J. Chem. SOC., Faraday Trans. 1, 1982, 78, 1603-1613 Initial Sintering of Magnesium Oxide in Carbon Dioxide BY TOMOYASU ITO Department of Chemistry, Faculty of Science, Tokyo Metropolitan University, Setagaya, Tokyo, Japan 158 Received 24th July, 198 1 Studies have been made of the initial sintering (surface area diminution and crystallite growth) of MgO freely dispersed in CO,. The sintering rate at 1 123 K was directly proportional to the CO, pressure in the range 0.67-93.1 kPa, and the apparent activation energy for sintering was 153 k 11 kJ mol-'. Increases in the cation vacancy concentration by doping with Mn ions resulted in virtually no variation in the sintering rate. From sintering measurements in 180-enriched CO,, the bulk l80 distribution in crystallites grown by sintering was determined and it was shown that most of the anions migrate only on the surface during sintering. Relative sintering rate constants at 1123 K were (3.4 f 0.3) x lo6, 38 f 8, 1.3 k 0.1 and 1 in H,O, CO,, 0, and Ar, respectively. It is concluded, on the basis of these facts, that sintering is enhanced by increased surface migration of 02- ions (the slower moving ions), caused by repetition of the adsorption-desorption cycle of CO, molecules (anion-exchange mechanism). Imperfections on the surface of polycrystalline oxide powders are greatly affected by the calcination conditions, and only oxide powders prepared under strictly controlled conditions show reproducible surface properties. Previously we have reported'?, the remarkable positive effect of H,O vapour on the initial sintering of freely dispersed MgO powders. This enhanced sintering has been explained by the anion-exchange mechanism, i.e. by increased surface migration of 02- ions caused by repetition of the adsorption-desorption cycle of H,O molecules. A similar effect may be expected for other oxygen-containing molecules. Carbon dioxide is advantageous for such a study because of its high thermal stability and known adsorption states. The main adsorbed species at 773 K has been proved by i.r. spectroscopy to be a bidentate carbonate i ~ n . ~ - ~ Although there have been no reports on the isotopic exchange reaction of oxygen atoms (i.e. the anion-exchange reaction) between CO, and MgO at high temperatures, this reaction proceeds even at 373 K.6 Tomizawa et al.7 have found some accelerating effects of CO, molecules on the initial sintering of MgO powders. However, the atmosphere used by them cannot be considered to be pure CO, because the effects of H,O vapour, evolved during preparation, were ignored. In addition, they proposed no sintering mechanism. In the present paper the initial sintering in CO, is studied and the mechanism is discussed with reference to: (1) the C0,-pressure dependence of the sintering rate, (2) the effect of Mn doping and (3) the I8O distribution in a MgO crystallite. The sintering rates were also measured in H,O, 0, and Ar. EXPERIMENTAL The apparatus and procedure for sintering and adsorption studies have been described previously.2 Carbon dioxide (> 99.99%, Takachiho Chemicals) was predried through a trap kept at 195 K. An isotopic exchange reaction of '*O between 180-enriched CO, and MgO was carried out in the same silica vessel as that used for the sintering study. The concentration of 1603 Published on 01 January 1982. Downloaded by Universidade Federal do Rio Grande do Sul on 06/09/2014 07:01:05. View Article Online / Journal Homepage / Table of Contents for this issue

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

J . Chem. SOC., Faraday Trans. 1, 1982, 78, 1603-1613
Initial Sintering of Magnesium Oxide in Carbon Dioxide
B Y TOMOYASU I T O
Department of Chemistry, Faculty of Science, Tokyo Metropolitan University, Setagaya, Tokyo, Japan 158
Received 24th July, 198 1
Studies have been made of the initial sintering (surface area diminution and crystallite growth) of MgO freely dispersed in CO,. The sintering rate at 1 123 K was directly proportional to the CO, pressure in the range 0.67-93.1 kPa, and the apparent activation energy for sintering was 153 k 11 kJ mol-'. Increases in the cation vacancy concentration by doping with Mn ions resulted in virtually no variation in the sintering rate. From sintering measurements in 180-enriched CO,, the bulk l80 distribution in crystallites grown by sintering was determined and it was shown that most of the anions migrate only on the surface during sintering. Relative sintering rate constants at 1123 K were (3.4 f 0.3) x lo6, 38 f 8, 1.3 k 0.1 and 1 in H,O, CO,, 0, and Ar, respectively.
It is concluded, on the basis of these facts, that sintering is enhanced by increased surface migration of 02- ions (the slower moving ions), caused by repetition of the adsorption-desorption cycle of CO, molecules (anion-exchange mechanism).
Imperfections on the surface of polycrystalline oxide powders are greatly affected by the calcination conditions, and only oxide powders prepared under strictly controlled conditions show reproducible surface properties. Previously we have reported'?, the remarkable positive effect of H,O vapour on the initial sintering of freely dispersed MgO powders. This enhanced sintering has been explained by the anion-exchange mechanism, i.e. by increased surface migration of 02- ions caused by repetition of the adsorption-desorption cycle of H,O molecules. A similar effect may be expected for other oxygen-containing molecules. Carbon dioxide is advantageous for such a study because of its high thermal stability and known adsorption states. The main adsorbed species at 773 K has been proved by i.r. spectroscopy to be a bidentate carbonate i ~ n . ~ - ~ Although there have been no reports on the isotopic exchange reaction of oxygen atoms (i.e. the anion-exchange reaction) between CO, and MgO at high temperatures, this reaction proceeds even at 373 K.6
Tomizawa et al.7 have found some accelerating effects of CO, molecules on the initial sintering of MgO powders. However, the atmosphere used by them cannot be considered to be pure CO, because the effects of H,O vapour, evolved during preparation, were ignored. In addition, they proposed no sintering mechanism.
In the present paper the initial sintering in CO, is studied and the mechanism is discussed with reference to: (1) the C0,-pressure dependence of the sintering rate, (2) the effect of Mn doping and (3) the I8O distribution in a MgO crystallite. The sintering rates were also measured in H,O, 0, and Ar.
E X P E R I M E N T A L The apparatus and procedure for sintering and adsorption studies have been described
previously.2 Carbon dioxide (> 99.99%, Takachiho Chemicals) was predried through a trap kept at 195 K. An isotopic exchange reaction of '*O between 180-enriched CO, and MgO was carried out in the same silica vessel as that used for the sintering study. The concentration of
1603
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online / Journal Homepage / Table of Contents for this issue

1604
l80 in CO, was measured by a mass spectrometer at prescribed time intervals. The initial '*O concentration in CO, was 4.48 atom %.
Magnesium oxide specimens were obtained by the thermal decomposition of two varieties of magnesium oxalate dihydrate (MO-6 and M0-9)., Unless otherwise stated, MO-6 was used. Magnesium oxide specimens prepared from MO-9 were usually preoxidized in 13 kPa 0, at 1123 K for 1 h before the sintering run in order to remove any organic contaminants. No essential change in sintering behaviour was produced by the oxygen pretreatment. Magnesium oxalate dihydrates doped with various concentrations of manganese ions were prepared by reaction of magnesium sulphate containing manganese ions with ammonium oxalate. Manganese concentrations in the oxalates, as determined by an atomic absorption spectrometer, were 8, 205, 760 and 1400 atom ppm.
I N I T I A L S I N T E R I N G O F MgO I N CO,
R E S U L T S
I N I T I A L S I N T E R I N G I N P U R E MgO The specific surface area, S, of MgO was 268 m2 g-l before the admission of CO,.
When MgO was sintered in 0.67-93.1 kPa of CO, at 1123 K, Sdecreased with sintering time, t , as shown in fig. 1. The initial sintering was evidently enhanced by the presence of CO,, but much less than with H,O vapour., The degree of secondary agglomeration between crystallites, S/SX,, where S, is an ideal specific surface area calculated from a crystallite size, was 0.85 k 0.1 throughout the sintering process. Little change in S / S , during the sintering indicates that the surface area reduction was a direct consequence of the crystallite growth. In such a case a kinetic equation
S m - S r = k, t (1)
is apparently applicable,, where So is S at t = 0, k, is a rate constant for surface area diminution and rn is a constant determined experimentally. Although there is some scatter, the curves in fig. 1 were best fitted to eqn (1) for rn = - 6.0 f 1 .O. The values of k , are shown in fig. 2 as a function of the CO, pressure, pco,; net values obtained by subtracting k, under evacuation (< Pa) from those in the presence of CO, are used. A resultant straight line with a slope of 1.06f0.07 means that k, is directly proportional to pco,.
The distribution of micropores in MgO specimens sintered in CO, was obtained by measuring an adsorption isotherm of 0, at 77 K. All the samples examined had no micropores below 1.2 nm diameter and showed the maximum distribution at 2.0-2.5 nm. This suggests that the sintering proceeded in all micropores, and not simply those of a particular diameter.
An Arrhenius plot of k , in the range of sintering temperature, T, of 973-1 123 K is shown in fig. 3. The slope gave an apparent activation energy of 153 k 1 1 kJ mol-l for the sintering rate, which seems rather larger than the value of 131 k 18 kJ mol-1 in H,O vapour.,
I N I T I A L S I N T E R I N G O F Mn-DOPED MgO In order to see whether the migration of cation vacancies, VM, is related to the
rate-determining step of the sintering process, a sintering study was also carried out on Mn-doped MgO. These specimens were preoxidized in 13 kPa 0, at 1123 K for 1 h before sintering in order to bring most manganese ions to the highest valence state. Values of k, in 20 kPa CO, at 1123 K are shown in fig. 4 as a function of Mn concentration, [Mn], and a least-squares fit gives a slope with 0.06f0.06. In spite of the large variation in [Mn] of 8-1400 atom ppm, k , is substantially constant.
E.s.r. spectra of the specimens after sintering showed weak Mn2+ signals. However,
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
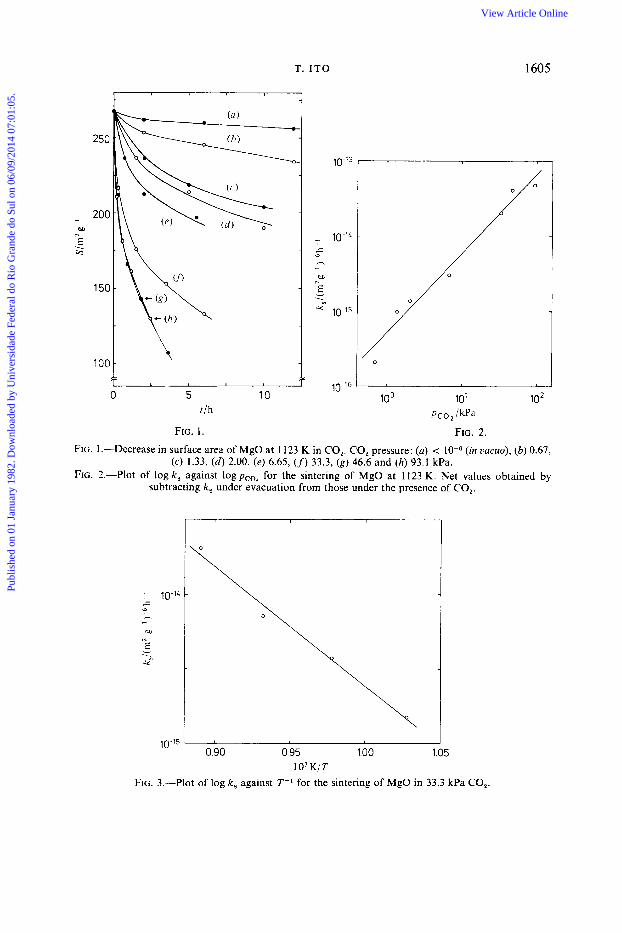
T. I T 0 1605
t
L I
0 5 10 t l h
FIG. 1.
0
10 O 10' lo2 Pco, I kPa
FIG. 2. FIG. 1 .-Decrease in surface area of MgO at 1 123 K in CO,. CO, pressure: (a) < (in vacuo), (b) 0.67,
(c) 1.33, ( d ) 2.00, (e) 6.65, (f) 33.3, ( g ) 46.6 and (h) 93.1 kPa. FIG. 2.-Plot of log k, against log pco, for the sintering of MgO at 1123 K. Net values obtained by
subtracting k , under evacuation from those under the presence of CO,.
-i 1O-'L -c h - I 0.0
N
E v . -ti
10-15 0.90 0.95 1.00 1.05
1 O 3 KIT
FIG. 3.-Plot of log k, against T-' for the sintering of MgO in 33.3 kPa CO,.
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
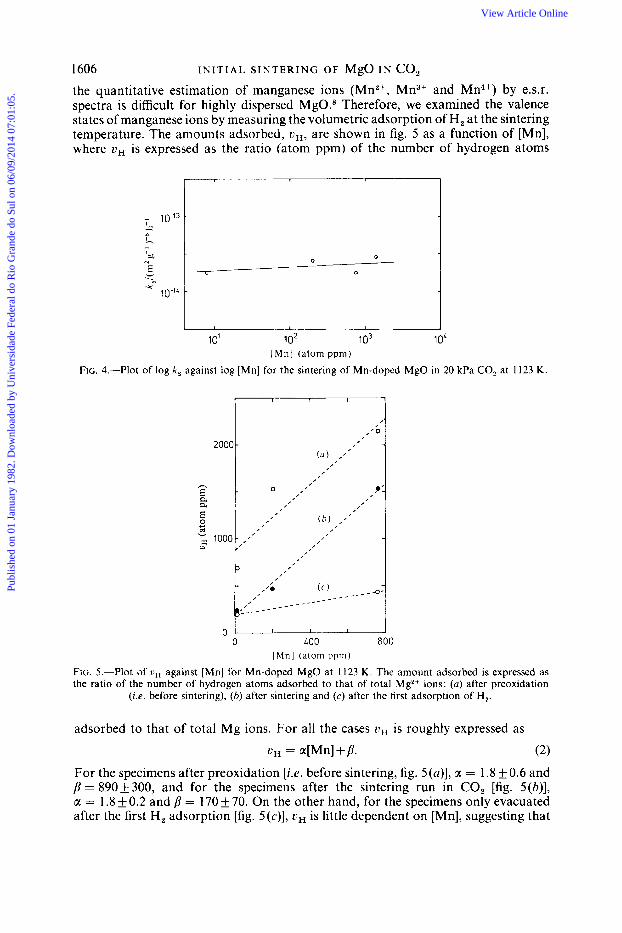
1606 I N I T I A L S I N T E R I N G OF MgO I N CO, the quantitative estimation of manganese ions (Mn2+, Mn3+ and Mn4+) by e.s.r. spectra is difficult for highly dispersed MgO.s Therefore, we examined the valence states of manganese ions by measuring the volumetric adsorption of H, at the sintering temperature. The amounts adsorbed, uH, are shown in fig. 5 as a function of [Mn], where uH is expressed as the ratio (atom ppm) of the number of hydrogen atoms
FIG. 4.-Plot of log k ,
0 0
u 0
I
10’ lo2 lo3 1 oL [ M n l (atom ppm)
against log [Mn] for the sintering of Mn-doped MgO in 20 kPa CO, at 1123 K.
I I I I 0 LOO 800
[ M n ] (atom ppm)
FIG. 5.-Plot of uH against [Mn] for Mn-doped MgO at 1123 K. The amount adsorbed is expressed as the ratio of the number of hydrogen atoms adsorbed to that of total Mg*+ ions: (a) after preoxidation
(Le. before sintering), (b) after sintering and (c) after the first adsorption of H,.
adsorbed to that of total Mg ions. For all the cases uH is roughly expressed as
uH = a[Mn]+p. (2)
For the specimens after preoxidation [i.e. before sintering, fig. 5(a)], a = 1.8 f 0.6 and p = 890k 300, and for the specimens after the sintering run in CO, [fig. 5(b) ] , a = 1.8 k 0.2 and p = 170 f 70. On the other hand, for the specimens only evacuated after the first H, adsorption [fig. 5(c)], uH is little dependent on [Mn], suggesting that
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
T. I T 0 1607 most of the manganese ions were in the bivalent state. Therefore, the slopes close to 2 in fig. 5 (a ) and (b) indicate that most of Mn ions were Mn4+ and that YM originating from Mn4+ ions was present during the sintering in a concentration nearly equal to [Mn]. The difference between fig. 5(a) and (b) may be due to some extrinsic adsorption other than manganese ions, and fig. 5(c) is due to the intrinsic H, adsorption.2
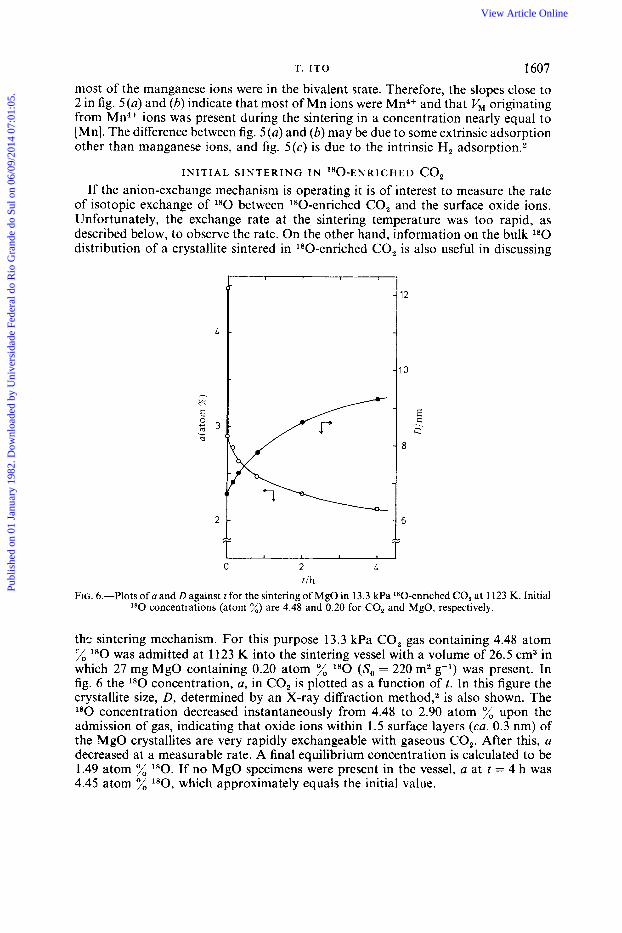
I N I T I A L S I N T E R I N G I N 1 8 0 - ~ ~ ~ ~ ~ ~ ~ ~ CO, If the anion-exchange mechanism is operating it is of interest to measure the rate
of isotopic exchange of l80 between 180-enriched CO, and the surface oxide ions. Unfortunately, the exchange rate at the sintering temperature was too rapid, as described below, to observe the rate. On the other hand, information on the bulk l80 distribution of a crystallite sintered in 180-enriched CO, is also useful in discussing
E
Q 1
- 8
1 I I I 1
0 2 4 1/11
FIG. 6.-Plots of a and D against t for the sintering of MgO in 13.3 kPa 180-enriched CO, at 1123 K. Initial '*O concentrations (atom %) are 4.48 and 0.20 for CO, and MgO, respectively.
ths sintering mechanism. For this purpose 13.3 kPa CO, gas containing 4.48 atom T/o l80 was admitted at 1123 K into the sintering vessel with a volume of 26.5 cm3 in which 27 mg MgO containing 0.20 atom % l80 (So = 220 m2 g-l) was present. In fig. 6 the l80 concentration, a, in CO, is plotted as a function o f t . In this figure the crystallite size, D, determined by an X-ray diffraction method,, is also shown. The l80 concentration decreased instantaneously from 4.48 to 2.90 atom % upon the admission of gas, indicating that oxide ions within 1.5 surface layers (ca. 0.3 nm) of the MgO crystallites are very rapidly exchangeable with gaseous CO,. After this, a decreased at a measurable rate. A final equilibrium concentration is calculated to be 1.49 atom % 1 8 0 . If no MgO specimens were present in the vessel, a at t = 4 h was 4.45 atom % l80, which approximately equals the initial value.
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
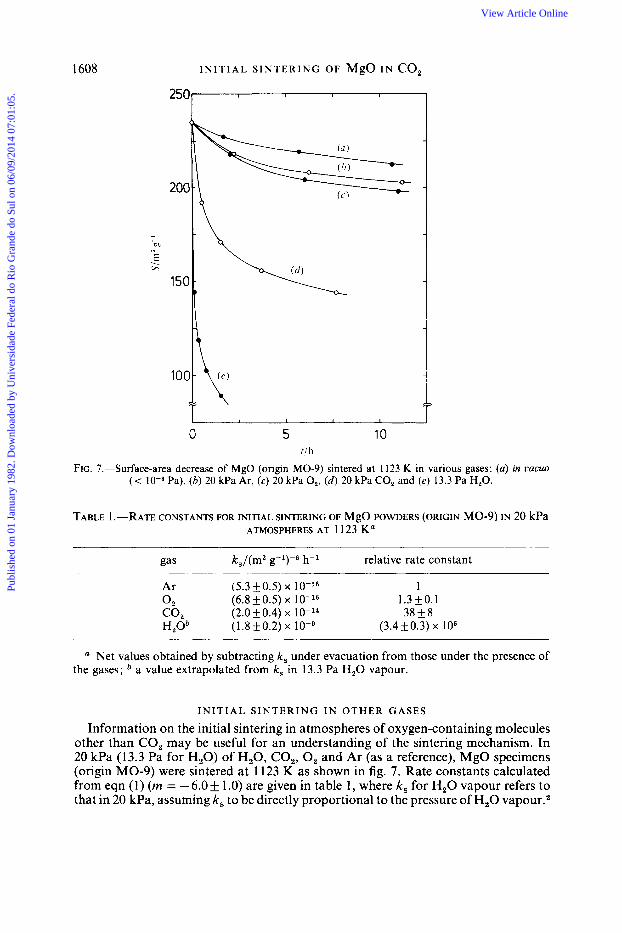
1608 INITIAL S I N T E R I N G OF MgO IN co,
250-
0 5 10 f i h
FIG. 7.-Surface-area decrease of MgO (origin MO-9) sintered at 1123 K in various gases: (a) in uucuo (< Pa), (b) 20 kPa Ar, (c) 20 kPa 0,, ( d ) 20 kPa CO, and (e) 13.3 Pa H,O.
TABLE 1 .-RATE CONSTANTS FOR INITIAL SINTERING OF MgO POWDERS (ORIGIN MO-9) IN 20 kPa ATMOSPHERES AT 1123 K a
gas k,/(m2 g-1)-6 h-l relative rate constant
Ar (5.3 k0.5) x 1 0 2 (6.8 k0.5) x 1.3k0.1 co2 (2.0 k 0.4) x 38f8 H20b (1.8k0.2) x (3.4k0.3) x lo6
a Net values obtained by subtracting k, under evacuation from those under the presence of the gases; a value extrapolated from k, in 13.3 Pa H 2 0 vapour.
INITIAL S I N T E R I N G I N OTHER GASES
Information on the initial sintering in atmospheres of oxygen-containing molecules other than CO, may be useful for an understanding of the sintering mechanism. In 20 kPa (13.3 Pa for H,O) of H,O, CO,, 0, and Ar (as a reference), MgO specimens (origin MO-9) were sintered at 1123 K as shown in fig. 7. Rate constants calculated from eqn (1) (rn = - 6.0 f 1 .O) are given in table 1 , where k, for H,O vapour refers to that in 20 kPa, assuming k, to be directly proportional to the pressure of H,O vapour.2
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online

T. I T 0 1609 All the rate constants in table 1 are net values obtained by subtracting k, under evacuation from values in the presence of the gases. Relative efficiencies for sintering are in the following order: H,O % CO, > 0, z Ar.
DISCUSSION
MECHANISM OF INITIAL SINTERING
The enhanced sintering is closely related to the adsorption of CO,. We have no reported data on the adsorption at the temperature used in the sintering measurements, but at 773 K volumetric and i.r. spectroscopic investigations have been reported by Evans et al.,3 Gregg et al.4 and Fukuda et al.5 Species adsorbed on MgO at 773 K have been proved by these authors to consist of bidentate carbonate ions
/o\ / \
Mg\ 0 P = O as a major component and simple carbonate ions
LO 0-cc’
\O as a minor component. Gregg et al.4 also found that at 773 K in 2 kPa CO, an adsorption equilibrium was reached within a few minutes with a surface coverage of ca. 0.06. These results suggest that, under the present sintering conditions, simple and bidentate carbonate species were also formed only in small amounts in a dynamic adsorption equilibrium with gaseous molecules.
Possible sintering mechanisms are : (1) a diffusion mechanism (surface, grain- boundary or bulk) ; (2) a viscous flow mechanism ; (3) an evaporation-condensation mechanism ; (4) an adsorption-induced space-charge mechanism and (5) an adsorption- desorption cycle mechanism (anion-exchange mechanism). Of these five, mechanisms (3) and (4) can be excluded., Mechanisms (1) and (2) both depend on the equilibrium adsorption amount, v , of CO, [in mechanism (1) the rate is proportional to v and in (2) to u2], and mechanism (5) depends on the frequency of the adsorption-desorption cycle of CO, molecules under a dynamic adsorption equilibrium.
We first look at mechanisms (1) and (2). The adsorption equilibrium of CO, on the surface sites, S’, is expressed as
K , S’+CO,eA (3)
where Kl is the equilibrium constant and A is the adsorbed species (non-dissociated).
(4) From eqn (3), we obtain
v = [A1 = K,[S’lP,o2. On the other hand, v can also be expressed as
where 8 is the surface coverage and where subscript 0 refers to a state before d In k, adsorption. Then we define A by
From eqn (4)-(6) we can obtain
A = d In Pco,’
A = 1-8
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online

1610 I N I T I A L S I N T E R I N G O F MgO I N CO,
for mechanism (l), since k, cc v. This equation implies that k, cc pe&?). Similarly, for mechanism (2) 3, = 2-26 i.e. k , c c p p ~ : ~ ) .
rate-determining, a reaction scheme Next we consider mechanism (5) . If, for example, an adsorption process is
K . \
S / + C 0 2 2 1 [ 1 k,
I + A (9)
is applied according to the absolute reaction rate theory, where I is an activated complex (non-dissociated) and K, and k , are the equilibrium constant and the rate constant, respectively. Then the frequency, R, of the adsorption-desorption cycle is
and we obtain 3,= 1-0 (1 1) for mechanism (9, since k , cc R. Eqn ( 1 1) also holds for the case of a desorption process being rate-determining.
The experimental values were 8 -g l 4 and 2 = 1.06 0.07 (fig. 2). Therefore, eqn (8) is not applicable to the present case, but either eqn (7) [mechanism (l)] or eqn (1 1) [mechanism (5 ) ] is valid.
For ionic oxides, sintering necessitates the migration of both cations and anions, and the sintering rate is determined by the migration rate of the slower of the two. The migration of Mg2+ ions on the surface and in the MgO bulk is not expected to be enhanced by the presence of CO, because non-dissociated adsorbed species (simple and bidentate carbonate ions) lead to no changes in the cation vacancy concentration or in the space charge distribution. This is supported by the sintering on Mn-doped MgO shown in fig. 4 and 5 , where k, is virtually constant against the wide range variation in [Mn], i.e. in VM. Thus cation migration (surface, grain-boundary or bulk) is not considered to be rate-determining. The enhanced sintering in CO, can therefore be attributed to the enhanced migration of the anions (the slower moving ions).
Mechanism (1) is unlikely to enhance anion migration since the carbonate ions formed are much larger and much heavier than the original 0,-. Therefore the remaining possible mechanism is (9, the adsorption-desorption cycle. In fact, the isotopic-exchange reaction of lSO between gaseous CO, and surface 0,- by this mechanism proceeds very rapidly (cf. fig. 6, the decrease in a from 4.48 to 2.90 atom % l80). This reaction scheme is represented, for example for a bidentate species, by
adsorption /’\ desorption C=O - Mg2+ +02-+ COO, (12)
Mg\o/ Mg2+ + 0;- + CO, -
where the subscript c refers to the 0 atom originally belonging to the oxide. The anion formed by the desorption of the COO, molecule will be in the site next to the original 0;-. This results in migration of the anion on the surface.
Guilliatt et a1.8 found that their Mn-doped MgO was sintered much faster in 0, than in H, or in vacuo, and they concluded that cation diffusion by the vacancy mechanism was rate-determining. In the present study, however, surface migration of anions has been proved to be rate-determining. This apparent difference is believed to result mainly from the difference in sintering temperature. Sintering in flowing 0,
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
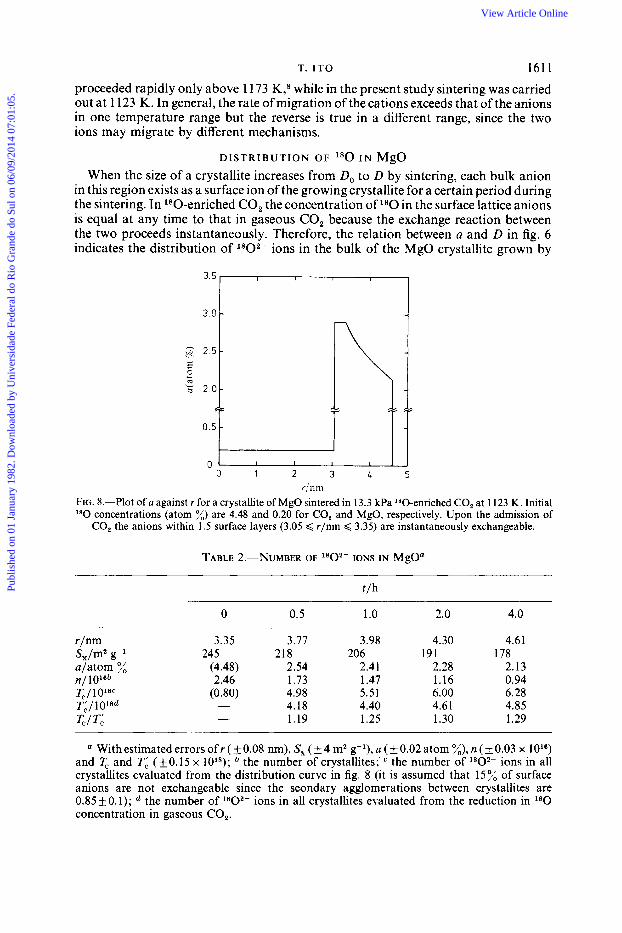
T. I T 0 161 1 proceeded rapidly only above 1 173 K,8 while in the present study sintering was carried out at 1 123 K. In general, the rate of migration of the cations exceeds that of the anions in one temperature range but the reverse is true in a different range, since the two ions may migrate by different mechanisms.
D I S T R I B U T I O N O F l80 I N MgO When the size of a crystallite increases from Do to D by sintering, each bulk anion
in this region exists as a surface ion of the growing crystallite for a certain period during the sintering. In 180-enriched CO, the concentration of l80 in the surface lattice anions is equal at any time to that in gaseous CO, because the exchange reaction between the two proceeds instantaneously. Therefore, the relation between a and D in fig. 6 indicates the distribution of 1802- ions in the bulk of the MgO crystallite grown by
3.0 1
0 1 2 3 4 5 r /nm
FIG. 8.-Plot of a against r for a crystallite of MgO sintered in 13.3 kPa I80-enriched CO, at 1123 K. Initial l80 concentrations (atom x) are 4.48 and 0.20 for CO, and MgO, respectively. Upon the admission of
CO, the anions within 1.5 surface layers (3.05 < r/nm < 3.35) are instantaneously exchangeable.
TABLE 2.-NUMBER OF 1802- IONS IN MgOa
0 0.5 1 .o 2.0 4.0
r/nm 3.35 3.77 3.98 4.30 4.61 Sx/m2 g-l 245 21 8 206 191 178 alatom % (4.48) 2.54 2.41 2.28 2.13 n/ 1 016b 2.46 1.73 1.47 1.16 0.94 q// 1 0lBC (0.80) 4.98 5.5 1 6.00 6.28 T, 1 1 0 led - 4.18 4.40 4.61 4.85 GlTd - 1.19 1.25 1.30 1.29
a With estimated errors of r ( f 0.08 nm), S, ( & 4 m2 g-l), a ( f 0.02 atom %), n (k 0.03 x 10l6) and T, and Td (f0.15 x lo1*); the number of crystallites: the number of 1802- ions in all crystallites evaluated from the distribution curve in fig. 8 (it is assumed that 15% of surface anions are not exchangeable since the seondary agglomerations between crystallites are 0.85 k 0.1); the number of 1 8 0 2 - ions in all crystallites evaluated from the reduction in l80 concentration in gaseous CO,.
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online

1612 I N I T I A L S I N T E R I N G O F MgO I N CO,
sintering if the bulk diffusion of anions is negligible. Here we assume cubic crystallites with a uniform size’ and define Y (= D / 2 ) as the distance from the centre of the cube to the surface plane. A plot of a against Y in a crystallite is shown in fig. 8, which assumes that anions within 1.5 surface layers are very easily exchangeable upon admission of CO,. If the l 8 0 distribution curve in fig. 8 reflects the real situation, the number of 1 8 0 2 - ions in all crystallites (equal to the number of 1 8 0 2 - ions in a crystallite mutiplied by the number of crystallites) evaluated from the distribution curve must be nearly equal at any time to that calculated from the reduction in l80 concentration in CO,. Calculated results are shown in table 2, which reveals that the ratios, ZJTL, of the values obtained by the two methods are 1.2-1.3 irrespective of t . This constancy and insignificant deviation from 1 suggest the approximate validity of the distribution curve in fig. 8. (If anions diffused freely in bulk, T,/ Ti would become 2.4 and 1.7 at t = 0.5 and 4.0 h, respectively, showing a non-constancy and larger deviations from 1 .) These facts indicate that the assumption of insignificant diffusivity of the bulk anions is valid and that most anions migrate only in a surface layer, which provides support for the anion-exchange mechanism in the initial sintering.
SINTERING R A T E IN VARIOUS GASES
As shown in table I the relative sintering rate constants are (3.4 0.3) x lo6, 38 f. 8 and 1.3 0. I for H,O, CO, and 0,, respectively. Since the sintering in H,O vapour also proceeds by an anion-exchange mechanism,, the isotopic exchange reaction rate of the surface oxide ions in H,O vapour should be ca. lo5 times that in CO,. Unfortunately the exchange rates were too rapid to determine the rate constants.2 In addition there are no reported studies of this subject except for the case of oxygen molecules around 750 K 9 7 lo The theoretical calculation of the exchange rate constant is fairly difficult because the detailed mechanism is obscure. However, the observed pressure dependence, A, of the sintering rate constant is 1 in both H , 0 2 and CO,. This suggests that a molecular adsorption process is rate-determining for the sintering reactions (as for the exchange reactions). In such a case, the exchange rate can be approximately predicted by the theory of absolute rates.ll For CO, an adsorption process is expressed by eqn (9) and the adsorption rate under a dynamic adsorption equilibrium is represented by
R=-- kT ‘I exp ( - E / k T ) [CO,] [S’] h qco* 4 s
where k is Boltzmann’s constant, h is Plank’s constant, E is the activation energy of adsorption at absolute zero and q is the molecular partition function per unit volume or unit area. For the simplest case qI and qsl may be equated to unity, and [S’] is assumed to be [S’], since 9 6 1. Using as E the apparent activation energy observed for sintering, R can be very roughly estimated. A similar calculation for H,O leads to the result that the adsorption rate for H,O under sintering conditions is ca. lo3 times that for CO,. Although semi-quantitative, this is consistent with the observed sintering rates.
In addition, the known properties of these gases indicate that the extents of chemical interactions with MgO surfaces at high temperatures are in the order H,O > CO, > 0,. For example, reported adsorption amounts in terms of apparent surface coverage are as follows: 0.1 512 in 0.8 kPa H,O vapour at 923 K and @.313 in 0.6 kPa H,O vapour at 773 K, 0.064 in 2.0 kPa CO, at 773 K and ca. 0.000214 in 0.008 kPa 0, at 773 K. The same order may be applicable to the isotopic exchange rate. These considerations may give support to the theory that sintering in these atmospheres proceeds by an anion-exc hange mechanism.
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online

T. I T 0 161 3
I thank Prof. Taneki Tokuda and his collaborators at the Tokyo Metropolitan University for helpful discussions and suggestions during the course of this work.
T. Ito and T. Tokuda, Nippon Kagaku Kaishi, 1974, 248. T. Ito, M. Fujita, M. Watanabe and T. Tokuda, Bull. Chem. SOC. Jpn, 1981, 54, 2412. J. V. Evans and T. L. Whateley, Trans. Faraday Soc., 1967, 63, 2769. S. J. Gregg and J. D. Ramsay, J . Chem. SOC. A, 1970, 2784. Y. Fukuda and K. Tanabe, Bull. Chem. SOC. Jpn, 1973, 46, 1616. 0. V. Krylov, Z. A. Markova, I. I. Tretyakov and E. A. Fokina, Kinet. Katal., 1965, 6, 128.
I . F. Guilliatt, and N. H. Brett, J. Chem. SOC., Faraday Trans. I , 1972, 68, 429. E. R. S. Winter, J. Chem. SOC. A, 1968, 2889.
A. Clark, The Theory of Adsorption and Catalysis (Academic Press, New York, 1970), p. 209.
London, 1957), vol. 2, p. 309.
' T. Tomizawan, H. Hashimoto and K. Moteki, Kogyo Kagaku Zasshi, 1966, 69, 2263.
lo J. Novakova, Catal. Rev., 1970, 4, 77.
l 2 A. G. Oblad, S. W. Weller and G. A. Mills, Proc. 2nd Znt. Congr. Surface Actiuity (Academic Press,
l3 T. Ito, K. Kanehon and T. Tokuda, Z . Phys. Chem. (N.F.), 1976, 103, 203. l4 R. J. Breakspere and L. A. R. Hassan, Aust. J. Chem., 1977, 30, 971.
(PAPER 1 / 1 178)
Publ
ishe
d on
01
Janu
ary
1982
. Dow
nloa
ded
by U
nive
rsid
ade
Fede
ral d
o R
io G
rand
e do
Sul
on
06/0
9/20
14 0
7:01
:05.
View Article Online
Related Documents











