Applied Catalysis A: General 217 (2001) 275–286 Study of the Co-VPO interaction in promoted n-butane oxidation catalysts C. Carrara, S. Irusta, E. Lombardo, L. Cornaglia ∗ Instituto de Investigaciones en Catálisis y Petroqu´ ımica – INCAPE (FIQ, UNL-CONICET), Santiago del Estero 2829, 3000 Santa Fe, Argentina Received 26 December 2000; received in revised form 5 April 2001; accepted 7 April 2001 Abstract Cobalt-impregnated VPO catalysts were prepared using different cobalt salts and impregnation methods. The catalytic tests showed that cobalt impregnation significantly increased the overall activity. The use of cobalt acetyl acetonate led to a more selective high loading catalyst. To investigate the origin of the cobalt effect, the solids were characterized using XRD, Raman spectroscopy, FT-IR, and XPS. The surface acidity was probed by adsorbing acetonitrile. No structural effects were detected through XRD. After 700 h on stream, the only phase detected in all cases was V(IV) vanadyl pyrophosphate. The surface oxidation state of vanadium was V(IV). The Co 2p XP spectrum showed an intense shoulder at 788 eV, indicating that Co(II) species were present. The concentration of very strong Lewis acid sites increased at higher cobalt loading, but its strength was unaffected. © 2001 Elsevier Science B.V. All rights reserved. Keywords: VPO; n-Butane oxidation; Maleic anhydride; Promoters; Cobalt; Lewis acidity 1. Introduction The VPO system exhibits a unique ability to activate and selectively oxidize alkanes. Vanadyl pyrophos- phate is commercially used to catalyze the selective oxidation of n-butane to maleic anhydride (MAN). It is generally agreed upon that the best catalyst precursor is VOHPO 4 ·0.5H 2 O which is converted to (VO) 2 P 2 O 7 [1] during activation. Besides, several VOPO 4 phases present in low proportion have been claimed as nec- essary for the catalytic act [2]. However, these phases have never been detected in the so-called equilibrated catalysts that have been on stream for 700 h [3]. ∗ Corresponding author. Tel.: +54-342-4536861; fax: +54-342-4536861. E-mail address: lmcornag@fiqus.unl.edu.ar (L. Cornaglia). A variety of cations have been added to vanadium phosphate catalysts to improve activity and selectiv- ity. It has been claimed that the addition of the first row transition metal produces an improvement in the maleic anhydride yield [4,5]. Several papers [4–10] refer to the effect of cobalt used as a promoter. Takita et al. [6] sustained that the additives were incorporated into the crystal lattice of vanadyl pyro- phosphate. Volta et al. [7] using in situ Raman spec- troscopy found that the incorporation of Co can change the V(V)/V(IV) balance during the activation period and can therefore change the catalytic perfor- mance in the steady state. Other authors [5,7,8] stud- ied the influence of cobalt addition on the composition of the (VO) 2 P 2 O 7 catalyst. The presence of cobalt induced surface phosphorus enrichment, which modi- fied the surface acidity. Zazhigalov et al. [8] proposed that cobalt stabilize the catalyst performance by 0926-860X/01/$ – see front matter © 2001 Elsevier Science B.V. All rights reserved. PII:S0926-860X(01)00615-9

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Applied Catalysis A: General 217 (2001) 275–286
Study of the Co-VPO interaction in promotedn-butane oxidation catalysts
C. Carrara, S. Irusta, E. Lombardo, L. Cornaglia∗Instituto de Investigaciones en Catálisis y Petroquı́mica – INCAPE (FIQ, UNL-CONICET),
Santiago del Estero 2829, 3000 Santa Fe, Argentina
Received 26 December 2000; received in revised form 5 April 2001; accepted 7 April 2001
Abstract
Cobalt-impregnated VPO catalysts were prepared using different cobalt salts and impregnation methods. The catalytic testsshowed that cobalt impregnation significantly increased the overall activity. The use of cobalt acetyl acetonate led to a moreselective high loading catalyst. To investigate the origin of the cobalt effect, the solids were characterized using XRD, Ramanspectroscopy, FT-IR, and XPS. The surface acidity was probed by adsorbing acetonitrile. No structural effects were detectedthrough XRD. After 700 h on stream, the only phase detected in all cases was V(IV) vanadyl pyrophosphate. The surfaceoxidation state of vanadium was V(IV). The Co 2p XP spectrum showed an intense shoulder at 788 eV, indicating that Co(II)species were present. The concentration of very strong Lewis acid sites increased at higher cobalt loading, but its strengthwas unaffected. © 2001 Elsevier Science B.V. All rights reserved.
Keywords: VPO; n-Butane oxidation; Maleic anhydride; Promoters; Cobalt; Lewis acidity
1. Introduction
The VPO system exhibits a unique ability to activateand selectively oxidize alkanes. Vanadyl pyrophos-phate is commercially used to catalyze the selectiveoxidation of n-butane to maleic anhydride (MAN). It isgenerally agreed upon that the best catalyst precursoris VOHPO4·0.5H2O which is converted to (VO)2P2O7[1] during activation. Besides, several VOPO4 phasespresent in low proportion have been claimed as nec-essary for the catalytic act [2]. However, these phaseshave never been detected in the so-called equilibratedcatalysts that have been on stream for 700 h [3].
∗ Corresponding author. Tel.: +54-342-4536861;fax: +54-342-4536861.E-mail address: [email protected] (L. Cornaglia).
A variety of cations have been added to vanadiumphosphate catalysts to improve activity and selectiv-ity. It has been claimed that the addition of the firstrow transition metal produces an improvement in themaleic anhydride yield [4,5]. Several papers [4–10]refer to the effect of cobalt used as a promoter.
Takita et al. [6] sustained that the additives wereincorporated into the crystal lattice of vanadyl pyro-phosphate. Volta et al. [7] using in situ Raman spec-troscopy found that the incorporation of Co canchange the V(V)/V(IV) balance during the activationperiod and can therefore change the catalytic perfor-mance in the steady state. Other authors [5,7,8] stud-ied the influence of cobalt addition on the compositionof the (VO)2P2O7 catalyst. The presence of cobaltinduced surface phosphorus enrichment, which modi-fied the surface acidity. Zazhigalov et al. [8] proposedthat cobalt stabilize the catalyst performance by
0926-860X/01/$ – see front matter © 2001 Elsevier Science B.V. All rights reserved.PII: S0 9 2 6 -8 6 0X(01 )00615 -9

276 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
forming cobalt phosphate, which improves its catalyticproperties.
According to Zazhigalov et al. [8], moderate sur-face acidity facilitates desorption of maleic anhydrideand prevents its complete oxidation to carbon oxides.They suggested that promoters play an importantrole in controlling the surface acidity of promotedVPO solids. Selective oxidation of n-butane requiresLewis–Brönsted acid site in combination with a sur-face vanadium redox site.
In a recent paper, we have studied impregnatedCo-VPO catalysts obtained using cobalt acetate [10].We concluded that on equilibrated catalysts, the ad-dition of cobalt increases the activity while slightlydecreasing the selectivity. This effect seems to be aconsequence of increased exposure of cobalt on thecatalyst surface. In this work, we have extended ourstudies using different salts and impregnation proce-dures. The rational was that cobalt acetyl acetonatecould allow us to change the cobalt dispersion on thesurface, while the high-temperature impregnation mayhelp intercalate the Co atoms between the VPO layers.Besides, in search of additional clues to understandthe role of Co on the surface of equilibrated solids, wehave now probed the surface acidity using acetonitrile.
2. Experimental
2.1. Catalyst preparation
The precursor was obtained by reduction of 5 g ofV2O5 with 30 ml of isobutanol and 20 ml of benzyl al-cohol under reflux for 3 h. Then orthophosphoric acid(100%) was added in the desired amount and the solu-tion refluxed for another 2 h. After completion of thereaction, the solid phase was recovered by filtrationand dried in air at 390 K overnight.
2.1.1. Promoted catalystThe impregnation was carried out using either
cobalt acetate or acetyl acetonate and following twoprocedures.
1. The impregnation method: the salt, acetate(VPCox-Ia) or acetyl acetonate of Co (VPCox-Iaawhere x represents wt.% of Co) was dissolvedin 30 ml of isobutanol. The previously preparedprecursor was added to this solution and the
suspension was heated to 330 K with continuousstirring. The solvent was evaporated and the wetsolid dried at 390 K. The final Co content of allof the preparations was determined by atomicabsorption.
2. The high-temperature impregnation method:cobalt acetyl acetonate was used in this case(VPCox-HTI). The salt was dissolved in a mixtureof 30 ml of isobutanol and 20 ml of benzyl alcohol,the precursor was added and refluxed overnight.The precipitate was then filtered and dried in airat 390 K.
2.2. Catalyst activation and testing
The promoted catalyst precursors and unpromotedVPO were activated in a fixed bed microreactor. Typ-ically, about 1 g of catalyst (screened 170–250 �mrange Tyler 60–80 mesh) was loaded into the reactorand covered with a layer of quartz wool.
The quartz reactor was 60 cm long and 1.0 cm i.d. atthe catalyst bed portion. It was mounted vertically ina tubular furnace. A programmable temperature con-troller was used to assure reproducibility of heatingstrategies. Mass flow controllers were used to feed thereactants in the right proportion. An on-line gas chro-matograph equipped with a FID detector was used toanalyze both the reactant and product streams. Separa-tion of C4H10 and maleic anhydride was accomplishedwith a 2 ft 1/8 in SS Chromosorb WAW AT-1200+1%H3PO4 80/100 column.
The activation of the catalyst was performed byheating from ambient temperature to 603 K, whileflowing a mixture of 0.75% n-butane in air, with a gashourly space velocity (GHSV) of 900 h−1. The tem-perature was kept constant at 603 K for 3 h. Those cat-alysts which only underwent this treatment are herecalled nonequilibrated catalysts. Then, the n-butaneconcentration was increased to 1.5% while still main-taining the GHSV at 900 h−1. The temperature wasraised until either 80% conversion or the maximumallowable temperature of 703 K was reached. Then,the GHSV was raised to 2500 h−1 and the temperaturewas slowly increased with the limitation that conver-sion never exceeded 80%. The catalytic data reportedhere were measured over solids that did not show anychange in activity and selectivity after at least 500 hon stream. At this point when the reaction temperature

C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286 277
was randomly varied up and down, reproducible val-ues of activities and selectivities were obtained. Thesesolids are called equilibrated catalysts.
2.3. Catalyst characterization
2.3.1. X-ray diffractionThe measurements were made with a Shimadzu
XD-D1 X-ray diffractometer, using nickel-filtered CuK� radiation with a scanning rate of 1◦ min−1.
2.3.2. Laser Raman spectroscopy (LRS)The Raman spectra were recorded with a Jasco
laser Raman spectrometer model TRS-600-SZ-P,equipped with a CCD (charge coupled device) withthe detector cooled to about 153 K using liquid N2.The excitation source was the 514.5 nm line of a Spec-tra 9000 Photometrics Ar ion laser. The laser powerwas set at 30 MW. All the spectra were recordedwith the samples under ambient conditions. The pow-dered solid was pressed into a thin wafer about 1 mmthick.
2.3.3. Infrared spectroscopyThe IR spectra were obtained using a Shimadzu
FT-IR 8101M spectrometer with a spectral resolutionof 4 cm−1. The solid samples were prepared in theform of pressed wafers (ca. 2 wt.% sample in KBr).The IR spectra of precursors were obtained using flu-orolube as a diluting agent in order to explore the highwave number region.
The samples for the adsorption experiments wereprepared by compressing the used catalysts at 9 tonnescm−2 in order to obtain a self-supporting wafer(50 mg, 10 cm diameter). They were mounted in atransportable infrared cell with CaF2 windows andexternal oven. The pretreatment was performed in ahigh-vacuum system. The sample was first outgassedat 723 K for 12 h in a dynamic vacuum of 7×10−4 Pa.After cooling to room temperature, a spectrum of thecatalyst wafer was taken. No bands were observed inthe 1600 cm−1 region, this is indicative that molec-ular water was eliminated. After that, 4.8 × 104 Paof acetonitrile was admitted into the cell and left incontact with the solid for 10 min, and then a spec-trum was recorded. No changes were observed inthe 1600 cm−1 region, therefore no water was intro-duced during acetonitrile adsorption. Spectra were
also recorded after evacuation of the cell for 10 minat 298, 353, and 423 K.
2.3.4. X-ray photoelectron spectroscopyThe XPS measurements were carried out using
an ESCA750 Shimadzu electron spectrometer. Non-monochromatic Al K� X-ray radiation was used. Theanode was operated at 8 kV and 30 mA and the pres-sure in the analysis chamber was about 2 × 10−6 Pa.The data were collected using an ESCAPAC 760computer interfaced to the spectrometer and analyzedwith the Googly software developed at the Universityof Pittsburgh.
The binding energies (BE) were referred to theC 1s signal (284.6 eV). Curve fitting was performedusing a Levenberg–Marquardt NLLSCF routine. Thebackground contribution was taken into account byassuming an integral type background which was in-cluded in the basic shape of each peak. Account hasbeen taken of the presence of K�3 and K�4 spectrallines of the large O 1s signal. These satellite peaksare 9.6 and 11.6 eV downshifted from the O 1s mainpeak and overlap the V 2p1/2 signal. Since the resolu-tion of the V 2p1/2 is poorer than that of the V 2p3/2level, we preferably used the latter BEs and widthsfor comparison with literature data. The V 2p and Co2p doublets were fitted using a Voigt function with20% Lorentzian character and 2p1/2/2p3/2 intensityratio = 0.5. Additional data for the curve fitting of theV 2p doublets include spin-orbit separation = 7.2 eV.
The surface P/V and Co/V atomic ratios werecalculated using the areas under the Co 2p3/2, P2p, and V 2p3/2 peaks, the Scotfield photoionizationcross-sections, the mean free paths of the electrons,and the instrumental function which was given by theESCA manufacturer.
3. Results and discussion
3.1. Catalytic behavior
Fig. 1A and B shows the catalytic results obtainedwith promoted and unpromoted solids. The impreg-nated solids exhibit a significantly higher conversionthan unpromoted VPO in the whole temperature range,with the exception of the VPCo1.5-Ia. For the pro-moted catalysts, the selectivity was nearly constant up
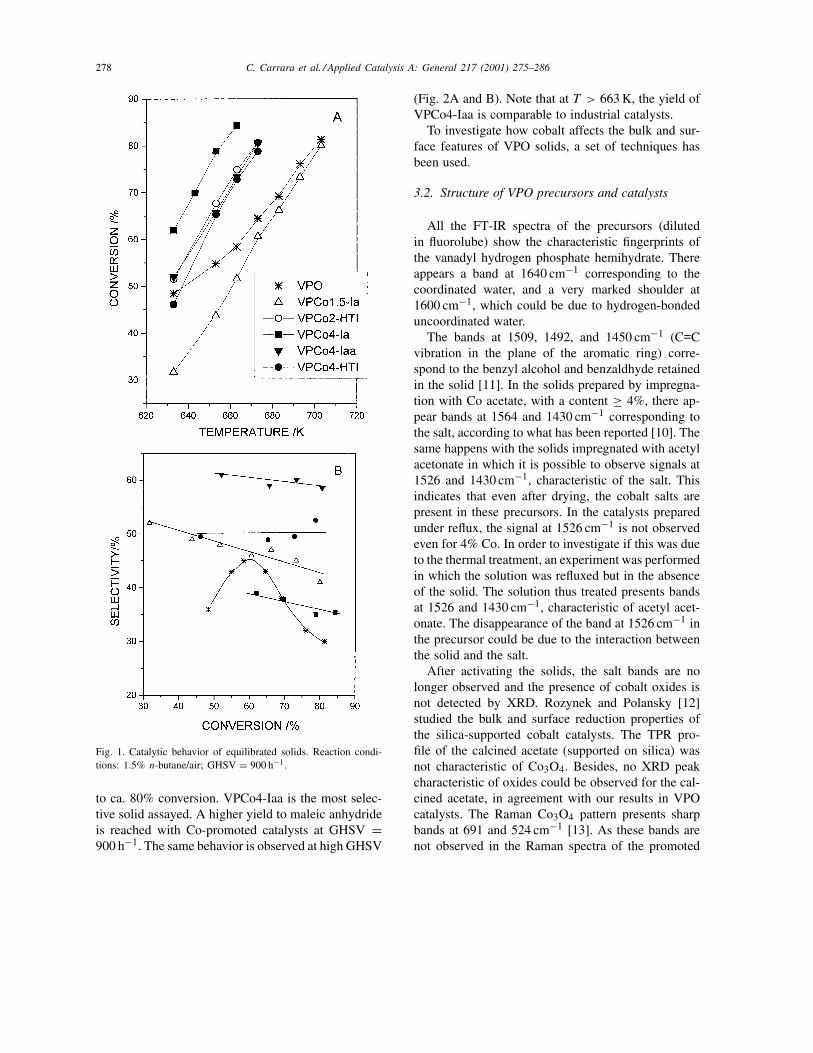
278 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
Fig. 1. Catalytic behavior of equilibrated solids. Reaction condi-tions: 1.5% n-butane/air; GHSV = 900 h−1.
to ca. 80% conversion. VPCo4-Iaa is the most selec-tive solid assayed. A higher yield to maleic anhydrideis reached with Co-promoted catalysts at GHSV =900 h−1. The same behavior is observed at high GHSV
(Fig. 2A and B). Note that at T > 663 K, the yield ofVPCo4-Iaa is comparable to industrial catalysts.
To investigate how cobalt affects the bulk and sur-face features of VPO solids, a set of techniques hasbeen used.
3.2. Structure of VPO precursors and catalysts
All the FT-IR spectra of the precursors (dilutedin fluorolube) show the characteristic fingerprints ofthe vanadyl hydrogen phosphate hemihydrate. Thereappears a band at 1640 cm−1 corresponding to thecoordinated water, and a very marked shoulder at1600 cm−1, which could be due to hydrogen-bondeduncoordinated water.
The bands at 1509, 1492, and 1450 cm−1 (C=Cvibration in the plane of the aromatic ring) corre-spond to the benzyl alcohol and benzaldhyde retainedin the solid [11]. In the solids prepared by impregna-tion with Co acetate, with a content ≥ 4%, there ap-pear bands at 1564 and 1430 cm−1 corresponding tothe salt, according to what has been reported [10]. Thesame happens with the solids impregnated with acetylacetonate in which it is possible to observe signals at1526 and 1430 cm−1, characteristic of the salt. Thisindicates that even after drying, the cobalt salts arepresent in these precursors. In the catalysts preparedunder reflux, the signal at 1526 cm−1 is not observedeven for 4% Co. In order to investigate if this was dueto the thermal treatment, an experiment was performedin which the solution was refluxed but in the absenceof the solid. The solution thus treated presents bandsat 1526 and 1430 cm−1, characteristic of acetyl acet-onate. The disappearance of the band at 1526 cm−1 inthe precursor could be due to the interaction betweenthe solid and the salt.
After activating the solids, the salt bands are nolonger observed and the presence of cobalt oxides isnot detected by XRD. Rozynek and Polansky [12]studied the bulk and surface reduction properties ofthe silica-supported cobalt catalysts. The TPR pro-file of the calcined acetate (supported on silica) wasnot characteristic of Co3O4. Besides, no XRD peakcharacteristic of oxides could be observed for the cal-cined acetate, in agreement with our results in VPOcatalysts. The Raman Co3O4 pattern presents sharpbands at 691 and 524 cm−1 [13]. As these bands arenot observed in the Raman spectra of the promoted

C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286 279
Fig. 2. Maleic anhydride yield of equilibrated catalysts: (A)GHSV = 900 h−1; (B) GHSV = 2500 h−1. Reaction conditions:1.5% n-butane/air.
catalyst, this is considered a confirmation of the XRDresults. Zazhigalov et al. [8] observed the appearanceof Co2P2O7 for the catalysts with Co/V > 0.15. Inour case (Co/V = 0.13), neither XRD nor Ramanspectroscopy indicate the presence of Co-containingphases.
In Table 2, the XRD data for nonequilibrated andequilibrated catalysts are shown. In all cases, vanadylpyrophosphate was the only crystalline phase detected.The decrease in the FWHMs of the (2 0 0) reflectionwith a long time-on-stream was observed. No struc-tural modifications (Table 1) were observed on impreg-nated solids through XRD. The FT-IR spectra of bothequilibrated and nonequilibrated solids show the fin-gerprints of vanadyl pyrophosphate between 700 and1700 cm−1, with the following assignments: ν(PO3) =1240 cm−1, ν(V=O) = 968 cm−1, ν(V–O–V) =797 cm−1, and ν(P–O–P) = 742 cm−1 [14].
In the cases where Co has been introduced duringthe phosphatization step, it was observed that theP–O–P vibration was modified [9]. The ν(P–O–P) at742 cm−1 has a similar intensity in the unpromotedand impregnated catalysts. This is substantiated by thehigh values (∼15 ± 1) of the 1245/742 intensity ratio(Table 1). It can be observed that the values obtainedfor the intensity ratio are lower in the case of thehigh-temperature Co-impregnated solids. This resultsfrom an increase in the band intensity correspondingto the P–O–P vibration. This increase is independentof both the Co content and the catalyst time-on-stream.This addition method of the Co promoter would alsobe introducing an increase in the number of P–O–Player linkages. The same effect was observed whencobalt was added during phosphatization (VPCo4-P).
The Raman spectra show that none of the equi-librated catalysts contain VOPO4 phases. This is atvariance with the findings of Hutchings et al. [7].They report that Co and Fe dopants can modify theVOPO4/(VO)2P2O7 dispersion. They concluded thatthe activity for maleic anhydride formation is a func-tion of the nature and concentration of the V(V) phasespresent. None of the three instrumental techniques al-lowed us to detect significant structural differencesin the catalysts with and without promoter addition,which could have explained their different catalyticbehavior. Let us see now if we can find “clues” onthe surface to explain this phenomenon. Presence ofV(V), Co oxidation state, P surface enrichment, and
280 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
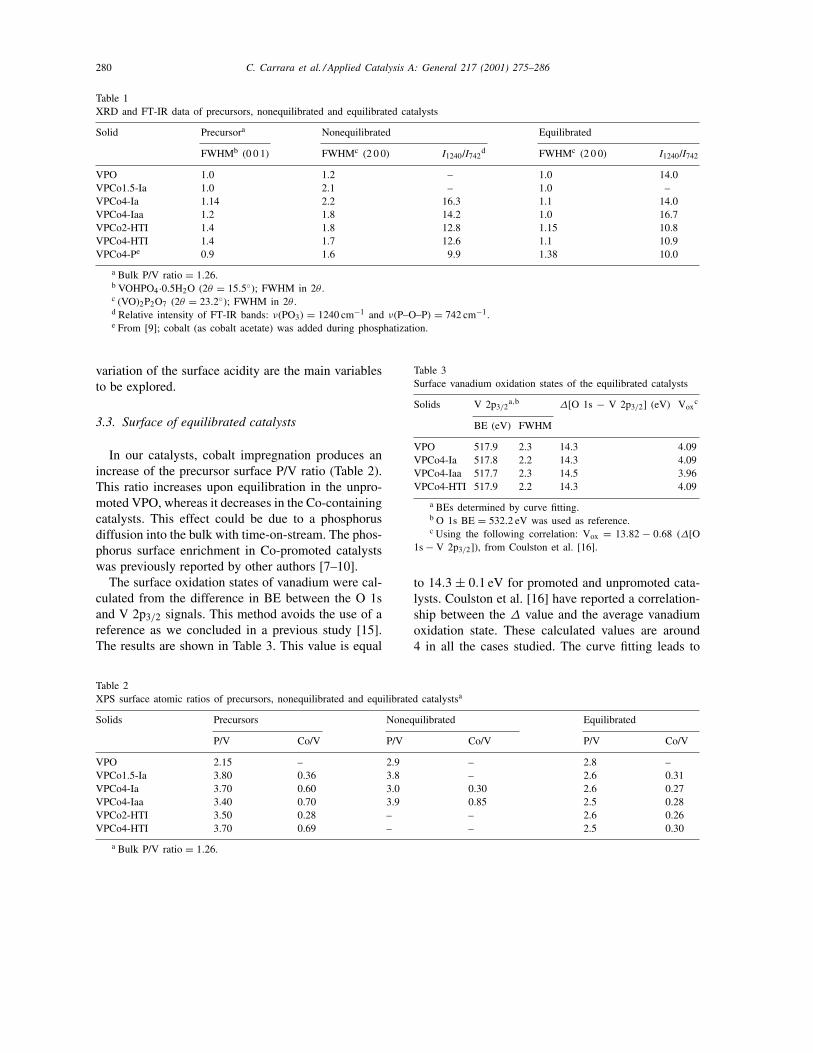
Table 1XRD and FT-IR data of precursors, nonequilibrated and equilibrated catalysts
Solid Precursora Nonequilibrated Equilibrated
FWHMb (0 0 1) FWHMc (2 0 0) I1240/I742d FWHMc (2 0 0) I1240/I742
VPO 1.0 1.2 – 1.0 14.0VPCo1.5-Ia 1.0 2.1 – 1.0 –VPCo4-Ia 1.14 2.2 16.3 1.1 14.0VPCo4-Iaa 1.2 1.8 14.2 1.0 16.7VPCo2-HTI 1.4 1.8 12.8 1.15 10.8VPCo4-HTI 1.4 1.7 12.6 1.1 10.9VPCo4-Pe 0.9 1.6 9.9 1.38 10.0
a Bulk P/V ratio = 1.26.b VOHPO4·0.5H2O (2θ = 15.5◦); FWHM in 2θ .c (VO)2P2O7 (2θ = 23.2◦); FWHM in 2θ .d Relative intensity of FT-IR bands: ν(PO3) = 1240 cm−1 and ν(P–O–P) = 742 cm−1.e From [9]; cobalt (as cobalt acetate) was added during phosphatization.
variation of the surface acidity are the main variablesto be explored.
3.3. Surface of equilibrated catalysts
In our catalysts, cobalt impregnation produces anincrease of the precursor surface P/V ratio (Table 2).This ratio increases upon equilibration in the unpro-moted VPO, whereas it decreases in the Co-containingcatalysts. This effect could be due to a phosphorusdiffusion into the bulk with time-on-stream. The phos-phorus surface enrichment in Co-promoted catalystswas previously reported by other authors [7–10].
The surface oxidation states of vanadium were cal-culated from the difference in BE between the O 1sand V 2p3/2 signals. This method avoids the use of areference as we concluded in a previous study [15].The results are shown in Table 3. This value is equal
Table 2XPS surface atomic ratios of precursors, nonequilibrated and equilibrated catalystsa
Solids Precursors Nonequilibrated Equilibrated
P/V Co/V P/V Co/V P/V Co/V
VPO 2.15 – 2.9 – 2.8 –VPCo1.5-Ia 3.80 0.36 3.8 – 2.6 0.31VPCo4-Ia 3.70 0.60 3.0 0.30 2.6 0.27VPCo4-Iaa 3.40 0.70 3.9 0.85 2.5 0.28VPCo2-HTI 3.50 0.28 – – 2.6 0.26VPCo4-HTI 3.70 0.69 – – 2.5 0.30
a Bulk P/V ratio = 1.26.
Table 3Surface vanadium oxidation states of the equilibrated catalysts
Solids V 2p3/2a,b ∆[O 1s − V 2p3/2] (eV) Vox
c
BE (eV) FWHM
VPO 517.9 2.3 14.3 4.09VPCo4-Ia 517.8 2.2 14.3 4.09VPCo4-Iaa 517.7 2.3 14.5 3.96VPCo4-HTI 517.9 2.2 14.3 4.09
a BEs determined by curve fitting.b O 1s BE = 532.2 eV was used as reference.c Using the following correlation: Vox = 13.82 − 0.68 (∆[O
1s − V 2p3/2]), from Coulston et al. [16].
to 14.3 ± 0.1 eV for promoted and unpromoted cata-lysts. Coulston et al. [16] have reported a correlation-ship between the ∆ value and the average vanadiumoxidation state. These calculated values are around4 in all the cases studied. The curve fitting leads to
C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286 281
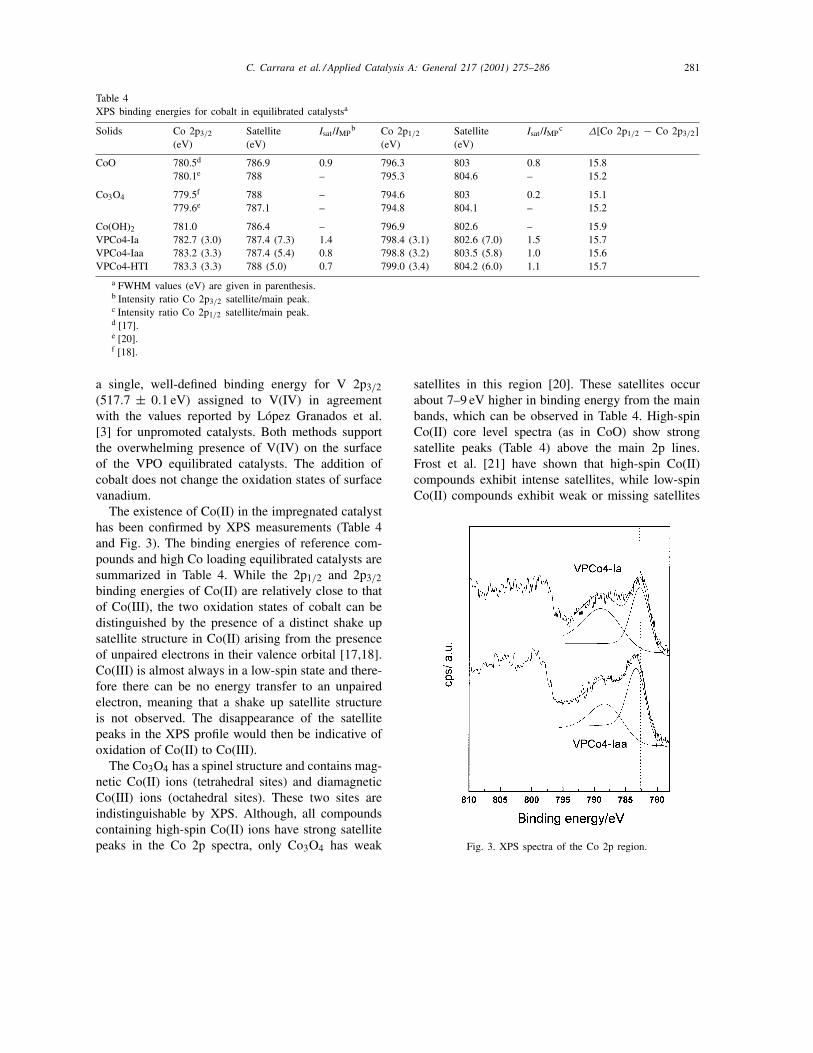
Table 4XPS binding energies for cobalt in equilibrated catalystsa
Solids Co 2p3/2
(eV)Satellite(eV)
Isat /IMPb Co 2p1/2
(eV)Satellite(eV)
Isat /IMPc ∆[Co 2p1/2 − Co 2p3/2]
CoO 780.5d 786.9 0.9 796.3 803 0.8 15.8780.1e 788 – 795.3 804.6 – 15.2
Co3O4 779.5f 788 – 794.6 803 0.2 15.1779.6e 787.1 – 794.8 804.1 – 15.2
Co(OH)2 781.0 786.4 – 796.9 802.6 – 15.9VPCo4-Ia 782.7 (3.0) 787.4 (7.3) 1.4 798.4 (3.1) 802.6 (7.0) 1.5 15.7VPCo4-Iaa 783.2 (3.3) 787.4 (5.4) 0.8 798.8 (3.2) 803.5 (5.8) 1.0 15.6VPCo4-HTI 783.3 (3.3) 788 (5.0) 0.7 799.0 (3.4) 804.2 (6.0) 1.1 15.7
a FWHM values (eV) are given in parenthesis.b Intensity ratio Co 2p3/2 satellite/main peak.c Intensity ratio Co 2p1/2 satellite/main peak.d [17].e [20].f [18].
a single, well-defined binding energy for V 2p3/2(517.7 ± 0.1 eV) assigned to V(IV) in agreementwith the values reported by López Granados et al.[3] for unpromoted catalysts. Both methods supportthe overwhelming presence of V(IV) on the surfaceof the VPO equilibrated catalysts. The addition ofcobalt does not change the oxidation states of surfacevanadium.
The existence of Co(II) in the impregnated catalysthas been confirmed by XPS measurements (Table 4and Fig. 3). The binding energies of reference com-pounds and high Co loading equilibrated catalysts aresummarized in Table 4. While the 2p1/2 and 2p3/2binding energies of Co(II) are relatively close to thatof Co(III), the two oxidation states of cobalt can bedistinguished by the presence of a distinct shake upsatellite structure in Co(II) arising from the presenceof unpaired electrons in their valence orbital [17,18].Co(III) is almost always in a low-spin state and there-fore there can be no energy transfer to an unpairedelectron, meaning that a shake up satellite structureis not observed. The disappearance of the satellitepeaks in the XPS profile would then be indicative ofoxidation of Co(II) to Co(III).
The Co3O4 has a spinel structure and contains mag-netic Co(II) ions (tetrahedral sites) and diamagneticCo(III) ions (octahedral sites). These two sites areindistinguishable by XPS. Although, all compoundscontaining high-spin Co(II) ions have strong satellitepeaks in the Co 2p spectra, only Co3O4 has weak
satellites in this region [20]. These satellites occurabout 7–9 eV higher in binding energy from the mainbands, which can be observed in Table 4. High-spinCo(II) core level spectra (as in CoO) show strongsatellite peaks (Table 4) above the main 2p lines.Frost et al. [21] have shown that high-spin Co(II)compounds exhibit intense satellites, while low-spinCo(II) compounds exhibit weak or missing satellites
Fig. 3. XPS spectra of the Co 2p region.

282 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
in their photoelectron spectra. Thus, it is not diffi-cult to differentiate high-spin Co(II) compounds fromCo2O3. Cobalt hydroxide has a strong satellite peakassociated with both the lines of the Co 2p region.
For the VPCo4-Iaa and VPCo4-Ia catalysts, the Co2p3/2 binding energies were different. The value of783.3 eV indicates that the presence of CoO on thesurface is unlikely. The Co 2p3/2 BE is 780.1 eV forCoO. For Co-promoted vanadyl pyrophosphate, thebinding energy is much higher. The Co 2p spectrumhas an intense shoulder at ∼788 eV. In addition, the2p1/2–2p3/2 spin-orbit splitting of 15.7 eV for this cat-alyst indicates that Co(II) species are present.
Fierro et al. [19] have proposed an interpretationfor cobalt, similar to that reported for copper. ForCo(II)-containing compounds, the main and the satel-lite peaks can be related, respectively, to 2p5 3d8L (Lmeans a ligand hole) and 2p5 3d7 electronic config-uration in the final state. One of the most importantconsequences of this theory is that useful informa-tion about the nature of the metal–ligand chemicalbonding can be simply gained by evaluating thesatellite/main peak intensity ratio and their energyseparation. They found that the energy separationincreases and the satellite/main peak intensity ratiodecreases with increasing covalent character of themetal–ligand bonding. On examining the XPS profileof the impregnated catalysts, the Co 2p satellite in-tensities approach satellite/main peak intensity ratiosobserved for bulk CoO, while the strong satellite linesare centered about 4.7–4.2 eV above the principalline. These observations indicate that Co(II) is notbeing oxidized to Co(III). Besides, the covalent char-acter of metal–ligand bonding is not affected neitherby the method nor the salt used for impregnation.
The high BEs (783.3–782.7 eV) might be related tostrong interactions between cobalt and other atoms ofthe solid matrix and/or to a highly dispersed cobalt.Binding energies of 783 eV were reported [22] forthe ion exchanged form of cobalt in zeolites. In thesesolids, the higher than normal binding energy is sug-gestive of cobalt in a highly oxidizing environment.Zazhigalov et al. [8] reported a similarly high bindingenergy of 783.0 eV for Co-promoted VPO catalysts.They compared their results with the value of 782.8found in cobalt molybdate or 782.2 and 783.0 forCoCl2 and CoF2, respectively, and concluded that themetal–ligand bonds are polarized in these reference
compounds and a similar structure may be expectedfor cobalt phosphates. They sustained that the highbinding energy values of 782.7–783 eV observed intheir catalysts can be considered as an additional indi-cation of the formation of cobalt phosphate (detectedby XRD). On the other hand, they suggested the exis-tence of strong interactions between cobalt atoms andphosphate groups with a shift of bonding electronstowards the phosphate anions.
In our catalysts, the Co/V nominal ratio is lowerthan 0.13, and no cobalt phosphates were detected byXRD, FT-IR, and Raman spectroscopy. In agreementwith our results, Hutchings and Higgins [5] have re-cently reported that the XRD patterns do not evenhint the formation of solid solutions at low promoterratios, M/V ≤ 0.12. Hutchings [4] proposes that thepromoters at high promoter/vanadium atomic ratiosact as phosphorus scavengers by either formation ofmetal phosphates or by formation of solid solutions.In our case, considering the high P/V surface ratio andthe fact that all the catalysts have similar Co/V sur-face ratios, it is possible that the high BE of Co 2p3/2be symptomatic of the presence of surface cobalt py-rophosphate. This surface compound does not havelong-range, three-dimensional order, because charac-teristic X-ray diffraction lines have not been observed.However, binding energies are not a sufficient crite-rion to confirm the presence of a given compound.
A difference in BE was observed between the twoimpregnated catalysts. This could be due to either dif-ferent surface Co(II) interactions or to a change of thecation distribution at the surface. Two different im-pregnating salts (cobalt acetate and acetyl acetonate)might produce catalysts with varying strengths in theinteraction between cobalt and VPO or different Codispersion.
3.4. Lewis acidity of equilibrated solids
To ascertain the effect of Co upon the Lewis acid-ity of the VPO formulations, acetonitrile was usedas a probe molecule. When this weak base is ad-sorbed on the VPO solids, the following bands areobserved: ν = 2252 cm−1 assigned to H-bondedand physisorbed molecules, ca. ν = 2270 cm−1 cor-responding to CN groups interacting with mediumstrong Lewis acid sites, ν = 2294 cm−1 associatedwith the Fermi resonance of the CN group (ν3 + ν4),

C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286 283
and a fourth band at 2322 cm−1 assigned to the in-teraction of the base with very strong Lewis acidcenters. A shift of 28 cm−1 is observed when thisbase is adsorbed on �-Fe2O3 [23]. Displacementsof ca. 75 cm−1 from the liquid phase frequency areobserved when acetonitrile is adsorbed on AlCl3 [24].
The band at 2252 cm−1 rapidly decreases with in-creasing temperature in all the equilibrated catalysts.A band at 2270 cm−1 (ν = 18 cm−1) appears in theVPO spectrum taken in the presence of the gas phase,and remains when the evacuation temperature is in-creased. This band is also observed in the VPCo4-Iaaand in VPCo4-HTI in the presence of the gas phase.But this band almost disappears upon temperatureincrease.
The analysis of the spectra obtained with the solidsthat were fast-activated (nonequilibrated), shows lessadsorption in general. However, it is also seen that allthe spectra show the signal assigned to the mediumstrong sites (2270 cm−1) when there is acetonitrile inthe gas phase. But these signals disappear after evac-uation at 298 K. It could be concluded that mediumstrong Lewis acid sites are present in all nonequi-librated catalysts; when the catalysts become equi-librated, these sites remain only in the unpromotedcatalyst and in the catalyst with low percentage of Co(1.5%).
The band at 2322 cm−1, attributed to very strongLewis acid sites, appears practically at the same fre-quency in all the solids (Fig. 4). It could accordingly beconcluded that the addition of Co in different amountsand by different methods does not introduce any mod-ification in the strength of the very strong Lewis acidsites. Table 5 also shows the intensity ratio of the bandat 2322 cm−1 with respect to that at 2297 cm−1. This
Table 5Acetonitrile adsorption on equilibrated catalysts
Catalyst Lewis site (cm−1) I2322/I2297a
Very strong Medium strong C/Fb 423 Kc
VPO 2322 2270 0.56 0.48VPCo1.5-Ia 2322 2270 0.80 0.53VPCo4-Ia 2322 – 0.60 1.89VPCo4-Iaa 2322 – 0.69 1.14VPCo4-HTI 2322 2270 0.53 1.75
a Relative intensity of FT-IR bands at ν = 2322 and 2297 cm−1.b Acetonitrile in the gas phase.c After evacuation at 423 K.
Fig. 4. FT-IR spectra of acetonitrile adsorption on promoted cat-alysts after evacuation at 423 K.
band is not influenced by the electronic environment[26], and it does not change with the catalyst acidity.Consequently, this ratio could be used to estimate thevery strong Lewis acid site concentration. After evac-uation at 423 K, the three catalysts with 4% Co containa higher concentration of very strong Lewis acid sitesthan those with 1.5% Co and the unpromoted VPO.According to Busca et al. [25], the very strong Lewisacid sites of the nonequilibrated organically preparedVPO are due to a stretching of the V–O–P linkage.This stretching is a consequence of the stacking folddisorder of the (2 0 0) planes. In our previous study[26], we have demonstrated that the ratio of very strongto medium strong Lewis sites significantly increaseswith time-on-stream, corresponding to a well-orderedstructure of the equilibrated catalysts.
3.5. Role of cobalt
The Co addition significantly increases the over-all activity. This seems to be a consequence of thepresence of cobalt on the equilibrated catalyst sur-face. Using cobalt acetyl acetonate for the precursor

284 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
impregnation produces a solid with higher selectivi-ties and performances comparable to commercial cat-alysts. In this complex structure, the promoter mightbe incorporated in the crystal lattice of (VO)2P2O7,as reported by Takita et al. [6]. They studied the in-corporation of promoter elements employing metalacetyl acetonates added during precursor synthesis.Incorporation of the promoter elements into the crys-tal lattice of (VO)2P2O7 brought about the shift in theIR absorption bands of the V=O stretching mode. Inour catalysts, the V=O vibration was not modified bythe presence of cobalt. They found the same behaviorin the case in which Zn was located at the surface ofvanadyl pyrophosphate. Guliants et al. [27] found alower Nb surface ratio of promoter, which probablyindicates higher solubility of the Nb promoter in theVPO matrix. Therefore, this suggested that promoterelements may partially form a solid solution. Anotherpromoting effect could be that the cobalt atoms affectthe layer linkages by becoming intercalated betweenthe layers of the (VO)2P2O7, as was reported forFe- and Cr-promoted catalysts [28]. However, the IRband at 742 cm−1, corresponding to the P–O–P vi-bration, has not significantly decreased in intensity inour Co-promoted catalysts.
Neither a volumetric cobalt phosphate phase norsolid solutions were detected in our catalysts. Thesolids showed, however, high Co/V and P/V surfaceratios plus a significant shift of the Co 2p BE. Allthis is consistent with the presence of a surface cobaltpyrophosphate in our catalysts.
Several authors [25–27,29,30] have recalled theimportance of the surface acidity for high activityand selectivity of the bulk VPO. The presence of acidsites should have an effect on the rate of activationof butane and on the rate of its oxidation to maleicanhydride. The acidity of the Co-promoted VPO cat-alyst was measured with NH3 TPD by Zazhigalovet al. [8]. It has been observed that adsorption ofammonia increases with increasing cobalt content.The ratio of the number of strong to weak adsorptionsites decreased with increasing cobalt content. Theyconcluded that the introduction of cobalt ions into theVPO catalyst is the main cause for the appearanceof weak acid sites, whereas the number of strongacid sites rises to a lesser extent and only on addi-tion of small amounts of cobalt (Co/V < 0.1 or 0.05depending on the preparation method).
Busca et al. [25] proposed a model of butane activa-tion in which it proceeds as a concerted simultaneousabstraction of two hydrogen atoms on an active center.This is composed of a strong Lewis acid–base V–Opair in which the empty d orbital of vanadium gener-ates the Lewis acidity, while the occupied 2p orbitalof oxygen is responsible for the Lewis basicity.
The equilibrated catalysts containing 4% Co show asignificant increase in the concentration of very strongLewis acid sites over both the base VPO and the onecontaining 1.5% Co. A model of Lewis acid sites invanadyl pyrophosphate was proposed by Trifirò et al.[1]. They indicated that the presence of defects in thestructure of vanadyl pyrophosphate is reflected on thesurface in an enhancement of Lewis acidity of surfaceunsaturated vanadium ions. According to this model,the appearance of very strong Lewis acid sites maybe related to a different polarization of the V–O–Pbonds, in our case induced by the presence of cobalt.The equilibrated VPCo4-Iaa solid is the most selec-tive one. It has, however, the lowest concentration ofvery strong Lewis sites among the impregnated solids
Fig. 5. Maleic anhydride selectivities as a function of very strongLewis acid sites concentration (data from Fig. 1B and Table 5).

C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286 285
with 4% Co. The correlation between selectivity andconcentration of very strong Lewis sites at differentconversions and space velocities is shown in Fig. 5.The presence of a maximum in selectivity might indi-cate that an optimum value exists for the concentrationof very strong Lewis sites. An excess of very strongLewis acid sites could decrease the rate of desorptionof MAN, decreasing the maleic anhydride selectivity.
Gleaves et al. [31] observed that the rate of desorp-tion of the olefinic intermediates and furan is low com-pared to MAN, which implies that MAN should berapidly desorbed to avoid its combustion. This wouldjustify the former assertion and might indicate that thepresence of very strong Lewis sites plays an importantrole regarding selectivity control. IR observations byKoyano et al. [32] of CO adsorbed al low temperatureindicated that n-butane preferably interacts at Lewisacid sites, and recalled the importance of these sitesto control selectivity. Note, however, that there couldbe other factors controlling the selectivity such as mi-crostructure, P/V ratio, or a defect-free structure.
4. Conclusions
They only apply to equilibrated catalysts.
1. As reported before [10], the addition of cobalt(≥2 wt.%) to VPO formulations significantlyincreases the catalytic activity (Fig. 1A).
2. When cobalt acetyl acetonate was used, the high-est selectivities to maleic anhydride were observed(Fig. 1B). Besides, these Co-VPO solids give thehighest yields (Fig. 2A and B). This ranking isalso maintained at T > 660 K if all the previouslystudied cobalt-containing catalysts were includedin Fig. 2 [9,10].
3. The XPS data is consistent with the presence ofCo-pyrophosphate in the first few surface layers(Table 4). However, bulk techniques (XRD, LRS,and FT-IR) could not detect the presence of cobaltphosphates.
4. The presence of cobalt does not modify the ox-idation state of vanadium at the surface layers.Only V(IV) is detected through XPS (Table 3), inagreement with our previous studies [9,10].
5. The presence of Co at the 4% level sharply in-creases the concentration of very strong Lewis
acid sites, although it does not affect their strength(Table 5). However, the best catalyst shows thelowest concentration among the catalysts con-taining 4% cobalt. This might indicate that anoptimum concentration of very strong Lewis acidsites is needed to maximize the selective oxidationto maleic anhydride (Fig. 5). More work is neededto elucidate this point.
Acknowledgements
The authors wish to acknowledge the financial sup-port received from UNL (CAI+D 96 Program). Theyare also grateful to the Japan International CooperationAgency (JICA) for the donation of the major instru-ments used in this study. They also acknowledge Prof.Elsa Grimaldi for the edition of the English paper.
References
[1] G. Centi, F. Trifirò, J. Ebner, V. Franchetti, Chem. Rev. 88(1988) 55.
[2] F. Ben Abdelouahab, R. Olier, M. Ziyad, J.C. Volta, J. Catal.134 (1992) 151.
[3] M. López Granados, J.L. Garcı́a Fierro, F. Cavani, A.Colombo, F. Giuntoli, F. Trifirò, Catal. Today 40 (1998) 251.
[4] G. Hutchings, Appl. Catal. 72 (1991) 1.[5] G. Hutchings, R. Higgins, J. Catal. 162 (1996) 153.[6] Y. Takita, K. Tanaka, S. Ichimaru, Y. Mizihara, Y. Abe, Y.
Ishihara, Appl. Catal. A 103 (1993) 281.[7] G. Hutchings, C. Kelly, M.T. Sananés-Schulz, A. Burrows,
J.C. Volta, Catal. Today 40 (1998) 273.[8] V.A. Zazhigalov, J. Haber, J. Stoch, A. Pyatnitzkaya, G.A.
Komashko, V.M. Belousov, Appl. Catal. A 96 (1993) 135.[9] L. Cornaglia, C. Carrara, J. Petunchi, E. Lombardo, Appl.
Catal. A 183 (1999) 177.[10] L. Cornaglia, C. Carrara, J. Petunchi, E. Lombardo, Catal.
Today 57 (2000) 313.[11] L. Cornaglia, C. Sánchez, E. Lombardo, Appl. Catal. A 95
(1993) 117.[12] M. Rozynek, C. Polansky, Appl. Catal. 73 (1991) 97.[13] H. Ohtuska, T. Tabata, O. Okada, L. Sabatino, G. Bellusi,
Catal. Lett. 44 (1997) 265.[14] G. Busca, F. Cavani, G. Centi, F. Trifirò, J. Catal. 99 (1986)
400.[15] L. Cornaglia, E.A. Lombardo, Appl. Catal. A 127 (1995) 125.[16] G.W. Coulston, E.A. Thompson, N. Herron, J. Catal. 163
(1996) 122.[17] A.S.K. Ainha, V. Shankar, Ind. Eng. Chem. Res. 32 (1993)
1061.[18] S.C. Petrosius, R.S. Drago, V. Young, G.C. Grunewald, J.
Am. Chem. Soc. 115 (1993) 6131.

286 C. Carrara et al. / Applied Catalysis A: General 217 (2001) 275–286
[19] G. Fierro, M. Lo Iacono, M. Inversi, R. Dragono, P. Porta,Topics Catal. 10 (2000) 39.
[20] B. Jit San, K. Klabunde, P. Sherwood, J. Am. Chem. Soc.113 (1991) 855.
[21] D.C. Frost, C.A. McDowell, I. Woosley, Chem. Phys. Lett.17 (1972) 320.
[22] J. Stencel, V. Rao, J. Diehl, K. Rhee, A. Dhere, R. DeAngelis,J. Catal. 84 (1983) 109.
[23] V. Lorenzelli, G. Busca, N. Sheppard, J. Catal. 66 (1980) 18.[24] K. Pursell, R. Drago, J. Am. Chem. Soc. 88 (1966) 919.[25] G. Busca, G. Centi, F. Trifirò, Appl. Catal. 25 (1986) 265.[26] L. Cornaglia, E. Lombardo, J. Anderson, J.L. Garcı́a Fierro,
Appl. Catal. A 100 (1993) 37.
[27] V. Guliants, J. Benziger, S. Sundaresan, I. Wachs, A. Hirte,Catal. Lett. 62 (1999) 87.
[28] R.L. McCormick, G.O. Alptekin, A.M. Herring, T.R. Ohno,F. Dec Steven, J. Catal. 172 (1997) 160.
[29] V. Zazhigalov, J. Haber, J. Stoch, I. Bacherikova, G. Komasko,A. Pyatnitskaya, Appl. Catal. 134 (1996) 225.
[30] S. Irusta, A. Boix, B. Pierini, C. Caspani, J. Petunchi, J.Catal. 187 (1999) 298.
[31] J.T. Gleaves, J. Ebner, T.C. Kuechler, Catal. Rev. Sci. Eng.30 (1998) 49.
[32] G. Koyano, T. Saito, M. Misono, J. Mol. Catal. A 155 (2000)31.
Related Documents

![The VPO Operator. [vpo_operator] 2 The VPO Operator Section Overview The role of the VPO operator Starting and stopping the Motif GUI The VPO Operator.](https://static.cupdf.com/doc/110x72/56649ea15503460f94ba4921/the-vpo-operator-vpooperator-2-the-vpo-operator-section-overview-the-role.jpg)

![The VPO Administrator. [vpo_administrator] 2 The VPO Administrator The VPO Administrator - Overview The role of the VPO Administrator Understand High.](https://static.cupdf.com/doc/110x72/56649e5e5503460f94b58169/the-vpo-administrator-vpoadministrator-2-the-vpo-administrator-the-vpo.jpg)







