Systemic Cardiac Amyloidoses Disease Profiles and Clinical Courses of the 3 Main Types Claudio Rapezzi, MD; Giampaolo Merlini, MD; Candida C. Quarta, MD; Letizia Riva, MD; Simone Longhi, MD; Ornella Leone, MD; Fabrizio Salvi, MD; Paolo Ciliberti, MD; Francesca Pastorelli, MD; Elena Biagini, MD; Fabio Coccolo, MD; Robin M.T. Cooke, MA; Letizia Bacchi-Reggiani, MSc, MStat; Diego Sangiorgi, MStat; Alessandra Ferlini, MD; Michele Cavo, MD; Elena Zamagni, MD; Maria Luisa Fonte, MD; Giovanni Palladini, MD; Francesco Salinaro, MD; Francesco Musca, MD; Laura Obici, MD; Angelo Branzi, MD; Stefano Perlini, MD Background—Most studies of amyloidotic cardiomyopathy consider as a single entity the 3 main systemic cardiac amyloidoses: acquired monoclonal immunoglobulin light-chain (AL); hereditary, mutated transthyretin-related (ATTRm); and wild-type transthyretin-related (ATTRwt). In this study, we compared the diagnostic/clinical profiles of these 3 types of systemic cardiac amyloidosis. Methods and Results—We conducted a longitudinal study of 233 patients with clear-cut diagnosis by type of cardiac amyloidosis (AL, n157; ATTRm, n61; ATTRwt, n15) at 2 large Italian centers providing coordinated amyloidosis diagnosis/management facilities since 1990. Average age at diagnosis was higher in AL than in ATTRm patients; all ATTRwt patients except 1 were elderly men. At diagnosis, mean left ventricular wall thickness was higher in ATTRwt than in ATTRm and AL. Left ventricular ejection fraction was moderately depressed in ATTRwt but not in AL or ATTRm. ATTRm patients less often displayed low QRS voltage (25% versus 60% in AL; P0.0001) or low voltage-to-mass ratio (1.10.5 versus 0.90.5; P0.0001). AL patients appeared to have greater hemodynamic impairment. On multivariate analysis, ATTRm was a strongly favorable predictor of survival, and ATTRwt predicted freedom from major cardiac events. Conclusions—AL, ATTRm, and ATTRwt should be considered 3 different cardiac diseases, probably characterized by different pathophysiological substrates and courses. Awareness of the diversity underlying the cardiac amyloidosis label is important on several levels, ranging from disease classification to diagnosis and clinical management. (Circulation. 2009;120:1203-1212.) Key Words: amyloid cardiomyopathy echocardiography electrocardiography myocardium myocytes, cardiac T he term amyloidosis describes a group of rare diseases characterized by extracellular accumulation of fibrillary proteins, leading to loss of normal tissue architecture. 1 Amy- loidosis may be systemic or localized and is currently classified according to the type of precursor protein. 2 The 3 most frequent and clinically challenging types of systemic amyloidosis are acquired monoclonal immunoglobulin light- chain amyloidosis (AL), characterized by clonal plasma cells in the bone marrow that produce the immunoglobulin lights chains of the fibrillary deposits; the hereditary, transthyretin (TTR)-related form (ATTRm), which can be caused by 100 mutations of TTR, a transport protein synthesized mainly by the liver; and wild-type (nonmutant) TTR-related amyloid- osis (ATTRwt) systemic “senile” amyloidosis, which af- fects mainly the hearts of elderly men. In all 3 forms, myocardial involvement is frequent and has major clinical implications. 3,4 Despite the intrinsic pathogenic heteroge- neity of cardiac amyloidosis, most available clinical/ instrumental studies address the disease as a single entity. Comparative studies are hampered by the wide variety of specialty settings in which amyloidosis patients can present. Correct recognition of cardiac amyloidosis and its Continuing medical education (CME) credit is available for this article. Go to http://cme.ahajournals.org to take the quiz. Received December 23, 2008; accepted July 17, 2009. From the Institute of Cardiology (C.R., C.C.Q., L.R., S.L., P.C., E.B., F.C., R.M.T.C., L.B.-R., D.S., A.B.), Department of Pathology (O.L.), and Institute of Hematology (M.C., E.Z.), University of Bologna, and S. Orsola-Malpighi Hospital, Bologna; Center for Amyloidosis, Fondazione IRCCS San Matteo and University of Pavia, Pavia (G.M., M.L.F., G.P., F. Salinaro, F.M., L.O., S.P.); Department of Neurology, Bellaria Hospital, Bologna (F. Salvi, F.P.); and Department of Diagnostic and Experimental Medicine, Section of Medical Genetics, University of Ferrara, Ferrara (A.F.), Italy. Correspondence to Professor Claudio Rapezzi, Istituto di Cardiologia, Policlinico S. Orsola-Malpighi, Via Massarenti n 9, 40125 Bologna, Italy. E-mail [email protected] © 2009 American Heart Association, Inc. Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.108.843334 1203 Heart Failure by guest on April 15, 2016 http://circ.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Systemic Cardiac AmyloidosesDisease Profiles and Clinical Courses of the 3 Main Types
Claudio Rapezzi, MD; Giampaolo Merlini, MD; Candida C. Quarta, MD; Letizia Riva, MD;Simone Longhi, MD; Ornella Leone, MD; Fabrizio Salvi, MD; Paolo Ciliberti, MD;
Francesca Pastorelli, MD; Elena Biagini, MD; Fabio Coccolo, MD; Robin M.T. Cooke, MA;Letizia Bacchi-Reggiani, MSc, MStat; Diego Sangiorgi, MStat; Alessandra Ferlini, MD;
Michele Cavo, MD; Elena Zamagni, MD; Maria Luisa Fonte, MD; Giovanni Palladini, MD;Francesco Salinaro, MD; Francesco Musca, MD; Laura Obici, MD;
Angelo Branzi, MD; Stefano Perlini, MD
Background—Most studies of amyloidotic cardiomyopathy consider as a single entity the 3 main systemic cardiacamyloidoses: acquired monoclonal immunoglobulin light-chain (AL); hereditary, mutated transthyretin-related (ATTRm);and wild-type transthyretin-related (ATTRwt). In this study, we compared the diagnostic/clinical profiles of these 3 typesof systemic cardiac amyloidosis.
Methods and Results—We conducted a longitudinal study of 233 patients with clear-cut diagnosis by type of cardiacamyloidosis (AL, n�157; ATTRm, n�61; ATTRwt, n�15) at 2 large Italian centers providing coordinated amyloidosisdiagnosis/management facilities since 1990. Average age at diagnosis was higher in AL than in ATTRm patients; allATTRwt patients except 1 were elderly men. At diagnosis, mean left ventricular wall thickness was higher in ATTRwtthan in ATTRm and AL. Left ventricular ejection fraction was moderately depressed in ATTRwt but not in AL orATTRm. ATTRm patients less often displayed low QRS voltage (25% versus 60% in AL; P�0.0001) or lowvoltage-to-mass ratio (1.1�0.5 versus 0.9�0.5; P�0.0001). AL patients appeared to have greater hemodynamicimpairment. On multivariate analysis, ATTRm was a strongly favorable predictor of survival, and ATTRwt predictedfreedom from major cardiac events.
Conclusions—AL, ATTRm, and ATTRwt should be considered 3 different cardiac diseases, probably characterized bydifferent pathophysiological substrates and courses. Awareness of the diversity underlying the cardiac amyloidosis labelis important on several levels, ranging from disease classification to diagnosis and clinical management. (Circulation.2009;120:1203-1212.)
Key Words: amyloid � cardiomyopathy � echocardiography � electrocardiography � myocardium� myocytes, cardiac
The term amyloidosis describes a group of rare diseasescharacterized by extracellular accumulation of fibrillary
proteins, leading to loss of normal tissue architecture.1 Amy-loidosis may be systemic or localized and is currentlyclassified according to the type of precursor protein.2 The 3most frequent and clinically challenging types of systemicamyloidosis are acquired monoclonal immunoglobulin light-chain amyloidosis (AL), characterized by clonal plasma cellsin the bone marrow that produce the immunoglobulin lightschains of the fibrillary deposits; the hereditary, transthyretin(TTR)-related form (ATTRm), which can be caused by �100
mutations of TTR, a transport protein synthesized mainly bythe liver; and wild-type (nonmutant) TTR-related amyloid-osis (ATTRwt) systemic “senile” amyloidosis, which af-fects mainly the hearts of elderly men. In all 3 forms,myocardial involvement is frequent and has major clinicalimplications.3,4 Despite the intrinsic pathogenic heteroge-neity of cardiac amyloidosis, most available clinical/instrumental studies address the disease as a single entity.Comparative studies are hampered by the wide varietyof specialty settings in which amyloidosis patients canpresent. Correct recognition of cardiac amyloidosis and its
Continuing medical education (CME) credit is available for this article. Go to http://cme.ahajournals.org to take the quiz.Received December 23, 2008; accepted July 17, 2009.From the Institute of Cardiology (C.R., C.C.Q., L.R., S.L., P.C., E.B., F.C., R.M.T.C., L.B.-R., D.S., A.B.), Department of Pathology (O.L.), and
Institute of Hematology (M.C., E.Z.), University of Bologna, and S. Orsola-Malpighi Hospital, Bologna; Center for Amyloidosis, Fondazione IRCCS SanMatteo and University of Pavia, Pavia (G.M., M.L.F., G.P., F. Salinaro, F.M., L.O., S.P.); Department of Neurology, Bellaria Hospital, Bologna (F. Salvi,F.P.); and Department of Diagnostic and Experimental Medicine, Section of Medical Genetics, University of Ferrara, Ferrara (A.F.), Italy.
Correspondence to Professor Claudio Rapezzi, Istituto di Cardiologia, Policlinico S. Orsola-Malpighi, Via Massarenti n 9, 40125 Bologna, Italy. [email protected]
© 2009 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.108.843334
1203
Heart Failure
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

various types remains a challenge, and the condition maybe vastly underdiagnosed.3 The aim of this study was tocompare the diagnostic/clinical profiles of different typesof systemic cardiac amyloidosis.
Clinical Perspective on p 1212
MethodsSetting and Study DesignWe conducted a multicenter longitudinal study of cardiac amyloid-osis based on data pooled from 2 large Italian centers. Since 1990,the cardiology, hematology, nephrology, neurology, and geneticsdepartments at our hospitals have provided coordinated centers forthe diagnosis and management of systemic amyloidosis. All data arecollated in centralized databases. Family screening is recommendedto all ATTRm patients.
We screened all patients diagnosed with systemic amyloidosiswho presented to the Bologna center from 1990 to May 2008 and tothe Pavia center from 2003 to June 2004. Consecutive patients withechocardiographically defined amyloidotic cardiomyopathy5 wereconsidered eligible for analysis.
We compared the 3 groups of patients (AL, ATTRwt, andATTRm) in terms of clinical/instrumental profiles at baseline (ie,first evaluation at either center) and clinical outcome. At presenta-tion, all patients provided informed consent for anonymous publica-tion of scientific data. In our country, formal ethics approval was notapplicable for this observational study involving only routinelyperformed procedures.
Diagnostic DefinitionsDiagnosis of systemic amyloidosis was defined by histologicaldocumentation of Congo Red staining and apple-green birefringenceunder cross-polarized light in at least 1 involved organ.6 Amyloidoticcardiomyopathy was defined echocardiographically as end-diastolicthickness of the interventricular septum �1.2 cm in the absence ofany other cause of ventricular hypertrophy.5 Clear-cut distinctionbetween TTR-related and AL amyloidosis was based on genotypingand/or immunohistochemistry.7,8 Diagnosis of familial ATTRm wasdefined by a documented TTR mutation at DNA analysis followingprocedures described elsewhere,9 ATTRwt by positive immunohis-tochemistry for TTR in the absence of any TTR mutation at DNAanalysis,10 and AL by the presence of monoclonal plasma cells in thebone marrow, plus both negative immunohistochemistry for TTRand the absence of any TTR mutation on DNA analysis.11,12
Kidney involvement was defined as the presence of 24-hour urineprotein excretion �0.5 g/d,5 and renal insufficiency was definedas glomerular filtration rate �60 mL/min. The definition ofperipheral nervous system involvement was based on character-istic neurological signs and symptoms (typical symmetric ascend-ing sensorimotor peripheral neuropathy). Autonomic impairmentwas defined by the presence of orthostatic hypotension, gastric-emptying disorder, pseudoobstruction, and voiding dysfunctionnot related to direct organ infiltration.5
Instrumental DefinitionsECG and echocardiographic measures were based on standarddefinitions.13,14 Left ventricular (LV) mass was calculated accordingto the method of Devereux et al15 and was classified as raised when�130 g/m2 in men and �110 g/m2 in women. LV restrictive fillingpattern was defined in terms of E-wave deceleration time �150 msaccompanied by E/A wave ratio �2.5 on pulsed Doppler echocar-diography.16 Voltage-to-mass ratio was calculated as Sokolow indexdivided by the cross-sectional area of the LV wall with the formuladefined by Carroll et al.17
Hemodynamic DataAt 1 center (Bologna), systematically collected hemodynamic datawere available for patients routinely submitted to myocardial biopsy
for pathogenic diagnosis or clinical evaluation at baseline. Availabledata included mean right atrial pressure (normal, �5 mm Hg), meanpulmonary capillary wedge pressure (normal, �12 mm Hg), cardiacindex (normal, between 2.5 and 4.2 L · min�1 · m�2), and dip-plateaumorphology of the right ventricle pressure tracing.
Histology and ImmunohistochemistryHistological documentation of amyloid deposition was obtainedeither from subcutaneous adipose tissue from abdominal fat or fromendomyocardial biopsies. All samples (5 per patient) were micro-wave fixed and processed, and multiple 2-�m sections were testedfor the presence of amyloid by Congo Red staining and apple-greenbirefringence under cross-polarized light microscopy. Amyloid lo-calization was described in terms of interstitial, vascular, andendocardial involvement.18 Immunohistochemical analysis was per-formed by the labeled streptavidin-biotin method with an antibodyagainst TTR (R.P. Linke, Max Plank Institute of Biochemistry,Germany) or by immunoelectron microscopy with specific antibodyproteins (DAKO, Ely, UK).
Follow-UpFollow-up visits were planned for every 6 months (or more fre-quently if clinically appropriate). Follow-up was closed in November2008; for patients who had not attended a visit in the last 6 months,vital status was ascertained by telephone and/or by contactingreferring physicians.
Statistical AnalysisSummary statistics are expressed as mean�SD, median (interquar-tile range), or numbers (percentages). We used 1-way ANOVA orthe Pearson �2 test for comparisons of baseline data between AL,ATTRwt, and ATTRm. The Bonferroni test was used to performpairwise comparisons for variables that reached global significance.We tested selected variables chosen for their potential pathophysio-logical relevance in a series of multivariate models constructed toassess independent associations with the type (reference category,AL). Dependent variables were treated continuously (with multivar-iate linear regression), except low QRS voltage, which was treated asa binary variable (in a logistic regression model) because of itsdiagnostic relevance. Analyses of hemodynamic and histologicaldata were restricted to the Bologna subpopulation. To explorefactors that could be associated with total mortality and majoradverse cardiac events (MACEs), we conducted multivariateanalysis of a set of variables that were selected a priori on thebasis of their potential clinical or pathophysiological relevance.Analyses were conducted with SPSS version 13 (SPSS Inc,Chicago, Ill) or STATA version 9 (Stata Corp, College Station,Tex). Values of P�0.05 were considered significant.
Results
Study PopulationOf the 500 patients diagnosed with systemic amyloidosis inour centers during the study period, 233 (47%: AL, n�157;ATTRm, n�61; ATTRwt, n�15) had echocardiographicevidence of cardiac amyloidosis and were included in thisanalysis. Of note, these patients represented 51% of all 307AL patients seen in the 2 centers during the study period (69patients first evaluated in Bologna, 88 seen in Pavia); 34% of178 ATTRm patients (Bologna, n�54; Pavia, n�7); and100% of 15 ATTRwt patients (Bologna, n�14; Pavia, n�1).All patients except 1 were of Italian descent (the family of 1ATTRm patient comes from the Republic of Macedonia).
1204 Circulation September 29, 2009
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from
Except for 1 ATTRm patient (with a Val30Met mutationrecognized 8 months before referral), amyloidosis was alwaysdiagnosed within our centers. In 150 cases, amyloidoticcardiomyopathy was detected at routine echocardiographicscreening after diagnosis of systemic amyloidosis (AL,n�102; ATTRm, n�48). In 3 cases (2 AL; 1 ATTRwt)diagnosis of cardiac systemic amyloidosis was incidental. Allthe remaining 80 patients were initially diagnosed as havinghypertrophic cardiomyopathy, heart failure, or arrhythmia ata secondary or tertiary cardiological center. In these cases,suspicion of amyloidotic cardiomyopathy always stemmedfrom critical appraisal of echocardiography and ECG (andsometimes clinical findings) before the final diagnosis of AL(n�53), ATTRm (n�13), or ATTRwt (n�14). In particular,echocardiographic signs suggesting possible cardiac amyloid-osis (in addition to increased LV wall thickness) weregranular sparkling appearance of ventricular myocardium,increased thickness of atrioventricular valves or interatrialseptum, and pericardial effusion.
TTR mutations were Glu89Gln (17 patients, 10 families, 5from Sicily), Val30Met (11 patients, 7 families, 5 from centralItaly), Ile68Leu (10 patients, 10 families, all from centraleastern/northeastern Italy), Thr49Ala (7 patients, 4 families),Glu54Lys (2 patients, 1 family), Ala36Pro (2 patients, 1 family),Arg34Thr (2 patients, 1 family), Ser23Asn (2 patients, 2 fami-lies), Thr59Lys (1 patient), His88Arg (1 patient), Phe64Leu (1patient), Gly47Ala (1 patient), Val30Ala (1 patient), Ser50Arg(1 patient), Phe33Val (1 patient), and Val14Leu (1 patient), anovel TTR mutation not previously described. Table 1 summa-
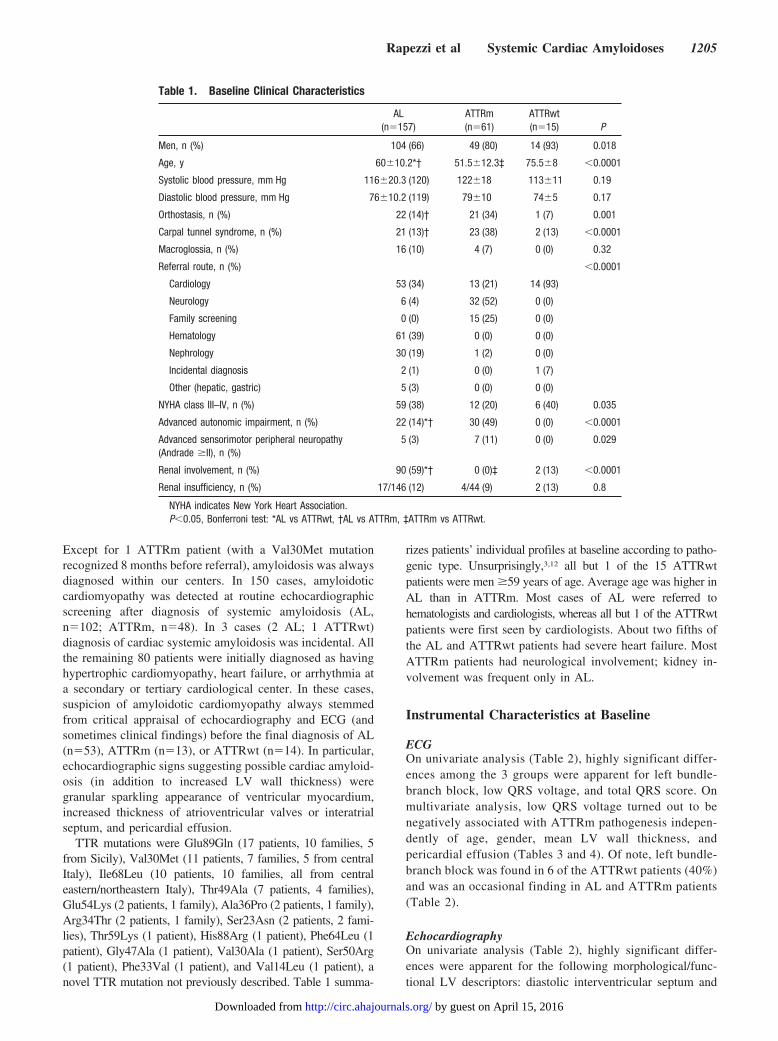
rizes patients’ individual profiles at baseline according to patho-genic type. Unsurprisingly,3,12 all but 1 of the 15 ATTRwtpatients were men �59 years of age. Average age was higher inAL than in ATTRm. Most cases of AL were referred tohematologists and cardiologists, whereas all but 1 of the ATTRwtpatients were first seen by cardiologists. About two fifths ofthe AL and ATTRwt patients had severe heart failure. MostATTRm patients had neurological involvement; kidney in-volvement was frequent only in AL.
Instrumental Characteristics at Baseline
ECGOn univariate analysis (Table 2), highly significant differ-ences among the 3 groups were apparent for left bundle-branch block, low QRS voltage, and total QRS score. Onmultivariate analysis, low QRS voltage turned out to benegatively associated with ATTRm pathogenesis indepen-dently of age, gender, mean LV wall thickness, andpericardial effusion (Tables 3 and 4). Of note, left bundle-branch block was found in 6 of the ATTRwt patients (40%)and was an occasional finding in AL and ATTRm patients(Table 2).
EchocardiographyOn univariate analysis (Table 2), highly significant differ-ences were apparent for the following morphological/func-tional LV descriptors: diastolic interventricular septum and
Table 1. Baseline Clinical Characteristics
AL(n�157)
ATTRm(n�61)
ATTRwt(n�15) P
Men, n (%) 104 (66) 49 (80) 14 (93) 0.018
Age, y 60�10.2*† 51.5�12.3‡ 75.5�8 �0.0001
Systolic blood pressure, mm Hg 116�20.3 (120) 122�18 113�11 0.19
Diastolic blood pressure, mm Hg 76�10.2 (119) 79�10 74�5 0.17
Orthostasis, n (%) 22 (14)† 21 (34) 1 (7) 0.001
Carpal tunnel syndrome, n (%) 21 (13)† 23 (38) 2 (13) �0.0001
Macroglossia, n (%) 16 (10) 4 (7) 0 (0) 0.32
Referral route, n (%) �0.0001
Cardiology 53 (34) 13 (21) 14 (93)
Neurology 6 (4) 32 (52) 0 (0)
Family screening 0 (0) 15 (25) 0 (0)
Hematology 61 (39) 0 (0) 0 (0)
Nephrology 30 (19) 1 (2) 0 (0)
Incidental diagnosis 2 (1) 0 (0) 1 (7)
Other (hepatic, gastric) 5 (3) 0 (0) 0 (0)
NYHA class III–IV, n (%) 59 (38) 12 (20) 6 (40) 0.035
Advanced autonomic impairment, n (%) 22 (14)*† 30 (49) 0 (0) �0.0001
Advanced sensorimotor peripheral neuropathy(Andrade �II), n (%)
5 (3) 7 (11) 0 (0) 0.029
Renal involvement, n (%) 90 (59)*† 0 (0)‡ 2 (13) �0.0001
Renal insufficiency, n (%) 17/146 (12) 4/44 (9) 2 (13) 0.8
NYHA indicates New York Heart Association.P�0.05, Bonferroni test: *AL vs ATTRwt, †AL vs ATTRm, ‡ATTRm vs ATTRwt.
Rapezzi et al Systemic Cardiac Amyloidoses 1205
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

posterior wall thicknesses, LV mass (in men), left atrialdiameter, LV end-systolic diameter, LV ejection fraction, andvoltage-to-mass ratio; differences in E-wave decelerationtime reached borderline significance. Frequency of increasedatrioventricular valve thickness ranged from 47% for AL to
as much as 67% for ATTRm (P�0.08). Independent associ-ations involving pathogenesis were recorded during multivar-iate analysis (Tables 3 and 4); increasing LV wall thicknesswas associated with ATTRwt, whereas voltage-to-mass ratiowas strongly associated with ATTRm.
Table 2. Baseline Instrumental Characteristics: ECG, Echocardiography, and Hemodynamic Evaluation (Bologna CenterOnly)
AL ATTRm ATTRwt P
ECG
n 157 61 15
Atrial fibrillation, n (%) 19 (12) 3 (5) 4 (27) 0.046
Pacemaker, n (%) 4 (3) 2 (3) 2 (13) 0.09
First-degree atrioventricular block, n/N (%) 26/137 (18) 15/60 (25) 5 (33) 0.34
Total QRS score, mV (n) 91.6�35.8*† (146) 112.3�33.5 (59) 119.7�37.3 (15) �0.0001
Low QRS voltage, n/N (%) 88/146 (60)† 15/60 (25) 6 (40) �0.0001
Right bundle-branch block, n/N (%) 28/146 (19) 7/60 (12) 2 (13) 0.39
Left bundle-branch block, n/N (%) 6/146 (4)* 4/60 (7)‡ 6 (40) �0.0001
Left anterior hemiblock, n/N (%) 42/146 (29) 18/60 (30) 3 (20) 0.74
LV hypertrophy (Sokolow �35 mm), n/N (%) 4/146 (3) 3/59 (5) 1 (6) 0.58
Presence of any infarct pattern, n/N (%) 101/146 (69) 41/59 (69) 10 (66) 0.98
“Ischemic pattern” (negative T waves), n/N (%) 70/146 (48) 25/59 (42) 6 (40) 0.69
QTc, ms (n) 453.1�56.6 (146) 454.6�49.4 (59) 465�37 (15) 0.72
Normal ECG, n (%) 7 (5) 6 (10) 0 (0) 0.19
Echocardiography
Diastolic interventricular septum thickness (mm) 15.8�2.8* 16.6�3.8‡ 19.7�4.1 �0.0001
Diastolic LV posterior wall thickness (mm) 14.6�2.9* 15.4�3‡ 17.9�3.8 �0.0001
Mean LV wall thickness (mm) 15.1�2.7* 16�3.2‡ 18.8�3.8 �0.0001
Left atrial diameter (mm) 46.4�7.3† 43.1�7.7‡ 49.5�6.6 0.002
LV ejection fraction (%) 52.5�13.1† 58�13‡ 44.2�15.4 �0.0001
LV ejection fraction �40%, n (%) 34 (22) 5 (8)‡ 6 (40) 0.009
LV end-diastolic diameter, mm 43.9�6.9 45.5�6.8 46.6�7.5 0.149
LV end-systolic diameter, mm (n) 29.9�6.3* (145) 31.2�8.2‡ 36.3�8.5 0.003
E-wave deceleration time, ms (n) 160.9�48.3† (89) 182.8�61.4 (55) 168.2�20.8 (15) 0.049
Restrictive filling pattern, n (%) 82/150 (55) 22/59 (37) 4/14 (29) 0.024
Pericardial effusion, n (%) 100 (64) 36 (59) 12 (80) 0.32
LV mass among men, g (n) 338.8�105.5* (84) 391.6�149.9‡ (40) 512.9�161.9 (14) �0.0001
LV mass among women, g (n) 290.8�96.3 (45) 137.6�42 (11) 281 (1) NA
Interatrial septum thickness, mm (n) 8.7�2.4 (45) 8.7�2.3 (35) 9.1�1.1(15) 0.814
Atrioventricular valve thickening, n (%) 31/66 (47) 36/54 (67) 7/14 (50) 0.08
Voltage/mass ratio (n) 0.9�0.5*† (151) 1.1�0.5‡ (60) 1.97�0.5 (15) �0.0001
Hemodynamic findings
n 43 38 12
Mean RA pressure, mm Hg 9.3�5.6§ 6.3�5.3 6.3�4.4 0.03
Raised mean RA pressure (�5 mm Hg), n (%) 31 (72)§ 14 (37) 6/12 (50) 0.006
Dip-plateau morphology of RV pressure tracing, n (%) 1 (2) 1 (3) 1/12 (8) 0.561
Mean PCWP, mm Hg 18.2�8.3§ 13�7.9 16.2�6.5 0.016
Raised mean PCWP (�12 mm Hg), n (%) 33 (77)§ 13 (34) 9 (75) �0.0001
Raised mean RA pressure and raised mean PCWP, n (%) 29 (67)§ 11 (29) 6 (50) 0.003
Cardiac index, L � min�1 � m�2 2.5�0.7 2.7�0.6 2.3�0.4 0.12
RA indicates right atrial; RV, right ventricular; and PCWP, pulmonary capillary wedge pressure.P�0.05 at Bonferroni test: *AL vs ATTRwt, †AL vs ATTRm, ‡ATTRm vs ATTRwt.§P�0.05 at Bonferroni test, AL vs ATTRm; P�0.05 at posthoc analysis.
1206 Circulation September 29, 2009
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

Hemodynamic MeasuresAnalysis of baseline hemodynamic measures was restricted toa single center (Bologna), where data were available for 43AL patients (62%), 38 ATTRm patients (70%), and 12 ATTRwtpatients (86%). Differences on univariate analysis (Table 2)among the 3 groups were apparent for mean and raised rightatrial pressure and mean and raised pulmonary capillary wedgepressure. Figure 1 reports frequencies of abnormal diastolicfunction measures (increased filling pressures, dip-plateau mor-phology, restrictive filling pattern at Doppler echocardiography).Frequency of the various abnormal findings varied considerably,ranging from 78% for at least 1 filling pressure abnormality to3% for dip-plateau morphology of the right ventricular pressuretracing (Figure 1A). Comparisons of the 3 pathogenic types(Figure 1B) showed differences for most of the single andcombined pressure measures; remarkably, abnormal values werealways most frequent in AL.
HistologyClinically driven biopsy findings were available for thesubpopulation of patients with available hemodynamicdata (see above). Frequency of vascular localization variedbetween pathogenic subtypes (AL, 34 of 43 [79%]; AT-TRm, 8 of 38 [21%]; ATTRwt, 0 of 12 [0%]; P�0.0001),being more common in AL. Frequency of inflammationalso appeared to vary (AL, 6 of 43 [14%]; ATTRm, 1 of 38[3%]; ATTRwt, 4 of 12 [33%]; P�0.014), being morecommon in ATTRwt.
OutcomeMedian duration of follow-up was 19 months (interquartilerange, 4 to 46 months) in AL, 26 months (interquartile range,
13 to 62 months) in ATTRm, and 19 months (interquartilerange, 10 to 40 months) in ATTRwt. All AL patients receivedmelphalan and/or desametazone, and 8 (5%) had high-dosechemotherapy with stem cell reperfusion; another patientreceived heart transplantation followed by stem cell reperfu-sion. Twenty-nine ATTRm patients (49%) had solid organtransplantation (orthotopic liver transplantation, n�20;planned heart-liver transplantation, n�9). The first recordedMACEs were as follows: death resulting from cardiovascularcauses in 31 AL patients (20%), 3 ATTRm patients (5%), and2 ATTRwt patients (13%); hospitalization for heart failure in48 AL patients (31%), 10 ATTRm patients (16%), and 4ATTRwt patients (27%); complete atrioventricular block in 5AL patients (3%), 3 ATTRm patients (5%), and 1 ATTRwtpatient (7%); stroke in 5 AL patients (3%); and atrialfibrillation/flutter in 17 AL patients (11%), 2 ATTRm pa-tients (3%), and 1 ATTRwt patient (7%).
Unadjusted overall survival at 2 years was 63% for AL,98% for ATTRm, and 100% for ATTRwt patients. Freedomfrom MACEs at 2 years was 51% for AL, 77% for ATTRm,and 69% for ATTRwt patients (Figures 2 and 3).
Table 5 reports the results of multivariate analysis. ATTRmand ATTRwt types were favorable predictors of overallsurvival, whereas severe heart failure (New York Heart Associ-ation class III to IV), increasing age, and mean LV wallthickness were unfavorable. With regard to freedom fromMACEs, ATTRwt turned out to be a strongly favorable predic-tor, whereas severe heart failure (New York Heart Associationclass III to IV), mean LV wall thickness, severe renal insuffi-ciency, and increasing age were unfavorable (restrictive fillingpattern did not reach significance, P�0.08).
DiscussionTo the best of our knowledge, this is the largest availablefollow-up study of cardiac amyloidosis. The results under-score profound differences between the 3 most frequentpathogenic types in terms of disease profile and long-termoutcome. Awareness of this heterogeneity may help orientaspects of the diagnostic workup of patients with suspectedcardiac amyloidosis and subsequent clinical management.
Relatively little is known about the relation between causesof amyloidosis and types and severity of cardiac involvement.Two studies from a single center focused on differencesbetween ATTRm and AL19 and between AL and ATTRwt.12
Table 3. Associations Between Baseline InstrumentalCharacteristics and Etiologic Type: Results of LogisticRegression
Low QRS Voltage
OR 95% CI P
Age (per 1 y) 0.98 0.95–1.00 0.097
Male gender 0.87 0.45–1.66 0.67
ATTRm 0.17 0.08–0.36 �0.0001
ATTRwt 0.52 0.15–1.78 0.29
Mean LV wall thickness (per 1 mm) 1.03 0.93–1.14 0.51
Pericardial effusion 1.63 0.88–3.01 0.12
CI indicates confidence interval.
Table 4. Associations Between Baseline Instrumental Characteristics and Etiologic Type: Results of Linear Regression
Increasing Mean LV Wall Thickness Increasing Voltage/Mass Ratio Decreasing LVEF
� 95% CI P � 95% CI P � 95% CI P
Age (per 1 y) 0.04 �0.009–0.08 0.117 �0.00 �0.007–0.006 0.98 �0.17 �0.39–0.06 0.16
Male gender 0.9 �0.22–2.02 0.113 �0.004 �0.16–0.15 0.96 �5.79 �11.45–0.13 0.04
ATTRm 0.87 �0.35–2.09 0.161 0.28 0.10–0.44 0.001 3.04 �2.96–9.04 0.32
ATTRwt 4.08 1.92–6.23 �0.0001 0.12 �0.17–0.42 0.81 �5.16 �15.88–5.56 0.34
Pericardial effusion 1.00 �0.05–2.06 0.06 �0.13 �0.28–0.013 0.075 0.25 �4.96–5.46 0.93
LVEF indicates LV ejection fraction; CI, confidence interval.
Rapezzi et al Systemic Cardiac Amyloidoses 1207
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

In both comparisons, AL was associated with worse progno-sis and more rapid progression of heart failure (despite greatermyocardial involvement in ATTRwt and lack of apparentdifferences in involvement between AL and ATTRm). In anindependent study focusing on survival analysis of patientswith amyloidotic cardiomyopathy, the few ATTRm patientsincluded in the series had a better outcome than the ALpatients.20 The present work describes patients with all 3 mainforms of systemic cardiac amyloidosis. The particular set-ting—2 large centers with coordinated facilities for diagnosisand management of systemic amyloidosis—facilitates thestudy of patients with each of the 3 main types of systemicamyloidosis satisfying a strict echocardiographic definition ofamyloidotic cardiomyopathy.
At presentation, the 3 groups of patients showed expected8
clinical differences (Table 1), including high prevalence of
neurological impairment in ATTRm, kidney involvement inAL, and heart failure in both AL and ATTRwt (a diseasethought to be confined to elderly men).10,12,21 The highprevalence of carpal tunnel syndrome in ATTRm supports theconcept that this condition frequently precedes diagnosis ofcardiomyopathy in these patients.22,23
Severity of cardiac amyloidosis is commonly describedin terms of ventricular wall thickness and systolic anddiastolic function. Remarkably, alterations in these 3indicators of cardiac involvement did not seem to go handin hand in the 3 subtypes (Table 2). Morphologically, LVwall thickness measures varied widely between the 3groups, with the ATTRwt group showing the highestaverage values (3 to 4 mm greater mean LV wall thicknessthan in AL or ATTRm). All patients had nondilated LV,but LV ejection fraction values varied considerably, tend-
3 (3%)
60 (65%)
55 (59%)
51 (55%)
46 (49%)
41 (44%)
73 (78%)
0 10 20 30 40 50 60 70 80 90 100
At least one marker
Either PCWP or RAP
PCWP
RAP
Both PCWP and RAP
RFP
Dip - plateau
35 (81%)
33 (77%)
31 (72%)
29 (67%)
21 (49%)
37 (86%)
1 (2%)
9 (75%)
3 (25%)
6 (50%)
6 (50%)
9 (75%)
9 (75%)
1 (8%)1 (3%)
27 (71%)
16 (42%)
13 (34%)
14 (37%)
11 (29%)
17 (47%)
0 10 20 30 40 50 60 70 80 90 100
At least one marker
Either PCWP or RAP
PCWP
RAP
Both PCWP and RAP
RFP
Dip - plateau
ATTRm (n = 38)ATTRwt (n = 12)AL (n = 43)
p=0.56
p=0.33
p=0.003
p=0.006
p<0.0001
p<0.0001
p=0.248
A
B
Figure 1. Frequencies of abnormal dia-stolic function measures within the entirestudy population (A) and in the 3 pathoge-netic types (B). RFP indicates restrictivefilling pattern; RAP, right atrial pressure;and PCWP, pulmonary capillary wedgepressure.
1208 Circulation September 29, 2009
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

ing to be normal in ATTRm, around lower normal limits inAL, and abnormally low in ATTRwt. In parallel, mean leftatrial diameter values tended to be normal in ATTRm,slightly increased in AL, and high in ATTRwt. Interest-ingly, increased AV valve thickness appeared to be partic-
ularly frequent in the 2 TTR-related forms. A plausible expla-nation for these morphological observations regards the durationof amyloid deposition, which seems to be much more protractedin the 2 TTR-related forms.
Cardiac amyloidosis is commonly considered a form ofrestrictive cardiomyopathy18 (ie, a myocardial disease withincreased parietal stiffness, causing precipitous rises in ven-tricular pressure accompanied by only small increases involume).18 Nevertheless, the majority of patients in each ofthe 3 groups did not display restrictive filling pattern, whichis traditionally considered the key noninvasive marker ofrestrictive pathophysiology. Subanalysis of baseline hemody-namic data (one of the largest such data sets currentlyavailable) suggests further insights into ventricular functionin the 3 pathogenic forms. As many as one fifth of thehemodynamically evaluated patients did not display anyabnormal findings (Figure 1). Furthermore, the 3 groupsshowed relevant hemodynamic differences, with AL patientsmost often displaying abnormal values in the different mea-sures of diastolic function. The higher frequency of hemody-namic impairment in the AL patients contrasts with theirlesser morphological involvement (in terms of LV wallthickness values). This mismatch could plausibly be attrib-
years7654310 2
Surv
ival
100
80
60
40
20
0
ALATTRm
at riskAL
ATTRm15059
6933
3619
149
years7654310 2
Surv
ival
100
80
60
40
20
0
ALATTRm
A
B
Figure 2. Overall survival (with 95% confidence intervals) ofpatients with ATTRm and AL before (A) and after (B) adjustmentfor renal involvement or insufficiency.
years7654310 2
MA
CE
–fr
ee s
urvi
val
100
80
60
40
20
0
ALATTRm
Figure 3. MACE-free survival (with 95% confidence intervals) inpatients with ATTRm and AL after adjustment for renal involve-ment or insufficiency.
Table 5. Multivariate Analysis of Risk of Death ResultingFrom Any Cause and MACEs*
HR 95% CI P
Death resulting from any cause
Age (each incremental 1 y) 1.03 1.002–1.06 0.031
Male gender 0.94 0.48–1.51 0.59
ATTRm vs AL 0.07 0.009–0.55 0.011
ATTRwt vs AL 0.08 0.018–0.41 0.002
LVEF (each incremental 1 %) 0.98 0.96–1.003 0.09
NYHA III-IV 3.44 1.96–6.05 �0.0001
Autonomic impairment 0.76 0.41–1.40 0.38
Mean LV wall thickness(each incremental 1 mm)
1.15 1.04–1.27 0.006
Restrictive filling pattern 1.18 0.67–2.09 0.58
Renal failure 1.43 0.83–2.45 0.2
MACEs
Age (each incremental 1 y) 1.04 1.01–1.06 �0.0001
Male gender 0.86 0.52–1.41 0.54
ATTRm vs AL 0.62 0.29–1.29 0.20
ATTRwt vs AL 0.31 0.11–0.89 0.029
LVEF (each incremental 1%) 0.99 0.97–1.009 0.31
NYHA class III–IV 3.44 2.13–5.55 �0.0001
Autonomic impairment 1.08 0.65–1.77 0.77
Mean LV wall thickness(each incremental 1 mm)
1.12 1.03–1.22 0.009
Restrictive filling pattern 1.51 0.95–2.41 0.08
Renal failure 1.64 1.03–2.62 0.038
Abbreviations as in Table 4, plus HR indicates hazard ratio; NHYA, New YorkHeart Association.
*MACEs include death resulting from from cardiovascular causes, hospital-ization for heart failure, complete atrioventricular block, atrial fibrillation/flutter,myocardial infarction, and stroke.
Rapezzi et al Systemic Cardiac Amyloidoses 1209
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

uted to the well-documented direct toxicity of the immuno-globulin circulating immunoglobulin light chains in AL,8,24
along with other plausible contributory factors. For instance,it is reasonable to hypothesize that a higher frequency ofvascular localization of amyloid deposition in AL could beresponsible for myocardial ischemia, contributing to ventric-ular dysfunction. Furthermore, gradual deposition in theTTR-related forms might allow the organism time to developlocal compensatory mechanisms (a rather less likely scenarioin the rapidly developing amyloidotic cardiomyopathy of ALpatients). Different types of amyloid substances could alsolead to different degrees of myocardial damage. Measurementof biochemical markers such as brain natriuretic protein andtroponin, which have provided useful insights into myocar-dial involvement in systemic amyloidosis,25–27 could shedlight on this possibility (number considerations precluded ameaningful analysis in the present study). Interestingly, thesingle currently available study comparing these biomarkersin different forms of amyloidotic cardiomyopathy suggestedthat brain natriuretic protein and troponin values are lower inATTRm than in AL.27,28
ECG is considered a key player to orient diagnostic suspicionof cardiac amyloidosis, with low QRS voltages providing aparticularly valuable noninvasive clue. Most biopsy-provenstudies of diagnostic accuracy have been based on seriescharacterized either by incomplete pathogenic diagnosis or bysmall numbers of ATTRm patients.29 The present study regardedpatients with clear-cut diagnosis by type, including apprecia-ble numbers with TTR-related disease (both ATTRm andATTRwt); in this context, the prevalence of low QRS voltages atthe time of diagnosis was somewhat lower than in other reports(�45% overall) and was particularly low in the ATTRm subset(25%) despite greater myocardial infiltration (as indicated bymean ventricular wall thickness values). Consequently, voltage-to-mass ratios were higher in TTR-related disease than in AL.Moreover, on multivariate analysis, ATTRm actuallyturned out to be negatively associated with low QRSvoltage. Pathophysiologically, these findings depict anintriguing scenario for an infiltrative disease: higher QRSvoltages in patients with greater wall thickness (ie, in the2 TTR-related forms). A possible explanation for thisfinding could be greater myocardial cellular damage (re-gardless of wall thickness) induced by light-chain toxicityin AL. Another interesting finding regards the relativelyfrequent occurrence of left bundle-branch block in ourpatients, especially those with TTR-related disease (up to40% in ATTRwt). Taken together, these observationsunderscore the importance of not excluding a diagnosis ofamyloidotic cardiomyopathy (especially of TTR-relatedforms) on the grounds of normal QRS voltage or leftbundle-branch block.
With regard to clinical outcome, we recorded substantialdifferences in overall and MACE-free survival between the 3groups that appear to contrast with degree of morphologicalinvolvement. In particular, the group with the least morpho-logical derangement (ie, AL) had a rather aggressive clinicalcourse. In contrast, the group that showed the greatest LV
wall thickness (ie, ATTRwt) seemed to have a less aggressivecourse despite the patients’ higher average age. These possi-ble discrepancies may depend on both cardiological andnoncardiological factors. Patients with AL may be penalizedby the apparently greater severity of hemodynamic impair-ment, as well as by disease burden in other organs. Of note,on multivariate analysis, LV wall thickness appeared to be apredictor of survival, along with age and severity of heartfailure (remarkably, only weak associations were apparent forrestrictive filling pattern and LVEF).
Study LimitationsDifferences in patients’ characteristics may have beeninfluenced by diagnosis in different phases of disease(lead-time bias). Nevertheless, we believe that the find-ings, when considered together, support the underlyinghypothesis that the 3 entities should be considered distinctforms of amyloidotic cardiomyopathy. For example, thekey cross-sectional finding that AL patients appear to havegreater functional impairment at diagnosis despite lessmorphological involvement cannot be explained in termsof lead-time bias alone.
It should be stressed that this study was primarily descrip-tive. We did not attempt to take into account correlated datafrom the various families. Furthermore, P values were notadjusted for multiple comparisons, and some detected differ-ences may be fortuitous.
Referral bias is another potential concern. However,despite the different characteristics of the 2 centers (eg,greater focus on AL in Pavia and on the TTR-related formsin Bologna), stratification by center did not reveal substan-tial differences in the main study findings (data notshown). Availability of baseline hemodynamic data inmany patients from 1 center was an important featureof this study. Although relevant minorities of AL andATTRm patients did not receive routine hemodynamicevaluation, it seems unlikely that the direction of therecorded differences between AL and the other 2 groupscould be wholly attributed to selection bias. When welooked at the baseline clinical, ECG, and echocardiograph-ic characteristics of patients with available hemodynamicdata, ATTRm (but not AL) patients appeared to have moresevere heart failure compared with patients lacking hemo-dynamic data (data not shown).
ATTRm is characterized by considerable allelic geneticheterogeneity linked to several factors, including specificmutation, geographic area, and type of aggregation (en-demic/nonendemic).30 –33 Our data on ATTRm derive froma nonendemic area with a high prevalence of mutationsother than Val30Met, and the results cannot automaticallybe generalized to other geographic settings or genotypemixes.
ConclusionsWithin a group of infiltrative cardiomyopathies that are tradi-tionally considered restrictive, the degree of infiltration (assessed
1210 Circulation September 29, 2009
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

by increased wall thickness) does not seem to be associated withthe severity of restrictive hemodynamic impairment. This studyalso supports the concept that cardiomyopathies resulting fromAL, ATTRm, and ATTRwt should be considered 3 differentcardiac diseases with different pathophysiological substrates andclinical courses. In particular, AL cardiomyopathy seems to beassociated with only slightly increased wall thickness, but itappears to show the highest frequencies of hemodynamic de-rangement (mainly because of diastolic dysfunction) and lowQRS voltages on ECG, and its clinical course may be ratheraggressive. ATTRm and especially ATTRwt seem to be asso-ciated with markedly increased wall thickness but less frequentlywith hemodynamic alterations; their clinical course appears to beless aggressive than that of AL despite the patients’ higheraverage age and greater morphological abnormalities. In the 2TTR-related cardiomyopathies, voltage-to-mass ratio tends to behigher than in AL patients (with the possibility of left bundle-branch block), and increased atrioventricular valve thicknessappears to be particularly frequent. Awareness of the diversityunderlying the shared label of cardiac amyloidosis is importanton several levels, ranging from disease classification to diagno-sis and clinical management.
Sources of FundingThe contributions of G.M., G.P., L.O. and S.P. were partiallysupported by: the EURAMY (“Systemic Amyloidoses in Europe”)project partially funded by the European Community’s Sixth Frame-work Program; CARIPLO (Fondazione Cassa di Risparmio delleProvincie Lombarde); NOBEL Project “Transcriptomics and Pro-teomics Approaches to Diseases of High Sociomedical Impact: ATechnology-Integrated Network”; Ricerca Finalizzata Malattie Rare,Ministero della Salute - Istituto Superiore di Sanita (526D/63);Ministero dell’Istruzione, dell’Universita e della Ricerca, Programmidi Ricerca Scientifica di Rilevante Interesse Nazionale 2007, ProjectNo 2007XY59ZJ_005 and No 2007AE8FX2_003. F.S. was partiallysupported by an investigator fellowship from Collegio Ghislieri,Pavia. C.R. was partially supported by Universita di Bologna, RFO(Ricerca Fondamentale Orientata) 2008.
DisclosuresNone.
References1. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl
J Med. 2003;349:583–596.2. Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda
S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid protein fibrilnomenclature. Amyloid. 2002;9:197–200.
3. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N EnglJ Med. 1997;337:898–909.
4. Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis: recognition, confir-mation, prognosis, and therapy. Mayo Clin Proc. 1999;74:490–494.
5. Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, HawkinsPN, Merlini G, Moreau P, Ronco P, Sanchorawala V, Sezer O, SolomonA, Grateau G. Definition of organ involvement and treatment response inimmunoglobulin light chain amyloidosis (AL): a consensus opinion fromthe 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol. 2005;79:319–328.
6. Arbustini E, Verga L, Concardi M, Palladini G, Obici L, Merlini G.Electron and immuno-electron microscopy of abdominal fat identifies andcharacterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid.2002;9:108–114.
7. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, GillmoreJD, Pepys MB, Hawkins PN. Misdiagnosis of hereditary amyloidosis asAL (primary) amyloidosis. N Engl J Med. 2002;6:346:1786–1791.
8. Falk RH. Diagnosis and management of the cardiac amyloidoses.Circulation. 2005;112:2047–2060.
9. Ferlini A, Fini S, Salvi F, Patrosso MC, Vezzoni P, Forabosco A.Molecular strategies in genetic diagnosis of transthyretin-relatedhereditary amyloidosis. FASEB J. 1992;6:2864–2866.
10. Kyle RA, Spittell PC, Gertz MA, Li CY, Edwards WD, Olson LJ,Thibodeau SN. The premortem recognition of systemic senile amy-loidosis with cardiac involvement. Am J Med. 1996;101:395–400.
11. Palladini G, Perfetti V, Merlini G. Therapy and management of systemicAL (primary) amyloidosis. Swiss Med Wkly. 2006;136:715–720.
12. Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemicamyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med. 2005;165:1425–1429.
13. Surawicz B, Knilans TK. Chou’s Electrocardiography in ClinicalPractice. 5th ed. Philadelphia, Pa: Saunders; 2001.
14. Cheitlin MD, Armstrong WF, Aurigemma GP, Beller GA, Bierman FZ,Davis JL, Douglas PS, Faxon DP, Gillam LD, Kimball TR, KussmaulWG, Pearlman AS, Philbrick JT, Rakowski H, Thys DM, Antman EM,Smith SC Jr, Alpert JS, Gregoratos G, Anderson JL, Hiratzka LF, HuntSA, Fuster V, Jacobs AK, Gibbons RJ, Russell RO; American College ofCardiology; American Heart Association; American Society of Echocar-diography. ACC/AHA/ASE 2003 guideline update for the clinical appli-cation of echocardiography: summary article: a report of the AmericanCollege of Cardiology/American Heart Association Task Force onPractice Guidelines (ACC/AHA/ASE Committee to Update the 1997Guidelines for the Clinical Application of Echocardiography). Circu-lation. 2003;108:1146–62.
15. Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I,Reichek N. Echocardiographic assessment of left ventricular hypertrophy:comparison to necropsy findings. Am J Cardiol. 1986;57:450–458.
16. Garcia MJ, Thomas JD, Klein AL. New Doppler echocardiographicapplications for the study of diastolic function. J Am Coll Cardiol.1998;32:865–875.
17. Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: char-acterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49:9–13.
18. McCarthy RE, Kasper EK. A review of the amyloidoses that infiltrate theheart. Clin Cardiol. 1998;21:547–552.
19. Dubrey SW, Cha K, Skinner M, LaValley M, Falk RH. Familial andprimary (AL) cardiac amyloidosis: echocardiographically similar diseaseswith distinctly different clinical outcomes. Heart. 1997;78:74–82.
20. Kristen AV, Perz JB, Schonland SO, Hegenbart U, Schnabel PA, KristenJH, Goldschmidt H, Katus HA, Dengler TJ. Non-invasive predictors ofsurvival in cardiac amyloidosis. Eur J Heart Fail. 2007;9:617–624.
21. Johansson B, Westermark P. Senile systemic amyloidosis: a clinico-pathological study of twelve patients with massive amyloid infiltration.Int J Cardiol. 1991;32:83–92.
22. Misu K, Hattori N, Nagamatsu M, Ikeda S, Ando Y, Nakazato M, Takei Y,Hanyu N, Usui Y, Tanaka F, Harada T, Inukai A, Hashizume Y, Sobue G.Late-onset familial amyloid polyneuropathy type I (transthyretin met30-associated familial amyloid polyneuropathy) unrelated to endemic focus inJapan: clinicopathological and genetic features. Brain. 1999;122:1951–1962.
23. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amy-loidotic polyneuropathy. Arch Neurol. 2005;62:1057–1062.
24. Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M,Apstein CS, Liao R. Human amyloidogenic light chains directly impaircardiomyocyte function through an increase in cellular oxidant stress.Circ Res. 2004;94:1008–1010.
25. Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, PerliniS, Obici L, Ascari E, d’Eril GM, Moratti R, Merlini G. Serum N-terminalpro-brain natriuretic peptide is a sensitive marker of myocardial dysfunctionin AL amyloidosis. Circulation. 2003;107:2440–2445.
26. Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, KimuraH. Amyloid peptides are toxic via a common oxidative mechanism. ProcNatl Acad Sci U S A. 1995;92:1989–1993.
27. Suhr OB, Anan I, Backman C, Karlsson A, Lindqvist P, Mörner S,Waldenström A. Do troponin and B-natriuretic peptide detect cardiomy-opathy in transthyretin amyloidosis? J Intern Med. 2008;263:294–301.
28. Palladini G, Saraiva MJ, Coelho T, Reis H, Obici L, Vezzoli M, VadaccaG, Merlini G, Perlini S. Differential effects on amyloid-induced myo-cardial dysfunction and N-terminal-pro brain natriuretic peptide release ofdifferent amyloidogenic proteins. Eur Heart J. 2004;25(suppl):36.Abstract.
Rapezzi et al Systemic Cardiac Amyloidoses 1211
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

29. Rahman JE, Helou EF, Gelzer-Bell R, Thompson RE, Kuo C, RodriguezER, Hare JM, Baughman KL, Kasper EK. Noninvasive diagnosis ofbiopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410–415.
30. Rapezzi C, Perugini E, Salvi F, Grigioni F, Riva L, Cooke RM, FerliniA, Rimessi P, Bacchi-Reggiani L, Ciliberti P, Pastorelli F, Leone O,Bartolomei I, Pinna AD, Arpesella G, Branzi A. Phenotypic andgenotypic heterogeneity in transthyretin-related cardiac amyloidosis:towards tailoring of therapeutic strategies? Amyloid. 2006;13:143–153.
31. Bittencourt PL, Couto CA, Clemente C, Farias AQ, Palacios SA, Mies S,Goldberg AC. Phenotypic expression of familial amyloid polyneuropathyin Brazil. Eur J Neurol. 2005;12:289–293.
32. Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-typeamyloid polyneuropathy in Japan: clinical and genetic heterogeneity.Neurology. 2002;58:1001–1007.
33. Planté-Bordeneuve V, Carayol J, Ferreira A, Adams D, Clerget-DarpouxF, Misrahi M, Said G, Bonay ti-Pellié G. Genetic study of transthyretinamyloid neuropathies: carrier risks among French and Portuguesefamilies. J Med Genet. 2003;40:e120.
CLINICAL PERSPECTIVEMost studies of amyloidotic cardiomyopathy consider as a single entity the 3 main systemic cardiac amyloidoses: acquiredmonoclonal immunoglobulin light-chain (AL); hereditary, mutated transthyretin-related (ATTRm); and wild-typetransthyretin-related (ATTRwt). We conducted a longitudinal study of 233 patients with clear-cut diagnosis by type ofcardiac amyloidosis (AL, n�157; ATTRm, n�61; ATTRwt, n�15) to compare clinical, morphological, and pathophys-iological profiles of these different types. Our results support the concept that cardiomyopathies resulting from AL,ATTRm, and ATTRwt should be considered 3 different cardiac diseases with different pathophysiological substrates andclinical courses. In particular, AL cardiomyopathy is associated with only slightly increased wall thickness, whereas itappears to show the highest frequencies of hemodynamic derangement (mainly resulting from diastolic dysfunction) andlow QRS voltages on ECG. The clinical course appears rather aggressive. In contrast, ATTRm and especially ATTRwt areassociated with markedly increased wall thickness but less frequently with hemodynamic alterations. In these 2transthyretin-related cardiomyopathies, the clinical course appears to be less aggressive than in AL despite the patients’higher average age and greater morphological abnormalities. Furthermore, the voltage-to-mass ratio tends to be higher thanin AL (with the possibility of left bundle-branch block), and increased AV valve thickness appears to be particularlyfrequent. Awareness of the diversity underlying the shared label of cardiac amyloidosis is important on several levels,ranging from disease classification to diagnosis and clinical management.
Go to http://cme.ahajournals.org to take the CME quiz for this article.
1212 Circulation September 29, 2009
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from

Musca, Laura Obici, Angelo Branzi and Stefano PerliniCavo, Elena Zamagni, Maria Luisa Fonte, Giovanni Palladini, Francesco Salinaro, Francesco Robin M.T. Cooke, Letizia Bacchi-Reggiani, Diego Sangiorgi, Alessandra Ferlini, MicheleLeone, Fabrizio Salvi, Paolo Ciliberti, Francesca Pastorelli, Elena Biagini, Fabio Coccolo,
Claudio Rapezzi, Giampaolo Merlini, Candida C. Quarta, Letizia Riva, Simone Longhi, OrnellaSystemic Cardiac Amyloidoses: Disease Profiles and Clinical Courses of the 3 Main Types
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2009 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.108.843334
2009;120:1203-1212; originally published online September 14, 2009;Circulation.
http://circ.ahajournals.org/content/120/13/1203World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on April 15, 2016http://circ.ahajournals.org/Downloaded from
Related Documents











