of April 9, 2019. This information is current as Facilitate Parasite Persistence as a Mechanism to Toxoplasma gondii by Stage-Specific Expression of Surface Antigens Seon-Kyeong Kim and John C. Boothroyd http://www.jimmunol.org/content/174/12/8038 doi: 10.4049/jimmunol.174.12.8038 2005; 174:8038-8048; ; J Immunol References http://www.jimmunol.org/content/174/12/8038.full#ref-list-1 , 20 of which you can access for free at: cites 58 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2005 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on April 9, 2019 http://www.jimmunol.org/ Downloaded from by guest on April 9, 2019 http://www.jimmunol.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

of April 9, 2019.This information is current as
Facilitate Parasite Persistence as a Mechanism toToxoplasma gondiiby
Stage-Specific Expression of Surface Antigens
Seon-Kyeong Kim and John C. Boothroyd
http://www.jimmunol.org/content/174/12/8038doi: 10.4049/jimmunol.174.12.8038
2005; 174:8038-8048; ;J Immunol
Referenceshttp://www.jimmunol.org/content/174/12/8038.full#ref-list-1
, 20 of which you can access for free at: cites 58 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

Stage-Specific Expression of Surface Antigens by Toxoplasmagondii as a Mechanism to Facilitate Parasite Persistence1
Seon-Kyeong Kim and John C. Boothroyd2
Toxoplasma persists in the face of a functional immune system. This success critically depends on the ability of parasites to activatea strong adaptive immune response during acute infection with tachyzoites that eliminates most of the parasites and to undergostage conversion to bradyzoites that encyst and persist predominantly in the brain. A dramatic change in antigenic compositionoccurs during stage conversion, such that tachyzoites and bradyzoites express closely related but antigenically distinct sets ofsurface Ags belonging to the surface Ag 1 (SAG1)-related sequence (SRS) family. To test the contribution of this antigenic switchto parasite persistence, we engineered parasites to constitutively express the normally bradyzoite-specific SRS9 (SRS9c) mutantsand tachyzoite-specific SAG1 (SAG1c) mutants. SRS9c but not wild-type parasites elicited a SRS9-specific immune responsemarked by IFN-� production, suggesting that stage-specificity of SRS Ags determines their immunogenicity in infection. Theinduction of a SRS9-specific immune response correlated with a continual decrease in the number of SRS9c cysts persisting in thebrain. In contrast, SAG1c mutants produced reduced brain cyst loads early in chronic infection, but these substantially increasedover time accompanying a hyperproduction of IFN-�, TNF-�, and IL-10, and severe encephalitis. We conclude that stage-specificexpression of SRS Ags is among the key mechanisms by which optimal parasite persistency is established and maintained. TheJournal of Immunology, 2005, 174: 8038–8048.
T oxoplasma gondii is an obligate intracellular, protozoanparasite highly prevalent in warm-blooded vertebrates (1).One route of infection is consumption of meat harboring
cysts that contain bradyzoites (BZs).3 Once in the gut, BZs rapidlyinvade and convert to tachyzoites (TZs) that proliferate and dis-seminate throughout the body (2). This acute infection with TZsactivates strong, long-lasting Ab and T cell responses that elimi-nate most of the parasites (3). Some parasites that survive undergostage conversion to BZs, a process that may be induced by theimmune response itself (4). BZs then encyst, establish a chronicinfection primarily in the brain and harmlessly persist for the lifeof an immunocompetent host.
A key feature of Toxoplasma persistence is the requirement of afunctional immune system to control the acute infection with TZs.In an immunodeficient host, uncontrolled TZ growth causes tissuedestruction, and persistent infection is not established (5). When achronically infected host later becomes immunodeficient, BZs re-activate to TZs causing severe neurological diseases (6). Anothercharacteristic of Toxoplasma persistence is that the parasite per-sists in the face of a long-lasting anti-Toxoplasma immune re-sponse. Toxoplasma succeeds in this daunting task by using a va-riety of strategies applied by microbial agents, which allow them
to persist until transmission to a new host (by carnivorism forToxoplasma) can be accomplished. Such strategies include: mod-ification of the intracellular environment within host cells to favorparasite survival (7); manipulation of the host immune response byproducing immunomodulatory molecules (8, 9); establishment ofchronic infection predominantly in the brain, where immune sur-veillance is controlled differently than in other peripheral tissues(10); and, the fact that BZs are metabolically quiescent and rela-tively nonproliferative during chronic infection (11).
Another immune-evasion mechanism commonly used in micro-bial persistence is the variation of antigenic composition that elim-inates epitopes that would otherwise be targeted by protective im-mune responses (12–14). Toxoplasma stage conversion involves adifferential expression of numerous genes in a stage-specific man-ner (4, 15), among which are the members of the surface Ag(SAG)1-related sequence (SRS) superfamily that encode GPI-an-chored surface proteins (�160 putative genes) (16). The prototypicSAG1 is the most abundant TZ SRS Ag (17). SRS Ags are struc-turally related (18), yet antigenically distinct, sharing 25–35%overall amino acid sequence identity and up to 50–90% identitywithin a given subfamily (16). The structure of SRS Ags suggeststheir possible role as cell adhesion molecules (18). In addition,extensive polymorphisms found among the three canonical lines ofToxoplasma imply that SRS Ags are under selective pressure fromthe immune response (19). A distinctive feature of the SRS anti-genic switch is that TZs and BZs express largely nonoverlappingsets of SRS Ags, but a single set of multiple SRS Ags is simulta-neously expressed by most if not all of the parasite population ina given developmental stage (16).
Because stage conversion and a dramatic change of the parasitesurface occur with the emergence of the adaptive immune re-sponse, it has been speculated that stage-specific expression ofSRS Ags has evolved as an immune-evasion mechanism that al-lows Toxoplasma to persist. Surface proteins are excellent targetsof neutralizing Abs and cell-mediated immune responses. In fact,SAG1 and SAG2A dominate the humoral response in the acuteinfection (20, 21), and SAG1 also activates T cells producing
Department of Microbiology and Immunology, Stanford University School of Med-icine, Stanford, CA 94305
Received for publication February 17, 2005. Accepted for publication April 13, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This research was supported by National Institutes of Health Grants AI41014 andAI21423.2 Address correspondence and reprint requests to Dr. John Boothroyd, Department ofMicrobiology and Immunology, Stanford University School of Medicine, 299 Cam-pus Drive, Stanford, CA 94305. E-mail address: [email protected] Abbreviations used in this paper: BZ, bradyzoite; TZ, tachyzoite; SRS, surface Ag1-related sequence; Pru, Prugniaud; HFF, human foreskin fibroblast; HPT, hypoxan-thine-xanthine-guanine phosphoribosyltransferase; dpi, days postinfection; WT, wildtype; SAG1, surface Ag 1.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

IFN-� (22, 23), the major cytokine mediating resistance againstToxoplasma (24). However, an immune response to TZ-specificAgs such as SAG1 and SAG2A is expected to be ineffectiveagainst BZs that do not express these molecules. Although parasiteAgs shared by both TZs and BZs can also activate an immuneresponse (22, 25), this is clearly insufficient to eliminate BZs. No-tably, immune responses to BZ-specific Ags, such as SAG2C/D (amember of the SRS family) and SAG4, LDH2, ENO1, and p-ATPase were undetectable in chronically infected humans (26). Inthis study (26), BAG1 and MAG1 were thought to be the onlyBZ-specific molecules immunogenic in infection. Recent data,however, demonstrated that MAG1 mRNA (15) and protein (27)are expressed in both the TZ and BZ stages, making it likely thata MAG1-specific response may have been elicited during acuteinfection with TZs. Because BAG1 shares a significant homologywith other small heat shock proteins across species (28), cross-reactivity of an immune response to other microorganisms remainsa distinct possibility. Thus, overall, BZ-specific Ags appear to bepoorly or not at all immunogenic in infection and this conditionmay be one of the mechanisms by which BZs escape immunesurveillance.
To explore the role of stage-specific expression of SRS Ags inToxoplasma persistence, we chose to focus on SAG1 and SRS9,two of the most abundant SRS Ags specific to TZs and BZs (15),respectively. A key aspect of our work is the use of mutant para-sites expressing SAG1 and SRS9 in a constitutive manner in boththe TZ and BZ stages, which allowed us to study the interactionbetween the host immune response and SRS Ags in relation totheir stage-specificity. We show that an immune response to SRS9,as well as to other BZ-specific SRS Ags, is lacking in naturalinfection and that stage-specific expression of SRS Ags has im-portant consequences regarding their immunogenicity in infectionand the ability of parasites to achieve optimal persistence in in-fected hosts.
Materials and MethodsToxoplasma culture
Prugniaud (Pru) strain parasites were maintained as TZs by passage inhuman foreskin fibroblast (HFF) monolayers cultured in DMEM supple-mented with 10% FCS, penicillin (100 U/ml), streptomycin (100 �g/ml),and L-glutamine (2 mM) (Invitrogen Life Technologies) in a humidified,5% CO2 incubator. Switching to BZs was induced by culturing infectedHFFs in HEPES-buffered RPMI 1640 (pH 8.1) supplemented with 2% FCSin an air incubator.
Recombinant SRS9 and polyclonal anti-SRS9 antisera
rSRS9 was produced in insect cells as a secreted protein using thepAcGP67.A baculovirus expression vector, as described for recombinant(rSAG1) production (18). SRS9 coding region lacking the N-terminal sig-nal sequence and C-terminal GPI anchor signal was PCR cloned from Prustrain genomic DNA using the following primers: 5�-ATCGGATCCTCTGCATGAAGGACTTCAGAG-3� and 5�-CTATCTAGACTAGTGATGGTGATGGTGATGCGCGTACGAAGCAGAACTG-3� (contains a 6-Histag). Highly pure (�98%) rSRS9 and rSAG1 were obtained by using nickelagarose beads. Mouse polyclonal anti-SRS9 antisera were raised inBALB/c mice (The Jackson Laboratory) by i.p. immunization with 20 �gof rSRS9 per mouse in 200 �l of Ribi adjuvant composed of purifiedmonophosphoryl lipid A and synthetic trehalose dicorynomycolate(Corixa). Mice were given four boosts spaced 3 wk apart with the same Agpreparations. Tail vein blood was allowed to coagulate overnight at 4°Cand spun for 5 min at 14,000 rpm in a microcentrifuge to collect serum.Rabbit polyclonal anti-SRS9 antisera were raised by intradermal immuni-zation (100 �g of rSRS9 in CFA) followed by four s.c. boosts spaced 3 wkapart (50 �g of rSRS9 in IFA; Covance Research Products).
Protein gel electrophoresis and Western blotting
TZs lysed out of HFFs were washed in PBS by centrifugation at 250 � gfor 10 min, resuspended in 0.4% SDS sample buffer containing 100 mMDTT, boiled for 5 min and subjected to a 12% SDS-PAGE. Proteins were
transferred to a nitrocellulose membrane, which was blocked in PBS/5%nonfat dry milk and incubated with rabbit anti-SAG1 or -SRS9 antiserafollowed by peroxidase-conjugated goat anti-rabbit IgG (Kirkegaard &Perry Laboratories). Detection was by ECL chemiluminescence reagents(Amersham Biosciences).
Immunofluorescence microscopy
Infected HFFs grown on cover slips were fixed in 3.7% formaldehyde andpermeabilized in 0.2% Triton X-100. Brain homogenates containing cystswere fixed in methanol. Samples were incubated in PBS/3% BSA withindicated anti-parasite Ag Abs followed by appropriate goat IgG coupledwith Alexa Fluor 488, Alexa Fluor 594, or Cascade Blue (MolecularProbes). Cover slips were mounted on a glass slide with Vectashield (Vec-tor Laboratories), and photographs were taken using the Image Pro Plussoftware and a 35 mm digital camera (model C4742-95; Hamamatsu) con-nected to an upright (model BX60; magnification, �1000; Olympus) or aninverted fluorescence microscope (model TE300; magnification, �200;Nikon).
Flow cytometry
TZs lysed from HFFs were incubated in PBS/2% FCS (106 TZ in 100 �lvolume) with indicated anti-parasite Ag Abs followed by appropriate goatIgG coupled with FITC or PE (BD Biosciences). Data acquisition andanalysis were by FACScan and CellQuest software (BD Biosciences).
Generation of SRS9-constitutive (SRS9c) and SAG1-constitutive(SAG1c) mutants
SRS9c and SAG1c were derived from Pru�hpt (hypoxanthine-xanthine-guanine phosphoribosyltransferase-deficient Pru strain), a gift from D. Sol-dati (University of Geneva, Geneva, Switzerland) (29). Upstream (pro-moter) sequences from the TZ-specific GRA1 (pGRA1) (30) and from theBZ-specific SRS9 (pSRS9) were used to express transgenic SRS9 andSAG1 in the TZ and BZ stage, respectively. The SRS9 promoter region(�1.5 kb) was PCR cloned from the Pru strain genomic DNA using thefollowing primers: 5�-GGGGAAGCTTTGTCACCGGTTCGGTGCACT-3� and 5�-GCCCATGCATTGTGTCGACCCGTGTGCACG-3�. Full-length SRS9 and SAG1 coding regions were cloned with the followingprimers: 5�-GGGCATGCATGAAAGGACAGGCAATATGCAG-3� and5�-GCCCTTAATTAATTACAATGAAGCAACAACGAACC-3� for SRS9;5�-GGGCATGCATTCGGTTTCGCTGCACCACTT-3� and 5�-GCCCTTAATTAATCACGCGACACAAGCTGCGA-3� for SAG1. PCR productswere digested with NsiI and PacI, and cloned between the indicated pro-moter and 3� GRA2 downstream sequence in a plasmid vector containinga copy of HPT gene driven by the dihydrofolate reductase promoter(pDHFR) (31) (see Fig. 3A). The plasmids were linearized with NotI andelectroporated into Pru�hpt as described (32). Clones were derived fromover two independent populations after selecting for HPT activity usingmycophenolic acid and xanthine (50 �g/ml each).
Mouse infection
Eight-week-old CBA/J female mice (The Jackson Laboratory) were in-fected i.p. with TZs. Infected HFFs were syringed-lysed using a 27-gaugeneedle to release TZs. TZs were washed in PBS by centrifugation at 250 �g for 10 min and counted with a hemacytometer. Mice were injected i.p.with 400 or 4000 TZs in 200 �l of PBS. We considered �3 wks postin-fection as the chronic phase of infection. All animal studies have beenreviewed and approved by Stanford University Administrative Panel forLaboratory Animal Care.
Recombinant SRS9 immunization and challenge infection
CBA/J females (8 wk of age) were immunized s.c. with rSRS9 (20 �g/mouse in a 100 �l volume) prepared in Ribi adjuvant or adjuvant alone intoone site at the back of the neck. Mice were boosted s.c. twice, 3 wk apart,with the same Ag preparations and challenged i.p. with 4000 wild-type(WT) TZs 7 days after the second boost.
Plaque assays to quantitate tissue parasite loads
Tissues were homogenized over 100-�m cell strainers. Aliquots of tissuehomogenates were syringe-lysed using 27-gauge needles to prevent intactmouse cells from serving as host cells and thus interfering with plaqueassays. Two-fold serial dilutions starting from 1% of the whole tissue wereadded to HFF monolayers in 12-well plates and cultured for 24 h inDMEM/3% FCS. Medium was replaced with DMEM/10% FCS the nextday. HFFs were fixed in methanol at 4 days postinfection (dpi) and stainedwith rabbit anti-SAG1 antisera followed by Alexa Fluor 488. Plaques were
8039The Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

counted using an inverted fluorescence microscope (magnification, �200).Parasite loads are expressed as PFU per entire organ.
Enumeration of parasite cysts and T cells in the brain
Brains were homogenized over 100-�m cell strainers. Cells were washedin 25 ml of PBS by centrifugation at 150 � g for 10 min and resuspendedin 20 ml of PBS/2% FCS. The 5% of the entire brain sample was stainedwith fluorescein-conjugated Dolichos biflorus agglutinin (Vector Labora-tories) to stain the cyst wall (33). After washing, samples were resuspendedin 500 �l of PBS/2% FCS, and 50 �l of aliquots were seeded in a flat-bottom 96-well plate. Cysts were counted in all 10 wells using an invertedfluorescence microscope (magnification, �200). To count T cells, 1% ofthe total brain homogenates preincubated with Fc block were stained withFITC-coupled anti-CD4 or anti-CD8 Abs (BD Biosciences). Serial dilu-tions of each sample were seeded in a flat-bottom 96-well plate and FITC�
cells were counted using an inverted fluorescence microscope (magnifica-tion, �200).
Histology and assessment of brain cyst size
Brains were fixed for �24 h in 3.7% formaldehyde solution. Sections(10-�m thick) were cut 100 �m apart and stained with H&E (Histotec).Photographs of H&E-stained sections were taken with a camera attached toa microscope (magnification, �200) and used to measure the diameter ofeach cyst as an arbitrary scale.
ELISA with serum and splenocyte culture supernatant
To quantitate Ag-specific IgG, serial dilutions of serum prepared from tailvein blood were added to ELISA plates coated with rSRS9 or rSAG1 (50ng/well). After incubation with peroxidase-coupled goat anti-rabbit IgGand its substrate (Kirkegaard & Perry Laboratories), plates were read colo-rimetrically at 450 nm. To detect cytokines secreted by cultured spleno-cytes, spleens were homogenized over 100-�m cell strainers. After lysingRBC (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA), splenocyteswere stimulated with indicated Ags in a 12-well plate (8 � 106 cells/wellin 2 ml of medium) using DMEM supplemented with 10% FCS, penicillin(100 U/ml), and streptomycin (100 �g/ml), L-glutamine (2 mM), and 2-ME(55 �M). At desired time points, 250 �l of supernatant were collected andanalyzed for IFN-�, TNF-�, and IL-10 levels using ELISA kits accordingto the manufacturer’s instructions (Pierce). Following incubation with per-oxidase-coupled detection Ab and its substrate, plates were read colori-metrically at 450 nm. Serum cytokine levels were determined using thesame ELISA kits.
Statistics
Student’s t test was performed to compare parasite loads in different ex-perimental groups.
ResultsSAG1 induces an IFN-�- and IL-10-producing immuneresponse, whereas SRS9 is not immunogenic in natural infection
SRS9 was first identified as an abundant, BZ-specific mRNA (15).To confirm that SRS9 is expressed as a BZ-specific surface pro-tein, we performed immunofluorescence assays using polyclonalantisera raised against rSRS9 produced by the baculovirus system.In vitro BZs were obtained by culturing in a high pH medium (34)HFFs infected with the Pru�hpt parasite engineered to expressGFP from the TZ-specific GRA1 promoter (pGRA1), a gift from G.Arrizabalaga (University of Idaho, Moscow, ID). We show that SRS9is expressed by GFPdim BZs but not by GFPbright TZs (Fig. 1A).SAG1, as expected, is expressed by TZs but not by BZs (Fig. 1B).BZs within brain cysts isolated from a Pru�hpt-infected mouse ex-press SRS9 but not SAG1 at the parasite surface (Fig. 1C).
To compare the nature of immune responses to SAG1 and SRS9in infection, CBA/J mice were infected i.p. with Pru�hpt TZs. Asexpected, a robust SAG1-specific serum IgG was detectable inchronic infection sera (6 wk postinfection) (Fig. 2A), and in vitrostimulation with rSAG1 of splenocytes from the same mice led tothe production of IFN-� (Fig. 2B). In addition to IFN-�, we foundthat the SAG1-specific T cells produced IL-10 (Fig. 2B), a potentantagonist of IFN-�.
In contrast, SRS9-specific serum IgG was conspicuously absentin the same infected mice (Fig. 2A), as well as in 12 differentchronically infected people who have most likely acquired infec-tion by ingesting BZ cysts (data not shown). The lack of detectableIgG in infection was not unique to SRS9 but was also true for otherBZ-specific SRS Ags, such as SAG2C, SAG2X, and SAG2Y, de-spite the fact that all these molecules were highly immunogenic inanimals when given along with an appropriate adjuvant (data notshown). A T cell response to SRS9 was also undetectable: spleno-cytes from the infected mice did not produce IFN-� or IL-10 whenstimulated with rSRS9 in vitro (Fig. 2B).
Generation of SAG1C and SRS9C mutants
The stage-specific expression of SRS Ags may be one of the rea-sons why BZs are not cleared by the immune system but persist.Because rSRS9 given with an adjuvant is highly immunogenic inanimals, we predicted that it should be immunogenic during aninfection if it could be expressed in the TZ stage and that theresulting immune response could eliminate BZs that subsequentlyappeared. Likewise, SAG1, if still expressed after TZ-to-BZ con-version, might continue to attract the robust SAG1-specific re-sponse leading to the elimination of BZs. To test these hypotheses,we engineered parasites to express SAG1 or SRS9 in a develop-mentally inappropriate, “constitutive” manner. SRS9c and SAG1c
mutants were derived from Pru�hpt by using the TZ-specificpGRA1 and BZ-specific pSRS9 promoters to drive SRS9 andSAG1 expression, respectively (Fig. 3A). To control for the pres-ence of the HPT gene in the expression vectors, WT parasites were
FIGURE 1. Stage-specific expression of SAG1 and SRS9. HFFs in-fected with Pru�hpt/pGRA1-GFP were grown on cover slips in a pH 8.1medium for 4 days to induce stage conversion. Parasite surface was stainedwith mouse anti-SRS9 antisera (A) or SAG1-specific mAb DG52 (B) (58),followed by Cascade Blue-coupled goat anti-mouse IgG. C, Brain cystsisolated from a Pru�hpt-infected CBA/J mouse (3 wk postinfection) werecostained with fluorescein-coupled Dolichos that binds to the cyst wall andrabbit anti-SRS9 or anti-SAG1 followed by Alexa Fluor 594-coupled goatanti-rabbit IgG. Ten-micron scale bars are shown.
8040 ROLE OF SRS Ags IN Toxoplasma PERSISTENCE
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

engineered by transforming Pru�hpt with the same vectors fromwhich the SAG1 or SRS9 coding region was removed (Fig. 3A).Clones were obtained from at least two independent populationsper construct after selecting for HPT activity.
Because WT TZs lack SRS9 expression, SRS9c mutants werereadily identified by Western blotting with TZ lysates (Fig. 3B), aswell as by flow cytometry with live TZs (Fig. 3C). TransgenicSRS9 reaches the parasite surface (Fig. 3C) and can be cleavedwith phosphatidylinositol phospholipase C indicating that it isGPI-anchored as strongly predicted from its sequence (data notshown). The relative abundance of SAG1 vs SRS9 in SRS9c mu-tants was estimated to be �5-fold (�178,000 SAG1 and �36,000SRS9 molecules per parasite) by comparing band intensities in aWestern blot performed with rabbit anti-SAG1, a gift from M.Grigg (University of British Columbia, Vancouver, BC, Canada)or anti-SRS9 antisera and serial dilutions of denatured recombi-nant proteins and TZ lysates (data not shown). Because of theTZ-specific nature of pGRA1 (Fig. 1), it is highly unlikely thatSRS9 is over-expressed in the BZ stage of SRS9c mutants com-pared with WT parasites.
SAG1c mutants were identified after infecting mice with indi-vidual clones and examining SAG1 expression in brain cysts byimmunofluorescence microscopy. Different mutant clones showedsimilar SAG1 expression levels in BZs (Fig. 3D), which appearedlower than in TZs (data not shown). Due to the difficulty in ob-taining a sufficient number of pure cysts from infected mice, theexact levels of SAG1 expression by SAG1c BZs vs TZs could notbe determined. In the TZ stage, SAG1 expression levels betweenWT and SAG1c mutants were indistinguishable (data not shown),as expected from the endogenous pSRS9 being active exclusivelyin the BZ stage (Fig. 1). Also, even in the context of the plasmidconstruct we used, pSRS9 is BZ-specific because the same con-struct containing the GFP gene instead of the SAG1 gene results inGFP expression only in the BZ stage (data not shown).
To avoid possible positional effects associated with random inte-gration of the expression vectors into the genome that could obscurethe interpretation of in vivo phenotypes, mutant clones from at least
FIGURE 2. Lack of a SRS9-specific immune response in natural infec-tion. A, Serum IgG titers specific for SAG1 or SRS9 determined by ELISAin Pru�hpt-infected CBA/J mice (6 wk postinfection; 400 TZs i.p.) andshown as absorbance readings at 450 nm. Each line represents a singlemouse. B, IFN-� and IL-10 production by splenocytes from the same mice.Cells were stimulated with rSAG1 or rSRS9 (20 �g/ml) for 48 h, andcytokines secreted into culture supernatants were measured by ELISA(lower limit of detection �30 pg/ml). The mean of duplicate determina-tions is shown.
FIGURE 3. Generation of SRS9c and SAG1c mutants. A, Plasmid con-structs used to create SRS9c, SAG1c, and WT parasites from Pru�hpt byelectroporation. B, Western blot with WT and SRS9c TZ lysates probed withmouse anti-SRS9 antisera and later reprobed with rabbit anti-SAG1 antisera. Atotal of 5 � 106 TZs per lane. SRS9c mutants S7 and S8 were cloned from apopulation distinct from the one K2 and K10 were cloned from. C, Flowcytometry with live WT and SRS9c TZs using SAG1-specific mAb DG52 andrabbit anti-SRS9 antisera. PE-coupled goat anti-mouse IgG and FITC-coupledgoat anti-rabbit IgG were used for detection. Dead host cell debris were gatedout based on forward light scatter and negative SAG1 staining. Contour plotsof SAG1 vs SRS9 expression were drawn to show that transgenic SRS9 ex-pression did not alter SAG1 expression levels at the SRS9c parasite surface. D,Brain cysts from WT- or SAG1c-infected mice (3 wk postinfection) fixed inmethanol and stained with rabbit anti-SAG1 antisera followed by Alexa Fluor488-coupled goat anti-rabbit IgG. SAG1c-C5 and -D7 were cloned from twoindependent populations.
8041The Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

two independent populations were used in all studies. WT parasiteswere maintained as either nonclonal or clonal populations, both ofwhich showed similar growth and dissemination rates in acute infec-tion (data not shown), as well as similar brain cyst loads in chronicinfection (Fig. 4, B and D). WT and SRS9c mutants showed similargrowth rates during coinfection of HFFs in vitro (data not shown).
Brain cyst loads continually decrease during chronic infectionwith SRS9c mutants
In CBA/J mice infected i.p. with 4000 TZs of WT and SRS9c
mutants, mortality usually began to occur 12–14 dpi, following thepeak of acute infection (Fig. 4A) and around the time most para-sites are found in the brain (35). With all infecting strains, mor-tality often occurred throughout the chronic phase (�3 wk postin-fection) with SRS9c-infected mice showing similar or slightlybetter (but not statistically significant) survival than WT-infectedmice (Fig. 4A). The number of cysts persisting in the brains ofWT-infected mice remained relatively constant during the chronicinfection (Fig. 4B). Brain cyst loads in SRS9c-infected mice weresimilar to or sometimes lower than those in WT infection in theearly phase of chronic infection (3 wk postinfection), but theysignificantly declined over time and were 17–50% of those in WT-infected brains by the late phase of chronic infection (6 wk postin-fection) (Fig. 4B).
When the inoculum size was reduced by 10-fold to 400 TZ permouse, the onset of death was generally delayed by more than aweek and the occurrence of mortality was still observed through-out the chronic phase (Fig. 4C). SRS9c-infected mice showed asimilar or marginally better survival during chronic infection thanWT-infected mice, which was not statistically significant but con-
sistently observed in all five independent experiments performed(data not shown). SRS9c-infected mice showed a better overallhealth status than WT-infected mice as they weighed more (27.6 �3.4 g; n � 7) than WT-infected mice (20.1 � 2.5 g; n � 6) at 9 wkpostinfection (age-matched, naive mice weighed 30.5 � 0.8 g; n �3) (data not shown). As with the higher inoculum and after initialparity, the brain cyst loads continually decreased in SRS9c-in-fected mice and were 20–30% of those in WT infection by 9 wkpostinfection (Fig. 4D). Notably, the 10-fold difference in inocu-lum size, which causes a clear difference in acute phase parasiteloads in the whole body (data not shown), yielded similar braincyst loads in chronic infection (Fig. 4, B and D). This implies thatthe number of persisting parasites is not simply determined by theinoculum size but may be controlled by complex factors including,e.g., the host response.
Because brain cyst loads in the early phase of chronic infectionwere similar between WT and SRS9c mutants, the subsequent re-duction in SRS9c cyst loads is unlikely due to a growth defect.Indeed, up to 15 dpi, both WT and SRS9c mutants showed similarparasite loads in the spleen (the major site of parasite replicationafter i.p. infection) (Fig. 5A) and in the brain (Fig. 5B), indicatingthat dissemination from the site of infection and subsequentgrowth in target tissues during the acute phase is comparable.Brain cyst counts at 15 dpi were also similar between WT (323 �140; n � 3) and SRS9c-K2 (300 � 46; n � 3) (data not shown).Moreover, there was no measurable difference in brain cyst sizebetween WT and SRS9c mutants measured in the H&E-stainedbrain sections (data not shown), suggesting that a defect in BZgrowth is not responsible for the decrease in SRS9c cyst loads.
FIGURE 4. Continual decrease in brain cyst loads in SRS9c infection. A, Survival curves of CBA/J mice infected i.p. with 4000 TZs of indicated parasitestrains (n � 12 per group). WT used is not a clone but a stable population. B, Brain cyst loads in representative mice from A determined at 3 and 6 wkpostinfection (wpi). C, Survival curves of mice infected i.p. with 400 TZs of indicated parasite strains (n � 13 per group). WT c3 and c28 are clonalpopulations. D, Brain cyst loads in representative mice from C determined at 4 and 9 wk postinfection (wpi). Each diamond represents a single mouse. Barsrepresent average cyst counts. �, Significant difference (p 0.05) in cyst loads between WT and SRS9c at the given week postinfection; #, significantdifference (p 0.05) in SRS9c cyst loads between the two time points.
8042 ROLE OF SRS Ags IN Toxoplasma PERSISTENCE
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

Another possible reason for observing decreased brain cystloads in SRS9c infection would be if the transgenic SRS9 expres-sion in TZs altered tissue tropism of the parasite such that theparasite preferentially located to and encysted in other tissues thanin the brain. This possibility is unlikely because parasite loadswere similar between WT- and SRS9c-infected brains up to 4 wkpostinfection (Figs. 5B, and 4, B and D). Also, regardless of theinfecting strain, cysts detectable after dolichos-fluorescein stainingwere present only in the brains and were absent in the lungs andlivers that are targets of the acute infection (35) (zero cysts in 5%of the total tissue homogenates, both at 4 and 9 wk postinfection;data not shown).
SRS9c mutants induce a SRS9-specific immune response ininfection
A SRS9-specific immune response that was absent in WT infectionwas readily detectable in SRS9c infection. Sera from SRS9c-in-fected mice (6 wk postinfection) had a high titer SRS9-specificIgG unlike WT-infected mice (Fig. 6A). As expected, SAG1-spe-cific IgG titers were high in all infected mice (data not shown).SRS9c infection also activated a SRS9-specific T cell responsemarked by IFN-� but not IL-10 production by cultured splenocytes(Fig. 6B). Although SAG1 clearly dominates the humoral responsein Toxoplasma infection, it is not known whether it also dominatesthe T cell response. In SRS9c infection, the amount of rSAG1-stimulated IFN-� production was substantially higher than that in-duced by rSRS9. Because SAG1 is the most abundant SRS Ag inTZs and expressed in �5-fold excess over SRS9 in SRS9c TZs, thesize of the T cell response to SAG1 is expected to be greater thanthat to SRS9 (and most other TZ-specific Ags for that matter).Neither T cell population produced detectable levels of TNF-�regardless of the infecting strain (data not shown). Generally, cy-tokine secretion in the absence of an antigenic stimulus (Fig. 6B)or in the presence of PMA and ionomycin (Fig. 6C) was higherwith splenocytes from infected mice than with cells from naivemice, indicating an infection-driven accumulation of immune cellsstill detectable in the spleen at 6 wk postinfection.
Mice immunized with rSRS9 show improved survival andreduced brain cyst loads following a challenge infection withWT parasite
To test whether the SRS9-specific immune response contributed tothe decrease in brain cyst loads in SRS9c infection, CBA/J mice wereimmunized s.c. with rSRS9 prepared in Ribi adjuvant or with adjuvantalone and then challenged i.p. with 4000 WT TZs. Following infec-
tion, rSRS9-immune mice showed better survival (Fig. 7A) and lowerbrain cyst loads (Fig. 7B) than the control group. This result suggeststhat the induction of a SRS9-specific immune response in SRS9c in-fection may have been a key mechanism that led to the clearance ofBZ cysts during chronic infection.
Brain cyst loads continually increase during chronic infectionwith SAG1c mutants
Because SAG1 dominates the immune response during the acuteinfection with TZs, one would anticipate that BZs still expressing
A
0
20000
40000
60000
80000
100000
120000
140000
SRS9c-K2SRS9c-S7WT
15d10d6d3d
PF
Us
per
sple
en
Days post-infection
PF
Us
per
brai
n
0
5000
10000
15000
20000
25000
30000
15d10d6d
SRS9c-K2WT
B
Days post-infection
FIGURE 5. Comparable growth of WT and SRS9c mutants in vivo. Par-asite loads determined by plaque assays at indicated day postinfection inthe spleen (A) and brain (B) of CBA/J mice infected with 400 TZs of WTor SRS9c mutants. Shown are mean PFU � SD (n � 3 per group per timepoint).
FIGURE 6. Induction of a SRS9-specific immune response in SRS9c
infection. A, Serum IgG titers specific for SRS9 determined by ELISA inCBA/J mice infected (6 wk postinfection) with 400 TZs of WT (n � 4) orSRS9c mutants (n � 2 per strain) and shown as absorbance readings at 450nm. Each line represents a single mouse. B, Cytokines produced by spleno-cytes from naive and infected (6 wk postinfection) mice cultured with noAg, rSAG1, or rSRS9 (20 �g/ml) for 6, 24, 48, or 72 h before supernatantswere analyzed in ELISA. For WT, mean � SD of two duplicate determi-nations from two mice is shown. For others, mean of duplicate determi-nations is shown. C, Same mean as in B except PMA (25 ng/ml) andionomycin (1 �g/ml) were used for stimulation. Lower limit of detectionwas �30 pg/ml for all cytokines tested.
8043The Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
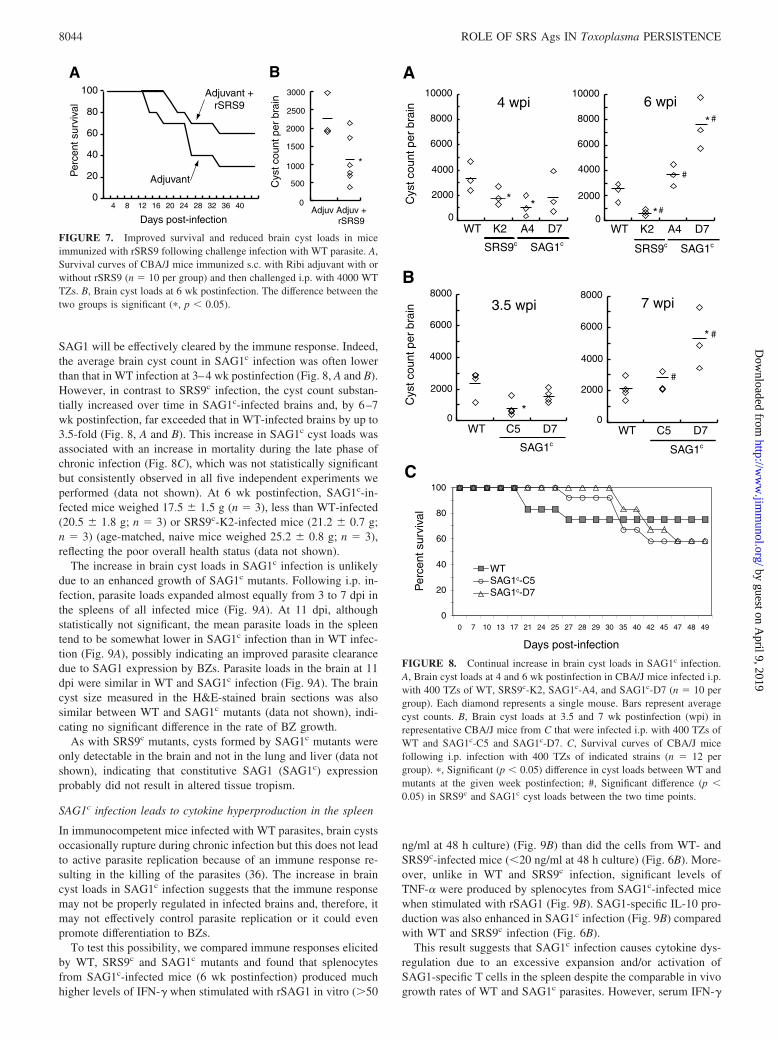
SAG1 will be effectively cleared by the immune response. Indeed,the average brain cyst count in SAG1c infection was often lowerthan that in WT infection at 3–4 wk postinfection (Fig. 8, A and B).However, in contrast to SRS9c infection, the cyst count substan-tially increased over time in SAG1c-infected brains and, by 6–7wk postinfection, far exceeded that in WT-infected brains by up to3.5-fold (Fig. 8, A and B). This increase in SAG1c cyst loads wasassociated with an increase in mortality during the late phase ofchronic infection (Fig. 8C), which was not statistically significantbut consistently observed in all five independent experiments weperformed (data not shown). At 6 wk postinfection, SAG1c-in-fected mice weighed 17.5 � 1.5 g (n � 3), less than WT-infected(20.5 � 1.8 g; n � 3) or SRS9c-K2-infected mice (21.2 � 0.7 g;n � 3) (age-matched, naive mice weighed 25.2 � 0.8 g; n � 3),reflecting the poor overall health status (data not shown).
The increase in brain cyst loads in SAG1c infection is unlikelydue to an enhanced growth of SAG1c mutants. Following i.p. in-fection, parasite loads expanded almost equally from 3 to 7 dpi inthe spleens of all infected mice (Fig. 9A). At 11 dpi, althoughstatistically not significant, the mean parasite loads in the spleentend to be somewhat lower in SAG1c infection than in WT infec-tion (Fig. 9A), possibly indicating an improved parasite clearancedue to SAG1 expression by BZs. Parasite loads in the brain at 11dpi were similar in WT and SAG1c infection (Fig. 9A). The braincyst size measured in the H&E-stained brain sections was alsosimilar between WT and SAG1c mutants (data not shown), indi-cating no significant difference in the rate of BZ growth.
As with SRS9c mutants, cysts formed by SAG1c mutants wereonly detectable in the brain and not in the lung and liver (data notshown), indicating that constitutive SAG1 (SAG1c) expressionprobably did not result in altered tissue tropism.
SAG1c infection leads to cytokine hyperproduction in the spleen
In immunocompetent mice infected with WT parasites, brain cystsoccasionally rupture during chronic infection but this does not leadto active parasite replication because of an immune response re-sulting in the killing of the parasites (36). The increase in braincyst loads in SAG1c infection suggests that the immune responsemay not be properly regulated in infected brains and, therefore, itmay not effectively control parasite replication or it could evenpromote differentiation to BZs.
To test this possibility, we compared immune responses elicitedby WT, SRS9c and SAG1c mutants and found that splenocytesfrom SAG1c-infected mice (6 wk postinfection) produced muchhigher levels of IFN-� when stimulated with rSAG1 in vitro (�50
ng/ml at 48 h culture) (Fig. 9B) than did the cells from WT- andSRS9c-infected mice (20 ng/ml at 48 h culture) (Fig. 6B). More-over, unlike in WT and SRS9c infection, significant levels ofTNF-� were produced by splenocytes from SAG1c-infected micewhen stimulated with rSAG1 (Fig. 9B). SAG1-specific IL-10 pro-duction was also enhanced in SAG1c infection (Fig. 9B) comparedwith WT and SRS9c infection (Fig. 6B).
This result suggests that SAG1c infection causes cytokine dys-regulation due to an excessive expansion and/or activation ofSAG1-specific T cells in the spleen despite the comparable in vivogrowth rates of WT and SAG1c parasites. However, serum IFN-�
FIGURE 7. Improved survival and reduced brain cyst loads in miceimmunized with rSRS9 following challenge infection with WT parasite. A,Survival curves of CBA/J mice immunized s.c. with Ribi adjuvant with orwithout rSRS9 (n � 10 per group) and then challenged i.p. with 4000 WTTZs. B, Brain cyst loads at 6 wk postinfection. The difference between thetwo groups is significant (�, p 0.05).
FIGURE 8. Continual increase in brain cyst loads in SAG1c infection.A, Brain cyst loads at 4 and 6 wk postinfection in CBA/J mice infected i.p.with 400 TZs of WT, SRS9c-K2, SAG1c-A4, and SAG1c-D7 (n � 10 pergroup). Each diamond represents a single mouse. Bars represent averagecyst counts. B, Brain cyst loads at 3.5 and 7 wk postinfection (wpi) inrepresentative CBA/J mice from C that were infected i.p. with 400 TZs ofWT and SAG1c-C5 and SAG1c-D7. C, Survival curves of CBA/J micefollowing i.p. infection with 400 TZs of indicated strains (n � 12 pergroup). �, Significant (p 0.05) difference in cyst loads between WT andmutants at the given week postinfection; #, Significant difference (p 0.05) in SRS9c and SAG1c cyst loads between the two time points.
8044 ROLE OF SRS Ags IN Toxoplasma PERSISTENCE
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

levels were similar among mice infected with WT, SRS9c, orSAG1c mutants: �2000 and �700 pg/ml at 4 and 6 wk postin-fection, respectively, compared with �300 pg/ml in naive mice(data not shown). Serum IL-10 and TNF-� were undetectable in allmice (data not shown). This result is perhaps not surprising giventhat the major site of parasite replication during chronic infectionis the brain. Cytokines locally produced in the brain may be morerelevant to the contrasting phenotypes of SRS9c and SAG1c
mutants.
Distinct immunological events occur in the brains infected withWT, SRS9c, and SAG1c mutants
In histologic examination of H&E-stained brain sections, menin-gitis and encephalitis were apparent in all infected mice (6 wkpostinfection) regardless of the infecting strain (Fig. 10, A–F).Mononuclear infiltrates were mostly found in the cerebral cortexwhere the great majority of cysts were localized. Compared withWT (Fig. 10B) and SRS9c infection (Fig. 10, C and D), SAG1c-
infected brains showed much severe inflammation with prominentareas of necrosis (Fig. 10, E and F).
Both CD4� and CD8� T cells are known to significantly con-tribute to Toxoplasma encephalitis (37), and were recruited to andaccumulated in the brains in WT, SRS9c, and SAG1c infection andgradually decreased in numbers during the chronic infection (Fig.10G). Given that T cells, especially IFN-�-secreting CD8� T cells,play a critical role in resistance to Toxoplasma (38, 39), it is no-table that more CD8� T cells were recruited to the brain in SAG1c
infection than in WT or SRS9c infection (Fig. 10G).In chronically infected brains, the majority of cysts were not
associated with mononuclear infiltrates (Fig. 10Ha). However,many sites of focal inflammation were found throughout the cere-bral cortex, some of which were seen to be intimately surroundingcysts (Fig. 10Hb) whereas others were not (Fig. 10Hc). The latter(Fig. 10Hc) might have been seen to associate with a cyst if sec-tions had been cut at different levels or may represent the situationafter a cyst had been cleared by the infiltrates. Counting these sitesof focal inflammation revealed that a significantly higher propor-tion of SAG1c cysts were closely associated with infiltrates thanWT cysts (Fig. 10H). This is not simply due to the increased num-bers of cysts and infiltrates present in SAG1c infection becauseSRS9c infection that caused moderate inflammation and producedfewer cysts than WT also had a higher proportion of cysts asso-ciated with infiltrates (Fig. 10H).
DiscussionToxoplasma surface proteins belonging to the SRS superfamily arestage-specifically expressed during TZ-to-BZ conversion concom-itant with the emergence of an adaptive immune response in acuteinfection. The stage conversion is critical in the establishment andmaintenance of a chronic infection, but it has not been previouslytested whether SRS antigenic switch indeed serves as an immune-evasion mechanism that allows parasites to persist. Using geneti-cally manipulated parasites expressing the normally stage-specificSAG1 and SRS9 Ags in a constitutive manner, we have demon-strated that altering parasite stage-specificity substantially affectsthe immune response they elicit in infection and interferes with theability of parasites to achieve optimal persistency.
BZ cysts persist despite the long-lasting immune response toTZ-specific SRS Ags, such as immunodominant (at least in termsof humoral response) SAG1 and SAG2A, as well as the responseto Ags shared by both TZs and BZs, suggesting that BZ-specificimmune responses are required to clear BZs. Somehow, Toxo-plasma avoids eliciting such a response because BZ-specific SRSAgs, such as SRS9, SAG2C, SAG2X, and SAG2Y, are not im-munogenic in infection. This result is true not only in mice infectedi.p. with TZs but also in humans who have most likely acquiredinfection through oral ingestion of BZ cysts. Therefore, the lack ofimmunogenicity of BZ SRS Ags appears to hold regardless of theinfection route (perhaps because BZs convert to TZs quickly uponreaching the gut before BZ Ags can be seen by the immune sys-tem). However, we found that constitutive SRS9 expression elic-ited a readily detectable SRS9-specific immune response and thisresponse correlated with a significant decrease in cyst loads per-sisting in the brain. This finding supports the hypothesis that theBZ-specific nature of SRS9 expression is one of the key factorscontributing to parasite persistence. The timing of SRS expressionlikely determines parasite immunogenicity in infection by regulat-ing the way they interact with the immune system: the amount ofSRS Ags available for presentation to the immune system is fargreater during acute infection with rapidly multiplying TZs than inchronic infection with metabolically quiescent BZs. Also, system-ically disseminating TZs may more readily encounter and activate
FIGURE 9. Cytokine hyperproduction in SAG1c infection despite com-parable in vivo growth of WT and SAG1c mutants. A, Parasite loads inspleen and brain determined by plaque assays at indicated day postinfec-tion in CBA/J mice infected i.p. with 400 TZs of indicated parasite strains.Shown are mean PFU � SD (n � 3 per group per time point). B, Cytokineproduction by splenocytes from mice uninfected or infected with indicatedparasite strains (6 wk postinfection) detected by ELISA after stimulatingcells in vitro with rSAG1 or rSRS9 (20 �g/ml) for up to 72 h. For WT,mean � SD of two duplicate determinations from two mice are shown. Forothers, means of duplicate determinations are shown. Lower limit of de-tection was �30 pg/ml for all cytokines tested.
8045The Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

immune cells than do BZs that encyst and predominantly reside inthe brain.
The contrasting phenotypes of SRS9c and SAG1c mutants mayexplain why evolution has led to the immunodominant SAG1 mol-ecule being expressed only in the TZ stage. Although SAG1c ex-pression effectively targets BZ cysts for clearance in the earlyphase of chronic infection, an over-stimulation of SAG1-specific Tcells seems to occur leading to severe encephalitis, and somehow,an increase in brain cyst loads over time. A similar phenomenonhas been seen in an attempt to obtain a protective immunity byimmunizing mice with SAG1: an i.p. immunization with purifiedSAG1 protein along with CFA resulted in an increase in mortalityand brain cyst burdens in mice subsequently challenged with avir-ulent Toxoplasma strain (40). This observation and the phenotypeof SAG1c in infected mice indicate that an excessive immune re-sponse to SAG1 can somehow lead to increased parasite growthand exacerbated disease in the chronic phase of infection. Thestudies with SRS9c and SAG1c mutants have demonstrated one ofmany purposes that may be served by stage-specific SRS expres-sion: BZ-specific expression avoids the induction of a potentiallyprotective immune response, whereas TZ-specific expressionavoids BZ clearance by an immune response and minimizes im-munopathology to the host.
The brain has been viewed as an immunoprivileged site due tosuch characteristics as the blood-brain barrier, lack of conventionallymphatics, better allograft acceptance, and low T cell trafficking(10). These features will undoubtedly contribute to the lack of
immunogenicity of BZ Ags in infection and facilitate BZ persis-tence in the brain. Recently, however, many studies have shownthat resident cells of the brain do have the ability to serve as potentAPCs that are quite capable of supporting the induction of immuneresponses (41). Moreover, Ags exclusively localized in the braincan be transported to the cervical lymph nodes and activate T cells,which are then recruited to the brain (42–44), and selectively acton target cells (45). Thus, it seems reasonable to speculate that BZAgs should be able to activate an immune response during chronicinfection of the brain and that the lack of immunogenicity of SRSAgs may be the result of complex mechanisms including theirlocalization in the brain.
The type of the immune response induced by TZ-specific SRSAgs may be one of the factors contributing to parasite persistenceby down-modulating the induction of an immune response to BZAgs. T cells are critical for controlling Toxoplasma replication andtheir protective effect is largely mediated by cytokines, especiallyIFN-� (24). We found that, in addition to IFN-� as previouslyshown (22, 23), SAG1-specific T cells produce IL-10. As a potentantagonist of IFN-�, IL-10 is a key regulator in preventing exces-sive inflammation in the brain and other tissues infected with Tox-oplasma (46–48). T cells simultaneously producing IFN-� andIL-10 have been frequently observed in persistent infections, in-cluding tuberculosis (49), malaria (50), leishmaniasis (51), andBorrelia burgdorferi infection (52). IL-10 produced by these Tcells limits the IFN-�-driven inflammation, and in doing so, hamperscomplete clearance of infectious agents. Previously, an elevated level
FIGURE 10. Distinct immunolog-ical events occurring in the brains inWT, SRS9c, and SAG1c infection.H&E stained brain sections fromCBA/J mice uninfected (A) and in-fected (B–F) (6 wk postinfection)with indicated parasite strains. Ar-rowheads indicate cysts. G, Numberof T cells per brain at indicated weekpostinfection (wpi). Shown aremean � SD (n � 3 per group). Whenexamined, all CD4� and CD8� Tcells were costained with anti-CD3Ab (data not shown). H, Proportionof brain cysts associated with im-mune response (6 wk postinfection)was calculated based on the numbersof lone cysts (a), cysts surrounded byinfiltrates (b), and inflammation fociapparently lacking cysts (c). Infil-trates around blood vessels or thosebroadly distributed over a large areaof the cortex were excluded from theanalysis.
8046 ROLE OF SRS Ags IN Toxoplasma PERSISTENCE
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

of IL-10 mediating immunosuppression was noted in the spleenof a mouse acutely infected with Toxoplasma (53). Conceiv-ably, a T cell response to the dominant SAG1 Ag may greatlycontribute to parasite clearance via IFN-� production and alsobe the major source of IL-10 that serves as a self-regulatorymechanism to turn off the IFN-�-driven Th1 type response.While preventing immunopathology in favor of host survival,IL-10 may also create an immunosuppressive environment thatis not ideal for eliciting primary responses to newly emergingBZ-specific Ags.
The pattern of changes in brain cyst loads and cellular infiltra-tion during chronic infections with SRS9c and SAG1c mutants isclearly different from that seen in WT infection. This finding in-dicates that mutant BZs are attracting an active immune responseto the brain that is greater than what is seen in WT infection re-sulting in BZ clearance in SRS9c infection and the emergence ofnew cysts in SAG1c infection. The fact that more brain cysts areintimately surrounded by immune infiltrates in the SAG1c andeven SRS9c infection than in WT infection argues for the possi-bility that some of these infiltrates may be T cells recruited to thebrain due to their specificity for SAG1 and SRS9. Alternatively, orin addition, the T cell response may be targeting an extracerebralparasite reservoir, thus changing the number of parasites coloniz-ing the brain during chronic infection. Understanding how SAG1-and SRS9-specific T cells are activated and maintained in the pe-riphery, how they are recruited to the brain and what their exacteffector functions are in dealing with persisting parasites will re-quire the ability to track Ag-specific T cells and parasites in vivoin a quantitative manner. Data available at the present time indicatethat the cytokine response induced by SRS9c and SAG1c mutantsin the periphery is qualitatively and quantitatively different: whileSRS9-specific T cells produce moderate levels of IFN-� in SRS9c
infection, IFN-�, TNF-�, and IL-10 are hyperproduced by SAG1-specific T cells in SAG1c infection. Dysregulated cytokine pro-duction has been linked to many immunologic diseases and shownto be a critical event in some cases (54, 55). Although it is difficultto fully appreciate exact mechanisms, a similar phenomenon maybe responsible for regulating parasite growth/differentiation andencephalitis seen in this study in the brains of mice infected withSRS9c and SAG1c mutants.
Another challenging issue in understanding the biology of theSRS superfamily and its role in parasite persistence is the possibleimmune-interference by presentation of altered peptide ligands(56, 57). Some SRS Ags may contain Ab and T cell epitopes thatcan interfere with immune responses to closely related SRS Ags.Such interference unlikely exists between SRS9 and SAG1, asimmune responses to both Ags were readily detectable in SRS9c
infection. However, SRS9 and SAG1 belong to different subgroupswithin the SRS superfamily sharing only �25% sequence identity.An altered peptide ligand phenomenon is more likely to be oper-ative among the SRS Ags of the same subgroup (in which se-quence identity can be up to 90%) and could be one of the mech-anisms contributing to the lack of immunogenicity of some of theSRS Ags in infection.
Overall, the results presented broaden our understanding of theimmunologic roles played by the developmentally regulated SRSAgs of Toxoplasma and clearly argue that the SRS antigenicswitch accompanying TZ-to-BZ stage conversion has evolved asone of the critical mechanisms that allow optimal parasite persis-tence in its intermediate host until such time as transmission to anew host becomes feasible.
AcknowledgmentsWe thank Drs. D. Soldati, G. Arrizabalaga, and M. Grigg for kindly pro-viding the Pru�hpt parasite, Pru�hpt/pGRA1-GFP parasite, and rabbit anti-SAG1 antisera, respectively.
DisclosuresThe authors have no financial conflict of interest.
References1. Tenter, A. M., A. R. Heckeroth, and L. M. Weiss. 2000. Toxoplasma gondii: from
animals to humans. Int. J. Parasitol. 30: 1217–1258.2. Dubey, J. P. 1997. Bradyzoite-induced murine toxoplasmosis: stage conversion,
pathogenesis, and tissue cyst formation in mice fed bradyzoites of different strainsof Toxoplasma gondii. J. Euk. Microbiol. 44: 592–602.
3. McLeod, R., R. G. Estes, D. G. Mack, and H. Cohen. 1984. Immune response ofmice to ingested Toxoplasma gondii: a model of toxoplasma infection acquiredby ingestion. J. Infect. Dis. 149: 234–244.
4. Lyons, R. E., R. McLeod, and C. W. Roberts. 2002. Toxoplasma gondiitachyzoite-bradyzoite interconversion. Trends Parasitol. 18: 198–201.
5. Beaman, M. H., F. G. Araujo, and J. S. Remington. 1994. Protective reconstitu-tion of the SCID mouse against reactivation of toxoplasmic encephalitis. J. Infect.Dis. 169: 375–383.
6. Luft, B. J., and J. S. Remington. 1992. Toxoplasmic encephalitis in AIDS. Clin.Infect. Dis. 15: 211–222.
7. Sinai, A. P., and K. A. Joiner. 1997. Safe haven: the cell biology of nonfusogenicpathogen vacuoles. Annu. Rev. Microbiol. 51: 415–462.
8. Denkers, E. Y., and B. A. Butcher. 2005. Sabotage and exploitation in macro-phages parasitized by intracellular protozoans. Trends Parasitol. 21: 35–41.
9. Aliberti, J., D. Jankovic, and A. Sher. 2004. Turning it on and off: regulation ofdendritic cell function in Toxoplasma gondii infection. Immunol. Rev. 201:26–34.
10. Fabry, Z., C. S. Raine, and M. N. Hart. 1994. Nervous tissue as an immunecompartment: the dialect of the immune response in the CNS. Immunol. Today15: 218–224.
11. Radke, J. R., M. N. Guerini, M. Jerome, and M. W. White. 2003. A change in thepremitotic period of the cell cycle is associated with bradyzoite differentiation inToxoplasma gondii. Mol. Biochem. Parasitol. 131: 119–127.
12. van der Woude, M. W., and A. J. Baumler. 2004. Phase and antigenic variationin bacteria. Clin. Microbiol. Rev. 17: 581–611.
13. Tortorella, D., B. E. Gewurz, M. H. Furman, D. J. Schust, and H. L. Ploegh. 2000.Viral subversion of the immune system. Annu. Rev. Immunol. 18: 861–926.
14. Kyes, S., P. Horrocks, and C. Newbold. 2001. Antigenic variation at the infectedred cell surface in malaria. Annu. Rev. Microbiol. 55: 673–707.
15. Cleary, M. D., U. Singh, I. J. Blader, J. L. Brewer, and J. C. Boothroyd. 2002.Toxoplasma gondii asexual development: identification of developmentally reg-ulated genes and distinct patterns of gene expression. Eukaryot. Cell 1: 329–340.
16. Jung, C., C. Y.-F. Lee, and M. E. Grigg. 2004. The SRS superfamily of Toxo-plasma surface proteins. Int. J. Parasitol. 34: 285–296.
17. Tomavo, S. 1996. The major surface proteins of Toxoplasma gondii: structuresand functions. Curr. Top. Microbiol. Immunol. 219: 45–54.
18. He, X.-L., M. E. Grigg, J. C. Boothroyd, and K. C. Garcia. 2002. Structure of theimmunodominant surface antigen from the Toxoplasma gondii SRS superfamily.Nat. Struct. Biol. 9: 606–611.
19. Grigg, M. E., S. Bonnefoy, A. B. Hehl, Y. Suzuki, and J. C. Boothroyd. 2001.Success and virulence in Toxoplasma as the result of sexual recombination be-tween two distinct ancestries. Science 294: 161–165.
20. Bessieres, M. H., S. Le Breton, and J. P. Seguela. 1992. Analysis by immuno-blotting of Toxoplasma gondii exo-antigens and comparison with somatic anti-gens. Parasitol. Res. 78: 222–228.
21. Partanen, P., H. J. Turunen, R. T. Paasivuo, and P. O. Leinikki. 1984. Immuno-blot analysis of Toxoplasma gondii antigens by human immunoglobulins G, M,and A antibodies at different stages of infection. J. Clin. Microbiol. 20: 133–135.
22. Prigione, I., P. Facchetti, L. Lecordier, D. Deslee, S. Chiesa, M.-F. Cesbron-Delauw, and V. Pistoia. 2000. T cell clones raised from chronically infectedhealthy humans by stimulation with Toxoplasma gondii excretory-secretory an-tigens cross-react with live tachyzoites: characterization of the fine antigenicspecificity of the clones and implications for vaccine development. J. Immunol.164: 3741–3748.
23. Khan, I. A., M. E. Eckel, E. R. Pfefferkorn, and L. H. Kasper. 1988. Productionof � interferon by cultured human lymphocytes stimulated with a purified mem-brane protein (P30) from Toxoplasma gondii. J. Infect. Dis. 157: 979–984.
24. Suzuki, Y., M. A. Orellana, R. D. Schreiber, and J. S. Remington. 1988. Inter-feron-�: the major mediator of resistance against Toxoplasma gondii. Science240: 516–518.
25. Fatoohi, A. F., G. J. Cozon, T. Greenland, J. Ferrandiz, J. Bienvenu, S. Picot, andF. Peyron. 2002. Cellular immune responses to recombinant antigens in pregnantwomen chronically infected with Toxoplasma gondii. Clin. Diagn. Lab. Immunol.9: 704–707.
26. Di Cristina, M., P. Del Porto, W. Buffolano, E. Beghetto, A. Spadoni,S. Guglietta, E. Piccolella, F. Felici, and N. Gargano. 2004. The Toxoplasmagondii bradyzoite antigens BAG1 and MAG1 induce early humoral and cell-mediated immune responses upon human infection. Microbes Infect. 6: 164–171.
27. Ferguson, D. J., and S. F. Parmley. 2002. Toxoplasma gondii MAG1 proteinexpression. Trends Parasitol. 18: 482.
8047The Journal of Immunology
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

28. Bohne, W., U. Gross, D. J. Ferguson, and J. Heesemann. 1995. Cloning andcharacterization of a bradyzoite-specifically expressed gene (hsp30/bag1) of Tox-oplasma gondii, related to genes encoding small heat-shock proteins of plants.Mol. Microbiol. 16: 1221–1230.
29. Donald, R. G., and D. S. Roos. 1998. Gene knock-outs and allelic replacementsin Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagen-esis. Mol. Biochem. Parasitol. 91: 295–305.
30. Kim, K., M. S. Eaton, W. Schubert, S. Wu, and J. Tang. 2001. Optimized ex-pression of green fluorescent protein in Toxoplasma gondii using thermostablegreen fluorescent protein mutants. Mol. Biochem. Parasitol. 113: 309–313.
31. Black, M. W., and J. C. Boothroyd. 1998. Development of a stable episomalshuttle vector for Toxoplasma gondii. J. Biol. Chem. 273: 3972–3979.
32. Soldati, D., and J. C. Boothroyd. 1993. Transient transfection and expression inthe obligate intracellular parasite Toxoplasma gondii. Science 260: 349–352.
33. Zhang, Y. W., S. K. Halonen, Y. F. Ma, M. Wittner, and L. M. Weiss. 2001.Initial characterization of CST1, a Toxoplasma gondii cyst wall glycoprotein.Infect. Immun. 69: 501–507.
34. Weiss, L. M., D. Laplace, P. M. Takvorian, H. B. Tanowitz, A. Cali, andM. Wittner. 1995. A cell culture system for study of the development of Toxo-plasma gondii bradyzoites. J. Eukaryot. Microbiol. 42: 150–157.
35. Zenner, L., A. Foulet, Y. Caudrelier, F. Darcy, B. Gosselin, A. Capron, andM. F. Cesbron-Delauw. 1999. Infection with Toxoplasma gondii RH and Prug-niaud strains in mice, rats and nude rats: kinetics of infection in blood and tissuesrelated to pathology in acute and chronic infection. Pathol. Res. Pract. 195:475–485.
36. Ferguson, D. J., W. M. Hutchison, and E. Pettersen. 1989. Tissue cyst rupture inmice chronically infected with Toxoplasma gondii: an immunocytochemical andultrastructural study. Parasitol. Res. 75: 599–603.
37. Suzuki, Y. 2002. Immunopathogenesis of cerebral toxoplasmosis. J. Infect. Dis.186(Suppl 2): S234–S240.
38. Shirahata, T., T. Yamashita, C. Ohta, H. Goto, and A. Nakane. 1994. CD8� Tlymphocytes are the major cell population involved in the early � interferonresponse and resistance to acute primary Toxoplasma gondii infection in mice.Microbiol. Immunol. 38: 789–796.
39. Khan, I. A., W. R. Green, L. H. Kasper, K. A. Green, and J. D. Schwartzman.1999. Immune CD8� T cells prevent reactivation of Toxoplasma gondii infectionin the immunocompromised host. Infect. Immun. 67: 5869–5876.
40. Kasper, L. H., K. M. Currie, and M. S. Bradley. 1985. An unexpected responseto vaccination with a purified major membrane tachyzoite antigen (P30) of Tox-oplasma gondii. J. Immunol. 134: 3426–3431.
41. Olson, J. K., and S. D. Miller. 2004. Microglia initiate central nervous systeminnate and adaptive immune responses through multiple TLRs. J. Immunol. 173:3916–3924.
42. Karman, J., C. Ling, M. Sandor, and Z. Fabry. 2004. Initiation of immune re-sponses in brain is promoted by local dendritic cells. J. Immunol. 173:2353–2361.
43. van der Most, R. G., K. Murali-Krishna, and R. Ahmed. 2003. Prolonged pres-ence of effector-memory CD8 T cells in the central nervous system after denguevirus encephalitis. Int. Immunol. 15: 119–125.
44. Marten, N. W., S. A. Stohlman, J. Zhou, and C. C. Bergmann. 2003. Kinetics ofvirus-specific CD8�-T-cell expansion and trafficking following central nervoussystem infection. J. Virol. 77: 2775–2778.
45. Cabarrocas, J., J. Bauer, E. Piaggio, R. Liblau, and H. Lassmann. 2003. Effectiveand selective immune surveillance of the brain by MHC class I-restricted cyto-toxic T lymphocytes. Eur. J. Immunol. 33: 1174–1182.
46. Lu, F., S. Huang, and L. H. Kasper. 2003. Interleukin-10 and pathogenesis ofmurine ocular toxoplasmosis. Infect. Immun. 71: 7159–7163.
47. Deckert-Schluter, M., C. Buck, D. Weiner, N. Kaefer, A. Rang, H. Hof,O. D. Wiestler, and D. Schluter. 1997. Interleukin-10 downregulates the intra-cerebral immune response in chronic Toxoplasma encephalitis. J. Neuroimmunol.76: 167–176.
48. Gazzinelli, R. T., M. Wysocka, S. Hieny, T. Scharton-Kersten, A. Cheever,R. Kuhn, W. Muller, G. Trinchieri, and A. Sher. 1996. In the absence of endog-enous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethalimmune response dependent on CD4� T cells and accompanied by overproduc-tion of IL-12, IFN-� and TNF-�. J. Immunol. 157: 798–805.
49. Gerosa, F., C. Nisii, S. Righetti, R. Micciolo, M. Marchesini, A. Cazzadori, andG. Trinchieri. 1999. CD4� T cell clones producing both interferon-� and inter-leukin-10 predominate in bronchoalveolar lavages of active pulmonary tubercu-losis patients. Clin. Immunol. 92: 224–234.
50. Plebanski, M., K. L. Flanagan, E. A. Lee, W. H. H. Reece, K. Hart, C. Gelder,G. Gillespie, M. Pinder, and A. V. Hill. 1999. Interleukin 10-mediated immuno-suppression by a variant CD4 T cell epitope of Plasmodium falciparum. Immunity10: 651–660.
51. Belkaid, Y., K. F. Hoffmann, S. Mendez, S. Kamhawi, M. C. Udey, T. A. Wynn,and D. L. Sacks. 2001. The role of interleukin (IL)-10 in the persistence ofLeishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194: 1497–1506.
52. Pohl-Koppe, A., K. E. Balashov, A. C. Steere, E. L. Logigian, and D. A. Hafler.1998. Identification of a T cell subset capable of both IFN-� and IL-10 secretionin patients with chronic Borrelia burgdorferi infection. J. Immunol. 160:1804–1810.
53. Khan, I. A., T. Matsuura, and L. H. Kasper. 1995. IL-10 mediates immunosup-pression following primary infection with Toxoplasma gondii in mice. ParasiteImmunol. 17: 185–195.
54. O’Garra, A. 1998. Cytokines induce the development of functionally heteroge-neous T helper cell subsets. Immunity 8: 275–283.
55. Abbas, A. K., K. M. Murphy, and A. Sher. 1996. Functional diversity of helperT lymphocytes. Nature 383: 787–793.
56. Plebanski, M., E. A. Lee, C. M. Hannan, K. L. Flanagan, S. C. Gilbert,M. B. Gravenor, and A. V. Hill. 1999. Altered peptide ligands narrow the rep-ertoire of cellular immune responses by interfering with T-cell priming. Nat.Med. 5: 565–571.
57. Sloan-Lancaster, J., and P. M. Allen. 1996. Altered peptide ligand-induced partialT cell activation: molecular mechanisms and role in T cell biology. Annu. Rev.Immunol. 14: 1–27.
58. Burg, J. L., D. Perelman, L. H. Kasper, P. L. Ware, and J. C. Boothroyd. 1988.Molecular analysis of the gene encoding the major surface antigen of Toxoplasmagondii. J. Immunol. 141: 3584–3591.
8048 ROLE OF SRS Ags IN Toxoplasma PERSISTENCE
by guest on April 9, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
Related Documents









![Reduced Expression of HLA Class I and II Antigens in Colon Cancer1 · [CANCER RESEARCH 50, 8023-8027, December 15, 1990] Reduced Expression of HLA Class I and II Antigens in Colon](https://static.cupdf.com/doc/110x72/5f5945785c4df2481d781bbc/reduced-expression-of-hla-class-i-and-ii-antigens-in-colon-cancer1-cancer-research.jpg)

