Experimental Physiology 1648 Exp Physiol 99.12 (2014) pp 1648–1662 Research Paper Research Paper Systemic oxidative–nitrosative–inflammatory stress during acute exercise in hypoxia; implications for microvascular oxygenation and aerobic capacity John D. S. Woodside 1 , Mariusz Gutowski 2 , Lewis Fall 3 , Philip E. James 4 , Jane McEneny 5 , Ian S. Young 5 , Shigehiko Ogoh 6 and Damian M. Bailey 3 1 Vascular Physiology Unit, Institute of Cardiovascular Science, University College London, London, UK 2 Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, Shanghai, China 3 Neurovascular Research Laboratory, Faculty of Life Sciences and Education, University of South Wales, Pontypridd, UK 4 Wales Heart Research Institute, Cardiff University School of Medicine, Heath Park, Cardiff, Pontypridd, UK 5 Centre for Public Health, Nutrition and Metabolism Group, Queen’s University Belfast, Belfast, UK 6 Department of Biomedical Engineering, Toyo University, Kawagoe-Shi, Saitama, Japan New Findings What is the central question of this study? Exercise performance is limited during hypoxia by a critical reduction in cerebral and skeletal tissue oxygenation. To what extent an elevation in systemic free radical accumulation contributes to microvascular deoxygenation and the corresponding reduction in maximal aerobic capacity remains unknown. What is the main finding and its importance? We show that altered free radical metabolism is not a limiting factor for exercise performance in hypoxia, providing important insight into the fundamental mechanisms involved in the control of vascular oxygen transport. Exercise performance in hypoxia may be limited by a critical reduction in cerebral and skeletal tissue oxygenation, although the underlying mechanisms remain unclear. We examined whether increased systemic free radical accumulation during hypoxia would be associated with elevated microvascular deoxygenation and reduced maximal aerobic capacity ( ˙ V O 2 max ). Eleven healthy men were randomly assigned single-blind to an incremental semi-recumbent cycling test to determine ˙ V O 2 max in both normoxia (21% O 2 ) and hypoxia (12% O 2 ) separated by a week. Continuous-wave near-infrared spectroscopy was employed to monitor concentration changes in oxy- and deoxyhaemoglobin in the left vastus lateralis muscle and frontal cerebral cortex. Antecubital venous blood samples were obtained at rest and at ˙ V O 2 max to determine oxidative (ascorbate radical by electron paramagnetic resonance spectroscopy), nitrosative (nitric oxide metabolites by ozone-based chemiluminescence and 3-nitrotyrosine by enzyme-linked immunosorbent assay) and inflammatory stress biomarkers (soluble intercellular/vascular cell adhesion 1 molecules by enzyme-linked immunosorbent assay). Hypoxia was associated with increased cerebral and muscle tissue deoxygenation and lower ˙ V O 2 max (P < 0.05 versus normoxia). Despite an exercise-induced increase in oxidative–nitrosative–inflammatory stress, hypoxia per se did not have an additive effect (P > 0.05 versus normoxia). Consequently, we failed to J. D. S. Woodside and D. M. Bailey contributed equally to this work. DOI: 10.1113/expphysiol.2014.081265 C 2014 The Authors. Experimental Physiology C 2014 The Physiological Society

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Expe
rim
enta
lPhy
siol
ogy
1648 Exp Physiol 99.12 (2014) pp 1648–1662
Research PaperResearch Paper
Systemic oxidative–nitrosative–inflammatory stress duringacute exercise in hypoxia; implications for microvascularoxygenation and aerobic capacity
John D. S. Woodside1, Mariusz Gutowski2, Lewis Fall3, Philip E. James4, Jane McEneny5, Ian S. Young5,Shigehiko Ogoh6 and Damian M. Bailey3
1Vascular Physiology Unit, Institute of Cardiovascular Science, University College London, London, UK2Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, Shanghai, China3Neurovascular Research Laboratory, Faculty of Life Sciences and Education, University of South Wales, Pontypridd, UK4Wales Heart Research Institute, Cardiff University School of Medicine, Heath Park, Cardiff, Pontypridd, UK5Centre for Public Health, Nutrition and Metabolism Group, Queen’s University Belfast, Belfast, UK6Department of Biomedical Engineering, Toyo University, Kawagoe-Shi, Saitama, Japan
New Findings� What is the central question of this study?
Exercise performance is limited during hypoxia by a critical reduction in cerebral andskeletal tissue oxygenation. To what extent an elevation in systemic free radical accumulationcontributes to microvascular deoxygenation and the corresponding reduction in maximalaerobic capacity remains unknown.
� What is the main finding and its importance?We show that altered free radical metabolism is not a limiting factor for exercise performancein hypoxia, providing important insight into the fundamental mechanisms involved in thecontrol of vascular oxygen transport.
Exercise performance in hypoxia may be limited by a critical reduction in cerebral and skeletaltissue oxygenation, although the underlying mechanisms remain unclear. We examined whetherincreased systemic free radical accumulation during hypoxia would be associated with elevatedmicrovascular deoxygenation and reduced maximal aerobic capacity (VO2max). Eleven healthymen were randomly assigned single-blind to an incremental semi-recumbent cycling test todetermine VO2max in both normoxia (21% O2) and hypoxia (12% O2) separated by a week.Continuous-wave near-infrared spectroscopy was employed to monitor concentration changesin oxy- and deoxyhaemoglobin in the left vastus lateralis muscle and frontal cerebral cortex.Antecubital venous blood samples were obtained at rest and at VO2max to determine oxidative(ascorbate radical by electron paramagnetic resonance spectroscopy), nitrosative (nitricoxide metabolites by ozone-based chemiluminescence and 3-nitrotyrosine by enzyme-linkedimmunosorbent assay) and inflammatory stress biomarkers (soluble intercellular/vascular celladhesion 1 molecules by enzyme-linked immunosorbent assay). Hypoxia was associated withincreased cerebral and muscle tissue deoxygenation and lower VO2max (P<0.05 versus normoxia).Despite an exercise-induced increase in oxidative–nitrosative–inflammatory stress, hypoxia perse did not have an additive effect (P > 0.05 versus normoxia). Consequently, we failed to
J. D. S. Woodside and D. M. Bailey contributed equally to this work.
DOI: 10.1113/expphysiol.2014.081265 C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1649Free radicals and oxygen transport
observe correlations between any metabolic, haemodynamic and cardiorespiratory parameters(P > 0.05). Collectively, these findings suggest that altered free radical metabolism cannotexplain the elevated microvascular deoxygenation and corresponding lower VO2max in hypoxia.Further research is required to determine whether free radicals when present in excess do indeedcontribute to the premature termination of exercise in hypoxia.
(Received 10 June 2014; accepted after revision 10 October 2014; first published online 17 October 2014)Corresponding author D. M. Bailey: Neurovascular Research Laboratory, Faculty of Life Sciences and Education,University of South Wales, South Wales CF37 4AT, UK. Email: [email protected]
Introduction
The origins of fatigue and precisely why maximalexercise performance is limited during acute exposure tohypoxia remain unclear. In combination with peripheralmechanisms that involve biochemical changes withinskeletal muscle (Allen et al. 2008), attention has beendrawn towards more central processes resulting fromfailure of the CNS to provide adequate drive to themotoneurones (i.e. central fatigue; Gandevia, 2001).Emerging evidence suggests that a critical, albeit undefinedreduction in the cerebral PO2 may constitute the upstreamsignal that serves to limit central motor command,resulting in the premature decision to terminate exercise(Noakes et al. 2001; Calbet et al. 2003; Amann et al. 2006;Subudhi et al. 2007, 2008).
Near-infrared spectroscopy (NIRS) is an opticaltechnique that provides continuous, non-invasivemonitoring of local tissue deoxygenation and thusreflects the balance between local tissue O2 delivery(Q O2T) and utilisation (VO2T) within a given regionof interest (Ferrari et al. 2011). Studies employingNIRS have consistently demonstrated a reduction inprefrontal cortical deoxygenation during submaximaland maximal whole-body exercise in hypoxic conditionscompared with normoxia (Verges et al. 2012). Morerecently, this was shown to be associated with lowerprefrontal cortical blood flow and a correspondingreduction in cortical oxygen delivery (Vogiatzis et al.2011), which may contribute to the lower (calculated)cerebral mitochondrial PO2 previously documented(Rasmussen et al. 2007; Bailey et al. 2011b). While currentinterests continue to focus primarily on the link toexercise performance per se, the fundamental mechanismsthat serve to limit tissue oxygenation remain to beestablished.
While acute exercise is an established pro-oxidantstimulus (Powers & Jackson, 2008), human studies havedemonstrated that hypoxia can equally serve to promotesystemic free radical accumulation due to independentcontributions from the muscle (Bailey et al. 2004b),brain (Bailey et al. 2009d, 2011b) and lungs (Baileyet al. 2010). Spin-trapping studies have shown hypoxiato compound exercise-induced lipid-derived alkoxyl
radical accumulation (Bailey et al. 2001; Davison et al.2006) despite a comparatively lower maximal (absolute)power output. We have reasoned that a reduction inmitochondrial PO2 even in the face of a comparativelylower O2 flux is potentially one of several as yetunidentified mechanisms, underpinning the synergistic(pro-oxidant) effects of hypoxic exercise (Bailey et al.2004b).
In terms of exercise performance, free radicals whenin excess have the potential to promote fatigue bydirectly interfering with the contractile components ofskeletal muscle (Ferreira & Reid, 2008; Powers & Jackson,2008) and increasing the central perception of fatigue(Mantovani et al. 2006). Furthermore, inflammatory stressin combination with nitrosative stress caused by theoxidative inactivation of the vasodilator molecule NO haveequal capacity to limit tissue oxygen transport subsequentto impaired vascular endothelial function (Bailey et al.2004a, 2006, 2013; Richardson et al. 2007; Eltzschig &Carmeliet, 2011). In support, a reduction in the vascularbioavailability of NO using L-NAME has been associatedwith a reduction in microvascular PO2 in contractingskeletal muscle (Ferreira et al. 2006).
In light of these findings, the present study wasdesigned to examine the potential relationships betweensystemic free radical metabolism, NIRS-derived indicesof local deoxygenation and exercise performance duringhypoxia. Given our previous findings (Bailey et al. 2001;Davison et al. 2006), we hypothesised that compared withnormoxia, hypoxia would compound exercise-inducedoxidative–nitrosative–inflammatory stress and that thiswould be associated with a more pronounced increasein microvascular deoxygenation and correspondingreduction in VO2max.
Methods
Ethics
The study was approved by the University of South WalesHuman Research Ethics Committee (UK). All procedureswere carried out in accordance with the Declaration ofHelsinki of the World Medical Association (Williams,
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

1650 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
2008), and written informed consent was obtained fromall participants.
Design
The present study adopted a randomised, single-blinddesign as outlined in Fig. 1. All measurements wereperformed in an environmental chamber (�120 m3)maintained at 21°C and 50% relative humidity (DesignEnvironmental, Ebbw Vale, UK). Participants attendedthe laboratory following a 12 h overnight fast and,following cannulation, rested in normoxia for 30 min ina semi-recumbent position to establish baseline controlNIRS measurements. Formal data collection commencedfollowing 30 min passive exposure and at the point ofvolitional exhaustion during a cycling test performed inboth normoxia (O2 = 21%) and (normobaric) hypoxia(O2 = 12%). For the hypoxia trial, an N2-rich gasmixture was delivered at a high flow rate from molecularsieves at the prevailing barometric pressure. Ambient O2
and CO2 concentrations were monitored continuouslyusing fast-responding paramagnetic and infraredanalysers (Servomex 1400 Series Analyser; Servomex,Crowborough, UK), respectively, and maintained via aservo-controlled system that allowed for the addition ofN2 or air through solenoid valves. Each trial was separatedby a 7 day recovery period.
Participants
Eleven healthy, physically active male participants wererecruited into the study. They were 23 ± 5 years old, witha body mass index of 26 ± 2 kg m−2. All participantswere sea-level residents, non-smokers and abstained fromtaking nutritional supplements, such as oral antioxidants,and anti-inflammatories. Participants were specificallyasked to refrain from physical activity, caffeine and alcoholfor a period of 48 h prior to formal experimentation,consistent with our previous approaches designed tominimise the biological variation associated with freeradical metabolism (Davison et al. 2012). They were alsoencouraged to follow a low-nitrate/nitrite (NO3
−/NO2−)
diet for 96 h prior to the study, with specific instructionsto avoid fruits, salads and cured meats (Wang et al. 1997).
Maximal exercise test
Following two familiarisation sessions, each participantwas seated on an electronically braked, semi-recumbentcycle ergometer (Corival; Lode BV, Groningen, TheNetherlands), with the backrest maintained at 70 deg. Thetest commenced with a 4 min (steady-state) workloadat 60 W (80 r.p.m.), before increasing by 30 W every2 min until volitional exhaustion. Each participant was
instructed to signal clearly to the investigators when theyconsidered they could continue at the specified poweroutput for no longer than 60 s, as previously described(Bailey et al. 2011a).
Cardiorespiratory measurements
Pulmonary gas exchange. Subjects wore a leak-free mask,and gas exchange was measured using a semi-automatedDouglas bag system. Expired gas fractions were measuredusing fast-responding paramagnetic O2 and infrared CO2
analysers (Servomex 1400 Series Analyser, Servomex,Crowborough, UK). The volume of expired gas fordetermination of ventilation (VE) was measured usinga dry gas meter (Harvard Apparatus, Ltd, Edenbridge,UK), and oxygen uptake (VO2 ) and carbon dioxide output(VCO2 ) were calculated via the Haldane equation.
Heart rate (HR) and arterial oxyhaemoglobin saturation(SaO2 ). Heart rate was recorded using ECG-calibratedbipolar telemetry (Vantage; Polar Electro, Oy, Finland)and SaO2 via finger-tip pulse oximetry (515C; NovametrixMedical Systems, Wallingford, CT, USA).
Metabolic measurements
Overnight fasted blood samples were obtained from acannula located in a forearm antecubital vein following30 min of rest and timed to coincide with the terminationof exercise. Blood was centrifuged at 600 g (4°C) for 10 minand the supernatant immediately snap-frozen and storedunder liquid nitrogen prior to batch analysis.
Oxidative stress: ascorbate free radical (A•−). Plasma(1 ml) was injected into a high-sensitivity multiple-boresample cell (AquaX; Bruker Daltonics Inc., Billerica,MA, USA) housed within a TM110 cavity of anelectron paramagnetic resonance (EPR) spectrometeroperating at X-band (9.87 GHz). Samples were recorded12 min after the end of plasma recovery by signalaveraging three scans with the following instrumentparameters: resolution, 1024 points; microwave power,20 mW; modulation amplitude, 0.65 G; receiver gain,2 × 105; time constant, 40.96 ms; scan rate, 0.25 G s−1
for scan width, 15 G. Spectra were filtered identically usingWINEPR (Version 2.11; Bruker, Karlsruhe, Germany), andthe double integral of each doublet was calculated usingOrigin software (OriginLabs, Northampton, MA, USA).The intra- and interassay coefficients of variation (CV)were both <5%.
Nitrosative stress: nitric oxide. Plasma NO metaboliteswere measured by ozone-based chemiluminescence.Samples (20 μl) were analysed for the total concentration
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1651Free radicals and oxygen transport
of NO [(NO3− + NO2
−) + S-nitrosothiols (RSNO)]by vanadium (III) reduction (Ewing & Janero, 1998).A separate sample (200 μl) was injected into tri-iodidereagent for the measurement of NO2
− + RSNO, and 5%acidified sulphanilamide was added and left to incubatein the dark at 21°C for 15 min to remove NO2
− for themeasurement of RSNO in a third parallel sample. PlasmaNO3
− was calculated as total NO – (NO2− + RSNO). All
calculations were performed using Origin/Peak Analysissoftware (OriginLabs). The intra- and interassay CVs forall NO metabolites were 7 and 10%, respectively.
Nitrosative stress: 3-nitrotyrosine (3-NT). Plasma 3-NTwas measured by enzyme-linked immunosorbentassay (ELISA; Hycult Biotechnology BV, Uden, TheNetherlands) with a lower detection limit of 2 nM andintra- and interassay CVs of <2 and <5%, respectively.
Inflammatory stress: soluble inter/vascular cellularadhesion molecules (sICAM-1/sVCAM-1). PlasmasICAM-1 and sVCAM-1 were measured by ELISA(Eli-pair; Diaclone, Besancon, France). The intra- andinterassay CVs were <6 and <12% for both analytes,respectively.
Microvascular deoxygenation indices
Pulsed continuous-wave NIRS (Oxymon Mk III; ArtinisMedical Systems BV, Zetten, The Netherlands) was usedto monitor changes in cerebral and skeletal muscleoxygenation at optical densities of 780 and 850 nm.A detailed overview of the theory underpinning thistechnique, including reliability and inherent limitations,has been published elsewhere (Rooks et al. 2010). Oneset of NIRS optodes (5.0 cm source–detector spacing,consistent with recent recommendations) was placed onthe skin over the left frontal prefrontal cortical regionof the forehead between Fp1 and F3, consistent with theanatomical landmarks of the International 10–20 systemfor EEG placement (Jasper, 1958). A second set of optodes(5.0 cm spacing) was attached to the shaved belly of theleft vastus lateralis �15 cm above the proximal borderof the patella and 5 cm lateral to the mid-line of thethigh (Subudhi et al. 2007). All optodes were secured inplace using double-sided adhesive tape and tight elasticbands. Concentration changes (�) in oxyhaemoglobin(O2Hb) and deoxyhaemoglobin (HHb) were calculatedusing the modified Beer–Lambert Law incorporatingdifferential pathlengths of 5.93 (Duncan et al. 1995) and4.95 (van der Zee et al. 1992) for cerebral and muscletissue, respectively. The parameter �[HHb] is closelyassociated with changes in venous O2 content and isthus less sensitive to the change in total haemoglobinconcentration (�[THb]) than to �[O2Hb]. Although
it is generally considered to be relatively insensitive toblood volume shifts during constant-load exercise, the factthat �[THb] is constantly changing under the optodesthroughout the course of incremental exercise cannot beignored (Quaresima & Ferrari, 2009). With this caveat inmind, we chose to focus on this signal as a relative estimateof changes in microvascular deoxygenation and O2
extraction reflecting the balance between Q O2T and VO2T
(DeLorey et al. 2003; Grassi et al. 2003). We also calculatedthe difference between �[O2Hb] and �[HHb] as asurrogate ‘oxygenation index’ (OI), consistent with NIRSstudies that have also employed similar continuous-wavedevices (Grassi et al. 2003). Finally, �[THb] was calculatedas the sum of �[O2Hb] and �[HHb] signals and used asa surrogate for changes in regional blood volume (VanBeekvelt et al. 2001). All NIRS signals were normalisedto reflect changes relative to a 60 s baseline control atrest in normoxia (arbitrarily defined as 0 μM) establishedimmediately prior to each of the experimental conditions,that is during both normoxia and hypoxia (see Fig. 1).Signals were recorded at 10 Hz, displayed in real timeand stored on a personal computer for offline analysis.Averages were calculated during the final 60 s of eachworkload and prior to volitional exhaustion.
Statistical analysis
Shapiro–Wilk W tests were employed to confirmdistribution normality. All data were analysed using atwo-way [condition (normoxia versus hypoxia) × state(rest versus maximal exercise) or intensity (rest versus30–100% maximal power output)] repeated-measuresANOVA. Bonferroni-corrected Student’s paired t testswere employed to locate differences when aninteraction effect was detected. Pearson product–momentcorrelations were used to determine relationships betweenselected variables. Significance was established at P < 0.05for all two-tailed tests, and values are presented as themeans ± SD.
Results
Cardiorespiratory responses
As anticipated, hypoxia was associated with a lower SaO2
and corresponding reduction in peak power output (from278 ± 34 to 232 ± 24 W, P < 0.05), time to exhaustion(from 1132 ± 129 to 941 ± 86 s, P < 0.05) and VO2max
(Table 1).
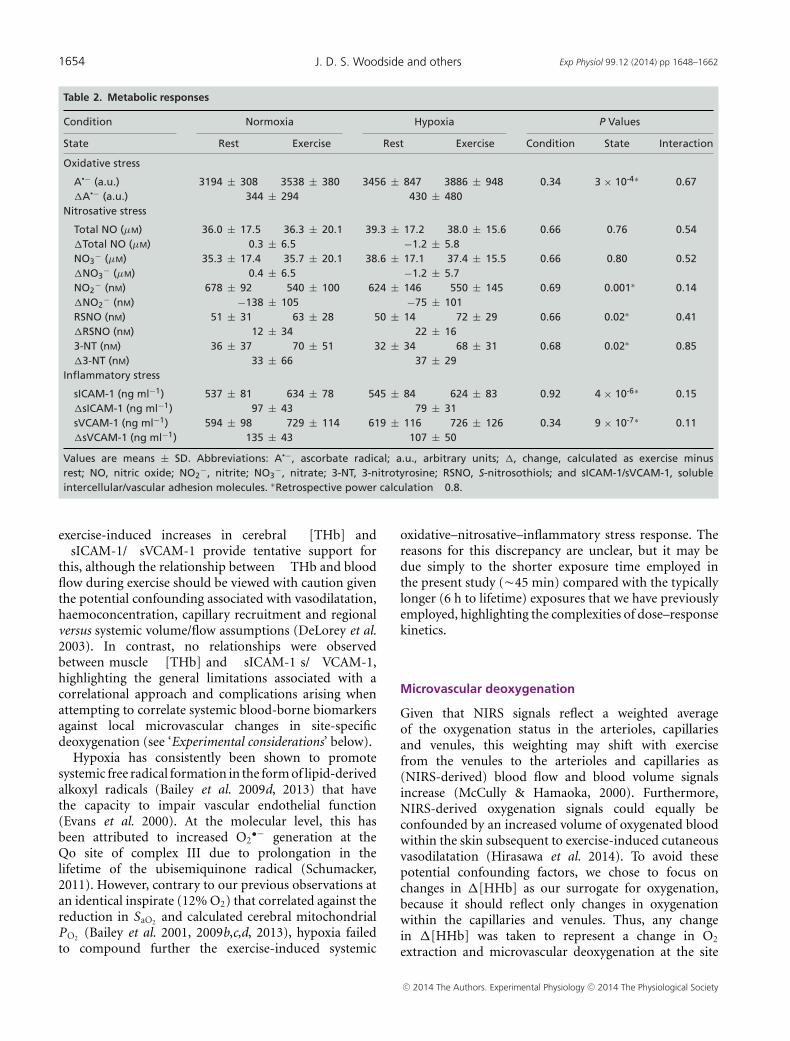
Metabolic responses
Exercise was associated with a general increase in A•¯(Table 2). Typical examples of the characteristic EPR
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

1652 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
doublet (aH4 = 1.76 G) obtained at rest and during
exercise in normoxia and hypoxia are illustrated inFig. 2. Likewise, exercise was shown to increase RSNO,3-NT, sICAM-1 and sVCAM-1, with a correspondingdecrease observed in NO2
−, indicative of elevatedoxidative–nitrosative–inflammatory stress. In contrast,hypoxia did not alter the metabolic response at rest orduring exercise.
Microvascular deoxygenation
Figure 3 illustrates the cerebral (left panels) and muscleNIRS responses (right panels) at rest and during exercise(relative to the respective normoxic baseline controls).These changes were expressed as a function of changesin relative exercise intensity, given as the percentage of themaximal power output achieved at each of the prevailinginspirates, although the qualitative outcome was identicalwhen expressed relative to changes in absolute exerciseintensity.
Cerebral. Compared with normoxia, hypoxia wasgenerally associated with ↓�[O2Hb], ↓OI and ↑�[HHb],whereas no differences were observed in �[THb]. Thesedifferences persisted throughout exercise, although thepatterns of change during normoxia and hypoxia weresimilar (↑�[O2Hb], ↑�[HHb], ↑�[THb] and ↓OI) asindicated by the lack of any interaction effects.
Muscle. Hypoxia was generally associated with↓�[O2Hb], ↓�[THb] and ↓OI and ↑�[HHb].Unlike the cerebral data, interaction effects were apparentfor all muscle NIRS variables, due in part to consistentlysmaller SD error bars and a corresponding increasein statistical power (retrospective calculation �0.8).As a consequence, progressive, stepwise decreases in�[O2Hb] and OI and increases in �[HHb] and �[THb]
were observed during exercise in hypoxia. Of note, theexercise-induced reduction in �[O2Hb] during normoxiawas apparent only towards the higher exercise intensities(90–100% VO2max), and �[THb] was shown to decrease(rather than plateau, as was the case for the cerebral data).
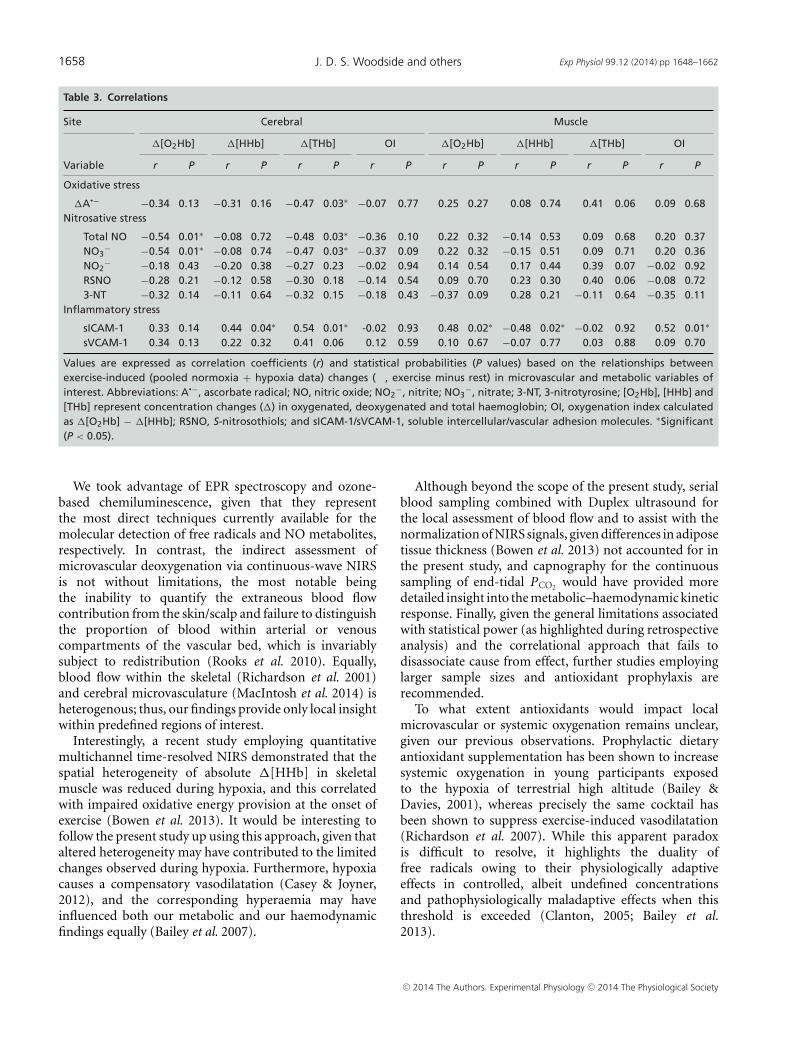
Correlations
Table 3 summarises the relationships observed betweenexercise-induced changes (pooled normoxia + hypoxiadata, given that a main effect for state and not conditionwas recorded) in microvascular and metabolic variables.Inverse relationships (P < 0.05) were observed betweencerebral �[O2Hb] and �total NO and �NO3
−, betweencerebral �[THb] and �A•−, �total NO and �NO3
−, andfinally, between muscle �[HHb] and �sICAM-1. Positiverelationships (P < 0.05) were observed between cerebral�[HHb]/cerebral �[THb]/muscle �[O2Hb]/muscle OIand �sICAM-1. In contrast, we failed to observe anyrelationships (P > 0.05) between the widened cerebraland muscle [HHb]DIFF (hypoxia minus normoxia values)observed at VO2max and �A•−DIFF relative to restingdifferences (Fig. 4). Finally, no relationships were observedbetween the hypoxia-induced reduction in SaO2 (pooledrest + exercise data) and changes in any of the metabolicvariables (r = −0.35 to 0.17, P > 0.05).
Discussion
The present study sought to determine whetheran elevation in systemic free radical accumulationis associated with a more pronounced increasein microvascular deoxygenation (↑�[HHb]/↓OI) andcorresponding reduction in VO2max during hypoxia. Whileexercise was consistently shown to increase systemicbiomarkers of oxidative–nitrosative–inflammatory stress,hypoxia failed to exert an additive effect. Thus,
N
30 min
H 7 days
Rest
30 min
Exercise Baseline Rest
Exercise Rest Baseline Rest
N N
~20 min 30 min 30 min ~15 min
Randomised, single-blinded, cross-over study
Figure 1. Experimental designLarge arrows indicate when venous blood samples were obtained. Abbreviations: H, hypoxia (O2 = 12%);and N, normoxia (O2 = 21%).
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1653Free radicals and oxygen transport
Table 1. Cardiorespiratory responses
Condition Normoxia Hypoxia P Values
State Rest Exercise Rest Exercise Condition State Interaction
SaO2 (%) 98 ± 1 96 ± 1∗ 85 ± 6† 77 ± 3∗,† 1 × 10-7 2 × 10-4 0.01‡
�SaO2 (%) −2 ± 1 −7 ± 5†
HR (beats min−1) 56 ± 8 187 ± 13∗ 67 ± 14† 185 ± 14∗ 0.01‡ <1 × 10-8 0.001‡
�HR (beats min−1) 131 ± 10 117 ± 9†
VE (l min−1) 10 ± 2 134 ± 18 11 ± 2 132 ± 11 0.80 <1 × 10-8 0.17�VE (l min−1) 125 ± 17 120 ± 11VO2 (l min−1) 0.32 ± 0.06 3.84 ± 0.29∗ 0.32 ± 0.04 2.97 ± 0.18∗,† 3 × 10-6 <1 × 10-8 03 × 10-8
�VO2 (l min−1) 3.52 ± 0.29 2.65 ± 0.18†
VCO2 (l min−1) 0.29 ± 0.09 4.21 ± 0.30∗ 0.32 ± 0.04 3.59 ± 0.21∗,† 8 × 10-5 <01 × 10-8 3 × 10-7
�VCO2 (l min−1) 3.92 ± 0.27 3.27 ± 0.21†
RER (a.u.) 0.91 ± 0.12 1.15 ± 0.02 1.02 ± 0.17 1.21 ± 0.02 0.001‡ 2 × 10-4 1.00�RER (a.u.) 0.19 ± 0.12 0.19 ± 0.16VE
/VO2 31 ± 5 35 ± 3 36 ± 6 44 ± 3 2 × 10-5 9 × 10-4 0.09
�VE/
VO2 4 ± 6 8 ± 6VE
/VCO2 35 ± 5 32 ± 3 36 ± 6 37 ± 3† 0.001‡ 0.63 0.03
�VE/
VCO2 −3 ± 7 1 ± 4†
Values are means ± SD. Abbreviations: a.u., arbitrary units; �, change, calculated as exercise minus rest; HR, heart rate; RER,respiratory exchange ratio; SaO2 , arterial oxyhaemoglobin saturation; VCO2 , carbon dioxide output; VE, ventilation; and VO2 , oxygenuptake. ∗Different versus rest for a given condition (normoxia or hypoxia; P < 0.05). †Different versus normoxia for a given state (restor exercise; P < 0.05). ‡Retrospective power calculation �0.8.
contrary to our original hypothesis, we did notobserve any relationships between the metabolic responseand the magnitude of microvascular deoxygenationwithin both muscle and cerebral tissue duringhypoxia. Given that our model of hypoxia failedto compound oxidative–nitrosative–inflammatory stress,further research employing alternative models, largersample sizes and antioxidant prophylaxis are warranted todetermine whether free radicals when in excess do indeedcontribute to increased microvascular deoxygenation andthe premature termination of exercise during hypoxia.
Oxidative–nitrosative–inflammatory stress
Given that the concentration of ascorbate in humanplasma is orders of magnitude higher than any oxidisingfree radical, combined with the low one-electronreduction potential (E º′ = 282 mV) associated withthe A•−/ascorbate monanion (AH−) couple (Williams &Yandell, 1982), any radical generated within the systemiccirculation has the potential to react endogenously withthis terminal small-molecule antioxidant to form thedistinctive A•− EPR doublet (R• + AH− → A•− + R-H;Buettner, 1993). Thus, the elevation in A•− provides directevidence for an increased systemic accumulation of freeradicals in response to maximal exercise.
To address the downstream consequences of increasedoxidative stress, we also chose to measure NO2
−/3-NT incombination with sICAM-1/sVCAM-1 as complementarybiomarkers of nitrosative and inflammatory stress,
respectively. Consistent with our previous observations,we observed an exercise-induced decrease in NO2
− andreciprocal rise in 3-NT, which was taken indirectly toreflect superoxide (O2
•−)-mediated inactivation of NOand corresponding formation of peroxynitrite (ONOO−;Gryglewski et al. 1986; Nauser & Koppenol, 2002), asfollows:
O2•− + NO
k=16−20×109 Ms−1→ ONOO− tyrosine nitration→ 3 − NT
However, contrary to our original hypothesis, we failedto observe any significant differences in total NO either asa function of exercise or of hypoxia.
Maximal exercise also invoked an inflammatory stressresponse, as indicated by the combined elevation insICAM-1 and sVCAM-1. It was unfortunate that we didnot measure additional complementary biomarkers ofinflammation, in particular tumour necrosis factor (TNF),given that it can act via the TNFR1 receptor subtype tostimulate cytosolic oxidant formation and depress skeletalmuscle contractile function (Reid & Moylan, 2011).
The appearance of cell adhesion molecules withinthe circulation is considered to reflect shedding causedby endothelial activation or damage triggering thetransmigration of leucocytes and adhesion of reticulocytesto the stimulated endothelium (Frenette & Wagner,1996). These findings are in agreement with thelimited studies conducted to date, with the increasesascribed, albeit speculatively, to elevated shear stresscaused by increased blood flow (Marsh & Coombes,2005). The linear relationships observed between the
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society
1654 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
Table 2. Metabolic responses
Condition Normoxia Hypoxia P Values
State Rest Exercise Rest Exercise Condition State Interaction
Oxidative stress
A•− (a.u.) 3194 ± 308 3538 ± 380 3456 ± 847 3886 ± 948 0.34 3 × 10-4∗ 0.67�A•− (a.u.) 344 ± 294 430 ± 480
Nitrosative stress
Total NO (μM) 36.0 ± 17.5 36.3 ± 20.1 39.3 ± 17.2 38.0 ± 15.6 0.66 0.76 0.54�Total NO (μM) 0.3 ± 6.5 −1.2 ± 5.8NO3
− (μM) 35.3 ± 17.4 35.7 ± 20.1 38.6 ± 17.1 37.4 ± 15.5 0.66 0.80 0.52�NO3
− (μM) 0.4 ± 6.5 −1.2 ± 5.7NO2
− (nM) 678 ± 92 540 ± 100 624 ± 146 550 ± 145 0.69 0.001∗ 0.14�NO2
− (nM) −138 ± 105 −75 ± 101RSNO (nM) 51 ± 31 63 ± 28 50 ± 14 72 ± 29 0.66 0.02∗ 0.41�RSNO (nM) 12 ± 34 22 ± 163-NT (nM) 36 ± 37 70 ± 51 32 ± 34 68 ± 31 0.68 0.02∗ 0.85�3-NT (nM) 33 ± 66 37 ± 29
Inflammatory stress
sICAM-1 (ng ml−1) 537 ± 81 634 ± 78 545 ± 84 624 ± 83 0.92 4 × 10-6∗ 0.15�sICAM-1 (ng ml−1) 97 ± 43 79 ± 31sVCAM-1 (ng ml−1) 594 ± 98 729 ± 114 619 ± 116 726 ± 126 0.34 9 × 10-7∗ 0.11�sVCAM-1 (ng ml−1) 135 ± 43 107 ± 50
Values are means ± SD. Abbreviations: A•−, ascorbate radical; a.u., arbitrary units; �, change, calculated as exercise minusrest; NO, nitric oxide; NO2
−, nitrite; NO3−, nitrate; 3-NT, 3-nitrotyrosine; RSNO, S-nitrosothiols; and sICAM-1/sVCAM-1, soluble
intercellular/vascular adhesion molecules. ∗Retrospective power calculation �0.8.
exercise-induced increases in cerebral �[THb] and�sICAM-1/�sVCAM-1 provide tentative support forthis, although the relationship between �THb and bloodflow during exercise should be viewed with caution giventhe potential confounding associated with vasodilatation,haemoconcentration, capillary recruitment and regionalversus systemic volume/flow assumptions (DeLorey et al.2003). In contrast, no relationships were observedbetween muscle �[THb] and �sICAM-1 s/�VCAM-1,highlighting the general limitations associated with acorrelational approach and complications arising whenattempting to correlate systemic blood-borne biomarkersagainst local microvascular changes in site-specificdeoxygenation (see ‘Experimental considerations’ below).
Hypoxia has consistently been shown to promotesystemic free radical formation in the form of lipid-derivedalkoxyl radicals (Bailey et al. 2009d, 2013) that havethe capacity to impair vascular endothelial function(Evans et al. 2000). At the molecular level, this hasbeen attributed to increased O2
•− generation at theQo site of complex III due to prolongation in thelifetime of the ubisemiquinone radical (Schumacker,2011). However, contrary to our previous observations atan identical inspirate (12% O2) that correlated against thereduction in SaO2 and calculated cerebral mitochondrialPO2 (Bailey et al. 2001, 2009b,c,d, 2013), hypoxia failedto compound further the exercise-induced systemic
oxidative–nitrosative–inflammatory stress response. Thereasons for this discrepancy are unclear, but it may bedue simply to the shorter exposure time employed inthe present study (�45 min) compared with the typicallylonger (6 h to lifetime) exposures that we have previouslyemployed, highlighting the complexities of dose–responsekinetics.
Microvascular deoxygenation
Given that NIRS signals reflect a weighted averageof the oxygenation status in the arterioles, capillariesand venules, this weighting may shift with exercisefrom the venules to the arterioles and capillaries as(NIRS-derived) blood flow and blood volume signalsincrease (McCully & Hamaoka, 2000). Furthermore,NIRS-derived oxygenation signals could equally beconfounded by an increased volume of oxygenated bloodwithin the skin subsequent to exercise-induced cutaneousvasodilatation (Hirasawa et al. 2014). To avoid thesepotential confounding factors, we chose to focus onchanges in �[HHb] as our surrogate for oxygenation,because it should reflect only changes in oxygenationwithin the capillaries and venules. Thus, any changein �[HHb] was taken to represent a change in O2
extraction and microvascular deoxygenation at the site
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1655Free radicals and oxygen transport
of O2 exchange, reflecting the balance between Q O2T andVO2T, a key determinant of the O2 driving pressure (i.e.PO2 ) required for diffusion between the capillary andmyocyte (Diederich et al. 2002).
In the present study, we specifically chose to normaliseNIRS signals relative to a baseline normoxic control periodthat preceded each of the experimental conditions in orderto determine to what extent hypoxia influenced bothresting and exercise-induced changes in microvascularoxygenation indices. We further chose to express thesesignals as a function of changes in relative exercise intensity(percentage of the maximal power output achieved)at each of the prevailing inspirates and not relativeto the maximal power output achieved in normoxiaas previously documented (Subudhi et al. 2007, 2008),given that changes in relative (and not absolute) exerciseintensity mediate the metabolic (and thus, presumably, theoxidative–nitrosative–inflammatory) response to exercise(Bouissou et al. 1986). Despite these subtle differences,our findings are consistent with previous studies (Subudhiet al. 2007, 2008) and indicate that incremental exerciseto volitional exhaustion was associated with more
pronounced cerebral and muscle deoxygenation duringhypoxia (↑�[HHb]/↓OI), tentatively taken to reflect amore pronounced increase in VO2 relative to Q O2T.
Traditionally, it has been suggested that a critical, albeitundefined reduction in cerebral oxygenation may posea central limitation to exercise, at least during severehypoxaemia (SaO2 �75%), by inhibiting cortical activationof efferent motoneurones (Noakes et al. 2001; Calbet et al.2003; Amann et al. 2006; Subudhi et al. 2007, 2008).This may serve to protect the cerebrovasculature againstexercise-induced ischaemic damage, which is entirelyreasonable given the vulnerability of the brain to energyfailure in light of its obligatory high rate of oxygenconsumption and limited glycolytic capacity (Baileyet al. 2009a). In support, increased prefrontal corticaloxygenation following the transition from hypoxia tohyperoxia has been associated with marked improvementsin exercise performance (Kayser et al. 1994; Amann et al.2007; Subudhi et al. 2008).
However, it is difficult to see how our findingssupport such a concept, given that the pattern ofcerebral deoxygenation (↑�[HHb]/↓OI) during exercise
Figure 2. Ascorbate radicalOxidation of the ascorbate monoanion (AH−) by an initiating species (R•) with a one electron reductionpotential (E º ′) greater than +282 mV yields the domesticated ascorbate radical (A•−). The unpairedelectron is delocalised over a highly conjugated tri-carbonyl π-system, rendering it resonance stabilisedand facilitating direct detection by electron paramagnetic resonance spectroscopy. Note the typicalincrease observed in the signal amplitude of A•− during physical exercise to volitional exhaustion innormoxia (O2 = 21%) and acute hypoxia (O2 = 12%).
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

1656 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
Figure 3. Changes in microvascular oxygenation as a function of relative exercise intensityValues are means ± SD expressed as concentration changes (�) in oxygenated ([O2Hb]), deoxygenated([HHb]) and total haemoglobin ([THb]) relative to control (baseline resting normoxic) values (stippledhorizontal line). Intensity reflects the percentage maximal power output achieved for each condition.Oxygenation index is calculated as �[O2Hb] − �[HHb]. ∗Different from preceding value for a givencondition (normoxia versus hypoxia). †Different between Conditions for a given intensity (rest to 100%maximal aerobic capacity). ‡Retrospective power calculation �0.8.
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1657Free radicals and oxygen transport
in hypoxia was comparable to that observed duringnormoxia, albeit ‘offset’ due primarily to changesincurred at rest. Indeed, the magnitude of increasein �[HHb] and decrease in OI at the point ofvolitional exhaustion during normoxia was still lower andhigher, respectively, than that observed at 30% VO2max
during hypoxia when participants still had significantcapacity to perform exercise. In contrast, other studiesemploying similar exercise protocols, hypoxic stimuliand continuous-wave NIRS devices have identified amore rapid exercise-induced increase in �[HHb] andcorresponding decline in �[O2Hb], cautiously taken toreflect evidence of central limitation (Subudhi et al.2007, 2008). Additional channels would have allowed forspatially resolved changes in more robust measures ofcerebral oxygenation, including the tissue oxygen index(Subudhi et al. 2011). Furthermore, more sophisticatedfrequency domain and time-resolved NIRS approaches forthe continuous measurement of absolute changes in tissuedeoxygenation would have provided improved sensitivityand quantitative insight into NIRS signals, given that theyeliminate potential confounding factors associated withunknown optical path length, absorption and scattering(Ferrari et al. 2011).
Based on the current literature, we originallyhypothesised that a more marked exercise-inducedincrease in the oxidative–nitrosative–inflammatory stressresponse during hypoxia would be associated withincreased microvascular deoxygenation. Given thatexercise in hypoxia is known to potentiate the reductionin prefrontal cortical blood flow, O2 delivery (Vogiatziset al. 2011) and (calculated) cerebral mitochondrialPO2 (Rasmussen et al. 2007; Bailey et al. 2011b),we believed that the potentiated free radical-mediatedreduction in vascular NO bioavailability would explainat least part of the variance by exacerbating tissuecapillary–mitochondrial diffusion limitation, the primaryresistance to vascular O2 transport (Wagner, 2010).Furthermore, when in excess, free radicals have thepotential to promote fatigue by directly impairingmuscular contractile and mitochondrial function(Ferreira & Reid, 2008; Powers & Jackson, 2008) andincreasing the central perception of fatigue (Mantovaniet al. 2006), with ergogenic benefits demonstratedfollowing dietary antioxidant prophylaxis (Richardsonet al. 2007; Powers & Jackson, 2008). However,given that the anticipated effects of hypoxia on theoxidative–nitrosative–inflammatory stress response failedto transpire, no such correlations were apparent, contraryto recent findings (Gatterer et al. 2013). Furthermore,we failed to observe any relationships between theexercise-induced (pooled normoxia + hypoxia values)increase in A•− and the magnitude of cerebral/muscledeoxygenation.
Experimental considerations
Contrary to our original hypothesis based on previousobservations (Bailey et al. 2001; Davison et al. 2006),the present model of hypoxia failed to compoundexercise-induced oxidative–nitrosative–inflammatorystress. Thus, we cannot unequivocally exclude thepossibility that free radicals when present in ‘excess’do indeed contribute to increased microvasculardeoxygenation and the premature termination ofexercise during hypoxia. Equally, it remains unclearto what extent the more distal systemic biomarkers ofoxidative–nitrosative–inflammatory stress reflect localformation at the microvascular level, where PO2 ispredicted to be the most reduced (Richardson et al.2006). This is likely to be underestimated, given theinverse relationship between tissue PO2 and free radicalformation (Bailey et al. 2004b, 2011b) and the potentialtime delay associated with systemic spillover, even thougha net output of comparable biomarkers has been shownto occur within a single arteriovenous transit across themusculoskeletal (Bailey et al. 2004b), cerebral (Bailey et al.2009d) and pulmonary circulation (Bailey et al. 2010).More invasive exchange studies of this nature will help toclarify additional sources of free radical formation, whichare likely to be multifactorial (Powers & Jackson, 2008).In addition to the afore-mentioned mitochondrialcontribution, alternative mechanisms involvingNADPH oxidase/xanthine oxidase/phospholipase A2
activation, haem auto-oxidation and liberation ofcatalytic iron with the capacity to promote Fenton andHaber–Weiss–mediated formation of the hydroxyl radicalcannot be excluded (Vollaard et al. 2005; Bailey et al. 2006;Clanton, 2007; Jackson, 2008). To what extent each ofthese independent sources and mechanisms contributesto the collective systemic oxidative stress response remainsto be established.
Furthermore, given the linear relationship known toexist between free radical-mediated lipid peroxidationand absolute power output in normoxia (Fogartyet al. 2010), the lower values invariably achieved inhypoxia may have led to further underestimation ofthe oxidative–nitrosative–inflammatory stress response.Although it was not our original intent to disassociaterelative from absolute exercise intensity, it would be ofinterest to match work rates across inspirates in follow-upstudies. Collectively, these experimental design limitationsmay have contributed towards the lack of correlationobserved between whole-body (metabolic stress) andlocal measures (microvascular deoxygenation). Thus, itwould be of interest to repeat the present study usingmicrodialysis techniques (Pattwell et al. 2003) combinedwith serial sampling to provide more local insight intomicrovascular free radical exchange kinetics.
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society
1658 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
Table 3. Correlations
Site Cerebral Muscle
�[O2Hb] �[HHb] �[THb] OI �[O2Hb] �[HHb] �[THb] OI
Variable r P r P r P r P r P r P r P r P
Oxidative stress
�A•− −0.34 0.13 −0.31 0.16 −0.47 0.03∗ −0.07 0.77 0.25 0.27 0.08 0.74 0.41 0.06 0.09 0.68Nitrosative stress
�Total NO −0.54 0.01∗ −0.08 0.72 −0.48 0.03∗ −0.36 0.10 0.22 0.32 −0.14 0.53 0.09 0.68 0.20 0.37�NO3
− −0.54 0.01∗ −0.08 0.74 −0.47 0.03∗ −0.37 0.09 0.22 0.32 −0.15 0.51 0.09 0.71 0.20 0.36�NO2
− −0.18 0.43 −0.20 0.38 −0.27 0.23 −0.02 0.94 0.14 0.54 0.17 0.44 0.39 0.07 −0.02 0.92�RSNO −0.28 0.21 −0.12 0.58 −0.30 0.18 −0.14 0.54 0.09 0.70 0.23 0.30 0.40 0.06 −0.08 0.72�3-NT −0.32 0.14 −0.11 0.64 −0.32 0.15 −0.18 0.43 −0.37 0.09 0.28 0.21 −0.11 0.64 −0.35 0.11
Inflammatory stress
�sICAM-1 0.33 0.14 0.44 0.04∗ 0.54 0.01∗ -0.02 0.93 0.48 0.02∗ −0.48 0.02∗ −0.02 0.92 0.52 0.01∗
�sVCAM-1 0.34 0.13 0.22 0.32 0.41 0.06 0.12 0.59 0.10 0.67 −0.07 0.77 0.03 0.88 0.09 0.70
Values are expressed as correlation coefficients (r) and statistical probabilities (P values) based on the relationships betweenexercise-induced (pooled normoxia + hypoxia data) changes (�, exercise minus rest) in microvascular and metabolic variables ofinterest. Abbreviations: A•−, ascorbate radical; NO, nitric oxide; NO2
−, nitrite; NO3−, nitrate; 3-NT, 3-nitrotyrosine; [O2Hb], [HHb] and
[THb] represent concentration changes (�) in oxygenated, deoxygenated and total haemoglobin; OI, oxygenation index calculatedas �[O2Hb] − �[HHb]; RSNO, S-nitrosothiols; and sICAM-1/sVCAM-1, soluble intercellular/vascular adhesion molecules. ∗Significant(P < 0.05).
We took advantage of EPR spectroscopy and ozone-based chemiluminescence, given that they representthe most direct techniques currently available for themolecular detection of free radicals and NO metabolites,respectively. In contrast, the indirect assessment ofmicrovascular deoxygenation via continuous-wave NIRSis not without limitations, the most notable beingthe inability to quantify the extraneous blood flowcontribution from the skin/scalp and failure to distinguishthe proportion of blood within arterial or venouscompartments of the vascular bed, which is invariablysubject to redistribution (Rooks et al. 2010). Equally,blood flow within the skeletal (Richardson et al. 2001)and cerebral microvasculature (MacIntosh et al. 2014) isheterogenous; thus, our findings provide only local insightwithin predefined regions of interest.
Interestingly, a recent study employing quantitativemultichannel time-resolved NIRS demonstrated that thespatial heterogeneity of absolute �[HHb] in skeletalmuscle was reduced during hypoxia, and this correlatedwith impaired oxidative energy provision at the onset ofexercise (Bowen et al. 2013). It would be interesting tofollow the present study up using this approach, given thataltered heterogeneity may have contributed to the limitedchanges observed during hypoxia. Furthermore, hypoxiacauses a compensatory vasodilatation (Casey & Joyner,2012), and the corresponding hyperaemia may haveinfluenced both our metabolic and our haemodynamicfindings equally (Bailey et al. 2007).
Although beyond the scope of the present study, serialblood sampling combined with Duplex ultrasound forthe local assessment of blood flow and to assist with thenormalization of NIRS signals, given differences in adiposetissue thickness (Bowen et al. 2013) not accounted for inthe present study, and capnography for the continuoussampling of end-tidal PCO2 would have provided moredetailed insight into the metabolic–haemodynamic kineticresponse. Finally, given the general limitations associatedwith statistical power (as highlighted during retrospectiveanalysis) and the correlational approach that fails todisassociate cause from effect, further studies employinglarger sample sizes and antioxidant prophylaxis arerecommended.
To what extent antioxidants would impact localmicrovascular or systemic oxygenation remains unclear,given our previous observations. Prophylactic dietaryantioxidant supplementation has been shown to increasesystemic oxygenation in young participants exposedto the hypoxia of terrestrial high altitude (Bailey &Davies, 2001), whereas precisely the same cocktail hasbeen shown to suppress exercise-induced vasodilatation(Richardson et al. 2007). While this apparent paradoxis difficult to resolve, it highlights the duality offree radicals owing to their physiologically adaptiveeffects in controlled, albeit undefined concentrationsand pathophysiologically maladaptive effects when thisthreshold is exceeded (Clanton, 2005; Bailey et al.2013).
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1659Free radicals and oxygen transport
Conclusion
In conclusion, altered free radical metabolismcannot explain the more pronounced microvasculardeoxygenation and corresponding lower VO2max observedin hypoxia. Given that the present model of hypoxiafailed to compound exercise-induced oxidative–nitrosative–inflammatory stress further, follow-upstudies employing alternative models, larger samplesizes, microvascular blood sampling and antioxidantprophylaxis are required to determine whether asystemic increase in free radicals does indeed contributeto increased microvascular deoxygenation and thepremature termination of exercise during hypoxia.
Figure 4. Lack of relationship between hypoxia-inducedchanges in microvascular deoxygenation and the systemicoxidative stress response during maximal exerciseValues were calculated as the differences (DIFF) between thenormoxic and hypoxic values observed at maximal exerciserelative to the resting control states for cerebral (A) andmuscle tissue (B). Abbreviation: �[HHb]/A•−, concentrationchanges in deoxygenated haemoglobin/ascorbate radical.
References
Allen DG, Lamb GD & Westerblad H (2008). Skeletal musclefatigue: cellular mechanisms. Physiol Rev 88, 287–332.
Amann M, Eldridge MW, Lovering AT, Stickland MK, PegelowDF & Dempsey JA (2006). Arterial oxygenation influencescentral motor output and exercise performance via effects onperipheral locomotor muscle fatigue in humans. J Physiol575, 937–952.
Amann M, Romer LM, Subudhi AW, Pegelow DF & DempseyJA (2007). Severity of arterial hypoxaemia affects the relativecontributions of peripheral muscle fatigue to exerciseperformance in healthy humans. J Physiol 581, 389–403.
Bailey D, Ainslie P, Jackson S, Richardson R & Ghatei M(2004a). Evidence against redox regulation of energyhomoeostasis in humans at high altitude. Clin Sci (Lond)107, 589–600.
Bailey DM, Bartsch P, Knauth M & Baumgartner RW (2009a).Emerging concepts in acute mountain sickness andhigh-altitude cerebral edema: from the molecular to themorphological. Cell Mol Life Sci 66, 3583–3594.
Bailey DM & Davies B (2001). Acute mountain sickness;prophylactic benefits of antioxidant vitaminsupplementation at high altitude. High Altitude Med Biol 2,21–29.
Bailey DM, Davies B & Young IS (2001). Intermittent hypoxictraining: implications for lipid peroxidation induced byacute normoxic exercise in active men. Clin Sci (Lond) 101,465–475.
Bailey DM, Dehnert C, Luks AM, Menold E, Castell C,Schendler G, Faoro V, Gutowski M, Evans KA, Taudorf S,James PE, McEneny J, Young IS, Swenson ER, Mairbaurl H,Bartsch P & Berger MM (2010). High-altitude pulmonaryhypertension is associated with a free radical-mediatedreduction in pulmonary nitric oxide bioavailability. J Physiol588, 4837–4847.
Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L,Gutowski M, Kewley E, McCord JM, Møller K & AinsliePN (2009b). Altered free radical metabolism in acutemountain sickness: implications for dynamic cerebralautoregulation and blood–brain barrier function. J Physiol587, 73–85.
Bailey DM, Evans KA, McEneny J, Young IS, Hullin DA, JamesPE, Ogoh S, Ainslie PN, Lucchesi C, Rockenbauer A, CulcasiM & Pietri S (2011a). Exercise-induced oxidative-nitrosativestress is associated with impaired dynamic cerebralautoregulation and blood–brain barrier leakage. Exp Physiol96, 1196–1207.
Bailey DM, Morris-Stiff G, McCord JM & Lewis MH (2007).Has free radical release across the brain after carotidendarterectomy traditionally been underestimated?:Significance of reperfusion hemodynamics. Stroke 38,1946–1948.
Bailey DM, Rimoldi SF, Rexhaj E, Pratali L, Salmon CS, VillenaM, McEneny J, Young IS, Nicod P, Allemann Y, Scherrer U &Sartori C (2013). Oxidative-nitrosative stress and systemicvascular function in highlanders with and withoutexaggerated hypoxemia. Chest 143, 444–451.
Bailey DM, Roukens R, Knauth M, Kallenberg K, Christ S,Mohr A, Genius J, Storch-Hagenlocher B, Meisel F,
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

1660 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
McEneny J, Young IS, Steiner T, Hess K & Bartsch P(2006). Free radical-mediated damage to barrier function isnot associated with altered brain morphology inhigh-altitude headache. J Cereb Blood Flow Metab 26,99–111.
Bailey DM, Taudorf S, Berg RM, Lundby C, Pedersen BK,Rasmussen P & Møller K (2011b). Cerebral formation of freeradicals during hypoxia does not cause structural damageand is associated with a reduction in mitochondrial PO2;evidence of O2-sensing in humans? J Cereb Blood Flow Metab31, 1020–1026.
Bailey DM, Taudorf S, Berg RMG, Jensen LT, Lundby C, EvansKA, James PE, Pedersen BK & Moller K (2009c).Transcerebral exchange kinetics of nitrite and calcitoningene-related peptide in acute mountain sickness: evidenceagainst trigeminovascular activation? Stroke 40,2205–2208.
Bailey DM, Taudorf S, Berg RMG, Lundby C, McEneny J,Young IS, Evans KA, James PE, Shore A, Hullin DA, McCordJM, Pedersen BK & Moller K (2009d). Increased cerebraloutput of free radicals during hypoxia: implications for acutemountain sickness? Am J Physiol Regul Integr Comp Physiol297, R1283–R1292.
Bailey DM, Young IS, McEneny J, Lawrenson L, Kim J, Barden J& Richardson RS (2004b). Regulation of free radical outflowfrom an isolated muscle bed in exercising humans. Am JPhysiol Heart Circ Physiol 287, H1689–H1699.
Bouissou P, Peronnet F, Brisson G, Helie R & Ledoux M (1986).Metabolic and endocrine responses to graded exercise underacute hypoxia. Eur J Appl Physiol Occup Physiol 55,290–294.
Bowen TS, Rossiter HB, Benson AP, Amano T, Kondo N,Kowalchuk JM & Koga S (2013). Slowed oxygen uptakekinetics in hypoxia correlate with the transient peak andreduced spatial distribution of absolute skeletal muscledeoxygenation. Exp Physiol 98, 1585–1596.
Buettner GR (1993). The pecking order of free radicals andantioxidants: lipid peroxidation, α-tocopherol, andascorbate. Arch Biochem Biophys 300, 535–543.
Calbet JA, Boushel R, Radegran G, Søndergaard H, Wagner PD& Saltin B (2003). Determinants of maximal oxygen uptakein severe acute hypoxia. Am J Physiol Regul Integr CompPhysiol 284, R291–R303.
Casey DP & Joyner MJ (2012). Compensatory vasodilatationduring hypoxic exercise: mechanisms responsible formatching oxygen supply to demand. J Physiol 590,6321–6326.
Clanton T (2005). Yet another oxygen paradox. J Appl Physiol99, 1245–1246.
Clanton TL (2007). Hypoxia-induced reactive oxygen speciesformation in skeletal muscle. J Appl Physiol 102,2379–2388.
Davison GW, Ashton T, McEneny J, Young IS, Davies B &Bailey DM (2012). Critical difference applied toexercise-induced oxidative stress: the dilemma ofdistinguishing biological from statistical change. J PhysiolBiochem 68, 377–384.
Davison GW, Morgan RM, Hiscock N, Garcia JM, Grace F,Boisseau N, Davies B, Castell L, McEneny J, Young IS, Hullin
D, Ashton T & Bailey DM (2006). Manipulation of systemicoxygen flux by acute exercise and normobaric hypoxia:implications for reactive oxygen species generation. Clin Sci(Lond) 110, 133–141.
DeLorey DS, Kowalchuk JM & Paterson DH (2003).Relationship between pulmonary O2 uptake kinetics andmuscle deoxygenation during moderate-intensity exercise. JAppl Physiol 95, 113–120.
Diederich ER, Behnke BJ, McDonough P, Kindig CA, BarstowTJ, Poole DC & Musch TI (2002). Dynamics ofmicrovascular oxygen partial pressure in contracting skeletalmuscle of rats with chronic heart failure. Cardiovasc Res 56,479–486.
Duncan A, Meek JH, Clemence M, Elwell CE, Tyszczuk L, CopeM & Delpy DT (1995). Optical pathlength measurements onadult head, calf and forearm and the head of the newborninfant using phase resolved optical spectroscopy. Phys MedBiol 40, 295–304.
Eltzschig HK & Carmeliet P (2011). Hypoxia andinflammation. N Engl J Med 364, 656–665.
Evans M, Anderson RA, Graham J, Ellis GR, Morris K, DaviesS, Jackson SK, Lewis MJ, Frenneaux MP & Rees A (2000).Ciprofibrate therapy improves endothelial function andreduces postprandial lipemia and oxidative stress in Type 2Diabetes Mellitus. Circulation 101, 1773–1779.
Ewing JF & Janero DR (1998). Specific S-nitrosothiol(thionitrite) quantification as solution nitrite aftervanadium(III) reduction and ozone-chemiluminescentdetection. Free Radic Biol Med 25, 621–628.
Ferrari M, Muthalib M & Quaresima V (2011). The use ofnear-infrared spectroscopy in understanding skeletal musclephysiology: recent developments. Philos Trans R Soc A MathPhys Eng Sci 369, 4577–4590.
Ferreira LF, Padilla DJ, Williams J, Hageman KS, Musch TI &Poole DC (2006). Effects of altered nitric oxide availabilityon rat muscle microvascular oxygenation duringcontractions. Acta Physiol (Oxf) 186, 223–232.
Ferreira LF & Reid MB (2008). Muscle-derived ROS andthiol regulation in muscle fatigue. J Appl Physiol 104,853–860.
Fogarty MC, Hughes CM, Burke G, Brown JC, Trinick TR,Duly E, Bailey DM & Davison GW (2010). Exercise-inducedlipid peroxidation: implications for deoxyribonucleic aciddamage and systemic free radical generation. Environ MolMutagen 52, 35–42.
Frenette PS & Wagner DD (1996). Adhesion molecules —blood vessels and blood cells. N Engl J Med 335, 43–45.
Gandevia SC (2001). Spinal and supraspinal factors in humanmuscle fatigue. Physiol Rev 81, 1725–1789.
Gatterer H, Greilberger J, Philippe M, Faulhaber M, Djukic R &Burtscher M (2013). Short-term supplementation withalpha-ketoglutaric acid and 5-hydroxymethylfurfural doesnot prevent the hypoxia induced decrease of exerciseperformance despite attenuation of oxidative stress. Int JSports Med 34, 1–7.
Grassi B, Pogliaghi S, Rampichini S, Quaresima V, Ferrari M,Marconi C & Cerretelli P (2003). Muscle oxygenation andpulmonary gas exchange kinetics during cycling exerciseon-transitions in humans. J Appl Physiol 95, 149–158.
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

Exp Physiol 99.12 (2014) pp 1648–1662 1661Free radicals and oxygen transport
Gryglewski RJ, Palmer RM & Moncada S (1986). Superoxideanion is involved in the breakdown of endothelium-derivedvascular relaxing factor. Nature 320, 454–456.
Hirasawa A, Yanagisawa S, Tanaka N, Funane T, Kiguchi M,Sørensen H, Secher NH & Ogoh S (2014). Influence of skinblood flow and source-detector distance on near-infraredspectroscopy-determined cerebral oxygenation in humans.Clin Physiol Funct Imaging, doi: 10.1111/cpf.12156.
Jackson MJ (2008). Free radicals generated by contractingmuscle: by-products of metabolism or key regulators ofmuscle function? Free Radic Biol Med 44,132–141.
Jasper HH (1958). The ten-twenty electrode system of theinternational federation. Electroencephalogr ClinNeurophysiol 10, 371–375.
Kayser B, Narici M, Binzoni T, Grassi B & Cerretelli P (1994).Fatigue and exhaustion in chronic hypobaric hypoxia:influence of exercising muscle mass. J Appl Physiol 76,634–640.
McCully KK & Hamaoka T (2000). Near-infrared spectroscopy:what can it tell us about oxygen saturation in skeletalmuscle? Exerc Sport Sci Rev 28, 123–127.
MacIntosh BJ, Crane DE, Sage MD, Rajab AS, Donahue MJ,McIlroy WE & Middleton LE (2014). Impact of a single boutof aerobic exercise on regional brain perfusion and activationresponses in healthy young adults. PLoS One 9, e85163.
Mantovani G, Maccio A, Madeddu C, Gramignano G, LussoMR, Serpe R, Massa E, Astara G & Deiana L (2006). Aphase II study with antioxidants, both in the diet andsupplemented, pharmaconutritional support, progestagen,and anti-cyclooxygenase-2 showing efficacy and safety inpatients with cancer-related anorexia/cachexia andoxidative stress. Cancer Epidemiol Biomarkers Prev 15,1030–1034.
Marsh SA & Coombes JS (2005). Exercise and the endothelialcell. Int J Cardiol 99, 165–169.
Nauser T & Koppenol WH (2002). The rate constant of thereaction of superoxide with nitrogen monoxide: approachingthe diffusion limit. J Phys Chem A 106, 4084–4086.
Noakes TD, Peltonen JE & Rusko HK (2001). Evidence that acentral governor regulates exercise performance duringacute hypoxia and hyperoxia. J Exp Biol 204, 3225–3234.
Pattwell D, Ashton T, McArdle A, Griffiths RD & Jackson MJ(2003). Ischemia and reperfusion of skeletal muscle lead tothe appearance of a stable lipid free radical in the circulation.Am J Physiol Heart Circ Physiol 284, H2400–H2404.
Powers SK & Jackson MJ (2008). Exercise-induced oxidativestress: cellular mechanisms and impact on muscle forceproduction. Physiol Rev 88, 1243–1276.
Quaresima V & Ferrari M (2009). Muscle oxygenation bynear-infrared-based tissue oximeters. J Appl Physiol 107, 371;author reply 372–373.
Rasmussen P, Dawson EA, Nybo L, van Lieshout JJ, Secher NH& Gjedde A (2007). Capillary-oxygenation-level-dependentnear-infrared spectrometry in frontal lobe of humans. JCereb Blood Flow Metab 27, 1082–1093.
Reid MB & Moylan JS (2011). Beyond atrophy: redoxmechanisms of muscle dysfunction in chronic inflammatorydisease. J Physiol 589, 2171–2179.
Richardson RS, Donato AJ, Uberoi A, Wray DW, Lawrenson L,Nishiyama S & Bailey DM (2007). Exercise-induced brachialartery vasodilation: role of free radicals. Am J Physiol HeartCirc Physiol 292, H1516–H1522.
Richardson RS, Duteil S, Wary C, Wray DW, Hoff J & CarlierPG (2006). Human skeletal muscle intracellularoxygenation: the impact of ambient oxygen availability. JPhysiol 571, 415–424.
Richardson RS, Haseler LJ, Nygren AT, Bluml S & Frank LR(2001). Local perfusion and metabolic demand duringexercise: a noninvasive MRI method of assessment. J ApplPhysiol 91, 1845–1853.
Rooks CR, Thom NJ, McCully KK & Dishman RK (2010).Effects of incremental exercise on cerebral oxygenationmeasured by near-infrared spectroscopy: a systematicreview. Progr Neurobiol 92, 134–150.
Schumacker PT (2011). Lung cell hypoxia: role ofmitochondrial reactive oxygen species signaling in triggeringresponses. Proc Am Thorac Soc 8, 477–484.
Subudhi AW, Dimmen AC & Roach RC (2007). Effects of acutehypoxia on cerebral and muscle oxygenation duringincremental exercise. J Appl Physiol 103, 177–183.
Subudhi AW, Lorenz MC, Fulco CS & Roach RC (2008).Cerebrovascular responses to incremental exercise duringhypobaric hypoxia: effect of oxygenation on maximalperformance. Am J Physiol Heart Circ Physiol 294,H164–H171.
Subudhi AW, Olin JT, Dimmen AC, Polaner DM, Kayser B &Roach RC (2011). Does cerebral oxygen delivery limitincremental exercise performance? J Appl Physiol 111,1727–1734.
Van Beekvelt MC, Colier WN, Wevers RA & Van Engelen BG(2001). Performance of near-infrared spectroscopy inmeasuring local O2 consumption and blood flow in skeletalmuscle. J Appl Physiol 90, 511–519.
van der Zee P, Cope M, Arridge SR, Essenpreis M, Potter LA,Edwards AD, Wyatt JS, McCormick DC, Roth SC, ReynoldsEO, Delpy DT (1992). Experimentally measured opticalpathlengths for the adult head, calf and forearm and thehead of the newborn infant as a function of inter optodespacing. Adv Exp Med Biol 316, 143–153.
Verges S, Rupp T, Jubeau M, Wuyam B, Esteve F, Levy P, PerreyS & Millet GY (2012). Cerebral perturbations during exercisein hypoxia. Am J Physiol Regul Integr Comp Physiol 302,R903–R916.
Vogiatzis I, Louvaris Z, Habazettl H, Athanasopoulos D,Andrianopoulos V, Cherouveim E, Wagner H, Roussos C,Wagner PD & Zakynthinos S (2011). Frontal cerebral cortexblood flow, oxygen delivery and oxygenation duringnormoxic and hypoxic exercise in athletes. J Physiol 589,4027–4039.
Vollaard NB, Shearman JP & Cooper CE (2005).Exercise-induced oxidative stress: myths, realities andphysiological relevance. Sports Med 35, 1045–1062.
Wagner PD (2010). The physiological basis of reduced VO2max
in Operation Everest II. High Altitude Med Biol 11, 209–215.Wang J, Brown MA, Tam SH, Chan MC & Whitworth JA
(1997). Effects of diet on measurement of nitric oxidemetabolites. Clin Exp Pharmacol Physiol 24, 418–420.
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society

1662 Exp Physiol 99.12 (2014) pp 1648–1662J. D. S. Woodside and others
Williams JR (2008). The Declaration of Helsinki and publichealth. Bull World Health Organ 86, 650–652.
Williams NH & Yandell JK (1982). Outer-sphereelectron-transfer reactions of ascorbic anions. Aust J Chem35, 1133–1144.
Additional information
Competing interests
None declared.
Author contributions
D.M.B. was the Principal Investigator and, in conjunction withJ.D.S.W. (PhD student), led the study conception/design and
acquisition/analysis/interpretation of data. D.M.B. wrote theoriginal manuscript and revisions thereof. M.G., L.F., P.E.J.,J.M. and I.S.Y. made substantial contributions to data analysis.S.O. revised the original manuscript critically for importantintellectual content. All authors gave final approval of the versionto be submitted.
Funding
This study was funded in part by the British Cycling Federation.
Acknowledgements
We appreciate the cheerful co-operation of study participants.
C© 2014 The Authors. Experimental Physiology C© 2014 The Physiological Society
Related Documents











