Stereocontrolled Synthesis and Rearrangement of Epoxides Hunsuk Chung A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy JULY 2007 1630440 Heilbron Laboratory Department of Chemistry Imperial College London London SW7 2AZ

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Stereocontrolled Synthesis and
Rearrangement of Epoxides
Hunsuk Chung
A thesis submitted in partial fulfilment of the requirements
for the degree of Doctor of Philosophy
JULY 2007
1630440
Heilbron Laboratory Department of Chemistry Imperial College London London SW7 2AZ

Copyright Notice
Imperial College of Science, Technology and Medicine Department Of Chemistry
Stereocontrolled Synthesis and Rearrangement of Epoxides
© 2007 Hunsuk Chung [email protected]
This publication may be distributed freely in its entirety and in its original form without the consent of the copyright owner.
Use of this material in any other published works must be appropriately referenced, and, if necessary, permission sought from the copyright owner.
Published by: Hunsuk Chung Chemistry Department Imperial College London South Kensington campus, London, SW7 2AZ, UK
29 August 2007
www.imperial.ac.uk

I conflrm that this report is my own work and where reference is made to otherresearch this is referenced in text.

Dedicated to my mum and dad for supporting me and encouraging me in fulfilling myacademic endeavour and ambitions.

Abstract
This thesis describes two related bodies of work, namely the epoxidation of
alkoxydihydropyrans and the synthesis of epoxidation catalysts. For clarity, separate
chapters comprising an introduction, results and discussion and conclusion and future work
are presented for each.
In Chapter 1, the diastereoselective formation of 4,5-c/s'-tetrahydrofuranones via the
oxidative rearrangement/Jones oxidation of 2-alkoxy-3,4-dihydro-2//-pyrans is described.
High levels of stereocontrol were observed with sterically demanding and branched
substituents. Furthermore, this oxidative rearrangement method was applied to the
synthesis of y-lactone-containing natural products such as a whisky lactone and a cognac
lactone. Additionally, in line with the rearrangement of the heterocycle series, we also
investigated an equally rapid route to carbocycles (cyclopentanes) by the cyclopropanation
of 2-alkoxy-3,4-dihydro-2//-pyrans using diazomalonate and a copper-based catalyst and a
mixture of insertion product and rearrangement product was obtained.
The enantioselective formation of tetrahydrofuranones from aryl-substituted
alkoxydihydropyrans using an asymmetric epoxidation catalyst (chiral manganese salen
catalyst) is presented in Chapter 2. Higher levels of stereocontrol were observed with more
sterically demanding and branched alkyl groups, or more electron-donating aryl groups.
In Chapter 3, we describe the synthesis of novel chiral ketone catalysts for the asymmetric
epoxidation of alkenes. An oxabicyclic ketone catalyst and an azabicyclic ketone catalyst
were synthesised and their reactivity and selectivity of epoxidation were investigated.
Finally, full experimental details and spectra are presented for all novel compounds
synthesised in the course of this work in Chapter 4.

Contents
Abstract........................................................................................... 5
Contents.......................................................................................... 6
Acknowledgements.............................................................................. 9
Abbreviations.................................................................................... 10
Chapter I.Diastereoselective oxidative rearrangement of 2-alkoxydihydropyrans................. 131. Introduction........................................................................... 141.1. Tetrahydrofiirans in natural products............................................... 141.2. Previous synthetic routes to tetrahydrofurans..................................... 171.2.1. Electrophilic cyclization strategies................................................. 171.2.1.1. Halocyclisation (E=halogen)......................................................... 171.2.1.2. Electrophilic cyclization via epoxyalcohol (E=O)................................ 201.2.1.3. Electrophilic cyclization via phenylsulfanyl migration (E=SPh)............... 251.2.1.4. Metal-catalysed cyclization (E=metal complex).................................. 261.2.2. Oxidation strategies................................................................... 311.2.2.1. Oxidation of 1,5-dienes............................................................... 311.2.2.2. Other oxidative strategies............................................................ 321.2.3. Reductive strategies.................................................................. 331.2.4. Strategies using allylic silanes and carbonyl compounds........................ 351.2.4.1. Cyclisation of allylic or propargylic silanes onto oxonium ion
intermediates.......................................................................... 351.2.4.2. Cyclisation onto silyl cations........................................................ 381.2.5. Radical reaction strategies........................................................... 401.2.5.1. C-C bond formation.................................................................. 401.2.5.2. C-O bond formation.................................................................. 441.2.6. C-C bond formation strategies...................................................... 491.2.6.1. Ene-type cyclisation.................................................................. 491.2.6.2. Other C-C bond formations.......................................................... 531.2.7. [3+2] cycloaddition strategy......................................................... 541.2.7.1. [3+2] cycloaddition using carbonyl ylides and alkenes.......................... 541.2.7.2. [3+2] cycloaddition using cyclopropanes and aldehydes........................ 561.2.7.3. Other [3+2] cycloaddition strategies................................................ 571.2.8. Miscellaneous strategies............................................................. 581.3. Background to the project........................................................... 622. Results and discussion............................................................... 662.1. Optimisation of the oxidative rearrangement...................................... 662.1.1. Preparation of dihydropyrans by hetero Diels-Alder reaction................... 66

2.1.2. Choice of oxidants for epoxidation................................................. 672.1.2.1. MCPBA................................................................................. 672.1.2.2. DMDO................................................................................. 732.1.2.3. DMDO generated in situ. ............................................................ 762.1.2.4. Methyl(trifluoromethyl)dioxirane generated in situ. ............................. 772.1.2.5. Hydrogenperoxide with MTO....................................................... 772.1.3. Product manipulation to simplify stereochemical analysis...................... 802.1.3.1. Acetal reduction....................................................................... 812.1.3.2. Acetal oxidation....................................................................... 822.2 . Application of oxidative rearrangement to dihydropyrans...................... 862.2.1. Preparation of dihydropyrans........................................................ 862.2.2. Diastereoselective formation of 2,3-ds-tetrahydrofurans........................ 912.2.3. Explanation of observed stereochemistry.......................................... 942.3. Synthesis of Quercus Lactone species............................................. 952.3.1. Proposed synthesis of Quercus lactones........................................... 952.3.2. Synthesis of tetrahydrofuranone precursors....................................... 972.3.3. Quercus lactones...................................................................... 992.4. Attempted Prevost toms-dihydroxylation.......................................... 1022.5. Preparation of cyclopentanes by cyclopropanation/rearrangement............ 1052.5.1. Previous work in this field........................................................... 1052.5.2. Attempted cyclopropanation with diazomalonate................................. 1073. Conclusions........................................................................... Ill
Chapter II. Enantioselective oxidative rearrangement of 2-alkoxydihydropyrans................ 1131. Introduction........................................................................... 1141.1. Asymmetric HDA reaction........................................................... 1151.1.1. Hetero Diels-Alder reaction under Cr3+ catalysis................................. 1151.1.2. Hetero Diels-Alder reaction of phenylsulfonyl enones under Ti4+
catalysis................................................................................. 1161.2. Asymmetric epoxidation methods.................................................. 1171.2.1. Fructose-derived chiral ketone catalyst by Shi.................................... 1171.2.2. Chiral manganese salen catalysts................................................... 1201.3. Concept of this project............................................................... 1222. Results and discussion.............................................................. 1232.1. Optimisation of enantioselective oxidative rearrangement...................... 1232.1.1. Enantioselective epoxidation by Shi's catalyst.................................... 1232.1.2. Enantioselective epoxidation by Jacobsen's catalyst............................. 1242.1. Enantioselective oxidative rearrangement by Jacobsen's catalyst............... 1262.2.1. Preparation of various phenyl enones.............................................. 1262.2.2. Preparation of various DHPs......................................................... 1302.2.3. Enantioselective formation of THFs................................................ 1322.2.4. Determination of product configuration .......................................... 1362.2.5. Attempted manipulation of DHPs by Baeyer-Villiger reaction................. 143

3. Conclusions and future work...................................................... 1483.1. Conclusions............................................................................ 1483.2. Future work............................................................................ 149
Chapter III.Synthesis of novel chiral ketone catalysts for enantioselective epoxidation ofalkenes........................................................................................................................... 1501. Introduction........................................................................... 1511.1. Previous chiral ketone catalyst in our group....................................... 1511.2. Concept of this project............................................................... 1552. Results and discussion.............................................................. 1562.1. Oxtf-bicyclic ketone catalyst......................................................... 1562.1.1. Synthesis of racemic oxa-bicyclic ketone.......................................... 1562.1.2. Epoxidation by racemic oxo-bicyclic ketone...................................... 1582.1.3. Synthesis of enantiomerically enriched oxcr-bicyclic ketone.................... 1602.1.4. Epoxidation by enantiomerically enriched oxcr-bicyclic ketone................ 1612.2. /Iztf-bicyclic ketone catalyst......................................................... 1642.2.1. Synthesis of racemic aza-bicyc\ic ketone catalyst................................ 1642.2.2. Epoxidation by racemic orza-bicyclic ketone catalyst............................ 1653. Conclusions and future work...................................................... 167
Chapter IV.Experimental procedures and data............................................................... 169General details.................................................................................... 170Nomenclature and numbering.................................................................. 1701. Compounds from Chapter 1......................................................... 1712. Compounds from Chapter 2......................................................... 2023. Compounds from Chapter 3......................................................... 224
Appendix.......................................................................................... 2361. NOESY spectra from Chapter 1..................................................... 2372. HPLC data from Chapter 2.......................................................... 2473. HPLC data from Chapter 3................................................................... 2564. NMR data for 0*tf-bicyclic catalyst from Chapter 3.............................. 260
References........................................................................................ 261
8

Acknowledgements
First of all, I would like to thank Professor Alan Armstrong for giving the opportunity and
for his encouragement, guidance and support over the past three years. I would also like to
acknowledge the financial support given by Dongbang FTL.
Further thanks go to all the members of the Armstrong group with whom I have shared a
lab and pub over the past three years, who have passed on their chemical expertise, and
made my three years in London so enjoyable. They are (in no particular order) Steve, Yunas,
James, Constantina, Chloe, Cas, Lee, Carl, Nigel, Tom, Nicky, Jamie, Richard, Lizzie,
James, Karen, Dave and Andrew. Specially, I would like to thank Yunas, Karen and James
for their kind proof-reading.
I would also like to acknowledge the Imperial College technical support.
Finally, I would like to thank my parents for their continued support throughout many years
as a student.

Abbreviations
'H Proton13C Carbon-13A Angstrom(s)Ac Acetylacac AcetylacetoneAIBN Azobisisobutyronitrileaq. AqueousAr Aryl substituentbpy 2,2'-Bipyridine'Bu wo-ButylnBu H-Butyl'Bu ter/-ButylBn BenzylBz BenzoylCat. CatalystCI Chemical ionisationcod 1,5-CyclooctadieneCone. ConcentratedCp CyclopentadieneCSA Camphorsulfonic acidcyHex Cyclohexyld Doubletdba DibenzylideneacetoneDBB di-tert-ButylbiphenylideDBMP 2,6-di-ter/-Butyl-4-methylpyridineDBU l,8-Diazabicyclo[5.4.0]undec-7-enedd Doublet of doubletsDET Diethyl tartrateDHP DihydropyranDFT Density functional theoryDMAP 4-(7V-Dimethylamino)pyridineDMDO DimethyldioxiraneDMF 7V,A/"-DimethylformamideDMSO Dimethyl sulfoxideDPE 1,2-Bis(diphenylphosphino)ethaneDPPBA 2-(Diphenylphosphino)benzaldehydedr Diastereoisomeric ratiodt Doublet of tripletsEDTA Ethylenediaminetetraacetateee Enantiomeric excessEPHP 1-Ethylpiperidine hydrophosphite
10

EqEtFODg hHDAHPLChuHzIDCPImIRJLDAmMMCBAMCPBAMemgMHzminmLmmolm.p.MSMTOm/zNBSNCSNHCNHPNISNMONMPNMRNOENOESYNuPCCpetrolPhPht
Equivalent(s)Ethyl6,6,7,7,8,8,8-Heptafluoro-2,2-dimethyl-3,5-octanedioneGram(s)Hour(s)Hetero Diels-AlderHigh-Performance Liquid ChromatographyIrradiation (photolysis)Hertzlodonium dicollidine perchlorateImidazoleInfraredCoupling constant (in Hz)Lithium di-z'so-propylamideMultipletMolarraeta-Chlorobenzoic acidmeto-Chloroperbenzoic acidMethylMilligram(s)MegahertzMinute(s)Millilitre(s)Millimole(s)Melting pointMolecular sievesMethyltrioxorheniumMass/charge ratio (in mass spectrometry)7V-BromosuccinimidejV-Chlorosuccinimide7V-Heterocyclic carbene7V-Hydroxyphthalimide7V-IodosuccinimideAf-Methylmorpholine-TV-OxidejV-Methyl pyrrolidinoneNuclear magnetic resonanceNuclear Overhauser effectCross-correlated NOE spectrumNucleophilePyridinium chloro chromatePetroleum ether (b.p. 40-60°C)PhenylPhthalimide
11

ppm PPTS iPr
PTABpy qrtssalensat.sexskewphostTBAFTBAITBDPSTBPAIBSTEATfTFATFAATHFTLCTMEDAIMSp-ToltriflateAf-TrisTsUHP
Part(s) per millionPyridinium /?-toluenesulfonateiso-Propy\Phenyltrimethylammonium tribromidePyridineQuartetRoom temperatureSingletAr,A^'-Bis(salicylidene)ethylenediamineSaturatedStrong cation exchange2,4-bis(diphenyl-phosphino)pentaneTripletTetra-w-butylammonium fluorideTetra-/7-butylammonium iodidetert-ButyldiphenylsilylTris(4-bromophenyl)aminiumter/-ButyldimethylsilylTriethylamineTrifluoromethanesulfonylTrifluoroacetic acidTrifluoroacetic anhydrideTetrahydrofiiranThin layer chromatographyN, N, jV',W-tetramethyl 1,2-ethanediamineTrimethylsilylp-ToluylTrifluoromethanesulfonate7V-2,4,6-Triisopropylbenzenesulfonylp-ToluenesulfonylUrea hydrogen peroxide
12

Chapter I. Diastereoselective oxidative rearrangement
of 2-alkoxydihydropyrans
13

1. Introduction1.1. Tetrahydrofurans in natural productsSubstituted tetrahydrofurans (THFs) are frequently encountered in various natural products 1
as well as being versatile synthetic intermediates. 2 Indeed, they feature predominantly in a
number of biologically significant molecules such as the polyether antibiotics, 13 lignans
and macrodiolides. 4
Polyether antibiotics, isolated from fermentation cultures ofStreptomyces, are distinguished
by a linear carbon framework containing THFs and tetrahydro-2//-pyrans, multiple centres
of asymmetry and a structure often terminating in a carboxylic acid. Representative
examples are shown in Fig. 1.
CO2 H Me Me Et
Isolasalocid
H Et H ~ Et
lonomycin
Fig. 1. Structure of representative polyether antibiotics.
In terms of biological activity, a carboxylate group and oxygen atoms of the antibiotics play
a role as ligands for the complexation of the most biologically significant polar cations such
as K+, Na+, Ca2+, Mg2+ and the biogenic amines. Such complexes are exceptionally
hydrophobic and the antibiotics can thus assist the transport of these cations across lipid
bilayers, inducing a range of biological responses that include ruminant growth promotion,
anticoccidial activity, and mammalian cardiovascular effects. Their features and synthesis
have been reviewed by Westley in 19825 and Huff in 2000. la
Lignans are a class of secondary plant metabolites produced by oxidative dimerization of
14

two phenylpropane units. Although their molecular backbone consists only of two
phenylpropane units, lignans show an enormous structural diversity. Some examples are
shown in Fig. 2. It is known that these compounds show potential anti-apoptotic activity6
or anti-cancer activity. 7 Such important bioactivities and the potential applications in
cancer chemotherapy have fuelled a growing interest in lignans and their synthetic
derivatives. 3
MeOOMe
erlangerins C
MeO OH
MeOAgastinol
OMe
OMe
OMe
phillygenol
Fig. 2. Structure of representative lignans.
Macrodiolides are an interesting class of natural products exhibiting many different
bioactivities. A wide range of total syntheses and biological activities of macrodiolides
were reviewed by Lee in 2005.4 Among the macrodiolides, this section focuses on only
amphidinolides bearing THFs because these are of particular interest to our group. These
amphidinolides were isolated from symbiotic marine dinoflagellates of the genus
Amphidinium sp. from inside cells of Okinawa marine flatworms. lb Since Yamasu and co-
workers found that Amphidinium show potent cytotoxic activity (70-90% inhibition at 3
yg-mL" 1 ) against murine lymphoma L1210 cells and human epidermoid carcinoma KB
cells in 1987,8 Amphidinium have been investigated worldwide; Kobayashi has provided a
comprehensive review of their isolation, properties and synthesis in 2004. lb Currently, our
group is interested in Amphidinolide C 1 and Tl 2 (Fig. 3). In terms of structure,
Amphidinolide C 1 is a 25-membered macrolide with two THF rings9 and amphidinolide
15

Tl 2 is a 19-membered macro lide with one THF ring. 10
OH '—'.. O''X
Amphidinolide C 1
OH
Amphidinolide T1 2
Fig. 3. Structure of representative amphidinolides.
16

1.2. Previous synthetic routes to tetrahydrofuransDue to the importance of the THF framework, enormous effort has been devoted towards
the development of methods for the synthesis of substituted THFs. There have been several
reviews to cover the previous methods reported up to 2001. 11 Thus this section will cover
key literature reports showing novel strategies or high levels of stereocontrol from 2002
onwards.
1.2.1. Electrophilic cyclization strategies.Electrophilic cyclization is one of the most common strategies to construct THF rings.
Generally, attack of an alcohol on the alkene moiety of unsaturated alcohols of general
structure 3 or 5 is promoted by an electrophile (E+) to form the THF 4 or 6 respectively
(Scheme 1). A wide range of electrophiles have been employed, including halogens, oxygen
(epoxide), sulfur or metals. This type of transformation has been widely used for the
synthesis of 2,5-disubstituted THFs. An important issue is often the control of exo- vesus
endo-ring closure.
HO, ^ ^ E* r°V_5-exo ^^ E
3 4
p+
5-endo
5
QE
6
Scheme 1. Formation of THF by electrophilic cyclization.
1.2.1.1. Halocyclisation (E=halogen)Since Harriett and co-workers reported the synthesis of 2,5-czs-disubstituted THFs by
halocyclization using a y,6-unsaturated alcohol and I2 in 1981, 12 it has been extensively
studied by several groups. For example, in 2002, Mootoo and co-workers showed the
formation of THF rings by iodocylisation using iodonium dicollidine perchlorate (IDCP) in
the synthesis of oligo-THFs (Scheme 2). 13 Here, it is noteworthy that the acetal moiety of
17

bis-aceta\ 7 is used as a nucleophilic oxygen source instead of the alcohol moiety and THF
8 is obtained as a single 2,5-trans isomer in 81% yield. The observed diastereoselectivity
could be explained by the cyclic transition state with minimised steric hindrance between
the iodoalkyl substituent of the eventual THF and the methyl group of the conformationally
restricted acetal. 14
Vb r
OH
Scheme 2. lodoetherification between acetal and alkene using IDCP; (a) IDCP, CH3 CN, 81%.
In 2005, Fujioka and co-workers reported a double iodoetherification of a symmetric a
diene acetal 9 (Scheme 3). 15 In terms of mechanism, hemiacetal intermediate 11 is obtained
via oxonium cation 10; a second intramolecular cyclization then occurs to give THF 12. In
this transformation, four stereogenic centres are defined in a single step and the major
isomer 12 is obtained with a diastereoisomeric ratio of 11 to 1 in 84% yield.
18

Ph
PhOJ?10 11
TBSO
12
Scheme 3. THF formation by double iodoetheriflcation of a symmetric diene acetal; (a) NIS, H2O, CH3 CN, dr 11:1 (major isomer to other isomers), 84%.
In spite of the widespread use of iodocyclisations, reagent-controlled enantioselective
versions of this strategy have rarely been investigated. In 2003, Kang and co-workers
reported enantioselective intramolecular iodocyclization of y-hydroxy-c/5-alkene 13 to form
2-substituted THFs 14 using chiral salen-Co complexes (Table 1). In this transformation,
they obtained THF in over 83% yield and with enantiomeric excesses ranging between 67%
and 90%. Higher enantiomeric excesses were observed for substrates with larger
substituents R.
Table 1. Enantioselective iodoetheriflcation using chiral salen-Co complexes; (a) I2 (1.5 eq), chiral salen-Co complex (0.3 eq), NCS (0.8 eq), CH2C12 .
13(R=alkyl)
RxO,
14
Q=K Co
BiM /h-a O-CTJ^'BU^Bu Bu 1
Chiral salen Co-complex
19

R
Me'Pr
"Pr
(CH2)3OTf
Yield (%)
96
83
85
89
ee (%)
67
73
85
90
Similarly, bromocyclisation strategies have been reported. For example, in 2002, Marko
and co-workers showed the synthesis of THF 16 by bromocyclisation of p,y-unsarurated
alcohol 15 (Scheme 4). 16 The key difference here is that the THF ring is formed by 5-endo-
cyclisation whereas all the others have been 5-exo-cyclisations because the silyloxy group
assists halocyclization by electron donation towards the bromonium ion. They obtained 16
as a single isomer in over 95% yield. The stereoselectivity is explained by stereospecific
anti-addition to the intermediate bromonium ion with the R substituent occupying a
pseudoequatorial position.
OHOTBS
TMS
15(R=alkyl)
a——————— ̂ - Rl
TMS
— £>H C .OTBS
\7BC;+
R/ '^°\
i?s.16
OTBS
TMS
Scheme 4. THF formation by bromocyclisation; (a) NBS, THF, 95%.
1.2.1.2. Electrophilic cyclization via epoxyalcohol (E=O).
Cyclisation of epoxyalcohols is another well established method for THF synthesis.
Traditionally, the epoxyalcohols were prepared by epoxidation of the appropriate
unsaturated alcohol. However, some recent examples demonstrate alternative methods for
epoxyalcohol generation. For example, as part of a total synthesis of ionomycin, Marshall
and co-workers showed the synthesis of 6/s-THFs via zinc-initiated triepoxide cascade
cyclization in the presence of tetrabutylammonium iodide (TBAI) (Scheme 5). 17 It is
20

notable that this polyepoxide cascade strategy is quite similar to the proposed biosynthesis
of THF-containing polyether antibiotics. 18 In terms of mechanism, the treatment of
terminal epoxy bromide 17 with zinc forms a transient allylic alkoxy zinc species 18. Two
sequential 5-exo-cyclisations then give bis-THF 19 in 62% yield.
OTBS
O
OTBS
17 19
OTBS
OBrZn OTBS
Scheme 5. Zinc-initiated triepoxide cascade cyclization; (a) Zn, TBAI, MeOH, 62%.
Furthermore, Forsyth and co-workers reported the synthesis of THFs via a one-pot polyol
cyclization cascade (Scheme 6). 19 Mechanistically, the terminal alcohol in 20 is selectively
activated by treatment with Af-2,4,6-triisopropylbenzenesulfonyl imidazole (7V-TrisIm).
Subsequently, the intramolecular nucleophilic substitution forms epoxide 21 and a second
intramolecular nucleophilic substitution gives the THF product 22 in 62% yield.
21

BnO
BnO
OH OH
20
OH OHOTris
BnO
BnO
Scheme 6. Synthesis of THFs via one-pot polyol cyclization cascade; (a) KOBu, BuOH, N-TrisIm, 62%.
As an alternative electrophile for cyclization, Borhan and co-workers in 2005 reported the
cyclization of 1,4,5-triols via an acetoxonium intermediate (Table 2). 20 The
enantiomerically enriched 1,4,5-triol is obtained by Sharpless asymmetric dihydroxylation.
The orthoester 24 is then obtained by reaction with trimethyl orthoacetate 23. Subsequent
ionisation of the orthoester 24 in the presence of Lewis acid gives a reactive acetoxonium
species 25 and THF 26 is then formed in up to 98% yield by intramolecular cyclization.
Table 2. Cyclization of 1,4,5-triol; (a) PPTS (cat), (b) BF3 -OEt2 .
OH
OMeOMe
OMe
23
OMe
24
AcO
R'
26
22

R Yield (%
"PentnBu
EtO2C
cyHex
81
82
62
50
Apart from the cyclization of y,5-epoxyalcohols, Karikomi and co-workers reported the
synthesis of THFs 28 by the cyclization of p,y-epoxyalcohols 27 using magnesium halide
(Scheme 7). 21 In contrast to the favoured formation of oxetane 29 by base-catalysed 4-exo
cyclization or the formation of 2,3-trans-THF 28b by acid catalysed 5-endo ring-closure,
this transformation by magnesium halide gives 2,3-c/s-THF 28a via two inversions; i) first
inversion in epoxide opening by halogen, ii) second inversion by intramolecular halogen
displacement by the alcohol. Here, 2,3-czs-THFs 28a were obtained as the major product
with 85:15 dr and up to 84% yield.
27inversion
inversion
HO
+Mg'O
29
H0v
inversion A/28a, 84% (dr 85:15)
.b28b
Scheme 7. Cyclization of 3,4-epoxyalcohols using magnesium halide; a) MgI2 , b) acid or base.
Bohran and co-workers reported an interesting new method for the formation of THFs via
cyclisation of epoxyalcohols 30a and 30b using a sulfoxonium ylide (Scheme 8). 22 It is
noteworthy that in this strategy the formation of THF is not obtained from direct
23

electrophilic cyclization of the starting 2,3-epoxyalcohol but involves Payne rearrangement
and nucleophilic substitution of the resulting isomeric epoxide with sulfoxonium ylide. In
this transformation, they obtained enantiomerically pure 2,3-cw-epoxyalcohol 30a and 2,3-
/nms-epoxyalcohol 30b from the Sharpless asymmetric epoxidation of allylic alcohols. The
reaction of these epoxyalcohols with trimethylsulfoxonium iodide affords the
corresponding 2,3-disubstituted THF in good yield with complete control of
stereochemistry. Thus, c/s-epoxide 30a gives cw-THF 31 a and /raws-epoxide 30b gives
trans-THF 31b. The selected results are shown in Scheme 8.
O OH0 rv-* ~~ H0\_/\ -
30a 31 a 30b 31 b
86% 91% 55% 38%
Scheme 8. Formation of THF via nucleophilic substitution of 2,3-epoxyalcohol using a sulfoxonium ylide; (a) NaH, DMSO, 80 °C.
Mechanistically, the reaction is postulated to proceed by an initial Payne rearrangement of
epoxyalcohol 30b to give the more sterically accessible terminal epoxide 32. This is then
attacked by sulfoxonium ylide to form fos-alkoxide 33 which subsequently cyclises to THF
31b by intramolecular nucleophilic substitution (Scheme 9).
24

O=SiCH2
base ' / 0
Payne (QV rearrangement 32
HQ
,bO
33 31 b
Scheme 9. Proposed mechanism of cyclization of 2,3-epoxyalcohol using a sulfoxonium ylide.
1.2.1.3. Electrophilic cyclization via phenylsulfanyl migration (E=SPh).
Electrophilic sulphur or selenium reagents can serve to activate alkenes for attack by
alcohols in an analogous manner to electrophilic halide. An alternative method for
generation of episulfonium ion intermediates starts from (3-hydroxysulfides. For example,
in 2002, Warren and co-workers reported the synthesis of enantiomerically pure THFs from
enantiomerically pure 2,4,5-triols 34a and 34b (Scheme 10). 23 They obtained 2,4-c/s-THF
36a and 2,4-trans-THF 36b as the major product, respectively, by 1,2-phenylsulfanyl
migration via the sulfonium ion intermediates 35a and 35b; stereoselectivity is decided by
the stereochemistry of starting triols 34a and 34b. The formation of tetrahydropyran
products via the attack of the primary alcohol on the sulfonium ion intermediate is
competitive to THF formation in this transformation and a mixture of THF and
tetrahydropyran is obtained when short reaction times (10 min) are used. However, only
THF is obtained (over 83% yield) as the thermodynamic product with longer reaction times
(48 h). The favoured formation of THF over that of the tetrahydropyran could be explained
by the Thorpe-Ingold effect.
25

SPh
OH OH 34a
SPh
OHOH OH
34b
,SPh
,SPh
PhS
36a
PhS
36bOH
Scheme 10. Synthesis of 2,4-substituted THF by phenylsulfonyl migration; (a) TsOH,CH2Cl 2 ,40 °C, 48 h.
1.2.1.4. Metal-catalysed cyclization (E=metal complex)Metals have been widely used for electrophilic cyclization. In this type of transformation,
the THF framework is constructed by the electrophilic attack of a hydroxyl group on a
metal-complexed olefln and the entire reaction is generally terminated by the protonolysis
of the metal-carbon bond. For example, in 2004, Widenhoefer and co-workers reported
platinum-catalysed intramolecular hydroalkoxylation of y-hydroxy alkene 37 to
regioselectively form THF 38 in up to 98% yield (Scheme II). 24 The reaction tolerates
substitution at the a, p and y-carbon atoms. Also, the reaction tolerates substitution at the
internal and terminal olefm carbons. When alcohol 37 was exposed to the reaction
condition, 2,3-trans THF 38 was obtained with the best dr of 8:1.
-Q
Ph
38
Scheme 11. Platinum-catalysed intramolecular hydroalkoxylation; (a) [PtCl 2(H :C=CH2 )]2 (2.5 mol°o), P(4- C6 H4CF3 )3 (5 mol%), C1 2 CHCHC1 2 , 70 °C.
26

The proposed mechanism is shown in Scheme 12. Initially, platinum activates the olefm 37
and the hydroxyl group attacks the platinum-complexed olefm 39 to form zwitterion 40.
Then, the corresponding THF 38 is obtained by proton transfer involving protonolysis of
Pt-C bond.
PtCI2 L
Ph 38
PtCI2L
Ph 39
Ph
H+
RCI2 L
Ph 40
Scheme 12. Proposed mechanism of platinum-catalysed intramolecular hydroalkoxylation.
Also, Kozmin and co-workers reported gold-catalysed double cyclization of 1,5-enynes 41
with a nucleophile alcohol substituent (Scheme 13). 25 They obtained bicyclic product 43 in
over 86% yield with high diastereoselectivity (dr >97:3). The cisltrans relationship between
C-2 and C-3 stems from the EIZ geometry of the initial alkene.
27

o
HPh
AuCU
42
protodemetallation
AuCU
AuCU
Scheme 13. Gold-catalysed double cyclization; (a) AuCl 3 (10 mol%), MeCN, 20 °C.
In terms of mechanism, cyclopropenyl gold intermediate 42 is opened by intramolecular
attack of oxygen nucleophile initiating a double cyclization; subsequent protodemetallation
gives the bicyclic product 43. In 2005, Dufiach and co-workers reported this type of
transformation using Sn(OTf)4 as a metal source and 2-monosubstituted THFs were
regioselectively obtained in up to 98% yield. 26
In the preparation of optically pure THFs, Kang and co-workers reported enantioselective
mercuriocyclisation of y-hydroxy-czs-alkenes using dimethylmalonate-derived
bisoxazoline-Hg(II) complexes.27 They obtained 2-monosubstituted THFs in up to 95% ee
and over 68% yield. In 2005, Gracza and co-workers reported the preparation of_ _ ** o _
enantiomerically pure THFs by a novel "pseudo-weso-trick" (Scheme 14). This Pd-
catalysed bicyclization of diastereoisomeric dihydroxyl alkene 44 furnished the €2
symmetrical product 45. In this transformation, a newly-formed C-O bond is
stereospecifically generated with a cis relationship to hydroxyl group at C-4. It is
remarkable that the enantiomerically pure bicyclic compound 45 is formed from
diastereoisomeric mixture 44 (dr 1:1) by the degeneration of the allylic stereogenic centre
because of the C2 symmetry of 45.
28

C2
BnCX 3 BnO
2 ,PdLnO'1 ""OH
45
Scheme 14. Preparation of THF by pseudo-meso-trick; (a) PdCl2 , CuCl : , AcONa, AcOH, 79%.
Wolfe and co-workers reported the Pd-catalysed reaction of aryl bromides with y-hydroxy
alkenes to prepare substituted tetrahydrofurans and a representative example is shown in''JQ ___ ___ ___
Scheme 15. The 2,5-trans-THY 47 was obtained from 46 as the major product with the
ratio over 20:1 and in over 60% yield.
.CX^Ph
46 47
Scheme 15. Pd-catalysed reaction of aryl bromide with y-hydroxy alkenes to prepare substituted tetrahydrofurans; (a) ArBr (2.0 eq), NaO'Bu (2.0 eq), Pd2(dba)3 (1 mol%), DPE-Phos (2 mol%), THF (0.25 M), 65 °C.
Two pathways have been proposed to explain the observed stereochemistry (Scheme 16).
Firstly, Pd(Ar)(OR) intermediate 48 is formed in path A and it could undergo insertion of
the olefin into the Pd-O bond. Then, a C-C bond is formed by reductive elimination to give
the product 47. Alternatively, Pd(Ar)(OR) intermediate 49 is formed in path B and it could
undergo insertion of the olefin into the Pd-C bond. Finally, the C-O bond is formed by
reductive elimination. The observed diastereoselectivity could be explained by the cyclic
transition state, where all substituents occupy pseudoequatorial positions.
29

PathB
R1
Ar
H HR2
47
Scheme 16. Pathways for Pd-catalysed reaction of aryl bromide with y-hydroxy alkenes.
In a similar manner, in 2002, Burke showed the construction of THF units by Pd-catalyzed
diastereoselective desymmetrisation of €2 diol acetate 50 in the total synthesis of
halichondrin B (Scheme 17). 30 Here, 2,4,5-trisubstituted THF 52 is obtained via palladium
7i-allyl complex 51 in 87% yield and the chiral ligand, DPPBA, controlled the
stereoselectivity of desymmetrisation.
AcO OAc
50
PdL
AcO51
OAc
52
Scheme 17. Pd-catalyzed desymmetrisation of C? diol acetate; (a) Pd2 (dba)3 , (/?,/?)-DPPBA, 0 °C, 87%.

1.2.2. Oxidation strategies. 1.2.2.1. Oxidation of 1,5-dienes.
Since Klein and Rojahn reported the oxidative cyclization of 1,5-dienes by KMnC>4 in
1965, the cyclization using metal oxides has been the most common oxidation strategy to
synthesise THFs. The general transformation from 1,5-dienes 53 to THF 54 is shown in
Scheme 18. This transformation usually shows high stereoselectivity although yields are
often low. However, it is noteworthy that this kind of cyclisation has been postulated as a
possible biosynthetic alternative to the polyepoxide cyclisation hypothesis. 18
Oxidation
HO53
O
54OH
Scheme 18. Oxidation of 1,5-diene.
In a recent example, in 2003, Donohoe and co-workers reported the cyclization of 1,5-T 'J
dienes by dihydroxylation using catalytic osmium tetroxide instead of KMnO4 (Table 3).
The initial dihydroxylation of the diene 55 furnishes an osmate ester 56 which then
undergoes cyclisation to give a single diastereoisomer, 2,5-c/si-THF 57, in 60-95% yield.
Also, for the example of polyTHF, in 2005, Piccialli and co-workers reported the oxidative
cyclization of polyalkene to pentafuranyl compound using RuO4 . 33
31
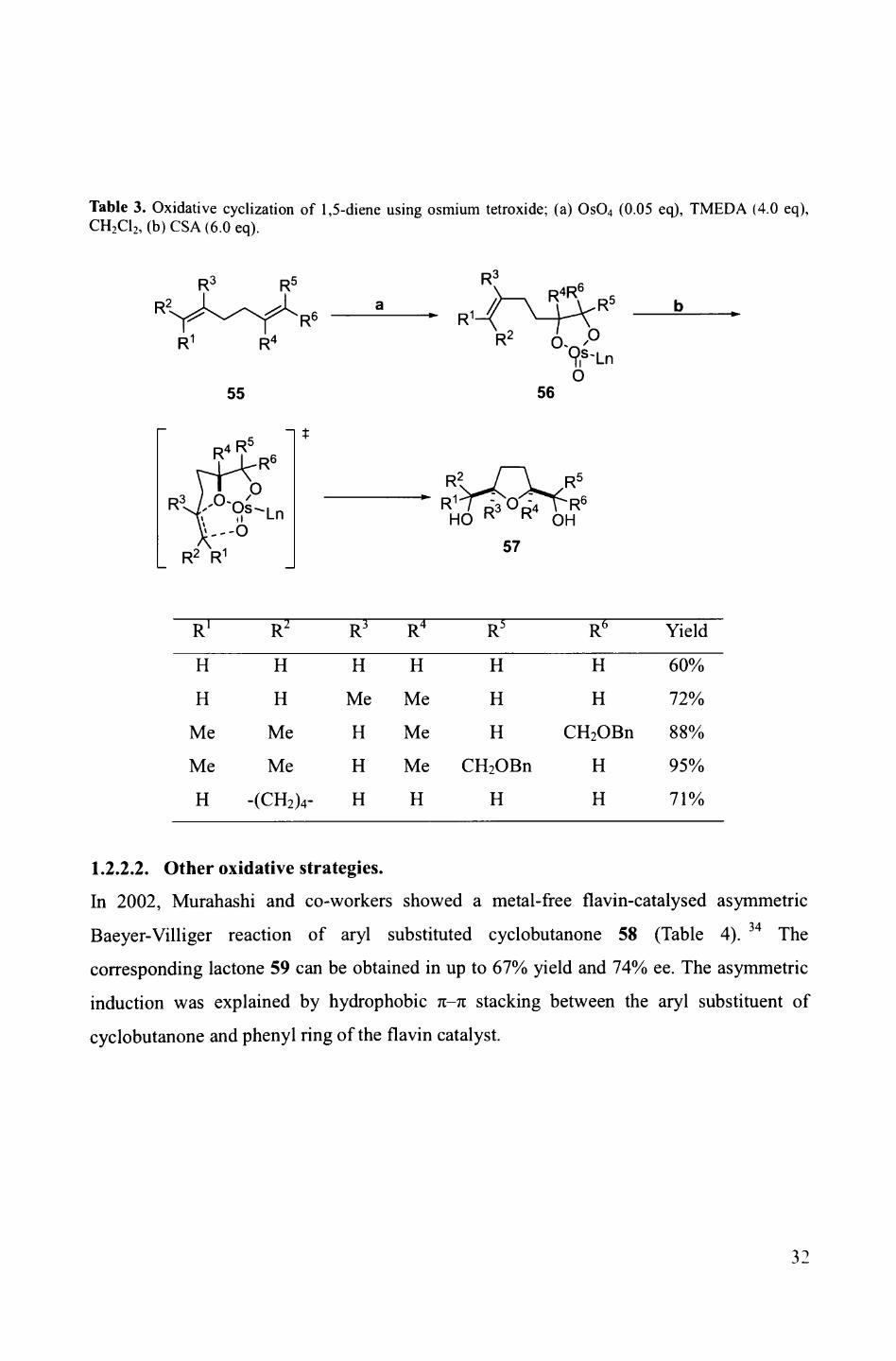
Table 3. Oxidative cyclization of 1,5-diene using osmium tetroxide; (a) OsO4 (0.05 eq), TMEDA (4.0 eq), CH2 C12 , (b) CSA (6.0 eq).
R5R2,
R6 R
R;
JR4R R£
R R
O55 56
vtR5LRQ
R3J''aOs^ Ln\\ 'A""0
R2 R 1
R 1
H
H
Me
Me
H
-
p2 T>3
H H
H Me
Me H
Me H
-(CH2)4- H
R4
H
Me
Me
Me
H
R2 / \ 1>^ >^
HO R3 R4
57
R3
H
H
H
CH2OBn
H
f.R6.
OH
R6
H
H
CH2OBn
H
H
Yield
60%
72%
88%
95%
71%
1.2.2.2. Other oxidative strategies.In 2002, Murahashi and co-workers showed a metal-free flavin-catalysed asymmetric
Baeyer-Villiger reaction of aryl substituted cyclobutanone 58 (Table 4). 34 The
corresponding lactone 59 can be obtained in up to 67% yield and 74% ee. The asymmetric
induction was explained by hydrophobic n-n stacking between the aryl substituent of
cyclobutanone and phenyl ring of the flavin catalyst.
32

Table 4. Bayer-Villiger reaction of cyclobutanone; (a) H2 O2 (1.5 eq), (S,S,pR,pR)-f\avin cat. (0.1 eq), AcONa (cat), CF 3CH2OH/MeOH/H2O.
58
o-Y,*
59
Ar
4-MeO-phenyl
4-MeO-phenyl
Ph
4-Br-phenyl
4-Cl-phenyl
00^k
B*'
:> . _ . 0
V Q ^r^N^ri HxN xkr^o' 1 1 +
-s>^ -^^
(S,S,pR,pR)-f\av'm catalyst
Yield (%)
67
53
67
28
34
ee (%)
61(+)
62(+)
63(5)
68(+)
66(5)
1.2.3. Reductive strategies.
Several strategies for THF synthesis have been reported which entail reductive cyclisation
of ketoalcohols 60 to THFs 61 (Scheme 19).
60R
61
Scheme 19. Synthesis of THF by reductive method.
Carreno and co-workers reported reductive cyclization of enantiomerically pure
hydroxylsulfinyl ketone 62 to give THF 63 (Scheme 20). 35 Here, they obtained 2,5-cw-
THF 63 as a major product with 6:1 dr in 71% yield. Activation of the carbonyl group of
the hydroxysulfmyl ketone 62 by TMSOTf favours the intramolecular nucleophilic addition
33

of the OH to give an intermediate mixed acetal precursor of the carboxonium intermediate
64. The axial approach of EtsSiH to 64, affording the czs-diastereomer, is favoured because
of the lower energy of the resulting cyclic transition state with all substituents in
pseudoequatorial positions
62
'"\SOp-Tol
Ph°cv» < ^-/"*
SOp-Tol
63(71%, dr 6:1)
Et3SiH (axial approach)
SOp-Tol
Ph0+
64
Scheme 20. Synthesis of THF by nucleophilic attack of alcohol moiety to ketone; (a) Et3 SiH, TMSOTf, CH2 C1 2,0 °C.
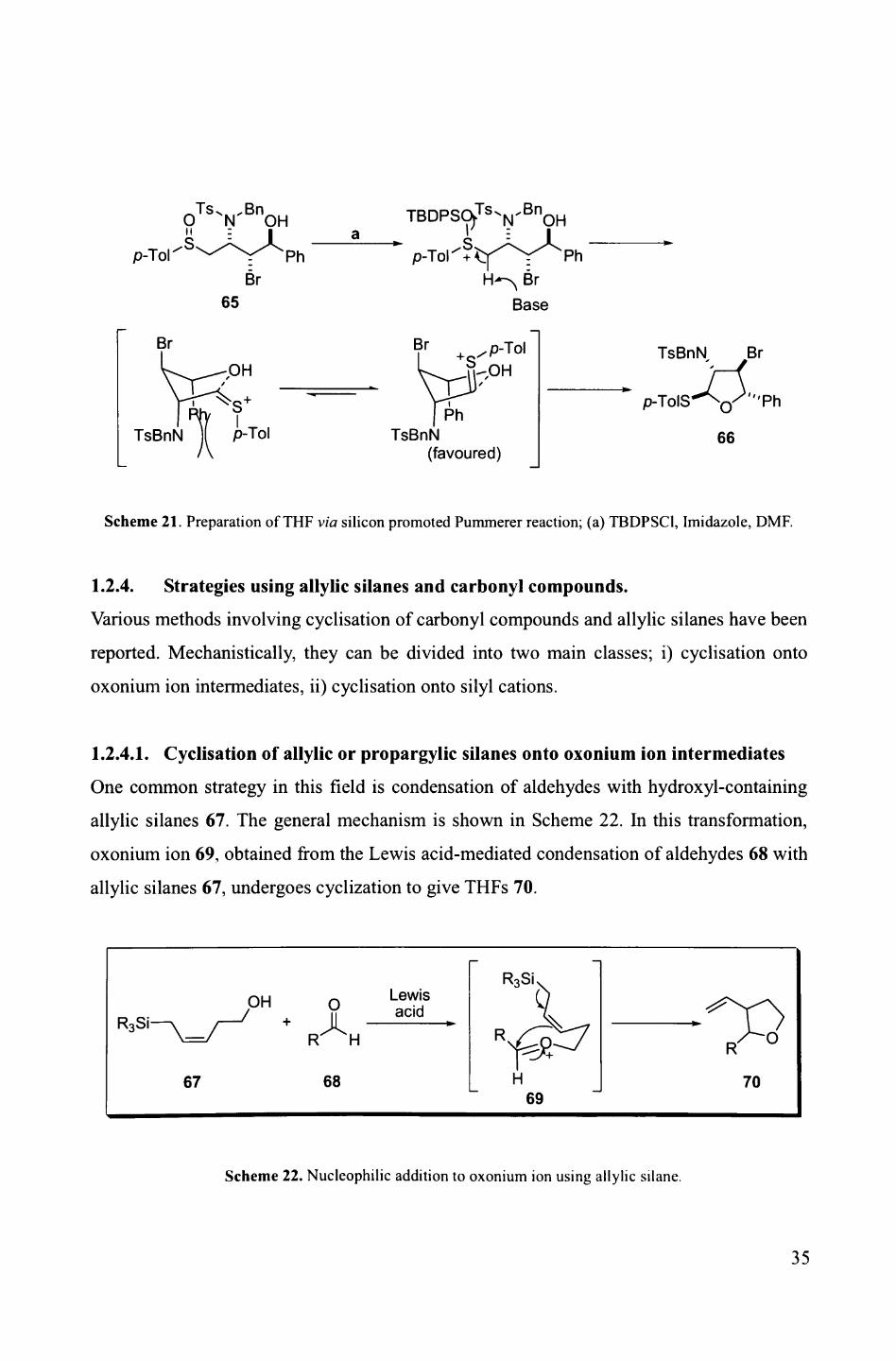
Also, in 2003, Raghavan and co-workers reported the use of sulfoxides instead of ketones
for this type of transformation. They prepared highly substituted THF 66 via silicon-
promoted Pummerer type reaction, proceeding in over 82% yield (Scheme 21). 36 In terms
of mechanism, the sulfoxide oxygen of 65 coordinates to silicon to give the sulfonium salt,
which yields THF 66 by the intramolecular attack of hydroxyl group. The observed
stereoselectivity is explained by the transition state with minimised steric interactions
between sulfonium moiety and NBnTs group.
34
Br65
TsBnN p-Tol
Base
Br
Ph TsBnN
(favoured)
TsBnN Br
^/'"
66
Scheme 21. Preparation of THF via silicon promoted Pummerer reaction; (a) TBDPSC1, Imidazole, DMF.
1.2.4. Strategies using allylic silanes and carbonyl compounds.
Various methods involving cyclisation of carbonyl compounds and allylic silanes have been
reported. Mechanistically, they can be divided into two main classes; i) cyclisation onto
oxonium ion intermediates, ii) cyclisation onto silyl cations.
1.2.4.1. Cyclisation of allylic or propargylic silanes onto oxonium ion intermediates
One common strategy in this field is condensation of aldehydes with hydroxyl-containing
allylic silanes 67. The general mechanism is shown in Scheme 22. In this transformation,
oxonium ion 69, obtained from the Lewis acid-mediated condensation of aldehydes 68 with
allylic silanes 67, undergoes cyclization to give THFs 70.
67
oR^H
68
RS
70
Scheme 22. Nucleophilic addition to oxonium ion using allylic silane.
35

For example, in 2004, Marsden and co-workers reported the construction of 2,3,4-
trisubstituted THFs via a Lewis acid-mediated condensation of aldehydes 72 with
allylsiloxanes 71 as a synthetic approach to lignans (Scheme 23). 37 Here, they obtained
2,3-c/s1 , 3,4-trans-isomGr 74a as a major product regardless of aldehyde substituent in the
presence of BF3-OEt2. In terms of mechanism, (£)-oxonium ion 73 obtained from the
condensation of aldehydes with allylsiloxanes undergoes cyclisation through chair-like
transition state with all substituents in an equatorial orientation.
72 (R=alkyl, aryl)
FMeoSi
73
-Ar
74a
Scheme 23. 2,3,4-Trisubstituted THF via a Lewis acid-mediated condensation of aldehydes with allylsiloxanes; (a) BF3 -OEt2, CH2 C12 , -78 °C, 8 h, then rt.
However, they obtained 2,3-trans-, 3,4-trans-isomer 74b via reversible ring-opening, when
the aldehyde contained electron-rich aryl substituents to stabilise the intermediate benzylic
cation 75 (Scheme 24).
-Ar
74a
-Ar-Ar
75 74b
Scheme 24. Formation of thermodynamic product via reversible ring-opening.
Furthermore, they showed the application of this method to the synthesis of aryltetralins38a
and virgatusin. 38b Also, in 2005, Garcia and co-workers showed the application of this
36
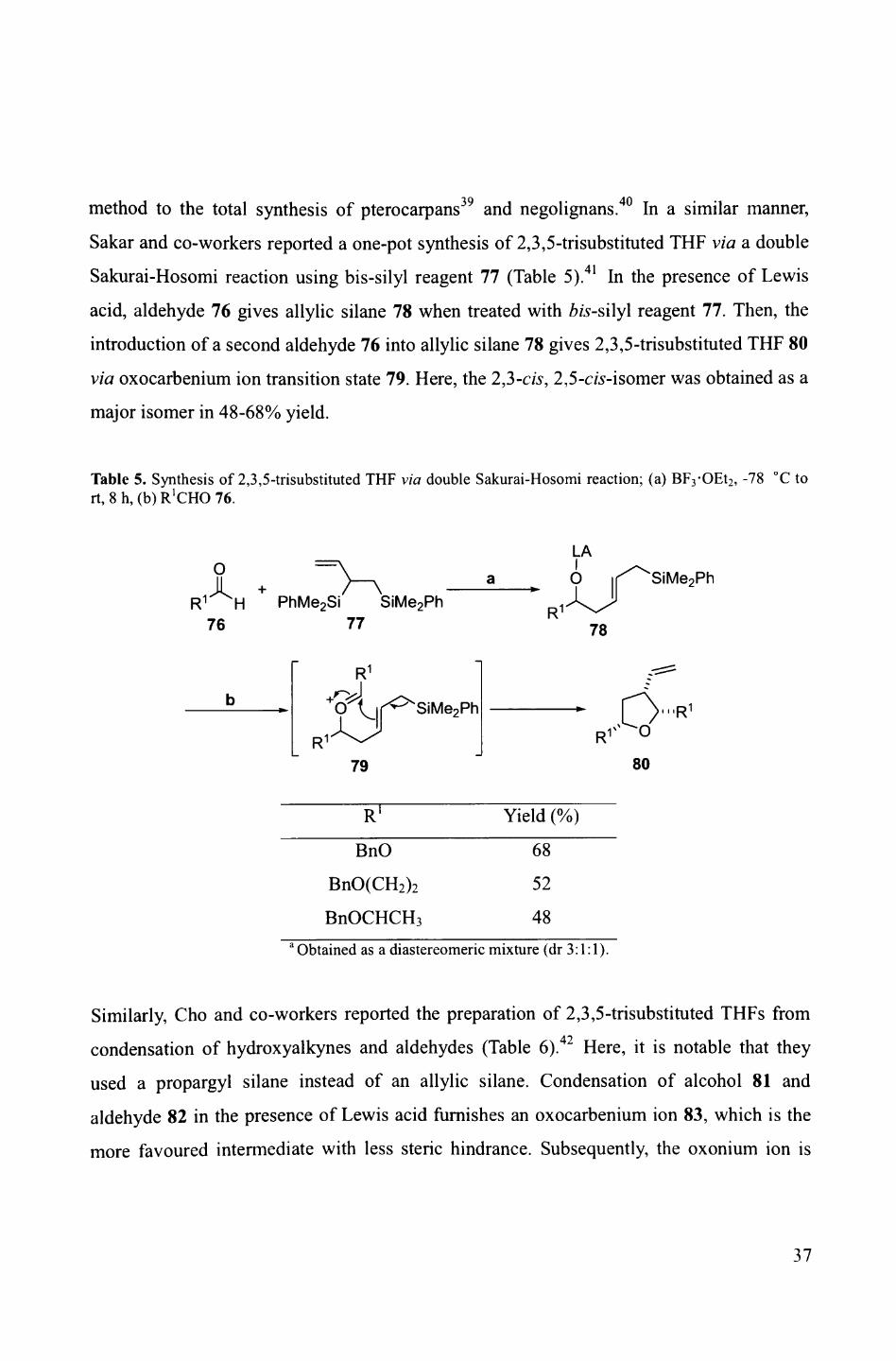
method to the total synthesis of pterocarpans39 and negolignans.40 In a similar manner,
Sakar and co-workers reported a one-pot synthesis of 2,3,5-trisubstituted THF via a double
Sakurai-Hosomi reaction using bis-silyl reagent 77 (Table 5). 41 In the presence of Lewis
acid, aldehyde 76 gives allylic silane 78 when treated with bis-silyl reagent 77. Then, the
introduction of a second aldehyde 76 into allylic silane 78 gives 2,3,5-trisubstituted THF 80
via oxocarbenium ion transition state 79. Here, the 2,3-cis, 2,5-cw-isomer was obtained as a
major isomer in 48-68% yield.
Table 5. Synthesis of 2,3,5-trisubstituted THF via double Sakurai-Hosomi reaction; (a) BF3 -OEt2 , -78 °C to rt, 8 h, (b) R'CHO 76.
oAu*76
PhMe2Si SiMe2 Ph 77
R1
SiMeoPh
,,R1
79 80
R Yield %
BnO
BnO(CH2)2
BnOCHCH3
68
52
48
a Obtained as a diastereomeric mixture (dr 3:1:1).
Similarly, Cho and co-workers reported the preparation of 2,3,5-trisubstituted THFs from
condensation of hydroxyalkynes and aldehydes (Table 6). 42 Here, it is notable that they
used a propargyl silane instead of an allylic silane. Condensation of alcohol 81 and
aldehyde 82 in the presence of Lewis acid furnishes an oxocarbenium ion 83, which is the
more favoured intermediate with less steric hindrance. Subsequently, the oxonium ion is
37

transformed to 2,3,5-trisubstituted THF 84 via a Prins-type cyclization. Here, the resulting
THFs are obtained in 63% to 93% yield and 2,5-c/s-selectivity is observed between Ph and
R (over 40:1 dr).
Table 6. Synthesis of 2,3,5-trisubstituted THF via Prins-type cyclization; (a) RCHO 82, TMSOTf, Et2 O.
TMS
HO
Ph81
I
f^-O*a v J ""^ /
\ / / LJ
WMS
R
Ph
cyHex
CH3 (CH2)4
CH3 COO(CH2)3
-*
Yield
91%
86%
71%
82%
rVV .j """^ /\ ^/ R to%^i^TMS
83
cisltrans
40:1
c/j only
cis only
c/'s only
Ph^/°\>R-qj
84
1.2.4.2. Cyclisation onto silyl cations.
Another common strategy in this field is Lewis acid-mediated annulation using allylic
silanes 86 and carbonyl compounds 85. This strategy uses the Lewis acid-complexed
alkoxide as a nucleophile to attack the intermediate silyl cation, resulting in a 1,2-shift of
the silyl moiety to give THF 87. The general mechanism is shown in Scheme 25.
oO-^^N. 0
R3 R2
85
(R 1 =alkyl,
^rv^/SiR
86
1k 3 -
PLewisacid
F+ i^XXXv
^1 3^' \__r
-i R 1 oSis2X R3
\A
\ _» / V/
\ /\
87
R2 ,R3 =alkyl, alkoxy)
Scheme 25. Nucleophilic substitution of Lewis acid-compexed alkoxide to silyl cation.
38

For example, Woerpel and co-workers used the SnCU-mediated cyclisation between
functionalised allylsilane 89 and a-ketoester 88 to prepare multisubstiruted THF 90 as a
single diastereoisomer in 85% yield (Scheme 26). 43
O SiMe,Ph PhMe2SL / OAc
C02 Et 88 89 90
Scheme 26. SnCl4 mediated [3+2] cyclisation between allylsilane and a-ketoester; (a) SnCl 4 , 85%.
As an alternative to the use of a Lewis acid-complexed alkoxide as oxygen nucleophile,
Angle and co-workers reported the reaction between aldehyde and allylsilane to prepare
3,5-cw-disubstituted THFs (12:1 cis to trans) in 83% yield (Scheme 27).44 Allylsilane 92
adds to the Lewis acid-activated aldehyde 91 to form silyl cation 93. Subsequently, the
more nucleophilic triethylsilyl ether participates in the cyclization to give THF 94. Here,
the major isomer shows cw-selectivity between the hydroxyl group at C-3 and the silyl
methylene substituent at C-5. Furthermore, they prepared various di-, tri- and tetra-
substituted THFs using a-substituted aldehydes and crotylsilanes. The observed
stereochemistry could be explained by facial selectivity (Felkin-Anh) of the addition of the
allylsilane to aldehyde.
39

Et3SiO/XCHO +
91
Et3SiO
H
92
H
/"""OHO'"( I \---V -SIR,
3,5-c/s-94 (Major, dr 12:1)
less nucleophilic oxygen LA
H
more nucleophilic , oxygen
H
93
Scheme 27. Reaction between aldehyde and allylsilane to prepare 3,5-c/s disubstituted THF; (a) BF3 -OEt2 , DBMP, CH2C12 ,-78 °C.
1.2.5. Radical reaction strategies.Various methods involving radical ring-closing reactions have been reported. There are two
main strategies for the synthesis of THFs; i) via C-C bond formation, ii) via C-O bond
formation.
1.2.5.1. C-C bond formationNumerous methods for the synthesis of THFs involving radical mediated C-C bond
formation have been reviewed.45 However, there appear to be two main strategies. Firstly,
the haloalkyl allylic ether 95 is used as a radical precursor and it cyclises via the formation
of C3-C4 bond in THF 96 by the treatment with a radical initiator (Scheme 28).
\fR O
95 96
Scheme 28. C-C bond formation by radical precursor with double bond.
40

In 2002, Roy and co-workers showed this type of C3-C4 bond formation via the C-O bond
cleavage of an epoxides in the synthesis of furano lignans (Scheme 29).46 The C-O bond of
the epoxide in radical precursor 97 is cleaved using Cp2TiCl to generate a p-alkoxy-radical.
The cyclisation product 99 was obtained as a mixture of diastereoisomers with ratio of 5:1
(3,4-cis to 3,4-trans) in up to 95% yield. They explained the observed stereoselectiviry by
suggesting that the chair-like transition state 98 is favoured in which Ar2 and the alkene
adopt pseudoequatorial positions.
OAr2
Ar1 O"
97
Ti lvO'\ H
98
OH Ar1
Ar' O 99
Scheme 29. C-C bond formation via epoxide cleavage; (a) i.Cp2TiCl, THF, rt, ii. H3 O+ .
In 2003, Oshima and co-workers reported this type of chemistry using gallium and indium
hydrides as radical initiator instead of toxic tin compounds.47 In 2004, Sibi and co-workers
reported the preparation of substituted THFs using a tandem radical addition-cyclisation
reaction (Scheme 30). 48 They used a doubly activated malonate-type p-oxygenated
acceptor 100 which underwent addition by a nucleophilic alkyl radical generated in situ
from a haloalkane. The intermediate malonyl radical 101 could then cyclise onto the
unactivated alkene. The product 102 was a diastereoisomeric mixture with 2,4-trans-
selectivity (up to 50: 1) and up to 79% yield.
E^ ^E
SO
R2 a
R3X^0X
R1 "
r^R2
T
2100 (E=ester, R\ R^= H, Me) 101,3^0
102
Scheme 30. C-C bond formation via radical addition-cyclization; (a) R3 X (alkyl halide), Bu3 SnH, Yb(OTf).i, Et3 B/02 ,-78 °C.
41

Similarly, in 2002, Kamimura and co-workers reported the preparation of trisubstituted
THFs via the C2-C3 bond formation from optically active radical precursor 103 (Scheme
31). In the presence of BusSnH and AIBN, the radical precursor smoothly gives a single
isomer 105 up to 86% yield via radical transition state 104. They again explained the high
stereoselectivity as arising from pseudoequatorial orientation of the substituents in a chair-
like transition state.
0
103(R 1 =alkyl, alkoxy,
R2=alkyl, aryl)
H
H
CO2Me
l-<o
104 105
Scheme 31. Synthesis of trisubstituted THF via C2-C3 bond formation; (a) Bu3 SnH, AIBN, toluene, 110 °C.
They also applied the method to the synthesis of tetrasubstituted THFs (Scheme 32). When
they used radical precursor 106a, they obtained a diastereoisomeric mixture of
tetrasubstituted THF 107a with the ratio of 3 to 1 (major isomer to other 3
diastereoisomers) in 98% yield (a, Scheme 32). The stereoselectivity was explained in the
way as the example in Scheme 31. Also, when they used radical precursor 106b, they
obtained a diastereoisomeric mixture of tetrasubstituted THF 107b with the ratio of 3 to 1
(major isomer to other 3 diastereoisomers) in 93% yield (b, Scheme 32). However, the
stereoselectivity was explained in different manner. When the methyl group at the radical
centre is located at a pseudoequatorial position in transition state, there would be
unfavoured steric repulsion between the Me group and 'butoxy ester group. However, when
the methyl group is located at pseudo-axial position in transition state 108, there would be
no steric repulsion and it would give the major THF 107b.
42

MeO
106 ab)
MeO2v
106b
Ph/,.
O
CO2Me
O'Bu
107a
COoMe
O'Bu 107b
Scheme 32. Synthesis of tetrasubstituted THF via C2-C3 bond formation; (a) Bu3 SnH, AIBN, toluene, 110°C.
In 2004, Lee and co-workers showed a similar C4-C5 bond formation via the radical
cyclization of p-alkoxyvinyl sulfoxides 109 (Scheme 33). 50 They obtained 2,5-cw-THF
110 as a major product with the a diastereoisomeric ratio over 92:8 and over 85% yield.
Furthermore, they showed the synthesis of allyl carbinols via Pummerer rearrangement and
subsequent allylation.
I
BnO>'=% Jo\ Jol
109 110
Scheme 33. C4-C5 bond formation via the radical cyclization of P-alkoxy vinyl sulfoxides; (a) Bu3 SnH, Et3B, toluene, -20°C.
The second main strategy uses haloalkyl propargyl ether 111 as a radical precursor and it
cyclises via the formation of C3-C4 bond in THF 112 (Scheme 34).
43

112
Scheme 34. C-C bond formation by radical precursor with triple bond.
In 2002, Roy and co-workers showed the radical-mediated cyclization of bromoalkyne 113 using 1-ethylpiperidine hypophosphite (EPHP) as a radical reducing agent and AIBN
(Scheme 35). 51 They obtained trisubstituted THF 115 with 2,3-/raws-relationship as a
single diastereoisomer in 75-89% yield, via the formation of radical 114. They applied this
method to the synthesis of (±)-dihydrosesamin. Similarly, the same authors reported the
radical cyclization of an a-bromocarbonyl compound using titanocene(III) chlorite as a
radical initiator in 200552 and Ranu reported the radical cyclization using indium(I) iodide
as a radical initiator in 2006. 53
Ar o
113(R=alkyl, alkoxy) 114 115
Scheme 35. Radical-mediated cyclization of bromoalkyne using EPHP and AIBN; (a) EPHP, AIBN, benzene, reflux.
1.2.5.2. C-O bond formationVarious methods for the synthesis of THFs involving radical-mediated C-O bond formation
have been reviewed. 54 Most of strategies use alkoxy radical precursor 116 to form the C-O
bond in THF 117 (Scheme 36).
44

-o-
116 117
Scheme 36. Formation of C-O bond via radical precursor.
In 2003, Ihara and co-workers reported the synthesis of THF 121 via alkoxy radical
cyclization of hydroxy vinyl bromide 118 using Bu3 SnH and AIBN (Scheme 37). 55 Firstly,
alkoxy radical 120 is generated from vinyl radical 119 and it leads to the formation of THF
121 in 55% yield. Here, the hydrogen atom of the hydroxyl group is selectively abstracted
in spite of the presence of two methyl groups.
AcO AcO'
XDHBr
118 119 120
121
Scheme 37. Synthesis of THF via alkoxy radical cyclization of hydroxy vinyl bromide; (a) Bu3 SnH, AIBN, benzene, reflux.
In 2003, Hartung and co-workers reported the synthesis of halogenated THFs 124 via
bromocyclisation of thione radical precursor 122 (Scheme 38). 56 Radical -CCls generated
in the reaction mixture induces homolytic cleavage of 122 to give the alkoxy radical 123;
this then undergoes a 5-exo-trig cyclisation followed by bromine capture to give THF 124.
45

Scheme 38. Bromocyclisation of thione radical precursor; (a) 250 W visible light discharge lamp, BrCCl3 , benzene, 15 °C.
When R1=Ph and R2 , R3=H, 2,5-trans-THF is obtained as a major product in a ratio of
72:28 (trans:cis) in 87% yield. When R 1 , R2=H and R3=Ph, 2,3-^^5-THF is also obtained
as a major product in a ratio of 98:2 (trans:cis) in 90% yield. However, when R 1 , R3 =H and
R2 is Ph, 2,4-ds-THF is obtained as the major product in a ratio of 68:32 (cis:trans) in 75%
yield. In 2004, Liu reported the synthesis of THFs from cation radical induced [3+2]
cycloaddition of electron-rich chalcone epoxide 125 to electron-rich alkene 126 initiated by
tris(4-bromophenyl)aminium hexachloroantimonate 128 (TBPA+SbC16~, Scheme 39). 57 In
this transformation, various multi-substituted THFs 127 were obtained as a mixture of 4
diastereoisomers in 61-82% yield.
O
PhAr
125
.R3
126(R 1 =MeorH, R2 =Ph or alkoxy R3= vinyl, Ph)
N+ -H SbCI6-
128TBPA+SbCI6-
Scheme 39. Synthesis of THF from cation radical induced [3+2] cycloaddition of chalcone epoxide; (a) TBPA+SbCl6 , CH 2Cl2 .
46

The reaction is believed to proceed via firstly, single electron transfer between epoxide 125
and TBPA+ 128 producing the cation radical 129 of epoxide 125 (Scheme 40).
Subsequently, selective cleavage of the Cp-O bond gives a new cation radical 130. Finally,
the addition of the cation radical 130 to electron-rich olefin 126 forms cation radical adduct
131, which undergoes a second electron transfer followed by ring-closure to give THF 127.
TBPA"1" electron transfer 1
TBPA
electron transfer 3
electron transfer 2
Ar
Ph
130
.R3
126
Scheme 40. Mechnism of cation radical induced [3+2] cycloaddition of chalcone epoxide and olefin.
A different example is the synthesis of THFs by C-O bond formation via homolytic
cleavage of the epoxide C-O bond reported by Gansauer and co-workers (Scheme 41). 58
Initial reaction between epoxide 132 and Cp2TiCl forms a Ti-O bond and a carbon-centred
radical which undergoes 5-exo-cyclisation to give 133. Homolytic cleavage of the Ti-O
bond then generates a new C-O bond to give THFs 134. The 3,4-c/s-THFs 134b were
47

obtained with high stereoselectivity in up to 73% yield. They explained this high
stereoselectivity by density functional theory (DFT) calculations which suggested that 3,4-
c/s-THFs 134b were the kinetically favoured products, in contrast to the
thermodynamically favoured 3,4-/r<ms-THFs 134a.
Et02CJ /=
CO2 Et
132
R
R
CICp2TiO
fJ. IEt02C J /==<
v^^ iC02Et
Rf
t
R
TiCp2CI
REt02C
TiCp2CI
EtO2CR
134a(thermodynamic
product)
•*• EtO2CR
RC02Et
134b(kinetic product)
Scheme 41. Synthesis of THF by the C-O bond formation via homolytic cleavage of Ti-O bond; (a) Mn, HC1, EtOAc.
Similar to this titanium-catalysed formation of THFs, Hilt and co-workers have reported the
iron-catalysed formation of THFs from the reaction between styrene oxide and unsaturated
alkenes via electron transfer steps. 59
48

1.2.6. C-C bond formation strategies.Various methods for THF formation involving C-C bond formation have been reported.
Ene-type cyclisations are prevalent.
1.2.6.1. Ene-type cyclisation.
Transition metal catalysed C-C bond formation from 1,6-enynes 135 is frequently used for
the formation of the C3-C4 bond in THFs 136 (Scheme 42). This type of transformation has
been described in several reviews. 60
MU
M= Pd or Rh135
Scheme 42. C-C bond formation from 1,6-enyne.
For example, in 2002, Zhang reported a Rh-catalysed Alder-ene reaction for the preparation
of a variety of chiral THFs using an air stable [{Rh(cod)Cl}2j precursor with BINAP ligand
(Scheme 43). 61 The functional! sed THFs 138 were obtained from enyne 137 in over 82%
yield and 99% ee for all substrates. Additionally, they reported a highly efficient kinetic
resolution of racemic 1,6-enynes bearing alkyl substituent at both the allylic positions.62
R 1 =Ar, alkyl, COMe, CO2 Et, CH 2OH R2=alkyl, OMe, OAc
Scheme 43. Preparation of THFs by Alder-ene reaction; (a) [{Rh(cod)Cl} 2], (5>BINAP, AgSbF6 .
49

In 2004, Mikami and co-workers showed ene-type carbocyclisation with skewphosrodium
complex (Scheme 44). 63 In contrast to Zhang's work, this carbocyclisation of enyne 139 is
accompanied by olefin migration, favouring the product 140b (44% yield, 91% ee) over the
product 140a (4% yield, 67% ee) at a high reaction temperature (80 °C). The high
temperature is necessary since it allows dramatically reduced reaction time (40 min), in
contrast to the longer reaction time (46 h) at room temperature.
MeO2C
_^ MeO2C MeO2C
Scheme 44. Ene-type carbocyclisation accompanying olefin migration; (a) [Rh((5r,5)-skewphos)] 2(SbF6)2 (5mol%), CH2C12 , 80 °C.
In 2005, Krishe and co-workers reported Rh-catalysed asymmetric cyclisation-
hydrogenolysis of 1,6-enynes 141 (Scheme 45). 64 In this transformation, they obtained
THF 143 in up to 85% yield and 98% ee. They suggested that the reaction proceeds via
intermediate 142.
142
-R2
hydrogenolysis
O 143
Scheme 45. Asymmetric hydrogenation of 1,6-enynes; (a) Rh(cod)2 OTf (3-5 mol%), chiral phosphine (3-5 mol%) CH2C12 , 25 °C, H 2 (1 atm), 2-3 h.
Similarly, Mikami and co-workers reported Pd-catalysed cyclization of 1,6-enynes 144 with
a new N,P-ligand bearing an achiral gew-dimethyloxazolidine (Scheme 46). 65 They
obtained THF 145 in 92% yield and 87% ee.
50

CO2 MeCO2 Me
Scheme 46. Pd-catalysed cyclization of 1,6-enynes; (a) [(MeCN)4Pd](BF4)2 (5 mol%), N,P-ligand (10 mol%), HCOOH(1.0eq),DMSO, 80 °C.
Lu and co-workers also reported Pd-catalysed cyclization of 1,6-enynes 146 initiated by
acetoxypalladation of the alkyne followed by alkene insertion and protonolysis of the
carbon palladium bond to give THFs 147 (Scheme 47). 66 When R*=Ph and R2=H, the
highest yield obtained was 96%. Also, they synthesised enantiomerically enriched THFs
using chiral oxazoline ligands in up to 65% yield and 77% ee.
R2OC
-O
146(R 1 , R2 =H, alkyl, Ph)
olefinic insertion
COR2 , n | frans-acetoxy-
palladation AcO
COR
R 1 ^protonolysis
AcO^O
147
Scheme 47. Cyclization via acetoxypalladation of 1,6-enynes; (a) Pd(OAc)2/bpy, HOAc, 80 °C.
The same authors also reported the synthesis of THFs via a divalent palladium-catalysed
three- component coupling (Scheme 48). 67 They used methylene malonate 148, propargyl
alcohol 149 and allyl chloride 150 for this one-pot synthesis. Initially, the reaction between
methylene malonate 148 and potassium propargylate generated from alcohol 149 by base
( lBuOK) gives malonate anion species 151. Subsequent cyclisation yields palladated THF
51

152 which then undergoes olefm insertion followed by p-elimination to produce THF 153
as a mixture of two diastereoisomers (2,5-cis and trans) in up to 75% yield.
MeO2Cx .CO2 Me Cl MeO2 C . MeO2C-J—f
EtO
148 149 150 153
MeO2C
151
MeCvEtfV-
152
Cl
MeO2C MeO2C
Pd
Ph
Scheme 48. Synthesis of THF via a divalent palladium-catalysed three component coupling; (a) 'BuOK, Pd(II)/LiCl, dr 1.1:1 (cis/trans), 75%.
Similarly, in 2004, Nakamura and co-workers reported the use of a Zn(II)/amine catalysed
coupling reaction between alkylidenemalonate and propargyl alcohol and they obtained_ AS
methylene THFs in 70-94% yield. In the same year, Zhang showed palladium-catalysed
cyclization using 1,2,7-trienes 154 instead of 1,6-enynes, followed by a Suzuki coupling
reaction (Scheme 49). 69 In this tandem process, they obtained 3,4-cw-THF 155 as a single
diastereoisomer in 59% yield.
H Ph
" Q155
Scheme 49. Palladium-catalysed cyclization using 1,2,7-trienes; (a) Aryl boronic acid, Pd(PPh,)4 , K3 PO4 -3H 2 O, toluene, 50 °C, 59%.
52

1.2.6.2. Other C-C bond formations.
As a different type of strategy for the C-C bond formation, Takacs and co-workers reported
a palladium-catalysed bisdiene carbocyclisation-nucleophilic trapping method in 2003
(Scheme 50). The palladium-mediated oxidative coupling of bisdiene 156 gives
palladacycle 157 and then treatment with nucleophilic trapping reagent, N-
hydroxyphthalimide (NHP), gives the THF 158 as a single diastereoisomer in up to 90%
yield.
156(R1 =alkyl, Bn, R2=alkyl)
H —
157
CH 2R 1
PdL
158
Scheme 50. Formation of THF by palladium-catalysed bisdiene carbocyclisation-nucleophilic trapping; (a) Pd2(dba)3 , (o-biphenyl)P('Bu)2 , NHP, 90%.
Another different strategy is based on olefm metathesis. In 2004, Evans and co-workers
demonstrated the preparation of the THF moiety in this way as part of their total synthesis
of Gaur acid (Scheme 51). 71 Olefin metathesis using the ruthenium based Grubbs N- heterocyclic carbene catalyst followed by hydrogenation converted diene 159 into THF 160
in 75% yield.
53

TBSO159 160
Scheme 51. Preparation of THF by ring-closing metathesis; (a) i. Grubbs NHC catalyst, DCE, 40 °C, ii. H2 , 70 °C, iii. HC1, MeOH.
Additionally, in 2005, Rychnovsky and co-workers reported the preparation of THF by
reductive cyclization (Scheme 52). 72 Reduction of nitrile 161 using lithium di-tert-
butylbiphenylide (LiDBB) generates an axial organolithium and the intramolecular
alkylation produces spiroacetal 162 as a single diastereoisomer in 63% yield.
162
Scheme 52. Preparation of THF by reductive cyclization; (a) LiDBB, THF, -78 °C, 63%.
1.2.7. [3+2] cycloaddition strategy.Various methods involving [3+2] cycloaddition have been reported. There are two main
strategies; i) [3+2] cycloaddition using carbonyl ylides and alkenes, ii) [3+2] cycloaddition
using cyclopropanes and aldehydes.
1.2.7.1. [3+2] cycloaddition using carbonyl ylides and alkenes.
A common strategy in this field is the Rh-catalysed [3+2] cycloaddition reaction between a
carbonyl ylide and an alkene. The general concept is shown in Scheme 53. Firstly, a
carbonyl ylide 165 is prepared from the reaction between carbonyl compound 163 and
diazo compound 164 and the THF 167 is formed by the annulation between dipolar ylide
165 and electron-deficient alkene 166.
54

0 No R2 R4JL
R 1 ^R2 R3j^ Rh2(OAc)4 __ _J^+ J< 0 + r,5/=\36
|\ iX \J l\
163 164 165 166
R2 R4
R^'/JR3
1 I 1+
)=LiL R5 R6 ]
R 3>R4
° \ -R6R^ / KR rY
R1 \5
167
Scheme 53. Rh-catalysed [3+2] annulation between carbonyl ylide and alkene.
For example, in 2002, Jamison and co-workers showed the three-component coupling
between diazo compound 169, alkene 170 and aldehyde 168 bearing a dicobalt
hexacarbonyl (Co2(CO)6) cluster (Scheme 54). 73 This transformation gives a single
diastereoisomer 171 in 74% yield. This type of transformation is usually limited to
electron-deficient alkenes, so it is notable that the use of the cobalt cluster-bearing aldehyde
increases the scope to encompass a wide range of alkenes. For example, when styrene was
employed, the corresponding THF was obtained in 46% yield.
(OC)3Coro(CO)3(OC)3Co^Co(CO)3
TH
168
I + MeO2C CO2Me-
169 170 171CO2Me
a range of alkenes: ^^Ph ^,^SiMe3
(46%,dr>20:1) (37%, dr>20:1) (11%, dr>20:1)
Scheme 54. Three-component coupling between diazo compound, alkene and aldehyde bearing cobalt cluster; (a) Rh2(OAc)4 , CH2 Cl 2 , 74%, single diastereoisomer.

In 2004, Hodgson and co-workers demonstrated the use of allenes for the cycloaddition, in
place of electron-deficient alkenes (Scheme 55). 74 They obtained cycloadduct 173 in 77%
yield from the reaction between allene and diazocarbonyl compound 172.
O
172O
O
173
Scheme 55. [3+2] cyclization using allene and carbonyl ylide; (a) Rh2(OAc)4, (b) allene.
1.2.7.2. [3+2] cycloaddition using cyclopropanes and aldehydes.
In the general process (Scheme 56), a cyclopropane 174 bearing diester and aryl
substituents generates a formal zwitterionic intermediate via Lewis acid-mediated ring-
opening which is then trapped by aryl aldehyde 175. In terms of stereochemistry, two Ar
groups (Ar 1 and Ar2) are located pseudoequatorially in the transition state, resulting in the
formation of 2,5-cw-THF 176.
^ COpMe o Lewis hx^ + JJ acidf "CC^Me Ar2 ^
174 175
Ar2 0=// \ OMe
MeO O'
MeC^C 176
Scheme 56. [3+2] annulation using cyclopropane and aldehyde.
Similarly, in 2005, Johnson and co-workers reported the tin-mediated formation of 2,5-c/s-
THFs 179 from aldehydes 178 and cyclopropanes 177 (Scheme 57). 75 Here, they obtained
2,5-ds-THFs from benzaldehyde and thienyl cyclopropane with diastereomer ratios of up
to 20:1 and in up to 97% yield. Also, they obtained 2,5-cw-THF from phenyl cyclopropane
56

and thienyl aldehyde (83:1 dr and up to 98% yield). However, when they used a,p-
unsaturated aldehydes or styrenyl cyclopropanes, they could obtained 2,5-ds-THFs with
low diastereoselectivity (<17:1), albeit in high yield (over 92%).
^rMeO2C
178 179
Me02C CO'Me Me02 C C°2Me
97% (dr 20:1) 98% (dr 83:1) 96% (dr 17:1)
Scheme 57. [3+2] Annulation using cyclopropane and aldehyde; (a) Sn(OTf)2 (5 mol%), CH2C1 2 .
In 2006, Yadav and co-workers reported scandium-mediated [3+2] cyclization of
silylmethyl-substituted cyclopropanes with aryl aldehydes and aryl ketones. 76 The
corresponding THFs were obtained with a diastereomeric ratio of 12.5:1 in up to 100%
yield. Also, Christie and co-workers have shown [3+2] annulation using aldehydes and
cyclopropanes bearing cobalt clusters instead of aryl substituents and a mixture of
diastereoisomeric THFs were obtained up to 85% yield. 77
1.2.7.3. Other [3+2] cycloaddition strategies.In 2004, Dulcere and co-workeres reported the formation of the THF unit via the
transformation of nitroalkyl allyl ether 180 into isoxazaolidine 182 (Scheme 58). 78 The
nitroalkyl allyl ether 180 first reacts with a trialkylchlorosilane and DBU to give
silylnitronate intermediate 181. Subsequently, an intramolecular [3+2] cycloaddition
constructs the THF moiety of isoxazaolidine 182. In this transformation, the 2,3,4-
trisubstituted THF 182 is obtained in 69-100% yield.
57

NO2 R1-
180
R3SiO + O
[3+2]
181
(R 1 =alkyl, R2 =alkyl or Ph, R3=alkyl)
Scheme 58. Transformation of silylnitronate into isozaolidine; (a) DBU, R3 SiCl, CH2 C1 2 .
1.2.8. Miscellaneous strategies.
Langer and co-workers have developed the synthesis of THFs using 1,3-dicarbonyl dianion
species. They have shown that the Lewis acid-mediated cyclization of epoxides with 1,3-
bis(trimethylsilyloxy)-l,3-butadienes as a neutral equivalent of 1,3-dicarbonyl dianion
generates 2-alkylidene THFs (Scheme 59). 79 Initial regioselective attack of the terminal
alkene of diene 183 on the epoxide 184 furnishes intermediate 185 with inversion of the
configuration. The subsequent TiCU-mediated conjugate addition of the epoxide-derived
hydroxy group onto the a,p-unsaturated ester moiety then gives intermediate 186. Finally,
2-alkylidene THF 187 is obtained by the elimination of silanolate. Thus, trans-epoxide
affords 4,5-cw-THF and c/s-epoxide affords 4,5-trans-TIiF. Also, £"-olefin geometry is
predominantly observed.
58

OTMS OTMS
OEtR
183
CI3TiO
184
185
187
CI3TiO
186
OEt
Scheme 59. Lewis acid-mediated cyclization of epoxides with l,3-b>is(trimethylsilyloxy)-l,3-butadienes to form 2-alkylidene THF; (a) TiCl4 (2.0 eq), CH2Cl2 , 4 A MS, -78 °C (5 h) then 20 °C (12 h).
Similarly, these authors reported the preparation of 2-alkylidene THFs via LDA mediated
generation of dianions from p-ketoesters and subsequent intermolecular cyclization with 1-
bromo-2-chloroethane. 80 Also, they showed the preparation of 2-alkylidene THFs by
LiCKVmediated cyclization of p-ketoesters with epibromohydrin. 81
In 2002, Ma^cosza and co-workers reported the preparation of 2,3-disubstituted THFs via
the formation of y-halocarbanions (Scheme 60). 82 Haloalkane 188 was deprotonated by the
treatment of 'BuOK and the resultant anion added to an aldehyde. The resulting y-
halocarbanion 189 cyclized to form 2,3-/r<msi-THF 190 as the major product in a
diastereoisomeric ratio of 69:31.
59

cr188
Y=CN, SO2 Ph, CO^Bu
+ H K+ "O-C-R
'Y
189 190
Scheme 60. 2,3-disubstituted THF via the formation of y-halocarbanion; (a) 'BuOK, -30 °C, THF, (b) RCHO (R-alkyl, aryl).
In 2004, Bode83 and Glorius84 independently developed the synthesis of butyrolactones by
direct annulation of enals and aldehydes catalyzed by Af-heterocyclic carbenes. For example,
Bode and co-workers prepared 4,5-c/si-butyrolactones by this type of transformation as a
major product (diastereoisomeric ratio of up to 5:1 and 41-81% yield) (Scheme 61).
Ar1,
191
Ar1
Ar2
192
193
Mes
HO
Mes'
194
Mes^ N^^N ^^- + \=J
193 (8 mol%)
192
""tVo
Ar2 °197
Mes
Scheme 61. Synthesis of butyrolactones by direct annulation of enals and aldehydes catalyzed by N- heterocyclic carbene; (a) DBU (7 mol%), 1:1 THF/'BuOH, 25 °C.
60

Initially, a,(3-unsaturated aldehyde 191 is attacked by 7V-heterocyclic carbene catalyst 193 to
give conjugated acyl anion equivalent 194, which then tautomerizes to homoenolate 195.
The subsequent addition of homoenolate 195 to aldehyde 192 gives alkoxide 196 which
then cyclises to afford lactone 197.
It can be seen from the above review that numerous methods have been developed for the
preparation of THFs. However, it is noteworthy that strategies allowing the synthesis of
2,3-ds-disubstituted THFs have rarely been reported. Therefore, the investigation of the
synthesis of 2,3-cw-disubstituted THFs is certainly desirable and necessary in this field.
61

1.3. Background to the projectThe idea for this project came from previous research undertaken in the Armstrong group.
During work towards the total synthesis of acremolactone A, our group found that the
oxidation of the 2-amino dihydropyran (DHP, 198), which had been expected to result in
epoxidation and simultaneous formation of the TV-oxide for subsequent Cope elimination,_ _ o c
instead resulted in the formation of THF 200 (Scheme 62). Presumably, following
epoxidation of DHP 198, the unstable epoxide intermediate 199 underwent ring opening to
give an iminium intermediate which then led to the THF 200.
H
198
H
199 200
Scheme 62. Oxidation of 2-amino DHP; (a) DMDO
After finding this unexpected rearrangement, our group became aware that Ireland had
reported a similar rearrangement in the synthesis of (±)-chalcogran. 86 Ireland showed that
the epoxidation of 2-alkoxy DHP 201 resulted in the formation of THF 202 (Scheme 63).
However, Ireland focused on only specific spiroketal systems.
62

•o o- 7/201 202
Scheme 63. Oxidative rearrangement of 2-alkoxy DHP by Ireland; (a) MCPBA (1.1 eq), MeOH, -5 °C (2 h) to rt, 82%.
On the basis of these two findings, our group investigated the aziridination of DHP 203
rather than epoxidation. The aziridination successfully effected an analogous
rearrangement; leading to the formation of pyrrolidine 204 (Scheme 64). 87
_ aziridination FT ^O^ "
203 x , 204
Scheme 64. Aminative rearrangement of 2-alkoxy DHPs by Armstrong.
In this research, our group showed that two different aziridination conditions were able to
control the relative stereochemistry of the resulting pyrrolidines; the combination of N-
bromosuccinimide (NBS) and Chloramine-T (TsNClNa) gave 2,3-/rarcs-pyrrolidine 206 and
the combination of Cu and PhlNTs gave 2,3-c/,s'-pyrrolidine 207 (Scheme 65). Using the
NBS reagent, DHP 205 was brominated on the less hindered face, trans to the isopropyl
substituent. Nucleophilic attack of TsNClNa resulted in inversion of configuration, leading
to the formation of 2,3-/ra7w-pyrrolidine 206. However, DHP 205 was directly aziridinated
on the less hindered face under the Cu-nitrene reagent system, leading to the formation of
2,3-c/s-pyrrolidine 207.
63

a, c
206
b,c
Ts
207
Scheme 65. Stereocontrolled aminative rearrangement of DHP; (a) NBS (20 mol%), TsNClNa (3.0 eq), CH3 CN, rt, 1 h, 64%; (b) Cu(MeCN)4PF6 (10 mol%), PhlNTs (1.1 eq), CH3 CN, 0 °C, 0.5 h, 52%; (c) Et3 SiH (3.0 eq), BF 3 -Et2 O (2.0 eq), -78 to 0 °C, 2 h, >90%.
In an analogous fashion, Hall also reported the epoxidation of DHP and ring contraction,
giving 2,5-disubstituted THF 208 (Scheme 66). 88 In this research, Hall suggested a
possible mechanism for the formation of THF 208 by ring-opening related to the
epoxidation. However, this study only focused on the formation of THFs with substitution
in the 6-position (R1=Me, H and R2=Et, 'Bu) and there was no further study of
stereoselectivity or alternative reaction conditions.
R
R 1 "CT "OR2O
O
208
OR2 R 1 = Me, H R2= Et, Bu
Scheme 66. Oxidative rearrangement of DHP by Hall; (a) MCPBA (1.0 eq), CH2C12 , 0-10 °C, 20-90%.
Consequently, we decided to investigate the possibility of using this oxidative
rearrangement for the Stereocontrolled synthesis of substituted THFs. In particular, we
would investigate the Stereocontrolled formation of 2,3-disubstituted THFs 209 and 210 by
this oxidative rearrangement because of the relative lack of methods for the synthesis of
2,3-disubstituted THFs (Scheme 67).
64

RO
209
OR2
Scheme 67. Stereocontrolled formation of 2,3-disubstituted THFs.
65

2. Results and discussionWe first planned to optimise the reaction conditions for the oxidative rearrangement by
screening several alternative oxidants. If we were able to optimise the conditions, we could
then investigate the diastereoselectivity of the rearrangement by application of the
conditions to various substituted DHPs. Furthermore, we could apply our method to natural
product synthesis.
2.1. Optimisation of the oxidative rearrangement2.1.1. Preparation of dihydropyrans by hetero Diels-Alder reactionThe hetero Diels-Alder (HDA) reaction of heterodienes with enol ethers using lanthanide
catalysts is a short and attractive route to make dihydropyrans. Moreover, the mildness of
the lanthanide catalyst is a notable feature which allows fragile but valuable functionality in
both the diene and the cycloadduct to survive. In addition to this, only a few mole percent
of catalyst suffices for reasonable reaction rates at room temperature. Because of these
attractions, the HDA was considered as a suitable method to prepare DHPs as a starting
material for the oxidative rearrangement.
Firstly, we prepared a simple DHP 211 by Danishefsky's method (Scheme 68). 89 The
excess of enol ether (7.0 eq) to diene (1.0 eq) was used because the enol ether played a role
as both the dieneophile and the solvent. A catalytic amount of YbFOD was sufficient to
give a reasonable yield (70%). However, if the pressure tube was opened during the
reaction for TLC analysis it caused a decrease of the yield from 70% to 40%.
O
(7.0 eq)211
HC
C(CH 3 ) 3
YbFOD
Scheme 68. HDA reaction of heterodiene; (a) YbFOD (5 mol%), 45 °C, pressure tube, 3 d, yield 70%
66

In terms of spectral data, the proton NMR spectrum for DHP 211 shows two characteristic
peaks for an OC//O proton and an alkene CH proton. Each appears to be a triplet, centred
at 8 4.96 ppm and 5 4.50 ppm respectively, with a vicinal coupling constant in both cases of
3.5 Hz. These two peaks were important to identify the oxidative rearrangement products in
the next stage as well as other DHPs by comparing the chemical shift and multiplicities of
these peaks. The V value of 3.5 Hz between two coupled protons at C-2 and C-3 suggests a
small dihedral angle and hence that the proton at C-2 is in an approximately
pseudoequatorial position (Fig. 4). Thus, the ethoxy group in C-2 is in a pseudoaxial
position as would be expected based on the anomeric effect.
H
(ps|eudoequatorial)
anomeric effect
(pseudoaxial)
211
Fig. 4. Suggested conformation of DHP 211.
2.1.2. Choice of oxidants for epoxidation
After the preparation of simple DHP 211, several oxidants such as MCPBA, DMDO,
methyl(trifluoromethyl)dioxirane and the combination of hydrogen peroxide with MTO
were tested in the desired epoxidation/rearrangement process.
2.1.2.1. MCPBA
Firstly, MCPBA was adopted for the rearrangement, since it can be regarded as a
"standard" reagent for alkene epoxidation. 90 Before use, commercial MCPBA (55%) was91washed with phosphate buffer (pH=7.5) to remove MCBA, followed by the iodometric
67

titration of MCPBA. 92 However, the reaction of DHP 211 with MCPBA (1.0 eq) gave the
unexpected five-membered ring lactone 212 (18%) as the only isolated product, after the
evaporation of solvent (Scheme 69).
OEt
212
Scheme 69. Oxidation of DHP by excess of MCPBA; (a) MCPBA (1.0 eq), CH 2C1 2 , 0 °C to it, 2 h, 18%.
The result was proved by the spectral data; for example, only 6 peaks were observed in the
13C NMR. Other experimental data such as IR spectral data93 and ! H NMR spectral data94
also matched literature values. A possible mechanism is shown in Scheme 70. Initial
formation of the desired rearrangement product 213 could be followed by Baeyer-Villiger
rearrangement to give 214 and subsequent peracid-mediated oxidation to the lactone 212.
To support this proposed mechanism, we reacted the lactol ether 213^ with MCPBA
eq) for 1 d and we obtained the lactone 212 in quantitive yield.
MCPBA (excess)
211
214
Scheme 70. Proposed mechanism of Baeyer-Villiger rearrangement.
The preparation of the lactol ether 213 (vide infra)
68

This formation of the lactone 212 may be occuring because the MCPBA had inadvertently
been used in excess. Therefore, we decided to use commercial MCPBA (77%) without any
prior purification and titration for this epoxidation, using 1.0 eq of MCPBA assuming that
the quoted 77% purity was correct (Scheme 71). Pleasingly, TLC analysis suggested that
the DHP 211 was completely consumed and NMR analysis indicated the formation of lactol
ether 213 and lactol 215. The lactol ether 213 and lactol 215 could be separated by column
purification. However, the diastereoisomers of the lactol ether 213 and the lactol 215 were
not separable by column purification in either case.
Scheme 71. Epoxidation of DHP by MCPBA; (a) MCPBA (^ 1.0 eq), CH2C12 , 45 °C, 2 h.
From the [ H NMR analysis, we found that the major product was a mixture of
diastereoisomeric lactol ethers 213 (2:1 ratio). The minor product was a mixture of
diastereoisomeric lactols 215 (1:1 ratio), presumably formed by hydrolysis of lactol ether
213 under the acidic reaction conditions. The structures of the rearrangement products are
supported by ! H NMR. The peak for the olefmic proton at C-5 in DHP 211 at 5 4.50 ppm is
now absent. A new dd at 5 4.45 ppm and a new triplet at 6 4.32 ppm suggest the formation
of lactol ether 213. The 13C NMR spectrum strongly supported the presence of the ketone
functional group because of the characteristic resonance at 6 170.3 ppm (Fig. 5). Also, the
formation of lactol 215 is supported by the absence of the peaks for the ethyl group in the
*HNMR spectrum.
69

4.32 and 4.45 ppm 4.50 ppn\
170.3 ppm
Fig. 5. Chemical shifts of DHP 211 and lactol ether 213.
Additionally, there was 1 H NMR evidence that lactol 215 existed as a mixture of lactol 215
and open-chain aldehyde form 216, with the former predominating (Fig. 6). 95 The
existence of open-chain form 216 can be found in the trace of aldehyde 1H (around 9 ppm)
in the H NMR spectrum and the low integration of this aldehyde proton proves that the
cyclic lactol 215 is a more stable form than open-chain 216.
Fig. 6. Equilibrium between cyclic lactol 215 and open-chain 216.
However, the isolated yield of the products was extremely low (14% for lactol ether 213
and 10% for lactol 215) and we suspected that the lactol ether 213 may be volatile. We
investigated this by placing the pure product 213 under high vacuum for 1 hour; the weight
of product was reduced from 74 mg to 6 mg. This indicated that DHP 211 was not an ideal
test substrate for the oxidative rearrangement because the rearrangement product 213 was
volatile. In order to solve the volatility problem, we decided to increase the product's
molecular weight by using a longer-chain ether. Thus, heavier DHPs 217 and 218 were also
prepared by the same HDA procedure as before (Table 7).
70

Table 7. HDA reaction of heterodiene and butyl vinyl ethers; (a) YbFOD (5 mol%), pressure tube.
O
O OR
Product
217
218
R<Bu
"Bu
Ratio3
1:1
3:1
2:1
Time
5d
8d
6d
6d
Temp.
70 °C
90 °C
90 °C
110 °C
Yield
5%
20%
55%
N/A
a The ratio of enol ether to diene.
The preparation of both DHPs 217 and 218 required more vigorous conditions (6-8 days
and 90 °C) than DHP 211 (3 days and 45 °C). Unfortunately, the yield of the synthesis of
DHP 217 was extremely poor (20%), in spite of the use of longer reaction time, higher
temperature and a large excess of dienophile. However, DHP 218 was obtained in a
reasonable yield (55%) at 90 °C while higher reaction temperature (110 °C) resulted in the
decomposition of the HDA product.
After the preparation of less volatile DHPs, they were used for the
epoxidation/rearrangement process. When DHP 217 was employed, the reaction with
MCPBA was completed in 4 hours by TLC analysis. The yield of the expected
rearrangement product, lactol ether 219, increased to 37% (Scheme 72). However, the yield
of the lactol 215 was reduced. The lactol 215 was only detected by TLC analysis and in the
*H crude NMR spectrum of the crude product. We could not isolate it by column
purification. The major rearrangement product, lactol ether 219, was obtained as
inseparable mixture of diastereoisomers in a 1:1 ratio.
71

O219(37%)
Scheme 72. Oxidative rearrangement of DHP 217; (a) MCBPA (55%, 1.0 eq), CH2 C1 2, 0 °C to rt, 4 h.
When DHP 218 was reacted with 1.0 eq of MCPBA, the reaction gave the anticipated lactol
ether 220 (Scheme 73). In this case, 'H NMR analysis of the crude reaction showed a 2:1
diastereomeric mixture of lactol ether 220 as the major product and a 1:1 mixture of lactol
215 as the minor product. In this case, the two pure isomers 220a (13%) and 220b (26%) of
unassigned relative configuration could be separated by column chromatography.
1•0 On Bu
218
a \ 57 \ 2—— ———— VV'"on iO
220a(13%) 220b (26%)
Scheme 73. Oxidative rearrangement of DHP 218; (a) MCBPA (55%, 1.0 eq), CH2 C1 2 , 0 °C to rt, 3 h.
In an attempt to reduce the reaction time, the number of equivalents of MCPBA was
increased from 1.0 to 1.5 (Scheme 74). However, it seemed that the use of excess MCPBA
caused Baeyer-Villiger rearrangement because the peaks for the proton at C-5 of the
tentatively assigned products (221 or 222) appear as two peaks at low field (55.15 and 5.47
ppm). Further isolation and characterisation were not undertaken for these compounds.
On Bu or
221 222
(tentatively assigned)
Scheme 74. Baeyer-Villiger rearrangement of DHP 218; (a) MCBPA (1.5 eq), CH2CI : , 0 °C to rt, 3 h.
72

The results of MCPBA oxidation are summarised in Table 8.
Table 8. Results of oxidative rearrangement by MCPBA; (a) MCBPA (l.Oeq), CH2C12 , 0 °C to rt.
^X)'
211: 217: 218:
DHP
211
217
218
OR
R=Et R=lBu R= nBu
Time
2h
2h
4h
3h
T Vo
213: 219: 220:
o^>212
Major product (yield, dr)
213(14%, 2: l a)
212b (18%,N/A)
219(37%, l:l a)
220(39%, 2: l c )
OR T V^ \o/ unO
R=Et 215 R=lBu R= n Bu
OEt
Minor product (yield, dr)
215(10%, l:l a)
N/A
215 (<5%, l:l a)
215 (<5%, l:l a)
a Isomers were inseparable. An excess of MCPBA (> 5.0 eq) was used. c Isomers were separable.
2.1.2.2. DMDO
The second choice for the epoxidation was DMDO and this was prepared by distillation
from a mixture of acetone and Oxone*. 6 After isolation, the solution of DMDO in acetone
was titrated using acetic acid, saturated KI solution and aqueous NazSiOs solution (0.002
M). The concentration of DMDO was found to be approximately 0.03 M. DHPs 211, 217
and 218 (as previously tested with MCPBA) were used for the oxidative rearrangement by
DMDO. For the reaction, 1 .0 eq of DMDO was reacted with the DHPs in DCM at 0 °C,
with the reaction being slowly allowed to warm to room temperature. The DHPs were
consumed completely within 2-4 h according to TLC analysis. The results are summarised
in Table 9.
73

Table 9. Results of oxidative rearrangement by DMDO; (a) DMDO (1.0 eq), CH2C1 2 , 0 °C to rt.
I . x^crxxoR211:R=Et 217: R=4 Bu 218: R=n Bu
3 . \X>F o o
213: R 220: R
1^0^
^OR + ^T^o OH1 1
O =Et 215=n Bu
On Bu
223
DHP
211
217
218
Time
3h
3h
3h
2d
Major product (yield, dr)
215(53%, l:l a)
215(72%, l:l a)
215(64%, l:l a)
215(32%, l:l a)
Minor product (yield, dr)
213 (N/A, 2:1 a )
N/A
220(9%, 2:1 b)
223(10%, N/A)
a Isomers were inseparable. Isomers were separable.
Interestingly, all reactions gave lactol 215 as major products, rather than the lactol ethers
213, 219 and 220. This result suggested the presence of water in the DMDO solution. The
formation of the lactols from the lactol ethers had already been observed in the previous
reaction with MCPBA. In terms of yield, the combined yield of lactols and lactol ethers
(213+215 = 53%, 215+219 = 72%, 215+220 = 73%) from DMDO reaction was notably
higher than the combined yield (213+215 = 24%, 215+219 <42%, 215+220 <44%) from
MCPBA reaction. During the optimisation, we obtained an interesting product when longer
reaction time (2 days) was used for the oxidation of DHP 218 with DMDO. Although the
major product was the lactol 215 (32%), the minor product was dihydropyranone 223 (10%,
found as one diastereoisomer) and there was no lactol ether 220 in the reaction mixture
(Scheme 75).
74

DMDO*^SL J.^On Bu
2d
Scheme 75. Proposed mechanism for formation of dihydropyranone 223.
Thus, we postulated that the lactol ether 220 converted into dihydropyranones 223 upon
prolonged exposure to the reaction conditions. Dihydropyranone 223 was detected by IR
analysis as the value of 1715 cm" 1 for the ketone functional group. The ! H NMR also
showed that a doublet at 5 1.32 ppm for the protons of the C-l methyl group, and a quartet
at 5 4.21 ppm for the proton at C-2. Regarding the relative configuration of the single
diastereoisomer 223, L H NMR suggested that 223 has "BuO in an axial orientation because
the V value of 3.0 Hz between two coupled protons in C-5 and C-6 suggests that H-6 is
equatorial (Fig. 7). Although this was not proven conclusively, it is likely that the Me group
will be in an equatorial position to avoid an unfavourable 1,3-diaxial interaction with the
alkoxy substituent; hence the /nms-diastereomer is preferred.
75

3.0 Hz
H
1,3-diaxial interaction
On Bu
(favoured)
Fig. 7. Expected configuration of dihydropyranone 223.
2.1.2.3. DMDO generated in situ
Acetone solutions of DMDO are useful and powerful oxidants. However, their use suffers
from disadvantages such as low concentration of oxidant and inefficient preparation. Thus,
the use of DMDO generated in situ was potentially an attractive alternative. Therefore we
tried the oxidative rearrangement of DHPs 211, 217 and 218 with DMDO generated from
the mixture of Oxone® and NaHCOs in situ at pH 7.5, based on a literature procedure. 97
The oxidative rearrangement by this method gave lactol ethers 213 and 220 as major
products and lactol 215 as a minor product (Table 10). Strangely, the oxidation of substrate
217 did not give lactol ethers 219 or lactol 215, showing the formation of a number of
minor products by TLC analysis. The combined yield (13%) of lactol ethers 213 and lactols
215 was considerably lower than the yield of the reaction using isolated DMDO (53%). The
low yield of reaction is due partly to the volatility of lactol ether 213. The combined yield
(76%) of lactol ether 220 and lactol 215 were slightly higher than the yield of DMDO
reaction (73%). However, this method required a much longer reaction time than other
methods. The results of the rearrangement using DMDO generated in situ are summarised
in Table 10.
76

Table 10. Results of oxidative rearrangement by DMDO generated in situ; (a) Oxone®/acetone, NaHCO3 , Na2EDTA (pH=7.5), CH2 C12 , 0 °C to rt, 1 d.
O OR
211:R=Et 213:R=Et 215 217: R=lBu 218: R=n Bu
DHP Major product (yield, d.r.) Minor product (yield, d.r.)
~211213(13%,2:l a) 215 (N/A, 1:1 a)
217 N/A
218 220(46%, 2:1 b) 215 (30 %, 1:1 a)
a Isomers were inseparable. Isomers were separable.
2.1.2.4. Methyl(trifluoromethyl)dioxirane generated in situ
This method was developed by Yang to overcome the low epoxidation rate of dioxiranes
generated in situ since methyl(trifluoromethyl)dioxirane formed from trifluoroacetone and
Oxone® is highly reactive. 98 Thus, we attempted this method with DHP 218 and the
substrate was consumed extremely quickly (<30 min). However, TLC analysis showed the
formation of a number of impurities and only small amounts of the desired product 220.
Therefore this chemistry was not investigated further.
2.1.2.5. Hydrogen peroxide with MTO
Initially, we tried the oxidative rearrangement of DHP 211 with only hydrogen peroxide (aq,
27% w/w). However, this resulted in formation of ketoaldehyde 224 in 20% yield. This is
effectively the product of hydrolysis of 211; a likely mechanism is shown in Scheme 76.
77

HO-H HO-H
211
OII
224
Scheme 76. Proposed mechanism for formation of ketoaldehyde 224; (a) H2O2 (27% w/w, 1.0 eq), CH3 CN, 15
h, 20%.
However, no oxidation/rearrangement product was observed. Thus, we explored the
methyltrioxorhenium(VII)-catalysed epoxidation with hydrogen peroxide." A generally
accepted catalytic cycle for the epoxidation using hydrogen peroxide with MTO is
described in Scheme 77. 99a
CH,i ":Re^
H202
H2O2 HoO
Scheme 77. Epoxidation of olefm by combination of hydrogen peroxide and MTO.
We also used pyridine as additive, which is known to effect ligand acceleration. 9913 When
DHP 211 was exposed to these conditions overnight, the reaction did not give the desired
78

rearrangement product. The ! H NMR spectrum of the crude mixture suggested the product
could be an epoxide 225, not the rearrangement product, due to the presence of a triplet at 5
4.34 ppm with a J value of 3.0 Hz which could conceivably correspond to the epoxide
proton (indicated by * in Fig. 8). It is possible that pyridine (which was still present by *H
NMR) prevented acid-mediated decomposition of the epoxide.
OEt ' CT "On Bu
Fig. 8. Tentatively assigned epoxide 225. Fig. 9. Tentatively assigned epoxide 226.
Additionally, TLC analysis showed this tentatively assigned epoxide was less polar than the
rearrangement products 213 and 215. To investigate the conversion of this putative epoxide
to the rearrangement product, we added a trace of CSA to the NMR tube and also tried to
heat the epoxide to effect the desired reaction. However, in neither case could we see the
formation of rearrangement product by ! H NMR. The oxidation of DHP 218 under
MTO/H2O2 conditions also gave the tentatively assigned epoxide 226. The epoxide proton
(indicated by * in Fig. 9) for the peak of epoxide proton appears as a triplet at 5 4.46 ppm
with a J value of 3.0 Hz. Interestingly, the reaction of DHP 218 gave a trace of
rearrangement product, lactol ether 220, as a minor product after column chromatography.
However, we could not effect transformation of the tentatively assigned epoxides 225 and
226 to the desired rearrangement products. Therefore, this chemistry was not investigated
further. The results of MTO oxidation with hydrogen peroxide are summarised in Table 11.
Table 11. Results of oxidative rearrangement by H2 O2 and MTO; (a) Method A: H2 O2 (1.0 eq, 27% w/w), CH3 CN, rt, 1 d, (b) Method B: MTO (0.5 mol%), pyridine (12 mol%), H2O2 (1.5 eq, 27% w/w), CH 2 C1 2 , rt, 1 d.
/"\Method A or B
OR211: R=Et 218: R=n Bu
79
DHP Method Product (yield)
211 A
211 B
218 B
224 (20%)
Tentatively assigned epoxide 225 (>95%a)
Tentatively assigned epoxide 226 (major, >95%) 220 (minor, N/A)
1 Crude yield.
Later work by a co-worker showed that the combination of urea hydrogenperoxide (UHP),
imidazole and MTO could give the rearrangement product 228 (Scheme 78). 100
Vx V^H
O Eti O OEt
Scheme 78. Oxidative rearrangement using MTO and UHP; (a) MTO (4 mol%), UHP (3.0 eq), ImH (0.5 eq), CH2C1 2, then acid, 55%
2.1.3. Product manipulation to simplify stereochemical analysis
Based on the results so far, we decided to use DMDO as the reagent of choice for the
oxidative rearrangement. The rearrangement by DMDO generally gave a mixture of lactol
and lactol ether as products, with the former predominating. Because the purpose of this
project was the investigation of stereocontrolled formation of 4,5-disubstituted THFs, we
needed to simplify the product mixture by converging the lactol and lactol ether to a
common product, and removing the lactol/lactol ether stereocentre. We considered two
approaches; reduction and oxidation (Scheme 79).
O b) OxidationO
a ) Reduction
R=H, Alkyl
O
Scheme 79. Two ideas to remove stereocentre.
80

2.1.3.1. Acetal reduction
In the related aminative rearrangement work, we had shown that it was possible to
selectively reduce the hemiacetal 229 in in the presence of the ketone with EtsSiH and
BF3 -Et2O (Scheme 80). 87
fr\\ Ts O
229
Scheme 80. Reduction of alkoxy pyrrolidine; (a) Et3 SiH (3.0 eq), BF3 -Et2O (2.0 eq), -78 to 0 °C, 2 h, >90%.
Therefore we initially attempted the reduction of lactol 215 under these conditions. If it
were successful, the other rearrangement product (lactol ether) could potentially be
hydrolyzed to the lactol and reduced in the same way, or reduced directly under the same
conditions. However, the reduction of lactol 215 with EtaSiH and BP3'Et2O did not give the
expected result: unwanted additional reduction of the ketone functional group was observed,
with consequent formation of a further stereocentre in the product 230 (Scheme 81).
'OH
Scheme 81. Attempted reduction of lactol 215; (a) Et3 SiH, BF3 -Et2 O, CH2C12 , -78 °C to rt, 4 h.
Evidence for this came from the ! H NMR spectrum, notably the disappearance of the
singlet for the methyl group next to the ketone and the appearance of a new Me-doublet.
Additionally, the IR spectrum showed disappearance of the C=O stretch at 1716 cm" 1 and a
new O-H stretch at 2936 cm" . This data confirmed the reduction of the ketone to the
81

secondary alcohol. In view of this result, we turned our attention to oxidation of the lactol
as an alternative.
2.1.3.2. Acetal oxidation
It was aimed to effect oxidation of the mixture of lactols and lactol ethers to a single
lactone product 231 (Scheme 82).
Oxidation '"OR
R=H, Alkyl
Scheme 82. Idea to remove stereocentre by oxidation.
Firstly, Dess Martin oxidation conditions 101 were tested for oxidation of lactol 215.
However, TLC analysis suggested formation of a complex product mixture. Next, we
investigated the use of PCC. Additionally, we adopted a phenyl substituted lactol in order to
facilitate TLC analysis under UV. To prepare phenyl substituted lactol 234, we prepared
DHP 232 by HDA reaction in 50% yield and DHP 232 was transformed by oxidative
rearrangement with DMDO (Scheme 83). In this case only the major product lactol 234
was isolated, in 80% yield.
Ph
232 233 234 (isolated)
Scheme 83. The preparation of lactol ether 233 and lactol 234; (a) YbFOD (5 mol%), pressure tube, 65 °C, 5 d, 50%, (b) DMDO (1.0 eq), CH 2 C1 2 , 0 °C to it, 80% (yield of 234).
The oxidation of phenyl substituted lactol 234 with PCC 102 showed an excellent

conversion to the corresponding lactone 235 as a single diastereoisomer in 88% yield
(Scheme 84).
235
Scheme 84. Oxidation of lactol 234 to lactone 235; (a) PCC (3.0 eq), CH2 C1: , rt, overnight, 88%.
Ideally we required an oxidant that could effect oxidation of both lactols and lactol ethers to
lactones. After the successful oxidation of the lactol 234, we considered the oxidation of
lactol ethers 233 under these conditions. The lactol ether 220 was employed alternatively,
because the lactol ether 233 was isolated in very low yield (<5%). However, the same PCC
oxidation could not oxidise lactol ether 220 to the corresponding lactone (Scheme 85).
Scheme 85. Attempted oxidation of lactol ether 220; (a) PCC (3.0 eq), CH2C12 , rt, overnight.
In order to oxidise lactol and lactol ether at the same time, we needed a different oxidant.
We decided to use another chromium based oxidant, Jones oxidant, 103 because literature
precedent showed the Jones oxidation of both lactols 104 and lactol ethers 105 gave the
corresponding lactones. So, we tried to oxidize simultaneously the mixture of lactol 215
and lactol ether 220 (7:1) under Jones oxidation conditions. Pleasingly, it gave the
corresponding lactone 231 as sole product in 60% yield (Scheme 86).
83

OnBu
231
Scheme 86. Convergence of rearrangement products; (a) Jones reagent (3.0 eq), acetone, 0 °C to rt, 60%.
In spite of the successful Jones oxidation, we needed to consider other cleaner oxidants
because the Jones reagent is based on toxic chromium. From a literature search, we were
able to find an oxidation method for conversion of lactol ethers into lactones using
hydrogen peroxide with aqueous HC1 as catalyst. 106 We assumed that this reaction may
proceed via initial hydrolysis to the lactol, and so would also be applicable to oxidation of
the lactol/lactol ether mixture. The mixture of lactol 215 and lactol ether 220 was therefore
tested under with these conditions (Scheme 87). However, TLC analysis suggested
formation of a complex product mixture.
231
Scheme 87. Attempted convergence of rearrangement products by HiC^ and HC1; (a) H2O2, HCl, rt, overnight.
Because only Jones oxidation had so far given the desired result, we decided to utilise it in
further studies. Furthermore, we wanted to combine our oxidative rearrangement and Jones
oxidation. For this, DHP 218 was selected as a model. After the rearrangement of DHP 218 to lactol ether 220 and lactol 215, the crude product mixture was simultaneously oxidized
by the Jones reagent to give lactone 231 in 69% yield over 2 steps (Scheme 88).
S4

OH OnBu
O O215 220
215 + 220O
231
Scheme 88. Model study to generalise the oxidative rearrangement; (a) DMDO (1.0 eq), CH:C1: , 0 °C to rt, 73%, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt, 94%.
After this successful model study we decided to apply our method to various substrates and
to investigate the stereoselectivity of the oxidative rearrangement.
85

2.2. Application of oxidative rearrangement to dihydropyrans 2.2.1. Preparation of dihydropyransTo apply our oxidative rearrangement to various DHPs and to investigate the
stereochemistry of the resulting products, various DHPs were synthesised using the HDA
reaction (Fig. 10).
BnO
Fig 10. Various DHP substrates.
Although most of the substrate enones were commercially available, (E)-5-benzyloxy-pent- 3-en-2-one 241 and (3E, 9Z)-dodecadien-2-one 242 were not. We prepared these enones by
Horner-Wadsworth-Emmons reaction of diethyl(2-oxopropyl) phosphonate and the
appropriate aldehydes. The olefination of benzyloxyacetaldehyde gave selectively (E)-5- benzyloxy-pent-3-en-2-one 241 in reasonable yield (Scheme 89).
OBnO-
OIIPv
BnO241
Scheme 89. Horner-Wadsworth-Emmons reaction of benzyloxyacetaldehyde; (a) LiCl (1.2 eq), DBU (1.0 eq), CH3CN/THF (1:1), rt, overnight, 59%.
86

The olefination of cw-6-nonenal also gave only (£)-isomer, (3E, 9Z)-dodecadien-2-one 242 (Scheme 90).
O O.P.
OEt OEt
242
Scheme 90. Horner-Wadsworth-Emmons reaction of c/s-6-nonenal; (a) LiCl (1.2 eq), DBU (1.0 eq), CH3 CN/THF(1:1), rt, overnight, 66%.
After we prepared the enones, the DHPs (232 and 236-240) were simply obtained in a reasonable yield (40-75%) from thermal HDA reaction (in a pressure tube) of the appropriate dienes and vinyl ethers in the presence of the Lewis acid catalyst YbFOD (Table 12)
Table 12. Preparation of various DHP by HDA reaction; (a) YbFOD (5 mol%), pressure tube, 3 d.
R 1I
r^X R2
LRI R2 R:
+ <s /-\p3 av ^
1 I^ ^^. „ + j+^
Ft2V
^ i .s^o vv—— / -0' "ORd ^ "0" "OR3endo adduct exo
R 1
Me'Pr
Me
Ph
CH2OBn
(CH2 )4CH=CHEt
R2
H
H
Me
H
H
H
Rj
nBu
nBu
nBu
Et
Et
Et
Ratio3
1:2
1:2
1:5
1:5
1:10
1:10———— c ———
Time
lOd
3d
5d
5d
Id
5d
adduct
Temp. Product (yield ) drc
50 °C 236(40%) 6:1
55 °C 237(75%) 6:1
100°C 238(54%) N/A
65 °C 232(50%) - d
55 °C 239(67%) - d
55 °C 240(56%) 4:1The ratio of heterodiene to enol ether. Combined yield of two diastereoisomers. "The ratio of en Jo
to exo determined by *H NMR. Endo product only .
S7

H NMR analysis showed that the HDA reactions generally afforded a mixture of two
diastereoisomers with one predominant (>4:1). Since it is known that these reactions are
generally endo selective,85 ' 89 the major isomers were assigned as the 2,4-cis isomers.
The conformations of these 4-substituted-2-alkoxydihydropyrans are of interest. The
dihydropyrans exist as rapidly inverting half-chair forms and the conformational
equilibrium seems to be governed by two factors 107 : (a) the anomeric effect which forces
the C-2 alkoxy group into the axial position, (b) the conformational free enthalpy of the C-4 substituent which prefers the pseudoequatorial position to minimise 1,3-interactions
(particularly when bulkier groups are involved). In order to identify the conformations
adopted, we investigated the coupling constants in the ! H NMR spectra. Particularly, the
interactions between C-2 proton and C-3 protons were studied. The conformation of the
major endo products and the corresponding coupling constants are shown in Table 13.
Table 13. Conformation of major endo isomers and coupling constants,
anomeric effect
F
DHPs
236
237
232
239
240
r — — v\.x / ———————— """
R1 Y i ^ hA OR2243^\
1,3-interactions
—— ==T~,U 9^w ^L f ^^ r>^
^^^L.^^ ^J ̂ ^
"* 1H
244
R' R2 ^C2-C3a (Hz) Jc2-C3 b (Hz)
Me "Bu
'Pr nBu
Ph Et
CH2OBn Et
(CH2)4CH=CHEt Et
7.0 3.0
9.5 2.0
9.0 2.0
7.5 1.0
9.0 2.0'di-axial coupling constants, axial/equatorial coupling constants.
The C-2 protons of the DHPs appear around 4.82-5.01 ppm as a doublet of doublets with
two coupling constants: (a) 7.0-9.5 Hz for the di-axial coupling between the Haxj a i at C-2
88

and the Haxiai at C-3, (b) 1.0-3.0 Hz for the axial/equatorial coupling between the Haxiai at C- 2 and the Hequatoriai at C-3. This suggests that the substituent at C-2 is located in a pseudoequatorial position. If the major endo products have a 2,4-cis relationship as reported
in the literature, the substituent at C-4 is also located in a pseudoequatorial position and the conformational equilibrium between conformer 243 and conformer 244 favours conformer
244. This suggests that the conformational equilibrium for the endo product is mainly governed by the 1,3-interactions. However, these di-axial coupling constants are not as large as could be expected because a small portion of conformer 243 still exists in this equilibrium, and because of the presence of sp2-centres in the ring.The conformation of the minor exo compounds for the DHPs (236, 237 and 240) was also investigated. The C-2 protons of these DHPs appear around 4.99-5.01 ppm as a triplet with a small coupling constant (2.5-3.0 Hz), signifying that the C-2 proton occupies an pseudoequatorial position. Since the minor exo products are believed to have the 2,4-trans relationship, the proton at C-4 would then be located in an axial position and the conformer 246 is favoured in the conformational equilibrium (Fig. 11).
anomeric effect
RH
245 246
Fig. 11. Conformation of minor exo isomers.
Interestingly, 1 H NMR analysis showed that the endo product 236a was epimerised to exo product 236b (6:1 to 1:3, Fig. 12), when the diastereomeric mixture was kept in CDCh at
room temperature for 1 d.
89

a) 1 H NMR spectrum of DHP 236.
*7C/r;O
j&^JAi
I r .o 5.0 4.0
b) ! H NMR spectrum of DHP 236 after 1 d.
C-2. C-5proton proton
<c-.x-o)
3.0
.O 5.0 O 3.0
Fig. 12. Epimerisation of DHP 236.
This epimerisation from 236a to 236b can be explained by the formation of acyclic oxonium species 247 and the subsequent ring closure (Scheme 91). This observation suggests that the DHP 236b is the thermodynamic isomer with a pseudoaxial substituent at C-2 preserved by the anomeric effect as well as the C4-substituent being in the preferred pseudoequatorial orientation.
90

H
H
236a
OH
+ OnBu
247
On Bu
236b
Scheme 91. Epimerization ofendo DHP 236a to exo DHP 236b.
2.2.2. Diastereoselective formation of 2,3-c/s-tetrahydrofuransAfter having prepared various DHPs (232 and 236-240), we applied our DMDO oxidative
rearrangement/Jones oxidation sequence. Because the initial oxidative rearrangement by
DMDO could potentially give a mixture of eight products (four diastereoisomeric lactols
and four diastereomeric lactol ethers), we generally effected Jones oxidation of the crude
product from the DMDO step without attempting to separate the intermediates.
DHP 236 was examined first. Oxidative rearrangement with DMDO followed by direct
Jones oxidation of the crude product mixture (Scheme 92) gave a 3:1 mixture of
diastereoisomeric lactones according to ! H NMR analysis. Column chromatography
allowed separation of lactone 250a (35% over the two steps from DHP 236) and 250b
(16%).
O O248(15%) 249(11%)
O O250a(35%) 250b(16%)
Scheme 92. Oxidative rearrangement of DHP 236; (a) DMDO (1.0 eq), CH 2 C1 2 , 0 °C to rt, (b) i. DMDO (1.0 eq), CH 2C1 2 , 0 °C to rt, ii. Jones reagent (3.0 eq), acetone, 0 °C to rt.

The identity of the lactone products was supported by the values of 1787, 1723 cm' 1 for the
carboxyl and carbonyl groups, respectively, in the IR spectra. The stereochemistry of each
diastereoisomer was identified by NOESY experiments, showing the key correlations
between the protons at C-4 and C-5 for 4,5-c/s-lactones 250a (Fig. 13) and the key
correlation between the proton at C-5 and the proton in methyl group at C-4 for 4,5-trans-
lactones 250b (Fig. 14).
Fig. 13. NOESY of 4,5-c/s-lactones 250a Fig. 14. NOESY of 4,5-mm?-lactones 250b
Therefore, the NOESY experiment proved the favoured formation of cis isomer between
4,5-c/s-lactones 250a and 4,5-frww-lactones 250b. After we obtained this result, we
investigated the possible epimerisation between lactone 250a and 250b to confirm that this
stereoselective process reflected a kinetic product ratio. Firstly, we re-submitted 250a and
250b respectively under the Jones oxidation conditions overnight. Secondly, we kept each
of the pure lactone in chloroform for one week at room temperature. However, neither
diastereoisomer showed epimerisation under these conditions.
Other DHPs (232 and 237-240) were also oxidized by DMDO, followed by direct Jones
oxidation to give the corresponding lactones (235 and 251-254) without the purification of
the rearrangement products. The results for the DMDO oxidative rearrangement followed
by Jones oxidation are summarised in Table 14.
92

Table 14. Diastereoselective oxidative rearrangement; (a) i. DMDO (1.0 eq), CH 2 C12 , 0 °C to rt, ii. Jones reagent (3.0 eq), acetone, 0 °C to rt.
DHP
218
236
237
238
232
239
240
R 1
H
Me'Pr
Me
Ph
CH2OBn
(CH2)4CH=CHEt
R2
H
H
H
Me
H
H
H
R3
"Bu
"Bu
"Bu
nBu
Et
Et
Et
Tetrahydrofuranone (yield3, drb)
231
250a, 250b
251a, 251b
252
235
253a, 253b
254
(69%, N/A)
(53%, 3:1)
(63%, 9:1)
(50%, N/A)
(65%, -c)
(48%, 95:5d)
(64%, -c)
a Yield over two steps. The ratio of 4,5-cw-lactone to 4,5-trans lactone (based on H NMR analysis). c 4,5- c/5-product only. d Estimated from the 'H NMR spectrum.
The yields over two steps were generally reasonable (48-69%). Additionally, lactone 253a
was isolated from the mixture of diastereoisomers whereas 253b could not be isolated
because of the low yield. It was notable that during the oxidative rearrangement and Jones
oxidation, the benzylic ether moiety in lactone 253 was tolerated and the DHP 240 bearing
an isolated olefm was selectively oxidised. In terms of stereoselectivity, product lactones
(235, 251, 254) showed excellent diastereoselectivity (dr >9:1) and higher levels of
stereocontrol were observed with more sterically demanding and branched R 1 substituents.
The ratio of the two isomers was determined from the [ H NMR of the mixture and NOESY
experiments indicated the favoured formation of 4,5-cw-lactones (235, 251a, 253a, 254).

2.2.3. Explanation of observed stereochemistryAfter we found excellent diastereoselectivity in our oxidative rearrangement method, it was
necessary to explain this observed stereoselectivity. It was already mentioned that higher
levels of stereocontrol were observed with more sterically demanding and branched R 1
substituents. Thus, we believed that the stereoselectivity would be related to the approach
of DMDO. A model for our explanation is shown in Scheme 93.
o:^O^OR2
256(unfavoured)
258(favoured)
R
O257
4,5-frans-lactones (Minor)
R
2594,5-c/s-lactones
(Major)
Scheme 93. Proposed explanation of observed stereochemistry.
As previously explained, the major cycloadduct from the HDA reaction is the endo DHP
with 2,4-pseudoequatorial substituents. In order to explain the observed stereochemistry,
this conformer 255 should be adopted. When the DMDO was delivered from top-face (a) of
the DHP 255, there would be steric hindrance between DMDO and the substituent R 1 of the
DHP. Therefore, the formation of epoxide 256 would be less favourable and the 4,5-trans-
lactone would be the minor product 257. However, when the DMDO was delivered from
the bottom-face (b) of the DHP 255, there would be no steric hindrance between DMDO
and substituent R 1 of the DHP. Therefore, the formation of epoxide 258 would be favoured
and it would undergo rearrangement to give the major 4,5-c/s-lactone 259.
94

2.3. Synthesis of Quercus Lactone speciesTo apply our oxidative rearrangement method to natural product synthesis, we focused on a
simple target molecule, Quercus lactone species. Structurally, Quercus lactones are 4,5-
disubstituted tetrahydrofuranones and so they were ideal for our purpose. Quercus lactones
consist of whisky lactone 260 (Fig. 15) and cognac lactone 261 (Fig. 16).
Fig. 15. 4,5-c/s-whisky lactone 260 Fig. 16. 4,5-c/s-cognac lactone 261
These lactones were isolated from different types of wood and they were identified as key
flavours of aged alcoholic beverages such as whisky, brandy, wine and cognac. 108
Especially, the cis diastereoisomer defines the "vanilla", "coconut", and "chocolate" notes
in Chardonnay and Cabernet Sauvignon wines. 109 Additionally, the Taylor group reported
the synthesis of the four possible stereoisomers of whisky lactone. 110
2.3.1. Proposed synthesis of Quercus lactonesA variety of syntheses of racemic whisky lactone 111 and cognac lactone 11Z have been
reported in the literature. Additionally, a few examples of the enantioselective synthesis of
c/s-Quercus lactones have been reported. 1111 ' 111 "' 113 Representative methods are shown in
Scheme 94. The Suzuki group prepared (45l,5 1S)-cw-whisky lactone by a route involving the
synthesis of THFs and manipulation of the side chain (A, Scheme 94). llln Also, the
Chevtchouk group reported the synthesis of both (4S,5S)-cis and (4S,5R)-trans isomers by
Baeyer-Villiger oxidation of cyclobutanones (B, Scheme 94). 113a
95

HO,, CO2 H - 1>,O
n Bu(B)
Scheme 94. Representative examples of previous Quercus lactone synthesis; (a) Benzene, heat, (b) i. NaNO2 , AcOH-H2 O, 0 °C, ii. H2 , Pd/C, EtOAc, (c) MCPBA.
Compared to previous synthetic approaches, it seemed that our oxidative rearrangement
method would give a relatively short synthetic route toward Quercus lactones. Our strategy is shown in Scheme 95. The target molecule, Quercus lactone 262, could be obtained from the reduction of the ketone functional group of disubstituted lactone 263 and, for this purpose, we considered Wolff-Kishner reduction or Barton-McCombie deoxygenation of the derived alcohols. The lactone 263 would be easily synthesised by the oxidative rearrangement of DHP 264 with DMDO and subsequent Jones oxidation. The DHP 264 also would be made from the HDA reaction of enones 265 and enol ether 266.
V^
262 263
R'For whisky lactone; R=n Bu, R'=n Pr, For cognac lactone; R=n Pent, R'=n Bu
265 266
Scheme 95. Retrosynthesis for Quercus lactones.

2.3.2. Synthesis of tetrahydrofuranone precursors
According to our synthetic strategy, we prepared heterodienes 269 and 270 by Swern
oxidation of the commercially available alcohols 267 and 268 (Scheme 96).
OH
R267 R= nPr 269 R=n Pr268 R= nBu 270 R=n Bu
Scheme 96. Typical Swern oxidation to prepare enone 269 and 270; (a) i. DMSO, CH2C12 , (COC1)2 (1.1 eq), 10 min, ii. Et3N, 30 min.
Having prepared the enones, we synthesised DHPs 271 and 272 by HDA reaction (Scheme
97).
OEt R^ ^CT ^OEt
269 R=n Pr 271 R= n Pr, 70%270 R=n Bu 272 R= n Bu, 65%
Scheme 97. Preparation of DHPs from enones 269 and 270; (a) YbFOD (5 mol%), pressure tube, 5 d.
DHP 271 was isolated in 70% yield and DHP 272 was isolated in 65% yield. Based on 1 U NMR analysis, the ratio of endo:exo compounds was 4:1 for in both cases. After the
preparation of the DHPs 271 and 272, we subjected them to the oxidative
rearrangement/Jones oxidation conditions (Scheme 98).
97

OEt
271 R=n Pr,272 R=n Bu
273a R=n Pr, 58% 274a R=n Bu, 48%
273bR=n Pr, 12% 274b R=n Bu, 6%
Scheme 98. Diastereoselective oxidative rearrangement of DHPs 271 and 272; (a) i. DMDO (1.0 eq), CH2 C12 , 0 °C to rt, ii. Jones reagent (3.0 eq), acetone, 0 °C to rt.
Both substrates afforded a mixture of two lactone diastereoisomers. ! H NMR analysis of
the crude reaction mixture showed a ratio of 5:1 for lactones 273a and 273b, and a ratio of
8:1 for lactones 274a and 274b. After column purification of the mixture 273a and 273b,
the lactone 273a was isolated in 58% yield and the lactone 273b was isolated in 12% yield.
Similarly, after column purification of the mixture 274a and 274b, the lactone 274a was
isolated in 48% yield and the lactone 274b was isolated in 6% yield. The stereochemistry of
each diastereoisomer was proved by NOESY experiments. 4,5-czs-Lactones 273a and 274a
showed the key NOE relationship between two protons at C-4 and C-5 and did not show
the NOE relationship between the proton at C-5 and the proton in methyl group of C-4 (Fig.
17). However, 4,5-/r<ms-lactones 273b and 274b showed the key NOE relationship
between the proton at C-5 and the proton in methyl group of C-4 and did not show the
relationship between two protons at C-4 and C-5 (Fig. 18).
Fig. 17. NOESY of 4,5-cw-lactones 273a (R=nPr) Fig. 18. NOESY of 4,5-/ra«s-lactones 273b (R=nPr) and 274a (R=nBu). and 274b (R=nBu).
98

2.3.3. Quercus lactones
To obtain the natural products, 4,5-c/s-Quercus lactones, it was necessary to reduce the
ketone functional group of lactones 273a and 274a. We considered two reduction method
methods, Wolff-Kishner reduction (route a in Scheme 99) and Barton-McCombie
deoxygenation of the derived alcohols (route b in Scheme 99).
Wolff-Kishner reduction
Route a)
Barton-McCombie deoxygenation
Scheme 99. Two ideas for reduction of ketone moiety.
From literature precedent, we found that a wide range of carbonyl functions could be
reduced by modified Wolff-Kishner conditions using NaBHaCN reduction of
tosylhydrazones. 114 We therefore attempted to form the tosylhydrazone of the 4,5-cis-
lactone 273a (Scheme 100).
*—273a NHTs
Scheme 100. Attempted Wolff-Kishner reduction; (a) TsNHNH2 , EtOH, it, 2 d.
99

However, the reaction showed little conversion even after 2 d at room temperature. Only a
trace of the hydrazone product was detected by the TLC and *H NMR analysis. Thus, we
moved our attention to Barton-McCombie deoxygenation. 115 Before the deoxygenation, we
needed to prepare thiocarbonyl compounds from the ketone 273a and 274a. The reduction
of the ketones 273a and 274a with NaBH4 successfully gave the alcohols 275 and 276 as a
1:1 mixture of diastereoisomers. Subsequently, the diastereoisomeric alcohols 275 and 276
were converted into the thiocarbonyl compounds 277 and 278 by treatment with
thiocarbonylimidazole at 80 °C (Scheme 101).
s273a R=nPr, 275 R=nPr (87%) 277 R=nPr (50%) 274a R=nBu 276 R=nBu (70%) 278 R=nBu, (77%)
Scheme 101. Preparation of thiocarbonyl compounds; (a) NaBH4 (1.0 eq), MeOH, it, overnight, (b) thiocarbonyldiimidazole (1.5 eq), DCE, 80-100 °C, overnight.
Finally, Barton-McCombie deoxygenation of thiocarbonyl compounds 277 and 278 gave
4,5-czs-whisky lactone 260 in 83% yield and cognac lactone 261 in 91% yield (Scheme 102) and the spectral data of two lactones 260 and 261 matched the literature data. 112f' 116
Thus, we had successfully applied our oxidative rearrangement method to synthesise 4,5- c/5-whisky lactone 260 and 4,5-ds-cognac lactone 261. Because of this successful result,
we were encouraged to try to widen the scope of our oxidative rearrangement method for
the preparation of 4,5-frww-THFs.
100

s277 R=n Pr,278 R=n Bu
260 R=n Bu (83%),261 R=n Pent(91%)
Scheme 102. Barton-McCombie deoxygenation; (a) "Bu3 SnH (1.1 eq), (0.1 eq), toluene, rt, overnight.
101

2.4. Attempted Prevost fraws-dihydroxylationIn spite of the successful synthesis of 4,5-c/s-tetrahydrofuranones by our novel
rearrangement method, it would be desirable to be able to synthesise 4,5-trans-
tetrahydrofuranones to make this method more universal. Because DMDO epoxidation
conditions gave the cis tetrahydrofuranone as a major product, we would need alternative
oxidation conditions. Towards the end of this project, we found that Sudalai et.al reported
Prevost trans dihydroxylation of olefms mediated by PhI(OAc)2 and LiBr (Scheme 103). 117
In this paper, they controlled the formation of syn diol 279 with NaIO4 and LiBr and anti diol 280 with PhI(OAc)2 and LiBr.
OH R2 R2b, c a, c
R
279 280
Scheme 103. Prevost-Woodward reaction; (a) PhI(OAc)2 (1.0 eq), LiBr (20 mol%), AcOH, 95 °C, 18 h, (b) NaIO4 (30 mol%), LiBr (20 mol%), AcOH, 95 °C, 18 h, (c) K2CO3 , MeOH, 25 °C, 24 h.
Mechanistically, the alkene reacts with bromine, generated in situ from alkali metal halides
by oxidation with NaIC>4 or PhI(OAc)2, and undergoes bromoacetoxylation via bromonium
ion 281. The intermediate species 283 from the acetal formation of trans-\,2-
bromoacetoxylation 282 gives the anti diol 280 by the ring opening with acetic acid
(Scheme 104). Also, the ring opening of intermediate species 283 by water gives the syn
diol 279 without the inversion of configuration.
102

OAc9 \
———— - ————— *\
yr°"R 1
283
i TT R2J . J, VI^ff ' + °V^ ''Or1 1 Br T br
281 282
R2 R2 . AcO, A. b HO./J',
y UAc y OHR1 R 1
284 280
Scheme 104. The formation of anti diol 280; (a) PhI(OAc)2 (1.0 eq), LiBr (20 mol%), AcOH, 95 °C, 18 h, (b) K2C03 , MeOH, 25 °C, 24 h.
Although this chemistry does not proceed via an epoxide, we speculated that intermediates
similar to 283, or the 1,2-diols, might be able to undergo a similar rearrangement leading to
a THF product. Moreover, the initial attack of bromine on the less hindered face followed
by opening of the brominium ion with inversion should lead to formation of the trans-THF,
by analogy to the NBS-mediated aziridination process developed earlier in the group. We
therefore undertook a test reaction with DHP 237 under these /rajw-dihydroxylation
conditions using PhI(OAc)2 and LiBr (Scheme 105).
x ' V^ + unidentified SO O nBu products
trace
Scheme 105. Attempted Prevost dihydroxylation; (a) PhI(OAc)2 (1.0 eq), LiBr (20 mol%), AcOH, 95 °C, 18 h, (b) K2CO3 , MeOH, 25 °C, 24 h.
Unfortunately, the reaction gave only a trace of rearrangement products along with several
other unidentified products. Additionally, we could not determine the stereochemistry
because of the complexity of the ! H NMR spectrum caused by the presence of three
103

stereocentres in the product. However, the formation of THFs at least to some extent was a
promising result and this process could potentially be optimised as part of future work.
104

2.5. Preparation of cyclopentanes by cyclopropanation/ rearrangement 2.5.1. Previous work in this fieldUsing the same rearrangement concept, we reasoned that reaction of DHPs with other electrophiles rather than oxygen and nitrogen could give a wide range of 5-membered ring products, and one possible reaction was the rearrangement of DHP by cyclopropanation leading to cyclopentanes. In fact, there was a literature precedent supporting this idea. Alonso and co-workers studied addition/rearrangement of dimethyl diazomalonate to DHPs (Scheme 106). 118 In this work they showed this reaction gave a diastereoisomeric mixture of rearrangement products 286 as minor products and insertion product 287 as a major product (1:2.5) and they proposed both products could arise from the ring opening of common cyclopropane intermediate.
CO2Me + N 2=(
QT ^OMe c02 Me285a
^
MeO2C-MeO2C QMe
286
CO2Me
OMe
287
Scheme 106. Addition/rearrangement of dimethyl diazomalonate to DHP 285a by Alonso; (a) Cu(F6 acac)2 , benzene, 1: 2.5 (the ratio of 286 to 287), 81% (total yield).
Also, Alonso and co-workers reported the influence of catalysts such as copper based catalysts and rhodium based catalysts on product composition 119 and selected representative examples are shown in Table 15. In this work, they found that the rearrangement product 286 was obtained as the major compound (52%, mixture of diastereoisomers) when Cu(F6acac)2 was used as a catalyst (entry 2) and other catalysts gave the rearrangement product as a minor product (entry 1 and 3).
105

Table 15. Influence of catalyst on composition of products in cyclopropanation; (a) ML,,, fluorobenzene.
285a
+ N2 =
Entry
1
2
3a For the
CO2Me
CO2 Me
MLn
Cu(acac)2
Cu(F6acac)2
Rh2OAc4
a
Time
8.5 h
4.0 h
2.5 h
1 cc° 1> MeO C A MeO2C-j — / + e 2MeO2C QMe '
286
Yield of 286a
5%
52%
12%
Yield of 287
75%
41%
39%mixture of diastereoisomers.
OMe
287
Apart from Alonso's work, there were some earlier preliminary studies in our group (Table 16). 120 Firstly, our group undertook the reaction of DHPs (285b and 211) with ethyl diazoacetate and catalytic Cu(acac)2 to give cyclopropanes (288 and 289) in a reasonable yield (67-72%). The resulting cyclopropanes were then treated with HC1 to promote the rearrangement and the cyclopropane 288 rearranged to the cyclopentane 290 in 68% yield. However, the cyclopropane 289 did not rearrange to give the cyclopentane and the ring system was opened to give 291 under these acidic conditions. Additionally, the unnecessary stereocentres at C-2 and C-7 in cyclopropanes (288, 289) and cyclopentane 290 caused difficulty in the stereochemical analysis and the reaction did not seem to be diastereoselective. Therefore, we decided to use dimethyl diazomalonate for the cyclopropanation as this would remove one unnecessary stereocentre at C-7. Furthermore, it was expected that two electron withdrawing ester moieties in diazomalonate might increase the reactivity of the cyclopropanation and the rearrangement to the cyclopentane.
106

Table 16. Cyclopropanation of DHP; (a) Cu(acac)2 (0.05 mol%), CH2 C12 , 90 °C, (b) 1M HC1 (aq), CH3 CN, rt, 20 min.
R'
285bR1 =H, R2 =Me 211 R 1 =Me, R2=Et
N2=<COOEt
H
R 1 'e
OEt OH
290R1 =H
O.
EtO
7/T5
288R1 =H, R2 =Me 289R1 =Me, R2 =Et
R 1
OEt
291 R 1 =Me
DHPs Cyclopropane (yield, dr) Rearrangement product (yield)
285b 288(67%, 1:1.3)
211 289 (72%, 1:1.4)
290 (68%)
291 (74%)
2.5.2. Attempted Cyclopropanation with diazomalonateIn contrast to ethyl diazoacetate, dimethyl diazomalonate 293 was not commercially
available and we therefore synthesised it using a known procedure (Scheme 107). 121
SO2CI+ NaN3
N
SO2N:
O O
292
O O 293
Scheme 107. Synthesis of dimethyl diazomalonate; (a) acetone/water (1:1), 40 °C, 3 h, (b) Diethylamine, Et2O,0 °C.
107

Firstly, p-toluenesulfonyl azide 292 was prepared via the reaction between p-
toluenesulfonyl chloride and sodium azide in 80% yield and it was identified by mass
spectroscopy (215 for M+NH4). Subsequently, p-toluenesulfonyl azide 292 reacted with
dimethyl malonate to give dimethyl diazomalonate 293 in 60% yield and the product's
spectroscopic data were in accordance with those in the literature. 12113 The diazomalonate
293 was then applied to the cyclopropanation of the commercially available DHP 285b (Scheme 108). The reaction conditions were exactly same as in the previous procedure (condition a, Table 16) except that diazomalonate was used.
COOMe a -. ,-F ^ + ... __/ _________^ unidentified.^x1^-... 2 \ *" products O OMe COOMe
285b 293
Scheme 108. Attempted cyclopropanation with diazomalonate; (a) Cu(acac)2 (0.05 mol%), CH2 C1 2 , 90 °C.
The reaction was monitored by TLC and DHP 285b was completely consumed within 4 h.
Although TLC analysis indicated the formation of four major products, only two of them could be isolated by chromatography. Unfortunately, *H and 13 C NMR spectral data were not clean. However, from ! H NMR analysis, we could see singlets at 3.74 ppm and 3.78 ppm indicating that the dimethyl malonate portion had been incorporated. Additionally, in mass spectroscopic analysis, we observed m/z at 245 (100%) for both fractions and this value matches to M+H for the 3 possible products in Fig. 19.
MeO2C QMe
Fig. 19. Three possible isomers from cyclopropanation.
108

Since Alonso showed the formation of cyclopentane from DHP 211 with Cu(p6acac)2 in
resonable yield (52%), we decided to change our reaction conditions (Scheme 109). DHP
285b and Cu(acac)2 were changed to DHP 211 and Cu(F6acac)2 .
CO2Me
295 (20%)
Scheme 109. Cyclopropanation of DHP 211 catalysed by Cu(F6acac)2; (a) diazomalonate (1.2 eq), Cu(F6acac)2 (0.05 mol%), 75 °C, 4 h.
The cyclopentane 294 was obtained as a mixture of stereoisomers (1:1) in ca. 20% yield
and the insertion product 295 was obtained in ca. 20% yield. The combined yield (ca. 40%)
might be increased by the use of more equivalents of diazomalonate because Alonso used
2.0 eq of diazomalonate to 1.0 eq of DHP and obtained a combined yield of 93%. We could
not obtain completely clean spectral data for compounds 294 and 295 even though they
were isolated as one single spot by TLC analysis. However, we could find some evidence
supporting their structures. For compound 294, two carboxyl group at 169.7 and 169.4 and
one carbonyl group at 167.1 were found in 13 C NMR analysis and 273 (M+H) was found in
the mass spectrum. For compound 295, a triplet with 7=3.5 Hz for the proton at C-2 was
found at 5.00 ppm and singlet for the proton at C-7 was found at 4.32 in ! H NMR analysis.
Moreover, two carbons in double bond at 147.1 and 109.8 and two carboxyl group at 169.2
and 168.9 were found in 13C NMR analysis and the mass spectrum again showed a peak at
273 (M+H). In fact, the ! H NMR spectrum of product 294 also showed a trace of aldehyde,
suggesting that the product 294 could be ring-opened under the slightly acidic CDCls
conditions.At this stage, we decided to suspend this cyclopropanation/rearrangement project because
of the difficulties in obtaining pure products and we therefore turned our attention to the
enantioselective oxidation/rearrangement. However, this process is potentially useful to
109

synthesise cyclopentanes and could merit further investigation. For example, the
modification of the substrate DHP may be effective to favour the formation of the
rearrangement product. In the cyclopropanation, the formation of the rearrangement
product would be competitive with the insertion product and we may need DHP 296
containing bulky substituent such as Ph (Scheme 110). Sterically bulky DHP 296 may
prevent the formation of addition product 297 by steric hindrance (pathway a) and would
favour the formation of rearrangement product 298 (pathway b). So, this cyclopropanation
of DHP containing bulky substituent should be pursued further for the future work.
COOMeMeOOC-^ H
Me02Q
R
pathway a
MeO2C\R
Me02C^
pathway b
O OEt297
Insertion product (unfavoured)
C09Me298
Rearrangement product (favoured)
Scheme 110. Idea to obtain rearrangement product 298.
110

3. Conclusions.The initial aim of this project was the preparation of THFs by the epoxidation of DHPs and
the subsequent rearrangement of epoxide intermediates. Fortunately, we were able to
optimise the reaction conditions and this oxidative rearrangement successfully gave THFs
in good yield using DMDO as oxidant (Scheme 111).
R
DMDO epoxidation
R 1R
OOR'
O
ring opening rearrangement
Scheme 111. Idea of oxidative rearrangement.
Furthermore, we investigated the diastereoselectivity of the oxidative rearrangement. For
this study, we needed to remove the unnecessary lactol stereocentre from the 2,5-
disubstituted THFs and this was achieved via Jones oxidation, giving 5-substituted
tetrahydrofuranones. After we established this typical procedure for the synthesis of
tetrahydrofuranones from DHPs, we were able to synthesise various 4.5-disubstituted
tetrahydrofuranones (Scheme 112).
1 eq DMDO
2 acetone /
Jones oxidation
Scheme 112. Diastereoselective synthesis of 4,5-cw-tetrahydrofuranone.
Higher levels of stereocontrol were observed with more sterically demanding and branched
111

R substituents and NOE studies indicated that the 4,5-c/si-tetrahydrofuranones were the
major product. This diastereoselectivity was explained by the size of substituent R 1 and the
direction of the approach of DMDO to the olefm. Finally, we applied our novel oxidative
rearrangement to the synthesis of natural products, the Quercus lactones. After the isolation
of 4,5-c/s-tetrahydrofuranones, we successfully synthesised whisky lactone (36% over 3
steps) and cognac lactone (49% over 3 steps) by the reduction of tetrahydrofuranones,
followed by Barton-McCombie deoxygenation of derived alcohols (Scheme 113).
1.NaBH4 , MeOH
2. (lm)2CS, DCE, 80 °C3. nBu3SnH, AIBN,
R=n Pr, n Bu toluene
Scheme 113. Synthesis of Quercus lactones.
Additionally, we attempted the synthesis of 4,5-trans-THF using Prevost dihydroxylation
method and we also adapted our rearrangement concept for the synthesis of cyclopentanes:
attempted cyclopropanation of DHPs gave mixtures of the insertion product and the
rearrangement product.
This whole section dealt with racemic material. Extension of the chemistry to
enantioselective synthesis was an important goal and this will be discussed in chapter II.
12

Chapter II. Enantioselective oxidative rearrangementof 2-alkoxydihydropyrans
113

1. IntroductionPreviously, we discussed the diastereoselective formation of tetrahydrofuranones using
oxidative rearrangement of DHPs. In this chapter, the enantioselective formation of
tetrahydrofuranones will be discussed. We considered two possible routes to synthesise
tetrahydrofuranones enantioselectively. Firstly, enantiomerically pure tetrahydrofuranones
could potentially be obtained by using an enantioselective HDA reaction, followed by the
established oxidative rearrangement method by DMDO (Scheme 114. route a). Secondly,
the enantiomerically pure tetrahydrofuranones could be obtained by using an
enantioselective epoxidation step after the formation of racemic DHPs (Scheme 114. route
b).
R'
enantioselective HDA
OR2
HDAR1 O
1) epoxidation
O OR^ 2)Jonesoxidation route (a)
1) enantioselective 1 epoxidation R
2)Jones Qoxidation route (b)
Scheme 114. Two ideas for enantioselective synthesis of tetrahydrofuranone.
114

1.1. Asymmetric HDA reactionIt is really attractive to synthesise chiral tetrahydrofuranones via the preparation of chiral
DHPs by asymmetric HDA because the chiral DHP precursors could afford disubstituted
tetrahydrofuranones containing two chiral centres in the oxidative rearrangement. From a
literature search, we found two asymmetric HDA reactions catalysed by chiral Lewis acids
which may be appropriate for our purpose.
1.1.1. Hetero Diels-Alder reaction under Cr3+ catalysisRecently, the Jacobsen group reported an enantioselective HDA reaction between ethyl
vinyl ether and a range of a,|3-unsaturated aldehydes affording the product DHP in 70-95%
yield and 89-98% ee using Cr3+ catalyst 299 (Scheme 115). 122
RR2
OEt
R 1 = H, Br, Me, R2=alkyl, Ar
'OEt
\O Cl
Cr3* catalyst 299
3+Scheme 115. Hetero-Diels-Alder reactions by Cr3+ catalysis; (a) CrJ+ catalyst (5 mol%), neat, rt, yield 70-95%, ee 89-98%.
However, the reaction was only performed with ct,|3-unsaturated aldehydes and we could
not find any examples for its application to a,p-unsaturated ketones. For the
diastereoselective oxidative rearrangement, most of our DHP substrates were prepared from
a,p~unsaturated ketones and some DHPs from a,p-unsaturated aldehydes were found not to
undergo the rearrangement. Thus, we would need to investigate the application of
Jacobsen's HDA reaction to a,p-unsaturated ketones, if we wanted to adopt this method
15

1.1.2. Hetero Diels-Alder reaction of phenylsulfonyl enones under Ti4+ catalysisThe Wada group reported an enantioselective HDA reaction between a range of vinyl ethers and a,p~unsaturated phenylsulfonyl ketones affording the product DHP in 77-96% yield and 59-97 % ee, using Ti4+ catalyst 300 (Scheme 116). 123
R 1
PhO2SO
R1 = Me, jPr, Ph, R2=alkyl
R 1
PhO2S CT ''OR2 XPh
Ti4+ catalyst 300: X=CI, Br
Scheme 116. Hetero Diels-Alder reactions by Ti4+ catalysis; (a) Ti4+ catalyst (5-10 mol%), MS 4A, CH2 C12 ,30, -50 or -78 °C, yield 77-96%, ee 59-97%.
The reaction was successfully performed with a,p-unsaturated ketones but the scope was limited to phenylsulfonyl ketones and it would be necessary to prepare phenylsulfonyl enones 302 via the reaction of dianion 301 with aldehydes (Scheme 117).
O ___a ,SO2 Ph ————OH O
S02 Ph SO2 Ph
301
OSO2Ph
302
Scheme 117. Preparation of phenylsulfonyl enone; (a) LDA (2.0 eq), 0 °C, THF, (b) RCHO, -78 °C, (c) p- TsOH, reflux, C6 H6 .
116

1.2. Asymmetric epoxidation methodsFor the enantioselective oxidative rearrangement, chiral tetrahydrofuranones could be still
obtained by asymmetric epoxidation method even though it would result in the formation of only one stereocentre, in contrast to the formation of two stereocentres by the asymmetric
HDA route. For the asymmetric epoxidation, we considered two representative catalysts for
the asymmetric epoxidation of DHP substrates. They were a fructose-derived chiral ketone catalyst by Shi 124 and a chiral salen manganese catalyst by Jacobsen 125 because both
catalysts are commercially available and show good reactivity and enantioselectivity with a variety of olefins.
1.2.1. Fructose-derived chiral ketone catalyst by Shi.In 1996, a fructose-derived ketone 304 was developed as an effective epoxidation catalyst
by the Shi group. 126 This catalyst is readily obtained from commercial sources or from very inexpensive D-fructose 303 by ketalisation and oxidation (Scheme 118).
^ OHr0vf-I !"•
HO^'S^OH
OH
D-fructose 303fructose-derived catalyst 304
Scheme 118. Preparation of fructose-derived ketone catalyst; (a) acetone, 0 °C, 53%, (b) PCC, CH2 C1 2 , rt,93%.
The structural feature of the catalyst is that stereocentres are close to the reacting centre allowing control of the approach of an olefin to the reacting dioxirane by sterically blocking
one face. The stereochemistry of epoxidation can then be explained by s/?/ro transition
states, in which 7i-electrons of the olefin attack the o -orbital of the dioxirane and the lone-
paired electrons of oxygen interact with the n -orbital of the olefin to give the epoxide (Fig. 20). l27
17

favoured unfavoured
Fig. 20. Two spiro transition states to explain stereochemistry of epoxidation.
Additionally, the fused ring and quaternary centre at the a-position to the ketone prevents
the epimerisation of the stereocentre and the inductively electron withdrawing substituent
increases the catalytic activity.
The catalyst's efficiency often shows high dependence on the reaction pH due to the
sensitivity of the catalyst to decomposition via Baeyer-Villiger reaction (Scheme 119),
control of pH (typically pH 7-8) can minimise this side-reaction. 128
HSO,
so42-
Baey.er-villi9er o\Jreaction
Hydrolysis
O
Scheme 119. Decomposition of catalyst via Baeyer-Villiger reaction. 12 '
118

Under controlled pH conditions, the fructose-derived ketone 304 shows excellent catalytic
activity and enantioselectivity in the epoxidation of a wide range of olefms. 129 Representative results are shown in Table 17.
Table 17. Representative result of epoxidation by fructose derived chiral ketone 304; (a) catalyst 304 (30 , Oxone, H2O/Me3 CN.
Entry
1
2
3
4
5
6
7
8
9
10
11
12
13
14
R 1
Ph
Ph
Ph
PhPh
C 10H21'Bu
Ph
Me
H
H
H
MeH
R 1
R3
R2
H
H
H
H
Me
Me
Me
Ph
C8Hn
R 1a >C
—————————— R2 X\-)
R IR3
RJ Yield (%)
Ph
Me
CH2OTBS
C2H4CO2Me
Me
Me
Me
(CH2)4
(CH2)4
H
H
iPr3 SiCH2 H
Ph H
(CH2)4
78
94
83
76
89
97
35
94
77
90
80
92
81
85
ee (%)
99
96
95
91
97
87
91
98
81
24
27
35
28
32
The catalyst works well with rnms-disubstituted olefms (entries 1-4) and trisubstituted
olefms (entries 5-9). Additionally, the reaction tolerates enol silylethers (entry 3),
ct,p-unsaturated esters (entry 4) and cyclic olefms (entries 8-9). However, the epoxidation
of c/s-disubstituted olefms (entries 10-13) and terminal olefms (entry 14) gave low ee.

1.2.2. Chiral manganese salen catalysts.
Since Jacobsen 130 and Katsuki 131 independently reported asymmetric epoxidation of
conjugated olefins catalysed by chiral salen manganese catalysts in 1990, various salen
manganese complexes (305-307) have been further improved (Fig. 21). The structural
features of the catalyst are C2-symmetry and two sp3 carbon stereocentres at the
ethylenediamine moiety. The catalysts show excellent catalytic activity and
enantioselectivity in the epoxidation of a wide range of olefins and representative examples
of the asymmetric epoxidation by chiral salen manganese catalysts from Jacobsen are
shown in Table 18.
Table 18. Representative results from epoxidation by chiral salen manganese catalyst; catalyst (<8 mol%), additives (0-20
Ph Phr~\/=N\ /N=\
R R305 R=H, 306 R-lBu
mol%), oxidant (NaOCl or ArlO).
R 1
RA
Entry catalyst
/~\_K_
/=\ Mn ^ — ̂ Bu1 — ̂ . ^ — QX NO — ̂ y— lBu
lBu Bu 1307
1
2
3
4
5
6
7
8
Fig. 21. Salen manganese catalysts. 910
B
C
A
A
B
B
C
C
C
C
1 R3
R 1
H
H
Ph
Ph
Ph
Ph
Ph
Ph
Ph
Ph
a
R1
Ph
Ph
H
H
H
Me
Me
Ph
Ph
R 1JL;
R3
Me
C02Et
Ph
Me
H
H
Ph
Ph
Me
(CH 2 ) 3
>Q
R3
Yield
73%
96%
63%
93%
75%
36%
69%
97%
91%
75%
ee
84%
93%
33%
20%
57%
30%
93%
92%
95%
86%
The catalyst works well with cis-disubstituted olefins (entries l' 30-2 132 ) and trisubstiruted
120

oleflns (entries 7-10). 133 The reaction also tolerates cyclic olefms (entry 10) and enoates
give a high level of enantioselectivity (entry 2). However, the epoxidation of trans-
disubstituted olefms is slow and a loss of selectivity is reported (entries 3-4) and also
terminal olefms (entries 5-6) are poor substrates. 130 Additionally, the epoxidation of various
chromene derivatives by salen manganese catalyst 307 shows excellent enantioselectivity (Scheme 120). 134
R2 R3R
R2R3
Scheme 120. Epoxidation of chromene derivatives by chiral salen manganese catalyst; (a) catalyst 307 (4 mol%, Fig. 2), NaOCl, yield 51-96%, ee 94-98%.
The enantioselectivity is induced through the interaction between the chiral salen ligand
and the incoming olefin (Fig. 22). In most cases, the observed stereoselectivities are
explained well by a side-on approach of the olefin (Fig. 22, a).
L S
Fig. 22. Plausible olefin approaches.
121

1.3. Concept of this projectFor the enantioselective oxidative rearrangement, we chose to study the asymmetric
epoxidation pathway to prepare chiral tetrahydrofuranones (Scheme 121) rather than the
asymmetric HDA reaction, because the epoxidation methodology was straightforward to
use, whereas the HDA reaction would either have to be extended (Cr3+ catalysis) or would
require substrate synthesis (Ti4+ catalysis).
1) enantioselective 1 epoxidation R\^^*O'
R1 O OR^ 2) Jonesoxidation
Scheme 121. Concept of enantioselective oxidative rearrangement via asymmetric epoxidation.
As mentioned above, the chiral ketone catalyst by Shi shows excellent catalytic activity and
ee for the epoxidation of trisubstituted cyclic olefins. The salen manganese catalyst also
gives good results with these substrates. Additionally, these catalysts are either
commercially available or easy to prepare. Thus, we decided to adopt these two catalysts
for our oxidative rearrangement.
122

2. Results and discussion
2.1. Optimisation of enantioselective oxidative rearrangement2.1.1. Enantioselective epoxidation by Shi's catalyst.Given the earlier successful oxidative rearrangement using DMDO, the use of chiral ketone catalysts for formation of chiral dioxiranes was an attractive possibility. The Armstrong group has developed several enantioselective chiral ketone catalysts, but earlier work has shown that these generally give very poor enantiomeric excesses for epoxidation of enol ethers. 135 Hence we elected to use Shi's catalyst 304 because of its ability to epoxidise enantioselectively various olefin systems, including enol ethers and esters. 126 ' 129 ' 136 Shi's catalyst was commercially available as a D-epoxone® from Alfa Aesar. The results from attempted epoxidation of DHPs with D-epoxone® is shown in Table 19. The DHP substrates were available from the previous diastereoselective oxidative rearrangement studies (DHP 211 and 237) or were easily synthesised using the HDA reaction (DHP 308). After the preparation of these DHPs, we tested epoxidation under two different pH conditions because Shi's catalyst is known to be sensitive to this parameter.
Table 19. Attempted epoxidation of DHPs with Shi's catalyst 304; (a) Oxone (5.0 eq), NaHCO 3 (15.5 eq), Shi's catalyst (3.0 eq), CH3CN, 0 °C, (b) Oxone® (1.4 eq), NaHCO3 (5.0 eq), Shi's catalyst (0.3 eq), nBu4NHSO3 (0.04 eq), CH3 CN, 0 °C.
conditions a or b——x——O' OR"
Entry
1
2
3
4
DHP Conditions3 R 1
211
211
237
308
a
bbb
Me
Me
Me
Ph
R2
H
H
H
H
Rj
H
H'Pr
H
R4
Et
Et"Bu
EtConditions a: pH~7, Conditions b: pH~10.5
Initially, we carried out the epoxidation of DHP under pH~7 (entry 1, conditions a).
123

However, this was not successful because TLC analysis showed largely starting material
with several unidentified weak spots (entry 1). In line with literature precedent, 129 we
assumed that catalyst decomposition by Baeyer-Villiger reaction at the pH employed was a
potential cause of the poor results. In an attempt to solve this problem, we initially
increased the amount of catalyst from 3.0 eq up to 5.0 eq and the reaction time up to
overnight. However, the desired product was still not observed. Thus, we changed the
reaction pH to 10.5-11.0 (entries 2-4, conditions b) because higher pH conditions are known to prevent the decomposition of the catalyst. 129 Unfortunately, higher pH conditions
still did not give any of the desired product, showing the same outcome on TLC inspection (entry 2). Other DHPs containing a tri-substituted olefin were also attempted (entries 3-4)
but they also did not give the expected products, although they had previously been epoxidised under DMDO conditions in good yield. Especially, it was disappointing that the
substrate with a phenyl-substituted alkene (entry 4) was not epoxidised, because aryl
alkenes are generally good substrates for Shi epoxidation. 129 In view of these disappointing
results, we decide to turn our attention to the Jacobsen epoxidation.
2.1.2. Enantioselective epoxidation by Jacobsen's catalyst.The Jacobsen chiral manganese salen catalyst is able to epoxidise a wide range of olefin
systems. 125 Both enantiomers of Jacobsen's catalyst are commercially available from Strem
and we adopted (/?,^)-Jacobsen's catalyst 307 for the epoxidation. Because we would
epoxidise a trisubstituted olefin in our DHPs, we followed the procedure for the epoxidation of trisubstituted olefins by Jacobsen. 133 1.5 eq of NaOCl was used as co-
oxidant and 0.2 eq of pyridine oxide was used as an additive (Table 20) because this
generally increased both ee and yield in Jacobsen's studies. Pleasingly, the oxidised
rearrangement products, tetrahydrofuranones (231, 252, 309), were obtained after the
epoxidation of DHPs with Jacobsen's catalyst followed by Jones oxidation. Perhaps not
surprisingly, the simple substrate 211 afforded essentially racemic product 231, as indicated
by its optical rotation. At this stage, we postulated that either the replacement of the proton
at C-4 with a sterically bulkier group or the replacement of the methyl at C-6 with an
124

aromatic group may lead to improved enantioselectivity. Firstly, we attempted the
epoxidation of DHP 238 bearing two methyl substituents at C-4 (entry 2). However, this still gave racemic product 252, as indicated by its optical rotation, as well as a low yield
(16%). In spite of this unfavourable result, we attempted the epoxidation of DHP 308 with a phenyl substituent at C-6. Pleasingly, we could obtain tetrahydrofuranone 309 with 38% ee
in 55% yield (entry 4). This result suggested the importance of the Ph substituent at C-6 which was adjacent to the olefm, rather than the bulkiness of substituent at C-4, which was one carbon away from the olefm.
Table 20. Epoxidation of DHPs with Jacobsen's catalyst 307; (a) 4-phenyl pyridine N-oxide (0.2 eq), (R,R)- Jacobsen's cat (0.05 eq), NaOCl (1.5 eq), CH2C1 2 , 0 °C to rt, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt.
D2 R3 N^A
a.b RiX(/ °
O
Entry DHP
1 211
2 238
3 308
R1
Me
Me
Ph
R2
H
Me
H
R3 R4 Tetrahydrofuranone (Yield3) ee
H
Me
H
Et"Bu
Et
231
252
309
(40%)
(16%)
(55%)
0%b
0%b
38%c
1 Over 2 steps. Based on the optical rotation observation. c Based on HPLC analysis.
125

2.2. Enantioselective oxidative rearrangement by Jacobsen's catalyst.After finding the importance of the phenyl substituent at C-6, we decided to investigate
more closely the effect of the DHP substituents on the product ee (Fig. 23). Specially, we
wished to explore the electronic effect of the aryl substituent at C-6 and the steric effect of
bulky substituents at C-4. For this goal, we first needed to synthesise various phenyl enones
to allow access to the DHP substrates via HDA chemistry.
R2̂ ,R3 ^-
i) tuning of aromatic group
i) tuning of alkyl group
Fig. 23. Idea for tuning of DHP.
2.2.1. Preparation of various phenyl enonesThe required aryl enones were synthesised through various routes such as Mukaiyama aldol
reaction, Friedel-Crafts acylation, the combination of Grignard reaction and oxidation and
Rupe rearrangement (Fig. 24).
QTMS O
R2 R3
Mukaiyama aldol reaction
R3 O
Friedel-Crafts acylation
Grignard reaction / MnO2 oxidation
Ph
Rupe rearrangement
Fig. 24. Various routes toward synthesis of various enones.
In the beginning, we were interested in the effect of the bulkiness of substituents at C-4 of
DHPs. From a literature search, we found that Mukaiyama aldol reaction gave various
126

bulky enone systems in a one-pot procedure 137 and we prepared enones 310-312 in 44-65%
yield (Table 21).
Table 21. Mukaiyama aldol reaction; (a) i. TiCl4 (1.0 eq), CH,C1 2 , rt, 2 h; ii. TFAA (1.0 eq), rt, 1 h; iii. TEA (2.0 eq), rt, 2 h, iv. H2O
R 1
EntryV 1
^R1 2
} 34
5
R 1
Me
Et
cyHex'Pr
Ph
Product (yield)
310 (44%)
311 (50%)
312 (65%)N/Aa
N/Aa
No reaction
However, extremely bulky substrates (entry 4 and 5) did not give the corresponding product.
For the synthesis of bulky substrate 314, Rupe rearrangement 138 provided an alternative
method. Commercially available tertiary a-acetylenic alcohol 313 was successfully
isomerised to a,(3-unsaturated carbonyl compound 314 via a 1,3-shift (Scheme 122).
O Ph
314
Scheme 122. Rupe Rearrangement; (a) H2 SO4 (1.5 eq), AcOH, 110 °C, 1 d, 55%.
After the preparation of phenyl enones (310-312 and 314), we needed a further set of
enones to investigate the electronic effect of various substituents on the aromatic ring.
However, neither Mukaiyama aldol reaction nor Rupe rearrangement seemed to be a good
choice because of the difficulty in the preparation of the requisite stating materials. At this
stage, we considered an alternative approach using Wittig olefmation (Table 22).
127

Table 22. Attempted Wittig olefmation; (a) CH3 PPh3 Br (3.0 eq), PhLi solution in Et2 O (1.80 M, 3.0 eq), THF, -78 °C to rt, overnight, (b) acetone (excess), toluene, 110 °C.
Cl a ^pph
Entry Ar Ylide (yield)
1 p-methoxyphenyl 315 (95%)
2 p-chlorophenyl 316(60%)
To prepare the representative examples of electron poor aryl enone and electron rich aryl
enones, we synthesised ylides 315 and 316 in reasonable yield from two different benzoyl
chlorides bearing aryl Cl and MeO substituents. Subsequently, the ylides were used in a
Wittig reaction with acetone. However, neither ylide underwent the desired reaction. To
examine optimal conditions, we used an excess of acetone as it was used as both solvent
and reactant (ketone). Additionally, the reaction temperature was increased from 0 °C to
50 °C and also longer reaction time was given up to 36 h. However, no product was
observed. Thus, the solvent was changed from acetone to toluene to allow the application of
higher reaction temperature (110 °C) and also the ketone was changed to pentan-3-one with
higher boiling point than acetone (Table 23). Unfortunately, none of the above attempts
gave any expected product from either ylide 315 or 316. TLC inspection showed that the
reaction mixture mainly consisted of starting materials.
Table 23. Attempted Wittig olefmation with pentan-3-one; (a) acetone (excess), toluene, 110 °C.
128

Entry Ylide Product
1 p-methoxyphenyl 315 N/Aa
2 /7-chlorophenyl 316 N/Aa
a No reaction
Thus, we needed to consider other alternatives and a literature search suggested the
application of Friedel-Crafts acylation to the synthesis of enones with electron rich phenyl
rings in the presence of the Lewis acid Aids. 139 According to literature procedures, we
synthesised enones with mono-, di- and tri-methoxy substituted phenyl moieties in good
yield (Table 24). However, the acylation did not work for the aryl system with a methyl
substituent (entry 5) nor with an electron withdrawing group (Cl, entry 4) at the para
position. It was suspected that these substrates might not be sufficiently electron-rich to
undergo the acylation.
Table 24. Friedel-Crafts acylation; (a) A1C1 3 , CS2 , 0 °C to rt, 1 d.
Cl
Entry
1
2
3
4
5
R 1
MeO
MeO
MeO
Cl
Me
R2
H
MeO
MeO
H
H
R3
H
H
MeO
H
H
Product (yield)
317 (75%)
318 (80%)
319 (80%)N/Aa
N/Aa
No reaction.
For the preparation of aryl enones with electron poor substituents, we employed Grignard
129

reaction between the appropriate aromatic aldehyde and a vinyl Grignard reagent, followed
by MnO2 oxidation of the resulting alcohol (Table 25).
Table 25. Grignard reaction and oxidation strategy; (a) 0.5 M Grignard reagent in THF (1.2 eq), Et2 O, rt, 5 h, (b) acetone, MnO2 (20.0 eq) rt.
a,b O
Entry
1
2
3
4
Ar
p-chloroxyphenyl
/7-nitrophenyl
/7-methylphenyl
2-Naph
Product (yield3)
320 (53%)
321 (13%)
322 (62%)
323 (69%)a Over two steps
For the MnC>2 oxidation, we used the crude mixture of the alcohol from the Grignard
reaction without purification. In addition to the synthesis of aryl enones with electron poor substituents such as chloro (320) and nitro (321), we prepared tolyl enone (322) and
naphthalenyl enone (323) in reasonable yield over 2 steps.
2.2.2. Preparation of various DHPsAfter having prepared a wide range of enones, we used them in the synthesis of various
DHPs (Table 26). At this stage, we adopted microwave technology to promote the HDA
reaction rather than using a pressure tube. In spite of its limitations for large scale reaction,
the use of microwaves dramatically reduced the reaction time from a few days to a few
hours.
130

Table 26. Preparation of various DHPs; (a) YbFOD (0.05 mol%), microwaves.
R2R2
Entry
1
2
34
5
6
7
8
9
10
11
12
A ^OR3
R 1
PhPhPhPh
Ph
p-MeO phenyl
w,p-MeO phenyl
m,m,p-MQO phenyl
Naph
p-Me phenyl
p-C\ phenyl
p-NOz phenyl
a
R2
H
Me
Et
cyHex
Ph
Me
Me
Me
Me
Me
Me
Me
- \R^
R3
Et
Et
Et
Et
Et/nBu
Et
Et/nBu
Et/nBu
Et
Et
Et
Et
"O^OR3
DHP (yield)
308 (48%)
324(41%)
325 (12%)
326 (30%)
N/A(0%)
327 (27%)
N/A (0%)
N/A(0%)
328(41%)
329 (40%)
330 (44%)
331 (58%)
From the results above, we found that bulky enone B-substituents slowed down the
cycloaddition (entries 1-5). The yield gradually decreased as the size of substituent R2
became larger. In the case of extremely bulky substrates, the reaction did not give any
cycloadduct and the product mixture was complex (entry 5). We assumed that the bulky R2
substituents would disfavour the s-cw-enone form which is required for cycloaddition, and
the enone would exist predominantly in the s-trans-enone form (Fig. 25).
131

s-c/s-enone s-frans-enone (disfavoured) (favoured)
Fig. 25. s-cw-enone and s-trans-enone.
We also found a trend caused by the electronic effect of the substituents in the aryl ring.
When the aryl group was electron-rich, the yield of cycloaddition was decreased (entries 6-
8 and 10). In the case of the reaction between aryl enones bearing dimethoxy (entry 7) and
trimethoxy substituents (entry 8) and an enol ether, we could not observe any cycloadduct and the reaction mixture was complex. However, we obtained the highest yield (58%) for
the p-NOz substituted phenyl enone (entry 12). These trends are as expected for reaction with an electron rich dienophile (the enol ether). Finally, we synthesised an interesting DHP
bearing a naphthalenyl moiety in 41% yield (entry 9).
Consequently, even though some of the HDA reactions gave a low yield or did not give the
cycloadduct, we had been able to prepare a wide range of DHPs for testing in the next step,
the oxidative rearrangement.
2.2.3. Enantioselective formation of THFsAfter the preparation of various DHPs, we synthesised the racemic tetrahydrofuranones via the previously established oxidative rearrangement method using DMDO followed by
Jones oxidation (Table 27). These racemic compounds were required in order to establish
conditions for eventual determination of enantiomeric excess by chiral HPLC. Conversion
was complete by TLC inspection and all DHPs were smoothly transformed to
tetrahydrofuranones in a reasonable yield over two steps (49-69%). After we obtained the
racemic tetrahydrofuranones, we were able in each case to resolve the two enantiomers by
chiral HPLC.
132

Table 27. Synthesis of racemic tetrahydrofuranones; (a) DMDO (1.0 eq), CH2C12 , 0 °C to rt, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt.
a,b
OEt
Entry
1
2
3
4
5
6
7
8
9
R 1
H
H
H
H
MeO
Me
Cl
NO2
Naph
R2 (=R3 ) Tetrahydrofuranone (yield3)
H
Me
Et
cyHex
Me
Me
Me
Me
Me
(±)-309 (50%)
(±)-332 (69%)
(±)-333 (59%)
(±)-334 (55%)
(±)-335 (50%)
(±)-336 (69%)
(±)-337 (53%)
(±)-338 (49%)
(±)-339 (53%)a Over 2 steps.
After the preparation of the racemic tetrahydrofuranones, we undertook the enantioselective
oxidative rearrangement via asymmetric epoxidation with Jacobsen's catalyst followed by
Jones oxidation. Firstly, we investigated the effect of substituent size at C-4 of the DHP in
the rearrangement (Table 28). The reactions were complete by TLC inspection and gave
tetrahydrofuranones in reasonable yield (40-55%). However, the yield gradually decreased
as the size of substituent R 1 and R2 became larger. In terms of enantiomeric excess, higher
levels of stereocontrol up to 80% ee (entry 4) were observed with more sterically
demanding and branched R 1 and R2 groups.
133

Table 28. Enantioselective synthesis of tetrahydrofuranones I; (a) 4-phenyl pyridine TV-oxide (0.2 eq), (R,R)- Jacobsen's catalyst 307 (0.05 eq), NaOCl (1.5 eq), CH2C12 , 0 °C to rt, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt.
,1 R2
pAA. a, b Ph v /JT '0
Entry R 1 (=R2) Tetrahydrofuranone (yield3)
1 H
2 Me
3 Et
4 cyHex
(S)-309 (55%)
(5)-332 (49%)
(5)-333 (45%)
(5)-334 (40%)
0 °
eeb
38%
58%
69%
80%a Over 2 steps, Determined by HPLC analysis)
Secondly, we investigated the electronic effect of the aryl substituent at C-6 of the DHP in
the rearrangement (Table 29).
Table 29. Enantioselective synthesis of tetrahydrofuranones II; (a) 4-phenyl pyridine N-oxide (0.2 eq), (R,R)- Jacobsen's catalyst 307 (0.05 eq), NaOCl (1.5 eq), CH2C12 , 0 °C to rt, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt.
Entry
1
2
3
4
5
6
fiAr O OEt
Ar
/7-MeO phenyl
Naph
p-Me phenyl
Ph
p-C\ phenyl
p-NO2 phenyl
v/_ a, b AN / V——— >•• v
O
Tetrahydrofuranone (yield3)
(5)-335 (35%)
(5)-339(41%)
(5)-336 (43%)
(S)-332 (49%)
(5)-337 (40%)
(5)-338 (64%)
•^s^
O
eeb
79%
71%
70%
58%
76%
46%Over 2 steps, Determined by HPLC analysis.
134

The reactions were again complete by TLC analysis and gave tetrahydrofuranones in
reasonable yield (40-64%) in most cases (entries 2-6). Additionally, we found that the yield
was generally higher when the aryl substituent was an electron-withdrawing group (entries
1, 3, 4, 6) In terms of enantiomeric excess, higher levels of stereocontrol (up to 79 % ee, entry 1) were generally observed with electron-donating aryl substituents (compare entries
1, 3, 6). In view of those results, the high ee observed for the oxidative rearrangement ofp- Cl-substituted aryl DHP (76% ee, entry 5) hinted that the electron-donating effect of the Cl- substituent by resonance rather than its inductive effect was a major influence on the enantioselectivity.
At this stage, we wondered whether the differing ee results could be partly due to differing
levels of background epoxidation for the various substrates. If NaOCl without Jacobsen's catalyst could epoxidise the DHPs, it would affect the product ee because of the competing formation of racemic products. Thus, we needed to check the background reaction without
catalyst. We chose three representative DHP examples and applied the same epoxidation conditions in the absence of Jacobsen's catalyst (Table 30).
Table 30. Background reaction without Jacobsen's catalyst 307; (a) 4-phenyl pyridine TV-oxide (0.2 eq), NaOCl (1.5 eq), CH2 C12 , 0 °C to rt.
OEt
Entry DHP (R) Time Product
1 327 (MeO) 3.5 h S.M. disappeared3 (no rearrangement product*3)
2 324 (H) 3.5 h S.M. disappeared3 (no rearrangement productb)
3 331 (NO2) 3.5 h No reaction3'13
"Determined by TLC analysis. b Determined by 'H NMR analysis.
However, we could not find any rearranged product by epoxidation from the background reactions of electron-rich aryl substituted DHP (entry 1), phenyl substituted DHP (entry 2)
.35

and electron poor aryl substituted DHP (entry 3). This suggested that the product ee values
would not be affected by background oxidation. Additionally, the result showed that the
electron-rich aryl substituted DHP (entry 1) is more reactive to NaOCl than the electron-
poor aryl substituted DHP (entry 3). This could explain the lower yields observed in the
epoxidation/rearrangement of the former substrates.
2.2.4. Determination of product configurationAfter observing promising levels of enantioselectivity in the enantioselective formation of
tetrahydrofuranones, it was necessary to determine and explain the observed product
configuration. Jacobsen reported that the enantioselectivity in the epoxidation can be
evaluated according to a general skewed side-on approach transition state model (Scheme
123). 133 The transition state 340 leads to severe steric repulsion between ligand and olefm
and the formation of the corresponding epoxide 341 will be disfavoured (Scheme 123, b).
However, such interactions are avoided in competing transition state 342 and it leads to the
formation of the expected epoxide 343 (Scheme 123, c).
(f?,R)-Jacobsen's Cat
Ph Me
Ph
(a) Side-on approach of Jacobsen's catalyst to trisubstituted olefin
Ph Me
O Ph 341
Me
PhPh
Ph MeVT<
O Ph 343
340
(b) Disfavoured transition state.
342
(c) Favoured transition state.
Scheme 123. Enantioselectivity explained by Jacobsen's model study.
136

On the basis of Jacobsen's model study, we expected the (^-configuration of the resulting
tetrahydrofuranone 345 from the formation of the favoured epoxide 344 (Scheme 124).
(R,ft)-Jacobsen'sCat
RPEt
Ar
R
OEt R
Ar
O
344
O
O s345
(a) Side-on approach of Jacobsen's catalyst to DHP. (b) (5)-Tetrahydrofuranone from favoured epoxide.
Scheme 124. Applcation of Jacobsen's model study to our DHP system.
In spite of our prediction for the observed configuration, we still needed evidence to confirm it. From a literature search, we found that Ghisalberti reported the optical rotation
value of enantiomerically pure (/?)-5-benzoyl-tetrahydrofuranone (/?)-309 as [(X]D= +4.2 in acetone and +9 in CHCl3 (Table 31). 140
Table 31. Comparison of (/?)-5-benzoyl-tetrahydrofuranone 309 and our result.
(R)-309 309
Ghisalberti's result (#)-309 Our observation 309
configuration (R)ee >99%
[a]D +4.2° in acetone (+9° in CHC13 )
(Unknown)
38%
0° inCHC!3
If our prediction was correct, our product should give a negative optical rotation value
because we expected our major product would have the (^-configuration. However, we
137

could not use optical rotation values to assign configuration because the observed optical
rotation for our sample of 38% ee was essentially zero ([ct] D=0 (c 0.03, CHCls)). This
would be due to the low ee (38%) of our product and the small magnitude of the optical
rotation for the enantiomerically pure compound. We therefore considered the comparison
of chiral HPLC retention times between enantiomerically pure benzoyl-tetrahydrofuranone
(7?)-309 and our product 309. However, to obtain chiral HPLC data for the optically pure
benzoyl-tetrahydrofuranone, we needed to synthesise it. From a literature search, we found
that Cahiez has reported a synthesis of (iS^-benzoyl-tetrahydrofuranone (5)-309 (Scheme 125). 140 ' m
o o o346 (S)-309
Scheme 125. Synthetic route toward (S)-5-benzoyl-dihydrofuranone by Cahiez; (a) (COC1)2 , DMF, CH2 C12, (b) PhMnCl, THF, -78 °C, 3h.
According to Cahiez's procedure, we synthesised (^-S-benzoyl-tetrahydrofuranone 309.
Firstly, crude acid chloride was obtained by the reaction of commercially available (S)-5-
oxo-tetrahydrofuran-2-carboxylic acid 346 and oxalyl chloride in the presence of DMF as a
catalyst. Subsequently, the addition of phenylmanganese chloride gave (5)-5-benzoyl-
tetrahydrofuranone (,S)-309 in 99% ee and 10% yield over 2 steps. We compared the chiral
HPLC data to those of our sample 309 (Fig. 26). This provided the evidence to support our
proposed configuration, showing that the major enantiomer from our Jacobsen's
epoxidation/rearrangement indeed had the (^-configuration
138

a) Racemic mixture from the oxidative rearrangement by DMDO.
VWD1 A iVrriirgtfi ~ ' nrr. [11C C3 0;
R
L__ _.___ ..__»______&_____
b) Chiral tetrahydrofuranone (38% ee) from the oxidative rearrangement by Jacobsen's catalyst.
R•^
c) Chiral tetrahydrofuranone (<99% ee) from ««.| Cahiez's synthesis.
VWO1 A Woveter^th-ZH nm (002-02O1 O)
\ S
_ __jo___._.„
E 8so-
Fig. 26. Comparison of HPLC data for 5-benzoyl-tetrahydrofuranone 309.
We next decided to examine more closely the observed electronic effect of the aryl substituent on enantioselectivity. From a literature search, we found previous studies on
electronic effects in asymmetric epoxidation using Jacobsen catalysts. Firstly, we found that Jacobsen and co-workers showed a linear Hammett relationship between the value of the substituent constant opara for the catalyst and the enantiomeric excess in the epoxidation of
(Z)-l,2-disubstituted alkene 348 (Scheme 126). 142
139

347a X=OMe, 347b X=Me, 347c X=H, 347d X=CI, 347e X=NO2
348
Scheme 126. Electronic tuning of Mn(salen) catalyst
Asymmetric epoxydation
In this study, they found that the simple change of the substituent (X) of the catalyst resulted in a dramatic change of the enantiomeric excess from 22% (347e, X=NO2) to 96% (347a, X=OMe). However, they only focussed on the tuning of the catalyst and they did not study the electronic tuning of the alkene substrate.Secondly, we found that Jacobsen and co-workers also found a good linear relationship between the electronic properties of (Z)-cinnamate esters (349a-349e) and the ratio of cis/trans-epoxides using a Hammett plot and the value of the substituent constant apara
(Scheme 127). 132
COoMe CO2Me CO2Me
349a X=OMe, c/s-epoxide frans-epoxide349b X=Me,349cX=H,349d X=CI,349e X=NO2
Scheme 127. Asymmetric epoxidation of (Z)-cinnamate esters; (a) (R,R)-Jacobsen's catalyst 307.
In this study, they found that changing the substituent (X) of the (Z)-cinnamate ester 349 resulted in a dramatic change of the ratio of cis/trans-epoxides from 0.3 (349e, X=NO:) to 11.7 (349a, X=OMe). Furthermore, they obtained facial selectivity (61-82%) by the equation (Eq. 1).
140

Facial selectivity = (eecis x %cis] + (eetrans x %trans) (Eq. 1)
However, this facial selectivity did not show a Hammett correlation to the substituent's
electronic properties. Additionally, Linde and co-workers showed a good relationship
between the electronic property of (Z)-stilbenes 350 and the ratio of c/s//ttmsi -epoxides
using a Hammett plot and the value of the modified substituent constant <Jpara+ (Scheme
128). 143
350a X=OMe, 350b X=H, 350c X=CI, 350d X=NO2
frans-epoxide
Scheme 128. Asymmetric epoxidation of (Z)-stilbenes 350; (a) (7?,/?)-Jacobsen's catalyst 307.
In this study, they found that changing the substituent (X) of (Z)-stilbenes 350 resulted in a
change of the ratio of cis/trans-Gpoxides from 0.2 (350d, X^NOz) to 2.2 (350a, X=OMe).
They also suggested that the modified substituent constant apara+ was more suitable to
explain the selectivity of cM//r<msi-epoxides than the substituent constant Opara in the
epoxidation of (Z)-stilbenes. However, Jacobsen and Linde focused on the correlation
between the electronic tuning of oleflns and the selectivity of cis/trans-epoxides. Their
study was therefore not appropriate to explain the epoxidation of our DHPs for two reasons.
Firstly, the epoxidation of the unique olefin in a rigid ring system (DHP) only gave the cis
epoxide and there was no issue of cis/trans-selectivity. Secondly, we were interested in the
correlation between the electronic tuning of olefins and the enantiomeric excess of the CM
epoxides. Thus, we decided to examine a Hammett plot based on our results.
For our own Hammett plots, we used the value of the substituent constant apara and the
141

modified substituent constant apara+ and log (S/R). Log (S/R) values were obtained from the
observed enantiomeric excess (Table 32).
Table 32. Values for Hammett plots; (a) i. oxidative rearrangement; ii. Jones oxidation. 144
OEt
Entry THF Ar 'para 'para ee %$• %/T \og(S/Kf
1 335 p-MeOphenyl -0.27 -0.78 79% 89.5 10.5 0.931
2 336 ^-Mephenyl -0.17 -0.31 70% 85.0 15.0 0.753
3 332 Ph 00 58% 79.0 21.0 0.575
4 337 ^-Clphenyl 0.23 0.11 75% 87.5 12.5 0.845
5 338 p-NO2 phenyl 0.78 0.79 46% 73.0 27.0 0.432
a % of S enantiomer. b % of S enantiomer. c S/R = (% of 5" enantiomer) / (% of/? enantiomer)
Firstly, we plotted log (S/R) against the substituent constant apara (Fig. 27). The black line
shows the best linear fit to the data produced by software*.
1
0.9
0.8
0.7
0.5
0.4
MeO
Me
y = -0.3616x + 0.7472 R 2 = 0.563
• NO;
-0.3 -0.2 -0.1 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8
-'para
Fig. 27. Correlation of the enantioselectivity of epoxidation against apara .
Microsoft Office Excel 2003
142

Clearly, these values did not show a reasonable correlation. So, we decided to plot log (S/R)
against the modified substituent constant apara+ and the correlation was indeed improved (a,
Fig. 28). Because the result with the chloro-substituent appeared to be furthest from the
best-fit line, we decided to examine the effect of omitting this data point. Indeed, this
modified data set now gave a reasonable straight line correlation (b, Fig. 28).
8
10.9
0.8
0.7
0.6
0.5
0.4
MeO y = -0.2937X + 0.6948
ClFT = 0.7096
§01 o
HNO-
1
0.9
0.8
0.7
0.6
0.5
-0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.80.4
. MeO y =-0.3189x +0.6486 R2 = 0.9607
Me
H
-0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
a) b)
Fig. 28. Correlation of the enantioselectivity of epoxidation against apara+ -
From our Hammett plots, we were able to see apara+ provided a better correlation to our
results than apara- The Qpara+ parameter is derived from a study of the rates of SN!
hydrolysis of substituted aryl halides, which proceed via a benzylic carbocation. A good
correlation of ee to this parameter suggests that the transition state leading to the major
enantiomer has a larger degree of benzylic cation character than the transition state leading
to the minor enantiomer.
2.2.5. Attempted manipulation of DHPs by Baeyer-Villiger reaction
Having established a method for the enantioselective synthesis of tetrahydrofuranones, it
would be desirable to apply our method to the synthesis of target molecules. From a
literature search, we found that tetradecenolide 351 (Fig. 29) would be a suitable target
molecule because of the structural similarity to our tetrahydrofuranones. This molecule was
known to have pheromone activity, stimulating oviposition of mosquitos. 145
143

C«H8 n 17
oo
351
Fig 29. Structure of tetradecenolide 351.
Another possible target was the THF 352, an intermediate in a synthesis of the C13-C29
fragment of caribenolide I (353) by Figadere and co-workers (Scheme 129). 146
caribenolide I 353
Scheme 129. Retrosynthesis of caribenolide I 353.
However, both of these targets would need substrates with no substituents at C-4 of the
tetrahydrofuranone and these could not be prepared in good enantiomeric excess by our
oxidative rearrangement method. To apply our method to the enantioselective synthesis of
various target molecules or various synthetic intermediates as shown above, we would need
to be able to manipulate the exocyclic ketone functional group. At this stage, we considered
Baeyer-Villiger oxidation to the corresponding ester which could be reduced to the
aldehyde functional group. The aldehyde functional group could then easily be transformed
144

into various functional groups and this would provide routes towards natural products or
useful synthetic intermediates (Scheme 130).
position a
Baeyer-Villiger oxidation
O OReduction
position b
Scheme 130. Idea for manipulation of tetrahydrofuranone.
However, there was an issue about the regiochemistry in the migration of the Baeyer-
Villiger reaction: we wanted to insert oxygen into between the aryl group and the ketone
(position a), not between the lactone and the ketone (position b). Initially, we attempted
Baeyer-Villiger oxidation of phenyl ketone (S)-309 with MCPBA at 80 °C (Scheme 131).
O
I O-°NvS)
(S)-354 (78%)
Scheme 131. Attempted Baeyer-Villiger reaction of phenyl ketone; (a) MCPBA (2.0 eq), C2H4C1 2 , 80 °C, 2 d.
However, NMR analysis showed the value of the chemical shift for the proton at C-5 had
moved from 5.79 ppm for the ketone (,S)-309 to 6.90 ppm in the product. Additionally, in
the 13 C NMR spectrum, C-5 moved from 78.3 ppm for the ketone (5)-309 to 95.8 ppm in
the product. These observations suggested that the product was ester 354 with the unwanted
regioselectivity because the observed large shifts downfield would not match with the
desired phenyl ester. We speculated that addition of electron donating substituents to the
145

phenyl group may favour the desired regioselectivity. Indeed, we could find literature
precedent for this tactic. Thus, Baeyer-Villiger of phenyl ketone 355 is reported to give the
ester 356, whereas the /?-methoxyphenyl variant 357 is reported to afford the aryl ester 358
(Scheme 132). 147 The strategy would be especially attractive because methoxy-substiruents
on the aryl ring had earlier been found to improve the enantioselectivity of the epoxidation.
OMe
R O
(a)
O
R O
357 358
(b)
OMe
Scheme 132. Effect of electronic tuning on Baeyer-Villiger oxidation; (a) MCPBA, rt, 91%, (b) MCPBA, 50 °C, 87%.
We therefore attempted the Baeyer-Villiger oxidation of the more electron-rich aryl ketone
359 bearing a MeO group (Scheme 133). Unfortunately, however, the electron rich ketone
did not react with MCPBA, even after 2 d at 80 °C. So, this reaction was not investigated
further at this stage.
146
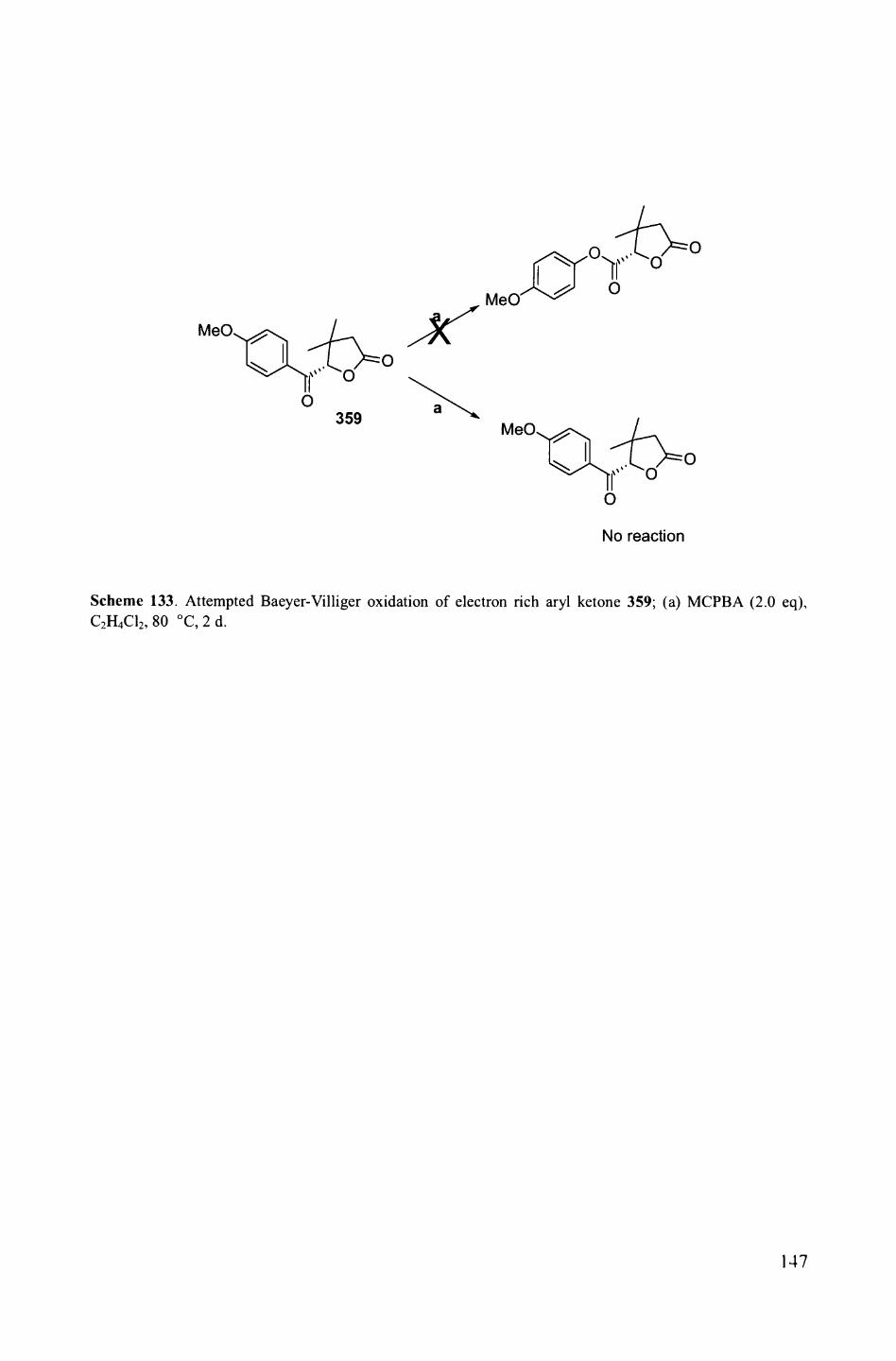
MeO
MeO
359 MeO
^ -O O
O
No reaction
Scheme 133. Attempted Baeyer-Villiger oxidation of electron rich aryl ketone 359; (a) MCPBA (2.0 eq), C2H4C12 , 80 °C, 2d.
147

3. Conclusions and future work3.1. ConclusionsIn this Chapter, we investigated the enantioselective oxidative rearrangement of various
aryl substituted DHPs to synthesise tetrahydrofuranones and this was achieved via
asymmetric epoxidation with Jacobsen's catalyst (Scheme 134). The best result (80% ee)
was obtained for tetrahydrofuranone 334 with a cyclohexyl moiety.
Ar O OEt
R R
R R
* Ar>y
O
=O
OEt
Scheme 134. Enantioselective oxidative rearrangement; (a) Asymmetric epoxidation by (^?,/?)-Jacobsen's catalyst 307, (b) Jones reagent (3.0 eq), acetone, 0 °C to rt.
We proved the configuration of our products by comparison to a known tetrahydrofuranone
and the observed enantioselectivity was in accordance with that observed previously for
Jacobsen epoxidation of trisubstituted alkenes. Additionally, we investigated the electronic
effect on the asymmetric epoxidation by Jacobsen's catalyst via tuning of the aryl moiety. A
Hammett plot using apara+ values suggested the importance of conjugation to the aryl
substituent in the asymmetric epoxidation. Furthermore, we attempted the manipulation of
the tetrahydrofuranone via Baeyer-Villiger oxidation. However, it did not give the desired
aryl ester.
148

3.2. Future workFuture work would include the investigation of alternative oxidants for Baeyer-Villiger
oxidation. If this were successful, we could prepare the corresponding aryl ester which
could then be reduced to the aldehyde functional group (Scheme 135). Furthermore, the
aldehyde functional group could then easily be transformed into various functional groups
by olefination or alkylation and this would provide routes towards natural product targets or
valuable synthetic intermediates.
Olefination
ArO
Baeyer-Villiger oxidation -O, Reduction
OO
Alkylation
Scheme 135. Manipulation of tetrahydrofuranones.
This whole section dealt with enantioselective oxidative rearrangement by metal-based
asymmetric epoxidation catalysis. In the next phase of the project, we explored the
development of a catalyst for metal-free asymmetric epoxidation and the results of this will
be discussed in Chapter III.
149

Chapter III. Synthesis of novel chiral ketone catalysts for
enantioselective epoxidation of alkenes
150

1. Introduction
1.1. Previous chiral ketone catalyst in our groupAsymmetric epoxidation is important in organic synthesis because epoxides can react with
various nucleophiles to provide ring-opened products stereospecifically. Use of
enantiomerically enriched epoxides allows control of the absolute configuration of the
resulting products to afford useful building blocks for asymmetric synthesis.
Enantioselective synthesis of epoxides by epoxidation of alkenes has been intensively
studied. Amongst non-metal mediated methods, epoxidation catalysed by chiral ketones
in the presence of Oxone® as an oxidant is particularly attractive. Shi's carbohydrate-
derived catalyst 304 is the best known chiral ketone to date (see Chapter II), but several
other classes of ketone have also been developed. In particular, new ketones that can be
used in low loadings and that will work with a wider range of alkene substitution patterns
are still required. Especially, the epoxidation of terminal alkenes with high
enantioselectivity and electron-poor alkenes such as unsaturated esters with fast conversion
are still remaining challenges. Shi and co-workers showed the improvement of ee in the
epoxidation of cis olefins and terminal olefins with modified carbohydrate-derived catalyst
360 by the modification of the spirocyclic substituent (Fig. 30). 136d ' 136e> 148 However, 1,1-
disubstituted alkenes still give poor ee.
NBoc
360
Ph
85% ee 71%ee 30% ee
Fig. 30. Epoxidation of alkenes with catalyst 360.
For the epoxidation of electron-poor alkenes, Shi and co-workers reported improved
151

catalytic reactivity and enantioselectivity by the replacement of the acetonide unit with
more electron-withdrawing ester groups (Fig. 31) because the electrophilicity of
electrophilic oxidant, dioxirane, can be increased by this replacement and it can prevent the
decomposition of the catalyst by Baeyer-Villiger reaction. 149 This ketone 362 gives very
good ee for a,p-trisubstituted unsaturated esters (361a-361c).
RCO2 Et
R2
361 a R1 =H, R2=Ph (96% ee), 361 b R1 =Me, R2=Ph (96% ee), 361 c R1 =R2=Me (82% ee) 362
Fig. 31. Epoxidation of enolates with catalyst 362.
Previously, the Armstrong group has synthesised various [3.2.1]-bicyclic ketone systems
and tested them as catalysts for the asymmetric epoxidation with Oxone (Fig 32). 150
X=NC02Et, O,Y=F, Cl, OAc, OCOR,Z=H
Fig. 32. Structure of [3.2.1] bicyclic ketones.
Much work has been done in the group on the effect of structure on the epoxidation
enantioselectivity. Firstly, the bridgehead atom (X, Fig. 32) plays an important role in
determining the activity and selectivity of the catalyst. 0jra-bicyclic ketones (X=O)

generally give higher enantiomeric excess than #z<3-bicyclic ketones (X=NCO2Et). 151
Secondly, the electron-withdrawing substituent at the a-position (Y, Fig. 32) makes the
ketone more electrophilic, activating it toward attack from Oxone®. The catalyst also
becomes more stable to possible side reaction such as the Baeyer-Villiger decomposition.
Thirdly, various a-monosubstituted ketones have been synthesised (Y = F, Cl, OAc, OCOR
and Z = H) and axial substituted derivatives represent the best catalysts to date in the series (93% ee for £-stilbene when X = O, Y = OAc and Z = H). 152 The observed stereochemistry can be explained by a spiro transition state (Scheme 136). The major enantiomers can be
obtained via the transition state 363 with less steric repulsion rather than the competing transition state 364 with severe steric repulsion between the olefin and substituent Y of the catalyst. Thus tram- and tri-substituted olefms are expected to give high enantioselectivity and terminal olefins are expected to give lower ee.
( a) Favoured spiro TS
'R2
(R,R) V'° "' (S' S) major
364
(b) Disfavoured spiro TS
Scheme 136. Explanation of stereochemistry from spiro transition state.
Very recently, our group has investigated the synthesis of 1,1-spirocyclic disubstituted ketones (365a-365d, Table 33) because Shi and co-workers found that the replacement of
the spiroketal unit in fructose-derived ketone 365a with an oxazolidinone gave improved enantioselectivity in the epoxidation of styrene. 148 Representative results are shown in
Table 33. 153 This replacement could provide enantioselectivity of up to 59% ee in the
epoxidation of styrene with catalyst 365b. However, in terms of conversion, the
oxazolidinone series (365b-365d) generally showed lower conversion than dioxolanone
365a.
153

Table 33. Epoxidation catalysed by ketones 365a-365d; Reaction conditions: alkene (1.0 eq), catalyst (0.1 eq), Oxone® (10.0 eq), NaHCO3 (15.5 eq), CH3 CN, aq. Na2EDTA (0.4 mM solution).
EtO2C O\ it
f ̂ \-^/ 365al^^\ 365b
^V 365c O 365d
365a 365bConv. 3 ee Conv. 3 ee
X=0, X=NPh, X=N i Pr, X=NMe
365c 365dConv. 3 ee Conv. 3 ee
£-Stilbene 100% 89% 76% 87% 90% 87% 85% 92%
Styrene 100% 46% 93% 59% 35% 54% 79% 58%
£-Ethylcinnamate 55% 74% 40% 82% N/A N/A 41% 80%3 Conversion (determined by ! H NMR)
154

1.2. Concept of this projectAs descrived above, the tropinone-derived spirocyclic catalysts (365a-365d) had given
promising results. Because previous work in the group had shown that oxcr-bicyclic
catalysts generally give higher enantioselectivities than the tropinone derivatives, it seemed
appropriate to study the effect of this modification on the spirocyclic catalysts. Therefore,
in this project, a new spiro-cyclic ketone (Fig. 33) with an oxygen bridgehead would be
synthesised and this catalyst would tested with various alkenes in the asymmetric
epoxidation process to allow comparison of the results with its tropinone counterpart.
X=O, NR
Fig. 33. Structure of new spirocyclic ketone system.
155

2. Results and discussion
2.1. 0*fl-bicyclic ketone catalyst2.1.1. Synthesis of racemic axa-bicyclic ketone.
The first target molecule was the oxa-bicyclic ketone 366 containing a 1,1-fused cyclic
carbonate (Fig. 34) using a synthetic strategy similar to that previously used for the tropinone series in our group. 153
366
Fig. 34. Oxa-bicyclic ketone catalyst 366.
To synthesise the target molecule, the parent oxa-bicylic ketone 369 was first synthesised using a known procedure (Scheme 137). Thus, 1,3-dipolar cycloaddition between furan and
1,1,3-trichloroacetone gave the o;ra-bicycle 367 and subsequent treatment with an excess of
zinc powder and copper (I) iodide effected complete dechlorination. The olefin in the
oxabicycle 368 was then reduced by catalytic hydrogenation.
156

oCl
Cl Cl
368 369
Scheme 137. Synthesis of parent oxa-bicyclic ketone; (a) CF3CH2OH, Et3N, 5 °C to rt, overnight, (b) Zn powder (2.1 eq), Cul (0.7 eq), benzene/MeOH, 0 °C to overnight, 45% (over 2 steps), (C) H2, 10% Pd/C, MeOH, rt, overnight, 98%.
After the preparation of parent ketone 369, the introduction of the exo-methylene unit on
the oxfl-bicyclic ketone 369 was successfully achieved using a Mannich reaction analogous
to that employed in the tropinone series (Scheme 138). 135 Thus, the ketone 369 was
deprotonated with LiHMDS at -78 °C and then treated with TMSC1. The resulting silyl
enol ether was then reacted with Eschenmoser's salt to give the Mannich base which was
then methylated with Mel. Elimination of trimethylamine was achieved by heating in the
presence of NaHCOa. Interestingly, the formation of dimethylene 371 was competitive with
the formation of desired methylene ketone 370, resulting in a moderate yield of methylene
ketone 370.
a, b, c, d, e
369O
370 (40%)O
371 (13%)
Scheme 138. Introduction of exo-methylene unit on oxa-bicyclic ketone by Mannich reaction; (a) 1.0 M LiHMDS (1.0 eq), THF, -78 °C, 30 min, (b) TMSC1 (1.5 eq), -78 to 0 °C, 30 min, (c) Eschenmoser's salt (1.0 eq), DMF, pressure tube, rt, 1.5 h, (d) Mel (5.0 eq), 50 °C, overnight, (e) NaHCO 3 (7.0 eq), DMF, 95 °C, overnight.
157

The exocyclic enone 370 was then dihydroxylated with a catalytic quantity of
K2Os(V2H2O and co-oxidant NMO in acetone/water using quinuclidine as the ligand
(Scheme 139). 135
372
Scheme 139. Dihydroxylation of methylene 370; (a) K2OsO4-2H2O (0.01 eq), quinuclidine (0.05 eq), NMO (1.9 eq), acetone/H2O, rt, 3 d, 71%.
We assumed that the reaction occurred stereoselectively on the less hindered exo-face, in
accordance with the stereochemical outcome in the tropinone series, which was proven by
NOESY studies. 153 The diol 372 was then cyclised with triphosgene to give the
dioxolanone 373 (Scheme 140).
^^
A a CI3CO OCCI3 —————
Scheme 140. Cyclisation of diol 372 to dioxolanone 373; (a) Pyridine, CH 2C1: , O °C to rt, 8 h, 72%.
2.1.2. Epoxidation by racemic oxa-bicyclic ketone
After we prepared racemic catalyst (±)-373, its potential catalytic efficiency towards the
epoxidation of (£)-stilbene was evaluated using the standard Yang one-phase conditions
(Table 34). Compared to tropinone derivative catalyst (entry 1), ojca-bicyclic ketone (±)-
373 clearly showed better catalytic reactivity. The epoxidation of (£)-stilbene was complete
15X

using only 10 mol% loading of catalyst in 10 minutes. The reaction also proved that the
catalyst was stable to the reaction conditions, since we were able to see the peaks for the catalyst in the 1 U NMR spectrum of the crude mixture.
Table 34. Epoxidation of £-stilbene catalysed by ketone (±)-373; Reaction conditions: alkene (1.0 eq), catalyst (0.1 eq), Oxone* (10.0 eq), NaHCO3 (15.5 eq), CH3CN, aq. Na2EDTA (0.4 mM solution).
Entry Alkene Catalyst Time Epoxide (conversion )
1 (£>Stilbene (±)-373 <10min (±)-374 (100%)
2 Tropinone 365a 30 min (±)-374 (86%) a Determined by crude 'H NMR.
As a consequence of this encouraging result, we investigated the epoxidation of various
olefins (Table 35).
Table 35. Epoxidation catalysed by ketone (±)-373; Reaction conditions: alkene (1.0 eq), catalyst (0.1 eq), Oxone® (10.0 eq), NaHCO3 (15.5 eq), CH3 CN, aq. Na2EDTA (0.4 mM solution).
Entry Alkene Catalyst Time Epoxide (conversion3)
1
2
3
4
5
6
7
8
9
10
Styrene
a-Methylstyrene
(^-p-Methylstyrene
Phenylcyclohexene
(£)-Ethylcinnamate
(±)-373 1 h
Tropinone 365a 1 h
(±)-373 1 h
Tropinone 365a 1 h
(±)-373 1 h
Tropinone 365a 1 h
(±)-373 1 h
Tropinone 365a 1 h
(±)-373 18 h
Tropinone 365a 18 h
(±)-375 (100%)
(±)-375(100%)
(±)-376(100%)
(±)-376(100%)
(±)-377(100%)
(±)-377(100%)
(±)-378(100%)
(±)-378(100%)
(±)-379 (33%)
(±)-379 (55%)' Determined by crude H NMR.
In all cases except (£>ethylcinnamate, 0*a-bicylic ketone (±)-373 showed excellent
159

catalytic reactivity. The epoxidation of olefins was complete with only 10 mol% loading of
catalyst in 1 h (entries 1-8). However, the ketone (±)-373 showed extremely low catalytic reactivity in the epoxidation of (F)-ethylcinnamate (entry 9). The epoxidation of (£)-
ethylcinnamate by ketone (±)-373 showed 33% conversion in 18 h and this represented even lower conversion than the 55% observed with the tropinone catalyst (entry 10).
2.1.3. Synthesis of enantiomerically enriched oxa-bicyc\ic ketoneSince racemic ketone (±)-373 generally showed excellent catalytic reactivity, we decide to investigate its synthesis in non-racemic form. In line with earlier studies in the group, 152 ' 154 we decided to employ desymmetrisation of parent ketone 369 via the formation of silyl enol ether with (/?,/?)-chiral amine 381 (Scheme 141). 155
369 (+)-380OTMS
Chiral amine 381
Scheme 141. Desymmetrisation of parent ketone (+)-369; (a) (+)-Bis[(7?)-a-methylbenzyl]amine 381 (1.5 eq), "BuLi (1.5 eq), TMSC1 (5.0 eq), THF, >40%.
The low yield of silyl enol ether 380 was caused by difficulties in removing the chiral
amine. The amine was not completely removed by washing with CuSC>4 (aq, sat) several times. Use of an SCX column successfully removed the amine, but resulted in some hydrolysis of 380 to starting ketone 369. After the desymmetrisation, we followed the reaction sequence previously employed for the synthesis of the racemic catalyst. Thus, Mannich reaction on (+)-380 followed by dihydroxylation of the resulting olefin 370 with K2OsO4 and NMO gave diol 372. Subsequently, reaction with triphosgene gave the chiral ketone catalyst (+)-373. To find out the enantiomeric purity of chiral catalyst (+)-373, we attempted to use chiral HPLC to separate the racemic ketone (±)-373 with OD(H), OJ(H),
OC, AD(H) and AS(H) columns with UV detection at 206 nm or 280 nm. However,
160

satisfactory resolution could not be achieved. As an alternative, we used 'H NMR
spectroscopy in the presence of the shift reagent Eu(hfc)3 . Pleasingly, when 30 mol% of the
shift reagent was used, a clear separation for the racemic ketone (±)-373 was detected
around 3.10-3.60 ppm by ! H NMR (b, Fig. 35). The same conditions were used for the chiral ketone (+)-373 and the enantiomeric excess of catalyst (+)-373 was evaluated as 68%
ee because the ratio of two enantiomers was 5.3 to 1.0 by the investigation of the corresponding integral in the ! H NMR spectrum (Fig. 35, c).
a)
Ab)
J ft.
c)
Fig 35. 'H NMR of catalyst 373; a) Racemic catalyst (±)-373 ; b) Racemic catalyst (±)-373 with 30 mol% shift reagent, c) Chiral catalyst (+)-373 with 30 mol% shift reagent.
2.1.4. Epoxidation by enantiomerically enriched oxa-bicyclic ketoneAfter having prepared enantioenriched chiral catalyst (+)-373, it was tested in the
asymmetric epoxidation of various olefms and the results were compared to the tropinone
161

series (Table 36).
Table 36. Epoxidation catalysed by chiral ketone (+)-373; Reaction conditions: alkene (1.0 eq), catalyst (0.1®eq), Oxone (10.0 eq), NaHCO 3 (15.5 eq), CH3 CN, aq. Na2EDTA (0.4 mM solution).
cC-
•\ II^L/0
A(+)-373
Entry
1
2
3
4
5
6
Alkene
(£)-Stilbene
Styrene
EtOOC O\ //
^^*X*^N
1^ V o/d^x 7^\(+)-365a
Catalyst
(+)-373
(-)-Tropinone 365a(+)-373
(-)-Tropinone 365a
a-Methylstyrene (+)-373(-)-Tropinone 365a
7 (£>p-Methylstyrene (+)-373
8
9
10
11
12
(-)-Tropinone 365a
Phenylcyclohexene (+)-373
(-)-Tropinone 365a
(£>Ethylcinnamate (+)-373
(-)-Tropinone 365a
O
*JJ-rCOOEti
^(-)-365a
ee0bsa eemaxb Epoxide0
62
86
28
46
1
19
48
63
55
73
46
74
91 (R,R)-314
N/A (S, S)-314
41 (R)-375N/A (5)-375
<1 (R)-316N/A (5)-376
71 (R,R)-311
N/A (S 5)-377
81 (R,R)-318N/A (5,5)-378
68 (25, 3/?)-379
N/A (2/? , 35)-379%, Determined by chiral HPLC, %, eemax = ee0bs x 100 / 68, c Configuration of major isomer.
Although we obtained the enantiomeric excess from the asymmetric epoxidation of various
olefins, it could not be directly compared to the enantiomeric excess from the epoxidation by the tropinone derivative (-)-365a because the chiral ketone catalyst (+)-373 was not
162

enantiomerically pure (ee = 68%) and the tropinone derivative had been used in
enantiomerically pure form. So, we assumed that there would be a linear relationship
between the enantiomeric excess of catalyst and the enantiomeric excess of the resulting
epoxides and we calculated the eemax by the equation; eemax = ee0bs x 1007 68. Previous
work in the group with ojra-bicylic chiral ketone catalysts has demonstrated that this linear
relationship is valid. 152 With the calculated eemax value, we compared our results to the
tropinone catalyst. For di- and trisubstituted aryl alkenes, the ojco-bicycle (+)-373 (entries 1,
7 and 9) did indeed generally afford higher enantioselectivity than its tropinone counterpart
(entries 2, 8 and 10). The highest enantiomeric excess was found for (^-stilbene (91%,
entry 1). However, ketone catalyst (+)-373 did not give any ee for the epoxidation of the
challenging substrate, a-methylstyrene (<1%, entry 5), and gave slightly lower ee than the
tropinone derivative (-)-365a for epoxidation of styrene and (£)-ethylcinnamate.
The observed stereoselectivity could be explained by the previously mentioned spiro transition state model. In general, the configuration of major enantiomers fits with with less
steric repulsion in one transition state (Scheme 142, 382) compared to the competing
transition state 383 with severe steric repulsion. Thus the tram- and trisubstituted olefms
(entries 1, 7, 9 and 11, Table 36) give high enantioselectivity whilst terminal olefins (entries
3 and 5, Table 36) give lower ee because they can more easily attack the dioxirane in two
different spiro transition states leading to both enantiomers.
(R.R)major
(a) Favoured spiro TS
.^\.Ph°' >Ph
(S.S,
383
(b) Disfavoured spiro TS
Scheme 142. Transition state for formation of both enantiomers of (£)-stilbene oxide.
163

2.2. Aza-bicyclic ketone catalystRecently, our group has reported a novel rearrangement sequence for the synthesis of aza-
bicyclic ketones (Scheme 143). 156 Alkoxy-pyrrolidine 384 (obtained by addition of a vinyl
Grignard reagent to the product of DHP aziridination/rearrangement) can give the 1-aza-
bicyclo[2.2.1]heptane ring system 385 via an unusual azfl-Prins-pinacol reaction mediated
by SnCU. On further exposure to Lewis acid, the [2.2.1]heptane ring expands to the isomeric [3.2.1]tropane 386.
Ts OMe ^ N
R 384 385
Scheme 143. Rearrangement of pyrrolidine 384 to azo-bicyclic ketone 386; (a) SnCl4 (1.0 M in heptane, 10.0 eq). CH2C12 , 0 °C to it, 16h.
We reasoned that the catalyst 386 could potentially be an interesting new class of ketone
catalyst which may display complementary reactivity and selectivity to our previous
bicyclic ketones. Thus, we decided to synthesise a representative ketone of this type and to
test it for the epoxidation of various olefins.
2.2.1. Synthesis of racemic aza-bicyclic ketone catalystFirstly, commercially available DHP 285 was aziridinated by Chloramine-T in the presence
of NBS to effect rearrangement to the corresponding pyrrolidine 387 (Scheme 144).
O OMe285 387
Scheme 144. Rearrangement of DHP 285 to pyrrolidine 387; (a) Chloramine-T (1.2 eq), NBS (0.2 eq). CH 3 CN, rt, 2 h, 50%.
164

Addition of the vinyl Grignard reagent 388 then gave alcohol 389 as a mixture of
diastereoisomers. Subsequent reaction with SnCU gave the aza-bicyc\ic ketone (±)-390a and (±)-390b as a separable mixture of isomers (Scheme 145). Both isomers were identified
by the comparison of 'H-NMR spectrum to the prevous results in the group. 157 The low
yield was caused by incomplete ring-expansion from [2.2.1]heptane ring to [3.2.1]tropane.
OMe + BrMg
387 388
O
(±)-390a(exo, 15%)
Ts N
O
(±)-390b(endo, 13%)
OMe
389
Scheme 145. Synthesis of azo-bicyclic ketone (±)-390a and (±)-390b; (a) Et2O, 0 °C to rt, 10 min, (b) SnCL, (1.0 M in heptane, 10.0 eq). CH2 C12 , 0 °C to rt, 16 h,
2.2.2. Epoxidation by racemic aza-bicyclic ketone catalystAfter the synthesis of racemic ketone (±)-390a and 390b, we tested the exo-aza-bicyc\ic ketone (±)-390a to investigate its catalytic activity for the epoxidation of olefms. Initially,
we adopted (^-stilbene as the olefin to be oxidised because it showed the highest reactivity in the epoxidation with oxa-bicyc\ic ketone catalyst (±)-373 (Table 37). However, exo-aza- bicyclic ketone catalyst (±)-390a showed inferior catalytic activity to oxa-bicylic ketone (±)-373 in all cases. The epoxidation of (£>stilbene showed extremely low conversion after
18 h (entry 1). Interestingly, the epoxidation of styrene by ketone catalyst (±)-390a still showed lower activity than oxa-bicy\ic ketone (±)-373. However, the reaction was
completed in 5 h (entry 4). Additionally, we attempted the epoxidation of (£>stilbene by
endo-aza-bicyclic ketone catalyst (±)-390b (entry 2). However, the catalytic activity of
165

endo-aza-bicyclic ketone (±)-390b was slightly inferior to the exo-aza-bicyc\ic ketone 390a
(entry 2). We assumed that the low catalytic activity for both might be caused by the methyl
substituent next to the ketone moiety because it could prevent the access of olefin to
dioxirane by steric hindrance. These preminary studies showed that these ketones can act as
catalysts, albeit less effectively than the earlier ones for stilbene epoxidation.
Table 37. Epoxidation of olefins by ketone (±)-390a; Reaction conditions: alkene (1.0 eq), catalyst (0.1 eq), Oxone®(10.0 eq), NaHCO3 (15.5 eq), CH3 CN, aq. Na2EDTA (0.4 mM solution).
Entry
1
2
3
4
5
Alkene Catalyst
(£)-Stilbene (±)-390a(±)-390b
(±)-373
Styrene (±)-390a(±)-373
time Conversion (%)a
18h
18h
<10min
5h
Ih
18
15
100>99
100'Determined by crude 'H NMR
166

3. Conclusions and future workIn this project, we synthesised several novel bicyclic ketone catalysts and we investigated
them as catalysts for alkene epoxidation using Oxone®. Firstly, oxor-bicyclic ketone catalyst
373 generally showed better catalytic reactivity and enantioselectivity than tropinone
derivative ketone catalyst 365a. However, this catalyst could not overcome the low
catalytic activity in the epoxidation of (£")-ethylcinnamate and the low enantioselectivity in
the epoxidation of a-methylstyrene. Secondly, a different class of bicyclic ketone 390 was
synthesised but it showed low catalytic activity in the epoxidation. Therefore, we would
still need to investigate different type of catalysts for the epoxidation of challenging olefms
such as (£)-ethylcinnamate and a-methylstyrene. For the future work, further manipulation
of the oxfl-bicyclic ketone catalyst 373 and the aza-bicyc\ic ketone catalyst 390 would be
desirable. Firstly, it is possible to replace the spiro ketal in oxa-bicyc\ic ketone 373 because
Shi and co-workers found that the replacement of the spiro ketal in fructose derivatives
with an oxazolidinone gave catalysts that provided very good ee in the epoxidation of
styrenes 149 and our group also found that oxazolidinones can give good selectivity (vide
ante). Thus, the spiro ketal moiety in oxor-bicyclic ketone 373 could be replaced with an
oxazolidinone (Fig. 36) to potentially improve the epoxidation enantioselectivity.
'' CT\
Fig 36. Qxa-bicyclic ketone bearing oxazolidinone moiety
Secondly, the replacement of the methyl group at the a-position of aza-bicyc\ic ketone 390
with other alkyl or aryl substituents would be interesting because the diverse property of
catalyst can be easily obtained by the tuning of a-substituent (Scheme 146). Especially,
167

electron-withdrawing substituents would be desirable because they should increase the reactivity.
HOOMe
Ts N
Ts N
O
•R R
O
Scheme 146. Tuning of a-substituent in aza-bicyc\ic ketone 390.
168

Chapter IV. Experimental procedures and data
169

General detailsSolvents were freshly distilled before use from sodium benzophenone (diethyl ether, THF,
toluene) or CaH2 (dichloromethane). Liquids reagents were distilled prior to use, while
other commercial solids were used as supplied. Reactions were run under a positive
pressure of nitrogen. Reaction temperatures were recorded as bath temperatures. Flash
column chromatography was performed using BDH ¥254 silica gel. Analytical thin layer
chromatography was performed on pre-coated Merck silica gel 60 ¥254 glass backed plates
and visualised by either UV light (254 nm) or reactive stain reagents as appropriate.
NMR analyses were performed on Bruker AC 250 MHz, AV 400 MHz or DX 400 MHz
instruments; Chemical shifts are quoted in ppm relative to TMS (as referenced to residual
CHCb 8H=7.26 or CDCls 8c=77.0), with coupling constants quoted in Hz. Splitting
patterns are abbreviated as follows: singlet (s), doublet (d), triplet (t), quartet (q), multiplet
(m) and combinations of the above. Infrared analyses were recorded on NaCl plates. A
Mattson Satellite FTIR spectrometer was employed in the absorption range of 4000-600
cm" 1 . Microwave reactions were carried out using a CEM Discover instrument. Chemical
lonisation Mass Spectrometry was carried out with ammonia reagent gas using a
Micromass Autospec-Q spectrometer at the Imperial College Mass Spectrometry Service.
Melting points were determined using a Reichert hot stage microscope apparatus. Optical rotations were recorded on an Optical-Activity AA-5 Polarimeter, with a path length of 10
cm in chloroform unless stated otherwise. [<X]D values are given in 10" 1 deg cm2 g" 1 .
Concentrations (c) are given in grams per 100 cm3 . Chiral HPLC was performed on
Hewlett Packard Series 1100 HPLC system with Chiralcel columns using 'PrOH/hexane as
eluent. For HPLC, retention times are quoted in minutes.
Nomenclature and numberingThe names of the compounds were determined according to IUPAC rules and Autonom
4.01.304 when applicable.
70

1. Compounds from Chapter 1.General procedure for hetero Diels-Alder reaction using pressure tube89
To vinyl ether (2.0-10.0 eq) at room temperature in the pressure tube was added diene (1.0
eq) and YbFOD (0.05 eq). The solution was allowed to stir at the corresponding
temperature (45-100 °C) for 1- 10 days. The reaction mixture allowed to room temperature.
Column chromatography eluting with a diethyl ether/petrol gave alkoxydihydropyrans.
Product
211
217
218
236
237
238
232
239
240
271
272
R 1
H
H
H
Me'Pr
Me
Ph
CH2OBn
(Z)-(CH2)4CH=CHEt
Me
Me
R2
H
H
H
H
H
Me
H
H
H
H
H
R3
Et'Bu
nBu
"Bu
nBu
"Bu
Et
Et
Et
Et
Et
R4
Me
Me
Me
Me
Me
Me
Me
Me
Me"Pr
"Bu
* C O
2-Ethoxy-6-methyl-3,4-dihydro-2H-pyran 211 : Prepared by the general procedure (1.0 eq
of diene, 8.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 45 °C, 3 d) to give 211 (2.35 g, 70%)
as a pale yellow oil. vmax (CHCy/cm' 1 2978 (CH), 2254, 1713, 1643 (C=C); 5 H (250 MHz,
CDC1 3 ) 4.96 (1H, t, J 3.5, OCHO), 4.50 (1H, t, J 3.5, C=CH), 3.83 (1H, dq, J 10.0, 7.0,
OC//H), 3.55 (1H, dq, J 10.0, 7.0, OCH//), 2.17-1.70 (4H, m, CH2 CH 2 ), 1.70 (3H, s,
171

CCH3 ), 1.19 (3H, t, J 7.0, CH2C//3 ); m/z (CI) 114 (100%), 143 (M//"), 160 (MNH4+\ Found: MH+, 143.1069. C 8H 14O2 requires MH+ , 143.1072.
2-tert-Butoxy-6-methyl-3,4-dihydro-2H-pyran 217: Prepared by the general procedure (1.0
eq of diene, 3.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 90 °C, 8 d) to give 217 (0.80 g,
20%) as a pale yellow oil. vmax (CHCy/cm' 1 1682 (C=C); 6H (250 MHz, CDC13 ) 5.18 (IH, t,
J 4.0, OCHO), 4.45 (IH, t, 74.0, C=CH), 2.18-1.81 (4H, m, CH2CH2), 1.64 (3H, s, CCH3 )
1.18 (9H, s, 3 x CCH3 ); 13 C could not be obtained due to the rapid decomposition of the
sample in CDC13 . m/z (CI) 171 (Mrf\ Found: MH+, 171.1383. C 10H 18O2 requires MH+, 171.1385.
2-Butoxy-6-methyl-3,4-dihydro-2H-pyrcm 218 159 : Prepared by the general procedure (1.0 eq
of diene, 2.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 90 °C, 6 d) to give 218 (2.80 g, 55%)
as a pale yellow oil. vmax (CHCl3 )/cm ! 2958-2936 (CH), 1721, 1687 (C=C); 5H (250 MHz,
CDC13 ) 4.97 (IH, t, J 3.5, OCHO), 4.52 (IH, t, J 3.5, C=CH), 3.78 (IH, dt, J 9.5, 6.5,
OC//H), 3.50 (IH, dt, J 9.5, 6.5, OCH#), 2.20-1.69 (4H, m, CH2CH2), 1.68-1.48 (2H, m, CH2), 1.42-1.24 (2H, m, CH2), 1.71 (3H, s, CCH3 ), 0.90 (3H, t, J 7.5, CH2C//3 ); 5C (125
MHz, CDC13 ) 147.4 (C), 97.4 (CH), 96.1 (CH), 67.9 (CH2), 31.8 (CH2), 26.4 (CH2), 19.9
(CH3 ), 19.3 (CH2), 17.0 (CH2), 13.8 (CH3 ); m/z (CI) 140, 171 (Mrf\ 197, Found: MH+ ,
171.1387. Ci 0H 18O2 requires MH+, 171.1385
(2S ,4S }-2-Butoxy-4,6-dimethyl-3,4-dihydro-2H-pyran 236: Prepared by the general
procedure (1.0 eq of diene, 2.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 50 °C, 10 d) to give
236 (1.90 g, 40%) in a 3:1 inseparable mixture as a colourless oil. vmax (CHCy/cirf 1 2960-
2873 (CH), 1719, 1674 (C=C); 5H (250 MHz, CDC13 ) 4.99 (lHmajor , t, J 3.0, OCHO), 4.88
(lHminor, dd, J 7.0, 3.0, OCHO), 4.43 (lHmajor, d, J 1.0, C=CH), 4.33 (lHminor, d, J 1.0,
C=CH), 3.82 (2Hboth, m, 2 x OC//H), 3.47 (2Hboth , m, 2 x OCHH}, 2.39 (2Hboth , m, 2 x
CH3C//), 1.92-1.25 (12Hboth, 6 x CH2 ), 1.72 (6Hboth , s, 2 x CCH3 ), 1.02-0.88 (6Hboth , m, 2 x
CHC//3 , 2 x CH 2C//3 ); 13 C 6C (100 MHz, CDC1 3 ) 147.5 (C)major , 146.1 (C)mmor , 103.1
172

(CH)minor, 102.3 (CH)major, 99.8 (CH)major, 96.8 (CH)minor, 68.4 (CH2)maJOr, 67.7 (CH2 )minor,
36.2 (CH)™^, 35.0 (CH)minor, 31.9 (CH2)minor, 31.8 (CH2 )major , 26.2(CH2 )both , 21.5
(CH3)major, 21.4 (CH 3)minor, 19.7 (CHaW, 19-5 (CH3 )major , 19.4 (CHzW, 19.2 (CH2 )maJOr,
13.7 (CH3 )both; m/z (CI) 185 (M//"), Found: MH+ , 185.1538. CnH20O2 requires MH+ , 185.1541.
(2S ,4R }-2-Butoxy-4-isopropyl-6-methyl-3,4-dihydro-2H-pyran 237: Prepared by the
general procedure (1.0 eq of diene, 2.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 55 °C, 3 d)
to give 237 (3.77 g, 75%) in a 6:1 inseparable mixture as a pale yellow oil. vmax
(CHCy/cm' 1 2958-2873 (CH), 1721, 1678 (C=C); 6H (250 MHz, CDCl3 )major 4.84 (1H, dd,
79.5, 2.0, OCHO), 4.36 (1H, d, J 1.5, OCH), 3.90 (1H, dt, J 9.5, 6.5, OCH//), 3.47 (1H,
dt, J 9.5, 6.5, OC//H), 2.08 (1H, dqq, J 12.5, 4.0, 1.5, (CH3)2C//), 1.86 (1H, ddt, J 12.5, 6.5,
1.7, (CH3)2CHC//), 1.71 (3H, s, CCH3 ), 1.64-1.25 (6H, m, 3 x CH2), 0.94-0.82 (9H, 3 x CH3 ); 6C (125 MHz, CDCl3 )major 146.8 (C), 99.9 (CH), 97.1 (CH), 67.8 (CH2), 33.1 (CH),
31.8 (CH), 31.7 (CH2), 31.5 (CH2), 19.8, 19.6 (2 x CH3 ), 19.2 (CH3), 19.1 (CH2), 13.8 (CH3 ); m/z (CI) 213 (MH", 100%), Found: MH+, 213.1860. Ci 3 H24O2 requires MH+,
213.1854.
2-Butoxy-4,4,6-trimethyl-3,4-dihydro-2H-pyran 23887 : Prepared by the general procedure
(1.0 eq of diene, 5.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 100 °C, 5 d) to give 238 (1.60
g, 54%) as apale yellow oil. vmax (CHCy/crn 1 2958-2872 (CH), 1676 (C=C); 5H (250 MHz,
CDC13 ) 4.88 (1H, dd, J 7.5, 3.0, OCHO), 4.32 (1H, s, C=CH), 3.88 (1H, dt, J 9.5, 6.5,
OC//H), 3.48 (1H, dt, J 9.5, 6.5, OCH//), 1.76-1.25 (6H, m, CH2 , CH2CH2), 1.71 (3H, s,
C=CCH3 ), 1.02 (3H, s, CH3 ), 1.00 (3H, s, CH3 ), 0.92 (3H, t, J7.5, CH2C//3);5C (63 MHz,
CDC1 3 ) 145.9 (C), 107.4 (CH), 98.4 (CH), 68.6 (CH2), 41.7 (CH2 ), 31.8 (CH2), 31.4 (CH3 ),
30.5 (CH3 ), 29.6 (C), 19.7 (CH3 ), 19.3 (CH2), 13.8 (CH3 ); m/z (CI) 199 (M//", 100%), Found: MH+ , 199.1695. Ci 2 H22O2 requires MH+, 199.1698.
(2S*,4R*}-2-Ethoxy-6-methyl-4-phenyl-3,4-dihydro-2H-pyran 23287 : Prepared by the
173

general procedure (1.0 eq of diene, 5.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 65 °C, 5 d)
to give 232 (210 mg, 50%) as a pale yellow oil. vmax (CHCy/cm' 1 3067-2872 (CH), 1677
(C=C); 5H (250 MHz, CDC13) 7.20-7.00 (5H, m, Ph), 4.82 (IH, dd, J 9.0, 2.0, OCHO), 4.50
(IH, m, C=CH), 3.34-3.93 (3H, m, OCH2 , C//CH3 ), 2.11-1.94 (2H, m, CHC//2), 1.77 (3H,
s, CCH3 ), 1.15 (3H, t,J 7.0 , CH2C//3 ); m/z (CI) 219 (M?/", 100%), Found: MH+ , 219.1384.
Ci4Hi 8O2 requires MH+, 219.1385.
(2S ,4S }-2-Benzyloxymethyl-2-ethoxy-6-methyl-3,4-dihydro-2H-pyran 23987 : Prepared by
the general procedure (1.0 eq of diene, 10.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 55 °C,
1 d) to give 239 (0.60 g, 67%) as a pale yellow oil. vmax (CHCl^/cm' 1 2976-2857 (CH),
1714, 1643 (C=C); 5H (250 MHz, CDC13 ) 7.38-7.23 (5H, Ph), 5.01 (IH, dd, J 7.5, 1.0,
OCHO), 4.55-4.51 (3H, CH3C=C/f, PhCH2), 4.00 (IH, dq, J 9.5, 7.0, OC//HCH3 ), 3.60
(IH, dq, J 9.5, 7.0, OCH//CH3 ), 3.43 (2H, d, J 7.0, BnOCH2), 2.73-2.55 (IH, m,
BnOCH2C//), 2.09 (IH, ddd, J 13.0, 6.5, 2.0, CHC//2CH), 1.77 (3H, s, CCH3 ), 1.67 (IH,
ddd, J 13.0, 6.5, 2.0, CHC//2CH), 1.26 (3H, t, J 7.0, CH2C//3 ); m/z (CI) 263 (Mff, 100%),
280 (MNH4+\ Found: MH+, 263.1652. Ci 6H22O3 requires MH+, 263.1647.
(2S*,4S\2-Ethoxy-6-methyl-4-(Z-octen-5-yl)-3,4-dihydro-2H-pyran 24087 : Prepared by the
general procedure (1.0 eq of diene, 10.0 eq of vinyl ether, 0.05 eq of Yb catalyst, 55 °C, 5
d) to give 240 (0.55 g, 56%) in a 4:1 inseparable mixture as a pale yellow oil. vmax
(CHCy/cnV 1 3002-2855 (CH), 1719, 1652 (C=C); 5H (250 MHz, CDC13 ) 5.40-5.24 (4Hboth,
m, 2 x CH=CH), 5.01 (lHminor, t, J3.0, OCHO), 4.88 (lHmajor, dd, J9.0, 2.0, OCHO), 4.51-
4.48 (lHminor, m, C=CH), 4.49-4.36 (lHmajor, m, C=CH), 4.01-3.77 (2Hboth , dt, J 9.5, 7.0, 2
x OC//H), 3.63-3.50 (2Hboth , 2 x OCH/f), 2.32-1.80 (10Hboth , m, 2 x C=CHC//CH2 , 2 x
CH2 , 2 x CH2 ), 1.72 (6Hboth , s, 2 x CH=CC//3), 1.52-1.12 (22Hboth , m, 8 x CH2 , 2 x
OCH2C//3 ), 0.94 (6Hboth , t, J 7.5, 2 x CH2C//3 ); m/z (CI) 253 (Mlf, 100%), 270 (MNH4\
100%), Found: MH+, 253.2172. C 16H28O2 requires MH+, 253.2168
(2S*,4S*}-2-Ethoxy-4-methyl-6-propyl-3,4-dihydro-2H-pyran 271: Prepared by the general
174

procedure (1.0 eq of diene, 5.0 eq of ethyl vinyl ether, 0.05 eq of Yb catalyst, 55 °C, 5 d) to
give 271 (0.55 g, 70%) in a 4:1 inseparable mixture as a colourless oil. vmax (CHCy/crrf 1
2961-2873 (CH), 1675 (OC); 6H (250 MHz, CDC13 ) 5.00 (IM,™, t, J 3.0, OCHO), 4.88
(IH™^, dd, J 8.5, 2.0, OCHO), 4.44 (lHminor , d, J 1.0, C=CH), 4.33 (lHmajor, d, J 1.0,
C=CH), 3.93 (IHmajor, dq, J 9.5, 7.0, OC//H), 3.56 (lHmajor, dq, J9.5, 7.0, OCH#), 2.48-
2.28 (2Hboth, m 2 x C//CH3 ), 2.03-1.90 (4Hboth, m, 2 x CH2), 1.55-1.14 (8Hboth , 2 x CH2 , 2 x
CH2), 1.23 (3Hmajor, t, J 7.0, OCH2C//3 ), 0.99 (3Hmajor , d, J 7.0, CHC//3), 0.89 (3Hmajor, t, J
7.5, CH2CH2C//3), 13C could not be obtained due to the rapid decomposition of the sample
in CDC13 ; m/z (CI) 185 (Mff,WQ%\ Found: MH+, 185.1550. CnH20O2 requires MH+,
185.1542.
(2S ,4S }-6-Butyl-2-ethoxy-4-methyl-3,4-dihydro-2H-pyran 212: Prepared by the general
HDA procedure (1.0 eq of diene, 5.0 eq of ethyl vinyl ether, 0.05 eq of Yb catalyst, 55 °C,
3h, microwave) to give 272 (1.01 g, 65%) in a 4:1 inseparable mixture as a colourless oil.
VmaxtCHCy/cm- 1 2957-2872 (CH), 1673 (C=C); 6H (250 MHz, CDC13) 5.02 (lHmajor, t, J
3.0, OCHO), 4.90 (lHmmor, dd, J 8.5, 2.0, OCHO), 4.45 (!Hmajor, d, J 0.5, C=CH), 4.35
(lHmmor, d, J 1.0, C=CH), 3.85 (lHmajor, dq, J9.5, 7.0, OC//H), 3.58 (IH^^, dq, J9.5, 7.0,
OCH//), 2.48-2.33 (lHmajor , m C//CH3 ), 2.09-1.95 (2Hmajor , m, CH2), 1.51-1.18 (18Hboth, m,
2 x CH2, 2 x CH2 , 2 x CH2> 2 x OCH2C//3 ), 1.01 (3Hminor , d, J7.0, CHC//3 ), 1.00 (3Hmajor, d,
J 7.0, CHC//3), 0.92 (3Hmajor, t, .77.0, CHC//3 ); 5C (125 MHz, CDC13 ) 151.2 (C)minor , 149.8
(C)major , 102.7 CH)major, 102.0 (CH)minor , 96.7 (CH)major, 99.8 (CH)mmor, 64.1 (CH2)minor, 63.5
(CH2)maj0r, 36.7 (CH2)minor, 35.2 (CH2)major, 33.7 (CH2)minor 33.4 (CH2)major , 29.2 (CH2)maj or ,
29.1 (CH2)minor, 26.4 (CH)minor, 22.1 (CH)major, 21.7 (CH,)^^, 21.6 (CH2)minor, 15.3
(CH3 )maj0r, 15.2 (CH3)major, 13.9 (CH3)major , 13.9 (CH3 )minor [22 out of 24 carbons observed];
m/z (CI) 199 (M?/f,100%) Found: MH+, 199.1697. C 12H22O2 requires MH+ , 199.1698.
175

Preparation of enone systems(E)-5-Benzyloxy-pent-3-en-2-one 241 16°:
=O __________
BnO241
To a solution of LiCl (0.30 g, 7.2 mmol) in MeCN (55 mL) at room temperature under N2
was added a solution of diethyl(2-oxopropyl)phosphonate (1.40 g, 7.2 mmol) in MeCN (10
mL). To the reaction mixture was added DBU (0.9 ml, 6.0 mmol) dropwise and then a
solution of benzyloxyacetaldehyde (0.9 g, 6.0 mmol) in THF/MeCN (1/1, 30 mL) was
added. After stirring overnight, the reaction mixture was quenched with water/brine (1/1,
total volume 200 mL). The organic phases were separated and extracted with EtOAc (2 x
50 mL). The combined organic layers were washed with brine (50 mL), dried over
MgSC>4 and concentrated to afford the crude product. Column chromatography eluting with
EtOAc/petrol (1/3) gave 241 (0.67 g, 59%) as a colourless oil. 5H (250 MHz, CDC13 ) 7.38-
7.30 (5H, m, Ph), 6.81 (1H, dt, J 16.0, 4.5, BnOCH2C//), 6.35 (1H, dt, J 16.0, 2.0,
C//C(O)CH3 ), 4.58 (2H, s, OCH2Ph), 4.21 (2H, dd, .74.5, 2.0, CH2OBn), 2.28 (3H, s, CH3 );
m/z (CI) 191 (MH*\ 208 (MNH4+, 100%) Found: MH+, 191.1063. C 12H 14O2 requires MH+,
191.1072.
(3E, 9Z)-Dodecadien-2-one 24287 :
To a solution of LiCl (0.50 g, 12.0 mmol) MeCN (90 mL) at room temperature under N 2
176

was added diethyl(2-oxopropyl)phosphonate (2.70 g, 12.0 mmol) in MeCN (20 mL). To the
reaction mixture was added DBU (1.50 g, 10.0 mmol) dropwise and then a solution of cis-
6-nonenal (1.40 g, 10.0 mmol) in THF:MeCN (1/1, 40 mL) was added. After stirring for
overnight, the reaction mixture was quenched with water/brine (1:1, total volume 200 mL).
The phases were separated and extracted with EtOAc (2 x 50 mL). The combined organic
layers were washed with brine (50 mL), dried over MgSO4 and concentrated to afford the
crude product. Column chromatography eluting with EtOAc/petrol (3/7) gave 242 (1.20 g,
66%) as a colourless oil. 5H (250 MHz, CDC13 ) 6.79 (IH, dt, J 16.0, 7.0, C//=CHC(O)CH3 ),
6.05 (IH, dt, J 16.0, 1.5, CH=C//C(O)CH3 ), 5.42-5.22 (2H, m, CH2C//=C#CH2), 2.26-
2.16 (2H, m, CH2), 2.22 (3H, s, COCH3 ), 2.08-1.95 (4H, m, 2 x CH2), 1.54-1.29 (4H, m, 2
x CH2), 0.94 (3H, t, .77.5, CH2C//3 ); m/z (CI) 181 (MH*\ 198 (MNH4+\
(E)-Hept-2-en-4-one 269 161 :
269
To a solution of DMSO in CH2C12 (25 mL) at -78 "C was added to a solution of 2.0 M of
oxalyl chloride in CH2C12 (5 mL) dropwise. After stirring for 20 min, a solution of alcohol
(1.00 g, 8.70 mmol) in CH2C12 (10 mL) was added dropwise. After stirring for 10 min, Et3N
was added dropwise and the reaction mixture allowed to warm to room temperature. After
stirring for 30 min, the reaction mixture was quenched by the addition of H2O. The organic
layer was extracted, dried over MgSC>4 and concentrated to give yellow oil. Column
chromatography eluting with EtO Ac/petrol (1/4) gave 269 (0.49 g, 50%) as yellow oil. 5H
(250 MHz, CDC1 3) 6.80 (IH, dq, J 13.0, 6.5, CH=C//CH3), 6.09 (IH, d, J 13.0,
C//=CHCH3 ), 2.47 (2H,t,J 7.5, C//2C(0)CH), 1.86 (3H, d, J 6.5, CHC//3 ), 1.68-1.51 (2H,
m, CH3 C//2 ), 0.90 (3H, t, J7.5, CH2C//3 ); m/z (CI) 113 (Mrf\ 130 (MNH4\ 100%).
177

General procedure for the oxidative rearrangement using MCPBA
215
Dihydropyrans
211
217
218
R
Et'Bu
nBu
Product
lactol ether
213
219
220
lactol
215
215
215
To a solution of 2-alkoxydihydropyrans (211, 217, 218, 1.0 eq) in CH2C12 , 77% MCPBA
(1.0 eq, max) in CH2 C12 at 0 U C under N2 was added dropwise. After stirring at room
temperature, the white precipitate was filtered off from the reaction mixture and then
phosphate buffer solution was added for the work-up. The reaction mixture was quenched
with aqueous saturated NaHCO3 solution, dried with MgSC>4 and filtered. Column
chromatography eluting with EtOAc/petrol (1/4-> 1/1) gave lactol ether (213, 217, 220) in a
mixture of diastereoisomers for the major product as a colourless oil and volatile lactol 215
in an inseparable mixture (1:1) for the minor product as a colourless oil.
sfc * %• #
(2R ,5R )-l-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone and (2R ,5S )-l-(5-hydroxy-
tetrahydro-furan-2-yl)-ethanone 21595a : vmax (CHCy/cm' 1 2959 (CH), 1716 (C=O), 1649;
6H (250 MHz, CDC13) 5.60 (!Hone , t, 72.5, C//OH), 5.53 (lHanother , t, J 2.5, C//OH), 5.11
(2Hboth, s, 2 x OH), 4.53 (lHone, dd, J 9.0, 5.0, C(O)CH), 4.36 (lHanother , t, J 8.0, C(O)CH),
2.53-1.72 (8Hboth, m, 4 x CH2), 2.15 (3Hone, s, CH3 ), 2.08 (3Hanother, s, CH3 ); 5C (125 MHz,
CDC13 ) 210.5, 209.2 (C), 99.4, 99.2 (CH), 84.0, 82.5 (CH), 33.3, 32.2 (CH 2 ), 26.5, 25.9
(CH3 ), 25.5, 25.1 (CH2 ); m/z (CI) 148 (MV///, 100%), Found: MNH4+ , 148.0975. C 6 H, ()O 3 requires MNH4+ , 148.0974.
178

(2R ,5R )-l-(5-Ethoxy-tetrahydro-furan-2-yl)-ethanone and (2R ,5S )-l-(5-ethoxy-
tetrahydro-furan-2-yl)-ethanone 21388 : Prepared by the general procedure (1.4 mmol of 2-
alkoxydihydropyran, 1.4 mmol MCPBA, 2 h) to give 213 (32 mg, > 14%) in an inseparable
mixture (2:1) as a colourless oil. vmax (CHCl3 )/cm- 1 2979 (CH), 2254, 1716 (OO); 8H (250
MHz, CDC13 ) 5.25 (1H™, t, J 3.5, OCHO), 5.18 (lHmajor , t, J 3.5, OCHO), 4.45 (lHmmor,
dd, J 9.0, 5.5, C(O)CH), 4.32 (lHmajor , t, J 8.0, C(O)CH), 3.85 (2Hboth, dq, J 9.5, 7.0, 2 x
OC//H), 3.46 (2Hboth, dq, J 9.5, 7.0, 2 x OCH//), 2.42-1.80 (8Hboth, m, 4 x CH2), 2.22
(3Hmajor, s, C(0)CH3), 2.18 (3H™, s, C(O)CH3), 1.17 (3Hminor, t, J 7.0, CH2C//3 ), 1.20
(3Hmajor , t, J 7.0, CH2CH3 ); 6C (125 MHz, CDC13 ) 105.0, 104.6 (CH), 84.7, 82.4 (CH), 63.2,
61.2 (CH2), 32.9, 31.6 (CH3), 26.9, 26.7 (CH2), 25.5, 19.1 (CH2), 15.0, 15.2 (CH3 ).
(2R ,5R )-l-(5-tert-Butoxy-tetrahydro-furan-2-yl)-ethanone and (2R ,5R )-l-(5-tert-butoxy-
tetrahydro-furan-2-yl)-ethanone 219: Prepared by the general procedure (1.2 mmol of 2-
alkoxydihydropyran, 1.2 mmol MCPBA, 4 h) to give 219 (82 mg, 37%) as an inseparable
mixture (2:1) in a pale yellow oil. v^tCHCy/cm' 1 2962-2875 (CH), 1714 (C=O), 1641;
5H (250 MHz, CDC13 ) 5.68 (lHminor, t, J 3.0, OCHO), 5.45 (lHmajor, t, J 3.0, OCHO), 4.50
(lHminor, t, J 8.0, C(O)CH), 4.18 (lHmajor, t, J 8.0, C(O)CH), 2.32-1.65 (14Hboth , m, 2 x
C(O)CH3 , 4 x CH2), 1.15 (18Hboth , s, 6 x CC//3 ); 6C (125 MHz, CDC13 ) 212.2 (C)major , 209.6
(C)minor, 99.8 (CH)major , 99.8 (CH)minor , 84.5 (CH)major , 82.0 (CH)mmor, 74.3 (C)both , 33.8
(CH3)major, 31.2 (CH3)minor , 28.8 (3 x CHaW, 28.7 (3 x CH3 )major, 27.2 (CH2)major, 27.0
(CH2)minor , 26.1(CH2)both ; m/z (CI) 204 (MNH4+\ Found: MNH/, 204.1595. CioH 18O3
requires MNH4+, 204.1560.
(2R*,5R*)-l-(5-Butoxy-tetrahydro-furan-2-yl)-ethanone and (2R ,5S )-l-(5-butoxy-
tetrahydro-furan-2-yl)-ethanone 220: Prepared by the general procedure (3.0 mmol of 2-
alkoxydihydropyran, 3.0 mmol MCPBA, 3h) to give major compounds 220a (74 mg, 13%)
and 220b (146 mg, 26%) as a separable mixture (2:1) in a colourless oil (220 mg,
220a+220b=39%).
220a (major): vmax (CHCl3 )/cm-' 2962-2876 (CH), 1722 (C=O), 1644; 6H (250 MHz,
79

CDC13 ) 5.28 (1H, t, J2.0, OCHO), 4.49 (1H, dd, J 9.0, 6.0, C(O)CH), 3.70 (1H, dt, .7 9.5, 6.5, OC//H), 3.42 (1H, dt, J 9.5, 6.5, OCH//), 2.40-1.79 (4H, m, CH2 , CH2), 2.19 (3H, s, CCH3 ), 1.62-1.42 (2H, m, CH2), 1.42-1.25 (2H, m, CH2), 0.91 (3H, t, y 7.5, CH2C//3 ); 5C (125 MHz, CDC13 ), 209.0 (C), 104.8 (CH), 82.4 (CH), 67.4 (CH2 ), 31.7 (CH2), 31.6 (CH2 ), 26.9 (CH2), 26.1 (CH3 ), 19.3 (CH2), 13.8 (CH3 ); m/z (CI) 171 (Mff\ Found: MNH4+, 204.1595. Ci 0H 18O3 requires MNH4+, 204.1560.
220b (minor): vmax (CHCy/cm' 1 2962-2876 (CH), 1721 (C=O), 1642; 5H (250 MHz,
CDC13 ) 5.19 (1H, dd, J4.5, 1.5, OCHO), 4.34 (1H, t, J8.0, C(O)CH), 3.80 (1H, dt, J 9.5, 6.5, OC//H), 3.43 (1H, dt, J 9.5, 6.5, OCH//), 2.25 (3H, s, CH3 ), 2.22-1.87 (4H, m, CH2 , CH2), 1.62-1.43 (2H, m, CH2), 1.42-1.22 (2H, m, CH2), 0.92 (3H, t, / 6.5, CH2C//3); 8C (125 MHz, CDC13 ), 211.0 (C), 105.3 (CH), 84.7 (CH), 67.7 (CH2 ), 32.8 (CH2), 31.7 (CH2), 26.7 (CH2), 25.5 (CH3), 19.4 (CH2), 13.9 (CH3); m/z (CI) 171 (Mrf\ Found: MNH4+, 204.1595. C 10H 18O3 requires MNH4+, 204.1560.
Oxidation of 2-ethoxy-6-methyl-3,4-dihydro-2H-pyran 211 using an excess of MCPBA 5-Ethoxy-dihydro-furan-2-one 212 ' :
OEt212
To a solution of 2-alkoxydihydropyran 211 (200 mg, 1.4 mmol) in CH2C12 (5 mL) at 0 °C
was added the excess of MCPBA. After stirring at room temperature under N2 for 2 h, the reaction mixture was filtered and the solvent removed under reduced pressure. Column chromatography eluting with EtOAc/Et3N/petrol (1/0.2/4) gave dihydrofuranone 212 (40 mg, 18%) as a pale yellow oil, vmax (CHCy/cm' 1 1783 (CO); 5H (270 MHz, CDC1 3 ) 5.55
(1H, d,73.5, OCHO), 3.86 (1H, dq, /9.5, 7.0, OC//H), 3.66 (1H, dq, J9.5, 7.0, OCH//), 2.75-2.05 (4H, m, CH2CH2 ,), 1.18 (3H, t, J 7.0, CH3); 6C (68 MHz, CDC1 3) 176.6 (C),
180

104.0 (CH), 64.0 (CH2), 28.8 (CH2), 26.8 (CH2), 14.9 (CH3 ); m/z (CI) 148 (AflW//, 100%),
Found: MH+, 131.0714. C6Hi 0O3 requires MH+, 131.0708.
Preparation of DMDO97
oA
A mixture of NaHCOs in acetone and water (3:4, 446 mL) was cooled down in ice bath.
Oxone® (120 g) was added portionwise (over 5 times, at 3 minutes intervals) with stirring.
After the last Oxone addition, the vacuum pump was slowly opened until the mixture
became foaming. The ice bath was removed. The distillation lasted until no more foaming
was observed under high vacuum pressure. DMDO in acetone was collected as a yellow
liquid by dry ice trap. The DMDO/acetone solution was dried with K2CO3 and filtered. For
the titration of DMDO, DMDO/acetone (1 mL) with the unknown concentration, the
mixture of acetic acid and acetone (3:2, 2 mL) and saturated KI solution (2 mL) was mixed
to give a brown solution. The mixture (1 mL) was titrated with aqueous Na2 S2O3 solution
(0.002 M). The concentration of DMDO was approximately 0.03 M.
splashhead
Oxone
to vacuum
CO2/acetone trap (CO2 piled around necl
ice bath (removed after Oxone addition)
181
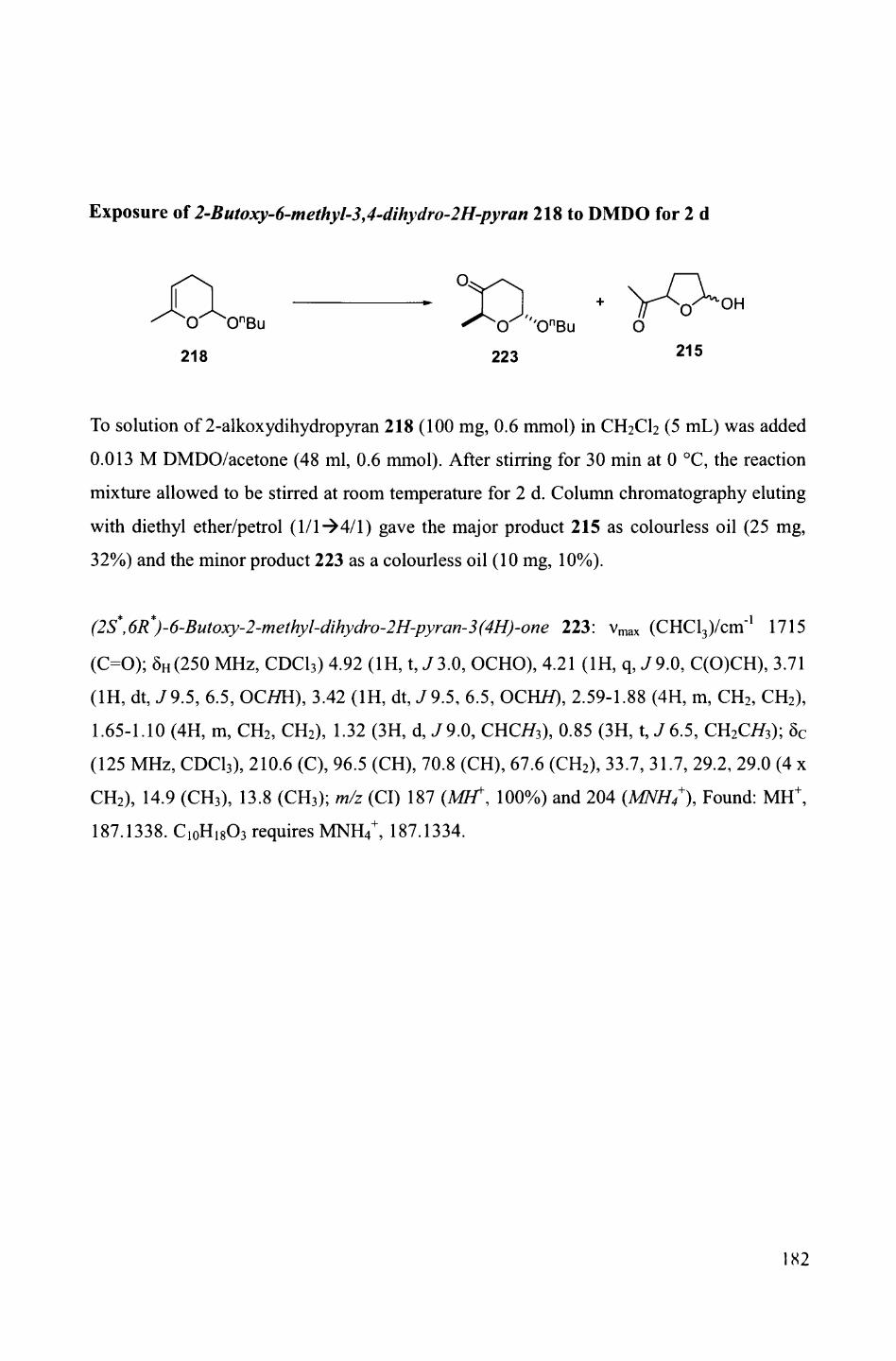
Exposure of 2-Butoxy-6-methyl-3,4-dihydro-2H-pyran 218 to DMDO for 2 d
"'On Bu
223 215
To solution of 2-alkoxydihydropyran 218 (100 mg, 0.6 mmol) in CH2 C12 (5 mL) was added
0.013 M DMDO/acetone (48 ml, 0.6 mmol). After stirring for 30 min at 0 °C, the reaction
mixture allowed to be stirred at room temperature for 2 d. Column chromatography eluting
with diethyl ether/petrol (!/!-> 4/1) gave the major product 215 as colourless oil (25 mg,
32%) and the minor product 223 as a colourless oil (10 mg, 10%).
(2S,6R)-6-Butoxy-2-methyl-dihydro-2H-pyran-3(4H)-one 223: vmax (CHCy/cm' 1715
(C=O); 6H (250 MHz, CDC13 ) 4.92 (IH, t, J 3.0, OCHO), 4.21 (IH, q, y 9.0, C(O)CH), 3.71
(IH, dt, y 9.5, 6.5, OC//H), 3.42 (IH, dt, J 9.5, 6.5, OCH#), 2.59-1.88 (4H, m, CH2 , CH2),
1.65-1.10 (4H, m, CH2 , CH2), 1.32 (3H, d, J 9.0, CHC//3 ), 0.85 (3H, t, J 6.5, CH2C//3); 5C
(125 MHz, CDC13 ), 210.6 (C), 96.5 (CH), 70.8 (CH), 67.6 (CH2 ), 33.7, 31.7, 29.2, 29.0 (4 x
CH2), 14.9 (CH3), 13.8 (CH3 ); m/z (CI) 187 (M/*, 100%) and 204 (MNH4+\ Found: MH+ ,
187.1338. Ci 0Hi 8O3 requires MNH4+, 187.1334.
182

General procedure for the oxidative rearrangement using DMDO/acetone
R 1
Et<Bu
nBu
"Bu
Et
R2
H
H
H
Me
Ph
Product
lactol ether
213
219
220
248
N/A
lactol
215
215
215
249
234
To solution of 2-alkoxydihydropyran in CtbCb at 0 °C was added a DMDO/acetone
solution. After stirring for 30 min, the reaction mixture was allowed to stir at room
temperature. Column chromatography eluting with diethyl ether/petrol gave lactols as
major products and lactol ethers as minor products. §
l-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone 215 and l-(5-ethoxy-tetrahydro-furan-2-yl)-
ethanone 213: Prepared by the general procedure (3.5 mmol of 2-alkoxydihydropyran, 0.04
M DMDO/acetone 90 mL, 3 h) to give 215 (263 mg, >53%) as a major product.
1-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone 215 and l-(5-tert-butoxy-tetrahydro-furan-
2-yl)-ethanone 219: Prepared by the general procedure (1.2 mmol of 2-alkoxydihydropyran,
0.03 M DMDO/acetone 40 mL, 3 h) to give 215 (109 mg, >72%) as a major product.
l-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone 215 and l-(5-butoxy-tetrahydro-furan-2-vl)-
§ For the spectral data of products 213, 215, 219, 220; see MCPB A reaction.
183

ethanone 220: Prepared by the general procedure (0.6 mmol of 2-alkoxydihydropyran,
0.013 M DMDO/acetone 48 mL, 3 h) to give 215 (50 mg, >64%) as a major product and
220(10mg,9%).
l-(5-Butoxy-3-methyl-tetrahydro-furan-2-yl)-ethanone 248 and l-(5-hydroxy-3-methyl-
tetrahydro-furan-2-yl) -ethanone 249: Prepared by the general procedure (1.1 mmol of 2-
alkoxydihydropyrans, 0.075 M DMDO/acetone 15 mL, 3 h) to give lactols 249 (15%) in a
mixture of 4 diastereoisomers as colourless oil and give lactol ethers 248 (17 mg, 11%) in a
mixture of 4 diastereoisomers as a colourless oil.
l-(5-Butoxy-3-methyl-tetrahydro-furan-2-yl)-ethanone 248: vmax (CHCl^/cm" 2962-2875
(CH), 1714 (C=O), 1641; 5H (250 MHz, CDCl3 )major 5.14 (IH, dd, J 5.0, 5.0, OCHO), 3.98
3.80 (IH, d, J9.5, C(O)CH), 3.86-3.35 (2H, m, OCH2), 2.52-1.80 (3H, m, CH, CH2), 2.24
(3H, s, COCH3 ), 1.70-1.31 (4H, m, CH2 , CH2), 1.13 (3H, d, J6.5, CHC//3 ), 0.92 (3H, t, J
7.5, CH2C//3 ); 6C (125 MHz, CDCl3 )major 211.1 (C), 104.7 (CH), 92.6 (CH), 67.7 (CH2),
41.5 (CH2), 35.6 (CH2), 31.7 (CH), 25.2 (CH3 ), 19.4 (CH2), 17.0 (CH3 ), 13.8 (CH3); m/z
(CI) 218 (MNH4+\ Found: MNH4+, 218.1753. CnH20O3 requires MNH4+, 218.1756.
l-(5-Hydroxy-3-methyl-tetrahydro-furan-2-yl)-ethanone 249: vmax (CHCy/cm' 1 3414 (OH) ,
2921-2850 (CH), 2091, 1644; 8H (250 MHz, CDCl3 )major 5.59 (IH, t, J4.5, C(O)CH), 3.97
(IH, d, J8.0, OCHCO), 2.57-2.03 (IH, m, CH3 C//), 2.27 (3H, s, COCH3), 1.77-1.53 (2H,
m, CH2), 1.21 (3H, d, J6.5, CHC//3 ); 5C (125 MHz, CDCl3)major 210.4 (C), 99.6 (CH), 90.7
(CH), 42.3 (CH2), 35.4 (CH), 25.8 (CH3 ), 17.9 (CH3); m/z (CI) 144 (M, 100%), 162
(MNH4+\ Found: MNH4+ , 162.1129. C7H 12O3 requires MNH4+, 162.1130.
l-(5-Hydroxy-3-phenyl-tetrahydro-furan-2-yl)-ethanone 234: Prepared by the general
procedure (0.7 mmol of 2-alkoxydihydropyran, 0.025 M DMDO/acetone 29 mL, 3 h) to
give major product, lactols 234 (80%), in a mixture of 4 diastereoisomers as a colourless oil.
vmax (CHCy/cirf 1 3445 (OH), 2944 (CH), 1780, 1714(CO), 1645; 6,, (250 MHz,
1S4

CDCl3 )major 7.38-7.11 (5H, Ph), 5.99 (1H, dd, J 5.0, 2.5, OC//OH), 4.96 (1H, d, J 7.5,
C(O)CH), 4.02-3.77 (1H, m, PhCH), 2.47-2.11 (2H, m, CH2 ), 1.64 (3H, s, C(O)CH3 ); 13 C NMR could not be recorded due to the complexity of spectrum; m/z (CI) 206 (M, 100%),
224 (MNH4+\ Found: MNH4+, 224.1281. Ci 2Hi 4O3 requires MNH4+, 224.1287.
185

General procedure for the oxidative rearrangement using DMDO generated in situ
' O OR
Dihydropyrans
211
218
IT o IT oo o215
R Product
lactol ether lactol
Et 213 215
"Bu 220 215
To a solution of acetone (> 20.0 eq) and 2-alkoxydihydropyrans (1.0 eq) in Cl^Cb at 0 °C
was added 0.4 mM Na2EDTA (aq). After the mixture of Oxone® (2.5 eq) and NaHCO3 (4.0
eq) was ground, it was added to the reaction mixture over 30 minutes. After stirring
vigorously at room temperature for overnight, the organic layer was separated by extraction
with aqueous saturated NaHCOs solution, dried with MgSO4 and filtered. Column
chromatography eluting with diethyl ether/petrol (3:2) gave lactol ether as a major
compound and give lactol as a minor compound.
l-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone 215 and l-(5-ethoxy-tetrahydro-furan-2-yl)-
ethanone 213: Prepared by the general procedure (60.0 mmol of acetone, 3.5 mmol of 2-
alkoxydihydropyran 211, 8.8 mmol of Oxone® and 14.0 mmol of NaHCOs) to give
lactolether 213 in a mixture of diastereoisomers (1:1) as a colourless oil (74 mg, 13%) and
give a minor compound 215.
l-(5-Hydroxy-tetrahydro-furan-2-yl)-ethanone 215 and l-(5-butoxy-tetrahydro-furan-2-vl)-
For the spectral data of lactol 215 and lactol ether 213 and 220; see the oxidative rearrangement using
MCPBA.
1X6

ethanone 220: Prepared by the general procedure (60.0 mmol of acetone, 1.8 mmol of 2-
alkoxydihydropyran 218, 4.5 mmol of Oxone* and 7.2 mmol of NaHCO 3 ) to give lactol
ether 220 in a mixture of diastereoisomers (1:1) as a colourless oil (74 mg, 13%) and give a minor compound 215.
Attempted oxidation of 2~ethoxy-6-methyl-3,4-dihydro-2H-pyran 211 using hydrogen
peroxide5-Oxo-hexanal224 }62 :
O
224
To solution of 2-alkoxydihydropyran 211 (200 mg, 1.4 mmol) in MeCN (3 mL) was added
a solution of 27 % hydrogen peroxide (176 mg, 1.4 mmol) in MeCN (3 mL). After the
reaction mixture stirred at room temperature for 1 5 h, saturated aqueous sodium sulflte was
used for the work-up. The reaction mixture was dried with MgSC>4, filtered and
concentrated. Column chromatography eluting with diethyl ether/petrol (4:1) to give a pale
yellow oil 224 (37 mg, 20%). vmax (CHCy/cm' 1 1720 (C=O), 1730 (C=O); 5H (500 MHz,
CDC13 ) 9.75 (IH, s, C(O)H), 2.55-2.35 (4H, 2 x CH2 ), 2.10 (3H, s, C(O)CH3 ), 1.86-1.80
(2H, m, CH2); 6C (125 MHz, CDC13 ) 208.0 (C), 43.0 (CH2 ), 42.3 (CH2), 30.0 (CH3 ), 16.0
(CH2) [5 out of 6 carbons observed]; m/z (CI) 115 (M), 131, 132 (MV///), 148 (100%),
Found: MNH4+ 132.1030 C6H 10O2 requires MNH4+ 132.1025.
187

General procedure for the epoxidation of dihydropyrans using hydrogen peroxide andMT099d
O OR ' X) OR
Tentatively assigned products
Dihydropyrans R Product
211 Et225
218 nBu 226
To the mixture of MTO (0.03 eq) and pyridine (0.6 eq) in CH2C12 was added 27 %
hydrogen peroxide (3.0 eq). To a solution of 2-alkoxydihydropyran (1.0 eq) in CH2 Cl2 was
added the solution of the oxidant dropwise at room temperature under N2 condition. After
the reaction mixture stirred for 8 h, saturated aqueous NaHCO3 was used for the work-up.
The reaction mixture was dried with MgSC>4, filtered and concentrated to give the crude as
a pale yellow oil.
Tentatively assigned product, 3-ethoxy-l-methyl-2,7-dioxa-bicyclo[4,l,0]heptane: Prepared
by the general procedure (0.02 mmol of MTO, 0.8 mmol of pyridine, 10.5 mmol of 27%
hydrogen peroxide and 3.5 mmol of 2-alkoxydihydropyran 211) to give the crude 225 as a
pale yellow oil. 6H (250 MHz, CDC13 ) 4.85 (1H, t, J 3.0, OCHO), 4.30 (1H, t, J 3.0,
CHepox,de), 3.71 (1H, dq, 79.5, 7.0, OC//H), 3.40 (1H, dq, 79.5, 7.0, OCR//), 2.05-1.35 (7H,
m, CCH3 , CH2 , CH2); 1.00 (3H, t, 7 7.0, CH2C//3 ); 13 C NMR could not be recorded due to
the rapid decomposition of the sample.
Tentatively assigned product, 3-butoxy-l-methyl-2,7-dioxa-bicyclo[4,l,0]heptane: Prepared
by the general procedure (0.02 mmol of MTO, 0.8 mmol of pyridine, 10.5 mmol of 27° o
hydrogen peroxide and 3.5 mmol of 2-alkoxydihydropyran 218) to give the crude 226 as a
188

pale yellow oil. 5 H (250 MHz, CDC13 ) 4.92 (1H, t, J 3.0, OCHO), 4.46 (1H, t, J 3.0,
CHepoxKie), 3.73 (1H, dt, 79.5, 6.5, OC//H), 3.43 (1H, dt,79.5, 6.5, OCH//), 2.10-1.19 (8H,
m, CH2 , CH2 , CH2 , CH2), 1.66 (3H, s, CCH3 ), 0.85 (3H, t, 77.5, CH2C//3 ); 13 C NMR could
not be recorded due to the rapid decomposition of the sample.
Oxidation of l-(5-Hydroxy-3-phenyl-tetrahydro-furan-2-yl)-ethanone using PCC
234
To a solution of l-(5-hydroxy-3-phenyl-tetrahydro-furan-2-yl)-ethanone 234 (116 mg, 0.56
mmol) in CH2 C12 (10 ml) was added a suspension of PCC (361 mg, 1.68 mmol) and neutral
alumina (815 mg, 8.9 mmol) in CH2C12 (20 mL). After stirring at room temperature for
overnight, the solution was filtered through a short pad of Florisil and the solid residue was
washed several times with diethyl ether. The solvent was removed under reduced pressure
to give a yellow solid. The colour of the solution turned into dark brown. Column
chromatography eluting with the EtOAc/petrol (3/7) gave (4R*,5R*)-5-acetyl-4-phenyl-
dihydro-furan-2-one 235 as a white solid (100 mg, 88%).n
The preparation of 3.0 M Jones oxidant 103
To a solution of CrO3 (67.0 g) in H2O (125 mL) was added carefully a solution of
concentrated H2 SC>4 (58 mL). The precipitated salts were dissolved by adding an additional
minimal quantity of water (total volume was 225 mL) to give 3.0 M Jones oxidant.
tt For the spectral data; see the rearrangement / Jones oxidation of alkoxydihydropyrans.
189

General procedure for rearrangement / Jones oxidation of alkoxydihydropyrans
R2 R3
R 1
Product
\/
^OR4
R 1 R2
231 Me H
250 Me Me251 Me *Pr
252 Me Me
235 Me Ph
253 Me CH2OBn254 Me (CH2)4CH=CHEt
273 "Pr Me
274 "Bu Me
R3R2T~Y
0
R3
H
H
H
Me
H
H
H
H
H
R4
"Bu
"Bu
"Bu
"Bu
Et
Et
Et
Et
Et
To solution of 2-alkoxydihydropyran in CHiCfe at 0 °C was added DMDO/acetone solution. After stirring for 30 min, the reaction mixture was allowed to warm to room temperature. After the crude was extracted with saturated aqueous NaHCOs solution, it was concentrated
to give the mixture of lactol and lactol ether. The crude mixture from oxidative rearrangement was dissolved into acetone at 0 °C and 3.0 M Jones reagent (3.0 eq to initial 2-alkoxydihydropyran) was added dropwise. After being stirred, the excess of oxidants was quenched by the addition of 2-propanol until the brown colour of the mixture turned to green. The reaction mixture was diluted with diethyl ether and the precipitated chromium salts were dissolved by the addition of saturated aqueous NH4C1 solution. The organic layer was separated and the aqueous layer was extracted with diethyl ether. Column chromatography gave 4,5-czs'-lactones as major products and 4,5-/ra/?s-lactones as minor
products. Structures of 4,5-c/s-isomers and 4,5-/nms-isomers were assigned by NOESY
190

experiment.**
5-Acetyl-dihydro-furan-2-one 231 163 : Prepared by the general procedure (0.9 mmol of 2-
alkoxydihydropyran, 0.013 M DMDO 70 mL, 3.0 M Jones reagent 0.9 mL, 3 h for DMDO
oxidation, 30 min for Jones oxidation) to give lactone 231 as a pale yellow oil (80 mg, 69%
over 2 steps). vmax (CHCl3 )/cm~' 1767 (lactone), 1723 (OO), 1634; 5H (250 MHz, CDC13 )
4.81 (1H, t, J 7.5, CH), 2.62-2.18 (4H, m, CH2CH2) 2.30 (3H, s, CH3 ); 6C (125 MHz,
CDC13 ), 205.4 (C), 175.7 (C), 82.0 (CH), 27.4 (CH2), 26.2 (CH3 ), 24.4 (CH2 ); m/z (CI) 146
(MNH4+, 100%) Found: MNH/, 146.0814. C6H8O3 requires MNH4+, 146.0817.
^c :£ * *
(4S ,5R )-5-Acetyl-4-methyl-dihydro-furcm-2-one 250a and (4S ,5R )-5-acetyl-4-methyl- dihydro-furan-2-one 250b 164 : Prepared by the general procedure (0.9 mmol of 2-
alkoxydihydropyran, 0.013 M DMDO 70 mL, 3.0 M Jones reagent 0.9 mL, 3 h for DMDO oxidation, 30 min for Jones oxidation) to give lactone 250 in a 3:1 mixture§§ ; 250a (45 mg,
35% over 2 steps) as a major product in pale yellow oil and 250b (20 mg, 16 % over 2
steps) as a minor product in a pale yellow oil.
(4R*,5R*)-5-Acetyl-4-methyl-dihydro-fitran-2-one 250a: vmax (CHCy/cm' 1 3058-2989 (CH),
1791 (lactone), 1720 (C=O), 1643; 5H (250 MHz, CDC13 ) 4.80 (1H, d, 77.0, OCH), 3.02-
2.82 (1H, m, C//CH3 ), 2.75 (1H, dd, J 17.0, 8.0, C//H), 2.29 (1H, dd, J 17.0, 8.0, CH#),
2.27 (3H, s, C(O)CH3 ), 1.02 (3H, d, J 7.0, CHCH3); 5C (125 MHz, CDC13 ), 205.7 (C),
175.4 (C), 85.1 (CH), 36.4 (CH2), 33.0 (CH), 28.5 (CH3 ), 14.8 (CH3 ); m/z (CI) 160 (MNH4\ 100%), Found: MNH4+, 160.0972. C7Hi 0O3 requires MNH4+, 160.0974.
(4S*,5R*)-5-Acetyl-4-methyl-dihydro-furan-2-one 250b: vmax (CHCy/cm" 1 1787 (lactone),
1723 (CO), 1613; 5H (250 MHz, CDC13) 4.33 (1H, d, J 7.0, OCH), 2.72 (1H, dd, J 17.0,
8.5, CH2 ), 2.23 (1H, dd, J 17.0, 8.5, CH2), 2.55 (1H, m, CH), 2.28 (3H, s, C(O)CH 3 ), 1.28
(3H, d, J 6.5, CHC//3 ); 6C (125 MHz, CDC13), 205.4 (C), 175.1 (C), 88.1 (CH), 35.9 (CH2),
33.6 (CH), 26.2 (CH3 ), 18.4 (CH3 ); m/z (CI) 160 (MV///, 100%), Found: MNH4+ ,
For NOESY spectra; see appendix. Ratio determined by 'H-NMR
191

160.0972. C7HioO3 requires MNH4+, 160.0974.
(4R ,5R )-5-Acetyl-4-isopropyl-dihydro-furan-2-one 251a and (4S*,5R*)-5-acetyl-4-
isopropyl-dihydro-furan-2-one 251b: Prepared by the general procedure (0.9 mmol of 2-
alkoxydihydropyran, 0.045 M DMDO 20 mL, 3.0 M Jones reagent 0.9 mL, 3 h for DMDO
oxidation, 30 min for Jones oxidation) to give an inseparable mixture of diastereoisomers,
251a and 251b in a 9:1 ratio (96 mg, 63% over 2 steps), as a colourless oil. vmax
(CHCy/cm 1 2965-2879 (CH), 1787 (lactone), 1720 (C=O), 1644; 5H (250 MHz, CDC13)
4.88 (lHmajor, d, J 7.0, OCR), 4.56 (lHminor , d, .7 5.0, OCR) 2.64-2.34 (6Hboth , m, 2 x CH2 , 2
x COCH), 2.29 (3Hmajor, s, C(O)CH3 ), 2.26 (3Hmmor, s, C(O)CH3 ), 1.90 (lHmmor, m,
C//CH3 ), 1.68 (lHmajor, m, C//CH3), 0.99 (3Hmajor , d, .7 6.5, CHC//3), 0.96 (3Hmmor, d, J 2.0,
CHC/ft), 0.94 (3Hminor, d, J 2.0, CHC//3 ), 0.87 (3Hmajor , d, J 6.5, CHC//3); 5C (125 MHz,
CDCl3 )major , 205.7 (C), 175.4 (C), 84.3 (CH), 46.4 (CH2 ), 31.3 (CH), 29.1 (CH), 27.6 (CH3 ), 21.4 (CH3 ), 20.0 (CH3); m/z (CI) 188 (MNH4+, 100%), Found: MNH4+ , 188.1289. C9H14O3
requires MNH4+, 188.1287.
5-Acetyl-4,4-dimethyl-dihydrofuran-2(3H)-one 252 165 : Prepared by the general procedure
(1.8 mmol of 2-alkoxydihydropyran, 0.035 M DMDO 50 mL, 3.0 M Jones reagent 2.3 mL,
3 h for DMDO oxidation, 30 min for Jones oxidation) to give lactone 252 as a colourless
oil (136 mg, 50% over 2 steps). vmax (CHCy/cm' 1 2969-2878 (CH), 2358, 1789 (lactone),
1714 (C=O), 1634; 5H (250 MHz, CDC13) 4.40 (1H, s, OCH), 2.43 (2H, d, .73.5, CH2), 2.27
(3H, s, C(O)CH3 ), 1.35 (3H, s, CH3), 1.05 (3H, s, CH3 ); 6C (125 MHz, CDC13), 206.1 (C),
174.7 (C), 89.7 (CH), 43.9 (CH2), 40.7 (C), 28.4, 26.2, 22.1 (3 x CH3); m/z (CI) 174
(MNH4+, 100%), Found: MNH4+, 174.1130. C 8Hi 2O3 requires MNH4+, 174.1130.
(4R*,5R*)-5-Acetyl-4-phenyl-dihydro-furan-2-one 235: Prepared by the general procedure
(0.9 mmol of 2-alkoxydihydropyran, 0.045 M DMDO 20 mL, 3.0 M Jones reagent 2.7 mL,
3 h for DMDO oxidation, 30 min for Jones oxidation) to give the only one diastereoisomer
lactone 235 as a white solid (120 mg, 65% over 2 steps), mp 92-93 °C;
192

3035-2877 (CH), 1772 (lactone), 1721 (C=O), 1650; 5H (250 MHz, CDC13 ) 7.35-7.13 (5H,
m, Ph), 5.09 (1H, d, J 8.0, OCR), 4.05 (1H, dt, J 8.0, 6.0, CH), 2.96 (1H, dd, J 17.5, 8.5,
C//H), 2.88 (1H, dd, J 17.5, 6.0, CH//), 1.64 (3H, s, CH3 ); 6C (125 MHz, CDC13 ), 205.0 (C),
175.6 (C), 135.8 (C), 129.2 (2 x CH), 128.4 (CH), 127.6 (2 x CH), 85.6 (CH), 44.0 (CH),
34.5 (CH2), 28.0 (CH3 ); m/z (CI) 222 (MNH4+, 100%), Found: MNH4+, 222.1127. C 12H 12O3
requires MNH4+, 222.1130
(4R ,5R )-5-Acetyl-4-(benzyloxymethyl)-dihydrofuran-2(3H)-one 253: Prepared by the
general procedure (0.8 mmol of 2-alkoxydihydropyran, 0.02 M DMDO 40 mL, 3.0 M
Jones reagent 2.4 mL, 3 h for DMDO oxidation, 30 min for Jones oxidation) to give the
only one diastereoisomer 253 (>95:5) as a colourless oil (96 mg, 48% over 2 steps), vmax
(CHCy/crrf 1 2874 (CH), 2089, 1780 (lactone), 1645; 5H (250 MHz, CDC13), 7.39-7.19 (5H,
m, Ph), 4.80 (1H, d, J 8.0, C(O)CH), 4.40 (1H, d, J 21.5, PhCH2), 4.36 (1H, d, J 21.5,
PhCH2), 3.53 (1H, dd, J 9.5, 3.0, BnOCH2), 3.38 (1H, dd, 79.5, 3.0, BnOCH2), 3.03 (1H,
dtt, J 8.0, 9.5, 3.0, BnOCH2C//), 2.72 (1H, dd, J 17.5, 9.5, C(O)CH2), 2.55 (1H, dd, J 17.5,
4.0, C(O)CH2), 2.23 (3H, s, C(O)CH3 ); 5C (125 MHz, CDC13 ) 206.5 (C), 175.5 (C), 136.9
(C), 128.4 (2 x CH), 127.9 (CH), 127.6 (2 x CH), 83.1 (CH), 73.1, 68.0 (2 x CH2), 38.1
(CH2), 32.2 (CH), 28.1 (CH3 ); m/z (CI) 266 (MNH4+, 100%), Found: MNH/, 266.1395.
Ci 4Hi 6O4 requires MNH4+, 266.1392.
(4S*,5R*,Z)-5-Acetyl-4-(oct-5-enyl)-dihydrofuran-2(3H)-one 254: Prepared by the general
procedure (1.2 mmol of 2-alkoxydihydropyran, 0.03 M DMDO 40 mL, 3.0 M Jones
reagent 2.5 mL, 3 h for DMDO oxidation, 30 min for Jones oxidation) to give the only one
diastereoisomer 254 as a colourless oil (130 mg, 64% over 2 steps). vmax (CHCy/cm" 1
3007-2858 (CH), 2358, 2330, 1790 (lactone), 1717 (C=O); 5H (250 MHz, CDC13 ) 5.47-5.14
(2H, m, CH=CH), 4.82 (1H, d, J 7.5, C(O)CH), 2.86-2.65 (1H, m, C(O)CHC//), 2.65 (1H,
dd, J 17.0, 8.0, C(O)C//2CH), 2.37 (1H, dd, J 17.0, 6.0, C(O)C//2CH), 2.26 (3H, s,
C(O)CH 3 ), 2.07-1.96 (2H, m, C//2 CH3 ), 1.52-1.02 (8H, m, 4 x CH 2 ), 0.94 (3H, t, J 7.5,
CH2C//3 ); 6C (125 MHz, CDC13 ) 205.8 (C), 175.5 (C), 132.2, 128.3 (2 x CH), 84.9 (CH),
193

38.7 (CH2 ), 33.6 (CH), 29.3, 28.8, 28.7, 27.1, 26.7 (5 x CH2 ), 20.5 (CH3 ), 14.3 (CH3 ); m/z
(CI) 256 (MNH4+, 100%), Found: MNH4+, 256.1907. Ci4H22O3 requires MNH4+, 256.1913 Structure of 2,3-c/s-254 was assigned by NOESY.
(4S*,5R*)-4-Methyl-5-pentanoyl-dihydrofuran-2(3H)-one 273a and (4R*,5 R*)-4-methyl-5-
pentanoyl-dihydrofuran-2(3H)-one 273b: Prepared by the general procedure (2.6 mmol of
2-alkoxydihydropyran, 0.03 M DMDO 90 mL, 3.0 M Jones reagent 3.0 mL, 3 h for DMDO
oxidation, 30 min for Jones oxidation) to give 273 in a 5:1 mixture***; 273a (290 mg, 58%)
and 273b (60 mg, 12%) as a pale yellow oil.
273a (major): vmax (CHCy/cm' 1 2255, 1788 (lactone), 1719 (C=O); 5H (250 MHz, CDC13 ),
4.80 (1H, d, J 7.0, OCR), 3.08-2.82 (1H, m, CH3 C#), 2.73 (1H, dd, J 17.0, 8.0,
C(O)C//2 CH), 2.29 (1H, dd, J 17.0, 4.5, C(O)C//2 CH), 2.78-2.32 (2H, m, CH2C//2CH3 ),
1.71-1.56 (2H, m, C//2CH2CH3 ), 1.00 (3H, d, y 7.0, CHC//3 ), 0.94 (3H, t, y 7.5, CH2C//3 );
6C (125 MHz, CDC13), 207.8 (C), 175.6 (C), 84.9 (CH), 42.9 (CH2), 36.4 (CH), 33.1, 16.0
(2 x CH2), 14.8, 13.6 (2 x CH3 ); m/z (CI) 188 (MNH4+, 100%) Found: MNH4+, 188.1289.
C9Hi4O3 requires MNH4+, 188.1286.
273b (minor): vmax (CHC\3)/cm l 2255, 1788 (lactone), 1719 (C=O); 6H (250 MHz, CDC13),
4.32 (1H, d, J 7.0, OCH), 2.68 (1H, dd, J 17.0, 8.5, C(O)CH2), 2.19 (1H, dd, J 17.0, 7.5,
C(O)CH2), 2.60-2.43 (3H, m, C//2CH2CH3 ,CH3 C//), 1.61 (2H, tq, J 11.0, 6.5,
CH2C//2CH3 ), 1.26 (3H, d, J 6.5, CHC#3), 0.91 (3H, t, J 6.5, CH2C//3 ); 5C (125 MHz,
CDC13 ), 207.8 (C), 175.0 (C), 87.9 (CH), 40.4, 35.8 (2 x CH2), 33.6 (CH), 18.5 (CH2), 16.2,
13.6 (2 x CH3); m/z (CI) 188 (MNH4+, 100%), Found: MNH/, 188.1284. C9H 14O3 requires
MNH4+, 188.1284.
(4S*,5R*)-4-Methyl-5-pentanoyl-dihydrofuran-2(3H)-one 274a and (4S*,5S*)-4-methyl-5-
pentanoyl-dihydrofuran-2(3H)-one 274b: Prepared by above general procedure (2.8 mmol
of 2-alkoxydihydropyran, 0.07 M DMDO 40 mL, 3.0 M Jones reagent 3.0 mL, 3 h for
Ratio determined by 'H-NMR
194

DMDO oxidation, 30 min for Jones oxidation) to give 274 in a 8:1 mixtureftt ; 274a (250
mg, 48%) and 274b (31 mg, 6%) in a 8:1 ratio in a pale yellow oil.
274a (major): vmax (CHCy/cm' 1 2254, 1789 (lactone), 1716 (C=O); 5H (250 MHz, CDC13 ),
4.80 (IH, d, J 10.0, OCH), 2.94 (IH, m, CH3C//), 2.72 (IH, dd, J 17.5, 8.0, C(O)C//2CH),
2.73-2.40 (2H, m, CH2 in nBu), 2.28 (IH, dd, J 17.5, 4.5, C(O)C//2CH), 1.62-1.54 (2H, m,
CH2 in nBu), 1.38-1.28 (2H, m, CH2 in "Bu), 0.99 (3H, d, J 7.0, CHC//3 ), 0.91 (3H, t, J 7.5,
CH2C//3 ); 6C (125 MHz, CDC13 ), 207.8 (C), 175.6 (C), 84.9 (CH), 40.7 (CH2 ), 36.4 (CH2),
36.4 (CH), 24.6, 22.2 (2 x CH2), 14.8, 13.8 (2 x CH3); m/z (CI) 202 (MNH4+, 100%),
Found: MNH4+, 202.1447. Ci 0Hi 6O3 requires MNH4+ , 202.1443.
274b (minor): vmax (CHCy/cm' 1 2254, 1789 (lactone), 1716 (C=O); 5H (250 MHz, CDC13 ),
4.33 (IH, d, J 9.5, OCH), 2.69 (IH, dd, J 17.5, 8.5, C(O)C//2CH), 2.62-2.48 (3H, m, CH3C//, CH2 in nBu), 2.20 (IH, dd, J 17.5, 4.5, C(O)C//2CH), 1.61-1.53 (2H, m, CH2 in
nBu), 1.37-1.26 (2H, m, CH2 in nBu), 1.27 (3H, d, J 6.5, CHC/fc), 0.90 (3H, t, J 7.5,
CH2C//3 ); 5C (125 MHz, CDC13 ), 207.4 (C), 175.3 (C), 88.0 (CH), 38.3, 35.8 (2 x CH2),
33.6 (CH), 24.8, 22.2 (2 x CH2), 18.5, 13.8 (2 x CH3 ); m/z (CI) 202 (MNH4+, 100%),
Found: MNH/, 202.1438. Ci 0Hi 6O3 requires MNH4+, 202.1443.
m Ratio determined by 'H-NMR
195

General procedure for the reduction of ketone using NaBH4 166
Ketones R Alcohols
273a ^Pr"275
274a nBu 276
To a solution of ketone (1.0 eq) in MeOH was added a solution of NaBH4 (1.0 eq) in EtOH. After stirring at room temperature under N2 for overnight, H2O was used for the workup. Column chromatography eluting with EtOAc/petrol (1/1) gave diastereoisomeric mixture of
alcohols as colourless oil.
(4S*,5R*)-5-(l-Hydroxybutyl)-4-methyl-dihydrofuran-2(3H)-one: Prepared by the general
procedure (148 mg of ketone 273a, 33 mg of NaBH4) to give alcohol 275 (2:1 diastereoisomeric mixture) as a colourless oil (130 mg, 87%). vmax (CHCl3)/cm" 1 3458 (OH),
2964 (CH), 2254, 1777 (lactone); 6H (250 MHz, CDCl3)major , 4.17 (1H, dd, J 8.5, 5.0, C//CHCH3 ), 3.80 (1H, dt, J 8.5, 3.0, HOC//), 2.78-2.68 (2H, m, CH3 C//, CH3 CHC//2), 2.24 (1H, dd, J5.5, 5.5, CH3 CHC//2), 1.81-1.38 (4H, m, 2 x CH2 in nPr), 1.13 (3H, d, J7.0,
CHC//5), 0.97 (3H, t, J 7.0, CH2C//3 ); 6C (125 MHz, CDClsWjor, 176.7 (C), 84.4 (CH), 69.5 (CH), 38.0, 36.8, 18.3 (3 x CH2 ), 32.0 (CH), 14.2, 14.0 (2 x CH3); m/z (CI) 190
(MW//, 100%), Found: MNH4+, 190.1441. C 9Hi 6O3 requires MNH4+, 190.1443.
(4S* ,5R*)-5-(l-Hydroxypentyl)-4-methyl-dihydrofuran-2(3H)-one: Prepared by the general
procedure (280 mg of ketone 274a, 69 mg of NaBH4) to give alcohol 276 (2:1
diastereoisomeric mixture) as a colourless oil (197 mg, 70%). vmax/cm' ] 3446 (OH), 2254,
1776 (lactone), 1642; 5H (250 MHz, CDCl3 )major , 4.28 (1H, dd, J8.5, 4.0, C//CHCH 3 ), 3.77
196

(1H, dt, J 8.5, 3.5, HOC//), 2.96-2.84 (1H, m, CH3 C//), 2.78-2.41 (4H, m, CH3 CHC//2,
HOCHC//2CH2), 1.92-1.25 (4H, m, 2 x CH2 in nBu), 1.15 (3H, d, J 7.0, CHC//3 ), 0.91 (3H,
t, J 6.5, CH2C//3); 5C (125 MHz, CDC1 3 ) 177.2 (C) major , 176.7 (C)minor , 84.8 (CH)major , 84.4
(CH)minor, 70.6 (CHUjo,, 69.6 (CH) mnoT, 38.06, 34.4, 27.2, 22.6 (4 x CH2 )minor , 37.1, 33.0,
27.6, 22.5 (4 x CH2)major, 32.5 (CH)major, 32.0 (CH)minor, 14.0, 13.9 (2 x CH3 )both [18 out of
20 carbons observed] m/z (CI) 204 (MNH4+, 100%) Found: MNH/, 204.1594. Ci 0Hi 8O3
requires MNH4+, 204.1599.
General procedure for the synthesis of thiocarbonylimidazolides 115c
RR, J. \^ ———————- _ V^o^°
HOInn
Alcohols R thiocarbonylimidazolides
275"Pr277
276 "Bu 278
To a solution of alcohol (1.0 eq) in DCE at room temperature under N2 was added
thiocarbonyldiimidazole (1.5 eq). The reaction mixture was stirred and the temperature was
gradually increased to 80-100 °C. After stirring overnight, H2O was used for the workup.
Column chromatography eluting with EtOAc/petrol (3:7) gave thiocarbonylimidazolides as
a diastereoisomeric mixture.***
O-1-((2R*,3S*)-3-Methyl-5-oxo-tetrahydrofuran-2-yl)butyl IH-imidazole-l-carbothioate:
Prepared by the general procedure (130 mg of alcohol, 203 mg of thiocarbonyldiimidazole,
NMR assignment; only for the major product.
197

80 °C) to give thiocarbonylimidazolide 277 (2:1 diastereoisomeric mixture) as a pale
yellow oil (80 mg, 50%). vmax (CHCy/cnf 1 3432, 2254, 1787 (lactone), 1714; 5H (250 MHz,
CDCl3)major 8.37, 7.66, 7.09 (3H, m, 3 x CH in Im), 5.81 (1H, dt, J 6.5, 4.0, Im(S)COCH),
4.74 (1H, dd, 76.5, 6.0, CH3CHC#), 2.90-1.37 (7H, m, CH3 C/f, CH3 CHC//2 , 2 x CH2 in
nPr), 1.12 (3H, d, J 6.5, CHC//3 ), 0.95 (3H, t, J 7.5, CH2C//3 ); 5C (125 MHz, CDCl3 )major
175.7 (C), 175.4 (C), 81.4, 81.3, 79.2 (3 x CH), 81.1 (CH), 80.6 (CH), 37.0, 32.5, 17.8 (3 x
CH2), 32.2 (CH3 ), 14.7, 14.0 (CH3 , CH3 ); m/z (CI) 283 (Mff, 100%), Found: MH+,
283.1116. Ci 3H 18N2O 3 S requires MH+, 283.1116.
O-1-((2R ,3S )-3-Methyl-5-oxo-tetrahydrofuran-2-yl)pentyl lH-imidazole-l-carbothioate\
Prepared by the general procedure (197 mg of alcohol, 267 mg of thiocarbonyldiimidazole,
100 °C) to give thiocarbonylimidazolide 278 (2:1 diastereoisomeric mixture) as a pale
yellow oil (250 mg, 77%). vmax (CHCy/cm' 1 2959, 2253, 1776 (lactone), 5H (250 MHz,
CDCl3 )maj0r, 8.45, 7.57, 7.09 (3H, 3 x CH in Im), 5.79 (1H, dt, J 6.5, 4.0, Im(S)COCH),
4.70 (1H, dd, 77.0, 4.0, CH3 CHC//), 2.92-2.76 (1H, m, CH3 C#), 2.00-1.84 (2H, m, CH2 in
nBu), 2.69 (1H, dd, 7 17.5, 8.5, CH3CHC//2), 2.18 (1H, dd, 7 17.5, 8.0, CH3CHC//2), 1.47-
1.29 (4H, m, 2 x CH2 in nBu), 1.12 (3H, d, 7 7.0, CHC//3 ), 0.92 (3H, t, 7 7.5, CH2C//3 ); 5C
(125 MHz, CDCl3 ) major, 182.2 (C), 175.6 (C), 137.0, 130.3, 117.6 (3 x CH), 81.0 (CH),
81.3 (CH), 36.6, 29.8, 27.0, 22.5 (4 x CH2), 32.2 (CH), 14.1, 13.8 (2 x CH3); m/z (CI) 297
Found: MH+, 297.1277. C 14H20N2O3 S requires MH+, 297.1279.
198

General procedure for the synthesis of Quercus lactones
Im
thiocarbonylimidazolides R Quercus lactones
277"Pr260
278 "Bu 261
To a refluxing solution of rc-BuSnH (1.1 eq) and AIBN (0.1 eq) in toluene at room
temperature under N2 was added dropwise a solution of thiocarbonylimidazolides (1.0 eq)
in toluene. After stirring for overnight, column purification eluting with EtOAc/petrol (1/1)
gave Quercus lactones.
(4S*,5S*)-5-Butyl-4-methyl-dihydrofuran-2(3H)-one 260 116 ' 167 : Prepared by the general
procedure (92 mg of «-BuSnH, 5 mg of AIBN, 80 mg of thiocarbonylimidazolide) to give
whisky lactone as a brown oil (37 mg, 83%). ! H NMR (250 MHz, CDC13 ) 4.44 (IH, dt, J
6.0, 4.5, CH3 CHC#), 2.70 (IH, dd, J 16.5, 8.0, C(O)C//H), 2.65-2.52 (IH, m, CH3 C#),
1.80-2.20 (IH, dd, J 16.5, 3.5, C(O)CHH), 1.80-1.27 (6H, m, 3 x CH2 in nBu), 1.01 (3H, d,
77.0, C//3 CH), 0.92 (3H, t, J 7.0, CH2C//3 ); 6C (125 MHz, CDC13 ) 176.9 (C), 83.7 (CH),
37.6 (CH2 ), 33.0 (CH), 29.6, 28.0, 22.5 (3 x CH2), 13.9, 13.8 (2 x CH3 ); m/z (CI) 174
(MNH4+, 100%), Found: MNH4+, 174.1493. C 9Hi 6O2 requires MNH4+, 174.1494.
(4S*,5S*)-4-Methyl-5-pentyl-dihydrofuran-2(3H)-one 261 168 : Prepared by the general
procedure (407 mg of w-BuSnH, 57 mg of AIBN, 205 mg of thiocarbonylimidazolide) to
give cognac lactone as a brown oil (108 mg, 91%). ! H NMR (250 MHz, CDC13 ) 4.40 (IH,
dd,76.0, 4.5, CH 3CHC//), 2.66 (IH, dd, J 17.0, 8.0, C(O)CH2 ), 2.60-2.50 (IH, m, CH 3 C//),
199

2.16 (1H, dd, J 17.0, 4.0, C(O)CH2), 1.69-1.26 (8H, m, 4 x CH2 in "Pent), 0.98 (3H, d, J 7.0,
C//3 CH), 0.87 (3H, t, 7 7.0, CH2C//3 ); 5C (125 MHz, CDC13 ) 176.8 (C), 83.6 (CH), 37.6
(CH2), 33.0 (CH), 31.6, 29.8, 25.6, 22.5 (4 x CH2 ), 25.1, 13.9 (2 x CH3 ); m/z (CI) 171
(Mtf\ 188 (MNH4\ 100%), Found: MNfV, 188.1648. Ci 0Hi 8O2 requires MNH4+, 188.1650.
Synthesis of Dimethyl diazomalonate
p-Toluenesulfonyl azide 292 121a :
SO2CI
292
To a solution of sodium azide (17.9 g, 0.28 mol) in the mixture of water (50 mL) and 90%
aqueous ethanol (100 mL) was added the solution of p-toluenesulfonyl chloride (47.6 g,
0.25 mol) in 99% ethanol (500 mL). After stirring at 40 °C for 3 h, the solvent was
removed at 40 °C under reduced pressure. The oily sulfonyl azide was washed with water
and organic layer was separated. It was dried over Na2 SC>4 and the solvent was removed at
40 °C under reduced pressure to give 292 as a colourless oil (38.0 g, 80%). 5H (400 MHz,
CDC13 ) 7.85 (2H, d, J 8.5, 2 x CH in Ph), 7.42 (2H, d, J 8.5, 2 x CH in Ph), 2.50 (3H, s,
CH3); m/z (CI) 189, 215 (MV7//).
200

Dimethyl diazomalonate 293 121b
^ ___ 0J$* 0o o II
293
To a solution of tosylazide (18.90 g, 96.0 mmol) and dimethyl malonate (11.4 mL, 0.10
mol) in benzene (85 mL) at room temperature was added triethylamine (13.7 mL, 0.10 mol).
After stirring for 15 h, the mixture was vacuum filtered and concentrated in vacuo. Column
chromatography eluting with EtOAc and hexane (1/3) gave 293 as a pale yellow oil (9.10 g,
60%). 5H (400 MHz, CDC13 ) 3.82 (6H, s, 2 x CH3).
Attempted cyclopropanation 118, 119
285b R 1 =H, R2=Me, 211 R1 =Me, R2=Et
To a solution of 6w(hexafluoroacetoacetonato)copper(II) (0.02 eq) in benzene at reflux
temperature (75 °C) under Ar was added dropwise (over 2 h) a solution of dimethyl
diazomalonate (1.05 eq) and 2-alkoxydihydropyran (1.00 eq) in benzene. After stirring at
75 °C for 4 h, the reaction mixture was cooled down to room temperature. The cold
reaction mixture was passed through a pad of neutral alumina to remove catalyst and
solvents were removed. Column chromatography eluting with EtOAc/hexane (3/7) gave
colourless oil. The ! H NMR spectrum indicated a complex product mixture.
201

2. Compounds from Chapter 2. Synthesis of enone systems/ -Phenylprop-2-en-l-one 1 69 :
To a mixture of CrOj, (0.04 g, 0.4 mmol) in CF^C^ (20 mL) was added sequentially aq
70 % 'BuOOH (3.00 g, 23.0 mmol) and l-phenylprop-2-en-l-ol (1.00 g, 7.5 mmol). The
reaction mixture was stirred vigorously under an air atmosphere for overnight. After
filtration on alumina and evaporation of solvent, column chromatography eluting with
diethyl ether/petrol (1/9) gave a pale yellow oil (0.41 g, 42%). 5H (250 MHz, CDC13 ) 7.97-
7.93 (2H, m, 2 x CH in Ph), 7.48-7.58 (3H, m, 3 x CH in Ph), 7.16 (IH, dd, J 17.0, 10.5,
C//=CH2), 6.43 (IH, dd, J 17.0, 2.0, CH=C//H), 5.93 (IH, dd, J 10.5, 2.0, CH=CH//); m/z
(CI) 133 (Mrf\ 150 (MNH4+, 100%).
138.l,3,3-Triphenylprop-2-en-l-one314
Ph O
Ph" ^ Ph 314
To a solution of propargyl alcohol (2.00 g, 7.0 mmol) in AcOH was added dropwise cone.
H2SO4 (1.03 g, 10.5 mmol). After stirring vigorously at 110 °C under N2 for overnight, the
reaction mixture was quenched by the addition of NaHCOs (sat, aq). The organic layer was
extracted by ether and water and recrystallization with MeOH gave 314 as a yellow solid
(1.10 g, 55%). mp 92-93 °C [lit. mp 86-87 °C 170]; 6H (250 MHz, CDC1 3 ) 7.94 (2H, d, J7.0,
2 x CH in PhCO), 7.50 (IH, t, J 7.5, CH in PhCO), 7.35-7.45 (7H, m, 7 x CH in Ph), 7.26-
202

7.32 (3H, m, 3 x CH in Ph), 7.18-7.24 (2H, m, 2 x CH in Ph), 7.15 (1H, s, CCH); m/z (CI) 285 (M/", 100%).
• _137General procedure for the preparation of enones by Mukaiyama-aldol reaction
RRo=< ———R
R
Me
Et
cyHex
//~\ /=\
O
Enones
310
311
312
To a solution of ketone and TiCU (1.0 eq) in Cl-bC^ was added dropwise the solution of
silyl enol ether (1.0 eq) in CFbCh. After stirring vigorously at r.t. under NI for 2 h, TFAA
(1.0 eq) was added dropwise. After stirring vigorously for an additional 1 h, TEA (2.0 eq)
was added. After stirring for an additional 2 h, the reaction was quenched by the addition of
HzO and the organic layer was extracted by ether and NfL^Cl (aq). Column chromatography
eluting with diethyl ether/petrol (1/9) gave enones.
3-Methyl-l-phenylbut-2-en-l-one 310 171 : Prepared by the general procedure (1.00 g of silyl
enol ether, 0.30 g of acetone) to give enone 310 (0.36 g, 44%) as a yellow oil. 5H(400 MHz,
CDC13 ) 7.92-7.94 (2H, m, 2 x CH in Ph), 7.41-7.56 (3H, m, 3 x CH in Ph), 6.76 (1H, s,
CCH), 2.22 (3H, s, CH3), 2.02 (3H, s, CH3 ); m/z (CI) 161 (Mf), 178 (MV///, 100%).
3-Ethyl-l-phenylpent-2-en-l-one 311 172 : Prepared by the general procedure (3.00 g of
silylenolether, 1.34 g of pentanone) to give enone 311 (1.47 g, 50%) as a brown oil. 5H (400
MHz, CDC1 3 ) 7.92-8.00 (2H, m, 2 x CH in Ph), 7.43-7.56 (3H, m, 3 x CH in Ph), 6.68 (1H,
203

s, CCH), 2.61 (2H, q, J 7.5, C#2CH3 ), 2.30 (2H, q, J 7.5, C//2CH3 ), 1.16 (3H, t, J 7.5,
CH2C//3), 1.13 (3H, t, y 7.5, CH2C//3 ); m/z (CI) 189 (M/"), 203, 220 (100%).
2-Cyclohexylidene-l-phenylethanone 312 173 : Prepared by the general procedure (3.00 g of
silylenolether, 1.53 g of cyclohexanone) to give enone 312 (2.04 g, 65%) as a brown oil. 5H
(400 MHz, CDC13 ) 7.93-8.00 (2H, m, 2 x CH in Ph), 7.43-7.58 (3H, m, 3 x CH in Ph), 5.58 (1H, s, CCH), 1.98-2.07 (4H, m, 2 x CH2 in CyHex), 1.53-1.68 (6H, m, 3 x CH2 in CyHex); m/z (CI) 201 (Mtf\ 215, 232 (100%).
General procedure for the preparation of ylides
PPh,
R
MeO
CI
Ylides
315
316
To a solution of CH3 PPh3 Br (3.0 eq) in THF at 0 °C under N2 was added PhLi solution in
Et2O (1.80 M, 3.0 eq). After allowing to warm up to room temperature over 2 h, the
reaction mixture (methylenetriphenylphosphorane) was cooled down to -78 °C. To the
reaction mixture was added a solution of benzoyl chloride (1.0 eq) in THF. After stirring vigorously for overnight, the reaction mixture was quenched by the addition of water and
the organic layer was extracted by EtOAc and NaCl (aq). Column chromatography eluting with EtOAc gave ylides.
/- (4-Methoxyphenyl)-2-(triphenylphosphanylidene)-ethanone 315 174 Prepared by the
general procedure (5.79 g of CH3 PPh3 Br, 9 mL of 1.80 M PhLi and 0.92 g of benzoyl
204

chloride) to give ylide 315 (2.11 g, 95%) as a white solid, mp 156-158 °C; 8H (400 MHz,
CDC13 ) 7.96 (2H, d, J 8.5, 2 x CH in Ph), 7.69-7.74 (6H, m, 6 x CH in Ph3 ), 7.41-7.53 (9H,
m, 9 x CH in Ph3), 6.86 (2H, d, J 8.5, 2 x CH in Ph), 4.40 (1H, s, P=CH), 3.76 (3H, s,
OCH3); m/z (CI) 263, 279 (100%), 411 (A47/1").
l-(4-Chlorophenyl)-2-(triphenylphosphanylidene)-ethanone 316 175 : Prepared by the general
procedure (5.79 g of CH3PPh3Br, 9 mL of 1.80 M PhLi and 0.94 g of benzoyl chloride) to give ylide 316 (1.35 g, 60%) as a white solid, mp 198-199 °C [lit. mp 196-198 °C 176]; 5H
(400 MHz, CDC13 ) 7.92 (2H, d, J 8.5, 2 x CH in Ph), 7.68-7.73 (6H, m, 6 x CH in Ph3), 7.44-7.58 (9H, m, 9 x CH in Ph3 ), 7.29 (2H, d, J 8.5, 2 x CH in Ph), 4.40 (1H, s, P=CH);
m/z (CI) 217, 279 (100%), 415 (MH*), 433 (MV7//).
General procedure for the preparation of enones by Friedel-Crafts acylation 177
X
MeO
MeO
MeO
Y
H
MeO
MeO
Z
H
H
MeO
Enones
317
318
319
To a solution of acid chloride in CS2 at 0 °C were added slowly A1C13 . After being stirred
for 5 min, benzene was added drop wise. After stirring at room temperature, CS2 was
removed in vacuo and the residue was decomposed by the addition of cold diluted HC1 (0.1
M). Column chromatography eluting with EtOAc/petrol (1/9) gave a corresponding product.
l-(4-Methoxyphenyl)-3-methylbut-2-en-l-one 317 178 : Prepared by the general procedure
205

(12 mL of anisole, 5.00 g of A1C13 , 4.00 g of acid chloride and 20 mL of CS2) to give a
corresponding product 317 as a yellow oil. 6H (400 MHz, CDC13 ) 7.95 (2H, d, J 8.5, 2 x CH
in Ph), 6.92 (2H, d, J 8.5, 2 x CH in Ph), 6.73 (1H, s, CH), 3.85 (3H, s, OCH5), 2.20 (3H, s,
CH5), 2.00 (3H, s, CH5); m/z (CI) 191 (Mf, 100%).
l-(3,4-Dimethoxyphenyl)-3-methylbut-2-en-l-one 318: Prepared by the general procedure
(12 mL of anisole, 5.00 g of A1C13 , 4.00 g of acid chloride and 20 mL of CS2) to give a
corresponding product 318 as a yellow oil. vmax (CHCy/cm" 1 2935 (CH), 1656 (a, (3-
unsaturated C=O); 6H (400 MHz, CDC13 ) 7.60 (1H, d, J 2.0, CH in Ph), 7.57 (1H, dd, J 8.0,
2.0, CH in Ph), 6.90 (1H, d, J 8.0, CH in Ph), 6.75 (1H, s, CH), 3.97 (6H, s, 2 x OCH3),
2.21 (3H, s, CH3 ), 2.04 (3H, s, CH3 ); 5C (100 MHz, CDC13 ) 190.2 (C), 155.5 (C), 152.7 (C),
149.0 (C), 132.3 (C), 122.6 (CH), 121.0 (CH), 110.5 (CH), 109.9 (CH), 56.1 (CH3 ), 56.0
(CH3 ), 28.0 (CH3 ), 21.1 (CH3 ); m/z (CI) 221 (Mff,\W%), Found: MH+ 221.1183,
C 13 H 16O3 requires MH+ 221.1178.
3-Methyl-l-(3,4,5-trimethoxyphenyl)but-2-en-l-one 319: Prepared by the general procedure
(12 mL of anisole, 5.00 g of A1C13 , 4.00 g of acid chloride and 20 mL of CS2) to give a
corresponding product 319 as a yellow oil. vmax (CHC^/cm" 1 2938 (CH), 1658 (a, 0-
unsaturated C=O); 6H (400 MHz, CDC13 ) 7.41 (1H, d, J 9.0, CH in Ph), 6.73 (1H, d, J 9.0,
CH in Ph), 6.67 (1H, s, CH), 3.91 (3H, s, OCH3 ), 3.90 (3H, s, OCH3 ), 3.89 (3H, s, OCH3 ),
2.22 (3H, s, CH3), 1.99 (3H, s, CH3 ); 5C (100 MHz, CDC13 ) 191.4 (C), 156.5(C), 154.6(C),
125.5(CH), 125.2(CH), 107.1 (CH), 61.9, 61.0, 56.1 (3 x CH3 ), 27.9, 27.2 (2 x CH3); m/z (CI) 250 (Mf",100%), Found: MH+ 251.1283, Ci 4H 18O4 requires MH+ 251.1283.

General procedure for the preparation of enones by Grignard reaction / Oxidation
o
Ar
/?-Me Phenyl
p-C\ phenyl
p-NC>2 phenyl
2-Naph
Enones
320
321
322
323
To a solution of aryl aldehyde (1.0 eq) in dried Et2O at room temperature under N2 was added 0.5 M (2-methylprop-l-enyl)magnesium bromide (in THF, 1.2 eq). After stirring for 5 h, the reaction was quenched with NH4C1 (aq). It was then extracted with Et2O and NaHCO3 (aq) followed by brine. The organic layer was dried by MgSO4 and the solvent was evaporated under reduced pressure. The crude product was dissolved in acetone and MnO2 (20.0 eq) was added to the mixture. After stirring at room temperature for overnight, Mn residues were removed by filtration and the resulting product was evaporated to give the crude product. Column chromatography eluting with Et2O/petrol (1/9) gave a
corresponding product.
3-Methyl-l-p-tolylbut-2-en-l-one 320: Prepared by the general procedure (40 mL of
Grignard reagent, 2.00 g of aldehyde and 29.00 g of MnO2) to give a corresponding product 320 as a yellow oil (1.80 g, 62%). vmax (CHCy/cirf 1 3030-2914 (CH), 1661 (a, 0-
unsaturated C=O), 6H (400 MHz, CDC13 ) 7.86 (2H, d, J 8.0, 2 x CH in Ph), 7.26 (2H, d, J 8.0, 2 x CH in Ph), 6.76 (1H, s, CH), 2.42 (3H, s, CH3 ), 2.22 (3H, s, CH3 ), 2.03 (3H, s, CH3 ); 5C (100 MHz, CDC13 ) 191.2 (C), 156.0 (CH), 143.0 (C), 136.7 (C), 129.1 (2 x CH), 128.3 (2 x CH), 121.3 (C), 28.0 (CH3 ), 21.6 (CH3 ), 21.1 (CH3 ); m/z (CI) 175 (A//T,100%),
207

Found: MH+ , 175.1122, Ci 2 H 14O requires MH+, 175.1123.
l-(4-Chlorophenyl)-3-methylbut-2-en-l-one 321: Prepared by the general procedure (34 mL
of Grignard reagent, 2.00 g of aldehyde and 30.00 g of MnO2 ) to give a corresponding
product 321 as a yellow oil (1.45 g, 53%). vmax (CHCy/cirT 1 2976-2913 (CH), 1662 (a, 0-
unsarurated C=O); 8H (400 MHz, CDC13 ) 7.89 (2H, d, J 8.5, 2 x CH in Ph), 7.43 (2H, d, J 8.5, 2 x CH in Ph), 6.72 (IH, s, CH), 2.23 (3H, s, CH 3), 2.04 (3H, s, CH3); 5C (100 MHz,
CDC13 ) 190.1 (C), 157.7 (CH), 138.6 (C), 137.6 (C), 129.6 (2 x CH), 128.7 (2 x CH),
120.7(C), 28.1 (CH3 ), 21.3 (CH3 ); m/z (CI) 195 (M/",100%), Found: MH+, 195.0577,
CiiHuOCl requires MH+, 195.0577.
l-(4-Nitrophenyl)-3-methylbut-2-en-l-one 322: Prepared by the general procedure (31 mL
of Grignard reagent, 2.00 g of aldehyde and 29.00 g of MnO2) to give a corresponding product 322 as a yellow solid (0.35 g, 13%); mp 114-116 °C; vmax (CHCy/cm' 1 2947-2903
(CH), 2853, 1656 (a, p-unsaturated C=O), 1613 (NO2); 5H (400 MHz, CDC13 ) 8.32 (2H, d,
79.0, 2 x CH in Ph), 8.08 (2H, d, 79.0, 2 x CH in Ph), 6.78 (IH, s, CH), 2.30 (3H, s, CH3 ), 2.10 (3H, s, CH3 ); 5C (100 MHz, CDC13 ) 189.3 (C), 160.4 (CH), 149.8 (C), 144.1 (C), 129.1
(2 x CH), 123.7 (2 x CH), 120.4 (C), 28.3 (CH3 ), 21.6 (CH3 ); m/z (CI) 206 (M7^,100%),
Found: MH+, 206.0816, CnHnNO3 requires MH+, 206.0817.
3-Methyl-l-(naphthalen-2-yl)but-2-en-l-one 323: Prepared by the general procedure (40
mL of Grignard reagent, 2.00 g of aldehyde and 32.00 g of MnO2) to give a corresponding
product 323 as a yellow solid (1.38 g, 69%). mp 102-104 °C; vmax (CHCy/cirf 1 3057-2911
(CH), 1658 (a, p-unsaturated C=O); 6H (400 MHz, CDC13 ) 8.45 (IH, s, CH in Naph), 8.07
(IH, d, 7 8.5, CH in Naph), 7.99 (IH, d, 78.0, CH in Naph), 7.91 (2H, t, 78.5, CH in Naph),
7.50-7.62 (2H, m, 2 x CH in Naph), 6.94 (IH, s, CH), 2.29 (3H, s, CH3 ), 2.10 (3H, s, CH 3 );
5C (100 MHz, CDC13 ) 191.5 (C), 156.7 (C), 136.6 (C), 135.3 (C), 132.6 (C), 129.5 (CH),
128.3 (CH), 128.1 (CH), 127.8 (CH), 126.6 (CH), 124.4 (CH), 121.3 (CH), 28.1 (CH 3 ),
21.3 (CH 3 ) [14 out of 15 carbons observed]; m/z (CI) 211 (M/MOO%) Found:
208

211.1128, Ci 5Hi4O requires MH+ , 211.1123
General procedure for Hetero Diels-Alder reaction using microwave
R 1
A, R^R 1
H
Me
Et
cyHex
Me
Me
Me
Me
Me
R2
Ph
Ph
Ph
Ph
2-Naph
p-MeO phenyl
/?-Me Phenyl
p-C\ phenyl
p-NO2 phenyl
v° OE<DHPs
308
324
325
326
328
327
329
330
331
To ethyl vinyl ether (5.0 eq) was added enone (1.0 eq) and YbFOD (0.05 eq). After the
reaction in microwave at the corresponding temperature (55-80 °C) for 2-10 h, Column
chromatography eluting with diethyl ether/petrol (5/95 to 1/9) gave 2-alkoxydihydropyrans.
2-Ethoxy-6-phenyl-3,4-dihydro-2H-pyran 308: Prepared by the general HDA procedure
(0.41 g of diene, 0.11 g of vinyl ether, 0.16 g of Yb catalyst, 55 °C, 2 h) to give
alkoxydihydropyran 308 (0.30 g, 48%) as a pale yellow oil. vmax (CHC\J/cm ] 2974 (CH),
1722, 1684 (C=C); §H (400 MHz, CDC13) 7.60 (2H, d, J 8.0, 2 x CH in Ph), 7.37-7.27 (3H,
m, 3 x CH in Ph), 5.45 (1H, dd,73.5, 4.5, OCHO), 5.26 (1H, t, 73.5, C=CH), 4.01 (1H, dq,
79.5, 7.0, OC//H), 3.72 (1H, dq, 7 9.5, 7.0, OCH//), 2.44-2.15 (2H, m, CH 2 ), 1.97-1.90
209

(2H, m, CH2), 1.28 (3H, t, y 7.0, CH3 ); 6C (125 MHz, CDC13 ) 148.1 (C), 136.0 (C), 128.1,
127.7, 124.3 (5 x CH), 98.0 (CH) 97.6 (CH), 63.8 (CH2), 26.5 (CH2), 17.6(CH2 ), 15.3
(CH3); m/z (CI) 205 (M/f,100%), Found: MH+ , 205.1225. C 13 H 16O2 requires MH+, 205.1228.
2-Ethoxy-4,4-dimethyl-6-phenyl-3,4-dihydro-2H-pyran 324: Prepared by the general
procedure (1.00 g of diene, 3.0 ml of vinyl ether, 0.33 g of Yb catalyst, 55 °C, 4 h) to give
alkoxydihydropyran 324 (0.60 g, 41%) as a yellow oil. v^ (CHCy/cm' 1 3058-2957 (CH),
2250, 1649 (C=C); 5H (500 MHz, CDC13 ) 7.62-7.65 (2H, m, 2 x CH in Ph), 7.28-7.39 (3H,
m, 3 x CH in Ph), 5.26 (IH, s, CCHC), 5.14 (IH, dd, J2.5, 8.0, OCHO), 4.15 (IH, dt, J9.5,
7.0, OC//H), 3.74 (IH, dt, 79.5, 7.0, OCH//), 1.85-1.89 (IH, m, CH2 ), 1.77-1.81 (IH, m,
CH2), 1.35 (3H, t, J7.0, CH2C//3 ), 1.21 (3H, s, CH3 ) 1.19 (3H, s, CH3 ); 5C (125 MHz,
CDC13 ) 146.6 (C), 135.7 (C), 128.1, 127.8, 124.6 (5 x CH), 108.6 (CH), 98.7 (CH), 64.6
(CH2), 42.0 (CH2 ), 31.5 (CH3), 30.6 (CH3 ), 30.5 (C), 15.4 (CH3 ); m/z (CI) 233 (MF^,100%),
Found: MH+ , 233.1537. Ci 5H20O2 requires MH+, 233.1542.
2-Ethoxy-4,4-diethyl-6-phenyl-3,4-dihydro-2H-pyran 325: Prepared by the general
procedure (1.20 g of diene, 2.30 g of vinyl ether, 0.34 g of Yb catalyst, 70-80 °C, 4 h) to
give alkoxydihydropyran 325 (0.20 g, 12%) as a pale yellow oil. vmax (CHCy/cm" 1 2968
(CH), 2254, 1713, 1650(C=C); 5H (400 MHz, CDC13 ) 7.58-7.61 (2H, m, 2 x CH in Ph),
7.24-7.35 (3H, m, 3 x CH in Ph), 5.20 (IH, s, CCHC), 5.06 (IH, dd, J 2.5, 8.5, OCHO),
4.14 (IH, dt, J9.5, 7.0, OC//H), 3.70 (IH, dt, J9.5, 7.0, OCH//), 1.68-1.80 (2H, m, CH2 ),
1.39-1.54 (4H, 2 x CC//2CH3 ), 1.32 (3H, t, J 7.0, OCH2C//3 ), 0.88 (3H, 1,77.5, CH3 ) 0.85
(3H, t, J 7.5, CH3); 5C (125 MHz, CDC13 ) 147.9 (C), 135.8 (C), 128.1, 127.9, 124.6 (5 x
CH), 106.1 (CH), 98.9 (CH), 64.6 (CH2), 37.5 (CH2), 36.7 (C), 33.6 (CH2), 31.5 (CH2 ),
15.4 (CH3 ), 8.3 (2 x CH3 ); m/z (CI) 261 (MF/f,100%) Found: MH+ , 261.1851. C 17 H24O2
requires MH+ , 261.1854.
4-Ethoxy-2-phenyl-3-oxa-spiro[5,5]undec-l-ene 326: Prepared by the general procedure
210

(1.85 g of diene, 3.31 g of vinyl ether, 0.49 g of Yb catalyst, 65 °C, 6 h) to give
alkoxydihydropyran 326 (0.75 g, 30%) as a pale yellow oil. vmax (CHCy/cirf 1 2927 (CH),
2251, 1648 (C=C); 8H (400 MHz, CDC13 ) 7.58-7.62 (2H, m, CH in Ph), 7.24-7.34 (3H, m,
CH in Ph), 5.38 (IH, s, CCHC), 5.07 (IH, dd, J 2.5, 8.5, OCHO), 4.12 (IH, dt, .7 9.5, 7.0,
OC//H), 3.69 (IH, dt, J 9.5, 7.0, OCH//), 2.00 (IH, d, J 13.0, IH in CH2 ), 1.38-1.68 (11H,
m, IH in CH2 , 5 x CH2 in CyHex), 1.31 (3H, t, J 7.0, CH2C//3 ); 5C (100 MHz, CDC13 )
146.8 (C), 135.8 (C), 128.6, 128.0, 124.6 (5 x CH), 107.1 (CH), 98.5 (CH), 64.5 (CH2 ),
39.4 (CH2), 39.3 (CH2), 36.6 (C), 33.6 (CH2), 26.0(CH2), 22.0 (CH2), 21.5 (CH2), 15.3
(CH3 ); m/z (CI) 273 (M/^,100%), Found: MH+, 273.1849. Ci 8H24O2 requires MH+, 273.1854.
2-Ethoxy-6-(4-methoxyphenyl)-4,4-dimethyl-3,4-dihydro-2H-pyran 327: Prepared by the
general procedure (1.00 g of diene, 1.91 g of vinyl ether, 0.29 g of Yb catalyst, 60 °C, 5 h) to give alkoxydihydropyran 327 (0.38 g, 27%) as a yellow oil. vmax (CHCy/cm" 1 3380,
2960 (CH), 2360, 2054, 1646 (C=C), 1622; 5H (400 MHz, CDC13 ) 7.55 (2H, d, J 8.5, 2 x
CH in Ph), 6.89 (2H, d, J8.5, 2 x CH in Ph), 5.11-5.13 (2H, m, CCHC, OCHO), 4.14 (IH,
dt, J 15.0, 7.5, IH in OC//H), 3.84 (3H s, OCH3 ), 3.73 (IH, dt, J 15.0, 7.5, IH in OCH//),
1.89-1.70 (2H, m, CH2), 1.34 (3H, t, J 7.0, CH2CH3 ), 1.18 (3H, s, CCH3 ) 1.16 (3H, s,
CCH3 ); 5C (100 MHz, CDC13 ) 159.4 (C), 146.4 (C), 128.4 (C), 125.9 (2 x CH), 113.5 (2 x
CH), 107.0 (CH), 98.7 (CH), 64.5 (CH2), 55.3 (CH3 ), 42.1 (CH2), 31.6 (CH3), 30.7 (CH3 ),
30.4 (C), 15.4 (CH3 ); m/z (CI) 263 (M//",100%), Found: MH+, 263.1642. Ci 6H22O3 requires
MH+, 263.1647.
2-Ethoxy-4,4-dimethyl-6-p-tolyl-3,4-dihydro-2H-pyran 329: Prepared by the general
procedure (2.00 g of diene, 4.10 g of vinyl ether, 0.61 g of Yb catalyst, 65 °C, 3 h) to give
alkoxydihydropyran 329 (1.13 g, 40%) as a yellow oil. v^tCHCy/cm' 1 2956-2926 (CH),
2867, 1649 (C=C); 5H (400 MHz, CDC13 ) 7.50 (2H, d, J8.0, 2 x CH in Ph), 7.15 (2H, d, J
8.0, 2 x CH in Ph), 5.19 (IH, s, C=CH), 5.11 (IH, dd, .72.5, 8.0, OCHO), 4.13 (IH, dt, J
9.5, 7.0, OC//H), 3.72 (IH, dt, J 9.5, 7.0, OCH//), 2.37 (3H, s, CH3 Ph), 1.73-1.86 (2H, m,
21

CH2), 1.33 (3H, t, J 7.0, CH2C//3), 1.18 (3H, s, CH3) 1.16 (3H, s, CH3); 5C (100 MHz,
CDC13 ) 146.6 (C), 137.6 (C), 132.9 (C), 128.8 (2 x CH), 124.4 (2 x CH), 107.8 (CH), 98.6
(CH), 64.5 (CH2), 42.0 (CH2), 31.5, 30.6 (2 x CH3 ), 30.5 (C), 21.2 (CH3 ), 15.4 (CH3 ); m/z
(CI) 247 (A^",100%), Found: MH+, 247.1689, C i6H22O2 requires MH+, 247.1685.
6-(4-chlorophenyl)-2-ethoxy-4,4-dimethyl-3,4-dihydro-2H-pyran 330: Prepared by the
general procedure (1.00 g of diene, 1.90 g of vinyl ether, 0.28 g of Yb catalyst, 65 °C, 5 h)
to give alkoxydihydropyran 330 (0.60 g, 44%) as a yellow oil. vmax (CHCy/cm' 1 2958-
2926 (CH), 2867, 1648 (C=C); 5H (400 MHz, CDC13 ) 7.54 (2H, d, J 8.5, 2 x CH in Ph),
7.31 (2H, d, .7 8.5, 2 x CH in Ph), 5.23 (IH, s, CCHC), 5.12 (IH, dd, .7 8.0, 2.5, OCHO),
4.11 (IH, dt, 7 9.5, 7.0, OC//H), 3.71 (IH, dt, J9.5, 7.0, OCH//), 1.72-1.87 (2H, m, CH2),
1.33 (3H, t, J 7.0, CH2C//3 ), 1.19 (3H, s, CCH3 ) 1.17 (3H, s, CCH3); 5C (100 MHz, CDC13 )
145.7 (C), 134.2 (C), 133.5 (C), 128.2 (2 x CH), 125.8 (2 x CH), 109.0 (CH), 98.8 (CH), 64.6 (CH2), 41.8 (CH2), 31.4 (CH3 ), 30.5 (C, CH3 ), 15.4 (CH3); m/z (CI) 267 (M/*",100%),
Found: MH+, 267.1149. Ci 5Hi 9O2Cl requires MH+, 267.1152.
6-(4-Nitrophenyl)-2-ethoxy-4,4-dimethyl-3,4-dihydro-2H-pyran 331: Prepared by the
general procedure (0.32 g of diene, 0.56 g of vinyl ether, 0.09 g of Yb catalyst, 65 °C, 5 h)
to give alkoxydihydropyran 331 (0.25 g, 58%) as a yellow oil. vmax (CHCy/cm" 1 3466,
2254, 1640 (C=C), 1518 (NO2); 6H (400 MHz, CDC13 ) 8.20 (2H, d, J 9.0, 2 x CH in Ph),
7.74 (2H, d, J 9.0, 2 x CH in Ph), 5.46 (IH, s, CCHC), 5.16 (IH, dd, J 7.5, 2.5, OCHO),
4.05-4.15 (IH, m, OC//H), 3.68-3.77 (IH, m, OCH//), 1.90-1.77 (2H, m, CH2), 1.32 (3H, t,
J 7.0, CH2C//3 ), 1.22 (3H, s, CH3 ) 1.19 (3H, s, CH3 ); 5C (100 MHz, CDC13) 147.1 (C),
145.0 (C), 141.8 (C), 125.0 (2 x CH), 123.5 (2 x CH), 112.8 (CH), 98.9 (CH), 64.8 (CH2 ),
41.5 (CH2 ), 31.2 (CH3 ), 30.6 (C), 30.2 (CH3 ), 15.3 (CH3 ); m/z (CI) 278 (Mff , 100%),
Found: MH+, 278.1405. Ci 5H 19NO4 requires MH+, 278.1392.
2-Ethoxy-4,4-dimethyl-6-(naphthalen-2-yl)-3,4-dihydro-2H-pyran 328: Prepared by the
general procedure (1.00 g of diene, 1.73 g of vinyl ether, 0.25 g of Yb catalyst, 65 °C, 5 h)

to give alkoxydihydropyran 328 (0.55 g, 41%) as a yellow oil. vmax (CHCy/cm' 1 3057-
2926 (CH), 2868, 1643 (C=C), 1626; 6H (400 MHz, CDC13 ) 8.09 (IH, s, CH in Naph),
7.79-7.90 (3H, m, 3 x CH in Naph), 7.71 (IH, dd, J 8.5, 1.5, CH in Naph), 7.45-7.51 (2H,
m, 2 x CH in Naph), 5.41 (IH, s, CCHC), 5.21 (IH, dd, J 2.5, 8.0, OCHO), 4.22 (IH, dt, J
9.5, 7.0, OC//H), 3.79 (IH, dt, .7 9.5, 7.0, OCH//), 1.82-1.94 (2H, m, CH2), 1.38 (3H, t, J
7.0, CH2C//3 ), 1.24 (3H, s, CH3 ) 1.21 (3H, s, CH3 ); 5C (100 MHz, CDC13 ) 146.6 (C), 133.3
(C), 133.0 (C), 132.9 (C), 128.4 (CH), 127.63 (CH), 127.55 (CH), 126.1 (CH), 125.9 (CH),
123.3 (CH), 122.8 (CH), 109.4 (CH) 98.8 (CH), 64.7 (CH2), 42.0 (CH2), 31.5 (CH3 ), 30.65
(CH3), 30.63 (C), 15.4 (CH3 ); m/z (CI) 283 (M/*",100%), Found: MH+, 283.1691. C 19H22O2
requires MH+ , 283.1698.
General procedure for rearrangement / Jones oxidation of alkoxydihydropyrans
RI
179
R2JLoJ
R 1
H
Me
Et
cyHex
Me
Me
Me
Me
Me
xOEt
R2
Ph
Ph
Ph
Ph
Naph
p-MeO phenyl
p-Me Phenyl
p-C\ phenyl
p-NOi phenyl
R2 / W-*- V^Vf °
o
Tetrahydrofuranones
(±)-309
(±)-332
(±)-333
(±)-334
(±)-339
(±)-335
(±)-336
(±)-337
(±)-338
213

To solution of 2-alkoxydihydropyran in CH2C12 at 0 °C was added DMDO/acetone solution.
After stirring for 30 min, the reaction mixture was allowed to be stirred at room
temperature. After the crude was extracted with saturated aqueous NaHCO3 solution, it was
concentrated to give the mixture of lactol and lactol ether. The crude mixture from
oxidative rearrangement was dissolved in acetone at 0 °C and 3.0 M Jones reagent (3.0 eq
to initial 2-alkoxydihydropyran) was added dropwise. After stirring, the excess of oxidants
was quenched by the addition of 2-propanol until the brown colour of the mixture turned to
be green. The reaction mixture was diluted with diethyl ether and the precipitated
chromium salts were dissolved by the addition of saturated aqueous NH4C1 solution. After
the organic layer was separated and the aqueous layer was extracted with diethyl ether,
column chromatography gave a corresponding lactone as a product.
(±}-5-Benzoyl-dihydrofuran-2(3H)-one 309 140 ' 141 : Prepared by the general procedure (4.0
mmol of 2-alkoxydihydropyran, 0.088 M DMDO 50 mL, 3.0 M Jones reagent 4 mL, 2 h for
DMDO oxidation, 1 h for Jones oxidation) to give lactone (±)-309 as a white crystal (380
mg, 50% over 2 steps), mp 79-80 °C [lit. mp 78-79 °C 180]; 5H (500 MHz, CDC13) 8.02-7.48
(5H, m, Ph), 5.82-5.76 (1H, m, CH2C//), 2.65-2.44 (4H, m, 2 x CH2); m/z (CI) 208 (MNH4\
100%), 398 Found: MNH4+, 208.0972 C n H 10O3 requires MNH4+, 208.0974.
(±}-5-Benzoyl-4,4-dimethyl-dihydrofuran-2(3H)-one 332: Prepared by the general
procedure (0.43 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 3.9 mL, 3.0 M Jones
reagent 0.7 mL, 2 h for DMDO oxidation, 2 h for Jones oxidation) to give lactone (±)-332
as colourless oil (65 mg, 69% over 2 steps). vmax (CHCy/cm' 1 2969 (CH), 2343, 1789
(lactone), 1690 (CO); 6H (500 MHz, CDC13) 7.96 (2H, d, J 8.5, 2 x CH in Ph), 7.67 (1H,
dd, J 8.5, 7.5, CH in Ph), 7.55 (2H, d, J 7.5, 2 x CH in Ph), 5.55 (1H, s, OCH), 2.60 (1H, d,
J 17.0, CH2), 2.36 (1H, d, J 17.0, CH2), 1.40 (3H, s, CCH3 ), 1.00 (3H, s, CCH 3 ); 5C (100
MHz, CDC13 ), 195.7 (C), 175.7 (C), 135.8 (C), 134.3 (CH), 129.1 (2 x CH), 128.6 (2 x CH)
85.5 (CH), 41.8 (CH2 ), 40.7 (C), 28.3, 23.5 (CH3 , CH3 ); m/z (CI) 236 (MNH4\ 100%)
Found: MNH4+, 236.1285. Ci 3 Hi 4O3 requires MNH4+ , 236.1287.
214

(±}-5-Benzoyl-4,4-diethyl-dihydrofuran-2(3H)-one 333: Prepared by the general procedure
(0.35 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 3 mL, 3.0 M Jones reagent 0.6 mL, 3
h for DMDO oxidation, 2 h for Jones oxidation) to give lactone (±)-333 as colourless oil
(51 mg, 59% over 2 steps). vmax (CHCy/cnV 1 3444, 2342, 1788 (lactone), 1649 (C=O); 5H
(500 MHz, CDC13 ) 7.96 (2H, d, J 8.5, 2 x CH in Ph), 7.68 (IH, dd, J 8.5, 7.5, CH in Ph),
7.55 (2H, d, J 7.5, 2 x CH in Ph), 5.71 (IH, s, OCH), 2.63 (IH, d, J 17.0, C(O)C//H), 2.30
(IH, d, J 17.0, C(O)CH//), 1.73-1.80 (IH, m, CMiCH3 ), 1.60-1.68 (IH, m, CH//CH3 ), 1.38- 1.46 (IH, m, C//HCH3 ), 1.12-1.22 (IH, m, CH//CH3 ), 1.09 (3H, s,77.5, CH3 ), 0.76
(3H, s, 7 7.5, CH3 ); 6C (125 MHz, CDC13 ), 196.6 (C), 176.3 (C), 136.0 (C), 134.3 (CH), 129.1 (2 x CH), 128.4 (2 x CH) 82.2 (CH), 48.0 (C), 38.7 (CH2), 28.8 (CH2), 26.2 (CH2), 9.1 (CH3), 8.4 (CH3 ); m/z (CI) 264 (MNH4+ , 100%) Found: MNH4+, 264.1596. Ci 5Hi 8O3 requires MNH4+, 264.1600.
(±}-l-Benzoyl-2-oxa-spiro[4,5]decan-3-one 334: Prepared by the general procedure (0.7
mmol of 2-alkoxydihydropyran, 0.12 M DMDO 6 mL, 3.0 M Jones reagent 1.2 mL, 3 h for
DMDO oxidation, 2 h for Jones oxidation) to give lactone (±)-334 as a colourless oil (100
mg, 55% over 2 steps). vmax (CHCy/cm' 1 3434, 2931 (CH), 2338, 1787 (lactone), 1685
(CO); 6H (400 MHz, CDC13) 7.94 (2H, d, J 9.0, 2 x CH in Ph), 7.65 (IH, dd, J 9.0, 7.5, CH in Ph), 7.53 (2H, d, .7 7.5, 2 x CH in Ph), 5.53 (IH, s, OCH), 2.55 (2H, s, CH2), 1.07-
1.89 (10H, m, 5 x CH2 in CyHex); 6C (100 MHz, CDC13 ), 196.2 (C), 175.9 (C), 136.2 (C),
134.2 (CH), 129.1 (2 x CH), 128.5 (2 x CH), 85.4 (CH), 45.0 (C), 37.3 (CH2 ), 36.3, 32.9, 25.2, 23.1, 22.2 (5 x CH2); m/z (CI) 276 (MNH4+, 100%), Found: MNH4+ , 276.1588. Ci 6Hi 8O3 requires MNH4+, 276.1600.
(±}-5-(4-Methoxybenzoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 335: Prepared by the
general procedure (0.76 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 7.0 mL, 3.0 M
Jones reagent 1.3 mL, 3 h for DMDO oxidation, 2 h for Jones oxidation) to give lactone
(±)-335 as colourless oil (95 mg, 50% over 2 steps). VmaxtCHCl^/cm' 1 2965-2931 (CH),
215

1788 (lactone), 1677 (C=O); 5H (400 MHz, CDC13 ) 8.00 (2H, d, J 9.0, 2 x CH in Ph), 7.01
(2H, d, J 9.0, 2 x CH in Ph), 5.48 (IH, s, OCH), 3.92 (3H, s, CH3 ), 2.62 (IH, d, J 17.0,
CH2), 2.33 (IH, d, J 17.0, CH2), 1.40 (3H, s, CCH3 ), 1.00 (3H, s, CCH3 ), 6C (100 MHz,
CDC13 ), 193.8 (C), 175.9 (C), 164.4 (C), 131.0 (2 x CH), 128.8 (C), 114.3 (2 x CH) 85.3
(CH), 55.6 (CH3 ), 41.8 (CH2 ), 40.6 (C), 28.4 (CH3 ), 23.5 (CH3 ); m/z (CI) 266 (MNH4\
100%) Found: MNH/, 266.1403. C 14Hi 6O4 requires MNH/, 266.1392.
(±)-4,4-Dimethyl-5-(4-methylbenzoyl)-dihydrofuran-2(3H)-one 336: Prepared by the
general procedure (0.62 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 6.0 mL, 3.0 M
Jones reagent 1.0 mL, 4 h for DMDO oxidation, 1 h for Jones oxidation) to give lactone
(±)-336 as white solid (102 mg, 69% over 2 steps), mp 134-136 °C; vmax (CHCy/cm' 1
2967-2931 (CH), 2874, 1788 (lactone), 1682 (C=O), 1606; 5H (400 MHz, CDC13) 7.86 (2H,
d, 78.0, 2 x CH in Ph), 7.34 (2H, d, 78.0, 2 x CH in Ph), 5.53 (IH, s, OCH), 2.61 (IH, d, 7
17.0, CH2), 2.46 (3H, s, CH3Ph), 2.34 (IH, d, 717.0, CH2), 1.40 (3H, s, CCH3 ), 0.99 (3H, s,
CCH3); 5C (100 MHz, CDC13 ), 195.2 (C), 175.9 (C), 145.5 (C), 133.3 (C), 129.8 (2 x CH),
128.7 (2 x CH), 85.4 (CH), 41.8 (CH2), 40.6 (C), 28.4, 23.5, 21.8 (3 x CH3 ); m/z (CI) 250
(MNHf, 100%), Found: MNH4+, 250.1432. Ci 4Hi 6O3 requires MNH4+ , 250.1443.
(±}-5-(4-Chlorobenzoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 337: Prepared by the
general procedure (0.75 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 7.0 mL, 3.0 M
Jones reagent 1.2 mL, 3 h for DMDO oxidation, 1 h for Jones oxidation) to give lactone
(±)-337 as white crystal (103 mg, 53% over 2 steps), mp 132-134 °C; vmax (CHCy/cnV 1
2963 (CH), 2874, 1789 (lactone), 1690 (C=O); 5H (400 MHz, CDC13 ) 7.92 (2H, d, 78.5, 2 x
CH in Ph), 7.53 (2H, d, 78.5, 2 x CH in Ph), 5.46 (IH, s, OCH), 2.59 (IH, d, 7 17.0, CH2 ),
2.38 (IH, d, 7 17.0, CH2), 1.45 (3H, s, CH3 ), 1.00 (3H, s, CH3 ); 5C (100 MHz, CDC1 3 ),
194.7 (C), 175.4 (C), 141.0 (C), 134.1 (C), 130.0 (2 x CH), 129.5 (2 x CH), 85.6 (CH), 41.9
(CH2 ), 40.8 (C), 28.2, 23.5 (2 x CH3 ); m/z (CI) 270 (MNH4+ , 100%) Found: MNH4+ , 270.0893. Ci 3 H, 3 O3 Cl requires MNH4+, 270.0897.
216

(±}-4,4-Dimethyl-5-(4-nitrobenzoyl)-dihydrofuran-2(3H)-one 338: Prepared by the general
procedure (0.43 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 4.0 mL, 3.0 M Jones
reagent 0.7 mL, 3 h for DMDO oxidation, 1 h for Jones oxidation) to give lactone (±)-338-ias colourless oil (55 mg, 49% over 2 steps). vmax (CHC^/crn 1 2980 (CH), 2939, 1790
(lactone), 1698 (C=O), 1603; 6H (400 MHz, CDC13 ) 8.39 (2H, d, 7 9.0, 2 x CH in Ph), 8.15
(2H, d, J 9.0, 2 x CH in Ph), 5.44 (IH, s, OCH), 2.57 (IH, d, J 17.0, CH2), 2.43 (IH, d, J
17.0, CH2), 1.44 (3H, s, CCH3 ), 1.02 (3H, s, CCH3); 5C (100 MHz, CDC13 ), 195.1 (C),
174.9 (C), 150.8 (C), 140.2 (C), 129.8 (2 x CH), 124.2 (2 x CH), 86.2 (CH), 42.0 (CH2),
41.1 (C), 27.9, 23.4 (2 x CH3 ); m/z (CI) 250 (MNH4+, 100%) Found: MNH4+, 281.1141.
Ci 3Hi 3NO5 requires MNH4+, 281.1137.
(±)-5-(2-Naphthoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 339: Prepared by the general
procedure (0.71 mmol of 2-alkoxydihydropyran, 0.12 M DMDO 7.0 mL, 3.0 M Jones
reagent 1.2 mL, 3 h for DMDO oxidation, 1 h for Jones oxidation) to give lactone (±)-339
as colourless oil (101 mg, 53% over 2 steps). vmax (CHCy/crrf 1 2962-2926 (CH), 1788
(lactone), 1681 (CO), 1627; 5H (400 MHz, CDC13) 8.48 (IH, s, CH in NapH), 7.93-8.04
(4H, m, 4 x CH in Naph), 7.62-7.71 (2H, 2 x CH in Naph), 5.71 (IH, s, OCH), 2.66 (IH, d,
J 17.0, CH2), 2.39 (IH, d, J 17.0, CH2), 1.46 (3H, s, CH3), 1.03 (3H, s, CH3); 5C (100 MHz,
CDC13 ), 195.6 (C), 175.8 (C), 136.0 (C), 133.2 (C), 132.4 (C), 130.7 (CH), 129.8 (CH),
129.3 (CH), 129.2 (CH), 127.9 (CH), 127.3 (CH), 123.7 (CH), 85.5 (CH), 41.9 (CH2 ), 40.8
(C), 28.4, 23.6 (2 x CH3 ); m/z (CI) 286 (MNH4+, 100%); Found: MNH4+, 286.1437.
C, 7Hi 6O3 requires MNH4+, 286.1443.
217

General procedure for enantioselective rearrangement / Jones oxidation of alkoxydihydropyrans using Jacobsen's catalyst
R1
o
R 1
H
Me
H
Me
Et
cyHex
Me
Me
Me
Me
Me
R2
Me
Me
Ph
Ph
Ph
Ph
Naph
/7-MeO phenyl
/7-Me Phenyl
p-C\ phenyl
p-NO2 phenyl
Tetrahydro furanones
231
252
(5)-309
(5)-332
(5)-333
(S)-334
(5)-339
(5)-335
(S)-336
(S)-337
(5)-338
A mixture of alkoxydihydropyran (1.0 eq) pre-dissolved in CHiCb, Jacobsen catalyst
(0.05-0.1 eq) and 4-phenyl pyridine vV-oxide (0.2 eq) was cooled down to 0 "C. Buffered
bleach (1.5-3.0 eq, pH~l 1.5) pre-cooled to 0 °C was added to the mixture and the reaction
was stirred at 0 °C. The reaction mixture was diluted with DCM, extracted with water and
dried with Na2SC>4. Short column purification was performed to remove Mn species with
the solvent system of EtOAc/petrol (1/1). After the reaction mixture was dissolved in acetone, Jones reagent (3.0 eq to initial 2-alkoxydihydropyran) was added dropwise. After stirring, the excess of oxidants was quenched by the addition of 2-propanol until the brown
218

colour of the mixture turned to green. The reaction mixture was diluted with diethyl ether.
After the organic layer was separated and the aqueous layer was extracted with diethyl ether, column chromatography gave a corresponding lactone as a product.
Acetyl-dihydro-furan-2-one 231: Prepared by the general procedure (300 mg of 2-
alkoxydihydropyran, 40 mg of Jacobsen's catalyst, 48 mg of pyridine TV-oxide, 0.7 ml of
NaOCl, 1.5 mL of 3.0 M Jones reagent, 10 h for epoxidation, 3 h for Jones oxidation) to
give lactone 231 as colourless oil (108 mg, 40% over 2 steps), [a] 2̂ 0 (c 0.03, CHC13);
6H (250 MHz, CDC13 ) 4.81 (1H, t, J 7.5, CH), 2.62-2.18 (4H, m, CH2CH2) 2.30 (3H, s, CH3).
Acetyl-4,4-dimethyl-dihydrofuran-2(3H)-one 252: Prepared by the general procedure (250 mg of 2-alkoxydihydropyran, 40 mg of Jacobsen's catalyst, 43 mg of pyridine TV-oxide, 1.0 mL of NaOCl, 1.5 mL of 3.0 M Jones reagent, 10 h for epoxidation, 3 h for Jones
oxidation) to give lactone 252 as colourless oil (32 mg, 16% over 2 steps), [af^ 0 (c 0.03,
CHC13 ); 5H (250 MHz, CDC13) 4.40 (1H, s, OCH), 2.43 (2H, d, J 3.5, CH2 ), 2.27 (3H, s, C(O)CH3 ), 1.35 (3H, s, CH3 ), 1.05 (3H, s, CH3).
(S)-5-Benzoyl-dihydrofuran-2(3H)-one 309: Prepared by the general procedure (276 mg of
2-alkoxydihydropyran, 30 mg of Jacobsen's catalyst, 51 mg of pyridine TV-oxide, 1.0 mL of NaOCl, 1.4 mL of 3.0 M Jones reagent, 6 h for epoxidation, 3 h for Jones oxidation) to give
lactone (5)-309 as white crystal (140 mg, 55 % over 2 steps), mp 189-191 °C; ee 38% (HPLC analysis: OD-H column, 254 nm, flow 1.0 ml/m, 'PrOH/hexane (5/95), tm=37.90
min, tM=46.15 min); [a] 2D° 0 (c 0.03, CHC13 ); 6H (500 MHz, CDC13 ) 8.02-7.48 (5H, m, Ph),
5.82-5.76 (1H, m, CH2C//), 2.65-2.44 (4H, m, 2 x CH2)
(+)-(S)-5-Benzoyl-4,4-dimethyl-dihydrofuran-2(3H)-one 332: Prepared by the general
procedure (300 mg of 2-alkoxydihydropyran, 80 mg of Jacobsen's catalyst, 50 mg of
pyridine TV-oxide, 2 mL of NaOCl, 2.2 mL of 3.0 M Jones reagent, 6 h for epoxidation, 3 h
219

for Jones oxidation) to give lactone (S)-332 as white crystal (140 mg, 49% over 2 steps),
mp 113-115 °C; ee 58% (HPLC analysis: OD column, 254 nm, flow 0.5 ml/m, 5 %
'PrOH/hexane (5/95), tM=53.24 min, tm=58.21 min); [a] 2D° +42.0 (c 0.01, CHC13); 5H (500
MHz, CDC13 ) 7.96 (2H, d, J 8.5, 2 x CH in Ph), 7.67 (1H, dd, J 8.5, 7.5, CH in Ph), 7.55
(2H, d, J7.5, 2 x CH in Ph), 5.55 (1H, s, OCH), 2.60 (1H, d, J 17.0, CH2), 2.36 (1H, d, J
17.0, CH2), 1.40 (3H, s, CCH3), 1.00 (3H, s, CCH3 ).
(+)-(S)-5-Benzoyl-4,4-diethyl-dihydrofuran-2(3H)-one 333: Prepared by the general
procedure (100 mg of 2-alkoxydihydropyran, 26 mg of Jacobsen's catalyst, 21 mg of
pyridine //-oxide, 1.2 mL of NaOCl, 0.7 mL of 3.0 M Jones reagent, 6 h for epoxidation, 3
h for Jones oxidation) to give lactone (5)-333 as colourless oil (44 mg, 45% over 2 steps),
ee 69% (HPLC analysis: OD-H column, 254 nm, flow 0.5 ml/m, ^rOH/hexane (2/98),
tM=49.18 min, tm=60.11 min); [a] 2D° +47.0 (c 0.01, CHC13 ); 6H (500 MHz, CDC13 ) 7.96 (2H,
d, J 8.5, 2 x CH in Ph), 7.68 (1H, dd, J 8.5, 7.5, CH in Ph), 7.55 (2H, d, J 7.5, 2 x CH in
Ph), 5.71 (1H, s, OCH), 2.63 (1H, d, J 17.1, C(O)CH2), 2.30 (1H, d, J 17.0, C(O)CH2),
1.73-1.80 (1H, m, C#HCH3 ), 1.60-1.68 (1H, m, CH//CH3 ), 1.38-1.46 (1H, m, C//HCH3 ),
1.12-1.22 (1H, m, CH//CH3 ),1.09 (3H, s, J1.5, CH3 ), 0.76 (3H, s, J 7.5, CH3 ).
(+)-(S)-l-Benzoyl-2-oxa-spiro[4,5]decan-3-one 334: Prepared by the general procedure
(300 mg of 2-alkoxydihydropyran, 70 mg of Jacobsen's catalyst, 50 mg of pyridine TV-oxide,
2 mL of NaOCl, 1.8 mL of 3.0 M Jones reagent, 6 h for epoxidation, 3 h for Jones
oxidation) to give lactone (5)-334 as colourless oil (120 mg, 40 % over 2 steps), ee 80%
(HPLC analysis: OD column, 254 nm, flow 0.5 ml/m, 'PrOH/hexane (5/95), tm=57.82 min,
tM=79.09 min); [a] 2D° +29.2 (c 0.01, CHC13); 5H (400 MHz, CDC13 ) 7.94 (2H, d, J 9.0, 2 x
CH in Ph), 7.65 (1H, dd, .7 9.0, 7.5, CH in Ph), 7.53 (2H, d, J7.5, 2 x CH in Ph), 5.53 (1H,
s, OCH), 2.55 (2H, s, CH2), 1.07-1.89 (10H, m, 5 x CH2 in CyHex).
(+)-(S)-5-(4-Methoxybenzoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 335: Prepared by the
general procedure (140 mg of 2-alkoxydihydropyran, 40 mg of Jacobsen's catalyst, 25 mg
220

of pyridine TV-oxide, 1.0 mL of NaOCl, 1.0 mL of 3.0 M Jones reagent, 6 h for epoxidation,
3 h for Jones oxidation) to give lactone (5)-335 as colourless oil (35 mg, 35% over 2 steps),
ee 79% (HPLC analysis: OD column, 254 nm, flow 0.5 ml/m, jPrOH/hexane (5/95),
tm=104.08 min, tM=l 19.72 min); [a] 2D° +56.0 (c 0.01, CHC13 ); 5H (400 MHz, CDC13 ) 8.00
(2H, d, J 9.0, 2 x CH in Ph), 7.01 (2H, d, J 9.0, 2 x CH in Ph), 5.48 (IH, s, OCH), 3.92 (3H,
s, CH3 ), 2.62 (IH, d, J 17.0, CH2), 2.33 (IH, d, J 17.0, CH2), 1.40 (3H, s, CCH3 ), 1.00 (3H, s, CCH3 ).
(+)-(S)-4,4-Dimethyl-5-(4-methylbenzoyl)-dihydrofuran-2(3H)-one 336: Prepared by the
general procedure (157 mg of 2-alkoxydihydropyran, 38 mg of Jacobsen's catalyst, 38 mg
of pyridine //-oxide, 1.3 mL of NaOCl, 1.1 mL of 3.0 M Jones reagent, 15 h for epoxidation,
1 h for Jones oxidation) to give lactone (5)-336 as white solid and oil (64 mg, 43% over 2
steps), mp 157-160 °C; ee 70% (HPLC analysis: OJ column, 254 nm, flow 1.2 ml/m,
^rOH/hexane (2/98), tm=33.14 min, tM=45.97 min); [a]2D° +37.0 (c 0.02, CHC13 ); 5H (400
MHz, CDC13 ) 7.86 (2H, d, J 8.0, 2 x CH in Ph), 7.34 (2H, d, J 8.0, 2 x CH in Ph), 5.53 (IH,
s, OCH), 2.61 (IH, d, J 17.0, CH2 ), 2.46 (3H, s, CH3 Ph), 2.34 (IH, d, J 17.0, CH2), 1.40
(3H, s, CCH3 ), 0.99 (3H, s, CCH3 ).
(+)-(S)-5-(4-Chlorobenzoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 337: Prepared by the
general procedure (200 mg of 2-alkoxydihydropyran, 51 mg of Jacobsen's catalyst, 39 mg
of pyridine TV-oxide, 2.0 mL of NaOCl, 1.2 mL of 3.0 M Jones reagent, 15 h for epoxidation,
1 h for Jones oxidation) to give lactone (S)-331 as white crystal (56 mg, 30% over 2 steps),
mp 155-158 °C; ee 76% (HPLC analysis: OD-H column, 254 nm, flow 0.5 ml/m,
jPrOH/hexane (5/95), tm=60.54 min, tM=74.56 min); [a] 2D° +25.0 (c 0.01, CHC13 ); 5H (400
MHz, CDC13 ) 7.92 (2H, d, J 8.5, 2 x CH in Ph), 7.53 (2H, d, J 8.5, 2 x CH in Ph), 5.46 (1 H,
s, OCH), 2.59 (IH, d, J 17.0, CH2 ), 2.38 (IH, d, J 17.0, CH2), 1.45 (3H, s, CH 3 ), 1.00 (3H,
s, CH3 ).
(+)-(S)-4,4-Dimethyl-5-(4-nitrobenzoyl)-dihydrofuran-2(3H)-one 338: Prepared by the
221

general procedure (90 mg of 2-alkoxydihydropyran, 19 mg of Jacobsen's catalyst, 17 mg of
pyridine Af-oxide, 1.0 mL of NaOCl, 0.5 mL of 3.0 M Jones reagent, 15 h for epoxidation, 3
h for Jones oxidation) to give lactone (5)-338 as colourless oil (54 mg, 64% over 2 steps),
ee 46% (HPLC analysis: OD-H column, 254 nm, flow 1.5 ml/m, 'PrOH/hexane (10/90),
tm=35.79 min, tM=43.31 min); [a] 2D° +14.0 (c 0.01, CHC13 ); 5H (400 MHz, CDC13 ) 8.39 (2H,
d, y 9.0, 2 x CH in Ph), 8.15 (2H, d, J 9.0, 2 x CH in Ph), 5.44 (IH, s, OCH), 2.57 (IH, d, J 17.0, CH2), 2.43 (IH, d, J 17.0, CH2), 1.44 (3H, s, CCH3 ), 1.02 (3H, s, CCH3 ).
(+)-(S)-5-(2-Naphthoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one 339: Prepared by the
general procedure (200 mg of 2-alkoxydihydropyran, 45 mg of Jacobsen's catalyst, 38 mg
of pyridine //-oxide, 2.0 mL of NaOCl, 1.2 mL of 3.0 M Jones reagent, 15 h for epoxidation,
1 h for Jones oxidation) to give lactone (S)-339 as colourless oil (78 mg, 41% over 2 steps).
ee 71% (HPLC analysis: AD-H column, 254 nm, flow 1.8 ml/m, 'PrOH/hexane (4/96),
tm=64.66 min, tM=69.32 min); [a] 2D° +31.0 (c 0.01, CHC13 ); 6H (400 MHz, CDC13 ) 8.48 (IH,
s, CH in NapH), 7.93-8.04 (4H, 4 x CH in Naph), 7.62-7.71 (2H, 2 x CH in Naph), 5.71
(IH, s, OCH), 2.66 (IH, d, J 17.0, CH2), 2.39 (IH, d, J 17.0, CH2), 1.46 (3H, s, CH3 ), 1.03
(3H, s, CH3 ).

Bayer-Villiger oxidation of 5-benzoyl-dihydrofuran-2(3H)-one l *la
5-Oxo-tetrahydrofuran-2-yl benzoate 354:
—————— ^ Q==0 fl ptrV N)'
354
To a solution of lactone (100 mg) in 1,2-dichloroethane (20 mL) at room temperature were
added NaH2PO4 (252 mg) and MCPBA (277 mg). After stirring at 80° C for 2 d, the
reaction mixture was quenched by sat. ^28263 (aq). The resulting mixture were extracted
with diethyl ether and washed with NaHCOs (aq) and brine. The organic layer was dried
over Na2 SO4 . After the evaporation of solvent, column chromatography eluting with
EtOAc/petrol (1/1) gave 354 as a colourless oil (84 mg, 78%). vmax (CHCy/cm' 1 3433,
2360, 1798 (lactone), 1733 (C=O), 1601; 5H (400 MHz, CDC13 ) 8.06 (2H, d, J7.0, 2x CH
in Ph), 7.63, (IH, t, J7.5, CH in Ph), 7.49 (2H, dd, 77.0, 7.5, 2 x CH in Ph), 6.90 (IH, dd, J
5.5, 1.0, OCHO), 2.40-2.89 (4H, m, C//2C#2); 5C (100 MHz, CDC13) 175.4 (C), 164,7 (C),
133.9 (CH), 129.9 (2 x CH), 128.8 (C), 128.6 (2 x CH), 95.8 (CH), 28.1, 25.9 (2 x CH2);
m/z (CI) 224 (MNH4+, 100 %); Found: MNH4+, 224.0922. CnHi 0O4 requires: MNH4+,
224.0923.
223

3. Compounds from Chapter 3.
Synthesis of «x0-bicyclic ketone catalyst(±)-8-Oxa-bicyclo[3.2. l]oct-6-en-3-one 368 181
To a rapidly stirring solution of furan (11.3 mL, 150.0 mmol) and trifluoroethanol (30 mL)
at 5 °C under Ar was added 1,1,3-trichloroacetone. To this stirring mixture was then added
triethylamine (8.6 mL, 62.0 mmol) dropwise. After stirring at room temperature for
overnight, the reaction was quenched by water (100 mL). The organic layer was separated
and the aqueous layer was extracted with CH2 C12 . The combined organic layer was dried
over Na2 SC>4 and concentrated under reduced pressure. Short column chromatography
eluting with diethyl ether and petrol (1/1) removed dark brown impurity.
To zinc powder (20.3 g, 310.0 mol) and copper (I) iodide (19.6 g, 100.0 mmol) was added
methanol (30 mL) and benzene (30 mL). After stirring for 20 min, the reaction mixture
from the previous step was then added dropwise at 0 °C to avoid a large exotherm. The
resulting reaction mixture was stirred for overnight under Ar, quenched by addition of
saturated Na2EDTA solution (30 mL) and then left for 1 hour. The slurry obtained was
filtered through a filter paper and washed with CH2C12 . The filtrate was dried over MgSO4
and concentrated under reduced pressure. Column chromatography eluting with
diethylether and petrol (1/1) gave 368 as a pale yellow solid (1.7 g, 45% over 2 steps), 8H
(400 MHz, CDC13 ) 5.95 (2H, s, 2 x CHbridge), 4.69 (2H, d, J 5.0, CH=CH), 2.39 (2H, dd, J
17.0, 5.0, 2 x CH2axC=O), 1.95 (2H, d, J 17.0, 2 x CH2eqC=O); m/z (CI) 125 (MH*\ 142
(MV///, 100%), 202, Found: MNH4+, 142.0869, C7H8O2 requires MNH4+ , 142.0868.
224

(± )-8-Oxa-bicyclo[3.2. lJoctan-3-one 369' 55 :
369
To a solution of oxabicycle 368 (200 mg, 1.0 mmol) in MeOH (5 mL) was added 10 %
Pd/C (20 mg). The reaction system was evacuated in the presence of hydrogen to leave a
positive pressure of hydrogen. After stirring for 24 h at room temperature, the reaction
mixture filtered through a short pad of Celite™ and the filtrate was evaporated to dryness
under reduced pressure to yield crude pale yellow oil. Column chromatography eluting with
diethylether and petrol (1/1) gave 369 as a pale yellow solid (198 mg, 98%), 8H(400 MHz,
CDC13 ) 4.67 (2H, s, 2 x CHbridge), 2.69 (2H, dd, J 15.5, 2.5, 2 x CH2axC=O), 2.23 (2 H, d, J 15.5, 2 x CH2eqC=O), 2.05-1.95 (2H, 2 x C//2eqCH2), 1.75-1.65 (2H, 2 x C//2axCH2).
( + )-2-Methylene-8-oxa-bicyclo[3.2.1Joctan-3-one 370 and 2,4-dimethylene-8-oxa-
bicyclo[3.2.l]octan-3-one 371:
(+)-370
To a stirred solution of the parent ketone (1.25 g, 9.9 mmol) in THF (15 mL) cooled to -
78 °C under Ar was added dropwise LiHMDS 1.0 M in THF (9.9 mL, 9.9 mmol). After
stirring at -78 °C for 30 min, Me3 SiCl (1.9 mL, 13.6 mmol) was added. After stirring for 30
min, the reaction mixture was quenched with a saturated solution of NH4C1 (50 mL) at 0 °C.
225

The organic layer was diluted with EtOAc (100 mL) and extracted with EtOAc (3 x 20 mL).
The combined organic layers were washed with a saturated solution of NaHCO3 (50 mL),
dried over MgSO4 and concentrated. To the solution of silyl enol ether in DMF (15 mL) in
a sealed tube under Ar was added Eschenmoser's salt (1.83 g, 9.9 mmol). After stirring at
room temperature for 1.5 h, Mel (2.9 mL, 47.5 mmol) was added. After stirring at 50 °C for
overnight, NaHCO3 (5.81 g, 69.2 mmol) was added and the reaction mixture diluted with
DMF (10 mL). After stirring at 95 °C for overnight, the reaction mixture cooled to room
temperature. The reaction mixture was diluted with EtOAc (100 mL), washed with H2O (50
mL), extracted with EtOAc (3 x 30 mL) and separated. The combined organics were dried
over MgSO4 and concentrated. Column chromatography eluting with EtOAc and Hexane
(1/1) gave methylene (±)-370 (0.54 g, 40% over 2 steps) as pale yellow oil and
dimethylene 371 (200 mg, 13% over 2 steps) as pale yellow oil.
(±)-2-Methylene-8-oxa-bicyclo[3.2.1]octan-3-one (±)-370: vmax (CHC^/cnV 1 2963 (CH),
1697 (C=O), 1629 (OC); 8H(400 MHz, CDC13) 5.95 (IH, s, C=CH2), 5.20 (IH, s, C=CH2),
5.01 (IH, d, J 6.5, OCHbridge), 4.77 (IH, t, J 6.5, OCHbridge), 2.81 (IH, dd, J 6.0, 18.0,
CH2axCO), 2.43 (IH, d, J 18.0, CHC//2eqCO), 2.18-2.31 (2H, m, CHC//2exoC//2exoCH),
1.75-1.93 (2H, m, CHC//2endoC//2endoCH); 6C (100 MHz, CDC13 ) 197.7 (C=O), 146.7 (C),
118.5 (CH2), 78.4 (CH) , 74.0 (CH), 48.1 (CH2), 32.1 (CH2), 29.3 (CH2); m/z (CI) 156
(MNH4+ , 100%), Found: MNH4+, 156.1026, C8Hi0O2 requires MNH4+, 156.1025.
2,4-Dimethylene-8-oxa-bicyclo[3.2.1]octan-3-one 371: vmax (CHCbycm" 3425, 2957,
1626 (C=C, C=O); 6H(400 MHz, CDC13 ) 6.15 (2 H, s, C=CH2), 5.32 (2 H, s, C=CH2 ), 5.01
(2H, dd, J 5.0, 3.0, OCHbridge), 2.36-2.27 (2H, m, CHC#2exo), 1.86 (2H, d, J 7.0,
CHC//2endoCH); 6C (100 MHz, CDC13 ) 186.1 (C=O), 146.2 (C), 120.0 (CH2), 78.8 (CH) ,
31.0 (CH2 ); m/z (CI) 168 (MNH4+, 100 %), Found: MNH4+, 168.1021, C 9H 10O2 requires
MNH4+, 168.1025
226

. * _ _ * _ _*(1R ,2R ,5S)-(±)-2-Hydroxy-2-(hydroxymethyl)-8-oxa-bicyclo[3.2.1]octan-3-one 372:
To a stirred suspension of K2 OsO4 -2H2 O (11 mg, 0.03 mmol), quinuclidine (22 mg, 0.15 mmol) and 7V-rnethylmorpholine-7V-oxide (680 mg, 5.8 mmol) in H2O (3 mL) was added a
solution of olefin (400 mg, 2.9 mmol) in acetone (6 mL). After stirring vigorously at room temperature for 3 d, solid sodium metabisulfite (-3.00 g) was added. After vigorous stirring
for a further hour, the reaction mixture was diluted with DCM (10 mL). The solid was
removed by filtration though a pad of Celite™ and washed with DCM (20 mL). The
combined filtrate and washings were evaporated to dryness. Column chromatography
eluting with methanol and dichloromethane (5/95) gave (±)-372 as a white solid (354 mg,
71%), mp (racemate) 108-110 °C; v^^CHC^/crn 1 3469 (OH), 2988 (CH), 1719 (C=O);
6H(400 MHz, CDC13) 4.75 (1H, t, y 5.5, OCHbridge), 4.50 (1 H, d, y 7.5, OCHbridge), 4.01 (1
H, d, J 12.5, CCH//OH), 3.59 (1H, d, J 12.5, CC//HOH), 3.16 (1H, dd, J 15.0, 5.5,
CHC//2axCO), 2.21 (1H, d, J 15.0, CHC//2eqCO), 2.05-2.13 (2H, m, CHC//2exoC//2exoCH),
1.70-1.78 (2H, m, CHC//2endoC//2endoCH); 6C (100 MHz, CDC13) 207.6 (CO), 80.2 (C),
78.4 (CH2), 75.6 (CH), 61.7 (CH), 46.3 (CH2), 28.6 (CH2), 23.4 (CH2); m/z (CI) 190
(MNH4+, 100%), Found: MNH4+, 190.1077, C 8H, 2O4 requires MNH4+, 190.1079.
(1R,2R,5S)-(-)-2-Hydroxy-2-(hydroxymethyl)-8-oxa-bicyclo[3.2.l]octan-3-one (-)-372: A
white solid, mp (non-racemic) 108-110 °C, [a] 2D° -39.7 (c 0.01, CHC13 ); 5H(400 MHz,
CDC1 3 ) 4.75 (1H, t, y 5.5, OCHbndge), 4.50 (1 H, d, J 7.5, OCHbndge), 4.01 (1 H, d, J 12.5,
CCH//OH), 3.59 (1H, d, J 12.5, CC//HOH), 3.16 (1H, dd, J 15.0, 5.5, CHC//2axCO), 2.21
(1H, d, J 15.0, CHC//2eqCO), 2.05-2.13 (2H, m, CHC//2exoC//2exoCH), 1.70-1.78 (2H, m,
CHC//2endoC//2endoCH).
227

,2R ,5S)-(±)-2',3-Dioxo-8-2'> 3-dioxo-8H-spiro [8-oxabicyclo[3.2.1]octane -2,4'-[l,3J dioxolane] 373:
To a stirred solution of diol (200 mg, 1.2 mmol) and pyridine (1.0 mL, 12.0 mmol) in DCM
(10 mL) at 0 °C under Ar was added a solution of triphosgene in DCM (5 mL). After
stirring between 0 °C and room temperature for 8 hours, it was quenched with H2 O (5 mL).
The organic phase was diluted with DCM, separated, dried over MgSO4 and concentrated.
Column chromatography eluting with ethylacetate and hexane (1/1) gave (±)-373 as a
white solid (171 mg, 72%), mp (racemate) 117-119 °C; vmax (CHC^/cnV 1 3449, 2254,
1813 (C=O), 1638 (C=O), 1265 (C-O); 6H(400 MHz, CDC13 ) 5.06 (IH, d, J9.0, CCH2O),
4.87 (IH, t, J 5.5, OCHbridge), 4.68 (IH, d, J 7.0, OCHbridge), 3.94 (IH, d, J 9.0, CCH2O),
3.17 (IH, dd, J 15.0, 5.5, CHC//2axCO), 2.51 (IH, d, J 15.0, CHC//2eqCO), 2.12-2.25 (2H,
m, CHC//2exoC//2eXoCH), 1.75 (IH, t, J9.0, CH2CHC//2endoCH2endoCH), 1.35 (IH, t, J9.0,
CH2CHCH2endoC//2endoCHC); 8C(100 MHz, CDC13) 199.0 (C=O), 152.7 (C-O), 84.3 (C),
79.8 (CH2), 76.0 (CH), 64.6 (CH), 47.5 (CH2 ), 28.2 (CH2 ), 23.6 (CH2); m/z (CI) 216
(MNH4+, 100 %), Found: MNH4+, 216.0877, C9Hi 0O5 requires MNH4+, 216.0872
(1R,2R, 5S)-(+)-2 ',3-Dioxo-8-2', 3-dioxo-8H-spiro [8-oxabicyclo[3.2.1] octane -2,4 '-[1,3]
dioxolane] (+)-373: A white solid, mp (chiral) 118-119 °C; [a] 2D° +15.7 (c 0.01, CHC13 ), ee
68% (Shift reagent Eu(hfc)3 ); 5H(400 MHz, CDC13 ) 5.06 (IH, d, 79.0, CCH2O), 4.87 (IH, t,
.7 5.5, OCHbrldge), 4.68 (IH, d, 77.0, OCHbndge), 3.94 (IH, d, 79.0, CCH2O), 3.17 (IH, dd, J
15.0, 5.5, CHC//2axCO), 2.51 (IH, d, J 15.0, CHC//2eqCO), 2.12-2.25 (2H, m,
CHC//2eXoC//2exoCH), 1.75 (IH, t, J 9.0, CH2CHC//2endoCH2endoCH), 1.35 (IH, t, J 9.0,
CH2CHCH2endoC//2endoCHC).
228

(+)-(8-Oxa-bicyclo[3.2. l]oct-2-en-3-yloxy)trimethylsilane 380 :
OTMS (+)-380
To a solution of the chiral amine (722 mg, 5.7 mmol) in THF at -78 °C under Ar was added
dropwise "BuLi (6.6 mL of 1.33 M solution in hexane, 8.7 mmol). After 5 min the solution
was allowed to warm to room temperature and then recooled to -78 °C. TMSCl (3.9 mL,
43.1 mmol) was added, and after 45 min at -78 °C the reaction was quenched with NaHCOs
(sat, aq), the two phase separated and the aqueous layer extracted with petrol. The organic
phase was washed with CuSC>4, dried and evaporated to give enol silane (+)-380 as
colourless oil (1200 mg, >95%), 8H(400 MHz, CDC1 3 ) 5.08 (IH, d, J5.0, CH=C), 4.58 (IH,
d, J 6.0, OCHbndge), 4.52 (IH, t, J 5.0, OCHbndge), 2.71 (IH, dd, J 5.0, 16.5, CH2axCO),
2.17-2.08 (IH, m, CHC//2eqCO), 2.03-1.88 (2H, m, CHC//2exoC//2exoCH), 1.78-1.69 (2H, m,
CHC//2end0C//2endoCH), 0.20 (9H, s, 3 x CH3); m/z (CI) 90, 199 (MH*\ 226, 216 (MNH4\
Found: MNH4+, 199.1153, C9H 18O2 requires MNH/, 199.1154
229

Synthesis of aza-bicyclic ketone catalyst
(2R , 5R )-5-Methoxy-l-tosylpyrrolidine-2-carbaldehyde and (2R , 5S )-5-methoxy-l- tosylpyrrolidine-2-carbaldehyde 38787 :
jT)—OMe
(f Ts
387
To a solution of dried Chloramine-T (13.65 g, 60 mmol, 1.2 eq) in acetonitrile (250 mL) at
room temperature was added the 2-alkoxydihydropyran (5.7 mL, 5.70 g 50 mmol, 1.0 eq).
After stirring for 5 min, NBS (1.78g, 10 mmol, 0.2 eq) was added. After stirring for 2 h, the
reaction mixture passed through a plug of Et3N-washed silica, flushing with EtOAc. The
resulting solution was concentrated in vacuo to give a dark orange oil. Column
chromatography eluting with ethylacetate and hexane (1/1) gave pale yellow oil 387 as a
mixture (6.99, 50 %) of 2 diastereisomers (1:1), 5H(400 MHz, CDC13 ) 9.70 (lHtrans, d, J 3.0,
CHO), 9.54 (lHcis, d, J 3.0, CHO), 7.77 (21^, d, J 8.5, ArH), 7.69 (2Hcis , d, J 8.0, ArH),
7.33 (2Hcis , d, J 8.0, ArH), 7.29 (2Htrans, d, J 8.5, ArH), 5.32 (IH^s, d, .74.0, NCH(OMe)),
5.12 (!Hcis , d, J 5.0, NCH(OMe)), 3.87-3.84 (lHtrans, m, C//CHO), 3.81 (1HC1S , td, J 8.5, 3.0,
C//CHO), 3.48 (3Hcis, s OMe), 3.28 (3Htrans , s, OMe), 2.43 (3Htrans , s, ArCH3 ), 2.41 (3HC1S , s,
ArCH3 ), 2.11-2.07 (4Hboth , m, 2 x C//HCHCHO), 1.96-1.94 (4Hboth, m, 2 x CH//CHCHO),
1.90-1.85 (4Hboth , m, 2 x C//HCH(OMe)), 1.29-1.21 (4Hboth, m, 2 x CH//CH(OMe)).
230

. 3S, 5S)-3-Methyl-8-(toluene-4-sulfonyl)-8-aza-bicyclo[3.2.1]octan-2-one 390b
and (±)-(!R*, 3R*, 5S*)-3-methyl-8-(toluene-4-sulfonyl)-8-aza-bicyclo[3.2.1]octan-2-one 390a:
[ V-OMeX^N \\ TsO
OMe
390b 390a
To a solution of aldehyde (4.63 g, 16.3 mmol, 1.0 eq) in Et2O (50 mL) at 0 "C was added
Grignard reagent (0.5 M in THF, 50.0 mL, 1.5 eq). The reaction was allowed to warm to
room temperature for 10 min before being quenched by the addition of saturated aqueous
NH4C1 solution (100 mL). The mixture was extracted into diethyl ether (2 \ 100 mL). The
combined organics washed with brine and then dried over Na2SC>4. Column
chromatography eluting with diethyl ether and petrol (2/3) gave the rearrangement
precursor as a mixture of diastereoisomers as a pale yellow oil (1.69 g). m/z (CI) 343
(MNH4 +\
To a solution of rearrangement precursor (1.69 g, 1.0 eq) in DCM (50 mL) at 0 °C was
added SnCU solution (1.0 M in heptanes, 10.4 mL). The mixture was allowed to warm to
room temperature and stirred for 16 h. The reaction mixture was quenched by the addition
of saturated aqueous NaHCOs (50 mL). The mixture was extracted into chloroform and
combined organics dried over Na2SC>4. Column chromatography eluting with diethyl ether
and petrol (1/1) gave isomeric tropanes endo 390b (0.20 g, 13 %) and exo 390a (0.23 g,
15 %) as pure compounds.
endo product 390b: 5R (400 MHz, CDC13 ) 7.74 (2H, d, J 8.0, 2 x SO2CCH), 7.30 (2H, d, J
8.0, ArH), 4.51-4.42 (1H, m, NC//(CH2)2), 4.38-4.32 (1H, m, NCHCO), 2.80-2.62 (1H, m,
C//CH3 ), 2.43 (3H, s, ArCH3 ), 1.83-0.86 (6H, m, CCH2C), 1.04 (3H, d, J 6.5, CMC//,); m/=
(CI) 294 (Mff), 311 (MV///, 100%), Found: MH+ , 294.1155, Ci5H 19NO3 S requires MH+ ,
231

294.1164.
exo product 390a: 8H (400 MHz, CDC13 ) 7.74 (2H, d, J 8.0, 2 x SO2CCH), 7.30 (2H, d, J
8.0, ArH), 4.48-4.40 (1H, m, NC//(CH2)2), 4.29-4.23 (1H, m, NCHCO), 2.51-2.34 (1H, m,
CHMe), 2.43 (3H, s, ArCH3 ), 2.03-1.66 (6H, m, CCH2C), 0.97 (3H, d, J 6.5, CHC#3 ); m/z
(CI) 294 (Mrf\ 311 (MNH4+ , 100%), Found: MNH4+ , 311.1432, C 15Hi 9NO3 S requires MNH4+, 311.1429.
General procedure for one-phase epoxidation system - Yang pH 7.5
To a solution of ketone catalyst (0.1 eq) and alkene (1.0 eq) in acetonitrile (6.0 ml) was
added aqueous Na2EDTA solution (4.0 ml of a 0.4 mM aqueous solution). Oxone® (10.0
eq) and NaHCOs (15.5 eq) were added in portions simultaneously over 30 minutes. The
reaction was stirred vigorously until completion (by TLC analysis) or for 18 hours, then
diluted with water. The reaction mixture was extracted into diethyl ether (30 mL x 3). The
combined organic extracts were dried over Na2SO4, filtered and evaporated to dryness
under reduced pressure. Column chromatography eluting with diethyl ether and hexane
previously washed with 1% triethylamine gave the relevant epoxides.
(2R*,3R*)-2,3-Diphenyloxirane 374 182 :
374
White solid; m.p. 68-70 °C; 5H (400 MHz, CDC13 ) 7.36-7.45 (10H, m, 2 x Ph), 3.91 (2H, s,
2 x CH); m/z (CI) 214 (MNH4+, 100 %); m/z (CI) 197 (M7/+), 214 (MNH4 + , 100%), Found:
MNH4+, 214.1239. Ci 4Hi 2O requires MNH4+, 214.1232.
(2R,3R)-2,3-Diphenyloxirane: white solid; m.p. 68-70 °C; eeobs 62%; eemax 91%; [a] 2D°
+60.0 (c 0.01, CHC13 ).
232

2-Phenyloxirane 375 183.
375
Colourless oil; 5H (400 MHz, CDC13 ) 7.40-7.28 (5H, m, Ph), 3.89 (IH, dd, J 2.5, 4.0, CH),
3.17 (IH, dd, 74.0, 5.5, C//H), 2.83 (IH, dd, 72.5, 5.5, CH//).
(R)-2-Phenyloxirane: colourless oil; eeobs 28%; eemax 41%; [a] 2D° +2.0 (c 0.01, CHC13 ).
184.2-Methyl-2-phenyloxirane 376 :
376
Colourless oil; 5H (400 MHz, CDC13 ) 7.48-7.28 (5H, m, Ph), 3.80 (IH, d, 7 11.0, C//H),
3.64 (IH, d, 7 11.0, CH//), 1.54 (3H, s, CH3); m/z (CI) 152 (MNH4+), 170, Found: MNH4+,
152.1077, C 9HioO requires MH+, 152.1075.
(R)-2-Methyl-2-phenyloxirane: colourless oil; ee0bs <1%; eemax <1%; [a] 2D° +20.0 (c 0.01,
CHC13 ).
233

.* ~ —*(2R ,3R )-2-Methyl-3-phenyloxiranellT*5 :
377
Colourless oil; 8H (400 MHz, CDC13 ) 7.39-7.26 (5H, m, Ph), 3.60 (IH, d, 72.0, CH), 3.06
(IH, dq, 72.0, 5.0, C//CH3 ), 1.48 (3H, d, 7 5.0, CH3 ); m/z (CI) 152 (MNH4+, 100 %),
Found: MNH4+, 152.1077, C9H 10O requires MH+, 152.1075.
(2R,3R)-2-Methyl-3-phenyloxirane: colourless oil; eeobs 48%; eemax 71%; [a]2D° +4.6 (c 0.02,
CHC13 ).
184.,6R )-l-Phenyl-7-oxa-bicyclo[4.l.O]heptane 378 IM :
378
Colourless oil, 5H (400 MHz, CDC13 ) 7.56-7.28 (5H, m, Ph), 4.03 (IH, d, 7 11.0, CH), 1.94-
1.37 (8H, 4 x CH2); m/z (CI) 175(MH+), 192, Found: MNH4+, 175.1124, C, 2Hi 4O2 requires
MH+, 175.1123.(1R,6R)-1-Phenyl-7-oxa-bicydo[4.1.0]heptane: colourless oil; ee0bs 55%; eemax 81%;
[a] 2D° +110.0 (c 0.01, CHC13).
234

,* ~ _*(2S ,3R )-Ethyl 3-phenyloxirane-2-carboxylate 379 186 :
OEt
379
Colourless oil; 7.42-7.30 (5H, m, Ph), 4.30 (2H, q, J 7.0, OCH2), 4.11 (IH, d, J 1.5, PhCH),
3.53 (IH, d, J 1.5, CH), 1.35 (3H, d, J 7.0, CH3); m/z (CI) 210 (MNH4+, 100 %), Found:
MNH4+, 210.1131, CnH 12O3 requires MH+, 210.1130.
(2S,3R)-ethyl 3-phenyloxirane-2-carboxylate: colourless oil; ee0bs 46%; eemax <68%; [a] 2£
+40.0 (c 0.01, CHC13 ).
235

Appendix
236

1. NOESY spectra from Chapter 1.
(4S*,5S*)-5-Acetyl-4-methyl-dihydrofuran-2(3H)-one250b
H Chung HC 165-1 in CDC13 ; noesy spectrum using DR ) ; Nov03/2 )
-
-
^
.1 llll A.. 1 A ..... ,
•
/
4
i4
• <••• '
»•
', '" *
:>i? "^ '•'*
•
•*•
1
»
£'•>•/': ': •'
*\-
^ *:r '
1
:
•':•:•'-1
-2
-3
-4
_ppra
ppra
237

II .
mdd"
E-
T-
1(1—IF^
UJdd . I
£/EOAON ' ^xdQ Bursn unloads Asaou : EIOQO ui g-gg; OH Bungo H
o
^

(4R ,5R )-5-Acetyl-4-isopropyl-dihydrofuran-2(3H)-one 251 a
o
H Chung HC 161 in CDC13 ; 1H spectrum using DRX400 ; /02/6
• » 01 *
-1
-2
-3
-4
-5
ppm
ppra 5
239

(4R ,5R )-5-Acetyl-4-phenyl-dihydrofuran-2(3H)-one 235
H Chung HC 164 ir
<
-4.
i COC13 ; noesy spectrum using DRX400 )ov02/2
XI 1 . JL
-
i
•
*
Kjf
• /
I
f
\
»
»
•
»
•
•
•
••)
•r$'
y. : »
»
«
'V
^/
1
-2
~-3
-4
-5
-7
•P
ppm
240

(4R*,5R*)-5-Acetyl-4-(benzyloxymethyl)-dihydrofuran-2(3H)-one253
OBn
S^oo
H Chung HC140-2 in CDC13 ; cosylspectrun using DRX400 : FeblO/431
-t *_—«JJLJLAI»i
**
• • * ••
• • *
-2
-6
-8
-PPB
241

(4S ,5R ,E)-5-Acetyl-4-(oct-5-enyl)-dihydrofuran-2(3H)-one 254
H Chung HC 159 in CDC13 ; noesy spectrum using DRX40C \Nov02/4
•ir
ppm 5
i.
••'' Wj..,..-.*..
-1
-2
-3
-4
ppm
242

(4St,5S*)-5-Butyryl-4-methyl-dihydrofuran-2(3H)-one273b
O
H. Chung - 1H NOESY of HC225A in CDC13 at 400MHz.
1
Nil
If! %.
; /
ppm
J?
< ii•H
]—i—i—i—r
i
-i—i—i—i—i—r
-1
-2
-3
-4
ppra
243

(4S*,5f?>5-Butyryl-4-methyl-dihydrofuran-2(3H)-one273a
V,Prn>
O
H. Chung - 1H NOESY of HC225B in CDC13 at 400MHz.
ii
-1
-3
ppra
ppra-i—i—i—r—T—r
3 ' .."• 2
244

Stc
uidd 0' T
g-j...
O'E---
9'2-
CTZ---
,,-..-4-
O'T---
uidd
S'T
.r
u n
"Fu
9'ZO'E
Illl
*»»!
S"-:O'f
u
IT
o

(4S,5f?)-4-Methyl-5-pentanoyl-dihydrofuran-2(3H)-one274a
x ilU Jn JU A A . . j
1)
1 II. . 1 —. — . — , — .—
.
. . . . J
----- -
.... . , . . 1
I.J.....
*
. • . i i . . . . i — *— , — . — . — t—— i
* %Li* Wjj jj
«
K<
• . • . .
w
(*
,jjrH
i . . . .
»*--*<r
.
1 • • i i •
, 9 i<~-r——-\
4 l
0.......-,
..J....J1
, ...J
»l
1
'.
»-•1 ft
f
Ijj
Tii
*.__.
hf-•
-i.
-i.
-2.
-2.
-3.
•3.
-4.
-4.
-5.
-6.
-6.
-7.
ppm
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 .1.5 1.0 ppm
246

2. HPLC chromatograms from Chapter 2.HPLC conditions and results for measuring the configurations.
5-Benzoyl-dihydrofuran-2(3H)-one 309
VWD1 A.
500 -
-*oo -
300 -
ZOO
1OO
,N/VVD1 A.
n/KLJ50O -
Column Conditions tm (configuration) tM (configuration)
Chiralcel OD-H 254; 5/95; 1.0 37.90 (R) 46.15(5)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
247

5-Benzoyl-4,4-dimethyl-dihydrofuran-2(3H)-one 332
WVD1 A.mAU
2500 -
nm (000-0101 .D>
Column Conditions tm (configuration) (configuration)
Chiralcel OD 254; 5/95; 0.5 58.21 (R) 53.24 (S)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
248

5-Benzoyl-4,4-diethyl-dihydrofuran-2(3H)-one333
VWD1 A. Wovalengur-2&4 nm (HCOO1-O2O1. D)
ITS -
1SO-
125-
1OO -
VVVC31 A, VVavclcngth—254 nm <MC\OO2-O3O1 -D)mAU
700
eoo
Column Conditions3 tm (configuration) tM° (configuration)
Chiralcel OD-H 254; 2/98; 0.5 60.11 (R) 49.18(5)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
249

1 -Benzoyl-2-oxa-spiro[4,5]decan-3-one 334
lAU
1-4OO
, <HC\001 0101 ID)
VWD1 A. Wa (HCVOO2 O3O1 D)
aoo
•700
300 -
2OO
1OO
o33
Column Conditions tm (configuration) tM (configuration)
Chiralcel OD 254; 5/95; 0.5 57.82 (K) 79.09 (S}a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
250

5-(4-Methoxybenzoyl)-4,4-dimethyl-dihydrofuran-2(3/-/)-one 335
WVD1 A. VVaveTehgtH*=:25-4 rTr
140
120
1OO
- KJ^ ~,.§Pi
VWD1 A. WssveienQtfi=2S-<l om <CMD3-O3O1 D)~nAU
120 -
Column Conditions tm (configuration) (configuration)
Chiralcel OD 254; 5/95; 0.5 104.08 (7?) 119.72(5)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
251

4,4-Dimethyl-5-(4-methylbenzoyl)-dihydrofuran-2(3H)-one336
Me,
WVD1 A. rtrrt (MC\MC393-DM D>
rim (t i C MH C39-4JA. D>
Column Conditions3 tm (configuration) IMC (configuration)
Chiralcel OJ 254; 5/95; 0.5 45.97 (R) 33.14(5)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
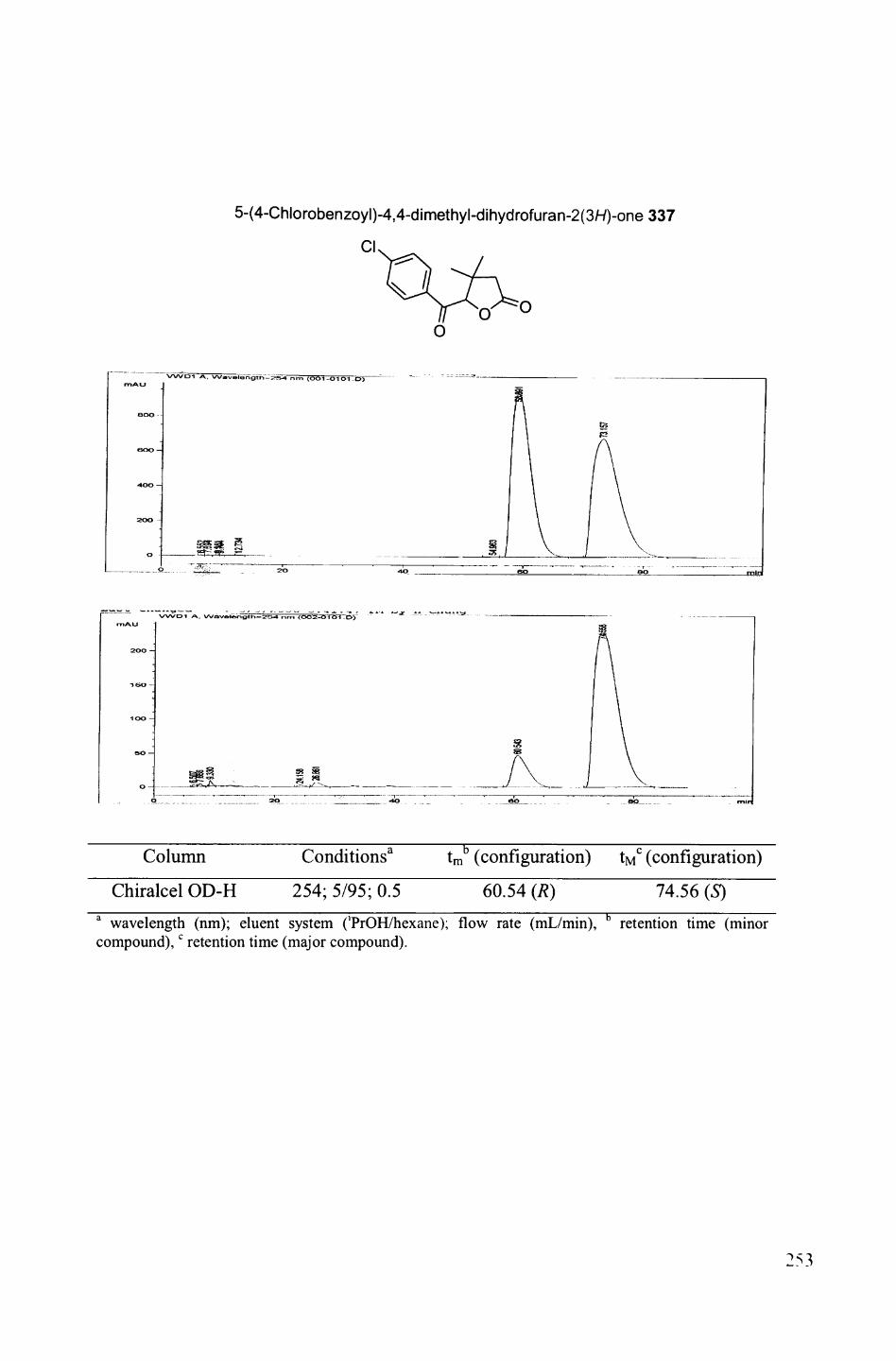
5-(4-Chlorobenzoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one337
CK
nm (OO1-O1O1.D)
WVD1 ^X, rtm (OO2-O1O1 -C>>-" JT. .±± „ >-* *«**>*_.
Si S3
Column Conditions tm (configuration) (configuration)
Chiralcel OD-H 254; 5/95; 0.5 60.54 (K) 74.56 (S)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
253

4,4-Dimethyl-5-(4-nitrobenzoyl)-dihydrofuran-2(3H)-one338
mAU
3SO -
"VWD1 A; W«v«l»ngtM-Z54 nm (MC\HC1O3 D)
Si s;
nnr» (HCM-IC4O4.D)
§ 1
Column Conditions3 tm (configuration) tM (configuration)
Chiralcel OD-H 254; 10/90; 1.5 35.79 (K) 43.31 (S)a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).

5-(2-Naphthoyl)-4,4-dimethyl-dihydrofuran-2(3H)-one339
X. W
§ g is S
mAU
1 4OO
S
Column Conditions tm (configuration) (configuration)
Chiralcel AD-H 254; 10/90; 1.5 35.79 43.31 OS)
a wavelength (nm); eluent system ('PrOH/hexane); flow rate (mL/min), retention time (minor compound), c retention time (major compound).
255

3. HPLC chromatograms from Chapter 3.HPLC conditions and results for measuring the enantiomeric excesses and the
configurations; common conditions for the HPLC; (a) column=chiralcel AS-H, (b) wave length=220 nm.
frans-2,3-Diphenyloxirane 374
Conditions t (configuration)
3/97; 1.0 5.29 (R) 5.72 (S)eluent system ('PrOH/hexane); flow rate (mL/min), retention time (min)
256

16
Conditions3
O
2-Phenyloxirane 375
0.1/99.9; 0.8
c»
.-1——•—J—
an
t (configuration)
17.53 (R) 21.47(5)eluent system ('PrOH/hexane); flow rate (mL/min), retention time (min)
2-Methyl-2-phenyloxirane 376
V
10 12 14
Conditions t (configuration)
7/93; 1.2 10.10 (R) 12.02 (5)eluent system ('PrOH/hexane); flow rate (mL/min), b retention time (min)
257

fr-ans-2-Methyl-3-phenyloxirane 377
Conditions t (configuration)
1/99; 1.0 6.36 (R,R) 8.41 (5,5)eluent system ('PrOH/hexane); flow rate (mL/min), retention time (min)
frans-Ethyl 3-phenyloxirane-2-carboxylate 379
s! I/'
6
Conditions3 t (configuration)
10/90; 1.0 6.23(5,5) 8.52 (R,R)
10
eluent system ('PrOH/hexane); flow rate (mL/min), retention time (min)
258

1-Phenyl-7-oxa-bicyclo[4.1 .OJheptane 378
a 10Conditions
7/93; 1.2
T-.
12 8 tO
t (configuration)
8.37 (2S, 3R) 10.84(2/^,35)
I12
eluent system ('PrOH/hexane); flow rate (mL/min), retention time (min)
259

4. NMR spectra for oxa-bicyclic catalyst from Chapter 3.(lR*,2S*,5S*)-(±)-2 I,3-Dioxo-8-2',3-dioxo-8H-spiro [8-oxabicyclo[3.2.1]octane-2,4'-[l,3]
dioxolane] 373
373
g—' ro
—4000C
— 3000C
— 2000C
—1000C
— 0
^ ' I8.0
ppm (11)
hc468
70\
60I
5 0I
4.0 30I
2.0I
1 0
200 1SO 160 140 120 100—i— 80
—i— 60 40
—i— 20 ppm
260

References
1 a) Paul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407-2474; b) Tsuda, M.; Kobayashi, J. Nat. Prod Rep. 2004, 21, 77-93.
2 a) Marshall, J.A.; Yu, R. H.; Perkins, J. F. J. Org. Chem. 1995, 60, 5500-5555; b) Ballini,
R.; Fiorini, D.; Gil, M. V.; Palmieri, A.; Roman, E.; Serrano, J. A. Tetrahedron Lett. 2003, 44, 2795-2797.
3 Saleem, M.; Kirn, H. J.; Ali, M. S.; Lee, Y. S. Nat. Prod. Rep. 2005, 22, 696-716.
4 Kang, E.; Lee, E. Chem. Rev. 2005, 105, 4348-4378.
5 Westley, J. W. Poly ether Antibiotics Vol 1-2; Marcel Dekker: New York, 1982.6 Lee, C.; Kirn, H.; Kho, Y. J. Nat. Prod. 2002, 65, 414-416.
7 Habtemariam, S. Toxicon 2003, 41, 723-727.
8 Yamasu, T.; Okazaki, A. Galaxea 1987, 6, 61-68.
9 Kobayashi, J.; Ishibashi, M.; Walchli, M. R.; Nakamura, H.; Hirata, Y.; Sasaki, T.;
Ohizumi, Y. J. Am. Chem. Soc. 1988, 110, 490-494.
10 Tsuda, M.; Endo, T.; Kobayashi, J. J. Org. Chem. 2000, 65, 1349-1352.
11 a) Boivin, T. L. B. Tetrahedron 1987, 43, 3309-3362; b) Figadere, B.; Harmange, J. Tetrahedron: Asymm. 1993, 4, 1711-1754; c) Elliott, M. C. Contemp. Org. Synth. 1994,
7, 457-474. d) Burns, C. J.; Middleton, D. S. Contemp. Org. Synth. 1996, 3, 229-242; e)
Elliott, M. C. Contemp. Org. Synth. 1997, 4, 238-259; e) Elliott, M. C. J. Chem. Soc.,
Perkin Trans. 1 1998, 4175^200; g) Elliott, M. C. J. Chem. Soc., Perkin Trans. 1 2000, 1291-1318; f) Elliott, M. C. J. Chem. Soc., Perkin Trans. 1 2001, 2303-2340; h) Elliott,
M. C. J. Chem. Soc., Perkin Trans. 1 2002, 2301-2323; i) Wolfe, J. P.; Hay, M. B.
Tetrahedron, 2007, 63, 261-290.
12 Rychnovsky, S. D.; Bartlett, P. A. J. Am. Chem. Soc. 1981, 103, 3963-3964.
13 Dabideen, D.; Ruan, Z. M.; Mootoo, D. R. Tetrahedron 2002, 58, 2077-2084.
14 Zhang, H. P.; Mootoo, D. R. J. Org. Chem. 1995, 60, 8134-8135.
15 Fujioka, H.; Ohba, Y.; Hirose, H.; Murai, K.; Kita, Y. Angew. Chem., Int. Ed. Engl. 2005, 44, 734-737.
261

16 Dumeunier, R.; Leclercq, C.; Marko, I. E. Tetrahedron Lett. 2002, 43, 2307-2311.
17 Marshall, J. A.; Mikowski, A. M. Org. Lett. 2006, 8, 4375-4378.
18 Cane, D. E.; Celmer, W. D.; Westiey, J. W. J. Am. Chem. Soc. 1983, 105, 3594-3600.
19 Dounay, A. B.; Florence, G. J.; Saito, A.; Forsyth, C. J. Tetrahedron 2002, 58, 1865-
1874.
20 Zheng, T.; Narayan, R. S.; Schomaker, J. M.; Borhan, B. J. Am. Chem. Soc. 2005, 727,
6946-6947.
21 Karikomi, M.; Watanabe, S.; Kimura, Y.; Uyehara, T. Tetrahedron Lett. 2002, 43,
1495-1498.
22 Schomaker, J. M.; Pulgam, V. R.; Borhan, B. J. Am. Chem. Soc. 2004, 126, 13600-
13601.
23 Caggiano, L.; Fox, D. J.; House, D.; Jones, Z. A.; Kerr, F.; Warren, S. J. Chem. Soc.,
Perkin Trans. I. 2002, 2634-2645.
24 Qian, H.; Han, X.; Widenhoefer, R. A. J. Am. Chem. Soc. 2004, 126, 9536-9537.
25 Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2005, 727, 6962-6963.
26 Coulombel, L.; Favier, L; Dunach, E. Chem. Commun. 2005, 2286-2288.
27 Kang, S.; Kirn, M. J. Am. Chem. Soc. 2003, 725, 4684-4685.
28 Matej, B.; L'ubos, R.; Ol'ga, K.; Tiber, G. Synlett 2005, 1609-1611.
29 Wolfe, J. P.; Rossi, M. A. J. Am. Chem. Soc. 2004, 126, 1620-1621.
30 Jiang, L.; Burke, S. D. Org. Lett. 2002, 4, 3411-3414.
31 Klein, E.; Rojahn, W. Tetrahedron 1965, 27, 2353-2358.
32 Donohoe, T. J.; Butterworth, S. Angew. Chem., Int. Ed. Engl. 2003, 42, 948-951.
33 Caserta, T.; Piccialli, V.; Gomez-Paloma, L.; Bifulco, G. Tetrahedron 2005, 61, 927-
939.34 Murahashi, S.; Ono, S.; Imada, Y. Angew., Chem. Int. Ed. Engl. 2002, 41, 2366-2368.
35 Carreno, M. C.; Mazery,R. D.; Urbano, A.; Colobert, F.; Solladie, G. J. Org. Chem.
2003, 68, 7779-7787.
36 Raghavan, S.; Rajender, A.; Rasheed, M. A.; Reddy, S. R. Tetrahedron Lett. 2003, 44,
8253-8256.
262

37 Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. J. Org. Chem. 2004, 69, 6874-6882.
38 a) Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. Chem. Commun.
2004, 2292-2293; b) Akindele, T.; Marsden, S. P.; Gumming, J. G. Org. Lett. 2005, 7, 3685-3688.
39 Jimenez-Gonzalez, L.; Alvarez-Corral, M.; Munoz-Dorado, M.; Rodriguez-Garcia, I.
Chem. Commun. 2005, 2689-2691.
40 Sergio, G.; Leticia, J.; Miriam, A.; Manuel, M.; Ignacio, R. Synlett 2005, 3011-3013.
41 Sarkar, T.; Haque, S. K.; Basak, A. Angew. Chem., Int. Ed Engl. 2004, 43, 1417-1419.
42 a) Shin, C.; Chavre, S. N.; Pae, A. N.; Cha, J. H.; Koh, H. Y.; Chang, M. H.; Choi, J.
H.; Cho, Y. S. Org. Lett. 2005, 7, 3283-3285; b) Chavre, S. N.; Choo, H.; Cha, J. H.;
Pae, A. N.; Choi, K. L; Cho, Y. S. Org. Lett. 2006, 8, 3617-3619.
43 Peng, Z.; Woerpel, K. A. Org. Lett. 2002, 4, 2945-2948.
44 Angle, S. R.; El-Said, N. A. J. Am. Chem. Soc. 2002, 124, 3608-3613.
45 a) Yet, L. Tetrahedron 1999, 55, 9349-9403; b) Bowman, W. R.; Cloonan, M. O.;
Krintel, S. L. J. Chem. Soc., Perkin. Trans. I 2001, 2885-2902; c) Majumdar, K. C.;
Basu, P. K.; Mukhopadhyay, P. P. Tetrahedron 2004, 60, 6239-6278; d) Srikanth,
G.S.C.; Castle, S. L. Tetrahedron 2005, 61, 10377-10441.
46 Roy, S. C.; Rana, K. K.; Guin, C. J. Org. Chem. 2002, 67, 3242-3248.
47 Takami, K.; Mikami, S.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Tetrahedron 2003,
59, 6627-6635.
48 Sibi, M. P.; Patil, K.; Rheault, T. R. Eur. J. Org. Chem. 2004, 372-384.
49 Kamimura, A.; Mitsudera, H.; Matsuura, K.; Omata, Y.; Shirai, M.; Yokoyama, S.;
Kakehi, A. Tetrahedron 2002, 58, 2605-2611.
50 Keum, G.; Kang, S. B.; Kirn, Y.; Lee, E. Org. Lett. 2004, 6, 1895-1897.
51 Roy, S. C.; Guin, C.; Rana, K. K.; Maiti, G. Tetrahedron 2002, 58, 2435-2439.
52 Jana, S.; Guin, C.; Roy, S. C. Tetrahedron Lett. 2005, 46, 1155-1157.
53 Ranu, B. C.; Mandal, T. Tetrahedron Lett. 2006, 47, 2859-2861.
54 Hartung, J.; Gottwald, T.; Spehar, K. Synthesis 2002, 1469-1498.
263

55 Yokota, M.; Toyota, M.; Ihara, M. Chem. Commun. 2003, 422-423.
56 Hartung, J.; Kneuer, R.; Laug, S.; Schmidt, P.; §pehar, K.; Svoboda, I.; Fuess, H. Eur,
J. Org. Chem. 2003, 4033-4052.
57 Congde, H.; Xiaodong, J.; Wei, Z.; Li, Y.; Jianming, L.; Zhong-Li, L. Synlett 2004,
251-254.
58 a) Gansauer, A.; Rinker, B.; Pierobon, M.; Grimme, S.; Gerenkamp, M.; Mttck-
Lichtenfeld, C. Angew. Chem., Int. Ed Engl. 2003, 42, 3687-3690; b) Gansauer, A.;
Rinker, B.; Ndene-Schiffer, N.; Pierobon, M.; Grimme, S.; Gerenkamp, M.; Miick-
Lichtenfeld, C. Eur. J. Org. Chem. 2004, 2337-2351.
59 Hilt, G.; Bolze, P.; Kieltsch, I. Chem. Commun. 2005, 1996-1998.
60 a) Fairlamb, I. J. S. Angew. Chem., Int. Ed. Engl. 2004, 43, 1048-1052; b) Tietze, L. F.;
Ila, H.; Bell, H. P. Chem. Rev. 2004, 104, 3453-3516.
61 Lei, A.; He, M.; Wu, S.; Zhang, X. Angew. Chem., Int. Ed. Engl. 2002, 41, 3457-3460.
62 Lei, A. W.; He, M. S.; Zhang, X. J. Am. Chem. Soc. 2003, 725, 11472-11473.
63 a) Mikami, K.; Yusa, Y.; Hatano, M.; Wakabayashi, K.; Aikawa, K. Chem. Commun.
2004, 98-99; b) Mikami, K.; Yusa, Y.; Hatano, M.; Wakabayashi, K.; Aikawa, K.
Tetrahedron 2004, 60, 4475-4480.
64 Jang, H.; Hughes, F. W.; Gong, H.; Zhang, J.; Brodbelt, J. S.; Krische, M. J. J. Am.
Chem. Soc. 2005, 727, 6174-6175.
65 Hatano, M.; Yamanaka, M.; Mikami, K. Eur. J. Org. Chem. 2003, 2552-2555.
66 Zhao, L.; Lu, X.; Xu, W. J. Org. Chem. 2005, 70, 4059-4063.
67 Liu, G.; Lu, X. Tetrahedron Lett. 2003, 44, 467-470.
68 Nakamura, M.; Liang, C.; Nakamura, E. Org. Lett. 2004, 6, 2015-2017.
69 Zhu, G.; Zhang, Z. Org. Lett. 2004, 6, 4041-4044.
70 Takacs, J. M.; Schroeder, S, D.; Han, J.; Gifford, M.; Jiang, X.; Saleh, T.; Vayalakkada,
S.; Yap, A. H. Org. Lett. 2003, 5, 3595-3598.
71 Evans, P. A.; Leahy, D. K.; Andrews, W. J.; Uraguchi, D. Angew. Chem., Int. Ed. Engl.
2004,43,4788-4791.
264

72 Takaoka, L. R.; Buckmelter, A. J.; LaCruz, T. R.; Rychnovsky, S. D. J. Am. Chem. Soc.
2005, 727, 528-529.
73 Skaggs, A. J.; Lin, E. Y.; Jamison, T. F. Org. Lett. 2002, 4, 2277-2280.
74 Hodgson, D. M; Strat, F. L. Chem. Commun. 2004, 822-823.
75 Pohlhaus, P. D.; Johnson, J. S. J. Org. Chem. 2005, 70, 1057-1059.
76 Gupta, A.; Yadav, V. K. Tetrahedron Lett. 2006, 47, 8043-8047.
77 Christie, S. D. R.; Davoile, R. J.; Elsegood, M. R. J.; Fryatt, R.; Jones, R. C. F.;
Pritchard, G. J. Chem. Commun. 2004, 2474-2475.
78 Roger, P.; Durand, A.; Rodriguez, J.; Dulcere, J. Org. Lett. 2004, 6, 2027-2029.
79 Langer, P.; Armbrust, H.; Eckardt, T.; Magull, J. Chem. Eur. J. 2002, 8, 1443-1454.
80 Langer, P.; Bellur, E. J. Org. Chem. 2003, 68, 9742-9746.
81 Freifeld, L; Holtz, E.; Dahmann, G.; Langer, P. Eur. J. Org. Chem. 2006, 3251-3258.
82 M9kosza, M.; Judka, M. Chem. Eur. J. 2002, 8, 4234-4240.
83 Sohn, S. S.; Rosen, E. L.; Bode, J. W. J. Am. Chem. Soc. 2004, 126, 14370-14371.
84 Burstein, C.; Glorius, F. Angew. Chem., Int. Ed. Engl. 2004, 43, 6205-6208.
85 Gumming, G. R. PhD thesis. Imperial College London, 2004.
86 a) Ireland, R. E.; Habich, D. Chem. Ber. 1981, 114, 1418-1427; b) Ireland, R. E.;
Habich, D. Tetrahedron Lett. 1980, 21, 1389-1392.
87 Armstrong, A.; Gumming, G. R.; Pike, K. Chem. Comm. 2004, 812-813.
88 Chernoff, H. C.; Hall, S. S. Chem. Ind. (London) 1970, 896-897.
89 a) Danishefsky, S.; Bednarsky, M., J. Am. Chem. Soc. 1983, 105, 3716-3717; b)
Danishefsky, S.; Bednarsky, M. Tetrahedron Lett. 1984, 25, 721-724.
90 Burke, S. D.; Danheiser, R. L. Handbook of Reagents for organic synthesis[2]
Oxidizing and Reducing Agents; John Wiley & Sons: New York, 1999, 84-89.
91 Nakayama, J.; Kamiyama, H. Tetrahedron Lett. 1992, 33, 7539-7542.
92 McDonald, R. N.; Steppel, R. N.; Dorsey, J. E. Org. Synth. 1970, 50, 15-18.
93 Tsybina, N. M.; Protopopova, T. V., Skoldinov, A. P., J. Org. Chem. USSR (Engl.
Transl.) 191Q, 6, 260-263.
94 Kawamura, T.; Takahashi, H. Bull. Chem. Soc. Jpn. 1969, 42, 1345-1350.
265

95 a) Baxter, E. W.; Reitz, A. B. J. Org. Chem. 1994, 59, 3175-3185; b) Kiely, D. E.;
Talhouk, J. W.; Riordan, J. M.; Gray, Kathy. J. Carbohyd. Chem. 1983, 2, 427-438.
96 Crandall, J. K.; Batal, D. J.; Sebesta, D. P.; Lin, F. J. Org. Chem. 1991, 56, 1153-1161.
97 Burke, S. D.; Danheiser, R. L. Handbook of Reagents for organic synthesis[2]
Oxidizing and Reducing Agents; John Wiley & Sons: New York, 1999, 149-153.
98 Yang, D.; Wong, M.; Yip, Y. J. Org. Chem. 1995, 60, 3887-3889.
99 a) Adam, W.; Mitchell, C. M., Angew. Chem., Int. Ed. Engl. 1996, 35, 533-535; b)
Rudolph, J.; Reddy, K. L.; Chiang, J. P.; Sharpless, K. B., J. Am. Chem. Soc. 1997,
119, 6189-6190; c) Vaino, A. R., J. Org. Chem. 2000, 65, 4210- 4212; d) Saladino, R.;
Neri, V.; Pelliccia, A. R.; Caminiti, R.; Sadun, C. J. Org. Chem. 2002, 67, 1323-1332.
100 Ashraff., C.; Armstrong, A. Unpublished Results.
101 Burke, S. D.; Danheiser R. L. Handbook of Reagents for organic synthesis[2]
Oxidizing and Reducing Agents; John Wiley & Sons: New York, 1999, 468-473.
102 a) Landais, Y.; Zekri, E. Eur. J. Org. Chem. 2002, 4037-4053; b) Nokami, J.; Ogawa,
H.; Miyamoto, S.; Mandai, T.; Wakabayashi, S.; Tsuji, J. Tetrahedron Lett. 1988, 29,
5181-5184.
103 Burke, S. D.; Danheiser, R. L. Handbook of Reagents for organic synthesis[2]
Oxidizing and Reducing Agents; John Wiley & Sons: New York, 1999, 100-102.
104 Craig, J. C.; Horning, E. C. J. Org. Chem. 1960, 25, 2098-2102.
105 a) Hackmann, C.; Schafer, H. J. Tetrahedron 1993, 49, 4559-4574; b) Vaupel, A.;
Knochel, P. J. Org. Chem. 1996, 61, 5743-5753; c) Wakabayashi, K.; Yorimitsu, H.,
Oshima, K. J. Am. Chem. Soc. 2001, 723, 5374-5375.
106 a) Takeda, T.; Watanabe, H.; Kitahara, T. Synlett 1997, 1149-1150; b) Masuzawa, Y.;
Tamogami, S.; Kitahara, T. Nat. Prod. Lett. 1999, 13, 239-246.
107 Desimoni, G; Astolfi, L.; Cambieri, M.; Gamba, A.; Tacconi, G. Tetrahedron 1973, 29,
2627-2634.
108 a) Maga, J. A. Food. Rev. Int. 1996, 72, 105-130; b) Pollnitz, A. P.; Jones, G. P.;
Sefton, M. A. J. Chromatogr. A. 1999, 857, 239-246.
109 Otsuka, K.; Zenibayashi, Y; Itoh, M.; Totsuka, A. Agric. Biol. Chem. 1974, 38, 485-
266

490.
110 Brown, R. C.; Taylor, D. K.; Elsey, G M. Org. Lett. 2006, 8, 463-466.Ilia) Nishikori, H.; Ito, K.; Katsuki, T. Tetrahedron: Asymm. 1998, 9, 1165-1170; b)
Tsuboi, S.; Sakamoto, J.; Yamashita, H.; Sakai, T.; Utaka, M. J. Org. Chem. 1998, 63,
1102-1108; c) Ito, K.; Yoshitake, M.; Katsuki, T. Tetrahedron 1996, 52, 3905-3920;
d) Taber, D. F.; Houze, J. B. J. Org. Chem. 1994, 59, 4004^006; e) Bloch, R.; Gilbert,
L. J. Org. Chem. 1987, 52, 4603^605; f) Sarmah, B. K.; Barua, N. C. Tetrahedron
1993, 49, 2253-2260; g) Zschage, O.; Hoppe, D. Tetrahedron 1992, 48, 5657-5666;
h) Miyata, O.; Shinada, T.; Kawakami, N.; Taji, K.; Ninomiya, I.; Naito, T.; Date, T.;
Okamura, K. Chem. Pharm. Bull. 1992, 40, 2579-2581; i) Casey, M.; Manage, A. C.;
Murphy, P. J. Tetrahedron Lett. 1992, 33, 965-968; j) Sharma, G. V.; Vepachdu, S.
R.; Chandrasekhar, S. Synth. Commun. 1990, 20, 3403-3410; k) Beckmann, M.;
Hildebrandt, H.; Winterfeldt, E. Tetrahedron: Asymm. 1990, 7, 335-345; 1) Marino, J.
P.; Pradilla, R. F. Tetrahedron Lett. 1985, 26, 5381-5384; m) Shengming, M.;
Zhangjie, S.; Zhanqian, Y. Tetrahedron 1999, 55, 12137-12148; n) Suzuki, Y.; Mori,
W.; Ishizone, H.; Naito, K.; Honda, T. Tetrahedron Lett. 1992, 33, 4931-4932; o)
Salaun, B.; Karkour, B.; Olliver, J. Tetrahedron 1989, 45, 3151-3162.
112 (a) Takahata, H.; Uchida, Y.; Momose, T. J. Org. Chem. 1995, 60, 5628-5633; (b) Pai,
Y.-C.; Fang, J.-M.; Wu, S. H. J. Org. Chem. 1994, 59, 6018-6025; (c) Ebata, T.;
Matsumoto, K.; Yoshikoshi, H.; Koseki, K.; Kawakami, H.; Okano, K.; Matsushita, H.
Heterocycles 1993, 36, 1017-1026; (d) Fukuzawa, S.; Seki, K.; Tatsuzawa, M.; Mutoh,
K. J. Am. Chem. Soc. 1997, 779, 1482-1483; (e) Ortuno, R. M.; Merce, R.; Font, J.
Tetrahedron 1987, 43, 4497-4506; (f) Rojo, J.; Garcfa, M.; Carretero, J. C.
Tetrahedron 1993, 49, 9787-9800; (g) Ha, H.-J.; Yoon, K.-N.; Lee, S.-Y.; Park, Y.-S.;
Lim, M.-S.; Yim, Y.-G. J. Org. Chem. 1998, 63, 8062-8066.
113 (a) Chevtchouk, T.; Ollivier, J.; Salaun, J. Tetrahedron: Asymm. 1997, 8, 1011-1014;
(b) Salaun, J.; Karkour, B. Tetrahedron Lett. 1988, 29, 1537-1540.
114 a) Hutchins, R. O.; Maryanof, B. E.; Milewski, C. A. J. Am. Chem. Soc. 1971, 93,
1793-1794; b) Nair', V.; Sinhababu, A. K. J. Org. Chem. 1978, 43, 5013-5017; c)
267

Miller, V. P.; Yang, D.; Weigel, T. M.; Han, O.; Liu, H. J. Org. Chem. 1989, 54, 4175-4188.
115 a) Crich, D.; Beckeith, A. L. J.; Chen, C.; Yao, Q.; Davison, I. G. E.; Longmore, R.
W.; Parrodi, C. A.; Quinte.; Jro-Cortes, L.; Sandoval-Ramirez, J. J. Am. Chem. Soc.
1995, 777, 8757-8768; b) Barton, D. H. R.; Crich, D.; Lobberding, A.; Zard, S. Z.
Tetrahedron 1986, 42, 2329-2338; c) Ito, K.; Fukuda, T.; Katsuki, T. Heterocycles 1997,46,401-411.
116 Reissig, H.; Angert, H.; Kunz, T.; Janowitz, A.; Handke, G; Bruce-Adjei. E. J. Org. Chem. 1993, 58, 6280-6285.
117 Emmanuvel, L.; Shaikh, T. M. A.; Sudalai, A. Org. Lett. 2005, 7, 5071-5074.
118 Alonso, M. E.; Garcia, M. C. J. Org. Chem. 1985, 50, 988-992.
119 Alonso, M. E.; Fernandez, R. Tetrahedron 1989, 45, 3313-3320.
120 Monta, A.; Armstrong, A. Unpublished Results.
121 a) Laszlo, P.; Polla, E. Tetrahedron Lett. 1984, 25, 3701-3704; b) Gajewski, J. J.;
Jurayj, J.; Kimbrough, D. R.; Gande, M. E.; Ganem, B.; Carpenter, B. K. J. Am. Chem.
Soc. 1987, 109, 1170-1186.
122 Gademann, K.; Chavez, D. E.; Jacobsen, E. N. Angew. Chem. Int. Ed. 2002, 41, 3059-
3061.
123 Wada, E.; Yasuoka, H.; Kanemasa, S. Chem. Lett. 1994, 1637-1640.
124 Frohn, M.; Shi, Y. Synthesis 2000, 1979-2000.
125 McGarrigle, E. M.; Gilheany, D. G. Chem. Rev. 2005, 105, 1563-1602.
126 Tu, Y.; Wang, Z.; Shi, Y. J. Am. Chem. Soc. 1996, 775, 9806-9807.
127 Cao, G. A.; Wang, Z. X.; Tu, Y.; Shi, Y. Tetrahedron Lett. 1998, 39, 4425-4428.
128 Wang, Z.; Tu, Y.; Frohn, M.; Shi, Y. J. Org. Chem. 1997, 62, 2328-2329.
129 Wang, Z.; Tu, Y; Frohn, M.; Zhang, J.; Shi, Y. J. Am. Chem. Soc. 1997, 779, 11224-
11235.
130 Zhang, W.; Loebach, J. L.; Wilson, S. R.; Jacobsen, E. N. J. Am. Chem. Soc. 1990, 112,
2801-2803.
268

131 Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N; Katsuki, T. Tetrahedron Lett. 1990, 31,
7345-7348.
132 Jacobsen, E. N.; Deng, L.; Furukawa, Y.; Martinez, L. E. Tetrahedron 1994, 50, 4323-
4334.
133 Brandes, B. D.; Jacobsen, E. N. J. Org. Chem. 1994, 59, 4378-4380.
134 Lee, N.; Muci, A. R.; Jacobsen, E. N. Tetrahedron Lett. 1991, 32, 5055-5058.
135 Armstrong, A.; Ahmed, G.; Dominguez-Fernandez, B.; Hayter, B. R.; Wailes, J. S. J.
Org. Chem. 2002, 67, 8610-8617.
136 a) Zhu, Y. M.; Tu, Y.; Yu, H. W.; Shi, Y. Tetrahedron Lett. 1998, 39, 7819-7822. b)
Tian, H. Q.; She, X. G.; Shu, L. H.; Yu, H. W.; Shi, Y. J. Am. Chem. Soc. 2000, 722,
11551-11552; c) Tian, H. Q.; She, X. G.; Xu, J. X.; Shi, Y. Org. Lett. 2001, 12, 1929-
1931.
137 Bouhlel, E.; Ben, H. B. Synth. Commun. 1992, 22, 2183-2186.
138 Hennion, G. F.; Davis, R. B.; Maloney, D. E. J. Am. Chem. Soc. 1949, 71, 2813-2814.
139 Kasturi, T. R.; Parvathi, S. J. Chem. Soc., Perkin Trans. I 1980, 448-456.
140 Hargreaves, J.; Park, J.; Ghisalberti, E. L.; Sivasithamparam, K.; Skelton, B. W.;
White, A. H. Aust. J. Chem. 2002, 55, 625-628.
141 Cahiez, G.; Metais, E. Tetrahedron: Asymm. 1997, 8, 1371-1376.
142 Jacobsen, E. N.; Zhang, W.; Guler, M. L. J. Am. Chem. Soc. 1991, 113, 6703-6704.
143 Linde, C.; Koliai, N.; Norrby, P. O.; Akermark, B. Chem. Eur. J. 2002, 8, 2568-2573.
144 Sykes, P. Mechanism in Organic Chemistry; 6th Ed.; Longman: London, 1986, 358-
395.145 a) Leal, W. S. J. Chem. Ecol. 1999, 25, 1055-1066; b) Nikonov, A. A.; Leal, W. S. J.
Chem. Ecol. 2002, 28, 1075-1090.
146 Jalce, G.; Franck, X.; Seon-Meniel, B.; Hocquemiller, R.; Figadere, B. Tetrahedron
Lett. 2006, 47, 5905-5908.
147 a) Matsunaga, S.; Yoshida, T.; Morimoto, H.; Kumagai, N.; Shibasaki, M. J. Am.
Chem. Soc. 2004, 726, 8777-8785; b) Andrus, M. B.; Hicken, E. J.; Stephens, J. C
269

Org. Lett. 2004, 6, 2289-2292; c) Kumagai, N.; Matsunaga, S.; Shibasaki, M. Org.
Lett. 2001,3,4251-4254.
148 Tian, H.; She, X.; Yu, H.; Shu, L.; Shi, Y. J. Org. Chem. 2002, 67, 2435-2446.
149 Wu, X.; She, X.; Shi, Y. J. Am. Chem. Soc. 2002, 124, 8792-8793.
150 Armstrong, A.; Hayter, B. R. Chem. Commun. 1998, 621-622.
151 Yang, D.; Yip, Y-C.; Chen, J.; Cheung, K-K. J. Am. Chem. Soc. 1998, 120, 7659-7660.
152 Reeves, J. R. PhD thesis, Imperial College London, 2002.
153 Bettati, M.; Armstrong, A. Unpublished Results.
154 a) Armstrong, A.; Moss, W. O.; Reeves, J. R. Tetrahedron: Asymm. 2001, 12, 2779-
2781; b) Armstrong, A.; Hayter, B. R.; Moss, W. O.; Reeves, J. R.; Wailes, J. S.
Tetrahedron: Asymm. 2000, 11, 2057-2061.
155 Bunn, B. J.; Cox, P. J.; Simpkins, N. S. Tetrahedron 1993, 49, 207-218.
156 Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335-1338.
157 Shanahan, S. E. PhD thesis, Imperial College London, 2006.
158 a) Jenner, G; Abdi-oskoui, H.; Rimmelin, J. Bull. Soc. Chim. Fr. 1979, 33-37; b)
Noecker, L. A.; Martino, J. A.; Foley, R J.; Rush, D. M.; Giuliano, R. M.; Villani Jr, F.
J. Tetrahedron: Asymm. 1988, 9, 203-212.
159 Morizur, J-P; Mercier, J.; Sarraf, M. Org. Mass Spectrum 1982, 327-330.
160 Hon, Y. S.; Lu, L.; Chang, R. C.; Lin, S. W.; Sun, P. P.; Lee, C. F. Tetrahedron 2000,
56, 9269-9279.
161 Bienvenue, A. J. Am. Chem. Soc. 1973, 95, 7345-7353.
162 Yamamoto, Y; Yatagai, H.; Saito, Y. J. Org. Chem. 1984, 49, 1096-1104.
163 Jack, K. C.; Elisa, R. Tetrahedron 2002, 58, 7027-7036.
164 Yates, P.; Jorgenson, M. J.; Singh, P. J. Am. Chem. Soc. 1969, 91, 4739-4748.
165 Bevinakatti, H. S.; Newadkar, R. V; Borude, D. P.; Mandal, A. K. J. Org. Chem. 1988,
53, 3843-3845.166 Ruegaer, H.; Stutz, S.; Goschke, R.; Spindler, F.; Maibaum, J. Tetrahedron Lett. 2000,
41, 10085-10089.
167 Moret, E.; Schlosser, M. Tetrahedron Lett. 1984, 25, 4491-4494.
270

168 a) Jefford, C. W.; Sledeski, A. W.; Boukouvalas, J. Helv. Chim. Acta 1989, 72, 1362-
1370; b) Rojo, J.; Garcia, M.; Carretero, J. C. Tetrahedron 1993, 49, 9787-9800.
169 a) Jacques, M.; Abdelaziz, N. A. Synthesis 1993, 785-787; b) Abdelkhalek, R.;
Fransoise, H.; Jacques, M. Tetrahedron Lett. 1999, 40, 2303-2036.
170 Mayr, H.; Bauml, E. Tetrahedron Lett. 1983, 24, 357-360.
171 Perez, I.; Sestelo, J. P.; Sarandeses, L. A. J. Am. Chem. Soc. 2001, 18, 4155-4160.
172 Trahanovsky, W. S.; Emeis, S. L. J. Am. Chem. Soc. 1975, 97, 3773-3777.
173 Balu, M. P.; Bhat, L.; Ila, H.; Junjappa, H. Ind. J. Chem. 1991, 30, 250-255.
174 a) Seno, M.; Tsuchiya, S.; Asahara, T. Chem. Lett. 1974, 405-406, b) Seno, M.;
Tsuchiya, S.; Kise, H.; Asahara, T. Bull. Chem. Soc. Jpn. 1975, 48, 2001-2005.
175 a) Kuchar, M.; Kakac, B.; Nemecek, O. Collect. Czech. Chem. Commun. 1972, 37,
3950-3955.
176 Biagi, G; Calderone, V.; Giorgi, L; Livi, O.; Martinotti, E.; Martelli, A.; Nardi, A. //
Farmaco, 2004, 59, 397-404.
177 Tirumalai, R. K.; Shankaranarayana P. J. Chem. Soc. Perkin Trans. 1 1980, 448-456.
178 a) Bhattacharyya, S.; Mukherjee, D. Synth. Commun. 1981, 11, 993-998. b) Watson, J.
M.; Irvine J. L.; Roberts, R. M. J. Am. Chem. Soc. 1973, 95, 3348-3354.
179 Armstrong, A.; Chung, H. Tetrahedron Lett. 2006, 47, 1617-1619.
180 Hou, R.; Wang, H.; Lin, Y.; Chen, L. Heterocycles, 2005, 65, 649-656.
181 Fohlisch, B.; Krimmer, D.; Gehrlach, E.; Kashammer, D. Chem. Ber. 1988, 727, 1585-
1593.182 Davoust, M.; Briere, J-R; Jaffres, P-A.; Metzner, P. J. Org. Chem. 2005, 10, 4166-
4169.183 Kurosaki, Y; Fukuda, T.; Iwao, M. Tetrahedron 2005, 61, 3289-3304.
184 Capriati, V; Florio, S.; Luisi, R.; Salomone, A. Org. Lett. 2002, 14, 2445-2448.
185 Shing, T. K. M.; Leung, G Y. C.; Luk, T. J. Org. Chem. 2005, 18, 7279-7289.
186 Denmark, S. E.; Matsuhashi, H. J. Org. Chem. 2002, W, 3479-3486.
271
Related Documents











