust-ees of AUB in 2014, so I was aware of some of the challenges the university faced but had not initially considered the position of President when I was first approached. However, after think-ing more about it and looking at what the university’s needs are and my skill set, although I’m not going to say it is a perfect fit, I liked what I could do and what I could contribute and thought it would be an important use of the sub-stantial number of remaining years in my career.
Several areas of importance stood out to me from my very first meeting to discuss this position. AUB is an in-stitution that has educated my great grandfathers, my grandfather, both of my parents, my wife’s grandfather, and her parents, and it is where I started, so its roots run deep in my family history.
What I saw in AUB is an institution that has an impact on its region like no other university I know of in the world. There are great universities in China, India, and the United States, and they counterbalance each other. I don’t mean any disrespect to the other great universities in Lebanon and in the re-gion, but there is no question that AUB casts a very large shadow and has a ma-jor impact on the political discourse in the region, the development of modern engineering and knowledge, and medi-cal technology and research.
Furthermore, AUB is a fundamental driver of the American liberal arts ethos in the region, and I thought I could con-tribute to strengthening its liberal arts curriculum with regard to my skill set
in building communities of trust that would be valuable for that institution and for that region. So I took the plunge and less than 7 weeks after that first in-terview, I was informed that I was the search committee’s and the Board lead-ership’s first choice for President.
Innovators in Research and Education
What will be your primary responsi-bilities as President?
My main goal is to ensure that the university continues to thrive and that the culture of progressive thought and innovation, service, and generosity to the community continues to be sup-ported. Another goal is that the deans of the six faculties that I’ll have the privilege of overseeing, including Ag-riculture and Food Sciences; Arts and Sciences; Engineering and Architec-ture; Health Sciences; Medicine; the Suliman S. Olayan School of Business;
as well as university interdisciplinary programs, all get supported to the de-gree they need. I also want to drive the university toward becoming a major research institution, which it was well into the 1980s and has been rebuilding since the end of the Lebanese Civil War in 1990.
In the Middle East, the amount of funding support for scientific research is relatively low. We don’t have the equiva-lent of the National Institutes of Health to provide funding, but you can make a strategic investment in scholarship in the arts, economics, political sciences, business, and medicine. So we are go-ing to have to be innovators in educa-tion and innovators in research without losing that liberal arts ethos that makes
AUB such a fundamental driver of the region’s moral compass.
Bold ChoicesWhat are your greatest challenges?The university has gone through
some periods of unrest, and this past year, students organized protests over increases in tuition. I would like to slow down the increase in student tuition, as I think the students have a point there, but we also have to show the value in the unique and high-quality education we provide.
We also need to invest more in mak-ing AUB a premier research institution. I’d like to be here long enough to pro-duce clear evidence that AUB is one of the 100 greatest and most valuable uni-versities in the world, which, from the perspective of an American institution in a country of 4 million people and close to 2 million refugees, is not going to be easy.
I think we can get it done because the quality of our faculty and students is stellar, but we are going to have to make some bold choices and not make as many mistakes as we could otherwise afford. Still, I want to emphasize that my goal is to develop an institutional cul-ture where people take risks, and even if there is an honest mistake, that mistake is celebrated and not castigated.
Mentor, Teacher, SupervisorWill you continue to see patients?I’ll be seeing patients at least a cou-
ple of times a month, but I’m not going to have a private clinic; I’ll have a teach-ing clinic. I have superb colleagues, including at least two excellent lung and aerodigestive physicians at AUB, and they will keep the private patients. I’d like to be a teaching attending with residents and fellows and with my col-leagues and still help mentor them to develop new trials and participate in global trials of new agents, new genom-ic drivers, and new treatment modali-ties like immunotherapy.
I would like to think that my role will be even more as a mentor, teacher, and supervisor than my work at Emory. I’ve always enjoyed teaching nurse practi-tioners, physician assistants, residents, fellows, medical students, and interna-tional faculty, so I will have more of a teaching role at AUB.
A Team of Stars at EmoryHow difficult was it to give up your re-
search program at Emory University?In one sense, it was very difficult, and
in another, it was somewhat rewarding.
Fadlo R. Khuri, MD, FACPcontinued from page 1
continued on page 66
The most surprising thing about medicine, and oncology in particular, is how much physicians
get back from their patients. We find inspiration, hope, and confidence in difficult times and the
ingenuity to figure out a solution when we can’t treat someone’s disease by the book.
—Fadlo R. Khuri, MD, FACP

PAGE 66 The ASCO Post | SEPTEMBER 10, 2015
Expert’s Corner
It’s very rewarding because we’ve grown a team of stars at Emory who are all very good citizens, outstanding scientists, and thought leaders in the biology and treatment of cancer.
For example, the lung cancer re-search program at Emory is superbly led by Haian Fu, PhD [Leader, Dis-covery and Development Therapeutics Program], and Suresh S. Ramalingam, MD [Director of Medical Oncology and the Lung Cancer Program]. Dr. Ramalingam is a major leader at ASCO and the Eastern Cooperative Oncology Group, and Dr. Fu is one of the leading cancer biologists focusing on protein-protein interactions. There are superb scientists such as Jing Chen, PhD [Co-Director of Experimental Therapeutics Program in Leukemia], Adam Marcus, PhD [Director, Integrated Cellular Im-aging Shared Resource], and others as well as great clinical investigators, such as Taofeek Owonikoko, MD, PhD [Associate Professor in the Depart-ment of Hematology and Medical On-cology], who was just accepted to the ASCO Leadership Development Pro-gram. The team has a lot of depth, and I’m very proud of them.
I’m actually happy to step back and see them succeed. But I will miss go-ing to their lab meetings and seeing the fabulous progress made by their undergraduate and graduate students, post-docs, and fellows. I am a per-
manent student and love science, so I will miss those interactions and the day-to-day, week-to-week accomplish-ments and delight in witnessing their discoveries. I love the idea of not just proving our scientific hypotheses but repudiating them as well and helping create new ones.
I’m realistic enough to know that I’m not going to have the bandwidth to help drive that kind of mission individ-ually at AUB. I’m going to have to help drive it collectively. But I am keeping my collaborations with Emory, and I’ll remain part of the program project as it goes forward.
Change for the Right ReasonsYou spent most of your youth in Leba-
non. Are you excited to be back there and in a position to make a major difference in the region?
I’m extremely excited. The real chal-lenge in life is about being able to rein-vent and redefine yourself every 10 to 15 years, and I think you should only do that if you are sure you are doing it for the right reasons. And to the best of my ability to look into my soul, I think I have made this change for the right reasons.
I want to help contribute to a new era of understanding and liberal think-ing and scientific renaissance as well as a renaissance in the humanities in the region. I was very fortunate to grow up in the Near East. I had a wonderful childhood and adolescence despite the
war. I learned a lot, and a lot of what I learned is because of this university, so it is time for me to help continue that tradition and maybe accelerate it a little and give back.
The most surprising thing about medicine, and oncology in particular, is how much physicians get back from their patients. We find inspiration, hope, and confidence in difficult times and the ingenuity to figure out a solu-tion when we can’t treat someone’s dis-ease by the book.
Medicine is a very rewarding two-way stream, so from that perspective, I think it would be great to be able to contribute in a meaningful way to AUB and to Lebanon because they
have contributed so much to me and to my growth and will continue to con-tribute to my life.
I enjoy being a member of ASCO and the American Association for Can-cer Research and will continue to be a member of both organizations. I will probably break my nearly perfect 20-year streak of attendance at the annual meet-ings but will likely come to each meeting every other year, and I will certainly at-tend the meetings online because it’s not just about the learning that is important to me. It is being part of the community of scholars and physicians that I do not want to lose touch with. n
Disclosure: Dr. Khuri reported no potential conflicts of interest.
Fadlo R. Khuri, MD, FACPcontinued from page 65
Don’t Miss these iMportant reports in this issue of The ASCO POST
Sagar Lonial, MD, on Elotuzumab and Lenalidomide/-Dexamethasone in Refractory Multiple Myeloma see page 1
Robert L. Ferris, MD, PhD, on HPV16 DNA in Post-Treatment Oropharyngeal Cancer see page 21
James O. Armitage, MD, FACP, FRCP, on Indolent Lymphoma see page 5
Julie Vose, MD, MBA, FASCO, on the Role of the Patient in Clinical Research see page 1
Daniel G. Haller, MD, on Treatments of Colorectal Liver Metastases see page 14
Steven Isakoff, MD, on Surgery and Adjuvant Radiotherapy in Breast Cancer see page 17
Andrew Roth, MD, and colleagues, on Risk of Suicide After Cancer see page 26
Jimmie Holland, MD, on Psychosocial Interventions for Older Patients With Cancer see page 27
Suresh S. Ramalingam, MD, on Targeted and Immunotherapeutic Approaches to Lung Cancer see page 3
Visit The ASCO Post online at ASCOPost.com

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 67
In MemoriamOncology Worldwide
Access to Cancer Medicines Not Uniform Across EuropeBy Caroline Helwick
A ccess to cancer medicines—in-cluding some old standbys—is
inconsistent across Europe, depriving many patients of treatments that are the standard of care elsewhere,1 ac-cording to Alexandru E. Eniu, MD, PhD, Chair of the European Soci-ety for Medical Oncology (ESMO) Emerging Countries Committee and Head of the Day Hospital Unit at the Cancer Institute Ion Chiricută, Cluj-Napoca, Romania.
“Inequalities exist in availability and patient costs, especially for newer, more expensive drugs, but drug short-ages also affect several essential, old and inexpensive drugs, and this should be unacceptable!” Dr. Eniu said at the ASCO/ESMO Joint Session on Global Perspective on Value at the 2015 ASCO Annual Meeting.
Disparities in Cancer Outcomes“For a number of years, we have
known there are important disparities in cancer outcomes across Europe,” he said. The recent EUROCARE-5 study documented the age-standardized in-cidence vs 5-year relative survival for breast cancer, prostate cancer, and mel-anoma by European region.2 Survival in Eastern Europe was generally low and below the European mean, particularly for cancers with good or intermediate prognosis, the study found.
Regarding breast cancer, for exam-ple, Eastern European countries had approximately half the risk of develop-ing the disease compared with North-ern European countries. However, their 5-year relative survival rate was only about 70%, vs 85% in Northern Europe.
Factors accounting for cancer outcome disparities are numerous, including the general population’s health and lifestyle, health-system infrastructure, cancer-care infrastruc-ture, stage at diagnosis, cancer “work-force,” and presence or absence of screening programs.
Factors Linked to Lack of Access
“Patient access and availability of cancer medication are also very impor-tant,” he said. “Without the drugs, you cannot influence the condition.”
He said the main contributors to the problem of access are the “dra-matically increased pricing” by phar-
maceutical companies and the “inco-herent reimbursement strategy” of the various national bodies, as they “face the problem of trying to accom-modate within budgets the new drugs and new indications.”
Health professionals also contrib-ute, in a way, by “not raising the bar high enough” in terms of the benefits demanded of expensive new drugs. “We have to ask more from ourselves,” he suggested.
Clinical Examples of Access Variability
The recent ESMO antineoplastic medicines survey shed light on the vari-ability in access to cancer medicines across Europe, including drugs listed on the World Health Organization (WHO) Essential Medicines list (www.who.int/medicines/publications/essentialmedi-cines/eml2015). The survey gathered information on new drug approval sta-tus, drug reimbursement and patient out-of-pocket costs, and actual avail-ability of drugs (drug shortages were a critical issue at the time) across the con-tinent, focusing on 14 solid tumor types.
Dr. Eniu used tamoxifen in adju-vant breast cancer as an example of the information gathered regarding its formulary inclusion and availabil-ity. “Tamoxifen was widely available across Europe, but when we ques-tioned how patients found the drug in the pharmacy, we found that it was not available in certain countries,” he said. “You can imagine this is a big is-sue. We showed that drug shortages affect several essential old and inex-pensive drugs, not just new ones, and this is not an issue of resources.”
Similarly, adjuvant trastuzumab (Herceptin) is widely available across Europe, usually at no cost to patients, but some countries require preap-proval, which causes weeks of delay in its use. Variability is also observed in the availability and out-of-pocket costs of drugs for metastatic breast
cancer. For example, for anti-HER2 therapy, the newer agents are not widely available or available only at full cost to patients.
A similar scenario is seen with tar-geted treatments for lung cancer, and for melanoma, many cutting-edge drugs are not available at all or only at full cost.
Search for Creative SolutionsAlthough the European Medicines
Agency (EMA) process is very trans-parent, and its decisions regarding safe-ty and efficacy of new drugs are valid for the entire European Union, there are 28 different countries with complicated reimbursement systems. The many na-
tional commissions and expert com-mittees often work with “weak data” and are “replicating at a lower level” the same assessment performed by the EMA, he said.
These groups, he added, are look-ing for “creative and desperate strat-egies.” In Dr. Eniu’s own country, Romania, it took almost 8 years to approve trastuzumab in the metastat-ic setting. “The problem is we cannot always accommodate the cost of new drugs,” he said.
Dr. Eniu further noted that the prices proposed by pharmaceutical companies are increasing substantially, that prices are often unrelated to their magnitude of benefit, and that there is little if any trans-parency in how these prices are set.
He added that many drugs ranked high on ESMO’s new Magnitude of Clinical Benefit Scale (MCBS)
[see The ASCO Post, August 10, 2015, page 5] may not be available in some countries. In fact, none of the top-ranked drugs—ipilimumab (Yervoy), vemurafenib (Zelboraf ), trametinib (Mekinist), or dabrafenib (Tafinlar)—is available in Romania.
Dr. Eniu and his team will be look-ing at their survey data in conjunction with the WHO Essential Medicines List and the Magnitude of Clinical Benefit Scale to better understand the availability of the most critical drugs per country, and they are expanding their survey internationally.
Solutions to problems related to ac-cess must be multimodal, he said. For one thing, health professionals must be more concerned about benefit and “raise the bar,” he suggested. “You wouldn’t buy a new car if it only drives 5 mph faster. You wouldn’t consider the investment worth it.”
The Magnitude of Clinical Benefit Scale can help improve access to cancer medications by informing the process of drug prioritization when resources are limited. National bodies should find ways to reimburse for “reasonable medicines” and apply these sound prin-ciples to public policy, he said. Finally, he added, the pharmaceutical industry should strive for “Justum pretium,” ie, “the just price.” n
Disclosure: Dr. Eniu has received honoraria from and served as a consultant or advisor to AstraZeneca, Novartis, and Roche. He also has received research funding from AstraZeneca, GlaxoSmithKline, Novartis, and Roche.
References1. Eniu AE: Disparities of cancer medi-
cine access in Europe. 2015 ASCO Annual Meeting. ASCO/European Society for Medical Oncology (ESMO) Joint Session: Global Perspective on Value.
2. De Angelis R, Sant M, Coleman MP, et al: Cancer survival in Europe 1999-2007 by country and age: Results of EURO-CARE-5—a population-based study. Lan-cet Oncol 15:23-34, 2014.
Disparities in Access to Cancer Medicines in the European Union
■ Across the 28 countries in the European Union, disparities exist in patient access to cancer medicines.
■ The recent ESMO antineoplastic medicines survey found that the problem is not limited to expensive new drugs.
■ In some countries, valuable drugs are not available or are only available at full cost to patients.
Access to Care
Inequalities exist in availability and patient costs, especially for newer, more expensive drugs, but
drug shortages also affect several essential, old and inexpensive drugs, and this should be unacceptable!
—Alexandru E. Eniu, MD, PhD

OS
PR
OB
AB
ILIT
Y
30% INCREASEIN MEDIAN OS
1.0
0.8
0.6
0.4
0.2
0.0
TIME FROM RANDOMIZATION (MONTHS)
330
335
308
294
267
241
228
180
185
143
148
109
116
81
78
64
60
47
41
30
24
22
13
13
6
5
1
2
0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28
0
Number at Risk
CYRAMZA + paclitaxelPlacebo+ paclitaxel
Hazard Ratio=0.81(0.68, 0.96); P=0.017
CYRAMZA+ paclitaxelPlacebo+ paclitaxel
9.6MONTHS
CYRAMZA+ paclitaxel
(8.5, 10.8)
7.4MONTHS
Placebo+ paclitaxel(6.3, 8.4)
VISIT WWW.CYRAMZAHCP.COM FOR MORE INFORMATION, INCLUDING CYRAMZA MONOTHERAPY TRIAL DATA
CYRAMZA® (ramucirumab) PLUS PACLITAXEL SIGNIFICANTLY EXTENDED OVERALL SURVIVAL (OS)1
• Median PFS with CYRAMZA plus paclitaxel was 4.4 months (95% CI: 4.2, 5.3) vs 2.9 months (95% CI: 2.8, 3.0) with placebo plus paclitaxel (hazard ratio 0.64 [95% CI: 0.54, 0.75]; P<0.001)1
- The percentage of events at the time of analysis was 85% (279 patients) and 88% (296 patients), respectively
• Significantly more patients responded to CYRAMZA combined with paclitaxel (28%; 95% CI: 23, 33) than to placebo plus paclitaxel (16%; 95% CI: 13, 20) (P<0.001)†1,2
CYRAMZA PLUS PACLITAXEL ALSO SIGNIFICANTLY DELAYED DISEASE PROGRESSION AND PROVIDED SIGNIFICANTLY GREATER ORR VS PLACEBO PLUS PACLITAXEL (SUPPORTIVE EFFICACY OUTCOME MEASURES)*1
CYRAMZA as a single agent, or in combination with paclitaxel, is indicated for the treatment of patients with advanced or metastatic gastric or gastroesophageal (GE) junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy.
2 FDA APPROVALSFor use both as monotherapy and in combination with paclitaxel
The phase III RAINBOW trial evaluated the efficacy and safety of CYRAMZA plus paclitaxel vs placebo plus paclitaxel in patients with locally advanced or metastatic gastric or GE junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- and platinum-containing chemotherapy. Major efficacy outcome measure was OS. Supportive efficacy outcome measures were progression-free survival (PFS) and objective response rate (ORR). All patients were Eastern Cooperative Oncology Group performance status 0 or 1. Prior to enrollment, 97% of patients had progressed during treatment or within 4 months after the last dose of first-line chemotherapy for metastatic disease. Twenty-five percent of patients had received anthracycline in combination with platinum/fluoropyrimidine therapy, while 75% did not. Patients were randomized 1:1 to CYRAMZA 8 mg/kg (n=330) or placebo (n=335) every 2 weeks (on days 1 and 15) of each 28-day cycle. Patients in both arms received paclitaxel 80 mg/m2 on days 1, 8, and 15 of each 28-day cycle.1,3
• The percentage of deaths at the time of analysis was 78% (256 patients) and 78% (260 patients) in the CYRAMZA plus paclitaxel and placebo plus paclitaxel treatment arms, respectively1
IMPORTANT SAFETY INFORMATION FOR CYRAMZA
CI=confidence interval. *Intent-to-treat (ITT) population.† ITT population. ORR was defined as complete plus partial response. Disease progression and tumor response were assessed by investigators in accordance with Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.2
RAINBOW OS: MEDIAN - MONTHS (95% CI)*1 MAJOR OUTCOME MEASURE
Warnings and PrecautionsHemorrhage • CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage,
including severe and sometimes fatal hemorrhagic events. In study 1, which evaluated CYRAMZA as a single agent in advanced gastric cancer, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In study 2, which evaluated CYRAMZA plus paclitaxel, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroid anti-inflammatory drugs (NSAIDs) were excluded from enrollment in studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Arterial Thromboembolic Events (ATEs)• Serious, sometimes fatal, ATEs including myocardial infarction, cardiac arrest,
cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.
Hypertension• An increased incidence of severe hypertension occurred in patients receiving
CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every 2 weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.
Infusion-Related Reactions (IRRs)• Prior to the institution of premedication recommendations across clinical
trials of CYRAMZA, IRRs occurred in 6 out of 37 patients (16%), including 2 severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for grade 3 or 4 IRRs.
Gastrointestinal Perforations• CYRAMZA is an antiangiogenic therapy that can increase the risk of
gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In study 2, the incidence of gastrointestinal perforations was also increased in patients who received CYRAMZA plus paclitaxel (1.2%) as compared to patients who received placebo plus paclitaxel (0.3%). Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing• Impaired wound healing can occur with antibodies inhibiting the VEGF pathway.
CYRAMZA has not been studied in patients with serious or nonhealing wounds. CYRAMZA, an antiangiogenic therapy, has the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume CYRAMZA following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.
Clinical Deterioration in Child-Pugh B or C Cirrhosis• Clinical deterioration, manifested by new onset or worsening encephalopathy,
ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)• RPLS has been reported at a rate of <0.1% in clinical studies with CYRAMZA.
Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
Proteinuria Including Nephrotic Syndrome• Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio for
the development of worsening of proteinuria during CYRAMZA therapy. Withhold CYRAMZA for urine protein levels that are ≥2 g over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to <2 g over 24 hours. Permanently discontinue CYRAMZA for urine protein levels >3 g over 24 hours or in the setting of nephrotic syndrome.
Thyroid Dysfunction• Monitor thyroid function during treatment with CYRAMZA. Embryofetal Toxicity• Based on its mechanism of action, CYRAMZA can cause fetal harm when
administered to pregnant women. Animal models link angiogenesis, VEGF, and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for at least 3 months after the last dose of CYRAMZA.
Most Common Adverse Reactions—Single Agent• The most commonly reported adverse reactions (all grades; grade 3/4) occurring
in ≥5% of patients receiving CYRAMZA and ≥2% higher than placebo in study 1 were hypertension (16% vs 8%; 8% vs 3%), diarrhea (14% vs 9%; 1% vs 2%), headache (9% vs 3%; 0% vs 0%), and hyponatremia (6% vs 2%; 3% vs 1%).
• The most common serious adverse events with CYRAMZA in study 1 were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients vs 8.7% of patients who received placebo.
• Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in study 1 were: neutropenia (4.7% vs 0.9%), epistaxis (4.7% vs 0.9%), rash (4.2% vs 1.7%), intestinal obstruction (2.1% vs 0%), and arterial thromboembolic events (1.7% vs 0%).
• Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and infusion-related reactions. In study 1, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria vs 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in study 1 was 0.8% and the rate of infusion-related reactions was 0.4%.
Most Common Adverse Reactions—Combination with Paclitaxel• The most commonly reported adverse reactions (all grades; grade 3/4)
occurring in ≥5% of patients receiving CYRAMZA plus paclitaxel and ≥2% higher than placebo plus paclitaxel in study 2 were fatigue/asthenia (57% vs 44%; 12% vs 6%), neutropenia (54% vs 31%; 41% vs 19%), diarrhea (32% vs 23%; 4% vs 2%), epistaxis (31% vs 7%; 0% vs 0%), hypertension (25% vs 6%; 15% vs 3%), peripheral edema (25% vs 14%; 2% vs 1%), stomatitis (20% vs 7%; 1% vs 1%), proteinuria (17% vs 6%; 1% vs 0%), thrombocytopenia (13% vs 6%; 2% vs 2%), hypoalbuminemia (11% vs 5%; 1% vs 1%), and gastrointestinal hemorrhage events (10% vs 6%; 4% vs 2%).
• The most common serious adverse events with CYRAMZA plus paclitaxel in study 2 were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients treated with CYRAMZA plus paclitaxel received granulocyte colony-stimulating factors.
• Adverse reactions resulting in discontinuation of any component of the CYRAMZA plus paclitaxel combination in 2% or more patients in study 2 were neutropenia (4%) and thrombocytopenia (3%).
• Clinically relevant adverse reactions reported in ≥1% and <5% of the CYRAMZA plus paclitaxel-treated patients in study 2 were sepsis (3.1% for CYRAMZA plus paclitaxel vs 1.8% for placebo plus paclitaxel) and gastrointestinal perforations (1.2% for CYRAMZA plus paclitaxel vs 0.3% for placebo plus paclitaxel).
Drug Interactions• No pharmacokinetic interactions were observed between ramucirumab
(CYRAMZA) and paclitaxel.
Use in Specific Populations• Pregnancy: Based on its mechanism of action, CYRAMZA can cause fetal harm.
Animal models link angiogenesis, VEGF, and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no available data on CYRAMZA use in pregnant women to inform any drug-associated risks. No animal studies have been conducted to evaluate the effect of ramucirumab on reproduction and fetal development. Advise females of reproductive potential of the potential risk for maintaining pregnancy, risk to the fetus, and risk to newborn and infant development, and to use effective contraception during CYRAMZA therapy and for at least 3 months following the last dose of CYRAMZA.
• Lactation: Because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, advise women that breastfeeding is not recommended during treatment with CYRAMZA.
• Females of Reproductive Potential: Advise females of reproductive potential that based on animal data CYRAMZA may impair fertility.
Please see Brief Summary of Prescribing Information for CYRAMZA, including Boxed Warnings for hemorrhage, gastrointestinal perforation, and impaired wound healing, on next page.
RB HCP ISI 24APR2015
References: 1. CYRAMZA (ramucirumab) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2015. 2. Wilke H, Muro K, Van Cutsem E, et al; for the RAINBOW Study Group. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224-1235. 3. Data on file, Eli Lilly and Company. ONC09302014b.
RB97116 05/2015 PRINTED IN USA © Lilly USA, LLC 2015. All rights reserved.
CYRAMZA is a trademark owned by or licensed to Eli Lilly and Company, its subsidiaries, or affiliates.
WARNING: HEMORRHAGE, GASTROINTESTINAL PERFORATION, AND IMPAIRED WOUND HEALINGHemorrhage: CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding. Gastrointestinal Perforation: CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.Impaired Wound Healing: Impaired wound healing can occur with antibodies inhibiting the VEGF pathway. Discontinue CYRAMZA therapy in patients with impaired wound healing. Withhold CYRAMZA prior to surgery and discontinue CYRAMZA if a patient develops wound healing complications.
CLIENT: Lilly FINISH SIZE: 21.75” wide x 14” high
JOB#: 29889-22 “Tabloid” ARTIST: MC
LOCATION: Mechanicals COLLECT DATE:
R|O|U|T|I|N|G| |S|T|A|G|E M04ED CW AD CPM ACD AE inserv
B:14.25 in
B:22 in
T:14 in
T:21.75 in
S:13 in
S:19 in

OS
PR
OB
AB
ILIT
Y
30% INCREASEIN MEDIAN OS
1.0
0.8
0.6
0.4
0.2
0.0
TIME FROM RANDOMIZATION (MONTHS)
330
335
308
294
267
241
228
180
185
143
148
109
116
81
78
64
60
47
41
30
24
22
13
13
6
5
1
2
0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28
0
Number at Risk
CYRAMZA + paclitaxelPlacebo+ paclitaxel
Hazard Ratio=0.81(0.68, 0.96); P=0.017
CYRAMZA+ paclitaxelPlacebo+ paclitaxel
9.6MONTHS
CYRAMZA+ paclitaxel
(8.5, 10.8)
7.4MONTHS
Placebo+ paclitaxel(6.3, 8.4)
VISIT WWW.CYRAMZAHCP.COM FOR MORE INFORMATION, INCLUDING CYRAMZA MONOTHERAPY TRIAL DATA
CYRAMZA® (ramucirumab) PLUS PACLITAXEL SIGNIFICANTLY EXTENDED OVERALL SURVIVAL (OS)1
• Median PFS with CYRAMZA plus paclitaxel was 4.4 months (95% CI: 4.2, 5.3) vs 2.9 months (95% CI: 2.8, 3.0) with placebo plus paclitaxel (hazard ratio 0.64 [95% CI: 0.54, 0.75]; P<0.001)1
- The percentage of events at the time of analysis was 85% (279 patients) and 88% (296 patients), respectively
• Significantly more patients responded to CYRAMZA combined with paclitaxel (28%; 95% CI: 23, 33) than to placebo plus paclitaxel (16%; 95% CI: 13, 20) (P<0.001)†1,2
CYRAMZA PLUS PACLITAXEL ALSO SIGNIFICANTLY DELAYED DISEASE PROGRESSION AND PROVIDED SIGNIFICANTLY GREATER ORR VS PLACEBO PLUS PACLITAXEL (SUPPORTIVE EFFICACY OUTCOME MEASURES)*1
CYRAMZA as a single agent, or in combination with paclitaxel, is indicated for the treatment of patients with advanced or metastatic gastric or gastroesophageal (GE) junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy.
2 FDA APPROVALSFor use both as monotherapy and in combination with paclitaxel
The phase III RAINBOW trial evaluated the efficacy and safety of CYRAMZA plus paclitaxel vs placebo plus paclitaxel in patients with locally advanced or metastatic gastric or GE junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- and platinum-containing chemotherapy. Major efficacy outcome measure was OS. Supportive efficacy outcome measures were progression-free survival (PFS) and objective response rate (ORR). All patients were Eastern Cooperative Oncology Group performance status 0 or 1. Prior to enrollment, 97% of patients had progressed during treatment or within 4 months after the last dose of first-line chemotherapy for metastatic disease. Twenty-five percent of patients had received anthracycline in combination with platinum/fluoropyrimidine therapy, while 75% did not. Patients were randomized 1:1 to CYRAMZA 8 mg/kg (n=330) or placebo (n=335) every 2 weeks (on days 1 and 15) of each 28-day cycle. Patients in both arms received paclitaxel 80 mg/m2 on days 1, 8, and 15 of each 28-day cycle.1,3
• The percentage of deaths at the time of analysis was 78% (256 patients) and 78% (260 patients) in the CYRAMZA plus paclitaxel and placebo plus paclitaxel treatment arms, respectively1
IMPORTANT SAFETY INFORMATION FOR CYRAMZA
CI=confidence interval. *Intent-to-treat (ITT) population.† ITT population. ORR was defined as complete plus partial response. Disease progression and tumor response were assessed by investigators in accordance with Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.2
RAINBOW OS: MEDIAN - MONTHS (95% CI)*1 MAJOR OUTCOME MEASURE
Warnings and PrecautionsHemorrhage • CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage,
including severe and sometimes fatal hemorrhagic events. In study 1, which evaluated CYRAMZA as a single agent in advanced gastric cancer, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In study 2, which evaluated CYRAMZA plus paclitaxel, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroid anti-inflammatory drugs (NSAIDs) were excluded from enrollment in studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Arterial Thromboembolic Events (ATEs)• Serious, sometimes fatal, ATEs including myocardial infarction, cardiac arrest,
cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.
Hypertension• An increased incidence of severe hypertension occurred in patients receiving
CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every 2 weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.
Infusion-Related Reactions (IRRs)• Prior to the institution of premedication recommendations across clinical
trials of CYRAMZA, IRRs occurred in 6 out of 37 patients (16%), including 2 severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for grade 3 or 4 IRRs.
Gastrointestinal Perforations• CYRAMZA is an antiangiogenic therapy that can increase the risk of
gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In study 2, the incidence of gastrointestinal perforations was also increased in patients who received CYRAMZA plus paclitaxel (1.2%) as compared to patients who received placebo plus paclitaxel (0.3%). Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing• Impaired wound healing can occur with antibodies inhibiting the VEGF pathway.
CYRAMZA has not been studied in patients with serious or nonhealing wounds. CYRAMZA, an antiangiogenic therapy, has the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume CYRAMZA following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.
Clinical Deterioration in Child-Pugh B or C Cirrhosis• Clinical deterioration, manifested by new onset or worsening encephalopathy,
ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)• RPLS has been reported at a rate of <0.1% in clinical studies with CYRAMZA.
Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
Proteinuria Including Nephrotic Syndrome• Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio for
the development of worsening of proteinuria during CYRAMZA therapy. Withhold CYRAMZA for urine protein levels that are ≥2 g over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to <2 g over 24 hours. Permanently discontinue CYRAMZA for urine protein levels >3 g over 24 hours or in the setting of nephrotic syndrome.
Thyroid Dysfunction• Monitor thyroid function during treatment with CYRAMZA. Embryofetal Toxicity• Based on its mechanism of action, CYRAMZA can cause fetal harm when
administered to pregnant women. Animal models link angiogenesis, VEGF, and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for at least 3 months after the last dose of CYRAMZA.
Most Common Adverse Reactions—Single Agent• The most commonly reported adverse reactions (all grades; grade 3/4) occurring
in ≥5% of patients receiving CYRAMZA and ≥2% higher than placebo in study 1 were hypertension (16% vs 8%; 8% vs 3%), diarrhea (14% vs 9%; 1% vs 2%), headache (9% vs 3%; 0% vs 0%), and hyponatremia (6% vs 2%; 3% vs 1%).
• The most common serious adverse events with CYRAMZA in study 1 were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients vs 8.7% of patients who received placebo.
• Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in study 1 were: neutropenia (4.7% vs 0.9%), epistaxis (4.7% vs 0.9%), rash (4.2% vs 1.7%), intestinal obstruction (2.1% vs 0%), and arterial thromboembolic events (1.7% vs 0%).
• Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and infusion-related reactions. In study 1, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria vs 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in study 1 was 0.8% and the rate of infusion-related reactions was 0.4%.
Most Common Adverse Reactions—Combination with Paclitaxel• The most commonly reported adverse reactions (all grades; grade 3/4)
occurring in ≥5% of patients receiving CYRAMZA plus paclitaxel and ≥2% higher than placebo plus paclitaxel in study 2 were fatigue/asthenia (57% vs 44%; 12% vs 6%), neutropenia (54% vs 31%; 41% vs 19%), diarrhea (32% vs 23%; 4% vs 2%), epistaxis (31% vs 7%; 0% vs 0%), hypertension (25% vs 6%; 15% vs 3%), peripheral edema (25% vs 14%; 2% vs 1%), stomatitis (20% vs 7%; 1% vs 1%), proteinuria (17% vs 6%; 1% vs 0%), thrombocytopenia (13% vs 6%; 2% vs 2%), hypoalbuminemia (11% vs 5%; 1% vs 1%), and gastrointestinal hemorrhage events (10% vs 6%; 4% vs 2%).
• The most common serious adverse events with CYRAMZA plus paclitaxel in study 2 were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients treated with CYRAMZA plus paclitaxel received granulocyte colony-stimulating factors.
• Adverse reactions resulting in discontinuation of any component of the CYRAMZA plus paclitaxel combination in 2% or more patients in study 2 were neutropenia (4%) and thrombocytopenia (3%).
• Clinically relevant adverse reactions reported in ≥1% and <5% of the CYRAMZA plus paclitaxel-treated patients in study 2 were sepsis (3.1% for CYRAMZA plus paclitaxel vs 1.8% for placebo plus paclitaxel) and gastrointestinal perforations (1.2% for CYRAMZA plus paclitaxel vs 0.3% for placebo plus paclitaxel).
Drug Interactions• No pharmacokinetic interactions were observed between ramucirumab
(CYRAMZA) and paclitaxel.
Use in Specific Populations• Pregnancy: Based on its mechanism of action, CYRAMZA can cause fetal harm.
Animal models link angiogenesis, VEGF, and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no available data on CYRAMZA use in pregnant women to inform any drug-associated risks. No animal studies have been conducted to evaluate the effect of ramucirumab on reproduction and fetal development. Advise females of reproductive potential of the potential risk for maintaining pregnancy, risk to the fetus, and risk to newborn and infant development, and to use effective contraception during CYRAMZA therapy and for at least 3 months following the last dose of CYRAMZA.
• Lactation: Because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, advise women that breastfeeding is not recommended during treatment with CYRAMZA.
• Females of Reproductive Potential: Advise females of reproductive potential that based on animal data CYRAMZA may impair fertility.
Please see Brief Summary of Prescribing Information for CYRAMZA, including Boxed Warnings for hemorrhage, gastrointestinal perforation, and impaired wound healing, on next page.
RB HCP ISI 24APR2015
References: 1. CYRAMZA (ramucirumab) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2015. 2. Wilke H, Muro K, Van Cutsem E, et al; for the RAINBOW Study Group. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224-1235. 3. Data on file, Eli Lilly and Company. ONC09302014b.
RB97116 05/2015 PRINTED IN USA © Lilly USA, LLC 2015. All rights reserved.
CYRAMZA is a trademark owned by or licensed to Eli Lilly and Company, its subsidiaries, or affiliates.
WARNING: HEMORRHAGE, GASTROINTESTINAL PERFORATION, AND IMPAIRED WOUND HEALINGHemorrhage: CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding. Gastrointestinal Perforation: CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.Impaired Wound Healing: Impaired wound healing can occur with antibodies inhibiting the VEGF pathway. Discontinue CYRAMZA therapy in patients with impaired wound healing. Withhold CYRAMZA prior to surgery and discontinue CYRAMZA if a patient develops wound healing complications.
CLIENT: Lilly FINISH SIZE: 21.75” wide x 14” high
JOB#: 29889-22 “Tabloid” ARTIST: MC
LOCATION: Mechanicals COLLECT DATE:
R|O|U|T|I|N|G| |S|T|A|G|E M04ED CW AD CPM ACD AE inserv
B:14.25 in
B:22 in
T:14 in
T:21.75 in
S:13 in
S:19 in

CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015 CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015
CYRAMZA RB-G HCP BS 29APR2015 9.25 x 13.25 PRINTER VERSION 1 OF 2
CYRAMZA® (ramucirumab) injectionBRIEF SUMMARY: For complete safety, please consult the full Prescribing Information.
WARNING: HEMORRHAGE, GASTROINTESTINAL PERFORATION, AND IMPAIRED WOUND HEALINGHemorrhage: CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Gastrointestinal Perforation: CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing: Impaired wound healing can occur with antibodies inhibiting the VEGF pathway. Discontinue CYRAMZA therapy in patients with impaired wound healing. Withhold CYRAMZA prior to surgery and discontinue CYRAMZA if a patient develops wound healing complications.
INDICATIONS AND USAGEGastric CancerCYRAMZA as a single agent, or in combination with paclitaxel, is indicated for the treatment of patients with advanced or metastatic, gastric or gastro-esophageal junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy.
CONTRAINDICATIONSNone.
WARNINGS AND PRECAUTIONSHemorrhageCYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 1, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In Study 2, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroid anti-inflammatory drugs (NSAIDs) were excluded from enrollment in Studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. In Study 3, the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDS or other antiplatelet therapy other than once daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore the risk of pulmonary hemorrhage in these groups of patients is unknown. In Study 4, the incidence of severe bleeding was 2.5% for CYRAMZA plus FOLFIRI and 1.7% for placebo plus FOLFIRI. Permanently discontinue CYRAMZA in patients who experience severe bleeding.Arterial Thromboembolic EventsSerious, sometimes fatal, arterial thromboembolic events (ATEs) including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.HypertensionAn increased incidence of severe hypertension occurred in patients receiving CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%), in patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%), and in patients receiving CYRAMZA plus FOLFIRI (11%) as compared to placebo plus FOLFIRI (3%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.Infusion-Related ReactionsPrior to the institution of premedication recommendations across clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including two severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.Gastrointestinal PerforationsCYRAMZA is an antiangiogenic therapy that can increase the risk of gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In Study 2, the incidence of gastrointestinal perforations was also increased in patients that received CYRAMZA plus paclitaxel (1.2%) as compared to patients receiving placebo plus paclitaxel (0.3%). In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel and 0.3% for placebo plus docetaxel. In Study 4, the incidence of gastrointestinal perforation was 1.7% for CYRAMZA plus FOLFIRI and 0.6% for placebo plus FOLFIRI. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.Impaired Wound HealingImpaired wound healing can occur with antibodies inhibiting the VEGF pathway. CYRAMZA has not been studied in patients with serious or non-healing wounds. CYRAMZA, an antiangiogenic therapy, has the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.Clinical Deterioration in Patients with Child-Pugh B or C CirrhosisClinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.Reversible Posterior Leukoencephalopathy Syndrome Reversible Posterior Leukoencephalopathy Syndrome (RPLS) has been reported with a rate of <0.1% in clinical studies with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.Proteinuria Including Nephrotic Syndrome In Study 4, severe proteinuria occurred more frequently in patients treated with CYRAMZA plus FOLFIRI compared to patients receiving placebo plus FOLFIRI. Severe proteinuria was reported in 3% of patients treated with CYRAMZA plus FOLFIRI (including 3 cases [0.6%] of nephrotic syndrome) compared to 0.2% of patients treated with placebo plus FOLFIRI. Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio for the development of worsening of proteinuria during CYRAMZA therapy. Withhold CYRAMZA for urine protein levels that are 2 or more grams over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to less than 2 grams over 24 hours. Permanently discontinue CYRAMZA for urine protein levels greater than 3 grams over 24 hours or in the setting of nephrotic syndrome.Thyroid Dysfunction Monitor thyroid function during treatment with CYRAMZA. In Study 4, the incidence of hypothyroidism reported as an adverse event was 2.6% in the CYRAMZA plus FOLFIRI treated patients and 0.9% in the placebo plus FOLFIRI treated patients.
Embryofetal Toxicity Based on its mechanism of action, CYRAMZA can cause fetal harm when administered to pregnant women. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for at 3 least months after the last dose of CYRAMZA.
ADVERSE REACTIONSClinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data are presented from two randomized, placebo controlled clinical trials in which patients received CYRAMZA: Study 1, a randomized (2:1), double-blind, clinical trial in which 351 patients received either CYRAMZA 8 mg/kg intravenously every two weeks or placebo every two weeks and Study 2, a double-blind, randomized (1:1) clinical trial in which 656 patients received paclitaxel 80 mg/m2 on days 1, 8, and 15 of each 28-day cycle plus either CYRAMZA 8 mg/kg intravenously every two weeks or placebo every two weeks. Both trials excluded patients with Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 2 or greater, uncontrolled hypertension, major surgery within 28 days, or patients receiving chronic anti-platelet therapy other than once daily aspirin. Study 1 excluded patients with bilirubin ≥1.5 mg/dL and Study 2 excluded patients with bilirubin >1.5 times the upper limit of normal.CYRAMZA Administered as a Single Agent Among 236 patients who received CYRAMZA (safety population) in Study 1, median age was 60 years, 28% were women, 76% were White, and 16% were Asian. Patients in Study 1 received a median of 4 doses of CYRAMZA; the median duration of exposure was 8 weeks, and 32 (14% of 236) patients received CYRAMZA for at least six months.In Study 1, the most common adverse reactions (all grades) observed in CYRAMZA-treated patients at a rate of ≥10% and ≥2% higher than placebo were hypertension and diarrhea. The most common serious adverse events with CYRAMZA were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients versus 8.7% of patients who received placebo.
Table 1: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients Receiving CYRAMZA in Study 1
Adverse Reactions (MedDRA)a
System Organ Class
CYRAMZA (8 mg/kg)N=236
PlaceboN=115
All Grades(Frequency %)
Grade 3-4(Frequency %)
All Grades(Frequency %)
Grade 3-4(Frequency %)
Gastrointestinal Disorders Diarrhea 14 1 9 2
Metabolism and Nutrition Disorders Hyponatremia 6 3 2 1
Nervous System Disorders Headache 9 0 3 0
Vascular Disorders Hypertension 16 8 8 3
aMedDRA Version 15.0.
Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in Study 1 were: neutropenia (4.7% CYRAMZA versus 0.9% placebo), epistaxis (4.7% CYRAMZA versus 0.9% placebo), rash (4.2% CYRAMZA versus 1.7% placebo), intestinal obstruction (2.1% CYRAMZA versus 0% placebo), and arterial thromboembolic events (1.7% CYRAMZA versus 0% placebo).Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including Grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and infusion-related reactions. In Study 1, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria versus 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in Study 1 was 0.8% and the rate of infusion-related reactions was 0.4%.CYRAMZA Administered in Combination with Paclitaxel Among 327 patients who received CYRAMZA (safety population) in Study 2, median age was 60 years, 31% were women, 63% were White, and 33% were Asian. Patients in Study 2 received a median of 9 doses of CYRAMZA; the median duration of exposure was 18 weeks, and 93 (28% of 327) patients received CYRAMZA for at least six months.In Study 2, the most common adverse reactions (all grades) observed in patients treated with CYRAMZA plus paclitaxel at a rate of ≥30% and ≥2% higher than placebo plus paclitaxel were fatigue, neutropenia, diarrhea, and epistaxis. The most common serious adverse events with CYRAMZA plus paclitaxel were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients treated with CYRAMZA plus paclitaxel received granulocyte colony-stimulating factors. Adverse reactions resulting in discontinuation of any component of the CYRAMZA plus paclitaxel combination in 2% or more patients in Study 2 were neutropenia (4%) and thrombocytopenia (3%).
Table 2: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients
Receiving CYRAMZA plus Paclitaxel in Study 2
Adverse Reactions (MedDRA) System Organ Class
CYRAMZA plus Paclitaxel(N=327)
Placebo plus Paclitaxel(N=329)
All Grades(Frequency %)
Grade ≥3(Frequency %)
All Grades(Frequency %)
Grade ≥3(Frequency %)
Blood and Lymphatic System DisordersNeutropenia 54 41 31 19
Thrombocytopenia 13 2 6 2
Gastrointestinal DisordersDiarrhea 32 4 23 2
Gastrointestinal hemorrhage events
10 4 6 2
Stomatitis 20 1 7 1
General Disorders and Administration Site Disorders
Fatigue/Asthenia 57 12 44 6
Peripheral edema 25 2 14 1
Metabolism and Nutrition Disorders
Hypoalbuminemia 11 1 5 1
Renal and Urinary Disorders
Proteinuria 17 1 6 0
Respiratory, Thoracic, and Mediastinal Disorders
Epistaxis 31 0 7 0
Vascular Disorder
Hypertension 25 15 6 3
CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015 CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015
CYRAMZA RB-G HCP BS 29APR2015 9.25 x 13.25 PRINTER VERSION 2 OF 2
Clinically relevant adverse reactions reported in ≥1% and <5% of the CYRAMZA plus paclitaxel treated patients in Study 2 were sepsis (3.1% CYRAMZA plus paclitaxel versus 1.8% placebo plus paclitaxel) and gastrointestinal perforations (1.2% CYRAMZA plus paclitaxel versus 0.3% for placebo plus paclitaxel).ImmunogenicityAs with all therapeutic proteins, there is the potential for immunogenicity. In 23 clinical trials, 86/2890 (3.0%) of CYRAMZA-treated patients tested positive for treatment-emergent anti-ramucirumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 14 of the 86 patients who tested positive for treatment-emergent anti-ramucirumab antibodies.The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to CYRAMZA with the incidences of antibodies to other products may be misleading.
DRUG INTERACTIONSNo pharmacokinetic interactions were observed between ramucirumab and paclitaxel.
USE IN SPECIFIC POPULATIONSPregnancyRisk SummaryBased on its mechanism of action, CYRAMZA can cause fetal harm. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no available data on CYRAMZA in pregnant women to inform any drug-associated risks. No animal studies have been conducted to evaluate the effect of ramucirumab on reproduction and fetal development. The background risk of major birth defects and miscarriage for the indicated populations are unknown. In the U.S. general population the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. Advise pregnant women of the potential risk to a fetus.Animal DataNo animal studies have been specifically conducted to evaluate the effect of ramucirumab on reproduction and fetal development. In mice, loss of the VEGFR2 gene resulted in embryofetal death and these fetuses lacked organized blood vessels and blood islands in the yolk sac. In other models, VEGFR2 signaling was associated with development and maintenance of endometrial and placental vascular function, successful blastocyst implantation, maternal and feto-placental vascular differentiation, and development during early pregnancy in rodents and non-human primates. Disruption of VEGF signaling has also been associated with developmental anomalies including poor development of the cranial region, forelimbs, forebrain, heart, and blood vessels.LactationRisk SummaryThere is no information on the presence of ramucirumab in human milk, the effects on the breast-fed infant, or the effects on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, advise women that breastfeeding is not recommended during treatment with CYRAMZA.Females and Males of Reproductive PotentialContraceptionFemalesBased on its mechanism of action, CYRAMZA can cause fetal harm. Advise females of reproductive potential to use effective contraception while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA.InfertilityFemalesAdvise females of reproductive potential that based on animal data CYRAMZA may impair fertility.Pediatric UseThe safety and effectiveness of CYRAMZA in pediatric patients have not been established. In animal studies, effects on epiphyseal growth plates were identified. In cynomolgus monkeys, anatomical pathology revealed adverse effects on the epiphyseal growth plate (thickening and osteochondropathy) at all doses tested (5-50 mg/kg). Ramucirumab exposure at the lowest weekly dose tested in the cynomolgus monkey was 0.2 times the exposure in humans at the recommended dose of ramucirumab as a single agent.Geriatric UseOf the 563 CYRAMZA-treated patients in two randomized gastric cancer clinical studies, 36% were 65 and over, while 7% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Renal ImpairmentNo dose adjustment is recommended for patients with renal impairment based on population pharmacokinetic analysis.Hepatic ImpairmentNo dose adjustment is recommended for patients with mild (total bilirubin within upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN, or total bilirubin >1.0-1.5 times ULN and any AST) or moderate (total bilirubin >1.5-3.0 times ULN and any AST) hepatic impairment based on population pharmacokinetic analysis. Clinical deterioration was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA.
DOSAGE AND ADMINISTRATION
Do not administer CYRAMZA as an intravenous push or bolus.
Recommended Dose and ScheduleThe recommended dose of CYRAMZA either as a single agent or in combination with weekly paclitaxel is 8 mg/kg every 2 weeks administered as an intravenous infusion over 60 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity. When given in combination, administer CYRAMZA prior to administration of paclitaxel.PremedicationPrior to each CYRAMZA infusion, premedicate all patients with an intravenous histamine H1 antagonist (e.g., diphenhydramine hydrochloride). For patients who have experienced a Grade 1 or 2 infusion-related reaction, also premedicate with dexamethasone (or equivalent) and acetaminophen prior to each CYRAMZA infusion.Dose ModificationsInfusion-Related Reactions (IRR)• Reduce the infusion rate of CYRAMZA by 50% for Grade 1 or 2 IRRs.• Permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.Hypertension• Interrupt CYRAMZA for severe hypertension until controlled with medical management.• Permanently discontinue CYRAMZA for severe hypertension that cannot be controlled with
antihypertensive therapy.Proteinuria• Interrupt CYRAMZA for urine protein levels ≥2 g/24 hours. Reinitiate treatment at a reduced dose
of 6 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. If the protein level ≥2 g/24 hours reoccurs, interrupt CYRAMZA and reduce the dose to 5 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours.
• Permanently discontinue CYRAMZA for urine protein level >3 g/24 hours or in the setting of nephrotic syndrome.
Wound Healing Complications• Interrupt CYRAMZA prior to scheduled surgery until the wound is fully healed.Arterial Thromboembolic Events, Gastrointestinal Perforation, or Grade 3 or 4 Bleeding• Permanently discontinue CYRAMZA.
For toxicities related to paclitaxel, refer to the current prescribing information.
PATIENT COUNSELING INFORMATION• Hemorrhage: Advise patients that CYRAMZA can cause severe bleeding. Advise patients to contact their health care provider for bleeding or symptoms of bleeding including lightheadedness]. • Arterial thromboembolic events: Advise patients of an increased risk of an arterial thromboembolic event. • Hypertension: Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms. • Gastrointestinal perforations: Advise patients to notify their health care provider for severe diarrhea, vomiting, or severe abdominal pain. • Impaired wound healing: Advise patients that CYRAMZA has the potential to impair wound healing. Instruct patients not to undergo surgery without first discussing this potential risk with their health care provider. • Pregnancy and fetal harm: Advise females of reproductive potential of the potential risk for maintaining pregnancy, risk to the fetus, and risk to postnatal newborn and infant development and to use effective contraception during CYRAMZA therapy and for at least 3 months following the last dose of CYRAMZA. • Lactation: Advise patients not to breastfeed during CYRAMZA treatment. • Infertility: Advise females of reproductive potential regarding potential infertility effects of CYRAMZA
Additional information can be found at www.CYRAMZAHCP.com.
Eli Lilly and Company, Indianapolis, IN 46285, USACopyright © 2015, Eli Lilly and Company. All rights reserved.
RB-G HCP BS 29APR2015

CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015 CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015
CYRAMZA RB-G HCP BS 29APR2015 9.25 x 13.25 PRINTER VERSION 1 OF 2
CYRAMZA® (ramucirumab) injectionBRIEF SUMMARY: For complete safety, please consult the full Prescribing Information.
WARNING: HEMORRHAGE, GASTROINTESTINAL PERFORATION, AND IMPAIRED WOUND HEALINGHemorrhage: CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Gastrointestinal Perforation: CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing: Impaired wound healing can occur with antibodies inhibiting the VEGF pathway. Discontinue CYRAMZA therapy in patients with impaired wound healing. Withhold CYRAMZA prior to surgery and discontinue CYRAMZA if a patient develops wound healing complications.
INDICATIONS AND USAGEGastric CancerCYRAMZA as a single agent, or in combination with paclitaxel, is indicated for the treatment of patients with advanced or metastatic, gastric or gastro-esophageal junction adenocarcinoma with disease progression on or after prior fluoropyrimidine- or platinum-containing chemotherapy.
CONTRAINDICATIONSNone.
WARNINGS AND PRECAUTIONSHemorrhageCYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 1, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In Study 2, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroid anti-inflammatory drugs (NSAIDs) were excluded from enrollment in Studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. In Study 3, the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDS or other antiplatelet therapy other than once daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore the risk of pulmonary hemorrhage in these groups of patients is unknown. In Study 4, the incidence of severe bleeding was 2.5% for CYRAMZA plus FOLFIRI and 1.7% for placebo plus FOLFIRI. Permanently discontinue CYRAMZA in patients who experience severe bleeding.Arterial Thromboembolic EventsSerious, sometimes fatal, arterial thromboembolic events (ATEs) including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.HypertensionAn increased incidence of severe hypertension occurred in patients receiving CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%), in patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%), and in patients receiving CYRAMZA plus FOLFIRI (11%) as compared to placebo plus FOLFIRI (3%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.Infusion-Related ReactionsPrior to the institution of premedication recommendations across clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including two severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.Gastrointestinal PerforationsCYRAMZA is an antiangiogenic therapy that can increase the risk of gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In Study 2, the incidence of gastrointestinal perforations was also increased in patients that received CYRAMZA plus paclitaxel (1.2%) as compared to patients receiving placebo plus paclitaxel (0.3%). In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel and 0.3% for placebo plus docetaxel. In Study 4, the incidence of gastrointestinal perforation was 1.7% for CYRAMZA plus FOLFIRI and 0.6% for placebo plus FOLFIRI. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.Impaired Wound HealingImpaired wound healing can occur with antibodies inhibiting the VEGF pathway. CYRAMZA has not been studied in patients with serious or non-healing wounds. CYRAMZA, an antiangiogenic therapy, has the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.Clinical Deterioration in Patients with Child-Pugh B or C CirrhosisClinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.Reversible Posterior Leukoencephalopathy Syndrome Reversible Posterior Leukoencephalopathy Syndrome (RPLS) has been reported with a rate of <0.1% in clinical studies with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.Proteinuria Including Nephrotic Syndrome In Study 4, severe proteinuria occurred more frequently in patients treated with CYRAMZA plus FOLFIRI compared to patients receiving placebo plus FOLFIRI. Severe proteinuria was reported in 3% of patients treated with CYRAMZA plus FOLFIRI (including 3 cases [0.6%] of nephrotic syndrome) compared to 0.2% of patients treated with placebo plus FOLFIRI. Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio for the development of worsening of proteinuria during CYRAMZA therapy. Withhold CYRAMZA for urine protein levels that are 2 or more grams over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to less than 2 grams over 24 hours. Permanently discontinue CYRAMZA for urine protein levels greater than 3 grams over 24 hours or in the setting of nephrotic syndrome.Thyroid Dysfunction Monitor thyroid function during treatment with CYRAMZA. In Study 4, the incidence of hypothyroidism reported as an adverse event was 2.6% in the CYRAMZA plus FOLFIRI treated patients and 0.9% in the placebo plus FOLFIRI treated patients.
Embryofetal Toxicity Based on its mechanism of action, CYRAMZA can cause fetal harm when administered to pregnant women. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for at 3 least months after the last dose of CYRAMZA.
ADVERSE REACTIONSClinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data are presented from two randomized, placebo controlled clinical trials in which patients received CYRAMZA: Study 1, a randomized (2:1), double-blind, clinical trial in which 351 patients received either CYRAMZA 8 mg/kg intravenously every two weeks or placebo every two weeks and Study 2, a double-blind, randomized (1:1) clinical trial in which 656 patients received paclitaxel 80 mg/m2 on days 1, 8, and 15 of each 28-day cycle plus either CYRAMZA 8 mg/kg intravenously every two weeks or placebo every two weeks. Both trials excluded patients with Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 2 or greater, uncontrolled hypertension, major surgery within 28 days, or patients receiving chronic anti-platelet therapy other than once daily aspirin. Study 1 excluded patients with bilirubin ≥1.5 mg/dL and Study 2 excluded patients with bilirubin >1.5 times the upper limit of normal.CYRAMZA Administered as a Single Agent Among 236 patients who received CYRAMZA (safety population) in Study 1, median age was 60 years, 28% were women, 76% were White, and 16% were Asian. Patients in Study 1 received a median of 4 doses of CYRAMZA; the median duration of exposure was 8 weeks, and 32 (14% of 236) patients received CYRAMZA for at least six months.In Study 1, the most common adverse reactions (all grades) observed in CYRAMZA-treated patients at a rate of ≥10% and ≥2% higher than placebo were hypertension and diarrhea. The most common serious adverse events with CYRAMZA were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients versus 8.7% of patients who received placebo.
Table 1: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients Receiving CYRAMZA in Study 1
Adverse Reactions (MedDRA)a
System Organ Class
CYRAMZA (8 mg/kg)N=236
PlaceboN=115
All Grades(Frequency %)
Grade 3-4(Frequency %)
All Grades(Frequency %)
Grade 3-4(Frequency %)
Gastrointestinal Disorders Diarrhea 14 1 9 2
Metabolism and Nutrition Disorders Hyponatremia 6 3 2 1
Nervous System Disorders Headache 9 0 3 0
Vascular Disorders Hypertension 16 8 8 3
aMedDRA Version 15.0.
Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in Study 1 were: neutropenia (4.7% CYRAMZA versus 0.9% placebo), epistaxis (4.7% CYRAMZA versus 0.9% placebo), rash (4.2% CYRAMZA versus 1.7% placebo), intestinal obstruction (2.1% CYRAMZA versus 0% placebo), and arterial thromboembolic events (1.7% CYRAMZA versus 0% placebo).Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including Grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and infusion-related reactions. In Study 1, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria versus 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in Study 1 was 0.8% and the rate of infusion-related reactions was 0.4%.CYRAMZA Administered in Combination with Paclitaxel Among 327 patients who received CYRAMZA (safety population) in Study 2, median age was 60 years, 31% were women, 63% were White, and 33% were Asian. Patients in Study 2 received a median of 9 doses of CYRAMZA; the median duration of exposure was 18 weeks, and 93 (28% of 327) patients received CYRAMZA for at least six months.In Study 2, the most common adverse reactions (all grades) observed in patients treated with CYRAMZA plus paclitaxel at a rate of ≥30% and ≥2% higher than placebo plus paclitaxel were fatigue, neutropenia, diarrhea, and epistaxis. The most common serious adverse events with CYRAMZA plus paclitaxel were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients treated with CYRAMZA plus paclitaxel received granulocyte colony-stimulating factors. Adverse reactions resulting in discontinuation of any component of the CYRAMZA plus paclitaxel combination in 2% or more patients in Study 2 were neutropenia (4%) and thrombocytopenia (3%).
Table 2: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients
Receiving CYRAMZA plus Paclitaxel in Study 2
Adverse Reactions (MedDRA) System Organ Class
CYRAMZA plus Paclitaxel(N=327)
Placebo plus Paclitaxel(N=329)
All Grades(Frequency %)
Grade ≥3(Frequency %)
All Grades(Frequency %)
Grade ≥3(Frequency %)
Blood and Lymphatic System DisordersNeutropenia 54 41 31 19
Thrombocytopenia 13 2 6 2
Gastrointestinal DisordersDiarrhea 32 4 23 2
Gastrointestinal hemorrhage events
10 4 6 2
Stomatitis 20 1 7 1
General Disorders and Administration Site Disorders
Fatigue/Asthenia 57 12 44 6
Peripheral edema 25 2 14 1
Metabolism and Nutrition Disorders
Hypoalbuminemia 11 1 5 1
Renal and Urinary Disorders
Proteinuria 17 1 6 0
Respiratory, Thoracic, and Mediastinal Disorders
Epistaxis 31 0 7 0
Vascular Disorder
Hypertension 25 15 6 3
CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015 CYRAMZA® (ramucirumab) injection RB-G HCP BS 29APR2015
CYRAMZA RB-G HCP BS 29APR2015 9.25 x 13.25 PRINTER VERSION 2 OF 2
Clinically relevant adverse reactions reported in ≥1% and <5% of the CYRAMZA plus paclitaxel treated patients in Study 2 were sepsis (3.1% CYRAMZA plus paclitaxel versus 1.8% placebo plus paclitaxel) and gastrointestinal perforations (1.2% CYRAMZA plus paclitaxel versus 0.3% for placebo plus paclitaxel).ImmunogenicityAs with all therapeutic proteins, there is the potential for immunogenicity. In 23 clinical trials, 86/2890 (3.0%) of CYRAMZA-treated patients tested positive for treatment-emergent anti-ramucirumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 14 of the 86 patients who tested positive for treatment-emergent anti-ramucirumab antibodies.The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to CYRAMZA with the incidences of antibodies to other products may be misleading.
DRUG INTERACTIONSNo pharmacokinetic interactions were observed between ramucirumab and paclitaxel.
USE IN SPECIFIC POPULATIONSPregnancyRisk SummaryBased on its mechanism of action, CYRAMZA can cause fetal harm. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no available data on CYRAMZA in pregnant women to inform any drug-associated risks. No animal studies have been conducted to evaluate the effect of ramucirumab on reproduction and fetal development. The background risk of major birth defects and miscarriage for the indicated populations are unknown. In the U.S. general population the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. Advise pregnant women of the potential risk to a fetus.Animal DataNo animal studies have been specifically conducted to evaluate the effect of ramucirumab on reproduction and fetal development. In mice, loss of the VEGFR2 gene resulted in embryofetal death and these fetuses lacked organized blood vessels and blood islands in the yolk sac. In other models, VEGFR2 signaling was associated with development and maintenance of endometrial and placental vascular function, successful blastocyst implantation, maternal and feto-placental vascular differentiation, and development during early pregnancy in rodents and non-human primates. Disruption of VEGF signaling has also been associated with developmental anomalies including poor development of the cranial region, forelimbs, forebrain, heart, and blood vessels.LactationRisk SummaryThere is no information on the presence of ramucirumab in human milk, the effects on the breast-fed infant, or the effects on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, advise women that breastfeeding is not recommended during treatment with CYRAMZA.Females and Males of Reproductive PotentialContraceptionFemalesBased on its mechanism of action, CYRAMZA can cause fetal harm. Advise females of reproductive potential to use effective contraception while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA.InfertilityFemalesAdvise females of reproductive potential that based on animal data CYRAMZA may impair fertility.Pediatric UseThe safety and effectiveness of CYRAMZA in pediatric patients have not been established. In animal studies, effects on epiphyseal growth plates were identified. In cynomolgus monkeys, anatomical pathology revealed adverse effects on the epiphyseal growth plate (thickening and osteochondropathy) at all doses tested (5-50 mg/kg). Ramucirumab exposure at the lowest weekly dose tested in the cynomolgus monkey was 0.2 times the exposure in humans at the recommended dose of ramucirumab as a single agent.Geriatric UseOf the 563 CYRAMZA-treated patients in two randomized gastric cancer clinical studies, 36% were 65 and over, while 7% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Renal ImpairmentNo dose adjustment is recommended for patients with renal impairment based on population pharmacokinetic analysis.Hepatic ImpairmentNo dose adjustment is recommended for patients with mild (total bilirubin within upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN, or total bilirubin >1.0-1.5 times ULN and any AST) or moderate (total bilirubin >1.5-3.0 times ULN and any AST) hepatic impairment based on population pharmacokinetic analysis. Clinical deterioration was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA.
DOSAGE AND ADMINISTRATION
Do not administer CYRAMZA as an intravenous push or bolus.
Recommended Dose and ScheduleThe recommended dose of CYRAMZA either as a single agent or in combination with weekly paclitaxel is 8 mg/kg every 2 weeks administered as an intravenous infusion over 60 minutes. Continue CYRAMZA until disease progression or unacceptable toxicity. When given in combination, administer CYRAMZA prior to administration of paclitaxel.PremedicationPrior to each CYRAMZA infusion, premedicate all patients with an intravenous histamine H1 antagonist (e.g., diphenhydramine hydrochloride). For patients who have experienced a Grade 1 or 2 infusion-related reaction, also premedicate with dexamethasone (or equivalent) and acetaminophen prior to each CYRAMZA infusion.Dose ModificationsInfusion-Related Reactions (IRR)• Reduce the infusion rate of CYRAMZA by 50% for Grade 1 or 2 IRRs.• Permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.Hypertension• Interrupt CYRAMZA for severe hypertension until controlled with medical management.• Permanently discontinue CYRAMZA for severe hypertension that cannot be controlled with
antihypertensive therapy.Proteinuria• Interrupt CYRAMZA for urine protein levels ≥2 g/24 hours. Reinitiate treatment at a reduced dose
of 6 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. If the protein level ≥2 g/24 hours reoccurs, interrupt CYRAMZA and reduce the dose to 5 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours.
• Permanently discontinue CYRAMZA for urine protein level >3 g/24 hours or in the setting of nephrotic syndrome.
Wound Healing Complications• Interrupt CYRAMZA prior to scheduled surgery until the wound is fully healed.Arterial Thromboembolic Events, Gastrointestinal Perforation, or Grade 3 or 4 Bleeding• Permanently discontinue CYRAMZA.
For toxicities related to paclitaxel, refer to the current prescribing information.
PATIENT COUNSELING INFORMATION• Hemorrhage: Advise patients that CYRAMZA can cause severe bleeding. Advise patients to contact their health care provider for bleeding or symptoms of bleeding including lightheadedness]. • Arterial thromboembolic events: Advise patients of an increased risk of an arterial thromboembolic event. • Hypertension: Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms. • Gastrointestinal perforations: Advise patients to notify their health care provider for severe diarrhea, vomiting, or severe abdominal pain. • Impaired wound healing: Advise patients that CYRAMZA has the potential to impair wound healing. Instruct patients not to undergo surgery without first discussing this potential risk with their health care provider. • Pregnancy and fetal harm: Advise females of reproductive potential of the potential risk for maintaining pregnancy, risk to the fetus, and risk to postnatal newborn and infant development and to use effective contraception during CYRAMZA therapy and for at least 3 months following the last dose of CYRAMZA. • Lactation: Advise patients not to breastfeed during CYRAMZA treatment. • Infertility: Advise females of reproductive potential regarding potential infertility effects of CYRAMZA
Additional information can be found at www.CYRAMZAHCP.com.
Eli Lilly and Company, Indianapolis, IN 46285, USACopyright © 2015, Eli Lilly and Company. All rights reserved.
RB-G HCP BS 29APR2015

PAGE 72 The ASCO Post | SEPTEMBER 10, 2015
Education in Oncology
The Flipped Classroom: Swapping the Traditional Lecture Hall for an Online VersionA Conversation With Charles Prober, MDBy Jo Cavallo
D espite enormous advances in modern medicine and the explo-
sion of biomedical information over the past century, the way medical education is taught in the United States is stuck in a format that does not optimize learn-ing, according to Charles Prober, MD, Senior Associate Dean for Medical Ed-ucation at Stanford School of Medicine in California.
The combination of the digitally em-powered learner, the rapid expansion of biomedical knowledge, and the increas-ing specialization within the practice of medicine is driving the need to rei-magine medical education, so lessons are more comprehensible and memo-rable, what Dr. Prober calls “stickier.” Dr. Prober believes that content should be more engaging, embrace a learning strategy that can be self-paced, and de-signed to make better use of the fixed amount of educational time available to train new doctors.
Rather than professors delivering a series of facts to students gathered in a lecture hall, in this “flipped classroom” model of education, students acquire basic knowledge on a specific subject through a series of short—10 minutes or less—videos presented online. The video sessions can be downloaded to a student’s computer or mobile device and viewed as often as necessary to master the content in preparation for in-depth interactive classroom discus-sions with the professors. In this flip-ping of the classroom model, lessons previously taught in class are learned at home, and “homework” is performed in the classroom, with small groups of students interacting with faculty.
The flipped classroom approach to teaching is part of the Stanford Medicine Interactive Learning Initiative (smili.stanford.edu), which was launched 2
years ago by the School of Medicine. One recently developed course using this model in microbiology/infectious diseases was created by Stanford in collaboration with four other medical schools, including the University of Cali-fornia, San Francisco; the University of Washington, Seattle; Duke University, Durham, North Carolina; and the Uni-versity of Michigan, Ann Arbor.
According to Dr. Prober, several medical school courses at Stanford have seen improved student attendance and course ratings since they migrated from poorly attended lectures as the primary mode of delivery to the flipped class-room approach.
Dr. Prober first described the use of the flipped classroom in medical educa-tion in 2012 in an article called “Lecture Halls Without Lectures—A Proposal for Medical Education” published in The New England Journal of Medicine.1
The model was further detailed a year later in an article he coauthored with Salman Khan, founder of the Khan Academy, Mountain View, California, called “Medical Education Reimag-ined: A Call to Action,” published in Academic Medicine.2
The ASCO Post talked with Dr. Prob-er about this new concept in higher learning and its impact on the educa-tion of medical students.
Growing Popularity Among Students
Stanford first used the flipped class-room concept in its biochemistry classes. How has it changed the way students’ learn, and what has been the reaction from students?
The core biochemistry class here was redesigned 4 years ago to follow the flipped classroom model. Historically, the course had not been well received,
as evidenced by very few students par-ticipating in organized lectures—only about 20% of students attended the lectures, and 80% found them not to be particularly compelling. Our faculty produced a series of short videos and, in parallel, designed richly interactive classroom sessions that became more patient-centered and problem-centered than the previous curriculum.
The students found the material much more relevant and compelling because it showed them why they needed to understand the informa-tion to take care of their future pa-tients. The course immediately be-came more popular, with up to 90% of the students attending the optional interactive sessions.
So the students are more engaged, the material is being delivered in a more efficient fashion, the course rat-ings have gone up substantially, and we believe that the students are retaining the material.
I am not suggesting that there is no room in medical education for the more conventional lecture-hall settings, but to be successful, those kinds of lectures would have to be special learning op-portunities for students.
The Sweet Spot for Delivering Videos
How did you determine that 10 min-utes is the optimal amount of time for stu-dents to grasp the material in the videos?
There is a fair amount of emerging data showing that 10 minutes of mate-rial is about right for a typical learner’s attention span. Some people even sug-gest that reducing the information to less than 10 minutes is beneficial, but there is a limit to how short you want to make these videos. So 6 to 10 min-utes is what we think is the sweet spot for delivering videos that have imbed-ded within them no more than two or three learning objectives as opposed to 20 learning objectives you might have in a conventional lecture.
Expanding ContentAre you producing these videos in a
variety of specialties, including medical oncology?
One of our faculty members who runs our introductory courses in molecular biology is an oncologist,
and he has created videos for some of the more difficult to understand concepts. For example, he devised one on the Kaplan-Meier survivor-ship curves. So far, we have content in biochemistry, epidemiology, bio-statistics, health policy, microbiolo-gy, immunology, infectious diseases, endocrinology, and cardiology. Our faculty is becoming more and more convinced that this is a strategy that may work for their students.
We are also producing videos for stu-dents in our clinical rotations. For exam-ple, one of our core clinical rotations is in surgery, and instead of presenting the in-formation in a lecture format, members of the surgical team have created short videos of the information. After watch-ing the videos, the students, who are in different hospitals doing their surgical rotations, then come together once a week in a central place to have an interac-tive classroom session about the material in the video.
Cultivating a Deeper Understanding
Why is the flipped classroom method effective in helping medical students retain knowledge?
GUEST EDITOR
Education in Oncology focuses on faculty development, med-
ical education curricula, fellow-ship training, and communication skills. The column is guest edited by Leora Horn, MD, MSc, Asso-ciate Professor of Medicine, Assis-tant Director of the Educator De-velopment Program, and Clinical Director of the Thoracic Oncology Program at Vanderbilt University School of Medicine, Nashville.
Leora Horn, MD, MSc
Professional Development
The combination of video material and the in-class activities provides students with a deeper understanding of the material and makes the course more relevant.
—Charles Prober, MD
continued on page 73

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 73
Announcements
There are several reasons. One is that the material is created to conform to a template, so there is consistency in the format regardless of the content. In a lecture situation, content can be de-livered via a variety of formats, which may leave the learner confused. For ex-ample, if you have 50 minutes to deliver a lecture, some professors use up to 150 slides. This may drive a delivery pace that is difficult for many students to fol-low. So I think format consistency in the video presentations automatically facilitates learning.
Also, the short video format allows
for efficient packaging of the informa-tion that the teacher wants to convey, and students can review the material as many times as they need to master the content. The videos allow students to learn at their own pace, on their own time. In a classroom setting, students have one shot at understanding the ma-terial being presented.
But what really allows the video material to be relevant and “to stick” is that the students can then put the principles they’ve learned online into practice in the classroom inter-acting with their peers and faculty. With this method, the combination of video material and the in-class
activities provides students with a deeper understanding of the material and makes the course more relevant; therefore, the information is more likely to stick with students for a lon-ger time.
One of the core elements of these videos are patient stories; students can learn some of the key concepts we are trying to teach about basic science and the pathogenesis of a specific disease. The patient stories are powerful and help students understand the impor-tance of learning this material.
The real test of the flipped class-room method, and it’s too soon to tell at the moment, is whether presenting
medical education in this new way pro-vides students with a superior ability to deal with clinical experiences than the traditional method of learning. So we are interested in tracking long-term outcomes. n
Disclosure: Dr. Prober reported no potential conflicts of interest.
References1. Prober CG, Heath C: Lecture halls
without lectures—A proposal for medical education. N Engl J Med 366:1657-1659, 2012.
2. Prober CG, Khan S: Medical educa-tion reimagined: A call to action. Acad Med 88:1407-1410, 2013.
W. Kimryn Rathmell, MD, PhD, Will Direct Division of Hematology and Oncology at Vanderbilt
W. Kimryn Rathmell, MD, PhD, Alexander Professor for Transla-
tional Science and Associate Director for Training and Education at the University of North Carolina’s Lineberger Compre-hensive Cancer Center, has been named Director of Vanderbilt’s Division of He-matology and Oncology. She began her new role as of September 1.
Dr. Rathmell is a physician-scientist whose research focuses on the genetic and molecular signals that drive renal cell carcinomas and who specializes in the treatment of patients with rare and complex kidney cancers, as well as pros-tate, bladder, and testicular cancers.
In her current research, Dr. Rath-mell and colleagues have identified fac-tors that are critical to transitions in the progression of kidney cancer. She has also led or participated in a number of the Cancer Genome Atlas projects.
At Vanderbilt, Dr. Rathmell will continue to care for patients with geni-tourinary malignancies, in addition to continuing research in her laboratory at Vanderbilt-Ingram Cancer Center (VICC).
“We are excited about the recruit-ment of Kim Rathmell, who has already demonstrated an impressive commit-ment to research excellence and train-ing. Her leadership of the Division of Hematology and Oncology will con-tribute significantly to VICC on many levels,” said Jennifer Pietenpol, PhD, B.F. Byrd Jr Professor of Oncology and Director of VICC.
Dr. Rathmell has been inducted into the Alpha Omega Alpha Honor Medical Society and the American So-ciety of Clinical Investigators, where she currently serves on the council as Secretary/Treasurer. She has also authored more than 80 articles, has served on the editorial boards for sev-eral scientific journals, and is an Asso-ciate Editor for the Journal of Clinical Investigation.
Dr. Rathmell earned her medical de-gree, as well as her doctorate (in biophys-ics), at Stanford University. Following an internship at the University of Chicago, she completed her residency and fellow-ship at the University of Pennsylvania. n
The Flipped Classroomcontinued from page 72
Jeffrey Rathmell, PhD, to Lead New Center for Immunobiology at Vanderbilt
Jeffrey Rathmell, PhD, has been recruited to Vanderbilt University
Medical Center (VUMC) to lead a new Center for Immunobiology, a structure supported by the Department of Pathol-ogy, Microbiology, and Immunology, the Department of Medicine, and Vander-bilt-Ingram Cancer Center (VICC). He took on his new role September 1.
Dr. Rathmell comes to Vanderbilt from Duke University Medical Center, where he was Associate Professor of Pharmacology and Cancer Biology, as well as of Immu-nology in the Duke Molecular Physiology Institute. He was additionally Director of Graduate Studies of Pharmacology. He has been named a Professor of Pathology, Microbiology, and Immunology and will serve as co-leader of the Host Tumor Inter-actions Research Program at VICC.
His work at Vanderbilt will focus on the field of immunometabolism and
how nutrient and metabolic pathways can influence immune responses.
“Vanderbilt has great immunologists across the medical school, and the main goals of the new Center for Immunobi-ology will be to foster basic immunol-ogy science, which touches on many different fields and diseases, and to help build the immunology community,” said Dr. Rathmell.
“We are delighted to welcome Jeff to VICC and look forward to his con-tributions and leadership as we grow cancer immunology from basic and translational research to clinical investi-gation,” said Jennifer Pietenpol, PhD, B.F. Byrd Jr Professor of Oncology and Director of VICC.
Dr. Rathmell has authored more than 90 research articles and serves as a member of the editorial board for sev-eral scientific journals.
He earned his doctorate in immu-nology at Stanford University and com-pleted a graduate fellowship with the National Science Foundation during his PhD training. He undertook postdoctor-al training at the Abramson Family Can-cer Research Institute at the University of Pennsylvania, where he earned fellow-ships from the Howard Hughes Medical Institute and the Irvington Institute for Immunological Research. n
W. Kimryn Rathmell, MD, PhD Jeffrey Rathmell, PhD
Visit The ASCO Post website at ASCOPost.com

PAGE 74 The ASCO Post | SEPTEMBER 10, 2015
Announcements
Iuliana Shapira, MD, Named Chief of Hematology and Oncology at SUNY Downstate
Iuliana Shapira, MD, has joined SUNY Downstate Medical Center
as Chief of the Division of Hematol-ogy and Oncology. In this position, Dr. Shapira will provide leadership in the clinical, academic, and administra-tive aspects of the program, as well as broaden the relationship with SUNY Downstate’s affiliated institutions.
Dr. Shapira was previously Associate Professor of Medicine at Hofstra North Shore-LIJ School of Medicine, where she served as Director of its Center for Cancer Genetics and Cancer Control and site Co-Investigator for numerous studies under a Cancer Community Oncology Program Grant from the Na-tional Institutes of Health.
She has served as Associate Editor of the Journal of Molecular Medicine since 2008.
Dr. Shapira is the 2016 President of the American Federation for Medi-cal Research (AFMR) Eastern Region, Councilor for AFMR’s National Coun-cil, and a member of the Federation of American Societies for Experimental Biology National Institutes of Health Research Enterprise Evaluation Sub-committee. She is also a member of the Audit Committee of the Alliance in Clinical Trials for Oncology, serving as National Cancer Institute Clinical Tri-als Monitoring Branch Auditor.
She served as Chairwoman for the Clinical Competency Committee and Site Program Director for the Hema-tology-Oncology Fellowship Program of North Shore University Hospital.
Dr. Shapira was the Jacobi Hospital Site Investigator for the Albert Einstein Mi-nority National Cancer Institute Com-munity Oncology Research Program Grant until March 2015.
Dr. Shapira graduated from the Car-ol Davila University of Medicine and Pharmacy in Bucharest, Romania. She trained in internal medicine at Saint Barnabas Medical Center in Livingston,
New Jersey, and completed a hema-tology-oncology fellowship at SUNY Downstate. She is board certified in in-ternal medicine, hematology, and medi-cal oncology. n
Iuliana Shapira, MD
Visit The ASCO Post
website at ASCOPost.com
REFERENCE: 1. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103-119.
Research has found that abnormal MAPK signaling may lead to increased or uncontrolled cell proliferation and resistance to apoptosis. Studies have shown that the MAPK pathway plays an important role in some cancers.1
Based on these findings, Genentech is investigating further ways to target the MAPK pathway.
Learn more at TargetMAPK.com.
IN ONCOLOGY, HAVE WE
MAXIMIZED THE POTENTIAL OF TARGETING THE MAPK PATHWAY?
© 2015 Genentech USA, Inc. All rights reserved. COB/092414/0002(1) Printed in USA.
79235ha_b.indd All Pages 5/19/15 4:31 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 75
Announcements
Andrew S. Chi, MD, PhD, Named Head of Neuro-Oncology at NYU’s Laura and Isaac Perlmutter Cancer Center
N ew York University (NYU) Lan-gone Medical Center announced
the appointment of physician-scientist and brain tumor specialist Andrew S. Chi, MD, PhD, as Chief of Neuro-On-
cology for its Laura and Isaac Perlmut-ter Cancer Center and Co-Director of the NYU Langone Brain Tumor Center.
In his new role, Dr. Chi will lead all neuro-oncology-related pro-
grams. In addition, he will co-lead the NYU Langone Brain Tumor Center in partnership with John G. Golfinos, MD, Chair of the Depart-ment of Neurosurgery.
Clinical and Investigative Expertise
Dr. Chi comes to NYU from Mas-sachusetts General Hospital, Dana-Faber/Harvard Cancer Center, and Harvard Medical School, where he held the title of Assistant Professor of Neurology. His clinical work at Massa-chuetts General Hospital included the neuro-oncologic treatment of primary brain tumors, metastatic tumors to the central nervous system, and neurologic complications of cancer.
In addition to his clinical expertise, Dr. Chi’s investigative work principally focuses on the identification of molecular genetic alterations that underlie the devel-opment, progression, and treatment resis-tance of brain tumors. By understanding the mechanism by which tumor genome alterations drive the growth of cancers, therapeutic strategies can be designed to improve outcomes for patients.
After completing his postdoctoral research at Massachusetts General Hos-pital, Dr. Chi began his research studies of the underlying genetic mechanisms of brain tumors in the laboratory on a team with Daniel P. Cahill, MD, PhD, and A. John Iafrate, MD, PhD, where they advanced the understanding of the molecular genetics of gliomas and un-covered novel molecular targets for the treatments for brain tumors.
Dr. Chi earned his medical degree and doctorate in biochemistry and molecular biology from Chicago Medical School. He completed his residency in neurol-ogy at Harvard Medical School, based at Massachusetts General Hospital and Brigham and Women’s Hospital, serving his final year as Chief Resident.
“I am thrilled to join NYU Langone and to work with the respected faculty at the Perlmutter Cancer Center,” said Dr. Chi. “We share a similar vision of inno-vation for the Cancer Center, and I am confident that as a result of these efforts, we will see major scientific and clinical advances against these horrible diseases in the coming years.” n
Andrew S. Chi, MD, PhD
REFERENCE: 1. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103-119.
Research has found that abnormal MAPK signaling may lead to increased or uncontrolled cell proliferation and resistance to apoptosis. Studies have shown that the MAPK pathway plays an important role in some cancers.1
Based on these findings, Genentech is investigating further ways to target the MAPK pathway.
Learn more at TargetMAPK.com.
IN ONCOLOGY, HAVE WE
MAXIMIZED THE POTENTIAL OF TARGETING THE MAPK PATHWAY?
© 2015 Genentech USA, Inc. All rights reserved. COB/092414/0002(1) Printed in USA.
79235ha_b.indd All Pages 5/19/15 4:31 PM

PAGE 76 The ASCO Post | SEPTEMBER 10, 2015
Women in Oncology
How Carolyn D. Runowicz, MD, FASCO, Is Shaping the Future of Gynecologic CancersBy Beth Howard
Carolyn D. Runowicz, MD, FASCO, has worn just about
every hat in the field of oncology—clinician, professor, researcher, ad-ministrator, and even cancer survivor. Currently the Executive Associate Dean for Academic Affairs and Profes-sor in the Department of Obstetrics and Gynecology at the Herbert Wert-heim College of Medicine at Florida International University in Miami, she has held numerous academic and professional leadership positions, con-ducted research in gynecologic oncol-ogy, and helped to blaze trails for other female oncologists.
A native of Willimantic, Con-necticut, Dr. Runowicz attended the University of Connecticut and then headed to Thomas Jefferson Medical College in Philadelphia for her medi-cal degree. The stage for her career in oncology was set in her first year
of medical school, when through an American Cancer Society fellowship, she “shadowed” gynecologic oncolo-gist George C. Lewis, MD.
“His passion for what he did was contagious,” said Dr. Runowicz. “He would get in early in the morning and stay until late at night, yet he never seemed tired. Every day was met with enthusiasm and vigor. I thought, wow, if I could find that same passion in my life, I would really be happy.” She also became enamored with the specialty of gynecologic oncology. “It was com-prehensive care of women with can-cer, from diagnosis to the end of life,” she said.
During her residency at Mount Si-nai Hospital in New York, from 1977 to 1981, Dr. Runowicz joined the Department of Obstetrics and Gyne-cology, guided by Saul B. Gusberg, MD [then Distinguished Service Pro-fessor and Chairman in the Depart-ment of Obstetrics and Gynecology at Mount Sinai School of Medicine], one of the founding fathers of gy-necologic oncology, and completed her fellowship in the specialty. (Also
important in her training was fellow-ship director Carmel Cohen, MD, Professor of Obstetrics, Gynecology, and Reproductive Medicine at Mount Sinai.)
A few years later, Dr. Runowicz was recruited to join Albert Einstein Col-lege of Medicine and Montefiore Med-ical Center to help develop a Division of Gynecologic Oncology and start a fellowship program. She had also be-gun conducting clinical trials in gyne-cologic cancers.
Fortuitous Timing It was an exciting time to be in the
field. “I was privileged to be at insti-
tutions where some of the new drugs were being developed—drugs that changed how cancer was treated,” said Dr. Runowicz. “I was at Mount Sinai when platinum-based chemothera-py came out, then at Einstein when Susan B. Horwitz, PhD, [Distin-guished Professor in the Department of Molecular Pharmacology] began to describe how paclitaxel worked. I had all these patients who had been on platinum-based chemotherapy, and
it stopped working. But we put them on paclitaxel and witnessed another miracle.”
Dr. Runowicz was also involved in researching cisplatin for the treat-ment of ovarian cancer. “It’s been amazing to see how these drugs abso-lutely changed how we treat cancer,” she said.
By now, Dr. Runowicz had found her own passion, but a diagnosis of breast cancer in 1992 threw her an unexpect-ed curve ball. Because the cancer had spread to her lymph nodes, treatment included chemotherapy, radiation, and tamoxifen.
The grueling regimen took 11 months to complete, but Dr. Runowicz never stopped working, even as she struggled with nausea and crushing fa-tigue. “I didn’t realize the cumulative ef-fect [of the treatment]. The first session of chemo was not that bad, the second was a little worse. With each time, it required an additional day to get over. And antinausea medications were al-most nonexistent then,” she said. “I like to think I was always empathetic with patients, but going through cancer gave me another look into what their lives were like.”
Hitting Her StrideIn 2002, Dr. Runowicz became
the Director of the Carole and Ray Neag Comprehensive Cancer Cen-ter at the University of Connecticut
Gynecologic Cancer
Carolyn D. Runowicz, MD, FASCO
I like to think I was always empathetic with patients, but going through cancer gave me another look
into what their lives were like. —Carolyn D. Runowicz, MD, FASCO
Ovarian Cancer Awareness Month

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 77
Women in Oncology
Health Center to rebuild its cancer program. She had also begun taking leadership roles in several profes-sional societies and accumulating several “firsts.” For instance, she was the first woman to head the tradi-tionally all-male Society of Gyne-cologic Oncologists. “I had to tread carefully,” she said. “But it was a great experience and privilege.”
In 2000, Dr. Runowicz proposed a name change for the group—to the Society of Gynecologic Oncology. “I had a vision of a much larger organi-zation that would no longer be about doctors but about the science,” she said. It took 10 years, but the group was finally renamed in 2011. “It was a name that indicated this was a serious professional organization focused on the science and not the doctors,” said Dr. Runowicz.
Dr. Runowicz was also the first gynecologic oncologist to serve on ASCO’s Board of Directors. Her other notable appointments include a term as President of the American Cancer Society from 2005 to 2006. In 2004, she was asked by President George W. Bush to serve as a member of the National Cancer Advisory Board for a 6-year appointment, which includ-ed two terms as its Chair.
At the same time, Dr. Runowicz managed to speak regularly on gyne-cologic cancers, menopause manage-ment, and breast cancer; to advocate for women’s health; and to write several books, including To Be Alive: A Woman’s Guide to a Full Life After Cancer (Henry Holt & Co, 1995), and coauthored with her husband Sheldon Cherry, MD, Associate Dean of Clinical Affairs and Professor of Obstetrics and Gynecology at Flor-ida International University’s Herbert Wertheim College of Medicine, The Menopause Book: A Guide to Women’s Health After 40 (Macmillan Publish-ers, 1994).
Enjoying New ChallengesAt Florida International Univer-
sity, Dr. Runowicz is enjoying the chal-lenges and rewards of helping guide the school’s fledgling medical program. The new medical school just graduated its third class. “We’re in the building phase,” said Dr. Runowicz. “There’s a lot of enthusiasm here.”
In addition to Dr. Runowicz’ aca-demic responsibilities at the univer-sity, she and her colleagues are inves-tigating a promising novel therapy for ovarian cancer: intraperitoneal delivery of paclitaxel via magneto-
electric nanoparticles, which may en-able researchers to overcome some of the barriers to the widespread use of intraperitoneal chemotherapy in ovarian cancer.
“The tumor opens up a pore, and the particle goes in and kills the cell,” she said. “We’ve cured ovarian can-cer in a mouse model. That’s a long
way from success in humans, but it’s a start.”
Dr. Runowicz also maintains a clinical practice in Miami and feels privileged to have had the opportu-nity to become an oncologist and to continue to have such a varied ca-reer, from taking care of patients and conducting research to being an aca-
demician. It is clear from her extraor-dinary list of past accomplishments in oncology care and her work today, Dr. Runowicz has found the same passion in her career she so admired in the career of her early mentor, Dr. Lewis. n
Disclosure: Dr. Runowicz reported no potential conflicts of interest.
The molecular alterations that lead to cancer are unique to each patient. At Foundation Medicine, our approach tests for all clinically relevant alterations driving a patient’s cancer.
As a result, a FoundationOne® comprehensive genomic profile can identify 3 times more targeted therapy options than traditional hot spot testing.1 We are more than just a test provider.
We help you access these therapies so you can deliver the best possible care for your patients.
Open up more possibilities of precision treatment with FoundationOne.
It’s what’s inside that counts.
The molecular alterations that lead to cancer are unique to each patient. At Foundation Medicine, our approach tests for all clinically relevant alterations driving a patient’s cancer.
As a result, a FoundationOne® comprehensive genomic profile can identify 3 times more targeted therapy options than traditional hot spot testing.1 We are more than just a test provider.
We help you access these therapies so you can deliver the best possible care for your patients.
Open up more possibilities of precision treatment with FoundationOne.
1. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023-1031.
©2015 Foundation Medicine, Inc. Foundation Medicine® and FoundationOne® are registered trademarks.
1. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023-1031.
©2015 Foundation Medicine, Inc. Foundation Medicine® and FoundationOne® are registered trademarks.
033858_fmdone_ad_5sizes_fa3.indd 1 5/18/15 8:34 AM

PAGE 78 The ASCO Post | SEPTEMBER 10, 2015
Through the Lens of Oncology History
The text and photographs on this page are excerpted from a four-volume series of books titled Oncology Tumors & Treatment: A Photographic History, by Stanley B. Burns, MD, FACS. The photos below are from the volume titled “The Anesthesia Era: 1845–1875.” To view additional photos from this series of books, visit burnsarchive.com.
A Century of Progress
“Café-au-lait colored spots with associated tumors of the nerves, skin, and bones that progress sometimes into
the giantism of an extremity or skin deformity.”
THE Anesthesia Era: 1845–1875
Hypertrophic Tumor of the Forehead, Philadelphia, Circa 1870
In 1869, the first periodical pro-duced with photographs appeared
in Europe. Just 1 year later, The Pho-tographic Review of Medicine and Sur-gery, with original photographs, was published in America. The publica-tion was edited by Philadelphia sur-geon Frank F. Maury, MD (1840–1879) and dermatologist Louis A. Duhring, MD (1843–1913). The journal was devoted to publishing monthly photographs and case re-ports of rare, unusual, or interesting patients.
Fascinating PhotographsOver a 2-year period, 1870 to
1872, 48 albumen prints were pub-lished. The images presented were the most fascinating pictures of the extremes of disease; they appeared for decades thereafter as illustrations in medical texts.
Ophthalmologist Richard J. Levis, MD (1827–1890) is remem-bered for introducing the wire loop used in cataract extraction. He pre-sented this showcased photograph with its case history entitled, “Hy-pertrophic Tumor of the Integument of Forehead, Eyelid & Eyebrow,” in 1870.
“The patient, a 30-year-old man, was noted at birth to have a small protuberance in the left temple. By
age 14, vision in his left eye was to-tally obscured. At age 30, the left eye has atrophied, and an entire quadrant of his face has been consumed by the persistent tumor. No treatment was advised.”
Progressive DeformitiesNeurofibromatosis, or von Reck-
linghausen’s disease, is a condition characterized by dermal, neural, and osseous deformities often progress-ing to severe and gross distortion. History’s most famous victim and ex-treme example of the disease is John Merrick, the “Elephant Man.” Typi-cally, the disease presents as a small elevation around the eye or temple which, left untreated, may steadily grow to enormous proportions, as seen in the photo at right.
In 1882, a student of Rudolf Virchow, MD (founder of cellular pathology), Friedrich Daniel von Recklinghausen, MD (1833–1910), gave the classic description of the dis-ease as a phakomatosis: “Café-au-lait colored spots with associated tumors of the nerves, skin, and bones that progress sometimes into the giantism of an extremity or skin deformity.”
The disease is genetically trans-mitted and there is no known cure for neurofibromatosis. Future thera-py lies in medical genetics. The map-
ping of the human genome and the study of inherited DNA sequences have made it possible to establish the genetic linkage and chromosome map location for neurofibromatosis. In 1987, Barker, et al detailed the
exact sequence (16220). n
Excerpted from Oncology Tumors & Treat-ment: A Photographic History, by Stanley B. Burns, MD. Photographs courtesy of Stan-ley B. Burns, MD, and The Burns Archive.

FLFL
ZYDELIG is a PI3Kδ inhibitor indicated for Relapsed FL after ≥2 systemic therapies Accelerated approval was granted for FL based on overall response rate. An
improvement in patient survival or disease related symptoms has not been established. Continued approval for this indication may be contingent upon verifi cation of clinical benefi t in confi rmatory trials.
FL=follicular B-cell non-Hodgkin lymphoma; PI3Kδ=phosphatidylinositol 3-kinase delta.
A fi rst-in-class selective inhibitor of PI3Kδ, a protein that is expressed in normal and malignant B cells
BECAUSE YOU CAN’T DO THIS TO FL…
THERE’S ZYDELIG®
IMPORTANT SAFETY INFORMATION BOXED WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, AND INTESTINAL PERFORATION
• Fatal and/or serious hepatotoxicity occurred in 14% of ZYDELIG-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue ZYDELIG as recommended
• Fatal and/or serious and severe diarrhea or colitis occurred in 14% of ZYDELIG-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue ZYDELIG as recommended
• Fatal and serious pneumonitis can occur. Monitor for pulmonary symptoms and bilateral interstitial infi ltrates. Interrupt or discontinue ZYDELIG as recommended
• Fatal and serious intestinal perforation can occur in ZYDELIG-treated patients. Discontinue ZYDELIG for intestinal perforation
Please see the following pages for additional Important Safety Information and Brief Summary of full Prescribing Information, including BOXED WARNING.
IMAGINE WHAT’S POSSIBLE
79649ha_c.indd 1 8/6/15 5:37 PM

POWERPOWERPOWERof responseof responseof response
DURATIONDURATIONDURATIONof responseof responseof responseIMPORTANT SAFETY INFORMATION (cont'd)
Contraindications• History of serious allergic reactions, including
anaphylaxis and toxic epidermal necrolysis (TEN)Warnings and Precautions
• Hepatotoxicity: Findings were generally observed within the fi rst 12 weeks of treatment and reversed with dose interruption. Upon rechallenge at a lower dose, ALT/AST elevations recurred in 26% of patients. In all patients, monitor ALT/AST every 2 weeks for the fi rst 3 months, every 4 weeks for the next 3 months, and every 1 to 3 months thereafter. If ALT/AST is >3× upper limit of normal (ULN), monitor for liver toxicity weekly. If ALT/AST is >5× ULN, withhold ZYDELIG and monitor ALT/AST and total bilirubin weekly until resolved. Discontinue ZYDELIG for recurrent hepatotoxicity. Avoid concurrent use with other hepatotoxic drugs
• Severe diarrhea or colitis: Grade 3+ diarrhea can occur at any time and responds poorly to antimotility agents. Avoid concurrent use with other drugs that cause diarrhea
• Pneumonitis: Evaluate for pneumonitis in patients presenting with pulmonary symptoms such as cough, dyspnea, hypoxia, interstitial infi ltrates on radiologic exam, or oxygen saturation decline by ≥5%
• Intestinal perforation: Advise patients to promptly report any new or worsening abdominal pain, chills, fever, nausea, or vomiting
• Severe cutaneous reactions: One case of TEN occurred in a study of ZYDELIG in combination with rituximab and bendamustine. Other severe or life-threatening (grade ≥3) cutaneous reactions have been reported. Monitor patients for the development
of severe cutaneous reactions and discontinue ZYDELIG if a reaction occurs
• Anaphylaxis: Serious allergic reactions including anaphylaxis have been reported. Discontinue ZYDELIG permanently and institute appropriate supportive measures if a reaction occurs
• Neutropenia: Treatment-emergent grade 3-4 neutropenia occurred in 31% of ZYDELIG-treated patients in clinical trials. In all patients, monitor blood counts ≥every 2 weeks for the fi rst 3 months. In patients with neutrophil counts <1.0 Gi/L, monitor weekly
• Embryo-fetal toxicity: ZYDELIG may cause fetal harm. Women who are or become pregnant while taking ZYDELIG should be apprised of the potential hazard to the fetus. Advise women to avoid pregnancy while taking ZYDELIG and to use e� ective contraception during and at least 1 month after treatment with ZYDELIGAdverse Reactions
• Most common adverse reactions (incidence ≥20%; all grades) were diarrhea, fatigue, nausea, cough, pyrexia, abdominal pain, pneumonia, and rash
• Most frequent serious adverse reactions (SAR) were pneumonia (15%), diarrhea (11%), and pyrexia (9%); SAR were reported in 50% of patients and 53% of patients discontinued or interrupted therapy due to adverse reactions
• Most common lab abnormalities (incidence ≥30%; all grades) were neutrophils decreased and ALT/AST elevations
Drug Interactions• CYP3A inducers: Avoid coadministration with
strong CYP3A inducers• CYP3A inhibitors: When coadministered with
strong CYP3A inhibitors, monitor closely for ZYDELIG toxicity
• CYP3A substrates: Avoid coadministration with CYP3A substratesDosage and Administration
• Adult starting dose: One 150 mg tablet twice daily, swallowed whole with or without food. Continue treatment until disease progression or unacceptable toxicity. The safe dosing regimen for patients who require treatment longer than several months is unknown
• Dose modifi cation: Consult the ZYDELIG full Prescribing Information for dose modifi cation and monitoring recommendations for the following specifi c toxicities: pneumonitis, ALT/AST elevations,
bilirubin elevations, diarrhea, neutropenia, and thrombocytopenia. For other severe or life-threatening toxicities, withhold ZYDELIG until toxicity is resolved and reduce the dose to 100 mg, twice daily, upon resuming treatment. If severe or life-threatening toxicities recur upon rechallenge, ZYDELIG should be permanently discontinued
Please see the following pages for Brief Summary of full Prescribing Information, including BOXED WARNING.
VISIT ZYDELIG.COM
• Most common adverse reactions (incidence ≥20%; all grades) were diarrhea, fatigue, nausea, cough, pyrexia, abdominal pain, pneumonia, and rash; 53% of patients discontinued or interrupted therapy due to adverse reactions
CI=confi dence interval; CR=complete response; DoR=duration of response; ORR=overall response rate; PR=partial response.
† Results of a single-arm, open-label trial of ZYDELIG (150 mg, twice daily) in patients with FL who failed to respond or relapsed after ≥2 prior therapies (which must have included both rituximab and an alkylating agent). Primary end point was ORR, as assessed by an independent review committee. ORR was defi ned as the proportion of subjects who achieved CR or PR. Secondary end point was DoR. DoR was measured from the onset of fi rst documented response (CR or PR) to disease progression or death.2
Demonstrated single-agent e� cacy in an open-label, pivotal, phase 2 trial2†
Powerful e� cacy, chemotherapy free
FDA approved in relapsed FL after ≥2 systemic therapies
Imagine what’s possible:ZYDELIG®—A fi rst-in-class PI3Kδ inhibitor
ZYDELIG is the FIRST AND ONLY KINASE INHIBITOR APPROVED IN FL.
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) RECOMMEND IDELALISIB MONOTHERAPY
AS AN OPTION for appropriate patients with relapsed/refractory FL.1*
ALT=alanine aminotransferase; AST=aspartate aminotransferase; NCCN®=National Comprehensive Cancer Network®.
* Please see the complete version of the NCCN Guidelines® for Non-Hodgkin’s Lymphomas available on NCCN.org for specifi c recommendations.
ZYDELIG in FL (n=72)
IMAGINE WHAT’S POSSIBLE
79649ha_c.indd 2-3 8/6/15 5:38 PM
POWERPOWERPOWERof responseof responseof response
DURATIONDURATIONDURATIONof responseof responseof responseIMPORTANT SAFETY INFORMATION (cont'd)
Contraindications• History of serious allergic reactions, including
anaphylaxis and toxic epidermal necrolysis (TEN)Warnings and Precautions
• Hepatotoxicity: Findings were generally observed within the fi rst 12 weeks of treatment and reversed with dose interruption. Upon rechallenge at a lower dose, ALT/AST elevations recurred in 26% of patients. In all patients, monitor ALT/AST every 2 weeks for the fi rst 3 months, every 4 weeks for the next 3 months, and every 1 to 3 months thereafter. If ALT/AST is >3× upper limit of normal (ULN), monitor for liver toxicity weekly. If ALT/AST is >5× ULN, withhold ZYDELIG and monitor ALT/AST and total bilirubin weekly until resolved. Discontinue ZYDELIG for recurrent hepatotoxicity. Avoid concurrent use with other hepatotoxic drugs
• Severe diarrhea or colitis: Grade 3+ diarrhea can occur at any time and responds poorly to antimotility agents. Avoid concurrent use with other drugs that cause diarrhea
• Pneumonitis: Evaluate for pneumonitis in patients presenting with pulmonary symptoms such as cough, dyspnea, hypoxia, interstitial infi ltrates on radiologic exam, or oxygen saturation decline by ≥5%
• Intestinal perforation: Advise patients to promptly report any new or worsening abdominal pain, chills, fever, nausea, or vomiting
• Severe cutaneous reactions: One case of TEN occurred in a study of ZYDELIG in combination with rituximab and bendamustine. Other severe or life-threatening (grade ≥3) cutaneous reactions have been reported. Monitor patients for the development
of severe cutaneous reactions and discontinue ZYDELIG if a reaction occurs
• Anaphylaxis: Serious allergic reactions including anaphylaxis have been reported. Discontinue ZYDELIG permanently and institute appropriate supportive measures if a reaction occurs
• Neutropenia: Treatment-emergent grade 3-4 neutropenia occurred in 31% of ZYDELIG-treated patients in clinical trials. In all patients, monitor blood counts ≥every 2 weeks for the fi rst 3 months. In patients with neutrophil counts <1.0 Gi/L, monitor weekly
• Embryo-fetal toxicity: ZYDELIG may cause fetal harm. Women who are or become pregnant while taking ZYDELIG should be apprised of the potential hazard to the fetus. Advise women to avoid pregnancy while taking ZYDELIG and to use e� ective contraception during and at least 1 month after treatment with ZYDELIGAdverse Reactions
• Most common adverse reactions (incidence ≥20%; all grades) were diarrhea, fatigue, nausea, cough, pyrexia, abdominal pain, pneumonia, and rash
• Most frequent serious adverse reactions (SAR) were pneumonia (15%), diarrhea (11%), and pyrexia (9%); SAR were reported in 50% of patients and 53% of patients discontinued or interrupted therapy due to adverse reactions
• Most common lab abnormalities (incidence ≥30%; all grades) were neutrophils decreased and ALT/AST elevations
Drug Interactions• CYP3A inducers: Avoid coadministration with
strong CYP3A inducers• CYP3A inhibitors: When coadministered with
strong CYP3A inhibitors, monitor closely for ZYDELIG toxicity
• CYP3A substrates: Avoid coadministration with CYP3A substratesDosage and Administration
• Adult starting dose: One 150 mg tablet twice daily, swallowed whole with or without food. Continue treatment until disease progression or unacceptable toxicity. The safe dosing regimen for patients who require treatment longer than several months is unknown
• Dose modifi cation: Consult the ZYDELIG full Prescribing Information for dose modifi cation and monitoring recommendations for the following specifi c toxicities: pneumonitis, ALT/AST elevations,
bilirubin elevations, diarrhea, neutropenia, and thrombocytopenia. For other severe or life-threatening toxicities, withhold ZYDELIG until toxicity is resolved and reduce the dose to 100 mg, twice daily, upon resuming treatment. If severe or life-threatening toxicities recur upon rechallenge, ZYDELIG should be permanently discontinued
Please see the following pages for Brief Summary of full Prescribing Information, including BOXED WARNING.
VISIT ZYDELIG.COM
• Most common adverse reactions (incidence ≥20%; all grades) were diarrhea, fatigue, nausea, cough, pyrexia, abdominal pain, pneumonia, and rash; 53% of patients discontinued or interrupted therapy due to adverse reactions
CI=confi dence interval; CR=complete response; DoR=duration of response; ORR=overall response rate; PR=partial response.
† Results of a single-arm, open-label trial of ZYDELIG (150 mg, twice daily) in patients with FL who failed to respond or relapsed after ≥2 prior therapies (which must have included both rituximab and an alkylating agent). Primary end point was ORR, as assessed by an independent review committee. ORR was defi ned as the proportion of subjects who achieved CR or PR. Secondary end point was DoR. DoR was measured from the onset of fi rst documented response (CR or PR) to disease progression or death.2
Demonstrated single-agent e� cacy in an open-label, pivotal, phase 2 trial2†
Powerful e� cacy, chemotherapy free
FDA approved in relapsed FL after ≥2 systemic therapies
Imagine what’s possible:ZYDELIG®—A fi rst-in-class PI3Kδ inhibitor
ZYDELIG is the FIRST AND ONLY KINASE INHIBITOR APPROVED IN FL.
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) RECOMMEND IDELALISIB MONOTHERAPY
AS AN OPTION for appropriate patients with relapsed/refractory FL.1*
ALT=alanine aminotransferase; AST=aspartate aminotransferase; NCCN®=National Comprehensive Cancer Network®.
* Please see the complete version of the NCCN Guidelines® for Non-Hodgkin’s Lymphomas available on NCCN.org for specifi c recommendations.
ZYDELIG in FL (n=72)
IMAGINE WHAT’S POSSIBLE
79649ha_c.indd 2-3 8/6/15 5:38 PM

© 2015 Gilead Sciences, Inc. All rights reserved. ZYDP0192 08/2015ZYDELIG, ZYDELIG logo, GILEAD and the GILEAD logo are trademarks of Gilead Sciences, Inc., or its related companies. All other marks are the property of their respective owners.
References: 1. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Hodgkin’s Lymphomas V.1.2015. © National Comprehensive Cancer Network, Inc 2015. All rights reserved. Accessed January 7, 2015. To view the most recent and complete version of the guideline, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc. 2. Gopal AK, Kahl BS, de Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008-1018.
IDEPU-53446_M1_BrfSmm_Kng_Ind_NoCode.indd12-16-2014 9:49 AM Suke Yawata / Dan Capobianco
Client CodeClient
LiveOverall TrimBleed
# of Colors
GILP0220 07/2014GILEAD
9.5” x 13”10” x 13.75”None
1C - Black
Colors Black
FontsHelvetica Neue LT Std (77 Bold Condensed, 57 Condensed, 77 Bold Condensed Oblique, 57 Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hssyawata5561 by
Printed At
None
Notes King Size Brief Summary-Indication
ZYDELIG® (idelalisib) tablets, for oral use
Brief Summary of full Prescribing Information. See full Prescribing Information. Rx Only.
INDICATIONS AND USAGE:
• ZYDELIG is indicated in combination with rituximab for the treatment of adults with relapsed chronic lymphocytic leukemia (CLL) for whom rituximab alone would be considered appropriate therapy due to other comorbidities.
• ZYDELIG is indicated for the treatment of adults with relapsed follicular B-cell non-Hodgkin lymphoma (FL) who have received ≥2 prior systemic therapies.
• ZYDELIG is indicated for the treatment of adults with relapsed small lymphocytic lymphoma (SLL) who have received ≥2 prior systemic therapies.
• Accelerated approval was granted for FL and SLL based on overall response rate. An improvement in patient survival or disease related symptoms has not been established. Continued approval for these indications may be contingent upon verification of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATION:
See Warnings and Precautions, Adverse Reactions, and Use in Specific Populations for additional information.
Adult Starting Dose: One 150 mg tablet taken orally twice daily (BID), swallowed whole with or without food. Continue treatment until disease progression or unacceptable toxicity. The optimal and safe dosing regimen for patients who required treatment longer than several months is unknown.
Dose Modifications:
• Pneumonitis: discontinue ZYDELIG for any symptomatic pneumonitis
• Hepatotoxicity: – ALT/AST >3 to 5x ULN or bilirubin >1.5 to 3x ULN: maintain ZYDELIG
dose; monitor weekly until ≤1x ULN – ALT/AST >5 to 20x ULN or bilirubin >3 to 10x ULN: withhold ZYDELIG;
monitor weekly until ≤1x ULN then resume ZYDELIG 100 mg BID – ALT/AST >20x ULN or bilirubin >10x ULN: permanently discontinue ZYDELIG
• Diarrhea: – Moderate (increase of 4-6 stools/day over baseline): maintain ZYDELIG
dose; monitor weekly until resolved – Severe (increase of ≥7 stools/day over baseline) or hospitalization:
withhold ZYDELIG; monitor weekly until resolved then resume ZYDELIG 100 mg BID
– Life-threatening: permanently discontinue ZYDELIG
• Neutropenia: – ANC 1 to <1.5 Gi/L: maintain ZYDELIG dose – ANC 0.5 to <1 Gi/L: maintain ZYDELIG dose; monitor weekly – ANC <0.5 Gi/L: withhold ZYDELIG; monitor weekly until ≥0.5 Gi/L then
resume ZYDELIG 100 mg BID
• Thrombocytopenia: – Platelets 50 to <75 Gi/L: maintain ZYDELIG dose – Platelets 25 to <50 Gi/L: maintain ZYDELIG dose; monitor weekly – Platelets <25 Gi/L: withhold ZYDELIG; monitor weekly until ≥25 Gi/L
then resume ZYDELIG 100 mg BID
• For other severe or life-threatening toxicities, withhold ZYDELIG until toxicity is resolved and reduce dose to 100 mg BID if resuming treatment. Permanently discontinue ZYDELIG if severe or life-threatening toxicities recur upon rechallenge.
CONTRAINDICATIONS:
History of serious allergic reactions including anaphylaxis and toxic epidermal necrolysis (TEN).
WARNINGS AND PRECAUTIONS:
Fatal and/or serious hepatotoxicity occurred in 14% of ZYDELIG-treated patients. ALT or AST >5x ULN have occurred, usually within the first 12 weeks of treatment and were reversible with dose interruption. Upon resuming
treatment at a lower dose, 26% of patients had recurrence of ALT and AST elevations. Discontinue ZYDELIG for recurrent hepatotoxicity. Avoid concurrent use of ZYDELIG with hepatotoxic drugs. In all patients, monitor ALT and AST every 2 weeks for the first 3 months, every 4 weeks for the next 3 months, then every 1 to 3 months thereafter. If ALT or AST >3x ULN, monitor weekly until elevation resolves; if ALT or AST >5x ULN, withhold ZYDELIG and monitor AST, ALT and total bilirubin weekly until elevation resolves [See Dosage and Administration].
Severe diarrhea or colitis (≥Grade 3) occurred in 14% of ZYDELIG-treated patients across clinical trials. ZYDELIG-induced diarrhea can occur at any time and responds poorly to antimotility agents. Median time to resolution ranged between 1 week and 1 month following ZYDELIG interruption with or without enteric or systemic corticosteroids. Avoid concurrent use of ZYDELIG with drugs that cause diarrhea. [See Dosage and Administration].
Fatal and serious pneumonitis occurred in ZYDELIG-treated patients. Patients taking ZYDELIG who present with pulmonary symptoms (e.g., cough, dyspnea, hypoxia, interstitial infiltrates, >5% decrease in oxygen saturation) should be evaluated for pneumonitis. If pneumonitis is suspected, withhold ZYDELIG until etiology of pulmonary symptoms has been determined. Patients thought to have ZYDELIG-induced pneumonitis were treated with ZYDELIG discontinuation and corticosteroids.
Fatal and serious intestinal perforation occurred in ZYDELIG-treated patients. At the time of perforation, some patients had moderate to severe diarrhea. Advise patients to promptly report any new or worsening abdominal pain, chills, fever, nausea, or vomiting. Permanently discontinue ZYDELIG in patients who experience intestinal perforation.
Severe Cutaneous Reactions: One case of TEN occurred in a study of ZYDELIG in combination with rituximab and bendamustine. Other severe or life-threatening (Grade ≥3) cutaneous reactions (dermatitis exfoliative, rash, rash erythematous, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic, exfoliative rash, skin disorder) have been reported. Monitor patients for severe cutaneous reactions and discontinue ZYDELIG.
Anaphylaxis: Serious allergic reactions including anaphylaxis have been reported in ZYDELIG-treated patients. Permanently discontinue ZYDELIG and institute appropriate supportive measures in patients who develop serious allergic reactions.
Neutropenia: Treatment-emergent neutropenia (Grade 3 or 4) occurred in 31% of ZYDELIG-treated patients across clinical trials. Monitor blood counts every 2 weeks for the first 3 months, and weekly when neutrophils are <1 Gi/L [See Dosage and Administration].
Embryo-fetal Toxicity: Idelalisib is teratogenic in rats and may cause fetal harm. Women who are or become pregnant while taking ZYDELIG should be apprised of the potential hazard to the fetus. Advise females of reproductive potential to avoid pregnancy during treatment and to use effective contraception during and for ≥1 month after treatment [See Use in Specific Populations].
ADVERSE REACTIONS:
See BOXED WARNING and Warnings and Precautions for additional serious adverse reactions.
Subjects with Relapsed CLL:
The safety assessment of ZYDELIG 150 mg BID + rituximab (up to 8 doses) is based on data from 110 adult subjects with relapsed CLL (Study 1). The median duration of exposure to ZYDELIG was 5 months.
• Adverse Reactions: Most common (≥2%) serious adverse reactions reported in 49% of subjects were pneumonia (17%), pyrexia (9%), sepsis (8%), febrile neutropenia (5%), and diarrhea (5%). Most common adverse reactions (incidence ≥5% and occurring at ≥2% higher incidence in ZYDELIG-treated subjects; all Grades) were pyrexia (35%), nausea (25%), pneumonia (23%), diarrhea (21%), chills (21%), rash (18%), vomiting (13%), headache (10%), sepsis (8%), sinusitis (8%), pain (7%), arthralgia (7%), GERD (6%), stomatitis (6%), bronchitis (6%), nasal congestion (5%), and urinary tract infection (5%). Most common adverse reactions leading to dose reductions in 15% of subjects were elevated transaminases, diarrhea or colitis, and rash. Most common adverse reactions leading to discontinuation in 10% of subjects were hepatotoxicity and diarrhea/colitis.
• Laboratory Abnormalities: Treatment emergent laboratory abnormalities (incidence ≥10% and occurring at ≥5% higher incidence in ZYDELIG-treated subjects; all Grades) were decreased neutrophils (60%), hypertriglyceridemia (56%), hyperglycemia (54%), increased ALT (35%), increased GGT (26%), increased lymphocytes (25%), increased AST (25%), decreased lymphocytes (20%), hyponatremia (20%), and hypoglycemia (11%).
Subjects with Indolent Non-Hodgkin Lymphoma (iNHL):
The safety assessment of ZYDELIG 150 mg BID is based on data from 146 adult subjects with iNHL. The median duration of exposure to ZYDELIG was 6.1 months (range: 0.3 to 26.4 months).
• Adverse Reactions: Most common serious adverse reactions reported in 50% of subjects were pneumonia (15%), diarrhea (11%), and pyrexia (9%). Most common adverse reactions (incidence ≥10%; all Grades) were diarrhea (47%), fatigue (30%), cough (29%), nausea (29%), pyrexia (28%), abdominal pain (26%), pneumonia (25%), rash (21%), dyspnea (17%), decreased appetite (16%), vomiting (15%), upper respiratory tract infection (12%), asthenia (12%), night sweats (12%), insomnia (12%), headache (11%), and peripheral edema (10%). Most common adverse reactions leading to dose interruption or discontinuation in 53% of subjects were diarrhea (11%), pneumonia (11%), and elevated transaminases (10%).
• Laboratory Abnormalities: Treatment emergent laboratory abnormalities (all Grades) were decreased neutrophils (53%), increased ALT (50%), increased AST (41%), decreased hemoglobin (28%), and decrease platelets (26%).
DRUG INTERACTIONS:
• CYP3A Inducers: Strong CYP3A inducers decreased idelalisib AUC by 75%. Avoid coadministration with strong CYP3A inducers (e.g., rifampin, phenytoin, St. John’s wort, carbamazepine).
• CYP3A Inhibitors: Strong CYP3A inhibitors increased idelalisib AUC 1.8-fold. Monitor for signs of ZYDELIG toxicity during coadministration and follow dose modifications for adverse reactions [See Dosage and Administration].
• CYP3A Substrates: ZYDELIG is a strong CYP3A inhibitor. Avoid coadministration with CYP3A substrates as AUC of sensitive CYP3A substrates increased 5.4-fold when coadministered.
USE IN SPECIFIC POPULATIONS:
Pregnancy: ZYDELIG is Pregnancy Category D and may cause fetal harm. In pregnant rats, embryo-fetal toxicities were observed, including decreased fetal weights, external malformations (short tail), skeletal variations (delayed ossification and/or unossification of the skull, vertebrae and sternebrae), urogenital blood loss, complete resorption, increased post-implantation loss, and malformations (vertebral agenesis with anury, hydrocephaly, microphthalmia/anophthalmia). Women who are or become pregnant during ZYDELIG treatment should be apprised of the potential hazard to the fetus [See Warnings and Precautions].
Nursing Mothers: It is not known whether idelalisib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ZYDELIG, a decision should be made whether to discontinue nursing or ZYDELIG, taking into account the importance of ZYDELIG to the mother.
Pediatric Use: Safety and effectiveness of ZYDELIG in children <18 years of age have not been established.
Geriatric Use: In clinical trials of ZYDELIG in patients with FL, SLL, and CLL, 63% of patients were ≥65 years old; no major differences in effectiveness were observed.
• In patients with iNHL: Compared to younger patients, older patients (≥65 years) had higher incidences of discontinuation due to adverse reaction (28% vs. 20%), serious adverse reactions (64% vs. 37%), and death (11% vs. 5%).
• In patients with CLL: Compared to younger patients, older patients (≥65 years) had higher incidences of discontinuation due to adverse reaction (11% vs. 5%), serious adverse reactions (51% vs. 43%), and death (3% vs. 0%).
Contraception in Females of Reproductive Potential: ZYDELIG may cause fetal harm. Advise females of reproductive potential to avoid pregnancy during treatment and to use effective contraception during and for ≥1 month after taking the last dose of ZYDELIG. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking ZYDELIG [See Warnings and Precautions].
Renal Impairment: No dose adjustment of ZYDELIG is necessary for patients with creatinine clearance ≥15 mL/min.
Hepatic Impairment: Idelalisib AUC increased up to 1.7-fold in subjects with ALT, AST, or bilirubin >ULN compared to healthy subjects with normal ALT, AST, or bilirubin. Safety and efficacy data are not available in patients with baseline ALT or AST >2.5x ULN or bilirubin >1.5x ULN as these patients were excluded from Studies 1 and 2. Monitor patients with baseline hepatic impairment for signs of ZYDELIG toxicity and follow dose modifications for adverse reactions [See Warnings and Precautions, Dosage and Administration].
205858-GS-000-PI July 2014
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, and INTESTINAL PERFORATION
• Fatal and/or serious hepatotoxicity occurred in 14% of ZYDELIG-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and/or serious and severe diarrhea or colitis occurred in 14% of ZYDELIG-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and serious pneumonitis can occur in ZYDELIG-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and serious intestinal perforation can occur in ZYDELIG-treated patients. Discontinue ZYDELIG for intestinal perforation [See Warnings and Precautions].
S:9.5”S:13”
T:10”T:13.75”
79649ha_c.indd 4 8/6/15 5:38 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 83
Reflections
Never Say NeverBy Grace Wang, MD
DK came to me in 1997 with dis-seminated breast cancer. We
talked about prognosis, and I told her that we’d never cured her disease. We discussed a clinical trial involving the experimental drug trastuzumab (Her-ceptin). She was a nurse and under-stood it was important for her to try the drug, not only for herself, but also for other patients.
“Dr. Wang! Your patient is having chills and shortness of breath,” said a nurse. It was DK. As I ran to the che-motherapy suite, I worried what could be happening. As physicians, our mot-to has always been, “First, do no harm.” Yet, when you have patients with dis-eases that are uniformly fatal, you be-
come more interested in research tri-als. There aren’t other choices.
I began my career nearly 4 decades ago as an assistant professor at the University of Miami Medical School treating breast cancer patients. It was a time when many more of my pa-tients presented with stage IV cancer. Thanks to breast cancer awareness and screening mammograms, many more patients are being cured today, and mortality rates have dropped 30%. However, one patient dying is one patient too many. When I came into private practice, I did clinical tri-als but not without some resistance.
At one point, we experimented with the natural (by this time, geneti-cally cloned) immune system modu-lator interleukin. It was, among lay-men anyway, the latest “magic bullet” for cancer. A reporter from the local newspaper interviewed my partner, Dr. Leonard Kalman, and me about the drug. I thought the interview had gone very well. Then to our dismay, we landed on the front page of the paper. We were compared to the La-etrile (a “wonder drug” supposedly distilled from apricot pits) clinic in Mexico that actor Steve McQueen went to for his cancer.
We were not under the umbrella of an academic institution, and, to be honest, interleukin had a lot of toxic-ity, including plummeting blood pres-sure, fever, and chills. It turned out that interleukin is now recognized as a drug that helps the immune system combat kidney cancer and melanoma. At the time, we hoped it would induce complete remissions and cure pa-tients with other stage IV cancers. We proceeded with our trials. I became reporter-shy for many years.
Improving Breast Cancer CareLet’s return to the story about DK,
the nurse with breast cancer who’d been coming to our clinic for years. In my heart, she was not cured, but even though she had diffuse bone metasta-ses, scan after scan showed she was in a complete remission. We finally stopped the trastuzumab in 2009. Today, DK is 75 and working as a hospice nurse. Trastuzumab was approved by the U.S. Food and Drug Administration in 1998, a landmark in the treatment of certain kinds of breast cancer.
I made the decision to leave my posi-tion at the University of Miami Medical School partly because I thought I would be bored treating only breast cancer pa-tients. Now that the research is so volu-minous and treatments are so targeted, I have come full circle. I’m never bored treating breast cancer patients, and I’m still doing clinical trials. Over my career, research has led to miracles. For exam-ple, thanks to the development of ima-tinib (Gleevec), we now see complete, long-term remissions in patients with chronic myeloid leukemia.
Everyone is becoming special-ized, most often as part of a disease-oriented team. We now have breast surgeons, breast plastic surgeons, and medical and radiation oncologists, all
focused on breast oncology. I go to breast cancer research conferences. The community physician is now part of research trials underway all over the world.
Medical ‘Miracles’To me DK is one of those medical
“miracles” that now occurs daily. When I reflect on my career, I remember other amazing patients like the one who went to dental school and had treatments for metastatic breast cancer that had spread to her bones. Today, she runs her own
dental practice, her children are gradu-ating from college, and she’s still in complete remission.
Another patient had 17 positive lymph nodes. She later adopted two babies, and she and her husband saw their children graduate from high school. She’s never had a relapse. And then there are the patients who devel-op new primary cancers that we pick up and are still cured because of good survivorship care.
When it comes to my patients and their prognoses, when I’m asked if there’s a cure for cancer, I’ve learned never to say never. n
Dr. Wang is a medical oncologist specializing in hematology/oncology in Miami, Florida.
When it comes to my patients and their prognoses, when I’m asked if there’s a cure for cancer, I’ve learned never to say never.
—Grace Wang, MD
The following essay by Grace Wang, MD, is adapted from The Big Casino: America’s Best Cancer Doctors Share Their Most Powerful Stories, which was coedited by Stan Winokur, MD, and Vincent Cop-pola and published in May 2014. The book is available on Amazon.com and thebigcasino.org.
Download The ASCO Post iPad App
FREE from iTunes today!© 2015 Gilead Sciences, Inc. All rights reserved. ZYDP0192 08/2015ZYDELIG, ZYDELIG logo, GILEAD and the GILEAD logo are trademarks of Gilead Sciences, Inc., or its related companies. All other marks are the property of their respective owners.
References: 1. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Hodgkin’s Lymphomas V.1.2015. © National Comprehensive Cancer Network, Inc 2015. All rights reserved. Accessed January 7, 2015. To view the most recent and complete version of the guideline, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc. 2. Gopal AK, Kahl BS, de Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008-1018.
IDEPU-53446_M1_BrfSmm_Kng_Ind_NoCode.indd12-16-2014 9:49 AM Suke Yawata / Dan Capobianco
Client CodeClient
LiveOverall TrimBleed
# of Colors
GILP0220 07/2014GILEAD
9.5” x 13”10” x 13.75”None
1C - Black
Colors Black
FontsHelvetica Neue LT Std (77 Bold Condensed, 57 Condensed, 77 Bold Condensed Oblique, 57 Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hssyawata5561 by
Printed At
None
Notes King Size Brief Summary-Indication
ZYDELIG® (idelalisib) tablets, for oral use
Brief Summary of full Prescribing Information. See full Prescribing Information. Rx Only.
INDICATIONS AND USAGE:
• ZYDELIG is indicated in combination with rituximab for the treatment of adults with relapsed chronic lymphocytic leukemia (CLL) for whom rituximab alone would be considered appropriate therapy due to other comorbidities.
• ZYDELIG is indicated for the treatment of adults with relapsed follicular B-cell non-Hodgkin lymphoma (FL) who have received ≥2 prior systemic therapies.
• ZYDELIG is indicated for the treatment of adults with relapsed small lymphocytic lymphoma (SLL) who have received ≥2 prior systemic therapies.
• Accelerated approval was granted for FL and SLL based on overall response rate. An improvement in patient survival or disease related symptoms has not been established. Continued approval for these indications may be contingent upon verification of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATION:
See Warnings and Precautions, Adverse Reactions, and Use in Specific Populations for additional information.
Adult Starting Dose: One 150 mg tablet taken orally twice daily (BID), swallowed whole with or without food. Continue treatment until disease progression or unacceptable toxicity. The optimal and safe dosing regimen for patients who required treatment longer than several months is unknown.
Dose Modifications:
• Pneumonitis: discontinue ZYDELIG for any symptomatic pneumonitis
• Hepatotoxicity: – ALT/AST >3 to 5x ULN or bilirubin >1.5 to 3x ULN: maintain ZYDELIG
dose; monitor weekly until ≤1x ULN – ALT/AST >5 to 20x ULN or bilirubin >3 to 10x ULN: withhold ZYDELIG;
monitor weekly until ≤1x ULN then resume ZYDELIG 100 mg BID – ALT/AST >20x ULN or bilirubin >10x ULN: permanently discontinue ZYDELIG
• Diarrhea: – Moderate (increase of 4-6 stools/day over baseline): maintain ZYDELIG
dose; monitor weekly until resolved – Severe (increase of ≥7 stools/day over baseline) or hospitalization:
withhold ZYDELIG; monitor weekly until resolved then resume ZYDELIG 100 mg BID
– Life-threatening: permanently discontinue ZYDELIG
• Neutropenia: – ANC 1 to <1.5 Gi/L: maintain ZYDELIG dose – ANC 0.5 to <1 Gi/L: maintain ZYDELIG dose; monitor weekly – ANC <0.5 Gi/L: withhold ZYDELIG; monitor weekly until ≥0.5 Gi/L then
resume ZYDELIG 100 mg BID
• Thrombocytopenia: – Platelets 50 to <75 Gi/L: maintain ZYDELIG dose – Platelets 25 to <50 Gi/L: maintain ZYDELIG dose; monitor weekly – Platelets <25 Gi/L: withhold ZYDELIG; monitor weekly until ≥25 Gi/L
then resume ZYDELIG 100 mg BID
• For other severe or life-threatening toxicities, withhold ZYDELIG until toxicity is resolved and reduce dose to 100 mg BID if resuming treatment. Permanently discontinue ZYDELIG if severe or life-threatening toxicities recur upon rechallenge.
CONTRAINDICATIONS:
History of serious allergic reactions including anaphylaxis and toxic epidermal necrolysis (TEN).
WARNINGS AND PRECAUTIONS:
Fatal and/or serious hepatotoxicity occurred in 14% of ZYDELIG-treated patients. ALT or AST >5x ULN have occurred, usually within the first 12 weeks of treatment and were reversible with dose interruption. Upon resuming
treatment at a lower dose, 26% of patients had recurrence of ALT and AST elevations. Discontinue ZYDELIG for recurrent hepatotoxicity. Avoid concurrent use of ZYDELIG with hepatotoxic drugs. In all patients, monitor ALT and AST every 2 weeks for the first 3 months, every 4 weeks for the next 3 months, then every 1 to 3 months thereafter. If ALT or AST >3x ULN, monitor weekly until elevation resolves; if ALT or AST >5x ULN, withhold ZYDELIG and monitor AST, ALT and total bilirubin weekly until elevation resolves [See Dosage and Administration].
Severe diarrhea or colitis (≥Grade 3) occurred in 14% of ZYDELIG-treated patients across clinical trials. ZYDELIG-induced diarrhea can occur at any time and responds poorly to antimotility agents. Median time to resolution ranged between 1 week and 1 month following ZYDELIG interruption with or without enteric or systemic corticosteroids. Avoid concurrent use of ZYDELIG with drugs that cause diarrhea. [See Dosage and Administration].
Fatal and serious pneumonitis occurred in ZYDELIG-treated patients. Patients taking ZYDELIG who present with pulmonary symptoms (e.g., cough, dyspnea, hypoxia, interstitial infiltrates, >5% decrease in oxygen saturation) should be evaluated for pneumonitis. If pneumonitis is suspected, withhold ZYDELIG until etiology of pulmonary symptoms has been determined. Patients thought to have ZYDELIG-induced pneumonitis were treated with ZYDELIG discontinuation and corticosteroids.
Fatal and serious intestinal perforation occurred in ZYDELIG-treated patients. At the time of perforation, some patients had moderate to severe diarrhea. Advise patients to promptly report any new or worsening abdominal pain, chills, fever, nausea, or vomiting. Permanently discontinue ZYDELIG in patients who experience intestinal perforation.
Severe Cutaneous Reactions: One case of TEN occurred in a study of ZYDELIG in combination with rituximab and bendamustine. Other severe or life-threatening (Grade ≥3) cutaneous reactions (dermatitis exfoliative, rash, rash erythematous, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic, exfoliative rash, skin disorder) have been reported. Monitor patients for severe cutaneous reactions and discontinue ZYDELIG.
Anaphylaxis: Serious allergic reactions including anaphylaxis have been reported in ZYDELIG-treated patients. Permanently discontinue ZYDELIG and institute appropriate supportive measures in patients who develop serious allergic reactions.
Neutropenia: Treatment-emergent neutropenia (Grade 3 or 4) occurred in 31% of ZYDELIG-treated patients across clinical trials. Monitor blood counts every 2 weeks for the first 3 months, and weekly when neutrophils are <1 Gi/L [See Dosage and Administration].
Embryo-fetal Toxicity: Idelalisib is teratogenic in rats and may cause fetal harm. Women who are or become pregnant while taking ZYDELIG should be apprised of the potential hazard to the fetus. Advise females of reproductive potential to avoid pregnancy during treatment and to use effective contraception during and for ≥1 month after treatment [See Use in Specific Populations].
ADVERSE REACTIONS:
See BOXED WARNING and Warnings and Precautions for additional serious adverse reactions.
Subjects with Relapsed CLL:
The safety assessment of ZYDELIG 150 mg BID + rituximab (up to 8 doses) is based on data from 110 adult subjects with relapsed CLL (Study 1). The median duration of exposure to ZYDELIG was 5 months.
• Adverse Reactions: Most common (≥2%) serious adverse reactions reported in 49% of subjects were pneumonia (17%), pyrexia (9%), sepsis (8%), febrile neutropenia (5%), and diarrhea (5%). Most common adverse reactions (incidence ≥5% and occurring at ≥2% higher incidence in ZYDELIG-treated subjects; all Grades) were pyrexia (35%), nausea (25%), pneumonia (23%), diarrhea (21%), chills (21%), rash (18%), vomiting (13%), headache (10%), sepsis (8%), sinusitis (8%), pain (7%), arthralgia (7%), GERD (6%), stomatitis (6%), bronchitis (6%), nasal congestion (5%), and urinary tract infection (5%). Most common adverse reactions leading to dose reductions in 15% of subjects were elevated transaminases, diarrhea or colitis, and rash. Most common adverse reactions leading to discontinuation in 10% of subjects were hepatotoxicity and diarrhea/colitis.
• Laboratory Abnormalities: Treatment emergent laboratory abnormalities (incidence ≥10% and occurring at ≥5% higher incidence in ZYDELIG-treated subjects; all Grades) were decreased neutrophils (60%), hypertriglyceridemia (56%), hyperglycemia (54%), increased ALT (35%), increased GGT (26%), increased lymphocytes (25%), increased AST (25%), decreased lymphocytes (20%), hyponatremia (20%), and hypoglycemia (11%).
Subjects with Indolent Non-Hodgkin Lymphoma (iNHL):
The safety assessment of ZYDELIG 150 mg BID is based on data from 146 adult subjects with iNHL. The median duration of exposure to ZYDELIG was 6.1 months (range: 0.3 to 26.4 months).
• Adverse Reactions: Most common serious adverse reactions reported in 50% of subjects were pneumonia (15%), diarrhea (11%), and pyrexia (9%). Most common adverse reactions (incidence ≥10%; all Grades) were diarrhea (47%), fatigue (30%), cough (29%), nausea (29%), pyrexia (28%), abdominal pain (26%), pneumonia (25%), rash (21%), dyspnea (17%), decreased appetite (16%), vomiting (15%), upper respiratory tract infection (12%), asthenia (12%), night sweats (12%), insomnia (12%), headache (11%), and peripheral edema (10%). Most common adverse reactions leading to dose interruption or discontinuation in 53% of subjects were diarrhea (11%), pneumonia (11%), and elevated transaminases (10%).
• Laboratory Abnormalities: Treatment emergent laboratory abnormalities (all Grades) were decreased neutrophils (53%), increased ALT (50%), increased AST (41%), decreased hemoglobin (28%), and decrease platelets (26%).
DRUG INTERACTIONS:
• CYP3A Inducers: Strong CYP3A inducers decreased idelalisib AUC by 75%. Avoid coadministration with strong CYP3A inducers (e.g., rifampin, phenytoin, St. John’s wort, carbamazepine).
• CYP3A Inhibitors: Strong CYP3A inhibitors increased idelalisib AUC 1.8-fold. Monitor for signs of ZYDELIG toxicity during coadministration and follow dose modifications for adverse reactions [See Dosage and Administration].
• CYP3A Substrates: ZYDELIG is a strong CYP3A inhibitor. Avoid coadministration with CYP3A substrates as AUC of sensitive CYP3A substrates increased 5.4-fold when coadministered.
USE IN SPECIFIC POPULATIONS:
Pregnancy: ZYDELIG is Pregnancy Category D and may cause fetal harm. In pregnant rats, embryo-fetal toxicities were observed, including decreased fetal weights, external malformations (short tail), skeletal variations (delayed ossification and/or unossification of the skull, vertebrae and sternebrae), urogenital blood loss, complete resorption, increased post-implantation loss, and malformations (vertebral agenesis with anury, hydrocephaly, microphthalmia/anophthalmia). Women who are or become pregnant during ZYDELIG treatment should be apprised of the potential hazard to the fetus [See Warnings and Precautions].
Nursing Mothers: It is not known whether idelalisib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ZYDELIG, a decision should be made whether to discontinue nursing or ZYDELIG, taking into account the importance of ZYDELIG to the mother.
Pediatric Use: Safety and effectiveness of ZYDELIG in children <18 years of age have not been established.
Geriatric Use: In clinical trials of ZYDELIG in patients with FL, SLL, and CLL, 63% of patients were ≥65 years old; no major differences in effectiveness were observed.
• In patients with iNHL: Compared to younger patients, older patients (≥65 years) had higher incidences of discontinuation due to adverse reaction (28% vs. 20%), serious adverse reactions (64% vs. 37%), and death (11% vs. 5%).
• In patients with CLL: Compared to younger patients, older patients (≥65 years) had higher incidences of discontinuation due to adverse reaction (11% vs. 5%), serious adverse reactions (51% vs. 43%), and death (3% vs. 0%).
Contraception in Females of Reproductive Potential: ZYDELIG may cause fetal harm. Advise females of reproductive potential to avoid pregnancy during treatment and to use effective contraception during and for ≥1 month after taking the last dose of ZYDELIG. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking ZYDELIG [See Warnings and Precautions].
Renal Impairment: No dose adjustment of ZYDELIG is necessary for patients with creatinine clearance ≥15 mL/min.
Hepatic Impairment: Idelalisib AUC increased up to 1.7-fold in subjects with ALT, AST, or bilirubin >ULN compared to healthy subjects with normal ALT, AST, or bilirubin. Safety and efficacy data are not available in patients with baseline ALT or AST >2.5x ULN or bilirubin >1.5x ULN as these patients were excluded from Studies 1 and 2. Monitor patients with baseline hepatic impairment for signs of ZYDELIG toxicity and follow dose modifications for adverse reactions [See Warnings and Precautions, Dosage and Administration].
205858-GS-000-PI July 2014
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, and INTESTINAL PERFORATION
• Fatal and/or serious hepatotoxicity occurred in 14% of ZYDELIG-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and/or serious and severe diarrhea or colitis occurred in 14% of ZYDELIG-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and serious pneumonitis can occur in ZYDELIG-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue ZYDELIG as recommended [See Dosage and Administration, Warnings and Precautions].
• Fatal and serious intestinal perforation can occur in ZYDELIG-treated patients. Discontinue ZYDELIG for intestinal perforation [See Warnings and Precautions].
S:9.5”
S:13”
T:10”
T:13.75”
79649ha_c.indd 4 8/6/15 5:38 PM

PAGE 84 The ASCO Post | SEPTEMBER 10, 2015
Book Review
A Cancer Handbook for Inquisitive Laypersons and Health-Care ProfessionalsBy Ronald Piana
“I was at a meeting in San Fran-cisco in 1978 and received a call
from my wife, Nancy: ‘Jack, I just came back from the doctor. He said I have a lump in my left breast.’” So begins The Cancer Solution, a new book by Jack C. Westman, MD, MS, Professor Emeri-tus of Psychiatry at the University of Wisconsin School of Medicine and Public Health, Madison. He has writ-ten more than 150 professional publica-tions and 12 books.
The lump in his wife’s breast turned out to be an adenocarcinoma, and Nancy Westman had a radical mastectomy. She was a survivor for 26 years, and then, in 2002, the cancer recurred. After a valiant and very dif-ficult struggle, she succumbed to the
disease in 2012. That personal strug-gle and loss were the seed for this book. From the day that Dr. West-man’s wife was first diagnosed with breast cancer in 1978, he began doing extensive research on the disease and the various treatment options.
Naturally, oncology has gone through a huge transformation since 1978, and Dr. Westman’s knowledge and views have also evolved during that period. His idea to construct an informative book that offers material interesting to both the lay and medical communities comes with considerable challenges, many of which, unfortu-nately, he doesn’t overcome.
For starters, a reader of The ASCO Post opening the book will be con-fronted by rather loosely constructed data detailing the main categories of cancer and a cursory look at cancer staging. There’s a description of con-
ventional treatments along with their various side effects. This common information won’t interest oncolo-gists, and its plodding format will turn off lay readers, even those with cancer or those caring for someone with cancer. Moreover, this kind of cancer information is best delivered in pamphlets or on any of the numer-ous well-vetted websites.
Unwarranted PotshotsMore problematic is the disorient-
ed segue Dr. Westman takes halfway through chapter 1, skipping into Lance Armstrong’s battle with metastatic testicular cancer and then into an off-handed critique of conventional cancer treatment, focusing on cisplatin, which, despite considerable side effects, had cured Lance Armstrong. “Overall, [cis-platin proved] of little long-term value, except against testicular cancer, illus-trating the fact that no single chemo-therapeutic agent in use at this time kills all kinds of cancer cells.” This statement is not a news flash, and it simply serves as a lead-up to the rest of the chapter, which takes unwarranted potshots at the oncology community.
Dr. Westman makes some valid points about much-needed cost con-trol, citing The New York Times article by Ezekiel Emanuel titled “A Plan to Fix Cancer Care.” However, he cannot resist the temptation to criticize the oncology community, stating, “The more doctors do for patients, the more reimbursement they receive.… On-cologists typically make more money if they use newly approved drugs and the latest radiation treatments than if they use cheaper, older alternatives that work just as well.” This is a broad and rather cheap accusation that commu-nity oncologists will surely find offen-sive. Moreover, there are no hard data to back it up.
The author then mentions ASCO’s Quality Oncology Practice Initiative (QOPI), giving a fairly good assess-ment of the project’s mission and goals. He also delivers a solid discussion about
the impending oncology workforce shortage. But once again, Dr. Westman then veers erratically into a soft-toned diatribe about oncologists’ salaries, drilling home his bone of contention, writing, “Separating cancer specialists’ income from treatment choices is im-portant.… Instead provider incentives should be aligned toward patient-cen-tered, coordinated care among cancer specialists….”
It becomes glaringly obvious that Dr. Westman did not spend time in busy community clinics when researching his book. Nor did he venture into the interior of the ultra-introspective on-cology community’s constant evolution toward patient-centered care, data col-lection, and patient communication. In these efforts, oncology serves as a mod-el for most other medical disciplines.
Important InformationIn the first half of chapter 3, “Navi-
gating the Cancer,” Dr. Westman pro-vides many simple-to-read instructions and hints for those newly entering the system, such as instructing patients how to track and maintain their own personal health record, offering sug-gestions and evaluations of several per-sonal electronic health chart systems on the market. Readers are also given brief summaries about living wills, do-not-resuscitate orders, and other tough but vital decision-making guides. This is important information, especially for caregivers.
In the second half of the chapter, Dr. Westman gets down to the nuts and bolts of cost, another reliable source of
information. But after a thorough trip through Medicaid tips to assessing the costs of nursing homes, Dr. Westman then wanders into thorny territory, advocating for “life panels,” which of course is a euphemism for the equally ridiculous term “death panels,” famous-ly coined by Sarah Palin in 2009. As the oncology community knows, high-quality palliative care is needed for can-cer patients facing end-of-life issues.
To make his argument for life pan-els, Dr. Westman uses a financial plan-ner named Bob Goldman, who told the story of his elderly mother whose life had become “more punishment than reward.” Evidently, she wanted out.
In what amounts to a disjointed ar-gument for physician-assisted suicide, Dr. Westman decries the costs of end-of-life care and seems to support Bob Goldman’s model for a life panel. He writes, “On a statutory life panel, a doc-tor would be held blameless. And Gold-man would have no problem adding a medical ethicist and a therapist.”
Central ProblemThe central problem with The Cancer
Solution, aside from its misleading title, is that the author tries too hard to be too many things: instructor, policy wonk, historian, and all-around muckraking curmudgeon. Thus, the book’s good sec-tions are muddled by the author’s strong opinions, which often come out of left field. Moreover, in pedantic fashion, the author assaults the reader with subtitled chapter sections such as “A Paradigm Shift is Needed” or “Models for Treat-ment Directed at Cancer Cells Them-
BookmarkTitle: The Cancer Solution: Taking Charge of Your Life With Cancer
Author: Jack C. Westman, MD, MS
Publisher: Archway Publishing
Publication date: January 15, 2015
Price: $20.00; paperback, 310 pages
The central problem with The Cancer Solution, aside from its misleading title, is that the author tries too hard to
be too many things: instructor, policy wonk, historian, and all-around muckraking curmudgeon.
Jack C. Westman, MD, MS
Please see Brief Summary of complete Prescribing Information
on adjacent pages.
Learn more about IRESSA at www.iressa-usa.com.IRESSA is a registered trademark of the AstraZeneca group
of companies.
©2015 AstraZeneca. All rights reserved. 3133300 7/15
For the treatment of metastatic NSCLC
A TKI for first-line use in EGFR mutation–positive patients
NOW APPROVED!
IndicationIRESSA is indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test.
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients with metastatic NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R) substitution mutations.
Important Safety Information • There are no contraindications for IRESSA
• Interstitial Lung Disease (ILD): ILD occurred in patients taking IRESSA. Withhold IRESSA for worsening of respiratory symptoms. Discontinue IRESSA if ILD is confirmed
• Hepatotoxicity: Obtain periodic liver function testing. Withhold IRESSA for Grade 2 or higher for alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) elevations. Discontinue for severe hepatic impairment
• Gastrointestinal Perforation: Discontinue IRESSA for gastrointestinal perforation
• Diarrhea: Withhold IRESSA for Grade 3 or higher diarrhea
• Ocular Disorders Including Keratitis: Withhold IRESSA for signs and symptoms of severe or worsening ocular disorders including keratitis. Discontinue for persistent ulcerative keratitis
• Bullous and Exfoliative Skin Disorders: Withhold IRESSA for Grade 3 or higher skin reactions or exfoliative conditions
• Embryo-fetal Toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception
• Advise women to discontinue breast-feeding during treatment with IRESSA
• The most commonly reported adverse drug reactions, reported in more than 20% of the patients and greater than placebo, were skin reactions and diarrhea
3133300/3148105_Iressa_ASCO Post.indd 1 8/11/15 9:54 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 85
Announcements
selves” and devotes a mere 80 or 90 words to these lofty goals. It feels rushed.
Dr. Westman saves the best writing for chapters 9, 10, and 11, during which he gives compelling descriptions of im-munotherapy, lifestyle issues, preven-tion, and complementary strategies. He also makes a solid case for more focus on discovering the etiology of a cancer and then developing ways to prevent disease or treat it at its earliest stage, as seen with successes in gastric cancer and cervical cancer.
During these sections, although they
are highly readable and interesting, Dr. Westman will certainly get challenged by his unwavering support of several eccentric researchers in the early 1900s who asserted, “the fundamental cause of cancer is a disturbance in cell metab-olism that makes a cell prefer fermenta-tion of glucose for energy production rather than the more efficient oxidative
metabolism.… Therefore, it may be that the uncontrolled growth of cancer cells arises when they are stuck in the fermentation process.”
Dr. Westman concludes his book with a “Where Do We Go From Here?” chapter that doesn’t really add much in the way of new material on this subject. If he maintained the same tempo and
interesting, if somewhat kooky, style and content as in his chapters on immu-notherapy and complementary strate-gies, this would have been a better read. As it is, The Cancer Solution may not be for all readers of The ASCO Post. That said, Dr. Westman lost his wife to can-cer, and wrote a book in her honor—a noble effort. n
Thomas J. Lynch Jr, MD, Leaves Yale Cancer Center
Thomas J. Lynch Jr, MD, Direc-tor of Yale Cancer Center and
Physician-in-Chief of Smilow Cancer Hospital at Yale-New Haven, left Yale in August to become Chairman and Chief Executive Officer of the Massachusetts General Physicians Organization.
Dr. Lynch joined Yale Cancer Cen-ter as Director in 2009 and assumed the role of inaugural Physician-in-Chief at Smilow, which opened that year. Under his leadership, more than 130 scientists and clinicians joined the institutions, new patient volume grew from 3,500 to 9,000 through key affiliations, and par-ticipation in therapeutic clinical trials grew by 325%.
Peter Schulam, MD, PhD, Profes-sor of Urology and Chair/Chief of Yale’s Department of Urology, will serve as the interim Director of Yale Cancer Center and Physician-in-Chief at Smilow.
Dr. Schulam joined in 2012 as inau-gural Chief of the Department of Urol-ogy at Yale-New Haven Hospital and Chair of the Department at Yale School of Medicine, where he has established a multidisciplinary team in urologic oncology. He has implemented a pro-gram for MRI-fusion–guided biopsy of prostate cancer, and leads a research program focused on prostate cancer im-aging. In addition, Dr. Schulam. n
Thomas J. Lynch Jr, MD
Please see Brief Summary of complete Prescribing Information
on adjacent pages.
Learn more about IRESSA at www.iressa-usa.com.IRESSA is a registered trademark of the AstraZeneca group
of companies.
©2015 AstraZeneca. All rights reserved. 3133300 7/15
For the treatment of metastatic NSCLC
A TKI for first-line use in EGFR mutation–positive patients
NOW APPROVED!
IndicationIRESSA is indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA-approved test.
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients with metastatic NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R) substitution mutations.
Important Safety Information • There are no contraindications for IRESSA
• Interstitial Lung Disease (ILD): ILD occurred in patients taking IRESSA. Withhold IRESSA for worsening of respiratory symptoms. Discontinue IRESSA if ILD is confirmed
• Hepatotoxicity: Obtain periodic liver function testing. Withhold IRESSA for Grade 2 or higher for alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) elevations. Discontinue for severe hepatic impairment
• Gastrointestinal Perforation: Discontinue IRESSA for gastrointestinal perforation
• Diarrhea: Withhold IRESSA for Grade 3 or higher diarrhea
• Ocular Disorders Including Keratitis: Withhold IRESSA for signs and symptoms of severe or worsening ocular disorders including keratitis. Discontinue for persistent ulcerative keratitis
• Bullous and Exfoliative Skin Disorders: Withhold IRESSA for Grade 3 or higher skin reactions or exfoliative conditions
• Embryo-fetal Toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception
• Advise women to discontinue breast-feeding during treatment with IRESSA
• The most commonly reported adverse drug reactions, reported in more than 20% of the patients and greater than placebo, were skin reactions and diarrhea
3133300/3148105_Iressa_ASCO Post.indd 1 8/11/15 9:54 PM

PAGE 86 The ASCO Post | SEPTEMBER 10, 2015
Announcements
Diane Simeone, MD, Named Upcoming Chair of PanCAN Scientific Board
Diane Simeone, MD, Director of the Pancreatic Cancer Center at
the University of Michigan, was recently named the Upcoming Chair of the Na-tional Scientific and Medical Advisory Board for the Pancreatic Cancer Action Network (PanCAN). Her 2-year term as
Chair runs from 2017 to 2019. Dr. Sime-one will serve as Chair-Elect until then.
Dr. Simeone is the Founding Direc-tor of the University of Michigan Pan-creatic Cancer Center. She also serves as the Director for the U-M Multidisci-plinary Pancreatic Cancer Clinic, and
leads the Translational Oncology Pro-gram at the University’s North Cam-pus Research Complex. She is Past President of the American Pancreatic Association and currently serves on the National Cancer Institute Pancre-atic Cancer Task Force. nDiane Simeone, MDTRIM: 8.125 X 10.875
IRESSA® (gefitinib) tablets for oral use
Brief Summary of Prescribing Information.
For complete prescribing information consult official package insert
INDICATIONS AND USAGEIRESSA is indicated for the first-line treatment of patients with metastatic non-small cell lung
cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or
exon 21 (L858R) substitution mutations as detected by an FDA-approved test [see Clinical Studies
(14) in the full Prescribing Information].
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients with metastatic
NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R)
substitution mutations [see Clinical Studies (14) in the full Prescribing Information].
DOSAGE AND ADMINISTRATIONPatient Selection Select patients for the first-line treatment of metastatic NSCLC with IRESSA based on the
presence of EGFR exon 19 deletion or exon 21 (L858R) substitution mutations in their tumor
[see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information].
Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at:
http://www.fda.gov/CompanionDiagnostics.
Recommended DoseThe recommended dose of IRESSA is 250 mg orally once daily with or without food until disease
progression or unacceptable toxicity.
Do not take a missed dose within 12 hours of the next dose.
Administration to Patients Who Have Difficulty Swallowing SolidsImmerse IRESSA tablets in 4 to 8 ounces of water by dropping the tablet in water, and stir for
approximately 15 minutes. Immediately drink the liquid or administer through a naso-gastric tube.
Rinse the container with 4 to 8 ounces of water and immediately drink or administer through the
naso-gastric tube.
Dose ModificationDose Modifications for Adverse Drug Reactions
Withhold IRESSA (for up to 14 days) for any of the following:
�� Acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever) [see Warnings and
Precautions (5.1) in the full Prescribing Information]
�� NCI CTCAE Grade 2 or higher in ALT and/or AST elevations [see Warnings and Precautions (5.2)
in the full Prescribing Information]
�� NCI CTCAE Grade 3 or higher diarrhea [see Warnings and Precautions (5.4) in the full Prescribing
Information]
�� Signs and symptoms of severe or worsening ocular disorders including keratitis [see Warnings
and Precautions (5.5) in the full Prescribing Information]
�� NCI CTCAE Grade 3 or higher skin reactions [see Warnings and Precautions (5.6) in the full
Prescribing Information]
Resume treatment with IRESSA when the adverse reaction fully resolves or improves to NCI CTCAE
Grade 1.
Permanently discontinue IRESSA for:
�� Confirmed interstitial lung disease (ILD) [see Warnings and Precautions (5.1) in the full
Prescribing Information]
�� Severe hepatic impairment [see Warnings and Precautions (5.2) in the full Prescribing
Information]
�� Gastrointestinal perforation [see Warnings and Precautions (5.3) in the full Prescribing
Information]
�� Persistent ulcerative keratitis [see Warnings and Precautions (5.5) in the full Prescribing
Information]
Dose Modifications for Drug Interactions
Strong CYP3A4 Inducers
Increase IRESSA to 500 mg daily in the absence of severe adverse drug reaction, and resume
IRESSA at 250 mg seven days after discontinuation of the strong CYP3A4 inducer [see Drug
Interactions (7) and Clinical Pharmacology (12.3) in the full Prescribing Information].
CONTRAINDICATIONSNone.
WARNINGS AND PRECAUTIONSInterstitial Lung Disease (ILD)ILD or ILD-like adverse drug reactions (e.g., lung infiltration, pneumonitis, acute respiratory
distress syndrome, or pulmonary fibrosis) occurred in 1.3% of the 2462 patients who received
IRESSA across clinical trials; of these, 0.7% were Grade 3 or higher and 3 cases were fatal.
Withhold IRESSA and promptly investigate for ILD in any patient who presents with worsening
of respiratory symptoms such as dyspnea, cough and fever. Permanently discontinue IRESSA if
ILD is confirmed [see Dosage and Administration (2.4) and Adverse Reactions (6.1) in the full
Prescribing Information].
HepatotoxicityIn patients who received IRESSA across clinical trials, 11.4% of patients had increased alanine
aminotransferase (ALT), 7.9% of patients had increased aspartate aminotransferase (AST), and
2.7% of patients had increased bilirubin. Grade 3 or higher liver test abnormalities occurred in
5.1% (ALT), 3.0% (AST), and 0.7% (bilirubin) of patients. The incidence of fatal hepatotoxicity
was 0.04%.
Obtain periodic liver function testing. Withhold IRESSA in patients with worsening liver function and
discontinue in patients with severe hepatic impairment [see Dosage and Administration (2.4), Adverse
Reactions (6.1), and Use in Specific Populations (8.7) in the full Prescribing Information].
Gastrointestinal PerforationGastrointestinal perforation occurred in three (0.1%) of the 2462 IRESSA-treated patients across
clinical trials [see Adverse Reactions (6.1) in the full Prescribing Information]. Permanently
discontinue IRESSA in patients who develop gastrointestinal perforation [see Dosage and
Administration (2.4) in the full Prescribing Information].
Severe or Persistent DiarrheaGrade 3 or 4 diarrhea occurred in 3% of 2462 IRESSA-treated patients across clinical trials.
Withhold IRESSA for severe or persistent (up to 14 days) diarrhea [see Dosage and Administration
(2.4) and Adverse Reactions (6.1) in the full Prescribing Information].
Ocular Disorders including KeratitisOcular disorders [keratitis (0.1%), corneal erosion and aberrant eyelash growth (0.2%), conjunctivitis,
blephritis and dry eye (6.7%)] occurred in the 2462 IRESSA-treated patients across clinical trials. The
incidence of Grade 3 ocular disorders was 0.1% [see Adverse Reactions (6.1) in the full Prescribing
Information]. Interrupt or discontinue IRESSA for severe, or worsening ocular disorders [see Dosage
and Administration (2.4) in the full Prescribing Information].
Bullous and Exfoliative Skin DisordersBullous conditions including toxic epidermal necrolysis, Stevens Johnson syndrome and erythema
multiforme have been reported from treatment with IRESSA. Erythema multiforme and dermatitis
bullous have been reported in two patients (0.08%) across NSCLC trials (Study 2, Study 3 and
Study 4). IRESSA treatment should be interrupted or discontinued if the patient develops severe
bullous, blistering or exfoliating conditions.
Embryo-fetal ToxicityBased on its mechanism of action and data from animal reproduction studies IRESSA can cause fetal
harm when administered to a pregnant woman. In animal reproductive studies, oral administration
of gefitinib from organogenesis through weaning resulted in fetotoxicity and neonatal death at
doses below the recommended human dose. Advise pregnant women of the potential risk to a
fetus. Advise females of reproductive potential to use effective contraception during treatment with
IRESSA and for at least two weeks following completion of therapy [see Use in Specific Populations
(8.1, 8.3) in the full Prescribing Information].
ADVERSE REACTIONS The following adverse drug reactions are discussed in more detail in other sections of the labeling:
�� Interstitial Lung Disease [see Warnings and Precautions (5.1) in the full Prescribing Information]
�� Hepatotoxicity [see Warnings and Precautions (5.2) in the full Prescribing Information]
�� Gastrointestinal Perforation [see Warnings and Precautions (5.3) in the full Prescribing
Information]
�� Severe or Persistent Diarrhea [see Warnings and Precautions (5.4) in the full Prescribing
Information]
�� Ocular Disorders including Keratitis [see Warnings and Precautions (5.5) in the full Prescribing
Information]
�� Bullous and Exfoliative Skin Disorders [see Warning and Precautions (5.6) in the full Prescribing
Information]
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates
observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of
another drug and may not reflect the rates observed in practice.
The safety of IRESSA is based on the data from 2462 patients with NSCLC who received IRESSA
250 mg daily monotherapy in three randomized clinical studies (Study 2, Study 3 and Study 4).
Patients with a history of interstitial lung disease, drug-induced interstitial disease, radiation
pneumonitis that required steroid treatment or any evidence of clinically active interstitial lung
disease were excluded from these studies.
Controlled Studies:
Study 2 was a randomized, multicenter, open-label trial in which 1217 patients were randomized to
receive first-line treatment for metastatic NSCLC; 607 patients received IRESSA 250 mg daily and
589 patients received carboplatin/paclitaxel. The median duration of treatment with IRESSA was 5.9
months. The study population characteristics were: median age 57 years, age less than 65 years
(73%), female (79%), Asian (100%), NSCLC adenocarcinoma histology (100%), never smoker
(94%), light ex-smoker (6%), ECOG PS 0 or 1 (90%).
Study 3 was a randomized, multicenter, double-blind, placebo-controlled trial in which 1692 patients
were randomized to receive second- or third-line treatment for metastatic NSCLC; of which 1126
patients received IRESSA 250 mg daily and 562 patients received placebo. The median duration of
treatment with IRESSA was 2.9 months. The study population characteristics were: median age
62 years, age less than 65 years (60%), female (33%), Caucasian (75%), Asian (21%), NSCLC
adenocarcinoma histology (48%), never smoker (22%), ECOG PS 0 or 1 (65%), PS 2 (29%), PS 3
(5%) and two or more prior therapies (51%).
Study 4 was a randomized, multicenter, open-label trial in which 1466 patients were randomized to
receive second-line treatment for metastatic NSCLC; 729 patients received IRESSA 250 mg daily and
715 patients received docetaxel. The median duration of treatment with IRESSA was 2.4 months. The
study population characteristics were: median age 61 years, age less than 65 years (61%), female
(36%), Caucasian (79%), Asian (21%), NSCLC adenocarcinoma histology (54%), never smoker
(20%), ECOG PS 0 or 1 (88%) and two or more prior therapies (16%).
The pooled safety database from the three randomized trials was used to evaluate for serious and
uncommon adverse drug reactions. Common adverse reactions were evaluated in Study 3. The
most frequent adverse reactions in Study 3 (incidence of >20% and greater than placebo) reported
in IRESSA-treated patients were skin reactions (47%) and diarrhea (29%). The most frequent fatal
adverse reactions in IRESSA-treated patients were respiratory failure (0.9%), pneumonia (0.8%),
and pulmonary embolism (0.5%).
Approximately 5% of IRESSA-treated patients and 2.3% of placebo-treated patients discontinued
treatment due to an adverse event. The most frequent adverse reactions that led to discontinuation in
patients treated with IRESSA were nausea (0.5%), vomiting (0.5%) and diarrhea (0.4%).
Table 1 – Selected Adverse Drug Reactions Occurring with an Incidence Rate ≥5% and an Increase of >2% of IRESSA-treated Patients in Study 3
Adverse Reaction
Percentage (%) of patientsIRESSA (N=1126) Placebo (N=562)
All Grades Grade 3 and 4 All Grades Grade 3 and 4Skin and subcutaneous tissue disordersSkin reactions1 47% 2% 17% 0.4%
Nail disorders2 5% 0.1% 0.7% 0%
Gastrointestinal disorders
Diarrhea3 29% 3% 10% 1%
Vomiting 14% 1.2% 10% 0.4%
Stomatitis4 7% 0.3% 4% 0.2%
Metabolism and nutrition disordersDecreased appetite 17% 2.3% 14% 2.0%
3133300/3148105_Iressa_ASCO Post.indd 2 8/11/15 9:54 PM
TRIM: 8.125 X 10.875
IRESSA® (gefitinib) tablets for oral use 2
Adverse Reaction
Percentage (%) of patientsIRESSA (N=1126) Placebo (N=562)
All Grades Grade 3 and 4 All Grades Grade 3 and 4Eye disordersConjunctivitis/blepharitis/dry eye5 6% 0% 3.2% 0%1 Includes Acne, Acne pustular, Dermatitis, Dermatitis acneiform, Dermatitis exfoliative, Drug eruption, Dry
skin, Erythema, Exfoliative rash, Folliculitis, Pruritus, Pruritus generalized, Rash, Rash erythematous, Rash
generalized, Rash macular, Rash maculo-papular, Rash papular, Rash pruritic, Rash pustular, Rash vesicular,
Skin exfoliation, Skin toxicity, Xeroderma2 Includes Ingrowing nail, Nail bed infection, Nail disorder, Nail infection, Onychoclasis, Onycholysis, Paronychia3 Includes Diarrhea, Feces soft, Frequent bowel movements4 Includes Aphthous stomatitis, Cheilitis, Glossodynia, Mouth ulceration, Mucosal inflammation, Oral mucosal
blistering, Stomatitis, Tongue disorder, Tongue ulceration5 Includes Blepharitis, Conjunctival hyperemia, Conjunctivitis, Dry eye, Eye irritation, Eye pruritus, Eye swelling,
Eyelid irritation, Eyelid edema, Eyelids pruritus
Table 2 – Treatment Emergent Laboratory Abnormalities Occurring More Frequently in IRESSA-Treated Patients in Study 3
Adverse Reaction
IRESSA PlaceboAll Grades
%Grade 3 and 4
%All Grades
%Grade 3 and 4
%Alanine aminotransferase increased1 38%2 2.4% 23%2 1.4%4
Aspartate aminotransferase increased1 40%3 2.0% 25%3 1.3%5
Proteinuria 35% 4.7% 31% 3.3%1 Patients were allowed to enter the clinical study with lab values of ALT or AST CTCAE grade 1 or 22 14% gefitinib patients and 10% placebo patients were CTC grade 1 or 2 ALT at baseline3 15% gefitinib patients and 12% placebo patients were CTC grade 1 or 2 AST at baseline4 0.2% of placebo patients were CTC grade 3 at baseline5 0.4% of placebo patients were CTC grade 3 at baseline
The following adverse reactions have been reported with IRESSA across NSCLC trials (Study 2,
Study 3 and Study 4) and are not listed elsewhere in Section 6: nausea (18%), asthenia (17%),
pyrexia (9%), alopecia (4.7%), hemorrhage (including epistaxis and hematuria) (4.3%), dry mouth
(2%), dehydration (1.8%), allergic reactions including angioedema and urticaria (1.1%), elevations
in blood creatinine (1.5%), and pancreatitis (0.1%).
Postmarketing ExperienceThe following adverse reactions have been identified during post-approval use of IRESSA. Because
these reactions are reported voluntarily from a population of uncertain size, it is not always possible
to reliably estimate their frequency or establish a causal relationship to drug exposure.
Renal and urinary disorders: cystitis, hemorrhagic cystitis
Skin and subcutaneous tissue disorders: cutaneous vasculitis
DRUG INTERACTIONSDrugs Affecting Gefitinib ExposureCYP3A4 Inducer
Drugs that are strong inducers of CYP3A4 increase the metabolism of gefitinib and decrease
gefitinib plasma concentrations. Increase IRESSA to 500 mg daily in patients receiving a strong
CYP3A4 inducer (e.g., rifampicin, phenytoin, or tricyclic antidepressant) and resume IRESSA at
250 mg 7 days after discontinuation of the strong inducer [see Dosage and Administration (2.4) and
Clinical Pharmacology (12.3) in the full Prescribing Information].
CYP3A4 Inhibitor
Drugs that are strong inhibitors of CYP3A4 (e.g., ketoconazole and itraconazole) decrease gefitinib
metabolism and increase gefitinib plasma concentrations. Monitor adverse reactions when
administering strong CYP3A4 inhibitors with IRESSA.
Drugs Affecting Gastric pH
Drugs that elevate gastric pH (e.g., proton pump inhibitors, histamine H2-receptor antagonists,
and antacids) may reduce plasma concentrations of gefitinib. Avoid concomitant use of IRESSA
with proton pump inhibitors, if possible. If treatment with a proton-pump inhibitor is required, take
IRESSA 12 hours after the last dose or 12 hours before the next dose of the proton-pump inhibitor.
Take IRESSA 6 hours after or 6 hours before an H2-receptor antagonist or an antacid [see Clinical
Pharmacology (12.3) in the full Prescribing Information].
Hemorrhage in Patients taking WarfarinInternational Normalized Ratio (INR) elevations and/or hemorrhage have been reported in some
patients taking warfarin while on IRESSA therapy. Patients taking warfarin should be monitored
regularly for changes in prothrombin time or INR.
USE IN SPECIFIC POPULATIONSPregnancyRisk Summary
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when
administered to a pregnant woman. In animal reproductive studies, oral administration of gefitinib
from organogenesis through weaning resulted in fetotoxicity and neonatal death at doses below the
recommended human dose (see Animal Data). Advise pregnant women of the potential hazard to a
fetus or potential risk for loss of the pregnancy.
The background risk of major birth defects and miscarriage for the indicated population is unknown;
however, the background risk in the U.S. general population of major birth defects is 2-4% and
miscarriage is 15-20% of clinically recognized pregnancies.
Data
Animal Data
A single dose study in rats showed that gefitinib crosses the placenta after an oral dose of
5 mg/kg (30 mg/m2, about 0.2 times the recommended human dose on a mg/m2 basis). When
pregnant rats were treated with 5 mg/kg from the beginning of organogenesis to the end of
weaning there was a reduction in the number of offspring born alive. This effect was more severe at
20 mg/kg (approximate the human clinical dose on a mg/m2 basis) and was accompanied by high
neonatal mortality soon after parturition. In rabbits, a dose of 20 mg/kg/day (240 mg/m2, about
twice the recommended dose in humans on a mg/m2 basis) caused reduced fetal weight.
LactationRisk Summary
It is not known whether IRESSA is excreted in human milk. Animal studies indicate the gefitinib
and its metabolites are present in rat milk at a concentration higher than those in maternal plasma.
Because of the potential for serious adverse reactions in nursing infants from IRESSA, advise
women to discontinue breast-feeding during treatment with IRESSA.
Data
Animal Data
Levels of gefitinib and its metabolites were 11-to-19-fold higher in milk than in blood, after oral
exposure of lactating rats to a dose of 5 mg/kg.
Females and Males of Reproductive PotentialContraception
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when administered
to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information].
Advise females of reproductive potential to use effective contraception during treatment with
IRESSA and for at least two weeks following completion of therapy.
Infertility
IRESSA may result in reduced fertility in females of reproductive potential [see Nonclinical
Toxicology (13.1) in the full Prescribing Information].
Pediatric UseThe safety and effectiveness of IRESSA in pediatric patients have not been established.
Geriatric UseOf the 823 patients enrolled in two randomized, active-controlled clinical trials 374 patients (45%)
were 65 years and older, and 93 patients (11%) were 75 years and older. No overall differences in
safety were observed between patients 65 years and older and those younger than 65 years. There
is insufficient information to assess for differences in efficacy between older and younger patients.
Renal ImpairmentLess than four percent (<4%) of gefitinib and its metabolites are excreted via the kidney. No clinical
studies were conducted with IRESSA in patients with severe renal impairment.
Hepatic ImpairmentThe systemic exposure of gefitinib was compared in patients with mild, moderate, or severe hepatic
impairment due to cirrhosis (according to Child-Pugh classification) and healthy subjects with
normal hepatic function (N=10/group). The mean systemic exposure (AUC0-�) was increased by
40% in patients with mild impairment, 263% in patients with moderate impairment, and 166% in
patients with severe hepatic impairment. Monitor adverse reactions when IRESSA is administered
to patients with moderate and severe hepatic impairment.
In a study comparing 13 patients with liver metastases and moderate hepatic impairment (addition
of CTC grade of baseline AST/SGOT, ALP, and bilirubin equals 3 to 5) to 14 patients with liver
metastases and normal hepatic function, the systemic exposure of gefitinib was similar [see
Warnings and Precautions (5.2) in the full Prescribing Information].
OVERDOSAGETwenty three patients were treated weekly with doses from 1500 mg to 3500 mg, and IRESSA
exposure did not increase with increasing dose. Adverse events were mostly mild to moderate in
severity, and were consistent with the known safety profile of IRESSA. In the event of suspected
overdose, interrupt IRESSA, institute supportive care, and observe until clinical stabilization. There
are no specific measures/treatments that should be taken following IRESSA overdosing.
PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labelling (Patient Information).
Interstitial Lung Disease: Advise patients to immediately contact their healthcare provider for new
onset or worsening of pulmonary symptoms such as dyspnea, cough and fever [see Warnings and
Precautions (5.1) in the full Prescribing Information].
Hepatotoxicity: Inform patients that they will need to undergo lab tests to monitor for liver function.
Advise patients to contact their healthcare provider to report any new symptoms indicating hepatic
toxicity [see Warnings and Precautions (5.2) in the full Prescribing Information].
Gastrointestinal Perforation: Advise patients that IRESSA can increase the risk of gastrointestinal
perforation and to seek immediate medical attention for severe abdominal pain [see Warnings and
Precautions (5.3) in the full Prescribing Information].
Severe or Persistent Diarrhea: Advise patients to contact their healthcare provider for severe or
persistent diarrhea [see Warnings and Precautions (5.4) in the full Prescribing Information].
Ocular Disorders including Keratitis: Advise patients promptly to contact their healthcare provider
if they develop eye symptoms, lacrimation, light sensitivity, blurred vision, eye pain, red eye or
changes in vision [see Warnings and Precautions (5.5) in the full Prescribing Information].
Bullous and Exfoliative Skin Disorders: Advise patients that IRESSA can increase the risk of bullous
and exfoliative skin disorders and to seek immediately medical attention for severe skin reactions
[see Warnings and Precautions (5.6) in the full Prescribing Information].
Embryo-fetal Toxicity: Advise pregnant women of the potential risk to a fetus or potential risk
for loss of the pregnancy [see Warnings and Precautions (5.7) and Use in Specific Populations
(8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective
contraception during treatment with IRESSA and for at least two weeks following completion of
therapy [see Use in Specific Populations (8.3) in the full Prescribing Information].
Lactation: Advise women to discontinue breast-feeding during treatment with IRESSA [see Use in
Specific Populations (8.2) in the full Prescribing Information].
IRESSA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2015
Manufactured for:
AstraZeneca Pharmaceuticals LP Wilmington, DE 19850
By: AstraZeneca UK Limited Macclesfield, Cheshire, England Product of Belgium
3148105 7/15 Issued: 07/15
Table 1 – Selected Adverse Drug Reactions Occurring with an Incidence Rate ≥5% and an Increase of >2% of IRESSA-treated Patients in Study 3 (cont'd.)
3133300/3148105_Iressa_ASCO Post.indd 3 8/11/15 9:54 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 87
Patient’s Corner
I’m Living a Full and Happy Life With Stage IV Breast CancerAfter 6 years on a new therapy, there is no evidence of cancer in my body.By Janet Klein, as told to Jo Cavallo
After coping with breast cancer for more than a decade, it is difficult
for me to put into words exactly how grateful I am to all the doctors, nurses,
and researchers whose efforts have kept me alive for all these years. And not just alive, but thriving.
A routine mammogram had dis-
covered two small tumors in my left breast, and a tissue biopsy confirmed stage I estrogen receptor–positive breast cancer. Even though the can-
cer was caught at such an early stage, and there was no evidence that it had spread, I wanted to have the best chance for a cure and opted for the most aggressive treatment my doctor offered: a contralateral prophylactic mastectomy followed by reconstruc-tive breast surgery.
After my surgery, my oncologist gave me the Oncotype DX genomic test to score, on a scale of 0 to 100, how likely it was that my cancer would recur, so we could evaluate whether adjuvant che-motherapy would be beneficial. I scored a 1, indicating that my risk of recurrence was low and that the potential harms of chemotherapy outweighed the benefits. And just to be extra cautious, because I was 49 years old and had not yet gone through menopause, my oncologist advised 5 years of tamoxifen. Unfortu-nately, 4 years later, my cancer recurred in the same breast and had spread to my iliac bone. Now, I was facing stage IV disease and possibly death.
The Clinical Trial That Is Saving My Life
My initial diagnosis didn’t come as a surprise to me. Both my mother and younger sister had breast cancer, and I always felt it was inevitable that I would be next in line. But when the cancer recurred, after all the indica-tions that it wouldn’t, I was devas-tated and concerned about having to go through additional surgery, plus chemotherapy and radiation therapy. Fortunately, before I had to make that decision, my oncologist told me about a new clinical trial investigating palbo-ciclib (Ibrance), a CDK4/6 inhibitor, in combination with letrozole.
Even though I did not test positive for the BRCA gene mutation, as an ex-tra precaution and to give myself the best shot at halting progression of my cancer, before I entered the trial, I de-cided to have my ovaries removed. After
continued on page 88
Janet Klein
TRIM: 8.125 X 10.875
IRESSA® (gefitinib) tablets for oral use
Brief Summary of Prescribing Information.
For complete prescribing information consult official package insert
INDICATIONS AND USAGEIRESSA is indicated for the first-line treatment of patients with metastatic non-small cell lung
cancer (NSCLC) whose tumors have epidermal growth factor receptor (EGFR) exon 19 deletions or
exon 21 (L858R) substitution mutations as detected by an FDA-approved test [see Clinical Studies
(14) in the full Prescribing Information].
Limitation of Use: Safety and efficacy of IRESSA have not been established in patients with metastatic
NSCLC whose tumors have EGFR mutations other than exon 19 deletions or exon 21 (L858R)
substitution mutations [see Clinical Studies (14) in the full Prescribing Information].
DOSAGE AND ADMINISTRATIONPatient Selection Select patients for the first-line treatment of metastatic NSCLC with IRESSA based on the
presence of EGFR exon 19 deletion or exon 21 (L858R) substitution mutations in their tumor
[see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information].
Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at:
http://www.fda.gov/CompanionDiagnostics.
Recommended DoseThe recommended dose of IRESSA is 250 mg orally once daily with or without food until disease
progression or unacceptable toxicity.
Do not take a missed dose within 12 hours of the next dose.
Administration to Patients Who Have Difficulty Swallowing SolidsImmerse IRESSA tablets in 4 to 8 ounces of water by dropping the tablet in water, and stir for
approximately 15 minutes. Immediately drink the liquid or administer through a naso-gastric tube.
Rinse the container with 4 to 8 ounces of water and immediately drink or administer through the
naso-gastric tube.
Dose ModificationDose Modifications for Adverse Drug Reactions
Withhold IRESSA (for up to 14 days) for any of the following:
�� Acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever) [see Warnings and
Precautions (5.1) in the full Prescribing Information]
�� NCI CTCAE Grade 2 or higher in ALT and/or AST elevations [see Warnings and Precautions (5.2)
in the full Prescribing Information]
�� NCI CTCAE Grade 3 or higher diarrhea [see Warnings and Precautions (5.4) in the full Prescribing
Information]
�� Signs and symptoms of severe or worsening ocular disorders including keratitis [see Warnings
and Precautions (5.5) in the full Prescribing Information]
�� NCI CTCAE Grade 3 or higher skin reactions [see Warnings and Precautions (5.6) in the full
Prescribing Information]
Resume treatment with IRESSA when the adverse reaction fully resolves or improves to NCI CTCAE
Grade 1.
Permanently discontinue IRESSA for:
�� Confirmed interstitial lung disease (ILD) [see Warnings and Precautions (5.1) in the full
Prescribing Information]
�� Severe hepatic impairment [see Warnings and Precautions (5.2) in the full Prescribing
Information]
�� Gastrointestinal perforation [see Warnings and Precautions (5.3) in the full Prescribing
Information]
�� Persistent ulcerative keratitis [see Warnings and Precautions (5.5) in the full Prescribing
Information]
Dose Modifications for Drug Interactions
Strong CYP3A4 Inducers
Increase IRESSA to 500 mg daily in the absence of severe adverse drug reaction, and resume
IRESSA at 250 mg seven days after discontinuation of the strong CYP3A4 inducer [see Drug
Interactions (7) and Clinical Pharmacology (12.3) in the full Prescribing Information].
CONTRAINDICATIONSNone.
WARNINGS AND PRECAUTIONSInterstitial Lung Disease (ILD)ILD or ILD-like adverse drug reactions (e.g., lung infiltration, pneumonitis, acute respiratory
distress syndrome, or pulmonary fibrosis) occurred in 1.3% of the 2462 patients who received
IRESSA across clinical trials; of these, 0.7% were Grade 3 or higher and 3 cases were fatal.
Withhold IRESSA and promptly investigate for ILD in any patient who presents with worsening
of respiratory symptoms such as dyspnea, cough and fever. Permanently discontinue IRESSA if
ILD is confirmed [see Dosage and Administration (2.4) and Adverse Reactions (6.1) in the full
Prescribing Information].
HepatotoxicityIn patients who received IRESSA across clinical trials, 11.4% of patients had increased alanine
aminotransferase (ALT), 7.9% of patients had increased aspartate aminotransferase (AST), and
2.7% of patients had increased bilirubin. Grade 3 or higher liver test abnormalities occurred in
5.1% (ALT), 3.0% (AST), and 0.7% (bilirubin) of patients. The incidence of fatal hepatotoxicity
was 0.04%.
Obtain periodic liver function testing. Withhold IRESSA in patients with worsening liver function and
discontinue in patients with severe hepatic impairment [see Dosage and Administration (2.4), Adverse
Reactions (6.1), and Use in Specific Populations (8.7) in the full Prescribing Information].
Gastrointestinal PerforationGastrointestinal perforation occurred in three (0.1%) of the 2462 IRESSA-treated patients across
clinical trials [see Adverse Reactions (6.1) in the full Prescribing Information]. Permanently
discontinue IRESSA in patients who develop gastrointestinal perforation [see Dosage and
Administration (2.4) in the full Prescribing Information].
Severe or Persistent DiarrheaGrade 3 or 4 diarrhea occurred in 3% of 2462 IRESSA-treated patients across clinical trials.
Withhold IRESSA for severe or persistent (up to 14 days) diarrhea [see Dosage and Administration
(2.4) and Adverse Reactions (6.1) in the full Prescribing Information].
Ocular Disorders including KeratitisOcular disorders [keratitis (0.1%), corneal erosion and aberrant eyelash growth (0.2%), conjunctivitis,
blephritis and dry eye (6.7%)] occurred in the 2462 IRESSA-treated patients across clinical trials. The
incidence of Grade 3 ocular disorders was 0.1% [see Adverse Reactions (6.1) in the full Prescribing
Information]. Interrupt or discontinue IRESSA for severe, or worsening ocular disorders [see Dosage
and Administration (2.4) in the full Prescribing Information].
Bullous and Exfoliative Skin DisordersBullous conditions including toxic epidermal necrolysis, Stevens Johnson syndrome and erythema
multiforme have been reported from treatment with IRESSA. Erythema multiforme and dermatitis
bullous have been reported in two patients (0.08%) across NSCLC trials (Study 2, Study 3 and
Study 4). IRESSA treatment should be interrupted or discontinued if the patient develops severe
bullous, blistering or exfoliating conditions.
Embryo-fetal ToxicityBased on its mechanism of action and data from animal reproduction studies IRESSA can cause fetal
harm when administered to a pregnant woman. In animal reproductive studies, oral administration
of gefitinib from organogenesis through weaning resulted in fetotoxicity and neonatal death at
doses below the recommended human dose. Advise pregnant women of the potential risk to a
fetus. Advise females of reproductive potential to use effective contraception during treatment with
IRESSA and for at least two weeks following completion of therapy [see Use in Specific Populations
(8.1, 8.3) in the full Prescribing Information].
ADVERSE REACTIONS The following adverse drug reactions are discussed in more detail in other sections of the labeling:
�� Interstitial Lung Disease [see Warnings and Precautions (5.1) in the full Prescribing Information]
�� Hepatotoxicity [see Warnings and Precautions (5.2) in the full Prescribing Information]
�� Gastrointestinal Perforation [see Warnings and Precautions (5.3) in the full Prescribing
Information]
�� Severe or Persistent Diarrhea [see Warnings and Precautions (5.4) in the full Prescribing
Information]
�� Ocular Disorders including Keratitis [see Warnings and Precautions (5.5) in the full Prescribing
Information]
�� Bullous and Exfoliative Skin Disorders [see Warning and Precautions (5.6) in the full Prescribing
Information]
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates
observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of
another drug and may not reflect the rates observed in practice.
The safety of IRESSA is based on the data from 2462 patients with NSCLC who received IRESSA
250 mg daily monotherapy in three randomized clinical studies (Study 2, Study 3 and Study 4).
Patients with a history of interstitial lung disease, drug-induced interstitial disease, radiation
pneumonitis that required steroid treatment or any evidence of clinically active interstitial lung
disease were excluded from these studies.
Controlled Studies:
Study 2 was a randomized, multicenter, open-label trial in which 1217 patients were randomized to
receive first-line treatment for metastatic NSCLC; 607 patients received IRESSA 250 mg daily and
589 patients received carboplatin/paclitaxel. The median duration of treatment with IRESSA was 5.9
months. The study population characteristics were: median age 57 years, age less than 65 years
(73%), female (79%), Asian (100%), NSCLC adenocarcinoma histology (100%), never smoker
(94%), light ex-smoker (6%), ECOG PS 0 or 1 (90%).
Study 3 was a randomized, multicenter, double-blind, placebo-controlled trial in which 1692 patients
were randomized to receive second- or third-line treatment for metastatic NSCLC; of which 1126
patients received IRESSA 250 mg daily and 562 patients received placebo. The median duration of
treatment with IRESSA was 2.9 months. The study population characteristics were: median age
62 years, age less than 65 years (60%), female (33%), Caucasian (75%), Asian (21%), NSCLC
adenocarcinoma histology (48%), never smoker (22%), ECOG PS 0 or 1 (65%), PS 2 (29%), PS 3
(5%) and two or more prior therapies (51%).
Study 4 was a randomized, multicenter, open-label trial in which 1466 patients were randomized to
receive second-line treatment for metastatic NSCLC; 729 patients received IRESSA 250 mg daily and
715 patients received docetaxel. The median duration of treatment with IRESSA was 2.4 months. The
study population characteristics were: median age 61 years, age less than 65 years (61%), female
(36%), Caucasian (79%), Asian (21%), NSCLC adenocarcinoma histology (54%), never smoker
(20%), ECOG PS 0 or 1 (88%) and two or more prior therapies (16%).
The pooled safety database from the three randomized trials was used to evaluate for serious and
uncommon adverse drug reactions. Common adverse reactions were evaluated in Study 3. The
most frequent adverse reactions in Study 3 (incidence of >20% and greater than placebo) reported
in IRESSA-treated patients were skin reactions (47%) and diarrhea (29%). The most frequent fatal
adverse reactions in IRESSA-treated patients were respiratory failure (0.9%), pneumonia (0.8%),
and pulmonary embolism (0.5%).
Approximately 5% of IRESSA-treated patients and 2.3% of placebo-treated patients discontinued
treatment due to an adverse event. The most frequent adverse reactions that led to discontinuation in
patients treated with IRESSA were nausea (0.5%), vomiting (0.5%) and diarrhea (0.4%).
Table 1 – Selected Adverse Drug Reactions Occurring with an Incidence Rate ≥5% and an Increase of >2% of IRESSA-treated Patients in Study 3
Adverse Reaction
Percentage (%) of patientsIRESSA (N=1126) Placebo (N=562)
All Grades Grade 3 and 4 All Grades Grade 3 and 4Skin and subcutaneous tissue disordersSkin reactions1 47% 2% 17% 0.4%
Nail disorders2 5% 0.1% 0.7% 0%
Gastrointestinal disorders
Diarrhea3 29% 3% 10% 1%
Vomiting 14% 1.2% 10% 0.4%
Stomatitis4 7% 0.3% 4% 0.2%
Metabolism and nutrition disordersDecreased appetite 17% 2.3% 14% 2.0%
3133300/3148105_Iressa_ASCO Post.indd 2 8/11/15 9:54 PM
TRIM: 8.125 X 10.875
IRESSA® (gefitinib) tablets for oral use 2
Adverse Reaction
Percentage (%) of patientsIRESSA (N=1126) Placebo (N=562)
All Grades Grade 3 and 4 All Grades Grade 3 and 4Eye disordersConjunctivitis/blepharitis/dry eye5 6% 0% 3.2% 0%1 Includes Acne, Acne pustular, Dermatitis, Dermatitis acneiform, Dermatitis exfoliative, Drug eruption, Dry
skin, Erythema, Exfoliative rash, Folliculitis, Pruritus, Pruritus generalized, Rash, Rash erythematous, Rash
generalized, Rash macular, Rash maculo-papular, Rash papular, Rash pruritic, Rash pustular, Rash vesicular,
Skin exfoliation, Skin toxicity, Xeroderma2 Includes Ingrowing nail, Nail bed infection, Nail disorder, Nail infection, Onychoclasis, Onycholysis, Paronychia3 Includes Diarrhea, Feces soft, Frequent bowel movements4 Includes Aphthous stomatitis, Cheilitis, Glossodynia, Mouth ulceration, Mucosal inflammation, Oral mucosal
blistering, Stomatitis, Tongue disorder, Tongue ulceration5 Includes Blepharitis, Conjunctival hyperemia, Conjunctivitis, Dry eye, Eye irritation, Eye pruritus, Eye swelling,
Eyelid irritation, Eyelid edema, Eyelids pruritus
Table 2 – Treatment Emergent Laboratory Abnormalities Occurring More Frequently in IRESSA-Treated Patients in Study 3
Adverse Reaction
IRESSA PlaceboAll Grades
%Grade 3 and 4
%All Grades
%Grade 3 and 4
%Alanine aminotransferase increased1 38%2 2.4% 23%2 1.4%4
Aspartate aminotransferase increased1 40%3 2.0% 25%3 1.3%5
Proteinuria 35% 4.7% 31% 3.3%1 Patients were allowed to enter the clinical study with lab values of ALT or AST CTCAE grade 1 or 22 14% gefitinib patients and 10% placebo patients were CTC grade 1 or 2 ALT at baseline3 15% gefitinib patients and 12% placebo patients were CTC grade 1 or 2 AST at baseline4 0.2% of placebo patients were CTC grade 3 at baseline5 0.4% of placebo patients were CTC grade 3 at baseline
The following adverse reactions have been reported with IRESSA across NSCLC trials (Study 2,
Study 3 and Study 4) and are not listed elsewhere in Section 6: nausea (18%), asthenia (17%),
pyrexia (9%), alopecia (4.7%), hemorrhage (including epistaxis and hematuria) (4.3%), dry mouth
(2%), dehydration (1.8%), allergic reactions including angioedema and urticaria (1.1%), elevations
in blood creatinine (1.5%), and pancreatitis (0.1%).
Postmarketing ExperienceThe following adverse reactions have been identified during post-approval use of IRESSA. Because
these reactions are reported voluntarily from a population of uncertain size, it is not always possible
to reliably estimate their frequency or establish a causal relationship to drug exposure.
Renal and urinary disorders: cystitis, hemorrhagic cystitis
Skin and subcutaneous tissue disorders: cutaneous vasculitis
DRUG INTERACTIONSDrugs Affecting Gefitinib ExposureCYP3A4 Inducer
Drugs that are strong inducers of CYP3A4 increase the metabolism of gefitinib and decrease
gefitinib plasma concentrations. Increase IRESSA to 500 mg daily in patients receiving a strong
CYP3A4 inducer (e.g., rifampicin, phenytoin, or tricyclic antidepressant) and resume IRESSA at
250 mg 7 days after discontinuation of the strong inducer [see Dosage and Administration (2.4) and
Clinical Pharmacology (12.3) in the full Prescribing Information].
CYP3A4 Inhibitor
Drugs that are strong inhibitors of CYP3A4 (e.g., ketoconazole and itraconazole) decrease gefitinib
metabolism and increase gefitinib plasma concentrations. Monitor adverse reactions when
administering strong CYP3A4 inhibitors with IRESSA.
Drugs Affecting Gastric pH
Drugs that elevate gastric pH (e.g., proton pump inhibitors, histamine H2-receptor antagonists,
and antacids) may reduce plasma concentrations of gefitinib. Avoid concomitant use of IRESSA
with proton pump inhibitors, if possible. If treatment with a proton-pump inhibitor is required, take
IRESSA 12 hours after the last dose or 12 hours before the next dose of the proton-pump inhibitor.
Take IRESSA 6 hours after or 6 hours before an H2-receptor antagonist or an antacid [see Clinical
Pharmacology (12.3) in the full Prescribing Information].
Hemorrhage in Patients taking WarfarinInternational Normalized Ratio (INR) elevations and/or hemorrhage have been reported in some
patients taking warfarin while on IRESSA therapy. Patients taking warfarin should be monitored
regularly for changes in prothrombin time or INR.
USE IN SPECIFIC POPULATIONSPregnancyRisk Summary
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when
administered to a pregnant woman. In animal reproductive studies, oral administration of gefitinib
from organogenesis through weaning resulted in fetotoxicity and neonatal death at doses below the
recommended human dose (see Animal Data). Advise pregnant women of the potential hazard to a
fetus or potential risk for loss of the pregnancy.
The background risk of major birth defects and miscarriage for the indicated population is unknown;
however, the background risk in the U.S. general population of major birth defects is 2-4% and
miscarriage is 15-20% of clinically recognized pregnancies.
Data
Animal Data
A single dose study in rats showed that gefitinib crosses the placenta after an oral dose of
5 mg/kg (30 mg/m2, about 0.2 times the recommended human dose on a mg/m2 basis). When
pregnant rats were treated with 5 mg/kg from the beginning of organogenesis to the end of
weaning there was a reduction in the number of offspring born alive. This effect was more severe at
20 mg/kg (approximate the human clinical dose on a mg/m2 basis) and was accompanied by high
neonatal mortality soon after parturition. In rabbits, a dose of 20 mg/kg/day (240 mg/m2, about
twice the recommended dose in humans on a mg/m2 basis) caused reduced fetal weight.
LactationRisk Summary
It is not known whether IRESSA is excreted in human milk. Animal studies indicate the gefitinib
and its metabolites are present in rat milk at a concentration higher than those in maternal plasma.
Because of the potential for serious adverse reactions in nursing infants from IRESSA, advise
women to discontinue breast-feeding during treatment with IRESSA.
Data
Animal Data
Levels of gefitinib and its metabolites were 11-to-19-fold higher in milk than in blood, after oral
exposure of lactating rats to a dose of 5 mg/kg.
Females and Males of Reproductive PotentialContraception
Based on its mechanism of action and animal data, IRESSA can cause fetal harm when administered
to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information].
Advise females of reproductive potential to use effective contraception during treatment with
IRESSA and for at least two weeks following completion of therapy.
Infertility
IRESSA may result in reduced fertility in females of reproductive potential [see Nonclinical
Toxicology (13.1) in the full Prescribing Information].
Pediatric UseThe safety and effectiveness of IRESSA in pediatric patients have not been established.
Geriatric UseOf the 823 patients enrolled in two randomized, active-controlled clinical trials 374 patients (45%)
were 65 years and older, and 93 patients (11%) were 75 years and older. No overall differences in
safety were observed between patients 65 years and older and those younger than 65 years. There
is insufficient information to assess for differences in efficacy between older and younger patients.
Renal ImpairmentLess than four percent (<4%) of gefitinib and its metabolites are excreted via the kidney. No clinical
studies were conducted with IRESSA in patients with severe renal impairment.
Hepatic ImpairmentThe systemic exposure of gefitinib was compared in patients with mild, moderate, or severe hepatic
impairment due to cirrhosis (according to Child-Pugh classification) and healthy subjects with
normal hepatic function (N=10/group). The mean systemic exposure (AUC0-�) was increased by
40% in patients with mild impairment, 263% in patients with moderate impairment, and 166% in
patients with severe hepatic impairment. Monitor adverse reactions when IRESSA is administered
to patients with moderate and severe hepatic impairment.
In a study comparing 13 patients with liver metastases and moderate hepatic impairment (addition
of CTC grade of baseline AST/SGOT, ALP, and bilirubin equals 3 to 5) to 14 patients with liver
metastases and normal hepatic function, the systemic exposure of gefitinib was similar [see
Warnings and Precautions (5.2) in the full Prescribing Information].
OVERDOSAGETwenty three patients were treated weekly with doses from 1500 mg to 3500 mg, and IRESSA
exposure did not increase with increasing dose. Adverse events were mostly mild to moderate in
severity, and were consistent with the known safety profile of IRESSA. In the event of suspected
overdose, interrupt IRESSA, institute supportive care, and observe until clinical stabilization. There
are no specific measures/treatments that should be taken following IRESSA overdosing.
PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labelling (Patient Information).
Interstitial Lung Disease: Advise patients to immediately contact their healthcare provider for new
onset or worsening of pulmonary symptoms such as dyspnea, cough and fever [see Warnings and
Precautions (5.1) in the full Prescribing Information].
Hepatotoxicity: Inform patients that they will need to undergo lab tests to monitor for liver function.
Advise patients to contact their healthcare provider to report any new symptoms indicating hepatic
toxicity [see Warnings and Precautions (5.2) in the full Prescribing Information].
Gastrointestinal Perforation: Advise patients that IRESSA can increase the risk of gastrointestinal
perforation and to seek immediate medical attention for severe abdominal pain [see Warnings and
Precautions (5.3) in the full Prescribing Information].
Severe or Persistent Diarrhea: Advise patients to contact their healthcare provider for severe or
persistent diarrhea [see Warnings and Precautions (5.4) in the full Prescribing Information].
Ocular Disorders including Keratitis: Advise patients promptly to contact their healthcare provider
if they develop eye symptoms, lacrimation, light sensitivity, blurred vision, eye pain, red eye or
changes in vision [see Warnings and Precautions (5.5) in the full Prescribing Information].
Bullous and Exfoliative Skin Disorders: Advise patients that IRESSA can increase the risk of bullous
and exfoliative skin disorders and to seek immediately medical attention for severe skin reactions
[see Warnings and Precautions (5.6) in the full Prescribing Information].
Embryo-fetal Toxicity: Advise pregnant women of the potential risk to a fetus or potential risk
for loss of the pregnancy [see Warnings and Precautions (5.7) and Use in Specific Populations
(8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective
contraception during treatment with IRESSA and for at least two weeks following completion of
therapy [see Use in Specific Populations (8.3) in the full Prescribing Information].
Lactation: Advise women to discontinue breast-feeding during treatment with IRESSA [see Use in
Specific Populations (8.2) in the full Prescribing Information].
IRESSA is a trademark of the AstraZeneca group of companies.
©AstraZeneca 2015
Manufactured for:
AstraZeneca Pharmaceuticals LP Wilmington, DE 19850
By: AstraZeneca UK Limited Macclesfield, Cheshire, England Product of Belgium
3148105 7/15 Issued: 07/15
Table 1 – Selected Adverse Drug Reactions Occurring with an Incidence Rate ≥5% and an Increase of >2% of IRESSA-treated Patients in Study 3 (cont'd.)
3133300/3148105_Iressa_ASCO Post.indd 3 8/11/15 9:54 PM

PAGE 88 The ASCO Post | SEPTEMBER 10, 2015
Technology in Oncology
just 9 months of being on the combina-tion therapy, all evidence of my cancer was gone. I have been in the trial for the past 6 years and remain cancer-free. My doctor has told me that as long as the treatment is effective for me, I will likely continue on it as maintenance therapy for the rest of my life.
Giving BackI know that with cancer there is no
guarantee remissions will last forever, especially when metastatic disease is involved, but I’m at peace. To be alive and living a full and satisfying life after a stage IV diagnosis is miraculous to me.
To express my appreciation to the oncology profession that has given me
so much and to contribute to impor-tant new research in breast cancer, I am participating in the Share the Jour-ney: Mind, Body, and Wellness After Breast Cancer clinical study (sharethe-journeyapp.org) [see more about the Share the Journey app above]. The study is investigating the symptoms women face after breast cancer treat-ment and what can be done to im-prove them. I’m happy to offer details
about my experience in the hope that they will provide researchers with the information needed to improve the lives of other women facing this dis-ease.
Being an Active Participant in Cancer Care
Having lived with breast cancer for so many years has also taught me the valuable lesson of how crucial it is to
be a full participant with my oncolo-gy team in my medical care, and it’s a message I repeat over and over to oth-er cancer survivors I meet. As won-derful and capable as oncologists and nurses are, they can’t fight a patient’s cancer by themselves; they need the patient’s help.
If patients think they can fight can-cer without being fully involved in their care, they are wrong. I have no doubt that the combination of having a wonderful oncology team providing me with the best medical guidance and being actively engaged in my treat-ment decisions has kept me alive for 10 years. And I fully expect it will keep me alive for many more. n
Ms. Klein lives in Sherman Oaks, California.
I have no doubt that the combination of having a wonderful oncology team providing me with the best medical guidance and being actively engaged in my treatment decisions has kept me alive for 10 years.
—Janet Klein
A Full and Happy Lifecontinued from page 87
‘Share the Journey’ Mobile App Aims to Understand the Different Experiences of Breast Cancer SurvivorsBy Jo Cavallo
In March 2015, Sage Bionetworks and Apple released “Share the Journey:
Mind, Body, and Wellness After Breast Cancer,” a patient-centered iPhone app that tracks five common consequences of breast cancer treatment, including fatigue, cognitive function, sleep distur-bances, mood changes, and a reduction in exercise performance. Users can also customize the app to include informa-tion about sexual dysfunction and other health concerns.
The Share the Journey app is an in-teractive research study that aims to understand why some breast cancer survivors recover more quickly than others, why their symptoms vary, and how their symptoms can be improved. The study is open to both healthy wom-en and breast cancer survivors between the ages of 18 and 80 who live in the United States.
Enrolling healthy women into the study is crucial to understanding the “human cost of breast cancer treat-ment,” said Patricia Ganz, MD, Direc-tor of Cancer Prevention and Control
Research at UCLA’s Jonsson Compre-hensive Cancer Center in Los Angeles and a collaborator on the development of the Share the Journey app.
“So often when I measure outcomes in women with breast cancer, the issue is how much is due to aging and how much is due to the cancer,” said Dr. Ganz. “The opportunity to have a large group of healthy women also complete the same instruments in a contempora-neous way may shed more light on how people either cope and improve their situation or who might be having more difficulty.”
The Share the Journey app uses an Apple open-source platform called Re-searchKit. Developed by the Jonsson Comprehensive Cancer Center, Penn Medicine, Dana-Farber Cancer Insti-tute, and Sage Bionetworks, who is the sponsor of the study, the app can be downloaded for free at the iTunes App Store (sharethejourneyapp.org). To en-sure participants’ confidentiality, each user is assigned a unique, random code for data collection by the researchers.
In addition to Dr. Ganz, Ann H. Partridge, MD, MPH, Clinical Direc-tor of the Breast Oncology Program at Dana-Farber Cancer Institute; Judy E. Garber, MD, MPH, Director, Cancer Risk and Prevention Clinic at Dana-Farber Cancer Institute; Kathryn H. Schmitz, PhD, MPH, Professor of Epi-demiology in Biostatistics and Epide-miology at the University of Pennsylva-nia Perelman School of Medicine; and Susan Love, MD, Medical Director of the Dr. Susan Love Research Founda-tion were also advisers on the develop-ment of the app.
Empowering SurvivorsShare the Journey is meant to pro-
vide women and their doctors, as well as the researchers monitoring the infor-mation, with a comprehensive, accurate accounting of participants’ daily health experiences. “This technology allows women to track the symptoms of their disease and what they think is making them feel better or worse,” said Andrew Trister, MD, PhD, MSE, Senior Physi-
cian at Sage Bionetworks and an inves-tigator of the research study. “The study is geared toward the idea that we will be able to turn what has previously been thought of as objective responses from people—also called anecdotes—about their disease into actionable, objective signals. Ultimately, we can use those signals to fuel further research that may lead to interventions to help people bet-ter live their lives after treatment. This study allows patients to see whether their health is getting better because they take on the role of being more of a manager of their symptoms and less of a voyeur.”
The app periodically asks women to answer questions about their cur-rent health and medical history; level of fatigue and mood; cognitive chang-es; and sleep and exercise patterns; it sends notices asking participants to complete certain tasks and surveys. The app then tracks and stores participants’ responses to the surveys; their journal entries about their symptoms; and data they input about various tasks they per-
Patricia Ganz, MD
Ann H. Partridge, MD, MPH
Susan Love, MD
Andrew Trister, MD, PhD, MSE

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 89
Technology in Oncology
formed, such as the amount of physical activity they completed in a day.
“This gives survivors the ability to know how they are doing in real time,” said Dr. Ganz. “We know from behav-ioral science research that very often people underestimate or overestimate how much they are walking or how much they are eating in a day. We built into this app the ability for users to monitor the number of steps they take and how many flights of stairs they climb daily. It also allows users to set goals for exercise.”
Having the ability to capture a vari-ety of information from patients, said Dr. Ganz, can provide clinicians with a strategy to personalize treatment. “About 25% to 30% of women have ongoing problems after breast cancer treatment ends, and that is the group we need to find better interventions for. We need to know who these women are and what their issues are. If we could iden-tify these women early on and/or dur-ing this post-treatment data collection
and learn the integrated characteristics that will cause them to have ongoing fa-tigue or cognitive difficulties, we might be able to personalize strategies to treat these patients upfront or follow up with early intervention,” Dr. Ganz explained.
Furthering Breast Cancer Research
The Share the Journey study will last about 6 months. Data captured in the app will then be processed and ana-lyzed by the study investigators and re-ported in scientific publications. Share the Journey participants can also opt to share their coded study data with re-searchers outside of this study.
“We think having data from this study shared broadly with the research community will help answer questions [about the after effects of breast cancer] from previous studies and help us gain insights from other research projects,” said Dr. Trister. “I also think this type of technology could lead to greater partici-pation in traditional clinical trials.”
Using Technology to Improve Care for Survivors
The Share the Journey app is not the first digital tool Dr. Ganz has helped de-velop. Several years ago, in response to the recommendations in the Institute of Medicine’s 2005 report, From Cancer Patient to Cancer Survivor: Lost in Tran-sition, which concluded that every can-cer survivor should have a coordinated survivorship care plan after treatment, Dr. Ganz collaborated with others to launch Journey Forward. Her collabo-rators included the National Coalition for Cancer Survivorship, Anthem, Inc., Genentech, and later the Oncology Nursing Society. The app was initially designed as a web-based care plan builder for professionals and has now expanded to include resource tools for patients and survivors in the form of a downloadable medical app.
The Journey Forward app allows sur-vivors and oncologists to create a post-treatment plan and includes a summary of the survivors’ cancer diagnosis and
treatment; a schedule for follow-up tests and surveillance; an area for psy-chosocial assessment; and information on managing ongoing symptoms and what to expect following treatment.
The app can be downloaded for free to both Apple iOS and Google Android mobile devices. The My Care Plan sec-tion of the app can also be downloaded in a printable PDF format. A Survivor-ship Library for Patients, where survi-vors can find information about their symptoms and care, is available on the Journey Forward website. To access the Survivorship Library for Patients and download the Journey Forward app, visit journeyforward.org.
“The information in the treatment and summary care plan in Journey For-ward can help survivors start a conver-sation with their health-care team,” said Dr. Ganz. “It really is a way to increase patient empowerment.” n
Disclosure: Dr Ganz is on the advisory board for InformedDNA. Dr. Trister reported no potential conflicts of interest.
Statement of Ownership, Management and Circulation (Requester Publication)1. Publication Title: The ASCO Post. 2. Publication Number: 2154-3283. 3. Filing Date: 8/31/15. 4. Issue Frequency: Bimonthly, except in January. 5. Number of Issues Pub-lished Annually: 23. 6. Annual Subscription Price (if any) $336.00. 7. Complete Mailing Address of Known Office of Publication: Harborside Press LLC, 37 Main St, Cold Spring Harbor, NY 11724-1423. Contact Person: John A. Gentile. Telephone: 631-935-7655. 8. Complete Mailing Address of Headquarters or General Business Office of Publisher: Harborside Press LLC, 37 Main Street, Cold Spring Harbor, NY 11724-1423. 9. Full Names and Complete Mailing Addresses of Publisher, Editor, and Managing Director: Publisher : John A. Gentile Jr, Harborside Press LLC, 37 Main Street, Cold Spring Harbor, NY 11724-1423. Editor: James O. Armitage, MD, Harborside Press LLC, 37 Main Street, Cold Spring Harbor, NY 11724-1423. Managing Editor: Cara Glynn, Harborside Press LLC, 37 Main St, Cold Spring Harbor, NY 11724-1423. 10. Owner: Full Name: Harborside Press LLC, John A. Gentile Jr (Principal), Anthony Cutrone (Principal), Conor Lynch (Principal). Complete Mailing Address: 37 Main Street, Cold Spring Harbor, NY 11724-1423. 11. Known Bondholders, Mortgagees, and Other Security Holders Owning or Holding 1 Percent or More of Total Amount of Bonds, Mortgages, or Other Se-curities: None. 12. Tax Status: N/A.13. Publication Title: The ASCO Post. 14. Issue Date for Circulation Data Below: September 10, 2015.15. Extent and Nature of Circulation - Average No. Copies Each Issue During Preceding 12 Months. (a) Total Number of Copies (Net press run): 29,774. (b) Legitimate Paid and/or Requested Distribution (By Mail and Outside the Mail) (1) Outside County Paid/Requested Mail Subscriptions stated on PS Form 3541: 12,559. (2) In-County Paid/Requested Mail Subscriptions stated on PS Form 3541: N/A. (3) Sales Through Dealers and Carriers, Street Vendors, Counter Sales, and other Paid or Requested Distribution Outside USPS®: N/A. (4) Requested Copies Distributed by Other Mail Classes Through the USPS (e.g. First-Class Mail®): 24. (c) Total Paid and/or Requested Circulation (sum of 15b (1), (2,), (3), and (4): 12,582. (d) Nonrequested Distribution (By Mail and Outside the Mail) (1) Outside County Nonrequested Copies Stated on PS Form 3541: 16,871. (2) In-County Nonrequested Copies Stated on PS Form 3541: N/A. (3) Nonrequested Copies Distributed Through the USPS by Other Classes of Mail: N/A. (4) Nonrequested Copies Distributed Outside the Mail: 143. (e) Total Nonrequested Distribution (Sum of 15d (1), (2), (3) and (4)) 17,014. (f) Total Distribution (Sum of 15c and e): 29,596. (g) Copies not Distributed: 178. (h) Total (Sum of 15f and g) 29,774. (i) Percent Paid and/or Requested Circulation: 43%15. Extent and Nature of Circulation - No. Copies of Single Issue Published Nearest to Filing Date. (a) Total Number of Copies (Net press run): 30,295. (b)Legitimate Paid and/or Requested Distribtuion (By Mail and Outside the Mail) (1) Outside County Paid/Requested Mail Subscriptions stated on PS Form 3541: 11,453. (2) In-County Paid/Requested Mail Subscriptions stated on PS Form 3541: N/A. (3) Sales Through Dealers and Carriers, Street Vendors, Counter Sales, and other Paid or Requested Distribution Outside USPS®: N/A. (4) Requested Copies Distributed by Other Mail Classes Through the USPS (e.g. First-Class Mail®): 23. (c) Total Paid and/or Requested Circulation (sum of 15b (1), (2,), (3), and (4): 11,476. (d) Nonrequested Distribution (By Mail and Outside the Mail) (1) Outside County Nonrequested Copies Stated on PS Form 3541: 18,598. (2) In-County Nonrequested Copies Stated on PS Form 3541: N/A. (3) Nonrequested Copies Distributed Through the USPS by Other Classes of Mail: N/A. (4) Nonrequested Copies Distributed Outside the Mail: 23. (e) Total Nonrequested Distribution (Sum of 15d (1), (2), (3) and (4)) 18,698 (f) Total Distribution (Sum of 15c and e): 30,174. g) Copies not Distributed: 121. (h) Total (Sum of 15f and g) 30,295. (i) Percent Paid and/or Requested Circulation: 38%16. Total circulation does not include electronic copies.17. Publication of Statement of Ownership for a Requester Publication is required and will be printed in the September 10, 2015 issue of this publication.18. Signature and Title of Editor, Publisher, Business Manager, or Owner: John A Gentile Jr, Publisher. Date: 8/31/15. I certify that all information furnished on this form is true and complete. I understand that anyone who furnishes false or misleading information on this form or who omits material or information requested on the form may be subject to criminal sanctions (including fines and imprisonment) and/or civil sanctions (including civil penalties).
PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PAID ADVERTISEMENT
32240_incjak_fa2_ascopst_advrt.indd All Pages 4/7/15 2:55 PM

PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PV
plus low-dose aspirin
• Poor compliance or tolerance to frequent phlebotomy• Symptomatic or progressive splenomegaly• High risk of thrombosis• Severe disease-related symptoms• Progressive myeloproliferation (leukocytosis or thrombocytosis)
Hydroxyurea (HU) or interferon-alpha as �rst-line cytoreductive therapy at any agea
Unmet need exists for a subset of patients not managed appropriately by current treatment
strategies (alone or in combination)
• Patients are intolerant of or resistant to HU
Hct, hematocrit.a All patients should be managed aggressively for their generic cardiovascular risk factors. HU should be used with caution in patients <40 years of age; busulfan may be considered in elderly patients (>70 years).
Phlebotomy to maintain Hct at <45%Polycythemia Vera: Are Some Patients at Increased Risk?
OverviewWhat Is Polycythemia Vera?Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) characterized by an overproduction of normal red blood cells, white blood cells and platelets that leads to an increased risk of thrombosis.1-4 Erythrocytosis (elevated red blood cell mass) is the most prominent clinical manifestation of PV, distinguishing it from other MPNs.5
PV may occur at any age but often presents later in life, with a median age at diagnosis of 60 years.6,7 Approximately 100,000 patients in the United States are living with PV.8
Clinical Presentation of PV Contributing to Its Diagnosis Janus kinases (JAKs) mediate cytokine signaling and growth factors.9,10 An important genetic discovery about a point mutation in the Janus kinase 2 (JAK2) gene has enhanced the understanding of PV.11
The speci� c JAK2V617F mutation is detected in >95% of patients with PV.5 Although the JAK2V617F mutation is the key driver of PV, an understanding of the clinical presentation of PV will help to facilitate a more accurate diagnosis.
PV is an elusive disease that may not be recognized for years. Diagnosis most frequently occurs by chance following a routine examination.12 Diagnosis may also occur after a thrombotic event or as a result of disease-related symptoms.12
The following important signs and symptoms warrant a prompt evaluation and suggest PV6,7:• Elevated hemoglobin or hematocrit levels• Thrombotic events • Splenomegaly (with or without thrombocytosis and/or leukocytosis)
Clinical Considerations in Managing PV Prognosis and Risk Factors In a large population-based study in more than 4,000 patients with PV, life expectancy was 36% lower than that of the general population.13
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.
Thrombotic and hemorrhagic complications are among the leading causes of morbidity and mortality associated with PV.14 Cancer and cardiovascular mortality are the frequent causes of deaths in PV.6,15
When assessing risks for morbidity and mortality in patients, consider the following3,14,16: • Elevated hematocrit levels • History of thrombosis • Advanced age (≥60 years) • Leukocytosis • Cardiovascular risk factors such as high cholesterol levels, hypertension, diabetes, obesity and smoking
According to data from a large, randomized, controlled clinical trial, the rate of death due to cardiovascular events or major thrombosis was four times higher in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained a hematocrit target of <45%2 (Figure 1).
Thrombosis, Splenomegaly and Other Disease-related Symptoms In PV, a wide spectrum of thrombotic manifestations exists that may occur before the disease is diagnosed.14 Palpable splenomegaly is an important physical � nding because increased spleen size is present in 30% to 40% of patients with PV.12 Additional signs and symptoms of PV, which may contribute to a substantial quality-of-life burden in patients with PV, include17: • Fatigue• Pruritus • Night sweats
CI = con� dence interval; Hct = hematocrit.
Figure 1. Kaplan-Meier curve for total cardiovascular events.
From The New England Journal of Medicine. Marchioli R, Finazzi, G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. Copyright ©2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Learn more about understanding the burden of Polycythemia Vera
at www.MPNConnect.com
The rate of death due to cardiovascular
events or major thrombosis was four times higher
in patients with elevated hematocrit levels of 45% to 50% compared with those who maintained
a hematocrit target of <45%.2
Hct, hematocrit; Hgb, hemoglobin.
Modi� ed from Barosi et al.19
Table 1. Assessment of hydroxyurea (HU) resistance and intolerance
HU Resistance HU Intolerance
After 12 weeks of HU at a total dose of ≥2 g/day or at the maximum tolerated dose, if <2 g/day
• Need for phlebotomy to maintain Hct level at <45% or
• Elevated platelet and white blood cell counts or
• <50% reduction in splenomegaly
At least 1 of the following:• Neutropenia (absolute
neutrophil count of <1.0 x 109/L)
• Platelet count of <100 x 109/L
• Hgb level of <10 g/dL• Leg ulcers or other
unacceptable nonhematologic HU-related toxicity
Clinical Need in PVTherapeutic approaches to PV focus on3,11:• Controlling and maintaining hematocrit levels at <45%• Treating complications of thrombosis and hemorrhage • Reducing thrombotic risk and minimizing the risk of leukogenic transformation • Managing splenomegaly and other disease-related symptoms
Phlebotomy is usually the starting point of treatment in patients with PV, in addition to therapy with low-dose aspirin.2,11 Low-dose aspirin has been shown to prevent both arterial and venous thrombotic complications in patients with PV.18
Cytoreductive therapy with hydroxyurea or interferon-alpha may also be helpful in patients who have dif� culty with phlebotomy, who have symptomatic or progressive splenomegaly or who experience severe symptoms.11 Although treatment with hydroxyurea may be tolerated by most patients, it is important to consider that approximately 25% of patients with PV developresistance to or intolerance of hydroxyurea (Table 1).19, 20
Despite current approaches, including phlebotomy, low-dose aspirin, interferon-alpha or cytoreductive therapy with hydroxyurea, some patients will not be able to gain and maintain hematocrit levels of <45%19,20 (Figure 2).
For some patients whose hematocrit levels remain elevated, and for those who continue to experience clinical signs and symptoms such as fatigue, pruritus, night sweats or splenomegaly, PV remains uncontrolled.11,20 Recently, standardized criteria for monitoring and assessing response in PV have been developed for clinical research. Evaluation of response includes such parameters as resolution of splenomegaly and other disease-related signs, hematocrit of <45%, blood count remission, absence of thrombotic events and bone marrow histology.21
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. CA Cancer J Clin. 2009;59:171-191. 2. Marchioli R, Finazzi G, Specchia G et al. N Engl J Med. 2013;368:22-33. 3. Tefferi A. Am J Hematol. 2013;88:507-516. 4. Spivak JL. Blood. 2002;100:4272-4290. 5. Spivak JL. Ann Intern Med. 2010;152:300-306. 6. Tefferi A, Rumi E, Finazzi G et al. Leukemia. 2013;27:1874-1881. 7. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995;123:656-664. 8. Data on � le. Incyte Corporation. 9. Verstovsek S. Postgrad Med. 2013;125:128-135.10. Staerk J, Kallin A, Demoulin JB et al. J Biol Chem. 2005;280:41893-41895.11. Barbui T, Barosi G, Birgegard G et al. J Clin Oncol. 2011;29:761-770.12. Passamonti F. Blood. 2012;120:275-284. 13. Hultcrantz M, Kristinsson SY, Andersson TML et al. J Clin Oncol.
2012;30:2995-3001.14. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program.
2012;2012:571-581. 15. Marchioli R, Finazzi G, Landol� R et al. J Clin Oncol. 2005;23:2224-2232. 16. De Stefano V, Za T, Rossi E et al. Haematologica. 2008;93:372-380.17. Mesa RA, Niblack J, Wadleigh M et al. Cancer. 2007;109:68-76.18. Landol� R, Marchioli R, Kutti J et al. N Engl J Med. 2004;350:114-124.19. Barosi G, Birgegard G, Finazzi G et al. Br J Haematol. 2010;148:961-963.20. Alvarez-Larrán A, Pereira A, Cervantes F et al. Blood. 2012;119:1363-1369.21. Barosi G, Mesa R, Finazzi G et al. Blood. 2013;121:4778-4781.
Figure 2. A practical management algorithm.11
©2015, Incyte Corporation. All rights reserved. EDU-1118i 03/15
PAID ADVERTISEMENT
32240_incjak_fa2_ascopst_advrt.indd All Pages 4/7/15 2:55 PM

PAGE 92 The ASCO Post | SEPTEMBER 10, 2015
Integrative Oncology
Coriolus versicolorBy Jyothirmai Gubili, MS Editor, Integrative Medicine Service, Memorial Sloan Kettering Cancer Center
Scientific name: Coriolus versicolorCommon names: Trametes versicolor, PSK, PSP, VPS, Turkey Tail, Yun Zhi, Kawaratake, Krestin
OverviewCoriolus versicolor, a mushroom
found throughout the world, belongs to the class Basidiomycetes. Mush-rooms are valued in traditional Ori-ental medicine for their health-pro-moting effects and are often combined
with herbs to build strength and stam-ina and to treat disease.
Coriolus is popularly known as a turkey tail mushroom because of its resemblance to the multicolored tail of a wild turkey. It is used not as food but as medicine and has been part of tradi-tional medicine as a tonic.
Polysaccharide K (PSK) and poly-saccharide peptide (PSP), compounds extracted from Coriolus versicolor, are thought to be responsible for its immu-nomodulatory and anticancer effects. Clinical data indicate that polysaccha-ride K increases the survival rates of patients with certain cancers (see below under “The Science”) without causing major adverse effects. It is commonly used in Asian countries to complement standard cancer treatments.
Versicolor polysaccharide (VPS), an-other extract obtained from the mush-room, is also under investigation for its potential anticancer effects.
Coriolus versicolor is available in health food stores and on the Inter-net in the form of capsules, extracts, or teas. Extracts, including polysac-charide peptide and versicolor poly-saccharide, are sold as dietary supple-ments in the United States.
The ScienceCoriolus versicolor acts as a biologic
response modifier. When used as an adjuvant, the proteoglycan polysac-charide K improved survival rates in patients with gastric1,2 and colorectal3-5
cancers. Polysaccharide peptide, used in conjunction with chemotherapy, has been shown to slow the progression of advanced non–small cell lung cancer.6
Coriolus extract by itself or in combination with other botanicals
exerted positive immunomodula-tory effects.7,8 However, studies of patients with breast cancer,9 hepato-cellular carcinoma,10 and leukemia11 produced mixed results.
In another study, versicolor poly-saccharide, a hot water extract of Coriolus, was found to enhance the development of large intestinal tu-mors in mice.12 More research is needed to determine the anticancer potential of Coriolus extracts.
Adverse EffectsDark-colored stools,13 darkening of the
fingernails,14 and low-grade hematologic and gastrointestinal toxicities have been reported following consumption of Cori-olus along with chemotherapeutic agents.3
For additional information, visit the “About Herbs” website at https://www.mskcc.org/cancer-care/integrative-medicine/herbs/coriolus-versicolor. n
Disclosure: Ms. Gubili reported no potential conflicts of interest.
The use of dietary supplements by patients with cancer has in-
creased significantly over the past 20 years despite insufficient evi-dence of safety and effectiveness. Finding reliable sources of infor-mation about dietary supplements can be daunting. Patients typically rely on family, friends, and the In-ternet, often receiving misleading information.
The ASCO Post’s Integrative On-cology series is intended to facilitate the availability of evidence-based information on integrative and complementary therapies com-monly used by patients with cancer. We chose Coriolus versicolor for this issue because of its increasing use by patients with cancer.
Compiled by Barrie R. Cassileth, PhD, and Jyothirmai Gubili, MS, Memorial Sloan Kettering Cancer Center. The free About Herbs website is managed by K. Simon Yeung, PharmD, MBA, LAc, Me-morial Sloan Kettering Cancer Center.
Visit the free About Herbs website at http://www.mskcc.org/
cancer-care/herb/maitake
Learn More About
Herbs, Botanicals, & Other Products
GUEST EDITOR
Integrative Oncology is guest edited by Barrie R. Cassileth, MS, PhD, Chief of the Integra-
tive Medicine Service and Laurance S. Rockefeller Chair in Integrative Medicine at Memorial Sloan Kettering Cancer Center, New York.
The Integrative Medicine Service at Memo-rial Sloan Kettering Cancer Center developed and maintains a free website—About Herbs (www.mskcc.org/aboutherbs)—that provides objective
and unbiased information about herbs, vitamins, minerals, and other dietary supplements, and unproved anticancer treatments. Each of the close to 300 and growing number of entries offer health-care professional and patient ver-sions, and entries are regularly updated with the latest research findings.
Barrie R. Cassileth, MS, PhD
Jyothirmai Gubili, MS

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 93
Integrative OncologyReferences
1. Nakazato H, Koike A, Saji S, et al: Efficacy of immunochemotherapy as ad-juvant treatment after curative resection of gastric cancer. Study Group of Immuno-chemotherapy with PSK for Gastric Can-cer. Lancet 343:1122-1126, 1994.
2. Niimoto M, Hattori T, Tamada R, et al: Postoperative adjuvant immunoche-motherapy with mitomycin C, futraful and PSK for gastric cancer: An analysis of data on 579 patients followed for five years. Jpn J Surg 18:681-686, 1988.
3. Ohwada S, Ikeya T, Yokomori T, et al: Adjuvant immunochemotherapy with oral Tegafur/Uracil plus PSK in patients with stage II or III colorectal cancer: A randomised controlled study. Br J Cancer 90:1003-1010, 2004.
4. Mitomi T, Tsuchiya S, Iijima N, et al: Randomized, controlled study on ad-juvant immunochemotherapy with PSK in curatively resected colorectal cancer. The Cooperative Study Group of Surgical Adjuvant Immunochemotherapy for Can-cer of Colon and Rectum (Kanagawa). Dis Colon Rectum 35:123-130, 1992.
5. Torisu M, Hayashi Y, Ishimitsu T, et al: Significant prolongation of disease-free period gained by oral polysaccharide K (PSK) administration after curative surgi-cal operation of colorectal cancer. Cancer Immunol Immunother 31:261-268, 1990.
6. Tsang KW, Lam CL, Yan C, et al: Cori-olus versicolor polysaccharide peptide slows progression of advanced non-small cell lung cancer. Respir Med 97:618-624, 2003.
7. Wong CK, Tse PS, Wong EL, et al:
Immunomodulatory effects of yun zhi and danshen capsules in health subjects—A randomized, double-blind, placebo-con-trolled, crossover study. Int Immunophar-macol 4:201-211, 2004.
8. Wong CK, Bao YX, Wong EL, et al: Immunomodulatory activities of Yunzhi and Danshen in post-treatment breast can-cer patients. Am J Chin Med 33:381-395, 2005.
9. Iino Y, Yokoe T, Maemura M, et al. Immunochemotherapies versus chemo-therapy as adjuvant treatment after cura-tive resection of operable breast cancer. Anticancer Res 15:2907-2911, 1995.
10. Suto T, Fukuda S, Moriya N, et al: Clinical study of biological response mod-ifiers as maintenance therapy for hepato-cellular carcinoma. Cancer Chemother
Pharmacol 33:S145-S148, 1994.11. Ohno R, Yamada K, Masaoka T, et
al: A randomized trial of chemoimmuno-therapy of acute nonlymphocytic leuke-mia in adults using a protein-bound poly-saccharide preparation. Cancer Immunol Immunother 18:149-154, 1984.
12. Toth B, Coles M, Lynch J: Effects of VPS extract of Coriolus versicolor on cancer of the large intestine using a serial sacrifice technique. In Vivo 20:341-346, 2006.
13. Shiu WCT, Leung TWT, Tao M: A clinical study of PSP on peripheral blood counts during chemotherapy. Phytothera-py Res 6:217-218, 1992.
14. Kidd PM: The use of mushroom glucans and proteoglycans in cancer treat-ment. Altern Med Rev 5:4-27, 2000.
To the One of the ManyBy Parvez Dara, MD, FACP, MBA, Toms River, New Jersey
(Using Shakespeare’s words to confront the plight of a Physician)
Give me that manThat is not passion’s slaveGive me that blanket that comforts and soothesFor in my heartThere was a fighting that would not let me sleep,Our indiscretion Sometimes serves us well.In those wakeful momentsWhen around a surgeon’s scalpel the blood congealsAnd time is spent to heal.What a piece of work is a manThe quintessence of dust.What is heWhose grief bears such emphasisSuch intricate complexity Of thought and action?How noble in reasonHow infinite in facultiesTo quell the cry of pain,How like an angelHow express and admirableTo drown the miseryAnd purge the disquietOf a thousand natural shocksThat flesh is heir to
And to take arms against a sea of troubleAnd by opposing, end them.Yet within the firmament of that reasonI could be bounded in a nutshell And count myself a king of infinite space, Were it not that I have bad dreams.These dreams, though this be madnessThere is method in’t.The vile mechanism feedsAnd eyes without feeling Feeling without sight,Cannot chart the course to reason.The spirit that I have seen, may be a devil And the devil hath power t’assume a pleasing shapeCleave the general ear with horrid speech, Make mad the guilty and appall the free.These clever studied orphans of untruthConfound the ignorant and amaze Indeed the very faculties of eyes and ears.They forget in their remarkable hypocrisy;This above all: to thine own self be true, And it must follow, as the night the day, Thou canst not then be false to any man.The power that exudes such tyrannyTis dangerous when the baser nature comes between The pass and fell incensed points of mighty opposites.
They know not what they doAs their power is fleetingAnd the unholy madness, a passing fancyA man may fish with the worm that hath eat of a king, And eat of the fish that hath fed of that wormThey thus find permanence in indignitywithin houses that last till doomsday.While chastising nobility, they cry andHumanity bleeds as one is lost to the many Eviscerating the noble cause of individualityThe chief good and market of this timeIs left wanting in art and science,Or somewhere in between.This warlike paragon of animalsAbuses me to damn me. As villainy though it have no tongue, Will speak with most miraculous organ!One day!Leaving in its vile dustThis beauty of the world,This noble of humansThis physician.In apprehension how like a god,I will wear him in my heart’s core, Ay, in my heart of heartsAs he grunts and sweats under the weary lifeBringing comfort through his discomfortTo the one of the many! n

Prevent bone complications longer
IMPORTANT SAFETY INFORMATION
Hypocalcemia• Pre-existing hypocalcemia must be corrected prior to
initiating therapy with XGEVA®. XGEVA® can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when XGEVA® is administered with other drugs that can also lower calcium levels. Advise patients to contact a healthcare professional for symptoms of hypocalcemia.
• An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.
Hypersensitivity• XGEVA® is contraindicated in patients with known
clinically signifi cant hypersensitivity to XGEVA®, including anaphylaxis that has been reported with use of XGEVA®. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically signifi cant allergic reaction occurs, initiate appropriate therapy and discontinue XGEVA® therapy permanently.
Drug Products with Same Active Ingredient• Patients receiving XGEVA® should not take Prolia® (denosumab).
Osteonecrosis of the Jaw• Osteonecrosis of the jaw (ONJ) can occur in patients
receiving XGEVA®, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure.
• Perform an oral examination and appropriate preventive dentistry prior to the initiation of XGEVA® and periodically during XGEVA® therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with XGEVA®.
• Patients who are suspected of having or who develop ONJ while on XGEVA® should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.
Atypical Subtrochanteric and Diaphyseal Femoral Fracture• Atypical femoral fracture has been reported with XGEVA®.
These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar fl are and are transverse or short oblique in orientation without evidence of comminution.
Median Time to First Bone Complication1
XGEVAVAV ®A®A 120 mg Q4W (n = 2,862)
zoledronic acid 4 mg Q4W (n = 2,861)
HR* = 0.83 (95% CI: 0.76-0.90) P < 0.001†
1 YEAR 2 YEARS
19.519.5months
2277..77months
Learn more at XGEVA.com
Data from a prespecifi ed integrated analysis of three international, phase 3, double-blind, double-dummy, active-controlled trials comparing XGEVA® with zoledronic acid for the prevention of bone complications in patients with bone metastases from solid tumors or multiple myeloma.1
*Hazard ratio (HR) is defi ned as the increase or decrease in likelihood of an event of interest (in this case a bone complication) for one group relative to that in a comparator group. †P value for superiority.
In a prespecifi ed integrated analysis of 3 pivotal trials (N = 5,723),XGEVA® was proven to delay the median time to fi rst bone complication by
XGEVA® is a convenient 120 mg subcutaneous injection administered onceevery 4 weeks.2
XGEVA® is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors. XGEVA® is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.
• RANK Ligand (RANKL) is produced by bone cells in the skeleton and is a key mediator of bone resorption4
• RANKL production is increased at sites of bone metastases, and stimulates osteoclasts to destroy bone4
• XGEVA® acts precisely to bind RANKL and inhibits osteoclast formation, function, and survival2
• Pre-existing hypocalcemia must be corrected prior to initiating therapy with XGEVA®2
©2014 Amgen Inc. All rights reserved. 04/14 80218-R1-V1 www.XGEVA.com
• Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During XGEVA® treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of XGEVA® therapy should be considered, pending a risk/benefi t assessment, on an individual basis.
Embryo-Fetal Toxicity• XGEVA® can cause fetal harm when administered to a
pregnant woman. Based on fi ndings in animals, XGEVA® is expected to result in adverse reproductive effects.
• Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of XGEVA®. Apprise the patient of the potential hazard to a fetus if XGEVA® is used during pregnancy or if the patient becomes pregnant while patients are exposed to XGEVA®.
Adverse Reactions• The most common adverse reactions in patients receiving
XGEVA® with bone metastasis from solid tumors were fatigue/asthenia, hypophosphatemia, and nausea. The most common serious adverse reaction was dyspnea. The most common adverse reactions resulting in discontinuation were osteonecrosis and hypocalcemia.
Please see brief summary of Prescribing Information on the following page.
REFERENCES: 1. Lipton A, Fizazi K, Stopeck AT, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48:3082-3092. 2. XGEVA® (denosumab) prescribing information, Amgen. 3. Brodowicz T, O’Byrne K, Manegold C. Bone matters in lung cancer. Ann Oncol. 2012;23:2215-2222. 4. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-1664.
months longer vs zoledronic acid1
months longer vs zoledronic acid 8.2
Bone complications, or skeletal-related events (SREs), are defi ned as radiation to bone, pathologic fracture, surgery to bone, and spinal cord compression.2,3
For patients with bone metastases from solid tumors
DOUS14CDNY3469_A_ONCAd_Tabloid_r11.indd 1 9/19/14 11:47 AM

Prevent bone complications longer
IMPORTANT SAFETY INFORMATION
Hypocalcemia• Pre-existing hypocalcemia must be corrected prior to
initiating therapy with XGEVA®. XGEVA® can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when XGEVA® is administered with other drugs that can also lower calcium levels. Advise patients to contact a healthcare professional for symptoms of hypocalcemia.
• An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.
Hypersensitivity• XGEVA® is contraindicated in patients with known
clinically signifi cant hypersensitivity to XGEVA®, including anaphylaxis that has been reported with use of XGEVA®. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically signifi cant allergic reaction occurs, initiate appropriate therapy and discontinue XGEVA® therapy permanently.
Drug Products with Same Active Ingredient• Patients receiving XGEVA® should not take Prolia® (denosumab).
Osteonecrosis of the Jaw• Osteonecrosis of the jaw (ONJ) can occur in patients
receiving XGEVA®, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure.
• Perform an oral examination and appropriate preventive dentistry prior to the initiation of XGEVA® and periodically during XGEVA® therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with XGEVA®.
• Patients who are suspected of having or who develop ONJ while on XGEVA® should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.
Atypical Subtrochanteric and Diaphyseal Femoral Fracture• Atypical femoral fracture has been reported with XGEVA®.
These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar fl are and are transverse or short oblique in orientation without evidence of comminution.
Median Time to First Bone Complication1
XGEVAVAV ®A®A 120 mg Q4W (n = 2,862)
zoledronic acid 4 mg Q4W (n = 2,861)
HR* = 0.83 (95% CI: 0.76-0.90) P < 0.001†
1 YEAR 2 YEARS
19.519.5months
2277..77months
Learn more at XGEVA.com
Data from a prespecifi ed integrated analysis of three international, phase 3, double-blind, double-dummy, active-controlled trials comparing XGEVA® with zoledronic acid for the prevention of bone complications in patients with bone metastases from solid tumors or multiple myeloma.1
*Hazard ratio (HR) is defi ned as the increase or decrease in likelihood of an event of interest (in this case a bone complication) for one group relative to that in a comparator group. †P value for superiority.
In a prespecifi ed integrated analysis of 3 pivotal trials (N = 5,723),XGEVA® was proven to delay the median time to fi rst bone complication by
XGEVA® is a convenient 120 mg subcutaneous injection administered onceevery 4 weeks.2
XGEVA® is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors. XGEVA® is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.
• RANK Ligand (RANKL) is produced by bone cells in the skeleton and is a key mediator of bone resorption4
• RANKL production is increased at sites of bone metastases, and stimulates osteoclasts to destroy bone4
• XGEVA® acts precisely to bind RANKL and inhibits osteoclast formation, function, and survival2
• Pre-existing hypocalcemia must be corrected prior to initiating therapy with XGEVA®2
©2014 Amgen Inc. All rights reserved. 04/14 80218-R1-V1 www.XGEVA.com
• Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During XGEVA® treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of XGEVA® therapy should be considered, pending a risk/benefi t assessment, on an individual basis.
Embryo-Fetal Toxicity• XGEVA® can cause fetal harm when administered to a
pregnant woman. Based on fi ndings in animals, XGEVA® is expected to result in adverse reproductive effects.
• Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of XGEVA®. Apprise the patient of the potential hazard to a fetus if XGEVA® is used during pregnancy or if the patient becomes pregnant while patients are exposed to XGEVA®.
Adverse Reactions• The most common adverse reactions in patients receiving
XGEVA® with bone metastasis from solid tumors were fatigue/asthenia, hypophosphatemia, and nausea. The most common serious adverse reaction was dyspnea. The most common adverse reactions resulting in discontinuation were osteonecrosis and hypocalcemia.
Please see brief summary of Prescribing Information on the following page.
REFERENCES: 1. Lipton A, Fizazi K, Stopeck AT, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48:3082-3092. 2. XGEVA® (denosumab) prescribing information, Amgen. 3. Brodowicz T, O’Byrne K, Manegold C. Bone matters in lung cancer. Ann Oncol. 2012;23:2215-2222. 4. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-1664.
months longer vs zoledronic acid1
months longer vs zoledronic acid 8.2
Bone complications, or skeletal-related events (SREs), are defi ned as radiation to bone, pathologic fracture, surgery to bone, and spinal cord compression.2,3
For patients with bone metastases from solid tumors
DOUS14CDNY3469_A_ONCAd_Tabloid_r11.indd 1 9/19/14 11:47 AM

Brief Summary: Consult package insert for complete Prescribing Information
INDICATIONS AND USAGE:Bone Metastasis from Solid Tumors. Xgeva is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors.Important Limitation of Use. Xgeva is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.DOSAGE AND ADMINISTRATION:Recommended Dosage. The recommended dose of Xgeva is 120 mg administered as a subcutaneous injection every 4 weeks in the upper arm, upper thigh, or abdomen. Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia.Preparation and Administration. Visually inspect Xgeva for particulate matter and discoloration prior to administration. Xgeva is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter. Prior to administration, Xgeva may be removed from the refrigerator and brought to room temperature (up to 25°C/77°F) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm Xgeva in any other way. Use a 27-gauge needle to withdraw and inject the entire contents of the vial. Do not re-enter the vial. Discard vial after single-use or entry.CONTRAINDICATIONS: Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with Xgeva.Hypersensitivity. Xgeva is contraindicated in patients with known clinically significant hypersensitivity to Xgeva.WARNINGS AND PRECAUTIONS: Drug Products with Same Active Ingredient. Xgeva includes the same active ingredient (denosumab) found in Prolia. Patients receiving Xgeva should not take Prolia.Hypersensitivity. Clinically significant hypersensitivity including anaphylaxis has been reported with use of Xgeva. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue Xgeva therapy permanently.Hypocalcemia. Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct pre-existing hypocalcemia prior to Xgeva treatment. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when Xgeva is administered with other drugs that can also lower calcium levels. In the postmarketing setting, severe symptomatic hypocalcemia has been reported. Advise patients to contact a healthcare professional for symptoms of hypocalcemia. An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.Osteonecrosis of the Jaw. Osteonecrosis of the jaw (ONJ) can occur in patients receiving Xgeva, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials, in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure (see Adverse Reactions). Seventy-nine percent of patients with ONJ had a history of tooth extraction, poor oral hygiene, or use of a dental appliance as a predisposing factor. Perform an oral examination and appropriate preventive dentistry prior to the initiation of Xgeva and periodically during Xgeva therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with Xgeva. Patients who are suspected of having or who develop ONJ while on Xgeva should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.Atypical Subtrochanteric and Diaphyseal Femoral Fracture. Atypical femoral fracture has been reported with Xgeva. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During Xgeva treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of Xgeva therapy should be considered, pending a risk/benefit assessment, on an individual basis.EMBRYO-FETAL TOXICITY: Xgeva can cause fetal harm when administered to a pregnant woman. Based on findings in animals, Xgeva is expected to result in adverse reproductive effects. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent peripheral lymph nodes, abnormal bone growth, and decreased neonatal growth (see Use in Specific Populations). Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after with the last dose of Xgeva. Apprise the patient of the potential hazard to a fetus if Xgeva is used during pregnancy or if the patient becomes pregnant while patients are exposed to Xgeva. Advise patients to contact their healthcare provider if they become pregnant or a pregnancy is suspected during this time.ADVERSE REACTIONS: The following adverse reactions are discussed below and elsewhere in the labeling:• Hypocalcemia• Osteonecrosis of the JawThe most common adverse reactions in patients receiving Xgeva (per-patient incidence greater than or equal to 25%) were fatigue/asthenia, hypophosphatemia, and nausea (see Table 1). The most common serious adverse reaction in patients receiving Xgeva was dyspnea. The most common adverse reactions resulting in discontinuation of Xgeva were osteonecrosis and hypocalcemia.Clinical Trials Experience. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice. The safety of Xgeva was evaluated in three randomized, double-blind, double-dummy trials in which a total of 2841 patients with bone metastasis from prostate cancer, breast cancer, or other solid tumors, or lytic bony lesions from multiple myeloma received at least one dose of Xgeva. In Trials 1, 2, and 3, patients were randomized to receive either 120 mg of Xgeva every 4 weeks as a subcutaneous injection or 4 mg (dose adjusted for reduced renal function) of zoledronic acid every 4 weeks by intravenous (IV) infusion. Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the
jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required. The median duration of exposure to Xgeva was 12 months (range: 0.1 – 41) and median duration on-study was 13 months (range: 0.1 – 41). Of patients who received Xgeva, 46% were female. Eighty-five percent were White, 5% Hispanic/Latino, 6% Asian, and 3% Black. The median age was 63 years (range: 18 – 93). Seventy-five percent of patients who received Xgeva received concomitant chemotherapy.Table 1. Per-patient Incidence of Selecteda Adverse Reactions of Any Severity (Trials 1, 2, and 3)
a Adverse reactions reported in at least 10% of patients receiving Xgeva in Trials 1, 2, and 3, and meeting one of the following criteria:
• At least 1% greater incidence in Xgeva-treated patients, or • Between-group difference (either direction) of less than 1% and more than
5% greater incidence in patients treated with zoledronic acid compared to placebo (US Prescribing Information for zoledronic acid)
b Laboratory-derived and below the central laboratory lower limit of normal [8.3 – 8.5 mg/dL (2.075 – 2.125 mmol/L) for calcium and 2.2 – 2.8 mg/dL (0.71 – 0.9 mmol/L) for phosphorus]
Severe Mineral/Electrolyte Abnormalities• Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less
than 1.75 mmol/L) occurred in 3.1% of patients treated with Xgeva and 1.3% of patients treated with zoledronic acid. Of patients who experienced severe hypocalcemia, 33% experienced 2 or more episodes of severe hypocalcemia and 16% experienced 3 or more episodes.
• Severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 15.4% of patients treated with Xgeva and 7.4% of patients treated with zoledronic acid.
Osteonecrosis of the JawIn the primary treatment phases of Trials 1, 2, and 3, ONJ was confirmed in 1.8% of patients in the Xgeva group (median exposure of 12.0 months; range 0.1 – 40.5) and 1.3% of patients in the zoledronic acid group. The trials in patients with breast (Trial 1) or prostate (Trial 3) cancer included an Xgeva open label extension treatment phase where patients were offered Xgeva 120 mg once every 4 weeks (median overall exposure of 14.9 months; range 0.1 – 67.2). The patient-year adjusted incidence of confirmed ONJ was 1.1% during the first year of treatment and 4.1% thereafter. The median time to ONJ was 20.6 months (range: 4 – 53).Atypical Subtrochanteric and Diaphyseal FractureAtypical femoral fracture has been reported with Xgeva.Postmarketing Experience. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.The following adverse reactions have been identified during post approval use of Xgeva:• Hypocalcemia: Severe symptomatic hypocalcemia, including fatal cases.• Hypersensitivity, including anaphylactic reactions.• Musculoskeletal pain, including severe musculoskeletal pain. Positive
rechallenge has been reported.Immunogenicity. As with all therapeutic proteins, there is potential for immunogenicity. Using an electrochemiluminescent bridging immunoassay, less than 1% (7/2758) of patients with osseous metastases treated with denosumab doses ranging from 30 – 180 mg every 4 weeks or every 12 weeks for up to 3 years tested positive for binding antibodies. No patient with positive binding antibodies tested positive for neutralizing antibodies as assessed using a chemiluminescent cell-based in vitro biological assay. There was no evidence of altered pharmacokinetic profile, toxicity profile, or clinical response associated with binding antibody development. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody (including neutralizing antibody) test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies to denosumab with the incidence of antibodies to other products may be misleading.DRUG INTERACTIONS: No formal drug-drug interaction trials have been conducted with Xgeva. In clinical trials in patients with breast cancer metastatic to bone, Xgeva was administered in combination with standard anticancer treatment. Serum denosumab concentrations at 1 and 3 months and reductions in the bone turnover marker uNTx/Cr (urinary N-terminal telopeptide corrected for creatinine) at 3 months were similar in patients with and without prior intravenous bisphosphonate therapy. There was no evidence that various anticancer treatments affected denosumab systemic exposure and pharmacodynamic effect. Serum denosumab concentrations at 1 and 3 months were not altered by concomitant chemotherapy and/or hormone therapy. The median reduction in uNTx/Cr from baseline to month 3 was similar between patients receiving concomitant chemotherapy and/or hormone therapy.USE IN SPECIFIC POPULATIONS:Pregnancy: Category D. Risk Summary: Xgeva can cause fetal harm when administered to a pregnant woman based on findings in animals. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent lymph nodes, abnormal bone growth and decreased neonatal growth. There are no adequate and well-controlled studies with Xgeva in pregnant women. Women should be advised not to become pregnant when taking Xgeva. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women who become pregnant during Xgeva treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. Patients or their physicians should call 1-800-77-AMGEN (1-800-772-6436) to enroll.Clinical Considerations: The effects of Xgeva are likely to be greater during the second and third trimesters of pregnancy. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. If the patient becomes pregnant during Xgeva therapy, consider the risks and benefits in continuing or discontinuing treatment with Xgeva.
Animal Data: The effects of denosumab on prenatal development have been studied in both cynomolgus monkeys and genetically engineered mice in which RANK ligand (RANKL) expression was turned off by gene removal (a “knockout mouse”). In cynomolgus monkeys dosed subcutaneously with denosumab throughout pregnancy at a pharmacologically active dose, there was increased fetal loss during gestation, stillbirths, and postnatal mortality. Other findings in offspring included absence of axillary, inguinal, mandibular, and mesenteric lymph nodes; abnormal bone growth, reduced bone strength, reduced hematopoiesis, dental dysplasia and tooth malalignment; and decreased neonatal growth. At birth out to one month of age, infants had measurable blood levels of denosumab (22-621% of maternal levels). Following a recovery period from birth out to 6 months of age, the effects on bone quality and strength returned to normal; there were no adverse effects on tooth eruption, though dental dysplasia was still apparent; axillary and inguinal lymph nodes remained absent, while mandibular and mesenteric lymph nodes were present, though small; and minimal to moderate mineralization in multiple tissues was seen in one recovery animal. There was no evidence of maternal harm prior to labor; adverse maternal effects occurred infrequently during labor. Maternal mammary gland development was normal. There was no fetal NOAEL (no observable adverse effect level) established for this study because only one dose of 50 mg/kg was evaluated. In RANKL knockout mice, absence of RANKL (the target of denosumab) also caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice showed altered maturation of the maternal mammary gland, leading to impaired lactation.Nursing Mothers. It is not known whether Xgeva is excreted into human milk. Measurable concentrations of denosumab were present in the maternal milk of cynomolgus monkeys up to 1 month after the last dose of denosumab (≤ 0.5% milk:serum ratio). Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Xgeva, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Maternal exposure to Xgeva during pregnancy may impair mammary gland development and lactation based on animal studies in pregnant mice lacking the RANK/RANKL signaling pathway that have shown altered maturation of the maternal mammary gland, leading to impaired lactation postpartum. However, in cynomolgus monkeys treated with denosumab throughout pregnancy, maternal mammary gland development was normal, with no impaired lactation. Mammary gland histopathology at 6 months of age was normal in female offspring exposed to denosumab in utero; however, development and lactation have not been fully evaluated.Pediatric Use. Xgeva is not recommended in pediatric patients. The safety and effectiveness of Xgeva in pediatric patients have not been established. Treatment with Xgeva may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Xgeva therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤ 10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates treated with denosumab at doses 5 and 25 times (10 and 50 mg/kg dose) higher than the recommended human dose of 120 mg administered once every 4 weeks, based on body weight (mg/kg), had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab. Cynomolgus monkeys exposed in utero to denosumab exhibited bone abnormalities, reduced hematopoiesis, tooth malalignment, decreased neonatal growth, and an absence of axillary, inguinal, mandibular, and mesenteric lymph nodes. Some bone abnormalities recovered once exposure was ceased following birth; however, axillary and inguinal lymph nodes remained absent 6 months post-birth.Geriatric Use. Of patients who received Xgeva in Trials 1, 2, and 3, 1260 (44%) were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Renal Impairment. Two clinical trials were conducted in patients without cancer and with varying degrees of renal function. In one study, patients (N=55) with varying degrees of renal function (ranging from normal through end-stage renal disease requiring dialysis) received a single 60 mg subcutaneous dose of denosumab. In a second study, patients (N=32) with severe renal dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis) were given two 120 mg subcutaneous doses of denosumab. In both studies, greater risk of developing hypocalcemia was observed with increasing renal impairment, and with inadequate/no calcium supplementation. Hypocalcemia was mild to moderate in severity in 96% of patients. Monitor calcium levels and, calcium and vitamin D intake.Females and Males of Reproductive Potential. ContraceptionFemales: Counsel patients on pregnancy planning and prevention. Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of Xgeva. Advise patients to contact their healthcare provider if they become pregnant, or a pregnancy is suspected, during treatment or within 5 months after the last dose of Xgeva. Males: The extent to which denosumab is present in seminal fluid is unknown. There is potential for fetal exposure to denosumab when a male treated with Xgeva has unprotected sexual intercourse with a pregnant partner. Advise males of this potential risk. OVERDOSAGE: There is no experience with overdosage of Xgeva. HOW SUPPLIED/STORAGE AND HANDLING: Xgeva is supplied in a single-use vial. Store Xgeva in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Once removed from the refrigerator, Xgeva must not be exposed to temperatures above 25°C/77°F or direct light and must be used within 14 days. Discard Xgeva if not used within the 14 days. Do not use Xgeva after the expiry date printed on the label. Protect Xgeva from direct light and heat. Avoid vigorous shaking of Xgeva.PATIENT COUNSELING INFORMATION:Advise patients to contact a healthcare professional for any of the following:• Symptoms of a hypersensitivity reaction, including rash, urticaria, pruritus,
lip swelling, shortness of breath, hypotension and respiratory tract edema• Symptoms of hypocalcemia, including paresthesias or muscle stiffness,
twitching, spasms, or cramps• Symptoms of ONJ, including pain, numbness, swelling of or drainage from
the jaw, mouth, or teeth• Persistent pain or slow healing of the mouth or jaw after dental surgery• Symptoms of atypical femoral fracture, including new or unusual thigh, hip,
or groin pain• Pregnancy or nursingAdvise patients of the need for:• Avoiding therapy with Xgeva if a serious allergic reaction occurred with prior
Xgeva or Prolia therapy• Proper oral hygiene and routine dental care• Informing their dentist that they are receiving Xgeva• Avoiding invasive dental procedures during treatment with Xgeva• The use of highly effective contraception during and for at least 5 months
after treatment with Xgeva for females of reproductive potentialAdvise patients that denosumab is also marketed as Prolia®. Patients should inform their healthcare provider if they are taking Prolia.
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.One Amgen Center DriveThousand Oaks, California 91320-1799©2010-2014 Amgen Inc. All rights reserved.Printed in USA.
Body SystemXgeva
n = 2841 %
Zoledronic Acid n = 2836
%GA STROINTESTINAL
Nausea Diarrhea
31 20
32 19
GE NERAL Fatigue/ Asthenia
45
46
IN VESTIGATIONS Hypocalcemiab
Hypophosphatemiab
18 32
9
20NE UROLOGICAL
Headache
13
14RE SPIRATORY
Dyspnea Cough
21 15
18 15
S:9.5”S
:13
”
DOUS14CDNY4736_XGEVA_Tabloid_BS_V10_8pt_r13.indd 1 8/11/14 11:56 AM
DOUS14CDNY3469_A_ONCAd_Tabloid_BS_r11.indd 1 9/19/14 11:45 AM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 97
In Memoriam
Breast Surgeon Carolyn Mary Kaelin, MD, MPH, FACS, Dies at 54
Carolyn Mary Kaelin, MD, MPH, FACS, died on July 28 at
the age of 54. A gifted and compassion-ate breast cancer surgeon, Dr. Kaelin was a surgical oncologist in the Wom-en’s Cancers Program at Dana-Farber Cancer Center and Director of the Breast Clinic at Brigham and Women’s Hospital.
Dr. Kaelin graduated from Smith College and Johns Hopkins School of Medicine. She earned her master’s degree from the Harvard School of Public Health. At 34, she became the Founding Director of the Comprehen-sive Breast Health Center at Brigham
and Women’s, at that time the young-est woman to hold a position of that distinction at a major Harvard teach-ing hospital. She quickly established herself as one of the nation’s premier breast cancer surgeons. Also active in research, Dr. Kaelin focused on how doctors and patients make medical de-cisions and on quality-of-life issues for breast cancer survivors, particularly the role of exercise. In 2001, Newsweek featured her as 1 of 15 “Women of the New Century.”
Patient and ProviderDr. Kaelin rode repeatedly in the
Pan-Mass Challenge, a 192-mile bi-cycle ride fundraiser for Dana-Far-ber. Shortly after a training ride in 2003, she noticed early signs of her own breast cancer.
A rare complication of breast can-cer therapy prevented Dr. Kaelin from returning to clinical practice. Instead, she redoubled her patient education and survivorship efforts, with an em-phasis on the underserved.
Dr. Kaelin initiated research on
the value of rowing for patients with postoperative lymphedema, with an assist from Olympian Holly Metcalf. She coauthored two award-winning books, Living Through Breast Cancer and The Breast Cancer Survivor’s Fit-ness Plan, and helped create an inno-
vative, exercise-centered breast can-cer recovery program for the YMCA.
She also established the Quality of Life Fund at Brigham and Wom-en’s to support breast cancer survi-vorship projects and launched the successful Knowledge, Strength, and Grace conference series for breast cancer patients and their families. In Aspen, Colorado, she cofounded the Quality of Life Cancer Fund along
with Barbara Berger. In 2010, Dr. Kaelin was diagnosed
with brain cancer unrelated to her pre-vious breast cancer. She had two brain surgeries and was a trailblazer on clini-cal trials testing medical treatments for brain cancer.
“We will miss her warmth, energy, intelligence, compassion, and humor,” said Eric Winer, MD, Director of the Breast Oncology Center in the Susan F. Smith Center. “She was uncompro-mising in her pursuit of truly outstand-ing care for each and every patient.”
Dr. Kaelin is survived by her hus-band William G. Kaelin, Jr, MD, and her children Kathryn Grace and Wil-liam (Tripp). n
Carolyn Mary Kaelin, MD, MPH, FACS
We will miss her warmth, energy, intelligence, compassion, and humor. She was uncompromising
in her pursuit of truly outstanding care for each and every patient.
—Eric Winer, MD
Chris Marshall, FRS, FMedSci, Dies at 66 of Colorectal Cancer
Chris Marshall, FRS, FMedSci, a rigorous scientist with a lasting leg-
acy of game-changing discoveries in can-cer research and generous support for his younger colleagues, has died at 66. The cause of death was colorectal cancer.
Professor Marshall was the Head of the Division for Cancer Biology at the Institute of Cancer Research in London and Professor of Cell Biology, and held a Cancer Research UK Gibb Life Fel-lowship. He established an internation-al reputation for his research into tumor cell signaling, with his major achieve-ments including the discovery of the NRAS oncogene and the finding that oncogenic Ras proteins drive cancer cell proliferation through overactiva-
tion of the MAP kinase-signaling path-way. His research paved the way for four new classes of cancer drugs to enter the clinic, two of which have been approved for use in patient treatment today.
Field-Changing DiscoveriesProfessor Marshall studied Natural
Sciences at Cambridge University, fol-lowed by a DPhil in cell biology at Ox-ford. His graduate studies were followed by postdoctoral work at the Imperial Cancer Research Fund (now Cancer Re-search UK) Lincoln’s Inn Fields Labora-tories, and Dana-Farber Cancer Institute.
In 1980, Professor Marshall moved to The Institute of Cancer Research and began studies to identify human cancer genes. This work, in collaboration with his colleague, the late Alan Hall, PhD, resulted in the identification of NRAS, a new human oncogene. Subsequent work from his laboratory showed that NRAS has important roles in leukemia, and oth-ers demonstrated the role of NRAS in melanoma.
Following the identification of NRAS, Dr. Marshall concentrated on studying how NRAS and the two other
RAS genes, HRAS and KRAS, act in cancer. His work in the field of cell sig-naling showed how RAS proteins are involved in transmitting signals from the outside of the cell to the cell nucle-us. This work laid the foundation for studies that showed the importance of the BRAF cancer gene in melanoma.
His laboratory is currently studying the cell-signaling mechanisms that al-low cancer cells to disseminate in the body. These studies are revealing how RHO family GTPase signaling path-ways determine different ways for tu-mor cells to migrate during invasion.
Innovator and MentorProfessor Marshall’s contributions
to science have been recognized by election to the European Molecular Bi-
ology Organisation, the Royal Society, and the Academy of Medical Sciences.
He was awarded the Sterling Med-al of the University of Pennsylvania (1997), the Novartis Medal of the Biochemical Society (1999), and the Buchanan Medal of the Royal Society (2008).
Both at the Institute of Cancer Research and in the wider scientific community, Professor Marshall was a highly valued colleague who went out of his way to help others. This willing-ness to mentor, support, and nurture talent has resulted in a cadre of suc-cessful scientists working in cancer research today.
His wife Lesley, his three children, and his four grandchildren survive Pro-fessor Marshall. n
Chris Marshall, FRS, FMedSci
Professor Marshall was a valued colleague who went out of his way to help others. This willingness to mentor,
support, and nurture talent has resulted in a cadre of successful scientists working in cancer research today.
Brief Summary: Consult package insert for complete Prescribing Information
INDICATIONS AND USAGE:Bone Metastasis from Solid Tumors. Xgeva is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors.Important Limitation of Use. Xgeva is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.DOSAGE AND ADMINISTRATION:Recommended Dosage. The recommended dose of Xgeva is 120 mg administered as a subcutaneous injection every 4 weeks in the upper arm, upper thigh, or abdomen. Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia.Preparation and Administration. Visually inspect Xgeva for particulate matter and discoloration prior to administration. Xgeva is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter. Prior to administration, Xgeva may be removed from the refrigerator and brought to room temperature (up to 25°C/77°F) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm Xgeva in any other way. Use a 27-gauge needle to withdraw and inject the entire contents of the vial. Do not re-enter the vial. Discard vial after single-use or entry.CONTRAINDICATIONS: Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with Xgeva.Hypersensitivity. Xgeva is contraindicated in patients with known clinically significant hypersensitivity to Xgeva.WARNINGS AND PRECAUTIONS: Drug Products with Same Active Ingredient. Xgeva includes the same active ingredient (denosumab) found in Prolia. Patients receiving Xgeva should not take Prolia.Hypersensitivity. Clinically significant hypersensitivity including anaphylaxis has been reported with use of Xgeva. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue Xgeva therapy permanently.Hypocalcemia. Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct pre-existing hypocalcemia prior to Xgeva treatment. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when Xgeva is administered with other drugs that can also lower calcium levels. In the postmarketing setting, severe symptomatic hypocalcemia has been reported. Advise patients to contact a healthcare professional for symptoms of hypocalcemia. An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.Osteonecrosis of the Jaw. Osteonecrosis of the jaw (ONJ) can occur in patients receiving Xgeva, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials, in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure (see Adverse Reactions). Seventy-nine percent of patients with ONJ had a history of tooth extraction, poor oral hygiene, or use of a dental appliance as a predisposing factor. Perform an oral examination and appropriate preventive dentistry prior to the initiation of Xgeva and periodically during Xgeva therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with Xgeva. Patients who are suspected of having or who develop ONJ while on Xgeva should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.Atypical Subtrochanteric and Diaphyseal Femoral Fracture. Atypical femoral fracture has been reported with Xgeva. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During Xgeva treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of Xgeva therapy should be considered, pending a risk/benefit assessment, on an individual basis.EMBRYO-FETAL TOXICITY: Xgeva can cause fetal harm when administered to a pregnant woman. Based on findings in animals, Xgeva is expected to result in adverse reproductive effects. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent peripheral lymph nodes, abnormal bone growth, and decreased neonatal growth (see Use in Specific Populations). Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after with the last dose of Xgeva. Apprise the patient of the potential hazard to a fetus if Xgeva is used during pregnancy or if the patient becomes pregnant while patients are exposed to Xgeva. Advise patients to contact their healthcare provider if they become pregnant or a pregnancy is suspected during this time.ADVERSE REACTIONS: The following adverse reactions are discussed below and elsewhere in the labeling:• Hypocalcemia• Osteonecrosis of the JawThe most common adverse reactions in patients receiving Xgeva (per-patient incidence greater than or equal to 25%) were fatigue/asthenia, hypophosphatemia, and nausea (see Table 1). The most common serious adverse reaction in patients receiving Xgeva was dyspnea. The most common adverse reactions resulting in discontinuation of Xgeva were osteonecrosis and hypocalcemia.Clinical Trials Experience. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice. The safety of Xgeva was evaluated in three randomized, double-blind, double-dummy trials in which a total of 2841 patients with bone metastasis from prostate cancer, breast cancer, or other solid tumors, or lytic bony lesions from multiple myeloma received at least one dose of Xgeva. In Trials 1, 2, and 3, patients were randomized to receive either 120 mg of Xgeva every 4 weeks as a subcutaneous injection or 4 mg (dose adjusted for reduced renal function) of zoledronic acid every 4 weeks by intravenous (IV) infusion. Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the
jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required. The median duration of exposure to Xgeva was 12 months (range: 0.1 – 41) and median duration on-study was 13 months (range: 0.1 – 41). Of patients who received Xgeva, 46% were female. Eighty-five percent were White, 5% Hispanic/Latino, 6% Asian, and 3% Black. The median age was 63 years (range: 18 – 93). Seventy-five percent of patients who received Xgeva received concomitant chemotherapy.Table 1. Per-patient Incidence of Selecteda Adverse Reactions of Any Severity (Trials 1, 2, and 3)
a Adverse reactions reported in at least 10% of patients receiving Xgeva in Trials 1, 2, and 3, and meeting one of the following criteria:
• At least 1% greater incidence in Xgeva-treated patients, or • Between-group difference (either direction) of less than 1% and more than
5% greater incidence in patients treated with zoledronic acid compared to placebo (US Prescribing Information for zoledronic acid)
b Laboratory-derived and below the central laboratory lower limit of normal [8.3 – 8.5 mg/dL (2.075 – 2.125 mmol/L) for calcium and 2.2 – 2.8 mg/dL (0.71 – 0.9 mmol/L) for phosphorus]
Severe Mineral/Electrolyte Abnormalities• Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less
than 1.75 mmol/L) occurred in 3.1% of patients treated with Xgeva and 1.3% of patients treated with zoledronic acid. Of patients who experienced severe hypocalcemia, 33% experienced 2 or more episodes of severe hypocalcemia and 16% experienced 3 or more episodes.
• Severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 15.4% of patients treated with Xgeva and 7.4% of patients treated with zoledronic acid.
Osteonecrosis of the JawIn the primary treatment phases of Trials 1, 2, and 3, ONJ was confirmed in 1.8% of patients in the Xgeva group (median exposure of 12.0 months; range 0.1 – 40.5) and 1.3% of patients in the zoledronic acid group. The trials in patients with breast (Trial 1) or prostate (Trial 3) cancer included an Xgeva open label extension treatment phase where patients were offered Xgeva 120 mg once every 4 weeks (median overall exposure of 14.9 months; range 0.1 – 67.2). The patient-year adjusted incidence of confirmed ONJ was 1.1% during the first year of treatment and 4.1% thereafter. The median time to ONJ was 20.6 months (range: 4 – 53).Atypical Subtrochanteric and Diaphyseal FractureAtypical femoral fracture has been reported with Xgeva.Postmarketing Experience. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.The following adverse reactions have been identified during post approval use of Xgeva:• Hypocalcemia: Severe symptomatic hypocalcemia, including fatal cases.• Hypersensitivity, including anaphylactic reactions.• Musculoskeletal pain, including severe musculoskeletal pain. Positive
rechallenge has been reported.Immunogenicity. As with all therapeutic proteins, there is potential for immunogenicity. Using an electrochemiluminescent bridging immunoassay, less than 1% (7/2758) of patients with osseous metastases treated with denosumab doses ranging from 30 – 180 mg every 4 weeks or every 12 weeks for up to 3 years tested positive for binding antibodies. No patient with positive binding antibodies tested positive for neutralizing antibodies as assessed using a chemiluminescent cell-based in vitro biological assay. There was no evidence of altered pharmacokinetic profile, toxicity profile, or clinical response associated with binding antibody development. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody (including neutralizing antibody) test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies to denosumab with the incidence of antibodies to other products may be misleading.DRUG INTERACTIONS: No formal drug-drug interaction trials have been conducted with Xgeva. In clinical trials in patients with breast cancer metastatic to bone, Xgeva was administered in combination with standard anticancer treatment. Serum denosumab concentrations at 1 and 3 months and reductions in the bone turnover marker uNTx/Cr (urinary N-terminal telopeptide corrected for creatinine) at 3 months were similar in patients with and without prior intravenous bisphosphonate therapy. There was no evidence that various anticancer treatments affected denosumab systemic exposure and pharmacodynamic effect. Serum denosumab concentrations at 1 and 3 months were not altered by concomitant chemotherapy and/or hormone therapy. The median reduction in uNTx/Cr from baseline to month 3 was similar between patients receiving concomitant chemotherapy and/or hormone therapy.USE IN SPECIFIC POPULATIONS:Pregnancy: Category D. Risk Summary: Xgeva can cause fetal harm when administered to a pregnant woman based on findings in animals. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent lymph nodes, abnormal bone growth and decreased neonatal growth. There are no adequate and well-controlled studies with Xgeva in pregnant women. Women should be advised not to become pregnant when taking Xgeva. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women who become pregnant during Xgeva treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. Patients or their physicians should call 1-800-77-AMGEN (1-800-772-6436) to enroll.Clinical Considerations: The effects of Xgeva are likely to be greater during the second and third trimesters of pregnancy. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. If the patient becomes pregnant during Xgeva therapy, consider the risks and benefits in continuing or discontinuing treatment with Xgeva.
Animal Data: The effects of denosumab on prenatal development have been studied in both cynomolgus monkeys and genetically engineered mice in which RANK ligand (RANKL) expression was turned off by gene removal (a “knockout mouse”). In cynomolgus monkeys dosed subcutaneously with denosumab throughout pregnancy at a pharmacologically active dose, there was increased fetal loss during gestation, stillbirths, and postnatal mortality. Other findings in offspring included absence of axillary, inguinal, mandibular, and mesenteric lymph nodes; abnormal bone growth, reduced bone strength, reduced hematopoiesis, dental dysplasia and tooth malalignment; and decreased neonatal growth. At birth out to one month of age, infants had measurable blood levels of denosumab (22-621% of maternal levels). Following a recovery period from birth out to 6 months of age, the effects on bone quality and strength returned to normal; there were no adverse effects on tooth eruption, though dental dysplasia was still apparent; axillary and inguinal lymph nodes remained absent, while mandibular and mesenteric lymph nodes were present, though small; and minimal to moderate mineralization in multiple tissues was seen in one recovery animal. There was no evidence of maternal harm prior to labor; adverse maternal effects occurred infrequently during labor. Maternal mammary gland development was normal. There was no fetal NOAEL (no observable adverse effect level) established for this study because only one dose of 50 mg/kg was evaluated. In RANKL knockout mice, absence of RANKL (the target of denosumab) also caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice showed altered maturation of the maternal mammary gland, leading to impaired lactation.Nursing Mothers. It is not known whether Xgeva is excreted into human milk. Measurable concentrations of denosumab were present in the maternal milk of cynomolgus monkeys up to 1 month after the last dose of denosumab (≤ 0.5% milk:serum ratio). Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Xgeva, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Maternal exposure to Xgeva during pregnancy may impair mammary gland development and lactation based on animal studies in pregnant mice lacking the RANK/RANKL signaling pathway that have shown altered maturation of the maternal mammary gland, leading to impaired lactation postpartum. However, in cynomolgus monkeys treated with denosumab throughout pregnancy, maternal mammary gland development was normal, with no impaired lactation. Mammary gland histopathology at 6 months of age was normal in female offspring exposed to denosumab in utero; however, development and lactation have not been fully evaluated.Pediatric Use. Xgeva is not recommended in pediatric patients. The safety and effectiveness of Xgeva in pediatric patients have not been established. Treatment with Xgeva may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Xgeva therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤ 10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates treated with denosumab at doses 5 and 25 times (10 and 50 mg/kg dose) higher than the recommended human dose of 120 mg administered once every 4 weeks, based on body weight (mg/kg), had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab. Cynomolgus monkeys exposed in utero to denosumab exhibited bone abnormalities, reduced hematopoiesis, tooth malalignment, decreased neonatal growth, and an absence of axillary, inguinal, mandibular, and mesenteric lymph nodes. Some bone abnormalities recovered once exposure was ceased following birth; however, axillary and inguinal lymph nodes remained absent 6 months post-birth.Geriatric Use. Of patients who received Xgeva in Trials 1, 2, and 3, 1260 (44%) were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Renal Impairment. Two clinical trials were conducted in patients without cancer and with varying degrees of renal function. In one study, patients (N=55) with varying degrees of renal function (ranging from normal through end-stage renal disease requiring dialysis) received a single 60 mg subcutaneous dose of denosumab. In a second study, patients (N=32) with severe renal dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis) were given two 120 mg subcutaneous doses of denosumab. In both studies, greater risk of developing hypocalcemia was observed with increasing renal impairment, and with inadequate/no calcium supplementation. Hypocalcemia was mild to moderate in severity in 96% of patients. Monitor calcium levels and, calcium and vitamin D intake.Females and Males of Reproductive Potential. ContraceptionFemales: Counsel patients on pregnancy planning and prevention. Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of Xgeva. Advise patients to contact their healthcare provider if they become pregnant, or a pregnancy is suspected, during treatment or within 5 months after the last dose of Xgeva. Males: The extent to which denosumab is present in seminal fluid is unknown. There is potential for fetal exposure to denosumab when a male treated with Xgeva has unprotected sexual intercourse with a pregnant partner. Advise males of this potential risk. OVERDOSAGE: There is no experience with overdosage of Xgeva. HOW SUPPLIED/STORAGE AND HANDLING: Xgeva is supplied in a single-use vial. Store Xgeva in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Once removed from the refrigerator, Xgeva must not be exposed to temperatures above 25°C/77°F or direct light and must be used within 14 days. Discard Xgeva if not used within the 14 days. Do not use Xgeva after the expiry date printed on the label. Protect Xgeva from direct light and heat. Avoid vigorous shaking of Xgeva.PATIENT COUNSELING INFORMATION:Advise patients to contact a healthcare professional for any of the following:• Symptoms of a hypersensitivity reaction, including rash, urticaria, pruritus,
lip swelling, shortness of breath, hypotension and respiratory tract edema• Symptoms of hypocalcemia, including paresthesias or muscle stiffness,
twitching, spasms, or cramps• Symptoms of ONJ, including pain, numbness, swelling of or drainage from
the jaw, mouth, or teeth• Persistent pain or slow healing of the mouth or jaw after dental surgery• Symptoms of atypical femoral fracture, including new or unusual thigh, hip,
or groin pain• Pregnancy or nursingAdvise patients of the need for:• Avoiding therapy with Xgeva if a serious allergic reaction occurred with prior
Xgeva or Prolia therapy• Proper oral hygiene and routine dental care• Informing their dentist that they are receiving Xgeva• Avoiding invasive dental procedures during treatment with Xgeva• The use of highly effective contraception during and for at least 5 months
after treatment with Xgeva for females of reproductive potentialAdvise patients that denosumab is also marketed as Prolia®. Patients should inform their healthcare provider if they are taking Prolia.
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.One Amgen Center DriveThousand Oaks, California 91320-1799©2010-2014 Amgen Inc. All rights reserved.Printed in USA.
Body SystemXgeva
n = 2841 %
Zoledronic Acid n = 2836
%GA STROINTESTINAL
Nausea Diarrhea
31 20
32 19
GE NERAL Fatigue/ Asthenia
45
46
IN VESTIGATIONS Hypocalcemiab
Hypophosphatemiab
18 32
9
20NE UROLOGICAL
Headache
13
14RE SPIRATORY
Dyspnea Cough
21 15
18 15
S:9.5”
S:1
3”
DOUS14CDNY4736_XGEVA_Tabloid_BS_V10_8pt_r13.indd 1 8/11/14 11:56 AM
DOUS14CDNY3469_A_ONCAd_Tabloid_BS_r11.indd 1 9/19/14 11:45 AM

PAGE 98 The ASCO Post | SEPTEMBER 10, 2015
Book Review
From Small-Town Mexico to Big Pharma, a Look at Opiates for Good and BadBy Ronald Piana
D espite growing awareness within the oncology community and the
emergence of the palliative care disci-pline, undertreatment of cancer pain in the United States remains a problematic issue. Study results have shown that pain in patients with cancer remains persis-tently undertreated, and the odds of un-dertreatment may be twice as high for minority patients. Poor communication skills between oncologists and their pa-tients may be at the heart of the issue in assessing and treating cancer pain.
Patient misperceptions about opi-oids—such as confusing dependence with addiction—contribute to the problem, as does the rise in illicit drug use. For instance, over the past decade, the highly publicized rise in prescrip-tion drug overdoses has led the Drug Enforcement Administration to initiate a rescheduling of hydrocodone com-bination products from schedule III to schedule II; this step dramatically increased the restrictions on doctors’ prescribing and dispensing practices for these necessary pain-relieving products.
In reaction to the recent reschedul-
ing of hydrocodone, many doctors who treat cancer pain have expressed con-cern that sensational coverage of illicit sales of opioid analgesics will lead to even tighter restrictions on pain medi-cations. Is our fear of illicit painkilling drugs overblown? Not according to award-winning journalist, Sam Qui-nones, who has just a penned a book called Dreamland: The True Tale of America’s Opiate Epidemic. With a great reporter’s narrative skill and the story-telling ability of a novelist, the author weaves together tales with hard data on pain medications, making it a compel-ling read for The ASCO Post audience.
A Revelatory AccountEpidemic is a scary word in medi-
cine, signifying an out-of-control spread of disease, and he boldly says that his book is a revelatory account of the corrosive threat facing America. The title of his book, Dreamland, is tak-en from a public swimming pool in the blue-collar town of Portsmouth, Ohio, a watery oasis in the heartland, one that over time became an idyllic gathering place for people of all stripes and eco-nomic backgrounds.
Then, according to Mr. Quinones, the prescription painkillers arrived, and the town declined. He uses one Matt Schoonover, an all-American boy, as the victim in his allegorical warning: Your town is next. “Across America, thousands of people like Matt Schoonover were dy-ing. Drug overdoses were killing more people every year than car accidents…. Now most of the fatal overdoses were from opiates: prescription painkillers or heroin,” writes Mr. Quinones.
However, you can’t build a nearly 400-page book on victimhood stories about good kids going wrong, falling into the trap of easy-to-access drugs, and bad guys peddling them. To that end, Mr. Quinones tries to balance the personal sagas of drug victims with enough history and facts to make his book interesting for readers looking for a deeper understanding of the drugs people use illicitly to get high and the patients with cancer who need drugs to relieve otherwise unendurable pain.
Pioneers in Pain ControlFor the readers of The ASCO Post,
there are several specific chapters. One, aptly called “The Pain,” gives a pretty good background of the long and diffi-cult history of pain control. He begins,
“Through most of the twentieth cen-tury, doctors treating the terminally ill faced attitudes that seemed medieval when it came to opiates.”
To drive home this arresting open-ing, he tends to drift into brief lapses of hyperbole and some very clunky writ-ing. As the author moves downstream in the chapter, the waters even out, as he gives an interesting background about the nascent years of hospice and the
early struggles to bring much needed awareness about unnecessary suffering.
Although the chapters devoted to the emergence of pain management should be the most interesting to the readers of The ASCO Post, they also present territory where the author seems un-steady. For instance, he describes how the World Health Organization’s pio-neering cancer chief, Jan Stjernswärd, MD, FRCP, changed pain management worldwide, creating the pain manage-ment ladder and finally making the dec-laration that freedom from pain was a universal human right.
He then segues into one of the true heroes of pain management, Kathleen Foley, MD, giving a fairly good account of her role in pain awareness and treat-ment. He ends his section on Dr. Foley mentioning that she argues that opi-ate treatment should not be confined to patients with cancer or postsurgical patients but should be for others with equally debilitating chronic pain. He seemed dangerously close to filling in the blanks for Dr. Foley, as if trying to make his point through her measured discussion on pain management.
A Closer Look at OxycodoneSince much of the brouhaha reported
in the lay press about fatal opioid over-doses has centered on oxycodone, Mr. Quinones’ next chapter put Purdue Pharma in his crosshairs. To his credit, he begins by giving a well-balanced dis-cussion of oxycodone’s history and effi-
cacy in pain management. Then he veers into what I would call slipshod nonsense that will drive the pain community mad. First, he never discusses the highly com-plex mechanisms that drive pain or the different kinds of pain, from somatic to neuropathic, nor does he talk about opi-oid receptors and the way these drugs actually assuage pain. Given he’s writing predominantly for a lay audience, that shortcoming may be forgiven.
What cannot be forgiven is his ap-parent ignorance on addiction. In an ef-fort to vilify oxycodone and the people who make and sell the drug, he launches into a half-baked theory about an adver-tising campaign that shadily promotes oxycodone as the “Holy Grail that had eluded researchers…a new time-release method that would lead to less addic-tion.” He continued, “OxyContin theo-retically parsed out oxycodone in a way that did not cause the intense highs and lows that cause addiction.”
The causes of psychological addiction to drugs are multifocal. Taking an opi-oid analgesic does not cause addiction or create an addictive personality. In ef-fect, by vilifying an inanimate substance, the author did to oxycodone what Peter Benchley did to the great white shark.
A Good StorytellerMr. Quinones is out of his depth
when he discusses pain, but he is a very good storyteller who has traveled the country gathering information about the scourge of illicit drug use. Howev-er, there is one message that he fails to deliver. Taking drugs for a recreational high comes with risk, sometimes lead-ing to death, but recreational drug us-ers make a choice to take these drugs. People with advanced cancer do not have the same choice; they need drugs to relieve their suffering. And as Russell Portenoy, MD, was quoted, “I believe in drugs. I think pharmaceuticals are a great gift to humankind.” n
Bookmark
Title: Dreamland: The True Tale of America’s Opiate Epidemic
Author: Sam Quinones
Publisher: Bloomsbury Press
Publication date: April 21, 2015
Price: $28.00; hardcover, 384 pages
Through most of the twentieth century, doctors treating the terminally ill faced attitudes that seemed medieval when it came to opiates.
—Sam Quinones

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 99
Announcements
ASCO and UICC Collaborate to Extend Reach of the Journal of Global Oncology
A SCO and the Union for Interna-tional Cancer Control (UICC)
announced their partnership to expand the reach of the Journal of Global Oncol-ogy ( JGO), a new open-access journal that will focus on cancer care, research, and care delivery issues unique to coun-
tries and settings with limited health-care resources.
It is estimated that today nearly two-thirds of all cancer deaths occur in low- and middle-income countries, and this is forecast to increase to 70% by 2030.1 While some journals publish
special issues and sections focused on global oncology, JGO will be the first peer- reviewed journal dedicated solely to this area of research.
The partnership between ASCO, which boasts more than 35,000 mem-bers around the world, and UICC,
whose membership represents more than 800 organizations in 155 coun-tries, will extend the reach of JGO to a broad international audience. ASCO and UICC will work together to solicit high-quality original research, secure funding, and promote JGO to their re-spective networks.
Through this collaboration, ASCO and UICC will meet a growing need for high-quality clinical cancer re-search in low- and middle-income countries by providing a peer-review platform focused on the challenges faced by researchers and care provid-ers in these countries.
“ASCO is committed to conquer-ing cancer through research, education, prevention, and delivery of high qual-ity patient care, and this aligns closely with UICC’s efforts to unite the cancer community to reduce the global cancer burden,” said David Kerr, MD, DSc, JGO’s founding Editor-in-Chief. “This partnership will amplify the impact of the Journal of Global Oncology in sup-port of both organizations’ missions and will give a voice to those clinicians who practice cancer medicine in chal-lenging conditions.”
JGO is funded through the Conquer Cancer Foundation of ASCO, with the support of the Doris Duke Charitable Foundation and Novartis Oncology. n
Reference1. International Agency for Research on
Cancer, GLOBOCAN, 2012.
David Kerr, MD, DSc
Like us on Facebook
facebook.com/TheASCOPost
The ASCO Post
Subscribe to THE ASCO POST
TODAY!To receive your complimentary print copy of THE ASCO POST,
go to www.ascopost.com/subscribe or call the Circulation Department at (631) 935-7651 to see if you qualify.
Get up-to-date information on:n Highly validated coverage of
cancer research & policy news
n Patient care & clinical practice issues
n Evidence-based research from peer-reviewed clinical journals
n Reports on major oncology meetings worldwide
n News from the National Institutes of Health, the National Cancer Institute, the US Food & Drug Administration and the US Congress

PAGE 100 The ASCO Post | SEPTEMBER 10, 2015
2015 Oncology Meetings 2015September
2015 Breast Cancer SymposiumSeptember 25-27 • San Francisco, CA For more information: http://breastcasym.org
European Cancer Congress (ECC 2015)September 25-29 • Vienna, Austria For more information: www.esmo.org/Conferences/European-Cancer-Congress-2015
17th Annual International Meeting of the Institute of Human VirologySeptember 27-30 • Baltimore, Maryland For more information: http://medschool.umaryland.edu/ihvmeeting/default.html
5th World Congress on Cancer TherapySeptember 28-30 • Atlanta, Georgia For more information: http://cancer.global-summit.com/americ
October
Advances in Cancer ImmunotherapyTM
October 2 • Nashville, Tennessee For more information: www.sitcancer.org/sitc-meetings/aci2015/tn
Institute for Clinical Immuno-Oncology (ICLIO) 1st Annual National ConferenceOctober 2 • Philadelphia, Pennsylvania For more information: http://accc-iclio.org/events/iclio-1st-annual-national-conference/
5th International Breast Cancer Prevention SymposiumOctober 2-3 • Le Gosier, Guadeloupe, French West Indies For more information: www.purdue.edu/breastcancer/
CAP ’15-The Pathologists’ MeetingTM (College of American Pathologists)October 4-7 • Nashville, Tennessee For more information: www.thepathologistsmeeting.org
American College of Surgeons Clinical CongressOctober 4-8 • Chicago, Illinois For more information: www.facs.org/meetings_events/future_congress/future
5th Annual Brain Tumor SymposiumOctober 5 • Philadelphia, Pennsylvania For more information: https://cme.jefferson.edu/content/5th-annual-brain-tumor-symposium
30th Anniversary Annual Critical Issues in Tumor Microenvironment: Angiogenesis, Metastasis, and Immunology October 5-8 • Cambridge, Massachusetts For more information: http://cmeregistration.hms.harvard.edu/events/30th-anniversary-annual-critical-issues-in-tumor-microenvironment-angiogenesis-metastasis-and-immuno/event-summary-567a0dd5664947b096f8a5fd33946e52.aspx
20th World Congress on Advances in Oncology and 18th International Symposium on Molecular Medicine October 8-10 • Athens, Greece For more information: www.spandidos-publications.com/pages/conference
Congress of the International Society of Pediatric OncologyOctober 8-11 • Cape Town, South Africa For more information: http://siop2015.kenes.com
Palliative Care in Oncology SymposiumOctober 9-10 • Boston, MA For more information: http://pallonc.org
Genetics and Genomics of Gynecologic CancersOctober 15-16 • New York, New York For more information: https://www.mskcc.org/event/genetics-and-genomics-gynecologic
National Comprehensive Cancer Network (NCCN) 10th Annual Congress: Hematologic Malignancies™October 16-17 • San Francisco, California For more information: www.nccn.org/professionals/meetings/hematological/default.aspx
ASTRO’s 57th Annual MeetingOctober 18-21 • San Antonio, Texas For more information: www.astro.org/Meetings-and-Events/2015-Annual-Meeting/Index.aspx
2015 International Cancer Education ConferenceOctober 21-23 • Tucson, Arizona For more information: http://2015.attendicec.org
ACCC 32nd National Oncology ConferenceOctober 21-24 • Portland, Oregon For more information: www.accc-cancer.org/meetings/calendar.asp
Lymphoma & Myeloma 2015: An International Congress on Hematologic MalignanciesOctober 22-24 • New York, New York For more information: http://www.imedex.com/lymphoma-myeloma-conference/index.asp
Cutaneous Oncology Symposium 2015October 23-24 • Fernandina Beach, FloridaFor more information: https://ce.mayo.edu/hematology-and-oncology/node/3306
4th Annual New Therapeutics in Oncology: The Road to Personalized MedicineOctober 23-25 • Los Angeles, CaliforniaFor more information: https://www.regonline.com/builder/site/?eventid=1727722
13th Annual West Coast Colorectal Cancer SymposiumOctober 23 • Seattle, Washington http://www.swedish.org/for-health-professionals/cme/conferences/colorectal-cancer-symposium
53rd Annual Meeting of the Japan Society of Clinical Oncology (JSCO)October 24-26 • Kyoto, Japan For more information: www.jsco.or.jp/english/index/page/id/73
ESGO 2015-International Meeting of the European Society of Gynaecological OncologyOctober 24-27 • Nice, France For more information: http://esgo2015.esgo.org
Modern Management of Urologic Cancers: A Multidisciplinary Approach (Memorial Sloan Kettering) October 29-31 • New York, New York For more information: http://www.themerzgroup.com/mskcc/mskcc-urologic-conference/
Lynn Sage Breast Cancer SymposiumOctober 29-November 1 • Chicago, Illinois For more information: www.lynnsagebreastcancer.org
continued on page 102

Learn more
GENB-24118 DCIS Print Ad_7-2_ASCO.indd 1 4/16/15 1:57 PM

PAGE 102 The ASCO Post | SEPTEMBER 10, 2015
2015 Oncology Meetings 2015Caring for the Caregivers XOctober 30 • Waltham, MA For more information: http://www.massmed.org Continuing-Education-and-Events/Event-Information/?code=CFC2015
NovemberNRCI Cancer ConferenceNovember 1-4 • Liverpool, United Kingdom For more information: http://conference.ncri.org.uk
3rd International Conference on Hematology & Blood Disorders November 2-4 • Atlanta, Georgia For more information: http://hematology.conferenceseries.com
33rd Annual Chemotherapy Foundation Symposium: Innovative Cancer Therapy for Tomorrow®November 4-6 • New York, New York For more information: http://www.chemotherapyfounda-tionsymposium.org/CMS/
Society for Immunotherapy of Cancer 30th Anniversary Annual MeetingNovember 4-8 • National Harbor, Maryland For more information: www.sitcancer.org/2015
JADPRO Live at APSHO for Advanced Practitioners in Oncology November 5-8, 2015 • Phoenix, Arizona JW Marriott Desert Ridge For more information: jadprolive.com
City of Hope Presents: Multidisciplinary Approaches to Cancer Symposium November 5-8 • Las Vegas, Nevada For mor information: https://cme.cityofhope.org/eventinfo_5980.html
Advanced Breast Cancer Third International Consensus Conference November 5-7 • Lisbon, Portugal For more information: www.abc-lisbon.org
13th Annual School of Breast Oncology November 5-7 • Atlanta, Georgia For more information: http://www.gotoper.com/conferences/sobo/meetings/13th-Annual-School-of-Breast-Oncology
AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics November 5-9 • Boston, Massachusetts For more information: http://www.aacr.org/Meetings/Pages/MeetingDetail
14th International Kidney Cancer SymposiumNovember 6-7 • Miami, Florida For more information: http://registeruo.niu.edu/iebms/wbe/wbe_p1_main.aspx?oc=40&cc=WBE4014167
ESMO Summit Americas 2015–Oncology Updates: From Evidence to PracticeNovember 6-8 • Miami, Florida For more information: www.esmo.org/Conferences/ESMO-Summit-Americas-2015
10th Annual New York Lung Cancer Symposium November 7 • New York, New York For more information: http://www.gotoper.com/conferences/nyl/meetings/10th-Annual-New-York-Lung-Cancer-Symposium
17th Annual Brain Tumor Update and 6th Annual International Symposium on Long-Term Control of Metastases to the Brain and Spine November 7-8 • Las Vegas, Nevada For more information: http://www.clevelandclinicmeded.com/live/courses/2015/brainmets15/.aspx?EventItemID=52&DetailItemID=196#.VSZlr_msXpU
Best of ASTRO November 13-14 • San Diego, California For more information: www.astro.org/Meetings-and-Events/2015-Best-of-ASTRO/Index.aspx
2015 Oncologic Emergency Medicine Conference November 13-14 • Houston, Texas For more information: http://www.mdanderson.org/education-and-research/education-and-training/schools-and-programs/cme-conference-management/conferences/d114243-2015-oncologic-emergency-medicine-conference.html
8th AACR Conference on the Science of Cancer Health Disparities in Racial/Ethnic Minorities and the Medically Underserved November 13-16 • Atlanta, Georgia For more information: http://www.aacr.org/Meetings/Pages/MeetingDetail.aspx?EventItemID=68#.Vba1EqTbJjq
12th International Conference of the Society for Integrative Oncology November 15-16 • Boston, MA For more information: www.integrativeonc.org/index.php/events
Society for Integrative Oncology 12th International Conference November 14-16 • Boston, Massachusetts For more information: http://www.integrativeonc.org/conference
Advances in Cancer ImmunotherapyTM
November 18 • San Francisco, California For more information: www.sitcancer.org/sitc-meetings/aci2015/casf
12th International Congress of the Society for Melanoma Research November 18-21 • San Francisco, California For more information: http://www.melanomacongress.com/
20th Annual Scientific Meeting of the Society for Neuro-OncologyNovember 19-22 • San Antonio, Texas For more information: www.soc-neuro-onc.org
ESMO Symposium on Immuno-Oncology November 20-21 • Lausanne, Switzerland For more information: www.esmo.org/Conferences/Immuno-Oncology-2015
December9th European Colorectal Congress (ECC)December 1-4 • St. Gallen, Switzerland For more information: www.colorectalsurgery.eu
Advances in Cancer ImmunotherapyTM
December 4 • New Orleans, Louisiana For more information: www.sitcancer.org/sitc-meetings/aci2015/la
10th Annual Practical Course in Dermoscopy & Update on Malignant Melanoma 2015 December 4-6 • Scottsdale, Arizona For more information: https://ce.mayo.edu/dermatology/node/2463
57th Annual ASH Meeting & ExpositionDecember 5-8 • Orlando, Florida For more information: www.hematology.org/
continued from page 100

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 103
Letters to the Editor
Unneccessary Complexity in Scientific Terminology?
In the highlighted quote in the article titled “Ado-Trastuzumab Emtansine
Fails to Replace Standard of Care in First-Line Metastatic Breast Cancer,” which appeared in on page 3 of the July 10 issue of The ASCO Post, a most re-markable sentence was constructed:
T-DM1 and T-DM1 plus pertuzumab demonstrated a noninferior progres-sion-free survival compared to trastu-zumab/taxane, but they were not found to be superior to it.
I’ve been retired for 15 years and am astounded by the advances made since then, especially the vast array of monoclonal antibody and genetically determined treatments. Astounding complexity! But some things are made more complex than they have to be.
When treatment A demonstrates a “noninferior progression-free survival” compared to treatment B, but [at the same time] is “not found to be superior” to it, one wonders what’s going on. Af-ter three or four readings, it dawns: The new treatment is just about the same as
the old, one way or the other.Please try to cut through the impe-
rious pretense of erudition. “Noninfe-rior,” for example, is a term that should be abandoned. n
—Charles P. Duvall MD, MACP Hilton Head Island, South Carolina
Mayo Clinic Receives $11 Million Grant from NCI to Study NHL Survivorship
Mayo Clinic has received a 5-year, $11 million grant from the
National Cancer Institute (NCI) to study survivorship in patients with non-Hodgkin lymphoma (NHL). The Lymphoma Epidemiology of Outcomes Cohort Study will enroll 12,000 patients with NHL. The study will follow these patients for long-term prognosis and survivorship.
“With an increasing number of Americans living with NHL, we need to find new and better ways to improve the length and quality of their lives,” said the study’s principal investiga-tor, James Cerhan, MD, PhD, Pro-fessor of Epidemiology in the Mayo Clinic College of Medicine and Chair of the Department of Health Sciences Research.
According to the NCI, about 70,000 cases of NHL will be diagnosed in the United States in 2015. The incidence of NHL has been increasing since 1950, although, over the past 2 decades, the rate of increase has slowed and survival rates have improved. These trends have led to an increasing number of NHL survivors—most recently estimated at 550,000.
The grant involves collaboration among multiple institutions, includ-ing lymphoma experts from Mayo Clinic, the University of Iowa, Em-ory University/Grady Health Sys-tem, MD Anderson Cancer Center, the University of Wisconsin, Cornell University, and the University of Mi-ami Health System/Jackson Memo-rial Hospital. n
NCCN 10th Annual Congress:
Hematologic Malignancies™
Reserve Your Seat Today at:
NCCN.org/hem
Sponsorship and exhibit opportunities available! For more information, e-mail [email protected]
This congress is approved for AMA PRA Category 1 Credit™ and is also certified for nurses and pharmacists. Learn more at NCCN.org/hem.
This activity is supported by educational grants from Actelion Pharmaceuticals US, Inc.; Astellas Scientific and Medical Affairs, Inc.; Celgene Corporation; Novartis Pharmaceuticals Corporation; Seattle Genetics; Spectrum Pharmaceuticals, Inc.; and Takeda Oncology.
October 16 – 17, 2015 | San Francisco Marriott Marquis | San Francisco, CA
NCCN.org/hemEarn CME/CNE/CPE Credit!
Attend 13 educational sessions featuring the latest advances in hematologic malignancies, plus 3 case-based panel discussions.
REGISTER NOW!
Friday, October 16, 2015
4:30 – 6:00 pm Registration, Exhibits, and Refreshments
6:00 – 8:35 pm Educational Sessions
Saturday, October 17, 2015
7:00 – 8:00 am Registration, Exhibits, and Breakfast
8:00 am – 5:00 pm Educational Sessions
Andrew D. Zelenetz, MD, PhDMemorial Sloan Kettering Cancer Center
Ranjana H. Advani, MDStanford Cancer Institute
Co-Chairs:
Reserve your seat by Friday, September 11, 2015 to receive the Early Bird Registration rate. As a benefit of membership, NCCN extends a 50% discount off the registration fee to staff employed at NCCN Member Institutions. Learn more at NCCN.org/hem.

PAGE 104 The ASCO Post | SEPTEMBER 10, 2015
In the News
Increased Interest in Simple Injection to Treat Women With Postmastectomy PainBy Charlotte Bath
After presenting results of a study showing that injecting a standard
analgesic combination into trigger points of pain along the inframamma-ry fold relieved postmastectomy pain, Laura J. Esserman, MD, MBA, Direc-tor of the Carol Franc Buck Breast Care Center and Co-Leader of the Breast Oncology Program at the University of California, San Francisco, received a lot of phone calls. Some of those phone calls were from fellow physicians inter-ested in possibly using the procedure with their own patients. Many more calls were from women who had read, heard, and/or seen media reports on the study.
“Our initial study was written up in The ASCO Post [February 15, 2014, volume 5, issue 3], and it generated an amazing response, mostly from women (or their families or friends) who were suffering and looking for a way to re-lieve their pain,” Dr. Esserman said in a recent interview. “We got so many calls from all over the country, from women who were feeling desperate” because of pain persisting weeks or more after their mastectomies were performed and interfering with their ability to wear a bra or to sleep.
That intense interest, along with ad-
ditional positive study results, led Dr. Esserman and colleagues at the Univer-sity of California, San Francisco, to pub-lish the study and to produce a video detailing how to determine a patient’s trigger points for neuropathic pain and to inject the analgesic. Results from 19 patients were presented at the 2013 San Antonio Breast Cancer Symposium,1 and results from 35 patients are in press (Annals of Surgical Oncology). The vid-eo has been posted at cancer.ucsf.edu/breastcancercenter. Recent renewed interest resulted from an article in The New York Times.2
Postmastectomy Pain Is Complex
Postmastectomy pain occurs in 20% to 40% of patients and “is complex,” Dr. Esserman said. The pain can be burning, shooting, or radiating. Much less com-monly, patients have intense itching.
“Although patients are often “sore and uncomfortable” following a mastectomy, “usually by 2 weeks, they are quite a bit better. Beyond 2 weeks, you may hear patients say, ‘I can’t wear my bra’ or ‘I have this intense point of pain that is re-ally too much,’” Dr. Esserman added.
“One of the causes of postmastectomy pain is irritation to the nerves that have
to be cut when you actually remove the breast,” Dr. Esserman said. These nerves “run along the chest wall going forward and backward,” she noted. “So, sometimes you can get this aching across the breast, and it may even reach into the back.”
Nerve pain, Dr. Esserman noted, “is not particularly responsive to narcotics.” Patients may have “pain that just can’t seem to get better or is not getting bet-ter with narcotics and is just persisting.” Such persistent pain led to the search for a more effective means of treatment.
Step-by-Step ApproachThe video presents a step-by-step
approach to alleviate neuropathic post-mastectomy pain, caused by damage to the cutaneous branches of the T4/T5 sensory nerves. The damage likely oc-
curs when the blood vessels (branches off the intercostal vessels)—which are accompanied by nerve branches (off the intercostal nerves)—are cut as the breast is being removed from the chest wall. When vessels bleed, they are then cauterized to control the bleeding. You have to deal with the bleeding, and the nerve branches are quite tiny and very difficult to see,” Dr. Esserman said, but cautery can lead to the formation of painful neuromas.
This approach to postmastectomy pain examines the inframammary fold to identify trigger points, which correlate with the egress of the T4/T5 cutaneous branches. Since neuro-pathic pain involves both inflamma-tory and immune mediators, which sensitize pain receptors, the trigger
Supportive Care
I am amazed at how well it has worked and how consistent that has been. The key is to know where these nerves are, and the surgeons should know where they are.
—Laura J. Esserman, MD, MBA
Delivered to your inbox every weekday evening. Visit ASCOPost.com to
learn more.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 105
In the News
points are injected with a combina-tion of a local anesthetic and a ste-roid—a 2-mL mixture of equal parts 0.5% bupivacaine and 4 mg/mL of dexamethasone. The mixture is mas-saged in, and for most patients, that is all that is needed, although roughly 20% of the women receiving this treatment will require two injections, and 10% may require three injections.
If the patient does not experience significant reduction in pain soon af-ter the first injection, “it is possible you are not in the right spot,” Dr. Es-serman said. In such cases, Dr. Esser-man would reassess the patient for a trigger point and try again. “You can try an ultrasound and look for a little neuroma,” she said. “If that doesn’t work, if someone got better, but not that much better, or got better and then a month later got worse, you go in and inject again. If you’ve done it three times and it is not working, you ought to try something different.”
Spreading the WordDr. Esserman has used this ap-
proach with patients whose mastec-tomies had been performed by her or other physicians. As mentioned in the video, the technique is effec-tive 90% to 95% of the time if trigger points can be identified.
“I am amazed at how well it has worked and how consistent that has been,” Dr. Esserman told The ASCO Post. This high success rate has led her to encourage other physicians to use
the technique. “That is why I did this video,” she said.
“The key is to know where these nerves are, and the surgeons should know where they are,” Dr. Esserman said. Then by watching the video, they should be able to successfully give the injections to patients with postmastec-tomy pain.
“Our pain management colleagues have identified ways in which you can be very successful in alleviating pain, and in fact they aren’t that mysterious. They are fairly easy to use, and one of the keys is to understand where the nerves are cauterized. These are things that are easy enough for us to learn and to integrate into our own day-to-day practice,” Dr. Esserman said.
Dr. Esserman has received calls about the treatment from interested physicians and sometimes initiates calls to colleagues to encourage them to try the procedure or at least to take a look at the video. “I’ve had some people e-mail me and say that they have done it. That’s exciting,” she added.
Dr. Esserman also has received calls from patients from other parts of the country who have heard about the procedure and are willing to trav-el great distances in the hope of be-coming free of postmastectomy pain. “Someone may call me up from New York and say, ‘I want to come out there,’ and I say, ‘Don’t do that. Let’s get your surgeon to take a look at this video.’” In some cases, Dr. Esserman refers patients to physicians in their
area who are already versed in the technique.
‘Not a Panacea’However, Dr. Esserman stressed,
“This is not a panacea for everything.” If the pain is not neuropathic or trigger points cannot be identified, “it is not go-ing to help,” she said, and in such cases, she would not go ahead with the treat-ment. “Occasionally, you have someone where that is not the cause of pain or even if it is, the technique isn’t working for some reason. I don’t have an answer for that.”
The technique has been tried a time or two in women who have pain follow-ing lumpectomies, Dr. Esserman added, but “it has not been very productive. I doubt that it is going to be the solution there, and we just have to keep looking” for other means of dealing with that type of postsurgical pain.
For some people with neuropathic pain, “the best thing to do is to try some of the medicines that block neuropathic pain, like Neurontin [gabapentin], and that can be the solution,” Dr. Esserman said. “You can use lidocaine patches. There are a number of things that peo-ple can try, and this should just be one of the tools.”
No More Surgery Than NeededKnowing that surgery and cautery
can cause postmastectomy pain “is one of the reasons I am also moti-vated not to do more surgery than is needed, because some people get per-
sistent pain,” Dr. Esserman said. And it can be very debilitating.
Working to prevent postmastecto-my pain “is part of our ongoing learn-ing and presents an opportunity,” Dr. Esserman said. “Nobody wants to cause pain or problems afterward. No one is doing it intentionally, but sometimes these things happen, and there is nothing we can do to stop it. But the more we understand it, the better we understand the source of pain and how to prevent long-term problems, we can even start to think about how to prevent the problems in the first place.”
For example, Dr. Esserman noted, “We are starting to work with our anes-thesia team to think about doing thorac-ic nerve blocks” to avoid causing injury or pain. “If you use the anesthetic first, you may reduce the chance of pain post-operatively, and patients may never ex-perience pain,” she noted. “There are lots of opportunities to continue to improve. That’s the great thing about medicine.” n
Disclosure: Dr. Esserman reported no potential conflicts of interest.
References1. Tang CJ, Eder SE, Lee DJ, Rabow
MW: A simple intervention to relieve chronic neuropathic post-mastectomy pain. 2013 San Antonio Breast Cancer Sympo-sium. Abstract P3-10-03. Presented De-cember 12, 2013.
2. Pfaff LG: When pain persists after breast cancer. The New York Times, June 8, 2015.
Expect Questions or Ask About Postmastectomy Pain
S ince postmastectomy pain oc-curs in an estimated 20% to
40% of patients undergoing the pro-cedure, physicians who treat these women can expect questions about the pain and how to deal with it. However, some patients may be re-luctant to ask about it.
“Sometimes, women don’t want to complain to their surgeons,” Laura J. Esserman, MD, MBA, said in an interview with The ASCO Post. “These women may feel like, ‘Hey, I’m cured. I shouldn’t be complain-ing.’ But people legitimately have problems with pain, so I think that
physicians should also ask patients about pain and make sure that they are not suffering.”
Dr. Esserman is Director of the Carol Franc Buck Breast Care Cen-ter and Co-Leader of the Breast On-cology Program at the University of California, San Francisco. She is also co-author of a study finding that injections of a combination of bupivacaine and dexamethasone into trigger points of pain identified along the inframammary fold pro-vided a safe and effective treatment option for neuropathic postmastec-tomy pain (in press, Annals of Surgi-cal Oncology).
The authors concluded, “This tech-nique should be added to the arma-mentarium of all surgeons who per-form breast surgery.” To help physicians learn about and practice the technique, Dr. Esserman presented a step-by-step
approach in a video available at cancer.ucsf.edu/breastcancercenter.
“Health-care providers should routinely screen their patients for the presence of postmastectomy pain syndrome,” the study authors added. This screening should include asking patients whether they have pain and whether the pain hinders their abil-ity to wear a bra or to sleep on the affected side. A focused examination should be done to ascertain whether there are trigger points of neuropathic pain caused by damage during surgery to the T4 and T5 sensory nerves as they exit the chest wall.
Listen and Look“You have to listen. You have to
look. You have to work with people to keep trying to find a solution to the pain,” Dr. Esserman said. “You can try things like lidocaine patches. Pain is
an important problem, and you have to try to pay attention to it, keep work-ing to try to figure out what is wrong.”
If it does turn out that the pain is caused by damage to the T4 and T5 sensory nerves and trigger points of pain can be identified, an injection of a 2-mL mixture of equal parts 0.5% bupivacaine and 4 mg/mL of dexamethasone can be a safe, sim-ple, and effective option for treating that pain.
As mentioned in the video, the technique is effective 90% to 95% of the time that trigger points can be identified. “I am amazed at how well it has worked and how consistent that has been,” Dr. Esserman stated. “This is something new that we have figured out that has made a really big difference.” n
Disclosure: Dr. Esserman reported no potential conflicts of interest.

IMBRUVICA® (ibrutinib) is the fi rst and only FDA-approved therapy for use in patients with Waldenström’s macroglobulinemia (WM)
DISCOVERING HOW FAR THERAPY CAN GO
IMBRUVICA® is indicated for the treatment of patients with
IMBRUVICA® is approved for use in 4 indications
Mantle cell lymphoma (MCL) who have received at least one prior therapy.Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verifi cation of clinical benefi t in confi rmatory trials.
Waldenström’s macroglobulinemia (WM).
Chronic lymphocytic leukemia (CLL) who have received at least one prior therapy.
Chronic lymphocytic leukemia with 17p deletion.
© Pharmacyclics LLC 2015© Janssen Biotech, Inc. 201506/15 PRC-01166
ADVERSE REACTIONSThe most common adverse reactions (≥25%) in patients with B-cell malignancies (MCL, CLL, WM) were thrombocytopenia* (57%, 52%, 43%), neutropenia* (47%, 51%, 44%), diarrhea (51%, 48%, 37%), anemia* (41%, 36%, 13%), fatigue (41%, 28%, 21%), musculoskeletal pain (37%, 28%†, NA‡), bruising (30%, 12%†, 16%†), nausea (31%, 26%, 21%), upper respiratory tract infection (34%, 16%, 19%), and rash (25%, 24%†, 22%†).
* Based on adverse reactions and/or laboratory measurements (noted as platelets, neutrophils, or hemoglobin decreased).
† Includes multiple ADR terms.‡ Not applicable; no associated ADRs.The most common Grade 3 or 4 non-hematological adverse reactions (≥5%) in MCL patients were pneumonia (7%), abdominal pain (5%), atrial fi brillation (5%), diarrhea (5%), fatigue (5%), and skin infections (5%).Approximately 6% (CLL), 14% (MCL), and 11% (WM) of patients had a dose reduction due to adverse events.Approximately 5% (CLL), 9% (MCL), and 6% (WM) of patients discontinued due to adverse events. Most frequent adverse
events leading to discontinuation were infections, subdural hematomas, and diarrhea in CLL patients and subdural hematoma (1.8%) in MCL patients.
DRUG INTERACTIONSCYP3A Inhibitors - Avoid co-administration with strong and moderate CYP3A inhibitors. If a moderate CYP3A inhibitor must be used, reduce the IMBRUVICA® dose.
CYP3A Inducers - Avoid co-administration with strongCYP3A inducers.
SPECIFIC POPULATIONSHepatic Impairment - Avoid use in patients with moderate or severe baseline hepatic impairment. In patients with mild impairment, reduce IMBRUVICA® dose.
Please review the Brief Summary of full Prescribing Information on the following pages.
WARNINGS AND PRECAUTIONSHemorrhage - Fatal bleeding events have occurred in patients treated with IMBRUVICA®. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria, and post-procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with IMBRUVICA®.
The mechanism for the bleeding events is not well understood. IMBRUVICA® may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies. Consider the benefi t-risk of withholding IMBRUVICA® for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding.
Infections - Fatal and non-fatal infections have occurred with IMBRUVICA® therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with IMBRUVICA®. Monitor patients for fever and infections and evaluate promptly.
Cytopenias - Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with IMBRUVICA®. Monitor complete blood counts monthly.
Atrial Fibrillation - Atrial fi brillation and atrial fl utter (range, 6 to 9%) have occurred in patients treated with IMBRUVICA®, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fi brillation. Periodically monitor patients clinically for atrial fi brillation. Patients who develop arrhythmic symptoms (eg, palpitations, lightheadedness) or new-onset dyspnea should have an ECG performed. If atrial fi brillation persists, consider the risks and benefi ts of IMBRUVICA® treatment and dose modifi cation.
Second Primary Malignancies - Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with IMBRUVICA®. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11%).
Tumor Lysis Syndrome - Tumor lysis syndrome has been reported with IMBRUVICA® therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden).
Embryo-Fetal Toxicity - Based on fi ndings in animals, IMBRUVICA® can cause fetal harm when administered to a pregnant woman. Advise women to avoid becoming pregnant while taking IMBRUVICA®. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
To learn more, visitwww.IMBRUVICA.com
IMPORTANT SAFETY INFORMATION
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P1-4_RL.indd 1-2 6/19/15 8:03 PM

IMBRUVICA® (ibrutinib) is the fi rst and only FDA-approved therapy for use in patients with Waldenström’s macroglobulinemia (WM)
DISCOVERING HOW FAR THERAPY CAN GO
IMBRUVICA® is indicated for the treatment of patients with
IMBRUVICA® is approved for use in 4 indications
Mantle cell lymphoma (MCL) who have received at least one prior therapy.Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verifi cation of clinical benefi t in confi rmatory trials.
Waldenström’s macroglobulinemia (WM).
Chronic lymphocytic leukemia (CLL) who have received at least one prior therapy.
Chronic lymphocytic leukemia with 17p deletion.
© Pharmacyclics LLC 2015© Janssen Biotech, Inc. 201506/15 PRC-01166
ADVERSE REACTIONSThe most common adverse reactions (≥25%) in patients with B-cell malignancies (MCL, CLL, WM) were thrombocytopenia* (57%, 52%, 43%), neutropenia* (47%, 51%, 44%), diarrhea (51%, 48%, 37%), anemia* (41%, 36%, 13%), fatigue (41%, 28%, 21%), musculoskeletal pain (37%, 28%†, NA‡), bruising (30%, 12%†, 16%†), nausea (31%, 26%, 21%), upper respiratory tract infection (34%, 16%, 19%), and rash (25%, 24%†, 22%†).
* Based on adverse reactions and/or laboratory measurements (noted as platelets, neutrophils, or hemoglobin decreased).
† Includes multiple ADR terms.‡ Not applicable; no associated ADRs.The most common Grade 3 or 4 non-hematological adverse reactions (≥5%) in MCL patients were pneumonia (7%), abdominal pain (5%), atrial fi brillation (5%), diarrhea (5%), fatigue (5%), and skin infections (5%).Approximately 6% (CLL), 14% (MCL), and 11% (WM) of patients had a dose reduction due to adverse events.Approximately 5% (CLL), 9% (MCL), and 6% (WM) of patients discontinued due to adverse events. Most frequent adverse
events leading to discontinuation were infections, subdural hematomas, and diarrhea in CLL patients and subdural hematoma (1.8%) in MCL patients.
DRUG INTERACTIONSCYP3A Inhibitors - Avoid co-administration with strong and moderate CYP3A inhibitors. If a moderate CYP3A inhibitor must be used, reduce the IMBRUVICA® dose.
CYP3A Inducers - Avoid co-administration with strongCYP3A inducers.
SPECIFIC POPULATIONSHepatic Impairment - Avoid use in patients with moderate or severe baseline hepatic impairment. In patients with mild impairment, reduce IMBRUVICA® dose.
Please review the Brief Summary of full Prescribing Information on the following pages.
WARNINGS AND PRECAUTIONSHemorrhage - Fatal bleeding events have occurred in patients treated with IMBRUVICA®. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria, and post-procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with IMBRUVICA®.
The mechanism for the bleeding events is not well understood. IMBRUVICA® may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies. Consider the benefi t-risk of withholding IMBRUVICA® for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding.
Infections - Fatal and non-fatal infections have occurred with IMBRUVICA® therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with IMBRUVICA®. Monitor patients for fever and infections and evaluate promptly.
Cytopenias - Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with IMBRUVICA®. Monitor complete blood counts monthly.
Atrial Fibrillation - Atrial fi brillation and atrial fl utter (range, 6 to 9%) have occurred in patients treated with IMBRUVICA®, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fi brillation. Periodically monitor patients clinically for atrial fi brillation. Patients who develop arrhythmic symptoms (eg, palpitations, lightheadedness) or new-onset dyspnea should have an ECG performed. If atrial fi brillation persists, consider the risks and benefi ts of IMBRUVICA® treatment and dose modifi cation.
Second Primary Malignancies - Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with IMBRUVICA®. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11%).
Tumor Lysis Syndrome - Tumor lysis syndrome has been reported with IMBRUVICA® therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden).
Embryo-Fetal Toxicity - Based on fi ndings in animals, IMBRUVICA® can cause fetal harm when administered to a pregnant woman. Advise women to avoid becoming pregnant while taking IMBRUVICA®. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
To learn more, visitwww.IMBRUVICA.com
IMPORTANT SAFETY INFORMATION
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P1-4_RL.indd 1-2 6/19/15 8:03 PM

Brief Summary of Prescribing Information for IMBRUVICA® (ibrutinib)IMBRUVICA® (ibrutinib) capsules, for oral useSee package insert for Full Prescribing Information
INDICATIONS AND USAGEMantle Cell Lymphoma: IMBRUVICA is indicated for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials [see Clinical Studies (14.1) in Full Prescribing Information].Chronic Lymphocytic Leukemia: IMBRUVICA is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy [see Clinical Studies (14.2) in Full Prescribing Information].Chronic Lymphocytic Leukemia with 17p deletion: IMBRUVICA is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) with 17p deletion [see Clinical Studies (14.2) in Full Prescribing Information].Waldenström’s Macroglobulinemia: IMBRUVICA is indicated for the treatment of patients with Waldenström’s macroglobulinemia (WM) [see Clinical Studies (14.3) in Full Prescribing Information].CONTRAINDICATIONSNoneWARNINGS AND PRECAUTIONSHemorrhage: Fatal bleeding events have occurred in patients treated with IMBRUVICA. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria and post procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with IMBRUVICA. The mechanism for the bleeding events is not well understood. IMBRUVICA may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies.Consider the benefit-risk of withholding IMBRUVICA for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding [see Clinical Studies (14) in Full Prescribing Information].Infections: Fatal and non-fatal infections have occurred with IMBRUVICA therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. [See Adverse Reactions]. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with IMBRUVICA. Monitor patients for fever and infections and evaluate promptly.Cytopenias: Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with IMBRUVICA.Monitor complete blood counts monthly. Atrial Fibrillation: Atrial fibrillation and atrial flutter (range, 6 to 9%) have occurred in patients treated with IMBRUVICA, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fibrillation. Periodically monitor patients clinically for atrial fibrillation. Patients who develop arrhythmic symptoms (e.g., palpitations, lightheadedness) or new onset dyspnea should have an ECG performed. If atrial fibrillation persists, consider the risks and benefits of IMBRUVICA treatment and dose modification [see Dosage and Administration (2.3) in Full Prescribing Information]. Second Primary Malignancies: Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with IMBRUVICA. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11 %).Tumor Lysis Syndrome: Tumor lysis syndrome has been reported with IMBRUVICA therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden). Embryo-Fetal Toxicity: Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. Ibrutinib caused malformations in rats at exposures 14 times those reported in patients with MCL and 20 times those reported in patients with CLL or WM, receiving the ibrutinib dose of 560 mg per day and 420 mg per day, respectively. Reduced fetal weights were observed at lower exposures. Advise women to avoid becoming pregnant while taking IMBRUVICA. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations].ADVERSE REACTIONSThe following adverse reactions are discussed in more detail in other sections of the labeling:• Hemorrhage [see Warnings and Precautions]• Infections [see Warnings and Precautions]• Cytopenias [see Warnings and Precautions]• Atrial Fibrillation [see Warnings and Precautions]• Second Primary Malignancies [see Warnings and Precautions]• Tumor Lysis Syndrome [see Warnings and Precautions]Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.Clinical Trials Experience: Mantle Cell Lymphoma: The data described below reflect exposure to IMBRUVICA in a clinical trial that included 111 patients with previously treated MCL treated with 560 mg daily with a median treatment duration of 8.3 months.The most commonly occurring adverse reactions (≥ 20%) were thrombo cytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, peripheral edema, upper respiratory tract infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting and decreased appetite (see Tables 1 and 2).The most common Grade 3 or 4 non-hematological adverse reactions (≥ 5%) were pneumonia, abdominal pain, atrial fibrillation, diarrhea, fatigue, and skin infections.Fatal and serious cases of renal failure have occurred with IMBRUVICA therapy. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 9% of patients.Adverse reactions from the MCL trial (N=111) using single agent IMBRUVICA 560 mg daily occurring at a rate of ≥ 10% are presented in Table 1.
Table 1: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with MCL (N=111)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaNauseaConstipationAbdominal painVomitingStomatitisDyspepsia
51312524231711
5005010
Infections and infestations
Upper respiratory tract infectionUrinary tract infectionPneumoniaSkin infectionsSinusitis
3414141413
03751
General disorders and administrative site conditions
FatiguePeripheral edemaPyrexiaAsthenia
41351814
5313
Table 1: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Mantle Cell Lymphoma (N=111) (continued)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Skin and subcutaneous tissue disorders
Bruising Rash Petechiae
302511
030
Musculoskeletal and connective tissue disorders
Musculoskeletal painMuscle spasmsArthralgia
371411
100
Respiratory, thoracic and mediastinal disorders
DyspneaCoughEpistaxis
271911
400
Metabolism and nutrition disorders
Decreased appetiteDehydration
2112
24
Nervous system disorders
DizzinessHeadache
1413
00
Table 2: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with MCL (N=111)
Percent of Patients (N=111)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 57 17Neutrophils Decreased 47 29Hemoglobin Decreased 41 9
* Based on laboratory measurements and adverse reactions
Ten patients (9%) discontinued treatment due to adverse reactions in the trial (N=111). The most frequent adverse reaction leading to treatment discontinuation was subdural hematoma (1.8%). Adverse reactions leading to dose reduction occurred in 14% of patients.Patients with MCL who develop lymphocytosis greater than 400,000/mcL have developed intracranial hemorrhage, lethargy, gait instability, and headache. However, some of these cases were in the setting of disease progression.Forty percent of patients had elevated uric acid levels on study including 13% with values above 10 mg/dL. Adverse reaction of hyperuricemia was reported for 15% of patients.Chronic Lymphocytic Leukemia: The data described below reflect exposure to IMBRUVICA in an open label clinical trial (Study 1) that included 48 patients with previously treated CLL and a randomized clinical trial (Study 2) that included 391 randomized patients with previously treated CLL or SLL.The most commonly occurring adverse reactions in Study 1 and Study 2 (≥ 20%) were thrombocytopenia, neutropenia, diarrhea, anemia, fatigue, musculo skeletal pain, upper respiratory tract infection, rash, nausea, and pyrexia.Approximately five percent of patients receiving IMBRUVICA in Study 1 and Study 2 discontinued treatment due to adverse events. These included infections, subdural hematomas and diarrhea. Adverse events leading to dose reduction occurred in approximately 6% of patients.Study 1: Adverse reactions and laboratory abnormalities from the CLL trial (N=48) using single agent IMBRUVICA 420 mg daily occurring at a rate of ≥ 10% are presented in Tables 3 and 4.
Table 3: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with CLL (N=48) in Study 1
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaConstipationNauseaStomatitisVomitingAbdominal painDyspepsia
63232121191513
4220200
Infections and infestations
Upper respiratory tract infectionSinusitisSkin infectionPneumoniaUrinary tract infection
4821171010
26680
General disorders and administrative site conditions
FatiguePyrexia Peripheral edemaAstheniaChills
3125231313
42040
Skin and subcutaneous tissue disorders
Bruising Rash Petechiae
542717
200
Respiratory, thoracic and mediastinal disorders
CoughOropharyngeal painDyspnea
191510
000
Musculoskeletal and connective tissue disorders
Musculoskeletal painArthralgiaMuscle spasms
272319
602
Nervous system disorders
DizzinessHeadachePeripheral neuropathy
211910
020
Metabolism and nutrition disorders
Decreased appetite 17 2
Neoplasms benign, malignant, unspecified
Second malignancies* 10* 0
Injury, poisoning and procedural complications
Laceration 10 2
Psychiatric disorders AnxietyInsomnia
1010
00
Vascular disorders Hypertension 17 8
*One patient death due to histiocytic sarcoma.
IMBRUVICA® (ibrutinib) capsules
Table 4: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with CLL (N=48) in Study 1
Percent of Patients (N=48)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 71 10Neutrophils Decreased 54 27Hemoglobin Decreased 44 0
* Based on laboratory measurements per IWCLL criteria and adverse reactions
Study 2: Adverse reactions and laboratory abnormalities described below in Tables 5 and 6 reflect exposure to IMBRUVICA with a median duration of 8.6 months and exposure to ofatumumab with a median of 5.3 months in Study 2.
Table 5: Non-Hematologic Adverse Reactions ≥ 10% Reported in Study 2
System Organ Class ADR Term
IMBRUVICA(N=195)
Ofatumumab(N=191)
All Grades(%)
Grade 3 or 4(%)
All Grades(%)
Grade 3 or 4(%)
Gastrointestinal disordersDiarrhea 48 4 18 2Nausea 26 2 18 0Stomatitis* 17 1 6 1Constipation 15 0 9 0Vomiting 14 0 6 1
General disorders and administration site conditions
Fatigue 28 2 30 2Pyrexia 24 2 15 1
Infections and infestationsUpper respiratory tract infection 16 1 11 2Pneumonia* 15 10 13 9Sinusitis* 11 1 6 0Urinary tract infection 10 4 5 1
Skin and subcutaneous tissue disorders
Rash* 24 3 13 0Petechiae 14 0 1 0Bruising* 12 0 1 0
Musculoskeletal and connective tissue disorders
Musculoskeletal Pain* 28 2 18 1Arthralgia 17 1 7 0
Nervous system disordersHeadache 14 1 6 0Dizziness 11 0 5 0
Injury, poisoning and procedural complications
Contusion 11 0 3 0Eye disorders
Vision blurred 10 0 3 0
Subjects with multiple events for a given ADR term are counted once only for each ADR term. The system organ class and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm.* Includes multiple ADR terms
Table 6: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Study 2
IMBRUVICA(N=195)
Ofatumumab(N=191)
All Grades(%)
Grade 3 or 4(%)
All Grades(%)
Grade 3 or 4(%)
Neutrophils Decreased 51 23 57 26Platelets Decreased 52 5 45 10Hemoglobin Decreased 36 0 21 0
* Based on laboratory measurements per IWCLL criteria
Waldenström’s MacroglobulinemiaThe data described below reflect exposure to IMBRUVICA in an open label clinical trial that included 63 patients with previously treated WM.The most commonly occurring adverse reactions in the WM trial (≥ 20%) were neutropenia, thrombocytopenia, diarrhea, rash, nausea, muscle spasms, and fatigue.Six percent of patients receiving IMBRUVICA in the WM trial discontinued treatment due to adverse events. Adverse events leading to dose reduction occurred in 11% of patients.Adverse reactions and laboratory abnormalities described below in Tables 7 and 8 reflect exposure to IMBRUVICA with a median duration of 11.7 months in the WM trial.
Table 7: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Waldenström’s Macroglobulinemia (N=63)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaNauseaStomatitis*Gastroesophageal reflux disease
37211613
0000
Skin and subcutaneous tissue disorders
Rash*Bruising* Pruritus
221611
000
Table 7: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Waldenström’s Macroglobulinemia (N=63) (continued)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
General disorders and administrative site conditions
Fatigue 21 0
Musculoskeletal and connective tissue disorders
Muscle spasms Arthropathy
2113
00
Infections and infestations
Upper respiratory tract infectionSinusitisPneumonia*Skin infection*
19191414
0062
Respiratory, thoracic and mediastinal disorders
EpistaxisCough
1913
00
Nervous system disorders
DizzinessHeadache
1413
00
Neoplasms benign, malignant, and unspecified (including cysts and polyps)
Skin cancer* 11 0
The system organ class and individual ADR terms are sorted in descending frequency order.* Includes multiple ADR terms.
Table 8: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with WM (N=63)
Percent of Patients (N=63)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 43 13Neutrophils Decreased 44 19Hemoglobin Decreased 13 8
* Based on laboratory measurements.
Postmarketing Experience: The following adverse reactions have been identified during post-approval use of IMBRUVICA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.Hypersensitivity reactions including anaphylactic shock (fatal), urticaria, and angioedema have been reported.
DRUG INTERACTIONSIbrutinib is primarily metabolized by cytochrome P450 enzyme 3A.CYP3A Inhibitors: In healthy volunteers, co-administration of ketoconazole, a strong CYP3A inhibitor, increased Cmax and AUC of ibrutinib by 29- and 24-fold, respectively. The highest ibrutinib dose evaluated in clinical trials was 12.5 mg/kg (actual doses of 840 – 1400 mg) given for 28 days with single dose AUC values of 1445 ± 869 ng • hr/mL which is approximately 50% greater than steady state exposures seen at the highest indicated dose (560 mg).Avoid concomitant administration of IMBRUVICA with strong or moderate inhibitors of CYP3A. For strong CYP3A inhibitors used short-term (e.g., antifungals and antibiotics for 7 days or less, e.g., ketoconazole, itraconazole, voriconazole, posaconazole, clarithromycin, telithromycin) consider interrupting IMBRUVICA therapy during the duration of inhibitor use. Avoid strong CYP3A inhibitors that are needed chronically. If a moderate CYP3A inhibitor must be used, reduce the IMBRUVICA dose. Patients taking concomitant strong or moderate CYP3A4 inhibitors should be monitored more closely for signs of IMBRUVICA toxicity [see Dosage and Administration (2.4) in Full Prescribing Information]. Avoid grapefruit and Seville oranges during IMBRUVICA treatment, as these contain moderate inhibitors of CYP3A [see Dosage and Administration (2.4), and Clinical Pharmacology (12.3) in Full Prescribing Information].CYP3A Inducers: Administration of IMBRUVICA with rifampin, a strong CYP3A inducer, decreased ibrutinib Cmax and AUC by approximately 13- and 10-fold, respectively.Avoid concomitant use of strong CYP3A inducers (e.g., carbamazepine, rifampin, phenytoin and St. John’s Wort). Consider alternative agents with less CYP3A induction [see Clinical Pharmacology (12.3) in Full Prescribing Information].
USE IN SPECIFIC POPULATIONSPregnancy: Pregnancy Category D [see Warnings and Precautions].Risk Summary: Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. If IMBRUVICA is used during pregnancy or if the patient becomes pregnant while taking IMBRUVICA, the patient should be apprised of the potential hazard to the fetus.Animal Data: Ibrutinib was administered orally to pregnant rats during the period of organogenesis at oral doses of 10, 40 and 80 mg/kg/day. Ibrutinib at a dose of 80 mg/kg/day was associated with visceral malformations (heart and major vessels) and increased post-implantation loss. The dose of 80 mg/kg/day in animals is approximately 14 times the exposure (AUC) in patients with MCL and 20 times the exposure in patients with CLL or WM administered the dose of 560 mg daily and 420 mg daily, respectively. Ibrutinib at doses of 40 mg/kg/day or greater was associated with decreased fetal weights. The dose of 40 mg/kg/day in animals is approximately 6 times the exposure (AUC) in patients with MCL administered the dose of 560 mg daily.Nursing Mothers: It is not known whether ibrutinib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from IMBRUVICA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric Use: The safety and effectiveness of IMBRUVICA in pediatric patients has not been established.Geriatric Use: Of the 111 patients treated for MCL, 63% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), infections (pneumonia and cellulitis) and gastrointestinal events (diarrhea and dehydration) occurred more frequently among elderly patients. Of the 391 patients randomized in Study 2, 61% were ≥ 65 years of age. No overall differences in effectiveness were observed between age groups. Grade 3 or higher adverse events occurred more frequently among elderly patients treated with IMBRUVICA (61% of patients age ≥ 65 versus 51% of younger patients) [see Clinical Studies (14.2) in Full Prescribing Information]. Of the 63 patients treated for WM, 59% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), and infections (pneumonia and urinary tract infection) occurred more frequently among elderly patients.
IMBRUVICA® (ibrutinib) capsules IMBRUVICA® (ibrutinib) capsules
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P1-4_RL.indd 3-4 6/19/15 8:03 PM

Brief Summary of Prescribing Information for IMBRUVICA® (ibrutinib)IMBRUVICA® (ibrutinib) capsules, for oral useSee package insert for Full Prescribing Information
INDICATIONS AND USAGEMantle Cell Lymphoma: IMBRUVICA is indicated for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials [see Clinical Studies (14.1) in Full Prescribing Information].Chronic Lymphocytic Leukemia: IMBRUVICA is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy [see Clinical Studies (14.2) in Full Prescribing Information].Chronic Lymphocytic Leukemia with 17p deletion: IMBRUVICA is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) with 17p deletion [see Clinical Studies (14.2) in Full Prescribing Information].Waldenström’s Macroglobulinemia: IMBRUVICA is indicated for the treatment of patients with Waldenström’s macroglobulinemia (WM) [see Clinical Studies (14.3) in Full Prescribing Information].CONTRAINDICATIONSNoneWARNINGS AND PRECAUTIONSHemorrhage: Fatal bleeding events have occurred in patients treated with IMBRUVICA. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria and post procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with IMBRUVICA. The mechanism for the bleeding events is not well understood. IMBRUVICA may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies.Consider the benefit-risk of withholding IMBRUVICA for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding [see Clinical Studies (14) in Full Prescribing Information].Infections: Fatal and non-fatal infections have occurred with IMBRUVICA therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. [See Adverse Reactions]. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with IMBRUVICA. Monitor patients for fever and infections and evaluate promptly.Cytopenias: Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with IMBRUVICA.Monitor complete blood counts monthly. Atrial Fibrillation: Atrial fibrillation and atrial flutter (range, 6 to 9%) have occurred in patients treated with IMBRUVICA, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fibrillation. Periodically monitor patients clinically for atrial fibrillation. Patients who develop arrhythmic symptoms (e.g., palpitations, lightheadedness) or new onset dyspnea should have an ECG performed. If atrial fibrillation persists, consider the risks and benefits of IMBRUVICA treatment and dose modification [see Dosage and Administration (2.3) in Full Prescribing Information]. Second Primary Malignancies: Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with IMBRUVICA. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11 %).Tumor Lysis Syndrome: Tumor lysis syndrome has been reported with IMBRUVICA therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden). Embryo-Fetal Toxicity: Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. Ibrutinib caused malformations in rats at exposures 14 times those reported in patients with MCL and 20 times those reported in patients with CLL or WM, receiving the ibrutinib dose of 560 mg per day and 420 mg per day, respectively. Reduced fetal weights were observed at lower exposures. Advise women to avoid becoming pregnant while taking IMBRUVICA. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations].ADVERSE REACTIONSThe following adverse reactions are discussed in more detail in other sections of the labeling:• Hemorrhage [see Warnings and Precautions]• Infections [see Warnings and Precautions]• Cytopenias [see Warnings and Precautions]• Atrial Fibrillation [see Warnings and Precautions]• Second Primary Malignancies [see Warnings and Precautions]• Tumor Lysis Syndrome [see Warnings and Precautions]Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.Clinical Trials Experience: Mantle Cell Lymphoma: The data described below reflect exposure to IMBRUVICA in a clinical trial that included 111 patients with previously treated MCL treated with 560 mg daily with a median treatment duration of 8.3 months.The most commonly occurring adverse reactions (≥ 20%) were thrombo cytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, peripheral edema, upper respiratory tract infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting and decreased appetite (see Tables 1 and 2).The most common Grade 3 or 4 non-hematological adverse reactions (≥ 5%) were pneumonia, abdominal pain, atrial fibrillation, diarrhea, fatigue, and skin infections.Fatal and serious cases of renal failure have occurred with IMBRUVICA therapy. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 9% of patients.Adverse reactions from the MCL trial (N=111) using single agent IMBRUVICA 560 mg daily occurring at a rate of ≥ 10% are presented in Table 1.
Table 1: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with MCL (N=111)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaNauseaConstipationAbdominal painVomitingStomatitisDyspepsia
51312524231711
5005010
Infections and infestations
Upper respiratory tract infectionUrinary tract infectionPneumoniaSkin infectionsSinusitis
3414141413
03751
General disorders and administrative site conditions
FatiguePeripheral edemaPyrexiaAsthenia
41351814
5313
Table 1: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Mantle Cell Lymphoma (N=111) (continued)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Skin and subcutaneous tissue disorders
Bruising Rash Petechiae
302511
030
Musculoskeletal and connective tissue disorders
Musculoskeletal painMuscle spasmsArthralgia
371411
100
Respiratory, thoracic and mediastinal disorders
DyspneaCoughEpistaxis
271911
400
Metabolism and nutrition disorders
Decreased appetiteDehydration
2112
24
Nervous system disorders
DizzinessHeadache
1413
00
Table 2: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with MCL (N=111)
Percent of Patients (N=111)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 57 17Neutrophils Decreased 47 29Hemoglobin Decreased 41 9
* Based on laboratory measurements and adverse reactions
Ten patients (9%) discontinued treatment due to adverse reactions in the trial (N=111). The most frequent adverse reaction leading to treatment discontinuation was subdural hematoma (1.8%). Adverse reactions leading to dose reduction occurred in 14% of patients.Patients with MCL who develop lymphocytosis greater than 400,000/mcL have developed intracranial hemorrhage, lethargy, gait instability, and headache. However, some of these cases were in the setting of disease progression.Forty percent of patients had elevated uric acid levels on study including 13% with values above 10 mg/dL. Adverse reaction of hyperuricemia was reported for 15% of patients.Chronic Lymphocytic Leukemia: The data described below reflect exposure to IMBRUVICA in an open label clinical trial (Study 1) that included 48 patients with previously treated CLL and a randomized clinical trial (Study 2) that included 391 randomized patients with previously treated CLL or SLL.The most commonly occurring adverse reactions in Study 1 and Study 2 (≥ 20%) were thrombocytopenia, neutropenia, diarrhea, anemia, fatigue, musculo skeletal pain, upper respiratory tract infection, rash, nausea, and pyrexia.Approximately five percent of patients receiving IMBRUVICA in Study 1 and Study 2 discontinued treatment due to adverse events. These included infections, subdural hematomas and diarrhea. Adverse events leading to dose reduction occurred in approximately 6% of patients.Study 1: Adverse reactions and laboratory abnormalities from the CLL trial (N=48) using single agent IMBRUVICA 420 mg daily occurring at a rate of ≥ 10% are presented in Tables 3 and 4.
Table 3: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with CLL (N=48) in Study 1
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaConstipationNauseaStomatitisVomitingAbdominal painDyspepsia
63232121191513
4220200
Infections and infestations
Upper respiratory tract infectionSinusitisSkin infectionPneumoniaUrinary tract infection
4821171010
26680
General disorders and administrative site conditions
FatiguePyrexia Peripheral edemaAstheniaChills
3125231313
42040
Skin and subcutaneous tissue disorders
Bruising Rash Petechiae
542717
200
Respiratory, thoracic and mediastinal disorders
CoughOropharyngeal painDyspnea
191510
000
Musculoskeletal and connective tissue disorders
Musculoskeletal painArthralgiaMuscle spasms
272319
602
Nervous system disorders
DizzinessHeadachePeripheral neuropathy
211910
020
Metabolism and nutrition disorders
Decreased appetite 17 2
Neoplasms benign, malignant, unspecified
Second malignancies* 10* 0
Injury, poisoning and procedural complications
Laceration 10 2
Psychiatric disorders AnxietyInsomnia
1010
00
Vascular disorders Hypertension 17 8
*One patient death due to histiocytic sarcoma.
IMBRUVICA® (ibrutinib) capsules
Table 4: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with CLL (N=48) in Study 1
Percent of Patients (N=48)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 71 10Neutrophils Decreased 54 27Hemoglobin Decreased 44 0
* Based on laboratory measurements per IWCLL criteria and adverse reactions
Study 2: Adverse reactions and laboratory abnormalities described below in Tables 5 and 6 reflect exposure to IMBRUVICA with a median duration of 8.6 months and exposure to ofatumumab with a median of 5.3 months in Study 2.
Table 5: Non-Hematologic Adverse Reactions ≥ 10% Reported in Study 2
System Organ Class ADR Term
IMBRUVICA(N=195)
Ofatumumab(N=191)
All Grades(%)
Grade 3 or 4(%)
All Grades(%)
Grade 3 or 4(%)
Gastrointestinal disordersDiarrhea 48 4 18 2Nausea 26 2 18 0Stomatitis* 17 1 6 1Constipation 15 0 9 0Vomiting 14 0 6 1
General disorders and administration site conditions
Fatigue 28 2 30 2Pyrexia 24 2 15 1
Infections and infestationsUpper respiratory tract infection 16 1 11 2Pneumonia* 15 10 13 9Sinusitis* 11 1 6 0Urinary tract infection 10 4 5 1
Skin and subcutaneous tissue disorders
Rash* 24 3 13 0Petechiae 14 0 1 0Bruising* 12 0 1 0
Musculoskeletal and connective tissue disorders
Musculoskeletal Pain* 28 2 18 1Arthralgia 17 1 7 0
Nervous system disordersHeadache 14 1 6 0Dizziness 11 0 5 0
Injury, poisoning and procedural complications
Contusion 11 0 3 0Eye disorders
Vision blurred 10 0 3 0
Subjects with multiple events for a given ADR term are counted once only for each ADR term. The system organ class and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm.* Includes multiple ADR terms
Table 6: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Study 2
IMBRUVICA(N=195)
Ofatumumab(N=191)
All Grades(%)
Grade 3 or 4(%)
All Grades(%)
Grade 3 or 4(%)
Neutrophils Decreased 51 23 57 26Platelets Decreased 52 5 45 10Hemoglobin Decreased 36 0 21 0
* Based on laboratory measurements per IWCLL criteria
Waldenström’s MacroglobulinemiaThe data described below reflect exposure to IMBRUVICA in an open label clinical trial that included 63 patients with previously treated WM.The most commonly occurring adverse reactions in the WM trial (≥ 20%) were neutropenia, thrombocytopenia, diarrhea, rash, nausea, muscle spasms, and fatigue.Six percent of patients receiving IMBRUVICA in the WM trial discontinued treatment due to adverse events. Adverse events leading to dose reduction occurred in 11% of patients.Adverse reactions and laboratory abnormalities described below in Tables 7 and 8 reflect exposure to IMBRUVICA with a median duration of 11.7 months in the WM trial.
Table 7: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Waldenström’s Macroglobulinemia (N=63)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
Gastrointestinal disorders
DiarrheaNauseaStomatitis*Gastroesophageal reflux disease
37211613
0000
Skin and subcutaneous tissue disorders
Rash*Bruising* Pruritus
221611
000
Table 7: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with Waldenström’s Macroglobulinemia (N=63) (continued)
System Organ Class Preferred Term All Grades (%)
Grade 3 or 4 (%)
General disorders and administrative site conditions
Fatigue 21 0
Musculoskeletal and connective tissue disorders
Muscle spasms Arthropathy
2113
00
Infections and infestations
Upper respiratory tract infectionSinusitisPneumonia*Skin infection*
19191414
0062
Respiratory, thoracic and mediastinal disorders
EpistaxisCough
1913
00
Nervous system disorders
DizzinessHeadache
1413
00
Neoplasms benign, malignant, and unspecified (including cysts and polyps)
Skin cancer* 11 0
The system organ class and individual ADR terms are sorted in descending frequency order.* Includes multiple ADR terms.
Table 8: Treatment-Emergent* Decrease of Hemoglobin, Platelets, or Neutrophils in Patients with WM (N=63)
Percent of Patients (N=63)All Grades
(%)Grade 3 or 4
(%)Platelets Decreased 43 13Neutrophils Decreased 44 19Hemoglobin Decreased 13 8
* Based on laboratory measurements.
Postmarketing Experience: The following adverse reactions have been identified during post-approval use of IMBRUVICA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.Hypersensitivity reactions including anaphylactic shock (fatal), urticaria, and angioedema have been reported.
DRUG INTERACTIONSIbrutinib is primarily metabolized by cytochrome P450 enzyme 3A.CYP3A Inhibitors: In healthy volunteers, co-administration of ketoconazole, a strong CYP3A inhibitor, increased Cmax and AUC of ibrutinib by 29- and 24-fold, respectively. The highest ibrutinib dose evaluated in clinical trials was 12.5 mg/kg (actual doses of 840 – 1400 mg) given for 28 days with single dose AUC values of 1445 ± 869 ng • hr/mL which is approximately 50% greater than steady state exposures seen at the highest indicated dose (560 mg).Avoid concomitant administration of IMBRUVICA with strong or moderate inhibitors of CYP3A. For strong CYP3A inhibitors used short-term (e.g., antifungals and antibiotics for 7 days or less, e.g., ketoconazole, itraconazole, voriconazole, posaconazole, clarithromycin, telithromycin) consider interrupting IMBRUVICA therapy during the duration of inhibitor use. Avoid strong CYP3A inhibitors that are needed chronically. If a moderate CYP3A inhibitor must be used, reduce the IMBRUVICA dose. Patients taking concomitant strong or moderate CYP3A4 inhibitors should be monitored more closely for signs of IMBRUVICA toxicity [see Dosage and Administration (2.4) in Full Prescribing Information]. Avoid grapefruit and Seville oranges during IMBRUVICA treatment, as these contain moderate inhibitors of CYP3A [see Dosage and Administration (2.4), and Clinical Pharmacology (12.3) in Full Prescribing Information].CYP3A Inducers: Administration of IMBRUVICA with rifampin, a strong CYP3A inducer, decreased ibrutinib Cmax and AUC by approximately 13- and 10-fold, respectively.Avoid concomitant use of strong CYP3A inducers (e.g., carbamazepine, rifampin, phenytoin and St. John’s Wort). Consider alternative agents with less CYP3A induction [see Clinical Pharmacology (12.3) in Full Prescribing Information].
USE IN SPECIFIC POPULATIONSPregnancy: Pregnancy Category D [see Warnings and Precautions].Risk Summary: Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. If IMBRUVICA is used during pregnancy or if the patient becomes pregnant while taking IMBRUVICA, the patient should be apprised of the potential hazard to the fetus.Animal Data: Ibrutinib was administered orally to pregnant rats during the period of organogenesis at oral doses of 10, 40 and 80 mg/kg/day. Ibrutinib at a dose of 80 mg/kg/day was associated with visceral malformations (heart and major vessels) and increased post-implantation loss. The dose of 80 mg/kg/day in animals is approximately 14 times the exposure (AUC) in patients with MCL and 20 times the exposure in patients with CLL or WM administered the dose of 560 mg daily and 420 mg daily, respectively. Ibrutinib at doses of 40 mg/kg/day or greater was associated with decreased fetal weights. The dose of 40 mg/kg/day in animals is approximately 6 times the exposure (AUC) in patients with MCL administered the dose of 560 mg daily.Nursing Mothers: It is not known whether ibrutinib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from IMBRUVICA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric Use: The safety and effectiveness of IMBRUVICA in pediatric patients has not been established.Geriatric Use: Of the 111 patients treated for MCL, 63% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), infections (pneumonia and cellulitis) and gastrointestinal events (diarrhea and dehydration) occurred more frequently among elderly patients. Of the 391 patients randomized in Study 2, 61% were ≥ 65 years of age. No overall differences in effectiveness were observed between age groups. Grade 3 or higher adverse events occurred more frequently among elderly patients treated with IMBRUVICA (61% of patients age ≥ 65 versus 51% of younger patients) [see Clinical Studies (14.2) in Full Prescribing Information]. Of the 63 patients treated for WM, 59% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), and infections (pneumonia and urinary tract infection) occurred more frequently among elderly patients.
IMBRUVICA® (ibrutinib) capsules IMBRUVICA® (ibrutinib) capsules
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P1-4_RL.indd 3-4 6/19/15 8:03 PM

PAGE 110 The ASCO Post | SEPTEMBER 10, 2015
Announcements
Joxel Garcia, MD, to Direct MD Anderson Moon Shots Program Prevention Efforts
The University of Texas MD Ander-son Cancer Center has appointed
former United States Public Health Service four-star Admiral Joxel Garcia, MD, as the inaugural Executive Direc-tor of the Cancer Prevention and Con-trol Platform, part of MD Anderson’s Moon Shots Program. He joined MD Anderson on August 31.
“Dr. Garcia is an internationally recognized health care leader with proven experience and success in a variety of health care settings,” said Giulio Draetta, MD, PhD, Professor of Genomic Medicine and Molecular & Cellular Oncology and co-leader of the Moon Shots Program. “We’re ex-cited to have someone of his caliber on board and know he will contribute a great deal to the platform and the program overall.”
The Cancer Prevention and Control Platform
The Moon Shots Program is an ef-fort to dramatically accelerate the pace of converting scientific discoveries into clinical and population-oriented advances that significantly reduce can-cer deaths. The Cancer Prevention and Control Platform implements and dis-seminates evidence-based, communi-ty-focused programs to advance cancer prevention, screening, early detection, and survivorship. The platform focuses on providing policy, education and services to achieve a measurable and sustainable reduction in the cancer burden, especially in the underserved population, for whom cancer and can-cer risk factors predominate. This ef-fort focuses on what’s known about diet, exercise, sun protection, tobacco avoidance, and human papillomavirus, among other topics.
“It’s estimated that as much as 50% of the cancer burden in the American population is preventable,” said Ernest Hawk, MD, Vice President and Divi-
sion Head of Cancer Prevention and Population Sciences and Coleader of the Cancer Prevention and Control Platform. “Cancer prevention and con-trol, practiced at both the individual and population levels, are critical to suc-cess in our mission of ending cancer.”
“I see myself as a catalyst to bring MD Anderson’s cancer control and pre-vention efforts to new heights in a sus-tainable and ever-growing way,” said Dr. Garcia.
A High-Level, Diverse CareerDr. Garcia began his medical career
as an obstetrician/gynecologist and then become the commissioner for the State of Connecticut Department of Public Health. After serving as the Deputy Director for the Pan American Health Organization/World Health Or-ganization, he moved into the corporate sector. There, he worked as Senior Vice President and Senior Medical Officer at Maximus Federal Services Inc.
President George W. Bush ap-pointed Dr. Garcia as the 13th U.S. Assistant Secretary for Health. At the same time, he was appointed as a four-star Admiral for the United States Public Health Service, and as the U.S. Representative to the World Health Organization. During this time, and as the highest ranking med-ical and public health official in the U.S., Dr. Garcia led more than 6,220 U.S. Public Service Commissioned Corps officers in the U.S. and in 88 countries for the protection, promo-tion, and advancement of health.
Following his government service, Dr. Garcia returned to his native Puerto Rico and served as the President and Dean of Medicine for Ponce School of Medicine and Health Sciences. In 2012, he returned to serve as the Director and Chief Medical Officer for the Washing-ton, D.C. Department of Health and as a Founding Partner with Aegis Health Security.
Dr. Garcia has served on several boards of nationally recognized health care organizations, including the U.S. Preventive Services Task Force and the National Dialogue on Cancer. He has received numerous awards and hon-ors including the Secretary of Defense Award for Exceptional Public Service and the U.S. Public Health Service Dis-tinguished Service Award. n
Joxel Garcia, MD
Renal Impairment: Less than 1% of ibrutinib is excreted renally. Ibrutinib exposure is not altered in patients with Creatinine clearance (CLcr) > 25 mL/min. There are no data in patients with severe renal impairment (CLcr < 25 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in Full Prescribing Information].Hepatic Impairment: Ibrutinib is metabolized in the liver. In a hepatic impairment study, data showed an increase in ibrutinib exposure. Following single dose administration, the AUC of ibrutinib increased 2.7-, 8.2- and 9.8-fold in subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe (Child-Pugh class C) hepatic impairment compared to subjects with normal liver function. The safety of IMBRUVICA has not been evaluated in patients with hepatic impairment. Monitor patients for signs of IMBRUVICA toxicity and follow dose modification guidance as needed. It is not recommended to administer IMBRUVICA to patients with moderate or severe hepatic impairment (Child-Pugh classes B and C) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3) in Full Prescribing Information].Females and Males of Reproductive Potential: Advise women to avoid becoming pregnant while taking IMBRUVICA because IMBRUVICA can cause fetal harm [see Use in Specific Populations].Plasmapheresis: Management of hyperviscosity in patients with WM may include plasmapheresis before and during treatment with IMBRUVICA. Modifications to IMBRUVICA dosing are not required.PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information).• Hemorrhage: Inform patients of the possibility of bleeding, and to report any signs or symptoms (blood in stools
or urine, prolonged or uncontrolled bleeding). Inform the patient that IMBRUVICA may need to be interrupted for medical or dental procedures [see Warnings and Precautions].
• Infections: Inform patients of the possibility of serious infection, and to report any signs or symptoms (fever,
chills, weakness, confusion) suggestive of infection [see Warnings and Precautions].• Atrial Fibrillation: Counsel patients to report any signs of palpitations, lightheadedness, dizziness, fainting, shortness
of breath, and chest discomfort [see Warnings and Precautions].• Second primary malignancies: Inform patients that other malignancies have occurred in patients who have been treated with
IMBRUVICA, including skin cancers and other carcinomas [see Warnings and Precautions].• Tumor lysis syndrome: Inform patients of the potential risk of tumor lysis syndrome and report any signs and symptoms
associated with this event to their healthcare provider for evaluation [see Warnings and Precautions].
• Embryo-fetal toxicity: Advise women of the potential hazard to a fetus and to avoid becoming pregnant [see Warnings
and Precautions].• Inform patients to take IMBRUVICA orally once daily according to their physician’s instructions
and that the capsules should be swallowed whole with a glass of water without being opened, broken, or chewed at approximately the same time each day [see Dosage and Administration (2.1) in Full Prescribing Information].
• Advise patients that in the event of a missed daily dose of IMBRUVICA, it should be taken as soon as possible on the same day with a return to the normal schedule the following day. Patients should not take extra capsules to make up the missed dose [see Dosage and Administration (2.5) in Full Prescribing Information].
• Advise patients of the common side effects associated with IMBRUVICA [see Adverse Reactions]. Direct the patient to a complete list of adverse drug reactions in PATIENT INFORMATION.
• Advise patients to inform their health care providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions].
• Advise patients that they may experience loose stools or diarrhea, and should contact their doctor if their diarrhea persists. Advise patients to maintain adequate hydration.
Active ingredient made in China.Distributed and Marketed by:Pharmacyclics LLCSunnyvale, CA USA 94085andMarketed by:Janssen Biotech, Inc.Horsham, PA USA 19044Patent http://www.imbruvica.comIMBRUVICA® is a registered trademark owned by Pharmacyclics LLC© Pharmacyclics LLC 2015© Janssen Biotech, Inc. 2015
IMBRUVICA® (ibrutinib) capsules
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P5HPV_RL.indd 1 6/19/15 8:02 PM

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 111
Clinical Trials
Clinical Trials Actively Recruiting Patients With Thyroid CancerCompiled by Liz Janetschek
PHASE I
Study Type: Phase I/Intervention-al/Single Group Assignment
Study Title: A Phase 1 Study of Dabrafenib in Combination With Lapa-tinib in BRAF Mutant Thyroid Cancer
Study Sponsor and Collaborators: National Cancer Institute
Purpose: To study the side effects and best dose of lapatinib ditosylate when given together with dabrafenib in treating patients with thyroid can-cer that cannot be removed by surgery and has not responded to previous treatment
Primary Outcome Measures: Maximum tolerated dose of lapatinib ditosylate, in combination with the es-tablished dose of dabrafenib, defined as the highest dose at which not more than 1/6 of the patients experience dose lim-iting toxicity [Time Frame: First 42 days of treatment]
Principal Investigator: Eric Sher-man, MD, Memorial Sloan Kettering Cancer Center; 646-888-4234, [email protected]
ClinicalTrials.gov Identifier: NCT01947023
Study Type: Phase I/Intervention-al/Single Group Assignment
Study Title: Evaluation of Thyroid Stunning From a Diagnostic Dose of I-123
Study Sponsor and Collaborators: University of Colorado, Denver
Purpose: To study if the small dose of radioiodine that is used for the do-simetry study on patients with differen-tiated thyroid cancer may stun the can-cer cells and make the thyroid cancer treatment less effective.
Primary Outcome Measures: Dif-ference in uptake of I-123 in the thyroid remnant in the neck in the two imaging studies. [Time Frame: 11 days]
Principal Investigator: Jennifer Kwak, MD, University of Colorado, Denver; contact Michelle Carr, CCRP, 720-848-6137, [email protected]
ClinicalTrials.gov Identifier: NCT02278198
PHASE I/II
Study Type: Phase I/II/Interven-tional/Single Group Assignment
Study Title: Administering Periph-eral Blood Lymphocytes Transduced With a Murine T-Cell Receptor Recog-nizing Human Thyroglobulin to People With Thyroglobulin Expressing Thy-roid Cancer
Study Sponsor and Collaborators: National Cancer Institute, National In-stitutes of Health Clinical Center
Purpose: To study an experimental therapy for treating patient with meta-static thyroid cancer that involves tak-ing white blood cells from the patient, growing them in the laboratory in large numbers, genetically modifying these specific cells with a type of virus (ret-rovirus) to attack only the tumor cells, and then giving the cells back to the patient. In this protocol, the patient’s white blood cells are being modified with a retrovirus that has the gene for antithyroglobulin incorporated.
Primary Outcome Measures: De-termine a safe dose of administration and determine if this approach will re-sult in an objective tumor regression. [Time Frame: Approximately 4 years]
Principal Investigator: James C.
Yang, MD, National Cancer Institute; 301-496-1574, [email protected]
ClinicalTrials.gov Identifier: NCT02390739
PHASE II
Study Type: Phase II/Intervention-al/Crossover Assignment
Study Title: A Randomized Phase 2 Study of Single Agent Dabrafenib (BRAFi) vs. Combination Regimen Dabrafenib (BRAFi) and Trametinib (MEKi) in Patients With BRAF Mu-tated Thyroid Carcinoma
Study Sponsor and Collaborators: National Comprehensive Cancer Net-work, Ohio State University Compre-hensive Cancer Center
Purpose: To study how well dab-rafenib works with or without tra-metinib in treating patients with recur-rent thyroid cancer.
Primary Outcome Measures: Overall objective response rate, defined as the proportion of patients who have a minor response, partial response, or complete response assessed accord-ing to RECIST. [Time Frame: up to 24 weeks]
Principal Investigator: Manisha Shah, MD, The Ohio State University Comprehensive Cancer Center; 614-293-4680, [email protected]
ClinicalTrials.gov Identifier: NCT01723202
Study Type: Phase II/Intervention-al/Parallel Assignment
Study Title: Central Neck Dissec-tion in Patients With Clinical Node Negative Thyroid Cancer
Study Sponsor and Collaborators: University of Wisconsin (Madison), National Cancer Institute, National In-stitutes of Health
Purpose: To study how well thyroid gland removal with or without central lymph node dissection works in treat-ing patients with thyroid cancer or suspected thyroid cancer that has not
spread to the lymph nodes.Primary Outcome Measures:
Postoperative serum calcium level, postoperative PTH, total calcium con-sumption, average episodes of hypocal-cemia (and symptom severity), and re-quirement for calcium/calcitriol [Time Frame: varies; see clinicaltrials.gov for more information]
Principal Investigator: Rebecca Sippel, MD, University of Wisconsin Hospital and Clinics; [email protected]
ClinicalTrials.gov Identifier: NCT02138214
Study Type: Phase II/Intervention-al/Single Group Assignment
Study Title: A Phase 2 Study of Trametinib in Combination With Ra-dioiodine (RAI) for RAS Mutant or RAS/RAF Wild-Type, RAI-Refractory Recurrent and/or Metastatic Thyroid Cancers
Study Sponsor and Collaborators: National Cancer Institute
Purpose: To study how well tra-metinib works in increasing tumoral iodine incorporation in patients with thyroid cancer that has come back or spread to another place in the body.
Primary Outcome Measures: Iodine incorporation in thyroid can-cer metastases to a predicted lesional absorbed radiation dose equal to or exceeding 2,000 cGy with the admin-istration of =< 300 mCi RAI (Cohort B), Progression free survival (Cohort A), Proportion of patients alive fol-lowing treatment with trametinib and I-124 (Cohort A) [Time Frame: At 6 months]
Principal Investigator: Alan Ho, MD, Memorial Sloan Kettering Cancer Center; 646-888-4235; [email protected]
ClinicalTrials.gov Identifier: NCT02152995 n
Editor’s Note: The clinical trials presented here do not represent all the trials listed on ClinicalTrials.gov. For the complete list, go to ClinicalTrials.gov.
The information contained in this Clinical Trials Resource
Guide includes actively recruiting clinical studies for patients with thyroid cancer. The trials are inves-tigating novel drug combinations; the effects of radioiodine; blood cell modification; biomarker-tar-geted therapies; and the effects of surgical intervention. All of the studies are listed on the National Institutes of Health website at Clin-icalTrials.gov.
Visit The ASCO Post website at ASCOPost.com
Renal Impairment: Less than 1% of ibrutinib is excreted renally. Ibrutinib exposure is not altered in patients with Creatinine clearance (CLcr) > 25 mL/min. There are no data in patients with severe renal impairment (CLcr < 25 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in Full Prescribing Information].Hepatic Impairment: Ibrutinib is metabolized in the liver. In a hepatic impairment study, data showed an increase in ibrutinib exposure. Following single dose administration, the AUC of ibrutinib increased 2.7-, 8.2- and 9.8-fold in subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe (Child-Pugh class C) hepatic impairment compared to subjects with normal liver function. The safety of IMBRUVICA has not been evaluated in patients with hepatic impairment. Monitor patients for signs of IMBRUVICA toxicity and follow dose modification guidance as needed. It is not recommended to administer IMBRUVICA to patients with moderate or severe hepatic impairment (Child-Pugh classes B and C) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3) in Full Prescribing Information].Females and Males of Reproductive Potential: Advise women to avoid becoming pregnant while taking IMBRUVICA because IMBRUVICA can cause fetal harm [see Use in Specific Populations].Plasmapheresis: Management of hyperviscosity in patients with WM may include plasmapheresis before and during treatment with IMBRUVICA. Modifications to IMBRUVICA dosing are not required.PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information).• Hemorrhage: Inform patients of the possibility of bleeding, and to report any signs or symptoms (blood in stools
or urine, prolonged or uncontrolled bleeding). Inform the patient that IMBRUVICA may need to be interrupted for medical or dental procedures [see Warnings and Precautions].
• Infections: Inform patients of the possibility of serious infection, and to report any signs or symptoms (fever,
chills, weakness, confusion) suggestive of infection [see Warnings and Precautions].• Atrial Fibrillation: Counsel patients to report any signs of palpitations, lightheadedness, dizziness, fainting, shortness
of breath, and chest discomfort [see Warnings and Precautions].• Second primary malignancies: Inform patients that other malignancies have occurred in patients who have been treated with
IMBRUVICA, including skin cancers and other carcinomas [see Warnings and Precautions].• Tumor lysis syndrome: Inform patients of the potential risk of tumor lysis syndrome and report any signs and symptoms
associated with this event to their healthcare provider for evaluation [see Warnings and Precautions].
• Embryo-fetal toxicity: Advise women of the potential hazard to a fetus and to avoid becoming pregnant [see Warnings
and Precautions].• Inform patients to take IMBRUVICA orally once daily according to their physician’s instructions
and that the capsules should be swallowed whole with a glass of water without being opened, broken, or chewed at approximately the same time each day [see Dosage and Administration (2.1) in Full Prescribing Information].
• Advise patients that in the event of a missed daily dose of IMBRUVICA, it should be taken as soon as possible on the same day with a return to the normal schedule the following day. Patients should not take extra capsules to make up the missed dose [see Dosage and Administration (2.5) in Full Prescribing Information].
• Advise patients of the common side effects associated with IMBRUVICA [see Adverse Reactions]. Direct the patient to a complete list of adverse drug reactions in PATIENT INFORMATION.
• Advise patients to inform their health care providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions].
• Advise patients that they may experience loose stools or diarrhea, and should contact their doctor if their diarrhea persists. Advise patients to maintain adequate hydration.
Active ingredient made in China.Distributed and Marketed by:Pharmacyclics LLCSunnyvale, CA USA 94085andMarketed by:Janssen Biotech, Inc.Horsham, PA USA 19044Patent http://www.imbruvica.comIMBRUVICA® is a registered trademark owned by Pharmacyclics LLC© Pharmacyclics LLC 2015© Janssen Biotech, Inc. 2015
IMBRUVICA® (ibrutinib) capsules
12562_Imbruvica_NowApproved_In4Indications_Ad_ASCO_071015_P5HPV_RL.indd 1 6/19/15 8:02 PM

Important Safety InformationWarning: Toxicity in hepatic impairment◗ IXEMPRA® (ixabepilone) in combination with
capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia- related death
◗ In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater in patients with hepatic impairment
◗ Caution should be used when using IXEMPRA as monotherapy in patients with AST or ALT >5 x ULN. Use of IXEMPRA in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended
◗ With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent in patients with hepatic impairment
Contraindications◗ IXEMPRA is contraindicated in patients: • with a known history of a severe (CTC grade 3/4)
hypersensitivity reaction to agents containing Cremophor® EL or its derivatives such as polyoxyethylated castor oil
• who have a baseline neutrophil count <1500 cells/mm3 or a platelet count <100,000 cells/mm3
Peripheral neuropathy◗ Peripheral neuropathy was common. Patients treated with
IXEMPRA should be monitored for symptoms of neuropathy, such as burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, or neuropathic pain
◗ Neuropathy occurred early during treatment; ~75% of new onset or worsening neuropathy occurred during the first 3 cycles. Patients experiencing new or worsening peripheral neuropathy may require changes in the dose or discontinuation of IXEMPRA
◗ Neuropathy was the most frequent cause of treatment discontinuation due to drug toxicity. Caution should be used when treating patients with diabetes mellitus or preexisting peripheral neuropathy
Myelosuppression◗ Myelosuppression is dose-dependent and primarily
manifested as neutropenia. ◗ Patients should be monitored for myelosuppression; frequent
peripheral blood cell counts are recommended for all patients receiving IXEMPRA
◗ Patients who experience severe neutropenia or thrombocytopenia should have their dose reduced. Neutropenia-related deaths occurred in 1.9% of 414 patients with normal hepatic function or mild hepatic impairment treated with IXEMPRA in combination with capecitabine. Neutropenia-related death occurred in 0.4% of 240 patients with IXEMPRA as monotherapy
Hypersensitivity reaction◗ Premedicate with an H1 and an H2 antagonist approximately
1 hour before IXEMPRA (ixabepilone) infusion and observe for hypersensitivity reactions (eg, flushing, rash, dyspnea, and bronchospasm)
◗ In case of severe hypersensitivity reactions, infusion of IXEMPRA should be stopped and aggressive supportive treatment (eg, epinephrine, corticosteroids) started
◗ Patients who experience a hypersensitivity reaction in one cycle of IXEMPRA must be premedicated in subsequent cycles with a corticosteroid in addition to the H1 and H2 antagonists, and extension of the infusion time should be considered
Pregnancy◗ Women should be advised not to become pregnant when
taking IXEMPRA. If this drug is used during pregnancy or the patient becomes pregnant, the patient should be apprised of the potential hazard to the fetus
Cardiac adverse reactions◗ Caution should be exercised in patients with a history of
cardiac disease. Discontinuation of IXEMPRA should be considered in patients who develop cardiac ischemia or impaired cardiac function due to reports of cardiovascular adverse reactions (eg, myocardial ischemia, supraventricular arrhythmia, and ventricular dysfunction). The frequency of cardiac adverse reactions (myocardial ischemia and ventricular dysfunction) was higher in the IXEMPRA in combination with capecitabine (1.9%) than in the capecitabine alone (0.3%) treatment group
Potential for cognitive impairment from excipients◗ IXEMPRA contains dehydrated alcohol USP. Consideration
should be given to the possibility of central nervous system and other effects of alcohol
Adverse reactions◗ The most common adverse reactions (≥20%) reported
by patients receiving IXEMPRA were peripheral sensory neuropathy, fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain. The following additional events occurred in ≥20% in combination treatment: palmar-plantar erythrodysesthesia (hand-foot) syndrome, anorexia, abdominal pain, nail disorder, and constipation. Drug-associated hematologic abnormalities (>40%) include neutropenia, leukopenia, anemia, and thrombocytopenia
Cremophor is a registered trademark of BASF AG.
AST = aspartate aminotransferase
ALT = alanine aminotransferase
ULN = upper limit of normal
CTC = common terminology criteria
Please see brief summary of Full Prescribing Information on adjacent pages.Reference: 1. IXEMPRA (ixabepilone) Prescribing Information; 2011.
For additional information, visit www.IXEMPRA.com.
Ixempra® is a registered trademark of R-Pharm US Operating LLC, a wholly owned subsidiary of R-Pharm US LLC. © 2015, R-PHARM US. All rights reserved. IXE-00009 9/15
In metastatic breast cancer patients whose disease is resistant to an anthracycline, a taxane, and capecitabine
Consider
IXEMPRA® (ixabepilone) monotherapy1
Indication◗ IXEMPRA® (ixabepilone) is indicated as monotherapy for the treatment
of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine
Warning: Toxicity in hepatic impairment1
◗ IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death
◗ In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater in patients with hepatic impairment
◗ Caution should be used when using IXEMPRA as monotherapy in patients with AST or ALT >5 x ULN. Use of IXEMPRA in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended
◗ With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent in patients with hepatic impairment
Please see Important Safety Information, including boxed WARNING regarding hepatic impairment, on next page.
34519_ccpmix_fa1_K_ASCOpst.indd 1-2 8/19/15 4:26 PM

Important Safety InformationWarning: Toxicity in hepatic impairment◗ IXEMPRA® (ixabepilone) in combination with
capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia- related death
◗ In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater in patients with hepatic impairment
◗ Caution should be used when using IXEMPRA as monotherapy in patients with AST or ALT >5 x ULN. Use of IXEMPRA in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended
◗ With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent in patients with hepatic impairment
Contraindications◗ IXEMPRA is contraindicated in patients: • with a known history of a severe (CTC grade 3/4)
hypersensitivity reaction to agents containing Cremophor® EL or its derivatives such as polyoxyethylated castor oil
• who have a baseline neutrophil count <1500 cells/mm3 or a platelet count <100,000 cells/mm3
Peripheral neuropathy◗ Peripheral neuropathy was common. Patients treated with
IXEMPRA should be monitored for symptoms of neuropathy, such as burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, or neuropathic pain
◗ Neuropathy occurred early during treatment; ~75% of new onset or worsening neuropathy occurred during the first 3 cycles. Patients experiencing new or worsening peripheral neuropathy may require changes in the dose or discontinuation of IXEMPRA
◗ Neuropathy was the most frequent cause of treatment discontinuation due to drug toxicity. Caution should be used when treating patients with diabetes mellitus or preexisting peripheral neuropathy
Myelosuppression◗ Myelosuppression is dose-dependent and primarily
manifested as neutropenia. ◗ Patients should be monitored for myelosuppression; frequent
peripheral blood cell counts are recommended for all patients receiving IXEMPRA
◗ Patients who experience severe neutropenia or thrombocytopenia should have their dose reduced. Neutropenia-related deaths occurred in 1.9% of 414 patients with normal hepatic function or mild hepatic impairment treated with IXEMPRA in combination with capecitabine. Neutropenia-related death occurred in 0.4% of 240 patients with IXEMPRA as monotherapy
Hypersensitivity reaction◗ Premedicate with an H1 and an H2 antagonist approximately
1 hour before IXEMPRA (ixabepilone) infusion and observe for hypersensitivity reactions (eg, flushing, rash, dyspnea, and bronchospasm)
◗ In case of severe hypersensitivity reactions, infusion of IXEMPRA should be stopped and aggressive supportive treatment (eg, epinephrine, corticosteroids) started
◗ Patients who experience a hypersensitivity reaction in one cycle of IXEMPRA must be premedicated in subsequent cycles with a corticosteroid in addition to the H1 and H2 antagonists, and extension of the infusion time should be considered
Pregnancy◗ Women should be advised not to become pregnant when
taking IXEMPRA. If this drug is used during pregnancy or the patient becomes pregnant, the patient should be apprised of the potential hazard to the fetus
Cardiac adverse reactions◗ Caution should be exercised in patients with a history of
cardiac disease. Discontinuation of IXEMPRA should be considered in patients who develop cardiac ischemia or impaired cardiac function due to reports of cardiovascular adverse reactions (eg, myocardial ischemia, supraventricular arrhythmia, and ventricular dysfunction). The frequency of cardiac adverse reactions (myocardial ischemia and ventricular dysfunction) was higher in the IXEMPRA in combination with capecitabine (1.9%) than in the capecitabine alone (0.3%) treatment group
Potential for cognitive impairment from excipients◗ IXEMPRA contains dehydrated alcohol USP. Consideration
should be given to the possibility of central nervous system and other effects of alcohol
Adverse reactions◗ The most common adverse reactions (≥20%) reported
by patients receiving IXEMPRA were peripheral sensory neuropathy, fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain. The following additional events occurred in ≥20% in combination treatment: palmar-plantar erythrodysesthesia (hand-foot) syndrome, anorexia, abdominal pain, nail disorder, and constipation. Drug-associated hematologic abnormalities (>40%) include neutropenia, leukopenia, anemia, and thrombocytopenia
Cremophor is a registered trademark of BASF AG.
AST = aspartate aminotransferase
ALT = alanine aminotransferase
ULN = upper limit of normal
CTC = common terminology criteria
Please see brief summary of Full Prescribing Information on adjacent pages.Reference: 1. IXEMPRA (ixabepilone) Prescribing Information; 2011.
For additional information, visit www.IXEMPRA.com.
Ixempra® is a registered trademark of R-Pharm US Operating LLC, a wholly owned subsidiary of R-Pharm US LLC. © 2015, R-PHARM US. All rights reserved. IXE-00009 9/15
In metastatic breast cancer patients whose disease is resistant to an anthracycline, a taxane, and capecitabine
Consider
IXEMPRA® (ixabepilone) monotherapy1
Indication◗ IXEMPRA® (ixabepilone) is indicated as monotherapy for the treatment
of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine
Warning: Toxicity in hepatic impairment1
◗ IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death
◗ In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater in patients with hepatic impairment
◗ Caution should be used when using IXEMPRA as monotherapy in patients with AST or ALT >5 x ULN. Use of IXEMPRA in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended
◗ With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent in patients with hepatic impairment
Please see Important Safety Information, including boxed WARNING regarding hepatic impairment, on next page.
34519_ccpmix_fa1_K_ASCOpst.indd 1-2 8/19/15 4:26 PM

IXEMPRA® Kit (ixabepilone) for Injection, for intravenous infusion onlyBrief Summary of Prescribing Information. For complete prescribing information consult official package insert.
WARNING: TOXICITY IN HEPATIC IMPAIRMENTIXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death [see Contraindications and Warnings and Precautions].
INDICATIONS AND USAGEIXEMPRA (ixabepilone) is indicated in combination with capecitabine for the treatment of patients with metastatic or locally advanced breast cancer resistant to treatment with an anthracycline and a taxane, or whose cancer is taxane resistant and for whom further anthracycline therapy is contraindicated. Anthracycline resistance is defined as progression while on therapy or within 6 months in the adjuvant setting or 3 months in the metastatic setting. Taxane resistance is defined as progression while on therapy or within 12 months in the adjuvant setting or 4 months in the metastatic setting.IXEMPRA is indicated as monotherapy for the treatment of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine.
CONTRAINDICATIONSIXEMPRA is contraindicated in patients with a history of a severe (CTC grade 3/4) hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) [see Warnings and Precautions].IXEMPRA is contraindicated in patients who have a neutrophil count <1500 cells/mm3 or a platelet count <100,000 cells/mm3 [see Warnings and Precautions].IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning and Warnings and Precautions].WARNINGS AND PRECAUTIONSPeripheral NeuropathyPeripheral neuropathy was common (see Table 1). Patients treated with IXEMPRA should be monitored for symptoms of neuropathy, such as burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, or neuropathic pain. Neuropathy occurred early during treatment; ~75% of new onset or worsening neuropathy occurred during the first 3 cycles. Patients experiencing new or worsening symptoms may require a reduction or delay in the dose of IXEMPRA [see Dosage and Administration (2.2) in Full Prescribing Information]. In clinical studies, peripheral neuropathy was managed through dose reductions, dose delays, and treatment discontinuation. Neuropathy was the most frequent cause of treatment discontinuation due to drug toxicity. In Studies 046 and 081, 80% and 87%, respectively, of patients with peripheral neuropathy who received IXEMPRA had improvement or no worsening of their neuropathy following dose reduction. For patients with grade 3/4 neuropathy in Studies 046 and 081, 76% and 79%, respectively, had documented improvement to baseline or grade 1, twelve weeks after onset.Table 1: Treatment-related Peripheral Neuropathy
IXEMPRA with capecitabine Study 046
IXEMPRA as monotherapyStudy 081
Peripheral neuropathy (all grades)a,b
Peripheral neuropathy (grades 3/4)a,b
Discontinuation due to neuropathyMedian number of cycles to onset of
grade 3/4 neuropathyMedian time to improvement of grade 3/4
neuropathy to baseline or to grade 1
67%23%21%
4
6.0 weeks
63%14%6%4
4.6 weeks
a Sensory and motor neuropathy combined.b 24% and 27% of patients in 046 and 081, respectively, had preexisting neuropathy (grade 1).
A pooled analysis of 1540 cancer patients treated with IXEMPRA indicated that patients with diabetes mellitus or preexisting peripheral neuropathy may be at increased risk of severe neuropathy. Prior therapy with neurotoxic chemotherapy agents did not predict the development of neuropathy. Patients with moderate to severe neuropathy (grade 2 or greater) were excluded from studies with IXEMPRA. Caution should be used when treating patients with diabetes mellitus or preexisting peripheral neuropathy.
MyelosuppressionMyelosuppression is dose-dependent and primarily manifested as neutropenia. In clinical studies, grade 4 neutropenia (<500 cells/mm3) occurred in 36% of patients treated with IXEMPRA in combination with capecitabine and 23% of patients treated with IXEMPRA monotherapy. Febrile neutropenia and infection with neutropenia were reported in 5% and 6% of patients treated with IXEMPRA in combination with capecitabine, respectively, and 3% and 5% of patients treated with IXEMPRA as monotherapy, respectively. Neutropenia-related death occurred in 1.9% of 414 patients with normal hepatic function or mild hepatic impairment treated with IXEMPRA in combination with capecitabine. The rate of neutropenia-related deaths was higher (29%, 5 out of 17) in patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN [see Boxed Warning, Contraindications, and Warnings and Precautions]. Neutropenia-related death occurred in 0.4% of 240 patients treated with IXEMPRA as monotherapy. No neutropenia-related deaths were reported in 24 patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN treated with IXEMPRA monotherapy. IXEMPRA must not be administered to patients with a neutrophil count <1500 cells/mm3. To monitor for myelosuppression, frequent peripheral blood cell counts are recommended for all patients receiving IXEMPRA. Patients who experience severe neutropenia or thrombocytopenia should have their dose reduced [see Dosage and Administration (2.2) in Full Prescribing Information].Hepatic ImpairmentPatients with baseline AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN experienced greater toxicity than patients with baseline AST or ALT ≤2.5 x ULN or bilirubin ≤1.5 x ULN when treated with IXEMPRA at 40 mg/m2 in combination with capecitabine or as monotherapy in breast cancer studies. In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater [see Warnings and Precautions]. With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent. The safety and pharmacokinetics of IXEMPRA as monotherapy were evaluated in a dose escalation study in 56 patients with varying degrees of hepatic impairment. Exposure was increased in patients with elevated AST or bilirubin [see Use in Specific Populations].IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity- and neutropenia-related death [see Boxed Warning, Contraindications, and Warnings and Precautions]. Patients who are treated with IXEMPRA as monotherapy should receive a reduced dose depending on the degree of hepatic impairment [see Dosage and Administration (2.2) in Full Prescribing Information]. Use in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended. Limited data are available for patients with AST or ALT >5 x ULN. Caution should be used when treating these patients [see Dosage and Administration (2.2) in Full Prescribing Information].Hypersensitivity ReactionsPatients with a history of a severe hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) should not be treated with IXEMPRA. All patients should be premedicated with an H1 and an H2 antagonist approximately 1 hour before IXEMPRA infusion and be observed for hypersensitivity reactions (eg, flushing, rash, dyspnea, and bronchospasm). In case of severe hypersensitivity reactions, infusion of IXEMPRA should be stopped and aggressive supportive treatment (eg, epinephrine, corticosteroids) started. Of the 1323 patients treated with IXEMPRA in clinical studies, 9 patients (1%) had experienced severe hypersensitivity reactions (including anaphylaxis). Three of the 9 patients were able to be retreated. Patients who experience a hypersensitivity reaction in one cycle of IXEMPRA must be premedicated in subsequent cycles with a corticosteroid in addition to the H1 and H2 antagonists, and extension of the infusion time should be considered [see Dosage and Administration (2.3) in Full Prescribing Information and Contraindications].PregnancyPregnancy Category D.IXEMPRA may cause fetal harm when administered to pregnant women. There are no adequate and well-controlled studies with IXEMPRA in pregnant women. Women should be advised not to become pregnant when taking IXEMPRA. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.Ixabepilone was studied for effects on embryo-fetal development in pregnant rats and rabbits given IV doses of 0.02, 0.08, and 0.3 mg/kg/day and 0.01, 0.03, 0.11, and 0.3 mg/kg/day, respectively. There were no teratogenic effects. In rats, an increase in resorptions and post-implantation loss and a decrease in the number of live fetuses and fetal weight was observed at the maternally
(Continued)
toxic dose of 0.3 mg/kg/day (approximately one-tenth the human clinical exposure based on AUC). Abnormalities included a reduced ossification of caudal vertebrae, sternebrae, and metacarpals. In rabbits, ixabepilone caused maternal toxicity (death) and embryo-fetal toxicity (resorptions) at 0.3 mg/kg/day (approximately one-tenth the human clinical dose based on body surface area). No fetuses were available at this dose for evaluation.Cardiac Adverse ReactionsThe frequency of cardiac adverse reactions (myocardial ischemia and ventricular dysfunction) was higher in the IXEMPRA (ixabepilone) in combination with capecitabine (1.9%) than in the capecitabine alone (0.3%) treatment group. Supraventricular arrhythmias were observed in the combination arm (0.5%) and not in the capecitabine alone arm. Caution should be exercised in patients with a history of cardiac disease. Discontinuation of IXEMPRA should be considered in patients who develop cardiac ischemia or impaired cardiac function.Potential for Cognitive Impairment from ExcipientsSince IXEMPRA contains dehydrated alcohol USP, consideration should be given to the possibility of central nervous system and other effects of alcohol [see Description (11) in Full Prescribing Information].ADVERSE REACTIONSThe following adverse reactions are discussed in greater detail in other sections. • Peripheral neuropathy [see Warnings and Precautions] • Myelosuppression [see Warnings and Precautions] • Hypersensitivity reactions [see Warnings and Precautions]Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.Unless otherwise specified, assessment of adverse reactions is based on one randomized study (Study 046) and one single-arm study (Study 081). In Study 046, 369 patients with metastatic breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 21 days, combined with capecitabine 1000 mg/m2 twice daily for 2 weeks followed by a 1-week rest period. Patients treated with capecitabine as monotherapy (n=368) in this study received 1250 mg/m2 twice daily for 2 weeks every 21 days. In Study 081, 126 patients with metastatic or locally advanced breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 3 weeks.The most common adverse reactions (≥20%) reported by patients receiving IXEMPRA were peripheral sensory neuropathy, fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain. The following additional reactions occurred in ≥20% in combination treatment: palmar-plantar erythrodysesthesia (hand-foot) syndrome, anorexia, abdominal pain, nail disorder, and constipation. The most common hematologic abnormalities (>40%) include neutropenia, leukopenia, anemia, and thrombocytopenia.Table 2 presents nonhematologic adverse reactions reported in 5% or more of patients. Hematologic abnormalities are presented separately in Table 3.
Table 2: Nonhematologic Drug-related Adverse Reactions Occurring in at Least 5% of Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126System Organ Classa/
Preferred TermTotal (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Infections and Infestations Upper respiratory tract infectionb 4 0 3 0 6 0Blood and Lymphatic SystemDisorders Febrile neutropenia 5 4c 1 1d 3 3d
Immune System Disorders Hypersensitivityb 2 1d 0 0 5 1d
Metabolism and Nutrition Disorders Anorexiab
Dehydrationb345
3d
2152
1d
<1d192
2d
1d
Psychiatric Disorders Insomniab 9 <1d 2 0 5 0Nervous System Disorders Peripheral neuropathy Sensory neuropathy b,e
Motor neuropathyb
Headache Taste disorderb Dizziness
65168128
215d
<1d
01d
16<1345
00001d
62101167
141d
000
Eye Disorders Lacrimation increased 5 0 4 <1d 4 0
Vascular Disorders Hot flushb 5 0 2 0 6 0Respiratory, Thoracic, andMediastinal Disorders Dyspneab
Coughb76
10
42
10
92
1d
0Gastrointestinal Disorders Nausea Vomitingb
Stomatitis/mucositisb
Diarrheab
Constipation Abdominal painb
Gastroesophageal reflux diseaseb
5339314422247
3d
4d
46d
02d
1d
402420396148
2d
23d
9<1d
1d
0
4229292216136
2d
1d
61d
2d
2d
0
Skin and Subcutaneous TissueDisorders Alopeciab
Skin rashb
Nail disorderb
Palmar-plantar erythrodysesthesia syndromeb,f
Pruritus Skin exfoliationb
Skin hyperpigmentationb
31172464
5511
01d
2d
18d
0<1d
0
371063
2314
00
<1d
17d
000
48998
622
02d
02d
1d
00
Musculoskeletal, Connective Tissue,and Bone Disorders Myalgia/arthralgiab
Musculoskeletal painb3923
8d
2d55
<1d
04920
8d
3d
General Disorders and AdministrationSite Conditions Fatigue/astheniab
Edemab
Pyrexia Painb
Chest painb
6081094
1601d
1d
1d
29542
<1
4<1d
000
569885
131d
1d
3d
1d
Table 2: Nonhematologic Drug-related Adverse Reactions Occurring in at Least 5% of Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126System Organ Classa/
Preferred TermTotal (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
a System organ class presented as outlined in Guidelines for Preparing Core Clinical Safety Information on Drugs by the Council for International Organizations of Medical Sciences (CIOMS). b A composite of multiple MedDRA Preferred Terms. c NCI CTC grading for febrile neutropenia ranges from Grade 3 to 5. Three patients (1%) experienced Grade 5 (fatal) febrile neutropenia. Other neutropenia-related deaths (9) occurred in the absence of reported febrile neutropenia [see Warnings and Precautions]. d No grade 4 reports. e Peripheral sensory neuropathy (graded with the NCI CTC scale) was defined as the occurrence of any of the following: areflexia, burning sensation, dysesthesia, hyperesthesia, hypoesthesia, hyporeflexia, neuralgia, neuritis, neuropathy, neuropathy peripheral, neurotoxicity, painful response to normal stimuli, paresthesia, pallanesthesia, peripheral sensory neuropathy, polyneuropathy, polyneuropathy toxic and sensorimotor disorder. Peripheral motor neuropathy was defined as the occurrence of any of the following: multifocal motor neuropathy, neuromuscular toxicity, peripheral motor neuropathy, and peripheral sensorimotor neuropathy. f Palmar-plantar erythrodysesthesia (hand-foot syndrome) was graded on a 1-3 severity scale in Study 046.
Table 3: Hematologic Abnormalities in Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126
Hematology ParameterGrade 3
(%)Grade 4
(%)Grade 3
(%)Grade 4
(%)Grade 3
(%)Grade 4
(%)
Neutropeniaa 32 36 9 2 31 23
Leukopenia (WBC) 41 16 5 1 36 13
Anemia (Hgb) 8 2 4 1 6 2
Thrombocytopenia 5 3 2 2 5 2a G-CSF (granulocyte colony stimulating factor) or GM-CSF (granulocyte macrophage colony stimulating factor) was used in 20% and 17% of patients who received IXEMPRA in Study 046 and Study 081, respectively.
The following serious adverse reactions were also reported in 1323 patients treated with IXEMPRA as monotherapy or in combination with other therapies in Phase 2 and 3 studies.Infections and Infestations: sepsis, pneumonia, infection, neutropenic infection, urinary tract infection, bacterial infection, enterocolitis, laryngitis, lower respiratory tract infectionBlood and Lymphatic System Disorders: coagulopathy, lymphopeniaMetabolism and Nutrition Disorders: hyponatremia, metabolic acidosis, hypokalemia, hypovolemiaNervous System Disorders: cognitive disorder, syncope, cerebral hemorrhage, abnormal coordination, lethargyCardiac Disorders: myocardial infarction, supraventricular arrhythmia, left ventricular dysfunction, angina pectoris, atrial flutter, cardiomyopathy, myocardial ischemiaVascular Disorders: hypotension, thrombosis, embolism, hemorrhage, hypovolemic shock, vasculitisRespiratory, Thoracic, and Mediastinal Disorders: pneumonitis, hypoxia, respiratory failure, acute pulmonary edema, dysphonia, pharyngolaryngeal painGastrointestinal Disorders: ileus, colitis, impaired gastric emptying, esophagitis, dysphagia, gastritis, gastrointestinal hemorrhageHepatobiliary Disorders: acute hepatic failure, jaundiceSkin and Subcutaneous Tissue Disorders: erythema multiformeMusculoskeletal, Connective Tissue, and Bone Disorders: muscular weakness, muscle spasms, trismusRenal and Urinary Disorders: nephrolithiasis, renal failureGeneral Disorders and Administration Site Conditions: chillsInvestigations: increased transaminases, increased blood alkaline phosphatase, increased gamma-glutamyltransferasePostmarketing ExperienceRadiation recall has been reported during postmarketing use of IXEMPRA. Because this reaction was reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.DRUG INTERACTIONSEffect of Other Drugs on IxabepiloneDrugs That May Increase Ixabepilone Plasma ConcentrationsCYP3A4 Inhibitors: Coadministration of ixabepilone with ketoconazole, a potent CYP3A4 inhibitor, increased ixabepilone AUC by 79% compared to ixabepilone treatment alone. If alternative treatment cannot be administered, a dose adjustment should be considered. The effect of mild or moderate inhibitors (eg, erythromycin, fluconazole, or verapamil) on exposure to ixabepilone has not been studied. Therefore, caution should be used when administering mild or moderate CYP3A4 inhibitors during treatment with IXEMPRA, and alternative therapeutic agents that do not inhibit CYP3A4 should be considered. Patients receiving CYP3A4 inhibitors during treatment with IXEMPRA should be monitored closely for acute toxicities (eg, frequent monitoring of peripheral blood counts between cycles of IXEMPRA) [see Dosage and Administration (2.2) in Full Prescribing Information].Drugs That May Decrease Ixabepilone Plasma ConcentrationsCYP3A4 Inducers: IXEMPRA is a CYP3A4 substrate. Coadministration of IXEMPRA with rifampin, a potent CYP3A4 inducer, decreased ixabepilone AUC by 43% compared to IXEMPRA treatment alone. Other strong CYP3A4 inducers (eg, dexamethasone, phenytoin, carbamazepine, rifabutin, and phenobarbital) may also decrease ixabepilone concentrations leading to subtherapeutic levels. Therefore, therapeutic agents with low enzyme induction potential should be considered for coadministration with IXEMPRA. St. John’s Wort may decrease ixabepilone plasma concentrations unpredictably and should be avoided. If patients must be coadministrated a strong CYP3A4 inducer, a gradual dose adjustment may be considered [see Dosage and Administration (2.2) in Full Prescribing Information]. Effect of Ixabepilone on Other DrugsIxabepilone does not inhibit CYP enzymes at relevant clinical concentrations and is not expected to alter the plasma concentrations of other drugs [see Clinical Pharmacology (12.3) in Full Prescribing Information].CapecitabineIn patients with cancer who received ixabepilone (40 mg/m2) in combination with capecitabine (1000 mg/m2), ixabepilone Cmax decreased by 19%, capecitabine Cmax decreased by 27%, and 5-fluorouracil AUC increased by 14%, as compared to ixabepilone or capecitabine administered separately. The interaction is not clinically significant given that the combination treatment is supported by efficacy data.
USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions].Nursing MothersIt is not known whether ixabepilone is excreted into human milk. Following intravenous administration of radiolabeled ixabepilone to rats on days 7 to 9 postpartum, concentrations of radioactivity in milk were comparable with those in plasma and declined in parallel with the plasma concentrations. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ixabepilone, a decision must be made whether to discontinue nursing or to discontinue IXEMPRA taking into account the importance of the drug to the mother.
Pediatric UseThe effectiveness of IXEMPRA (ixabepilone) in pediatric patients has not been established. IXEMPRA was evaluated in one Phase 1 and one Phase 2 trial. The pediatric patients had a safety profile consistent with that seen in adults, and no new safety signals were identified.In the Phase 1 open-label, dose-finding trial, the safety of IXEMPRA was evaluated in 19 pediatric patients with advanced or refractory solid tumors and 2 with acute leukemias. IXEMPRA was administered as a one-hour IV infusion daily for the first five days of a 21-day cycle at one of 5 dose levels, ranging from 3 to 10 mg/m2. Among the 21 patients, 12 ranged in age from 2 to 12 years and 9 ranged from 13 to 18 years. The maximum tolerated dose was 8 mg/m2 IV daily for 5 days every 21 days. No significant activity was observed. The pharmacokinetics of ixabepilone were characterized by population pharmacokinetic analysis of data for 16 patients from this trial, who were aged 2 to 18 years (median 12 years). The pharmacokinetic parameters of ixabepilone in these pediatric patients were compared to the corresponding parameters of 130 adult patients enrolled in clinical trials using the same dosing schedule. The median BSA normalized clearance of ixabepilone in pediatric patients (17 L/h/m2) was similar to that in adult patients (20 L/h/m2).In the Phase 2 trial of 59 patients with advanced or refractory solid tumors, 28 ranged in age from 3 to 12 years and 19 ranged in age from 13 to 18 years. Twelve additional patients over the age of 18 were treated in this trial. IXEMPRA was administered at a dose of 8 mg/m2 IV daily for 5 days every 21 days. This trial was terminated early due to lack of efficacy.Geriatric UseClinical studies of IXEMPRA did not include sufficient numbers of subjects aged sixty-five and over to determine whether they respond differently from younger subjects.Forty-five of 431 patients treated with IXEMPRA in combination with capecitabine were ≥65 years of age and 3 patients were ≥75. Overall, the incidence of grade 3/4 adverse reactions was higher in patients ≥65 years of age versus those <65 years of age (82% versus 68%) including grade 3/4 stomatitis (9% versus 1%), diarrhea (9% versus 6%), palmar-plantar erythrodysesthesia syndrome (27% versus 20%), peripheral neuropathy (24% versus 22%), febrile neutropenia (9% versus 3%), fatigue (16% versus 12%), and asthenia (11% versus 6%). Toxicity-related deaths occurred in 2 (4.7%) of 43 patients ≥65 years with normal baseline hepatic function or mild impairment.Thirty-two of 240 breast cancer patients treated with IXEMPRA as monotherapy were ≥65 years of age and 6 patients were ≥75. No overall differences in safety were observed in these patients compared to those <65 years of age.Hepatic ImpairmentIXEMPRA was evaluated in 56 patients with mild to severe hepatic impairment defined by bilirubin levels and AST levels. Compared to patients with normal hepatic function (n=17), the area under the curve (AUC0-infinity) of ixabepilone increased by: • 22% in patients with a) bilirubin >1 – 1.5 x ULN or b) AST >ULN but bilirubin <1.5 x ULN; • 30% in patients with bilirubin >1.5 – 3 x ULN and any AST level; and • 81% in patients with bilirubin >3 x ULN and any AST level.Doses of 10 and 20 mg/m2 as monotherapy were tolerated in 17 patients with severe hepatic impairment (bilirubin >3 x ULN).IXEMPRA in combination with capecitabine must not be given to patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning, Contraindications, and Warnings and Precautions]. Dose reduction is recommended when administering IXEMPRA as monotherapy to patients with hepatic impairment [see Dosage and Administration (2.3) in Full Prescribing Information]. Because there is a need for dosage adjustment based upon hepatic function, assessment of hepatic function is recommended before initiation of IXEMPRA and periodically thereafter.Renal ImpairmentIxabepilone is minimally excreted via the kidney. No controlled pharmacokinetic studies were conducted with IXEMPRA in patients with renal impairment. IXEMPRA in combination with capecitabine has not been evaluated in patients with calculated creatinine clearance of <50 mL/min. IXEMPRA as monotherapy has not been evaluated in patients with creatinine >1.5 times ULN. In a population pharmacokinetic analysis of IXEMPRA as monotherapy, there was no meaningful effect of mild and moderate renal insufficiency (CrCL >30 mL/min) on the pharmacokinetics of ixabepilone.
OVERDOSAGEExperience with overdose of IXEMPRA is limited to isolated cases. The adverse reactions reported in these cases included peripheral neuropathy, fatigue, musculoskeletal pain/myalgia, and gastrointestinal symptoms (nausea, anorexia, diarrhea, abdominal pain, stomatitis). The highest dose mistakenly received was 100 mg/m2 (total dose 185 mg).There is no known antidote for overdosage of IXEMPRA. In case of overdosage, the patient should be closely monitored and supportive treatment should be administered. Management of overdose should include supportive medical interventions to treat the presenting clinical manifestations.
NONCLINICAL TOXICOLOGYCarcinogenesis, Mutagenesis, Impairment of FertilityCarcinogenicity studies with ixabepilone have not been conducted. Ixabepilone did not induce mutations in the microbial mutagenesis (Ames) assay and was not clastogenic in an in vitro cytogenetic assay using primary human lymphocytes. Ixabepilone was clastogenic (induction of micronuclei) in the in vivo rat micronucleus assay at doses ≥0.625 mg/kg/day.There were no effects on male or female rat mating or fertility at doses up to 0.2 mg/kg/day in both males and females (approximately one-fifteenth the expected human clinical exposure based on AUC). The effect of ixabepilone on human fertility is unknown. However, when rats were given an IV infusion of ixabepilone during breeding and through the first 7 days of gestation, a significant increase in resorptions and pre- and post-implantation loss and a decrease in the number of corpora lutea was observed at 0.2 mg/kg/day. Testicular atrophy or degeneration was observed in 6-month rat and 9-month dog studies when ixabepilone was given every 21 days at intravenous doses of 6.7 mg/kg (40 mg/m2) in rats (approximately 2.1 times the expected clinical exposure based on AUC) and 0.5 and 0.75 mg/kg (10 and 15 mg/m2) in dogs (approximately 0.2 and 0.4 times the expected clinical exposure based on AUC).Animal ToxicologyOverdoseIn rats, single intravenous doses of ixabepilone from 60 to 180 mg/m2 (mean AUC values ≥8156 ng•h/mL) were associated with mortality occurring between 5 and 14 days after dosing, and toxicity was principally manifested in the gastrointestinal, hematopoietic (bone-marrow), lymphatic, peripheral-nervous, and male-reproductive systems. In dogs, a single intravenous dose of 100 mg/m2 (mean AUC value of 6925 ng•h/mL) was markedly toxic, inducing severe gastrointestinal toxicity and death 3 days after dosing.
PATIENT COUNSELING INFORMATION[see FDA-Approved Patient Labeling in Full Prescribing Information]Peripheral NeuropathyPatients should be advised to report to their physician any numbness and tingling of the hands or feet [see Warnings and Precautions].Fever/NeutropeniaPatients should be instructed to call their physician if a fever of 100.5° F or greater or other evidence of potential infection such as chills, cough, or burning or pain on urination develops [see Warnings and Precautions].Hypersensitivity ReactionsPatients should be advised to call their physician if they experience urticaria, pruritus, rash, flushing, swelling, dyspnea, chest tightness or other hypersensitivity-related symptoms following an infusion of IXEMPRA [see Warnings and Precautions].PregnancyPatients should be advised to use effective contraceptive measures to prevent pregnancy and to avoid nursing during treatment with IXEMPRA [see Warnings and Precautions and Use in Specific Populations].Cardiac Adverse Reactions Patients should be advised to report to their physician chest pain, difficulty breathing, palpitations or unusual weight gain [see Warnings and Precautions].IXEMPRA® (ixabepilone) for injection Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, GermanyDILUENT for IXEMPRA Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, GermanyDistributed by Bristol-Myers Squibb Company, Princeton, NJ 08543 USA
1236925A7 5645-0006 Rev October 2011RPH-00001
(Continued)
34519_ccpmix_fa1_K_ASCOpst.indd 4 8/19/15 4:26 PM

IXEMPRA® Kit (ixabepilone) for Injection, for intravenous infusion onlyBrief Summary of Prescribing Information. For complete prescribing information consult official package insert.
WARNING: TOXICITY IN HEPATIC IMPAIRMENTIXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death [see Contraindications and Warnings and Precautions].
INDICATIONS AND USAGEIXEMPRA (ixabepilone) is indicated in combination with capecitabine for the treatment of patients with metastatic or locally advanced breast cancer resistant to treatment with an anthracycline and a taxane, or whose cancer is taxane resistant and for whom further anthracycline therapy is contraindicated. Anthracycline resistance is defined as progression while on therapy or within 6 months in the adjuvant setting or 3 months in the metastatic setting. Taxane resistance is defined as progression while on therapy or within 12 months in the adjuvant setting or 4 months in the metastatic setting.IXEMPRA is indicated as monotherapy for the treatment of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine.
CONTRAINDICATIONSIXEMPRA is contraindicated in patients with a history of a severe (CTC grade 3/4) hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) [see Warnings and Precautions].IXEMPRA is contraindicated in patients who have a neutrophil count <1500 cells/mm3 or a platelet count <100,000 cells/mm3 [see Warnings and Precautions].IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning and Warnings and Precautions].WARNINGS AND PRECAUTIONSPeripheral NeuropathyPeripheral neuropathy was common (see Table 1). Patients treated with IXEMPRA should be monitored for symptoms of neuropathy, such as burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, or neuropathic pain. Neuropathy occurred early during treatment; ~75% of new onset or worsening neuropathy occurred during the first 3 cycles. Patients experiencing new or worsening symptoms may require a reduction or delay in the dose of IXEMPRA [see Dosage and Administration (2.2) in Full Prescribing Information]. In clinical studies, peripheral neuropathy was managed through dose reductions, dose delays, and treatment discontinuation. Neuropathy was the most frequent cause of treatment discontinuation due to drug toxicity. In Studies 046 and 081, 80% and 87%, respectively, of patients with peripheral neuropathy who received IXEMPRA had improvement or no worsening of their neuropathy following dose reduction. For patients with grade 3/4 neuropathy in Studies 046 and 081, 76% and 79%, respectively, had documented improvement to baseline or grade 1, twelve weeks after onset.Table 1: Treatment-related Peripheral Neuropathy
IXEMPRA with capecitabine Study 046
IXEMPRA as monotherapyStudy 081
Peripheral neuropathy (all grades)a,b
Peripheral neuropathy (grades 3/4)a,b
Discontinuation due to neuropathyMedian number of cycles to onset of
grade 3/4 neuropathyMedian time to improvement of grade 3/4
neuropathy to baseline or to grade 1
67%23%21%
4
6.0 weeks
63%14%6%4
4.6 weeks
a Sensory and motor neuropathy combined.b 24% and 27% of patients in 046 and 081, respectively, had preexisting neuropathy (grade 1).
A pooled analysis of 1540 cancer patients treated with IXEMPRA indicated that patients with diabetes mellitus or preexisting peripheral neuropathy may be at increased risk of severe neuropathy. Prior therapy with neurotoxic chemotherapy agents did not predict the development of neuropathy. Patients with moderate to severe neuropathy (grade 2 or greater) were excluded from studies with IXEMPRA. Caution should be used when treating patients with diabetes mellitus or preexisting peripheral neuropathy.
MyelosuppressionMyelosuppression is dose-dependent and primarily manifested as neutropenia. In clinical studies, grade 4 neutropenia (<500 cells/mm3) occurred in 36% of patients treated with IXEMPRA in combination with capecitabine and 23% of patients treated with IXEMPRA monotherapy. Febrile neutropenia and infection with neutropenia were reported in 5% and 6% of patients treated with IXEMPRA in combination with capecitabine, respectively, and 3% and 5% of patients treated with IXEMPRA as monotherapy, respectively. Neutropenia-related death occurred in 1.9% of 414 patients with normal hepatic function or mild hepatic impairment treated with IXEMPRA in combination with capecitabine. The rate of neutropenia-related deaths was higher (29%, 5 out of 17) in patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN [see Boxed Warning, Contraindications, and Warnings and Precautions]. Neutropenia-related death occurred in 0.4% of 240 patients treated with IXEMPRA as monotherapy. No neutropenia-related deaths were reported in 24 patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN treated with IXEMPRA monotherapy. IXEMPRA must not be administered to patients with a neutrophil count <1500 cells/mm3. To monitor for myelosuppression, frequent peripheral blood cell counts are recommended for all patients receiving IXEMPRA. Patients who experience severe neutropenia or thrombocytopenia should have their dose reduced [see Dosage and Administration (2.2) in Full Prescribing Information].Hepatic ImpairmentPatients with baseline AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN experienced greater toxicity than patients with baseline AST or ALT ≤2.5 x ULN or bilirubin ≤1.5 x ULN when treated with IXEMPRA at 40 mg/m2 in combination with capecitabine or as monotherapy in breast cancer studies. In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity-related deaths was greater [see Warnings and Precautions]. With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent. The safety and pharmacokinetics of IXEMPRA as monotherapy were evaluated in a dose escalation study in 56 patients with varying degrees of hepatic impairment. Exposure was increased in patients with elevated AST or bilirubin [see Use in Specific Populations].IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity- and neutropenia-related death [see Boxed Warning, Contraindications, and Warnings and Precautions]. Patients who are treated with IXEMPRA as monotherapy should receive a reduced dose depending on the degree of hepatic impairment [see Dosage and Administration (2.2) in Full Prescribing Information]. Use in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended. Limited data are available for patients with AST or ALT >5 x ULN. Caution should be used when treating these patients [see Dosage and Administration (2.2) in Full Prescribing Information].Hypersensitivity ReactionsPatients with a history of a severe hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) should not be treated with IXEMPRA. All patients should be premedicated with an H1 and an H2 antagonist approximately 1 hour before IXEMPRA infusion and be observed for hypersensitivity reactions (eg, flushing, rash, dyspnea, and bronchospasm). In case of severe hypersensitivity reactions, infusion of IXEMPRA should be stopped and aggressive supportive treatment (eg, epinephrine, corticosteroids) started. Of the 1323 patients treated with IXEMPRA in clinical studies, 9 patients (1%) had experienced severe hypersensitivity reactions (including anaphylaxis). Three of the 9 patients were able to be retreated. Patients who experience a hypersensitivity reaction in one cycle of IXEMPRA must be premedicated in subsequent cycles with a corticosteroid in addition to the H1 and H2 antagonists, and extension of the infusion time should be considered [see Dosage and Administration (2.3) in Full Prescribing Information and Contraindications].PregnancyPregnancy Category D.IXEMPRA may cause fetal harm when administered to pregnant women. There are no adequate and well-controlled studies with IXEMPRA in pregnant women. Women should be advised not to become pregnant when taking IXEMPRA. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.Ixabepilone was studied for effects on embryo-fetal development in pregnant rats and rabbits given IV doses of 0.02, 0.08, and 0.3 mg/kg/day and 0.01, 0.03, 0.11, and 0.3 mg/kg/day, respectively. There were no teratogenic effects. In rats, an increase in resorptions and post-implantation loss and a decrease in the number of live fetuses and fetal weight was observed at the maternally
(Continued)
toxic dose of 0.3 mg/kg/day (approximately one-tenth the human clinical exposure based on AUC). Abnormalities included a reduced ossification of caudal vertebrae, sternebrae, and metacarpals. In rabbits, ixabepilone caused maternal toxicity (death) and embryo-fetal toxicity (resorptions) at 0.3 mg/kg/day (approximately one-tenth the human clinical dose based on body surface area). No fetuses were available at this dose for evaluation.Cardiac Adverse ReactionsThe frequency of cardiac adverse reactions (myocardial ischemia and ventricular dysfunction) was higher in the IXEMPRA (ixabepilone) in combination with capecitabine (1.9%) than in the capecitabine alone (0.3%) treatment group. Supraventricular arrhythmias were observed in the combination arm (0.5%) and not in the capecitabine alone arm. Caution should be exercised in patients with a history of cardiac disease. Discontinuation of IXEMPRA should be considered in patients who develop cardiac ischemia or impaired cardiac function.Potential for Cognitive Impairment from ExcipientsSince IXEMPRA contains dehydrated alcohol USP, consideration should be given to the possibility of central nervous system and other effects of alcohol [see Description (11) in Full Prescribing Information].ADVERSE REACTIONSThe following adverse reactions are discussed in greater detail in other sections. • Peripheral neuropathy [see Warnings and Precautions] • Myelosuppression [see Warnings and Precautions] • Hypersensitivity reactions [see Warnings and Precautions]Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.Unless otherwise specified, assessment of adverse reactions is based on one randomized study (Study 046) and one single-arm study (Study 081). In Study 046, 369 patients with metastatic breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 21 days, combined with capecitabine 1000 mg/m2 twice daily for 2 weeks followed by a 1-week rest period. Patients treated with capecitabine as monotherapy (n=368) in this study received 1250 mg/m2 twice daily for 2 weeks every 21 days. In Study 081, 126 patients with metastatic or locally advanced breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 3 weeks.The most common adverse reactions (≥20%) reported by patients receiving IXEMPRA were peripheral sensory neuropathy, fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain. The following additional reactions occurred in ≥20% in combination treatment: palmar-plantar erythrodysesthesia (hand-foot) syndrome, anorexia, abdominal pain, nail disorder, and constipation. The most common hematologic abnormalities (>40%) include neutropenia, leukopenia, anemia, and thrombocytopenia.Table 2 presents nonhematologic adverse reactions reported in 5% or more of patients. Hematologic abnormalities are presented separately in Table 3.
Table 2: Nonhematologic Drug-related Adverse Reactions Occurring in at Least 5% of Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126System Organ Classa/
Preferred TermTotal (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Infections and Infestations Upper respiratory tract infectionb 4 0 3 0 6 0Blood and Lymphatic SystemDisorders Febrile neutropenia 5 4c 1 1d 3 3d
Immune System Disorders Hypersensitivityb 2 1d 0 0 5 1d
Metabolism and Nutrition Disorders Anorexiab
Dehydrationb345
3d
2152
1d
<1d192
2d
1d
Psychiatric Disorders Insomniab 9 <1d 2 0 5 0Nervous System Disorders Peripheral neuropathy Sensory neuropathy b,e
Motor neuropathyb
Headache Taste disorderb Dizziness
65168
128
215d
<1d
01d
16<1345
00001d
62101167
141d
000
Eye Disorders Lacrimation increased 5 0 4 <1d 4 0
Vascular Disorders Hot flushb 5 0 2 0 6 0Respiratory, Thoracic, andMediastinal Disorders Dyspneab
Coughb76
10
42
10
92
1d
0Gastrointestinal Disorders Nausea Vomitingb
Stomatitis/mucositisb
Diarrheab
Constipation Abdominal painb
Gastroesophageal reflux diseaseb
5339314422247
3d
4d
46d
02d
1d
402420396
148
2d
23d
9<1d
1d
0
4229292216136
2d
1d
61d
2d
2d
0
Skin and Subcutaneous TissueDisorders Alopeciab
Skin rashb
Nail disorderb
Palmar-plantar erythrodysesthesia syndromeb,f
Pruritus Skin exfoliationb
Skin hyperpigmentationb
31172464
55
11
01d
2d
18d
0<1d
0
37
1063
23
14
00
<1d
17d
000
48998
622
02d
02d
1d
00
Musculoskeletal, Connective Tissue,and Bone Disorders Myalgia/arthralgiab
Musculoskeletal painb3923
8d
2d55
<1d
04920
8d
3d
General Disorders and AdministrationSite Conditions Fatigue/astheniab
Edemab
Pyrexia Painb
Chest painb
608
1094
1601d
1d
1d
29542
<1
4<1d
000
569885
131d
1d
3d
1d
Table 2: Nonhematologic Drug-related Adverse Reactions Occurring in at Least 5% of Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126System Organ Classa/
Preferred TermTotal (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
Total (%)
Grade 3/4 (%)
a System organ class presented as outlined in Guidelines for Preparing Core Clinical Safety Information on Drugs by the Council for International Organizations of Medical Sciences (CIOMS). b A composite of multiple MedDRA Preferred Terms. c NCI CTC grading for febrile neutropenia ranges from Grade 3 to 5. Three patients (1%) experienced Grade 5 (fatal) febrile neutropenia. Other neutropenia-related deaths (9) occurred in the absence of reported febrile neutropenia [see Warnings and Precautions]. d No grade 4 reports. e Peripheral sensory neuropathy (graded with the NCI CTC scale) was defined as the occurrence of any of the following: areflexia, burning sensation, dysesthesia, hyperesthesia, hypoesthesia, hyporeflexia, neuralgia, neuritis, neuropathy, neuropathy peripheral, neurotoxicity, painful response to normal stimuli, paresthesia, pallanesthesia, peripheral sensory neuropathy, polyneuropathy, polyneuropathy toxic and sensorimotor disorder. Peripheral motor neuropathy was defined as the occurrence of any of the following: multifocal motor neuropathy, neuromuscular toxicity, peripheral motor neuropathy, and peripheral sensorimotor neuropathy. f Palmar-plantar erythrodysesthesia (hand-foot syndrome) was graded on a 1-3 severity scale in Study 046.
Table 3: Hematologic Abnormalities in Patients with Metastatic or Locally Advanced Breast Cancer Treated with IXEMPRA
Study 046 Study 081IXEMPRA with capecitabine
n=369
Capecitabine
n=368
IXEMPRA monotherapy
n=126
Hematology ParameterGrade 3
(%)Grade 4
(%)Grade 3
(%)Grade 4
(%)Grade 3
(%)Grade 4
(%)
Neutropeniaa 32 36 9 2 31 23
Leukopenia (WBC) 41 16 5 1 36 13
Anemia (Hgb) 8 2 4 1 6 2
Thrombocytopenia 5 3 2 2 5 2a G-CSF (granulocyte colony stimulating factor) or GM-CSF (granulocyte macrophage colony stimulating factor) was used in 20% and 17% of patients who received IXEMPRA in Study 046 and Study 081, respectively.
The following serious adverse reactions were also reported in 1323 patients treated with IXEMPRA as monotherapy or in combination with other therapies in Phase 2 and 3 studies.Infections and Infestations: sepsis, pneumonia, infection, neutropenic infection, urinary tract infection, bacterial infection, enterocolitis, laryngitis, lower respiratory tract infectionBlood and Lymphatic System Disorders: coagulopathy, lymphopeniaMetabolism and Nutrition Disorders: hyponatremia, metabolic acidosis, hypokalemia, hypovolemiaNervous System Disorders: cognitive disorder, syncope, cerebral hemorrhage, abnormal coordination, lethargyCardiac Disorders: myocardial infarction, supraventricular arrhythmia, left ventricular dysfunction, angina pectoris, atrial flutter, cardiomyopathy, myocardial ischemiaVascular Disorders: hypotension, thrombosis, embolism, hemorrhage, hypovolemic shock, vasculitisRespiratory, Thoracic, and Mediastinal Disorders: pneumonitis, hypoxia, respiratory failure, acute pulmonary edema, dysphonia, pharyngolaryngeal painGastrointestinal Disorders: ileus, colitis, impaired gastric emptying, esophagitis, dysphagia, gastritis, gastrointestinal hemorrhageHepatobiliary Disorders: acute hepatic failure, jaundiceSkin and Subcutaneous Tissue Disorders: erythema multiformeMusculoskeletal, Connective Tissue, and Bone Disorders: muscular weakness, muscle spasms, trismusRenal and Urinary Disorders: nephrolithiasis, renal failureGeneral Disorders and Administration Site Conditions: chillsInvestigations: increased transaminases, increased blood alkaline phosphatase, increased gamma-glutamyltransferasePostmarketing ExperienceRadiation recall has been reported during postmarketing use of IXEMPRA. Because this reaction was reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.DRUG INTERACTIONSEffect of Other Drugs on IxabepiloneDrugs That May Increase Ixabepilone Plasma ConcentrationsCYP3A4 Inhibitors: Coadministration of ixabepilone with ketoconazole, a potent CYP3A4 inhibitor, increased ixabepilone AUC by 79% compared to ixabepilone treatment alone. If alternative treatment cannot be administered, a dose adjustment should be considered. The effect of mild or moderate inhibitors (eg, erythromycin, fluconazole, or verapamil) on exposure to ixabepilone has not been studied. Therefore, caution should be used when administering mild or moderate CYP3A4 inhibitors during treatment with IXEMPRA, and alternative therapeutic agents that do not inhibit CYP3A4 should be considered. Patients receiving CYP3A4 inhibitors during treatment with IXEMPRA should be monitored closely for acute toxicities (eg, frequent monitoring of peripheral blood counts between cycles of IXEMPRA) [see Dosage and Administration (2.2) in Full Prescribing Information].Drugs That May Decrease Ixabepilone Plasma ConcentrationsCYP3A4 Inducers: IXEMPRA is a CYP3A4 substrate. Coadministration of IXEMPRA with rifampin, a potent CYP3A4 inducer, decreased ixabepilone AUC by 43% compared to IXEMPRA treatment alone. Other strong CYP3A4 inducers (eg, dexamethasone, phenytoin, carbamazepine, rifabutin, and phenobarbital) may also decrease ixabepilone concentrations leading to subtherapeutic levels. Therefore, therapeutic agents with low enzyme induction potential should be considered for coadministration with IXEMPRA. St. John’s Wort may decrease ixabepilone plasma concentrations unpredictably and should be avoided. If patients must be coadministrated a strong CYP3A4 inducer, a gradual dose adjustment may be considered [see Dosage and Administration (2.2) in Full Prescribing Information]. Effect of Ixabepilone on Other DrugsIxabepilone does not inhibit CYP enzymes at relevant clinical concentrations and is not expected to alter the plasma concentrations of other drugs [see Clinical Pharmacology (12.3) in Full Prescribing Information].CapecitabineIn patients with cancer who received ixabepilone (40 mg/m2) in combination with capecitabine (1000 mg/m2), ixabepilone Cmax decreased by 19%, capecitabine Cmax decreased by 27%, and 5-fluorouracil AUC increased by 14%, as compared to ixabepilone or capecitabine administered separately. The interaction is not clinically significant given that the combination treatment is supported by efficacy data.
USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions].Nursing MothersIt is not known whether ixabepilone is excreted into human milk. Following intravenous administration of radiolabeled ixabepilone to rats on days 7 to 9 postpartum, concentrations of radioactivity in milk were comparable with those in plasma and declined in parallel with the plasma concentrations. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ixabepilone, a decision must be made whether to discontinue nursing or to discontinue IXEMPRA taking into account the importance of the drug to the mother.
Pediatric UseThe effectiveness of IXEMPRA (ixabepilone) in pediatric patients has not been established. IXEMPRA was evaluated in one Phase 1 and one Phase 2 trial. The pediatric patients had a safety profile consistent with that seen in adults, and no new safety signals were identified.In the Phase 1 open-label, dose-finding trial, the safety of IXEMPRA was evaluated in 19 pediatric patients with advanced or refractory solid tumors and 2 with acute leukemias. IXEMPRA was administered as a one-hour IV infusion daily for the first five days of a 21-day cycle at one of 5 dose levels, ranging from 3 to 10 mg/m2. Among the 21 patients, 12 ranged in age from 2 to 12 years and 9 ranged from 13 to 18 years. The maximum tolerated dose was 8 mg/m2 IV daily for 5 days every 21 days. No significant activity was observed. The pharmacokinetics of ixabepilone were characterized by population pharmacokinetic analysis of data for 16 patients from this trial, who were aged 2 to 18 years (median 12 years). The pharmacokinetic parameters of ixabepilone in these pediatric patients were compared to the corresponding parameters of 130 adult patients enrolled in clinical trials using the same dosing schedule. The median BSA normalized clearance of ixabepilone in pediatric patients (17 L/h/m2) was similar to that in adult patients (20 L/h/m2).In the Phase 2 trial of 59 patients with advanced or refractory solid tumors, 28 ranged in age from 3 to 12 years and 19 ranged in age from 13 to 18 years. Twelve additional patients over the age of 18 were treated in this trial. IXEMPRA was administered at a dose of 8 mg/m2 IV daily for 5 days every 21 days. This trial was terminated early due to lack of efficacy.Geriatric UseClinical studies of IXEMPRA did not include sufficient numbers of subjects aged sixty-five and over to determine whether they respond differently from younger subjects.Forty-five of 431 patients treated with IXEMPRA in combination with capecitabine were ≥65 years of age and 3 patients were ≥75. Overall, the incidence of grade 3/4 adverse reactions was higher in patients ≥65 years of age versus those <65 years of age (82% versus 68%) including grade 3/4 stomatitis (9% versus 1%), diarrhea (9% versus 6%), palmar-plantar erythrodysesthesia syndrome (27% versus 20%), peripheral neuropathy (24% versus 22%), febrile neutropenia (9% versus 3%), fatigue (16% versus 12%), and asthenia (11% versus 6%). Toxicity-related deaths occurred in 2 (4.7%) of 43 patients ≥65 years with normal baseline hepatic function or mild impairment.Thirty-two of 240 breast cancer patients treated with IXEMPRA as monotherapy were ≥65 years of age and 6 patients were ≥75. No overall differences in safety were observed in these patients compared to those <65 years of age.Hepatic ImpairmentIXEMPRA was evaluated in 56 patients with mild to severe hepatic impairment defined by bilirubin levels and AST levels. Compared to patients with normal hepatic function (n=17), the area under the curve (AUC0-infinity) of ixabepilone increased by: • 22% in patients with a) bilirubin >1 – 1.5 x ULN or b) AST >ULN but bilirubin <1.5 x ULN; • 30% in patients with bilirubin >1.5 – 3 x ULN and any AST level; and • 81% in patients with bilirubin >3 x ULN and any AST level.Doses of 10 and 20 mg/m2 as monotherapy were tolerated in 17 patients with severe hepatic impairment (bilirubin >3 x ULN).IXEMPRA in combination with capecitabine must not be given to patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning, Contraindications, and Warnings and Precautions]. Dose reduction is recommended when administering IXEMPRA as monotherapy to patients with hepatic impairment [see Dosage and Administration (2.3) in Full Prescribing Information]. Because there is a need for dosage adjustment based upon hepatic function, assessment of hepatic function is recommended before initiation of IXEMPRA and periodically thereafter.Renal ImpairmentIxabepilone is minimally excreted via the kidney. No controlled pharmacokinetic studies were conducted with IXEMPRA in patients with renal impairment. IXEMPRA in combination with capecitabine has not been evaluated in patients with calculated creatinine clearance of <50 mL/min. IXEMPRA as monotherapy has not been evaluated in patients with creatinine >1.5 times ULN. In a population pharmacokinetic analysis of IXEMPRA as monotherapy, there was no meaningful effect of mild and moderate renal insufficiency (CrCL >30 mL/min) on the pharmacokinetics of ixabepilone.
OVERDOSAGEExperience with overdose of IXEMPRA is limited to isolated cases. The adverse reactions reported in these cases included peripheral neuropathy, fatigue, musculoskeletal pain/myalgia, and gastrointestinal symptoms (nausea, anorexia, diarrhea, abdominal pain, stomatitis). The highest dose mistakenly received was 100 mg/m2 (total dose 185 mg).There is no known antidote for overdosage of IXEMPRA. In case of overdosage, the patient should be closely monitored and supportive treatment should be administered. Management of overdose should include supportive medical interventions to treat the presenting clinical manifestations.
NONCLINICAL TOXICOLOGYCarcinogenesis, Mutagenesis, Impairment of FertilityCarcinogenicity studies with ixabepilone have not been conducted. Ixabepilone did not induce mutations in the microbial mutagenesis (Ames) assay and was not clastogenic in an in vitro cytogenetic assay using primary human lymphocytes. Ixabepilone was clastogenic (induction of micronuclei) in the in vivo rat micronucleus assay at doses ≥0.625 mg/kg/day.There were no effects on male or female rat mating or fertility at doses up to 0.2 mg/kg/day in both males and females (approximately one-fifteenth the expected human clinical exposure based on AUC). The effect of ixabepilone on human fertility is unknown. However, when rats were given an IV infusion of ixabepilone during breeding and through the first 7 days of gestation, a significant increase in resorptions and pre- and post-implantation loss and a decrease in the number of corpora lutea was observed at 0.2 mg/kg/day. Testicular atrophy or degeneration was observed in 6-month rat and 9-month dog studies when ixabepilone was given every 21 days at intravenous doses of 6.7 mg/kg (40 mg/m2) in rats (approximately 2.1 times the expected clinical exposure based on AUC) and 0.5 and 0.75 mg/kg (10 and 15 mg/m2) in dogs (approximately 0.2 and 0.4 times the expected clinical exposure based on AUC).Animal ToxicologyOverdoseIn rats, single intravenous doses of ixabepilone from 60 to 180 mg/m2 (mean AUC values ≥8156 ng•h/mL) were associated with mortality occurring between 5 and 14 days after dosing, and toxicity was principally manifested in the gastrointestinal, hematopoietic (bone-marrow), lymphatic, peripheral-nervous, and male-reproductive systems. In dogs, a single intravenous dose of 100 mg/m2 (mean AUC value of 6925 ng•h/mL) was markedly toxic, inducing severe gastrointestinal toxicity and death 3 days after dosing.
PATIENT COUNSELING INFORMATION[see FDA-Approved Patient Labeling in Full Prescribing Information]Peripheral NeuropathyPatients should be advised to report to their physician any numbness and tingling of the hands or feet [see Warnings and Precautions].Fever/NeutropeniaPatients should be instructed to call their physician if a fever of 100.5° F or greater or other evidence of potential infection such as chills, cough, or burning or pain on urination develops [see Warnings and Precautions].Hypersensitivity ReactionsPatients should be advised to call their physician if they experience urticaria, pruritus, rash, flushing, swelling, dyspnea, chest tightness or other hypersensitivity-related symptoms following an infusion of IXEMPRA [see Warnings and Precautions].PregnancyPatients should be advised to use effective contraceptive measures to prevent pregnancy and to avoid nursing during treatment with IXEMPRA [see Warnings and Precautions and Use in Specific Populations].Cardiac Adverse Reactions Patients should be advised to report to their physician chest pain, difficulty breathing, palpitations or unusual weight gain [see Warnings and Precautions].IXEMPRA® (ixabepilone) for injection Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, GermanyDILUENT for IXEMPRA Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, GermanyDistributed by Bristol-Myers Squibb Company, Princeton, NJ 08543 USA
1236925A7 5645-0006 Rev October 2011RPH-00001
(Continued)
34519_ccpmix_fa1_K_ASCOpst.indd 4 8/19/15 4:26 PM

PAGE 116 The ASCO Post | SEPTEMBER 10, 2015
In the Literature
Emerging Clinical Data on Cancer Management
COLORECTAL CANCER
Long-Term Use of Thyroid Hormone Replacement Linked to Decreased Risk of Colorectal Cancer
Long-term thyroid hormone re-placement was associated with a de-creased risk of colorectal cancer, but hyperthyroidism and untreated hy-pothyroidism were associated with a modestly elevated risk, according to a study using a large population-based medical records database from the United Kingdom.
A total of 20,990 colorectal cancer patients and 82,054 control patients, exclusive of those with familial colorec-tal cancer syndromes or inflammatory bowel disease, were identified for this nested case-control study. Every colorec-tal cancer case patient was matched with four eligible control patients on age, sex, practice site, and duration of follow-up. “As expected, case patients were more likely to have a medical history of diabe-tes mellitus and more likely to be former or current smokers and alcohol users,” the investigators noted.
Significant ResultsThe results were reported in the
Journal of the National Cancer Insti-tute. Yu-Xiao Yang, MD, of the Perel-man School of Medicine, University of Pennsylvania, Philadelphia, served as the corresponding author of this article.
Patients with clinical or subclinical hypothyroidism (thyroid-stimulating hormone [TSH] levels > 4 mg/dL) who were treated with thyroid hor-mone replacement “had lower risk of colorectal cancer, with an adjusted odds ratio of 0.92 (95% CI = 0.86–0.98, P = .009),” the researchers reported. In further analysis, thyroid hormone re-placement for more than 1 year was as-sociated with a lower colorectal cancer risk, and this protective association was stronger with increasing time.
The adjusted odds ratios for colorec-tal cancer associated with thyroid hor-mone replacement were 0.88 (95% CI = 0.79–0.99, P = .03) for treatment initiat-ed 5 to 10 years before the index date and 0.68 (95% CI = 0.55–0.83, P < .001) for treatment initiated more than 10 years before. The protective effect of thyroid hormone replacement was also higher for patients who underwent colectomy and was more evident among women.
Among patients who did not receive thyroid hormone replacement, those
with clinical or subclinical hypothy-roidism had a modestly higher risk for colorectal cancer than those without documented thyroid dysfunction, with an adjusted odds ratio of 1.16 (95% CI = 1.08–1.24, P < .001). Patients with hyperthyroidism (TSH < 4 mg/dL) also had an increased risk of colorectal cancer, with an adjusted odds ratio of 1.21 (95% CI = 1.08–1.36, P = .001).
Potential Impact“If confirmed, results from this study
may help health care providers decide between early vs late treatment with thyroid hormone replacement in asymp-tomatic subclinical hypothyroidism or alternatively increased colonic screening in those individuals,” the investigators concluded. “Further research evaluating the chemopreventive potential of thy-roid hormone replacement and the ex-act biological mechanism for this effect might enable more specific chemopre-ventive drugs in the future.”
This study was supported by the Na-tional Center for Research Resources and the National Center for Advancing Translational Sciences, National Insti-tutes of Health.
Boursi B, et al: J Natl Cancer Inst 107:djv084, 2015.
HODGKIN LYMPHOMA
Increased Lifetime Risk of Cardiovascular Disease for Patients Treated for Hodgkin Lymphoma
Survivors of Hodgkin lymphoma treated as adolescents or adults are at increased risk of cardiovascular diseas-es throughout their lives, according to results of a retrospective cohort study of 2,524 Dutch patients followed for a median of 20 years. “Treating physi-cians and patients should be aware of the persistently increased risk of car-diovascular diseases throughout life, and the results of our study may direct guidelines for follow-up of patients with Hodgkin lymphoma,” Frederika A. van Nimwegen, MSc, of the Neth-erlands Cancer Institute, Amsterdam, advised in JAMA Internal Medicine.
All patients were diagnosed with Hodgkin lymphoma when younger than 52 years (median age, 27.3 years), had been treated from 1965 through 1995, and had survived for 5 years since their diagnosis. Among the 2,524 pa-tients, 2,052 individuals (81.3%) had
received mediastinal radiation, and 773 patients (30.6%) had received anthra-cycline-containing chemotherapy.
At a median follow-up of 20.3 years (range, 5–47 years), the researchers iden-tified 1,713 cardiovascular events in 797 patients, with 410 patients having two or more events. The most frequently occur-ring cardiovascular disease was coronary heart disease, with 401 patients devel-oping it as their first event, followed by valvular heart disease (374 events) and heart failure (140 events). The median intervals between Hodgkin lymphoma treatment and first cardiovascular dis-ease events were 18 years for coronary heart disease, 24 years for valvular heart disease, and 19 years for heart failure.
Cumulative RisksThe cumulative risk of any type of
cardiovascular disease was 50% at 40 years after diagnosis, and 51% of pa-tients with a cardiovascular disease had multiple events. For patients treated before age 25, the cumulative risk at 60 years of age or older was 20% for coronary heart disease, 31% for valvular heart disease, and 11% for heart failure as first events.
“Compared with the general popula-tion, four- to sevenfold increased risks of coronary heart disease or heart fail-ure are observed 35 years or more after Hodgkin lymphoma treatment, resulting in 857 excess cardiovascular events per 10,000 person-years,” the researchers re-ported. Although patients treated before 25 years of age had the highest relative risks, “substantial absolute excess risks were also observed for patients treated at older ages,” the authors added.
“Adjusted for year of Hodgkin lym-phoma diagnosis, sex, and ever smoking, mediastinal radiotherapy and anthra-cycline-containing chemotherapy were associated with an increased risk of any cardiovascular disease,” the investiga-tors noted. Mediastinal radiotherapy increased the risk of coronary heart dis-ease, valvular heart disease, and heart failure, whereas anthracycline-contain-ing chemotherapy increased the risks of valvular heart disease and heart failure as first events compared with patients who did not receive those cancer treat-ments. “Radiation below the diaphragm or vincristine-containing chemotherapy did not influence the risks of any cardio-vascular disease,” the researchers stated.
“No interactions on a multiplicative scale were found between mediastinal radiotherapy and anthracycline dose or
mediastinal radiotherapy and smoking; the joint effects of these outcomes ap-peared to be additive rather than multi-plicative,” the authors wrote.
Accompanying Commentary“This work by van Nimwegen et al
can specifically help physicians identify their highest-risk patients: those with a history of Hodgkin lymphoma who were treated at a younger age and those who are the longest from treatment,” Emily Tonorezos, MD, MPH, of the Weill Cornell Medical College, New York, and Linda Overholser, MD, of the University of Colorado Denver School of Medicine, Aurora, wrote in a related commentary.
“For most encounters, starting by asking a few key cancer history questions will help identify these patients: (1) What kind of cancer did you have? (2) How old were you when your lympho-ma was diagnosed? (3) Did you receive chest radiotherapy? (4) Did you receive doxorubicin (many patients know it by the brand name Adriamycin [the red medicine])? Our clinical experience has been that patients typically know the an-swers to these basic questions, and these responses will go a long way toward identifying at-risk patients. Nonetheless, the future of good care for cancer survi-vors will require establishment of the ev-idence-based best practices for this pop-ulation,” the commentary concluded.
van Nimwegen FA, et al: JAMA Intern Med 175:1007-1017, 2015.
Tonorezos E, Overholser L: JAMA Intern Med 175:1017-1018, 2015.
HIV INFECTION
Determining Why Patients With HIV Infection and Non–AIDS-Defining Cancers Are Less Likely to Receive Cancer Treatment
A survey sent to medical and ra-diation oncologists to identify factors contributing to observed disparities in cancer treatment between patients in-fected with HIV and those not infected “found that a substantial proportion of physicians (21%) would alter their treatment recommendations based on HIV status,” Gita Suneja, MD, of the University of Salt Lake City, Utah, and colleagues reported in the Journal of Oncology Practice. “The likelihood of offering standard treatment was associ-
continued on page 118

NOVEMBER 5-8, 2015JW Marriott Desert Ridge
Phoenix, Arizona
Advanced Practitioners
Learn more at jadprolive.com
This CE/CME/CEU accredited conference is jointly provided by:
in Oncology
A CE EVENT FOR
REGISTER TODAY!Tell Your NP & PA to

PAGE 118 The ASCO Post | SEPTEMBER 10, 2015
In the Literature
ated with concerns about toxicity, effi-cacy, and comfort level with discussing cancer treatment adverse effects and prognosis,” the investigators stated.
Before the advent of highly active an-tiretroviral therapy improved survival in HIV-infected individuals, “cancers in HIV-infected individuals were largely AIDS-defining cancers related to severe immu-nosuppression, such as Kaposi sarcoma,” the researchers wrote. With improved sur-vival in HIV-infected individuals, cancers considered non–AIDS-defining cancers, “such as lung, colorectal, and anal cancers, have become an increasingly important cause of mortality in the HIV-infected population. Despite improvements in the management of HIV, HIV-infected patients with cancer have worse survival compared with uninfected counterparts,” the authors stated. This lower survival, they noted, could be due in part to a dis-parity in receiving cancer treatment, as seen in recent population-based studies.
The survey was mailed to 500 medi-cal and radiation oncologists randomly selected from the American Medical Association Physician Masterfile, with greater sampling in areas of highest HIV prevalence. Recipients had the option of responding by mail or online.
Among 273 physicians who respond-ed, 79% indicated they would provide standard cancer treatment to HIV-infected patients, but 69% disagreed with the state-ment, “Sufficient guidelines are available to aid in treatment decision-making for HIV-infected patients with non–AIDS-defining malignancies.” In addition, 45% never or rarely discussed their cancer man-agement plan with an HIV specialist; 20% were not comfortable discussing cancer
treatment adverse effects with their HIV-infected patients with cancer, and 15% were uncomfortable discussing prognosis.
Multivariable Analysis“In multivariable analysis, physi-
cians comfortable discussing adverse effects and prognosis were more likely to provide standard cancer treatment (adjusted odds ratio, 1.52; 95% CI, 1.12–2.07). Physicians with concerns about toxicity and efficacy of treatment were significantly less likely to provide standard cancer treatment (adjusted odds ratio, 0.67; 95% CI, 0.53–0.85),” the investigators stated.
Among medical oncologists, 77% scored as providing standard therapy to HIV-infected patients. This was as-sessed by responses to three specialty-specific management questions, with 18% indicating they would not use stan-dard chemotherapy agents, 48% indi-cating that they would use lower doses and fewer cycles, and 51% indicating that they would discontinue therapy if adverse effects occurred when treating HIV-infected patients with cancer.
Among radiation oncologists, 80% scored as providing standard therapy to HIV-infected patients. Responses to three specialty-specific management questions showed that 20% would use lower radiation doses, 27% would treat with smaller fields, and 31% would dis-continue therapy if adverse effects oc-curred when treating HIV-infected pa-tients with cancer.
The authors noted that concerns cited regarding safety and efficacy of can-cer treatment in HIV-infected patients “are not surprising, given the dearth of high-quality data and resulting lack of evidence-based guidelines specific to
HIV-infected patients with non–AIDS-defining cancers. Clinical trial data are available to inform the management of HIV-infected patients with non-Hodgkin lymphoma and anal cancer but not most other non–AIDS-defining cancers. This is because HIV-infected patients have historically been excluded from clinical trials, so randomized trial data regarding treatment outcomes are largely unavail-able.” The authors added that the National Cancer Institute and other organizations have initiated steps to address this issue.
“Inclusion of HIV-infected patients in cancer clinical trials, development of cancer treatment guidelines specific to HIV-infected patients, and enhanced care coordination between oncologists and HIV specialists may reduce cancer treat-ment disparities for HIV-infected patients with cancer,” the investigators concluded.
Suneja G, et al: J Oncol Pract 11:e380-e387, 2015.
HEPATOCELLULAR CARCINOMA
More Than One-Third of Those Diagnosed With Hepatocellular Carcinoma as Outpatients Have Diagnostic Delays of 3 or More Months
Nearly 20% of patients with hepa-tocellular carcinoma “wait more than 3 months from presentation to diag-nosis, which can contribute to interval tumor growth,” Nishant Patel, MD, and colleagues concluded in the Journal of the National Comprehensive Cancer Network. They based their conclusions on a review of records of consecutive patients with cirrhosis and hepatocel-lular carcinoma at Parkland Memorial Health and Hospital System, a large ur-ban safety net hospital in Dallas, be-tween January 2005 and July 2012.
“Diagnostic delays are particularly common among outpatients, occurring in more than one-third,” the researchers found. “These delays may be related to several potential issues, including provid-ers failing to recognize positive surveillance tests, patients missing radiology appoint-ments, and insensitive diagnostic tests. Al-though we did not find any difference in re-ceipt of hepatocellular carcinoma–directed treatment, diagnostic delays were associ-ated with potential interval tumor growth in nearly one-fifth of patients.”
Study DetailsAmong 457 patients with cirrho-
sis and hepatocellular carcinoma, 231 were inpatients, and 226 (49.5%) were diagnosed as outpatients. Although the time from presentation to diagnosis was
less than 1 week for more than 90% of inpatients, the median time to diagnosis was 2.2 months for outpatients, with 87 patients (38.5%) experiencing a diag-nostic delay, defined as a time to diag-nosis exceeding 3 months.
Higher rates of diagnostic delays were observed among those diagnosed as outpatients who had hepatic en-cephalopathy (56% vs 35%). “Among 49 patients with mass-forming hepato-cellular carcinoma and diagnostic delay, 18% had interval tumor growth of 2 cm or greater,” the investigators stated.
The median age of the patients was 56 years, and more than 75% were men. “Our population was racially diverse, with 36% African Americans, 30% His-panic Caucasians, and 26% non-Hispan-ic Caucasians,” the researchers noted.
Lower rates of diagnostic delays were observed in patients diagnosed as outpa-tients who presented after implementa-tion of a comprehensive electronic medi-cal record system, 26% vs 60% for those presenting before electronic medical records. The authors noted, “diagnostic delays may be more common in hospital systems without an electronic medical record, because of higher rates of un-recognized positive surveillance tests.” Patients presenting with an abnormal ultrasound, with or without an elevated alpha-fetoprotein level, also had lower rates of diagnostic delay, 27% vs 50%.
Outcome DisparitiesThe investigators noted that the in-
cidence of hepatocellular carcinoma is increasing because of a growing num-ber of cases of nonalcoholic fatty liver disease and hepatitis C virus. Curative options are “only available for those di-agnosed at an early stage. Patients with early hepatocellular carcinoma achieve 5-year survival rates near 70% with re-section or liver transplantation, whereas those with advanced hepatocellular car-cinoma have a median survival of less than 1 year,” the authors added.
“Despite the availability of efficacious surveillance tests, only 40% of hepato-cellular carcinoma cases are diagnosed at an early stage nationally,” the research-ers wrote. “Tumor stage at diagnosis can be impacted by several factors in clinical practice, including low surveillance rates and delays in follow-up of abnormal screening tests. These issues may be par-ticularly prevalent among racial minori-ties and socioeconomically disadvan-taged patients, potentially contributing to racial and socioeconomic disparities in cancer outcomes.”
Patel N, Yopp AC, Singal AG: J Natl Compr Canc Netw 13:543-549, 2015.© George Booth/The New Yorker Collection/www.cartoonbank.com
Emerging Clinical Datacontinued from page 116

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 119
In the Literature
COMORBIDITIES
Cancer Diagnosis Among Patients With Diabetes Reduced Adherence to Evidence-Based Medications
“A cancer diagnosis among patients with diabetes reduced adherence with evidence-based medications, particular-ly if patients’ life expectancy was short,” according to a study among Medicare beneficiaries reported in the Journal of Oncology Practice. “These findings em-phasize the vulnerability of individuals faced with a new cancer diagnosis and the difficulty in maintaining ongoing therapy for comorbid conditions, which may have potentially important conse-quences for longer-term survivorship,” concluded Bruce C. Stuart, PhD, of the University of Maryland School of Phar-macy, Baltimore, and colleagues.
The study compared 4,348 patients with newly diagnosed cancer between January and December 2007 with 28,507 cancer-free controls “assigned a pseudo-diagnosis date,” the researchers explained, so that the distribution of index months was the same for the patients with cancer and the controls. All patients were select-ed from a 5% random sample of Medicare beneficiaries with diabetes enrolled in Medicare Part D in 2007 and 2008.
The patients with cancer were older, had higher rates of comorbid conditions, and were less likely to receive Medicare Part D low-income subsidies.
Key FindingsAdherence was determined by track-
ing for 6 months before and 6 months after the diagnosis (or pseudodiagnosis) date the proportion of days covered with oral hypoglycemic agents, renin-angio-tensin-aldosterone system inhibitors, and statins. Overall, the patients with cancer had “relatively larger declines in medication adherence” of between 3% and 5% (P < .001) than did the con-trols, the researchers reported. Patients with cancer who had sharper declines of between 8% and 11% (P < .001) had a short life expectancy, defined by the investigators as death occurring 7 to 12 months after the index date.
For longer-term survivors, the re-searchers noted, “the declines in pro-portion of days covered values were relatively small but still appear larger for patients with cancer than for controls.”
The largest declines in adherence to evidence-based medications recom-mended in diabetes treatment guide-lines “were observed for patients who died in the 6 months after our observa-
tion period, suggesting that poor cancer prognosis was at least partly responsible. The declines in treatment associated with a cancer diagnosis persisted even when limited to persons with longer-term survival, suggesting that not all of the decline can be attributed to short life expectancy,” the investigators observed.
“We did not find a differential effect
on diabetes medication adherence as-sociated with Part D low-income sub-sidy enrollment, suggesting that new financial burdens associated with cancer treatment were not associated with the observed declines in adherence to these drug classes after a cancer diagnosis,” the researchers reported. “Many of the medications recommended for individu-
als with diabetes during this period were generic, and copayments for generic drugs, which were already low, decreased further during the study period. Recent evidence indicates that the financial bur-den associated with adherence to typical diabetes drug regimens is not large.” n
Stuart BC, Davidoff AJ, Erten MZ: J On-col Pract. June 23, 2015 (early release online).
A complimentary CME-webcast
Management of Locally Advanced HER2-Positive Breast Cancer
In collaboration with
TUMOR BOARD SERIES
This activity has been approved for AMA PRA Category 1 Credit™
EDUCATIONAL NEEDSThis webcast provides expert insight on optimizing treatment of patients with HER2-positive early-stage/locally advanced breast cancer. It will consist of a case presentation of a patient with HER2-positive early-stage/locally advanced disease. Suggested treatment options based on best practices, current treatment guidelines, and data from relevant recent clinical trials will be discussed. Expert faculty will present various aspects of the case, including radiologic imaging, pathologic findings (particularly HER2 testing), and options for neoadjuvant therapy with HER2-targeted agents. Recommendations regarding surgical resection, options for adjuvant therapy with chemotherapy and targeted agents, and radiation therapy post surgery also will be addressed.
AUDIENCEThis activity is designed for health care practitioners including medical oncologists, surgical oncologists and general surgeons, radiation oncologists, radiologists, pathologists, and other health care professionals who manage patients with breast cancer.
www.ccfcme.org/tumorboards
LEARNING OBJECTIVES• Assess the role of neoadjuvant
chemotherapy in HER2-positive breast cancer, including use of HER2-targeted agents (trastuzumab, pertuzumab, ado-trastuzumab emtansine).
• Discuss FDA approval of HER2-targeted agents for breast cancer, based on neoadjuvant data and use of pathologic complete response (pCR) as a surrogate marker.
• Review HER2-testing guidelines (ASCO-CAP) for patients with breast cancer.
• State the indications for breast magnetic resonance imaging (MRI).
• Appraise recent and ongoing clinical trials of adjuvant therapy in HER2-positive breast cancer.
• Assess local treatment after neoadjuvant surgery.
• Summarize key phase 3 trials of HER2-targeted neoadjuvant therapy including TRYPHENA, NEOSPHERE, NeoALTTO (lapatinib, trastuzumab, or combination in addition to paclitaxel) and ongoing NSABP B-50-I phase 3 trial (KATHERINE) of trastuzumab versus ado-trastuzumab emtansine as adjuvant therapy.
The Cleveland Clinic Center for Continuing Education acknowledges an educational grant for support of this activity from Genentech, Inc.
FACULTYBenjamin C. Calhoun, MD, PhDStephen R. Grobmyer, MD Halle Moore, MD Mikkael Sekeres, MD, MSLaura Shepardson, MD Rahul Tendulkar, MD Cleveland Clinic, Cleveland, Ohio
Eleftherios (Terry) P. Mamounas, MD, MPH, FACS UF Health Cancer Center at Orlando Health, Orlando, Florida
PROGRAM CHAIRJame Abraham, MD Cleveland Clinic, Cleveland, Ohio
NOW AVAILABLE

PAGE 120 The ASCO Post | SEPTEMBER 10, 2015
Announcements
The American Society of Clinical Oncology (ASCO) is seeking an accomplished leader in clinical oncology to serve as their next Chief Executive O� cer (CEO). ASCO is a professional society committed to conquering cancer through research, education, prevention, and delivery of high quality patient care. Founded in 1964, ASCO today counts more than 36,000 members worldwide and has an annual budget of $100M. The CEO works with a distinguished Board of Directors, a talented professional sta� of more than 350 individuals, and hundreds of dedicated volunteers to envision and implement a strategic plan that engages and serves all physicians as they work to � ght cancer. The CEO provides leadership and vision on the execution of strategic plans and oversees the � scal health of the organization while building and supporting a highly committed and capable management team. The CEO must ensure that ASCO’s current major initiatives are successful and also guide a conversation about new opportunities for ASCO to support the � eld in a dynamic healthcare environment. All of these e� orts will be sustained by responsible management of internal resources and diligent philanthropic e� orts.
The successful candidate for this prestigious position will have experience as a leader in clinical oncology with a deep passion for, commitment to, and track record of accomplishments in cancer clinical medicine. The candidate should have working knowledge of and some experience in the many areas in which ASCO plays an important role in improving cancer care, including clinical and translational research, physician and public education and communication, cancer care delivery, governmental a� airs and policy, and global health. A prior leadership role with ASCO in a volunteer or board capacity is strongly desired as is an in-depth understanding of the current trends and issues a� ecting oncology clinical practice and clinical research today. A medical degree is required. The ideal candidate must have a demonstrated track record of managing and leading a large and complex organization with a comparably sized budget and a highly educated, high intellect professionals. The CEO also serves as CEO of the a� liated Conquer Cancer Foundation, and experience with philanthropy is bene� cial.
This position is based at ASCO headquarters in Alexandria, Virginia. Candidates from all oncology disciplines and sub-specialties, domestic and international, are encouraged to apply. ASCO is proud to be an Equal Opportunity Employer (EOE).
Chief Executive O� cer
Isaacson, Miller has been retained for this important recruitment and all nominations, inquiries, and applications should be directed in con� dence to: www.imsearch.com/5499.
James Welsh, MD, FACRO, Elected President of the American College of Radiation Oncology
Loyola University Medical Center radiation oncologist James Welsh,
MD, FACRO, was recently elected president of the American College of Radiation Oncology (ACRO).
Dr. Welsh, installed as president during ACRO’s Annual Meeting in Washington, DC, will serve a 2-year term as President. In 2017, Dr. Welsh will transition to Chairman of the Board for ACRO.
ACRO is the essential professional society for success in the day-to-day practice of radiation oncology. It fo-cuses on ensuring the highest quality of care for radiation therapy patients and promoting success in the practice of radiation oncology through education, responsible socioeconomic advocacy, and integration of science and technol-ogy into clinical practice.
In addition to ACRO, Dr. Welsh has actively and extensively served in other professional organizations. Dr. Welsh recently concluded an
8-year term as Advisor to the Nuclear Regulatory Commission’s Advisory Committee on the Medical Uses of Isotopes. He formerly served as Chair of the Radiological Society of North America’s Radiation Oncology Scien-tific Program Subcommittee and as an Alternate Councilor in the Ameri-can College of Radiology. Dr. Welsh held or holds several leadership posi-tions within the American Society for Radiation Oncology.
Dr. Welsh is Professor, Medical Di-rector, and Director of Clinical and Translational Research in the Depart-ment of Radiation Oncology of Loyola University Chicago Stritch School of Medicine. He also is Chief of Radiation Oncology at Edward Hines Jr VA Hos-pital. Dr. Welsh is a fellow of the Ameri-can College of Radiation Oncology.
Dr. Welsh earned his medical degree from the State University of New York at Stony Brook and a master’s degree in molecular biophysics and biochem-
istry from Yale University. Dr. Welsh completed a residency in radiation on-cology at Johns Hopkins Hospital and
completed a fellowship in helical to-motherapy while at the University of Wisconsin-Madison. n
Arno Mundt, MD, outgoing President of the American College of Radiation Oncology, passes the gavel to incoming President James Welsh, MD, FACRO. Photo courtesy of Loyola University of Chicago Stritch School of Medicine and the American College of Radiation Oncology.

ASCOPost.com | SEPTEMBER 10, 2015 PAGE 121
Perspective
Mr. Collender is so right. All new cancer drugs must go through the rigors of clinical trial investigations to ensure their safety and efficacy, but unless we can significantly improve the current paltry rate of just 3% adult participa-tion in clinical studies—participation is even lower among minority patients and patients aged 65 and older—we will not be able to advance cancer care for all patients in the future. In fact, a study of genitourinary cancer clinical trials found that 1 in 10 adult clinical trials ends prematurely due to poor ac-crual,2 which can unnecessarily doom a potentially effective drug from ever making it to the marketplace.
Barriers to Trial ParticipationThe barriers to patient participa-
tion in clinical trials are many. Patients may fear a reduced quality of life, have concerns about not receiving the ex-perimental drug, worry about potential treatment side effects and loss of con-trol over treatment decisions, and have anxiety about the inconvenience a trial might cause.
Physicians hamper the process, too, by not encouraging patients to join studies or not referring them to appro-priate trials. Excessive administrative and regulatory burdens and decreased federal funding for clinical trials are big contributing factors as well.
Federally funded research through the National Institutes of Health (NIH) has played a critical role in every ma-jor advance in cancer treatment over the past 50 years. But this funding has remained flat for more than a decade, leaving NIH federal research funds with a 23% loss when adjusted for bio-medical inflation. Promising research is now going unfunded, and new studies are being scaled back, leaving fewer pa-tients with the opportunity to partici-pate in clinical trials.
Recently, the U.S. House of Rep-resentatives passed the 21st Century Cures Act, which includes more than $9 billion in additional funding for the NIH and the U.S. Food and Drug Administration (FDA). The increased funding will strengthen both agencies and the medical community as a whole. I applaud this effort and urge the Senate to pass the bill.
All of these challenges are occurring at a pivotal point in cancer research, and ASCO is taking steps to help mitigate the problem of low patient participa-tion in clinical studies. In July, ASCO published a position statement, “Im-
proving the Evidence Base for Treat-ing Older Adults With Cancer,” which calls for federal agencies to broaden clinical trials to include older patients,3 and members of ASCO’s Cancer Re-search Committee published “Modern-izing Eligibility Criteria for Molecu-larly Driven Trials,” which examines the need to redefine eligibility criteria for
participation in clinical studies investi-gating molecularly targeted agents.4
Including Older PatientsAlthough more than 60% of cancers
in the United States occur in people aged 65 and older—a population that is expected to double by midcentury5—the evidence base for treating these pa-
tients is lacking because older adults are underrepresented in clinical trials, and trials designed specifically for this age group are rare. The result is that we have less evidence on how to treat this popu-lation of patients and less knowledge regarding the risk of toxicity and key endpoints of importance than we do for
Julie M. Vose, MD, MBA, FASCOcontinued from page 1
continued on page 122
TRIPLE YOUR IMPACT!
If you donate to the Conquer Cancer Foundation during September, CCF Board Member Raj Mantena
will match your gift 2-to-1, up to $1 million.
Help us take down cancer with three times your support.
Donors can participate in this matching gift challenge at conquercancerfoundation.org/match or by mailing a check to the Conquer Cancer Foundation at: P.O. Box 896076, Charlotte, NC 28289-6076.
Mail donations must be postmarked by September 30, 2015 to qualify for the 2-to-1 match.
conquercancerfoundation.org/match
$50 $100
$100 $200
$500 $1,000
$150$300 $1,500

PAGE 122 The ASCO Post | SEPTEMBER 10, 2015
Perspective
younger adult and pediatric patients. To make clinical trials more inclu-
sive and expand the number of older patients in clinical trials, ASCO is pro-posing five recommendations:1. Use clinical trials to improve the evi-
dence base for treating older adults.2. Leverage research designs and infra-
structure to improve the evidence base for treating older adults.
3. Increase FDA authority to incentiv-ize and require research on older adults with cancer.
4. Increase clinicians’ recruitment of older adults with cancer into clinical trials.
5. Utilize journal policies to incentivize researchers to consistently report on the age distribution and health-risk profiles of research participants.In the paper, ASCO also outlines 16
specific action items to facilitate imple-mentation of its recommendations. These measures include asking regula-tory agencies, research funders, and researchers to consider whether evi-dence exists to support eligibility crite-ria based on the three primary reasons older adults are excluded from clinical studies: age, performance status, and comorbid health conditions.
Molecularly Driven TrialsIn ASCO’s second position paper,
members of ASCO’s Cancer Research Committee propose the organization
of a public workshop, with input from regulatory bodies and key stakehold-ers, to develop a streamlined algorith-mic approach to determining eligibil-ity criteria for study protocols in this era of molecularly driven therapy. ASCO is working with collaborators, including the FDA and Friends of Cancer Research, toward a meeting in spring 2016.
This is an important first step as trials of molecularly targeted cancer therapies increase in number and our understanding of the molecular driv-ers of cancer advances. “Although eligibility criteria are necessary to define the population under study and conduct trials safely, excessive requirements may severely restrict the population available for study,” wrote the paper’s authors.
Hopefully, fewer eligibility restric-tions will enable more patients to par-ticipate in clinical trials and speed our understanding of the risks and benefits
of investigational targeted therapies based on the molecular characteristics of cancer.
Advancing CuresIn addition to the steps ASCO is tak-
ing on a national level to boost clinical trial participation, there are also ap-proaches on the community level that can improve patient recruitment and
retention. For example, you can budget time during an office visit to provide all eligible patients with clear information about appropriate clinical studies at your institution or refer them to a study at another cancer center. At this time, you can answer any questions patients may have regarding potential risks and benefits, time involved, and any costs associated with the trial.
I commend Mr. Collender for his courage in speaking out about the need for all of us— clinicians, scientists, and public officials—to step up our efforts to encourage more patients to join
clinical trials. Our patients are the true heroes of cancer research, and without their participation in clinical studies, we will not make the progress necessary to find cures for cancer.
We owe it to Mr. Collender and to the millions of other patients with can-cer to advance scientific discovery into more effective therapies and improved outcomes for all patients. n
Disclosure: Dr. Vose reported no potential conflicts of interest
References1. Collender S: Clinical trials need cancer
patients. The New York Times. June 19, 2015. Available at www.nytimes.com/2015/06/19/opinion/clinical-trials-need-cancer-patients.html. Accessed July 28, 2015.
2. Stensland KD, McBride R, Wisniv-esky JP, et al: Premature termination of gen-itourinary cancer clinical trials. 2014 ASCO Annual Meeting. Abstract 288.
3. Hurria A, Levit LA, Dale W, et al: Im-proving the evidence base for treating older adults with cancer: American Society of Clinical Oncology statement. J Clin Oncol. July 20, 2015 (early release online).
4. Kim ES, Bernstein D, Hilsenbeck SG, et al: Modernizing eligibility criteria for molecularly driven trials. J Clin Oncol. July 20, 2015 (early release online).
5. West LA, Cole S, Goodkind D, et al: 65+ in the United States: 2010. U.S. Cen-sus Bureau. June 2014. Available at www.census.gov/content/dam/Census/library/publications/2014/demo/p23-212.pdf. Accessed July 28, 2015.
Julie M. Vose, MD, MBA, FASCOcontinued from page 121
Unless we can significantly improve the current paltry rate of just 3% adult participation in clinical studies … we will not be able to advance cancer care for all patients in the future.
—Julie M. Vose, MD, MBA, FASCO
The ASCO Post Wants to Hear From YouWe encourage readers to share their opinions and thoughts on issues of interest to the oncology community.
Write to The ASCO Post at [email protected]
Harborside Press 37 Main Street
Cold Spring Harbor, NY 11724
Phone: 631.692.0800 Fax: 631.692.0805
www.ASCOPost.com

Quality Cancer Care: Pursuing Excellence
ARE YOU READY TO BEGIN?
Delivering high quality care is your #1 goal. The best way to accurately assess the
quality of care you deliver is by participating in QOPI (Quality Oncology Practice Initiative).
Developed by ASCO, QOPI is your resource for established quality measures based on
published guidelines and expert consensus. QOPI provides a standardized process to
routinely assess care, a secure web-based data submission and reporting system, and
access to special improvement projects.
PARTICIPATE IN QOPI TO:
• Understand the care you provide relative to care guidelines and national benchmarks
• Gather reliable information to guide improvement activities
• Meet external quality reporting requirements• Move towards QOPI Certification
Visit qopi.asco.org for fall round updates.
The pursuit of excellence STARTS WITH QOPI
THE FALL ROUND WILL OPEN IN
MID-SEPTEMBER.

Our Roots Go DeepTake a deeper look at our reliability and qualityvisit biotechnologybyamgen.comDownload the LAYAR app on your smartphone and scan this page.
At Amgen, we pour commitment, passion, and a drivefor perfection into every biologic medicine we make.
From innovative biotechnology to extensive experience in biologic manufacturing, see how Amgen strives to deliver on its commitment to your patients.
©2014 Amgen Inc. All rights reserved. 80012-R2-V1
our roots go deepFor reliability and quality,
Related Documents











