Purdue University Purdue e-Pubs Open Access Dissertations eses and Dissertations Spring 2015 Structural and biophysical analysis of the proteasomal deubiquitinase, UCH37 Marie Elizabeth Morrow Purdue University Follow this and additional works at: hps://docs.lib.purdue.edu/open_access_dissertations Part of the Biochemistry Commons , and the Biophysics Commons is document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] for additional information. Recommended Citation Morrow, Marie Elizabeth, "Structural and biophysical analysis of the proteasomal deubiquitinase, UCH37" (2015). Open Access Dissertations. 522. hps://docs.lib.purdue.edu/open_access_dissertations/522

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Purdue UniversityPurdue e-Pubs
Open Access Dissertations Theses and Dissertations
Spring 2015
Structural and biophysical analysis of theproteasomal deubiquitinase, UCH37Marie Elizabeth MorrowPurdue University
Follow this and additional works at: https://docs.lib.purdue.edu/open_access_dissertations
Part of the Biochemistry Commons, and the Biophysics Commons
This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] foradditional information.
Recommended CitationMorrow, Marie Elizabeth, "Structural and biophysical analysis of the proteasomal deubiquitinase, UCH37" (2015). Open AccessDissertations. 522.https://docs.lib.purdue.edu/open_access_dissertations/522

PURDUE UNIVERSITY GRADUATE SCHOOL
Thesis/Dissertation Acceptance
To the best of my knowledge and as understood by the student in the Thesis/Dissertation Agreement, Publication Delay, and Certification/Disclaimer (Graduate School Form 32), this thesis/dissertation adheres to the provisions of Purdue University’s “Policy on Integrity in Research” and the use of copyrighted material.
Marie Elizabeth Morrow
STRUCTURAL AND BIOPHYSICAL ANALYSIS OF THE PROTEASOMALDEUBIQUITINASE, UCH37
Doctor of Philosophy
Chittaranjan Das
Andrew D. Mesecar
Christine A. Hrycyna
Chittaranjan Das
Kavita Shah
R. E. Wild 04/15/2015


STRUCTURAL AND BIOPHYSICAL ANALYSIS OF THE PROTEASOMAL
DEUBIQUITINASE, UCH37
A Dissertation
Submitted to the Faculty
of
Purdue University
by
Marie Elizabeth Morrow
In Partial Fulfillment of the
Requirements for the Degree
of
Doctor of Philosophy
May 2015
Purdue University
West Lafayette, Indiana

ii
ACKNOWLEDGEMENTS
To my parents, Jamie and Christine, thank you for everything and for
supporting my dreams. To my siblings, Thomas and K-Kolbz, you kids are the
best, I love you.
To Missy Ray (Kennedy) who made me love chemistry first. I want to be
as intimidating as you were back then.
Thanks to everyone at the University of Florida (GO GATORS!), especially
Carrie Haskell-Luevano who gave me the opportunity to do research in her lab,
and Erica Haslach-Meckes and Anamika Singh who introduced me to research
and inspired me to continue doing this, I’m so lucky to have had the both of you
as mentors.
Thanks to everyone at Purdue who helped me in classes, trained me or
showed me a new technique, let me borrow chemicals and instruments, or just
made me laugh on bad days. First, to my advisor, Chitta Das, who convinced me
to pursue protein crystallography even though I swore I never would get near it. I
have been lucky to be able to learn from you and I appreciate every fight we’ve
had. Seriously. Second, to my committee members who have provided me with
tons of guidance and support, amazing letters of recommendation, and a few
memorable parties: Andy Mesecar, Chris Hrycyna, and Kavita Shah. Thanks to

iii
the entire Hrycyna group for all of their help over the years, especially the great
Karen Olsen, Patty Wiley, and Kelsey Bohn. To Lake Paul for the biophysics help
and the abuse. To my Purdue friends for all our adventures outside of lab: Sarah
St. John, Chris Clark, Chris Collins, Renata Everett, Chad Keyes, Ian Klein,
Vanessa Klein, Ruth Campbell, Karen Olsen and Walter Bosley, Patty Wiley,
Kelsey Bohn, Judy Ronau, and Mike Sheedlo. Special thanks of course to all
former and current members of the Das lab: David Boudreaux, Joe Chaney,
Chris Davies, Myung-Il Kim, Judith Ronau, Mike Sheedlo, Rashmi Shrestha, Amy
Bueno, Raj Babar, and Cameron Wade. Judy, you are the craziest person I’ve
ever met, but grad school has been so much better because of that. I appreciate
all your help in my project and your awesome friendship. Rashmi, you don’t
deserve all the teasing we do, but you’ve grown up a lot since the first time we
met and I’m so glad we’ve been labmates and friends. Mike, we’ve done this
whole thing together, luckily for both of us. Now we finally get to have separate
projects! Thanks for all of the help, BFF.
The last acknowledgment goes to Alex Riehm: I love you!!! The past 5
years haven’t been easy, and I couldn’t have done this without your support,
every day.

iv
TABLE OF CONTENTS
Page LIST OF TABLES ................................................................................................ vii LIST OF FIGURES ............................................................................................. viii ABSTRACT ......................................................................................................... xi CHAPTER 1: INTRODUCTION ............................................................................ 1
1.1 Ubiquitination .............................................................................................. 1 1.2 Deubiquitination .......................................................................................... 4
1.2.1 UCH Family ..................................................................................... 7 1.3 The 26S Proteasome .................................................................................. 8
1.3.1 Deubiquitination at the 26S Proteasome ....................................... 13 1.4 References ............................................................................................... 17
CHAPTER 2: STRUCTURE OF TSUCH37cat-UBVME ....................................... 26
2.1 Introduction ............................................................................................... 26 2.2 Materials and Methods ............................................................................. 27
2.2.1 Synthesis of Ubiquitin Vinyl Methyl Ester ...................................... 27 2.2.2 Cloning, Expression, and Protein Purification of TsUCH37cat ........ 30 2.2.3 Complexation of TsUCH37cat with UbVME .................................... 31 2.2.4 Selenomethionine-labeled Protein Purification .............................. 32 2.2.5 Crystallization and Structure Solution ............................................ 33
2.3 Results ..................................................................................................... 37 2.3.1 Structure of TsUCH37cat-UbVME ................................................... 37 2.3.2 Active Site Binding ......................................................................... 39 2.3.3 Distal Site Binding ......................................................................... 40 2.3.4 Crossover Loop ............................................................................. 41
2.4 Discussion ................................................................................................ 43 2.5 References ............................................................................................... 45

v
Page
CHAPTER 3: KINETIC AND BIOPHYSICAL CHARACTERIZATION OF TSUCH37 ........................................................................................................... 47
3.1 Introduction ............................................................................................... 47 3.2 Materials and Methods ............................................................................. 50
3.2.1 Cloning, Expression, and Protein Purification ................................ 50 3.2.2 Analytical Ultracentrifugation ......................................................... 51 3.2.3 Ubiquitin-AMC Hydrolysis .............................................................. 51 3.2.4 Isothermal Titration Calorimetry ..................................................... 52
3.3 Results ..................................................................................................... 53 3.3.1 Analysis of Crystallographic Dimerization of TsUCH37cat-UbVME 53 3.3.2 Kinetic Characterization of TsUCH37cat and TsUCH37FL .............. 54 3.3.3 Analysis of UCH37 Oligomeric State ............................................. 57 3.3.4 Analysis of UCH37 Binding to Rpn13 ............................................ 58
3.4 Discussion ................................................................................................ 59 3.5 References ............................................................................................... 62
CHAPTER 4: ANALYSIS OF THE TSUCH37∆C46–UBVME STRUCTURE AND THE ROLE OF THE ULD ................................................................................... 64
4.1 Introduction ............................................................................................... 64 4.2 Materials and Methods ............................................................................. 67
4.2.1 Molecular Dynamics Simulations ................................................... 67 4.2.2 Site-directed Mutagenesis and Protein Purification ....................... 67 4.2.3 Ubiquitin-AMC Hydrolysis Assays ................................................. 68 4.2.4 Synthesis of Asymmetric Triubiquitin Substrate and Assays ......... 69
4.3 Results ..................................................................................................... 71 4.3.1 Analysis of Molecular Dynamics Simulations ................................. 71 4.3.2 Ubiquitin-AMC Hydrolysis by UCH37 ULD Mutants ....................... 73 4.3.3 Triubiquitin Cleavage by ULD Mutants .......................................... 75
4.4 Discussion ................................................................................................ 80 4.5 References ............................................................................................... 81
CHAPTER 5: RECENT FINDINGS ON THE STRUCTURE AND ACTIVATION OF UCH37 AND OTHER DEUBIQUITINASES .................................................. 82
5.1 Introduction ............................................................................................... 82 5.2 Analysis of UCH37-Rpn13-Ub and UCH37-NFRKB-Ub structures ........... 82
5.2.1 Crossover loop .............................................................................. 84 5.2.2 NFRKB Mode of Inhibition ............................................................. 85
5.3 Analysis of Kinetic Findings in Vanderlinden et. al and Sahtoe et. al ....... 87 5.4 Small Structures Effect Large Changes: A Review of Deubiquitinases .... 87
5.4.1 Unproductive Active Sites .............................................................. 88

vi
Page 5.4.2 Insertions and the JAMM Domain .................................................. 91 5.4.3 Substrate Filtering Loops ............................................................... 93 5.4.4 Substrate-occluding Loops ............................................................ 95
5.5 Conclusions .............................................................................................. 96 5.6 References ............................................................................................... 98
APPENDIX ....................................................................................................... 102 VITA ................................................................................................................. 117 PUBLICATIONS ............................................................................................... 118

vii
LIST OF TABLES
Page Table 2.1 Table of crystallographic statistics ................................................................ 38 3.1 Dissociation constants of proteasome-associated DUBs and their binding
partners ........................................................................................................ 60 4.1 Lys48-Glu51 distances in ubiquitin-bound PDB structures ........................... 66 4.2 ULD mutations introduced into human UCH37 ............................................. 73

viii
LIST OF FIGURES
Figure Page 1.1 Scheme of mechanisms of ubiquitination and deubiquitination ...................... 2 1.2 Scheme of the types of ubiquitination ............................................................. 3 1.3 Scheme of directionally specific deubiquitination ............................................ 5 1.4 Domain diagrams of UCH family deubiquitinases ........................................... 7 1.5 Structure of the 26S proteasome .................................................................... 9 1.6 Structure of the 20S and AAA ATPases ....................................................... 10 1.7 Scheme of deubiquitination/degradation at the 26S proteasome ................. 14 2.1 Synthesis of UbVME ..................................................................................... 29 2.2 Generation of the TsUCH37cat-UbVME complex .......................................... 32 2.3 Crystals of the TsUCH37cat-UbVME complex ............................................... 34 2.4 Diffraction patterns ....................................................................................... 35 2.5 Active site of TsUCH37 ................................................................................ 39 2.6 Distal site of TsUCH37 ................................................................................. 41 2.7 Crossover loop of TsUCH37cat-UbVME structure ......................................... 42 3.1 Domain diagram of UCH37 and Rpn13 ........................................................ 48

ix
Figure Page 3.2 Structure of human UCH37 .......................................................................... 49 3.3 Analytical ultracentrifugation of the TsUCH37cat-UbVME complex ............... 53 3.4 Michaelis Menten kinetics for TsUCH37cat UbAMC hydrolysis ..................... 55 3.5 Analytical ultracentrifugation of human UCH37 and Rpn13 .......................... 56 3.6 Isothermal titration calorimetry of UCH37 and Rpn13 binding ...................... 58 4.1 Structure of TsUCH37∆C46-UbVME ............................................................... 65 4.2 Mutant triubiquitin synthesis ......................................................................... 70 4.3 Molecular dynamics simulations of TsUCH37-UbVME ................................. 72 4.4 UbAMC hydrolysis by UCH37 ULD mutants ................................................. 74 4.5 HPLC/MS separations of mutant ubiquitin monomers .................................. 76 4.6 Cleavage of mutant triubiquitin by UCH37 in the presence of Rpn13 ........... 78 4.7 Cleavage of varying length K48 polyubiquitin chains by UCH37 .................. 79 5.1 Conformational changes in the ULD domain ................................................ 83 5.2 Activation of UCH37 by crossover loop binding to Rpn13 ............................ 84 5.3 Mode of NFRKB inhibition of UCH37 ............................................................ 86 5.4 Misaligned active sites of deubiquitinating enzymes .................................... 90 5.5 Insertion domains of JAMM metalloproteases .............................................. 92 5.6 Crossover loops for ubiquitin-bound UCH DUBs .......................................... 93 5.7 Occluding loops of USP14 ............................................................................ 95

x
Appendix Figure Page A 6.1 Scheme of Ubl-UIM purification method of endogenous proteasomes ... 103 A 6.2 Polyubiquitination of GFP-titin-cyclinPY substrate .................................... 107 A 6.3 Silver-stained SDS PAGE gel of rabbit 26S proteasome ........................ 109 A 6.4 Activity of UbVME-treated proteasome ................................................... 110 A 6.5 Activity of UbVME- and MG132-treated proteasome .............................. 112

xi
ABSTRACT
Morrow, Marie Elizabeth. Ph.D., Purdue University, May 2015. Structural and Biophysical Analysis of the Proteasomal Deubiquitinase, UCH37. Major Professor: Chittaranjan Das.
Ubiquitin carboxyl-terminal hydrolase 37, or UCH37, is a deubiquitinating
enzyme associated with the 26S proteasome, the primary protein degradation
machinery in eukaryotic cells. UCH37 is responsible for the disassembly of
polymeric ubiquitin chains, or polyubiquitin, which have been ligated onto
proteins in order to target them for degradation. The 26S utilizes two associated
deubiquitinating enzymes, UCH37 and USP14, and one intrinsic, Rpn11, to
remove polyubiquitin chains from substrate proteins as they are unfolded and
translocated into the proteolytic core of the proteasome, where proteins are
cleaved into small peptides and then released for recycling by the cell. UCH37
associates with the proteasome via binding of its C-terminal KEKE motif to the C-
terminus of Rpn13, a proteasomal ubiquitin receptor which ensnares
polyubiquitinated prey for degradation. UCH37 is known to be catalytically
activated upon binding to Rpn13, allowing cleavage of Lys48-linked polyubiquitin
chains from their distal end, an exo-specific deubiquitination. However, free
UCH37 cleaves polyubiquitin poorly and is believed to be autoinhibited by its C-

xii
terminal UCHL5-like domain, or ULD, which may also be responsible for its
oligomerization in solution. This work examines the structural, biophysical, and
catalytic characteristics of UCH37 in order to elucidate its mechanism of
activation by Rpn13, assess its biophysical assembly with Rpn13 within the
greater proteasomal context, and ascertain its mechanism of exo-specificity
despite the proteasome’s processing of a variety of polyubiquitinated substrates.
To this end, a 1.7 Å resolution X-ray crystal structure was solved of the
catalytic domain of a UCH37 homolog from Trichinella spiralis in complex with
ubiquitin vinyl methyl ester (UbVME), a suicide inhibitor substrate. Our structure,
in combination with another solved of a longer construct of TsUCH37 in complex
with UbVME, provided structural insights into the ability of UCH37 to process
polyubiquitin, namely that its C-terminal UCHL5-like domain (ULD) is responsible
for its exo-specific activity due to a network of interactions with ubiquitin’s Lys48.
Through biophysical and kinetic characterization, we have affirmed the
poor activity of UCH37 alone, but do not ascribe it to autoinhibition because it
does not oligomerize as previously thought, rather we find that it sediments in a
monomer-dimer equilibrium in analytical ultracentrifugation experiments. We
have characterized its binding and activation by Rpn13, finding that UCH37 binds
to Rpn13 with a 22 nM dissociation constant and that mutations to UCH37’s ULD
render it unable to be activated by Rpn13. Interestingly, we have found that while
Rpn13 activates UCH37 for ubiquitin-AMC cleavage, a monoubiquitin fluorogenic
substrate, it appears to slow the enzyme’s processing of Lys48-linked
polyubiquitin chains in our assays.

xiii
Altogether, we have confirmed that UCH37 exists primarily as a monomer
which binds tightly to its proteasomal subunit, Rpn13, and can exo-specifically
cleave Lys48-linked polyubiquitin chains. However, UCH37 may not be activated
as was previously thought, by Rpn13 alone, and likely requires full association
with the 26S proteasome.

1
CHAPTER 1: INTRODUCTION
1.1 Ubiquitination
Ubiquitination occurs through a coordinated enzymatic cascade ending in
the attachment of ubiquitin’s C-terminal glycine (Gly76) to an acceptor lysine
residue via an isopeptide bond. This is achieved through sequential ubiquitin
activation (E1 enzymes), conjugation (E2 enzymes), and ligation (E3 enzymes).
The E1 enzyme, of which there are only two in humans, binds both ubiquitin and
ATP-Mg2+, forms an adenylated ubiquitin intermediate, and then its catalytic
cysteine attacks this adenylated ubiquitin to form a ubiquitin-charged E1,
connected by a high energy thioester bond 1. Ubiquitin is then passed on to one
of about 40 E2 enzymes by attack of their catalytic cysteine to form a charged E2
2. Subsequently, the charged E2 binds to one of hundreds of E3 enzymes, which
then permits ubiquitin ligation onto a target protein either through direct transfer
from the E2 onto the substrate, or by E2 hand-off to the E3 enzyme, which itself
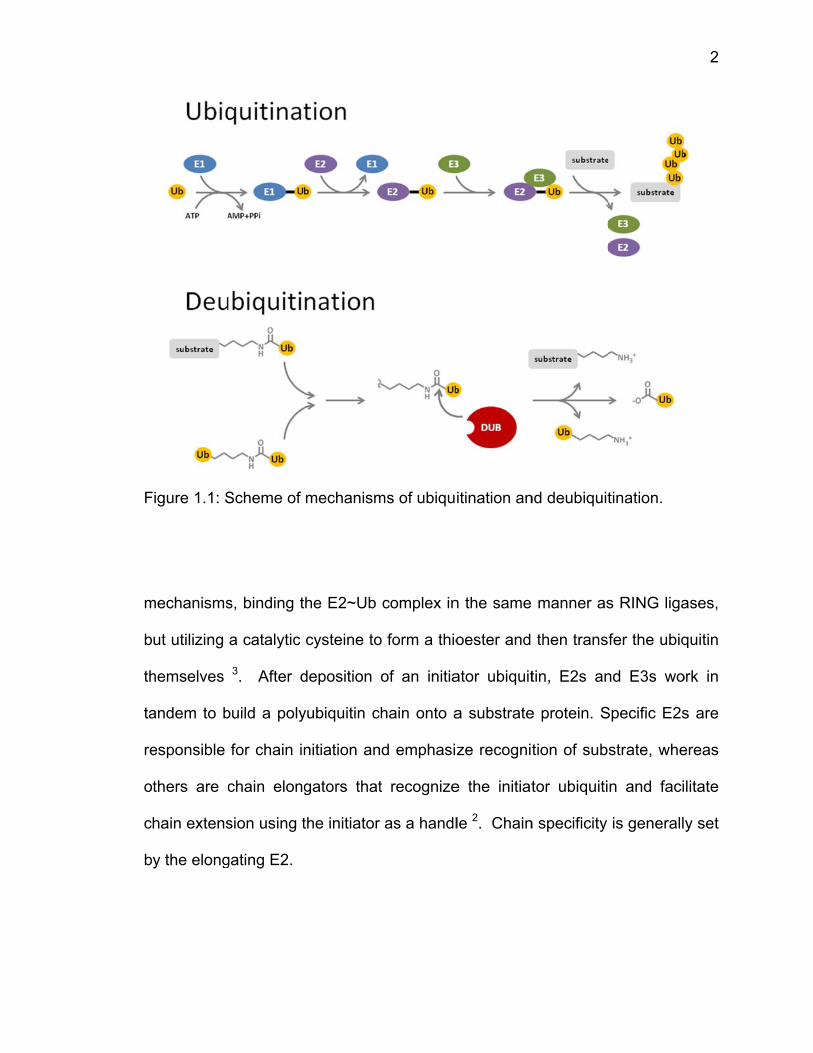
ligates the ubiquitin onto an acceptor lysine (Fig. 1.1) 2. The determinant of either
of these two mechanisms is inherent in the E3 enzyme; RING/U-box ligases
mediate direct E2 transfer, while HECT ligases form a thioester with ubiquitin and
transfer it themselves. RBR ligases (RING in-between RING) act by combining
both
F
m
b
th
ta
re
o
c
b
Figure 1.1: S
mechanisms
ut utilizing
hemselves
andem to b
esponsible
thers are c
hain extens
y the elong
Scheme of
s, binding t
a catalytic
3. After d
build a poly
for chain in
chain elong
sion using t
gating E2.
mechanism
he E2~Ub
cysteine to
deposition
yubiquitin c
nitiation an
gators that
the initiator
ms of ubiqu
complex in
o form a thio
of an initia
hain onto a
d emphasi
t recognize
as a handl
itination an
n the same
oester and
ator ubiquit
a substrate
ze recognit
e the initiat
le 2. Chain
nd deubiquit
manner as
then trans
tin, E2s an
e protein. S
tion of sub
tor ubiquiti
n specificity
tination.
s RING liga
fer the ubiq
nd E3s wo
Specific E2s
strate, whe
n and faci
is generall
2
ases,
quitin
ork in
s are
ereas
litate
y set

F
K
p
h
m
p
a
p
a
Figure 1.2: S
As ub
K63), its mo
olyubiquitin
omotypic c
most well-st
rotein degr
protein de
rogression
ttributed to
Scheme of
biquitin con
onomers ca
n (Fig 1.2).
chain linkag
tudied and
radation at
gradation s
through de
lysosomal
the types o
ntains seve
an be ligate
This host
ges, all res
classical p
the 26S pr
signal, but h
egradation
trafficking
of ubiquitina
en lysines i
ed together
of accepto
sponsible f
polyubiquiti
roteasome.
have also b
of cell cycle
through the
ation.
tself (K6, K
r to form po
or lysines p
for a variet
n chain is
K11-linked
been found
e regulators
e ESCRT p
K11, K27, K
olymeric ch
provides a
ty of cellul
K48-linked
d chains ar
to be cruc
s4-9. K63-lin
pathway, N
K29, K33,
hains, know
wide varie
lar events.
d, the signa
re also prim
cial for cell c
nked chains
F-κB activa
3
K48,
wn as
ety of
The
al for
marily
cycle
s are
ation,

4
and DNA damage repair. Ubiquitin can also be linked through its start methionine
to form linear ubiquitin chains, which are involved in NF-κB activation as well as
cell death 10. Additionally, monoubiquitination serves as a signal for a variety of
cellular events, notably transcriptional regulation and degradation of membrane
proteins 11-13. Currently, little is known about the biological function of chains
linked through K6, K27, K29, and K33 14. Adding further complexity to the
system, ubiquitin chains can be heterotypic, either through mixed ubiquitin chain
linkages that may be “branched” (mixed chain type) or “forked” (two ubiquitin
chains stemming from one monomer) chains, or as mixed ubiquitin-SUMO
chains, all of which are in their early stages of biological characterization 9,15-17.
The mechanisms by which E2s and E3s recognize, bind, initiate, and elongate
ubiquitin chains of varying topologies is still under investigation, as well as
identification of their specific substrates.
1.2 Deubiquitination
In opposition to ubiquitination lies deubiquitination, the hydrolysis of the
isopeptide bond (or Met1-linked amide bonds in linear polyubiquitin) and
subsequent release of ubiquitin from its substrate (Fig. 1.1). This is achieved by a
~100-membered group of enzymes called deubiquitinases, or DUBs. They are
further broken down into mechanistic families, the cysteine proteases and the
metallo-proteases. Cysteine DUBs hydrolyze isopeptide bonds utilizing catalytic
Cys, His, and Asp triads, as well as an oxyanion-stabilizing Gln residue. Metallo-

D
m
w
T
h
p
z
m
u
m
ty
h
(a
F
DUBs have
molecule. T
with the cat
The cystein
ydrolases
roteases (O
inc metall
metalloenzy
biquitin an
monomers.
ypes, direc
ighly spec
associated
igure 1.3: Sc
a zinc ion
his Zn is st
talytic wate
ne proteas
(UCHs), u
OTUs), and
oproteases
ymes (JAMM
d a substr
DUBs hav
ctional chai
cific for on
molecule o
cheme of dir
n responsib
tabilized by
er stabilized
ses include
ubiquitin-sp
d the Mach
s have o
Ms). DUBs
rate protein
ve additiona
n cleavage
ne chain
of the SH3 d
rectionally s
ble for cata
y coordinat
d by hydrog
e four sub
pecific prot
hado Josep
only one
can hydro
n’s lysine re
al specificit
e, and sub
type, for
domain of S
pecific deub
alysis by a
tion to two
gen bonds
bfamilies:
teases (US
phin domai
subfamily,
olyze both i
esidue, as
ties for par
bstrate pref
example
STAM) can
biquitination.
n active si
His and on
to a nearb
the ubiqu
SPs), the
in protease
the JAB
isopeptide
well as be
rticular poly
ference. So
the metal
only cleav
te-bound w
ne Asp res
by Glu res
uitin C-term
ovarian tu
es (MJDs).
B1/MPN/MO
bonds betw
etween two
yubiquitin c
ome DUBs
lo-DUB AM
e Lys63-lin
5
water
idue,
idue.
minal
umor
The
OV34
ween
o Ub
chain
s are
MSH
ked

6
polyubiquitin chains. Other DUBs have specificity for the substrate which has
been ubiquitinated. DUBs that are responsible for chain cleavage have further
specificity for the directionality of their cleavage activity: some remove whole
chains from the site of attachment to a substrate, called en bloc cleavage; some
cleave in the middle of a chain, or endo specificity; and the third group cleaves
from the furthest end of the chain (distal monomer) and removes monomers
sequentially, exo-specific cleavage (Fig. 1.3) 18.
In addition to their DUB domains, many deubiquitinases contain ubiquitin
binding domains (UBDs) which either provide additional stabilization to ubiquitin
binding or confer specificity. Typically, these domains bind monoubiquitin,
sometimes polyubiquitin, with weak affinity in the high micromolar range. They
are most efficient at improving ubiquitin binding when multiple UBDs are found in
one DUB, or if a DUB within a larger complex binds to other proteins containing
UBDs 18,19. Examples of some of the most frequently-occurring UBDs are UBAs
(ubiquitin associated domains), UIMs (ubiquitin interacting motifs), and ZnFs
(zinc finger ubiquitin binding domains) 19. UBDs are crucial for the activity of
many deubiquitinases and are also critical regulators of ubiquitin binding across
the entire proteome.

m
U
p
fa
k
p
re
c
N
F
The
members in
UCHL3, of
roteasome
amily memb
nown to
olyubiquitin
eferred to
atalytic clef
N-terminal U
igure 1.4: D
smallest fa
n humans:
unknown f
; and BAP
bers, UCHL
cleave sm
n chains. T
as the cr
fts 29,30. The
UCH domai
omain diagr
1.
amily of D
UCHL1, k
function; U
P1, which d
L1 and UC
mall substr
his is believ
rossover lo
e other two
n as well as
rams of UCH
2.1 UCH F
DUBs is th
known to b
CH37, invo
deubiquitina
HL3, are si
rates from
ved to be d
oop, which
family mem
s extra C-te
H family deub
amily
he UCHs,
be involved
olved in de
ates histon
ingle-doma
the C-te
due to the p
sterically
mbers, UCH
erminal dom
biquitinases
composed
d in Parkin
eubiquitinat
e H2A 20-2
ain proteins
erminus of
presence o
controls a
H37 and BA
mains (Fig
.
of four fa
nson’s dise
tion at the
8. The first
s which are
ubiquitin,
of a gating
access to
AP1, conta
1.4). UCH3
7
amily
ease;
26S
t two
only
not
loop,
their
in an
37’s

8
C-terminal extension was named the ULD, or UCHL5-like domain, which is
believed to autoinhibit the enzyme’s catalytic activity. At the end lies its KEKE
motif, a region responsible for its binding to the 26S proteasome through the
proteasomal subunit Rpn13, which has a complementary KEKE motif of its own
21,22,31,32. BAP1 has a putative ULD domain, by sequence similarity, which has yet
to be characterized 24,33. BAP1 additionally has a nuclear localization signal at its
far C-terminal end responsible for its cellular localization 24. Both UCH37 and
BAP1 are known to process larger substrates than UCHL1 and UCHL3; UCH37
disassembles polyubiquitin chains at the 26S proteasome, while BAP1
deubiquitinates histone H2A as part of the Polycomb repressor DUB complex
(PR-DUB) 25,26,34,35. UCH37 has been found within the assembly of another
macromolecular complex, the Ino80 chromatin remodeling complex, where it
exists in a generally inactive form, the role of which has yet to be explored 36.
This study focuses on the activity of UCH37, especially as it relates to its role at
the 26S proteasome.
1.3 The 26S Proteasome
The 26S proteasome is a 2.5 MDa proteolytic machine responsible for
degrading the majority of cellular proteins 37-39. It consists of a 20S core particle
composed of proteolytic enzymes and a 19S regulatory particle responsible for
capturing and feeding ubiquitinated proteins into the mouth of the 20S. The 20S
is made up of 4 stacked heptameric rings of structurally similar, but not identical,
subunits 39-42. The external rings contain seven α subunits while the internal rings

c
th
lik
o
th
e
b
FhdA
ontain seve
he proteoly
ke (β2), an
f sequence
hreonine fo
nsures clea
y the α sub
Figure 1.5: ighlighted ieubiquitina
Adapted from
en β subun
tic chambe
d chymotry
e sites 39.
or catalytic
avage into
bunits; the “
Structure in blue, 20
ase in red,m PDB ID 4
nits. The ex
er, where β
ypsin-like (β
The three
activity 39,
small pepti
“closed” for
of the 26SS core par, and ubiq4CR2.
xternal α su
subunits u
β5) activitie
β-subunit
,43. Passag
des, provid
m of the ch
S proteasorticle in yellquitin recep
ubunits act
utilize their
es to cleave
proteases
ge of prote
ded they ca
hannel is on
ome. The 1low, AAA Aptors Rpn1
as the gat
caspase-lik
e proteins a
all rely on
eins through
an enter. En
nly 9 Å wide
19S regulaATPases in10 and Rp
ed channe
ke (β1), try
at a wide va
n an N-term
h this cham
ntry is regu
e, only allow
atory particn purple, Rppn13 in gr
9
l into
psin-
ariety
minal
mber
lated
wing
cle is pn11 reen.

e
to
p
te
th
m
w
m
FcA1
ntry by sma
o the “open
olypeptide
ermini of R
he 20S α su
In ord
must be un
whether un
mounting to
Figure 1.6: rystallized
ATPases, RRYP.
aller peptid
n” form at ~
chain (Fig
pts 2, 3, an
ubunits 42,45
der to trans
folded into
nfolding an
support the
Structure in an ope
Rpts 1-6, are
es, rather t
~20 Å wide
. 1.6) 44. T
nd 5 of the
5-51.
slocate thro
o a linear p
nd transloc
e theory tha
of the 20en and close shown at
than entire
e, the core
This openin
19S AAA
ough the po
polypeptide
cation are
at unfolding
0S and Ased gate (t right. Ada
globular pr
particle ac
ng is facilita
ATPases i
ore of the 2
e chain; the
sequentia
g is coupled
AAA ATPas(left) and tpted from P
roteins 44. W
ccommodat
ated by do
nto the bin
20S core p
ere is som
al, howeve
d to transloc
ses. The 2the heterohPDB ID 4C
When conve
tes an unfo
cking of th
ding pocke
particle, pro
e debate a
er evidenc
cation. This
20S has bhexameric
CR2, 1G0U,
10
erted
olded
he C-
ets of
oteins
as to
ce is
s
been AAA
, and

11
event is achieved by the 19S regulatory particle’s AAA ATPase subunits, Rpts 1-
6, a heterohexameric motor which utilizes ATP hydrolysis to pull polypeptide
chains into the 20S (Fig. 1.6). These Rpts dock to the outer α rings of the 20S
and serve as the base of the 19S RP. Studies of other AAA unfoldases,
especially ClpXP, a bacterial unfoldase, has suggested that translocation and
unfolding are simultaneously achieved through bursts of mechanical force 52,53.
Both ClpXP and the φ29 DNA packaging motor have been shown to exist 90% of
the time in a dwell state, with only 10% of its time spent in a burst of activity 53,54.
This has yet to be confirmed in the 26S Rpts, but cryoEM structures of the Rpts
engaged and disengaged with substrate suggest this may be the case 48-50,55,56.
In addition to Rpts 1-6, the base of the 19S regulatory particle contains
two scaffolding proteins, Rpn1 and Rpn2, as well as the two constitutive ubiquitin
receptors, Rpn10 and Rpn13 39,57. Rpn1 and Rpn2 act to recruit associated
proteins and shuttle factors to the 19S. Through interactions with Ubl (ubiquitin-
like) domains, Rpn1 acts as a docking site for shuttle factors which bring
polyubiquitinated proteins to the proteasome, such as Rad23B and Dsk2 58-61.
Rpn1 is also responsible for recruitment of one of the proteasome’s associated
deubiquitinating enzymes, USP14, discussed below. Thus far, Rpn2 is only
known to anchor one of the intrinsic proteasomal ubiquitin receptors, Rpn13, to
the proteasome, no other shuttle factors or associating ubiquitin receptors 57,61-64.
Rpn10 and Rpn13 are the intrinsic ubiquitin receptors at the proteasome,
although shuttle factors and some temporarily-associating ubiquitin receptors
(Rad23B, Dsk2, Dss1, Ddi1, AIRAP) also bind polyubiquitin and transport it to the

12
proteasome 58,65,66. Interestingly, deletion of these receptors and shuttle factors
(currently known ones) does not impair growth of yeast 60,64. Rpn10 and Rpn13
bind tightly to the proteasome, whereas the other shuttle factors bind weakly and
transiently 61,63. It is possible that there are even more shuttle factors or receptors
to be discovered that may rescue protein degradation upon deletion of this set.
Rpn10, or S5a in humans, utilizes two ubiquitin interacting motifs (UIMs) to bind
polyubiquitin avidly and can also recruit the shuttle factor Rad23B 57,58,67-72. It has
an additional N-terminal von Willebrand A (VWA) domain of unknown function.
Rpn13 contains an N-terminal pleckstrin homology domain referred to as the
pleckstrin-like receptor for ubiquitin (Pru) domain, which binds ubiquitin in a novel
mode compared to other ubiquitin binding domains 21-23,62-64. Rpn13’s C-terminal
domain is responsible for binding UCH37, the second proteasome-associated
deubiquitinase. Rpn10 and Rpn13 lie on the outer edge of the 19S, at opposite
ends, affording polyubiquitin chains a broad surface area for binding as well as
the flexibility of multiple conformations and chain branching (Fig. 1.5) 50,56,57.
Wrapping around and above the 19S base complex lies its lid complex,
one of the least understood components of the 26S. Functions have not been
assigned for its 9 subunits except Rpn11, the proteasome’s constitutive
deubiquitinase. Rpns 3, 5, 6, 7, 9, and 12 contain a proteasome cyclosome
initiation factor (PCI) domain, but the function of these proteins is currently
unknown, aside from acting as scaffolds for other components 73. Rpn11, a
JAMM metallo-DUB, requires dimerization with Rpn8, which contains an inactive
MPN domain, to form its active deubiquitinating module 55,74-80. The lid sits above

13
and around the opening pore of the AAA ATPases, with Rpn11 poised
immediately adjacent to the access point of polyubiquitinated substrates 50,55.
1.3.1 Deubiquitination at the 26S Proteasome
After polyubiquitinated proteins are brought to the 26S proteasome via
shuttle factors and transient ubiquitin receptors, they bind to the proteasome’s
intrinsic ubiquitin receptors, Rpn13 and Rpn1021,23,57,58,62,81. As substrates are
unfolded and translocated into the interior of the core particle, the metallo-
deubiquitinase, Rpn11, cleaves off whole ubiquitin chains from the substrate
protein, releasing them back into the cellular pool of ubiquitin 32,75-77. Rpn11
utilizes a catalytic zinc ion bound by two histidines and an aspartate to cleave
polyubiquitin chains in an en bloc fashion, that is, the entire chain is removed
from its acceptor lysine on a substrate protein74,79. From cryo-EM structures of
the 26S engaged and free of ubiquitinated substrates, it is known that Rpn11
initially exists in an occluded state that is misaligned with the central pore and
ATPases, which subsequently undergoes a dramatic conformational change
upon substrate binding and engagement 50,55. This conformational change aligns
the active site of Rpn11 immediately above the central pore and ATPase ring
opening, which then allows it to cleave entire polyubiquitin chains from an
engaged substrate protein 50,55. Rpn11 is a highly promiscuous DUB capable of
cleaving many different chain types and possibly having endopeptidase activity
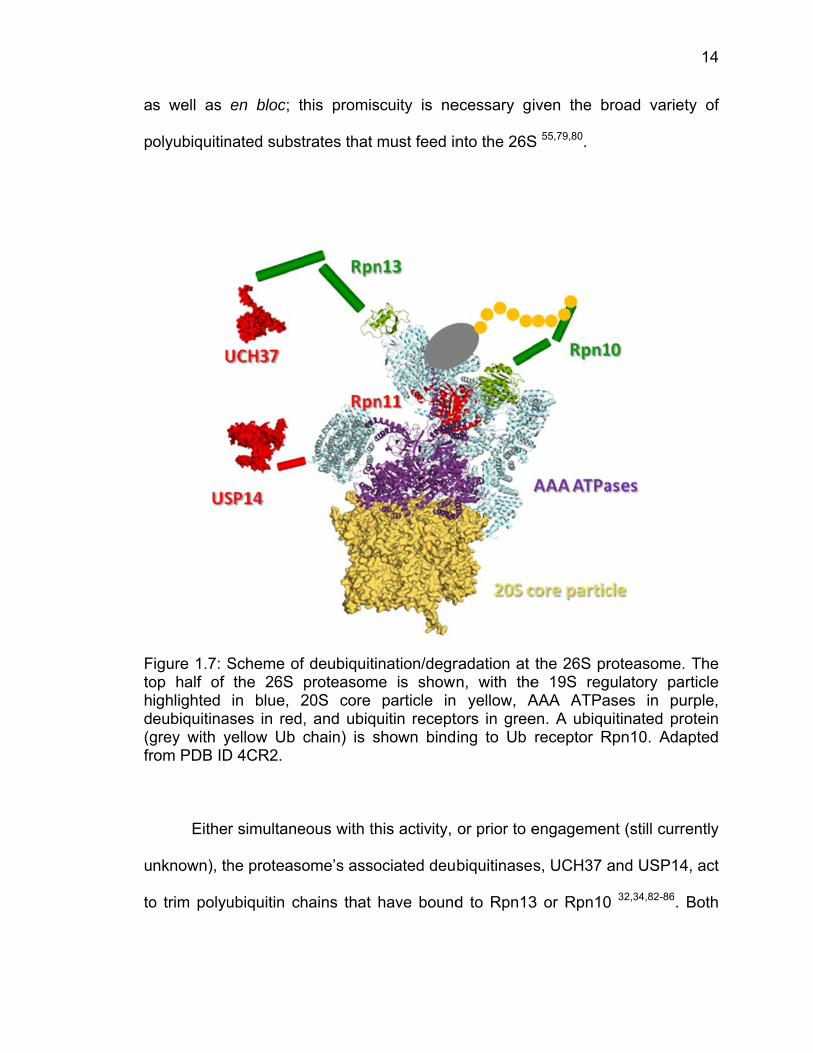
a
p
Ftohd(gfr
u
to
s well as
olyubiquitin
Figure 1.7: Sop half of ighlighted eubiquitinagrey with yrom PDB ID
Eithe
nknown), th
o trim polyu
en bloc; th
nated subst
Scheme of the 26S in blue, 2
ases in red,yellow Ub cD 4CR2.
r simultane
he proteaso
ubiquitin ch
his promis
trates that m
f deubiquitinproteasome20S core , and ubiquchain) is s
eous with th
ome’s asso
hains that h
cuity is ne
must feed in
nation/degre is showparticle in
uitin recepthown bind
his activity,
ociated deu
have bound
ecessary g
nto the 26S
radation at n, with the yellow, A
tors in greeing to Ub
or prior to e
biquitinase
d to Rpn13
iven the b
S 55,79,80.
the 26S pre 19S regAAA ATPaen. A ubiqureceptor R
engagemen
s, UCH37 a
3 or Rpn10
broad varie
roteasome.gulatory paases in puuitinated prRpn10. Ada
nt (still curr
and USP14
0 32,34,82-86.
14
ty of
The rticle
urple, otein
apted
rently
4, act
Both

15
UCH37 and USP14 are cysteine protease DUBs which cleave polyubiquitin
chains exo-specifically, that is from the furthest monomer (distal) from the
substrate protein and working their way inwards. USP14 associates with the
proteasome through its Ubl domain, which binds to Rpn1, a known docking point
for other Ubl domain-containing proteins. UCH37, however, binds to an ubiquitin
receptor, Rpn13, through matching KEKE motifs within both of their C-terminal
domains. USP14 and UCH37 have poor basal levels of deubiquitinase activity
alone, but become significantly activated upon recruitment to the 26S
proteasome21,78,84,87,88. They are generally thought to be present in
substoichiometric amounts at the 26S, especially USP14 due to the fact that its
binding partner, Rpn1, is known to bind to multiple proteins at that same site.
Currently it is believed that UCH37 is specific for Lys48-linked chains and that
USP14 may process other chain types, however, the variety of ubiquitinated
species brought to the proteasome indicates that these DUBs are probably more
promiscuous than first thought.
A few theories exist as to what role these associated DUBs play in
proteasome degradation: (1) they recycle monoubiquitin, for further use by the
cell 89,90; (2) they may allow dissociation of chains prior to substrate commitment
for degradation, and in turn rescue a small portion of proteins slated for
degradation that may be inappropriately labeled 34,35; or (3) after a polyubiquitin
chain has been freed from its substrate by Rpn11, the two associated DUBs
sequentially remove ubiquitin monomers until the affinity of the polyubiquitin
chain for Rpn10 or Rpn13 is poor enough to dissociate from the 26S, allowing

16
“resetting” of the proteasome for another round of degradation 39,91. Their
inhibition has been shown to accelerate proteasomal degradation, however,
further work is needed to clarify the biological role of proteasome-associated
deubiquitination 86,92.
Within this work, we present the X-ray crystal structure of a UCH37
homolog bound to ubiquitin, as well as biophysical and kinetic data, which
provides a better structural understanding of the specificity and activation of this
proteasome-bound DUB. Despite the broad spectrum of ubiquitinated
proteasomal substrates, this DUB appears to maintain a limited specificity. We
hope that these studies of UCH37 will obtain a better picture of how
deubiquitinating enzymes in general balance a need for specificity in the face of a
plethora of ubiquitinated proteins, as well as how these enzymes are kept in
inactive/active states by cellular protein partners.

17
1.4 References
1 Schulman, B. A. & Harper, J. W. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nature reviews. Molecular cell biology 10, 319-331, doi:10.1038/nrm2673 (2009).
2 Meyer-Schwesinger, C. et al. A new role for the neuronal ubiquitin C-
terminal hydrolase-L1 (UCH-L1) in podocyte process formation and podocyte injury in human glomerulopathies. J Pathol 217, 452-464 (2009).
3 Smit, J. J. & Sixma, T. K. RBR E3-ligases at work. EMBO reports 15, 142-
154, doi:10.1002/embr.201338166 (2014). 4 Kirkpatrick, D. S. et al. Quantitative analysis of in vitro ubiquitinated cyclin
B1 reveals complex chain topology. Nature cell biology 8, 700-710, doi:10.1038/ncb1436 (2006).
5 Jin, L., Williamson, A., Banerjee, S., Philipp, I. & Rape, M. Mechanism of
ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133, 653-665, doi:10.1016/j.cell.2008.04.012 (2008).
6 Matsumoto, M. L. et al. K11-linked polyubiquitination in cell cycle control
revealed by a K11 linkage-specific antibody. Molecular cell 39, 477-484, doi:10.1016/j.molcel.2010.07.001 (2010).
7 Rape, M. Assembly of k11-linked ubiquitin chains by the anaphase-
promoting complex. Sub-cellular biochemistry 54, 107-115, doi:10.1007/978-1-4419-6676-6_9 (2010).
8 Wickliffe, K. E., Williamson, A., Meyer, H. J., Kelly, A. & Rape, M. K11-
linked ubiquitin chains as novel regulators of cell division. Trends in cell biology 21, 656-663, doi:10.1016/j.tcb.2011.08.008 (2011).
9 Meyer, H. J. & Rape, M. Enhanced protein degradation by branched
ubiquitin chains. Cell 157, 910-921, doi:10.1016/j.cell.2014.03.037 (2014). 10 Iwai, K., Fujita, H. & Sasaki, Y. Linear ubiquitin chains: NF-kappaB
signalling, cell death and beyond. Nature reviews. Molecular cell biology 15, 503-508, doi:10.1038/nrm3836 (2014).
11 Mukhopadhyay, D. & Riezman, H. Proteasome-independent functions of
ubiquitin in endocytosis and signaling. Science 315, 201-205, doi:10.1126/science.1127085 (2007).

18
12 Henry, K. W. et al. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes & development 17, 2648-2663, doi:10.1101/gad.1144003 (2003).
13 Zhang, Y. Transcriptional regulation by histone ubiquitination and
deubiquitination. Genes & development 17, 2733-2740, doi:10.1101/gad.1156403 (2003).
14 Komander, D. & Rape, M. The ubiquitin code. Annu Rev Biochem 81, 203-
229 (2012). 15 Kim, H. T. et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and
ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. The Journal of biological chemistry 282, 17375-17386, doi:10.1074/jbc.M609659200 (2007).
16 Kim, H. T., Kim, K. P., Uchiki, T., Gygi, S. P. & Goldberg, A. L. S5a
promotes protein degradation by blocking synthesis of nondegradable forked ubiquitin chains. The EMBO journal 28, 1867-1877, doi:10.1038/emboj.2009.115 (2009).
17 Guzzo, C. M. & Matunis, M. J. Expanding SUMO and ubiquitin-mediated
signaling through hybrid SUMO-ubiquitin chains and their receptors. Cell cycle 12, 1015-1017, doi:10.4161/cc.24332 (2013).
18 Komander, D., Clague, M. J. & Urbe, S. Breaking the chains: structure and
function of the deubiquitinases. Nature reviews. Molecular cell biology 10, 550-563, doi:10.1038/nrm2731 (2009).
19 Husnjak, K. & Dikic, I. Ubiquitin-binding proteins: decoders of ubiquitin-
mediated cellular functions. Annual review of biochemistry 81, 291-322, doi:10.1146/annurev-biochem-051810-094654 (2012).
20 Maraganore, D. M. et al. UCHL1 is a Parkinson's disease susceptibility
gene. Ann Neurol 55, 512-521 (2004). 21 Yao, T. et al. Proteasome recruitment and activation of the Uch37
deubiquitinating enzyme by Adrm1. Nature cell biology 8, 994-1002 (2006).
22 Hamazaki, J. et al. A novel proteasome interacting protein recruits the
deubiquitinating enzyme UCH37 to 26S proteasomes. The EMBO journal 25, 4524-4536 (2006).

19
23 Qiu, X. B. et al. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. The EMBO journal 25, 5742-5753, doi:10.1038/sj.emboj.7601450 (2006).
24 Ventii, K. H. et al. BRCA1-associated protein-1 is a tumor suppressor that
requires deubiquitinating activity and nuclear localization. Cancer Res 68, 6953-6962 (2008).
25 Stone, M. et al. Uch2/Uch37 is the major deubiquitinating enzyme
associated with the 26S proteasome in fission yeast. Journal of molecular biology 344, 697-706 (2004).
26 Scheuermann, J. C. et al. Histone H2A deubiquitinase activity of the
Polycomb repressive complex PR-DUB. Nature 465, 243-247 (2010). 27 Kurihara, L. J., Semenova, E., Levorse, J. M. & Tilghman, S. M.
Expression and functional analysis of Uch-L3 during mouse development. Molecular and cellular biology 20, 2498-2504 (2000).
28 Dennissen, F. J. et al. Mutant ubiquitin (UBB+1) associated with
neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3). FEBS letters 585, 2568-2574, doi:10.1016/j.febslet.2011.06.037 (2011).
29 Misaghi, S. et al. Structure of the ubiquitin hydrolase UCH-L3 complexed
with a suicide substrate. The Journal of biological chemistry 280, 1512-1520 (2005).
30 Boudreaux, D. A., Maiti, T. K., Davies, C. W. & Das, C. Ubiquitin vinyl
methyl ester binding orients the misaligned active site of the ubiquitin hydrolase UCHL1 into productive conformation. Proceedings of the National Academy of Sciences of the United States of America 107, 9117-9122 (2010).
31 Qiu, X. B. et al. hRpn13/ADRM1/GP110 is a novel proteasome subunit
that binds the deubiquitinating enzyme, UCH37. Embo J 25, 5742-5753 (2006).
32 Koulich, E., Li, X. & DeMartino, G. N. Relative structural and functional
roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell 19, 1072-1082 (2008).
33 Sanchez-Pulido, L., Kong, L. & Ponting, C. P. A common ancestry for
BAP1 and Uch37 regulators. Bioinformatics 28, 1953-1956 (2012).

20
34 Lam, Y. A., Xu, W., DeMartino, G. N. & Cohen, R. E. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737-740 (1997).
35 Lam, Y. A., DeMartino, G. N., Pickart, C. M. & Cohen, R. E. Specificity of
the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. The Journal of biological chemistry 272, 28438-28446 (1997).
36 Yao, T. et al. Distinct modes of regulation of the Uch37 deubiquitinating
enzyme in the proteasome and in the Ino80 chromatin-remodeling complex. Molecular cell 31, 909-917 (2008).
37 Voges, D., Zwickl, P. & Baumeister, W. The 26S proteasome: a molecular
machine designed for controlled proteolysis. Annual review of biochemistry 68, 1015-1068 (1999).
38 Coux, O., Tanaka, K. & Goldberg, A. L. Structure and functions of the 20S
and 26S proteasomes. Annual review of biochemistry 65, 801-847 (1996). 39 Finley, D. Recognition and processing of ubiquitin-protein conjugates by
the proteasome. Annual review of biochemistry 78, 477-513 (2009). 40 Lupas, A., Koster, A. J. & Baumeister, W. Structural features of 26S and
20S proteasomes. Enzyme & protein 47, 252-273 (1993). 41 Groll, M. et al. A gated channel into the proteasome core particle. Nature
structural biology 7, 1062-1067, doi:10.1038/80992 (2000). 42 Smith, D. M. et al. Docking of the proteasomal ATPases' carboxyl termini
in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell 27, 731-744, doi:10.1016/j.molcel.2007.06.033 (2007).
43 Seemuller, E. et al. Proteasome from Thermoplasma acidophilum: a
threonine protease. Science 268, 579-582 (1995). 44 Rabl, J. et al. Mechanism of gate opening in the 20S proteasome by the
proteasomal ATPases. Mol Cell 30, 360-368, doi:10.1016/j.molcel.2008.03.004 (2008).
45 Gillette, T. G., Kumar, B., Thompson, D., Slaughter, C. A. & DeMartino, G.
N. Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J Biol Chem 283, 31813-31822, doi:10.1074/jbc.M805935200 (2008).

21
46 da Fonseca, P. C. & Morris, E. P. Structure of the human 26S proteasome: subunit radial displacements open the gate into the proteolytic core. J Biol Chem 283, 23305-23314, doi:10.1074/jbc.M802716200 (2008).
47 Unverdorben, P. et al. Deep classification of a large cryo-EM dataset
defines the conformational landscape of the 26S proteasome. Proc Natl Acad Sci U S A 111, 5544-5549, doi:10.1073/pnas.1403409111 (2014).
48 Sledz, P. et al. Structure of the 26S proteasome with ATP-gammaS bound
provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc Natl Acad Sci U S A 110, 7264-7269, doi:10.1073/pnas.1305782110 (2013).
49 Beck, F. et al. Near-atomic resolution structural model of the yeast 26S
proteasome. Proc Natl Acad Sci U S A 109, 14870-14875, doi:10.1073/pnas.1213333109 (2012).
50 Lander, G. C. et al. Complete subunit architecture of the proteasome
regulatory particle. Nature 482, 186-191, doi:10.1038/nature10774 (2012). 51 Tian, G. et al. An asymmetric interface between the regulatory and core
particles of the proteasome. Nature structural & molecular biology 18, 1259-1267, doi:10.1038/nsmb.2147 (2011).
52 Maillard, R. A. et al. ClpX(P) generates mechanical force to unfold and
translocate its protein substrates. Cell 145, 459-469, doi:10.1016/j.cell.2011.04.010 (2011).
53 Sen, M. et al. The ClpXP protease unfolds substrates using a constant
rate of pulling but different gears. Cell 155, 636-646, doi:10.1016/j.cell.2013.09.022 (2013).
54 Nyquist, K. & Martin, A. Marching to the beat of the ring: polypeptide
translocation by AAA+ proteases. Trends in biochemical sciences 39, 53-60, doi:10.1016/j.tibs.2013.11.003 (2014).
55 Matyskiela, M. E., Lander, G. C. & Martin, A. Conformational switching of
the 26S proteasome enables substrate degradation. Nature structural & molecular biology 20, 781-788, doi:10.1038/nsmb.2616 (2013).
56 Lasker, K. et al. Molecular architecture of the 26S proteasome
holocomplex determined by an integrative approach. Proceedings of the National Academy of Sciences of the United States of America 109, 1380-1387 (2012).

22
57 Sakata, E. et al. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc Natl Acad Sci U S A 109, 1479-1484, doi:10.1073/pnas.1119394109 (2012).
58 Elsasser, S., Chandler-Militello, D., Muller, B., Hanna, J. & Finley, D.
Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. The Journal of biological chemistry 279, 26817-26822, doi:10.1074/jbc.M404020200 (2004).
59 Elsasser, S. et al. Proteasome subunit Rpn1 binds ubiquitin-like protein
domains. Nature cell biology 4, 725-730, doi:10.1038/ncb845 (2002). 60 Elsasser, S. & Finley, D. Delivery of ubiquitinated substrates to protein-
unfolding machines. Nature cell biology 7, 742-749, doi:10.1038/ncb0805-742 (2005).
61 Rosenzweig, R., Bronner, V., Zhang, D., Fushman, D. & Glickman, M. H.
Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. The Journal of biological chemistry 287, 14659-14671, doi:10.1074/jbc.M111.316323 (2012).
62 Schreiner, P. et al. Ubiquitin docking at the proteasome through a novel
pleckstrin-homology domain interaction. Nature 453, 548-552 (2008). 63 Chen, X., Lee, B. H., Finley, D. & Walters, K. J. Structure of proteasome
ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Molecular cell 38, 404-415 (2010).
64 Husnjak, K. et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor.
Nature 453, 481-488, doi:10.1038/nature06926 (2008). 65 Wilkinson, C. R. et al. Proteins containing the UBA domain are able to
bind to multi-ubiquitin chains. Nature cell biology 3, 939-943, doi:10.1038/ncb1001-939 (2001).
66 Paraskevopoulos, K. et al. Dss1 is a 26S proteasome ubiquitin receptor.
Molecular cell 56, 453-461, doi:10.1016/j.molcel.2014.09.008 (2014). 67 Wilkinson, C. R. et al. Analysis of a gene encoding Rpn10 of the fission
yeast proteasome reveals that the polyubiquitin-binding site of this subunit is essential when Rpn12/Mts3 activity is compromised. The Journal of biological chemistry 275, 15182-15192 (2000).

23
68 Zhang, D. et al. Together, Rpn10 and Dsk2 can serve as a polyubiquitin chain-length sensor. Molecular cell 36, 1018-1033, doi:10.1016/j.molcel.2009.11.012 (2009).
69 Riedinger, C. et al. Structure of Rpn10 and its interactions with
polyubiquitin chains and the proteasome subunit Rpn12. The Journal of biological chemistry 285, 33992-34003, doi:10.1074/jbc.M110.134510 (2010).
70 Walters, K. J., Kleijnen, M. F., Goh, A. M., Wagner, G. & Howley, P. M.
Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41, 1767-1777 (2002).
71 Wang, Q., Young, P. & Walters, K. J. Structure of S5a bound to
monoubiquitin provides a model for polyubiquitin recognition. Journal of molecular biology 348, 727-739, doi:10.1016/j.jmb.2005.03.007 (2005).
72 Zhang, N. et al. Structure of the s5a:k48-linked diubiquitin complex and its
interactions with rpn13. Molecular cell 35, 280-290, doi:10.1016/j.molcel.2009.06.010 (2009).
73 Ciechanover, A. & Stanhill, A. The complexity of recognition of
ubiquitinated substrates by the 26S proteasome. Biochimica et biophysica acta 1843, 86-96, doi:10.1016/j.bbamcr.2013.07.007 (2014).
74 Verma, R. et al. Role of Rpn11 metalloprotease in deubiquitination and
degradation by the 26S proteasome. Science 298, 611-615 (2002). 75 Maytal-Kivity, V., Reis, N., Hofmann, K. & Glickman, M. H. MPN+, a
putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC biochemistry 3, 28 (2002).
76 Yao, T. & Cohen, R. E. A cryptic protease couples deubiquitination and
degradation by the proteasome. Nature 419, 403-407 (2002). 77 Guterman, A. & Glickman, M. H. Complementary roles for Rpn11 and
Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem 279, 1729-1738, doi:10.1074/jbc.M307050200 (2004).
78 Koulich, E., Li, X. & DeMartino, G. N. Relative structural and functional
roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Molecular biology of the cell 19, 1072-1082 (2008).

24
79 Worden, E. J., Padovani, C. & Martin, A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nature structural & molecular biology 21, 220-227, doi:10.1038/nsmb.2771 (2014).
80 Mansour, W. et al. Disassembly of lys11 and mixed linkage polyubiquitin
conjugates provides insights into function of proteasomal deubiquitinases rpn11 and ubp6. The Journal of biological chemistry 290, 4688-4704, doi:10.1074/jbc.M114.568295 (2015).
81 Janse, D. M., Crosas, B., Finley, D. & Church, G. M. Localization to the
proteasome is sufficient for degradation. The Journal of biological chemistry 279, 21415-21420, doi:10.1074/jbc.M402954200 (2004).
82 Lee, M. J., Lee, B. H., Hanna, J., King, R. W. & Finley, D. Trimming of
ubiquitin chains by proteasome-associated deubiquitinating enzymes. Molecular & cellular proteomics : MCP 10, R110 003871, doi:10.1074/mcp.R110.003871 (2011).
83 Crosas, B. et al. Ubiquitin chains are remodeled at the proteasome by
opposing ubiquitin ligase and deubiquitinating activities. Cell 127, 1401-1413, doi:10.1016/j.cell.2006.09.051 (2006).
84 Leggett, D. S. et al. Multiple associated proteins regulate proteasome
structure and function. Molecular cell 10, 495-507 (2002). 85 Hanna, J. et al. Deubiquitinating enzyme Ubp6 functions noncatalytically
to delay proteasomal degradation. Cell 127, 99-111 (2006). 86 Lee, B. H. et al. Enhancement of proteasome activity by a small-molecule
inhibitor of USP14. Nature 467, 179-184 (2010). 87 Hu, M. et al. Structure and mechanisms of the proteasome-associated
deubiquitinating enzyme USP14. The EMBO journal 24, 3747-3756 (2005).
88 Jiao, L. et al. Mechanism of the Rpn13-induced activation of Uch37.
Protein & cell 5, 616-630, doi:10.1007/s13238-014-0046-z (2014). 89 Hanna, J., Meides, A., Zhang, D. P. & Finley, D. A ubiquitin stress
response induces altered proteasome composition. Cell 129, 747-759, doi:10.1016/j.cell.2007.03.042 (2007).

25
90 Chernova, T. A. et al. Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. The Journal of biological chemistry 278, 52102-52115, doi:10.1074/jbc.M310283200 (2003).
91 Thrower, J. S., Hoffman, L., Rechsteiner, M. & Pickart, C. M. Recognition
of the polyubiquitin proteolytic signal. The EMBO journal 19, 94-102, doi:10.1093/emboj/19.1.94 (2000).
92 Lee, M. J., Lee, B. H., Hanna, J., King, R. W. & Finley, D. Trimming of
ubiquitin chains by proteasome-associated deubiquitinating enzymes. Molecular & cellular proteomics : MCP (2010).

26
CHAPTER 2: STRUCTURE OF TSUCH37CAT-UBVME
2.1 Introduction
Structural approaches to studying ubiquitination/deubiquitination
machinery has yielded extensive information about its detailed mechanisms,
providing vital understanding of these proteins’ ability to recognize either highly
specific chain types or to be grossly promiscuous for any ubiquitinated molecule
available. This approach has given the field incredible insight into the biological
significance of ubiquitination. The structures of many deubiquitinases have been
solved alone and in complex with ubiquitin or a ubiquitin variant. Generally, DUBs
bind monoubiquitin quite poorly, especially if they act as polyubiquitin chain
trimmers in cells. Therefore, in order to capture a DUB-ubiquitin bound state,
covalent linkage of monoubiquitin is required, to prevent dissociation during
crystallography. For this end, a handful of suicide inhibitor ubiquitin variants are
used in structural biology. One of these is ubiquitin vinyl methyl ester (UbVME),
used in this study, which seems to have the highest reactivity with UCH family
DUBs. Here, I have solved the X-ray crystal structure of a UCH37 homolog from
Trichinella spiralis in complex with ubiquitin vinyl methyl ester. This structure
highlights the similarities of UCH-family DUB binding to ubiquitin, as many
contacts are conserved with UCHL1 and UCHL3. However, the active site

27
crossover loop, a structural feature common to UCH enzymes, is not resolved in
the TsUCH37cat-UbVME structure due to a high amount of flexibility that is not
abrogated upon ubiquitin binding, an unexpected result that hints at UCH37’s
mechanism of activation.
2.2 Materials and Methods
2.2.1 Synthesis of Ubiquitin Vinyl Methyl Ester
The synthesis of glycine vinyl methyl ester (GlyVME) has been previously
published, but was modified in our hands (Fig 2.1) 1-3. For the Boc protection
reaction, 8 grams (88 mmol) of 3-amino-1,2-propanediol was dissolved in 150 mL
water, then cooled on ice in order to add 23 grams (105 mmol) of di-tert-butyl
dicarbonate (Boc anhydride), after which the reaction was returned to room
temperature. Then the reaction was brought to pH 10.5 by addition of sodium
hydroxide and the reaction was allowed to run overnight at room temperature.
The reaction was diluted with 100 mL of ethyl acetate, cooled on ice, and then
brought to pH 2.5 with hydrochloric acid. The product was then extracted out with
8 x 50 mL ethyl acetate. The organic layer was washed with NaHSO4 and brine,
dried over sodium sulfate, and then rotovapped down and stored at -20 C. For
the oxidation reaction, 7-8 g of Boc-propanediol was dissolved in 125 mL water,
to which 1.4 molar equivalents of NaIO4 were added. The reaction was stirred for
2-12 hours. The product was extracted out with 3 x 100 mL ethyl acetate, dried
over sodium sulfate, and then rotovapped down. The aldehyde product was used

28
within a day and stored at -20 C. For the Horner Wadsworth Emmons reaction, 1
equivalent of sodium hydride (60% suspension in mineral oil) was added to a
flame-dried round bottom and immediately suspended in 40 mL dry THF, then
purged with N2. The sodium hydride was washed 3 x 30 mL dry THF and then 1
equivalent of trimethyl phosphonoacetate was added over 1 hr on ice. Additional
THF was added as needed to keep the reaction in solution. The Boc-aldehyde
was dissolved in minimal THF and added to the reaction over 1 hr on ice. After
addition, the reaction was allowed to warm to room temperature and run from 6-
12 hrs. The reaction was quenched with 200 mL water and then THF was
removed by rotovapping. The product, Boc-GlyVME, was extracted out with 3 x
50 mL chloroform and the organic layer was washed once with 50 mL of 2%
hydrochloric acid and once with 50 mL saturated sodium carbonate. The organic
layer was dried over sodium sulfate and then rotovapped down and stored until
purification. Boc-GlyVME was purified by silica flash chromatography using a
gradient of 0-20% ethyl acetate in hexanes, pooling only fractions containing the
E isomer. Solvent was rotovapped off, the product was washed 2 x with DCM,
and then rotovapped down again. For Boc deprotection and crystallization of the
final product, 2 molar equivalents of p-toluenesulfonic acid was dissolved in 100-
200 mL diethyl ether, dried over sodium sulfate, and decanted off. Boc-GlyVME
was dissolved in minimal ether and added to the pTSA solution. GlyVME tosyl
salt crystallized out overnight, was filtered out, and stored at -20 C until reaction
with UbMESNa.
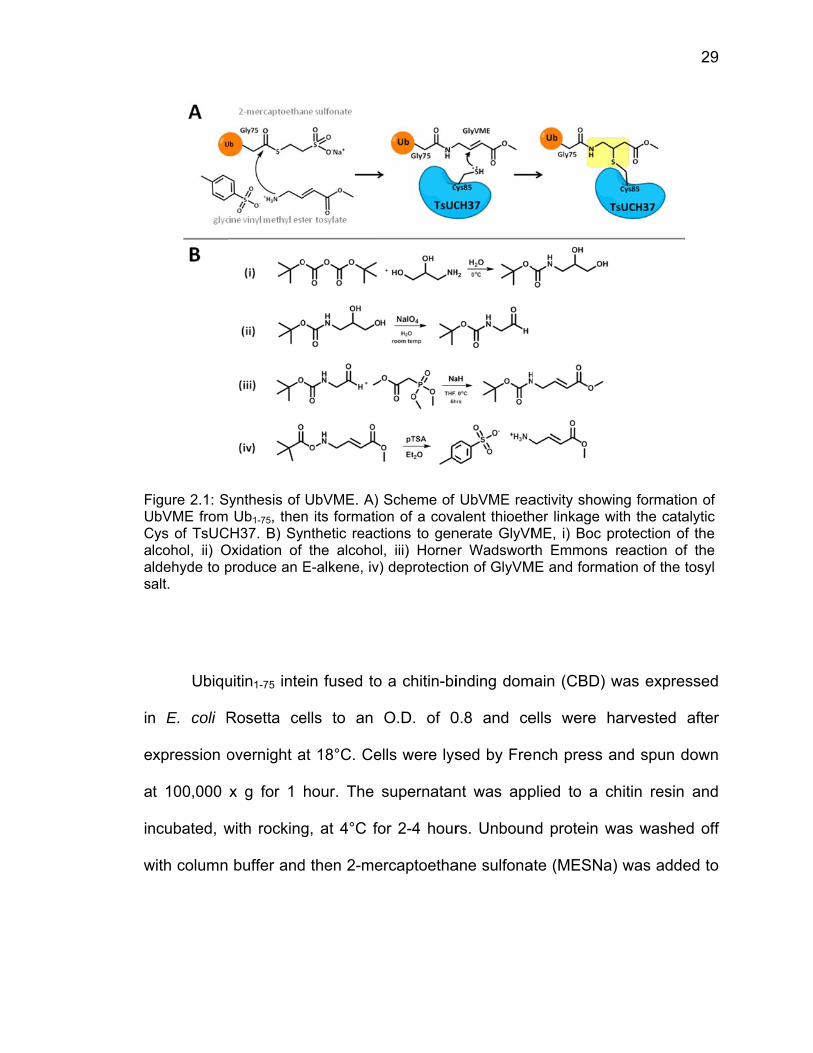
in
e
a
in
w
FUCaasa
Ubiqu
n E. coli R
xpression o
t 100,000
ncubated, w
with column
igure 2.1: SUbVME fromCys of TsUC
lcohol, ii) Oldehyde to palt.
uitin1-75 inte
Rosetta ce
overnight a
x g for 1 h
with rocking
buffer and
Synthesis of Ub1-75, then
CH37. B) SyOxidation of produce an E
ein fused to
ells to an
at 18°C. Ce
hour. The
g, at 4°C fo
d then 2-me
UbVME. A) n its formationthetic reacthe alcoho
E-alkene, iv)
o a chitin-bi
O.D. of 0
ells were lys
supernatan
or 2-4 hour
ercaptoetha
Scheme of on of a covations to genl, iii) Horne) deprotectio
inding dom
0.8 and ce
sed by Fre
nt was app
rs. Unboun
ane sulfona
UbVME reaalent thioethnerate GlyVMer Wadsworton of GlyVM
main (CBD)
ells were h
nch press
plied to a c
nd protein w
ate (MESNa
activity showher linkage wME, i) Boc pth Emmons
ME and forma
was expre
harvested
and spun d
chitin resin
was washe
a) was adde
wing formatiowith the cataprotection o
reaction ofation of the
29
ssed
after
down
and
ed off
ed to
on ofalyticf thef thetosyl

30
the column to displace Ub1-75 from the intein group by incubating overnight at
37°C. The eluate was collected and concentrated down to 1.5 mL.
In order to generate UbVME, UbMESNa was incubated with 200 mg
GlyVME and 125 mg NHS dissolved in 1 M NaHCO3 at pH 8 overnight at room
temperature. After incubation, UbVME was dialyzed into 50 mM NaOAc pH 4.5
for 4 hours, then applied to a Mono S cation exchange column for purification
from UbMESNa or hydrolyzed Ub1-75. Fractions were tested for reactivity with
UCHL3 and the most reactive fractions were pooled, concentrated down, and
flash frozen and stored at -80°C.
2.2.2 Cloning, Expression, and Protein Purification of TsUCH37cat
Full-length Trichinella spiralis (Ts) UCH37 in the pET28a vector was sent
from the lab of Katerina Artavanis-Tsakonas, who had previously identified the
enzyme as a UCH family member and confirmed it to be UCH37 by co-
immunoprecipitations and pull-downs of proteasomal subunits 4. Following
standard cloning protocols, the catalytic domain of TsUCH37, residues 1-226,
was subcloned into the pGEX 6P1 vector between BamHI and XhoI digestion
sites. The protein was expressed in E. coli Rosetta DE3 cells to an O.D. of 1.0
and the cells were harvested after expression overnight at 18°C. Cells were lysed
by French press and spun down at 100,000 x g for 1 hour. The supernatant was
applied to glutathione sepharose beads and unbound proteins were washed off
with column buffer (1 x PBS, 400 mM KCl). GST-fused TsUCH37cat was eluted
with reduced glutathione and incubated with PreScission Protease (GE

31
Biosciences) overnight at 4°C. TsUCH37cat was run back over the glutathione
beads to capture GST, and then the pure protein was concentrated down and run
on a HiLoad Superdex 75 for further purification. Pure fractions were
concentrated down, flash frozen, and stored at -80°C.
2.2.3 Complexation of TsUCH37cat with UbVME
Test reactions to complex TsUCH37cat with UbVME were set up in 12 uL
scale to determine the ideal concentration to push complexation to completion.
Three tests were done at 37°C for 3 hours at 29 mg/mL, 14.4 mg/mL, and 9.6
mg/mL TsUCH37cat (Fig. 2.2). For the final scale up reaction, 14.4 mg/mL was
chosen. The scale-up reaction was composed of 600 uL of 14.4 mg/mL
TsUCH37cat, 600 uL UbVME, and 70 uL 1M Tris pH 8.0, for a total volume of 1.9
mL (Fig. 2.2). After 3 hours at 37°C, the reaction was diluted to 4 mL and run on
a Superdex 75 for further purification, but an unexpected higher molecular weight
species was not purified, so all fractions from this step were pooled, buffer
exchanged, and run on a MonoQ anion exchange column in 0-40% 50 mM Tris
pH 7.6, 1 M NaCl, 1 mM DTT over 45 column volumes. The pure complex eluted
at 17% 1 M NaCl (Fig. 2.2). Pure fractions were pooled, concentrated down to 3-
5 mg/mL, flash frozen, and stored at -80°C.
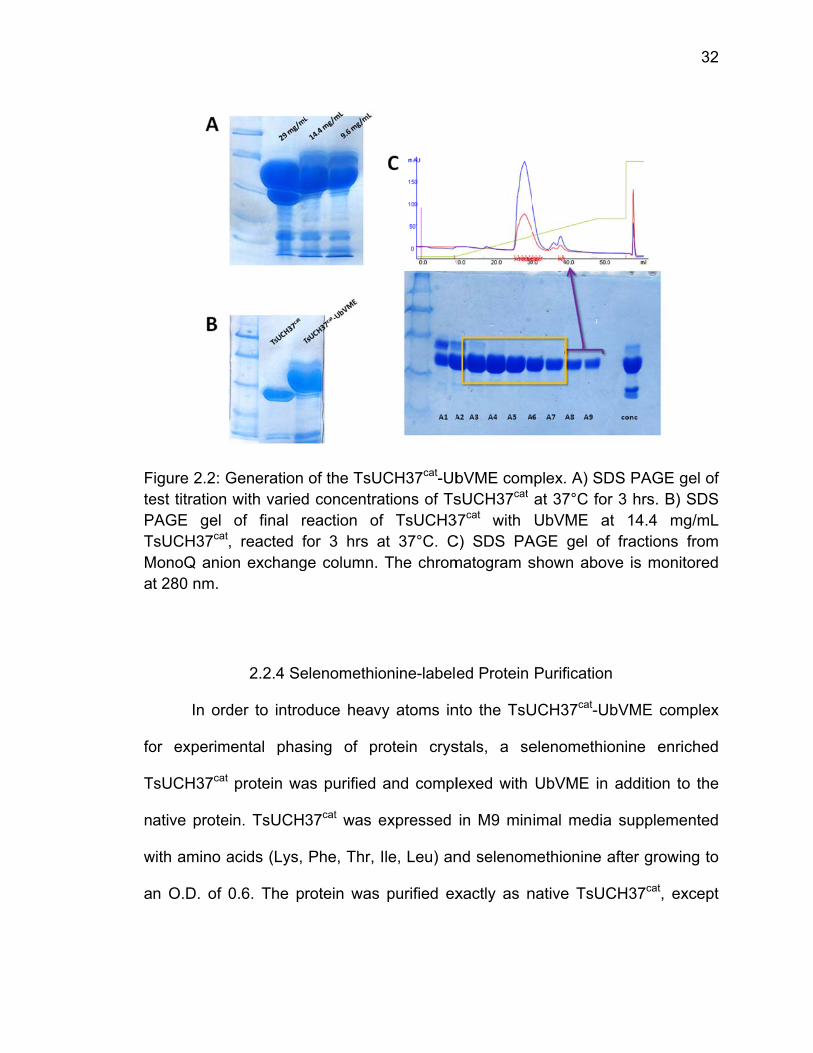
FtePTMa
fo
T
n
w
a
Figure 2.2: Gest titration
PAGE gel TsUCH37cat
MonoQ aniot 280 nm.
In ord
or experim
TsUCH37cat
ative prote
with amino a
n O.D. of
Generation with variedof final r
, reacted fon exchang
2.2.4 Se
der to intro
mental phas
protein wa
ein. TsUCH
acids (Lys,
0.6. The p
of the TsUd concentrareaction offor 3 hrs age column.
elenomethio
oduce heav
sing of pr
as purified
37cat was e
Phe, Thr,
rotein was
UCH37cat-Ubations of Tsf TsUCH3at 37°C. CThe chrom
onine-label
vy atoms in
rotein crys
and compl
expressed
Ile, Leu) an
purified ex
bVME comsUCH37cat
37cat with C) SDS PAmatogram s
ed Protein
nto the TsU
stals, a se
lexed with
in M9 min
nd selenom
xactly as n
mplex. A) SDat 37°C forUbVME a
AGE gel ofshown abov
Purification
UCH37cat-U
elenomethio
UbVME in
imal media
methionine a
native TsUC
DS PAGE gr 3 hrs. B)
at 14.4 mgf fractions ve is monit
n
bVME com
onine enri
addition to
a suppleme
after growin
CH37cat, ex
32
gel of SDS g/mL from
tored
mplex
ched
o the
ented
ng to
xcept

33
that each buffer was supplemented with 2-5 mM DTT to keep selenomethionine
in a reducing environment. Mass spectrometry of SeMet TsUCH37cat by protein
MALDI confirmed that all four methionines in the protein were enriched with
SeMet, an M+1 molecular weight of 26238.6 Da and M+2 of 13117.1 Da, with a
calculated molecular weight of 26238 Da. SeMet TsUCH37cat was complexed
with UbVME and purified by MonoQ anion exchange chromatography. Pure
fractions were pooled, concentrated down, and flash frozen and stored at -80°C.
Yields for the SeMet protein were reduced; therefore the SeMet complex was
lower concentration than the original complex.
2.2.5 Crystallization and Structure Solution
Native TsUCH37cat-UbVME was screened at 3 mg/mL in ~700
crystallographic conditions by sitting drop vapor diffusion. A hit was identified in
the Hampton Research Ammonium Sulfate grid screen, composed of 3 M
ammonium sulfate, 0.1 M bicine pH 9 at room temperature after 2 days by
hanging drop vapor diffusion. However, rather than single, 3D crystals, the initial
hit appeared to be stacks of 2D plate crystals. In anticipation of poor data due to
multi-latticed crystals, the initial hit was optimized by additive screening. Single
3D crystals appeared with the addition of 2 mM glutathione (mixture of oxidized
and reduced). Crystallization attempts with the SeMet complex in the same
mother liquor composition as the native hit did not yield any crystals, therefore
microseeding with native crystals was done to induce SeMet complex
c
m
fr
o
g
w
c
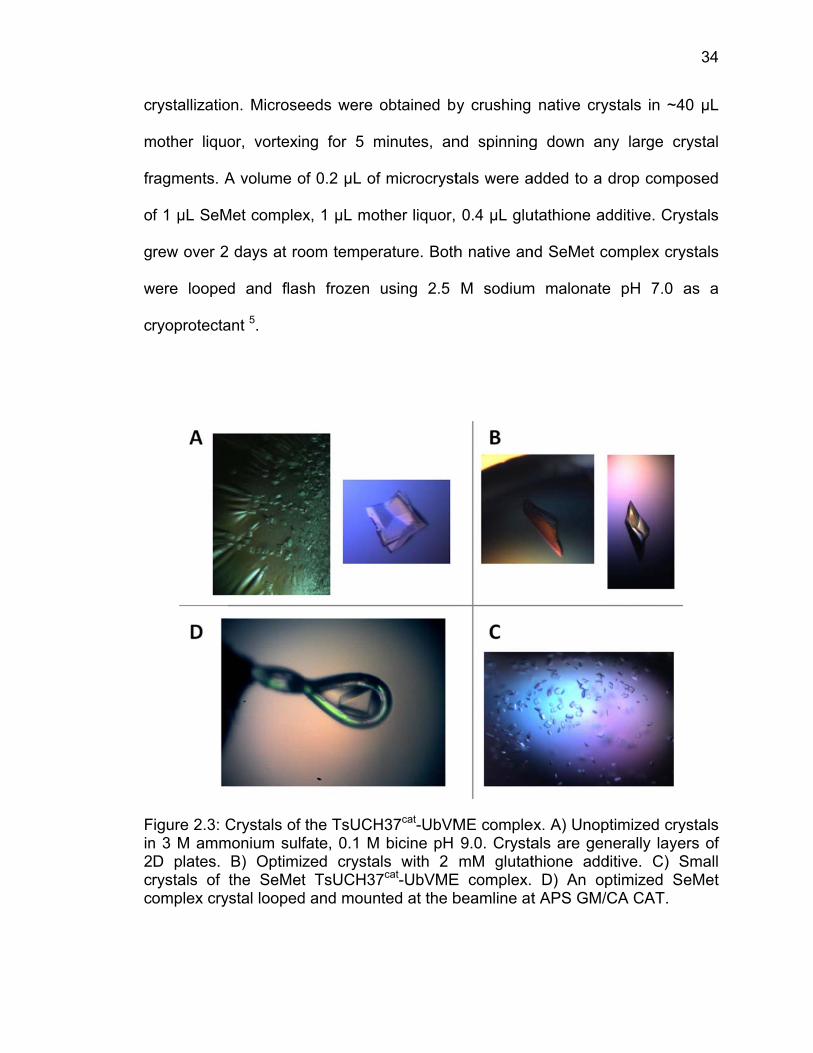
Fin2cc
rystallizatio
mother liquo
ragments. A
f 1 µL SeM
rew over 2
were looped
ryoprotecta
Figure 2.3: Cn 3 M ammD plates. rystals of omplex cry
on. Microse
or, vortexin
A volume o
Met complex
days at ro
d and flas
ant 5.
Crystals of monium sulf
B) Optimizthe SeMet
ystal looped
eeds were o
ng for 5 m
of 0.2 µL of
x, 1 µL mot
oom temper
sh frozen
the TsUCHfate, 0.1 M zed crystat TsUCH37d and moun
obtained by
minutes, an
f microcryst
ther liquor,
rature. Both
using 2.5
H37cat-UbVMbicine pH
ls with 2 7cat-UbVMEted at the b
y crushing
nd spinning
tals were a
0.4 µL glu
h native an
M sodium
ME comple9.0. CrystamM glutat
E complex. beamline at
native cry
g down an
added to a d
utathione ad
nd SeMet co
m malonate
ex. A) Unopals are genthione add D) An opt APS GM/C
ystals in ~4
ny large cr
drop comp
dditive. Cry
omplex cry
e pH 7.0 a
ptimized crynerally layeditive. C) Sptimized SeCA CAT.
34
40 µL
rystal
osed
ystals
ystals
as a
ystals ers of Small eMet

L
6.
c
d
w
a
w
Fca
Data
aboratory o
. Native cry
ollected at
ispersion (
was collecte
dditive. Th
which was u
Figure 2.4: rystals anddditives. Co
was colle
on a Mar30
ystal data w
t the sele
SAD) phas
ed on som
e diffractio
unable to be
Diffraction d B) the Seollected at
ected at
00 CCD det
was collect
nium peak
sing with an
me of the
n pattern in
e indexed (
patterns. eMet TsUCthe ID-B be
the 23-ID-
tector (Mar
ed up to 1
k, 0.979 Å
n f’ of -8.74
unoptimize
ndicated a
Fig. 2.4).
From A) thCH37cat-UbVeamline of G
-B beamlin
r USA) and
.9 Å at 1.0
Å for singl
4 and an f
ed crystals
multi-lattic
he unoptimVME crystaGM/CA CA
ne at Arg
d processed
033 Å and
le-waveleng
f’’ of 7.12.
s, lacking
ce or multip
mized TsUCals after op
AT at Argon
gonne Nat
d with HKL2
1.7 Å data
gth anoma
Diffraction
the glutath
ple-crystal d
CH37cat-UbVptimization ne’s APS.
35
tional
2000
was
alous
data
hione
data,
VME with

36
The initial model was obtained from the Phenix AutoSol wizard using
selenium SAD phases with an input of 8 Se sites (from Matthews coefficient,
determined to be a dimer in the asymmetric unit). The initial model was given a
FOM (figure of merit) of 0.338, and initial Rwork of 0.3695 and Rfree of 0.3884. Its
sequence was built in using the Phenix AutoBuild wizard with additional manual
model building in Coot 7,8. Two copies of the complex were found in the
asymmetric unit, having a space group of C2. Refinement of the structure was
done in Phenix using some TLS refinement (entire asymmetric unit considered to
be one TLS group) and optimized weighting for stereochemical restraints 7.
Overall completeness of the data was poor, at 88.5%, but this can be credited to
poor completeness in the highest resolution shells (42%), which did not prevent
structure solution or refinement. The final model had an R factor of 17.4% and an
Rfree of 21% with <0.2% of residues in the disallowed region of the
Ramachandran plot and scoring a 98% in assessment by Molprobity 9. The
structure was deposited in the Protein Databank (PDB) under the entry 4I6N 10.

37
2.3 Results
2.3.1 Structure of TsUCH37cat-UbVME
TsUCH37cat-UbVME crystallized in the C2 space group with two copies of
the complex in the asymmetric unit. The final model had an R factor of 17.4%
and an Rfree of 21% (Table 2.1).
The first structural element to come to our attention was the presence of
electron density for a disulfide bond between Cys71 of each TsUCH37cat
monomer, leading to disulfide-mediated dimerization in the asymmetric unit.
Human UCH37 was previously thought to oligomerize in solution through its C-
terminal domain, therefore this result was unexpected. In order to determine if
this dimerization has biological relevance, the TsUCH37cat-UbVME complex and
TsUCH37cat alone were both subjected to analytical ultracentrifugation, the
results of which are discussed in Section 3.3. We concluded that this disulfide
bond formation was a crystallographic artifact rather than a biologically significant
event. It likely arose as a result of the introduction of glutathione as an additive
and may have assisted crystal packing into a better form than the initial 2D plate
crystals. The two copies of the complex have an RMSD of 0.39, indicating very
few differences between them. Analysis of the complex, for the purpose of this
document, will focus on Chains A and B in the PDB file rather than the copy,
Chains C and D.

T
aNbRincRdRreeOa
Table 2.1: Ta
Numbers in Rmerge =Σ|Ih - ntensity. Rwork = Σ ||Fo
Rfree is the saeflections thaOrdered resind Gln152 to
able of crysta
parentheses<Ih>|/Σ Ih , w
obs|-k|Fcal||/ Σame as Robs at was not inidues: Pro-3o Gln225 in
allographic s
s refer to datwhere Ih is th
Σ|Fobs| for a selecte
ncluded in pr3 to Gly141 a
Chain A.
statistics.
ta in the highhe observed
ed subset (5rior refinemeand Lys153 t
hest resolutiintensity an
5% and 9%, ent calculatioto Asp224 in
ion shell. nd <Ih> is the
respectivelyons. n Chain C; P
e average
y) of the
Pro-3 to Gly1
38
141

fo
(P
P
c
te
h
b
FcoCsu
The s
our other U
PDB ID
Plasmodium
atalytic clef
erminal tail
ydrogen b
etween the
igure 2.5: Aompared to
C-terminal tauperposed i
structure o
UCH family
1CMX), U
m falciparum
ft (Fig. 2.5)
of ubiquitin
bond netwo
e backbone
Active site human UCHil of UbVMEn purple. All
2.
f TsUCH37
ubiquitin-bo
UCHL3-UbV
m UCHL3-
) bind ubiqu
n, residues
ork within
of TsUCH3H37 (purple)E (orange) a contacts ar
3.2 Active
7cat-UbVME
ound struct
VME (1XD
-UbVME (2
uitin in a hi
70-75, is t
the cataly
37. A) Activ) upon ubiquand TsUCH3re 2.7 – 3.2 Å
Site Bindin
E shows ve
tures in the
D3), UCHL
2WDT). Its
ighly conse
thoroughly
ytic cleft,
ve site archuitin binding.37cat (teal), wÅ.
g
ery similar
e PDB: Yuh
L1-UbVME
s active sit
erved mann
stabilized b
notably th
hitecture of . B) Interactiwith human
features to
h1-Ub alde
(3KW5),
te and gen
ner 11-13. Th
by an exten
rough con
TsUCH37 ions betweeUCH37 res
39
o the
hyde
and
neral
he C-
nsive
tacts
(teal) en the idues

40
of the tail as well as Arg72 and Arg74’s side chains. The active site tetrad,
composed of Cys85, Asp176, His161, and the oxyanion-stabilizing residue
Gln79, is arranged in a canonical orientation for the UCH family, which is seen in
papain-like cysteine proteases as well. The catalytic Cys85 of TsUCH37 has
flipped about 90° compared to Cys88 of the unbound human enzyme (PDB
3IHR) upon binding to GlyVME, a mimic of the acyl-enzyme intermediate during
catalysis. This phenomenon is also seen in the ubiquitin-bound and unbound
structures of UCHL1, and is believed to be a conformational switch from an
unproductive form of the enzyme that may exist as a protective mechanism 12.
Some deubiquitinases operate within an oxidative environment, and this
conformational change may protect the enzyme against cysteine oxidation 14.
2.3.3 Distal Site Binding
Stabilization of ubiquitin’s C-terminal tail is the primary mode of ubiquitin
binding by UCH family enzymes, with the second-most important being its distal
site interactions with ubiquitin’s Leu8, Thr7, and Thr9 as well as ubiquitin’s Ile44
patch. Ubiquitin-interacting residues from the Ts to human UCH37 are not highly
conserved compared to its catalytic cleft residues. The distal site of TsUCH37
utilizes different hydrophobic groups than the human enzyme for ubiquitin
binding, such as replacement of Ser37 with Leu36 and substitution of the large
Trp36 with Val34 and Val35 (Fig 2.6). Additionally, comparing the distal pockets
of the human unbound enzyme versus bound TsUCH37, there appears to be a

c
L
FTusu
w
c
st
fa
a
st
b
onformation
8-T9 hairpi
igure 2.6: DTsUCH37 (te
biquitin’s Ileuperposed i
The c
which varies
left of the U
tabilizing it
amily memb
ll make co
tructures o
elieved tha
nal change
n turn.
Distal site oeal) comparee44 patch (on purple.
crossover l
s in length
UCH doma
ts C-termin
bers crysta
ontacts with
f the enzym
at the UCH
e that enclos
of UCH37. Aed to humanorange) and
2
oop is a st
among the
ain and bind
nal tail furth
llized in com
h ubiquitin
mes, are no
crossover
ses the dis
A) Ubiquitinn UCH37 (pud TsUCH37
2.3.4 Cross
tructural fe
e family me
ds to ubiqu
her for cata
mplex with
via their
ot resolved
loop is lock
stal residues
n (orange) burple) unbou
7cat (teal), w
sover Loop
ature comm
embers 13.
uitin when i
alysis 11,12.
an ubiquiti
crossover
d due to hig
ked into a s
s tighter aro
binding to tund. B) Inter
with human
mon to all
It lies acro
t is bound
The singl
n variant fo
loops, whi
gh flexibility
specific con
ound ubiqu
the distal siractions betwUCH37 res
UCH enzy
oss the cata
to the enzy
e domain
ound in the
ich in unbo
y (Fig. 2.7).
nformation u
41
uitin’s
ite of ween idues
mes,
alytic
yme,
UCH
PDB
ound
. It is
upon
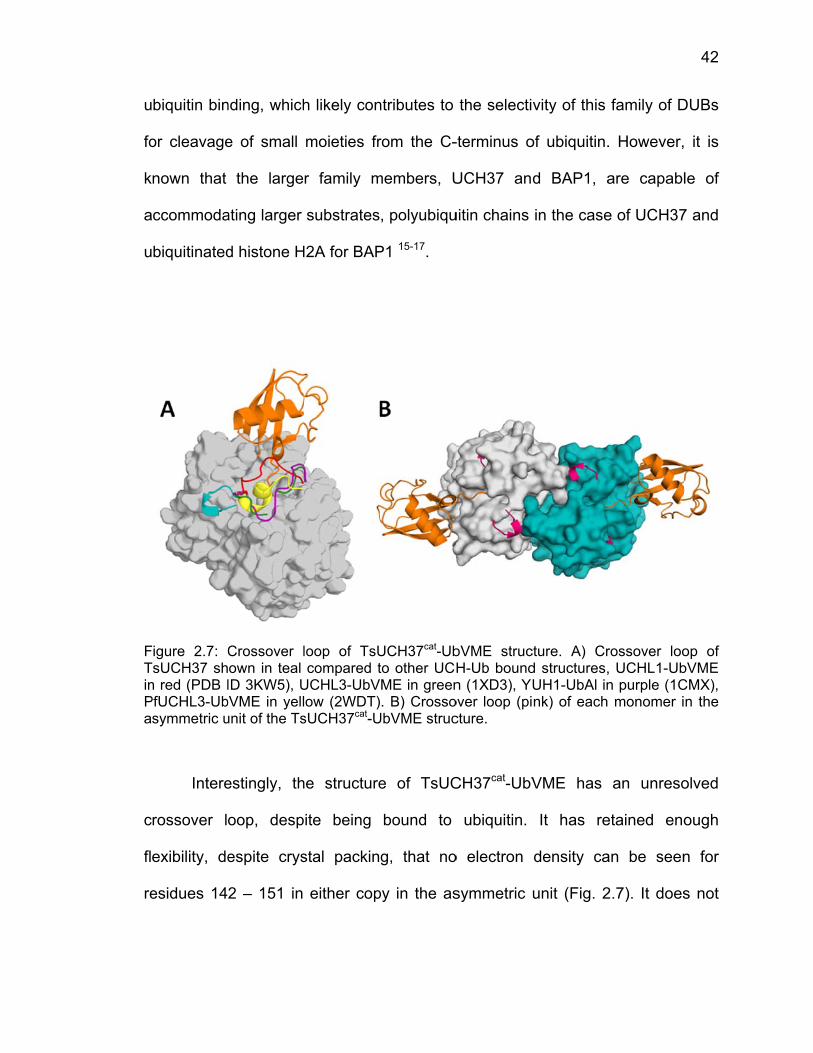
u
fo
k
a
u
FTinPa
c
fl
re
biquitin bin
or cleavage
nown that
ccommoda
biquitinated
igure 2.7: CTsUCH37 shn red (PDB IPfUCHL3-Ub
symmetric u
Intere
rossover l
exibility, de
esidues 14
nding, which
e of small
the large
ating larger
d histone H
Crossover lown in teal ID 3KW5), U
bVME in yellunit of the Ts
estingly, th
oop, desp
espite crys
2 – 151 in
h likely con
moieties fr
r family m
substrates
H2A for BAP
loop of TsUcompared t
UCHL3-UbVow (2WDT)
sUCH37cat-U
he structure
ite being
stal packin
either cop
ntributes to
rom the C-
members, U
, polyubiqu
P1 15-17.
UCH37cat-Ubto other UCHME in green. B) Crosso
UbVME struc
e of TsUC
bound to
g, that no
py in the as
the selecti
-terminus o
UCH37 an
uitin chains
bVME strucH-Ub boundn (1XD3), Y
over loop (picture.
CH37cat-UbV
ubiquitin.
o electron
symmetric
vity of this
of ubiquitin.
d BAP1, a
in the case
cture. A) Crd structures,UH1-UbAl innk) of each
VME has
It has re
density ca
unit (Fig. 2
family of D
. However,
are capabl
e of UCH37
rossover loo, UCHL1-Ubn purple (1C monomer i
an unreso
etained eno
an be seen
2.7). It does
42
DUBs
it is
le of
7 and
op of bVME CMX), n the
olved
ough
n for
s not

43
make any contacts with ubiquitin in the situation of this complex: UCH37 –
monoubiquitin. This leads one to believe that an additional protein binding event
would be required to stabilize the crossover loop, that it may require a different
minimal substrate (diubiquitin, triubiquitin, etc) or that the crossover loop binds to
another protein regulator. We speculate that this other protein may be Rpn13,
and that this binding event may be the source of activation of UCH37’s catalytic
activity.
2.4 Discussion
Here we have presented the structure of TsUCH37cat bound to ubiquitin
vinyl methyl ester, which has provided some valuable insights into the
mechanism of this UCH family deubiquitinase. The enzyme relies on a complex
network of interactions around ubiquitin’s C-terminal tail for substrate
stabilization, which is highly conserved between TsUCH37 and the other yeast
and human homologs of UCH enzymes. Additionally, TsUCH37 utilizes distal site
binding to recognize ubiquitin’s Ile44 patch and Leu8-Thr9 motif. However, the
residues responsible for distal site binding are not as conserved as those in the
catalytic cleft, compared to human UCH37. This lack of conservation may impact
the affinity of ubiquitin binding, which will be explored in Part 2 through
comparison of the enzyme’s catalytic activity compared to the human enzyme.
This region of the enzyme may confer selectivity among UCH family enzymes,
distinguishing each from one another, as their catalytic clefts are nearly identical.

44
The most significant structural difference between TsUCH37cat and the
other UCH family structures is that its crossover loop has not gained sufficient
stabilization upon ubiquitin binding to be visualized in its X-ray crystal structure.
The crossover loop is a structural element of UCH enzymes which is responsible
for substrate filtering and binding, which appears to not play a role in ubiquitin
binding for TsUCH37, and likely human UCH37 as well. We speculate that the
crossover loop would be resolved in the structure if it was satisfying all its
necessary contacts, which probably requires binding to an additional partner. We
further hypothesize that this binding partner may be Rpn13, the proteasomal
subunit which anchors UCH37 to the 26S proteasome. It seems therefore that
the crossover loop in UCH37 may be a key element in the regulation of UCH37’s
catalytic activity through protein-protein contacts. Further examination of the
crossover loop in binding studies should confirm our hypothesis.

45
2.5 References
1 Borodovsky, A. et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chemistry & biology 9, 1149-1159 (2002).
2 Borodovsky, A. et al. A novel active site-directed probe specific for
deubiquitylating enzymes reveals proteasome association of USP14. The EMBO journal 20, 5187-5196 (2001).
3 Liu, S. & Hanzlik, R. P. Structure-activity relationships for inhibition of
papain by peptide Michael acceptors. Journal of medicinal chemistry 35, 1067-1075 (1992).
4 White, R. R. et al. Characterisation of the Trichinella spiralis
deubiquitinating enzyme, TsUCH37, an evolutionarily conserved proteasome interaction partner. PLoS Negl Trop Dis 5, e1340 (2011).
5 Holyoak, T. et al. Malonate: a versatile cryoprotectant and stabilizing
solution for salt-grown macromolecular crystals. Acta crystallographica. Section D, Biological crystallography 59, 2356-2358 (2003).
6 Otwinowski, Z. a. M., W. Processing of X-ray Diffraction Data Collected in
Oscillation Mode. Vol. 276 (Academic Press (New York), 1997). 7 Adams, P. D. et al. PHENIX: a comprehensive Python-based system for
macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography 66, 213-221 (2010).
8 Emsley, P. & Cowtan, K. Coot: model-building tools for molecular
graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126-2132 (2004).
9 Chen, V. B. et al. MolProbity: all-atom structure validation for
macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography 66, 12-21, doi:10.1107/S0907444909042073 (2010).
10 Morrow, M. E. et al. Stabilization of an Unusual Salt Bridge in Ubiquitin by
the Extra C-Terminal Domain of the Proteasome-Associated Deubiquitinase UCH37 as a Mechanism of Its Exo Specificity. Biochemistry, doi:10.1021/bi4003106 (2013).

46
11 Misaghi, S. et al. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. The Journal of biological chemistry 280, 1512-1520 (2005).
12 Boudreaux, D. A., Maiti, T. K., Davies, C. W. & Das, C. Ubiquitin vinyl
methyl ester binding orients the misaligned active site of the ubiquitin hydrolase UCHL1 into productive conformation. Proceedings of the National Academy of Sciences of the United States of America 107, 9117-9122 (2010).
13 Johnston, S. C., Riddle, S. M., Cohen, R. E. & Hill, C. P. Structural basis
for the specificity of ubiquitin C-terminal hydrolases. The EMBO journal 18, 3877-3887 (1999).
14 Kulathu, Y. et al. Regulation of A20 and other OTU deubiquitinases by
reversible oxidation. Nature communications 4, 1569, doi:10.1038/ncomms2567 (2013).
15 Lam, Y. A., Xu, W., DeMartino, G. N. & Cohen, R. E. Editing of ubiquitin
conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737-740 (1997).
16 Lam, Y. A., DeMartino, G. N., Pickart, C. M. & Cohen, R. E. Specificity of
the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. The Journal of biological chemistry 272, 28438-28446 (1997).
17 Scheuermann, J. C. et al. Histone H2A deubiquitinase activity of the
Polycomb repressive complex PR-DUB. Nature 465, 243-247 (2010).

47
CHAPTER 3: KINETIC AND BIOPHYSICAL CHARACTERIZATION OF TSUCH37
3.1 Introduction
The biophysical characteristics of UCH37 keenly regulate its kinetic
activity as well as biological association with its proteasomal binding partner,
Rpn13. Its ULD, or UCHL5-like domain, has been shown to alter its activity and
ability to bind to the 26S proteasome. Within this ULD lies the KEKE motif, a
region spanning the final 20-30 amino acids of the protein, which is responsible
for its binding to the proteasomal subunit Rpn13. Rpn13 harbors a
complementary C-terminal KEKE motif, which binds to UCH37 (Fig 3.1).
Interestingly, the ULD of UCH37 is also thought to play two additional roles within
the enzyme: (1) regulation of its oligomeric state and (2) autoinhibition of the
enzyme’s catalysis1-5. The oligomerization of UCH37 was explained by
tetramerization of the human enzyme in its X-ray crystal structure (PDB ID 3IHR)
as well as in-solution higher order oligomers observed during size-exclusion
chromatography (Fig 3.2) 5. Autoinhibition has been seen by multiple groups in
the context of purified protein, by deletion of the ULD and comparison of its
activity versus that of the full-length enzyme against ubiquitin 7-amino-4-
methylcoumarin, a fluorogenic monoubiquitin substrate standard in the DUB field,
but limited in that it does not address the processing of polyubiquitin 1,2,5-9.

U
o
a
d
w
c
U
a
o
p
a
F
UCH37 has
wn, yet it
ppears rath
ismantling
when Rpn13
leavage, w
ULD autoinh
ctivated up
n the prote
resence of
t the time o
Figure 3.1: D
never bee
has good a
her advanc
its proteas
3 has been
which has le
hibition thro
pon binding
easome1. E
f UbAMC is
of these exp
Domain dia
en shown to
activity aga
ced, when t
somal subs
added to U
ed others t
ough KEKE
to the entir
xamination
s a limited a
periments.
gram of UC
o cleave po
ainst UbAM
he enzyme
strate, poly
UCH37, it i
to speculat
E motif bind
re 19S regu
n of the enz
approach,
CH37 and R
olyubiquitin
MC 3,4. The
e has not b
yubiquitin.
s slightly m
te that Rpn
ing1. Ultima
ulatory part
zyme witho
but was th
Rpn13.
chains ap
e concept o
een definiti
To add to
more active
n13 relieves
ately, UCH
ticle, where
ut the 19S
e system w
ppreciably o
of autoinhib
ively capab
the confu
in polyubiq
s the effec
37 is maxim
e Rpn13 res
and only in
within our r
48
on its
bition
ble of
sion,
quitin
cts of
mally
sides
n the
each

U
c
th
b
Fa
h
a
T
a
Our g
ULD: does i
onfer its au
his ultimate
e responsib
Figure 3.2: symmetric
Herei
uman UCH
utoinhibitio
Through kin
utoinhibited
goals for th
t mediate o
utoinhibitory
ely relate to
ble for all th
Structure unit. The c
in, the kine
H37 are pr
n, activatio
netic analy
d in its full-
is compone
oligomeriza
y effect, or
o binding to
hree activiti
of human atalytic Cys
etic and b
robed, in o
on, and pr
ysis of TsU
length form
ent of the p
ation in solu
r is this a s
the protea
es.
UCH37 (Ps residue is
iophysical
order to be
roteasomal
UCH37, w
m, but desp
project were
ution? If so
separate ev
asome, as t
PDB ID 3IHs shown in y
properties
etter unders
l associatio
we find tha
ite investig
e to dissec
, does this
vent? And h
this one reg
HR) as a tyellow sphe
of TsUCH
stand the
on still elu
at the enz
ations into
ct the role o
oligomeriz
how does a
gion appea
tetramer ineres.
H37 as we
mechanism
uding the
yme is ind
the biophy
49
of the
ation
all of
ars to
n the
ell as
ms of
field.
deed
ysical

50
characteristics of the event, we are still unclear about how the enzyme transitions
between its basal and activated states.
3.2 Materials and Methods
3.2.1 Cloning, Expression, and Protein Purification
TsUCH37cat and TsUCH37cat-UbVME were purified as described
previously in Part 2.2.2. TsUCH37FL with an N-terminal 6xHis tag in pET28a+
was expressed in E. coli by Dr. Myung-Il Kim as described in Morrow et. al, 2013
10. For isothermal titration calorimetry, human Rpn13 was expressed and purified
by Dr. Judith Ronau from E. coli on glutathione beads and subsequently by size
exclusion chromatography. Human UCH37 proteins used for isothermal titration
calorimetry were wild-type and an E284A mutant (discussed further in Section
4.3.2), both expressed from a pET28a+ plasmid in E. coli, purified on Ni-NTA
beads using a 50 – 500 mM imidazole gradient (purification buffer of 20 mM
sodium phosphate pH 7.0, 300 mM NaCl, ). Due to an engineered HRV 3C
protease site, Prescission Protease (GE Biosciences) was added to remove the
6xHis tag and linker, which was subsequently removed by incubation with
glutathione beads. Second step purification was done on both wild-type and
UCH37 E284A on a Sephadex S200 size exclusion column (GE Biosciences)
and pure fractions were pooled, concentrated down, and flash frozen as aliquots.

51
3.2.2 Analytical Ultracentrifugation
TsUCH37cat and TsUCH37cat-UbVME were both dialyzed extensively
against 50 mM Tris pH 7.4, 200 mM NaCl, and 1 mM DTT. Samples were run at
concentrations a range of concentrations: 8, 16, and 32 µM for TsUCH37cat and
10, 18, and 31 µM for TsUCH37cat-UbVME to determine oligomeric states at high
concentrations. Samples were run on a Beckman-Coulter XLA analytical
ultracentrifuge at 50,000 rpm and monitored at 280 nm for 150 scans.
Sedimentation coefficient distributions were analyzed by SEDFIT (v. 13.0b) 11.
For analysis of human UCH37 and Rpn13, proteins were both extensively
dialyzed against 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP. For analysis of
individual oligomerization states, UCH37 was run at 8, 16, and 32 µM and Rpn13
was run at 13.5, 27, and 54 µM. Analysis of the UCH37-Rpn13 complex was run
at concentrations of 4 and 8 µM, 4 and 16 µM, and 4 and 32 µM of UCH37 and
Rpn13, respectively. Samples were run and analyzed by the same methods as
TsUCH37cat and TsUCH37cat-UbVME, above.
3.2.3 Ubiquitin-AMC Hydrolysis
Cleavage of 7-amino-4-methylcoumarin from the C-terminus of
monoubiquitin, or UbAMC cleavage, was monitored in a reaction buffer
containing 50 mM Tris pH 7.6, 0.5 mM EDTA, 0.1% bovine serum albumin, and 5
mM DTT. TsUCH37cat and TsUCH37FL were diluted in reaction buffer to 7 nM
final reaction concentration and preincubated at 30°C for 5 minutes prior to the
reaction. Reactions were initiated by addition of UbAMC (Boston Biochem) and

52
were measured on a Tecan fluorescence plate reader (Männedorf, Switzerland)
with 380 nm excitation wavelength and 465 nm emission wavelength at 30°C for
1 hr. Progress curves and Michaelis-Menten kinetics were plotted and fit in
SigmaPlot (Systat Software).
3.2.4 Isothermal Titration Calorimetry
For isothermal titration calorimetry, wild-type UCH37, UCH37 E284A, and
Rpn13 were dialyzed extensively together against 50 mM Tris pH 7.5, 50 mM
NaCl, 1 mM TCEP. ITC experiments were done using a MicroCal ITC200 (GE
Biosciences). For determination of the Kd of UCH37 wild-type and Rpn13
binding, two experiments were averaged together: 20 µM UCH37 in the cell with
228 µM Rpn13 injected, and 10 µM UCH37 in the cell with 100 µM Rpn13
injected. For UCH37 E284A, 10 µM E284A was in the cell and 100 µM Rpn13
was injected. The data was analyzed and fit to a single binding site model in
SEDPHAT 11.

c
C
d
c
b
Fc
fr
A
3.3.1 A
As d
rystallized
Cys71 of ea
imerization
rystallizatio
y itself was
Figure 3.3: Aompared to
rom the sam
AUC was ru
Analysis of
discussed
as a dimer
ach TsUCH
n was a b
on, the oligo
s determine
Analytical uo TsUCH37
me batch o
un on the co
Crystallogr
in Sectio
r, with dime
H37 protein
biologically
omeric state
ed by analyt
ultracentrifu7cat alone (A
f the TsUC
omplex as w
3.3 Resul
raphic Dime
on 2.3.1,
erization me
n. In order
y relevant
e of the com
tical ultrace
ugation of tA). C) Table
CH37cat-UbV
well as the
lts
erization of
the TsUC
ediated by
to determi
process o
mplex as w
entrifugation
the TsUCHe of sedime
VME compl
catalytic d
TsUCH37c
CH37cat-Ub
a disulfide
ine if TsUC
or merely
well as the c
n (AUC). Ta
H37cat-UbVMentation coe
lex used fo
omain of T
cat-UbVME
bVME com
e bond betw
CH37cat-UbV
an artifac
catalytic do
aking aliquo
ME complexefficients.
r crystalliza
TsUCH37 a
53
mplex
ween
VME
ct of
main
ots
x (B)
ation,
lone.

54
At concentrations higher than that in cells (8 – 32 µM), neither TsUCH37cat nor
the TsUCH37cat-UbVME complex were found to exist in solution as dimers. Both
are monomeric, with sedimentation coefficients (S20,w) of 3.3 for the complex and
2.8 for the catalytic domain (Fig. 3.3). Therefore, the dimerization event observed
in the crystal structure is an artifact of crystal packing, mediated by disulfide bond
formation resulting from oxidative conditions prevailing in the crystallization buffer
(glutathione additive).
3.3.2 Kinetic Characterization of TsUCH37cat and TsUCH37FL
In order to characterize the catalytic activity of TsUCH37, its activity
against a standard DUB substrate, a fluorogenic monoubiquitin derivative called
UbAMC, was assessed. The original goal of studying TsUCH37 previously was
for drug targeting12, therefore, it was of interest to examine its catalytic
mechanism compared to that of human UCH37 and the other UCH family DUBs.
Compared to the catalytic domain of human UCH37, TsUCH37cat has about a 20-
fold lower KM, indicating an improvement in substrate binding, however, the kcat
was 100-fold lower, yielding an overall 5-fold decrease in efficiency of the
enzyme (Fig 3.4) 9. It would appear that TsUCH37’s catalytic domain binds
substrate tighter, but that may also impair its ability to dissociate product for
another round of catalysis. Not surprisingly, TsUCH37cat’s KM is about 14-fold
higher than UCHL3 and 23-fold higher than that of UCHL1, both of which bind
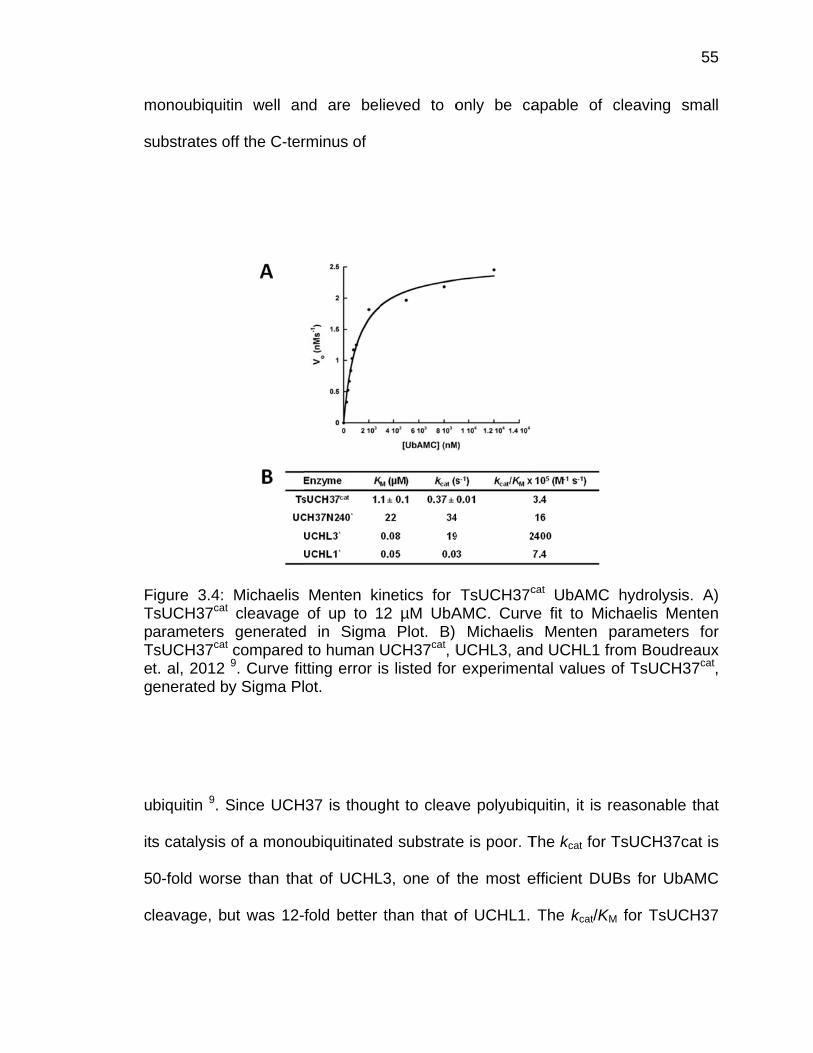
m
s
FTpTeg
u
it
5
c
monoubiquit
ubstrates o
Figure 3.4: TsUCH37cat
arameters TsUCH37cat
t. al, 2012 enerated b
biquitin 9. S
s catalysis
0-fold wors
leavage, bu
tin well an
off the C-ter
Michaelis cleavage generatedcompared 9. Curve fity Sigma Pl
Since UCH
of a monou
se than tha
ut was 12-
nd are bel
rminus of
Menten kiof up to 1
d in Sigmato human
tting error iot.
37 is thoug
ubiquitinate
at of UCHL
fold better
lieved to o
inetics for 12 µM UbAa Plot. B) UCH37cat, Us listed for
ght to cleav
ed substrate
L3, one of
than that o
only be ca
TsUCH37c
AMC. CurvMichaelis
UCHL3, anr experimen
ve polyubiq
e is poor. T
the most e
of UCHL1.
apable of
cat UbAMCve fit to Mis Menten nd UCHL1 fntal values
quitin, it is r
The kcat for
efficient DU
The kcat/K
cleaving s
C hydrolysischaelis Meparametersfrom Boudrof TsUCH3
reasonable
TsUCH37c
UBs for UbA
KM for TsUC
55
small
s. A) enten s for eaux 37cat,
e that
cat is
AMC
CH37
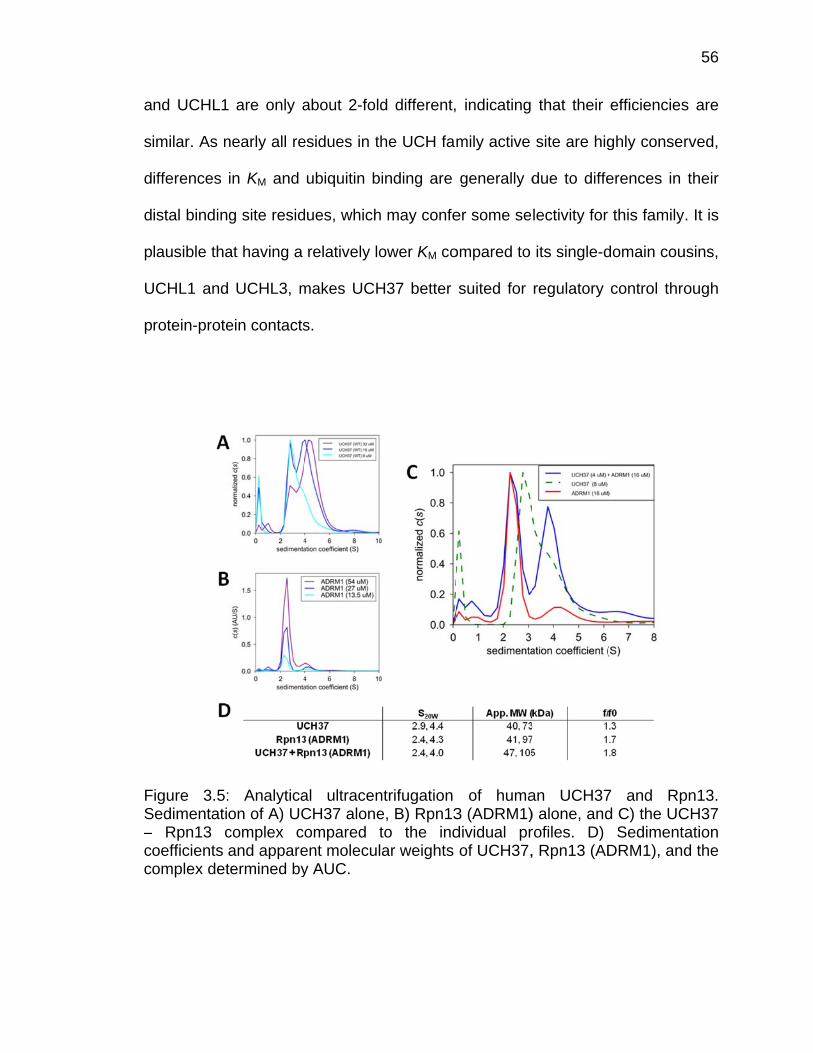
a
s
d
d
p
U
p
FS–cc
nd UCHL1
imilar. As n
ifferences
istal bindin
lausible tha
UCHL1 and
rotein-prote
Figure 3.5:Sedimentati– Rpn13 coefficients omplex det
are only a
nearly all re
in KM and
g site resid
at having a
UCHL3, m
ein contacts
: Analyticaon of A) UCcomplex cand apparetermined by
about 2-fold
esidues in t
ubiquitin b
dues, which
relatively lo
makes UCH
s.
al ultracenCH37 aloneompared tent moleculy AUC.
d different,
he UCH fam
inding are
h may confe
ower KM co
H37 better
ntrifugation e, B) Rpn13to the indar weights
indicating
mily active
generally d
er some se
ompared to
suited for
of huma3 (ADRM1)dividual prof UCH37,
that their
site are hig
due to diffe
lectivity for
its single-d
regulatory
an UCH37) alone, androfiles. D) , Rpn13 (AD
efficiencies
ghly conser
erences in
this family
domain cou
control thro
7 and Rpd C) the UC
SedimentDRM1), and
56
s are
rved,
their
. It is
usins,
ough
pn13. CH37 ation d the

57
3.3.3 Analysis of UCH37 Oligomeric State
In order to probe whether the previously proposed model of UCH37
tetramerization or higher order oligomerization was possible, we examined its
oligomerization by analytical ultracentrifugation (Fig 3.5) 5. Additionally, the
stoichiometry of the binding of UCH37 to Rpn13, its proteasomal binding partner,
was determined. Analytical ultracentrifugation of UCH37 alone at 8, 16, and 32
µM yielded data indicating that the enzyme primarily exists as a monomer at
lower concentrations, but is capable of a concentration-dependent rapid
monomer-dimer equilibrium, which is seen most prominently in the 32 µM
concentration sample. Higher order oligomers were not detected at that those
concentrations, which does not rule out the possibility, but indicates that at
cellular concentrations, the enzyme is likely monomeric.
As for the UCH37-Rpn13 complex, first the solution state of Rpn13 was
determined alone at 13.5, 27, and 54 µM. Rpn13 primarily exists as a monomer
with a small population of higher order oligomers or aggregates, however this
proportion is quite small. The UCH37-Rpn13 complex was run at three different
concentrations of Rpn13 (8, 16, and 32 µM), but with UCH37 fixed at 4 µM. The
complex exists in a 1:1 stoichiometry, which is not a surprise given that Rpn13
only has one recognition motif for UCH37 to bind. These results do not appear to
support the theory that Rpn13 may relieve UCH37 of its autoinhibition through
binding its ULD to change the oligomeric state of the enzyme. Both proteins are
predominantly monomeric in solution and form a 1:1 complex at the proteasome.

k
th
cy
tw
re
p
h
w
3
FoR
Ultim
nowledge o
he binding
ysteine DU
wo ubiquitin
elationship
urified full-
ave looked
were studied
.6). The dis
Figure 3.6: f binding c
Rpn13. C) T
3.3.
ately, the
of UCH37
affinities o
UB that ass
n receptors
with Rpn1
-length hum
d at the ye
d for their b
ssociation c
Isothermal urves of A)
Table of the
.4 Analysis
most imp
is its bindi
of the pro
ociates wit
s, Rpn10 an
3 has bee
man UCH3
ast homolo
binding to R
constant wa
titration ca) wild-type rmodynam
of UCH37
portant biop
ng to Rpn1
teasome s
th the prote
nd Rpn13,
n less exp
7 and full-
ogs. For ou
Rpn13: wild
as determin
alorimetry oUCH37 anic paramete
Binding to
physical p
13. Previou
subunit Rp
easome, as
for ubiquiti
plored. For
-length hum
ur experime
-type and a
ned by aver
of UCH37 and Rpn13 aers.
Rpn13
arameter m
us studies
n1 for US
s well as th
in13-15. How
this experi
man Rpn13
ents, two U
a ULD muta
raging two e
and Rpn13and B) UCH
missing in
have exam
P14, the o
he affinity o
wever, UCH
iment, we
3; most stu
UCH37 pro
ant, E284A
experiment
binding. HH37 E284A
58
our
mined
other
of the
H37’s
used
udies
teins
A (Fig
ts for
Heats A and

59
wild-type, one with 20 µM UCH37 in the cell and 228 µM Rpn13 as the titrant,
and a second with 10 µM UCH37 in the cell and 100 µM Rpn13 as the titrant.
The average Kd was 22 ± 6 nM. UCH37 E284A was only run as a single
experiment, with 10 µM E284A in the cell and 100 µM Rpn13 injected, which
yielded a Kd of 18.5 ± 7 nM. Although UCH37 in cells is known to exist as a
population of free enzyme, not bound to the 26S proteasome and can associate
with the Ino80 chromatin remodeling complex, these dissociation constants
suggest very tight binding between this DUB and its proteasomal anchor, Rpn13
2. This interaction is known to be abolished upon deletion of UCH37’s KEKE
motif, and it is clear that even though the ULD mutation E284A impairs activation
of the enzyme (Section 4.3.2), the ULD region likely does not contribute
significantly to binding to Rpn13 1,6-8. Additionally, as the Kd of UCH37-Rpn13
binding is so low, it further disproves the possibility that UCH37 oligomerizes in
cells.
3.4 Discussion
Thorough characterization of UCH37’s kinetic and biophysical properties
is necessary to dissect its cellular association with the 26S proteasome and
potential autoinhibition. These studies have shed more light on the role of its ULD
in catalysis and binding, but more work is still needed to understand its
activation. We have confirmed that dimerization of the TsUCH37cat-UbVME
complex in its crystal structure is a crystallographic artifact of tight packing. Our
studies of the kinetic activities of TsUCH37cat and TsUCH37FL, and our analysis

o
p
c
c
U
(T
b
a
Tb
m
u
f the oligo
revious lite
ellular conc
omparing a
UCH37 bind
Table 3.1).
e a const
ssociating
Table 3.1: inding part
measured 1
ndoubtedly
omerization
erature, in th
centrations
activity to th
ds to Rpn1
This data s
itutive mem
subunit. Its
Dissociationers. Value
12 nM diss
y constituti
state of h
hat we do n
, but we sti
he catalytic
3 in a 1:1
seems to in
mber of th
s binding is
on constantes for * are
sociation c
ve membe
human UCH
not see dim
ll observe t
c domain al
ratio, and
ndicate tha
he 26S pro
within rang
ts of protefrom ref 13
constant fo
ers18. In c
H37, both
merization o
the autoinh
lone 5,9,16,17
with a 22
at UCH37, i
oteasome
ge of the pre
easome-asswhile those
or the Rpn
comparison
support an
of the full-le
hibition phe
7. We have
nM dissoc
n contrast
rather tha
eviously
sociated De under # ar
n2-Rpn13 i
n, the yea
nd conflict
ngth enzym
nomenon w
e confirmed
ciation con
to USP14,
n a transi
DUBs and re from ref
interaction,
ast homolo
60
with
me at
when
d that
stant
may
ently
their 18.
two
og of

61
USP14, Ubp6, has two possible binding sites on Rpn1, one a tighter 62 nM Kd
site, the other much weaker at nearly 2 µM13. Rpn1 is known to bind to shuttle
factors and other Ubl domain-containing proteins, therefore, USP14 is not always
bound to it. These numbers would suggest that UCH37 is more frequently found
in a proteasomal context than USP14 and may play a more significant biological
role.
However, these investigations still leave open the question of how UCH37
is activated at the proteasome, if it occurs merely through association with a
conformationally-accessible Rpn13, or if another binding partner is required. A
UCH37 mutant, E284A, which could not be activated by Rpn13 during ubiquitin-
AMC hydrolysis (Section 4.3.2) bound to Rpn13 with nearly the same Kd as the
wild-type enzyme. This mutation isolates Rpn13’s activation of UCH37 to an
event independent of simple binding. Further studies of this mutant in the
presence of di- or tri-ubiquitin, as well as in the presence of Rpn2, the
proteasomal subunit which binds Rpn13’s N-terminus, may provide the key to
UCH37’s mode of activation.

62
3.5 References
1 Yao, T. et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nature cell biology 8, 994-1002 (2006).
2 Yao, T. et al. Distinct modes of regulation of the Uch37 deubiquitinating
enzyme in the proteasome and in the Ino80 chromatin-remodeling complex. Molecular cell 31, 909-917 (2008).
3 Lam, Y. A., DeMartino, G. N., Pickart, C. M. & Cohen, R. E. Specificity of
the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. The Journal of biological chemistry 272, 28438-28446 (1997).
4 Lam, Y. A., Xu, W., DeMartino, G. N. & Cohen, R. E. Editing of ubiquitin
conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737-740 (1997).
5 Burgie, S. E., Bingman, C. A., Soni, A. B. & Phillips, G. N., Jr. Structural
characterization of human Uch37. Proteins (2011). 6 Qiu, X. B. et al. hRpn13/ADRM1/GP110 is a novel proteasome subunit
that binds the deubiquitinating enzyme, UCH37. The EMBO journal 25, 5742-5753, doi:10.1038/sj.emboj.7601450 (2006).
7 Hamazaki, J. et al. A novel proteasome interacting protein recruits the
deubiquitinating enzyme UCH37 to 26S proteasomes. The EMBO journal 25, 4524-4536 (2006).
8 Koulich, E., Li, X. & DeMartino, G. N. Relative structural and functional
roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Molecular biology of the cell 19, 1072-1082 (2008).
9 Boudreaux, D. A., Chaney, J., Maiti, T. K. & Das, C. Contribution of active
site glutamine to rate enhancement in ubiquitin C-terminal hydrolases. The FEBS journal 279, 1106-1118 (2012).
10 Morrow, M. E. et al. Stabilization of an Unusual Salt Bridge in Ubiquitin by
the Extra C-Terminal Domain of the Proteasome-Associated Deubiquitinase UCH37 as a Mechanism of Its Exo Specificity. Biochemistry, doi:10.1021/bi4003106 (2013).

63
11 Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical journal 78, 1606-1619 (2000).
12 White, R. R. et al. Characterisation of the Trichinella spiralis
deubiquitinating enzyme, TsUCH37, an evolutionarily conserved proteasome interaction partner. PLoS Negl Trop Dis 5, e1340 (2011).
13 Chen, X., Lee, B. H., Finley, D. & Walters, K. J. Structure of proteasome
ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Molecular cell 38, 404-415 (2010).
14 Riedinger, C. et al. Structure of Rpn10 and its interactions with
polyubiquitin chains and the proteasome subunit Rpn12. The Journal of biological chemistry 285, 33992-34003, doi:10.1074/jbc.M110.134510 (2010).
15 Husnjak, K. et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor.
Nature 453, 481-488 (2008). 16 Zhou, Z. R., Zhang, Y. H., Liu, S., Song, A. X. & Hu, H. Y. Length of the
active-site crossover loop defines the substrate specificity of ubiquitin C-terminal hydrolases for ubiquitin chains. The Biochemical journal 441, 143-149 (2012).
17 Jiao, L. et al. Mechanism of the Rpn13-induced activation of Uch37.
Protein & cell 5, 616-630, doi:10.1007/s13238-014-0046-z (2014). 18 Rosenzweig, R., Bronner, V., Zhang, D., Fushman, D. & Glickman, M. H.
Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. The Journal of biological chemistry 287, 14659-14671, doi:10.1074/jbc.M111.316323 (2012).

64
CHAPTER 4: ANALYSIS OF THE TSUCH37∆C46-UBVME STRUCTURE AND THE ROLE OF THE ULD
4.1 Introduction
Although the TsUCH37cat-UbVME structure provided valuable insight into
the mechanism of UCH37 and its ability to recognize and bind monoubiquitin, we
are still lacking information about the role of the ULD in catalysis, binding, and
activation. However, another group member, Dr. Myung-Il Kim, was able to
crystallize and solve the structure of a longer construct of TsUCH37 in complex
with UbVME for us to glean information regarding the ULD, hereafter referred to
as TsUCH37∆C46-UbVME (Fig 4.1). Due to cleavage of the protein during
purification, only a portion of the ULD was shown in the structure, but it provided
important clues regarding ubiquitin recognition by the enzyme. Contacts between
the catalytic domain of TsUCH37∆C46 and ubiquitin are identical to that of
TsUCH37cat, including a lack of ordered density for the crossover loop residues.
However, contacts between ubiquitin and the ULD are seen, making this an
additional ubiquitin binding interface. These contacts involve hydrogen bonds
and salt bridge interactions between TsUCH37’s Arg261 and Tyr262 with
ubiquitin’s Gln49 and Lys48, which forces a salt bridge interaction between

Fw
L
b
U
w
b
s
L
w
T
p
Figure 4.1: with the ULD
ys48 and G
ound struc
UCH37 beha
works its wa
elieve that
pecificity; U
ys48 will n
with the ULD
This mecha
olyubiquitin
Structure oD.
Glu51 of ub
ctures in th
aves exosp
ay inward
the TsUC
UCH37 is o
ot be enga
D. This rec
anism expl
n, however
of TsUCH3
biquitin, an
he PDB (T
pecifically, t
to the prox
CH37∆C46-Ub
only capab
aged in an
cognition of
lains the e
r, it does n
37∆C46-UbV
event here
Table 4.1).
that it only c
ximal mono
bVME acco
ble of cleav
isopeptide
Lys48 wou
enzyme’s
not provide
VME with in
etofore unse
It has bee
cleaves fro
omer of a
ounts for th
ving the dis
bond and
uld only pe
specificity
e structural
nset of Lys
een in any
en previou
om the dista
polyubiquit
he structur
stal monom
will be free
ermit exosp
in cleavin
clues as
s48 interac
other ubiq
usly shown
al monomer
tin chain 1
ral basis of
mer becaus
e for intera
ecific cleav
ng Lys48-li
to its abili
65
ctions
uitin-
that
r and
. We
f this
se its
cting
vage.
nked
ty to

c
u
Te
leave other
tilizes addit
Table 4.1: Lntries indic
r chain top
tional intera
Lys48-Glu5cate multiple
pologies. Th
actions by t
51 distancee copies of
his chapter
the ULD to
es in ubiquUb in the c
r explores t
confer spe
uitin-bound crystallogra
the possibi
cificity and
PDB strucaphic asymm
lity that UC
bind ubiqu
ctures. Mumetric unit.
66
CH37
itin.
ultiple

67
4.2 Materials and Methods
4.2.1 Molecular Dynamics Simulations
A starting model of human UCH37-UbVME was made by modeling of full-
length TsUCH37 using the human UCH37 structure (PDB ID 3IHR) as the search
model in the SwissModel homology modeling server 2. The final 46 residues
missing in the TsUCH37∆C46-UbVME were appended from the homology model
in Coot, which then underwent one round of refinement in Phenix 3,4. Professor
Markus Lill (Purdue University) then utilized this model for molecular dynamics
simulations, methods described in Morrow et. al, 2013 5. From the 2 ns
simulation, snapshots were examined for specific residues’ proximities to
ubiquitin. The majority of potential interactions could be seen at the 1.3 ns
snapshot.
4.2.2 Site-directed Mutagenesis and Protein Purification
Based on interactions seen in the MD simulations described above, a list
of Ub-interacting ULD residues were generated from the Ts enzyme and
corresponding residues in the human enzymes were mutated to Ala. Site-
directed mutagenesis was performed using the AccuPower PCR PreMix
(Bioneer) and mutations were confirmed by sequencing. Proteins were
expressed in Rosetta2 DE3 E. coli expression cells and purified by Ni NTA
beads. After cleavage of the 6xHis tag by Prescission Protease (GE
Biosciences), proteins were passed over GSH beads to remove the tag and

68
protease. Proteins were further purified by size-exclusion chromatography on a
Superdex 200 HiLoad column (GE Biosciences). Pure fractions were pooled,
concentrated down, and flash frozen as aliquots. Concentrations were
determined by UV/Vis. Human Rpn13FL was provided by Dr. Judith Ronau.
4.2.3 Ubiquitin-AMC Hydrolysis Assays
Cleavage of 7-amino-4-methylcoumarin from the C-terminus of
monoubiquitin, or UbAMC cleavage, was monitored in a reaction buffer
containing 50 mM Tris pH 7.6, 0.5 mM EDTA, 0.1% bovine serum albumin, and 5
mM DTT. UCH37 wild-type and mutants were pre-incubated with Rpn13 on ice
for 1 hr, and then diluted in reaction buffer to final reaction concentrations of 0.5
nM and 15 nM, respectively, and warmed to 30°C for 5 minutes prior to the
reaction. Reactions were initiated by addition of UbAMC (Boston Biochem) and
were measured on a Tecan fluorescence plate reader (Männedorf, Switzerland)
with 380 nm excitation wavelength and 465 nm emission wavelength at 30°C for
1 hr. Progress curves were plotted in Kaleidagraph.

69
4.2.4 Synthesis of Asymmetric Triubiquitin Substrate and Assays
Site-directed mutagenesis was used to introduce a Gly76Val mutation into
a UbW77 construct in the pGEX-6P-1, which was subsequently confirmed by
sequencing. The double mutant UbG76V W77 was expressed in Rosetta2 DE3 E.
coli cells and purified on glutathione beads. After treatment with Prescission
Protease (GE Biosciences) to remove its N-terminal GST tag, the protein was run
back over glutathione beads to remove the tag and protease. UbG76V W77 was
further purified by size exclusion chromatography on a Sephadex 75 HiLoad
column (GE Biosciences) and pure fractions were pooled, concentrated down,
and flash frozen as aliquots.
Untagged ubiquitin in the pRSET vector was expressed in Rosetta2 DE3
E. coli cells, spun down, resuspended in purification buffer A (50 mM sodium
acetate pH 4.5, 2 mM DTT), lysed by French press, heated to 80°C for 10
minutes, then spun down at 30,000xg for 30 minutes. The supernatant was
brought to pH 4.5 by 1N HCl and then was purified by cation exchange
chromatography on SP sepharose beads (GE Biosciences) by gradient elution
with purification buffer B (same as A, but with 1 M NaCl). Pure fractions were
pooled and concentrated down, then further buffer exchanged and purified by
size exclusion chromatography on a Sephadex 75 HiLoad column (GE
Biosciences) into 50 mM Tris pH 7.6, 50 mM NaCl, 1 mM DTT. Pure fractions
were pooled, concentrated down, and flash frozen as aliquots.
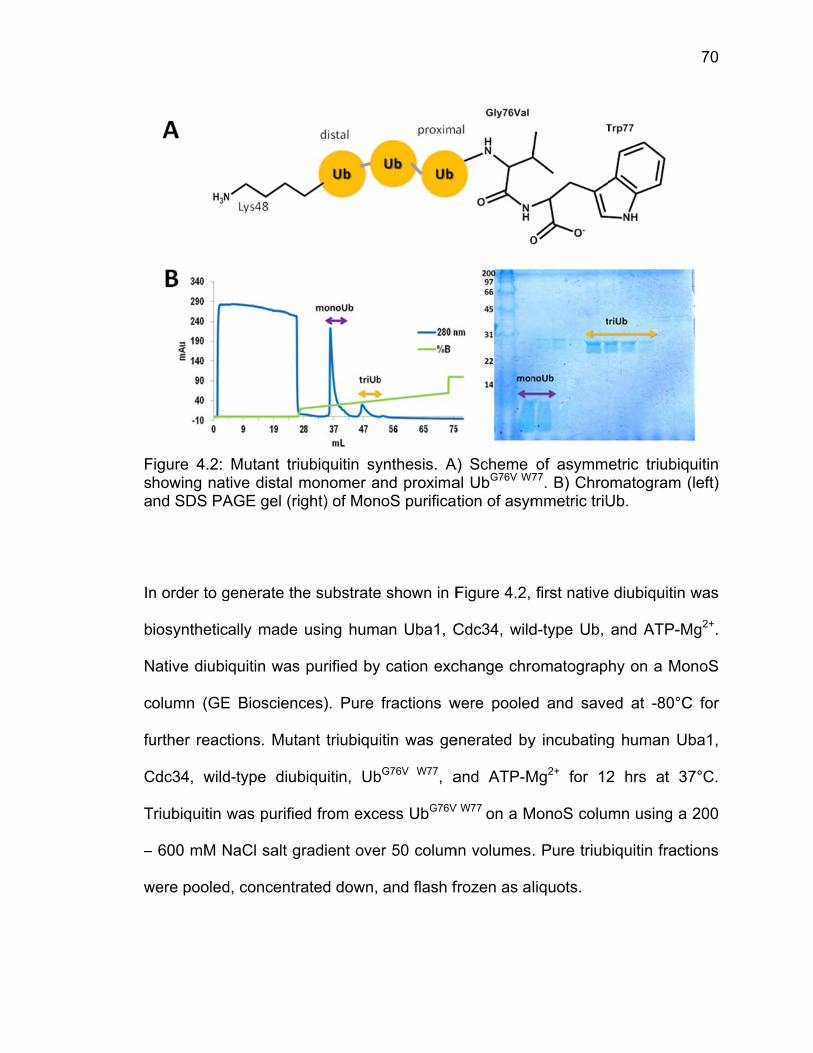
Fsa
In
b
N
c
fu
C
T
–
w
Figure 4.2: howing natnd SDS PA
n order to g
iosynthetic
Native diubi
olumn (GE
urther react
Cdc34, wild
Triubiquitin w
– 600 mM N
were pooled
Mutant triutive distal mAGE gel (rig
generate the
ally made
quitin was
E Bioscienc
tions. Muta
d-type diub
was purified
NaCl salt gr
d, concentra
ubiquitin symonomer aght) of Mon
e substrate
using hum
purified by
ces). Pure
ant triubiqui
biquitin, Ub
d from exce
radient ove
ated down,
ynthesis. And proximaoS purifica
e shown in F
an Uba1, C
cation exc
fractions w
itin was ge
bG76V W77, a
ess UbG76V
r 50 colum
and flash f
A) Scheme al UbG76V W7
tion of asym
Figure 4.2,
Cdc34, wild
change chro
were pooled
enerated by
and ATP-M
V W77 on a M
mn volumes
frozen as a
of asymm77. B) Chrommetric triU
first native
d-type Ub,
omatograp
d and save
y incubating
Mg2+ for 12
MonoS colu
. Pure triub
liquots.
etric triubiqomatogram Ub.
e diubiquitin
and ATP-M
hy on a Mo
ed at -80°C
g human U
2 hrs at 3
mn using a
biquitin frac
70
quitin (left)
n was
Mg2+.
onoS
C for
Uba1,
37°C.
a 200
ctions

71
For polyubiquitin cleavage assays, wild-type K48-linked di-, tri-, and tetra-
ubiquitin were generated biosynthetically in the same manner as mutant tri
UbG76V W77. 1.5 µM wild-type or E284A UCH37 was incubated for 1 hour on ice
with 50 µM GST-Rpn13 (for triUbG76V W77 assays) or 5 µM untagged Rpn13 (for
wild-type polyubiquitin assays) in buffer containing 50 mM Tris pH 7.6, 0.5 mM
EDTA, 0.1% bovine serum albumin, and 5 mM DTT. Reactions were started with
the addition of 15 µM triUbG76V W77, tetraubiquitin, wild-type triubiquitin, or
diubiquitin and time points were quenched with SDS PAGE buffer. Reactions
were run on 15% SDS PAGE gels and stained with Coomassie.
4.3 Results
4.3.1 Analysis of Molecular Dynamics Simulations
After Professor Markus Lill (Purdue University) generated a 2 ns molecular
dynamics simulation for full-length TsUCH37-UbVME, each frame was analyzed
for potential interactions between the ULD of TsUCH37, the mobile element, and
ubiquitin, which was held stationary. The majority of interactions were seen at a
1.3 ns snapshot, showing potential interactions between many ubiquitin-facing
ULD residues and ubiquitin (Fig. 4.3). Although some interactions are only within
van der Waals or salt bridge distances, some possible hydrogen bonds were also
observed. The majority of the residues at this interface are also highly conserved
from yeast up to human UCH37, therefore, the most conserved residues were

Fcyhh
d
in
d
m
Figure 4.3: yan, grey, ighlight intighlighted i
deemed to
ntroduced in
etermine if
more likely,
Molecular and orang
eractions fn red.
be candida
n a handful
f these resid
its exospec
dynamics sge). TsUCHfrom a 1.3
ates for solu
of these h
dues are re
cificity.
simulationsH37∆C46-Ub
ns snapsh
ution studie
ighly-conse
esponsible
s of full-lenbVME is shhot, with h
es. Single p
erved residu
for UCH37
ngth TsUCHhown in olhighly cons
point mutati
ues (Table
7’s activatio
H37-UbVMlive. (i) andserved resi
ons to Ala w
4.2) in orde
on by Rpn1
72
E (in d (ii) dues
were
er to
3 or,

Tw
a
sy
p
b
w
w
a
c
m
Table 4.2. Uwere unable
To as
nd cleave
ystem exa
resence an
inding and
would abrog
were pre-in
ssociation
omplexes
monitored a
ULD mutatioe to be purif
4.3.2 Ubiq
ssess the e
ubiquitin c
mining the
nd absence
activation
gate activa
cubated w
of the two
were warm
nd plotted a
ons introdufied, due to
uitin-AMC
effects of U
hains withi
e efficiency
e of Rpn13
of the enz
ation. For th
with 30-fold
o proteins.
med to 30°
as progress
uced into hu poor solub
Hydrolysis
ULD mutati
n the prote
y of these
3. If ULD in
zyme, pres
he UbAMC
d excess R
Immediate
°C in reac
s curves, sh
uman UCHbility or insta
by UCH37
ons on the
easome, w
mutants in
nteractions
sumably mu
C assay, U
Rpn13 at
ly prior to
ction buffer
hown in Fig
37. Mutatioability.
ULD Muta
e ability of
we utilized a
n cleaving
are neces
utations wi
LD mutatio
4°C for 1
addition of
r. UbAMC
gure 4.4.
ons listed w
nts
UCH37 to
a more min
UbAMC in
ssary for Rp
thin this re
ons (Table
hour to a
f substrate
hydrolysis
73
with *
bind
nimal
n the
pn13
egion
4.2)
allow
, the
was

FU
U
w
c
m
m
to
e
R
d
Figure 4.4: UCH37 with
All U
UbAMC hyd
with previou
hange6,7. T
mutation ha
merely catal
o human U
ither in bin
Rpn13, or b
issect its r
UbAMC h and withou
LD mutant
drolysis ap
usly publish
The lack of
as somehow
lytic activat
CH37, whic
nding and/o
binding and
ole within t
hydrolysis but Rpn13.
ts assayed
proximately
ed activatio
activation
w abolishe
ion of the e
ch would in
or catalysis
d/or deactiv
the context
by UCH37
were activ
y 2-fold, ex
on of wild-t
of E284A
ed either R
enzyme. E2
ndicate tha
s of polyub
vation to pa
t of Rpn13
ULD mut
vated in th
xcept E284
type UCH37
was intrigu
Rpn13’s ab
284 is highly
t it must ha
biquitin, bin
artners with
3 activation
tants. Prog
he presenc
4A. These
7 in the ran
uing, thoug
ility to bind
y conserve
ave functio
nding and/o
hin the Ino8
and bindin
gress curve
e of Rpn13
results co
nge of a 10
h, because
d to UCH3
d from yea
nal signific
or activatio
80 complex
ng, we pur
74
es of
3 for
onflict
0-fold
e this
37 or
st up
ance
on by
x. To
sued

75
isothermal titration calorimetry (ITC) to determine the binding affinity of E284A
with Rpn13 compared to wild-type UCH37. The results, shown in Section 3.3.4,
indicate that binding to Rpn13 is not impaired by this mutation as its Kd is close to
that of wild-type UCH37. Therefore, this mutation may specifically inhibit the
mechanism of activation of UCH37 by Rpn13, specifically.
Interestingly, ULD mutations near E284 do not impair activation within the
context of the UbAMC assay, such as R280 and Y281. These two residues are
not as conserved as E284; R280 is substituted with Met, Lys, or Leu, and Y281 is
replaced by Trp in lower organisms. Perhaps these residues are more important
for ubiquitin recognition, rather than activation.
4.3.3 Triubiquitin Cleavage by ULD Mutants
In order to assess directional cleavage by UCH37, an asymmetric
polyubiquitin substrate was needed. To this end, UbW77, a construct utilized for
studies of the activity of UCHL1, was given an additional mutation, Gly76Val, by
site-directed mutagenesis in order to render its Trp77 non-cleavable by UCH37 8.
This double mutant monomer can be detected by an HPLC/MS assay due
to changes in its biochemical properties: 1) increased hydrophobicity and 2)
increased molecular weight. If ULD mutations abrogated exospecificity, equal
proportions of Ubwt and UbG76V W77 would be cleaved from either end of the
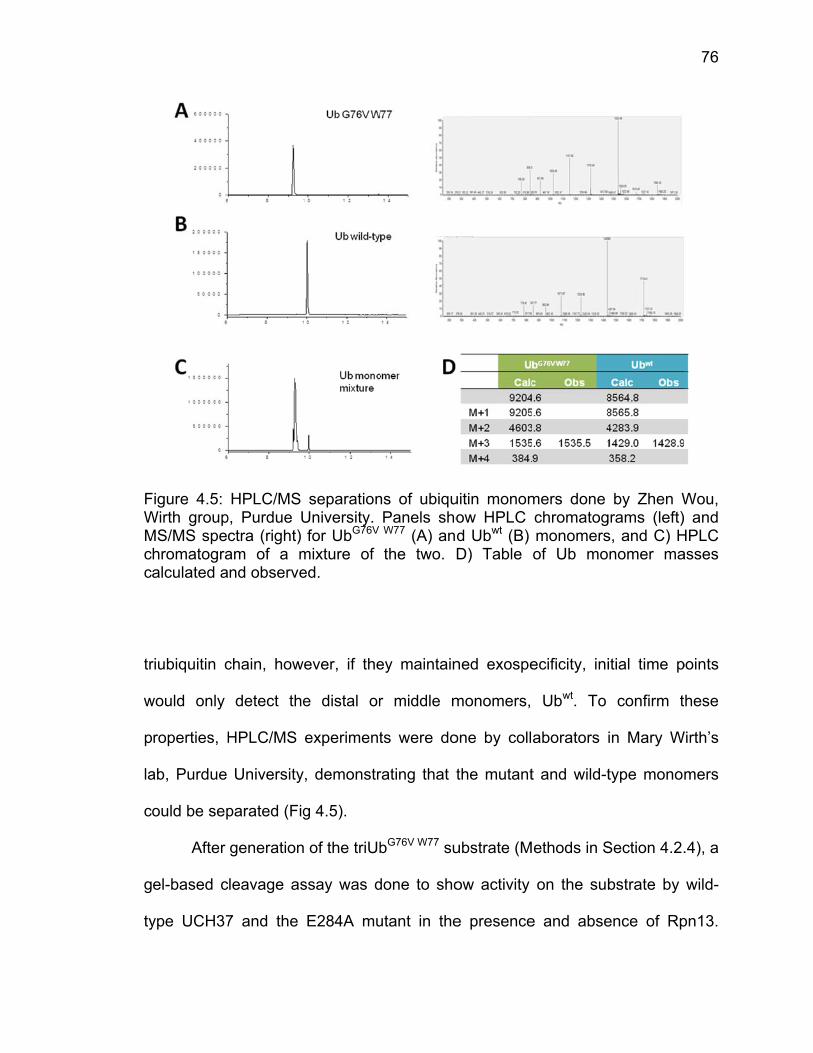
FWMcc
tr
w
p
la
c
g
ty
Figure 4.5: Wirth groupMS/MS spechromatograalculated a
riubiquitin c
would only
roperties, H
ab, Purdue
ould be sep
After
el-based c
ype UCH37
HPLC/MS p, Purdue Uctra (right) am of a mnd observe
chain, how
detect the
HPLC/MS
University
parated (Fig
generation
leavage as
7 and the
separationUniversity. for UbG76V
mixture of ed.
ever, if the
e distal or
experiment
, demonstr
g 4.5).
of the triUb
ssay was d
E284A mu
ns of ubiquPanels shoW77 (A) anthe two.
ey maintain
r middle m
ts were do
rating that t
bG76V W77 su
done to sho
utant in the
uitin monomow HPLC d Ubwt (B) D) Table
ned exospe
monomers,
one by coll
the mutant
ubstrate (M
ow activity
e presence
mers done chromatogmonomersof Ub mo
ecificity, ini
Ubwt. To
aborators i
t and wild-t
ethods in S
on the sub
e and abse
by Zhen Wgrams (left)s, and C) Hnomer ma
itial time p
confirm t
in Mary W
type monom
Section 4.2.
bstrate by
ence of Rp
76
Wou, and
HPLC asses
points
hese
Wirth’s
mers
.4), a
wild-
pn13.

77
Interestingly, Rpn13 appears to slow processing of triUb for both the wild-type
and E284A UCH37 (Fig 4.6). In the presence of Rpn13, almost no monoubiquitin
is generated by the two hour timepoint but the monoubiquitin band at the two
hour timepoint for UCH37 alone is about eight times more intense. Additionally,
after two hours, UCH37 + Rpn13 still has a significant amount of triubiquitin to
cleave, whereas UCH37 alone has cleaved almost all of its triubiquitin down to
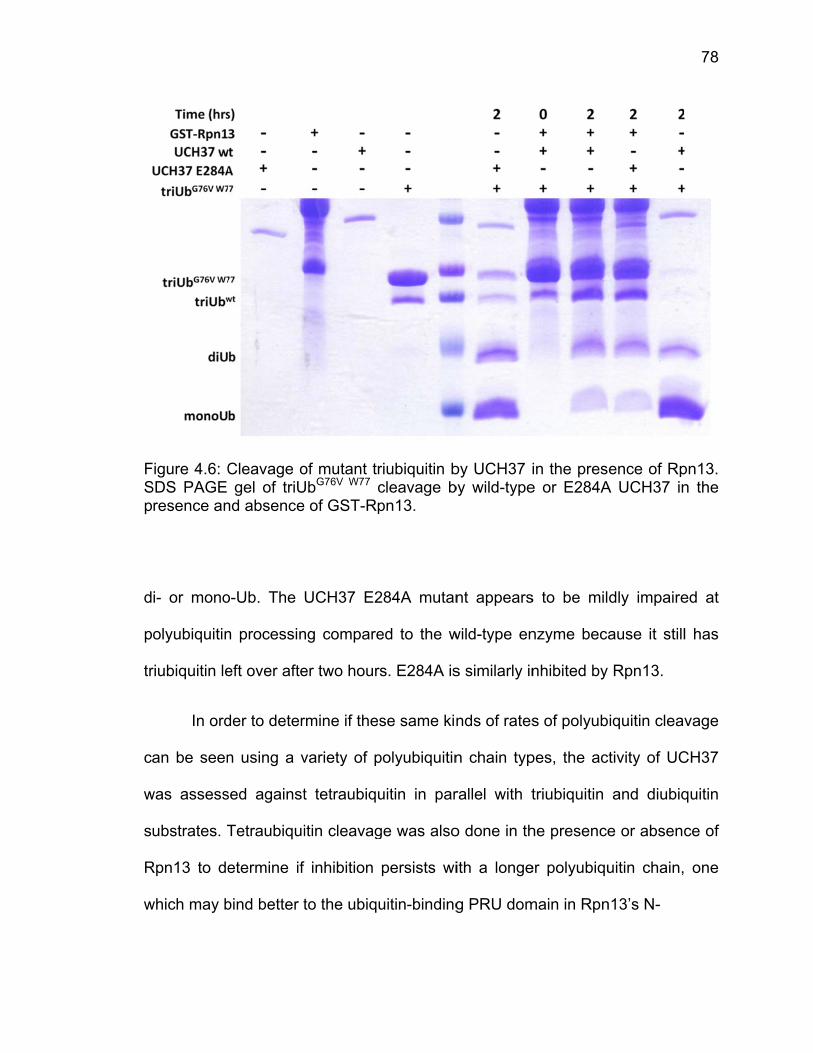
FSp
d
p
tr
c
w
s
R
w
Figure 4.6: CSDS PAGE
resence an
i- or mono
olyubiquitin
riubiquitin le
In ord
an be seen
was assess
ubstrates. T
Rpn13 to de
which may b
Cleavage ogel of triU
nd absence
o-Ub. The
n processin
eft over afte
der to deter
n using a v
sed against
Tetraubiqui
etermine if
bind better t
of mutant trUbG76V W77 ce of GST-Rp
UCH37 E2
ng compare
er two hours
rmine if thes
variety of p
t tetraubiqu
itin cleavag
f inhibition
to the ubiqu
riubiquitin bcleavage bpn13.
284A mutan
ed to the w
s. E284A is
se same kin
polyubiquitin
uitin in par
ge was also
persists wi
uitin-binding
by UCH37 iby wild-type
nt appears
wild-type en
s similarly in
nds of rates
n chain typ
rallel with t
o done in th
ith a longe
g PRU dom
in the prese or E284A
s to be mil
nzyme beca
nhibited by
s of polyub
pes, the ac
triubiquitin
he presence
er polyubiqu
main in Rpn
ence of RpA UCH37 in
dly impaire
ause it still
Rpn13.
iquitin cleav
tivity of UC
and diubiq
e or absenc
uitin chain,
n13’s N-
78
pn13. n the
ed at
l has
vage
CH37
quitin
ce of
one
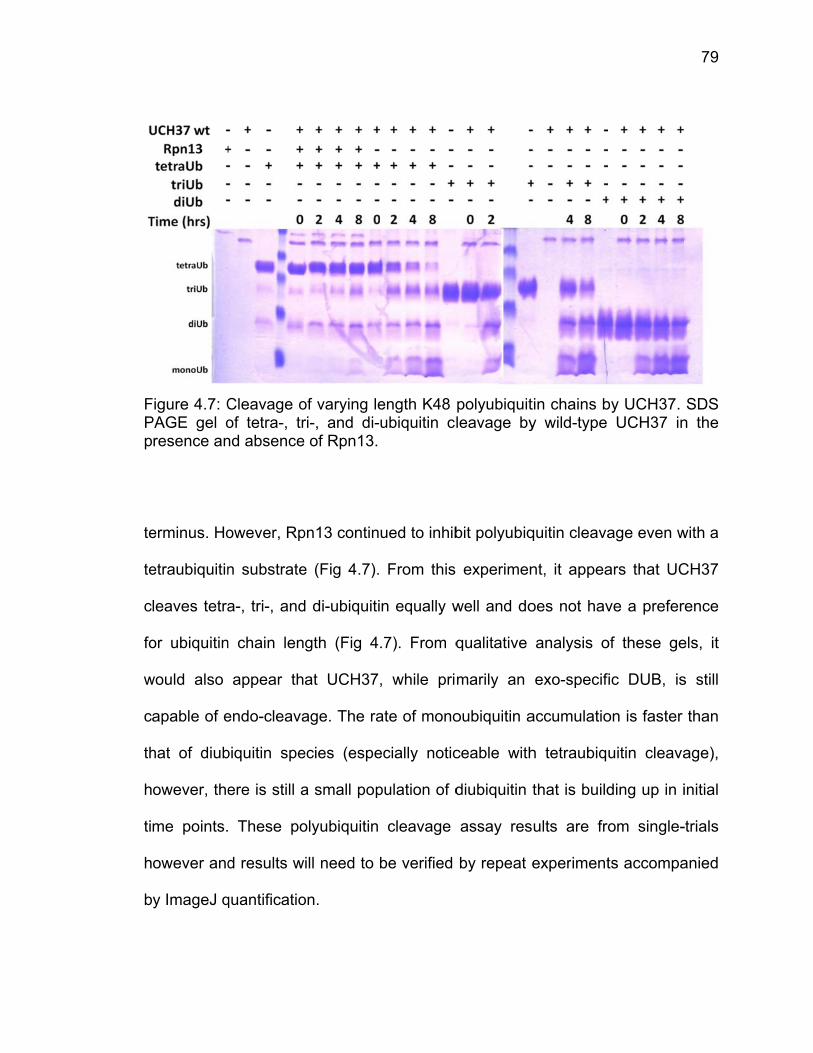
FPp te
te
c
fo
w
c
th
h
ti
h
b
Figure 4.7: CPAGE gel o
resence an
erminus. Ho
etraubiquitin
leaves tetra
or ubiquitin
would also
apable of e
hat of diub
owever, the
me points.
owever and
y ImageJ q
Cleavage oof tetra-, trnd absence
owever, Rp
n substrate
a-, tri-, and
n chain len
appear th
endo-cleava
biquitin spe
ere is still a
These po
d results w
quantificatio
of varying leri-, and di-
e of Rpn13.
pn13 contin
e (Fig 4.7).
d di-ubiquiti
gth (Fig 4
at UCH37,
age. The ra
ecies (espe
a small pop
olyubiquitin
ill need to b
on.
ength K48 ubiquitin cl
ued to inhib
From this
n equally w
.7). From q
, while pri
ate of mono
ecially notic
pulation of d
cleavage
be verified
polyubiquitleavage by
bit polyubiq
experimen
well and do
qualitative
marily an
oubiquitin a
ceable with
diubiquitin t
assay res
by repeat e
tin chains by wild-type
quitin cleava
nt, it appea
oes not hav
analysis o
exo-specif
accumulatio
h tetraubiqu
that is build
ults are fro
experiment
by UCH37. UCH37 in
age even w
ars that UC
ve a prefer
of these ge
ic DUB, is
on is faster
uitin cleava
ding up in i
om single-
ts accompa
79
SDS n the
with a
CH37
rence
els, it
s still
than
age),
initial
-trials
anied

80
4.4 Discussion
We have analyzed the contribution of UCH37’s ULD and found it to
provide 1) exo-specificity through binding to ubiquitin’s Lys48 and 2) a means of
activation of the enzyme through interactions with Rpn13. After analysis of
ubiquitin-AMC cleavage by ULD mutants in the presence and absence of Rpn13,
we have identified Glu284 as a critical regulator of Rpn13’s activation, in that
when it is mutated to Ala, activation is lost.
We have generated a novel polyubiquitin for the study of directional-
specific cleavage, triUbG76V W77, which allows detection of a monoubiquitin variant
by differences in molecular weight, hydrophobicity, and molar absorptivity. We
have not utilized this triubiquitin for exo-specificity assays yet, but have analyzed
UCH37’s ability to process tetra-, tri-, and di-ubiquitin in the presence and
absence of Rpn13. It initially appears that Rpn13 slows polyubiquitin cleavage by
UCH37, which has been noted by others but not fully explored9. The investigation
into the mechanism of activation of UCH37 has yet to be completely exhausted,
especially as it pertains to polyubiquitin cleavage. However, our novel substrate
has broader uses for other directional-specific deubiquitinating enzymes that
have sufficient rates of polyubiquitin cleavage and may be a useful tool within this
field.

81
4.5 References
1 Lam, Y. A., DeMartino, G. N., Pickart, C. M. & Cohen, R. E. Specificity of the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. The Journal of biological chemistry 272, 28438-28446 (1997).
2 Kiefer, F., Arnold, K., Kunzli, M., Bordoli, L. & Schwede, T. The SWISS-
MODEL Repository and associated resources. Nucleic acids research 37, D387-392 (2009).
3 Emsley, P. & Cowtan, K. Coot: model-building tools for molecular
graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126-2132 (2004).
4 Adams, P. D. et al. PHENIX: a comprehensive Python-based system for
macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography 66, 213-221 (2010).
5 Morrow, M. E. et al. Stabilization of an Unusual Salt Bridge in Ubiquitin by
the Extra C-Terminal Domain of the Proteasome-Associated Deubiquitinase UCH37 as a Mechanism of Its Exo Specificity. Biochemistry, doi:10.1021/bi4003106 (2013).
6 Yao, T. et al. Proteasome recruitment and activation of the Uch37
deubiquitinating enzyme by Adrm1. Nature cell biology 8, 994-1002 (2006).
7 Jiao, L. et al. Mechanism of the Rpn13-induced activation of Uch37.
Protein & cell 5, 616-630, doi:10.1007/s13238-014-0046-z (2014). 8 Luchansky, S. J., Lansbury, P. T., Jr. & Stein, R. L. Substrate recognition
and catalysis by UCH-L1. Biochemistry 45, 14717-14725 (2006). 9 VanderLinden, R. T. et al. Structural Basis for the Activation and Inhibition
of the UCH37 Deubiquitylase. Molecular cell 57, 901-911, doi:10.1016/j.molcel.2015.01.016 (2015).

82
CHAPTER 5: RECENT FINDINGS ON THE STRUCTURE AND ACTIVATION OF UCH37 AND OTHER DEUBIQUITINASES
5.1 Introduction
As of the writing of this document, recent work by two groups has
uncovered two novel structures of UCH37: (1) bound to its activator, Rpn13, and
ubiquitin and (2) bound to a fragment of NFRKB, its deactivator and a component
of the Ino80 complex, both done by Vanderlinden et. al, 2015 and Sahtoe et. al,
2015 1,2. These papers confirm some of the findings of this work, as well as leave
some questions open still open about how UCH37 is regulated. This chapter will
encompass an analysis of the new structures of UCH37 followed by a review of
deubiquitinating enzymes whose specificity and activation rely on small structural
elements, such as loops, in the same manner as UCH37.
5.2 Analysis of UCH37-Rpn13-Ub and UCH37-NFRKB-Ub Structures
Both structures reveal dramatic conformational changes on the part of
UCH37’s ULD domain (Fig 5.1) The ULD of apo UCH37 involves a helix-turn-
helix (α9 and α10) followed by a shorter helix, α11, and ending with a short
unstructured loop, as the structure lacks density for the final 18 amino acids of

Fa
U
b
a
a
st
U
a
b
a
Figure 5.1: Cs labeled.
UCH37’s C
inding to b
bout 120°
pproximate
tructure wit
ULD, with he
bove the ca
inding to R
ctivation.
Conformati
-terminus.
both Rpn13
towards he
ely 90° tow
thout ubiqu
elix α10 kin
atalytic dom
Rpn13 and
onal chang
However,
3 and NFRK
elix α10 an
wards the ca
uitin, even g
nking at its c
main1. Thes
NFRKB as
ges in the U
this region
KB, in whic
nd a new h
atalytic dom
greater con
center, pos
se structure
s well as pr
ULD domai
n dramatic
ch helix α1
helix, α12,
main (Fig 5
nformationa
sitioning the
es all show
rovide a str
in. From se
cally remod
11 of UCH3
is resolved
5.1). In the
al changes
e rest of α10
a direct rol
ructural bas
even structu
dels itself u
37 rotates
d, which is
NFRKB-bo
are seen in
0, α11, and
le of the UL
sis for UCH
83
ures,
upon
back
bent
ound
n the
d α12
LD in
H37’s

re
w
a
e
u
in
R
Flo
Surpr
esolved in
was proven
ctivator an
nough flex
nresolved)
n these stru
Rpn13, a str
Figure 5.2: Aoop is in pin
risingly, ou
x-ray crysta
n incorrect
nd substrat
ibility that it
1,2. Howeve
uctures (Fig
ructural val
Activation onk, with key
5.2.
r prediction
al structure
by these
e, the cros
t could not
er, importa
g 5.2). Both
idation for R
of UCH37 by residues h
1 Crossove
n that the c
es upon UC
recent st
ssover loop
be entirely
nt interactio
h Met148 a
Rpn13’s ac
by crossovehighlighted.
er Loop
crossover
CH37 bindin
tructures.
p (residues
y resolved (
ons betwee
and Phe149
ctivation me
er loop bind. From PDB
loop would
ng to ubiqu
Despite th
s 143-163)
(residues 1
en it and R
9 make dire
echanism. R
ing to Rpn1B ID 4UEL.
d be compl
uitin and Rp
he presenc
) still main
154-159 are
Rpn13 are
ect contact
Rpn13’s
13. Crossov
84
etely
pn13
ce of
tains
e still
seen
t with
ver

85
stabilization of an open conformation of the crossover loop likely allows improved
substrate binding and catalysis, especially for the proximal ubiquitin monomer not
seen in these structures. Mutations to Met148 and Phe149 render the enzyme
unable to be activated by Rpn13 in UbAMC assays1,2. It can be predicted that the
rest of the crossover loop would be visualized in a UCH37-diubiquitin-Rpn13
structure, and that more of the residues in this region would contribute to
substrate stabilization at the active site, especially the isopeptide bond.
Alternatively, it is possible that the cross over loop maintains its dynamic
character throughout catalytic steps of the enzyme, independent of substrate
binding.
5.2.2 NFRKB Mode of Inhibition
The structures of UCH37 bound to the Ino80 component, NFRKB,
illuminate the way in which the DUB is inhibited catalytically through both its
active and distal sites. NFRKB hijacks the distal region of UCH37 that binds to
the Leu8-Thr9 hairpin of ubiquitin, a key motif within ubiquitin for binding. NFRKB
buries its own Phe100 and Arg101 within the distal pocket, occluding ubiquitin
binding (Fig 5.3). Additionally, the large helix of NFRKB that lies across the active
site face of UCH37 induces small conformational strains that lead to an
unproductive orientation of the active site. The catalytic His has rotated and now
lies an unproductive 6.3 Å from the catalytic cysteine. All of these small changes
can be utilized for small-molecule targeting of UCH37, as they directly occupy
binding sites of ubiquitin.
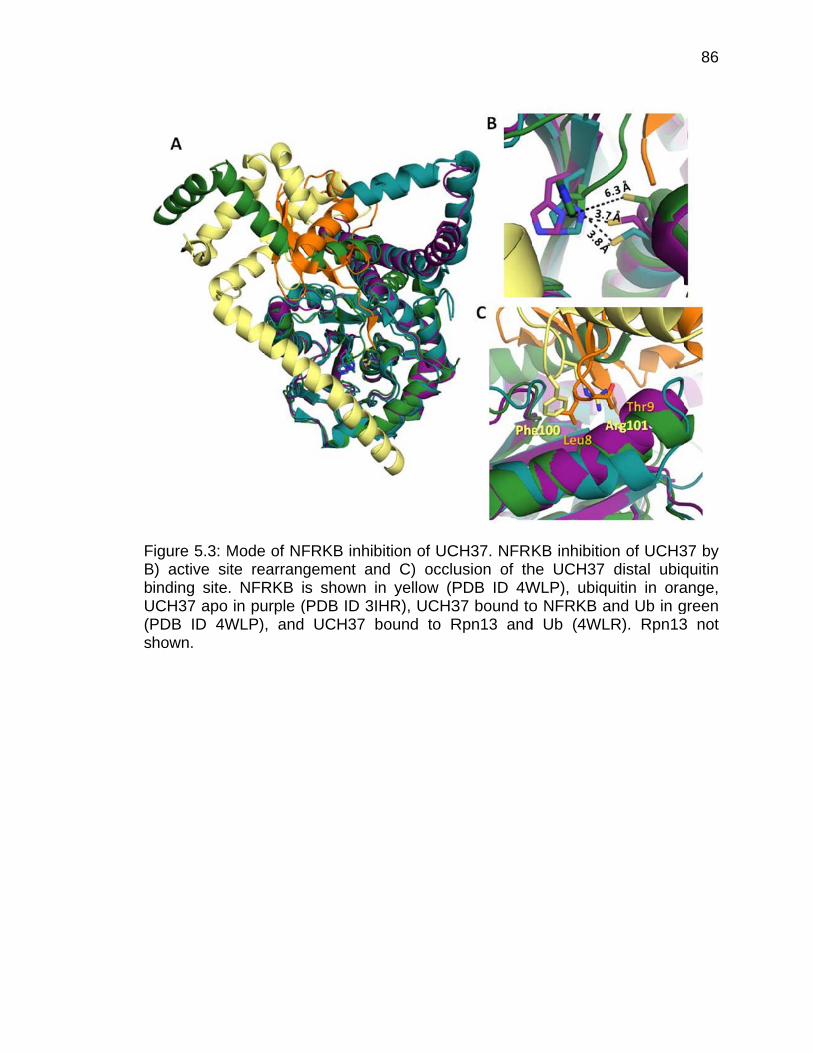
FBbU(Ps
Figure 5.3: MB) active si
inding siteUCH37 apo PDB ID 4Whown.
Mode of NFite rearrang. NFRKB iin purple (
WLP), and
FRKB inhibgement ans shown in(PDB ID 3IH UCH37 b
bition of UCnd C) occlun yellow (PHR), UCH3bound to R
CH37. NFRKusion of thPDB ID 4W37 bound toRpn13 and
KB inhibitiohe UCH37 WLP), ubiqo NFRKB ad Ub (4WL
on of UCH3distal ubiq
uitin in oraand Ub in gLR). Rpn13
86
37 by quitin ange, green 3 not

87
5.3 Analysis of Kinetic Findings in Vanderlinden et. al and Sahtoe et. al
Interestingly, these groups studied the activation of the enzyme in UbAMC
hydrolysis assays using one of the ULD mutants discussed earlier, E284A in my
studies, but numbered E283A in the isoform these groups used. They found that
Rpn13 lowers the KM of UCH37 for ubiquitin 5-fold, but that the KM of the E283A
mutant in the presence of Rpn13 is only 1.5-fold improved compared to UCH37
alone, indicating that this residue may be essential to its activation mechanism,
similar to the results presented previously (Section 4.3.2)2. These results validate
our earlier findings, that E284 is essential to the activation mechanism of UCH37.
5.4 Small Structures Effect Large Changes: A Review of Deubiquitinases
Among the ~100 deubiquitinating enzymes in the human genome, a little
less than half of these do not contain auxiliary ubiquitin binding domains or
ubiquitin-like domains beyond their active sites3. These deubiquitinases must rely
on conformational movement and binding pockets inherent in their active sites
alone, or utilize non-canonical interactions with ubiquitin to bind substrate. There
have been many thorough reviews of the various ubiquitin binding and ubiquitin
like domains; however, little focus has been drawn to the smaller dynamic
movements that significantly contribute to deubiquitinating enzyme catalysis4-8.

88
5.4.1 Unproductive Active Sites
One of the simplest, but integral, conformational changes within
deubiquitinases (DUBs) is the reorientation of catalytic residues from an
unproductive form in the apo enzyme to a productive conformation upon ubiquitin
binding. This has been seen in structures of the cysteine protease DUBs and
frequently involves misaligned catalytic cysteines or histidines within their papain-
like active sites, less often their catalytic aspartic acid or stabilizing oxyanion
glutamine residues (Fig 5.4).
Catalytic rearrangement occurs upon ubiquitin binding for the UCH family
members UCHL19,10 and UCH371,2,11-14. In the apo UCHL1 active site, the
catalytic histidine resides 8.2 Å away from the catalytic cysteine and is turned 90°
from the typical orientation of a papain-like active site, an unproductive distance
for catalysis9. Upon ubiquitin binding, the histidine swings 90° to lay in-plane with
the catalytic Cys and has moved to a productive 4 Å distance10. In the apo active
site of UCH37, the catalytic His is in a productive orientation, but its catalytic
cysteine is rotated inwards, toward the His residue rather than towards the rest of
the catalytic cleft where ubiquitin will bind11-13. Upon ubiquitin binding, the Cys
rotates 70° to face Gly76 of ubiquitin, an appropriate conformation1,2,14.
Unproductive active sites have been found in the active sites of OTU-
domain containing DUBs as well, both OTU1B, K48-specific, and OTULIN, a
linear polyubiquitin-cleaving DUB. Upon binding to ubiquitin, OTUB1 has a
similar conformational change to UCHL1 and UCH37; its His rotates down 90° to
lock in plane with the catalytic Cys, and its Cys flips inward to face Gly76 of

89
ubiquitin, altogether moving the two residues closer by 3 Å into a catalytically-
productive conformation15,16 (Fig 5.4). The structure of OTULIN’s active site has
partial occupancy for two distinct orientations: one in which the catalytic His and
Cys are in appropriate conformations, and one in which the catalytic His is flipped
to occupy the space which Gly76 resides in the linear diubiquitin-bound
structure17.
HAUSP/USP7, one of the most well-characterized USP-family DUBs also
contains a misaligned active site, wherein its catalytic Cys is positioned 10 Å
away from the catalytic His (Fig 5.4). Upon ubiquitin binding, its catalytic Cys,
His, and Asp move closer together, to a 3.7 Å distance between the Cys and His
and a 2.7 Å distance between the His and Asp.
The current theory as to why these DUBs prefer a catalytically-
unproductive active site orientation in absence of ubiquitin is that it may protect
against oxidation18. Some deubiquitinases have been found to be highly
susceptible to oxidation, which can lead to irreversible modification (sulphinic or
sulphonic acid) of their catalytic cysteines, rendering the enzyme catalytically
dead. In the seminal work describing DUB oxidation, the OTU A20 was capable
of an initial state of reversible oxidation, which would attain irreversibility upon
continued exposure to the oxidant18. A20 does not have a misaligned active site,
however it is believed that DUBs with misoriented cysteines may induce
protective interactions with nearby residues, keeping the cysteine shielded from
oxidants.
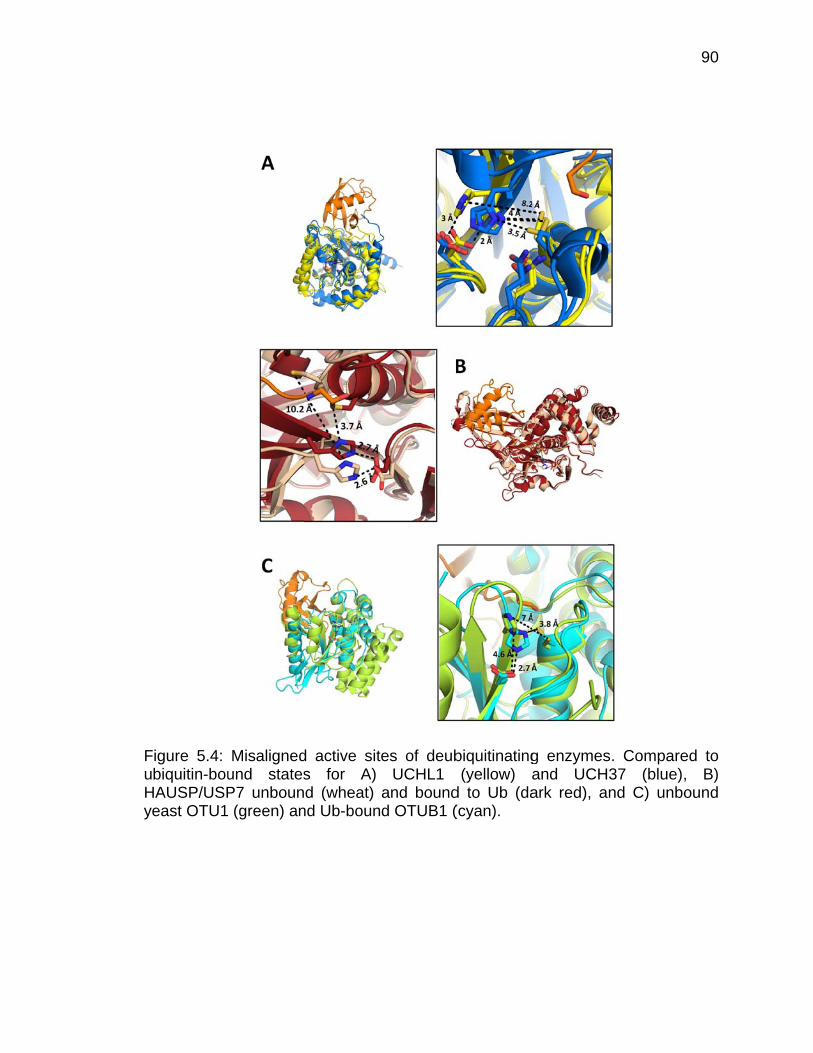
FuHy
Figure 5.4: biquitin-bou
HAUSP/USPeast OTU1
Misalignedund statesP7 unboun (green) an
d active sits for A) d (wheat)
nd Ub-boun
tes of deubUCHL1
and boundd OTUB1 (
biquitinating(yellow) a
d to Ub (da(cyan).
g enzymesand UCHark red), a
s. Compare37 (blue),nd C) unbo
90
ed to , B) ound

91
5.4.2 Insertions and the JAMM Domain
Regulation of JAMM domain containing deubiquitinating enzymes is
mainly held by their insertion domains, numbered Ins-1 and Ins-219. The
insertions act as substrate stabilizers and confer specificity, as seen in the
structure of AMSH-LP bound to diubiquitin19 (Fig 5.5). AMSH-LP uses the sheets
of Ins-1 to bind the distal ubiquitin monomer and stabilize the isopeptide bond for
cleavage, and Ins-2 for additional isopeptide stabilization and binding of the
proximal ubiquitin monomer. Superposition of the structures of human AMSH,
another JAMM family DUB, and the AMSH-like S. pombe orthologue Sst2 on the
AMSH-LP structure shows similar modes of binding and stabilization by their
highly-conserved insertion domains. These small domains provide significant
contribution to catalysis; when mutated, they can render the enzyme catalytically
impaired. However, mutations to Ins-2 do not contribute to substrate binding as
they only affect kcat, not KM20,21. The isopeptide contacts by the insertions also
maintain specificity, in that AMSH-LP, AMSH, and Sst2 are all highly specific for
K63-linked polyubiquitin chains.
In contrast, two structures were recently solved for Rpn11, the JAMM DUB
resident in the 26S proteasome responsible for en bloc cleavage of ubiquitin
chains from proteasomal substrates, in which only Ins-1 was utilized for substrate
recognition and catalysis22-24. Ins-2 does not contribute to substrate catalysis;
rather, it assists docking Rpn11 to the proteasomal subunit Rpn2, as predicted

Faaoin
b
c
s
c
p
Figure 5.5: nd Ins-2 tond the morthologue S
n pink, teal,
y the rece
atalytic eng
pecificity27.
omplete re
rotein22,28-3
Insertion doo ubiquitin bodels bounSst2, and D light grey,
ent cryoEM
gagement
It is know
moval of po
0.
omains of binding in And to diUbD) Rpn11. Jand green,
M maps of
of Ins-2 p
wn to cleav
olyubiquitin
JAMM metA) the strucb of B) AMJAMM dom, respective
the 26S p
rovides a
ve entire ch
n from the l
talloproteascture of AMMSH, C) t
mains are inely.
proteasome
structural b
hains in an
ysine of att
ses. ContribSH-LP bouthe AMSH-n dark grey
e24-26 (Fig
basis for R
n en bloc f
tachment o
butions of und to K63--like S. poy with inser
5.5). This
Rpn11’s lac
fashion, tha
onto a subs
92
Ins-1 -diUb ombe rtions
non-
ck of
at is,
strate

ty
m
te
a
d
Fin(r
Memb
ypes of ub
members (e
ermed the c
bsence of
ensity is no
Figure 5.6: Cn grey surfared), yeast
bers of the
biquitin sub
except BAP
crossover l
ubiquitin, th
ot resolved.
Crossover ace, ubiquiYUH1 (pur
5.4.3 Su
e UCH fam
strates the
P1) contain
oop, to filte
hese DUBs
. Upon ubiq
loops for utin in orangple), UCHL
ubstrate-filte
mily of DUB
ey act upon
n a flexible
er out large
s’ crossove
quitin bindin
biquitin-bouge, and croL3 (green),
ering Loops
Bs utilize g
n. Structur
loop span
er substrate
r loops are
ng, nearly e
und UCH Dossover looand UCH37
s
ating loops
res of all t
nning their
es for cleav
e flexible an
every family
DUBs. The ops highligh7 (teal).
s to restric
he UCH fa
catalytic c
vage31-33. In
nd their elec
y member
UCH domahted for UC
93
ct the
amily
clefts,
n the
ctron
ain is CHL1

94
displays an ordered crossover loop that contributes some interactions to
ubiquitin’s C-terminal tail, buried within the active site (Fig 5.6). Only UCH37 still
lacks an ordered crossover loop, even in the presence of ubiquitin and its
activator, the proteasomal subunit Rpn131,2.
Through diubiquitin cleavage assays and protein chimeras altering the
length of the crossover loop, it is apparent that this loop is responsible for
substrate specificity by steric filtering32,33. Generally, the UCH family is believed
to only cleave small moieties from the C-terminus of ubiquitin, not processing of
polyubiquitin. The smaller family members, human UCHL1, human UCHL3, and
yeast YUH1, which contain solely a UCH domain, have the shortest crossover
loops and are not capable of polyubiquitin cleavage, only cleavage of ubiquitin-
AMC, a fluorogenic ubiquitin substrate with only the small AMC fluorescent group
attached to its C-terminus. The two larger UCH family members, UCH37 and
BAP1, have C-terminal extensions beyond their catalytic domains and contain
longer crossover loops than the other family members, which is believed to allow
them to cleave polyubiquitin. A Drosophila homolog of BAP1, Calypso, has been
shown to deubiquitinate histone H2B34 and UCH37 is proteasome-bound, and
therefore must process the variety of polyubiquitinated prey captured by the 26S.
The substrate-filtering theory was proven by an elegant experiment in which the
crossover loop of UCH37 was expanded by a poly-glycine insertion32. This
chimeric DUB was capable of both K48- and K63-polyubiquitin chain cleavage32.
Within that same study, the crossover loop of wild-type UCHL3 was
biochemically nicked, but this damage did not inhibit the DUB’s cleavage of
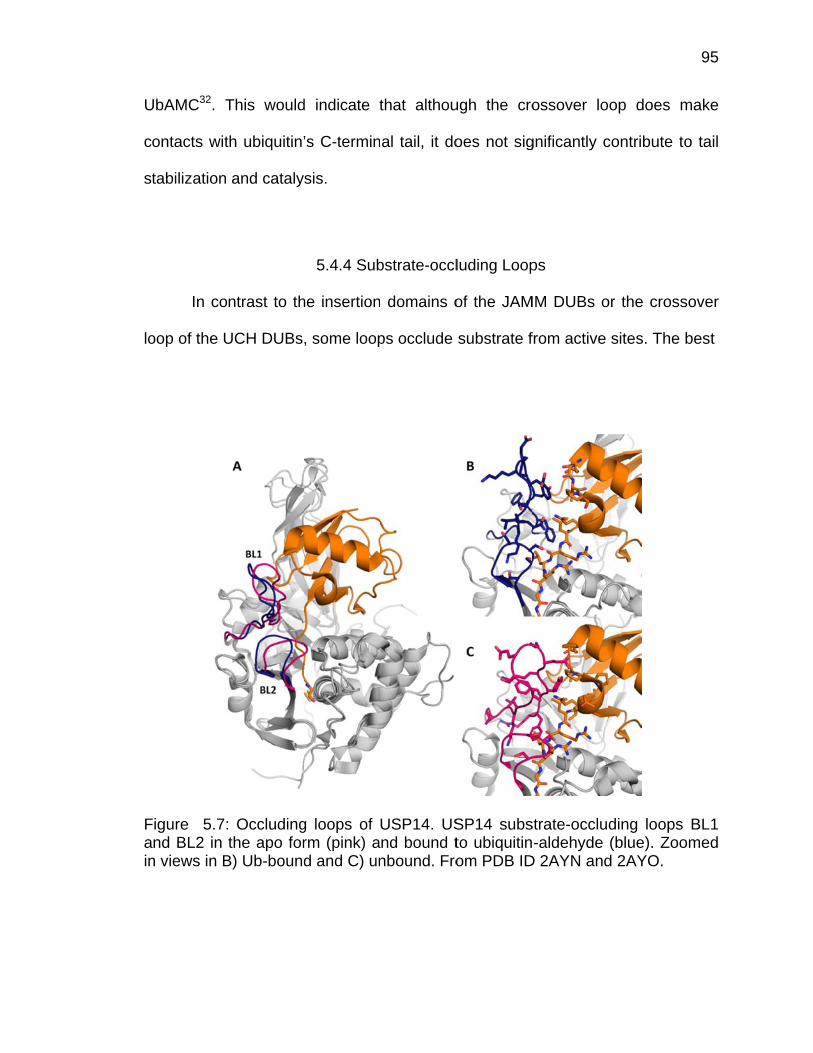
U
c
st
lo
Fain
UbAMC32. T
ontacts wit
tabilization
In co
oop of the U
Figure 5.7:nd BL2 in
n views in B
This would
th ubiquitin’
and cataly
ntrast to th
UCH DUBs
Occludingthe apo for
B) Ub-bound
d indicate t
’s C-termin
sis.
5.4.4 Sub
he insertion
, some loop
g loops of rm (pink) ad and C) un
that althou
al tail, it do
bstrate-occl
domains o
ps occlude
USP14. USnd bound tnbound. Fro
ugh the cro
oes not sig
luding Loop
of the JAM
substrate f
SP14 substo ubiquitinom PDB ID
ossover lo
gnificantly c
ps
MM DUBs o
from active
strate-occlu-aldehyde
D 2AYN and
op does m
contribute to
or the cross
sites. The
ding loops (blue). Zoo
d 2AYO.
95
make
o tail
sover
best
BL1 omed

96
known case of this is seen in the structures of USP14 alone and bound to
ubiquitin-aldehyde35. In its apo form, USP14’s active site is blocked by two loops,
BL1 and BL2, which occupy a portion of the space where ubiquitin’s C-terminal
tail would bind in order to access its catalytic cysteine (Fig 5.7). Upon binding to
ubiquitin, the entirety of these loops, as well as individual side chains, open up to
allow ubiquitin binding. Oddly, many of the residues within these loops are also
responsible for ubiquitin binding, such as Tyr333 and Phe331. These loops are
attributed to USP14’s poor reactivity with ubiquitin probes, namely ubiquitin-vinyl
sulfone, but that it becomes activated upon association with the 26S proteasome,
potentially through conformational restrains of these loops into a more open
form35. Although many other USP family DUBs contain loops within this region,
such as HAUSP/USP7, they do not sterically block those USPs active sites. It is
believed that the length and conformation of these loops confer USP14 with
unique reactivity compared to other USP family DUBs.
5.5 Conclusions
Through examination of the recent UCH37 activating and deactivating x-
ray crystal structures as well as an assessment of other small conformational
contributions to the regulation of deubiquitinating enzymes, we have a newfound
appreciation for the layers of specificity and mechanisms of action of DUBs, a
class of enzymes which process an incredible variety of cellular substrates. Only
~100 DUBs specifically recognize monoubiquitinated substrates, 8 homotypic

97
chain types, and a startling number of mixed linkage chain possibilities. Even
more shocking is that only half of them require additional ubiquitin binding
domains beyond their catalytic core in order to attain specificity and improve
substrate binding. DUBs containing only a catalytic domain rely on small
structures within themselves to restrict absolute specificity, as in the case of the
JAMM DUBs AMSH and AMSH-LP, or to allow processing of a broad spectrum
of substrates at a highly specific location and under certain conditions, as in the
case of the proteasomal DUBs Rpn11, USP14, and UCH37.
Although we have uncovered significant information regarding the binding
and catalysis of ubiquitin, as well as the binding and activation of Rpn13, to
UCH37, many questions still remain regarding its ability to process polyubiquitin
at the 26S proteasome. Despite our structural findings regarding its exo-specific
recognition of Lys48-linked chains, it is highly implausible that UCH37 would
have such limited polyubiquitin processing skills at the proteasome, with
increasing reports of proteasome processing of non-canonical ubiquitin
signals36,37. Further structural and biochemical studies of UCH37 within the
context of the 26S proteasome are needed to understand its role in the coupling
of deubiquitination and degradation.

98
5.6 References
1 Sahtoe, D. D. et al. Mechanism of UCH-L5 Activation and Inhibition by DEUBAD Domains in RPN13 and INO80G. Molecular cell 57, 887-900, doi:10.1016/j.molcel.2014.12.039 (2015).
2 VanderLinden, R. T. et al. Structural Basis for the Activation and Inhibition
of the UCH37 Deubiquitylase. Molecular cell 57, 901-911, doi:10.1016/j.molcel.2015.01.016 (2015).
3 Komander, D., Clague, M. J. & Urbe, S. Breaking the chains: structure and
function of the deubiquitinases. Nature reviews. Molecular cell biology 10, 550-563, doi:10.1038/nrm2731 (2009).
4 Faesen, A. C., Luna-Vargas, M. P. & Sixma, T. K. The role of UBL
domains in ubiquitin-specific proteases. Biochemical Society transactions 40, 539-545, doi:10.1042/BST20120004 (2012).
5 Husnjak, K. & Dikic, I. Ubiquitin-binding proteins: decoders of ubiquitin-
mediated cellular functions. Annual review of biochemistry 81, 291-322, doi:10.1146/annurev-biochem-051810-094654 (2012).
6 Dikic, I., Wakatsuki, S. & Walters, K. J. Ubiquitin-binding domains - from
structures to functions. Nature reviews. Molecular cell biology 10, 659-671, doi:10.1038/nrm2767 (2009).
7 Grabbe, C. & Dikic, I. Functional roles of ubiquitin-like domain (ULD) and
ubiquitin-binding domain (UBD) containing proteins. Chemical reviews 109, 1481-1494, doi:10.1021/cr800413p (2009).
8 Harper, J. W. & Schulman, B. A. Structural complexity in ubiquitin
recognition. Cell 124, 1133-1136, doi:10.1016/j.cell.2006.03.009 (2006). 9 Das, C. et al. Structural basis for conformational plasticity of the
Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proceedings of the National Academy of Sciences of the United States of America 103, 4675-4680 (2006).
10 Boudreaux, D. A., Maiti, T. K., Davies, C. W. & Das, C. Ubiquitin vinyl
methyl ester binding orients the misaligned active site of the ubiquitin hydrolase UCHL1 into productive conformation. Proceedings of the National Academy of Sciences of the United States of America 107, 9117-9122 (2010).

99
11 Burgie, S. E., Bingman, C. A., Soni, A. B. & Phillips, G. N., Jr. Structural characterization of human Uch37. Proteins (2011).
12 Nishio, K. et al. Crystal structure of the de-ubiquitinating enzyme UCH37
(human UCH-L5) catalytic domain. Biochemical and biophysical research communications 390, 855-860 (2009).
13 Maiti, T. K. et al. Crystal structure of the catalytic domain of UCHL5, a
proteasome-associated human deubiquitinating enzyme, reveals an unproductive form of the enzyme. The FEBS journal 278, 4917-4926 (2011).
14 Morrow, M. E. et al. Stabilization of an Unusual Salt Bridge in Ubiquitin by
the Extra C-Terminal Domain of the Proteasome-Associated Deubiquitinase UCH37 as a Mechanism of Its Exo Specificity. Biochemistry, doi:10.1021/bi4003106 (2013).
15 Messick, T. E. et al. Structural basis for ubiquitin recognition by the Otu1
ovarian tumor domain protein. The Journal of biological chemistry 283, 11038-11049, doi:10.1074/jbc.M704398200 (2008).
16 Edelmann, M. J. et al. Structural basis and specificity of human otubain 1-
mediated deubiquitination. The Biochemical journal 418, 379-390, doi:10.1042/BJ20081318 (2009).
17 Keusekotten, K. et al. OTULIN antagonizes LUBAC signaling by
specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312-1326, doi:10.1016/j.cell.2013.05.014 (2013).
18 Kulathu, Y. et al. Regulation of A20 and other OTU deubiquitinases by
reversible oxidation. Nature communications 4, 1569, doi:10.1038/ncomms2567 (2013).
19 Sato, Y. et al. Structural basis for specific cleavage of Lys 63-linked
polyubiquitin chains. Nature 455, 358-362, doi:10.1038/nature07254 (2008).
20 Davies, C. W., Paul, L. N., Kim, M. I. & Das, C. Structural and
thermodynamic comparison of the catalytic domain of AMSH and AMSH-LP: nearly identical fold but different stability. Journal of molecular biology 413, 416-429, doi:10.1016/j.jmb.2011.08.029 (2011).

100
21 Shrestha, R. K. et al. Insights into the mechanism of deubiquitination by JAMM deubiquitinases from cocrystal structures of the enzyme with the substrate and product. Biochemistry 53, 3199-3217, doi:10.1021/bi5003162 (2014).
22 Verma, R. et al. Role of Rpn11 metalloprotease in deubiquitination and
degradation by the 26S proteasome. Science 298, 611-615, doi:10.1126/science.1075898 (2002).
23 Pathare, G. R. et al. Crystal structure of the proteasomal deubiquitylation
module Rpn8-Rpn11. Proceedings of the National Academy of Sciences of the United States of America 111, 2984-2989, doi:10.1073/pnas.1400546111 (2014).
24 Worden, E. J., Padovani, C. & Martin, A. Structure of the Rpn11-Rpn8
dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nature structural & molecular biology 21, 220-227, doi:10.1038/nsmb.2771 (2014).
25 Matyskiela, M. E., Lander, G. C. & Martin, A. Conformational switching of
the 26S proteasome enables substrate degradation. Nature structural & molecular biology 20, 781-788, doi:10.1038/nsmb.2616 (2013).
26 Lander, G. C. et al. Complete subunit architecture of the proteasome
regulatory particle. Nature 482, 186-191 (2012). 27 Mansour, W. et al. Disassembly of Lys11- and mixed-linkage polyubiquitin
conjugates provide insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J Biol Chem, doi:10.1074/jbc.M114.568295 (2014).
28 Yao, T. & Cohen, R. E. A cryptic protease couples deubiquitination and
degradation by the proteasome. Nature 419, 403-407 (2002). 29 Cooper, E. M. et al. K63-specific deubiquitination by two JAMM/MPN+
complexes: BRISC-associated Brcc36 and proteasomal Poh1. The EMBO journal 28, 621-631, doi:10.1038/emboj.2009.27 (2009).
30 Koulich, E., Li, X. & DeMartino, G. N. Relative structural and functional
roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell 19, 1072-1082, doi:10.1091/mbc.E07-10-1040 (2008).
31 Misaghi, S. et al. Structure of the ubiquitin hydrolase UCH-L3 complexed
with a suicide substrate. J Biol Chem 280, 1512-1520 (2005).

101
32 Popp, M. W., Artavanis-Tsakonas, K. & Ploegh, H. L. Substrate filtering by the active site crossover loop in UCHL3 revealed by sortagging and gain-of-function mutations. The Journal of biological chemistry 284, 3593-3602 (2009).
33 Zhou, Z. R., Zhang, Y. H., Liu, S., Song, A. X. & Hu, H. Y. Length of the
active-site crossover loop defines the substrate specificity of ubiquitin C-terminal hydrolases for ubiquitin chains. The Biochemical journal 441, 143-149 (2012).
34 Scheuermann, J. C. et al. Histone H2A deubiquitinase activity of the
Polycomb repressive complex PR-DUB. Nature 465, 243-247 (2010). 35 Hu, M. et al. Structure and mechanisms of the proteasome-associated
deubiquitinating enzyme USP14. The EMBO journal 24, 3747-3756 (2005).
36 Mansour, W. et al. Disassembly of lys11 and mixed linkage polyubiquitin
conjugates provides insights into function of proteasomal deubiquitinases rpn11 and ubp6. The Journal of biological chemistry 290, 4688-4704, doi:10.1074/jbc.M114.568295 (2015).
37 Meyer, H. J. & Rape, M. Enhanced protein degradation by branched
ubiquitin chains. Cell 157, 910-921, doi:10.1016/j.cell.2014.03.037 (2014).

APPENDIX

102
APPENDIX
Inhibition of Uch37 and Usp14 at the 26s Proteasome and Its Effects on Degradation
6.1 Introduction
Although UCH37 is capable of activation by Rpn13 alone, its
activity is further enhanced within the context of the entire 26S proteasome, a
mechanism of activation which remains a mystery1-3. Due to UCH37’s poor
cleavage of di- or polyubiquitin substrates alone and in the presence of Rpn13, it
became clear that activity assays would not succeed without the entire 26S
proteasome present as an activator. To this end, we pursued purification of
endogenous 26S from rabbit tissue using an affinity-tag method developed by the
Goldberg group4,5. Using this proteasome, we hoped to study the roles of the
deubiquitinases UCH37 and USP14 within overall protein degradation and
whether their activities are coupled to the rate of degradation of polyubiquitinated
substrates.
This purification method relies on the affinity of the Rpn1 and Rpn10
subunits of the 26S proteasome for the Ubl domain of Rad23B, a shuttle factor

Fp
c
b
re
G
c
a
w
re
m
Figure A roteasome
apable of b
inding dom
eceptor. Fo
GST-tag, a
aptured by
nd then the
which binds
emoved by
more gentle
6.1: Sches.
binding to e
main of Rp
or purificati
allowing im
y this GST-
e proteasom
s to the GS
a subtract
e purificatio
eme of U
either of th
n1 or to o
on of the
mmobilizatio
-Ubl, non-s
me is elute
ST-Ubl and
ion step ov
n method t
bl-UIM pu
ose subun
ne of the
proteasome
on on glu
specifically
ed by additi
d releases
ver Ni NTA
than the tra
urification
its6,7. Its U
UIM doma
e, the Ubl
utathione b
binding pr
on of a 6xH
the protea
beads. Th
aditional su
method o
bl binds to
ains of Rpn
of Rad23B
beads. Pro
roteins are
His-tagged
some. Exc
his method
ucrose or g
of endoge
either the
n10, a ubiq
B is fused
oteasomes
washed a
UIM of Rp
cess His-UI
is consider
lycerol gra
103
nous
Ubl-
quitin
to a
are
away,
pn10,
IM is
red a
dient

104
centrifugation method or the TAP- or FLAG-tagged Rpn11 method, allowing
association of transient factors and ensuring the presence of the associated
deubiquitinases, UCH37 and USP144,5. This purification method is outlined in
Figure 6.1.
The goal of this purification method is to investigate the contribution of
UCH37 and USP14 to proteasomal degradation. There is evidence for the role of
USP14 from purified S. cerevisiae proteasomes, however, these lack UCH378,9.
The goal of the following work is to understand how inhibition of UCH37 and
USP14, achieved by incubation with the suicide inhibitor UbVME, affects
substrate degradation and deubiquitination.
6.2 Methods
6.2.1 Purification of Rabbit 26S Proteasomes
Following the method established by Besche et. al, we purified
endogenous levels of 26S proteasome from rabbit tissue (Pel-freez)4,5. This
affinity-tag method relied on the affinity of the proteasomal shuttle factor,
Rad23B, specifically its Ubl domain, for either Rpn10’s UIM domain or Rpn1’s
Ubl-binding site6,7. 2-4 grams of rabbit muscle tissue were homogenized on ice in
proteasome purification buffer (25 mM HEPES pH 7.5, 50 mM NaCl, 5 mM
MgCl2, 10% glycerol), PB, to which 1 mM ATP and 1 mM DTT were added, and
then were spun down at 100,000xg for 1 hour. 2 mg of GST-tagged Rad23B Ubl,
which had been previously purified recombinantly from E. coli, was immobilized

105
on glutathione beads and any excess washed off with PB. Rabbit lysate was
rocked with immobilized GST-Ubl for 2 hours at 4°C. Beads were collected in an
empty glass column and unbound proteins were allowed to flow through. 20
column volumes of PB with added ATP and DTT were used to wash the beads. 2
mg of 6xHis-tagged Rpn10 UIM was added to the beads and was rocked
overnight at 4°C to induce elution of pure proteasomes. Proteasomes were then
run over Ni NTA beads to re-capture the His-UIM. Pure proteasomes were
concentrated down and flash frozen as aliquots.
6.2.2 20S Activity Assays
To confirm the presence of the 20S core particle and test its activity, the
hydrolysis of succinate-Leu-Leu-Val-Tyr-AMC, a known 20S substrate, was
measured in the presence of rabbit 26S proteasomes. Suc-LLVY-AMC was
dissolved in DMSO. 5 nM 26S proteasome was diluted in AMC assay buffer
containing 50 mM Tris pH 7.6, 0.5 mM EDTA, 0.1% bovine serum albumin, and 5
mM DTT and allowed to reach 30°C. 100 µM Suc-LLVY-AMC was added to 5 nM
proteasome in assay buffer and AMC hydrolysis was measured on a Tecan plate
reader at 380 nm excitation wavelength and 465 nm emission wavelength at
30°C for 1 hr. Progress curves were plotted in Kaleidagraph.

106
6.2.3 Ubiquitin-AMC Hydrolysis Assays
To test the deubiquitinating activity of endogenous UCH37 and USP14,
hydrolysis of UbAMC was measured. Proteasomes were diluted to 5 nM in
reaction buffer, 50 mM Tris pH 7.6, 0.5 mM EDTA, 0.1% bovine serum albumin,
and 5 mM DTT. Reactions were initiated by addition of UbAMC (Boston
Biochem) and were measured on a Tecan fluorescence plate reader (Männedorf,
Switzerland) with 380 nm excitation wavelength and 465 nm emission
wavelength at 30°C for 1 hr. Progress curves were plotted in Kaleidagraph.
6.2.4 Synthesis and Degradation of GFP-Titin-CyclinPY Substrate
In order to measure rates of degradation by the rabbit 26S, a
polyubiquitinated proteasomal substrate was needed. In the literature, there are a
handful of substrates, however, each is limited in scope and/or synthetic
simplicity. We utilized a substrate developed by the Martin group at UC Berkeley,
a GFP-tagged unstructured protein, titin, fused to cyclin, a known proteasomal
substrate, with an engineered PY motif, a degron which signals E3 ligases for
polyubiquitination10,11. After expression and purification from E. coli, the GFP-
titin-cyclinPY was polyubiquitinated by the E3 ligase Rsp5, also bacterially
expressed.

Fle
(E
D
in
m
w
o
e
w
a
c
Figure A 6.2eft, SDS PA
A mix
E3 enzyme
DTT in buffe
ncubated fo
mixture was
which point
f 40 µM GF
For d
ither 1 µM
were run w
dded to the
ontained 10
2: PolyubiquAGE gel of u
xture of 2.3
e), 0.5 mM
er containin
or 3-6 hours
s aliquoted
the substra
FP-titin-cycl
degradation
UbVME o
ith one tim
e t=0 time
00 nM 26S
uitination ofubiquitinatio
3 µM E1 en
ubiquitin, 4
ng 50 mM H
s at 37°C (
out, flash f
ate was use
linPY-Ubn.
n reactions
r buffer co
me point pe
point to pr
S proteasom
f GFP-titin-on reaction
nzyme, 5 µ
40 µM GFP
HEPES pH
Fig 6.2). Af
frozen, and
ed directly w
s, rabbit 26
ntrol for 2
er tube, an
revent any
me (+/- UbV
-cyclinPY sun shown at
µM Ubc4 (E
P-titin-cyclin
7.5, 50 mM
fter the fina
d stored at
with an ass
6S proteas
hours on i
d with 5xS
proteasom
VME), 2 µM
bstrate. Scright.
E2 enzyme
nPY, 5 mM A
M NaCl, 10
al time poin
-80°C unti
sumed stoc
some was
ce. Degrad
SDS PAGE
mal degrada
M GFP-titin-
cheme show
e), 11 µM R
ATP, and 1
0% glycerol
nt, the subs
l further us
ck concentr
incubated
dation reac
E buffer alr
ation. Reac
-cyclinPY-Ub
107
wn at
Rsp5
mM
was
strate
se, at
ation
with
ctions
eady
ctions
bn, 1

108
mM ATP, 1 mM DTT, and an ATP recycling system (creatine phosphokinase,
inorganic pyrophosphate, creatine phosphate) in 50 mM HEPES pH 7.5, 50 mM
NaCl, 10% glycerol. The reactions were run at 25 °C and were quenched with 5x
SDS PAGE buffer.
6.3 Results
6.3.1 Impact of Deubiquitinase Inhibition on Proteasome Degradation
The 26S proteasome was purified from rabbit muscle tissue using the Ubl-
UIM method developed by the Goldberg group and can be seen in Fig 6.34,5.
This method did not purify a large amount of proteasomes and retained the GST-
Ubl and His-UIM proteins despite subtraction over Ni-NTA beads. However, there
was sufficient to study the effects of inhibition of the associated deubiquitinases,
UCH37 and USP14, by UbVME treatment. To this end, proteasomes were
treated with and without 1 µM UbVME for 2 hours on ice to catalytically inactivate
the endogenous associated deubiquitinases. Then each sample was assessed
for its deubiquitinating activity by UbAMC hydrolysis assays and for its 20S core
particle activity using a fluorogenic peptide substrate, succinate-Leu-Leu-Val-Tyr-
AMC, or SucLLVY-AMC. Not surprisingly, in the presence or absence of UbVME,
the proteasome’s 20S activity was not changed (Fig 6.4). However,

Fu
d
(F
d
b
cy
a
in
S
s
Figure A 6.3sing the Ub
eubiquitina
Fig 6.4). U
egrade a p
y Rsp5, a
yclin, a kno
largely uns
n the prese
SDS PAGE
ubstrate. U
3: Silver-stabl-UIM meth
ating activity
bVME-inhib
proteasoma
system de
own protea
structured p
nce and ab
E gels sh
UbVME inhib
ained SDShod.
y was com
bited protea
l substrate,
eveloped by
somal subs
protein idea
bsence of U
howing dis
bition of the
S PAGE ge
mpletely abo
asomes we
, a GFP-titin
y the Marti
strate, with
al for degra
UbVME we
sappearanc
e proteasom
el of rabbit
olished upo
ere next as
n-cyclinPY f
n group10,1
an engine
adation by t
ere determin
ce of the
me resulted
26S protea
on treatme
ssessed fo
fusion prote
1. This sub
eered degro
he 26S. De
ned by Ima
highly po
d in about h
asome. Pu
nt with UbV
r their abili
ein ubiquitin
bstrate con
on fused to
egradation r
ageJ analys
olyubiquitin
half as slow
109
rified
VME
ity to
nated
tains
titin,
rates
sis of
nated
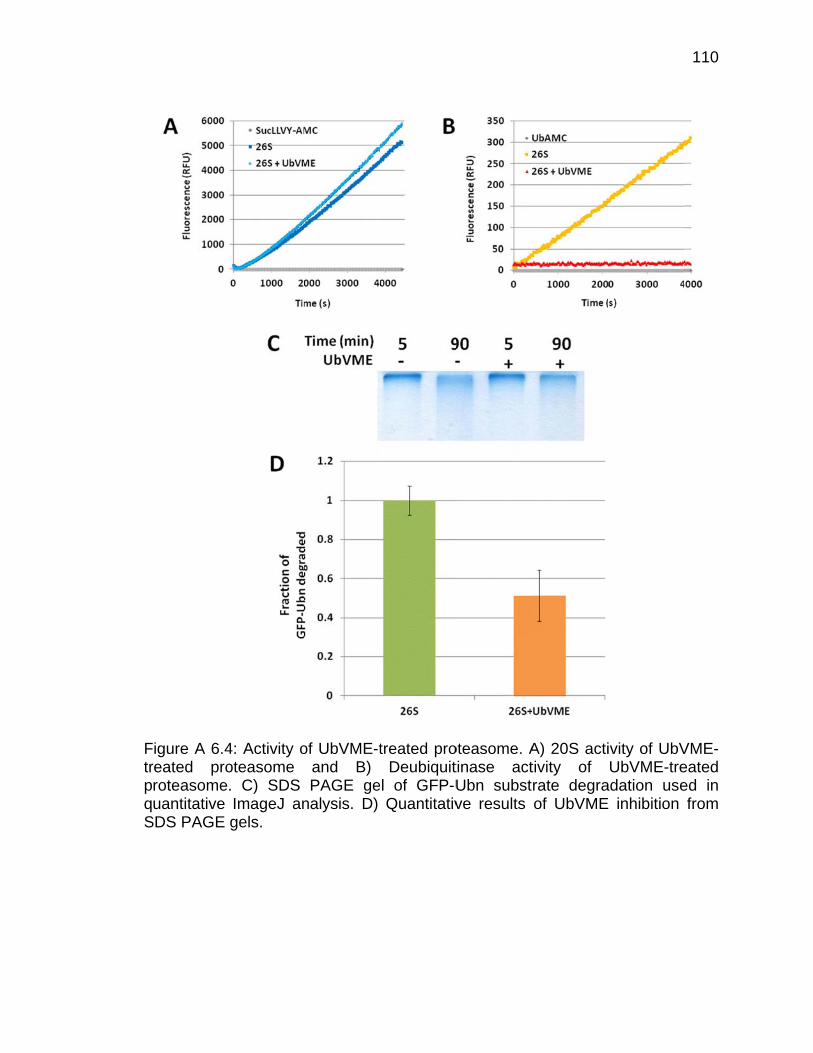
FtrpqS
Figure A 6.4reated proroteasomeuantitative
SDS PAGE
4: Activity ooteasome . C) SDS ImageJ angels.
of UbVME-tand B) PAGE ge
nalysis. D)
treated proDeubiquit
l of GFP-UQuantitativ
oteasome. Atinase actUbn substve results
A) 20S actitivity of rate degraof UbVME
ivity of UbVUbVME-tre
adation use inhibition
110
VME-eated ed in from

111
degradation compared to uninhibited proteasomes, despite activity of the 20S
core particle being unchanged in the presence of UbVME (Fig 6.4). This would
suggest that deubiquitination by UCH37 and USP14 are coupled to degradation,
a theory still under investigation.
Due to this result, we were curious about the effect of simultaneous
deubiquitinase and 20S inhibition, achieved by the addition of UbVME and the
20S inhibitor MG132. MG132 inhibits the β5 subunit of the 20S core particle,
thereby slowing its proteolytic activities by inhibiting one of the proteolytic
subunits12,13. We were curious as to the contribution of UbVME in slowing
proteasomal degradation compared to MG132, a well-characterized inhibitor.
Incubation of the 26S with inhibitors was done on ice for 2 hours in the presence
of either MG132 or a DMSO control, or UbVME or a buffer control. First, we
tested 20S and deubiquitinase activity by SucLLVY-AMC and UbAMC hydrolysis
(Fig 6.5). As expected, MG132 inhibits 20S activity but not deubiquitinating
activity, and UbVME inhibits deubiquitinating activity but not 20S activity. 20S
activity was slightly enhanced in the presence of UbVME, a phenomenon
previously observed by the Goldberg group (using ubiquitin-aldehyde), but
explained as an increase in AAA ATPase activity upstream14. Interestingly,
MG132 seems to slightly enhance deubiquitinating activity, but is within error,
therefore was not further investigated. These results indicate appropriate levels
of inhibition of the 20S and deubiquitinases by their respective inhibitors, so
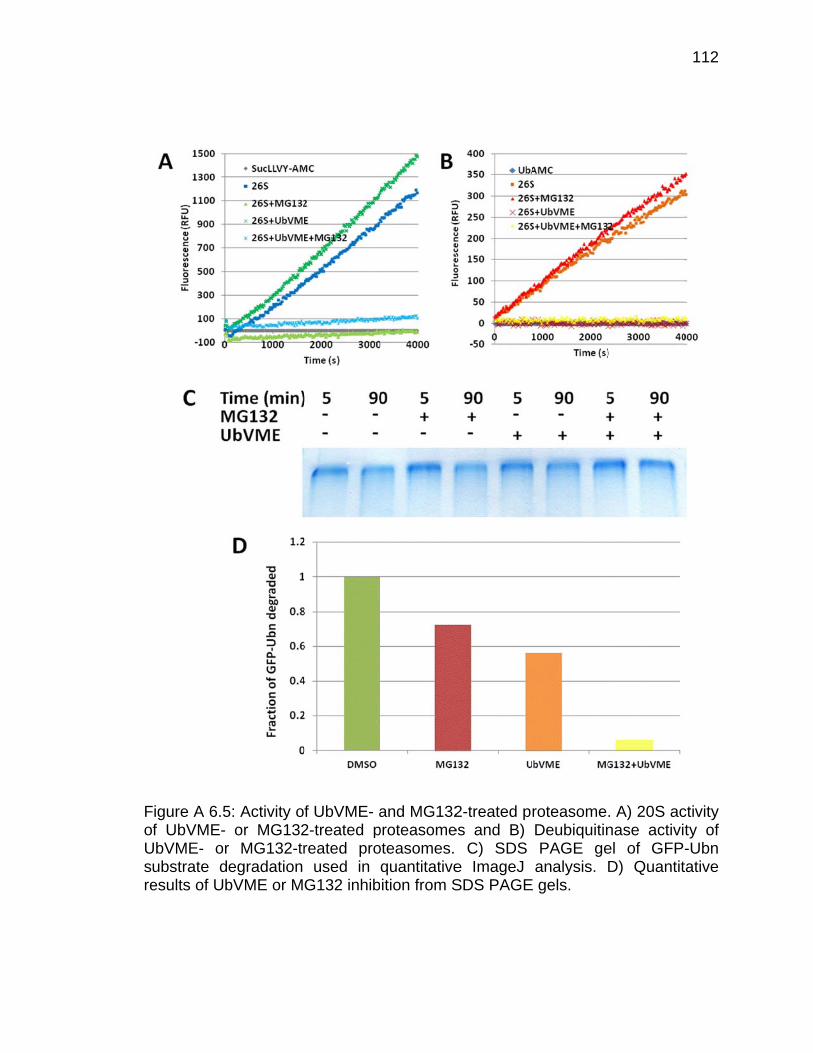
FoUsre
Figure A 6.5f UbVME-
UbVME- orubstrate desults of Ub
5: Activity oor MG132
r MG132-tregradation bVME or M
f UbVME- a2-treated preated pro
used in G132 inhib
and MG132proteasomeoteasomes. quantitativeition from S
2-treated pres and B)
C) SDS e ImageJ SDS PAGE
roteasomeDeubiquitinPAGE geanalysis. D
E gels.
. A) 20S acnase activiel of GFPD) Quantit
112
ctivity ty of -Ubn tative

113
these samples were used to test degradation of the GFP-titin-cyclinPY-Ubn
substrate (Fig 6.5).
The rates of degradation of the GFP-Ubn substrate were measured by
running time points on SDS PAGE gels to show disappearance of the heavily
polyubiquitinated band, which were subsequently quantified by ImageJ analysis.
MG132 alone appears to only decrease degradation by about 25% compared to
the uninhibited sample, which is understandable because it only inhibits one of
the catalytic subunits of the 20S, rather than all three. UbVME alone shows a
decrease of 50% in degradation, similar to the results shown above in Figure 6.5.
However, the combination of MG132 and UbAMC slows degradation by >90%
compared to the uninhibited sample. As this amount is even greater than the
additive 75% of the two inhibitors alone, this result indicates that significant
inhibition of degradation is occurring in a coupled deubiquitination-degradation
mechanism.
6.4 Further Directions
Investigation into the coupling of UCH37/USP14 deubiquitination and
degradation by the 26S proteasome could prove vital to our understanding of all
protein degradation, but especially how these deubiquitinases may be the first
step in regulation of this cellular machine. The experiments addressed here
indicate that deubiquitination may be coupled to degradation, however, it is
necessary to separate out the effects of deubiquitination by Rpn11 before any
conclusions can be made. This could be achieved by incubation of proteasomes

114
with 1,10-phenanthroline, a known inhibitor of Rpn1112,15-17. It is necessary to
determine if deubiquitination by the cysteine protease deubiquitinases has a
separate function from that of Rpn11 and which level of deubiquitination
contributes most to slowing the rate of proteasomal degradation.
Additionally, the assays described here rely on SDS PAGE gel analysis of
the disappearance of a band of highly polyubiquitinated GFP substrates,
however, disappearance does not isolate deubiquitination from degradation.
These experiments are currently being pursued by a labmate, Michael Sheedlo,
using a T7 probe and polyubiquitinated Sic1, another proteasome substrate, to
determine the contributions of deubiquitination vs degradation. More specific
answers to these questions are needed before we can definitively say that
deubiquitination by UCH37 and USP14 are indeed coupled to and a contributing
factor during proteasomal degradation.

115
6.5 References
1 Lam, Y. A., DeMartino, G. N., Pickart, C. M. & Cohen, R. E. Specificity of the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. The Journal of biological chemistry 272, 28438-28446 (1997).
2 Lam, Y. A., Xu, W., DeMartino, G. N. & Cohen, R. E. Editing of ubiquitin
conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737-740 (1997).
3 Yao, T. et al. Proteasome recruitment and activation of the Uch37
deubiquitinating enzyme by Adrm1. Nature cell biology 8, 994-1002 (2006).
4 Besche, H. C. & Goldberg, A. L. Affinity purification of mammalian 26S
proteasomes using an ubiquitin-like domain. Methods in molecular biology 832, 423-432, doi:10.1007/978-1-61779-474-2_29 (2012).
5 Besche, H. C., Haas, W., Gygi, S. P. & Goldberg, A. L. Isolation of
mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48, 2538-2549, doi:10.1021/bi802198q (2009).
6 Hiyama, H. et al. Interaction of hHR23 with S5a. The ubiquitin-like domain
of hHR23 mediates interaction with S5a subunit of 26 S proteasome. The Journal of biological chemistry 274, 28019-28025 (1999).
7 Elsasser, S. et al. Proteasome subunit Rpn1 binds ubiquitin-like protein
domains. Nature cell biology 4, 725-730, doi:10.1038/ncb845 (2002). 8 Koulich, E., Li, X. & DeMartino, G. N. Relative structural and functional
roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Molecular biology of the cell 19, 1072-1082 (2008).
9 Lee, B. H. et al. Enhancement of proteasome activity by a small-molecule
inhibitor of USP14. Nature 467, 179-184, doi:10.1038/nature09299 (2010). 10 Beckwith, R., Estrin, E., Worden, E. J. & Martin, A. Reconstitution of the
26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nature structural & molecular biology 20, 1164-1172, doi:10.1038/nsmb.2659 (2013).

116
11 Matyskiela, M. E., Lander, G. C. & Martin, A. Conformational switching of the 26S proteasome enables substrate degradation. Nature structural & molecular biology 20, 781-788, doi:10.1038/nsmb.2616 (2013).
12 Bush, K. T., Goldberg, A. L. & Nigam, S. K. Proteasome inhibition leads to
a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 272, 9086-9092 (1997).
13 Stein, M. L. et al. Systematic comparison of peptidic proteasome inhibitors
highlights the alpha-ketoamide electrophile as an auspicious reversible lead motif. Angewandte Chemie 53, 1679-1683, doi:10.1002/anie.201308984 (2014).
14 Peth, A., Kukushkin, N., Bosse, M. & Goldberg, A. L. Ubiquitinated
proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. The Journal of biological chemistry 288, 7781-7790, doi:10.1074/jbc.M112.441907 (2013).
15 Cooper, E. M. et al. K63-specific deubiquitination by two JAMM/MPN+
complexes: BRISC-associated Brcc36 and proteasomal Poh1. The EMBO journal 28, 621-631, doi:10.1038/emboj.2009.27 (2009).
16 Verma, R. et al. Role of Rpn11 metalloprotease in deubiquitination and
degradation by the 26S proteasome. Science 298, 611-615, doi:10.1126/science.1075898 (2002).
17 Guterman, A. & Glickman, M. H. Complementary roles for Rpn11 and
Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem 279, 1729-1738, doi:10.1074/jbc.M307050200 (2004).

VITA

117
VITA
Marie Morrow was born in Winston-Salem, North Carolina to Jamie and
Christine Morrow. She grew up in Jacksonville, Florida and went to Stanton
College Preparatory School where she first came to love chemistry in Missy
Ray’s AP/IB Chemistry class. She went on to study chemistry at the University of
Florida where she did undergraduate research in the lab of Carrie Haskell-
Luevano, developing peptide inhibitors for melanocortin receptors. After
graduating with her Bachelor’s degree, she went to Purdue University to pursue
her Ph.D in chemistry. In Chitta Das’s group, she has studied the structure and
biophysical/biochemical properties of the proteasomal deubiquitinase UCH37.
After graduating, she will start a post-doctoral research position in the lab of
Cynthia Wolberger, Johns Hopkins University School of Medicine, studying the
structure and function of ubiquitination/deubiquitination machinery.

PUBLICATIONS

Stabilization of an Unusual Salt Bridge in Ubiquitin by the ExtraC‑Terminal Domain of the Proteasome-Associated DeubiquitinaseUCH37 as a Mechanism of Its Exo SpecificityMarie E. Morrow,† Myung-Il Kim,† Judith A. Ronau,† Michael J. Sheedlo,† Rhiannon R. White,‡
Joseph Chaney,† Lake N. Paul,§ Markus A. Lill,∥ Katerina Artavanis-Tsakonas,‡ and Chittaranjan Das*,†
†Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, United States‡Division of Cell and Molecular Biology, Imperial College London, Sir Alexander Fleming Building, Imperial College Road, LondonSW7 2AZ, U.K.§Bindley Biosciences Center, Purdue University, West Lafayette, Indiana 47907, United States∥Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 575 Stadium Mall Drive, West Lafayette,Indiana 47907, United States
*S Supporting Information
ABSTRACT: Ubiquitination is countered by a group ofenzymes collectively called deubiquitinases (DUBs); ∼100 ofthem can be found in the human genome. One of the mostinteresting aspects of these enzymes is the ability of somemembers to selectively recognize specific linkage typesbetween ubiquitin in polyubiquitin chains and their endoand exo specificity. The structural basis of exo-specificdeubiquitination catalyzed by a DUB is poorly understood.UCH37, a cysteine DUB conserved from fungi to humans, is aproteasome-associated factor that regulates the proteasome bysequentially cleaving polyubiquitin chains from their distal ends, i.e., by exo-specific deubiquitination. In addition to the catalyticdomain, the DUB features a functionally uncharacterized UCH37-like domain (ULD), presumed to keep the enzyme in aninhibited state in its proteasome-free form. Herein we report the crystal structure of two constructs of UCH37 from Trichinellaspiralis in complex with a ubiquitin-based suicide inhibitor, ubiquitin vinyl methyl ester (UbVME). These structures show thatthe ULD makes direct contact with ubiquitin stabilizing a highly unusual intramolecular salt bridge between Lys48 and Glu51 ofubiquitin, an interaction that would be favored only with the distal ubiquitin but not with the internal ones in a Lys48-linkedpolyubiquitin chain. An inspection of 39 DUB−ubiquitin structures in the Protein Data Bank reveals the uniqueness of the saltbridge in ubiquitin bound to UCH37, an interaction that disappears when the ULD is deleted, as revealed in the structure of thecatalytic domain alone bound to UbVME. The structural data are consistent with previously reported mutational data on themammalian enzyme, which, together with the fact that the ULD residues that bind to ubiquitin are conserved, points to a similarmechanism behind the exo specificity of the human enzyme. To the best of our knowledge, these data provide the only structuralexample so far of how the exo specificity of a DUB can be determined by its noncatalytic domain. Thus, our data show that,contrary to its proposed inhibitory role, the ULD actually contributes to substrate recognition and could be a major determinantof the proteasome-associated function of UCH37. Moreover, our structures show that the unproductively oriented catalyticcysteine in the free enzyme is aligned correctly when ubiquitin binds, suggesting a mechanism for ubiquitin selectivity.
The ubiquitin proteasome system (UPS), present in alleukaryotes, is responsible for the majority of controlled
degradation and recycling of proteins within the cell.1−5
Polyubiquitinated, and to some extent monoubiquitinated,proteins are recognized and degraded by the 26S proteasome, a2.5 MDa self-compartmentalizing proteolytic complex.6−13 It iscomposed of two major units: the 20S core particle (CP)consisting of 28 subunits and the 19S regulatory particle (RP)containing 19 subunits in yeast. The proteolytic active sites arehoused within the luminal chamber of the barrel-shaped CP,capped on both ends by the RP, which contains ubiquitinreceptors and enzymes that prepare substrates for degradation.
Entry of substrates into the CP is regulated by the RP, primarilyby opening and closing of the substrate translocation channel.Before the substrate is translocated into the narrow channelleading to the lumen of the CP, it is obligatorily deubiquitinatedwith the help of the RP-resident JAMM metalloproteaseRpn1114−16 and unfolded by Rpt subunits that sit within thebase subcomplex of the RP.7,9,14 However, additional regulation
Received: March 11, 2013Revised: April 25, 2013Published: April 25, 2013
Article
pubs.acs.org/biochemistry
© 2013 American Chemical Society 3564 dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−3578
118

is performed by proteasome-associated deubiquitinatingenzymes, whose underlying mechanism is still poorly under-stood.7,17
Attachment of ubiquitin to a lysine residue(s) on targetproteins is catalyzed by the sequential action of three enzymaticsystems: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating),and E3 (ubiquitin-ligating) enzymes.18,19 Usually, ubiquitina-tion of a target protein results in the attachment of apolyubiquitin chain in which successive ubiquitin moieties areattached to one of the seven lysines, or the N-terminal aminogroup of the preceding monomer, to generate a homopolymericstructure.18,20 Polyubiquitin chains of a distinct topology arethus generated depending on which amino group of ubiquitin isused for chain extension (lysines 6, 11, 27, 29, 33, 48, and 63 orthe amino group of Met1). A polyubiquitin chain of a specifictopology is meant for a specific type of functional out-come.20−25 For example, a Lys48 (K48)-linked chain usuallyserves as the signal for proteasomal degradation, whereas K63chains signal other types of functions such as endocytosis, DNArepair, and NF-κB signaling.24,26
Ubiquitination works as a reversible post-translationalmodification, like phosphorylation. Deubiquitinating enzymes,or DUBs, can hydrolytically remove ubiquitin from proteinadducts, thereby opposing the action of ubiquitin conjugatingmachinery.27−33 Consequently, DUBs have been found to playimportant regulatory roles in numerous ubiquitin-dependentcellular processes.32−35 In mechanistic terms, these enzymescan be categorized into two main groups: cysteine proteasesand zinc metalloproteases. The zinc metalloproteases consist ofonly one family, the JAB1/MPN/MOV34 metalloenzymes(JAMMs). The cysteine proteases are further broken down intofour families based on the structure of their catalytic domain:ubiquitin carboxyl-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs),and Machado-Josephin domain proteases (MJDs).32
UCH37 (also known as UCHL5) is a 37 kDa DUB of theUCH family and is one of the two proteasome-associatedDUBs, the other being USP14 (Ubp6 in yeast), known toregulate protein degradation by the mammalian protea-some.36−40 These associated DUBs, along with Rpn11, aconstitutive member of the RP, conduct deubiquitination at theproteasome. However, the activities of the three enzymes aredistinct. Rpn11 is responsible for en-bloc removal ofpolyubiquitin chains prior to (or concurrent with) unfoldingand translocation of the substrate into the CP, an activity thatappears to be coupled to substrate degradation.15−17 USP14and UCH37 on the other hand are known to have chain-trimming functions.17,37,41 The importance of these associatedDUBs to proteasome function was revealed throughpharmacological inhibition of these enzymes. A small-moleculeinhibitor of USP14 appears to accelerate proteasomaldegradation of certain substrates, whereas UCH37 inhibitioncan stall proteolysis, consistent with distinct functional rolesplayed by the two enzymes.42−44
UCH37 was first identified as the PA700 isopeptidase, thecysteine DUB tightly associated with the RP, also known asPA700.38,45,46 Like other UCH family members, it contains aconserved catalytic triad of a cysteine, a histidine, and anaspartate. UCH37 has a canonical UCH domain that is 45%similar to UCHL1 and 49% similar to UCHL3, its single-domain family members.47−50 It also has an additional C-terminal tail domain responsible for its interaction with theRpn13 subunit of the RP.51−54 Proteasome-bound UCH37 is
thought to behave as an “editor”, relieving poorly ubiquitinatedsubstrates from degradation by sequentially dismantling theirK48-linked polyubiquitin chains from the very distal end,removing one ubiquitin at a time.37,38,45 Such a type of chaindisassembling activity can be termed as an exo cleavage activityin contrast to the endo activity, which leads to dismantling ofchains by cleavage between internal ubiquitins. Although it hasrespectable UbAMC (ubiquitin aminomethylcoumarin) hydrol-ysis activity in its unbound form, UCH37 has been shown torequire association with the proteasome to cleave diubiquitin(and polyubiquitin) chains.37 Additionally, its UbAMChydrolysis activity is enhanced upon binding with Rpn13.37,54
Interestingly, UCH37 also associates in the nucleus with thehuman Ino80 chromatin remodeling complex, where it is heldin an inactive state compared to the free enzyme.55 It thusserves as an example of a DUB whose catalytic activity is bothpositively and negatively regulated by binding to specificprotein partners, making it an attractive target for structuralstudies. Crystal structures have been determined for both thecatalytic domain and full-length human UCH37;56−58 however,the mechanism of its catalytic regulation upon binding toassociated protein factors is not known. Any mechanisticunderstanding of its regulation must require structuralinformation about UCH37 and its catalytic domain bound toubiquitin, which has yet to be reported.TsUCH37 is a recently characterized lower-organism
homologue of UCH37 from Trichinella spiralis (Ts), aninfectious helminth found nearly worldwide. TsUCH37 wasidentified by White et al. by incubation of the whole-cell lysateof Ts larvae with the HA-UbVME probe (HA, thehemagglutinin epitope, fused with the N-terminus of ubiquitinvinyl methyl ester), an epitope-tagged irreversible inhibitor ofcysteine DUBs.59 Its structural and functional homology withhuman UCH37 was then confirmed by sequence analysis, co-immunoprecipitation with proteasomal subunits, and UbAMChydrolysis assays. TsUCH37 is 45% identical to its humanhomologue and was shown to pull down TsADRM1, thecorresponding Rpn13 homologue, by co-immunoprecipita-tion.59 The sequence and functional conservation betweenthe Ts and human enzymes implies a similar chain-editing roleof the former at the proteasome. To understand themechanisms associated with UCH37, we have crystallized twoconstructs of TsUCH37 bound to ubiquitin vinyl methyl ester.The structures illuminate the mode of ubiquitin recognition inthe enzyme by revealing binding interactions with the catalyticdomain, which are conserved among UCH enzymes, andinteractions unique to UCH37, notably ubiquitin binding bythe ULD, providing further explanation of the proteasome-associated exo-specific deubiquitination activity of the DUB.
■ MATERIALS AND METHODSCloning, Expression, and Purification. TsUCH37cat.
TsUCH37cat (residues 1−226) was subcloned from the full-length construct (residues 1−309) in pET28a(+) into pGEX-6P-1 (GE Biosciences) using BamHI and XhoI restriction sites.The protein was expressed in Escherichia coli Rosetta cells(Novagen) grown at 37 °C in LB medium containing 100 μg/Lampicillin to an OD600 of 1.0 and then induced with 0.5 mMisopropyl β-D-thiogalactoside (IPTG) at 18 °C for 16 h.Harvested cells were resuspended in lysis buffer (1× phosphatebuffered saline and 400 mM KCl) and lysed with a Frenchpress. The lysate was then purified on a glutathione S-transferase (GST) column (GE Biosciences) followed by
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783565
119

cleavage of the GST tag by PreScission Protease (GEBiosciences) per the manufacturer’s instructions. It was furtherpurified by size exclusion chromatography on a Superdex 75column (GE Biosciences). Intein-fused ubiquitin1−75 in pTXB1was expressed in E. coli Rosetta cells and purified on chitinbeads (New England Biosciences). Ubiquitin vinyl methyl ester(UbVME) was synthesized by overnight incubation of Ub1−75-MESNa (MESNa, sodium mercaptoethanesulfonate) withglycine vinyl methyl ester and then purified on a MonoScation exchange column (GE Biosciences). Glycine vinylmethyl ester was synthesized by a modified, previouslypublished procedure.60 TsUCH37cat was reacted withUbVME for 4 h, followed by purification on a MonoQ anionexchange column (GE Biosciences) to separate any unreactedTsUCH37cat. Selenomethionine TsUCH37cat protein (SeMetTsUCH37cat) was grown in M9 minimal medium supple-mented with selenomethionine, reacted with UbVME, andpurified as described above.TsUCH37ΔC46. TsUCH37FL was subcloned previously into
pET28a(+) with an N-terminally fused His tag (Novagen).TsUCH37FL was expressed in E. coli Rosetta cells, grown at 37°C in LB medium containing 10 μg/L kanamycin to an OD600of 0.8, and then induced with 0.5 mM IPTG at 18 °C for 16 h.Harvested cells were resuspended in lysis buffer [50 mM Tris-HCl (pH 7.6), 200 mM NaCl, and 3 mM β-mercaptoethanol]and lysed with a French press. His-tagged TsUCH37FL waspurified by immobilized metal affinity chromatography (IMAC)and eluted with lysis buffer including 500 mM imidazole. Elutedproteins were further purified by size exclusion chromatography(SEC) on a Superdex 75 column (GE Biosciences) in 50 mMHEPES (pH 7.6) and 3 mM dithiothreitol (DTT). SDS−PAGE on the fractions indicated a cleavage of the full-lengthprotein, so the construct described is actually a proteolyticcleavage product of the full-length protein. The crystal structure(described below) lacks density for the last 46 amino acids fromthe C-terminus; therefore, this construct will hereafter bedescribed as TsUCH37ΔC46. Fractions containing the targetprotein were pooled, concentrated, and reacted with UbVME.UbVME was synthesized and reacted with purifiedTsUCH37ΔC46 as was done with TsUCH37cat. To separateunreacted TsUCH37ΔC46, the complex was further purified bySEC on a Superdex 75 column (GE Biosciences).Crystallization and Structure Determination.
TsUCH37cat−UbVME Complex. The TsUCH37cat−UbVMEcomplex was concentrated to 3 mg/mL in 50 mM Tris (pH7.6), 200 mM NaCl, and 1 mM DTT. Crystals were grown in 2days at room temperature by hanging drop vapor diffusion in 3M ammonium sulfate and 0.1 M bicine (pH 9.0) with 2 mM L-glutathione (mixture of oxidized and reduced) additive.Crystals were cryoprotected in 2.5 M sodium malonate andflash-frozen in liquid nitrogen.61 Diffraction data were collectedon a Mar300 CCD detector (Mar USA) at the 23-ID-Bbeamline at Argonne National Laboratory (Argonne, IL). Dataup to 1.7 Å were collected on SeMet TsUCH37cat−UbVMEcrystals at the selenium peak (0.979 Å) for SAD (single-wavelength anomalous dispersion) phasing. Data wereprocessed with HKL2000.62
The initial model was obtained by Se-SAD phasing in thePhenix AutoSol wizard.63 Its sequence was built in using thePhenix AutoBuild wizard, as well as manual model building inCoot.63,64 Structural refinement was conducted in Phenix usingTLS refinement (with the entire asymmetric unit taken as oneTLS group), as well as optimized weighting for stereochemical
restraints.63 The data were run through Phenix Xtriage, whichconfirmed the chosen space group, C2, and did not detectevidence of crystal twinning.63 The completeness of thecrystallographic data for the TsUCH37cat−UbVME complexwas less than ideal (see Table 2); however, this did not hinderthe determination of the structure or the generation of thestructural model presented herein and can be ascribed to poorcompleteness in the highest-resolution shells.
TsUCH37ΔC46−UbVME Complex. The TsUCH37ΔC46−UbVME complex was concentrated to 5 mg/mL in 50 mMHEPES (pH 7.6) and 2 mM DTT. Crystals were grown in 60days at room temperature in 0.2 M ammonium chloride (pH5.8) and 18% PEG3350. Crystals were cryoprotected inethylene glycol and flash-frozen in liquid nitrogen. Diffractiondata were collected on a Mar300 CCD detector (Mar USA) atthe 23-ID-B beamline at Argonne National Laboratory. Data upto 2.0 Å were collected on TsUCH37ΔC46−UbVME crystals at1.033 Å. Data were processed with HKL2000.62
The initial model was obtained by molecular replacementusing the Phenix AutoMR wizard, with a monomer of theTsUCH37cat−UbVME complex as the search model.63 Manualmodel building was conducted in Coot, and structuralrefinement was conducted initially in Refmac using TLSrefinement and then using simulated annealing and individual Bfactor refinement in Phenix.63,64 The data were run throughPhenix Xtriage, which confirmed the chosen space group, R3,and did not detect any evidence of crystal twinning.63
UbAMC Hydrolysis Assay. TsUCH37cat was diluted inreaction buffer [50 mM Tris (pH 7.6), 0.5 mM EDTA, 0.1%bovine serum albumin, and 5 mM DTT] to a final reactionconcentration of 7 nM and preincubated at 30 °C for 5 minprior to the addition of the UbAMC substrate (BostonBiochem). UbAMC cleavage was measured on a Tecan(Mannedorf, Switzerland) fluorescence plate reader with 380nm excitation and 465 nm emission wavelengths at 30 °C. Datawere fit to Michaelis−Menten kinetics in SigmaPlot (SystatSoftware, San Jose, CA).
Analytical Ultracentrifugation. Sedimentation velocityexperiments were conducted with the Beckman-Coulter XLAanalytical ultracentrifuge. The sample was extensively dialyzedagainst 50 mM Tris-HCl, 200 mM NaCl, and 1 mM DTT (pH7.4). The TsUCH37cat and TsUCH37cat−UbVME complexconcentration ranged from 10 to 32 μM. The samples werecentrifuged at 50000 rpm using a two-sector 1.2 cm path-lengthcarbon-filled Epon centerpiece. The experiments were con-ducted on an An-50 Ti rotor at 20 °C. The density and relativeviscosity of the buffers were calculated using SEDNTERPversion 1.09 (http://www.rasmb.bbri.org/rasmb/windows/sednterp-philo): 1.0079 g/mL and 0.01036 P, respectively.The partial specific volume (vbar) of the protein was alsocalculated from the protein sequence using SEDNTERP(0.7340 mL/g for TsUCH37cat and 0.7317 mL/g for theTsUCH37cat−UbVME complex). The samples were monitoredat 280 nm with a 4 min delay and 150 scans. The c(s)distributions were analyzed using SEDFIT version 13.0b.65
Molecular Dynamics Simulations. A model of full-lengthTsUCH37 was generated by the SwissModel homologymodeling server using the structure of the full-length humanprotein as a template.66 Missing ULD residues produced by themodel were appended to the TsUCH37ΔC46−UbVMEstructure in Coot, and a single round of refinement wasconducted in Phenix, to produce a final model hereafter termed“the system”.63,64 The system was solvated in a box of TIP3P
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783566
120
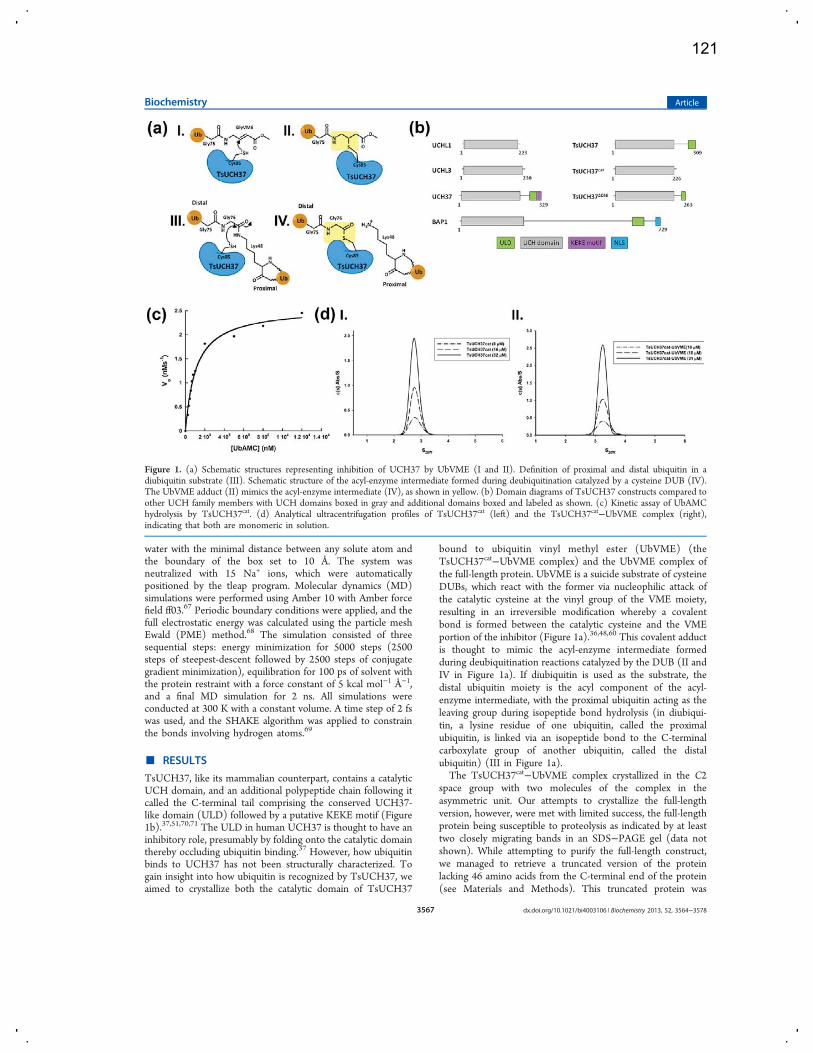
water with the minimal distance between any solute atom andthe boundary of the box set to 10 Å. The system wasneutralized with 15 Na+ ions, which were automaticallypositioned by the tleap program. Molecular dynamics (MD)simulations were performed using Amber 10 with Amber forcefield ff03.67 Periodic boundary conditions were applied, and thefull electrostatic energy was calculated using the particle meshEwald (PME) method.68 The simulation consisted of threesequential steps: energy minimization for 5000 steps (2500steps of steepest-descent followed by 2500 steps of conjugategradient minimization), equilibration for 100 ps of solvent withthe protein restraint with a force constant of 5 kcal mol−1 Å−1,and a final MD simulation for 2 ns. All simulations wereconducted at 300 K with a constant volume. A time step of 2 fswas used, and the SHAKE algorithm was applied to constrainthe bonds involving hydrogen atoms.69
■ RESULTS
TsUCH37, like its mammalian counterpart, contains a catalyticUCH domain, and an additional polypeptide chain following itcalled the C-terminal tail comprising the conserved UCH37-like domain (ULD) followed by a putative KEKE motif (Figure1b).37,51,70,71 The ULD in human UCH37 is thought to have aninhibitory role, presumably by folding onto the catalytic domainthereby occluding ubiquitin binding.37 However, how ubiquitinbinds to UCH37 has not been structurally characterized. Togain insight into how ubiquitin is recognized by TsUCH37, weaimed to crystallize both the catalytic domain of TsUCH37
bound to ubiquitin vinyl methyl ester (UbVME) (theTsUCH37cat−UbVME complex) and the UbVME complex ofthe full-length protein. UbVME is a suicide substrate of cysteineDUBs, which react with the former via nucleophilic attack ofthe catalytic cysteine at the vinyl group of the VME moiety,resulting in an irreversible modification whereby a covalentbond is formed between the catalytic cysteine and the VMEportion of the inhibitor (Figure 1a).36,48,60 This covalent adductis thought to mimic the acyl-enzyme intermediate formedduring deubiquitination reactions catalyzed by the DUB (II andIV in Figure 1a). If diubiquitin is used as the substrate, thedistal ubiquitin moiety is the acyl component of the acyl-enzyme intermediate, with the proximal ubiquitin acting as theleaving group during isopeptide bond hydrolysis (in diubiqui-tin, a lysine residue of one ubiquitin, called the proximalubiquitin, is linked via an isopeptide bond to the C-terminalcarboxylate group of another ubiquitin, called the distalubiquitin) (III in Figure 1a).The TsUCH37cat−UbVME complex crystallized in the C2
space group with two molecules of the complex in theasymmetric unit. Our attempts to crystallize the full-lengthversion, however, were met with limited success, the full-lengthprotein being susceptible to proteolysis as indicated by at leasttwo closely migrating bands in an SDS−PAGE gel (data notshown). While attempting to purify the full-length construct,we managed to retrieve a truncated version of the proteinlacking 46 amino acids from the C-terminal end of the protein(see Materials and Methods). This truncated protein was
Figure 1. (a) Schematic structures representing inhibition of UCH37 by UbVME (I and II). Definition of proximal and distal ubiquitin in adiubiquitin substrate (III). Schematic structure of the acyl-enzyme intermediate formed during deubiquitination catalyzed by a cysteine DUB (IV).The UbVME adduct (II) mimics the acyl-enzyme intermediate (IV), as shown in yellow. (b) Domain diagrams of TsUCH37 constructs compared toother UCH family members with UCH domains boxed in gray and additional domains boxed and labeled as shown. (c) Kinetic assay of UbAMChydrolysis by TsUCH37cat. (d) Analytical ultracentrifugation profiles of TsUCH37cat (left) and the TsUCH37cat−UbVME complex (right),indicating that both are monomeric in solution.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783567
121

purified by Ni affinity chromatography and reacted withUbVME, and the complex was purified using ion-exchangechromatography. This complex, hereafter termed theTsUCH37ΔC46−UbVME complex (TsUCH37 missing the last46 residues), crystallized in the R3 space group with onecomplex in the asymmetric unit.The catalytic activity of TsUCH37cat was measured with a
UbAMC hydrolysis assay (Figure 1c), which yieldedMichaelis−Menten parameters as shown in Table 1. Compared
to the catalytic domain of human UCH37, TsUCH37cat has anapproximately 20-fold lower KM, indicating a higher affinity forthis substrate compared to that of the human protein, but a100-fold lower kcat, a substantially lower turnover number.Consequently, TsUCH37cat is nearly 5-fold less efficient thanthe UCH domain of human UCH37.Crystals of the TsUCH37cat−UbVME complex diffracted to
1.7 Å. The structure was determined by single-wavelengthanomalous dispersion (SAD) using anomalous scattering fromselenium (TsUCH37cat was labeled with selenium). Manualmodel building using Coot, followed by multiple rounds ofrefinement using Phenix, produced a final model with an Rfactor of 17.4% and an Rfree of 21% (see Table 2 forcrystallographic and refinement parameters).63,64 The finalrefined model corresponding to the asymmetric unit consists oftwo copies of the TsUCH37cat−UbVME complex, composed ofTsUCH37cat, residues 1−226, covalently connected via athioether bond linking the catalytic cysteine with the VMEgroup of UbVME (residues 1−75 of ubiquitin attached toGlyVME as the 76th residue, which is modeled as methyl 4-amino butanoate). The refined model was of high stereo-chemical quality, with <0.2% of residues in the disallowedregion of the Ramachandran plot and scoring in the upper 98%according to Molprobity evaluation.72 The structure of theTsUCH37ΔC46−UbVME complex (2.0 Å resolution) wasdetermined by molecular replacement using theTsUCH37cat−UbVME structure as the search model (Table2). The final refined model with good stereochemical quality(<0.2% of residues in the disallowed region of theRamachandran plot and Molprobity score of 63%) has aminoacids 5−263 of the protein and one UbVME linked via athioether bond to the catalytic cysteine. The structures of theUCH domain in the two constructs are very similar, except fortwo loop regions (see below), with Cα root-mean-squaredeviations (rmsds) of 0.32 Å between the two (the loop regionswere excluded from the calculation of the rmsd). Whendiscussing the structure of the UCH domain alone or itsinteraction with UbVME, we will therefore use the structure ofthe TsUCH37cat−UbVME complex because its resolution ishigher while specifically mentioning any structural feature thatis different in the UCH domain of TsUCH37ΔC46−UbVMEcomplex.Initial analysis of the structure revealed that the two copies in
the asymmetric unit of TsUCH37cat−UbVME crystals are
linked by a disulfide bond between Cys71 of the twoTsUCH37cat chains (Figure 2a). It is possible that the disulfidebond forms because the protein exists as a dimer in solution,bringing the cysteines into proximity of each other, or is a resultof crystallographic packing. To determine if this disulfide is acrystallographic artifact or a biologically relevant association, wedetermined the oligomerization state of complexed anduncomplexed (apo) TsUCH37cat by sedimentation velocityanalytical ultracentrifugation (AUC). We found that both thecomplex and the apo protein exist as monomers in solutionwith sedimentation coefficients (S20,w) of 3.3 and 2.8,respectively (Figure 1d), indicating that this disulfide is likelya result of crystal packing. TsUCH37 is expected to bepredominantly localized to the cytosol, a reducing environment,and therefore should not rely on disulfide-mediated dimeriza-tion for catalytic activity. Moreover, the observation that the
Table 1. Kinetic Parameters for TsUCH37cat
enzyme KM (nM) kcat (s−1) kcat/KM (×105 M−1 s−1)
TsUCH37cat 1085 0.37 3.4UCH37N240a 21493 34 16UCHL3a 77.1 19 2414UCHL1a 47.0 0.03 7.4
aKinetic parameters previously determined, from ref 75.
Table 2. Crystallographic and Refinement Statisticsa
SeMet TsUCH37cat−UbVME
TsUCH37ΔC46−UbVME
Data Collectionspace group C121 R3cell dimensions
a, b, c (Å) 171.2, 55.8, 73.9 147.4, 147.4, 40.5α, β, γ (deg) 90, 113.4, 90 90, 90, 120
wavelength (Å) 0.979 1.033resolution (Å) 50.00−1.70 (1.73−1.70) 50−2.0 (2.03−2.00)Rsym or Rmerge
b (%) 8.7 (50.0) 8.5 (83.8)I/σI 15.9 (3.0) 4.9 (4.1)completeness (%) 88.5 (42.0) 100.0 (100.0)redundancy 6.8 (3.5) 5.8 (5.7)
Refinementresolution (Å) 27.9−1.7 38.6−2.0no. of uniquereflections
62326/3126 22270/2002
Rworkc/Rfree
d 17.4/21.1 19.3/24.0no. of atoms
proteine 4650 2428ligand 24 8water 437 100
average B factor(Å2)
protein 36.2 43.4ligand 36.5 32.4water 44.0 44.3
rmsdbond lengths(Å)
0.013 0.009
bond angles(deg)
1.48 1.07
Ramachandran plot(%)
favored 98.1 97.7allowed 1.6 1.0outliers 0.4 1.3
aNumbers in parentheses refer to data in the highest-resolution shell.bRmerge = ∑|Ih − ⟨Ih⟩|/∑Ih, where Ih is the observed intensity and ⟨Ih⟩is the average intensity. cRwork = ∑||Fobs| − k|Fcal||/∑|Fobs|.
dRfree is thesame as Robs for a selected subset (5 and 9%) of the reflections thatwas not included in prior refinement calculations. eOrdered residues:Pro3−Gly141 and Lys153−Asp224 in chain C and Pro3−Gly141 andGln152−Gln225 in chain A of the SeMet TsUCH37cat−UbVMEstructure and Gly4−Lys57, Thr72−Gly141, and Glu157−Ala263 ofthe TsUCH37ΔC46−UbVME structure.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783568
122

TsUCH37ΔC46−UbVME complex is a monomer in theasymmetric unit and that the segment of residues 57−71,which is used as a part of the dimer interface in the crystals ofthe TsUCH37cat−UbVME complex, is disordered in theTsUCH37ΔC46−UbVME structure (Figures 2 and 3) supports
the notion that the dimer observed in the TsUCH37cat−UbVME structure is a crystallographic dimer and may not existin solution. The two copies of the complex in the dimerobserved in the crystals of the TsUCH37cat−UbVME complexhave very similar structures with an rmsd of 0.39 Å between Cαatoms. We will therefore focus on one of them in discussionspresented below.Overall Structure of the UCH Domain of TsUCH37.
The overall structure of the TsUCH37 catalytic domain issimilar to that of other structurally characterized UCHenzymes.49,50,73 It has the classical αβα fold, in which a centralsix-stranded β-sheet is surrounded by six α-helices, five on oneside (α1−α5) and one on the other (α6) (Figure 3). The
overall architecture of TsUCH37cat can be seen as bilobal, withone of the lobes comprising helices α1−α5 and the othercomprising the β-sheets and helix α6. The active site is locatedat the interface of the two lobes, with Cys85 from helix α2 inone lobe and His161 from β3 in the other forming the catalyticCys-His pair. An adjacent loop provides the third member ofthe triad, Asp176. Most of the secondary structural elementsseen in TsUCH37cat are conserved in UCHL1 and UCHL3,with the only noticeable difference being the conformation of asegment following β2, residues 57−71. This segment is a helixin UCHL1 and UCHL3 and is in somewhat of an extendedlooplike conformation in human UCH37 (hUCH37) but isfairly ordered; in the structure of the various constructs ofhuman UCH37 determined so far, this loop has been found tobe in a similar conformation regardless of crystallographicpacking (Figure S1 of the Supporting Information).56−58,74 Incontrast, this segment appears to be flexible in TsUCH37 and isvisualized only in the TsUCH37cat−UbVME structure, in whichit forms the dimer interface between the two subunits in theasymmetric unit. In the TsUCH37ΔC46−UbVME complex, acrystallographic monomer, this loop is disordered (Figure 3).Although the possibility that its binding can influence the loopdynamics cannot be ruled out, it is unlikely that UbVME hasanything to do with the dynamic behavior of the loop because itdoes not bind to it. We therefore propose that the loop isintrinsically flexible in TsUCH37 but can become orderedunder certain circumstances, such as under the constraints ofcrystallographic packing.It is possible that the corresponding loop segment in
hUCH37 is somewhat dynamic as well, but it appears to besignificantly more flexible in TsUCH37. The significance of thisdifference in dynamics between the two proteins is not clear atthe moment. Intriguingly, the loop’s dynamic behavior appearsto have an effect on the conformation of a tryptophan residue(Trp55) adjacent to the active site (Figure S2 of the SupportingInformation). This tryptophan is conserved among Schizo-saccharomyces pombe (Sp), Ts, and human UCH37 (Figure S3
Figure 2. Crystal structures of TsUCH37 constructs bound to UbVME. (a) Dimeric structure of TsUCH37cat bound to UbVME (orange) incrystals. Monomers are colored teal (chain A) and gray (chain C). The inset shows the disulfide bridge that links the two subunits via Cys71. Theelectron density is rendered from the 2Fo − Fc map contoured at 1σ. (b) Monomer of the TsUCH37cat−UbVME structure. (c) Structure of theTsUCH37ΔC46−UbVME complex, with TsUCH37ΔC46 colored olive and UbVME orange.
Figure 3. Secondary structures of TsUCH37 constructs.TsUCH37ΔC46−UbVME and TsUCH37cat−UbVME complexes aresuperposed with α-helices and 310-helices colored pale yellow, β-sheetsblue, loops green, and UbVME orange. Arrows indicate where theTsUCH37ΔC46−UbVME structure lacks density, compared to theTsUCH37cat−UbVME structure, from residue 57 to 71.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783569
123

of the Supporting Information). In the TsUCH37ΔC46−UbVME complex, Trp55 makes contact with the OMe groupof the suicide inhibitor, which in the actual substrate (aubiquitinated protein or the diubiquitin motif of a polyubiquitinchain) would be replaced by the hydrocarbon portion of theisopeptide-linked lysine side chain (Figure 1a). The sameresidue in the TsUCH37cat−UbVME complex shows a differentorientation with respect to the OMe group and appears to haveadopted a more open position for interaction with theisopeptide unit (Figure S2 of the Supporting Information).Therefore, Trp55 not only may provide important contactswith the isopeptide link to hold it in place near the active sitebut also may confer a certain plasticity to the active site ofUCH37, which may be useful for an induced-fit type ofengagement with the substrate.As stated before, in the TsUCH37ΔC46−UbVME structure,
we are able to visualize 40 additional amino acids after theUCH domain, the first 41 amino acids (residues 223−263) ofthe ULD in TsUCH37. The polypeptide chain, after emergingfrom the C-terminus of the UCH domain, adopts a helicalstructure of six turns (α7), takes a U-turn, and then continuesas a helix (α8). α7 and α8 are arranged as a helix−turn−helixmotif with a number of interhelical contacts, and this motifadopts a similar orientation with respect to the UCH domain asobserved in hUCH37 (Figures 3 and 4b).57 The only differencein this motif between TsUCH37 and hUCH37 is that it issomewhat shorter in the former. The ULD in TsUCH37appears to have a proteolytically susceptible region afterAla263, perhaps immediately following it, producing the C-terminal truncation we are observing here. When we model themissing part of the ULD, using the structure of hUCH37 as atemplate (see Materials and Methods), it is apparent that α8could have continued on after the cleavage site (Figure 4b)almost as a long helix all the way up to residue 285, except foran interruption at Arg268 where four successive residues,
including the arginine, adopt nonhelical dihedral anglesproducing a kink (a kink featuring equivalent residues is alsoseen in the template structure). As expected from the hUCH37structure, the model shows that after the interruption, the helixwould terminate at or near amino acid 285 (Figure 4), wherethe polypeptide chain reverses its direction as a turn segmentthat appears to cap the C-terminus of the helix. The putativeKEKE motif was not modeled because it is absent in thetemplate structure. Interestingly, the structure of theTsUCH37ΔC46−UbVME complex reveals side chains from α8making contact with ubiquitin, specifically with its Lys48residue, an interaction that may explain the distal end specificitydisplayed by UCH37 (discussed in more detail below). Also,the side chains from the modeled part of the ULD, missing inour structure, appear to present themselves for additionalcontacts with ubiquitin. Indeed, the two most conservedresidues in the ULD, Glu265 and Asn272, are facing ubiquitinand lie within contact distances (Figure 4c). Thus, it is possiblethat they may actually bind to ubiquitin. Alternatively, incontrast to what is predicted by the model, these residues maybe used for making contact with Rpn13, explaining why theyare conserved.
Active-Site Geometry. The catalytic triad in this cysteineprotease assumes a canonical arrangement in the ubiquitin-bound complex. The distance between the catalytic cysteineand histidine is 3.9 Å (Nδ−Sγ distance) in both structures, andthat between the histidine and aspartate is 2.8 Å (Nε−Oδ) inthe TsUCH37cat−UbVME complex and 2.9 Å in theTsUCH37ΔC46−UbVME complex. The distance between theCεH group of the catalytic histidine and the side chain carbonyloxygen of the oxyanion stabilizing glutamine (Gln79) is 3.3 Åin the TsUCH37cat−UbVME complex and 3.1 Å in theTsUCH37ΔC46−UbVME complex, suggesting a significantCH···O interaction between them, an interaction seen inother cysteine proteases as well.75 We were unable to crystallize
Figure 4. ULD−ubiquitin interactions. (a) Sequence alignment of the ULD of UCH37 highlighting conserved residues in UCH37 homologues.Glu265 and Asn272 (according to Ts numbering) are absolutely conserved (highlighted in red). (b) Superposition of the TsUCH37ΔC46−UbVMEcomplex (ULD colored olive and UbVME orange), human UCH37 (ULD colored purple, PDB entry 3IHR), and TsUCH37 with the entire ULDmodeled (cyan) based on the structure of the ULD in human UCH37. The model was generated using SwissModel and MD simulation (please seeMaterials and Methods). This model is taken from a snapshot collected at 1.3 ns during a 2 ns MD simulation run. (c) Structure of the TsUCH37−ubiquitin complex with the entire ULD modeled as shown in panel b, showing that the conserved residues of the ULD could make additionalcontacts with ubiquitin. The regions marked i and ii are expanded in the panels below. The UCH domain is colored gray.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783570
124

the apo form of either TsUCH37cat or TsUCH37ΔC46. In itsplace, we use the structure of apo human UCH37 to gaininsight into structural changes in the active-site region that mayoccur upon ubiquitin binding.57,58 Comparison with thestructures of human UCH37 reveals that the catalytic cysteinehas changed its orientation, going from the apo form to theubiquitin-bound form, adopting a more productive orientationin the latter, an orientation in which the catalytic cysteine’s sidechain faces the catalytic cleft (Figure 5g). This analysis suggeststhat UCH37 exists in an unproductive form in the absence of
ubiquitin, with the catalytic thiol facing the interior of theprotein rather than the open space in the catalytic cleft,58 but isinduced to adopt a more productive form upon its binding.Thus, UCH37 may offer yet another example of a UCH DUBthat undergoes substrate-induced reorganization to a moreproductive form.48,76
Crossover Loop Flexibility. A common structural featurepresent in all UCH enzymes is the crossover loop, which inTsUCH37 spans residues 141−157 (connecting α5 with β3). Itstraddles the active-site cleft as a flexible loop and is known to
Figure 5. Recognition of ubiquitin by TsUCH37. (a) Surface rendering of TsUCH37cat (cyan) with ubiquitin binding regions highlighted. The distalsite is colored yellow and the active-site cysteine red, and resolved portions of the crossover loop are colored pink. (b) Surface rendering ofTsUCH37ΔC46 (green) with ubiquitin binding regions highlighted as in panel a, except with additional C-terminal tail ubiquitin binding residuescolored blue. (c) Interactions near the active-site cleft with the C-terminal hexapeptide tail of ubiquitin. UbVME residues are colored orange,TsUCH37 residues teal, and human UCH37 residues purple. (d) Interactions of Arg72 of ubiquitin with surrounding residues of TsUCH37cat.Density from the 2Fo − Fc map is contoured at 1σ (blue mesh). (e) UCH37 distal-site binding residues, with TsUCH37 colored teal and humanUCH37 purple. (f) Ile44 patch interacting residues, with UbVME colored orange, TsUCH37 teal, and human UCH37 purple. Waters involved inbinding are also shown, enveloped with density from the 2Fo − Fc map contoured at 1σ. Sequence alignment of this region in TsUCH37 comparedto human UCH37 is shown as an inset. (g) Active site of TsUCH37 (teal), showing the catalytic residues, compared to human UCH37 (purple),with UbVME colored orange.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783571
125

provide steric constraint, limiting the size of the leaving groupat the C-terminus of ubiquitin.74,77 Accordingly, UCH enzymes,such as UCHL1 and UCHL3, can cleave only small leavinggroups from the C-terminus of ubiquitin, not large proteins oranother ubiquitin.47 However, UCH37 is known to cleavediubiquitin (and polyubiquitin chains), but only when it isassociated with the RP, being activated upon binding to itsprotein cofactor, Rpn13.37 All previously determined structuresof UCH enzymes bound to ubiquitin have shown a resolvedcrossover loop, which makes contact with at least one residuefrom the C-terminal tail of ubiquitin. In the apo form ofUCHL3, the closest homologue of UCH37, the loop isdisordered but becomes ordered when ubiquitin is bound.48,50
The ubiquitin-bound structures of PfUCHL3 and the yeastubiquitin hydrolase Yuh1 show an ordered crossover loopmaking contacts with side chains on the C-terminal tail ofubiquitin.73,78 In contrast, the structures of the TsUCH37−UbVME constructs present the only examples so far of a UCHDUB in which the crossover loop is still disordered even afterubiquitin is bound, indicating that the loop is flexible and doesnot contribute to ubiquitin binding. A small network of van derWaals interactions and hydrogen bonds seem to stabilize part ofthe crossover loop (residues 152−157) in a short helicalconformation in the structure of the TsUCH37cat−UbVMEcomplex, but the same segment in the TsUCH37ΔC46−UbVMEstructure is disordered and hence not visible, supportingdynamic sampling of conformations by this loop. Theobservation that the crossover loop is flexible despite thebound ubiquitin may be related to its activation by itsproteasome cofactor Rpn13.37 By not engaging with ubiquitin,the loop is available to freely interact with the cofactor, whichmay stabilize it in a conformation that leaves the active sitemaximally open to accommodate the isopeptide bond betweentwo ubiquitins or between ubiquitin and an acceptor protein.Interactions with Ubiquitin. The interaction of UbVME
with the TsUCH37cat UCH domain buries a total of 2355 Å2 ofsolvent accessible surface area, a value comparable to theamount buried in other UCH domain ubiquitin complexes (theburied accessible surface area in the TsUCH37ΔC46−UbVMEcomplex is 2479 Å2).48,76 The interaction is predominantlylocalized at two areas on TsUCH37, the active-site cleft and thedistal site (Figure 5a,b) The active-site cleft engages the C-terminal hexapeptide segment, Leu71ArgLeuArgGly-Gly76VME,of UbVME with numerous intermolecular contacts that includevan der Waals, hydrogen bonding, electrostatic, and water-mediated interactions (Figure 5c). This segment sits in theactive-site cleft with an extended conformation to maximizeinteractions with both backbone and side chain atoms of nearbyresidues of the enzyme. As seen in other UCH structures, thenarrowest part of the active-site cleft surrounds the terminalGly-Gly motif, with the last Gly (GlyVME in this case) beingplaced immediately adjacent to the Sγ atom of the catalyticcysteine, precisely located for nucleophilic attack on the scissilepeptide bond (Figure 5a,b). It is interesting to note that Arg72of UbVME is engaged in at least three major interactions(Figure 5d), suggesting that it contributes significantly tostabilizing the enzyme−substrate complex. The interactionswith Arg72 imply that TsUCH37 will find NEDD8 (neuralprecursor cell expressed, developmentally downregulated 8, astructurally similar ubiquitin-like protein modifier with asequence that is 60% identical with that of ubiquitin) as apoorer substrate because this arginine is replaced with alaninein NEDD8. Indeed, TsUCH37 does not cleave NEDD8-AMC
(see Figure S4 of the Supporting Information). Many of theactive-site interactions observed in the ubiquitin-boundstructures of UCHL1, UCHL3, PfUCHL3, and Yuh1 areconserved in both TsUCH37 structures. Additionally, thoseresidues surrounding the C-terminal hexapeptide tail ofubiquitin are strongly conserved between the Ts and humanprotein (Figure 5c).The interactions at the active-site cleft appear to be necessary
for precise cleavage at the terminal glycine residue of ubiquitin,while the distal site provides additional interaction to stabilizethe enzyme−substrate complex (Figure 5e,f). The distal siteengages the N-terminal β-hairpin of ubiquitin, which docks byutilizing interactions primarily involving the two-residue β-turnsegment, Leu8 and Thr9 of ubiquitin. These interactions aremostly hydrophobic in nature, involving van der Waals contactof Leu8 and Thr9 with Val35, Leu36, Ile206, Phe216, andLeu218, residues that constitute the surface-exposed hydro-phobic crevice that is the distal site. Leu36, Ile206, Phe216, andLeu218 are conserved among Sp, Ts, Pf, and human UCH37(Figure S3 of the Supporting Information), suggesting theimportance of distal-site binding in enzyme−substrate recog-nition.Ile44 of ubiquitin, a residue widely used in recognition by
ubiquitin-binding proteins, including DUBs, is seen making vander Waals contacts with Val34 on a greasy loop in TsUCH37,residues 34−36 (residues Val35 and Leu36 extend into thedistal-site pocket) (Figure 5f). A similar motif is used in otherUCH enzymes to bind to Ile44 of ubiquitin. Val34 ofTsUCH37 also makes contacts with His68 and Val70, which,together with Ile44 and Leu8 from the N-terminal β-hairpinturn, form the so-called Ile44 patch on ubiquitin. Thus, thebinding potential of the Ile44 patch on ubiquitin appears to befully satisfied in structures of the two complexes presented here,with each residue in the patch making at least one contact withthe enzyme. The structural data presented here are supportedby previously reported mutational analysis of the PA700isopeptidase. Replacing Ile44 and Leu8 from the Ile44 patchwith alanine in the distal ubiquitin of a diubiquitin substrateresults in significantly impaired catalysis with no detectablehydrolysis product.45 Val34 and Val35 are replaced withtryptophan and serine, respectively, going from Ts to humanUCH37 (Figure 5f) (Val34 provides additional contacts withVal70 of UbVME). These residues also show variability amongother UCH family members. Subtle differences in the Ile44patch-binding residues could be one of the contributing factorsin the difference in KM between human and Ts UCH37,especially as most of the residues at the active site are conservedbetween the two.There appear to be no striking conformational changes
between the ubiquitin-bound form of TsUCH37 and apohUCH37 except for the aforementioned reorientation of thecatalytic cysteine. However, we cannot rule out the possibilitythat significant conformational changes might have occurred asa result of ubiquitin binding in the Ts enzyme because we couldnot crystallize its apo form.
Ubiquitin Binding by the ULD. As mentioned earlier, theULD of hUCH37 was thought to have an inhibitory role,presumably by folding onto the catalytic domain andobstructing substrate binding.37 In contrast, the structure ofthe TsUCH37ΔC46−UbVME complex provides crystallographicevidence that the ULD can actually contribute to ubiquitinbinding and therefore can play a productive role in catalysis.Arg261 and Tyr262 on α8 of the ULD approach ubiquitin to
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783572
126

engage in van der Waals contact with of three of its side chains,Lys48 (with Arg261) and Gln49 and Arg72 (both with Tyr262)(Figure 6). Most notably, Arg261 is oriented in such a way toengage in close van der Waals contact with the hydrocarbonportion of the Lys48 side chain, forcing it to adopt an unusualconformation that allows an intramolecular salt bridgeinteraction with Glu51. This interaction is not observed inany of the 39 other ubiquitin-bound DUB structures currentlyfound in the PDB, catalogued in Table 3; the Lys48−Glu51distance is greater than 5.8 Å in all. Figure 6b shows theorientation of the same lysine in the TsUCH37cat−UbVMEcomplex. Clearly, the orientation is different in this structure,and the intramolecular salt bridge in ubiquitin is absent,suggesting that Arg261 of the ULD plays a role in inducing theunusual orientation of Lys48 of ubiquitin. Arg261 is conservedamong Sp, Ts, and human UCH37 (Figure 7) but is replacedwith leucine in PfUCH37 (also known as PfUCH54). Tyr262 isconserved in human and Ts forms but is substituted withtryptophan in Sp and PfUCH37. Inspection of the structurereveals that the van der Waals contact with Lys48 is still feasiblewith leucine in place of arginine and tryptophan canconservatively replace tyrosine as well. Thus, it is likely that
ULD binding with Lys48 and subsequent formation of theintramolecular salt bridge we are observing here are conservedfeatures of UCH37 in general.UCH37, as a part of PA700, is known to selectively cleave
polyubiquitin chains from the very distal end, sequentiallyremoving one ubiquitin at a time.38 The structural basis of thisexo cleavage specificity is not yet known. The uniqueorientation of Lys48 stabilized by Arg261 leading to theintramolecular salt bridge may explain this selectivity. Wepropose that although a similar type of interaction betweenArg261 and ubiquitin’s Lys48 is possible with an internalubiquitin, the intramolecular salt bridge will be absent in thiscase because the amino group of the lysine is acylated andhence not charged. Thus, it is the lack of an additionalinteraction with an internal ubiquitin that makes binding toLys48 of the terminal ubiquitin more favored, hence the exoselectivity.
■ DISCUSSION
UCH37 is a proteasome-associated UCH DUB known to havepolyubiquitin chain-editing function. It preferentially cleavesthe chain from its very distal tip.38 Such a function might rescue
Figure 6. ULD of TsUCH37 binding to ubiquitin. (a) TsUCH37ΔC46 (olive) ULD residues interacting with UbVME (orange). The inset showsinteractions of Arg261 and Tyr262 with UbVME, as well as the intramolecular salt bridge formed between Lys48 and Glu51 of ubiquitin. Densityrendered from the 2Fo − Fc map contoured to 0.7σ. (b−e) Comparison of the Lys48−Glu51 distances in ubiquitin observed in other DUB−ubiquitin structures. (b) Lys48 and Glu51 form a 3.7 Å salt bridge in the TsUCH37ΔC46−UbVME structure (olive), but not in the TsUCH37cat−UbVME structure (9.9 Å). (c) The same distance in all other UCH−ubiquitin structures is ≥9 Å: UCHL3−UbVME (yellow, PDB entry 1XD3),UCHL1−UbVME (red, PDB entry 3KW5), Yuh1−Ubal (pink, PDB entry 1CMX), PfUCHL3−UbVME (purple, PDB entry 2WDT), andTsUCH37cat−UbVME (teal). (d) The same distance is 8.7 Å in the Otu1−ubiquitin structure (dark red, PDB entry 3BY4) and 6.0 Å in the DUBA−Ubal structure (pale yellow, PDB entry 3TMP). (e) The same distance is 10 Å in the HAUSP/USP7−Ubal structure (blue, PDB entry 1NBF) and10.9 Å in the USP14−Ubal structure (brown, PDB entry 2AYO).
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783573
127

certain substrates from being committed to further downstreamaction of the proteasome.38 It is also possible that certainsubstrates carry inappropriate polyubiquitin tags that are notoptimal for their degradation. The chain-editing function mightbe essential for releasing these substrates to clear up ubiquitinreceptors for binding to productive substrates. A regulator ofproteasome function, it is itself regulated by binding to theproteasome: UCH37 is activated upon binding to Rpn13, asubunit of PA700 (the 19S proteasome or RP), the mechanism
of which is not understood. We report here the structure of twoconstructs of UCH37 from the infectious helminth T. spiralis(Ts) bound to the suicide inhibitor UbVME. This workconstitutes the first structural analysis of a ubiquitin pathwayprotein in the organism showing how ubiquitin is recognized bythis UCH family DUB in Ts. The structures reveal strikingconservation of the ubiquitin binding mode among UCHDUBs, from lower eukaryotes to human (Figure S5 of theSupporting Information). It also shows important structuraldifferences between other UCH DUBs, such as UCHL1 andUCHL3, some of which could be used for the specializedfunction of UCH37. While revealing interesting differences, theTs structures provide a number of details that may also holdtrue for the human enzyme, advancing our understanding ofUCH37 in general.The active-site cysteine may undergo ubiquitin-mediated
reorientation to a more productive form (Figure 5g), makingUCH37 yet another example of a UCH DUB that showsregulation of activity by ubiquitin, a feature that may provideselectivity to this group of cysteine proteases. Structures of thetwo constructs reveal invariant parts of the enzyme, likely lessdynamic parts, while also revealing parts that are more dynamicin nature, such as the segment of residues 57−71 and Trp55.Future studies should reveal the role of such dynamic parts incatalysis or regulation thereof.Importantly, the structure of the construct with the
additional 40 amino acids after the UCH domain reveals thatthe ULD could contribute to ubiquitin binding (Figures 4 and6), an unexpected finding because it was thought to beinhibitory in the human enzyme.37 The interaction of Arg261on the ULD appears to engage Lys48 of the distal ubiquitin in away that would be energetically most favored with the veryterminal ubiquitin in a polyubiquitin chain, possibly explainingthe exo specificity displayed by mammalian UCH37. Thesestructural data are consistent with previously reported muta-tional analysis probing substrate specificity of the PA700isopeptidase: mutation of Lys48 to cysteine on the distalubiquitin of a diubiquitin substrate results in severely impairedcatalysis.45 Apart from the broad agreement with theaforementioned experimental work, this observation of theintramolecular Lys48−Glu51 salt bridge in the distal ubiquitin,apparently induced by Arg261, is purely crystallographic at thispoint, although it seems unlikely that lattice forces haveanything to do with it. Even if the opposite is true, the fact thatsuch interactions are physiologically relevant cannot be ignored.The lack of an intramolecular Lys48−Glu51 salt bridge in anyother ubiquitin-bound DUB structures to date (Table 3) makesthis unusual interaction more intriguing, and worth additionalstudy. This observation therefore lays the structural ground-work for future mutational analysis aimed at validating theirexistence in solution and their role in substrate specificity.It is interesting to note that a salt bridge interaction, albeit an
intermolecular one, involving Lys48 of ubiquitin and an acidicside chain of the enzyme is also seen in the structure of USP7bound to ubiquitin aldehyde (the Lys48 side chain of the distalubiquitin is interacting with Asp305 and Glu308 of USP7).79
Such bifurcated salt bridges will perhaps contribute substan-tially to the binding of the enzyme to distal ubiquitin in a K48-linked chain, based on which one may predict that USP7 willalso exhibit exo specificity. This needs to be examined.Preferential cleavage from the very distal tip of a Lys48-linkedpolyubiquitin chain may be a feature common to DUBs thatwork on chains of this topology. Lys48-linked chains are known
Table 3. Lys48−Glu51 Distances for All DUB−UbiquitinComplexes
PDBentry DUB−ubiquitin complex
Lys48−Glu51distancea (Å)
UCH Family1XD3 UCHL3−UbVME 9.1, 11.51CMX YUH1−Ubal 10.82WDT PfUCHL3−UbVME 7.3, 9.63IFW UCHL1 S18Y−UbVME 8.73KVF UCHL1 I93M−UbVME 12.33KW5 UCHL1−UbVME 13.1
USP Family3TMP DUBA−Ubal 8.32Y5B USP21−linear diUbal 7.8, 10.8, 6.61NBF HAUSP−Ubal 10.0, 10.72AYO USP14−Ubal 10.93MHS SAGA complex (UBP8)−Ubal 9.22HD5 USP2, Ub 9.02G45 IsoT, Ub 8.6, 8.92J7Q M48 USP−UbVME 9.9, 10.83V6E USP2, Ub variant 7.03V6C USP2, Ub variant 7.43IHP USP5, Ub covalent 9.6, 6.33MTN USP21, ubiquitin-based USP21-specific
inhibitor8.5
3IT3 USP21, Ub covalent 7.4, 7.5, 7.4, 7.43N3K USP8, covalent Ub-like variant 10.43NHE USP2a, Ub 7.42IBI USP2, Ub covalent 7.6
OTU Family4IUM arterivirus papain-like protease 2, Ub 7.43ZNH CCHF viral, Ub-propargyl 9.54I6L OTUB1, Ub 9.13PT2 viral OTU, Ub 10.74HXD Nairovirus viral OTU, Ub 8.3, 10.13BY4 OTU, Ub 6.03PRM CCHF viral OTU, Ub 9.5, 10.03PRP CCHF viral OTU, Ub 10.3, 10.93C0R OTU, Ub 8.44DHZ h/ceOTUB1-ubiquitin aldehyde-
UBC13∼Ub9.2, 8.8
4DHJ ceOTUB1 ubiquitin aldehydeUBC13∼Ub complex
11.4, 8.5, 6.9, 11.8, 9.8,10.1
4DDI OTUB1/UbcH5b∼Ub/Ub 7.5, 12.6, 12.6, 7.5,12.6, 7.5
4DDG OTUB1/UbcH5b∼Ub/Ub 11.3, 7.1, 8.03PHW OTU domain of CCHF virus, Ub 7.6, 7.2, 9.0, 5.8
MJD Family3O65 Ataxin-3-like, Ub 9.6, 11.4, 12.8, 11.52JRI Ataxin 3, Ub 12.3, 12.9
JAMM Family2ZNV AMSH-LP, Lys63-linked diubiquitin 9.0, 8.3, 11.9, 11.3
aMultiple distance entries refer to those in the other subunits of thecrystallographic asymmetric unit.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783574
128
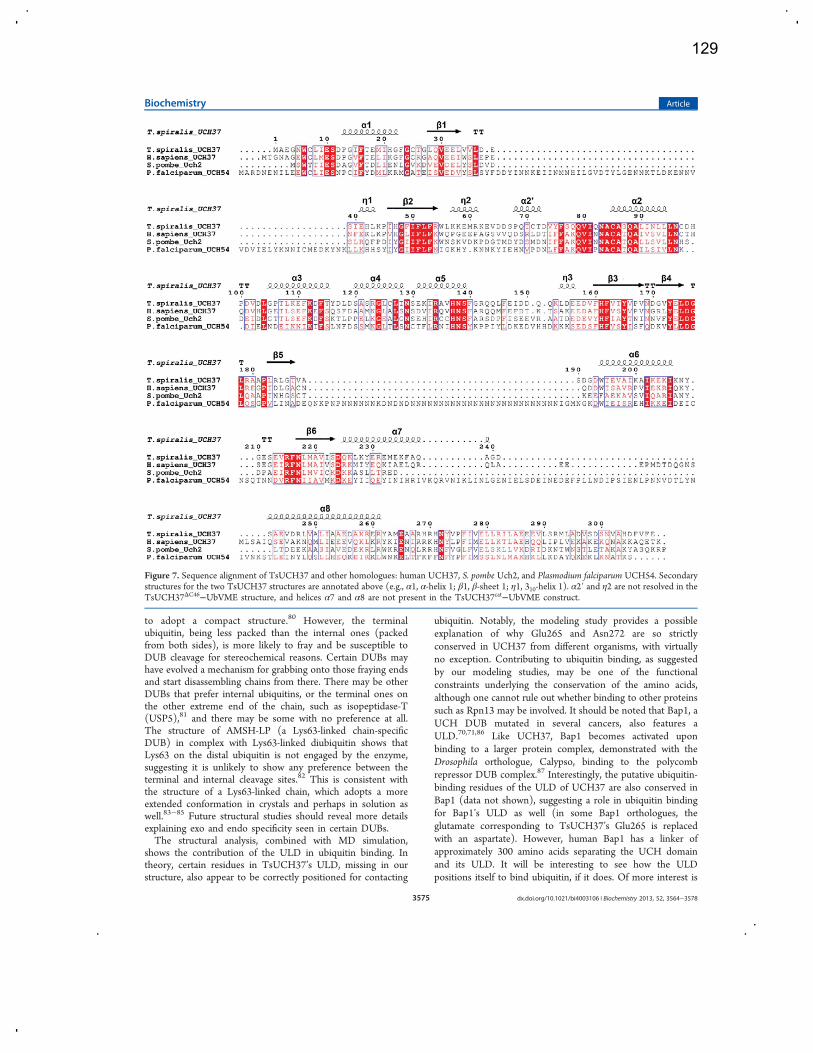
to adopt a compact structure.80 However, the terminalubiquitin, being less packed than the internal ones (packedfrom both sides), is more likely to fray and be susceptible toDUB cleavage for stereochemical reasons. Certain DUBs mayhave evolved a mechanism for grabbing onto those fraying endsand start disassembling chains from there. There may be otherDUBs that prefer internal ubiquitins, or the terminal ones onthe other extreme end of the chain, such as isopeptidase-T(USP5),81 and there may be some with no preference at all.The structure of AMSH-LP (a Lys63-linked chain-specificDUB) in complex with Lys63-linked diubiquitin shows thatLys63 on the distal ubiquitin is not engaged by the enzyme,suggesting it is unlikely to show any preference between theterminal and internal cleavage sites.82 This is consistent withthe structure of a Lys63-linked chain, which adopts a moreextended conformation in crystals and perhaps in solution aswell.83−85 Future structural studies should reveal more detailsexplaining exo and endo specificity seen in certain DUBs.The structural analysis, combined with MD simulation,
shows the contribution of the ULD in ubiquitin binding. Intheory, certain residues in TsUCH37’s ULD, missing in ourstructure, also appear to be correctly positioned for contacting
ubiquitin. Notably, the modeling study provides a possibleexplanation of why Glu265 and Asn272 are so strictlyconserved in UCH37 from different organisms, with virtuallyno exception. Contributing to ubiquitin binding, as suggestedby our modeling studies, may be one of the functionalconstraints underlying the conservation of the amino acids,although one cannot rule out whether binding to other proteinssuch as Rpn13 may be involved. It should be noted that Bap1, aUCH DUB mutated in several cancers, also features aULD.70,71,86 Like UCH37, Bap1 becomes activated uponbinding to a larger protein complex, demonstrated with theDrosophila orthologue, Calypso, binding to the polycombrepressor DUB complex.87 Interestingly, the putative ubiquitin-binding residues of the ULD of UCH37 are also conserved inBap1 (data not shown), suggesting a role in ubiquitin bindingfor Bap1’s ULD as well (in some Bap1 orthologues, theglutamate corresponding to TsUCH37’s Glu265 is replacedwith an aspartate). However, human Bap1 has a linker ofapproximately 300 amino acids separating the UCH domainand its ULD. It will be interesting to see how the ULDpositions itself to bind ubiquitin, if it does. Of more interest is
Figure 7. Sequence alignment of TsUCH37 and other homologues: human UCH37, S. pombe Uch2, and Plasmodium falciparum UCH54. Secondarystructures for the two TsUCH37 structures are annotated above (e.g., α1, α-helix 1; β1, β-sheet 1; η1, 310-helix 1). α2′ and η2 are not resolved in theTsUCH37ΔC46−UbVME structure, and helices α7 and α8 are not present in the TsUCH37cat−UbVME construct.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783575
129

knowing whether the ULD has independent ability to bind toubiquitin.
■ ASSOCIATED CONTENT*S Supporting InformationSupporting figures and NEDD8 hydrolysis data (Figure S4).This material is available free of charge via the Internet athttp://pubs.acs.org.Accession CodesCoordinates and structure factors have been deposited in theProtein Data Bank as entries 4I6N and 4IG7.
■ AUTHOR INFORMATIONCorresponding Author*Brown Laboratory of Chemistry, 560 Oval Dr., WestLafayette, IN 47907. E-mail: [email protected]. Phone: (765)494-5478. Fax: (765) 494-0239.Author ContributionsM.E.M. and M.-I.K. contributed equally to this work.FundingFinancial support from the National Institutes of Health(1R01RR026273, C.D.) is gratefully acknowledged. TheGeneral Medicine and Cancer Institutes Collaborative AccessTeam (GM/CA CAT) of the Advanced Photon Source atArgonne National Laboratory has been funded in whole or inpart with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General MedicalSciences (Y1-GM-1104).NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge Venugopalan Nagarajan, Ruslan Sanishvili,and Craig Ogata at beamline 23-ID-B of the Advanced PhotonSource for assistance with data collection. Use of the AdvancedPhoton Source was supported by the U.S. Department ofEnergy, Basic Energy Sciences, Office of Science, underContract DE-AC02-06CH11357. We thank Emma DeWaltand Garth Simpson from the Department of Chemistry, PurdueUniversity, for their assistance with SONICC imaging of initialcrystals.
■ ABBREVIATIONSSDS−PAGE, sodium dodecyl sulfate−polyacrylamide gelelectrophoresis; DTT, dithiothreitol; IPTG, isopropyl β-D-1-thiogalactopyranoside; UbVME, ubiquitin vinyl methyl ester;UbAMC, ubiquitin aminomethylcoumarin; UCH37, ubiquitincarboxyl-terminal hydrolase 37; DUB, deubiquitinating enzymeor deubiquitinase; PDB, Protein Data Bank.
■ REFERENCES(1) Ciechanover, A. (2005) Proteolysis: From the lysosome toubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79−87.(2) Ciechanover, A., and Schwartz, A. L. (2002) Ubiquitin-mediateddegradation of cellular proteins in health and disease. Hepatology 35,3−6.(3) Varshavsky, A. (1997) The ubiquitin system. Trends Biochem. Sci.22, 383−387.(4) Wilkinson, K. D. (2000) Ubiquitination and deubiquitination:Targeting of proteins for degradation by the proteasome. Semin. CellDev. Biol. 11, 141−148.(5) Goldberg, A. L. (2003) Protein degradation and protectionagainst misfolded or damaged proteins. Nature 426, 895−899.
(6) Pickart, C. M., and Cohen, R. E. (2004) Proteasomes and theirkin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 5, 177−187.(7) Finley, D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477−513.(8) Baumeister, W., Walz, J., Zuhl, F., and Seemuller, E. (1998) Theproteasome: Paradigm of a self-compartmentalizing protease. Cell 92,367−380.(9) Matyskiela, M. E., and Martin, A. (2012) Design principles of auniversal protein degradation machine. J. Mol. Biol. 425, 199−213.(10) Lander, G. C., Estrin, E., Matyskiela, M. E., Bashore, C.,Nogales, E., and Martin, A. (2012) Complete subunit architecture ofthe proteasome regulatory particle. Nature 482, 186−191.(11) Lasker, K., Forster, F., Bohn, S., Walzthoeni, T., Villa, E.,Unverdorben, P., Beck, F., Aebersold, R., Sali, A., and Baumeister, W.(2012) Molecular architecture of the 26S proteasome holocomplexdetermined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A.109, 1380−1387.(12) Goldberg, A. L. (2007) Functions of the proteasome: Fromprotein degradation and immune surveillance to cancer therapy.Biochem. Soc. Trans. 35, 12−17.(13) Demartino, G. N., and Gillette, T. G. (2007) Proteasomes:Machines for all reasons. Cell 129, 659−662.(14) Guterman, A., and Glickman, M. H. (2004) Deubiquitinatingenzymes are IN/(trinsic to proteasome function). Curr. Protein Pept.Sci. 5, 201−211.(15) Yao, T., and Cohen, R. E. (2002) A cryptic protease couplesdeubiquitination and degradation by the proteasome. Nature 419,403−407.(16) Verma, R., Aravind, L., Oania, R., McDonald, W. H., Yates, J. R.,III, Koonin, E. V., and Deshaies, R. J. (2002) Role of Rpn11metalloprotease in deubiquitination and degradation by the 26Sproteasome. Science 298, 611−615.(17) Lee, M. J., Lee, B. H., Hanna, J., King, R. W., and Finley, D.(2011) Trimming of ubiquitin chains by proteasome-associateddeubiquitinating enzymes. Mol Cell Proteomics. 10, R110.003871.(18) Pickart, C. M. (2001) Mechanisms underlying ubiquitination.Annu. Rev. Biochem. 70, 503−533.(19) Schulman, B. A. (2011) Twists and turns in ubiquitin-likeprotein conjugation cascades. Protein Sci. 20, 1941−1954.(20) Pickart, C. M. (2000) Ubiquitin in chains. Trends Biochem. Sci.25, 544−548.(21) Haglund, K., and Dikic, I. (2005) Ubiquitylation and cellsignaling. EMBO J. 24, 3353−3359.(22) Ikeda, F., and Dikic, I. (2008) Atypical ubiquitin chains: Newmolecular signals. ‘Protein Modifications: Beyond the Usual Suspects’review series. EMBO Rep. 9, 536−542.(23) Fushman, D., and Wilkinson, K. D. (2011) Structure andrecognition of polyubiquitin chains of different lengths and linkage.F1000 Biol. Rep. 3, 26.(24) Komander, D., and Rape, M. (2012) The ubiquitin code. Annu.Rev. Biochem. 81, 203−229.(25) Kulathu, Y., and Komander, D. (2012) Atypical ubiquitylation:The unexplored world of polyubiquitin beyond Lys48 and Lys63linkages. Nat. Rev. Mol. Cell Biol. 13, 508−523.(26) Chen, Z. J., and Sun, L. J. (2009) Nonproteolytic functions ofubiquitin in cell signaling. Mol. Cell 33, 275−286.(27) Komander, D., Clague, M. J., and Urbe, S. (2009) Breaking thechains: Structure and function of the deubiquitinases. Nat. Rev. Mol.Cell Biol. 10, 550−563.(28) Komander, D. (2010) Mechanism, specificity and structure ofthe deubiquitinases. Subcell. Biochem. 54, 69−87.(29) Amerik, A. Y., and Hochstrasser, M. (2004) Mechanism andfunction of deubiquitinating enzymes. Biochim. Biophys. Acta 1695,189−207.(30) Wilkinson, K. D. (1997) Regulation of ubiquitin-dependentprocesses by deubiquitinating enzymes. FASEB J. 11, 1245−1256.(31) Wilkinson, K. D. (2009) DUBs at a glance. J. Cell Sci. 122,2325−2329.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783576
130

(32) Nijman, S. M., Luna-Vargas, M. P., Velds, A., Brummelkamp, T.R., Dirac, A. M., Sixma, T. K., and Bernards, R. (2005) A genomic andfunctional inventory of deubiquitinating enzymes. Cell 123, 773−786.(33) Love, K. R., Catic, A., Schlieker, C., and Ploegh, H. L. (2007)Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat.Chem. Biol. 3, 697−705.(34) Reyes-Turcu, F. E., Ventii, K. H., and Wilkinson, K. D. (2009)Regulation and cellular roles of ubiquitin-specific deubiquitinatingenzymes. Annu. Rev. Biochem. 78, 363−397.(35) Tsou, W. L., Sheedlo, M. J., Morrow, M. E., Blount, J. R.,McGregor, K. M., Das, C., and Todi, S. V. (2012) Systematic analysisof the physiological importance of deubiquitinating enzymes. PLoSOne 7, e43112.(36) Borodovsky, A., Kessler, B. M., Casagrande, R., Overkleeft, H. S.,Wilkinson, K. D., and Ploegh, H. L. (2001) A novel active site-directedprobe specific for deubiquitylating enzymes reveals proteasomeassociation of USP14. EMBO J. 20, 5187−5196.(37) Yao, T., Song, L., Xu, W., DeMartino, G. N., Florens, L.,Swanson, S. K., Washburn, M. P., Conaway, R. C., Conaway, J. W., andCohen, R. E. (2006) Proteasome recruitment and activation of theUch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 8, 994−1002.(38) Lam, Y. A., Xu, W., DeMartino, G. N., and Cohen, R. E. (1997)Editing of ubiquitin conjugates by an isopeptidase in the 26Sproteasome. Nature 385, 737−740.(39) Hanna, J., Hathaway, N. A., Tone, Y., Crosas, B., Elsasser, S.,Kirkpatrick, D. S., Leggett, D. S., Gygi, S. P., King, R. W., and Finley,D. (2006) Deubiquitinating enzyme Ubp6 functions noncatalyticallyto delay proteasomal degradation. Cell 127, 99−111.(40) Hu, M., Li, P., Song, L., Jeffrey, P. D., Chenova, T. A.,Wilkinson, K. D., Cohen, R. E., and Shi, Y. (2005) Structure andmechanisms of the proteasome-associated deubiquitinating enzymeUSP14. EMBO J. 24, 3747−3756.(41) Koulich, E., Li, X., and DeMartino, G. N. (2008) Relativestructural and functional roles of multiple deubiquitylating proteinsassociated with mammalian 26S proteasome. Mol. Biol. Cell 19, 1072−1082.(42) Lee, B. H., Lee, M. J., Park, S., Oh, D. C., Elsasser, S., Chen, P.C., Gartner, C., Dimova, N., Hanna, J., Gygi, S. P., Wilson, S. M., King,R. W., and Finley, D. (2010) Enhancement of proteasome activity by asmall-molecule inhibitor of USP14. Nature 467, 179−184.(43) D’Arcy, P., Brnjic, S., Olofsson, M. H., Fryknas, M., Lindsten, K.,De Cesare, M., Perego, P., Sadeghi, B., Hassan, M., Larsson, R., andLinder, S. (2011) Inhibition of proteasome deubiquitinating activity asa new cancer therapy. Nat. Med. 17, 1636−1640.(44) D’Arcy, P., and Linder, S. (2012) Proteasome deubiquitinases asnovel targets for cancer therapy. Int. J. Biochem. Cell Biol. 44, 1729−1738.(45) Lam, Y. A., DeMartino, G. N., Pickart, C. M., and Cohen, R. E.(1997) Specificity of the ubiquitin isopeptidase in the PA700regulatory complex of 26 S proteasomes. J. Biol. Chem. 272, 28438−28446.(46) Stone, M., Hartmann-Petersen, R., Seeger, M., Bech-Otschir, D.,Wallace, M., and Gordon, C. (2004) Uch2/Uch37 is the majordeubiquitinating enzyme associated with the 26S proteasome in fissionyeast. J. Mol. Biol. 344, 697−706.(47) Larsen, C. N., Krantz, B. A., and Wilkinson, K. D. (1998)Substrate specificity of deubiquitinating enzymes: Ubiquitin C-terminal hydrolases. Biochemistry 37, 3358−3368.(48) Misaghi, S., Galardy, P. J., Meester, W. J., Ovaa, H., Ploegh, H.L., and Gaudet, R. (2005) Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J. Biol. Chem. 280, 1512−1520.(49) Das, C., Hoang, Q. Q., Kreinbring, C. A., Luchansky, S. J.,Meray, R. K., Ray, S. S., Lansbury, P. T., Ringe, D., and Petsko, G. A.(2006) Structural basis for conformational plasticity of the Parkinson’sdisease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci.U.S.A. 103, 4675−4680.
(50) Johnston, S. C., Larsen, C. N., Cook, W. J., Wilkinson, K. D.,and Hill, C. P. (1997) Crystal structure of a deubiquitinating enzyme(human UCH-L3) at 1.8 Å resolution. EMBO J. 16, 3787−3796.(51) Hamazaki, J., Iemura, S., Natsume, T., Yashiroda, H., Tanaka, K.,and Murata, S. (2006) A novel proteasome interacting protein recruitsthe deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J.25, 4524−4536.(52) Husnjak, K., Elsasser, S., Zhang, N., Chen, X., Randles, L., Shi,Y., Hofmann, K., Walters, K. J., Finley, D., and Dikic, I. (2008)Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453,481−488.(53) Schreiner, P., Chen, X., Husnjak, K., Randles, L., Zhang, N.,Elsasser, S., Finley, D., Dikic, I., Walters, K. J., and Groll, M. (2008)Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548−552.(54) Qiu, X. B., Ouyang, S. Y., Li, C. J., Miao, S., Wang, L., andGoldberg, A. L. (2006) hRpn13/ADRM1/GP110 is a novelproteasome subunit that binds the deubiquitinating enzyme,UCH37. EMBO J. 25, 5742−5753.(55) Yao, T., Song, L., Jin, J., Cai, Y., Takahashi, H., Swanson, S. K.,Washburn, M. P., Florens, L., Conaway, R. C., Cohen, R. E., andConaway, J. W. (2008) Distinct modes of regulation of the Uch37deubiquitinating enzyme in the proteasome and in the Ino80chromatin-remodeling complex. Mol. Cell 31, 909−917.(56) Nishio, K., Kim, S. W., Kawai, K., Mizushima, T., Yamane, T.,Hamazaki, J., Murata, S., Tanaka, K., and Morimoto, Y. (2009) Crystalstructure of the de-ubiquitinating enzyme UCH37 (human UCH-L5)catalytic domain. Biochem. Biophys. Res. Commun. 390, 855−860.(57) Burgie, S. E., Bingman, C. A., Soni, A. B., and Phillips, G. N.(2012) Structural characterization of human Uch37. Proteins 80, 649−654.(58) Maiti, T. K., Permaul, M., Boudreaux, D. A., Mahanic, C.,Mauney, S., and Das, C. (2011) Crystal structure of the catalyticdomain of UCHL5, a proteasome-associated human deubiquitinatingenzyme, reveals an unproductive form of the enzyme. FEBS J. 278,4917−4926.(59) White, R. R., Miyata, S., Papa, E., Spooner, E., Gounaris, K.,Selkirk, M. E., and Artavanis-Tsakonas, K. (2011) Characterisation ofthe Trichinella spiralis deubiquitinating enzyme, TsUCH37, anevolutionarily conserved proteasome interaction partner. PLoSNeglected Trop. Dis. 5, e1340.(60) Borodovsky, A., Ovaa, H., Kolli, N., Gan-Erdene, T., Wilkinson,K. D., Ploegh, H. L., and Kessler, B. M. (2002) Chemistry-basedfunctional proteomics reveals novel members of the deubiquitinatingenzyme family. Chem. Biol. 9, 1149−1159.(61) Holyoak, T., Fenn, T. D., Wilson, M. A., Moulin, A. G., Ringe,D., and Petsko, G. A. (2003) Malonate: A versatile cryoprotectant andstabilizing solution for salt-grown macromolecular crystals. ActaCrystallogr. D59, 2356−2358.(62) Otwinowski, Z., and Minor, W. (1997) Processing of X-rayDiffraction Data Collected in Oscillation Mode.Methods Enzymol. 276,307−326.(63) Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I.W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., and Grosse-Kunstleve, R. W. (2010) PHENIX: A comprehensive Python-basedsystem for macromolecular structure solution. Acta Crystallogr. D66,213−221.(64) Emsley, P., and Cowtan, K. (2004) Coot: Model-building toolsfor molecular graphics. Acta Crystallogr. D60, 2126−2132.(65) Schuck, P. (2000) Size-distribution analysis of macromoleculesby sedimentation velocity ultracentrifugation and lamm equationmodeling. Biophys. J. 78, 1606−1619.(66) Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006) TheSWISS-MODEL workspace: A web-based environment for proteinstructure homology modelling. Bioinformatics 22, 195−201.(67) Case, D., Darden, T., Cheatham, T., III, Simmerling, C., Wang,J., Duke, R., Luo, R., Walker, R., Zhang, W., and Merz, K. (2012)AMBER 12, University of California, San Francisco.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783577
131

(68) Darden, T., York, D., and Pedersen, L. (1993) Particle meshEwald: An N·log (N) method for Ewald sums in large systems. J.Chem. Phys. 98, 10089.(69) Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. (1977)Numerical integration of the cartesian equations of motion of a systemwith constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23,327−341.(70) Misaghi, S., Ottosen, S., Izrael-Tomasevic, A., Arnott, D.,Lamkanfi, M., Lee, J., Liu, J., O’Rourke, K., Dixit, V. M., and Wilson, A.C. (2009) Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol.Cell. Biol. 29, 2181−2192.(71) Sanchez-Pulido, L., Kong, L., and Ponting, C. P. (2012) Acommon ancestry for BAP1 and Uch37 regulators. Bioinformatics 28,1953−1956.(72) Chen, V., Arendall, W., Headd, J., Keedy, D., Immormino, R.,Kapral, G., Murray, L., Richardson, J., and Richardson, D. (2010)MolProbity: All-atom structure validation for macromolecularcrystallography. Acta Crystallogr. D66, 12−21.(73) Johnston, S. C., Riddle, S. M., Cohen, R. E., and Hill, C. P.(1999) Structural basis for the specificity of ubiquitin C-terminalhydrolases. EMBO J. 18, 3877−3887.(74) Zhou, Z. R., Zhang, Y. H., Liu, S., Song, A. X., and Hu, H. Y.(2012) Length of the active-site crossover loop defines the substratespecificity of ubiquitin C-terminal hydrolases for ubiquitin chains.Biochem. J. 441, 143−149.(75) Boudreaux, D. A., Chaney, J., Maiti, T. K., and Das, C. (2012)Contribution of active site glutamine to rate enhancement in ubiquitinC-terminal hydrolases. FEBS J. 279, 1106−1118.(76) Boudreaux, D. A., Maiti, T. K., Davies, C. W., and Das, C.(2010) Ubiquitin vinyl methyl ester binding orients the misalignedactive site of the ubiquitin hydrolase UCHL1 into productiveconformation. Proc. Natl. Acad. Sci. U.S.A. 107, 9117−9122.(77) Popp, M. W., Artavanis-Tsakonas, K., and Ploegh, H. L. (2009)Substrate filtering by the active site crossover loop in UCHL3 revealedby sortagging and gain-of-function mutations. J. Biol. Chem. 284,3593−3602.(78) Artavanis-Tsakonas, K., Weihofen, W. A., Antos, J. M., Coleman,B. I., Comeaux, C. A., Duraisingh, M. T., Gaudet, R., and Ploegh, H. L.(2010) Characterization and structural studies of the Plasmodiumfalciparum ubiquitin and Nedd8 hydrolase UCHL3. J. Biol. Chem. 285,6857−6866.(79) Hu, M., Li, P., Li, M., Li, W., Yao, T., Wu, J. W., Gu, W., Cohen,R. E., and Shi, Y. (2002) Crystal structure of a UBP-familydeubiquitinating enzyme in isolation and in complex with ubiquitinaldehyde. Cell 111, 1041−1054.(80) Eddins, M. J., Varadan, R., Fushman, D., Pickart, C. M., andWolberger, C. (2007) Crystal structure and solution NMR studies ofLys48-linked tetraubiquitin at neutral pH. J. Mol. Biol. 367, 204−211.(81) Reyes-Turcu, F. E., Shanks, J. R., Komander, D., and Wilkinson,K. D. (2008) Recognition of polyubiquitin isoforms by the multipleubiquitin binding modules of isopeptidase T. J. Biol. Chem. 283,19581−19592.(82) Sato, Y., Yoshikawa, A., Yamagata, A., Mimura, H., Yamashita,M., Ookata, K., Nureki, O., Iwai, K., Komada, M., and Fukai, S. (2008)Structural basis for specific cleavage of Lys 63-linked polyubiquitinchains. Nature 455, 358−362.(83) Datta, A. B., Hura, G. L., and Wolberger, C. (2009) Thestructure and conformation of Lys63-linked tetraubiquitin. J. Mol. Biol.392, 1117−1124.(84) Komander, D., Reyes-Turcu, F., Licchesi, J. D., Odenwaelder, P.,Wilkinson, K. D., and Barford, D. (2009) Molecular discrimination ofstructurally equivalent Lys 63-linked and linear polyubiquitin chains.EMBO Rep. 10, 466−473.(85) Tenno, T., Fujiwara, K., Tochio, H., Iwai, K., Morita, E. H.,Hayashi, H., Murata, S., Hiroaki, H., Sato, M., Tanaka, K., andShirakawa, M. (2004) Structural basis for distinct roles of Lys63- andLys48-linked polyubiquitin chains. Genes Cells 9, 865−875.
(86) Ventii, K. H., Devi, N. S., Friedrich, K. L., Chernova, T. A.,Tighiouart, M., Van Meir, E. G., and Wilkinson, K. D. (2008) BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinat-ing activity and nuclear localization. Cancer Res. 68, 6953−6962.(87) Scheuermann, J. C., de Ayala Alonso, A. G., Oktaba, K., Ly-Hartig, N., McGinty, R. K., Fraterman, S., Wilm, M., Muir, T. W., andMuller, J. (2010) Histone H2A deubiquitinase activity of thePolycomb repressive complex PR-DUB. Nature 465, 243−247.
Biochemistry Article
dx.doi.org/10.1021/bi4003106 | Biochemistry 2013, 52, 3564−35783578
132

research papers
J. Synchrotron Rad. (2013). 20, 531–540 doi:10.1107/S0909049513007942 531
Journal of
SynchrotronRadiation
ISSN 0909-0495
Received 12 October 2012
Accepted 22 March 2013
# 2013 International Union of Crystallography
Printed in Singapore – all rights reserved
Integrated nonlinear optical imaging microscopefor on-axis crystal detection and centering at asynchrotron beamline
Jeremy T. Madden,a Scott J. Toth,a Christopher M. Dettmar,a Justin A. Newman,a
Robert A. Oglesbee,a Hartmut G. Hedderich,a R. Michael Everly,a Michael Becker,b
Judith A. Ronau,a Susan K. Buchanan,c Vadim Cherezov,d Marie E. Morrow,a
Shenglan Xu,b Dale Ferguson,b Oleg Makarov,b Chittaranjan Das,a
Robert Fischettib and Garth J. Simpsona*
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47906, USA,bGM/CA@APS, Advanced Photon Source, Argonne National Laboratory, 9700 South Cass Avenue,
Argonne, IL 60439, USA, cNIDDK, National Institutes of Health, Building 50, Room 4503, 50 South
Drive, Bethesda, MD 20814, USA, and dDepartment of Molecular Biology, The Scripps Research
Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: [email protected]
Nonlinear optical (NLO) instrumentation has been integrated with synchrotron
X-ray diffraction (XRD) for combined single-platform analysis, initially
targeting applications for automated crystal centering. Second-harmonic-
generation microscopy and two-photon-excited ultraviolet fluorescence micro-
scopy were evaluated for crystal detection and assessed by X-ray raster
scanning. Two optical designs were constructed and characterized; one
positioned downstream of the sample and one integrated into the upstream
optical path of the diffractometer. Both instruments enabled protein crystal
identification with integration times between 80 and 150 ms per pixel,
representing a �103–104-fold reduction in the per-pixel exposure time relative
to X-ray raster scanning. Quantitative centering and analysis of phenylalanine
hydroxylase from Chromobacterium violaceum cPAH, Trichinella spiralis
deubiquitinating enzyme TsUCH37, human �-opioid receptor complex kOR-
T4L produced in lipidic cubic phase (LCP), intimin prepared in LCP, and �-cellulose samples were performed by collecting multiple NLO images. The
crystalline samples were characterized by single-crystal diffraction patterns,
while �-cellulose was characterized by fiber diffraction. Good agreement was
observed between the sample positions identified by NLO and XRD raster
measurements for all samples studied.
Keywords: XRD; NLO; SHG; SONICC; centering; protein; TPE-UVF; microscopy;LCP; two-photon.
1. Introduction
The high photon flux and energy tunability of synchrotron
radiation sources have made them indispensable tools for
X-ray analysis, with applications spanning protein structure
determination through materials science and nanotechnology
(Rasmussen et al., 2011; Moukhametzianov et al., 2008; Bates
et al., 2006; Berger et al., 2010; Dauter, 2006; Ihee et al., 2010;
le Maire et al., 2011; Parker et al., 2006; Riekel et al., 2005).
The increasing drive toward tighter focusing has enabled
structure determination on ever-smaller crystals and sub-
domains within materials, but presents growing challenges for
reliable crystal centering. These challenges are particularly
relevant for protein crystal diffraction, in which the drive
toward fully automated X-ray diffraction analysis at
synchrotron sources has introduced bottlenecks in sample
positioning (Andrey et al., 2004; Moukhametzianov et al., 2008;
Pothineni et al., 2006; Aishima et al., 2010; Cherezov et al.,
2009; Stepanov et al., 2011a). Diffraction-quality protein
crystals are typically obtained through crystallization screen-
ings, followed by optimization, and then are placed into cryo-
loops, which are flash-cooled in liquid nitrogen to reduce
X-ray damage and aid in sample handling (Dobrianov et al.,
1999; Karain et al., 2002). High-throughput methods for
automated crystal positioning are frustrated by complications
of reliable centering of smaller and smaller protein crystals
within more complex and turbid matrices. The current most
reliable methods for crystal centering involve rastering the
133

sample using a focused X-ray beam (Accardo et al., 2010;
Hilgart et al., 2011; Cherezov et al., 2009; Stepanov et al., 2011a;
Aishima et al., 2010; Song et al., 2007). From the resulting
X-ray diffraction images recorded as a function of sample
position in the beam, protein crystals are centered based on
the locations of strongest Bragg-like diffraction. X-ray fluor-
escence raster is also relatively fast, but it requires a conve-
nient X-ray fluorescent element to be present in the crystal
(Stepanov et al., 2011a).
While generally successful, X-ray raster scanning suffers
from several limitations. First, the method is relatively slow,
often utilizing >2 s per pixel (raster cell), corresponding to
analysis times from several minutes up to an hour depending
on the number of cells in the raster grid and on the exposure
time (Aishima et al., 2010). Rastering is commonly performed
first with a coarse grid, and then a finer grid, to minimize the
number of cells, and to increase speed. The total pixel number
is in turn dependent on the size of the X-ray beam, the speed
of the detector and analysis, as well as the scanned size of the
cryo-loop and the crystal itself (Cherezov et al., 2009; Song
et al., 2007). Recent advances in diffraction image read times
using single-photon-counting arrays (pixel array detectors)
(Broennimann et al., 2006), allowing integration times as low
as 2 ms per image (Aishima et al., 2010), can significantly
reduce the time frame for raster scanning measurements.
However, the time required for raster scanning will still ulti-
mately be limited by the collective times required to obtain
sufficient signal to noise (S/N) in a given pixel, to translate the
sample through the X-ray source, and to reconstruct the
crystal positions based on automated analysis of the compiled
diffraction images. Diffraction is a relatively inefficient process
with far more X-ray photons absorbed or inelastically scat-
tered than detected for diffraction analysis, contributing to
sample damage, even under the cryogenic conditions typically
utilized. With small crystals or beams, incident X-ray inten-
sities must be increased accordingly to achieve diffracted
intensities equivalent to those for large crystals, thereby
increasing absorbed X-ray dose and exacerbating damage.
Alternative methods for automated loop centering based on
optical imaging include bright-field image analysis and ultra-
violet fluorescence (UVF) microscopy, which takes advantage
of intrinsic fluorescent properties of protein crystals (Jain &
Stojanoff, 2007; Vernede et al., 2006; Pohl et al., 2004; Andrey
et al., 2004; Pothineni et al., 2006). However, algorithms for
protein crystal centering (e.g. based on crystal edge-finding
algorithms) are error-prone for microcrystals and turbid
matrices, such as lipidic cubic phase (LCP). Methods opti-
mized for analysis within the mother liquor often prove
unreliable for a loop-mounted crystal, in part because algo-
rithms often cannot easily distinguish between the loop,
features in the cryo-cooled mother-liquor and the crystal.
Furthermore, both bright-field and UVF imaging are chal-
lenging to reliably implement in turbid matrices, where optical
scattering frustrates reliable crystal imaging. UVF also has
a potential disadvantage of inducing UV photodamage to
samples from long exposures, or in highly labile proteins, but
the exposure times required for imaging are typically short
enough to minimize such effects (Vernede et al., 2006; Chen et
al., 2009; Nanao & Ravelli, 2006).
More recently, nonlinear optical imaging (NLO) methods
such as second-harmonic generation (SHG) and two-photon-
excited UV fluorescence (TPE-UVF) have emerged as viable
alternatives for high-contrast crystal visualization (Kissick et
al., 2010; Madden et al., 2011). SHG, or the frequency doubling
of light, is symmetry forbidden in disordered media (e.g.
amorphous protein aggregates or proteins in solution) but is
allowed for certain classes of crystals (Haupert & Simpson,
2011). Fortuitously, the chirality intrinsic to proteins typically
results in the adoption of SHG-active crystal classes. Recent
quantum chemical calculations suggest an SHG coverage of
approximately 84% of protein crystals in the Protein Crystal
Database using an optimized instrument (Haupert et al.,
2012). TPE-UVF provides a complimentary method to SHG
for protein crystal detection, with contrast dependent on the
presence of aromatic side-chains (primarily tryptophan),
independent of crystallinity. Crystals that are weakly active to
SHG imaging but contain fluorescent amino acid residues can
be detected (Madden et al., 2011). Furthermore, TPE-UVF
can aid in distinguishing SHG-active small-molecule and salt
crystals from protein crystals.
The high selectivity for crystals and negligible background
from disordered protein aggregates typically produces high-
contrast SHG images, which are highly compatible with
automated image analysis algorithms designed for protein
crystal detection and centering (Haupert & Simpson, 2011).
SHG measurements have recently enabled crystal detection
for diffraction centering using off-line instrumentation
(Kissick et al., 2013), in which protein crystals were first
imaged under cryogenic conditions with an SHG microscope,
and then manually compared with diffraction images obtained
by X-ray raster scanning with good agreement. A major
benefit of NLO instruments is the reduction in time required
to determine crystal locations with high contrast, as
measurements for an entire loop can be obtained in as little as
a few seconds, compared with tens of minutes routinely
required for X-ray raster imaging. The spatial resolution of
NLO instruments is also high (�1–2 mm), whereas X-ray
diffraction (XRD) rastering with this type of resolution would
take substantially longer to scan an area equivalent to that of
the entire NLO image (>72 h at 1 s per pixel for a 512 � 512
pixel image). Furthermore, reducing the reliance on X-ray
raster imaging would minimize X-ray-induced sample damage
(Hilgart et al., 2011; Ravelli & Garman, 2006).
By integrating SHG and TPE-UVF imaging directly into a
synchrotron X-ray diffraction beamline, the robotic controls,
automated positioning capabilities, cryogenics and other
beamline utilities of high-throughput synchrotron facilities can
be leveraged. However, the spatial constraints of a typical
synchrotron X-ray experimental hutch represent a nontrivial
hurdle for development of compatible NLO instrumentation.
Typical research NLO instruments occupy a large footprint
(an optical table approximately 120 cm � 300 cm), far greater
than the space available on a typical beamline. In this
work, two complementary prototypes for an on-line compa-
research papers
532 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope J. Synchrotron Rad. (2013). 20, 531–540
134

tible instrument combining synchrotron
XRD and NLO imaging are described.
Assessment of these systems was
performed by direct comparisons
between NLO images and those
obtained by X-ray diffraction rastering.
2. Experimental methods
Two separate instruments were
designed and constructed for inte-
grating XRD and NLO imaging, each
with its own advantages and limitations.
The upstream version introduced the
incident light coaxial and parallel with
the direction of the X-ray beam path,
while the downstream system was
coaxial and anti-parallel. The upstream
version was designed to fully integrate
with the existing optical path, while the
downstream version was optimized for
high flexibility and compatibility with
diverse beamline configurations. Both
systems were rated as Class I laser
systems on-site, with enclosed beam
paths, shutters and interlocks to ensure
no exposed collimated optical radiation.
The integrated NLO microscopes were
installed at beamlines 23-ID-B and 23-
ID-D at the Advanced Photon Source
(APS) at Argonne National Laboratory
in Argonne, IL, USA. A basic schematic
of the instruments and beam paths as they were installed on
the synchrotron beamline can be seen in Fig. 1. Detailed
descriptions and photographs are provided.
2.1. Integrated nonlinear optical microscope designs
The upstream illumination NLO system was designed to sit
above the existing instrumentation at GM/CA beamline 23-
ID-B at the APS, and couple directly into the existing optical
path. A Fianium FemtoPower 1060 ultrafast fiber laser was
utilized, producing �160 fs pulses centered around 1060 nm,
with a 50 MHz repetition rate, maximum power of 1.5 W,
allowing for a maximum power of �140 mW at the sample,
with 80% of the overall loss arising from the objective. The
Fianium source was composed of an oscillator coupled via a
1.5 m fiber to a dispersion compensator and free-space coupler
unit, with dimensions of approximately 15 cm� 13 cm� 8 cm.
A heated doubling crystal (Newlight Photonics Inc.,
SHG1663-IM, HTS 85141000) was permanently assembled in
the beam path, with the fundamental beam focused into the
crystal with a plano-convex lens ( f = 35 mm) and collimated
with another plano-convex lens ( f = 100 mm) after the
doubling crystal. The efficiency of SHG from the doubling
crystal was controlled by either introducing or removing a
1064 nm zero-order half-wave plate using a flip mount (New
Focus, 8892-K). The scanning assembly consisted of a
galvanometer mirror (Cambridge Technology, 6210H) and
resonant scanning mirror (Cambridge Technology, 1-003-
3002509), controlling the beam position on the horizontal
slow-scan and vertical fast-scan axes, respectively. The beam
was directed into a telocentric lens pair consisting of two
plano-convex lenses ( f = 75 mm and f = 250 mm) leading to an
additional 3.3� beam expansion after the scan head. The
incident light then reflected off a dichroic mirror stack
(Semrock, PBP01-529/23-25x36 and Chroma, 900dcsp)
designed to reflect 1060 nm and s-polarized 530 nm incident
light. The p-polarized component of the returning 530 nm light
was transmitted by this same dichroic for epi-detected SHG
(i.e. SHG detected in the backward direction through the same
objective as the incident light). High-reflectivity dichroic
mirrors for both 1060 nm and 530 nm light (Semrock, FF550-
Di01-25x36) delivered both wavelengths to the back aperture
of the 10� objective (Optem, 28-21-10), which was modified
with a �1.2 mm hole bored through the center to allow
X-ray access. In epi, the p-polarized SHG returning through
the dichroic mirror was passed through a bandpass filter
set (Chroma, HQ530/30m and CVI, 03FCG567/KG3) and into
a compact photomultiplier tube (PMT) module (Hamamatsu,
H10722-10). SHG and TPE-UVF were collected in the
transmission direction by a plano-convex lens ( f = 25.4 mm)
research papers
J. Synchrotron Rad. (2013). 20, 531–540 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope 533
Figure 1(a) Schematic of the downstream NLO microscope; (b) schematic of the upstream NLOmicroscope; (c) close-up view of the downstream NLO microscope, with the solid arrowrepresenting incident laser propagation (red, 1060 nm) and dashed arrows representing thefrequency-doubled signal (green, SHG at 530 nm); (d) close-up view of the upstream NLOmicroscope, with solid arrows representing incident laser propagation (red, 1060 nm; green, 530 nm)and dashed arrows representing the measured signal (green, SHG at 530 nm; blue, TPE-UVF).
135

affixed to a right-angle prism using optical epoxy (Norland
Optical Adhesive 63). Another plano-convex lens ( f =
25.4 mm) coupled the detected light into a near-UV-compa-
tible liquid light guide (Oriel Instruments, 77554) collimated
with a plano-convex lens ( f = 25.4 mm) into the detection
assembly. Both the SHG and TPE-UVF were then reflected
off a primary dichroic beam splitter (Semrock, FF555-Di03-
25x36), then separated at a second dichroic beam splitter
(Chroma, z1064rdc-sp) for selective detection of SHG
(through Chroma, HQ530/30m and CVI, 03FCG567/KG3
filters) and TPE-UVF (through Semrock, SP01-532RS-25 and
FF01-440/SP-25 filters). Both the SHG and TPE-UVF were
focused onto the faces of the PMT modules (Hamamatsu,
H10722-10) by a plano-convex lens ( f = 60 mm) positioned
between the primary and secondary dichroic beam splitters.
Backlight illumination was achieved using an LED (ThorLabs,
MCWHL2) passing through the primary dichroic beam
splitter and into the liquid light guide. The illumination light
was then focused through the trans-SHG/TPE-UVF collection
optics and onto the sample.
The downstream NLO system was also designed with the
optical axis of the objective co-axial with the axis of X-ray
propagation [Figs. 1(a) and 1(b)], using a similar laser source.
The size constraints associated with this beamline, specifically
the restrictions imposed by the support structure of the
beamline and the area and instruments surrounding the
sample, limited the available footprint of the NLO system to
39 cm � 19 cm. The scanning assembly was composed of dual
galvanometers (Cambridge Technologies, 6210HSM40B),
mounted in a two-dimensional galvo 30 mm cage cube
(Thorlabs, GCM002), with each scanning mirror rotating
along either the x or y axis. With the scan head inducing a 90�
turn into the beam path, the incident light was directed
through a telocentric lens pair, mounted in a 30 mm cage cube,
and composed of an aspheric lens ( f = 10 mm) and a plano-
convex lens ( f = 50 mm), leading to a 5� beam expansion. The
incident light was then focused onto the sample by a long-
working-distance IR 10� objective (Mitutoyo, NT46-403)
generating SHG at 530 nm. Up to 650 mW of 1064 nm light
could be delivered to the sample with this system with the use
of the IR objective (compared with 140 mWwith the upstream
system). The SHG was detected in the epi-direction, collected
through the incident objective and reflected through a filter set
and onto a compact PMT module (Hamamatsu, H10722-10)
by a dichroic mirror (Omega Optical, 580DCLP) centered
around 532 nm and mounted in a rotatable kinematically
controlled cage cube platform. The SHG signal was detected
through a filter set composed of a KG3 (Thorlabs, FGS900)
and 530 nm filter (Chroma, z532/10x). Bright-field images
were also collected in the epi-direction using a module
composed of an aspheric lens ( f = 20 mm) and a CMOS
camera (Thorlabs, DCC1645C), manually inserted when
bright-field images were desired. Including the laser source,
the total footprint of the microscope was 25 cm � 15 cm �15 cm. The microscope was translated to the sample, at a
height of 1.4 m, to perform SHG detection and centering
measurements. The foundation of the microscope was a
high-precision long-travel translation stage (Newport,
M-IMS300V), and its electronics box (Newport, ESP 300,
three-axis motion controller), capable of translating the laser
pulse-compressor/output coupler, the microscope and the
support structure to and from the sample between X-ray
measurements, corresponding to approximately 20 cm of
travel, with an absolute accuracy of 2 mm.
The electronics package was designed and constructed in
collaboration with the Jonathan Amy Facility for Chemical
Instrumentation at Purdue University (JAFCI). The electro-
nics package integrated the electronics associated with the
microscope, including the power supplies, control boards and
data acquisition card (National Instruments), into a compact
housing for easy mounting and transport, with a footprint of
46 cm� 61 cm � 31 cm. Data were acquired as photon counts
using a gated multi-scalar card (Becker & Hickl, PMS-400a),
controlled using a custom-designed Labview program, which
was also written in collaboration with JAFCI. Data recon-
struction and imaging were completed through ImageJ (NIH,
2011).
2.2. X-ray raster scan scheme
XRD analysis and NLO images were acquired on all
samples studied on 23-ID-B. Diffraction of kOR-T4L was
acquired with a 5 mm-diameter X-ray beam, 5� 5 mm cell size,
12.0 keV X-ray beam, with 1 s exposure times, a photon flux
of 2.7 � 1010 photons s�1 (full unattenuated beam) and a
detector distance of 300 mm. Diffraction of TsUCH37 was
acquired with a 10 mm-diameter X-ray beam, a 10 � 10 mmcell, a photon flux of 1.3 � 1010 photons s�1 (10-fold
attenuation) and detector distance of 300 mm. Diffraction of
�-cellulose was acquired with a 10 mm-diameter X-ray beam,
a 10 � 10 mm X-ray beam with a photon flux of 2.7 �109 photons s�1 (50-fold attenuation) and detector distance of
300 mm. The resulting NLO images and XRD raster
measurements were compared using ImageJ and JBluIce
(Hilgart et al., 2011), which employs DISTL (Zhang et al.,
2006), to assess the degree of correlation of the sample posi-
tion within the loop. The boundaries of the raster grids and
raster cell sizes were defined using the software GUI JBluIce
(Stepanov et al., 2011b). Bragg candidates, which estimate the
number of well-ordered reflections, were generated for each
X-ray diffraction image; they are shown color-coded in the
figures as unsmoothed XRD raster images. The X-ray beam
size was adjusted using a mini-beam collimator (Fischetti et al.,
2009).
3. Sample materials
Phenylalanine hydroxylase from Chromobacterium violaceum
(cPAH) was purified as a glutathione s-transferase (GST)
fusion protein. The GST tag was cleaved with PreScission
protease (GE Biosciences). For crystallization, cPAH was
concentrated to 10 mg ml�1 in a solution of 5 mMHEPES, pH
7.4. Crystals of cPAH were obtained at ambient temperature
utilizing hanging-drop vapor diffusion from solution 43 of
research papers
534 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope J. Synchrotron Rad. (2013). 20, 531–540
136

Hampton Research’s PEG/Ion 2 screen [0.1 M Na-HEPES,
pH 7.0, 0.01 M magnesium chloride hexahydrate, 0.005 M
nickel (II) chloride hexahydrate and 15% w/v PEG 3350] with
8.3 mM hexammine cobalt (III) chloride and 8.3 mM guani-
dine hydrochloride as additives. Crystals were briefly soaked
in 25% ethylene glycol and then flash-cooled in liquid
nitrogen.
Crystals of human �-opioid receptor in complex with an
antagonist JDTic were obtained as described by Wu et al.
(2012). Briefly, the human �-opioid receptor sequence was
modified by fusing T4 lysozyme (T4L) into intracellular loop 3
(Gly261–Arg263), performing N/C-terminal truncations
(�Glu2Ala42, �Arg359Val380) and introducing a single
point mutation Ile1353.29Leu. The resulting construct kOR-
T4L was expressed in baculovirus infected sf9 insect cells.
Receptor was extracted from isolated membranes using
dodecylmaltoside/cholesterol hemisuccinate detergent
mixture, purified by metal-affinity chromatography, and
concentrated to 40 mg ml�1. Lipidic cubic phase crystal-
lization was performed as previously described (Caffrey &
Cherezov, 2009; Cherezov et al., 2004), by mixing protein
solution with 10% cholesterol in monoolein at 2/3 protein
solution/lipid ratio, and dispensing 50 nL protein laden LCP
boluses overlaid with 800 nL precipitant solutions in a 96-well
glass sandwich plate (Marienfeld) (Cherezov & Caffrey, 2003)
using a NT8-LCP crystallization robot (Formulatrix). Crystals
were obtained in 100 mM sodium citrate pH 5.8–6.4, 28–32%
(v/v) PEG 400, 350–450 mM potassium nitrate, and were
harvested directly from LCP matrix using MiTeGen micro-
mounts and flash-cooled in liquid nitrogen.
The catalytic domain of Trichinella spiralis deubiquitinating
enzyme UCH37 was expressed in E. coli as a GST-fused
construct, purified on a glutathione-agarose column,
complexed with ubiquitin vinyl methyl ester (UBVME), and
subsequently purified by ion-exchange chromatography.
Crystals of this complex, hereafter referred to simply as
TsUCH37-UbVME complex, were grown by hanging-drop
vapor diffusion in 3 M ammonium sulfate, 0.1 M bicine pH 9.0,
and 2 mM l-glutathione (mixture of reduced and oxidized)
over two days at room temperature.
The �-cellulose was prepared from pulpwood that under-
went both the Kraft process and subsequent mercerization
(Sixta et al., 2004; Takai & Colvin, 1978).
A construct encoding the membrane domain of E. coli
O157:H7 intimin was expressed, purified and crystallized as
described previously (Fairman et al., 2012). Briefly, Int208-449
was expressed in the outer membranes of E. coli BL21(DE3)
cells, extracted with the detergent Elugent (Calbiochem), and
purified by Ni-NTA affinity and anion-exchange chroma-
tography using buffers containing dodecyl maltoside
(Anatrace). Size-exclusion chromatography was used as a final
purification step and served to exchange the detergent to
lauryl dimethyl amine oxide (LDAO, Anatrace) using a buffer
containing 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 0.01%
NaN3 and 0.05% LDAO. The protein was concentrated to
20 mg ml�1, heptanetriol was added at 3% w/v, and the solu-
tion was mixed with monoolein at a 2/3 protein-to-lipid ratio.
A Mosquito LCP robot (TTP Labtech) was used to dispense
100 nL protein–lipid droplets, overlaid with 750 nL well
solutions. Intimin crystals grew from 100 mM sodium citrate,
pH 4.5–5.5, 50–100 mM NaCl, 100–150 mM MgCl2 and 30–
34% PEG 400. Crystals were mounted directly from the LCP
mixture and flash-cooled in liquid nitrogen.
4. Results and discussion
Data were acquired with both downstream and upstream
versions of the NLO instrument, and schematic representa-
tions along with photographs of the beam paths are shown
in Fig. 1.
Fig. 2 (acquired via the upstream system) shows a large
TsUCH37-UbVME crystal. Both the presence and position of
the crystal can be independently confirmed with bright-field
imaging (a), NLOmicroscopy and XRDmeasurements. Signal
intensities of the corresponding epi-SHG (b), transmission-
SHG (c) and TPE-UVF (d) were measured and processed in
ImageJ. Although the crystal is visible using conventional
optical imaging approaches, NLO microscopy produced
substantial improvements in contrast compared with bright-
field imaging. An X-ray diffraction raster was acquired (e) and
a representative diffraction image is shown ( f).
Intimin protein crystals in LCP were examined using the
upstream NLO system. In Fig. 3 the bright-field image is
shown in (a), with the corresponding trans-SHG image (b),
and X-ray raster acquired with a 5 � 5 mm beam, confirming
the presence of a protein crystal (c), with the spot having
greatest protein-like diffraction circled and the resulting
diffraction pattern provided (d). All protein crystals identified
by SHG and XRD were accurate for absolute position within
the resolution of the 5 mm X-ray beam.
In Fig. 4 (acquired via the upstream system) a bright-field
image of a kOR-T4L crystal within frozen lipidic cubic phase
is shown (a). As often arises with lipidic mesophase crystal-
lizations, the looped droplets exhibited high optical scattering
upon freezing that frustrated conventional bright-field
imaging approaches for crystal positioning. Transmission SHG
(b) and TPE-UVF (c) images were acquired, exhibiting loca-
lized areas (�2–5 mm) of signal within the loop, suggesting the
presence of a crystal. Crystals were confirmed via a 5 mm-
diameter X-ray beam and 5 � 5 mm cell X-ray raster scan (d),
in which several pixels exhibit weak, but detectable, diffrac-
tion with Bragg analysis consistent with the presence of a
protein crystal. Diffraction patterns for the brightest spot are
shown in Fig. 4(e). However, signal is observed in the trans-
SHG and TPE-UVF images that does not correspond to areas
of protein-like diffraction in the X-ray raster image. This
signal discrepancy is tentatively attributed to protein crystals
that are too small to produce Bragg peaks by XRD, or to the
presence of other ordered materials arising in a false positive.
False negatives for particular focal planes were also observed,
in which analysis of the diffraction patterns obtained from the
raster image indicates the presence of protein-like diffraction
located in areas that did not exhibit substantial SHG or TPE-
UVF due to the finite depth of field (�25 mm). However,
research papers
J. Synchrotron Rad. (2013). 20, 531–540 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope 535
137

acquisition of multiple focal planes through samples has been
observed to recover crystal locations more quantitatively (not
shown).
In SHG measurements the possibility of false positives
exists from other SHG-active structures. Most notably, some
salts commonly used in crystallization screening can adopt
non-centrosymmetric SHG-active lattices and produce bright
SHG. Alternatively, noncrystalline structures exhibiting
molecular ordering over distances significantly greater than
the wavelength of light can also potentially produce false
positives for SHG. An example of a false positive, from a
noncentrosymmetric vanadate salt crystal, is shown in Fig. S1
of the supplementary information1 in which a cryo-loop
containing a crystal grown in LCP was examined with the
upstream NLO instrument, and yielded substantial signal in
the epi- and transmission-SHG directions. X-ray raster scans
suggested the presence of salt-like diffraction, in addition to
ice diffraction, as there was ice present on the sample loop.
Key signatures for an SHG-active salt were found to be bright
epi-SHG and little to no detectable TPE-UVF. These salt
crystal signatures can be exploited to reduce the likelihood of
false positives. False positives can arise
using TPE-UVF if there is protein
aggregate located within the loop
because TPE-UVF probes the presence
of aromatic residues and is not crystal
specific. Salt crystals and protein
aggregates are common occurrences
with protein crystal growth, generating
false positives for SHG and TPE-UVF
measurements, respectively. Fortu-
nately, most simple salts adopt SHG-
inactive centrosymmetric structures.
Complementary use of these two tech-
niques can significantly reduce the
likelihood of false positives and false
negatives.
Combined NLO imaging and XRD
was also applied to studies of �-cellu-lose, which exhibits fiber-like diffrac-
tion. NLO measurements performed
on loop-mounted cellulose generated
moderate S/N for multiple fibers within
the sample loop (Fig. 5, acquired via
the upstream system). Although fiber
diffraction was evident from the cellu-
lose samples, the DISTL algorithm used
in raster scanning, which searches for
discrete Bragg reflections or spots and
not fiber diffraction, does not indicate
these areas, but rather seems to show
that no measurable sample is present.
Manual inspection of the individual
diffraction patterns was performed to discern the presence of
fiber diffraction.
cPAH crystals ranging in size from 50 mm to 200 mm in
length were imaged with both the downstream instrument
with epi-only detection and X-ray raster scanning [Fig. S2
(supplementary information)]. The locations of intense
protein-like Bragg diffraction typically agreed well with those
of brightest epi-SHG for both large and small cPAH crystals
(e.g. Fig. S2). However, departures between the two were also
observed. Several explanations for the differences were
considered. First, the presence of multiple crystalline domains
within the crystal (e.g. from twinning) may cause the diffrac-
tion spot total to deviate from indicating optimal protein
ordering. Second, inhomogeneous optical scattering of the
incident or detected light can potentially impact the contrast
through effects unrelated to the crystal SHG activity.
However, bright-field images do not suggest substantial
differences in optical transmissivity across the crystal that
might have influenced contrast. Finally, NLO measurements
probe a much narrower depth of field than X-ray diffraction,
which is penetrating. If a particular crystal was not positioned
within the depth of field of the beam-scanning NLO micro-
scope, the SHG efficiency will be substantially reduced or
entirely absent within the detection limits of the instrument.
Despite the quantitative discrepancies, the presence of SHG
research papers
536 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope J. Synchrotron Rad. (2013). 20, 531–540
Figure 2(a) Bright-field image of a T. spiralis UCH37 1-226/UbVME complex crystal (�100 mm thick) andthe corresponding (b) epi-SHG, (c) trans-SHG, (d) TPE-UVF and (e) X-ray raster scan within the300 � 300 mm box. ( f ) X-ray diffraction of a representative 10 mm-diameter area from (e). X-rayenergy: 12 keV; exposure time: 1 s; photon flux: 2.7 � 109 photons s�1 (10-fold attenuation);detector distance: 300 mm; maximum theoretical resolution: 2.25 A. The large difference in the epi-and trans-SHG signals is expected for thick samples owing to the difference in the forward andbackward coherence length. The intensities of the two directions will approach equality as thesample thickness approaches the backwards coherence length (�100 nm). Scale bars are 100 mm.(Three darkened spots, apparent in this figure, arose from separate X-ray ‘burn tests’ to assess X-raydamage, the results of which will be published in a future study.)
1 Supplementary data for this paper are available from the IUCr electronicarchives (Reference: WA5051). Services for accessing these data are describedat the back of the journal.
138

signals above the background correlated with the areas of
the crystal generating a detectable protein-like diffraction,
providing preliminary confirmation of the ability of the
downstream instrument to rapidly generate information for
crystal position as a complement to X-ray raster scanning.
The polyimide loops (MiTeGen) were found to undergo
noticeable deformation with less than 100 mW incident power
using the downstream system, whereas the nylon loops were
more robust, and were not damaged at these powers. No
noticeable damage could be induced in either loop types using
the upstream system during either SHG or TPE-UVF
measurements (120 mW and 90 mW, respectively). Several
mechanisms were considered for the observed laser-induced
damage to the polyimide loops when measured with the
downstream system. Previous studies suggest that damage
from multi-photon absorption and plasma formation was
found to be an important, if not dominant, mechanism for
damage in biological NLO imaging (Sacconi et al., 2006).
However, those measurements were performed under condi-
tions of tight focusing [high numerical aperture (NA)] and
on live cells/tissues. However, alternative mechanisms may
dominate in the present low-NA studies of purified protein
crystals maintained under cryogenic conditions. Local heating
was also considered as a possible damage mechanism, arising
from either one- or two-photon absorption of the incident
beam. The marked difference in damage susceptibilities
between the upstream and downstream systems is consistent
with this mechanism, differing notably in the use of a resonant
research papers
J. Synchrotron Rad. (2013). 20, 531–540 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope 537
Figure 4(a) Bright-field image of a membrane protein (human �-opioid receptorcomplex) crystal in lipidic cubic phase and the corresponding (b) trans-SHG and (c) TPE-UVF, with (d) an X-ray raster summary overlayshowing corrected Bragg-like reflection counts. (e) X-ray diffraction ofthe 5 mm-diameter area corresponding to the red circles in each image.X-ray energy: 12.0 keV; exposure time: 1 s; photon flux: 2.7 �1010 photons s�1 (unattenuated beam); sample-to-detector distance:300 mm; maximum theoretical resolution: 2.25 A. Scale bars are 20 mm.Cross-hairs were added to (b) and (c) to assist in orienting the fields ofview with respect to the bright-field and diffraction raster images.
Figure 3(a) Bright-field for an intimin protein crystal generated in LCP withcorresponding (b) trans-SHG and (c) X-ray raster summary overlayshowing corrected Bragg-like reflection counts. (d) X-ray diffraction ofthe 5 mm-diameter area corresponding to the red circles in each image,with X-ray energy 12.0 keV, exposure time 1 s, photon flux 2.7 �1010 photons s�1 (unattenuated beam), sample-to-detector distance of300 mm, resulting in a maximum theoretical resolution of 2.25 A. Scalebars are 50 mm. Cross-hairs were added to (a) and (b) to assist in orientingthe field of view with respect to the diffraction raster images.
139

8 kHz scan mirror for the upstream system and a galvan-
ometer-driven mirror operating at 200 Hz on the downstream
system. Rapid beam-scanning using a resonant scanner
combined with long-wavelength (>1 mm) incident light was
shown previously to have no detectable effect on crystal
diffraction quality using a variety of protein crystals, including
myoglobin crystals containing heme groups exhibiting strong
visible light absorption (Kissick et al., 2013). Myoglobin was
specifically chosen, as the color center was anticipated to be
highly susceptible to light-induced perturbation (Banerjee et
al., 1969). However, no statistically significant structural
changes to the lattice were observed in laser-exposed versus
unexposed regions of single crystals (Kissick et al., 2013).
The susceptibility for damage using the polyimide loops
increased notably for TPE-UVF, as the optical transparency
was substantially reduced at 530 nm. Whereas loop absorption
is negligible at 1 mm for SHG, roughly 30% of the incident
530 nm light for TPE-UVF is absorbed by the standard
yellow-tinted polyimide loop material (MiTeGen, http://www.
mitegen.com/). By positioning the loop to avoid the outer
turning points of the fast-scan mirror or blocking the beam at
those locations, no noticeable damage could be induced in the
polyimide loops during TPE-UVF imaging.
Both of the NLO imaging systems presented in this paper
have strengths and limitations, and either could be utilized as
a method for locating and centering protein crystals on a
synchrotron beamline. With a small footprint and the ability to
insert and remove the instrument, there is potential for a
single design of the downstream instrument to be utilized on a
variety of different beamlines. However, the time required for
translating the entire microscope to and from the sample
increases the total time for collecting SHG images and XRD
of the protein. Indeed, the microscope positioning required
substantially more time (�2 min) than the sample imaging
(�40 s). Furthermore, the absolute accuracy of the translation
stage (in this case,�2 mm) can ultimately dictate the precision
in crystal positioning. In addition, the downstream instrument
did not have transmission-SHG detection capabilities. For
protein crystals, detection in transmission provides substantial
improvements in detection limits for weakly SHG-active
proteins, as thickness greater than the crystals’ coherence
lengths can decrease the overall SHG intensity in the epi
direction (Boyd, 2009; Kestur et al., 2012). The absence of
transmission detection could potentially be remedied by
introducing additional optics or integrating into existing
optical paths.
The direct integration of the upstream system eliminated
the need for a translation stage for inserting the microscope,
as was used with the downstream system. This significantly
reduced the time between imaging and XRD, which allowed
for a marked improvement on throughput of data collection.
The upstream system did still require the transmission detec-
tion optics to translate in and out for XRD collection in
transmission, but epi-detected SHG can be performed
concurrently with X-ray diffraction, with only a factor of three
reduction in signal intensity with the mini-beam collimator
in place. The positioning of the collection optics does not,
however, require precise realignment allowing for a significant
improvement on the translation time, as compared with the
downstream instrument, where the entire microscope requires
translation with high precision. The upstream system had
some design trade-offs to accommodate the existing optical
path, which in part accounted for the lower infrared (IR)
throughput and available power in the upstream system. The
biggest losses came from the incident objective in which 80%
of the IR power was lost from reflections because it was not
designed for IR incident light. Choosing optics with a more
broadband anti-reflective coating (ARC) will significantly
improve the power throughput. Testing performed in-house,
with an IR-ARC objective, resulted in a doubling of the
IR transmittance, corresponding to an anticipated four-fold
improvement in signal at the sample (unpublished). The
multiple imaging modes (SHG and TPE-UVF), as well as both
epi and transmission detection, improves the ability of the
upstream system to detect protein crystals that could other-
wise be missed on the downstream system.
research papers
538 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope J. Synchrotron Rad. (2013). 20, 531–540
Figure 5(a) Bright-field image of �-cellulose fibers and the corresponding (b) epi-SHG and (c) trans-SHG images, all 300� 300 mm. (d) X-ray diffraction ofa 10 mm-diameter area within the red circle of each image. X-ray energy:12.0 keV; exposure time: 1 s; photon flux: 2.7 � 1010 photons s�1
(unattenuated beam); sample-to-detector distance: 300 mm; maximumtheoretical resolution: 2.25 A. Scale bars are 100 mm. Cross-hairs wereadded to (b) and (c) to assist in orienting the fields of view with respect tothe bright-field image.
140

Based on these combined results, integrating a NLO
microscope with a synchrotron XRD instrument complements
stand-alone X-ray raster scanning for crystal centering in three
key respects. First, it is expected to minimize radiation-
induced sample damage compared with X-ray raster techni-
ques for X-ray labile crystals or small crystals difficult to
quickly detect at low X-ray flux (Kissick et al., 2013). Second,
NLO microscopy significantly increases the spatial resolution
and reduces the total acquisition time for the determination of
crystal location. For a large sample area (150 � 150 mm)
scanned with a small beam size (5 � 5 mm), X-ray raster
images for the protein crystals typically required approxi-
mately 30 min to acquire with a 1 s X-ray exposure time. For
NLO measurements on identical samples, the acquisition time
for the collection of each image was typically <10 s. The
downstream NLO system allows 512 � 512 pixel images with
40 s acquisitions, and the upstream system allows 150 � 150
pixel images with 1 s acquisitions, which is roughly a >104-fold
reduction in the per-pixel acquisition time compared with the
X-ray raster acquisition time per cell (�3 s per pixel, corre-
sponding to a 1 s exposure, with 2 s of dead-time between
pixel acquisitions). The theoretical resolution of the objective
was 1.6 mm with 2 mm measured spatial resolution. The
downstream NLO system required a total time of 2.5 min for
translation of the microscope from its resting position to the
sample and then back to the resting position following NLO
measurements, resulting in a total acquisition time for each
sample of the order of 3 min, which is still significantly faster
and of higher resolution compared with X-ray raster scan
measurements performed on the same sample. In the
upstream system, no dead-time was required for epi-detection
(in fact, SHG imaging can be performed while acquiring
diffraction measurements), and only a few seconds of trans-
lation time were required to raise and lower the collection
optics in transmission. Third, for weakly diffracting systems
where rapid automated diffraction scoring is challenging,
NLO measurements may significantly increase the ability to
locate protein crystals.
5. Conclusion
Two different designs of integrated NLO instruments were
constructed and characterized targeting applications for
automated sample positioning. The systems were evaluated
using protein crystals (TsUCH37-UbVME, kOR-T4L, cPAH,
Intimin) and fibers (�-cellulose). Both NLO and XRD
exhibited good agreement for crystal positioning, consistent
with previous off-line measurements specifically targeting
protein crystals (Kissick et al., 2013). The integrated NLO and
synchrotron XRD instrument was found to enable precise
centering of �-cellulose samples for fiber diffraction without
requiring the development of an application-specific analysis
algorithm. The NLO instrument produced images with <10 s
image acquisition times, compared with 3–60 min for X-ray
rastering performed at much lower spatial resolution. By
nature of the higher resolution of NLO image acquisition, the
per-pixel raw data acquisition time was approximately five
orders of magnitude faster than X-ray raster scanning. Once
fully developed, NLO imaging may serve to identify regions of
interest for targeted X-ray scanning, or ultimately serve as the
sole or primary method for precise automated crystal posi-
tioning, such that all of the X-rays striking the crystal are
dedicated to structure elucidation.
Despite these successes, a relatively small variety of crystals
were used to characterize the instruments in this initial study.
Further studies on a greater diversity of protein crystals will
help define the scope of use for NLO methods in automated
centering. Additionally, the present study focused exclusively
on the hardware for visualization, and not on subsequent
algorithms for image analysis and automated crystal posi-
tioning. Higher contrast afforded by NLO imaging has the
potential to significantly improve the reliability of such algo-
rithms if the combined techniques of SHG and TPE-UVF
provide sufficient protein crystal coverage for general-purpose
use.
These studies provided a foundation for future efforts
combining NLO measurements with synchrotron X-ray
diffraction. The data presented here support the use of the
NLO microscopy for automated or manual crystal centering
prior to or in lieu of raster scanning. Potential scope of use
where all optical crystal positioning would be preferred
includes the analysis of smaller crystals (<5 mm), where the
low crystal volume may present challenges for rapid crystal
positioning by X-ray raster scanning. SHG also enables posi-
tioning of fibrous material exhibiting fiber diffraction, such as
cellulose, collagen, chitin etc. Further potential applications
include defect studies, X-ray damage studies and studies of
active pharmaceutical ingredients.
The authors acknowledge Huixian Wu, Victoria J. Hall,
Emma L. DeWalt, Valerie Pye, Martin Caffrey, Nicholas
Noinaj and James W. Fairman for aiding in sample prepara-
tion. Instrumentation development was supported in part by
the Center for Direct Catalytic Conversion of Biomass to
Biofuels (C3Bio), an Energy Frontier Research Center funded
by the US Department of Energy, Office of Science, Office of
Basic Energy Sciences, Award No. DE-SC0000997, and by the
NIH-NIGMS through the R01GM-103401. GM/CA@APS has
been funded in whole or in part with federal funds from the
National Cancer Institute (Y1-CO-1020) and the National
Institute of General Medical Sciences (Y1-GM-1104). Use
of the Advanced Photon Source was supported by the US
Department of Energy, Basic Energy Sciences, Office of
Science, under contract No. DE-AC02-06CH11357. Support is
also acknowledged from the NIH Common Fund in Structural
Biology, grant P50 GM073197. SKB is supported by the
Intramural Research Program of the NIH, National Institute
of Diabetes and Digestive and Kidney Diseases.
References
Accardo, A., Gentile, F., Mecarini, F., De Angelis, F., Burghammer,M., Di Fabrizio, E. & Riekel, C. (2010). Langmuir, 26, 15057–15064.
research papers
J. Synchrotron Rad. (2013). 20, 531–540 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope 539
141

Aishima, J., Owen, R. L., Axford, D., Shepherd, E., Winter, G., Levik,K., Gibbons, P., Ashton, A. & Evans, G. (2010). Acta Cryst. D66,1032–1035.
Andrey, P., Lavault, B., Cipriani, F. & Maurin, Y. (2004). J. Appl.Cryst. 37, 265–269.
Banerjee, R., Alpert, Y., Leterrier, F. & Williams, R. J. (1969).Biochemistry, 8, 2862–2867.
Bates, S., Zografi, G., Engers, D., Morris, K., Crowley, K. & Newman,A. (2006). Pharm. Res. 23, 2333–2349.
Berger, M. A., Decker, J. H. & Mathews, I. I. (2010). J. Appl. Cryst.43, 1513–1518.
Boyd, R. (2009). J. Biomed. Opt. 14, 029902.Broennimann, Ch., Eikenberry, E. F., Henrich, B., Horisberger, R.,Huelsen, G., Pohl, E., Schmitt, B., Schulze-Briese, C., Suzuki, M.,Tomizaki, T., Toyokawa, H. & Wagner, A. (2006). J. SynchrotronRad. 13, 120–130.
Caffrey, M. & Cherezov, V. (2009). Nat. Protoc. 4, 706–731.Chen, J., Callis, P. R. & King, J. (2009). Biochemistry, 48, 3708–3716.Cherezov, V. & Caffrey, M. (2003). J. Appl. Cryst. 36, 1372–1377.Cherezov, V., Hanson, M. A., Griffith, M. T., Hilgart, M. C., Sanishvili,R., Nagarajan, V., Stepanov, S., Fischetti, R. F., Kuhn, P. & Stevens,R. C. (2009). J. R. Soc. Interface, 6, S587–S597.
Cherezov, V., Peddi, A., Muthusubramaniam, L., Zheng, Y. F. &Caffrey, M. (2004). Acta Cryst. D60, 1795–1807.
Dauter, Z. (2006). Acta Cryst. D62, 1–11.Dobrianov, I., Caylor, C., Lemay, S., Finkelstein, K. & Thorne, R.(1999). J. Cryst. Growth, 196, 511–523.
Fairman, J. W., Dautin, N., Wojtowicz, D., Liu, W., Noinaj, N.,Barnard, T. J., Udho, E., Przytycka, T. M., Cherezov, V. &Buchanan, S. K. (2012). Structure, 20, 1233–1243.
Fischetti, R. F., Xu, S., Yoder, D. W., Becker, M., Nagarajan, V.,Sanishvili, R., Hilgart, M. C., Stepanov, S., Makarov, O. & Smith,J. L. (2009). J. Synchrotron Rad. 16, 217–225.
Haupert, L. M., DeWalt, E. L. & Simpson, G. J. (2012). Acta Cryst.D68, 1513–1521.
Haupert, L. M. & Simpson, G. J. (2011). Methods, 55, 379–386.Hilgart, M. C., Sanishvili, R., Ogata, C. M., Becker, M., Venugopalan,N., Stepanov, S., Makarov, O., Smith, J. L. & Fischetti, R. F. (2011).J. Synchrotron Rad. 18, 717–722.
Ihee, H., Wulff, M., Kim, J. & Adachi, S. (2010). Int. Rev. Phys. Chem.29, 453–520.
Jain, A. & Stojanoff, V. (2007). J. Synchrotron Rad. 14, 355–360.Karain, W. I., Bourenkov, G. P., Blume, H. & Bartunik, H. D. (2002).Acta Cryst. D58, 1519–1522.
Kestur, U. S., Wanapun, D., Toth, S. J., Wegiel, L. A., Simpson, G. J. &Taylor, L. S. (2012). J. Pharm. Sci. 101, 4201–4213.
Kissick, D. J., Dettmar, C. M., Becker, M., Mulichak, A. M., Cherezov,V., Ginell, S. L., Battaile, K. P., Keefe, L. J., Fischetti, R. F. &Simpson, G. J. (2013). Acta Cryst. D69, 843–851.
Kissick, D. J., Gualtieri, E. J., Simpson, G. J. & Cherezov, V. (2010).Anal. Chem. 82, 491–497.
Madden, J. T., DeWalt, E. L. & Simpson, G. J. (2011). Acta Cryst.D67,839–846.
Maire, A. le, Gelin, M., Pochet, S., Hoh, F., Pirocchi, M., Guichou,J.-F., Ferrer, J.-L. & Labesse, G. (2011). Acta Cryst. D67, 747–755.
Moukhametzianov, R., Burghammer, M., Edwards, P. C., Petit-demange, S., Popov, D., Fransen, M., McMullan, G., Schertler,G. F. X. & Riekel, C. (2008). Acta Cryst. D64, 158–166.
Nanao, M. H. & Ravelli, R. B. (2006). Structure, 14, 791–800.NIH (2011). ImageJ, http://rsbweb.nih.gov/ij/.Parker, S., Kenney, C., Gnani, D., Thompson, A., Mandelli, E.,Meddeler, G., Hasi, J., Morse, J. & Westbrook, E. (2006). IEEETrans. Nucl. Sci. 53, 1676–1688.
Pohl, E., Ristau, U., Gehrmann, T., Jahn, D., Robrahn, B., Malthan,D., Dobler, H. & Hermes, C. (2004). J. Synchrotron Rad. 11, 372–377.
Pothineni, S. B., Strutz, T. & Lamzin, V. S. (2006). Acta Cryst. D62,1358–1368.
Rasmussen, S. G., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y.,Kobilka, T. S., Thian, F. S., Chae, P. S., Pardon, E., Calinski, D.,Mathiesen, J. M., Shah, S. T., Lyons, J. A., Caffrey, M., Gellman,S. H., Steyaert, J., Skiniotis, G., Weis, W. I., Sunahara, R. K. &Kobilka, B. K. (2011). Nature (London), 477, 549–555.
Ravelli, R. B. & Garman, E. F. (2006). Curr. Opin. Struct. Biol. 16,624–629.
Riekel, C., Burghammer, M. & Schertler, G. (2005). Curr. Opin.Struct. Biol. 15, 556–562.
Sacconi, L., Dombeck, D. A. & Webb, W. W. (2006). Proc. Natl Acad.Sci. USA, 103, 3124–3129.
Sixta, H., Harms, H., Dapia, S., Parajo, J., Puls, J., Saake, B., Fink, H. &Roder, T. (2004). Cellulose, 11, 73–83.
Song, J., Mathew, D., Jacob, S. A., Corbett, L., Moorhead, P. & Soltis,S. M. (2007). J. Synchrotron Rad. 14, 191–195.
Stepanov, S., Hilgart, M., Yoder, D. W., Makarov, O., Becker, M.,Sanishvili, R., Ogata, C. M., Venugopalan, N., Aragao, D., Caffrey,M., Smith, J. L. & Fischetti, R. F. (2011a). J. Appl. Cryst. 44, 772–778.
Stepanov, S., Makarov, O., Hilgart, M., Pothineni, S. B., Urakhchin,A., Devarapalli, S., Yoder, D., Becker, M., Ogata, C., Sanishvili, R.,Venugopalan, N., Smith, J. L. & Fischetti, R. F. (2011b). Acta Cryst.D67, 176–188.
Takai, M. & Colvin, J. R. (1978). J. Polym. Sci. 16, 1335–1342.Vernede, X., Lavault, B., Ohana, J., Nurizzo, D., Joly, J., Jacquamet, L.,Felisaz, F., Cipriani, F. & Bourgeois, D. (2006). Acta Cryst. D62,253–261.
Wu, H., Wacker, D., Mileni, M., Katritch, V., Han, G. W., Vardy, E.,Liu, W., Thompson, A. A., Huang, X. P., Carroll, F. I., Mascarella,S. W., Westkaemper, R. B., Mosier, P. D., Roth, B. L., Cherezov, V.& Stevens, R. C. (2012). Nature (London), 485, 327–332.
Zhang, Z., Sauter, N. K., van den Bedem, H., Snell, G. & Deacon,A. M. (2006). J. Appl. Cryst. 39, 112–119.
research papers
540 Jeremy T. Madden et al. � Integrated nonlinear optical imaging microscope J. Synchrotron Rad. (2013). 20, 531–540
142
Related Documents











