Sorption reversibility kinetics in the ternary system radionuclide–bentonite colloids/nanoparticles–granite fracture filling material F. Huber a,b,⇑ , P. Kunze a , H. Geckeis a , T. Schäfer a,b,⇑ a Karlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany b Institute of Geological Sciences, Department of Earth Sciences, Freie Universität Berlin, Berlin, Germany article info Article history: Received 8 August 2010 Accepted 5 August 2011 Available online 24 August 2011 abstract The kinetics of radionuclide desorption from bentonite colloids and subsequent sorption onto fracture filling material can influence colloid-facilitated radionuclide migration in ground water. To shed light on the significance of these issues batch-type experiments using a cocktail of strong and weak sorbing radionuclides as well as FEBEX bentonite colloids in the presence of fracture filling material from Grimsel (Switzerland) under Grimsel ground water conditions have been conducted. Results show that tri- and tetravalent radionuclides, 232 Th(IV), 242 Pu(IV) and 243 Am(III) are clearly colloid associated in contrast to 233 U(VI), 237 Np(V) and 99 Tc(VII). Concentrations of colloid-borne 232 Th(IV), 242 Pu(IV) and 243 Am(III) decrease after 100 h showing desorption from bentonite colloids while 233 U(VI) and 99 Tc(VII) concen- trations remain constant over the entire experimental time of 7500 h thus showing no interaction either to colloids or to the fracture filling material. 232 Th(IV) and 242 Pu(IV) data yield a slower dissociation from colloids compared to 243 Am(III) indicating stronger RN–colloid interaction. In the case of 237 Np(V), a decrease in concentration after 300 h is observed which can be explained either by slow reduction to Np(IV) and subsequent sorption to mineral surfaces in accordance with the evolution of pe/pH and/or by a slow sorption onto the fracture filling material. No influence of the different fracture filling material size fractions (0.25–0.5 mm, 0.5–1 mm and 1–2 mm) can be observed implying reaction independence of the mineral surface area and mineralogical composition. The driving force of the observed metal ion desorption from colloids is binding to fracture filling material surfaces being in excess of the available colloid surface area (76:1, 55:1 and 44:1 for the 0.25–0.5 mm, 0.5–1 mm and 1–2 mm size fraction of the FFM, respectively). Ó 2011 Elsevier Ltd. All rights reserved. 1. Introduction The migration of radionuclides out of a nuclear waste disposal into the environment is a potential threat to nature and mankind due to their radio- and chemo-toxicity even in trace concentra- tions. It is, therefore, of great importance to gain insight into the various chemical and physical processes including colloid genera- tion, stability and mobility governing radionuclide (RN) migration behavior to assure the development of sound risk assessment strategies (Schäfer and Noseck, 2010). Much effort has been spent in the past and is still ongoing today to shed light on the various complex geochemical processes and their impact on RN mobility, among them, sorption of radionuclides onto colloids/nanoparticles. For detailed discussion on the current knowledge gaps concerning montmorillonite colloid- radionuclide interaction, including inter alia radionuclide desorption rates the reader is referred to Wold (2010) and Schäfer and Noseck (2010). In general colloids/nano- particles range between 1 nm and 1000 nm in size consisting of a huge ensemble of various inorganic and/or organic matter such as e.g. mineral precipitates, rock forming mineral fragments, humic and fulvic acids and microorganisms (McCarthy and Zachara, 1989). Colloids/nanoparticles are ubiquitous in natural surface and subsurface waters and their mineralogy often is closely related to the host rock formation. Furthermore, some radionuclides, espe- cially the tri- and tetravalent actinides (Th, Pu, Am) show strong hydrolysis and subsequent polymerization and are, therefore, prone to the formation of so called ‘‘eigencolloids’’ (Kim et al., 1984; Rothe et al., 2009). The importance of colloids for migration of contaminants has been proven by various laboratory and field studies carried out in the past (Kersting et al., 1999), among them the colloid and radionuclide retardation (CRR) experiments conducted at the Grimsel test site (Möri et al., 2003; Geckeis et al., 2004). These experiments revealed under the given hydro-geochemical condi- tions (mean fracture residence time of 88 min) that in particular the transport of tri- and tetravalent radionuclides, Am(III) and 0883-2927/$ - see front matter Ó 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.apgeochem.2011.08.005 ⇑ Corresponding authors at: Karlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany. Tel.: +49 7247822384; fax: +49 7247824308. E-mail addresses: fl[email protected] (F. Huber), [email protected] (T. Schäfer). Applied Geochemistry 26 (2011) 2226–2237 Contents lists available at SciVerse ScienceDirect Applied Geochemistry journal homepage: www.elsevier.com/locate/apgeochem

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Applied Geochemistry 26 (2011) 2226–2237
Contents lists available at SciVerse ScienceDirect
Applied Geochemistry
journal homepage: www.elsevier .com/ locate /apgeochem
Sorption reversibility kinetics in the ternary system radionuclide–bentonitecolloids/nanoparticles–granite fracture filling material
F. Huber a,b,⇑, P. Kunze a, H. Geckeis a, T. Schäfer a,b,⇑a Karlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germanyb Institute of Geological Sciences, Department of Earth Sciences, Freie Universität Berlin, Berlin, Germany
a r t i c l e i n f o a b s t r a c t
Article history:Received 8 August 2010Accepted 5 August 2011Available online 24 August 2011
0883-2927/$ - see front matter � 2011 Elsevier Ltd. Adoi:10.1016/j.apgeochem.2011.08.005
⇑ Corresponding authors at: Karlsruhe Institute of TNuclear Waste Disposal (INE), P.O. Box 3640,Tel.: +49 7247822384; fax: +49 7247824308.
E-mail addresses: [email protected] (F. Hube(T. Schäfer).
The kinetics of radionuclide desorption from bentonite colloids and subsequent sorption onto fracturefilling material can influence colloid-facilitated radionuclide migration in ground water. To shed lighton the significance of these issues batch-type experiments using a cocktail of strong and weak sorbingradionuclides as well as FEBEX bentonite colloids in the presence of fracture filling material from Grimsel(Switzerland) under Grimsel ground water conditions have been conducted. Results show that tri- andtetravalent radionuclides, 232Th(IV), 242Pu(IV) and 243Am(III) are clearly colloid associated in contrastto 233U(VI), 237Np(V) and 99Tc(VII). Concentrations of colloid-borne 232Th(IV), 242Pu(IV) and 243Am(III)decrease after �100 h showing desorption from bentonite colloids while 233U(VI) and 99Tc(VII) concen-trations remain constant over the entire experimental time of 7500 h thus showing no interaction eitherto colloids or to the fracture filling material. 232Th(IV) and 242Pu(IV) data yield a slower dissociation fromcolloids compared to 243Am(III) indicating stronger RN–colloid interaction. In the case of 237Np(V), adecrease in concentration after �300 h is observed which can be explained either by slow reduction toNp(IV) and subsequent sorption to mineral surfaces in accordance with the evolution of pe/pH and/orby a slow sorption onto the fracture filling material. No influence of the different fracture filling materialsize fractions (0.25–0.5 mm, 0.5–1 mm and 1–2 mm) can be observed implying reaction independence ofthe mineral surface area and mineralogical composition. The driving force of the observed metal iondesorption from colloids is binding to fracture filling material surfaces being in excess of the availablecolloid surface area (76:1, 55:1 and 44:1 for the 0.25–0.5 mm, 0.5–1 mm and 1–2 mm size fraction ofthe FFM, respectively).
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
The migration of radionuclides out of a nuclear waste disposalinto the environment is a potential threat to nature and mankinddue to their radio- and chemo-toxicity even in trace concentra-tions. It is, therefore, of great importance to gain insight into thevarious chemical and physical processes including colloid genera-tion, stability and mobility governing radionuclide (RN) migrationbehavior to assure the development of sound risk assessmentstrategies (Schäfer and Noseck, 2010). Much effort has been spentin the past and is still ongoing today to shed light on the variouscomplex geochemical processes and their impact on RN mobility,among them, sorption of radionuclides onto colloids/nanoparticles.For detailed discussion on the current knowledge gaps concerningmontmorillonite colloid- radionuclide interaction, including inter
ll rights reserved.
echnology (KIT), Institute for76021 Karlsruhe, Germany.
alia radionuclide desorption rates the reader is referred to Wold(2010) and Schäfer and Noseck (2010). In general colloids/nano-particles range between 1 nm and 1000 nm in size consisting of ahuge ensemble of various inorganic and/or organic matter suchas e.g. mineral precipitates, rock forming mineral fragments, humicand fulvic acids and microorganisms (McCarthy and Zachara,1989). Colloids/nanoparticles are ubiquitous in natural surfaceand subsurface waters and their mineralogy often is closely relatedto the host rock formation. Furthermore, some radionuclides, espe-cially the tri- and tetravalent actinides (Th, Pu, Am) show stronghydrolysis and subsequent polymerization and are, therefore,prone to the formation of so called ‘‘eigencolloids’’ (Kim et al.,1984; Rothe et al., 2009).
The importance of colloids for migration of contaminants hasbeen proven by various laboratory and field studies carried outin the past (Kersting et al., 1999), among them the colloid andradionuclide retardation (CRR) experiments conducted at theGrimsel test site (Möri et al., 2003; Geckeis et al., 2004). Theseexperiments revealed under the given hydro-geochemical condi-tions (mean fracture residence time of 88 min) that in particularthe transport of tri- and tetravalent radionuclides, Am(III) and

F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237 2227
Pu(IV), is facilitated in the presence of colloidal phases leading toan un-retarded breakthrough whereas non colloidal-associatedradionuclides like U(VI) or Np(V) exhibit slight retardation due tofracture surface interaction. In parallel, a laboratory program fo-cused on short-term desorption kinetics to link this data to thefield experiments. Therefore, these studies focused on desorptiontimes 6340 h (Geckeis et al., 2004).
Variation of ground water flow rates in laboratory columnmigration experiments revealed an attachment/filtration of col-loids/nanoparticles onto the rock matrix even under geochemical/electrostatic conditions favoring the stability of colloids in the bulksolution (Schäfer et al., 2004; Missana et al., 2008). Reasons for thelatter findings were attributed to chemical and physical heteroge-neity of the fracture surface or flow path geometry. Overall, theresults mentioned above clearly show the complexity of the ternarysystem RN–nanoparticle–collector (fracture filling/surface).
Another key aspect regarding the potential influence of colloids/nanoparticles on contaminant mobility is sorption reversibility/irreversibility and the kinetics involved. Probably one of the mostextensively investigated systems is Cs sorption reversibility ontoclay (especially illite) (Comans, 1987; Comans et al., 1991; De Kon-ing and Comans, 2004). Reversibility of trace metal sorption inbatch-type studies has often been found to be incomplete, whichis interpreted by some authors as partially ‘‘irreversible’’ binding(e.g. Bellenger and Staunton, 2008; Galunin et al., 2009, after1 day desorption time). Isotopic exchange studies on Sm(III) andYb(III) (Coppin et al., 2003) and spectroscopic work using time-resolved laser fluorescence spectroscopy (TRLFS) (Rabung et al.,2005), however, indicate the formation of inner-sphere surfacecomplexes at pH > 5 for Eu(III)/Cm(III) in the form � S—O—Eu=CmðOHÞð2�xÞ
x ðH2OÞ5�x which are expected to be slow but fullyreversible. Also work of Latrille et al. (2006) on Sn(IV) and Pu(IV)with Callovo-Oxfordian argillite has revealed sorption reversibility.The apparent lack of reversibility might, therefore, be due to theshort desorption experiment duration in comparison to the slowdesorption kinetics as found e.g. for Cd(II) (Comans, 1987) or Th(IV)(Bouby et al., 2009). The desorption kinetics of colloid-boundradionuclides and a possible subsequent sorption onto the rockmatrix might have a strong impact on the colloid-facilitated trans-port depending on the rate of advection (residence time) (Turneret al., 2006). Therefore, reliable data on radionuclide desorptionrates are needed to be included in reactive transport models. Ignor-ing reversibility kinetics can lead to an over-estimation of radionu-clide mobility and travel distances in ground water systems.
The aim of this study is to investigate the sorption and desorp-tion process of 99Tc(VII), 232Th(IV), 233U(VI), 237Np(V), 242Pu(IV) and243Am(III) onto FEBEX bentonite colloids under Grimsel groundwater conditions in the presence of fracture filling material (FFM)from the Grimsel Test Site (GTS) for a long (weeks–months) equi-librium time to extend the already existing datasets for short con-tact times (Missana and Geckeis, 2006). The selected radionuclidesand their oxidation states provide a good representation of nuclearwaste constituents and their possible forms. Therefore, batch-typestudies were conducted regarding the following aspects: Impact of(i) 237Np(V), 242Pu(IV) and 243Am(III) concentration, (ii) influence ofavailable surface area and (iii) contact time on RN sorption andreversibility processes.
2. Materials and methods
2.1. Fracture filling material (FFM) characterization
Fracture filling material originates from the Grimsel Test Site(GTS), Switzerland. The so called Grimsel granodiorite representsa medium to coarse grained crystalline rock possessing several
joint and fracture systems induced by a relatively cold and,therefore, brittle deformation history (Missana and Geckeis,2006). The Grimsel materials consist mainly of plagioclase(29–33 vol.%), quartz (27–28 vol.%), K-feldspar (12–24 vol.%) andbiotite (7–11 vol.%). As accessory minerals muscovite/sericite, apa-tite, sphene, epidote, zircon, chlorite, calcite and opaque mineralswere identified (Alexander et al., 2001). For the batch type sorptionstudies fracture filling material (FFM) was crushed, sieved andfreeze dried under atmospheric conditions. Three different sizefractions, namely 0.25–0.5 mm, 0.5–1 mm and 1–2 mm wereseparated. The main part of the batch-type experiments was onthe 1–2 mm size fraction. Fracture filling material was character-ized by powder X-ray diffraction (XRD), scanning electron micros-copy (SEM), energy dispersive X-ray microanalysis (EDX), specificsurface area analysis (N2-BET) and X-ray fluorescence analysis(XFR) prior to the experiments.
Powder X-ray diffraction was applied to examine the mineralcomposition of the three different FFM size fractions used in thisstudy. XRD patterns were recorded from 5� 2h to 100� 2h, using0.01� 2h steps, and 2s counting time per step with a Bruker AXSD8 powder diffractometer equipped with a BSI (Baltic ScientificInstrument) Si(Li) solid detector and Cu Ka radiation. For evalua-tion of the spectra the software DiffacPlus from Bruker-AXS wasused in combination with the database of the International Centrefor Diffraction Data (PDF). All spectra show a very similar mineral-ogical composition independent of the grain size. Prevailing min-eral phases are quartz, plagioclase (anorthite/albite), K-feldspar,biotite and muscovite.
Scanning electron microscopy (SEM) of the Grimsel FFM grainsshows an angular morphology with rough heterogeneous grainsurfaces. Grain sizes for the three different size fractions, namely0.25–0.5 mm, 0.5–1 mm and 1–2 mm were validated by SEM.EDX spectra yield mainly K-feldspar, quartz and element signa-tures for sheet-silicates, most likely of biotite-type. The specificsurface areas of the different FFM size fractions were determinedby BET N2-adsorption. The samples were heated to 300 �C and de-gassed for 6 h. The determination of the surface area was done viamultiple point analysis and a subsequent fit with the BET isotherm.In all BET measurements care was taken to have sufficient totalsurface (>1 m2) for reliable measurements. BET measurements ofthe three size fractions yielded values of 0.238 m2 g�1,0.187 m2 g�1 and 0.153–0.166 m2 g�1 for 0.25–0.5 mm, 0.5–1 mmand 1–2 mm size fraction, respectively. Full adsorption isothermsof the particles have been measured to detect a possible micro-porosity. The observed hysteresis in the isotherms corroboratesthe presence of a meso-/macro-porosity potentially offering addi-tional surface area for sorption of the radionuclides accessible viadiffusion processes. Pore sizes are heterogeneously distributedcovering a broad range between �1.5 and �210 nm with a slightpredominance between �3 and �10 nm.
Chemical rock composition analysis by XRF was conducted todetermine the bulk major and trace element distribution in thefracture filling material. As expected for a granodiorite, major ele-mental composition is dominated by SiO2, Al2O3, Na2O and K2Orepresenting the main mineral elements for quartz and K-feld-spar/albite which is in accordance with the EDX analysis. Interest-ingly, the total Fe content (2.96 wt.% Fe2O3ðtÞ) is mainly composedof Fe(II) with 1.87 wt.% FeO, which gives a hint to minor oxidationeffects through the separation and preparation of the FFM size frac-tions. The complete major and trace elemental analysis is given inTable 1.
2.2. FEBEX bentonite colloid characterization
For the experiments FEBEX (Full-scale Engineered BarrierEXperiment) bentonite from the deposit of Cabo de Gata, Almería
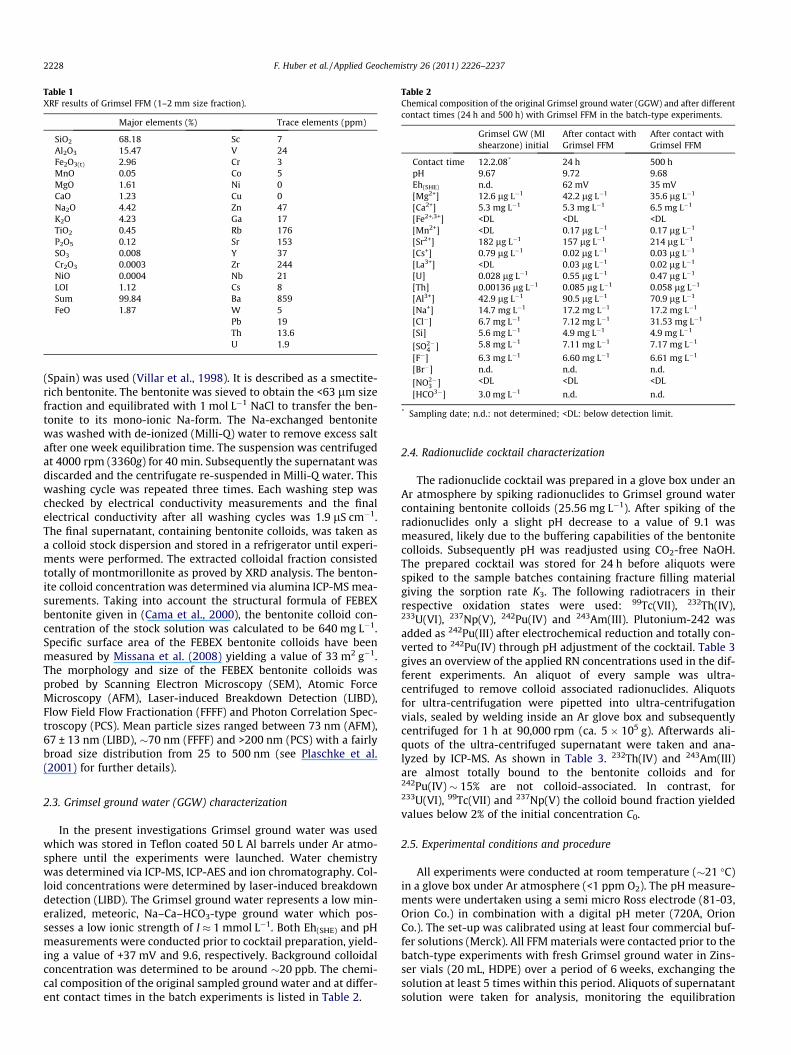
Table 1XRF results of Grimsel FFM (1–2 mm size fraction).
Major elements (%) Trace elements (ppm)
SiO2 68.18 Sc 7Al2O3 15.47 V 24Fe2O3(t) 2.96 Cr 3MnO 0.05 Co 5MgO 1.61 Ni 0CaO 1.23 Cu 0Na2O 4.42 Zn 47K2O 4.23 Ga 17TiO2 0.45 Rb 176P2O5 0.12 Sr 153SO3 0.008 Y 37Cr2O3 0.0003 Zr 244NiO 0.0004 Nb 21LOI 1.12 Cs 8Sum 99.84 Ba 859FeO 1.87 W 5
Pb 19Th 13.6U 1.9
Table 2Chemical composition of the original Grimsel ground water (GGW) and after differentcontact times (24 h and 500 h) with Grimsel FFM in the batch-type experiments.
Grimsel GW (MIshearzone) initial
After contact withGrimsel FFM
After contact withGrimsel FFM
Contact time 12.2.08* 24 h 500 hpH 9.67 9.72 9.68Eh(SHE) n.d. 62 mV 35 mV[Mg2+] 12.6 lg L�1 42.2 lg L�1 35.6 lg L�1
[Ca2+] 5.3 mg L�1 5.3 mg L�1 6.5 mg L�1
[Fe2+,3+] <DL <DL <DL[Mn2+] <DL 0.17 lg L�1 0.17 lg L�1
[Sr2+] 182 lg L�1 157 lg L�1 214 lg L�1
[Cs+] 0.79 lg L�1 0.02 lg L�1 0.03 lg L�1
[La3+] <DL 0.03 lg L�1 0.02 lg L�1
[U] 0.028 lg L�1 0.55 lg L�1 0.47 lg L�1
[Th] 0.00136 lg L�1 0.085 lg L�1 0.058 lg L�1
[Al3+] 42.9 lg L�1 90.5 lg L�1 70.9 lg L�1
[Na+] 14.7 mg L�1 17.2 mg L�1 17.2 mg L�1
[Cl�] 6.7 mg L�1 7.12 mg L�1 31.53 mg L�1
[Si] 5.6 mg L�1 4.9 mg L�1 4.9 mg L�1
[SO2�4 ] 5.8 mg L�1 7.11 mg L�1 7.17 mg L�1
[F�] 6.3 mg L�1 6.60 mg L�1 6.61 mg L�1
[Br�] n.d. n.d. n.d.
[NO2�3 ] <DL <DL <DL
[HCO3�] 3.0 mg L�1 n.d. n.d.
* Sampling date; n.d.: not determined; <DL: below detection limit.
2228 F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237
(Spain) was used (Villar et al., 1998). It is described as a smectite-rich bentonite. The bentonite was sieved to obtain the <63 lm sizefraction and equilibrated with 1 mol L�1 NaCl to transfer the ben-tonite to its mono-ionic Na-form. The Na-exchanged bentonitewas washed with de-ionized (Milli-Q) water to remove excess saltafter one week equilibration time. The suspension was centrifugedat 4000 rpm (3360g) for 40 min. Subsequently the supernatant wasdiscarded and the centrifugate re-suspended in Milli-Q water. Thiswashing cycle was repeated three times. Each washing step waschecked by electrical conductivity measurements and the finalelectrical conductivity after all washing cycles was 1.9 lS cm�1.The final supernatant, containing bentonite colloids, was taken asa colloid stock dispersion and stored in a refrigerator until experi-ments were performed. The extracted colloidal fraction consistedtotally of montmorillonite as proved by XRD analysis. The benton-ite colloid concentration was determined via alumina ICP-MS mea-surements. Taking into account the structural formula of FEBEXbentonite given in (Cama et al., 2000), the bentonite colloid con-centration of the stock solution was calculated to be 640 mg L�1.Specific surface area of the FEBEX bentonite colloids have beenmeasured by Missana et al. (2008) yielding a value of 33 m2 g�1.The morphology and size of the FEBEX bentonite colloids wasprobed by Scanning Electron Microscopy (SEM), Atomic ForceMicroscopy (AFM), Laser-induced Breakdown Detection (LIBD),Flow Field Flow Fractionation (FFFF) and Photon Correlation Spec-troscopy (PCS). Mean particle sizes ranged between 73 nm (AFM),67 ± 13 nm (LIBD), �70 nm (FFFF) and >200 nm (PCS) with a fairlybroad size distribution from 25 to 500 nm (see Plaschke et al.(2001) for further details).
2.3. Grimsel ground water (GGW) characterization
In the present investigations Grimsel ground water was usedwhich was stored in Teflon coated 50 L Al barrels under Ar atmo-sphere until the experiments were launched. Water chemistrywas determined via ICP-MS, ICP-AES and ion chromatography. Col-loid concentrations were determined by laser-induced breakdowndetection (LIBD). The Grimsel ground water represents a low min-eralized, meteoric, Na–Ca–HCO3-type ground water which pos-sesses a low ionic strength of I � 1 mmol L�1. Both Eh(SHE) and pHmeasurements were conducted prior to cocktail preparation, yield-ing a value of +37 mV and 9.6, respectively. Background colloidalconcentration was determined to be around �20 ppb. The chemi-cal composition of the original sampled ground water and at differ-ent contact times in the batch experiments is listed in Table 2.
2.4. Radionuclide cocktail characterization
The radionuclide cocktail was prepared in a glove box under anAr atmosphere by spiking radionuclides to Grimsel ground watercontaining bentonite colloids (25.56 mg L�1). After spiking of theradionuclides only a slight pH decrease to a value of 9.1 wasmeasured, likely due to the buffering capabilities of the bentonitecolloids. Subsequently pH was readjusted using CO2-free NaOH.The prepared cocktail was stored for 24 h before aliquots werespiked to the sample batches containing fracture filling materialgiving the sorption rate K3. The following radiotracers in theirrespective oxidation states were used: 99Tc(VII), 232Th(IV),233U(VI), 237Np(V), 242Pu(IV) and 243Am(III). Plutonium-242 wasadded as 242Pu(III) after electrochemical reduction and totally con-verted to 242Pu(IV) through pH adjustment of the cocktail. Table 3gives an overview of the applied RN concentrations used in the dif-ferent experiments. An aliquot of every sample was ultra-centrifuged to remove colloid associated radionuclides. Aliquotsfor ultra-centrifugation were pipetted into ultra-centrifugationvials, sealed by welding inside an Ar glove box and subsequentlycentrifuged for 1 h at 90,000 rpm (ca. 5 � 105 g). Afterwards ali-quots of the ultra-centrifuged supernatant were taken and ana-lyzed by ICP-MS. As shown in Table 3. 232Th(IV) and 243Am(III)are almost totally bound to the bentonite colloids and for242Pu(IV) � 15% are not colloid-associated. In contrast, for233U(VI), 99Tc(VII) and 237Np(V) the colloid bound fraction yieldedvalues below 2% of the initial concentration C0.
2.5. Experimental conditions and procedure
All experiments were conducted at room temperature (�21 �C)in a glove box under Ar atmosphere (<1 ppm O2). The pH measure-ments were undertaken using a semi micro Ross electrode (81-03,Orion Co.) in combination with a digital pH meter (720A, OrionCo.). The set-up was calibrated using at least four commercial buf-fer solutions (Merck). All FFM materials were contacted prior to thebatch-type experiments with fresh Grimsel ground water in Zins-ser vials (20 mL, HDPE) over a period of 6 weeks, exchanging thesolution at least 5 times within this period. Aliquots of supernatantsolution were taken for analysis, monitoring the equilibration
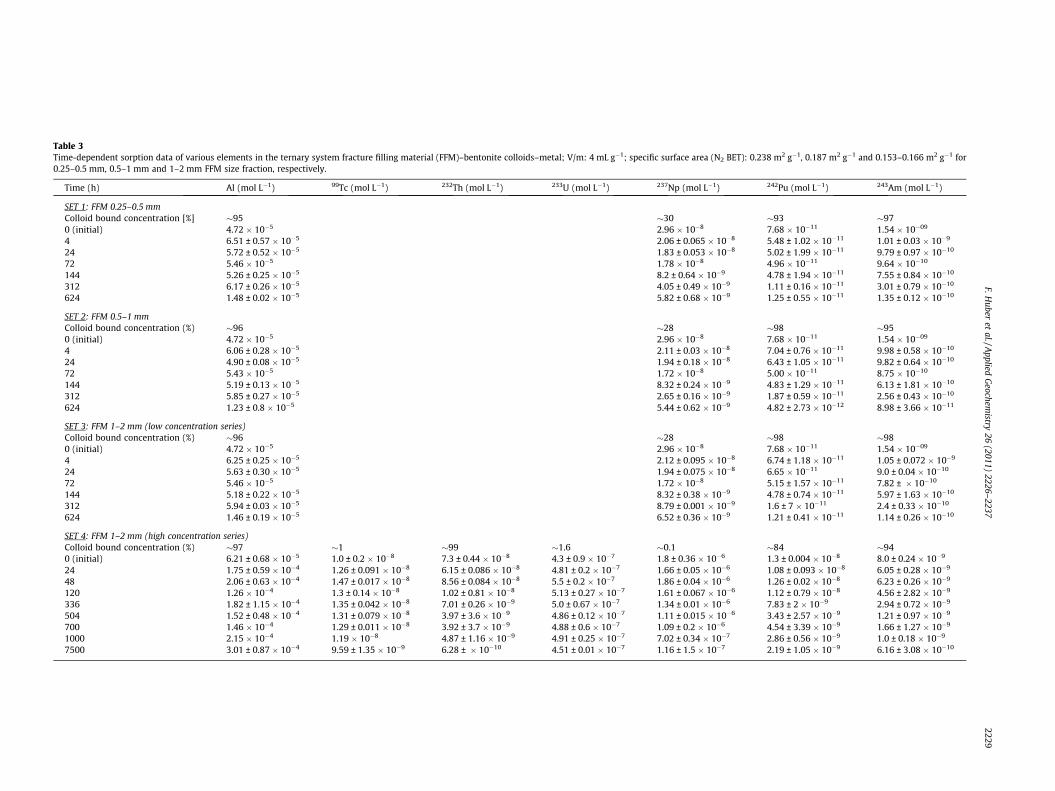
Table 3Time-dependent sorption data of various elements in the ternary system fracture filling material (FFM)–bentonite colloids–metal; V/m: 4 mL g�1; specific surface area (N2 BET): 0.238 m2 g�1, 0.187 m2 g�1 and 0.153–0.166 m2 g�1 for0.25–0.5 mm, 0.5–1 mm and 1–2 mm FFM size fraction, respectively.
Time (h) Al (mol L�1) 99Tc (mol L�1) 232Th (mol L�1) 233U (mol L�1) 237Np (mol L�1) 242Pu (mol L�1) 243Am (mol L�1)
SET 1: FFM 0.25–0.5 mmColloid bound concentration [%] �95 �30 �93 �970 (initial) 4.72 � 10�5 2.96 � 10�8 7.68 � 10�11 1.54 � 10�09
4 6.51 ± 0.57 � 10�5 2.06 ± 0.065 � 10�8 5.48 ± 1.02 � 10�11 1.01 ± 0.03 � 10�9
24 5.72 ± 0.52 � 10�5 1.83 ± 0.053 � 10�8 5.02 ± 1.99 � 10�11 9.79 ± 0.97 � 10�10
72 5.46 � 10�5 1.78 � 10�8 4.96 � 10�11 9.64 � 10�10
144 5.26 ± 0.25 � 10�5 8.2 ± 0.64 � 10�9 4.78 ± 1.94 � 10�11 7.55 ± 0.84 � 10�10
312 6.17 ± 0.26 � 10�5 4.05 ± 0.49 � 10�9 1.11 ± 0.16 � 10�11 3.01 ± 0.79 � 10�10
624 1.48 ± 0.02 � 10�5 5.82 ± 0.68 � 10�9 1.25 ± 0.55 � 10�11 1.35 ± 0.12 � 10�10
SET 2: FFM 0.5–1 mmColloid bound concentration (%) �96 �28 �98 �950 (initial) 4.72 � 10�5 2.96 � 10�8 7.68 � 10�11 1.54 � 10�09
4 6.06 ± 0.28 � 10�5 2.11 ± 0.03 � 10�8 7.04 ± 0.76 � 10�11 9.98 ± 0.58 � 10�10
24 4.90 ± 0.08 � 10�5 1.94 ± 0.18 � 10�8 6.43 ± 1.05 � 10�11 9.82 ± 0.64 � 10�10
72 5.43 � 10�5 1.72 � 10�8 5.00 � 10�11 8.75 � 10�10
144 5.19 ± 0.13 � 10�5 8.32 ± 0.24 � 10�9 4.83 ± 1.29 � 10�11 6.13 ± 1.81 � 10�10
312 5.85 ± 0.27 � 10�5 2.65 ± 0.16 � 10�9 1.87 ± 0.59 � 10�11 2.56 ± 0.43 � 10�10
624 1.23 ± 0.8 � 10�5 5.44 ± 0.62 � 10�9 4.82 ± 2.73 � 10�12 8.98 ± 3.66 � 10�11
SET 3: FFM 1–2 mm (low concentration series)Colloid bound concentration (%) �96 �28 �98 �980 (initial) 4.72 � 10�5 2.96 � 10�8 7.68 � 10�11 1.54 � 10�09
4 6.25 ± 0.25 � 10�5 2.12 ± 0.095 � 10�8 6.74 ± 1.18 � 10�11 1.05 ± 0.072 � 10�9
24 5.63 ± 0.30 � 10�5 1.94 ± 0.075 � 10�8 6.65 � 10�11 9.0 ± 0.04 � 10�10
72 5.46 � 10�5 1.72 � 10�8 5.15 ± 1.57 � 10�11 7.82 ± � 10�10
144 5.18 ± 0.22 � 10�5 8.32 ± 0.38 � 10�9 4.78 ± 0.74 � 10�11 5.97 ± 1.63 � 10�10
312 5.94 ± 0.03 � 10�5 8.79 ± 0.001 � 10�9 1.6 ± 7 � 10�11 2.4 ± 0.33 � 10�10
624 1.46 ± 0.19 � 10�5 6.52 ± 0.36 � 10�9 1.21 ± 0.41 � 10�11 1.14 ± 0.26 � 10�10
SET 4: FFM 1–2 mm (high concentration series)Colloid bound concentration (%) �97 �1 �99 �1.6 �0.1 �84 �940 (initial) 6.21 ± 0.68 � 10�5 1.0 ± 0.2 � 10�8 7.3 ± 0.44 � 10�8 4.3 ± 0.9 � 10�7 1.8 ± 0.36 � 10�6 1.3 ± 0.004 � 10�8 8.0 ± 0.24 � 10�9
24 1.75 ± 0.59 � 10�4 1.26 ± 0.091 � 10�8 6.15 ± 0.086 � 10�8 4.81 ± 0.2 � 10�7 1.66 ± 0.05 � 10�6 1.08 ± 0.093 � 10�8 6.05 ± 0.28 � 10�9
48 2.06 ± 0.63 � 10�4 1.47 ± 0.017 � 10�8 8.56 ± 0.084 � 10�8 5.5 ± 0.2 � 10�7 1.86 ± 0.04 � 10�6 1.26 ± 0.02 � 10�8 6.23 ± 0.26 � 10�9
120 1.26 � 10�4 1.3 ± 0.14 � 10�8 1.02 ± 0.81 � 10�8 5.13 ± 0.27 � 10�7 1.61 ± 0.067 � 10�6 1.12 ± 0.79 � 10�8 4.56 ± 2.82 � 10�9
336 1.82 ± 1.15 � 10�4 1.35 ± 0.042 � 10�8 7.01 ± 0.26 � 10�9 5.0 ± 0.67 � 10�7 1.34 ± 0.01 � 10�6 7.83 ± 2 � 10�9 2.94 ± 0.72 � 10�9
504 1.52 ± 0.48 � 10�4 1.31 ± 0.079 � 10�8 3.97 ± 3.6 � 10�9 4.86 ± 0.12 � 10�7 1.11 ± 0.015 � 10�6 3.43 ± 2.57 � 10�9 1.21 ± 0.97 � 10�9
700 1.46 � 10�4 1.29 ± 0.011 � 10�8 3.92 ± 3.7 � 10�9 4.88 ± 0.6 � 10�7 1.09 ± 0.2 � 10�6 4.54 ± 3.39 � 10�9 1.66 ± 1.27 � 10�9
1000 2.15 � 10�4 1.19 � 10�8 4.87 ± 1.16 � 10�9 4.91 ± 0.25 � 10�7 7.02 ± 0.34 � 10�7 2.86 ± 0.56 � 10�9 1.0 ± 0.18 � 10�9
7500 3.01 ± 0.87 � 10�4 9.59 ± 1.35 � 10�9 6.28 ± � 10�10 4.51 ± 0.01 � 10�7 1.16 ± 1.5 � 10�7 2.19 ± 1.05 � 10�9 6.16 ± 3.08 � 10�10
F.Huber
etal./A
ppliedG
eochemistry
26(2011)
2226–2237
2229
2230 F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237
process. The solid to liquid ratio was 1:4 (g m L�1) for all experi-ments. After this equilibration phase the supernatant was ex-changed with the radionuclide cocktail. The influence of the FFMon the initial Grimsel ground water element concentrations wasmonitored by ICP-MS measurements (Table 2). Triplicates of everysample were prepared: 2 of the 3 identical samples with 2 g solidand 8 mL of cocktail solution and one sample with 4 g solid and16 mL of solution. First aliquots were taken 1 h after spiking ofthe samples, the next aliquots after 24 h, 48 h, 120 h, 336 h,504 h, 700 h, 1000 h and 7500 h, respectively. An aliquot of everysample (�4 mL) was ultra-centrifuged (Beckman XL-90, rotor type90Ti) at 90,000 rpm (centrifugal force of about 5 � 105 g) for60 min to remove colloid associated radionuclides. This ultra-cen-trifugation procedure has been proven to be suitable to effectivelyremove bentonite colloids as well as Th(IV) ‘‘eigencolloids’’ fromsolution (Altmaier et al., 2004). The remaining supernatant wassubsequently analyzed by ICP-MS as described in detail above.
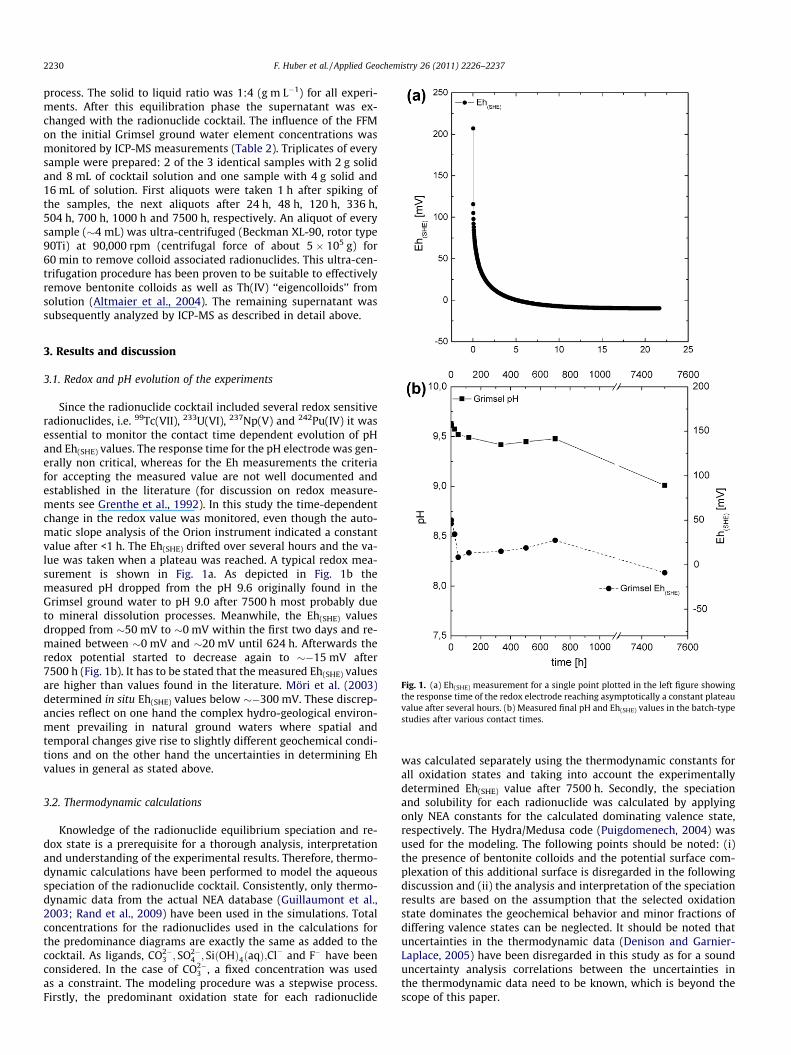
Fig. 1. (a) Eh(SHE) measurement for a single point plotted in the left figure showingthe response time of the redox electrode reaching asymptotically a constant plateauvalue after several hours. (b) Measured final pH and Eh(SHE) values in the batch-typestudies after various contact times.
3. Results and discussion
3.1. Redox and pH evolution of the experiments
Since the radionuclide cocktail included several redox sensitiveradionuclides, i.e. 99Tc(VII), 233U(VI), 237Np(V) and 242Pu(IV) it wasessential to monitor the contact time dependent evolution of pHand Eh(SHE) values. The response time for the pH electrode was gen-erally non critical, whereas for the Eh measurements the criteriafor accepting the measured value are not well documented andestablished in the literature (for discussion on redox measure-ments see Grenthe et al., 1992). In this study the time-dependentchange in the redox value was monitored, even though the auto-matic slope analysis of the Orion instrument indicated a constantvalue after <1 h. The Eh(SHE) drifted over several hours and the va-lue was taken when a plateau was reached. A typical redox mea-surement is shown in Fig. 1a. As depicted in Fig. 1b themeasured pH dropped from the pH 9.6 originally found in theGrimsel ground water to pH 9.0 after 7500 h most probably dueto mineral dissolution processes. Meanwhile, the Eh(SHE) valuesdropped from �50 mV to �0 mV within the first two days and re-mained between �0 mV and �20 mV until 624 h. Afterwards theredox potential started to decrease again to ��15 mV after7500 h (Fig. 1b). It has to be stated that the measured Eh(SHE) valuesare higher than values found in the literature. Möri et al. (2003)determined in situ Eh(SHE) values below ��300 mV. These discrep-ancies reflect on one hand the complex hydro-geological environ-ment prevailing in natural ground waters where spatial andtemporal changes give rise to slightly different geochemical condi-tions and on the other hand the uncertainties in determining Ehvalues in general as stated above.
3.2. Thermodynamic calculations
Knowledge of the radionuclide equilibrium speciation and re-dox state is a prerequisite for a thorough analysis, interpretationand understanding of the experimental results. Therefore, thermo-dynamic calculations have been performed to model the aqueousspeciation of the radionuclide cocktail. Consistently, only thermo-dynamic data from the actual NEA database (Guillaumont et al.,2003; Rand et al., 2009) have been used in the simulations. Totalconcentrations for the radionuclides used in the calculations forthe predominance diagrams are exactly the same as added to thecocktail. As ligands, CO2�
3 ; SO2�4 ; SiðOHÞ4ðaqÞ;Cl� and F� have been
considered. In the case of CO2�3 , a fixed concentration was used
as a constraint. The modeling procedure was a stepwise process.Firstly, the predominant oxidation state for each radionuclide
was calculated separately using the thermodynamic constants forall oxidation states and taking into account the experimentallydetermined Eh(SHE) value after 7500 h. Secondly, the speciationand solubility for each radionuclide was calculated by applyingonly NEA constants for the calculated dominating valence state,respectively. The Hydra/Medusa code (Puigdomenech, 2004) wasused for the modeling. The following points should be noted: (i)the presence of bentonite colloids and the potential surface com-plexation of this additional surface is disregarded in the followingdiscussion and (ii) the analysis and interpretation of the speciationresults are based on the assumption that the selected oxidationstate dominates the geochemical behavior and minor fractions ofdiffering valence states can be neglected. It should be noted thatuncertainties in the thermodynamic data (Denison and Garnier-Laplace, 2005) have been disregarded in this study as for a sounduncertainty analysis correlations between the uncertainties inthe thermodynamic data need to be known, which is beyond thescope of this paper.
F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237 2231
Technicium-99 is mainly present as pertechnetate Tc(VII)O)4� un-der the experimental conditions. A reduction to the tetravalent TcO2
begins at values below ��100 mV for a pH around 9.7 which is ingood agreement with calculations published in Neck et al. (1999).As the solubility limiting solid phase TcO2(cr) was selected, whichhas a solubility in the range of log(c) � �5 to �4 between pH 9and 10, respectively. As the initial Tc concentration is at log(c) = �8no precipitation of TcO2(cr) is expected in the experiments.
The redox stable 232Th occurs in the tetravalent form in naturalaquatic systems. Thermodynamic calculations yield Th(OH)4(aq) asthe predominant solution species over a broad pH range includingpH 9.7 and the solution is under-saturated with respect to Th(O-H)4(am) under the experimental conditions.
Results for U yield the uranyl–hydroxo-complex UO2ðOHÞ�3 asdominant species under the prevailing pH/Eh conditions. Reduc-tion of hexavalent U to amorphous U(IV)O2 is thermodynamicallyfeasible only at Eh values lower than �210 mV between pH 9and 10. As solubility limiting solid phases both UO2(OH)2(am)and UO2CO3(cr) have been chosen yielding no over-saturation.The recently discovered Ca2UO2(CO3)3 (Bernhard et al., 2001) com-plex is not included in the thermodynamic calculations since thiscomplex has not yet been selected by the NEA review.
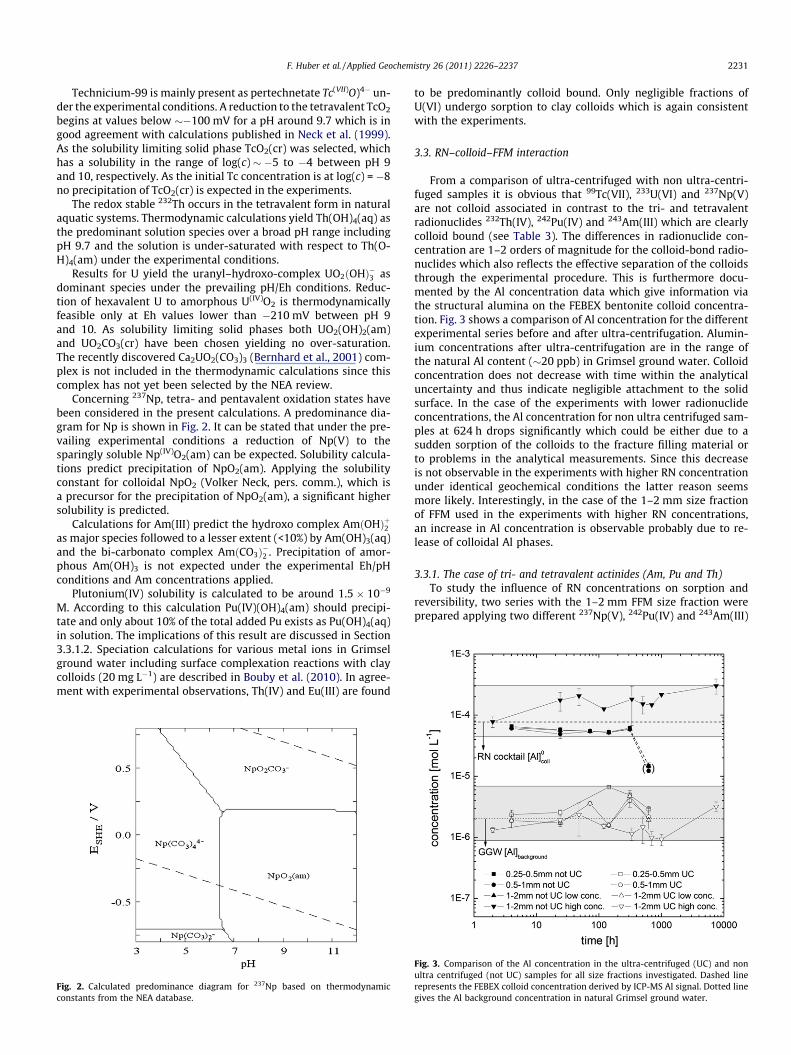
Concerning 237Np, tetra- and pentavalent oxidation states havebeen considered in the present calculations. A predominance dia-gram for Np is shown in Fig. 2. It can be stated that under the pre-vailing experimental conditions a reduction of Np(V) to thesparingly soluble Np(IV)O2(am) can be expected. Solubility calcula-tions predict precipitation of NpO2(am). Applying the solubilityconstant for colloidal NpO2 (Volker Neck, pers. comm.), which isa precursor for the precipitation of NpO2(am), a significant highersolubility is predicted.
Calculations for Am(III) predict the hydroxo complex AmðOHÞþ2as major species followed to a lesser extent (<10%) by Am(OH)3(aq)and the bi-carbonato complex AmðCO3Þ�2 . Precipitation of amor-phous Am(OH)3 is not expected under the experimental Eh/pHconditions and Am concentrations applied.
Plutonium(IV) solubility is calculated to be around 1.5 � 10�9
M. According to this calculation Pu(IV)(OH)4(am) should precipi-tate and only about 10% of the total added Pu exists as Pu(OH)4(aq)in solution. The implications of this result are discussed in Section3.3.1.2. Speciation calculations for various metal ions in Grimselground water including surface complexation reactions with claycolloids (20 mg L�1) are described in Bouby et al. (2010). In agree-ment with experimental observations, Th(IV) and Eu(III) are found
Fig. 2. Calculated predominance diagram for 237Np based on thermodynamicconstants from the NEA database.
to be predominantly colloid bound. Only negligible fractions ofU(VI) undergo sorption to clay colloids which is again consistentwith the experiments.
3.3. RN–colloid–FFM interaction
From a comparison of ultra-centrifuged with non ultra-centri-fuged samples it is obvious that 99Tc(VII), 233U(VI) and 237Np(V)are not colloid associated in contrast to the tri- and tetravalentradionuclides 232Th(IV), 242Pu(IV) and 243Am(III) which are clearlycolloid bound (see Table 3). The differences in radionuclide con-centration are 1–2 orders of magnitude for the colloid-bond radio-nuclides which also reflects the effective separation of the colloidsthrough the experimental procedure. This is furthermore docu-mented by the Al concentration data which give information viathe structural alumina on the FEBEX bentonite colloid concentra-tion. Fig. 3 shows a comparison of Al concentration for the differentexperimental series before and after ultra-centrifugation. Alumin-ium concentrations after ultra-centrifugation are in the range ofthe natural Al content (�20 ppb) in Grimsel ground water. Colloidconcentration does not decrease with time within the analyticaluncertainty and thus indicate negligible attachment to the solidsurface. In the case of the experiments with lower radionuclideconcentrations, the Al concentration for non ultra centrifuged sam-ples at 624 h drops significantly which could be either due to asudden sorption of the colloids to the fracture filling material orto problems in the analytical measurements. Since this decreaseis not observable in the experiments with higher RN concentrationunder identical geochemical conditions the latter reason seemsmore likely. Interestingly, in the case of the 1–2 mm size fractionof FFM used in the experiments with higher RN concentrations,an increase in Al concentration is observable probably due to re-lease of colloidal Al phases.
3.3.1. The case of tri- and tetravalent actinides (Am, Pu and Th)To study the influence of RN concentrations on sorption and
reversibility, two series with the 1–2 mm FFM size fraction wereprepared applying two different 237Np(V), 242Pu(IV) and 243Am(III)
Fig. 3. Comparison of the Al concentration in the ultra-centrifuged (UC) and nonultra centrifuged (not UC) samples for all size fractions investigated. Dashed linerepresents the FEBEX colloid concentration derived by ICP-MS Al signal. Dotted linegives the Al background concentration in natural Grimsel ground water.
2232 F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237
concentrations. In both sample sets the same solid to solution ratio(1:4 g m L�1) has been used. For the experiments with higher radio-nuclide concentration two samples have been prepared for longterm measurements after 7500 h. In addition to Pu(IV), Th(IV) waschosen as a homologue for tetravalent actinides. Fig. 4 shows the
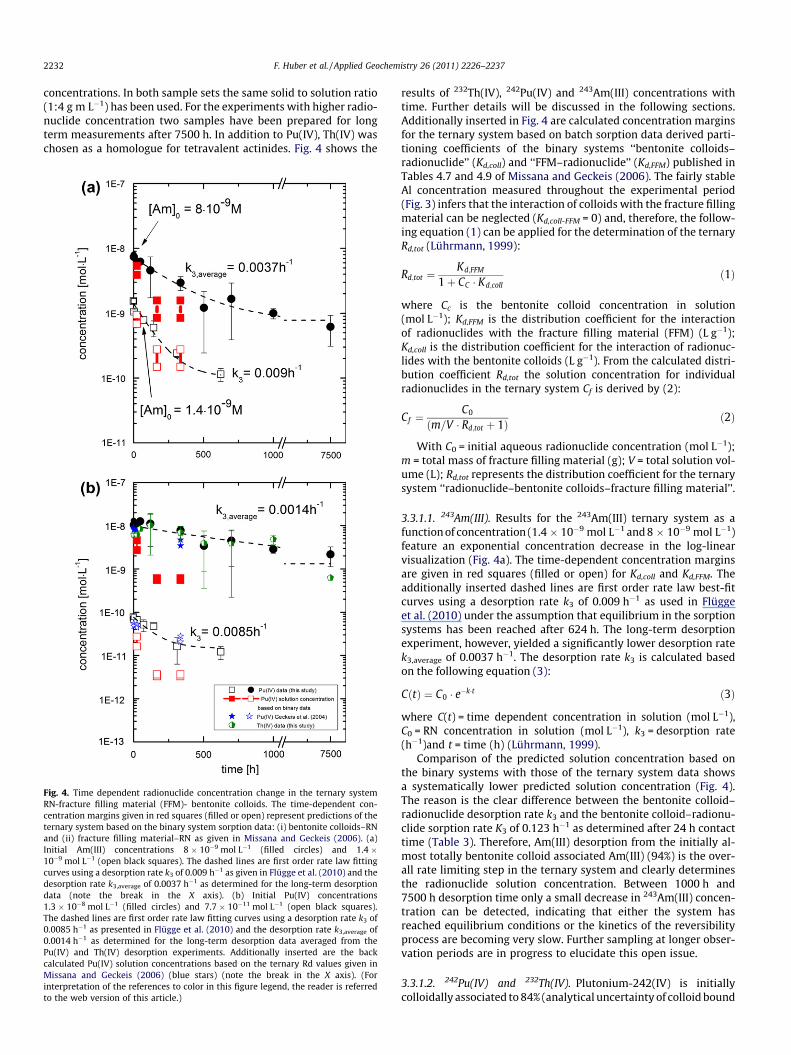
Fig. 4. Time dependent radionuclide concentration change in the ternary systemRN-fracture filling material (FFM)- bentonite colloids. The time-dependent con-centration margins given in red squares (filled or open) represent predictions of theternary system based on the binary system sorption data: (i) bentonite colloids–RNand (ii) fracture filling material–RN as given in Missana and Geckeis (2006). (a)Initial Am(III) concentrations 8 � 10�9 mol L�1 (filled circles) and 1.4 �10�9 mol L�1 (open black squares). The dashed lines are first order rate law fittingcurves using a desorption rate k3 of 0.009 h�1 as given in Flügge et al. (2010) and thedesorption rate k3,average of 0.0037 h�1 as determined for the long-term desorptiondata (note the break in the X axis). (b) Initial Pu(IV) concentrations1.3 � 10�8 mol L�1 (filled circles) and 7.7 � 10�11 mol L�1 (open black squares).The dashed lines are first order rate law fitting curves using a desorption rate k3 of0.0085 h�1 as presented in Flügge et al. (2010) and the desorption rate k3,average of0.0014 h�1 as determined for the long-term desorption data averaged from thePu(IV) and Th(IV) desorption experiments. Additionally inserted are the backcalculated Pu(IV) solution concentrations based on the ternary Rd values given inMissana and Geckeis (2006) (blue stars) (note the break in the X axis). (Forinterpretation of the references to color in this figure legend, the reader is referredto the web version of this article.)
results of 232Th(IV), 242Pu(IV) and 243Am(III) concentrations withtime. Further details will be discussed in the following sections.Additionally inserted in Fig. 4 are calculated concentration marginsfor the ternary system based on batch sorption data derived parti-tioning coefficients of the binary systems ‘‘bentonite colloids–radionuclide’’ (Kd,coll) and ‘‘FFM–radionuclide’’ (Kd,FFM) published inTables 4.7 and 4.9 of Missana and Geckeis (2006). The fairly stableAl concentration measured throughout the experimental period(Fig. 3) infers that the interaction of colloids with the fracture fillingmaterial can be neglected (Kd,coll-FFM = 0) and, therefore, the follow-ing equation (1) can be applied for the determination of the ternaryRd,tot (Lührmann, 1999):
Rd;tot ¼Kd;FFM
1þ CC � Kd;collð1Þ
where Cc is the bentonite colloid concentration in solution(mol L�1); Kd,FFM is the distribution coefficient for the interactionof radionuclides with the fracture filling material (FFM) (L g�1);Kd,coll is the distribution coefficient for the interaction of radionuc-lides with the bentonite colloids (L g�1). From the calculated distri-bution coefficient Rd,tot the solution concentration for individualradionuclides in the ternary system Cf is derived by (2):
Cf ¼C0
ðm=V � Rd;tot þ 1Þ ð2Þ
With C0 = initial aqueous radionuclide concentration (mol L�1);m = total mass of fracture filling material (g); V = total solution vol-ume (L); Rd,tot represents the distribution coefficient for the ternarysystem ‘‘radionuclide–bentonite colloids–fracture filling material’’.
3.3.1.1. 243Am(III). Results for the 243Am(III) ternary system as afunction of concentration (1.4 � 10�9 mol L�1 and 8 � 10�9 mol L�1)feature an exponential concentration decrease in the log-linearvisualization (Fig. 4a). The time-dependent concentration marginsare given in red squares (filled or open) for Kd,coll and Kd,FFM. Theadditionally inserted dashed lines are first order rate law best-fitcurves using a desorption rate k3 of 0.009 h�1 as used in Flüggeet al. (2010) under the assumption that equilibrium in the sorptionsystems has been reached after 624 h. The long-term desorptionexperiment, however, yielded a significantly lower desorption ratek3,average of 0.0037 h�1. The desorption rate k3 is calculated basedon the following equation (3):
CðtÞ ¼ C0 � e�k�t ð3Þ
where C(t) = time dependent concentration in solution (mol L�1),C0 = RN concentration in solution (mol L�1), k3 = desorption rate(h�1)and t = time (h) (Lührmann, 1999).
Comparison of the predicted solution concentration based onthe binary systems with those of the ternary system data showsa systematically lower predicted solution concentration (Fig. 4).The reason is the clear difference between the bentonite colloid–radionuclide desorption rate k3 and the bentonite colloid–radionu-clide sorption rate K3 of 0.123 h�1 as determined after 24 h contacttime (Table 3). Therefore, Am(III) desorption from the initially al-most totally bentonite colloid associated Am(III) (94%) is the over-all rate limiting step in the ternary system and clearly determinesthe radionuclide solution concentration. Between 1000 h and7500 h desorption time only a small decrease in 243Am(III) concen-tration can be detected, indicating that either the system hasreached equilibrium conditions or the kinetics of the reversibilityprocess are becoming very slow. Further sampling at longer obser-vation periods are in progress to elucidate this open issue.
3.3.1.2. 242Pu(IV) and 232Th(IV). Plutonium-242(IV) is initiallycolloidally associated to 84% (analytical uncertainty of colloid bound
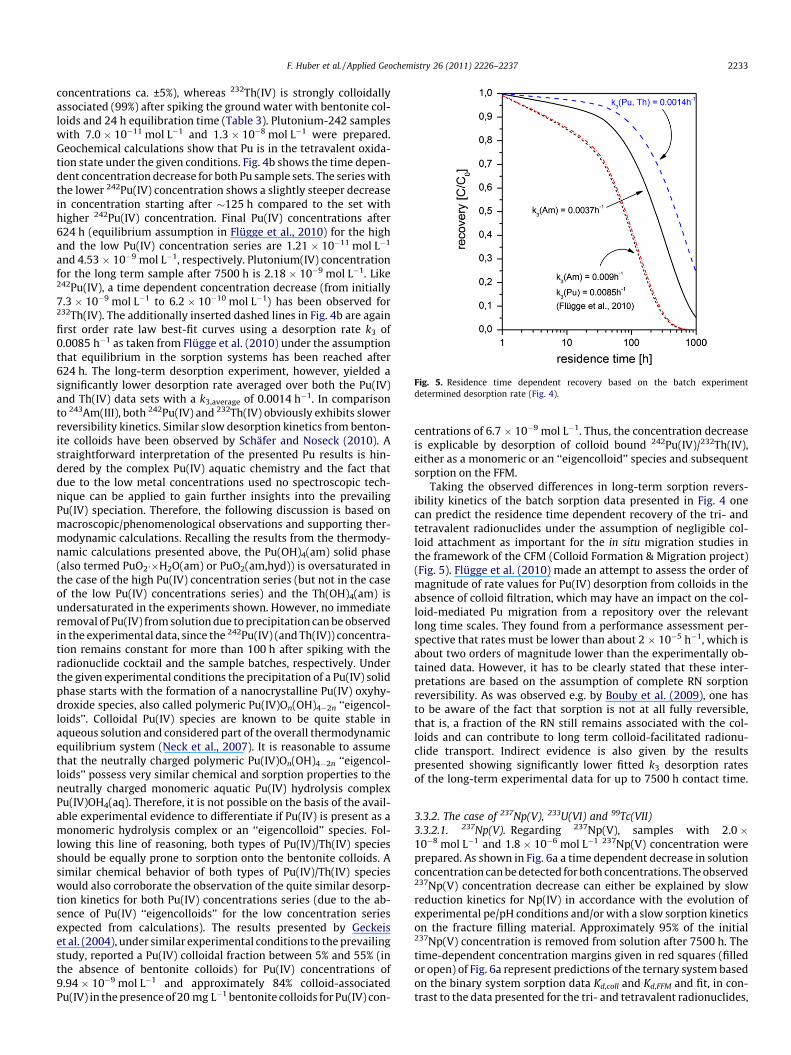
Fig. 5. Residence time dependent recovery based on the batch experimentdetermined desorption rate (Fig. 4).
F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237 2233
concentrations ca. ±5%), whereas 232Th(IV) is strongly colloidallyassociated (99%) after spiking the ground water with bentonite col-loids and 24 h equilibration time (Table 3). Plutonium-242 sampleswith 7.0 � 10�11 mol L�1 and 1.3 � 10�8 mol L�1 were prepared.Geochemical calculations show that Pu is in the tetravalent oxida-tion state under the given conditions. Fig. 4b shows the time depen-dent concentration decrease for both Pu sample sets. The series withthe lower 242Pu(IV) concentration shows a slightly steeper decreasein concentration starting after �125 h compared to the set withhigher 242Pu(IV) concentration. Final Pu(IV) concentrations after624 h (equilibrium assumption in Flügge et al., 2010) for the highand the low Pu(IV) concentration series are 1.21 � 10�11 mol L�1
and 4.53 � 10�9 mol L�1, respectively. Plutonium(IV) concentrationfor the long term sample after 7500 h is 2.18 � 10�9 mol L�1. Like242Pu(IV), a time dependent concentration decrease (from initially7.3 � 10�9 mol L�1 to 6.2 � 10�10 mol L�1) has been observed for232Th(IV). The additionally inserted dashed lines in Fig. 4b are againfirst order rate law best-fit curves using a desorption rate k3 of0.0085 h�1 as taken from Flügge et al. (2010) under the assumptionthat equilibrium in the sorption systems has been reached after624 h. The long-term desorption experiment, however, yielded asignificantly lower desorption rate averaged over both the Pu(IV)and Th(IV) data sets with a k3,average of 0.0014 h�1. In comparisonto 243Am(III), both 242Pu(IV) and 232Th(IV) obviously exhibits slowerreversibility kinetics. Similar slow desorption kinetics from benton-ite colloids have been observed by Schäfer and Noseck (2010). Astraightforward interpretation of the presented Pu results is hin-dered by the complex Pu(IV) aquatic chemistry and the fact thatdue to the low metal concentrations used no spectroscopic tech-nique can be applied to gain further insights into the prevailingPu(IV) speciation. Therefore, the following discussion is based onmacroscopic/phenomenological observations and supporting ther-modynamic calculations. Recalling the results from the thermody-namic calculations presented above, the Pu(OH)4(am) solid phase(also termed PuO2��H2O(am) or PuO2(am,hyd)) is oversaturated inthe case of the high Pu(IV) concentration series (but not in the caseof the low Pu(IV) concentrations series) and the Th(OH)4(am) isundersaturated in the experiments shown. However, no immediateremoval of Pu(IV) from solution due to precipitation can be observedin the experimental data, since the 242Pu(IV) (and Th(IV)) concentra-tion remains constant for more than 100 h after spiking with theradionuclide cocktail and the sample batches, respectively. Underthe given experimental conditions the precipitation of a Pu(IV) solidphase starts with the formation of a nanocrystalline Pu(IV) oxyhy-droxide species, also called polymeric Pu(IV)On(OH)4�2n ‘‘eigencol-loids’’. Colloidal Pu(IV) species are known to be quite stable inaqueous solution and considered part of the overall thermodynamicequilibrium system (Neck et al., 2007). It is reasonable to assumethat the neutrally charged polymeric Pu(IV)On(OH)4�2n ‘‘eigencol-loids’’ possess very similar chemical and sorption properties to theneutrally charged monomeric aquatic Pu(IV) hydrolysis complexPu(IV)OH4(aq). Therefore, it is not possible on the basis of the avail-able experimental evidence to differentiate if Pu(IV) is present as amonomeric hydrolysis complex or an ‘‘eigencolloid’’ species. Fol-lowing this line of reasoning, both types of Pu(IV)/Th(IV) speciesshould be equally prone to sorption onto the bentonite colloids. Asimilar chemical behavior of both types of Pu(IV)/Th(IV) specieswould also corroborate the observation of the quite similar desorp-tion kinetics for both Pu(IV) concentrations series (due to the ab-sence of Pu(IV) ‘‘eigencolloids’’ for the low concentration seriesexpected from calculations). The results presented by Geckeiset al. (2004), under similar experimental conditions to the prevailingstudy, reported a Pu(IV) colloidal fraction between 5% and 55% (inthe absence of bentonite colloids) for Pu(IV) concentrations of9.94 � 10�9 mol L�1 and approximately 84% colloid-associatedPu(IV) in the presence of 20 mg L�1 bentonite colloids for Pu(IV) con-
centrations of 6.7 � 10�9 mol L�1. Thus, the concentration decreaseis explicable by desorption of colloid bound 242Pu(IV)/232Th(IV),either as a monomeric or an ‘‘eigencolloid’’ species and subsequentsorption on the FFM.
Taking the observed differences in long-term sorption revers-ibility kinetics of the batch sorption data presented in Fig. 4 onecan predict the residence time dependent recovery of the tri- andtetravalent radionuclides under the assumption of negligible col-loid attachment as important for the in situ migration studies inthe framework of the CFM (Colloid Formation & Migration project)(Fig. 5). Flügge et al. (2010) made an attempt to assess the order ofmagnitude of rate values for Pu(IV) desorption from colloids in theabsence of colloid filtration, which may have an impact on the col-loid-mediated Pu migration from a repository over the relevantlong time scales. They found from a performance assessment per-spective that rates must be lower than about 2 � 10�5 h�1, which isabout two orders of magnitude lower than the experimentally ob-tained data. However, it has to be clearly stated that these inter-pretations are based on the assumption of complete RN sorptionreversibility. As was observed e.g. by Bouby et al. (2009), one hasto be aware of the fact that sorption is not at all fully reversible,that is, a fraction of the RN still remains associated with the col-loids and can contribute to long term colloid-facilitated radionu-clide transport. Indirect evidence is also given by the resultspresented showing significantly lower fitted k3 desorption ratesof the long-term experimental data for up to 7500 h contact time.
3.3.2. The case of 237Np(V), 233U(VI) and 99Tc(VII)3.3.2.1. 237Np(V). Regarding 237Np(V), samples with 2.0 �10�8 mol L�1 and 1.8 � 10�6 mol L�1 237Np(V) concentration wereprepared. As shown in Fig. 6a a time dependent decrease in solutionconcentration can be detected for both concentrations. The observed237Np(V) concentration decrease can either be explained by slowreduction kinetics for Np(IV) in accordance with the evolution ofexperimental pe/pH conditions and/or with a slow sorption kineticson the fracture filling material. Approximately 95% of the initial237Np(V) concentration is removed from solution after 7500 h. Thetime-dependent concentration margins given in red squares (filledor open) of Fig. 6a represent predictions of the ternary system basedon the binary system sorption data Kd,coll and Kd,FFM and fit, in con-trast to the data presented for the tri- and tetravalent radionuclides,
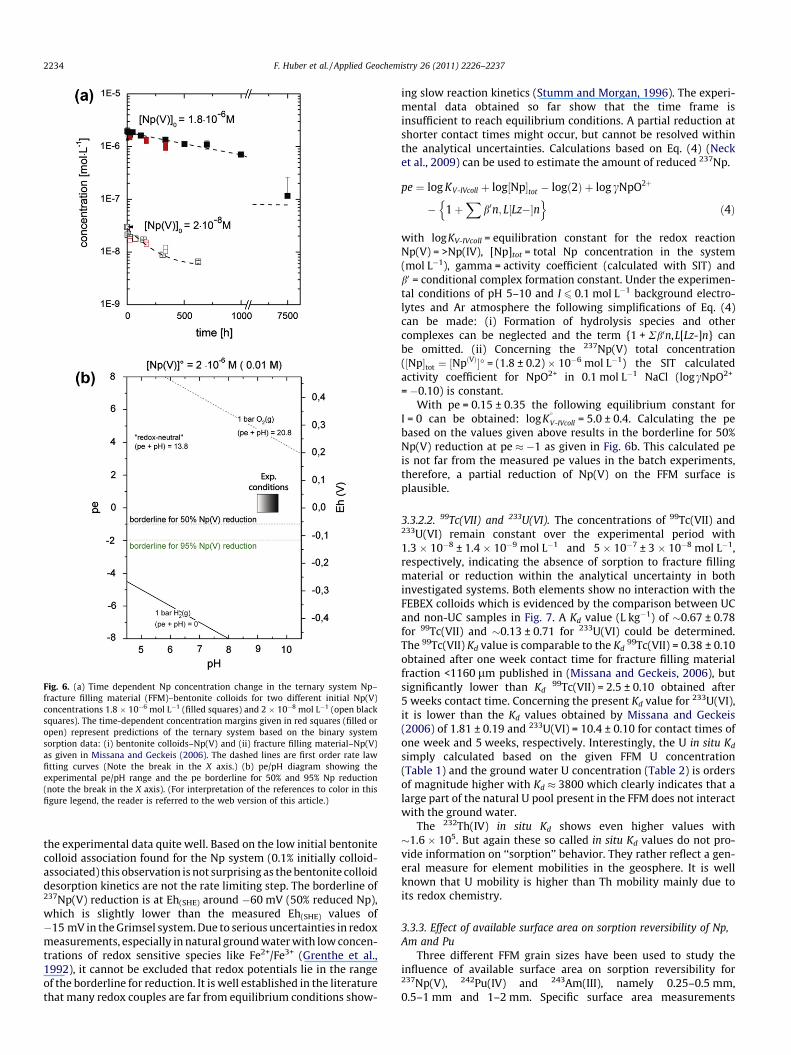
Fig. 6. (a) Time dependent Np concentration change in the ternary system Np–fracture filling material (FFM)–bentonite colloids for two different initial Np(V)concentrations 1.8 � 10�6 mol L�1 (filled squares) and 2 � 10�8 mol L�1 (open blacksquares). The time-dependent concentration margins given in red squares (filled oropen) represent predictions of the ternary system based on the binary systemsorption data: (i) bentonite colloids–Np(V) and (ii) fracture filling material–Np(V)as given in Missana and Geckeis (2006). The dashed lines are first order rate lawfitting curves (Note the break in the X axis.) (b) pe/pH diagram showing theexperimental pe/pH range and the pe borderline for 50% and 95% Np reduction(note the break in the X axis). (For interpretation of the references to color in thisfigure legend, the reader is referred to the web version of this article.)
2234 F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237
the experimental data quite well. Based on the low initial bentonitecolloid association found for the Np system (0.1% initially colloid-associated) this observation is not surprising as the bentonite colloiddesorption kinetics are not the rate limiting step. The borderline of237Np(V) reduction is at Eh(SHE) around �60 mV (50% reduced Np),which is slightly lower than the measured Eh(SHE) values of�15 mV in the Grimsel system. Due to serious uncertainties in redoxmeasurements, especially in natural ground water with low concen-trations of redox sensitive species like Fe2+/Fe3+ (Grenthe et al.,1992), it cannot be excluded that redox potentials lie in the rangeof the borderline for reduction. It is well established in the literaturethat many redox couples are far from equilibrium conditions show-
ing slow reaction kinetics (Stumm and Morgan, 1996). The experi-mental data obtained so far show that the time frame isinsufficient to reach equilibrium conditions. A partial reduction atshorter contact times might occur, but cannot be resolved withinthe analytical uncertainties. Calculations based on Eq. (4) (Necket al., 2009) can be used to estimate the amount of reduced 237Np.
pe ¼ log KV-IVcoll þ log½Np�tot � logð2Þ þ log cNpO2þ
� 1þX
b0n; L½Lz��nn o
ð4Þ
with logKV-IVcoll = equilibration constant for the redox reactionNp(V) = >Np(IV), [Np]tot = total Np concentration in the system(mol L�1), gamma = activity coefficient (calculated with SIT) andb0 = conditional complex formation constant. Under the experimen-tal conditions of pH 5–10 and I 6 0.1 mol L�1 background electro-lytes and Ar atmosphere the following simplifications of Eq. (4)can be made: (i) Formation of hydrolysis species and othercomplexes can be neglected and the term {1 + Rb0n,L[Lz-]n} canbe omitted. (ii) Concerning the 237Np(V) total concentration(½Np�tot ¼ ½NpðVÞ�� = (1.8 ± 0.2) � 10�6 mol L�1) the SIT calculatedactivity coefficient for NpO2+ in 0.1 mol L�1 NaCl (logcNpO2+
= �0.10) is constant.With pe = 0.15 ± 0.35 the following equilibrium constant for
I = 0 can be obtained: logK
V-IVcoll = 5.0 ± 0.4. Calculating the pebased on the values given above results in the borderline for 50%Np(V) reduction at pe � �1 as given in Fig. 6b. This calculated peis not far from the measured pe values in the batch experiments,therefore, a partial reduction of Np(V) on the FFM surface isplausible.
3.3.2.2. 99Tc(VII) and 233U(VI). The concentrations of 99Tc(VII) and233U(VI) remain constant over the experimental period with1.3 � 10�8 ± 1.4 � 10�9 mol L�1 and 5 � 10�7 ± 3 � 10�8 mol L�1,respectively, indicating the absence of sorption to fracture fillingmaterial or reduction within the analytical uncertainty in bothinvestigated systems. Both elements show no interaction with theFEBEX colloids which is evidenced by the comparison between UCand non-UC samples in Fig. 7. A Kd value (L kg�1) of �0.67 ± 0.78for 99Tc(VII) and �0.13 ± 0.71 for 233U(VI) could be determined.The 99Tc(VII) Kd value is comparable to the Kd
99Tc(VII) = 0.38 ± 0.10obtained after one week contact time for fracture filling materialfraction <1160 lm published in (Missana and Geckeis, 2006), butsignificantly lower than Kd
99Tc(VII) = 2.5 ± 0.10 obtained after5 weeks contact time. Concerning the present Kd value for 233U(VI),it is lower than the Kd values obtained by Missana and Geckeis(2006) of 1.81 ± 0.19 and 233U(VI) = 10.4 ± 0.10 for contact times ofone week and 5 weeks, respectively. Interestingly, the U in situ Kd
simply calculated based on the given FFM U concentration(Table 1) and the ground water U concentration (Table 2) is ordersof magnitude higher with Kd � 3800 which clearly indicates that alarge part of the natural U pool present in the FFM does not interactwith the ground water.
The 232Th(IV) in situ Kd shows even higher values with�1.6 � 105. But again these so called in situ Kd values do not pro-vide information on ‘‘sorption’’ behavior. They rather reflect a gen-eral measure for element mobilities in the geosphere. It is wellknown that U mobility is higher than Th mobility mainly due toits redox chemistry.
3.3.3. Effect of available surface area on sorption reversibility of Np,Am and Pu
Three different FFM grain sizes have been used to study theinfluence of available surface area on sorption reversibility for237Np(V), 242Pu(IV) and 243Am(III), namely 0.25–0.5 mm,0.5–1 mm and 1–2 mm. Specific surface area measurements
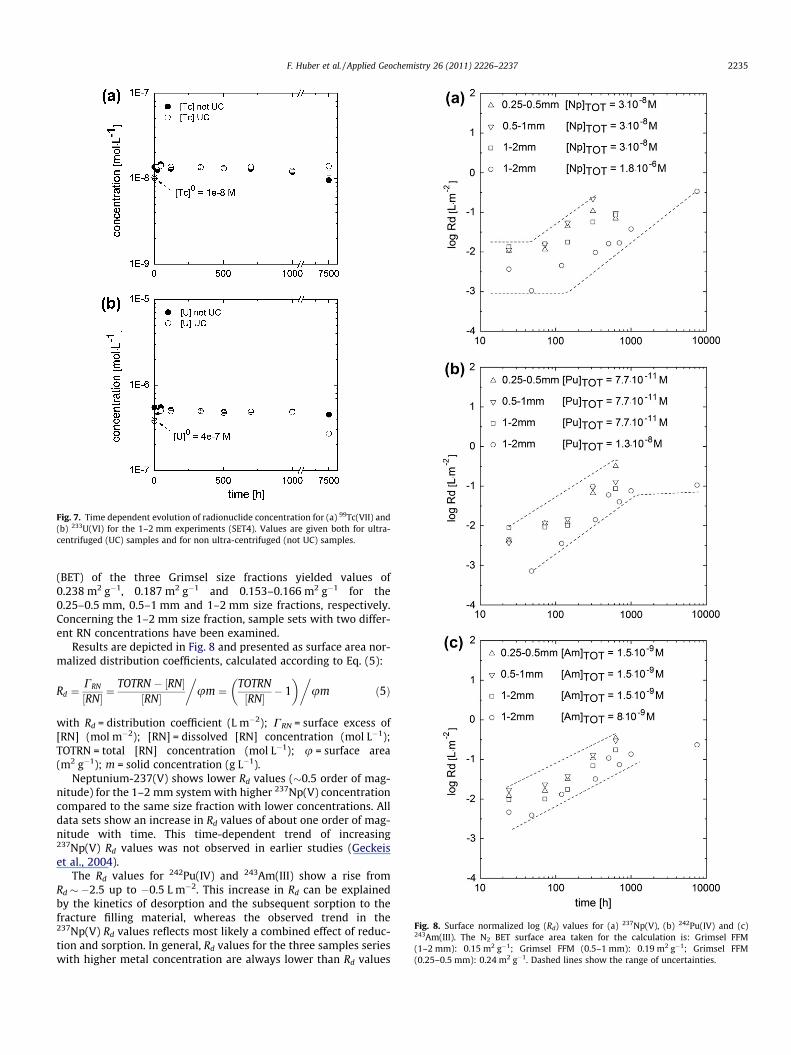
Fig. 8. Surface normalized log (Rd) values for (a) 237Np(V), (b) 242Pu(IV) and (c)243Am(III). The N2 BET surface area taken for the calculation is: Grimsel FFM(1–2 mm): 0.15 m2 g�1; Grimsel FFM (0.5–1 mm): 0.19 m2 g�1; Grimsel FFM(0.25–0.5 mm): 0.24 m2 g�1. Dashed lines show the range of uncertainties.
Fig. 7. Time dependent evolution of radionuclide concentration for (a) 99Tc(VII) and(b) 233U(VI) for the 1–2 mm experiments (SET4). Values are given both for ultra-centrifuged (UC) samples and for non ultra-centrifuged (not UC) samples.
F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237 2235
(BET) of the three Grimsel size fractions yielded values of0.238 m2 g�1, 0.187 m2 g�1 and 0.153–0.166 m2 g�1 for the0.25–0.5 mm, 0.5–1 mm and 1–2 mm size fractions, respectively.Concerning the 1–2 mm size fraction, sample sets with two differ-ent RN concentrations have been examined.
Results are depicted in Fig. 8 and presented as surface area nor-malized distribution coefficients, calculated according to Eq. (5):
Rd ¼CRN
½RN� ¼TOTRN � ½RN�
½RN�
�um ¼ TOTRN
½RN� � 1� ��
um ð5Þ
with Rd = distribution coefficient (L m�2); CRN = surface excess of[RN] (mol m�2); [RN] = dissolved [RN] concentration (mol L�1);TOTRN = total [RN] concentration (mol L�1); u = surface area(m2 g�1); m = solid concentration (g L�1).
Neptunium-237(V) shows lower Rd values (�0.5 order of mag-nitude) for the 1–2 mm system with higher 237Np(V) concentrationcompared to the same size fraction with lower concentrations. Alldata sets show an increase in Rd values of about one order of mag-nitude with time. This time-dependent trend of increasing237Np(V) Rd values was not observed in earlier studies (Geckeiset al., 2004).
The Rd values for 242Pu(IV) and 243Am(III) show a rise fromRd � �2.5 up to �0.5 L m�2. This increase in Rd can be explainedby the kinetics of desorption and the subsequent sorption to thefracture filling material, whereas the observed trend in the237Np(V) Rd values reflects most likely a combined effect of reduc-tion and sorption. In general, Rd values for the three samples serieswith higher metal concentration are always lower than Rd values

2236 F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237
for the three samples series with lower metal concentration. Thisobservation may a commonly observed non-linear sorption behav-ior. Rd values often increase at decreasing metal concentrationswhich is usually assigned to a weak and strong sorption process.The envelope curves given in Fig. 8 give an uncertainty marginfor the kinetics of actinide dissociation from FEBEX bentonite col-loid that can be used in reactive transport modeling (Noseck andKlotz, 2002). The surface normalized Rd values reflect no system-atic trend in terms of the available surface area and/or the miner-alogy of the FFM. The latter observation was expected since themineralogical composition of the three different FFM size fractionsis very similar based on the XRD results stated above.
4. Conclusions
Batch type experiments studying the reversibility of radionu-clide binding to bentonite colloids in the presence of fracture fill-ing material from Grimsel, of different grain sizes spanning arange from 0.25 to 2 mm, have been conducted using a cocktailof radionuclides spiked to natural ground water from the Grimseltest site. Technicium-99(VII), 233U(VI) and 237Np(V) are not colloi-dally associated under the experimental conditions. Concentra-tions of 99Tc(VII) and 233U(VI) remain constant over the wholeexperiment duration (7500 h) showing both no interaction withthe fracture filling material due to sorption effects and no reduc-tion to sparingly soluble phases. In contrast, the tri- and tetrava-lent radionuclides 243Am(III), 232Th(IV) and 242Pu(IV) and arealmost totally associated with bentonite colloids. The three col-loid-associated RNs show slow desorption form colloids whichbecomes detectable after about 100 h contact time with the frac-ture filling material and proceeds for up to at least 7500 h. Thereason for the observed desorption of 232Th(IV), 242Pu(IV) and243Am(III) in the presence of FFM could be attributed to the high-er surface area of the FFM available for radionuclide sorptioncompared to the bentonite surface area under the given experi-mental conditions. Results for 243Am(III) slightly differ from242Pu(IV) and 232Th(IV) in terms slower desorption kinetics. Over-all, it has to be stated that the experimental duration of almost1 year was not sufficiently long to fully establish sorption equilib-rium unequivocally. Further studies at longer time scales are inprogress to shed light on this issue.
In the case of 237Np(V) a decrease in concentration could be ex-plained by both slow sorption to fracture filling material and, morelikely, by reduction to 237Np(IV). The latter assumption is corrobo-rated by means of geochemical modeling, which also demonstratesagreement with the experimental results that no reduced speciesof 233U(VI) and 99Tc(VII) occur under the prevailing geochemicalconditions of the experiments. Overall, the results show that theRN desorption kinetics from bentonite colloids are quite indepen-dent of (i) the grain size or respective surface area of the investi-gated material, at least in the size fraction range 0.25–2 mm and(ii) the concentration, in the case of 237Np(V), 242Pu(IV) and243Am(III).
The findings show the importance of the kinetics of RN–colloidbinding for colloid facilitated radionuclide migration. Theseresults need to be included in codes for simulating colloid-facilitated radionuclide transport to reduce the degree of uncer-tainty in input parameters leading to an improvement in thequality of the simulation results. RN dissociation from colloidshas to be considered when results from migration experimentsperformed in the laboratory and in the field are transferred to as-sess the long-term colloid impact on radionuclide mobility in anuclear waste repository.
Acknowledgments
We are indebted to Nora Groschopf (University of Mainz,Geoscience Department) for the XRF analysis of the fracture fillingmaterial. This work is partly funded by the Federal Ministry ofEconomics and Technology (BMWi) under the joint FZK-INE, GRSresearch project ‘‘KOLLORADO & KOLLORADO 2’’ and the SKB/KTH ‘‘Colloid project’’ as well as by the European Atomic EnergyCommunity Seventh Framework Program [FP7/2007–2013] underGrant Agreement No. 212287, Collaborative Project RECOSY. Wealso want to thank all organizations contributing to the fundingof the Colloid Formation and Migration (CFM) project Phase I,namely: ANDRA (France), BMWi (Germany), JAEA (Japan), CRIEPI(Japan), AIST (Japan) and NAGRA (Switzerland).
References
Alexander, W.R., Ota, K., Frieg, B., 2001. The NAGRA-JNC in situ study of safetyrelevant radionuclide retardation in fractured crystalline rock II: the RRP projectmethodology development, field and laboratory tests. NAGRA Technical ReportSeries NTB 00-06, Wettingen, Switzerland.
Altmaier, M., Neck, V., Fanghänel, Th., 2004. Solubility and colloid formation ofTh(IV) in concentrated NaCl and MgCl2 solution. Radiochim. Acta 92, 537–543.
Bellenger, J.-P., Staunton, S., 2008. Adsorption and desorption of 85Sr and 137Cs onreference minerals, with and without inorganic and organic surface coatings. J.Environ. Radioact. 99, 831–840.
Bernhard, G., Geipel, G., Reich, T., Brendler, V., Amayri, S., Nitsche, H., 2001.Uranyl(VI) carbonate complex formation: validation of the Ca2UO2 (CO3)(3)(aq.)species. Radiochim. Acta 89, 511–518.
Bouby, M., Geckeis, H., Lützenkirchen, J., Mihai, S., Schäfer, T., 2009. Interaction ofbentonite colloids with Eu and Th in presence of humic acid: a flow field–flowfractionation study. In: 4th Ann. Workshop Proc. Integrated ProjectFundamental Processes of Radionuclide Migration – 6th EC FP IP FUNMIG,FZKA Report 7461, pp. 271–279.
Bouby, M., Geckeis, H., Lützenkirchen, J., Mihai S., Schäfer, T., 2010. Interaction ofbentonite colloids with Eu, Th and U in presence of humic acid: a flow field–flow fractionation study. Geochim. Cosmochim. Acta, submitted.
Cama, J., Ganor, J., Ayora, C., Lasaga, A., 2000. Smectite dissolution kinetics at 80 �Cand pH 8 8. Geochim. Cosmochim. Acta 64, 2701–2717.
Comans, R.N.J., 1987. Adsorption, desorption and isotopic exchange of cadmium onillite: evidence for complete reversibility. Water Res. 21, 1573–1576.
Comans, R.N.J., Haller, M., Depreter, P., 1991. Sorption of cesium on illite –nonequilibrium behavior and reversibility. Geochim. Cosmochim. Acta 55, 433–440.
Coppin, F., Castet, S., Berger, G., Loubet, M., 2003. Microscopic reversibility of Smand Yb sorption onto smectite and kaolinite: experimental evidence. Geochim.Cosmochim. Acta 67, 2515–2527.
De Koning, A., Comans, R.N.J., 2004. Reversibility of radiocaesium sorption on illite.Geochim. Cosmochim. Acta 68, 2815–2823.
Denison, F.H., Garnier-Laplace, J., 2005. The effects of database parameteruncertainty on uranium(VI) equilibrium calculations. Geochim. Cosmochim.Acta 69, 2183–2191.
Flügge, J., Küntzel, M., Schäfer, T., Gaus, I., Noseck, U., 2010. Modeling colloid-boundradionuclide transport at the Grimsel Test Site. In: International GroundwaterSymp. IAHR. 2010. Valencia, Spain.
Galunin, E., Alba, M.D., Avilés, M.A., Santos, M.J., Vidal, M., 2009. Reversibility of Laand Lu sorption onto smectites: implications for the design of engineeredbarriers in deep geological repositories. J. Hazard. Mater. 172, 1198–1205.
Geckeis, H., Schäfer, T., Hauser, W., Rabung, T., Missana, T., Degueldre, C., Möri, A.,Eikenberg, J., Fierz, T., Alexander, W.R., 2004. Results of the Colloid andRadionuclide Retention experiment (CRR) at the Grimsel Test Site (GTS),Switzerland – impact of reaction kinetics and speciation on radionuclidemigration. Radiochim. Acta 92, 765–774.
Grenthe, I., Stumm, W., Laaksuharju, M., Nilsson, A.C., Wikberg, P., 1992. Redoxpotentials and redox reactions in deep ground water systems. Chem. Geol. 98,131–150.
Guillaumont, R., Fanghänel, T., Fuger, J., Grente, I., Neck, V., Palmer, D.A., Rand, M.H.,2003. Update on the Chemical Thermodynamics of Uranium, Neptunium,Plutonium, Americium and Technetium. Elsevier, Amsterdam.
Kersting, A.B., Efurd, D.W., Finnegan, D.L., Rokop, D.J., Smith, D.K., Thompson, J.L.,1999. Migration of plutonium in ground water at the Nevada Test Site. Nature397, 56–59.
Kim, J.I., Buckau, G., Baumgärtner, F., Moon, H., Lux, D., 1984. Colloid generation andthe actinide migration in Gorleben ground waters. Mater. Res. Soc. Symp. Proc.26, 31–40.

F. Huber et al. / Applied Geochemistry 26 (2011) 2226–2237 2237
Latrille, C., Ly, J., Herbette, M., 2006. Retention of Sn(IV) and Pu(IV) onto fourargillites from the Callovo–Oxfordian level at Bure (France) from eightequilibrated sedimentary waters. Radiochim. Acta 94, 421–427.
Lührmann, L., 1999. Modellierung des kolloidgetragenen Schadstofftransports unterBerücksichtigung von Sorptions- und Filtrationsprozessen. PhD Thesis. Univ.Erlangen-Nürnberg.
McCarthy, J.F., Zachara, J.M., 1989. Subsurface transport of contaminants: mobilecolloids in the subsurface environment may alter the transport of contaminants.Environ. Sci. Technol. 23, 497–502.
Missana, T., Geckeis, H. (Eds.), 2006. Grimsel Test Site – Investigation Phase V. TheCRR Final Project Report Series II: Supporting Laboratory Experiments withRadionuclides and Bentonite Colloids NAGRA Technical Report Series NTB 03-02, Wettingen, Switzerland.
Missana, T., Alonso, U., Garcia-Gutierrez, M., Mingarro, M., 2008. Role of bentonitecolloids on europium and plutonium migration in a granite fracture. Appl.Geochem. 23, 1484–1497.
Möri, A., Alexander, W.R., Geckeis, H., Hauser, W., Schäfer, T., Eikenberg, J., Fierz, T.,Degueldre, C., Missana, T., 2003. The colloid and radionuclide retardationexperiment at the Grimsel Test Site: influence of bentonite colloids onradionuclide migration in a fractured rock. Colloids Surf. A – Physicochem.Eng. Aspects 217, 33–47.
Neck, V., Altmaier, M., Fanghänel, Th., 2007. Solubility of plutonium hydroxides/hydrous oxides under reducing conditions and in the presence of oxygen. CRChim. 10, 959–977.
Neck, V., Altmaier, M., Fellhauer, D., Runke, J., Fanghänel, T., 2009. Quantification ofthe redox potential for the reduction of Np(V) in non-complexing aqueoussolutions at pH 5–10. In: 1st Ann. Workshop Proc. 7th EC FP – Recosy CP,Barcelona 10–12th
Neck, V., Fanghänel, Th., Kim, J.I., 1999. Thermodynamische Modellierung vonTechnetium in natürlichen aquatischen Systemen. FZKA 6340,Forschungszentrum Karlsruhe, Karlsruhe.
Noseck, U., Klotz, D., 2002. Modelling of colloid-facilitated contaminant transportwith the computer code TRAPIC: theoretical basis and application. RadionuclideRetention in Geologic Media. Workshop Proc. pp. 231–239.
Plaschke, M., Schäfer, T., Bundschuh, T., Manh, T.N., Knopp, R., Geckeis, H., Kim, J.I.,2001. Size characterization of bentonite colloids by different methods. Anal.Chem. 73, 4338–4347.
Puigdomenech, I., 2004. HYDRA and MEDUSA chemical equilibrium software.Software and documentation. <http://web.telia.com/~u15651596/>.
Rabung, T., Pierret, M.C., Bauer, A., Geckeis, H., Bradbury, M.H., Baeyens, B., 2005.Sorption of Eu(III)/Cm(III) on Ca–montmorillonite and Na–illite. Part 1: Batchsorption and time-resolved laser fluorescence spectroscopy experiments.Geochim. Cosmochim. Acta 69, 5393–5402.
Rand, M., Fuger, J., Grenthe, I., Neck, V., Rai, D., 2009. Chemical Thermodynamics ofThorium. OECD, Nuclear Energy Agency. OECD Publishing.
Rothe, J., Walther, C., Brendebach, B., Büchner, S., Fuss, M., Denecke, M.A., Geckeis,H., 2009. A combined XAFS, ESI TOF-MS and LIBD study on the formation ofpolynuclear Zr(IV), Th(IV) and Pu(IV) species. J. Phys.: Conf. Ser., 190 012188.
Schäfer, T., Noseck, U. (Eds), 2010. Colloid/ Nanoparticle formation and mobility inthe context of deep geological nuclear waste disposal. Project KOLLORADO-1;Final report. FZKA 7515, Forschungszentrum Karlsruhe, Karlsruhe.
Schäfer, T., Geckeis, H., Bouby, M., Fanghänel, T., 2004. U, Th, Eu and colloid mobilityin a granite fracture under near-natural flow conditions. Radiochim. Acta 92,731–737.
Stumm, W., Morgan, J., 1996. Aquatic Chemistry: Chemical Equilibria and Rates inNatural Waters, 3rd ed. John Wiley and Sons, Inc., New York.
Turner, N.B., Ryan, J.N., Saiers, J.E., 2006. Effect of desorption kinetics on colloid-facilitated transport of contaminants: cesium, strontium, and illite colloids.Water Resour. Res. 42, W12509. doi:10.1029/2006WR004972.
Villar, M.V., Martin, P.L., Pelayo, M., Ruiz, B., Rivas, P., Alonso, E., Lloret, A., Pintado,X., Gens, A., Linares, J., Huertas, F., Caballero, E., Jimenz de Cisneros, C., Obis, J.,Perez, A., Velasco, J., 1998. FEBEX bentonite: origin, properties and fabrication ofblocks (full-scale engineered barriers experiment in crystalline host rock). 05/98, Enresa (empresa nacional de residuos radiactivos, s.a.), Madrid, Spain.
Wold, S., 2010. Sorption of prioritized elements on montmorillonite colloids andtheir potential to transport radionuclides. SKB Technical Report TR-10-20Swedish Nuclear Fuel and Waste Management Co (SKB), Stockholm, Sweden.
Related Documents











