Molecular Cell, Vol. 6, 1473–1484, December, 2000, Copyright 2000 by Cell Press Structure of the AAA ATPase p97 et al., 1998). In mammals, p97, along with NSF and the Xiaodong Zhang, 1,8 Anthony Shaw, 1,8 Paul A. Bates, 1 Golgi t-SNARE syntaxin 5, mediates the fusion of Golgi Richard H. Newman, 1 Brent Gowen, 2 Elena Orlova, 2 membranes (Rabouille et al., 1995; Kondo et al., 1997). Michael A. Gorman, 1 Hisao Kondo, 1,5 Pawel Dokurno, 1,6 Although p97 and NSF are highly homologous, they ap- John Lally, 1 Gordon Leonard, 3 Hemmo Meyer, 1,7 pear to act in distinct fusion events, presumably due to Marin van Heel, 2 and Paul S. Freemont 1,2,4 additional specific cofactors; p97 requires the cofactor 1 Molecular Structure and Function, Biomolecular p47 for interactions with syntaxin 5 (Kondo et al., 1997; Modelling, and Cell Biology Laboratories Rabouille et al., 1998), whereas NSF requires SNAPs Imperial Cancer Research Fund for SNARE interactions and disassembly (Sollner et al., London WC2A 3PX 1993a, 1993b). 2 Centre for Structural Biology Homologs of p97 are found in archaea, which do not Department of Biochemistry contain internal membranes, suggesting roles for p97 Wolfson Laboratories other than membrane fusion as well as indicating that Imperial College of Science, Technology, and Medicine p97 is the more ancient form of the two membrane fusion South Kensington, London SW7 2AY ATPases. p97/Cdc48p has also been implicated in pro- United Kingdom teasomal protein degradation since it binds to Ufd2p/ 3 Joint Structural Biology Group E4 and Ufd3p, which are components of ubiquitin-medi- European Synchrotron Radiation Facility ated proteolysis (Ghislain et al., 1996; Koegl et al., 1999). Grenoble Cedex Recently, p97 has been implicated in protein degrada- France tion by its interaction with UFD1, a component of the ubiquitin fusion degradation pathway (Meyer et al., 2000). p97 is also a target of tyrosine phosphorylation Summary during T cell activation, although the significance of this is not clear (Egerton et al., 1992). p97, an abundant hexameric ATPase of the AAA family, The AAA proteins are characterized by the presence is involved in homotypic membrane fusion. It is thought of one or two conserved AAA domains (230–250 resi- to disassemble SNARE complexes formed during the dues) that contain the Walker A and B motifs. Members process of membrane fusion. Here, we report two struc- of this family are involved in a myriad of cellular pro- tures: a crystal structure of the N-terminal and D1 cesses as diverse as membrane fusion (p97 and NSF), ATPase domains of murine p97 at 2.9 A ˚ resolution, organelle biogenesis (PAS1p), motor proteins (TorsinA), and a cryoelectron microscopy structure of full-length DNA helicases (RuvB), proteolysis (Lon and Clp), and rat p97 at 18 A ˚ resolution. Together, these structures transcriptional regulation (SUG1 and TBP1; for reviews, show that the D1 and D2 hexamers pack in a tail-to- see Patel and Latterich, 1998; Neuwald et al., 1999). tail arrangement, and that the N domain is flexible. A The contrast between the functional diversity of AAA comparison with NSF D2 (ATP complex) reveals possi- proteins and the fact that the AAA cassette is conserved ble conformational changes induced by ATP hydroly- from archaea to modern eukaryotes suggest that it plays sis. Given the D1 and D2 packing arrangement, we a fundamental role in all life forms. One prevalent hy- propose a ratchet mechanism for p97 during its ATP pothesis for the mode of action of AAA proteins is that hydrolysis cycle. they perform nucleotide-dependent chaperone-like func- tions and aid in the assembly, disassembly, or functional Introduction operation of protein complexes (Confalonieri and Du- guet, 1995; Morgan and Burgoyne, 1995; Leonhard et p97 and NSF belong to the AAA (ATPases associated al., 1999; Neuwald et al., 1999). Very little is known, with various cellular activities) family of proteins (Neu- however, about how changes in nucleotide binding and/ wald et al., 1999). p97/VAT/CDC48p are abundant homo- or hydrolysis are coupled to AAA function, although the hexameric (97 kDa subunit) Mg 21 -dependent ATPases Walker A and B boxes are essential for ATPase activity. that catalyze certain homotypic membrane fusion events. The role of the AAA minimal consensus is also not imme- In budding yeast, the p97 homolog, Cdc48p, fuses ER diately obvious, although mutations in this region can membranes together utilizing the t-SNARE Ufe1p (Patel abolish the enzymatic activity of FtsH, an ATP-depen- dent protease found in prokaryotic cells, mitochondria, and chloroplasts (Karata et al., 1999). p97 and NSF comprise two AAA domains (D1 and 4 To whom correspondence should be addressed (e-mail: p.freemont@ D2) and an z200 residue N-terminal substrate/adaptor ic.ac.uk). 5 Present address: Cambridge Institute for Medical Research, Uni- molecule recognition domain (N). The D1 and D2 do- versity of Cambridge, Wellcome Trust/MRC Building, Hills Road, mains of p97 have high sequence identity (40%), sug- Cambridge CB2 2XY, United Kingdom. gesting that p97 may have evolved from gene duplica- 6 Present address: Section of Structural Biology, Institute of Cancer tion of a AAA domain. Subsequent evolutionary events Research, London SW7 3RP, United Kingdom. may have led to the emergence of the p97 homolog 7 Present address: Department of Cell Biology, Yale University Medi- NSF, to perform more specialized functions in mem- cal School, New Haven, Connecticut 06520. 8 These authors contributed equally to this work. brane fusion pathways. From electron microscopy stud-

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Molecular Cell, Vol. 6, 1473–1484, December, 2000, Copyright 2000 by Cell Press
Structure of the AAA ATPase p97
et al., 1998). In mammals, p97, along with NSF and theXiaodong Zhang,1,8 Anthony Shaw,1,8 Paul A. Bates,1
Golgi t-SNARE syntaxin 5, mediates the fusion of GolgiRichard H. Newman,1 Brent Gowen,2 Elena Orlova,2
membranes (Rabouille et al., 1995; Kondo et al., 1997).Michael A. Gorman,1 Hisao Kondo,1,5 Pawel Dokurno,1,6
Although p97 and NSF are highly homologous, they ap-John Lally,1 Gordon Leonard,3 Hemmo Meyer,1,7
pear to act in distinct fusion events, presumably due toMarin van Heel,2 and Paul S. Freemont1,2,4
additional specific cofactors; p97 requires the cofactor1 Molecular Structure and Function, Biomolecularp47 for interactions with syntaxin 5 (Kondo et al., 1997;Modelling, and Cell Biology LaboratoriesRabouille et al., 1998), whereas NSF requires SNAPsImperial Cancer Research Fundfor SNARE interactions and disassembly (Sollner et al.,London WC2A 3PX1993a, 1993b).2 Centre for Structural Biology
Homologs of p97 are found in archaea, which do notDepartment of Biochemistrycontain internal membranes, suggesting roles for p97Wolfson Laboratoriesother than membrane fusion as well as indicating thatImperial College of Science, Technology, and Medicinep97 is the more ancient form of the two membrane fusionSouth Kensington, London SW7 2AYATPases. p97/Cdc48p has also been implicated in pro-United Kingdomteasomal protein degradation since it binds to Ufd2p/3 Joint Structural Biology GroupE4 and Ufd3p, which are components of ubiquitin-medi-European Synchrotron Radiation Facilityated proteolysis (Ghislain et al., 1996; Koegl et al., 1999).Grenoble CedexRecently, p97 has been implicated in protein degrada-Francetion by its interaction with UFD1, a component of theubiquitin fusion degradation pathway (Meyer et al.,2000). p97 is also a target of tyrosine phosphorylationSummaryduring T cell activation, although the significance of thisis not clear (Egerton et al., 1992).p97, an abundant hexameric ATPase of the AAA family,
The AAA proteins are characterized by the presenceis involved in homotypic membrane fusion. It is thoughtof one or two conserved AAA domains (230–250 resi-to disassemble SNARE complexes formed during thedues) that contain the Walker A and B motifs. Membersprocess of membrane fusion. Here, we report two struc-of this family are involved in a myriad of cellular pro-tures: a crystal structure of the N-terminal and D1cesses as diverse as membrane fusion (p97 and NSF),ATPase domains of murine p97 at 2.9 A resolution,organelle biogenesis (PAS1p), motor proteins (TorsinA),and a cryoelectron microscopy structure of full-lengthDNA helicases (RuvB), proteolysis (Lon and Clp), andrat p97 at 18 A resolution. Together, these structurestranscriptional regulation (SUG1 and TBP1; for reviews,show that the D1 and D2 hexamers pack in a tail-to-see Patel and Latterich, 1998; Neuwald et al., 1999).
tail arrangement, and that the N domain is flexible. AThe contrast between the functional diversity of AAA
comparison with NSF D2 (ATP complex) reveals possi-proteins and the fact that the AAA cassette is conserved
ble conformational changes induced by ATP hydroly- from archaea to modern eukaryotes suggest that it playssis. Given the D1 and D2 packing arrangement, we a fundamental role in all life forms. One prevalent hy-propose a ratchet mechanism for p97 during its ATP pothesis for the mode of action of AAA proteins is thathydrolysis cycle. they perform nucleotide-dependent chaperone-like func-
tions and aid in the assembly, disassembly, or functionalIntroduction operation of protein complexes (Confalonieri and Du-
guet, 1995; Morgan and Burgoyne, 1995; Leonhard etp97 and NSF belong to the AAA (ATPases associated al., 1999; Neuwald et al., 1999). Very little is known,with various cellular activities) family of proteins (Neu- however, about how changes in nucleotide binding and/wald et al., 1999). p97/VAT/CDC48p are abundant homo- or hydrolysis are coupled to AAA function, although thehexameric (97 kDa subunit) Mg21-dependent ATPases Walker A and B boxes are essential for ATPase activity.that catalyze certain homotypic membrane fusion events. The role of the AAA minimal consensus is also not imme-In budding yeast, the p97 homolog, Cdc48p, fuses ER diately obvious, although mutations in this region canmembranes together utilizing the t-SNARE Ufe1p (Patel abolish the enzymatic activity of FtsH, an ATP-depen-
dent protease found in prokaryotic cells, mitochondria,and chloroplasts (Karata et al., 1999).
p97 and NSF comprise two AAA domains (D1 and4 To whom correspondence should be addressed (e-mail: p.freemont@D2) and an z200 residue N-terminal substrate/adaptoric.ac.uk).
5 Present address: Cambridge Institute for Medical Research, Uni- molecule recognition domain (N). The D1 and D2 do-versity of Cambridge, Wellcome Trust/MRC Building, Hills Road, mains of p97 have high sequence identity (40%), sug-Cambridge CB2 2XY, United Kingdom. gesting that p97 may have evolved from gene duplica-6 Present address: Section of Structural Biology, Institute of Cancer
tion of a AAA domain. Subsequent evolutionary eventsResearch, London SW7 3RP, United Kingdom.may have led to the emergence of the p97 homolog7 Present address: Department of Cell Biology, Yale University Medi-NSF, to perform more specialized functions in mem-cal School, New Haven, Connecticut 06520.
8 These authors contributed equally to this work. brane fusion pathways. From electron microscopy stud-
Molecular Cell1474
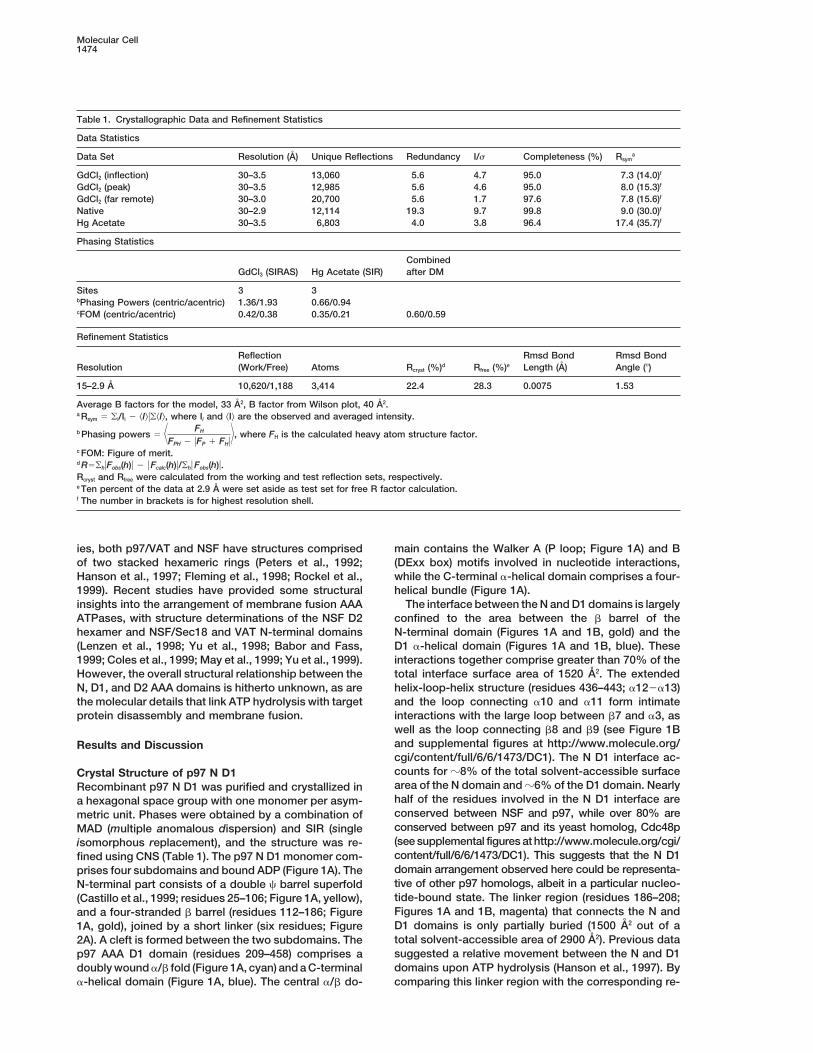
Table 1. Crystallographic Data and Refinement Statistics
Data Statistics
Data Set Resolution (A) Unique Reflections Redundancy I/s Completeness (%) Rsyma
GdCl2 (inflection) 30–3.5 13,060 5.6 4.7 95.0 7.3 (14.0)f
GdCl2 (peak) 30–3.5 12,985 5.6 4.6 95.0 8.0 (15.3)f
GdCl2 (far remote) 30–3.0 20,700 5.6 1.7 97.6 7.8 (15.6)f
Native 30–2.9 12,114 19.3 9.7 99.8 9.0 (30.0)f
Hg Acetate 30–3.5 6,803 4.0 3.8 96.4 17.4 (35.7)f
Phasing Statistics
CombinedGdCl3 (SIRAS) Hg Acetate (SIR) after DM
Sites 3 3bPhasing Powers (centric/acentric) 1.36/1.93 0.66/0.94cFOM (centric/acentric) 0.42/0.38 0.35/0.21 0.60/0.59
Refinement Statistics
Reflection Rmsd Bond Rmsd BondResolution (Work/Free) Atoms Rcryst (%)d Rfree (%)e Length (A) Angle (8)
15–2.9 A 10,620/1,188 3,414 22.4 28.3 0.0075 1.53
Average B factors for the model, 33 A2, B factor from Wilson plot, 40 A2.a Rsym 5 oi/Ii 2 kIluokIl, where Ii and kIl are the observed and averaged intensity.
b Phasing powers 5 7 FH
FPH 2 uFP 1 FHu8, where FH is the calculated heavy atom structure factor.
c FOM: Figure of merit.d R5ohuFobs(h)u 2 u Fcalc(h)u/ohu Fobs(h)u.Rcryst and Rfree were calculated from the working and test reflection sets, respectively.e Ten percent of the data at 2.9 A were set aside as test set for free R factor calculation.f The number in brackets is for highest resolution shell.
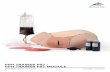
ies, both p97/VAT and NSF have structures comprised main contains the Walker A (P loop; Figure 1A) and B(DExx box) motifs involved in nucleotide interactions,of two stacked hexameric rings (Peters et al., 1992;
Hanson et al., 1997; Fleming et al., 1998; Rockel et al., while the C-terminal a-helical domain comprises a four-helical bundle (Figure 1A).1999). Recent studies have provided some structural
insights into the arrangement of membrane fusion AAA The interface between the N and D1 domains is largelyconfined to the area between the b barrel of theATPases, with structure determinations of the NSF D2
hexamer and NSF/Sec18 and VAT N-terminal domains N-terminal domain (Figures 1A and 1B, gold) and theD1 a-helical domain (Figures 1A and 1B, blue). These(Lenzen et al., 1998; Yu et al., 1998; Babor and Fass,
1999; Coles et al., 1999; May et al., 1999; Yu et al., 1999). interactions together comprise greater than 70% of thetotal interface surface area of 1520 A2. The extendedHowever, the overall structural relationship between the
N, D1, and D2 AAA domains is hitherto unknown, as are helix-loop-helix structure (residues 436–443; a122a13)and the loop connecting a10 and a11 form intimatethe molecular details that link ATP hydrolysis with target
protein disassembly and membrane fusion. interactions with the large loop between b7 and a3, aswell as the loop connecting b8 and b9 (see Figure 1Band supplemental figures at http://www.molecule.org/Results and Discussioncgi/content/full/6/6/1473/DC1). The N D1 interface ac-counts for z8% of the total solvent-accessible surfaceCrystal Structure of p97 N D1area of the N domain and z6% of the D1 domain. NearlyRecombinant p97 N D1 was purified and crystallized inhalf of the residues involved in the N D1 interface area hexagonal space group with one monomer per asym-conserved between NSF and p97, while over 80% aremetric unit. Phases were obtained by a combination ofconserved between p97 and its yeast homolog, Cdc48pMAD (multiple anomalous dispersion) and SIR (single(see supplemental figures at http://www.molecule.org/cgi/isomorphous replacement), and the structure was re-content/full/6/6/1473/DC1). This suggests that the N D1fined using CNS (Table 1). The p97 N D1 monomer com-domain arrangement observed here could be representa-prises four subdomains and bound ADP (Figure 1A). Thetive of other p97 homologs, albeit in a particular nucleo-N-terminal part consists of a double c barrel superfoldtide-bound state. The linker region (residues 186–208;(Castillo et al., 1999; residues 25–106; Figure 1A, yellow),Figures 1A and 1B, magenta) that connects the N andand a four-stranded b barrel (residues 112–186; FigureD1 domains is only partially buried (1500 A2 out of a1A, gold), joined by a short linker (six residues; Figuretotal solvent-accessible area of 2900 A2). Previous data2A). A cleft is formed between the two subdomains. Thesuggested a relative movement between the N and D1p97 AAA D1 domain (residues 209–458) comprises adomains upon ATP hydrolysis (Hanson et al., 1997). Bydoubly wound a/b fold (Figure 1A, cyan) and a C-terminal
a-helical domain (Figure 1A, blue). The central a/b do- comparing this linker region with the corresponding re-
Structure of p97 ATPase1475
Figure 1. Crystal Structure of p97 N D1 Domain
(A) Ribbon representation of N D1 protomer structure. The domains are colored individually, with the N-terminal double c barrel domain(yellow), b barrel domain (gold), the D1 a/b domain (cyan), and the C-terminal a-helical domain (blue). Also highlighted are the Walker A (Ploop) motif (black), Walker B (DExx box) motif (mauve), sensor loop (red), and the proposed hinge region between the N and D1 domains(magenta). Bound ADP nucleotide is shown as balls and sticks. For clarity, only the secondary structure elements discussed in the text arelabeled.(B) A close-up view of the N D1 interface (boxed region in [A]). A small rotation was applied to optimize the view of the interface.(C) The N D1 hexamer forms a wheel-like structure of z160 A in diameter and a central hole z15 A in diameter. One protomer is colored asin (A). The a/b domain fold (cyan) forms the spokes of the wheel, while the helical domain (blue) forms the outer rim. N-terminal domains(yellow) are oriented counterclockwise off the main body of the wheel. Bound ADP moieties (brown) drawn as Van der Waal spheres arelocated between protomers. The arrow indicates the ADP binding pocket.(D) The N D1 hexamer is rotated 908 (relative to Figure 1C) so that the amino-terminal side is on top. The thickness of N D1 is z20–40 A. TheN domain lies in the same plane as D1 and spans a similar thickness. Note the ADP moieties (brown spheres) bound in deep pockets.(E) A close-up view of the hexamer interface (boxed in [C]). For clarity, only secondary structure elements discussed in the text are labeled.The dashed line indicates the protomer–protomer interface. The sensor residues (Asn-348, yellow; Arg-359, blue) are shown as sticks nearthe interface.
gion in the NSF D2 structure (Lenzen et al., 1998; Yu ture-based sequence alignment reveals two highly con-served glycines at the N and C termini of the linker (Gly-et al., 1998), significant conformational differences are
observed, indicating that the linker region could be 186 and Gly-208, respectively; Figure 2C, left box). It isplausible that these glycines act as pivot points, allowingflexible (Figure 2B; p97, magenta; NSF, gold). A struc-

Molecular Cell1476
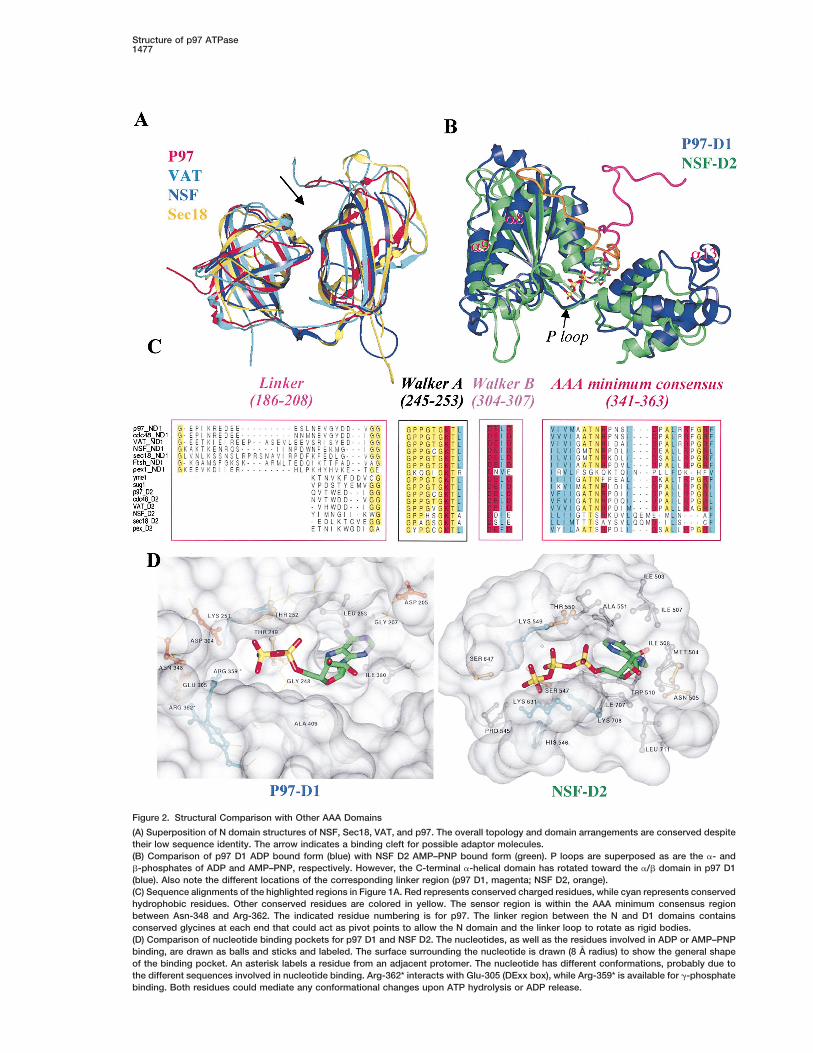
relative movements between N and D1 domains upon but unlike NSF D2, the ADP adopts anti instead of synATP hydrolysis similar to the relay helix region in myosin conformation. For p97, the adenine ring is sandwiched(Fisher et al., 1995). between Leu-253 and Ile-380 (Figure 2D). Both residues
p97 and recombinant N D1 exist as homohexamers are highly conserved within the AAA family except inin solution (Peters et al., 1992; Kondo et al., 1997; data NSF D2, where they are both replaced by Ala. In NSFnot shown). In the N D1 crystal structure, the 6-fold D2, the adenine ring is bound between Ile-507 and Ile-crystallographic axis is coincident with the molecular 707 (Lenzen et al., 1998), corresponding to Val-206 andsymmetry axis. Full-length endogenous p97 also crystal- Gly-408 in p97 D1. These and other sequence differ-lizes in a hexagonal lattice, supporting a symmetric p97 ences result in a distinctly differently shaped adeninehexamer packing arrangement (our unpublished data). binding pocket (Figure 2D), possibly explaining the ob-The N D1 hexamer forms a ring structure of a diameter served conformations adopted by bound nucleotides.of z160 A with a thickness between z20 and 40 A, and Interestingly, the adenine ring also forms hydrogenan inner hole of z15 A (Figures 1C and 1D). Looking bonds with residues of the N D1 linker region (Asp-205toward the N terminus, the hexamer forms a wheel-like [O] and Gly-207 [N, O]), suggesting a possible connec-structure with N-terminal domains positioned counter- tion between the conformation of the N domain withclockwise in between two adjacent a/b domains. The respect to D1 and bound nucleotide. The adenine ringsurface of the hexamer is flat except for the deep nucleo- also forms hydrogen bonds with Thr-249 (O) and Gly-tide binding pocket that lies between protomers (Figures 250 (N), two highly conserved residues within the Walker1C and 1D). Looking from the C terminus, the surface A motif (P loop). The a-phosphate interacts with Thr-is markedly more rugged, with a central funnel-shaped 252 (N, O), which is one of the few absolutely conservedopening. The diameter of the funnel ranges from z15 residues within the AAA family (Figure 2C). The b-phos-to 70 A to form in part a cage-like structure. A side view phate interacts with Gly-248 (N), Thr-249 (N), Lys-251shows the N-terminal domain in the same level and of (eN), and Thr-252 (O), all of which are highly conservedsimilar height to the D1 hexamer (Figure 1D, yellow). and form part of the Walker A motif. The ADP b-phos-Figure 1D also illustrates the flat top and more rugged phate also hydrogen bonds with Asp-304 of the WalkerC-terminal surface of the N D1 hexamer. The interpro- B motif (DExx motif) via an ordered water molecule.tomer contacts within the hexameric ring are formed However, we observe no Mg21 in our electron densityprimarily through interactions between adjacent a/b and maps despite the presence of 10 mM Mg21 in crystalliza-C-terminal a-helical domains (Figure 1E). Each protomer tion mixtures, which we attribute to the high salt con-buries z11% (1500 A2) of the total solvent-accessible centration (4 M formate) used for N D1 crystallization.surface area for D1. Two interaction regions are particu- Interestingly, the Walker B motif is also involved in inter-larly notable: a7 and the preceding loop interact with protomer contacts, since Glu-305 (DExx) forms a salta9 from an adjacent protomer near the central opening bridge with Arg-362 from a neighboring protomer (Figurein the hexamer; a5 and the surrounding region form 2D). Arg-362 is part of the AAA minimal consensus se-interactions with the a-helical domain at the outer rim quence and is highly conserved in a number of AAAof the hexamer (the extended helix-loop-helix structure proteins apart from, notably, NSF/Sec18 (Figure 2C).comprising a12 and a13; Figure 1E). It is notable that These interactions therefore couple Mg21-ATP/ADPa5 has few interactions with the rest of the a/b domain, binding to interprotomer contacts, and could thus medi-suggesting that it may be a flexible region in terms of ate any conformational changes induced by ATP hydro-any possible rigid body movement of the a/b domain. lysis or ADP release, including rearrangements of pro-We can speculate that the interactions between a5 and tomers within the hexamer. Furthermore, these residuesthe extended helix-loop-helix at the outer rim of the are highly conserved in the AAA family, suggesting thathexamer are required to maintain hexamerization during the ADP and associated interprotomer interactions ob-any conformational changes induced by ATP hydrolysis. served for p97 D1 are representative of other AAA pro-Interestingly, a13 is conserved in most other D2 domains
teins.within the AAA family but is notably absent in NSF/Sec18(see Figure 2B and supplemental figures at http://www.
Cryo-EM Reconstruction of Full-Length p97molecule.org/cgi/content/full/6/6/1473/DC1). SinceIn order to obtain a more complete structural descriptionNSF D2 has little ATPase activity (Matveeva et al., 1997),of p97, a single-particle analysis was undertaken usingthis loop may not be required. A structure-based se-cryo-EM images of purified rat liver cytosol p97 (99.9%quence alignment shows that z70% of the residuessequence identity to murine p97) in the presence ofinvolved in interprotomer contacts are conserved be-Mg21-ADP. As a result, we have obtained the structuretween the D1 domains of p97, VAT, and Cdc48p (seeof full-length p97 at z18 A resolution (Figure 3). The p97supplemental figures at http://www.molecule.org/cgi/molecule consists of two distinct layers, clearly con-content/full/6/6/1473/DC1). In terms of D2 domains,nected via well-defined densities, which together formnearly half of the interface residues are also conserveda cage-like structure with overall dimensions of z145 Ain p97, VAT, and Cdc48p (see supplemental figures atin diameter and z110 A in height (Figures 3A and 3B).http://www.molecule.org/cgi/content/full/6/6/1473/DC1),These dimensions are similar to earlier lower resolutionsuggesting a similar interprotomer interface for both theEM studies of NSF and p97 (Hanson et al., 1997) andD1 and D2 hexamers, with the exception of NSF/Sec18.VAT (Rockel et al., 1999). The 3D reconstruction revealsVery few interprotomer contact residues are conservedan internal cavity and openings on the lateral surfacebetween p97 D1 and NSF D2, although the overall topol-of the molecule (Figure 3). Interestingly, the p97 EMogy and hexameric arrangement are clearly similar (seestructure appears to have pseudo D6 symmetry, with aFigures 2 and 5).
In the N D1 crystal structure, we observe bound ADP, pseudo 2-fold axis in the middle of the molecule perpen-
Structure of p97 ATPase1477
Figure 2. Structural Comparison with Other AAA Domains
(A) Superposition of N domain structures of NSF, Sec18, VAT, and p97. The overall topology and domain arrangements are conserved despitetheir low sequence identity. The arrow indicates a binding cleft for possible adaptor molecules.(B) Comparison of p97 D1 ADP bound form (blue) with NSF D2 AMP–PNP bound form (green). P loops are superposed as are the a- andb-phosphates of ADP and AMP–PNP, respectively. However, the C-terminal a-helical domain has rotated toward the a/b domain in p97 D1(blue). Also note the different locations of the corresponding linker region (p97 D1, magenta; NSF D2, orange).(C) Sequence alignments of the highlighted regions in Figure 1A. Red represents conserved charged residues, while cyan represents conservedhydrophobic residues. Other conserved residues are colored in yellow. The sensor region is within the AAA minimum consensus regionbetween Asn-348 and Arg-362. The indicated residue numbering is for p97. The linker region between the N and D1 domains containsconserved glycines at each end that could act as pivot points to allow the N domain and the linker loop to rotate as rigid bodies.(D) Comparison of nucleotide binding pockets for p97 D1 and NSF D2. The nucleotides, as well as the residues involved in ADP or AMP–PNPbinding, are drawn as balls and sticks and labeled. The surface surrounding the nucleotide is drawn (8 A radius) to show the general shapeof the binding pocket. An asterisk labels a residue from an adjacent protomer. The nucleotide has different conformations, probably due tothe different sequences involved in nucleotide binding. Arg-362* interacts with Glu-305 (DExx box), while Arg-359* is available for g-phosphatebinding. Both residues could mediate any conformational changes upon ATP hydrolysis or ADP release.

Molecular Cell1478
Figure 3. Cyro-EM Reconstruction of Rat Liver Cytosol p97 at 18 A
(A) Side view of p97, perpendicular to the 6-fold molecular axis, shows a truncated barrel-like structure with side holes. The main body ofthe molecule shows pseudo 2-fold symmetry along an axis perpendicular to the 6-fold, although there are protruding densities at one end(top). The overall molecular dimensions are indicated. The 145 A diameter of the EM map is less than the 160 A observed in the N D1 crystalstructure, which we attribute to the ill-defined N domain in the EM reconstruction.(B) Cut-open view of (A) reveals a cage-like structure formed from the two-ring layers corresponding to the N D1 (blue) and D2 hexamers(green).(C) Top view, down the 6-fold axis, corresponding to the N D1 hexamer (blue), reveals a large annulus, surface pockets, and disconnecteddensities, and is different in appearance to the D2 layer. The black arrow indicates the pocket that coincides with the nucleotide bindingpocket indicated by the arrow in Figure 1C.(D) Bottom view, corresponding to the D2 hexamer (green), shows a closed annulus and several distinctive surface features.
dicular to the 6-fold axis (see Figure 3B). The top surface structed based on the crystal structure of D1 using theprogram JIGSAW (Bates and Sternberg, 1999). The sec-of the structure has an annulus diameter of z34 A,
whereas the bottom appears closed at the present reso- ondary structures are all conserved between D1 andD2, though there are differences in two loop regions.lution (Figures 3C and 3D). There are also pockets on
both surfaces, with those on the top surface particularly One of them corresponds to a flexible loop close to the6-fold axis, with three residues inserted in D2. This loopdeep (Figures 3C and 3D). The threshold of surface ren-
dering was chosen such that the corresponding volume is also partially disordered in the D1 crystal structure.The other one corresponds to the loop between a12matches the 550 kDa molecular mass of p97. Extra den-
sities observed around the outside rim of the top, more and a13 on the edge of the hexamer interface, with oneresidue deleted in D2. Overall, the D2 model suggestsopen, ring (Figure 3C), are reliable as they do not disap-
pear at the higher threshold of surface rendering, though a very similar hexamer interface as well as interactionswith bound nucleotide to that of D1. The N D1 crystalthey are diffused at very low thresholds. We attribute
this unconnected density to flexibility of that part of the structure and the D2 hexamer model were then dockedinto the full-length p97 EM density map (Figure 4). Thestructure. The 145 A diameter of the EM map is less
than the 160 A observed in the N D1 crystal structure, best agreement places the N D1 crystal structure intowhich we attribute to the ill-defined N domain in the EM the top, more open, ring, with N-terminal domains ac-reconstruction. counting for the disconnected density on the outside of
the rim (Figure 4A). However, in the EM reconstruction,this density is shifted upward relative to the plane ofFitting of N D1 Crystal Structure into EM Density
In order to obtain a model of full-length p97, a homology the D1 ring in the X-ray structure (Figure 4C) and isdisconnected from the main body. This would requiremodel of D2 (40% sequence identity to D1) was con-

Structure of p97 ATPase1479
Figure 4. Comparison and Fitting of the D1 Crystal Structure and the D2 Homology Model with the p97 EM Structure
The D1/D2 model is in yellow, fitted into the EM reconstruction (blue).(A) Top view, showing the overall dimension of the D1 model agrees with the main body of the EM density. The partially disconnected EMdensity on the outer rim accounts for the flexible N domain that is not shown for the sake of clarity.(B) Bottom view, showing good agreement between the D2 model and EM reconstruction.(C) Side view of the p97 D1/D2 hexamer model fitted into EM density. The width and height are in agreement, though there are extra densitiesin the EM map between the D1 and D2 rings that could correspond to the C-terminal 73 residues. The locations of the a-helical domains areindicated by long arrows.(D) Tilted side view highlighting one p97 D1/D2 monomer model fitted into the EM density. The C-terminal a-helical domains from both theD1 and D2 protomers are located on the surface of the molecule near the 2-fold axis and could form part of an interhexamer interface. Apossible linker region between D1 and D2 is indicated as a dashed yellow line. The short arrow indicates the location of the C termini of p97 D2.
a rearrangement in the position of the N domain relative arrangement between D1 and D2, with a distance ofz48 A between them (Figures 4B and 4C). This placesto D1 and reflects the flexible nature of the N domain,
with crystallization of N D1 capturing a specific confor- the nucleotide binding pockets of D2 onto the otherouter surface of the molecule resulting in a tail-to-tailmation. However, at the present resolution, we are un-
able to accurately place the N domain into the EM map, arrangement for the D1 and D2 hexamers (Figures 4Band 4C). Such a fitting places the C-terminal a-helicaland it is therefore omitted from our fitted models (Figure
4). To provide an accurate fit of the crystal structure into domains of both the D1 and D2 rings in close proximityto each other in the middle of the molecule (Figure 4C,the EM map, we have used a systematic and conserva-
tive approach, using the program ESSENS (Kleywegt arrows), and places the ATP binding sites on the ex-posed surfaces of the protein. This fit is consistent withand Jones, 1997) and the D1 hexamer as a search model.
The best solution places the nucleotide binding pockets the central cage-like features of the EM density, giventhat both the D1 and D2 hexamers have convex surfacesof D1 into the deep pockets observed on the top surface
of the EM map (see arrows in Figures 1C and 3C). Subse- on their respective C-terminal sides. The length of thelinker (26 residues) is adequate to link D1 and D2, andquent fitting of the D2 hexamer into the bottom closed
ring using the same approach results in a pseudomirror would be positioned on the surface in this current model

Molecular Cell1480
with minimal overlap with the rest of the p97 molecule lography or NMR (Babor and Fass, 1999; Coles et al.,1999; May et al., 1999; Yu et al., 1999). Here, we also(Figure 4D). A surface linker is also consistent with prote-
ase sensitivity studies of NSF, which shows a cleavage report the structure of an N domain, but unlike previousstudies the N domain is linked to its subsequent D1pattern that results in three distinct fragments: N, D1,
and D2 (Hanson et al., 1997). The observed difference domain. Superposition of all four N domains reveals asurprising similarity in both domain topology and inter-in size of the D1 annulus between the EM and crystal
structures (Figures 3C and 4A) may reflect either differ- domain arrangement (Figure 2A), despite the low se-quence identity or structure determination method (se-ences in bound nucleotide or interactions between the
D1 and D2 domains in full-length p97. However, it should quence ID range: 8%–25%; 50% of residues can besuperposed ,1 A rmsd on Ca). Thus, N domains can bebe noted that our final D1/D2 model represents a conser-
vative fit of the p97 D1 crystal structure (and D2 homol- considered as single structural elements comprising twoconserved subdomains that pack similarly in a numberogy model) into the EM reconstruction, as no adjust-
ments have been made to the crystallographic hexamer of different proteins. Further support for this notioncomes from one of the reported NSF N domain crystalto either improve the fit or take into account any confor-
mational flexibility within the hexamer. structures (May et al., 1999), where three independentcopies of the N-terminal domain all adopt the sameA number of other packing arrangements for the D1
and D2 hexamers is possible, such as head-to-tail, subdomain interactions and arrangement.A superposition of the double C barrels of NSF/Sec18where D1 and D2 stack on top of each other in the
same orientation, or head-to-head, where the nucleotide and p97/VAT/Cdc48p reveals detailed differences. Thefirst C loop in p97/VAT/Cdc48p (residues 32–37) is sig-binding pockets for each hexamer face inside the mole-
cule. To check these and other packing arrangements, nificantly shorter when compared to NSF/Sec18, wherethe C loop is replaced with a two-turn a helix. Studieswe took our N D1/D2 hexamer model and then adjusted
each hexamer separately by flipping the D1 and D2 on other double C barrel structures suggest that C loopsare typically involved in substrate binding or catalyticmodels 1808 relative to their hexamer planes. Correla-
tion scores for each new fit were then calculated using interactions (Castillo et al., 1999). The fact that p97 hasa shorter and more rigid surface C loop makes it tempt-ESSENS (Kleywegt and Jones, 1997). The score ob-
tained from placing D1 in the up position (see Figure 4) ing to speculate that the C loop is involved in bindingto the p97 adaptor protein p47. Coles et al. (1999) haveis significantly better than the best score obtained from
the down position (score ratio of 1.3). A similar result shown that the VAT N domain can bind polypeptides,catalyze the refolding of certain substrates, and preventwas obtained by placing D2 in the down position relative
to the up position, supporting a tail-to-tail arrangement protein aggregation. They propose a binding site thatlies between the C loops of the amino-terminal doubleas the best fit. Our current packing arrangement thus
results in pseudo C2 symmetry between the D1 and D2 C barrel and the flexible loops of the C-terminal b barrelthat corresponds to a band of positive charge acrossrings, and an z208 rotation of one ring relative to the
other. the upper face of the molecule. Babor and Fass (1999)and Yu et al. (1999) both propose similar SNAP bindingA number of previous studies have suggested a head-
to-tail packing arrangement for the D1 and D2 hexamer sites for NSF between the two subdomains in a regionrings (Hanson et al., 1997; Lenzen et al., 1998; Rockel of positive charge. For p97, the large b-turn-b structureet al., 1999). These studies have been based on lower (residues 153–157) in the b barrel subdomain, togetherresolution EM data (negative stain and quick freeze/ with the first C loop, forms a hydrophobic cleft that maydeep etch), and crystal lattice packing for NSF D2. Len- provide another p47 binding site (the p97–p47 complexzen et al. (1998), for example, suggested a possible is stable up to 0.4 M salt; H. K., unpublished data).head-to-tail arrangement for NSF based on the NSF Further studies are in progress to address these issues.D2 crystal structure, the charge complementarity, and p97 D1 and NSF D2 have similar topologies and do-crystal lattice packing arrangements. However, the au- main arrangements (Figure 2B). A superposition of indi-thors also noted that a substantial cavity between D1 vidual protomers results in 28% of all residues havingand D2 would minimize any stabilizing effects due to rmsd ,1 A (Ca). However, more than half the residuescharge complementarity (Lenzen et al., 1998). In the p97 in the a/b domain have rmsd ,1 A (Ca), which is dueN D1 crystal lattice, both tail-to-tail and head-to-head to a small rotation of the C-terminal a-helical domainarrangements are observed. In the VAT EM study relative to the a/b domain (Figure 2B). In p97 D1, the cleft(Rockel et al., 1999), the NSF D2 crystal structure was formed between these two domains is slightly closedfitted into the negatively stained EM density suggesting compared to that of NSF D2 (Figure 2B). This domaina head-to-tail arrangement of D1/D2 domains, though movement may reflect structural differences in nucleo-it could be argued that the resolution of the EM recon- tide binding states between the NSF D2 and p97 D1struction would make it difficult to distinguish one orien- structures, namely AMP–PNP versus ADP (see below).tation from the other. Our reconstruction of p97 mole- In p97 D1, the N D1 linker (Figures 2B and 5A, magenta)cules in a hydrated environment, combined with higher is pushed outward, while the corresponding linker regionresolution and carefully evaluated fitting, now provides in NSF D2 (Figures 2B and 5A, orange) is positioneda more accurate conformational arrangement for the D1 between the a/b domain and the a-helical domain, inand D2 rings. between protomers. Interestingly, if this orientation is
applied to p97, the N domains would be positioned ontop of the hexamer ring, as proposed previously for NSFStructure Comparisons with Other
AAA Family Members (May et al., 1999).A superposition of both hexamers shows the P loopsThe structures of N domains from NSF, Sec18, and VAT
have previously been determined using either crystal- to have similar conformations (0.3 A rmsd [Ca]). In addi-

Structure of p97 ATPase1481
Figure 5. Speculative Model of p97 Acting as a Molecular Ratchet
(A) Superposition of p97 D1 in an ADP state (blue) with NSF D2 in an ATP state (green) on their respective P loops. A small clockwise rotationaround the hexamer axis is observed between the green and blue monomers. ADP moieties are drawn as brown spheres. Also shown arethe corresponding linker regions between p97 N D1 (magenta) and NSF D1/D2 (orange).(B) Possible ratchet mechanism for p97 with D1 and D2 rings acting as interdependent and counter-balanced parts in the ATP hydrolysiscycle. Top left, the D1 and D2 rings are shown as ATP- and ADP-bound states, respectively. ATP hydrolysis of D1 would cause a clockwiserotation of D1. Top right, the rotation of D1 causes a conformational change in D2 to allow ADP release, resulting in an ATP-ready or emptystate, while D1 returns to an ADP-bound state. Bottom right, ATP hydrolysis in D2 would cause a counterclockwise motion. Bottom left, therotation caused by D2 would allow the release of ADP in D1, resulting in an ATP-ready state, while D2 returns to an ADP-bound state. Thebinding of ATP by D1 would initiate another hydrolysis cycle (top left).
tion, residues involved in g-phosphate binding in NSF possible link between ATP hydrolysis and interprotomerrearrangements mediated partly by the DExx box is in-D2 (Lys-557 and Asp-612) are conserved in p97 D1 (Lys-
251 and Asp-305), with the exception of Lys-639. In triguing. The p97 N D1 structure reported here is oneof the first structural descriptions for an active ATPasep97, Arg-359 from an adjacent protomer is at a position
similar to Lys-639, suggesting that Arg-359 could bind AAA domain and is thus more representative of otherAAA family proteins.the g-phosphate in p97 D1. Furthermore, residues in-
volved in Mg21 binding in NSF D2 (Thr-557, Asp-611,and Asp-612) are also conserved in p97 D1 (Thr-252, Speculative Conformational Changes upon ATP
Hydrolysis and Possible Ratchet Mechanism of p97Asp-304, and Asp-305, respectively), as well as in thewhole AAA family. It is plausible, therefore, that p97 N In order to assess any possible conformational changes
upon ATP hydrolysis, we attempted to grow and/or soakD1 will bind ATP (or AMP–PNP) using similar interactionsto those observed for NSF D2. If the P loops are used crystals in ADP, AMP–PNP, or ATP–gS. However, we
observed only bound ADP, which we attribute to theto superpose both hexamers, the a- and b-phosphatesof ADP and AMP–PNP align structurally (0.7 A rmsd; formation of a stable N D1–ADP complex prior to purifi-
cation. We were unable to remove the bound ADP inFigures 2B and 5A). Such a superposition is relevant,since it is known that in other ATPases the P loop is nondenaturing conditions, suggesting that the N D1–
ADP complex is highly stable. In order to obtain insightsgenerally static upon nucleotide hydrolysis (Smith andRayment, 1996). From these structural comparisons, it is into any possible conformational changes upon ATP
hydrolysis, we compared the p97 D1–ADP hexamer withnot unreasonable to assume that the NSF D2 AMP–PNPcomplex is representative of a p97 D1–ATP complex, that of NSF–ATP (AMP–PNP; Lenzen, et al., 1998; Yu et
al., 1998). We note that such a comparison betweenalthough further experimental evidence is required toconfirm this issue, namely a p97–ATP complex structure. two different proteins sharing limited sequence identity
in different nucleotide-bound states can be misleading,The AAA family in general is characterized by a num-ber of well-defined sequence motifs, such as the P loop and thus the following discussion should be considered
as purely speculative.(Walker A), DExx box (Walker B), and the AAA minimalconsensus sequence (Neuwald et al., 1999). It is well A superposition of the p97 D1 and NSF D2 hexamers,
based on their respective P loops, shows that althoughestablished that P loops are involved in nucleotide bind-ing, while the DExx box is involved in Mg21 binding and the general protomer contacts are maintained, a small
apparent clockwise rotation is observed for p97 D1 pro-indirectly in nucleotide binding. The p97 N D1 structurenow shows that the DExx box is also involved in interpro- tomers with respect to NSF (Figure 5A; p97, blue; NSF,
green). This clockwise rotation of the p97 hexamer withtomer contacts, and by inference, hexamerization. A

Molecular Cell1482
temperature by hanging drop vapor diffusion after mixing equalrespect to the NSF hexamer (using fixed P loops) centersvolumes of 16 mg/ml N D1 with reservoir solutions containing 3.8–4.2around the surface helix a8, and might reflect the confor-M sodium formate, 10% glycerol, and 5% PEG600 (pH 5.5–6.0). Deriv-mational shift between the ATP and ADP states for p97atives were prepared by soaking crystals in mother liquor containing
D1. Based on this structural comparison, we also pro- 50 mM GdCl3 for 16 hr and 10 mM mercuric acetate for 4 hr. Crystalspose that for p97 D1, residues Asn-348 to Arg-359 act were obtained in the presence of 10 mM MgCl2 and 10 mM ADP,
AMP–PNP, or ATP-g-S, and in the absence of added nucleotide andas g-phosphate sensors (Figures 1A and 1E, red), withMgCl2. All crystal forms were subsequently found to contain onlyArg-359 directly contacting the ATP g-phosphate boundADP. Attempts to produce nucleotide-free forms of N D1 usingto an adjacent protomer, while Asn-348 from the adja-multiple steps of exhaustive dialysis against EDTA followed by fur-cent protomer could be positioned on the other sidether dialyses against activated charcoal produced only ADP-bound
of the g-phosphate (see Figure 1E, near the protomer crystals, suggesting that a highly stable N D1–ADP complex isinterface; Asn-348, yellow; Arg-359, blue). This region formed before purification.is within the conserved AAA minimum consensus se-quence and has previously been proposed as a g-phos- Data Collection, Phasing, and Refinement
Diffraction data were collected using crystals flash frozen at –1108Kphate sensor for the family (Neuwald et al., 1999).directly from the mother liquor, and then annealed for 5 s beforeThe fitting of the D1/D2 structure/model into the cryo-being refrozen. Data for the mercury acetate derivative were mea-EM density of a full-length p97–ADP complex suggestssured using a Rigaku RU200 rotating anode source equipped withtail-to-tail packing with their respective nucleotide bind-Yale mirrors and an RAXIS-II imaging plate. Native data were col-
ing sites at opposite ends of the molecule. The EM lected at X11 beamline of the DESY synchrotron in Hamburg, usingreconstruction also suggests that D1 and D2 of p97 a MAR 345 mm image plate. Gadolinium MAD data were collected
on the BM14 beamline at ESRF Grenoble, using a MAR CCD detec-could exist in different nucleotide-bound states. This,tor. The space group is P622 with unit cell dimensions a 5 b 5combined with the possible conformational changes146.0 A, c 5 84.7 A with one monomer per asymmetric unit. Datadiscussed above, allows us to speculate on a concep-processing was carried out using HKL (Otwinowski and Minor, 1997),tual model for p97 acting as a molecular ratchet, withand the final statistics are summarized in Table 1. The structure
the D1 and D2 hexameric rings forming two intercon- was solved by MAD methods combined with single isomorphousnected but negatively cooperating parts. In this model, replacement (SIR). Diffraction data were collected at three wave-
lengths around the Gd LIII absorption edge on GdCl2-derivatizedATP hydrolysis within one hexameric ring (e.g., D1)crystals along with a far remote data set collected at 0.973 A. Threewould promote ADP release in the D2 ring resulting inGd sites were identified using anomalous differences measured atan ATP-ready (empty) conformation for D2 (Figure 5B,the peak wavelength as input into the program SHELXS (Sheldrick,top row). Given that the D1 and D2 rings are related1990). These were then refined and phases obtained using the pro-
by pseudo C2 symmetry, subsequent ATP binding and gram SHARP (de La Fortelle and Bricogne, 1997), with the besthydrolysis by D2 would promote ADP release from the statistics being obtained when including only the far remote and
inflection point (1.71 A) data sets. These two wavelength MADD1 ring to form an ATP-ready/empty state (Figure 5B,phases were then combined with the phases from the mercury ace-bottom row). In support of this speculative model istate derivative using the CCP4 programs (CCP4, 1994). The phasingthe observation that site-specific mutations that abolishstatistics are summarized in Table 1. The resulting maps at 3.0 AATPase activity within one hexamer affects the ATPaseallowed a trace of most of the backbone apart from the N-terminal
activity in the other (H. K., unpublished data), consistent region and some surface loops that were missing. After each cyclewith the notion that both hexameric rings are coupled of tracing and rebuilding using O (Jones et al., 1991), the partial
model was subjected to an energy minimization refinement with afor full catalytic activity. However, experimental supportmaximum likelihood target using CNS (Brunger et al., 1998), andof the proposed model requires a full-length p97 struc-phases were combined with experimental phases to produce im-ture and further mutagenesis studies. Nevertheless, theproved maps that enabled further model building. After building thecombined X-ray crystallography and cryo-EM structuresbackbone of residues 21–430, the model was refined using a native
presented here provide some insights into the possible data set to 2.9 A resolution, and side chains were gradually addedmechanism of p97, and now form the basis for further after careful inspections of 2Fo–Fc maps. Simulated annealing as
implemented in CNS was used for subsequent refinement, using aexperiments aimed at explaining the finer details of con-starting temperature of 25008 K. A random 10% of the starting dataformational changes upon ATP hydrolysis, adaptor bind-was set aside for cross validation (free R factor calculation). Duringing, and possible SNARE disassembly.refinement, clear continuous density was observed within the activesite region corresponding to ADP. Subsequent refinement of crys-Experimental Procedurestals formed in the presence of different nucleotides always revealeddensity corresponding to ADP. Simulated annealing omit maps alsoExpression, Purification, and Crystallizationconfirmed the presence of ADP. A number of ordered water mole-DNA encoding residues 2–458 (N and D1 domains) of murine p97cules was also added during the refinement, and the final modelwas amplified by PCR from a full-length p97 cDNA and clonedcontains residues 21–458 of murine p97, one ADP, and 51 waterinto pPRO-EX HTb (Life Technologies). Recombinant protein wasmolecules resulting in a free R factor of 28.3% and an R factor ofexpressed in E. coli ER2566 (New England Biolabs) by induction of22.4%. Refinement statistics are also listed in Table 1. In the finala midlog-phase culture with 1 mM IPTG. Cells were resuspendedmodel, 20 residues at the N terminus that are not visible in ourin 50 mM Tris (pH 8.0), 20 mM imidazole, and 500 mM NaCl, andelectron density maps are missing, as are the side chains for 23lysed in a French pressure cell or by sonication. Recombinant N D1surface charge residues (replaced by Ala). The loop regions betweenwas eluted from a HiTrap chelating column (equilibrated with nickelresidues 337–339, 312–316, and 432–437 also have poor electronsulfate) with a linear gradient of 20–500 mM imidazole in 50 mM Trisdensity. All residues lie in allowed regions in the Ramachandran(pH 8.0) and 500 mM NaCl, and the His tag cleaved with rTEV prote-plot (82.4% in most favored regions).ase as per the manufacturer’s (Life Technologies) instructions.
Cleaved N D1 was dialyzed into 20 mM HEPES (pH 7.5), 10 mMMgCl2, 600 mM KCl, and 2 mM b-mercaptoethanol, and concen- Cryomicroscopy and 3D Image Processing
Full-length rat liver p97 was purified as described (Kondo et al.,trated to 16 mg/ml as determined by the Bradford (Bio-Rad) assaybefore adding nucleotide (ADP, ATP-g-S, or AMP–PNP) for a final 1997), and vitrified specimens were prepared for cryo-EM as de-
scribed (Dubochet et al., 1988) and transferred to a CM200 FEGconcentration of 10 mM. Crystals were obtained in 24–72 hr at room

Structure of p97 ATPase1483
cryomicroscope using a Gatan side entry cryo holder. Images were Dube, P., Tavares., P., Lurz, R., and van Heel, M. (1993). Bacterio-phage SPP1 portal protein: a DNA pump with 13-fold symmetry.taken under low-dose conditions, at 38,000-fold magnification and
2.5 6 0.5 mm defocus. The micrographs were digitized using an EMBO J. 12, 1303–1309.Image Science GmbH (Berlin, Germany) patchwork densitometer at Dube, P., Orlova, E.V., Zemlin, F., van Heel, M., Harris, J.R., anda step size of 2.8 A on the specimen scale. The contrast transfer Markl, J. (1995). Three-dimensional structure of keyhole limpet he-function of the microscope (CTF) was determined by finding zero mocyanin by cryoelectron microscopy and angular reconstitution.positions of Thon rings (Erickson and Klug, 1970) for each micro- J. Struct. Biol. 115, 226–232.graph separately. Correction of CTF was done by reversing phases
Dubochet, J., Adrian, M., Chang, J.J., Homo, J.C., Lepault, J., Mc-of odd rings; amplitudes were not corrected. Since images were
Dowell, A.W., and Schultz, P. (1988). Cryo-electron microscopy oftaken using a range of different defocus values, the structure was
vitrified specimens. Q. Rev. Biophys. 21, 129–228.not affected by zeroes in the CTF.
Egerton, M., Ashe, O.R., Chen, D., Druker, B.J., Burgess, W.H., andMolecular images were selected interactively. Image processingSamelson, L.E. (1992). VCP, the mammalian homolog of cdc48, iswas performed using the IMAGIC-5 software (van Heel et al., 1996)tyrosine phosphorylated in response to T cell antigen receptor acti-on DEC workstations running UNIX. The images were subjected tovation. EMBO J. 11, 3533–3540.multireference alignment (van Heel, 1989) and automated classifica-
tion (Dube et al., 1993). Relative orientations of characteristic views Erickson, H.P., and Klug, A. (1970). Measurement and compensation(van Heel and Stoffler-Meilicke, 1985) were defined by angular re- of defocusing and aberrations by Fourier processing of electronconstitution (van Heel, 1987), assuming C6 symmetry. The 3D distri- micrographs. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 261, 105–118.bution of molecular densities were reconstructed using the exact Fisher, A.J., Smith, C.A., Thoden, J.B., Smith, R., Sutoh, K., Holden,filter back projection algorithm (van Heel and Harauz, 1986; Rader- H.M., and Rayment, I. (1995). X-ray structures of the myosin motormacher, 1988), and then iteratively refined (Schatz et al., 1995). The domain of Dictyostelium discoideum complexed with MgADP.BeFxfinal 3D reconstruction was calculated from 150 class averages that and MgADP.AlF4-. Biochemistry 34, 8960–8972.included z3100 original molecular images. The handedness of the
Fleming, K.G., Hohl, T.M., Yu, R.C., Muller, S.A., Wolpensinger, B.,hexamer was determined by comparison with the N D1 crystal struc-
Engel, A., Engelhardt, H., Brunger, A.T., Sollner, T.H., and Hanson,ture. The contour level used for rendering of the 3D maps was
P.I. (1998). A revised model for the oligomeric state of the N-ethylma-taken at 1s above the level of the average density in the map. The
leimide-sensitive fusion protein, NSF. J. Biol. Chem. 273, 15675–resolution of the resulting maps was assessed using FSC criteria
15681.(van Heel and Harauz, 1986). We have taken into account that the
Ghislain, M., Dohmen, R.J., Levy, F., and Varshavsky, A. (1996).structure has 6-fold symmetry, meaning that the threshold has beenCdc48p interacts with Ufd3p, a WD repeat protein required for ubi-increased by √6 (Dube et al. 1995). Surface rendering was performedquitin-mediated proteolysis in Saccharomyces cerevisiae. EMBO J.using the Iris Explorer. The fitting of the X-ray structure into the EM15, 4884–4899.map was performed using ESSENS (Kleywegt and Jones, 1997) andHanson, P.I., Roth, R., Morisaki, H., Jahn, R., and Heuser, J.E. (1997).inspected in O (Jones et al., 1991).Structure and conformational changes in NSF and its membranereceptor complexes visualized by quick-freeze/deep-etch electronAcknowledgmentsmicroscopy. Cell 90, 523–535.
We thank David Frith and Arnold Coffer for rat liver p97 preparations. Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. (1991).We are indebted to Suhail Islam for assistance with graphics using Improved methods for the building of protein models in electronhis program PREPI (S. Islam, ICRF). We also thank Mike Sternberg density maps and the location of errors in these models. Acta Crys-for informative discussions. We are particularly grateful to Graham tallogr. A 47, 110–119.Warren for his early encouragement and helpful discussions.
Karata, K., Inagawa, T., Wilkinson, A.J., Tatsuta, T., and Ogura, T.(1999). Dissecting the role of a conserved motif (the second region
Received October 10, 2000; revised November 13, 2000.of homology) in the AAA family of ATPases. Site-directed mutagene-sis of the ATP-dependent protease FtsH. J. Biol. Chem. 274, 26225–
References26232.
Kleywegt, G.J., and Jones, T.A. (1997). Template convolution toBabor, S.M., and Fass, D. (1999). Crystal structure of the Sec18penhance or detect structural features in macromolecular electron-N-terminal domain. Proc. Natl. Acad. Sci. USA 96, 14759–14764.density maps. Acta Crystallogr. D 53, 179–185.
Bates, P.A., and Sternberg, M.J.E. (1999). Model building by compar-Koegl, M., Hoppe, T., Schlenker, S., Ulrich, H.D., Mayer, T.U., andison at CASP3: using expert knowledge and computer automation.Jentsch, S. (1999). A novel ubiquitination factor, E4, is involved inProteins Suppl. 3, 47–54.multiubiquitin chain assembly. Cell 96, 635–644.
Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P.,Kondo, H., Rabouille, C., Newman, R., Levine, T.P., Pappin, D., Free-Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M.,mont, P., and Warren, G. (1997). p47 is a cofactor for p97-mediatedPannu, N.S., et al. (1998). Crystallography & NMR system. Actamembrane fusion. Nature 388, 75–78.Crystallogr. D 54, 905–921.Lenzen, C.U., Steinmann, D., Whiteheart, S.W., and Weis, W.I. (1998).Castillo, R.M., Mizuguchi, K., Dhanaraj, V., Albert, A., Blundell, T.L.,Crystal structure of the hexamerization domain of N-ethylmaleimide-and Murzin, A.G. (1999). A six-stranded double-psi beta barrel issensitive fusion protein. Cell 94, 525–536.shared by several protein superfamilies. Structure 7, 227–236.Leonhard, K., Stiegler, A., Neupert, W., and Langer, T. (1999). Chap-CCP4 (Collaborative Computational Project 4) (1994). The CCP4erone-like activity of the AAA domain of the yeast Yme1 AAA prote-suite: programs for protein crystallography. Acta Crystallogr. D 50,ase. Nature 398, 348–351.760–763.
Matveeva, E.A., He, P., and Whiteheart, S.W. (1997). N-ethylmalei-Coles, M., Diercks, T., Liermann, J., Groger, A., Rockel, B., Baumeis-mide-sensitive fusion protein contains high and low affinity ATP-ter, W., Koretke, K.K., Lupas, A., Peters, J., and Kesseler, H. (1999).binding sites that are functionally distinct. J. Biol. Chem. 272, 26413–The solution structure of VAT-N reveals a ‘missing link’ in the evolu-26418.tion of complex enzymes from a simple babb element. Curr. Biol.
9, 1158–1168. May, A.P., Misura, K.M.S., Whiteheart, S.W., and Weis, W.I. (1999).Crystal structure of the amino-terminal domain of N-ethylmaleimide-Confalonieri, F., and Duguet, M. (1995). A 200-amino acid ATPasesensitive fusion protein. Nat. Cell. Biol 1, 175–182.module in search of a basic function. Bioessays 17, 639–650.
de La Fortelle, E., and Bricogne, G. (1997). Maximum likelihood Meyer, H.H., Shorter, J.G., Seemann, J., Pappin, D., and Warren, G.(2000). A complex of mammalian ufd1 and npl4 links the AAA-heavy-atom parameter refinement for the multiple isomorphous re-
placement and multiwavelength anomalous diffraction methods. ATPase, p97, to ubiquitin and nuclear transport pathways. EMBOJ. 19, 2181–2192.Methods Enzymol. 276, 472–494.

Molecular Cell1484
Morgan, A., and Burgoyne, R.D. (1995). A role for soluble NSF attach-ment proteins (SNAPs) in regulated exocytosis in adrenal chromaffincells. EMBO J. 14, 232–239.
Neuwald, A.F., Aravind, L., Spouge, J.L., and Koonin, E.V. (1999).AAA1: a class of chaperone-like ATPases associated with the as-sembly, operation, and disassembly of protein complexes. GenomeRes. 9, 27–43.
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffractiondata collected in oscillation mode. Methods Enzymol. 276, 307–326.
Patel, S., and Latterich, M. (1998). The AAA team: related ATPaseswith diverse functions. Trends Cell. Biol. 8, 65–67.
Patel, S.K., Indig, F.E., Olivieri, N., Levine, N., and Latterich, M.(1998). Organelle membrane fusion: a novel function for the syntaxinhomolog Ufe1p in ER membrane fusion. Cell 92, 611–620.
Peters, J.-M., Harris, J.R., Lustig, A., Muller, S., Engel, A., Volker,S., and Franke, W.W. (1992). Ubiquitous soluble Mg(21)-ATPasecomplex. A structural study. J. Mol. Biol. 223, 557–571.
Rabouille, C., Levine, T.P., Peters, J.M., and Warren, G. (1995). AnNSF-like ATPase, p97, and NSF mediate cisternal regrowth frommitotic Golgi fragments. Cell 82, 905–914.
Rabouille, C., Kondo, H., Newman, R., Hui, N., Freemont, P., andWarren, G. (1998). Syntaxin 5 is a common component of the NSF-and p97-mediated reassembly pathways of Golgi cisternae frommitotic Golgi fragments in vitro. Cell 92, 603–610.
Radermacher, M. (1988). Three-dimensional reconstruction of singleparticles from random and nonrandom tilt series. J. Electron Mi-crosc. Tech. 9, 359–394.
Rockel, B., Walz, J., Hegerl, R., Peters, J., Typke, D., and Baumeister,W. (1999). Structure of VAT, a CDC48/p97 ATPase homolog fromthe archaeon Thermoplasma acidophilum as studied by electrontomography. FEBS Lett. 451, 27–32.
Schatz, M., Orlova, E.V., Dube, P., Jager, J., and van Heel, M. (1995).Structure of Lumbricus terrestris hemoglobin at 30 A resolutiondetermined using angular reconstitution. J. Struct. Biol. 114, 28–40.
Sheldrick, G.M. (1990). Phase annealing in SHELX-90: direct meth-ods for larger structures. Acta Crystallogr. A 46, 467–473.
Smith, C.A., and Rayment, I. (1996). Active site comparisons high-light structural similarities between myosin and other P loop pro-teins. Biophys. J. 70, 1590–1602.
Sollner, T., Bennett, M.K., Whiteheart, S.W., Scheller, R.H., and Roth-man, J.E. (1993a). A protein assembly-disassembly pathway in vitrothat may correspond to sequential steps of synaptic vesicle docking,activation, and fusion. Cell 75, 409–418.
Sollner, T., Whiteheart, S.W., Brunner, M., Erdjumentbromage, H.,Geromanos, S., Tempst, P., and Rothman, J.E. (1993b). SNAP recep-tors implicated in vesicle targeting and fusion. Nature 362, 318–324.
van Heel, M. (1987). Angular reconstitution: a posteriori assignmentof projection directions for 3D reconstruction. Ultramicroscopy 21,111–124.
van Heel, M. (1989). Classification of very large electron microscopialimage data sets. Optik 82, 114–126.
van Heel, M., and Harauz, G. (1986). Resolution criteria for threedimensional reconstructions. Optik 73, 119–122.
van Heel, M., and Stoffler-Meilicke, M. (1985). Characteristic viewsof E. coli and B. stearothermophilus 30S ribosomal subunits in theelectron microscope. EMBO J. 4, 2389–2395.
van Heel, M., Harauz, G., Orlova, E.V., Schmidt, R., and Schatz, M.(1996). A new generation of the IMAGIC image processing system.J. Struct. Biol. 116, 17–24.
Yu, R.C., Hanson, P.I., Jahn, R., and Brunger, A.T. (1998). Structureof the ATP-dependent oligomerization domain of N-ethylmaleimidesensitive factor complexed with ATP. Nat. Struct. Biol. 5, 803–811.
Yu, R.C., Jahn, R., and Brunger, A.T. (1999). NSF N-terminal domaincrystal structure: models of NSF function. Mol. Cell 4, 97–107.
Protein Data Bank Accession Number
Coordinates have been deposited with accession code 1E32.
Related Documents