Synthesis and transformation of iron-based layered double hydroxides C. Ruby a, ⁎, M. Usman a , S. Naille a , K. Hanna a , C. Carteret a , M. Mullet a , M. François b , M. Abdelmoula a a Laboratoire de Chimie Physique et Microbiologie pour l'Environnement (LCPME), UMR7564 CNRS-Nancy Université, 405, Rue de Vandœuvre 54600 Villers-Lès-Nancy, France b Institut Jean Lamour, Département P2M, Matériaux-Métallurgie-Nanosciences-Plasmas-Surfaces, UMR 7198-CNRS-Nancy-Université-UPV-Metz, Faculté des Sciences, BP 239, 54506 Villers-lès-Nancy, France abstract article info Article history: Received 16 July 2009 Received in revised form 15 October 2009 Accepted 13 November 2009 Available online 26 November 2009 Keywords: LDH Green rust Pyroaurite Hydrotalcite Fougerite Mössbauer spectroscopy Iron-based layered double hydroxides (LDHs) have the general formula [M II (1-x) M III x (OH) 2 ] x+ ·[(x/n) A n− , m H 2 O] x− and contain a molar fraction of iron, i.e. Fe II or Fe III situated in the cationic layers, higher than 50%. LDHs containing Fe II species are interesting materials for several applications such as the reduction of anionic pollutants or the degradation of organic pollutants. They are mostly prepared either by coprecipitation of dissolved species or by oxidation of hydroxylated Fe II species. The synthesis routes were visualised in binary and ternary mass-balance diagrams. The LDH[Fe II –Fe III ] particles were well crystallised hexagonal plates, the size of which diminishes rapidly when divalent or trivalent cations substitute ions. The LDH[Fe II –Fe III –CO 3 ] transformed into a mixture of magnetite Fe 3 O 4 and siderite FeCO 3 in alkaline conditions, but the adsorption of silicate or phosphate anions on the lateral faces of the crystal prevented this decomposition. In oxic conditions, two mechanism of transformation were identified: (i) a fast in situ oxidation within the solid, (ii) dissolution–precipitation of the LDH forming ferric oxyhydroxides such as goethite (α-FeOOH). © 2009 Elsevier B.V. All rights reserved. 1. Introduction Mixed M II –M III layered double hydroxides (LDHs) are described by the general formula [M II (1-x) M III x (OH) 2 ] x+ ·[(x/n) A n− , m H 2 O] x− where M II and M III are metal cations in the brucite-type layers Mg (OH) 2 , A n− are the intercalated anions and x is the molar fraction of the trivalent cation and also the electrostatic charge of both the brucite-type layers and the anionic interlayers. Most commonly, the values of x are found in the range of 0.2–0.33 (Khan and O'Hare, 2002). Recently, the interest for the synthesis and the characterisation of LDHs increased since these materials can be used for many potential applications. For example, LDHs are well known for their capacity to exchange various types of inorganic anions (Miyata, 1983; Khan and O'Hare, 2002). LDHs can easily incorporate organic and bioorganic molecules such as ascorbic acid (Aisawa et al., 2007), urease (Vial et al., 2008), DNA (Oh et al., 2006) and even ferritin (Clemente-León et al., 2008). A particular type of LDHs which contains only iron as a cation in the brucite-type layers is called Green Rust (GR), i.e. the LDH[Fe II –Fe III ]. GR is an intermediate compound that forms during the Fe II species oxidation in solution (Feitknecht and Keller, 1950). For example, carbonated GR was observed as a corrosion product in water drain by Stampfl (1969). In 1996, a natural sample found in a hydromorphic soil in the forest of Fougères (Brittany, France) was identified to be homologous to green rust by using Mössbauer and Raman spectrocopies (Trolard et al. 1997). This mineral was then named fougerite (IMA 2003-057). More recently, three new occurrences were found: one in coal mine drainage sediment in South Wales (Bearcock et al., 2006) and two others in Denmark (Christiansen et al., 2009). In this last study, an X-ray diffraction pattern that corresponds to synthetic carbonated GR was presented for the first time. The properties of GR, i.e. the LDH[Fe II – Fe III ], were also studied because of its ability to reduce several anionic pollutants such as nitrate (Hansen, 2001), chromate (Loyaux-Law- niczak et al., 2000; Bond and Fendorf, 2003; Legrand et al., 2004) and seleniate (Refait et al., 2000, 2005a; Hayashi et al., 2009). Finally, GR was shown to promote the reduction/oxidation of an azo dye and therefore its removal from water (Kone et al., 2009) and the Fenton- like oxidation of phenol at neutral pH (Hanna et al., 2010). In this paper, we will focus on the description of the synthesis routes and transformation of LDHs that contain a minimum of 50 % of iron atoms in the brucite-like layers, i.e. compound for which the ratio n(Fe)/{n(M II )+n(M III )} is N 50%. Similar to the terminology common- ly used for metallic alloys, these compounds are called iron-based LDHs. The partial substitution of the Fe II or Fe III cations by other divalent or trivalent species lead to ternary systems such as LDH[{Fe II – M II }–Fe III ] or LDH[Fe II –{Fe III –M III }. To our knowledge, three types of partial substitutions were studied: Fe II by Ni II (Refait and Génin, 1993; Refait et al., 2005b), Fe II by Mg II (Refait et al., 2001) and more recently Fe III by Al III (Ruby et al., 2008). Fully substituted compounds that correspond to the minerals pyroaurite Mg 6 Fe 2 (OH) 16 CO 3, 4H 2 O (Allmann, 1968) and reevesite Ni 18 Fe 6 (OH) 48 (CO 3 ) 3 ,12H 2 O, (De Waal and Viljoen, 1971) are not strico sensu iron-based LDHs, and the synthesis of such compounds will not be considered here. We will Applied Clay Science 48 (2010) 195–202 ⁎ Corresponding author. Tel.: +33 3 83 68 52 53. E-mail address: [email protected] (C. Ruby). 0169-1317/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.clay.2009.11.017 Contents lists available at ScienceDirect Applied Clay Science journal homepage: www.elsevier.com/locate/clay

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Applied Clay Science 48 (2010) 195–202
Contents lists available at ScienceDirect
Applied Clay Science
j ourna l homepage: www.e lsev ie r.com/ locate /c lay
Synthesis and transformation of iron-based layered double hydroxides
C. Ruby a,⁎, M. Usman a, S. Naille a, K. Hanna a, C. Carteret a, M. Mullet a, M. François b, M. Abdelmoula a
a Laboratoire de Chimie Physique et Microbiologie pour l'Environnement (LCPME), UMR7564 CNRS-Nancy Université, 405, Rue de Vandœuvre 54600 Villers-Lès-Nancy, Franceb Institut Jean Lamour, Département P2M, Matériaux-Métallurgie-Nanosciences-Plasmas-Surfaces, UMR 7198-CNRS-Nancy-Université-UPV-Metz, Faculté des Sciences,BP 239, 54506 Villers-lès-Nancy, France
⁎ Corresponding author. Tel.: +33 3 83 68 52 53.E-mail address: [email protected]
0169-1317/$ – see front matter © 2009 Elsevier B.V. Aldoi:10.1016/j.clay.2009.11.017
a b s t r a c t
a r t i c l e i n f oArticle history:Received 16 July 2009Received in revised form 15 October 2009Accepted 13 November 2009Available online 26 November 2009
Keywords:LDHGreen rustPyroauriteHydrotalciteFougeriteMössbauer spectroscopy
Iron-based layered double hydroxides (LDHs) have the general formula [MII(1-x)M
IIIx(OH)2]
x+·[(x/n) An−, mH2O]x− and contain a molar fraction of iron, i.e. FeII or FeIII situated in the cationic layers, higher than 50%.LDHs containing FeII species are interesting materials for several applications such as the reduction of anionicpollutants or the degradation of organic pollutants. They are mostly prepared either by coprecipitation ofdissolved species or by oxidation of hydroxylated FeII species. The synthesis routes were visualised in binaryand ternary mass-balance diagrams. The LDH[FeII–FeIII] particles were well crystallised hexagonal plates, thesize of which diminishes rapidly when divalent or trivalent cations substitute ions. The LDH[FeII–FeIII–CO3]transformed into a mixture of magnetite Fe3O4 and siderite FeCO3 in alkaline conditions, but the adsorptionof silicate or phosphate anions on the lateral faces of the crystal prevented this decomposition. In oxicconditions, two mechanism of transformation were identified: (i) a fast in situ oxidation within the solid, (ii)dissolution–precipitation of the LDH forming ferric oxyhydroxides such as goethite (α-FeOOH).
(C. Ruby).
l rights reserved.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
MixedMII–MIII layered double hydroxides (LDHs) are described bythe general formula [MII
(1-x)MIIIx(OH)2]x+·[(x/n) An−, m H2O]x−
where MII and MIII are metal cations in the brucite-type layers Mg(OH)2, An− are the intercalated anions and x is the molar fraction ofthe trivalent cation and also the electrostatic charge of both thebrucite-type layers and the anionic interlayers. Most commonly, thevalues of x are found in the range of 0.2–0.33 (Khan and O'Hare,2002). Recently, the interest for the synthesis and the characterisationof LDHs increased since these materials can be used for manypotential applications. For example, LDHs are well known for theircapacity to exchange various types of inorganic anions (Miyata, 1983;Khan and O'Hare, 2002). LDHs can easily incorporate organic andbioorganic molecules such as ascorbic acid (Aisawa et al., 2007),urease (Vial et al., 2008), DNA (Oh et al., 2006) and even ferritin(Clemente-León et al., 2008). A particular type of LDHswhich containsonly iron as a cation in the brucite-type layers is called Green Rust(GR), i.e. the LDH[FeII–FeIII]. GR is an intermediate compound thatforms during the FeII species oxidation in solution (Feitknecht andKeller, 1950). For example, carbonated GR was observed as acorrosion product in water drain by Stampfl (1969). In 1996, anatural sample found in a hydromorphic soil in the forest of Fougères(Brittany, France) was identified to be homologous to green rust byusing Mössbauer and Raman spectrocopies (Trolard et al. 1997). This
mineral was then named fougerite (IMA 2003-057). More recently,three new occurrences were found: one in coal mine drainagesediment in South Wales (Bearcock et al., 2006) and two others inDenmark (Christiansen et al., 2009). In this last study, an X-raydiffraction pattern that corresponds to synthetic carbonated GR waspresented for the first time. The properties of GR, i.e. the LDH[FeII–FeIII], were also studied because of its ability to reduce several anionicpollutants such as nitrate (Hansen, 2001), chromate (Loyaux-Law-niczak et al., 2000; Bond and Fendorf, 2003; Legrand et al., 2004) andseleniate (Refait et al., 2000, 2005a; Hayashi et al., 2009). Finally, GRwas shown to promote the reduction/oxidation of an azo dye andtherefore its removal from water (Kone et al., 2009) and the Fenton-like oxidation of phenol at neutral pH (Hanna et al., 2010).
In this paper, we will focus on the description of the synthesisroutes and transformation of LDHs that contain a minimum of 50 % ofiron atoms in the brucite-like layers, i.e. compound for which the ration(Fe)/{n(MII)+n(MIII)} is N50%. Similar to the terminology common-ly used for metallic alloys, these compounds are called iron-basedLDHs. The partial substitution of the FeII or FeIII cations by otherdivalent or trivalent species lead to ternary systems such as LDH[{FeII–MII}–FeIII] or LDH[FeII–{FeIII–MIII}. To our knowledge, three types ofpartial substitutions were studied: FeII by NiII (Refait and Génin, 1993;Refait et al., 2005b), FeII by MgII (Refait et al., 2001) and more recentlyFeIII by AlIII (Ruby et al., 2008). Fully substituted compoundsthat correspond to the minerals pyroaurite Mg6Fe2(OH)16CO3,4H2O(Allmann, 1968) and reevesite Ni18Fe6(OH)48(CO3)3,12H2O, (DeWaaland Viljoen, 1971) are not strico sensu iron-based LDHs, and thesynthesis of such compounds will not be considered here. We will
196 C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
report synthesis routes of iron-based LDH and transformation modesat both oxic and anoxic conditions.
2. Synthesis and characterisation
2.1. Formation paths: General description
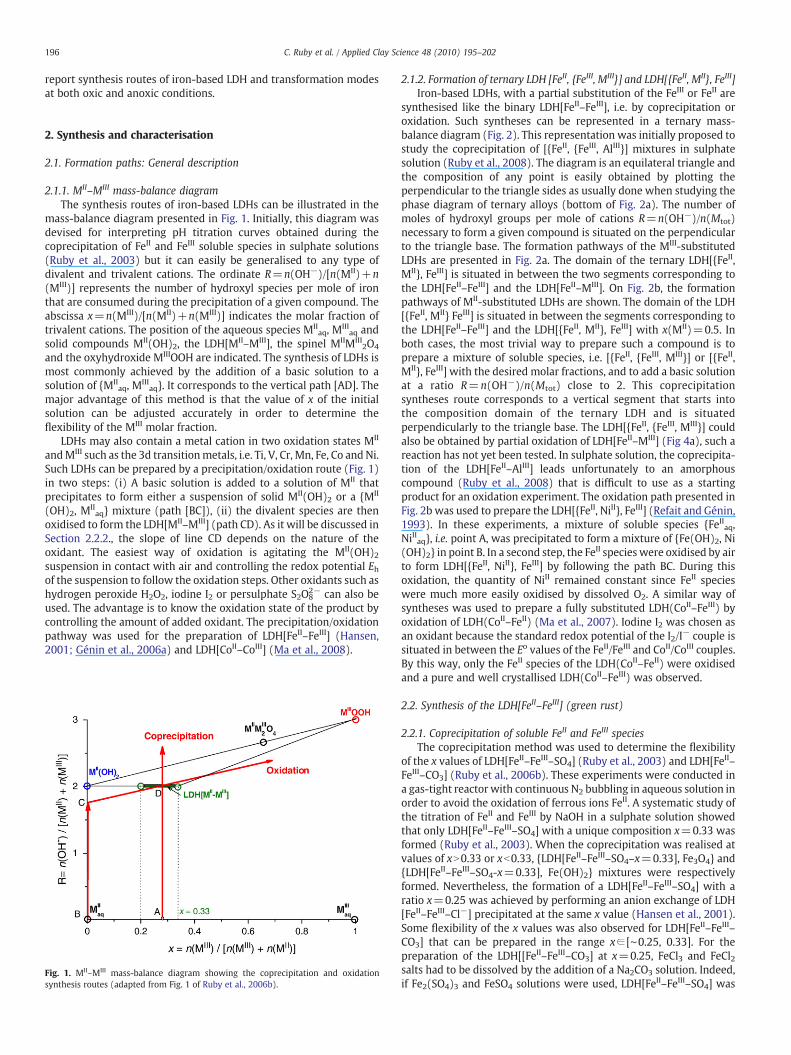
2.1.1. MII–MIII mass-balance diagramThe synthesis routes of iron-based LDHs can be illustrated in the
mass-balance diagram presented in Fig. 1. Initially, this diagram wasdevised for interpreting pH titration curves obtained during thecoprecipitation of FeII and FeIII soluble species in sulphate solutions(Ruby et al., 2003) but it can easily be generalised to any type ofdivalent and trivalent cations. The ordinate R=n(OH−)/[n(MII)+n(MIII)] represents the number of hydroxyl species per mole of ironthat are consumed during the precipitation of a given compound. Theabscissa x=n(MIII)/[n(MII)+n(MIII)] indicates the molar fraction oftrivalent cations. The position of the aqueous species MII
aq, MIIIaq and
solid compounds MII(OH)2, the LDH[MII–MIII], the spinel MIIMIII2O4
and the oxyhydroxide MIIIOOH are indicated. The synthesis of LDHs ismost commonly achieved by the addition of a basic solution to asolution of {MII
aq, MIIIaq}. It corresponds to the vertical path [AD]. The
major advantage of this method is that the value of x of the initialsolution can be adjusted accurately in order to determine theflexibility of the MIII molar fraction.
LDHs may also contain a metal cation in two oxidation states MII
andMIII such as the 3d transitionmetals, i.e. Ti, V, Cr, Mn, Fe, Co and Ni.Such LDHs can be prepared by a precipitation/oxidation route (Fig. 1)in two steps: (i) A basic solution is added to a solution of MII thatprecipitates to form either a suspension of solid MII(OH)2 or a {MII
(OH)2, MIIaq} mixture (path [BC]), (ii) the divalent species are then
oxidised to form the LDH[MII–MIII] (path CD). As it will be discussed inSection 2.2.2., the slope of line CD depends on the nature of theoxidant. The easiest way of oxidation is agitating the MII(OH)2suspension in contact with air and controlling the redox potential Ehof the suspension to follow the oxidation steps. Other oxidants such ashydrogen peroxide H2O2, iodine I2 or persulphate S2O8
2− can also beused. The advantage is to know the oxidation state of the product bycontrolling the amount of added oxidant. The precipitation/oxidationpathway was used for the preparation of LDH[FeII–FeIII] (Hansen,2001; Génin et al., 2006a) and LDH[CoII–CoIII] (Ma et al., 2008).
Fig. 1. MII–MIII mass-balance diagram showing the coprecipitation and oxidationsynthesis routes (adapted from Fig. 1 of Ruby et al., 2006b).
2.1.2. Formation of ternary LDH [FeII, {FeIII, MIII}] and LDH[{FeII, MII}, FeIII]Iron-based LDHs, with a partial substitution of the FeIII or FeII are
synthesised like the binary LDH[FeII–FeIII], i.e. by coprecipitation oroxidation. Such syntheses can be represented in a ternary mass-balance diagram (Fig. 2). This representation was initially proposed tostudy the coprecipitation of [{FeII, {FeIII, AlIII}] mixtures in sulphatesolution (Ruby et al., 2008). The diagram is an equilateral triangle andthe composition of any point is easily obtained by plotting theperpendicular to the triangle sides as usually done when studying thephase diagram of ternary alloys (bottom of Fig. 2a). The number ofmoles of hydroxyl groups per mole of cations R=n(OH−)/n(Mtot)necessary to form a given compound is situated on the perpendicularto the triangle base. The formation pathways of the MIII-substitutedLDHs are presented in Fig. 2a. The domain of the ternary LDH[{FeII,MII}, FeIII] is situated in between the two segments corresponding tothe LDH[FeII–FeIII] and the LDH[FeII–MIII]. On Fig. 2b, the formationpathways of MII-substituted LDHs are shown. The domain of the LDH[{FeII, MII} FeIII] is situated in between the segments corresponding tothe LDH[FeII–FeIII] and the LDH[{FeII, MII}, FeIII] with x(MII)=0.5. Inboth cases, the most trivial way to prepare such a compound is toprepare a mixture of soluble species, i.e. [{FeII, {FeIII, MIII}] or [{FeII,MII}, FeIII] with the desired molar fractions, and to add a basic solutionat a ratio R=n(OH−)/n(Mtot) close to 2. This coprecipitationsyntheses route corresponds to a vertical segment that starts intothe composition domain of the ternary LDH and is situatedperpendicularly to the triangle base. The LDH[{FeII, {FeIII, MIII}] couldalso be obtained by partial oxidation of LDH[FeII–MIII] (Fig 4a), such areaction has not yet been tested. In sulphate solution, the coprecipita-tion of the LDH[FeII–AlIII] leads unfortunately to an amorphouscompound (Ruby et al., 2008) that is difficult to use as a startingproduct for an oxidation experiment. The oxidation path presented inFig. 2b was used to prepare the LDH[{FeII, NiII}, FeIII] (Refait and Génin,1993). In these experiments, a mixture of soluble species {FeIIaq,NiIIaq}, i.e. point A, was precipitated to form a mixture of {Fe(OH)2, Ni(OH)2} in point B. In a second step, the FeII species were oxidised by airto form LDH[{FeII, NiII}, FeIII] by following the path BC. During thisoxidation, the quantity of NiII remained constant since FeII specieswere much more easily oxidised by dissolved O2. A similar way ofsyntheses was used to prepare a fully substituted LDH(CoII–FeIII) byoxidation of LDH(CoII–FeII) (Ma et al., 2007). Iodine I2 was chosen asan oxidant because the standard redox potential of the I2/I− couple issituated in between the Eo values of the FeII/FeIII and CoII/CoIII couples.By this way, only the FeII species of the LDH(CoII–FeII) were oxidisedand a pure and well crystallised LDH(CoII–FeIII) was observed.
2.2. Synthesis of the LDH[FeII–FeIII] (green rust)
2.2.1. Coprecipitation of soluble FeII and FeIII speciesThe coprecipitation method was used to determine the flexibility
of the x values of LDH[FeII–FeIII–SO4] (Ruby et al., 2003) and LDH[FeII–FeIII–CO3] (Ruby et al., 2006b). These experiments were conducted ina gas-tight reactor with continuous N2 bubbling in aqueous solution inorder to avoid the oxidation of ferrous ions FeII. A systematic study ofthe titration of FeII and FeIII by NaOH in a sulphate solution showedthat only LDH[FeII–FeIII–SO4] with a unique composition x=0.33 wasformed (Ruby et al., 2003). When the coprecipitation was realised atvalues of xN0.33 or xb0.33, {LDH[FeII–FeIII–SO4–x=0.33], Fe3O4} and{LDH[FeII–FeIII–SO4-x=0.33], Fe(OH)2} mixtures were respectivelyformed. Nevertheless, the formation of a LDH[FeII–FeIII–SO4] with aratio x=0.25 was achieved by performing an anion exchange of LDH[FeII–FeIII–Cl−] precipitated at the same x value (Hansen et al., 2001).Some flexibility of the x values was also observed for LDH[FeII–FeIII–CO3] that can be prepared in the range x∈ [∼0.25, 0.33]. For thepreparation of the LDH[[FeII–FeIII–CO3] at x=0.25, FeCl3 and FeCl2salts had to be dissolved by the addition of a Na2CO3 solution. Indeed,if Fe2(SO4)3 and FeSO4 solutions were used, LDH[FeII–FeIII–SO4] was

Fig. 2. Ternary mass-balance diagrams showing the domains and syntheses routes of MIII (a) and MII (b) substituted iron-based LDHs.
197C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
formed before LDH[FeII–FeIII–CO3] and only the LDH[x=0.33] wasobtained.
2.2.2. Oxidation of Fe(OH)2 and {Fe(OH)2, FeII} mixture
Another easy way to prepare LDH[FeII–FeIII] is oxidation by air of asuspension that contains Fe(OH)2 precipitates. In sulphate andchloride solutions (Génin et al., 2006a), only suspensions containinga specific amount of soluble FeII species led to the formation of thepure LDH. For example in sulphate solution, this excess of FeIIaq isdescribed by the following chemical reaction:
5FeIIðOHÞ2 þ Fe
2þaq þ SO
2−4 þ 1=2O2 þ H2O⇔Fe
II4 Fe
III2 ðOHÞ12SO4 ð1Þ
Therefore, the initial FeIIaq solution was partially precipitated byadding a NaOH solution with a ratio r=n(OH−)/n(FeII) of exactly 5/3.If FeII species were precipitated with a ratio r equal to 2, pure Fe(OH)2was formed without any excess of FeIIaq and the oxidation yieldedpure magnetite. For 5/3b rb2, the formation of {LDH[FeII–FeIII–SO4],Fe3O4} mixtureswas observed. Other experimentswere carried out byusing persulphate ions S2O8
2− as oxidant (Ruby et al., 2006a). In thiscase, the oxidation of pure Fe(OH)2 led to the formation of pure LDH[FeII–FeIII] according to the following reaction:
5FeIIðOHÞ2 þ S2O
2−8 þ 1=2O2 þ H2O⇔Fe
II4 Fe
III2 ðOHÞ12SO4 ð2Þ
The results with several oxidants (Ruby et al., 2006a) indicatedthat the thermodynamically favourable reactions were pH indepen-dent, as in the case of reactions (1) and (2). If the reduction of theoxidant corresponds to the general half reaction:
ðOxÞ þ ne−⇔ðRedÞ þ νOH− ð3ÞPreparing the LDH with a FeIII molar fraction x requires a ratio
r=n(OH−)/n(FeII) of the initial solution (Ruby et al., 2006a):
r ¼ 2−ðν=nÞx ð4ÞFor example, for the redox couple O2/OH−, the half reaction is O2+
H2O+2e−+2 OH− and (ν/n)=1. For preparing LDH[FeII–FeIII] at anx value of 1/3, an r value of 5/3 is obtained according to Eq. (4) whichis in good agreement with the experimental observations.
2.2.3. Reactivity of FeII species with FeIII oxyhydroxidesLDH[FeII–FeIII] can also be formed by reaction of FeII species with
FeIII oxyhydroxides such as lepidocrocite γ-FeOOH (Tamaura, 1985)
or ferrihydrite (Hansen, 2001). In these experiments, soluble FeII
species were added to a suspension that contains the ferricoxyhydroxide at a pH close to 7. This pH allows a sufficienthydroxylation rate of the FeII species necessary to activate thereaction. LDH[FeII–FeIII–SO4] can also be prepared by fixing thehydroxylation rate R=n(OH−)/[n(FeII)+n(FeIII)] at exactly 7 with abasic solution such as NaOH (Ruby et al., 2003). This ratio correspondsto the formation of a mixture {FeOOH, 2 Fe(OH)2} that washypothesised to be the precursor of the formation of the LDH[FeII–FeIII–SO4]. A heterogeneous reaction at the surface of the ferricoxyhydroxide particles was described by Ruby et al. (2006b).
2.3. Characterisation of iron-based layered double hydroxides
2.3.1. Morphology of the crystalsNon-substituted LDH[FeII–FeIII] prepared at room temperature
either by coprecipitation or oxidation showedwell-defined hexagonalplates with a lateral size D in the range of 100–500 nm and a thicknessd of ∼10–50 nm as observed by TEM (Fig. 3a). The ratio D/d was closeto 10 for LDH[FeII–FeIII−SO4
2−] and close to 5 for LDH[FeII–FeIII−CO32−]
(Ruby et al., 2006b). In this case, many LDH[FeII–FeIII−CO32−] crystals
were seen with their basal plane situated perpendicularly to the TEMgrid (Fig 3a). Diffraction of isolated crystals along the [001] zone axiswas performed and allowed us to determine the parameter a of thehexagonal cell (a∼3.2 Å).
The substitution of the trivalent or the divalent cation by anothercation rapidly decreased the crystal size (Fig. 3b, c). For example, thesubstitution of half of the FeIII ions by AlIII in LDH[FeII–FeIII–SO4]decreased the lateral crystal size to ∼10 nm (Fig. 3b). A more drasticeffect was observed if the FeII species were fully substituted by NiII
(Fig. 3c). In this case, the LDH consisted of nano-crystals and onlyheating in the range of 100 °C–180 °C formed larger crystals with ahexagonal shape (not shown). Note that heating to these tempera-tures decomposed iron-based LDH into {Fe3O4, Fe(OH)2} or {Fe3O4,FeCO3} mixtures. Therefore, heating is of no use for increasing thecrystal size of iron-based LDHs.
2.3.2. Crystallographical structureThe structure of LDH[FeII–FeIII–SO4], LDH[FeII–FeIII–AlIII–SO4] and
LDH[FeII–FeIII–CO3] were recently investigated by high resolution X-ray diffraction experiments performed at the European SynchrotronRadiation Facility (ESRF) (Aïssa et al., 2006; Ruby et al., 2008).
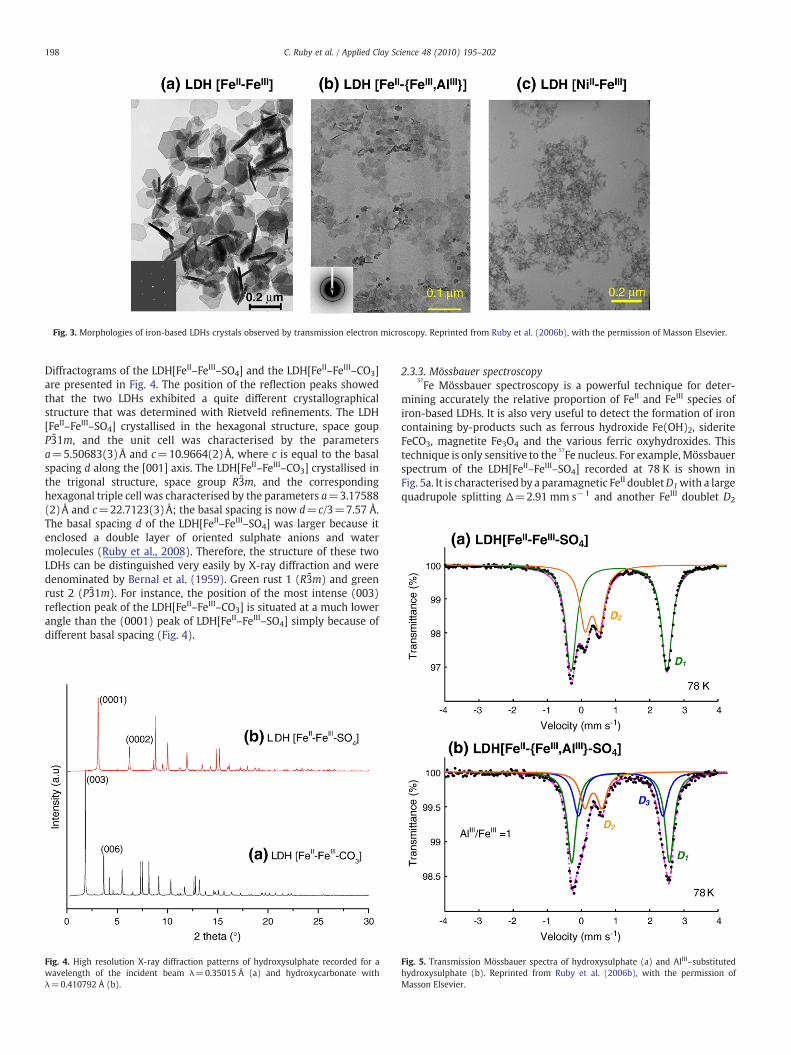
Fig. 3. Morphologies of iron-based LDHs crystals observed by transmission electron microscopy. Reprinted from Ruby et al. (2006b), with the permission of Masson Elsevier.
198 C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
Diffractograms of the LDH[FeII–FeIII–SO4] and the LDH[FeII–FeIII–CO3]are presented in Fig. 4. The position of the reflection peaks showedthat the two LDHs exhibited a quite different crystallographicalstructure that was determined with Rietveld refinements. The LDH[FeII–FeIII–SO4] crystallised in the hexagonal structure, space goupP3 ̄1m, and the unit cell was characterised by the parametersa=5.50683(3)Å and c=10.9664(2)Å, where c is equal to the basalspacing d along the [001] axis. The LDH[FeII–FeIII–CO3] crystallised inthe trigonal structure, space group R3̄m, and the correspondinghexagonal triple cell was characterised by the parameters a=3.17588(2)Å and c=22.7123(3)Å; the basal spacing is now d=c/3=7.57 Å.The basal spacing d of the LDH[FeII–FeIII–SO4] was larger because itenclosed a double layer of oriented sulphate anions and watermolecules (Ruby et al., 2008). Therefore, the structure of these twoLDHs can be distinguished very easily by X-ray diffraction and weredenominated by Bernal et al. (1959). Green rust 1 (R3 ̄m) and greenrust 2 (P3̄1m). For instance, the position of the most intense (003)reflection peak of the LDH[FeII–FeIII–CO3] is situated at a much lowerangle than the (0001) peak of LDH[FeII–FeIII–SO4] simply because ofdifferent basal spacing (Fig. 4).
Fig. 4. High resolution X-ray diffraction patterns of hydroxysulphate recorded for awavelength of the incident beam λ=0.35015 Å (a) and hydroxycarbonate withλ=0.410792 Å (b).
2.3.3. Mössbauer spectroscopy57Fe Mössbauer spectroscopy is a powerful technique for deter-
mining accurately the relative proportion of FeII and FeIII species ofiron-based LDHs. It is also very useful to detect the formation of ironcontaining by-products such as ferrous hydroxide Fe(OH)2, sideriteFeCO3, magnetite Fe3O4 and the various ferric oxyhydroxides. Thistechnique is only sensitive to the
57Fe nucleus. For example, Mössbauer
spectrum of the LDH[FeII–FeIII–SO4] recorded at 78 K is shown inFig. 5a. It is characterised by a paramagnetic FeII doubletD1with a largequadrupole splitting Δ=2.91 mm s−1 and another FeIII doublet D2
Fig. 5. Transmission Mössbauer spectra of hydroxysulphate (a) and AlIII–substitutedhydroxysulphate (b). Reprinted from Ruby et al. (2006b), with the permission ofMasson Elsevier.
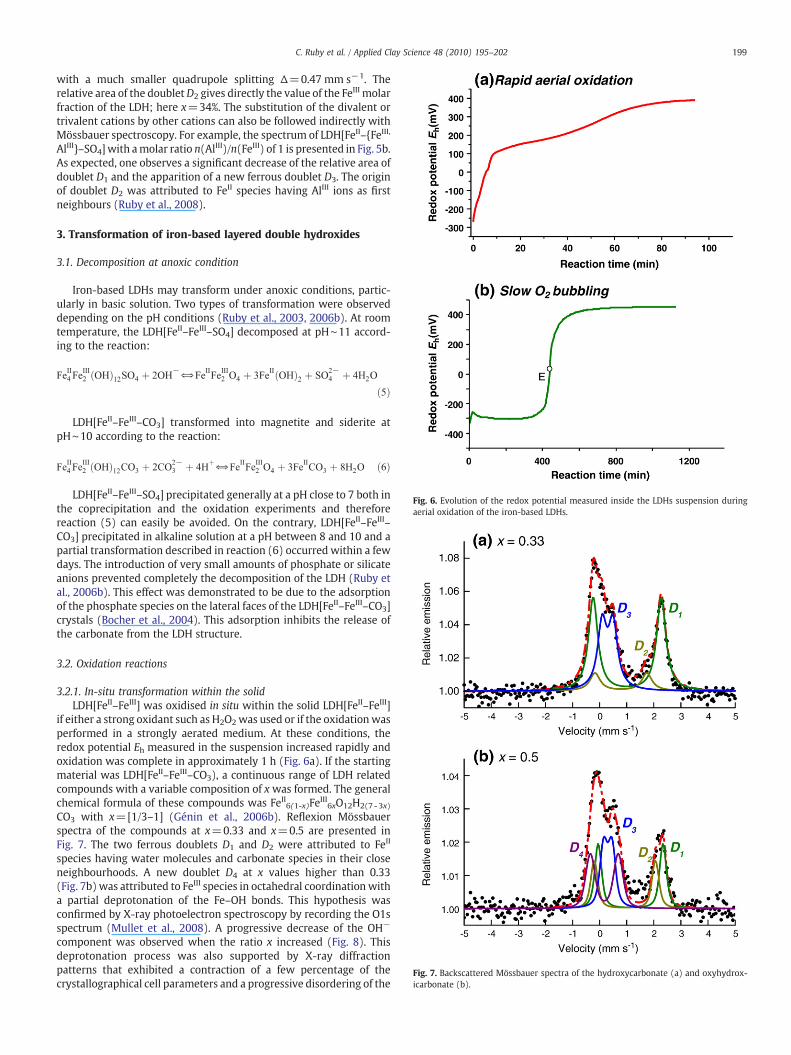
Fig. 6. Evolution of the redox potential measured inside the LDHs suspension duringaerial oxidation of the iron-based LDHs.
Fig. 7. Backscattered Mössbauer spectra of the hydroxycarbonate (a) and oxyhydrox-icarbonate (b).
199C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
with a much smaller quadrupole splitting Δ=0.47 mm s−1. Therelative area of the doubletD2 gives directly the value of the FeIII molarfraction of the LDH; here x=34%. The substitution of the divalent ortrivalent cations by other cations can also be followed indirectly withMössbauer spectroscopy. For example, the spectrum of LDH[FeII–{FeIII,
AlIII}–SO4]with amolar ratio n(AlIII)/n(FeIII) of 1 is presented in Fig. 5b.As expected, one observes a significant decrease of the relative area ofdoublet D1 and the apparition of a new ferrous doublet D3. The originof doublet D2 was attributed to FeII species having AlIII ions as firstneighbours (Ruby et al., 2008).
3. Transformation of iron-based layered double hydroxides
3.1. Decomposition at anoxic condition
Iron-based LDHs may transform under anoxic conditions, partic-ularly in basic solution. Two types of transformation were observeddepending on the pH conditions (Ruby et al., 2003, 2006b). At roomtemperature, the LDH[FeII–FeIII–SO4] decomposed at pH∼11 accord-ing to the reaction:
FeII4 Fe
III2 ðOHÞ12SO4 þ 2OH
−⇔FeIIFe
III2 O4 þ 3Fe
IIðOHÞ2 þ SO2−4 þ 4H2O
ð5Þ
LDH[FeII–FeIII–CO3] transformed into magnetite and siderite atpH∼10 according to the reaction:
FeII4 FeIII2 ðOHÞ12CO3 þ 2CO2−
3 þ 4Hþ⇔FeIIFeIII2 O4 þ 3FeIICO3 þ 8H2O ð6Þ
LDH[FeII–FeIII–SO4] precipitated generally at a pH close to 7 both inthe coprecipitation and the oxidation experiments and thereforereaction (5) can easily be avoided. On the contrary, LDH[FeII–FeIII–CO3] precipitated in alkaline solution at a pH between 8 and 10 and apartial transformation described in reaction (6) occurred within a fewdays. The introduction of very small amounts of phosphate or silicateanions prevented completely the decomposition of the LDH (Ruby etal., 2006b). This effect was demonstrated to be due to the adsorptionof the phosphate species on the lateral faces of the LDH[FeII–FeIII–CO3]crystals (Bocher et al., 2004). This adsorption inhibits the release ofthe carbonate from the LDH structure.
3.2. Oxidation reactions
3.2.1. In-situ transformation within the solidLDH[FeII–FeIII] was oxidised in situ within the solid LDH[FeII–FeIII]
if either a strong oxidant such as H2O2was used or if the oxidationwasperformed in a strongly aerated medium. At these conditions, theredox potential Eh measured in the suspension increased rapidly andoxidation was complete in approximately 1 h (Fig. 6a). If the startingmaterial was LDH[FeII–FeIII–CO3), a continuous range of LDH relatedcompounds with a variable composition of x was formed. The generalchemical formula of these compounds was FeII6(1-x)FeIII6xO12H2(7 -3x)
CO3 with x=[1/3–1] (Génin et al., 2006b). Reflexion Mössbauerspectra of the compounds at x=0.33 and x=0.5 are presented inFig. 7. The two ferrous doublets D1 and D2 were attributed to FeII
species having water molecules and carbonate species in their closeneighbourhoods. A new doublet D4 at x values higher than 0.33(Fig. 7b) was attributed to FeIII species in octahedral coordinationwitha partial deprotonation of the Fe–OH bonds. This hypothesis wasconfirmed by X-ray photoelectron spectroscopy by recording the O1sspectrum (Mullet et al., 2008). A progressive decrease of the OH−
component was observed when the ratio x increased (Fig. 8). Thisdeprotonation process was also supported by X-ray diffractionpatterns that exhibited a contraction of a few percentage of thecrystallographical cell parameters and a progressive disordering of the
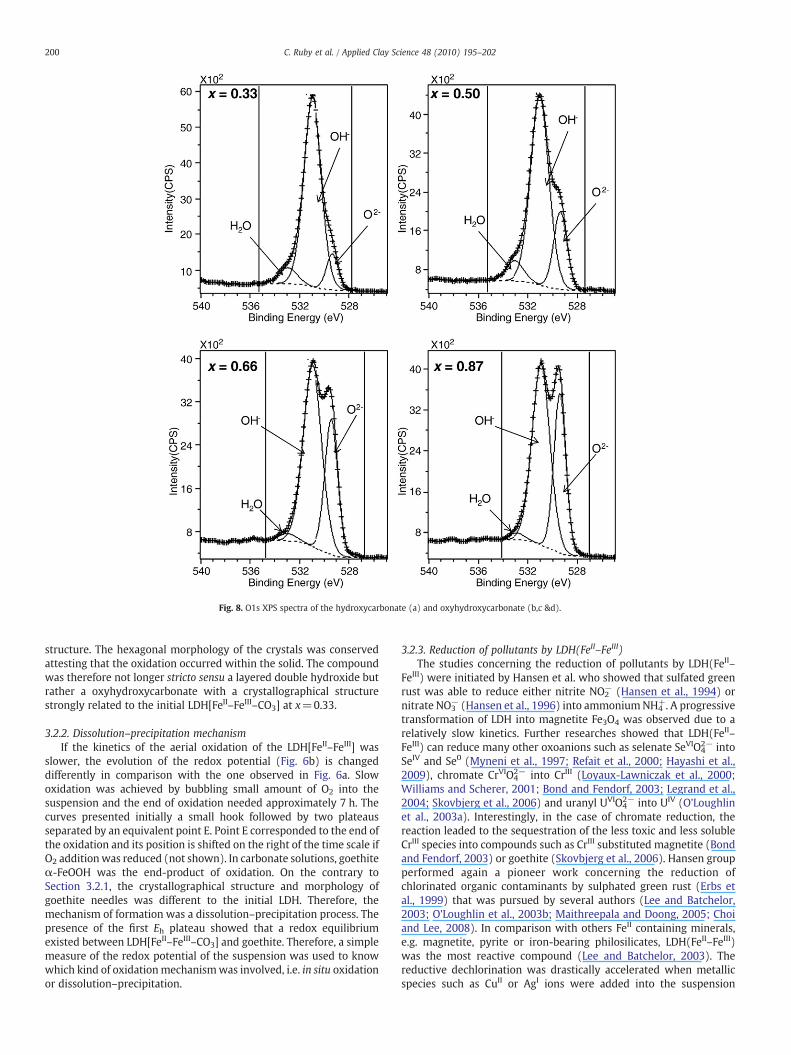
Fig. 8. O1s XPS spectra of the hydroxycarbonate (a) and oxyhydroxycarbonate (b,c &d).
200 C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
structure. The hexagonal morphology of the crystals was conservedattesting that the oxidation occurred within the solid. The compoundwas therefore not longer stricto sensu a layered double hydroxide butrather a oxyhydroxycarbonate with a crystallographical structurestrongly related to the initial LDH[FeII–FeIII–CO3] at x=0.33.
3.2.2. Dissolution–precipitation mechanismIf the kinetics of the aerial oxidation of the LDH[FeII–FeIII] was
slower, the evolution of the redox potential (Fig. 6b) is changeddifferently in comparison with the one observed in Fig. 6a. Slowoxidation was achieved by bubbling small amount of O2 into thesuspension and the end of oxidation needed approximately 7 h. Thecurves presented initially a small hook followed by two plateausseparated by an equivalent point E. Point E corresponded to the end ofthe oxidation and its position is shifted on the right of the time scale ifO2 additionwas reduced (not shown). In carbonate solutions, goethiteα-FeOOH was the end-product of oxidation. On the contrary toSection 3.2.1, the crystallographical structure and morphology ofgoethite needles was different to the initial LDH. Therefore, themechanism of formation was a dissolution–precipitation process. Thepresence of the first Eh plateau showed that a redox equilibriumexisted between LDH[FeII–FeIII–CO3] and goethite. Therefore, a simplemeasure of the redox potential of the suspension was used to knowwhich kind of oxidationmechanismwas involved, i.e. in situ oxidationor dissolution–precipitation.
3.2.3. Reduction of pollutants by LDH(FeII–FeIII)The studies concerning the reduction of pollutants by LDH(FeII–
FeIII) were initiated by Hansen et al. who showed that sulfated greenrust was able to reduce either nitrite NO2
− (Hansen et al., 1994) ornitrate NO3
− (Hansen et al., 1996) into ammoniumNH4+. A progressive
transformation of LDH into magnetite Fe3O4 was observed due to arelatively slow kinetics. Further researches showed that LDH(FeII–FeIII) can reduce many other oxoanions such as selenate SeVIO4
2− intoSeIV and Se0 (Myneni et al., 1997; Refait et al., 2000; Hayashi et al.,2009), chromate CrVIO4
2− into CrIII (Loyaux-Lawniczak et al., 2000;Williams and Scherer, 2001; Bond and Fendorf, 2003; Legrand et al.,2004; Skovbjerg et al., 2006) and uranyl UVIO4
2− into UIV (O'Loughlinet al., 2003a). Interestingly, in the case of chromate reduction, thereaction leaded to the sequestration of the less toxic and less solubleCrIII species into compounds such as CrIII substituted magnetite (Bondand Fendorf, 2003) or goethite (Skovbjerg et al., 2006). Hansen groupperformed again a pioneer work concerning the reduction ofchlorinated organic contaminants by sulphated green rust (Erbs etal., 1999) that was pursued by several authors (Lee and Batchelor,2003; O'Loughlin et al., 2003b; Maithreepala and Doong, 2005; Choiand Lee, 2008). In comparison with others FeII containing minerals,e.g. magnetite, pyrite or iron-bearing philosilicates, LDH(FeII–FeIII)was the most reactive compound (Lee and Batchelor, 2003). Thereductive dechlorination was drastically accelerated when metallicspecies such as CuII or AgI ions were added into the suspension

201C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
(O'Loughlin et al., 2003b; Maithreepala and Doong, 2005; Choi andLee, 2008). The key role of added metallic cations was explained byO'Loughlin et al. (2003b) and Suzuki et al. (2008) who clearlydemonstrated that CuII and AgI species were transformed into Cu0 andAg0. These metals were proposed to be localised at the surface of theLDH(FeII–FeIII) and allowed an acceleration of the reaction by acting asa galvanic couple with the iron species.
4. Conclusion
Iron-based layered double hydroxides LDHs[{FeII–MII},{FeIII–MIII }]that contain an iron molar fraction N50% were synthesised either bycoprecipitation of dissolved cations or by the oxidation of MII
hydroxides. Non-substituted LDH[FeII–FeIII], commonly called greenrust, prepared at room temperature by both methods exhibited wellcrystallised hexagonal crystals. Partial substitution of FeII or FeIII byother cations decreased crystal size. LDH[FeII–FeIII–SO4] contained abi-layer of oriented sulphate anions and water molecules in theinterlayer space. LDH[FeII–FeIII–CO3] contained only one layer of CO3
2−
and water molecules inside the interlayer space. Heating or alkalineconditions (pHN9) activated the decomposition of iron-based LDHsinto biphasic mixtures. Adsorption of small quantities of phosphate orsilicate anions on the lateral faces of the crystals slowed down thistransformation in the case of LDH[FeII–FeIII–CO3]. At oxic conditions,fast reactions caused an in situ transformation of LDH that transformsinto an oxyhydroxy-salt by deprotonation of hydroxyl groups. On thecontrary to most of the other, iron-based LDHs had a flexibility of the xvalues in the range of 1/3–1.When oxidationwas slower, the LDHwastransformed into a ferric oxyhydroxide by a dissolution–precipitationmechanism. The structural characterisation of iron-based LDHs wasinvestigated by several techniques such as high resolution X-raydiffraction, transmission electron microscopy and X-ray photoelec-tron andMössbauer spectroscopies. Future works could be devoted touse iron-based LDHs for reducing anionic pollutants in conditionscloser to field conditions, e.g. column reactors. Therefore theformation and reactivity of such compounds as coatings at the surfaceof minerals (e.g. quartz or clays) could be the subject of furtherstudies.
Acknowledgments
The authors would like to thank Prof. Gerhard Lagaly (University ofKiel, Germany) for his remarks and corrections of the manuscript.
References
Aisawa, S., Higashiyama, N., Takahashi, S., Hirahara, H., Ikematsu, D., Kondo, H.,Nakayama, H., Narita, E., 2007. Intercalation behavior of l-ascorbic acid into layereddouble hydroxides. Appl. Clay Sci. 35, 146–154.
Aïssa, R., Francois, M., Ruby, C., Fauth, F., Medjahdi, G., Abdelmoula, M., Génin, J.-M.R.,2006. Formation and crystallographical structure of hydroxysulphate and hydro-xycarbonate green rusts synthetised by coprecipitation. J. Phys. Chem. Solids 67,1016–1019.
Allmann, R., 1968. Crystal structure of pyroaurite. Acta Crystallogr., B Struct. Crystallogr.Cryst. Chem. 24, 972–977.
Bearcock, J.M., Perkins, W.T., Dinelli, E., Wade, S.C., 2006. Fe(II)/Fe(III) ‘green rust’developed within ochreous coal mine drainage sediment in South Wales, UK.Mineral. Mag. 70, 731–741.
Bernal, J.D., Dasgupta, D., Mackay, A.L., 1959. The oxides and hydroxides of iron andtheir structural inter-relationships. Clay Miner. Bull. 4, 15–30.
Bocher, F., Géhin, A., Ruby, C., Ghanbaja, J., Abdelmoula, M., Génin, J.-M.R., 2004.Coprecipitation of Fe(II–III) hydroxycarbonate green rust stabilised by phosphateadsorption. Solid State Sci. 6, 117–124.
Bond, D.L., Fendorf, S., 2003. Kinetics and structural constraints of chromate reductionby green rusts. Environ. Sci. Technol. 37, 2750–2757.
Choi, J., Lee, W., 2008. Enhanced degradation of tetrachloroethylene by green rusts withplatinum. Environ. Sci. Technol. 42, 3356–3362.
Christiansen, B.C., Balic-Zunic, T., Dideriksen, K., Stipp, S.L.S., 2009. Identification ofgreen rust in groundwater. Environ. Sci. Technol. 43, 3436–3441.
Clemente-León, M., Coronado, E., Primo, V., Ribera, A., Soriano-Portillo, A., 2008. Hybridmagnetic materials formed by ferritin intercalated into a layered double hydroxide.Solid State Sci. 10, 1807–1813.
De Waal, S.A., Viljoen, E.A., 1971. Nickel minerals from Barberton, South Africa. IV.Reevesite, a member of the hydrotalcite group. Am. Mineral. 56, 1077–1081.
Erbs, M., Hansen, H.C.B., Olsen, C.E., 1999. Reductive dechlorination of carbontetrachloride using iron(II)iron(III) hydroxide sulfate (green rust). Environ. Sci.Technol. 33, 307–311.
Feitknecht, W., Keller, G., 1950. Über die dunkelgrünen hydroxyverbindungen deseisens. Z. Anorg. Allg. Chem. 262, 61–68.
Génin, J.-M.R., Ruby, C., Géhin, A., Refait, P., 2006a. Synthesis of green rusts by oxidationof Fe(OH)2, their products of oxidation and reduction of ferric oxyhydroxides; Eh–pH Pourbaix diagrams. C. R. Geosci. 338, 433–446.
Génin, J.-M.R., Ruby, C., Upadhyay, C., 2006b. Structure and thermodynamics of ferrous,stoichiometric and ferric oxyhydroxycarbonate green rusts; redox flexibility andfougerite mineral. Solid State Sci. 8, 1330–1343.
Hanna, K., Kone, T., Ruby, C., 2010. Fenton-like oxidation and mineralizationof phenol using synthetic Fe(II)–Fe(III) green rusts. Environ. Sci. Pollut. Res. 17,124–134.
Hansen, H.C.B., 2001. Environmental chemistry of iron(II)–iron(III) LDHs (green rusts).In: Rives, V. (Ed.), Layered Double Hydroxides: Present and Future. Nova SciencePublishers, Huntington, N.Y., pp. 469–493.
Hansen, H.C.B., Borggaard, O.K., Sorensen, J., 1994. Evaluation of the free energy offormation of Fe(II)–Fe(III) hydroxide-sulfate (green rust) and its reduction ofnitrite. Geochim. Cosmochim. Acta 58, 2599–2608.
Hansen, H.C.B., Koch, C.B., Nancke-Krogh, H., Borggaard, O.K., Sorensen, J., 1996. Abioticnitrate reduction to ammonium: key role of green rust. Environ. Sci. Technol. 30,2053–2056.
Hansen,H.C.B., Guldberg, S., Erbs,M., Koch, C.B., 2001.Kineticsofnitrate reductionbygreenrusts-effects of interlayer anion and Fe(II):Fe(III) ratio. Appl. Clay Sci. 18, 81–91.
Hayashi, H., Kanie, K., Shinoda, K., Muramatsu, A., Suzuki, S., Sasaki, H., 2009. pH-dependence of selenate removal from liquid phase by reductive Fe(II)–Fe(III)hydroxysulfate compound, green rust. Chemosphere 76, 638–643.
Khan, A.I., O'Hare, D., 2002. Intercalation chemistry of layered double hydroxides:recent developments and applications. J. Mater. Chem. 12, 3191–3198.
Kone, T., Hanna, K., Abdelmoula, M., Ruby, C., Carteret, C., 2009. Reductivetransformation and mineralization of an azo dye by hydroxysulphate green rustpreceding oxidation using H2O2 at neutral pH. Chemosphere 75, 212–219.
Lee, W., Batchelor, B., 2003. Reductive capacity of natural reductants. Environ. Sci.Technol. 37, 535–541.
Legrand, L., Figuigui, A.E., Mercier, F., Chausse, A., 2004. Reduction of aqueous chromateby Fe(II)/Fe(III) carbonate green rust: kinetic and mechanistic studies. Environ. Sci.Technol. 38, 4587–4595.
Loyaux-Lawniczak, S., Refait, Ph., Ehrhardt, J.-J., Lecomte, P., Génin, J.-M.R., 2000.Trapping of Cr by formation of ferrihydrite during reduction of chromate ions byFe(II)–Fe(III) hydroxysalt green rusts. Environ. Sci. Technol. 34, 438–443.
Ma, R., Liu, Z., Takada, K., Iyi, N., Bando, Y., Sasaki, T., 2007. Synthesis and exfoliation ofCo2+–Fe3+ layered double hydroxides: an innovative topochemical approach.J. Am. Chem. Soc. 129, 5257–5263.
Ma, R., Takada, K., Fukuda, K., Iyi, N., Bando, Y., Sasaki, T., 2008. Topochemical synthesisof monometallic (Co2+–Co3+) layered double hydroxide and its exfoliation intopositively charged Co(OH)2 nanosheets. Angew. Chem. 120, 92–95.
Maithreepala, R.A., Doong, R.-A., 2005. Enhanced dechlorination of chlorinatedmethanes and ethenes by chloride green rust in the presence of copper(II).Environ. Sci. Technol. 39, 4082–4090.
Miyata, S., 1983. Anion-exchange properties of hydrotalcite-like compounds. Clays ClayMiner. 31, 305–311.
Mullet, M., Guillemin, Y., Ruby, C., 2008. Oxidation and deprotonation of synthetic FeII–FeIII (oxy)hydroxycarbonate Green Rust: an X-ray photoelectron study. J. SolidState Chem. 181, 81–89.
Myneni, S.C.B., Tokunaga, T.K., Brown Jr., G.E., 1997. Abiotic selenium redoxtransformations in the presence of Fe(II, III) oxides. Science 278, 1106–1109.
Oh, J.-M., Kwak, S.-Y., Choy, J.-H., 2006. Intracrystalline structure of DNA moleculesstabilized in the layered double hydroxide. J. Phys. Chem. Solids 67, 1028–1031.
O'Loughlin, E.J., Kelly, S.D., Cook, R.E., Csencsits, R., Kemner, K.M., 2003a. Reduction ofuranium(VI) by mixed iron(II)/iron(III) hydroxide (green rust): formation of UO2
nanoparticles. Environ. Sci. Technol. 37, 721–727.O'Loughlin, E.J., Kelly, S.D., Kemner, K.M., Csencsits, R., Cook, R.E., 2003b. Reduction of AgI,
AuIII, CuII, andHgII by FeII/FeIII hydroxysulfate green rust. Chemosphere 53, 437–446.Refait, P., Génin, J.-M.R., 1993. The oxidation of nickel(II)–iron(II) hydroxides in
chloride-containing aqueous media. Corros. Sci. 34, 2059–2070.Refait, P., Simon, L., Génin, J.-M.R., 2000. Reduction of SeO4
2− anions and anoxicformation of iron(II)–iron(III) hydroxy-selenate green rust. Environ. Sci. Technol.34, 819–825.
Refait, P., Abdelmoula, M., Trolard, F., Génin, J.-M.R., Bourrié, G., 2001. Mössbauer andXAS study of a green rust mineral; the partial substitution of Fe2+ by Mg2+. Am.Mineral. 86, 731–739.
Refait, P., Abdelmoula, M., Simon, L., Génin, J.-M.R., 2005a. Mechanisms of formationand transformation of Ni–Fe layered double hydroxides in SO3
2− and SO42−
containing aqueous solutions. J. Phys. Chem. Solids 66, 911–917.Refait, Ph., Drissi, S.H., Abdelmoula, M., Jeannin, M., Reffass, M., Génin, J.-M.R., 2005b.
Mechanisms of formation and transformation of Ni–Fe hydroxycarbonates.AIP Conference Proceedings, 765 (Industrial Applications of the Mössbauer Effect),pp. 79–84.
Ruby, C., Géhin, A., Abdelmoula, M., Génin, J.-M.R., Jolivet, J.-P., 2003. Coprecipitation ofFe(II) and Fe(III) cations in sulphated aqueous medium and formation ofhydroxysulphate green rust. Solid State Sci. 5, 1055–1062.
Ruby, C., Géhin, A., Aïssa, R., Génin, J.-M.R., 2006a. Mass-balance and Eh–pH diagrams ofFeII–III green rust in aqueous sulphated solution. Corros. Sci. 48, 3824–3837.

202 C. Ruby et al. / Applied Clay Science 48 (2010) 195–202
Ruby, C., Aïssa, R., Géhin, A., Cortot, J., Abdelmoula, M., Génin, J.-M.R., 2006b. Green rustssynthesis by coprecipitation of FeII–FeIII ions and mass-balance diagram. C. R.Geosci. 338, 420–432.
Ruby, C., Abdelmoula, M., Aïssa, R., Medjahdi, G., Brunelli, M., François, M., 2008.Aluminium substitution in iron(II–III)-layered double hydroxides: formation andcationic order. J. Solid State Chem. 181, 2285–2291.
Skovbjerg, L.L., Stipp, S.L.S., Utsunomiya, S., Ewing, R.C., 2006. The mechanisms ofreduction of hexavalent chromium by green rust sodium sulphate: formation of Cr-goethite. Geochim. Cosmochim. Acta 70, 3582–3592.
Stampfl, P.P., 1969. Ein basiches eisen-II–III-karbonat in rost. Corros. Sci. 9, 185–187.Suzuki, S., Shinoda, K., Sato, M., Fujimoto, S., Yamashita, M., Konishi, H., Doi, T., Kamimura,
T., Inoue, K., Waseda, Y., 2008. Changes in chemical state and local structure of greenrust by addition of copper sulphate ions. Corros. Sci. 50, 1761–1765.
Tamaura, Y., 1985. Ferrite formation from the intermediate, green rust II, in thetransformation reaction of ferric hydroxide oxide, γ-FeO(OH), in aqueoussuspension. Inorg. Chem. 24, 4363–4366.
Trolard, F., Génin, J.-M.R., Abdelmoula, M., Bourrié, G., Humbert, B., Herbillon, A., 1997.Identification of a green rust mineral in a reductomorphic soil by Mossbauer andRaman spectroscopies. Geochim. Cosmochim. Acta 61, 1107–1111.
Vial, S., Prevot, V., Leroux, F., Forano, C., 2008. Immobilization of urease in ZnAl LayeredDouble Hydroxides by soft chemistry routes. Microporous Mesoporous Mater. 107,190–201.
Williams, A.G.B., Scherer, M.M., 2001. Kinetics of Cr(VI) reduction by carbonate greenrust. Environ. Sci. Technol. 35, 3488–3494.
Related Documents











