UNIVERSITÉ PARIS-SUD 11 FACULTÉ DES SCIENCES D'ORSAY ECOLE DOCTORALE : INNOVATION THÉRAPEUTIQUE : DU FONDAMENTAL A L’APPLIQUÉ PÔLE : INGENIERIE DES PROTEINES ET CIBLES THERAPEUTIQUES ANNÉE 2009 - 2010 SÉRIE DOCTORAT N° THÈSE Présentée À L’UNITÉ DE FORMATION ET DE RECHERCHE FACULTE DES SCIENCES D'ORSAY UNIVERSITÉ PARIS-SUD 11 pour l’obtention du grade de DOCTEUR DE L’UNIVERSITÉ PARIS-SUD 11 par M. Julien HENRI ETUDES STRUCTURALES ET FONCTIONNELLES D’ACTEURS DE LA TERMINAISON DE LA TRADUCTION ET DE LA STABILITE DES ARN MESSAGERS EUCARYOTES soutenue le : jeudi 30 septembre 2010 JURY : Dr Annie MOUGIN Dr. Sébastien FRIBOURG Dr. Yves MECHULAM Dr. Laurent CHAVATTE Dr. Marc GRAILLE Pr. Herman VAN TILBEURGH

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

UNIVERSITÉ PARIS-SUD 11
FACULTÉ DES SCIENCES D'ORSAY
ECOLE DOCTORALE : INNOVATION THÉRAPEUTIQUE : DU FONDAMENTAL A L’APPLIQUÉ
PÔLE : INGENIERIE DES PROTEINES ET CIBLES THERAPEUTIQUES
ANNÉE 2009 - 2010 SÉRIE DOCTORAT N°
THÈSE
Présentée
À L’UNITÉ DE FORMATION ET DE RECHERCHE
FACULTE DES SCIENCES D'ORSAY
UNIVERSITÉ PARIS-SUD 11
pour l’obtention du grade de DOCTEUR DE L’UNIVERSITÉ PARIS-SUD 11
par M. Julien HENRI
ETUDES STRUCTURALES ET FONCTIONNELLES D’ACTEURS DE LA TERMINAISON DE LA TRADUCTION ET DE LA STABILITE
DES ARN MESSAGERS EUCARYOTES
soutenue le : jeudi 30 septembre 2010 JURY : Dr Annie MOUGIN
Dr. Sébastien FRIBOURG
Dr. Yves MECHULAM
Dr. Laurent CHAVATTE
Dr. Marc GRAILLE
Pr. Herman VAN TILBEURGH


1
Sommaire
Introduction .............................................................................................................................. 4
Avant-propos : l'information génétique...........................................................................................4 1. Etapes initiales de l'expression génique .......................................................................................8
1.1. Transcription.............................................................................................................................8 1.2. Maturation.................................................................................................................................8 1.3. Translocation ..........................................................................................................................13
2. La traduction ................................................................................................................................16
2.1. Acteurs de la traduction ..........................................................................................................16 2.2. Mécanisme moléculaire de la traduction eucaryote................................................................19
3. La terminaison de la traduction..................................................................................................25
3.1. Mécanisme moléculaire de la terminaison..............................................................................25 3.2. Facteurs de terminaison de classe I : spécificité, structure et mode de reconnaissance. ........27 3.3. Facteurs de terminaison de classe II : rôle et structure. ..........................................................32 3.4. Structure et mécanisme d'action du complexe de terminaison eRF1-eRF3 ...........................37 3.5. Protéines influençant l’efficacité de terminaison : .................................................................40
4. Stabilité des ARN messagers et dégradation .............................................................................51
4.1. Dégradation normale...............................................................................................................52 4.2. Dégradation ciblée spécifique.................................................................................................58
5. Couplage des mécanismes de l'expression génique ...................................................................71 6. Présentation de la thématique de recherche ..............................................................................73
Stratégie expérimentale ......................................................................................................... 75
1. Expression et purification des protéines ....................................................................................75
1.1. Clonage ...................................................................................................................................75 1.2. Surexpression en système bactérien .......................................................................................76 1.3. Purification..............................................................................................................................76
2. Caractérisation du matériel protéique .......................................................................................78
2.1. Contrôles de routine...............................................................................................................78 2.2. Diffusion de la lumière laser à différents angles (MALLS) ...................................................78 2.3. Dichroïsme circulaire (CD) ....................................................................................................79
3. Cristallisation................................................................................................................................79

2
Résultats .................................................................................................................................. 81 1. Tpa1 et Ett1, nouveaux acteurs de la terminaison de la traduction ........................................81
1.1. Tpa1, structure et fonction ......................................................................................................81 Article 1 : Structural and functional insights into S. cerevisiae Tpa1, a putative prolyl hydroxylase influencing translation termination and transcription .........................................113
1.2. Structure de Nro1, orthologue d'Ett1 ....................................................................................117 Article 2 : Structural and functional study of Nro1/Ett1, a protein involved in translation termination in S. cerevisiae and in the control of gene expression in response to O2 levels in S. pombe. ..............................................................................................................................................129
1.3. Etude biochimique de Tpa1-Ett1 ..........................................................................................130 Conclusions, Perspectives............................................................................................................142
2. Dom34-Hbs1, complexe central du No-Go Decay et du Non-functional rRNA Decay .........149
2.1. Procédures expérimentales ...................................................................................................150 2.2. Détermination de la structure : Résultats ..............................................................................157 2.3. Caractérisations fonctionnelles .............................................................................................159
Article 3 : Structural and functional dissection of the Dom34-Hbs1 complex reveals independent functions in NGD and NRD.....................................................................................166
Conclusions, Perspectives............................................................................................................167 Conclusion générale ............................................................................................................. 172 Bibliographie......................................................................................................................... 174

3

4
Introduction
Avant-propos : l'information génétique
Une cellule vivante est un système délimité dans l'espace qui peut assurer de façon
autonome sa conservation, sa reproduction et sa régulation. La fonction d'une cellule, sa
forme et ses capacités d'adaptation et d'évolution sont déterminées par l'information contenue
dans son génome.
Il contient l'ensemble des informations nécessaires pour garantir le fonctionnement
normal et l'homéostasie d'une cellule. Les expériences d'Avery, MacLeod et MacCarty ont
révélé que les acides nucléiques sont le support physique de l'information génétique. L'acide
desoxyribonucléique (ADN) est le support génétique des bactéries, des archées et des
eucaryotes ainsi que de certains virus. La molécule d'ADN est un enchaînement linéaire des
nucléosides adénosine, thymidine, guanosine et cytidine reliés par des liaisons
phosphodiester; les bases nucléosidiques sont complémentaires deux à deux : adénine et
thymine d’une part, guanine et cytosine d’autre part; deux brins d'ADN antiparallèles
s'associent en formant une double hélice régulière. L'ordre d'enchaînement des nucléotides sur
un brin constitue la séquence de l'ADN. La complémentarité des bases confère la possibilité
de créer deux copies de séquences identiques d'une molécule d'ADN lors de la réplication

5
(Meselson and Stahl, 1958). Le génome est ainsi transmis aux cellules-filles issues d'une
division et conservé d'une génération à la suivante, de telle sorte qu'une cellule-fille contient
la copie de l'ADN de la cellule-mère dont elle est issue : l'information génétique portée par
l'ADN est héréditaire.
Le génome est organisé en gènes, des segments d'ADN contenant l'information
nécessaire pour produire les protéines. Celles-ci remplissent les fonctions générales de
structure, transport, régulation, signalisation et motricité de toute cellule vivante. De multiples
étapes sont nécessaires pour exprimer une protéine à partir d'un gène : l'ADN est transcrit en
une molécule d'ARN messager (ARNm), puis l'ARNm sert de modèle pour la production de
la protéine (Figure 1) (Crick, 1970).
L'ARNm est un polymère linéaire de ribonucléotides dont la séquence est issue de l'ADN
et qui code pour la séquence d'acides aminés d'un polypeptide. Il sert de convoyeur
d'information entre l'information héréditaire (ADN) et l'information fonctionnelle (protéines)
au niveau de sa séquence codante : la suite des triplets de nucléotides dicte la séquence
primaire des polypeptides traduits selon le code génétique.
L'ARNm est une copie en séquence de l'ADN d'un gène, avec des caractéristiques
distinctes importantes :
- la molécule d'ARNm étant plus courte que l'ADN, elle peut être transportée dans la
cellule à travers la membrane séparant le noyau du cytoplasme;
- les nucléotides composant l'ARNm portent un hydroxyl (-OH) en 2' de leur ribose (ce
sont des ribonucléotides), ce qui confère à la molécule à la fois une plus grande flexibilité
mais aussi une plus grande sensibilité à l'hydrolyse;
- les nucléosides de l'ARNm sont l'adénosine, la cytidine, la guanosine et l'uridine, cette
dernière remplaçant la thymidine de l'ADN.

6
L'ARNm est un simple brin d'acide nucléique soumis comme tel aux lois de
l'appariement des bases de Chargaff. Si la séquence des bases représente la structure primaire
de l'information portée, l'ARNm adopte également des structures tridimensionnelles,
dépendantes de la présence de séquences complémentaires voisines dans la molécule. Ces
séquences s'apparient pour former des doubles brins et induisent l'apparition de structures
secondaires dont les tiges-boucles. Les structures secondaires de l'ARNm peuvent lui conférer
une fonction particulière de par leur forme (site alternatif de traduction) ou leurs propriétés
catalytiques (ribozymes).
L'expression génique est une fonction essentielle à toute cellule vivante. Son niveau, sa
fidélité et son efficacité sont précisément contrôlés. Au cours de ma thèse je me suis
concentré sur les évènements finaux de l'expression génique, particulièrement la traduction de
l'ARNm en protéines, et sur les régulations prenant place autour de la molécule d'ARNm à
cette étape. Nous avons ciblé des acteurs récemment identifiés de la traduction et de la
stabilité des ARNm et avons entrepris pour chacun une caractérisation structurale par
cristallographie aux rayons X et une étude fonctionnelle dans le cadre d’une collaboration
avec l’équipe de Bertrand Séraphin.
Le travail réalisé est focalisé sur l'ARNm en tant qu'acteur central de l'expression
génique. Sauf indication contraire, nous avons privilégié la levure ascomycète Saccharomyces
cerevisiae comme organisme eucaryote modèle.

7
Figure 1 : L'ARN messager est au centre de l'expression génique : l'ADN est transcrit en ARNm qui, après maturation, est exporté vers le cytoplasme où il est traduit en protéine (Crick, 1970).
m7G
D’après www.genome.gov
AAAAA…AAAAA

8
1. Etapes initiales de l'expression génique
Synthèse, maturation et transport de l'ARNm
1.1. Transcription
L'expression génique débute par la transcription, production d'une copie ARN messager
des séquences d'ADN encadrée par un promoteur et un terminateur (Kornberg, 2007). La
transcription des ARNm est catalysée par l'ARN polymérase II; elle est soumise à un strict
contrôle spatio-temporel.
D'une part l'ADN ne peut être transcrit que si les séquences promotrices de recrutement
de l'ARN polymérase sont accessibles. Ainsi, le surenroulement de l'hélice d'ADN et sa
structuration en nucléosomes par des protéines de compaction, les histones, le rendent inapte
à la transcription. La méthylation de bases cytosine de l'ADN réprime également la
transcription. D'autre part des effecteurs protéiques jouent un rôle activateur ou inhibiteur en
se fixant sur des séquences spécifiques de régulation; ils ajustent le niveau de la transcription
en fonction des besoins métaboliques de la cellule.
1.2. Maturation
Dès sa transcription l'ARN messager subit des modifications chimiques groupées sous le
terme de maturation, lui conférant ses fonctions ultérieures et assurant sa stabilité contre la
dégradation. Un ARNm subit successivement trois étapes de maturation : protection par une
coiffe en 5', protection par une queue poly(A) en 3' et épissage des introns.

9
1.2.1. Capping (ajout d'une coiffe) en 5'
Les pré-ARNm nouvellement transcrits sont la cible d'une modification en 5', en amont
de la séquence codante. Une série de trois activités enzymatiques successives résulte en l'ajout
d'une guanosine méthylée en N7 (appelée m7G) reliée au premier nucléotide (5'-pppN) de
l'ARNm par une liaison inversée 5'-5' (Figure 2) (Cowling; Gu and Lima, 2005). Les enzymes
de capping sont recrutées par l'ARN polymérase II après la synthèse de 25 à 30 nucléotides
du transcrit ARNm; c'est le domaine C-terminal (CTD) phosphorylé de l’ARN polymérase II
qui recrute directement la machinerie de capping (McCracken et al., 1997).
La première étape du capping est l'hydrolyse du premier nucléotide 5'-pppN du pré-
ARNm en 5'-ppN par une ARN triphosphatase : Cet1 chez la levure S. cerevisiae (Tsukamoto
et al., 1997) ou Mce1 chez les eucaryotes supérieurs. La guanylyl-transférase (Ceg1 chez
Saccharomyces cerevisiae (Shibagaki et al., 1992)) transfère ensuite un GTP sur le 5'-ppN en
passant par un intermédiaire covalent enzyme-(lysyl-N)-GMP, puis en fixant le GMP sur
l'ARNm formant ainsi le Gppp-ARNm. Les guanylyl-transférases contiennent un domaine
nucléotidyl-transférase (NT) et un domaine à repliement OB. Chez les eucaryotes supérieurs,
l'ARN-triphosphatase et la guanylyl-transférase sont associées en une seule protéine à
domaines séparés tandis que chez la levure S. cerevisiae, les protéines Cet1 et Ceg1 forment
un complexe hétérodimérique. En dernière étape du capping, la guanine-N7-méthyltransférase
(Abd1 chez S. cerevisiae (Wang and Shuman, 1997)) transfère un groupement méthyl de la S-
adénosyl-méthionine au Gppp-ARNm, formant le m7Gppp-ARNm.
Cette structure particulière protège l'ARNm contre les dégradations exonucléolytiques
5'3' en empêchant les exonucléases d'accéder à une séquence hydrolysable. La dégradation
de la coiffe est par conséquent une étape cruciale lors de la dégradation des ARNm matures,
comme nous le verrons plus loin.
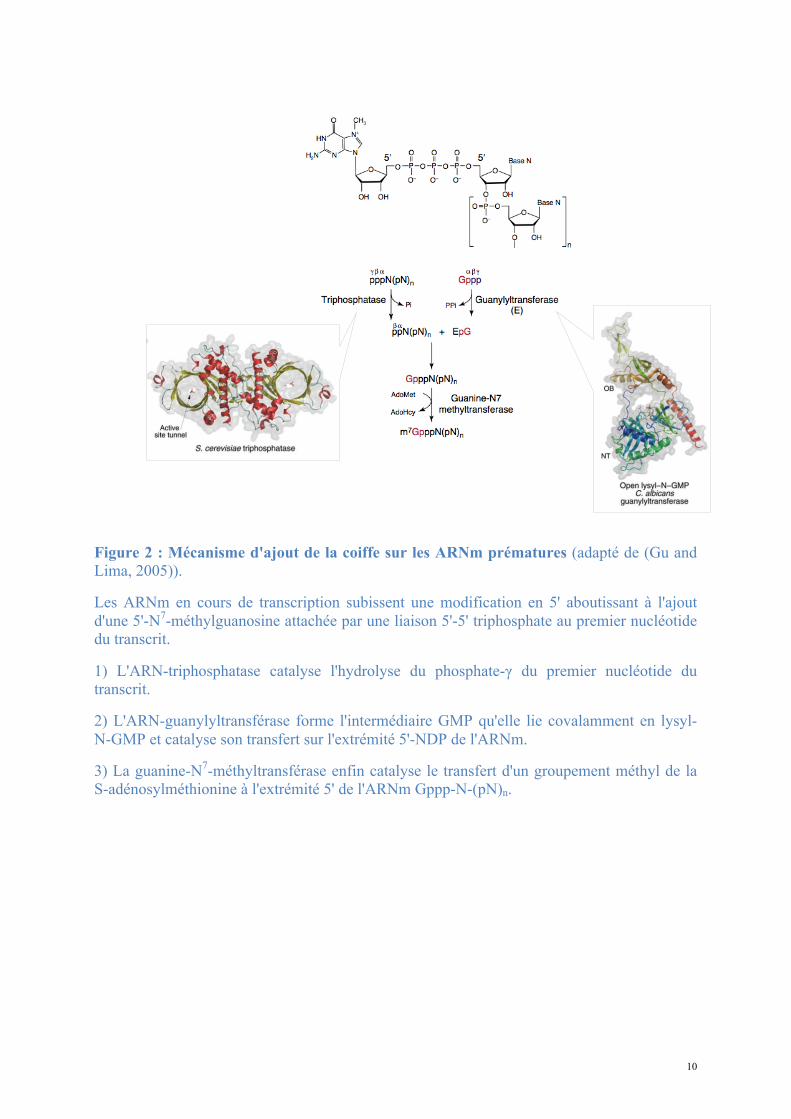
10
Figure 2 : Mécanisme d'ajout de la coiffe sur les ARNm prématures (adapté de (Gu and Lima, 2005)).
Les ARNm en cours de transcription subissent une modification en 5' aboutissant à l'ajout d'une 5'-N7-méthylguanosine attachée par une liaison 5'-5' triphosphate au premier nucléotide du transcrit.
1) L'ARN-triphosphatase catalyse l'hydrolyse du phosphate-γ du premier nucléotide du transcrit.
2) L'ARN-guanylyltransférase forme l'intermédiaire GMP qu'elle lie covalamment en lysyl-N-GMP et catalyse son transfert sur l'extrémité 5'-NDP de l'ARNm.
3) La guanine-N7-méthyltransférase enfin catalyse le transfert d'un groupement méthyl de la S-adénosylméthionine à l'extrémité 5' de l'ARNm Gppp-N-(pN)n.

11
1.2.2. Polyadénylation en 3'
A l'exception de ceux codant pour certaines histones, tous les ARNm eucaryotes
subissent une modification en deux étapes de leur extrémité 3' : clivage endonucléolytique
suivi par l’addition d'une série d'adénosines appelée queue poly(A) (Figure 3) (Danckwardt et
al., 2008; Millevoi and Vagner, 2010; Zhao et al., 1999).
Deux signaux canoniques en aval de la séquence codante de l'ARNm assurent le
recrutement de la machinerie protéique de clivage :
- La séquence AAUAAA est reconnue par le complexe CPSF, déterminant le clivage 10 à
30 nucléotides en aval, généralement avant un motif CA (Murthy and Manley, 1995).
- La séquence riche en G/U ou U située 30 nucléotides après le site de clivage CA et
appelée DSE (downstream sequence element) recrute le facteur hétérotrimérique CstF
stimulant le clivage (Takagaki et al., 1990).
D'autres facteurs accessoires peuvent être recrutés sur des séquences USEs placées en
amont du site de clivage (upstream sequence elements) et jouent également un rôle activateur
sur le complexe de clivage-polyadénylation. Un motif UGUA, également accessoire et
présent en une ou plusieurs copies à des distances variables du site CA, représente un mode
alternatif de recrutement de la machinerie de clivage. Après assemblage des complexes de
clivage, la coupure serait catalysée par Cpsf73 (Kolev et al., 2008). L'extrémité 3' ainsi
générée sert de substrat à l'addition d'environ 250 adénosines par la poly(A) polymérase PAP
(Proudfoot and O'Sullivan, 2002).
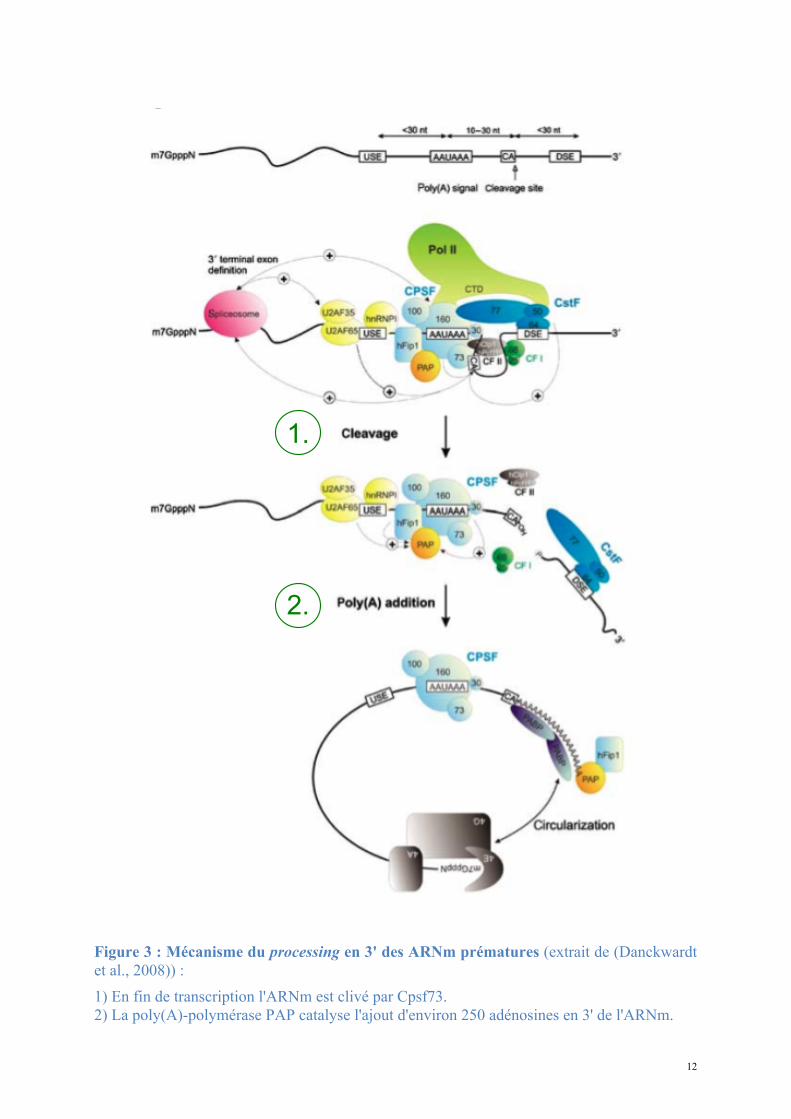
12
Figure 3 : Mécanisme du processing en 3' des ARNm prématures (extrait de (Danckwardt et al., 2008)) : 1) En fin de transcription l'ARNm est clivé par Cpsf73. 2) La poly(A)-polymérase PAP catalyse l'ajout d'environ 250 adénosines en 3' de l'ARNm.
1.
2.

13
1.2.3. Epissage
L'ARNm transcrit porte l'information codant pour la protéine exclusivement au niveau de
certaines séquences transcrites : les exons. Les exons sont séparés par des séquences non
codantes appelées introns, qui doivent être éliminées pour la production normale de protéines.
L'épissage consiste en une excision des introns et une ligation des exons restants. Les
réactions d'épissage sont catalysées par les ribonucléoprotéines du complexe d'épissage (U1,
U2, U5, U6) et par l'ARNm lui-même qui adopte une structure caractéristique en lasso. Ceci
résulte en un raccourcissement de la séquence de l'ARNm.
Un intron n'est pas systématiquement considéré comme tel et pourra parfois être conservé
dans la séquence finale au même titre qu'un exon; de même, un exon pourra être excisé dans
certains cas. L'épissage est alors dit alternatif et l'ARNm après maturation pourra avoir des
séquences différentes appelées variants d'épissage. Ainsi à partir d'un gène on obtiendra
éventuellement plusieurs ARNm différents, ceux-ci codant pour des protéines de différentes
longueurs voire de différentes séquences. L'épissage alternatif est considéré comme la plus
grande source de diversité des protéines chez les Vertébrés (Nilsen and Graveley, 2010).
Après ligation des exons, les sites d'épissages sont marqués chez les eucaryotes supérieurs par
la présence d'un complexe protéique de jonction des exons (EJC) qui y remplace le complexe
d'épissage.
1.3. Translocation
Chez les eucaryotes, l'ADN est contenu dans un compartiment cellulaire délimité par une
membrane : le noyau. La composition du noyau est différente de celle du reste de la cellule (le
cytoplasme). Les échanges de matières entre le noyau et le cytoplasme se font
majoritairement par les pores nucléaires, des structures cylindriques octamériques jouant le

14
rôle de tunnel (Strambio-De-Castillia et al., 2010). Le passage à travers le pore nucléaire est
non-spécifique et non-orienté pour un objet de petite taille (moins de 30kDa), capable de
diffuser passivement : c'est le cas des molécules d'eau, des ions et de certains métabolites. Ce
passage sera assisté par un mécanisme actif couplé à une consommation d'énergie pour les
molécules de plus grande taille. La taille des molécules d'ARNm les contraint à traverser le
pore nucléaire par cette voie active et à être convoyées jusqu'au cytoplasme lorsque les étapes
de maturation sont accomplies (Kelly and Corbett, 2009) (Figure 4).
Le transport de l'ARNm à travers le pore nucléaire est strictement orienté du noyau vers
le cytoplasme où a lieu la traduction des protéines. Chez S. cerevisiae des protéines
adaptatrices sont déposées sur les ARNm pendant la transcription et la maturation. Elles
recrutent des facteurs d'export nucléaire, couplant ainsi les étapes initiales de la production
d'ARNm à son transport ultérieur.
(i) C'est d'abord le complexe protéique THO qui est recruté sur l'ARNm pendant la
transcription, puis THO et le domaine CTD de l'ARN-polymérase II recrutent les protéines
Hrp1, Npl3, Sub2, Yra1 et Nab2. Le principal facteur d'export nucléaire est le complexe
Mex67-Mtr2 (Santos-Rosa et al., 1998), dont le recrutement déplace Sub2 et Yra1 lorsque la
transcription et la maturation en 3' sont résolues.
(ii) Les complexes ribonucléoprotéiques (mRNPs) quittent alors le noyau à travers le
complexe du pore nucléaire. Ce dernier est formé par un assemblage de protéines appelées
nucléoporines (Nups) dont certaines contiennent des répétitions de motifs phénylalanine-
glycine (FG); les FG-Nups tapissent la paroi intérieure du pore et assurent le passage contrôlé
des molécules vers le cytoplasme ou vers le noyau. Les FG-Nups interagissent faiblement
avec Mex67 pour faciliter le transport rapide et sélectif des mRNPs (Strasser et al., 2000).
(iii) Dans le cytoplasme, l'hélicase Dbp5 remodèle les mRNPs et dissocie les facteurs
d'export Mex67 et Nab2 des ARNm. Dbp5 est activée par son partenaire Gle1, elle-même

15
activée par l'hexakisphosphate (Alcazar-Roman et al., 2010).
Dans le cytoplasme, le facteur protéique eIF4E se fixe sur le m7G en 5’ des ARNm et la
protéine Pab1 se fixe sur la queue poly(A) en 3’ des ARNm. Ces deux protéines interagissent
entre elles via eIF4G pour former une structure circulaire fermée. A ce stade l'ARNm mature
dans le cytoplasme est directement prêt pour entrer dans l'étape finale de l'expression.
Figure 4 : Export nucléo-cytoplasmique de l'ARNm (extrait de (Kelly and Corbett, 2009) :
Un ensemble de protéines assurent le transport rapide et efficace de l'ARNm nouvellement transcrit et maturé du noyau vers le cytoplasme.

16
2. La traduction
Synthèse ARN-dépendante des protéines
L'ARNm mature sert de matrice à la production de polypeptides au cours de l'étape de
traduction qui a lieu dans le cytoplasme. La traduction est une réaction de polymérisation des
acides aminés catalysée par un complexe macromoléculaire de grande taille, le ribosome, et
des ARN spécialisés, les ARN de transfert (ARNt). La description présentée ici est axée sur
les systèmes eucaryotes.
2.1. Acteurs de la traduction
2.1.1. L'ARN messager
L'ARNm mature dans le cytoplasme est présent sous une forme circularisée et associée à
de nombreuses protéines sous la forme d'assemblages ribonucléoprotéiques de grande taille,
les mRNP. Une visualisation directe par microscopie électronique confirme l'organisation
circulaire des ARNm traduits (Kopeina et al., 2008; Wells et al., 1998). Les ARNm sont
traduits simultanément par plusieurs ribosomes, les assemblages complets étant nommés
polyribosomes.
2.1.2. Les ARN de transfert
Ce sont de courtes molécules d'ARN (70 à 95 nucléotides) fortement modifiées et
structurées (D. Peter Snustad, 2010). Ils présentent deux caractéristiques essentielles
déterminant leur fonction :
(i) une boucle anticodon de trois nucléotides capables de s'apparier de manière

17
complémentaire avec la séquence de trois nucléotides d'un codon de l'ARNm;
(ii) un site de fixation covalente d'un acide aminé en 3'.
Selon les organismes, il existe 30 à 46 ARNt différents qui vont reconnaître les 61 codons
sens selon les correspondances du code génétique. Les ARNt servent ainsi de « décodeurs » et
d'adaptateurs entre les codons de l'ARNm et les acides aminés du polypeptide en formation.
Les ARNt interagissent avec le ribosome.
2.1.3. Le ribosome
C’est un complexe macromoléculaire de protéines et d'ARN ribosomiques (ARNr)
synthétisant les protéines dans toute cellule vivante (D. Peter Snustad, 2010). Le ribosome est
un assemblage moléculaire de grande taille (25nm et 2,5MDa chez les procaryotes, 4,2MDa
chez les eucaryotes), fonctionnellement assimilable à une aminoacyl polymérase ARNm- et
ARNt-dépendante. Le centre catalytique est composé d'ARNr. La catalyse par le ribosome
consomme de l'énergie et augmente la vitesse de la traduction 106 fois, par un effet
exclusivement entropique de positionnement des ARNt, d'orientation et de protection du site
actif de l'effet du solvant. Le ribosome assure également le contrôle de la précision de la
traduction (Rodnina et al., 2005; Rodnina and Wintermeyer, 2009).
La détermination de la structure à haute résolution du ribosome procaryote par les
équipes de Ramakrishnan, Steitz et Yonath au début des années 2000 a dévoilé l'organisation
atomique du complexe et ouvert la voie à une compréhension du mécanisme moléculaire de la
traduction (Schmeing and Ramakrishnan, 2009; Williamson, 2009). Ce travail a été
récompensé par le prix Nobel de chimie en 2009.
Le ribosome est constitué d'une petite sous-unité et d'une grande sous-unité,
l'organisation étant globalement la même chez tous les organismes. Chez les procaryotes, le

18
ribosome a une taille totale de 70S, la petite sous-unité (30S) est constituée de l'ARNr 16S et
de 21 protéines associées, la grande sous-unité (50S) des ARNr 5S et 23S et de 31 protéines
associées. Chez les eucaryotes, il a une taille de 80S et est constitué d'une petite sous-unité
(40S) contenant un ARNr de 18S et 33 protéines associées et d'une grande sous-unité (60S)
contenant les ARNr 5,8S, 5S et 28S et 49 protéines associées. La sous-unité 40S regroupe les
sites de décodage de l'ARNm : c'est là que la reconnaissance codon-anticodon a lieu. La sous-
unité 60S est responsable de la catalyse de la formation de la liaison peptidique. Le ribosome
contient trois sites de fixation des ARNt : le site A (Aminoacyl), P (Peptidyl) et E (Exit).
L'ARNm se fixe sur un sillon de la petite sous-unité, autour d'une protubérance (neck); il peut
se déplacer séquentiellement codon par codon.
Au cours de la traduction le ribosome a trois fonctions catalytiques essentielles (Rodnina
et al., 2005):
(i) la reconnaissance codon-anticodon facilitée par la sous-unité 40S et la promotion d'un
aminoacyl-ARNt complémentaire d'un codon au niveau du site A, induisant la fermeture de la
petite sous-unité (mécanisme ratchet) et l'accommodation de l'aminoacyl-ARNt dans le site
A;
(ii) la formation des liaisons peptidiques entre les acides aminés successifs au niveau du
PTC (peptidyl-transferase center);
(iii) la translocation des ARNt du site A au site P, puis du site P au site E et le
déplacement de l'ARNm au niveau du site de décodage, d’un codon à son suivant en
respectant un pas de trois nucléotides.
Le ribosome augmente la vitesse des réactions chimiques de la traduction (vingt liaisons
peptidiques formées par seconde) et favorise leur spécificité, diminuant les erreurs
d'incorporation d'acides aminés à 1 incorporation erronée sur 100 000.

19
2.2. Mécanisme moléculaire de la traduction eucaryote
Le rôle du ribosome consiste à synthétiser avec la plus haute précision possible des
polymères d’acides aminés (ou protéines) en utilisant comme support de l’information
génétique des polymères de nucléotides : les ARNm. Pour cela, il utilise comme molécule de
décodage des ARNt couplés selon la correspondance universelle du code génétique à un
groupement aminoacyl spécifique (aminoacyl-ARNt) et qui reconnaissent spécifiquement les
codons. Ainsi, les aminoacyl-ARNt vont reconnaître les 61 codons sens et permettre
l’incorporation des vingt acides aminés constituant les protéines. Plusieurs ARNt
reconnaissent des codons différents mais codant pour un même résidu : le code génétique est
redondant.
Le ribosome facilite le recrutement spécifique de l’aminoacyl-ARNt compatible avec le
codon de l’ARNm présent au niveau de son site A et catalyse la formation d'une liaison
peptidique entre l'acide aminé porté par cet ARNt et la chaîne polypeptidique associée à
l’ARNt présent au site P. Le ribosome lit l'ARNm par pas de trois nucléotides, polymérisant
ainsi une suite d'acides aminés en un polypeptide. Par conséquent, cette étape nécessite une
haute fidélité pour que la séquence linéaire d'acides aminés du polypeptide corresponde à la
séquence de codons d'un ARNm donné.
La traduction se déroule en quatre étapes successives, strictement contrôlées par
l'intervention coopérative de facteurs protéiques.
2.2.1. Initiation
L'étape initiale de la traduction est centrée sur le recrutement du ribosome et du premier
aminoacyl-ARNt sur le codon initiateur. C'est une étape lente, déterminant la vitesse de
traduction globale de l'ARNm. L'initiation commence par la formation d'un complexe de

20
préinitiation 43S entre la petite sous-unité du ribosome et les facteurs d'initiation eIF1A, eIF1,
eIF2-GTP, eIF3 et eIF5; eIF2-GTP est chargé par un méthionyl-ARNt (Jackson et al., 2010)
(Figure 5). Le complexe 43S est recruté sur l'ARNm qui est présent sous une forme circulaire
stable (Amrani et al., 2008). Celle-ci est formée par l’interaction entre les deux extrémités de
l’ARNm via l’association de la protéine eIF4G, sous-unité du complexe eIF4F (avec
également eIF4E et eIF4A) fixé au niveau de la coiffe en 5’ et la protéine Pab1 qui interagit
avec la queue poly(A) en 3’.
Lorsque l'interaction 43S-ARNm est établie, le 43S « scanne » la séquence 5' non-
codante de l'ARNm jusqu'à ce que le premier codon traduit (AUG - méthionine), appelé
codon initiateur, entre dans le site de reconnaissance de la sous-unité 40S. Ceci induit
l'hydrolyse du GTP par eIF2 et la formation subséquente du complexe 48S. Les facteurs eIF2-
GDP, eIF5, eIF1 et eIF3 sont alors libérés et remplacés sur le 48S par la sous-unité 60S en
complexe avec eIF5B-GTP. L'hydrolyse du GTP par eIF5B et sa libération simultanée à celui
d'eIF1A laissent sur l'ARNm le ribosome 80S complet avec un ARNt-Met sur le codon
initiateur dans le site P du ribosome et un site A libre pour l'entrée de l'aminoacyl ARNt
suivant. Le ribosome est prêt à entrer en élongation.
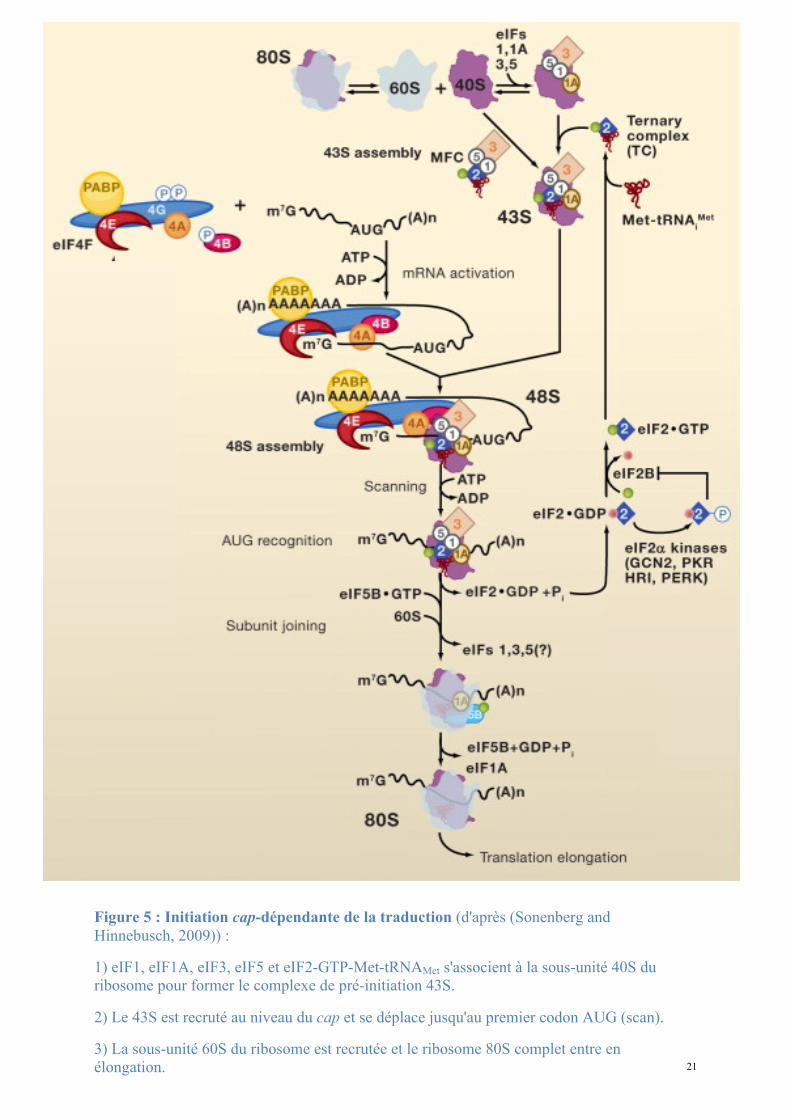
21
Figure 5 : Initiation cap-dépendante de la traduction (d'après (Sonenberg and Hinnebusch, 2009)) :
1) eIF1, eIF1A, eIF3, eIF5 et eIF2-GTP-Met-tRNAMet s'associent à la sous-unité 40S du ribosome pour former le complexe de pré-initiation 43S.
2) Le 43S est recruté au niveau du cap et se déplace jusqu'au premier codon AUG (scan).
3) La sous-unité 60S du ribosome est recrutée et le ribosome 80S complet entre en élongation.

22
2.2.2. Elongation
C'est la réaction de synthèse du polypeptide du premier au dernier acide aminé (Rodnina
and Wintermeyer, 2009) (Figure 6). L'addition des acides aminés est séquentielle, c'est une
étape extrêmement rapide de la traduction. L'élongation nécessite l'interaction des aminoacyl-
ARNt avec eEF-1A:GTP.
Dans un premier temps l'aminoacyl-ARNt:eEF-1A est recruté au site A du ribosome; le
positionnement correct de l'aminoacyl-ARNt requiert l'hydrolyse du GTP par eEF-1A. Si
l'interaction codon-anticodon est respectée l'acide aminé méthionine (Met) du site P est
transféré de l'ARNt0 initiateur à l’acide aminé (AA1) associé à l’ARNt présent au niveau du
site A (ARNt1). Le site peptidyl-transférase de la sous-unité 60S du ribosome catalyse la
formation de cette première liaison peptidique Met-AA1; cette réaction est accompagnée de la
libération d'eEF-1A:GDP. L'ARNt0 qui portait la méthionine initiatrice est à présent déacylé.
Dans un deuxième temps le ribosome se déplace d'un codon sur l'ARNm, l'ARNt0
initiateur rejoint le site E tandis que le Met-AA1-ARNt1 entre dans le site P, libérant le site A
qui contient le codon 2. La translocation est assistée par eEF-2:GTP qui hydrolyse son GTP.
Le ribosome est alors en étape de post-initiation. La formation d'une nouvelle liaison
peptidique entre Met-AA1 et le nouvel acide aminé (AA2) est couplée au départ de l'ARNt0
initiateur du site E. eEF-2:GTP catalyse la translocation des ARNt1 et ARNt2 des sites P et A
vers les sites E et P respectivement, libérant une nouvelle fois le site A pour le codon suivant
(codon 3).
Ce processus est répété pour chaque codon entrant dans le site A du ribosome et pouvant
s’apparier avec son aminoacyl-ARNt. L'élongation a une vitesse de réaction du site peptidyl-
transfert de 160s-1 (Johansson et al., 2008).

23
"DNA-RNA-Protein". Nobelprize.org. 15 Jun 2010 http://nobelprize.org/educational/medicine/dna/a/translation/elongation.html
Figure 6 : Elongation de la traduction :
1) Les aminoacyl-ARNt s'associent à eEF1-GTP.
2) Un aa-ARNt-eEF1-GTP entre dans le site A du ribosome.
3) La boucle anticodon de l'ARNt interagit avec le codon de l'ARNm au site A; si l'interaction est spécifique, l'aa-ARNt-eEF1-GTP reste dans le site A, dans le cas contraire le complexe est évacué et remplacé par un nouvel aa-ARNt-eEF1-GTP.
4) Lorsque le bon aa-ARNt est recruté le ribosome catalyse l'association par une liaison peptidique de l'acide aminé au polypeptide présent dans le site P.
5) Le ribosome se déplace d'un codon vers l'extrémité 3' de l'ARNm.
6) Le peptidyl-ARNt est déplacé du site A vers le site P, l'ARNt précédent est déplacé du site P vers le site E et le site A est libéré pour l'entrée d'un nouvel aa-ARNt-eEF1-GTP.

24
2.2.3. Terminaison
La terminaison intervient en fin d'élongation après l'ajout du dernier acide aminé et se
conclut par l'hydrolyse de la dernière liaison peptide-ARNt et la libération de la chaîne
polypeptidique nouvellement synthétisée (Kisselev et al., 2003; Kisselev, 2001; Kisselev and
Buckingham, 2000; Petry et al., 2008). L'ajout répétitif d'acides aminés à la chaîne
polypeptidique en formation s'arrête lorsque le site A du ribosome est occupé par un codon
stop, non reconnu par les aminoacyl-ARNt. Les facteurs de terminaison sont des protéines
reconnaissant spécifiquement ces codons stop et induisant la libération du polypeptide
nouvellement formé. Cette étape est détaillée dans la partie suivante (3. La terminaison de la
traduction).
2.2.4. Recyclage
A la fin de la terminaison le ribosome 80S se trouve à l'extrémité 5' de la séquence
codante avec un ARNt dans son site P et les facteurs de terminaison dans son site A. Le
recyclage correspond aux libérations successives du complexe eRF1-eRF3 du site A, de la
sous-unité 60S, de l'ARNm et de l'ARNt (Jackson et al., 2010). Encore mal définie, cette
étape requiert l'intervention des facteurs d'élongation eIF1A, eIF1 et eIF3 qui se fixent sur la
sous-unité 40S en vue de reformer un complexe de pré-initiation 43S fonctionnel.
L'intervention de la protéine ABCE1 a été récemment mise en évidence chez l’homme pour
induire la séparation des sous-unités du ribosome entre elles et des facteurs qui y sont attachés
et donc la dissociation des ribosomes en post-terminaison (Pisarev et al., 2010). Ce
mécanisme nécessite l'hydrolyse de NTP par ABCE1.

25
3. La terminaison de la traduction
3.1. Mécanisme moléculaire de la terminaison
L'élongation de la traduction implique la reconnaissance spécifique des codons présents
au niveau du site A du ribosome par les ARNt. Or, parmi la combinaison de 64 codons
possibles seuls 61 sont complémentaires à un ARNt et sont supports de l'élongation. La
terminaison est déclenchée par l'entrée d'un des 3 codons stop UAA, UAG ou UGA
(également appelés codons ocre, ambre et opale respectivement) dans le site A du ribosome
(Rospert et al., 2005) (Figure 7). Au lieu d'être reconnus par l'un des aminoacyl-ARNt, les
codons stop sont reconnus par des protéines : les facteurs de terminaison de la traduction.
Ceux-ci se fixent au niveau du site A du ribosome par interaction directe avec les codons stop
(Chavatte et al., 2003; Chavatte et al., 2002; Kervestin et al., 2001). Ils induisent directement
l’hydrolyse de la liaison entre le dernier ARNt et le peptide nouvellement synthétisé.
Il existe deux classes de facteurs de terminaison que nous allons décrire successivement.
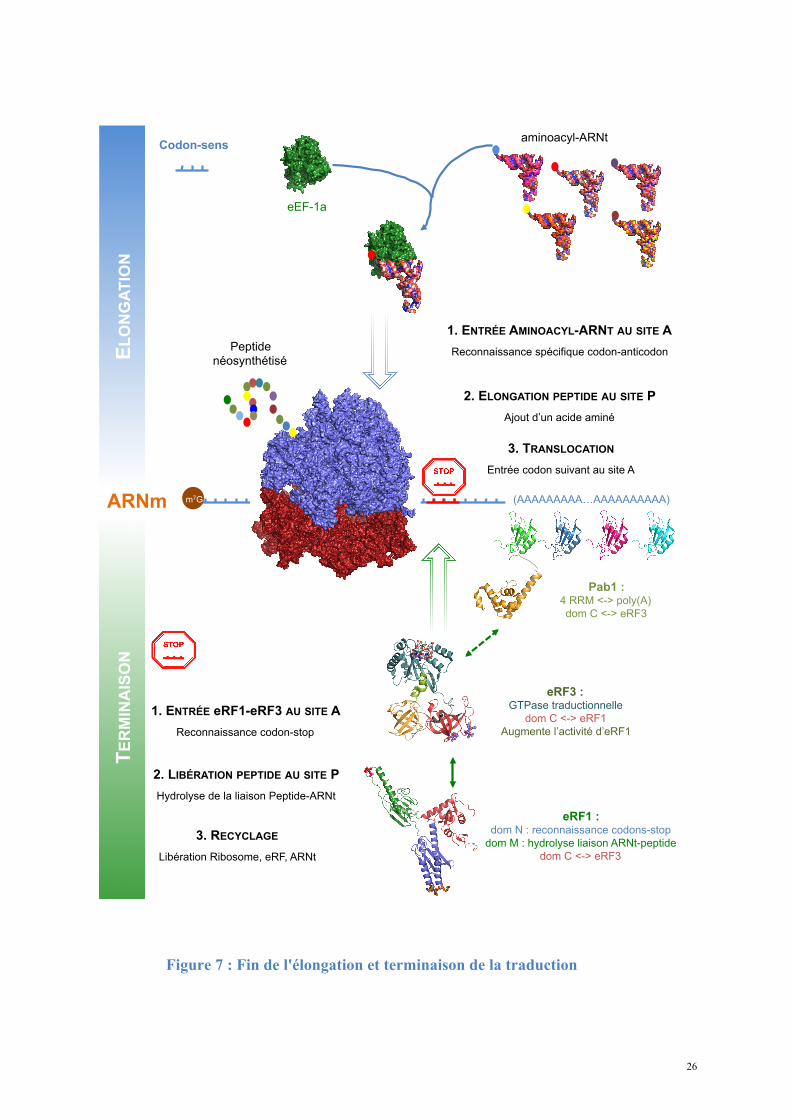
26
EL
ON
GA
TIO
N
ARNm (AAAAAAAAA…AAAAAAAAAA) m7G
1. ENTRÉE AMINOACYL-ARNT AU SITE A
Reconnaissance spécifique codon-anticodon
2. ELONGATION PEPTIDE AU SITE P
Ajout d’un acide aminé
eEF-1a
aminoacyl-ARNt
Peptide
néosynthétisé
Pab1 : 4 RRM <-> poly(A)
dom C <-> eRF3
TE
RM
INA
ISO
N
Codon-sens
eRF3 : GTPase traductionnelle
dom C <-> eRF1
Augmente l’activité d’eRF1
eRF1 : dom N : reconnaissance codons-stop
dom M : hydrolyse liaison ARNt-peptide
dom C <-> eRF3
1. ENTRÉE eRF1-eRF3 AU SITE A
Reconnaissance codon-stop
2. LIBÉRATION PEPTIDE AU SITE P
Hydrolyse de la liaison Peptide-ARNt
3. RECYCLAGE
Libération Ribosome, eRF, ARNt
3. TRANSLOCATION
Entrée codon suivant au site A
Figure 7 : Fin de l'élongation et terminaison de la traduction

27
3.2. Facteurs de terminaison de classe I : spécificité, structure
et mode de reconnaissance.
3.2.1. Fonction des facteurs de terminaison de classe I
Les facteurs de terminaison de classe I (RF1 et RF2 chez les procaryotes, aRF1 chez les
archées et eRF1 chez les eucaryotes) reconnaissent directement les codons stop en se fixant au
site A du ribosome et catalysent l'hydrolyse de la liaison ester connectant le polypeptide
nouvellement synthétisé au dernier ARNt présent dans le site P; ils permettent ainsi la
libération du polypeptide et de l'ARNt séparément (Caskey et al., 1971; Craigen and Caskey,
1987; Goldstein et al., 1970). Les facteurs de terminaison procaryotes RF1 et RF2 ont des
spécificités de reconnaissance différentes : RF1 catalyse la terminaison aux codons UAG et
UAA; RF2 catalyse la terminaison aux codons UGA et UAA. L'interaction RF-codon stop est
déterminée par des motifs tripeptidiques : PAT sur RF1 et SPF sur RF2. La structure du
ribosome procaryote entier co-cristallisé avec RF1 ou RF2 confirme l'interaction directe et
son positionnement propice à l'hydrolyse de la liaison ARNt-peptide au site P (Korostelev et
al., 2008; Laurberg et al., 2008; Weixlbaumer et al., 2008).
Bien qu'aucune similarité de séquence ne soit observable entre eRF1/aRF1 et
RF1/RF2/mtRF1, que le mode de reconnaissance des codons stop et leur mode d'interaction
avec les facteurs de terminaison de classe II soient distincts dans les différents domaines du
vivant, un triplet d'acides aminés GGQ est retrouvé chez tous les facteurs de terminaison de
classe I (Frolova et al., 1999). Sur eRF1 il est porté sur une boucle exposée au solvant au
niveau du domaine M. Il a été proposé (Song et al., 2000) que la glutamine de ce motif serait
impliquée dans la coordination d'une molécule d'eau au site P du ribosome; l'attaque
nucléophile par cette molécule d'eau hydrolyserait la liaison ester entre le polypeptide et le

28
dernier ARNt. Cependant la substitution de la glutamine n'abolit pas totalement la
terminaison, alors que les mutations des glycines la suppriment (Frolova et al., 1999). Le
mécanisme moléculaire de la catalyse et le rôle précis de la glutamine restent à déterminer.
A la différence des procaryotes, les eucaryotes ne possèdent qu'un facteur de terminaison
de classe I, eRF1, dont la capacité de reconnaissance est omnipotente vis-à-vis des trois
codons stop (Drugeon et al., 1997).
3.2.2. Structure du facteur de terminaison de classe I eucaryote eRF1
Au niveau moléculaire, la terminaison de la traduction est prise en charge par deux
fonctions distinctes du facteur de terminaison de classe I : la reconnaissance spécifique des
codons stop et l'hydrolyse de la liaison polypeptide-ARNt. La détermination de la structure
cristallographique d'eRF1 de H. sapiens (Song et al., 2000) (Figure 8) a apporté les premières
informations sur le mécanisme moléculaire de la terminaison chez les eucaryotes. La protéine
est organisée en trois domaines indépendants (Figure 8). La structure globale d'eRF1
ressemble par sa forme et ses dimensions à un ARNt et les structures de ces deux molécules
peuvent être superposées avec de fortes correspondances (Figure 9).
Le domaine 1 (N-terminal) est impliqué dans la reconnaissance du codon stop via les
motifs NIKS et YxxCxxxF (Chavatte et al., 2002; Kolosov et al., 2005). Ces motifs sont donc
fonctionnellement homologues à la boucle anti-codon des ARNt. Le domaine 2 (domaine
central) porte le motif GGQ universellement conservé qui va interagir au niveau du site PTC
du ribosome. Le domaine 3 (C-terminal) est responsable de l’interaction avec eRF3
(Merkulova et al., 1999), et également avec la phosphatase PP2A (Andjelkovic et al., 1996).
Une structure du domaine C de H. sapiens, récemment déterminée par RMN (Mantsyzov et
al., 2010), révèle l'organisation d'un sous-domaine peu visible dans la structure
cristallographique. Les mesures de dynamique de la chaîne carbonée montrent une grande

29
flexibilité des résidus 414 à 437 correspondant à ce sous-domaine. Remarquablement, les
auteurs mettent en évidence une conversion entre deux conformations limites, dont l'une place
le minidomaine à proximité du site N-terminal de reconnaissance des codons stop. La
mutation de ces résidus diminue effectivement l'efficacité de reconnaissance des codons stop,
qui impliquerait par conséquent des résidus des domaines N et C.
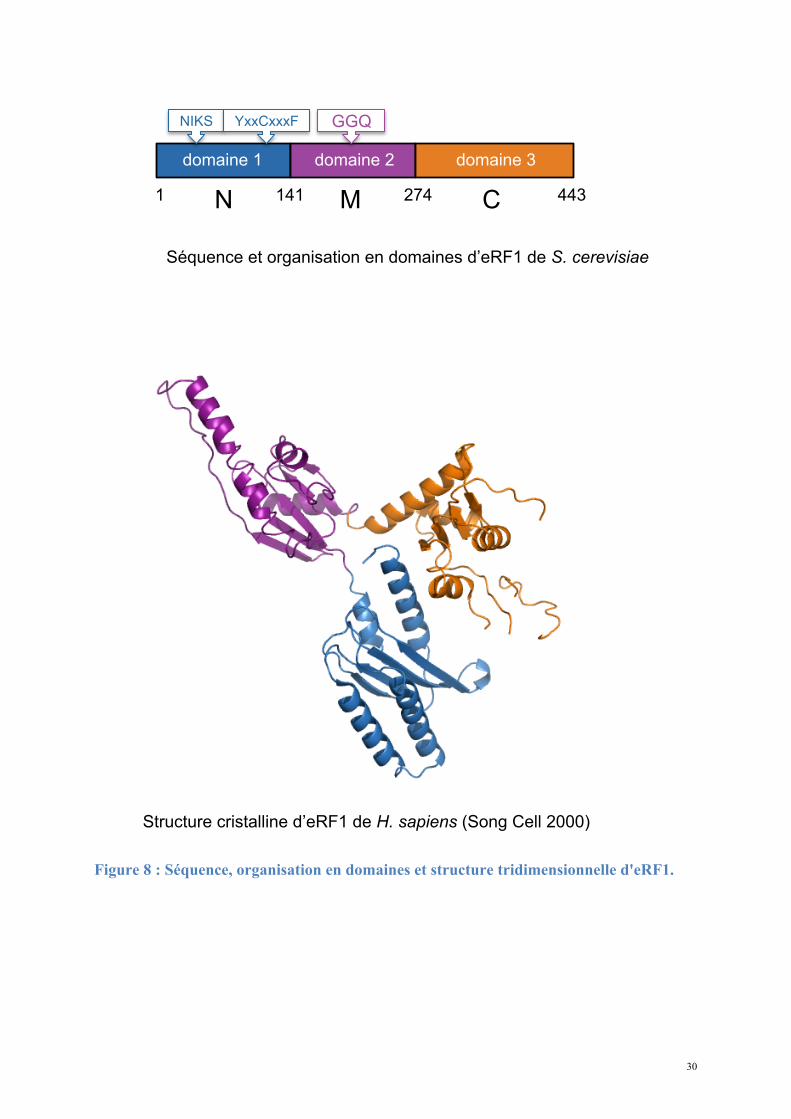
30
Figure 8 : Séquence, organisation en domaines et structure tridimensionnelle d'eRF1.
Séquence et organisation en domaines d’eRF1 de S. cerevisiae
1 N 141 M 274 C 443
Structure cristalline d’eRF1 de H. sapiens (Song Cell 2000)
domaine 1 domaine 2 domaine 3
GGQ NIKS YxxCxxxF
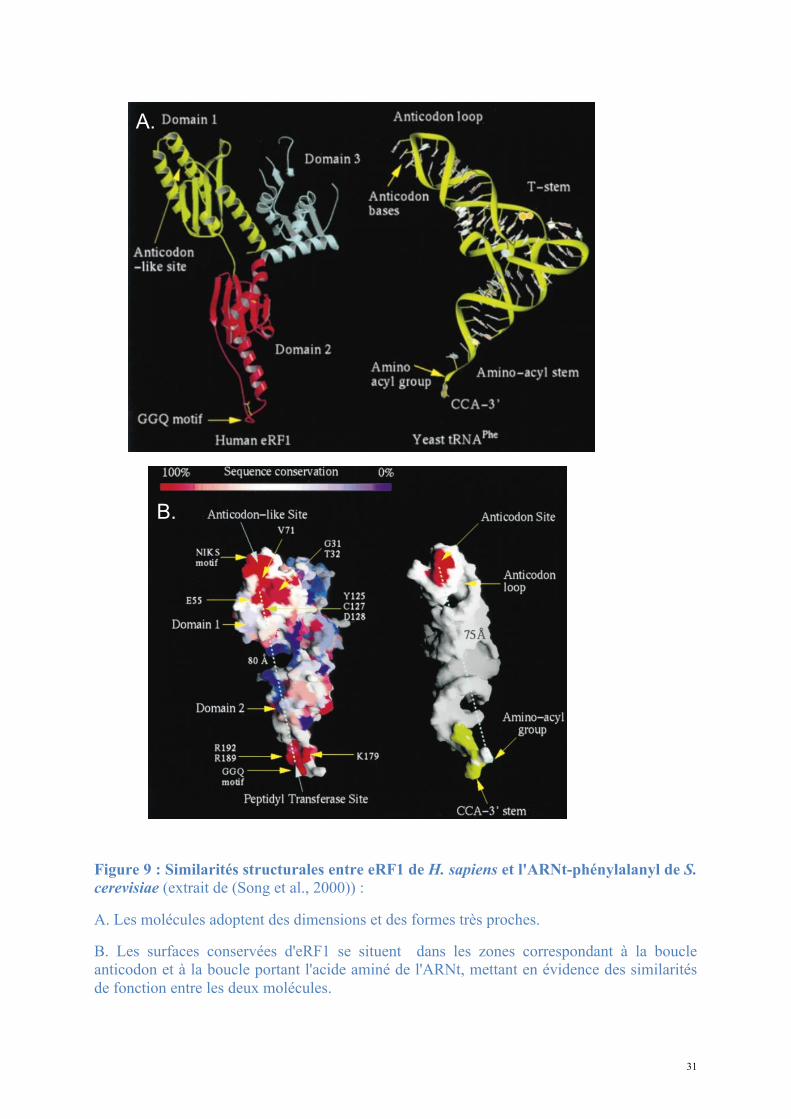
31
Figure 9 : Similarités structurales entre eRF1 de H. sapiens et l'ARNt-phénylalanyl de S. cerevisiae (extrait de (Song et al., 2000)) :
A. Les molécules adoptent des dimensions et des formes très proches.
B. Les surfaces conservées d'eRF1 se situent dans les zones correspondant à la boucle anticodon et à la boucle portant l'acide aminé de l'ARNt, mettant en évidence des similarités de fonction entre les deux molécules.
A.
B.

32
3.3. Facteurs de terminaison de classe II : rôle et structure.
3.3.1. Fonction des facteurs de terminaison de classe II
L'activité de terminaison centrée sur les facteurs de terminaison de classe I est stimulée
par les facteurs de terminaison de classe II RF3 (procaryotes), aRF3 (archées) ou eRF3
(eucaryotes) (Tuite and Stansfield, 1994; Zhouravleva et al., 1995). Leur présence n'est pas
strictement nécessaire pour déclencher la terminaison in vitro, mais ces protéines sont
essentielles pour la survie des cellules. Les facteurs de terminaison de classe II sont des
GTPases dont l'activité dépend du ribosome. Les protéines procaryotes et eucaryotes ont des
fonctions différentes. Chez les procaryotes, RF3 intervient après la libération du peptide et
facilite la dissociation de RF1, permettant un recyclage efficace du facteur de terminaison de
classe I (Gao et al., 2007; Zavialov et al., 2001). Chez les eucaryotes eRF3 stimule l'hydrolyse
de la liaison peptide-ARNt au site P et requiert pour cela une activité GTPase dépendant du
ribosome et d'eRF1 (Zhouravleva et al., 1995). L'activité GTPase couple la reconnaissance du
codon stop par eRF1 à la libération efficace du peptide (Alkalaeva et al., 2006; Salas-Marco
and Bedwell, 2004). In vitro, la fixation d'eRF1, eRF3 et de GTP sur un complexe à l'étape
précédent la terminaison conduit à un changement de positionnement d'eRF1 dans le site A et
à l'hydrolyse rapide de la liaison peptidyl-ARNt.
3.3.2. Structure du facteur de terminaison de classe II eucaryote eRF3
La protéine eRF3 est organisée en plusieurs domaines de conservation variable : les
domaines N et M sont faiblement conservés et ne sont pas essentiels pour la viabilité de la
levure S. cerevisae. Ainsi ils ne semblent pas directement impliqués dans la terminaison de la
traduction; le domaine C au contraire est bien conservé chez tous les eucaryotes et est

33
fonctionnellement impliqué dans l'interaction avec eRF1, l'hydrolyse du GTP et la
terminaison.
La structure à haute résolution de la partie C-terminale d'eRF3 de S. pombe (Kong et al.,
2004) (Figure 10) révèle une organisation en trois sous-domaines distincts. La structure de
chacun est homologue aux domaines des facteurs d'élongation eucaryote eEF1α et procaryote
EF-Tu : eRF3 appartient à la famille des GTPases impliquées dans la traduction (EF-Tu, EF-
G, IF2 chez les procaryotes; eIF2, eEF1, eEF2 chez les eucaryotes).
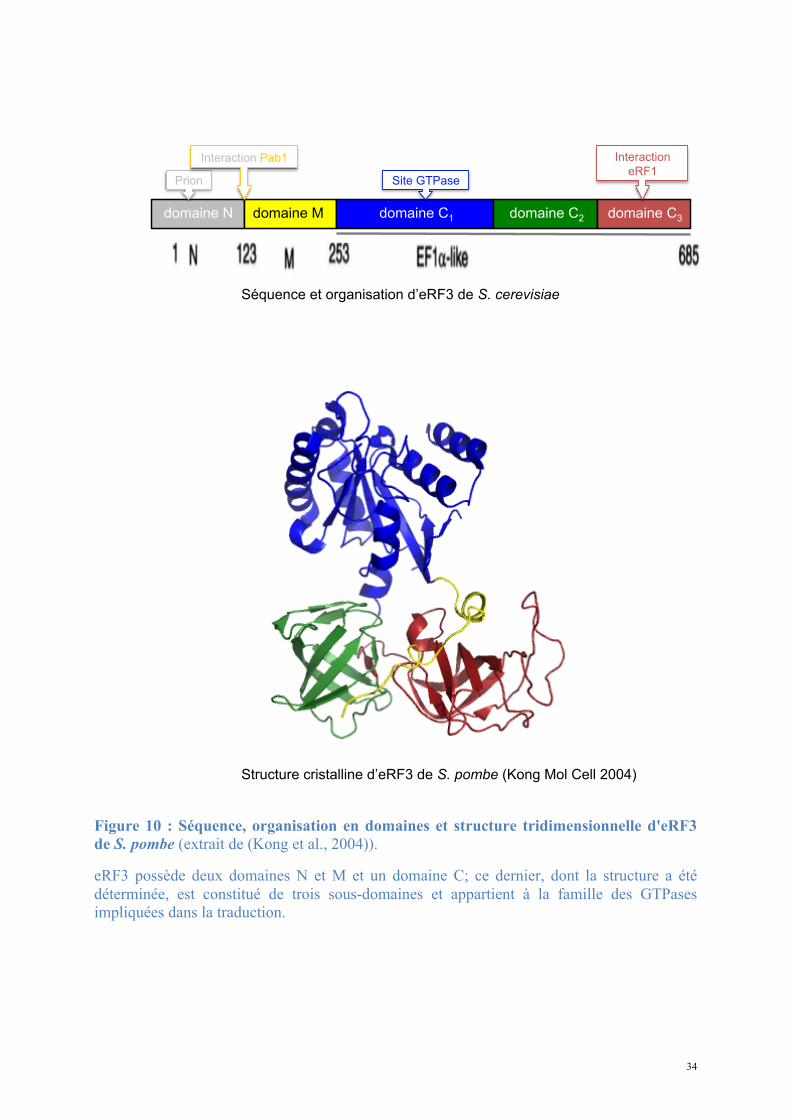
34
Figure 10 : Séquence, organisation en domaines et structure tridimensionnelle d'eRF3 de S. pombe (extrait de (Kong et al., 2004)).
eRF3 possède deux domaines N et M et un domaine C; ce dernier, dont la structure a été déterminée, est constitué de trois sous-domaines et appartient à la famille des GTPases impliquées dans la traduction.
Structure cristalline d’eRF3 de S. pombe (Kong Mol Cell 2004)
Séquence et organisation d’eRF3 de S. cerevisiae
domaine N domaine M domaine C1 domaine C2 domaine C3
Interaction
eRF1 Site GTPase Prion
Interaction Pab1

35
- Le domaine 1 est un domaine GTPase. Le nucléotide est fixé par un réseau de résidus
portés par quatre boucles : G1/P, G2, G3 et G4. La base est située dans une poche hydrophobe
entre les boucles G3 et G4. La boucle P porte la séquence consensus GXXXXGK(S/T) et fixe
les phosphates α et β du nucléotide. Les boucles G2 et G3, également appelées switchs I et II
respectivement, subissent un important changement de conformation lors de la fixation des
nucléotides. Le switch I est impliqué dans la discrimination GDP/GTP via l'interaction d'une
Thréonine avec le phosphate γ; le switch II complète cette discrimination par l'intervention
d'une Glycine.
La fixation des nucléotides guanine par eRF3 a été caractérisée par des mesures
cinétiques (Pisareva et al., 2006). L'affinité d'eRF3 pour les nucléotides est de 70µM pour le
GTP et de 1µM pour le GDP, et les deux se dissocient rapidement de la protéine (koff de 3,3 et
2,4s-1 respectivement). La protéine eRF1 change significativement l'équilibre de fixation des
nucléotides en ralentissant fortement la dissociation du GTP mais pas celle du GDP; la vitesse
de dissociation du GTP est réduite à 0,14s-1 et l'affinité augmente à 0,7µM en présence
d’eRF1. L'effet est accentué grâce à la présence de Mg2+ qui renforce la spécificité vis-à-vis
du nucléotide en présence d’eRF1. La protéine eRF1 jouerait donc le rôle d'un inhibiteur de la
dissociation du GTP. Les résultats montrent également que la fixation du GDP suit un modèle
simple de fixation en une seule étape (one-step) tandis que la fixation du GTP procède en
deux étapes successives, généralement caractéristiques d'un changement de conformation du
site de fixation le rendant propre à accommoder le substrat.
Les mesures d'affinité d'eRF3 pour les nucléotides GTP et GDP (Kong et al., 2004)
montrent que le Mg2+ joue un rôle opposé pour la fixation du GDP (diminue l'affinité) et pour
la fixation du GTP (augmente l'affinité). Ce mode d'échange de nucléotide expliquerait
l'absence de protéine facilitant le remplacement du GDP par le GTP (facteur d'échange de

36
guanosine, GEF) pour eRF3, qui assurerait ce mécanisme seule avec l'aide du Mg2+
disponible dans la cellule.
- Les domaines 2 et 3 (C-terminal) adoptent chacun un repliement de type tonneau-β.
Une région très conservée et chargée négativement est exposée à la surface du domaine 3; elle
porte le motif GRFTLRD dont la substitution par mutagenèse montre son importance pour la
viabilité cellulaire et la fixation d'eRF1 (Stansfield et al., 1996).
3.3.3. eRF3 de S. cerevisiae est un prion
Indépendamment de son rôle dans la terminaison de la traduction via son interaction avec
la protéine Pab1 (voir 3.4.1.), le domaine N-terminal spécifique de la protéine eRF3 de S.
cerevisiae est le déterminant du phénotype prion [PSI+] (Doel et al., 1994). La séquence de ce
domaine N est très peu conservée chez les orthologues d’eRF3 et ce domaine n'est pas
essentiel à la survie des cellules.
Un prion est une protéine pouvant adopter plusieurs conformations différentes, et dont
l'une des conformations peut être transmise par "contamination", lors d'une interaction directe
protéine-protéine (Shkundina and Ter-Avanesyan, 2007); les protéines prions sont agrégées
sous une forme inactive. Le phénotype [PSI+] est associé à l'inactivation d'eRF3 et à une
efficacité de terminaison diminuée, pouvant conduire à une translecture des codons stop et à
une poursuite anormale de l'élongation. Les 114 résidus N-terminaux de la protéine ont la
capacité de maintenir la forme prion de la protéine dans une cellule (Shkundina et al., 2006).
La possibilité que le phénotype prion ne soit pas un défaut mais puisse avoir une fonction
régulatrice de l'activité d'eRF3 fait encore débat.

37
3.4. Structure et mécanisme d'action du complexe de
terminaison eRF1-eRF3
La protéine eRF1 présente quatre fonctions essentielles et distinctes : liaison au ribosome,
reconnaissance du codon stop, interaction avec les facteurs de terminaison de classe II et
interaction avec le site peptidyl-transférase du ribosome (Frolova et al., 2000). La fonction de
terminaison est portée par les domaines N et M, seuls nécessaires pour la libération du peptide
(Bertram et al., 2000). L'interaction avec les facteurs de terminaison de classe II est médiée
par le domaine C mais l'effet activateur sur eRF3 implique également les domaines N et M.
Contrairement aux facteurs de terminaison procaryotes, eRF1 et eRF3 forment un
complexe stable en l'absence de ribosome. L’activité GTPase d’eRF3 n’est détectée qu’en
présence d’eRF1 et du ribosome et elle stimule la libération efficace du peptide nouvellement
synthétisé par hydrolyse de la liaison peptide-ARNt au site P du ribosome.
La détermination de la structure à basse résolution (3,8Å) du complexe eRF1-eRF3 chez
S. pombe et H. sapiens a mis en évidence les zones d'interaction précises de chaque protéine
(Cheng et al., 2009) (Figure 11). La formation du complexe induit un changement de
conformation global d'eRF1 dont le domaine M contenant le motif GGQ se déplace de 44Å.
La modélisation du complexe à basse résolution en utilisant les données de diffusion des
rayons X aux petits angles (SAXS) suggère de plus que ce mouvement du domaine M d'eRF1
le positionne à proximité immédiate du domaine GTPase d'eRF3, absent dans la structure
cristalline. Dans ce modèle, la chaîne latérale du résidu Arg192 se situerait alors près du site
de fixation du GTP par eRF3 de la même manière que le domaine régulateur de RhoGDI avec
la GTPase Cdc42. Le domaine M d’eRF1 participerait à la fixation du nucléotide par eRF3 en
interagissant avec les phosphates; il peut également jouer un rôle dans le positionnement des
boucles switch I et switch II. Ce modèle est renforcé par l'importance de l'Arg192 pour

38
l'activité de terminaison et pour le mécanisme proposé d'activité coopérative d'eRF1-eRF3
(Alkalaeva et al., 2006). Le fait que l'Arg192 joue un rôle dans l'interaction eRF1-eRF3 et
qu'elle soit placée dans le domaine M à une très courte distance du motif GGQ (distance Cα-
Cα : R192-Q185 11,8Å) relie la fonction catalytique d'eRF1 à la fonction GTPase d'eRF3.
Ainsi l'action coopérative d'eRF1-eRF3 et l'hydrolyse du GTP se révèlent importantes pour
l'activité de terminaison elle-même.
Le complexe a une structure compacte similaire à celle du complexe d'élongation EF-
Tu:ARNt. Il semble adopter une structure appropriée pour une entrée dans le site A du
ribosome contenant le codon stop. Le triplet GGQ du domaine M d'eRF1 se positionnerait
alors à proximité de la boucle amino-acyl de l'ARNt, et se trouverait proche du PTC du
ribosome où il hydrolyserait la liaison peptidyl-ARNt. Le mode de fixation d'eRF1 dans le
ribosome serait alors similaire à celui de RF1/RF2 chez les procaryotes.
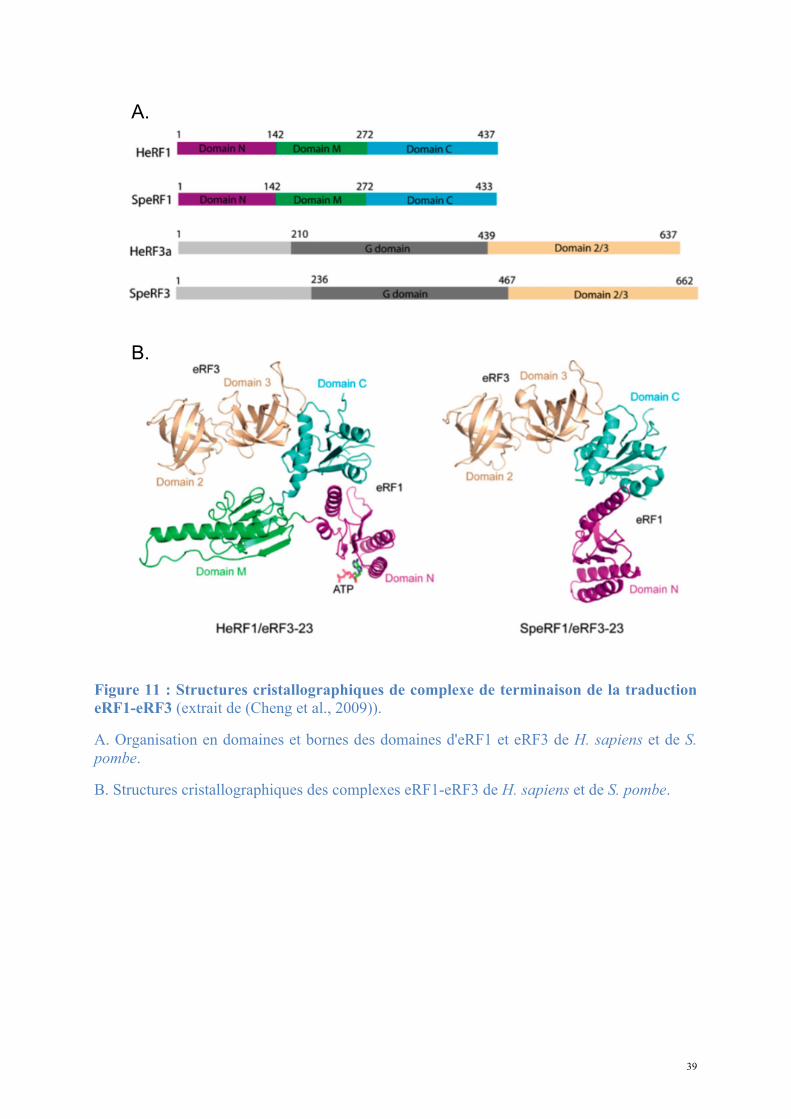
39
Figure 11 : Structures cristallographiques de complexe de terminaison de la traduction eRF1-eRF3 (extrait de (Cheng et al., 2009)).
A. Organisation en domaines et bornes des domaines d'eRF1 et eRF3 de H. sapiens et de S. pombe.
B. Structures cristallographiques des complexes eRF1-eRF3 de H. sapiens et de S. pombe.
A.
B.
B.

40
3.5. Protéines influençant l’efficacité de terminaison :
Pab1, forme prion d’eRF3, Gle1, Npl3, Dbp5, Rli1, Mtq2-Trm112, Tpa1,
Ett1,…
La terminaison de la traduction est dépendante de la reconnaissance des codons stop
entrant au site A du ribosome. Les facteurs de terminaison de classe I et II sont nécessaires et
suffisants pour induire la libération aux codons stop du peptide néoformé, comme observé in
vitro à partir de protéines purifiées. Cependant, bien qu'aucun ARNt ne présente de boucle
anticodon complémentaire à un codon stop, des aminoacyl-ARNt de complémentarité voisine
(near-cognate) peuvent être incorporés (Rospert et al., 2005). Par conséquent, l'efficacité de
terminaison de la traduction dépend de la compétition entre ces deux évènements. La
fréquence de cet évènement de lecture du ribosome à travers le codon stop (ou translecture)
dépend d'un équilibre entre l'activité de terminaison et l'activité d'élongation anormale. La
fidélité de terminaison de la traduction est directement modulée :
- par la nature du nucléotide directement en aval du codon stop (Bonetti et al., 1995).
- en terme de quantité de facteurs de terminaison disponibles, par la régulation du niveau
de la transcription des gènes codant pour les eRF mais aussi par le phénomène [PSI+] décrit
précédemment qui diminue la quantité d’eRF3 actif disponible dans la cellule (Gasch et al.,
2000);
- par des modifications post-traductionnelles : méthylation du motif GGQ d'eRF1
(Heurgue-Hamard et al., 2005);
- par des facteurs protéiques agissant en trans : des études d'interaction à grande échelle
complétées par des caractérisations de phénotypes de délétions géniques mettent en évidence
des interactions protéine-protéine liées à l'efficacité de terminaison (von der Haar and Tuite,
2007) (Figure 12).

41
Ces protéines modulent l'efficacité de reconnaissance des codons stop, facilitent le
recyclage du ribosome ou assurent le contrôle de la stabilité des ARNm. Certaines peuvent
favoriser la translecture du codon stop et contrôler la traduction des séquences en aval du
cadre de lecture ouvert. La majorité de ces protéines ont été décrites comme interagissant
avec eRF1 et eRF3 et sont indirectement impliquées dans la traduction (Itt1, Tpa1, Pab1,
Mtq2, Rli1, Gle1, Dbp5, Npl3).
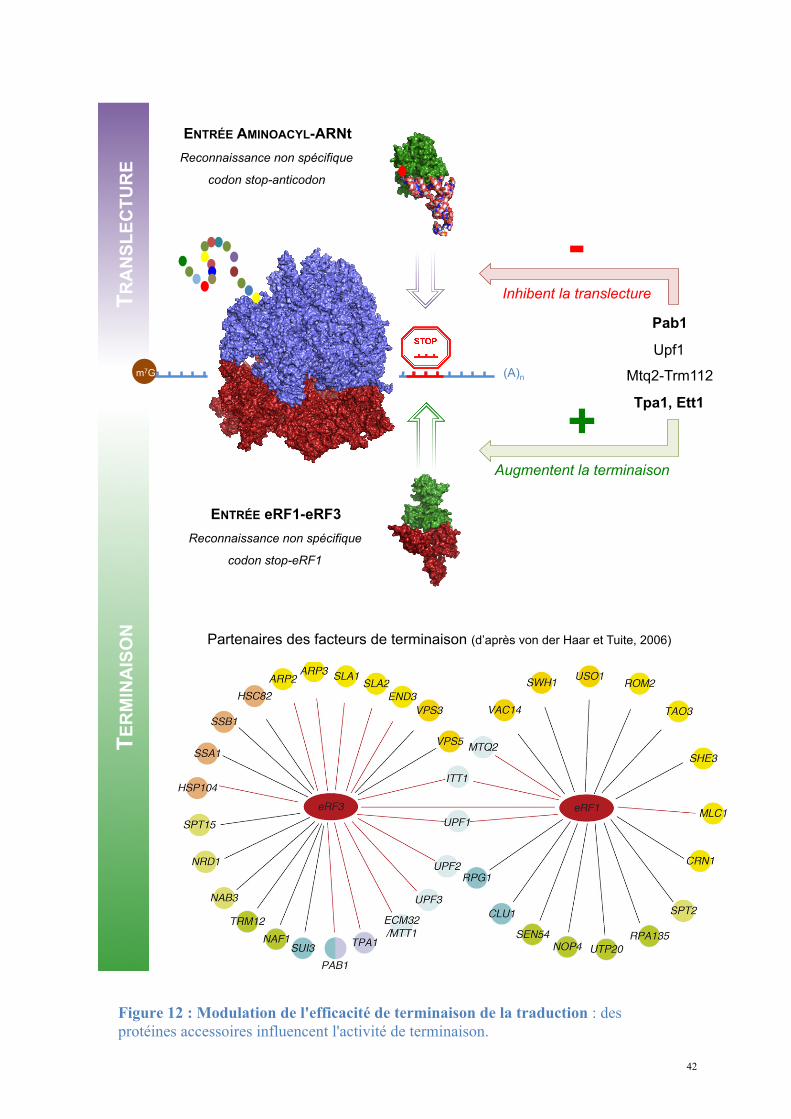
42
+ Augmentent la terminaison
TR
AN
SL
EC
TU
RE
(A)n m7G
ENTRÉE AMINOACYL-ARNt
Reconnaissance non spécifique
codon stop-anticodon
ENTRÉE eRF1-eRF3
Reconnaissance non spécifique
codon stop-eRF1
TE
RM
INA
ISO
N
Pab1
Upf1
Mtq2-Trm112
Tpa1, Ett1
Partenaires des facteurs de terminaison (d’après von der Haar et Tuite, 2006)
- Inhibent la translecture
adenylic acid residues, this triggers remodelling of proteincomplexes bound to the mRNA. This, in turn, renders themRNA a target for degradation.
Recent results suggest that translation terminationcould exert direct control over the rate of mRNA dead-enylation. eRF3 (Sup35p) binds to Pab1p [61,62], and yeaststrains in which this interaction is impaired show reducedrates of mRNA deadenylation and increased mRNA stabi-lity [45]. Moreover, deadenylation seems to be dependenton normal translation termination because mutations ineRF3 that decrease termination efficiency (but show noreduced binding to Pab1p) also stabilize mRNAs [8]. Thefact that eRF3 binding to Pab1p prevents the oligomeriza-tion of Pab1p on poly(A) tails [62] suggests a possiblemolecular mechanism by which contacts between thesetwo proteins during a translation termination eventrelease Pab1p from the poly(A) tail, thus providing accessfor the deadenylation factors. eRF3 also interacts with asecond deadenylation factor, Tpa1p, and defects in the
deadenylase complex produce a stop-codon readthroughphenotype [63]. A tight connection therefore existsbetween translation termination and deadenylation andmRNA turnover.
Alterations of mRNA levels through surveillancepathwaysSeveral surveillance pathways exist in eukaryotic cells toidentify aberrant mRNAs and rapidly destroy them.mRNAs that contain a premature stop codon are recognizedby the nonsense-mediated decay (NMD) machinery, whichinvolves the canonical translation termination factors inconjunction with dedicated NMD factors (see earlier;reviewed in ref. [32]). A second pathwayoperates onmRNAsthat contain no in-frame stop codon, on which ribosomestranslate through to the 30-end of the mRNA and stall. Theresulting dead-end complexes are recognized and rapidlydegradedby thenon-stopdecay (NSD)pathway (reviewed inRef. [64]). mRNAs on which a ribosome has stalled during
Figure 3. The putative interacting partners for eRF1 and eRF3 cluster into functional categories. Physical interactions for yeast eRF1 and eRF3 reported in the literature wereretrieved from the BIOGrid database [57]. Circles represent the interaction partners, and are colour-coded according to functional category (see colour key in figure).Connecting lines between partners represent the interaction: interactions that have been reported in several studies and corroborated by different experimental techniquesare in red; large-scale interaction studies that have not yet been confirmed by independent experimental approaches (reported in one study and by one technique only) arein black. Interactions are shown in detail where several functionally related proteins are reported to interact with the eRFs; other reported interacting partners are also listed.A large number of interactions with functionally related proteins indicate that the eRFs might have important non-translational roles in the respective process, or that theprocess in question might exert control over the efficiency of translation termination.
Review TRENDS in Microbiology Vol.15 No.2 83
www.sciencedirect.com
Figure 12 : Modulation de l'efficacité de terminaison de la traduction : des protéines accessoires influencent l'activité de terminaison.

43
3.4.1. Pab1, acteur secondaire de la terminaison
3.4.1.1. Pab1 est impliquée dans la terminaison de la traduction.
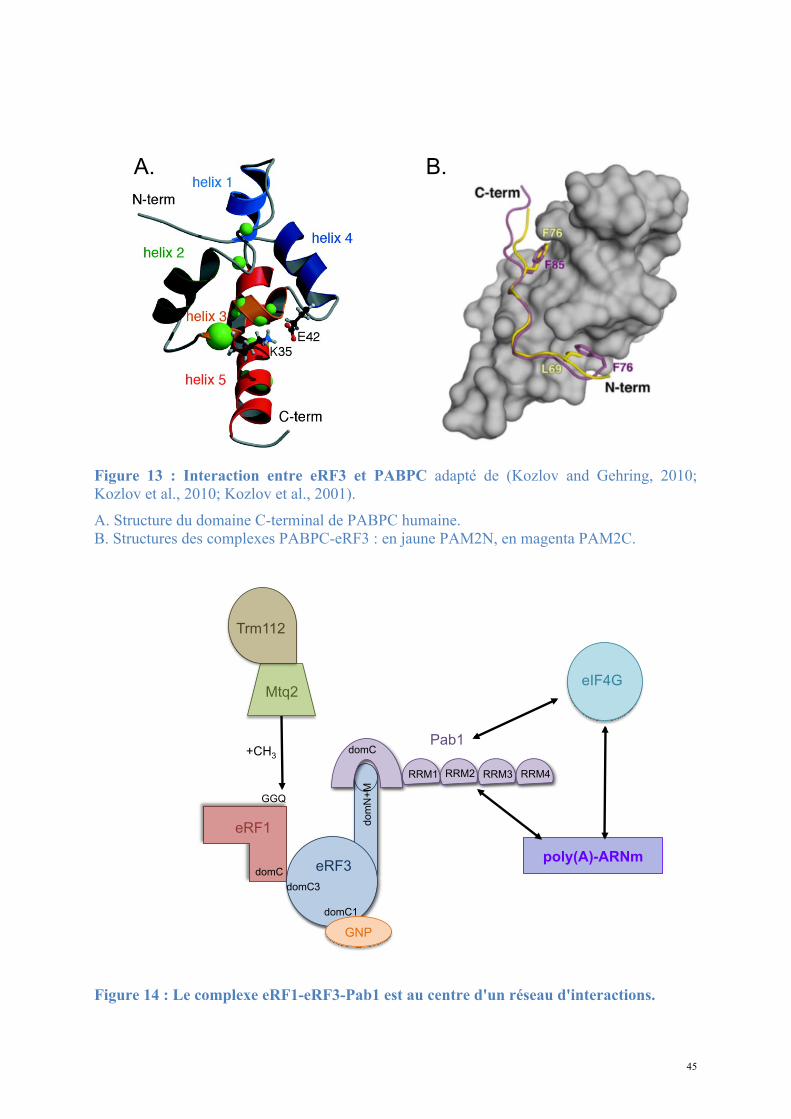
Pab1 interagit directement avec eRF3 in vitro et in vivo (Cosson et al., 2002). Plus
précisément, ce sont les résidus 473 à 577 du domaine C-terminal de Pab1 qui interagissent
avec les résidus 1 à 239 du domaine N+M d'eRF3 (Figure 13). Ces domaines d'interaction
sont largement conservés puisque Pab1 de S. cerevisiae peut fixer eRF3 NM de H. sapiens.
La surexpression de Pab1 augmente l'efficacité de terminaison, ceci étant lié à son interaction
avec eRF3 mais pas à un effet indirect sur la formation de prions [PSI+].
3.4.1.2. Structure de Pab1.
Pab1 est constituée de quatre domaines N-terminaux reliés à un domaine C-terminal par
une région peu conservée.
- Les quatre domaines N-terminaux sont des RRM (RNA Recognition Motifs) impliqués
dans la reconnaissance de l'ARNm et dont les séquences sont fortement conservées entre les
espèces. L'affinité et la spécificité de l'interaction avec l'ARN in vitro est principalement
assurée par les RRM1 et 2, tandis que les RRM3 et 4 jouent un rôle accessoire dans la fixation
(Kuhn and Pieler, 1996). Les RRM1 et 2 contiennent également les sites de fixation d'eIF4G.
- Une région connectrice (ou linker) est riche en prolines et méthionines. Son rôle n'est
pas caractérisé mais il serait important pour l’interaction de Pab1 avec certains
partenaires dont Pbp1 (Mangus et al., 2003). De plus, la délétion de cette région affecte
également la stabilité des ARNm en les rendant plus sensibles à la dégradation (Simon and
Seraphin, 2007).
- Le domaine C-terminal MLLE montre un repliement en fuseau d'hélices-α (Kozlov et
al., 2001). Sa structure et ses fonctions d'interaction ont surtout été étudiées chez H. sapiens,
dont la protéine Pab1 est appelée PABPC1 et son domaine C-terminal PABC. Ce domaine est

44
impliqué dans l’interaction avec différentes protéines qui ont en commun le motif PAM2 de
douze résidus : 1xx[P/F/L]NxxAxEFxP12. Le site de fixation aux peptides PAM2 est porté par
deux cavités séparées par l'hélice-α3. La signature KITGMLLE chez les eucaryotes supérieurs
ou KITGMILD chez la levure S. cerevisiae ainsi que deux motifs LGExLF/Y et VxEA,
conservés chez toutes les Pab, sont en interaction avec les résidus L3N4A7E9F10P12 des motifs
PAM2. Parmi les protéines fixant le domaine MLLE de PABC via le motif PAM2 se trouvent
eRF3 (deux motifs se recouvrant partiellement) (Figure 13), Tob, Pan3, NF-X1, Paip2 et
l'Ataxin-2. La présence de deux motifs indépendants sur eRF3 amène les auteurs de l'étude
(Kozlov et al., 2010) à proposer un mécanisme coopératif augmentant l'affinité et prévenant la
fixation de plus d'une protéine eRF3 par Pab1.
3.4.1.3. Pab1 est une protéine clef du métabolisme de l'ARNm et de la traduction.
- Pab1 régule la longueur de la queue poly(A) en limitant son élongation (Amrani et al.,
1997), en inhibant la poly(A)-polymérase Pap1 (Lingner et al., 1991) et contrôle la vitesse de
déadénylation par la poly(A)-nucléase Pan2 (Brown and Sachs, 1998); par ailleurs elle
protège l'ARNm de la dégradation en 3' (Ford et al., 1997).
- Pab1 intervient dans l'initiation de la traduction, via son interaction avec eIF4G (Tarun
and Sachs, 1996) (Hentze, 1997) (Wells et al., 1998); elle a un effet anti-suppresseur par son
interaction avec eRF3, révélant son rôle dans la terminaison.
45
Figure 13 : Interaction entre eRF3 et PABPC adapté de (Kozlov and Gehring, 2010; Kozlov et al., 2010; Kozlov et al., 2001). A. Structure du domaine C-terminal de PABPC humaine. B. Structures des complexes PABPC-eRF3 : en jaune PAM2N, en magenta PAM2C.
Figure 14 : Le complexe eRF1-eRF3-Pab1 est au centre d'un réseau d'interactions.
A. B.
C.
B.
D.
eRF1
eRF3
Pab1
poly(A)-ARNm
Mtq2
Trm112
+CH3
GNP
eIF4G
GGQ
domC
domC3
domC1
dom
N+
M
domC
RRM1 RRM2 RRM3 RRM4

46
3.4.2. Mtq2-Trm112 méthyle la glutamine du motif GGQ d'eRF1
La chaîne latérale de la glutamine du motif GGQ du facteur de terminaison de classe I
subit une méthylation post-traductionnelle chez tous les organismes étudiés (Heurgue-Hamard
et al., 2005). Cette méthylation est nécessaire pour une terminaison efficace chez E. coli
(Mora et al., 2007). En revanche, l'importance de cette modification n'a pour le moment pas
été déterminée chez les eucaryotes.
Chez la levure S. cerevisiae, cette modification post-traductionnelle est assurée par le
complexe hétérodimérique Mtq2-Trm112. Dans ce complexe, la protéine Mtq2 est la sous-
unité catalytique à activité méthyltransférase dépendante du SAM. Son partenaire Trm112
aurait plutôt un rôle de stabilisation et d’activation. Le complexe Mtq2-Trm112 méthyle la
glutamine du motif GGQ d'eRF1 lorsque celle-ci est en complexe sous forme eRF1-
eRF3:GTP (Heurgue-Hamard et al., 2006). L'étude des conditions d'activité de Mtq2-Trm112
montre une spécificité de substrat pour eRF1-eRF3:GTP/GMPPNP mais pas pour eRF1-
eRF3:GDP dans les mêmes conditions de mesure, ce qui est en faveur d’un changement
conformationnel significatif du complexe de terminaison lors de la fixation du GTP, comme
cela a été proposé à partir du modèle à basse résolution d'eRF1-eRF3:GTP (Cheng et al.,
2009).
Trm112 est formée de deux domaines : un motif doigt de zinc composé des extrémités N-
et C-terminales de la protéine et un domaine central en hélices α qui n’est présent que chez les
protéines eucaryotes (Heurgue-Hamard et al., 2006). Trm112 seule est homodimérique en
solution mais exclusivement hétérodimérique en solution avec Mtq2, ce qui suggère que les
interfaces d'interaction de Trm112 avec elle-même ou avec Mtq2 se recouvrent au moins
partiellement. Trm112 s'associe avec d'autres protéines in vivo, dont les ARNt-
méthyltransférases Trm11 (Purushothaman et al., 2005) et Trm9 (Mazauric et al., 2010) et la

47
saccharopine déshydrogénase Lys9 (Storts and Bhattacharjee, 1987). Dans chaque cas,
Trm112 semble participer indirectement à l'activité catalytique de son partenaire en
améliorant sa solubilité. Trm112 étant indépendamment nécessaire aux activités de ces
protéines distinctes, sa disponibilité limitée permettrait d'établir un équilibre entre les
différentes fonctions (Studte et al., 2008).
3.4.3. Tpa1, régulateur de l’efficacité de terminaison de la traduction
3.4.3.1. Tpa1 est impliquée dans la terminaison de la traduction.
Chez la levure S. cerevisiae, le gène YER049w code pour une protéine de fonction
inconnue qui a été annotée Tpa1 (pour Termination and PolyAdenylation) car la délétion de
ce gène entraîne l'apparition de trois phénotypes notables (Keeling et al., 2006) :
- Diminution de l'efficacité de terminaison de la traduction par augmentation de la
translecture des codons stop. Cet effet est observé chez les levures normales comme chez les
[PSI+].
- Augmentation de la longueur des queues poly(A) des ARNm.
- Augmentation de la stabilité des ARNm.
De plus, Tpa1 a été coimmunoprécipitée à partir d'extraits cellulaires avec les facteurs de
terminaison eRF1-eRF3 et la protéine Pab1. L'ensemble formerait un complexe mRNP fixé à
l'extrémité 3' des ARNm. Enfin, Tpa1 a été identifiée parmi les protéines ayant une affinité
pour le complexe de pore nucléaire (Rout et al., 2000).
L’analyse bioinformatique de la séquence primaire de Tpa1 indique que cette protéine
contient un domaine DSBH (double-stranded β-helix) caractéristique des dioxygénases
dépendant du Fe2+ (Aravind and Koonin, 2001). Ce repliement se caractérise par deux
feuillets β en sandwich entourés d'un ensemble d'hélices α. Le centre catalytique contient une

48
triade d'acides aminés Hx(D/E)...H qui fixent un cation métallique. Ces résidus se situent dans
une cavité définie par les deux feuillets β, et dans laquelle les substrats peuvent se fixer. Les
Fe2+-dioxygénases sont une classe d'enzymes très variées trouvées chez les procaryotes et les
eucaryotes. Elles catalysent l'oxydation par l'oxygène moléculaire d'un substrat organique.
Elles sont impliquées dans des voies extrêmement diverses : modifications des histones,
métabolisme des lipides, réparation de l'ADN ou de l'ARN, biosynthèse du collagène,
biosynthèse des antibiotiques, régulation de la réponse cellulaire à l'hypoxie (Clifton et al.,
2006). La prédiction du repliement de Tpa1 n'apporte pas d'éléments évidents pour interpréter
son implication dans la terminaison de la traduction et la stabilité des ARNm.
L'orthologue de Tpa1 chez S. pombe est la protéine Ofd1 qui est impliquée dans la
régulation de l'expression de gènes de réponse à l'hypoxie via le facteur de transcription Sre1
qui est ancré à la membrane du réticulum endoplasmique (Hughes and Espenshade, 2008; Lee
et al., 2009). Lorsque la concentration en O2 est faible, Sre1 est activée par clivage
protéolytique et la translocation du domaine N-terminal de Sre1 (Sre1N) dans le noyau permet
l'expression des gènes d'adaptation à l'hypoxie. Au contraire, à partir d'une certaine
concentration en O2, le domaine Sre1N est dégradé par un mécanisme dépendant d’Ofd1,
prévenant son rôle d'activateur de la transcription. De part sa ressemblance avec des
dioxygénases, Ofd1 pourrait jouer le rôle de senseur de la concentration en O2 et
d'interrupteur de l'adaptation de la transcription à un métabolisme hypoxique.
Le lien direct, s'il existe, entre l'implication d'Ofd1 dans la régulation de la réponse
cellulaire à l'hypoxie et le rôle de son orthologue Tpa1 dans la stabilité des ARNm et
l'efficacité de terminaison de la traduction chez S. cerevisiae n’est pour l’heure pas évident.
En effet, les phénotypes observés sont significativement différents. De plus il n’existe pas
d'orthologue de Sre1 chez S. cerevisiae et l'effet de la délétion du gène codant pour Ofd1 sur
l'efficacité de terminaison n'a pas été mesuré.

49
3.4.3.2. Tpa1/Ofd1 interagit avec Ett1/Nro1.
Nro1 est un régulateur positif de la stabilité de Sre1N par inhibition directe d'Ofd1 (Lee et
al., 2009). Les auteurs de cette étude montrent une fixation de Nro1 avec Ofd1 en absence
d'oxygène et proposent que cette interaction soit le déterminant de la stabilité de Sre1N et du
niveau de transcription des gènes de réponse à l'hypoxie.
En parallèle, des études à grande échelle des complexes protéiques de S. cerevisiae par
purification TAP suivie d'une identification par MALDI-MS et LC-MS-MS (Krogan et al.,
2006) proposent un ensemble de partenaires potentiels pour Tpa1. Parmi ceux-ci se trouve la
protéine Ett1, produit du gène YOR051c et qui est l’orthologue chez S. cerevisiae de Nro1.
Ett1, tout comme Tpa1, a été inventoriée dans une liste de protéines enrichies lors de la
purification du complexe de pore nucléaire (Rout et al., 2000). Cependant sa localisation
exclusivement nucléaire écarte la possibilité qu'elle soit elle-même intégrée au pore nucléaire
mais indique plutôt qu'elle serait en interaction transitoire avec les nucléoporines qui le
constituent.
Elle a également été identifiée parmi les protéines inhibant la réplication du virus BMV
dans la levure (Kushner et al., 2003) : la délétion de YOR051c provoque l'augmentation de
l'expression d'un gène rapporteur placé sous le contrôle transcriptionnel d'un promoteur du
BMV. Dans la mesure où la majorité des protéines identifiées dans cette étude interviennent
dans les trois fonctions cellulaires suivantes : métabolisme des ARNm, régulation du niveau
d'expression des facteurs de réplication et régulation de la composition de la membrane
cellulaire, il est probable que la protéine Ett1 intervienne dans l’une de ces trois fonctions. Le
rôle précis d'Ett1 reste inconnu, mais sa possible implication dans le métabolisme des ARNm
est à mettre en relation avec son interaction avec Tpa1.
La compréhension du rôle de Tpa1 et d'Ett1 dans la terminaison de la traduction nécessite

50
davantage de recherche.
Les cellules consacrent une quantité significative d'énergie à la synthèse des protéines.
Un tiers du poids sec de la plupart des cellules est du à des protéines impliquées dans
l'expression (Snustad D. Peter, 2010). Chez Escherichia coli les ribosomes représentent un
quart du poids sec de la cellule . L'efficacité de l'expression génique est nécessaire pour
limiter les pertes d'énergie de ce processus, optimiser le métabolisme cellulaire et s'adapter
aux conditions environnementales.
La régulation des mécanismes de transcription, maturation, export et traduction par les
facteurs canoniques (ribosomes, ARNt, facteurs d'élongation et de terminaison) est complétée
par des protéines supplémentaires (parmi lesquelles Pab1, Mtq2-Trm112, Tpa1 et Ett1)
modulant l'efficacité de l'expression. Ceci aboutit au final à l'augmentation ou la diminution
du nombre possible de protéines produites et permet une adaptation fine et optimale de
l'expression génique aux conditions du milieu.

51
4. Stabilité des ARN messagers et dégradation
L'ARN messager est une molécule intermédiaire située au centre de l'expression génique
chez tous les êtres vivants. Il apparaît comme un relai d'information entre les gènes et les
protéines, en servant à la fois à copier la séquence de l'ADN et à la transférer dans le
cytoplasme en vue de sa traduction.
L'ARN messager est une molécule complexe dont la durée de vie est limitée.
L'abondance de la molécule est déterminée par l'équilibre entre sa production (transcription)
et son élimination (dégradation) (Houseley and Tollervey, 2009).
Les cellules eucaryotes possèdent des mécanismes dédiés à la dégradation contrôlée et
spécifique des ARNm. Lorsque l'ARNm est destiné à être dégradé il ne sert plus de support à
la traduction et est rapidement éliminé de la cellule, interrompant l'expression à sa dernière
étape (Tharun S., 2001). L'existence de l'ARNm en tant qu'intermédiaire a permis l'apparition
et l'évolution de mécanismes de régulations post-transcriptionnelles : en ajustant la quantité
de protéines produites lors de la traduction, ils augmentent ainsi la précision du niveau
d'expression génique pour chaque gène transcrit. Ce contrôle en dernière étape peut assurer
aussi l'adaptation rapide aux conditions environnementales de la cellule.
La traduction est une étape limitante de l'expression génique. Tous les organismes
eucaryotes étudiés possèdent des mécanismes prenant place lors de la traduction et contrôlant
non seulement la quantité mais aussi la qualité des ARNm. Ces mécanismes de contrôle
interviennent dans différents cas de figure et permettent d'améliorer la qualité des ARNm
disponibles en éliminant les molécules non fonctionnelles (Doma and Parker, 2007). Les
ARNm portant des erreurs dues à un défaut de la machinerie de transcription ou de maturation
ou bien apparues aléatoirement au cours du temps seront éliminés.

52
Les mécanismes décrits ici sont spécifiques de la dégradation des ARNm dans le
cytoplasme. Il existe des voies de dégradation spécifiques du noyau qui ne sont pas citées.
4.1. Dégradation normale
Contrôle de la quantité d'ARNm disponible pour la traduction
La majeure partie des fonctions cellulaires étant assurée par des protéines, les cellules
produisent des milliers d'ARNm codant pour autant de protéines différentes. La quantification
des transcrits chez S. cerevisiae révèle qu'une cellule contient environ 60600 ARNm codés
par 4665 gènes et il est estimé qu’il y a de 0,3 à 200 copies d’un même ARNm dans la cellule
(Velculescu et al., 1997). Ainsi une molécule d'ARNm de l'actine sera traduite 1000 fois pour
produire autant de protéines (Futcher et al., 1999). L'ARNm étant le substrat de la traduction,
la production de chaque protéine est directement reliée à la quantité d'ARNm disponible et à
sa vitesse de traduction (Tharun S., 2001). Par conséquent la régulation précise des
concentrations de chaque ARNm est cruciale pour un fonctionnement normal des cellules. La
stabilité des ARNm est un facteur essentiel de l'expression génique.
Les molécules d'ARNm sont en renouvellement permanent dans la cellule (turnover), la
vitesse de renouvellement étant déterminée par le rapport entre le temps de production et le
temps de dégradation. La dégradation ajuste la quantité d'ARNm disponible pour la
traduction. Le temps de demi-vie (t1/2) varie grandement dans les cellules eucaryotes, allant de
quelques minutes à plusieurs heures ou plusieurs jours. Le temps de dégradation des ARNm
d'une cellule varie d'un ARNm à l'autre, et pour un ARNm donné le temps de dégradation
dépendra des conditions physiologiques et environnementales.

53
La voie de dégradation majeure chez la levure est initiée par une première étape de
dégradation de la queue poly(A) présente à l’extrémité 3’ des ARNm : la déadénylation
(Muhlrad and Parker, 1992). Elle est suivie par la dégradation quasi irréversible de la coiffe
m7G en 5’ (décoiffage ou décapping) (Coller and Parker, 2004). L’ARNm non protégé subit
des dégradations exonucléolytiques 5'3' et 3’5’. Une dégradation efficace nécessite
l'exposition de sites de digestion par les exonucléases : soit une déprotection en 3', soit une
déprotection en 5', soit une coupure endonucléolytique et l'exposition de nouvelles extrémités
3' et 5' (Mitchell and Tollervey, 2000a; Tharun S., 2001). Les vitesses de déadénylation et de
decapping varient d'un ARNm à l'autre et déterminent la durée de vie de l'ARNm.
4.1.1. Déadénylation en 3'
Tous les ARNm, qu'ils soient instables ou normaux, subissent une dégradation
déadénylation-dépendante (Decker and Parker, 1993). Au cours de la phase de déadénylation
initiale, les queues poly(A) sont raccourcies lentement. Pendant la deuxième phase, la
déadénylation s'accélère jusqu'à une longueur correspondant à un oligo(A) de 3 à 21
nucléotides. Enfin une troisième phase de raccourcissement de la queue oligo(A) peut avoir
lieu, mais l'ARNm sera de toute façon à la fin de la deuxième étape soumis à une dégradation
globale rapide.
Il existe plusieurs complexes de déadénylation chez les eucaryotes (Parker and Song,
2004) (Figure 15). Une première déadénylase formée par les protéines Pan2 et Pan3 (où Pan2
est la sous-unité catalytique et Pan3 la sous-unité activatrice) intervient pour raccourcir la
queue poly(A) (environ 50 A chez l’homme) (Brown and Sachs, 1998). Le complexe Ccr4-
Pop2 associé à des protéines accessoires (dont Not1-5) intervient ensuite pour dégrader le
reste de la queue poly(A) (Denis and Chen, 2003; Tucker et al., 2002; Tucker et al., 2001).

54
Chez les mammifères une troisième nucléase intervient : la poly(A)-ribonucléase PARN
(Korner and Wahle, 1997) qui dégrade notamment les ARNm possédant des éléments
d'instabilité riches en AU (ARE). La PARN n'est cependant pas présente chez S. cerevisiae ni
D. melanogaster.
Chez l’Homme, la dégradation 3'5' est contrôlée par les facteurs de terminaison de la
traduction (Funakoshi et al., 2007). La surexpression du facteur de terminaison eRF3 inhibe la
déadénylation et la dégradation conséquente de l'ARNm. Chez l’Homme, eRF3, Tob et Pan3
possèdent un domaine PAM2 d'interaction avec le domaine MLLE de PABPC1 (orthologue
humain de Pab1) (Figure 12). Les complexes eRF1-eRF3, Ccr4-Pop2 (via Tob) et Pan2-Pan3
interagissent de façon compétitive avec la protéine PABPC1 liée à la queue poly(A). Le
modèle proposé fait intervenir une compétition entre le complexe eRF1-eRF3 et les
complexes de déadénylation pour fixer PABPC1 lors de la terminaison de la traduction. La
fixation de PABPC1 par les complexes de déadénylation active la dégradation de la queue
poly(A).
Les ARNm sont déadénylés à un rythme lent et régulier au fur et à mesure des
traductions, avant d'être rapidement dégradés lorsque la longueur de la queue poly(A) passe
en-dessous d'un seuil critique, que Pab1 n'interagit plus et que la protection face aux
exonucléases 3'5’ est levée.

55
Figure 15 : Les différents complexes eucaryotes de déadénylation (d'après (Parker and Song, 2004)). Les complexes de déadénylation sont affectés différemment par Pab1 et la coiffe.
4.1.2. Decapping en 5'
Suite à la déadénylation, la coiffe m7GpppN présente à l’extrémité 5’ est hydrolysée par
le complexe enzymatique Dcp1-Dcp2 (Beelman et al., 1996; Dunckley and Parker, 1999; Gu
and Lima, 2005) (Figure 16). Dcp2 catalyse la libération du m7GDP et crée ainsi une
extrémité 5’ phosphate au niveau des ARNm. Dcp1 assiste Dcp2 et augmente son activité par
une régulation allostérique. Cependant, l’activité intrinsèque de ce complexe reste faible et
nécessite des facteurs accessoires pour être optimale. Parmi ceux-ci figurent les protéines
Lsm1-7 formant des complexes multimériques autour de l'ARN (Boeck et al., 1998) et les
protéine Dhh1 (Coller and Parker, 2004; Coller et al., 2001), Pat1 (Hatfield et al., 1996) et
Stm1 (Balagopal and Parker, 2009b). Le decapping est une étape quasi irréversible, destinant
définitivement un ARNm à la dégradation.
56
Figure 16 : Mécanisme moléculaire du decapping (d'après (Gu and Lima, 2005) et (Parker and Song, 2004))
a. Dcp1-Dcp2 est le complexe majeur de decapping (Beelman et al., 1996; Dunckley and Parker, 1999); DcpS est l'enzyme de decapping dite scavenger (charognard), ciblant les ARN cappés de petite taille (Nuss et al., 1975) : elle cible notamment les ARNm après deadénylation et digestion par l'exosome, ainsi que le m7GDP produit par Dcp1-Dcp2.
b. Le complexe majeur de decapping Dcp1-Dcp2 peut être recruté sur différents substrats ARNm selon les complexes protéiques qui y sont associés : - le decapping normal fait intervenir le complexe Lsm1-7 interagissant avec l'ARNm, la protéine Pat1 et l'hélicase Dhh1; - lors de la dégradation des ARNm aberrant par la voie NMD (voir plus loin) Upf1 recrute directement Dcp1-Dcp2; - Rps28a reconnaît une structure tige-boucle de l'ARNm et interagit avec Edc3 qui recrute Dcp1-Dcp2 (Badis et al., 2004).
rally distinct families: the divalent-cation-dependentRNA triphosphatases of protozoa, eukaryotic virusesand fungi, and the divalent-cation-independent RNAtriphosphatases of metazoa and plant [12,13]. In yeast,the triphosphatase and guanylyltransferase polypeptidesare encoded by different genes, whereas mammalianspecies contain a single bifunctional polypeptide com-
posed of an N-terminal triphosphatase domain and a C-terminal guanylyltransferase domain [3].
The divalent-cation-dependent RNA triphosphatasefamily was exemplified by previous studies of the Sac-charomyces cerevisiae RNA triphosphatase Cet1, a structurethat revealed a dimeric architecture and two paralleltunnels, which are each formed by eight antiparallel bstrands (Figure 2a). The tunnel center includes severalcharged and hydrophilic sidechains that bind a manga-nese ion and a sulfate ion. The latter is in a position thatprobably approximates the position of the g-phosphate ofthe triphosphate-terminated pre-mRNA before hydroly-sis [12].
The metal-independent class of triphosphatase enzymewas exemplified by the RNA triphosphatase domain frommouse capping enzyme [Mce1(1–210)] (Figure 2b) [13].The structure is unrelated to that of the yeast RNAtriphosphatase Cet1, but shares significant similarity withmembers of the cysteine phosphatase superfamily. TheMce1 active site includes the HCxxxxxR(S/T) signaturemotif, which encompasses a phosphate-binding loop (P-loop) and active site cysteine (Cys126). Specificity for pre-RNA is apparently achieved by maintaining an active sitethat is sufficient for interaction with 50 triphosphate-terminated pre-RNA, but too deep to allow 50 diphos-phate-terminated pre-RNA access to the active sitecysteine.
Structure and mechanism of the RNAguanylyltransferase enzymesGuanylyltransferases consist of two structural domains: anN-terminal nucleotidyl transferase (NT) domain and aC-terminal OB-fold domain [14]. The overall fold of RNAguanylyltransferases resembles that of DNA and RNAligases, and structure-based alignment of the amino acidsequences of several family members reveals conservedresidues and motifs that localize to the nucleotide-binding site and NT motifs [15]. Guanylyltransferasesutilize GTP in a two-step reaction that involves a covalentenzyme-(lysyl-N)–GMP intermediate before GMP trans-fer to the diphosphate-terminated pre-mRNA. RNA gua-nylyltransferase structures have been determined forChlorella virus PBCV-1 in open and closed configurationsbound to GTP, as a closed enzyme–guanylate intermedi-ate and in an open complex with the cap analog GpppG[14,16]. More recently, structures of a cellular RNAguanylyltransferase from Candida albicans (Cgt1) weredetermined in open GTP-bound and open enzyme–guanylate configurations (Figure 2c–e) [17!!].
Based on these structures, and on structures of DNA andRNA ligase family members, a general mechanism forphosphoryl transfer by the nucleotidyltransferase familyhas been proposed [15]. In the first step, magnesium andGTP bind to the open form of the enzyme (Figure 2c).
100 Protein–nucleic acid interactions
Figure 1
HN
N
N+
O
H2N
N
OP
CH3
O
O P
O
O P
O
O– O– O–
O–
OO
O
OH OH
PO OO
Base N
Base N
5!5!
n
ppN(pN)n + EpG
pppN(pN)n
" #$ $# "
#$
Gppp
GpppN(pN)n
DcpS
Pi
Dcp1–Dcp2 DcpS
Guanine-N7
methyltransferase
Guanylyltransferase(E)
Triphosphatase
O OH
O OH
(a)
(b)
Pi PPi
AdoHcy
AdoMet
m7GpppN(pN)n
pN(pN)n ppN(pN)n
+ +
m7Gpp m7Gp
Current Opinion in Structural Biology
mRNA cap structure and its metabolism. (a) Chemical structure ofthe mRNA cap. The cap consists of N7-methyl guanosine linkedby an inverted 50-50 triphosphate bridge to the first nucleosideof the mRNA chain (base N can be adenine, guanine, cytosineor uracil). (b) Enzymatic synthesis and degradation of the mRNA cap.The cap is formed on nascent RNA by the sequential action ofthree enzymes: RNA triphosphatase, RNA guanylyltransferase andguanine-N7 RNA methyltransferase. RNA guanylyltransferasefirst forms a covalent lysyl-N–GMP adduct before transfer of theGMP to the 50 diphosphate RNA end. Degradation of the RNA capin the 50-30 decay pathway occurs through hydrolysis by theDcp2–Dcp1 complex in a reaction that generates m7GDP and50 monophosphate-terminated mRNA. Hydrolysis of the RNA capin the 30-50 decay pathway is catalyzed by DcpS in a reaction thatgenerates m7GMP and diphosphate-terminated RNA. DcpS isalso able to hydrolyze m7GDP to release m7GMP and phosphate.
Current Opinion in Structural Biology 2005, 15:99–106 www.sciencedirect.com

57
4.1.3. Dégradation exonucléolytique 5’3’ et 35’
Lorsque l'ARNm perd ses protections en 3' et/ou 5', sa forme circulaire est déstabilisée, il
n'est plus traduit efficacement et devient accessible aux exonucléases. La dégradation
exonucléolytique est processive et donc extrêmement rapide. Les ARNm destinés à la
dégradation sont accumulés dans des loci cytoplasmiques appelés P-bodies (Balagopal and
Parker, 2009a). La dégradation 5'3' des ARNm cytoplasmiques est assurée par
l’exonucléase Xrn1 (Larimer et al., 1992; Stevens and Maupin, 1987). La dégradation 3'5'
des ARNm déadénylés est réalisée par l'exosome cytoplasmique (Butler, 2002; Schmid and
Jensen, 2008; van Hoof and Parker, 1999). L'exosome eucaryote est un assemblage
multiprotéique complexe (Mitchell and Tollervey, 2000b). Il contient un cœur de neuf sous-
unités RNase PH extrêmement conservées et essentielles pour la viabilité de la cellule. Chez
la levure et l'homme, ces sous-unités sont inactives et jouent un rôle structural en recrutant les
protéines accessoires dont les protéines à activité nucléase : Rrp44/Dis3 dans le cytoplasme a
une activité processive sur les ARN structurés ou non. D'autres cofacteurs tels que Ski7,
Rrp47 et Mpp6 régulent le recrutement de l'exosome pour des dégradations ciblées. Ski7
recrute le complexe Ski (formé par les protéines Ski2, Ski3 et Ski8) à activité régulatrice
(Anderson and Parker, 1998). L'exosome présente également une activité endonucléase via le
domaine PIN de sa sous-unité Dis3 (Lebreton et al., 2008).
La dégradation est un mécanisme impliquant de nombreux facteurs qui interagissent
précisément, manifestant un niveau de contrôle élevé. La durée de vie d'un ARNm sera
principalement dictée par la longueur de sa queue poly(A) et son niveau de traduction. Ainsi
le m7G et Pab1-poly(A) jouent à la fois le rôle de protecteurs des ARNm et d’activateurs de
l’initiation de la traduction. La dégradation rapide des ARNm nécessite le plus souvent la

58
dégradation de ces éléments. Cependant un certain nombre de mécanismes ciblés spécifiques
vont accélérer la dégradation de l'ARNm dans certaines conditions, réduisant son temps de
demi-vie et son niveau d'expression.
4.2. Dégradation ciblée spécifique
Contrôle-qualité des ARNm en cours de traduction
Un aspect fondamental de la biogenèse et de la fonction des ARNm eucaryotes est
l'existence de systèmes de contrôle-qualité qui reconnaissent et dégradent les ARNm non
fonctionnels. Il existe plusieurs mécanismes de dégradation ciblée de l'ARNm (Doma and
Parker, 2007; Jacobson A., 2007; LaRiviere et al., 2006; Vasudevan et al., 2002). Ils ciblent
des défauts inhérents à l'ARNm lui-même, mais affectant aussi le ribosome. Parmi les
mécanismes possibles nous nous intéressons plus particulièrement à ceux qui sont liés à la
terminaison de la traduction. Les ARNm sur lesquels la traduction s'arrête trop tôt, ne s'arrête
pas ou s'arrête au cours de l'étape d'élongation sont pris en charge par des machineries
spécifiques induisant leur dégradation.
Quatre voies de contrôle qualité de l'ARN et dépendantes de la traduction ont été
caractérisées et sont décrites ci-dessous: la NMD, la NSD, la NGD et la NRD.
4.2.1. NMD, nonsense mediated mRNA decay
La voie NMD (Figure 17) est un mécanisme de surveillance assurant la dégradation
rapide des ARNm contenant des codons stop en amont du site normal de terminaison ou

59
codon stop précoce chez les eucaryotes (Baker and Parker, 2004; Conti and Izaurralde, 2005;
Isken and Maquat, 2008; Lejeune et al., 2003). L'ARNm portant un codon stop précoce est dit
aberrant. La voie NMD prévient ainsi la synthèse de protéines tronquées et potentiellement
non fonctionnelles. Elle contrôle également l'expression de 10 à 20% des transcrits non
aberrants de la cellule (Holbrook et al., 2004; Mendell et al., 2004) et est essentielle au
développement normal chez les vertébrés (McIlwain et al., 2010; Medghalchi et al., 2001;
Wittkopp et al., 2009).
La présence d'un codon stop précoce peut s'expliquer par une mutation ponctuelle
substitutive non-sens dans la séquence codante du gène (erreur de l'ADN polymérase au cours
de la réplication), une mutation similaire dans l'ARNm (erreur de l'ARN polymérase II), un
défaut d'épissage laissant un intron entre le codon initiateur et le codon stop naturel ou un
décalage de cadre de lecture par le ribosome.
L'existence de mutations non-sens homozygotes sur un gène unique du génome explique
16% des cas de mucoviscidose : le gène codant pour le transporteur membranaire CFTR
présente une substitution non-sens sur le triplet codant pour la phénylalanine 508. Il a pour
conséquence la production d'ARNm codant pour des protéines tronquées des 972 derniers
résidus (Fanen, 2001). La voie NMD dégrade l'ARNm aberrant codant pour la CFTR
tronquée. Celle-ci est pourtant partiellement fonctionnelle et la recherche d'inhibiteurs de la
voie NMD a mené à la découverte du PTC124 (ataluren). Cette molécule induit la production
de la protéine entière chez le modèle murin en favorisant l’incorporation d’un acide aminé au
niveau des codons stop précoces, améliorant significativement l'état des malades de ce type de
mucoviscidose (Kerem et al., 2008).
Les erreurs d'épissage consistent en l'apparition de variants d'épissage contenant des
introns dans le cadre de lecture. Les introns ne sont normalement pas traduits, par conséquent

60
l'apparition aléatoire de codons stop dans les introns n'est pas contre-sélectionnée au cours de
l'évolution. La probabilité de rencontrer un codon stop dans une séquence aléatoire s'élève à
3/64, soit 4,68%. Dans une répartition normale des codons d'une séquence non sélectionnée,
un codon stop sera présent statistiquement tous les 20 codons environ. Pour un intron de taille
moyenne de 1113 nucléotides (Deutsch and Long, 1999) il est extrêmement probable de
rencontrer un codon stop précoce.
Enfin un décalage du cadre de lecture du ribosome sur l'ARNm en cours de traduction
changera la séquence traduite, substituant à une séquence sélectionnée au cours de l'évolution
contre la présence de codons stop, une séquence non sélectionnée de même type que les
introns. Pour une séquence de plus de 100 nucléotides il y aura probablement un codon stop
précoce.
Si les conséquences des événements de terminaison précoce de la traduction sont directes
et ont un impact sur la fonction des protéines, on constate cependant que ces effets
n'apparaissent que dans maximum 25% des cas, montrant une grande efficacité de la voie
NMD in vivo (Linde et al., 2007). Ainsi un ARNm aberrant ne produira généralement aucune
protéine à l'étape de la traduction. Le système de contrôle qualité cible les ARNm aberrants
grâce à l'action coordonnée d'un ensemble de facteurs permettant de discriminer un codon
stop normal d'un codon stop précoce. Chez les eucaryotes étudiés (S. cerevisiae, C. elegans,
D. melanogaster, M. musculus, H. sapiens), des différences significatives apparaissent dans
les mécanismes de discrimination codon stop normal / codon stop précoce (Rehwinkel et al.,
2006). La NMD se déroule en trois étapes clefs (Baker and Parker, 2004; Conti and
Izaurralde, 2005) : la reconnaissance des codons stop par les facteurs de terminaison de la
traduction, la discrimination entre un codon stop normal et un codon stop précoce et enfin la
dégradation de l'ARNm s'il est aberrant (Figure 17).
61
ARNm NORMAL ARNm ABERRANT
Upf1-Upf2-Upf3
Complexe de surveillance
Exosome
Dégradation 3’-5’ processive
EJC
Marqueur site d’épissage
Smg5-Smg6-Smg7
Reconnaissance Upf1-P
Structures des acteurs de la voie NMD :
Figure 17 : Mécanisme et acteurs de la voie NMD adapté de (Baker and Parker, 2004)

62
1. Terminaison de la traduction.
La NMD prend place lorsqu'un codon stop précoce entre dans le ribosome. D'un point de
vue moléculaire, il s'agit d'un arrêt de l'étape d'élongation et du recrutement du complexe de
terminaison eRF1-eRF3 au site A du ribosome.
2. Discrimination entre un codon stop normal et un codon stop précoce
Le devenir de l'ARNm lors de l'étape de terminaison dépendra entièrement de la position
du codon stop :
(i) un codon stop normal permet une terminaison efficace et un recyclage des facteurs de
terminaison et du ribosome, laissant l'ARNm stable pour de nouvelles traductions;
(ii) un codon stop précoce détermine une réduction de l'expression de l'ARNm et le
destine à la dégradation rapide.
Le mode de discrimination des codons stop normaux et précoces, et ainsi des ARNm
normaux et aberrants, a fait l'objet de nombreuses recherches. Le mécanisme de
discrimination est basé sur l'interaction du complexe de terminaison eRF1-eRF3 avec les
facteurs de surveillance Upf1, Upf2, Upf3, la protéine Pab1 ainsi que les protéines Smg et
l'EJC chez certains organismes.
- Upf1-Upf2-Upf3 forment le cœur du complexe de surveillance
La délétion des gènes codant pour les protéines Upf1, 2 ou 3 résulte dans la stabilisation
des ARNm aberrants (Atkin et al., 1997; Cui et al., 1995; He et al., 1993; Kunz et al., 2006;
Leeds et al., 1992; Lelivelt and Culbertson, 1999; Maderazo et al., 2000; Peltz et al., 1993;
Wittmann et al., 2006). Upf3 est une protéine nucléaire s'associant au complexe de jonction
des exons (EJC) sur les ARNm matures. Upf2 est recrutée par Upf3 et l'EJC lors de l'export

63
nucléo-cytoplasmique. Upf1 est majoritairement cytoplasmique et interagit avec les facteurs
de terminaison eRF1 et eRF3 d'une part, et Upf2 et Upf3 d'autre part, reliant la traduction à la
surveillance (He et al., 1996, 1997; Schell et al., 2003). De plus, Upf1 est une
phosphoprotéine dont l’état de phosphorylation détermine l'activation de la voie NMD
(Ohnishi et al., 2003).
- Modes de reconnaissance du codon stop précoce
Le modèle principal proposé (faux-UTR) et soutenu par des expériences menées chez la
levure, la drosophile et l'homme (Amrani et al., 2004; Muhlemann et al., 2008) propose qu’un
ARNm aberrant est distingué d’un ARNm normal par la distance séparant le codon stop de la
queue poly(A) en 3’. Dans le cas d’un ARNm normal, la proximité du codon stop et de la
queue poly(A) permet une interaction directe entre eRF3 présent au niveau du codon stop via
son association avec eRF1 et le domaine C-terminal de Pab1, fixée à la queue poly(A). En
revanche, dans le cas d’un codon stop précoce, l’éloignement de celui-ci par rapport à la
queue poly(A) empêcherait l’interaction eRF3-Pab1. C’est cette absence de contexte optimal
de terminaison qui permettrait le recrutement du complexe de surveillance Upf1-Upf2-Upf3
au niveau du codon stop précoce, conduisant à l'activation de la voie NMD (Amrani et al.,
2004; Muhlemann, 2008).
Une étape de régulation supplémentaire existerait chez l’homme où l'EJC interviendrait
comme un élément de la discrimination codon stop normal/précoce (Le Hir et al., 2001). En
effet, si le ribosome arrive à un codon stop placé en amont d'une jonction exon-exon sur
laquelle un EJC est déposé, Upf1 est recrutée au complexe de terminaison eRF1-eRF3; son
interaction avec Upf2-Upf3 en interaction avec l'EJC conduit à l'assemblage du complexe de
surveillance complet Upf1-Upf2-Upf3 et à l'activation de la voie NMD.

64
3. Dégradation de l'ARNm aberrant reconnu
Chez les métazoaires, la phosphorylation d'Upf1 serait l’élément déclenchant la cascade
de réactions conduisant à la dégradation des ARNm aberrants (Kashima et al., 2006; Ohnishi
et al., 2003; Yamashita et al., 2009). Après assemblage du complexe de surveillance au codon
stop précoce, Upf1 phosphorylée recrute Smg7 (Ohnishi et al., 2003), et l'ARNm est adressé
dans des zones ponctuelles du cytoplasme, les P-bodies, où la traduction est réprimée et la
dégradation initiée. Les enzymes de la voie générale de dégradation des ARNm,
précédemment décrites sont alors recrutées. Le raccourcissement des queues poly(A) par les
déadénylases, puis le decapping en 5' et la digestion 5'3' par Xrn1 ou la dégradation 3'5'
par l'exosome et le complexe Ski terminent la dégradation de l'ARNm aberrant.
Initialement identifiée chez D. melanogaster (Gatfield and Izaurralde, 2004) une
dégradation initiée par coupure endonucléolytique dans le voisinage du codon stop précoce
est également observée chez les mammifères (Eberle et al., 2009). La coupure
endonucléolytique est catalysée par la protéine Smg6. Les fragments 5' et 3' générés sont
dégradés par les exonucléases classiques et le complexe exosome-Ski. La voie NMD peut
donc être indépendante des déadénylases et des enzymes de decapping.

65
4.2.2. NSD, non-stop decay
La NSD cible les ARNm ne portant aucun codon stop par suite d'une mutation de l'ADN,
de l'ARN ou d'une erreur de l'ARN-PolII, d'une terminaison de la transcription au niveau de la
séquence codante ou d'une dégradation 3'5' d'un ARNm interrompue (van Hoof et al.,
2002). Les ARNm sans codon stop causent un blocage des ribosomes sous une forme non
recyclée en 3' de la séquence codante.
Ils sont instables chez les eucaryotes, dégradés par l'activité 3'5' exonucléase de
l'exosome. La dégradation fait intervenir la protéine accessoire de l'exosome Ski7, un
homologue du facteur d'élongation eEF-1A et du facteur de terminaison eRF3. La partie N-
terminale de Ski7 est nécessaire pour la dégradation des ARNm normaux tandis que la partie
C-terminale joue un rôle spécifique dans la reconnaissance des ARNm non-stop. La partie C-
terminale de Ski7 contient un domaine GTPase, suggérant que l'interaction entre le ribosome
et le domaine GTPase de Ski7 déclenche la dégradation de l'ARNm par l'exosome. Ski7
interagit directement avec des éléments de l'exosome cytoplasmique via sa partie N-terminale
(Araki et al., 2001).
Cependant, il faut noter que cette voie a été mise en évidence par une approche
artificielle, chez la levure S. cerevisiae, seul organisme possédant la protéine Ski7 (Atkinson
et al., 2008). L’existence de ce mécanisme chez d’autres organismes ainsi que sa pertinence
biologique restent donc à confirmer.
4.2.3. NGD, no-go decay
4.2.3.1. Mécanisme
La voie NGD cible des ARNm sur lesquels les ribosomes en cours d'élongation sont

66
longuement arrêtés par une structure stable de type tige-boucle et cela indépendamment de la
présence d’un codon stop dans le site A. Les ARNm causant une pause de l'élongation par le
ribosome sont reconnus et dégradés par une étape initiale de clivage endonucléolytique à
proximité de la tige-boucle insérée, suivie d'une dégradation 5'3' par Xrn1 et une
dégradation 3'5' par le complexe Ski et l'exosome. Le clivage est dépendant de la traduction
et fait au moins intervenir les protéines Dom34 et Hbs1, des homologues des facteurs de
terminaison eRF1 et eRF3 (Doma and Parker, 2006) (Figure 18).
La voie NGD a été initialement observée sur des ARNm portant une tige-boucle
artificielle stable insérée dans le cadre de lecture, mais elle cible également les ARNm
dépurinés (site abasique, élongation arrêtée) (Gandhi et al., 2008). La voie NGD constitue
donc un moyen d’éliminer des complexes de traduction bloqués, qui peuvent être la
conséquence de dommages sur l'ARN ou servir de mécanismes de régulation post-
transcriptionnelle. Sans la voie NGD, les transcrits bloquant l'élongation sans faire intervenir
de codon stop ségrégeraient un ensemble de ribosomes fonctionnels.
4.2.3.2. Structure de Dom34
Les structures des protéines Dom34 eucaryote (S. cerevisiae, (Graille et al., 2008)) et
archée (Thermoplasma acidophilum, également appelée Pelota (Lee et al., 2007)) ont été
résolues. Dom34 est organisée en trois domaines formant un Y et dont les domaines M et C
partagent des homologies structurales significatives avec les domaines M et C d'eRF1.
- Le domaine N est formé d'un feuillet β courbé de sept brins antiparallèles et de deux
hélices α. Le repliement est proche de la famille des Sm-folds, qui est retrouvé dans de
nombreuses protéines qui s'associent en heptamères pour interagir avec l'ARN.
- Le domaine M est formé d'un feuillet β entouré de trois hélices α. L'unique différence
structurale entre les domaines M de Dom34 et d'eRF1 est significative puisqu'elle concerne la

67
boucle exposant les résidus essentiels GGQ d'eRF1, qui n'est pas présente chez Dom34.
Sachant que le triplet GGQ est le déterminant de l'hydrolyse de la liaison peptidyl-ARNt lors
de la terminaison de la traduction, cela suggère fortement que Dom34 ne porte pas cette
activité. Les protéines en cours de synthèse sur un ARNm cible de la NGD resteraient alors
covalamment lié à l’ARNt présent dans le site P. Leur devenir reste à l’heure actuelle
inconnu.
- Le domaine C adopte également un repliement centré sur un feuillet β entouré de deux
hélices α de chaque côté. Les trois séquences d'interaction d'eRF1 avec eRF3 (résidus 281-
305, motif 411GILRY415 et une séquence C-terminale acide) sont conservées dans la structure
de Dom34. Il est donc très probable que la même région d’eRF1 et de Dom34 soit impliquée
dans l’interaction avec leurs partenaires : eRF3 et Hbs1, respectivement.
Les structures des domaines de Dom34 et de Pelota sont très proches du point de vue des
domaines isolés malgré une homologie de séquence faible (21%). Cependant l'organisation
globale des domaines montre de grands changements de conformation : si l'on superpose les
domaines N, on observe une rotation de 90° du domaine M et 30° du domaine C de Dom34
par rapport à la structure de Pelota.
Une étude initiale suggérait un rôle direct du domaine N-terminal de Dom34 dans
l'activité endonucléase initiant la voie NGD et avait identifié certains résidus importants pour
l’activité enzymatique (Lee et al., 2007). Cependant, cette hypothèse a été écartée par des
expériences contradictoires qui ont en revanche montré que la substitution des résidus
présents dans trois boucles conservées de Dom34 diminue fortement l'activité de dégradation
des cibles de la NGD (Passos et al., 2009). Parmi ces boucles, deux sont localisées au niveau
du domaine N (boucle A :49SKLD53 et boucle B : 87TVTDES92) et l’autre au niveau du
domaine M (boucle C : 174KKK176).

68
4.2.3.3. Rôle et activité d'Hbs1
Dom34 interagit avec Hbs1, une protéine dont la séquence est homologue à la partie C-
terminale d'eRF3 (Wallrapp et al., 1998). Hbs1 est une GTPase de la famille de eEF-1A
(Carr-Schmid et al., 2002). Elle fixe le GTP et le GDP et son affinité pour le GTP mais pas
pour le GDP est augmentée en présence de Dom34, présentant ainsi une analogie
fonctionnelle avec les protéines eRF1 et eRF3 (Graille et al., 2008).
Les homologies structurales et fonctionnelles observées sur Dom34 et Hbs1 attribuent au
complexe Dom34-Hbs1 un rôle similaire à eRF1-eRF3 dans la reconnaissance des ribosomes
arrêtés et leur libération par hydrolyse d'une liaison chimique. Cependant des différences
significatives de la structure de Dom34 (absence de mode de reconnaissance des codons stop
et absence du motif GGQ) montrent la spécificité du complexe. Les cellules eucaryotes
auraient donc acquis au cours de l'évolution ces deux systèmes de libération du ribosome
assurant des activités de terminaison différentes.
4.2.4. NRD, non-fonctional rRNA decay
Dom34 et Hbs1 ont été récemment impliquées dans une voie de contrôle qualité des
ARNr non-fonctionnels de la petite sous-unité ribosomale (18S NRD, (Cole et al., 2009;
LaRiviere et al., 2006)) (Figure 18).
Le ribosome est un assemblage ribonucléoprotéique très stable (Meselson et al., 1964),
les ARNr ayant eux-même une longue durée de vie (estimée à 5 jours dans des cellules de
foie de rat (Loeb et al., 1965)). Pourtant, des mutations sur les ARNr 25S et 18S affectant le
fonctionnement des ribosomes entraînent l'élimination rapide des sous-unités ribosomiques
les contenant. Ces ARNr non fonctionnels pouvant apparaître suite à une mutation de la

69
séquence codante de l'ADN ou de dommages mutationnels induits par des agents alkylants,
un stress oxydatif ou des radiations UV sont éliminés par la voie NRD. Comme pour la
structure tige-boucle de la NGD, les expériences présentées pour la voie NRD sont basées sur
des altérations de l'ARNr 18S, le rendant artificiellement non fonctionnel. La pertinence
biologique des voies NGD et NRD, telles qu’elles ont été mises en évidence, reste discutable.
Cependant, une étude récente montre que des sous-unités ribosomales immatures et non
fonctionnelles entrant en traduction sont dégradées par un mécanisme impliquant Dom34 et
Hbs1 (Soudet et al., 2010). Une autre étude in vivo dans des cellules de plantes infectées par
un virus montre qu'un ARNm présentant un site apurinique dans son cadre de lecture est
substrat d'une voie de dégradation accélérée faisant intervenir Dom34 et Hbs1 (Gandhi et al.,
2008). Ces deux études rationalisent le rôle biologique et cellulaire de Dom34 et Hbs1 en tant
que protéines ciblant les ribosomes arrêtés en cours d'élongation, sans codon stop au site A.
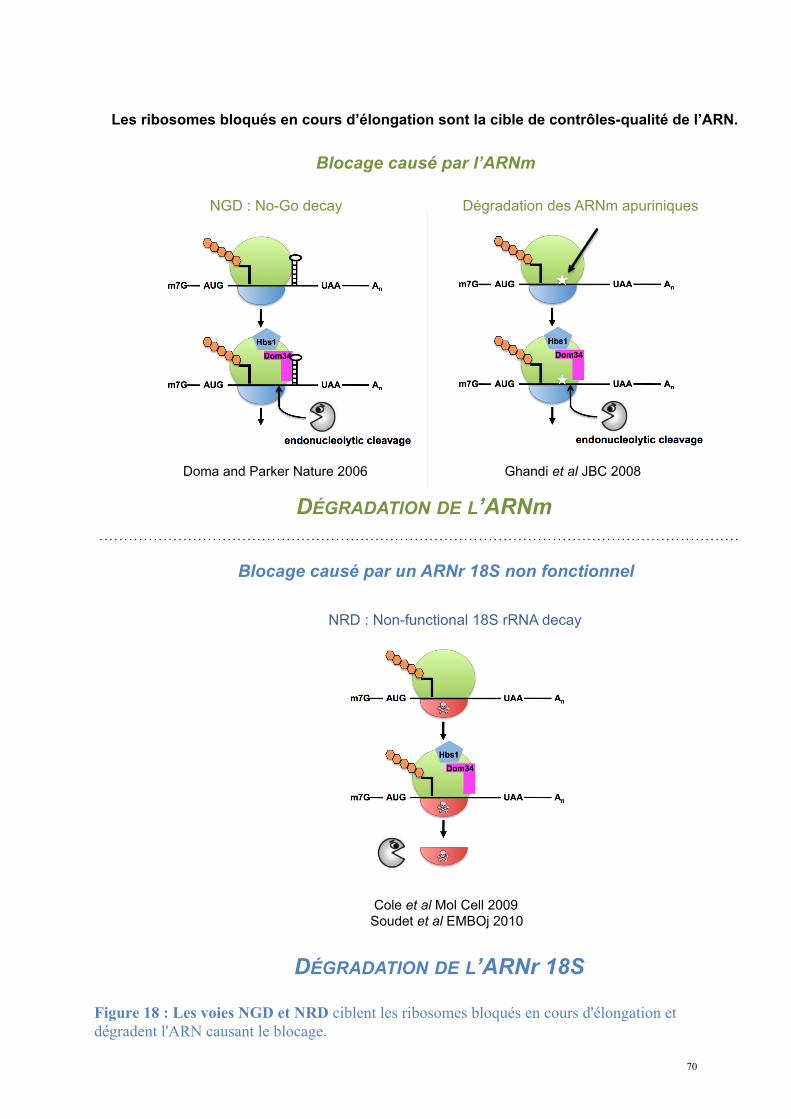
70
Blocage causé par l’ARNm
NGD : No-Go decay
Doma and Parker Nature 2006
Dégradation des ARNm apuriniques
Ghandi et al JBC 2008
DÉGRADATION DE L’ARNm
NRD : Non-functional 18S rRNA decay
Cole et al Mol Cell 2009
Soudet et al EMBOj 2010
Blocage causé par un ARNr 18S non fonctionnel
DÉGRADATION DE L’ARNr 18S
Les ribosomes bloqués en cours d’élongation sont la cible de contrôles-qualité de l’ARN.
Figure 18 : Les voies NGD et NRD ciblent les ribosomes bloqués en cours d'élongation et dégradent l'ARN causant le blocage.

71
5. Couplage des mécanismes de l'expression
génique
Les mécanismes de l'expression se recoupent, les acteurs de l'expression ont
des rôles multiples
Les processus de transcription, traduction et dégradation sont étroitement interconnectés.
Ils contribuent à différents niveaux à l'efficacité et à la fidélité de l'expression génique (Moore
and Proudfoot, 2009). En effet, si l'expression génique suit le schéma linéaire allant de l'ADN
à l'ARN via la transcription, puis de l'ARN aux protéines par la traduction, il apparaît que
plusieurs complexes moléculaires responsables de chaque étape interagissent entre eux. A
chaque étape de l'expression génique, des protéines multifonctionnelles facilitent la
communication entre les évènements précoces et les évènements tardifs et garantissent ainsi
une coordination de la « chaîne de montage », sous la forme d'un réseau de régulation d'une
grande complexité.
A titre d'exemple, de nombreux facteurs ont été décrits comme intervenants à plusieurs
niveaux de l'expression génique.
(1) Le facteur de surveillance central de la voie NMD Upf1 sert d'interrupteur
déclenchant la dégradation des ARNm portant un codon stop précoce. Cette activité est
déterminée par la phosphorylation contrôlée d'Upf1 par Smg1; or, Upf1 phosphorylée
interagit avec eIF3 de manière à bloquer les évènement de réinitiation, empêchant au final de
nouvelles traductions des ARNm aberrants (Isken et al., 2008).
(2) Les protéines Dbp5 et Gle1 contrôlent le transport unidirectionnel de l'ARNm mature
à travers le pore nucléaire en remodelant la structure du mRNP. Ces deux protéines sont

72
également nécessaires pour une terminaison efficace de la traduction chez S. cerevisiae
(Alcazar-Roman et al.; Dossani et al., 2009; Gross et al., 2007). De plus, Gle1 est également
liée à l'initiation de la traduction à travers son interaction avec le facteur eIF3 (Bolger et al.,
2008).
(3) La protéine Pab1 joue un rôle ambivalent en assurant à la fois la protection des
ARNm de la digestion rapide par les exonucléases 3'5' telles que l'exosome, mais
également en recrutant le complexe de dégradation partielle de la queue poly(A) Pan2-Pan3 et
entraînant in fine la dégradation des ARNm fonctionnels (Boeck et al., 1996).
(4) Le complexe de terminaison de la traduction eRF1-eRF3 enfin, qui en reconnaissant
les codons stop va contribuer à la stabilisation ou à la dégradation des ARNm selon qu’ils
sont normaux ou aberrants, respectivement.

73
6. Présentation de la thématique de recherche
L'expression génique suit en première approximation un schéma global simple et très
bien caractérisé. De nombreuses voies de contrôle garantissent l'efficacité et la spécificité du
système. Si plusieurs de ces voies ont été identifiées ces dernières années, certaines étapes
restent manifestement mystérieuses quant à leur mode de déclenchement et au contrôle de
leur bon déroulement. C'est le cas notamment de l'export nucléo-cytoplasmique et du
couplage ribosome-dépendant entre la traduction et la dégradation. Les facteurs identifiés
n'ont pas toujours un mode d'action clair et il est probable que des acteurs soient encore à
identifier. S'il est avéré que la stabilité des ARNm varie grandement d'une molécule à l'autre,
il n'est pas facile d'expliquer pourquoi des ARNm fonctionnels peuvent avoir des temps de
demi-vie différents. De même les conditions métaboliques et environnementales ne semblent
pas jouer directement sur les niveaux de stabilité de l'ARNm, alors qu'il s'agit d'un mode de
régulation essentiel de la transcription.
L'objectif de mon travail de thèse était d'apporter des éléments de réponse concernant :
(i) Le rôle de facteurs nouvellement impliqués dans les mécanismes de terminaison de la
traduction et de dégradation de l'ARNm. Nous avons étudié le cas des protéines eucaryotes
Tpa1/Ofd1 et Ett1/Nro1. Les questions que nous nous posions étaient les suivantes :
- Quelle est la fonction de Tpa1 dans la terminaison de la traduction et la stabilité
des ARNm?
- Peut-on relier les phénotypes attribués à Ofd1 chez S. pombe et ceux attribués à
Tpa1 chez S. cerevisiae ?
- Quelle est la relation entre Tpa1/Ofd1 et Ett1/Nro1 ?
- Quelle est la cible de l'activité dioxygénase proposée pour Tpa1 ?

74
- Quel est le rôle d'Ett1/Nro1 ?
(ii) Le mécanisme moléculaire responsable du No-Go Decay et de la voie de dégradation
des ARNr 18S non fonctionnels.
- Quelles sont les bases moléculaires de l’interaction entre Dom34 et Hbs1 ?
- Comment fonctionne le complexe Dom34-Hbs1 ?
- Quel est le rôle de Hbs1 dans le complexe ?
- Quel est le rôle de Dom34 vis-à-vis de l'activité de Hbs1 ?
- Quelles sont les similarités structurales avec eRF1-eRF3 et comment les expliquer
en terme de fonction et de spécificité ?
La détermination de la structure à haute résolution des protéines peut apporter des
réponses à ces questions (Cantor C. R., 1980; Ducruix A., 1992; Janin J., 1999). Pour cela,
chaque protéine cible a été exprimée, purifiée et cristallisée. La détermination des structures à
haute résolution par diffraction des rayons X, en complément des analyses biochimiques et
biophysiques in vitro a fourni des bases pour proposer et attribuer des fonctions biologiques
précises. Ce travail a été mené en étroite collaboration avec l'équipe de Bertrand Séraphin au
CGM (FRE 3144, Gif-sur-Yvette) puis à l'IGBMC (UMR 7104-U596, Illkirch) de manière à
employer les outils de la génétique moléculaire pour une validation de nos modèles in vivo
chez la levure S. cerevisiae.
Les différents résultats obtenus suggèrent l'existence d'une nouvelle voie de contrôle de
l'efficacité de la terminaison et apportent une meilleure connaissance des mécanismes de la
NGD et de la NRD. La stratégie expérimentale, les résultats obtenus et les conclusions
apportées pour chaque projet seront présentés et discutés ici autour des articles auxquels ils
ont conduits.

75
Stratégie expérimentale
L'objectif du travail était la caractérisation structurale et fonctionnelle in vitro de
molécules biologiques ciblées. Cette étude exige de disposer de quantités importantes
(quelques dizaines de milligrammes) d'un produit très pur. L'étape préliminaire de chaque
projet était l'expression et la purification de chaque protéine.
Les expériences de routine ont été réalisées sur la plateforme du laboratoire de
Génomique Structurale de la Levure à partir des protocoles mis en place et optimisés au cours
du projet de génomique structurale de la Levure (Quevillon-Cheruel et al., 2004).
1. Expression et purification des protéines
La surexpression du gène codant pour la protéine cible dans une bactérie permet d'obtenir
rapidement une grande quantité de protéines.
1.1. Clonage
Construction d'un vecteur d'expression
La séquence codant pour la protéine cible chez S. cerevisiae est amplifiée à partir de
l'ADN génomique de la Levure (souche S288c) par réaction de polymérisation en chaîne
(PCR). Le produit de PCR est inséré dans le site de clonage multiple d'un plasmide
d'expression bactérien.
Les plasmides de routine du laboratoire sont des variants des vecteurs commerciaux
pET9, pET21 et pET28; ils portent en phase avec le cadre de lecture de la séquence codante

76
insérée des séquences supplémentaires codant pour une étiquette (tag) polypeptidique
facilitant la purification : hexa-histidine (His6) ou streptavidine (Trp-Ser-His-Pro-Gln-Phe-
Glu-Lys).
1.2. Surexpression en système bactérien
Les plasmides de surexpression sont introduits dans des bactéries E. coli BL21-DE3 Gold
ou Rosetta pLysS (Novagen) compétentes. Les bactéries sont mises en préculture dans 20mL
de milieu riche (2YT Bio101-Inc.) sélectif contenant l'antibiotique correspondant à la
résistance portée par le plasmide (ampicilline/kanamycine à 50µg/mL, chloramphénicol à
25µg/mL); la préculture arrivée à saturation est transférée dans 800mL de 2YT-antibiotique.
La croissance des bactéries est contrôlée par mesure de la densité optique à 600nm. Lorsque
la DO680nm atteint 0,8, l'ajout d'IPTG (50µg/mL) dans le milieu de culture induit la
surexpression de la protéine-cible. La température d'induction est optimisée pour une
expression maximale de protéine soluble (37°C, 28°C, 23°C ou 15°C); l'induction dure 3h à
37°C, 20h aux températures inférieures.
Les bactéries sont collectées par centrifugation 20min à 4000g et resuspendues dans
25mL d'un tampon optimal pour la solubilité de la protéine cible. Les culots cellulaires sont
stockés à -20°C jusqu'à l'étape de purification.
1.3. Purification
1.3.1. Préparation des extraits cellulaires
Les culots cellulaires sont décongelés et soumis à des cycles de sonication (Branson
Sonifier 250, 3 cycles de 30s à 30% de puissance), les extraits cellulaires totaux sont
centrifugés 20 minutes à 20000g et le surnageant est récupéré pour la purification. La
stratégie de purification combine plusieurs des techniques suivantes.

77
1.3.2. Chromatographie d'affinité
1.3.2.1. Chromatographie d'affinité sur un métal immobilisé (IMAC)
Les protéines-cibles portant le tag hexa-histidine sont purifiées sur une résine greffée
avec des ions Ni2+ (4mL NiNTA-Agarose Qiagen par culot cellulaire) et éluées par
compétition par l'imidazole (concentrations croissantes en imidazole : 100mM, 200mM et
400mM). Le protocole IMAC simple peut être complété par une étape de lavage à 1M de
NaCl avant les étapes d’élution.
1.3.2.2. Chromatographie d'affinité sur StrepTactin
Les protéines portant un tag Streptavidine sont retenues spécifiquement sur un analogue
de la biotine greffé à un support solide (StrepTactin GE Healthcare) et éluées par la biotine.
1.3.3. Chromatographie d'échange d'ions (IEX)
La chromatographie d'échange d'ions sépare les protéines selon leur charge de surface.
L'élution de la protéine est déclenchée par une augmentation graduelle de la concentration en
sel (de 50mM à 1M NaCl) à un débit de 1mL/min. Des fractions de 1,5mL en sortie de
colonne sont collectées.
1.3.4. Chromatographie d'exclusion de taille (SEC)
La chromatographie d'exclusion de taille sépare les protéines selon leur taille par passage
à travers un tamis moléculaire de billes poreuses. La solution contenant la protéine est
concentrée par centrifugation sur membrane (Vivaspin) à un volume de 5 ou 2mL, puis
injectée sur le tamis moléculaire (S75 ou S200 16/60 HL, GE Healthcare) à un débit de
1mL/min. Des fractions de 1,5mL en sortie de colonne sont collectées.

78
Les chromatographies d'échange d'ions et d'exclusion de taille sont réalisées sur un
système automatisé FPLC Purifier ou Xpress (GE Healthcare).
2. Caractérisation du matériel protéique
2.1. Contrôles de routine
Les techniques de base de caractérisation des protéines en solution sont utilisées : SDS-
PAGE, dosage spectrophotométrique UV-visible et spectrométrie de masse MALDI-TOF.
2.2. Diffusion de la lumière laser à différents angles (MALLS)
Le couplage de la chromatographie d'exclusion de taille analytique (Superdex 200 HR
10/30 GE Healthcare) avec un système de détection triple en sortie de colonne (SEC-TDA302
Viscotek, logiciel OmniSEC) permet une estimation précise du poids moléculaire et du
volume hydrodynamique des protéines en solution. L'appareil couple un réfractomètre
(estimation de la concentration de la solution éluée), un détecteur de la diffusion de lumière
aux petits angles et aux grands angles (estimation du rayon hydrodynamique) et un
viscosimètre. L'ensemble des données est intégré pour calculer le poids moléculaire apparent
de l'espèce éluée. Par comparaison au poids moléculaire théorique, la méthode sert à
discriminer entre les états monomériques et multimériques des protéines et à caractériser la
stœchiométrie des complexes.

79
2.3. Dichroïsme circulaire (CD)
Le dichroïsme circulaire caractérise la différence d'absorption de la lumière polarisée
circulairement à droite ou à gauche par des molécules optiquement active. Les protéines sont
optiquement actives et la longueur d'onde de l'effet CD est spécifique d'une structure
secondaire donnée. Cette technique est employée pour la validation de l'état de structuration
d'une protéine purifiée. Les expériences ont été réalisées sur un spectropolarimètre Jasco JB10
entre 190 et 250nm (trajet optique 1mm, accumulation 50nm/min) sur une solution de
protéine diluée à 1-10µM de liaison peptidique.
3. Cristallisation
La cristallisation des protéines a été réalisée sur la plateforme de génomique structurale
mise en place au laboratoire (Leulliot et al., 2005; Quevillon-Cheruel et al., 2004).
L'utilisation de robots de micropipetage (Cartesian Nanodrop, Tecan Genesis) a permis
l'automatisation de la recherche de conditions de cristallisation des protéines purifiées. Les
conditions initiales de cristallogenèse sont déterminées sur des cribles larges d'agents
précipitants commerciaux (Qiagen : kits Classics, MBI, MBII, Nucleix, ProComplex, MPD).
Ces expériences sont réalisées suivant la méthode de diffusion de vapeur dans des boîtes de
96 puits. Un mélange de 0,1µL de précipitant et de 0,1µL de protéine est déposé en goutte
assise dans un support séparé du réservoir. L'ensemble est scellé par un adhésif transparent et
stocké à 20°C. Les gouttes sont observées quotidiennement à la loupe binoculaire.
Lorsque les premiers cristaux apparaissent dans l'une des conditions testées et dans le but
d'augmenter la taille et la régularité des cristaux, la composition de la solution de

80
cristallisation est optimisée sur le robot de pipetage Tecan en gouttes assises de 0,5+0,5µL en
faisant varier les paramètres suivants:
(i) pH : préparation de solutions de cristallisation en présence d'un des 48 tampons pH
présents dans le kit développé au laboratoire;
(ii) concentration de chaque agent précipitant : préparation d'une matrice à deux
dimensions faisant varier la concentration de deux précipitants simultanément,
(iii) agents précipitants additionnels : sur les conditions optimisées en pH et en
concentration, recherche d'additifs, de sels, de molécules cryoprotectantes favorisant la
cristallisation.
Enfin, lorsque la composition de la solution de cristallisation est jugée optimale le
volume des gouttes est augmenté jusqu'à un mélange de 1 à 2µL de protéine et de 1 à 2µL de
précipitant, déposés à la main en goutte suspendue. A ce stade le microseeding est testé en
vue de l'amélioration de la taille et de la qualité des cristaux. Les meilleurs cristaux sont
congelés à l'aide de cryo-boucles en nylon et trempés dans un mélange de la solution de
cristallisation et d'un cryoprotectant (15 puis 30% de glycérol ou d'éthyène glycol en routine)
puis congelés et stockés dans l'azote liquide.
Les essais de diffraction ont été réalisés en utilisant le faisceau de rayons X produit par
les sources de rayonnement synchrotron ESRF (Grenoble) ou SOLEIL (Saint-Aubin).
Les données collectées sont traitées avec les programmes XDS (Kabsch, 2010a) et
XSCALE (Diederichs, 2006). Les structures sont résolues grâce aux programmes SHELXD
(Sheldrick, 2008) et SHARP (Bricogne et al., 2003) puis reconstruites et affinées grâce aux
suites de programmes CCP4 (Collaborative Computational Project, 1994), PHENIX (Adams
et al., 2002) et COOT (Emsley and Cowtan, 2004).


81
Résultats
1. Tpa1 et Ett1, nouveaux acteurs de la
terminaison de la traduction
1.1. Tpa1, structure et fonction
Dans le but d'éclaircir le rôle de Tpa1 dans la terminaison de la traduction et la stabilité
des ARNm et d'élucider son mode de fonctionnement une partie de mon travail de thèse a été
consacré à la détermination de la structure à haute résolution de Tpa1.
1.1.1. Procédures expérimentales
A. Clonage, expression et purification
Le plasmide pBS3077 permettant de surexprimer cette protéine chez E. coli nous a été
fourni par notre collaborateur Bertrand Séraphin. Il s’agit d’un plasmide dérivé du pET24
dans lequel la séquence codante pour la protéine Tpa1 a été clonée en phase avec la séquence
nucléotidique codant pour une étiquette hexa-histidines en 5’ du gène. La protéine a été
exprimée à 23°C en utilisant la souche Rosetta DE3 pLysS (Novagen) d’E. coli transformée
par le plasmide pBS3077, en milieu 2YT-kanamycine. L’expression de Tpa1 a été induite par
ajout d’IPTG à 50µg/mL.
Les cellules ont été collectées par centrifugation, resuspendues en tampon A (20mM Tris
pH 7,5, 200mM NaCl, 5mM β-mercaptoethanol) et cassées par ultrasonication puis

82
centrifugées. Le surnageant de culture est déposé sur résine de NiNTA (Qiagen), et soumis à
un premier lavage avec du tampon A contenant également 10mM d’imidazole pH 7 (W10)
puis un second lavage avec du tampon A contenant 50mM d'imidazole pH 7 (W50).
L'éluat obtenu en tampon A contenant 200mM d'imidazole pH 7 (E200) est concentré et
soumis à une chromatographie d'exclusion de taille Superdex 200HL 16/60 en tampon A. A
l’issue de cette étape de purification, la protéine est pure et homogène (Figure 19). Les
spectres de dichroïsme circulaire indiquent la présence d’éléments de structure secondaire de
type hélices α et brins β.
La substitution des méthionines par des séléno-méthionines dans le milieu de culture a
permis la production de protéine marquée qui a été purifiée selon le même protocole que la
protéine native. Le marquage Séléno-méthionine (Se-Met) permet de faciliter la détermination
de la structure, comme décrit plus loin.
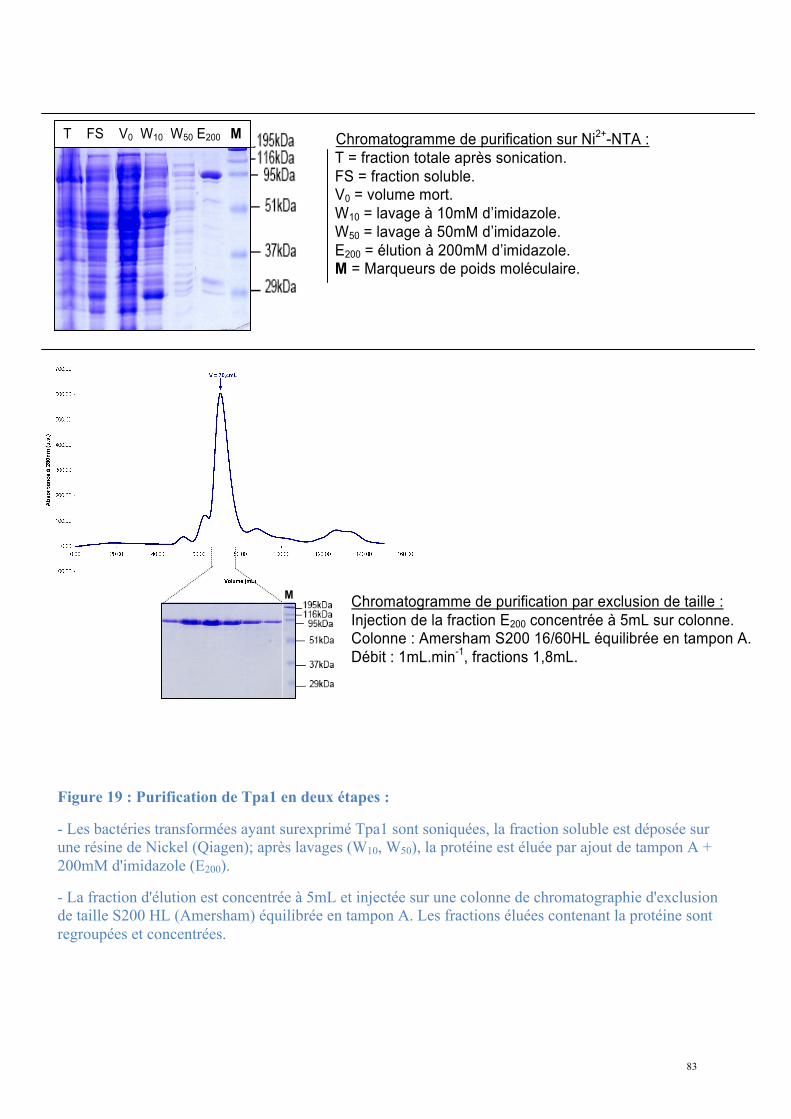
83
Chromatogramme de purification sur Ni2+-NTA :
T = fraction totale après sonication.
FS = fraction soluble. V0 = volume mort.
W10 = lavage à 10mM d’imidazole.
W50 = lavage à 50mM d’imidazole.
E200 = élution à 200mM d’imidazole. M = Marqueurs de poids moléculaire.
Chromatogramme de purification par exclusion de taille :
Injection de la fraction E200 concentrée à 5mL sur colonne. Colonne : Amersham S200 16/60HL équilibrée en tampon A.
Débit : 1mL.min-1, fractions 1,8mL.
T FS V0 W10 W50 E200 M
M
Figure 19 : Purification de Tpa1 en deux étapes :
- Les bactéries transformées ayant surexprimé Tpa1 sont soniquées, la fraction soluble est déposée sur une résine de Nickel (Qiagen); après lavages (W10, W50), la protéine est éluée par ajout de tampon A + 200mM d'imidazole (E200).
- La fraction d'élution est concentrée à 5mL et injectée sur une colonne de chromatographie d'exclusion de taille S200 HL (Amersham) équilibrée en tampon A. Les fractions éluées contenant la protéine sont regroupées et concentrées.

84
B. Cristallisation
La protéine purifiée selon le protocole ci-dessus est concentrée à 15mg/mL (Vivaspin 20,
3kDa MWCO, 5000g). La solution protéique est déposée avec un volume équivalent de
solution de cristallisation sur les kits de criblage en goutte assise (0,1µL+0,1µL, kits Classics,
MB class I, ProComplex et MPD de Qiagen) par le robot de pipetage Cartesian Nanodrop.
Au bout de quatre jours, des cristaux apparaissent :
- dans la condition 22 du kit Classics (forme 1) : 0,1M Hepes pH 7,5; 70%
méthylpentanediol (MPD). Ce sont des bâtonnets polycristallins de petite taille et de forme
irrégulière.
- dans la condition 64 du kit MPD (forme 2) : 0,1M Hepes pH 7; 40% MPD. Ce sont des
cubes de petite taille.
Les cristaux de la forme 2 ont été optimisés en taille par l'ajout de 80mM de NaF. Les
cristaux de la forme 1 ont été significativement optimisés par l'utilisation d'un gradient à deux
dimensions faisant varier la concentration en Hepes pH7,5 (0-0,15M) et en MPD (40-80%).
La concentration optimale en MPD est de 44%; la concentration en Hepes pH7,5 a été
maintenue à 0,1M. Une possibilité supplémentaire d'amélioration de la condition de
cristallisation est explorée par utilisation de screens d'additifs et de sels; l'ajout de 0,1M de
Mg2(NO3)2 augmentait significativement la taille et la régularité des cristaux. La
concentration de MPD et de Mg2(NO3)2 est alors optimisée par un nouveau gradient en deux
dimensions pour amener à la composition optimale de la solution de cristallisation de Tpa1 :
0,1M Hepes pH7,5, 44% MPD, 30mM Mg2(NO3)2.
L'utilisation d'une boîte « Low Profile » a permis d'augmenter le volume des gouttes tout
en conservant le principe de la goutte assise et en utilisant les robots de cristallisation.
Différents rapports de volumes du mélange protéine-précipitant ont été testés : 1µL+1µL,

85
1,5µL+1,5µL, 2µL+2µL, 1,5µL+1µL, 1µL+1,5µL; ce dernier rapport a permis d'obtenir les
cristaux de plus grande taille. L'ensemencement des gouttes de cristallisation par ajout de
microcristaux issus des conditions initiales (streak-seeding) a également été testé et a permis
une amélioration significative de la qualité apparente des cristaux (Figure 22).
Les cristaux poussent très lentement : les meilleurs ont été obtenus après quelques
semaines à quelques mois d'équilibration. Les cristaux de protéine marquée Se-Met ont été
obtenus dans les mêmes conditions.
En parallèle de la caractérisation structurale de Tpa1 seule sous forme entière d'autres
tentatives de cristallisation ont été expérimentées :
- L’analyse bioinformatique de la séquence primaire de Tpa1 indique que cette protéine
contient un domaine DSBH (double-stranded β-helix) de dioxygénase dépendant du Fe2+ et
du 2-oxoglutarate (Aravind and Koonin, 2001). Nous avons émis l'hypothèse que l'ajout de
Fer dans la solution de Tpa1 après purification pourrait favoriser l'homogénéité de la
préparation et ainsi améliorer la cristallogenèse. J'ai mélangé la protéine purifiée concentrée à
200µM à 1mM d'un mélange FeCl2-Ascorbate. L'apparition rapide d'un précipité se confirme
après 30 minutes d'équilibration : la concentration de protéine dans le surnageant après
centrifugation diminue très fortement, prévenant les essais de cristallogenèse dans les
conditions mises au point.
- L'incubation de la solution purifiée de Tpa1 concentrée à 200µM avec une solution de
2-oxoglutarate à 1mM pendant 30 minutes n'a pas eu d'effet remarquable sur la solubilité de la
protéine. Le mélange a été utilisé pour des essais de co-cristallogenèse, produisant des
cristaux dans la condition optimisée pour la protéine seule.

86
- L'analyse de la séquence primaire de Tpa1 suggère que sa partie N-terminale est
constituée d'un domaine DSBH (Aravind and Koonin, 2001). L'étude de l'orthologue Ofd1
confirme la présence d'un domaine dioxygénase en N-terminal et attribue la fonction
régulatrice de la protéine à un domaine C-terminal indépendant (Hughes and Espenshade,
2008). Nous avons donc envisagé la résolution de la structure cristallographique par une
approche par domaines séparés et j'ai entrepris le clonage des deux constructions Tpa11-268
(Tpa1N) et Tpa1269-645 (Tpa1C) dans les vecteurs d'expression bactérien pET21a et pET28a
respectivement, en fusion avec un tag hexa-histidine. La purification de Tpa1N par NiNTA-
SEC en tampon A n'a pas permis l'obtention de cristaux dans les screens utilisés en routine; la
purification de Tpa1C n'a quant à elle pas abouti à une solution pure et homogène du domaine
dans les conditions testées (Figure 20).
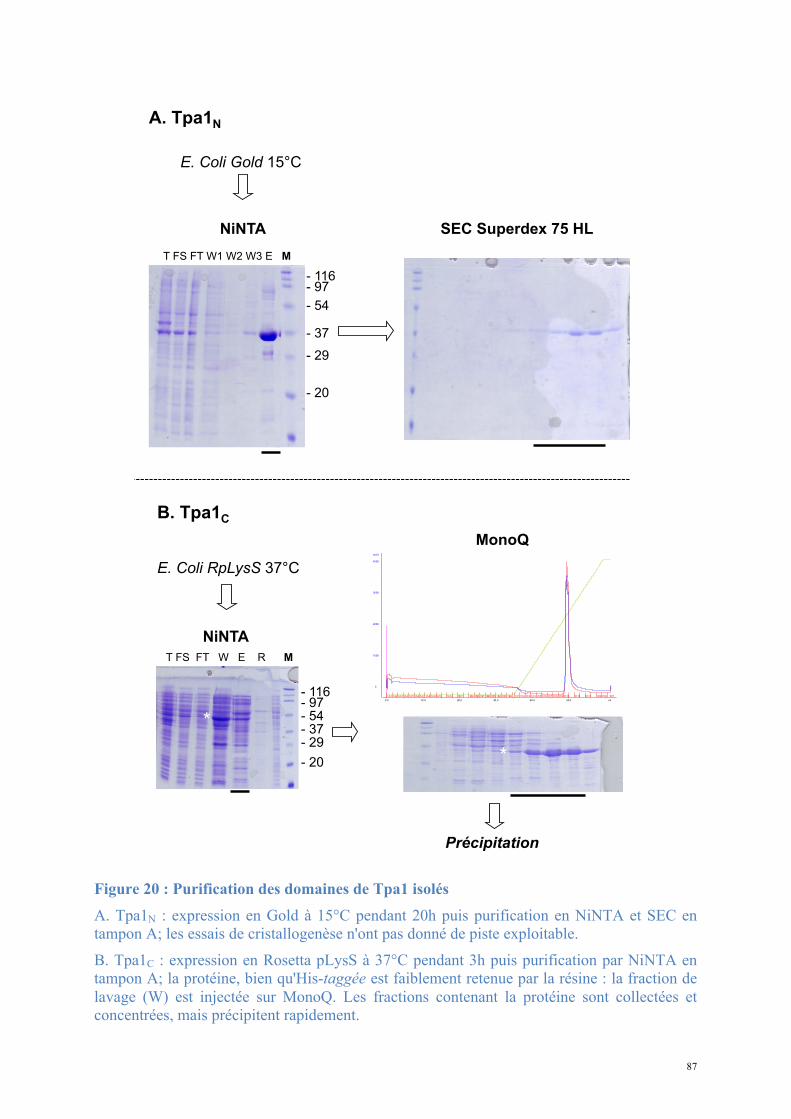
87
Figure 20 : Purification des domaines de Tpa1 isolés
A. Tpa1N : expression en Gold à 15°C pendant 20h puis purification en NiNTA et SEC en tampon A; les essais de cristallogenèse n'ont pas donné de piste exploitable.
B. Tpa1C : expression en Rosetta pLysS à 37°C pendant 3h puis purification par NiNTA en tampon A; la protéine, bien qu'His-taggée est faiblement retenue par la résine : la fraction de lavage (W) est injectée sur MonoQ. Les fractions contenant la protéine sont collectées et concentrées, mais précipitent rapidement.
SEC Superdex 75 HL
*
E. Coli Gold 15°C
NiNTA
T FS FT W1 W2 W3 E M
NiNTA
T FS FT W E R M
MonoQ
E. Coli RpLysS 37°C
*
*
A. Tpa1N
Précipitation
B. Tpa1C
-! 54
-! 116
-! 20
-! 29
-! 37
-! 97
-! 54
-! 116
-! 20
-! 29
-! 37
-! 97

88
- La cristallogenèse requiert une protéine homogène et stable. La présence de séquences
désordonnées diminue les chances de cristallisation et la qualité des cristaux éventuellement
obtenus. Considérant les difficultés rencontrées lors de la production des cristaux nous avons
souhaité mettre en évidence de possibles zones particulièrement stables et structurées dans la
protéine. J'ai réalisé une digestion de la protéine purifiée par différentes protéases placées
dans des conditions de faible activité (protéolyse ménagée). 1µM à 20µM des enzymes
suivantes ont été ajoutées à Tpa1 purifiée en tampon A concentrée à 200µM : trypsine,
chymotrypsine, pepsine, papaïne; après 5, 30 ou 180 minutes d'incubation à 20°C les
solutions sont analysées sur SDS-PAGE.
Les digestions par la trypsine, la chymotrypsine et la papaïne aux deux concentrations,
ainsi que la digestion à la pepsine à 20µM sont complètes dès 5 minutes d'incubation. La
digestion par la pepsine concentrée à 1µM pendant 5 et 30 minutes produit plusieurs
intermédiaires de dégradation de Tpa1 qui sont digérés au-delà (180 minutes) (Figure 21); ces
fragments sont des cibles intéressantes pour la cristallogenèse, puisque résistant à cette
condition de protéolyse. Nous n'avons pas eu besoin d'exploiter ce résultat, mais
l'identification de ces fragments de Tpa1 par séquençage protéique couplé à la spectrométrie
de masse aurait pu permettre de définir une séquence à sous-cloner pour de nouveaux essais
de cristallogenèse.

89
Figure 21 : Résultats de la protéolyse ménagée de Tpa1 par la pepsine (1µM pendant 5, 30 et 180 minutes).
Les flèches verte et bleue indiquent des produits de protéolyse d'environ 50 et 35kDa respectivement. La digestion est complète au bout de 180 minutes.
- La résolution de la structure nécessite la détermination des phases. En complément de
l'approche classique utilisant le signal anomal du Sélénium (MAD ou SAD, voir plus loin)
nous avons envisagé l'utilisation d'atomes lourds pour un phasage par remplacement multiple
isomorphe (MIR) (Dauter, 2002). Des cristaux de protéine native ont été trempés 5 à 30
minutes dans la solution de cristallisation mélangée à différentes solutions contenant des
atomes lourds (concentration finale 1mM) susceptibles de se fixer sur des sites spécifiques
(kits Hampton et Jenna Bioscience) : Osmium (OsCl3, K2OsCl6), Mercure (HgCl2, mersalyl-
Hg, Hg-acétate, phényl-Hg-acétate, méthyl-HgCl), Platine (K2PtCl6, K2Pt(SCN)6, K2PtCl4),
Or (KAuCl4, AuCl3). Les cristaux résistaient bien au traitement et ne présentaient aucun
dommage apparent suite au traitement (Figure 22).
5’ 30’ 180’ M
-! 116
-! 97
-! 54
-! 37
-! 29
-! 20
>
>

90
Protéine native :
Résolution 9Å
Fo
rme
1
Résolution 2,8Å
Protéine Séléno-méthionylée
Résolution 3,5Å
40% MPD - 100mM Hepes pH7,5 40% MPD - 100mM Hepes pH7,5 - 80mM NaF
Fo
rme
2
Pas de diffraction
Trempages métaux lourds
Pas de diffraction
Figure 22 : Cristallisation de Tpa1 native et Se-Met.
Tpa1 cristallise dans deux conditions différentes. La forme 1 a été optimisée et a permis l'obtention de jeux de données de diffraction, puis la détermination de la structure.

91
C. Diffraction et collecte des données
Les cristaux sont directement montés dans une boucle de nylon et plongés dans l'azote
liquide, le MPD jouant le rôle d'agent cryoprotectant aux concentrations utilisées lors des
expériences de cristallisation. Le cristal est placé dans le faisceau de rayons X d'une ligne de
lumière dédiée à la biocristallographie au synchrotron ESRF.
Les faisceaux diffractés par le cristal sont collectés et enregistrés sur un détecteur (Figure
23). L'intensité des faisceaux diffractés contient l'information sur le contenu de la maille
cristalline. Le réseau réciproque étant la transformée de Fourier du réseau direct défini par la
maille, les taches de diffraction correspondent à l'intersection du réseau réciproque et de la
sphère d'Ewald. A un vecteur du réseau réciproque s = ha*+kb*+lc* correspond une tache de
diffraction repérée par le triplet d'indices de Miller (h,k,l). La position des réflexions est
déterminée et permet réciproquement de déterminer la maille et le système cristallin.
Un jeu de données de diffraction consiste en un ensemble de clichés de diffraction.
Chaque cliché est mesuré pendant la rotation de 1° du cristal dans le faisceau et pendant un
temps variable selon la puissance de la ligne (1s en routine).
Aucune diffraction n'a été observée pour les cristaux de la forme 2. En revanche, les
cristaux de la forme 1 optimisés diffractaient jusqu'à 2,8Å pour la protéine native. Les
cristaux de protéine Se-Met présentaient généralement une importante anisotropie de
diffraction, fournissant des données jusqu'à 3Å dans une direction mais seulement jusqu'à 6Å
dans une autre. Néanmoins deux jeux de données ont pu être obtenus à 3,7Å et 3,4Å de
résolution à partir de cristaux de protéine marquée à la Séléno-méthionine. Les données ont
permis de résoudre la structure par SAD (diffusion anomale à une longueur d'onde simple) en
utilisant le signal du Sélénium.

92
Les données collectées sont utilisées pour la première étape de la détermination de la
structure. Le traitement initial des données procède en trois étapes successives :
- détermination des paramètres de maille et du groupe d'espace caractérisant le réseau
cristallin ;
- mesure et intégration des intensités caractérisant le motif dans le réseau cristallin
- mise à l'échelle et regroupement des données résultantes.
Les programmes XDS (Kabsch, 2010a, b) ou MOSFLM (Leslie, 2006) ont été utilisés
pour traiter les données.
Les mises à l'échelle ont été réalisées par XSCALE (Diederichs, 2006) ou SCALA
(Evans, 2006).
Protéine native
Saccharomyces cerevisiae Tpa1 diffraction
Protein Séléno-methionylée
Exemples de clichés de diffraction des cristaux de Tpa1
Protéine native Protein Séléno-methionylée
Groupe d’espace, paramètres de maille et statistiques des jeux collectés
Figure 23 : Exemples de clichés de diffraction de cristaux de Tpa1
Les cercles concentriques représentent les limites de résolution.

93
a. Détermination des paramètres de maille et du réseau de Bravais
La maille est l'unité délimitée par les nœuds du réseau de Bravais permettant de définir
l'ensemble du cristal par des opérations de translations. L'indexation des données correspond à
la détermination des vecteurs a*, b* et c* compatibles avec la position des taches, et
permettant de déterminer le réseau direct. Les modules et les angles de ces vecteurs
définissent les paramètres de la maille. Le programme XDS propose une liste des différents
réseaux de Bravais possibles pour les réflexions mesurées, chaque solution étant assortie d'un
score de pénalité proportionnel à la différence entre les paramètres de maille estimés et les
contraintes géométriques des paramètres de maille d’un réseau de Bravais idéal donné. La
solution correcte combine le système cristallin de plus haute symétrie à la pénalité la plus
faible possible (généralement inférieure à 20). Pour chaque réseau de Bravais il peut exister
plusieurs groupes de Laue (groupes d'espace) selon les symétries internes de la maille.
Les cristaux de protéine native appartiennent au réseau cristallin orthorhombique centré
(oC), avec les paramètres de maille suivants : a=81,2 Å, b=104,8 Å, c=205,6 Å, α=β=γ=90°
(Figure 24). Deux groupes d'espace énantiomorphes sont possibles : C222 ou C2221. Les
cristaux de protéine Se-Met appartiennent au réseau cristallin primitif orthorhombique (oP),
avec les paramètres de maille suivants : a=105,7Å, b=168,2Å, c=197,3Å, α=β=γ=90°. Deux
jeux de données ont été collectés à l’ESRF (Grenoble) sur deux cristaux Se-Met différents. Le
premier jeu de données a été obtenu sur la ligne BM30-A, le second sur la ligne ID23-EH1.
Les groupes d'espace étaient identiques et les paramètres de maille très proches. La maille de
ces cristaux présente un doublement d'une longueur par rapport à celle des cristaux de
protéine native (bSeMet=2anative), révélant une symétrie interne supplémentaire dans les jeux de
cristaux de protéine marquée.
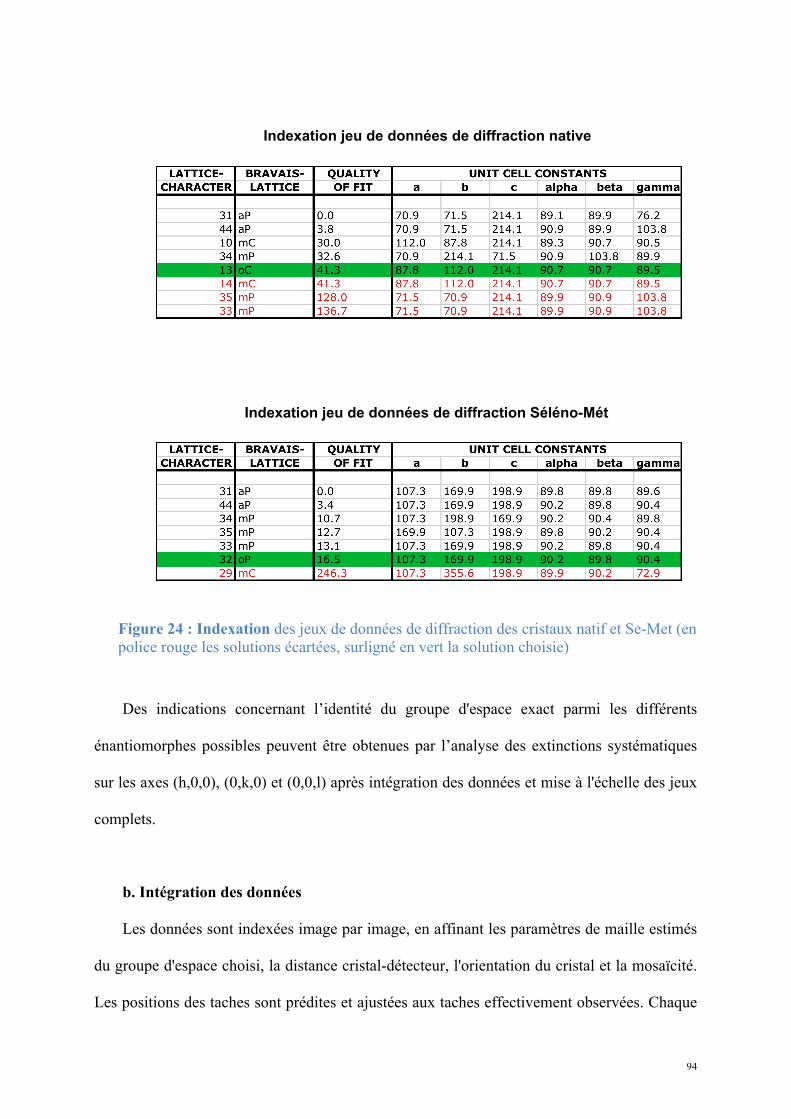
94
Des indications concernant l’identité du groupe d'espace exact parmi les différents
énantiomorphes possibles peuvent être obtenues par l’analyse des extinctions systématiques
sur les axes (h,0,0), (0,k,0) et (0,0,l) après intégration des données et mise à l'échelle des jeux
complets.
b. Intégration des données
Les données sont indexées image par image, en affinant les paramètres de maille estimés
du groupe d'espace choisi, la distance cristal-détecteur, l'orientation du cristal et la mosaïcité.
Les positions des taches sont prédites et ajustées aux taches effectivement observées. Chaque
Indexation jeu de données de diffraction native
Indexation jeu de données de diffraction Séléno-Mét
Figure 24 : Indexation des jeux de données de diffraction des cristaux natif et Se-Met (en police rouge les solutions écartées, surligné en vert la solution choisie)

95
tache est indexée en h, k et l. L'intensité de chaque tache est mesurée en appliquant un masque
autour de la tache : le centre de la couronne correspond à la surface intégrée, la couronne
externe correspond au bruit de fond; chaque intensité est ainsi corrigée contre le bruit de fond.
c. Mise à l'échelle et conversion des intensités en amplitudes.
Les images collectées sur le cristal contiennent chacune une fraction de l'ensemble des
réflexions possibles, obtenues par la rotation complète du cristal dans le faisceau. Afin de
constituer un jeu complet de réflexions, les images sont assemblées. La complétude du jeu est
estimée selon le nombre de réflexions observées parmi toutes les réflexions possibles. Les
jeux natif et Se-Met étaient complets à 97% (Figure 25).
La mise à l'échelle permet la correction des variations d'intensités liées à l'expérience :
- variation de la puissance d'irradiation au cours de la collecte;
- diminution du pouvoir de diffraction du cristal au cours de l'irradiation;
- différence de vitesse de passage des nœuds à travers la sphère d'Ewald.
Ces facteurs de correction sont appliqués en prenant en compte les réflexions
équivalentes reliées par la symétrie du cristal et enregistrées plusieurs fois. Parmi les
équivalences de réflexions pouvant être prises en compte à la mise à l'échelle se trouve la
correspondance entre les paires de Friedel : à une réflexion (h,k,l) correspond une réflexion
(-h,-k,-l).
La qualité du jeu de données est évaluée par :
(i) la comparaison des intensités des réflexions équivalentes : le Rsym évalue cette
équivalence;

96
où Ih,i est l'intensité de la ième mesure d'une réflexion et Îh la moyenne des intensités des
mesures sur les réflexions équivalentes;
(ii) le rapport signal sur bruit : I / [σ(I)], où σ(I) est l'écart-type de l'intensité. La valeur de
ce rapport diminue quand la résolution augmente, l'intensité des taches diminuant à haute
résolution;
Ces deux paramètres définissent la résolution maximale à laquelle les données sont
exploitables. Pour ma part, j’ai considéré que la résolution limite est atteinte lorsque le Rsym
dépassait une valeur de 60% et le signal I / [σ(I)] était inférieur à 2 dans une même tranche de
résolution.
Le jeu natif collecté à 2,8Å de résolution contenait 97% des réflexions possibles, avec un
I / [σI] global de 6,8. Le Rsym vaut 15,3%, 50% dans la plus forte tranche de résolution
(Figure 25).
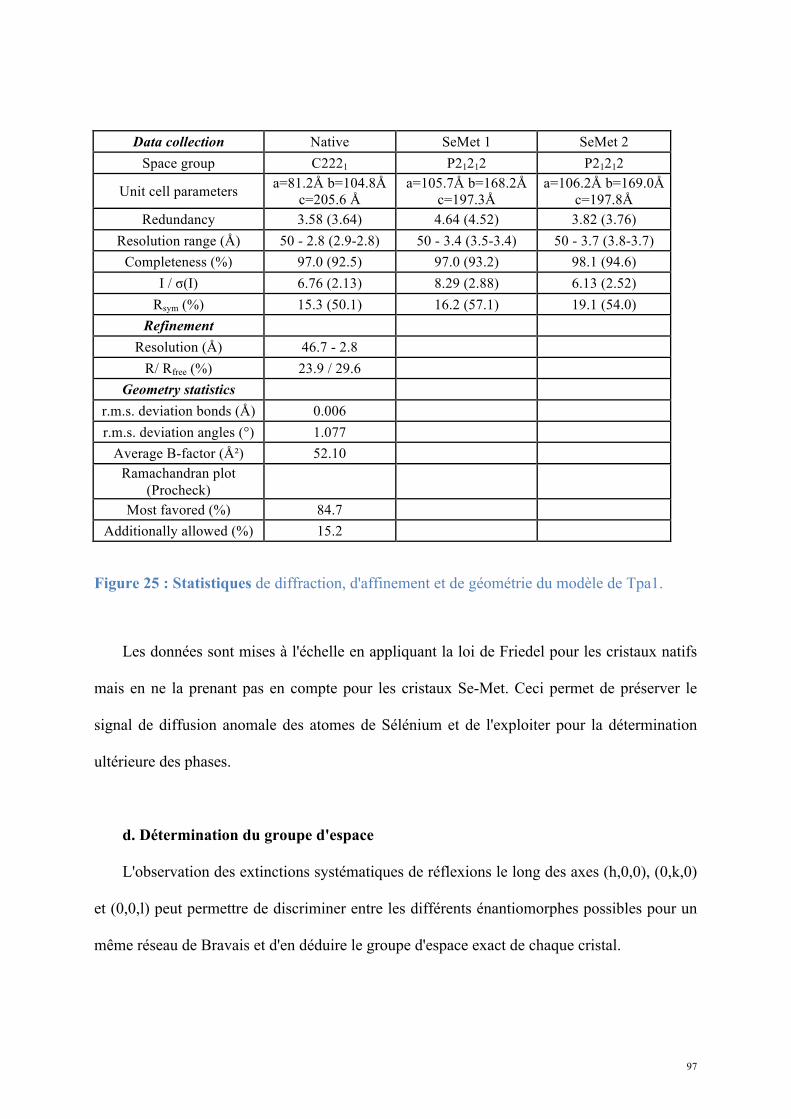
97
Data collection Native SeMet 1 SeMet 2 Space group C2221 P21212 P21212
Unit cell parameters a=81.2Å b=104.8Å c=205.6 Å
a=105.7Å b=168.2Å c=197.3Å
a=106.2Å b=169.0Å c=197.8Å
Redundancy 3.58 (3.64) 4.64 (4.52) 3.82 (3.76) Resolution range (Å) 50 - 2.8 (2.9-2.8) 50 - 3.4 (3.5-3.4) 50 - 3.7 (3.8-3.7)
Completeness (%) 97.0 (92.5) 97.0 (93.2) 98.1 (94.6) I / σ(I) 6.76 (2.13) 8.29 (2.88) 6.13 (2.52)
Rsym (%) 15.3 (50.1) 16.2 (57.1) 19.1 (54.0) Refinement
Resolution (Å) 46.7 - 2.8 R/ Rfree (%) 23.9 / 29.6
Geometry statistics r.m.s. deviation bonds (Å) 0.006 r.m.s. deviation angles (°) 1.077
Average B-factor (Ų) 52.10 Ramachandran plot
(Procheck)
Most favored (%) 84.7 Additionally allowed (%) 15.2
Figure 25 : Statistiques de diffraction, d'affinement et de géométrie du modèle de Tpa1.
Les données sont mises à l'échelle en appliquant la loi de Friedel pour les cristaux natifs
mais en ne la prenant pas en compte pour les cristaux Se-Met. Ceci permet de préserver le
signal de diffusion anomale des atomes de Sélénium et de l'exploiter pour la détermination
ultérieure des phases.
d. Détermination du groupe d'espace
L'observation des extinctions systématiques de réflexions le long des axes (h,0,0), (0,k,0)
et (0,0,l) peut permettre de discriminer entre les différents énantiomorphes possibles pour un
même réseau de Bravais et d'en déduire le groupe d'espace exact de chaque cristal.

98
Les extinctions systématiques des réflexions en (2n+1) sur l'axe (0,0,l) montrent que le
groupe d'espace du cristal de protéine native est C2221 (et non C222) (Figure 26). Les
extinctions systématiques (2n+1) sur les trois axes pour les cristaux de protéine Se-Met
suggèrent fortement que le groupe d'espace correct est P212121 (et non P222, P2221 ou
P21212) (Figure 27).
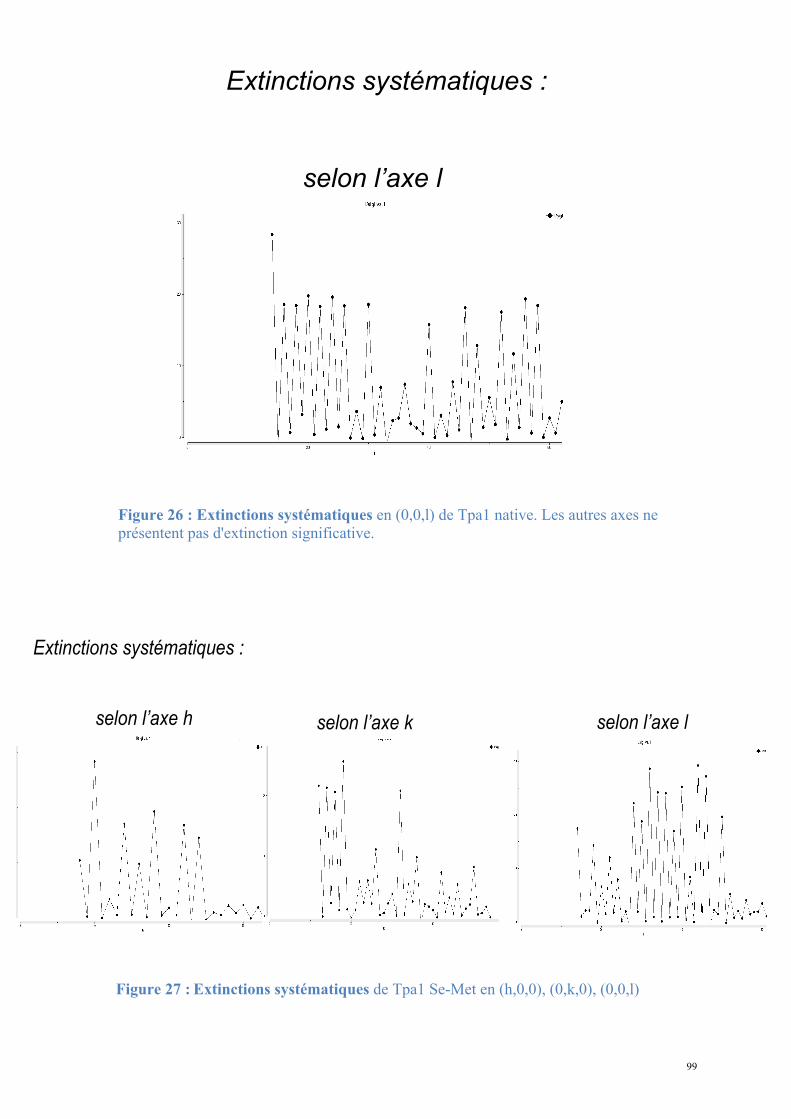
99
En fonction de la résolution (Å) :
Complétude (%)
RSym
I / !I
Extinctions systématiques :
selon l’axe h
selon l’axe k
selon l’axe l
Statistiques de jeux de données des cristaux de Tpa1 Se-Met
Figure 26 : Extinctions systématiques en (0,0,l) de Tpa1 native. Les autres axes ne présentent pas d'extinction significative.
Figure 27 : Extinctions systématiques de Tpa1 Se-Met en (h,0,0), (0,k,0), (0,0,l)
En fonction de la résolution (Å) :
Complétude (%)
RSym
I / !I
Extinctions systématiques :
selon l’axe l
Saccharomyces cerevisiae Tpa1 diffraction Statistiques de jeux de données des cristaux de Tpa1 natifs
En fonction de la résolution (Å) :
Complétude (%)
RSym
I / !I
Extinctions systématiques :
selon l’axe h
selon l’axe k
selon l’axe l
Statistiques de jeux de données des cristaux de Tpa1 Se-Met
En fonction de la résolution (Å) :
Complétude (%)
RSym
I / !I
Extinctions systématiques :
selon l’axe h
selon l’axe k
selon l’axe l
Statistiques de jeux de données des cristaux de Tpa1 Se-Met

100
e. Détermination du contenu de l'unité asymétrique
L'unité asymétrique peut contenir plusieurs molécules, non reliées par des opérations de
symétrie cristallographique. Connaissant le volume de l'unité asymétrique et le poids
moléculaire de la protéine étudiée (74,4kDa pour Tpa1), on peut estimer le nombre de
protéines dans l'unité asymétrique en calculant le volume massique ou coefficient de
Matthews (Kantardjieff and Rupp, 2003) :
où V est le volume de l'unité asymétrique, M la masse moléculaire de la protéine et Z le
nombre de molécules dans l'unité asymétrique. Une valeur moyenne du VM de 2,69 a été
calculée pour les cristaux de protéines, correspondant à un taux de solvant de 50%.
Le coefficient de Matthews a été calculé pour chaque jeu. Pour le jeu natif C2221, une
valeur de 2,94 est obtenue pour une seule molécule ce qui correspond à 58% de solvant dans
l'unité asymétrique. Pour le jeu Se-Met P212121, le coefficient de Matthews ne permet pas de
trancher entre 4 ou 5 molécules dans l'unité asymétrique. Cependant, les relations de
paramètres de maille entre le jeu natif et les jeux Se-Met permettent de déduire le contenu de
l'unité asymétrique en P212121.
En effet, dans le cas des cristaux natifs, l’unité asymétrique en C2221 contient une
molécule, une maille C2221 contient 8 unités asymétriques et donc 8 protéines au total. Dans
le cas des cristaux de protéine marquée au Se-Met, l’unité asymétrique en P212121 contient n
molécules, une maille contient 4 unités asymétriques et donc 4n protéines. La maille P212121
étant 2 fois plus grande que la maille C2221 (bSe-Met=2anative), elle contiendra 16 molécules soit
VM =
V
M Z

101
4 dans chaque unité asymétrique. Le coefficient de Matthews correspondant à la présence de
4 molécules de Tpa1 dans l’unité asymétrique du groupe d’espace P212121 indique une
proportion de solvant de 59%. Ces 4 molécules sont reliées par des symétries non
cristallographiques.
D. Résolution de la structure
La collecte des données fournit un jeu complet d'intensités des rayons diffractés;
cependant les faisceaux de rayons X en tant qu'ondes sont aussi définis par une phase, qui
reste spécifique à chaque atome et à chaque tache de diffraction. La localisation et
l’attribution de la nature des atomes dans l'unité asymétrique se calculent à partir du facteur
de diffusion atomique qui ne peut être obtenu qu'en disposant de l'information complète
d'intensité et de phase du faisceau diffracté.
La détermination de la phase a été réalisée par la méthode de diffusion anomale du
Sélénium (Se-SAD). Dans ce cas, la loi de Friedel est abolie pour les réflexions des atomes de
Sélénium, c’est à dire que les réflexions (h,k,l) et (-h,-k,-l) ne sont plus équivalentes. En effet,
à la longueur d'onde d'absorption du diffuseur anomal, des photons incidents sont absorbés
par l'atome et réémis avec un décalage de phase (phénomène de diffusion inélastique,
diffusion "anomale"). Le facteur de dispersion atomique acquiert une composante réelle f' et
une composante irréelle f'' dépendant de la longueur d'onde. Des scans de fluorescence sont
mesurés sur les cristaux de protéine Se-Met pour confirmer la présence du Sélénium et pour
déterminer la longueur d'onde du faisceau de rayons X à laquelle la diffusion anomale du
sélénium est maximale. A partir de ce scan, un faisceau à une énergie de 12,659keV
correspondant à une longueur d’onde λ de 0,9794Å a été utilisé pour irradier les cristaux de
protéines Se-Met (Figure 28).
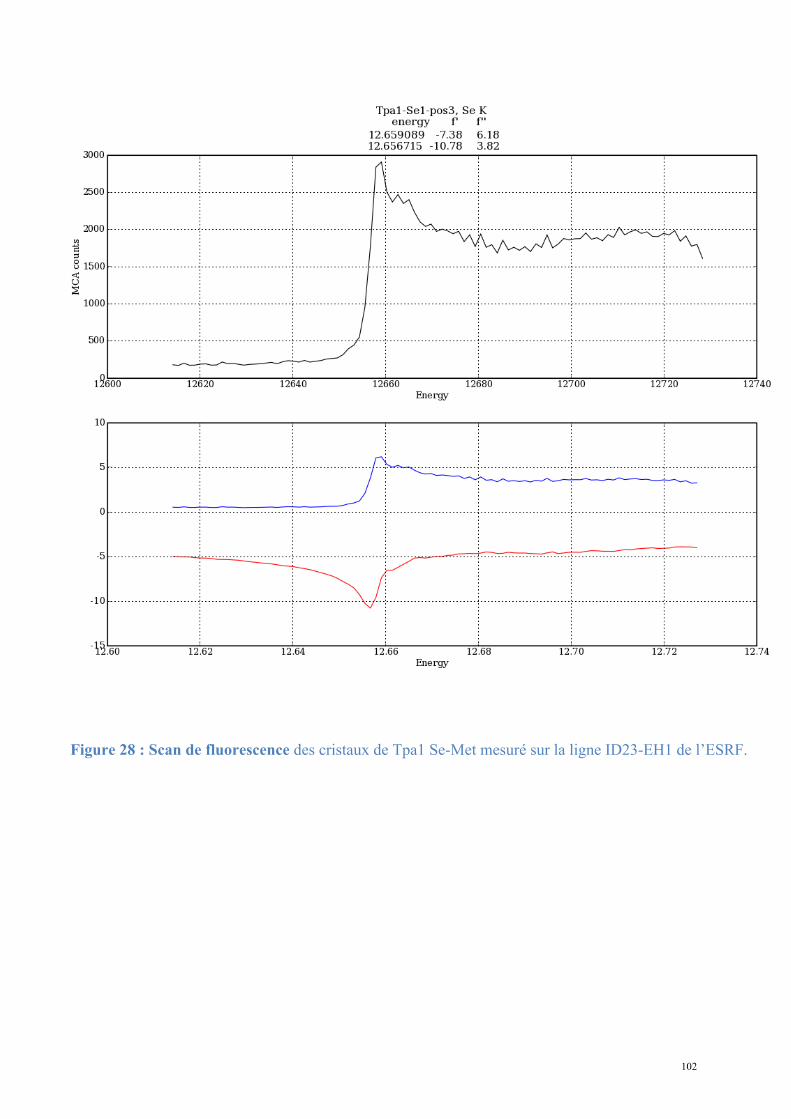
102
Figure 28 : Scan de fluorescence des cristaux de Tpa1 Se-Met mesuré sur la ligne ID23-EH1 de l’ESRF.

103
a. Recherche des sites Sélénium :
Le jeu de données collectées sur la ligne ID23-EH1 à la longueur d'onde de diffusion du
Sélénium sur le cristal de Tpa1 marquée en Se-Met est utilisé pour localiser les sites de
Sélénium. La séquence primaire de la protéine indique que chaque monomère de Tpa1
contient 644 résidus dont 9 méthionines en incluant la méthionine initiatrice. Considérant que
l'unité asymétrique contient 4 molécules, nous avons recherché 36 sites Se dans une gamme
de résolution de 30 à 5,5Å. Le programme SHELXD propose 32 sites qui présentent des
statistiques satisfaisantes.
b. Détermination des phases :
Les coordonnées des 32 sites localisés par le programme SHELXD sont affinées par le
programme SHARP qui va optimiser les coordonnées x, y et z de ces sites, leur occupation et
leur facteur B. Ce programme va également éliminer les sites les moins probables et
rechercher de nouveaux sites, le cas échéant. Dans le cas de la protéine Tpa1, les jeux de
données collectés n’étant pas de bonne qualité. Différents essais de détermination des phases
ont été menés et les meilleurs résultats ont été obtenus lorsque les deux jeux de données Se-
Met collectés sur les lignes BM30-A et ID23-EH1 sont utilisés pour affiner les coordonnées
des sites Se et pour déterminer les phases. Le programme SHARP après plusieurs cycles
d’affinement propose 36 sites affinés et indique un pouvoir de phasage supérieur à 0,75
jusqu’à 6 Å de résolution (Figure 29). Compte tenu de la présence de quatre copies de la
protéine dans l’unité asymétrique, le programme RESOLVE (Wang et al., 2004) a été utilisé
pour améliorer le jeu de phases expérimentales déterminées par le programme SHARP en
réalisant les étapes suivantes : extension des phases de 6 Å (résolution où le pouvoir de
phasage reste significatif) jusqu’à 3,2 Å, recherche de symétries non cristallographiques à
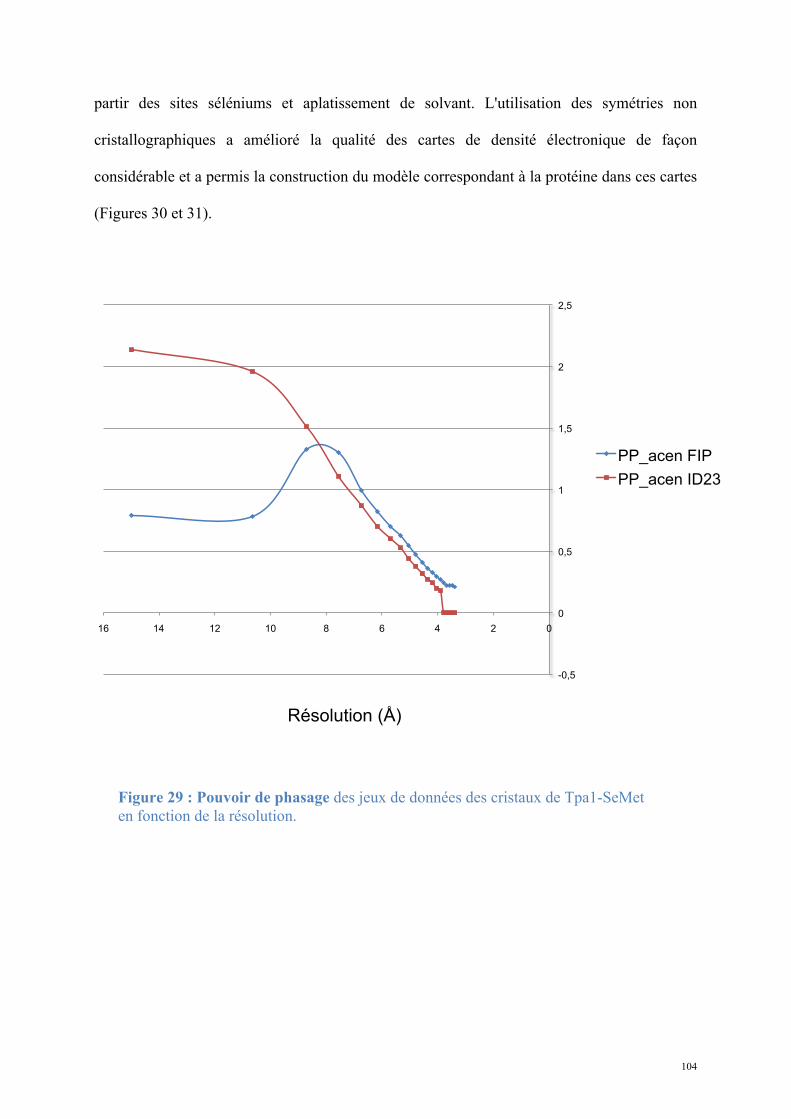
104
partir des sites séléniums et aplatissement de solvant. L'utilisation des symétries non
cristallographiques a amélioré la qualité des cartes de densité électronique de façon
considérable et a permis la construction du modèle correspondant à la protéine dans ces cartes
(Figures 30 et 31).
-0,5
0
0,5
1
1,5
2
2,5
0 2 4 6 8 10 12 14 16
PP_acen FIP
PP_acen ID23
Résolution (Å)
Pouvoir de phasage des jeux de cristaux de protéine SeMet
à la longueur d’onde du pic de diffusion anomale
Figure 29 : Pouvoir de phasage des jeux de données des cristaux de Tpa1-SeMet en fonction de la résolution.

105
Contribution des NCS au phasage de Tpa1
Sans NCS Avec NCS
4 groupes de sites Se sont reliés par des symétries non-crystallographiques (Resolve)
Cartes électroniques après phasage avec ou sans NCS (Resolve)
Figure 30 : Cartes de densité électronique après phasage par RESOLVE, en prenant ou en ne prenant pas en compte les symétries non cristallographiques (NCS).
Les cartes 2Fo-Fc sont contourées à 1σ. Le squelette de Carbones α du modèle final est représenté par les traits bleus.
Figure 31 : Carte de densité électronique après le dernier affinement contourée à 1σ au niveau du site de fixation du Fer.

106
c. Construction du modèle :
La qualité des cartes de densité électronique expérimentale à 3,2 Å de résolution ainsi
obtenues ne permettait pas une construction automatique du modèle complet de la protéine.
Le modèle a par conséquent été construit manuellement avec le programme COOT, puis
affiné avec le programme REFMAC (Murshudov et al., 1997).
Une fois le modèle satisfaisant, il est utilisé pour un remplacement moléculaire dans les
données natives à meilleure résolution par le programme MOLREP (Vagin and Teplyakov,
2010). La résolution d'une structure par remplacement moléculaire consiste à essayer de
positionner dans la maille cristalline une molécule de structure voisine connue. Le
positionnement de la molécule est un problème à six dimensions qui sont inconnues (trois
angles de rotations et trois vecteurs de translation). Les orientations potentielles sont calculées
par des fonctions de rotation, puis à partir de chaque orientation retenue sont calculées les
translations possibles par des fonctions de translation. L'évaluation des solutions obtenues se
fait par comparaison des fonctions de Patterson (vecteurs interatomiques) calculées à partir
des données de diffraction et des fonctions de Patterson calculées à partir du modèle. Si la
fonction de rotation a été correctement choisie, la fonction de translation a un maximum très
marqué. La solution obtenue est soumise à validation par analyse de la qualité de
l’empilement cristallin et à un affinement par REFMAC, qui améliore significativement et
spécifiquement les facteurs R et Rfree.
Une nouvelle série de cycles de reconstruction-affinement en utilisant les programmes
COOT (Emsley and Cowtan, 2004) et PHENIX.REFINE (Adams et al., 2010) ainsi que les
données des jeux natifs a abouti au modèle final. Les reconstructions-affinements sont arrêtés
lorsque :

107
- la corrélation entre les facteurs de structures calculés et observés est satisfaisante; le
facteur R est calculé comme suit :
Une partie des données de diffraction n'est pas utilisée pour l'affinement, mais réservé au
calcul d'un Rfree, indépendant des valeurs affinées. Après le dernier affinement, les valeurs de
R et Rfree pour la structure de Tpa1 à 2,8 Å de résolution s'élèvent à 23.9 et 29.6.
- les cartes de densités électroniques 2Fobs-Fcalc et Fobs-Fcalc sont satisfaisantes : toute la
densité est occupée par le modèle et le modèle n'est présent que dans la densité;
- la stéréochimie du modèle est respectée, à savoir :
. les longueurs et les angles des liaisons. Notre modèle final ne dévie pas
significativement des valeurs canoniques : rms deviationlongueurs=0,006Å, rms
deviationangles=1,077°.
. la conformation de la chaîne principale des acides aminés. Celle-ci est caractérisée
par trois angles φ, ψ et ω; les contraintes exercées sur la liaison peptidique et les structures
secondaires de la protéine limitent les combinaisons de valeurs favorables énergétiquement
pour ces angles. Le diagramme de Ramachandran permet de visualiser en deux dimensions les
valeurs possibles des angles φ et ψ. Le modèle final de Tpa1 contient 99,7% des résidus dans
les zones autorisées (Figure 32).
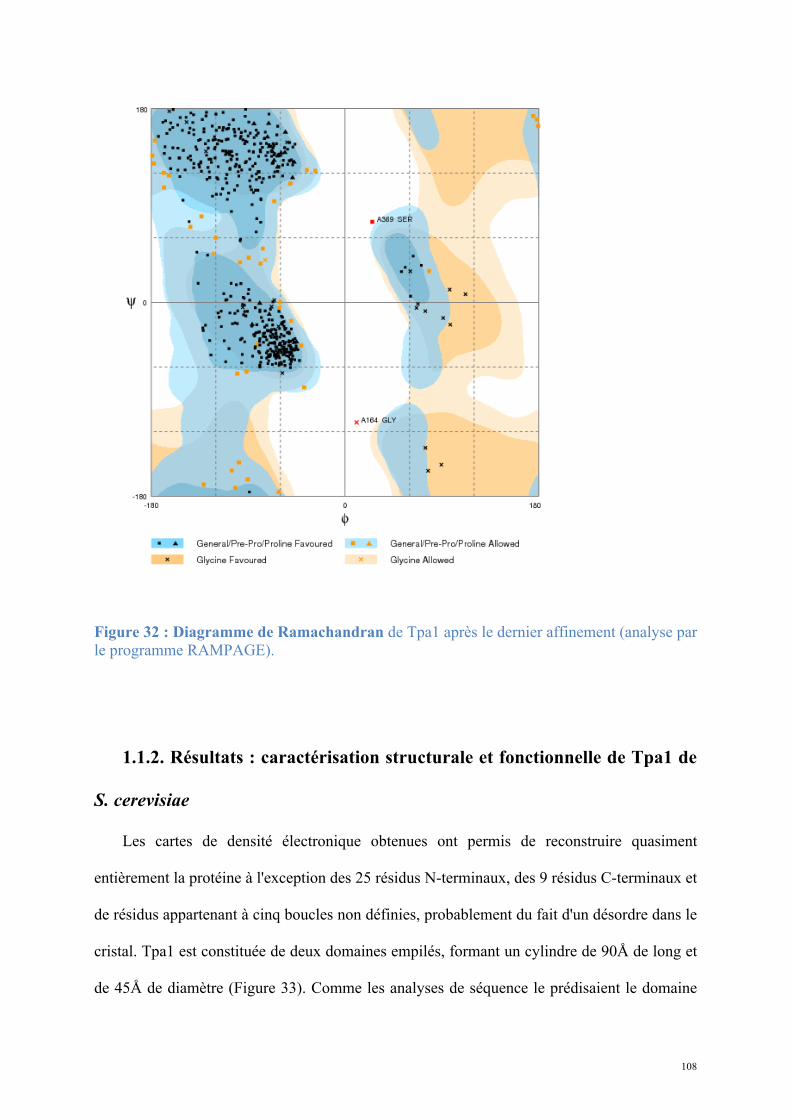
108
Figure 32 : Diagramme de Ramachandran de Tpa1 après le dernier affinement (analyse par le programme RAMPAGE).
1.1.2. Résultats : caractérisation structurale et fonctionnelle de Tpa1 de
S. cerevisiae
Les cartes de densité électronique obtenues ont permis de reconstruire quasiment
entièrement la protéine à l'exception des 25 résidus N-terminaux, des 9 résidus C-terminaux et
de résidus appartenant à cinq boucles non définies, probablement du fait d'un désordre dans le
cristal. Tpa1 est constituée de deux domaines empilés, formant un cylindre de 90Å de long et
de 45Å de diamètre (Figure 33). Comme les analyses de séquence le prédisaient le domaine

109
N-terminal adopte un repliement α/β de type double-stranded β-helix (DSBH) : un double
feuillet central de quatre brins-β antiparallèles, complété par de trois brins-β accessoires et
plusieurs hélices-α. La recherche d'homologues structuraux par comparaison de structures
secondaires (EBI-SSM (Krissinel and Henrick, 2004)) confirme l'appartenance de Tpa1N à la
famille des dioxygénases à domaine DSBH. Plus précisément, Tpa1N adopte un repliement
très proche de la prolyl-hydroxylase PHD2 de H. sapiens (rmsd=2,6 sur 171 Cα alignés). La
fonction de PHD2 a été attribuée à l'adaptation cellulaire à l'hypoxie par régulation de la
stabilité du facteur de transcription HIF-1α (Chowdhury et al., 2009; McDonough et al.,
2006). L'homologie structurale suggère que Tpa1 jouerait un rôle comparable de régulation
d'une protéine-cible par hydroxylation d'une proline.
Après le dernier affinement une densité résiduelle est présente au fond de la cavité
délimitée par les deux feuillets-β; sa forme sphérique et sa taille, ainsi que sa proximité de
trois résidus H...D...H (Figure 31), caractéristiques des dioxygénases à domaine DSBH,
suggèrent qu'il s'agit d'un ion métallique impliqué dans le mécanisme catalytique. Cette
densité résiduelle est située dans une poche très conservée de ce domaine. L'analyse
élémentaire réalisée sur la protéine purifiée (Service Central d'Analyse du CNRS, Lyon)
révèle la présence d'une quantité importante de Fer dans l'échantillon : 1 Fer pour 2 protéines;
PHD2 fixe également un ion Fer dans son site catalytique. La densité supplémentaire a été
attribuée à un Fe2+ et deux molécules d'eau et l'affinement ultérieur montrait une occupation
satisfaisante par ces molécules.
Les essais de co-cristallisation avec du 2-oxoglutarate dans les conditions décrites ont
permis d'obtenir des cristaux et de résoudre la structure de Tpa1 entière par remplacement
moléculaire. Cependant aucune densité supplémentaire n'était remarquable dans les cartes,
révélant que le 2-oxoglutarate ne s'est pas fixé en quantité significative sur la protéine.
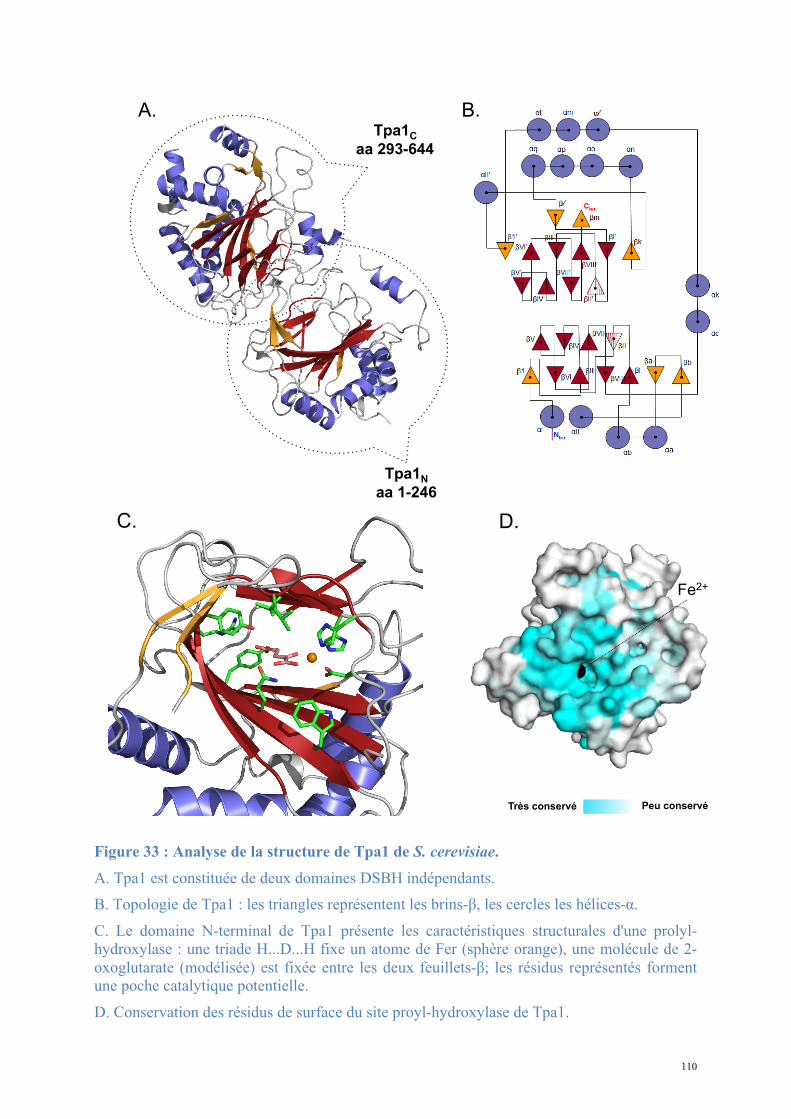
110
Figure 33 : Analyse de la structure de Tpa1 de S. cerevisiae. A. Tpa1 est constituée de deux domaines DSBH indépendants. B. Topologie de Tpa1 : les triangles représentent les brins-β, les cercles les hélices-α.
C. Le domaine N-terminal de Tpa1 présente les caractéristiques structurales d'une prolyl-hydroxylase : une triade H...D...H fixe un atome de Fer (sphère orange), une molécule de 2-oxoglutarate (modélisée) est fixée entre les deux feuillets-β; les résidus représentés forment une poche catalytique potentielle.
D. Conservation des résidus de surface du site proyl-hydroxylase de Tpa1.
Tpa1N
aa 1-246
Tpa1C
aa 293-644
Très conservé Peu conservé
Fe2+
A. B.
C. D.
Tpa1N
aa 1-246
Tpa1C
aa 293-644
Très conservé Peu conservé
Fe2+
A. B.
C. D.
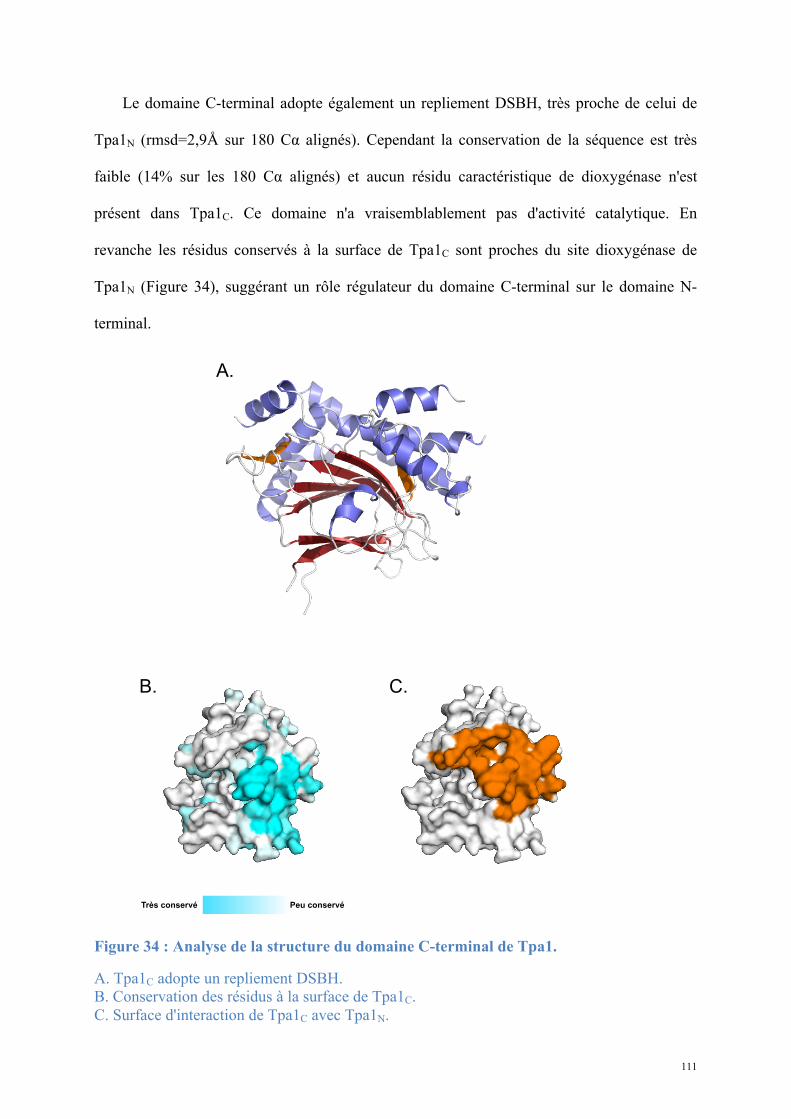
111
Le domaine C-terminal adopte également un repliement DSBH, très proche de celui de
Tpa1N (rmsd=2,9Å sur 180 Cα alignés). Cependant la conservation de la séquence est très
faible (14% sur les 180 Cα alignés) et aucun résidu caractéristique de dioxygénase n'est
présent dans Tpa1C. Ce domaine n'a vraisemblablement pas d'activité catalytique. En
revanche les résidus conservés à la surface de Tpa1C sont proches du site dioxygénase de
Tpa1N (Figure 34), suggérant un rôle régulateur du domaine C-terminal sur le domaine N-
terminal.
Figure 34 : Analyse de la structure du domaine C-terminal de Tpa1.
A. Tpa1C adopte un repliement DSBH. B. Conservation des résidus à la surface de Tpa1C. C. Surface d'interaction de Tpa1C avec Tpa1N.
Très conservé Peu conservé
A.
B. C.

112
La résolution de la structure et l'étude génétique réalisée en parallèle dans l'équipe de
Bertrand Séraphin ont fait l'objet d'une publication dans le Journal of Biological Chemistry.
L'article (Article 1) est présenté sous la forme publiée ci-après.

113
Article 1 : Structural and functional insights into
S. cerevisiae Tpa1, a putative prolyl hydroxylase
influencing translation termination and
transcription

Structural and functional insights into S. cerevisiae Tpa1, a putative prolyl hydroxylase influencing translation termination and transcription
Julien Henri1*, Delphine Rispal2,3*, Emilie Bayart2, Herman van Tilbeurgh1, Bertrand Séraphin2,3 and Marc Graille1.
1. Institut de Biochimie et Biophysique Moléculaire et Cellulaire (IBBMC). CNRS UMR8619 Bat 430 Université Paris Sud 91405 Orsay Cedex. France 2. Centre de Génétique Moléculaire (CGM). CNRS, 1 avenue de la Terrasse, 91198 Gif-sur-Yvette Cedex. France 3. IGBMC, 1 rue Laurent Fries, BP 10142, 67404 Illkirch Cedex, France * Both authors contributed equally. Running head: Structural and functional study of Tpa1, an O2 sensor.
To whom correspondence should be addressed: Bertrand Séraphin, IGBMC, 1 rue Laurent Fries, BP 10142, 67404 Illkirch Cedex, France. Phone: 33 388653356; Fax: 33 388653200. E-mail: [email protected] Marc Graille, IBBMC, CNRS UMR8619 Bat 430 Université Paris Sud 91405 Orsay Cedex. France. Phone: 33 169153157; Phone: 33 169853715. E-mail: [email protected] Abstract. Efficiency of translation termination relies on the specific recognition of the three stop codons by the eukaryotic translation termination factor eRF1. To date, only few proteins are known to be involved in translation termination in eukaryotes. S. cerevisiae Tpa1, a largely conserved but uncharacterized protein, has been described to associate with a mRNP complex located at the 3’ end of mRNAs that contains at least eRF1, eRF3 and Pab1. Deletion of the TPA1 gene results in a decrease of translation termination efficacy, an increase in mRNAs half-lives and longer mRNA poly(A) tails. In parallel, S. pombe Ofd1, a Tpa1 ortholog, and its partner Nro1 have been implicated in the regulation of the stability of a transcription factor that regulates genes essential for the cell response to hypoxia. To gain insight into Tpa1/Ofd1 function, we have solved the crystal structure of the S. cerevisiae Tpa1 protein. This protein is composed of two equivalent domains with the double-stranded β-helix (DSBH) fold. The N-terminal domain displays a highly conserved active site with strong similarities with prolyl-4-hydroxylases. Further functional studies show that the integrity of Tpa1 active site as well as the presence of YOR051c (the S. cerevisiae Nro1 ortholog) are essential for correct translation termination. In parallel, we show that Tpa1 represses the expression of genes regulated by Hap1, a transcription factor involved in the response to levels of heme and oxygen. Altogether, our results support that
Tpa1 is a prolyl-4-hydroxylase acting as an oxygen sensor and influencing several distinct regulatory pathways. Introduction.
Protein synthesis is a complex process performed by ribosomes, translation factors and amino-acyl tRNAs that act synergistically to translate mRNAs into corresponding proteins. The mechanism of translation of mRNA into protein can be divided into three major steps: initiation, elongation and termination. In eukaryotes, translation initiation relies on the formation of a stable closed-loop structure bringing the 5’ m7G cap and the 3’ poly(A) tail of a single mRNA in proximity and requires at least eleven initiation factors (eIFs) (1,2). During elongation, the mRNA codon present in the ribosomal A site is recognized by a cognate amino-acyl tRNA associated with elongation factor (eEF) 1A. The nascent polypeptide chain is then transferred from the tRNA present in the P-site to the amino acid of the A-site tRNA. Subsequently, eEF2 induces translocation of peptidyl-tRNA from the A to the P-site allowing the next elongation step to proceed. The entrance of one of the three stop codons (UAA, UAG, or UGA) in the A-site triggers translation termination. In contrast with other codons, stop codons are not recognized by cognate tRNAs but by proteins known as class I release factors (RF1 and RF2 in bacteria and eRF1 in eukaryotes). These are also directly involved in the hydrolysis of the ester bond

connecting the newly synthesized polypeptide chain to the tRNA located in the ribosomal P-site (3-5). The activity of class I RFs is stimulated by the class II RFs GTPases: RF3 in bacteria and eRF3 in eukaryotes (3-6). The eRF1 and eRF3 proteins form a stable complex in the absence of the ribosome (7,8). The GTP hydrolysis activity of eRF3 is dependent on ribosome-bound eRF1 (9) but eRF3’s precise role remained obscure until recently. In vitro reconstitution of eukaryotic translation system indicated that binding of the eRF1-eRF3-GTP ternary complex to pre-termination complexes causes a structural rearrangement of the ribosome. This results in GTP hydrolysis by eRF3 followed by eRF1-induced hydrolysis of peptidyl-tRNA (6).
Translation termination relies on the efficient and specific recognition of stop codons (UGA, UAA and UAG) by eRF1 despite the presence of near-cognate tRNAs (such as the tRNATrp that recognizes the UGG codon) that could induce amino acid misincorporation and the synthesis of C-terminal extensions. The efficiency of stop codon recognition is primarily influenced by the stop codon itself and the first downstream nucleotide, commonly referred to as the tetranucleotide termination signal (10). Several trans-acting factors influencing the accuracy of this process have also been identified (11). Among these, the interaction of eRF3 with the poly(A)-binding protein (Pab1p), which itself associates with the 3'-poly(A) tail present on almost all eukaryotic mRNAs has been proposed to participate to normal translation termination and to mRNA stabilization (12,13). The glutamine side chain from the strictly conserved GGQ motif from class I release factors has also been shown to be post-translationally modified both in prokaryotes and in eukaryotes, thereby affecting the efficiency of translation termination in vivo, at least in E. coli (14-18). More recently, the deletion of the uncharacterized S. cerevisiae open reading frame YER049W has been shown to result in increased readthrough of stop codons, longer poly(A) tails and increased mRNAs half-lives (19). The YER049W gene product, named Tpa1, was co-immunoprecipitated with translation termination factors eRF1 and eRF3 as well as with the poly(A) binding protein Pab1p, suggesting that it could be part of a messenger ribonucleoprotein (mRNP) complex associated
at the mRNA 3’ end. Sequence analysis suggested that Tpa1 belongs to the 2 oxoglutarate-Fe(II) dioxygenase family (20). Dioxygenase enzymes catalyze oxidation reactions involving dioxygen and use 2-oxoglutarate (2OG) as co-substrate and ferrous ion (Fe2+) as cofactor. They are involved in various biological processes such as fatty acid metabolism, antibiotic biosynthesis, nucleic acid repair, collagen biosynthesis, histone Lys/Arg demethylation and transcriptional regulation of the hypoxic response (21,22). Recently, the Ofd1 protein, a Tpa1 ortholog from S. pombe, has been implicated with Nro1 in the regulation of low oxygen gene expression (23,24). In S. pombe, the Sre1 transcription factor regulates the expression of genes involved in cholesterol and lipid biosynthesis (25,26). Under low oxygen levels, the membrane bound Sre1 protein is proteolytically cleaved, thus releasing its N-terminal transcription factor domain (27,28). As a consequence, this latter enters the nucleus and activates the expression of genes essential for hypoxic growth. In the absence of oxygen, the Ofd1 C-terminal domain was reported to interact with Nro1 and the Sre1 N-terminal domain is stabilized. In the presence of oxygen, the Ofd1 N-terminal dioxygenase domain would act as an oxygen sensor, activating its own C-terminal domain and preventing its interaction with Nro1, thus enhancing degradation of the Sre1 N-terminal domain.
Based on these two independent studies realized in S. cerevisiae and S. pombe, the precise Tpa1/Ofd1 function remains obscure. On the one hand, no clear ortholog of S. pombe Sre1 protein (homologous to human SREBP) has been found in S. cerevisiae. On the other hand, a S. cerevisiae ortholog of Nro1 (YOR051cp) does exist and was reported to interact with Tpa1 (29). Therefore, Tpa1/Odf1 may have a role that is organism dependent and/or it may act on several distinct pathways. We present here the results of structural, biochemical and genetic studies that improve our understanding of the function of Tpa1, a protein conserved from fungi to human. Experimental procedures Cloning, Expression, and Purification of Tpa1 To over-express Tpa1 in E. coli, its coding sequence was amplified from genomic DNA with oligonucleotides OBS2151 and OBS2152

(Supplementary Table 1) and inserted in the BspE1 site of a modified pET vector, yielding pBS3077. The protein has been expressed using E. coli Rosetta (DE3) pLysS strain (Novagen) at 23°C in 2xYT medium (BIO101 Inc.) supplemented with kanamycin at 50µg/mL. At OD600 = 0.8, the transcription of the plasmid was induced during 20h by the addition of 50µg/mL IPTG. Cells were harvested by centrifugation, resuspended in 35mL of 20mM Tris-HCl pH 7.5, 200mM NaCl, 5mM β-mercaptoethanol (buffer A) and stored at -20°C. Cell lysis was performed by sonication. The His-tagged protein was purified on a Ni-NTA column (Qiagen) followed by gel filtration chromatography on a SuperdexTM 200 HL 16/60 column (Amersham Biosciences) equilibrated in buffer A. Protein labeling with SeMet was conducted as described (30). The SeMet protein was purified using the same protocol as the native protein. Size-exclusion chromatography – Multi-angle laser light scattering measurements Sample injection, chromatography and detection was carried out using a Triple Detector Array (TDA302) system coupled in line to a GPCmax chromatographic system (Viscotek). A sample of 100µL of Tpa1 (6.2mg/mL) was injected at a flow rate of 0.5mL/min on a SuperdexTM 200 HR 10/30 column (Amersham Biosciences) equilibrated in buffer A. Elution was followed by a UV-Visible spectrophotometer, a differential refractometer, a 7° Low Angle Light Scattering detector, a 90° Right Angle Light Scattering detector and a differential pressure viscometer. The data were collected and processed with the program OmniSEC (Viscotek). Mw was directly calculated from the absolute light scattering measurements. The dn/dc value (0.168) for Tpa1 has been determined experimentally using the refractive index at four different concentrations of Tpa1. Structure determination Crystals of the native and SeMet protein were grown from a mixture in a 1:1 ratio of 15mg/mL protein solution in buffer A and crystallization liquor containing 0.1M Hepes pH 7.5, 44% methylpentanediol (MPD) and 0.1M Mg(NO3)2, at 18°C over 2 months. The crystals were directly flash cooled in liquid nitrogen.
Native crystal (crystal 1) diffracted to 2.8Å resolution on beam line ID14-EH2 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). This crystal belongs to space group C2221 with one molecule in the asymmetric unit (a = 81.2Å, b = 104.8Å, c = 205.6Å, α = β = γ = 90°). SeMet crystals suffered from serious diffraction anisotropy (6Å in one direction and 3.5-2.5Å in the other one) but datasets could be recorded at ESRF (Grenoble, France) to 4Å resolution on beam line ID23-EH1 (crystal 2) and 3.3Å on beam line BM30A (crystal 3). The diffraction data were collected at the selenium edge (λ = 0.9796Å). These crystals belong to space group P212121 with four molecules in the asymmetric unit (a = 105.7Å, b = 167.9Å, c = 196.9Å, α = β = γ = 90°). The structure was determined by the single wavelength anomalous dispersion method using the anomalous signal of selenium atoms. Data were processed with the XDS package (31). The program SHELXD was used to find an initial set of 32 Se sites (40 were expected according to the protein sequence and cell content) from the crystal 2 dataset in the 30-5.5Å resolution range (32). Refinement of the Se sites and phasing were carried out with the program SHARP using the SAD datasets collected on crystals 2 and 3 (33). Additional sites were included in the refinement after inspection of residual maps. The final substructure model comprises 36 Se atoms. Phase refinement and extension were carried out via density modification and NCS averaging with the program RESOLVE (34). The experimental phases allowed initial automatic building by BUCCANEER (35). Iterative cycles of manual rebuilding using COOT (36) followed by refinement with REFMAC (37) led to a first almost complete model. This model was then used for molecular replacement using native data collected on crystal 1 with MOLREP and finally submitted to a new series of manual rebuilding with COOT and refinement with PHENIX (38). The statistics for data collection and refinement are summarized in Table 1. Due to the absence of electron density, the following regions were omitted from the final model: 1-25, 94-110, 269-275, 306-330, 455-459, 562-584 and 635-644. The atomic coordinates and structure factors have been deposited into the Brookhaven Protein Data Bank under accession numbers (XXXX).

Strain and media Saccharomyces cerevisiae strains YJW618 (MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade1-14 PSI[+]) and YJW619 (MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade1-14 tpa1::LEU2 [PSI+]) were obtained from D. Bedwell (19). YOR051C was deleted in these strains with a Candida glabrata HIS3 cassette amplified with OBS3866/OBS3867, giving strains BSY2236 and BSY2237, respectively. Cells were grown at 30°C in standard media. The plasmid, pBS3820, containing the wild type TPA1 gene was constructed by amplifying the TPA1 ORF with upstream and downstream sequences from genomic DNA using oligonucleotides OBS3527 and OBS3528 and inserting it between the BamH1 and Not1 sites of the pRS424 (TRP1, 2µ) vector (39). The H159A/D161N allele was constructed by site directed mutagenesis with primers OBS3687 and OBS3688 (Supplementary Table 1) yielding pBS3793. Domain deletions, pBS 3794 and pBS 3795, were constructed by PCR and cloning using oligonucleotides OBS3738-OBS3741. The TAP tag was introduced by cloning using oligonucleotides OBS3822-OBS3824, OBS3831, OBS3832 and OBS3844 yielding pBS3796-3799. Stop codon readthrough measurements Yeast strains were transformed with these reporters containing the coding sequences of the Renilla and firefly luciferase separated either by a stop codon (TAA) or a sense codon (CAA) as described (19). Three transformants were pooled and grown in selective liquid media to OD600 0,5-0,8 for enzymatic assays. 10 µL of yeast culture was used to measure the luminescence with the dual luciferase reporter assay reagent (Promega) in a Berthold luminometer. The percent readthrough is calculated from the ratio of firefly luciferase produced by the stop codon containing construct to the firefly luciferase by the CAA containing construct, normalized, in each case to the level of Renilla luciferase produced in the same cells. Three biological experiments were performed with, in each case, each one technical duplicates.
β-galactosidase assay
Reporters, provided by B. Guiard (40), contained Hap1 driven UAS elements from the CYC1 or CYB2 genes driving expression of β-galactosidase. Yeast strains were transformed with these reporters and 3 transformants were pooled to perform β-galactosidase assays. Cells were grown to an OD600 of 0,5-0,8. 1mL of culture is used. Assays were performed as previously described with three biological replicate and for each assay one technical duplicate (41). Protein analyses Proteins extracts (42) were fractionated on 8% SDS PAGE and TAP TAG proteins were detected with PAP (Sigma) and an ECL kit (GE Healthcare). Results.
The structure of full-length Saccharomyces cerevisiae Tpa1 has been solved using the single-wavelength anomalous diffraction signal of SeMet substituted protein crystals and refined to 2.8Å resolution using diffraction data from native protein crystals. The resulting density maps allowed us to rebuild 567 amino acids out of 644. The missing residues correspond to the 25 N-terminal residues, the 9 C-terminal residues or belong to five loops for which no electron density was observed. The native protein crystallized in space group C2221 with one molecule in the asymmetric unit while the SeMet labeled protein crystals grown in the same conditions belong to space group P21212 with four Tpa1 copies in the asymmetric unit (one unit cell parameter is twice longer than for the native crystals). As a consequence, the crystal packing is identical between both crystal forms and the Tpa1 protomers are organized as two identical dimers (buried surface 2,000Å2). This suggests that Tpa1 can form homodimers in solution (see below).
Tpa1 is composed of two stacked domains, forming a cylinder of approximately 90Å length and 45Å diameter (Fig. 1A). The N-terminal domain (Tpa1N) encompasses amino acid 26 to 260 (residues 1 to 25 are not visible in the electron density maps) while the C-terminal domain (Tpa1C) encompasses amino acid 293 to 635 (residues 636-644 could not be modeled due to the lack of electron density). Both domains are packed together and connected by a linker (amino acids 261-292) that partially folds as α-helix (αL).

The N-terminal catalytic domain
Sequence analysis has suggested that S. cerevisiae Tpa1 contains a N-terminal 2-oxoglutarate (2OG) – Fe(II) dioxygenase domain from the Double-Stranded Beta-Helix (DSBH) family based on the conservation of three characteristic motifs in the N-terminal part of the protein: a HXD dyad (where X is any amino acid) near the N-terminus, a His towards the C-terminus as well as an Arg or Lys further downstream (20). Basically, the DSBH fold consists of eight β-strands forming a β-sandwich structure comprised of two four-stranded antiparallel sheets (21). The β-sandwich is partitioned between a minor (β-strands 2 if present, 7, 4 and 5) and a major sheet (β-strands 1, 8, 3 and 6) that very often contains additional strands. As proposed, the Tpa1N domain adopts a DSBH fold consisting of a β-sandwich of two antiparallel β-sheets with five α-helices packed onto the major sheet (Fig. 1B). This latter is composed of five β-strands (order 1, 8, 3, 6 and A). Two additional strands (βB and βC) are located at the entrance of the β-sandwich cavity.
The DSBH fold is shared by a wide variety of Fe2+/2OG dependent dioxygenases and many crystal structures have been described for this architecture (21,43). Structural alignments with Tpa1N reveal similarity with prolyl-4-hydroxylases (rmsd of 2.3-2.5Å over 150-170 Cα atoms and 15-19% sequence identity), phytanoyl CoA dioxygenase (rmsd of 2.6Å over 140 Cα atoms and 15% sequence identity), DNA/RNA repair proteins AlkB/ABH3 (rmsd of 2.6Å over 135-140 Cα atoms and 10-15% sequence identity) and isopenicilin N synthase (rmsd of 2.7-2.9Å over 150 Cα atoms and 10% sequence identity). However, the closest structural homolog is human PHD2cat, a prolyl-4-hydroxylase that acts as an oxygen-sensing component and post-translationally hydroxylates the hypoxia-inducible transcription factor HIF1α in the presence of oxygen, thus leading to its degradation by the proteasome (rmsd of 2.5Å over 153 Cα atoms and 19% sequence identity (44)). Analysis of the amino acid side chains strictly conserved between Tpa1N and PHD2cat reveals that most are clustered within the cavity located between both β-sheets that corresponds to PHD2cat enzyme active site (Fig. 1C). First of all, the
HXD…H triad (positions 159, 161 and 227 in Tpa1, respectively) superposes perfectly with the characteristic HXD…H Fe2+ binding motif observed in PHD2cat. The H159XD161 dyad is located within the loop connecting strands β2 and β3 while the second histidine (His227) originates from strand β7. In typical DSBH enzymes, these three residues form a triad, leaving three coordination sites on the octahedral Fe(II) centre for the binding of 2OG and dioxygen. During refinement, a strong residual electron density peak was observed in close proximity to H159, D161 and H227 of Tpa1, exactly at the position corresponding to the iron atom in PHD2cat structure. Element analysis confirmed the presence of iron in the protein samples (ratio 1 iron atom for 2 Tpa1, data not shown). This metal is present at the entrance of a highly conserved cavity known to bind the 2OG co-substrate. All but two residues forming the 2OG binding pocket are strictly conserved between Tpa1N and PHD2cat (Fig. 1C, differences are Leu156 and Gln242 in Tpa1N versus Tyr310 and Thr387 in PHD2cat). At the bottom of this cavity, the positively charged side chain from Arg238 from β8, as well as Tyr173, are well positioned to interact with the carboxyl side chain of the 2OG co-substrate. Similarly, the side chains from Tyr150, Ile171, Leu189 and Val229 of Tpa1N form the wall of the 2OG binding site and structurally match with identical residues from PHD2cat. Altogether, these elements strongly suggest that the Tpa1N domain displays a putative active site configuration that is close to that from PHD2cat.
Further inspection of amino acid sequences of the Tpa1/Ofd1 family clearly shows that the iron atom is located in the middle of a highly conserved groove delineated by side chains from Asp144, Ser146, Asp160, Ile163 and Arg166 on one side and from the β-hairpin made by strands βB and βC (Lys82, Asp86, Ile87, Tyr88 and Gln92) on the other side (Fig. 1D-E). This groove might be extended by residues 94 to 102 (connecting strand βC to helix α4), which are well conserved in fungi, but undefined in electron density maps and hence absent from the final model. It is very likely that this loop will become ordered upon substrate binding. In addition, the rather negative electrostatic potential of this crevice suggests that it could

interact with a molecule or peptide containing positive charges (Fig. 1F).
In summary, the strong structural similarity between Tpa1N and human HIF prolyl-4-hydroxylase combined with the characteristics of the substrate binding groove suggest that Tpa1N has a prolyl-4-hydroxylase activity on Pro and Arg/Lys containing peptides that remain to be identified. The C-terminal domain and its association with Tpa1N
Contrary to the Tpa1N domain, amino acid sequence analysis did not provide convincing predictions for the 3D structure of the Tpa1C domain. Unexpectedly, this domain (residues 293-635) adopts a DSBH very similar to Tpa1N (rmsd of 2.5Å over 153 Cα atoms despite only 13% sequence identity; Fig. 2A), suggesting an internal duplication event. The minor sheet of Tpa1C is only made of 3 strands (β7’, 4’ and 5’) as the antiparallel hydrogen bond pairing of residues corresponding to strand β2’ and the neighboring strand β7’ are not maintained (Fig. 1A-B). The major sheet is composed of seven antiparallel β strands (order A’, 6’, 3’, 8’, 1’, C’ and B’). Seven of the eight α-helices are packed against one face of the major sheet. Two helices (αA’ and αB’) are specific of this domain while the remaining five (α1’ to α5’) are structural matches of the corresponding helices (α1 to α5) from Tpa1N. Despite its structural resemblance with Tpa1N, the HXD…H catalytic triad is absent in Tpa1C and is replaced by residues Thr515, Cys517 and Glu610, which are not conserved in Tpa1 orthologs (Fig. 2A and 3). In addition, the cavity of the Tpa1C domain that corresponds to the active site in enzymes with a DSBH fold is not lined by conserved residues and is largely occluded by the linker region (residues 261-290) connecting both Tpa1 domains. Altogether, these elements argue that this domain is very unlikely to be associated with any enzymatic activity.
Tpa1C has a large and highly conserved surface area that mainly contacts Tpa1N domain (buried surface area of 1,200Å2; Fig. 2B-C) and hence is not accessible. Contrary to Tpa1C, the region from Tpa1N engaged in interdomain interaction does not display significant sequence conservation. Both domains interact via orthogonal packing
of their minor sheets (Fig. 1A-B). This interaction positions the α-helical layers from both domains at the opposite sides of Tpa1. Tpa1C is located below the entrance of the Tpa1N active site and contributes to the formation of the conserved groove that runs across the Tpa1N active site (Fig. 1D-E). In addition, one face of Tpa1C displays a negative electrostatic potential and is juxtaposed to the Tpa1N conserved and negatively charged groove (Fig. 1F). This results in a large surface with negative potential that could be implicated in binding to a positively charged partner.
As stated above, analysis of the crystal packing reveals a tightly packed crystallographic dimer that buries 2,000 Å2 surface area, suggesting that Tpa1 forms dimers (Fig. 2D-E). Dimer formation occurs between Tpa1C domains and is mediated by poorly conserved residues from helices αA’, α1’, αC’, α4’ and α5’, strand β1’ and from the loops connecting αL to αA’, αB’ to αC’ and β1’ to β2’. The core of this interface is mainly hydrophobic with some polar residues involved in its periphery. Analysis of the quaternary structure of Tpa1 in solution using size exclusion chromatography coupled online to a triple detection array (TDA, Viscotek) indicated a molecular mass of 143.3 kDa corresponding to the value expected for the homodimer (150 kDa compared to 75 kDa for the monomer, Fig. 2F). The presence of Tpa1 homodimers in vivo remains to be determined. However, it is noteworthy that among the 3 residues from S. cerevisiae Tpa1 that have been identified by in-depth phosphoproteome analysis (45), one (Ser293) is located within a loop (connection between αL to αA’) involved in this homodimeric interface. In our structure, this residue is not modified but the protein has been produced in E. coli. Hence, we cannot exclude that the phosphorylation of Ser294 from Tpa1 will affect its quaternary structure and potentially its function.
To our knowledge, this is the first example of a Fe(II)-2OG dioxygenase family with two DSBH domains in tandem. Cupin superfamily members (phosphomannose isomerase (46), oxalate decarboxylase (47), quercetin-2,3-dioxygenase (48) and the human transcription cofactor pirin (49)) also have repeated DSBH domains but they are not 2OG dependent enzymes. Tpa1 differs from these

proteins as Tpa1 domains are independent while in pirin and related proteins, the domains are connected to each other by a strand swapping mechanism: N-terminal fragment contributing a β-strand to the C-terminal domain and vice versa. Modeling of Tpa1 bound to a “substrate” peptide. The strong structural similarity of Tpa1 with prolyl-4-hydroxylases strongly suggests Tpa1 is an enzyme that post-translationally modifies proline residues in the presence of oxygen. Among these enzymes, some display broad substrate specificity as exemplified by collagen and algal prolyl-4-hydroxylases that modify the central proline of the X-Pro-Gly motif at several positions within collagen and proteins from the lumen of plant endoplasmic reticulum (50,51). On the contrary, the human HIF1α prolyl-4-hydroxylase (PHD2 or EGLN1) selectively modifies Pro564 from hypoxia-inducible transcription factor (HIF1α) in the presence of oxygen, thereby leading to its degradation by the proteasome (52). Very recently, the mode of substrate binding to prolyl-4-hydroxylases was revealed by two high-resolution crystal structures. The first one corresponds to the complex between algal prolyl-4-hydroxylase bound to a peptide corresponding to the motif Ser-Pro repeated 5 times (rmsd with Tpa1N of 2.4 Å over 152 Cα atoms, 15% sequence identity; (53)). The second is the crystal structure of PHD2cat in complex with a 19-residues peptide (region 556-574 containing the targeted Pro564) from the C-terminal oxygen degradation domain (CODD) from HIF1α (rmsd with Tpa1N of 2.5 Å over 170 Cα atoms, 19% sequence identity; (54)). The CODD peptide binds in an extended form to a well-defined cleft of the PHD2cat active site. Using these complexes as templates, we have modeled peptides within the Tpa1N putative active site (Fig. 2G). In both models (with the exception of the C-terminal part of the CODD peptide), no major steric hindrance between the peptides and Tpa1 is observed. The peptides fit into the highly conserved long groove containing the putative Tpa1N active site and the hydroxylated Proline is positioned close to the metal from the active site (distance between the Pro C4 atoms and the iron atom ≈ 4-5Å). In both cases, the peptides have the
same directionality within the active site groove (i.e. the N-terminal extremity from both peptides is located at the same end of the groove). The main difference between the Tpa1-peptide model structure and the prolyl-4-hydroxylase - substrate peptide complexes resides in the conformation of the β-hairpin made by strands βB and βC. In Tpa1, this region (residues 81-91) is in an open conformation and forms one side of the conserved groove (Fig. 2H). It is also engaged in a large interaction with Tpa1C. In the PHD2cat apo-structure, this region has an open conformation as observed for Tpa1N, while upon CODD peptide binding, it folds back onto the substrate and clamps Pro564 in the active site. This region undergoes the largest conformational changes upon substrate binding (54). Similar conformational changes occur upon peptide binding for the corresponding β-hairpin of algal prolyl-4-hydroxylase (53). Whether rearrangements of the Tpa1 β-hairpin take place upon substrate binding is hard to predict in absence of any information on Tpa1 substrates. Rearrangements of the β-hairpin very likely would affect the interface between both Tpa1 domains. This would result in a different orientation of these domains thereby exposing to solvent part of the highly conserved region of Tpa1C that interacts with Tpa1N. Mutation of conserved residues in the putative Tpa1 catalytic site promote stop codon readthrough
The implication of Tpa1 in translational readthrough, mRNA decay and poly(A) tail length control (19) offered a possibility to test whether the putative catalytic residues of Tpa1 are important for its function. Similarly, the two domains of Ofd1, the Schizosaccharomyces pombe homolog of Tpa1, were shown to have different functions in the oxygen dependent degradation of the active form of the Sre1 transcription factor (23). It was thus also interesting to test the role(s) of the two domains of Tpa1. For this purpose, we concentrated on the level of trans-translation of stop codons using reporter plasmids encoding two luciferases (Renilla and firefly) separated either by a UAA stop codon or by CAA encoding glutamine (Fig. 4A, (19)). These constructs were introduced in a wild-type yeast strain or a strain deleted for TPA1

(Δtpa1). In addition, these plasmids were introduced in the Δtpa1 strain simultaneously with a plasmid encoding either the wild-type TPA1 gene or mutant alleles. Stop codon readthrough was assayed by measuring the relative activities of the two types of luciferase produced by these cells, with 100% readthrough being taken as the ratio observed for the construct with a CAA codon. Consistent with the previous report (19), deletion of the TPA1 gene resulted in a roughly 3-fold increase of the readthrough of the UAA stop codon (Fig. 4B and 4C). Introduction of a plasmid borne copy of the wild type TPA1 gene essentially corrected this defect, restoring low readthrough of stop codon (Fig. 4B). To inactivate the catalytic activity of Tpa1, we substituted simultaneously H159 in alanine and D161 in asparagine. Substitution of either one of the equivalent residues in the related protein hABH3 totally abrogates its DNA repair activity (55). Interestingly, a plasmid encoding the H159A/D161N allele of the TPA1 gene failed to complement the deletion of TPA1 in the stop codon readthrough assay (Fig. 4B). Similarly, plasmids encoding either the Tpa1N or Tpa1C domains failed to restore trans-translation to a wild type level (Fig. 4C). To exclude that mutation of the putative Tpa1 catalytic center or deletion of one or the other of its domains, destabilized the resulting protein (which would have provided a straightforward explanation for their apparent lack of activity), the levels of the wild-type and mutant proteins fused to an epitope tag were measured by western blot (Fig. 4D). This demonstrated that the three mutants were expressed to a level similar to the wild type protein and were equally stable (even though a slightly lower level of the protein containing Tpa1C alone was detected). We conclude that the observed phenotypes do not result from lack of Tpa1. Thus it appears that both domains of Tpa1, as well as its putative catalytic activity, are required to favor stop codon recognition in yeast cells. The S. cerevisiae Nro1 homolog also affects stop codon readthrough
Recently, a factor, Nro1, was shown to interact with Ofd1 in S. pombe and proposed to inhibit the ability of Ofd1 to degrade the active form of the Sre1 transcription factor (24). It is noteworthy that, using TAP purification (56),
the Nro1 homolog in S. cerevisiae, Yor051c, was independently found to interact with Tpa1 (homologous to Ofd1) (29). To strengthen the parallel between these distantly related yeast species, we constructed a S. cerevisiae strain lacking the YOR051c gene and a strain in which both the TPA1 and YOR051c genes had been simultaneously inactivated. Both strains were transformed with the reporter plasmids and the level of stop codon trans-reading was assayed (Fig. 5). This demonstrated that, similarly to Tpa1, Yor051c deletion increased stop codon readthrough. Moreover, the effects of Tpa1 and Yor051c were not additive suggesting that they were acting in the same pathway. Altogether, these data support the idea that, like for Ofd1 and Nro1 in S. pombe, Yor051c and Tpa1 activities are linked in S. cerevisiae. However, while Nro1 was suggested to antagonize Ofd1 activity, we observed similar, rather than opposite effects of Tpa1 and Yor051C on stop codon trans-reading. Tpa1 deletion affects expression of some genes responsive to oxygen concentration
Because Ofd1 regulates expression of a transcription factor mediating the response to oxygen concentration in S. pombe (23), and given the structural evidence implicating Tpa1 in an oxygen-dependent reaction, we asked whether expression of genes known to be affected by oxygen concentration is influenced by the presence of Tpa1. Genes under the control of Mga2 (57) and Hap1 (58) were tested for this purpose. While we did not detect a significant difference of expression of a β-galactosidase reporter driven by a Mga2 sensitive promoter between wild type and Δtpa1 strains (data not shown), we observed significant changes of expression of reporters which transcription was driven by activating sequences binding Hap1, such as the Cyc1 UAS1 and the Cyb2 UAS1 (Fig. 6). In both cases, β-galactosidase levels were increased roughly 4 fold in the strain lacking Tpa1 compared to the wild type isogenic control. Because Mga2-dependent promoters were not affected in this assay, this result suggests that Tpa1 represses specifically Hap1 activity by an unknown mechanism. Discussion.
In eukaryotes, the detection of the three mRNA stop codons in the A-site of the

ribosome is ensured by two interacting translation termination factors: eRF1 and eRF3. Upon binding to the ribosome, this complex triggers hydrolysis of the ester bond connecting the polypeptide chain to the tRNA located in the ribosomal P-site, thereby releasing the newly synthesized protein. Accurate translation termination requires efficient recognition of stop codons (UAA, UAG, or UGA) by eRF1. Tpa1, a previously uncharacterized protein was recently shown to affect this process (19). In parallel, Ofd1, the Schizosaccharomyces pombe homolog of Tpa1, was implicated in the oxygen-dependent regulation of Sre1, the sterol regulatory element binding protein from fission yeast (23). Due to the fact that independent studies in different organisms on the role of the Tpa1/Ofd1 orthologs yielded non-overlapping results, no unifying view on the Tpa1/Ofd1system has yet been proposed. To progress on this issue, we initiated a structure-based functional analysis of S. cerevisiae Tpa1. We have solved the crystal structure of Tpa1 to 2.8Å resolution. As suggested for S. pombe Ofd1, Tpa1 is composed of two domains. Interestingly, both domains adopt the same Double-Stranded beta-helix (DSBH) fold found in 2OG-Fe(II) dioxygenases (Fig. 1A-B & 2A; (43)). The N-terminal domain (Tpa1N) harbors the catalytic residues (HXD…H triad) present in all members of this class of enzymes (Fig. 1C & 2A) while they are absent in the structurally very similar C-terminal domain. The three invariant residues are localized in the middle of a highly conserved groove of the N-terminal domain (Fig. 1E). The conservation of both fold and catalytically important residues between Tpa1 and prolyl-4-hydrolases (such as human PHD2 and algal prolyl-4-hydroxylase (44,53); Fig. 1C) strongly suggests that Tpa1 possesses a prolyl-4-hydroxylase enzymatic activity. Tpa1 and Yor051c are involved in correct translation termination
We further investigated the involvement of Tpa1 in the control of stop codon readthrough, poly(A) tail length and mRNA stability (19). In our hands, we were not able to detect significant effects of Tpa1 deletion on poly(A) tail length and mRNA stability. However, consistent with the previous results of Keeling and coworkers (19), the deletion of TPA1 gene resulted in a
roughly 3-fold increase of the readthrough of the UAA stop codon (Fig. 4B-C). This effect was suppressed by transformation of the Δtpa1 strain with a plasmid encoding wild-type Tpa1. Next, we tested whether Tpa1 active site is involved in translation termination efficiency by replacing the invariant HXD motif from the triad involved in metal binding by the AXN sequence. This mutant did not complement the Δtpa1 to restore translation termination efficiency, proving that the integrity of Tpa1 putative active site is necessary for this process, at least at UAA stop codons. As Hughes et al suggested that the N-terminal domain of Ofd1 regulates, in an oxygen-dependent manner, the activity of its C-terminal domain on the degradation of Sre1N (23), we tested the role of the respective deletions of the Tpa1N and Tpa1C domains on stop codon readthrough. Both Tpa1 domains are required for translation termination to occur at a wild type level (Fig. 4C). Finally, in S. pombe, Nro1 was shown to interact with Ofd1 leading to the inhibition of its effect on Sre1N degradation (24). Interestingly, large-scale comprehensive purification in yeast S. cerevisiae using TAP purification identified the Nro1 homolog (Yor051c) as a Tpa1 partner (29). As observed for TPA1 deletion, disruption of the YOR051c gene results in a 3-fold increase of UAA stop codon readthrough compared to wild-type cells. In addition, the combined deletion of both TPA1 and YOR051c genes did not further diminish translation termination efficiency compared to individual deletions, supporting a concomitant action. These data support the idea that Yor051c and Tpa1 are acting in the same pathway in S. cerevisiae. The involvement of Tpa1 in correct translation termination sustains co-immunoprecipitation results showing that Tpa1 interacts with translation termination factors eRF1 and eRF3 as well as with poly(A) binding protein Pab1 in S. cerevisiae (19). Tpa1 is very likely to be a prolyl-4-hydroxylase acting as an oxygen sensor (52) and the integrity of its active site is crucial to reduce stop codon readthrough. Therefore, we suspect that in the presence of oxygen, Tpa1 hydroxylates a proline residue in (a) specific target protein(s). As it needs dioxygen as co-substrate, Tpa1 should be unable to hydroxylate its substrate during hypoxia. As a consequence, this could increase the level of inaccurate translation termination. Whether

Tpa1 affects readthrough by modifying directly eRF1, eRF3 or Pab1, or another protein controlling these factors, remains to be investigated.
Yor051c, the S. cerevisiae Nro1 ortholog, has been identified in a genetic-wide screen aimed at identifying genes from S. cerevisiae that affect replication of a positive-strand RNA virus (Brome mosaic virus or BMV) (59). Among the nearly 100 genes whose deletion either enhanced or reduced viral replication, many are involved in transcription, RNA processing, translation and protein degradation. Deletion of YOR051c gene resulted in enhancement of viral replication, suggesting that Yor051c inhibits replication. This is not the first time that proteins from yeast but also human are identified to affect both RNA virus replication and translation. This is for instance the case for yeast proteins Pat1, Lsm1 and Lsm6, functioning in mRNA decapping. These proteins are involved in BMV RNA translation and are recruited from translation complexes to RNA replication complexes (60,61). Similarly, in human cells, translation and replication of hepatitis C virus (HCV) RNA depends on Pat1, Rck/P54 (Dhh1 in yeast) and the Lsm1-7 complex (62). However, it remains to be determined whether or not there is a functional link between the roles of Yor051c in inhibition of BMV replication and in enhancement of stop codon recognition or whether the effect of Yor051c on BMV results from altered termination of translation of a specific BMV replication factor. Tpa1 is involved in regulation of Hap1 transcription factor activity. The precise identification of the cellular function of Tpa1 is complicated by the observation that the orthologous Ofd1 is implicated in an apparently completely different process i.e. the oxygen-dependent regulation of Sre1, the fission yeast sterol regulatory element binding protein (or SREBP; (23)). The S. pombe SREBP transcription factor is anchored at the endoplasmic reticulum (ER) membrane, but upon sterol depletion, it is cleaved and its N-terminal domain enters the nucleus to activate transcription of genes encoding sterol biosynthesis enzymes, as observed for its mammalian counterpart (25,28). In addition, in response to low oxygen levels, Sre1 stimulates transcription of genes
required for adaptation to hypoxia. In budding yeast, several transcription factors (Upc2, Ecm22, Mga2, Hap1, Rox1, …) have been implicated in the response to sterol depletion and/or hypoxia but none present significant sequence homology with members of the SREBP family (57,58,63,64). We have therefore investigated the effect of the deletion of TPA1 on genes controlled by Mga2 and Hap1, as representative of transcription factor affecting the response to oxygen concentration. Mga2 is an ER membrane protein activated during hypoxia by processing of the full-length protein into a smaller soluble transcription factor domain that is addressed to the nucleus similarly to S. pombe Sre1 and mammalian SREBP (57,64,65). Hap1 is a transcription factor involved in the complex regulation of gene expression in response to levels of heme and oxygen (66,67). Our choice to study Hap1 was also motivated by the observation that the deletion of TPA1 gene was shown to rescue the synthetic lethality phenotype of a Δhap1Δupc2Δecm22 triple mutant (68). No effect of the deletion of TPA1 on the expression of reporters regulated by the Mga2 transcription factor was detected (data not shown). In contrast, deletion of TPA1 generated significant changes (4-fold increase) in transcription of two reporters driven by the Cyc1 UAS1 or the Cyb2 UAS1 (Fig. 6). Expression of both CYC1 and CYB2 is stimulated by these sequences in the presence of oxygen through the Hap1 transcription factor (69-71). Our analysis therefore suggests that Tpa1 represses specifically Hap1 activity by an unknown mechanism.
The functional link between Tpa1 and Hap1 is reminiscent of that between Ofd1 and Sre1N (23). Interestingly, another prolyl-4-hydroxylase, human PHD2, has been implicated in the control of the activity of a family of transcription factors: HIF (for hypoxia-inducible factors) by hydroxylation of conserved proline residues that targets HIF factors to ubiquitin mediated degradation (for a review, see (72)). We have experimental evidence that in S. cerevisiae, Tpa1 also influences the activity of a transcription factor: Hap1. This latter is structurally unrelated to SREBPs and HIF. However, Hap1 is degraded into smaller fragments in the absence of oxygen while only the unprocessed form is observed in the presence of oxygen (58). We have observed that Tpa1 represses the activity

of Hap1 (Fig. 6). Tpa1 functions very likely as an oxygen sensor through its N-terminal PHD domain but the role of Tpa1 in modulating the activity of Hap1 remains to be clarified. Conclusion The resolution of the crystal structure of Tpa1 revealed that this protein contains two DSBH domains and that it is a putative prolyl-4-hydroxylase whose active site is located in its N-terminal domain. Tpa1 probably acts as an oxygen sensor system and our genetic experiments show that it is involved in two
unrelated cellular processes. First, Tpa1 reduces stop codon readthrough and its enzymatic activity is critical for this function. Second, Tpa1 participates in the regulation of the transcription of genes by the Hap1 transcription factor and represses this latter. Whether the implication of Tpa1 in these two pathways is mediated by a unique substrate protein that will further act on distinct proteins involved in these two processes or by at least two substrates implicated in each process, remains to be elucidated.
References. 1. Gallie, D. R. (1991) Genes Dev 5, 2108-2116 2. Pestova, T., Lorsch, J. R., and Hellen, C. U. T. (2007) The mechanism of translation initiation
in eukaryotes. in Translational Control in Biology and Medecine (Mathews, M. B., Sonenberg, N., Hershey, J.W.B. ed.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor. pp 87-128
3. Frolova, L., Le Goff, X., Rasmussen, H. H., Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A. L., Celis, J. E., Philippe, M., and et al. (1994) Nature 372, 701-703
4. Kisselev, L., Ehrenberg, M., and Frolova, L. (2003) Embo J 22, 175-182 5. Kisselev, L. L., and Buckingham, R. H. (2000) Trends Biochem Sci 25, 561-566 6. Alkalaeva, E. Z., Pisarev, A. V., Frolova, L. Y., Kisselev, L. L., and Pestova, T. V. (2006)
Cell 125, 1125-1136 7. Stansfield, I., Jones, K. M., Kushnirov, V. V., Dagkesamanskaya, A. R., Poznyakovski, A. I.,
Paushkin, S. V., Nierras, C. R., Cox, B. S., Ter-Avanesyan, M. D., and Tuite, M. F. (1995) Embo J 14, 4365-4373
8. Zhouravleva, G., Frolova, L., Le Goff, X., Le Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995) Embo J 14, 4065-4072
9. Frolova, L., Le Goff, X., Zhouravleva, G., Davydova, E., Philippe, M., and Kisselev, L. (1996) Rna 2, 334-341
10. Bonetti, B., Fu, L., Moon, J., and Bedwell, D. M. (1995) J Mol Biol 251, 334-345 11. von der Haar, T., and Tuite, M. F. (2007) Trends Microbiol 15, 78-86 12. Amrani, N., Ganesan, R., Kervestin, S., Mangus, D. A., Ghosh, S., and Jacobson, A. (2004)
Nature 432, 112-118 13. Cosson, B., Couturier, A., Chabelskaya, S., Kiktev, D., Inge-Vechtomov, S., Philippe, M., and
Zhouravleva, G. (2002) Mol Cell Biol 22, 3301-3315 14. Graille, M., Heurgue-Hamard, V., Champ, S., Mora, L., Scrima, N., Ulryck, N., van
Tilbeurgh, H., and Buckingham, R. H. (2005) Mol Cell 20, 917-927 15. Heurgue-Hamard, V., Champ, S., Engstrom, A., Ehrenberg, M., and Buckingham, R. H.
(2002) Embo J 21, 769-778 16. Heurgue-Hamard, V., Champ, S., Mora, L., Merkulova-Rainon, T., Kisselev, L. L., and
Buckingham, R. H. (2005) J Biol Chem 280, 2439-2445 17. Heurgue-Hamard, V., Graille, M., Scrima, N., Ulryck, N., Champ, S., van Tilbeurgh, H., and
Buckingham, R. H. (2006) J Biol Chem 281, 36140-36148 18. Mora, L., Heurgue-Hamard, V., de Zamaroczy, M., Kervestin, S., and Buckingham, R. H.
(2007) J Biol Chem 282, 35638-35645 19. Keeling, K. M., Salas-Marco, J., Osherovich, L. Z., and Bedwell, D. M. (2006) Mol Cell Biol
26, 5237-5248 20. Aravind, L., and Koonin, E. V. (2001) Genome Biol 2, RESEARCH0007 21. Clifton, I. J., McDonough, M. A., Ehrismann, D., Kershaw, N. J., Granatino, N., and
Schofield, C. J. (2006) J Inorg Biochem 100, 644-669 22. Chang, B., Chen, Y., Zhao, Y., and Bruick, R. K. (2007) Science 318, 444-447

23. Hughes, B. T., and Espenshade, P. J. (2008) Embo J 27, 1491-1501 24. Lee, C. Y., Stewart, E. V., Hughes, B. T., and Espenshade, P. J. (2009) Embo J 28, 135-143 25. Espenshade, P. J., and Hughes, A. L. (2007) Annu Rev Genet 41, 401-427 26. Todd, B. L., Stewart, E. V., Burg, J. S., Hughes, A. L., and Espenshade, P. J. (2006) Mol Cell
Biol 26, 2817-2831 27. Hughes, A. L., Lee, C. Y., Bien, C. M., and Espenshade, P. J. (2007) J Biol Chem 282, 24388-
24396 28. Hughes, A. L., Todd, B. L., and Espenshade, P. J. (2005) Cell 120, 831-842 29. Krogan, N. J., Cagney, G., Yu, H., Zhong, G., Guo, X., Ignatchenko, A., Li, J., Pu, S., Datta,
N., Tikuisis, A. P., Punna, T., Peregrin-Alvarez, J. M., Shales, M., Zhang, X., Davey, M., Robinson, M. D., Paccanaro, A., Bray, J. E., Sheung, A., Beattie, B., Richards, D. P., Canadien, V., Lalev, A., Mena, F., Wong, P., Starostine, A., Canete, M. M., Vlasblom, J., Wu, S., Orsi, C., Collins, S. R., Chandran, S., Haw, R., Rilstone, J. J., Gandi, K., Thompson, N. J., Musso, G., St Onge, P., Ghanny, S., Lam, M. H., Butland, G., Altaf-Ul, A. M., Kanaya, S., Shilatifard, A., O'Shea, E., Weissman, J. S., Ingles, C. J., Hughes, T. R., Parkinson, J., Gerstein, M., Wodak, S. J., Emili, A., and Greenblatt, J. F. (2006) Nature 440, 637-643
30. Hendrickson, W. A., Horton, J. R., and LeMaster, D. M. (1990) Embo J 9, 1665-1672 31. Kabsch, W. (1993) J. Appl. Cryst. 26, 795-800 32. Schneider, T. R., and Sheldrick, G. M. (2002) Acta Crystallogr D Biol Crystallogr 58, 1772-
1779 33. Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003) Acta
Crystallogr D Biol Crystallogr 59, 2023-2030 34. Terwilliger, T. C. (1999) Acta Crystallogr D Biol Crystallogr 55, 1863-1871 35. Cowtan, K. (2006) Acta Crystallogr D Biol Crystallogr 62, 1002-1011 36. Emsley, P., and Cowtan, K. (2004) Acta Crystallogr D Biol Crystallogr 60, 2126-2132 37. Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr D Biol
Crystallogr 53, 240-255 38. Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty,
N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., and Terwilliger, T. C. (2002) Acta Crystallogr D Biol Crystallogr 58, 1948-1954
39. Christianson, T. W., Sikorski, R. S., Dante, M., Shero, J. H., and Hieter, P. (1992) Gene 110, 119-122
40. Ramil, E., Agrimonti, C., Shechter, E., Gervais, M., and Guiard, B. (2000) Mol Microbiol 37, 1116-1132
41. Dziembowski, A., Ventura, A. P., Rutz, B., Caspary, F., Faux, C., Halgand, F., Laprevote, O., and Seraphin, B. (2004) Embo J 23, 4847-4856
42. Kushnirov, V. V. (2000) Yeast 16, 857-860 43. Hausinger, R. P. (2004) Crit Rev Biochem Mol Biol 39, 21-68 44. McDonough, M. A., Li, V., Flashman, E., Chowdhury, R., Mohr, C., Lienard, B. M., Zondlo,
J., Oldham, N. J., Clifton, I. J., Lewis, J., McNeill, L. A., Kurzeja, R. J., Hewitson, K. S., Yang, E., Jordan, S., Syed, R. S., and Schofield, C. J. (2006) Proc Natl Acad Sci U S A 103, 9814-9819
45. Albuquerque, C. P., Smolka, M. B., Payne, S. H., Bafna, V., Eng, J., and Zhou, H. (2008) Mol Cell Proteomics 7, 1389-1396
46. Cleasby, A., Wonacott, A., Skarzynski, T., Hubbard, R. E., Davies, G. J., Proudfoot, A. E., Bernard, A. R., Payton, M. A., and Wells, T. N. (1996) Nat Struct Biol 3, 470-479
47. Just, V. J., Stevenson, C. E., Bowater, L., Tanner, A., Lawson, D. M., and Bornemann, S. (2004) J Biol Chem 279, 19867-19874
48. Steiner, R. A., Kalk, K. H., and Dijkstra, B. W. (2002) Proc Natl Acad Sci U S A 99, 16625-16630
49. Pang, H., Bartlam, M., Zeng, Q., Miyatake, H., Hisano, T., Miki, K., Wong, L. L., Gao, G. F., and Rao, Z. (2004) J Biol Chem 279, 1491-1498
50. Cassab, G. I. (1998) Annu Rev Plant Physiol Plant Mol Biol 49, 281-309 51. Myllyharju, J. (2003) Matrix Biol 22, 15-24 52. Schofield, C. J., and Ratcliffe, P. J. (2004) Nat Rev Mol Cell Biol 5, 343-354

53. Koski, M. K., Hieta, R., Hirsila, M., Ronka, A., Myllyharju, J., and Wierenga, R. K. (2009) J Biol Chem 284, 25290-25301
54. Chowdhury, R., McDonough, M. A., Mecinovic, J., Loenarz, C., Flashman, E., Hewitson, K. S., Domene, C., and Schofield, C. J. (2009) Structure 17, 981-989
55. Sundheim, O., Vagbo, C. B., Bjoras, M., Sousa, M. M., Talstad, V., Aas, P. A., Drablos, F., Krokan, H. E., Tainer, J. A., and Slupphaug, G. (2006) Embo J 25, 3389-3397
56. Puig, O., Caspary, F., Rigaut, G., Rutz, B., Bouveret, E., Bragado-Nilsson, E., Wilm, M., and Seraphin, B. (2001) Methods (San Diego, Calif 24, 218-229
57. Jiang, Y., Vasconcelles, M. J., Wretzel, S., Light, A., Martin, C. E., and Goldberg, M. A. (2001) Mol Cell Biol 21, 6161-6169
58. Hon, T., Dodd, A., Dirmeier, R., Gorman, N., Sinclair, P. R., Zhang, L., and Poyton, R. O. (2003) J Biol Chem 278, 50771-50780
59. Kushner, D. B., Lindenbach, B. D., Grdzelishvili, V. Z., Noueiry, A. O., Paul, S. M., and Ahlquist, P. (2003) Proc Natl Acad Sci U S A 100, 15764-15769
60. Diez, J., Ishikawa, M., Kaido, M., and Ahlquist, P. (2000) Proc Natl Acad Sci U S A 97, 3913-3918
61. Noueiry, A. O., Diez, J., Falk, S. P., Chen, J., and Ahlquist, P. (2003) Mol Cell Biol 23, 4094-4106
62. Scheller, N., Mina, L. B., Galao, R. P., Chari, A., Gimenez-Barcons, M., Noueiry, A., Fischer, U., Meyerhans, A., and Diez, J. (2009) Proc Natl Acad Sci U S A 106, 13517-13522
63. Davies, B. S., and Rine, J. (2006) Genetics 174, 191-201 64. Hoppe, T., Matuschewski, K., Rape, M., Schlenker, S., Ulrich, H. D., and Jentsch, S. (2000)
Cell 102, 577-586 65. Jiang, Y., Vasconcelles, M. J., Wretzel, S., Light, A., Gilooly, L., McDaid, K., Oh, C. S.,
Martin, C. E., and Goldberg, M. A. (2002) Eukaryot Cell 1, 481-490 66. Pfeifer, K., Kim, K. S., Kogan, S., and Guarente, L. (1989) Cell 56, 291-301 67. Zhang, L., and Hach, A. (1999) Cell Mol Life Sci 56, 415-426 68. Valachovic, M., Bareither, B. M., Shah Alam Bhuiyan, M., Eckstein, J., Barbuch, R.,
Balderes, D., Wilcox, L., Sturley, S. L., Dickson, R. C., and Bard, M. (2006) Genetics 173, 1893-1908
69. Pfeifer, K., Arcangioli, B., and Guarente, L. (1987) Cell 49, 9-18 70. Pfeifer, K., Prezant, T., and Guarente, L. (1987) Cell 49, 19-27 71. Lodi, T., and Guiard, B. (1991) Mol Cell Biol 11, 3762-3772 72. Kaelin, W. G. (2005) Annu Rev Biochem 74, 115-128 Acknowledgments We are indebted to K. Blondeau for her help with SeMet labeling and V. Henriot for some cloning. We thank D.M. Bedwell, C.E. Martin, M. Globerg and B. Guiard for plasmids and yeast strains. The work in the group of B. Séraphin was supported by La Ligue contre le Cancer ‘Equipe Labellisée 2008’ and the CNRS. BS, HvT and MG were supported by the European Union 6th Framework programs ‘3D-Repertoire’ (LSHG-CT-2005-512028). MG thanks the CNRS and the Agence Nationale pour la Recherche (Grant ANR-06-BLAN-0075-02). JH and DR are recipients of PhD fellowships from Université Paris Sud-11. Legends to figures. Figure 1. S. cerevisiae Tpa1 structure. A. Ribbon representation of the Tpa1 protein. B. Topology diagram. The same color code as in panel A is used. C. Stereo view of Tpa1N active site (light brown) superposed on PHD2cat (yellow). The 2OG co-substrate has been modeled into the active site cavity by superposing the structure of a DSBH protein obtained in the presence of 2OG onto the Tpa1N domain. Black and red spheres depict the iron atom and water molecules observed in the crystal structure, respectively. D. Surface representation of the Tpa1 protein. Tpa1N (wheat) and Tpa1C (slate) are joined by a linker (grey). Iron atom is represented as a black sphere.

E. Surface representation of residue conservation mapped at the surface of Tpa1. Coloring is from blue (highly conserved) to grey (low conservation). F. Representation of electrostatic potential at the Tpa1 surface. Figure 2. A. Stereo view representation of the superposition of Tpa1N (light brown) on Tpa1C (blue) B. Surface representation of residue conservation mapped at the surface of the Tpa1C domain. The same color code as in Fig. 1E is used. C. Tpa1C interacting surface with Tpa1N D. Ribbon representation of the Tpa1 homodimer observed in the crystal. One monomer is shown in blue and the other one in green. Tpa1N and Tpa1C are in light and dark colors, respectively. E. Surface representation of residue conservation mapped at the surface of Tpa1 homodimer. The same color code as in Fig. 1E is used. F. Size-exclusion chromatography as followed by a triple detector array of Tpa1. For clarity, only, the UV absorption (280nm) for the eluted sample (left y axis) and molecular mass calculated from light scattering (right y axis, logarithmic scale) are shown. G. Surface representation of Tpa1 active site (same color code as Fig. 1E) with the HIFα CODD (yellow) and (Ser-Pro)5 (pink) peptides shown in ribbon. The hydroxylated proline is shown in sticks. The iron atom is shown as a black sphere. H. Same representation as panel G. Tpa1N and Tpa1C are colored grey and beige, respectively. The βB-βC β-hairpin is shown in green. Figure 3. Multiple sequence alignment of Tpa1 Eukaryotic orthologs of Tpa1 were aligned using ClustalW. Strictly conserved residues are in white on a black background. Partially conserved amino acids are boxed. Secondary structure elements assigned from the S. cerevisiae Tpa1 structure are indicated above the alignment. Figure 4: Requirement of both Tpa1 domain and of conserved residues in its putative catalytic center for stop codon readthrough. A. Structure of the reporter used to measure stop codon readthrough. Synthesis of mRNAs encoding both the Renilla and firefly luciferases is driven by a PGK1 promoter (P PGK). The two ORFs are separated either by a CAA codon encoding glutamine, of by the UAA stop codon. Percent of stop codon readthrough is calculated as the ratio of firefly luciferase to Renilla luciferase for the stop codon containing construct divided by the ratio of firefly luciferase to Renilla luciferase for the construct encoding glutamine x 100. B. Percent of UAA readthrough in wild type (PSI+) cells, or in strain deleted for tpa1 carrying either an empty vector, a plasmid encoding wild type TPA1 gene, or the tpa1 H159A/D161N is plotted. Standard deviation from 3 experiments is indicated. C. Same as in B, except that plasmids encoding either Tpa1N or Tpa1C were used. D. Western blot analysis of total cellular protein extracted from cells expressing TPA1-TAP, tpa1N-TAP, tpa1-H159A/D161N-TAP or tpa1C-TAP. Figure 5: Yor051c affects stop codon readthrough The readthrough of stop codon in the wild type strain, in a stain lacking Tpa1, in a strain lacking Yor051c, or in a strain lacking both factors was measured as described above (Fig. 4). Figure 6: TPA1 is required for full activity of Hap1-dependent promoters A. Structure of the two reporters used for this assay. B. The levels of β-galactosidase activity measured for the reporter with the Cyc1 UAS1 or the Cyb2 UAS in either the wild type or the Δtpa1 strains are plotted with the standard deviation from 3 experiments.
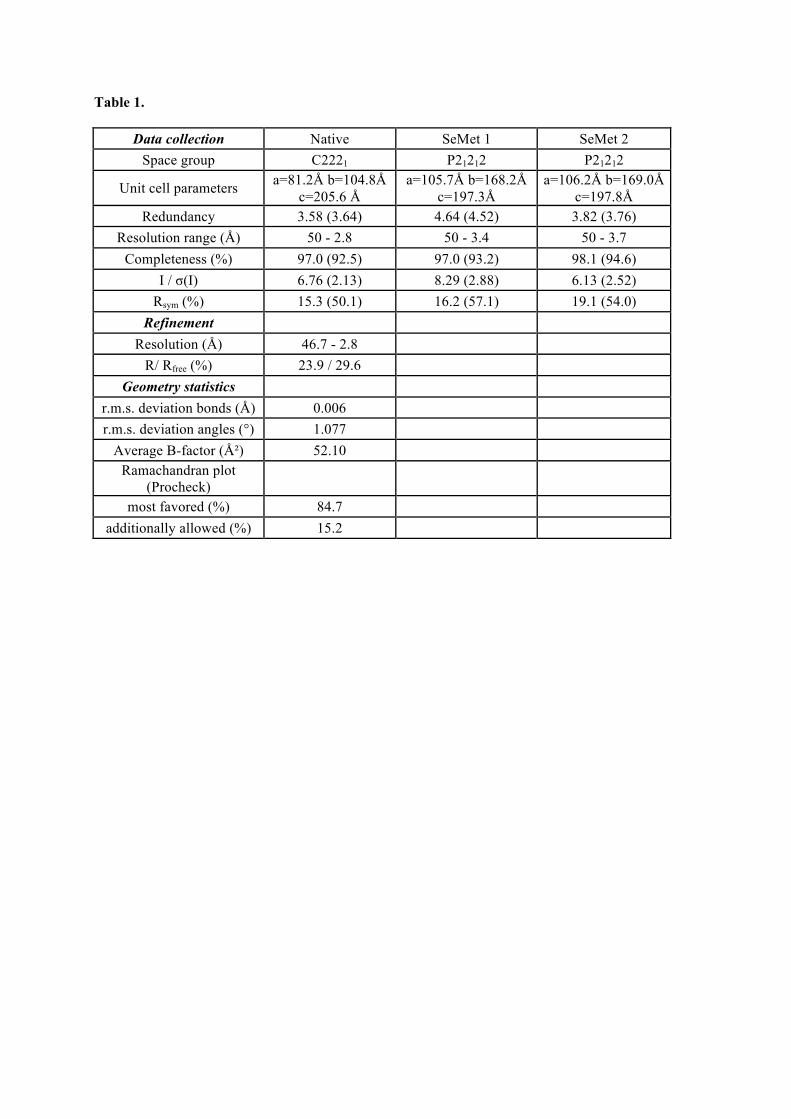
Table 1.
Data collection Native SeMet 1 SeMet 2 Space group C2221 P21212 P21212
Unit cell parameters a=81.2Å b=104.8Å c=205.6 Å
a=105.7Å b=168.2Å c=197.3Å
a=106.2Å b=169.0Å c=197.8Å
Redundancy 3.58 (3.64) 4.64 (4.52) 3.82 (3.76) Resolution range (Å) 50 - 2.8 50 - 3.4 50 - 3.7
Completeness (%) 97.0 (92.5) 97.0 (93.2) 98.1 (94.6) I / σ(I) 6.76 (2.13) 8.29 (2.88) 6.13 (2.52)
Rsym (%) 15.3 (50.1) 16.2 (57.1) 19.1 (54.0) Refinement
Resolution (Å) 46.7 - 2.8 R/ Rfree (%) 23.9 / 29.6
Geometry statistics r.m.s. deviation bonds (Å) 0.006 r.m.s. deviation angles (°) 1.077
Average B-factor (Ų) 52.10 Ramachandran plot
(Procheck)
most favored (%) 84.7 additionally allowed (%) 15.2
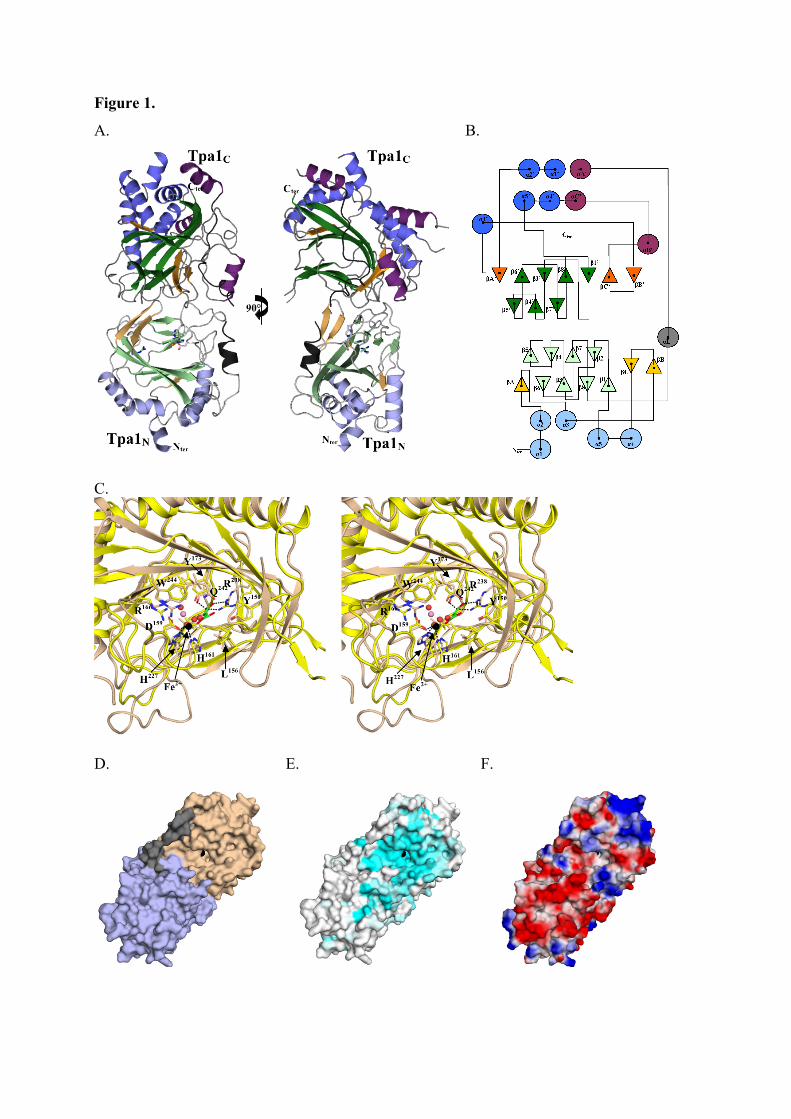
Figure 1.
A. B.
C.
D. E. F.
90°
Tpa1C Tpa1C
Tpa1N Tpa1N Nter Nter
Cter Cter
H161
D159
Q242
L156
R238
Y173
W244
Y150
R
166
Fe2+
H
227
H161
D159
Q242
L156
R238
Y173
W244
Y150
R
166
Fe2+
H
227
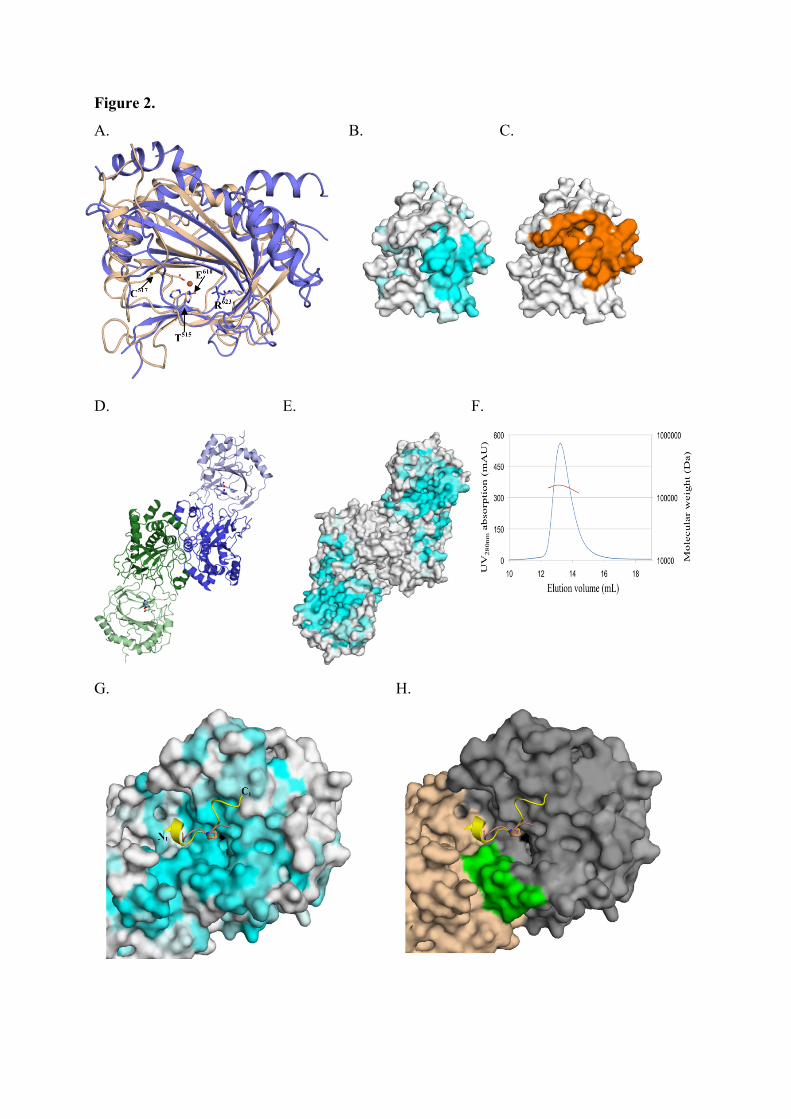
Figure 2.
A. B. C.
D. E. F.
G. H.
C517
E610
T515
R623
Nt
Ct

Figure 3.
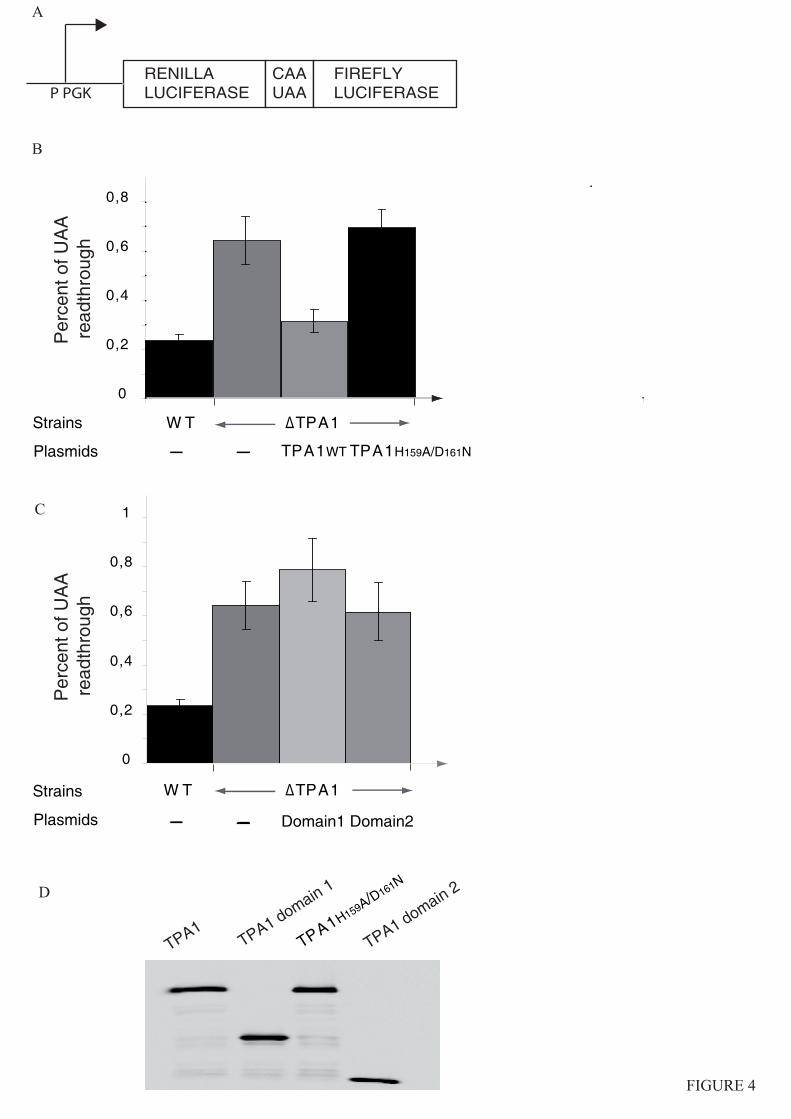
B
FIGURE 4
C
D
TPA1 TPA1 domain 1
TPA1H159A/D161N
TPA1 domain 2
W T ΔTPA1
0
0,2
0,4
0,6
0,8
1
RENILLALUCIFERASE
CAAUAAP PGK
A
Strains
0
0,2
0,4
0,6
0,8
Plasmids TPA1WT TPA1H159A/D161N
Strains
Plasmids
W T ΔTPA1
Domain1 Domain2
Per
cent
of U
AA
read
thro
ugh
Per
cent
of U
AA
read
thro
ugh
FIREFLYLUCIFERASE
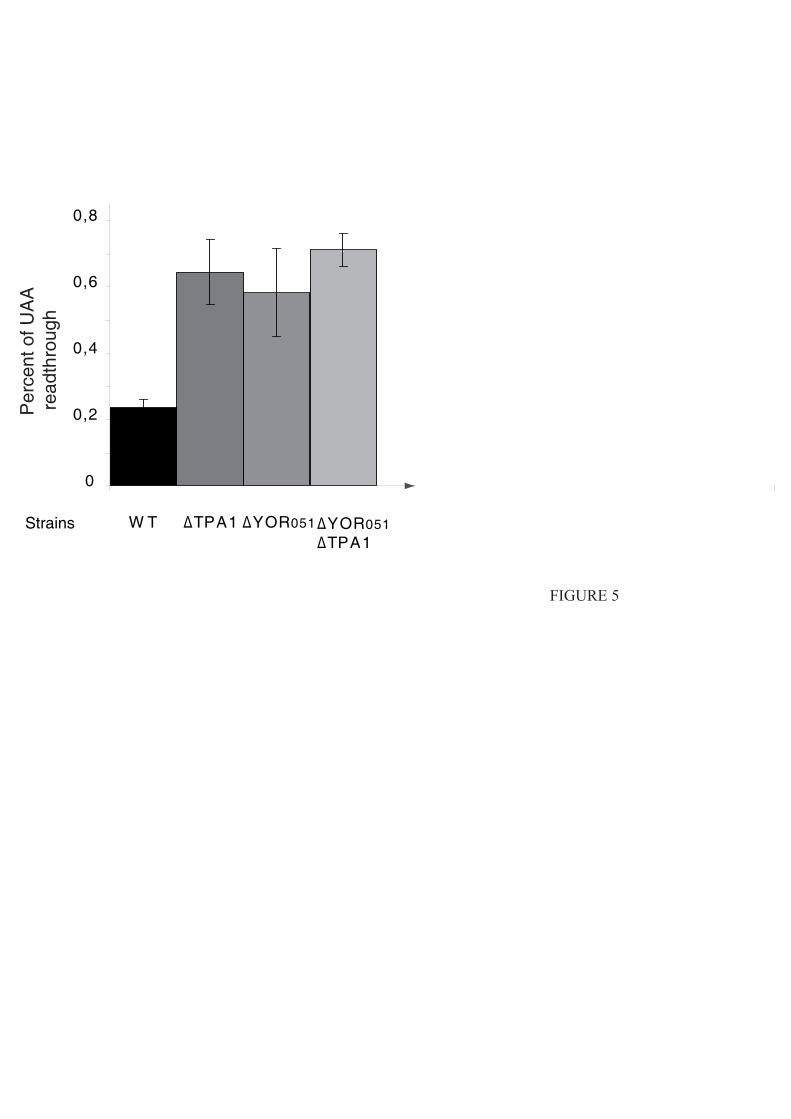
0
0,2
0,4
0,6
0,8
W T ΔTPA1 ΔYOR051
FIGURE 5
Per
cent
of U
AA
read
thro
ugh
Strains ΔYOR051ΔTPA1
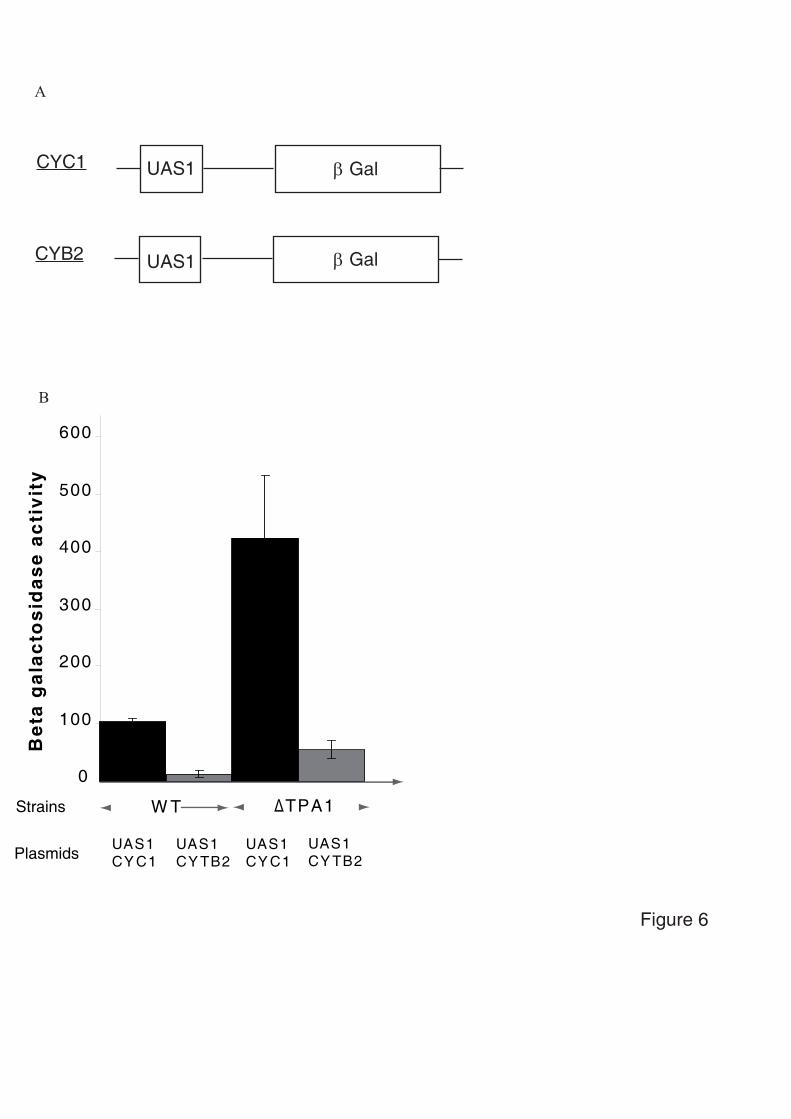
0
100
200
300
400
500
600
UAS1 CY C1
UAS1 CY TB2
W T ΔTPA1Strains
Plasmids
Bet
a g
alac
tosi
das
e ac
tivi
ty
β GalUAS1CYC1
β GalUAS1CYB2
A
B
UAS1 CY C1
UAS1 CY TB2
Figure 6


114
Pendant la rédaction de notre article une autre équipe a publié l'analyse de la structure
cristallographique de Tpa1 de S. cerevisiae en complexe avec le Fer ou avec le Fer et le 2-
oxoglutarate (Kim et al., 2010). Les auteurs de l'étude ont utilisé une version tronquée des 20
premiers résidus (invisibles dans notre structure car trop flexibles), taggée en C-terminal; la
protéine est exprimée à partir d'un vecteur pET21 à une température d'induction de 30°C. La
purification est réalisée en tampon Tris PH7,9 50mM, NaCl 500mM, Glycérol 10%, en deux
étapes : une NiNTA suivie d'une chromatographie d'exclusion de taille S200 HL. Ces
différences de méthode par rapport à notre approche peuvent expliquer les différences
obtenues en cristallogenèse (solution de cristallisation pour la protéine native : 100mM Tris
pH8,5, 25% PEG 3350, 200mM Li(SO4)2, 0,14mM Fe-ascorbate) et en cristallographie
(groupe d'espace P321, résolution maximale de 2,5Å et 1,8Å pour les structures de protéine
native). Les auteurs signalent qu'une amélioration significative de la diffraction a été obtenue
par ajout de Fe-ascorbate dans la solution cryoprotectante, un paramètre d'optimisation que
nous n'avons pas testé.
La comparaison de nos coordonnées avec les leurs montre une grande similarité
(rmsd=0,621Å pour 517 Cα) (Figure 35). La fixation du 2-oxoglutarate dans le site
dioxygénase n'induit pas de changement notable de la structure.
Leur structure inclut 28 résidus qui ne sont pas définis dans nos cartes, dont une longue
boucle (acides aminés 93 à 110) à proximité du site dioxygénase; du fait de sa position et de
sa flexibilité apparente cette boucle pourrait réguler l'accessibilité du site actif.
Bien que le groupe d'espace de nos cristaux et des leurs soient différents (P212121 ou
C2122 et P321) le mode d'empilement des molécules dans le cristal est identique : dans tous
les cas Tpa1 cristallise en formant les mêmes contacts. La formation du dimère a été évaluée

115
in vitro par des mesures d'ultracentrifugation à l'équilibre qui ont confirmé la formation du
dimère.
Les auteurs de l'étude ont construit 4 ions sulfates sur la face opposée au site actif. Les
résidus Lys152, Lys182, Arg190 et Lys236, impliqués dans la fixation des sulfates, sont
faiblement conservés. Les auteurs proposent néanmoins que ces résidus font partie d'une
surface chargée positivement susceptible d'interagir avec l'ARNm. Cette hypothèse a été
testée par des expériences de retard sur gel de poly-r(A) mis en présence de quantités
croissantes de Tpa1. Les résultats montrent une interaction directe de Tpa1 avec le poly-r(A)
selon un mode qui reste à préciser.
116
Figure 35 : Comparaison des structures de Tpa1.
Alignement des structures de Tpa1 de notre étude (Henri et al., 2010) (en bleu) sur la structure de l'équipe de Kim et collaborateurs (en blanc). Les coordonnées des atomes sont très proches. Les résidus supplémentaires de leur structure sont colorés en vert.
y - 90°

117
1.2. Structure de Nro1, orthologue d'Ett1
Notre étude a mis en évidence un effet de la délétion d'Ett1 sur la terminaison de la
traduction, de façon similaire à la délétion de Tpa1 : Ett1 est nécessaire pour une terminaison
de la traduction efficace, et ce dans la même voie que Tpa1 (Henri, 2010).
Ett1 est également prédite nucléaire, et interagit avec le pore nucléaire, à l’instar de Tpa1
(Rout et al., 2000). Ett1 inhibe la réplication du Brome Mosaic Virus (BMV) lors de la
transfection de S. cerevisiae (Kushner et al., 2003). Les protéines connues pour avoir le même
effet sur la réplication du BMV sont impliquées dans le métabolisme de l'ARNm (Lsm1/6,
Pat1, Dhh1), dans la réplication (facteurs d'adressage au protéasome) et dans le maintien de la
composition membranaire (Ole1, Acb1, Scs2).
La fonction d'Ett1 chez S. cerevisiae est obscure. Nous avons envisagé une
caractérisation structurale mais nous n'avons pas pu obtenir de cristaux de cette protéine.
L'homologue d'Ett1 chez S. pombe est Nro1 et l'implication de celle-ci dans l'adaptation
cellulaire à l'hypoxie a fait l'objet d'une étude détaillée. Nro1 est une protéine non essentielle
de 393 acides aminés également prédite pour interagir avec le complexe du pore nucléaire
(Chen et al., 2004). J'ai exprimé et purifié Nro1, puis résolu sa structure.
1.2.1. Procédure expérimentale
A. Expression et purification de Nro1
a. Design de la séquence d'expression
L'analyse de la répartition des résidus hydrophobes et polaires le long de la séquence de
la protéine Nro1 de S. pombe par le programme HCA montre que peu de résidus hydrophobes
sont présents au niveau des 54 résidus N-terminaux de Nro1 suggérant l’absence de structure
secondaire et une forte flexibilité (Figure 36). Nous avons par conséquent exprimé une

118
version tronquée de ces résidus (Nro1ΔN54); le vecteur de clonage utilisé a été synthétisé par
un fournisseur (GenScript) de manière à coder pour Nro1ΔN54-His6 en tandem avec son
partenaire Ofd1-Strep.
b. Expression
L'expression dans la souche E. coli Rosetta pLysS induite 20h à 15°C permet de produire
les deux protéines Ofd1 et Nro1 en quantités importantes. Après cassage des cellules en
tampon A, la purification procède en quatre étapes : NiNTA, chromatographie d'exclusion de
taille Superdex 200 16/60 HiLoad, puis chromatographie d'échange d'ion MonoQ; cette
dernière n'est pas assez résolutive pour séparer Nro1 des autres protéines de la solution : les
fractions contenant Nro1 sont mélangées et injectées une nouvelle fois sur la colonne dans les
mêmes conditions, permettant cette fois une séparation complète de Nro1 des derniers
contaminants. La protéine Nro1 éluée pure en deuxième MonoQ est dialysée en tampon A,
puis concentrée pour les expériences de cristallisation (Figure 37).
Prédiction structures secondaires par HCA (hydrophobic cluster analysis)
Les résidus colorés en vert sont hydrophobes et susceptibles de former des structures secondaires
Les groupes verticaux prédisent des brins !, les groupes horizontaux des hélices "
hcarequest Drawhca by Luc Canard
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360
M
I
R
R
Q
L
R
A
A
A
L
K
K
Q
Q
Q
L
E
K
Q
K
Q
E
A
Y
E
LN
K
E
NE
N
V
D
N
E
M
E
D
E
M
L
V
Y
E
E
D
N
I
Q
L
W
L
Y
E
M
R
E
K
L
E
N
D
D
I
D
A
V
L
V
FI
H
E
A
D
R
I
L
R
N
E
D
I
L
K
D
F
H
A
A
Y
A
L
L
A
V
E
L
F
E
I
A
Q
K
R
L
K
E
N
E
E
Y
I
D
A
A
I
E
R
A
Q
L
L
D
AN
E
R
L
F
L
A
L
A
R
A
Y
L
E
K
V
R
V
L
V
W
R
H
D
N
E
E
L
A
N
I
V
Q
L
V
N
Y
I
E
K
A
I
Q
Y
L
R
L
A
Q
DE
Y
F
D
A
LD
L
R
L
Y
I
LY
L
F
Q
F
D
Q
F
E
A
F
L
L
D
V
C
I
I
A
L
W
L
K
V
V
D
NA
Y
Y
K
L
I
A
Q
E
A
V
L
N
N
YF
A
E
Y
Y
M
D
L
L
D
N
E
N
V
D
D
L
I
N
K
AW
L
N
N
V
D
W
N
V
I
Y
L
D
KE
R
L
L
K
L
A
D
I
K
M
D
L
A
Q
I
V
Q
D
E
A
Q
D
N
Y
L
KM
I
R
R
Q
L
R
A
A
A
L
K
K
Q
Q
Q
L
E
K
Q
K
Q
E
A
Y
E
LN
K
E
NE
N
V
D
N
E
M
E
D
E
M
L
V
Y
E
E
D
N
I
Q
L
W
L
Y
E
M
R
E
K
L
E
N
D
D
I
D
A
V
L
V
FI
H
E
A
D
R
I
L
R
N
E
D
I
L
K
D
F
H
A
A
Y
A
L
L
A
V
E
L
F
E
I
A
Q
K
R
L
K
E
N
E
E
Y
I
D
A
A
I
E
R
A
Q
L
L
D
AN
E
R
L
F
L
A
L
A
R
A
Y
L
E
K
V
R
V
L
V
W
R
H
D
N
E
E
L
A
N
I
V
Q
L
V
N
Y
I
E
K
A
I
Q
Y
L
R
L
A
Q
DE
Y
F
D
A
LD
L
R
L
Y
I
LY
L
F
Q
F
D
Q
F
E
A
F
L
L
D
V
C
I
I
A
L
W
L
K
V
V
D
NA
Y
Y
K
L
I
A
Q
E
A
V
L
N
N
YF
A
E
Y
Y
M
D
L
L
D
N
E
N
V
D
D
L
I
N
K
AW
L
N
N
V
D
W
N
V
I
Y
L
D
KE
R
L
L
K
L
A
D
I
K
M
D
L
A
Q
I
V
Q
D
E
A
Q
D
N
Y
L
K
370 380 390
N
Y
L
K
E
A
C
N
A
I
K
E
A
QV
E
LD
Y
V
E
F
V
E
A
Y
AN
Y
L
K
E
A
C
N
A
I
K
E
A
QV
E
LD
Y
V
E
F
V
E
A
Y
A
hcarequest Drawhca by Luc Canard
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360
M
I
R
R
Q
L
R
A
A
A
L
K
K
Q
Q
Q
L
E
K
Q
K
Q
E
A
Y
E
LN
K
E
NE
N
V
D
N
E
M
E
D
E
M
L
V
Y
E
E
D
N
I
Q
L
W
L
Y
E
M
R
E
K
L
E
N
D
D
I
D
A
V
L
V
FI
H
E
A
D
R
I
L
R
N
E
D
I
L
K
D
F
H
A
A
Y
A
L
L
A
V
E
L
F
E
I
A
Q
K
R
L
K
E
N
E
E
Y
I
D
A
A
I
E
R
A
Q
L
L
D
AN
E
R
L
F
L
A
L
A
R
A
Y
L
E
K
V
R
V
L
V
W
R
H
D
N
E
E
L
A
N
I
V
Q
L
V
N
Y
I
E
K
A
I
Q
Y
L
R
L
A
Q
DE
Y
F
D
A
LD
L
R
L
Y
I
LY
L
F
Q
F
D
Q
F
E
A
F
L
L
D
V
C
I
I
A
L
W
L
K
V
V
D
NA
Y
Y
K
L
I
A
Q
E
A
V
L
N
N
YF
A
E
Y
Y
M
D
L
L
D
N
E
N
V
D
D
L
I
N
K
AW
L
N
N
V
D
W
N
V
I
Y
L
D
KE
R
L
L
K
L
A
D
I
K
M
D
L
A
Q
I
V
Q
D
E
A
Q
D
N
Y
L
KM
I
R
R
Q
L
R
A
A
A
L
K
K
Q
Q
Q
L
E
K
Q
K
Q
E
A
Y
E
LN
K
E
NE
N
V
D
N
E
M
E
D
E
M
L
V
Y
E
E
D
N
I
Q
L
W
L
Y
E
M
R
E
K
L
E
N
D
D
I
D
A
V
L
V
FI
H
E
A
D
R
I
L
R
N
E
D
I
L
K
D
F
H
A
A
Y
A
L
L
A
V
E
L
F
E
I
A
Q
K
R
L
K
E
N
E
E
Y
I
D
A
A
I
E
R
A
Q
L
L
D
AN
E
R
L
F
L
A
L
A
R
A
Y
L
E
K
V
R
V
L
V
W
R
H
D
N
E
E
L
A
N
I
V
Q
L
V
N
Y
I
E
K
A
I
Q
Y
L
R
L
A
Q
DE
Y
F
D
A
LD
L
R
L
Y
I
LY
L
F
Q
F
D
Q
F
E
A
F
L
L
D
V
C
I
I
A
L
W
L
K
V
V
D
NA
Y
Y
K
L
I
A
Q
E
A
V
L
N
N
YF
A
E
Y
Y
M
D
L
L
D
N
E
N
V
D
D
L
I
N
K
AW
L
N
N
V
D
W
N
V
I
Y
L
D
KE
R
L
L
K
L
A
D
I
K
M
D
L
A
Q
I
V
Q
D
E
A
Q
D
N
Y
L
K
370 380 390
N
Y
L
K
E
A
C
N
A
I
K
E
A
QV
E
LD
Y
V
E
F
V
E
A
Y
AN
Y
L
K
E
A
C
N
A
I
K
E
A
QV
E
LD
Y
V
E
F
V
E
A
Y
A
Figure 36 : Prédiction de structures secondaires par analyse des clusters hydrophobes (HCA)
Les résidus colorés en vert sont hydrophobes et susceptibles de former des structures secondaires.
Les groupes verticaux prédisent des brins β, les groupes horizontaux des hélices α.
Les 54 résidus N-terminaux (encadrés en rouge) sont prédits peu structurés, justifiant de travailler sur une troncation ΔN54 de la protéine.
119
Expression par polycistron en Rosetta 15°C
Nro1!N54-His6 + Ofd1-Strep
Figure 37 :
(en haut) Purification de Nro1ΔN54 en quatre étapes : NiNTA-SEC-MonoQ-MonoQ; Nro1ΔN54 est encadrée en blanc dans les fractions éluées; la protéine éluée pure en sortie de deuxième MonoQ est dyalisée en tampon A et concentrée.
(en bas) Cristallisation de Nro1ΔN54 : premières pistes et cristaux optimisés.

120
B. Cristallisation
La protéine concentrée à 13mg/mL est utilisée pour la recherche de conditions initiales de
cristallisation sur des screens larges (Classics, MBI, Nucleix, ProComplex). Plusieurs
conditions contenant du sulfate d'ammonium favorisent la nucléation mais les cristaux sont
toujours nombreux et de petite taille.
La condition retenue pour optimisation contenait 50mM d'Hepes pH 7, 1,6M de sulfate
d’ammonium et 10mM de MgCl2 (Figure 37). Les autres conditions initiales de cristallisation
n'ont pas permis l'obtention de formes cristallines exploitables.
Dans un premier temps, j'ai diminué la concentration de la protéine purifiée à 7,2mg/mL
de manière à ralentir la croissance des cristaux après nucléation. Les cristaux restaient
irréguliers (cassures) et maclés (multiples cristaux à partir d'un unique point de nucléation).
La recherche d'agents de cristallisation supplémentaires sur un screen d’additifs a permis
d’identifier deux précipitants améliorant l’aspect macroscopique des cristaux : le 1,4-
butanediol (4%) et la polyvinylpyrrolidone (PVP, 3%). Les cristaux obtenus étaient des pavés
épais et réguliers (forme 2). J'ai en parallèle augmenté le volume des gouttes de cristallisation
à partir de la condition de départ et obtenu une nouvelle forme cristalline ressemblant à une
double pyramide à base hexagonale (forme 1). Le streak-seeding a favorisé l'apparition de
quelques cristaux de grande taille.
La protéine marquée par des Séléno-Méthionines a été exprimée, purifiée et cristallisée
selon le même protocole.
C. Diffraction et collecte des données
Les cristaux obtenus ont été incubés dans une solution contenant le mélange de
cristallisation et des concentrations croissantes de glycérol ou d'éthylène glycol (15 puis
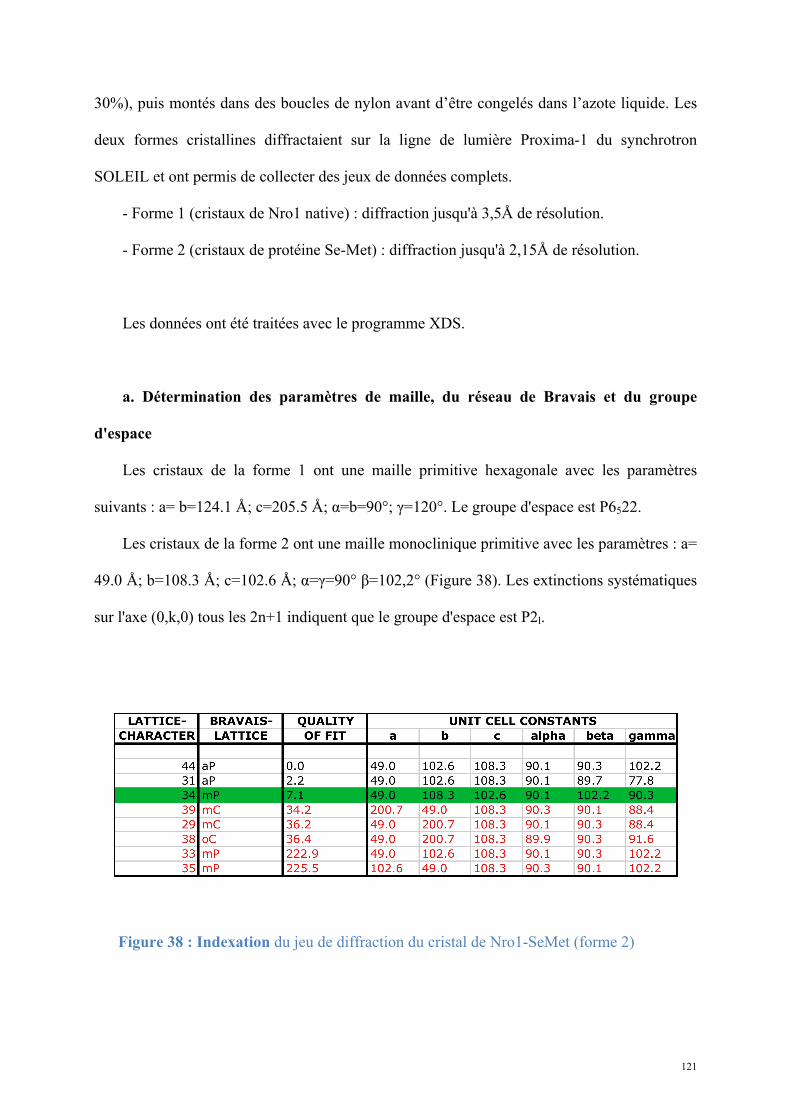
121
30%), puis montés dans des boucles de nylon avant d’être congelés dans l’azote liquide. Les
deux formes cristallines diffractaient sur la ligne de lumière Proxima-1 du synchrotron
SOLEIL et ont permis de collecter des jeux de données complets.
- Forme 1 (cristaux de Nro1 native) : diffraction jusqu'à 3,5Å de résolution.
- Forme 2 (cristaux de protéine Se-Met) : diffraction jusqu'à 2,15Å de résolution.
Les données ont été traitées avec le programme XDS.
a. Détermination des paramètres de maille, du réseau de Bravais et du groupe
d'espace
Les cristaux de la forme 1 ont une maille primitive hexagonale avec les paramètres
suivants : a= b=124.1 Å; c=205.5 Å; α=b=90°; γ=120°. Le groupe d'espace est P6522.
Les cristaux de la forme 2 ont une maille monoclinique primitive avec les paramètres : a=
49.0 Å; b=108.3 Å; c=102.6 Å; α=γ=90° β=102,2° (Figure 38). Les extinctions systématiques
sur l'axe (0,k,0) tous les 2n+1 indiquent que le groupe d'espace est P2l.
Figure 38 : Indexation du jeu de diffraction du cristal de Nro1-SeMet (forme 2)

122
b. Intégration des données, mise à l'échelle et conversion des intensités en
amplitudes
Les taches de diffractions sont indexées en h, k et l; chaque intensité est mesurée et
corrigée contre le bruit de fond. Les images sont assemblées et mises à l'échelle par le
programme XSCALE. Les statistiques de ces jeux de données sont résumées dans la figure
39.
Dans le cas des cristaux de protéine marquée Se-Met, quatre jeux ont été collectés : un
pour exploiter les données à haute résolution et trois pour déterminer les phases de la structure
par diffusion anomale du Sélénium à plusieurs longueurs d'onde (MAD). Les trois jeux
collectés pour l’expérience MAD sont collectés à trois longueurs d'onde différents : pic de
diffusion anomale (0,9791Å), inflexion (0,97946Å) et remote (diffusion normale, 0,95373Å).
Ces trois jeux ont été traités en ignorant la loi de Friedel, de manière à conserver le signal
anomal du Sélénium.
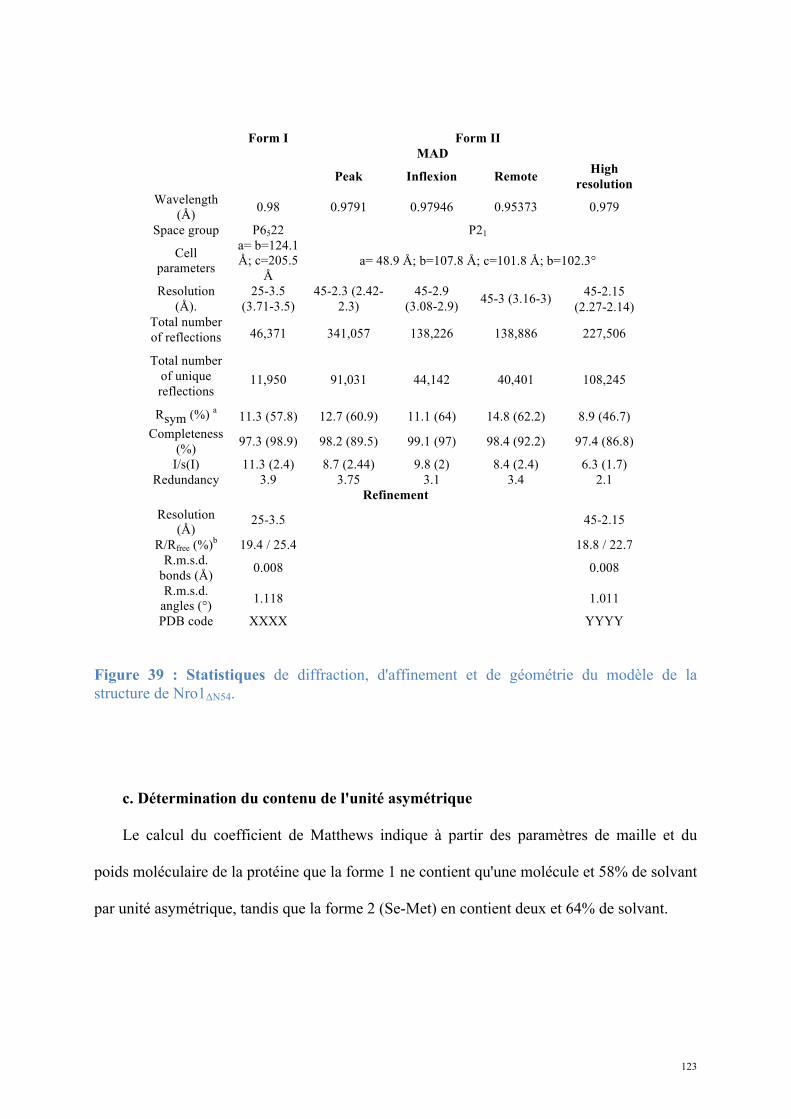
123
Form I Form II MAD
Peak Inflexion Remote High resolution
Wavelength (Å) 0.98 0.9791 0.97946 0.95373 0.979
Space group P6522 P21
Cell parameters
a= b=124.1 Å; c=205.5
Å a= 48.9 Å; b=107.8 Å; c=101.8 Å; b=102.3°
Resolution (Å).
25-3.5 (3.71-3.5)
45-2.3 (2.42-2.3)
45-2.9 (3.08-2.9) 45-3 (3.16-3) 45-2.15
(2.27-2.14) Total number of reflections 46,371 341,057 138,226 138,886 227,506
Total number of unique reflections
11,950 91,031 44,142 40,401 108,245
Rsym (%) a 11.3 (57.8) 12.7 (60.9) 11.1 (64) 14.8 (62.2) 8.9 (46.7) Completeness
(%) 97.3 (98.9) 98.2 (89.5) 99.1 (97) 98.4 (92.2) 97.4 (86.8)
I/s(I) 11.3 (2.4) 8.7 (2.44) 9.8 (2) 8.4 (2.4) 6.3 (1.7) Redundancy 3.9 3.75 3.1 3.4 2.1
Refinement Resolution
(Å) 25-3.5 45-2.15
R/Rfree (%)b 19.4 / 25.4 18.8 / 22.7 R.m.s.d.
bonds (Å) 0.008 0.008
R.m.s.d. angles (°) 1.118 1.011
PDB code XXXX YYYY
Figure 39 : Statistiques de diffraction, d'affinement et de géométrie du modèle de la structure de Nro1ΔN54.
c. Détermination du contenu de l'unité asymétrique
Le calcul du coefficient de Matthews indique à partir des paramètres de maille et du
poids moléculaire de la protéine que la forme 1 ne contient qu'une molécule et 58% de solvant
par unité asymétrique, tandis que la forme 2 (Se-Met) en contient deux et 64% de solvant.

124
D. Résolution
a. Recherche des sites Sélénium
Les données collectées sur les cristaux de protéine marquée ont été mises à l'échelle par
XSCALE, puis utilisées pour une recherche des 8 sites de Sélénium (4 par molécule) par le
programme SHELXD. Celui-ci semble localiser 6 sites sur les 8 attendus. Cela peut
s'expliquer par une flexibilité trop importante de l’une des méthionines de chacune des deux
copies présentes dans l’unité asymétrique.
b. Détermination des phases
Ces sites ont été affinés avec le programme SHARP comme cela avait été réalisé pour la
structure de la protéine Tpa1. L'utilisation des données collectées à trois longueurs d'onde
pour l’expérience de MAD s'est avérée nécessaire du fait du faible nombre de Sélénium (6
séléniums détectés pour un total de 694 résidus dans l’unité asymétrique soit un rapport de 1
Se pour 116 acides aminés). La bonne qualité des cartes obtenues (Figures 40 et 41) a permis
la reconstruction automatique de la majorité de la protéine par le programme ARP-WARP
(Perrakis et al., 2001), complétée par des cycles de reconstruction manuelle par COOT et par
des affinements par PHENIX.REFINE.
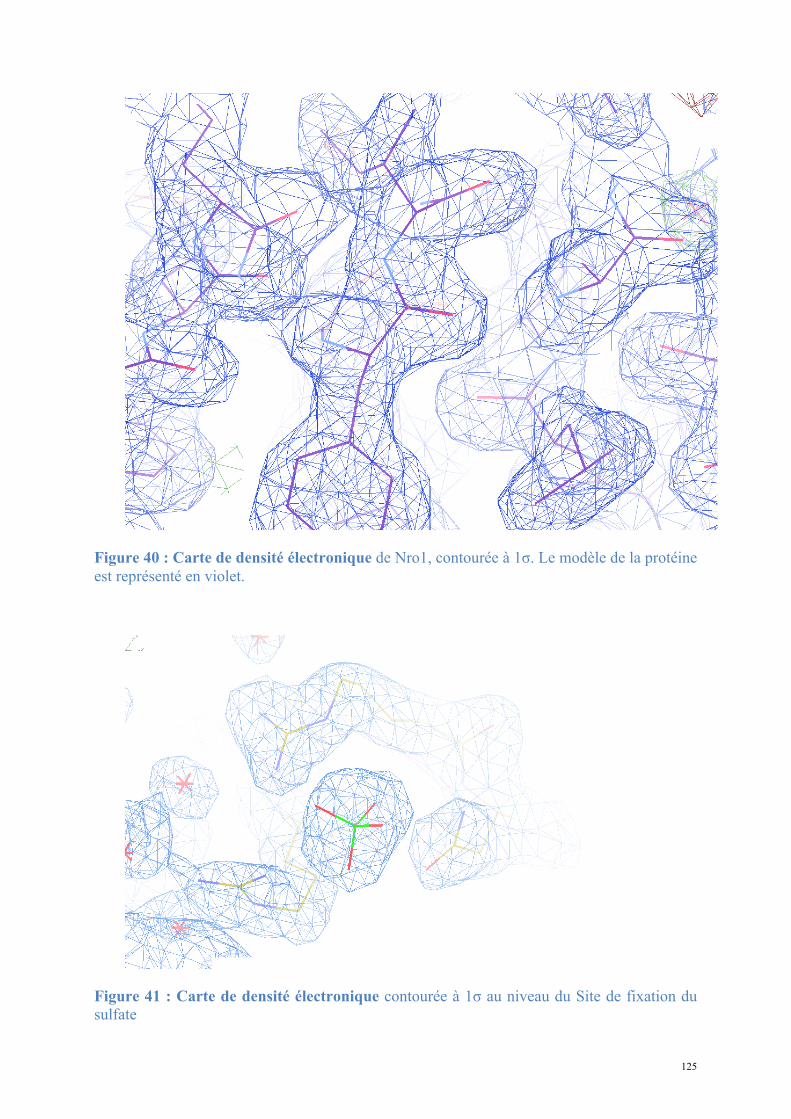
125
Figure 40 : Carte de densité électronique de Nro1, contourée à 1σ. Le modèle de la protéine est représenté en violet.
Figure 41 : Carte de densité électronique contourée à 1σ au niveau du Site de fixation du sulfate

126
Le modèle final est de bonne qualité, comme en attestent les statistiques après le dernier
affinement : R / Rfree = 18,8% / 22,7%. Tous les résidus adoptent des couples de valeurs des
angles phi/psi autorisées dans le diagramme de Ramachandran. La boucle comprise entre les
résidus Asn303 et Val308 n'a pas pu être modélisée du fait de l'absence de densité électronique.
Le modèle obtenu à partir des données à 2,15Å de P21 a été utilisé pour un remplacement
moléculaire dans les données à 3,5Å de P6522. L'affinement a révélé des densités
supplémentaires correspondant à la boucle de la protéine précédemment non construite et à
une partie du tag histidines. Les modèles obtenus à partir des deux groupes d'espace sont
extrêmement proches : rmsd de 0,57Å calculé sur 327 Cα. L'empilement cristallin est
identique, avec un axe de pseudo-symétrie d'ordre 2 supplémentaire dans le P6522. L'unité
asymétrique en P21 contient deux molécules identiques (rmsd=0,349Å sur 328 Cα). Les
monomères partagent une surface enfouie de 1122Å2, mais les expériences de SEC-MALLS
montrent que Nro1ΔN54 est monomérique en solution (Mapparent=39,7kDa, Mthéorique=39,5kDa).
1.2.2. Résultats principaux : analyse de la structure
Nro1ΔN54 est exclusivement constituée d'hélices-α : 13 hélices regroupées en 6 paires et
une hélice indépendante en C-terminal (Figure 42). Chaque paire d'hélices reliées par une
courte boucle (turn) est caractéristique du repliement TPR (TetratricoPeptide Repeat)
(D'Andrea and Regan, 2003). Les 6 motifs TPR de Nro1ΔN54 s'empilent avec une rotation de
quelques degrés entre chaque motif, l'ensemble adoptant une structure hélicoïdale.
Nro1ΔN54 est un homologue structural de diverses protéines à repliement TPR, avec une
conservation de séquence extrêmement faible : avec la protéine PilF de P. aeruginosa, rmsd
de 4,1Å sur 200 Cα alignés mais seulement 8% d'identité de séquence. Le repliement TPR a

127
été décrit dans des voies très diverses (régulation transcriptionnelle, contrôle du cycle
cellulaire, épissage, prolyl-isomérisation), avec comme point commun une implication dans
les interactions protéine-protéine ou protéine-peptide. Nro1ΔN56 jouerait ainsi probablement
un rôle dans la reconnaissance d'un substrat protéique.
Nro1 présente deux surfaces conservées. L'une d'elles possède trois résidus chargés
exposés au solvant et bien conservés : RxLR, à proximité de laquelle une densité non
attribuée à la protéine était observable après le dernier affinement (Figure 40). Le
positionnement d'un ion SO42- à cet endroit se justifiait par la forme sphérique de la densité et
la composition de la solution de cristallisation (1,6M de sulfate d'ammonium). Une seconde
densité non attribuée était observable à proximité, mais nous n'avons pour l'instant pas pu
définir la nature de la molécule qui en est responsable.
L'analyse structurale de Nro1 amène des éléments pour préciser la fonction d'Ett1 chez S.
cerevisiae. Le rôle des zones conservées et des résidus fixant le sulfate et la molécule non
identifiée sont en cours d'évaluation in vivo. L'identification de la molécule inconnue est en
cours.
La résolution de la structure de Nro1 et les résultats de l'analyse qui en découle
devraient faire l'objet d'une publication qui est en cours de rédaction. Le manuscrit tel qu'il
est rédigé à ce jour (Article 2) est présenté ci-après.
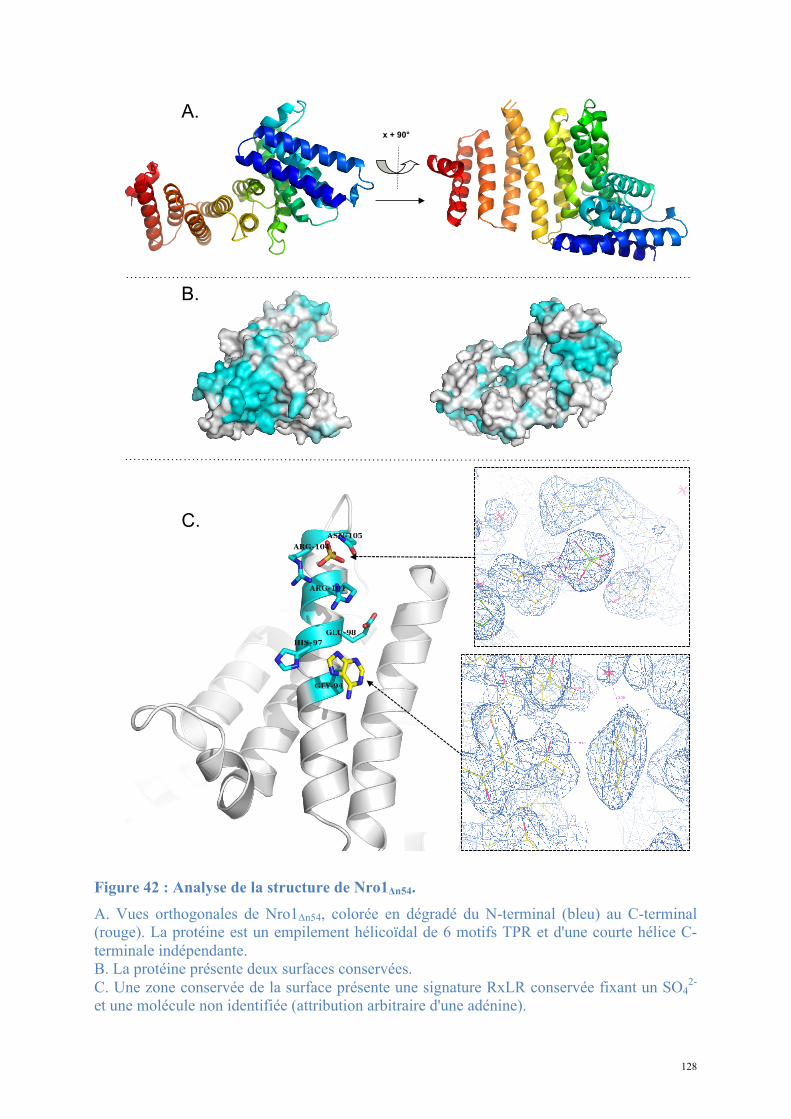
128
Figure 42 : Analyse de la structure de Nro1Δn54. A. Vues orthogonales de Nro1Δn54, colorée en dégradé du N-terminal (bleu) au C-terminal (rouge). La protéine est un empilement hélicoïdal de 6 motifs TPR et d'une courte hélice C-terminale indépendante. B. La protéine présente deux surfaces conservées. C. Une zone conservée de la surface présente une signature RxLR conservée fixant un SO4
2- et une molécule non identifiée (attribution arbitraire d'une adénine).
A.
x + 90°
B.
C.

129
Article 2 : Structural and functional study of
Nro1/Ett1, a protein involved in translation
termination in S. cerevisiae and in the control of gene
expression in response to O2 levels in S. pombe.

Structural and functional study of Nro1/Ett1,
a protein involved in translation termination in S. cerevisiae
and in the control of gene expression in response to O2 levels in S. pombe.
Introduction
Protein synthesis is the last step of gene expression and relies on a multi-step process that
requires the synergistic interplay between mRNA, ribosomes, translation factors and
aminoacyl-tRNAs. The mechanism of translation proceeds in four successive and highly
regulated operations: initiation, elongation, termination and ribosome recycling. During
elongation, specific aminoacyl-tRNAs bound to an elongation factor (eEF-1A in eukaryotes)
recognize their corresponding codons present in the ribosomal A-site with respect to the
anticodon-codon cognate interaction. Translation termination that leads to the release of the
newly synthesized proteins, does not rely on the recognition of the three stop codons (UAA,
UAG, or UGA) by specific tRNAs but by proteins called class I release factors: RF1 and RF2
in prokaryotes and eRF1 in eukaryotes (Kisselev, Ehrenberg et al. 2003). The eRF1 protein
mimics a tRNA molecule in shape and recognizes the stop codon present in the ribosomal A-
site via a strongly conserved NIKS motif located within its N-terminal domain (Song,
Mugnier et al. 2000)(Frolova, Seit-Nebi et al. 2002). Eukaryotic eRF1 interacts with the eRF3
GTPase, the class II release factor, to form a stable heterodimeric complex in the absence of
the ribosome (Zhouravleva, Frolova et al. 1995)(Stansfield, Jones et al. 1995). Binding of the
eRF1-eRF3-GTP ternary complex to a pre-termination ribosome complex promotes a
rearrangement of this complex and leads to GTP hydrolysis followed by rapid hydrolysis of
the ester bond connecting the newly synthesized polypeptide chain to the last tRNA present in
the ribosomal P-site (Salas-Marco and Bedwell 2004)(Alkalaeva, Pisarev et al. 2006). This
last step is catalyzed by the universally conserved Gly-Gly-Gln motif (GGQ) from eRF1
central domain (Zavialov, Mora et al. 2002). This motif dives into the peptidyl transferase
center of the ribosome and is proposed to coordinate a water molecule for nucleophilic attack
of the ester bond via the Gln side chain (Jin, Kelley et al. 2010)(Trobro and Aqvist 2007).
Once newly synthesized proteins are released, the ribosomes are dissociated and recycled for
a new round of translation by the combined action of eRF1, eRF3 and ABCE1 factors
(Pisarev, Skabkin et al. 2010).
Translation termination efficiency depends on the specific and rapid recognition of stop
codons (UGA, UAA and UAG) by eRF1 despite the presence of near-cognate tRNAs (such as

the tRNATrp that recognizes the UGG codon) that could induce amino acids misincorporation
and thereby production of longer proteins. The efficiency of stop codon recognition is
primarily influenced by the stop codon itself and the first downstream nucleotide (Bonetti, Fu
et al. 1995) and secondly by several trans-acting factors whose number has recently increased
significantly. First, the bacterial PrmC and eukaryotic Mtq2-Trm112 heterodimer, two SAM-
dependant methyltransferases that catalyze the post-translational modification of the Gln side
chain from the GGQ motif from class I release factors impacts the efficiency of translation
termination in vivo (i.e. decreases the readthrough of stop codons), at least in Escherichia coli
(Heurgue-Hamard, Graille et al. 2006)(Mora, Heurgue-Hamard et al. 2007). Second, the
poly(A) binding protein (Pab1 in yeast S. cerevisiae) interacts via its C-terminal domain with
eRF3 and formation of this complex has an anti-suppressor effect in yeast (Cosson, Couturier
et al. 2002). Third, S. cerevisiae Rli1 (for Rnase L Inhibitor 1), a highly conserved ATP-
binding cassette protein orthologous to human ABCE1, which is involved in ribosome
recycling, interacts physically with eRF1 and eRF3 and is required for proper stop codon
recognition (Khoshnevis, Gross et al. 2010). Fourth, many proteins (such as Npl3, Dbp5, Gle1
as well the inositol hexakisphosphate IP6 molecule) implicated in nuclear export of mRNAs
promote translation termination accuracy and some of them interact with translation
termination factors eRF1 and eRF3 (Gross, Siepmann et al. 2007; Bolger, Folkmann et al.
2008; Estrella, Wilkinson et al. 2009; Alcazar-Roman, Bolger et al. 2010; Khoshnevis, Gross
et al. 2010).
Finally, Saccharomyces cerevisiae Tpa1 protein is involved in translation termination
efficiency and reduces stop-codon readthrough (Keeling, Salas-Marco et al. 2006; Henri,
Rispal et al. 2010). In addition, this protein interacts with Pab1, eRF1 and eRF3 and hence
may be part of an mRNP complex associated at the mRNA 3’ end (Keeling, Salas-Marco et
al. 2006). Structure and sequence analysis strongly support that Tpa1 is a prolyl hydroxylase
(PHD) 2-oxoglutarate-Fe(II) dioxygenase, an enzyme catalyzing the O2-dependant
hydroxylation of proline side chain. However, this activity has not been demonstrated, mainly
because no clear substrate has been identified to date (Henri, Rispal et al. 2010). Tpa1 is
composed of a N-terminal putative PHD domain harbouring the HXD…H signature found in
this family of proteins and a C-terminal domain. We have shown that both Tpa1 domains as
well as the integrity of Tpa1 putative active site are required for correct translation
termination. In addition, Tpa1 turns out to act as an O2-sensor that represses the expression of
genes regulated by Hap1, a transcription factor involved in the response to levels of heme and
oxygen. In parallel, a Tpa1 ortholog from Schizosaccharomyces pombe, the Ofd1 protein has

been implicated in the O2-dependent regulation of the stability of the Sre1 transcription factor
that regulates the expression of genes involved in cholesterol and lipid biosynthesis (Hughes
and Espenshade 2008). As observed for Tpa1, the integrity of Ofd1 putative prolyl
hydroxylase active site is crucial for this process (Hughes and Espenshade 2008). Under low
oxygen levels, the membrane bound Sre1 protein is proteolytically cleaved, thus releasing its
N-terminal domain that enters the nucleus and activates the expression of genes essential for
hypoxic growth (Hughes, Todd et al. 2005; Todd, Stewart et al. 2006; Hughes, Lee et al.
2007). In the presence of oxygen, Ofd1 may act as an oxygen sensor that enhances the
degradation of Sre1N (Hughes and Espenshade 2008). In addition, the same group has
identified S. pombe Nro1, a poorly characterized protein known to interact with the nuclear
pore complex as a negative regulator of Ofd1 (Chen, Du et al. 2004; Lee, Stewart et al. 2009).
From these studies, they propose the following mechanism in which Ofd1 acts as an O2-
sensor. Under normoxia, the Ofd1 N-terminal dioxygenase domain could prevent binding of
Nro1 to Ofd1 C-terminal domain, which accelerates the degradation of Sre1N. On the
contrary, under hypoxia, the Ofd1 dioxygenase domain would be inactive and Nro1 would be
able to bind to Ofd1 C-terminal domain, resulting in stabilization of Sre1N domain.
Interestingly, the S. cerevisiae YOR051C gene encodes for a Nro1 ortholog that has
been show to interact with Tpa1 by high-throughput tandem-affinity purification (Krogan,
Cagney et al. 2006) and to be co-purified together with Tpa1 with yeast nuclear pore complex
(Rout, Aitchison et al. 2000). This protein also inhibits the replication of Brome mosaic virus
in S. cerevisiae similarly to genes coding for proteins involved in mRNA metabolism (Ski
complex and Ski7), transcription elongation or nucleocytoplasmic transport (Mog1, Nup170)
(Kushner, Lindenbach et al. 2003). Finally, this protein copurifies with ribosomal proteins
together with Stm1 (Van Dyke, Nelson et al. 2004), a protein modulating mRNA decay
(Balagopal and Parker 2009) and involved in TOR (target of rapamycin) signalling as well as
with Ykt6, a vesicle membrane protein (v-SNARE). Based on the link observed between
Ofd1 and Nro1 in S. pombe and on the effect of Tpa1 on translation termination efficiency,
we have tested the effect of YOR051C deletion on stop codon readthrough in S. cerevisiae.
The deletion of YOR051C in vivo induced the same phenotype as TPA1 gene deletion (i.e. a
significant increase of stop-codon readthrough) and we named this gene product Ett1 (for
Enhancer of translation termination 1; (Henri, Rispal et al. 2010)). Contrary to the inhibitory
effect of Nro1 on Ofd1 in S. pombe, the effect of ETT1 deletion is comparable to that of TPA1
deletion, thus indicating that both proteins are acting in the same pathway and that Ett1 does
not inhibit Tpa1. Altogether, these studies establish a functional link between Tpa1 and Ett1

in S. cerevisiae as well as between Ofd1 and Nro1 in S. pombe. However, it remains to be
established how these orthologous proteins are involved in these two different pathways. To
gain a mechanistic insight into Ett1/Nro1 function, we have solved the crystal structure of a
fragment from S. pombe Nro1 and performed structure-based in vivo studies in yeast S.
cerevisiae.
Material and methods
Cloning, expression and purification
The coding sequence of S. pombe Nro1 deleted from its 54 N-terminal residues and fused to a
N-terminal hexahistidine tag obtained by de novo gene synthesis (GenScript Corporation,
Piscataway, NJ, USA) was further subcloned into a bacterial pET21-a expression vector,
using NcoI and XhoI sites. The protein has been expressed using E. coli Rosetta pLysS strain
at 23°C in 2xYT medium supplemented with ampicillin at 50 g/mL. At OD600 = 0.8 the
transcription of the plasmid was induced for 20h by the addition of 50 g/mL IPTG. Cells
were harvested by centrifugation, resuspended in 25mL of 20mM Tris-HCl pH 7.5, 200mM
NaCl, 5mM -mercaptoethanol (buffer A) and stored at -20°C. Cell lysis was performed by
sonication. The His-tagged protein was purified on an Ni-NTA column (Qiagen) followed by
gel filtration chromatography on a SephadexTM 200 HL 16/60 column (Amersham
Biosciences) equilibrated in buffer A. Two successive anion exchange chromatographies on a
MonoQ HF column (Amersham Biosciences) were used as a final step, and the protein eluted
at 400mM NaCl was dialyzed against buffer A. Protein labelling was conducted as previously
described (Hendrickson, Horton et al. 1990) and the SeMet protein was purified using the
same protocol as the native protein.
Structure determination
Crystals of the native protein (Form I) were grown from a mixture in a 1:1 ratio of 7.2mg/mL
protein solution in buffer A and crystallization liquor containing 0.1M Hepes pH 7.0, 1.6M
Ammonium Sulfate and 10mM MgCl2, in 2 L hanging drops. Crystals of the SeMet protein
(Form II) were grown in the same conditions except for the addition of 3%
polyvinylpyrrolidone as an additive, in 1 L sitting drops. The crystals were cryo-protected by
transfer into the crystallization condition with progressively higher ethylene glycol
concentration up to 30% and then flash-cooled in liquid nitrogen.

Crystal form I yielded diffraction up to 3.5Å resolution on beam line Proxima-1 at SOLEIL
synchrotron (Saint-Aubin, France). This crystal belongs to space group P6522 with one
molecule in the asymmetric unit. Crystal form II diffracted up to 2.15 Å resolution but the
high resolution limit decreased with time. This crystal belongs to space group P21 with two
molecules in the asymmetric unit (a = 48.9Å, b = 107.8Å, c = 101.8Å, = 102.3°).
The structure was determined from Se-Met labelled protein crystals by the multi-wavelength
anomalous dispersion method using the anomalous signal of selenium atoms. Data were
processed with the XDS package (Kabsch 1993). Considering the cell parameters and the
molecular mass of the protein, the Matthews coefficient suggested that 2 molecules and 70%
solvent were present in the asymmetric unit. The program SHELXD was used to find an
initial set of the 6 expected Se sites (3 per molecule) (Schneider and Sheldrick 2002).
Refinement of the Se sites and phasing were carried out with the program SHARP (Bricogne,
Vonrhein et al. 2003). The obtained electron density maps were of excellent quality and
allowed automatic building by ARP-WARP. Iterative cycles of manual rebuilding using
COOT (Emsley and Cowtan 2004) followed by refinement with PHENIX (Adams, Grosse-
Kunstleve et al. 2002) led to an almost complete model containing two Nro1 molecules, 618
water molecules and 2 SO42- in the asymmetric unit. The loop between Asn303 and Val308
could not be built due to lack of electron density.
This model was then used for molecular replacement with MOLREP using the native low
resolution data and finally submitted to a new series of manual rebuilding with COOT and
refinement with PHENIX. This allowed us to build the N-terminal His6-tag and the Asn303-
Val308 loop, thereby leading to a complete model of the crystallized protein.
The statistics for data collection and refinement are summarized in Table 1. The atomic
coordinates and structure factors have been deposited into the Brookhaven Protein Data Bank
under accession numbers (XXXX and YYY).

Results
Nro1 structure resolution
The structure of Schizosaccharomyces pombe Nro1 deleted from its 54 N-terminal residues
that were predicted to be poorly folded by bioinformatics analysis, has been solved using the
multi-wavelength anomalous diffusion method from SeMet substituted protein crystals. The
protein crystallized in two different space groups (P21 and P6522) with no significant
difference between the two models after complete rebuilding and refinement (root mean
square deviation or rmsd=0.55 Å over 322 aligned residues).
The model obtained from the highest-resolution dataset (P21) contained two molecules in the
asymmetric unit. No significant differences are observed between both molecules that are
virtually identical (rmsd=0.35 Å). These two molecules interact via a large surface of 2,250
Å2 raising the possibility that this protein associates as homodimers. However, a size-
exclusion chromatography coupled to Multi-angle laser light scattering (MALLS) experiment
yielded to a molecular weight of 39.7kDa, very close to the theoretical value expected for a
monomer (39.5kDa; data not shown). We thus conclude that Nro1 N54 is monomeric in
solution and that the observed dimerization more likely results from a crystallization artefact.
Nro1 belongs to the Tetratrico Peptide Repeat (TPR) superfamily.
The Nro1 N54 monomer is composed of 13 -helices organized as 6 pairs of antiparallel
helices related by an angle of approximately 20° and a short C-terminal helix (hC) stacked
onto the last helical hairpin motif (Fig.1A-B). Helix hC shields from the solvent hydrophobic
residues from the last helix pair (Leu340, Met347, Leu363 and Ile370) through residues located on
one face of helix hC (Val378, Leu380, Tyr384, Phe387, Val388 and Tyr391). The residues located on
the other face of helix hC are polar or charged residues (Ser381, Asp383, Glu386, Glu389) and are
exposed to the solvent, allowing helix hC to act as a solvating cap that stabilizes the Nro1
protein. The pairs of helices are packed against each other’s to form a right-handed
superhelical molecule as observed in the structures of members of Tetratrico Peptide Repeat
(TPR) superfamily. Hence, based on its structure, Nro1 can be considered as a new member
of this TPR family that includes bacterial and eukaryotic proteins. Such classification could
not be predicted solely from sequence analysis.
The TPR motif is composed of 3 to 16 tandem repeats of degenerate 34 amino acids that
adopt a helix-turn-helix antiparallel arrangement with interacting helices (namely helix A and
helix B) separated by a short loop (D'Andrea and Regan 2003). In general, TPR motifs are

stacked together so as that helix A from TPRn is packed between helix B from TPRn and helix
A from TPRn+1. In Nro1, the 12 helices forming the 6 TPR motifs are organized as follows
from N-terminus to C-terminus: TPR1A, TPR1
B, TPR2A, TPR2
B, TPR3A, TPR3
B, TPR4A,
TPR4B, TPR5
A, TPR5B, TPR6
A, TPR6B
with the thirteenth C-terminal helix (hC) running above
the 6th TPR motif with an angle of approximately 45° with TPR6A and TPR6
B (Fig.1B). These
TPRs are longer (50 residues) than canonical TPR motifs (34 amino acids long) and are
organized into two subdomains: TPR-N and TPR-C. TPR-N is composed of TPR 1 to 3 and
within this domain, consecutive TPR units are related by a 34° rotation. TPR-C is formed by
TPR 4 to 6 and in this case, consecutive TPR units are related by a rotation of 15°. These
Nro1-N and Nro1-C subdomains are similar in structure (rmsd of 2.8Å over 97 C atoms;
10% sequence identity). The C-terminal TPR unit from TPR-N (TPR3) and the N-terminal
TPR from TPR-C (TPR4) are related by a rotation of 50°. Hence, Nro1 does not form a highly
regular right-handed molecule compared to S. cerevisiae Sec17, a TPR protein involved in
vesicular transport (Rice and Brunger 1999).
Nro1 binds SO42-
via a conserved RxLR signature
After the last step of refinement of the model using the high resolution (2.15Å) dataset, two
strong residual electron density peaks were observed in the vicinity of helix TPR1B. The first
density was spherical and close to Arg101, Arg104 and Asn105 (Fig.1C). As the crystals were
grown in 1.6M ammonium sulphate as a precipitant, we modelled this density with a SO42-
ion. It has to be noticed that a sulphate ion is found in the same position of the two Nro1
molecules present in the asymmetric unit. The second unexplained residual electron density
lies at a crystal packing site. This density is planar and reminiscent of an aromatic molecule
such as indole or purine moieties (Fig.1C). As none of the protein buffer and crystallization
solution components could fit within this density, this ligand is very likely to be a molecule
co-purified with Nro1 from E. coli. This unknown ligand stacks onto Gly94 from helix TPR1B
and is in the vicinity from Leu72, His97, Glu98 and Arg101.
This unidentified ligand is only 10 Å away from the sulphate ion and both molecules are
mainly interacting with residues from TPR1B. Interestingly, with the exception of Asn105, all
these residues are strictly conserved within Nro1/Ett1 orthologs, strongly supporting a role of
this region in Nro1/Ett1 function (Fig1D & S1). Additional strictly conserved and solvent-
exposed residues (Gln68, Leu72, Asp100, Tyr120 and Leu124) are surrounding this region and

contribute to form a large conserved region. This region is further surrounded by highly
conserved residues such as Asp64, Ile96, His117, Tyr149, Ala150, Glu153 and Arg154.
As this conserved region is located close to the N-terminal part of the crystallized Nro1
fragment, we cannot exclude that it is partly or entirely masked by the 54 residues deleted in
our structural study. As our structure revealed that the crystallized fragment from Nro1 is
made of 6 TPR motifs, we analyzed the sequence of S. pombe Nro1 using the bioinformatics
tool TPRPred (MPI Tübingen, http://toolkit.tuebingen.mpg.de/tprpred). This server indeed
predicts the 6 TPR motifs observed in the structure. Another TPR motif (TPR0) is predicted
between Arg5 and Pro38, a region deleted in the Nro1 fragment used in this study. As a
consequence, full-length Nro1 would be constituted of 7 TPR motifs plus a C-ter
supplementary helix. This represents a usual architecture since 20% of the TPR-containing
proteins in S. cerevisiae are expected to contain 7 repeats (D'Andrea and Regan 2003).
However, as in TPR fold, TPRn is packed against helix A from TPRn+1, this would imply that
this conserved region mainly formed by residues from helix B from TPR1 should not be
buried by the N-terminal part that is absent in our crystal structure.
Hence, we propose that this highly conserved region located in the convex side of the N-
terminal part of the helical Nro1 monomer is fully accessible to the solvent. In addition, this
region looks like an ideal binding site for ligands containing negative charges (mimicked by
the sulphate ion from the crystallization condition) such as nucleotides or phosphopeptides.
Comparison with other TPR proteins.
As the biochemical function of Nro1 is still largely obscure, searches within the Protein Data
Bank (PDB) of structurally homologous proteins may bring some clues about Nro1 activity.
Numerous TPR containing proteins have been described from a structural point of view so
far. This TPR fold is ubiquitous and largely involved in various protein-protein or protein-
peptide interactions with different functions: transcriptional regulation, protein translocation,
cell cycle control, pre-mRNA splicing, peptidyl prolyl isomerase, … (D'Andrea and Regan
2003). Hence, Nro1 fold suggests that the function of this protein may directly be linked with
its ability to interact with a protein or peptide partner. A common theme found in these TPR
proteins is the binding of a peptide within the concave groove of these helical proteins. This is
indeed the case for cochaperone proteins such as cyclophilin 40 (Cyp40), Hsp70/Hsp90
organizing protein (Hop), FK506 binding proteins (FKBPs), which all have in common to
bind the acidic C-terminal MEEVD peptide from Hsp90 (Riggs, Roberts et al. 2003; Wu, Li
et al. 2004).

Searches for Nro1 N54 structural homologues using the DALI server (Holm, Kaariainen et al.
2008) identify approximately one hundred different protein structures with Z-score ranging
from 12 to 5. However, with the exception of bacterial PilF protein from P. aeruginosa (rmsd
of 4.1 Å over 200 C atoms ; 8% sequence identity (Kim, Oh et al. 2006)), these homologues
possess high rmsd values with the entire crystallized fragment (rmsd of 7-9Å for structural
alignment of 200-230 residues). Some of these homologues are : i) gamma-SNAP from
zebrafish (Z-score of 11.8; rmsd of 8.6Å over 216 C atoms; (Bitto, Bingman et al. 2008))
and Sec17 from yeast S. cerevisiae (Z-score of 9; rmsd of 7.9Å over 200 C atoms; (Rice and
Brunger 1999)), two proteins involved in vesicular transport; ii) the Cdc26 subunit of the
anaphase-promoting complex (Z-score of 9.4; rmsd of 6.6Å over 220 C atoms; (Wang, Dye
et al. 2009)).
As from our structural analysis, Nro1 monomer could be divided into two subdomains (Nro1-
N and Nro1-C), we have performed the same analysis using the coordinates of each of these
subdomains. Strikingly, the same strong structural homologues are found for both Nro1-N and
Nro1-C domains (Z-scores of 9-11 and rmsd ranging from 2.5Å to 4Å over 90-120 C atoms;
sequence identity between 5 and 14%). Among these, some are of interest in light of the
involvement of Nro1/Ett1 in the same pathways as the putative prolyl-4-hydroxylases Ofd1
from S. pombe and Tpa1 from S. cerevisiae (Lee, Stewart et al. 2009)(Henri, Rispal et al.
2010).
First, the TPR domain from the subunit of human collagen prolyl 4-hydroxylase (C-P4H)
2 2 enzyme tetramer displays rmsd values of 2.9Å over 90 C atoms (5% sequence identify)
and 2.7Å over 81 C atoms (5% sequence identity) with Nro1-N and Nro1-C domains,
respectively (Pekkala, Hieta et al. 2004). This domain is involved in the binding of the X-Pro-
Gly triplets from collagen and enhances the activity of the C-P4H catalytic dioxygenase
domain (also located within this subunit) responsible for hydroxylation of the Pro side
chain. This domain has a higher affinity for peptides containing unmodified Pro side chains
than for peptides containing hydroxylated Pro (Hyp). Its peptide-binding site is located in the
concave surface formed by helices A from contiguous TPR units and is lined by several Tyr
side chains important for peptide binding. These Tyr side chains are not conserved in Nro1
orthologues but the region corresponding to this peptide binding groove in Nro1-C can be
considered as the second highest conserved patch present at the surface of Nro1 (Fig.2A-B;
after the region involved in sulphate binding).

Second, some members of the immunophilin superfamily are also close structural homologues
of Nro1. Immunophilins are peptidyl-prolyl isomerases (PPIases) inhibited by the
cyclosporine A, FK506 and rapamycin immunosuppressors. They assist protein folding by
catalysing the cis-trans isomerisation of proline imidic peptide bonds in proteins. Some of
these such as Cyp40, FKBP51 and FKPB52 are composed of one or two N-terminal PPIase
domains followed by a C-terminal TPR domain composed of 3 TPR units (rmsd values of
3.3Å over 90 C atoms with 9% sequence identify and 3.7Å over 125 C atoms with 7%
sequence identity with Nro1-N (Fig.2C) and Nro1-C domains, respectively; (Sinars, Cheung-
Flynn et al. 2003; Wu, Li et al. 2004)). FKBP51, FKBP52 and Cyp40 are involved in the
maturation of mammalian steroid receptors and at least one of these is present in the mature
receptors in combination with Hsp90. These proteins are associated via their TPR domain
with the C-terminal MEEVD sequence motif from Hsp90 (Riggs, Roberts et al. 2003).
Structural comparison of both Nro1-N and Nro1-C domains with complex of FKBP52 bound
to the Hsp90 MEEVD peptide clearly shows that the FKBP52 residues involved in Hsp90
binding are not conserved in Nro1. In addition, the concave faces from Nro1-N and to a lesser
extend Nro1-C that may be involved in peptide binding are not strongly conserved. Hence, we
cannot exclude that Nro1 will bind a peptide in its Nro1-C groove but the sequence of this
peptide is probably not as conserved as the Hsp90 MEEVD motif recognized by FKBP51,
FKBP52, Cyp40 but also Hop, protein phosphatase 5 …
Although many structures of TPR domains have been solved to date, it is difficult to predict
with some certainty the function of Nro1 from the analysis of its structure. The identification
of the role of S. pombe Nro1 in the O2-dependent regulation of Sre1 stability and of S.
cerevisiae Ett1 in the regulation of translation termination efficiency will need more studies.
Discussion
The entrance of one of the three stop codons (UGA, UAA and UAG) in the ribosomal A-site
is the initial signal triggering translation termination and release of newly synthesized proteins
(Kisselev and Buckingham 2000). Contrary to the other codons, these three nonsense codons
are not recognized by specific aminoacyl tRNAs but by the class I translation termination
factor eRF1 in eukaryotes (Frolova, Le Goff et al. 1994). However, fidelity of stop codon
recognition does not rely only on eRF1 ability to recognize stop codons more stringently than
the competing near-cognate tRNAs (such as the tRNATrp that recognizes the UGG codon)

present in cells but is also influenced by many additional proteins (von der Haar and Tuite
2007).
Among these factors, we have recently focused on two proteins from S. cerevisiae: Tpa1 and
Ett1. Surprisingly, while Tpa1 and Ett1 are important in translation termination efficiency
(Keeling, Salas-Marco et al. 2006)(Henri, Rispal et al. 2010), their orthologues from S.
pombe, Ofd1 and Nro1, respectively, are involved in an apparently unrelated pathway, i.e. the
regulation of the stability of Sre1, a transcription factor controlling expression of genes
involved in cholesterol and lipid biosynthesis (Hughes and Espenshade 2008; Lee, Stewart et
al. 2009). Two other important differences between these proteins stand in 1) the inhibitory
effect exerted by Nro1 on Ofd1 while Ett1 does not inhibit Tpa1 and 2) the requirement of
Tpa1 but not Ofd1 active site integrity in the regulation of these phenomena. Here, we present
the crystal structure of a S. pombe Nro1 fragment missing the 54 N-terminal residues. This
structure reveals that this protein adopts a TPR fold, which is ubiquitous and largely involved
in various protein-protein or protein-peptide interactions with different functions. In addition,
the structure highlights a highly conserved region that binds a sulphate ion as well as an
unidentified ligand in our structure. This site is very like to be of functional importance and
most probably acts as a putative ligand/protein binding site.
However, despite these studies, the precise role of these two proteins in the control of
translation termination accuracy or the O2-dependent regulation of Sre1 stability remains
obscure. A first possibility was that Tpa1 and Ett1 are involved in mRNA export. This was
based first on the observation that these two proteins in S. cerevisiae as well as Nro1 in S.
pombe copurify with nuclear pore complex (Chen, Du et al. 2004)(Rout, Aitchison et al.
2000) and second that many proteins involved in mRNA export (such as Dbp5, Npl3 and
Gle1) also influence translation termination (Gross, Siepmann et al. 2007; Bolger, Folkmann
et al. 2008; Estrella, Wilkinson et al. 2009). However, deletion of either TPA1 or NRO1 genes
does not affect mRNA export in budding and fission yeasts, respectively, ruling out this
assumption (Keeling, Salas-Marco et al. 2006)(Chen, Du et al. 2004). Another possibility is
that Ett1/Nro1 assists Tpa1/Nro1 to perform its enzymatic activity, i.e. most likely
hydroxylation of prolyl residues. S. cerevisiae Ett1 is functionally linked with Tpa1, a
structural homolog of collagen P4H (C-P4H) 2-oxoglutarate dioxygenase domain (Henri,
Rispal et al. 2010). Interestingly, the enzymatic activity of this C-P4H dioxygenase domain is
enhanced by a peptide-substrate binding domain of collagen prolyl-4-hydroxylase, which
adopts a TPR fold similar to Ett1/Nro1 (Fig.2A; (Pekkala, Hieta et al. 2004)). The C-P4H
TPR domain displays a hydrophobic groove lined by exposed tyrosine side chains that are

important for the interaction with the [X-Pro-Gly]n peptide-substrate of collagen. In Nro1, no
tyrosine residues are present at the corresponding position but this region displays some
degree of sequence conservation suggesting that it could mediate binding to a peptide with a
conserved signature different from the [X-Pro-Gly]n motif found in collagen (Fig.2B). Based
on this C-P4H model, Ett1/Nro1 could influence Tpa1 enzymatic activity by assisting Tpa1 in
the recognition of its substrate. It is also noteworthy that the coupling of dioxygenase and
TPR domains within a single polypeptide chain is not unique to C-P4H but is also observed in
leprecan-like proteins 1 and 2, which are prolyl-hydroxylases also acting on collagen (Jarnum,
Kjellman et al. 2004), in human Asp/Asn beta-hydroxylase (Korioth, Gieffers et al. 1994) and
in histone demethylases specific for H3K27me3/me2 (Cloos, Christensen et al. 2008).
Another aspect that could be considered is the structural homology between Nro1 TPR
subdomains and the TPR domain from FK506-binding proteins: FKBP51 and FKBP52
(Sinars, Cheung-Flynn et al. 2003; Wu, Li et al. 2004). Interestingly, FKBP38, which is
composed of a peptidyl prolyl isomerase (PPIase) domain and three TPR repeats interacts
with PHD2, a prolyl hydroxylase controlling the stability of the HIF transcription factor
(Barth, Nesper et al. 2007). In this complex, FKBP38 is not substrate for PHD2 but acts as a
PHD2 cofactor. Another example of synergy between PPIase and prolyl hydroxylase domains
occurs in the stabilization of collagen triple helix. Hydroxylation of collagen prolyl residues is
catalyzed by C-P4H on the nascent polypeptide chain during translation and the subsequent
cis/trans isomerisation reaction catalyzed by cyclophilin is the rate-limiting step for the
formation of collagen triple helix (Steinmann, Bruckner et al. 1991). In the case of Ett1, this
protein has been shown to co-purify with the ribosome together with Stm1 (Van Dyke,
Nelson et al. 2004). Hence, Ett1 together with Tpa1 may be recruited to translating ribosomes
to perform hydroxylation of proline residues on some nascent proteins. This could explain the
initial observation that Tpa1 interacts with Pab1, eRF1 and eRF3 (Keeling, Salas-Marco et al.
2006). In addition, we have observed that the S. pombe Nro1 N54 protein copurifies with E.
coli FKBP (data not shown), a protein that is only composed by the PPIase domain. As no cis
proline residues are observed in our 2.15Å resolution crystal structure of Nro1 N54, this
suggests that Nro1 N54 may not be a FKBP substrate but rather a potential partner. From the
superposition of FKBP52 crystal structure onto Nro1-N TPR subdomain, the PPIase domain
is close from the highly conserved region of Nro1 that is involved in binding the sulphate ion
from the crystallization conditions (Fig.2C-D). Therefore, we cannot exclude that this region
is involved in binding of FKBPs from yeast S. cerevisiae. To date, four FKBPs named Fpr1-4
have been described in baker’s yeast and none of them has clearly identified TPR repeats

(Arevalo-Rodriguez, Wu et al. 2004). Binding of Ett1 to any of these FKBP has not been
described in S. cerevisiae but this will be the subject of future studies.
Further studies will be needed to clarify the interconnection between Ett1/Nro1 and
Tpa1/Ofd1 and to identify the mechanistic differences that allow Nro1 but not Ett1 to inhibit
Ofd1/Tpa1 activity. Similarly, while Tpa1/Ofd1 have a clear ortholog in human genome, we
did not detect any Ett1/Nro1 human ortholog. Hence, this aspect will also have to be
investigated.
LEGENDS TO FIGURES
Figure 1. Ribbon representation of Nro1 N54 structure.
A. The monomer is colored from N-terminus (blue) to C-terminus (red).
B. Each TPR unit is represented with different colors. Helices A and B are in light and
dark colors, respectively. Same orientation as panel A.
C. Detailed view of the ligand binding sites. Strictly conserved residues are shown as
sticks. The C atom from Gly94 is shown as a sphere. The Fo-Fc electron density map
is contoured at 3 . The SO42- ion originating from crystallization conditions is shown
as sticks. To identify the residual Fo-Fc electron density close to Gly94 and to give an
idea of its size, a putative and unidentified ligand is indicated in the figure. This
should not be considered as informative on the identity of the ligand.
D. Mapping of the residues conservation at the surface of Nro1 N54. Color coding is from
white (no conservation) to cyan (high conservation). The orientation is similar to panel
A.
Figure 2. Comparison with TPR domains from C-P4H and FKBP52.
A. Ribbon representation of the superimposition of the C-P4H TPR domain (pink) onto
the Nro1-C subdomain (green).
B. Mapping of the residues conservation at the surface of Nro1-C subdomain. Color
coding is from white (no conservation) to cyan (high conservation). The orientation is
similar to panel A. For clarity, the C-P4H TPR domain has been omitted.
C. Ribbon representation of the superimposition of the FKBP52 (TPR domain in blue and
PPIase domain in green) onto the Nro1-N subdomain (gold). The ligands bound to
Nro1-N are shown as balls.

D. Mapping of the residues conservation at the surface of Nro1-N subdomain. Color
coding is from white (no conservation) to cyan (high conservation). The orientation is
similar to panel C. For clarity, the FKBP52 TPR domain has been omitted.
ACKNOWLEDGMENTS
We are indebted to K. Blondeau for her help with SeMet labeling and A. Doizy for technical
assistance. We thank M. Argentini and D. Cornu for mass spectrometry analysis (SiCaps,
IMAGIF platform, Gif/Yvette, France). This work was supported by the European Union 6th
Framework programs ‘3D-Repertoire’ (LSHG-CT-2005-512028), the CNRS and the Agence
Nationale pour la Recherche (Grant ANR-06-BLAN-0075-02). JH is a recipient of PhD
fellowship from Université Paris Sud-11.
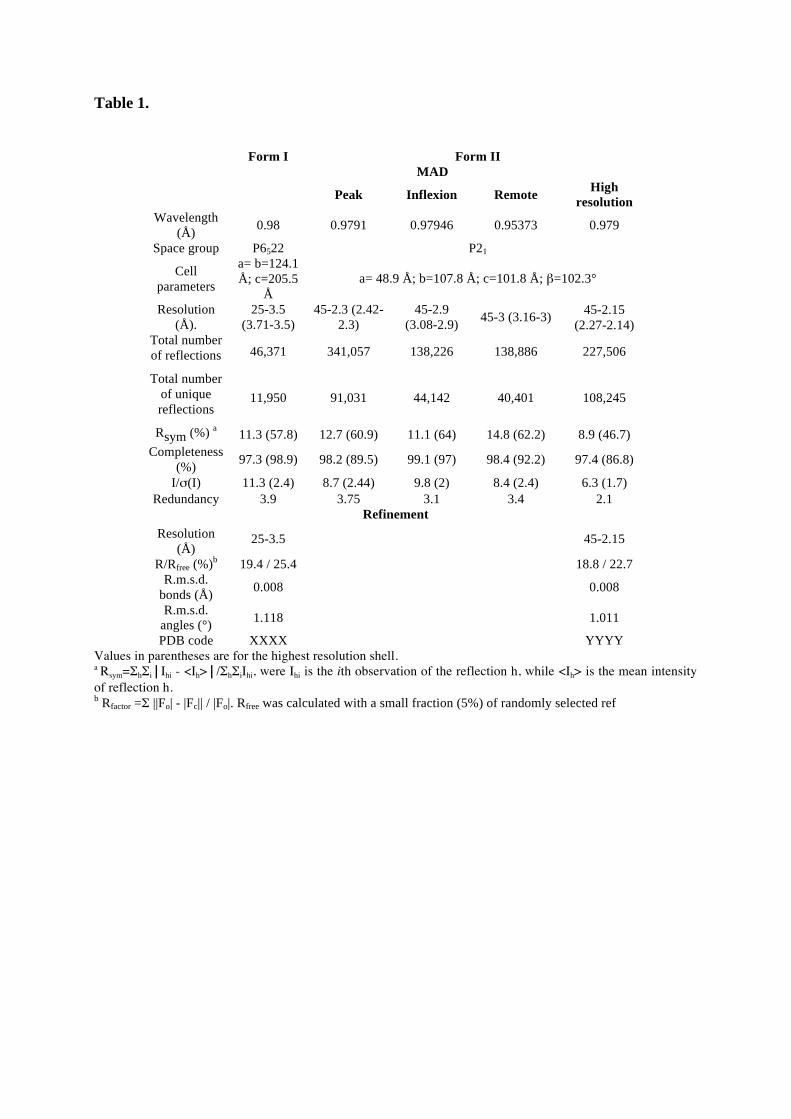
Table 1.
Form I Form II
MAD
Peak Inflexion Remote High
resolution
Wavelength (Å)
0.98 0.9791 0.97946 0.95373 0.979
Space group P6522 P21
Cell parameters
a= b=124.1 Å; c=205.5
Å a= 48.9 Å; b=107.8 Å; c=101.8 Å; =102.3°
Resolution (Å).
25-3.5 (3.71-3.5)
45-2.3 (2.42-2.3)
45-2.9 (3.08-2.9)
45-3 (3.16-3) 45-2.15
(2.27-2.14) Total number of reflections 46,371 341,057 138,226 138,886 227,506
Total number of unique reflections
11,950 91,031 44,142 40,401 108,245
Rsym (%) a 11.3 (57.8) 12.7 (60.9) 11.1 (64) 14.8 (62.2) 8.9 (46.7)
Completeness (%)
97.3 (98.9) 98.2 (89.5) 99.1 (97) 98.4 (92.2) 97.4 (86.8)
I/ (I) 11.3 (2.4) 8.7 (2.44) 9.8 (2) 8.4 (2.4) 6.3 (1.7) Redundancy 3.9 3.75 3.1 3.4 2.1
Refinement
Resolution (Å)
25-3.5 45-2.15
R/Rfree (%)b 19.4 / 25.4 18.8 / 22.7 R.m.s.d.
bonds (Å) 0.008 0.008
R.m.s.d. angles (°)
1.118 1.011
PDB code XXXX YYYY Values in parentheses are for the highest resolution shell. a Rsym= h i Ihi - <Ih> / h iIhi, were Ihi is the ith observation of the reflection h, while <Ih> is the mean intensity of reflection h. b Rfactor = ||Fo| - |Fc|| / |Fo|. Rfree was calculated with a small fraction (5%) of randomly selected ref

References.
Adams, P. D., R. W. Grosse-Kunstleve, et al. (2002). "PHENIX: building new software for automated crystallographic structure determination." Acta Crystallogr D Biol Crystallogr 58(Pt 11): 1948-54.
Alcazar-Roman, A. R., T. A. Bolger, et al. (2010). "Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1." J Biol Chem 285(22): 16683-92.
Alkalaeva, E. Z., A. V. Pisarev, et al. (2006). "In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3." Cell 125(6): 1125-36.
Arevalo-Rodriguez, M., X. Wu, et al. (2004). "Prolyl isomerases in yeast." Front Biosci 9: 2420-46.
Balagopal, V. and R. Parker (2009). "Stm1 modulates mRNA decay and Dhh1 function in Saccharomyces cerevisiae." Genetics 181(1): 93-103.
Barth, S., J. Nesper, et al. (2007). "The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability." Mol Cell Biol 27(10): 3758-68.
Bitto, E., C. A. Bingman, et al. (2008). "Structure and dynamics of gamma-SNAP: insight into flexibility of proteins from the SNAP family." Proteins 70(1): 93-104.
Bolger, T. A., A. W. Folkmann, et al. (2008). "The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation." Cell 134(4): 624-33.
Bonetti, B., L. Fu, et al. (1995). "The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae." J Mol Biol 251(3): 334-45.
Bricogne, G., C. Vonrhein, et al. (2003). "Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0." Acta Crystallogr D Biol Crystallogr 59(Pt 11): 2023-30.
Chen, X. Q., X. Du, et al. (2004). "Identification of genes encoding putative nucleoporins and transport factors in the fission yeast Schizosaccharomyces pombe: a deletion analysis." Yeast 21(6): 495-509.
Cloos, P. A., J. Christensen, et al. (2008). "Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease." Genes Dev 22(9): 1115-40.
Cosson, B., A. Couturier, et al. (2002). "Poly(A)-binding protein acts in translation termination via eukaryotic release factor 3 interaction and does not influence [PSI(+)] propagation." Mol Cell Biol 22(10): 3301-15.
D'Andrea, L. D. and L. Regan (2003). "TPR proteins: the versatile helix." Trends Biochem Sci 28(12): 655-62.
Emsley, P. and K. Cowtan (2004). "Coot: model-building tools for molecular graphics." Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1): 2126-32.
Estrella, L. A., M. F. Wilkinson, et al. (2009). "The shuttling protein Npl3 promotes translation termination accuracy in Saccharomyces cerevisiae." J Mol Biol 394(3): 410-22.
Frolova, L., X. Le Goff, et al. (1994). "A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor." Nature 372(6507): 701-3.
Frolova, L., A. Seit-Nebi, et al. (2002). "Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1." Rna 8(2): 129-36.
Gross, T., A. Siepmann, et al. (2007). "The DEAD-box RNA helicase Dbp5 functions in translation termination." Science 315(5812): 646-9.

Hendrickson, W. A., J. R. Horton, et al. (1990). "Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure." Embo J 9(5): 1665-72.
Henri, J., D. Rispal, et al. (2010). "Structural and functional insights into S. cerevisaie Tpa1, a putative prolyl hydroxylase influencing translation termination and transcription." J Biol Chem in press.
Heurgue-Hamard, V., M. Graille, et al. (2006). "The zinc finger protein Ynr046w is plurifunctional and a component of the eRF1 methyltransferase in yeast." J Biol Chem 281(47): 36140-8.
Holm, L., S. Kaariainen, et al. (2008). "Searching protein structure databases with DaliLite v.3." Bioinformatics 24(23): 2780-1.
Hughes, A. L., C. Y. Lee, et al. (2007). "4-Methyl sterols regulate fission yeast SREBP-Scap under low oxygen and cell stress." J Biol Chem 282(33): 24388-96.
Hughes, A. L., B. L. Todd, et al. (2005). "SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast." Cell 120(6): 831-42.
Hughes, B. T. and P. J. Espenshade (2008). "Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member." Embo J 27(10): 1491-501.
Jarnum, S., C. Kjellman, et al. (2004). "LEPREL1, a novel ER and Golgi resident member of the Leprecan family." Biochem Biophys Res Commun 317(2): 342-51.
Jin, H., A. C. Kelley, et al. (2010). "Structure of the 70S ribosome bound to release factor 2 and a substrate analog provides insights into catalysis of peptide release." Proc Natl Acad Sci U S A 107(19): 8593-8.
Kabsch, W. (1993). "Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants." J. Appl. Cryst. 26: 795-800.
Keeling, K. M., J. Salas-Marco, et al. (2006). "Tpa1p is part of an mRNP complex that influences translation termination, mRNA deadenylation, and mRNA turnover in Saccharomyces cerevisiae." Mol Cell Biol 26(14): 5237-48.
Khoshnevis, S., T. Gross, et al. (2010). "The iron-sulphur protein RNase L inhibitor functions in translation termination." EMBO Rep 11(3): 214-9.
Kim, K., J. Oh, et al. (2006). "Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa." Biochem Biophys Res Commun 340(4): 1028-38.
Kisselev, L. L. and R. H. Buckingham (2000). "Translational termination comes of age." Trends Biochem Sci 25(11): 561-6.
Kisselev, L. L., M. Ehrenberg, et al. (2003). "Termination of translation: interplay of mRNA, rRNAs and release factors?" Embo J 22(2): 175-82.
Korioth, F., C. Gieffers, et al. (1994). "Cloning and characterization of the human gene encoding aspartyl beta-hydroxylase." Gene 150(2): 395-9.
Krogan, N. J., G. Cagney, et al. (2006). "Global landscape of protein complexes in the yeast Saccharomyces cerevisiae." Nature 440(7084): 637-43.
Kushner, D. B., B. D. Lindenbach, et al. (2003). "Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus." Proc Natl Acad Sci U S A 100(26): 15764-9.
Lee, C. Y., E. V. Stewart, et al. (2009). "Oxygen-dependent binding of Nro1 to the prolyl hydroxylase Ofd1 regulates SREBP degradation in yeast." Embo J 28(2): 135-43.
Mora, L., V. Heurgue-Hamard, et al. (2007). "Methylation of bacterial release factors RF1 and RF2 is required for normal translation termination in vivo." J Biol Chem 282(49): 35638-45.

Pekkala, M., R. Hieta, et al. (2004). "The peptide-substrate-binding domain of collagen prolyl 4-hydroxylases is a tetratricopeptide repeat domain with functional aromatic residues." J Biol Chem 279(50): 52255-61.
Pisarev, A. V., M. A. Skabkin, et al. (2010). "The role of ABCE1 in eukaryotic posttermination ribosomal recycling." Mol Cell 37(2): 196-210.
Rice, L. M. and A. T. Brunger (1999). "Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex disassembly." Mol Cell 4(1): 85-95.
Riggs, D. L., P. J. Roberts, et al. (2003). "The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo." Embo J 22(5): 1158-67.
Rout, M. P., J. D. Aitchison, et al. (2000). "The yeast nuclear pore complex: composition, architecture, and transport mechanism." J Cell Biol 148(4): 635-51.
Salas-Marco, J. and D. M. Bedwell (2004). "GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination." Mol Cell Biol 24(17): 7769-78.
Schneider, T. R. and G. M. Sheldrick (2002). "Substructure solution with SHELXD." Acta Crystallogr D Biol Crystallogr 58(Pt 10 Pt 2): 1772-9.
Sinars, C. R., J. Cheung-Flynn, et al. (2003). "Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes." Proc Natl Acad Sci U S A 100(3): 868-73.
Song, H., P. Mugnier, et al. (2000). "The crystal structure of human eukaryotic release factor eRF1--mechanism of stop codon recognition and peptidyl-tRNA hydrolysis." Cell 100(3): 311-21.
Stansfield, I., K. M. Jones, et al. (1995). "The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae." Embo J 14(17): 4365-73.
Steinmann, B., P. Bruckner, et al. (1991). "Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase." J Biol Chem 266(2): 1299-303.
Todd, B. L., E. V. Stewart, et al. (2006). "Sterol regulatory element binding protein is a principal regulator of anaerobic gene expression in fission yeast." Mol Cell Biol 26(7): 2817-31.
Trobro, S. and J. Aqvist (2007). "A model for how ribosomal release factors induce peptidyl-tRNA cleavage in termination of protein synthesis." Mol Cell 27(5): 758-66.
Van Dyke, M. W., L. D. Nelson, et al. (2004). "Stm1p, a G4 quadruplex and purine motif triplex nucleic acid-binding protein, interacts with ribosomes and subtelomeric Y' DNA in Saccharomyces cerevisiae." J Biol Chem 279(23): 24323-33.
von der Haar, T. and M. F. Tuite (2007). "Regulated translational bypass of stop codons in yeast." Trends Microbiol 15(2): 78-86.
Wang, J., B. T. Dye, et al. (2009). "Insights into anaphase promoting complex TPR subdomain assembly from a CDC26-APC6 structure." Nat Struct Mol Biol 16(9): 987-9.
Wu, B., P. Li, et al. (2004). "3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex." Proc Natl Acad Sci U S A 101(22): 8348-53.
Zavialov, A. V., L. Mora, et al. (2002). "Release of peptide promoted by the GGQ motif of class 1 release factors regulates the GTPase activity of RF3." Mol Cell 10(4): 789-98.
Zhouravleva, G., L. Frolova, et al. (1995). "Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3." Embo J 14(16): 4065-72.
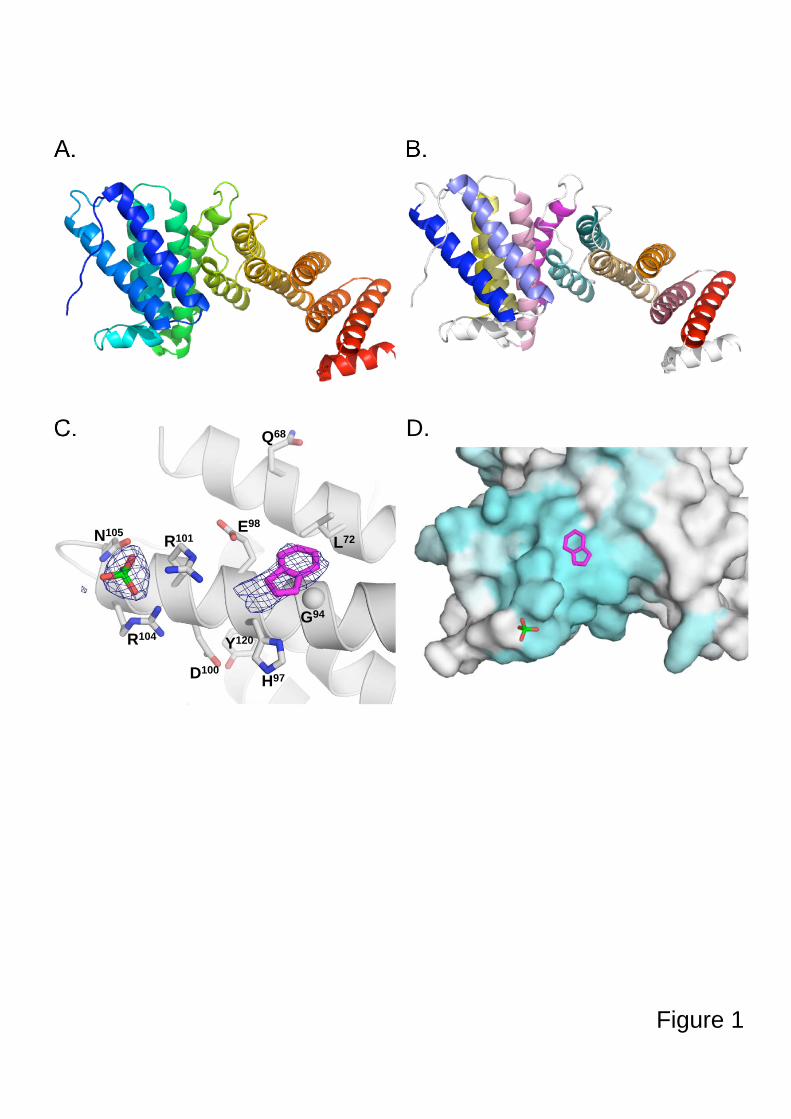
A.
B.
C. D.
Figure 1
Q68
L72
E98
H97
Y120
D100
R101
R104
N105
G94
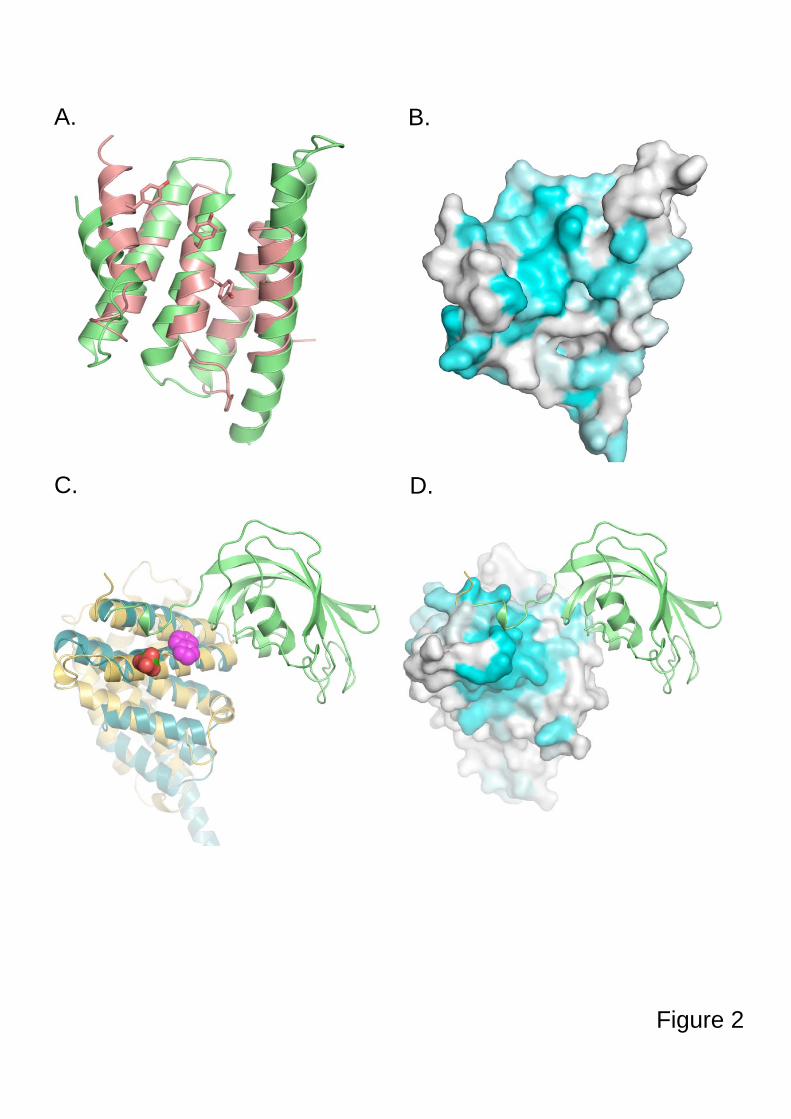
A. B.
Figure 2
C. D.
Legend to figure S1. Multiple sequence alignment of Nro1
Eukaryotic orthologs of Nro1 were aligned using ClustalW. Strictly conserved residues are in white on a black background. Partially conserved amino acids are boxed. Secondary structure elements assigned from the S. pombe Nro1 N54 structure are indicated above the alignment.


130
1.3. Etude biochimique de Tpa1-Ett1
A. Activité enzymatique dioxygénase in vitro
Tpa1 possède un domaine fortement homologue à la prolyl-hydroxylase PHD2 (Figure
33), et présentant généralement toutes les caractéristiques d'une 2OG-FeII dioxygénase. J'ai
souhaité déterminer si cette activité proposée pouvait être effectivement mesurée in vitro, et
s'il était possible de trouver un substrat pour l'enzyme. Considérant le phénotype de la
délétion de Tpa1 sur l'efficacité de terminaison et la co-purification de Tpa1 avec les
protéines eRF1, eRF3 et Pab1, nous avons émis l'hypothèse que Tpa1 modifiait l'une de ces
protéines en présence éventuelle du poly(A). D'autre part, l'homologue Ofd1 de S. pombe
cible Sre1 pour faciliter sa dégradation; Sre1 n'a pas été identifié chez S. cerevisiae, mais
d'autres facteurs de transcription sont connus pour réguler la réponse à l'hypoxie. En
particulier, Hap1 est une protéine impliquée dans la réponse à la carence en stérol, dont
l'activité dépend de la disponibilité de l'O2 et de l'hème. Le gène codant pour Hap1 est un
synthétique létal de gènes impliqués dans la voie des stérols (Upf2, Ecm22) et la délétion du
gène TPA1 dans une souche Dhap1/Dupc2/Decm22 supprime cette létalité (Valachovic et al.,
2006). Enfin, Hap1 présente une séquence conservée contenant une proline et plusieurs
résidus chargés positivement dans une zone peu structurée (King et al., 1999). Hap1 apparaît
donc comme un candidat intéressant pour l'activité dioxygénase de Tpa1.
Les 2OG-FeII dioxygénases oxydent un substrat en convertissant le 2OG et l'O2 en
succinate et CO2 (Figure 43). Pour les prolyl-4-hydroxylases, le substrat est le tripeptide
XP(A/G), dont un atome d’hydrogène de l’atome C4 de la proline est substitué par un
groupement hydroxyl OH (Chowdhury et al., 2009). L'apparition de succinate peut être
détectée par un système d'enzymes rapporteurs issus du cycle de Krebs : la succinyl-CoA-
synthase convertit le succinate en succinyl-CoA, qui est transformé par la pyruvate kinase en

131
pyruvate, qui est lui-même transformé en lactate dans une troisième étape par la lactate
déshydrogénase; cette dernière réaction est particulièrement intéressante puisqu'elle est
couplée à l'oxydation du NADH en NAD+. La disparition de NADH est proportionnelle à la
production de succinate initiale, et peut être suivie par mesure spectrophotométrique à 340nm,
longueur d’onde à laquelle seul le NADH absorbe (méthode décrite dans l'article (Luo et al.,
2006) et utilisée dans l'article (McCusker and Klinman, 2009)).

132
Figure 43 : Mesure de l'activité dioxygénase/prolyl-hydroxylase de Tpa1.
Le système d'enzymes rapporteurs utilisé couple la production de succinate à l'oxydation du NADH en NAD+. La diminution de l'absorbance à 340nm, spécifique du NADH dans ce mélange, est directement proportionnelle à la quantité de succinate produite (Luo et al., 2006).
Les dioxygénases ont une activité basale faible en présence de dioxygène et de 2-oxoglutarate, sans substrat protéique.
J'ai réalisé ces mesures enzymatiques indirectes pour Tpa1 seule, Ofd1 seule, Tpa1-Pab1-
poly(A), Tpa1-Pab1-poly(A)-eRF1-eRF3, avec un système mis en place au laboratoire ou un
système commercial (Megazyme). Alors que l'activité rapportée était observable pour du
succinate seul (contrôle positif), la différence d'absorbance à 340nm n'était significative pour
aucun mélange réactionnel testé. Enfin, j'ai réalisé ces mesures enzymatiques sur un domaine
de la protéine Hap1 purifiée contenant le peptide candidat, et sur le peptide lui-même sans
pouvoir mettre en évidence une activité spécifique. Afin de garantir les meilleures conditions
d'activité du site dioxygénase, j'ai répété les expériences à 5 pH différents (6,5; 7,0; 7,5; 8,0;
8,5) et avec divers métaux (Fe3+, Mn2+, Ni2+, Co2+), sans observer d'amélioration; les
XP(G/A) + O2 + 2-oxoglutarate XP(G/A) + CO2 + succinate
OH
Activité enzymatique rapportée :
suivi de l’absorbance à 340nm

133
domaines de Tpa1 exprimés et purifiés séparément n'avaient pas davantage d'activité, seuls ou
en combinaison équimolaire.
Les mesures in vitro de l'activité de Tpa1/Ofd1 par le système d'enzymes rapporteurs ont
été répétées en présence d'Ett1 et de Nro1ΔN54 respectivement. Comme pour la protéine seule,
je n'ai pas détecté d'activité dioxygénase dans les conditions testées. Le substrat et les
conditions d'activité restent donc à déterminer.
B. Interactions
a. Tpa1-Ett1 / Ofd1-Nro1ΔN54
Des études à grande échelle menées chez S. cerevisiae ont montré une interaction entre le
produit du gène YOR051c, la protéine Ett1 et Tpa1 (Krogan et al., 2006). C'est une approche
ciblant globalement les complexes protéiques qui a été employée : purification des protéines
par affinité en tandem (TAP) suivie d'une caractérisation par spectrométrie de masse MALDI-
TOF ou chromatographie en phase liquide. Dans notre article (Henri et al., 2010) nous
montrons que la délétion de YOR051c cause sur la terminaison de la traduction un effet
comparable et non additif à la délétion du gène codant pour Tpa1, suggérant que Tpa1 et Ett1
agissent dans la même voie. En parallèle, l'étude du système de régulation de Sre1 chez S.
pombe montre qu'Ofd1 agit de façon coordonnée avec Nro1 (negative regulator of Ofd1) (Lee
et al., 2009); les auteurs de l'article montrent une interaction directe entre les deux protéines
(Figure 44). Cependant les conditions appliquées pour mettre en évidence l'interaction sont
particulières, les auteurs ayant recours à un agent pontant (DSP) et en concentrant 10 fois les
fractions retenues (bound) pour les immunoprécipitations.

134
Figure 44 : Mise en évidence de l'interaction directe entre Ofd1 et Nro1 (extrait de (Lee et al., 2009).
A. Ofd1 et Nro1 co-localisent dans le noyau des cellules (détection de Myc-Nro1 par anticorps anti-Myc et d'Ofd1 par anticorps anti-Ofd1).
B. Nro1 co-immunoprécipite avec Ofd1 dans des extraits cellulaires en présence d'un agent pontant (DSP). Le western blot montre la détection de Nro1 et d'Ofd1 par leurs anticorps respectifs. C. L'interaction directe entre Nro1 et Ofd1 est observable in vitro en fixant Ofd1-GST sur des billes d'anticorps anti-GST. Nro1 n'est pas retenue par le GST seul. D. Nro1 fixe Ofd1 entière ou son domaine C-terminal, mais pas son domaine N-terminal.

135
Dans le but d'évaluer l'hypothèse de l'interaction entre Tpa1 et Ett1 nous avons cloné,
surexprimé et purifié Ett1 en fusion avec un tag streptavidine. La protéine montre une forte
stabilité aux températures élevées (une étape initiale de purification par chauffage 10 minutes
à 65°C) et bien que non His-taggée, elle est fortement retenue sur résine de NiNTA.
Cependant, la purification n'a pu être réalisée qu'à pH acide (50mM NaCitrate pH5,6, 200mM
NaCl, 5mM β-mercaptoethanol : tampon C) tandis que Tpa1 n'est stable qu'à pH neutre ou
faiblement basique. Il a été difficile de tester l'interaction entre les deux protéines mais je n'ai
pas observé de co-purification de Tpa1-His6 et Ett1-Strep sur NiNTA ou StrepTactin (Figure
45). De même, l'injection du mélange équimolaire sur chromatographie d'exclusion de taille
ne permet pas de coéluer les deux protéines. De plus, nos collaborateurs n’ont pas réussi à
mettre en évidence l’existence de ce complexe par TAP-tag sur des extraits de levure.
Avec la même approche l'interaction entre Ofd1 et Nro1 a été évaluée après co-
expression en polycistron des deux protéines. Ofd1 est alors Strep-taggée et Nro1ΔN54 His-
taggée, le surnageant après sonication est soumis à une purification par affinité sur résine
NiNTA. Comme cela était attendu Nro1ΔN54-His6 est retenue en grande quantité sur la résine;
Ofd1-Strep est également observée dans la fraction d'élution mais en moindre quantité : les
deux protéines semblent interagir faiblement. La faiblesse apparente de l'interaction se
confirme par chromatographie d'exclusion de taille : les deux protéines sont clairement
séparées lors du passage sur la colonne (Figure 46). L'interaction Ofd1-Nro1 est faible,
transitoire ou requiert des conditions particulières qui ne sont pas réunies.
Nous pouvons donc en conclure que soit cette interaction est très faible ou très rapide,
soit elle requiert des conditions particulières que nous n’avons pas testées : hypoxie,
partenaires supplémentaires.
136
Figure 45 : Essais de mise en évidence de l'interaction entre Tpa1 et Ett1.
(A gauche) Les culots de bactéries ayant surexprimé Tpa1 His-taggée (flèche orange) ou Ett1 Strep-taggée (flèche jaune) sont mélangés et soumis à une sonication, une centrifugation puis une purification d'affinité sur StrepTactin. Après le 4ème lavage (W4) Tpa1 est totalement absente des fractions éluées tandis qu'Ett1 est éluée seule.
(A droite) Chaque protéine est purifiée séparément par chromatographie d'affinité puis chromatographie d'exclusion de taille. Le mélange équimolaire Tpa1-Ett1 est soumis à une chromatographie d'exclusion de taille analytique (chromatogramme bleu); pour comparaison, le chromatogramme de Tpa1 injectée seule dans les mêmes conditions est superposé (chromatogramme marron). Les protéines sont éluées en deux pics distincts se recouvrant partiellement : le complexe, attendu à un volume d'élution inférieur aux deux espèces séparées, n'est pas observable.
StrepTactin
I FT W1 W2 W3W4 W5 E1 E2 E3 E4 E5 E6 R M
Mélange des culots
Expression Tpa1-His6 et Ett1-Strep Purification
de chaque protéine
Co-injection sur SEC
S200 HR 10/30
-! 75 -! 50
-! 250
-! 25
> >
<
<
75 -
50 -
250 -
25 -
A. Tpa1-Ett1
B. Ofd1-Nro1
Co-expression en polycistron
d’Ofd1-Strep et Nro1-His6 SEC S200 HL 16/60
M
<
< NiNTA
T FS FT W1 W2 E50 E100 E200 E400 E400’ R M
>
*
> -! 75
-! 50
-! 250
-! 25
Inj
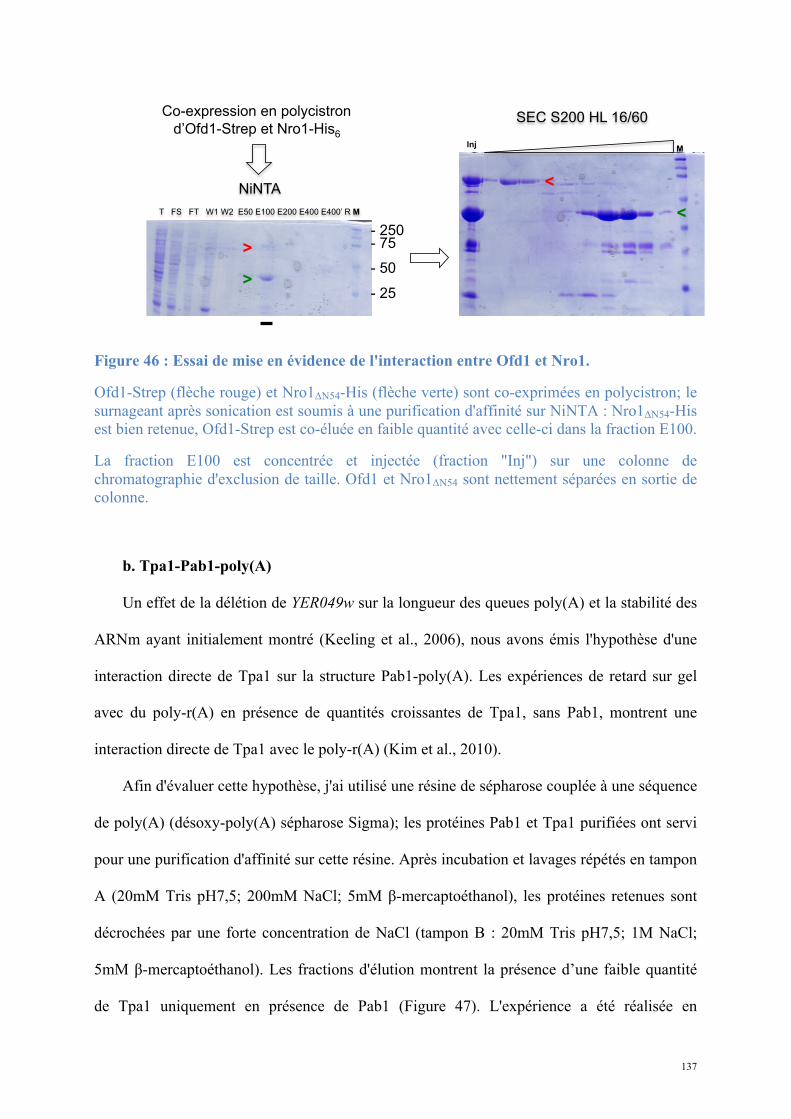
137
Figure 46 : Essai de mise en évidence de l'interaction entre Ofd1 et Nro1.
Ofd1-Strep (flèche rouge) et Nro1ΔN54-His (flèche verte) sont co-exprimées en polycistron; le surnageant après sonication est soumis à une purification d'affinité sur NiNTA : Nro1ΔN54-His est bien retenue, Ofd1-Strep est co-éluée en faible quantité avec celle-ci dans la fraction E100.
La fraction E100 est concentrée et injectée (fraction "Inj") sur une colonne de chromatographie d'exclusion de taille. Ofd1 et Nro1ΔN54 sont nettement séparées en sortie de colonne.
b. Tpa1-Pab1-poly(A)
Un effet de la délétion de YER049w sur la longueur des queues poly(A) et la stabilité des
ARNm ayant initialement montré (Keeling et al., 2006), nous avons émis l'hypothèse d'une
interaction directe de Tpa1 sur la structure Pab1-poly(A). Les expériences de retard sur gel
avec du poly-r(A) en présence de quantités croissantes de Tpa1, sans Pab1, montrent une
interaction directe de Tpa1 avec le poly-r(A) (Kim et al., 2010).
Afin d'évaluer cette hypothèse, j'ai utilisé une résine de sépharose couplée à une séquence
de poly(A) (désoxy-poly(A) sépharose Sigma); les protéines Pab1 et Tpa1 purifiées ont servi
pour une purification d'affinité sur cette résine. Après incubation et lavages répétés en tampon
A (20mM Tris pH7,5; 200mM NaCl; 5mM β-mercaptoéthanol), les protéines retenues sont
décrochées par une forte concentration de NaCl (tampon B : 20mM Tris pH7,5; 1M NaCl;
5mM β-mercaptoéthanol). Les fractions d'élution montrent la présence d’une faible quantité
de Tpa1 uniquement en présence de Pab1 (Figure 47). L'expérience a été réalisée en
StrepTactin
I FT W1 W2 W3W4 W5 E1 E2 E3 E4 E5 E6 R M
Mélange des culots
Expression Tpa1-His6 et Ett1-Strep Purification
de chaque protéine
Co-injection sur SEC
S200 HR 10/30
-! 75 -! 50
-! 250
-! 25
> >
<
<
75 -
50 -
250 -
25 -
A. Tpa1-Ett1
B. Ofd1-Nro1
Co-expression en polycistron
d’Ofd1-Strep et Nro1-His6 SEC S200 HL 16/60
M
<
< NiNTA
T FS FT W1 W2 E50 E100 E200 E400 E400’ R M
>
*
> -! 75
-! 50
-! 250
-! 25
Inj

138
remplaçant le poly(A) par un poly(U) (désoxy-poly(U) agarose Sigma). Dans ce cas, seule
Pab1 est retenue, Tpa1 ne l'est plus. Ce résultat est un argument en faveur d'une interaction
directe spécifique entre Tpa1 et la structure 3' des ARNm en présence de Pab1. Des
expériences complémentaires seront nécessaires pour approfondir cette étude.
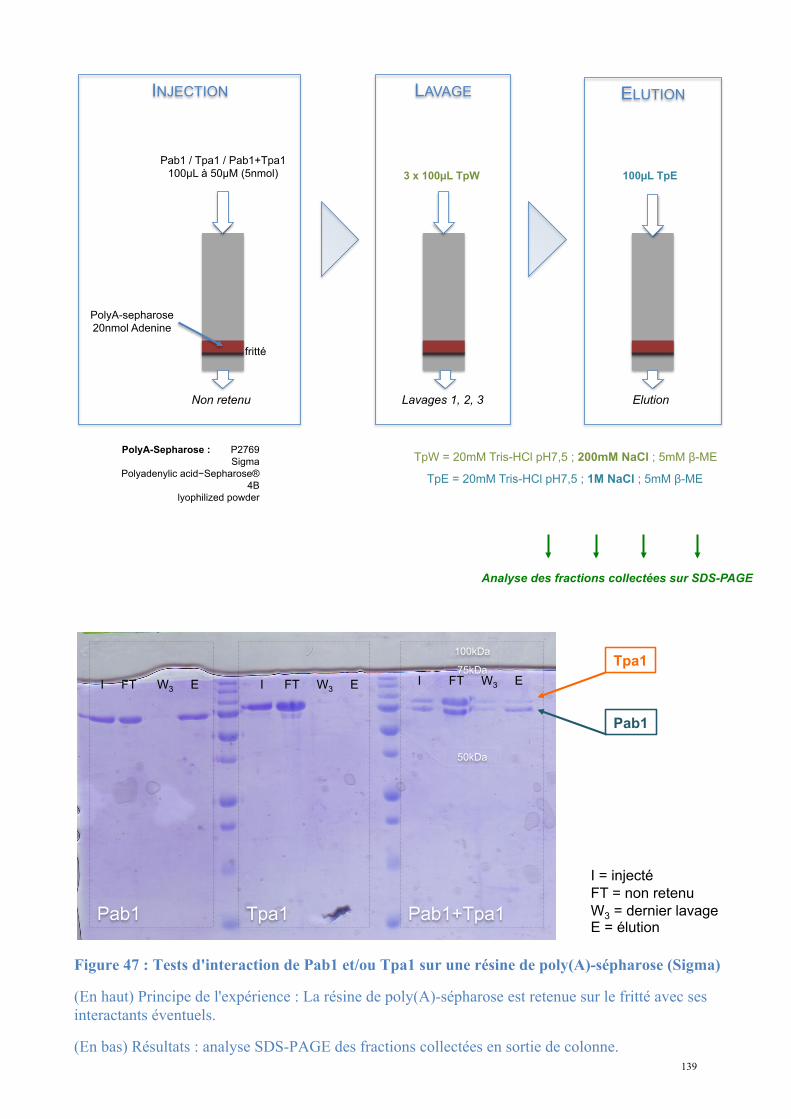
139
PolyA-sepharose
20nmol Adenine
INJECTION
Pab1 / Tpa1 / Pab1+Tpa1
100!L à 50!M (5nmol)
fritté
LAVAGE
3 x 100!L TpW
ELUTION
100!L TpE
TpW = 20mM Tris-HCl pH7,5 ; 200mM NaCl ; 5mM "-ME
TpE = 20mM Tris-HCl pH7,5 ; 1M NaCl ; 5mM "-ME
Non retenu Lavages 1, 2, 3 Elution
P2769
Sigma
Polyadenylic acid#Sepharose®
4$
lyophilized powder
PolyA-Sepharose :
Analyse des fractions collectées sur SDS-PAGE
Figure 47 : Tests d'interaction de Pab1 et/ou Tpa1 sur une résine de poly(A)-sépharose (Sigma)
(En haut) Principe de l'expérience : La résine de poly(A)-sépharose est retenue sur le fritté avec ses interactants éventuels.
(En bas) Résultats : analyse SDS-PAGE des fractions collectées en sortie de colonne.
100kDa
75kDa
50kDa
Pab1 Tpa1 Pab1+Tpa1
I FT W3 E I FT W3 E I FT W3 E
I = injecté
FT = non retenu
W3 = dernier lavage E = élution
Tpa1
Pab1

140
Je n'ai pas pu mettre en évidence d'interaction significative entre Tpa1 et Ett1, ni entre Ofd1 et
Nro1 in vitro. J'ai néanmoins utilisé l'expérience de fixation de Tpa1 sur Pab1-poly(A) pour
déterminer si Ett1 pouvait être retenue par le complexe Tpa1-Pab1-poly(A). Les résultats ne
montrent pas de fixation d’Ett1 sur Tpa1-Pab1-poly(A) (Figure 48). L'effet d'Ett1 sur
l'efficacité de terminaison ne semble pas lié à une interaction directe de la protéine sur Tpa1-
Pab1-poly(A) mais il pourrait nécessiter la présence de d'autres partenaires.
Figure 48 : Test d'interaction de Tpa1-Pab1-poly(A)-sépharose avec ou sans Ett1.
L'expérience précédente (Figure 47) est répétée en présence d'Ett1.
I = injecté
FT = non retenu
E = élution
Tpa1
Pab1
Ett1
100kDa
75kDa
50kDa
Pab1
+Tpa1
+Ett1
I FT E
Pab1
+Tpa1
I FT E

141
c. Tpa1-eRF1-eRF3-Pab1
Les conclusions des travaux de Keeling et ses collaborateurs proposent que Tpa1 fait
partie d'un complexe ribonucléoprotéique en 3' des ARNm reliant eRF3, Pab1 et Pan2-Pan3
(Keeling et al., 2006). Afin de déterminer si Tpa1 interagit effectivement avec eRF3 et Pab1
in vitro j'ai fixé eRF3-GST sur une résine de glutathion-sépharose et incubé les partenaires
potentiels avant des lavages répétés et une analyse des protéines retenues.
Parmi les conditions testées (eRF3entière, eRF1, Tpa1 / eRF3NM, Pab1, Tpa1 / eRF3C,
eRF1, Tpa1), je n’ai jamais pu mettre en évidence d’interaction entre Tpa1 et ces différentes
protéines. Si l'interaction existe, elle semble trop faible ou trop transitoire pour être observée.
La formation du complexe pourrait également requérir la présence de l'ARNm, ce qui n’a pas
été testé pour l’instant.

142
Conclusions, Perspectives
L'étude de Tpa1 révèle d'une part qu'elle joue un rôle dans la terminaison de la traduction
et l'adaptation cellulaire à l'hypoxie, et d'autre part que son fonctionnement est lié à un site
actif dioxygénase. L'activité de Tpa1 cible un ou plusieurs substrats, dont la modification
post-traductionnelle vraisemblablement par hydroxylation va affecter le fonctionnement. La
régulation de l'activité d'un substrat par modification post-traductionnelle est un mode
d'activation ou d'inactivation rapide et réversible d'une voie dans laquelle ce facteur est
impliqué. Ainsi en fonction des conditions environnementales, Tpa1 jouerait le rôle à la fois
de senseur et d'interrupteur sur des voies liées à la terminaison et à la réponse à l'hypoxie.
Nous pouvons postuler l'existence d'une protéine responsable de la réduction d'un substrat
oxydé par Tpa1, contrant ou compensant l'effet de cette dernière lorsque les conditions
environnementales changent et permettant ainsi le retour à un état normal.
La caractérisation de Tpa1 requiert encore l’identification d'un substrat direct de l'activité
prolyl-4-hydroxylase proposée. L'interaction avec le substrat selon le modèle de PHD2-HIF
suggère la fixation d'un peptide peu structuré. La seule contrainte forte étant le
positionnement d'une proline à proximité du centre [H...D...H]-Fe2+, un grand nombre de
protéines peuvent être la cible de Tpa1. De même, on peut supposer l'existence de substrats
multiples de la protéine, justifiant son apparente versatilité selon les organismes. Connaissant
des protéines cibles de son activité il y aura intérêt à comprendre le mode de reconnaissance
au niveau moléculaire en entreprenant la co-cristallisation de Tpa1 avec son substrat
peptidique ou entier. L'utilisation du mutant non catalytique [A...D...N] pourra être mise à
profit pour stabiliser le complexe enzyme-substrat sans affecter directement l'affinité.

143
Si le défaut de substrat spécifique semble être la cause évidente de l'inactivité de Tpa1 in
vitro, il est possible que Tpa1 ne soit active que lorsqu'elle subit des modifications post-
traductionnelles. Tpa1 exprimée chez E. coli ne présenterait logiquement pas les
modifications qu'elle pourrait subir in vivo dans la levure. Il sera ainsi informatif de
caractériser la masse de Tpa1 purifiée à partir d'extraits de levure. Si des modifications
apparaissent alors, nous pouvons émettre l'hypothèse qu'elles régulent l'activité dioxygénase
ou l'interaction avec le substrat en fonction des conditions environnementales.
A partir des données structurales de Tpa1 et de l'effet direct de son inactivation sur
l'efficacité de terminaison, on peut imaginer l'existence d'une voie de régulation de
l'expression en fonction des conditions environnementales. La compréhension du rôle de
Tpa1 dans la terminaison de la traduction serait facilitée si l'on parvient à établir un lien entre
l'hypoxie et l'efficacité de terminaison, la longueur des queues poly(A) et la stabilité des
ARNm. Plusieurs autres facteurs accessoires de la traduction pourraient être identifiés dans
cette voie potentielle d'adaptation par limitation des dépenses énergétiques liées à l'expression
génique globale.
Une piste intéressante pour l'interprétation de la fonction de Tpa1 se trouve dans l'étude
de la protéine Mod5 de S. cerevisiae (Benko et al., 2000) (Figure 49). Cette protéine catalyse
la modification post-transcriptionnelle d'un ARNt à partir de diméthylallyl pyrophosphate
(DMAPP). Une fois modifié, cet ARNt favorise la translecture des codons stop par insertion
d'une tyrosine. Cependant, le DMAPP est également le substrat d'Erg20, enzyme initiatrice de
la voie de biosynthèse des stérols. Les auteurs montrent qu'un niveau normal d'activité de
Mod5 et d'Erg20 assure une consommation équilibrée de DMAPP et la production normale
d'ARNt-modifié et de stérols. Cependant, lorsque l'activité d'Erg20 augmente, elle entre en

144
compétition avec Mod5 pour l'utilisation du substrat commun, entraînant une diminution de la
modification des ARNt. La présence d'ARNt non maturé entraîne une diminution de la
translecture des codons stop, augmentant l'efficacité de terminaison et conférant un effet
indirect à Erg20 dans l'efficacité de terminaison.
Ofd1, l’orthologue de Tpa1 se trouve indirectement impliquée dans le contrôle de la
synthèse des stérols via son effet sur Sre1 dans S. pombe. Ainsi, son effet sur l'efficacité de
terminaison pourrait être indirect. En effet, nous avons montré que la protéine Tpa1 réprime le
facteur de transcription Hap1 en présence d’O2. En condition d’hypoxie, Hap1 réprime
l’expression de gènes impliqués dans la voie de biosynthèse des stérols tels que Erg2, Erg5 et
Erg11 (Hickman MolCellBiol 2007). On peut ainsi imaginer le processus suivant. En
présence d’O2 Tpa1 réprime directement ou indirectement Hap1. Au contraire lors de
l’hypoxie, état mimé partiellement par la délétion du gène TPA1, le facteur de transcription
Hap1 n’est plus réprimé et va donc pouvoir réprimer les gènes de la voie de biosynthèse des
stérols. Par conséquent, plus de substrat sera disponible pour la protéine Mod5, générant ainsi
davantage d’ARNt suppresseur et donc davantage de translecture des codons stop. En résumé,
la délétion du gène TPA1 devrait conduire selon cette hypothèse à l’augmentation de la
translecture des codons stop, phénomène initialement observé par Keeling et collaborateurs
(Keeling et al., 2006) et dans notre étude (Henri, 2010). L’effet de Tpa1 sur la terminaison de
la traduction serait alors indirect. Cette hypothèse est à explorer en recherchant de nouvelles
cibles directes (prolyl-4-hydroxylation) et indirectes (facteurs de transcription) de Tpa1/Ofd1
sur la biosynthèse des stérols.
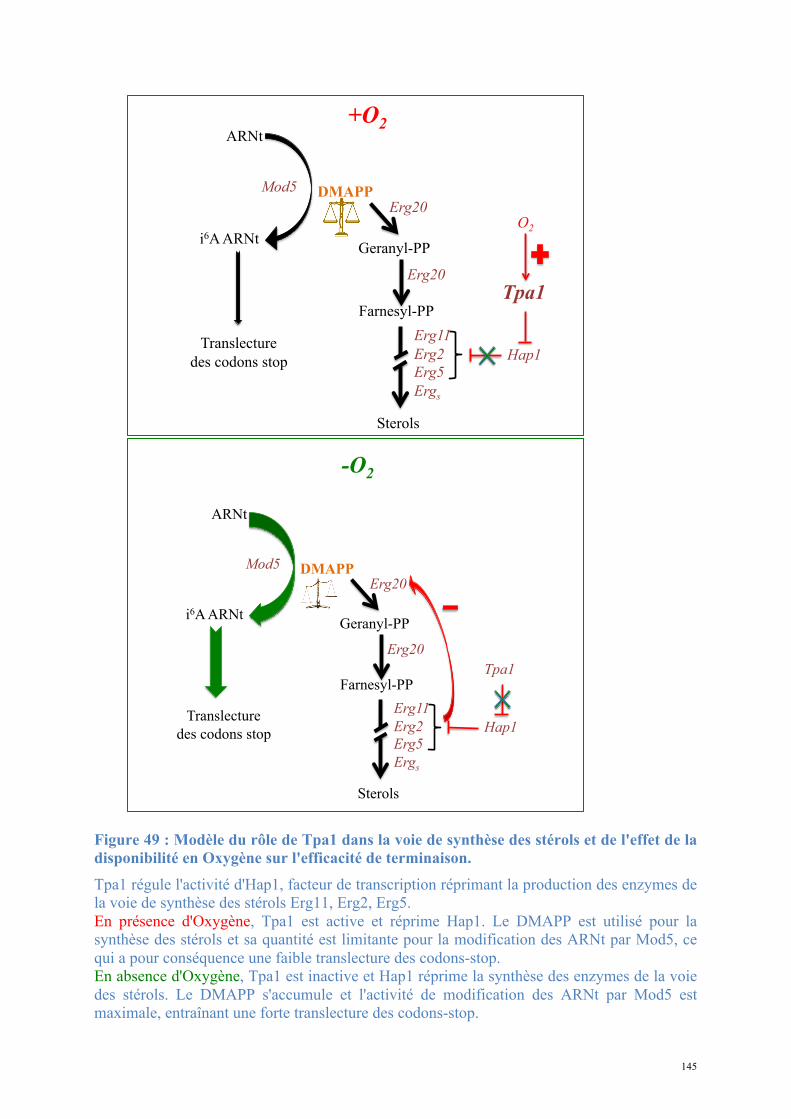
145
Figure 49 : Modèle du rôle de Tpa1 dans la voie de synthèse des stérols et de l'effet de la disponibilité en Oxygène sur l'efficacité de terminaison. Tpa1 régule l'activité d'Hap1, facteur de transcription réprimant la production des enzymes de la voie de synthèse des stérols Erg11, Erg2, Erg5. En présence d'Oxygène, Tpa1 est active et réprime Hap1. Le DMAPP est utilisé pour la synthèse des stérols et sa quantité est limitante pour la modification des ARNt par Mod5, ce qui a pour conséquence une faible translecture des codons-stop. En absence d'Oxygène, Tpa1 est inactive et Hap1 réprime la synthèse des enzymes de la voie des stérols. Le DMAPP s'accumule et l'activité de modification des ARNt par Mod5 est maximale, entraînant une forte translecture des codons-stop.
Erg20 DMAPP
Geranyl-PP
ARNt
i6A ARNt
Mod5
Erg20
Farnesyl-PP
Erg11
Erg2
Erg5
Ergs
Sterols
Translecture
des codons stop Hap1
Tpa1
+O2
O2
Erg20 DMAPP
Geranyl-PP
ARNt
i6A ARNt
Mod5
Erg20
Farnesyl-PP
Erg11
Erg2
Erg5
Ergs
Sterols
Translecture
des codons stop Hap1
Tpa1
-O2

146
La résolution de la structure de Nro1 de S. pombe, orthologue d'Ett1 de S. cerevisiae, a
mis en évidence son repliement TPR caractéristique des protéines clefs des interactions
protéine-protéine et protéine-peptide. La protéine est une superhélice monomérique. Elle
présente une surface très conservée contenant une signature RXLR fixant un SO42- et une
molécule non identifiée. Cette région pourrait être impliquée dans l’interaction avec un
ligand. L'homologie structurale de Tpa1 et Ett1 avec les domaines catalytiques et de fixation
du substrat de la Collagène-prolyl-4-hydroxylase suggère un mode de fonctionnement
similaire. Cependant l'opposition apparente entre Ofd1, négativement régulée par Nro1, et
Tpa1, ayant un effet nécessitant la présence d'Ett1, reste sans réponse. Il est probable que les
modes d'interaction entre Nro1/Ett1 et les substrats soient différents.
L'identification d'un substrat d'activité d'Ofd1/Tpa1 est ici encore nécessaire pour une
caractérisation plus précise du système. Il sera alors envisageable de déterminer si ce substrat
est également fixé par Nro1/Ett1, si l'activité dioxygénase est favorisée ou inhibée par cette
fixation, et de tenter une co-cristallisation de la protéine avec le peptide.
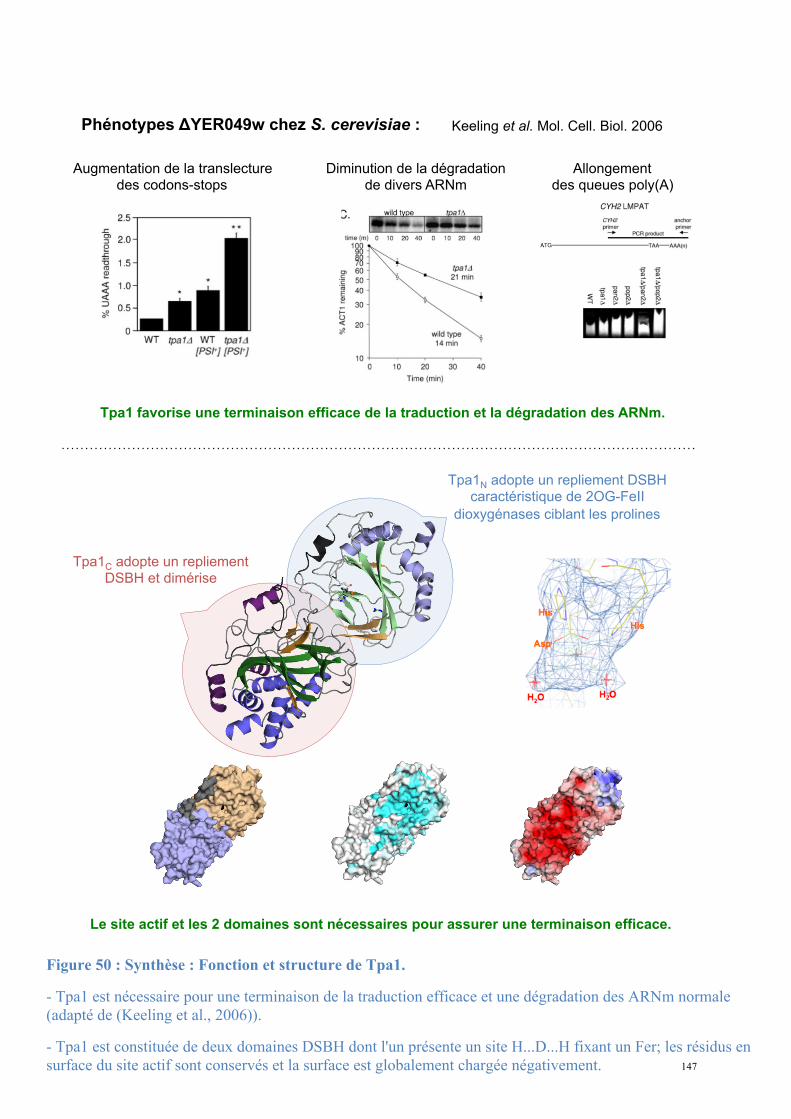
147
Keeling et al. Mol. Cell. Biol. 2006 Phénotypes !YER049w chez S. cerevisiae :
Tpa1 favorise une terminaison efficace de la traduction et la dégradation des ARNm.
Augmentation de la translecture des codons-stops
Diminution de la dégradation de divers ARNm
Allongement des queues poly(A)
Tpa1N adopte un repliement DSBH caractéristique de 2OG-FeII
dioxygénases ciblant les prolines
Tpa1C adopte un repliement DSBH et dimérise
Le site actif et les 2 domaines sont nécessaires pour assurer une terminaison efficace.
Figure 50 : Synthèse : Fonction et structure de Tpa1.
- Tpa1 est nécessaire pour une terminaison de la traduction efficace et une dégradation des ARNm normale (adapté de (Keeling et al., 2006)).
- Tpa1 est constituée de deux domaines DSBH dont l'un présente un site H...D...H fixant un Fer; les résidus en surface du site actif sont conservés et la surface est globalement chargée négativement.
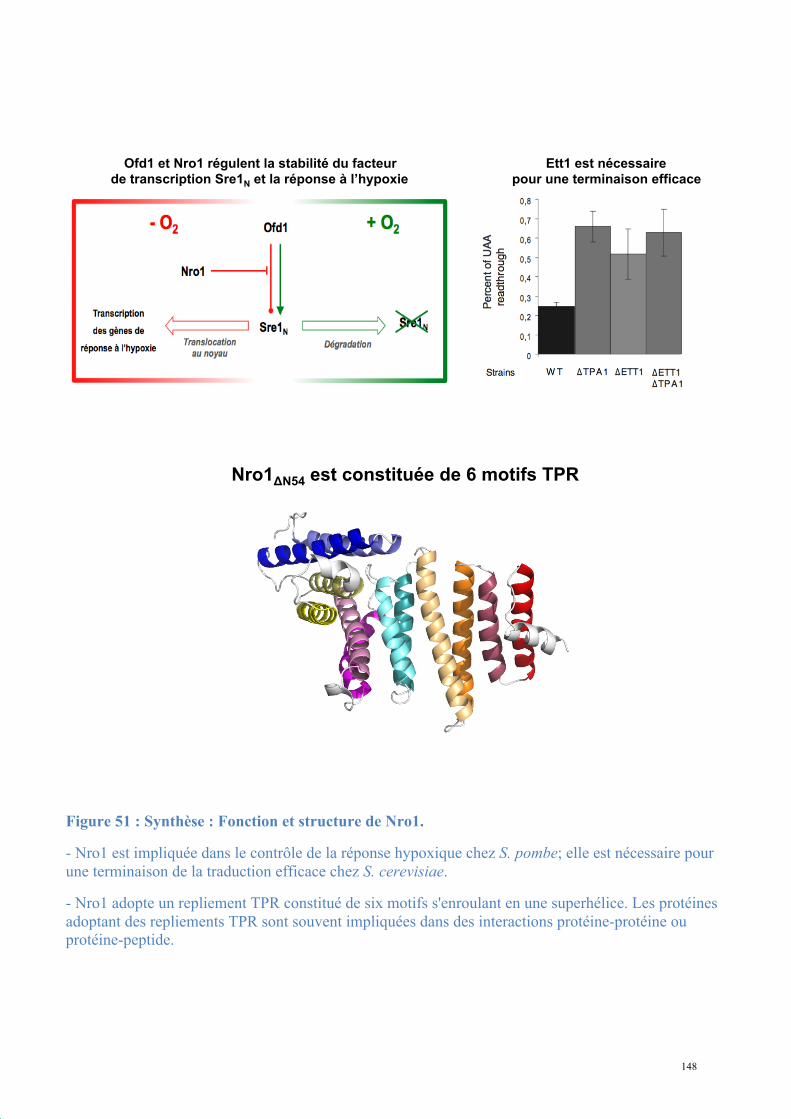
148
B. 1. 2.
3. 4.
A. 1. 2.
3. 4.
Ett1 est nécessaire
pour une terminaison efficace
Nro1!N54 est constituée de 6 motifs TPR
Ofd1 et Nro1 régulent la stabilité du facteur
de transcription Sre1N et la réponse à l’hypoxie
B. 1. 2.
3. 4.
A. 1. 2.
3. 4.
Ett1 est nécessaire
pour une terminaison efficace
Nro1!N56 est constituée de 6 motifs TPR
Ofd1 et Nro1 régulent la stabilité du facteur
de transcription Sre1N et la réponse à l’hypoxie
Figure 51 : Synthèse : Fonction et structure de Nro1.
- Nro1 est impliquée dans le contrôle de la réponse hypoxique chez S. pombe; elle est nécessaire pour une terminaison de la traduction efficace chez S. cerevisiae.
- Nro1 adopte un repliement TPR constitué de six motifs s'enroulant en une superhélice. Les protéines adoptant des repliements TPR sont souvent impliquées dans des interactions protéine-protéine ou protéine-peptide.


149
2. Dom34-Hbs1, complexe central du No-Go Decay et
du Non-functional rRNA Decay
Au cours de l'expression génique des altérations de l'ARN peuvent réduire l'efficacité de
l'élongation de la traduction. Les cellules eucaryotes possèdent des voies de reconnaissance
des ribosomes en pause. Cet arrêt peut être causé par :
- l'ARNm : présence dans le cadre de lecture d'une tige-boucle (NGD, (Doma and Parker,
2006)) ou d'un nucléotide dépuriné et donc non décodé par le ribosome (Gandhi et al., 2008);
- l'ARNr lui-même : la NRD cible les ribosomes non fonctionnels (soit du à un ARNr
18S non fonctionnel soit du fait d'un défaut de biogénèse de la petite-sous-unité du ribosome)
entrés en élongation (LaRiviere et al., 2006; Soudet et al., 2010).
Dans tous les cas, ces pauses du ribosome induisent une dégradation de l'ARN fautif. Les
protéines Dom34 et Hbs1, conservées chez tous les eucaryotes, ont des séquences et des
structures homologues aux facteurs de terminaison eRF1 et eRF3, respectivement. Comme
eRF1-eRF3, elles forment un complexe 1:1 qui fixe les nucléotides GTP et GDP. Elles jouent
un rôle central dans la NGD, la dégradation des ARNm apuriniques et la NRD, en étant
nécessaires au clivage de l'ARNm ou à la dégradation de l'ARNr 18S.
La structure de Dom34 a été résolue précédemment au laboratoire (Graille et al., 2008).
Afin de compléter notre compréhension du système Dom34-Hbs1 dans la dégradation des
ARN induisant des pauses dans l'élongation de la traduction nous avons entrepris la
caractérisation structurale et biochimique d'Hbs1 et du complexe Hbs1-Dom34.

150
2.1. Procédures expérimentales
2.1.1. Expression et purification de Dom34 et Hbs1
Comme préalablement mis au point, Dom34 s'exprime en grandes quantités en E. coli
BL21 DE3 Gold à 15°C. La purification en deux étapes par NiNTA-SEC en tampon A donne
de bons résultats, même si la protéine n'est pas stable au-delà de quelques heures.
La protéine Hbs1 s'exprime également très bien dans les mêmes conditions, et la
purification est satisfaisante en NiNTA-SEC en tampon A. Cependant aucune piste de
cristallisation exploitable n'a été obtenue avec la protéine entière. La structure primaire
d'Hbs1 indique une organisation en domaine du même type que celle d'eRF3 : un domaine N-
terminal peu conservé, un domaine GTPase et deux domaines C-terminaux repliés en
tonneaux-β. Afin d'éliminer les parties non structurées de la protéine et de faciliter la
cristallogenèse une série de séquences codant pour Hbs1 tronquée en N-terminal ont été
construites (ΔN109, ΔN134, ΔN162). Les deux premières constructions conservent leur
capacité d'interaction avec Dom34. Seul le fragment Hbs1ΔN134 a permis d'obtenir des cristaux
exploitables pour résoudre la structure de cette protéine.
2.1.2. Cristallisation
La protéine Hbs1ΔN134-apo (seule) ou pré-incubée avec du GDP donne des cristaux
réguliers de grande taille dès les conditions initiales des criblages larges en 0,1M Tris pH8,5,
12% PEG-6000, 150mM NaCl (Figure 52). La protéine marquée par des Séléno-méthionines,
produite dans les mêmes conditions, cristallise seule en 0,1M Hepes pH7, 15% PEG-4000,
0,1M MgCl2. Les cristaux sont des prismes réguliers de grande taille dans les gouttes des
screens, suffisant pour envisager des essais de diffraction. Les cristaux sont congelés dans
l'azote liquide après cryoprotection en 15 puis 30% d'éthylène glycol en complément de la
solution de cristallisation du réservoir.

151
Des cristaux de Hbs1 pré-incubée avec du GTPγS ont également été obtenus dans des
conditions proches, mais ne diffractaient pas au-delà de 8Å malgré diverses tentatives
d'optimisation.
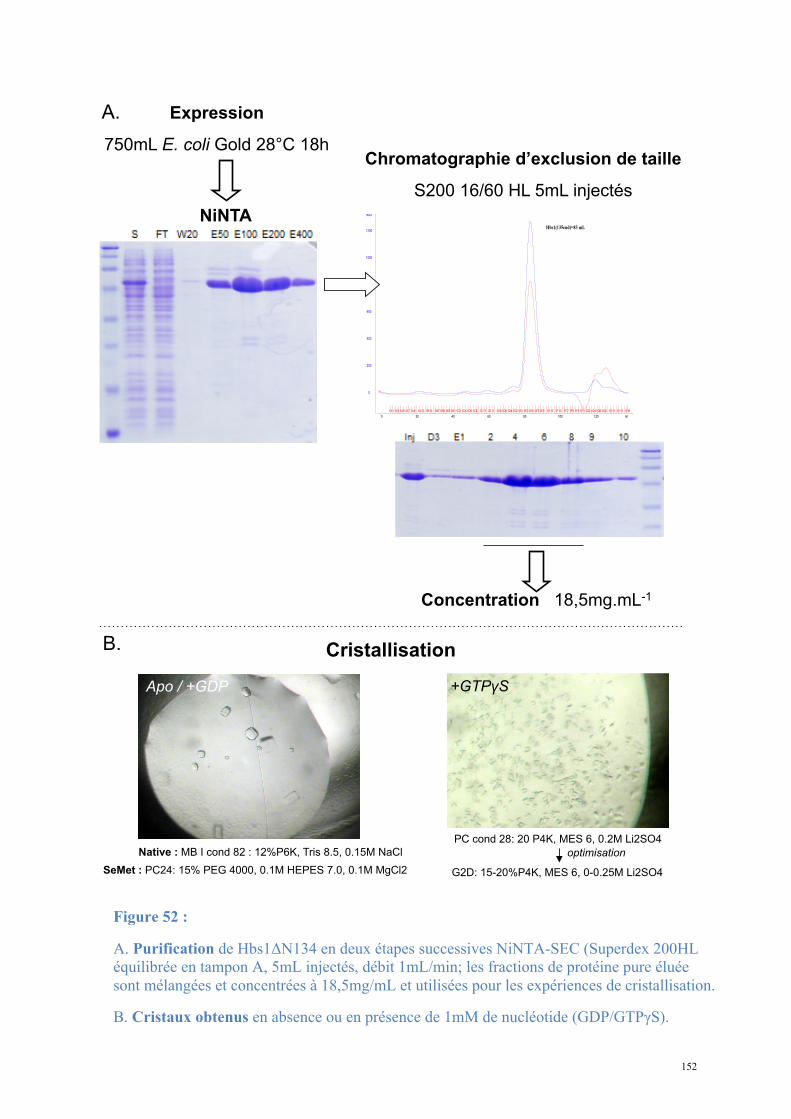
152
Expression
750mL E. coli Gold 28°C 18h
NiNTA
Chromatographie d’exclusion de taille
S200 16/60 HL 5mL injectés
Concentration 18,5mg.mL-1
PC cond 28: 20 P4K, MES 6, 0.2M Li2SO4
G2D: 15-20%P4K, MES 6, 0-0.25M Li2SO4
optimisation
+GTP!S
Native : MB I cond 82 : 12%P6K, Tris 8.5, 0.15M NaCl
SeMet : PC24: 15% PEG 4000, 0.1M HEPES 7.0, 0.1M MgCl2
Apo / +GDP
Cristallisation
A.
B.
Figure 52 :
A. Purification de Hbs1ΔN134 en deux étapes successives NiNTA-SEC (Superdex 200HL équilibrée en tampon A, 5mL injectés, débit 1mL/min; les fractions de protéine pure éluée sont mélangées et concentrées à 18,5mg/mL et utilisées pour les expériences de cristallisation.
B. Cristaux obtenus en absence ou en présence de 1mM de nucléotide (GDP/GTPγS).
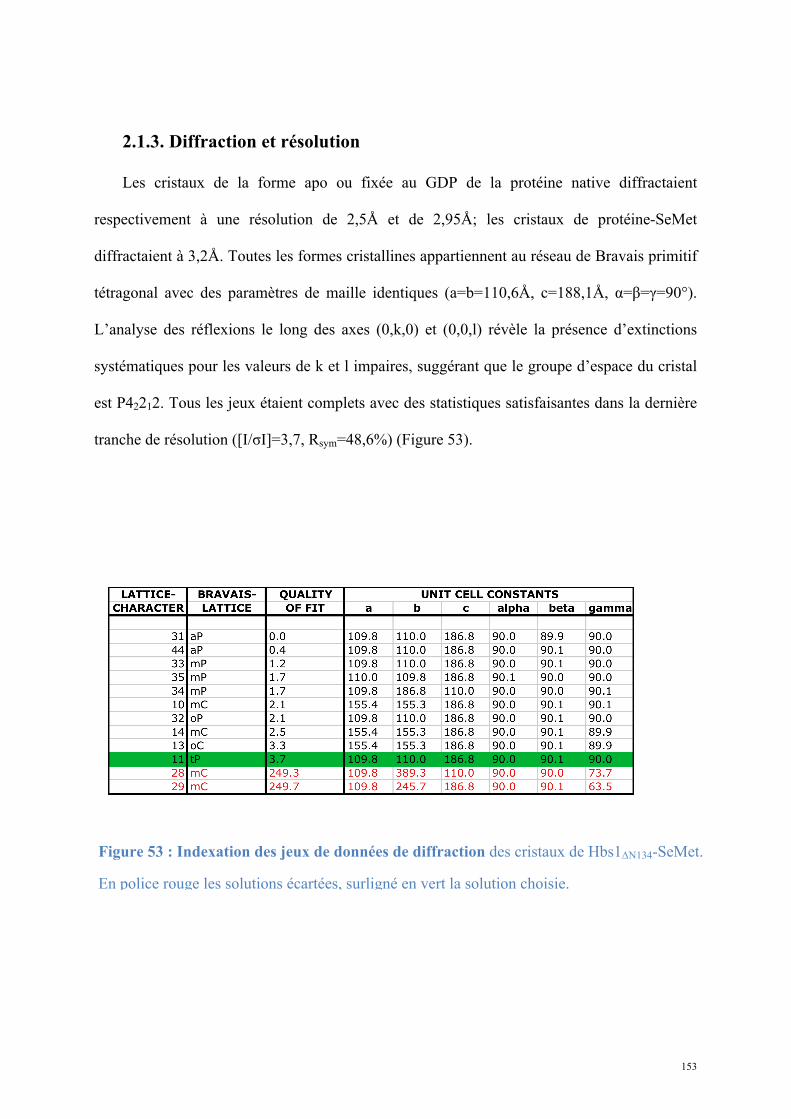
153
2.1.3. Diffraction et résolution
Les cristaux de la forme apo ou fixée au GDP de la protéine native diffractaient
respectivement à une résolution de 2,5Å et de 2,95Å; les cristaux de protéine-SeMet
diffractaient à 3,2Å. Toutes les formes cristallines appartiennent au réseau de Bravais primitif
tétragonal avec des paramètres de maille identiques (a=b=110,6Å, c=188,1Å, α=β=γ=90°).
L’analyse des réflexions le long des axes (0,k,0) et (0,0,l) révèle la présence d’extinctions
systématiques pour les valeurs de k et l impaires, suggérant que le groupe d’espace du cristal
est P42212. Tous les jeux étaient complets avec des statistiques satisfaisantes dans la dernière
tranche de résolution ([I/σI]=3,7, Rsym=48,6%) (Figure 53).
Indexation jeu de données de diffraction Se-Met
Figure 53 : Indexation des jeux de données de diffraction des cristaux de Hbs1ΔN134-SeMet.
En police rouge les solutions écartées, surligné en vert la solution choisie.

154
Le signal anomal du sélénium enregistré à la longueur d'onde du pic de diffusion anomale
sur les cristaux SeMet a permis de déterminer les phases et de construire un premier modèle
complet d'Hbs1ΔN134-apo. J'ai utilisé les programmes SOLVE-RESOLVE pour détecter les
sites sélénium, déterminer les phases, les optimiser par aplatissement de solvant et calculer les
cartes de densité électronique. La construction automatique du modèle a été réalisée par le
programme BUCCANEER (Cowtan, 2006) puis complétée par des cycles de reconstruction
manuelle et d'affinement avec les programmes COOT et PHENIX.REFINE, respectivement
(Figure 54). La structure de la forme Hbs1-GDP a été résolue par remplacement moléculaire
avec la structure Hbs1-apo comme modèle en utilisant le programme MOLREP.
Les structures finales présentent les statistiques suivantes (Figure 54) :
- Forme apo : Rwork / Rfree = 0,216/0,271, 100% de résidus dans les zones acceptées du
diagramme de Ramachandran, rmsdbond=0,006Å, rmsdangles=1,02°.
- Forme GDP : Rwork / Rfree = 0,214/0,277, 100% de résidus dans les valeurs acceptées du
diagramme de Ramachandran, rmsbond=0,011Å, rmsangles=1,358°. La structure d'Hbs1
complexée au GDP est très proche de la structure apo (rmsd = 0,4Å sur 430 Cα) : le GDP
n'induit pas de changement majeur dans la structure globale (Figure 55).
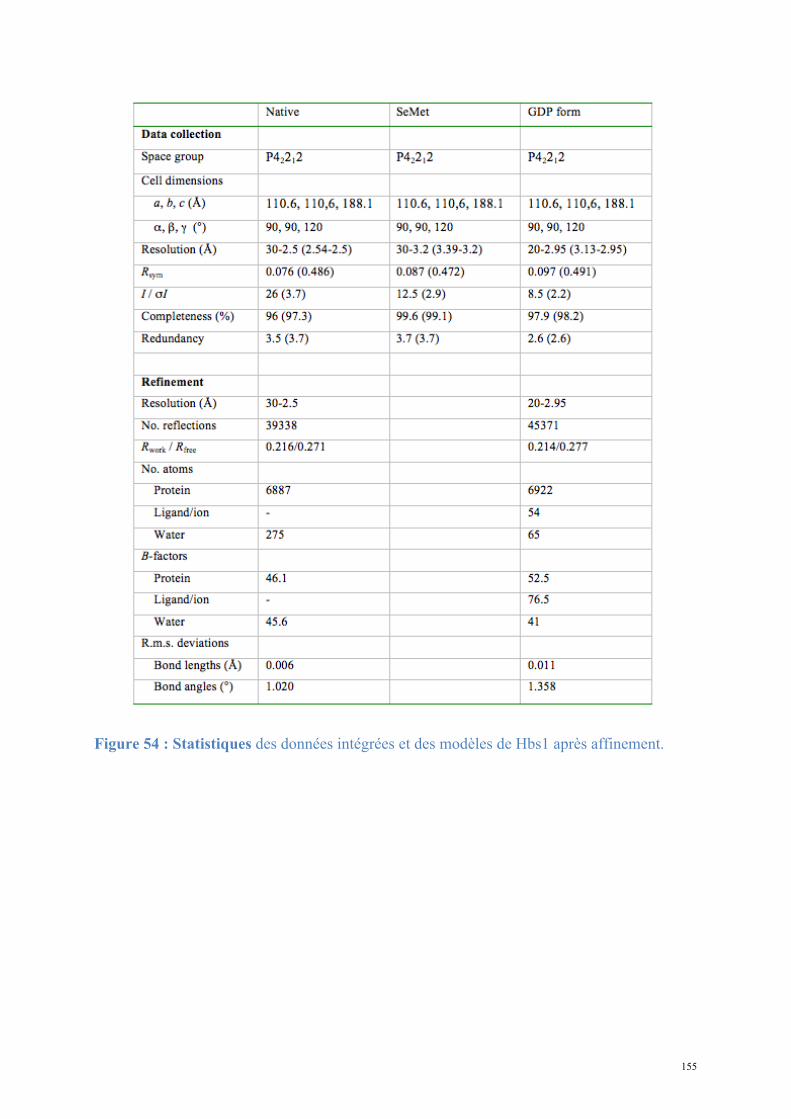
155
Figure 54 : Statistiques des données intégrées et des modèles de Hbs1 après affinement.

156
Figure 55 : Carte de densité électronique contourée à 1σ de Hbs1 après le dernier affinement; la protéine est colorée en jaune.
Figure 56 : Carte de densité électronique contourée à 1σ au niveau du site de fixation du GTP/GDP : (à gauche) forme apo, (à droite) forme GDP.

157
2.2. Détermination de la structure : Résultats
J'ai résolu la structure de Hbs1ΔN134 seule à 2,5Å et à 2,95Å en complexe avec du GDP.
La protéine forme un dimère dans l'unité asymétrique. Cependant le profil d'élution de SEC et
les propriétés de diffusion des rayons X aux petits angles (SAXS) montrent que la protéine est
monomérique en solution.
Hbs1 forme un prisme triangulaire de 50Å de côté et 30Å de profondeur (Figure 57 A.).
Comme les alignements de séquence le laissaient penser, la protéine est constituée des trois
domaines caractéristiques des GTPases impliquées dans la traduction dont eRF3 et EF-Tu
font partie : un domaine GTPase central (résidus 165 à 375) et deux tonneaux-β en C-terminal
(domaines II, résidus 399 à 498, et III, résidus 499 à 610); une extension en N-terminal est
largement non-repliée (résidus 140 à 164), mais interagit avec le domaine II.
Hbs1 adopte une structure fermée : le domaine GTPase est très proche des domaines II et
III. Ceci la rapproche d'EF-Tu mais la différencie d'eRF3 dont le domaine GTPase est
indépendant des deux autres, dans une forme globalement ouverte. Des mesures de SAXS sur
Hbs1 montrent que la structure en solution correspond bien à la forme fermée observée dans
le cristal et non une forme ouverte de type eRF3 (Figure 57 C.).
Le domaine GTPase d'Hbs1 adopte le repliement classique des GTPases EF-Tu, eRF3 et
Ras (rmsd=2,8-3Å sur 195 Cα). Un feuillet β central est entouré de sept hélices α. Deux
segments flexibles conservées sont responsables de la fixation du nucléotide : les switchs I et
II (Figure 57). La région switch I n'apparait pas dans les cartes de la forme apo mais devient
partiellement ordonnée en présence du nucléotide GDP. Celui-ci est fixé par quatre séquences
: les switchs I et II, la boucle G4 et la boucle P. Les résidus Val176, Asp177, Lys180, Ser181,
Thr182, His255, Asn313, Lys314, Asp316 et Phe354 sont impliqués dans la fixation du nucléotide
par Hbs1 (Figure 57 B.).
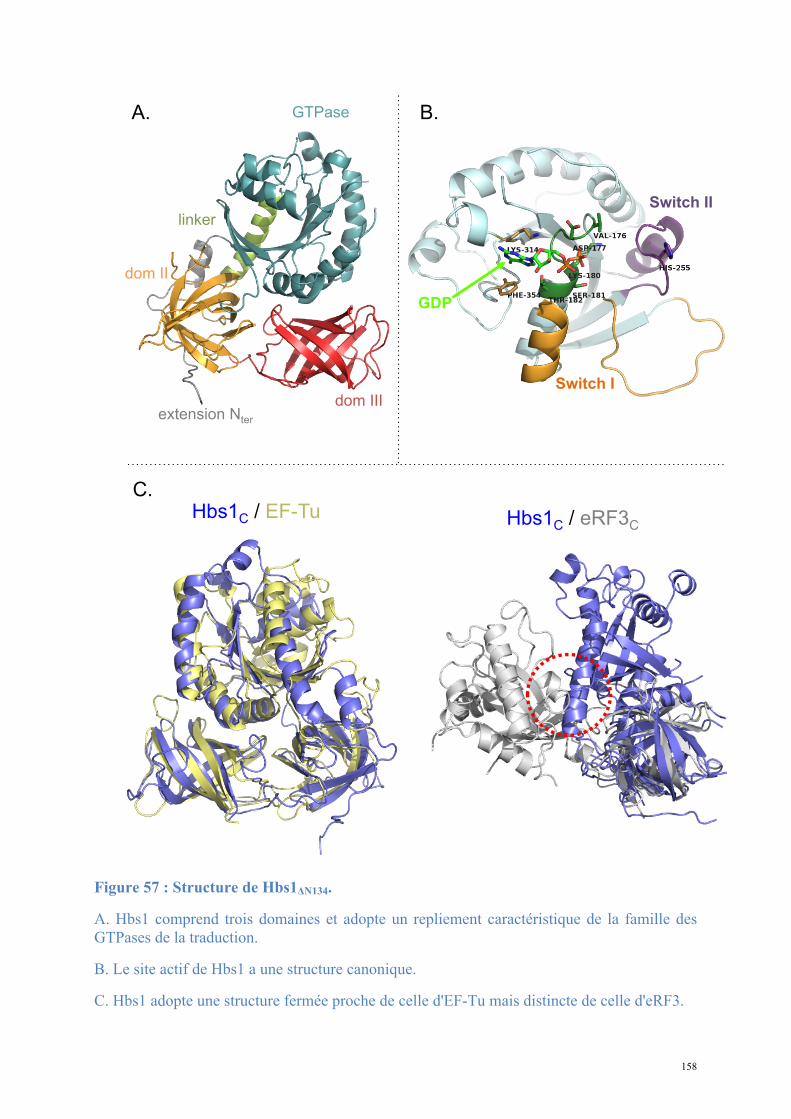
158
Figure 57 : Structure de Hbs1ΔN134.
A. Hbs1 comprend trois domaines et adopte un repliement caractéristique de la famille des GTPases de la traduction.
B. Le site actif de Hbs1 a une structure canonique.
C. Hbs1 adopte une structure fermée proche de celle d'EF-Tu mais distincte de celle d'eRF3.
Hbs1C / eRF3
C Hbs1
C / EF-Tu
GTPase
dom II
dom III extension N
ter
linker
Switch I
Switch II
GDP
A. B.
C.

159
Disposant des structures de Dom34 et Hbs1, ainsi que de la structure du complexe entre
leurs homologues eRF1-eRF3, nous avons construit des modèles du complexe Dom34-Hbs1
et testé leur validité en solution par diffusion des rayons X aux petits angles (SAXS, en
collaboration avec Dominique Durand de l'équipe de Conformation des macromolécules
biologiques en solution, à l'IBBMC) (Article 3 Figure 2.).
2.3. Caractérisations fonctionnelles
2.3.1. Affinité pour le GDP et le GTP
L'identification du site de fixation du GDP sur Hbs1 grâce à cette structure permet
d'identifier un ensemble de résidus impliqués dans l'interaction. Parmi les résidus cités, deux
ont déjà été identifiés comme essentiels pour la complémentation de la délétion d'Hbs1 in
vivo; ce sont les résidus Val176 et His255. Le résidu Lys180 de la boucle P qui est critique pour
la fixation des nucléotides par les protéines G intervient également dans la fixation.
Nous avons généré des mutants ponctuels pour chacun de ces résidus : Val176 substituée
par une Glycine, His255 substitué par un Glutamate et Lys180 substituée par une Alanine. J'ai
sous-cloné les mutants d'un plasmide d'expression de levure (fourni par nos collaborateurs)
vers le plasmide d'expression bactérien pET21a en fusion avec un tag hexa-histidine et
procédé à la purification comme décrit pour la protéine sauvage. Les rendements de
purification, bien que plus faibles, permettaient de produire suffisamment de matériel pour les
mesures d'affinité pour le GDP et le GTP.
Dans un premier temps, le repliement et la stabilité des protéines sauvages et mutantes
ont été évalués par dichroïsme circulaire (CD) et par calorimétrie différentielle à balayage
(DSC) (Figure 58). Les spectres CD des protéines sauvage et mutantes sont très similaires; les
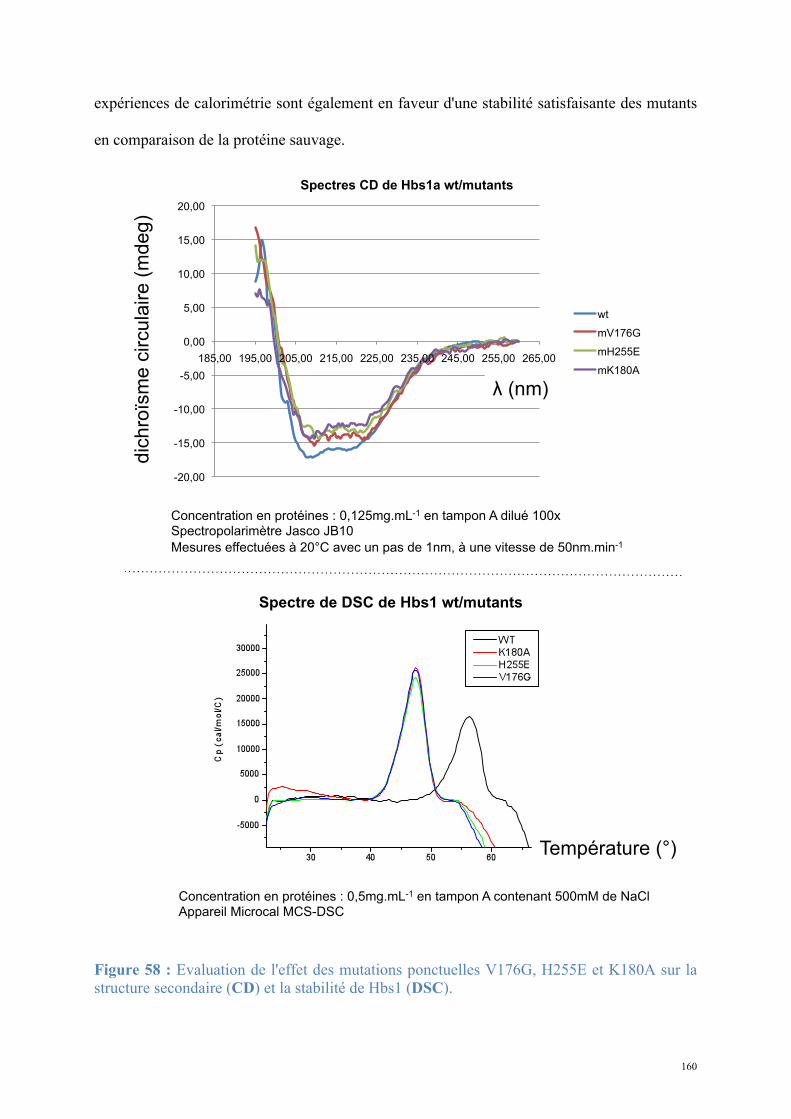
160
expériences de calorimétrie sont également en faveur d'une stabilité satisfaisante des mutants
en comparaison de la protéine sauvage.
Figure 58 : Evaluation de l'effet des mutations ponctuelles V176G, H255E et K180A sur la structure secondaire (CD) et la stabilité de Hbs1 (DSC).
-20,00
-15,00
-10,00
-5,00
0,00
5,00
10,00
15,00
20,00
185,00 195,00 205,00 215,00 225,00 235,00 245,00 255,00 265,00
Spectres CD de Hbs1a wt/mutants
wt
mV176G
mH255E
mK180A
dic
hro
ïsm
e c
ircu
laire
(m
de
g)
Concentration en protéines : 0,125mg.mL-1 en tampon A dilué 100x Spectropolarimètre Jasco JB10
Mesures effectuées à 20°C avec un pas de 1nm, à une vitesse de 50nm.min-1
! (nm)
Spectre de DSC de Hbs1 wt/mutants
Concentration en protéines : 0,5mg.mL-1 en tampon A contenant 500mM de NaCl Appareil Microcal MCS-DSC
Température (°)

161
Ensuite, j’ai utilisé deux méthodes pour évaluer l’aptitude de ces mutants à fixer les
nucléotides GDP et GTP.
Les expériences initiales de différence de fluorescence utilisaient des dérivés fluorescents
des nucléotides GDP et GTP (les nucléotides sont couplés au méthylanthraniloyl ;
λexmax=349nm, λém
max=458nm). Les expériences ont été réalisées à 25°C en tampon A
+2,5mM MgCl2 et 10% Glycérol. La fixation du nucléotide fluorescent par la protéine change
l'environnement immédiat de celui-ci, entraînant une diminution de la fluorescence. Cet effet
est utilisé pour déterminer l'affinité des formes sauvages ou mutantes d'Hbs1 (2µM) pour le
GDP ou le GTP, par ajouts successifs de nucléotides (2µM) jusqu'à stabilisation (saturation)
de la différence de fluorescence. La valeur de la différence de fluorescence à demi-saturation
correspond au Kd de la protéine pour le ligand. Les résultats de l'expérience montrent une
diminution significative de la fixation pour toutes les mutations des résidus : Val176Gly,
His255Glu et Lys180Ala nécessaires pour la fixation du GDP ou du GTP par Hbs1.
Cependant, malgré la répétition des expériences de fluorescence dans les mêmes
conditions (au moins trois mesures par mutants), les résultats montraient une dispersion forte
à chaque point de mesure. Ceci est du au fait que l’on fait varier la concentration de la
molécule dont on suit le signal de fluorescence, introduisant ainsi un fort bruit de fond. Par
conséquent, les barres d’erreur sur les valeurs de Kd étaient trop importantes.
Nous avons donc de nouveau purifié les protéines pour mesurer l’affinité par
microcalorimétrie par titration isotherme (ITC), équipement disponible sur la plate-forme de
biophysique de l’IBBMC (collaboration avec Magali Aumont-Nicaise de l'équipe de
Modélisation et d'Ingénierie des Protéines). Pour cela, 20 injections successives de 2µL des
nucléotides GDP et GTP (250µM) dans une solution contenant la protéine Hbs1 sauvage ou

162
les mutants (20µM en tampon A) ont été réalisées. La fixation de nucléotide sur la protéine
cause une consommation d'énergie sous forme de chaleur, mesurable pour la protéine sauvage
mais non mesurable pour les mutants.
Les résultats confirment donc les expériences antérieures avec une plus grande précision.
Le Kd d'Hbs1 sauvage pour le GDP s'élève à 1,3µM, tandis qu'il est très élevé (non calculable)
pour chaque mutant, prouvant une perte très significative de l'affinité. De plus, les
expériences d'ITC ont révélé que la protéine Hbs1 sauvage est plus affine pour le GDP en
absence de Mg2+ qu’en présence de Mg2+. Ce résultat est en accord avec l'effet du Mg2+ sur la
fixation de GDP par eRF3 précédemment mesuré (Kong et al., 2004; Pisareva et al., 2006).
2.3.2. Interactions
L'interaction entre Dom34 et Hbs1 est directement observable par purification d'affinité
ciblant spécifiquement l'une des deux protéines : les deux protéines sont co-éluées dans une
stoechiométrie 1:1. De même, l'injection du mélange sur une colonne de chromatographie
d'exclusion de taille des deux protéines purifiées séparément conduit à la co-élution en
complexe équimolaire (Graille et al., 2008).
Afin de déterminer la constante d'affinité du complexe Dom34-Hbs1 et de calculer ainsi
la proportion de dimère en solution pour les expériences de SAXS, nous avons envisagé des
expériences de microcalorimétrie par titration isotherme. Cependant les mesures réalisées ne
montraient aucun échange de chaleur mesurable, indiquant que la formation du complexe est
globalement athermique.
La résonance plasmonique de surface (Biacore 2000, GE Healthcare) permet de mesurer
directement la fixation d'une espèce moléculaire de grande taille (analyte) sur une surface de

163
taille réduite couplée à un partenaire potentiel (ligand). La méthode ne requiert pas de
marquage préalable ni de caractéristique particulière de l'interaction. J'ai fixé covalamment
Dom34 (ligand) sur une puce CM5 par couplage chimique au EDC-NHS, inactivé les sites
non occupés par l'éthanolamine, puis injecté des solutions de concentrations croissantes de
Hbs1 (analyte).
La fixation cause un changement de l'indice de réfraction de la surface proportionnel à la
quantité de matière fixée, et mesurable sur l'intensité d'un faisceau de lumière incident. On
observe effectivement une variation de l'indice de réfraction lors de l'injection, qui atteint un
plateau, puis revient à la valeur de base lors de l'arrêt de l'injection (Figure 59). C'est une
courbe caractéristique de fixation-dissociation, dont l'interprétation nous apporte deux
informations importantes quant à l'affinité :
- la valeur atteinte au plateau est proportionnelle à la concentration de ligand injecté,
jusqu'à un niveau maximal de réponse : la saturation de l'interaction est spécifique de la
concentration de l'analyte; la demi-saturation permet d'obtenir le Kd;
- les vitesses d'association et de dissociation permettent de calculer respectivement le kon
et le koff de l'interaction, et d'en déduire indépendamment le Kd.
La valeur calculée du Kd de l'interaction Dom34:Hbs1 est d'environ 8µM. Elle est
inférieure d’un facteur 10 à l'affinité calculée d'eRF1:eRF3 (Kononenko et al., 2008). Les
résultats sont validés par l'expérience inversée où Hbs1 est le ligand et Dom34 l'analyte. De
plus, ces expériences révèlent que Dom34 a une affinité pour lui-même (homodimérisation)
de l'ordre de 25µM.
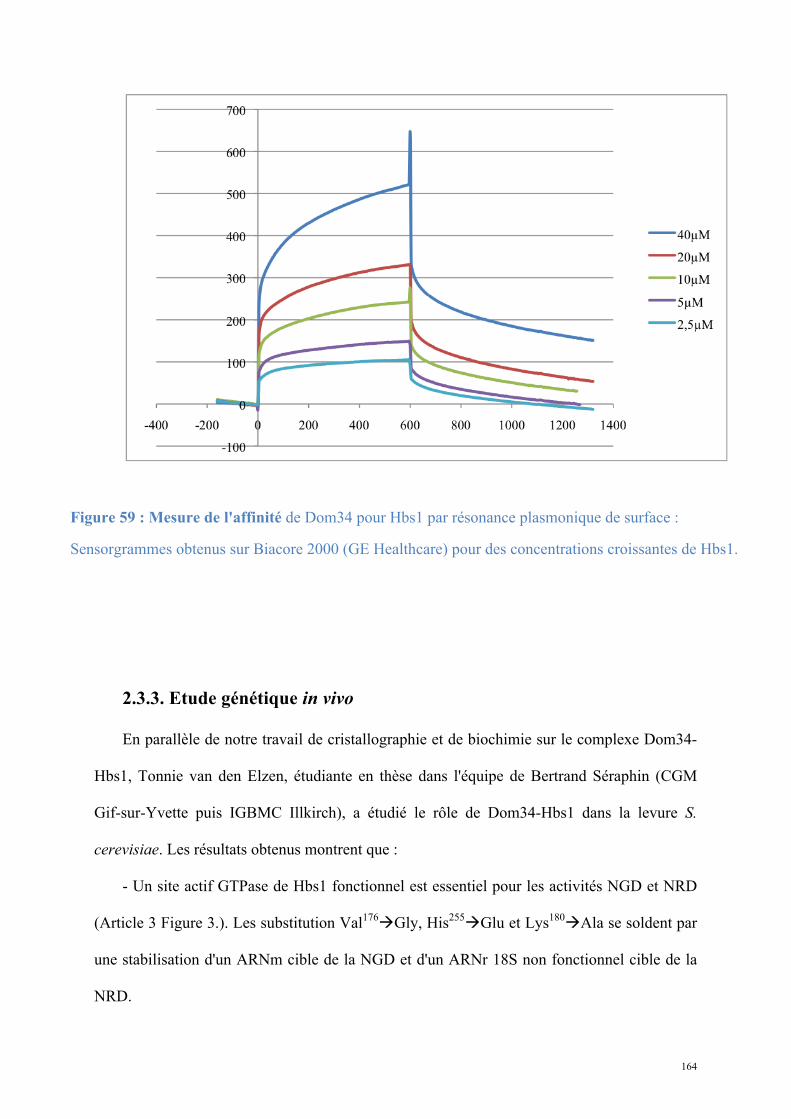
164
2.3.3. Etude génétique in vivo
En parallèle de notre travail de cristallographie et de biochimie sur le complexe Dom34-
Hbs1, Tonnie van den Elzen, étudiante en thèse dans l'équipe de Bertrand Séraphin (CGM
Gif-sur-Yvette puis IGBMC Illkirch), a étudié le rôle de Dom34-Hbs1 dans la levure S.
cerevisiae. Les résultats obtenus montrent que :
- Un site actif GTPase de Hbs1 fonctionnel est essentiel pour les activités NGD et NRD
(Article 3 Figure 3.). Les substitution Val176Gly, His255Glu et Lys180Ala se soldent par
une stabilisation d'un ARNm cible de la NGD et d'un ARNr 18S non fonctionnel cible de la
NRD.
Figure 59 : Mesure de l'affinité de Dom34 pour Hbs1 par résonance plasmonique de surface :
Sensorgrammes obtenus sur Biacore 2000 (GE Healthcare) pour des concentrations croissantes de Hbs1.

165
- L'interaction entre Dom34 et Hbs1 est essentielle à l'activité NGD mais pas à l'activité
NRD (Article 3 Figure 4.). La mutation des résidus identifiés dans l'interface de notre modèle
de Dom34-Hbs1 entraîne une stabilisation de la cible NGD mais n'affecte que peu (surface
d'interaction de Dom34) voire pas du tout (surface d'interaction d'Hbs1) la cible NRD. La
NGD et la NRD sont dissociées d'un point de vue génétique, suggérant qu'une activité de
Hbs1 nécessaire à la NGD ne l’est pas pour la NRD.
Les résultats de cette étude structure-fonction ont fait l'objet d'un article soumis pour
publication dans la revue Nature Structural and Molecular Biology. L'article (Article 3), en
cours de révision, est présenté ci-après.

166
Article 3 : Structural and functional dissection of
the Dom34-Hbs1 complex reveals independent
functions in NGD and NRD

1
Structural and functional dissection of the Dom34-Hbs1 complex reveals independent functions
in NGD and NRD
Antonia M.G. van den Elzen1,2,*, Julien Henri3,*, Noureddine Lazar3, Maria Gas-Lopez1,2, Dominique
Durand3, François Lacroute1, Herman van Tilbeurgh3, Bertrand Séraphin1,2,# and Marc Graille3,#.
1. Equipe Labellisée La Ligue, Centre de Génétique Moléculaire (CGM). CNRS 1 avenue de la
Terrasse 91198 Gif-sur-Yvette Cedex. France
2. and, IGBMC CNRS UMR7104/INSERM U964/Université de Strasbourg, 1 rue Laurent Fries, BP
10142, 67404 Illkirch Cedex, France
3. Institut de Biochimie et Biophysique Moléculaire et Cellulaire (IBBMC). CNRS UMR8619 Bat 430
Université Paris Sud 91405 Orsay Cedex. France
* Both authors contributed equally.
# Correspondance should be addressed to : [email protected] or [email protected]

2
Abstract.
Eukaryotic cells have evolved several quality control pathways relying on translation to detect
defective RNAs and target them to degradation. The Dom34 and Hbs1 proteins, related to translation
termination factors, are involved in No-Go decay (NGD) and non-functional rRNA decay (NRD),
pathways that eliminate RNAs responsible for strong ribosomal stalls. We have solved the crystal
structure of Hbs1+/-GDP and obtained a low-resolution model of the Dom34-Hbs1 complex. This
complex mimics elongation factor-tRNA and translation termination eRF1-eRF3 complexes
supporting that it binds to ribosomal A-site. We further show that nucleotide binding by Hbs1 is
essential for NGD and NRD. Surprisingly, mutagenesis of Hbs1 residues predicted to be located at the
Dom34-Hbs1 interface strongly impairs NGD but has almost no effect on NRD. Hence, NGD and
NRD can be genetically dissociated suggesting that mRNA and rRNA present in a stalled translation
complex may not always be simultaneously degraded.

3
The transfer of genetic information from DNA into functional RNAs requires a multi-step
combination of transcription and processing in eukaryotes. These processes are sources of errors that
potentially affect the overall accuracy of gene expression. To minimize these defaults that in fine may
lead to cell death or diseases, eukaryotic cells have developed several quality-control mechanisms
aimed at detecting and degrading these non-functional RNAs 1,2.
For example nonsense-mediated decay (NMD) targets for degradation faulty mRNAs
harboring in-frame premature termination codons, that if translated could produce deleterious
truncated proteins 2,3. NMD functions through the interplay between the translational apparatus
(ribosome, release factors eRF1 and eRF3…) and specific NMD factors (Upf1-3, Smg1, Smg5-7)
which recruit the mRNA degradation machineries. Aberrant mRNAs that are devoid of in-frame stop
codons are degraded by the non-stop decay pathway (NSD). NSD facilitates the recycling of the
ribosomes stalled at the 3’ end of these mRNAs and requires Ski factors and the exosome 4,5.
More recently, two novel RNA surveillance pathways that turned out to involve identical
factors have been described in yeast. First, Doma & Parker described that mRNAs containing an
engineered stable stem loop that forces the ribosome to pause during elongation are rapidly degraded.
Endonuclease cleavage in the vicinity of the stem loop is followed by 5’3’ degradation of the 3’
cleavage product by the exonuclease Xrn1 and 3’5’ degradation of the 5’ cleavage product
dependent on Ski7 and the Ski complex that in turn recruit the exosome 6. This pathway, named No-
Go Decay or NGD, also targets depurinated mRNAs, a chemical damage that also induces ribosome
stalling 7. NGD involves at least the proteins Dom34 and Hbs1 6 that are related to translation
termination factors eRF1 and eRF3 respectively.
In parallel, Dom34 and Hbs1 have been implicated in the 18S NRD pathway 8,9. This
surveillance mechanism degrades non-functional 18S rRNAs that are able to associate into ribosomal
subunits but incapable of supporting efficient translation. This pathway also targets ribosomes
containing immature 20S pre-rRNAs that engage in translation 10. Neither Dom34 nor Hbs1 are
required for efficient growth in the yeast Saccharomyces cerevisiae. However, deletion of HBS1 or
DOM34 have been demonstrated to cause severe growth defects in strains containing a null allele for
one out of several genes encoding proteins of the small ribosomal subunit (RPS30A, RPS30B,
RPS14A) 11
. These strains are still viable as pairs of genes encode the corresponding ribosomal
proteins. However, in deleted strains the remaining gene copy is probably insufficient to maintain a
proper balance of the corresponding ribosomal protein. This would lead to the accumulation of
incomplete/unstable small ribosomal subunit predisposed to stalling. Hence, growing evidence points
towards a role of Dom34 and Hbs1 proteins in clearing cells from RNAs inducing translational stalls,
i.e. poorly functional small ribosomal subunits, mRNAs with in-frame stable stem loops and
depurinated RNAs.
Studies in yeast, fruit fly and mouse have shown that Dom34 (also known as Pelota in higher
eukaryotes) is required for correct mitotic and meiotic cell division 12-15. Dom34/Pelota is structurally

4
related to the translation termination factor eRF1 with the exception of its N-terminal domain that
adopts a Sm-fold, commonly found in proteins involved in RNA recognition or degradation 16,17. This
domain was proposed to exhibit an in vitro divalent metal ions endonuclease activity 17 but this finding
was recently challenged 18. First, the in vitro nuclease activity previously observed for the yeast
Dom34 protein could not be reproduced. Second, over-expression of the Rps30a protein was sufficient
to restore some mRNA cleavage in the absence of Dom34.
Dom34 associates with Hbs1, a member of the family of eEF-1A-like GTPases including
translation elongation factors eEF-1A and EF-Tu that deliver aminoacyl-tRNAs to the ribosomal A-
site, eRF3, and Ski7 11,16,19,20. Initially, Hbs1 was identified as Hsp70 subfamily B suppressor. In yeast,
deletion of both SSB1 and SSB2 genes (encoding Hsp70 chaperones interacting with the nascent
polypeptide chain at the exit of the ribosome channel during protein synthesis) results in slower cell
growth, in the decrease of the number of translating ribosomes and in slower translation 21. This
phenotype is suppressed by over-expression of Hbs1 suggesting that Hbs1 may help in stop codon-
independent peptide release from ribosomes stalled by the absence of Hsp70-mediated nascent
polypeptide channeling, a function closely related to the one proposed for Hbs1 and Dom34 in both
NGD and 18S NRD 22. Hbs1 is a GTP binding protein whose affinity for GTP but not GDP is
enhanced by Dom34, similarly to the effect of eRF1 on eRF3 binding to GTP 11,16,23.
To gain insight into the mechanism of the Dom34-Hbs1 complex in these quality control
pathways, we have solved the crystal structure of yeast Hbs1 and obtained a low-resolution model of
the Dom34-Hbs1complex by SAXS. This allowed a precise study of the role of Hbs1 nucleotide
binding site and interaction with Dom34 in NGD and NRD, which demonstrated, interestingly, that
these two processes could be genetically separated.

5
Results
Overall structure
As the N-terminal region of S. cerevisiae Hbs1 is predicted to be poorly folded, we have
purified several Hbs1 fragments starting at different positions. Diffracting crystals could only be
obtained for a fragment encompassing residues 135-610 (hereafter named Hbs1dN134). Its structure
was solved using the SAD method (see Fig.S1 for experimental electron density maps). The structures
of the apo and GDP bound forms were further refined to 2.5Å and 2.95Å resolution, respectively. As
expected from sequence analysis, Hbs1 is a member of the eEF-1A protein family. Hbs1dN134 is
composed of a N-terminal GTPase domain (residues 165 to 375) and two 6-stranded -barrel domains
(domain II from residues 399 to 498 and domain III from residues 499 to 610; Fig.1A). The GTPase
domain is preceded by a long N-terminal loop (residues 140 to 164) partially folded as an α-helix
( A), which packs against domain II. The GTPase domain adopts the classical α/β structure common
to GTPases such as EF-Tu, eRF3 and Ras 24,25. It is connected to domain II by a long helix ( 7;
residues 376 to 398). Both domains II and III adopt a fold similar to the corresponding domains from
EF-Tu and eRF3 24,26. In all structures of bacterial and eukaryotic members from the eEF-1A
superfamily, domains II and III are held together in the same relative orientation but this is not true for
the GTPase domain. In the structure of S. pombe eRF3, there are no interdomain contacts between the
GTPase domain and domains II and III 24 (Fig.S2A). In the case of Hbs1dN134, the GTPase domain
packs against the domains II and III in a manner highly reminiscent of the EF-Tu-GDPNP complex.
Small-angle X-ray scattering experiments (SAXS) in solution on Hbs1dN134 show that the Hbs1
conformation observed in our crystal structure fully agrees with the conformation of the protein in
solution and hence is not induced by crystal packing (see supplementary data, Fig.S2)
The nucleotide binding site
Similarly to the other members of the eEF-1A family, Hbs1 is a GTP binding protein 11,16. We
have solved the structure of Hbs1dN134 either in its apo-form or in the presence of GDP. As observed
for the apo and nucleotide bound forms of archaeal SelB elongation factor and eukaryotic release
factor eRF3 24,27, no large scale domain movements are observed between the apo and GDP bound
forms of Hbs1dN134 (rmsd of 0.4 Å over 430 C atoms).
GTPases are characterized by the presence of a P-loop and two highly flexible and conserved
segments called switch regions I and II that are directly involved in nucleotide binding (Fig.1B and
S3). They adopt radically different conformations depending on which nucleotide is bound (GDP or
GTP). Among GTPases, the structure of the switch regions is highly variable for the GDP forms but
invariant for the GTP forms 25. In the Hbs1dN134 apo-form, switch I (residues 194-234) is mostly
disordered (absence of electron density for the region 205-228) although its N-terminal part folds as a
helix ( 2). Upon GDP binding, a portion from this switch (residues 217-228) becomes structured and

6
folds as a small -helix ( 2’, residues 217-223) antiparallel to helix 2 (Fig.1B). Switch II (residues
251-268) forms an -helix ( 3) connected to strand 3 by a loop. The orientation of helix 3 remains
constant between the apo and GDP bound forms but the 3- 3 loop (residues 251-258) exhibits some
flexibility (Fig.1B). Most of Hbs1 residues contacting GDP structurally match with the corresponding
residues in the E. coli EF-Tu – GDP complex 28 (Fig.1C). The P-loop coordinates the GDP phosphate
groups via the main chain atoms from residues Val176 to Thr182 and side chains from polar residues
(Asp177, Ser181 and Thr182). The guanine base is stacked between the hydrophobic moieties from
Lys314 and Phe354. In the EF-Tu GDP complex, switch II contacts the nucleotide. This is not the case
for the Hbs1dN134 GDP complex probably because of the absence of Mg2+ bound to GDP in our
structure compared to EF-Tu – GDP.
Two point mutants of the GTP binding site (Val176Gly from the P-loop and His255Glu of the
switch II region) were previously shown not to complement a HBS1 deletion, suggesting that GTP
binding is required for Hbs1 function 11. As stated above, Val176 is part of the P-loop and is involved
in phosphate binding. In the Hbs1dN134-GDP complex, His255 is not directly in contact with GDP
but it may play a crucial role for GTP binding and/or hydrolysis. To characterize the effect of these
mutations on nucleotide binding, we have produced three single point mutants (Val176Gly, His255Glu
as well as Lys180Ala from the P-loop). We then tested their ability to bind in vitro to fluorescent
derivatives of GDP and GTP as previously shown for Hbs1 wild-type protein 16. No significant GDP
or GTP binding to the mutants could be observed compared to the wild-type protein (Fig.S4). This was
probably not due to folding defects as demonstrated by comparison of circular dichroism spectra of
WT and mutant constructs (data not shown). These experiments clearly show that the substitutions
introduced completely abolish both GDP and GTP binding.
Low-resolution model of the Dom34-Hbs1 complex.
Hbs1 and Dom34, as is the case for eRF3 and eRF1, physically interact together 11,16. The
recent crystal structure of the complex between eRF1 and an eRF3 fragment lacking the GTPase
domain confirmed that the eRF1-eRF3 interface is located between the eRF1 C-terminal domain and
the eRF3 domain III 29. We suggest that the formation of the Hbs1-Dom34 complex very likely occurs
via the corresponding domains. Superimposition of these Dom34 and Hbs1 domains onto the structure
of the eRF1-eRF3 complex strongly argues in favor of the involvement in complex formation of
residues from loops connecting strands A’ to B’ and C’ to D’ from Hbs1 (Fig.1D). In this model,
these loops face residues from the C-terminal part of helix 8, from 11 as well as strand 13 from
Dom34.
To validate our Dom34-Hbs1 interaction model, we have assessed the effect of introducing
domain deletion in Dom34 (C-terminally Strep-tagged) on the interaction with Hbs1 (C-terminally
His6-tag) after co-expression in E. coli. Strep-Tactin sepharose affinity purification demonstrates that

7
Hbs1 is retained on the column only when co-expressed with Dom34 (Fig.S5A-B). Deletion of the N-
terminal or central domain does not strongly affect complex formation (Fig.S5C-D). In contrast,
deletion of Dom34 C-terminal domain almost completely abolished complex formation, indicating that
this domain is crucial for the interaction with Hbs1, similar to the role of eRF1 C-terminal domain in
eRF3 binding (Fig.S5E; 29).
In order to get further insight into how Hbs1 and Dom34 might interact, we have performed
SAXS measurements on the Dom34-Hbs1dN134 complex (Fig.2A and Fig.S6A, supplementary data).
From the crystal structures of yeast and archaeal Dom34 (comparison of these structures revealed
large conformational changes) and yeast Hbs1dN134 and of the eRF1-eRF3 complex 16,17,29, we have
generated two initial models (yeast-like and archaeal-like) for the Dom34-Hbs1dN134 complex and
compared the corresponding experimental and calculated scattering curves (see supplementary files
for details on the different models and Fig.S6B-C for a representation of the initial models). The best
agreement with the experimental scattering curve is obtained for the archaeal-like model (Fig.2A;
value of 5.9 vs 13.4 for the yeast-like model). In addition to the contacts between the Dom34 C-
terminal domain and the Hbs1 domain III, this model highlights contacts between the Dom34 central
domain and the GTPase domain from Hbs1 in this model. We have performed rigid-body modelling
with the program SASREF 30 to improve the fit between calculated and experimental curves (Fig.2A-
B, values ≤ 1.7, see supplementary files for details). The resulting models show that in the Dom34-
Hbs1 complex, Dom34 adopts a tRNA-shape with its N-terminal and central domains matching with
the anticodon and amino acyl acceptor arms of a tRNA molecule, respectively, as observed in the EF-
Tu – tRNA complex (Fig.2B-C; 26). This further suggests that Dom34-Hbs1 binds into the ribosomal
A site.
Nucleotide binding by Hbs1 is crucial for NRD and NGD.
Several studies on members of the eEF-1A family have already demonstrated the critical role of GTP
binding for their function. Consistently, two point mutations within Hbs1 GTP binding site
(Val176Gly and His255Glu), which we have shown abolish nucleotide binding (Fig.S4), do not
complement a HBS1 deletion 11. To investigate further the importance of nucleotide binding by Hbs1
in NGD and NRD, we have tested the physiological importance of Hbs1 mutations affecting
nucleotide binding (Val176Gly and His255Glu and Lys180Ala, Fig.S4) by examining the effect of
these point mutants on NGD, NRD and growth in strains deleted for small ribosomal subunit genes in
S. cerevisiae.
We first constructed a protein A tagged version of Hbs1 that allowed us to assess expression of wild-
type and mutant proteins. The wild-type Hbs1-ProtA fusion was shown to be indistinguishable from
wild type in all assays (see below) and mutants Val176Gly, Lys180Ala and His255Glu were produced
normally in yeast cells (Fig.S7). It has been shown in a ski7 context that the 5’ endonucleolytic
cleavage product of NGD substrate PGK1-SL accumulates in a manner dependent upon the presence

8
of both Dom34 and Hbs1 6,18. In Hbs1 mutants abolishing nucleotide binding, production of this
degradation intermediate was reduced (Fig.3A lanes 4-6 and C). The ratio of degradation intermediate
to full-length reporter mRNA was reduced to 40% of the ratio observed in presence of wild type Hbs1,
a level similar to the one detected in absence of Hbs1.
The rapid degradation of the NRD substrate 18S A1492C has been shown to depend at least partly on
the presence of both Dom34 and Hbs1 9. In absence of one or the other of these factors, 18S A1492C
steady state levels were increased (Fig.4A and B, compare lanes 1 to lanes 2 and 3). Hbs1 mutants
unable to bind nucleotide stabilized the 18S A1492C reporter (Fig.4A lane 4-6 and C), leading to
steady state levels 3 to 4 fold higher than in presence of wild type Hbs1 (lane 2,3), a level similar to
the one observed in absence of Hbs1 (lane 1).
Screening for mutations generating a synthetic growth phenotype in a dom34 background identified
mutations in genes encoding the Rps28 proteins of the small ribosomal subunit (data not shown). This
finding is consistent with previous observations of a severe growth defect observed when deletion of
DOM34 or HBS1 were combined with deletion of one out of several 40S subunit protein encoding
genes 11. In accordance with these earlier data, the Val176Gly and His255Glu mutants could not
complement the slow growth of rps28A∆hbs1∆ strain (Fig.4E, compare with strain expressing wild-
type Hbs1). The Lys180Ala mutant proved also to be non-functional in this assay (Fig.4E).
In conclusion, Hbs1 mutants that abolish nucleotide binding are functionally inactive in the three
assays that we used. We conclude that Hbs1 nucleotide binding is essential for NGD endonucleolytic
cleavage, 18S rRNAs NRD and for the function of Hbs1 required in strains lacking a gene encoding a
ribosomal protein of the small subunit.
Role of the Dom34-Hbs1 interaction in NGD and NRD.
Our SAXS and deletion analysis are consistent with the idea that the Dom34 and Hbs1 interaction is
mediated mainly through the C-terminal domain of Dom34 and domain III of Hbs1, in a way similar
to the eRF1- eRF3 interaction. To test the functional importance of the Dom34-Hbs1 interaction, we
designed several mutants targeting residues of the proposed interaction surface, namely:
Tyr300Ala/Glu361Ala, Glu361Arg and Glu361Ala/Gln364Ala in Dom34 and Arg517Glu, Leu520Arg
and Arg557Ala/His558Ala in Hbs1 (Fig.1D). According to our Dom34-Hbs1 interaction model that
fits best with results of SAXS measurements on the Dom34-Hbs1dN134 complex, the Dom34 central
domain also contacts the GTPase domain from Hbs1. To address the functional importance of this
interaction we also mutated this hypothetical interaction surface. In the Dom34 central domain, a
conserved loop rich in basic residues (174KKKR177) was mutated into alanines (KKKR mutant). While
this work was in progress, the same mutation was independently shown to strongly reduce NGD 18.
Mutations of similarly positioned basic residues in eRF1 affect eRF3 GTPase activity, eRF1-eRF3
interaction and function 29. In our model this loop could contact the -phosphate of a GTP molecule
bound to Hbs1. We also mutated a highly conserved 212SPGF215 motif in the central domain of Dom34

9
into AAGA (SPGF mutant). Mutation of the PGF peptide or of the proline alone was independently
shown to reduce NGD 18. In our structural model the SPGF motif could face a conserved 256RDF258
motif from Hbs1 GTPase domain, which we also mutated into alanines (RDF mutant, Fig.S8).
To verify whether mutants are expressed in S. cerevisiae, Dom34 mutants were introduced in a
construct carrying a C-terminal 3HA-tag. As described above, Hbs1 mutants contained a C-terminal
protein A-tag. Western blot analysis demonstrated that all mutants were expressed in yeast (Fig.S7).
All mutants of the hypothetical interaction surfaces caused a reduction in NGD (Fig.3A lanes 7-10, B,
C and D). The ratio of degradation intermediate to full-length reporter mRNA was decreased to the
same extent as observed in strains lacking Hbs1 or Dom34 (Fig.3A and B lanes 1). Surprisingly, none
of the Hbs1 mutants affected NRD as the level of 18S A1492C was identical to the one observed in
the presence of wild type Hbs1, i.e., 3-4 fold lower than in strain lacking Hbs1 (Fig.4A lanes 7-10, C).
In contrast, all Dom34 mutants caused some stabilization of the 18S A1492C NRD substrate (Fig.4B
and D), even though not to the same extent as in absence of Dom34. Interestingly, all of these Dom34
and Hbs1 mutants complemented the slow growth defect observed in dom34∆rps28A∆ and
hbs1 rps28A∆ strains, respectively (Fig.4E). These results indicate that the interaction between
Dom34 and Hbs1 is critical for efficient NGD but has a more reduced effect on NRD and growth in
the context of a strain lacking a gene encoding a ribosomal protein of the small subunit.

10
Discussion.
Eukaryotic cells have developed several highly sophisticated surveillance pathways dedicated to the
detection of non-functional or damaged molecules (DNA, RNA but also proteins), thereby leading to
their repair or rapid degradation. These mechanisms and their dysfunction play crucial roles in cellular
functions and disease. In this study, we focused our attention on Hbs1 and Dom34, two interacting
proteins implicated in two RNA quality control pathways: NGD and NRD 6,7,9,10. These surveillance
mechanisms have in common to specifically detect and degrade RNA molecules (mRNAs or rRNAs)
that cannot correctly undergo translation elongation by eukaryotic ribosomes. However, the precise
mechanisms of these pathways are largely unclear.
The structure of Hbs1dN134 reported here displays the same domain organization as other members of
eEF-1A family members such as eRF3, eEF-1A, Ski7 and bacterial EF-Tu. In the structure of Hbs1
and EF-Tu, the GTPase domain is tightly packed onto domains II and III (Fig.1A) but for eRF3 the
structure is “open” as the GTPase domain makes only a few contacts with domains II and III 24. Our
SAXS data (Fig.S2) for Hbs1 as well as mutant analysis of S. pombe eRF3 24 support, however, the
existence of a conserved “closed” conformation corresponding to our crystal structure that is likely to
be necessary for correct function of members of the eEF-1A family.
Like other eEF-1A factors, Hbs1 binds GTP and GDP through its GTPase domain. Hbs1 is thus likely
to cycle between GTP bound, GDP bound and free forms. We observe that nucleotide binding is
essential for all the Hbs1 functions that we tested namely: NGD, NRD and growth complementation.
In eRF3 GTP binding (stimulated by eRF1) promotes a rearrangement of the termination complex
before peptide release 23,31-33. This leads to GTP hydrolysis followed by rapid hydrolysis of peptidyl-
tRNA, peptide release, tRNA liberation and subunit dissociation 32,34,35. Similarly, GTP association
with Hbs1 (stimulated by Dom34 16) is likely to promote rearrangement of the ribosome. Our mutants
of Hbs1 impairing GTP binding, thereby also preventing GTP hydrolysis, are likely to impede
remodeling of the stalled ribosome and/or stabilize Dom34 present in the A-site. The phenotype
observed for Hbs1 mutant unable to bind nucleotides suggest that such rearrangements are a
prerequisite to initiate the degradation of the small ribosomal subunit or bound mRNA.
Based on the available crystal structures of eRF1-eRF3 complex, Hbs1 and Dom34 (from yeast and
archaea that display radically different orientation of the central domain relative to the N and C-
terminal domains), we have build two Hbs1dN134-Dom34 models by superimposing the Hbs1 and
Dom34 C-terminal domains onto the corresponding regions of the eRF1-eRF3 complex (Fig.S6B-C).
SAXS experiments (Fig.2A) show that the model where yeast Dom34 adopt the conformation
observed in the archaeal Dom34 structure 17, rather than the cognate structure 16, is the most likely to
represent the complex in solution. Interestingly, Hbs1 homologues are absent from archaea 19
supporting the idea that in eukaryotes Hbs1 might push Dom34 into an active conformation. Our
Dom34-Hbs1 model is not only reminiscent of the eRF1-eRF3 complex but also displays strong

11
analogy to the bacterial EF-Tu – tRNA complex, involved in the translation elongation step 26.
Comparison of Hbs1-Dom34 model and EF-Tu-tRNA complex (Fig.2B-C) shows that Dom34 and the
tRNA have the same shape and localization relative to the GTPase domain of their respective partners.
The N-ter, central and C-ter domains from Dom34 match with the anticodon stem, the amino acyl
acceptor arm and the T stem from the tRNA, respectively. Thus, the N-terminal domain of Dom34 is
likely to bind close to the mRNA codon in the A-site of stalled ribosome. This domain is structurally
unrelated to eRF1 N-ter domain and adopts an Sm-fold 36,37. Two loops (residues 49-53 and 87-92)
from this Dom34 domain that are functionally important for NGD pathway in yeast 18 occupy
approximately the same position as the tRNA anticodon loop in our model and hence could directly
contact the mRNA codon, probably in a sequence independent manner.
Our model suggests that the interaction between Hbs1 and Dom34 occurs through two surfaces:
binding of Hbs1 domain III with Dom34 C-terminal domain and contacts between the Hbs1 GTPase
and the Dom34 central domains. Our binding data indicate that the former is essential for interaction
while the second probably plays a regulatory role. Consistently, in human, binding of the central
domain of eRF1 to eRF3 stimulates the GTP binding property and GTPase activity of the latter 31. It
has been proposed that this involves stabilization of the eRF3 switch region by eRF1 Arg192 29,
therefore explaining the increase in the affinity of eRF3 for GTP in the presence of eRF1 23,33. GTP
association with Hbs1 is similarly stimulated by Dom34 16.
Interestingly, our results indicate that the proposed interaction between Dom34 and Hbs1 is essential
for NGD, but less crucial for NRD or growth in a rps28 strain. Moreover, the interaction surface
present in Dom34 appears more critical for NRD than the one of Hbs1. While we cannot exclude that
this results from a low residual activity of Hbs1 mutants, such residual activity would anyway
discriminate between NGD and NRD. This indicates that for NGD a function of Hbs1 is required that
is not essential for NRD to occur. As the presence of Dom34 and Hbs1 is essential for both NGD and
NRD, a possible explanation for our results is that an additional factor stabilizes the Dom34-Hbs1
interaction during NRD but not during NGD. Alternatively, NGD may require a longer interaction
with Dom34/Hbs1 than NRD, and thus be more affected by a weakly interacting Hbs1. It is also
possible that Dom34 interacts with another factor specifically required for NGD through the
interaction surface used by Hbs1, explaining the asymmetry of our mutant phenotypes. While the
corresponding mechanistic details remain to be elucidated, our data indicate importantly that NGD and
NRD can be genetically dissociated. Thus, in contrast to earlier proposal 9, mRNA and rRNA presents
in a stalled translation complex may not always be simultaneously degraded. Interestingly, the
observation that growth phenotype assayed in the absence of Rps28a correlates perfectly with their
effects on NRD, but not on NGD, allows us also to conclude that in these cells NRD has become
critical. It can be proposed that the absence of NRD may be quantitatively more detrimental to cells
than defective NGD: in the first case, a small fraction of ribosomes stalled may quickly have a general

12
impact on translation by sequestering functional ribosome in dead polysomes, while in the second
case, only a few mRNA and ribosomes would be affected.
Overall, our data show that eukaryotic cells use the Hbs1-Dom34 complex as mimicry of
tRNA-elongation factor complex to perform RNA quality control mechanisms. The rules governing
the specific recruitment of this complex to the A-site of stalled ribosomes remain to be clarified. This
binding contributes to NGD and NRD. However, our data indicate that these two processes may be
functionally separated. The mechanisms controlling the outcome of Dom34-Hbs1 binding to stalled
ribosome, i.e., degradation of the substrate mRNA and/or destruction of the small ribosomal subunit
remain to be deciphered. Another point to be clarified is the issue of the incomplete proteins produced
by these stalled ribosomes. During translation termination, the eRF1 central domain contributes to
protein release through a universally conserved GGQ motif 38. As this GGQ motif is not present in
Dom34, it still remains to be determined whether hydrolysis of peptidyl-tRNA and release/degradation
of truncated peptides occur in NGD and/or NRD.

13
Acknowledgments
We are indebted to Dr K. Blondeau, B. Faivre, J. Cicolari, D. Lebert, Y. Billier, B. Bonneau and D.
Rentz for technical assistance. We thank M. Moore and R. Parker for plasmid gifts and Dr P. Vachette
for helpful discussion. This work was supported by the Agence Nationale pour la Recherche (grants
ANR-06-BLAN-0075-02 and ANR-07-BLAN-0093), the Association Française contre les Myopathies
(AFM), La Ligue contre le Cancer Equipe Labellisée 2008, the CNRS, the ESF EUROCORES RNA
Quality and the EU “3D-Repertoire” program (LSHG-CT-2005-512028). J. Henri and T. van den
Elzen hold pre-doctoral grants from the Université Paris-Sud 11 and Université de Strasbourg,
respectively. M. Gas-Lopez is supported by the Spanish Ministry of Research. We acknowledge
SOLEIL for provision of synchrotron radiation facilities and we would like to thank Dr Andrew
Thompson and Dr Javier Perez for assistance with beamlines Proxima-1 and SWING, respectively.
Author contributions.
J.H., A.M.G.vdE., M.G., B.S. designed experiments. J.H., A.M.G.vdE, N.L., D.D., F.L. and M.G.L.
performed experiments. J.H., A.M.G.vdE, D.D., HvT., M.G. and B.S. analyzed data and wrote the
paper.

14
Legends to figures.
Figure 1. Hbs1 structure representation.
A. Ribbon representation of the Hbs1dN134 structure. For domains GTPase, II and III from Hbs1, the
rmsd values with the corresponding domains from EF-Tu and eRF3 are ≈ 2.8-3 Å over 175-190 C
atoms (25-38% sequence identity), ≈1-1.3Å over 80-90 C atoms (20-30% sequence identity) and ≈
1.4-1.7Å over 100-110 C atoms (18-27% sequence identity), respectively.
B. Conformational changes of switch regions upon GDP binding. For both apo and GDP bound form
of Hbs1dN134, the crystal asymmetric unit contains two molecules organized as a non
crystallographic dimer, yielding to 4 sets of coordinates for this Hbs1 monomer. These 4 copies are
virtually identical (rmsd ≈ 0.4-0.5Å) except for the switch regions that adopt different conformations.
For clarity, only the secondary structure elements for Hbs1dN134 bound to GDP are shown (beige).
The P-loop and switches I and II in the Hbs1-GDP complex are colored in yellow, red and green,
respectively. The conformation of these regions in the Hbs1dN134 apo forms I and II are in blue and
pink, respectively. GDP is shown as balls and sticks.
C. Comparison of the GDP binding mode as observed in Hbs1dN134 (same color code as panel B) and
EF-Tu (grey). The GDP bound to EF-Tu is shown as black sticks.
D. Superimposition of Dom34 C-terminal domain (green) and Hbs1 domain III (orange) from yeast
proteins onto the corresponding domain from human eRF1 and eRF3 (grey). Mutated residues are
shown as sticks.
Figure 2. SAXS analysis of the Dom34-Hbs1dN134 complex.
A. Comparison of the scattering curves calculated from the coordinates of the yeast-like (blue),
archaeal-like (green), and SASREF Dom34-Hbs1dN134 models (red) with the experimental SAXS
curve (black).
B. Ribbon representation of the optimized Dom34-Hbs1dN134 model. Dom34 N-terminal, central and
C-terminal domains are coloured in light green, green and dark green, respectively. Hbs1 GTPase, II
and III domains are coloured in light blue, cyan and dark blue, respectively. Loops A, B and C from
Dom34 that have been shown to be functionally important in NGD 18, are indicated in yellow, orange
and red, respectively.
C. Ribbon representation of the bacterial EF-Tu-tRNA complex 26. For clarity, EF-Tu and Hbs1 are
shown with the same orientation. EF-Tu GTPase, II and III domains are coloured in pink, magenta and
purple, respectively.
Figure 3. Effects of Hbs1 and Dom34 mutants on NGD.

15
A. Northern analysis of the steady state levels of NGD substrate PGK1-SL degradation intermediates
in a ski7∆hbs1∆ S. cerevisiae strain transformed with Hbs1 mutants. The PGK1-SL reporter was
expressed from the plasmid pRP1251 and detected with the oligonucleotide oRP132 6.
B. Northern analysis of the steady state levels of PGK1-SL reporter degradation intermediates in a
ski7∆dom34∆ S. cerevisiae strain transformed with Dom34 mutants.
C. and D. Quantification of panels A and B respectively. For each mutant the ratio of 5’ intermediate
over full length RNA (endogenous PGK1 and PGK1-SL) was calculated and then standardized for the
ratios calculated for Hbs1-proteinA or Dom34-3HA. Average ± SD of 3 biological replicas are shown.
Figure 4. Effects of Hbs1 and Dom34 mutants on NRD and growth in ribosomal protein deletion
context.
A. Northern analysis of the steady state levels of NRD substrate 18S A1492C in a hbs1∆ S. cerevisiae
strain transformed with Hbs1 mutants. ScR1 RNAs are presented as loading controls. The 18S
A1492C reporter was expressed from the plasmid pWL160-A1492C and detected by the
oligonucleotide FL125 8.
B. Analysis of the steady state levels of NRD substrate 18S A1492C in a dom34∆ S. cerevisiae strain
transformed with Dom34 mutants. ScR1 RNAs are presented as loading controls.
C. and D. Quantification of A and B respectively. For each mutant the ratio of 18S A1492C over scR1
signal was calculated and then standardized for the ratios calculated for Hbs1-proteinA or Dom34-
3HA. Average ± SD of 3 biological replicas are shown.
E. Strains deleted for both small ribosomal subunit protein Rps28A and Hbs1 or Dom34
(rps28A∆hbs1∆ and rps28A∆dom34∆) transformed with Hbs1 mutants or Dom34 mutants were
subcloned on YPDA plates and grown at 30°C (3 days).

16
Materials and methods.
Cloning, Expression, and Purification of Hbs1 proteins
The truncated version of the HBS1 gene (YKR0084c) deleted for the first 134 residues was amplified
by PCR from S. cerevisiae S288C genomic DNA. An additional His6 tag was introduced downstream
of the 3’ coding sequence. The PCR products were cloned into a modified pET28 vector between EagI
and NotI restriction sites. The protein was expressed and purified as previously described for the full-
length protein 16. A seleno-methionine (Se-Met) derivative of Hbs1dN134 was expressed in Se-Met-
supplemented minimal medium using the same expression system as for the native protein. The Se-
Met protein was purified with the same method as for the native protein.
The full-length mutated versions (V176G, H255E, K180A) of the HBS1 gene (YKR0084c) was
amplified by PCR from S. cerevisiae expression plasmids and inserted in an ampicillin resistant
pET21a vector. An additional His6 tag was introduced downstream of the 3’ coding sequence between
BamHI and NotI restriction sites. The mutants were expressed and purified using the same protocol as
the Hbs1dN134 protein. See further details in Supplementary Material and Methods.
Yeast strains and plasmids
All yeast strains were constructed by standard methods and are derivative of BMA64 (Table S2).
Plasmids are listed in Table S3 while oligonucleotides used in plasmid construction are listed in Table
S4.
Dom34 and upstream flanking sequence were amplified from genomic DNA using primers OBS2597
and OBS2598 and cloned between the XhoI and XbaI sites of pRS415, giving pBS3217. Hbs1 with its
flanking sequences was amplified from genomic DNA using primers OBS3474 and OBS3475 and
cloned between the SmaI and HindIII sites of pRS415, generating pBS3611. C-terminal tags (3HA for
Dom34 and ProtA for Hbs1) were introduced by fusion PCR. (For primers and templates used see
Table S5.) The Dom34-3HA product digested by HindIII and NheI was cloned into pBS3217,
generating pBS3685. The Hbs1-ProtA product digested by NdeI and HindIII was cloned into
pBS3611, generating pBS3614.
Dom34 and Hbs1 point mutations were introduced in pBS3685 or pBS3614 respectively by
QuickChange mutagenesis, except for the KKKR mutant, which was produced by fusion PCR,
digested by HindIII and XhoI and cloned into pBS3685 (see Table S5 for primers and templates).
Dom34∆domain1 (aminoacids 1-136 deleted) and Dom34∆domain2 (aminoacids 141-275 replaced by
(Gly)4) domain deletion mutants were produced by fusion PCR (see Table S5). The resulting
fragments were digested by XhoI and NheI and cloned into pBS3217. Dom34∆domain3 (aminoacids
276-386 deleted) was produced by PCR with OBS3396 and OBS3397 using pBS3217 as template.
The resulting fragment digested by BglII/NheI was cloned into pBS3217. Dom34 domain deletion
mutants were amplified with the introduction of a C-terminal strep-tag using OBS3440 and OBS3171

17
(Dom34∆domain1), OBS3170 and OBS3171 (Dom34∆domain2) or OBS3170 and OBS3443
(Dom34∆domain3), digested with NdeI and KpnI and cloned into a pACYC-LIC+ vector. All
constructs were verified by sequencing.
RNA analysis
Yeast cultures were grown in CSM-Leu-Ura (NGD assay) or CSM-Leu-Trp (NRD assay) containing
2% galactose at 30 °C until OD600 0.6-1.2. Total RNA was obtained by hot phenol extraction,
separated on 1.5 % agarose- 6.3% formaldehyde gel (15 µg for NGD, 10 µg for NRD) and transferred
to a HybondTM-XL membrane (GE Healthcare) as described earlier 39. Probes oRP132 and FL125 were
5’ labeled with T4 polynucleotide kinase (Fermentas) and γ-32P ATP. For detection of endogenous
scR1 probes were made by a random priming reaction on a PCR product obtained with primers
OBS1884 and OBS1885 using the Neblot® kit (New England Biolabs) and α-32P dCTP.
Prehybridization and hybridization of the membranes with the labeled probes was performed in
Church buffer at 42 °C and 40 °C respectively (except for scR1: both at 55 °C). Signals were
visualized with a Typhoon 8600 Variable Mode Imager (Molecular Dynamics) and quantified using
ImageQuant 5.2 software.

18
References.
1. Doma, M.K. & Parker, R. RNA quality control in eukaryotes. Cell 131, 660-8 (2007).
2. Isken, O. & Maquat, L.E. Quality control of eukaryotic mRNA: safeguarding cells from
abnormal mRNA function. Genes Dev 21, 1833-56 (2007).
3. Amrani, N., Sachs, M.S. & Jacobson, A. Early nonsense: mRNA decay solves a translational
problem. Nat Rev Mol Cell Biol 7, 415-25 (2006).
4. Frischmeyer, P.A. et al. An mRNA surveillance mechanism that eliminates transcripts lacking
termination codons. Science 295, 2258-61 (2002).
5. van Hoof, A., Frischmeyer, P.A., Dietz, H.C. & Parker, R. Exosome-mediated recognition and
degradation of mRNAs lacking a termination codon. Science 295, 2262-4 (2002).
6. Doma, M.K. & Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in
translation elongation. Nature 440, 561-4 (2006).
7. Gandhi, R., Manzoor, M. & Hudak, K.A. Depurination of Brome mosaic virus RNA3 in vivo
results in translation-dependent accelerated degradation of the viral RNA. J Biol Chem 283,
32218-28 (2008).
8. LaRiviere, F.J., Cole, S.E., Ferullo, D.J. & Moore, M.J. A late-acting quality control process
for mature eukaryotic rRNAs. Mol Cell 24, 619-26 (2006).
9. Cole, S.E., LaRiviere, F.J., Merrikh, C.N. & Moore, M.J. A convergence of rRNA and mRNA
quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol
Cell 34, 440-50 (2009).
10. Soudet, J., Gelugne, J.P., Belhabich-Baumas, K., Caizergues-Ferrer, M. & Mougin, A.
Immature small ribosomal subunits can engage in translation initiation in Saccharomyces
cerevisiae. Embo J 29, 80-92 (2010).
11. Carr-Schmid, A., Pfund, C., Craig, E.A. & Kinzy, T.G. Novel G-protein complex whose
requirement is linked to the translational status of the cell. Mol Cell Biol 22, 2564-74 (2002).
12. Adham, I.M. et al. Disruption of the pelota gene causes early embryonic lethality and defects
in cell cycle progression. Mol Cell Biol 23, 1470-6 (2003).
13. Davis, L. & Engebrecht, J. Yeast dom34 mutants are defective in multiple developmental
pathways and exhibit decreased levels of polyribosomes. Genetics 149, 45-56 (1998).
14. Eberhart, C.G. & Wasserman, S.A. The pelota locus encodes a protein required for meiotic
cell division: an analysis of G2/M arrest in Drosophila spermatogenesis. Development 121,
3477-86 (1995).
15. Xi, R., Doan, C., Liu, D. & Xie, T. Pelota controls self-renewal of germline stem cells by
repressing a Bam-independent differentiation pathway. Development 132, 5365-74 (2005).
16. Graille, M., Chaillet, M. & van Tilbeurgh, H. Structure of yeast Dom34: a protein related to
translation termination factor Erf1 and involved in No-Go decay. J Biol Chem 283, 7145-54
(2008).
17. Lee, H.H. et al. Structural and functional insights into Dom34, a key component of No-Go
mRNA decay. Mol Cell 27, 938-50 (2007).
18. Passos, D.O. et al. Analysis of Dom34 and Its Function in No-Go Decay. Mol Biol Cell
(2009).
19. Atkinson, G.C., Baldauf, S.L. & Hauryliuk, V. Evolution of nonstop, no-go and nonsense-
mediated mRNA decay and their termination factor-derived components. BMC Evol Biol 8,
290 (2008).
20. Wallrapp, C. et al. The product of the mammalian orthologue of the Saccharomyces cerevisiae
HBS1 gene is phylogenetically related to eukaryotic release factor 3 (eRF3) but does not carry
eRF3-like activity. FEBS Lett 440, 387-92 (1998).
21. Nelson, R.J., Ziegelhoffer, T., Nicolet, C., Werner-Washburne, M. & Craig, E.A. The
translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 71, 97-
105 (1992).
22. Inagaki, Y., Blouin, C., Susko, E. & Roger, A.J. Assessing functional divergence in EF-1alpha
and its paralogs in eukaryotes and archaebacteria. Nucleic Acids Res 31, 4227-37 (2003).
23. Pisareva, V.P., Pisarev, A.V., Hellen, C.U., Rodnina, M.V. & Pestova, T.V. Kinetic analysis

19
of interaction of eukaryotic release factor 3 with guanine nucleotides. J Biol Chem 281,
40224-35 (2006).
24. Kong, C. et al. Crystal structure and functional analysis of the eukaryotic class II release factor
eRF3 from S. pombe. Mol Cell 14, 233-45 (2004).
25. Vetter, I.R. & Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions.
Science 294, 1299-304 (2001).
26. Nissen, P. et al. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP
analog. Science 270, 1464-72 (1995).
27. Leibundgut, M., Frick, C., Thanbichler, M., Bock, A. & Ban, N. Selenocysteine tRNA-
specific elongation factor SelB is a structural chimaera of elongation and initiation factors.
Embo J 24, 11-22 (2005).
28. Song, H., Parsons, M.R., Rowsell, S., Leonard, G. & Phillips, S.E. Crystal structure of intact
elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05 A resolution. J
Mol Biol 285, 1245-56 (1999).
29. Cheng, Z. et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev
23, 1106-18 (2009).
30. Petoukhov, M.V. & Svergun, D.I. Global rigid body modelling of macromolecular complexes
against small-angle scattering data. Biophys J (2005).
31. Kononenko, A.V. et al. Role of the individual domains of translation termination factor eRF1
in GTP binding to eRF3. Proteins 70, 388-93 (2008).
32. Alkalaeva, E.Z., Pisarev, A.V., Frolova, L.Y., Kisselev, L.L. & Pestova, T.V. In vitro
reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and
eRF3. Cell 125, 1125-36 (2006).
33. Hauryliuk, V., Zavialov, A., Kisselev, L. & Ehrenberg, M. Class-1 release factor eRF1
promotes GTP binding by class-2 release factor eRF3. Biochimie 88, 747-57 (2006).
34. Pisarev, A.V. et al. The role of ABCE1 in eukaryotic posttermination ribosomal recycling.
Mol Cell 37, 196-210 (2010).
35. Salas-Marco, J. & Bedwell, D.M. GTP hydrolysis by eRF3 facilitates stop codon decoding
during eukaryotic translation termination. Mol Cell Biol 24, 7769-78 (2004).
36. Seraphin, B. Sm and Sm-like proteins belong to a large family: identification of proteins of the
U6 as well as the U1, U2, U4 and U5 snRNPs. Embo J 14, 2089-98 (1995).
37. Wilusz, C.J. & Wilusz, J. Eukaryotic Lsm proteins: lessons from bacteria. Nat Struct Mol Biol
12, 1031-6 (2005).
38. Song, H. et al. The crystal structure of human eukaryotic release factor eRF1--mechanism of
stop codon recognition and peptidyl-tRNA hydrolysis. Cell 100, 311-21 (2000).
39. Prouteau, M., Daugeron, M.C. & Seraphin, B. Regulation of ARE transcript 3' end processing
by the yeast Cth2 mRNA decay factor. Embo J 27, 2966-76 (2008).
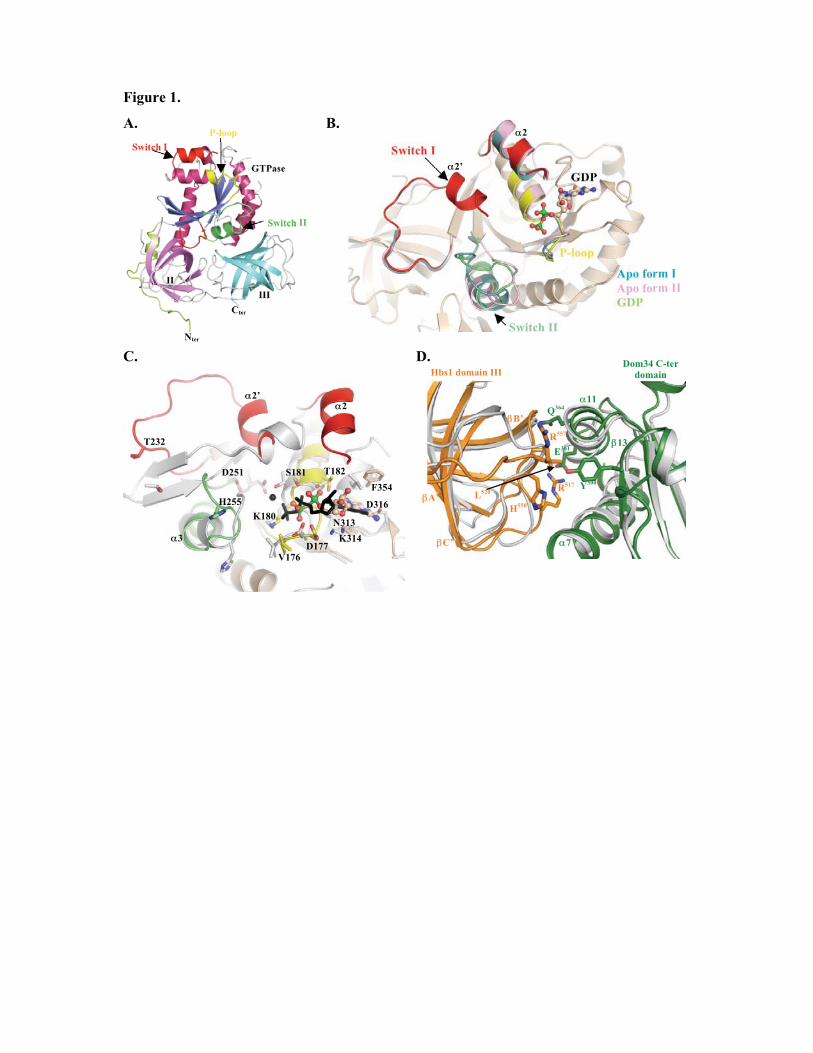
Figure 1.
A. B.
C. D.
III
II
GTPase
Nter
Cter
Switch I
Switch II
P-loop
Apo form I
Apo form II
GDP
Switch I
Switch II
P-loop
GDP
T232
H255
D251
D316
K314
N313
V176
D177
K180
S181 T182
F354
�2 �2’
�3
Dom34 C-ter
domain Hbs1 domain III
�2
�2’
H558
R557
R517
L
520
Q364
E361
Y300
�7
�11
�13
�B’
�A’
�C’
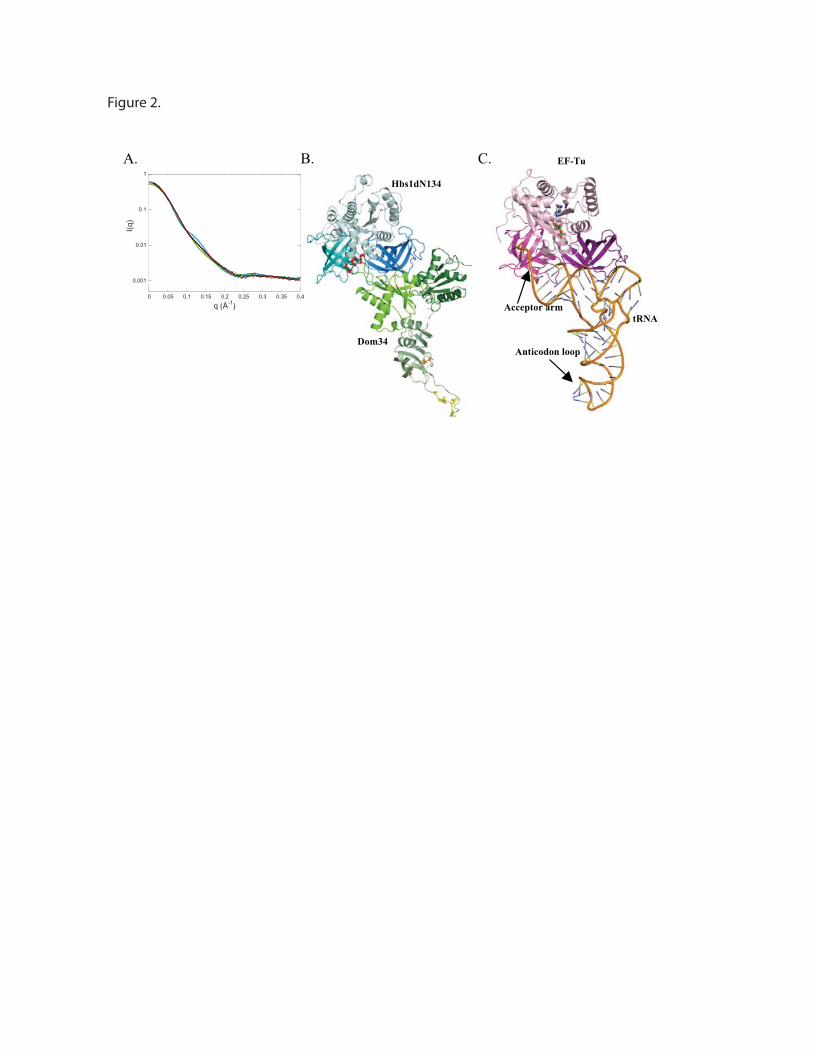
.C .B .A
EF-Tu
tRNA
Anticodon loop
Acceptor arm
Hbs1dN134
Dom34
0.001
0.01
0.1
1
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4
I(q
)
q (Å-1)
Figure 2.
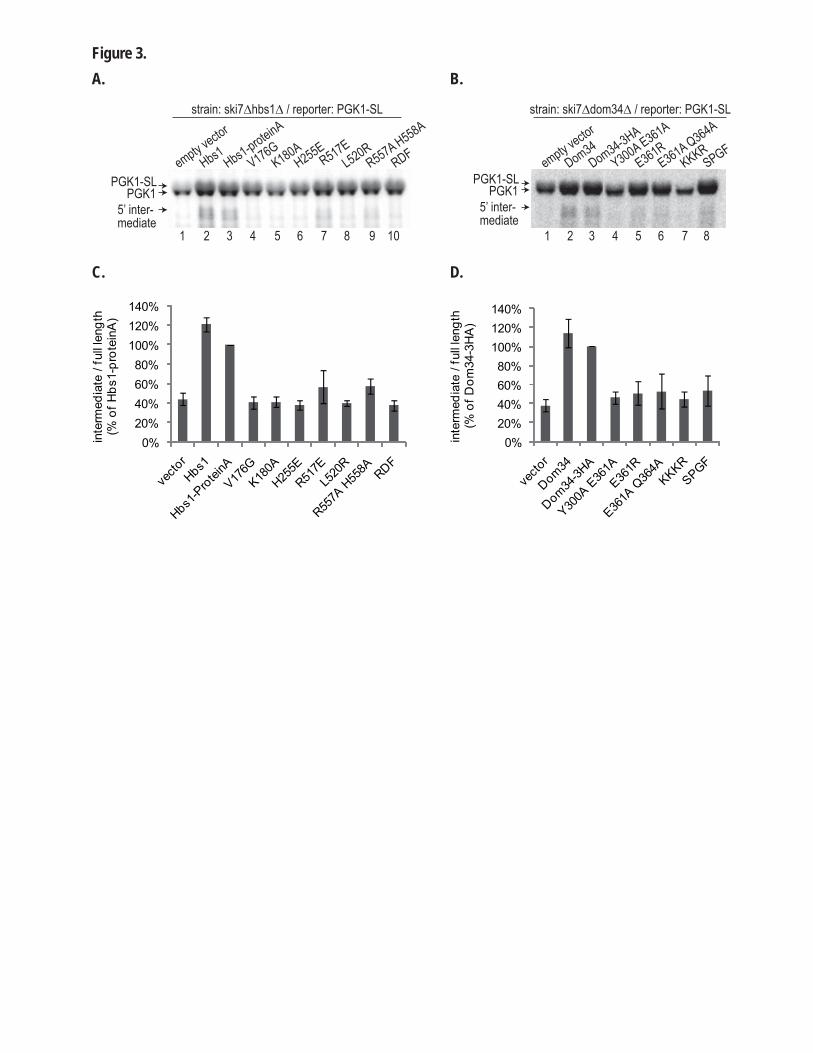
empty vector
Hbs1 Hbs1-proteinA
V176GK180A
H255ER517E
L520RR557A H558A
RDF empty v
ector
Dom34 Dom34-3HA
Y300A E361A
E361RE361A Q364A
KKKRSPGF
strain: ski7�hbs1� / reporter: PGK1-SL
PGK1-SL PGK1-SLPGK1 PGK1
5’ inter-mediate
5’ inter-mediate
1 98765432 10 1 8765432
strain: ski7�dom34� / reporter: PGK1-SL
0%20%40%60%80%
100%120%140%
inte
rmed
iate
/ fu
ll le
ngth
(% o
f Hbs
1-pr
otei
nA)
0%20%40%60%80%
100%120%140%
inte
rmed
iate
/ fu
ll le
ngth
(% o
f Dom
34-3
HA
)
Figure 3.A. B.
C. D.
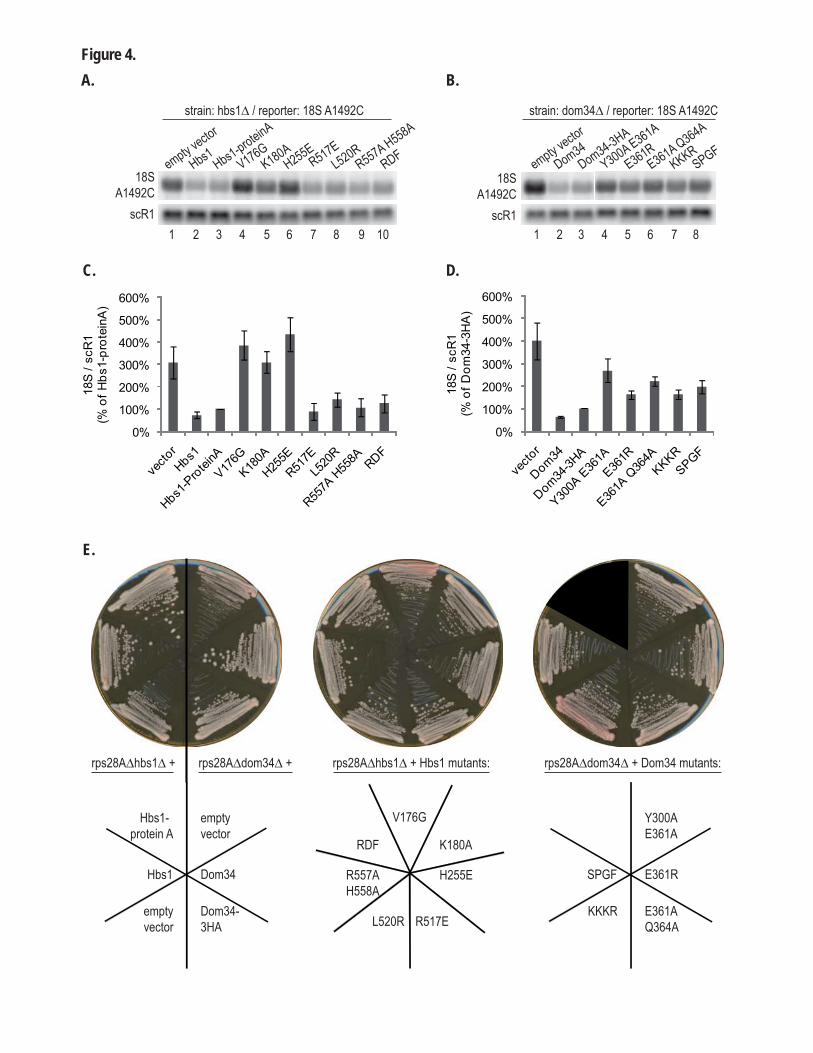
empty vector
Hbs1 Hbs1-proteinA
V176GK180A
H255ER517E
L520RR557A H558A
RDF empty v
ector
Dom34 Dom34-3HA
Y300A E361A
E361RE361A Q364A
KKKRSPGF
strain: hbs1� / reporter: 18S A1492C
1 98765432 10 1 8765432
Figure 4.A. B.
strain: dom34� / reporter: 18S A1492C
C. D.
18S A1492C
scR1
18S A1492C
scR1
0%
100%
200%
300%
400%
500%
600%
18S
/ sc
R1
(% o
f Hbs
1-pr
otei
nA)
0%
100%
200%
300%
400%
500%
600%
18S
/ sc
R1
(% o
f Dom
34-3
HA
)
Hbs1-protein A
Hbs1
empty vector
empty vector
Dom34
Dom34-3HA
V176G
K180A
H255E
R517EL520R
R557A H558A
RDF
SPGF
KKKR
Y300A E361A
E361R
E361A Q364A
rps28A�hbs1� + Hbs1 mutants: rps28A�dom34� + Dom34 mutants: rps28A�hbs1� + rps28A�dom34� +
E.

Supplementary Information
Supplementary Material and methods
Crystallization and Structure determination
Crystals of the native protein were grown from a mixture in a 1:1 ratio of 15mg/mL protein solution in
buffer A (20mM Tris-HCl pH 7.5, 200mM NaCl, 10mM �-mercaptoethanol) and crystallization liquor
containing 0.1M Tris pH 8.5, 12% PEG 6000 and 0.15M NaCl, at 18°C. Crystallization liquor for the
Se-Met protein contained 0.1M Hepes pH 7.0, 15% PEG 4000 and 0.1M MgCl2. The crystals were
flash cooled in liquid nitrogen after addition of 15% and 30% of ethyleneglycol as a crytoprotectant.
Native and Se-Met crystals of the apo-protein diffracted respectively to 2.5Å and 3.2Å resolution on
beamline Proxima1 at SOLEIL synchrotron (Saint-Aubin, France) at 100 K. The crystals belong to
space group P42212 with two molecules in the asymmetric unit (a = b = 110.6Å, c = 188.1Å). The Se-
Met crystal diffraction data were collected at the selenium edge (� = 0.9796 Å). The structure was
determined by the single wavelength anomalous dispersion (SAD) method using the anomalous signal
of selenium atoms. Data were processed with the XDS package. The SOLVE/RESOLVE package was
used to find selenium atom sites in the 30-3.2Å resolution range, to calculate experimental electron
density maps and to improve them by NCS averaging, solvent flattening and phase extension using the
2.5 Å resolution native dataset 1,2. The quality of the experimental phases allowed automatic building
of a partial model with the program RESOLVE (Fig.S1). This model was completed by iterative
cycles of manual rebuilding using COOT followed by refinement with PHENIX.REFINE 3,4. All
residues display main chain dihedral angles that fall within allowed regions of the Ramachandran plot
as defined by the program PROCHECK 5.
In parallel, we have been able to obtain crystals of the HbsdN134 GDP bound form. A 2.95 Å
resolution dataset was recorded from a crystal grown by mixing 0.2�l of SeMet labelled protein
(18mg/ml and incubated with 1mM GDP in the absence of Mg2+ ion) with an equal volume of the
following crystallization condition: 12% PEG 4,000 (w/v), 20% glycerol, 50mM MOPS pH 7.0, 0.5M
KCl. The crystals were flash cooled in liquid nitrogen after transfer into a crystallization condition
containing 30% glycerol. Data were collected on beamline Proxima1 at SOLEIL synchrotron (Saint-
Aubin, France) at 100K (� = 0.9796 Å).
The statistics for data collection and refinement are summarized in Table S1. The atomic coordinates
and structure factors have been deposited into the Brookhaven Protein Data Bank under accession
number (XXXX and YYYY).
SAXS studies
The Dom34-Hbs1dN134 complex was prepared by mixing equimolar amounts of both purified
Dom34 and Hbs1dN134 proteins in buffer A. SAXS experiments on the Hbs1-Dom34 complex were
performed using the Nanostar instrument at IBBMC in Orsay. The X-rays were produced by a rotating

anode (Cu K�, wavelength � = 1.54 Å), and the scattered X-rays were collected using a two-
dimensional position sensitive detector (Vantec) positioned at 662 mm from the sample. The scattering
vector range was 0.011 < q < 0.40 Å-1 where q = 4�sin�/� and 2� is the scattering angle. Experiments
on Hbs1 were performed on the beamline SWING, at the Synchrotron SOLEIL. The incident beam
energy was 12 keV, and the sample to detector (Aviex CCD) distance was set to 1843 mm. The
scattering vector range was 0.008 < q < 0.5 Å-1. Several successive frames (typically 20) of 4 s each
were recorded for both sample and pure solvent. We checked that X rays did not cause irradiation
damage by comparing the successive frames, before calculating the average intensity and experimental
error. In both cases scattering from the buffer was measured and subtracted from the corresponding
protein spectra. Intensities were scaled using the scattering of water. For each protein, a range of
concentrations was explored. For the Hbs1-Dom34 complex the concentration of the solution was
maintained above 3 g.L-1 to avoid dissociation. No significant effect of the concentration was
observed. Calculations of the molar mass of each protein from the intensity extrapolated to q � 0 gave
values that were within 15 % of the theoretical mass.
Scattering patterns from crystal structures were calculated using the program CRYSOL 6. The relative
position of some domains (see Supplementary Results) was refined using the rigid-body modelling
program SASREF 7, which uses simulated annealing to find an optimal configuration of the domains
by fitting the SAXS curve. An ultimate adjustment was performed using the program CRYSOL. The
goodness of fit was characterized by the following parameter,
where N is the number of experimental points, c is a scaling factor, and Icalc(qj) and �(qj) are the
calculated intensity and the experimental error at the scattering vector qj, respectively.
Fluorescence studies.
Methylanthraniloyl (Mant)-GTP and Mant-GDP were purchased from Jena Biosciences. Excitation
was performed at 349 nm and emission scanned from 400 to 500 nm with a Cary Eclipse
fluorospectrophotometer (Varian). Measurements were performed as previously described 8. Binding
of ligands to Hbs1 was quantified by measuring fluorescence difference of Mant-guanine nucleotide at
448nm against ligand concentration. Experiments were performed in triplicate.
Strep-purification
BL21(DE3) cells containing C-terminally Strep-tagged Dom34 and/or C-terminally His-tagged Hbs1
expressing plasmid were grown in 200 ml ZYM-5052 autoinduction medium 9 containing
chloramphenicol 25 �g/ml and/or kanamycine 50 �g/ml at 37 °C for 7 hours, then at 25 °C for 14

hours. Cells were harvested by spinning 6000 rpm 10 min 4 °C, washed in 20 ml PBS (4 °C) followed
by centrifugation at 5000 rpm 8 min 4 °C, and then resuspended in 10 ml buffer W (100 mM Tris
pH8; 1 mM EDTA; 150 mM NaCl). Cells were lysed by passing them through a Cell Disruptor
(Constant Systems) at 1.55 kbar, followed by rinsing with 5 ml buffer W. The resulting 15 ml cell
lysates were spun down at 12000 rpm 30 min 4 °C. Strep-purification was performed on the
supernatant with the Strep-tag� / Strep-tactin� affinity purification kit (IBA Biotechnology), following
the manufacturers’ instructions and using a 200 �l column volume. Samples from induced culture,
supernatant and pellet after cell lysis and flowthrough (1:40000 of total volume) and of elution
fraction E3 and E4 (1:560 of total elution volume) were analyzed on 12 % SDS-PAGE and visualized
by Coomassie staining.

Supplementary Data
Hbs1 overall structure.
The structure of Hbs1dN134 has been solved by SAD. Hbs1dN134 is composed of three
domains, forming a triangular prism of approximately 50Å side length and 30Å depth. The GTPase
domain adopts the classical �/� structure common to GTPases. such as EF-Tu, eRF3 and Ras. It is
composed of a central six-stranded �-sheet (strands order �6, �5, �4, �1, �3 and �2, which is
antiparallel to the others) flanked by 7 �-helices (�1, �2, �6 and �7 on one side of the sheet and �3-5
on the other; Fig. 1A). The GTPase domain is connected to domain II by the long helix �7 (residues
376 to 398) and domain II is linked to domain III by a short extended stretch of peptides. Both
domains II and III adopt a 6-stranded �-barrel fold similar to the corresponding domains from archaeal
elongation factor 1-� (aEF-1A), bacterial elongation factor (EF-Tu) and eukaryotic class II release
factor Although they belong to the same SCOP superfamily, domains II and III adopt radically
different topologies (strands order �A, �B, �E, �D, �C and �F for domain II and �A’, �D’, �C’, �B’,
�E’ and �F’ for domain III).
For both the apo and GDP bound form of Hbs1dN134, the crystal asymmetric unit contains two
molecules organized as a non crystallographic dimer, yielding to 4 sets of coordinates for this Hbs1
monomer. These 4 copies are virtually identical (rmsd 0.4-0.5Å over 430 residues) except for the
switch regions that adopt different conformations. Despite the presence of 2 copies per asymmetric
unit, both size exclusion chromatography coupled online to a triple detection array and small-angle X-
ray scattering (SAXS) indicated that the protein is monomeric in solution. Among these four copies of
Hbs1dN134, the switch II region is disordered in one GDP bound copy and adopts 3 different
conformations in the others.
Hbs1 conformation.
Several structures of GTPases involved in translation have now been solved, revealing important
differences in the orientation of the GTPase domain relative to domains II-III. In the structures of
bacterial EF-Tu, the GTPase domain undergoes a substantial rotation with respect to domains II-III
depending on the bound nucleotide (GDP or the GTP non-hydrolyzable analog GDPNP; 10-13). On the
opposite, the structures of archaeal SelB elongation factor obtained in the absence of nucleotide or in
the presence of GDP or GDPNP by co-crystallization, are virtually the same and correspond to the
conformation adopted by EF-Tu in the presence of GTP 14. Finally, the conformation observed in the
crystal structure of S. pombe class II release factor eRF3 is radically different from that of EF-Tu-GTP
and nucleotide (GDP or GDPNP) binding does not affect its conformation 15 (Fig.S2A). Whether this
reflects a functional property of eRF3 remains to be determined.
To investigate the conformation adopted by Hbs1dN134 in solution, we have performed small-angle
X-ray scattering analysis on this fragment in the absence of nucleotide. As a first approach, we have

compared the diffusion curves calculated from two different Hbs1dN134 conformations to the final
experimental scattering pattern. The first conformation corresponds to that observed in the crystal
while the second is based on the conformation of S. pombe eRF3. As shown in Fig.S2B, the best fit is
observed for the conformation corresponding to the Hbs1 crystal structure (� value of 2.8 compared to
18.5 for the eRF3 based model). However, there is some discrepancy at q values of 0.15-0.2 Å-1 with
the final experimental scattering pattern. As in our crystal structure of Hbs1dN134, most of the switch
I region was missing due to intrinsic disorder, we have first modelled this region using the coordinates
of the corresponding region of eEF1A in the structure of the eEF1A-eEF1B� complex 16. The location
of this switch I region was further adjusted by superimposing residues 35-73 from eEF1A switch I
region onto helices �2 and �2’ from the Hbs1-GDP complex to yield the model used to calculate the
initial diffusion curve. As switch I region from G proteins is known to be highly flexible in the
absence of nucleotide, we decided to move this switch I region (residues 176 to 235) as a rigid body
group using the SASREF program 7. Movement of this switch I region by 10Å is sufficient to obtain a
model with an excellent agreement with the SAXS curve (Fig.S2B, � value of 1.6).
Altogether, these experiments performed in solution show that the Hbs1dN134 conformation trapped
in the crystal is similar to that in solution and radically different from the conformation observed for
eRF3 15
Generation of Dom34-Hbs1 models for SAXS.
To obtain more information on the Dom34-Hbs1 complex from S. cerevisiae, we have performed
SAXS measurements on this complex and compared diffusion curves calculated from different models
to the experimental one. To build these models, we used the known crystal structures of isolated yeast
and archaeal Dom34 and yeast Hbs1dN134 and of the complex between eRF1 and an eRF3 fragment
lacking the GTPase domain 8,17,18. In these models, Dom34 C-terminal domain and Hbs1 domain III
were superimposed onto the corresponding domains from human eRF1 and eRF3, respectively 17. Due
to the absence of the eRF3 GTPase domain in the crystal structure of eRF1-eRF3 complex, we
assumed that Hbs1 conformation in the complex is similar to that of the protein alone in the crystal
and in solution. This is further supported by site directed mutagenesis experiments on eRF3 that
identified residues from domain III (His582, Arg642, Arg646, Val654 and Lys656 from S. pombe
eRF3 corresponding to Phe528, Arg591, Arg595, Ala603 and Lys605 from Hbs1, respectively) as
critical for cell growth 15. In the Hbs1 structure, these highly conserved residues are located at the
interface between domain III and the GTPase domain, this strongly suggest that the integrity of this
domain interface is essential for eRF3, and by inference Hbs1, activities. In addition, the substitution
by Ala of two eRF3 residues from domain III (Arg642 and Arg646) results in loss of complementation
of eRF3 deletion in S. pombe and substantially weaker eRF1 binding activity 15. These residues are
located outside eRF1-eRF3 interface 17 and would directly interact with the eRF3 GTPase domain in a
so-called eRF3 “close” conformation that would be similar to EF-Tu and Hbs1 structures. This implies

that this “close” conformation is necessary for correct function of eRF3 and by extension to members
of the eEF-1A family. We therefore used the coordinates of the Hbs1dN134 fragment previously
determined by combining the Hbs1dN134 crystal structure and modelling of the switch I region with
SASREF. As a consequence, the only difference between these models is the position of Dom34
domains relative to each other. We have previously observed that S. cerevisiae and archaeal Pelota
proteins adopt radically different configuration with rotation by roughly 20° and 70° relative to C-
terminal domain for N-terminal and central domains, respectively 8. This allowed us to build two
models based on either yeast Dom34 conformation (yeast-like model, Fig.S4A) or archaeal Pelota
conformation (archaeal-like model, Fig.S4B).
From the comparison of the scattering curves calculated from both models with the SAXS curve
(Fig.4A), it turned out that the archaeal-like model gave the best agreement and could serve as a
starting point model for further improvement using SASREF. In this archaeal-like model, the
conserved and positively charged loop C from Dom34 (residues 174-176) that has been shown to be
important for NGD in yeast 19 is located within the central domain and is in close proximity of the
Hbs1 GTP binding site. This loop could hence contact the GTP �-phosphate and be responsible for the
higher affinity of Hbs1 for GTP in the presence of Dom34. During the rigid body refinement process
using SASREF, we therefore decided to move only the N-terminal and central domains from Dom34
as the comparison of yeast Dom34 and archaeal Pelota crystal structures clearly highlighted intrinsic
flexibility of these domains 8. However, as we anticipate that Dom34 loop C may play an active role in
the increase of Hbs1 affinity for GTP in the presence of Dom34, we forced the C� atom from residues
174-177 of this loop to be within less than 15 Å away from the C� atoms of Val176 and His255 from
Hbs1. We obtain a set of models providing calculated scattering curves compatible with the
experimental curve (�1.7). All these models have similar conformations differing mainly in the
position of Dom34 N-terminal domain, which can rotate on itself. The SAXS measurements cannot
discriminate the different positions adopted by this domain.
Table S1. Data collection and refinement statistics
Native SeMet GDP form
Data collection
Space group P42212 P42212 P42212
Cell dimensions
a, b, c (Å) 110.6, 110,6, 188.1 110.6, 110,6, 188.1 110.6, 110,6, 188.1
�, �, � (°) 90, 90, 120 90, 90, 120 90, 90, 120
Resolution (Å) 30-2.5 (2.54-2.5) 30-3.2 (3.39-3.2) 20-2.95 (3.13-2.95)
Rsym 0.076 (0.486) 0.087 (0.472) 0.097 (0.491)
I / �I 26 (3.7) 12.5 (2.9) 8.5 (2.2)
Completeness (%) 96 (97.3) 99.6 (99.1) 97.9 (98.2)
Redundancy 3.5 (3.7) 3.7 (3.7) 2.6 (2.6)
Refinement
Resolution (Å) 30-2.5 20-2.95
No. reflections 39338 45371
Rwork / Rfree 0.216/0.271 0.214/0.277
No. atoms
Protein 6887 6922
Ligand/ion - 54
Water 275 65
B-factors
Protein 46.1 52.5
Ligand/ion - 76.5
Water 45.6 41
R.m.s. deviations
Bond lengths (Å) 0.006 0.011
Bond angles (°) 1.020 1.358
Table S2. Saccharomyces cerevisiae strains
Strain Genotype
BMA64 MAT �, ura3-1, trp1�, ade2-1, leu2-3,112, his3-11,15
BSY1970 MAT �, ura3-1, trp1�, ade2-1, leu2-3,112, his3-11,15, dom34�::HIS3
BSY2029 MAT a, ura3-1, trp1�, ade2-1, leu2-3,112, his3-11,15, dom34�::HIS3 ski7�::Kan R
BSY2051 MAT a, ura3-1, trp1�, ade2-1, leu2-3,112, his3-11,15, dom34�::HIS3 rps28A�::Kan R
BSY2145 MAT a, ura3-1, trp1�, ade 2-1, leu2-3,112, his3-11,15, hbs1�::KanR
BSY2204 Mat a, ura3-1, trp1�, ade2-1, leu2-3, 112, his3-11, 15, ski7�::KanR, hbs1�::KanR
BSY2218 Mat a, ura3-1, trp1�, ade2-1, leu2-3,112, his3-11,15, rps28A�::KanR, hbs1�::KanR
Table S3. Plasmids
������� �� � ����� ����� ��
�����
�����
������� ����������� ��� ��
�����
�����
��� ��������������� ���� ��
������� ������ ���������������� ��� ����� ��!
������� ������ �������������� ��� ����� ��!
������� ������ ������������������������ ��� ����� ��!
�����"� ������ �������������������� ��� ����� ��!
Table S4. Oligonucleotides
���� ���� ���
������
#���""� $�$$$ �$$$ � �$��$ $
#���""� �$$��� $$ � � ����
#������ � �$ ����$ $�$�$ ���$� � ���� ������������$�$$�
#�����" �� ��� $ $�$� $ � $$�$�� $�� ������ �� ��$����� ��
#������ $$����� � �$ $$�� �� $���$ $$
#������ � $$� ���� ������$ ��$�$$$�$$���� ������ �� ��$����� �
�
#������ $���������$ $�$�$ ���$
#������ � $$�$�� $�� ������ �� ��$���
#������ $�$$�$���$ ��� � ���� � �����$�� �$ � $$ �$�
#������ ���� �� � $� � �$$ $� � �� $ � ��$�
#������ �����$$ $� ��� �� �� �� ���$ ��� � ���$ � �� � $$�
#������ ��$ $� � �� $$�$$�$$�$$� � ���� $ �� �$$�
#������ ��$$ $$ � $$ $ ���$��� ��$��� ��
#������ $� $�� $�� $� ����$� $����$ $�
#������ $$����� � �$$ $� � �� $ � ��$�
#������ � $$� ���� ������$ ��$�$$$�$$���� $� ����$� $����$ $
�
#������ ������$$$$$ �$�� � � � � � �
#������ ��� $���$$$� � ��$���$ �� �$ $
#������ $ $��� ��$�$$�����$
#������ ����$$$�� $$��$ �������$�$$
#������� $$ ���� � �$�� ��$$� ��� $����$
#������ � $ $��$�$$����$ $�����$ $�� ����$$ � ���� ���$�
#���"�� � ��$���� �$$$�� ��� � $$$�
#���"�� � $�� $$ � ��$� �$$$� ������ �� ��$����� ��
#���"�� �$ �$$�$ $$ $� ��� � �$ �$�����$ ��
#���"�� $� $�� $�� $�$� ���$$ �$�� � �$$ � $$ ���
#���"�� $� $��$��$��$��$��$$� � �� � ��� ������$���� � $
#���"�� �$�� $� $�$$� $�$ �� ��$ �$� ��$ $���$ �$
������ $$��$$� $� $�
%��� �$ $$ ��� $$����
Table S5. Fusion PCR primers and templates
����� �����
��&���'(� ����� ������
�������� #���"�� #���"�� #���"��
�������� #���"�� #���"�� #���"��
)������*
����+,�� ������� �% �,��* �*���-.��
/�&0��&�����,+12����"3
�
�������� #������ #������ #������
�������� #������ #������ #������
*4���
������&
����+,�� ������� ��������
/��0,'�����,+12�����3
�
�������� #���"�� #���"�� #������
�������� #������ #������ #������
)����� 555��
�'�,&�
����+,�� ������� ������� �
�������� #������ #������ #������
�������� #������ #������ #������
)����
67��,�&�
����+,�� ������� ������� �
�������� #������ #������ #������
�������� #������ #������ #������
)����
67��,�&�
����+,�� ������� ������� �

Figure S1. Experimental electron density map.
Stereo view representation of the experimental electron density map obtained by phasing at 3.2 Å
resolution followed by phase extension up to 2.5Å resolution. The map is contoured at 1�. The final
model fitted into the electron density map is represented as sticks.
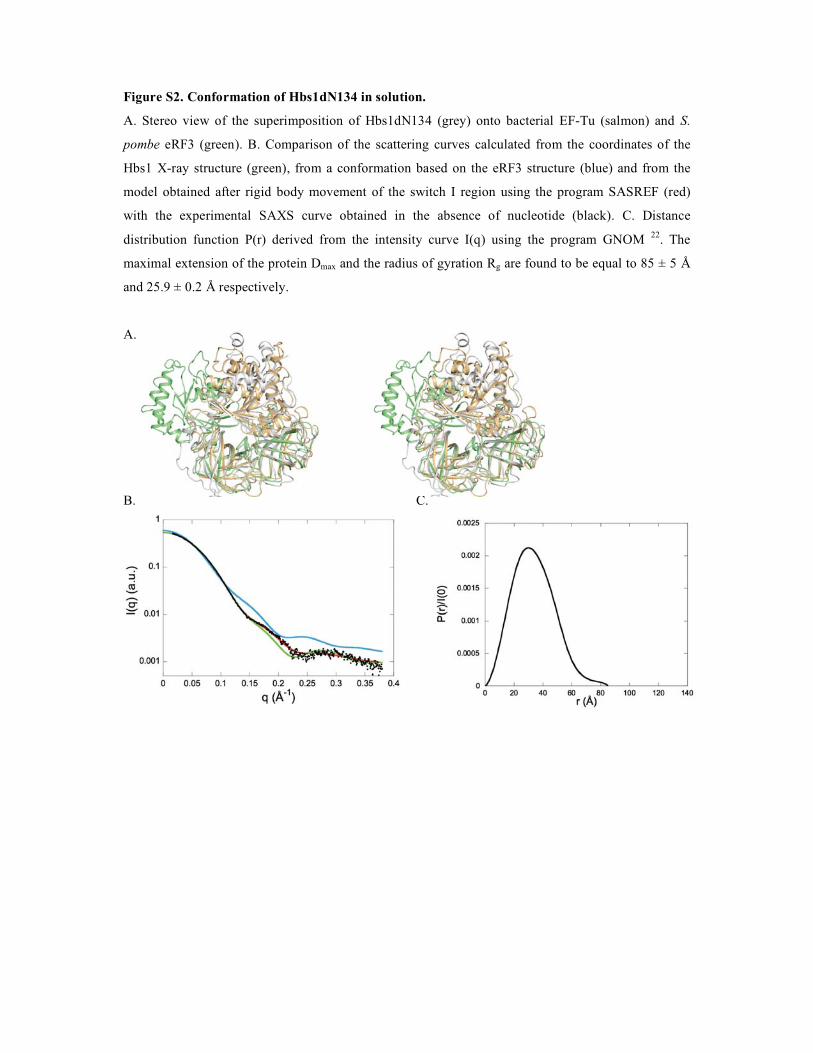
Figure S2. Conformation of Hbs1dN134 in solution.
A. Stereo view of the superimposition of Hbs1dN134 (grey) onto bacterial EF-Tu (salmon) and S.
pombe eRF3 (green). B. Comparison of the scattering curves calculated from the coordinates of the
Hbs1 X-ray structure (green), from a conformation based on the eRF3 structure (blue) and from the
model obtained after rigid body movement of the switch I region using the program SASREF (red)
with the experimental SAXS curve obtained in the absence of nucleotide (black). C. Distance
distribution function P(r) derived from the intensity curve I(q) using the program GNOM 22. The
maximal extension of the protein Dmax and the radius of gyration Rg are found to be equal to 85 ± 5 Å
and 25.9 ± 0.2 Å respectively.
A.
B. C.
C
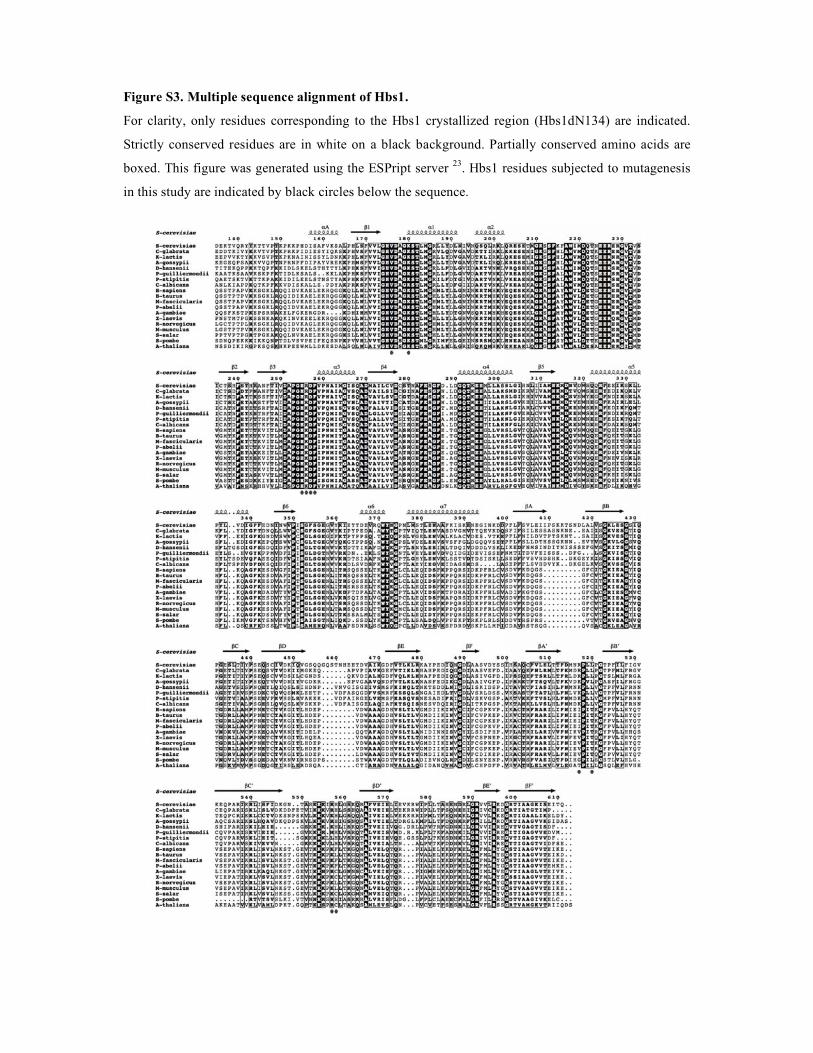
Figure S3. Multiple sequence alignment of Hbs1.
For clarity, only residues corresponding to the Hbs1 crystallized region (Hbs1dN134) are indicated.
Strictly conserved residues are in white on a black background. Partially conserved amino acids are
boxed. This figure was generated using the ESPript server 23. Hbs1 residues subjected to mutagenesis
in this study are indicated by black circles below the sequence.
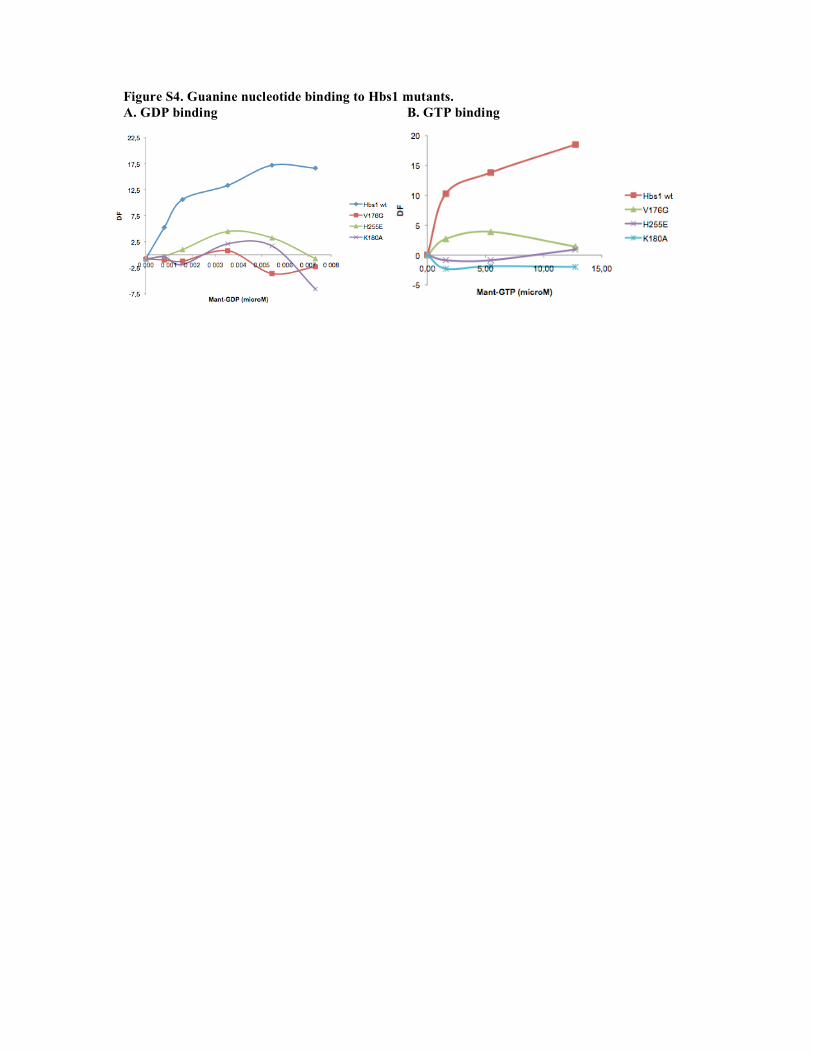
Figure S4. Guanine nucleotide binding to Hbs1 mutants. A. GDP binding B. GTP binding

Figure S5. Involvement of Dom34 domains in Dom34-Hbs1 interaction
C-terminally Strep-tagged Dom34 and/or C-terminally His-tagged Hbs1 were co-expressed in
BL21(DE3) cells. Protein purification with Strep-tactin sepharose was performed on the soluble
extract obtained after cell lysis. Results were analysed on a 12% SDS-PAGE and visualized by
Coomassie staining.
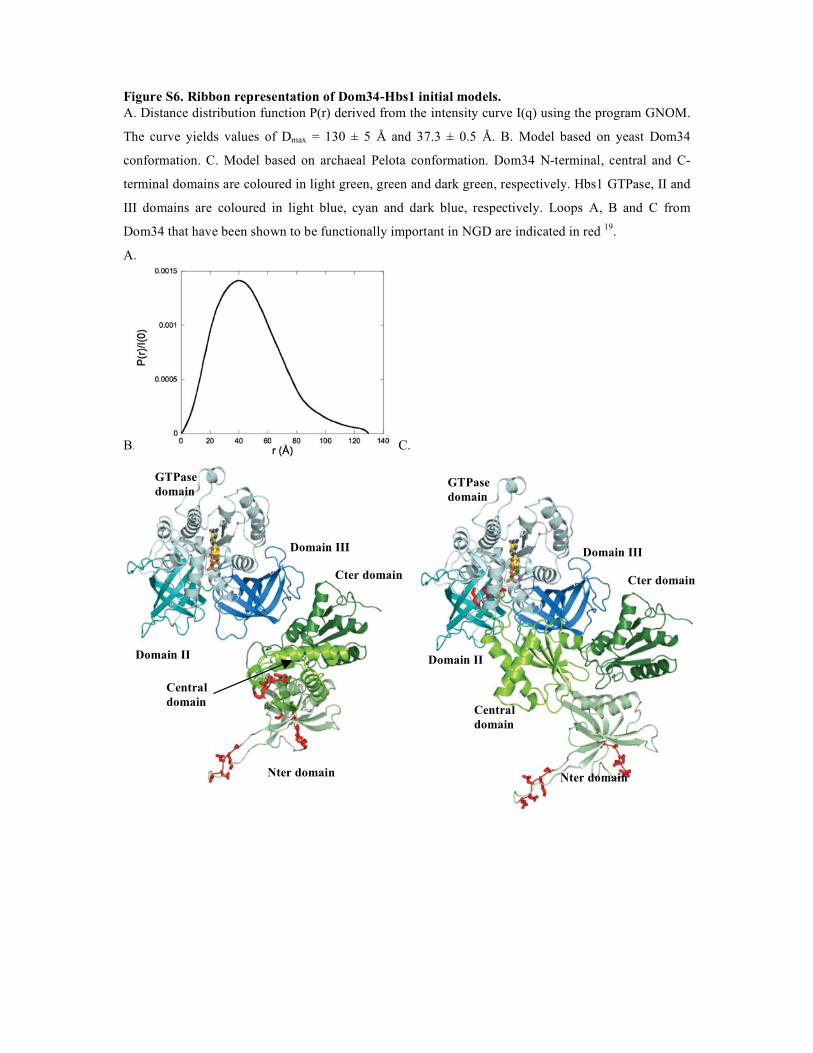
Figure S6. Ribbon representation of Dom34-Hbs1 initial models. A. Distance distribution function P(r) derived from the intensity curve I(q) using the program GNOM.
The curve yields values of Dmax = 130 ± 5 Å and 37.3 ± 0.5 Å. B. Model based on yeast Dom34
conformation. C. Model based on archaeal Pelota conformation. Dom34 N-terminal, central and C-
terminal domains are coloured in light green, green and dark green, respectively. Hbs1 GTPase, II and
III domains are coloured in light blue, cyan and dark blue, respectively. Loops A, B and C from
Dom34 that have been shown to be functionally important in NGD are indicated in red 19.
A.
B. C.
GTPase domain
Domain II
Domain III
Nter domain
Cter domain
Central domain
GTPase domain
Domain II
Domain III
Nter domain
Cter domain
Central domain
.

Figure S7. Expression of Dom34 and Hbs1 mutants
Mutant expression was verified by protein analysis on protein extract 24 from the strains used for the
analysis of the effect of mutants on NRD. Dom34-3HA and its mutants were detected with anti-HA
antibody and a secondary antibody goat-anti-mouse antibody (Pierce), Hbs1-protein A and its mutants
were detected with PAP (Sigma).
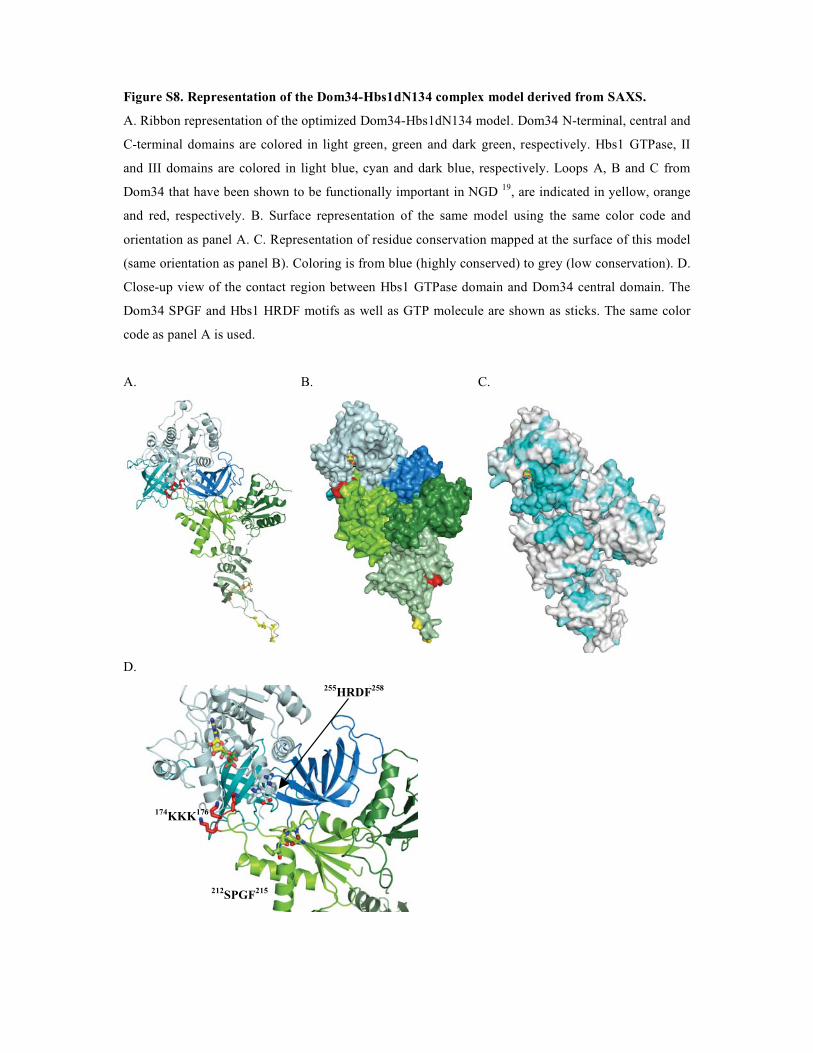
Figure S8. Representation of the Dom34-Hbs1dN134 complex model derived from SAXS.
A. Ribbon representation of the optimized Dom34-Hbs1dN134 model. Dom34 N-terminal, central and
C-terminal domains are colored in light green, green and dark green, respectively. Hbs1 GTPase, II
and III domains are colored in light blue, cyan and dark blue, respectively. Loops A, B and C from
Dom34 that have been shown to be functionally important in NGD 19, are indicated in yellow, orange
and red, respectively. B. Surface representation of the same model using the same color code and
orientation as panel A. C. Representation of residue conservation mapped at the surface of this model
(same orientation as panel B). Coloring is from blue (highly conserved) to grey (low conservation). D.
Close-up view of the contact region between Hbs1 GTPase domain and Dom34 central domain. The
Dom34 SPGF and Hbs1 HRDF motifs as well as GTP molecule are shown as sticks. The same color
code as panel A is used.
A. B. C.
D.
174KKK176
255HRDF258
212SPGF215

Supplementary references
1. Terwilliger, T.C. Reciprocal-space solvent flattening. Acta Crystallogr D Biol Crystallogr 55, 1863-71 (1999).
2. Terwilliger, T.C. & Berendzen., J. Automated MAD and MIR structure solution Acta Cryst.
D 55, 849-861 (1999). 3. Adams, P.D. et al. PHENIX: building new software for automated crystallographic structure
determination. Acta Crystallogr D Biol Crystallogr 58, 1948-54 (2002). 4. Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr
D Biol Crystallogr 60, 2126-32 (2004). 5. Laskowski, R.A., MacArthur, M.W., Moss, D.S. & Thornton, J.M. PROCHECK: a program to
check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283-91 (1993). 6. Svergun, D.I., Barberato, C. & Koch, M.H.J. CRYSOL - a program to evaluate X-ray solution
scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768-773 (1995).
7. Petoukhov, M.V. & Svergun, D.I. Global rigid body modelling of macromolecular complexes against small-angle scattering data. Biophys J (2005).
8. Graille, M., Chaillet, M. & van Tilbeurgh, H. Structure of yeast Dom34: a protein related to translation termination factor Erf1 and involved in No-Go decay. J Biol Chem 283, 7145-54 (2008).
9. Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein
Expr Purif 41, 207-34 (2005). 10. Andersen, G.R., Nissen, P. & Nyborg, J. Elongation factors in protein biosynthesis. Trends
Biochem Sci 28, 434-41 (2003). 11. Kjeldgaard, M., Nissen, P., Thirup, S. & Nyborg, J. The crystal structure of elongation factor
EF-Tu from Thermus aquaticus in the GTP conformation. Structure 1, 35-50 (1993). 12. Noble, C.G. & Song, H. Structural studies of elongation and release factors. Cell Mol Life Sci
65, 1335-46 (2008). 13. Song, H., Parsons, M.R., Rowsell, S., Leonard, G. & Phillips, S.E. Crystal structure of intact
elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05 A resolution. J
Mol Biol 285, 1245-56 (1999). 14. Leibundgut, M., Frick, C., Thanbichler, M., Bock, A. & Ban, N. Selenocysteine tRNA-
specific elongation factor SelB is a structural chimaera of elongation and initiation factors. Embo J 24, 11-22 (2005).
15. Kong, C. et al. Crystal structure and functional analysis of the eukaryotic class II release factor eRF3 from S. pombe. Mol Cell 14, 233-45 (2004).
16. Andersen, G.R., Valente, L., Pedersen, L., Kinzy, T.G. & Nyborg, J. Crystal structures of nucleotide exchange intermediates in the eEF1A-eEF1Balpha complex. Nat Struct Biol 8, 531-4 (2001).
17. Cheng, Z. et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev 23, 1106-18 (2009).
18. Lee, H.H. et al. Structural and functional insights into Dom34, a key component of No-Go mRNA decay. Mol Cell 27, 938-50 (2007).
19. Passos, D.O. et al. Analysis of Dom34 and Its Function in No-Go Decay. Mol Biol Cell (2009).
20. Doma, M.K. & Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561-4 (2006).
21. LaRiviere, F.J., Cole, S.E., Ferullo, D.J. & Moore, M.J. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell 24, 619-26 (2006).
22. Svergun, D.I. Determination of the regularization parameter in indirect transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495 - 503 (1992).
23. Gouet, P., Courcelle, E., Stuart, D.I. & Metoz, F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305-8 (1999).
24. Kushnirov, V.V. Rapid and reliable protein extraction from yeast. Yeast 16, 857-60 (2000).


167
Conclusions, Perspectives
Cette étude a permis de mettre en évidence le rôle de Hbs1 dans les voies NGD et NRD.
Hbs1 appartient à la famille des GTPases de la traduction et son activité de fixation du
nucléotide est nécessaire à la NGD et à la NRD. En revanche, si la formation du complexe est
critique pour une NGD normale, elle est moins importante pour la NRD.
Le complexe Dom34-Hbs1 tel que nous l'avons modélisé et validé par SAXS est très
similaire au complexe d'élongation EF-Tu:ARNt et au complexe de terminaison eRF1-eRF3,
ce qui est cohérent avec tous les points communs fonctionnels précédemment établis. Tout
cela suggère que le complexe Dom34-Hbs1 entre dans le site A des ribosomes bloqués en
cours d'élongation.
Les cellules eucaryotes disposent ainsi de deux complexes ayant évolué pour cibler les
ribosomes en pause :
- eRF1-eRF3 catalyse l'hydrolyse de la liaison peptidyl-ARNt lorsque le ribosome arrive
à un codon stop;
- Dom34-Hbs1 résout le blocage des ribosomes en pause en participant à l’élimination de
l'ARN fautif. Dom34-Hbs1 joue par conséquent un rôle central dans le contrôle qualité des
ARN.
Des questions restent cependant sans réponse dans les mécanismes NGD-NRD :
- Quelle est la nucléase assurant la dégradation de l'ARN ? S’agit-il du ribosome ?
- Quel est le mécanisme spécifique à la NRD qui le différencie de la NGD ?
- Comment l'ARNr 18S non fonctionnel est-il pris en charge et destiné à la dégradation
après fixation de Dom34-Hbs1 ?

168
- D'une manière générale les voies NGD et NRD font-elles intervenir des facteurs
supplémentaires ? En temps que voies de contrôle qualité, sont-elles liées à d'autres
évènements prenant place sur l'ARNm ?
- Quelles sont les conditions biologiques concrètes dans lesquelles les voies NGD et
NRD sont appliquées ? Quelle est l'importance de ces mécanismes pour le développement,
l'adaptation et la survie des cellules ? Le complexe Dom34-Hbs1 aurait-t-il un rôle dans le
vieillissement cellulaire, au cours duquel l'accumulation de mutations peut affecter voire
mettre un terme au fonctionnement de la cellule ?
A ce titre, Dom34 n'est pas une protéine essentielle chez la levure, mais la délétion de
dom34 conduit à un retard de développement et une incapacité à sporuler (Davis and
Engebrecht, 1998). De manière similaire, la délétion du gène codant pour Dom34/Pelota
cause un défaut de développement des gamètes chez la Drosophile (Eberhart and Wasserman,
1995) et un défaut létal de développement des embryons chez la Souris (Adham et al., 2003).
Ces résultats suggèrent que Dom34/Pelota est nécessaire pour la progression normale de la
mitose et de la méiose. Une étude récente montre que Dom34 interagit avec les
microfilaments d'actine chez les mammifères (Burnicka-Turek et al., 2010). Chez les
métazoaires Dom34 joue un rôle essentiel lié au cycle cellulaire.
La différence d'importance de Dom34 entre les différents organismes est à mettre en
parallèle avec les expériences réalisées sur Upf1/Rent1 : accessoire chez la Levure (Amrani et
al., 2006), celle-ci est essentielle au développement de la Souris (Medghalchi et al., 2001;
Weischenfeldt et al., 2008). En plus de son rôle dans l'élimination des ARNm aberrants, Upf1
intervient dans la régulation d'ARNm normaux (Nicholson et al., 2010). Nous pouvons donc
supposer qu'en plus de son rôle d'élimination d'ARN anormaux, Dom34 joue elle aussi un rôle
plus global dans la régulation de l'expression génique. Il serait intéressant de valider cette
hypothèse en évaluant les effets liés à Hbs1.

169
Dom34 a probablement un rôle régulateur sur l'activité GTPase de Hbs1, qu'il serait utile
de caractériser. Les nombreux points communs entre Dom34-Hbs1 et eRF1-eRF3 suggèrent
que les mesures d'activité devraient nécessiter la présence du ribosome eucaryote purifié.
La structure du complexe Dom34-Hbs1, déterminée chez une équipe concurrente,
permettra la vérification des hypothèses que nous avons émises dans notre étude. La co-
cristallisation avec du GDP ou du GTP et/ou du Mg2+ permettrait de déterminer les bases
moléculaires de l'affinité de Hbs1 pour les nucléotides.
Les apports de la co-cristallisation des facteurs de terminaison avec le ribosome bactérien
ont été significatifs pour la compréhension au niveau moléculaire de la terminaison de la
traduction. Disposer de la structure à haute résolution du ribosome eucaryote apporterait
certainement une contribution considérable et permettrait d'envisager la détermination de sa
structure en complexe avec eRF1-eRF3 et Dom34-Hbs1.
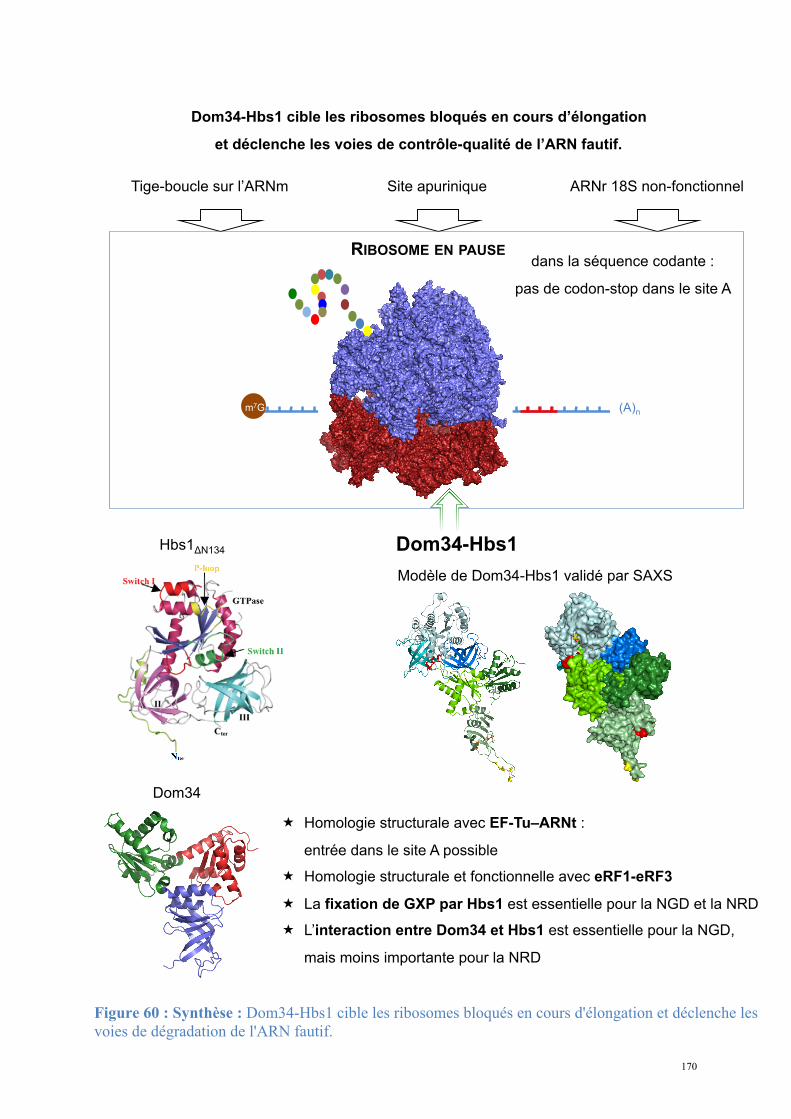
170
m7G (A)n
Dom34-Hbs1 cible les ribosomes bloqués en cours d’élongation
et déclenche les voies de contrôle-qualité de l’ARN fautif.
Tige-boucle sur l’ARNm Site apurinique ARNr 18S non-fonctionnel
RIBOSOME EN PAUSE dans la séquence codante :
pas de codon-stop dans le site A
Hbs1!N134
Dom34
Modèle de Dom34-Hbs1 validé par SAXS
!! Homologie structurale avec EF-Tu–ARNt :
entrée dans le site A possible
!! Homologie structurale et fonctionnelle avec eRF1-eRF3
!! La fixation de GXP par Hbs1 est essentielle pour la NGD et la NRD
!! L’interaction entre Dom34 et Hbs1 est essentielle pour la NGD,
mais moins importante pour la NRD
Dom34-Hbs1
Figure 60 : Synthèse : Dom34-Hbs1 cible les ribosomes bloqués en cours d'élongation et déclenche les voies de dégradation de l'ARN fautif.

171

172
Conclusion générale
Le travail réalisé au cours de ma thèse apporte des éléments de compréhension sur des
systèmes contrôlant l'efficacité de la terminaison et assurant le contrôle qualité des ARN chez
les eucaryotes. La résolution des structures de protéines impliquées dans ces voies dont l’une
a été nouvellement identifiée, couplée à des caractérisations biochimiques et fonctionnelles, a
amené des éléments d'explication sur leur rôle et leur fonctionnement.
Tpa1 et Ett1 sont individuellement nécessaires à une terminaison efficace de la
traduction. Tpa1 a une structure très similaire à des dioxygénases hydroxylant les prolines et
son site actif est nécessaire pour une terminaison normale. Ett1 est formée de sept motifs
TPR, un repliement généralement impliqué dans des interactions protéine-protéine et
protéine-peptide et retrouvé dans certains cas fusionné à un domaine prolyl-hydroxylase. Des
études complémentaires restent nécessaires pour mettre en évidence l’activité enzymatique de
Tpa1 et identifier son ou ses substrats. Ceci devrait permettre de mieux comprendre le mode
de fonctionnement de ces protéines et leurs relations avec le mécanisme de régulation de la
stabilité d’un facteur de transcription des gènes de réponse à l’hypoxie. Si cette relation est
établie, il serait alors intéressant d’essayer de mettre en évidence s’il existe un intérêt pour les
cellules de diminuer l’efficacité de terminaison de la traduction en conditions d’hypoxie et
d’étudier par une approche protéomique la présence de protéine présentant une extension C-
terminale lorsque les levures sont confrontées à une absence d’O2. Enfin, pour l’heure, tandis
qu’un orthologue de Tpa1 est présent chez l’homme, aucun orthologue d’Ett1 n’est

173
détectable. Si cela se confirme, il serait intéressant d’étudier le rôle et la régulation de
l’orthologue humain de Tpa1.
Dom34-Hbs1 est le complexe central de la dégradation des ARNm et des ARNr 18S non-
fonctionnels induisant des pauses dans l'élongation. Le complexe est homologue au complexe
de terminaison eRF1-eRF3. Sa forme le rend apte à entrer dans le site A du ribosome en
pause. La fixation du nucléotide par le domaine GTPase d'Hbs1 est nécessaire à la NGD et à
la NRD, mais l'interaction entre les deux protéines n'est nécessaire que pour la NGD.
La caractérisation des acteurs des voies NGD et NRD met une nouvelle fois en évidence
les liens étroits entre la traduction et le contrôle qualité des ARN. Le fonctionnement normal
de ces voies garantit une expression génique efficace et une réponse rapide aux défauts
apparaissant sur les ARN. Les fonctions connues de Dom34 et les phénotypes associés à sa
délétion montrent la possibilité d'une fonction plus générale de la NGD et/ou de la NRD dans
le développement. Les résultats obtenus sur le plan moléculaire gagneraient à être mis en
perspective d'un point de vue biologique plus global.
Le nombre de mécanismes de contrôle qualité prenant place autour de la traduction met
en évidence la complexité du métabolisme de l'ARN, une molécule centrale de l'expression
génique. Il est tentant de conjecturer l'existence de nouvelles voies de contrôle ciblant l'ARN
en cours de traduction. La recherche et la caractérisation de nouveaux acteurs de la
terminaison et de la stabilité des ARN restent ouvertes.


174
Bibliographie
Adams, P.D., Afonine, P.V., Bunkoczi, G., Chen, V.B., Davis, I.W., Echols, N., Headd, J.J., Hung, L.W., Kapral, G.J., Grosse-Kunstleve, R.W., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213-221. Adams, P.D., Grosse-Kunstleve, R.W., Hung, L.W., Ioerger, T.R., McCoy, A.J., Moriarty, N.W., Read, R.J., Sacchettini, J.C., Sauter, N.K., and Terwilliger, T.C. (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948-1954. Adham, I.M., Sallam, M.A., Steding, G., Korabiowska, M., Brinck, U., Hoyer-Fender, S., Oh, C., and Engel, W. (2003). Disruption of the pelota gene causes early embryonic lethality and defects in cell cycle progression. Mol Cell Biol 23, 1470-1476. Alcazar-Roman, A.R., Bolger, T.A., and Wente, S.R. (2010). Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. J Biol Chem 285, 16683-16692. Alkalaeva, E.Z., Pisarev, A.V., Frolova, L.Y., Kisselev, L.L., and Pestova, T.V. (2006). In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 125, 1125-1136. Amrani, N., Ganesan, R., Kervestin, S., Mangus, D.A., Ghosh, S., and Jacobson, A. (2004). A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432, 112-118. Amrani, N., Ghosh, S., Mangus, D.A., and Jacobson, A. (2008). Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453, 1276-1280. Amrani, N., Minet, M., Le Gouar, M., Lacroute, F., and Wyers, F. (1997). Yeast Pab1 interacts with Rna15 and participates in the control of the poly(A) tail length in vitro. Mol Cell Biol 17, 3694-3701. Amrani, N., Sachs, M.S., and Jacobson, A. (2006). Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7, 415-425. Anderson, J.S., and Parker, R.P. (1998). The 3' to 5' degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3' to 5' exonucleases of the exosome complex. EMBO J 17, 1497-1506. Andjelkovic, N., Zolnierowicz, S., Van Hoof, C., Goris, J., and Hemmings, B.A. (1996). The catalytic subunit of protein phosphatase 2A associates with the translation termination factor eRF1. EMBO J 15, 7156-7167. Araki, Y., Takahashi, S., Kobayashi, T., Kajiho, H., Hoshino, S., and Katada, T. (2001). Ski7p G protein interacts with the exosome and the Ski complex for 3'-to-5' mRNA decay in yeast. EMBO J 20, 4684-4693. Aravind, L., and Koonin, E.V. (2001). The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol 2, RESEARCH0007.

175
Atkin, A.L., Schenkman, L.R., Eastham, M., Dahlseid, J.N., Lelivelt, M.J., and Culbertson, M.R. (1997). Relationship between yeast polyribosomes and Upf proteins required for nonsense mRNA decay. J Biol Chem 272, 22163-22172. Atkinson, G.C., Baldauf, S.L., and Hauryliuk, V. (2008). Evolution of nonstop, no-go and nonsense-mediated mRNA decay and their termination factor-derived components. BMC Evol Biol 8, 290. Badis, G., Saveanu, C., Fromont-Racine, M., and Jacquier, A. (2004). Targeted mRNA degradation by deadenylation-independent decapping. Mol Cell 15, 5-15. Baker, K.E., and Parker, R. (2004). Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol 16, 293-299. Balagopal, V., and Parker, R. (2009a). Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol 21, 403-408. Balagopal, V., and Parker, R. (2009b). Stm1 modulates mRNA decay and Dhh1 function in Saccharomyces cerevisiae. Genetics 181, 93-103. Beelman, C.A., Stevens, A., Caponigro, G., LaGrandeur, T.E., Hatfield, L., Fortner, D.M., and Parker, R. (1996). An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature 382, 642-646. Benko, A.L., Vaduva, G., Martin, N.C., and Hopper, A.K. (2000). Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc Natl Acad Sci U S A 97, 61-66. Bertram, G., Bell, H.A., Ritchie, D.W., Fullerton, G., and Stansfield, I. (2000). Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA 6, 1236-1247. Boeck, R., Lapeyre, B., Brown, C.E., and Sachs, A.B. (1998). Capped mRNA degradation intermediates accumulate in the yeast spb8-2 mutant. Mol Cell Biol 18, 5062-5072. Boeck, R., Tarun, S., Jr., Rieger, M., Deardorff, J.A., Muller-Auer, S., and Sachs, A.B. (1996). The yeast Pan2 protein is required for poly(A)-binding protein-stimulated poly(A)-nuclease activity. J Biol Chem 271, 432-438. Bolger, T.A., Folkmann, A.W., Tran, E.J., and Wente, S.R. (2008). The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell 134, 624-633. Bonetti, B., Fu, L., Moon, J., and Bedwell, D.M. (1995). The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol 251, 334-345. Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003). Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr 59, 2023-2030. Brown, C.E., and Sachs, A.B. (1998). Poly(A) tail length control in Saccharomyces cerevisiae occurs by message-specific deadenylation. Mol Cell Biol 18, 6548-6559. Burnicka-Turek, O., Kata, A., Buyandelger, B., Ebermann, L., Kramann, N., Burfeind, P., Hoyer-Fender, S., Engel, W., and Adham, I.M. (2010). Pelota interacts with HAX1, EIF3G and SRPX and the resulting protein complexes are associated with the actin cytoskeleton. BMC Cell Biol 11, 28. Butler, J.S. (2002). The yin and yang of the exosome. Trends Cell Biol 12, 90-96. Cantor C. R., S.P.R. (1980). Biophysical chemistry, part II : techniques for the study of biological structure and function (Freeman and co). Carr-Schmid, A., Pfund, C., Craig, E.A., and Kinzy, T.G. (2002). Novel G-protein complex whose requirement is linked to the translational status of the cell. Mol Cell Biol 22, 2564-2574.

176
Caskey, C.T., Beaudet, A.L., Scolnick, E.M., and Rosman, M. (1971). Hydrolysis of fMet-tRNA by peptidyl transferase. Proc Natl Acad Sci U S A 68, 3163-3167. Chavatte, L., Kervestin, S., Favre, A., and Jean-Jean, O. (2003). Stop codon selection in eukaryotic translation termination: comparison of the discriminating potential between human and ciliate eRF1s. EMBO J 22, 1644-1653. Chavatte, L., Seit-Nebi, A., Dubovaya, V., and Favre, A. (2002). The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J 21, 5302-5311. Chen, X.Q., Du, X., Liu, J., Balasubramanian, M.K., and Balasundaram, D. (2004). Identification of genes encoding putative nucleoporins and transport factors in the fission yeast Schizosaccharomyces pombe: a deletion analysis. Yeast 21, 495-509. Cheng, Z., Saito, K., Pisarev, A.V., Wada, M., Pisareva, V.P., Pestova, T.V., Gajda, M., Round, A., Kong, C., Lim, M., et al. (2009). Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev 23, 1106-1118. Chowdhury, R., McDonough, M.A., Mecinovic, J., Loenarz, C., Flashman, E., Hewitson, K.S., Domene, C., and Schofield, C.J. (2009). Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 17, 981-989. Clifton, I.J., McDonough, M.A., Ehrismann, D., Kershaw, N.J., Granatino, N., and Schofield, C.J. (2006). Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J Inorg Biochem 100, 644-669. Cole, S.E., LaRiviere, F.J., Merrikh, C.N., and Moore, M.J. (2009). A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell 34, 440-450. Collaborative Computational Project, N. (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760-763. Coller, J., and Parker, R. (2004). Eukaryotic mRNA decapping. Annu Rev Biochem 73, 861-890. Coller, J.M., Tucker, M., Sheth, U., Valencia-Sanchez, M.A., and Parker, R. (2001). The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7, 1717-1727. Conti, E., and Izaurralde, E. (2005). Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol 17, 316-325. Cosson, B., Berkova, N., Couturier, A., Chabelskaya, S., Philippe, M., and Zhouravleva, G. (2002). Poly(A)-binding protein and eRF3 are associated in vivo in human and Xenopus cells. Biol Cell 94, 205-216. Cowling, V.H. (2009). Regulation of mRNA cap methylation. Biochem J 425, 295-302. Cowtan, K. (2006). The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 62, 1002-1011. Craigen, W.J., and Caskey, C.T. (1987). The function, structure and regulation of E. coli peptide chain release factors. Biochimie 69, 1031-1041. Crick, F. (1970). Central dogma of molecular biology. Nature 227, 561-563. Cui, Y., Hagan, K.W., Zhang, S., and Peltz, S.W. (1995). Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature translational termination codon. Genes Dev 9, 423-436. D. Peter Snustad, M.J.S. (2010). Principles of genetics, 5th edn (John Wiley and Sons). D'Andrea, L.D., and Regan, L. (2003). TPR proteins: the versatile helix. Trends Biochem Sci 28, 655-662. Danckwardt, S., Hentze, M.W., and Kulozik, A.E. (2008). 3' end mRNA processing: molecular mechanisms and implications for health and disease. EMBO J 27, 482-498.

177
Dauter, Z. (2002). New approaches to high-throughput phasing. Curr Opin Struct Biol 12, 674-678. Davis, L., and Engebrecht, J. (1998). Yeast dom34 mutants are defective in multiple developmental pathways and exhibit decreased levels of polyribosomes. Genetics 149, 45-56. Decker, C.J., and Parker, R. (1993). A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev 7, 1632-1643. Denis, C.L., and Chen, J. (2003). The CCR4-NOT complex plays diverse roles in mRNA metabolism. Prog Nucleic Acid Res Mol Biol 73, 221-250. Deutsch, M., and Long, M. (1999). Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res 27, 3219-3228. Diederichs, K. (2006). Some aspects of quantitative analysis and correction of radiation damage. Acta Crystallogr D Biol Crystallogr 62, 96-101. Doel, S.M., McCready, S.J., Nierras, C.R., and Cox, B.S. (1994). The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 137, 659-670. Doma, M.K., and Parker, R. (2006). Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561-564. Doma, M.K., and Parker, R. (2007). RNA quality control in eukaryotes. Cell 131, 660-668. Dossani, Z.Y., Weirich, C.S., Erzberger, J.P., Berger, J.M., and Weis, K. (2009). Structure of the C-terminus of the mRNA export factor Dbp5 reveals the interaction surface for the ATPase activator Gle1. Proc Natl Acad Sci U S A 106, 16251-16256. Drugeon, G., Jean-Jean, O., Frolova, L., Le Goff, X., Philippe, M., Kisselev, L., and Haenni, A.L. (1997). Eukaryotic release factor 1 (eRF1) abolishes readthrough and competes with suppressor tRNAs at all three termination codons in messenger RNA. Nucleic Acids Res 25, 2254-2258. Ducruix A., G.R. (1992). Crystallization of nucleic acids and proteins, a practical approach (Oxford University Press). Dunckley, T., and Parker, R. (1999). The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J 18, 5411-5422. Eberhart, C.G., and Wasserman, S.A. (1995). The pelota locus encodes a protein required for meiotic cell division: an analysis of G2/M arrest in Drosophila spermatogenesis. Development 121, 3477-3486. Eberle, A.B., Lykke-Andersen, S., Muhlemann, O., and Jensen, T.H. (2009). SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16, 49-55. Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-2132. Evans, P. (2006). Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72-82. Fanen, P., Hasnain A. (2001). Mucoviscidose et gène CFTR. Atlas Genet Cytogenet Oncol Haematol. Ford, L.P., Bagga, P.S., and Wilusz, J. (1997). The poly(A) tail inhibits the assembly of a 3'-to-5' exonuclease in an in vitro RNA stability system. Mol Cell Biol 17, 398-406. Frolova, L.Y., Merkulova, T.I., and Kisselev, L.L. (2000). Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA 6, 381-390. Frolova, L.Y., Tsivkovskii, R.Y., Sivolobova, G.F., Oparina, N.Y., Serpinsky, O.I., Blinov, V.M., Tatkov, S.I., and Kisselev, L.L. (1999). Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA 5, 1014-1020.

178
Funakoshi, Y., Doi, Y., Hosoda, N., Uchida, N., Osawa, M., Shimada, I., Tsujimoto, M., Suzuki, T., Katada, T., and Hoshino, S. (2007). Mechanism of mRNA deadenylation: evidence for a molecular interplay between translation termination factor eRF3 and mRNA deadenylases. Genes Dev 21, 3135-3148. Futcher, B., Latter, G.I., Monardo, P., McLaughlin, C.S., and Garrels, J.I. (1999). A sampling of the yeast proteome. Mol Cell Biol 19, 7357-7368. Gandhi, R., Manzoor, M., and Hudak, K.A. (2008). Depurination of Brome mosaic virus RNA3 in vivo results in translation-dependent accelerated degradation of the viral RNA. J Biol Chem 283, 32218-32228. Gao, H., Zhou, Z., Rawat, U., Huang, C., Bouakaz, L., Wang, C., Cheng, Z., Liu, Y., Zavialov, A., Gursky, R., et al. (2007). RF3 induces ribosomal conformational changes responsible for dissociation of class I release factors. Cell 129, 929-941. Gasch, A.P., Spellman, P.T., Kao, C.M., Carmel-Harel, O., Eisen, M.B., Storz, G., Botstein, D., and Brown, P.O. (2000). Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11, 4241-4257. Gatfield, D., and Izaurralde, E. (2004). Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature 429, 575-578. Goldstein, J.L., Beaudet, A.L., and Caskey, C.T. (1970). Peptide chain termination with mammalian release factor. Proc Natl Acad Sci U S A 67, 99-106. Graille, M., Chaillet, M., and van Tilbeurgh, H. (2008). Structure of yeast Dom34: a protein related to translation termination factor Erf1 and involved in No-Go decay. J Biol Chem 283, 7145-7154. Gross, T., Siepmann, A., Sturm, D., Windgassen, M., Scarcelli, J.J., Seedorf, M., Cole, C.N., and Krebber, H. (2007). The DEAD-box RNA helicase Dbp5 functions in translation termination. Science 315, 646-649. Gu, M., and Lima, C.D. (2005). Processing the message: structural insights into capping and decapping mRNA. Curr Opin Struct Biol 15, 99-106. Hatfield, L., Beelman, C.A., Stevens, A., and Parker, R. (1996). Mutations in trans-acting factors affecting mRNA decapping in Saccharomyces cerevisiae. Mol Cell Biol 16, 5830-5838. He, F., Brown, A.H., and Jacobson, A. (1996). Interaction between Nmd2p and Upf1p is required for activity but not for dominant-negative inhibition of the nonsense-mediated mRNA decay pathway in yeast. RNA 2, 153-170. He, F., Brown, A.H., and Jacobson, A. (1997). Upf1p, Nmd2p, and Upf3p are interacting components of the yeast nonsense-mediated mRNA decay pathway. Mol Cell Biol 17, 1580-1594. He, F., Peltz, S.W., Donahue, J.L., Rosbash, M., and Jacobson, A. (1993). Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1- mutant. Proc Natl Acad Sci U S A 90, 7034-7038. Henri (2010). Structural and functional insights into S. cerevisiae Tpa1, a putative prolyl hydroxylase influencing translation termination and transcription. Journal of Biological Chemistry. Henri, J., Rispal, D., Bayart, E., van Tilbeurgh, H., Seraphin, B., and Graille, M. (2010). Structural and functional insights into S. cerevisiae Tpa1, a putative prolyl hydroxylase influencing translation termination and transcription. J Biol Chem. Hentze, M.W. (1997). eIF4G: a multipurpose ribosome adapter? Science 275, 500-501. Heurgue-Hamard, V., Champ, S., Mora, L., Merkulova-Rainon, T., Kisselev, L.L., and Buckingham, R.H. (2005). The glutamine residue of the conserved GGQ motif in Saccharomyces cerevisiae release factor eRF1 is methylated by the product of the YDR140w gene. J Biol Chem 280, 2439-2445.

179
Heurgue-Hamard, V., Graille, M., Scrima, N., Ulryck, N., Champ, S., van Tilbeurgh, H., and Buckingham, R.H. (2006). The zinc finger protein Ynr046w is plurifunctional and a component of the eRF1 methyltransferase in yeast. J Biol Chem 281, 36140-36148. Holbrook, J.A., Neu-Yilik, G., Hentze, M.W., and Kulozik, A.E. (2004). Nonsense-mediated decay approaches the clinic. Nat Genet 36, 801-808. Houseley, J., and Tollervey, D. (2009). The many pathways of RNA degradation. Cell 136, 763-776. Hughes, B.T., and Espenshade, P.J. (2008). Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member. EMBO J 27, 1491-1501. Isken, O., Kim, Y.K., Hosoda, N., Mayeur, G.L., Hershey, J.W., and Maquat, L.E. (2008). Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133, 314-327. Isken, O., and Maquat, L.E. (2008). The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet 9, 699-712. Jackson, R.J., Hellen, C.U., and Pestova, T.V. (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11, 113-127. Jacobson A., I.E. (2007). Nonsense-mediated mRNA Decay : From Yeast to Metazoans. In Translational Control in Biology and Medicine, C. Press, ed., pp. 655-687. Janin J., D.M. (1999). Biologie structurale, principes et méthodes biophysiques (Paris: Hermann). Johansson, M., Bouakaz, E., Lovmar, M., and Ehrenberg, M. (2008). The kinetics of ribosomal peptidyl transfer revisited. Mol Cell 30, 589-598. Kabsch, W. (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133-144. Kabsch, W. (2010b). Xds. Acta Crystallogr D Biol Crystallogr 66, 125-132. Kantardjieff, K.A., and Rupp, B. (2003). Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci 12, 1865-1871. Kashima, I., Yamashita, A., Izumi, N., Kataoka, N., Morishita, R., Hoshino, S., Ohno, M., Dreyfuss, G., and Ohno, S. (2006). Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20, 355-367. Keeling, K.M., Salas-Marco, J., Osherovich, L.Z., and Bedwell, D.M. (2006). Tpa1p is part of an mRNP complex that influences translation termination, mRNA deadenylation, and mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol 26, 5237-5248. Kelly, S.M., and Corbett, A.H. (2009). Messenger RNA export from the nucleus: a series of molecular wardrobe changes. Traffic 10, 1199-1208. Kerem, E., Hirawat, S., Armoni, S., Yaakov, Y., Shoseyov, D., Cohen, M., Nissim-Rafinia, M., Blau, H., Rivlin, J., Aviram, M., et al. (2008). Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 372, 719-727. Kervestin, S., Frolova, L., Kisselev, L., and Jean-Jean, O. (2001). Stop codon recognition in ciliates: Euplotes release factor does not respond to reassigned UGA codon. EMBO Rep 2, 680-684. Kim, H.S., Kim, H.L., Kim, K.H., Kim do, J., Lee, S.J., Yoon, J.Y., Yoon, H.J., Lee, H.Y., Park, S.B., Kim, S.J., et al. (2010). Crystal structure of Tpa1 from Saccharomyces cerevisiae, a component of the messenger ribonucleoprotein complex. Nucleic Acids Res 38, 2099-2110. King, D.A., Zhang, L., Guarente, L., and Marmorstein, R. (1999). Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat Struct Biol 6, 64-71.

180
Kisselev, L., Ehrenberg, M., and Frolova, L. (2003). Termination of translation: interplay of mRNA, rRNAs and release factors? EMBO J 22, 175-182. Kisselev, L.L. (2001). Translation termination in eukaryotes: from simplicity to complexity. Folia Biol (Praha) 47, 1-4. Kisselev, L.L., and Buckingham, R.H. (2000). Translational termination comes of age. Trends Biochem Sci 25, 561-566. Kolev, N.G., Yario, T.A., Benson, E., and Steitz, J.A. (2008). Conserved motifs in both CPSF73 and CPSF100 are required to assemble the active endonuclease for histone mRNA 3'-end maturation. EMBO Rep 9, 1013-1018. Kolosov, P., Frolova, L., Seit-Nebi, A., Dubovaya, V., Kononenko, A., Oparina, N., Justesen, J., Efimov, A., and Kisselev, L. (2005). Invariant amino acids essential for decoding function of polypeptide release factor eRF1. Nucleic Acids Res 33, 6418-6425. Kong, C., Ito, K., Walsh, M.A., Wada, M., Liu, Y., Kumar, S., Barford, D., Nakamura, Y., and Song, H. (2004). Crystal structure and functional analysis of the eukaryotic class II release factor eRF3 from S. pombe. Mol Cell 14, 233-245. Kononenko, A.V., Mitkevich, V.A., Dubovaya, V.I., Kolosov, P.M., Makarov, A.A., and Kisselev, L.L. (2008). Role of the individual domains of translation termination factor eRF1 in GTP binding to eRF3. Proteins 70, 388-393. Kopeina, G.S., Afonina, Z.A., Gromova, K.V., Shirokov, V.A., Vasiliev, V.D., and Spirin, A.S. (2008). Step-wise formation of eukaryotic double-row polyribosomes and circular translation of polysomal mRNA. Nucleic Acids Res 36, 2476-2488. Kornberg, R.D. (2007). The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A 104, 12955-12961. Korner, C.G., and Wahle, E. (1997). Poly(A) tail shortening by a mammalian poly(A)-specific 3'-exoribonuclease. J Biol Chem 272, 10448-10456. Korostelev, A., Asahara, H., Lancaster, L., Laurberg, M., Hirschi, A., Zhu, J., Trakhanov, S., Scott, W.G., and Noller, H.F. (2008). Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci U S A 105, 19684-19689. Kozlov, G., and Gehring, K. (2010). Molecular basis of eRF3 recognition by the MLLE domain of poly(A)-binding protein. PLoS One 5, e10169. Kozlov, G., Menade, M., Rosenauer, A., Nguyen, L., and Gehring, K. (2010). Molecular determinants of PAM2 recognition by the MLLE domain of poly(A)-binding protein. J Mol Biol 397, 397-407. Kozlov, G., Trempe, J.F., Khaleghpour, K., Kahvejian, A., Ekiel, I., and Gehring, K. (2001). Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc Natl Acad Sci U S A 98, 4409-4413. Krissinel, E., and Henrick, K. (2004). Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60, 2256-2268. Krogan, N.J., Cagney, G., Yu, H., Zhong, G., Guo, X., Ignatchenko, A., Li, J., Pu, S., Datta, N., Tikuisis, A.P., et al. (2006). Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637-643. Kuhn, U., and Pieler, T. (1996). Xenopus poly(A) binding protein: functional domains in RNA binding and protein-protein interaction. J Mol Biol 256, 20-30. Kunz, J.B., Neu-Yilik, G., Hentze, M.W., Kulozik, A.E., and Gehring, N.H. (2006). Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 12, 1015-1022. Kushner, D.B., Lindenbach, B.D., Grdzelishvili, V.Z., Noueiry, A.O., Paul, S.M., and Ahlquist, P. (2003). Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc Natl Acad Sci U S A 100, 15764-15769.

181
Larimer, F.W., Hsu, C.L., Maupin, M.K., and Stevens, A. (1992). Characterization of the XRN1 gene encoding a 5'-->3' exoribonuclease: sequence data and analysis of disparate protein and mRNA levels of gene-disrupted yeast cells. Gene 120, 51-57. LaRiviere, F.J., Cole, S.E., Ferullo, D.J., and Moore, M.J. (2006). A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell 24, 619-626. Laurberg, M., Asahara, H., Korostelev, A., Zhu, J., Trakhanov, S., and Noller, H.F. (2008). Structural basis for translation termination on the 70S ribosome. Nature 454, 852-857. Le Hir, H., Gatfield, D., Izaurralde, E., and Moore, M.J. (2001). The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 20, 4987-4997. Lebreton, A., Tomecki, R., Dziembowski, A., and Seraphin, B. (2008). Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 456, 993-996. Lee, C.Y., Stewart, E.V., Hughes, B.T., and Espenshade, P.J. (2009). Oxygen-dependent binding of Nro1 to the prolyl hydroxylase Ofd1 regulates SREBP degradation in yeast. EMBO J 28, 135-143. Lee, H.H., Kim, Y.S., Kim, K.H., Heo, I., Kim, S.K., Kim, O., Kim, H.K., Yoon, J.Y., Kim, H.S., Kim do, J., et al. (2007). Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol Cell 27, 938-950. Leeds, P., Wood, J.M., Lee, B.S., and Culbertson, M.R. (1992). Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol 12, 2165-2177. Lejeune, F., Li, X., and Maquat, L.E. (2003). Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12, 675-687. Lelivelt, M.J., and Culbertson, M.R. (1999). Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol Cell Biol 19, 6710-6719. Leslie, A.G. (2006). The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62, 48-57. Leulliot, N., Tresaugues, L., Bremang, M., Sorel, I., Ulryck, N., Graille, M., Aboulfath, I., Poupon, A., Liger, D., Quevillon-Cheruel, S., et al. (2005). High-throughput crystal-optimization strategies in the South Paris Yeast Structural Genomics Project: one size fits all? Acta Crystallogr D Biol Crystallogr 61, 664-670. Linde, L., Boelz, S., Neu-Yilik, G., Kulozik, A.E., and Kerem, B. (2007). The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur J Hum Genet 15, 1156-1162. Lingner, J., Radtke, I., Wahle, E., and Keller, W. (1991). Purification and characterization of poly(A) polymerase from Saccharomyces cerevisiae. J Biol Chem 266, 8741-8746. Loeb, J.N., Howell, R.R., and Tomkins, G.M. (1965). Turnover of ribosomal RNA in rat liver. Science 149, 1093-1095. Luo, L., Pappalardi, M.B., Tummino, P.J., Copeland, R.A., Fraser, M.E., Grzyska, P.K., and Hausinger, R.P. (2006). An assay for Fe(II)/2-oxoglutarate-dependent dioxygenases by enzyme-coupled detection of succinate formation. Anal Biochem 353, 69-74. Maderazo, A.B., He, F., Mangus, D.A., and Jacobson, A. (2000). Upf1p control of nonsense mRNA translation is regulated by Nmd2p and Upf3p. Mol Cell Biol 20, 4591-4603. Mangus, D.A., Evans, M.C., and Jacobson, A. (2003). Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4, 223. Mantsyzov, A.B., Ivanova, E.V., Birdsall, B., Alkalaeva, E.Z., Kryuchkova, P.N., Kelly, G., Frolova, L.Y., and Polshakov, V.I. (2010). NMR solution structure and function of the C-terminal domain of eukaryotic class 1 polypeptide chain release factor. FEBS J.

182
Mazauric, M.H., Dirick, L., Purushothaman, S.K., Bjork, G.R., and Lapeyre, B. (2010). Trm112p is a 15-kDa zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in yeast. J Biol Chem 285, 18505-18515. McCracken, S., Fong, N., Rosonina, E., Yankulov, K., Brothers, G., Siderovski, D., Hessel, A., Foster, S., Shuman, S., and Bentley, D.L. (1997). 5'-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev 11, 3306-3318. McCusker, K.P., and Klinman, J.P. (2009). Modular behavior of tauD provides insight into the origin of specificity in alpha-ketoglutarate-dependent nonheme iron oxygenases. Proc Natl Acad Sci U S A 106, 19791-19795. McDonough, M.A., Li, V., Flashman, E., Chowdhury, R., Mohr, C., Lienard, B.M., Zondlo, J., Oldham, N.J., Clifton, I.J., Lewis, J., et al. (2006). Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci U S A 103, 9814-9819. McIlwain, D.R., Pan, Q., Reilly, P.T., Elia, A.J., McCracken, S., Wakeham, A.C., Itie-Youten, A., Blencowe, B.J., and Mak, T.W. (2010). Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci U S A 107, 12186-12191. Medghalchi, S.M., Frischmeyer, P.A., Mendell, J.T., Kelly, A.G., Lawler, A.M., and Dietz, H.C. (2001). Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet 10, 99-105. Mendell, J.T., Sharifi, N.A., Meyers, J.L., Martinez-Murillo, F., and Dietz, H.C. (2004). Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36, 1073-1078. Merkulova, T.I., Frolova, L.Y., Lazar, M., Camonis, J., and Kisselev, L.L. (1999). C-terminal domains of human translation termination factors eRF1 and eRF3 mediate their in vivo interaction. FEBS Lett 443, 41-47. Meselson, M., Nomura, M., Brenner, S., Davern, C., and Schlessinger, D. (1964). Conservation of Ribosomes during Bacterial Growth. J Mol Biol 9, 696-711. Meselson, M., and Stahl, F.W. (1958). The replication of DNA. Cold Spring Harb Symp Quant Biol 23, 9-12. Millevoi, S., and Vagner, S. (2010). Molecular mechanisms of eukaryotic pre-mRNA 3' end processing regulation. Nucleic Acids Res 38, 2757-2774. Mitchell, P., and Tollervey, D. (2000a). mRNA stability in eukaryotes. Curr Opin Genet Dev 10, 193-198. Mitchell, P., and Tollervey, D. (2000b). Musing on the structural organization of the exosome complex. Nat Struct Biol 7, 843-846. Moore, M.J., and Proudfoot, N.J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688-700. Mora, L., Heurgue-Hamard, V., de Zamaroczy, M., Kervestin, S., and Buckingham, R.H. (2007). Methylation of bacterial release factors RF1 and RF2 is required for normal translation termination in vivo. J Biol Chem 282, 35638-35645. Muhlemann, O. (2008). Recognition of nonsense mRNA: towards a unified model. Biochem Soc Trans 36, 497-501. Muhlemann, O., Eberle, A.B., Stalder, L., and Zamudio Orozco, R. (2008). Recognition and elimination of nonsense mRNA. Biochim Biophys Acta 1779, 538-549. Muhlrad, D., and Parker, R. (1992). Mutations affecting stability and deadenylation of the yeast MFA2 transcript. Genes Dev 6, 2100-2111.

183
Murshudov, G.N., Vagin, A.A., and Dodson, E.J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240-255. Murthy, K.G., and Manley, J.L. (1995). The 160-kD subunit of human cleavage-polyadenylation specificity factor coordinates pre-mRNA 3'-end formation. Genes Dev 9, 2672-2683. Nicholson, P., Yepiskoposyan, H., Metze, S., Zamudio Orozco, R., Kleinschmidt, N., and Muhlemann, O. (2010). Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67, 677-700. Nilsen, T.W., and Graveley, B.R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457-463. Nuss, D.L., Furuichi, Y., Koch, G., and Shatkin, A.J. (1975). Detection in HeLa cell extracts of a 7-methyl guanosine specific enzyme activity that cleaves m7GpppNm. Cell 6, 21-27. Ohnishi, T., Yamashita, A., Kashima, I., Schell, T., Anders, K.R., Grimson, A., Hachiya, T., Hentze, M.W., Anderson, P., and Ohno, S. (2003). Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell 12, 1187-1200. Parker, R., and Song, H. (2004). The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol 11, 121-127. Passos, D.O., Doma, M.K., Shoemaker, C.J., Muhlrad, D., Green, R., Weissman, J., Hollien, J., and Parker, R. (2009). Analysis of Dom34 and its function in no-go decay. Mol Biol Cell 20, 3025-3032. Peltz, S.W., Brown, A.H., and Jacobson, A. (1993). mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev 7, 1737-1754. Perrakis, A., Harkiolaki, M., Wilson, K.S., and Lamzin, V.S. (2001). ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr 57, 1445-1450. Petry, S., Weixlbaumer, A., and Ramakrishnan, V. (2008). The termination of translation. Curr Opin Struct Biol 18, 70-77. Pisarev, A.V., Skabkin, M.A., Pisareva, V.P., Skabkina, O.V., Rakotondrafara, A.M., Hentze, M.W., Hellen, C.U., and Pestova, T.V. (2010). The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell 37, 196-210. Pisareva, V.P., Pisarev, A.V., Hellen, C.U., Rodnina, M.V., and Pestova, T.V. (2006). Kinetic analysis of interaction of eukaryotic release factor 3 with guanine nucleotides. J Biol Chem 281, 40224-40235. Proudfoot, N., and O'Sullivan, J. (2002). Polyadenylation: a tail of two complexes. Curr Biol 12, R855-857. Purushothaman, S.K., Bujnicki, J.M., Grosjean, H., and Lapeyre, B. (2005). Trm11p and Trm112p are both required for the formation of 2-methylguanosine at position 10 in yeast tRNA. Mol Cell Biol 25, 4359-4370. Quevillon-Cheruel, S., Liger, D., Leulliot, N., Graille, M., Poupon, A., de La Sierra-Gallay, I.L., Zhou, C.Z., Collinet, B., Janin, J., and Van Tilbeurgh, H. (2004). The Paris-Sud yeast structural genomics pilot-project: from structure to function. Biochimie 86, 617-623. Rehwinkel, J., Raes, J., and Izaurralde, E. (2006). Nonsense-mediated mRNA decay: Target genes and functional diversification of effectors. Trends Biochem Sci 31, 639-646. Rodnina, M.V., Beringer, M., and Bieling, P. (2005). Ten remarks on peptide bond formation on the ribosome. Biochem Soc Trans 33, 493-498. Rodnina, M.V., and Wintermeyer, W. (2009). Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol 21, 435-443.

184
Rospert, S., Rakwalska, M., and Dubaquie, Y. (2005). Polypeptide chain termination and stop codon readthrough on eukaryotic ribosomes. Rev Physiol Biochem Pharmacol 155, 1-30. Rout, M.P., Aitchison, J.D., Suprapto, A., Hjertaas, K., Zhao, Y., and Chait, B.T. (2000). The yeast nuclear pore complex: composition, architecture, and transport mechanism. J Cell Biol 148, 635-651. Salas-Marco, J., and Bedwell, D.M. (2004). GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol 24, 7769-7778. Santos-Rosa, H., Moreno, H., Simos, G., Segref, A., Fahrenkrog, B., Pante, N., and Hurt, E. (1998). Nuclear mRNA export requires complex formation between Mex67p and Mtr2p at the nuclear pores. Mol Cell Biol 18, 6826-6838. Schell, T., Kocher, T., Wilm, M., Seraphin, B., Kulozik, A.E., and Hentze, M.W. (2003). Complexes between the nonsense-mediated mRNA decay pathway factor human upf1 (up-frameshift protein 1) and essential nonsense-mediated mRNA decay factors in HeLa cells. Biochem J 373, 775-783. Schmeing, T.M., and Ramakrishnan, V. (2009). What recent ribosome structures have revealed about the mechanism of translation. Nature 461, 1234-1242. Schmid, M., and Jensen, T.H. (2008). The exosome: a multipurpose RNA-decay machine. Trends Biochem Sci 33, 501-510. Sheldrick, G.M. (2008). A short history of SHELX. Acta Crystallogr A 64, 112-122. Shibagaki, Y., Itoh, N., Yamada, H., Nagata, S., and Mizumoto, K. (1992). mRNA capping enzyme. Isolation and characterization of the gene encoding mRNA guanylytransferase subunit from Saccharomyces cerevisiae. J Biol Chem 267, 9521-9528. Shkundina, I.S., Kushnirov, V.V., Tuite, M.F., and Ter-Avanesyan, M.D. (2006). The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 172, 827-835. Shkundina, I.S., and Ter-Avanesyan, M.D. (2007). Prions. Biochemistry (Mosc) 72, 1519-1536. Simon, E., and Seraphin, B. (2007). A specific role for the C-terminal region of the Poly(A)-binding protein in mRNA decay. Nucleic Acids Res 35, 6017-6028. Snustad D. Peter, S.M.G. (2010). Principles of genetics, 5th edn (Wiley). Sonenberg, N., and Hinnebusch, A.G. (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731-745. Song, H., Mugnier, P., Das, A.K., Webb, H.M., Evans, D.R., Tuite, M.F., Hemmings, B.A., and Barford, D. (2000). The crystal structure of human eukaryotic release factor eRF1--mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell 100, 311-321. Soudet, J., Gelugne, J.P., Belhabich-Baumas, K., Caizergues-Ferrer, M., and Mougin, A. (2010). Immature small ribosomal subunits can engage in translation initiation in Saccharomyces cerevisiae. EMBO J 29, 80-92. Stansfield, I., Eurwilaichitr, L., Akhmaloka, and Tuite, M.F. (1996). Depletion in the levels of the release factor eRF1 causes a reduction in the efficiency of translation termination in yeast. Mol Microbiol 20, 1135-1143. Stevens, A., and Maupin, M.K. (1987). A 5'----3' exoribonuclease of Saccharomyces cerevisiae: size and novel substrate specificity. Arch Biochem Biophys 252, 339-347. Storts, D.R., and Bhattacharjee, J.K. (1987). Purification and properties of saccharopine dehydrogenase (glutamate forming) in the Saccharomyces cerevisiae lysine biosynthetic pathway. J Bacteriol 169, 416-418. Strambio-De-Castillia, C., Niepel, M., and Rout, M.P. (2010). The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 11, 490-501.

185
Strasser, K., Bassler, J., and Hurt, E. (2000). Binding of the Mex67p/Mtr2p heterodimer to FXFG, GLFG, and FG repeat nucleoporins is essential for nuclear mRNA export. J Cell Biol 150, 695-706. Studte, P., Zink, S., Jablonowski, D., Bar, C., von der Haar, T., Tuite, M.F., and Schaffrath, R. (2008). tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol Microbiol 69, 1266-1277. Takagaki, Y., Manley, J.L., MacDonald, C.C., Wilusz, J., and Shenk, T. (1990). A multisubunit factor, CstF, is required for polyadenylation of mammalian pre-mRNAs. Genes Dev 4, 2112-2120. Tarun, S.Z., Jr., and Sachs, A.B. (1996). Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J 15, 7168-7177. Tharun S., P.R. (2001). Turnover of mRNA In Eukaryotic Cells. In RNA, Pergamon, ed., pp. 245-257. Tsukamoto, T., Shibagaki, Y., Imajoh-Ohmi, S., Murakoshi, T., Suzuki, M., Nakamura, A., Gotoh, H., and Mizumoto, K. (1997). Isolation and characterization of the yeast mRNA capping enzyme beta subunit gene encoding RNA 5'-triphosphatase, which is essential for cell viability. Biochem Biophys Res Commun 239, 116-122. Tucker, M., Staples, R.R., Valencia-Sanchez, M.A., Muhlrad, D., and Parker, R. (2002). Ccr4p is the catalytic subunit of a Ccr4p/Pop2p/Notp mRNA deadenylase complex in Saccharomyces cerevisiae. EMBO J 21, 1427-1436. Tucker, M., Valencia-Sanchez, M.A., Staples, R.R., Chen, J., Denis, C.L., and Parker, R. (2001). The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104, 377-386. Tuite, M.F., and Stansfield, I. (1994). Termination of protein synthesis. Mol Biol Rep 19, 171-181. Vagin, A., and Teplyakov, A. (2010). Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66, 22-25. Valachovic, M., Bareither, B.M., Shah Alam Bhuiyan, M., Eckstein, J., Barbuch, R., Balderes, D., Wilcox, L., Sturley, S.L., Dickson, R.C., and Bard, M. (2006). Cumulative mutations affecting sterol biosynthesis in the yeast Saccharomyces cerevisiae result in synthetic lethality that is suppressed by alterations in sphingolipid profiles. Genetics 173, 1893-1908. van Hoof, A., Frischmeyer, P.A., Dietz, H.C., and Parker, R. (2002). Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295, 2262-2264. van Hoof, A., and Parker, R. (1999). The exosome: a proteasome for RNA? Cell 99, 347-350. Vasudevan, S., Peltz, S.W., and Wilusz, C.J. (2002). Non-stop decay--a new mRNA surveillance pathway. Bioessays 24, 785-788. Velculescu, V.E., Zhang, L., Zhou, W., Vogelstein, J., Basrai, M.A., Bassett, D.E., Jr., Hieter, P., Vogelstein, B., and Kinzler, K.W. (1997). Characterization of the yeast transcriptome. Cell 88, 243-251. von der Haar, T., and Tuite, M.F. (2007). Regulated translational bypass of stop codons in yeast. Trends Microbiol 15, 78-86. Wallrapp, C., Verrier, S.B., Zhouravleva, G., Philippe, H., Philippe, M., Gress, T.M., and Jean-Jean, O. (1998). The product of the mammalian orthologue of the Saccharomyces cerevisiae HBS1 gene is phylogenetically related to eukaryotic release factor 3 (eRF3) but does not carry eRF3-like activity. FEBS Lett 440, 387-392. Wang, J.W., Chen, J.R., Gu, Y.X., Zheng, C.D., and Fan, H.F. (2004). Direct-method SAD phasing with partial-structure iteration: towards automation. Acta Crystallogr D Biol Crystallogr 60, 1991-1996.

186
Wang, S.P., and Shuman, S. (1997). Structure-function analysis of the mRNA cap methyltransferase of Saccharomyces cerevisiae. J Biol Chem 272, 14683-14689. Weischenfeldt, J., Damgaard, I., Bryder, D., Theilgaard-Monch, K., Thoren, L.A., Nielsen, F.C., Jacobsen, S.E., Nerlov, C., and Porse, B.T. (2008). NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22, 1381-1396. Weixlbaumer, A., Jin, H., Neubauer, C., Voorhees, R.M., Petry, S., Kelley, A.C., and Ramakrishnan, V. (2008). Insights into translational termination from the structure of RF2 bound to the ribosome. Science 322, 953-956. Wells, S.E., Hillner, P.E., Vale, R.D., and Sachs, A.B. (1998). Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2, 135-140. Williamson, J.R. (2009). The ribosome at atomic resolution. Cell 139, 1041-1043. Wittkopp, N., Huntzinger, E., Weiler, C., Sauliere, J., Schmidt, S., Sonawane, M., and Izaurralde, E. (2009). Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol Cell Biol 29, 3517-3528. Wittmann, J., Hol, E.M., and Jack, H.M. (2006). hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol Cell Biol 26, 1272-1287. Yamashita, A., Izumi, N., Kashima, I., Ohnishi, T., Saari, B., Katsuhata, Y., Muramatsu, R., Morita, T., Iwamatsu, A., Hachiya, T., et al. (2009). SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev 23, 1091-1105. Zavialov, A.V., Buckingham, R.H., and Ehrenberg, M. (2001). A posttermination ribosomal complex is the guanine nucleotide exchange factor for peptide release factor RF3. Cell 107, 115-124. Zhao, J., Hyman, L., and Moore, C. (1999). Formation of mRNA 3' ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63, 405-445. Zhouravleva, G., Frolova, L., Le Goff, X., Le Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995). Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 14, 4065-4072.


L'expression génique est une fonction essentielle dont le niveau, la fidélité et l'efficacité sont
précisément contrôlés. Au cours de ma thèse je me suis concentré sur un acteur de l'efficacité
de terminaison de la traduction Tpa1, sur son partenaire Ett1 et sur le complexe Dom34-Hbs1
impliqué dans les voies de contrôle qualité de l'ARN, NGD et NRD. Nous avons entrepris une
caractérisation structurale par cristallographie aux rayons X de chacune de ces protéines,
complétée par une étude fonctionnelle.
Mots-clefs : terminaison de la traduction, dégradation des ARNm, contrôle qualité des
ARN, hypoxie, dioxygénase, DSBH, TPR, GTPase
STRUCTURAL AND FUNCTIONAL STUDIES OF ACTORS OF TRANSLATION TERMINATION
EFFICIENCY AND MESSENGER RNA STABILITY IN EUKARYOTES
Gene expression is an essential function which level, fidelity and efficiency are tightly
controlled. During my thesis I have focused on the actor of translation termination efficiency
Tpa1, on its partner Ett1 and on the complex Dom34-Hbs1 involved in the RNA quality-
control mechanisms NGD and NRD. We have solved the crystal structure of each of these
proteins and realized a biochemical functional characterization.
Keywords : translation termination, mRNA degradation, RNA quality-control, hypoxia,
dioxygenase, DSBH, TPR, GTPase
LABORATOIRE DE RATTACHEMENT :
Institut de biochimie et biophysique moléculaire et cellulaire (IBBMC)
Equipe de Génomique Structurale de la Levure
UMR CNRS 8619 bâtiment 430 Université Paris XI 91405 Orsay Cedex
PÔLE : Ingénierie des protéines
UNIVERSITÉ PARIS XI UFR «FACULTÉ DE PHARMACIE DE CHATENAY-
MALABRY »
5, rue Jean Baptiste Clément 92296 CHÂTENAY-MALABRY Cedex
Related Documents











