JOURNAL OF MOLECULAR SPECTROSCOPY 185, 158–172 (1997) ARTICLE NO. MS977367 Sub-Doppler Infrared Spectra and Torsion – Rotation Energy Manifold of Methanol in the CH-Stretch Fundamental Region Li-Hong Xu,* Xiaoliang Wang,² Thomas J. Cronin,² David S. Perry,² Gerald T. Fraser,‡ and Alan S. Pine‡ * Department of Physical Sciences, University of New Brunswick, Saint John, New Brunswick, Canada E2L 4L5; ² Department of Chemistry, University of Akron, Akron, Ohio 44325; and ‡Optical Technology Division, National Institute of Standards and Technology, Gaithersburg, Maryland 20899 Received May 6, 1997 The infrared spectrum of jet-cooled methanol in the CH-stretch fundamental region has been investigated by two sub-Doppler laser techniques: optothermally detected molecular-beam electric resonance and direct-absorption slit-jet spectroscopy. With the aid of microwave-infrared double resonance and ground state combination differences, 27 subbands in the frequency range 2967 to 3027 cm 01 have been assigned to the n 2 fundamental. Perturbation systems in the K * Å 0 E , 01 E , and 02 E symmetry subbands have been analyzed to yield matrix elements of 0.013, 0.041, and 0.75 cm 01 , respectively. The A–E torsional tunneling splitting for J Å 0 of the n 2 vibration of 03.26 cm 01 is of opposite sign and a factor of three smaller in magnitude than the ground state value of /9.12 cm 01 . q 1997 Academic Press I. INTRODUCTION ( n 7 , A * and n 11 , A 9 ) are located close to the strong CO- stretch band and create difficulties for systematic assign- ment. There is evidence ( 29–32 ) to suggest that the in-plane This work reports molecular-beam investigations of the ( A * ) CH 3 rock is in Coriolis resonance with the CO stretch infrared (IR) absorption spectrum of CH 3 OH in the 3 mm and has a torsional potential barrier approximately 50% region of the CH-stretching fundamental bands. Methanol higher than the ground state value. has been of interest to high-resolution spectroscopists for To gain more information on the interactions of the low many years, yet still remains a challenge to understand be- amplitude vibrations with the torsion, Lees et al. have under- cause of the large amplitude hindered internal rotation and taken high-resolution studies of the OH-bend band ( n 6 , A * ) its interaction with the small amplitude vibrations (Fig. 1). ( 33 ) . The observed OH-bend torsion – rotation energy-level Of the 12 normal fundamental modes, the torsion ( n 12 ,A 9 pattern is inverted in comparison with the ground state, pos- symmetry in C s ) is the lowest in frequency and has been sibly because of interaction with the first excited torsion of studied extensively in the microwave (MW), millimeter- the CO stretch as suggested from the calculated vibration– wave (MMW) ( 1–7 ), and far-infrared (FIR) ( 8–14 ) re- torsion – rotation energy. Also, the OH-stretch fundamental gions of the spectrum. This body of work has been an im- ( n 1 , A * ) near 3672 cm 01 has been investigated extensively portant testing ground for internal-rotor models ( 1, 3, 15– ( 34–36 ) under both room temperature and molecular-beam 20 ). Recently this region has been revisited by Xu and conditions. The effective torsional barrier from a global fit Hougen ( 21, 22 ) using a one-dimensional torsional Hamilto- to the OH-stretch levels ( 36 ) was 439(13) cm 01 , or about nian ( 3 ) with an extended internal axis method (IAM). An 18% higher than the ground state barrier ( 22 ) of extensive set of pure rotation and rotation – torsion transitions 373.594(14) cm 01 . (The numbers in parentheses refer an have been fit to a global Hamiltonian to within experimental expanded uncertainty with a coverage factor k Å 2 in units precision, establishing the groundwork for the spectroscopic of the least significant digit.) The anharmonic extension of studies of the excited vibrational states. the OH bond might be expected to decrease the effective The CO-stretch fundamental ( n 8 , A * symmetry ) , centered barrier ; so the increase was attributed to a possible nonlinear near 1033 cm 01 , has been studied extensively ( 23–28 ) be- coupling between the OH stretch and torsion ( 35 ) or a har- cause of interest in the rich optically pumped FIR laser emis- monic mixing of the displacement coordinates of other vibra- sion observed from methanol. An additional question of in- tions of the same symmetry ( 36 ). terest is the interaction between the CO-stretch fundamental Similar to the OH stretch, the CH-stretch modes ( n 2 , A *, and the large amplitude torsion. Even though n 8 is the second n 3 , A *, and n 9 , A 9 ) have fundamentals in the 3 mm region lowest frequency mode, the observed CO-stretch torsion – and might be extensively mixed with ‘‘dark’’ background rotation energy structure is still not properly accounted for by existing models. The weak CH 3 -rock fundamental bands bath states arising from numerous lower-lying vibrations and 158 0022-2852/97 $25.00 Copyright q 1997 by Academic Press All rights of reproduction in any form reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF MOLECULAR SPECTROSCOPY 185, 158–172 (1997)ARTICLE NO. MS977367
Sub-Doppler Infrared Spectra and Torsion–Rotation Energy Manifoldof Methanol in the CH-Stretch Fundamental Region
Li-Hong Xu,* Xiaoliang Wang,† Thomas J. Cronin,† David S. Perry,† Gerald T. Fraser,‡ and Alan S. Pine‡
*Department of Physical Sciences, University of New Brunswick, Saint John, New Brunswick, Canada E2L 4L5;†Department of Chemistry, University of Akron, Akron, Ohio 44325; and ‡Optical Technology Division,
National Institute of Standards and Technology, Gaithersburg, Maryland 20899
Received May 6, 1997
The infrared spectrum of jet-cooled methanol in the CH-stretch fundamental region has been investigated by twosub-Doppler laser techniques: optothermally detected molecular-beam electric resonance and direct-absorption slit-jetspectroscopy. With the aid of microwave-infrared double resonance and ground state combination differences, 27subbands in the frequency range 2967 to 3027 cm01 have been assigned to the n2 fundamental. Perturbation systems inthe K* Å 0 E , 01 E , and 02 E symmetry subbands have been analyzed to yield matrix elements of 0.013, 0.041, and0.75 cm01 , respectively. The A–E torsional tunneling splitting for J Å 0 of the n2 vibration of 03.26 cm01 is of oppositesign and a factor of three smaller in magnitude than the ground state value of /9.12 cm01 . q 1997 Academic Press
I. INTRODUCTION (n7 , A* and n11 , A9) are located close to the strong CO-stretch band and create difficulties for systematic assign-ment. There is evidence (29–32) to suggest that the in-plane
This work reports molecular-beam investigations of the(A*) CH3 rock is in Coriolis resonance with the CO stretch
infrared (IR) absorption spectrum of CH3OH in the 3 mmand has a torsional potential barrier approximately 50%region of the CH-stretching fundamental bands. Methanolhigher than the ground state value.has been of interest to high-resolution spectroscopists for
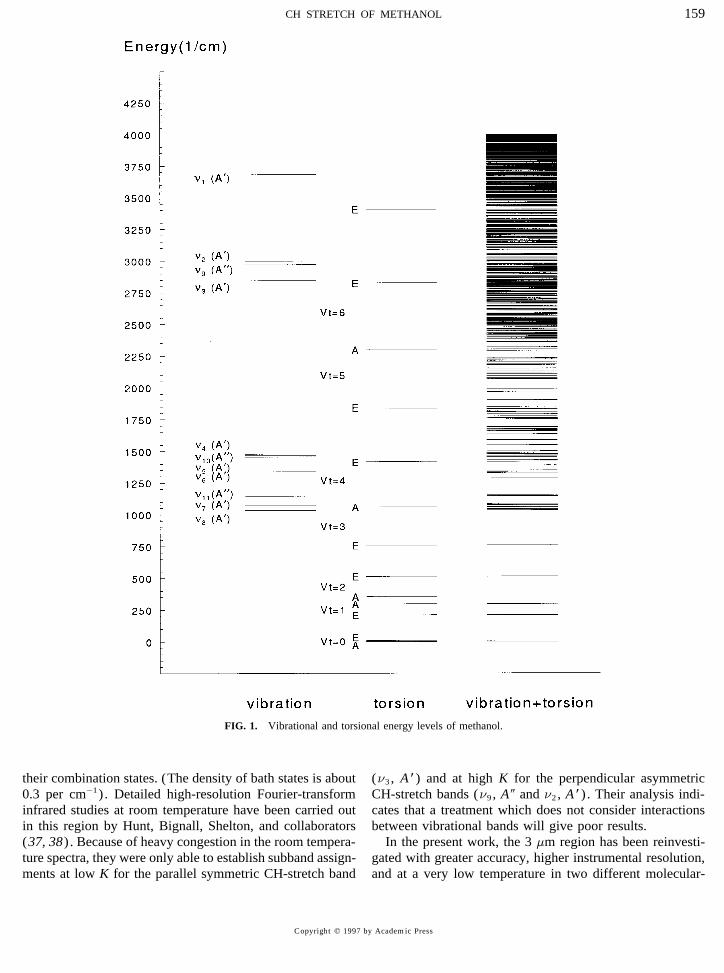
To gain more information on the interactions of the lowmany years, yet still remains a challenge to understand be-amplitude vibrations with the torsion, Lees et al. have under-cause of the large amplitude hindered internal rotation andtaken high-resolution studies of the OH-bend band (n6 , A *)its interaction with the small amplitude vibrations (Fig. 1) .(33) . The observed OH-bend torsion–rotation energy-levelOf the 12 normal fundamental modes, the torsion (n12 , A 9pattern is inverted in comparison with the ground state, pos-symmetry in Cs) is the lowest in frequency and has beensibly because of interaction with the first excited torsion ofstudied extensively in the microwave (MW), millimeter-the CO stretch as suggested from the calculated vibration–wave (MMW) (1–7) , and far-infrared (FIR) (8–14) re-torsion–rotation energy. Also, the OH-stretch fundamentalgions of the spectrum. This body of work has been an im-(n1 , A *) near 3672 cm01 has been investigated extensivelyportant testing ground for internal-rotor models (1, 3, 15–(34–36) under both room temperature and molecular-beam20) . Recently this region has been revisited by Xu andconditions. The effective torsional barrier from a global fitHougen (21, 22) using a one-dimensional torsional Hamilto-to the OH-stretch levels (36) was 439(13) cm01 , or aboutnian (3) with an extended internal axis method (IAM). An18% higher than the ground state barrier (22) ofextensive set of pure rotation and rotation–torsion transitions373.594(14) cm01 . (The numbers in parentheses refer anhave been fit to a global Hamiltonian to within experimentalexpanded uncertainty with a coverage factor k Å 2 in unitsprecision, establishing the groundwork for the spectroscopicof the least significant digit.) The anharmonic extension ofstudies of the excited vibrational states.the OH bond might be expected to decrease the effectiveThe CO-stretch fundamental (n8 , A * symmetry) , centeredbarrier ; so the increase was attributed to a possible nonlinearnear 1033 cm01 , has been studied extensively (23–28) be-coupling between the OH stretch and torsion (35) or a har-cause of interest in the rich optically pumped FIR laser emis-monic mixing of the displacement coordinates of other vibra-sion observed from methanol. An additional question of in-tions of the same symmetry (36) .terest is the interaction between the CO-stretch fundamental
Similar to the OH stretch, the CH-stretch modes (n2 , A*,and the large amplitude torsion. Even though n8 is the secondn3 , A*, and n9 , A9) have fundamentals in the 3 mm regionlowest frequency mode, the observed CO-stretch torsion–and might be extensively mixed with ‘‘dark’’ backgroundrotation energy structure is still not properly accounted for
by existing models. The weak CH3-rock fundamental bands bath states arising from numerous lower-lying vibrations and
1580022-2852/97 $25.00Copyright q 1997 by Academic PressAll rights of reproduction in any form reserved.
AID JMS 7367 / 6t1f$$$241 09-09-97 22:35:14 mspas
CH STRETCH OF METHANOL 159
FIG. 1. Vibrational and torsional energy levels of methanol.
their combination states. (The density of bath states is about (n3 , A *) and at high K for the perpendicular asymmetricCH-stretch bands (n9 , A 9 and n2 , A *) . Their analysis indi-0.3 per cm01) . Detailed high-resolution Fourier-transform
infrared studies at room temperature have been carried out cates that a treatment which does not consider interactionsbetween vibrational bands will give poor results.in this region by Hunt, Bignall, Shelton, and collaborators
(37, 38) . Because of heavy congestion in the room tempera- In the present work, the 3 mm region has been reinvesti-gated with greater accuracy, higher instrumental resolution,ture spectra, they were only able to establish subband assign-
ments at low K for the parallel symmetric CH-stretch band and at a very low temperature in two different molecular-
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$242 09-09-97 22:35:14 mspas

XU ET AL.160
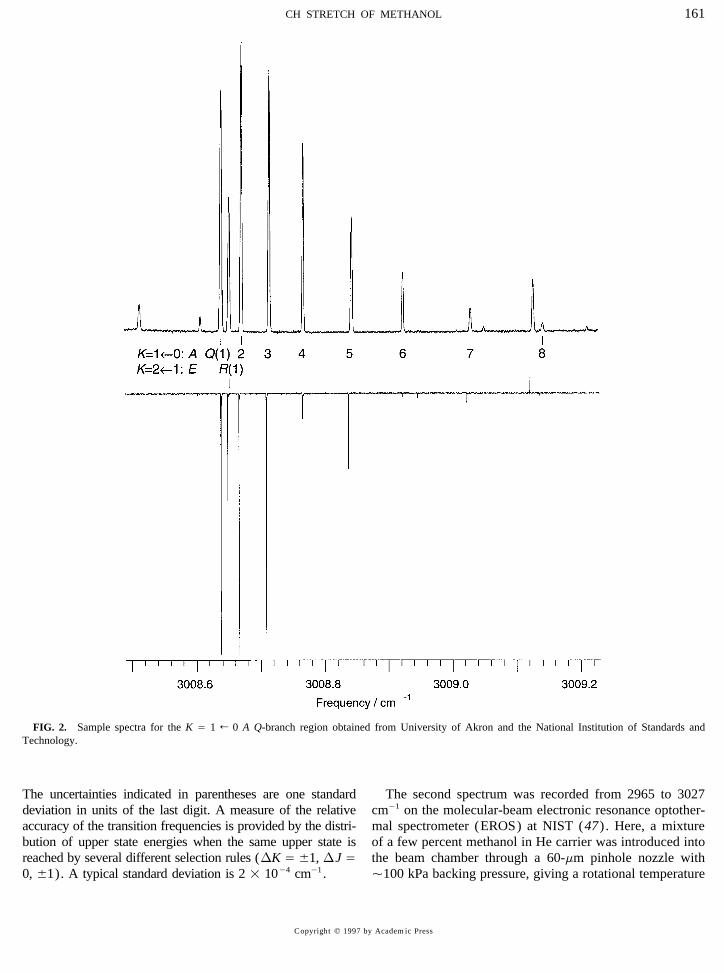
beam instruments. Motivation for this work comes from and one at the University of Akron. Figure 2 shows a segmentof sample spectra in a similar spectral region obtained fromthree perspectives:the two spectrometers. The initial work was carried out inde-
(i) From the theoretical point of view, the interactions pendently, but the data have now been combined for morebetween the torsion and the low-amplitude vibrations are of comprehensive analysis. Both spectrometers employ a color-fundamental interest (39) . Spectra in the excited CH-stretch center laser pumped byÇ1.6 W of 647-nm light from a kryptonregion are part of the effort to build up a comprehensive ion laser to produce 8 to 9 mW of single frequency tunableenergy-level pattern for methanol which will assist in under- infrared radiation in the 3 mm region. In both cases the methanolstanding the molecular dynamics and torsion–vibration in- was cooled in a nozzle expansion, and spectra were recordedteractions. at sub-Doppler resolution. Nonetheless, as indicated below, the
(ii ) Infrared-assisted photofragment spectroscopy (IR- detection techniques were rather different and the two spectraLAPS) experiments in the nOH Å 2 through 6 regions of contain complementary information.methanol have revealed a hierarchy of intramolecular relax- The first spectrum was recorded from 2977 to 3027 cm01
ation pathways and time scales (40) . The strongest interac- at the University of Akron using direct absorption detection.tion is a one-to-one resonance between the OH and CH The apparatus included a pulsed 2 1 0.01 cm slit nozzlestretches (40, 41) . At nOH Å 5, the OH stretch is anharmoni- system (45) and a multi-reflection cell (46) to enhance thecally shifted to lower frequency to bring the nOH Å 5 state absorption of the infrared radiation by the free jet. A mixture(5 n1) into resonance with the nOH Å 4 plus nCH Å 1 combi- of about 6% by volume methanol (Aldrich, 99.9%) in argonnation (4 n1 / n2) . Detailed information on the CH-stretch (Union Caribide, 99.997%) was expanded through the slitfundamental is essential in order to build an understanding nozzle operated piezoelectrically at 34.5 Hz with a pulseof the structure of the lower regions of the torsion–vibration duration of 400 msec and a stagnation pressure of 65 kPa.energy manifold which might be extended to the highly ex- The laser beam crossed through the jet about 1.5 cm fromcited vibrational states. the nozzle 25 times. The use of two matched InSb infrared
(iii ) From the astrophysical point of view, methanol is detectors enabled noise reduction by baseline subtraction.an important species widespread in interstellar clouds (42) The regular pattern of absorption intensities correspondedwhich has also recently been observed in comets in both the to a 17 K rotational temperature. In the planar expansion,microwave and the 3 mm regions (43, 44) . The cometary the residual Doppler linewidth was about 75 MHz. A furthercommunity requires data that will permit simulation of the 3 reduction in the linewidth could be obtained under moremm methanol emission spectrum under cometary conditions. vigorous expansion conditions but this would result in a
lower rotational temperature with a corresponding loss ofThe present paper reports sub-Doppler spectra of jet-
spectral information. Only a few lines were blended undercooled methanol covering mainly the n2 , A *, CH-stretch
the experimental conditions employed. The overview of thefundamental band. Altogether, about 19 b-type (DK Å {1)
spectrum, shown in Fig. 3, is a composite spliced togetherand 8 a-type (DK Å 0) n2 subbands have been identified
from many short sections.involving K * up to 3 for A and from 03 to 4 for E . Three
Three signals were recorded simultaneously: ( i) the trans-subbands of the n9 , A 9, CH-stretch fundamental band have
mitted laser intensity through the jet, ( ii ) the transmittedalso been identified. In contrast to the n1 OH stretch and the
intensity through a reference gas cell containing ethylenen3 symmetric CH stretch which are normal in terms of the
for absolute frequency calibration, and (iii ) the transmittedordering of the A and E levels, we find the n2 asymmetric
intensity through a 150 MHz marker etalon (BurleighCH stretch to have a torsion– K–rotation energy-level pat-
CFT100P) for relative frequency calibration. The spectrumtern that is inverted relative to the ground state.
was recorded in 0.15 cm01 sections which were spliced uti-lizing marker etalon fringes. The digitized signal from atemperature-controlled and pressure-sealed 7.5 GHz (Bur-II. EXPERIMENTAL1
leigh FCL975) spectrum analyzer provided a check on theidentity of the marker etalon fringes at which splices wereThe results reported here are based on separate molecular-made. A strong methanol line at 3001.0249 cm01 was usedbeam spectra recorded on two different spectrometers, one atas the reference line for a daily reproducibility check of linethe National Institute of Standards and Technology (NIST)intensities and frequency calibration. The frequency calibra-tion was accomplished by fitting 139 ethylene lines (46) to
1 Certain commercial instruments and materials are identified in this paper the marker etalon fringe number to obtainin order to specify adequately the experimental procedure. In no case doessuch identification imply recommendation or endorsement by the National frequency Å 0.004989832(12)Institute of Standards and Technology, nor does it imply that the instrumentsor materials identified are necessarily the best available for the purpose. 1 fringe number / 2976.9924(5).
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$242 09-09-97 22:35:14 mspas
CH STRETCH OF METHANOL 161
FIG. 2. Sample spectra for the K Å 1 R 0 A Q-branch region obtained from University of Akron and the National Institution of Standards andTechnology.
The uncertainties indicated in parentheses are one standard The second spectrum was recorded from 2965 to 3027cm01 on the molecular-beam electronic resonance optother-deviation in units of the last digit. A measure of the relative
accuracy of the transition frequencies is provided by the distri- mal spectrometer (EROS) at NIST (47) . Here, a mixtureof a few percent methanol in He carrier was introduced intobution of upper state energies when the same upper state is
reached by several different selection rules (DK Å {1, DJ Å the beam chamber through a 60-mm pinhole nozzle withÇ100 kPa backing pressure, giving a rotational temperature0, {1). A typical standard deviation is 2 1 1004 cm01 .
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$242 09-09-97 22:35:14 mspas
XU ET AL.162
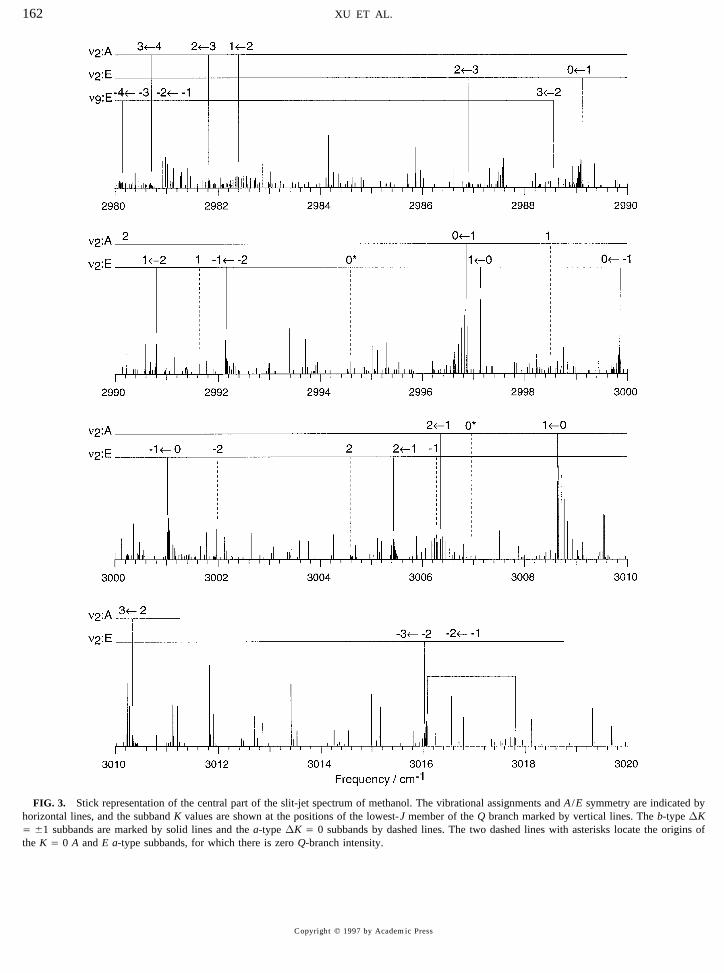
FIG. 3. Stick representation of the central part of the slit-jet spectrum of methanol. The vibrational assignments and A /E symmetry are indicated byhorizontal lines, and the subband K values are shown at the positions of the lowest-J member of the Q branch marked by vertical lines. The b-type DKÅ {1 subbands are marked by solid lines and the a-type DK Å 0 subbands by dashed lines. The two dashed lines with asterisks locate the origins ofthe K Å 0 A and E a-type subbands, for which there is zero Q-branch intensity.
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$7367 09-09-97 22:35:14 mspas

CH STRETCH OF METHANOL 163
near 10 K. The molecular beam was focused onto a liquid- coordinate and tunneling between the three equivalent posi-tions of the methyl group, the G6 molecular symmetry group,He-cooled bolometer using quadrupole focusing in the flight
chamber between the skimmer and the detector. The use with representations G Å A1 , A2 , and E , is used. The rigidrotor basis functions and all the normal modes other thanof the quadrupole focusing/defocusing field enhances the
detection sensitivity, but the observed intensities are more the torsion are either A1 (A *) or A2 (A 9) . Like the vibrationalground state, the n2 vibration is split by torsional tunnelingcomplicated than the transition absorption strengths since
they also reflect the Stark tuning of the probed levels which into A1 and E components.The rotational levels of the A tunneling component canmay vary both in magnitude and sign as shown in the bottom
part of the Fig. 2. A few spectral gaps were encountered in be categorized as A1 and A2 and these symmetries can berelated to the traditional labels, A/ and A0 (16) . All of thewhich H2O absorption obscured the methanol spectrum and
spoiled control of the laser scan. Calibration methane traces rotational levels of the E component have E symmetry butare often labeled for convenience by E1 (for K § 0) or E2were recorded at the beginning and end of a day to minimize
errors due to gaps and drifts. On a given day the calibration (for K õ 0) according to the relative orientations of thetorsional and K-rotational angular momenta (1) . It is im-interferometer drift was typically less thanÇ10 MHz, defin-
ing the overall precision. Separations of close lines in a portant to note that E1 and E2 levels, having the same symme-try, are coupled by the asymmetry of the molecule and maysingle scan should have precision better than 1 MHz. The
internal consistency in frequency calibration between the be mixed by perturbations. In this paper, we will mainly usethe signed K notation for E levels, but will occasionallyAkron and NIST spectra was typically Ç0.0005 cm01 .
A comparison of the two spectra showed the EROS employ E1 and E2 when convenient.An alternative notation often used denotes a torsion–rota-method to have very high peak sensitivity that yielded more
lines in a given spectral region than did the direct absorption tion–vibration level as (nttK , J)n where nt is the torsionalquantum number, t is the torsional index associated withmethod. However, the complicated intensity behavior from
the Stark focusing caused many lines to be missing in the the torsional symmetry, and n is the quantum number of avibration of interest other than the torsion. In this notation,EROS experiment that were quite intense in the direct ab-
sorption experiment. Frequently, the EROS method pro- K , the projection of the overall rotational angular momentumJ along the molecular symmetry axis, is indicated withoutduced a strong Q branch without showing the corresponding
P-branch and R-branch lines. a sign. K and t may be combined to obtain A , E1 , or E2
according to the rule: mod(K / t) /3 Å 0 is E1 ; 1 is A ;In addition to the spectra of jet-cooled methanol, high-resolution Fourier-transform (FT) spectra in the CH-stretch and 2 is E2 . The torsional index is particularly useful for
examining the dependence of the torsional energy on K .region (2700–3150 cm01) were recorded. Traces were takenat 0.003 cm01 resolution at both room temperature and 190 Kon a modified DA8.002 Bomem spectrometer at the Steacie A. Infrared–Microwave Double ResonanceInstitute for Molecular Sciences of the National Research
The initial assignments for four of the subbands wereCouncil of Canada in Ottawa. Although these FT spectramade with the aid of infrared–microwave (IR–MW) three-are extremely crowded, they give a broad spectral view andlevel double-resonance experiments on the EROS machinehave provided some checks on the assignments. The FTat NIST (48) . In this case, infrared radiation was tuned towork will be the subject of a separate communication.coincide with a strong feature in the NIST spectrum, andthen a microwave synthesizer was scanned through a seriesIII. ASSIGNMENTSof known ground state transition frequencies. When the in-frared signal varied due to a resonance with a specific micro-The molecular-beam spectra between 2965 and 3027 cm01
wave transition, that transition was recorded in the computer.cover mainly the n2 , A* asymmetric CH-stretch fundamental.To confirm that the double resonance indeed involved theThe predominant subbands are those with perpendicular b-lower infrared level, the infrared radiation was masked totype selection rules, but a number of weaker parallel a-typecheck that some absorption due to the ground state micro-subbands are found as well. Some initial line assignmentswave transition still remained. The relevant level schemeswere established by infrared–microwave double resonanceare shown in Fig. 4 and the assignments are discussed indi-(IR–MW DR); others were found by pattern recognitionvidually below.and polynomial fits of the line positions within individual
subbands. Assignments were then confirmed using the strin- (a) K Å 1 R 0 A system (Fig. 4a). The infrared radia-tion was first tuned to the strong transition at 3008.6389gent combination loop tests detailed below.
If methanol were constrained to be a rigid molecule, its cm01 , which was clearly identified as the first line of a Qbranch. To search for infrared–microwave double reso-energy levels could be categorized in the Cs point group as A*
or A 9. However, to account for the large amplitude torsional nance, a microwave synthesizer was stepped through the
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$242 09-09-97 22:35:14 mspas

XU ET AL.164
FIG. 4. Infrared–microwave double-resonance schemes (a) K Å 1 R 0 A , (b) K Å 0 R 1 A , (c) K Å 2 R 1 A , and (d) K Å 2 R 1 E .
known ground state transitions and a strong double-reso- the schemes of Figs. 4b and 4c with knowledge of the generalnance signal was found at the 48 372.456 MHz frequency energy level structure, it was deduced that the previous Qof the J Å 1 R 0, K Å 0 A ground state a-type microwave branch around 2996.87 cm01 belonged to the K Å 0 R 1transition. This result indicates that the infrared transition is A/
R A0 subband and the present Q branch around 3006.27the Q(1) member of the K Å 1 R 0 A subband. cm01 to one of the two components of the K Å 2 R 1 A
subband. To further confirm these assignments, the micro-(b) K Å 0 R 1 A system (Fig. 4b). Another Q branchwave source was then scanned to search for the upper state Karound 2996.87 cm01 was examined with the double-reso-Å 2 asymmetry doublets, and indeed three double-resonancenance experiment. In this case, the infrared radiation wassignals were found at frequencies of 29.60, 88.79, and 206.68set to coincide with the first line of the Q branch atMHz in resonance with Q(3) at 3006.2334 cm01 , Q(4) at2996.8701 cm01 and the microwave synthesizer was scanned3006.1848 cm01 , and Q(5) at 3006.1257 cm01 , respectively.through ground state transitions. A double-resonance signalThe expected quadrupole focusing behavior for these K-was found at 834.267 MHz, corresponding to the J Å 1, Kdoublet systems suggested that this stronger Q-branch shad-Å 1 A0
R A/ transition across the levels of the lowest K Å 1ing to low frequency corresponds to the K Å 2 R 1 A/
Rground-state asymmetry doublet. The observation of double-A0 component, while a weaker observed Q branch movingresonance signals at 2502.778 and 5005.321 MHz for theto higher frequency is the K Å 2 R 1 A0
R A/ component.second and third Q lines respectively confirmed that theR-, P- and Q-branch assignments for the above three sys-lower state of this infrared Q branch is K Å 1 A .
tems were then extended by using ground state combination(c) K Å 2 R 1 A system (Fig. 4c). The third Q branch
differences.for which a double-resonance search was performed is near(d) K Å 2 R 1 E system (Fig. 4d). A strong infrared3006.27 cm01 . The first two members of the Q branch gave
absorption at 3013.5259 cm01 was examined with the dou-double-resonance signals with microwave frequencies ofble-resonance technique and found to give a double-reso-2502.778 and 5005.321 MHz. These were clearly the same Jnance signal with the second harmonic of the microwaveÅ 2 and 3, K Å 1 ground state asymmetry doublet transitionssynthesizer set to 12 466.74 MHz. This corresponds to theas for the previous system above, implying the lower state ofJ Å 4 member of the K Å 2 R 1 E ground state Q branchthe Q branch was K Å 1 A and that the upper state had to be
K Å 2 from the absence of a J Å 1 line. Then, by combining at 24 933.47 MHz, as shown in Fig. 4d.
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$243 09-09-97 22:35:14 mspas

CH STRETCH OF METHANOL 165
B. Subband Assignments Using Ground State n(K / 1 R K) IR 0 n(K / 1 R K / 2) IR
Combination Differences Å n(K / 2 R K)0 ,
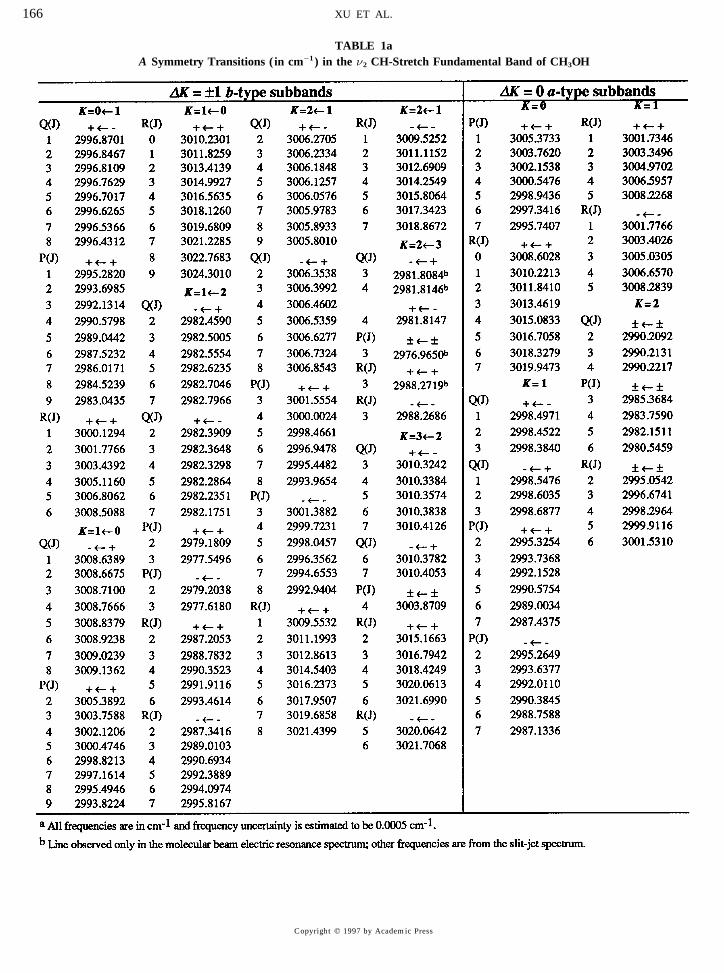
Altogether, 19 DK Å {1 b-type subbands for 0 £ K £3 for A and 03 £ K £ 4 for E have been identified using where n(K* R K9)IR refers to the transition K* R K9 goingground state energies calculated from the parameters re- from the ground state to the n2 excited vibrational state, andported in Ref. (22) . All of these assignments are attributed n(K/ 2 R K)0 refers to an energy difference within the groundto the n2 , A * asymmetric CH-stretch fundamental. This vi- state. As the n(K / 2 R K)0 values are extremely sensitive tobrational identification is based on lower resolution spectra K and t, this test puts each subband assignment on a solidof methanol and its isotopomers (49, 50) . Table 1 lists all footing. Because only a few K states were populated in thethe assigned transitions arranged according to K * R K 9 sub- cold molecular beam, some of the ‘‘higher’’ K subbands, suchbands and is divided into two parts according to A /E symme- as K* Å 3 or 4, were confirmed with a second (K* R K9)IR
try. The majority of the lines were first recognized as series subband taken from the Fourier-transform spectra.and classified into R , P , and Q subbranches by performing (c) a-type transitions in the n2 band. In addition to thepolynomial fits in m (m Å J / 1, J , and 0J for R-, Q-, and b-type transitions discussed above, systematic searches forP-branch transitions, respectively) . The subbranches were a-type transitions were performed and eight a-type subbandsthen linked into subbands and the full quantum number as- were found with ÉK *maxÉ Å 2 for both A and E torsionalsignments determined by applying two stringent checks de- symmetries. The a-type transition frequencies are calculatedscribed separately below. precisely from the observed b-type transitions. The ratios of
Not included in Table 1 are a great many unassigned the observed b-type to a-type subband intensities vary be-lines. The number is particularly great in the molecular beam tween 5 and 10 to 1, suggesting an a /b hybrid transitionelectric resonance spectrum because it extends further on moment for the n2 asymmetric CH-stretch vibrational mode.the low frequency side and because it picks up many lines
(d) Transitions in the n9 band. Three DK Å {1 sub-not seen in the slit jet spectrum. Below 3000 cm01 , manybands attributable to the n9 A2 (A 9) asymmetric CH stretchof the unassigned lines probably belong to the n9 band.were also assigned as described in Section (a) above andAbove 3000 cm01 , the most intense unassigned lines in theare centered at frequencies below 2989 cm01 . These sub-slit jet spectrum are about 5% of the largest peak in Fig. 3.bands are marked in Fig. 3 but an additional investigation
(a) Confirmation of J 9, K 9, and t9 for a single subband. of the n9 region is planned and these results will be presentedR , P , and Q transitions within a subband originating from in a subsequent publication.a (K9, t9) lower state must satisfy the ground state combina-tion differences
IV. PERTURBATIONS
R(J) 0 Q(J / 1) Å Q(J) 0 P(J / 1) Several J-localized perturbations were observed in the n2
state in the subbands terminating on the K* Å 0 E andÅ n(J / 1 R J)0 ,01 E levels and confirmed by ground state combinationdifferences. For K* Å 0 E , the J* Å 1 line is shifted away
where n(J / 1 R J)0 is a ground state a-type frequency. from its expected position by Ç0.009 cm01 while for K ÅThe latter are accurately known from ground-state studies 01 E , the J * Å 5 line is shifted away from its expected(21, 22) and are generally sufficiently sensitive to K 9 and position by Ç0.033 cm01 . In both cases, the intensity bor-t9 to distinguish clearly between different possible assign- rowing of the perturbing (‘‘dark’’) state is sufficient to allowments. Thus, the infrared combination difference formulas its observation.could be used not only to connect the R-, P-, and Q-branch A stronger perturbation was found in the K Å 02 R 01series but also in most cases to assign t9 for a subband. For E subbands. The K* Å 02 E upper states are split by É1.7an assignment to be considered verified, the combination cm01 so the K Å 02 R 01 E transitions appear in Fig. 3 asrelations had to be satisfied to within the experimental uncer- two complete subbands near 3016.0 and 3017.8 cm01 , eachtainty. Sometimes the a-type frequencies are similar for dif- with P , Q , and R branches. The combination differences offerent K 9 and t9 possibilities, hence caution must be exer- the type described above in Section B(a) were sufficient tocised in such cases. establish that the lower state is K9 Å 01 E for both of these
subbands. The combination differences of the type described(b) Confirmation of K* and t* for two subbands con-nected to the same upper state. Additional combination- in Section B(b) then confirmed that K* Å 02 E is the upper
state of the 3016.1 cm01 subband. Unfortunately, the K Ådifference verification of assignments is obtained by noticingfor an upper state (K * Å K / 1, t*) 02 R 03 E transitions corresponding to the 3017.8 cm01
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$243 09-09-97 22:35:14 mspas
XU ET AL.166
TABLE 1aA Symmetry Transitions ( in cm01 ) in the n2 CH-Stretch Fundamental Band of CH3OH
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$7367 09-09-97 22:35:14 mspas

CH STRETCH OF METHANOL 167
TABLE 1bE Symmetry Transitions ( in cm01 ) in the n2 CH-Stretch Fundamental Band of CH3OH
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$7367 09-09-97 22:35:14 mspas

XU ET AL.168
dark states must be less than or equal to 1 and 5, respectively;( ii ) from the assignments for each subband associated withthe perturbed levels, we find that the line series seem regularonce we move away from the perturbing region, suggestingthat the perturbation mechanism involves an anharmonic in-teraction rather than Coriolis forces. If this is the case, onewould expect the interacting states to carry the same K value.The stronger interaction at K* Å 02 E has a matrix elementof about 0.75 cm01 which is approximately independent ofJ*. Therefore, it could be anharmonic or possibly z-typeCoriolis in origin but definitely not x- or y-type Coriolis.
V. TORSIONAL AND ROTATIONAL STRUCTUREOF THE n2 VIBRATION
In this section, it will be shown that, with the exceptionFIG. 5. Deperturbation scheme. The bright and dark zeroth-order energy of the perturbations noted above, the energy levels of the n2levels are represented as dashed lines and the observed energy levels are
vibration follow an apparently regular pattern. However, thesolid lines.pattern is not the one anticipated by the Hamiltonian (22)which has been so successful in fitting the ground and firsttorsionally excited states.
subband were too weak to be observed. It could be shownthat the 3017.8 cm01 subband is not a DK Å 3 subband, i.e.,
TABLE 2K Å /2 R 01 E , since the K * Å /2 upper state energiesPerturbations in the n2 Bandare known from other transitions. Since the 3017.8 cm01
subband represents the only group of moderately intenselines found in the slit jet spectra above 3000 cm01 whichhave not otherwise been assigned, the K Å 02 R 01 Eassignment is reasonable.
Each of the perturbed systems has been treated as a two-state interaction with a 2 1 2 secular determinant containingan interaction matrix element W12 . From the observed per-turbed energy separation de and intensity ratio R for theallowed and the perturbation-induced satellite transitions(Fig. 5) , we can obtain the unperturbed energy separationde* and off-diagonal matrix element W12 using the relations
de* Å (R 0 1)de(R / 1)
and W12 Å√ÉdeÉ2 0 Éde*É2
2.
Table 2 collects all the transitions associated with the pertur-bation systems along with the calculated unperturbed energyseparations and off-diagonal matrix elements.
It is difficult at this stage to firmly identify the perturbingstates, because as seen in Fig. 1 the density of vibrationalstates near 3000 cm01 is significant and current methanolHamiltonian models cannot give sufficiently reliable predic-tions to assign possible background perturbing states. How-ever, for the two localized perturbation systems, the follow-ing information can be deduced about the dark backgroundstates: ( i ) since the perturbations occur at J Å 1 for K Å 0E and JÅ 5 for KÅ01 E , the K values of the two perturbing
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$244 09-09-97 22:35:14 mspas

CH STRETCH OF METHANOL 169
TABLE 3K Å 1 A and 2 A Asymmetry Splittings in the n2 Excited CH-Stretch Vibrational State and Splitting
Constants, D Å [ (J / K ) ! / (J 0 K ) ! ] [S / TJ (J / 1)] , for Methanol ( in MHz)
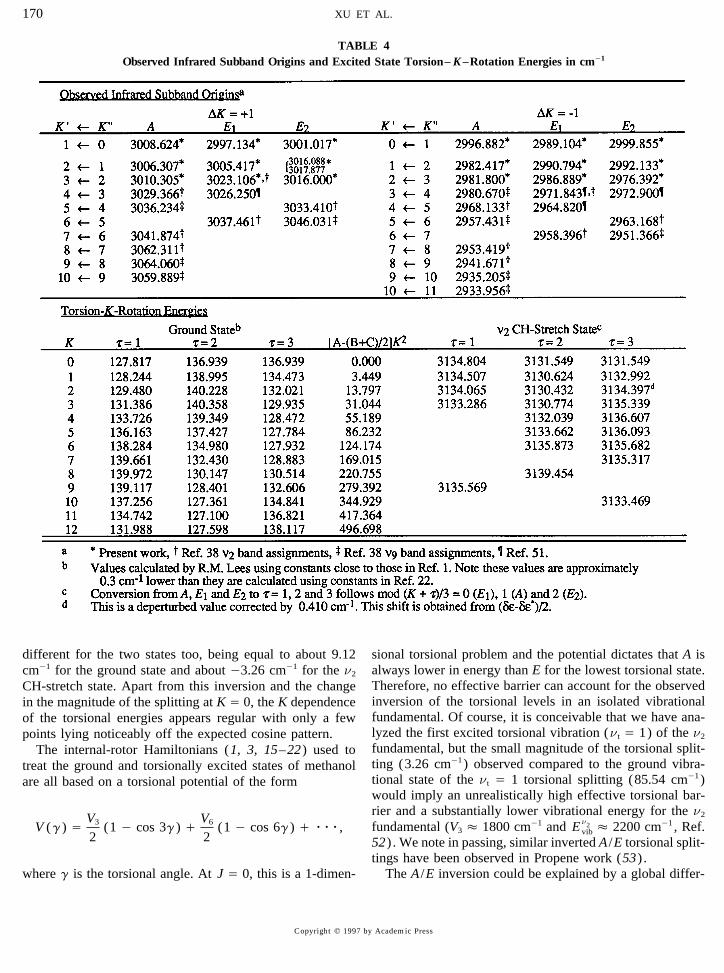
A. Rotational Level Structure state energies, we subtract [A0 (B/ C) /2]K 2 using groundstate rotation constants. Table 4 summarizes the relevant
The new information on the asymmetry splittings for Kinformation used to plot torsion– K–rotation energy curvesÅ 1 and 2 A doublets in the n2 excited CH-stretch state isfor each t value for the ground and n2 excited CH-stretch
shown in the upper portion of Table 3. These experimentalstates as functions of K in Fig. 6. Table 4 also includes
asymmetry splittings allowed a determination of splittingthe subband origins reported in Ref. (38) . It is somewhat
parameters S and T from the relationsurprising to see that although the latter were assigned toboth the n2 and n9 vibrational bands, in fact all fall on extrap-olations of our t curves along the threefold cosine functionsD Å (J / K)!
(J 0 K)![S / TJ(J / 1)] .
shown in Fig. 6. Therefore, it is likely that all of the upperstates represented in Fig. 6, including those previously as-
In the lower portion of Table 3, the asymmetry splitting signed as n9 , can be attributed to the single vibrationallyparameters, S and T , for the n2 state are compared to those excited state designated as n2 in previous low-resolutionfor the ground, CO-stretch, and OH-bend states. The K Å studies (49, 50) . This attribution is supported by our assign-1 A asymmetry splittings for the n2 CH-stretch state are ments of three clearly different subbands (Fig. 3) about 30comparable with those of the ground state, but the K Å 2 A cm01 lower in frequency than their n2 counterparts, whichasymmetry splittings are significantly larger, indicating a is indeed the approximate shift expected for the n9 band.possible interaction with one or more dark states. Comparison of the two sets of torsion– K–rotation curves
for the ground and the n2 excited CH-stretch states revealsB. Torsional Level Structure an interesting yet puzzling energy structure for the n2 CH-
stretch state. The torsional oscillation pattern in the excitedTo study the torsion– K–rotation energy structure in thestate seems regular and follows a threefold cosine functionn2 excited CH-stretch state, we first fit the K subbands tofor all three t curves with a slight alteration of oscillationJ(J / 1) Taylor series expansions in order to extract the J-period possibly due to a difference in rotational structuresindependent subband origins which are collected in the topfor the two states. However, the sign of the torsional split-part of Table 4. The observed infrared subband origins inting, namely the ordering of the E /A levels for K Å 0, isTable 4 allow one to map the torsion– K–rotation energyreversed in going from the ground state to the n2 excitedstructure in the n2 excited CH-stretch state. To remove the
rigid-rotor contribution to the K dependence of the upper CH-stretch state. The magnitude of the E /A splitting is quite
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$244 09-09-97 22:35:14 mspas
XU ET AL.170
TABLE 4Observed Infrared Subband Origins and Excited State Torsion– K–Rotation Energies in cm01
different for the two states too, being equal to about 9.12 sional torsional problem and the potential dictates that A isalways lower in energy than E for the lowest torsional state.cm01 for the ground state and about 03.26 cm01 for the n2
Therefore, no effective barrier can account for the observedCH-stretch state. Apart from this inversion and the changeinversion of the torsional levels in an isolated vibrationalin the magnitude of the splitting at K Å 0, the K dependencefundamental. Of course, it is conceivable that we have ana-of the torsional energies appears regular with only a fewlyzed the first excited torsional vibration (nt Å 1) of the n2points lying noticeably off the expected cosine pattern.fundamental, but the small magnitude of the torsional split-The internal-rotor Hamiltonians (1, 3, 15–22) used toting (3.26 cm01) observed compared to the ground vibra-treat the ground and torsionally excited states of methanoltional state of the nt Å 1 torsional splitting (85.54 cm01)are all based on a torsional potential of the formwould imply an unrealistically high effective torsional bar-rier and a substantially lower vibrational energy for the n2
fundamental (V3 É 1800 cm01 and En2vib É 2200 cm01 , Ref.V (g) Å V3
2(1 0 cos 3g) / V6
2(1 0 cos 6g) / rrr,
52) . We note in passing, similar inverted A /E torsional split-tings have been observed in Propene work (53) .
The A /E inversion could be explained by a global differ-where g is the torsional angle. At J Å 0, this is a 1-dimen-
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$244 09-09-97 22:35:14 mspas

CH STRETCH OF METHANOL 171
FIG. 6. Torsion– K–rotation energy diagram for the ground and the n2 excited states.
of Energy Research, U.S. Department of Energy under Grant No. DE-ential perturbation of the n2 levels by background state(s)FG02-90ER14151. Support of this work does not constitute endorsementwith torsional components nt § 1. Since the K subbandby the DOE of views expressed in this paper.
origins are shifted, anharmonic or parallel Coriolis interac-tions, but not perpendicular Coriolis, are involved; so the
REFERENCESoverall perturbing combination vibration must have A* sym-metry. A simple linear combination model indicates a 13% 1. R. M. Lees and J. G. Baker, J. Chem. Phys. 48, 5299–5318 (1968).
2. H. M. Pickett, E. A. Cohen, D. E. Brinza, and M. M. Schaefer, J. Mol.mixing arising from such a background state would beSpectrosc. 89, 542–547 (1981).enough to change the observed n2 fundamental to an inverted
3. E. Herbst, J. K. Messer, F. C. DeLucia, and P. Helminger, J. Mol.A /E pattern. An alternative explanation of the A /E inversionSpectrosc. 108, 42–57 (1984).
involves torsionally induced coupling among the CH 4. K. V. L. N. Sastry, R. M. Lees, and F. C. DeLucia, J. Mol. Spectrosc.stretches (54) . In either case, the present work suggests that 103, 486–494 (1984).
5. F. C. DeLucia, E. Herbst, T. Anderson, and P. Helminger, J. Mol.to properly account for the observed n2 fundamental energy-Spectrosc. 134, 395–411 (1989).level pattern, some kind of interacting band analysis will be
6. T. Anderson, F. C. DeLucia, and E. Herbst, Ap. J. Suppl. Series 72,required. Such approaches are now under investigation and797–814 (1990).
will be reported in future papers. 7. O. I. Baskakov and M. A. O. Pashaev, J. Mol. Spectrosc. 151, 282–291 (1992).
8. F. Matsushima, K. M. Evenson, and L. R. Zink, J. Mol. Spectrosc. 164,ACKNOWLEDGMENTS517–530 (1994).
9. H. Odashima, F. Matsushima, K. Nagai, S. Tsunekawa, and K. Takagi,One of us (LHX) is greatly indebted to Drs. J. T. Hougen and R. M.Lees for numerous fruitful discussions and valuable comments on the manu- J. Mol. Spectrosc. 173, 404–422 (1995).
10. S. P. Belov, G. Winnewisser, and E. Herbst, J. Mol. Spectrosc. 174,script. This work was financially supported in part by the Natural Sciencesand Engineering Research Council of Canada, the University of New Bruns- 253–269 (1995).
11. G. Moruzzi, F. Strumia, P. Carnesecchi, B. Carli, and M. Carlotti,wick Research Fund, the NASA Upper Atmosphere Research Program, andthe Division of Chemical Sciences, Office of Basic Energy Sciences, Office Infrared Phys. 29, 47–86 (1989).
12. G. Moruzzi, P. Riminucci, F. Strumia, B. Carli, M. Carlotti, R. M.of University Research, U.S. Department of Energy under Grant No. DE-AI02-94ER14411. The work at the University of Akron was supported by Lees, I. Mukhopadhyay, J. W. C. Johns, B. P. Winnewisser, and M.
Winnewisser, J. Mol. Spectrosc. 144, 139–200 (1990).the Division of Chemical Sciences, Office of Basic Energy Sciences, Office
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$244 09-09-97 22:35:14 mspas

XU ET AL.172
13. G. Moruzzi, F. Strumia, J. C. S. Moraes, R. M. Lees, I. Mukhopadhyay, Colloquium on High Resolution Molecular Spectroscopy, Dijon,France, Sept., 1995, paper Q30.J. W. C. Johns, B. P. Winnewisser, and M. Winnewisser, J. Mol. Spec-
34. R. G. Lee, R. H. Hunt, E. K. Plyler, and D. M. Dennison, J. Mol.trosc. 153, 511–577 (1992).Spectrosc. 57, 138–154 (1975).14. N. Ioli, G. Moruzzi, P. Riminucci, F. Strumia, J. C. S. Moraes, B. P.
35. P. Carrick, R. F. Curl, M. Dawes, E. Koester, K. K. Murray, M. Petri,Winnewisser, and M. Winnewisser, J. Mol. Spectrosc. 171, 130–144and M. L. Richnow, J. Mol. Struct. 223, 171–184 (1990).(1995).
36. I. Kleiner, G. T. Fraser, J. T. Hougen, and A. S. Pine, J. Mol. Spectrosc.15. D. G. Burkhard and D. M. Dennison, Phys. Rev. 84, 408–417 (1951).147, 155–172 (1991).16. E. V. Ivash and D. M. Dennison, J. Chem. Phys. 21, 1804–1816
37. R. H. Hunt, W. N. Shelton, W. B. Cook, O. N. Bignall, J. W. Mirick,(1953).and F. A. Flaherty, J. Mol. Spectrosc. 149, 252–256 (1991).17. K. T. Hecht and D. M. Dennison, J. Chem. Phys. 26, 48–69 (1957).
38. O. N. Bignall, R. H. Hunt, and W. N. Shelton, J. Mol. Spectrosc. 166,18. Y. Y. Kwan and D. M. Dennison, J. Mol. Spectrosc. 43, 291–319137–146 (1994).(1972).
39. J. T. Hougen, J. Mol. Spectrosc. 181, 287–296 (1997).19. B. Kirtman, J. Chem. Phys. 37, 2516–2539 (1962).40. L. Lubich, O. V. Boyarkin, R. D. F. Settle, D. S. Perry, and T. R. Rizzo,20. K. Nakagawa, S. Tsunekawa, and T. Kojima, J. Mol. Spectrosc. 126,
Faraday Disc. Chem. Soc. 102, 167–178 (1995).329–340 (1987).41. Lauri Halonen, J. Chem. Phys. 106, (19) 7931–7945 (1997).21. Li-Hong Xu and J. T. Hougen, J. Mol. Spectrosc. 169, 396–40942. F. J. Lovas, J. Phys. Chem. Ref. Data 21, 181–272 (1992). [see(1995).
references therein]22. Li-Hong Xu and J. T. Hougen, J. Mol. Spectrosc. 173, 540–55143. D. Bockelee-Morvan, J. Crovisier, P. Colom, and D. Despois, Astron.(1995).
Astrophys. 287, 647–665 (1994).23. R. M. Lees, J. Chem. Phys. 57, 2249–2252 (1972).44. D. Bockelee-Morvan, T. Y. Brooke, and J. Crovisier, Icarus 116, 18–24. J. P. Sattler, T. L. Worchesky, and W. A. Riessler, Infrared Phys. 18,
39 (1995).521–528 (1978).45. G. A. Bethardy and D. S. Perry, J. Phys. Chem. 98, 6651 (1993).
25. J. P. Sattler, W. A. Riessler, and T. L. Worchesky, Infrared Phys. 19,46. D. Kaur, A. M. de Souza, J. Wanna, S. A. Hammad, L. Mercorelli, and
217–224 (1979).D. S. Perry, Appl. Opt. 29, 119 (1990).
26. J. O. Henningsen, J. Mol. Spectrosc. 85, 282–300 (1981).47. A. S. Pine and G. T. Fraser, J. Chem. Phys. 89, 100–109 (1988).
27. H. Rudolph, J. Avery, and J. O. Henningsen, J. Mol. Spectrosc. 117, 48. Li-Hong Xu, A. M. Andrews, and G. T. Fraser, J. Chem. Phys. 103,38–45 (1986). 14–19 (1995).
28. G. Moruzzi, F. Strumia, P. Carnesecchi, R. M. Lees, I. Mukhopadhyay, 49. A. Serrallach, R. Meyer, and Hs. Gunthard, J. Mol. Spectrosc. 52, 94–and J. W. C. Johns, Infrared Phys. 29, 583–606 (1989). 129 (1974).
29. R. M. Lees, J. Chem. Phys. 57, 824–826 (1972). 50. F. C. Cruz, A. Scalabrin, D. Pereira, P. A. M. Vazquez, Y. Hase, and30. J. O. Henningsen, J. Mol. Spectrosc. 83, 70–93 (1980). F. Strumia, J. Mol. Spectrosc. 156, 22–38 (1992).31. R. M. Lees, M. A. Walton, and J. O. Henningsen, J. Mol. Spectrosc. 51. Li-Hong Xu, A. S. Smith, R. M. Lees, and Z. F. Lu, unpublished results.
88, 90–94 (1981). 52. Li-Hong Xu and J. T. Hougen, unpublished results.32. J. O. Henningsen, J. Mol. Spectrosc. 91, 430–457 (1982). 53. A. Ainetschian, G. T. Fraser, J. Ortigoso, and B. H. Pate, J. Chem.33. R. M. Lees, Li-Hong Xu, and Z. F. Lu, ‘‘Fourier Transform Spectros- Phys. 100, 729 (1994).
54. X.-L. Wang and D. S. Perry, manuscript in preparation.copy of the CH3-Rocking and OH-Bending Bands of CH3OH,’’ XIV
Copyright q 1997 by Academic Press
AID JMS 7367 / 6t1f$$$245 09-09-97 22:35:14 mspas
Related Documents