Synthesis of arylpyrazole linked benzimidazole conjugates as potential microtubule disruptors Ahmed Kamal a,⇑ , Anver Basha Shaik a , Sowjanya Polepalli b , G. Bharath Kumar a , Vangala Santhosh Reddy a , Rasala Mahesh a , Srujana Garimella b , Nishant Jain b a Medicinal Chemistry and Pharmacology, Hyderabad 500007, India b Centre for Chemical Biology, CSIR—Indian Institute of Chemical Technology, Hyderabad 500007, India article info Article history: Received 31 October 2014 Revised 1 January 2015 Accepted 2 January 2015 Available online 8 January 2015 Keywords: Anticancer Caspase-3 Molecular modeling Pyrazole–benzimidazole conjugates Tubulin polymerization abstract In an attempt to develop potent and selective anticancer agents, a series of twenty arylpyrazole linked benzimidazole conjugates (10a–t) were designed and synthesized as microtubule destabilizing agents. The joining of arylpyrazole to the benzimidazole moiety resulted in a four ring (A, B, C and D) molecular scaffold that comprises of polar heterocyclic rings in the middle associated with rotatable single bonds and substituted aryl rings placed in the opposite directions. These conjugates were evaluated for their ability to inhibit the growth of sixty cancer cell line panel of the NCI. Among these some conjugates like 10a, 10b, 10d, 10e, 10p and 10r exhibited significant growth inhibitory activity against most of the cell lines ranging from 0.3 to 13 lM. Interestingly, the conjugate 10b with methoxy group on D-ring expressed appreciable cytotoxic potential. A549 cells treated with some of the potent conjugates like 10a, 10b and 10d arrested cells at G2/M phase apart from activating cyclin-B1 protein levels and disrupt- ing microtubule network. Moreover, these conjugates effectively inhibited tubulin polymerization with IC 50 values of 1.3–3.8 lM. Whereas, the caspase assay revealed that they activate the casepase-3 leading to apoptosis. Particularly 10b having methoxy substituent induced activity almost 3 folds higher than CA- 4. Furthermore, a competitive colchicine binding assay and molecular modeling analysis suggests that these conjugates bind to the tubulin successfully at the colchicine binding site. These investigations reveal that such conjugates having pyrazole and benzimidazole moieties have the potential in the devel- opment of newer chemotherapeutic agents. Ó 2015 Published by Elsevier Ltd. 1. Introduction Microtubules are very important structural components of cytoskeleton present in the cell cytoplasm and made up of poly- merized a- and b-tubulin heterodimers. Microtubules involved in broad range of cellular functions such as cell division, maintenance of cell structure, cell signaling, cell migration and intracellular transport are critical to the cell survival. 1–3 Moreover, these com- ponents express dynamic instability where the tubulin dimers polymerize progressively to generate long cylindrical microtubules that subsequently depolymerizes very rapidly resulting individual tubulin units. 4,5 This dynamic lengthening and shortening of spin- dle microtubules decides largely the shape of the mitotic spindle and promotes the appropriate alignment of chromosomes at the spindle centre. 6 Therefore, microtubule dynamics is one of the most attractive strategies for the development of cancer therapeu- tics. A variety of compounds that bind to tubulin-microtubule have been part of the pharmacopoeia of anticancer therapy for decades. In this connection, small molecules that particularly perturb microtubule network and cause mitotic arrest are of immense interest in current cancer chemotherapy. 7 Microtubule targeting drugs are well known to interact with tubulin through at least by one of the following binding sites such as laulimalide, taxane/epothilone, vinca alkaloid, and colchicine sites. 3 However, colchicine binding site small molecule inhibitors such as colchicine (1), combretastatin A-4 (2) and nocodazole (3) exert their vital biological effects by inhibiting tubulin assembly and perturb dynamic stability (Fig. 1A). 8 In addition, some of the natural products and their designed analogues that distinguish col- chicine-binding site have received considerable attention towards the discovery of cancer therapeutics. The use of colchicine has been inadequate in the treatment of cancer because of its toxicity, how- ever it can be considered as a significant hallmark for the genera- tion of potent chemotherapeutics. 9 Hence, potential attention has risen towards the discovery and development of novel molecules http://dx.doi.org/10.1016/j.bmc.2015.01.004 0968-0896/Ó 2015 Published by Elsevier Ltd. ⇑ Corresponding author. Tel.: +91 40 27193157; fax: +91 40 27193189. E-mail address: [email protected] (A. Kamal). Bioorganic & Medicinal Chemistry 23 (2015) 1082–1095 Contents lists available at ScienceDirect Bioorganic & Medicinal Chemistry journal homepage: www.elsevier.com/locate/bmc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Bioorganic & Medicinal Chemistry 23 (2015) 1082–1095
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc
Synthesis of arylpyrazole linked benzimidazole conjugates aspotential microtubule disruptors
http://dx.doi.org/10.1016/j.bmc.2015.01.0040968-0896/� 2015 Published by Elsevier Ltd.
⇑ Corresponding author. Tel.: +91 40 27193157; fax: +91 40 27193189.E-mail address: [email protected] (A. Kamal).
Ahmed Kamal a,⇑, Anver Basha Shaik a, Sowjanya Polepalli b, G. Bharath Kumar a,Vangala Santhosh Reddy a, Rasala Mahesh a, Srujana Garimella b, Nishant Jain b
a Medicinal Chemistry and Pharmacology, Hyderabad 500007, Indiab Centre for Chemical Biology, CSIR—Indian Institute of Chemical Technology, Hyderabad 500007, India
a r t i c l e i n f o a b s t r a c t
Article history:Received 31 October 2014Revised 1 January 2015Accepted 2 January 2015Available online 8 January 2015
Keywords:AnticancerCaspase-3Molecular modelingPyrazole–benzimidazole conjugatesTubulin polymerization
In an attempt to develop potent and selective anticancer agents, a series of twenty arylpyrazole linkedbenzimidazole conjugates (10a–t) were designed and synthesized as microtubule destabilizing agents.The joining of arylpyrazole to the benzimidazole moiety resulted in a four ring (A, B, C and D) molecularscaffold that comprises of polar heterocyclic rings in the middle associated with rotatable single bondsand substituted aryl rings placed in the opposite directions. These conjugates were evaluated for theirability to inhibit the growth of sixty cancer cell line panel of the NCI. Among these some conjugates like10a, 10b, 10d, 10e, 10p and 10r exhibited significant growth inhibitory activity against most of the celllines ranging from 0.3 to 13 lM. Interestingly, the conjugate 10b with methoxy group on D-ringexpressed appreciable cytotoxic potential. A549 cells treated with some of the potent conjugates like10a, 10b and 10d arrested cells at G2/M phase apart from activating cyclin-B1 protein levels and disrupt-ing microtubule network. Moreover, these conjugates effectively inhibited tubulin polymerization withIC50 values of 1.3–3.8 lM. Whereas, the caspase assay revealed that they activate the casepase-3 leadingto apoptosis. Particularly 10b having methoxy substituent induced activity almost 3 folds higher than CA-4. Furthermore, a competitive colchicine binding assay and molecular modeling analysis suggests thatthese conjugates bind to the tubulin successfully at the colchicine binding site. These investigationsreveal that such conjugates having pyrazole and benzimidazole moieties have the potential in the devel-opment of newer chemotherapeutic agents.
� 2015 Published by Elsevier Ltd.
1. Introduction
Microtubules are very important structural components ofcytoskeleton present in the cell cytoplasm and made up of poly-merized a- and b-tubulin heterodimers. Microtubules involved inbroad range of cellular functions such as cell division, maintenanceof cell structure, cell signaling, cell migration and intracellulartransport are critical to the cell survival.1–3 Moreover, these com-ponents express dynamic instability where the tubulin dimerspolymerize progressively to generate long cylindrical microtubulesthat subsequently depolymerizes very rapidly resulting individualtubulin units.4,5 This dynamic lengthening and shortening of spin-dle microtubules decides largely the shape of the mitotic spindleand promotes the appropriate alignment of chromosomes at thespindle centre.6 Therefore, microtubule dynamics is one of themost attractive strategies for the development of cancer therapeu-
tics. A variety of compounds that bind to tubulin-microtubule havebeen part of the pharmacopoeia of anticancer therapy for decades.In this connection, small molecules that particularly perturbmicrotubule network and cause mitotic arrest are of immenseinterest in current cancer chemotherapy.7
Microtubule targeting drugs are well known to interact withtubulin through at least by one of the following binding sites suchas laulimalide, taxane/epothilone, vinca alkaloid, and colchicinesites.3 However, colchicine binding site small molecule inhibitorssuch as colchicine (1), combretastatin A-4 (2) and nocodazole (3)exert their vital biological effects by inhibiting tubulin assemblyand perturb dynamic stability (Fig. 1A).8 In addition, some of thenatural products and their designed analogues that distinguish col-chicine-binding site have received considerable attention towardsthe discovery of cancer therapeutics. The use of colchicine has beeninadequate in the treatment of cancer because of its toxicity, how-ever it can be considered as a significant hallmark for the genera-tion of potent chemotherapeutics.9 Hence, potential attention hasrisen towards the discovery and development of novel molecules
N NH NH
N
H3CO
OCH3
H3CO RA
B C D
Area of tubulin binding
Bridge
Cytotoxic activity
R = OCH3 > H > Cl > (Cl)2 > F > CF3
Figure 1B. Structure and activity relationship of arylpyrazole linked benzimidazoleconjugates: The compounds that possessing 3,4,5-trimethoxyphenyl as A-ring andvarious substituents on the D-ring showed significant anticancer activity. 3,4,5-trimethoxyphenyl ring is a hall mark for tubulin binding. Whereas, the substituentson the D-ring that alter their cytotoxic activity is represented as R group.
OO
O
OCH3
O
NH
O
N
HN
NHO
OOS
3
H3CO
H3COOCH3 OCH3
R=OH CA4R=OPO3Na2 CA4PR=NHSer, HCl AVE8062
2
4
N
N
R2
R1
ABI
BA
N NH
R1
NH
NR2
N NH NH
N
H3COOCH3
H3CO
OCH3
10a-t 10b
R
1
O
C R3
Figure 1A. Structures of some anticancer molecules: colchicine (1), combretastatin (2), nocodazole (3), 2-aryl-4-benzoyl-imidazole series (ABI) (4), arylpyrazole inkedbenzimidazole conjugates (10a–t) and lead conjugate (10b).
A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1083
that affect tubulin assembly through impeding multi-drug resis-tance and acute cytotoxicity.
The pyrazole linked benzimidazoles were synthesized keepingin mind both the pyrazole as wellas benzimidazole basic units;benzimidazole containing nocodazole (3) a widely employed anti-neoplastic agent and a cell cycle synchronizing agent to inducemitotic arrest (Fig. 1A).10,11 Benzimidazole scaffold is the mostprivileged structure in the field of medicinal chemistry and thisresidue is a constituent of vitamin B12 that further supports itspotential for their development as therapeutic agents (Fig. 1A).12
Another important benzimidazole derivative is (PPTMB) an effec-tive anticancer agent that significantly disrupts microtubuledynamics, leading to mitotic arrest and JNK activation, conse-quently induces mitochondria-related apoptotic cell death of pros-tate cancer cells.13 In an endeavor to develop potential anticanceragents, recently we reported terphenyl benzimidazoles as inhibi-tors of tubulin polymerization that arrest the cells in G2/M phaseof the cell cycle.14 Some other heterocyclic compounds, like pyra-zoles have received considerable attention owing to their diversechemotherapeutic properties including versatile antineoplasticactivities.15 In addition, some pyrazoles exert their anticancerpotential by inhibiting various enzymes and proteins that play acritical role in the cell division.16 Additionally, tri- and tetra-substi-tuted pyrazole derivatives are also known to exhibit potent anti-cancer efficacy by the inhibition of p38a MAP kinase.17
Structural combinations of heterocyclic rings in certain ordersimpart potent anticancer activity to the designed molecules.18
Since benzimidazole is a basic building block for various biologi-cally active compounds including tubulin inhibitors, some 2-aryl-4-benzoyl-imidazole (ABI-III) (4) (Fig. 1A) congeners with basicthree ring scaffold (A, B and C) was designed as highly active tubu-lin polymerization inhibitors.19 We previously reported that acombination of two heterocyclic moieties as a single molecular
scaffold could show enhanced cell growth arrest by preventingtubulin polymerization.20,21 Herein, we designed a molecular scaf-fold that comprises of four rings (A, B, C, D) with a benzimidazoleas well as pyrazole basic units and tested for their cytotoxic poten-tial and antitubulin activity. The A (aryl) and D (fused benzene ringof benzimidazole) rings were decorated with carefully selectedsubstituents (–OCH3, –OCH2O–, –Cl, –F and –CF3) with a view togenerate potent tubulin inhibitors (Fig. 1B). The results of ourinvestigations, which validated the underlying hypothesis ispresented in the following sections.
2. Results and discussion
2.1. Chemistry
The synthesis of pyrazole linked benzimidazole conjugates10a–t described in this study was depicted in Scheme 1. The finalcompounds were successfully achieved by oxidative cyclization of

OR1
R2R3
N NH
R3R2
R1
O O
O
O
R3R2
R1N NH O
R3R2
R1O
N NH O
R3R2
R1H
i ii
iii
ivv
5(a-d)6(a-d) 7(a-d)
9(a-d)10(a-t)
NH
N R4
R5
a = R1(OCH3),R2(OCH3),R3(OCH3)5, 6, 7, 8, 9(a-d)
b = R1(OCH3),R2(OCH3),R3(H)c = R1(H),R2(OCH3),R3(H)d = R1,R2(-OCH2O-),R3(H)
R1 R2 R3 R4 R510a OCH3 OCH3 OCH3 H H10b OCH3 OCH3 OCH3 H OCH310c OCH3 OCH3 OCH3 H F10d OCH3 OCH3 OCH3 H Cl10e OCH3 OCH3 OCH3 Cl Cl10f OCH3 OCH3 OCH3 H CF3
10i H OCH3 OCH3 Cl Cl10h H OCH3 OCH3 H F10g H OCH3 OCH3 H H
10j H OCH3 H H H10k H OCH3 H H OCH310l H OCH3 H H F10m H OCH3 H H Cl
10o H OCH3 H H CF310p -OCH2O- H H H
10n H OCH3 H Cl Cl
10q -OCH2O- H H OCH310r -OCH2O- H H F10s -OCH2O- H Cl Cl10t -OCH2O- H H CF3
R1 R2 R3 R4 R5
N NH OH
R3R2
R1
8(a-d)
Scheme 1. Synthesis of pyrazole linked benzimidazole conjugate. Reagents and conditions: (i) NaOEt, diethyl oxalate, EtOH, 0 �C to rt, 4 h; (ii) NH2–NH2.2HCl, EtOH, reflux3 h; (iii) LiAlH4, THF, 0 �C to rt, 1–2 h; (iv) IBX, dry DMSO, rt, 1 h; (v) Na2S2O5, EtOH, o-phenylenediamines, rt, 3–4 h.
1084 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
various o-phenylenediamines with substituted phenyl pyrazolecarbaldehydes (9a–d) in the presence of sodium metabisulfite inrefluxing ethanol.14 Different acetophenones were utilized innucleophilic substitution reaction with diethyl oxalate in the pres-ence of sodium ethoxide to afford diketo esters (6a–d) which upontreatment with NH2–NH2�2HCl in ethanol produced the corre-sponding pyrazole esters (7a–d) in good yields.20 These pyrazoleesters underwent reduction by LiAlH4 to yield (3-substitutedaryl-1H-pyrazol-5-yl)methanols (8a–d) and subsequently these alco-hols were selectively oxidized by IBX in DMSO to produce pyrazolecarbaldehyde intermediates (9a–d) in excellent yields. All the tar-get compounds were characterized by IR, 1H NMR, 13C NMR, massand HRMS spectral data (See Table 1).
2.2. Biological activity
2.2.1. In vitro cytotoxic activityTo understand the structure activity relationship (SAR) we syn-
thesized twenty pyrazole linked benzimidazole conjugates havingdifferent substituents on the A and D-rings. A-ring constituted theelectron donating group like (OCH3)3, OCH3)2, OCH3), and 3,4-(OCH2O) whereas the D-ring possessed both the electron donat-ing as well as withdrawing groups like OCH3, H, F, Cl, Cl2 and CF3.All the synthesized conjugates were evaluated for their anticanceractivity against a panel of sixty human cancer cell lines of nine
cancer types (leukemia, non-small cell lung, colon, CNS, melanoma,ovarian, renal, prostate and breast cancer) as per the NCI protocol.Among the twenty conjugates most of them (10a, 10b, 10d, 10e,10g, 10j, 10k, 10l, 10n, 10o, 10p, 10q, 10r, 10s) were taken upfor the single dose (10 lM) screening. The mean growth values(percentage) are summarized in the Supporting information(Table S1). Some of these conjugates like 10a, 10b, 10d, 10e, 10pand 10r showed profound inhibitory effect in the primary screen-ing with the mean growth percentage range of 3.04–60.54%. Subse-quently, these conjugates were evaluated in five dose screening(0.01, 0.1, 1.0, 10, 100 lM) to examine their potency. These conju-gates exhibited a broad spectrum of cytotoxic activity against var-ious cancer cell lines with the GI50 value ranging from 0.34 to10 lM and their GI50 values (lM) are represented in the Support-ing information (Table S2). Moreover, these results suggest thatconjugates like 10a, 10b, 10d and 10e with trimethoxy phenylgroup as A-ring display profound potency than 10p and 10r thatpossess 3,4-(methylenedioxy) group on the same A-ring. One ofthe most promising compound 10b that has trimethoxy substitu-ents on the A-ring and monomethoxy on the D-ring manifestedpotent antiproliferative activity with the GI50 values <1 lM, partic-ularly against T-47D, MDA-MB-435, BT-549 (breast) MOLT-4,RPMI-8226, in (leukemia), NCI-H226 (lung) and SK-MEL-5 (mela-noma) cancer cell lines. Whereas, in case of 10a which is devoidof any substitution on the D-ring showed notable activity against
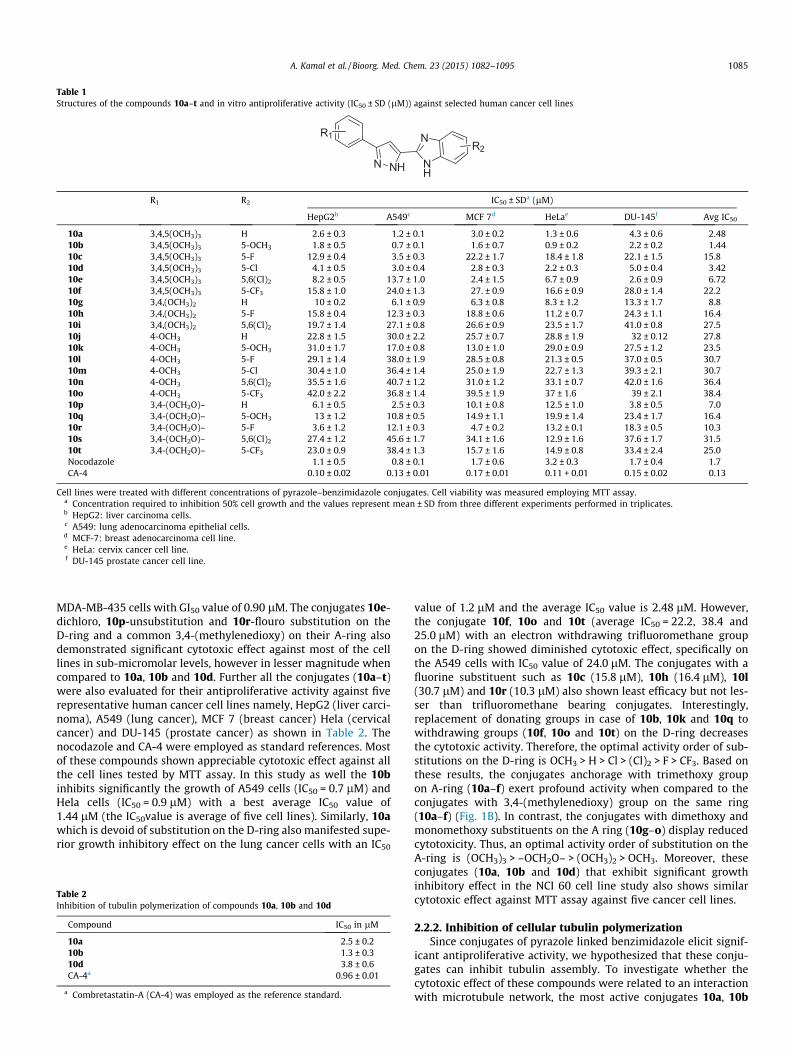
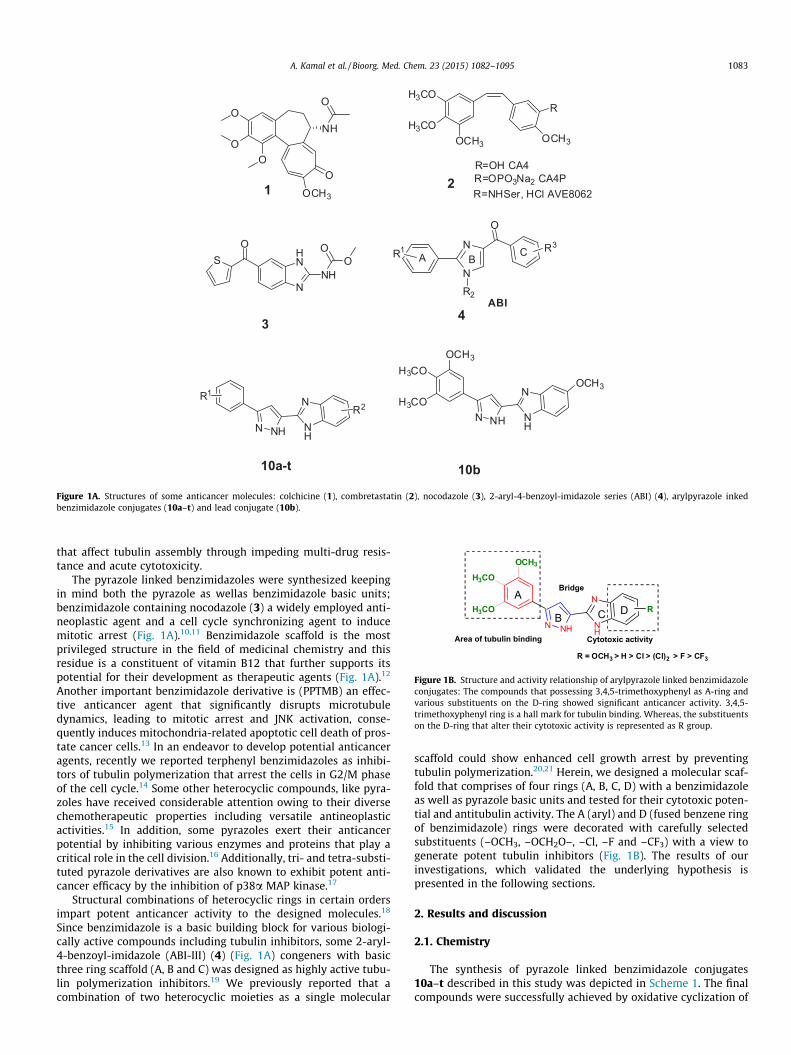
Table 1Structures of the compounds 10a–t and in vitro antiproliferative activity (IC50 ± SD (lM)) against selected human cancer cell lines
N NH
R1
NH
NR2
R1 R2 IC50 ± SDa (lM)
HepG2b A549c MCF 7d HeLae DU-145f Avg IC50
10a 3,4,5(OCH3)3 H 2.6 ± 0.3 1.2 ± 0.1 3.0 ± 0.2 1.3 ± 0.6 4.3 ± 0.6 2.4810b 3,4,5(OCH3)3 5-OCH3 1.8 ± 0.5 0.7 ± 0.1 1.6 ± 0.7 0.9 ± 0.2 2.2 ± 0.2 1.4410c 3,4,5(OCH3)3 5-F 12.9 ± 0.4 3.5 ± 0.3 22.2 ± 1.7 18.4 ± 1.8 22.1 ± 1.5 15.810d 3,4,5(OCH3)3 5-Cl 4.1 ± 0.5 3.0 ± 0.4 2.8 ± 0.3 2.2 ± 0.3 5.0 ± 0.4 3.4210e 3,4,5(OCH3)3 5,6(Cl)2 8.2 ± 0.5 13.7 ± 1.0 2.4 ± 1.5 6.7 ± 0.9 2.6 ± 0.9 6.7210f 3,4,5(OCH3)3 5-CF3 15.8 ± 1.0 24.0 ± 1.3 27. ± 0.9 16.6 ± 0.9 28.0 ± 1.4 22.210g 3,4,(OCH3)2 H 10 ± 0.2 6.1 ± 0.9 6.3 ± 0.8 8.3 ± 1.2 13.3 ± 1.7 8.810h 3,4,(OCH3)2 5-F 15.8 ± 0.4 12.3 ± 0.3 18.8 ± 0.6 11.2 ± 0.7 24.3 ± 1.1 16.410i 3,4,(OCH3)2 5,6(Cl)2 19.7 ± 1.4 27.1 ± 0.8 26.6 ± 0.9 23.5 ± 1.7 41.0 ± 0.8 27.510j 4-OCH3 H 22.8 ± 1.5 30.0 ± 2.2 25.7 ± 0.7 28.8 ± 1.9 32 ± 0.12 27.810k 4-OCH3 5-OCH3 31.0 ± 1.7 17.0 ± 0.8 13.0 ± 1.0 29.0 ± 0.9 27.5 ± 1.2 23.510l 4-OCH3 5-F 29.1 ± 1.4 38.0 ± 1.9 28.5 ± 0.8 21.3 ± 0.5 37.0 ± 0.5 30.710m 4-OCH3 5-Cl 30.4 ± 1.0 36.4 ± 1.4 25.0 ± 1.9 22.7 ± 1.3 39.3 ± 2.1 30.710n 4-OCH3 5,6(Cl)2 35.5 ± 1.6 40.7 ± 1.2 31.0 ± 1.2 33.1 ± 0.7 42.0 ± 1.6 36.410o 4-OCH3 5-CF3 42.0 ± 2.2 36.8 ± 1.4 39.5 ± 1.9 37 ± 1.6 39 ± 2.1 38.410p 3,4-(OCH2O)– H 6.1 ± 0.5 2.5 ± 0.3 10.1 ± 0.8 12.5 ± 1.0 3.8 ± 0.5 7.010q 3,4-(OCH2O)– 5-OCH3 13 ± 1.2 10.8 ± 0.5 14.9 ± 1.1 19.9 ± 1.4 23.4 ± 1.7 16.410r 3,4-(OCH2O)– 5-F 3.6 ± 1.2 12.1 ± 0.3 4.7 ± 0.2 13.2 ± 0.1 18.3 ± 0.5 10.310s 3,4-(OCH2O)– 5,6(Cl)2 27.4 ± 1.2 45.6 ± 1.7 34.1 ± 1.6 12.9 ± 1.6 37.6 ± 1.7 31.510t 3,4-(OCH2O)– 5-CF3 23.0 ± 0.9 38.4 ± 1.3 15.7 ± 1.6 14.9 ± 0.8 33.4 ± 2.4 25.0Nocodazole 1.1 ± 0.5 0.8 ± 0.1 1.7 ± 0.6 3.2 ± 0.3 1.7 ± 0.4 1.7CA-4 0.10 ± 0.02 0.13 ± 0.01 0.17 ± 0.01 0.11 + 0.01 0.15 ± 0.02 0.13
Cell lines were treated with different concentrations of pyrazole–benzimidazole conjugates. Cell viability was measured employing MTT assay.a Concentration required to inhibition 50% cell growth and the values represent mean ± SD from three different experiments performed in triplicates.b HepG2: liver carcinoma cells.c A549: lung adenocarcinoma epithelial cells.d MCF-7: breast adenocarcinoma cell line.e HeLa: cervix cancer cell line.f DU-145 prostate cancer cell line.
A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1085
MDA-MB-435 cells with GI50 value of 0.90 lM. The conjugates 10e-dichloro, 10p-unsubstitution and 10r-flouro substitution on theD-ring and a common 3,4-(methylenedioxy) on their A-ring alsodemonstrated significant cytotoxic effect against most of the celllines in sub-micromolar levels, however in lesser magnitude whencompared to 10a, 10b and 10d. Further all the conjugates (10a–t)were also evaluated for their antiproliferative activity against fiverepresentative human cancer cell lines namely, HepG2 (liver carci-noma), A549 (lung cancer), MCF 7 (breast cancer) Hela (cervicalcancer) and DU-145 (prostate cancer) as shown in Table 2. Thenocodazole and CA-4 were employed as standard references. Mostof these compounds shown appreciable cytotoxic effect against allthe cell lines tested by MTT assay. In this study as well the 10binhibits significantly the growth of A549 cells (IC50 = 0.7 lM) andHela cells (IC50 = 0.9 lM) with a best average IC50 value of1.44 lM (the IC50value is average of five cell lines). Similarly, 10awhich is devoid of substitution on the D-ring also manifested supe-rior growth inhibitory effect on the lung cancer cells with an IC50
Table 2Inhibition of tubulin polymerization of compounds 10a, 10b and 10d
Compound IC50 in lM
10a 2.5 ± 0.210b 1.3 ± 0.310d 3.8 ± 0.6CA-4a 0.96 ± 0.01
a Combretastatin-A (CA-4) was employed as the reference standard.
value of 1.2 lM and the average IC50 value is 2.48 lM. However,the conjugate 10f, 10o and 10t (average IC50 = 22.2, 38.4 and25.0 lM) with an electron withdrawing trifluoromethane groupon the D-ring showed diminished cytotoxic effect, specifically onthe A549 cells with IC50 value of 24.0 lM. The conjugates with afluorine substituent such as 10c (15.8 lM), 10h (16.4 lM), 10l(30.7 lM) and 10r (10.3 lM) also shown least efficacy but not les-ser than trifluoromethane bearing conjugates. Interestingly,replacement of donating groups in case of 10b, 10k and 10q towithdrawing groups (10f, 10o and 10t) on the D-ring decreasesthe cytotoxic activity. Therefore, the optimal activity order of sub-stitutions on the D-ring is OCH3 > H > Cl > (Cl)2 > F > CF3. Based onthese results, the conjugates anchorage with trimethoxy groupon A-ring (10a–f) exert profound activity when compared to theconjugates with 3,4-(methylenedioxy) group on the same ring(10a–f) (Fig. 1B). In contrast, the conjugates with dimethoxy andmonomethoxy substituents on the A ring (10g–o) display reducedcytotoxicity. Thus, an optimal activity order of substitution on theA-ring is (OCH3)3 > –OCH2O– > (OCH3)2 > OCH3. Moreover, theseconjugates (10a, 10b and 10d) that exhibit significant growthinhibitory effect in the NCI 60 cell line study also shows similarcytotoxic effect against MTT assay against five cancer cell lines.
2.2.2. Inhibition of cellular tubulin polymerizationSince conjugates of pyrazole linked benzimidazole elicit signif-
icant antiproliferative activity, we hypothesized that these conju-gates can inhibit tubulin assembly. To investigate whether thecytotoxic effect of these compounds were related to an interactionwith microtubule network, the most active conjugates 10a, 10b

Figure 3. Western blot analysis of Cyclin-B1: Treatment of A549 cells with 5 lMconcentrations of 10a, 10b and 10d for 24 h resulted an increase in cyclin B1. Thepotent compound 10b show an expression of cyclin B1 levels significantly. Tubulinwas employed as loading control. CA-4, colchicine (col.) were used as positivecontrol and DMSO as negative control.
1086 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
and 10d were examined for their effect on the inhibition of tubulinassembly. Thus, we incubated tubulin with varying concentrationsof the potential conjugates to determine the IC50 values for anti-tubulin activity. The results of tubulin polymerization inhibitionassay are summarized in Table 2. All the lead conjugates exhibitremarkable activity comparable to combretastatin-A4. Interest-ingly the conjugate 10b that showed substantial increase in anti-proliferative activity also inhibits tubulin polymerizationsignificantly with an IC50 value of 1.3 lM. However, the othertwo promising conjugates 10a and 10d inhibited tubulin assemblywith IC50 values of 2.5 lM and 3.8 lM, respectively (Table 2).
2.2.3. Analysis of cell cycle distributionFlow cytometry is routinely employed to distinguish the popu-
lation of cells in different phases of cell cycle based on the DNAcontent of the cells.22 The antiproliferative activity of the pyrazolelinked benzimidazole conjugates correlates to the cell cycle arrestas analyzing by the cell cycle distribution. Since conjugates 10a,10b and 10d inhibit tubulin polymerization apart from promisingcytotoxicity, A549 cells were treated with 10a, 10b and 10d at5 lM for 24 h and observed that the cells accumulated significantlyin G2/M phase of the cell cycle. Cells treated with potent conjugate10b showed 79% arrest of cells in G2/M phase. Whereas, 10a and10d results in increase of mitotic arrest by 77% and 70%, respec-tively, however the lower population of cells were observed inthe S phase. In contrast, DMSO treated cells showed a majority ofthe cell content in G1 phase (67.7%) of the cell cycle (Fig. 2).
2.2.4. Effect on cellular cyclin-B1 levels by immunoblot analysisTo obtain further insight into the results of tubulin inhibitory
activity, we investigated the effect of these conjugates on cyclin-B1. This protein is one of the important regulatory proteins ofmitosis and accumulation of cyclin-B1 is an indication for G2/Marrest.23 Thus, we treated A549 cells with lead conjugates at5 lM concentrations for 24 h and performed immunoblot analysisfor cyclin-B1. For comparison, combretastatin A-4 and colchicinewere included as positive controls and tubulin as loading control.
Figure 2. Anti-mitotic effects of 10a, 10b and 10d by FACS analysis: Induction of cell cytreatment at 5 lM for 24 h. Flow cytometric analysis of DNA content demonstrates that 10effective in the assay, followed by 10a (77.32%) and then 10d (70.23%). DMSO treatequantitatively measured.
Interestingly, compounds like 10a and 10b that show potent anti-tubulin activity, considerably induced cyclin-B1 protein levels likecolchicine as well as CA-4 treated cells. However, 10d exhibitednegligible effect on the cyclin-B1 protein. Therefore, the accumula-tion of cyclin-B1 levels suggests that these conjugates demonstratecytotoxic effect through cell cycle arrest at mitosis (Fig. 3).
2.2.5. Effect of 10a, 10b and 10d on microtubule networkThe collection of spindle microtubules, microtubule-associated
proteins (MAPs) and the microtubule organizing centres (MTOC)is called as mitotic spindle. These are key components of the cellthat play a critical role in the segregation of chromosomes duringcell division.24 Occurrence of irregular spindle fibres due to dis-rupted microtubule network is a hallmark of cells treated withantitubulin agents. The inhibitors of tubulin assembly cause severeperturbation in the microtubule dynamics leading to irregularmorphology. Since some of the pyrazole linked benzimidazoles sig-nificantly inhibit the growth of cells, tubulin polymerization andaccumulate the cells in G2/M phase of the cell cycle. Therefore, itwas considered of interest to understand the effect of these conju-gates on the microtubule network. In order to determine if the cellcycle arrest is due to spindle abnormality, A549 cells were treatedwith 5 lM concentrations of 10a, 10b and 10d were and stainedwith tubulin antibody. Immunofluorescence analysis reveals that
cle G2/M arrest by compounds 10a, 10b and 10d. A549 cells were harvested aftera, 10b and 10d arrest A549 cancer cells in G2/M phase and 10b (79.04%) is the most
d cells served as control. The percentage of cells in each phase of cell cycle was
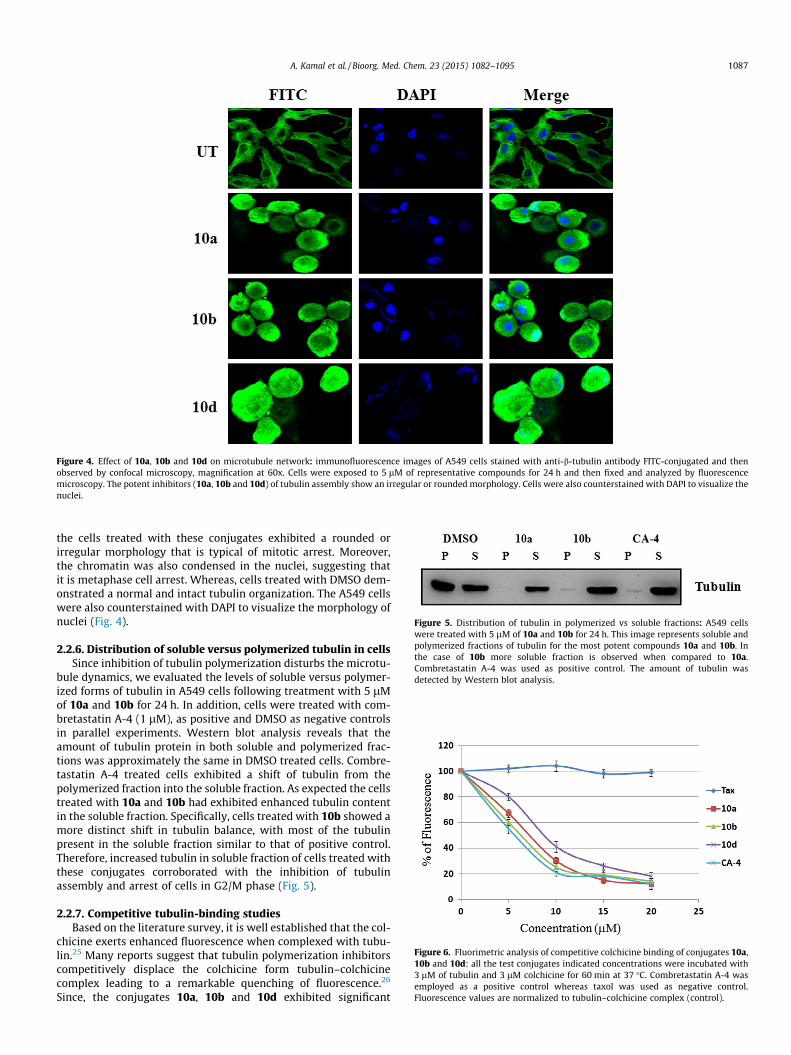
Figure 4. Effect of 10a, 10b and 10d on microtubule network: immunofluorescence images of A549 cells stained with anti-b-tubulin antibody FITC-conjugated and thenobserved by confocal microscopy, magnification at 60x. Cells were exposed to 5 lM of representative compounds for 24 h and then fixed and analyzed by fluorescencemicroscopy. The potent inhibitors (10a, 10b and 10d) of tubulin assembly show an irregular or rounded morphology. Cells were also counterstained with DAPI to visualize thenuclei.
Figure 6. Fluorimetric analysis of competitive colchicine binding of conjugates 10a,10b and 10d: all the test conjugates indicated concentrations were incubated with3 lM of tubulin and 3 lM colchicine for 60 min at 37 �C. Combretastatin A-4 wasemployed as a positive control whereas taxol was used as negative control.Fluorescence values are normalized to tubulin–colchicine complex (control).
Figure 5. Distribution of tubulin in polymerized vs soluble fractions: A549 cellswere treated with 5 lM of 10a and 10b for 24 h. This image represents soluble andpolymerized fractions of tubulin for the most potent compounds 10a and 10b. Inthe case of 10b more soluble fraction is observed when compared to 10a.Combretastatin A-4 was used as positive control. The amount of tubulin wasdetected by Western blot analysis.
A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1087
the cells treated with these conjugates exhibited a rounded orirregular morphology that is typical of mitotic arrest. Moreover,the chromatin was also condensed in the nuclei, suggesting thatit is metaphase cell arrest. Whereas, cells treated with DMSO dem-onstrated a normal and intact tubulin organization. The A549 cellswere also counterstained with DAPI to visualize the morphology ofnuclei (Fig. 4).
2.2.6. Distribution of soluble versus polymerized tubulin in cellsSince inhibition of tubulin polymerization disturbs the microtu-
bule dynamics, we evaluated the levels of soluble versus polymer-ized forms of tubulin in A549 cells following treatment with 5 lMof 10a and 10b for 24 h. In addition, cells were treated with com-bretastatin A-4 (1 lM), as positive and DMSO as negative controlsin parallel experiments. Western blot analysis reveals that theamount of tubulin protein in both soluble and polymerized frac-tions was approximately the same in DMSO treated cells. Combre-tastatin A-4 treated cells exhibited a shift of tubulin from thepolymerized fraction into the soluble fraction. As expected the cellstreated with 10a and 10b had exhibited enhanced tubulin contentin the soluble fraction. Specifically, cells treated with 10b showed amore distinct shift in tubulin balance, with most of the tubulinpresent in the soluble fraction similar to that of positive control.Therefore, increased tubulin in soluble fraction of cells treated withthese conjugates corroborated with the inhibition of tubulinassembly and arrest of cells in G2/M phase (Fig. 5).
2.2.7. Competitive tubulin-binding studiesBased on the literature survey, it is well established that the col-
chicine exerts enhanced fluorescence when complexed with tubu-lin.25 Many reports suggest that tubulin polymerization inhibitorscompetitively displace the colchicine form tubulin–colchicinecomplex leading to a remarkable quenching of fluorescence.26
Since, the conjugates 10a, 10b and 10d exhibited significant
0 2 4 6 8
10 12
Flou
rese
nce
Uni
tsCaspase-3 Assay
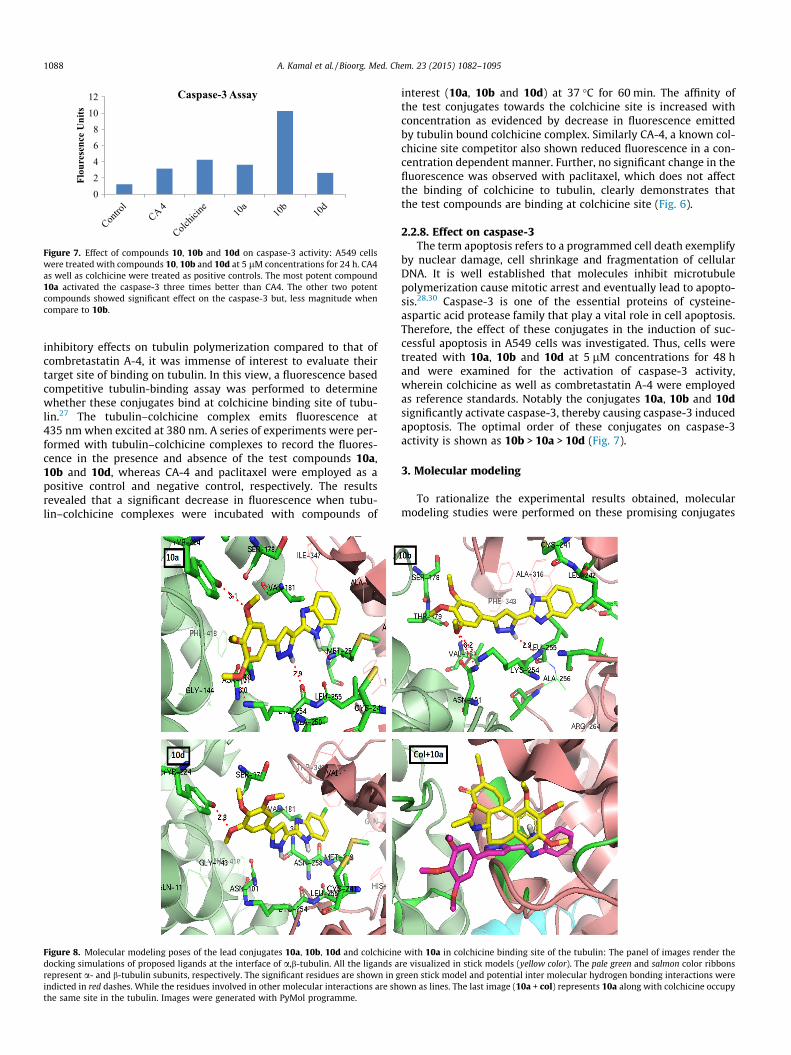
Figure 7. Effect of compounds 10, 10b and 10d on caspase-3 activity: A549 cellswere treated with compounds 10, 10b and 10d at 5 lM concentrations for 24 h. CA4as well as colchicine were treated as positive controls. The most potent compound10a activated the caspase-3 three times better than CA4. The other two potentcompounds showed significant effect on the caspase-3 but, less magnitude whencompare to 10b.
1088 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
inhibitory effects on tubulin polymerization compared to that ofcombretastatin A-4, it was immense of interest to evaluate theirtarget site of binding on tubulin. In this view, a fluorescence basedcompetitive tubulin-binding assay was performed to determinewhether these conjugates bind at colchicine binding site of tubu-lin.27 The tubulin–colchicine complex emits fluorescence at435 nm when excited at 380 nm. A series of experiments were per-formed with tubulin–colchicine complexes to record the fluores-cence in the presence and absence of the test compounds 10a,10b and 10d, whereas CA-4 and paclitaxel were employed as apositive control and negative control, respectively. The resultsrevealed that a significant decrease in fluorescence when tubu-lin–colchicine complexes were incubated with compounds of
Figure 8. Molecular modeling poses of the lead conjugates 10a, 10b, 10d and colchicinedocking simulations of proposed ligands at the interface of a,b-tubulin. All the ligands arepresent a- and b-tubulin subunits, respectively. The significant residues are shown in gindicted in red dashes. While the residues involved in other molecular interactions are shthe same site in the tubulin. Images were generated with PyMol programme.
interest (10a, 10b and 10d) at 37 �C for 60 min. The affinity ofthe test conjugates towards the colchicine site is increased withconcentration as evidenced by decrease in fluorescence emittedby tubulin bound colchicine complex. Similarly CA-4, a known col-chicine site competitor also shown reduced fluorescence in a con-centration dependent manner. Further, no significant change in thefluorescence was observed with paclitaxel, which does not affectthe binding of colchicine to tubulin, clearly demonstrates thatthe test compounds are binding at colchicine site (Fig. 6).
2.2.8. Effect on caspase-3The term apoptosis refers to a programmed cell death exemplify
by nuclear damage, cell shrinkage and fragmentation of cellularDNA. It is well established that molecules inhibit microtubulepolymerization cause mitotic arrest and eventually lead to apopto-sis.28,30 Caspase-3 is one of the essential proteins of cysteine-aspartic acid protease family that play a vital role in cell apoptosis.Therefore, the effect of these conjugates in the induction of suc-cessful apoptosis in A549 cells was investigated. Thus, cells weretreated with 10a, 10b and 10d at 5 lM concentrations for 48 hand were examined for the activation of caspase-3 activity,wherein colchicine as well as combretastatin A-4 were employedas reference standards. Notably the conjugates 10a, 10b and 10dsignificantly activate caspase-3, thereby causing caspase-3 inducedapoptosis. The optimal order of these conjugates on caspase-3activity is shown as 10b > 10a > 10d (Fig. 7).
3. Molecular modeling
To rationalize the experimental results obtained, molecularmodeling studies were performed on these promising conjugates
with 10a in colchicine binding site of the tubulin: The panel of images render there visualized in stick models (yellow color). The pale green and salmon color ribbonsreen stick model and potential inter molecular hydrogen bonding interactions wereown as lines. The last image (10a + col) represents 10a along with colchicine occupy

A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1089
(10a, 10b and 10d). Autodock4 was employed and these com-pounds were docked in the colchicine binding site of the tubulin(PDB code: 3E22).29 The colchicine binding site is generally locatedat the interface of a,b-tubulin heterodimers.30 The a,b-subunits areshown as light green and salmon colored ribbons, respectively(Fig. 8). The amino acid residues that interacting with these conju-gates are depicted as green sticks while other residues are shownas lines and the hydrogen bonds are represented as red dashes.All the promising conjugates docked with tubulin in a similar man-ner at the colchicine site of tubulin. The trimethoxyphenyl A-ring isburied in the a-subunit of unsubstituted is placed in the b-subunit.Although, trimethoxy phenyl group is a hallmark for the tubulinbinding at colchicine site, these conjugates are built with trimeth-oxy substituent on A-ring. The most potent conjugate 10b with amethoxy substitution on the D-ring occupies the b-subunit, animportant hydrogen bonding interactions were observed betweenO atom of methoxy group and OH of btyr202 (O---H). Moreover,trimethoxyphenyl group present as A-ring of 10b show polar inter-actions with aAsn101 and bLys254 (O---HN). Additionally, thepyrazole NH of the same conjugate formed a hydrogen bond withNH of bLeu255 (NH---NH). Whereas, benzimidazole NH develops aweak electrostatic interactions with bCys241. It is indicated thatthe presence of methoxy group on the D-ring of 10b favored tooccupy the trimethoxy substituent of A-ring at the interface ofa,b-subunits. On the other hand the binding pose of 10a alsoexhibited similar type of interactions comparable to 10b and sand-wiched at the interface of a,b-subunits of tubulin. A strong hydro-gen bond was found between O of trimethoxy group of A-ring andNH of bLys254 (O---HN). Interestingly, the O atom of the sameresidue forms a strong hydrogen bond with NH of the pyrazole(NH---O). In addition to these interactions a weak hydrogen bondwas observed between methoxy O of the A-ring and OH of thetyr224 (O---OH). Moreover, some weak electrostatic interactionswere identified with amino acid residues bval181, bmet259 andbcys241. Among the three potent conjugates 10d with a chlorineatom on the D-ring showed diminished antitubulin activity, whichmight be unsuccessful docking. Only a few hydrogen bondinginteractions were observed with tyr224 of a-subunit and Asn258b-subunit whereas, some hydrophobic interactions were also iden-tified with blys254, met259, leu255 and bcys241 of b-subunitexplaining that loss in activity of compound 10d when comparedto 10a and 10b. However, B and C-rings are buried at the interfaceof the two subunits of the tubulin. Specifically, 10b is a promisingtubulin polymerization inhibitor is sandwiched between thebLeu248, bLys254 and bcys241 of b-subunit and aAsn101 andaSer178 of a-subunit, exhibited successful molecular interactionswith tubulin. A pose of cocrystalized structure colchicine superim-posed with 10a also visualized to gain further insight of ourfindings (Fig. 8).
4. Conclusion
In summary, a series of arylpyrazole linked benzimidazole con-jugates (10a–t) comprising of A, B, C and D ring system were syn-thesized and evaluated for their cytotoxic activity against differenthuman cancer cell lines. Among them some representative conju-gates like 10a, 10b, 10d, 10e, 10p and 10r were tested throughNCI five dose screen. The most potent conjugates 10a, 10b and10d that possess trimethoxy phenyl group as A-ring exhibited pro-found cytotoxicity in most of the cancer cell lines at submicromo-lar concentrations. All the synthesized compounds (10a–t) alsoshowed significant antiproliferative activity against five cancer celllines such as HepG2 (liver carcinoma), A549 (lung cancer), MCF 7(breast cancer) Hela (cervical cancer) and DU-145 (prostate cancer)with the IC50 values ranging from 0.7 to 42 lM. A549 cells treated
with some of the potent conjugates like 10a, 10b and 10d causedaccumulation of the cells at G2/M phase of the cell cycle, activatecyclin-B1 protein levels and disrupt microtubule system. Ashypothesized, the lead compounds 10a, 10b and 10d elicit theircytotoxic activity by inhibition of tubulin polymerization withthe IC50 values 1.3 lM, 2.5 lM and 3.8 lM, respectively. These rep-resentative conjugates also activate the caspase-3 leading to induc-tion of apoptotic cell death. Furthermore, molecular modelinganalysis revealed that these conjugates considerably occupied col-chicine binding site of the tubulin. Finally, it is established suchconjugates containing pyrazole and benzimidazole moieties couldbe effective inhibitors of tubulin polymerization that are furtheramenable for the development of related newer molecules aspotential chemotherapeutic drugs.
5. Experimental section
5.1. General
All the chemicals and reagents were purchased from Aldrich(Sigma–Aldrich, St. Louis, MO, USA), Lancaster (Alfa Aesar, JohnsonMatthey Company, Ward Hill, MA, USA), or Spectrochem Pvt. Ltd(Mumbai, India). Progresses of all the reactions were monitoredby TLC performed on silica gel glass plates containing 60 GF-254,and visualizations were achieved by UV light or iodine indicator.Purification of the compounds has done by the application of Col-umn chromatography with Merck 60–120 mesh silica gel. 1H NMRspectra were recorded on Bruker UXNMR/XWIN-NMR (300 MHz)or Inova Varian-VXR-unity (400, 500 MHz) instruments. 13C NMRspectra were recorded on Bruker UXNMR/XWIN-NMR (75 MHz)instrument. Chemical shifts (d) are reported in ppm downfieldfrom an internal TMS standard. ESI spectra were recorded on aMicro mass Quattro LC using ESI+ software with capillary voltage3.98 kV and ESI mode positive ion trap detector. High-resolutionmass spectra (HRMS) were recorded on a QSTAR XL Hybrid MS–MS mass spectrometer. Melting points were determined with anElectro thermal melting point apparatus and are uncorrected.
5.2. General procedures
5.2.1. Synthesis of ethyl 2,4-dioxo-4-(substitutedphenyl)butanoates 6(a–d)
To the freshly prepared sodium ethoxide added an appropriateamount of diethyl oxalate (1.0 mol) at 0 �C. After proper dissolu-tion, different acetophenones 5(a–d) (1.0 mol) were chargedslowly in small portions by maintaining the temperature at 0 �C.The stirring was continued at room temperature for 4 h and com-pletion of reaction was confirmed by TLC. The reaction mixturewas neutralized with diluted H2SO4 and extracted with ethyl ace-tate to afford diketo esters 6(a–d) in good yields (85–90%). Thesecompounds were taken as such in the next step for the preparationof phenyl pyrazole esters.
5.2.2. Synthesis of ethyl 3-substituted phenyl-1H-pyrazole-5-carboxylates 7(a–d)
To each of the di keto esters 6(a–d) (1.0 mol) obtained in theearlier step was added NH2–NH2�2HCl (1.5 mol) in ethanol andthe reaction mixture heated to reflux for 3 h. Reactions were mon-itored by TLC using ethyl acetate and hexane as mobile phase. Thesolvent was removed under vacuum then added water to the res-idue followed by extracted with ethyl acetate (50 mL � 4). Theorganic layer so obtained was dried on anhydrous NaSO4 and evap-orated the solvent to yield crude products. Subsequently thesecompounds were purified by column chromatography using ethyl

1090 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
acetate and hexane. The pure compounds 7(a–d) were eluted at30–40% of ethyl acetate with good yields (70–80%).
5.2.2.1. Ethyl 3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-car-boxylate (7a). This compound was prepared employing themethod described above using trimethoxy acetophenone as start-ing material. Yellow solid; (yield 75.0%): Rf = 0.5 (50% ethyl ace-tate/hexane); 1H NMR (400 MHz, CDCl3); d 1.33–1.40 (t, 3H,J1 = 6.7 Hz, J2 = 7.5 Hz, –CH3), 3.88 (s, 3H, –OCH3), 3.95 (s, 6H, –OCH3) 4.33–4.45 (q, 2H, J1 = 6.7 Hz, J2 = 7.5 Hz, CH2), 7.01 (s, 2H,ArH), 7.05 (s, 1H, ArH) ppm; MS (ESI) m/z 307 [M+H].
5.2.2.2. Ethyl 3-(3,4-dimethoxyphenyl)-1H-pyrazole-5-carboxyl-ate (7b). This compound was prepared employing the methoddescribed above using 3,4-dimethoxy acetophenone as startingmaterial. Pale yellow solid; (yield 78.0%): Rf = 0.3 (30% ethyl ace-tate/hexane); 1H NMR (300 MHz, CDCl3); d 1.23–1.31 (t, 3H,J1 = 7.1 Hz, –CH3), 3.85 (s, 3H, –OCH3), 3.90 (s, 3H, –OCH3) 4.19–4.32 (q, 2H, J1 = 7.1 Hz, –CH2), 6.88 (d, 1H, J1 = 7.1 Hz, –ArH), 6.94(s, 1H, ArH), 7.21–7.28 (m, 1H, ArH) 7.29–7.34 (m, 1H, ArH) 9.67(br s, 1H, –NH) ppm; MS (ESI) m/z 277 [M+H].
5.2.2.3. Ethyl 3-(4-methoxyphenyl)-1H-pyrazole-5-carboxylate(7c). This compound was prepared employing the methoddescribed above using trimethoxy acetophenone as starting mate-rial. Yellow solid; yellow solid; (yield 80.0%): Rf = 0.3 (30% ethylacetate/hexane); 1H NMR (300 MHz, CDCl3); d 1.23–1.37 (t, 3H,J1 = 6.7 Hz, J2 = 7.5 Hz, –CH3), 3.81 (s, 3H, –OCH3), 4.17–4.36 (q,2H, J1 = 6.7 Hz, J2 = 7.5 Hz, CH2), 6.8 (s, 1H, ArH), 6.90 (d, 2H,J = 2.2 Hz, ArH), 7.62 (d, 2H, J = 9.0 Hz, ArH) ppm; MS (ESI) m/z247 [M+H].
5.2.2.4. Ethyl 3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-car-boxylate (7d). This compound was prepared employing themethod described above using trimethoxy acetophenone as start-ing material. Pale yellow colored solid; (yield 75.0%): Rf = 0.3(40% ethyl acetate/hexane); 1H NMR (500 MHz, CDCl3); d 1.33–1.40 (t, 3H, J1 = 6.7 Hz, J2 = 7.5 Hz, –CH3), 4.33–4.45 (q, 2H,J1 = 6.7 Hz, J2 = 7.5 Hz, CH2), 6.0 (s, 2H, OCH2O), 6.86 (d, 1H,J = 8.3 Hz, ArH)7.05 (s, 1H, ArH), 7.21–7.28 (m, 2H, ArH) ppm; MS(ESI) m/z 261 [M+H].
5.2.3. Synthesis of (3-substitutedphenyl-1H-pyrazol-5-yl)meth-anols 8(a–d)
To the each pyrazole carboxylates 7(a–d) produced in the pre-vious step was added LiAH4 (0.5 mol) in dry THF at 0 �C and contin-ued the stirred for 1 h at room temperature. After completion thereaction, added saturated NH4Cl solution drop wise to quenchthe LiAlH4. The solvent THF was removed under vacuum thenextracted with ethyl acetate (100 mL � 4). The organic layers weredried on anhydrous Na2SO4 and evaporated ethyl acetate to yieldcolor less solid products of (3-substitutedphenyl-1H-pyrazol-5-yl)methanols 8(a–d) (yield 70–80%). The pyrazole alcohols soproduced in this step were pure, and taken as such for theoxidation.
5.2.4. Synthesis of 3-subtitutedphenyl-1H-pyrazole-5-carbaldehydes 9(a–d)
To each (3-substitutedphenyl-1H-pyrazol-5-yl)methanols 9(a–d)produced in the earlier step was added IBX (1.2 mol) in DMSOand stirred for 1 h at room temperature. After completion of thereaction added ice cold water to the reaction mixture andextracted with ethyl acetate (50 mL � 4). The organic layers weredried on anhydrous Na2SO4 and evaporated the ethyl acetate toobtain pure compounds of 3-subtitutedphenyl-1H-pyrazole-5-car-
baldehydes 9(a–d) in good yields (80–85%). These pyrazole carbal-dehydes were as such taken in the next step for the preparation ofpyrazole linked benzimidazole conjugates (10a–t).
5.2.4.1. 3-(3,4,5-Trimethoxyphenyl)-1H-pyrazole-5-carbalde-hyde (9a). 3-(3,4,5-Trimethoxyphenyl)-1H-pyrazole-5-carbalde-hyde 9a was prepared using above method by the addition of(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)methanol 8a (2.64 g10 mmol) IBX (3.36 g 1.2 mmol). Yellow colored solid; (2.09 g yield80%): Rf = 0.3 (40% ethyl acetate/hexane); 1H NMR (400 MHz,CDCl3); d 3.82 (s, 3H, –OCH3), 3.92 (s, 6H, –OCH3), 6.85–7.25 (m,3H, ArH), 9.96 (s, 1H, CHO) ppm; MS (ESI) m/z 263 [M+H].
5.2.4.2. 3-(3,4-Dimethoxyphenyl)-1H-pyrazole-5-carbaldehyde(9b). 3-(3,4-Dimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9bwas prepared using above method by the addition of (3-(3,4-dime-thoxyphenyl)-1H-pyrazol-5-yl)methanol 8b (2.34 g 10 mmol) IBX(3.36 g 1.2 mmol). Yellow colored solid; (1.85 g yield 82%):Rf = 0.4 (40% ethyl acetate/hexane); 1H NMR (300 MHz, CDCl3); d3.87 (s, 3H, –OCH3), 3.91 (s, 3H, –OCH3), 6.93–7.04 (m, 1H, ArH),7.27–7.47 (m, 2H, ArH), 7.86–8.10 (m, 1H, ArH) 9.93 (s, 1H, CHO)ppm; MS (ESI) m/z 233 [M+H].
5.2.4.3. 3-(4-Methoxyphenyl)-1H-pyrazole-5-carbaldehyde(9c). 3-(4-Methoxyphenyl)-1H-pyrazole-5-carbaldehyde 9c wasprepared using above method by the addition of (3-(4-methoxy-phenyl)-1H-pyrazol-5-yl)methanol 8c (2.04 g 10 mmol) IBX(3.36 g 1.2 mmol). Yellow colored solid; (1.71 g yield 85%):Rf = 0.3 (40% ethyl acetate/hexane); 1H NMR (400 MHz, CDCl3); d3.85 (s, 3H, –OCH3), 7.78–7.82 (m, 1H, ArH), 7.86–7.93 (m, 2H,ArH), 7.95–8.03 (m, 2H, ArH) 9.95 (s, 1H, CHO) ppm; MS (ESI)m/z 203 [M+H].
5.2.4.4. 3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbalde-hyde (9d). 3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbalde-hyde 9d was prepared using above method by the addition of(3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)methanol 8d (2.18 g10 mmol) IBX (3.36 g 1.2 mmol). Yellow colored solid; (1.83 g yield80%): Rf = 0.3 (40% ethyl acetate/hexane); 1H NMR (300 MHz,CDCl3); d 6.03 (s, 2H, –OCH2O), 7.35–7.45 (m, 1H, ArH), 7.87–7.94(m, 2H, ArH), 8.10–8.17 (m, 1H, ArH) 10.2 (s, 1H, CHO ppm; MS(ESI) m/z 217 [M+H].
5.2.5. General procedure for synthesis of pyrazole linkedbenzimidazole conjugates 10(a–t)
To the each 3-subtitutedphenyl-1H-pyrazole-5-carbaldehydes(9a–d) (1.0 mmol) obtained in the above step was added varioussubstituted o-phenylenediamines (1.0 mmol) and catalytic amountof Na2S2O5 (5 mg). The reaction mixture was stirred at a tempera-ture of 85 �C for 3–4 h and the completion of reaction was con-firmed by TLC. The reaction mixture was cooled to roomtemperature and extracted with ethyl acetate (50 � 4 mL). Thecombined organic layer was washed with water, brine solutionand dried over anhydrous Na2SO4. Evaporation of the solvent undervacuum yielded crude product and further this was purified by col-umn chromatography using ethyl acetate/hexane to afford purecompounds of pyrazole linked benzimidazole conjugates 10(a–t)in good yields (75–85%).
5.2.5.1. 2-(3-(3,4,5-Trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10a). This compound was synthesizedemploying the same procedure described above by the additionof 3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a(262 mg, 1.0 mmol) and o-phenylenediamine (108 mg, 1.0 mmol).The compound was obtained as yellow solid. Yield: (285 mg,81.4%); mp: 184–185 �C; 1H NMR (400 MHz, CDCl3); d 3.76

A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1091
(s, 6H, –OCH3), 3.84 (s, 3H, –OCH3), 6.83 (s, 2H, ArH), 7.17–7.29 (m,3H, ArH), 7.59 (s, 2H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d54.8, 59.3, 100.3, 101.6, 120.9, 124.3, 136.6, 145.4, 152.2 ppm; IR(KBr) (mmax, cm�1): m = 3471, 3063, 1691, 1588, 1506, 1427, 1309,1272, 1243, 1309, 1272, 1243, 1187, 999 cm�1; MS (ESI) m/z 351[M+H]; HR-MS (ESI) m/z for C19H19N4O3 calculated m/z:351.1451, found m/z: 351.1451.
5.2.5.2. 5-Methoxy-2-(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10b). This compound was pre-pared using the procedure described above by the addition of3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a(262 mg, 1.0 mmol) and 4-methoxybenzene-1,2-diamine(138 mg, 1.0 mmol). The compound obtained as yellow coloredsolid. Yield: (305 mg, 80.1%); mp: 190–192 �C; 1H NMR(400 MHz, CDCl3); d 3.87 (s, 6H, –OCH3), 3.93 (s, 6H, –OCH3), 6.87(d, 1H, J = 8.8 Hz, ArH), 7.08 (s, 2H, ArH), 7.23 (s, 1H, ArH), 7.47–7.54 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 55.1,55.6, 59.8, 100.8, 102.5, 111.1, 125.0, 137.4, 145.7, 152.9,155.6 ppm; IR (KBr) (mmax, cm�1): m = 3069, 2936, 2832, 1737,1587, 1505, 1464, 1420, 1271, 1126, 1017, 994 cm�1; MS (ESI) m/z 381 [M+H]; HR-MS (ESI) m/z for C20H21N4O4 calculated m/z:381.1557, found m/z: 381.1559.
5.2.5.3. 5-Fluoro-2-(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10c). This compound was preparedusing the procedure described above by the addition of 3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a (262 mg,1.0 mmol) and 4-fluorobenzene-1,2-diamine (126 mg 1.0 mmol).The compound obtained as brown solid. Yield: (285 mg, 77.4%);mp: 180–183 �C; 1H NMR (400 MHz, CDCl3); d 3.86 (s, 3H, –OCH3), 3.93 (s, 6H, –OCH3), 6.90–7.01 (m, 1H, ArH), 7.08 (m, 2H,ArH), 7.22 (s, 1H, ArH), 7.59 (s, 2H, ArH); 13C NMR (75 MHz,CDCl3 + DMSO-d6): d 54.8, 59.3, 100.2, 101.7, 108.8, 109.1, 131.6,136.7, 146.7, 152.2, 156.3, 159.4 ppm; IR (KBr) (mmax, cm�1):m = cm�1; MS (ESI) m/z 369 [M+H]; HR-MS (ESI) m/z for C19H18N4-
O3F calculated m/z: 369.1357, found m/z: 369.1357.
5.2.5.4. 5-Chloro-2-(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10d). This compound was preparedusing the procedure described above by the addition of 3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a (262 mg,1.0 mmol) and 4-chlorobenzene-1,2-diamine (142 mg 1.0 mmol).The compound obtained as yellow solid. Yield: 310 mg (80.5%);mp: 204–206 �C; 1H NMR (300 MHz, CDCl3); d 3.86 (s, 3H, –OCH3), 3.94 (s, 6H, –OCH3), 7.08 (s, 2H, ArH), 7.18 (d, 1H,J = 8.4 Hz, ArH), 7.24 (s, 1H, ArH), 7.51–7.62 (m, 1H, ArH), 7.64 (s,1H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 54.2, 58.3,100.0, 101.1, 120.3, 124.7, 126.1, 128.3, 135.9, 138.7, 151.5 ppm;IR (KBr) (mmax, cm�1): m = 3280, 3170, 3165, 2956, 1740, 1596,1508, 1483, 1440, 1326, 1248, 1136, 998 cm�1; MS (ESI) m/z 385[M+H]; HR-MS (ESI) m/z for C19H18N4O3Cl calculated m/z:385.1061, found m/z: 385.1062.
5.2.5.5. 5,6-Dichloro-2-(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10e). This compound was pre-pared using the procedure described above by the addition of3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a(262 mg, 1.0 mmol) and 4,5-dichlorobenzene-1,2-diamine(175 mg 1.0 mmol). The compound obtained as yellow coloredsolid. Yield: 310 mg (74%); mp: 218–220 �C; mp: 190–192 �C; 1HNMR (300 MHz, CDCl3); d 3.85 (s, 3H, –OCH3), 3.93 (s, 6H, –OCH3), 7.07 (s, 2H, ArH), 7.22 (s, 1H, ArH), 7.69 (s, 2H, ArH), 12.7(br s, 1H, –NH), 13.3 (br s, 1H, –NH); 13C NMR (75 MHz, CDCl3 +DMSO-d6): d 55.0, 59.5, 100.7, 101.9, 124.3, 137.0, 152.4 ppm; IR(KBr) (mmax, cm�1): m = 3227, 3068, 2933, 1708, 1630, 1591, 1509,
1477, 1454, 1412, 1303, 1238, 1125, 996 cm�1; MS (ESI) m/z 419[M+H]; HR-MS (ESI) m/z for C19H17N4O3Cl2 calculated m/z:419.0672, found m/z: 419.0666.
5.2.5.6. 5-(Trifluoromethyl)-2-(3-(3,4,5-trimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10f). This compoundwas prepared using the procedure described above by the additionof 3-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9a(262 mg, 1.0 mmol) and 4-(trifluoromethyl)benzene-1,2-diamine(176 mg 1.0 mmol). The compound obtained as light brown col-ored crystal. Yield: 320 mg (76.5%); mp: 188–190 �C; 1H NMR(300 MHz, CDCl3); d 3.70 (s, 6H, –OCH3), 3.83 (s, 3H, –OCH3), 6.78(s, 2H, ArH), 7.17 (s, 1H, ArH), 7.41 (d, 1H, J = 8.3 Hz, ArH), 7.50–7.61 (m, 1H, ArH), 7.82 (s, 1H. ArH); 13C NMR (75 MHz, CDCl3 +DMSO-d6): d 54.9, 59.4, 100.7, 101.8, 117.7, 122.0, 122.4, 122.8,124.03, 125.6, 136.8, 144.4, 148.1, 152.3 ppm; IR (KBr) (mmax,cm�1): m = 3147, 2942, 1591, 1505, 1469, 1418, 1332, 1247, 1164,1126, 1051, 1002, 938 cm�1; MS (ESI) m/z 419 [M+H]; HR-MS(ESI) m/z for C20H18N4O3F3 calculated m/z: 419.1325, found m/z:419.1319.
5.2.5.7. 2-(3-(3,4-Dimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10g). This compound was prepared usingthe procedure described above by the addition of 3-(3,4-dime-thoxyphenyl)-1H-pyrazole-5-carbaldehyde 9b (232 mg 1.0 mmol)and o-phenylenediamine (108 mg, 1.0 mmol). The compoundobtained as yellow colored solid. Yield: 240 mg (74.9%); mp:196–198 �C; 180–182 �C; 1H NMR (300 MHz, CDCl3); d 3.91 (s,3H, –OCH3), 3.94 (s, 3H, –OCH3), 6.97 (d, 1H, J = 8.4 Hz, ArH),7.19–7.24 (m, 3H, ArH), 7.33–7.36 (m, 1H, ArH), 7.40 (s, 1H, ArH),7.58–7.68 (m, 2H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d54.5, 99.6, 107.8, 110.5, 113.6, 116.8, 120.8, 137.4, 145.0,147.7 ppm; IR (KBr) (mmax, cm�1): m = 3450, 3128, 2957, 1753,1622, 1588, 1562, 1491, 1445, 1329, 1295, 1245, 1126, 1041,937 cm�1; MS (ESI) m/z 321 [M+H]; HR-MS (ESI) m/z forC18H17N4O2 calculated m/z: 321.1346, found m/z: 321.1343.
5.2.5.8. 2-(3-(3,4-Dimethoxyphenyl)-1H-pyrazol-5-yl)-5-fluoro-1H-benzo[d]imidazole (10h). This compound was preparedusing the procedure described above by the addition of 3-(3,4-dimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9b (232 mg1.0 mmol) and 4-fluorobenzene-1,2-diamine (126 mg 1.0 mmol).The compound obtained as brown colored solid. Yield: (265 mg,78.3%); mp: 179–181 �C; 1H NMR (300 MHz, CDCl3); d 3.84 (s,3H, –OCH3), 3.89 (s, 3H, –OCH3), 6.95–7.07 (m, 2H, ArH), 7.22 (s,1H, ArH), 7.25–7.34 (m, 1H, ArH), 7.38 (d, 1H, J = 8.9 Hz, ArH),7.44 (s, 1H, ArH), 7.54 (s, 1H, ArH), 8.21 (br s, 1H, –NH); 13C NMR(75 MHz, CDCl3 + DMSO-d6): d 54.9, 100.1, 108.2, 110.9, 114.1,117.2, 121.3, 122.0, 137.8, 144.6, 145.7, 148.2 ppm; IR (KBr) (mmax,cm�1): m = 3421, 3108, 2956, 2836, 1691, 1576, 1545, 1496, 1414,1286, 1190, 1169, 1076, 947 cm�1; MS (ESI) m/z 339 [M+H]; HR-MS (ESI) m/z for C18H17N4O3F calculated m/z: 339.1257, foundm/z: 339.1256.
5.2.5.9. 5,6-Dichloro-2-(3-(3,4-dimethoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10i). This compound was preparedusing the procedure described above by the addition of 3-(3,4-dimethoxyphenyl)-1H-pyrazole-5-carbaldehyde 9b (232 mg1.0 mmol) and 4,5-dichlorobenzene-1,2-diamine (175 mg1.0 mmol). The compound obtained as brown colored solid. Yield:(295 mg, 75.8%); mp: 192–194 �C; 1H NMR (400 MHz, CDCl3); d3.91 (s, 3H, –OCH3), 3.94 (s, 3H, –OCH3), 6.96 (d, 1H, J = 8.0 Hz,ArH), 7.18 (s, 1H, ArH), 7.34 (d, 1H, J = 8.0 Hz, ArH), 7.38 (s, 1H,ArH), 7.62 (s, 1H, ArH), 7.71 (s, 1H, ArH), 13.25 (br s, 1H, NH);13C NMR (75 MHz, CDCl3 + DMSO-d6): d 54.4, 54.5, 99.8, 107.7,110.3, 116.8, 123.7, 147.7 ppm; IR (KBr) (mmax, cm�1): m = 3422,

1092 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
3258, 2924, 2859, 1615, 1589, 1508, 1441, 1409, 1257, 1171, 1021,969 cm�1; MS (ESI) m/z 390 [M+H]; HR-MS (ESI) m/z forC19H19N4O3 calculated m/z: 389.0572, found m/z: 390.0568.
5.2.5.10. 2-(3-(4-Methoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10j). This compound was prepared using the proce-dure described above by the addition of 3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg, 1.0 mmol) and o-phen-ylenediamine (108 mg, 1.0 mmol). The compound obtained as yel-low colored solid. Yield: (266 mg, 78% yield); mp: 174–176 �C; 1HNMR (300 MHz, CDCl3); d 3.87 (s, 3H, –OCH3), 6.99 (d, 1H,J = 8.4 Hz, ArH), 7.18–7.28 (m, 4H, ArH), 7.53 (s, 1H, ArH), 7.64 (s,1H, ArH), 7.73 (d, 2H, J = 8.1 Hz, ArH); 13C NMR (75 MHz, CDCl3 +DMSO-d6): d 54.6, 100.1, 113.7, 121.4, 126.2, 159.0 ppm; IR (KBr)(mmax, cm�1): m = 3326, 3162, 3010, 1725, 1655, 1610, 1580, 1525,1476, 1468, 1270, 1215, 1162, 1115, 1080, 1040, 975 cm�1; MS(ESI) m/z 291 [M+H]; HR-MS (ESI) m/z for C17H15N4O calculatedm/z: 291.1240, found m/z: 291.1238.
5.2.5.11. 5-Methoxy-2-(3-(4-methoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10k). This compound was preparedusing the procedure described above by the addition of 3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg,1.0 mmol) and 4-methoxybenzene-1,2-diamine (138 mg,1.0 mmol). The compound obtained as yellow colored solid. Yield:(265 mg, 82.8%); mp: 188–189 �C; 1H NMR (400 MHz, CDCl3); d3.83 (s, 6H, –OCH3), 6.82 (s, 1H, ArH), 6.96–7.07 (m, 2H, ArH),7.09–7.16 (m, 1H, ArH), 6.41–7.51 (m, 1H, ArH), 7.71–7.81 (m,2H, ArH), 7.13–8.21 (m, 1H, ArH); 13C NMR (75 MHz, CDCl3 +DMSO-d6): d 53.7, 54.1, 98.9, 110.1, 112.8, 125.3, 126.4, 129.0,130.7, 139.3, 154.6, 158.0 ppm; IR (KBr) (mmax, cm�1): m = 3613,3238, 2951, 2833, 1617, 1537, 1504, 1458, 1251, 1199, 1157,1027 cm�1; MS (ESI) m/z 321 [M+H]; HR-MS (ESI) m/z forC18H17N4O2 calculated m/z: 321.1346, found m/z: 321.1343.
5.2.5.12. 5-Fluoro-2-(3-(4-methoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10l). This compound was preparedusing the procedure described above by the addition of 3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg,1.0 mmol) and 4-fluorobenzene-1,2-diamine (126 mg 1.0 mmol).The compound obtained as yellow colored solid. Yield: (225 mg,72.9%); mp: 196–198 �C; 1H NMR (400 MHz, CDCl3); d 3.87 (s,3H, –OCH3), 6.96–7.01 (m, 2H, ArH), 7.16 (s, 1H, ArH), 7.49–7.55(m, 3H, ArH), 7.71 (d, 2H, J = 8.3 Hz, ArH); 13C NMR (75 MHz,CDCl3 + DMSO-d6): d 53.3, 98.8, 112.5, 124.9, 155.4, 157.6,158.5 ppm; IR (KBr) (mmax, cm�1): m = 3618, 3242, 2961, 1615,1534, 1497, 1459, 1347, 1302, 1259, 1179, 1137, 1107, 1024,960 cm�1; MS (ESI) m/z 309 [M+H]; HR-MS (ESI) m/z forC17H14FN4O calculated m/z: 309.1151, found m/z: 309.1158.
5.2.5.13. 5-Chloro-2-(3-(4-methoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10m). This compound was preparedusing the procedure described above by the addition of3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg,1.0 mmol) and 4-fluorobenzene-1,2-diamine (126 mg 1.0 mmol)and 4-chlorobenzene-1,2-diamine (142 mg 1.0 mmol). The com-pound obtained as yellow colored solid. Yield: (250 mg, 76.9%);mp: 184–186 �C; 1H NMR (400 MHz, CDCl3); d 3.85 (s, 3H,–OCH3), 6.99 (d, 1H, J = 8.6 Hz, ArH), 7.11–7.20 (m, 2H, ArH),7.49–7.69 (m, 2H, ArH), 7.72 (d, 2H, J = 8.4 Hz, ArH), 7.81 (s, 1H,ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 53.5, 99.0, 112.6,120.4, 125.0, 157.8 ppm; IR (KBr) (mmax, cm�1): m = 3614, 3230,3060, 2923, 2853, 1615, 1533, 1502, 1458, 1413, 1254, 1176,1026 cm�1; MS (ESI) m/z 325 [M+H]; HR-MS (ESI) m/z for C17H18
N4OCl calculated m/z: 325.0856, found m/z: 325.0841.
5.2.5.14. 5,6-Dichloro-2-(3-(4-methoxyphenyl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10n). This compound was preparedusing the procedure described above by the addition of 3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg,1.0 mmol) and 4,5-dichlorobenzene-1,2-diamine (175 mg1.0 mmol). The compound obtained as yellow colored solid. Yield:(270 mg, 75.2%); mp: 189–191 �C; 1H NMR (500 MHz, CDCl3); d3.87 (s, 3H, –OCH3), 6.99 (d, 1H, J = 8.3 Hz, ArH), 7.15 (s, 1H, ArH),7.53 (s, 3H, ArH), 7.67–7.76 (m, 2H, ArH); 13C NMR (75 MHz,CDCl3 + DMSO-d6): d 53.5, 99.2, 112.6, 123.1, 125.1, 132.5, 137.9,148.0, 157.8 ppm; IR (KBr) (mmax, cm�1): m = 3620, 3236, 3121,2926, 2857, 1685, 1620, 1582, 1536, 1476, 1448, 1323, 1265,1150, 1120, 1100, 1092, 1025, 966 cm�1; MS (ESI) m/z 360[M+H]; HR-MS (ESI) m/z for C17H13N2OCl2 calculated m/z:360.0466, found m/z: 360.0463.
5.2.5.15. 2-(3-(4-Methoxyphenyl)-1H-pyrazol-5-yl)-5-(trifluoro-methyl)-1H-benzo[d]imidazole (10o). This compound was pre-pared using the procedure described above by the addition of3-(4-methoxyphenyl)-1H-pyrazole-5-carbaldehyde (9c) (202 mg,1.0 mmol) and 4-(trifluoromethyl)benzene-1,2-diamine (176 mg1.0 mmol). The compound obtained as brown colored solid. Yield:265 mg (74.0%); mp: 203–205 �C; 1H NMR (500 MHz, CDCl3); d3.86 (s, 3H, –OCH3), 7.00 (d, 2H, J = 8.6 Hz, ArH), 7.17 (s, 1H, ArH),7.45 (d, 1H, J = 8.3 Hz, ArH), 7.73 (d, 1H, J = 8.3 Hz, ArH), 7.89 (s,3H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 54.7, 100.1,109.3, 109.6, 113.8, 126.3, 156.8, 159.0, 160.0 ppm; IR (KBr) (mmax,cm�1): m = 3619, 3233, 2968, 2847, 1617, 1535, 1507, 1463, 1442,1332, 1265, 1161, 1122, 1027, 963 cm�1; MS (ESI) m/z 359[M+H]; HR-MS (ESI) m/z for C18H14N4OF3 calculated m/z:359.1114, found m/z: 359.1114.
5.2.5.16. 2-(3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)-1H-benzo[d]imidazole (10p). This compound was prepared usingthe procedure described above by the addition of 3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbaldehyde 9d (216 mg1.0 mmol) and o-phenylenediamine (108 mg, 1.0 mmol). The com-pound obtained as light yellow colored solid. Yield: 195 mg (75%);mp: 206–208 �C; 1H NMR (400 MHz, CDCl3); d 6.04 (s, 2H, –OCH2
O–), 6.89 (d, 1H, J = 8.6 Hz, ArH), 7.16 (s, 1H, ArH), 7.19–7.24 (m,1H, ArH), 7.30 (s, 1H, ArH), 7.58–7.68 (m, 4H, ArH); IR (KBr) (mmax,cm�1): m = 3142, 2916, 1660, 1602, 1575, 1482, 1436, 1383, 1341,1280, 1215, 1145, 1090, 960 cm�1; MS (ESI) m/z 305 [M+H]; HR-MS (ESI) m/z for C17H13N4O2 calculated m/z: 305.1033, foundm/z: 305.1030.
5.2.5.17. 2-(3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)-5-methoxy-1H-benzo[d]imidazole (10q). This compound wasprepared using the procedure described above by the addition of3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbaldehyde 9d(216 mg 1.0 mmol) and 4-methoxybenzene-1,2-diamine (138 mg,1.0 mmol). The compound obtained as light yellow colored solid.Yield: (260 mg, 77.8%); mp: 186–188 �C; 1H NMR (300 MHz,CDCl3); d 3.87 (s, 3H, –OCH3), 6.01 (s, 2H, –OCH2O–), 6.89 (d, 1H,J = 8.4 Hz, ArH), 7.13 (s, 1H, ArH), 7.26–7.31 (m, 1H, ArH), 7.49 (s,4H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 53.8, 98.2,98.9, 99.5, 103.9, 104.1, 106.9, 109.7, 117.0, 117.5, 130.4, 145.7,146.2, 154.2 ppm; IR (KBr) (mmax, cm�1): m = 3420, 3261, 3101,1690, 1665, 1630, 1545, 1412, 1326, 1300, 1248, 1205, 1185,1165, 1025, 992 cm�1; MS (ESI) m/z 335 [M+H]; HR-MS (ESI) m/zfor C18H15N4O3 calculated m/z: 335.1138, found m/z: 335.1136.
5.2.5.18. 2-(3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)-5-flu-oro-1H-benzo[d]imidazole (10r). This compound was preparedusing the procedure described above by the addition of 3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbaldehyde 11d (150 mg

A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1093
0.0694 mmol) and 4-fluorobenzene-1,2-diamine (126 mg1.0 mmol). The compound obtained as light brown colored solid.Yield: (240 mg, 74.5%); mp: 190–192 �C; 1H NMR (500 MHz,CDCl3); 6.03 (s, 2H, –OCH2O–), 6.89 (d, 1H, J = 8.6 Hz, ArH), 6.92–6.98 (m, 1H, ArH), 7.12 (s, 1H, ArH), 7.24–7.34 (m, 2H, ArH), 7.59(s, 1H, ArH), 7.63 (s, 1H, ArH); 13C NMR (75 MHz, CDCl3 + DMSO-d6): d 100.4, 100.7, 105.4, 108.1, 118.8, 147.0, 147.5 ppm; IR(KBr) (mmax, cm�1): m = 3132, 3016, 2920, 1612, 1586, 1540, 1425,1412, 1365, 1280, 1251, 1140, 1026, 975, 940 cm�1; MS (ESI) m/z323 [M+H]; HR-MS (ESI) m/z for C17H12FN4O2 calculated m/z:323.09443, found m/z: 323.09399.
5.2.5.19. 2-(3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)-5,6-dichloro-1H-benzo[d]imidazole (10s). This compound wasprepared using the procedure described above by the addition of3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbaldehyde 11d(150 mg 0.0694 mmol) and 4,5-dichlorobenzene-1,2-diamine(175 mg 1.0 mmol). The compound as brown colored solid. Yield:(290 mg, 77.7%); mp: 180–182 �C; 1H NMR (400 MHz, CDCl3); d6.04 (s, 2H, –OCH2O–), 6.90 (d, 1H, J = 8.4 Hz, ArH), 7.12 (s, 1H,ArH), 7.24–7.32 (m, 1H, ArH), 7.70 (s, 1H, ArH), 7.75 (s, 2H, ArH);13C NMR (75 MHz, CDCl3 + DMSO-d6): d 99.8, 104.4, 104.5, 107.2,111.2, 117.9, 121.6, 123.5, 132.5, 134.0, 139.7, 141.8, 146.2,146.6 ppm; IR (KBr) (mmax, cm�1): m = 3129, 2909, 1609, 1575,1499, 1467, 1422, 1480, 1416, 1251, 1100, 1041, 971, 938 cm�1;MS (ESI) m/z 373 [M+H]; HR-MS (ESI) m/z for C17H11N4O2Cl2calcu-lated m/z: 373.0253, found m/z: 373.0256.
5.2.5.20. 2-(3-(Benzo[d][1,3]dioxol-5-yl)-1H-pyrazol-5-yl)-5-(trifluoromethyl)-1H-benzo[d]imidazole (10t). This compoundwas prepared using the procedure described above by the additionof 3-(benzo[d][1,3]dioxol-5-yl)-1H-pyrazole-5-carbaldehyde 11d(150 mg 0.0694 mmol) and 4-(trifluoromethyl)benzene-1,2-dia-mine (176 mg 1.0 mmol). The compound obtained as yellow col-ored solid. Yield: (265 mg, 71.2%); mp: 178–180 �C; 1H NMR(300 MHz, CDCl3); d 6.04 (s, 2H, –OCH2O–), 6.90 (d, 1H, J = 8.3 Hz,ArH), 7.18 (s, 1H, ArH), 7.30 (s, 1H, ArH), 7.51 (s, 4H, ArH); IR(KBr) (mmax, cm�1): m = 3220, 3120, 2936, 1665, 1620, 1572, 1566,1470, 1428, 1383, 1245, 1160, 1125, 1104, 1092, 1010, 956 cm�1;MS (ESI) m/z 373 [M+H]; HR-MS (ESI) m/z for C18H12N4O2F3 calcu-lated m/z: 373.0259, found m/z: 373.0256.
5.3. Cell Cultures, maintenance and antiproliferative evaluation
All cell lines used in this study were purchased from the Amer-ican Type Culture Collection (ATCC, United States). A549, MCF-7,and HeLa were grown in Dulbecco’s modified Eagle’s medium (con-taining 10% FBS in a humidified atmosphere of 5% CO2 at 37 �C).HepG2 and DU145 cells were cultured in Eagle’s minimal essentialmedium (MEM) containing non-essential amino acids, 1 mMsodium pyruvate, 10 mg/mL bovine insulin, and 10% FBS. Cellswere trypsinized when sub-confluent from T25 flasks/60 mmdishes and seeded in 96-wel plates. The synthesized test com-pounds were evaluated for their in vitro antiproliferative in fourdifferent human cancer cell lines. A protocol of 48 h continuousdrug exposure was used, and a MTT cell proliferation assay wasused to estimate cell viability or growth. The cell lines were grownin their respective media containing 10% fetal bovine serum andwere seeded into 96-well microtiter plates in 200 lL aliquots atplating densities depending on the doubling time of individual celllines. The microtiter plates were incubated at 37 �C, 5% CO2, 95%air, and 100% relative humidity for 24 h prior to addition of exper-imental drugs. Aliquots of 2 lL of the test compounds were addedto the wells already containing 198 lL of cells, resulting in therequired final drug concentrations. For each compound, four con-centrations (0.01, 0.1, 1, 10, and 100 lM) were evaluated, and each
was done in triplicate wells. Plates were incubated further for 48 h,and the assay was terminated by the addition of 10 lL of 5% MTTand incubated for 60 min at 37 �C. Later, the plates were air-dried.Bound stain was subsequently eluted with 100 lL of DMSO, andthe absorbance was read on an multimode plate reader (TecanM200) at a wavelength of 560 nm. Percent growth was calculatedon a plate by plate basis for test wells relative to control wells. Theabove determinations were repeated thrice. The growth inhibitoryeffects of the compounds were analyzed by generating doseresponse curves as a plot of the percentage surviving cells versuscompound concentration. The sensitivity of the cancer cells tothe test compound was expressed in terms of IC50, a value definedas the concentration of compound that produced 50% reduction ascompared to the control absorbance. IC50 values are indicated asmeans ± SD of three independent experiments. 31
5.4. Dot-blot assay
Cells were trypsinized when sub-confluent from T25 flasks/60 mm dishes and seeded in 6-well plates. The pyrazole-oxadiaz-ole conjugates were evaluated for their activity against Cyclin B1.A549 cells were treated with 5 lM concentrations of (10a, 10band 10d) for 24 h. Subsequently, cells were harvested and proteinswere quantified using Amido Black followed by densitometry anal-ysis. Equal amount of protein were blotted on nitrocellulose mem-brane using Bio-Dot SF microfiltration apparatus (Bio-Rad). Briefly,nitrocellulose membrane and 3 filters papers (Whatmann 3) weresoaked in IX TBS solution for 10 min. Later, the filter papers, mem-brane were arranged in the apparatus and connected to vacuumpump (Millipore). The membranes were rehydrated using 100 lLof 1� TBS by vacuum filtration. Subsequently, 50 lL volumes ofequal protein samples were blotted on the membrane and washedwith 200 lL of 1� TBS through application of vacuum. The blot wasblocked with 5% blotto for 1 h at room temperature. Immunoblotanalysis was performed as described previously using UVP, bio-spectrum 810 imaging system.32
5.5. Analysis of cell cycle
A549 cells were grown in 60 mm dishes and were incubated for24 h in the presence or absence of test compounds 10a, 10b and10d at 5 lM 10 lM concentrations. Cells were harvested usingTrypsin-EDTA, fixed with ice-cold 70% ethanol at 4 �C for 30 min,ethanol was removed by centrifugation and cells were stained with1 mL of DNA staining solution [0.2 mg of propidium Iodide (PI), and2 mg RNase A] for 30 min in dark at 37 �C as described earlier. TheDNA contents of 20,000 events were measured by flow cytometer(BD FACSCanto II). Histograms were analyzed using FCS express 4plus.32
5.6. Tubulin polymerization assay
An in vitro assay for monitoring the time-dependent polymeri-zation of tubulin to microtubules was performed employing a fluo-rescence-based tubulin polymerization assay kit (BK011,Cytoskeleton, Inc.) according to the manufacturer’s protocol. Thereaction mixture in a final volume of 50 lL in PEM buffer(80 mM PIPES, 0.5 mM EGTA, 2 mM MgCl2, pH 6.9) in 384 wellplates contained 2 mg/mL bovine brain tubulin, 10 lM fluorescentreporter, 1 mM GTP in the presence or absence of test compoundsat 37 �C. Tubulin polymerization was followed by monitoring thefluorescence enhancement due to the incorporation of a fluores-cence reporter into microtubules as polymerization proceeds. Fluo-rescence emission at 420 nm (excitation wavelength is 360 nm)was measured for 1 h at 1-min intervals in a multimode platereader (Tecan M200). To determine the IC50 values of the

1094 A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095
compounds against tubulin polymerization, the compounds werepre-incubated with tubulin at varying concentrations (0.01, 0.1,1, 10 and 20 lM). Assays performed under similar conditions asemployed for polymerization assays as described above.33
5.7. Immunohistochemistry of tubulin and analysis of nuclearmorphology
A549 cells were seeded on glass cover slip, incubated for 24 h inthe presence or absence of test compounds 10a, 10b and 10d at aconcentration of 5 lM. Cells grown on coverslips were fixed in 3.5%formaldehyde in phosphate-buffered saline (PBS) pH 7.4 for 10 minat room temperature. Cells were permeabilized for 6 min in PBScontaining 0.5% Triton X-100 (Sigma) and 0.05% Tween-20 (Sigma).The permeabilized cells were blocked with 2% BSA (Sigma) in PBSfor 1 h. Later, the cells were incubated with primary antibody fortubulin from (sigma) at (1:200) diluted in blocking solution for4 h at room temperature. Subsequently the antibodies wereremoved and the cells were washed thrice with PBS. Cells werethen incubated with FITC labeled anti-mouse secondary antibody(1:500) for 1 h at room temperature. Cells were washed thrice withPBS and mounted in medium containing DAPI. Images were cap-tured using the Olympus confocal microscope FLOW VIEW FV1000 series and analyzed with FV10ASW 1.7 series software.
5.8. Western blot analysis of soluble versus polymerized tubulin
A549 cells were seeded in 12-well plates at 1 � 105 cells perwell in complete growth medium. Following treatment of cellswith respective compounds 10a, 10b and 10d for duration of24 h, cells were washed with PBS and subsequently soluble andinsoluble tubulin fractions were collected. To collect the solubletubulin fractions, cells were permeabilized with 200 lL of pre-warmed lysis buffer [80 mM Pipes-KOH (pH 6.8), 1 mM MgCl2,1 mM EGTA, 0.2% Triton X-100, 10% glycerol, 0.1% protease inhibi-tor cocktail (Sigma-Aldrich)] and incubated for 3 min at 30 �C. Lysisbuffer was gently removed, and mixed with 100 lL of 3 � Lae-mmli’s sample buffer (180 mM Tris–Cl pH 6.8, 6% SDS, 15% glyc-erol, 7.5% b-mercaptoethanol and 0.01% bromophenol blue).Samples were immediately heated to 95 �C for 3 min. To collectthe insoluble tubulin fraction, 300 lL of 1 � Laemmli’s sample buf-fer was added to the remaining cells in each well, and the sampleswere heated to 95 �C for 3 min. Equal volumes of samples were runon an SDS-10% polyacrylamide gel and were transferred to a nitro-cellulose membrane employing semidry transfer at 50 mA for 1 h.Blots were probed with mouse anti-human a-tubulin diluted1:2000 mL (Sigma) and stained with rabbit anti-mouse secondaryantibody coupled with horseradish peroxidase, diluted 1:5000 mL(Sigma). Bands were visualized using an enhanced Chemilumines-cence protocol (Pierce) and radiographic film (Kodak).34
5.9. Competitive tubulin binding assay
The various concentrations (5 lM, 10 lM, 15 lM and 20 lM) ofconjugates 10a, 10b and 10d were coincubated with 3 lM colchi-cines in 30 mM Tris buffer containing 3 lM tubulin at 37 �C for60 min. The standard CA-4 was employed as a positive controlwhereas peclitaxel was used as the negative control. After incuba-tion, the fluorescence of tubulin–colchicine complex was measuredby using Tecan multimode reader at excitation wavelength of380 nm and emission wavelength of 435 nm; whereas 30 mM Trisbuffer was used as a blank. The raw fluorescence values were nor-malized first by subtracting the fluorescence of the buffer and thensetting the fluorescence of 3 lM tubulin with 3 lM colchicine to100%. Values represented were ± SD of at least three independentexperiments.
5.10. Caspase-3 assay
A549 cells were seeded in 12 well plates as mentioned aboveand were treated with different compounds 10a, 10b and 10d at5 lM concentration. After 24 h of incubation with the compound,cells were lysed using lysis buffer and incubated at 4 �C for10 min which then collected and spun at 4 �C, 10,000 rpm for10 min. The supernatant was collected which contains the protein.The protein was quantified employing Bradford Assay. Equalamount of proteins were added with assay buffer and caspase sub-strate which gives fluorescence units thereby calculated the activ-ity against caspase.34
6. Molecular modeling
AutoDock4 was employed to dock lead pyrazole linked benzim-idazoles in colchicine binding site of tubulin.35,36 Initial Cartesiancoordinates for the protein–ligand complex structure were derivedfrom crystal structure of tubulin (PDB ID: 3E22). The protein tar-gets were prepared for molecular docking simulation by removingwater molecules, bound ligands. Hydrogen atoms and Kollmancharges were added to each protein atom. Auto-Dock Tools-1.5.6(ADT) was used to prepare and analyze the docking simulationsfor the AutoDock4. Coordinates of each compound were generatedusing Chemdraw11.0 followed by MM2 energy minimization. Theinteraction of protein and ligands in binding pocket for Autodock4was defined by a Grid box. The grid box was created with 60 pointsequally in each direction of x, y, and z. AutoGrid4 was used to pro-duce grid maps for AutoDock4 calculations where the search spacesize utilized grid points of 0.375 Å. The Lamarckian genetic algo-rithm was opted to search for the best conformers. Each dockingexperiment was performed 100 times, yielding 100 docked confor-mations. Parameters used for the docking were as follows: popula-tion size of 150; random starting position and conformation;maximal mutation of 2 Å in translation and 50 degrees in rota-tions; elitism of 1; mutation rate of 0.02 and crossover rate of0.8; and local search rate of 0.06. Simulations were performed witha maximum of 1.5 million energy evaluations and a maximum of50,000 generations. Final docked conformations were clusteredusing a tolerance of 1.0 Å root mean square deviation. The bestmodel was picked based on the best stabilization energy. Finalfigures for molecular modeling were visualized by using PyMol.37
Acknowledgements
A.B.S, G.B.K thanks CSIR, New Delhi for the award of seniorresearch fellowships. We also thank CSIR for financial supportunder the 12th Five Year plan project ‘Affordable Cancer Therapeu-tics’ (CSC0301) and ‘Small Molecules in Lead Exploration (SMiLE)’(CSC0111).
Supplementary data
Supplementary data (mean growth percentage for inhibition(Table S1), GI50 values for NCI selected compounds (Table S2), fivedose results of selected compounds, 1H NMR and 13C NMR spectraof final compounds are provided) associated with this article canbe found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.01.004.
References and notes
1. Perez, E. A. Mol. Cancer Ther. 2009, 8, 2086.2. Dumontet, C.; Jordan, M. A. Nat. Rev. Drug Disc. 2010, 10, 790. Review. Erratum
in: Nat. Rev. Drug Disc. 2010, 9, 897.3. Jordan, A.; Wilson, L. Nat. Rev. Cancer 2004, 4, 253.

A. Kamal et al. / Bioorg. Med. Chem. 23 (2015) 1082–1095 1095
4. Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D. D. Pharm. Res. 2012, 29, 2943.5. Jordan, A.; Kamath, K. Curr. Cancer Drug Targets 2007, 7, 730.6. Mitchison, T.; Kirschner, M. Nature 1984, 312, 237.7. Bacher, G.; Beckers, T.; Emig, P.; Klenner, T.; Kutscher, B. Pure Appl. Chem. 2001,
73, 1459.8. Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Med. Res. Rev. 2008, 28,
155.9. Jain, N.; Yada, D.; Shaik, T. B.; Vasantha, G.; Reddy, P. S.; Kalivendi, S. V.;
Sreedhar, B. Chem. Med. Chem. 2011, 6, 859.10. Attia, S. M. J. Appl. Toxicol. 2013, 33, 426.11. Samson; Donoso, J. A.; Heller-Bettinger, I.; Watson, D.; Himes, R. H. J.
Pharmacol. Exp. Ther. 1979, 208, 411.12. Hörig, J. A.; Renz, P. Eur. J. Biochem. 1980, 105, 587.13. Chang, L.; Chang, C. S.; Chiang, P. C.; Ho, Y. F.; Liu, J. F.; Chang, K. W.; Guh, J. H.
Br. J. Pharmacol. 2010, 160, 1677.14. Kamal, A.; Reddy, M. K.; Shaik, T. B.; Rajender; Srikanth, Y. V.; Reddy, V. S.;
Kumar, G. B.; Kalivendi, S. V. Eur. J. Med. Chem. 2012, 50, 9.15. Shen, L.; Zhu, J.; Li, M.; Zhao, B. X.; Miao, J. Y. Eur. J. Med. Chem. 2012, 54, 287.16. El-Dakdouki, H.; Adamski, N.; Foster, L.; Hacker, M. P.; Erhardt, P. W. J. Med.
Chem. 2011, 54, 8224.17. Abu Thaher, B.; Arnsmann, M.; Totzke, F.; Ehlert, J. E.; Kubbutat, M. H.;
Schächtele, C.; Zimmermann, M. O.; Koch, P.; Boeckler, F. M.; Laufer, S. A. J. Med.Chem. 2012, 55, 961.
18. Li, C. M.; Wang, Z.; Lu, Y.; Ahn, S.; Narayanan, R.; Kearbey, J. D.; Parke, D. N.; Li,W.; Miller, D. D.; Dalton Cancer Res. 2011, 71, 216.
19. Chen, J.; Wang, Z.; Li, C. M.; Lu, Y.; Vaddady, P. K.; Meibohm, B.; Dalton, J. T.;Miller, D. D.; Li, W. J. Med. Chem. 2010, 53, 7414.
20. Kamal, A.; Shaik, A. B.; Jain, N.; Kishor, C.; Nagabhushana, A.; Supriya, B.;Kumar, G. B.; Chourasiya, S. S.; Suresh, Y.; Mishra, R. K.; Addlagatta, A. Eur. J.Med. Chem. 2013. http://dx.doi.org/10.1016/j.ejmech.2013.10.077.
21. Kamal, A.; Reddy, V. S.; Karnewar, S.; Chourasiya, S. S.; Shaik, A. B.;Nagabhushana, A.; Kumar, G. B.; Kishor, C.; Addlagatta, A.; Kotamraju, S.Chem. Med. Chem. 2015, 2013, 8.
22. Frisa, S.; Jacobberger, J. W. PLoS ONE 2009, 4, e7064.23. Hwang, A.; Maity, A.; McKenna, W. G.; Muschel, R. J. J. Biol. Chem. 1995, 270,
28419.24. Walczak, E.; Heald, R. Int. Rev. Cytol. 2008, 265, 111.25. Bhattacharyya; Wolff, J. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 2627.26. Bhattacharyya; Howard, R.; Maity, S. N.; Brossi, A.; Sharma, P. N.; Wolff, J. Proc.
Natl. Acad. Sci. U.S.A. 1986, 83, 2052.27. Risinger, L.; Westbrook, C. D.; Encinas, A.; Mülbaier, M.; Schultes, C. M.;
Wawro, S.; Lewis, J. D.; Janssen, B.; Giles, F. J.; Mooberry, S. L. J. Pharmacol. Exp.Ther. 2011, 336, 652.
28. Beale, M.; Allwood, D. M.; Bender, A.; Bond, P. J.; Brenton, J. D.; Charnock-Jones,D. S.; Ley, S. V.; Myers, R. M.; Shearman, J. W.; Temple, J.; Unger, J.; Watts, C. A.;Xian, J. ACS Med. Chem. Lett. 2012, 19, 177.
29. Nitesh, S.; Vaibhav, J.; Ranjan, P.; Somnath, K.; Sarita, D.; Neha, T.; Purusottam,M.; Garima, P.; Maneesh, K.; Dipon, D.; Shakti, R.; Satapathy; Sumit, S.; Sankar,K.; Guchhait, N. K.; Chanakya; Bharatam, P. V. Med. Chem. Commun. 2014, 5,766.
30. Kamal, A.; Sultana, F.; Ramaiah, M. J.; Srikanth, Y. V.; Viswanath, A.; Kishor, C.;Sharma, P.; Pushpavalli, S. N.; Addlagatta, A.; Pal-Bhadra, M. ChemMedChem2012, 6, 292.
31. Kamal, A.; Rao, M. P.; Swapna, P.; Srinivasulu, V.; Bagul, C.; Shaik, A. B.;Mullagiri, K.; Kovvuri, J.; Reddy, V. S.; Vidyasagar, K.; Nagesh, N. Org. Biomol.Chem. 2014, 12, 2370.
32. Kamal, A.; Shaik, A. B.; Polepalli, S.; Reddy, V. S.; Kumar, G. B.; Gupta, S.;Krishna, K. V.; Nagabhushana, A.; Mishra, R. K.; Jain, N. Org. Biomol. Chem. 2014,12, 7993.
33. Reddy, M. A.; Jain, N.; Yada, D.; Kishore, C.; Vangala, J. R.; Surendra, R. P.;Addlagatta, A.; Kalivendi, S. V.; Sreedhar, B. J. Med. Chem. 2011, 54, 6751.
34. Vangala, R.; Dudem, S.; Jain, N.; Kalivendi, S. V. J. Biol. Chem. 2014, 289, 12612.35. AutoDock, version4.0; http://www.scripps.edu/mb/olson/doc/autodock/.36. DeLano, W. L. Scientific, San Carlos, CA, USA, http://www.pymol.org., 2002.37. Ravelli, R. B.; Gigant, B.; Curmi, P. A.; Jourdain, I.; Lachkar, S.; Sobel, A.;
Knossow, M. Nature 2004, 428, 198.
Related Documents











