Designed Monomers and Polymers 11 (2008) 473–482 www.brill.nl/dmp Synthesis, Characterization and Thermally-Activated Curing of Azobenzene-Containing Benzoxazines Bar˙ ıs K˙ ıskan, Fatma Dogan, Yasem˙ ın Yuksel Durmaz and Yusuf Yagc˙ ı ∗ Istanbul Technical University, Department of Chemistry, Maslak 34469, Istanbul, Turkey Abstract Two new benzoxazine monomers with aliphatic and aromatic substituents, as well as azobenzene moieties were synthesized and characterized by spectroscopic methods. Their thermal curing behavior in the absence of any catalyst was investigated by differential scanning calorimetry. The thermal properties of the cross- linked structures were also studied by thermogravimetric analysis. While the monomeric forms gave some indication of thermally-induced isomerization, the azo groups were shown to be fixed in the disordered form in the densely cross-linked matrix. © Koninklijke Brill NV, Leiden, 2008 Keywords Thermoset, polybenzoxazine, azobenzene, thermal curing, benzoxazine 1. Introduction Polybenzoxazines are a relatively new class of versatile materials that can be used in many fields, such as electronics and aerospace industries, because they have a good combination of attractive properties. These include nearly zero shrinkage upon curing [1], thermal stability [2, 3] and chemical resistance [4]. Polybenzoxazines formed by thermally-activated ring-opening of the corresponding benzoxazines without any catalyst and without generating any by-products [4, 5] (Scheme 1). Moreover, benzoxazine monomers can be prepared simply from inexpensive and commercially available phenols, primary amines and formaldehyde [6]. Therefore, the chemistry of benzoxazine synthesis offers a wide range of molecular design flexibility by using appropriate starting materials and polybenzoxazine properties can be tailored. For specific applications, properties of polybenzoxazines can be improved in several ways. For example, allyl [7], acetylene [8], nitrile [9], propargyl [10] and maleimide [11] functional benzoxazines were synthesized in order to insert addi- tional cross-linking sites for thermal curing. The mechanical and thermal properties * To whom correspondence should be addressed. E-mail: [email protected] © Koninklijke Brill NV, Leiden, 2008 DOI:10.1163/156855508X328167

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Designed Monomers and Polymers 11 (2008) 473–482www.brill.nl/dmp
Synthesis, Characterization and Thermally-ActivatedCuring of Azobenzene-Containing Benzoxazines
Barı̇s Kı̇skan, Fatma Dogan, Yasemı̇n Yuksel Durmaz and Yusuf Yagcı̇ ∗
Istanbul Technical University, Department of Chemistry, Maslak 34469, Istanbul, Turkey
AbstractTwo new benzoxazine monomers with aliphatic and aromatic substituents, as well as azobenzene moietieswere synthesized and characterized by spectroscopic methods. Their thermal curing behavior in the absenceof any catalyst was investigated by differential scanning calorimetry. The thermal properties of the cross-linked structures were also studied by thermogravimetric analysis. While the monomeric forms gave someindication of thermally-induced isomerization, the azo groups were shown to be fixed in the disordered formin the densely cross-linked matrix.© Koninklijke Brill NV, Leiden, 2008
KeywordsThermoset, polybenzoxazine, azobenzene, thermal curing, benzoxazine
1. Introduction
Polybenzoxazines are a relatively new class of versatile materials that can be used inmany fields, such as electronics and aerospace industries, because they have a goodcombination of attractive properties. These include nearly zero shrinkage uponcuring [1], thermal stability [2, 3] and chemical resistance [4]. Polybenzoxazinesformed by thermally-activated ring-opening of the corresponding benzoxazineswithout any catalyst and without generating any by-products [4, 5] (Scheme 1).
Moreover, benzoxazine monomers can be prepared simply from inexpensive andcommercially available phenols, primary amines and formaldehyde [6]. Therefore,the chemistry of benzoxazine synthesis offers a wide range of molecular designflexibility by using appropriate starting materials and polybenzoxazine propertiescan be tailored.
For specific applications, properties of polybenzoxazines can be improved inseveral ways. For example, allyl [7], acetylene [8], nitrile [9], propargyl [10] andmaleimide [11] functional benzoxazines were synthesized in order to insert addi-tional cross-linking sites for thermal curing. The mechanical and thermal properties
* To whom correspondence should be addressed. E-mail: [email protected]
© Koninklijke Brill NV, Leiden, 2008 DOI:10.1163/156855508X328167

474 B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482
Scheme 1. Thermally-activated ring-opening reaction of bisbenzoxazine monomers.
of polybenzoxazines can be improved with the formation of blends [4, 12, 13],co-polymers [14] and polybenzoxazine-based organic–inorganic nanocomposites[15–17]. The use of benzoxazine functional macromonomers of polystyrene [18],poly(ε-caprolactone) [19], poly(propylene oxide) [20] and poly(methyl methacry-late) [21] has recently been introduced as an alternative approach to modify theproperties of polybenzoxazines.
Recently, ‘stimuli-responsive’ materials have received increasing attention. Var-ious types of stimuli, e.g., temperature, pH, ionic strength, solvent vapors, light, oran electrical field, can affect properties and conformation of polymers [22–30].
Polymers-containing azobenzene units are of special interest with respect to thereversibility of the photo-orientation and thermal cis to trans isomerization [31, 32].Interconversions between trans and higher energy cis isomeric states can be effectedboth photo-chemically (trans ↔ cis) and thermally (cis → trans) with a high degreeof efficiency and an absence of competing side reactions. That one configurationalisomer is furnished photo-chemically while the other is favored thermally makes itpossible to effectively drive or ‘switch’ azobenzene-modified species into a desiredgeometry and polarity. Thus, incorporation of azobenzene moieties into polymersmight bring remarkable photo- and thermo-regulated behavior when subjected tochanges in incident light or heat [33–38].
In this work, we synthesized two benzoxazine monomers with aliphatic andaromatic substituents as well as azobenzene moieties, and then studied thermalcuring behavior in the absence of any catalyst. We further searched if the result-ing networks would exhibit cis–trans isomerization. The thermal properties of thecross-linked structures were also investigated.
2. Experimental
2.1. Chemicals
Phenol (Aldrich, 97%), paraformaldehyde (Acros, 96%), sodium hydroxide (Acros,97+%), n-butylamine (Acros, 98%), 1,4-dioxane (Aldrich, �99%) , sodium nitrite(Acros, 99%), sodium sulfate (Acros, 99%), hydrochloric acid (Acros, ca. 37%),aniline (Acros, 99.8%), chloroform (Acros, 99+%), diethylether (Acros, 99+%)and ethanol (Acros, 99%) were used as received.

B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482 475
2.2. Characterization
1H-NMR spectra were recorded in CDCl3 with Si(CH3)4 as an internal standard,using a Bruker AC250 instrument at a proton frequency of 250.133 MHz. FT-IRspectra were recorded on a Perkin-Elmer FT-IR Spectrum One spectrometer viaattenuated total reflectance technique with 20 scans for each sample. Differentialscanning calorimetry (DSC) was performed on a Perkin-Elmer Diamond DSC witha heating rate of 10◦C/min under nitrogen flow (20 ml/min). Thermogravimetricanalysis (TGA) was performed on Perkin-Elmer Diamond TA/TGA with a heatingrate of 10◦C/min under nitrogen flow (200 ml/min). UV-Vis spectra were recordedon a Shimadzu UV-1601 spectrometer.
2.3. Synthesis of 4-(Phenyldiazenyl)phenol
In a flask, concentrated HCl (8 ml) and water (8 ml) were added. This solution wascooled to 0◦C and aniline (2.5 g, 27 mmol) was added. Then, NaNO2 (2 g, 29 mmol)in 10 ml water was added to the cooled solution slowly with heat control, keepingthe solution temperature below 10◦C. After addition, solution was stirred 20 minin ice-bath. Then, separately, phenol (2.5 g, 27 mmol) was dissolved in a 25 ml of10% NaOH solution. This prepared solution was added slowly to the diazonium saltcontaining solution under stirring, keeping the temperature below 15◦C. Stirringwas continued for 45 min. The formed yellow-orange solid was filtered and washedwith water. The raw solid was crystallized with a ethanol/water mixture. Yield 80%.
2.4. Synthesis of 3-Phenyl-6-(phenyldiazenyl)-3,4-dihydro-2H-benzo[e][1,3]oxazine
In a 250-ml flask, paraformaldehyde (0.65 g, 22 mmol) in 100 ml dioxane wascooled in an ice bath. To this solution, aniline (1 g, 11 mmol) in 25 ml dioxane wasadded portion-wise. The solution was kept stirring for 15 min below 5◦C. There-after, a solution of 4-(phenyldiazenyl)phenol (2 g, 10 mmol) in 25 ml dioxane wasadded. The solution was refluxed at 110◦C for 6 h. Removal of the solvent in arotary evaporator gave a viscous residue that was dissolved in 100 ml diethyletherand washed several times with 0.1 M NaOH solution and finally with distilled wa-ter. Then, the ether solution was dried with anhydrous sodium sulfate, followed byevaporation of ether under vacuum to afford pale orange solid. Yield 60%.
2.5. Synthesis of 3-Butyl-3,4-dihydro-2H-benzo[e][1,3]oxazine
The same procedure as mentioned above for benzoxazine was used. Yield 63%.
3. Results and Discussion
Azobenzene-containing benzoxazines were synthesized simply from 4-(phenyl-diazenyl)phenol using the method described in the literature (see Scheme 2) [35].
The chemical structure of benzene and butyl substituted compounds, namely3-phenyl-6-(phenyldiazenyl)-3,4-dihydro-2H-benzo[e][1,3]oxazine (AzoP-a) and

476 B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482
Scheme 2. Synthesis of AzoP-a and AzoP-b.
Figure 1. FT-IR spectra of (a) AzoP-a and (b) AzoP-b.
3-butyl-3,4-dihydro-2H-benzo[e][1,3]oxazine (AzoP-b) was confirmed by both FT-IR and 1H-NMR spectroscopy. The FT-IR spectrum presented in Fig. 1a showscharacteristic absorptions of benzoxazine structure for AzoP-a at 1228 cm−1
(asymmetric stretching of C–O–C), 1366 cm−1 (CH2 wagging), and 919, 931and 1493 cm−1 (tri-substituted benzene ring). Absorption bands of azo groupare typically located at 1456 cm−1 (stretching of trans N=N). The butyl substi-tuted benzoxazine compound AzoP-b exhibits similar absorptions with slight shifts(Fig. 1b); benzoxazine C–O–C at 1232 cm−1, –CH2 at 1375 cm−1, benzene ringstretching at 913 cm−1 and 1491 cm−1, and stretching of trans N=N absorptionband at 1468 cm−1.
The 1H-NMR spectra shown in Figs 2 and 3 also establish the structures of bothmonomers. The two singlets at 4.73 and 5.43 ppm are typical for the protons ofAr–CH2–N and O–CH2–N in the oxazine ring of AzoP-a, respectively. The aro-matic protons appeared between at 6.90–7.86 ppm (Fig. 2). In Fig. 3, the 1H-NMRspectrum of AzoP-b shows the similar oxazine Ar–CH2–N and O–CH2–N singletat 4.09 and 5.02 ppm. In this case, butyl group signals are clearly detectable.

B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482 477
Figure 2. 1H-NMR spectrum of AzoP-a.
Figure 3. 1H-NMR spectrum of AzoP-b.
The thermally-activated cure behavior of azobenzene monomers was studied byDSC and compared with that of the model monomer B-a (without azobenzenemoiety). The results are summarized in Table 1. As stated previously, benzox-azine groups are expected to undergo ring-opening polymerization. This exothermicevent was detected for both monomers (Figs 4 and 5). AzoP-a exhibited an onsetat 222◦C and a maximum at 229◦C with an exotherm energy of 413 J/g. Similarly,for AzoP-b, the onset of the exotherm started at about 224◦C with a maximum at241◦C, and 173 J/g as the heat of polymerization.

478 B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482
Table 1.DSC characteristics of AzoP-a, AzoP-b and B-a monomer
Monomer Melting point Onset of curing Maximum curing Heat of exotherm(◦C) (◦C) (◦C) (J/g)
B-a 97 239 252 198AzoP-a 116 222 229 413AzoP-b 91 224 241 173
DSC experiments were performed at a heating rate of 10◦C/min under nitrogen flow.
Figure 4. DSC thermogram of AzoP-a.
Figure 5. DSC thermogram of AzoP-b.
B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482 479
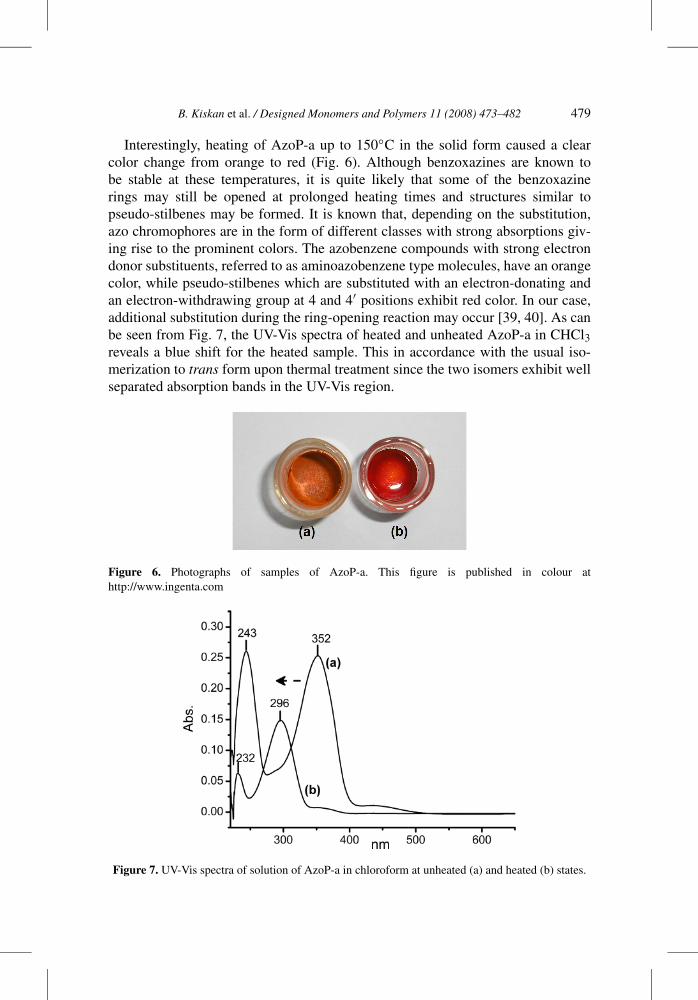
Interestingly, heating of AzoP-a up to 150◦C in the solid form caused a clearcolor change from orange to red (Fig. 6). Although benzoxazines are known tobe stable at these temperatures, it is quite likely that some of the benzoxazinerings may still be opened at prolonged heating times and structures similar topseudo-stilbenes may be formed. It is known that, depending on the substitution,azo chromophores are in the form of different classes with strong absorptions giv-ing rise to the prominent colors. The azobenzene compounds with strong electrondonor substituents, referred to as aminoazobenzene type molecules, have an orangecolor, while pseudo-stilbenes which are substituted with an electron-donating andan electron-withdrawing group at 4 and 4′ positions exhibit red color. In our case,additional substitution during the ring-opening reaction may occur [39, 40]. As canbe seen from Fig. 7, the UV-Vis spectra of heated and unheated AzoP-a in CHCl3reveals a blue shift for the heated sample. This in accordance with the usual iso-merization to trans form upon thermal treatment since the two isomers exhibit wellseparated absorption bands in the UV-Vis region.
Figure 6. Photographs of samples of AzoP-a. This figure is published in colour athttp://www.ingenta.com
Figure 7. UV-Vis spectra of solution of AzoP-a in chloroform at unheated (a) and heated (b) states.

480 B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482
The cured samples did not show any indication of isomerization, niether ther-mally nor photo-chemically. This may be due to the highly dense cross-linkedstructure observed with benzoxazines.
Thermal stability of the cured AzoP-a and AzoP-b was investigated by TGA un-der nitrogen exposure. The TGA and derivate profiles of cured AzoP-a (a), (ad) andcured AzoP-b (b), (bd), respectively, are shown in Fig. 8 and the results are summa-rized in Table 2. It can be seen that the char yield at 800◦C of the cured AzoP-a issignificantly higher than cured AzoP-b benzoxazine. This behavior can be attributedto the constructive effect of the extra phenyl ring of AzoP-a on the thermal stabil-ity. Also, the initial weight loss temperature of cured AzoP-a is slightly higher thanthe cured AzoP-b benzoxazine. Both cured materials exhibited mainly two mainweight loss events before 300◦C and at around 400◦C (Fig. 8). The first degrada-tion is assigned to the amine evaporation and is the consequence of the Mannichbase cleavage and the initial degradation. The second and third major weight losscan be ascribed to the phenol degradation.
Figure 8. TGA thermograms and their derivatives of cured AzoP-a (a), (ad) and cured AzoP-b (b),(bd), respectively.
Table 2.TGA analysis of cured AzoP-a and AzoP-b
Cured product T5%a (◦C) T10%
b (◦C) Td,max (◦C) Ycc at 800◦C (%)
AzoP-a 267 308 283, 446 58AzoP-b 225 245 258, 365 38
TGA analysis was performed at a heating rate of 10◦C/min under nitrogen flow (200 ml/min).

B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482 481
In conclusion, benzoxazines with azobenzene chromophoric groups have beensynthesized and characterized. These monomers have been shown to readily un-dergo thermal ring-opening reaction in the absence of added catalyst to formcross-linked polymer networks. The benzoxazines cured this way did not exhibitthermally- and photo-chemically-induced isomerization and azobenzenes are in thedisordered form in highly dense cross-linked network. However, the cured prod-ucts have high thermal stability and it was oberserved that AzoP-a exhibited higherthermal stability because of the additional aromatic group. Further studies on thepreparation of benzoxazines with azogroups yielding less dense cross-linked struc-tures are now in progress.
References
1. H. Ishida and D. J. Allen, J. Polym. Sci. Part B: Polym. Phys. 34, 1019 (1996).2. H. Y. Low and H. Ishida, Polym. Degrad. Stabil. 91, 805 (2006).3. B. Kiskan, Y. Yagci, E. Sahmetlioglu and L. Toppare, J. Polym. Sci. Part A: Polym. Chem. 45, 999
(2007).4. N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci. 32, 1344 (2007).5. X. Ning and H. Ishida, J. Polym. Sci. Part A: Polym. Chem. 32, 1121 (1994).6. W. J. Burke, J. Am. Chem. Soc. 71, 609 (1949).7. D. F. Pei, Y. Gu and X. X. Cai, Acta Polym. Sin. 5, 595 (1998).8. H. J. Kim, Z. Brunovska and H. Ishida, Polymer 40, 1815 (1999).9. Z. Brunovska and H. Ishida, J. Appl. Polym. Sci. 73, 2937 (1999).
10. T. Agag and T. Takeichi, Macromolecules 34, 7257 (2001).11. Y. L. Liu, J. M. Yu and C. I. Chou, J. Polym. Sci. Part A: Polym. Chem. 42, 5954 (2004).12. H. Ishida and Y. H. Lee, J. Appl. Polym. Sci. 81, 1021 (2001).13. H. Ishida and Y. H. Lee, Polymer 42, 6971 (2001).14. B. Kiskan, Y. Yagci and H. Ishida, J. Polym. Sci. Part A: Polym. Chem. 46, 414 (2008).15. H. Ishida and S. Heights, US Patent 6,323,270 (2001).16. Y. H. Liu, W. Zhang, Y. Chen and S. X. Zheng, J. Appl. Polym. Sci. 99, 927 (2006).17. J. Zhang, R. Xu and D. Yu, Eur. Polym. J. 43, 743 (2007).18. B. Kiskan, D. Colak, A. E. Muftuoglu, I. Cianga and Y. Yagci, Macromol. Rapid Commun. 26,
819 (2005).19. B. Kiskan and Y. Yagci, Polymer 46, 11690 (2005).20. A. Yildirim, B. Kiskan, A. L. Demirel and Y. Yagci, Eur. Polym. J. 42, 3006 (2006).21. M. A. Tasdelen, B. Kiskan and Y. Yagci, Macromol. Rapid Commun. 27, 1539 (2006).22. Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stoever, Macromolecules 38, 5937 (2005).23. H. Zhang and J. Ruehe, Macromolecules 38, 4855 (2005).24. M. O. Gallyamov, B. Tartsch, A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and
M. Moeller, Macromol. Rapid Commun. 25, 1703 (2004).25. M. O. Gallyamov, B. Tartsch, A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and
M. Moeller, J. Microsc. 215, 245 (2004).26. M. O. Gallyamov, B. Tartsch, A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and
M. Moeller, Chem. Eur. J. 10, 4599 (2004).27. T. Tanaka, H. Ogino and M. Iwamoto, Langmuir 23, 11417 (2007).

482 B. Kı̇skan et al. / Designed Monomers and Polymers 11 (2008) 473–482
28. K. Namiki, A. Sakamoto, M. Murata, S. Kume and H. Nishihara, Chem. Commun. 44, 4650(2007).
29. V. E. Campbell, P. Paoprasert, J. D. Mykietyn, I. In, D. J. Mcgee and P. Gopalan, J. Polym. Sci.Part A: Polym. Chem. 45, 3166 (2007).
30. A. Natansohn and P. Rochon, Chem. Rev. 102, 4139 (2002).31. T. Asano, T. Yano and T. Okada, J. Am. Chem. Soc. 104, 4900 (1982).32. T. Asano and T. Okada, J. Org. Chem. 49, 4387 (1984).33. H. Durr and H. B. Laurent (Eds), in: Photochromism — Molecules and Systems, p. 172. Elsevier,
New York, NY (1990).34. G. S. Kumar and D. C. Neckers, Chem. Rev. 89, 1915 (1989).35. G. S. Kumar, in: Azo Functional Polymers: Functional Group Approach in Macromolecular De-
sign, p. 91. Technomic Publishing, Lancaster, PA (1992).36. M. Irie, Advances in Polymer Science, Vol. 94. Springer, Berlin (1990).37. M. S. Beattie, C. Jacksont and G. D. Jaycox, Polymer 39, 2597 (1998).38. H. Lee, J. Pietrasik and K. Matyjaszewski, Macromolecules 39, 3914 (2006).39. C. J. Barrett, J. Mamiya, K. G. Yager and T. Ikeda, Soft Matter 3, 1249 (2007).40. Y. Yu and T. Ikeda, Macromol. Chem. Phys. 206, 1705 (2005).
Related Documents











