
Sources Page & Holmes
Jan 08, 2016
Sources Page & Holmes Vladimir Likic presentation: http ://science.marshall.edu/murraye/Clearer%20Matrix%20slide% 20show.pdf Wikipedia Lecture at : http:// cs.njit.edu / usman /courses/bnfo601_fall08/ AffineGap.pdf. - PowerPoint PPT Presentation
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


Sources
• Page & Holmes• Vladimir Likic presentation: http
://science.marshall.edu/murraye/Clearer%20Matrix%20slide%20show.pdf
• Wikipedia• Lecture at :
http://cs.njit.edu/usman/courses/bnfo601_fall08/AffineGap.pdf

Homoplasy – structural or DNA resemblance due to parallelism or convergent evolution rather than to common ancestry

Which are homoplasious?

Problem: which base positions share common descent?
agtggtcttgctacattgctagctaaatcgatcatgatcgatgattcaggtagctaaatcgatcatgatcgatgattcaggcgatgtcatgactgatcagtacattgctagctaaatcgatcatgatcgatgattcaggcgatgtcatgagatcatgatcgatgattcaggcgatgtcatgactgatcagggatgatgat
Alignment – residue to residue correspondence between 2 or more sequences such that the order of residues in each sequence is preserved.agtggtcttgctacattgctagctaaatcgatcatgatcgatgattcagg tagctaaatcgatcatgatcgatgattcaggcgatgtcatgactgatcag tacattgctagctaaatcgatcatgatcgatgattcaggcgatgtcatga gatcatgatcgatgattcaggcgatgtcatgactgatcagggatgatgat
agtggtcttgctacattgctagctaaatcgatcatgatcgatgattcagg tagctaaatcgatcatgatcgatgattcaggcgatgtcatgactgatcag tacattgctagctaaa----tcatgatcgatgattcaggcgatgtcatga gatcatgatcgatgattcaggcgat------actgatcagggatgatgat
Indels make alignment trickier

Assembly – (from ensembl) - When the genome of a species is to be sequenced, the chromosomes from many cells are broken at random positions into small fragments, which are sequenced, and reassembled into long sequences (contigs). Contigs may be assembled into longer sequences called scaffolds and sometimes, if the depth of sequencing is high enough, there may be enough information to assemble most of the scaffolds into chromosomes. The resulting collection of sequences after assembly is called a genome assembly.
Alignment problems (examples) 1) different sequences of the same allele from the same
locus within the same individual2) sequences of different alleles from the same locus within
the same individual3) same locus from different individuals

Alignment Methods• Dot plot – qualitative• Sequence alignment – quantitative;
constructing the best alignment using a scoring scheme
Types of Alignment• Global – best alignment over the entire length• Local – best alignment in small region; used when
comparing sequences of different lengths• Multiple – beyond pairwise
cagcacttggattctgg & cagcgtgg
Localcagca-cttggattctgg---cagcgtgg-------
Global (best depending on gap penalties)cagcacttggattctggcagc----g—t----gg

Gaps
• residue to nothing match that can be inserted in either sequence
• are not part of the DNA sequence, only a construct for alignment
• Gap to gap match is meaningless and not allowed

Dot plots – heuristic; make matrix, place dots; find diagonals

Alignment with scoring schemes
• score to select the best possible alignment given scoring scheme
Scoring scheme • A set of rules that assigns a score to a particular alignment
between two sequences• Goal is to maximize score• Score is sum of residue substitution scores and gap penalties

atggcgt +1+1+1-1+1+1 = 4atg-agt
+1 for match-1 for mismatch No gap penalty
atggcgt +1-1+1-1+1+1 = 2a-tgagt
Substitution matrix: c t a gc 1 -1 -1 -1t -1 1 -1 -1a -1 -1 1 -1g -1 -1 -1 1
Substitution matrix: c t a gc 2 1 -1 -1T 1 2 -1 -1a -1 -1 2 1g -1 -1 1 2
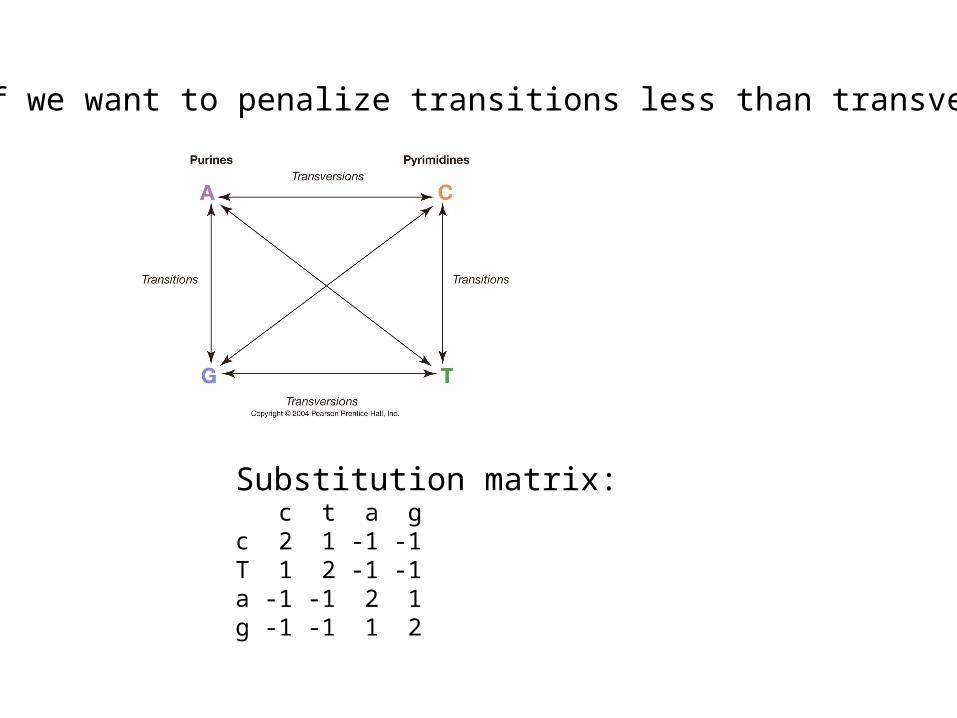
What if we want to penalize transitions less than transversions?

Protein substitution matrices• More complex than DNA scoring matrices.• Proteins are composed of twenty amino acids, and
physical-chemical properties of individual amino acids vary considerably.
• can be based on any property of amino acids: size, polarity, charge, hydrophobicity.
• Evolutionary substitution matrices – empirically derived by assessment of frequencies of changes at particular levels of divergence

Evolutionary substitution matrices• PAM ("point accepted mutation") family
PAM250, PAM120, etc.• BLOSUM ("Blocks substitution matrix") family
BLOSUM62, BLOSUM50, etc.• The BLOSUM matrices were developed more
recently and considered better.

Blosum62
Blosum80 is used for less divergent sequencesBlosum45 is used for more divergent sequencesEtc.

• Because gaps often result in radical protein changes (frame shifts, premature stop), the penalty for a gap is usually several times greater than the penalty for a mutation.
• Once created, gaps of more than one residue might be less expensive than a completely new gap - in other words gap opening penalties and gap extension penalties are often defined separately
Gaps

Wi=g+h*i(for i>= 1, where i = gap length )
•g: gap opening penalty•h: gap extension penalty•The ratio between gand h determines the relative weight for opening versus extension
–Small g, Large h: gap length more important–Large g, Small h: gap length less important
Affine gap penalty function W(i)

ATGTAGTGTATAGTACATGCAATGTAG-------TACATGCA
ATGTAGTGTATAGTACATGCAATGTA--G--TA---CATGCA
Wi=g+h*i
G = -3H = -1
Substitution matrix: c t a gc 2 1 -1 -1T 1 2 -1 -1a -1 -1 2 1g -1 -1 1 2
26 – 3 – 1(7) = 16
26 – 3 (3) – 1(7) =10

How do we find the best alignment?
Brute-force approach:Generate the list all possible alignments between two sequences, score them, select the alignment with the best score
The number of possible global alignments between two sequences of length N is
For two sequences of 250 residues this is ~10149

Needleman-Wunsch and Smith-Waterman are both algorithms that find the best alignment through breaking the problem down into sub problems using dynamic programming
…however, it is only the best based on the scoring matrix and the gap opening and extension penalities
These methods are computationally expensive

BLAST – Basic Local Alignment Search Tool
- Tries to find the highest scoring ungapped local alignment between a query and a database
- Uses a word length (w) and scans for matches with a higher threshold (T) when aligned with words in the query
- The local alignment is then extended in both directions until the score falls below the best score reached so far.
- Many types of blast can be found at http://blast.ncbi.nlm.nih.gov/Blast.cgi
Related Documents











