Structural study of ribonucleotide reductase inhibitor hydrazones. Synthesis and X-ray diffraction analysis of a copper(II)-benzoylpyridine-2-quinolinyl hydrazone complex Gabriella Tamasi a , Luisa Chiasserini b , Luisa Savini b, * , Alessandro Sega b , Renzo Cini a, * a Department of Chemical and Biosystem Sciences and Technologies, University of Siena, Via Aldo Moro 2, I-53100 Siena, Italy b Dipartimento Farmaco Chimico Tecnologico, University of Siena, Via Aldo Moro 2, I-53100 Siena, Italy Received 4 November 2004; received in revised form 7 March 2005; accepted 11 March 2005 Abstract Single crystals as yellow needles of N-(4,8-dimethyl-quinolin-2-yl)-N 0 -(1-pyridin-2-yl-ethylidene)-hydrazine, HL 1 , 1, and N-(4- methyl-quinolin-2-yl)-N 0 -(phenyl-pyridin-2-yl-methylene)-hydrazine, HL 2 , 2, were obtained from methanol (MeOH) and analyzed via X-ray diffraction (XRD). HL 2 reacted with copper(II) acetate to produce a dark red powder that gave single crystals of [Cu(L 2 )(OOCCH 3 )] Æ 0.9C 6 H 5 CH 3 , 3 Æ 0.9C 6 H 5 CH 3 when recrystallized from toluene. The conformation of the N(quinolinyl,q)– C(q)–N(H)–N(imine,i)–C–C(pyridine,p)–N(p) grouping is trans,trans,trans,trans or tttt, and ttcc for 1 and 2, respectively, at the solid state, as revealed via single crystal X-ray diffraction. Thus, the structure of 1 has the methyl (hydrazone) group syn to the N–H bond and syn to the N(q) and N(p) atom. On the other side, the structure of 2 is stabilized by a strong intra-molecular N–H...N hydrogen bond which involves the pyridyl nitrogen atom. The molecule 1 is almost planar, the torsion angles do not deviate more than 4° from the idealized values of 0° and 180°. In the structure of 2 the pyridyl ring is almost coplanar with the N(q)–C(q)–NH–N(i)–C system, whereas the phenyl (Ph) ring is twisted by ca. 55°. The structure of 3 has the L 2 ligand as deprotonated at the N–N function and in a cttc conformation as opposite to the ttcc one found for pure 2. The metal center is coordinated through N(q), N(i), N(p) and through an oxygen atom from a carboxylate anion. The molecular modeling analysis of 1 and 2 (semi-empirical molecular orbital at ZernerÕs intermediate neglect of differential overlap (ZINDO/1) level and density functional theory (DFT) methods) gave good agreement with the X-ray structures. Semi-empirical quantum mechanics analysis of 3 allowed to assign the UV–Vis spectrum that is characterized by strong absorptions in the visible, UVA and UVB regions. Owing to the ribonucleotide reductase inhibitory activity of the ligand, to the ascertained anticancer activity shown previously by related copper(II)-hydrazone complexes, and to the oxygen radical scavenger activity of several copper(II)-complexes, 3 is potentially anticancer and anti-inflammatory. Ó 2005 Elsevier Inc. All rights reserved. Keywords: Hydrazones; Copper(II) complex; DFT; Molecular orbital; X-ray diffraction 1. Introduction It has been reported that ribonucleotide reductase (RR) of mammalians has low activity in resting cells, high activity in rapidly growing normal cells, and very high activity in malignant cells [1]. Inhibitors of this enzyme are therefore potential anticancer agents. The RR macro- molecule consists of two subunits M1 and M2; this latter has been studied in detail and M2 inhibitors may be metal ion chelators and radical scavengers. One of the metal chelating agents and radical scavengers that has been reported as promising inhibitors of RR is N-(phenyl- pyridin-2-yl-methylene)-N 0 -pyridin-2-yl-hydrazine (see Scheme 1, hereafter HL 4 , 4) which is reported to inhibit 0162-0134/$ - see front matter Ó 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.jinorgbio.2005.03.009 * Corresponding authors. Tel.: +39 577 244368; fax: +39 577 234177. E-mail address: [email protected] (R. Cini). www.elsevier.com/locate/jinorgbio Journal of Inorganic Biochemistry 99 (2005) 1347–1359 JOURNAL OF Inorganic Biochemistry

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF
www.elsevier.com/locate/jinorgbio
Journal of Inorganic Biochemistry 99 (2005) 1347–1359
InorganicBiochemistry
Structural study of ribonucleotide reductase inhibitorhydrazones. Synthesis and X-ray diffraction analysis of a
copper(II)-benzoylpyridine-2-quinolinyl hydrazone complex
Gabriella Tamasi a, Luisa Chiasserini b, Luisa Savini b,*, Alessandro Sega b, Renzo Cini a,*
a Department of Chemical and Biosystem Sciences and Technologies, University of Siena, Via Aldo Moro 2, I-53100 Siena, Italyb Dipartimento Farmaco Chimico Tecnologico, University of Siena, Via Aldo Moro 2, I-53100 Siena, Italy
Received 4 November 2004; received in revised form 7 March 2005; accepted 11 March 2005
Abstract
Single crystals as yellow needles of N-(4,8-dimethyl-quinolin-2-yl)-N 0-(1-pyridin-2-yl-ethylidene)-hydrazine, HL1, 1, and N-(4-methyl-quinolin-2-yl)-N 0-(phenyl-pyridin-2-yl-methylene)-hydrazine, HL2, 2, were obtained from methanol (MeOH) and analyzedvia X-ray diffraction (XRD). HL2 reacted with copper(II) acetate to produce a dark red powder that gave single crystals of[Cu(L2)(OOCCH3)] Æ 0.9C6H5CH3, 3 Æ 0.9C6H5CH3 when recrystallized from toluene. The conformation of the N(quinolinyl,q)–C(q)–N(H)–N(imine,i)–C–C(pyridine,p)–N(p) grouping is trans,trans,trans,trans or tttt, and ttcc for 1 and 2, respectively, at the solidstate, as revealed via single crystal X-ray diffraction. Thus, the structure of 1 has the methyl (hydrazone) group syn to the N–H bondand syn to the N(q) and N(p) atom. On the other side, the structure of 2 is stabilized by a strong intra-molecular N–H. . .N hydrogenbond which involves the pyridyl nitrogen atom. The molecule 1 is almost planar, the torsion angles do not deviate more than 4� fromthe idealized values of 0� and 180�. In the structure of 2 the pyridyl ring is almost coplanar with the N(q)–C(q)–NH–N(i)–C system,whereas the phenyl (Ph) ring is twisted by ca. 55�. The structure of 3 has the L2 ligand as deprotonated at the N–N function and in acttc conformation as opposite to the ttcc one found for pure 2. The metal center is coordinated through N(q), N(i), N(p) and throughan oxygen atom from a carboxylate anion. The molecular modeling analysis of 1 and 2 (semi-empirical molecular orbital at Zerner�sintermediate neglect of differential overlap (ZINDO/1) level and density functional theory (DFT) methods) gave good agreement withthe X-ray structures. Semi-empirical quantummechanics analysis of 3 allowed to assign the UV–Vis spectrum that is characterized bystrong absorptions in the visible, UVA and UVB regions. Owing to the ribonucleotide reductase inhibitory activity of the ligand, tothe ascertained anticancer activity shown previously by related copper(II)-hydrazone complexes, and to the oxygen radical scavengeractivity of several copper(II)-complexes, 3 is potentially anticancer and anti-inflammatory.� 2005 Elsevier Inc. All rights reserved.
Keywords: Hydrazones; Copper(II) complex; DFT; Molecular orbital; X-ray diffraction
1. Introduction
It has been reported that ribonucleotide reductase(RR) ofmammalians has low activity in resting cells, highactivity in rapidly growing normal cells, and very high
0162-0134/$ - see front matter � 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.jinorgbio.2005.03.009
* Corresponding authors. Tel.: +39 577 244368; fax: +39 577234177.
E-mail address: [email protected] (R. Cini).
activity in malignant cells [1]. Inhibitors of this enzymeare therefore potential anticancer agents. The RRmacro-molecule consists of two subunits M1 and M2; this latterhas been studied in detail andM2 inhibitors may bemetalion chelators and radical scavengers. One of the metalchelating agents and radical scavengers that has beenreported as promising inhibitors of RR is N-(phenyl-pyridin-2-yl-methylene)-N 0-pyridin-2-yl-hydrazine (seeScheme 1, hereafter HL4, 4) which is reported to inhibit

1
2
345
6
78
4a
8a9
1011
N NN
H
H
HHH
H
H
H
H HH
H
HH
NH
H
H
H
12
18
191'
2'
3'
4'5'
6'
1
2
345
6
78
4a
8a 910
11N N
N
H
H
HHH
H
H
H
H
N H
H
H
H
H
H
H
H
H
18
1'
2'
3'
4'5'
6'
1213
14
1516
17
HL1, 1 HL2, 2N-(4,8-dimethyl-quinolin-2-yl)-N’-(1-pyridin- N-(4-methyl-quinolin-2-yl)-N’-(phenyl- 2-yl-ethylidene)-hydrazine pyridin-2-yl-methylene)-hydrazine
1
2
34
910
11
18
1'
2'
3'
4'5'
6'
1213
14
1516
17
N NN
H
HHH
H
N H
H
H
H
H
H
H
H
H
H
H
5
6
H
1
2
34
910
11
18
1'2'
3'
4'5'
6'
2" 1"
6"
5"4"
3"
N NN
H
HHH
H
N H
H
H
H
N
H
H
H
H
H
H
5
6
H
HL4, 4 HL5, 5N-(phenyl-pyridin-2-yl-methylene)-N’- N-(di-pyridin-2-yl-methylene)-N’- pyridin-2-yl-hydrazine pyridin-2-yl-hydrazine
Scheme 1. The drawings represent the structural formula of the HL1, 1, and HL2, 2, ligand molecules prepared in this work, with the numbering ofthe atoms used throughout the manuscript. The molecules of HL4, 4, [1] and HL5, 5 [34,35] are also represented. Several conformers for each ligandcan be drawn on the basis of the rotations around the N1–C2–N9–N10 (/1), C2–N9–N10–C11 (/2), N9–N10–C11–C2 0 (/3), N10–C11–C2 0–N1 0 (/4),N9–N10–C11–C200 (/5) and N10–C11–C200–N100 (/6) (this latter for HL5, 5, only) torsion angles. The conformers here depicted are as follows(trans = t, cis = c): HL1, 1, ttcc; HL2, 2, ttcc; HL4, 4, ttcc; HL5, 5, ttcct00t00.
1348 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
also ‘‘the proliferation of the hormone dependent humanmammary tumor cell line MDA-MB231’’ [1]. However,even corresponding 2 0-quinolinyl-hydrazones haveshown good inhibitory effects on proliferation of Bur-kitt�s lymphoma cells with IC50 in the range 0.01–0.04 lM [1]. The research efforts on RR enzymes bothfrom structural and catalytic activity standpoints havemuch intensified during the last years. In fact, the search‘‘Ribonucleotide Reductase’’ performed in the ResearchCollaboratory for Structural Bioinformatics – ProteinData Bank (RCSB-PDB) structural data base [2] forthe period 1997–May 2004, shows 38 structures of en-zyme systems studied via NMR and/or XRD techniques.To cite just one of the most recent XRD-structures, thatof a RR from Escherichia coli, contains iron ions coordi-nated by two histidine and four carboxylate ligands [3].
It is well known that metal complexes of certain metalions (Cu2+/+, Mn3+/2+, Fe3+/2+) have peculiar affinitiestowards highly reactive radical species. The role of cop-per in CuZn-superoxide dismutase (CuZn-SOD) en-zymes has been extensively investigated during the pastdecades by enzymologists and bio-inorganic chemists[4,5]. Metal complexes that might act via a SOD-likemechanism for the cure of inflammatory and rheumaticdiseases have been designed and prepared. If metal ionsare combined with active drugs whose beneficial effectshave been ascertained through a long time and wideadministration to humans and other animals, new pow-
erful, multitherapic agents may be obtained as in thecase of ([Cu2(Indomethacin)4(N,N-dimethylformam-ide)2]) [5].
It happened to some of us finding routes for preparingseries of metal complexes of non-steroidal anti-inflamma-tory drugs (NSAIDs) from the ‘‘oxicam’’ family [6–13]and that the Cu2+-complexes had promising oxygen rad-ical scavenger activity [11] and promising anti-prolifera-tive activities against selected tumor cell lines (NationalCancer Institute, USA, NCI, DTP Compound #624662) [13]. On another side it happened also that someof us were able to prepare and to evaluate biologicalproperties of series of hydrazones [14–18].
On the basis of these findings and owing to the pau-city of structure determinations on this class of com-pounds reported in the literature, we thought tosynthesize and characterize some hydrazone derivatives,and to investigate their physico-chemical properties aswell as their reactivity towards Cu2+ ions. We wish to re-port here on the selected results from this work.
2. Experimental
2.1. Starting materials
Solvents and starting compounds for all the prepara-tions of the ligands were as previously reported by some

G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1349
of us [14–18]. Diaquatetrakis(acetate)copper(II) was re-agent grade from Carlo Erba (Milan).
2.2. Synthesis
2.2.1. Preparation of the ligandsHL1, 1, and HL2, 2, were synthesized according to
previously described methods [19]. Single crystals of 1
and 2 were obtained on slowly concentrating (via spon-taneous evaporation at 25 �C) solutions of the ligands inmethanol (MeOH) previously dried on molecular sieves4 A (Merck, Darmstadt). The purity of the two com-pounds was tested via elemental analysis. HL1, 1.Found: C, 73.90; H, 6.38; N, 19.45%. Calcd. forC18H18N4 (Fw, 290.37): C, 74.46; H, 6.25; N, 19.30%.HL2, 2. Found: C, 78.40; H, 5.20; N, 16.28%. Calcd.for C22H18N4 (Fw, 338.41): C, 78.08; H, 5.36; N,16.56%. Further analysis of purity consisted of 1HNMR analysis (see below).
2.3. Preparation of the metal complex
[Cu{N(q), N(i), N(p)-L2-(-H9)}(O-OOCCH3)] Æ0.9C6H5CH3, 3 Æ 0.9C6H5CH3. Thirty-four mg HL2
(0.10 mmol) were dissolved in 8 ml MeOH, whereas20 mg Cu(CH3COO)2 Æ H2O (0.11 mmol) were dissolvedin 3 ml MeOH (with gentle heating). The two warmsolutions were mixed under stirring and the final mixturewas heated at ca. 50 �C for 15 min; the solvent was par-tially evaporated till a final volume of ca. 2 ml. Fifty mlof Et2O were added to the resulting deep-red mixture.No precipitate formed on mixing. The solution was con-centrated to ca. 5 ml. A crystalline precipitate formed; itwas filtered off, washed with Et2O and dried in the air.The mother liquor was mixed with ca. 50 ml Et2O andthe solution was again concentrated under suction.Some more precipitate formed, that was collected as justabove reported. The overall crude precipitate wasrecrystallized from toluene. Tufts of dark prisms formedwithin two days by slowly evaporating a clear toluenesolution stored at 25 �C. Yield 20 mg. Anal. Found. C,66.30%; H, 4.97; N, 10.60. Calcd. [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)] Æ 0.9C6H5CH3, C30.3H27.2Cu-N4O2 (Fw, 542.92), C, 67.03; H, 5.05; N, 10.32%.
2.4. X-ray crystallography
2.4.1. Data collections
Pale yellow prisms of dimension 0.20 · 0.15 · 0.10,HL1, 1 and 0.30 · 0.20 · 0.15 mm3, HL2, 2, and adark-red plate of dimensions 0.30 · 0.50 · 0.10mm3, [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)] Æ0.9C6H5CH3, 3 Æ 0.9C6H5CH3 were selected under thepolarizing microscope and then mounted on glass fi-bers. The data collections were performed through aSiemens P4 automatic four circle diffractometer with
a graphite monochromatized Mo Ka radiation (k,0.71073 A). The accurate cell constant determinationswere obtained via the least-squares method appliedto the values of 22, 26 and 30 randomly selectedstrong reflections for 1, 2 and 3 Æ 0.9C6H5CH3, respec-tively. The intensities were corrected for Lorentz-polarization effects (XSCANS [20]), and absorptioneffects (via the w-scan technique based on at least threereflections for 1 and 2, XEMP [21], and via DIFABS[22] for 3 Æ 0.9C6H5CH3). The extinction correction wasapplied to the three sets of data and the extinctioncoefficient converged to 0.00217, 0.01945 and 0.00000for 1, 2 and 3 Æ 0.9C6H5CH3, respectively. The selectedcrystallographic data are listed in Table 1.
2.4.2. Structure solutions and refinements
The structure solution for 1, 2 and 3 Æ 0.9C6H5CH3
were performed through the direct methods ofSHELXS-86 [23], SHELX-97 [24], and SIR-92 [25],respectively, as implemented in the WinGX package[26,27], and subsequent difference-Fourier maps andleast-squares cycles. All the non-hydrogen atoms for 1,2 and 3 were easily located through this procedurewhereas all the hydrogen atoms were located in calcu-lated position through the HFIX and AFIX options ofSHELX-97 [24]. The Fourier-difference map for 3
showed four peaks around an inversion center, threeof them were interpreted as three carbon atoms of thearomatic ring of a toluene molecule T1. The forth onewas interpreted as the carbon atom of the methyl substi-tuent of toluene. The ring carbon atoms were assigned asite occupation factor (SOF) of 1, whereas the methylcarbon was assigned SOF 0.5. Thus, this toluene mole-cule is affected by a statistical disorder that simulatesthe inversion center for the co-crystallized solvent mole-cule (see Section 3, for further information on co-crys-tallized toluene). Once the structure of T1 was solvedand partially refined, the subsequent Fourier-differencemap showed the presence of five new peaks around acrystallographically independent inversion center. Thosepeaks were interpreted as five carbon atoms of a seconddisordered co-crystallized toluene molecule T2. Draw-ings of disordered T1 and T2 are reported in SchemeS1 (Supporting Material). The occupancies for tolueneatoms were those that optimize the thermal parametersand the agreement with the analytical data (C,H,N) col-lected from the crystals in the same specimen of the crys-tal used for the XRD analysis. C1T2 and C2T2 wereconsidered as the sites of two carbon atoms and assignedSOF 0.8 (Scheme S1). The other three carbon atoms,namely C3T2, C4T2, C5T2, were given SOF 0.4. Thenot-hydrogen atoms were refined as anisotropic whereasthe hydrogen atoms were treated as isotropic. Themethyl groups of 1, 2 and 3 are affected by a rotationaldisorder around the C–C bond so that two sets of threehydrogen atoms each at SOF 0.5 were refined. The

Table 1Selected crystallographic data for HL1, 1, HL2, 2, and [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)] Æ 0.9C6H5CH3, 3 Æ 0.9C6H5CH3
Parameter Value
1 2 3 Æ 0.9C6H5CH3
Empirical formula C18H18N4 C22H18N4 C30.3H27.2CuN4O2
Formula weight 290.37 338.41 542.92Temperature/K 295(2) 293(2) 293(2)Wavelength/A 0.71073 0.71073 0.71073Crystal system, space group Monoclinic, C2/c Triclinic, P�1 Triclinic, P�1Unit cell dimensions
a/A 22.493(2) 8.201(1) 9.143(2)b/A 7.983(1) 8.520(1) 11.207(1)c/A 16.633(3) 13.754(1) 13.621(2)a/� 90 74.01(1) 76.00(1)b/� 91.98(1) 76.41(1) 79.52(1)c/� 90 74.52(1) 87.99(1)
Volume/A3 2984.9(7) 876.7(2) 1331.6(4)Z, calculated density/Mg m�3 8, 1.292 2, 1.282 2, 1.354Absorption coefficient/mm�1 0.080 0.078 0.854Reflections collected/unique 3258/2596 [Rint = 0.0429] 3726/3028 [Rint = 0.0256] 5539/4598 [Rint = 0.0302]Data/restraints/parameters 2596/0/200 3028/0/233 4598/6/361Goodness-of-fit on F2 0.984 1.002 1.078Final R indices [I > 2r(I)] R1 = 0.0633, wR2 = 0.1269 R1 = 0.0563, wR2 = 0.1133 R1 = 0.0659, wR2 = 0.1409R indices (all data) R1 = 0.1569, wR2 = 0.1642 R1 = 0.1383, wR2 = 0.1437 R1 = 0.1175, wR2 = 0.1658Largest differential peak and hole (e A�3) 0.226 and �0.186 0.177 and �0.118 0.647 and �0.503
1350 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
hydrogen atoms for the four ring carbon atoms and forthe methyl group of T1 of 3 Æ 0.9C6H5CH3 were set incalculated position via HFIX/AFIX options ofSHELX-97 package. The hydrogen atom linked to thecarbon in para position to the methyl group was not in-cluded in the refinement. The hydrogen atoms of T2were not included at all owing to the severe disorder thataffects that molecule. Some ring C–C bond distances forthe second toluene molecule of 3 Æ 0.9C6H5CH3 were re-strained at 1.40 ± 0.02 A, whereas the C–CH3 bond dis-tance was restrained at 1.50 ± 0.02 A. The final R1 andwR2 agreement factors were 0.0633, 0.0563 and 0.0659for 1, 2 and 3 Æ 0.9C6H5CH3, respectively. The calcula-tions relevant to the structure refinements were per-formed through the SHELX-97 package [24], whereasthe molecular geometry and the molecular graphics werecomputed by using the PARST-97 [28], and ORTEP-3[29] packages.
2.5. Spectroscopy
The IR spectra were recorded by using the Nujol mulland KBr pellet techniques and a Perkin–Elmer model1600 Fourier-transform spectrometer. UV spectra wererecorded by using a Perkin–Elmer EZ-201 spectropho-tometer. 1H and 13C NMR spectra of compound HL2,were recorded on a Bruker Avance 600 MHz spectrom-eter. 2D NMR experiments, correlated spectroscopy(COSY), heteronuclear correlation (HETCOR) and nu-clear overhauser effect spectroscopy (NOESY), were
performed on a Bruker Avance AMX 400 MHzspectrometer.
2.6. Elemental analysis and chromatography
CHN elemental analyses were performed at Mikro-analytisches Labor Pascher, An der Pulvermuhle 1,D-53424 Remagen-Bandorf (Germany). The gas-chro-matographic analysis of the toluene solvent was carriedout through a Carlo Erba instrument MFC 500 (ovenHRGC 5300; detector, EL 480; mega series integrator),equipped with capillary column Supelco SPB5(30 m · 0.23 mm, film 0.25 lm).
2.7. Computational methods
2.7.1. Semi-empirical methods
All the calculations were performed using the Hyper-Chem 5.1 package [30]. The molecular structures forseveral conformers and protonation forms for 1, 2, 3and related model molecules were optimized at the gasphase by using the ZINDO/1 level of approximation[31], and the restricted Hartree–Fock wave function,RHF. The convergence criterion for the geometry opti-mizations (without any restraint for all the molecules,unless otherwise specified) was 0.01 kJ mol�1.
The molecules optimized were HLn-tttt, -tttc, -ttct,-cttt, -ttcc, -cttc, -ctct, -ctcc for n = 1, 2, 4; Ln-(-H9)-tttt,-tttc, -ttct, -cttt, -ttcc, -cttc, -ctct, -ctcc for n = 1, 2, 4; L5-ttcct00t00, -ctttc00t00, -ttcct00c00, -ttctt00c00, -ttctt00t00; L5-(-H9)-ttcct00t00, -ctttc00t00, -ttcct00c00, -ttctt00c00, -ttctt00t00.

Fig. 2. Ortep drawing for the molecule of HL2, 2. The ellipsoidsenclose 50% probability.
G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1351
The UV spectra were computed by using single pointcalculations at the ZINDO/S [31] singly excited interac-tion configuration level on the molecules previouslyoptimized through the ZINDO/1 method. The r–rand p–p overlap weighting factors were those imple-mented in the program [31].
Density functional (DF) and ab initio methods. Thecalculations were carried out through the GAUSS-IAN-98 package [32] by using the IBM SP4 machinesof CINECA (North East Inter-University Consortiumfor Advanced Computing, Casalecchio di Reno, Italia).The DF computations were performed through theBecke3LYP hybrid functional (hereafter namedB3LYP) [33] and using the standard 6-31+G* basis set[33]. The structure optimization were considered endedwhen the maximum atomic displacement and rootmean-square RMS of atomic displacements were smal-ler than 0.0018 and 0.0012 A, and when the maximumforce and RMS of forces were smaller than 0.00045and 0.0003 mdyne. The molecules investigated were 4
and 5 (Scheme 1), and the starting structures were thosefound at the solid state via X-ray diffraction from the lit-erature ([1] and [34,35], respectively) or from this work,or from previous optimization at the semi-empiricalmethods.
3. Results and discussion
3.1. X-ray crystallography
3.1.1. Structures of the ligands
The molecules of HL1, 1, and HL2, 2, are depicted inFigs. 1 and 2, respectively, whereas the selected bonddistances and angles are listed in Tables 2 and 3.
3.1.1.1. HL1. All the atoms of 1 are almost co-planaras shown by the selected torsion angles N1–C2–N9–N10 (�177.4(3)�), C2–N9–N10–C11 (�178.0(3)�),N9–N10–C11–C2 0 (179.4(3)�) and N10–C11–C2 0–N1 0
(176.5(3)�) and by the largest deviation from theleast-square planes. The molecule has a tttt conforma-
Fig. 1. Ortep drawing for the molecule of HL1, 1. The ellipsoidsenclose 50% probability.
tion (Scheme 1). As regards the plane defined by endo-cyclic atoms of quinoline the largest deviations isshown by C12 (�0.182(4) A), that deviates by0.111(4) A even from the plane defined by the pyridylgroup. The dihedral angles between the quinoline sys-tem and the C(q)–N(H)–N–C–C(p) least square planesis 4.6(2)�, whereas the dihedral angle between the quin-oline and pyridyl planes is 3.6(1)�. The structure of 1
has the same conformation as 5 [34,35] around theN–N group, however the arrangement of the C(p)–N(H)–N–C–C(p) chain has larger deviations from pla-narity for 5; C–N(H)–N–C and N(H)–N–C–C torsionangles being �178.0(5) (/2, Scheme 1) and�179.4(5)� for 1, and �169.6(5) (/2), 0.5(5) (/3) and�179.9(5)� (/5) for 5. One of the pyridyl rings of 5
is tilted with respect to the N(H)–N–C plane by ca.42�. The values of the torsion angles show also that1 has the anti-E configuration in agreement with thesolid state structure for a similar a-N-quinolyl-hydra-zone (methoxyacetyl in the place of a-pyridyl) [17].The analysis of the bond distances is in agreement withthe Lewis representation depicted in Scheme 1 as re-gards the N-(4,8-dimethyl-quinolin-2-yl)-hydrazinemoiety. The N1–C2 bond distance 1.316(4) A, shorterthan C2–N9 (1.377(4) A) and than N1 0–C2 0
(1.344(4) A), is in agreement with a significant doublebond character. The N10–C11 bond length(1.293(4) A) is in agreement with a double bond char-acter, too. The trend of the bond lengths discussed issimilar to that found previously for 5.
Intramolecular hydrogen bond type interactions rea-sonably take place between C14–H and N1 (C. . .N,2.809(5) A), C3–H and N10 (C. . .N, 2.792(4) A), C3 0-H and N10 (C. . .N, 2.769(4) A), and C12–H and N1 0
(2.839(5) A). Extensive intermolecular hydrogen bondinteractions link the molecules in the crystal. Selectedexamples are as follows. Two molecules are paired via

Table 2Selected bond lengths (A) for HL1, 1, HL2, 2, [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)], 3
Vector Length
1 2 3
Cu–O(1) 1.952(4)Cu. . .O(2) 2.559(5)Cu–N(1) 1.982(4)Cu–N(10) 1.943(4)Cu–N(10) 2.001(4)O(1)–C(19) 1.284(6)O(2)–C(19) 1.223(6)C(19)–C(20) 1.505(8)N(1)–C(2) 1.316(4) 1.312(3) 1.363(7)N(1)–C(8A) 1.378(4) 1.377(3) 1.397(6)N(9)–N(10) 1.366(4) 1.357(3) 1.347(6)N(9)–C(2) 1.377(4) 1.381(3) 1.367(6)N(10)–C(11) 1.293(4) 1.302(3) 1.311(6)N(1 0)–C(20) 1.344(4) 1.348(3) 1.356(7)N(1 0)–C(60) 1.333(4) 1.329(3) 1.335(7)C(2)–C(3) 1.419(4) 1.412(4) 1.424(7)C(3)–C(4) 1.354(5) 1.362(4) 1.351(7)C(4)–C(4A) 1.432(5) 1.421(4) 1.416(8)C(11)–C(20) 1.483(5) 1.481(4) 1.458(8)C(11)–C(12) 1.493(4) 1.488(3) 1.490(7)C(4A)–C(8A) 1.415(4) 1.411(4) 1.421(8)C(20)–C(30) 1.391(4) 1.385(4) 1.392(7)C(30)–C(40) 1.367(5) 1.373(3) 1.385(8)C(40)–C(50) 1.376(5) 1.369(3) 1.359(8)C(50)–C(60) 1.364(5) 1.364(4) 1.385(8)
Table 3Selected bond angles (�) for HL1, 1, HL2, 2, [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)], 3
Vectors Angle
1 2 3
O(1)–Cu–N(1) 107.9(2)O(1)–Cu–N(10) 169.7(2)O(1)–Cu–N(1 0) 92.4(2)N(1)–Cu–N(10) 79.7(2)N(1)–Cu–N(10) 158.9(2)N(10)–Cu–N(10) 80.9(2)Cu–O(1)–C(19) 103.3(3)Cu–N(1)–C(2) 110.0(3)Cu–N(10)–N(9) 119.0(3)Cu–N(10)–C(11) 117.5(4)Cu–N(10)–C(20) 112.9(4)C(2)–N(1)–C(8A) 117.6(3) 116.8(3) 118.2(5)N(10)–N(9)–C(2) 119.4(3) 119.5(2) 108.8(5)C(11)–N(10)–N(9) 118.2(3) 119.4(2) 123.5(5)C(60)–N(10)–C(20) 117.1(3) 117.7(3) 119.2(5)N(1)–C(2)–N(9) 114.6(3) 114.3(3) 121.4(5)N(1)–C(2)–C(3) 123.8(3) 124.3(3) 121.0(5)N(9)–C(2)–C(3) 121.6(3) 121.4(2) 117.6(5)C(4)–C(3)–C(2) 119.5(3) 119.9(3) 121.6(6)C(3)–C(4)–C(4A) 119.1(3) 118.1(3) 118.7(5)N(10)–C(11)–C(20) 115.2(3) 128.1(2) 113.7(5)N(10)–C(11)–C(12) 124.2(3) 113.2(2) 124.9(5)C(20)–C(11)–C(12) 120.5(3) 118.7(2) 121.3(4)N(1 0)–C(20)–C(30) 121.8(3) 121.3(3) 120.3(5)N(1 0)–C(20)–C(11) 116.3(3) 117.6(3) 115.0(4)C(30)–C(20)–C(11) 121.9(3) 121.1(3) 124.7(5)C(40)–C(30)–C(20) 118.9(4) 119.2(3) 119.4(6)C(30)–C(40)–C(50) 119.5(2) 119.49(17) 119.8(6)C(60)–C(50)–C(40) 118.0(2) 118.04(18) 118.5(6)N(1 0)–C(60)–C(50) 124.9(4) 124.2(3) 122.7(6)
Fig. 3. The diagram shows the packing of HL1, 1, as viewed down theb axis.
1352 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
two N9–H (�x, y, �z + 0.5). . .N1 (N. . .N, 3.165(6) A)interactions. The two molecules are not coplanar andthe dihedral angle between the least square planes is62(1)�. It has to be noted that these two hydrogen bondsmay contribute for ca. �8 kcal mol�1 each to the forma-tion of the pair. This estimation comes after the compu-tation from this laboratory of the formation energy of(imidazole)N1–H. . .N3 0(imidazole) from electronicenergies computed at B3LYP/and MP2/6-31+G** level[36].
The crystal is stabilized by extensive networks ofstacking interactions. In fact the quinoline system haslarge overlap regions with the pyridyl system, the short-est interatomic distance being C8A. . .C4 0 (�x, �y, �z)(3.395(5) A). On examining the cell along the b axisthe molecules of HL1 are placed almost on the diagonalof the ac plane forming parallel strips that consist ofpairs of molecules (Fig. 3).
3.1.1.2. HL2. The molecule of 2, HL2, has a ttcc confor-mation and is almost planar except the phenyl ring thatis significantly twisted with respect to the C–C(Ph)bond. The HN–N(i)–C–C(Ph) and N(i)–C–C(Ph)–C(Ph) torsion angles are 177.5(2) and 55.2(3)�, respec-tively. The pyridyl ring is almost coplanar with theHN–N–C–C(Ph) chain (torsion angle, 10.4(5)�) andthe nitrogen (pyridyl) atom is oriented in a way to form
a strong intramolecular H-bond with the (N)H function(N. . .N, 2.637(4) A; N–H. . .N, 132(1)�). The N(q)–C–N(H)–N@C–C(p) chain has a ttc conformation and

G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1353
the N1–C2–N9–N10 (/1) and C2–N9–N10–C11 (/2)torsion angles are 166.8(5) and �171.2(5)�. The p-elec-tron delocalization is significant as confirmed by theN1–C2, C2–N9, N9–N10 and N10–C11 bond lengths1.312(3), 1.381(4), 1.357(3) and 1.302(4) A. These valuescompare well with the corresponding ones found for 1,HL1 and 5 [34,35].
The crystal packing is stabilized by extensive andstrong stacking interactions which involve the quino-line system, the C–NH–N@C chain and the pyridylring (Fig. 4). The interatomic contact distances beingas short as 3.494(3) A (N1. . .C4 0 (�x, �y + 1,�z + 1)). So, each N–N-quinolinyl group is sand-wiched between a symmetry related group and a pyr-idyl ring. Strips of HL2 molecules are placed alongthe c axis and the quinoline planes are almost perpen-dicular to the a axis. N1 is the hydrogen acceptor fromC13–H (�x, �y + 1, �z + 1) (C. . .N, 3.854(7) A; C–H. . .N, 158(1)�). It has to be noted that for 2 the pres-ence of a bulkier Ph group with respect to Me (as in 1)does not allow the pairing of molecules via two N9–H. . .N1 hydrogen-bonds (provided a tttt conformationis assumed). This could be a reasonable explanation forthe differences of conformations for 1 and 2 at the solidstate. A search for a free 2-benzoylpyridine-2-pyridylor -2-quinolinyl hydrazone derivative molecules, as car-ried out on the Cambridge Structural Data Base(CSDB) [37] did not give any hints. On the other handonly the structure of 2-methanalpyridine-2-pyridylhydrazone [38] was found at CSDB, when the searchfor non-hindered hydrazones was carried out. This lat-ter molecule has again a tttt conformation as 1 andalso a strong pairing via two N–H. . .N hydrogenbonds (N. . .N, 3.079 A).
Fig. 4. The diagram shows the packing of HL2, 2, as viewed down thea axis.
3.1.2. Structure of 3 Æ 0.9C6H5CH3
The structure of the complex molecule is representedin Fig. 5 whereas the selected bond distances and anglesare listed in Tables 2 and 3.
The Cu2+ cation has a 4 + 1 coordination mode beinglinked to the quinolinyl, imine and pyridyl nitrogenatoms from the L2-(-H9) ligand and from an oxygenatom from the acetato anion in the equatorial plane.The carbonyl type oxygen atom from acetato is roughlyapical with respect to the equatorial donors and weaklybound to the metal center. The L2-(-H9) anionic ligandhas the cttc conformation as compared to the ttcc onefound for the neutral ligand 2 HL2 at the solid state.The analysis of the structural effects upon this changeare discussed below, see computational methods. TheCu–N1, –N10 and –N1 0 bond distances are 1.982(4),1.943(4) and 2.001(4) A, respectively, showing that theimine nitrogen is the most effective donor. The Cu–O(equatorial, eq) and –O(apical, ap) bond distancesare 1.952(4) and 2.560(5) A respectively, showing thatthe coordination is mostly planar with just a weak apicalinteraction. The two coordination bond angles that in-volve the L2-(-H9) ligand are 79.7(2) and 80.9(2)�,respectively. Noteworthy, the O(eq)–Cu–N1 bond angle(107.9(2)�) is much larger than the O(eq)–Cu–N1 0 one(92.4(2)�).
The Cu2+ center deviates just by 0.0111(7) A from theleast-squares plane defined by the equatorial donors andpoints outwards of the apical oxygen atom from carb-oxylato. Interestingly, the O(eq)–Cu–N(imine) bond an-gle is 169.7(2)�. This coordination arrangement suggeststhat the region around Cu2+ trans to O(ap) is crowdedby some other particles. In fact, a co-crystallized toluenemolecule is located close to the metal atom and the pyr-idyl system, and is almost parallel to the coordinationleast-squares plane (dihedral angle 8.0(1)�) and to thepyridyl least-squares plane (dihedral angle 5.6(1) or15.3(1)�).
Fig. 5. Ortep drawing of the complex molecule [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)], 3. The ellipsoids enclose 50% probability.

1354 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
Interestingly, one of the toluene-ring carbon atoms isrelatively close to the metal center, the Cu. . .C2T1 con-tact distance being 3.277(6) A. It has to be noted that thesum of the Van der Waals radii for Cu and C is 3.10 A[39], so that a linkage between the metal center and acarbon atom of toluene or the p-system has to be ex-cluded in favor of a stacking-type interaction betweenp-systems.
The N1–C2, C2–N9 and N9–N10 bond lengths forL2-(-H9) ligand are 1.363(7), 1.368(6) and 1.347(6) Arespectively showing a significant increase of the N1–C2 bond length upon metal ligation whereas C2–N9and N9–N10 bond distances are not much affected.The N10–C11, C11–C2 0 and C2 0–N1 0 bond distancesfor 3 are 1.311(6), 1.458(8), and 1.356(7) A respectively;thus C11–C2 0 only has a significant change upon coordi-nation to metal (decrease by 0.024 A, six times the esd).The orientation of the phenyl ring with respect to thehydrazone chain changes significantly upon coordina-tion; in fact, the smallest N10–C11–C12–C torsion angleis 55.2(5)� for HL2 and 67.4(5)� for 3.
Crystal packing. The complex molecules are piled al-most along the b axis and leave holes that are filled bythe cocrystallized toluene molecules (Fig. 6). This pack-ing arrangement is in agreement with the behavior of thecrystal under heating. In fact, at 95–100 �C the crystalsopacize when observed at the polarizing microscope.The quinolinyl systems related via an inversion center(1 1/2 1/2) stack each other, the overlap surface is largeeven though the interatomic distances are relativelylarge, ca. 3.7 A. The pyridyl rings related via inversioncenter (1/2 0 1) stack at ca. 3.6 A. Interestingly the tolu-
Fig. 6. The diagram shows the packing of 3 Æ 0.9C6H5CH3 as viewedalmost down the a axis.
ene molecules play a significant role in the stacking net-work. T1 molecules are piled between complexmolecules and are disordered so to simulate an inversioncenter located inside the six-member ring. It looks like apara-xylene molecule. Several observations are in favorof co-crystallized toluene instead of xylene. The refine-ment of the model with site occupation factor, SOF, 1for the methyl carbon increases significantly the conven-tional R1 agreement factor; the elemental analysis of thecrystals has a better agreement with the presence of tol-uene instead of xylene; the gas-chromatographic analy-sis of the toluene solvent used for crystal growthprocedure shows a very small content of para-xylene(<0.01%).
The T1 molecule overlaps with Cu–pyridyl system ofcomplex molecule. Short contact distances involveC6 0. . .C1T1 (�x + 1, �y + 1, �z + 2) 3.420(8) A andCu1. . .C2T1 (see above). Molecule T2 is not signifi-cantly involved in p–p stacking interactions; instead itis anchored by two symmetry related (inversion centerat 1/2 1 1/2) methyl groups from the acetato ligands.The distance of a hydrogen atom (H204) from the clos-est carbon atom namely C4T2, is 2.76(1) A that suggeststhe presence of C–H. . .p (toluene) interactions [40].
Acetato ligand. The analysis of the C19–O1 and C19–O2 bond distances, 1.284(6) and 1.223(6) A, respec-tively, shows that the covalent character of the Cu–Olinkage is relatively large, ca. 30%, on the basis of thetheory by Hocking and Hambley [41], even though aweak Cu. . .O(t) interaction exists to the second oxygenatom of the acetato ligand.
3.2. Spectroscopy
3.2.1. NMR
Assignments of the proton and carbon resonances ofcompound HL2, 2 were done from analysis of corre-sponding 1D NMR spectrum and COSY and HET-COR experiments. Protons H5 and H8 were assignedthrough nuclear overhauser effect (NOE) experiments;irradiation of methyl protons gave a NOE value onthe doublet at 7.92 ppm, thus this signal correspondsto H5 and the doublet at 7.68 ppm corresponds toH8. The listing of 1H NMR and 13C NMR data is re-ported in Table 4. Information on the conformationwas obtained from a NOESY study done on a0.01 M solution of 2 in DMSO-d6 (d6 deuterated di-methyl sulfoxide). The only non-trivial NOE detectedis between protons H6 0 and H8. This NOE howeveris of key importance since it shows that the conforma-tion in solution is close to those found in the solid stateand by ZINDO/1 calculations. On the basis of theH6 0. . .H8 distance one can possibly say that the con-formation in solution is closer to ZINDO/1(H6 0. . .H8, 5.14 A) than to the solid state (H6 0. . .H8,6.06 A) conformation.
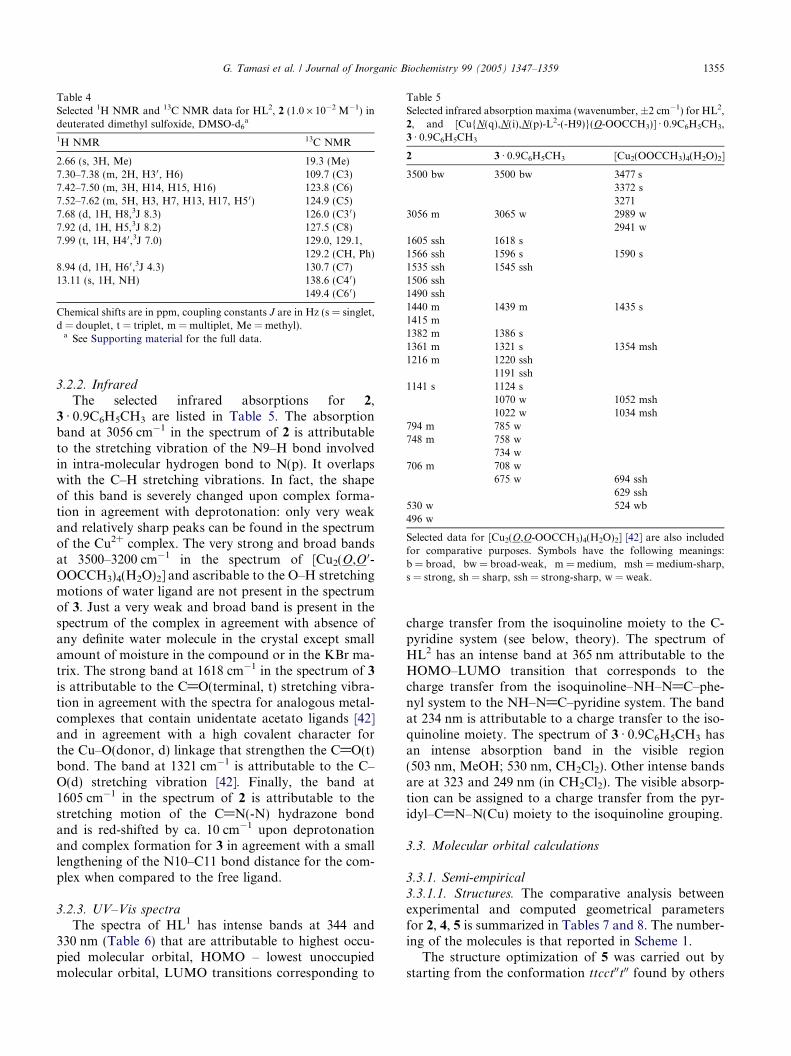
Table 4Selected 1H NMR and 13C NMR data for HL2, 2 (1.0 · 10�2 M�1) indeuterated dimethyl sulfoxide, DMSO-d6
a
1H NMR 13C NMR
2.66 (s, 3H, Me) 19.3 (Me)7.30–7.38 (m, 2H, H30, H6) 109.7 (C3)7.42–7.50 (m, 3H, H14, H15, H16) 123.8 (C6)7.52–7.62 (m, 5H, H3, H7, H13, H17, H5 0) 124.9 (C5)7.68 (d, 1H, H8,3J 8.3) 126.0 (C30)7.92 (d, 1H, H5,3J 8.2) 127.5 (C8)7.99 (t, 1H, H4 0,3J 7.0) 129.0, 129.1,
129.2 (CH, Ph)8.94 (d, 1H, H6 0,3J 4.3) 130.7 (C7)13.11 (s, 1H, NH) 138.6 (C4 0)
149.4 (C60)
Chemical shifts are in ppm, coupling constants J are in Hz (s = singlet,d = douplet, t = triplet, m = multiplet, Me = methyl).a See Supporting material for the full data.
Table 5Selected infrared absorption maxima (wavenumber, ±2 cm�1) for HL2,2, and [Cu{N(q),N(i),N(p)-L2-(-H9)}(O-OOCCH3)] Æ 0.9C6H5CH3,3 Æ 0.9C6H5CH3
2 3 Æ 0.9C6H5CH3 [Cu2(OOCCH3)4(H2O)2]
3500 bw 3500 bw 3477 s3372 s3271
3056 m 3065 w 2989 w2941 w
1605 ssh 1618 s1566 ssh 1596 s 1590 s1535 ssh 1545 ssh1506 ssh1490 ssh1440 m 1439 m 1435 s1415 m1382 m 1386 s1361 m 1321 s 1354 msh1216 m 1220 ssh
1191 ssh1141 s 1124 s
1070 w 1052 msh1022 w 1034 msh
794 m 785 w748 m 758 w
734 w706 m 708 w
675 w 694 ssh629 ssh
530 w 524 wb496 w
Selected data for [Cu2(O,O-OOCCH3)4(H2O)2] [42] are also includedfor comparative purposes. Symbols have the following meanings:b = broad, bw = broad-weak, m = medium, msh = medium-sharp,s = strong, sh = sharp, ssh = strong-sharp, w = weak.
G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1355
3.2.2. Infrared
The selected infrared absorptions for 2,3 Æ 0.9C6H5CH3 are listed in Table 5. The absorptionband at 3056 cm�1 in the spectrum of 2 is attributableto the stretching vibration of the N9–H bond involvedin intra-molecular hydrogen bond to N(p). It overlapswith the C–H stretching vibrations. In fact, the shapeof this band is severely changed upon complex forma-tion in agreement with deprotonation: only very weakand relatively sharp peaks can be found in the spectrumof the Cu2+ complex. The very strong and broad bandsat 3500–3200 cm�1 in the spectrum of [Cu2(O,O 0-OOCCH3)4(H2O)2] and ascribable to the O–H stretchingmotions of water ligand are not present in the spectrumof 3. Just a very weak and broad band is present in thespectrum of the complex in agreement with absence ofany definite water molecule in the crystal except smallamount of moisture in the compound or in the KBr ma-trix. The strong band at 1618 cm�1 in the spectrum of 3is attributable to the C@O(terminal, t) stretching vibra-tion in agreement with the spectra for analogous metal-complexes that contain unidentate acetato ligands [42]and in agreement with a high covalent character forthe Cu–O(donor, d) linkage that strengthen the C@O(t)bond. The band at 1321 cm�1 is attributable to the C–O(d) stretching vibration [42]. Finally, the band at1605 cm�1 in the spectrum of 2 is attributable to thestretching motion of the C@N(-N) hydrazone bondand is red-shifted by ca. 10 cm�1 upon deprotonationand complex formation for 3 in agreement with a smalllengthening of the N10–C11 bond distance for the com-plex when compared to the free ligand.
3.2.3. UV–Vis spectra
The spectra of HL1 has intense bands at 344 and330 nm (Table 6) that are attributable to highest occu-pied molecular orbital, HOMO – lowest unoccupiedmolecular orbital, LUMO transitions corresponding to
charge transfer from the isoquinoline moiety to the C-pyridine system (see below, theory). The spectrum ofHL2 has an intense band at 365 nm attributable to theHOMO–LUMO transition that corresponds to thecharge transfer from the isoquinoline–NH–N@C–phe-nyl system to the NH–N@C–pyridine system. The bandat 234 nm is attributable to a charge transfer to the iso-quinoline moiety. The spectrum of 3 Æ 0.9C6H5CH3 hasan intense absorption band in the visible region(503 nm, MeOH; 530 nm, CH2Cl2). Other intense bandsare at 323 and 249 nm (in CH2Cl2). The visible absorp-tion can be assigned to a charge transfer from the pyr-idyl–C@N–N(Cu) moiety to the isoquinoline grouping.
3.3. Molecular orbital calculations
3.3.1. Semi-empirical
3.3.1.1. Structures. The comparative analysis betweenexperimental and computed geometrical parametersfor 2, 4, 5 is summarized in Tables 7 and 8. The number-ing of the molecules is that reported in Scheme 1.
The structure optimization of 5 was carried out bystarting from the conformation ttcct00t00 found by others
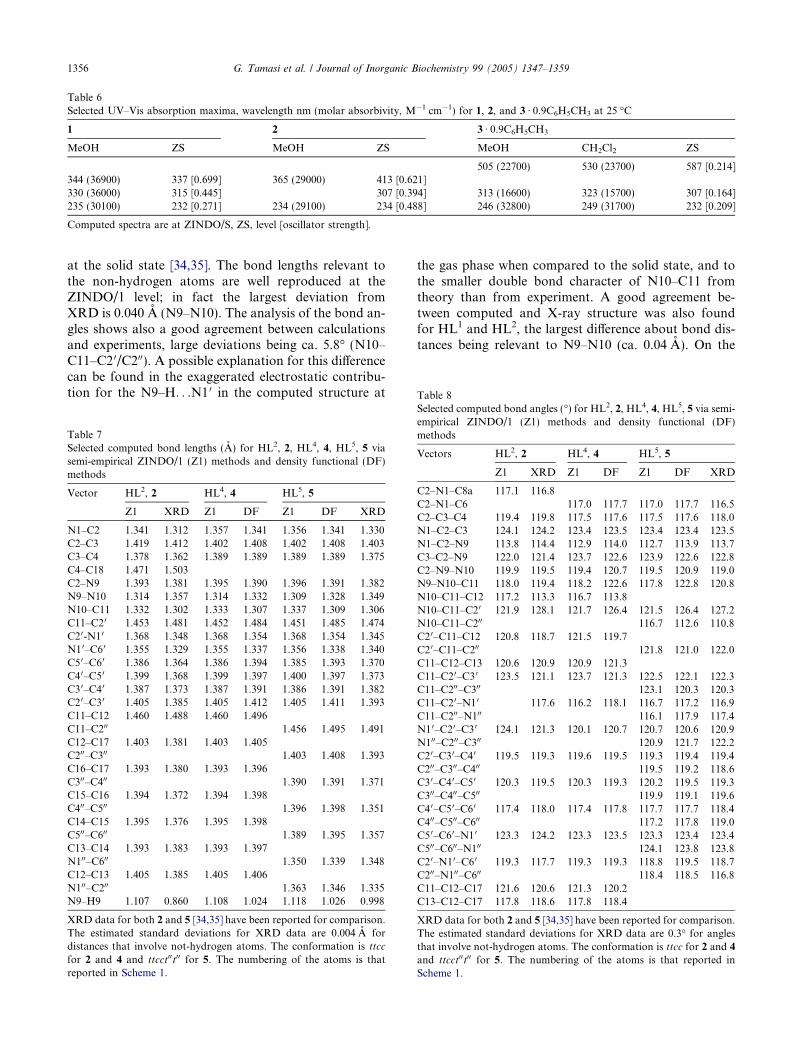
Table 6Selected UV–Vis absorption maxima, wavelength nm (molar absorbivity, M�1 cm�1) for 1, 2, and 3 Æ 0.9C6H5CH3 at 25 �C
1 2 3 Æ 0.9C6H5CH3
MeOH ZS MeOH ZS MeOH CH2Cl2 ZS
505 (22700) 530 (23700) 587 [0.214]344 (36900) 337 [0.699] 365 (29000) 413 [0.621]330 (36000) 315 [0.445] 307 [0.394] 313 (16600) 323 (15700) 307 [0.164]235 (30100) 232 [0.271] 234 (29100) 234 [0.488] 246 (32800) 249 (31700) 232 [0.209]
Computed spectra are at ZINDO/S, ZS, level [oscillator strength].
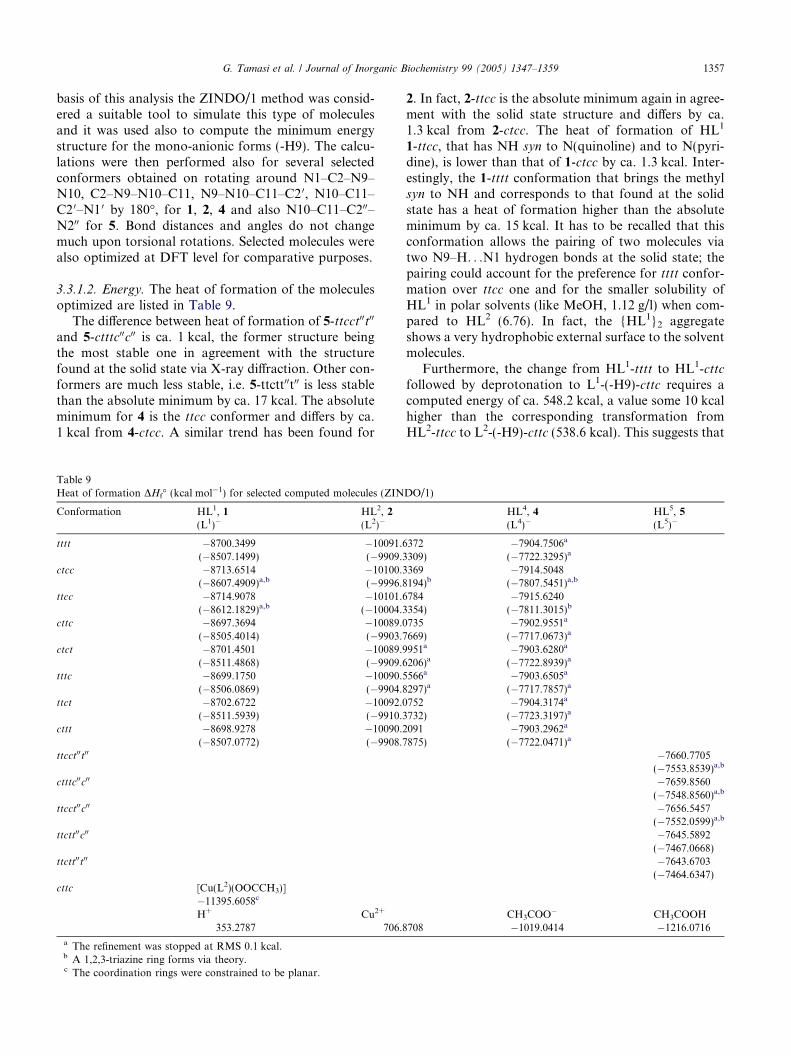
Table 8
1356 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
at the solid state [34,35]. The bond lengths relevant tothe non-hydrogen atoms are well reproduced at theZINDO/1 level; in fact the largest deviation fromXRD is 0.040 A (N9–N10). The analysis of the bond an-gles shows also a good agreement between calculationsand experiments, large deviations being ca. 5.8� (N10–C11–C2 0/C200). A possible explanation for this differencecan be found in the exaggerated electrostatic contribu-tion for the N9–H. . .N1 0 in the computed structure at
Table 7Selected computed bond lengths (A) for HL2, 2, HL4, 4, HL5, 5 viasemi-empirical ZINDO/1 (Z1) methods and density functional (DF)methods
Vector HL2, 2 HL4, 4 HL5, 5
Z1 XRD Z1 DF Z1 DF XRD
N1–C2 1.341 1.312 1.357 1.341 1.356 1.341 1.330C2–C3 1.419 1.412 1.402 1.408 1.402 1.408 1.403C3–C4 1.378 1.362 1.389 1.389 1.389 1.389 1.375C4–C18 1.471 1.503C2–N9 1.393 1.381 1.395 1.390 1.396 1.391 1.382N9–N10 1.314 1.357 1.314 1.332 1.309 1.328 1.349N10–C11 1.332 1.302 1.333 1.307 1.337 1.309 1.306C11–C20 1.453 1.481 1.452 1.484 1.451 1.485 1.474C20-N10 1.368 1.348 1.368 1.354 1.368 1.354 1.345N1 0–C6 0 1.355 1.329 1.355 1.337 1.356 1.338 1.340C50–C60 1.386 1.364 1.386 1.394 1.385 1.393 1.370C40–C50 1.399 1.368 1.399 1.397 1.400 1.397 1.373C30–C40 1.387 1.373 1.387 1.391 1.386 1.391 1.382C20–C30 1.405 1.385 1.405 1.412 1.405 1.411 1.393C11–C12 1.460 1.488 1.460 1.496C11–C200 1.456 1.495 1.491C12–C17 1.403 1.381 1.403 1.405C200–C300 1.403 1.408 1.393C16–C17 1.393 1.380 1.393 1.396C300–C400 1.390 1.391 1.371C15–C16 1.394 1.372 1.394 1.398C400–C500 1.396 1.398 1.351C14–C15 1.395 1.376 1.395 1.398C500–C600 1.389 1.395 1.357C13–C14 1.393 1.383 1.393 1.397N100–C600 1.350 1.339 1.348C12–C13 1.405 1.385 1.405 1.406N100–C200 1.363 1.346 1.335N9–H9 1.107 0.860 1.108 1.024 1.118 1.026 0.998
XRD data for both 2 and 5 [34,35] have been reported for comparison.The estimated standard deviations for XRD data are 0.004 A fordistances that involve not-hydrogen atoms. The conformation is ttcc
for 2 and 4 and ttcct00t00 for 5. The numbering of the atoms is thatreported in Scheme 1.
the gas phase when compared to the solid state, and tothe smaller double bond character of N10–C11 fromtheory than from experiment. A good agreement be-tween computed and X-ray structure was also foundfor HL1 and HL2, the largest difference about bond dis-tances being relevant to N9–N10 (ca. 0.04 A). On the
Selected computed bond angles (�) for HL2, 2, HL4, 4, HL5, 5 via semi-empirical ZINDO/1 (Z1) methods and density functional (DF)methods
Vectors HL2, 2 HL4, 4 HL5, 5
Z1 XRD Z1 DF Z1 DF XRD
C2–N1–C8a 117.1 116.8C2–N1–C6 117.0 117.7 117.0 117.7 116.5C2–C3–C4 119.4 119.8 117.5 117.6 117.5 117.6 118.0N1–C2–C3 124.1 124.2 123.4 123.5 123.4 123.4 123.5N1–C2–N9 113.8 114.4 112.9 114.0 112.7 113.9 113.7C3–C2–N9 122.0 121.4 123.7 122.6 123.9 122.6 122.8C2–N9–N10 119.9 119.5 119.4 120.7 119.5 120.9 119.0N9–N10–C11 118.0 119.4 118.2 122.6 117.8 122.8 120.8N10–C11–C12 117.2 113.3 116.7 113.8N10–C11–C20 121.9 128.1 121.7 126.4 121.5 126.4 127.2N10–C11–C200 116.7 112.6 110.8C2 0–C11–C12 120.8 118.7 121.5 119.7C2 0–C11–C200 121.8 121.0 122.0C11–C12–C13 120.6 120.9 120.9 121.3C11–C20–C3 0 123.5 121.1 123.7 121.3 122.5 122.1 122.3C11–C200–C300 123.1 120.3 120.3C11–C20–N10 117.6 116.2 118.1 116.7 117.2 116.9C11–C200–N100 116.1 117.9 117.4N1 0–C20–C3 0 124.1 121.3 120.1 120.7 120.7 120.6 120.9N100–C200–C300 120.9 121.7 122.2C2 0–C30–C40 119.5 119.3 119.6 119.5 119.3 119.4 119.4C200–C300–C400 119.5 119.2 118.6C3 0–C40–C50 120.3 119.5 120.3 119.3 120.2 119.5 119.3C300–C400–C500 119.9 119.1 119.6C4 0–C50–C60 117.4 118.0 117.4 117.8 117.7 117.7 118.4C400–C500–C600 117.2 117.8 119.0C5 0–C60–N10 123.3 124.2 123.3 123.5 123.3 123.4 123.4C500–C600–N100 124.1 123.8 123.8C2 0–N10–C6 0 119.3 117.7 119.3 119.3 118.8 119.5 118.7C200–N100–C600 118.4 118.5 116.8C11–C12–C17 121.6 120.6 121.3 120.2C13–C12–C17 117.8 118.6 117.8 118.4
XRD data for both 2 and 5 [34,35] have been reported for comparison.The estimated standard deviations for XRD data are 0.3� for anglesthat involve not-hydrogen atoms. The conformation is ttcc for 2 and 4
and ttcct00t00 for 5. The numbering of the atoms is that reported inScheme 1.
G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1357
basis of this analysis the ZINDO/1 method was consid-ered a suitable tool to simulate this type of moleculesand it was used also to compute the minimum energystructure for the mono-anionic forms (-H9). The calcu-lations were then performed also for several selectedconformers obtained on rotating around N1–C2–N9–N10, C2–N9–N10–C11, N9–N10–C11–C2 0, N10–C11–C2 0–N1 0 by 180�, for 1, 2, 4 and also N10–C11–C200–N200 for 5. Bond distances and angles do not changemuch upon torsional rotations. Selected molecules werealso optimized at DFT level for comparative purposes.
3.3.1.2. Energy. The heat of formation of the moleculesoptimized are listed in Table 9.
The difference between heat of formation of 5-ttcct00t00
and 5-ctttc00c00 is ca. 1 kcal, the former structure beingthe most stable one in agreement with the structurefound at the solid state via X-ray diffraction. Other con-formers are much less stable, i.e. 5-ttctt00t00 is less stablethan the absolute minimum by ca. 17 kcal. The absoluteminimum for 4 is the ttcc conformer and differs by ca.1 kcal from 4-ctcc. A similar trend has been found for
Table 9Heat of formation DHf� (kcal mol�1) for selected computed molecules (ZIN
Conformation HL1, 1 HL2, 2(L1)� (L2)�
tttt �8700.3499 �10091.6(�8507.1499) (�9909.3
ctcc �8713.6514 �10100.3(�8607.4909)a,b (�9996.8
ttcc �8714.9078 �10101.6(�8612.1829)a,b (�10004.3
cttc �8697.3694 �10089.0(�8505.4014) (�9903.7
ctct �8701.4501 �10089.9(�8511.4868) (�9909.6
tttc �8699.1750 �10090.5(�8506.0869) (�9904.8
ttct �8702.6722 �10092.0(�8511.5939) (�9910.3
cttt �8698.9278 �10090.2(�8507.0772) (�9908.7
ttcct00t00
ctttc00c00
ttcct00c00
ttctt00c00
ttctt00t00
cttc [Cu(L2)(OOCCH3)]�11395.6058c
H+ Cu2+
353.2787 706.8
a The refinement was stopped at RMS 0.1 kcal.b A 1,2,3-triazine ring forms via theory.c The coordination rings were constrained to be planar.
2. In fact, 2-ttcc is the absolute minimum again in agree-ment with the solid state structure and differs by ca.1.3 kcal from 2-ctcc. The heat of formation of HL1
1-ttcc, that has NH syn to N(quinoline) and to N(pyri-dine), is lower than that of 1-ctcc by ca. 1.3 kcal. Inter-estingly, the 1-tttt conformation that brings the methylsyn to NH and corresponds to that found at the solidstate has a heat of formation higher than the absoluteminimum by ca. 15 kcal. It has to be recalled that thisconformation allows the pairing of two molecules viatwo N9–H. . .N1 hydrogen bonds at the solid state; thepairing could account for the preference for tttt confor-mation over ttcc one and for the smaller solubility ofHL1 in polar solvents (like MeOH, 1.12 g/l) when com-pared to HL2 (6.76). In fact, the {HL1}2 aggregateshows a very hydrophobic external surface to the solventmolecules.
Furthermore, the change from HL1-tttt to HL1-cttcfollowed by deprotonation to L1-(-H9)-cttc requires acomputed energy of ca. 548.2 kcal, a value some 10 kcalhigher than the corresponding transformation fromHL2-ttcc to L2-(-H9)-cttc (538.6 kcal). This suggests that
DO/1)
HL4, 4 HL5, 5(L4)� (L5)�
372 �7904.7506a
309) (�7722.3295)a
369 �7914.5048194)b (�7807.5451)a,b
784 �7915.6240354) (�7811.3015)b
735 �7902.9551a
669) (�7717.0673)a
951a �7903.6280a
206)a (�7722.8939)a
566a �7903.6505a
297)a (�7717.7857)a
752 �7904.3174a
732) (�7723.3197)a
091 �7903.2962a
875) (�7722.0471)a
�7660.7705(�7553.8539)a,b
�7659.8560(�7548.8560)a,b
�7656.5457(�7552.0599)a,b
�7645.5892(�7467.0668)�7643.6703(�7464.6347)
CH3COO� CH3COOH708 �1019.0414 �1216.0716

1358 G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359
the complex formation from HL1 is predictably less fa-vored than that from HL2 even in the case the steric hin-drance due to the methyl group at C8 of the quinolinylgroup is not taken into account.
Finally, the barrier for the rotation around 1- and 2-ttcc N10–C11 is high (ca. 120 kcal) and this is related tothe high double bond character for N10–C11 and to thelack of the strong intramolecular N9–H. . .N1 0.
3.3.1.3. UV–Vis spectra. The computed spectrum forHL1, 1, has absorption bands at 337 nm (oscillatorstrength 0.699; HOMO–LUMO transition, MolecularOrbitals, MO # 55–56), 315 (0.445; MO # 55–57), 232(0.271; MO # 53–56) and 213 (0.645; MO # 54–59).The computed spectrum for L1-(-H9)-cttc has the fol-lowing selected absorption bands: 467 nm (oscillatorstrength 0.955) that corresponds to a mono-electronjump from HOMO (MO # 55) to LUMO (# 56) andto a charge shift from phenyl grouping to the N–Nand quinoline system; 362 nm (0.255) that correspondsto a transition from HOMO to LUMO + 2; 262 nm(0.327) that corresponds to a transition from HOMO-1 to LUMO, i.e. to a charge shift from quinoline to-wards pyridine. The computed spectrum for HL2, 2,has selected peaks at 413 nm (0.621; HOMO (# 63)–LUMO (# 64)), 307 (0.394; # 63–65), 234 (0.488; #63–69), whereas that of L2-(-H9)-cttc has peaks at453 nm (0.856; HOMO–LUMO), 354 (0.295; HOMO–LUMO + 2) and 212 (0.725; HOMO-3-LUMO).
The computed spectrum of 3 at ZINDO/S level withr–r and p–p constants of 1.267 and 0.585 has peaks at587 nm (0.214), 307 (0.164), 232 (0.209) in better agree-ment with those found in CH2Cl2 (this solvent is consid-ered as not coordinating towards copper(II)) than withthat found in MeOH.
3.3.2. Density functional
The computed structure of 5-ttcct00t00 (Tables 7 and 8)has a very good agreement with the X-ray structure[34,35]. Even the bond distances and angles at thebis(pyridyl)hydrazone moiety are very well reproduced,the difference relevant to N9–N10 bond length being0.021 A. Thus the DFT methods is able to reproducethe X-ray structures of this type of molecules better thansemi-empirical ZINDO/1 methods at least in the casethe expanded basis set 6-31+G* are used. However ithas to be noted that time and the computational costsare much higher at DFT than at ZINDO/1 level. Similargood results have been obtained on optimizing HL4, 4,in the ttcc conformation.
4. Conclusion
This work reports on the synthesis and the struc-tural characterization of the first copper complex ever
studied via X-ray diffraction with a ribonucleotidereductase RR inhibitor and an effective anti-prolifera-tive agent from the family of 2-benzoylpyridine-2 0-quinolinyl-hydrazones. The metal compound, a potentialanticancer and oxygen radical scavenger by itself, hasthe metal linked to three nitrogen atoms (quinolinyl,hydrazone, pyridyl) from the N-deprotonated ligandand to an acetate anion. The ability of the ligand toform strong tri-coordinate metal-species, confirms thehypothesis that the inhibition of RR passes throughthe chelation of the enzyme�s metal centers. The com-plex strongly absorbs in the visible region (ca. 500 nmand absorbivity ca. 23000 M�1 cm�1); this suggests toinvestigate the possible activity of such a type of com-plexes as photo-therapeutic agents. It is well knownthat photodynamic therapies (PDTs) are an approachfor the treatment of superficial tumors and not-malig-nant diseases [43,44]. The anticancer molecules HL1
and HL2 are strong light absorbers in the UVA regionand their photochemical properties and possibly theirusage as PDT agents could be worth of testing. Infact, previously published works showed that certainhydrazones have interesting photo-reactivity [45]. Fur-thermore, this work shows that the deprotonation andmetal coordination of such ligands shift the absorptionof light into the visible region, i.e. toward a safer radi-ation for healthy tissues. Thus, copper(II)-complexesof the type prepared in this work are much powerfulpotential photodynamic agents than the free ligands,provided they have a suitable photochemistry in thebiological environment and they are selectively ab-sorbed by the target tissue. Finally, the coordinatingmode of HL1 probably is different from that of HL2.In fact, metal ions could not link to N(q), becauseof the presence of a bulky Me group at C8, close toN1. It has to be noted that a search on the CSDBfor a group that consists of a Cu2+ ion linked toN(q) and N10 of a 8-methyl-2-(N9–N10)-quinolinylfragment, gave no hints.
Acknowledgements
The authors acknowledge the invaluable suggestionsby the late Prof. Cesare Pellerano.
Thanks are expressed to Dr. F. Berrettini for the X-ray data collections at CIADS (Centro Interdiparti-mentale di Analisi e Determinazioni Strutturalidell�Universita di Siena). Prof. C. Rossi and Dr. A.Tognazzi, Department of Chemical and Biosystem Sci-ences and Technologies, University of Siena, areacknowledged for the gas-chromatographic analysis.The work was supported by the University of Siena,and Ministero dell�Istruzione, Universita e Ricerca(MIUR, Roma).

G. Tamasi et al. / Journal of Inorganic Biochemistry 99 (2005) 1347–1359 1359
Appendix A. Supplementary data
Crystallographic data for the structure analysis havebeen deposited (as CIF formatted files) with the Cam-bridge Crystallographic Data Center, CCDC Nos.254757-254759 for 1, 2, 3 Æ 0.9C6H5CH3. Copies of thisinformation may be obtained free of charge from theDirector, CCDC, 12 Union Road, Cambridge, CB21EZ, UK (fax: +44 1223 336033; e-mail: [email protected] or www: http://www.ccdc.cam.ac.uk). Tablesof atomic coordinates for the computed structures viaZINDO/1 and DF theory methods, a Scheme relatedto the treatment of disorder for the toluene moleculesand a table for the NMR have been deposited as sup-porting material. Supplementary data associated withthis article can be found, in the online version atdoi:10.1016/j.jinorgbio.2005.03.009.
References
[1] J. Easmon, G. Heinisch, G. Purstinger, T. Langer, J.K. Osterrei-cher, H.H. Grunicke, J. Hofmann, J. Med. Chem. 40 (1997) 4420–4425, and references cited therein.
[2] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat,H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein DataBank, Nucleic Acid Res. 28 (2000) 235–242.
[3] W.C. Voegtli, M. Sommerhalter, L. Saleh, J. Baldwin, J.M.Bollinger Jr., A.C. Rosenzweig, J. Am. Chem. Soc. 125 (2003)15822–15830.
[4] I.N. Zelko, T.J. Mariani, R.J. Folz, Free Radic. Biol. Med. 33(2002) 337–349, and references cited therein.
[5] S. Ramadan, T.W. Hambley, B.J. Kennedy, P.A. Pay, Inorg.Chem. 43 (2004) 2943–2946, and references cited therein.
[6] S. Defazio, R. Cini, Polyhedron 22 (2003) 1355–1366.[7] S. Defazio, R. Cini, J. Chem. Soc., Dalton Trans. (2002) 1888–
1897.[8] R. Cini, Comments Inorg. Chem. 22 (2000) 151–186.[9] D. Di Leo, F. Berrettini, R. Cini, J. Chem. Soc., Dalton Trans.
(1998) 1993–2000.[10] R. Cini, J. Chem. Soc., Dalton Trans. (1996) 111–116.[11] R. Cini, R. Pogni, R. Basosi, A. Donati, C. Rossi, L. Sabadini,
L. Rollo, S. Lorenzini, R. Gelli, R. Marcolongo, Metal BasedDrugs 2 (1995) 43–56.
[12] R. Cini, G. Giorgi, A. Cinquantini, C. Rossi, M. Sabat, Inorg.Chem. 29 (1990) 5197–5200.
[13] The National Cancer Institute, USA, The Developmental Ther-apeutics Program NCI/NIH. Available from: http://dtp.nci.nih.gov/docs/dtp_search.html, Compound # 624662.
[14] L. Savini, P. Massarelli, L. Chiasserini, A. Sega, C. Pellerano, A.Barzi, G. Nocentini, Eur. J. Med. Chem. 30 (1995) 547–552.
[15] L. Savini, P. Massarelli, L. Chiasserini, C. Nencini, C. Pellerano,Il Farmaco, Ed. Sc. 52 (1997) 609–613.
[16] C. Pellerano, L. Savini, L. Selvolini, Boll. Chim. Farm. 117 (1978)721–730.
[17] G. Giorgi, F. Ponticelli, L. Savini, L. Chiasserini, C. Pellerano, J.Chem. Soc., Perkin Trans. 2 (2000) 2259–2264.
[18] G. Giorgi, L. Savini, L. Chiasserini, C. Nencini, C. Pellerano,Polyhedron 17 (1998) 3851–3858.
[19] C. Pellerano, L. Savini, P. Massarelli, Il Farmaco, Ed. Sc. 40(1985) 645–654.
[20] XSCANS User Manual; Siemens Analytical X-ray InstrumentsInc., Madison, WI, 1994.
[21] XEMP Empirical Absorption Correction Program; SiemensAnalytical X-ray Instruments Inc., Madison, WI, 1994.
[22] N. Walker, D. Stuart, Acta Cryst. A39 (1983) 158–166.[23] G.M. Sheldrick, SHELXS-86, Program for the Solution of Crystal
Structures, University of Gottingen, 1986.[24] G.M. Sheldrick, SHELX-97, Program for the Solution and
Refinement of Crystal Structures, University of Gottingen, 1997.[25] A. Altomare, M.C. Burla, M. Camalli, G.L. Castarano, C.
Giacovazzo, A. Guagliardi, A.G.C. Moliterni, G. Polidori, B.Spagna, SIR-92, A package for crystal structure solution by directmethods and refinement, J. Appl Cryst. 32 (1999) 115–119.
[26] L.J. Farrugia, WinGX an Integrated System of WindowsPrograms for the Solution, Refinement and Analysis of SingleCrystal X-ray Diffraction Data, Version 1.64.05, University ofGlasgow, 1999.
[27] L.J. Farrugia, J. Appl. Cryst. 32 (1999) 837–838.[28] M. Nardelli, PARST-97, A System of Computer Routines for
Calculating Molecular Parameters from Results of Crystal Struc-ture Analyses, University of Parma, 1997.
[29] C.K. Johnson, M.N. Burnett, ORTEP-3 for Windows, Oak RidgeNational Laboratory, 1998. 32-bit Implementation by Farrugia,L.J. University of Glasgow.
[30] HyperChem�, Molecular Modeling System, Release 5.1 Pro forWindows, Hypercube Inc., Gainesville, FL, 1997.
[31] HyperChem�, Reference Manual, Hypercube Inc., Gainesville,FL, 1997.
[32] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A.Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery Jr.,R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam, A.D.Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V.Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz, A.G.Baboul, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I.Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith,M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M.Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong,J.L. Andres, C. Gonzalez, M. Head-Gordon, E.S. Replogle,J.A. Pople, GAUSSIAN-98, Revision A.7, Gaussian Inc.,Pittsburgh PA, 1998.
[33] A. Frisch, M.J. Frisch, GAUSSIAN-98, User�s Reference, seconded., Gaussian Inc., Pittsburgh, PA, 1998.
[34] C.F. Ishak, R.T. Pflaum, N.C. Baenziger, Acta Cryst. C40 (1984)2047–2049.
[35] T. Taya, T. Sakamoto, K. Doi, M. Otomo, Bull. Chem. Soc. Jpn.66 (1993) 3652–3656.
[36] S. Defazio, G. Tamasi, R. Cini, Comptes Rendus Chimie, in press.[37] The Cambridge Crystallographic Data Center, The Cambridge
Crystallographic Data Base, Version 5.24, November 2002.[38] A.T. Casey, B.F. Hoskins, I.P. Traverso, Aust. J. Chem. 37 (1984)
739–744.[39] A. Bondi, J. Phys. Chem. 68 (1964) 441–446.[40] M. Nishio, Cryst. Eng. Comm. 6 (2004) 130–137.[41] R.K. Hocking, T.W. Hambley, Inorg. Chem. 42 (2003) 2833–
2835.[42] K. Nakamoto, Infrared and Raman Spectra of Coordination
Compounds, fourth ed., J. Wiley & Sons, New York, 1987.[43] M.J. McKeage, L. Maharaj, S.J. Bernes-Price, Coord. Chem.
Rev. 232 (2002) 127–132.[44] A. Massey, Y.Z. Xu, P. Karran, Curr. Biol. 11 (2001) 1142–1145.[45] R.S. Becker, F. Chagneau, J. Am. Chem. Soc. 114 (1992) 1373–
1381.
Related Documents











