Systolic blood pressure peak during maximal exercise testing: A possible determinant of endothelial turnover in healthy subjects Michele M. Ciulla, 1 * Carola Gianni, 1 Pietro Broglia, 1,2 Silvia Lonati, 3 Ilaria Silvestris, 3 Roberta Paliotti, 1 Fabrizio Giofre `, 1 Erica Rampoldi, 4 Agostino Cortelezzi, 3 and Fabio Magrini 1,2 Previous studies have suggested that physical exercise may have an effect on the turnover of the endothelial compartment. Following a maximal exercise testing (Bruce protocol), a prompt and significant increase in the number of circulating endothelial cells (CECs) was detected (D 1 50% vs. basal; P 5 0.0001) in 12 healthy volunteers, without significant changes in the marker of myocardial ischemia; the frequency of CECs correlated significantly with systolic blood pres- sure (SBP) and rate-pressure product at peak exercise (r 5 0.78, P 5 0.003, and r 5 0.64, P 5 0.03, respectively). These results support the role of peak SBP during maximal exercise possibly as mechanical fac- tor facilitating the detachment of CECs and the endothelial turnover. The endothelium is a dynamic structure maintained by a continuous self- renewal of the endothelial cells (ECs) that in basal conditions accounts for 0.1% replications per day [1]; this rate could be affected by several physio- logical and pathological conditions. As the number of vital and apoptotic CECs detached from the endothelium is related to the turnover of endothe- lial progenitor cells (EPCs) in physiological conditions [2], the number of peripheral blood (PB) ECs has been proposed as diagnostic, therapeutic, or prognostic marker of vascular injury and neovascularization [3]. Unfortu- nately, PB EPCs are extremely rare, and their accurate detection and enu- meration is a technical challenge especially when high sensitive techniques are used, such as flow cytometry [4,5]. In patients with cardiovascular diseases, physical exercise may increase the release, mobilization, and number of PB EPCs [6,7]. In healthy subjects, dynamic exercise [8], altitude [9], or simulated [10] hypoxia seem to increase the number of PB EPCs, but the mechanism underlying this increase has not yet been clarified, although myocardial ischemia has been convincingly ruled out [11,12]. The objective of this study is to clarify whether changes in blood pressure (BP) during a single session of intense physical exercise could affect the endothelial turnover in healthy subjects. The sample size was defined a priori to detect a reasonable experimental effect with an adequate power. We enrolled 12 healthy nonsmokers, non- obese (BMI 24.7 ± 2.1 kg/m 2 ), normotensive, adult volunteer males (mean age 37 ± 12 years), not currently on any medication, and who had not resided at high altitudes (>3,000 m) or performed an intense physical exer- cise in the 7 days preceding the test. Eligibility for the study was determined by an oral questionnaire; included subjects gave an informed consent. All study subjects underwent the same maximal exercise testing procedure according to Bruce [13], using a cyclergometer (Ec1000, Custo Med, Otto- brunn, Germany). The protocol involved a minute of unloaded exercise, fol- lowed by a progressive increase in the load (25 W every 2 min), until the achievement of 85% of the predicted maximum heart rate (220-age). During the test, the following parameters were recorded: systolic and diastolic BP (SBP, DBP, with an aneroid sphygmomanometer at the end of each stage of exercise), and heart rate (HR, with an electrocardiogram continuous recording, Custo Card M, Custo Med, Ottobrunn, Germany). Positivity of the test for elec- trocardiography and/or symptoms was considered as exclusion criteria. In each subject, two samples of venous blood from an antecubital vein were collected immediately before and after the exercise (about 5 ml each; total of 20 ml) and processed to determine the serum levels of cardiac Tro- ponin I (cTnI), the hematocrit, and the number of EPCs and CECs. Plasma levels of cTnI were determined by an immunoenzymatic method using a specific analyzer (Dimension RxL, Dade-Behring, Milton Keynes, Bucking- hamshire, UK). CECs and EPCs were defined according to Biguzzi et al. [14], respectively, as cells CD452/CD1461/CD311 and cells CD452/ CD341/KDR1, and were quantified by Flow Cytometry (FACScan, Becton Dickinson, San Jose, CA) according to a previously described procedure collecting 500,000 events per sample (Figure 1) [15]. Data were analyzed using SPSS – Rel 13 (SPSS Inc., Chicago, IL). All quantitative variables were tested for Gaussian distribution with the Kolmogorov-Smirnov test. Changes in any of the studied variables at each time intervals were tested by ANOVA. The relationship between PB EPCs changes and other varia- bles was tested by regression analysis. In all cases, P < 0.05 was consid- ered significant. All study subjects completed the exercise testing reaching the 85% of pre- dicted maximum heart rate (mean 165 ± 15 b/min) at a load of 165 ± 36 W with neither symptoms nor ECG changes suggestive of reduced coronary reserve. Serum cTnI levels after the test remained in the normal range (<0.15 ng/ml). At peak of exercise, a significant increase in BP was observed (SBP from 123 ± 15 to 182 ± 20 mmHg, P < 0.0001; DBP from 84 ± 11 to 95 ± 13 mmHg, P 5 0.0067); a summary of studied parameters is reported in Table I. The number of CECs increased significantly from 16.3 (range, 10.8–23.8) to 24.6 (range, 20.1–43.6) cells per micoliter (P < 0.001; mean differences 95% CI 211.15, from 215.64 to 26.65). At maximal exer- cise, a direct correlation between SBP, rate-pressure product (RPP), and the number of CECs measured after the exercise testing was found (r 5 0.7795, P < 0.001; r 5 0.6364, P < 0.05, respectively). The absolute change in CECs after the exercise significantly correlated with peak SBP and RPP (r 5 0.6163, P < 0.05; r 5 0,6044, P < 0.05, respectively). The number of PB EPCs showed an upward tendency, but this increase did not reach statistical significance. In physiological conditions, EPCs are responsible for the maintenance of a complex network of vessels: their mobilization is generally proportional to the number of vital and apoptotic CECs detached from the endothelium. The results of our study support the role of peak SBP during maximal exercise for the renewal of ECs, possibly by facilitating the detachment of CECs and, TABLE I. Main Studied Variables at Rest and at Peak Exercise Rest Peak exercise P Subjects (n8) 12 – Age (years) 37 ± 12 – BMI (kg/m 2 ) 24.7 ± 2.1 – Load (W) – 165 ± 36 – Load duration (MM:SS) – 12:47 ± 02:55 – SBP (mmHg) 123 ± 15 182 ± 20 <0.0001*** DBP (mmHg) 84 ± 11 95 ± 13 0.0067** MBP (mmHg) 97 ± 11 124 ± 10 <0.0001*** HR (1 per min) 96 ± 18 165 ± 15 <0.0001*** RPP (mmHg/min) 11,915 ± 2734 30,082 ± 4520 <0.0001*** cTnI (ng/ml) 0.03 ± 0.03 0.03 ± 0.03 0.72 CECs (n/ml) (range) 16.3 (10.8–23.8) 24.6 (20.1 – 43.6) 0.0001*** BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pres- sure; MBP, mean blood pressure; HR, heart rate; RPP, rate-pressure product; cTnI, cardiac troponin I; CECs, circulating endothelial cells. P < 0.05 was consid- ered significant. 00 5 **, 000 5 ***. Letter V V C 2009 Wiley-Liss, Inc. American Journal of Hematology 449 http://www3.interscience.wiley.com/cgi-bin/jhome/35105

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Systolic blood pressure peak during maximal exercisetesting: A possible determinant of endothelial turnoverin healthy subjects
Michele M. Ciulla,1* Carola Gianni,1 Pietro Broglia,1,2 Silvia Lonati,3 Ilaria Silvestris,3 Roberta Paliotti,1
Fabrizio Giofre,1 Erica Rampoldi,4 Agostino Cortelezzi,3 and Fabio Magrini1,2
Previous studies have suggested that physical exercise may have an
effect on the turnover of the endothelial compartment. Following a
maximal exercise testing (Bruce protocol), a prompt and significant
increase in the number of circulating endothelial cells (CECs) was
detected (D 1 50% vs. basal; P 5 0.0001) in 12 healthy volunteers,
without significant changes in the marker of myocardial ischemia; the
frequency of CECs correlated significantly with systolic blood pres-
sure (SBP) and rate-pressure product at peak exercise (r 5 0.78, P 5
0.003, and r 5 0.64, P 5 0.03, respectively). These results support the
role of peak SBP during maximal exercise possibly as mechanical fac-
tor facilitating the detachment of CECs and the endothelial turnover.
The endothelium is a dynamic structure maintained by a continuous self-
renewal of the endothelial cells (ECs) that in basal conditions accounts for
0.1% replications per day [1]; this rate could be affected by several physio-
logical and pathological conditions. As the number of vital and apoptotic
CECs detached from the endothelium is related to the turnover of endothe-
lial progenitor cells (EPCs) in physiological conditions [2], the number of
peripheral blood (PB) ECs has been proposed as diagnostic, therapeutic, or
prognostic marker of vascular injury and neovascularization [3]. Unfortu-
nately, PB EPCs are extremely rare, and their accurate detection and enu-
meration is a technical challenge especially when high sensitive techniques
are used, such as flow cytometry [4,5].
In patients with cardiovascular diseases, physical exercise may increase
the release, mobilization, and number of PB EPCs [6,7]. In healthy subjects,
dynamic exercise [8], altitude [9], or simulated [10] hypoxia seem to increase
the number of PB EPCs, but the mechanism underlying this increase has
not yet been clarified, although myocardial ischemia has been convincingly
ruled out [11,12].
The objective of this study is to clarify whether changes in blood pressure
(BP) during a single session of intense physical exercise could affect the
endothelial turnover in healthy subjects.
The sample size was defined a priori to detect a reasonable experimental
effect with an adequate power. We enrolled 12 healthy nonsmokers, non-
obese (BMI 24.7 ± 2.1 kg/m2), normotensive, adult volunteer males (mean
age 37 ± 12 years), not currently on any medication, and who had not
resided at high altitudes (>3,000 m) or performed an intense physical exer-
cise in the 7 days preceding the test. Eligibility for the study was determined
by an oral questionnaire; included subjects gave an informed consent. All
study subjects underwent the same maximal exercise testing procedure
according to Bruce [13], using a cyclergometer (Ec1000, Custo Med, Otto-
brunn, Germany). The protocol involved a minute of unloaded exercise, fol-
lowed by a progressive increase in the load (25 W every 2 min), until the
achievement of 85% of the predicted maximum heart rate (220-age). During
the test, the following parameters were recorded: systolic and diastolic BP
(SBP, DBP, with an aneroid sphygmomanometer at the end of each stage of
exercise), and heart rate (HR, with an electrocardiogram continuous recording,
Custo Card M, Custo Med, Ottobrunn, Germany). Positivity of the test for elec-
trocardiography and/or symptoms was considered as exclusion criteria.
In each subject, two samples of venous blood from an antecubital vein
were collected immediately before and after the exercise (about 5 ml each;
total of 20 ml) and processed to determine the serum levels of cardiac Tro-
ponin I (cTnI), the hematocrit, and the number of EPCs and CECs. Plasma
levels of cTnI were determined by an immunoenzymatic method using a
specific analyzer (Dimension RxL, Dade-Behring, Milton Keynes, Bucking-
hamshire, UK). CECs and EPCs were defined according to Biguzzi et al.
[14], respectively, as cells CD452/CD1461/CD311 and cells CD452/
CD341/KDR1, and were quantified by Flow Cytometry (FACScan, Becton
Dickinson, San Jose, CA) according to a previously described procedure
collecting 500,000 events per sample (Figure 1) [15]. Data were analyzed
using SPSS – Rel 13 (SPSS Inc., Chicago, IL). All quantitative variables
were tested for Gaussian distribution with the Kolmogorov-Smirnov test.
Changes in any of the studied variables at each time intervals were tested
by ANOVA. The relationship between PB EPCs changes and other varia-
bles was tested by regression analysis. In all cases, P < 0.05 was consid-
ered significant.
All study subjects completed the exercise testing reaching the 85% of pre-
dicted maximum heart rate (mean 165 ± 15 b/min) at a load of 165 ± 36 W
with neither symptoms nor ECG changes suggestive of reduced coronary
reserve. Serum cTnI levels after the test remained in the normal range
(<0.15 ng/ml). At peak of exercise, a significant increase in BP was
observed (SBP from 123 ± 15 to 182 ± 20 mmHg, P < 0.0001; DBP from
84 ± 11 to 95 ± 13 mmHg, P 5 0.0067); a summary of studied parameters
is reported in Table I. The number of CECs increased significantly from 16.3
(range, 10.8–23.8) to 24.6 (range, 20.1–43.6) cells per micoliter (P < 0.001;
mean differences 95% CI 211.15, from 215.64 to 26.65). At maximal exer-
cise, a direct correlation between SBP, rate-pressure product (RPP), and the
number of CECs measured after the exercise testing was found (r 5
0.7795, P < 0.001; r 5 0.6364, P < 0.05, respectively). The absolute
change in CECs after the exercise significantly correlated with peak SBP
and RPP (r 5 0.6163, P < 0.05; r 5 0,6044, P < 0.05, respectively). The
number of PB EPCs showed an upward tendency, but this increase did not
reach statistical significance.
In physiological conditions, EPCs are responsible for the maintenance of
a complex network of vessels: their mobilization is generally proportional to
the number of vital and apoptotic CECs detached from the endothelium. The
results of our study support the role of peak SBP during maximal exercise
for the renewal of ECs, possibly by facilitating the detachment of CECs and,
TABLE I. Main Studied Variables at Rest and at Peak Exercise
Rest Peak exercise P
Subjects (n8) 12 –Age (years) 37 ± 12 –BMI (kg/m2) 24.7 ± 2.1 –Load (W) – 165 ± 36 –Load duration (MM:SS) – 12:47 ± 02:55 –SBP (mmHg) 123 ± 15 182 ± 20 <0.0001***DBP (mmHg) 84 ± 11 95 ± 13 0.0067**MBP (mmHg) 97 ± 11 124 ± 10 <0.0001***HR (1 per min) 96 ± 18 165 ± 15 <0.0001***RPP (mmHg/min) 11,915 ± 2734 30,082 ± 4520 <0.0001***cTnI (ng/ml) 0.03 ± 0.03 0.03 ± 0.03 0.72CECs (n/ml) (range) 16.3
(10.8–23.8)24.6
(20.1 – 43.6)0.0001***
BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pres-sure; MBP, mean blood pressure; HR, heart rate; RPP, rate-pressure product;cTnI, cardiac troponin I; CECs, circulating endothelial cells. P < 0.05 was consid-ered significant. 00 5 **, 000 5 ***.
Letter
VVC 2009 Wiley-Liss, Inc.
American Journal of Hematology 449 http://www3.interscience.wiley.com/cgi-bin/jhome/35105

therefore, accelerating the endothelial turnover. Although the increase in EPCs
number in our study did not reach statistical significance, it is a well-known
fact that PB EPCs are extremely rare and their mobilization takes place hours
after the application of the stimulus, whereas in this study the blood was drawn
after about 10 min after the goal of the exercise test was achieved.
Our observations on the effects of exercise on the CECs may help to
explain the benefits that exercise has on the cardiovascular system, sug-
gesting a role of mechanical factors such as BP on the endothelial renewal
in physiological conditions.
1Department of Respiratory and Cardiovascular Disease, Centro di FisiologiaClinica e Ipertensione, Laboratory of Cardiovascular Imaging,
Universita di Milano, Milan, Italy2Unita Operativa di Medicina ad Indirizzo Cardiovascolare, Fondazione IRCCS
Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena, Milan, Italy3Dipartimento di Scienze Mediche, Universita di Milano, Milan, Italy4Unita Operativa Laboratorio Centrale di Analisi Chimico-Cliniche e
Microbiologiche e Orientamento dei Laboratori Specialistici, Fondazione IRCCSOspedale Maggiore Policlinico, Mangiagalli e Regina Elena, Milan, Italy*Correspondence to: Michele M. Ciulla, Department of Respiratory and
Cardiovascular Disease, Centro di Fisiologia Clinica e Ipertensione, IRCCSOspedale Maggiore Policlinico Mangiagalli e Regina Elena, Via F. Sforza 35
20122 Milano, Italy. Email: [email protected] online 31 March 2009 in Wiley InterScience
(www.interscience.wiley.com).DOI: 10.1002/ajh.21424
Conflict of interest: Nothing to report.
References1. Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology
and in the pathophysiology of vascular disorders. Blood 1998;91:3527–3561.
2. Hunting CB, Noort WA, Zwaginga JJ. Circulating endothelial (progenitor)cells reflect the state of the endothelium: Vascular injury, repair and neovas-cularization. Vox Sang 2005;88:1–9.
3. Hristov M, Erl W, Weber PC. Endothelial progenitor cells: Mobilization,differentiation, and homing. Arterioscler Thromb Vasc Biol 2003;23:1185–1189.
4. Ozdogu H, Sozer O, Boga C, et al. Flow cytometric evaluation of circulatingendothelial cells: A new protocol for identifying endothelial cells at severalstages of differentiation. Am J Hematol 2007;82:706–11.
5. Khan SS, Solomon MA, McCoy JP Jr. Detection of circulating endothelialcells and endothelial progenitor cells by flow cytometry. Cytometry B2006;70:104–105.
6. Laufs U, Werner N, Link A, et al. Physical training increases endothelialprogenitor cells, inhibits neointima formation, and enhances angiogenesis.Circulation 2004;109:220–226.
7. Sandri M, Adams V, Gielen S, et al. Effects of exercise and ischemia onmobilization and functional activation of blood-derived progenitor cells inpatients with ischemic syndromes: Results of 3 randomized studies. Circula-tion 2005;111:3391–3399.
8. Rehman J, Li J, Parvathaneni L, et al. Exercise acutely increases circulatingendothelial progenitor cells and monocyte/macrophage-derived angiogeniccells. J Am Coll Cardiol 2004;43:2314–2318.
9. Ciulla MM, Giorgetti A, Lazzari L, et al. High-altitude trekking in the Hima-layas increases the activity of circulating endothelial cells. Am J Hematol2005;79:76–78.
10. Ciulla MM, Cortiana M, Silvestris I, et al. Effects of simulated altitude (nor-mobaric hypoxia) on cardiorespiratory parameters and circulating endothelialprecursors in healthy subjects. Respir Res 2007;8:58–65.
11. Tian Y, Nie J, Tong TK, et al. Changes in serum cardiac troponins followinga 21-km run in junior male runners. J Sports Med Phys Fitness2006;46:481–488.
12. Neumayr G, Gaenzer H, Pfister R, et al. Plasma levels of cardiac troponin Iafter prolonged strenuous endurance exercise. Am J Cardiol 2001;87:369–371.
13. Bruce RA. Clinical exercise testing. A review of personal and communitypractice experience. Prim Care 1994;21:405–414.
14. Biguzzi E, Mancuso P, Franchi F, et al. Circulating endothelial cells(CECs)and Progenitors (CEPs) in severe haemophiliacs with different clinical phe-notype. Br J Haematol 2009;144:803–805.
15. Ciulla MM, Giorgetti A, Silvestris I, et al. Endothelial colony forming capacityis related to C-reactive protein levels in healthy subjects. Curr NeurovascRes 2006;3:99–106.
Figure 1. A: Gate used to exclude necrotic/dead cell fragments and debris. B:Gate used to depict CD452 (nonhematopoietic) cells. C: Negative control. D, E: Pre-maximal and postmaximal exercise testing CECs, respectively. [Color figure can beviewed in the online issue, which is available at www.interscience.wiley.com.]
letter
450 American Journal of Hematology
Familial sideroblastic anemia associated with cardiacatrial septal defect
Masaki Mori,1,2* Shu Nakamoto,2 Youichi Akifuji,2 Takayuki Tanaka,2
Norio Komatsu,3 Kiyohiko Hatake,4 and Keiya Ozawa1
Sideroblastic anemia (SA) is defined by the presence of ringed sidero-
blasts in the bone marrow, and may be due to both hereditary and
acquired causes. The most common hereditary form is X-linked SA
(XLSA), which is due to mutations in the erythroid-specific 5-aminole-
vulinate synthase gene (ALAS2) [1,2] and occurs predominantly in
men [3]. Another form of XLSA, X-linked SA and ataxia, is due to muta-
tions in the mitochondrial ATP binding cassette transporter ABCB7
[4,5]. Other syndromic forms are inherited in an autosomal recessive
manner (thiamine-responsive megaloblastic anemia with diabetes and
deafness [6]; mitochondrial myopathy, lactic acidosis and SA [7,8]) or
result from sporadic congenital defects in mitochondrial DNA (Pearson
marrow pancreas syndrome) [9].
A 41-year-old man first came to our attention in 1990 for the evaluation
and surgical correction of an atrial septal defect (ASD). He had more than
30-year history of mild anemia, which had been observed without treatment.
Hematological assessment (Table I) showed a red blood cell count of 2.79
3 1012/L, hemoglobin of 9.1 g/dL, hematocrit of 26.7%, mean corpuscular
volume of 96 fL, and reticulocytes of 0.03 3 1012/L. The serum iron of 199
mg/dL, the total iron-binding capacity of 205 mg/dL with a transferrin satura-
tion of 97%, and the serum ferritin of 350 ng/mL indicated mild iron over-
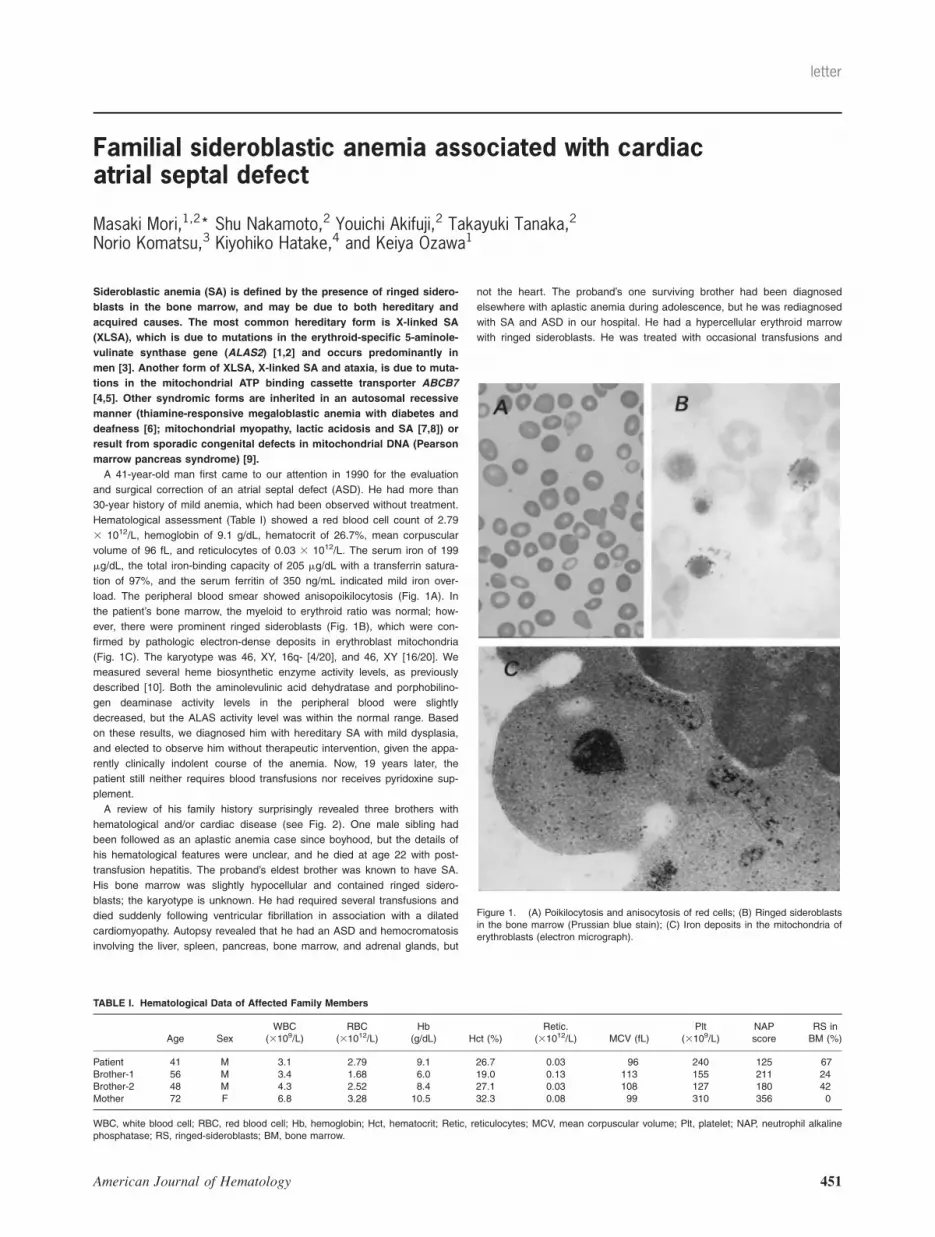
load. The peripheral blood smear showed anisopoikilocytosis (Fig. 1A). In
the patient’s bone marrow, the myeloid to erythroid ratio was normal; how-
ever, there were prominent ringed sideroblasts (Fig. 1B), which were con-
firmed by pathologic electron-dense deposits in erythroblast mitochondria
(Fig. 1C). The karyotype was 46, XY, 16q- [4/20], and 46, XY [16/20]. We
measured several heme biosynthetic enzyme activity levels, as previously
described [10]. Both the aminolevulinic acid dehydratase and porphobilino-
gen deaminase activity levels in the peripheral blood were slightly
decreased, but the ALAS activity level was within the normal range. Based
on these results, we diagnosed him with hereditary SA with mild dysplasia,
and elected to observe him without therapeutic intervention, given the appa-
rently clinically indolent course of the anemia. Now, 19 years later, the
patient still neither requires blood transfusions nor receives pyridoxine sup-
plement.
A review of his family history surprisingly revealed three brothers with
hematological and/or cardiac disease (see Fig. 2). One male sibling had
been followed as an aplastic anemia case since boyhood, but the details of
his hematological features were unclear, and he died at age 22 with post-
transfusion hepatitis. The proband’s eldest brother was known to have SA.
His bone marrow was slightly hypocellular and contained ringed sidero-
blasts; the karyotype is unknown. He had required several transfusions and
died suddenly following ventricular fibrillation in association with a dilated
cardiomyopathy. Autopsy revealed that he had an ASD and hemocromatosis
involving the liver, spleen, pancreas, bone marrow, and adrenal glands, but
not the heart. The proband’s one surviving brother had been diagnosed
elsewhere with aplastic anemia during adolescence, but he was rediagnosed
with SA and ASD in our hospital. He had a hypercellular erythroid marrow
with ringed sideroblasts. He was treated with occasional transfusions and
TABLE I. Hematological Data of Affected Family Members
Age SexWBC
(3109/L)RBC
(31012/L)Hb
(g/dL) Hct (%)Retic.
(31012/L) MCV (fL)Plt
(3109/L)NAPscore
RS inBM (%)
Patient 41 M 3.1 2.79 9.1 26.7 0.03 96 240 125 67Brother-1 56 M 3.4 1.68 6.0 19.0 0.13 113 155 211 24Brother-2 48 M 4.3 2.52 8.4 27.1 0.03 108 127 180 42Mother 72 F 6.8 3.28 10.5 32.3 0.08 99 310 356 0
WBC, white blood cell; RBC, red blood cell; Hb, hemoglobin; Hct, hematocrit; Retic, reticulocytes; MCV, mean corpuscular volume; Plt, platelet; NAP, neutrophil alkalinephosphatase; RS, ringed-sideroblasts; BM, bone marrow.
Figure 1. (A) Poikilocytosis and anisocytosis of red cells; (B) Ringed sideroblastsin the bone marrow (Prussian blue stain); (C) Iron deposits in the mitochondria oferythroblasts (electron micrograph).
letter
American Journal of Hematology 451

underwent repair of a central type of ASD. The patient and his brothers had
no gastrointestinal symptoms, and they were not prescribed pyridoxine in
compliance with their wishes to be observed without medication. A sister
died in infancy of unknown causes.
The clinical and hematologic features of these individuals do not fit with
recognized causes of congenital SA. Although the ASD may represent an
unrelated genetic abnormality in this family, the strong association of the SA
and ASD is suggestive of a novel, inherited syndromic SA that may be
revealed by studies at the molecular level.
Acknowledgments
The authors thank Dr. Masao Kondo for giving valuable advice and techni-
cal support regarding the measurement of Heme biosynthesis.
1Division of Hematology, Department of MedicineJichi Medical University, Tochigi, Japan
2Department of Medicine, Tottori Prefectural HospitalTottori, Japan
3Department of Hematology, Yamanashi UniversityYamanashi, Japan
4Division of Medical Oncology/Hematology, Cancer ChemotherapyCenter, Japanese Foundation for Cancer Research, Tokyo, Japan
*Correspondence to: Masaki Mori, Division of Hematology,Department of Medicine, Jichi Medical University, 3311-1 Yakushiji,
Shimotsuke, Tochigi 329-0498, Japan.E-mail: [email protected]
Published online 31 March 2009 in Wiley InterScience(www.interscience.wiley.com).
DOI: 10.1002/ajh.21425Conflict of interest: Nothing to report.
References1. Cotter PD, Baumann M, Bishop DF. Enzymatic defect in ‘‘X-linked’’ sidero-
blastic anemia: Molecular evidence for erythroid d-aminolevulinate synthasedeficiency. Proc Natl Acad Sci USA 1992;89:4028–4032.
2. Cotter PD, Rucknagel DL, Bishop DF. X-linked sideroblastic anemia: Identifi-cation of the mutation in the erythroid-specific d-aminolevulinate synthasegene (ALAS2) in the original family described by Cooley. Blood 1994;84:3915–3924.
3. Bottomley SS. Sideroblastic anemias. In: Greer JP, Foerster J, Rodgers GM,et al., editors. Wintrobe’s Clinical Hematology, 12th ed. Philadelphia: Lippin-cott Williams and Wilkins; 2008. pp 835–856.
4. Allikmets R, Raskind WH, Hutchinson A, et al. Mutation of a putative mito-chondrial iron transporter gene (ABC7) in X-linked siderobalstic anemia andataxia (ALSA/A). Hum Mol Genet 1999;8:743–749.
5. Pondarre C, Campagna DR, Antiochos B, et al. Abcb7, the gene responsiblefor X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis.Blood 2007;109:3567–3569.
6. Fleming JC, Tartaglini E, Steinkamp MP, et al. The gene mutated in thi-amine-responsive anaemia with diabetes and deafness (TRMA) encoding afunctional thiamine transporter. Nat Genet 1999;22:305–308.
7. Casas KA, Fischel-Ghodsian N. Mitochondrial myopathy and sideroblasticanemia. Am J Med Genet 2004;125A:201–204.
8. Bykhovskaya Y, Casas K, Mengesha E, et al. Missense mutation in psudour-idine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblasticanemia (MLASA). Am J Hum Genet 2004;74:1303–1308.
9. Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hema-tol 2002;39:270–281.
10. Kondo M, Ohe M, Mizuguchi M. Decreased leukocyte ferrochelatase activityin erythropoietic protoporphyria. J Dermatol 1989;16:116–121.
Figure 2. Family history of the patient with both sideroblastic anemia and anatrial septal defect. HT, hypertension; CI, cerebral infarction; GC, gastric cancer;SA, sideroblastic anemia; DCM, dilated cardiomyopathy; ASD, atrial septal defect;AA, aplastic anemia.
Essential thrombocythemia in patients with platelet counts below600x109/L: Applicability of the 2008 World Health Organizationdiagnostic criteria revision proposal
Mi Kwon, Santiago Osorio, Carolina Munoz, Jose Manuel Sanchez, Ismael Buno, andJose Luis Dıez-Martın
The World Health Organization (WHO) diagnostic criteria as well as the
Polycythemia Vera Study Group (PVSG) criteria define platelet counts
above 600x109/L as the threshold for essential thrombocythemia (ET)
diagnosis [1,2]. It has been argued that such threshold excludes a
number of patients with actual ET with platelet counts below 600 3
109/L [3–5]. Recently, a proposal for revision of the WHO diagnostic
criteria for ET has been published, which includes the combination of
histological bone marrow study and testing of the JAK2 mutation to
facilitate the diagnosis of ET with borderline thrombocytosis [6,7]. The
aim of this study was to evaluate the applicability of the proposal of
the WHO revised diagnostic criteria in patients presumed to have ET
with platelet counts below 600 3 109/L. Additionally, clinical and labo-
ratory features of this group were compared to the group with platelet
counts above 600 3 109/L to assess any differences between both
groups. Finally, clinical and laboratory features of JAK2 positive
patients were compared to JAK2 negative patients to confirm in our
series the differences previously described in the literature [8,9].
In this retrospective study, we included 92 nonconsecutive patients with a
presumptive diagnosis of ET made between June 1989 and February 2008
in a single institution, and who received follow-up between 2006 and 2008.
Diagnosis of ET was made following classic 2001 WHO criteria [1], excluding
patients with polycythemia vera and patients who showed iron deficiency.
Cases with primary myelofibrosis (PMF) and myelodysplastic syndrome
(MDS) were excluded according to WHO criteria as well. A group of patients
with platelet counts between 425 and 600 3 109/L were included in the
analysis. The presumption of ET diagnosis in this group of patients was
based on compatible bone marrow histology, the presence of JAK2 mutation
or/and persistence of thrombocytosis for more than 2 years without evidence
of an alternative cause. The new proposed 2008 WHO criteria were eval-
uated in those cases who did not fulfill the prior criteria due to platelet
letter
452 American Journal of Hematology
counts below 600 3 109/L, and this subgroup was analyzed separately.
Patients were stratified in high, intermediate and low thrombosis risk groups
according to previously published criteria (Table I) [10]. The V617F mutation
detection in the JAK2 gene was performed by polymerase chain reaction
and probe dissociation (melting curve) analysis, using a previously published
method [11]. Mutations of exon 12 were not analyzed for this study.
Similar to previously published data, in our series of 92 patients, ET diagno-
sis was more frequent in women (64%) (Table II). The median age of presen-
tation was 51 years (range 19–84), and the median platelet count at diagnosis
was 693 3 109/L (range 424–2,777). Fifty one percent of patients showed
JAK2 V617F mutation. There were no significant differences between JAK2
positive patients and JAK2 negative patients regarding demographic features.
JAK2 positive patients showed higher Hb and hematocrit values compared
with JAK2 negative patients (P 5 0.03 and P 5 0.007, respectively). In addi-
tion, the JAK2 positive group tended to have lower erythropoietin levels (P 5
0.05). Thrombotic events were almost twice as frequent in JAK2 positive
patients (23 vs. 11%), although with no statistical significance. Therefore,
supporting previous reports [8,9], JAK2 positive patients showed a phenotype
closer to polycythemia vera patients with higher Hb and hematocrit values,
lower erythropoietin values and more frequent thrombotic events.
The main clinical and laboratory characteristics of patients showing plate-
let counts below 600 3 109/L (n 5 30) are compared to the group with pla-
telet counts above 600 3 109/L (n 5 62) in Table III. There were no signifi-
cant differences between both groups in demographic, clinical and labora-
tory features. The median age of the borderline platelet count group was 51
years (range 19–83) and 20 were female (70%). At diagnosis their median
platelet count was 527 3 109/L (range 424–597). Fifteen patients (50%)
showed the presence of JAK2 mutation.
From the 30 patients who did not fulfill the 2001 WHO criteria due to low
platelet counts, 25 (83%) did fulfill the modified criteria allowing ET diagnosis.
Among them, one patient showed an alternative cause of thrombocytosis;
however, JAK2 mutation was positive confirming the primary cause of the dis-
TABLE I. Thrombotic Risk Groups*
Low risk Age <60 years and no history of thrombosis,extreme thrombocytosis (platelet count>1,500x109/L), or cardiovascular risk factors.
High risk Age >60 years or history of thrombosis.Intermediate risk Neither low risk nor high risk.
*Ref 8: A Tefferi, S. Murphy. Blood Reviews. 2001;15:121.
TABLE II. Clinical, Laboratory, and Demographic Features in 92 Patientswith ET
TotalJAK2positive
JAK2negative P*
Number 92 47 (51%) 45 (49%) NSFemale(number, %)
59(64%)
29(61%)
30(67%)
NS
Age(years,median, range)
51(19–84)
55(30–84)
47(19–82)
NS
Plateletesx109/L(median,range)
693(424–2,777)
696(429–1,634)
682(424–2,777)
NS
Hb g/dL(median, range)
14.5(11–18)
14.7(11.8–16.8)
14(11–18)
0.03
Hct %(median,range)
43(31–52)
44(35–53)
41(31–52)
0.007
Leucocytes3109/L(median,range)
8.5(3.6–24.2)
8.8(3.6–24.2)
7.7(5.2–15)
NS
LDH UI/L(median,range)
380(39–1413)
390(39–893)
346(190–1,413)
NS
Erythropoietin(median, range)
5.7(0–76)
4.2(0.5–76)
8.7(0–25)
0.05
Splenomegaly(number, %)
16(17%)
9(19%)
7(16%)
NS
Bone MarrowCelullarity >3.5
(number,available, %)
30/88(34%)
18/46(38%)
12/43(26%)
NS
Compatiblecytology(number,available, %)
77/87(88%)
43/46(91%)
39/41(86%)
NS
Compatibletephrine(number,available, %)
79/89(86%)
42/45(89%)
39/45(86%)
NS
Symptoms(number, %)
13 (14%) 7 (15%) 6 (13%) NS
Thrombotic event(number, %)
16 (18%) 11 (23%) 5 (11%) NS
Art / Ven 12 / 5 8 / 3 4 / 1 NSHaemorrhagicevent(number, %)
3 (4%) 2 (4%) 1 (2%) NS
Risk (number, %)Low 26 (28%) 11 (23%) 15 (33%) NSIntermediate 21(22%) 10 (21%) 11 (24%) NSHigh 45 (48%) 26 (55%) 19 (42%) NS
Outcome (number, %)AML 0 0 0 NSFibrosis 5 (5%) 2 (4%) 3 (7%) NSPV 0 0 0 NS
Further thromboticevent (number, %)
6 (7%) 2 (4%) 4 (9%) NS
Comparison between JAK2 positive versus JAK2 negative patients.*P value corresponds to comparison between JAK2 positive and JAK2 negativepatients.
TABLE III. Clinical, Laboratory, and Demographic Features of ET Patientswith Platelet Counts >600 3 109/L Compared with Those with PlateletCounts <600 3 109/L
Plateletcount
>600 3 109/L
Plateletcount
<600 3 109/L P
Number 62 30Female (number, %) 39 (63%) 20 (70%) NSMale (number, %) 23 (37%) 10 (30%) NSAge (years,median, range)
51 (21–84) 51 (19–83) NS
Mayorcardiovascularrisk factors
36 (58%) 17 (57%) NS
Risk category (number, %)Low 18 (29%) 8 (26%) NSIntermediate 15 (24%) 6 (19%) NSHigh 29 (46%) 16 (53%) NS
Platelets 3109/L(median, range)
801 (600–2,777) 527 (424–597)
Hb g/dL(median, range)
14.5 (11.8–18) 14.3 (11–16.8) NS
Hct % (median, range) 43 (34–52) 43 (31–50) NSLeucocytes 3109/L(median, range)
8.6 (3.6–24.2) 8.5 (5.2–13.8) NS
LDH UI/L (median, range) 390 (190–1,413) 337 (39–938) NSErytropoietin U/L(median, range)
7.7 (0.5–76) 4.7 (0–24) NS
Splenomegaly(number, %)
11 (18%) 5 (17%) NS
Bone marrowCellularity >3,5
(number, available, %)22/58 (38%) 8/28 (26%) NS
Compatible histology(number, available, %)
60/62 (96%) 25/29 (83%) NS
Abnormal cytogenetics(number, available, %)
2/40 (5%) 0/24 (0%) NS
JAK2 mutation (number, %) 32 (51%) 15 (50%) NSClinical featuresSymptoms (number, %) 9 (14%) 4 (13%) NSThrombotic events %/pt/yr 4.7 5.2 NSHaemorrhagic events
(number, %)3 (4%) 0 NS
Follow-up, years(median, range)
4.6 (0.33–14.8) 2.7 (0.33–18.7)
letter
American Journal of Hematology 453

order. Five patients remained not fulfilling the new criteria due to insufficient
bone marrow sample or incompatible histology. However, one of these
patients showed JAK2 mutation, without features of MDS or PMF, confirming
ET. The remaining four patients had no second bone marrow sample avail-
able. One of them was older than 60 years with mayor cardiovascular risk fac-
tors, and no alternative causes of thrombocytosis, therefore, considered at
high risk of thrombosis and managed as ET although bone marrow sample
was insufficient for a definitive diagnosis. The other three cases showed a
bone marrow histology not definitive for ET diagnosis, however platelet counts
remained higher than 4003109/L during a median follow-up of 2 years in
these patients with no evidence of alternative causes of thrombocytosis mak-
ing ET diagnosis very likely. A second pathology revision of these bone mar-
row histologies confirmed absence of PMF or prefibrotic PMF features.
Remarkably, 74% of the patients belonged to high and intermediate risk
groups at diagnosis. The median time of follow-up was 2.75 years (range
0.33–18.7). During follow-up, 27 out of 30 patients were treated with antiag-
gregating drugs (mainly aspirin), three with antithrombotic therapy, and 20
with myelosuppressive therapy. None of the 10 patients who did not receive
myelosuppressive therapy showed a spontaneous decrease of platelet
counts to normal values remaining with platelet counts above 400 3 109/L,
and only two of them exceeded 600 3 109/L. Furthermore, transformation
from ET to myelofibrosis was observed in two patients, both JAK2 negative,
supporting the diagnosis of ET.
The diagnosis of ET has been based on exclusion of other chronic myelo-
proliferative disease and secondary thrombocytosis. The WHO and the
PVSG criteria consider a platelet count above 600 3 109/L as an absolute
requirement for a diagnosis of ET [1,2]. Recently, a proposal of WHO criteria
modification lowering the platelet counts threshold to 450 3 109/L and
including the detection of JAK2 mutation has been published [6].
Although in our study the new criteria have been applied retrospectively,
in patients presumed to have ET, we observed that a high proportion of
patients with platelet counts between 400 and 600 3 109/L (25 out of 30)
can be diagnosed as having ET following these criteria. Although the new
criteria propose 450 3 109/L as the cut off of platelet counts for ET diagno-
sis, we included only one case with platelet counts below that threshold
(424 3 109/L) since the rest of the clinical data supported the diagnosis.
The rest of the included cases showed platelet counts above 450 3 109/L.
The detection of the JAK2 mutation in this setting enables accurate ET diag-
nosis not only in cases with borderline thrombocytosis but more importantly
in cases with alternative potential causes of thrombocytosis and also in
cases where a bone marrow sample is not available or is not fully consistent
with ET diagnosis. This observation raises the question of bone marrow
morphology as a subjective diagnostic tool in ET diagnosis. Its reliability
depends on the observer training and experience, and it is subject to inter-
observer variability. A recent study showed substantial interobserver variabil-
ity regarding histopathology assessment in the diagnosis of ET subtypes,
particularly for overall diagnosis and individual cellular characteristics such
as megakaryocyte morphology [12]. In our study, we found one patient with
maintained high platelet counts, no evident causes of secondary thrombocy-
tosis, presence of JAK2 mutation which supports ET diagnosis, but a bone
marrow histology not fully diagnostic. In our opinion, ET is the likely cause
of the thrombocytosis in this case. On the other hand, in JAK2 negative
patients with platelet counts <600 3 109/L, the maintenance of high platelet
counts over subsequent follow-up in the absence of therapy, and without
causes of secondary thrombocytosis supports ET diagnosis. Furthermore,
some of these cases transformed to myelofibrosis. In our study, four JAK2
negative patients showed a bone marrow histology not compatible or insuffi-
cient for ET at diagnosis, making ET diagnosis particularly challenging. This
shows the not uncommon presentation of probable early phase ET cases in
clinical practice, which remains not fulfilling the complete criteria for definite
diagnosis. Testing for other clonal markers which are currently being studied
such as MPL mutations could be useful in this setting [13].
Finally, we did not find any significant differences from the comparison
between patients with platelet counts above and patients with counts below
600 3 109/L regarding clinical presentation, laboratory profile including JAK2
mutation frequency, and outcome. In our opinion, this finding indirectly sug-
gests that ET is the most likely diagnosis in these patients with platelet
counts below 600 3 109/L.
In conclusion, the modified WHO diagnostic criteria enable the clinician to
make an early an accurate diagnosis of ET in patients with platelet counts
below 600 3 109/L. Moreover, a high proportion of these patients may be at
high risk of vascular complications, and may benefit from being correctly
diagnosed and treated in earlier phases of the disease. Prospective studies
using these new criteria in all patients suspected to have ET with platelet
counts between 450 and 600 3 109/L will provide more information about
their validation in the investigation of ET.
Acknowledgments
The authors are indebted to Isabel Perez-Sanchez, Victor Echeverrıa,
Monica Ballesteros, Magdalena Mayayo, Javier Menarguez, Antonio Escu-
dero Soto. This work has been partially supported by grant PI05-2505 from
the Spanish Ministry of Health (FIS-ISCIII).
Department of HematologyGregorio Maranon G. U. Hospital
Madrid, SpainPublished online 31 March 2009 in Wiley InterScience
(www.interscience.wiley.com).DOI: 10.1002/ajh.21428
Conflict of interest: Nothing to report.
References1. Vardiman JW, Harris NL, Brunning RD. The World Health Organization
(WHO) classification of the myeloid neoplasms. Blood.. 2002;100:2292–2302.
2. Murphy S, Peterson P, Iland H, et al. Experience of the Polycythemia VeraStudy Group with essential thrombocythemia: A final report on diagnostic crite-ria, survival, and leukemic transition by treatment. Semin Hematol 1997;34:29–39.
3. Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormalcomplete blood cell count in adults. Mayo Clin Proc 2005;80: 923–936.
4. Sacchi S, Vinci G, Gugliotta L, et al. Diagnosis of essential thrombocythemiaat platelet counts between 400 and 600 3 10(9)/L. Gruppo Italiano MalattieMeloproliferative Croniche (GIMMC). Haematologica 2000;85:492–495.
5. Lengfelder E, Hochhaus A, Kronawitter U, et al. Should a platelet limit of600 3 10(9)/l be used as a diagnostic criterion in essential thrombocythae-mia? An analysis of the natural course including early stages. Br J Haematol1998;100:15–23.
6. Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of theWorld Health Organization diagnostic criteria for polycythemia vera, essen-tial thrombocythemia, and primary myelofibrosis: Recommendations from anad hoc international expert panel. Blood 2007;110:1092–1097.
7. Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferativeneoplasms: The 2008 World Health Organization criteria and point-of-carediagnostic algorithms. Leukemia 2008;22:14–22.
8. Speletas M, Katodritou E, Daiou C, et al. Correlations of JAK2-V617F muta-tion with clinical and laboratory findings in patients with myeloproliferativedisorders. Leuk Res 2007;31:1053–1062.
9. Wolanskyj A, Lasho T, Schwager S, et al. JAK2V617F mutation in essentialthrombocythaemia: Clinical associations and long-term prognostic relevance.Br J Haematol 2005;131:208–213.
10. Tefferi A, Murphy S. Current opinion in essential thrombocythemia: Patho-genesis, diagnosis, and management. Blood Rev 2001;15:121–131.
11. Lay M, Mariappan R, Gotlib J, et al. Detection of the JAK2 V617F mutationby LightCycler PCR and probe dissociation analysis. J Mol Diagn 2006;8:330–334.
12. Wilkins BS, Erber WN, Bareford D, et al. Bone marrow pathology in essen-tial thrombocythemia: Interobserver reliability and utility for identifying dis-ease subtypes. Blood 2008;111:60–70.
13. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferativedisorders: analysis of the PT-1 cohort. Blood 2008;112:141–149.
letter
454 American Journal of Hematology

Reappearance of acute myeloid leukemia after almost23 years of continuous complete remission
Sung Ho Lee1, Lool Abebe2, Elisabeth Paietta1, Avi Einzig,3 and Peter H. Wiernik1*
Despite major advances in the treatment of acute myeloid leukemia
(AML) in adults over the last 3 decades, most patients with other than
acute promyelocytic leukemia (APL) still succumb to the disease. For
young adults (<60 years of age), death during initial treatment has
become the exception rather than the rule it once was, primarily due
to major improvements in supportive care, and approximately 70% will
achieve a complete remission (CR) with appropriate treatment. How-
ever, even among young adults with non-APL AML, only about 25%
are cured with present-day therapy, and most relapse and die well
within several years of obtaining a first CR. Relapse is usually
assumed to be the result of subclinical disease that persisted through-
out initial treatment. However, occasional reports of very late relapses
of AML suggest that other mechanisms, such as the development of a
secondary leukemia may be operative in at least some patients. We
report here an extremely late reappearance of AML and discuss the
implications of this observation.
In July 1984, a previously healthy 19-year-old Hispanic male was referred
for further evaluation of a right preauricular mass, and a white blood cell
count (WBC) of 2,000 per ml with 4% polymorphonuclear neutrophils and
96% mature lymphocytes. He had a nonintentional 16 lbs weight loss asso-
ciated with mild anorexia and increased fatigability. He had no history of
serious illness or relevant environmental exposure and was taking no medi-
cation. He had no personal or family history of blood dyscrasias. Two years
before, the patient had a routine blood test that demonstrated normal blood
counts and a normal WBC differential.
At presentation, he had several 1 3 1 cm cervical lymph nodes and a 3
3 3 cm nontender mobile, rubbery right preauricular lymph node as well as
several 2 3 1.5 cm left axillary lymph nodes. There were no hepatospleno-
megaly, gingival hypertrophy, ecchymoses, or petechiae. His tonsils were
enlarged bilaterally.
His WBC count was 2,200 cells per microliter, hemoglobin 12.3 g/dl, hem-
atocrit 37.1%, and platelet count 195,000 cells per microliter. His peripheral
blood smear showed granulocytopenia with the majority of WBC being
immature leukocytes. A bone marrow biopsy demonstrated a hypercellular
marrow largely replaced by blast forms diagnostic of AML FAB-type M4.
Immunophenotyping of the marrow aspirate was consistent with a diagnosis
of immature monocytic leukemia (Table I, Fig. 1). Cytogenetic studies
revealed 45, XY, 221 in all 20 metaphases analyzed. The patient received
induction chemotherapy with a standard regimen of daunorubicin, 45 mg/M2
daily intravenously for 3 days together with a continuous 7-day intravenous
infusion of cytarabine at the rate of 100 mg/M2/day. By treatment day 23,
the patient had achieved a bone marrow CR by standard morphologic crite-
ria and a normal karyotype in all 20 metaphases studied, and his lympha-
denopathy had resolved. One month later, when the patient’s bone marrow
biopsy still demonstrated morphologic CR and normal cytogenetics, consoli-
dation therapy with two daily injections of daunorubicin and a 5-day continu-
ous infusion of cytarabine was given. Both drugs were given at the same
daily doses given initially. A second identical consolidation course was given
1 month after recovery from the first. After hematologic recovery from that
treatment, the patient was started on a program of intensive maintenance
therapy [3], which consisted of intravenous cytarabine bolus injections, 100
mg/M2 every 12 hr and oral 6-thioguanine at the same dose and schedule.
Both drugs were given for a variable number of days every 3 months until
marrow aplasia was achieved. His treatments were not complicated by seri-
ous infections and this regimen was continued for 3 years. The last bone
marrow cytogenetic evaluation at our center in September 1990 still revealed
a normal karyotype.
The patient functioned normally without significant illness and with normal
blood counts until March 2007 when, at the age of 41, he developed left hip
discomfort. A bone marrow biopsy was performed elsewhere and revealed a
hypercellular marrow with 80% blasts with monocytic morphologic features,
similar to those seen at his initial presentation, and compatible with relapse
of AML FAB-type M4. Immunocytochemistry of biopsy sections confirmed
acute myelomonocytic leukemia. Cytogenetic evaluation revealed a complex
karyotype, 47, XY, 14, 27, 111, in nine of 20 metaphases examined (Table
I, Fig. 1). A satisfactory explanation for his hip discomfort was never found.
In April 2007, he received a standard induction course of idarubicin and
cytarabine. A postinduction bone marrow biopsy approximately 3 weeks later
revealed residual leukemia (9% myeloblasts) with trilineage dysplasia and
he received a second induction course identical to the first. The patient toler-
ated the treatment well, and his left hip pain improved. He was then referred
for allogenic bone marrow transplantation and his sole sibling was tested for
histocompatibility. Before the compatibility results were available, a bone
marrow biopsy in May 2007 showed residual leukemia with 11.7% blasts,
and 4 days later another marrow biopsy showed a hypercellular marrow with
80% blasts. The patient expired 2 weeks later at the end of May 2007,
approximately 22 years and 10 months after his initial diagnosis of AML.
Relapses of AML are most frequent during the first 2–3 years of CR, with
the majority occurring in the first year [4]. Recurrences of AML after more
than 5 years of CR are rare and account for only approximately 3% of all
relapses [5,6]. Table II summarizes late relapses that have been reported in
the literature, and our case [5,7–9]. Among the 23 cases in Table II, 5 had
M3 and 11 had monocytic (FAB M4/5) morphologic features at initial diagno-
sis. FAB subtypes were unchanged at relapse in 8/8 cases for which data
are available (5 with M3, 2 with M4, 1 with M1). More interestingly, cytoge-
netic data were identical at relapse in 13 of the 14 patients in whom relapse
studies had been performed. Our patient is the only one in whom a change
in karyotype was observed. However, one other patient without initial data
demonstrated a deletion of the long arm of chromosome 5 at relapse, an
aberration commonly observed in secondary leukemias [10]. It is remarkable
that all six late relapses in patients with APL failed to demonstrate significant
karyotypic changes. Zompi et al. [11] reported two APL cases who relapsed
after 29 and 23 months of first CR, and relapsed with other than APL. Cyto-
genetic changes at relapse in both patients suggested therapy-related AML,
with monosomies 5 and 7 and trisomy 11 in one patient, and monosomy 7
and del(5q) in the other. Both patients had been consolidated with daunoru-
bicin and cytarabine, followed by maintenance therapy with 6-mercaptopur-
ine and methotrexate, plus all-trans retinoic acid in one case. A literature
review at that time [11] suggested that in APL, relapses after 2–4 years of
CR commonly present with cytogenetic features of secondary leukemia, fre-
quently involving chromosome 7. This observation suggests that very late
relapses in APL are caused by true recurrence of latent disease, whereas
relapses in primary APL with short latency are therapy-induced secondary
AMLs. This conclusion may also apply to the majority of very late relapses
of non-M3 AML, given that only one patient in Table II, aside from our case,
relapsed with a karyotype suggestive of secondary disease. The data sug-
gest that the late-appearing AML in our patient is not a late relapse, but a
secondary AML. However, he did not receive drugs that have been com-
monly associated with secondary AML, nor was he aware of relevant envi-
ronmental exposure. Furthermore, his reappearance of AML occurred more
than 19 years after his last exposure to chemotherapy.
Generally, patients with relapsed AML have a poor prognosis with less
likelihood of achieving CR than de novo patients, and postrelapse survival
uncommonly exceeds 3–12 months [7,12]. The shorter the initial remission,
letter
American Journal of Hematology 455

the less likely a second remission will be achieved. In addition, there is a
direct relationship between the duration of the first and second remission.
Therefore, late relapse patients might be expected to have a relatively good
prognosis. Such was the case in one series of late relapse patients in which
a second CR rate of 87% was observed in 15 patients [5].
The biologic mechanism of very late relapse is not known. It has been
suggested that initial therapy may selectively spare the leukemic stem cell.
Over time, a preleukemic clone may acquire further mutations resulting in
relapsed leukemia [1]. Alternatively, local inflammation in the bone marrow
microenvironment may cause residual leukemia cells to escape from dor-
mancy [13]. Another possibility is that an etiologic agent has persisted dur-
ing CR in patients who relapse, even those with late relapses. Some viruses
are known to remain dormant in humans for decades only to cause disease
at a later date (i.e., Herpes zoster), and viruses are known to cause acute
leukemia in many vertebrate species. Persistence of an etiologic agent might
explain relapse in donor cells after an allogeneic bone marrow transplant as
well [14].
The present patient with a reappearance of AML after 22 years and 8
months of CR is the patient with the longest reported interval between two
presentations of AML reported to date. His chemoresistant and aggressive
disease at relapse, which led to his demise within 2 months and his karyo-
type all suggest that he had developed a secondary AML. Although monos-
omy 7 is a frequent finding in secondary leukemias following alkylating
agents [10], trisomy 4 and 11 are less common, but well-documented. Tris-
omy 4 as a single karyotypic abnormality may develop as a secondary event
following chemo- or radiotherapy, or long-term antibiotic treatment, and is
commonly associated with myelomonocytic or monocytic leukemia [rev. in
15]. On the other hand, several cases of de novo AML with trisomy 4 have
been reported [15,16]. Of interest, one of the published cases with very late
AML relapse had initially presented and relapsed with trisomy 4, albeit in
combination with der(13;14)(q10;q10) [9]. Trisomy 11 has been reported in
Figure 1. Top Plate 1 and 2—Bone marrow biopsy done on patients admitted onJuly 31, 1984, showed diffuse blast infiltrate with monocytic features and no differ-entiation, replacing the entire marrow. Flow cytometry at that time revealed thatthe blasts were positive for myelomonocytic markers (S3.13–80%; VIM-2, 25%;VIM-8 30%) (12) plate 1, magnification: 31,000; plate 2, magnification: 3400. Bot-tom 3 plates with immunohistochemical stains performed on bone marrow biopsyspecimen done on March 22, 2007, showed marrow blast cells staining strongly forc-kit, CD33, and CD 68 confirming the myelomonocytic features as those seen inthe original bone marrow biopsy in 1984 (magnification: 3400).
TABLE I. Bone Marrow Data
Initial July 30, 1984 Relapse March 22, 2007
Bone marrowbiopsy
Hypercellular marrowlargely replaced byimmature cells ofmyeloid series
Hypercellular marrowwith 80% blasts ofmyeloid series
Karyotype 45, XY,221 [20] 47, XY, 14, 27, 111 [9]/46,XY [11] (FISH forETO/AML1 Negative)
Immunophenotype S3.131 [1], CD11b1,CD151, CD65s1a
CD331, CD1171, CD681b
FAB type M4 M4
aFlow cytometry on 1984 [1].
bImmunostain was done on paraffin block, Flow cytometry was not available.S3.13 is a precursor antigen with similar distribution on hematopoietic cells asCD34 [2].
TABLE II. Reported Cases of Very Late Relapse AML (>5 Years)
ReferenceSex/age at
initial diagnosisInitial durationin remission
FAB Cytogenetics
Initial Relapse Initial Relapse
Ustun et al. [7] F/4 18 years M1 M1 46, XX, t(18;22) (q23;q11.2) 46, XX, t(18;22) (q23;q11.2)Latagliata et al. [8] F/16 12 years 11 months M3 M3 t(15;17) T(15;17)Medeiros et al. [5] F/52 11 years 8 months M4 NA NA NormalMedeiros et al. [5] M/41 11 years 2 months M4 NA NA NAMedeiros et al. [5] F/55 10 year 8 months M4 NA Normal NormalMedeiros et al. [5] F/35 9 years 8 months M2 NA NA 5q2Medeiros et al. [5] M/43 9 years 8 months M1 NA NA NormalMedeiros et al. [5] F/48 9 years 3 months M1 NA 46, del(1), 213, 1mar NAMedeiros et al. [5] M/50 9 years M1 NA NA NAMedeiros et al. [5] M/13 8 years 8 months M4 NA Normal NormalLatagliata et al. [8] F/30 8 years 5 months M3 M3 t(15;17) t(15;17)Medeiros et al. [5] F/22 8 years 3 months M3 NA 46, XX, t(15;17) 46, XX, t(15;17)Medeiros et al. [5] F/47 8 years 1 months M4 NA Normal NormalMeloni et al. [9] F/19 8 years M4 M4 46, XX, der(13;14) (q10;q10), 14 46, XX, der(13;14) (q10;q10), 14Medeiros et al. [5] F/63 6 years 1 months M5 NA NA NormalLatagliata et al. [8] F/16 5 years 11 months M3 M3 46, XX, t(15;17) 46, XX, t(15;17)Medeiros et al. [5] M/63 5 years 7 months M4 NA NA NormalMedeiros et al. [5] M/77 5 years 5 months M4 NA Normal NormalMedeiros et al. [5] M/22 5 years 4 months M1 NA NA 46, XY, t(15;17)Medeiros et al. [5] M/53 5 years 4 months M4 NA Normal NormalLatagliata et al. [8] M/16 5 year 1 month M3 M3 46, XY, t(15;17) 46, XY, t(15;17)Latagliata et al. [8] M/22 5 year M3 M3 46, XY, t(15;17) 46, XY, t(15;17)Present case M/19 22 years 8 months M4 M4 45, XY, 221 [20] 47, XY, 14, 27, 111 [9]/46, XY [11]
letter
456 American Journal of Hematology

de novo as well as secondary AML [17]. In summary, contrary to most
instances of very late relapses in AML (Table II), our patient’s cytogenetic
anomalies at relapse point to a secondary AML, although the chemotherapy
to which he was initially exposed, and the 19-year interval between his most
recent presentation with AML and his previous exposure to chemotherapy
suggest yet another mechanism for his second presentation with leukemia.
1Cancer Center, Montefiore Medical Center North Division2Department of Pathology, Montefiore Medical
Center North Division, Bronx, New York3Department of Medicine, Albert Einstein College of Medicine
Bronx, New York*Correspondence to: Peter H. Wiernik, Cancer Center
Montefiore Medical Center North Division, 600 East 233rd StreetBronx, New York. E-mail: [email protected]
Published online 9 April 2009 in Wiley InterScience(www.interscience.wiley.com).
DOI: 10.1002/ajh.21431Conflict of interest: Nothing to report.
References1. Konrad M, Metzler M, Panzer S, et al. Late relapses evolve from slow-
responding subclones in t(12;21)-positive acute lymphoblastic leukemia:Evidence for the persistence of a preleukemic clone. Blood 2003;101:3635–3640.
2. Ferraro D, Gabbianelli M, Peschel C, et al. Surface phenotypes of humanprogenitor cells defined by monoclonal antibodies. Blood 1985;66:496–501.
3. Dutcher JP, Wiernik PH, Markus S, et al. Intensive maintenance therapyimproves survival in adult acute nonlymphocytic leukemia: An eight-year fol-low-up. Leukemia 1988;2:413–419.
4. Schiffer CA, Dodge R, Larson RA. Long-term follow-up of cancer and leukemiaGroup B studies in acute myeloid leukemia. Cancer 1997;80:2210–2214.
5. Medeiros BC, Minden MD, Schuh AC, et al. Characteristics and outcomes of
acute myelogenous leukemia patients with very late relapse (> 5 years).Leuk Lymphoma 2007;48:65–71.
6. Mulronney DA, Dover DC, Li S, et al. Twenty years of follow-up among survi-vors of childhood and adult acute myeloid leukemia. Cancer 2008;112:2071–2079.
7. Ustun C, Kalla A, Bollag RJ, et al. Relapsed acute myelogenous leukemiaoccurring after 18 years with recurrent novel chromosomal abnormalityt(12;22)(q23;q11.2). Cancer Genet Cytogenet 2007;117:135–138.
8. Latagliata R, Carmosino I, Breccia M, et al. Late relapses in acute promyelo-cytic leukemia. Acta Haematol 2007;117:106–108.
9. Meloni G, Mancini M, Gianfelici V, et al. Late relapse of acute myeloid leuke-mia with mutated NPM1 after eight years: Evidence of NPM1 mutationstability. Haematologica 2009;94:298–300.
10. Pedersen-Bjergaard J, Pedersen M, Roulston D, et al. Different genetic path-ways in leukemogenesis for patients presenting with therapy-related myelodys-plasia and therapy-related acute myeloid leukemia. Blood 1995;86:3542–3552.
11. Zompi S, Legrand O, Bouscary D, et al. Therapy-related acute myeloid leu-kemia after successful therapy for acute promyelocytic leukemia witht(15;17): A report of two cases and a review of the literature. Br J Haematol2000;110:610–613.
12. Kantarjian HM, Keating MJ, Walters RS, et al. The characteristics and out-come of patients with late relapse acute myelogenous leukemia. J ClinOncol 1998;6:232–238.
13. Indraccolo S, Stievano L, Minusso S, et al. Interruption of tumor dormancyby transient angiogenic burst within tumor microenvironment. Proc NatlAcad Sci USA 2006;103:4216–4221.
14. Witherspoon RP, Schubach W, Neiman P, et al. Donor cell leukemia devel-oping six years after marrow grafting for acute leukemia. Blood 1985;65:1172–1174.
15. Weber E, Nowotny H, Haas OA, et al. Trisomy 4: A specific karyotype anomalyin primary and secondary acute myeloid leukemia. Leukemia 1990;4:219–221.
16. Kwong YL, Liang R, Chan LC. Trisomy 4 in acute myeloid leukemia. Leuke-mia 1991;5:354–355.
17. Heinonen K, Mrozek K, Lawrence D, et al. Clinical characteristics of patientswith de novo acute myeloid leukemia and isolated trisomy 11: A Cancer andLeukemia Group B study. 1998;101:513–520.
Gemcitabine-based combination chemotherapyas salvage treatment for refractory or relapsingaggressive non-Hodgkin’s lymphoma
Shih-Hung Yang,1,2 Zhong-Zhe Lin,2,3 Sung-Hsin Kuo,1,2,3,4* and Ann-Lii Cheng2,3,4
Although CHOP (cyclophosphamide, adriamycin, vincristine, and pre-
dnisolone) or more intensive chemotherapy regimens with or without
rituximab can cure around 50% of advanced-stage aggressive non-
Hodgkin’s lymphoma (NHL), a substantial proportion of the patients
develop refractory or relapsing diseases. However, high-dose chemo-
therapy followed by autologous stem cell transplantation usually res-
cues a limited number of patients. Historically, traditional chemother-
apy regimens, including ESHAP (etoposide, methylprednisolone, high-
dose Ara-C, and cisplatin), ICE (ifosfamide, carboplatin, and etopo-
side), DHAP (high-dose Ara-C, cisplatin, and dexamethasone), and
EPOCH (etoposide, doxorubicin, vincristine, cyclophosphamide, and
prednisone) are used for salvage treatment in refractory or relapsing
NHL [1]; however, the use of these regimens is often limited by the rel-
atively severe toxicities. For example, high-dose Ara-C-containing regi-
mens (ESHAP and DHAP) have significant hematological, skin, con-
junctival, and mucosal toxicities [2,3]. Accumulated cardiac toxicity
would be a major problem of anthracycline-containing regimens
(EPOCH) for patients after standard first-line CHOP-based regimens
[4]. Previous studies using ICE for a salvage chemotherapy regimen
usually enrolled transplant-eligible patients, and the hematological
toxicity remained significant, although the aggressive prophylactic
granulocyte colony stimulating factor (G-CSF) was used [5]. Therefore,
a safe and effective salvage chemotherapy regimen is needed for
the treatment of relapsing or refractory aggressive NHL, irrespective
of the initial response to chemotherapy, patients’ age, or patients’
comorbidities.
Gemcitabine is another nucleoside analog with easily manageable toxic-
ities. A phase II trial of gemcitabine for refractory or relapsing aggressive
NHL has demonstrated an overall response rate (RR) of 19%, with accept-
able hematological toxicities [6]. The median response duration was 6
months [6]. The result indicated that gemcitabine is safe and has good activ-
ity for refractory or relapsing NHL. To increase the RR and survival in refrac-
tory or relapsing NHL, it is reasonable to combine gemcitabine with other
effective salvage chemotherapy drugs. In fact, cisplatin (an active agent
used in heavily pretreated patients with NHL) has shown in vitro synergy
with gemcitabine in some cancer cell lines [7], and etoposide (a topoisomer-
ase inhibitor with single-agent activity) has shown synergy with cisplatin in
heavily pretreated patients with aggressive NHL [8]. The combination of eto-
poside, cisplatin, and Ara-C in patients with refractory NHL had an RR of
32% and a high incidence (66%) of myelosuppression [9]. Based on these
findings, we investigated the efficacy and safety of a gemcitabine-based sal-
vage regimen (gemcitabine in combination with etoposide, cisplatin, and ste-
roid, GEPS) by retrospectively evaluating the clinical outcome of 15 patients
with refractory or relapsing aggressive NHL.
letter
American Journal of Hematology 457

Between January 2001 and January 2008, our study enrolled 15 patients
with relapsing or refractory aggressive NHL treated at the National Taiwan
University Hospital with GEPS, mainly a 4-week cycle of gemcitabine 800
mg/m2 intravenously for 30 min on days 1 and 8, etoposide 40 mg/m2/day
intravenously for 1 hr on days 1–3, cisplatin 40 mg/m2 intravenously for 24
hr on day 1, and methylprednisolone 135 mg/m2/day on days 1–4. Three
patients were treated with modified regimens (without steroid in two patients
and without etoposide in one patient). Seven patients were not given the
day 8 dose of gemcitabine to avoid potentially severe neutropenia. Rituxi-
mab (R) was used with GEPS in nine patients, but seven of them had been
given prior rituximab-containing chemotherapy. No patients had prior stem
cell transplants before the gemcitabine-based salvage regimen.
The clinicopathologic features, chemotherapy responses, and clinical out-
comes are summarized in Table I. The median age was 63 years (44–89
years). About two-thirds (n 5 11) of patients at diagnosis or before GEPS had
stage III or IV. The median international prognostic index (IPI) score at diagno-
sis was 3. The median ECOG performance statuses at diagnosis and before
GEPS were both 1. Nearly all patients (n 5 14, 93.3%) had initial extranodal
involvement, and three had bone marrow involvement. Diffuse large B cell lym-
phoma (DLBCL) was present in 11 patients (73%), anaplastic lymphoma kin-
ase-negative anaplastic large cell lymphoma in two, mantle cell lymphoma in
one, and peripheral T-cell lymphoma in one. The median time from diagnosis
of NHL to salvage with GEPS-based regimens was 12 months (1.9–39.1
months). Two-thirds had failed prior anthracycline-based chemotherapy.
The objective RR was 60% (six complete responses [CR] and three partial
responses [PR]). For patients in first relapse, the RR was 50%. However,
four of five patients in subsequent relapses had CR. In patients receiving
concomitant rituximab, CR occurred in four, PR occurred in two (RR 66%),
and progressive disease (PD) occurred in three. Of the three patients with
DLBCL treated without concurrent rituximab, the same RR (one CR, one PR
and one PD) was observed. In the present study, nine patients received at
least one salvage chemotherapy regimen (one regimen in three patients,
two regimens in four patients, and more than two regimens in two patients,
respectively) after the failure of the GEPS-based regimen. None of the
patients were treated with autologous stem cell transplantation after the
GEPS-based regimen because of older age (median age: 63 years for the
group, and 58 years for the responders). The exception, the youngest case
(Case# 12, 44 years), achieved CR after R-GEPS, however, she experi-
enced early CNS relapse and had rapid deterioration of conditions 2 months
later. At the median follow-up of 7.5 months (1–60 months) after initiation of
the GEPS-based regimen, 10 patients were dead, and all deaths were attrib-
uted to progressive NHL. The median progression-free survival (PFS) and
overall survival (OS) after initiation of GEPS were 3.6 months (95% confi-
dence interval [CI], 1.3–5.9 months) and 10.2 months (95% CI, 0–29.5
months), respectively.
Table II lists the incidence of the main toxicities graded according to the
Common Terminology Criteria for Adverse Events v3.0. In total, 52 cycles of
chemotherapy were administered, and the median number of cycles given
per patient was three (1–8 cycles). One treatment-related death because of
Candida tropicalis fungemia was observed. Grade 3–4 anemia, neutropenia,TABLEI.
PatientCharacteristicsandOutcomes
No.
Age
Sex
Histology
ECOG
IPI
Stagea
Initialextranodal
invo
lvement
Prior
regim
ens
GEPS
cycle
Response
Regim
en
PFS(m
onths)
Regim
ens
afterGEPS
orR-G
EPSb
#1
56
FPTCL
21
II/IV
–1
1PD
GEPS
0.7,death
byPD
0#2
53
MDLBCL
13
IV/II
parotidgland,intestines,
stomach
24
CR
R-G
EPS
34.7,alivewithoutrecurrence
0#3
89
MDLBCL
11
II/III
cecum
11
PD
R-G
EPS
0.7,death
byPD
1#4
73
MDLBCL
14
IV/IV
stomach,intestines,
bonemarrow
15
PR
R-G
EPS
3.8,death
byPD
1#5
64
MDLBCL
11
II/II
testis
17
CR
R-G
EPS
31.4,alivewithoutrecurrence
0#6
84
FDLBCL
14
IV/I
lung,intestines
12
PR
GEPS
2.2,death
byPD
0#7
72
FDLBCL
25
IV/IV
bonemarrow
11
PD
R-G
EPS
1.2,lossoffollow
up
2#8
58
MALCL-
01
III/III
skin
44
CR
GEPS
4.1,death
byPD
>2
ALK(-)
#9
57
FALCL-
00
III/III
skin
52
PD
GEPS
2.4,death
byPD
>2
ALK(-)
#10
66
FDLBCL
04
IV/IV
stomach,pancreas,
adrenalgland
18
CR
GEP
59.7,alivewithoutrecurrence
0#11
67
FDLBCL
24
IV/IV
stomach,ascites
13
PD
GEP
2.3,death
byPD
1#12
44
FDLBCL
10
I/IV
breast
24
CR
R-G
PS
3.6,death
byPD
2#13
63
MDLBCL
04
IV/IV
stomach,liver,pancreas,
spleen
12
PD
R-G
EPS
1.5,death
byPD
2#14
57
MMCL
01
IV/I
stomach,bonemarrow
26
CR
R-G
EPS
16.4,alivewithoutrecurrence
0#15
55
FDLBCL
03
III/III
stomach
12
PR
R-G
EPS
1.6,death
byPD
2
Abbreviations:M,male;F,
female;DLBCL,diffuselargeB-celllymphoma;ALCL,anaplasticlargecelllymphoma;ALK,anaplasticlymphomakinase;MCL,mantlecelllymphoma;IPI,internationalprognosticindex;GEPS,gemcitabine,eto-
poside,cisplatin,andmethylprednisolone;CR,complete
response;PR,partialresponse;PD,progressivedisease;R,rituxim
ab;PFS,progression-freesurvival.
aStage:atdiagnosis/before
GEPS.
bRegim
ensafterGEPSorR-G
EPS:Noneofpatients
hadbeentreatedwithautologousstem
celltransplantation;0,nosalvageregim
en;1,onesalvageregim
en;2,twosalvageregim
ens;>2,more
thantwosalvageregim
ens.
TABLE II. Toxicity Profiles
Maximum toxicity grade (n)
0 1–2 3 4 5
Hematologic 1 5 3 6 0
Lung 11 2 2 0 0Liver 15 0 0 0 0Renal 15 0 0 0 0Cardiac 13 2 0 0 0Neurological 12 1 2 0 0Dermatological 12 2 1 0 0Metabolic 2 9 3 1 0Constitution 9 6 0 0 0Infection 9 2 2 1 1Hemorrhage 13 1 1 0 0GI 5 8 2 0 0Coagulation 14 0 1 0 0
letter
458 American Journal of Hematology

and thrombocytopenia were noted in four (26.7%), 7 (46.7%), and 4 (26.7%)
patients, respectively. However, these hematological toxicities were mostly of
short duration and easily manageable. G-CSF (5 mg/kg) had been used in
12 patients (80%), but only on an average for 2.3 days (0–9 days) per cycle
of chemotherapy. Grade 3–4 nonhematological toxicities (mostly metabolic
effects [26.7%] and infections [20%]) were infrequent, including abnormal
liver aminotransferases, hyponatremia, hypokalemia, hyperglycemia, urinary
tract infection, and pneumonia. Only one patient had more than Grade 2
vomiting. Peripheral neurotoxicity (Grade I numbness) occurred in only one
patient after two cycles of chemotherapy.
This retrospective analysis showed the first attempt to assess the combi-
nation of not only gemcitabine, cisplatin, and steroid, but also of etoposide
to treat relapsing or refractory aggressive NHL. Synergism between the
gemcitabine-cisplatin and cisplatin-etoposide has been demonstrated in vitro
or in vivo [7,8]. Although the results of testing gemcitabine plus cisplatin and
steroid therapy in patients with relapsed or refractory NHL were promising
[10–12], relatively high doses of gemcitabine plus cisplatin were significantly
more myelosuppressive. Compared to the previous studies, our study used
much lower gemcitabine and cisplatin doses. Incorporating a relatively low
dose of etoposide did not significantly increase the incidence of grade 3–4
hematological toxicities. Additionally, cisplatin, which was given intravenously
for 24 hr, caused only minimal nausea and vomiting. Indeed, reduced cispla-
tin-related gastrointestinal toxicity when cisplatin is administered for 24 hr
has been reported previously [13].
In this retrospective study, the efficacy of the GEPS-based regimen was com-
parable with that of the previously published platinum and, or etoposide-contain-
ing regimens with or without gemcitabine [2,3,5,10–12]. The high percentage of
patients in our cohort with poor prognostic factors, such as older age, advanced
stage, high LDH, more extranodal involvement, and high IPI score may account
for their worse PFS and OS. Notably, four of our patients (26.7%) who received
GEPS or R-GEPS lived and remained relapse-free for more than 1 year. These
findings suggest that GEPS with or without rituximab is a novel and feasible
combination salvage therapy for patients with advanced refractory or relapsing
aggressive NHL, and may be an adjuvant treatment for older patients and
patients who are not candidates for high-dose chemotherapy. Additional pro-
spective studies examining the efficacy of GEPS-based salvage regimens for
refractory or relapsing aggressive NHL are warranted.
1Department of Oncology, National Taiwan University HospitalYun-Lin Branch, Yunlin, Taiwan
2Department of Oncology, National Taiwan, University Hospital and NationalTaiwan University, College of Medicine Taipei, Taiwan3Cancer Research Center, National Taiwan University
College of Medicine, Taipei, Taiwan4Department of Internal Medicine, National Taiwan University
Hospital, Taipei, Taiwan*Correspondence to: Sung-Hsin Kuo, Department of Oncology
National Taiwan University Hospital, Yun-Lin Branch, Yunlin, TaiwanE-mail: [email protected]
or Ann-Lii Cheng, Department of OncologyNational Taiwan University Hospital, Taipei, Taiwan
E-mail: [email protected]
Published online 16 April 2009 in Wiley InterScience(www.interscience.wiley.com).
DOI: 10.1002/ajh.21436Conflict of interest: Nothing to report.
Contract grant sponsor: National Science Council, Taiwan;Contract grant numbers: NSC96-2321-B-002-013, NSC96-2321-B-002-014,
NSC96-2314-B-002-164MY3;Contract grant sponsor: Department of Health, Taiwan
Contract grant number: DOH96-DT-B-111-001;Contract grant sponsor: National Taiwan University Hospital, Taiwan;
Contract grant number: NTUH 96-S626.
References1. Gisselbrecht C, Mounier N. Improving second-line therapy in aggressive
non-Hodgkin’s lymphoma. Semin Oncol 2004;31:12–16.2. Velasquez WS, Cabanillas F, Salvador P, et al. Effective salvage therapy for
lymphoma with cisplatin in combination with high-dose Ara-C and dexame-thasone (DHAP). Blood 1988;71:117–122.
3. Velasquez WS, McLaughlin P, Tucker S, et al. ESHAP—An effective chemo-therapy regimen in refractory and relapsing lymphoma: A 4-year follow-upstudy. J Clin Oncol 1994;12:1169–1176.
4. Gutierrez M, Chabner BA, Pearson D, et al. Role of a doxorubicin-containingregimen in relapsed and resistant lymphomas: An 8-year follow-up study ofEPOCH. J Clin Oncol 2000;18:3633–3642.
5. Moskowitz CH, Bertino JR, Glassman JR, et al. Ifosfamide, carboplatin, andetoposide: A highly effective cytoreduction and peripheral-blood progenitor-cell mobilization regimen for transplant-eligible patients with non-Hodgkin’slymphoma. J Clin Oncol 1999;17:3776–3785.
6. Fossa A, Santoro A, Hiddemann W, et al. Gemcitabine as a single agent inthe treatment of relapsed or refractory aggressive non-Hodgkin’s lymphoma.J Clin Oncol 1999;17:3786–3792.
7. Bergman AM, Ruiz van Haperen VW, Veerman G, et al. Synergistic interac-tion between cisplatin and gemcitabine in vitro. Clin Cancer Res 1996;2:521–530.
8. Judson IR, Wiltshaw E. Cis-dichlorodiammineplatinum (cis-platinum) andetoposide (VP-16) in malignant lymphoma—An effective salvage regimen.Cancer Chemother Pharmacol 1985;14:258–261.
9. O’Donnell MR, Forman SJ, Levine AM, et al. Cytarabine, cisplatin, and eto-poside chemotherapy for refractory non-Hodgkin’s lymphoma. Cancer TreatRep 1987;71:187–189.
10. Chau I, Harries M, Cunningham D, et al. Gemcitabine, cisplatin andmethylprednisolone chemotherapy (GEM-P) is an effective regimen inpatients with poor prognostic primary progressive or multiply relapsedHodgkin’s and non-Hodgkin’s lymphoma. Br J Haematol 2003;120:970–977.
11. Crump M, Baetz T, Couban S, et al. Gemcitabine, dexamethasone, andcisplatin in patients with recurrent or refractory aggressive histology B-cell non-Hodgkin lymphoma: A Phase II study by the National CancerInstitute of Canada Clinical Trials Group (NCIC-CTG). Cancer 2004;101:1835–1842.
12. Ng M, Waters J, Cunningham D, et al. Gemcitabine, cisplatin and methyl-prednisolone (GEM-P) is an effective salvage regimen in patients withrelapsed and refractory lymphoma. Br J Cancer 2005;92:1352–1357.
13. Salem P, Hall SW, Benjamin RS, et al. Clinical phase I-II study of cis-dichloro-diammineplatinum(II) given by continuous iv infusion. Cancer TreatRep 1978;62:1553–1555.
Successful treatment of cyclic thrombocytopenia withthrombopoietin-mimetic agents: A report of two patients
Prithviraj Bose,1 Khader K. Hussein,2 Deirdra R. Terrell,1 Dietmar Berger,3
Lawrence Rice,4 and James N. George1*
Cyclic thrombocytopenia (CTP), characterized by periodic oscillations
of the platelet count with cycles of 20–40 days, is a rare disorder that
has demographic and clinical features similar to immune thrombocyto-
penic purpura (ITP) [1]. Like ITP, the pathogenesis can involve both
accelerated platelet destruction and insufficient platelet production [1].
However, CTP appears to be less responsive to conventional therapies
used for ITP [1]. Thrombopoietin (TPO)-mimetic agents have recently
been documented to be effective for most patients with ITP [2–5]. We
letter
American Journal of Hematology 459

present two women with CTP who have been effectively treated with
TPO-mimetic agents following failure of multiple treatments for ITP.
The first report is an 11 year follow-up of a patient whose initial 1 year
of treatment with recombinant pegylated human megakaryocyte
growth and development factor (PEG-rHuMGDF) has been previously
reported [6]. She was successfully treated with PEG-rHuMGDF for 12
years, and is currently treated with romiplostim. The second patient
has been successfully treated with romiplostim for 3 years. TPO-mim-
etic agents can provide effective long-term treatment for patients with
severe and symptomatic CTP.
CTP is a rare disorder, which is usually first diagnosed and treated as ITP.
There is no consensus definition of CTP that establishes a clear distinction
from ITP. A systematic review in 2004 documented only 51 reported patients
who were described as having CTP [1]. The cyclic nature of these patients’
thrombocytopenia was only recognized after a median of 2 years (range, 1–
420 months), following failure of multiple treatments for ITP [1]. The patho-
genesis of CTP is unclear and may involve both cyclic accelerated platelet
destruction, as well as cyclic failure of platelet production [1]. Multiple mech-
anisms of thrombocytopenia have been documented in patients with CTP,
including autoimmune platelet destruction, megakaryocytic hypoplasia that
may also be immune-mediated, and hormonal factors, since platelet count
cycles in pre-menopausal women may be linked to the menstrual cycle [1].
CTP is a chronic, persistent disorder. Follow-up of the 51 reported cases for
a median of 31 months demonstrated that two (4%) patients had died from
hemorrhage, one (2%) died of another cause with continuing CTP, 14 (27%)
were in remission, and 34 (67%) continued to have CTP [1]. The remarkable
failure rate of CTP to treatment with corticosteroids, intravenous immunoglo-
bulin (IVIg), and splenectomy [1] may be inherent to CTP, or may represent
selection of reported patients who were recognized to have CTP only after
failure of the treatment for ITP.
There are multiple reasons why treatment with TPO-mimetic agents, such
as romiplostim [3,7] and eltrombopag [2] that were approved in 2008 for
treatment of chronic ITP, may be more effective for patients with CTP than
standard therapies used for ITP [1]. Conventional treatments for ITP focus
on control of the increased platelet destruction. Ineffective platelet produc-
tion may be a more prominent cause of thrombocytopenia in CTP than in
ITP, and TPO-mimetic agents may provide more effective correction of this
abnormality [2–5]. Since CTP is a chronic disorder and complete remissions
appear to be uncommon, TPO-mimetic agents can provide continuous sup-
port over a long duration [7], without the danger of corticosteroid side effects
and risk of immunosuppression associated with standard treatments of ITP
[3,8]. The role of granulocyte-colony stimulating factor (G-CSF) as the stand-
ard treatment for cyclic neutropenia for over 20 years [9–11] provides a
precedent for the hematopoietic growth factor treatment of CTP. To support
the principle that TPO-mimetic agents are effective long-term treatment for
patients CTP, we report two patients who have been successfully treated for
12 and 3 years.
Patient 1 is a 78-year-old white woman. Her initial diagnosis with severe
and symptomatic ITP in 1951, recognition of CTP in 1986, her failure to
respond to many treatments (corticosteroids, splenectomy, IVIg, vincristine,
azathioprine, alpha interferon, colchicine, cyclosporine, plasma exchange,
protein A column immunoadsorption, and estrogens), and her successful
treatment with PEG-rHuMGDF for 1 year through 1998 have been previ-
ously reported [6]. Prior to the initiation of PEG-rHuMGDF treatment, 13
measurements of endogenous TPO during one 28-day cycle documented
an inverse relationship between the platelet counts and TPO levels [6].
Although the development of PEG-rHuMGDF by Amgen was stopped in
1998 because of the development of anti-PEG-rHuMGDF antibodies that
cross-reacted with native TPO in 13 of 535 subjects, [5,12] it was continued
to be provided for this patient, because it was her only effective treatment.
She was maintained on self-subcutaneous injections of PEG-rHuMGDF
(1.5–2.3 mg/kg), two to three times per week through October 2008.
Although her platelet counts continued to cycle, treatment with PEG-
rHuMGDF increased her nadir platelet counts from less than 10 3 109/L,
which was accompanied by purpura and mucosal bleeding, to about 60 3
109/L; peak platelet counts were increased from 30 to 300 3 109/L. For the
past 7 years, platelet counts less than 10 3 109/L, associated with minimal
purpura, have been recorded only four times. She never had severe bleed-
ing that required hospitalization or red cell transfusions since beginning on
PEG-rHuMGDF. No adverse effects related to PEG-rHuMGDF have
occurred. Throughout her course, peripheral blood smears have been exam-
ined by her primary hematologist (L.R.) at 1–3 month intervals. Her hemo-
globin and white blood cell counts have remained normal; she has not devel-
oped leukoerythroblastic changes suggestive of marrow fibrosis; she never
developed antibodies to PEG-rHuMGDF. On November 3, 2008, her treat-
ment was changed to romiplostim following its FDA approval. She began
with a weekly subcutaneous dose of 1 mg/kg that was gradually increased to
4 mg/kg after 8 weeks. During her 22 weeks of treatment with romiplostim,
weekly platelet counts have demonstrated continued cycling, with nadirs
occurring at 3–4 week intervals (platelet counts at nadir, 4–95 3 109/L
[median 42 3 109/L] and at their peak, 196–706 3 109/L [median 289 3
109/L]).
Patient 2 is an 84-year-old white woman who was initially diagnosed with
ITP in December 2003, when she presented with purpura, lower gastrointes-
tinal bleeding, and a platelet count of 16 3 109/L. She was healthy, except
for hypertension requiring atenolol for 2 years and hypothyroidism treated
with synthroid for 8 years. Bone marrow biopsy was normal. Her course
over the next 2 years is illustrated in Fig. 1A. She responded rapidly to pre-
dnisone, but because of intermittent severe and symptomatic thrombocyto-
penia, apparently coinciding with attempts to taper the prednisone dose, she
had a splenectomy in April 2004. For the next 15 months, she had fluctua-
tions of her platelet count in spite of treatment with azathioprine, rituximab,
and danazol. Throughout this time she continued to receive prednisone and
also was given IVIg and intravenous methylprednisolone intermittently for
severe thrombocytopenia accompanied by extensive purpura, oral blood blis-
ters, and occasionally also overt rectal bleeding. Colonoscopy was normal.
Figure 1. Data are presented for the course of Patient 2. A documents hercourse from diagnosis in December 2003 till the initiation of romiplostim treatmentin February 2006. Treatments in addition to prednisone indicated at the top were:1, splenectomy; 2, azathioprine; 3, rituximab; and 4, danazol. B documents hercourse since initiation of romiplostim treatment in February 2006. [Color figure canbe viewed in the online issue, which is available at www.interscience.wiley.com.]
letter
460 American Journal of Hematology

In July 2005 she was referred for a clinical trial of romiplostim, however she
failed to qualify multiple times because she never maintained a platelet
count less than 30 3 109/L throughout the screening period. A drug-induced
etiology, related to the intermittent use of acetaminophen or diazepam was
considered, but was excluded when all medications except atenolol and
synthroid were stopped for 4 weeks. Finally, in January 2006, CTP was
recognized when it was noted that her episodes of severe thrombocytopenia
occurred in cycles of approximately 3 weeks.
In February 2006, Patient 2 was enrolled in a single-patient romiplostim
protocol that did not require consistent thrombocytopenia as an inclusion cri-
terion. Her course during romiplostim treatment over the next 3 years is
illustrated in Fig. 1B. Initially she was treated weekly, according to the stand-
ard protocol for romiplostim [3]. Doses were escalated from 2 to 13 mg/kg
over a period of 4 months with no apparent platelet count response and only
slight platelet count cycles. In July 2006, 5 months after beginning romiplos-
tim, prednisone was re-initiated, and the dose of romiplostim was gradually
decreased as the amplitude of platelet count cycles increased. Then, the
interval between romiplostim doses was increased to avoid extreme throm-
bocytosis. Prednisone was stopped in March 2008. She is currently main-
tained on 1.5 mg/kg of romiplostim every 3–4 weeks. Throughout her course,
peripheral blood smears have been examined by her primary hematologist
(K.K.H.) or his technologist at weekly intervals. For the past year, she has
felt well, symptoms related to corticosteroid side effects have resolved, and
her weekly platelet counts have not been less than 30 3 109/L for 2 years.
Platelet count cycling has appeared to persist, although the amplitude of the
cycles has appeared to diminish. The presence of CTP, rather than truly
chronic refractory ITP, is supported by the extreme amplitude of the platelet
count cycles when response to romiplostim began.
We have no explanation for the initial lack of response to very high doses
of romiplostim followed by increasing sensitivity to low doses of romiplostim,
administered at 3–4 week intervals. It is possible that the addition of a low
dose of prednisone augmented the effect of romiplostim. Although her
response may have represented a spontaneous modification of disease
severity, the role of romiplostim in the response is supported by the extreme
thrombocytosis when a response occurred, and the ability of romiplostim to
maintain her platelet count in an asymptomatic range for the past year with-
out additional treatment.
These two patients demonstrate the value of two different TPO-mimetic
agents for CTP, a chronic and refractory disorder. These patients also dem-
onstrate the long-term value of TPO-mimetic agents, as they have been
treated for 3 and 12 years, and they continue to be treated. Patient 2 illus-
trates the need to adjust the romiplostim regimen to achieve maximal bene-
fit, similar to the experience with romiplostim treatment of patients with ITP,
in whom platelet count fluctuations on a stable dose of romiplostim are com-
monly observed [4]. Although patients with ITP are treated weekly, as is
Patient 1, Patient 2 has required romiplostim only once every 2–5 weeks
during the past year. The basis for her changing responsiveness to romi-
plostim is not known.
The response of CTP to TPO-mimetic agents is analogous to the
response of cyclic neutropenia to G-CSF [9–11]. Although cyclic neutropenia
is a genetic disorder and CTP often appears to be an acquired autoimmune
disorder, both respond to growth factors by increasing the amplitude of the
oscillations with an increased cell count at the nadir of the cycle [9–11].
The long-term safety of TPO-mimetic agents remains uncertain as data
from clinical trials is currently limited to 3 years [7], and widespread clinical
use is only now beginning. Patient 1 in this report represents the longest
experience with any TPO-mimetic agent and Patient 2 is among the patients
with the longest duration of treatment with romiplostim. Neither has devel-
oped any peripheral blood abnormalities, suggesting increased marrow retic-
ulin, a dose-dependent and reversible effect of TPO [5]. An increased occur-
rence of thromboembolic events is a potential risk of TPO-mimetic agents
[3,5,7]. Patient 1 had one episode of exertional chest pain during her second
week of PEG-rHuMGDF treatment in 1997, when her platelet count was
1,400 3 109/L [6]. Thrombotic complications remain a concern for Patient 2
because of her age, hypertension, and intermittently high platelet counts.
The experience of these two patients illustrates the ineffectiveness of con-
ventional ITP treatments for patients with CTP, and emphasizes the need to
consider the diagnosis of CTP. Their experience also supports the effective-
ness of long-term treatment with TPO-mimetic agents for CTP. Although
cycling of the platelet count appears to persist, the nadir platelet count is
increased and bleeding symptoms are prevented.
Acknowledgments
The investigational agents were provided and the patient studies were
supported by Amgen Inc., Thousand Oaks, CA.
1Department of Medicine, University of Oklahoma Health Sciences CenterOklahoma City, Oklahoma
2Central Oklahoma Cancer Center, Oklahoma City, Oklahoma3Amgen Inc., Thousand Oaks, California
4Department of Medicine, The Methodist HospitalWeill Cornell Medical College, Houston, Texas
Correspondence to: James N. George, The University ofOklahoma Health Sciences Center, Room CHB 358
801 NE 13th Street, Oklahoma City, OK 73104E-mail: [email protected]
Published online 16 April 2009 in Wiley InterScience(www.interscience.wiley.com).
DOI: 10.1002/ajh.21435Conflict of interest: Authors K.K.H., D.R.T., L.R., and J.N.G. have received
research support from Amgen Inc.; J.N.G. and D.R.T. have served as consultantsfor Amgen Inc.; L.R. is a member of the Speakers’ Bureau for Amgen and
GlaxoSmithKline; andD.B. is an employee of Amgen.
References1. Go RS. Idiopathic cyclic thrombocytopenia. Blood Rev 2004;19:53–59.2. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of
chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–2347.
3. Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patientswith chronic immune thrombocytopenic purpura: A double-blind randomizedcontrolled trial. Lancet 2008;371:395–403.
4. George JN, Terrell DR. Novel thrombopoietic agents: A new era for patientswith thrombocytopenia. Haematologica 2008;93:1445–1449.
5. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment ofthrombocytopenia. Annu Rev Med 2009;60:193–206.
6. Rice L, Nichol JL, McMillan R, Roskos LK, Bacile M. Cyclic immune throm-bocytopenia responsive to thrombopoietic growth factor therapy. Amer JHematol 2001;68:210–214.
7. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-termtreatment with romiplostim in thrombocytopenic patients with chronic ITP.Blood 2009;113:2161–2171.
8. Portielje JEA, Westendorp RGJ, Kluin-Nelemans HC, Brand A. Morbidityand mortality in adults with idiopathic thrombocytopenic purpura. Blood2001;97: 2549–2554.
9. Hammond WP, Price TH, Souza LM, Dale DC. Treatment of cyclic neutrope-nia with granulocyte colony-stimulating factor. New Eng J Med 1989;320:1306–1311.
10. Bonilla MA, Dale DC, Zeidler C, et al. Long-term safety of treatment withrecombinant human granulocyte colony-stimulating factor (r-metHuG-CSF) inpatients with severe congenital neutropenias. Br J Haematol 1994;88:723–730.
11. Haurie C, Dale DC, Mackey MC. Cyclic neutropenia and other periodic hem-atological disorders: A review of mechanisms and mathematical models.Blood Rev 1998;92:2629–2640.
12. Li JZ, Yang C, Xia YP, et al. Thrombocytopenia caused by the developmentof antibodies to thrombopoietin. Blood 2001;98:3241–3248.
letter
American Journal of Hematology 461

HLA type and risk of alloimmunization in sickle cell disease
Carolyn Hoppe,1* William Klitz,2,3 Elliott Vichinsky,1 and Lori Styles1
Red blood cell (RBC) transfusions are frequently required to treat
patients with sickle cell disease (SCD) [1]. One of the most serious
complications of repeat transfusion is alloimmunization to RBC anti-
gens [2]. Because human leukocyte antigen (HLA) genes mediate the
response to foreign antigens, particular HLA alleles may predispose to
the development of alloimmunization in patients with SCD who receive
multiple transfusions. We conducted a case-control study to determine
if particular HLA alleles are associated with alloimmunization and
whether HLA homozygosity influences the risk of developing RBC
alloantibodies. High-resolution HLA genotyping was performed on
DNA samples from 159 multiply transfused patients with SCD. HLA
allele frequencies were compared between alloantibody-positive and
alloantibody-negative groups. The HLA-DRB1*1503 allele was associ-
ated with an increased risk (P = 0.039), while HLA-DRB1*0901 con-
ferred protection from alloimmunization (P = 0.008). HLA Class II locus
homozygosity was more frequently observed in the alloantibody-nega-
tive group (P = 0.01). These preliminary findings suggest that particu-
lar HLA-DRB1 alleles and overall homozygosity at HLA class II loci are
associated with alloimmunization risk in SCD. If confirmed, HLA type
may serve as a useful genetic predictor of alloimmunization risk, and
permit a targeted approach to the use of phenotypically matched
blood.
The prevalence of alloimmunization in multiply transfused patients with
sickle cell disease (SCD) ranges between 30 and 47% when full or partial
red blood cell (RBC) antigen matching is not performed [1,3]. Although multi-
factorial in etiology, this high rate of alloimmunization is partly due to RBC
antigen disparity between racially mismatched blood donors and recipients
[3,4].
Once alloimmunized, patients with SCD are at risk for subsequent delayed
hemolytic transfusion reactions and development of additional RBC allo- and
autoantibodies [2,5]. Unfortunately, pretransfusion tests that can reliably pre-
dict which patients will develop RBC alloantibodies are lacking. Partial typing
of donor erythrocytes for the Rh C, E, and Kell antigens have been shown
to dramatically decrease alloimmunization and subsequent hemolytic trans-
fusion reactions [6,7]. However, difficulty in locating matched donors and the
considerable cost of providing phenotypically matched RBCs have precluded
widespread acceptance of this approach [8,9]. Early identification of patients
likely to develop alloantibodies could lead to focused, cost-effective transfu-
sion strategies by limiting the use of phenotypically matched RBCs to those
at greatest risk.
Because the ability to respond to foreign antigens is mediated by immuno-
logic factors, the human leukocyte antigen (HLA) genes undoubtedly play a
role in predisposition to alloimmunization. In multiply transfused patients with
SCD, limited studies based on serotypic data previously demonstrated HLA
associations with alloimmunization [10,11]. Recent studies in other trans-
fused populations have documented a relationship between the development
of specific RBC antibodies and the expression of HLA class II alleles [12–
14]. We carried out this study to determine whether similar HLA allelic asso-
ciations with alloimmunization exist in patients with SCD who underwent
transfusion.
A total of 159 patients were included in this study; all were homozygous
for the sickle cell mutation (HbSS) with a mean age of 14.8 years (range, 4
to 47 years). There were 59 alloantibody-positive and 100 alloantibody-nega-
tive patients, with no differences in age or sex between these two groups
(Table I). Of the alloantibody-positive patients, 34 (58%) had developed a
single RBC alloantibody, while 13 (22%) had multiple alloantibodies. Data on
specific alloantibodies were not available for 10 (17%) of the alloimmunized
patients. The most commonly reported alloantibodies were anti-E and anti-
K, in 15 (25%) and 14 (24%) of alloimmunized patients, respectively. Anti-
bodies to C (6%), M (7%), e (7%), D (5%), Lea (7%), Fya (5%), CW (5%),
Jka (5%), Fyb (2%), Lua (7%), S (5%), c (2%), V (2%), H (2%), and Cha
(2%) antigens were less frequently observed.
Although global tests comparing the alloimmunized and the nonalloim-
munized groups found no differences in HLA allele frequency distributions
between the two groups, particular HLA-DRB1 alleles were associated
with alloimmunization (Table II). HLA-DRB1*1503 was observed more fre-
quently in the alloantibody-positive group (34%) than in the alloantibody-
negative group (20%) (OR 5 2.02, P 5 0.039) while HLA-DRB1*0901 was
found exclusively in the alloantibody-negative group (11%) (OR 5 0.13,
P 5 0.008). We found an overrepresentation of HLA-DQB1*0502 and
HLA-A*3001 and an underrepresentation of HLA-DPB1*1701 in the alloan-
tibody-positive group, but these associations did not reach significance.
Neither locus-wide nor individual allelic effects were found at the HLA-C
and HLA-B loci. Homozygosity for HLA class I alleles was rare, but nomi-
nally more common in the alloantibody-negative group. For the HLA class
II loci, a greater degree of homozygosity (9%) was observed in the alloan-
tibody-negative group compared to the alloantibody-positive group (1%)
(Table III). The effect of homozygosity could not be localized to individual
HLA alleles.
Autoantibodies were more common in alloantibody-positive patients (29%)
compared to alloantibody-negative patients (9%) (Pexact 5 0.0007). No HLA
associations were found with the development of autoantibodies in the
alloimmunized group. However, HLA-DRB1*0301 was strongly associated
with the development of autoantibodies in alloantibody-negative individuals
(Pexact 5 0.0052).
Previous studies have indicated that patients with SCD represent a sub-
population of transfused patients who are at much higher risk of alloimmu-
nization than the general transfused population [1,3]. It is unclear why
some individuals mount strong alloantibody responses after initial transfu-
sions, while others are unresponsive despite repeated transfusions. In the
SCD population, racial differences between recipient and donor expressed
RBC antigens are known to contribute to the increased risk of alloimmuni-
zation in patients with SCD, but predictors of individual response have not
been identified. Patient age, sex, malignancy, diabetes, transplantation,
autoimmune disease, and cumulative RBC transfusion history have been
linked to alloimmunization in the general population [15]. Others have sug-
gested that the risk of alloimmunization is primarily determined by genetic
factors [16].
As antigen-specific recognition by the immune system is largely regulated
by HLA genes, variation in HLA alleles may contribute to the risk of alloim-
munization in multiply transfused individuals. Our results showing a risk-
conferring effect of HLA-DRB1*1503 and a protective effect of HLA-
DRB1*0901 with alloimmunization suggest that particular HLA alleles may
modulate the immunological response to alloantigens and are supported by
recent studies linking specific HLA-DRB1 alleles to RBC alloimmunization
risk in other transfused populations. In one of these studies, Reviron et al.
found an increased risk of alloimmunization to the Jka blood group antigens
in patients carrying any of the three HLA-DRB1*0101, DRB1*0102, and
DRB1*1001 alleles shared by a common HLA-DRB1 sequence [14]. Simi-
larly, Chiaroni et al. found a higher frequency of HLA-DRB1*11 and HLA-
DRB1*13 alleles in European patients with anti-K antibodies [12]. These
studies suggest that RBC alloimmunization may occur in response to pre-
sentation of RBC antigens by specific HLA-DR molecules. As our study
included patients with SCD with various RBC alloantibodies, examination of
HLA allelic associations with alloimmunization to individual RBC antigens
was limited by small patient numbers in each group. Nonetheless, among
the 13 patients in our study who had anti-K alloantibodies, we found a simi-
lar excess of HLA-DRB1*11 and HLA-DRB1*13 alleles (70%) as compared
letter
462 American Journal of Hematology
to nonalloimmunized controls (50%). As HLA-DRB1*11 and HLA-DRB1*13
share epitope sequences implicated in peptide binding, this selective
response to the K antigen could be due to HLA restriction in T cell
response. A larger study in patients with SCD alloimmunized to K antigen
is needed to confirm this potential HLA association.
Because of the very high allelic polymorphism and relatively even allele
frequencies in our study population, homozygotes at one or more HLA loci
were uncommon, limiting power to detect allele-specific homozygosity
effects. However, we found that patients homozygous for at least two alleles
at any of the three HLA class II loci were less likely to have alloantibodies
compared to patients who were homozygous for one or no alleles. In studies
investigating the association of HLA and HIV disease, HLA homozygosity
and reduced antigen disparity were shown to increase the rate of HIV pro-
gression [17,18]. As suggested by these studies, homozygosity for HLA
alleles may reduce the ability to mount an immunogenic response because
of ineffective recognition and presentation of a diverse array of foreign anti-
gens. Our preliminary findings require replication in a much larger study to
confirm this hypothesis.
The development of autoantibodies in association with RBC alloimmuniza-
tion has become increasingly recognized as a serious problem in patients
with hemoglobinopathies [5,19]. Our results showing an increased proportion
of patients with autoantibodies in the alloimmunized group (29%) as com-
pared to the nonalloimmunized group (9%) agree with previous studies [2,5]
and suggest that alloimmunized patients have heightened humoral sensitiv-
ity, with increased propensity to form autoantibodies. This is supported by
data showing that type 2 immunity is up-regulated by allogeneic transfusion
[20,21]. These humoral responses appear to be reduced when WBC reduc-
tion of RBC transfusions is employed [22]. Others have speculated that stim-
ulation of autoantibody production results from the conformational changes
in antigenic epitopes that occur after alloantibodies have bound to trans-
fused RBCs [5,23]. The development of autoantibodies in patients with SCD
may be regulated by yet other immunomodulatory genes, including HLA,
although no specific genetic associations have been reported. Notably, HLA-
DRB1*0301, an allele commonly associated with autoimmune disorders
such as type I diabetes, was overrepresented in the group of nonalloimmu-
nized patients who developed isolated autoantibodies (78%) compared to
alloimmunized patients who developed autoantibodies (11%).
The practical implications of these findings, if confirmed, could be signifi-
cant in terms of benefit to patients and cost reduction to the transfusion
service. Even if an alloimmunized patient does not develop clinically signifi-
cant hemolysis, blood banks often need to determine the specificities of anti-
bodies directed against the offending RBCs and locate RBC units lacking
the corresponding antigens for transfusion. The unit price of RBCs may be
doubled or even tripled if extensive antigen testing is required [2]. Thus,
transfusion services must balance the high cost of recruiting and collecting
RBCs from the <1% of random donors with the required phenotype with the
potential costs of assessing and managing a small fraction of patients who
develop delayed hemolytic transfusion reactions.
Taken together, our results suggest that patients with SCD bearing spe-
cific HLA alleles carry an increased risk of alloimmunization with repeated
RBC transfusions. In addition, overall homozygosity at the HLA locus may
offer protection from alloimmunization. If confirmed, these findings could
have a significant clinical impact on patients with SCD, as early identification
of those most likely to develop alloantibodies would allow for selective use of
extended matched RBC units in genetically susceptible patients.
Methods
One hundred fifty-nine patients with SCD who underwent transfusion, for
whom alloimmunization data were available, were included in this study.
Eighty-four patients received transfusions at our institution and 75 children
were transfused either prior to, or as part of, enrollment in the Stroke Pre-
vention Trial for Sickle Cell Disease (STOP) study. Age for local patients
was documented at the time of medical record review. Age for patients
enrolled in STOP was documented at the end of data collection for the trial.
Transfusion histories and alloimmunization data on local patients were
obtained from institutional blood bank and medical records. Informed con-
sent was obtained from local patients before the collection of samples for
HLA typing. The transfusion guidelines were previously reported: in brief,
transfusions were hemoglobin S negative and WBC reduced; red cell pheno-
type was determined before transfusion. With the onset of the STOP study,
patients received C-, E-, and Kell- matched units. Alloimmunization data and
genomic DNA were collected and stored on patients participating in the
STOP study and have been described previously [7,24]. This study was
reviewed and approved by the Institutional Review Board at Children’s Hos-
pital & Research Center Oakland.
Screening for allo- and autoantibodies was performed before each transfu-
sion using standard methods, including gel and antiglobulin techniques [25].
If the screening test was positive, the antibody was identified by means of
RBC antigen panels with gel and other antibody enhancement techniques
[26,27].
High-resolution HLA genotyping was performed for HLA class I (A, C, and
B) and class II (DRB1, DQB1, and DPB1) loci using established DNA-based
methods [28]. Briefly, DNA was amplified using PCR and analyzed using
sequence-specific oligonucleotide probes (SSOP). This method utilizes an
immobilized SSO probe format, whereby sequence-specific probes are
immobilized onto a nylon membrane support and the amplified PCR product
hybridized to the probe array. Class I typing methodology included amplifica-
tion of exons 2 and 3 separately in a multiplex reaction, followed by hybrid-
ization to probes for exons 2 and 3 on optimized immobilized probe strips.
For allelic resolution, some sample types required group-specific amplifica-
tions followed by immobilized probe analysis to distinguish them. Computer
software programs were used to identify genotypes from the probe hybrid-
ization patterns. HLA alleles were classified according to the nomenclature
defined by the World Health Organization [29].
HLA allele frequency distributions were compared between alloantibody-
positive and alloantibody-negative patient samples using tests of independ-
ence with the log-likelihood ratio or G-statistic for each of the six HLA loci
TABLE II. Differences in HLA Allele Frequency DistributionsBetween Alloimmunized and Nonalloimmunized Patients with SCD
HLA locus HLA allele Pexact ORa (95% CI)
Class IA A*3001 0.08C No difference NsB No difference Ns
Class IIDRB1 DRB1*0901 0.008 0.13 (0, 0.82)
DRB1*1503 0.039 2.02 (1.07, 3.91)DQB1 DQB1*0502 0.07DPB1 DPB1*1701 0.10
OR, odds ratio; 95% CI, 95% confidence interval.aOR and 95% CI presented for HLA alleles reaching significance (Pexact � 0.05).
TABLE I. Demographic and Transfusion Data in Alloimmunizedand Nonalloimmunized Patients with SCD
Alloantibody-positive
Alloantibody-negative All patients
Number of patients 59 100 159Mean age in years (range) 15.2 (5–45) 14.4 (4–47) 14.8 (4–47)Proportion males 48% 50% 50%Number with >10 priortransfusionsa
54 (93%) 92 (91%) 146 (92%)
Number with autoantibodies 17 (29%) 10 (9%) 27 (17%)
aSeparate packed RBC transfusion episodes.
TABLE III. Degree of Class II HLA Homozygosity and Alloimmunization
Number of homozygousHLA allelesa
Alloantibody-positive (n 5 59)
Alloantibody-negative (n 5 100)
0–1 36% 54%2–3 1% 9%
P(one-sided exact test) 5 0.011.aNumber of homozygous alleles present at the HLA class II loci (DRB1, DQB1,DPB1).
letter
American Journal of Hematology 463

examined [30]. This approach uses a strategy often taken in population
genetics and overcomes the problem of multiple testing inherent in testing for
the presence or absence of an allele in individuals. It gives both an overall
P-value for a table and a P-value for the individual contribution of each allele,
expressible as its g2 contribution to the total and as an odds ratio (OR),
respectively. Rare alleles (those observed less than five times in the alloanti-
body-positive and -negative patients combined) were excluded from testing.
Global tests were summarized with a P-value to demonstrate differences
between allelic distributions at a locus for the alloantibody-positive and -neg-
ative groups. The associations of specific alleles with antibody status for
each HLA locus were reported as an OR with its associated significance
value. Homozygosity at all six HLA loci was examined and a ranked score
based on locus homozygosity was assigned by comparing one group of indi-
viduals who were homozygous for less than two HLA alleles to a second
group of individuals homozygous for two or more HLA alleles. The effect of
HLA homozygosity on alloimmunization risk was examined using a g(2) test.
HLA associations with autoimmunization in the alloantibody-positive and
alloantibody-negative groups were examined with a 2 3 4 test of heterogene-
ity and adjustments for multiple testing were made using the Bonferroni cor-
rection.
1Department of Hematology/Oncology, Children’s Hospital and Research CenterOakland, Oakland, California
2School of Public Health, University of California, Berkeley, California3Public Health Institute, Oakland, California
*Correspondence to: Carolyn Hoppe, Department of Hematology/Oncology,Children’s Hospital and Research Center Oakland, 747, 52nd Street,
Oakland, CA 94609, E-mail: [email protected] online 21 April 2009 in Wiley InterScience
(www.interscience.wiley.com).DOI: 10.1002/ajh.21442
Conflict of interest: Nothing to report.Contract grant sponsor: Doris Duke Charitable Foundation, Clinical Scientist
Development Award; Contract grant sponsor: National Institutes of Health grants;Contract grant numbers: NS40292, HL64556-01, M01RR01271.
References1. Rosse WF, Gallagher D, Kinney TR, et al. Transfusion and alloimmunization
in sickle cell disease. The Cooperative Study of Sickle Cell Disease. Blood1990;76:1431–1437.
2. Aygun B, Padmanabhan S, Paley C, Chandrasekaran V. Clinical significanceof RBC alloantibodies and autoantibodies in sickle cell patients who receivedtransfusions. Transfusion 2002;42:37–43.
3. Vichinsky EP, Earles A, Johnson RA, et al. Alloimmunization in sickle cellanemia and transfusion of racially unmatched blood [see comments]. N EnglJ Med 1990;322:1617–1621.
4. Luban NL. Variability in rates of alloimmunization in different groups of chil-dren with sickle cell disease: Effect of ethnic background. Am J PediatrHematol Oncol 1989;11:314–319.
5. Castellino SM, Combs MR, Zimmerman SA, et al. Erythrocyte autoantibod-ies in paediatric patients with sickle cell disease receiving transfusion ther-apy: Frequency, characteristics and significance. Br J Haematol 1999;104:189–194.
6. Ambruso DR, Githens JH, Alcorn R, et al. Experience with donors matchedfor minor blood group antigens in patients with sickle cell anemia who arereceiving chronic transfusion therapy. Transfusion 1987;27:94–98.
7. Vichinsky EP, Luban NL, Wright E, et al. Prospective RBC phenotypematching in a stroke-prevention trial in sickle cell anemia: A multicentertransfusion trial. Transfusion 2001;41:1086–1092.
8. Castro O, Sandler SG, Houston-Yu P, Rana S. Predicting the effect of trans-fusing only phenotype-matched RBCs to patients with sickle cell disease:Theoretical and practical implications. Transfusion 2002;42:684–690.
9. Tahhan HR, Holbrook CT, Braddy LR, et al. Antigen-matched donor blood inthe transfusion management of patients with sickle cell disease. Transfusion1994;34:562–569.
10. Alarif L, Castro O, Ofosu M, et al. HLA-B35 is associated with red cellalloimmunization in sickle cell disease. Clin Immunol Immunopathol 1986;38:178–183.
11. Reisner EG, Kostyu DD, Phillips G, et al. Alloantibody responses in multiplytransfused sickle cell patients. Tissue Antigens 1987;30:161–166.
12. Chiaroni J, Dettori I, Ferrera V, et al. HLA-DRB1 polymorphism is associatedwith Kell immunisation. Br J Haematol 2006;132:374–378.
13. Noizat-Pirenne F, Tournamille C, Bierling P, et al. Relative immunogenicity ofFya and K antigens in a Caucasian population, based on HLA class IIrestriction analysis. Transfusion 2006;46:1328–1333.
14. Reviron D, Dettori I, Ferrera V, et al. HLA-DRB1 alleles and Jk(a) immuniza-tion. Transfusion 2005;45:956–959.
15. Bauer MP, Wiersum-Osselton J, Schipperus M, et al. Clinical predictors ofalloimmunization after red blood cell transfusion. Transfusion 2007;47:2066–2071.
16. Higgins JM, Sloan SR. Stochastic modeling of human RBC alloimmuniza-tion: Evidence for a distinct population of immunologic responders. Blood2008;112:2546–2553.
17. Gao X, Nelson GW, Karacki P, et al. Effect of a single amino acid change inMHC class I molecules on the rate of progression to AIDS. N Engl J Med2001;344:1668–1675.
18. Tang J, Costello C, Keet IP, et al. HLA class I homozygosity accelerates dis-ease progression in human immunodeficiency virus type 1 infection. AIDSRes Hum Retroviruses 1999;15:317–324.
19. Singer ST, Wu V, Mignacca R, et al. Alloimmunization and erythrocyte auto-immunization in transfusion-dependent thalassemia patients of predomi-nantly asian descent. Blood 2000;96:3369–3373.
20. Babcock GF, Alexander JW. The effects of blood transfusion on cytokineproduction by TH1 and TH2 lymphocytes in the mouse. Transplantation1996;61:465–468.
21. Kirkley SA, Cowles J, Pellegrini VD Jr, et al. Cytokine secretion after alloge-neic or autologous blood transfusion. Lancet 1995;345:527.
22. Blumberg N, Heal JM, Gettings KF. WBC reduction of RBC transfusions isassociated with a decreased incidence of RBC alloimmunization. Transfu-sion 2003;43:945–952.
23. Young PP, Uzieblo A, Trulock E, et al. Autoantibody formation after alloim-munization: Are blood tranfusions a risk factor for autoimmune hemolyticanemia? Transfusion 2004;44:67–72.
24. Adams R, McKie V, Hsu L, et al. Prevention of a first stroke by transfusionsin children with sickle cell anemia and abnormal results on transcranial dop-pler ultrasonagraphy. N Engl J Med 1998;339:5–11.
25. American Association of Blood Banks. AABB Technical Manual, 14th ed.Bethesda: American Association of Blood Banks; 2002.
26. Mollison PL, editor. Blood Transfusion in Clinical Medicine, 9th ed. Oxford,UK: Blackwell; 1993.
27. Weisbach V, Kohnhauser T, Zimmermann R, et al. Comparison of the per-formance of microtube column systems and solid-phase systems and thetube low-ionic-strength solution additive indirect antiglobulin test in thedetection of red cell alloantibodies. Transfus Med 2006;16:276–284.
28. Trachtenberg EA, Bugawan TL, Apple R, et al. Efficient typing of the HLA-Blocus using the PCR and immobilized oligonucleotide probes. Human Immu-nol 1994;40:43.
29. Bodmer JG, Marsh SG, Albert ED, et al. Nomenclature for factors of theHLA system, 1996. Tissue Antigens 1997;49:297–321.
30. Sokal R, Rohlf F. Biometry: The Principles and Practice of Statistics in Bio-logical Research. San Francisco: W.H. Freeman; 1995.
letter
464 American Journal of Hematology
Related Documents











