CHAPTER SIX Structure of b-Adrenergic Receptors Florian Brueckner * , Chayne L. Piscitelli * , Ching-Ju Tsai * , Jörg Standfuss * , Xavier Deupi * ,† , Gebhard F.X. Schertler * ,1 * Laboratory of Biomolecular Research, Paul Scherrer Institut, Villigen PSI, Switzerland † Condensed Matter Theory, Paul Scherrer Institut, Villigen PSI, Switzerland 1 Corresponding author: e-mail address: [email protected] Contents 1. Introduction 118 2. Toward the Structures of b-Adrenergic Receptors 119 2.1 Engineering b-adrenergic receptors for structural studies 119 2.2 Expression of b-adrenergic receptors 123 2.3 Purification of b-adrenergic receptors 125 2.4 Crystallization of b-adrenergic receptors 134 2.5 Crystallography of b-adrenergic receptors 137 3. Lessons from the Structures of b-Adrenergic Receptors 141 3.1 Ligand binding modes of pharmacologically relevant drugs 141 3.2 Ligand selectivity 144 3.3 Insights into the activation mechanism 144 4. Outlook 146 Acknowledgments 147 References 147 Abstract b-Adrenergic receptors (bARs) control key physiological functions by transducing sig- nals encoded in catecholamine hormones and neurotransmitters to activate intracellu- lar signaling pathways. As members of the large family of G protein-coupled receptors (GPCRs), bARs have a seven-transmembrane helix topology and signal via G protein- and arrestin-dependent pathways. Until 2007, three-dimensional structural information of GPCRs activated by diffusible ligands, including bARs, was limited to homology models that used the related photoreceptor rhodopsin as a template. Over many years, several labs have developed strategies that have finally allowed the structures of the turkey b 1 AR and the human b 2 AR to be determined experimentally. The challenges to overcome included heterologous receptor overexpression, design of stabilized and crystallizable modified receptor constructs, ligand-affinity purification of active re- ceptor and the development of novel techniques in crystallization and micro- crystallography. The structures of bARs in complex with inverse agonists, antagonists, Methods in Enzymology, Volume 520 # 2013 Elsevier Inc. ISSN 0076-6879 All rights reserved. http://dx.doi.org/10.1016/B978-0-12-391861-1.00006-X 117

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

CHAPTER SIX
Structure of b-AdrenergicReceptorsFlorian Brueckner*, Chayne L. Piscitelli*, Ching-Ju Tsai*,Jörg Standfuss*, Xavier Deupi*,†, Gebhard F.X. Schertler*,1*Laboratory of Biomolecular Research, Paul Scherrer Institut, Villigen PSI, Switzerland†Condensed Matter Theory, Paul Scherrer Institut, Villigen PSI, Switzerland1Corresponding author: e-mail address: [email protected]
Contents
1.
MetISShttp
Introduction
hods in Enzymology, Volume 520 # 2013 Elsevier Inc.N 0076-6879 All rights reserved.://dx.doi.org/10.1016/B978-0-12-391861-1.00006-X
118
2. Toward the Structures of b-Adrenergic Receptors 1192.1
Engineering b-adrenergic receptors for structural studies 119 2.2 Expression of b-adrenergic receptors 123 2.3 Purification of b-adrenergic receptors 125 2.4 Crystallization of b-adrenergic receptors 134 2.5 Crystallography of b-adrenergic receptors 1373.
Lessons from the Structures of b-Adrenergic Receptors 141 3.1 Ligand binding modes of pharmacologically relevant drugs 141 3.2 Ligand selectivity 144 3.3 Insights into the activation mechanism 1444.
Outlook 146 Acknowledgments 147 References 147Abstract
b-Adrenergic receptors (bARs) control key physiological functions by transducing sig-nals encoded in catecholamine hormones and neurotransmitters to activate intracellu-lar signaling pathways. As members of the large family of G protein-coupled receptors(GPCRs), bARs have a seven-transmembrane helix topology and signal via G protein-and arrestin-dependent pathways. Until 2007, three-dimensional structural informationof GPCRs activated by diffusible ligands, including bARs, was limited to homologymodels that used the related photoreceptor rhodopsin as a template. Over many years,several labs have developed strategies that have finally allowed the structures of theturkey b1AR and the human b2AR to be determined experimentally. The challengesto overcome included heterologous receptor overexpression, design of stabilizedand crystallizable modified receptor constructs, ligand-affinity purification of active re-ceptor and the development of novel techniques in crystallization and micro-crystallography. The structures of bARs in complex with inverse agonists, antagonists,
117

118 Florian Brueckner et al.
and agonists have revealed the binding mode of ligands with different efficacies, haveallowed to obtain insights into ligand selectivity, and have provided better templates fordrug design. Also, the structures of b2AR in complex with a G protein and a G protein-mimicking nanobody have provided important insights into the mechanism of receptoractivation and G protein coupling. This chapter summarizes the strategies and methodsthat have been successfully applied to the structural studies of bARs. These are exem-plified with detailed protocols toward the structure determination of stabilized turkeyb1AR–ligand complexes. We also discuss the spectacular insights into adrenergic recep-tor function that were obtained from the structures.
1. INTRODUCTION
Adrenergic receptors (ARs) belong to the amine receptor cluster of
the rhodopsin-like family of G protein-coupled receptors (GPCRs)
(Fredriksson, Lagerstrom, Lundin, & Schioth, 2003). These receptors are
present on almost all peripheral tissues and on many neuronal populations
within the central and sympathetic nervous system where they activate in-
tracellular signaling cascades in response to the binding of the endogenous
catecholamines adrenaline and noradrenaline. As hormones and neurotrans-
mitters, adrenaline and noradrenaline act as agonists on ARs and thus control
a variety of physiological functions, including heart and lung function, blood
pressure, and a variety of metabolic and central nervous system functions.
ARs constitute important drug targets; for instance, bAR inverse agonists
and antagonists are widely used to treat hypertension and heart disease, while
b2AR agonists are important anti-asthma medicines.
The first evidence for the existence of multiple types of ARs came from
experiments that demonstrated two different rank orders of potency of cat-
echolamines toward different physiological effects, thereby establishing an
“a” and a “b” class of catecholamine-induced cellular responses
(Ahlquist, 1948). Subsequent analyses based on radioligand binding assays
identified pharmacologically distinct classes of ARs based on selectivity to
various exogenous compounds, substantiating the notion of multiple AR
subtypes. With the advent of molecular cloning, the genetic structure of
the AR family was finally elucidated. The first AR to be cloned was the
hamster b2AR, and its sequence revealed a striking similarity to the photo-
receptor rhodopsin, including seven-transmembrane (7TM) domains
predicted from hydropathy plots and conserved residues in the transmem-
brane domains (Dixon et al., 1986). Today, a total of nine AR subtypes have

119Structure of b-Adrenergic Receptors
been identified and classified into three major types based on pharmacolog-
ical and molecular evidence, namely, a1, a2 and bARs, with three subtypes
in each class (Bylund et al., 1994). As more receptors were cloned and se-
quenced, it became clear that ARs, as well as rhodopsin and other hormone
and neurotransmitter receptors, were part of a family of 7TM receptors
(Dohlman, Caron, & Lefkowitz, 1987). Experiments with chimeric recep-
tors led to the determination of structural domains involved in ligand bind-
ing and G protein coupling (Kobilka et al., 1988; Wong, Parker, & Ross,
1990). bARs couple to the stimulatory G protein Gs and thereby activate
adenylyl cyclase. In addition, coupling to the inhibitory G protein Gi and
G protein-independent signaling has also been observed (reviewed by
Patel, Noor, & Rockman, 2010). Differential activation of these signaling
pathways can be modulated by biased ligands.
The dim-light photoreceptor rhodopsin was the first GPCR to have its
three-dimensional structure determined, first by electron crystallography
and then by X-ray crystallography (reviewed in Schertler, 2005). The next
GPCR structures to be solved were the human b2- and the turkey b1ARs,
and thus they were the first structures of GPCRs activated by diffusible
ligands. This chapter recapitulates the ingenious strategies that have been
necessary to overcome the challenges of their structure determination as well
as some of the important insights gained from these structures.
2. TOWARD THE STRUCTURES OF b-ADRENERGICRECEPTORS
2.1. Engineering b-adrenergic receptors for structuralstudies
Wild-type rhodopsin can be purified in large quantities from native sources
and directly used in structural studies (Edwards et al., 2004). In contrast,
GPCRs activated by diffusible ligands, such as bARs, express only in very
low abundance in native materials and, therefore, need to be recombinantly
produced in heterologous expression systems. In addition, the wild-type
forms of most GPCRs are not suitable for structural studies due to their
low stability and high inherent conformational heterogeneity (Peleg,
Ghanouni, Kobilka, & Zare, 2001). It is thus generally required to engineer
GPCRs in order to optimize expression, increase stability, remove disor-
dered or flexible regions and heterogeneous posttranslational modifications
and improve crystallizability (Tate & Schertler, 2009). To overcome these
difficulties, three completely different approaches have been used so far to

120 Florian Brueckner et al.
obtain crystal structures of bARs: thermostabilization by site-directed mu-
tagenesis of b1AR, cocrystallization of b2AR with a Fab fragment and cre-
ation of fusion chimeras of b2AR with T4-lysozyme (T4L).
The wild-type turkey b1AR is expressed to higher levels and is inher-
ently more stable than the human b1AR (Serrano-Vega & Tate, 2009),
which made it more suitable as a starting point for receptor engineering.
In an early work, Ross and coworkers discovered that the Cys116(3.27)L
(numbers in parenthesis refer to the Ballesteros–Weinstein general number-
ing scheme for GPCRs (Ballesteros &Weinstein, 1995; see Table 6.1) point
mutation and C-terminal truncations substantially improve receptor expres-
sion in insect cells (Parker, Kameyama, Higashijima, &Ross, 1991; Parker &
Ross, 1991). A receptor construct further optimized for expression and
purification was later developed, which included additionally an N-terminal
truncation (Warne, Chirnside, & Schertler, 2003). The homogeneity of the
purified receptor was also improved, since the N-terminal truncation
removed an N-glycosylation site and eliminated proteolysis that had been
observed at the N-terminus. Subsequently, six thermostabilizing point
mutations were identified by a systematic alanine scanning approach that
increased the apparent melting temperature by 21 �C and improved stability
in the harsh small-micelle detergents ideal for crystallization (Serrano-Vega,
Magnani, Shibata, & Tate, 2008).
While flexible or unstructured regions ofGPCRs are generally implicated
in the interaction with regulatory and signaling proteins, they often impede
crystallization. For GPCRs, in particular, these flexible regions constitute a
significant portion of the surface area of the receptor outside the transmem-
brane region and are thus likely to form crystal contacts. In order to improve
the crystallizability of the turkey b1AR, in addition to the N-terminal trun-
cation mentioned above, the C-terminus (after helix 8) and part of intra-
cellular loop (ICL) 3 were also truncated (Warne, Serrano-Vega, Tate, &
Schertler, 2009). The palmitoylation site Cys358(8.59) was mutated to
alanine to reduce potential heterogeneity. The resulting final construct,
referred to as b1AR36-m23 (Fig. 6.1A), yielded crystals diffracting to high
resolution that allowed structure determination (Warne et al., 2008). The
related construct b1AR44-m23, in which the ICL3 truncation is two amino
acids shorter, has been used to obtain crystal structures with bound agonists
(Warne et al., 2011). Recently, Tate and colleagues sought to further
stabilize the turkey b1AR and succeeded by introducing three more muta-
tions including one that created an engineered salt bridge present in the
b2AR, resulting in a receptor thatwas 31 �Cmore thermostable than thewild
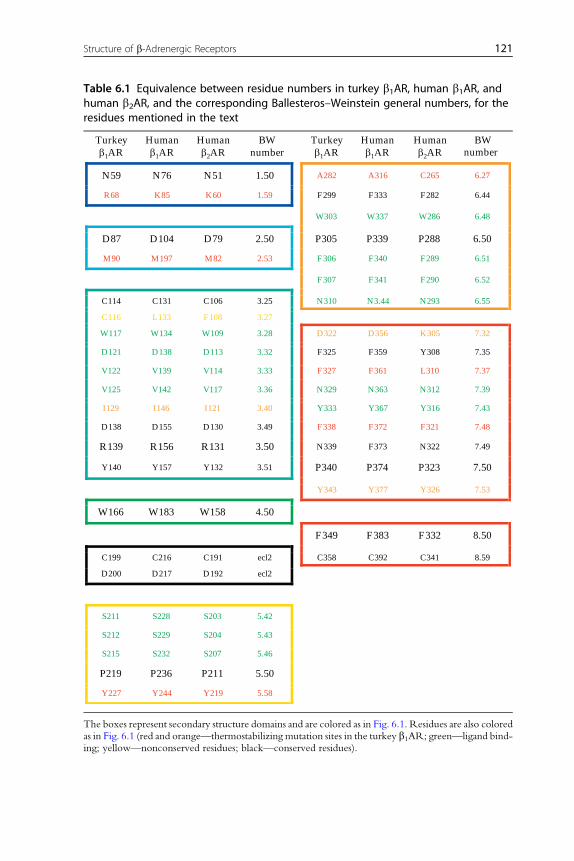
Table 6.1 Equivalence between residue numbers in turkey b1AR, human b1AR, andhuman b2AR, and the corresponding Ballesteros–Weinstein general numbers, for theresidues mentioned in the text
Turkeyb1AR
Humanb1AR
Humanb2AR
BWnumber
Turkeyb1AR
Humanb1AR
Humanb2AR
BW number
N59 N76 N51 1.50 A282 A316 C265 6.27
R68 K85 K60 1.59 F299 F333 F282 6.44
W303 W337 W286 6.48
D87 D104 D79 2.50 P305 P339 P288 6.50
M90 M197 M82 2.53 F306 F340 F289 6.51
F307 F341 F290 6.52
C114 C131 C106 3.25 N310 N3.44 N293 6.55
C116 L133 F108 3.27
W117 W134 W109 3.28 D322 D356 K305 7.32
D121 D138 D113 3.32 F325 F359 Y308 7.35
V122 V139 V114 3.33 F327 F361 L310 7.37
V125 V142 V117 3.36 N329 N363 N312 7.39
I129 I146 I121 3.40 Y333 Y367 Y316 7.43
D138 D155 D130 3.49 F338 F372 F321 7.48
R139 R156 R131 3.50 N339 F373 N322 7.49
Y140 Y157 Y132 3.51 P340 P374 P323 7.50
Y343 Y377 Y326 7.53
W166 W183 W158 4.50
F349 F383 F332 8.50
C199 C216 C191 ecl2 C358 C392 C341 8.59
D200 D217 D192 ecl2
S211 S228 S203 5.42
S212 S229 S204 5.43
S215 S232 S207 5.46
P219 P236 P211 5.50
Y227 Y244 Y219 5.58
The boxes represent secondary structure domains and are colored as in Fig. 6.1. Residues are also coloredas in Fig. 6.1 (red and orange—thermostabilizing mutation sites in the turkey b1AR; green—ligand bind-ing; yellow—nonconserved residues; black—conserved residues).
121Structure of b-Adrenergic Receptors
C
C
L
C1163.27Limproves expression
RemovespalmitoylationC358A
cys-bridge
Salt bridge
SN
WV
V
D
W
FF YSS
N
F
FS
V
A
L
A
M
DK
V
L
R68S
M90V
Y227A
A282L
F327A
F338M
D200
D322K
I129V
Y343L
D
Y
N
A
N DR
W PP
P
N-term deletion (3–32)
β1AR36: deletion (243–270 + 277–278)β1AR44: deletion (243–270)
ICL3
deletion (369–483) + 6x His tag
Turkey b1-adrenergic receptor (2VT4)
Human b2-adrenergic receptor (2RH1)
Removes glycosylationN187E
cys-bridge
Salt bridge
A282L
233260
C
C
SN
WV
V
D
W
FF YSS
N
YD
Y
N
Y
N DR
W PP
P
T4-Lys
E
F TEVcleavage site
FLAGtag
HA signalsequence
IS
D192
K
F
F
A
B
deletion (365–413)
C
C-term
C-term
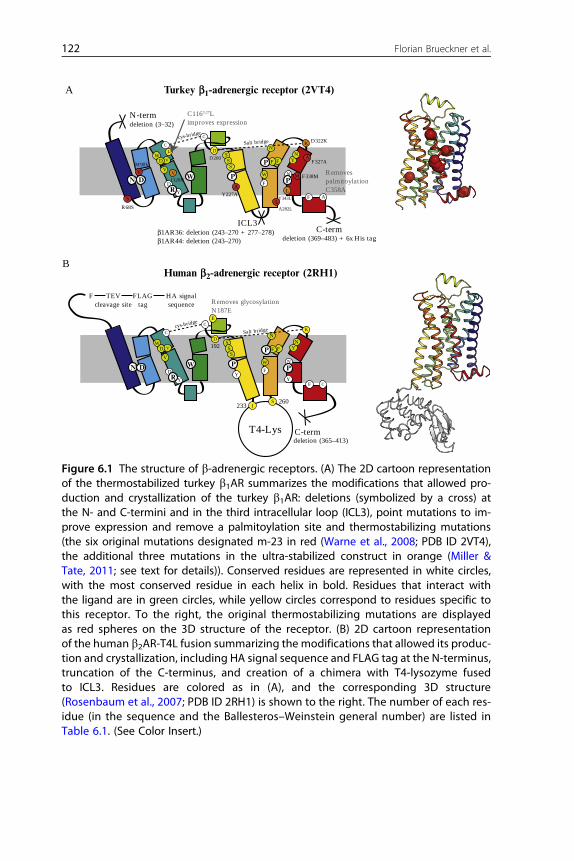
Figure 6.1 The structure of b-adrenergic receptors. (A) The 2D cartoon representationof the thermostabilized turkey b1AR summarizes the modifications that allowed pro-duction and crystallization of the turkey b1AR: deletions (symbolized by a cross) atthe N- and C-termini and in the third intracellular loop (ICL3), point mutations to im-prove expression and remove a palmitoylation site and thermostabilizing mutations(the six original mutations designated m-23 in red (Warne et al., 2008; PDB ID 2VT4),the additional three mutations in the ultra-stabilized construct in orange (Miller &Tate, 2011; see text for details)). Conserved residues are represented in white circles,with the most conserved residue in each helix in bold. Residues that interact withthe ligand are in green circles, while yellow circles correspond to residues specific tothis receptor. To the right, the original thermostabilizing mutations are displayedas red spheres on the 3D structure of the receptor. (B) 2D cartoon representationof the human b2AR-T4L fusion summarizing the modifications that allowed its produc-tion and crystallization, including HA signal sequence and FLAG tag at the N-terminus,truncation of the C-terminus, and creation of a chimera with T4-lysozyme fusedto ICL3. Residues are colored as in (A), and the corresponding 3D structure(Rosenbaum et al., 2007; PDB ID 2RH1) is shown to the right. The number of each res-idue (in the sequence and the Ballesteros–Weinstein general number) are listed inTable 6.1. (See Color Insert.)
122 Florian Brueckner et al.

123Structure of b-Adrenergic Receptors
type, referred to as b1AR-JM3 (Miller & Tate, 2011). A similar approach to
thermostabilization bymutagenesis has also been successfully applied to crys-
tallize the adenosine A2A receptor in an inverse agonist- (Dore et al., 2011)
and agonist-bound conformation (Lebon et al., 2011).
Structural studies of the human b2AR have used strategies different
than thermostabilization by mutagenesis. The first structure was obtained
with a C-terminally truncated b2AR in complex with a Fab fragment
(Rasmussen et al., 2007), albeit only at medium resolution (3.4–3.7 A). A
high-resolution structure could be obtained with a construct where, in
addition to truncating the C-terminus, part of ICL3 was replaced with
the small (18 kDa) soluble protein T4L (Rosenbaum et al., 2007). This chi-
mera had a reduced structural flexibility and improved crystallization prop-
erties by providing additional hydrophilic surface for crystal contacts. Since
this initial success, the T4L-fusion strategy has been successfully applied to
obtain crystal structures of several additional GPCRs (Chien et al., 2010;
Haga et al., 2012; Hanson et al., 2012; Jaakola et al., 2008; Kruse et al.,
2012; Shimamura et al., 2011; Wu et al., 2010). A thermostabilizing muta-
tion (E122W) discovered in b2AR (Roth, Hanson, & Stevens, 2008)
allowed a simplified purification scheme without a ligand-affinity step and
resulted in an increased yield of functionally active receptor (Hanson
et al., 2008). Equivalent mutations in CXCR4 and dopamine D3 receptors
have been applied in the course of their structure determination (Chien
et al., 2010; Wu et al., 2010). Heterogeneous glycosylation in b2AR has
been removed by enzymatic deglycosylation and the PNGaseF-inaccessible
site at asparagine 187 was mutated to glutamate (Rosenbaum et al., 2007).
2.2. Expression of b-adrenergic receptorsThe b1AR was first isolated from turkey erythrocytes (Shorr, Strohsacker,
Lavin, Lefkowitz, & Caron, 1982), and the b2AR from frog erythrocytes
(Shorr, Lefkowitz, & Caron, 1981) and mammalian lung tissue (Benovic,
Shorr, Caron, & Lefkowitz, 1984). Cloning of the genes of turkey b1AR(Yarden et al., 1986) and human b2AR (Kobilka et al., 1987) opened the
way to heterologous overexpression of these proteins of low natural
abundance. For both receptors, the expression levels could be dramatically
increased by using the baculovirus expression system in insect cells (Parker
et al., 1991). The expression of the turkey b1AR was further increased by
receptor engineering as described in the previous section. Functional
expression of the human b2AR was enhanced by adding a cleavable signal
0 50 100 150 2000
5
10
15
3H-DHA concentration (nM)
Spec
ific
lig
and
bind
ing
(pm
ol/m
g)
Bmax = 14.1 ± 0.5 pmol/mg
KD = 19.1 ± 2.5 nM
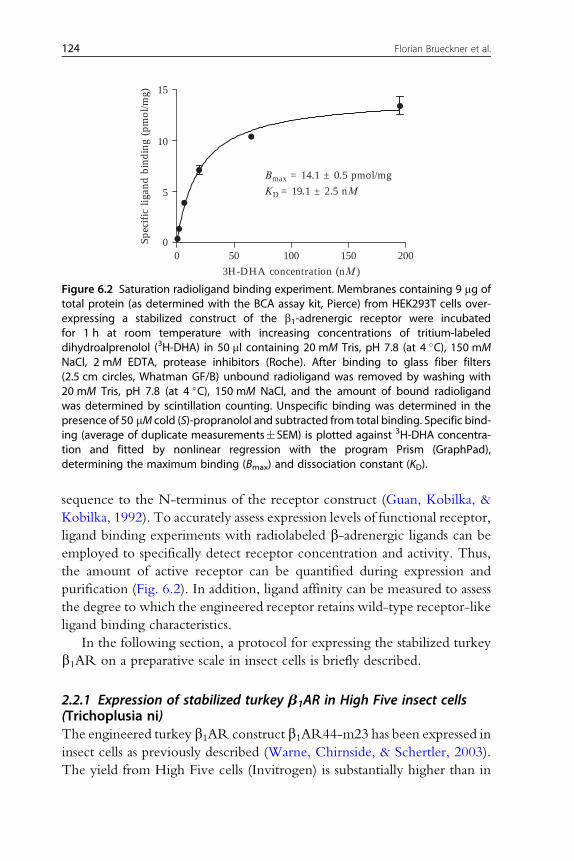
Figure 6.2 Saturation radioligand binding experiment. Membranes containing 9 mg oftotal protein (as determined with the BCA assay kit, Pierce) from HEK293T cells over-expressing a stabilized construct of the b1-adrenergic receptor were incubatedfor 1 h at room temperature with increasing concentrations of tritium-labeleddihydroalprenolol (3H-DHA) in 50 ml containing 20 mM Tris, pH 7.8 (at 4 �C), 150 mMNaCl, 2 mM EDTA, protease inhibitors (Roche). After binding to glass fiber filters(2.5 cm circles, Whatman GF/B) unbound radioligand was removed by washing with20 mM Tris, pH 7.8 (at 4 �C), 150 mM NaCl, and the amount of bound radioligandwas determined by scintillation counting. Unspecific binding was determined in thepresence of 50 mM cold (S)-propranolol and subtracted from total binding. Specific bind-ing (average of duplicate measurements�SEM) is plotted against 3H-DHA concentra-tion and fitted by nonlinear regression with the program Prism (GraphPad),determining the maximum binding (Bmax) and dissociation constant (KD).
124 Florian Brueckner et al.
sequence to the N-terminus of the receptor construct (Guan, Kobilka, &
Kobilka, 1992). To accurately assess expression levels of functional receptor,
ligand binding experiments with radiolabeled b-adrenergic ligands can be
employed to specifically detect receptor concentration and activity. Thus,
the amount of active receptor can be quantified during expression and
purification (Fig. 6.2). In addition, ligand affinity can be measured to assess
the degree to which the engineered receptor retains wild-type receptor-like
ligand binding characteristics.
In the following section, a protocol for expressing the stabilized turkey
b1AR on a preparative scale in insect cells is briefly described.
2.2.1 Expression of stabilized turkey b1AR in High Five insect cells(Trichoplusia ni)The engineered turkey b1AR construct b1AR44-m23 has been expressed in
insect cells as previously described (Warne, Chirnside, & Schertler, 2003).
The yield from High Five cells (Invitrogen) is substantially higher than in

125Structure of b-Adrenergic Receptors
Sf9 cells. Insect cells are grown in suspension culture at 27 �C with shaking
at 150 rpm. High Five cells are cultured in Ex-cell 405 media (SAFC Bio-
sciences) supplemented with 5% (v/v) FBS (Gibco) and 1% CD lipid
concentrate (Gibco). Expression is performed in 4–8 L culture volume with
maximum 500 mL per 2-L flask. It has been observed that the expression
level of b1AR decreases when the High Five cells have been kept in culture
for several weeks (Tony Warne, personal communication).
1. Produce high titer virus stocks of 2–4�108 pfu/mL in Sf9 cells.
2. Grow High Five cells to 1.5–2.0�106 cells/mL.
3. Dilute with an equal volume of fresh prewarmed media.
4. Add 20 mL virus stock per liter of diluted cell suspension (corresponds to
a multiplicity of infection of 5–10).
5. From this point on, the cell density should not increase substantially if the
virus titer was high enough.
6. Harvest the cells 40–48 h after infection by centrifugation at 2500� g for
5 min.
7. Resuspend in a total volume of 25 mL per liter of culture with 20 mM
Tris, pH 7.8, 1 mM EDTA, 1�Complete EDTA-free protease inhib-
itor cocktail tablets (Roche).
8. Freeze the cell suspension in liquid nitrogen and store at �80 �C. One
freeze-thaw cycle is necessary in any case to break the cells.
2.3. Purification of b-adrenergic receptorsPurification of bARs from native material was enabled by the development
of a ligand-affinity chromatography resin with the antagonist alprenolol
covalently linked to sepharose (Fig. 6.3) (Caron et al., 1979). Using a
combination of alprenolol ligand-affinity chromatography (ALAC) and
size exclusion chromatography (SEC) the turkey b1AR had been purified
>15,000-fold from turkey erythrocytes (Shorr et al., 1982). The engineered
turkey b1AR expressed in insect cells for structural studies is routinely
purified with immobilizedmetal-affinity chromatography (IMAC) followed
by ALAC (Warne et al., 2009) and is described in more detail below. The
purification of human b2AR for crystallization differed slightly from the
regime used for b1AR purification. Instead of IMAC, an N-terminal M1
Flag tag was employed for first-pass isolation of receptor from the solubilized
membrane fraction, followed by ALAC to isolate a highly purified func-
tional receptor population (Rosenbaum et al., 2007). A stabilized variant
of the engineered human b2AR-T4L construct was subsequently identified
Sepharose 4B
OO S
OH
O
N
HO
H
O
OH
Linker Alprenolol
Alprenolol ligand-affinitychromatography (ALAC) resin
IMACelution
kDa
Marker ALACLoad
ALAC elution fraction
20
25
37
50
75
11 12 13 14 15
0
20
40
60
80
100
120
Abs
orba
nce
at 2
80 n
m (m
Au)
0.0 10.0 20.0 30.0 40.0 50.0Elution volume (ml)
1 2 3 4 5 7 9 11 13 15 17 19 21
ALAC load ALAC wash ALAC elution
Fractionnumber
A
B
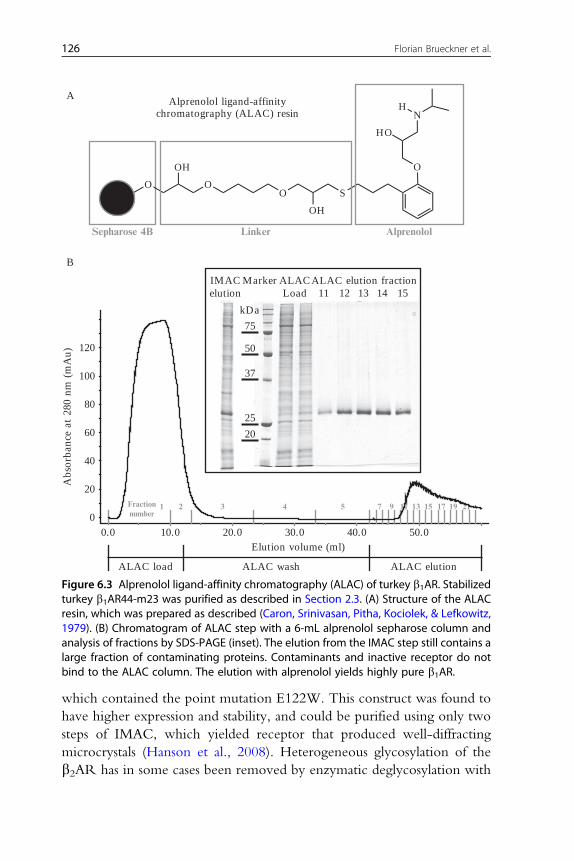
Figure 6.3 Alprenolol ligand-affinity chromatography (ALAC) of turkey b1AR. Stabilizedturkey b1AR44-m23 was purified as described in Section 2.3. (A) Structure of the ALACresin, which was prepared as described (Caron, Srinivasan, Pitha, Kociolek, & Lefkowitz,1979). (B) Chromatogram of ALAC step with a 6-mL alprenolol sepharose column andanalysis of fractions by SDS-PAGE (inset). The elution from the IMAC step still contains alarge fraction of contaminating proteins. Contaminants and inactive receptor do notbind to the ALAC column. The elution with alprenolol yields highly pure b1AR.
126 Florian Brueckner et al.
which contained the point mutation E122W. This construct was found to
have higher expression and stability, and could be purified using only two
steps of IMAC, which yielded receptor that produced well-diffracting
microcrystals (Hanson et al., 2008). Heterogeneous glycosylation of the
b2AR has in some cases been removed by enzymatic deglycosylation with

127Structure of b-Adrenergic Receptors
PNGaseF. More details about purification strategies used for the individual
structure determinations of b1AR and b2AR are included in Tables 6.2 and
6.3, respectively.
Below, a detailed preparative purification scheme of stabilized turkey
b1AR is provided, including membrane preparation, solubilization, IMAC,
and ALAC.
2.3.1 Membrane preparation from High Five cells for b1AR purificationPreparation of membranes from insect cells is an initial crude purification
step, which breaks the cells and removes soluble and loosely membrane-
associated biomolecules. All membrane preparation steps are performed at
4 �C or on ice and in presence of protease inhibitors (Complete EDTA-free,
Roche).
1. Thaw cell suspension from 4 L culture and resuspend by stirring in a final
volume of 360 mL with 20 mM Tris–HCl, pH 8.0, 1 mM EDTA,
0.5�protease inhibitors (TE buffer).
2. Centrifuge at 150,000� g for 2 h
3. Remove the supernatant including a floating white layer that may form
4. Resuspend the membrane pellets in a final volume of 240 mL in TE
buffer. This can be conveniently achieved with an electric disperser
(e.g., ULTRA-TURRAX, IKA) at low speed and with the shortest pos-
sible use.
5. Centrifuge at 150,000� g for 1.5 h.
6. Resuspend the membrane pellets in a final volume of 90 mL 20 mM
Tris–HCl, pH 7.8 (e.g., with an electric disperser). This will result in
a total protein concentration of approximately 10–20 mg/mL (as deter-
mined by the BCA assay)
7. Freeze in aliquots of 30 mL (corresponding to 1.3 L of the original cul-
ture) in liquid nitrogen and store at �80 �C
2.3.2 Solubilization of stabilized turkey b1ARMembrane lipids and integral membrane proteins including turkey b1ARcan be solubilized with the mild detergent decylmaltoside (DM). All steps
are performed at 4 �C or on ice and in the presence of protease inhibitors
(Complete EDTA-free, Roche).
1. Thawmembranes from an aliquot corresponding to 1.3 L of the original
culture (30 mL suspension in this example) and dilute to 5–10 mg/mL
total protein concentration (approx. 80 mL final volume) in 20 mM
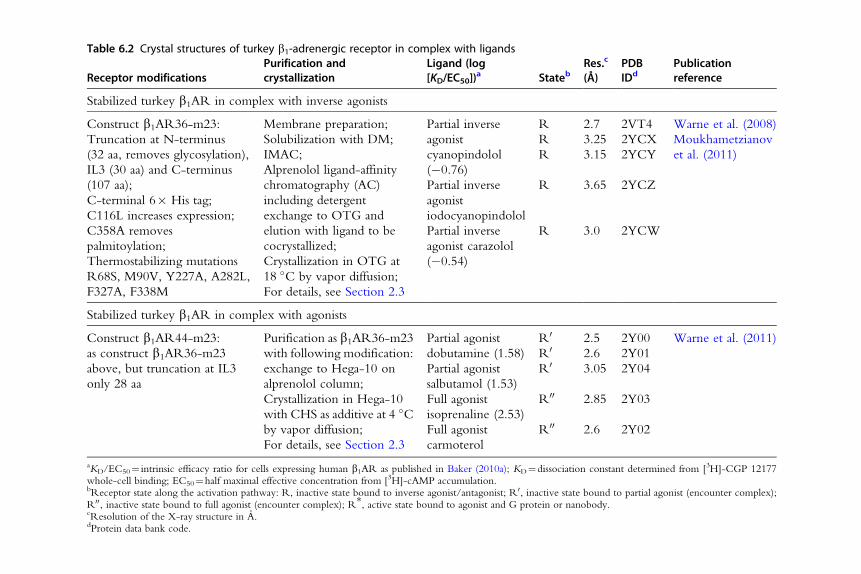
Table 6.2 Crystal structures of turkey b1-adrenergic receptor in complex with ligands
Receptor modificationsPurification andcrystallization
Ligand (log[KD/EC50])
a StatebRes.c
(Å)PDBIDd
Publicationreference
Stabilized turkey b1AR in complex with inverse agonists
Construct b1AR36-m23:
Truncation at N-terminus
(32 aa, removes glycosylation),
IL3 (30 aa) and C-terminus
(107 aa);
C-terminal 6� His tag;
C116L increases expression;
C358A removes
palmitoylation;
Thermostabilizing mutations
R68S, M90V, Y227A, A282L,
F327A, F338M
Membrane preparation;
Solubilization with DM;
IMAC;
Alprenolol ligand-affinity
chromatography (AC)
including detergent
exchange to OTG and
elution with ligand to be
cocrystallized;
Crystallization in OTG at
18 �C by vapor diffusion;
For details, see Section 2.3
Partial inverse
agonist
cyanopindolol
(�0.76)
R 2.7 2VT4 Warne et al. (2008)
R 3.25 2YCX Moukhametzianov
et al. (2011)R 3.15 2YCY
Partial inverse
agonist
iodocyanopindolol
R 3.65 2YCZ
Partial inverse
agonist carazolol
(�0.54)
R 3.0 2YCW
Stabilized turkey b1AR in complex with agonists
Construct b1AR44-m23:
as construct b1AR36-m23
above, but truncation at IL3
only 28 aa
Purification as b1AR36-m23
with following modification:
exchange to Hega-10 on
alprenolol column;
Crystallization in Hega-10
with CHS as additive at 4 �Cby vapor diffusion;
For details, see Section 2.3
Partial agonist
dobutamine (1.58)
R0 2.5 2Y00 Warne et al. (2011)
R0 2.6 2Y01
Partial agonist
salbutamol (1.53)
R0 3.05 2Y04
Full agonist
isoprenaline (2.53)
R00 2.85 2Y03
Full agonist
carmoterol
R00 2.6 2Y02
aKD/EC50¼ intrinsic efficacy ratio for cells expressing human b1AR as published in Baker (2010a); KD¼dissociation constant determined from [3H]-CGP 12177whole-cell binding; EC50¼half maximal effective concentration from [3H]-cAMP accumulation.bReceptor state along the activation pathway: R, inactive state bound to inverse agonist/antagonist; R0, inactive state bound to partial agonist (encounter complex);R00, inactive state bound to full agonist (encounter complex); R*, active state bound to agonist and G protein or nanobody.cResolution of the X-ray structure in A.dProtein data bank code.
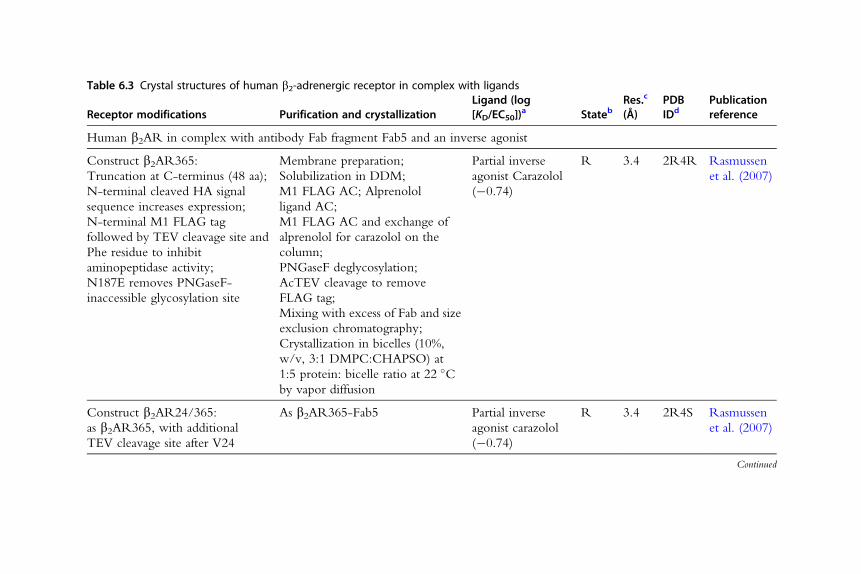
Table 6.3 Crystal structures of human b2-adrenergic receptor in complex with ligands
Receptor modifications Purification and crystallizationLigand (log[KD/EC50])
a StatebRes.c
(Å)PDBIDd
Publicationreference
Human b2AR in complex with antibody Fab fragment Fab5 and an inverse agonist
Construct b2AR365:
Truncation at C-terminus (48 aa);
N-terminal cleaved HA signal
sequence increases expression;
N-terminal M1 FLAG tag
followed by TEV cleavage site and
Phe residue to inhibit
aminopeptidase activity;
N187E removes PNGaseF-
inaccessible glycosylation site
Membrane preparation;
Solubilization in DDM;
M1 FLAG AC; Alprenolol
ligand AC;
M1 FLAG AC and exchange of
alprenolol for carazolol on the
column;
PNGaseF deglycosylation;
AcTEV cleavage to remove
FLAG tag;
Mixing with excess of Fab and size
exclusion chromatography;
Crystallization in bicelles (10%,
w/v, 3:1 DMPC:CHAPSO) at
1:5 protein: bicelle ratio at 22 �Cby vapor diffusion
Partial inverse
agonist Carazolol
(�0.74)
R 3.4 2R4R Rasmussen
et al. (2007)
Construct b2AR24/365:
as b2AR365, with additional
TEV cleavage site after V24
As b2AR365-Fab5 Partial inverse
agonist carazolol
(�0.74)
R 3.4 2R4S Rasmussen
et al. (2007)
Continued
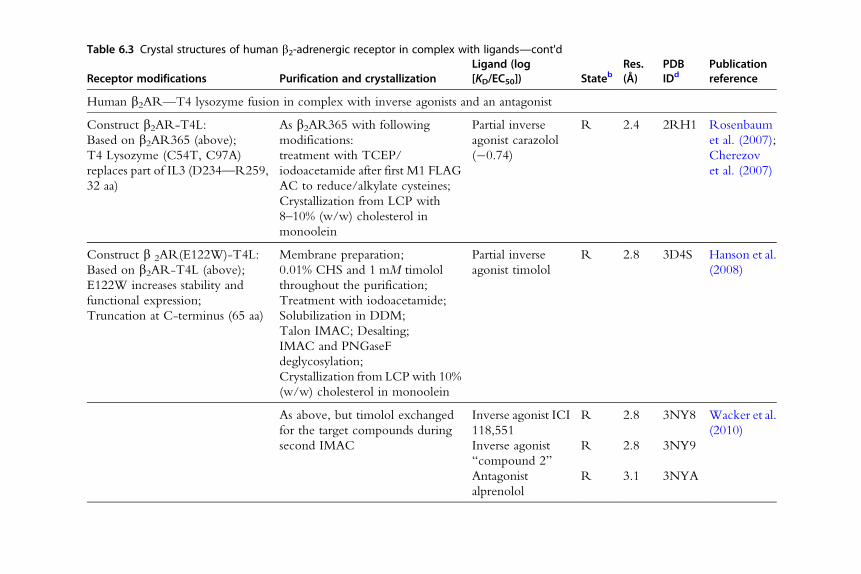
Table 6.3 Crystal structures of human b2-adrenergic receptor in complex with ligands—cont'd
Receptor modifications Purification and crystallizationLigand (log[KD/EC50]) Stateb
Res.(Å)
PDBIDd
Publicationreference
Human b2AR—T4 lysozyme fusion in complex with inverse agonists and an antagonist
Construct b2AR-T4L:
Based on b2AR365 (above);
T4 Lysozyme (C54T, C97A)
replaces part of IL3 (D234—R259,
32 aa)
As b2AR365 with following
modifications:
treatment with TCEP/
iodoacetamide after first M1 FLAG
AC to reduce/alkylate cysteines;
Crystallization from LCP with
8–10% (w/w) cholesterol in
monoolein
Partial inverse
agonist carazolol
(�0.74)
R 2.4 2RH1 Rosenbaum
et al. (2007);
Cherezov
et al. (2007)
Construct b 2AR(E122W)-T4L:
Based on b2AR-T4L (above);
E122W increases stability and
functional expression;
Truncation at C-terminus (65 aa)
Membrane preparation;
0.01% CHS and 1 mM timolol
throughout the purification;
Treatment with iodoacetamide;
Solubilization in DDM;
Talon IMAC; Desalting;
IMAC and PNGaseF
deglycosylation;
Crystallization from LCPwith 10%
(w/w) cholesterol in monoolein
Partial inverse
agonist timolol
R 2.8 3D4S Hanson et al.
(2008)
As above, but timolol exchanged
for the target compounds during
second IMAC
Inverse agonist ICI
118,551
R 2.8 3NY8 Wacker et al.
(2010)
Inverse agonist
“compound 2”
R 2.8 3NY9
Antagonist
alprenolol
R 3.1 3NYA

Human b2AR—T4L fusion in complex with a covalently bound agonist
Construct b2AR(H93C)-T4L:
Based on b2AR-T4L;
TEV cleavage site after D23;
H93C for covalent ligand binding;
Truncation at C-terminus (65 aa);
C-terminal 6� His tag
As for b2AR365, but with the
following modifications:
Exchange of ligand to FAUC50
and detergent to MNG-3
amphiphile in the second M1
FLAG AC step;
Alkylation with iodoacetamide
after second M1 FLAC AC step;
Cleavage of N-terminal 23 aa with
AcTEV; IMAC;
Crystallization from LCPwith 10%
(w/w) cholesterol in monoolein
Covalently bound
agonist FAUC50
R 3.5 3PDS Rosenbaum
et al. (2011)
Human b2AR—T4L fusion in complex with G protein-mimicking nanobody Nb80
Construct b2AR-T4L:
as described above
As for b2AR365 but with
following modifications:
Exchange of ligand to BI-167107
and detergent to MNG-3 in the
second M1 FLAG AC step;
PNGaseF deglycosylation;
Mix with nanobody Nb80;
Crystallization from LCPwith 10%
(w/w) cholesterol in monoolein
Agonist
BI-167107
R* 3.5 3POG Rasmussen,
Choi, et al.
(2011)
Continued
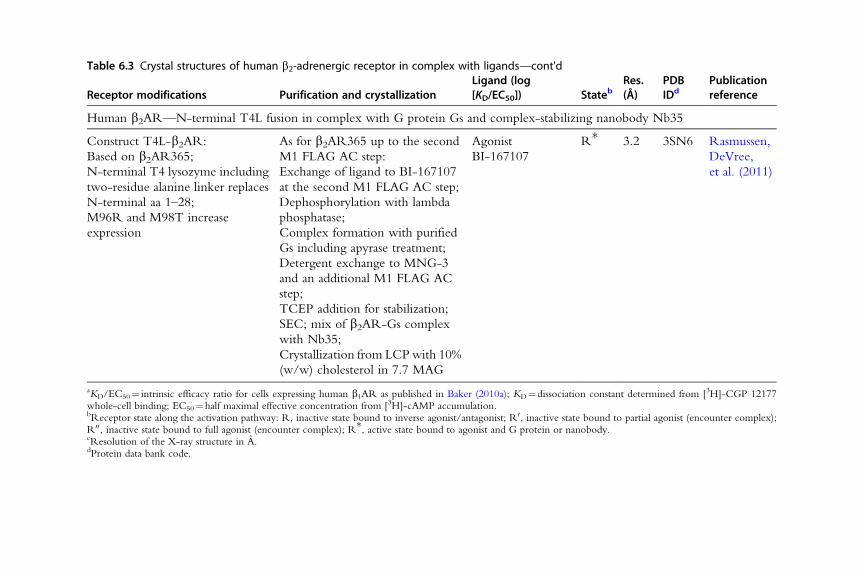
Table 6.3 Crystal structures of human b2-adrenergic receptor in complex with ligands—cont'd
Receptor modifications Purification and crystallizationLigand (log[KD/EC50]) Stateb
Res.(Å)
PDBIDd
Publicationreference
Human b2AR—N-terminal T4L fusion in complex with G protein Gs and complex-stabilizing nanobody Nb35
Construct T4L-b2AR:
Based on b2AR365;
N-terminal T4 lysozyme including
two-residue alanine linker replaces
N-terminal aa 1–28;
M96R and M98T increase
expression
As for b2AR365 up to the second
M1 FLAG AC step:
Exchange of ligand to BI-167107
at the second M1 FLAG AC step;
Dephosphorylation with lambda
phosphatase;
Complex formation with purified
Gs including apyrase treatment;
Detergent exchange to MNG-3
and an additional M1 FLAG AC
step;
TCEP addition for stabilization;
SEC; mix of b2AR-Gs complex
with Nb35;
Crystallization from LCPwith 10%
(w/w) cholesterol in 7.7 MAG
Agonist
BI-167107
R* 3.2 3SN6 Rasmussen,
DeVree,
et al. (2011)
aKD/EC50¼ intrinsic efficacy ratio for cells expressing human b1AR as published in Baker (2010a); KD¼dissociation constant determined from [3H]-CGP 12177whole-cell binding; EC50¼half maximal effective concentration from [3H]-cAMP accumulation.bReceptor state along the activation pathway: R, inactive state bound to inverse agonist/antagonist; R0, inactive state bound to partial agonist (encounter complex);R00, inactive state bound to full agonist (encounter complex); R*, active state bound to agonist and G protein or nanobody.cResolution of the X-ray structure in A.dProtein data bank code.

133Structure of b-Adrenergic Receptors
Tris–HCl, pH 7.8, 400 mM NaCl, 2.5 mM imidazole and 1�protease
inhibitors (final concentrations).
2. Add DM to a final concentration of 1.5% (w/v) from a 20% (w/v) stock
solution and stir briefly.
3. Centrifuge at 150,000� g for 1 h to remove insoluble material.
2.3.3 Immobilized metal-affinity chromatographyThe turkey b1AR can be substantially enriched by IMAC through a
C-terminal 6�His tag. All steps are performed at 4 �C or on ice and in pres-
ence of protease inhibitors (Complete EDTA-free, Roche). Run the IMAC
column at a flow rate of 2 mL/min.
1. Add6 mLofNi2þ-loadedChelating Sepharose Fast Flow (GEHealthcare)
equilibrated in IMAC buffer A (20 mM Tris, pH 7.8, 350 mM NaCl,
2.5 mM imidazole, 0.2�protease inhibitors, 0.15% (w/v) DM) to the su-
pernatant of the previous step and mix gently for 1–2 h (e.g., in 50 mL
tubes on a roller).
2. Wash resin with 60 mL IMAC buffer A.
3. Transfer and pack IMAC resin to a column which already contains 4 mL
of the same type of resin equilibrated in IMAC buffer A.
4. Wash column with 20 mL IMAC buffer A
5. Apply a linear gradient 0–10% IMAC buffer B (IMAC buffer A con-
taining 250 mM Imidazole) in 50 mL
6. Wash with 125 mL 10% IMAC buffer B
7. Apply a linear gradient 10–100% IMACbuffer B in 20 mL and then con-
tinue washing with 100% IMAC buffer B to elute His-tagged b1AR8. Pool elution fractions as judged by UV monitoring (approx. 50 mL) and
analyze by SDS-PAGE. A band for b1AR should clearly be visible at an
apparent molecular weight slightly above 25 kDa (based on soluble
protein markers), but many bands of contaminating proteins will usually
also still be present at this step (Fig. 6.3B).
2.3.4 Alprenolol ligand-affinity chromatographyIn the final purification step, only correctly folded b1AR binds to the
alprenolol ligand-affinity resin, while incorrectly folded receptor and most
contaminating proteins are not retained, resulting in a highly pure prepara-
tion (Fig. 6.3). The detergent can be exchanged during this step, and the
receptor can be eluted with a ligand of choice from the column. All steps
are performed at 4 �C or on ice.

134 Florian Brueckner et al.
1. Equilibrate a 6 mL alprenolol sepharose columnwith alprenolol sepharose
wash buffer (20 mM Tris–HCl, pH 7.8, 350 mM NaCl, 1 mM EDTA,
0.1% (w/v) DM).
2. Load the IMAC elution fractions at 0.2 mL/min.
3. Wash at 0.2 mL/min with 60 mL alprenolol sepharose wash buffer
containing the detergent of choice. For vapor diffusion crystallization,
exchange the detergent to 0.35% (w/v) HEGA-10 (for crystalli-
zation with stabilizing antagonists/inverse agonists, 0.35% (w/v)
octylthioglucoside (OTG) will also work).
4. Elute receptor at 0.3 mL/min with alprenolol sepharose elution buffer
(20 mM Tris–HCl, pH 7.8, 350 mM NaCl, 0.2 mM EDTA, detergent)
supplemented with a competing ligand of choice, for example, 30 mMhigh-affinity antagonist/inverse agonist or 0.2 mM agonist.
5. Analyze elution fractions by SDS-PAGE as judged by UV absorption
monitoring or using a protein assay (e.g., Bradford, BCA). Note that
many b-adrenergic ligands absorb at 280 nm and therefore the elution
buffer will result in an elevated baseline in the UV-monitored chromato-
gram. A major band should be visible slightly above 25 kDa for the pu-
rified b1AR, while most of the contaminating proteins should have been
removed by now (Fig. 6.3B). Pool elution fractions containing pure
b1AR.
6. The thermal stability of the purified receptor can be determined by
a thermal shift assay with the fluorescent dye CPM (Alexandrov,
Mileni, Chien, Hanson, & Stevens, 2008). In contrast to a radioligand
binding assay, measurement of the thermal shift allows to compare the
stability of the purified receptor with different bound ligands without
the requirement of having each of them radiolabeled (Fig. 6.4).
2.4. Crystallization of b-adrenergic receptorsMembrane proteins can be crystallized by vapor diffusion methods directly
from detergent solution, which often yields type II crystals, in which protein
molecules form hydrophilic crystal contacts, while detergent is bound to the
hydrophobic protein surface (Michel, 1983). Alternatively, purified mem-
brane proteins can be reconstituted into a lipid bilayer forming a lipidic cubic
phase (LCP), and subsequently crystallized with a batch method (Caffrey,
2008). In the LCP method, type I crystals are usually obtained, which are
stacked sheets of protein in a lipid bilayer. Here, the protein molecules
can form lateral hydrophobic crystal contacts in the lipid bilayer and

30 40 50 60 70 80200
300
400
500
Temperature (°C)
Flu
ores
cenc
e in
tens
ity
(a.u
.)
(a) 1 μM alprenolol(b) 30 μM alprenolol(c) 10 μM cyanopindolol
a
b
c
B
ON
CH3
CH3
CH3
CH3
CH3
O
N
O
O
ON
CH3
O
N
O
O
S
Protein
+ HS-protein(exposed cysteine)
pH 6–8CPM Fluorescent CPM-thiol adduct
A
lexcitation = 387 nmlemission = 463 nm
Figure 6.4 Fluorescent thermal stability assay of the turkey b1AR. (A) Thermal unfoldingof proteins exposes free cysteine residues, previously buried inside the protein core. Thisprocess canbedetected in thepresenceofN-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), which form strongly fluorescent thiol adducts with free cyste-ines. (B) Thermal denaturation of stabilized turkey b1AR44-m23 in presence of (a) 1 mMalprenolol, (b) 30 mM alprenolol, and (c) 10 mM cyanopindolol monitored by measuringthe fluorescence of the CPM-thiol adduct at 463 nm. The data (gray curves) clearly showan increase of fluorescence with increasing temperature. The drop at higher tempera-tures is most likely due to protein precipitation. Data were fitted by nonlinear regressionwith Prism (GraphPad), depicted by the black curves. The transitionmidpoints determineapparent melting temperatures of (a) 54.1 �C, (b) 58.8 �C, and (c) 66.4 �C. Thus, the sta-bilityof theb1AR ishighlydependenton theconcentrationandthe typeof ligandpresent.
135Structure of b-Adrenergic Receptors
hydrophilic contacts between sheets. While the formation of type II crystals
requires small-micelle detergents in order to expose enough hydrophilic
protein surface for the formation of crystal contacts, larger and less
destabilizing detergents can be used for the LCP method, since detergent
is replaced by lipid during reconstitution and therefore does not interfere
by masking potential crystal contact areas.

136 Florian Brueckner et al.
To date, all reported crystal structures of the turkey b1AR have been
obtained by classical vapor diffusion methods from receptor in detergent
micelles. The crystals do not exactly correspond to type I or type II crystals,
since hydrophilic as well as hydrophobic crystal contacts are present (Warne
et al., 2008). To facilitate lattice formation, the choice of detergent used in
crystallization screens can have an enormous impact on the success rate for
obtaining well-diffracting crystals. Typically, shorter-chain detergents that
form more compact micelles are less likely to interfere with crystal packing.
However, shorter-chain detergents tend to be less stabilizing than longer-
chain detergents. Thus, the engineering of a highly stable receptor that
can withstand the use of shorter detergents is invaluable to increase the
chances for crystallization success.
The above-described thermostabilization of the receptor and the use of
the strongly stabilizing ligand cyanopindolol also increased the stability in
harsher small-micelle detergents (Warne et al., 2009; Fig. 6.4). As described
above, the turkey b1AR is initially extracted and purified gently using DM
and is exchanged into shorter-chain harsher detergents in the final purification
step. The first structure of the b1AR in complex with cyanopindolol (Warne
et al., 2008) and also a structure with iodocyanopindolol (Moukhametzianov
et al., 2011) were obtained in OTG. For the cocrystallization with less
stabilizing ligands, including agonists, the milder detergent HEGA-10
was successfully used in combination with addition of cholesteryl
hemisuccinate (CHS) and a lower crystallization temperature (Warne et al.,
2011). Another important factor for the success in crystallization was the
removal of flexible or unstructured regions in ICL3 and the C-terminus,
since crystals could not be obtained without these truncations (Warne
et al., 2009).
Different methods have been used for crystallizing the human b2AR. In
one case, a Fab fragment recognizing the ICL3 region forms a stable com-
plex with the receptor, thereby providing additional hydrophilic surface for
crystal contacts. The complex was cocrystallized by vapor diffusion and
allowed the first nonrhodopsin GPCR crystal structure to be determined,
albeit only at medium resolution (Rasmussen et al., 2007). In another case,
replacing part of ICL3 with T4L reduced conformational heterogeneity and
also provided additional hydrophilic surface for crystal contacts. Crystalliza-
tion of the b2AR ICL3-T4L fusion from LCP yielded crystals diffracting to
high resolution (Rosenbaum et al., 2007). The LCP method was also used
for cocrystallizing the b2AR-T4L fusion with a G protein-mimicking
nanobody (Rasmussen, Choi, et al., 2011) and for cocrystallizing an

137Structure of b-Adrenergic Receptors
N-terminal T4L fusion in complex with the G protein Gs and a complex-
stabilizing nanobody (Rasmussen, DeVree, et al., 2011).
The following section describes a protocol for cocrystallizing the stabilized
turkey b1ARwith orthosteric ligands as also described inWarne et al. (2011).
2.4.1 Cocrystallization of the stabilized turkey b1-adrenergic receptorwith inverse agonists, antagonists, and agonistsThepurified turkeyb1ARcanbecocrystallizedwithmanydifferentorthosteric
ligands by vapor diffusion using the detergent HEGA-10 and CHS as an addi-
tive. All steps are performed at 4 �C or on ice.
1. Concentrate the ligand-affinity eluate using centrifugal ultrafiltration de-
vices with 50,000 molecular weight cutoff to a final protein concentra-
tion of 15–20 mg/mL. During concentration, exchange the buffer to
10 mM Tris, pH 7.7, 100 mM NaCl, 0.1 mM EDTA, 0.35% (w/v)
HEGA-10, 1.0 mM inverse agonist, antagonist, or agonist.
2. Addition of 0.45–1.8 mg/mL CHS and an increase in the HEGA-10
concentration to 0.5–0.65% (w/v) may improve crystallization, partic-
ularly for cocrystallization with agonists. For this purpose, a 10 mg/
mL CHS stock solution in 2% (w/v) HEGA-10 can be prepared.
3. Centrifuge receptor preparation at 130,000� g for 10 min to remove
aggregates.
4. Set up sitting drop vapor diffusion crystallization experiments mixing
200 nL of protein solution and 200 nL precipitant solution containing
21–32% PEG 600 and 0.1 M bicine, pH 9.0.
5. Incubate at 4 �C until the usually needle-shaped crystals reach a size of at
least 5–10 mm in the shortest dimension.
2.5. Crystallography of b-adrenergic receptorsCrystals of membrane proteins including GPCRs are usually small in size and
often display inhomogeneous diffraction properties. The technological
advances in microcrystallography and the development of dedicated micro-
focus beamlines at synchrotrons generating X-ray beams of micrometer-sized
focal spot and high flux substantially improve quality of X-ray diffraction data
collected from weakly diffracting small or inhomogeneous crystals (Evans,
Axford, Waterman, & Owen, 2011; Riekel, Burghammer, & Schertler,
2005). The small beam diameter allows matching the beam to the crystal
size, which reduces background scattering and radiation damage and
allows selecting regions of an inhomogeneous crystal with more favorable

138 Florian Brueckner et al.
diffraction properties. From crystals that are larger in one or two dimensions,
diffraction data can be collected from different positions, which maximizes
data completeness and at the same timeminimizes radiation damage. The high
flux of microfocus beamlines increases the signal-to-noise ratio of weakly
diffracting crystals.
The usefulness of microcrystallography for GPCR crystals has first been
demonstrated with crystals of thermostabilized recombinant rhodopsin. A
complete dataset was collected with a 5-mm beam from several positions of
a single needle-shaped crystal (Standfuss et al., 2007). The human b2AR in
complex with a Fab fragment produced thin weakly diffracting crystals. Only
with microbeams could data with favorable signal-to-noise ratio be obtained
from several positions on single crystals (Rasmussen et al., 2007). A high-
resolution dataset of b2AR was obtained by merging data collected with a
10-mm beam from 27 microcrystals grown in LCP (Cherezov et al., 2007).
A similar strategy was used for the b2AR-Gs complex (Rasmussen,
DeVree, et al., 2011). For the structure determination of b1AR and its many
ligand complexes, microfocus beams were essential for locating the best
diffracting parts of single crystals, as well as allowing several wedges to be
collected from different positions (Warne et al., 2008; 2011).
The first GPCR X-ray crystal structure, that of bovine rhodopsin, was
determined by multiwavelength anomalous dispersion from a mercury-
derivatized crystal (Palczewski et al., 2000). All subsequent GPCR structure
determinations, including those of bARs, could be solved by molecular re-
placement, since the overall structure of GPCRs is sufficiently similar.
Below, a general procedure for X-ray diffraction data collection of b1ARcrystals is described and an example for the crystallographic structure deter-
mination of a b1AR–ligand complex from our lab is presented (Fig. 6.5).
2.5.1 X-ray diffraction data collection of turkey b1-adrenergic receptor1. Soak the crystals briefly in synthetic mother liquor containing an in-
creased PEG600 concentration of 50–70% (w/v) before cryocooling
in liquid nitrogen. This apparently improves the diffraction properties
by dehydrating the crystal but also introduces non-isomorphism.
2. Using a microfocus synchrotron beamline, test the diffraction properties
of different parts of the crystal.
3. Collect diffraction data from the best diffracting parts of the crystal.
4. So far, various different crystal forms of b1AR crystals have been ob-
served in the space groups P1,C2, and P21. However, similar antiparallel
dimers of the receptor are observed in all of the crystal forms.
139Structure of b-Adrenergic Receptors
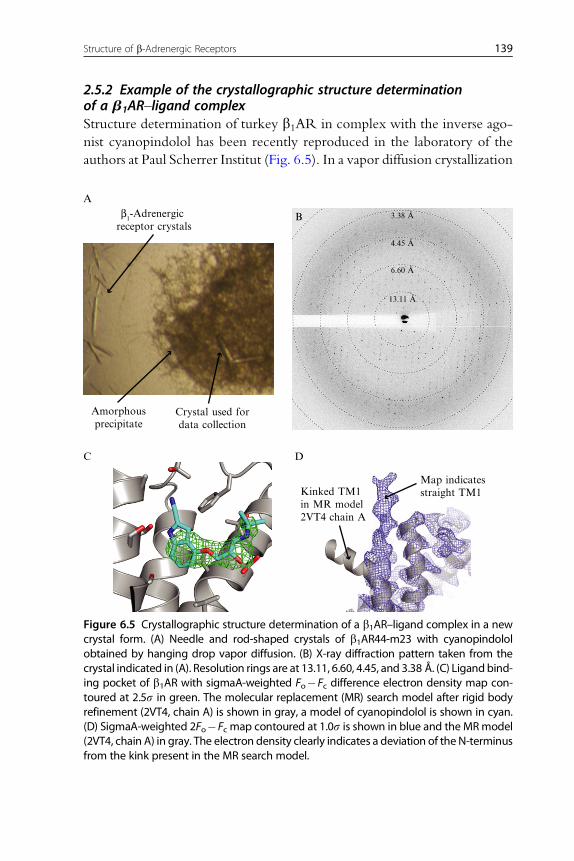
2.5.2 Example of the crystallographic structure determinationof a b1AR–ligand complexStructure determination of turkey b1AR in complex with the inverse ago-
nist cyanopindolol has been recently reproduced in the laboratory of the
authors at Paul Scherrer Institut (Fig. 6.5). In a vapor diffusion crystallization
A
Bβ1-Adrenergic receptor crystals
Amorphousprecipitate
Crystal used fordata collection
C D
Kinked TM1in MR model2VT4 chain A
Map indicatesstraight TM1
3.38 Å
4.45 Å
6.60 Å
13.11 Å
Figure 6.5 Crystallographic structure determination of a b1AR–ligand complex in a newcrystal form. (A) Needle and rod-shaped crystals of b1AR44-m23 with cyanopindololobtained by hanging drop vapor diffusion. (B) X-ray diffraction pattern taken from thecrystal indicated in (A). Resolution rings are at 13.11, 6.60, 4.45, and 3.38 Å. (C) Ligandbind-ing pocket of b1AR with sigmaA-weighted Fo�Fc difference electron density map con-toured at 2.5s in green. The molecular replacement (MR) search model after rigid bodyrefinement (2VT4, chain A) is shown in gray, a model of cyanopindolol is shown in cyan.(D) SigmaA-weighted 2Fo�Fc map contoured at 1.0s is shown in blue and the MRmodel(2VT4, chain A) in gray. The electron density clearly indicates a deviation of the N-terminusfrom the kink present in the MR search model.

140 Florian Brueckner et al.
experiment at 22 �C, 1 mL of a 3 mg/mL protein solution (measured by the
Bradford assay) in 10 mMTris–HCl, pH 8.0, at 4 �C, 50 mMNaCl, 0.1 mM
EDTA, 0.35% OTG, and 1 mM cyanopindolol was mixed with 1 mL reser-
voir solution containing 0.1 M ADA, pH 7.5, and 32% (v/v) PEG 600. The
crystal that was used for structure determination was harvested after 8 days
and had a size of approximately 200�50�20 mm (Fig. 6.5A). It was trans-
ferred to a drop with the same conditions as the crystallization drop, but with
a higher PEG 600 concentration of 55% (v/v). After 5 min the crystal was
cryocooled by plunging into liquid nitrogen. The diffraction quality of 25
crystals was tested at the microfocus diffractometer MD2 at beamline S06SA
of the Swiss Light Source with a focused spot size of 25�5 mm. The best
crystal gave rise to visible spots to about 3.5 A (Fig. 6.5B). A complete
dataset was collected from seven different positions along the crystal in order
to minimize radiation damage. It consists of 402 0.5�-oscillation images,
each exposed 1.0 s with X-rays of 12.4 keV (1.0 A) and at a flux of approx-
imately 1�1012 photons/s. Good data statistics were obtained to 3.8 A res-
olution (Table 6.4). A molecular replacement solution was obtained with
Phaser using the turkey b1AR without ligand (PDB ID 2VT4, monomer
A) as a search model. Four molecules were found in the asymmetric unit
Table 6.4 X-ray data collection statistics of turkey b1-adrenergic receptor in complexwith cyanopindolol in a new crystal form
Space group P21
Unit cell a, b, c (A) 113, 56, 141
Unit cell b (�) 96
Resolution (A) 50–3.8 (4.0–3.8)a
Rmeas (%) 24.8 (74.7)a
I/s 4.6 (2.1)a
Completeness (%) 98.0 (97.2)a
Multiplicity 3.7 (3.7)a
Average mosaicity (�) 0.99
Solvent content (%) 60.2
Number of protein molecules in asymmetric unit 4
aValues for highest resolution shell are given in brackets.

141Structure of b-Adrenergic Receptors
of a new crystal form in space group P21. After rigid body refinement with
Refmac5, a strong feature was observed in the Fo�Fc difference electron
density map in the orthosteric ligand binding pocket, which is consistent
with the presence of the ligand cyanopindolol (Fig. 6.5C). Although the
molecular replacement model has a kink in the N-terminus of TM1 (pre-
sumably due to a crystal packing artifact), the 2Fo�Fc electron density
clearly indicates that all four molecules in the new crystal form have a straight
conformation of TM1 (Fig. 6.5D).
3. LESSONS FROM THE STRUCTURES OF b-ADRENERGICRECEPTORS
Different strategies have been successful in overcoming the hurdles to-
ward the structures of bARs. Inactive states of the human b2AR have been
obtained in complex with a Fab fragment and in form of T4L fusion chi-
meras. Cocrystallization in complex with nanobodies and in complex with
the G protein Gs has allowed obtaining structures of the active state. In con-
trast, the turkey b1AR has been stabilized by mutagenesis in the inactive
state, which allowed cocrystallization with agonists. The sophisticatedmeth-
odologies developed in the course of determining these bAR structures have
meanwhile proved instrumental in determining the crystal structures of sev-
eral additional 7TM receptors from different families. In this section, we
summarize some of the most relevant information that these bAR structures
have provided.
3.1. Ligand binding modes of pharmacologically relevantdrugsIn the period 2007–2011, bARs have been cocrystallized with 13 different
ligands, including inverse agonists, antagonists, and partial/full agonists
(Tables 6.2 and 6.3; Fig. 6.6). The turkey b1AR has been crystallized
in complex with the pharmacologically relevant agonists dobutamine,
salbutamol, isoproterenol, and carmoterol (Warne et al., 2011). As expected
from extensive mutagenesis studies (Liapakis et al., 2000, Strader, Candelore,
Hill, Sigal, & Dixon, 1989; Strader et al., 1988; Wieland, Zuurmond, Krasel,
Ijzerman, & Lohse, 1996), these agonists bind in a small binding pocket
between TM3, TM5, TM6, and TM7with a virtually identical bindingmode
(Fig. 6.6A). These crystal structures have revealed that agonists induce or
stabilize specific rotamer conformational changes of Ser 212(5.43) and Ser
215(5.46) and a small contraction of the binding pocket. While homology

A
2VT4, Cyanopindolol2YCW, Carazolol
Turkey b1-adrenergic receptor
Human b2-adrenergic receptor
2Y01, Dobutamine
S215
S211
S212
N310
TM3TM5 TM2
TM7
TM6
B
2Y04, Salbutamol
F306
N329
Y333
V122 D121 W117 S5.46
S5.42
V3.33 D3.32 W3.28W117
W330L101
V102
Y7.43N7.39
F6.51S5.43
N6.55F7.35
S207 V114 D113W109
Y316
N312F289
S204
S203
N293
TM5TM3
TM2 W109
TM7
TM6
3NY9, Novel inverse agonist 3NYA, Alprenolol 3P0G, BI-167107
2RH1, Carazolol 3D4S, Timolol 3NY8, ICI 118,551
Y308
I309(7.36)
V326(7.36)F325
2Y03, Isoprenaline 2Y02, Carmoterol
Figure 6.6 Ligand binding to b-adrenergic receptors. The binding pockets in ligandcomplex crystal structures of (A) turkey b1AR and (B) human b2AR viewed from theextracellular side. The Protein Data Bank code and the name of the ligand are notedabove the structures. The protein backbone is depicted as gray ribbons, side chainsin the binding pockets as pale green sticks (approx. within 4 Å from the ligand), and
142 Florian Brueckner et al.

143Structure of b-Adrenergic Receptors
modeling was relatively successful in defining this binding mode (see, for in-
stance, Swaminath et al., 2005; Xhaard, Rantanen, Nyronen, & Johnson,
2006; Krystek, Kimura, & Tebben, 2006) Asn293(6.55) had been predicted
to interact with the b-hydroxyl of catecholamines, based on its influence on
the enantiomeric selectivity (Wieland et al., 1996). However, in all the ago-
nist-bound crystal structures, this residue interacts instead with the aromatic
ring system of the ligand. It is thus likely that the chirality of the b-hydroxylinfluences the position of the aromatic ring system, thus altering its interaction
with Asn293(6.55).
While the strength of the ligand–receptor interactions is likely linked to
the efficacy of the ligand, these small local changes do not translate into the
stabilization of an active conformation in the thermostabilized b1AR. Thus,
these structures are likely to represent an “encounter complex,” that is, a low-
affinity nonsignaling state of the receptor formed on initial agonist binding
that cannot proceed through the regular activation mechanism. Until now,
the only structures of ARs in an active state have been obtained with the
human b2AR in complex with the high affinity (Kd¼84 pM) and extremely
low off-rate (150 h�1) full agonist BI-167107 (Fig. 6.6B). This agonist is also
not able to stabilize a crystallizable active conformationby itself. This required
the presence of a nanobody (Rasmussen, Choi, et al., 2011) or of a G protein
(Rasmussen, DeVree, et al., 2011), which presumably reduce the energy
barrier of the transition to an active state (Deupi & Kobilka, 2010). In sum-
mary, the currently available structures suggest that agonist binding alone is
not sufficient to stabilize a G protein-binding active conformation.
the ligand in pale yellow sticks, with nitrogen atoms blue, oxygen red, and the sulfuratom in timolol in yellow. The numbering of transmembrane helices (TMs) involvedin the binding pocket is shown. The numbering of amino acid side chains in the bindingpocket and in the extended binding pocket in case of the larger ligands dobutamineand BI-167107 is also shown (in the sequence and the Ballesteros–Weinstein generalnumber). Side chains of b1AR/b2AR in the binding pocket in TM3 (T118/T110 andT126/T118), at the N-terminus of TM5 (Y207/Y199 and A208/A200), in extracellular loop2 (F201/F193 and T203/T195) and in the extended binding pocket of carmoterol andBI-167107 in extracellular loop 2 (C199/C191 and D200/D192) are not shown for clarity.Homologous amino acids that are different in b1AR and b2AR are shown in italics. Po-tential hydrogen bonds between the protein side chains and the ligands are depicted asorange dashed lines. SigmaA-weighted 2Fo�Fc electron density maps were obtainedfrom the Electron Density Server (Kleywegt et al., 2004) or calculated from depositedstructure factors with Babinet's bulk solvent type scaling using the program REFMAC5and are shown with a blue mesh contoured at 1.0s. (See Color Insert.)

144 Florian Brueckner et al.
The structures of the complexes between bARs and pharmacologically
relevant ligands provide a valuable tool for drug design. For instance, the
structure of inactive, inverse-agonist-bound b2AR has been used to perform
an exhaustive in silico screening of drug-like molecules, which resulted in the
discovery of new chemotypes for inhibitors (Kolb et al., 2009). It is likely
that similar studies on agonist-bound structures of human ARs will bring
forth discovery of novel agonist molecules.
3.2. Ligand selectivityThe availability of the structures of the two closely related b1AR and b2ARallowed, for the first time, a detailed study of the molecular basis of ligand
selectivity in GPCRs. The strong similarity of their binding pockets most
likely indicates that selectivity does not only arise from the ligand binding
modes observed in the crystal structures. On the other hand, the sequence
similarity in the extracellular regions is lower, and the second extracellular
loop has been shown to influence ligand binding (Bokoch et al., 2010; Klco,
Wiegand, Narzinski, & Baranski, 2005; Shi & Javitch, 2004). Thus, it is
plausible that extracellular regions can have an impact on the different phar-
macological properties between subtypes. Two independent computational
studies (Dror et al., 2011; Gonzalez, Perez-Acle, Pardo, & Deupi, 2011)
have used molecular dynamics simulations to suggest the presence of sec-
ondary binding sites, located in the extracellular loops 2 and 3 and TM7,
that act as transient binding sites during ligand entry. The lower sequence
similarity between subtypes in these secondary sites might partially encode
ligand selectivity. In addition, the b-adrenergic structures have also revealedthat larger ligands extend beyond the outside of the catecholamine binding
pocket toward a secondary cavity between the extracellular ends of TM1, 2,
3, and 7. This “minor binding pocket“ has nonconserved residues that may
be responsible for subtype selectivity for larger compounds (Fig. 6.6) (Warne
et al., 2011) and also has been proposed to be involved in biased signaling
(Rosenkilde, Benned-Jensen, Frimurer, & Schwartz, 2010).
3.3. Insights into the activation mechanismThe b2AR has been extensively used as a model system to study the confor-
mational changes that accompany GPCR activation. For instance, a number
of fluorescence spectroscopy approaches have shown that activation pro-
ceeds through a sequence of conformational intermediates that can be spe-
cifically stabilized by agonists (see Kobilka, 2007 for a review). Similar results

145Structure of b-Adrenergic Receptors
have been also obtained through completely different approaches, as for
instance quantitative mass spectrometry (Kahsai et al., 2011) or NMR spec-
troscopy (Bokoch et al., 2010; Liu, Horst, Katritch, Stevens, & Wuthrich,
2012). However, the active state structures of the b2AR have finally allowed
the observation in molecular detail of the conformational changes that lead
to the stabilization of an active state. In addition, the structures of complexes
between bAR and agonists have provided insight into how ligand binding is
translated into receptor activation.
Themajor rearrangement during activation consists in the stabilization of
an opening in the cytoplasmic side of the receptor, mainly by the movement
of TM6 and, to a lesser extent, TM5 and TM7, that allows the binding of
the cytoplasmic partner. These large-scale movements are accompanied by a
number of smaller local structural changes near the binding site. Binding of
the catechol (or catechol-like) group of agonists to the conserved Ser resi-
dues in TM5 (Fig. 6.1) translates into rotamer conformational changes of
Ser204(5.43) and Ser207(5.46) (Warne et al., 2011; Rasmussen, Choi,
et al., 2011; Rasmussen, DeVree, et al., 2011; Fig. 6.6).
These small local structural changes result in (1) a small inward movement
of TM5 around Pro211(5.50) that is propagated toward the cytoplasmic
region of TM5 (Sansuk et al., 2011) and (2) relocation of Phe282(6.44) toward
Ile121(3.40) (Rasmussen,Choi, et al., 2011;Rasmussen,DeVree, et al., 2011),
which is themostnoticeablechange in thevicinityof thebindingpocket.These
seemingly small changes alter the packing at the TM3–TM5–TM6 interface
and result in a small rotation of TM6 around Phe282(6.44) that is amplified
to a large movement of the cytoplasmic end of TM6 due to the Pro-induced
distortion in this helix (Deupi&Standfuss, 2011). Importantly, agonist binding
alone isnot able to trigger thiskey transmissionswitchefficiently.Thus,binding
of an intracellular partner may be required to overcome the energy barrier
related to this structural change.
Despite the small number of interactions between agonists and TM7
(Figs. 6.1 and 6.6), the structure of the b2AR–Gs complex reveals a notice-
able rearrangement of TM7 around Asn318(7.45). Together with the small
rearrangement of the Met82(2.45) side chain and the larger movement of
TM6 near the G protein-binding site, this results in a change of the packing
at the TM2–TM3–TM6–TM7 interface, similar to rhodopsin (Deupi &
Standfuss, 2011; Deupi, Standfuss, & Schertler, 2012). These structural
changes are probably involved in the relocation of Tyr326(7.53) toward
the protein core, where it participates in the intrahelical interactions that
stabilize the active state.

146 Florian Brueckner et al.
In summary, agonist binding stabilizes a series of local structural changes
that result in a contraction of the binding pocket. Binding of an intracellular
partner stabilizes the active conformation of key transmission switches
that link agonist binding to the stabilization of the active state, resulting
in the larger rearrangements observed in the active state crystal structures
of the b2AR. These structural changes can be categorized into three intra-
molecular activation pathways through TM5/TM3, TM6, and TM7/TM2
(Deupi et al., 2012). Ligands with different chemical properties may differ-
ently activate these pathways, thus stabilizing slightly different active states
of the receptor with specific signaling properties, as observed by 19F NMR
in the human b2AR (Liu et al., 2012). This simple mechanism could help
to explain the complex pharmacology of ARs, for example, biased
signaling.
4. OUTLOOK
Recent development and establishment of new strategies and tech-
niques in protein biochemistry and engineering have allowed for the more
efficient production of highly purified, stable constructs of bARs amenable
to crystallization. This, combined with advances in the techniques of crys-
tallization and microcrystallography, has permitted the determination of the
structures of the turkey b1AR and the human b2AR in complex with a panel
of inverse agonists, antagonists, and agonists. These structures have revealed
the binding mode of medically relevant drugs and have generated new hy-
potheses regarding ligand selectivity and ligand-induced receptor activation.
Importantly, the techniques pioneered in bARs are being applied to obtain
structures of other GPCRs.
The turkey b1AR is currently the best template for drug design directed
to the human b1AR. However, although these two receptors are highly ho-
mologous (76% sequence identity in the transmembrane bundle), they have
distinct pharmacological profiles (Baker, 2010b). Thus, obtaining a crystal
structure of the human b1AR is key to study the structural basis of its phar-
macological properties. Furthermore, to establish the molecular basis for li-
gand selectivity, it is essential to obtain structures of the other members of
the AR family (the b3AR, three a1ARs and three a2ARs). This information
can be used to develop subtype-selective drugs with fewer side effects and to
unravel the molecular mechanisms that determine functional selectivity of
biased ligands of bARs, that is, how biased ligands bind to the receptor
and how this translates into selective activation of signaling pathways.

147Structure of b-Adrenergic Receptors
ACKNOWLEDGMENTSWe thank TonyWarne and Christopher G. Tate for advice. We thank Dmitry B. Veprintsev
for critical reading of the chapter. We thank our colleagues at the Laboratory of Bio-
molecular Research for advice and the staff of beamline S06SA at the Swiss Light Source
for support. F. B. is supported by a Marie Curie Intra European Fellowship and an
EMBO long-term fellowship. C. L. P. is supported by the ETH Zurich Postdoctoral
Fellowship program and the Marie Curie Actions for People COFUND program.
Research related to this chapter in our lab is supported by the Swiss National Science
Foundation (grant 31003A_132815), and the National Center for Competence in
Research in the Structural Biology Program of ETH Zurich.
REFERENCESAhlquist, R. P. (1948). A study of the adrenotropic receptors. The American Journal of Phys-
iology, 153, 586–600.Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A., & Stevens, R. C. (2008).
Microscale fluorescent thermal stability assay for membrane proteins. Structure, 16,351–359.
Baker, J. G. (2010a). The selectivity of beta-adrenoceptor agonists at human beta1-, beta2-and beta3-adrenoceptors. British Journal of Pharmacology, 160, 1048–1061.
Baker, J. G. (2010b). A full pharmacological analysis of the three turkey b-adrenoceptors andcomparison with the human b-adrenoceptors. PLoS One, 5, e15487.
Ballesteros, J., & Weinstein, H. (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations inG protein-coupled receptors. Methods Neuroscience, 25, 366–428.
Benovic, J. L., Shorr, R. G., Caron, M. G., & Lefkowitz, R. J. (1984). The mammalian beta2-adrenergic receptor: Purification and characterization. Biochemistry, 23, 4510–4518.
Bokoch, M. P., Zou, Y., Rasmussen, S. G. F., Liu, C.W., Nygaard, R., Rosenbaum, D.M.,et al. (2010). Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature, 463, 108–112.
Bylund, D. B., Eikenberg, D. C., Hieble, J. P., Langer, S. Z., Lefkowitz, R. J.,Minneman, K. P., et al. (1994). International Union of Pharmacology nomenclatureof adrenoceptors. Pharmacological Reviews, 46, 121–136.
Caffrey, M. (2008). Crystallizing membrane proteins for structure determination: Use oflipidic mesophases. Annual Review of Biophysics, 38, 29–51.
Caron, M. G., Srinivasan, Y., Pitha, J., Kociolek, K., & Lefkowitz, R. J. (1979). Affinitychromatography of the beta-adrenergic receptor. The Journal of Biological Chemistry,254, 2923–2927.
Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G. F., Thian, F. S.,Kobilka, T. S., et al. (2007). High-resolution crystal structure of an engineered humanbeta2-adrenergic G protein-coupled receptor. Science, 318, 1258–1265.
Chien, E. Y. T., Liu,W., Zhao, Q., Katritch, V.,WonHan, G., Hanson,M. A., et al. (2010).Structure of the human dopamine D3 receptor in complex with a D2/D3 selectiveantagonist. Science, 330, 1091–1095.
Deupi, X., & Kobilka, B. (2010). Energy landscapes as a tool to integrate GPCR structure,dynamics, and function. Physiology, 25, 293.
Deupi, X., & Standfuss, J. (2011). Structural insights into agonist-induced activation ofG-protein-coupled receptors. Current Opinion in Structural Biology, 21, 541–551.
Deupi, X., Standfuss, J., & Schertler, G. (2012). Conserved activation pathways in G protein-coupled receptors. Biochemical Society Transactions, 40, 383–388.

148 Florian Brueckner et al.
Dixon, R. A., Kobilka, B. K., Strader, D. J., Benovic, J. L., Dohlman, H. G., Frielle, T., et al.(1986). Cloning of the gene and cDNA for mammalian beta-adrenergic receptor andhomology with rhodopsin. Nature, 321, 75–79.
Dohlman, H. G., Caron, M. G., & Lefkowitz, R. J. (1987). A family of receptors coupled toguanine nucleotide regulatory proteins. Biochemistry, 26, 2657–2664.
Dore, A. S., Robertson, N., Errey, J. C., Ng, I., Hollenstein, K., Ben Tehan, et al. (2011).Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthinesXAC and caffeine. Structure, 19, 1283–1293.
Dror, R. O., Pan, A. C., Arlow, D. H., Borhani, D.W., Maragakis, P., Shan, Y., et al. (2011).Pathway and mechanism of drug binding to G-protein-coupled receptors. Proceedings of theNational Academy of Sciences of the United States of America, 108, 13118–13123.
Edwards, P. C., Li, J., Burghammer, M., McDowell, J. H., Villa, C., Hargrave, P. A., et al.(2004). Crystals of native and modified bovine rhodopsins and their heavy atom deriv-atives. Journal of Molecular Biology, 343, 1439–1450.
Evans, G., Axford, D., Waterman, D., & Owen, R. L. (2011). Macromolecular micro-crystallography. Crystallography Reviews, 17, 105–142.
Fredriksson, R., Lagerstrom, M. C., Lundin, L.-G., & Schioth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis,paralogon groups, and fingerprints. Molecular Pharmacology, 63, 1256–1272.
Gonzalez, A., Perez-Acle, T., Pardo, L., & Deupi, X. (2011). Molecular basis of ligand dis-sociation in b-adrenergic receptors. PLoS One, 6, e23815.
Guan, X. M., Kobilka, T. S., & Kobilka, B. K. (1992). Enhancement of membrane insertionand function in a type IIIb membrane protein following introduction of a cleavable signalpeptide. The Journal of Biological Chemistry, 267, 21995–21998.
Haga, K., Kruse, A. C., Asada, H., Yurugi-Kobayashi, T., Shiroishi, M., Zhang, C., et al.(2012). Structure of the human M2 muscarinic acetylcholine receptor bound to an an-tagonist. Nature, 482, 547–551.
Hanson, M. A., Cherezov, V., Griffith, M. T., Roth, C. B., Jaakola, V.-P., Chien, E. Y. T.,et al. (2008). A specific cholesterol binding site is established by the 2.8 A structure of thehuman beta2-adrenergic receptor. Structure, 16, 897–905.
Hanson, M. A., Roth, C. B., Jo, E., Griffith, M. T., Scott, F. L., Reinhart, G., et al. (2012).Crystal structure of a lipid G protein-coupled receptor. Science, 335, 851–855.
Jaakola, V.-P., Griffith, M. T., Hanson, M. A., Cherezov, V., Chien, E. Y. T., Lane, J. R.,et al. (2008). The 2.6 angstrom crystal structure of a human A2A adenosine receptorbound to an antagonist. Science, 322, 1211–1217.
Kahsai,A.W.,Xiao,K.,Rajagopal,S.,Ahn,S., Shukla,A.K.,Sun, J., et al. (2011).Multiple ligand-specific conformations of the b2-adrenergic receptor.Nature Chemical Biology, 7, 692–700.
Klco, J. M., Wiegand, C. B., Narzinski, K., & Baranski, T. J. (2005). Essential role for thesecond extracellular loop in C5a receptor activation.Nature Structural &Molecular Biology,12, 320–326.
Kleywegt, G. J., Harris, M. R., Zou, J. Y., Taylor, T. C., Wahlby, A., & Jones, T. A. (2004).The Uppsala Electron-Density Server. Acta Cryst D, 60, 2240–2249.
Kobilka, B. K. (2007). G protein coupled receptor structure and activation. Biochimica etBiophysica Acta, 1768, 794–807.
Kobilka, B. K., Dixon, R. A., Frielle, T., Dohlman, H. G., Bolanowski, M. A., Sigal, I. S.,et al. (1987). cDNA for the human beta 2-adrenergic receptor: A protein withmultiple membrane-spanning domains and encoded by a gene whose chromosomallocation is shared with that of the receptor for platelet-derived growth factor. Proceedingsof the National Academy of Sciences of the United States of America, 84, 46–50.
Kobilka, B. K., Kobilka, T. S., Daniel, K., Regan, J. W., Caron, M. G., & Lefkowitz, R. J.(1988). Chimeric alpha 2-, beta 2-adrenergic receptors: Delineation of domains involvedin effector coupling and ligand binding specificity. Science, 240, 1310–1316.

149Structure of b-Adrenergic Receptors
Kolb, P., Rosenbaum, D. M., Irwin, J. J., Fung, J. J., Kobilka, B. K., & Shoichet, B. K.(2009). Structure-based discovery of beta2-adrenergic receptor ligands. Proceedings ofthe National Academy of Sciences of the United States of America, 106, 6843–6848.
Kruse, A. C., Hu, J., Pan, A. C., Arlow, D. H., Rosenbaum, D. M., Rosemond, E., et al.(2012). Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature,482, 552–556.
Krystek, S. R., Kimura, S. R., & Tebben, A. J. (2006). Modeling and active site refinementfor G protein-coupled receptors: Application to the beta-2 adrenergic receptor. Journal ofComputer-Aided Molecular Design, 20, 463–470.
Lebon, G., Warne, T., Edwards, P. C., Bennett, K., Langmead, C. J., Leslie, A. G. W., et al.(2011). Agonist-bound adenosine A2A receptor structures reveal common features ofGPCR activation. Nature, 474, 521–525.
Liapakis, G., Ballesteros, J. A., Papachristou, S., Chan, W. C., Chen, X., & Javitch, J. A.(2000). The forgotten serine. A critical role for Ser-2035.42 in ligand binding to andactivation of the beta 2-adrenergic receptor. The Journal of Biological Chemistry, 275,37779–37788.
Liu, J. J., Horst, R., Katritch, V., Stevens, R. C., & Wuthrich, K. (2012). Biased signalingpathways in b2-adrenergic receptor characterized by 19F-NMR. Science, 335,1106–1110.
Michel,H. (1983).Crystallization ofmembrane proteins.Trends inBiochemical Sciences,8, 56–59.Miller, J. L., & Tate, C. G. (2011). Engineering an ultra-thermostable b1-adrenoceptor.
Journal of Molecular Biology, 413, 628–638.Moukhametzianov, R., Warne, T., Edwards, P. C., Serrano-Vega, M. J., Leslie, A. G. W.,
Tate, C. G., et al. (2011). Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proceedings of the National Academy ofSciences of the United States of America, 108, 8228–8232.
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., et al.(2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science, 289,739–745.
Parker, E. M., Kameyama, K., Higashijima, T., & Ross, E. M. (1991). Reconstitutivelyactive G protein-coupled receptors purified from baculovirus-infected insect cells.The Journal of Biological Chemistry, 266, 519–527.
Parker, E. M., & Ross, E. M. (1991). Truncation of the extended carboxyl-terminal domainincreases the expression and regulatory activity of the avian beta-adrenergic receptor.TheJournal of Biological Chemistry, 266, 9987–9996.
Patel, C. B., Noor, N., & Rockman, H. A. (2010). Functional selectivity in adrenergic andangiotensin signaling systems. Molecular Pharmacology, 78, 983–992.
Peleg, G., Ghanouni, P., Kobilka, B. K., & Zare, R. N. (2001). Single-molecule spectros-copy of the beta(2) adrenergic receptor: Observation of conformational substates in amembrane protein. Proceedings of the National Academy of Sciences of the United States ofAmerica, 98, 8469–8474.
Rasmussen, S. G. F., Choi, H.-J., Fung, J. J., Pardon, E., Casarosa, P., Chae, P. S., et al.(2011). Structure of a nanobody-stabilized active state of the b(2) adrenoceptor. Nature,469, 175–180.
Rasmussen, S. G. F., Choi, H.-J., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S.,Edwards, P. C., et al. (2007). Crystal structure of the human beta2 adrenergic G-protein-coupled receptor.Nature, 450, 383–387.
Rasmussen, S. G. F., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S.,et al. (2011). Crystal structure of the b2 adrenergic receptor-Gs protein complex.Nature,477, 549–555.
Riekel, C., Burghammer, M., & Schertler, G. (2005). Protein crystallography micro-diffraction. Current Opinion in Structural Biology, 15, 556–562.

150 Florian Brueckner et al.
Rosenbaum, D. M., Cherezov, V., Hanson, M. A., Rasmussen, S. G. F., Thian, F. S.,Kobilka, T. S., et al. (2007). GPCR engineering yields high-resolution structural insightsinto beta2-adrenergic receptor function. Science, 318, 1266–1273.
Rosenbaum, D. M., Zhang, C., Lyons, J. A., Holl, R., Aragao, D., Arlow, D. H., et al.(2011). Structure and function of an irreversible agonist-b(2) adrenoceptor complex.Nature, 469, 236–240.
Rosenkilde, M. M., Benned-Jensen, T., Frimurer, T. M., & Schwartz, T. W. (2010). Theminor binding pocket: A major player in 7TM receptor activation. Trends in Pharmaco-logical Sciences, 31, 567–574.
Roth, C. B., Hanson, M. A., & Stevens, R. C. (2008). Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41),a critical residue in GPCR structure. Journal of Molecular Biology, 376, 1305–1319.
Sansuk, K., Deupi, X., Torrecillas, I. R., Jongejan, A., Nijmeijer, S., Bakker, R. A., et al.(2011). A structural insight into the reorientation of transmembrane domains 3 and 5during family A G protein-coupled receptor activation. Molecular Pharmacology, 79,262–269.
Schertler, G. F. X. (2005). Structure of rhodopsin and the metarhodopsin I photo-intermediate. Current Opinion in Structural Biology, 15, 408–415.
Serrano-Vega, M. J., Magnani, F., Shibata, Y., & Tate, C. G. (2008). Conformationalthermostabilization of the 1-adrenergic receptor in a detergent-resistant form. Proceedingsof the National Academy of Sciences, 105, 877–882.
Serrano-Vega, M., & Tate, C. (2009). Transferability of thermostabilizing mutationsbetween beta-adrenergic receptors. Molecular Membrane Biology, 26, 385–396.
Shi, L., & Javitch, J. A. (2004). The second extracellular loop of the dopamine D2 receptorlines the binding-site crevice. Proceedings of the National Academy of Sciences of the UnitedStates of America, 101, 440–445.
Shimamura, T., Shiroishi, M., Weyand, S., Tsujimoto, H., Winter, G., Katritch, V., et al.(2011). Structure of the human histamine H1 receptor complex with doxepin. Nature,475, 65–70.
Shorr, R. G., Lefkowitz, R. J., & Caron, M. G. (1981). Purification of the beta-adrenergicreceptor. Identification of the hormone binding subunit. The Journal of Biological Chem-istry, 256, 5820–5826.
Shorr, R. G., Strohsacker, M. W., Lavin, T. N., Lefkowitz, R. J., & Caron, M. G. (1982).The beta 1-adrenergic receptor of the turkey erythrocyte. Molecular heterogeneity rev-ealed by purification and photoaffinity labeling. The Journal of Biological Chemistry, 257,12341–12350.
Standfuss, J., Xie, G., Edwards, P. C., Burghammer, M., Oprian, D. D., & Schertler, G. F. X.(2007). Crystal structure of a thermally stable rhodopsin mutant. Journal of MolecularBiology, 372, 1179–1188.
Strader, C. D., Candelore, M. R., Hill, W. S., Sigal, I. S., & Dixon, R. A. (1989). Identi-fication of two serine residues involved in agonist activation of the beta-adrenergicreceptor. The Journal of Biological Chemistry, 264, 13572–13578.
Strader, C. D., Sigal, I. S., Candelore, M. R., Rands, E., Hill, W. S., & Dixon, R. A. (1988).Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have differ-ent roles in receptor function. The Journal of Biological Chemistry, 263, 10267–10271.
Swaminath, G., Deupi, X., Lee, T. W., Zhu, W., Thian, F. S., Kobilka, T. S., et al. (2005).Probing the beta2 adrenoceptor binding site with catechol reveals differences in bindingand activation by agonists and partial agonists. The Journal of Biological Chemistry, 280,22165–22171.
Tate, C. G., & Schertler, G. F. X. (2009). Engineering G protein-coupled receptors tofacilitate their structure determination.Current Opinion in Structural Biology, 19, 386–395.

151Structure of b-Adrenergic Receptors
Wacker, D., Fenalti, G., Brown, M. A., Katritch, V., Abagyan, R., Cherezov, V., et al.(2010). Conserved binding mode of human beta2 adrenergic receptor inverse agonistsand antagonist revealed by X-ray crystallography. Journal of the American Chemical Society,132, 11443–11445.
Warne, T., Chirnside, J., & Schertler, G. (2003). Expression and purification of truncated,non-glycosylated turkey beta-adrenergic receptors for crystallization. Biochimica etBiophysica Acta, 1610, 133–140.
Warne, T., Moukhametzianov, R., Baker, J. G., Nehme, R., Edwards, P. C.,Leslie, A. G. W., et al. (2011). The structural basis for agonist and partial agonist actionon a b(1)-adrenergic receptor. Nature, 469, 241–244.
Warne, T., Serrano-Vega, M. J., Baker, J. G., Moukhametzianov, R., Edwards, P. C.,Henderson, R., et al. (2008). Structure of a beta1-adrenergic G-protein-coupled recep-tor. Nature, 454, 486–491.
Warne, T., Serrano-Vega, M. J., Tate, C. G., & Schertler, G. F. X. (2009). Development andcrystallization of a minimal thermostabilised G protein-coupled receptor. Protein Expres-sion and Purification, 65, 204–213.
Wieland, K., Zuurmond, H. M., Krasel, C., Ijzerman, A. P., & Lohse, M. J. (1996). Involve-ment of Asn-293 in stereospecific agonist recognition and in activation of the beta2-adrenergic receptor. Proceedings of the National Academy of Sciences of the United Statesof America, 93, 9276–9281.
Wong, S. K., Parker, E. M., & Ross, E. M. (1990). Chimeric muscarinic cholinergic: Beta-adrenergic receptors that activate Gs in response to muscarinic agonists. The Journal ofBiological Chemistry, 265, 6219–6224.
Wu, B., Chien, E. Y. T., Mol, C. D., Fenalti, G., Liu, W., Katritch, V., et al. (2010). Struc-tures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antag-onists. Science, 330, 1066–1071.
Xhaard, H., Rantanen, V.-V., Nyronen, T., & Johnson, M. S. (2006). Molecular evolutionof adrenoceptors and dopamine receptors: Implications for the binding of catechol-amines. Journal of Medicinal Chemistry, 49, 1706–1719.
Yarden, Y., Rodriguez, H., Wong, S. K., Brandt, D. R., May, D. C., Burnier, J., et al.(1986). The avian beta-adrenergic receptor: Primary structure and membrane topology.Proceedings of the National Academy of Sciences of the Unite States of America, 83, 6795–6799.
Related Documents











